investigating the behavioural and antiapoptotic effects of
TRANSCRIPT

Investigating the behavioural and antiapoptotic effects of Deep
Brain Stimulation in a model of moderate-to-severe Traumatic
Brain Injury
by
Faiza Azreen Mahmud
A thesis submitted in conformity with the requirements
for the degree of Master of Applied Science
Institute of Biomedical Engineering
University of Toronto
© Copyright by Faiza Azreen Mahmud (2021)

ii
Investigating the behavioural and antiapoptotic effects of Deep
Brain Stimulation in moderate-to-severe Traumatic Brain Injury
Abstract
Traumatic brain injury (TBI) is a neurological event where an external mechanical force
to the brain can lead to devastating effects on physical, cognitive, and/or behavioural function in
humans. Currently, no therapeutic approach is substantially efficient in treating prolonged
secondary injury from TBI. Deep brain stimulation (DBS) is a neurosurgical procedure used to
manage debilitating symptoms in motor disorders. While DBS is demonstrated to modulate
targeted brain circuitry, its underlying mode of action is yet to be determined. This thesis
hypothesizes that high-frequency acute DBS delivered to the anterior thalamic nucleus (ANT)
will lead to recovery of memory deficits and reduce anxiety-type behaviour in a well-established
TBI rodent model, through a decrease in apoptosis. Salient findings suggest: (i) Acute
stimulation of the ANT improved spatial memory in rats with moderate-to-severe TBI, compared
to rats receiving no DBS; however, (ii) anti-apoptotic effects after ANT-DBS, as assessed via
caspase-3 measurements, were inconclusive.

iii
Acknowledgments
I would first like to express gratitude and my appreciation to Dr. Clement Hamani for his
unwavering support for the past 2 years. I am not sure what I would be doing right now without
your mentorship throughout my journey as an aspiring researcher. Thank you for granting me the
opportunity to join your lab as a technician at Sunnybrook shortly after I had completed my
bachelor’s degree. I am forever grateful for your kindness, and for helping me retain my research
aspirations in Neuroscience to this day. The past year has been difficult in terms of staying
motivated to continue my research, but I still want to pursue a path in this field because of your
guidance and ambition for all our projects. I would also like to thank Dr. Milos Popovic for giving
me this opportunity to pursue a career in Biomedical Engineering, and for allowing me to extend
my reach into (somewhat) unfamiliar territory, without hesitation. I am grateful to my committee
members, Dr. Jose Zariffa, and Dr. Suneil Kalia for their expertise and constructive criticism that
have helped me refine my thought process for this thesis. It is unfortunate that we were only able
to interact twice, and only during committee meetings. I hope that we will have more chances to
meet in the future.
I would also like to extend my deepest gratitude to Mustansir Diwan, for helping me throughout
the past 2 years and refining my technical skills in the lab with an extraordinary degree of patience.
Thank you for providing insightful advice, indulging in tangents we would go into during one of
our discussions, and the humour and jokes that followed. You have made my time in the lab a
wonderful learning experience and I have immense gratitude for all your support, even on days
when I had felt incredibly disheartened.
Thank you to Ying Meng and our amazing post-docs, Darryl Gidyk and Flavia Gouveia for all
their invaluable experimental help, especially under these current circumstances. Thank you for
teaching me with utmost patience, especially when learning stereotaxic surgery. I am sad that we
have not been able to interact with one another for half of my thesis, due to the pandemic and
current social distancing measures. I have learned a great deal from all of you about behavioural
neuroscience, and I hope to grow to be as passionate and dedicated a scientist as you all are. Thanks
to Esther Silk for being such an awesome lab-mate. It was always a pleasure hanging out and
working together when we were in each other’s presence.
Lastly, I am forever and always grateful to my beloved husband, Aadiyat. We have both come a
long way with our ambitions since high school and you are my greatest strength. I consider myself
to be very lucky to have you as my support system. You have constantly nurtured my interests and
brought me back up when my spirits were low. In both the best and worst of times, you were all
that I have had. Thank you for holding my hand throughout undergrad and our marriage. I thrive
to be a better person because we’re in this together.

iv
Table of Contents
Contents
Chapter 1 – Introduction ..................................................................................................................1
1.1 Traumatic Brain Injury (TBI) ..............................................................................................1
1.1.1 Definition and Classifying TBI ................................................................................1
1.1.2 Epidemiology ...........................................................................................................2
1.1.3 Neuropathophysiology following Acute TBI ..........................................................4
1.2 Models of Traumatic Brain Injury .......................................................................................6
1.2.1 Controlled Cortical Impact (CCI) ............................................................................8
1.2.2 Weight-drop models.................................................................................................9
1.2.3 Fluid Percussion Injury models (FPI) ......................................................................9
1.2.4 Blast and Penetrative injury models ......................................................................11
1.3 Deep Brain Stimulation......................................................................................................14
1.3.1 Overview ................................................................................................................14
1.3.2 Proposed Mechanisms of DBS ..............................................................................15
1.3.3 Deep Brain Stimulation in Traumatic Brain Injury ...............................................18
1.4 Rationale, objectives, and hypothesis ................................................................................21
Chapter 2 – General methods & materials .....................................................................................23
2.1 Animals and Surgical procedures .......................................................................................23
2.1.1 Animals .....................................................................................................................23
2.1.2. DBS electrode implantation ...................................................................................24
2.1.3 Fluid percussion injury (FPI) and acute Deep Brain Stimulation ..........................25
2.2. Behavioural assessments ....................................................................................................27
2.2.1. Novel location and novel object recognition Habituation: Open field testing .......28
2.2.2. Marble Burying ......................................................................................................28

v
2.2.3. NLR/NOR: Familiarization, novel location and novel object recognition
testing .....................................................................................................................30
2.2.4. Barnes maze ...........................................................................................................31
2.2.5 Cresyl violet staining .............................................................................................33
2.3. Neurochemical experiments...............................................................................................34
2.3.1. Caspase-3 Enzyme-linked immunosorbent assay (ELISA) ...................................35
2.4. Statistical Analyses ............................................................................................................36
Chapter 3 – Results ........................................................................................................................37
3.1 Study 1: To characterize behavioural deficits in the moderate-to-severe TBI fluid
percussion injury (FPI) model in rats.................................................................................37
3.2 Study 2: To investigate the effects of acute ANT-DBS in the FPI model through a
battery of behavioural tests associated with learning, memory and anxiety-like
behaviour............................................................................................................................44
3.3 Study 3: To test whether ANT-DBS reduces hippocampal apoptosis in FPI-exposed
rats. .....................................................................................................................................51
Chapter 4 – Discussion ..................................................................................................................53
4.1. ANT-DBS ..........................................................................................................................55
4.2. Caspase-3 ...........................................................................................................................58
4.3. Limitations and future perspectives ...................................................................................60
4.4. Conclusions ........................................................................................................................64

vi
List of Abbreviations
AD Alzheimer's Disease
ADn Anterodorsal nucleus
ALS Amyotropic Lateral Sclerosis
AM Anteromedial nucleus
ANOVA Analysis of Variance
ANT Anterior Nucleus of the Thalamus
APOE Apolipoprotein E gene
APP Amyloid Precursor Protein
AUP Animal Use Protocol
AV Anteroventral nucleus
BBB Blood-Brain Barrier
BDNF Brain-Derived Neurotrophic Factor
BUNS Barnes maze Unbiased Strategy
Ca2+ Calcium ion
CCI Controlled Cortical Impact
CHIMERA Closed-Head Impact Model of Engineered Rotational Acceleration
CT Computerized Tomography
DAI Diffuse Axonal Injury
DBS Deep Brain Stimulation
DGC Granular cells of the Dentate Gyrus
DN Cerebellar Dentate Nucleus
DNA Deoxyribonucleic Acid
DTI Diffusion Tensor Imaging
DWI Diffusion Weighted Imaging
EC Entorhinal Cortex
ELISA Enzyme-linked Immunosorbent Assay
FPI Fluid Percussion Injury
GABA Gamma-aminobutyric Acid
GCS Glasgow Coma Scale

vii
Gpi Globus Pallidus internus
HFS High Frequency Stimulation
K+ Potassium ion
LCN Lateral Cerebellar Nucleus
MB Marble Burying
MCS Minimally Conscious State
MRI Magnetic Resonance Imaging
mTBI Mild Traumatic Brain Injury
MSN Medial Septal Nucleus
Na+ Sodium ion
NMDA N-Methyl-D-Aspartate
NLR Novel Object Location Recognition
NOR Novel Object Recognition
OR Operating Room
PBBI Penetrating Ballistic-like Blast Injury
PD Parkinson's Disease
PPTg Pedunculopontine tegmental nucleus
PTSD Post-Traumatic Stress Disorder
REB Research Ethics Board
SRI Sunnybrook Research Institute
STN Subthalamic Nucleus
TBI Traumatic Brain Injury
TDP-43 Transactive Response DNA-binding protein 43
TNF Tumor Necrosis Factor
TUNEL Terminal deoxynucleotidyltransferase-mediated dUTP nick end-
labelling
Vim Ventral intermediate nucleus of the thalamus

viii
List of Figures
Figure 1: Typical experimental set-up in TBI animal models for Fluid Percussion Injury (FPI)..
....................................................................................................................................................... 10
Figure 2: Timeline for experiments performed for both studies 1 (TBI groups; no electrode
implantation surgery was performed) and 2 (DBS-TBI groups) .................................................. 27
Figure 3: Open field arrangement for DBS-TBI animals ............................................................. 28
Figure 4: (A) Typical setup of marble burying (MB) testing cages before placing animal for
behavioural protocol. (B) MB cage after 30 minutes of behavioural testing. (C) Side-view of
buried marbles for animal ID’d as ‘Fz042’................................................................................... 29
Figure 5: (A) Novel location recognition (NLR). (B) Novel object recognition (NOR) .............. 31
Figure 6: Barnes maze .................................................................................................................. 33
Figure 7: Defensive marble burying test for TBI, sham-TBI (craniotomy, no FPI) and naïve (no
surgical procedure) groups ............................................................................................................ 38
Figure 8: Novel object recognition (NOR) in TBI, sham-TBI and naïve groups ......................... 39
Figure 9: Memory deficits observed in TBI animals compared to other groups in the novel
location recognition (NLR) test .................................................................................................... 40
Figure 10: Barnes maze data for TBI treatment and control groups displaying changes in
performance across all variables, between train and test days ...................................................... 42
Figure 11: Average Barnes maze performance in all variables (latencies, search strategy and
error count) for TBI treatment and control groups ....................................................................... 43
Figure 12: Rat brains recovered 24h post-Barnes maze testing .................................................... 44
Figure 13: Average number of buried marbles in the DBS treatment group demonstrate no
significant improvement in anxiety type behaviour, compared to sham DBS-TBI, and TBI only
groups ............................................................................................................................................ 45

ix
Figure 14: NOR performance in DBS-TBI groups demonstrate no significant difference in novel
object exploration (A) or preference (B) ...................................................................................... 46
Figure 15: NLR performance in DBS-TBI groups demonstrate no significant difference in novel
location exploration (A) or preference (B), in comparison to sham DBS-TBI, or TBI only groups.
....................................................................................................................................................... 47
Figure 16: Barnes maze data for DBS-TBI, sham DBS-TBI and TBI groups displaying changes
in performance across all variables, between train and test days ................................................. 48
Figure 17: Average Barnes maze performance across all variables (latencies, search strategy and
error count) in DBS-TBI, sham DBS-TBI and TBI only groups .................................................. 49
Figure 18: Electrode location ........................................................................................................ 51
Figure 19: Caspase-3 measurements obtained using an enzyme immunosorbent assay (ELISA)
for TBI and non-TBI hemispheres ................................................................................................ 52

1
Chapter 1 – Introduction
1.1 Traumatic Brain Injury (TBI)
1.1.1 Definition and Classifying TBI
Traumatic brain injury (TBI) is defined as a neurological event whereby damage to the
head and brain tissue results from external mechanical forces, such as those caused by rapid
acceleration or deceleration of motor vehicles, blast waves from explosive devices, or impact
from a penetrating object1,2. Due to the physical, psychosocial and cognitive impairments not
immediately visible in patients suffering TBI, it is often referred to as a ‘silent epidemic’3,4. TBI
currently remains a critically significant worldwide public health issue and is the leading cause
of mental health disorders, death and disabilities associated with trauma3. Consequently, due to
the varying degrees of head trauma, survivors of TBI suffer from long-term neuropsychiatric
sequelae of behavioural disorders, post-traumatic stress disorder, anxiety, depression, and
cognitive dysfunction5,6. Head injury, when left without proper treatment, is also reported to be a
high risk factor for developing Alzheimer’s disease (AD)7. Amyloid β-protein accumulation, a
pathological trait implicated in AD, is also found in individuals who carry the e4 allele of the
APOE gene, which is increased in expression following head injury7,8. TBI is characterized by
cellular and structural deficits as well as neurological dysfunction, including, but not limited to,
neuroinflammation, vascular injury, cortical contusions and clinical endophenotypes, as well as
diffuse axonal injury (DAI)9. In humans, cognitive impairments and pathophysiological events
following TBI are largely dependent on injury severity1.
Extensive care and rehabilitation are necessary for TBI survivors and currently, there are
no therapeutic interventions sufficiently effective in improving the outcomes and complexities of
clinical TBI10. Both primary and secondary injury mechanisms are responsible for subsequent
structural and functional damage following TBI11,12. Primary injury is associated with direct
mechanical disruptions in brain tissue following the initial injury impact. This includes blood
vessel ruptures or hemorrhaging, focal contusions, hematomas, axonal shearing and DAI13.
Secondary injury, which occurs over several hours to months following TBI, includes non-
mechanical damage that lead to complex cellular, molecular and metabolic cascading events that

2
can further exacerbate the injury and eventually lead to atrophy and neuronal cell death2,13.
Immediate cell death caused by the initial impact of trauma on the brain can be irreversible and
difficult to treat; however, current treatment interventions focusing on interrupting cascading
events caused by secondary injury may provide neuroprotection and a significant improvement
in TBI incidences1. To date, there are no known or well-established neuroprotective interventions
for treating TBI2.
Severity of injury is often used to classify TBI in clinical settings, from mild to moderate
and severe11,14. Traditionally, clinical TBI is assessed using the Glasgow Coma Scale (GCS),
where the degree of injury severity is scored out of 15 points, based on eye, verbal and motor
movements6,14,15. Mild TBI (mTBI), the least severe and most common form of TBI3,16, is
diagnosed in patients usually with a non-penetrating closed-head trauma, and receives a GCS
score of 13-159,15. Moderate and severe TBI are evaluated by scores of 9-12 and ≤8,
respectively14, and require intensive care and neurosurgical interventions11. In addition to the
GCS, clinical guidelines for evaluating the magnitude of TBI also factor in structural imaging
(e.g. computed tomography or magnetic resonance imaging scans), severity and duration of loss
of consciousness, and other neurological symptoms, such as headaches, seizures, amnesia,
attention and/or memory impairment13.
Concussion is a term sometimes interchangeably used with mTBI. While most patients
generally recover quickly from mTBI, persistent symptoms have been reported, such as post-
traumatic amnesia, slower response times, disorientation, impaired learning and other memory
deficits9,16. Diagnosis and management of clinical mTBI can be challenging due to variability in
the extent of cell death and grey matter damage, that are not easily detectable by conventional
brain imaging techniques9,17. Currently, there are no clear models available for characterizing
and validating known biomarkers and long-term cognitions associated with clinical mTBI9,11.
Further investigation is needed using different approaches, including the use of experimental TBI
models, in order to obtain comprehensive interventions to translate into the clinical setting10.
1.1.2 Epidemiology
TBI is a major cause of death and disability across different age groups. Globally, it is
estimated to cause an annual incidence of approximately 10 million deaths and/or
hospitalizations in industrialized countries3,4. Each year, an estimated 57-69 million individuals

3
have or will have experienced TBI, of which 81% are mild and 11% are moderate in severity of
injury1,3. By utilizing statistical models and available national databases, current global data
estimated nearly 60% of TBIs annually are due to road traffic collisions; 20-30% are due to fall
incidents, while the remaining 10-20% are due to war, violence, or a combination of
occupational and sports injuries3,4. The highest incidences of TBI per capita were reported in the
United States, Canada and in European regions, while South-east Asian and Western Pacific
regions demonstrated the highest overall individual and systemic burden of TBI3.
In the United States alone, it is estimated that 1.7 million people sustain a TBI each year,
leading to 52,000 deaths and 275,000 hospital admissions5,6. In addition, >40% of participants in
a state-wide population-based survey reported having at least one TBI in their lifetime, while the
US Centers for Disease Control and Prevention had reported approximately 500-800 new cases
of TBI for every 100,000 people per year between 2000-20103,11. In 2013, 2.8 million TBI-
related deaths and hospitalizations were reported in the United States, of which 79% elderly
adults and 54% children suffered from fall-related accidents, which was found to be a primary
cause of TBI, especially amongst the elderly5,6. American/Alaskan Natives and African-
Americans had previously been reported to have a 4 times higher susceptibility rate to TBI due to
violence than white males4. Differences in gender have also been reported, where males were
consistently shown to have a higher incidence of TBI (1.5 times more likely) compared to
women4,6; older adult groups >60 years demonstrated higher occurrences of TBI compared to
younger adults4,11. This increase in TBI amongst the elderly can be explained by their longer life
expectancy with increased complex comorbidities and slower hospital recovery time from injury,
compared to younger adults5. In 2015, the US Defense and Veterans Brain Injury Center had
reported more than 22,000 military service members who had sustained a TBI from previous
combat and military training; 82% of these incidences were categorized as mTBI11.
In Canada, 23,000 hospitalizations per year were due to TBI, with 8% of these
individuals succumbing to their injuries5. In Ontario, a majority of TBI from work-place injuries
were found in manufacturing and government service sectors5,18. A retrospective chart review
showed that 57.8% of work-place related TBI incidences occurred in men, with the highest
incidence of occupational TBI found in the transportation and storage industry, at 81.5 per
100,000 individuals18. Trauma from being struck by or against an object was a major mechanism
of occupational TBI, followed by fall-related injuries18.While current epidemiological data have

4
demonstrated the significant disease burden of TBI, reports from middle- and low-income
regions may be limited in accurate estimates, in comparison to reports from high-income regions,
resulting in a large disparity in global TBI incidences3,11. This is largely due to the lack of quality
and robustness in data collection from low- and middle-income regions, where health care
systems have fewer resources and greater disease burden3. Further action and changes in research
policy are necessary to gather more reliable estimates of TBI occurrences in regions with limited
resources3,6.
1.1.3 Neuropathophysiology following Acute TBI
Though mild forms of TBI are the most prevalent, the clinical impact, mortality and socio-
economic burden of moderate-to-severe TBI comprise an enormous healthcare problem.
Characterizing and validating neuropathologies associated with moderate-to-severe TBI in
animal studies would allow for further advancement of our knowledge for underlying
physiological and neurochemical responses of TBI in humans and potentially lead to the
development of novel treatment modalities11,19.
Specific features of numerous TBI animal models have supported our ongoing
investigation of the sequelae that follow acute TBI in different brain regions11,20–22. Often,
neuropathological changes of TBI would consistently result in similar effects, and can be
categorized as either focal or diffuse injury that are followed by primary and secondary phases of
neurological damage22–24. Primarily, due to severe impact caused by direct blunt force or
penetrative trauma to the brain surface, focal injuries, such as discrete cortical and subcortical
lacerations and/or contusions, intracranial bleeding, subdural hematoma and subarachnoid
hemorrhages occur22. Pathologies caused by focal injury tend to have variable results, depending
on the type of focal injury, and the neuroanatomical location in which they had appeared23. On
the other hand, diffuse injury tends to display consistent pathological events, and is not
necessarily caused by similar neurological insults; rather, there would be shearing and stretching
of brain tissue due to rotational or sudden changes in acceleration induced forces11,22. A notable
form of diffuse injury is referred to as DAI, while microvascular and hypoxic-ischemic injuries
also affect several anatomical regions of the brain24. Unlike mTBI, that is characterized by more
diffuse injuries, as had previously been denoted in both animal models of closed-head trauma
and in clinical settings16,22,25,26, most acute moderate-to-severe TBIs are heterogeneous, having

5
both focal and diffuse injuries22,24. Investigating the neurochemistry and neuropathology behind
acute moderate-to-severe TBI implicates animal models where typically, a craniotomy is
performed, and the exposed skull is subjected to direct crush impact by a well-established
impactor device (e.g. fluid percussion27, controlled cortical impact28,29, further discussed in
section 1.2).
Rapid shearing and acute disruption of axons lead to DAI, one of the most commonly
observed neuropathology features of TBIs22,30,31. Owing to its higher white to gray matter
volume ratio, large mass, size, and characteristic gyri and sulci, the structure of the human brain
is susceptible to damage caused by linear acceleration/deceleration, or rotational forces24,32. DAI
severity corresponds to the amount of deceleration force impacting on the brain22; however,
while DAI is characteristic of TBI neuropathology, challenges arise in their detection and
identification using conventional imaging techniques, such as computerized tomography (CT) or
magnetic resonance imaging (MRI) scans of affected brain regions33. Histological techniques and
novel MRI sequences and techniques, such as diffusion weighted imaging (DWI) and diffusion
tensor imaging (DTI), have clearly identified changes in axonal integrity following DAI in
patients, shortly after experiencing trauma22. Any disruption in linear microtubular and
neurofilament structure would render axons highly susceptible to axonal swellings due to the
accumulation of membrane organelles and disassembled cytoskeletal components; subsequent
interruption in axonal transport would lead to secondary neuronal disconnection between
synapses, and eventually, the initiation of Wallerian degeneration 31,34. This active process of
degeneration is characterized by cytoskeletal pathology, displaying significant calcium influx,
due to the lack of regulation of calpain-mediated proteolysis of cytoskeletal proteins24. This
excessive calcium ion influx precedes microtubule disassembly, dysfunction in the mitochondria,
and eventually prepare the mechanically injured axons for excessive stretching, cell death, as
well as gradual axotomy24,31,34. Patients that experience a progressive decline in cognitive and
motor abilities several years after the initial TBI continue to display swellings within the
damaged axons, as well as further neuronal disconnection35
Hemorrhages within the corpus callosum and rostral brainstem can be observed in
moderate-to-severe TBI, while axonal injury and neuropathology can be detected with
histological and immunostaining techniques, marking for neurofilament and amyloid precursor
proteins (APP), shortly after TBI24.

