introduction to computational chemistry. its.unc.edu 2 outline introduction methods in...
Post on 21-Dec-2015
282 views
TRANSCRIPT

Introduction to Introduction to Computational Computational
Chemistry Chemistry
Introduction to Introduction to Computational Computational
Chemistry Chemistry Shubin Liu, Ph.D.
Research Computing Center
University of North Carolina at Chapel Hill

its.unc.edu 2
OutlineOutline
Introduction
Methods in Computational Chemistry
•Ab Initio
•Semi-Empirical
•Density Functional Theory
•New Developments (QM/MM)
Hands-on ExercisesThe PPT format of this presentation is available here:
http://its2.unc.edu/divisions/rc/training/scientific//afs/isis/depts/its/public_html/divisions/rc/training/scientific/short_courses/
its.unc.edu 3
About UsAbout Us
ITS – Information Technology Services
• http://its.unc.edu
• http://help.unc.edu
• Physical locations: 401 West Franklin St. 211 Manning Drive
• 10 Divisions/Departments Information Security IT Infrastructure and Operations
Research Computing Center Teaching and Learning
User Support and Engagement Office of the CIO
Communication Technologies Communications
Enterprise Applications Finance and Administration

its.unc.edu 4
Research ComputingResearch Computing
Where and who are we and what do we do?• ITS Manning: 211 Manning Drive
• Website
http://its.unc.edu/research-computing.html
• Groups
Infrastructure -- Hardware
User Support -- Software
Engagement -- Collaboration

its.unc.edu 5
About MyselfAbout Myself
Ph.D. from Chemistry, UNC-CH
Currently Senior Computational Scientist @ Research Computing Center, UNC-CH
Responsibilities:
• Support Computational Chemistry/Physics/Material Science software
• Support Programming (FORTRAN/C/C++) tools, code porting, parallel computing, etc.
• Offer short courses on scientific computing and computational chemistry
• Conduct research and engagement projects in Computational Chemistry Development of DFT theory and concept tools
Applications in biological and material science systems

its.unc.edu 6
About YouAbout You
Name, department, research interest?
Any experience before with high performance computing?
Any experience before with computational chemistry research?
Do you have any real problem to solve with computational chemistry approaches?

its.unc.edu 7
Think BIG!!!Think BIG!!!
What is not chemistry?• From microscopic world, to nanotechnology, to daily life, to
environmental problems
• From life science, to human disease, to drug design
• Only our mind limits its boundary
What cannot computational chemistry deal with?• From small molecules, to DNA/proteins, 3D crystals and
surfaces
• From species in vacuum, to those in solvent at room temperature, and to those under extreme conditions (high T/p)
• From structure, to properties, to spectra (UV, IR/Raman, NMR, VCD), to dynamics, to reactivity
• All experiments done in labs can be done in silico
• Limited only by (super)computers not big/fast enough!

its.unc.edu 8
Central Theme of Computational Chemistry
Central Theme of Computational Chemistry
DYNAMICS
REACTIVITY
STRUCTURE CENTRAL DOGMA OF MOLECULAR BIOLOGY
SEQUENCE
STRUCTURE
DYNAMICS
FUNCTION
EVALUTION

its.unc.edu 9
Multiscale Hierarchy of Modeling
Multiscale Hierarchy of Modeling

its.unc.edu 10
What is Computational Chemistry?
What is Computational Chemistry?
Application of computational methods and algorithms in chemistry
• Quantum Mechanicali.e., via Schrödinger Equation
also called Quantum Chemistry
• Molecular Mechanical i.e., via Newton’s law F=ma
also Molecular Dynamics
• Empirical/Statisticale.g., QSAR, etc., widely used in clinical and medicinal chemistry
Focus TodayFocus Today
Ht
i ˆ
Ht
i ˆ

its.unc.edu 11
How Big Systems Can We Deal with?
How Big Systems Can We Deal with?
Assuming typical computing setup (number of CPUs, memory, disk space, etc.)
Ab initio method: ~100 atoms
DFT method: ~1000 atoms
Semi-empirical method: ~10,000 atoms
MM/MD: ~100,000 atoms

its.unc.edu 12
ij
n
1i ij
n
1i
N
1 i
2i
2
r
1
r
Z-
2m
h- H
n
ij
n
1i ij
n
1i r
1ih H
Starting Point: Time-Independent Schrodinger
Equation
Starting Point: Time-Independent Schrodinger
Equation
EH
Ht
i ˆ
Ht
i ˆ
its.unc.edu 13
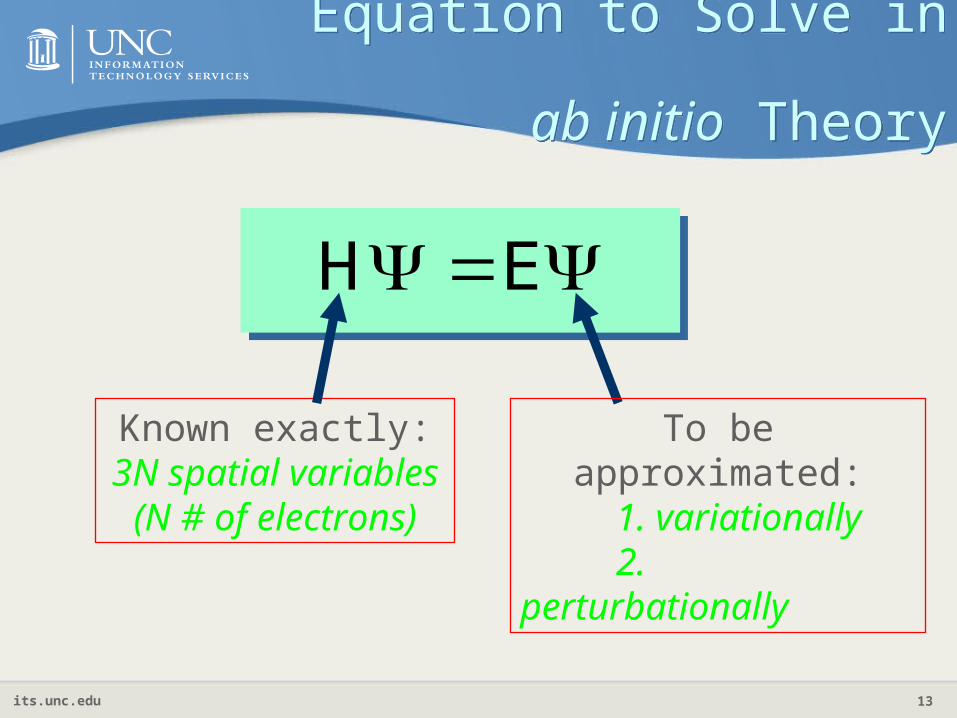
Equation to Solve in ab initio Theory
Equation to Solve in ab initio Theory
EH
Known exactly:3N spatial variables
(N # of electrons)
To be approximated:1. variationally2. perturbationally

its.unc.edu 14
Hamiltonian for a Molecule
Hamiltonian for a Molecule
kinetic energy of the electrons kinetic energy of the nuclei electrostatic interaction between the electrons
and the nuclei electrostatic interaction between the electrons electrostatic interaction between the nuclei
nuclei
BA AB
BAelectrons
ji ij
nuclei
A iA
Aelectrons
iA
nuclei
A Ai
electrons
i e
R
ZZe
r
e
r
Ze
mm
22
22
22
2
22ˆ H
nuclei
BA AB
BAelectrons
ji ij
nuclei
A iA
Aelectrons
iA
nuclei
A Ai
electrons
i e
R
ZZe
r
e
r
Ze
mm
22
22
22
2
22ˆ H

its.unc.edu 15
Ab Initio Methods
Ab Initio Methods
Accurate treatment of the electronic distribution using the full Schrödinger equation
Can be systematically improved to obtain chemical accuracy
Does not need to be parameterized or calibrated with respect to experiment
Can describe structure, properties, energetics and reactivity
What does “ab intio” mean?
• Start from beginning, with first principle Who invented the word of the “ab initio” method?
• Bob Parr of UNC-CH in 1950s; See Int. J. Quantum Chem. 37(4), 327(1990) for details.

