introduction to drug discovery -...
TRANSCRIPT

CHAPTER 1
Introduction to Drug Discovery

Introduction :
Cheminformatics or Chemo informatics is another method to reach the
destiny of drug discovery. Chemoinformatics is an interface science aimed
primarily at discovering novel chemical entities that will ultimately result in the
development of novel treatments for unmet medical needs, although these same
methods are also applied in other fields that ultimately design new molecules. The
field combines expertise from, among others, chemistry, biology, physics,
biochemistry, statistics, mathematics, and computer science. Cheminformatics
consists of several in-silico techniques which are widely used in pharmaceutical
companies in the process of drug discovery. The discovery of new medical
treatments to meet unmet medical needs is one of the most important endeavors in
humanity. The process is time consuming, expensive, and fraught with many
challenges.
Drug discovery is perhaps the area of research in which
chemoinformatics has found the greatest application. Traditional pharmaceutical
industry would require 12-14 years and costing upto $1.2 - $1.4 billion to bring a
drug from discovery to market, in this approach drugs were discovered by
synthesizing compounds in a time-consuming multi-step processes with failures
attributed to poor pharmacokinetics (39%), lack of efficacy (30%), animal toxicity
(11%), adverse effects in humans (10%) and various commercial and
miscellaneous factors. Today, the process of drug discovery has been
revolutionized with the advent of genomics, proteomics, bioinformatics and
efficient technologies like, combinatorial chemistry, cheminformatics, high
throughput screening (HTS), virtual screening, de novo design, in vitro, in silico
ADMET screening, Quantitative structure-activity relationship (QSAR) and

structure-based drug design. The process of finding a new drug against a chosen
target for a particular disease usually involves high-throughput screening (HTS),
wherein large libraries of chemicals are tested for their ability to modify the target.
The primary application of cheminformatics is in the storage of information related
to the drug molecules and the efficient presentation of such stored information
during the process of lead optimization. A range of parameters can be used to
assess the quality of a compound, or a series of compounds, as proposed in the
Lipinski's Rule of Five. Such parameters include calculated properties such as
cLogP to estimate lipophilicity, rotatable bonds to estimate molecular flexibility,
molecular weight, Hydrogen Bond Acceptors and Hydrogen Bond Donors to
estimate Pharmacophoric Properties, polar surface area and measured properties,
such as potency, in-vitro measurement of enzymatic clearance etc. Some
descriptors such as ligand efficiency (LE) and lipophilic efficiency (LiPE) combine
such parameters to assess drug likeness.
Role of Bio and Cheminformatics in Drug Discovery:
Bio and Cheminformatics tools, plays a major role in identifying target
molecule, which could be a potential drug. Not all small molecules can be drugs,
and not all proteins can be drug targets. A small molecule must have certain
properties, and a protein must contain a binding site that is complementary with
these properties.
The majority of successful drugs show their response by competing for a
binding site on a protein with and endogenous small molecule. Target
identification is one of the most important areas for in silico research. The current

dominant paradigm in pharmaceutical drug discovery seeks to find a particular
small molecule inhibitor to bind to a specific receptor, a macromolecular target.
However our ability is depending on the detection of efficient methods of
manipulation of organic small molecules and discovery of the novel biological
targets. Several approaches and databases can be applied for the discovery of drug
targets.
One important use of these databases in target discovery is to infer
relative gene expression levels, simply by counting how often a given EST
sequence appears in a given cell or tissue. Gene expression levels are important
because the phenotype is determined by the small portion of genes that are
expressed at any given time in a cell or tissue type, and changes in gene expression
can be associated with disease.
Thus, by comparing levels of gene expression in normal and disease
states, novel drug targets can be identified by in silico methods .Knowledge of
prokaryotic and viral genomes supports identification of targets for drugs against
infectious disease. Metabolic pathways and proteins associated to them and
specific to micro-organism are of particular interest.
A drug affecting such a target is less likely to interact with a human
homologue. Proteins with sequences similar across bacterial clades offer the
possibility of broad spectrum antibiotics. A drug-discovery program typically starts
with the identification of suitable drug targets and ends with the candidate drug
production and sells in the market after clinical trials.

Fig 1: Milestones of Drug Discovery.
Market facts towards modern drug discovery:
The majority of pharmaceutical drug discovery programs currently,
begins with a known macromolecular targets, and seek to identify a suitable small
molecule modulator (Ratti and Trist,2001;Dean,Zanders, and Bailey,2001). The
process of optimizing the lead molecule into a candidate drug is usually the longest
and most expensive stage in the drug discovery process. Following the selection of
the candidate molecule from limited chemical space of analogs of original lead
compounds, to the drug development, scientists develop large scale production

methods, and conduct the preclinical animal safety studies. Over 90% of the
compounds entering clinical trials fail to make it to market (Venkatesh and
Lipper,1999). The average cost to bring new chemical entity (NCE) to market is
estimated to be $770 million (Kettler, 1999). In this research article we are mainly
focused on to identify a lead molecule by using cheminformatics tools and docking
study using parallel computing. It will help chemists at the stage where lead
optimization takes place.
The thesis is divided in to seven chapters.