6
Initial acute trauma to the brain has shown to initiate a multifaceted series of
neurochemical changes24. DAI and its resultant damage to cell membrane integrity causes a
disruption in ion transport. For example, a high influx of calcium ions and efflux of potassium
from ion channels result in increased excitatory glutamate neurotransmitter release, which binds
to N-methyl-D-aspartate (NMDA) receptors, causing excess depolarization of neurons; this
disruption in ion balance across cell membranes leads to increased activity to restore homeostasis
and the subsequent impairment to oxidative metabolism and depletion of energy stores22–24. In
addition to the axons undergoing secondary chemical effects following DAI, moderate-to-severe
TBI consistently leads to a cascading series of neuroinflammatory events that gradually
accumulate, contributing to a higher risk for long-term neurodegenerative diseases such as
Alzheimer’s23, Parkinson’s disease (PD) as well as amyotrophic lateral sclerosis (ALS)24. The
heterogeneity of TBI has been characterized not only by secondary injury cascades involving
neuronal cell death, neuroinflammation, degradation of white matter, and microglial activation
following initial trauma, but also the presence of neuropathological markers, such as an increase
in caspase-3, APP, phosphorylated tau, or TAR DNA-binding protein 43 (TDP-43), all of which
contribute to the late onset of neurodegenerative diseases that significantly impact the quality of
life for patients who had experienced TBI23,24,31.
1.2 Models of Traumatic Brain Injury
In order to further unravel and explore the pathophysiology of TBI in humans, elucidate
causal disease mechanisms, and develop preliminary therapeutic interventions, it is necessary to
have valid animal models1,9. For any animal model being implemented, specific principles are
required to be met to avoid miscommunication, poor representation, and replication of results in
our disease or endo-phenotype of interest. Such principles include i) determining a clear
objective with respect to utilizing an animal model; ii) having based our model on established
clinical attributes that define the condition; iii) the biofidelity or the degree of accuracy between
key features of the animal model and the human condition of interest; iv) displaying a correlation
between clinical results in humans and the efficacy of pre-clinical data obtained from the animal
model to verify its validity9,36. Numerous models have been developed for replicating the various
aspects of human TBI to provide adequate paradigms for testing treatment approaches.
Validation of said models is dependent on resemblance to the human condition in terms of
behavioural deficits, pathophysiology, causation, as well as response to therapeutic strategies36,37.

7
TBI in humans is known for its considerable heterogeneity exhibited by diverse modes of
primary injury, complemented with corresponding complexities from various secondary insults
in underlying cellular pathways38. In a practical setting, due to the highly heterogenous nature of
human TBI, current reproducible experimental animal models focus on partial aspects of injury.
For every available TBI model, only a given number of processes and mechanistic properties of
TBI can be accurately replicated1,9,38. Therefore, no single TBI animal model in the current
literature is capable of reproducing all processes of TBI presented in a clinical setting38.
However, despite the absence of an all-encompassing TBI model, currently available pre-clinical
models are capable of accurately mimicking various aspects and severity of TBI, which are
instrumental in understanding their mechanisms37. Successful TBI models include weight-drop39–
41, closed cortical impact (CCI)10,28, fluid percussion injury (FPI)27,42,43, blast injury, which may
consist of diffuse1,44,45 or more focal injuries, as depicted by the penetrating ballistic-like blast
injury (PBBI) model46–48. In addition, in vitro models have also been generated to provide less
expensive and more environmentally isolated TBI experiments, having been found to predict
some in vivo results49. Both chemical and mechanical injury have been applied to immortalized
cell lines49–51 (Table 1). Despite their ease of access and repeatable characteristics, however, in
vitro TBI models still face a challenge in encompassing the many complexities of TBI,
particularly the events that follow a primary blast injury49,52, which have been well-characterized
by most in vivo models.
An extensive body of literature in pre-clinical TBI research had been acquired using the
CCI and FPI models in rodents and large animals to determine behavioural and sensorimotor
impairments, as well as underlying histopathological responses, such as increased intracranial
pressure, neuroinflammation and localized grey and white matter damage51,53–55. Hence, models
of TBI are widely discussed due to their sensitive and highly reproducible properties and will be
relevant to the experiments presented in this thesis. However, regardless of the injury model
protocol implemented in experimental TBI studies, common technical attributes, such as the
frequency of anaesthetic exposure in animal models, could potentially compromise the outcome
of most TBI models, that typically require the use of anesthesia9. There is also concern over the
lack of sensitivity and robustness of previous mild TBI models, as histological or behavioural
data demonstrate variance across mild TBI studies, and will need to be addressed with further
refinement and characterization17,37,56. Below we will briefly describe the most commonly used

8
in vivo TBI models, namely controlled cortical impact, fluid percussion injury and blast and
preventative injury. Table 1 is then provided with further details on these models as well as
additional preparations.
1.2.1 Controlled Cortical Impact (CCI)
Controlled cortical impact (CCI) injury is produced by a device consisting of a
pressurized gas (i.e. pneumatic)29 or an electromagnetically-driven10 piston that accelerates a rod
over the surgically exposed dural surface10. Key parameters of injury are controlled by
characteristics of the impactor, such as the velocity, dwell time and depth, which can be adjusted
to produce varying degrees of TBI. In a closed-head variant of the model, repetitive and mild
TBI paradigms are used with no dural exposure10,57. Alongside widespread changes in
morphology and pathology (e.g. neuroinflammation, contusion, axonal injury, cortical tissue
loss), functional deficits have been observed, including overall motor and frontal lobe function,
as well as cognitive impairments in learning and memory28,29,58–61. These deficits have been
detected in widely accepted and reliable behavioural tests, including the Barnes maze26,62, Morris
water maze63, rotarod26,64 and elevated plus maze61,65, among others. Progressive declines in
cerebral blood flow have also been reported in rats, persisting even 1 year post-CCI injury66.
Although CCI is known for producing significant cortical contusions, CCI impact can lead to
post-traumatic apnea and tissue necrosis near the injury site57. Varying one or more of the
impactor characteristics could make an injury parameter change from minor to very severe.
Hence, pilot studies are often required to establish a suitable TBI paradigm10. Due to its focal
nature, one caveat of CCI is the inability to model diffuse forms of TBI67.
Like most pre-clinical TBI models described in this chapter, CCI is limited to replicating
one or more aspects of human TBI, as opposed to the entire event10,54. Some forms of clinical
TBI occur after a rapid impact to the skull68. CCI is more useful to mimicking these types of
head injury scenarios 25,69. Another variant of the CCI model, referred to as the CHIMERA
(Closed-head impact model of engineered rotational acceleration), utilizes rotational acceleration
forces on a closed skull. This mimics closed head injury and does not require surgical
intervention to deliver the impact69. The CHIMERA-TBI model has been shown to display key
features of human TBI. These include deficits in motor activity, cognitive and behavioural tasks

9
(e.g. increased impulsivity) 25, as well as diffuse axonal injury, microgliosis, increased levels of
inflammatory markers (e.g. TNF-α) and hyperphosphorylation of the tau protein69.
1.2.2 Weight-drop models
Weight-drop models typically involve an injury caused by a guided free-falling weighted
object dropped onto either the intact skull or exposed dura of an animal38,70. Mass of the weight
and the height at which it falls can be altered to produce different levels of injury severity.
Marmarou’s widely-recognized impact acceleration model has been proposed to mimic diffuse
TBI observed in humans, and utilized a weight dropped over a plate fastened onto a rat’s
cranium39. Diffuse axonal injury is commonly observed in this model, along with widespread
bilateral damage to neurons, as well as hemorrhages and astrogliosis39,71. Other variants of this
model delivered a focal type of injury, such as those described by Feeney et al41, involving an
impact to an exposed, intact dura, or Shohami et al, whose model involved delivering a focal
blunt injury over one side of the unprotected rodent skull40. Both models were shown to induce
cortical contusions, activation of microglia and astrocytes, and damage to the blood-brain barrier
(BBB)2. While weight-drop models are easy to implement and tend to closely mimic clinical
conditions observed in human TBI, the disadvantage lies in their variability of injury severity
and high mortality rate due to respiratory depression1,39.
1.2.3 Fluid Percussion Injury models (FPI)
Fluid percussion injury (FPI) is one of the most commonly used TBI models and will be
the one implemented in the current study. First introduced as a rabbit model in 196672, FPI
involves the usage of a fluid-filled cylinder in contact with an adjustable metal pendulum, that
exerts hydraulic pressure to induce an injury that resembles blunt-force trauma9. The model
requires a craniectomy so that dura mater is exposed. Injury is induced in the form of a rapid
injection of fluid into the exposed dura. The amount of intra-cranial pressure exerted by the fluid
is controlled by adjusting the height of the pendulum (Figure 1)1,9,27,73. Following injury, FPI
yields a combination of both diffuse neuronal injury and focal cortical contusions in targeted and
surrounding regions of the brain1,16,54. Histological staining has indicated inflammation, neuronal
death and diffuse axonal injury where FPI was implemented27,73–75.
FPI has been characterized to generate either middle/midline or lateral FPI. The former is
applied closer to the sagittal suture of the skull73,76,77, whereas lateral FPI42,55 is applied to more

10
lateral cortical regions, being the most widely used paradigm1. Despite the popularity of lateral
FPI in studying mechanisms of neuronal cell death, midline FPI is recently becoming a more
preferred model for brain injury in sports-related concussions and blast-induced TBI78,79. FPI has
been practiced across multiple species, being successfully demonstrated to induce TBI pathology
and neurobehavioural deficits in rats, mice, rabbits, dogs, pigs, sheep and cats16,27,43,73,76,80–82. It
allows good replication of mild to moderate TBI but does not involve rotational or rapid
acceleration-deceleration forces, as observed in some axonal pathology of human TBI9,67. As
described above, a disadvantage of the model is the requirement of a craniectomy, which does
not mitigate a scenario commonly observed during closed-head injury83.
Figure 1: Typical experimental set-up in TBI animal models for Fluid Percussion Injury (FPI),
which rapidly exerts fluid pulse at the target site when struck by the pendulum. Adapted and
reprinted with permission from ref 1.

11
1.2.4 Blast and Penetrative injury models
Brain injuries affiliated with blast and explosive shock waves, often seen in military
personnel45, are caused by objects rapidly propelled and dispersed primarily by strong blast,
rotational and acceleration/deceleration forces, as well as chemical emissions and thermal
energy54,70,84. Blast-induced TBIs are characterized by clinically relevant neuropathological (e.g.
cerebral brain edema, phosphorylated tauopathies, chronic neuroinflammation), and cognitive
(e.g. spatial learning and memory impairments up to 1 month post-injury) responses45,65. Models
of blast-induced TBI have previously been demonstrated, where anesthetized rodents were
exposed to controlled detonations of an explosive tube, or compressed air at a fixed
distance44,84,85. Several models have been generated to cover either single or multiple
components of a blast injury. Despite their biomechanical relevance to military warfare, varied
results have been reported, which can be difficult to compare across different laboratories1,86.
Further standardization of experimental designs are still required to replicate blast models for a
better understanding of the mechanisms and biomarkers associated with blast injury45,86.
Additionally, penetrating TBI caused by firearms are also prevalent in the military and
civilians, particularly in young adults6,46. Projectile objects travelling with high energy can lead
to cavities formed in the brain87 and experimental studies have replicated this using missile48 and
modified air rifle models46,47. As penetrating ballistic injury models form large, temporary
cavities, they are notable for their acute and delayed neuroinflammatory responses, and extensive
intracerebral hemorrhage at the lesion site87. The pathological nature of a penetrating injury
model can be influenced by the path at which the projectile travels, the magnitude and energy
transferred46,47,70.

12
Table 1: Animal models of TBI (adapted and modified from refs 1,2,50,51).
Models
and
subtypes
Primary
type of
Injury Species Strengths Limitations
Refer
ences Weight-Drop
Marmarou's Diffuse
rat, mouse
Well-characterized model; Biomechanics of injury mechanism
Difficult to reproduce;
1,2,39
resemble that of human TBI
High mortality rate due to apnea
Maryland's Diffuse rat Biomechanics of injury mechanism
Not enough replication, further
71
resemble that of human TBI
characterization needed
Feeney's Focal rat Biomechanics of injury mechanism
Craniotomy required; High mortality rate due to
41
resemble that of human TBI
apnea and variable skull fracture response
Shohami's Focal rat, mouse User-friendly device; Difficult to reproduce
40,88,8
9
severity of injury is adjustable
Controlled Cortical Focal
rat, mouse, ferret,
Severity of injury is adjustable; highly reproducible; Craniotomy required
10,28,2
9,57,58
,90 Impact (CCI)
swine, monkey low mortality rate
Blast Diffuse
rat, mouse, swine
Biomechanics of injury mechanism
High variability between injuries;
44,45,8
6
resemble that of military TBI
standardization required
Fluid Percussion Injury
Middle Mixed
rat, rabbit, cat,
Severity of injury is adjustable;
Craniotomy required; variability in injury severity
43,73,7
6,91,92
dog, sheep, swine highly reproducible
between experiments; high mortality rate due to apnea

13
Lateral Mixed
rat, mouse, swine
Severity of injury is adjustable;
Craniotomy required; variability in injury severity
27,42,8
2
highly reproducible between experiments; high mortality rate due to apnea
Repeated mild-TBI Diffuse
rat, mouse, swine
Biomechanics of injury mechanism
Further characterization required; variability in
56,84,9
3
resemble that of sports TBI
optimal number and frequency of applied injury
Penetrating ballistic- Focal
rat, cat, sheep
Biomechanics of injury mechanism
Standardization required
46–48 like brain injury
resemble that of penetrating TBI in humans
Cryogenic brain lesion Focal
rat, mouse
Severity of injury is adjustable; highly reproducible;
Replicates human TBI in very specific conditions
94,95
can be easily quantified In vitro Static mechanical injury Mixed
Various cell lines/
Severity of injury is adjustable; inexpensive and user-
Specific mechanical variables at impact cannot be
49–51
cultures (e.g. human,
friendly technique; easily quantifiable; prospect of
easily measured and estimated
mouse, rat, drosophila)
studying secondary injury pathways
(e.g. force, strain); tissue injury may be
difficult to verify (e.g. with Barotrauma injury)
Dynamic mechanical injury Mixed
Various cell lines/
Biomechanics of injury mechanism resemble that of
Tissue deformation cannot be easily measured;
cultures (e.g. human,
human TBI caused by rapid acceleration/deceleration
potential inconsistency in injury modelling due to
49–51
mouse, rat, drosophila)
forces; severity of injury is adjustable; highly reproducible
limited strain rate measurements; some models
can replicate secondary brain injury
are expensive and not user-friendly (e.g. cell -stretch
mechanical injury)

14
Chemical injury Mixed
Various cell lines/
Can be studied in isolation or in conjunction with other No limitations cited
cultures (e.g. human,
in vitro models (e.g. glutamate with cell-stretch injury model);
50,51
mouse, rat, drosophila)
prospect of studying secondary injury pathways
(e.g. excitotoxicity, oxidative stress)
1.3 Deep Brain Stimulation
1.3.1 Overview
The use of electrical stimulation for the treatment of neuropsychiatric disorders was
initially described as early as the late-1940s to early 1950s96. As conducted to date, deep brain
stimulation (DBS) was implemented as an alternative therapeutic technique to conventional
surgical ablation or lesioning of target brain tissue for the treatment of conditions such as
dystonia, essential tremor, and Parkinson’s disease97. Due to its adjustable and reversible
properties, DBS had rapidly superseded the need for lesioning and was found to be an effective
surgical treatment option for movement disorders98, particularly when introduced to subcortical
brain structures, such as the subthalamic nucleus (STN), ventral intermediate nucleus of the
thalamus (Vim), and globus pallidus internus (GPi))99–102. DBS comprises the use of electrodes
implanted at specific brain targets involved in the neurocircuitry of a given disease, from which
electrical stimulation is delivered. These electrodes or leads are typically connected to a pulse
generator (via extension wires), which is placed beneath the skin, near the clavicle to deliver the
desired current. The pulse generator can be programmed with the necessary stimulation
parameters, from a carefully selected combination of a wide array of frequencies, pulse widths,
and currents or voltages98,103. The adjustable property of DBS allows modifications to these
parameters, providing neuromodulatory interventions in accordance with underlying
neuropathological processes98. In addition, the stimulation-induced side effects of DBS can be
attenuated by the adjustment of stimulation parameters or discontinuing the therapy. Upon
reinstatement, therapeutic effects of DBS can frequently be regained19.

15
Following its successful adaption into movement disorders, DBS has since expanded its
application to other neurological and psychiatric disorders, including (but not limited to)
Alzheimer’s disease104–106, epilepsy107, anorexia nervosa108, Tourette’s syndrome19,109,110, chronic
pain111, obsessive-compulsive disorder (OCD)103,112,113, aggressiveness114, treatment-resistant and
major depressive disorder115, post-traumatic stress disorder (PTSD)116, stroke117, as well as
traumatic brain injury118, amongst others. Despite its clinical benefits and success, the
physiological mechanisms by which DBS exerts its effects are still poorly understood. A variety
of hypotheses have been suggested to explain the exact mechanism of action, in addition to
conflicting evidence as to whether DBS results in excitation or inhibition of local axonal
projections and neuronal elements102,119. Further understanding and conflation of ideas to draw
mechanisms for DBS will likely benefit in making precise and improved modifications in order
to efficiently treat other neurological disorders102.
1.3.2 Proposed Mechanisms of DBS
Single pulses of electrical stimulation often depolarize cells and axons near the electrode
site; However, in clinical settings, DBS is often delivered at frequencies above 100Hz (high
frequency stimulation; HFS). As described above in our overview, it was previously thought that
HFS mimicked the effects of targeted ablation120–122, suggesting that DBS behaves as a reversible
lesioning technique123. Stimulation parameters typically employed were initially found to have
an inhibitory effect on target nuclei, where DBS led to a decrease in spontaneous firing of
neuronal populations19. However, the precise mechanism of action for DBS and its therapeutic
impact on a stimulated target is not well understood and is dependent on several factors (e.g.,
duration of stimulation, physiology of the targeted region, composition of neural elements,
frequency, amplitude, pulse width of stimulation). These factors interact with one another to
modulate biological processes at varying subcellular compartments19,98. More recently,
hypotheses beyond the simple excitation/inhibition of target regions and its neuronal elements
have been investigated.
There was an initial belief that DBS has therapeutic benefits resembling that of lesion
therapy due to inhibition of target neuronal activity98,102. Cessation of local firing of neurons has
been induced by STN-, thalamic- and GPi-DBS along with a reduction in abnormal firing
patterns in target sites, leading to a reduction in motor symptoms102. One of the earliest proposed

16
mechanisms was the so-called depolarization block98,102. This occurs when high frequency
stimulation leads to a gradual inactivation of voltage-gated Na+ and Ca2+ channels, and an
increase in extracellular K+ concentrations, likely mediated by glial cells, which further reduces
depolarization of cell membranes124–127. This resultant silencing of neighboring neuronal
populations in the vicinity of DBS electrodes contributed to the early hypothesis of DBS acting
as a reversible surgical lesion in PD patients99,100,123,128,129 and in pre-clinical Parkinsonian
models130–133. Additionally, neurotransmitter release from astrocytes134 following HFS is said to
play a role in suppressing neurotransmitter responses135,136.
Another hypothesis postulated for the local inhibitory effects of DBS at high frequencies is
the disruption of information flow at the stimulation site due to the dissociation of input and
output signals, thereby producing a “jamming” signal102. This may occur when HFS depresses
synaptic function, as stimulation pulses generate action potentials directly onto the efferent axon,
while the soma is silenced. This ‘decoupling’ hypothesis would result in regular spiking output,
and reversal of an otherwise irregular or pathological firing pattern, promoting the abolition of
pathological frequencies137,138. DBS of the subthalamic nucleus in parkinsonian states has
previously demonstrated suppression of motor symptom-affiliated beta-band frequencies during
HFS, as well as shortly after HFS is no longer applied139–141. Subsequently, pathological
oscillatory activity is replaced with more regular rhythms within the STN, which correlate with
improvements in rigidity and bradykinesia142–145. Chiken and Nambu102 postulated that
disruption of information flow through the GPi due to DBS was invoked by spontaneous
discharges due to strong GABAergic inhibition. In addition, STN-DBS was previously found to
block transmission of signals through both hyperdirect and indirect basal ganglia pathways, but
not the direct pathway. This was evident by significantly reduced or diminishing of excitatory
signals but retention of cortically evoked inhibitory signals146. Overall, parkinsonian motor
symptoms can be suppressed through STN- or GPi-DBS, by disrupting abnormal firing rates and
patterns in major basal ganglia pathways, prior to this information flow being transmitted to the
thalamus and finally the motor cortex, from which motor symptoms can be expressed102. This
hypothesis of DBS disrupting pathological oscillatory patterns and signal transmission may
consolidate a more plausible explanation for its bimodal, yet therapeutic effects; DBS regulates
and potentially corrects for inappropriate firing patterns and rates, as well as the dynamic activity
that are responsible for motor symptoms in PD102.