its.unc.edu 16
Three Approximations Three Approximations
Born-Oppenheimer approximation
• Electrons act separately of nuclei, electron and nuclear coordinates are independent of each other, and thus simplifying the Schrödinger equation
Independent particle approximation
• Electrons experience the ‘field’ of all other electrons as a group, not individually
• Give birth to the concept of “orbital”, e.g., AO, MO, etc.
LCAO-MO approximation
• Molecular orbitals (MO) can be constructed as linear combinations of atom orbitals, to form Slater determinants

its.unc.edu 17
Born-Oppenheimer Approximation
Born-Oppenheimer Approximation
the nuclei are much heavier than the electrons and move more slowly than the electrons
freeze the nuclear positions (nuclear kinetic energy is zero in the electronic Hamiltonian)
calculate the electronic wave function and energy
E depends on the nuclear positions through the nuclear-electron attraction and nuclear-nuclear repulsion terms
E = 0 corresponds to all particles at infinite separation
nuclei
BA AB
BAelectrons
ji ij
nuclei
A iA
Aelectrons
ii
electrons
i eel r
ZZe
r
e
r
Ze
m
2222
2
2ˆ H
nuclei
BA AB
BAelectrons
ji ij
nuclei
A iA
Aelectrons
ii
electrons
i eel r
ZZe
r
e
r
Ze
m
2222
2
2ˆ H
d
dEE
elel
elelel
elelel *
* ˆ,ˆ
HH
d
dEE
elel
elelel
elelel *
* ˆ,ˆ
HH

its.unc.edu 18
Approximate Wavefunctions
Approximate Wavefunctions
Construction of one-electron functions (molecular orbitals, MO’s) as linear combinations of one-electron atomic basis functions (AOs) MO-LCAO approach.
Construction of N-electron wavefunction as linear combination of anti-symmetrized products of MOs (these anti-symmetrized products are denoted as Slater-determinants).
down)-(spin
up)-(spin ;
1
iiu ik
N
kklil rq
down)-(spin
up)-(spin ;
1
iiu ik
N
kklil rq

its.unc.edu 19
The Slater DeterminantThe Slater Determinant
zcbazcba
zzzz
cccc
bbbb
aaaa
n
zcbazcban
zcba
n
n
n
n
n
nn
n
321
321
321
321
321
312321
321 Α
!1
!1
zcbazcba
zzzz
cccc
bbbb
aaaa
n
zcbazcban
zcba
n
n
n
n
n
nn
n
321
321
321
321
321
312321
321 Α
!1
!1

its.unc.edu 20
The Two Extreme Cases
The Two Extreme Cases
One determinant: The Hartree–Fock method.
All possible determinants: The full CI method.
NN 321 321HF NN 321 321HF
There are N MOs and each MO is a linear combination of N AOs. Thus, there are nN coefficients ukl, which are determined by making stationary the functional:
The ij are Lagrangian multipliers.
N
lkijljklki
N
jiij uSuHE
1,
*
1,HFHFHF ˆ
N
lkijljklki
N
jiij uSuHE
1,
*
1,HFHFHF ˆ

its.unc.edu 21
The Full CI MethodThe Full CI Method
The full configuration interaction (full CI) method expands the wavefunction in terms of all possible Slater determinants:
There are possible ways to choose n molecular orbitals from a set of 2N AO basis functions.
The number of determinants gets easily much too large. For example:
n
N2
1ˆ ;
2
1,CICICI
2
1CI
cScHEc
n
N
*n
N
1ˆ ;
2
1,CICICI
2
1CI
cScHEc
n
N
*n
N
91010
40
91010
40
Davidson’s method can be used to find one or a few eigenvalues of a matrix of rank 109.

its.unc.edu 22
NN 321 321HF NN 321 321HF
N
lkijljklki
N
jiij uSuHE
1,
*
1,HFHFHF ˆ
N
lkijljklki
N
jiij uSuHE
1,
*
1,HFHFHF ˆ
N
ilikikl
N
lkklmn
N
nmmn uuPnlmkPhPEH
1
*
1,21
1,nucHFHF ; ˆ
N
ilikikl
N
lkklmn
N
nmmn uuPnlmkPhPEH
1
*
1,21
1,nucHFHF ; ˆ
0HF
Euki
0HF
Euki
Hartree–Fock equations
The Hartree–Fock MethodThe Hartree–Fock Method

its.unc.edu 23
|S Overlap integral
|
2
1|PHF
ii
occ
i
cc2PDensity Matrix
SF iii cc
The Hartree–Fock Method
The Hartree–Fock Method

its.unc.edu 24
1. Choose start coefficients for MO’s
2. Construct Fock Matrix with coefficients
3. Solve Hartree-Fock-Roothaan equations
4. Repeat 2 and 3 until ingoing and outgoing
coefficients are the same
Self-Consistent-Field (SCF)
Self-Consistent-Field (SCF)
SF iii cc

its.unc.edu 25
Semi-empirical methods(MNDO, AM1, PM3, etc.)
Semi-empirical methods(MNDO, AM1, PM3, etc.)
Full CIFull CI
perturbational hierarchy(CASPT2, CASPT3)
perturbational hierarchy(CASPT2, CASPT3)
perturbational hierarchy(MP2, MP3, MP4, …)
perturbational hierarchy(MP2, MP3, MP4, …)
excitation hierarchy(MR-CISD)
excitation hierarchy(MR-CISD)
excitation hierarchy(CIS,CISD,CISDT,...)
(CCS, CCSD, CCSDT,...)
excitation hierarchy(CIS,CISD,CISDT,...)
(CCS, CCSD, CCSDT,...)
Multiconfigurational HF(MCSCF, CASSCF)
Multiconfigurational HF(MCSCF, CASSCF)
Hartree-Fock(HF-SCF)
Hartree-Fock(HF-SCF)
Ab Initio MethodsAb Initio Methods

its.unc.edu 26
Who’s Who

its.unc.edu 27
Size vs AccuracySize vs Accuracy
Number of atoms
0.1
1
10
1 10 100 1000
Acc
urac
y (k
cal/m
ol) Coupled-cluster,
Multireference
Nonlocal density functional,Perturbation theory
Local density functional,Hartree-Fock
Semiempirical Methods
Full CI

its.unc.edu 28
ROO,e= 291.2 pm 96.4 pm
95.7 pm 95.8 pm
symmetry: Cs
Equilibrium structure of (HEquilibrium structure of (H22O)O)22
W.K., J.G.C.M. van Duijneveldt-van de Rijdt, and W.K., J.G.C.M. van Duijneveldt-van de Rijdt, and
F.B. van Duijneveldt, F.B. van Duijneveldt, Phys. Chem. Chem. Phys.Phys. Chem. Chem. Phys. 22, 2227 (2000)., 2227 (2000).
Experimental [J.A. Odutola and T.R. Dyke, J. Chem. Phys 72, 5062 (1980)]: ROO
2 ½ = 297.6 ± 0.4 pm
SAPT-5s potential [E.M. Mas et al., J. Chem. Phys. 113, 6687 (2000)]: ROO
2 ½ – ROO,e= 6.3 pm ROO,e(exptl.) = 291.3 pm
AN EXAMPLE
its.unc.edu 29
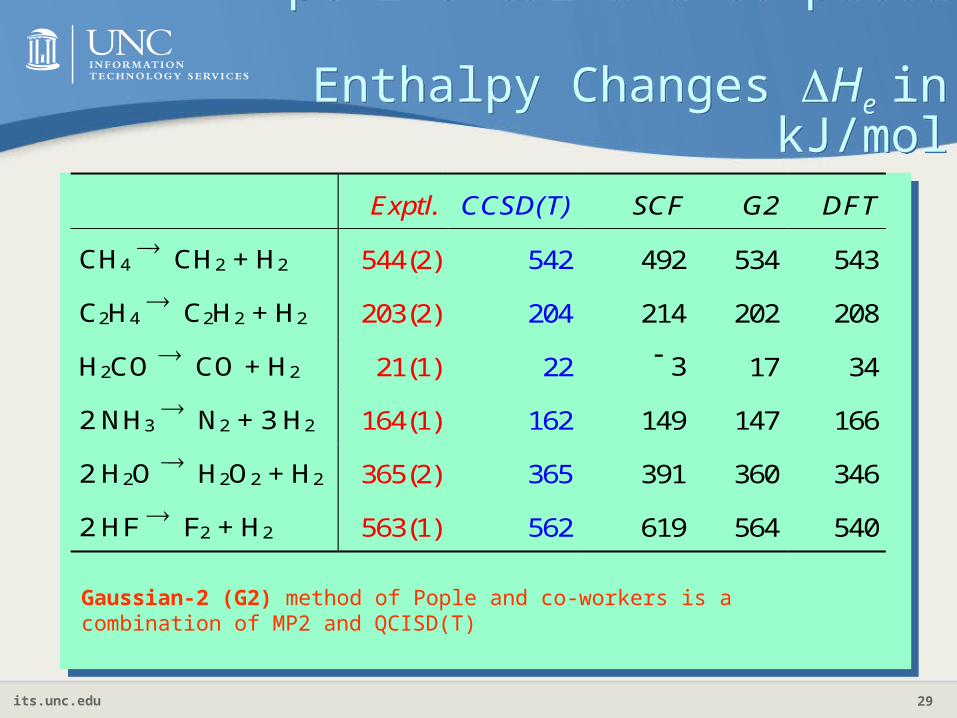
Experimental and Computed Enthalpy Changes He in
kJ/mol
Experimental and Computed Enthalpy Changes He in
kJ/mol
Exptl. CCSD(T) SCF G2 DFT
CH4 CH2 + H2 544(2) 542 492 534 543
C2H4 C2H2 + H2 203(2) 204 214 202 208
H2CO CO + H2 21(1) 22 3 17 34
2 NH3 N2 + 3 H2 164(1) 162 149 147 166
2 H2O H2O2 + H2 365(2) 365 391 360 346
2 HF F2 + H2 563(1) 562 619 564 540
Exptl. CCSD(T) SCF G2 DFT
CH4 CH2 + H2 544(2) 542 492 534 543
C2H4 C2H2 + H2 203(2) 204 214 202 208
H2CO CO + H2 21(1) 22 3 17 34
2 NH3 N2 + 3 H2 164(1) 162 149 147 166
2 H2O H2O2 + H2 365(2) 365 391 360 346
2 HF F2 + H2 563(1) 562 619 564 540
Gaussian-2 (G2) method of Pople and co-workers is a combination of MP2 and QCISD(T)