1.Introduction to Drug Discovery:
Highlights the importance of the research work and objectives of the
present investigations. Various cheminformatics tools used during the drug
discovery process are also reviewed in this chapter. The idea that effect of drug in
human body are mediated by specific interactions of the drug molecule with
biological macromolecules, (proteins or nucleic acids in most cases) led scientists
to the conclusion that individual chemicals are required for the biological activity
of the drug. This made for the beginning of the modern era in pharmacology, as
pure chemicals, instead of crude extracts, became the standard drugs. Examples of
drug compounds isolated from crude preparations are morphine, the active agent in
opium, and digoxin, a heart stimulant originating from Digitalis lanata. Organic
chemistry also led to the synthesis of many of the co-chemicals isolated from
biological sources.
Computational methodologies have become a crucial component of
many drug discovery programs, from hit identification to lead optimization and
beyond, and approaches such as ligand or structure based virtual screening
techniques are widely used in many discovery efforts. One key methodology to be
considered here docking of small molecules to protein binding sites, it was
pioneered during the early 1980s, and remains a highly active area of research.
When only the structure of a target and its active or binding site is available, high-
throughput docking is primarily used as a hit identification tool. However, similar
calculations are often also used later on during lead optimization, when
modifications to known active structures can quickly be tested in computer models
before compound synthesis. At a broader sense drug designing is classified in to

two major areas, The first is referred to as ligand-based drug design and the
second, structure-based drug design.
Ligand-based drug design (or indirect drug design) relies on knowledge
of other molecules that bind to the biological target of interest. These other
molecules may be used to derive a pharmacophore model that defines the
minimum necessary structural characteristics a molecule must possess in order to
bind to the target. In other words, a model of the biological target may be built
based on the knowledge of what binds to it, and this model in turn may be used to
design new molecular entities that interact with the target. Alternatively, a
quantitative structure-activity relationship (QSAR), in which a correlation between
calculated properties of molecules and their experimentally determined biological
activity, may be derived. These QSAR relationships in turn may be used to predict
the activity of new analogs.
Structure-based drug design (or direct drug design) relies on knowledge
of the three dimensional structure of the biological target obtained through
methods such as x-ray crystallography or NMR spectroscopy. If an experimental
structure of a target is not available, it may be possible to create a homology model
of the target based on the experimental structure of a related protein. Using the
structure of the biological target, candidate drugs that are predicted to bind with
high affinity and selectivity to the target may be designed using interactive
graphics and the intuition of a medicinal chemist. Alternatively various automated
computational procedures may be used to suggest new drug candidates. As
experimental methods such as X-ray crystallography and NMR, the amount of
information concerning 3D structures of biomolecular targets has increased

dramatically. In parallel, information about the structural dynamics and electronic
properties about ligands has also increased. This has encouraged the rapid
development of the structure-based drug design. Current methods for structure-
based drug design can be divided roughly into two categories. The first category is
about “finding” ligands for a given receptor, which is usually referred as database
searching. In this case, a large number of potential ligand molecules are screened
to find those fitting the binding pocket of the receptor. This method is usually
referred as ligand-based drug design. The key advantage of database searching is
that it saves synthetic effort to obtain new lead compounds. Another category of
structure-based drug design methods is about “building” ligands, which is usually
referred as receptor-based drug design. In this case, ligand molecules are built up
within the constraints of the binding pocket by assembling small pieces in a
stepwise manner. These pieces can be either individual atoms or molecular
fragments. The key advantage of such a method is that novel structures, not
contained in any database, can be suggested.
1.1 Active site identification:
Active site identification is the first step in this program. It analyzes the
protein to find the binding pocket, derives key interaction sites within the binding
pocket, and then prepares the necessary data for Ligand fragment link. The basic
inputs for this step are the 3D structure of the protein and a pre-docked ligand in
PDB format, as well as their atomic properties. Both ligand and protein atoms need
to be classified and their atomic properties should be defined, basically, into four
atomic types:
1.1.1 Hydrophobic atom: All carbons in hydrocarbon chains or in aromatic
groups.

1.1.2 H-bond donor: Oxygen and nitrogen atoms bonded to hydrogen atom(s).
1.1.3 H-bond acceptor: Oxygen and sp2 or sp hybridized nitrogen atoms with
lone electron pair(s).
1.1.4 Polar atom: Oxygen and nitrogen atoms that are neither H-bond donor
nor H-bond acceptor, sulfur, phosphorus, halogen, metal, and carbon atoms
bonded to hetero-atom(s).
The space inside the ligand binding region would be studied with
virtual probe atoms of the four types above so the chemical environment of all
spots in the ligand binding region can be known. Hence we are clear what kind of
chemical fragments can be put into their corresponding spots in the ligand binding
region of the receptor.
Structure-based drug design attempts to use the structure of proteins as a
basis for designing new ligands by applying accepted principles of molecular
recognition. The basic assumption underlying structure-based drug design is that a
good ligand molecule should bind tightly to its target. Thus, one of the most
important principles for designing or obtaining potential new ligands is to predict
the binding affinity of a certain ligand to its target and use it as a criterion for
selection.
One early method was developed by Böhm to develop a general-
purposed empirical scoring function in order to describe the binding energy. The
following “Master Equation” was derived:

where:
• desolvation – enthalpic penalty for removing the ligand from solvent
• motion – entropic penalty for reducing the degrees of freedom when a ligand
binds to its receptor
• configuration – conformational strain energy required to put
"active" conformation
• interaction – enthalpic gain for "resolvating" the ligand with its receptor
The basic idea is that the overall binding free energy can be
decomposed into independent components that are known to b
binding process. Each component reflects a certain kind of free energy alteration
during the binding process between a ligand and its target receptor. The Master
Equation is the linear combination of these components. According to Gibbs
energy equation, the relation between dissociation equilibrium constant, K
the components of free energy was built.
Various computational methods are used to estimate each of the
components of the master equation. For exampl
upon ligand binding can be used to estimate the desolvation energy. The number of
rotatable bonds frozen upon ligand binding is proportional to the motion term. The
configurational or strain energy can be estimated using
calculations. Finally the interaction energy can be estimated using methods such as
enthalpic penalty for removing the ligand from solvent
entropic penalty for reducing the degrees of freedom when a ligand
conformational strain energy required to put the ligand in its
"active" conformation
enthalpic gain for "resolvating" the ligand with its receptor
The basic idea is that the overall binding free energy can be
decomposed into independent components that are known to be important for the
binding process. Each component reflects a certain kind of free energy alteration
during the binding process between a ligand and its target receptor. The Master
Equation is the linear combination of these components. According to Gibbs
energy equation, the relation between dissociation equilibrium constant, K
the components of free energy was built.
Various computational methods are used to estimate each of the
components of the master equation. For example, the change in polar surface area
upon ligand binding can be used to estimate the desolvation energy. The number of
rotatable bonds frozen upon ligand binding is proportional to the motion term. The
configurational or strain energy can be estimated using molecular mechanics
calculations. Finally the interaction energy can be estimated using methods such as
enthalpic penalty for removing the ligand from solvent
entropic penalty for reducing the degrees of freedom when a ligand
the ligand in its
enthalpic gain for "resolvating" the ligand with its receptor
The basic idea is that the overall binding free energy can be
e important for the
binding process. Each component reflects a certain kind of free energy alteration
during the binding process between a ligand and its target receptor. The Master
Equation is the linear combination of these components. According to Gibbs free
energy equation, the relation between dissociation equilibrium constant, Kd, and
Various computational methods are used to estimate each of the
e, the change in polar surface area
upon ligand binding can be used to estimate the desolvation energy. The number of
rotatable bonds frozen upon ligand binding is proportional to the motion term. The
molecular mechanics
calculations. Finally the interaction energy can be estimated using methods such as