17
In contrast to the inactivation of neuronal cell bodies, axons can still be activated following
high frequency DBS. This may lead to the excitation or inhibition of structures at a distance from
the target, depending on firing patterns, rates and whether the stimulation of axonal projections
releases GABA or glutamate102,147. In addition, with a complex interplay of principal neurons
and interneurons receiving projections from stimulating targets, the consequences of delivering
HFS in structures at a distance are difficult to predict. For example, despite receiving excitatory
glutamatergic inputs from the STN, GABAergic inhibition in the GPi likely overrides these
excitatory drive, thus inducing the activation of inhibitory afferents133,148. Additionally, through
the activation of both glutamatergic and GABAergic afferent projections149, both excitatory and
inhibitory postsynaptic potentials may be generated in neural populations of the STN, suggesting
that DBS can not only generate both excitation signals that are distal from the target region, but
locally inhibit neuronal soma within populations surrounding the electrode102,147. Another
example that is very relevant to this thesis is the inhibitory effects of high frequency, high
current stimulation of the anterior thalamic nucleus (ANT) over dentate gyrus hippocampal cells.
In a series of studies in which ANT-DBS was used to mitigate seizure activity or modulate
memory performance, stimulation was found to reduce the firing rate of dentate gyrus granule
cells150 as well as hippocampal excitability151. This is of interest because the ANT is an
important hub in the limbic circuitry152–154and has direct and indirect connections with the
hippocampal formation155,156. Finally, of great importance are the chronic consequences of DBS,
since this therapy is applied to humans continuously for years. Plastic changes such as long-term
potentiation and increases in neurotrophin levels and neurogenesis have all been described after
electrical stimulation and may underlie some of the behavioral consequences of DBS19.
In summary, DBS at high frequencies in known neuropathological substrates, particularly
within the basal ganglia, has demonstrated inhibition of neuronal activity near the electrodes
through mechanisms that potentially involve a reduced glutamate release and the release of local
GABA via the excitation of GABAergic terminals, and adenosine to promote synaptic inhibition
of afferents, as well as alterations within voltage-gated Na+/K+ channels. On the other hand,
through synaptic modulation, DBS of target structures is also shown to be capable of increasing
axonal firing in efferent projections and axonal fibers within the vicinity of the implanted DBS
electrodes, while concurrently inhibiting or decoupling somatic activity157,158. Ultimately, the
resultant stimulation of any target neuronal population will vary due to the cumulative effects of

18
DBS parameters, and the composition of excitatory and inhibitory neural elements in any given
neuropathological target site98. Therefore, further investigating is required to explain how DBS
can have a similar mechanistic approach when extrapolated to other neurological or
neuropsychiatric disorders, where a diverse range of neural circuitries are engaged and affected
to generate their respective pathologies.
1.3.3 Deep Brain Stimulation in Traumatic Brain Injury
Depending on the severity and condition of brain injury, TBI is notorious for initiating a
cascade of secondary damage, involving neuropathological processes that may lead to cognitive
dysfunction, changes in consciousness as well as a considerable amount of sensorimotor
damage22. Regulation of secondary damage is one of the key means of intervention, following
the onset of injury to help restore loss of physical and cognitive function; to date, therapeutic
strategies to manage this cascade of neurological events is still under development, with widely
varying reported outcomes118. In cases of motor dysfunction, a combination of neuromodulation
and rehabilitative training has been implemented to help promote recovery of motor circuits.
DBS has become a conventional treatment option for addressing movement disorders, especially
when affiliated motor symptoms no longer respond to pharmacotherapy. As mentioned, DBS of
the GPi and STN was shown to effectively treat motor symptoms, dyskinesias and fluctuations in
patients with PD98. However, the limited knowledge on the relationship between behavioural
changes following TBI and the corresponding impacted neuroanatomical structures and neural
circuitry has restrained our ability to propose effective treatment options for addressing TBI
recovery159. Chan et al118 had previously demonstrated improved behavioural and motor recovery
when DBS was delivered in a post-stroke rodent model, and further extended these findings to
support the anti-inflammatory and neuroprotective properties of this treatment modality.
Stimulation of the lateral cerebellar nucleus (LCN), along with motor training in rats promoted
long term potentiation, and functional cortical reorganization, following contralateral unilateral
FPI118; findings of this study further support DBS of the cerebellar dentate nucleus (DN) at
frequencies that mimic neuronal firing rates to activate glutamatergic efferents, thus broadly
influencing thalamocortical activity in patients with acquired brain injury.
Deficits in executive functioning in both pre-clinical models and clinical populations may
be significantly improved through stimulation of the thalamus, regions of the limbic (Papez)

19
circuit as well as frontal cortical areas and subcortical structures119,160. Distorted and
desynchronized hippocampal oscillations following TBI in rodent models resulted in cognitive
dysfunction, including learning, memory, and spatial navigation160; subsequently, DBS following
TBI may assist in realignment of oscillatory activity. In particular, targeting regions such as the
medial septal nucleus (MSN) to improve hippocampal function, had previously demonstrated
normalized object recognition, and shorter latency periods in spatial navigation during Barnes
maze in TBI rats161. Furthermore, acute HFS (either 6 hours, or 7 days after injury) of the median
and dorsal raphe nuclei in the midbrain of a rodent FPI model had improved memory in
behavioural tasks, likely due to the suggested neuroprotective effects of serotonin160; however
one caveat that study had noted was the lack of significant differences between treatment groups
following day 3 of a Morris water maze task, likely due to underlying spontaneous recovery162.
Other targets of stimulation to improve memory include the fornix, and central thalamus which
in turn was shown to increase activity in the dentate gyrus to encourage better regulation of
emotions, and executive functioning in patients with TBI, as displayed by greater performance in
cognitive and functional scores160.
Additionally, a small population of TBI patients with varying levels of consciousness,
including of minimally conscious states (MCS) have reported differing benefits in behaviour and
environmental awareness following DBS163. Unfortunately, a lack of control for targeted
stimulation and the overall heterogeneous nature of head injury acquired by patients create
additional complications in finding efficient and novel treatment options to approach the
challenges faced by patients with MCS163. Optimizing for patient-specific symptoms, the
temporal properties of administering DBS in TBI, neuromodulation, as well as rehabilitation
training conditions may improve therapeutic efficacy in reducing injury-induced motor and
cognitive deficits164. Finally, there is growing evidence to suggest that DBS confers
neuroprotection from excitotoxicity and progressive neuronal loss, thereby improving the
likelihood of recovering from neurodegeneration119. Criteria for considering a therapeutic
intervention to have neuroprotective effects include a decrease in progression of neurological
symptoms, a slowing of neuropathogenesis, as well as attenuation of neuronal cell death119. For
example, evidence from pre-clinical AD models using DBS propose neuroprotective effects by
increasing hippocampal neurogenesis, promotion of synaptic plasticity as well as neurotrophic
factors, and by facilitating clearance of misfolded proteins117,165. However, McKinnon et al119

20
note that acute stimulation parameters are used in these pre-clinical studies, hence not directly
translatable in clinical settings, where chronic stimulation parameters are employed in AD
patients.
More relevant to this thesis, DBS of the ANT resulted in an inhibition of GPi activity, and
a rescuing of neurons in the dorsal striatum, leading to an anti-epileptic effect in a pre-clinical
model of drug-resistant temporal lobe epilepsy166. Excitotoxicity reduction thus normalizing
function in the basal ganglia-limbic circuitry may be a mechanism by which ANT-DBS exerts
neuroprotection166. In terms of memory, ANT-DBS in rats has been shown to improve memory
performance in the Morris Water Maze, possibly via the induction of hippocampal
neurogenesis167–169. ANT-DBS was also found to have a far-reaching neuroprotective effect in
hippocampal regions170,171, through regulating inflammation and promoting adenosine
production166,172. In addition to its anticonvulsant effect in pre-clinical models, data acquired
from human studies, notably in the multicenter, randomized, double-blind, bilateral Stimulation
of the ANT for Epilepsy (SANTE) trial (n = 110) demonstrated the efficacy and safety of DBS in
the treatment of refractory epilepsy, and has since been granted approval as an adjunctive
therapy by the U.S. Food and Drug Administration173,174. Towards the end of the blinded phase,
patients in the treatment group experienced 40.4% reduction in median seizure frequency relative
to baseline, while the control group experienced 14.5%173. A 2-year follow-up by Fisher et al173
noted a 56% seizure reduction rate in patients, while Salanova et al175 reported a median 69%
reduction in seizure frequency in 74 patients, 5 years after the initial SANTE trial. In spite of
these findings, significant differences in neuropsychological test scores for mood and cognition
were not observed between patients in treatment and control groups173, although some patients
reported impairments in memory and depression, following ANT-DBS (majority of these
findings were reported by patients with a previous history of memory impairment and/or
depression)175. These responses were not deemed to be serious and resolved spontaneously.
Long-term follow-up studies of the treatment group demonstrated improvements in executive
function, attention, and subjective cognitive function174–176.
Despite converging evidence for the efficacy of ANT-DBS, it is still difficult to evaluate
the mechanism of action by which ANT-DBS confers neuroprotective effects in patients, notably
due to timing in which DBS is considered an option. Thus, disease progression may have
advanced to irreparable stages119. This is particularly relevant in TBI patients, as numerous

21
factors, including severity, place of injury as well as the stage of neurodegeneration in which
DBS is deployed. Continuous and follow-up studies of DBS in validated animal models are still
necessary to corroborate stimulation parameters and target regions in which the most favourable
neuroprotective effects may be achieved119.
1.4 Rationale, objectives, and hypothesis
While DBS is now a widely accepted treatment option for the management of motor
symptoms in Parkinson’s disease, tremor and dystonia177, there is currently no conclusive
evidence suggesting that DBS will lead to significant outcomes in a moderate-to-severe acute
TBI model. A study using c-Fos and behavioural analyses was able to demonstrate that rats
undergoing stimulation of the ANT (i.e., 130 Hz, 400 µA, 90 µs) experienced decreased activity
in the hippocampus, with a reduction in glutamate release and therefore, less excitotoxicity107.
Animals that underwent contextual fear conditioning tests following high current stimulation of
the ANT displayed impaired memory likely related to a decreased hippocampal recruitment150.
In addition, acute stimulation of the ANT for 6 hours during status epilepticus in the rat
pilocarpine seizure model was shown to have an anticonvulsant effect, while reducing
hippocampal neuroinflammation and apoptosis107. Overall, the ANT and its relayed projections
toward the hippocampus are critical in facilitating contextual and spatial memory150. Given that
neuroprotective effects were demonstrated after stimulation, the ANT will be our target for
improving memory deficits in moderate-to-severe TBI in rats.
To study the effects of acute ANT-DBS in providing neuroprotection and improving
memory performance following moderate-to-severe TBI, this thesis will consist of 3 aims:
Aim 1: To characterize behavioural deficits in the moderate-to-severe TBI fluid percussion
injury (FPI) model in rats.
Aim 2: To investigate the effects of acute ANT-DBS in the FPI model through a battery of
behavioural tests associated with learning, memory and anxiety-like behaviour.
Aim 3: To test whether ANT-DBS reduces hippocampal apoptosis in FPI-exposed rats.

22
Given the DBS effects described above and the relevance of the ANT in memory
processes, this project will test the following hypotheses:
1. Moderate-to-severe TBI will induce memory deficits and anxiety-type responses in rats,
as measured by poorer outcome of performance in a battery of behavioural tests,
compared to non-TBI groups.
2. Conversely, increased performance outcomes in our rodent behavioural paradigm
following ANT-DBS will demonstrate improved memory function, and reduced anxiety-
type behaviour in rats exposed to acute moderate-to-severe TBI.
3. ANT-DBS will decrease levels of the biomarker caspase-3, thus demonstrating reduced
apoptosis signaling in the hippocampus of rats in a moderate-to-severe TBI model.

23
Chapter 2 – General methods & materials
2.1 Animals and Surgical procedures
2.1.1 Animals
The following animal use protocol (AUP) was approved by the research ethics board
(REB) at Sunnybrook Research Institute (SRI). Animals were cared for and used in accordance
with the guidelines outlined by the Canadian Council on Animal Care. Adult, male outbred
Sprague-Dawley rats (225-250g, Charles River Laboratories, Quebec), were individually housed
within the animal care facility at SRI and were acclimatized to 12-hour light-dark cycles (7:00
a.m. – 7:00 p.m.) upon arrival to the facility. The animals were fed in their home cages with a
standard laboratory rat diet and given water on an ad libitum basis. All surgical procedures took
place within operating rooms (OR1, OR2, OR3) located in the SRI animal care facility. All tools
and equipment were sterilized prior to commencement. Handling took place after all surgical
procedures were completed, due to a short acclimatization period prior to surgery.
New cohorts of animals were utilized for each study. These cohorts were applied for
different objectives (i.e., study 1 for validating lateral FPI TBI model using behavioural testing
and future histological analyses; study 2 for ANT-DBS effects in behaviour and histology; study
3 for neurochemical measurements of caspase-3 24 hr post-TBI). Table 2 outlines the number of
animals used per group, across all 3 studies described in this thesis.

24
Table 2: Animal cohorts and corresponding number of animals across experiments 1-3. Brain
recovery for the animals in experiment 3 was performed 24hr post-TBI for neurochemical
analysis:
2.1.2. DBS electrode implantation
A fully assembled stereotaxic frame (Model 900, David Kopf instruments, Tujunga, CA)
with appropriate ear and bite bars was set up. Each animal was anesthetized in a closed induction
chamber with 4% isofluorane (500 ml/min room air) for up to 3-5 minutes. Once the animal was
fully anesthetized, an electric hair clipper was used to shave their head. The animal was then
inserted into a pre-assembled stereotaxic frame. Ear bar distance and nose cone height was
adjusted such that their head was firmly centered and positioned flat in the antero-posterior
plane. Prior to making an incision, eye lubricant (BNP ophthalmic ointment) was applied to the
animal’s eyes. The animal’s body was covered with extra drapes or towels to retain body
warmth. The exposed scalp was sanitized with betadine and 70% ethanol. Ketoprofen was
Naïve TBISham TBI (burr
holes only)DBS-TBI Sham DBS-TBI Control (burr holes-TBI)
2
Effects of acute
ANT-DBS in the
FPI model
through
behavioural
testing
2 - - - 10 9 13 32 Y
3
Testing whether
ANT-DBS
reduces
hippocampal
apoptosis in FPI-
exposed rats.
3 - - - 8 7 8 23 N
-
Y
Total no. of
animals/co
hort
1
13 10 12 - -
No. of animals/group
35
Cohort #
Characterizing
behavioural
deficits in the
moderate-to-
severe TBI (FPI)
model in rats
1
Behavioural
testing?
(Y/N)
Experiment
#Study objective

25
subcutaneously administered (ANAFEN, 10mg/ml). A midline sagittal skin incision from rostral
to caudal was made with a 15 blade so that the anterior region of the skull was exposed. Excess
bleeding and debris were controlled using sterile saline solution (0.9% NaCl) compression with
gauze and/or cotton swabs. At specific stereotaxic coordinates (obtained from Paxinos and
Watson’s rat brain atlas178) to locate the ANT bilaterally (antero-posterior -1.50; medio-lateral
+/- 1.50), holes of the necessary diameters were made to the skull using a drill and the
appropriate drill bits. The dura mater was exposed with a fine tip 25G needle. Two additional
holes were made over anterior skull regions for the insertion of stainless-steel screws used to
anchors the cap. Insulated stainless steel electrodes (250 µm in diameter; Plastics One model
E363) covered with polyamide with a partially exposed tip (~0.5-0.75 mm) were then lowered
bilaterally to a depth of 6.00mm into the ANT and later used as cathodes. The anode was a
stainless-steel electrode (125 µm in diameter; Plastics One model E363) peeled from the varnish
and wrapped around anchoring screws. Both ANT electrodes and the anode were connected to a
plastic pedestal (Plastics One model MS363) using acrylic cement. Acrylic dental cement paste
(dental acrylic powder and liquid curing solvent, Lang) was also used to further seal and secure
the implanted electrode, as well as creating a skull cap. Sham-DBS animals underwent the same
electrode implantation, without receiving stimulation. Another control group (control surgery)
was included that had burr holes drilled into their skull at the above coordinates but did not have
electrodes implanted. Following the procedure, animals were returned to their home facility,
where they would recover for one week, during which they were closely monitored.
2.1.3 Fluid percussion injury (FPI) and acute Deep Brain Stimulation
One week after electrode implantation, animals were retrieved from their home facility and
brought to an impromptu TBI surgery room, where the fluid percussion injury device was
located. Each animal was anesthetized with 4% isoflurane, prepared, and positioned to the
stereotaxic frame, as described for DBS implantation. An incision extending from the DBS cap
to the occipital region was then made such that the sagittal suture is exposed. Once the exposed
surface was cleaned of connective tissue and blood, a craniotomy (5mm in diameter) was drilled
using a trephine drill bit (Meisinger 227RF-050-HP trephine, stainless steel, ID 4.0mm) near the

26
sagittal sutures, between the lambda and bregma near the right parietal cortex. Care was exerted
so that the dura mater was not penetrated. An injury ‘hub’ was temporarily secured over the
craniotomy using acrylic dental cement (Lang) in preparation for lateral fluid percussion injury.
Injury hubs were made by cutting the plastic ends of 25-gauge disposable needles (BD
PrecisionGlide, #2538-CABD305175) at a height and elevated angle suitable for the given
craniotomy. Once the cement solidified, the temporary seal was checked for leaks by injecting a
small pool of saline into the secure hub and observing whether it decreases in volume. This was
to prevent dampening of the seal formed between the liquid in the hub and high-pressure liquid
from the FPI device. Rats in the TBI group were connected to the lateral FPI device (Model
FP302; Amscien Instruments, Richmond, Virginia, USA)27 via the hub, to induce moderate
traumatic brain injuries, with impact pressures of 2.5-3.0 atm. These values were defined based
on pilot experiments conducted in our laboratory demonstrating injury severity. In addition,
sham control animals were also included, where animals underwent the same procedure, but no
FPI is induced. Following FPI, the injury hub with the temporary seal was removed, and the
initial scalp incision was sutured before placing the animals back in their home cages. Sham-TBI
animals were drilled with shallow burr holes as a control group undergoing all surgical
procedures, but without lateral FPI.
Immediately after undergoing TBI, animals were transferred to the ante room of the TBI
behavioural suite, where plexi-glass boxes (Toronto Plastics, 25 cm L x 25 cm W x 45 cm H)
padded with clean bedding were set up for acute stimulation. Before the animal regains
consciousness from the anesthesia, electrodes were connected to a 6-channel SL6C commutator
(Plastics One), via connection cables. Using extended 6-channel cables (Plastics One; 363-340/6,
50 cm) the commutator was connected to a stimulator (ANS, model# 3510). Once connected,
animals were given DBS bilaterally for 6 hours at parameters in the range of those associated
with antiapoptotic and anti-inflammatory effects in our previous studies (130 Hz, 90
microseconds, 300 µA)150. All stimulated animals were periodically monitored to observe any
changes in behaviour or discomfort, and to ensure that DBS electrode caps had not broken off.
Following acute stimulation, animals were disconnected from their stimulation boxes, returned to
their respective cages, and taken back to their home facility to rest and recover.

27
2.2. Behavioural assessments
Prior to testing, animals were closely monitored for 4 days post-surgery for any major
motor impairments (e.g., gait or balancing issues) or abnormal behaviour (e.g., excess grooming,
lethargy, loss of appetite). During the 4-day rest time, animals were consistently handled for up
to 2 minutes each, within their housing facility. Behavioural testing for all groups took place
over the course of 5 days (including 1 rest day) for spatial learning and memory, including a
novel location and object recognition test (NLR/NOR)179,180, marble burying 181 and Barnes maze
182. The following behavioural protocol was designed and optimized, based on literature where
these tasks were previously proposed and described (Figure 2).
Figure 2: Timeline for experiments performed for both studies 1 (TBI groups; no electrode
implantation surgery was performed) and 2 (DBS-TBI groups). Following habituation and
routine handling of Sprague-Dawley rats, DBS electrodes were implanted and FPI was
implemented approximately 7 days later at ~2.5-3.0 atm. DBS-TBI animals received acute
stimulation for 6 h following TBI (day 1). After a fixed interval, the animals underwent a series
of behavioural tests to measure anxiety type behaviour (Marble burying; MB) and memory
(Novel location recognition; NLR, novel object recognition; NOR, and Barnes maze). The
animals were sacrificed, and brains were recovered 24 h after the last day of behavioural testing.