its.unc.edu 30
LCAO Basis FunctionsLCAO Basis Functions
’s, which are atomic orbitals, are called basis functions
usually centered on atoms
can be more general and more flexible than atomic orbital functions
larger number of well chosen basis functions yields more accurate approximations to the molecular orbitals
c
c

its.unc.edu 31
Basis FunctionsBasis Functions
Slaters (STO)
Gaussians (GTO)
Angular part *
Better behaved than Gaussians
2-electron integrals hard
2-electron integrals simpler
Wrong behavior at nucleus
Decrease too fast with r
r)exp( r)exp(
2nml rexp*zyx 2nml rexp*zyx

its.unc.edu 32
Contracted Gaussian Basis Set
Contracted Gaussian Basis Set
Minimal
STO-nG
Split Valence: 3-21G,4-31G,6-31G
• Each atom optimized STO is fit with n GTO’s
• Minimum number of AO’s needed
• Each atom optimized STO is fit with n GTO’s
• Minimum number of AO’s needed
• Contracted GTO’s optimized per atom• Doubling of the number of valence AO’s
• Contracted GTO’s optimized per atom• Doubling of the number of valence AO’s

its.unc.edu 33
Polarization / Diffuse Functions
Polarization / Diffuse Functions
Polarization: Add AO with higher angular momentum (L) to give more flexibility
Example: 3-21G*, 6-31G*, 6-31G**, etc.
Diffusion: Add AO with very small exponents for systems with very diffuse electron densities such as anions or excited statesExample: 6-31+G*, 6-311++G**

its.unc.edu 34
Correlation-Consistent Basis Functions
Correlation-Consistent Basis Functions
a family of basis sets of increasing size
can be used to extrapolate to the basis set limit
cc-pVDZ – DZ with d’s on heavy atoms, p’s on H
cc-pVTZ – triple split valence, with 2 sets of d’s and one set of f’s on heavy atoms, 2 sets of p’s and 1 set of d’s on hydrogen
cc-pVQZ, cc-pV5Z, cc-pV6Z
can also be augmented with diffuse functions (aug-cc-pVXZ)

its.unc.edu 35
Pseudopotentials, Effective Core Potentials
Pseudopotentials, Effective Core Potentials
core orbitals do not change much during chemical interactions
valence orbitals feel the electrostatic potential of the nuclei and of the core electrons
can construct a pseudopotential to replace the electrostatic potential of the nuclei and of the core electrons
reduces the size of the basis set needed to represent the atom (but introduces additional approximations)
for heavy elements, pseudopotentials can also include of relativistic effects that otherwise would be costly to treat

its.unc.edu 36
Correlation EnergyCorrelation Energy
HF does not include correlations anti-parallel electrons
Eexact – EHF = Ecorrelation
Post HF Methods:
• Configuration Interaction (CI, MCSCF, CCSD)
• Møller-Plesset Perturbation series (MP2, MP4)
Density Functional Theory (DFT)

its.unc.edu 37
Configuration-Interaction (CI)
Configuration-Interaction (CI)
In Hartree-Fock theory, the n-electron wavefunction is approximated by one single Slater-determinant, denoted as:
This determinant is built from n orthonormal spin-orbitals. The spin-orbitals that form are said to be occupied. The other orthonormal spin-orbitals that follow from the Hartree-Fock calculation in a given one-electron basis set of atomic orbitals (AOs) are known as virtual orbitals. For simplicity, we assume that all spin-orbitals are real.
In electron-correlation or post-Hartree-Fock methods, the wavefunction is expanded in a many-electron basis set that consists of many determinants. Sometimes, we only use a few determinants, and sometimes, we use millions of them:
In this notation, is a Slater-
determinant that is obtained by replacing a certain number of
occupied orbitals by virtual ones.
Three questions: 1. Which determinants should we include? 2. How do we determine the expansion coefficients? 3. How do we evaluate the energy (or other properties)?
HF
HF
cHFCI

its.unc.edu 38
Truncated configuration interaction: CIS, CISD, CISDT, etc.
Truncated configuration interaction: CIS, CISD, CISDT, etc.
We start with a reference wavefunction, for example the Hartree-Fock determinant.
We then select determinants for the wavefunction expansion by substituting orbitals of the reference determinant by orbitals that are not occupied in the reference state (virtual orbitals).
Singles (S) indicate that 1 orbital is replaced, doubles (D) indicate 2 replacements, triples (T) indicate 3 replacements, etc., leading to CIS, CISD, CISDT, etc.
NNkji 321HF NNkji 321HF
etc. ,321 ,321 NN NkbaabijNkja
ai etc. ,321 ,321 NN Nkba
abijNkja
ai
its.unc.edu 39
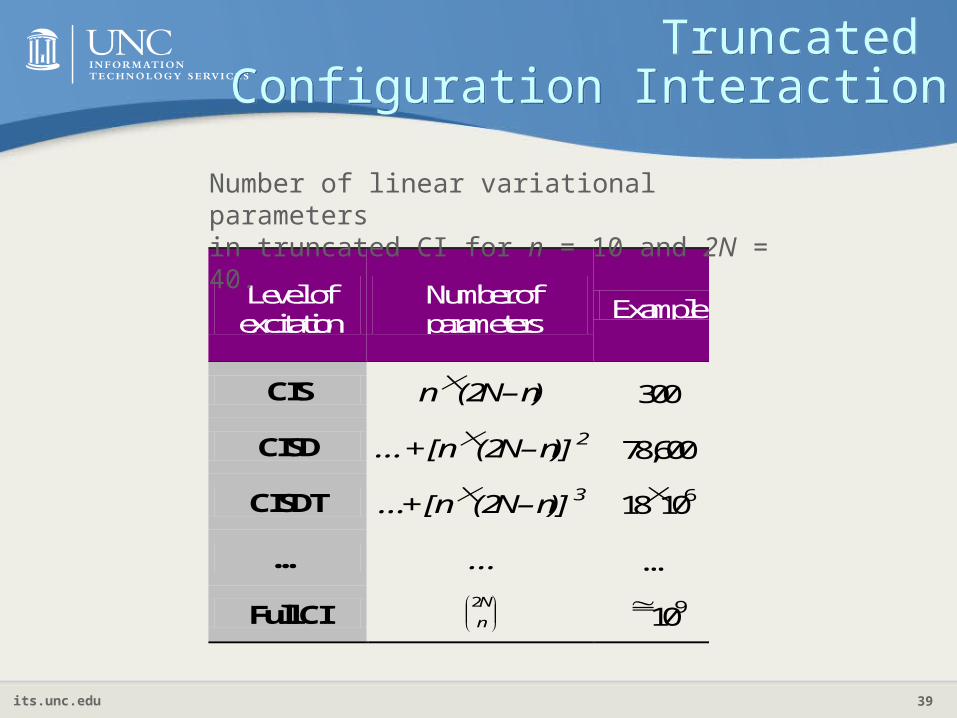
Truncated Configuration Interaction
Truncated Configuration Interaction
Level of excitation
Number of parameters
Example
CIS n (2N – n) 300
CISD … + [n (2N – n)] 2 78,600
CISDT …+ [n (2N – n)] 3 18106
… … …
Full CI
n
N2 109
Number of linear variational parametersin truncated CI for n = 10 and 2N = 40.