the change in non polar surface, statistically derived potentials of mean force, the
number of hydrogen bonds formed, etc. In practice, the components of the master
equation are fit to experimental data using multiple linear regression. This can be
done with a diverse training set including many types of ligands and receptors to
produce a less accurate but more general "global" model or a more restricted set of
ligands and receptors to produce a more accurate but less general "local" model.
1.2 Rational drug discovery
In contrast to traditional methods of drug discovery, which rely on trial-
and-error testing of chemical substances on cultured cells or animals, and matching
the apparent effects to treatments, rational drug design begins with a hypothesis
that modulation of a specific biological target may have therapeutic value. In order
for a biomolecule to be selected as a drug target, two essential pieces of
information are required. The first is evidence that modulation of the target will
have therapeutic value. This knowledge may come from, for example, disease
linkage studies that show an association between mutations in the biological target
and certain disease states. The second is that the target is "drugable". This means
that it is capable of binding to a small molecule and that its activity can be
modulated by the small molecule.
Once a suitable target has been identified, the target is normally cloned
and expressed. The expressed target is then used to establish a screening assay. In
addition, the three-dimensional structure of the target may be determined.

The search for small molecules that bind to the target is begun by
screening libraries of potential drug compounds. This may be done by using the
screening assay (a "wet screen"). In addition, if the structure of the target is
available, a virtual screen may be performed of candidate drugs. Ideally the
candidate drug compounds should be "drug-like", that is they should possess
properties that are predicted to lead to oral bioavailability, adequate chemical and
metabolic stability, and minimal toxic effects. Several methods are available to
estimate drug likeness such as Lipinski's Rule of Five and a range of scoring
methods such as Lipophilic efficiency. Several methods for predicting drug
metabolism have been proposed in the scientific literature, and a recent example is
SPORCalc. Due to the complexity of the drug design process, two terms of interest
are still serendipity and bounded rationality. Those challenges are caused by the
large chemical space describing potential new drugs without side-effects.
1.3 Computer-aided drug design
Computer-aided drug design uses computational chemistry to discover,
enhance, or study drugs and related biologically active molecules. The most
fundamental goal is to predict whether a given molecule will bind to a target and if
so how strongly. Molecular mechanics or molecular dynamics are most often used
to predict the conformation of the small molecule and to model conformational
changes in the biological target that may occur when the small molecule binds to it.
Semi-empirical, ab initio quantum chemistry methods, or density functional theory
are often used to provide optimized parameters for the molecular mechanics
calculations and also provide an estimate of the electronic properties (electrostatic

potential, polarizability, etc.) of the drug candidate that will influence binding
affinity.
Molecular mechanics methods may also be used to provide semi-
quantitative prediction of the binding affinity. Also, knowledge-based scoring
function may be used to provide binding affinity estimates. These methods use
linear regression, machine learning, neural nets or other statistical techniques to
derive predictive binding affinity equations by fitting experimental affinities to
computationally derived interaction energies between the small molecule and the
target.
Ideally the computational method should be able to predict affinity
before a compound is synthesized and hence in theory only one compound needs to
be synthesized. The reality however is that present computational methods are
imperfect and provide at best only qualitatively accurate estimates of affinity.
Therefore in practice it still takes several iterations of design, synthesis, and testing
before an optimal molecule is discovered. On the other hand, computational
methods have accelerated discovery by reducing the number of iterations required
and in addition have often provided more novel small molecule structures.
Drug design with the help of computers may be used at any of the following stages
of drug discovery:
1. hit identification using virtual screening (structure- or ligand-based design)
2. hit-to-lead optimization of affinity and selectivity (structure-based design,
QSAR, etc.)