28
2.2.1. Novel location and novel object recognition Habituation: Open field testing
On Day 1 (five days after TBI), animals underwent the habituation phase of the
NLR/NOR tasks where each freely explored an open field arena for 5 minutes (Figure 3). Each
open field consisted of a large, transparent plexi-glass box (Toronto Plastics, 45cm L x 45cm W
x 40cm H), with no other objects or bedding within the field. Each trial was recorded using a
camcorder (Sony Handycam® HDR-CX455) mounted to the ceiling and controlled via
smartphone wi-fi. The animals were then removed from the arena and kept in their respective
home cages, placed in the anteroom of the behavioural suite, until the next behavioural task
commenced.
Figure 3: Open field arrangement for DBS-TBI animals. Each animal was placed in individual
plexi-glass boxes for 5 minutes. No other objects or bedding was included in the open field.
After 5 minutes of exploration, the animals were returned to their respective home cages. Opaque
barriers were placed between open field arenas to prevent animals from seeing each other during
recording sessions.
2.2.2. Marble Burying
Open field habituation was followed approximately 60 minutes later by marble burying
(MB). This test was conducted in rat cages set up with clean bedding, evenly levelled and

29
pressed flat with ~5 cm in depth. 15 brightly coloured glass marbles were placed parallelly on
the surface of the bedding and spaced evenly, in a 3x5 arrangement (Figure 4a). The animals
were then retrieved from their home cages in the anteroom and each animal was positioned at the
center of each MB cage. As soon as all the animals were placed in their MB cages, they
proceeded to roam freely for 30-minute sessions, while these cages were covered with lids.
Throughout the session, behavioural changes were noted for each animal. At the end of the
session, animals were removed and taken back to their respective home cages. Without
disrupting the arrangement of the marbles or the bedding, bird’s-eye and side-view images (e.g.,
Figure 4b, c) were captured for analysis. The number of marbles buried above 50% of their
volume was blindly counted from the images. Used bedding was then discarded and the animals
in their home cages were returned to their housing facility. In this test, a high number of buried
marbles represent anxiety-type behaviour.
Figure 4: (A) Typical setup of marble burying (MB) testing cages before placing animal for
behavioural protocol. (B) MB cage after 30 minutes of behavioural testing. (C) Side-view of
buried marbles for animal ID’d as ‘Fz042’. 15 brightly coloured marbles were lined up in a 3x5
arrangement over clean bedding. ID’d animals were placed in their corresponding test cages and
observed for 30 minutes before returning to their respective home cages.

30
2.2.3. NLR/NOR: Familiarization, novel location and novel object recognition
testing
Day 2 (6 days after TBI) involved the following phases – familiarization, novel location
test, and novel object test phases, each of which was performed in 5-minute sessions per animal.
During familiarization, each animal was placed in an arena containing two identical objects
(objects A + A; Figure 5a). Open field boxes from the previous day were used as the arena for
the above-mentioned phases. Animals were then removed after familiarization and temporarily
placed in a holding cage. They were then replaced in the arena, where one of the identical objects
was relocated to a different corner to establish a novel location for said object. Finally, the same
animals were each tested in 5-minute NOR sessions, where the newly relocated object A was
replaced with a novel object B (i.e., A + B; Figure 5b). The objects utilized included 2 identical
towers made of Lego bricks (i.e., object A), and 2 large falcon cell culture media flasks filled
with clean bedding (i.e., object B). The arena and the objects were regularly cleaned and
sterilized with Clidox®, as well as 70% ethanol, such that the next animal did not detect the
odour of the previously tested animal. At the end of these sessions, animals were returned to their
home cages and allowed to rest for at least 24 hours before proceeding to the final behavioural
task. Apart from the familiarization test, all test sessions were recorded using the overhead video
recording device, as was previously used for the open field sessions. Scoring was conducted
blindly. Since rodents have an innate preference for novelty, animals that recognize the familiar
object and location will prefer to spend more time at the novel object and location183. Variables
measured were the time exploring in the novel or old location or with the novel and old object.
The percent time exploring the novel object or location was calculated as the time exploring the
novel object or location/total exploration time. The novel object or location index was calculated
as time exploring the novel object or location – time exploring the old object or location/ total
exploration time.

31
Figure 5: (A) Novel location recognition (NLR). Each animal was placed in individual plexi-
glass boxes for 5 minutes with two identical objects, one of which was moved to a new location.
While not pictured, the animals underwent familiarization sessions prior to exploring newly
located objects. Each animal was randomly assigned to either objects A (flasks) or objects B
(Lego tower) placed diagonally from each other (white square = previous location) and allowed
to explore for 5 minutes. (B) Novel object recognition (NOR). Similar to NLR, animals were
replaced in their randomly assigned arena, only the newly located object was replaced with a
novel object, unfamiliar to the animal.
2.2.4. Barnes maze
On the 4th and 5th days of testing (8 and 9 days after TBI), visuo-spatial learning and memory
were evaluated using the Barnes maze paradigm182,184. The maze consisted of an elevated
circular arena, with 20 circular holes at equal diameter and spacing (122cm platform diameter,
20 holes, 10cm hole diameter; Figure 6). To encourage animals to quickly locate the escape box,
aversive cues were added, including an overhead centre lightbulb of 100W-equivalent wattage,
and a high-powered table fan to induce high-speed winds across the maze. All other light sources

32
were blocked with dark covers, such that the overhead 100W lightbulb was the only source of
light. Visual cues were made up of cross, square, circular and triangular shapes, cut out from
black construction paper and placed on each wall surrounding the arena, within the animal’s eye
level. The escape box (35.0cm L x 11.5cm W x 10.0cm D) was placed below one of these
circular holes, near a visual cue to indicate the location for each animal to escape, while all other
holes were concealed with false bottoms. The entire arena was surrounded by dark curtains,
behind which the experimenter would observe each trial, and to prevent distracting them during
testing. All animals were kept in the anteroom of the behavioural suite, and one at a time, each
animal was brought in for testing. Each trial had a duration of 3 minutes, after which the animal
was removed, should they fail to locate the escape route. All animals completed a given set of
trials before proceeding to subsequent trials for Barnes maze. The first day of behavioural testing
in the Barnes maze was assigned for acquisition and habituation trials, during which the animals
were trained to familiarize themselves with the task. On the second day, the animals were
exposed once again to the test and scored for their visuospatial memory, by following the same
steps as described above. The following variables were recorded: Time to locate the escape
route, time to enter the escape route, error count (number of times the animals erroneously select
the escape box), latency to the first nose poke, total latency to find the escape route, and the
search strategy. The search strategy is ranked based on a cognitive score scale from the Barnes
maze unbiased strategy (BUNS) classification algorithm, as previously described by Illouz et al
(2016): The animals' ability to locate the escape box was ranked on a scale of 0 to 1, with 0.25
increments, where 0 is considered random (little to no searching), 0.25 = serial (animals search
each consecutive hole around the circumference of the maze), 0.5 = focused search/long
correction (animals search within one quadrant nearest to or away from the escape box,
respectively), 0.75 = correction (animals make 1-2 errors within the quadrant nearest to the
escape box before locating it) and 1 = direct (no errors when locating escape box). A higher
average value on the cognitive scale implies a relatively more direct navigational approach in
locating the escape route of the maze. We note that the Barnes maze in our protocol was
purposely made more difficult with animals undergoing only a few training sessions. This was
because we noticed that TBI animals exposed to more prolonged Barnes maze testing (two or
three days of training followed by probe testing) do not show performance deficits.

33
Figure 6: Barnes maze. 8 days after the initial injury, animals were allowed 3 minutes (180 sec)
to explore, locate and enter the correct escape box within a circular Barnes maze. Aversive cues
were implemented to motivate the need to escape the maze. These included one 100-W light
source affixed above the maze, as well as a high-speed fan. Visual cues (not pictured) consisting
of black geometric shapes made of construction paper were glued onto the walls of the testing
room. Experimenter observed each trial behind dark curtains and animals were retrieved from the
maze if they fail to enter the escape box within the 3-minute duration. Animals who successfully
entered the escape box could remain in there for at least 30 seconds before returning them to
their home cages. Animals were tested for two trials per day, for 2 days, as per a modified
untraining protocol. All parameters were scored manually through video recordings.
2.2.5 Cresyl violet staining
Following the last day of behavioural testing, animals were anesthetized with 5%
isoflurane and sacrificed for brain recovery. Fresh, non-perfused brains were removed from the
skull and stored at -80° C for histology. To assess electrode location, 20-µm thick coronal
sections covering the injury, were obtained using a cryostat (Leica biosystems) and mounted on
labelled glass slides (Fisher Scientific) which were then prepared for cresyl violet staining. For

34
tissue fixation, slides mounted with fresh tissue slices were placed in an incubating chamber
overnight, that was heated at 60°C, and containing a large dish with 500 ml 10% formalin, made
with 37% formaldehyde solution (Sigma Aldrich, cat#F8775). 0.2% cresyl violet stain was
prepared by mixing 2g cresyl violet acetate (Sigma Aldrich, cat#C5042) and 1000ml deionized
water, and filtered overnight. Four drops of glacial acetic acid (Sigma Aldrich, cat#695092) were
added to every 100ml of cresyl violet stain used for the fixed slides.
Within the next 24 hours, fixed slides were removed from incubation, and initially placed
into staining dishes containing deionized water for up to 3 minutes, to wash off any excess
formalin vapour. Subsequent steps involved dehydrating the fixed tissue in incremental
concentrations of ethanol solution (70%, 95%, 100% ethanol) and xylenes (MedStore, #534056),
followed by rehydration of tissue slices by placing the slides in a reversed sequence of the series
of ethanol concentrations (i.e., 100%, 95%, 70%) and deionized water, to allow more efficient
diffusion of the cresyl violet stain within the fresh tissue slices. After approximately 2-3 minutes
of staining, the slides were rinsed with deionized water, subsequently followed by further
dehydration in 70%, 95%, and 100% ethanol solutions and xylenes. Glacial acetic acid was
added to the 95% ethanol step to aid in cellular differentiation within each stained coronal tissue
slice. Finally, each slide was cover slipped using 50x24mm cover glasses (Fisher Scientific, #12-
548-5M) and Eukitt® quick-hardening mounting medium (Sigma Aldrich, cat#03989) and
allowed to dry overnight before viewing under a microscope to confirm the location of our DBS
electrodes.
2.3. Neurochemical experiments
A new cohort of animals (n = 23) prepared for neurochemistry, underwent electrode
implantation/control surgery along with TBI protocols, as described above. Twenty-four hours
following TBI (and subsequent acute stimulation for DBS-TBI groups), DBS/sham DBS animals
were anesthetized with 5% isoflurane and sacrificed through decapitation. Brains were removed
from the skull, divided in two hemispheres, and stored at -80°C for neurochemical analyses. TBI
exposed animals had one TBI and one Non-TBI hemisphere with no electrodes implanted. Sham
DBS-TBI exposed animals had bilateral electrodes implanted but received no stimulation in one
TBI and one Non-TBI hemisphere. DBS-TBI exposed animals had bilateral electrodes implanted
and received stimulation in one TBI and one non-TBI hemisphere.

35
2.3.1. Caspase-3 Enzyme-linked immunosorbent assay (ELISA)
Tissue samples were prepared by first dissecting out the hippocampus and the pericortical
regions from both, the ipsilateral (TBI-induced) and the contralateral hemispheres of the rat brain
using the 1.5mm and 1.0mm WPI biopsy punch tools (#WPKP1018) respectively. The
hippocampus dissected was the region directly below the TBI-induced cortex. The pericortical
region chosen was 1.0mm medial, lateral, anterior and posterior of the TBI-induced cortex. The
dissected tissue was collected in a 2.0mL pre-weighed SARSTED cryotube and homogenized in
250μL of RIPA lysis buffer that included one protease-inhibitor cocktail tablet (ROCHE) per
10mL of buffer solution. Lysates were then sonicated once for 15 sec and centrifuged at 11,000
G for 15 min at 4°C to remove cell debris. The supernatant was collected in a separate tube and
stored at -80°C until further processing.
Total protein concentration was quantified using the bicinchoninic acid assay (BCA,
Thermo Scientific, USA). Rat Casp-3 ELISA kits were purchased from Biomatik (#EKC40528,
Cambridge, Ontario) to measure caspase-3 protein levels in the separate frozen sample aliquots
from each subject. This assay employs a quantitative sandwich enzyme immunoassay technique
that quantifies the target antigen between two layers of antibodies using a pre-coated 96-well
microplate provided by the manufacturer.
Standard and samples (in duplicates) were added to the appropriate microplate wells that
were pre-coated with antibody specific to Casp-3. Any Casp-3 present would bind to the
immobilized antibody. After removing any unbound substances, a biotin-conjugated antibody
specific to Casp-3 was added to all samples, followed by multiple washings, and the addition of
avidin conjugated Horseradish Peroxidase (HRP). Following a wash to remove any unbound
avidin-enzyme reagent, a substrate solution was added to the wells that would result in colour
development in proportion to the amount of Casp-3 bound in the initial step. The colour
development was terminated by addition of sulphuric acid solution and the intensity of the colour
was measured spectrophotometrically at a wavelength of 450nm, with a correction wavelength
set to 570nm. The plates were read on a Synergy H1 microplate reader (Biotek, Winooski,
Vermont, US) and a four-parameter logistic regression curve was used for plotting the standard
curve and data extrapolation. The concentration of Casp-3 in the samples was determined by

36
comparing the optical density of the samples to the standard curve. The coefficient of variance
among the duplicates was less than 15%.
2.4. Statistical Analyses
One-way ANOVA was performed for the comparison of behavioural data in
experiments 1 (naïve, sham-TBI and TBI groups) and 2 (TBI, Sham DBS-TBI and TBI groups),
followed by Tukey’s post-hoc analysis for multiple comparisons. Caspase-3 results were
analyzed with 2-way ANOVA with TBI (TBI, non- TBI) and DBS (no electrode, sham DBS,
DBS) as independent factors. All statistical analyses were completed using Prism 5.0 for
windows (GraphPad Software, San Diego California). Statistical significance was set at p ≤
0.05. In the graphs, results are presented as mean ± standard error.

37
Chapter 3 – Results
3.1 Study 1: To characterize behavioural deficits in the moderate-to-severe TBI
fluid percussion injury (FPI) model in rats
We first conducted experiments to validate our TBI model and behavioural tests. Three
groups were considered: TBI (n=10), Sham-TBI (n=12), and naïve animals not exposed to TBI
(n=13). Our hypothesis was that the group of animals exposed to TBI would present increased
anxiety in the marble burying test and memory deficits in the NOR/NLR and Barnes maze tests.
All rats fed normally, gained weight, and did not present any gross locomotion abnormalities
after lateral FPI.
In the defensive burying test, no significant differences were found across groups (F=1.52;
p=0.23), though animals exposed to sham-TBI and TBI had a 25 and a 32% reduction in the
number of buried marbles compared to the naïve group, respectively (Figure 7).

38
Figure 7: Defensive marble burying test for TBI, sham-TBI (craniotomy, no FPI) and naïve
(no surgical procedure) groups. Animals do not show a significant difference in performance
based on number of marbles buried across groups.
In the novel object recognition test, we found a significant effect for percent time spent
with the novel object (F=3.3, p=0.05) and a trend towards significance for the index to explore
the novel object (F=3.0, p=0.06). Compared to the naïve group, the percent time exploring the
novel object was 21% lower in sham-TBI rats (p=0.04) and 17% lower after TBI exposure.
Similarly, the index exploring the novel object was 63% lower in the sham-TBI group and 50%
lower in TBI exposed animals compared to the naïve group (Figure 8A, B).
Naiv
e
Sham T
BI
TBI
0
2
4
6
8
Marble Burying
Bu
ried
Marb
les

39
Figure 8: Novel object recognition (NOR) in TBI, sham-TBI and naïve groups. The animals’
preference for the novel vs. familiar object was determined by the % time spent with the novel
object (A) and the discriminatory index (B), which utilizes the differences in exploration time for
the familiar object, divided by the total exploration time of the novel and familiar objects.
Rodents have a spontaneous tendency to explore novel objects, and the general trend shows that
TBI animals have less preference for the novel object compared to naïve animals. Significant
differences denoted by ‘*’. One-way ANOVA was applied to compare behavioural data,
followed by Tukey’s post-hoc. Lines over the bars indicate groups in which differences were
found to be significant.
As for the novel location recognition test, we found a trend towards significance for the
percent time spent in the novel location (F=2.6, p=0.08), and significant differences across
groups in the novel location index (F=3.78, p=0.03). Compared to naïve rats, the percent time
exploring the novel location was 12% and 31% lower in sham-TBI and TBI exposed groups,
respectively. The index exploring the novel location was 64% lower in sham-TBI rats and 145%
lower in TBI exposed animals (p=0.03) compared to naïve controls (Figure 9A, B).

40
Figure 9: Memory deficits observed in TBI animals compared to other groups in the novel
location recognition (NLR) test. Like NOR, the animals’ preference for the newly located
object vs. familiar location was determined by the % time spent in the novel location (A) and the
discriminatory index (B), which utilizes the differences in exploration time for the familiar
location, divided by the total amount of exploration of the novel and familiar locations. Values
≥1 demonstrate a preference for the novel location, while a negative value ≤ -1 shows preference
for the old/familiar location. The innate tendency for rodents to prefer exploring and recognizing
a novelly-located object more than the familiar object is equated to better performance in spatial
memory and cognition179. Significant differences denoted by ‘*’. One-way ANOVA was applied
to compare behavioural data, followed by Tukey’s post-hoc. Lines over the bars indicate groups
in which differences were found to be significant.
Barnes maze was conducted for two days, a shorter version of the 4 days test usually used
in preclinical work. As described in the methods section, undertraining was explicitly desired in
our experiments so that TBI animals would present a more striking deficit that could be rescued
by DBS in the second aim of our thesis. In the first day of training (average of two sessions), no

41
significant differences were observed in the most commonly scored Barnes maze variables
across groups, that is the total latency period (F=0.54; p=0.60) and error count (F=1.46; p= 0.25).
Most importantly, during the day of testing naïve animals had a decrease in the time to locate and
enter the escape route, error count, latency to the first nose poke, total latency and an increase in
the search strategy compared to the training sessions (Figure 10). This suggested that animals
learned the task and performed as expected. In contrast, animals exposed to TBI had a worse
performance in every variable scored, suggesting a learning deficit with the established
conditions (Figure 11). The Sham-TBI group performed in between naïve and TBI exposed
animals, but never reached the deficits observed in the latter group.
In the testing day, significant differences across groups were found for the time to locate
the escape route (F=9.45, p=0.0006), time to enter the escape route (F=8.34, p=0.001), primary
latency to the first nose poke (F=10.08, p=0.0004), and the total latency (F=8.56, p=0.01). A
trend towards significance was found for the error count (F=3.05, p=0.06), while no-significant
differences were observed for search strategy (F=1.56, p=0.22). Figure 11 and Table 2 show that
both TBI and Sham-TBI exposed rats had a worse performance than naïve animals in almost all
variables. In most measures, scores in TBI animals were worse than in Sham-TBI rats.
Table 3: Barnes maze results in Aim 1- statistical data comparing Naïve, Sham-TBI and TBI
exposed animals.
Variable ANOVA Naïve vs Sham-TBI
Naïve vs TBI
TBI vs Sham-TBI
Time to locate the escape route
F=9.45, p=0.0006* p=0.26 p=0.0004* p=0.02*
Time to enter the escape route F=8.34, p=0.001* p=0.02* p=0.001* p=0.44
Error count F=3.05, p=0.06 p=0.51 p=0.05* p=0.36
Latency to the first nose poke F=10.08, p=0.0004* p=0.23 p=0.0003* p=0.02*
Total latency F=8.56, p=0.01* p=0.02* p=0.001* p=0.44
Search strategy F=1.56, p=0.22 p=0.92 p=0.22 p=0.39

42
Figure 10: Barnes maze data for TBI treatment and control groups displaying changes in
performance across all variables, between train and test days. TBI animals tend to make
more errors and take longer durations to explore and locate the escape box, compared to naïve
animals. Search strategies were scored on a scale of 0-1 where values closer to 0 = more random
exploration, while values closer to 1 = more direct navigation to escape the maze.
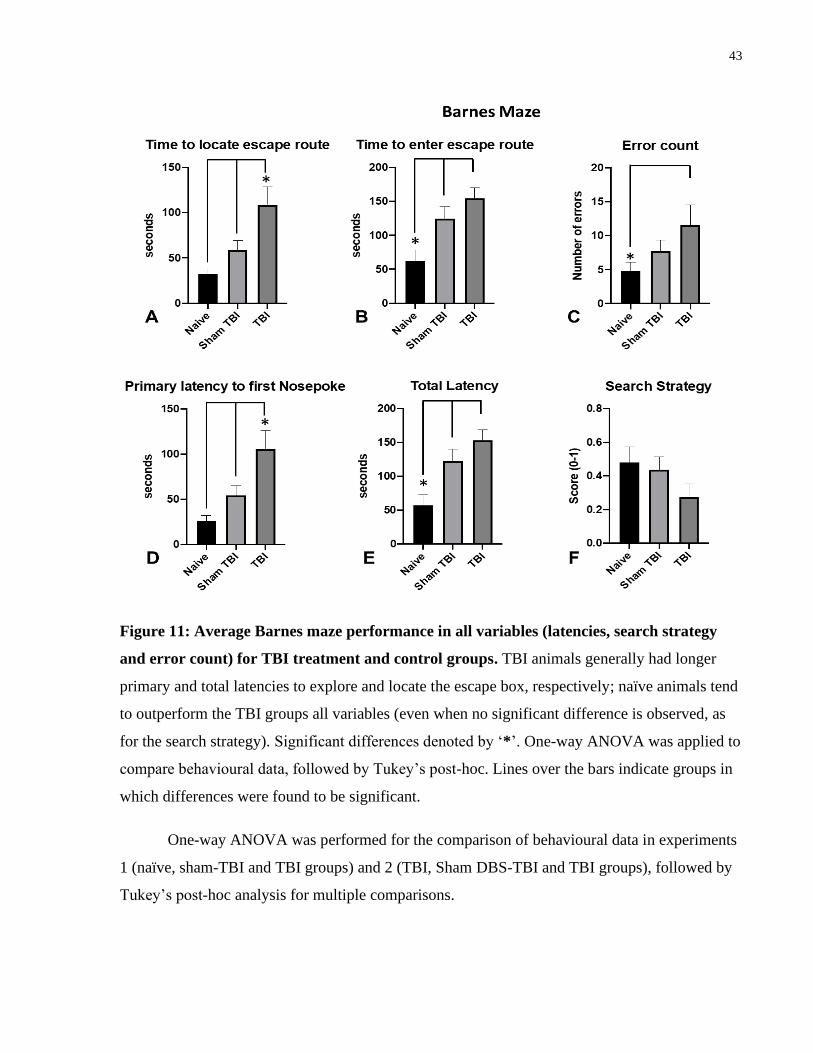
43
Figure 11: Average Barnes maze performance in all variables (latencies, search strategy
and error count) for TBI treatment and control groups. TBI animals generally had longer
primary and total latencies to explore and locate the escape box, respectively; naïve animals tend
to outperform the TBI groups all variables (even when no significant difference is observed, as
for the search strategy). Significant differences denoted by ‘*’. One-way ANOVA was applied to
compare behavioural data, followed by Tukey’s post-hoc. Lines over the bars indicate groups in
which differences were found to be significant.
One-way ANOVA was performed for the comparison of behavioural data in experiments
1 (naïve, sham-TBI and TBI groups) and 2 (TBI, Sham DBS-TBI and TBI groups), followed by
Tukey’s post-hoc analysis for multiple comparisons.