its.unc.edu 40
Multi-Configuration Self-Consistent Field (MCSCF)
Multi-Configuration Self-Consistent Field (MCSCF)
The MCSCF wavefunctions consists of a few selected determinants or CSFs. In the MCSCF method, not only the linear weights of the determinants are variationally optimized, but also the orbital coefficients.
One important selection is governed by the full CI space spanned by a number of prescribed active orbitals (complete active space, CAS). This is the CASSCF method. The CASSCF wavefunction contains all determinants that can be constructed from a given set of orbitals with the constraint that some specified pairs of - and -spin-orbitals must occur in all determinants (these are the inactive doubly occupied spatial orbitals).
Multireference CI wavefunctions are obtained by applying the excitation operators to the individual CSFs or determinants of the MCSCF (or CASSCF) reference wave function.
kCCck
kkk )ˆˆ(CISD-MR 21 k
kk
kk kdCkCc 21ˆ)ˆ(MRCI-IC
Internally-contracted MRCI:

its.unc.edu 41
Coupled-Cluster Theory
Coupled-Cluster Theory
System of equations is solved iteratively (the convergence is accelerated by utilizing Pulay’s method, “direct inversion in the iterative subspace”, DIIS).
CCSDT model is very expensive in terms of computer resources. Approximations are introduced for the triples: CCSD(T), CCSD[T], CCSD-T.
Brueckner coupled-cluster (e.g., BCCD) methods use Brueckner orbitals that are optimized such that singles don’t contribute.
By omitting some of the CCSD terms, the quadratic CI method (e.g., QCISD) is obtained.

its.unc.edu 42
Møller-Plesset Perturbation Theory
Møller-Plesset Perturbation Theory
The Hartree-Fock function is an eigenfunction of the
n-electron operator .
We apply perturbation theory as usual after decomposing the Hamiltonian into two parts:
More complicated with more than one reference determinant (e.g., MR-PT, CASPT2, CASPT3, …)
F
FHH
FH
HHH
ˆˆˆ
ˆˆ
ˆˆ
1
0
10
FHH
FH
HHH
ˆˆˆ
ˆˆ
ˆˆ
1
0
10
MP2, MP3, MP4, …etc.number denotes order to which energy is computed (2n+1 rule)

its.unc.edu 43
Semi-Empirical MethodsSemi-Empirical Methods
These methods are derived from the Hartee–Fock model, that is, they are MO-LCAO methods.
They only consider the valence electrons. A minimal basis set is used for the valence shell. Integrals are restricted to one- and two-center integrals and
subsequently parametrized by adjusting the computed results to experimental data.
Very efficient computational tools, which can yield fast quantitative estimates for a number of properties. Can be used for establishing trends in classes of related molecules, and for scanning a computational poblem before proceeding with high-level treatments.
A not of elements, especially transition metals, have not be parametrized

its.unc.edu 44
Semi-Empirical MethodsSemi-Empirical Methods
Number 2-electron integrals () is n4/8, n = number of basis
functions
Treat only valence electrons explicit
Neglect large number of 2-electron integrals
Replace others by empirical parameters
Models:
• Complete Neglect of Differential Overlap (CNDO)
• Intermediate Neglect of Differential Overlap (INDO/MINDO)
• Neglect of Diatomic Differential Overlap (NDDO/MNDO, AM1, PM3)

its.unc.edu 45
AB
ABVUH
AB
ABVUH Ufrom atomic spectraVvalue per atom pair
0H 0H on the same atom
SH AB SH AB BAAB 21 BAAB 21
One parameter per element
Approximations of 1-e integrals
Approximations of 1-e integrals
its.unc.edu 46
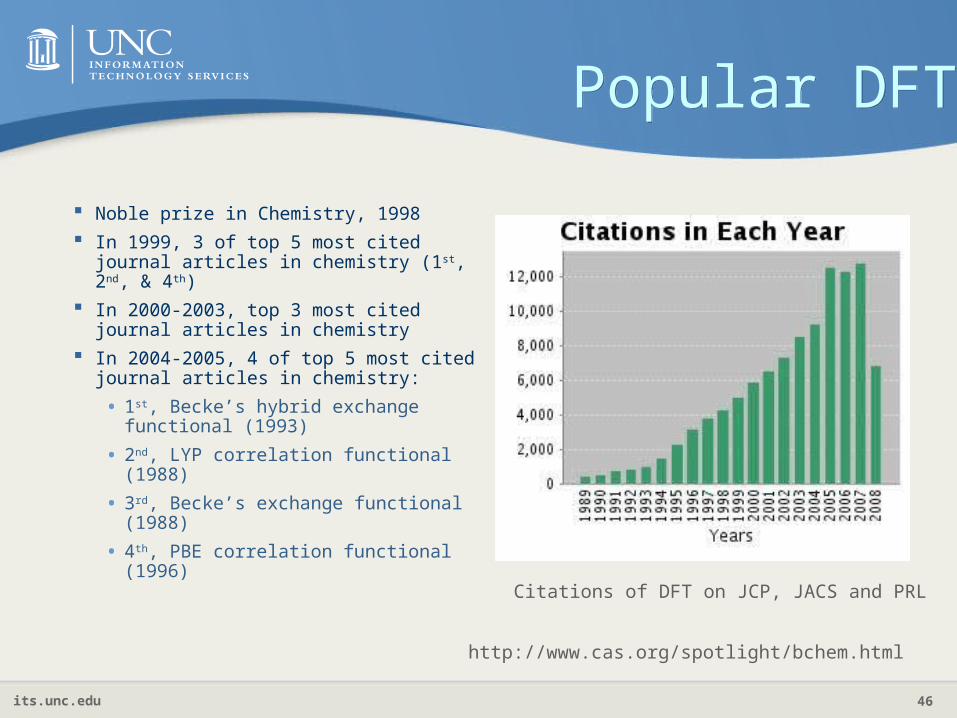
Popular DFTPopular DFT
Noble prize in Chemistry, 1998
In 1999, 3 of top 5 most cited journal articles in chemistry (1st, 2nd, & 4th)
In 2000-2003, top 3 most cited journal articles in chemistry
In 2004-2005, 4 of top 5 most cited journal articles in chemistry:
• 1st, Becke’s hybrid exchange functional (1993)
• 2nd, LYP correlation functional (1988)
• 3rd, Becke’s exchange functional (1988)
• 4th, PBE correlation functional (1996)
http://www.cas.org/spotlight/bchem.html
Citations of DFT on JCP, JACS and PRL

its.unc.edu 47
Brief History of DFTBrief History of DFT
First speculated 1920’
•Thomas-Fermi (kinetic energy) and Dirac (exchange energy) formulas
Officially born in 1964 with Hohenberg- Kohn’s original proof
GEA/GGA formulas available later 1980’
Becoming popular later 1990’
Pinnacled in 1998 with a chemistry Nobel prize

its.unc.edu 48
What could expect from DFT?What could expect from DFT?
LDA, ~20 kcal/mol error in energy
GGA, ~3-5 kcal/mol error in energy
G2/G3 level, some systems, ~1kcal/mol
Good at structure, spectra, & other properties predictions
Poor in H-containing systems, TS, spin, excited states, etc.

its.unc.edu 49
Density Functional TheoryDensity Functional Theory
Two Hohenberg-Kohn theorems:
•“Given the external potential, we know the ground-state energy of the molecule when we know the electron density ”.
•The energy density functional is variational.
E
HEnergy

its.unc.edu 50
But what is E[]?But what is E[]?
How do we compute the energy if the density is known? The Coulombic interactions are easy to compute:
But what about the kinetic energy TS[] and exchange-correlation energy Exc[]?
,][ , ][ ,][ 2
1extnenn rr
rr
rrrrr
ddJdVEr
ZZE
nuclei
BA AB
BA
E[] = TS[] + Vne[] + J[] + Vnn[] + Exc[]

its.unc.edu 51
Kohn-Sham SchemeKohn-Sham Scheme
,|)(|)(
,)(
,||
)()(
,||
)(
,2
1
and
)()()(ˆ
where
,ˆ
2
3
2
nknknk
xcxc
ee
a a
ane
xceene
nknknk
rfr
ErV
rdrr
rrV
Rr
ZrV
K
rVrVrVKH
H
The Only Unknown
• Suppose, we know the exact density.
• Then, we can formulate a Slater determinant that generates this exact density (= Slater determinant of system of N non-interacting electrons with same density ).
• We know how to compute the kinetic energy Ts exactly from a Slater determinant.
• Then, the only thing unknown is to calculate Exc[].

its.unc.edu 52
All about Exchange-Correlation
Energy Density Functional
All about Exchange-Correlation
Energy Density Functional
LDA – f(r) is a function of (r) only
GGA – f(r) is a function of (r) and |∇(r)|
Mega-GGA – f(r) is also a function of ts(r), kinetic energy density
Hybrid – f(r) is GGA functional with extra contribution from Hartree-Fock exchange energy
rrrr dfQXC ,,, 2
Jacob's ladder for the five generation of DFT functionals, according to the vision of John Perdew with indication of some of the most common DFT functionals within each rung.