3. lead optimization optimization of other pharmaceutical properties while
maintaining affinity
In order to overcome the insufficient prediction of binding affinity calculated by
recent scoring functions, the protein-ligand interaction and compound 3D structure
information are used to analysis. For structure-based drug design, several post-
screening analysis focusing on protein-ligand interaction has been developed for
improving enrichment and effectively mining potential candidates:
1.3.1 Consensus scoring
• Selecting candidates by voting of multiple scoring functions
• May lose the relationship between protein-ligand structural
information and scoring criterion
1.3.2 Geometric analysis
• Comparing protein-ligand interactions by visually inspecting
individual structures
• Becoming intractable when the number of complexes to be
analyzed increasing
1.3.3 Cluster analysis
• Represent and cluster candidates according to protein-ligand 3D
information
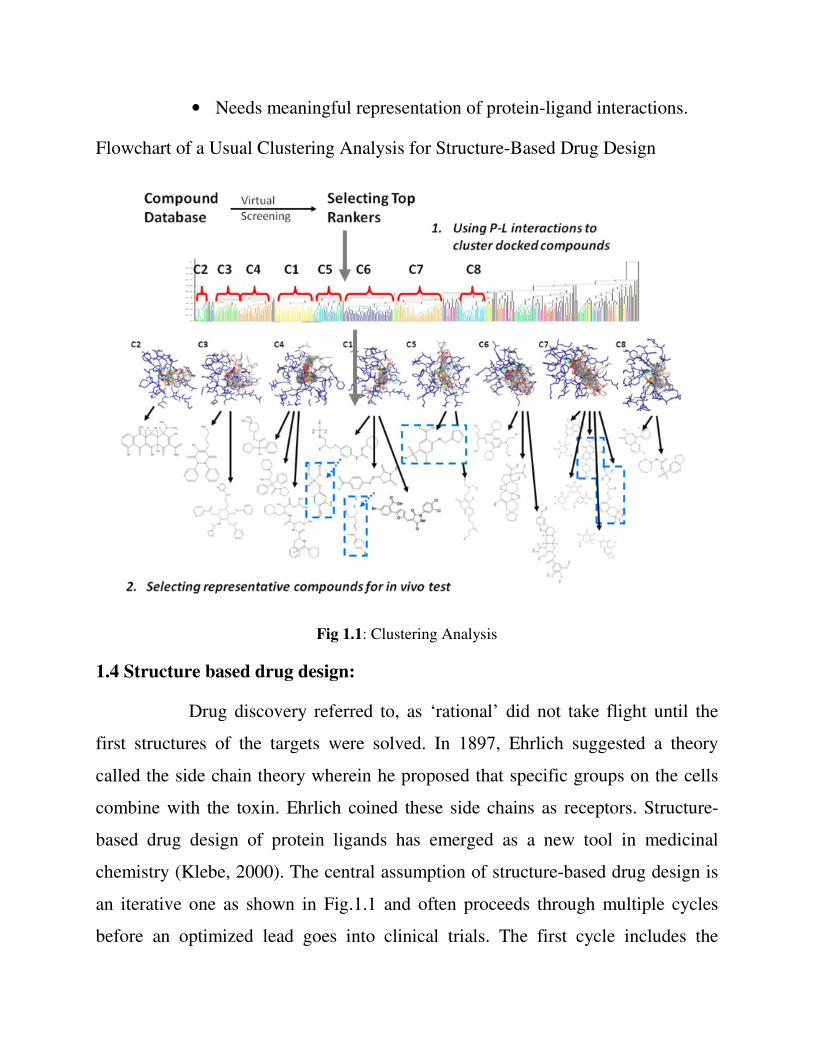
• Needs meaningful representation of protein-ligand interactions.
Flowchart of a Usual Clustering Analysis for Structure-Based Drug Design
Fig 1.1: Clustering Analysis
1.4 Structure based drug design:
Drug discovery referred to, as ‘rational’ did not take flight until the
first structures of the targets were solved. In 1897, Ehrlich suggested a theory
called the side chain theory wherein he proposed that specific groups on the cells
combine with the toxin. Ehrlich coined these side chains as receptors. Structure-
based drug design of protein ligands has emerged as a new tool in medicinal
chemistry (Klebe, 2000). The central assumption of structure-based drug design is
an iterative one as shown in Fig.1.1 and often proceeds through multiple cycles
before an optimized lead goes into clinical trials. The first cycle includes the

cloning, purification and structure determination of the target protein or nucleic
acid by one of three principal methods: X-ray crystallography, NMR or
comparative modeling. Using computer algorithms, compounds or fragments of
compounds from a database are positioned into a selected region of the structure.
These compounds are scored and ranked based on their steric and electrostatic
interactions with the target site and the best compounds are tested further with
biochemical assays. In the second cycle, structure determination of the target in
complex with a promising lead from the first cycle, one with at least micromolar
inhibition in vitro, reveals sites on the compound that can be optimized to increase
potency. Additional cycles include synthesis of the optimized lead, structure
determination of the new target: lead complex, and further optimization of the lead
compound. After several cycles of the drug design process, the optimized
compounds usually show marked improvement in binding, and often, specificity
for the target.
1.5 Evaluating a protein structure for structure based drug design:
Once a target has been identified, it is necessary to obtain accurate
structural information. There are three primary methods for structure determination
that are useful for drug-design: X-ray crystallography, NMR, and homology
modeling. High-resolution crystal structures are the most common desired source
of structural information for drug design, particularly for proteins that range in size
from a few amino acids to 998kD (Pellecchia et al., 2002). Another advantage of
crystallography is that ordered water molecules are visible in the experimental data
and are often useful in drug design. A crystal structure should be evaluated for the
resolution of the diffracted amplitudes (often simply called resolution); reliability,
or R factors; coordinate error; temperature factors; and chemical correctness.

Fig. 1.2 Steps involved in structure based drug design through in silico approach.
Typically, crystal structures determined with data extending above 2.5
A0 are acceptable for drug design purposes since they have a high data to
parameter ratio, and the placement of residues in the electron density map is
unambiguous. The R factor and R free reported for a model are measures for the
correlation between the model and experimental data. The R free value should be
below 28 % and ideally below 25 %, and the R factor should be well below 25 %
in order to use the structure in drug design. If the only structure available for a
particular target does not meet the resolution or R factor results should be judged
carefully. In principle, the drug discovery process involves three pre-clinical stages
before clinical trials, namely target selection, lead identification, and clinical
candidate selection.

Target identification
Target Validation
Lead Identification
Lead molecule optimization
Clinical trails
Due to rapid advances in structural biology and computer
technology, structurebased computer-aided drug design (CADD) using docking
techniques, virtual screening and library design, along with target/structure
focusing combinatorial chemistry, has become a powerful tool in the multi-step
process of drug discovery. As an emerging technology, CADD accelerates drug
development by making use of the accumulated information of existing drugs
and diseases, combined with inter-disciplinary inputs from other fields. This
process extensively uses mathematical models and simulation tools based on the
evaluation of potential risks from drug safety and the experimental design of
new trials (Kapetanovic, 2008). Fast expansion in this area has been made
possible by advances in software and hardware computational power and
sophistication, identification of molecular targets, and an increasing database of
publicly available target protein structures. CADD is being utilized to identify
hits (active drug candidates), select leads (most likely candidates for further
evaluation), and optimize leads i.e. transform biologically active compounds
into suitable drugs by improving their physicochemical, pharmaceutical,
ADMET/PK pharmacokinetic) properties (Pozzan, 2006). The term biological
target is frequently used in pharmaceutical research to describe the native
protein in the body whose activity is modified by a drug resulting in a desirable