44
Figure 12: Rat brains recovered 24hr post-Barnes maze testing. Contusion on TBI (right)
animals observed where fluid percussion injury (FPI) was exerted on the right hemisphere, in
comparison to Sham-TBI (left).
3.2 Study 2: To investigate the effects of acute ANT-DBS in the FPI model
through a battery of behavioural tests associated with learning, memory and
anxiety-like behaviour.
After defining TBI-induced memory deficits, we have treated groups of TBI exposed rats
with DBS (n=10), Sham-DBS (n=9), or control surgery (no electrodes implanted; n=13). All
animals in the DBS and Sham-DBS groups had electrodes implanted within the ANT.
No significant differences were found across groups in the defensive burying test (F=0.79,
p=0.46; Figure 13). Similarly, no significant differences across groups were observed for both
the percent time (F=2.50, p=0.1) and index (F=2.50, p=0.1) in the novel object recognition test
(Figure 14A, B). Differences across groups were also not found to be significant for the percent
time (F=0.38, p=0.69) or index (F=0.38, p=0.69) in the novel location recognition test (Figure
15A, B).

45
Figure 13: Average number of buried marbles in the DBS treatment group demonstrate no
significant improvement in anxiety type behaviour, compared to sham DBS-TBI, and TBI
only groups. Animals did not show a significant difference in performance based on number of
marbles buried across groups after acute ANT-DBS. This suggests that acute ANT-DBS does not
improve anxiety-type behaviour in animals with moderate-to-severe TBI. One-way ANOVA was
applied to compare behavioural data, followed by Tukey’s post-hoc.
TBI
Sham D
BS T
BI
DBS T
BI
0
2
4
6
Marble Burying
Bu
ried
Mar
ble
s

46
Figure 14: NOR performance in DBS-TBI groups demonstrate no significant difference in novel
object exploration (A) or preference (B). This suggests that acute stimulation of the ANT has no
significant therpaeutic effect on non-spatial or working memory following moderate-to-severe
TBI. One-way ANOVA was applied to compare behavioural data, followed by Tukey’s post-
hoc.

47
Figure 15: NLR performance in DBS-TBI groups demonstrate no significant difference in novel
location exploration (A) or preference (B), in comparison to sham DBS-TBI, or TBI only groups.
One-way ANOVA was applied to compare behavioural data, followed by Tukey’s post-hoc.
In the Barnes maze, no significant differences were observed in the first day of training
for the total latency period (F=2.57; p=0.09) and error count (F=0.53; p= 0.59). Similar to
experiments in aim 1, TBI-exposed rats had worse or unaltered memory performance in every
variable scored compared in testing sessions, relative to the training sessions, suggesting a
learning deficit in this group of animals (Figure 16). TBI-exposed animals given DBS had a
decrease in the time to locate and enter the escape route, error count, latency to the first nose
poke, total latency and an increase in the search strategy compared to the training sessions,
suggesting an expected pattern of learning. Sham DBS-TBI animals implanted with electrodes
that received no DBS showed signs of memory improvement compared to TBI, though less
pronounced than those recorded in the DBS-TBI group.

48
Figure 16: Barnes maze data for DBS-TBI, Sham DBS-TBI and TBI groups displaying
changes in performance across all variables, between train and test days. TBI animals
tended to make more errors and take longer durations to explore and locate the escape box, while
TBI animals receiving acute ANT-DBS had an improvement in performance by making fewer
errors, with a shorter duration in exploring and escaping the maze, which suggest an
improvement in spatial memory in moderate-to-severe TBI.
DBS was largely found to improve memory performance in the Barnes maze. Significant
differences across groups were noted in the time to enter the escape route (F=8.54, p=0.001),
error count (F=3.97, p=0.03), total latency (F=8.75, p=0.001), and search strategy (F=5.75,
p=0.008). In contrast, results were not found to be significant for the time to locate the escape
route (F=2.43, p=0.11) and the latency to first nose-poke (F=2.44, p=0.10). As can be
appreciated in Figure 17 and Table 3, we found significant improvements in TBI rats given DBS
compared to those exposed to TBI alone. In some variables (e.g., time to enter the escape route,
total latency), TBI animals given DBS also fared better than those receiving sham stimulation. In
others (time to locate the escape route, latency to the first nose poke), sham stimulation exerted a
modest effect, with a mild improvement in memory performance. Finally, the mere insertion of
electrodes in the absence of stimulation was found to be part of the therapy, as observed in the

49
number of errors and search strategy. When these variables were considered, the magnitude of
stimulation effects was similar to that of non-stimulated animals, suggesting that electrode
insertion played a role in the therapeutic response of DBS.
Figure 17: Average Barnes maze performance across all variables (latencies, search
strategy and error count) in DBS-TBI, Sham DBS-TBI and TBI only groups. In general,
DBS animals demonstrated an improvement in performance across all variables measured,
suggesting that ANT-DBS may facilitate recovery from spatial memory deficits. Significant
differences denoted by ‘*’. One-way ANOVA was applied to compare behavioural data,
followed by Tukey’s post-hoc. Lines over the bars indicate groups in which differences were
found to be significant.

50
Table 4: Barnes maze results in Aim 2 - statistical data comparing controls (TBI Ctl), Sham
DBS and DBS-treated animals, all of which were exposed to TBI.
Variable ANOVA TBI Ctl vs TBI Sham DBS
TBI Ctl vs TBI DBS
TBI Sham DBS vs TBI DBS
Time to locate the escape route
F=2.43, p=0.11 p=0.79 p=0.09 p=0.36
Time to enter the escape route F=8.54, p=0.001* p=0.97 p=0.002* p=0.007*
Error count F=3.97, p=0.03* p=0.07 p=0.05* p=0.99
Latency to the first nose poke F=2.44, p=0.10 p=0.84 p=0.09 p=0.33
Total latency F=8.75, p=0.001* p=0.99 p=0.002* p=0.005*
Search strategy F=5.75, p=0.008* p=0.04* p=0.01* p=0.93
Additionally, histological sections stained with cresyl violet revealed that electrodes in DBS and
sham treated animals were all within the boundaries of the anterior thalamic nucleus (Figure 18).

51
Figure 18: Electrode location. Schematic representation of atlas plates showing the tip of the
electrodes in TBI animals undergoing DBS (black circles; n=10) or sham-treatment (grey circles;
n=9). Note that electrodes were located within the boundaries of the anterior nuclear complex.
While electrodes were bilaterally implanted, they were represented unilaterally in this schematic
figure. Numbers below the plates denote distance from bregma. Plates adapted from Paxinos and
Watson’s rat brain atlas178.
3.3 Study 3: To test whether ANT-DBS reduces hippocampal apoptosis in FPI-
exposed rats.
Once we have defined that DBS induced significant memory improvements in TBI
exposed animals, we prepared new batches of rats for neurochemical analysis. Animals

52
undergoing TBI (n=8), DBS-TBI (n=8) or Sham DBS-TBI (n=7) were sacrificed 24 hours after
lateral FPI, and their brains processed with ELISA for the measurement of caspase-3 in the
perilesional cortex and dorsal hippocampus (Figure 19). In the perilesional cortex, 2-way
ANOVA revealed no significant TBI effect (F(1,40) = 0.18; p=0.67), DBS effect (F(2,40) =
0.39; p= 0.68) or TBI vs DBS interaction (F(2,40) = 0.02; p=0.98). Similarly, 2-way ANOVA
revealed no significant TBI effect (F(1,40) = 0.11; p=0.73), DBS effect (F(2,40) = 0.39; p= 0.68)
or TBI vs DBS interaction (F(2,40) = 0.01; p=0.99) in the hippocampus beneath the lesion.
Figure 19: Caspase-3 measurements obtained using an enzyme immunosorbent assay
(ELISA) for TBI and non-TBI hemispheres. Animals were sacrificed 24hrs after their
respective interventions. Levels of caspase-3 were measured relative to protein content in the
hippocampus and perilesional cortex (tissue samples obtained within 1.0mm radius of TBI-
induced cortex). We report no significant differences across groups, notably, no substantial
increase in caspase levels within TBI animals. 2-way ANOVA applied with TBI (TBI, non- TBI)
and DBS (no electrode, sham DBS, DBS) as independent factors.

53
Chapter 4 – Discussion
The overall outcome of our paradigm generated a reduction in memory deficits for
animals with DBS compared to non-stimulated treatment groups, when tested with the Barnes
maze, on most behavioural metrics. However, DBS groups had not gained a significant change in
behaviour, or memory deficits when taking results of the NOR/NLR or the defensive marble
burying test into account. Marble burying performances also led to non-significant differences
between sham-TBI and TBI groups (i.e., 25% sham-TBI vs. 32% TBI reduction in marble
burying). In both novel object and novel location tests, TBI animals demonstrated worse
performance compared to naïve animals, as indicated by their exploration time and
discrimination of the novel stimuli. However, behavioural deficits in sham-TBI animals were
comparable to TBI groups (i.e., 21% sham-TBI vs. 17% TBI in NOR) during NOR, which may
likely be attributed to surgical methodology, such as heat induced while drilling a sham
craniotomy or the volume of anesthesia implemented. Despite observing similar results in novel
object discrimination and exploration for both sham-TBI and TBI animals, the latter fared worse
in performance overall.
Another aspect of our paradigm to note is the undertraining protocol for Barnes maze,
which had involved two days in total. In a pilot study, we had employed a standard Barnes maze
protocol, consisting of a habituation phase 24 hours prior to training to reduce the likelihood of
anxiety behaviour185, and allow the animals to habituate to the maze as well as the escape box.
This was followed by a 3-day acquisition/training phase (2 trials, each 180s per animal). Finally,
an acquisition probe phase was implemented 48 hours after the last training session to assess
memory retention, ascertain the search strategies of the animals and whether or not distal cues
were also involved186. The different stages of Barnes maze phases evaluate spatial reference
memory, working memory, retrieval of memory, and in some cases, (where a reversal learning
phase is included) cognitive flexibility182,186,187. Training and probe trials evaluate spatial
learning and memory, and hippocampal functioning182,185, while cognitive flexibility associated
with reversal learning, involves the frontal cortex188. However, during our pilot studies involving
the TBI treatment and naïve control groups, the standard Barnes maze protocol did not present
any notable deficits in behaviour or spatial memory for TBI animals compared to naïve groups,

54
after the first 1-2 days of acquisition. Therefore, we had modified the standard protocol, such that
the animals had undergone 2 training trials in total, over the span of 1 day, followed by testing on
the next day. This is similar to previous studies in different preclinical models. For example, one
study by Attar et al. (2013) assessing spatial memory in a strain of AD mice had reported
significant cognitive deficits during the probe trial, when a similar, shorter (5 training trials)
Barnes maze paradigm was implemented, in comparison to a longer protocol189. By doing so,
they had successfully evaluated spatial memory in a more difficult, yet cost-effective approach
within AD transgenic mice189. Undertraining was also used as a strategy to show the behavioural
effects of entorhinal cortex DBS on memory performance in rodents exposed to the Morris water
maze190.
Our decision of undertraining for Barnes maze demonstrated significant differences
across groups for both primary and total latencies, as well as the number of errors made prior to
locating the escape box. Therefore, we believed that this variant of the Barnes maze protocol best
fitted our experimental aims. Furthermore, unlike a spatial reference memory and learning test
like the Morris water maze, the Barnes maze allows for memory and cognitive assessment
without the added anxiety associated with its aquatic aspect189. However, the Barnes maze still
consists of modest aversive stimuli, which makes this test relatively less motivating compared to
the Morris water maze185. In our case, we found that animals demonstrated high exploration,
where, despite knowing the location of the escape box, they would not reach or enter to safety as
quickly as during the initial trials. This prolonged exploring resulted in an increase in primary
latency times, which is not uncommon for this behavioural test185. Additional weak aversive
stimuli were also introduced to our paradigm (e.g. overhead bright light source, high speed fan),
so that the animals respond to their innate agoraphobia191 and are compelled to reach the escape
box without additional delays. Conversely, taking caution in implementing too many aversive
stimuli in the Barnes maze is important, as they can potentially distract animals from focusing on
their task of seeking immediate shelter, but acclimatizing to the environment would reduce this
confounding variable185. O’ Leary & Brown (2012)192 had concluded that the overall learning
and memory performance in rodents was not affected by the number of habituation/shaping
trials, but the authors had recommended a habituation phase when there is no knowledge of any
motor deficiencies or anxiety level in our test subjects192,193. We had extensively handled the rats
prior to testing and motor abilities were also assessed to reduce the effects of confounding

55
anxiety levels or motor deficits. Overall, TBI, followed by sham-TBI animals consistently
performed worse than naïve animals.
Our TBI model was intended to replicate a scenario in which stimulation would be
delivered to patients affected by moderate-to-severe TBI shortly after the impact. In addition to
our selected TBI model, our modified behavioural protocol was more likely to reveal significant
deficits in learning and memory. Subsequently, our main goal was to rescue any possible
memory deficits with acute DBS, hypothesized to occur by providing some degree of
neuroprotection within a short recovery window, as examined in Aim 2. Stimulation parameters
for DBS in our animal model were derived from work in animal models of epilepsy107 and
approximated to reflect clinically relevant settings meant for therapeutic intervention in humans.
4.1. ANT-DBS
Reports of DBS of subcortical targets are far and few between, especially in the case of
TBI studies160. Direct structural and secondary damage from TBI that follows the initial insult
and nearby distal regions, have previously been targeted with DBS to alleviate disorders of
consciousness160. In particular, thalamic nuclei and associated white matter tracts necessary for
attention and arousal are targeted in patients with moderate-to-severe TBI; however, because of
the heterogeneity of structural and functional damage caused by TBI, patients are also likely to
develop comorbidities of movement disorders, depression and memory deficits160. Studies testing
for memory and cognitive function in TBI models or patients are limited, but cumulative
findings all have directed to components of the Papez circuit, anterior thalamus and frontal
cortical regions as primary targets for attenuating memory deficits and improving higher
executive function in the TBI subpopulation160
This study was designed to evaluate the efficacy of DBS in the anterior nucleus of the
thalamus, testing whether reductions in anxiety-like behaviour and recovery from memory
deficits could be observed. Known for its significance in learning and memory processes, the
ANT is a key component of the limbic circuit of Papez194. Specific subnuclei of the ANT make
up three parallel hippocampal-ANT circuits195,196; crucial for relaying emotional and visceral
information to the pre-frontal cortex, thus being involved in higher executive function and
cognition (anteromedial; AM)197; firing rhythmic theta activity to the hippocampal formation, in
a return-loop system (anteroventral; AV)197; and finally, the anterodorsal nucleus (AD), which is

56
involved in both spatial navigation (via head direction cells198) and non-spatial forms of
memory195,196. Several behavioural experiments in animals have alluded to performance
disruption in a wide range of spatial learning tasks, when lesioning the ANT195,199,208,200–207,
particularly those involved in spatial alternation150,195, as well as disruption in temporal order
discrimination during non-spatial tasks209. In addition, clinical studies have previously
demonstrated a disturbance in mnemonic processes, contributing to diencephalic amnesia196.
Furthermore, while a series of reciprocal and interconnected brain regions support different types
of spatial learning207, lesions in the ANT may disrupt a range of fundamental body signals
necessary for effectively processing spatial information210.
During our behavioural paradigm following acute DBS, the defensive marble burying
trials demonstrated no significant differences. The high variability across treatment groups may
have occurred due to chance, where some marbles may not be considered buried if the animal
had clearly stepped on them during exploration. Though defensive burying paradigms have been
proposed to measure anxiety-type responses, we note this is an innate behaviour, where rodents
tend to bury objects that are unfamiliar or may be perceived as dangerous. That said, defensive
burying is well validated and routinely used. As no changes in buried marbles were found
between TBI and sham-TBI groups, it is not unexpected that ANT-DBS did not rescue this
behaviour. With negative data, however, whether ANT-DBS delivered immediately after TBI
improves anxiety-type behaviour remains elusive.
Treatment and control groups during both novel object and novel location recognition
tests garnered no significant differences in both percent time spent with novel stimuli, and
discrimination between familiar and novel stimuli. Readily-learned spatial alternation tests (e.g.
T-maze, radial arm maze) are known to be particularly sensitive to ANT lesioning in
rodents183,206; however, not only is working memory affected by ANT lesions, but tasks
involving the need for reference memory are also affected (e.g. finding the location of the escape
box on the Barnes maze, or locating the submerged platform of a Morris water maze (e.g. Wolff
et al. (2008)211). Deficits in place learning were also found following ANT lesions where, for
instance, behavioural tasks with rats placed in a Morris water maze arena show an increase in
latencies to locate the hidden escape platform, as well as greater inaccuracies in search strategy
on removal of the escape platform211,212. In addition, proactive interference and temporal
discrimination are likely to be impaired and play a role in alternation deficits, as demonstrated by

57
behavioural studies where objects or odours are presented in a continuous sequence of
information183,209, suggesting that spatial/non-spatial tasks with a highly demanding memory
requirement would be challenging for rats with ANT lesions. As theta activity in the AV nucleus
contributes to the processing and distinction of temporal information via synchronous oscillation
of AV neurons with hippocampal theta rhythms, lesions in the ANT could interfere with this
synchronous activity183,213. Consequently, this theta disruption would affect a rodent’s ability to
temporally organize consecutive stimuli on exposure to a continuous stream of information
during memory tasks. However, when testing for memory during object-recognition tasks in
discretely parsed trials, ANT lesion studies have demonstrated no direct effect on the ability to
recognize a novel stimulus in between-block trials205,210. Furthermore, significant damage to the
hippocampus in rats would suggest loss of both parsing/chunking of information, as well as
making finer temporal distinctions when presented with continuous sets of stimuli183. Overall,
complexities in spatial alternation deficits affiliated with ANT damage can be explained by one
or both of the following factors: (i) Damage to the three major nuclei of ANT would lead to
impairments in spatial processing, as these nuclei comprise a diverse range of spatial functions;
and (ii) unintentional damage to sites adjacent or relatively close to the ANT during surgical
procedures may disrupt neural plasticity and activity, thus amassing behavioural deficits in both
spatial and non-spatial memory210. Resultant combined lesions of all three nuclei would increase
severity of behavioural impairments during highly complex spatial learning tasks, compared to
confined lesioning of selective ANT subnuclei210,214. As moderate-to-severe TBI can be a
combination of both focal and diffuse injuries to the brain, it is not surprising to find that our
lateral FPI near the hippocampus would prompt deficits for animals in a wide range of spatial
and non-spatial behavioural tasks, with each deficit ascribed to one or several components of the
limbic circuitry. Like the Morris water maze, successful escaping of the Barnes maze also
involves effectively acquiring both navigational skills and spatial cues. While our protocol was
relatively shorter than the regular Barnes maze paradigm, consecutive and repetitive trials on
both days for TBI animals did not necessarily improve their latencies to locate the escape box.
Contrary to our findings in NOR and Marble burying, DBS had a significant impact on
TBI rats during the Barnes maze trials, particularly in the number of errors prior to entering the
escape box, total latency period and search strategy; however, primary latencies to locate the
escape box were not significant across groups. Of the subcomponents of the anterior thalamus,