its.unc.edu 53
LDA FunctionalsLDA Functionals
Thomas-Fermi formula (Kinetic) – 1 parameter
Slater form (exchange) – 1 parameter
Wigner correlation – 2 parameters
3/223/5 310
3, FFTF CdCT rr
3/13/23/13/4 438
3, XX
SX CdCE rr
rr
r
db
aEWC 3/11

its.unc.edu 54
Popular Functional: BLYP/B3LYPPopular Functional: BLYP/B3LYP
Two most well-known functionals are the Becke exchange functional Ex[] with 2 extra parameters &
The Lee-Yang-Parr correlation functional Ec[] with 4 parameters a-d
Together, they constitute the BLYP functional:
The B3LYP functional is augmented with 20% of Hartree-Fock exchange:
rrrr dedeEEE cxcxxc , , LYPBLYPBBLYP
3/4
2
2
23/4 ,1
LDA
XBX EE
rdettCbd
aE cWWF
LYPc
3/123/53/23/1 18
1
9
12
1
1
nlkmPPbEEaEN
lkkl
N
nmmncxxc
1,1,
LYPBB3LYP

its.unc.edu 55
Density FunctionalsDensity Functionals
LDAlocal density
GGAgradient corrected
Meta-GGAkinetic energy density included
Hybrid“exact” HF exchange component
Hybrid-meta-GGA
VWN5
BLYPHCTHBP86
TPSSM06-L
B3LYPB97/2MPW1K
MPWB1KM06
Better scaling with system size
Allow density fitting for even better scaling
Meta-GGA is “bleeding edge” and therefore largely untested (but better in theory…)
Hybrid makes bigger difference in cost and accuracy
Look at literature if somebodyhas compared functionals forsystems similar to yours!
Incr
easi
ng q
ualit
y a
nd c
om
puta
tional co
st

its.unc.edu 56
Percentage of occurrences of the names of the several functionals indicated in Table 2, in journal titles and abstracts, analyzed from the ISI Web of Science (2007).
S.F. Sousa, P.A. Fernandes and M.J. Ramos, J. Phys. Chem. A 10.1021/jp0734474 S1089-5639(07)03447-0
Density FunctionalsDensity Functionals

its.unc.edu 57
Problems with DFTProblems with DFT
ground-state theory only
universal functional still unknown
even hydrogen atom a problem: self-interaction correction
no systematic way to improve approximations like LDA, GGA, etc.
extension to excited states, spin multiplets, etc., though proven exact in theory, is not trivial in implementation and still far from being generally accessible thus far

its.unc.edu 58
DFT DevelopmentsDFT Developments
Theoretical• Extensions to excited states, etc.
• Better functionals (mega-GGA), etc
• Understanding functional properties, etc.
Conceptual • More concepts proposed, like electrophilicity, philicity, spin-
philicity, surfaced-integrated Fukui fnc
• Dynamic behaviors, profiles, etc.
Computational • Linear scaling methods
• QM/MM related issues
• Applications
its.unc.edu 59
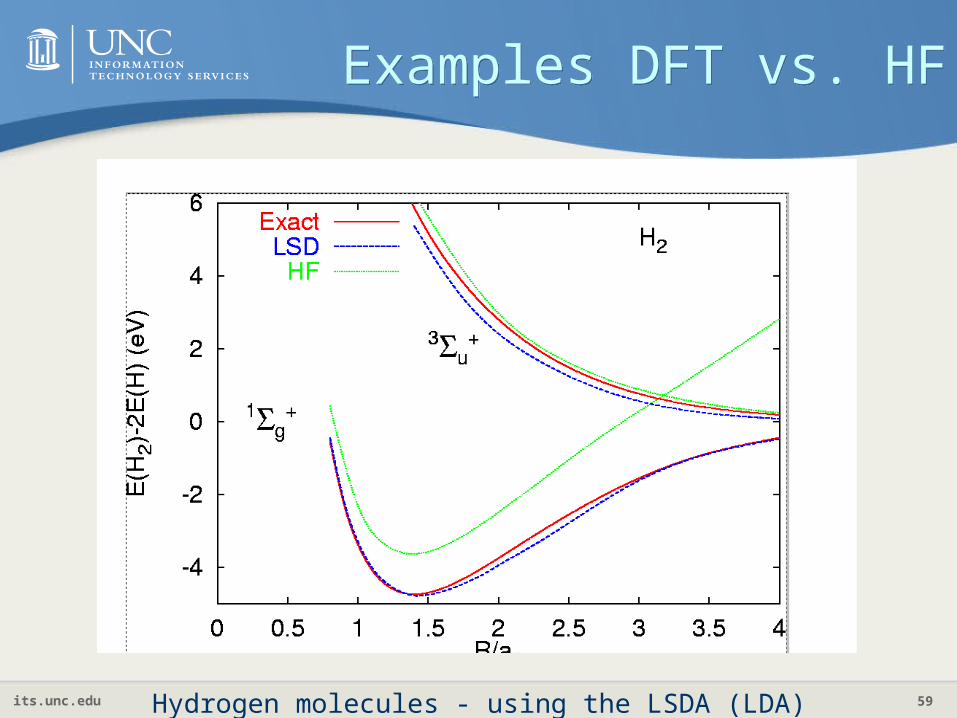
Examples DFT vs. HFExamples DFT vs. HF
Hydrogen molecules - using the LSDA (LDA)

its.unc.edu 60
Chemical Reactivity TheoryChemical Reactivity Theory
Chemical reactivity theory quantifies the reactive propensity of isolated species through the introduction of a set of reactivity indices or descriptors. Its roots go deep into the history of chemistry, as far back as the introduction of such fundamental concepts as acid, base, Lewis acid, Lewis base, etc. It pervades almost all of chemistry.
Molecular Orbital Theory• Fukui’s Frontier Orbital (HOMO/LUMO) model• Woodward-Hoffman rules• Well developed: Nobel prize in Chemistry, 1981• Problem: conceptual simplicity disappears as computational
accuracy increases because it’s based on the molecular orbital description
Density Functional Theory (DFT)• Conceptual DFT, also called Chemical DFT, DF Reactivity
Theory• Proposed by Robert G. Parr of UNC-CH, 1980s• Still in development
-- Morrel H. Cohen, and Adam Wasserman, J. Phys. Chem. A 2007, 111,2229

its.unc.edu 61
DFT Reactivity TheoryDFT Reactivity Theory
General Consideration• E E [N, (r)] E []
• Taylor Expansion: Perturbation resulted from an external attacking agent leading to changes in N and (r), N and (r),
''2!
,,
22
2
2
rrrrr
rrr2
1
rrr
rrr
2
ddE
dNE
NN
N
E
dE
NN
E
NENNEE
NN
N
Assumptions: existence and well-behavior of all above partial/functional derivatives

its.unc.edu 62
Conceptual DFTConceptual DFT
Basic assumptions
•E E [N, (r)] E []
•Chemical processes, responses, and changes expressible via Taylor expansion
•Existence, continuous, and well-behavedness of the partial derivatives