therapeutic effect. In this context, the biological target is often referred to as a
drug target. The most common drug targets of currently marketed drugs include
as below (Overington et al., 2006)
• Enzymes
• ligand-gated ion channels
• voltage-gated ion channels
• G protein-coupled receptors
• nuclear hormone receptors
• Transporters
• DNA, Integrins, Miscellaneous
1.6 Species-specific genes as drug targets:
Comparative analysis of the complete genome sequences of bacterial
pathogens available in the public databases offers the first insights into drug
discovery approaches of the near future (Galperin and Koonin, 1999). An
interesting approach to the prediction of potential drug targets designated as the
differential genome display has been proposed by Huynen and co-workers
(Huynen et al., 1997). This approach relies on the fact that genome of parasitic
microorganisms are generally much smaller and code for fewer proteins than the
genomes of free-living organisms. The genes that are present in the genome of a
parasitic bacterium but absent in a closely related genome of free pathogenecity
can be considered as potential drug targets. Exhaustive comparison of H.influenzae
and E.coli gene products identified 40 H. influenzae genes that have been
exclusively found in pathogens and thus constitute potential drug targets.
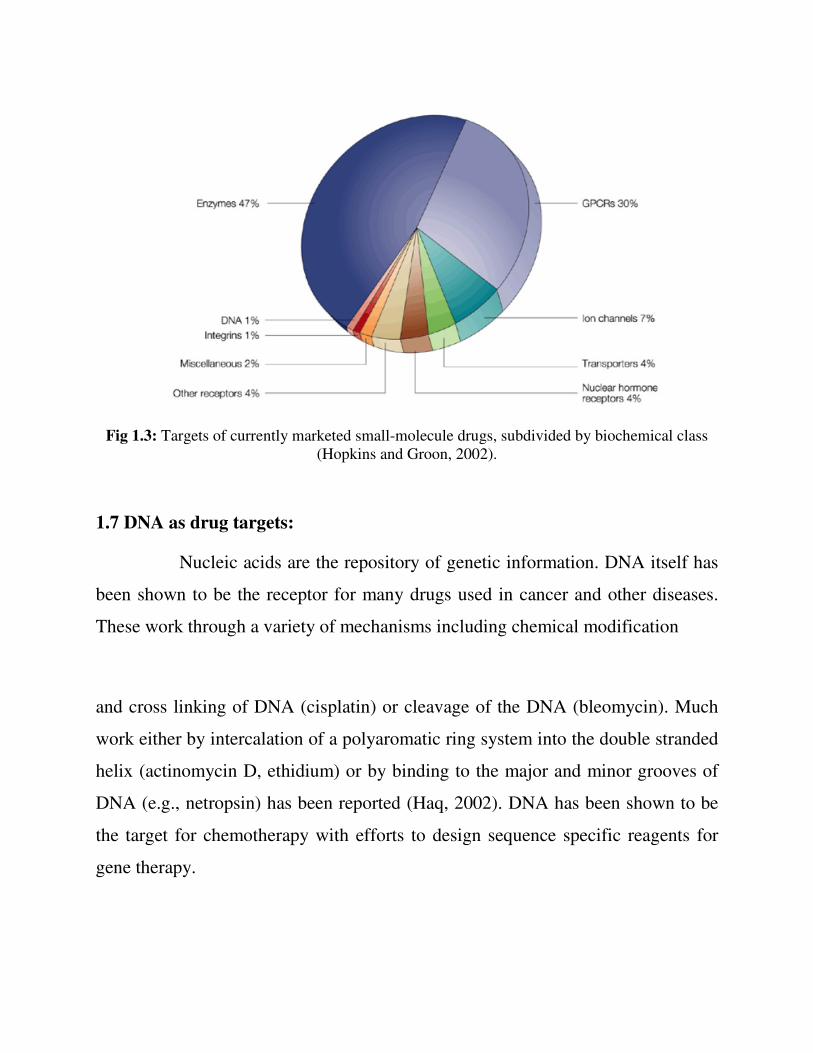
Fig 1.3: Targets of currently marketed small-molecule drugs, subdivided by biochemical class (Hopkins and Groon, 2002).
1.7 DNA as drug targets:
Nucleic acids are the repository of genetic information. DNA itself has
been shown to be the receptor for many drugs used in cancer and other diseases.
These work through a variety of mechanisms including chemical modification
and cross linking of DNA (cisplatin) or cleavage of the DNA (bleomycin). Much
work either by intercalation of a polyaromatic ring system into the double stranded
helix (actinomycin D, ethidium) or by binding to the major and minor grooves of
DNA (e.g., netropsin) has been reported (Haq, 2002). DNA has been shown to be
the target for chemotherapy with efforts to design sequence specific reagents for
gene therapy.

1.8 RNA as drug target:
Recent advances in the determination of RNA structure and function
have led to new opportunities that will have a significant impact on the
pharmaceutical industry. RNA, which, among other functions, serves as a
messenger between DNA and proteins, was thought to be an entirely flexible
molecule without significant structural complexity. However, recent studies have
revealed a surprising intricacy in RNA structure. This observation unlocks
opportunities for the pharmaceutical industry to target RNA with small molecules.
Perhaps more importantly, drugs that bind to RNA might produce effects that
cannot be achieved by drugs that bind to proteins (Ecker and Griffey, 1999). Proof
of the principle has already been provided by success of several classes of drugs
obtained from natural sources that bind to RNA or RNA-protein complexes.
1.9 Membranes as drug targets:
Membranes are significant structural elements, both in defining the
boundaries of a cell as well as providing interior compartments within the cell
associated with particular functions. Cell membranes themselves can also act as
targets for molecular recognition. An understanding of the structural and dynamic
functions of the membranes (e.g., plasma membranes and intercellular
membranes) may add to a more rational design of drug molecules with improved
permeation characteristics or specific membrane effects. Many general anesthetics
are believed to work by their physical effects when dissolved in membranes.
Several classes of antibiotics like gramicidin A, antifungals like alamethicin and
toxins such as mellitin found in bee venoms have direct effects on planar lipid
bilayers, causing transmembrane pores.