58
AM is responsible for processing temporal information and response flexibility, AD provides
directional signals while AV allows for place learning and discrimination195. Animals with a
lesioned ANT may fail to achieve regular patterns of accuracy during behavioural tasks, as
observed during Barnes maze trials, compared to their naïve counterparts. DBS used at high, but
still clinically relevant stimulation current has led to a decrease in spontaneous neuronal firing,
as well as excitation of axonal projections around the electrodes placed at our target region19.
Conversely, the frequency range of stimulation may also have an effect on overall behaviour and
cognition, as suggested by theta frequency-dependent restorative effects on cognitive
performance of DBS in TBI rats215. Our Barnes maze data may suggest overall improvement in
memory following DBS in TBI rats; however, whether this was an immediate consequence of
stimulation on oscillatory activity, or changes in cell firing in the ANT or structures at a distance
from target remain to be demonstrated, as DBS was applied several days prior to behavioural
testing. Based on our previous findings of significant decrease in hippocampal caspase activity in
a rodent model of status epilepticus107, we hypothesized that one possible explanation for the
improvement in spatial memory performance observed after DBS could be that of a reduced
hippocampal apoptosis following TBI.
4.2. Caspase-3
A large family of proteins, known as caspases is the primary mediator for the regulated
destruction of cells, or apoptosis216. Initiation of apoptosis directs a sequence of biochemical
changes (e.g. nuclear fragmentation, cell shrinkage)217, gradually leading to programmed cell
death. Axonal injury following head trauma, neuronal and glial cell deaths all contribute to the
overall neuropathology of TBI, and these cells observed within contusions tend to display
apoptotic morphologies, within the first 24 hours and up to one week post-injury218. Furthermore,
apoptotic oligodendrocytes and neurons exhibiting nuclear DNA strand breaks have previously
been identified in situ using the terminal deoxynucleotidyltransferase-mediated dUTP nick end-
labelling (TUNEL) technique. These TUNEL+ cells thus confirmed the presence of apoptotic
neuronal cell death in human white matter tracts following TBI218. In addition, not only have
apoptotic and necrotic cells been detected in TBI models at the site of injury post-trauma, but
regions that are remote to the site of injury are also affected up to weeks following head
trauma.218

59
Caspase-3 is the most abundant cysteine protease in the brain and is crucial for DNA
fragmentation and chromatin condensation processes during programmed cell death218. It is also
an essential protein for brain development219,220. Numerous studies have consolidated neuronal
cell death as a common event occurring in neurological disorders, including ischemic
stroke117,220 and epilepsy107,221. An upregulation in caspase-3 had been reported in both clinical222
and pre-clinical CCI TBI studies223,224. The caspase-3 pathway contributes to the accumulation of
proteolytic activity leading to the production of cleaved substrates, such as isoforms of tau, a
notable biomarker of neurodegenerative disorders224. Conversely, inhibiting this caspase-3-
mediated pathway following moderate insults may be a potential therapeutic approach to
TBI220,224. In pre-clinical rodent models, an upregulation of caspase-3 activity is usually observed
24-72 hours following TBI, with maximal expression 7 days following the initial insult107.
Furthermore, one study has previously demonstrated chronic, prolonged upregulation of caspase-
3 for a least 3 months post-TBI224. DBS has been proposed to reduce neuronal apoptosis through
the upregulation of anti-apoptotic and down-regulation of pro-apoptotic genes and inflammatory
responses, thus providing a neuroprotective effect; however, finding a direct causal relationship
between DBS and the proposed neuroprotective processes is still poorly understood225,226.
Our current study measured caspase-3 levels from rat hippocampal and perilesional tissue
recovered 24 hours post-TBI, with treatment groups receiving acute ANT-DBS or sham
stimulation one week prior to experimental TBI. Our reasoning for this timeframe was to
determine whether we could observe a remarkable difference in caspase-3 activity, with acute
ANT-DBS leading to a significant reduction in caspase-3 levels compared to TBI-only groups, as
its neuroprotective effects could be facilitated. Our decision for collecting brain samples 24
hours post-TBI, as opposed to 3-7 days (optimal time interval to detect caspase-3 changes) was
based on previous work suggesting that at this shorter timeframe, caspase-3 concentrations are
several-fold higher compared to control brain tissue227. Moreover, the 24-hour window would
allow us to collect samples for measuring other substrates, including glutamate transporter
proteins, cytokines, and microglial markers to be analyzed in future studies. While caspase-3 is
altered at 24 hours post-TBI, longer time intervals might have not generated measurable
differences for the other substrates mentioned above. However, our findings demonstrated that
acute stimulation of ANT led to no significant DBS effects on caspase-3 activity within tissue
obtained after the 24-hour post-TBI period and compared to animals in control groups. There are

60
a few limitations to this study that should be noted: ELISA did not discriminate the cell types
responsible for the detection of caspase levels. We have only measured caspase 3 and not its
cleaved products. Given that caspase-3 and its cleaved bi-products have prolonged damaging
effects to neuronal cell survival beyond 24 hours post-TBI107,224, samples obtained at longer
timeframes (e.g. 72-hours post-TBI, 7 days post-TBI) for each group would likely have provided
more compelling evidence for neuroprotection by acute DBS. Future work remains to be
conducted measuring additional markers of apoptosis (TUNEL, electron microscopy studies) at
the optimal timeframe to detect the actual effects of ANT-DBS on apoptosis in TBI models.
4.3. Limitations and future perspectives
In our first study, we have investigated a reasonable number of animals but some of our
findings only yielded trends towards significance. Sham-TBI involves drilling the bone, which
may have generated heat damage in cortical areas, thus may have influenced behaviour. As TBI
rats were significantly different from naïve groups, we have decided to pursue experiments in
aim 2. Another potential control would have been animals exposed to anesthesia, opening of
superficial skin/subcutaneous planes but without bone drilling.
Whether acute ANT-DBS is sufficient in providing significant neuroprotective effects in a
clinical setting is still unclear, especially given that TBI can affect several brain regions, and lead
to long-term neuropathology that may not be as obvious shortly after the initial insult. One
criticism for our DBS-TBI model was the sequence in which we had performed the surgeries,
where implantation of DBS electrodes occurred prior to utilizing our FPI model, as opposed to
the converse sequence of events. Translation of this model into clinical settings may be
misleading, suggesting a pre-treatment paradigm, where TBI had occurred in a patient with
previously implanted electrodes. We had chosen our sequence of DBS implantation followed by
trauma, to reduce the likelihood of electrodes missing target regions during implantation, due to
heterogeneous anatomical distortion caused by lateral FPI. While TBI was exerted some distance
away from our DBS targets, the FPI device may cause diffuse injury, extensively disrupting the
anatomical position of target sites. Moreover, our arrangement of surgical procedures was
suitable for our behavioural paradigm. If the electrodes were implanted immediately after FPI,
the electrodes themselves would have a potential effect on the animals receiving stimulation,
during behavioural testing (e.g., insertional effect). Stereotaxic surgeries already require

61
prolonged anesthesia protocols to locate anatomical landmarks for implanting electrodes.
Consequently, to reduce prolonged sedation, post-anesthetic neurological impact on behaviour,
we chose not to anesthetize animals for extended periods228. Should we have chosen to stimulate
the animals with transiently implanted electrodes that could later be removed, animals would
have needed to be under anesthesia for approximately 7 hours (surgery + 6h of DBS). Another
limitation for our experiments is that we did not have sufficient data from the literature to
estimate an effect size and calculate statistical power before the study. This did not allow us to
determine the appropriate number of animals per treatment group prior to the study. All ‘n’
values were estimated based on previous work conducted in epilepsy animals (e.g., Amorim et
al, 2015107). As behavioral analyses were found to be significant, our sample size was sufficient
to demonstrate an improvement in memory performance after DBS.
In our model, acute stimulation was advantageous in isolating TBI effects on spatial
memory in behavioural testing. Chronic intermittent stimulation extending for several days or
weeks, typically used in clinical settings, may further elucidate neuroprotection via ANT-DBS.
Future pre-clinical studies may consider chronic DBS before determining its efficacy in
moderate-to-severe TBI. Previous clinical findings have suggested that long-term DBS in PD
patients demonstrated significant structural and global functional changes in connectivity and
subsequently have an influence on neuroplasticity229; this would likely add to the rationale of
chronic stimulation in moderate-to-severe TBI patients for longer sessions for its therapeutic
effects to apply. Unfortunately, highly variable, and conflicting results have been demonstrated
for DBS implemented on TBI, whether on a well-characterized rodent model, or in human
studies160. Case studies of utilizing electrical stimulation on memory tend to report conflicting
results, for which cautionary interpretation is required19. Rather than facilitating memory and
cognitive function, high-current ANT stimulation led to impairments in rats150; these memory
deficits were also consistent in epilepsy patients receiving ANT stimulation173. Epileptic activity
has been shown to increase aberrant adult hippocampal neurogenesis, of which the newborn
granular cells display abnormal morphology and irregular patterns of integration into
hippocampal circuits230; however, a low-current acute stimulation did not promote aberrant
neurogenesis190. While the ANT is a crucial part of the limbic circuit, it is also possible that acute
stimulation of another limbic region, such as the entorhinal cortex (EC) would have yielded a
different outcome. In contrast, Encinas et al. (2011)168 had demonstrated high frequency

62
stimulation of the ANT promoted adult neurogenesis within the subgranular zone, leading to
proliferation and survival of in vivo adult-generated granular cells of the dentate gyrus (DGC);
however, at the time, its effect on memory and cognitive function was yet to be determined. In
addition, Stone et al. (2011)190 for instance, demonstrated acute DBS supported the proliferation,
differentiation, survival and maturation of new adult-generated DGCs, while subsequently
integrating and contributing to functions of hippocampal circuitry. This was confirmed by the
formation of spatial memory in mice during a Morris water maze task within 6 weeks of EC
bilateral acute stimulation190. Conversely, the authors also noted that facilitating spatial memory
formation occurred several weeks after DBS, as opposed to treatment groups receiving
stimulation within 1 week before or after behavioural training190. This finding may inform our
own behavioural testing results, particularly in the variability of primary latencies across groups
during Barnes maze trials. Although our behavioural data suggest an improvement in spatial
memory in DBS groups acutely stimulated ~ 2 weeks before training, it does not clarify whether
acute stimulation preceding the behavioural task by several weeks would have maximized the
functional integration of new DGCs, thus establishing activity-dependent hippocampal
connectivity. Training an additional group of DBS-TBI animals in a similar behavioural
paradigm, albeit occurring after a prolonged period following acute stimulation (e.g., 5-6 weeks
post-acute DBS), may have provided further information on hippocampal-dependent spatial
memory following Barnes maze. Moreover, detailed analyses of primary path lengths before the
animal locates the escape box may have provided additional evidence of whether a higher
frequency of spatially precise search strategies were being used by stimulated rats, suggesting
improved spatial memory. Further investigation of the neurocircuitry modulated by DBS
(whether acute or chronic) is likely necessary, for example via immediate early genes, as well as
additional imaging (e.g., through retroviral labelling of newly generated adult neurons;
fluorescent anterograde tracing).
In our neurochemical study, only caspase-3 levels were measured. Our decision for
quantifying capase-3 activity using ELISA, as opposed to opting for other assays (e.g., western
blot, immunohistochemistry) was made due to its high sensitivity and reliability231 in detecting
rodent caspase-3 in a mixed cell population within homogenized brain tissue. It is important to
note that all caspase-3 sample concentrations were well within the detection range of our kit (i.e.,
0.312 ng/ml-20 ng/ml). We were disappointed to find no notable difference in measurements of

63
caspase-3 in brain regions affected by TBI, in relation to total protein concentration in our
homogenized brain tissue, and compared to control samples (i.e., tissue from hemisphere
contralateral to TBI region). Despite the high sensitivity rate (i.e., 0.078 ng/ml) of our ELISA kit,
as well as accuracy within the affiliated standard curve, future studies using other assays more
suitable for measuring apoptotic activity, should be considered. Other measurements of apoptotic
markers using the TUNEL assay, followed by electron microscopy could yield different results;
however, cautionary measurement of neurons exhibiting caspase-3 activation using TUNEL is
recommended. This is due to its non-specific aspect, in that TUNEL will label all forms of DNA
fragments with free 3’-hydroxyl termini, regardless of what molecular mechanisms led to these
free termini232. Hence, careful interpretation is required when differentiating apoptotic cells from
non-apoptotic cells (e.g., necrotic cells, cells undergoing gene transcription or DNA repair).
Another limitation is that we did not quantify cleaved caspase-3, a well-studied biomarker of
apoptosis and a known effector of caspase-mediated pathways. This is due to notable factors,
including the lack of reliable and widely available ELISA kits for measuring rodent-specific
cleaved caspase-3. Furthermore, significant upregulation of cleaved caspase-3-immunopositive
cells was previously observed at chronic time intervals, beginning from one month after
experimental TBI was performed in rats, persisting for up to 3 months after TBI233. Given that
our timeline of experimentation occurred within acute timepoints (i.e., caspase-3 measurement
24h post-FPI), it is likely that we would not detect measurable concentrations of cleaved
caspase-3 in our paradigm. In conjunction with chronic stimulation, performing
immunohistochemical analyses of coronal brain sections affected by TBI, using cleaved caspase-
3 specific antibodies may render further insights of whether DBS confers neuroprotection in
moderate-to-severe TBI models. Necrotic processes may contribute to changes in caspase levels
within 24 hours post-TBI227. Furthermore, we have yet to measure concentrations of other
contributing substrates (e.g., cytokines, microglial markers, glutamate transporter proteins) to
neuroinflammation. The neurotrophic factor BDNF is known to be highly active in regions of the
hippocampus, and functions in growth and differentiation of newly developed synapses and
neurons, as well as supporting existing neurons. Survival of hippocampal cells following TBI
may be facilitated by neuroprotection induced by BDNF, through the active suppression of
caspase-3 expression234. DBS has previously been shown to augment levels of BDNF235,236, in
regions such as the pedunculopontine tegmental nucleus (PPTg-DBS), with the hippocampus
also involved in neuroprotection following rat PPTg-DBS234. BDNF may also contribute to anti-

64
anhedonic-like effects following chronic stimulation of the ventromedial prefrontal cortex237.
Thus, it is possible that increases in trophic factors rather than a reduction in neuroinflammation
are involved in DBS effects on improving memory.
Finally, whether ANT-DBS follows a similar mechanism of increased BDNF levels to
inhibit caspase-3 activity is yet to be investigated. Quantitative measurements of BDNF may be
considered in parallel to caspase-3 activity, which could potentially provide a coherent
understanding of how effectively neuroprotection is facilitated by acute DBS in TBI rats.
4.4. Conclusions
In summary, this thesis presents validation of a moderate-to-severe TBI rodent model,
which demonstrated impaired cognition and memory, within which acute ANT-DBS stimulation
improved some aspects of memory, in particular, spatial navigation in TBI rats. Our findings
remain inconclusive for determining mechanism of memory improvement from moderate-to-
severe TBI using acute ANT-DBS. Although further evidence of neuroprotective properties of
DBS (whether we consider acute or chronic stimulation) may be resolved from investigating
additional neuroinflammatory markers, findings of this thesis reiterate that neuromodulation of
the ANT may improve deficits in memory and cognition, when implemented shortly after
moderate-to-severe TBI.

65
References
1. Xiong, Y., Mahmood, A. & Chopp, M. Animal models of traumatic brain injury. Nat. Rev.
Neurosci. 14, 128–142 (2013).
2. Albert-Weissenberger, C. & Sirén, A. L. Experimental traumatic brain injury. Exp. Transl.
Stroke Med. 2, (2010).
3. Dewan, M. C. et al. Estimating the global incidence of traumatic brain injury. J.
Neurosurg. 130, 1080–1097 (2019).
4. Hyder, A. A., Wunderlich, C. A., Puvanachandra, P., Gururaj, G. & Kobusingye, O. C.
The impact of traumatic brain injuries: A global perspective. NeuroRehabilitation 22,
341–353 (2007).
5. Fu, T. S., Jing, R., McFaull, S. R. & Cusimano, M. D. Recent trends in hospitalization and
in-hospital mortality associated with traumatic brain injury in Canada: A nationwide,
population-based study. J. Trauma Acute Care Surg. 79, 449–455 (2015).
6. Ma, X., Aravind, A., Pfister, B. J., Chandra, N. & Haorah, J. Animal models of traumatic
brain injury and assessment of injury severity. Mol. Neurobiol. 56, 5332–5345 (2019).
7. Fleminger, S., Oliver, D. L., Lovestone, S., Rabe-Hesketh, S. & Giora, A. Head injury as a
risk factor for Alzheimer’s disease: The evidence 10 years on; a partial replication. J.
Neurol. Neurosurg. Psychiatry 74, 857–862 (2003).
8. Guo, Z. et al. Head injury and the risk of AD in the MIRAGE study. Neurology (2000).
9. Wojnarowicz, M. W., Fisher, A. M., Minaeva, O. & Goldstein, L. E. Considerations for
experimental animal models of concussion, traumatic brain injury, and chronic traumatic
encephalopathy-these matters matter. Front. Neurol. 8, 240 (2017).
10. Osier, N. D. & Dixon, C. E. The controlled cortical impact model: Applications,
considerations for researchers, and future directions. Frontiers in Neurology 7, 134
(2016).
11. Blennow, K. et al. Traumatic brain injuries. Nat. Rev. Dis. Prim. 2, 16084 (2016).

66
12. Davis, A. E. Mechanisms of traumatic brain injury: Biomechanical, structural and cellular
considerations. Crit. Care Nurs. Q. 23, 1–13 (2000).
13. Najem, D. et al. Traumatic brain injury: classification, models, and markers. Biochem.
Cell Biol. 96, 391–406 (2018).
14. Lu, J., Marmarou, A., Lapane, K., Turf, E. & Wilson, L. A method for reducing
misclassification in the extended Glasgow Outcome Score. J. Neurotrauma 27, 843–852
(2010).
15. Teasdale, G. & Jennett, B. Assessment of coma and impaired consciousness. Lancet 304,
81–84 (1974).
16. Spain, A. et al. Mild fluid percussion injury in mice produces evolving selective axonal
pathology and cognitive deficits relevant to human brain injury. J. Neurotrauma 27,
1429–1438 (2010).
17. Angoa-Pérez, M. et al. Animal models of sports-related head injury: Bridging the gap
between pre-clinical research and clinical reality. J. Neurochem. 129, 916–931 (2014).
18. Colantonio, A. et al. Examining occupational traumatic brain injury in Ontario. Can. J.
public Heal. Rev. Can. santé publique 101 Suppl, 58–62 (2010).
19. Hamani, C. & Temel, Y. Deep brain stimulation for psychiatric disease: Contributions and
validity of animal models. Sci. Transl. Med. 4, (2012).
20. DeKosky, S. T., Blennow, K., Ikonomovic, M. D. & Gandy, S. Acute and chronic
traumatic encephalopathies: pathogenesis and biomarkers. Nat. Rev. Neurol. 9, 192–200
(2013).
21. Shin, S. S., Dixon, C. E., Okonkwo, D. O. & Richardson, R. M. Neurostimulation for
traumatic brain injury. J. Neurosurg. 121, 1219–1231 (2014).
22. Blennow, K., Hardy, J. & Zetterberg, H. The neuropathology and neurobiology of
traumatic brain injury. Neuron 76, 886–899 (2012).
23. Villapol, S. Neuropathology of Traumatic Brain Injury and Its Role in the Development of

67
Alzheimer’s Disease. in Amyloid Diseases (IntechOpen, 2019).
doi:10.5772/intechopen.81945
24. Mckee, A. C. & Daneshvar, D. H. The neuropathology of traumatic brain injury. in
Handbook of Clinical Neurology 127, 45–66 (Elsevier B.V., 2015).
25. Vonder Haar, C. et al. Repetitive closed-head impact model of engineered rotational
acceleration (CHIMERA) injury in rats increases impulsivity, decreases dopaminergic
innervation in the olfactory tubercle and generates white matter inflammation, tau
phosphorylation and degener. Exp. Neurol. 317, 87–99 (2019).
26. Mouzon, B. et al. Repetitive mild traumatic brain injury in a mouse model produces
learning and memory deficits accompanied by histological changes. J. Neurotrauma 29,
2761–2773 (2012).
27. McIntosh, T. K. et al. Traumatic Brain Injury in the rat: Characterization of a lateral fluid-
percussion model. Neuroscience 28, 233–244 (1989).
28. Dixon, C. E., Clifton, G. L., Lighthall, J. W., Yaghmai, A. A. & Hayes, R. L. A controlled
cortical impact model of traumatic brain injury in the rat. J. Neurosci. Methods 39, 253–
262 (1991).
29. Lighthall, J. W. Controlled Cortical Impact: A new experimental brain injury model. J.
Neurotrauma 5, 1–15 (1988).
30. Meythaler, J. M., Peduzzi, J. D., Eleftheriou, E. & Novack, T. A. Current concepts:
Diffuse axonal injury-associated traumatic brain injury. Arch. Phys. Med. Rehabil. 82,
1461–1471 (2001).
31. Johnson, V. E., Stewart, W. & Smith, D. H. Axonal pathology in traumatic brain injury.
Exp. Neurol. 246, 35–43 (2013).
32. Meaney, D. F. et al. Biomechanical Analysis of Experimental Diffuse Axonal Injury. J.
Neurotrauma 12, 689–694 (1995).
33. Kim, J. J. & Gean, A. D. Imaging for the diagnosis and management of traumatic brain

68
injury. Neurotherapeutics 8, 39–53 (2011).
34. Armstrong, R. C., Mierzwa, A. J., Marion, C. M. & Sullivan, G. M. White matter
involvement after TBI: Clues to axon and myelin repair capacity. (2015).
doi:10.1016/j.expneurol.2015.02.011
35. Smith, D. H., Hicks, R. & Povlishock, J. T. Therapy development for diffuse axonal
injury. Journal of Neurotrauma 30, 307–323 (2013).
36. Van Dam, D. & De Deyn, P. P. Drug discovery in dementia: The role of rodent models.
Nat. Rev. Drug Discov. 5, 956–970 (2006).
37. Stewart, A. M. & Kalueff, A. V. Developing better and more valid animal models of brain
disorders. Behavioural Brain Research 276, 28–31 (2015).
38. Morales, D. M. et al. Experimental models of traumatic brain injury: Do we really need to
build a better mousetrap? Neuroscience 136, 971–989 (2005).
39. Marmarou, A. et al. A new model of diffuse brain injury in rats. Part I: Pathophysiology
and biomechanics. J. Neurosurg. 80, 291–300 (1994).
40. Shohami, E., Shapira, Y. & Cotev, S. Experimental closed head injury in rats:
Prostaglandin production in a noninjured zone. Neurosurgery 22, 859–863 (1988).
41. Feeney, D. M., Boyeson, M. G., Linn, R. T., Murray, H. M. & Dail, W. G. Responses to
cortical injury: I. Methodology and local effects of contusions in the rat. Brain Res. 211,
67–77 (1981).
42. Carbonell, W. S., Maris, D. O., Mccall, T. & Grady, M. S. Adaptation of the fluid
percussion injury model to the mouse. J. Neurotrauma 15, 217–229 (1998).
43. Millen, J. E., Glauser, F. L. & Fairman, R. P. A comparison of physiological responses to
percussive brain trauma in dogs and sheep. J. Neurosurg. 62, 587–591 (1985).
44. Cernak, I. et al. Involvement of the central nervous system in the general response to
pulmonary blast injury. J. Trauma 40, S100-4 (1996).