its.unc.edu 63
DFT Reactivity IndicesDFT Reactivity Indices
Electronegativity (chemical potential)
Hardness / Softness
Maximum Hardness Principle (MHP)
HSAB (hard and Soft Acid and Base) Principle
/1,22
12
2
SN
E HOMOLUMO
2LUMOHOMO
N
E
its.unc.edu 64
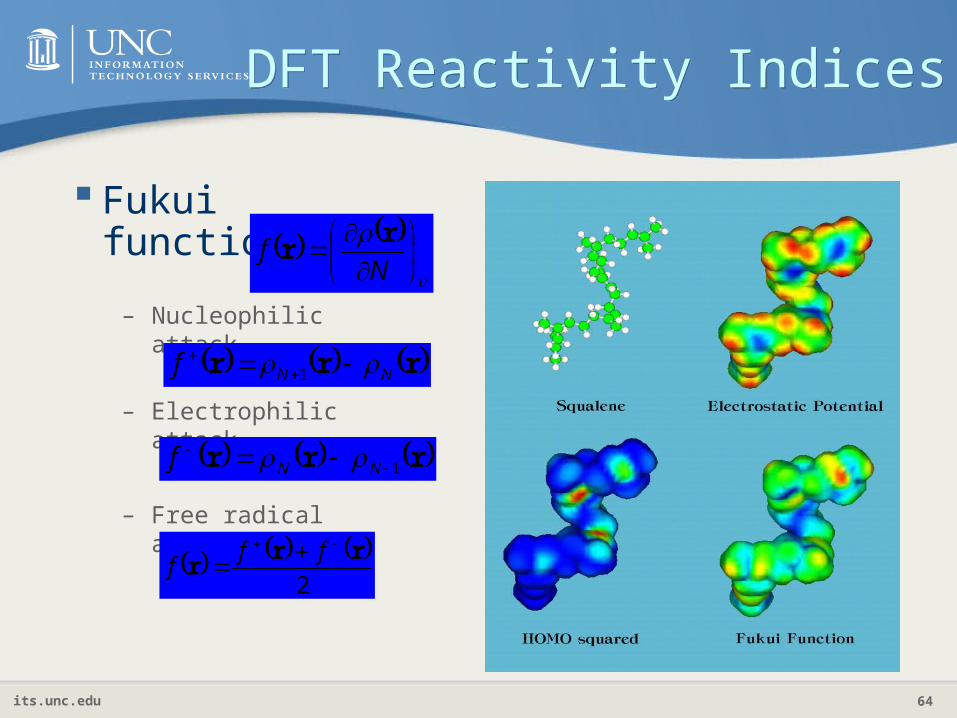
DFT Reactivity IndicesDFT Reactivity Indices
Fukui function
N
fr
r
– Nucleophilic attack
rrr NNf
1
– Electrophilic attack
rrr 1 NNf
– Free radical activity
2
rrr
fff
its.unc.edu 65
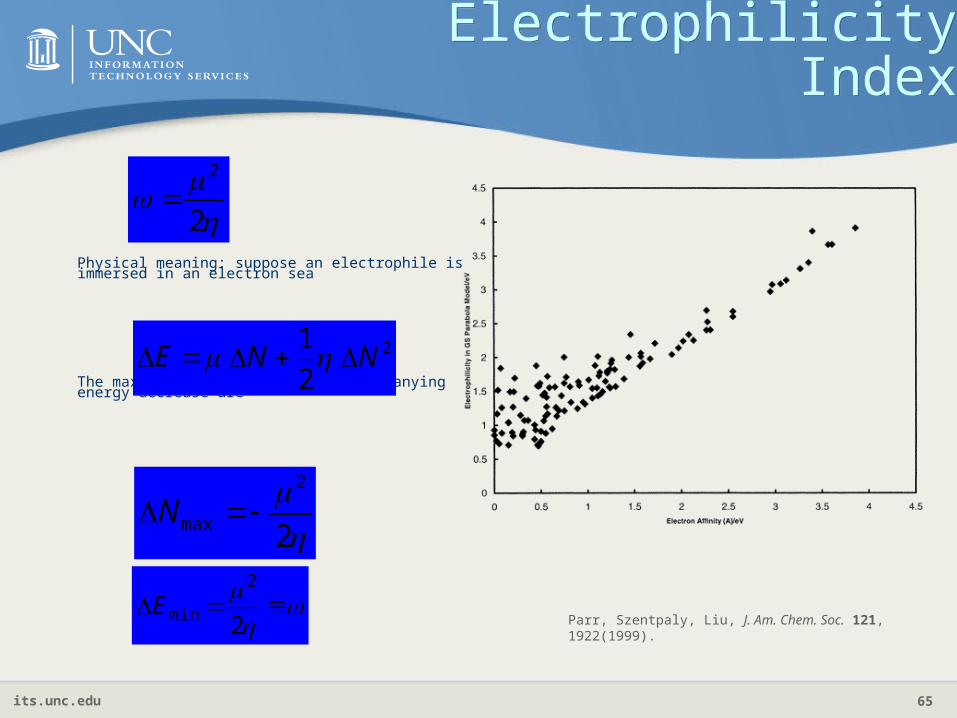
Electrophilicity IndexElectrophilicity Index
Physical meaning: suppose an electrophile is immersed in an electron sea
The maximal electron flow and accompanying energy decrease are
2
2
1NNE
2
2
max N
2
2
2
2
minEParr, Szentpaly, Liu, J. Am. Chem. Soc. 121, 1922(1999).
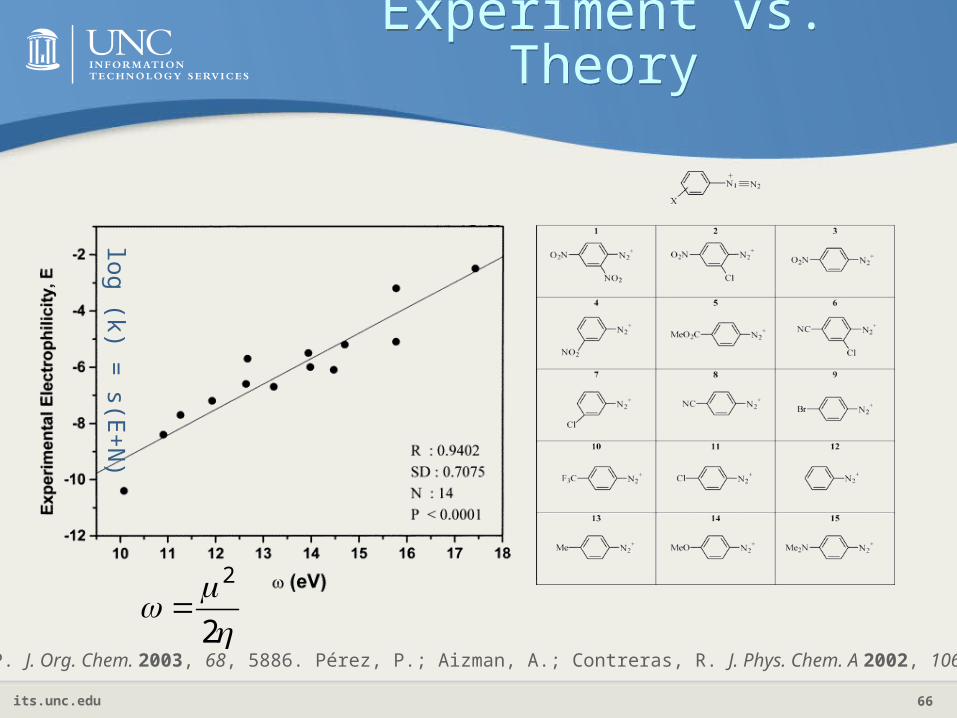
its.unc.edu 66
Experiment vs. Theory
Experiment vs. Theory
Pérez, P. J. Org. Chem. 2003, 68, 5886. Pérez, P.; Aizman, A.; Contreras, R. J. Phys. Chem. A 2002, 106, 3964.
2
2
log (k) = s(E
+N
)

its.unc.edu 67
Minimum Electrophilicity PrincipleMinimum Electrophilicity Principle
Analogous to the maximum hardness principle (MHP)
Separately proposed by Noorizadeh and Chattaraj
Concluded that “the natural direction of a chemical reaction is toward a state of minimum electrophilicity.”
Noorizadeh, S. Chin. J. Chem. 2007, 25, 1439.Noorizadeh, S. J. Phys. Org. Chem. 2007, 20, 514.Chattaraj, P.K. Ind. J. Phys. Proc. Ind. Natl. Sci. Acad. Part A 2007, 81, 871.
non-
LA
1 2 3 4 5 6 7
Aa -0.091 -
0.085
-0.093 -0.093 -
0.088
-0.087 -0.083 -0.090
Bb -0.089 -
0.084
-0.088 -0.089 -
0.087
-0.087 -0.0842 -
0.0892
Aa -0.172 -
0.247
-0.230 -0.220 -
0.218
-0.226 -0.2518 -
0.2161
Bb -0.171 -
0.246
-0.247 -0.233 -
0.221
-0.226 -0.2506 -
0.2157
Yue Xia, Dulin Yin, Chunying Rong, Qiong Xu, Donghong Yin, and Shubin Liu, J. Phys. Chem. A, 2008, 112, 9970.
its.unc.edu 68
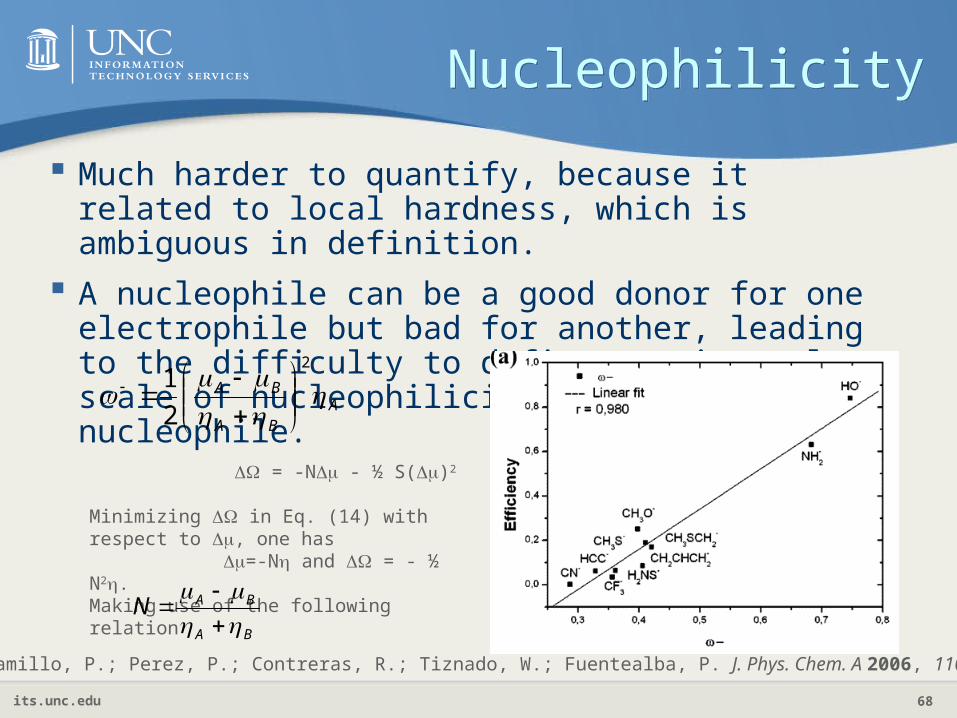
NucleophilicityNucleophilicity
Much harder to quantify, because it related to local hardness, which is ambiguous in definition.
A nucleophile can be a good donor for one electrophile but bad for another, leading to the difficulty to define a universal scale of nucleophilicity for an nucleophile.
ABA
BA
2
2
1
Jaramillo, P.; Perez, P.; Contreras, R.; Tiznado, W.; Fuentealba, P. J. Phys. Chem. A 2006, 110, 8181.
= -N - ½ S()2
Minimizing in Eq. (14) with respect to , one has =-N and = - ½ N2.Making use of the following relation
BA
BAN