1.10 Proteins as drug targets:
Proteins continue to assume significant attention from the
pharmaceutical and biotechnology industries as a valuable source of potential drug
targets. Proteins provide the critical link between genes and disease, and as such
are the key to the understanding of basic biological processes including disease
pathology, diagnosis, and treatment. Researchers have discovered many potential
therapeutic targets, and there are currently more than 700 products in various
phases of development. However, translating the study of proteins into validated
drug targets poses substantial challenges. Genome sequences instruct cells on
how and when to make proteins. The proteins in turn are the active players in the
cell. Proteins form the machinery of cells, allow cells to communicate, and can
control growth or death of an organism. Because of their role in cells, most of the
drug targets are proteins. Drugs work by binding specifically to a protein.
Extensive knowledge about the function of a protein can guide the selection of
targets for pharmaceutical chemists. Studying the complex domain of 200,000-
300,000 distinct and interactive proteins poses substantial challenges.
Most target proteins for drug evelopment participate in key regulatory
steps in the human body or in an infectious organism. As such, they tend to be
present in few copies only and often within specific cells. Their isolation and
purification using traditional preparative biochemical means and in quantities
required for routine assays has been a formidable challenge. This situation has
been radically changed by the ability to clone and express proteins. Thus many key
target proteins are now becoming available in sufficient amounts to make them
amenable not only to biological assays but also to NMR studies in solution and to

crystallization for X-ray analysis. The number of protein structures solved using X-
ray or NMR has begun to rise sharply and more than 60,230 proteinthree-
dimensional structures have been deposited in the Protein Data Bank till date (Feb,
2013) (www. rcsb.org/pdb/) shown in
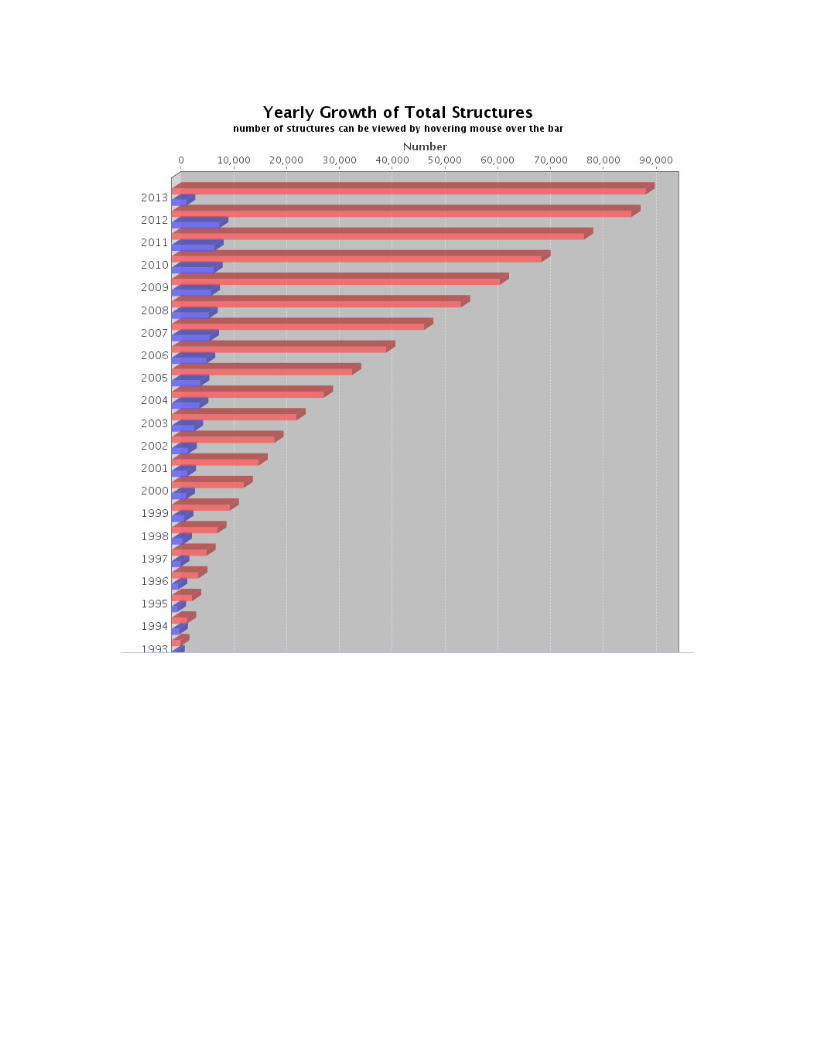
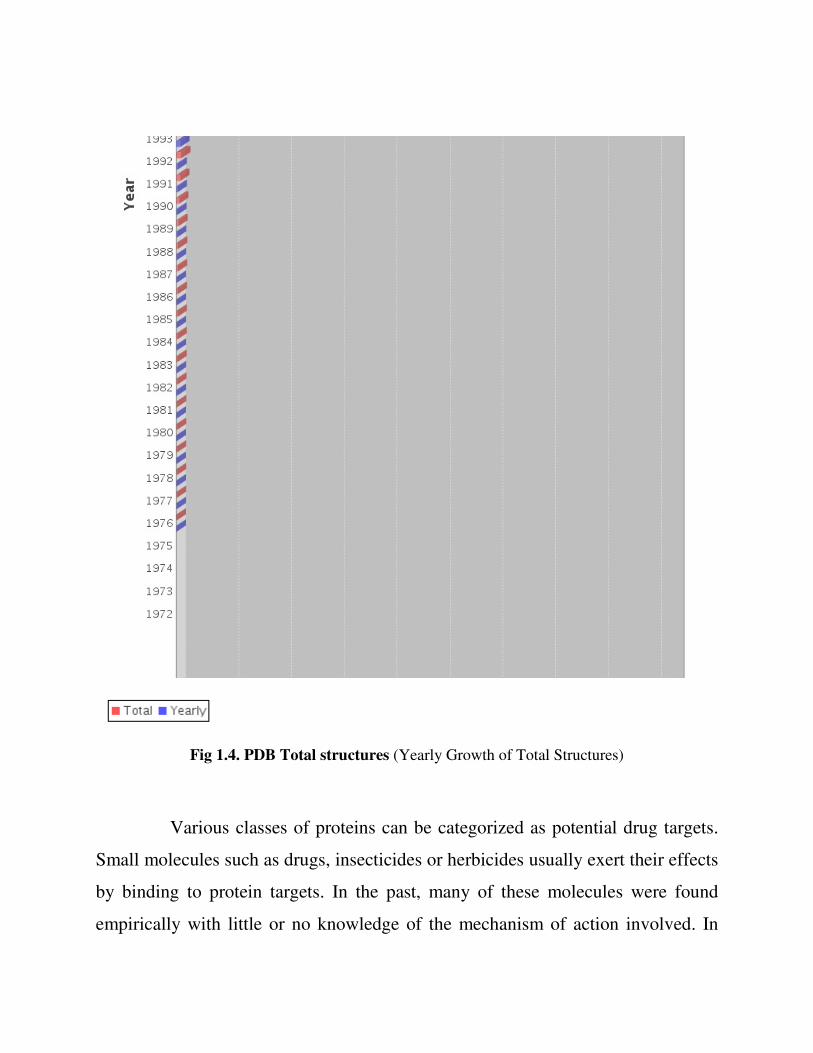
Fig 1.4. PDB Total structures (Yearly Growth of Total Structures)
Various classes of proteins can be categorized as potential drug targets.
Small molecules such as drugs, insecticides or herbicides usually exert their effects
by binding to protein targets. In the past, many of these molecules were found
empirically with little or no knowledge of the mechanism of action involved. In