69
45. Goldstein, L. E. et al. Chronic traumatic encephalopathy in blast-exposed military
veterans and a blast neurotrauma mouse model. Sci. Transl. Med. 4, (2012).
46. Williams, A. J., Hartings, J. A., Lu, X. C. M., Rolli, M. L. & Tortella, F. C. Penetrating
ballistic-like brain injury in the rat: Differential time courses of hemorrhage, cell death,
inflammation, and remote degeneration. J. Neurotrauma 23, 1828–1846 (2006).
47. Williams, A. J. et al. Characterization of a new rat model of penetrating ballistic brain
injury. J. Neurotrauma 22, 313–331 (2005).
48. Carey, M. E., Sarna, G. S. & Farrell, J. B. Brain edema following an experimental missile
wound to the brain. J. Neurotrauma 7, 13–20 (1990).
49. Morrison, B., Elkin, B. S., Dollé, J.-P. & Yarmush, M. L. In vitro models of traumatic
brain injury. Annu. Rev. Biomed. Eng. 13, 91–126 (2011).
50. Kumaria, A. In vitro models as a platform to investigate traumatic brain injury. ATLA
Altern. to Lab. Anim. 45, 201–211 (2017).
51. Estrada-Rojo, F. et al. Models used in the study of traumatic brain injury. Rev. Neurosci.
29, 139–149 (2018).
52. Chen, Y. C., Smith, D. H. & Meaney, D. F. In-vitro approaches for studying blast-induced
traumatic brain injury. J. Neurotrauma 26, 861–876 (2009).
53. Katz, P. S. & Molina, P. E. A lateral fluid percussion injury model for studying traumatic
brain injury in rats. in Methods in Molecular Biology 1717, 27–36 (Humana Press, New
York, NY, 2018).
54. Osier, N. D., Carlson, S. W., DeSana, A. & Dixon, C. E. Chronic histopathological and
behavioral outcomes of experimental traumatic brain injury in adult male animals. J.
Neurotrauma 32, 1861–1882 (2015).
55. Thompson, H. J. et al. Lateral fluid percussion brain injury: A 15-year review and
evaluation. J. Neurotrauma 22, 42–75 (2005).
56. De Ross, A. L. et al. Multiple head injuries in rats; effects on behavior. J. Trauma 52,

70
708–714 (2002).
57. Yu, S. et al. Severity of controlled cortical impact traumatic brain injury in rats and mice
dictates degree of behavioral deficits. Brain Res. 1287, 157–163 (2009).
58. Smith, D. H. et al. A model of parasagittal controlled cortical impact in the mouse:
cognitive and histopathologic effects. J. Neurotrauma 12, 169–178 (1995).
59. Lighthall, J. W., Goshgarian, H. G. & Pinderski, C. R. Characterization of axonal injury
produced by controlled cortical impact. J. Neurotrauma 7, 65–76 (1990).
60. Fox, G. B., Fan, L., Levasseur, R. A. & Faden, A. I. Sustained sensory/motor and
cognitive deficits with neuronal apoptosis following controlled cortical impact brain injury
in the mouse. J. Neurotrauma 15, 599–614 (1998).
61. Washington, P. M. et al. The effect of injury severity on behavior: A phenotypic study of
cognitive and emotional deficits after mild, moderate, and severe controlled cortical
impact injury in mice. J. Neurotrauma 29, 2283–2296 (2012).
62. Fox, G. B., Fan, L., Levasseur, R. A. & Faden, A. I. Effect of traumatic brain injury on
mouse spatial and nonspatial learning in the Barnes circular maze. J. Neurotrauma 15,
1037–1046 (1998).
63. Hamm, R. J. et al. Cognitive deficits following traumatic brain injury produced by
controlled cortical impact. J. Neurotrauma 9, 11–20 (1992).
64. Monaco, C. M. et al. Environmental enrichment promotes robust functional and
histological benefits in female rats after controlled cortical impact injury. Exp. Neurol.
247, 410–418 (2013).
65. Bondi, C. O. et al. Found in translation: Understanding the biology and behavior of
experimental traumatic brain injury. Neurosci. Biobehav. Rev. 58, 123–146 (2015).
66. Kochanek, P. M. et al. Cerebral blood flow at one year after controlled cortical impact in
rats: Assessment by magnetic resonance imaging. J. Neurotrauma 19, 1029–1037 (2002).
67. Weber, M. T., Arena, J. D., Xiao, R., Wolf, J. A. & Johnson, V. E. CLARITY reveals a

71
more protracted temporal course of axon swelling and disconnection than previously
described following traumatic brain injury. Brain Pathol. 29, 437–450 (2019).
68. Yu, S. et al. Severity of controlled cortical impact traumatic brain injury in rats and mice
dictates degree of behavioral deficits. Brain Res. 1287, 157–163 (2009).
69. Namjoshi, D. R. et al. Merging pathology with biomechanics using CHIMERA (Closed-
Head Impact Model of Engineered Rotational Acceleration): a novel, surgery-free model
of traumatic brain injury. Mol. Neurodegener. 9, 55 (2014).
70. Smith, D. H. et al. Pre-clinical traumatic brain injury common data elements: toward a
common language across laboratories. J. Neurotrauma 32, 1725–1735 (2015).
71. Kilbourne, M. et al. Novel model of frontal impact closed head injury in the rat. J.
Neurotrauma 26, 2233–2243 (2009).
72. Lindgren, S. & Rinder, L. Experimental studies in head injury - II. Pressure propagation in
‘percussion concussion’. Biophysik 3, 174–180 (1966).
73. McIntosh, T. K., Noble, L., Andrews, B. & Faden, A. I. Traumatic brain injury in the rat:
characterization of a midline fluid-percussion model. Cent. Nerv. Syst. Trauma 4, 119–134
(1987).
74. Shultz, S. R., MacFabe, D. F., Foley, K. A., Taylor, R. & Cain, D. P. A single mild fluid
percussion injury induces short-term behavioral and neuropathological changes in the
Long-Evans rat: Support for an animal model of concussion. Behav. Brain Res. 224, 326–
335 (2011).
75. Brodhun, M. et al. Immunomorphological sequelae of severe brain injury induced by
fluid-percussion in juvenile pigs - Effects of mild hypothermia. Acta Neuropathol. 101,
424–434 (2001).
76. Hayes, R. L. et al. A new model of concussive brain injury in the cat produced by
extradural fluid volume loading: II. Physiological and neuropathological observations.
Brain Inj. 1, 93–112 (1987).

72
77. Dixon, C. E. et al. A fluid percussion model of experimental brain injury in the rat. J.
Neurosurg. 67, 110–119 (1987).
78. Lifshitz, J., Kelley, B. J. & Povlishock, J. T. Perisomatic thalamic axotomy after diffuse
traumatic brain injury is associated with atrophy rather than cell death. J. Neuropathol.
Exp. Neurol. 66, 218–229 (2007).
79. Cao, T., Thomas, T. C., Ziebell, J. M., Pauly, J. R. & Lifshitz, J. Morphological and
genetic activation of microglia after diffuse traumatic brain injury in the rat. Neuroscience
225, 65–75 (2012).
80. Sullivan, H. G., Martinez, J. & Becker, D. P. Fluid percussion model of mechanical brain
injury in the cat. J. Neurosurg. 45, 520–534 (1976).
81. Fritz, H. G. et al. A pig model with secondary increase of intracranial pressure after severe
traumatic brain injury and temporary blood loss. J. Neurotrauma 22, 807–821 (2005).
82. Armstead, W. M. Vasopressin-induced protein kinase C-dependent superoxide generation
contributes to ATP-sensitive potassium channel but not calcium-sensitive potassium
channel function impairment after brain injury. Stroke 32, 1408–1414 (2001).
83. Morganti-Kossmann, M. C., Yan, E. & Bye, N. Animal models of traumatic brain injury:
Is there an optimal model to reproduce human brain injury in the laboratory? Injury 41,
S10–S13 (2010).
84. Wang, Y. et al. Tightly coupled repetitive blast-induced traumatic brain injury:
Development and characterization in mice. J. Neurotrauma 28, 2171–2183 (2011).
85. Dewitt, D. S. & Prough, D. S. Blast-induced brain injury and posttraumatic hypotension
and hypoxemia. J. Neurotrauma 26, 877–887 (2009).
86. Bauman, R. A. et al. An introductory characterization of a combat-casualty-care relevant
swine model of closed head injury resulting from exposure to explosive blast. J.
Neurotrauma 26, 841–860 (2009).
87. Williams, A. J., Wei, H. H., Dave, J. R. & Tortella, F. C. Acute and delayed

73
neuroinflammatory response following experimental penetrating ballistic brain injury in
the rat. J. Neuroinflammation 4, (2007).
88. Chen, Y., Constantini, S., Trembovler, V., Weinstock, M. & Shohami, E. An experimental
model of closed head injury in mice: Pathophysiology, histopathology, and cognitive
deficits. J. Neurotrauma 13, 557–568 (1996).
89. Flierl, M. A. et al. Mouse closed head injury model induced by a weight-drop device. Nat.
Protoc. 4, 1328–1337 (2009).
90. King, C. et al. Brain temperature profiles during epidural cooling with the ChillerPad in a
monkey model of traumatic brain injury. J. Neurotrauma 27, 1895–1903 (2010).
91. Härtl, R., Medary, M., Ruge, M., Arfors, K. E. & Ghajar, J. Blood-brain barrier
breakdown occurs early after traumatic brain injury and is not related to white blood cell
adherence. Acta Neurochir. Suppl. 70, 240–2 (1997).
92. Pfenninger, E. G., Reith, A., Breitig, D., Grunert, A. & Ahnefeld, F. W. Early changes of
intracranial pressure, perfusion pressure, and blood flow after acute head injury. Part 1:
An experimental study of the underlying pathophysiology. J. Neurosurg. 70, 774–779
(1989).
93. Friess, S. H. et al. Repeated traumatic brain injury affects composite cognitive function in
piglets. J. Neurotrauma 26, 1111–1121 (2009).
94. Pappius, H. M. Local cerebral glucose utilization in thermally traumatized rat brain. Ann.
Neurol. 9, 484–491 (1981).
95. Tengvar, C. & Olsson, Y. Uptake of macromolecules into neurons from a focal vasogenic
cerebral edema and subsequent axonal spread to other brain regions - A preliminary study
in the mouse with horseradish peroxidase as a tracer. Acta Neuropathol. 57, 233–235
(1982).
96. Miocinovic, S., Somayajula, S., Chitnis, S. & Vitek, J. L. History, applications, and
mechanisms of deep brain stimulation. JAMA Neurology 70, 163–171 (2013).

74
97. Laitinen, L. V., Bergenheim, A. T. & Hariz, M. I. Ventroposterolateral pallidotomy can
abolish all parkinsonian symptoms. in Stereotactic and Functional Neurosurgery 58, 14–
21 (Stereotact Funct Neurosurg, 1992).
98. Lozano, A. M., Hutchison, W. D. & Kalia, S. K. What have we learned about movement
disorders from functional neurosurgery? (2017). doi:10.1146/annurev-neuro-070815
99. Benabid, A. L. et al. Long-term suppression of tremor by chronic stimulation of the
ventral intermediate thalamic nucleus. Lancet 337, 403–406 (1991).
100. Benabid, A. L. et al. Acute and long-term effects of subthalamic nucleus stimulation of
Parkinson’s disease. in Stereotactic and Functional Neurosurgery 62, 76–84 (Stereotact
Funct Neurosurg, 1994).
101. Benabid, A. L. et al. Chronic electrical stimulation of the ventralis intermedius nucleus of
the thalamus as a treatment of movement disorders. J. Neurosurg. 84, 203–214 (1996).
102. Chiken, S. & Nambu, A. Mechanism of deep brain stimulation: inhibition, excitation, or
disruption? Neuroscientist 22, 313–322 (2016).
103. Goodman, W. K. & Alterman, R. L. Deep brain stimulation for intractable psychiatric
disorders. Annu. Rev. Med. 63, 511–524 (2012).
104. Senova, S., Fomenko, A., Gondard, E. & Lozano, A. M. Anatomy and function of the
fornix in the context of its potential as a therapeutic target. J Neurol Neurosurg Psychiatry
91, 547–559 (2020).
105. Laxton, A. W. et al. A phase I trial of deep brain stimulation of memory circuits in
Alzheimer’s disease. Ann. Neurol. 68, 521–534 (2010).
106. Jakobs, M., Lee, D. J. & Lozano, A. M. Modifying the progression of Alzheimer’s and
Parkinson’s disease with deep brain stimulation. (2019).
doi:10.1016/j.neuropharm.2019.107860
107. Amorim, B. O. et al. Deep brain stimulation induces antiapoptotic and anti-inflammatory
effects in epileptic rats. J. Neuroinflammation 12, (2015).

75
108. Lipsman, N. et al. Subcallosal cingulate deep brain stimulation for treatment-refractory
anorexia nervosa: A phase 1 pilot trial. Lancet 381, 1361–1370 (2013).
109. Kuhn, J. et al. Deep brain stimulation for psychiatric disorders. Dtsch. Arzteblatt Int. 107,
105–13 (2010).
110. Xu, W. et al. Deep brain stimulation for Tourette’s syndrome. Translational
Neurodegeneration 9, 1–19 (2020).
111. Boccard, S. G. J., Pereira, E. A. C. & Aziz, T. Z. Deep brain stimulation for chronic pain.
Journal of Clinical Neuroscience 22, 1537–1543 (2015).
112. Freire, R. C., Cabrera-Abreu, C. & Milev, R. Neurostimulation in anxiety disorders, post-
traumatic stress disorder, and obsessive-compulsive disorder. in Advances in Experimental
Medicine and Biology 1191, 331–346 (Springer, 2020).
113. Davidson, B. et al. Magnetic resonance-guided focused ultrasound capsulotomy for
treatment-resistant psychiatric disorders. Oper. Neurosurg. (2020).
doi:10.1093/ons/opaa240
114. Gouveia, F. V. & Hamani, C. Commentary: Amygdala and hypothalamus: historical
overview with a focus on aggression. Neurosurgery (2019). doi:10.1093/neuros/nyz045
115. Hamani, C., Diwan, M., Isabella, S., Lozano, A. M. & Nobrega, J. N. Effects of different
stimulation parameters on the antidepressant-like response of medial prefrontal cortex
deep brain stimulation in rats. J. Psychiatr. Res. 44, 683–687 (2010).
116. Gouveia, F. et al. Neuromodulation strategies in post-traumatic stress disorder: from
preclinical models to clinical applications. Brain Sci. 9, 45 (2019).
117. Gondard, E. et al. Deep brain stimulation rescues memory and synaptic activity in a rat
model of global ischemia. J. Neurosci. 39, 2430–2440 (2019).
118. Chan, H. H. et al. Lateral cerebellar nucleus stimulation promotes motor recovery and
suppresses neuroinflammation in a fluid percussion injury rodent model. Brain Stimul. 11,
1356–1367 (2018).

76
119. McKinnon, C. et al. Deep brain stimulation: potential for neuroprotection. Ann. Clin.
Transl. Neurol. (2018). doi:10.1002/acn3.682
120. Bergman, H., Wichmann, T. & Delong, M. R. Reversal of Experimental Parkinsonism by
Lesions of the Subthalamic Nucleus. New Series 249, (1990).
121. Aziz, T. Z., Peggs, D., Sambrook, M. A. & Crossman, A. R. Lesion of the subthalamic
nucleus for the alleviation of 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP)‐
induced parkinsonism in the primate. Mov. Disord. 6, 288–292 (1991).
122. Gill, S. S. & Heywood, P. Bilateral dorsolateral subthalamotomy for advanced
Parkinson’s disease. Lancet 350, 1224 (1997).
123. Filali, M., Hutchison, W. D., Palter, V. N., Lozano, A. M. & Dostrovsky, J. O.
Stimulation-induced inhibition of neuronal firing in human subthalamic nucleus. Exp.
Brain Res. 156, 274–281 (2004).
124. Beurrier, C., Bioulac, B., Audin, J. & Hammond, C. High-frequency stimulation produces
a transient blockade of voltage-gated currents in subthalamic neurons. J. Neurophysiol.
85, 1351–1356 (2001).
125. Bikson, M. et al. Suppression of epileptiform activity by high frequency sinusoidal fields
in rat hippocampal slices. J. Physiol. 531, 181–191 (2001).
126. Do, M. T. H. & Bean, B. P. Subthreshold sodium currents and pacemaking of subthalamic
neurons: Modulation by slow inactivation. Neuron 39, 109–120 (2003).
127. Shin, D. S. et al. High frequency stimulation or elevated K+ depresses neuronal activity in
the rat entopeduncular nucleus. Neuroscience 149, 68–86 (2007).
128. Benazzouz, A., Gross, C., Féger, J., Boraud, T. & Bioulac, B. Reversal of rigidity and
improvement in motor performance by subthalamic high‐frequency stimulation in MPTP‐
treated monkeys. Eur. J. Neurosci. 5, 382–389 (1993).
129. Welter, M. L. et al. Effects of high-frequency stimulation on subthalamic neuronal activity
in Parkinsonian patients. Arch. Neurol. 61, 89–96 (2004).

77
130. Boraud, T., Bezard, E., Bioulac, B. & Gross, C. High frequency stimulation of the internal
Globus Pallidus (GPi) simultaneously improves parkinsonian symptoms and reduces the
firing frequency of GPi neurons in the MPTP-treated monkey. Neurosci. Lett. 215, 17–20
(1996).
131. Tai, C.-H. et al. Electrophysiological and metabolic evidence that high‐frequency
stimulation of the subthalamic nucleus bridles neuronal activity in the subthalamic nucleus
and the substantia nigra reticulata. FASEB J. 17, 1820–1830 (2003).
132. Shi, L. H., Luo, F., Woodward, D. J. & Chang, J. Y. Basal ganglia neural responses during
behaviorally effective deep brain stimulation of the subthalamic nucleus in rats
performing a treadmill locomotion test. Synapse 59, 445–457 (2006).
133. Chiken, S. & Nambu, A. High-frequency pallidal stimulation disrupts information flow
through the pallidum by GABAergic inhibition. (2013). doi:10.1523/JNEUROSCI.4144-
11.2013
134. Tawfik, V. L. et al. Deep brain stimulation results in local glutamate and adenosine
release: Investigation into the role of astrocytes. Neurosurgery 67, 367–375 (2010).
135. Cunha, R. A. Neuroprotection by adenosine in the brain: From A1 receptor activation to
A2A receptor blockade. Purinergic Signalling 1, 111–134 (2005).
136. Bekar, L. et al. Adenosine is crucial for deep brain stimulation-mediated attenuation of
tremor. Nat. Med. 14, 75–80 (2008).
137. Montgomery, E. B. & Baker, K. B. Mechanisms of deep brain stimulation and future
technical developments. Neurol. Res. 22, 259–266 (2000).
138. Kang, G. & Lowery, M. M. Interaction of oscillations, and their suppression via deep
brain stimulation, in a model of the cortico-basal ganglia network. IEEE Trans. Neural
Syst. Rehabil. Eng. 21, 244–253 (2013).
139. Eusebio, A. et al. Deep brain stimulation can suppress pathological synchronisation in
parkinsonian patients. J. Neurol. Neurosurg. Psychiatry 82, 569–573 (2011).