its.unc.edu 69
Philicity and FugalityPhilicity and Fugality
Philicity: defined as ·f(r)
• Chattaraj, Maiti, & Sarkar, J. Phys. Chem. A 107, 4973(2003)
• Still a very controversial concept, see JPCA 108, 4934(2004); Chattaraj, et al. JPCA, in press.
Spin-Philicity: defined same as but in spin resolution
• Perez, Andres, Safont, Tapia, & Contreras. J. Phys. Chem. A 106, 5353(2002)
Nuclofugality & Electrofugality
2
)( 2 AEn
2
)( 2 IEe
Ayers, P.W.; Anderson, J.S.M.; Rodriguez, J.I.; Jawed, Z. Phys. Chem. Chem. Phys. 2005, 7, 1918.Ayers, P.W.; Anderson, J S.M.; Bartolotti, L.J. Int. J. Quantum Chem. 2005, 101, 520.
its.unc.edu 70
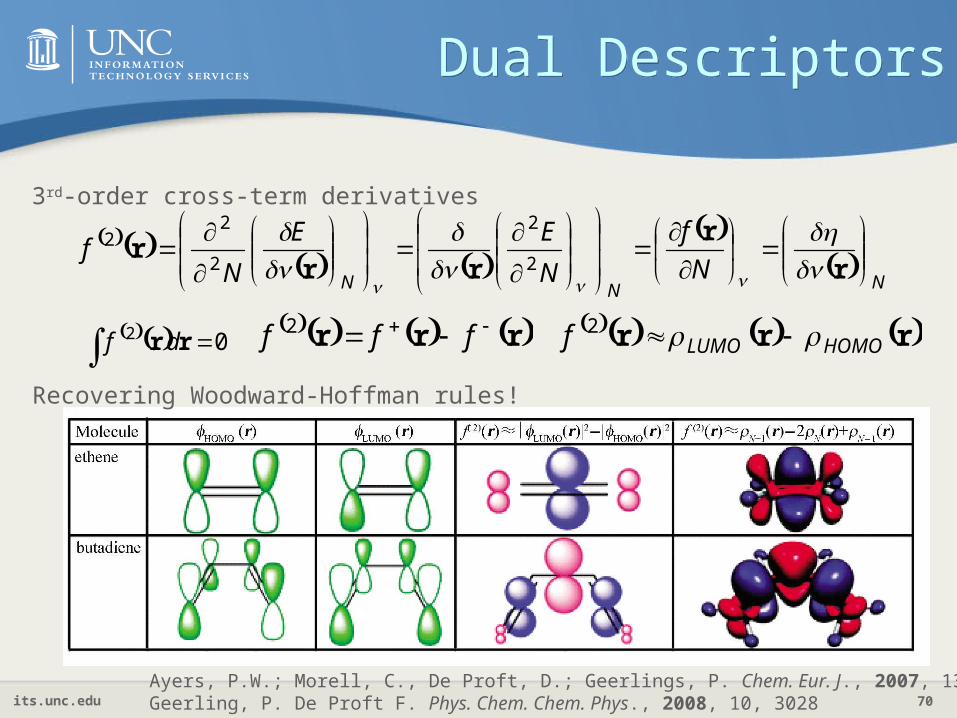
Dual DescriptorsDual Descriptors
NNN N
f
N
EE
Nf
r
r
rrr
2
2
2
22
3rd-order cross-term derivatives
02 rr df rrr fff 2 rrr HOMOLUMOf 2
Recovering Woodward-Hoffman rules!
Ayers, P.W.; Morell, C., De Proft, D.; Geerlings, P. Chem. Eur. J., 2007, 13, 8240 Geerling, P. De Proft F. Phys. Chem. Chem. Phys., 2008, 10, 3028

its.unc.edu 71
Steric EffectSteric Effect
one of the most widely used concepts in chemistry
originates from the space occupied by atom in a molecule
previous work attributed to the electron exchange correlation
Weisskopf thought of as “kinetic energy pressure”
Weisskopf, V.F., Science 187, 605-612(1975).

its.unc.edu 72
Steric effect: a DFT descriptionSteric effect: a DFT description
Assume
since
we have
E[] ≡ Es[] + Ee[] + Eq[]
E[] = Ts[] + Vne[] + J[] + Vnn[] + Exc[]
Ee[] = Vne[] + J[] + Vnn[] Eq[] = Exc[] + EPauli[] = Exc[] + Ts[] - Tw[]
Es[] ≡ E[] - Ee[] - Eq[] = Tw[]
r
r
rdTW
2
8
1
S.B. Liu, J. Chem. Phys. 2007, 126, 244103.S.B. Liu and N. Govind, J. Phys. Chem. A 2008, 112, 6690.S.B. Liu, N. Govind, and L.G. Pedersen, J. Chem. Phys. 2008, 129, 094104.M. Torrent-Sucarrat, S.B. Liu and F. De Proft, J. Phys. Chem. A 2009, 113, 3698.
its.unc.edu 73
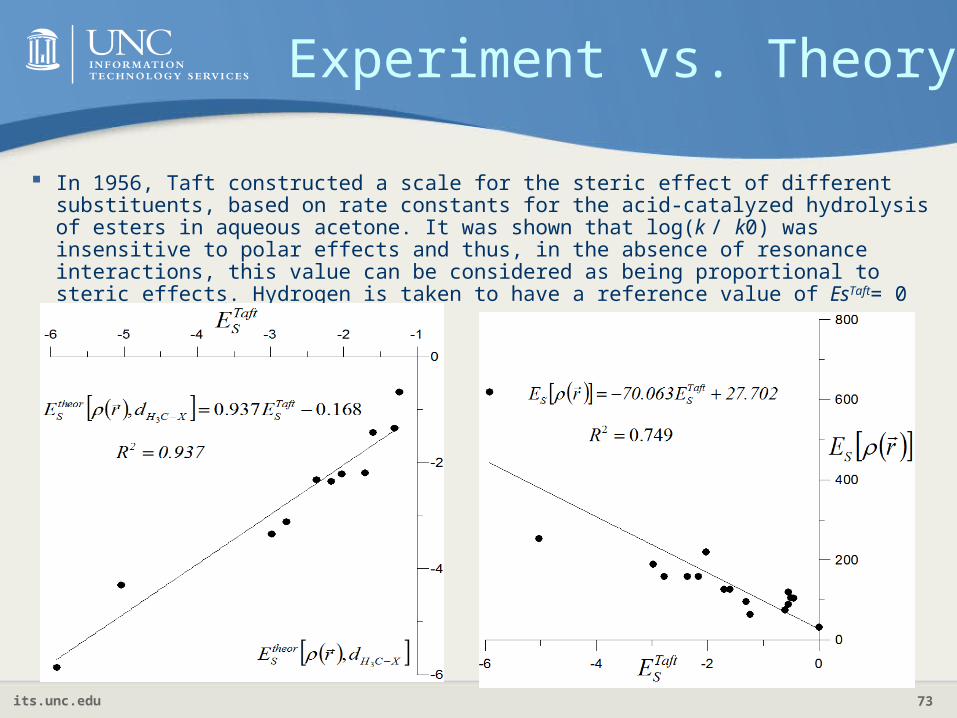
In 1956, Taft constructed a scale for the steric effect of different substituents, based on rate constants for the acid-catalyzed hydrolysis of esters in aqueous acetone. It was shown that log(k / k0) was insensitive to polar effects and thus, in the absence of resonance interactions, this value can be considered as being proportional to steric effects. Hydrogen is taken to have a reference value of EsTaft= 0
Experiment vs. Theory

its.unc.edu 74
QM/MM Example: Triosephosphate Isomerase (TIM)
QM/MM Example: Triosephosphate Isomerase (TIM)
494 Residues, 4033 Atoms, PDB ID: 7TIM
Function: DHAP (dihydroxyacetone phosphate) GAP (glyceraldehyde 3-phosphate)
GAP
DHAPH2O