many cases, the targets that are modified by these substances were identified in
retrospect. Interestingly, the majority of drugs currently in use modulate either
enzymes or receptors, most of them G-protein-coupled receptors.
1.10.1 Enzymes :
The macromolecule responsible for the catalysis of biochemical reactions
are an obvious target when a disease state is associated with production of a
biologically active species. Enzymes are a classic target for therapeutic
intervention and numerous well-studied examples exist. Traditional medicinal
chemistry enzyme targets include kinases, phosphodiesterases, proteases and
phosphotases. Some of the examples of drug targeted against enzymes are listed in
Table 1.1
1.10.2 G-proteins
G-protein–coupled receptors are a super family of seven transmembrane
spanning proteins that are activated by a wide range of extracellular ligands and are
expressed in virtually all tissues. Signaling through receptors regulate a wide
variety of physiological processes such as neurotransmission, chemotaxis,
inflammation, cell proliferation, cardiac and smooth muscle contraction as well as
visual and chemosensory perception.
In view of their widespread distribution and importance in health
and disease, it is not surprising that GPCRs are the most successful class of target
proteins for drug discovery research (Cacace et al., 2003). The sequencing of
human genome has led to the prediction of as many as 1000 GPCRs, of which 400
are nonchemosensory receptors and can therefore be considered as potential drug-
targets (Venter et al., 2001). It has been estimated that up to 50 % of all marketed

drugs directly target this family of receptors (Drews, 2000), some of which are
listed in Table 1.2.
The goal in developing drugs against the targets listed is often to
modulate the function of the human protein while the goal in developing drugs
against pathogenic organisms is total inhibition, leading to the death of the
pathogen. Antimicrobial drugs should be essential to the pathogen, have a unique
function in the pathogen, be present only in the pathogen, and be able to be
inhibited by a small molecule. The target should be essential, in that it is a part of a
crucial cycle in the cell, and its elimination should lead to the pathogen’s death.
Tab 1
Few currently marketed drugs that target GPCRs
GPCR Indication(s) Drug(s)
Histamine Allergies, ulcers Cimetidine, Ranitidine,
Terfenadine

β -adrenergic Hypertension, asthma Atenolol, Albuterol,
Salmeterol
α -adrenergic Benign
prostatichypertrophy
Terazosin, Doxazosin
Dopamine Psychosis, Parkinson’s Aripiprazole, Ropinerole
Serotonin Migraine, anxiety Zolmitriptan, Clozapine,
Buspirone
Opoid Pain Butarphanol
Angiotensin Hypertension Losartan, Eprosartan
Muscarinic acetylcoline Alzheimer’s disease Bethanechol,Dicyclomine
Leukotriene Asthma Pranlukast
The target should be unique; no other pathway should be able to
supplement the function of the target and overcome the presence of the inhibitor. If
the macromolecule satisfies all the outlined criteria to be a drug target but
functions in healthy human cells as well as in a pathogen, specificity can often be
engineered into the inhibitor by exploiting structural or biochemical differences
between the pathogenic and human forms. Finally, the target molecule should be
capable of inhibition by binding of a small molecule. Enzymes are often excellent
drug targets because compounds are designed to fit within the active site pocket.

Structures determined by nuclear magnetic resonance, using a
concentrated protein or nucleic acid in solution are also valuable sources for drug
design (Pellecchia et al., 2002). Since the target is in solution it is sometimes
possible to interpret the dynamics of the target from the data. If no experimentally
determined structure is available, a homology model can be used for drug design
(Enyedy et al., 2001). To evaluate a homology model, SWISS MODEL outputs a
confidence factor per residue that reflects the amount of structural information
used to create that portion of the model. Using the structural information obtained
through the above techniques, the structure is then prepared for drug design
programs.
1.11 Present state of the art: Computer-aided drug design:
Given the vast size of organic chemical space drug discovery cannot be
reduced to a simple “synthesize and test” drudgery (Kuntz, 1992). There is an
urgent need to identify and/ or design drug-like molecules from the vast expanse of
what could be synthesized. In silico methods have the potential to reduce both time
and cost in developing suggestions on drug/ lead-like molecules. Computational
tools have the advantage for delivering new lead candidate more quickly and at
lower cost. Drug discovery in the 21st century is expected to be different in at least
two distinct ways: development of individualized medicine departing from
genomic information and extensive use of in silico simulations to facilitate target
identification, structure prediction and lead/drug discovery.