78
140. Brittain, J.-S., Sharott, A. & Brown, P. The highs and lows of beta activity in cortico-basal
ganglia loops. Eur. J. Neurosci. 39, 1951–1959 (2014).
141. Khanna, P., Carmena, J. M., Battaglia, F. P. & Schnitzer, M. J. Neural oscillations: beta
band activity across motor networks. Curr. Opin. Neurobiol. 32, 60–67 (2015).
142. Hashimoto, T., Elder, C. M., Okun, M. S., Patrick, S. K. & Vitek, J. L. Stimulation of the
subthalamic nucleus changes the firing pattern of pallidal neurons. J. Neurosci. 23, 1916–
1923 (2003).
143. Kühn, A. A. et al. High-frequency stimulation of the subthalamic nucleus suppresses
oscillatory β activity in patients with Parkinson’s disease in parallel with improvement in
motor performance. J. Neurosci. 28, 6165–6173 (2008).
144. Xu, W., Russo, G. S., Hashimoto, T., Zhang, J. & Vitek, J. L. Subthalamic nucleus
stimulation modulates thalamic neuronal activity. J. Neurosci. 28, 11916–11924 (2008).
145. Cleary, D. R. et al. Deep brain stimulation entrains local neuronal firing in human globus
pallidus internus. J. Neurophysiol. 109, 978–987 (2013).
146. Maurice, N., Thierry, A. M., Glowinski, J. & Deniau, J. M. Spontaneous and evoked
activity of substantia nigra pars reticulata neurons during high-frequency stimulation of
the subthalamic nucleus. J. Neurosci. 23, 9929–9936 (2003).
147. Lozano, A. M. & Lipsman, N. Probing and Rregulating dysfunctional circuits using deep
brain stimulation. Neuron 77, 406–424 (2013).
148. Deniau, J. M., Degos, B., Bosch, C. & Maurice, N. Deep brain stimulation mechanisms:
Beyond the concept of local functional inhibition. Eur. J. Neurosci. 32, 1080–1091
(2010).
149. Lee, K. H., Chang, S. Y., Roberts, D. W. & Kim, U. Neurotransmitter release from high-
frequency stimulation of the subthalamic nucleus. J. Neurosurg. 101, 511–517 (2004).
150. Hamani, C. et al. Anterior thalamus deep brain stimulation at high current impairs
memory in rats. Exp. Neurol. 225, 154–162 (2010).

79
151. Covolan, L., De Almeida, A.-C., Amorim, B., Cavarsan, C. & Miranda, M. F. Effects of
anterior thalamic nucleus deep brain stimulation in chronic epileptic rats. PLoS One 9,
97618 (2014).
152. Morgane, P. J., Galler, J. R. & Mokler, D. J. A review of systems and networks of the
limbic forebrain/limbic midbrain. Prog. Neurobiol. 75, 143–160 (2005).
153. Hescham, S. et al. Behavioral effects of deep brain stimulation of different areas of the
Papez circuit on memory-and anxiety-related functions. Behav. Brain Res. 292, 353–360
(2015).
154. Grodd, W., Kumar, V. J., Schüz, A., Lindig, T. & Scheffler, K. The anterior and medial
thalamic nuclei and the human limbic system: tracing the structural connectivity using
diffusion-weighted imaging. Sci. Rep. 10, (2020).
155. Robertson, R. T. & Kaitz, S. S. Thalamic connections with limbic cortex. I.
Thalamocortical projections. J. Comp. Neurol. 195, 501–525 (1981).
156. Mathiasen, M. L., Louch, R. C., Nelson, A. D., Dillingham, C. M. & Aggleton, J. P.
Trajectory of hippocampal fibres to the contralateral anterior thalamus and mammillary
bodies in rats, mice, and macaque monkeys. Brain Neurosci. Adv. 3, 239821281987120
(2019).
157. McIntyre, C. C., Savasta, M., Kerkerian-Le Goff, L. & Vitek, J. L. Uncovering the
mechanism(s) of action of deep brain stimulation: Activation, inhibition, or both. Clinical
Neurophysiology 115, 1239–1248 (2004).
158. Chiken, S. & Nambu, A. Disrupting neuronal transmission: Mechanism of DBS?
Frontiers in Systems Neuroscience 8, 33 (2014).
159. Hofer, A. S. & Schwab, M. E. Enhancing rehabilitation and functional recovery after brain
and spinal cord trauma with electrical neuromodulation. Curr. Opin. Neurol. 32, 828–835
(2019).
160. Kundu, B., Brock, A. A., Englot, D. J., Butson, C. R. & Rolston, J. D. Deep brain
stimulation for the treatment of disorders of consciousness and cognition in traumatic

80
brain injury patients: A review. Neurosurg. Focus 45, E14 (2018).
161. Lee, D. J. et al. Medial septal nucleus theta frequency deep brain stimulation improves
spatial working memory after traumatic brain injury. J. Neurotrauma 30, 131–139 (2013).
162. Gonzalez, M. M. C., Blaya, M. O., Alonso, O. F., Bramlett, H. M. & Hentall, I. D.
Midbrain raphe stimulation improves behavioral and anatomical recovery from fluid-
percussion brain injury. J. Neurotrauma 30, 119–130 (2013).
163. Rezaei Haddad, A., Lythe, V. & Green, A. L. Deep brain stimulation for recovery of
consciousness in minimally conscious patients after traumatic brain injury: a systematic
review. Neuromodulation 22, 373–379 (2019).
164. Clayton, E., Kinley-Cooper, S. K., Weber, R. A. & Adkins, D. L. Brain stimulation:
Neuromodulation as a potential treatment for motor recovery following traumatic brain
injury. Brain Res. 1640, 130–138 (2016).
165. Hescham, S. et al. Fornix deep brain stimulation enhances acetylcholine levels in the
hippocampus. Brain Struct. Funct. 221, 4281–4286 (2016).
166. Du, T. et al. Deep brain stimulation of the anterior nuclei of the thalamus relieves basal
ganglia dysfunction in monkeys with temporal lobe epilepsy. CNS Neurosci. Ther. (2020).
doi:10.1111/cns.13462
167. Toda, H., Hamani, C., Fawcett, A. P., Hutchison, W. D. & Lozano, A. M. The regulation
of adult rodent hippocampal neurogenesis by deep brain stimulation: Laboratory
investigation. J. Neurosurg. 108, 132–138 (2008).
168. Encinas, J. M., Hamani, C., Lozano, A. M. & Enikolopov, G. Neurogenic hippocampal
targets of deep brain stimulation. J. Comp. Neurol 519, 6–20 (2011).
169. Hamani, C., Stone, S. S., Garten, A., Lozano, A. M. & Winocur, G. Memory rescue and
enhanced neurogenesis following electrical stimulation of the anterior thalamus in rats
treated with corticosterone. (2011). doi:10.1016/j.expneurol.2011.08.023
170. Liang, S. et al. Neuroprotective effect of electric conduction treatment on hippocampus

81
cell apoptosis in KA induced acute temporal lobe epileptic rats. Brain Stimul. 9, 933–939
(2016).
171. Yang, A. C. et al. Potential protective effects of chronic anterior thalamic nucleus
stimulation on hippocampal neurons in epileptic monkeys. Brain Stimul. 8, 1049–1057
(2015).
172. Chen, Y. C. et al. Deep brain stimulation of the anterior nucleus of the thalamus reverses
the gene expression of cytokines and their receptors as well as neuronal degeneration in
epileptic rats. Brain Res. 1657, 304–311 (2017).
173. Fisher, R. et al. Electrical stimulation of the anterior nucleus of thalamus for treatment of
refractory epilepsy. Epilepsia 51, 899–908 (2010).
174. Salanova, V. Deep brain stimulation for epilepsy. Epilepsy Behav. 88, 21–24 (2018).
175. Salanova, V. et al. Long-term efficacy and safety of thalamic stimulation for drug-
resistant partial epilepsy. Neurology 84, 1017–1025 (2015).
176. Oh, Y. S. et al. Cognitive improvement after long-term electrical stimulation of bilateral
anterior thalamic nucleus in refractory epilepsy patients. Seizure 21, 183–187 (2012).
177. Milosevic, L. et al. Neuronal inhibition and synaptic plasticity of basal ganglia neurons in
Parkinson’s disease. doi:10.1093/brain/awx296
178. Paxinos, G. & Watson, C. The Rat Brain in Stereotaxic Coordinates. (Elsevier Science,
2008).
179. Antunes, M. & Biala, G. The novel object recognition memory: Neurobiology, test
procedure, and its modifications. Cognitive Processing 13, 93–110 (2012).
180. Leger, M. et al. Object recognition test in mice. Nat. Protoc. 8, 2531–2537 (2013).
181. Angoa-Pérez, M., Kane, M. J., Briggs, D. I., Francescutti, D. M. & Kuhn, D. M. Marble
burying and nestlet shredding as tests of repetitive, compulsive-like behaviors in mice. J.
Vis. Exp. 50978 (2013). doi:10.3791/50978

82
182. Barnes, C. A. Memory deficits associated with senescence: A neurophysiological and
behavioral study in the rat. J. Comp. Physiol. Psychol. 93, 74–104 (1979).
183. Dumont, J. R. & Aggleton, J. P. Dissociation of recognition and recency memory
judgments after anterior thalamic nuclei lesions in rats. Behav. Neurosci. 127, 415–431
(2013).
184. O’Leary, T. P., Savoie, V. & Brown, R. E. Learning, memory and search strategies of
inbred mouse strains with different visual abilities in the Barnes maze. Behav. Brain Res.
216, 531–542 (2011).
185. Gawel, K., Gibula, E., Marszalek-Grabska, M., Filarowska, J. & Kotlinska, J. H.
Assessment of spatial learning and memory in the Barnes maze task in rodents—
methodological consideration. Naunyn. Schmiedebergs. Arch. Pharmacol. 392, 1–18
(2019).
186. Paul, C.-M., Magda, G. & Abel, S. Spatial memory: Theoretical basis and comparative
review on experimental methods in rodents. Behav. Brain Res. 203, 151–164 (2009).
187. Gawel, K., Gibula, E., Marszalek-Grabska, M., Filarowska, J. & Kotlinska, J. H.
Assessment of spatial learning and memory in the Barnes maze task in rodents—
methodological consideration. Naunyn. Schmiedebergs. Arch. Pharmacol. 392, 1–18
(2019).
188. Chawla, A., Cordner, Z. A., Boersma, G. & Moran, T. H. Cognitive impairment and gene
expression alterations in a rodent model of binge eating disorder. Physiol. Behav. 180, 78–
90 (2017).
189. Attar, A. et al. A shortened Barnes maze protocol reveals memory deficits at 4-months of
age in the triple-transgenic mouse model of Alzheimer’s disease. PLoS One 8, (2013).
190. Stone, S. S. D. et al. Stimulation of entorhinal cortex promotes adult neurogenesis and
facilitates spatial memory. J. Neurosci. 31, 13469–13484 (2011).
191. Pitts, M. W. Barnes maze procedure for spatial learning and memory in mice. 8, (2018).

83
192. O’Leary, T. P. & Brown, R. E. The effects of apparatus design and test procedure on
learning and memory performance of C57BL/6J mice on the Barnes maze. J. Neurosci.
Methods 203, 315–324 (2012).
193. O’Leary, T. P. & Brown, R. E. Optimization of apparatus design and behavioral measures
for the assessment of visuo-spatial learning and memory of mice on the Barnes maze.
Learn. Mem. 20, 85–96 (2013).
194. Żakowski, W. Neurochemistry of the anterior thalamic nuclei. Mol. Neurobiol. 54, 5248–
5263 (2017).
195. Aggleton, J. P. et al. Hippocampal-anterior thalamic pathways for memory: Uncovering a
network of direct and indirect actions. Eur. J. Neurosci. 31, 2292–2307 (2010).
196. Jankowski, M. M. et al. The anterior thalamus provides a subcortical circuit supporting
memory and spatial navigation. Front. Syst. Neurosci. 7, 45 (2013).
197. Bouwens van der Vlis, T. A. M. et al. Deep brain stimulation of the anterior nucleus of the
thalamus for drug-resistant epilepsy. Neurosurgical Review 42, 287–296 (2019).
198. Taube, J. S. Head direction cells recorded in the anterior thalamic nuclei of freely moving
rats. J. Neurosci. 15, 70–86 (1995).
199. Aggleton, J. P. & Sahgal, A. The contribution of the anterior thalamic nuclei to
anterograde amnesia. Neuropsychologia 31, 1001–1019 (1993).
200. Aggleton, J. P., Neave, N., Nagle, S. & Hunt, P. R. A comparison of the effects of anterior
thalamic, mamillary body and fornix lesions on reinforced spatial alternation. Behav.
Brain Res. 68, 91–101 (1995).
201. Byatt, G. & Dalrymple-Alford, J. C. Both anteromedial and anteroventral thalamic lesions
impair radial-maze learning in rats. Behav. Neurosci. 110, 1335–1348 (1996).
202. Mitchell, A. S. & Dalrymple-Alford, J. C. Lateral and anterior thalamic lesions impair
independent memory systems. Agglet. Brown (1996). doi:10.1101/lm.122206
203. Sziklas, V. & Petrides, M. The effects of lesions to the anterior thalamic nuclei on object-

84
place associations in rats. Eur. J. Neurosci. 11, 559–566 (1999).
204. Van Groen, T., Kadish, I. & Wyss, J. M. Role of the anterodorsal and anteroventral nuclei
of the thalamus in spatial memory in the rat. Behav. Brain Res. 132, 19–28 (2002).
205. Mitchell, A. S. & Dalrymple-Alford, J. C. Dissociable memory effects after medial
thalamus lesions in the rat. Eur. J. Neurosci. 22, 973–985 (2005).
206. Loukavenko, E. A., Ottley, M. C., Moran, J. P., Wolff, M. & Dalrymple-Alford, J. C.
Towards therapy to relieve memory impairment after anterior thalamic lesions: Improved
spatial working memory after immediate and delayed postoperative enrichment. Eur. J.
Neurosci. 26, 3267–3276 (2007).
207. Taube, J. S. The head direction signal: origins and sensory-motor integration. Annu. Rev.
Neurosci. 30, 181–207 (2007).
208. Dumont, J. R., Wright, N. F., Pearce, J. M. & Aggleton, J. P. The impact of anterior
thalamic lesions on active and passive spatial learning in stimulus controlled
environments: Geometric cues and pattern arrangement. Behav. Neurosci. 128, 161–177
(2014).
209. Wolff, M., Gibb, S. J. & Dalrymple-Alford, J. C. Beyond spatial memory: The anterior
thalamus and memory for the temporal order of a sequence of odor cues. J. Neurosci. 26,
2907–2913 (2006).
210. Aggleton, J. P. & Nelson, A. J. D. Why do lesions in the rodent anterior thalamic nuclei
cause such severe spatial deficits? Neurosci. Biobehav. Rev. 54, 131–144 (2015).
211. Wolff, M., Loukavenko, E. A., Will, B. E. & Dalrymple-Alford, J. C. The extended
hippocampal-diencephalic memory system: Enriched housing promotes recovery of the
flexible use of spatial representations after anterior thalamic lesions. Hippocampus 18,
996–1007 (2008).
212. Moreau, P. H. et al. Lesions of the anterior thalamic nuclei and intralaminar thalamic
nuclei: Place and visual discrimination learning in the water maze. Brain Struct. Funct.
218, 657–667 (2013).

85
213. Tsanov, M. et al. Oscillatory entrainment of thalamic neurons by theta rhythm in freely
moving rats. J. Neurophysiol. 105, 4–17 (2011).
214. Aggleton, J. P., Hunt, P. R., Nagle, S. & Neave, N. The effects of selective lesions within
the anterior thalamic nuclei on spatial memory in the rat. Behav. Brain Res. 81, 189–198
(1996).
215. Lee, D. J. et al. Septohippocampal neuromodulation improves cognition after traumatic
brain injury. J. Neurotrauma 32, 1822–1832 (2015).
216. Hengartner, M. O. The biochemistry of apoptosis. Nature 407, 770–776 (2000).
217. Hiebert, J. B., Shen, Q., Thimmesch, A. R. & Pierce, J. D. Traumatic brain injury and
mitochondrial dysfunction. Am. J. Med. Sci. 350, 132–138 (2015).
218. Raghupathi, R. Cell death mechanisms following traumatic brain injury. in Brain
Pathology 14, 215–222 (International Society of Neuropathology, 2004).
219. Porter, A. G. & Jänicke, R. U. Emerging roles of caspase-3 in apoptosis. Cell Death and
Differentiation 6, 99–104 (1999).
220. Jayakumar, T. et al. Hinokitiol, a natural tropolone derivative, offers neuroprotection from
thromboembolic stroke in vivo. Evidence-based Complement. Altern. Med. 2013, (2013).
221. Chen, N. et al. High-frequency stimulation of the hippocampus protects against seizure
activity and hippocampal neuronal apoptosis induced by kainic acid administration in
macaques. Neuroscience 256, 370–378 (2013).
222. Lorente, L. et al. Serum caspase-3 levels and mortality are associated in patients with
severe traumatic brain injury. BMC Neurol. 15, (2015).
223. Zhang, X., Chen, Y., Jenkins, L. W., Kochanek, P. M. & Clark, R. S. B. Bench-to-bedside
review: Apoptosis/programmed cell death triggered by traumatic brain injury. Critical
Care 9, 66–75 (2005).
224. Glushakova, O. Y. et al. Role of caspase-3-mediated apoptosis in chronic caspase-3-
cleaved tau accumulation and blood-brain barrier damage in the corpus callosum after

86
traumatic brain injury in rats. J. Neurotrauma 35, 157–173 (2018).
225. Meng, D. W., Liu, H. G., Yang, A. C., Zhang, K. & Zhang, J. G. Stimulation of anterior
thalamic nuclei protects against seizures and neuronal apoptosis in hippocampal CA3
Region of kainic acid‑induced epileptic rats. Chin. Med. J. (Engl). 129, 960–966 (2016).
226. Huang, C. et al. The neuroprotective effect of deep brain stimulation at nucleus basalis of
Meynert in transgenic mice with Alzheimer’s disease. (2018).
doi:10.1016/j.brs.2018.08.015
227. Lopez-Meraz, M. L., Niquet, J. & Wasterlain, C. G. Distinct caspase pathways mediate
necrosis and apoptosis in subpopulations of hippocampal neurons after status epilepticus.
in Epilepsia 51, 56–60 (Epilepsia, 2010).
228. Hüske, C. et al. Towards optimized anesthesia protocols for stereotactic surgery in rats:
Analgesic, stress and general health effects of injectable anesthetics. A comparison of a
recommended complete reversal anesthesia with traditional chloral hydrate
monoanesthesia. Brain Res. 1642, 364–375 (2016).
229. Van Hartevelt, T. J. et al. Neural plasticity in human brain connectivity: The effects of
long term deep brain stimulation of the subthalamic nucleus in Parkinson’s disease. PLoS
One 9, e86496 (2014).
230. Jessberger, S. et al. Seizure-associated, aberrant neurogenesis in adult rats characterized
with retrovirus-mediated cell labeling. J. Neurosci. 27, 9400–9407 (2007).
231. Leng, S. X. et al. ELISA and multiplex technologies for cytokine measurement in
inflammation and aging research. Journals Gerontol. - Ser. A Biol. Sci. Med. Sci. 63, 879–
884 (2008).
232. Mirzayans, R. & Murray, D. Do TUNEL and other apoptosis assays detect cell death in
preclinical studies? Int. J. Mol. Sci. 21, 1–12 (2020).
233. Glushakov, A. O. et al. Chronic upregulation of cleaved-caspase-3 associated with chronic
myelin pathology and microvascular reorganization in the thalamus after traumatic brain
injury in rats. Int. J. Mol. Sci. 19, 3151 (2018).

87
234. Rajneesh, C. P. et al. Deep brain stimulation of the pedunculopontine tegmental nucleus
renders neuroprotection through the suppression of hippocampal apoptosis: An
experimental animal study. Brain Sci. 10, (2020).
235. Dandekar, M. P. et al. Medial forebrain bundle deep brain stimulation reverses anhedonic-
like behavior in a chronic model of depression: importance of BDNF and inflammatory
cytokines. Mol. Neurobiol. 56, 4364–4380 (2019).
236. Do-Monte, F. H., Rodriguez-Romaguera, J., Rosas-Vidal, L. E. & Quirk, G. J. Deep brain
stimulation of the ventral striatum increases BDNF in the fear extinction circuit. Front.
Behav. Neurosci. 7, (2013).
237. Hamani, C. et al. Deep brain stimulation reverses anhedonic-like behavior in a chronic
model of depression: Role of serotonin and brain derived neurotrophic factor. Biol.
Psychiatry 71, 30–35 (2012).