its.unc.edu 75
Glu 165 (the catalytic base), His 95 (the proton shuttle)
DHAP GAP
TIM 2-step 2-residue Mechanism
TIM 2-step 2-residue Mechanism
its.unc.edu 76
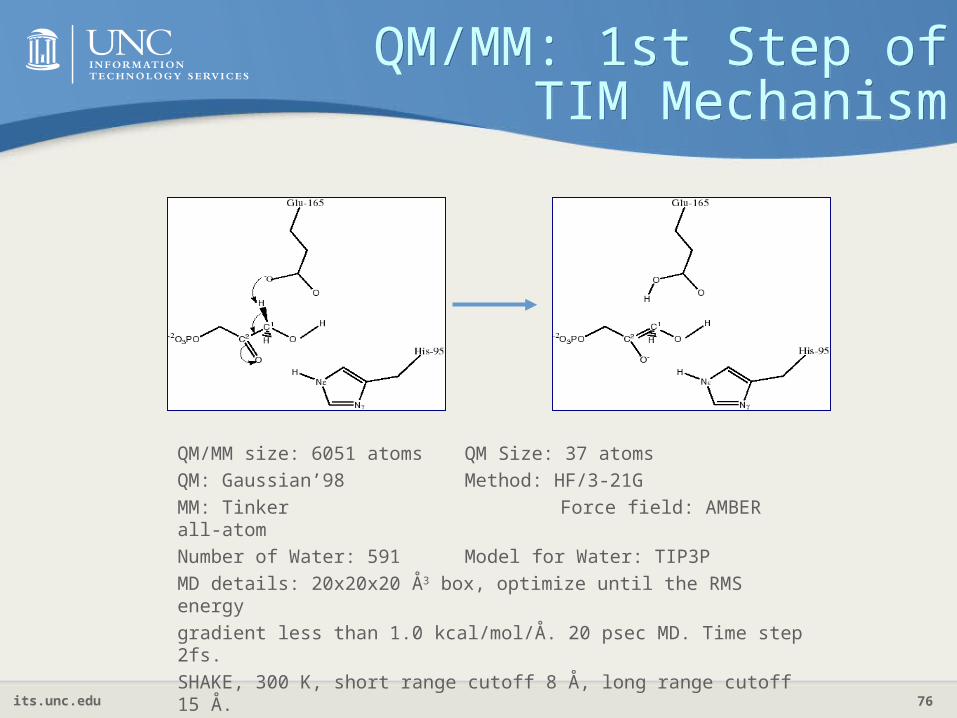
QM/MM: 1st Step of TIM Mechanism
QM/MM: 1st Step of TIM Mechanism
QM/MM size: 6051 atoms QM Size: 37 atoms
QM: Gaussian’98 Method: HF/3-21G
MM: Tinker Force field: AMBER all-atom
Number of Water: 591 Model for Water: TIP3P
MD details: 20x20x20 Å3 box, optimize until the RMS energy
gradient less than 1.0 kcal/mol/Å. 20 psec MD. Time step 2fs.
SHAKE, 300 K, short range cutoff 8 Å, long range cutoff 15 Å.
its.unc.edu 77
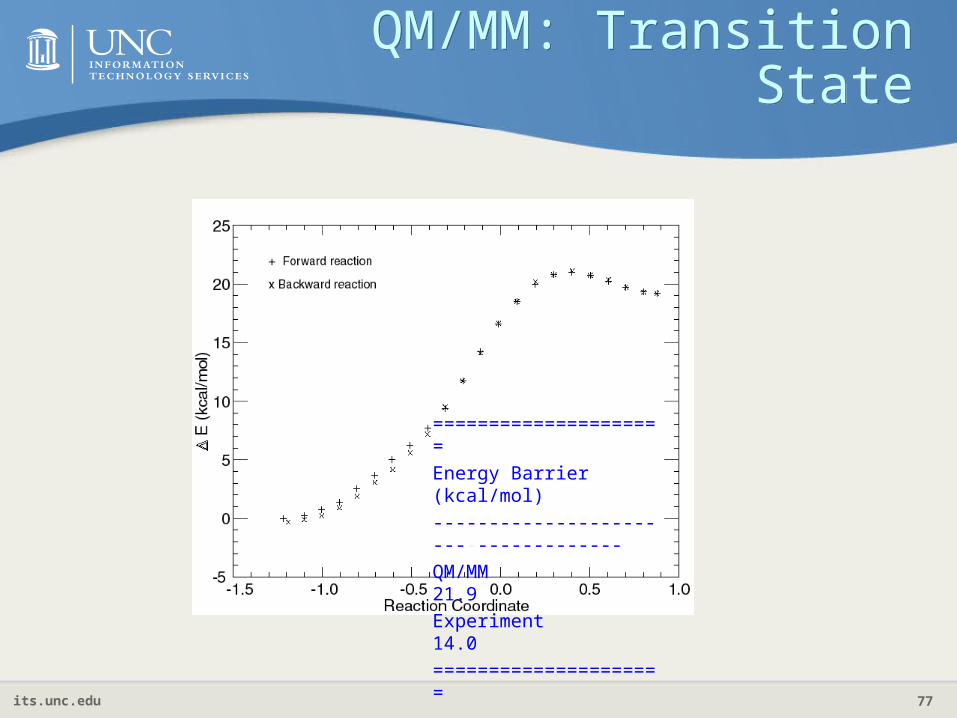
QM/MM: Transition State
QM/MM: Transition State
=====================
Energy Barrier (kcal/mol)
-------------------------------------
QM/MM 21.9
Experiment 14.0
=====================

its.unc.edu 78
What’s New: Linear Scaling O(N) MethodWhat’s New: Linear
Scaling O(N) Method Numerical Bottlenecks:
• diagonalization ~N3
• orthonormalization ~N3
• matrix element evaluation ~N2-N4
Computational Complexity: N log N
Theoretical Basis: near-sightedness of density matrix or orbitals
Strategy:
• sparsity of localized orbital or density matrix
• direct minimization with conjugate gradient
Models: divide-and-conquer and variational methods
Applicability: ~10,000 atoms, dynamics
0
10
20
30
40
50
60
70
80
90
100
0 100 200 300 400 500 600 700 800 900
Atoms
CPU
seco
nds
per C
G st
epOLMO
NOLMO
Diagonalization

its.unc.edu 79
What Else … ? What Else … ?
Solvent effect
•Implicit model vs. explicit model
Relativity effect
Transition state
Excited states
Temperature and pressure
Solid states (periodic boundary condition)
Dynamics (time-dependent)

its.unc.edu 80
Limitations and Strengths of ab initio
quantum chemistry
Limitations and Strengths of ab initio
quantum chemistry

its.unc.edu 81
Popular QM codesPopular QM codes
Gaussian (Ab Initio, Semi-empirical, DFT)
Gamess-US/UK (Ab Initio, DFT)
Spartan (Ab Initio, Semi-empirical, DFT)
NWChem (Ab Initio, DFT, MD, QM/MM)
MOPAC/2000 (Semi-Empirical)
DMol3/CASTEP (DFT)
Molpro (Ab initio)
ADF (DFT)
ORCA (DFT)

its.unc.edu 82
Reference BooksReference Books
Computational Chemistry (Oxford Chemistry Primer) G. H. Grant and W. G. Richards (Oxford University Press)
Molecular Modeling – Principles and Applications, A. R. Leach (Addison Wesley Longman)
Introduction to Computational Chemistry, F. Jensen (Wiley)
Essentials of Computational Chemistry – Theories and Models, C. J. Cramer (Wiley)
Exploring Chemistry with Electronic Structure Methods, J. B. Foresman and A. Frisch (Gaussian Inc.)
its.unc.edu 83
Questions & Comments Questions & Comments
Please direct comments/questions about research computing to
E-mail: [email protected]
Please direct comments/questions pertaining to this presentation to
E-Mail: [email protected]
Please direct comments/questions about research computing to
E-mail: [email protected]
Please direct comments/questions pertaining to this presentation to
E-Mail: [email protected]
The PPT format of this presentation is available here:http://its2.unc.edu/divisions/rc/training/scientific/
/afs/isis/depts/its/public_html/divisions/rc/training/scientific/short_courses/

its.unc.edu 84
Hands-on: Part IHands-on: Part I
Purpose: to get to know the available ab initio and semi-empirical methods in the Gaussian 03 / GaussView package
• ab initio methods Hartree-Fock
MP2
CCSD
• Semiempirical methods AM1
The WORD .doc format of this hands-on exercises is available here: http://its2.unc.edu/divisions/rc/training/scientific/
/afs/isis/depts/its/public_html/divisions/rc/training/scientific/short_courses/labDirections_compchem_2009.doc

its.unc.edu 85
Hands-on: Part IIHands-on: Part II
Purpose: To use LDA and GGA DFT methods to calculate IR/Raman spectra in vacuum and in solvent. To build QM/MM models and then use DFT methods to calculate IR/Raman spectra
• DFT LDA (SVWN)
GGA (B3LYP)
• QM/MM