The expectations from computational methods for reliable and
expeditious protocols for developing suggestions on potential leads are
continuously on the increase. Several conceptual and methodological concerns
remain before an automation of drug design in silico could be contemplated.
Computational methods are needed to exploit the structural information to
understand specific molecular recognition events and to elucidate the function of
the target macromolecule (Fig. 1.4). This information should ultimately lead to the
design of small molecule ligands for the target, which will block/activate its
normal function and thereby act as improved drugs.
As structural genomics, bioinformatics, and computational power
continue to explode with new advances, further successes in structure-based drug
design are likely to follow. Each year, new targets are being identified; structures
of those targets are being determined at an amazing rate, and capability to capture a
quantitative picture of the interactions between macromolecules and ligands is
accelerating.
1.12 Success of computer-assisted molecular design:
The greatest success of computer-aided structure-based drug design to
date is the HIV-1 protease inhibitors that have been approved by the United States
Food and Drug Administration and reached the market (Wlodawer and Vondrasek,
1998). An example of the success story is that of SAR work carried out on
antibacterial agent, Norfloxacin showed 6-fluro derivative of norfloxacin being
500 fold more potent over nalidixic acid. Other examples of drugs that were
developed using computer –assisted drug design include Captopril
(antihypertensive), Crixican (anti- HIV) (Greer et al., 1994), Teveten

(antihypertensive) (Keenan et al., 1993), Aricept(for Alzheimers disease).
1.13 Utility of Homology Models in the Drug Discovery Process:
Advances in bioinformatics and protein modeling algorithms, in addition
to the enormous increase in experimental protein structure information, have aided
in generation of databases that comprise homology models of a significant portion
of known genomic protein sequences. Currently, 3D structure information can be
generated for up to 56 % of all known proteins. However, there is considerable
controversy concerning the real value of homology models for drug design.
Despite the numerous uncertainties that are associated with homology modeling,
recent research has shown that this approach can be used to significant advantage
in the identification and validation of drug targets, as well as for the identification
and optimization of lead compounds. Homology modelbased drug design has been
applied to epidermal growth factor receptor tyrosine (Kawakami et al., 1996),
Trusopt (for Glaucoma) (Geer et al., 1994) and Zomig (for migraine) (Glen et al.,
1995).

Fig 1.4. Holistic approach in an in silico method of drug discovery
Drug target
identification In silico
generation of
candidates
Molecular
modeling
targets with
candidates
Filters to
asses
drug
likeness
Lead like
compounds Affinity
based
virtual
screening
Further
optimizatio
n
Leads

kinase protein (Ghosh et al., 2001), Bruton’s tyrosine kinase (Mahajan
et al.,1999), Janus kinase 3 (sudbeck et al., 1999) and human aurora 1 and 2
kinases (Venkayalapati et al., 2003). In the thesis, focus is on the application of
homology model of hHH2R of GPCR family used to design new drug molecule.
Thus, it can be said that pharmaceutical and biotechnology research has undergone
great change. Traditionally, the crucial impasse in the industry’s search for new
drug targets was the availability of biological data. Now with the advent of human
genomic sequence, bioinformatics offers several approaches for the prediction of
structure and function of proteins on the basis of sequence and structural
similarities.
The protein sequence to structure and to function relationship is well
established and reveals that the structural details at atomic level help understand
molecular function of proteins. Impressive technological advances in areas such as
structural characterization of biomacromolecules, computer sciences and molecular
biology have made rational drug design feasible and present a holistic approach.
Drug discovery is perhaps the area of research in which
chemoinformatics has found the greatest scope. Drug discovery generally follows a
set of common stages as depicted below.

Fig 1.6: An illustration of the typical workflow of a drug discovery endeavor
First, a biological target is identified that is screened against many
thousands of molecules in parallel. The results from these screens are referred to as
hits. A number of the hits will be followed up as leads with various profiling
analyses to determine whether any of these molecules are suitable for the target of
interest. The leads can then be converted to candidates by optimizing on the
biological activity and other objectives of interest, such as the number of synthetic
steps. Once a suitable candidate has been designed, the candidate enters preclinical
development. Chemoinformatics is involved from initially designing screening
libraries, to determining hits to take forward to lead optimization and the
determination of candidates by application of a variety of tools. The lifecycle of
drug discovery can take many years and hundreds of millions of dollars to achieve.
Recent estimates are leaning toward an average drug discovery that lasts at least a
decade (10–15 years) and costs almost a billion US dollars ($897 million) in
bringing a single drug to market, with many avenues necessarily being explored
throughout the process [DiMasi et al. 2003]. However these figures are only
estimates, and there is no doubt that the discovery of new medical treatments is a
very complex process and the field of chemoinformatics is assisting in allowing us
to explore new areas that we have so far been unable to access.

In this research article the tool that has been majorly used is
Discovery Studio 1.5 from Accelrys. Discovery Studio facilitates modeling and
simulation solutions for protein modeling and computational chemistry. Discovery
Studio provides the most advanced software solutions for life science researchers
available today. From project conception to lead optimization, Discovery Studio
includes a diverse collection of sophisticated software applications to take your
research to the next level, all conveniently packaged into a single, easy-to use
Linux- or Windows-based environment. Because Discovery Studio is built upon
Pipeline Pilot™, Accelrys' scientific operating platform, any software that you
need can be integrated into the research environment, whether it's software from
Accelrys, in-house developers, or other vendors.

Fig 1.7: Discovery Studio facilitates modeling and simulation solutions for protein modeling and computational chemistry.