information theoretic approach to whole genome phylogenies david burstein igor ulitsky tamir tuller...
Post on 20-Dec-2015
213 views
TRANSCRIPT

Information Theoretic Approach to Whole
Genome Phylogenies
David Burstein Igor Ulitsky Tamir Tuller Benny Chor
School Of Computer ScienceTel Aviv University
School Of Computer ScienceTel Aviv University

Tree of Life“I believe it has been with the tree of life, which fills with its dead and broken branches the crust of the earth, and covers the surface with its ever branching and beautiful ramifications"... Charles Darwin, 1859

Accepted Evolutionary Model: Trees Initial period: Primordial soup, where “you
are what you eat”. Recombination events. Horizontal transfers.
Formation of distinct taxa. Speciation events induce a tree-like evolution.

Accepted Evolutionary Model: Trees
Reconstructing this phylogenetictree is the major challengein evolutionary biology.
But…

Phylogenetic Trees Based on What?1. Morphology2. Single genes3. Whole genomes

Whole Genome Phylogenies: Motivation Cons for single genes trees
Require preprocessing Gene duplications Often too sensitive
Pros for whole genomes trees Fully automatic More information Seems essential in viruses
What about proteomes trees? Less “noise”, but do require preprocessing

Whole Genome Phylogenies: Biological Motivation Recently (last 2-4 years) it was discovered (in laboratories) that ~60% of the genome transcribes to RNA, but this RNA does not translate to proteins. We are in the dark as to what this non-coding RNA does. But we should not ignore it and
concentrate just on 3% coding parts!

Whole Genome Phylogenies: Availability Due to sequencing techniques that
were unthinkable just 15 years ago, we now have the complete genome sequences of hundreds of species, from all ranks and sizes of life.
These sequences are publicly available. They are a true treasure for analysis.

Whole Genome Phylogenies: Challenges Very large inputs: Up to 5G bp long Extreme length variability (5G to 1M bp) No meaningful alignment Different segments experienced different
evolutionary processes

Previous Approaches Genome rearrangements (Hannanelly & Pevzner 1995,…) Gene/domain contents (Snel et al. 1999,…)
Li et al (2001) – “Kolmogorov complexity” Otu et al (2003) – “Lempel Ziv compression” “IT” Qi et al (2004) – Composition vectors
Common approach (ours too): Compute pairwise distances Build a tree from distance matrix (e.g. using
Neighbor Joining, Saitou and Nei 1987)
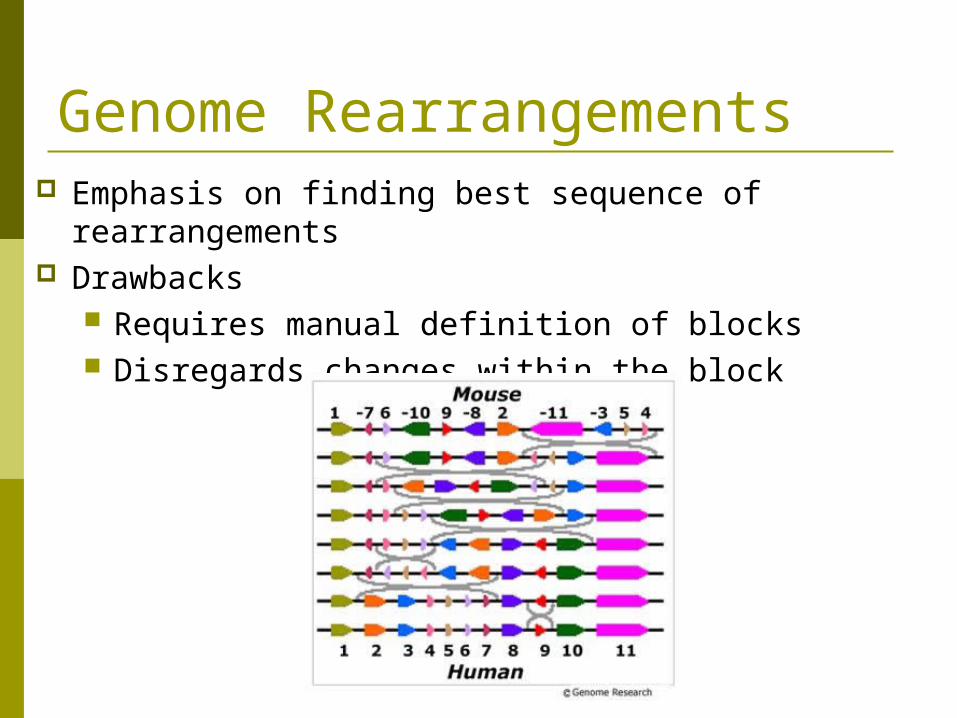
Genome Rearrangements Emphasis on finding best sequence of rearrangements Drawbacks
Requires manual definition of blocks Disregards changes within the block

Gene/Domain Content Genome equi length Boolean vector Various tree construction methods The drawback
Requires gene/domain definition/knowledge Disregards most of the genetic information

“Information Theoretic” Approaches

Ming Li et al.- “Kolomogorov Complexity” Kolmogorov Complexity is a wonderful
measure But … it is not computable “Approximate” KC by compression Drawbacks
Justification of the “approximation” Reportedly slow.

Otu et al.: “Lempel-Ziv Distance”
Run LZ compression on genome A. Use Genome A dictionary to compress Genome B. Log compression ratio (B given A vs. B given B)
≈ distance (B, A) Easy to implement Linear running time Drawback:
Dictionary size effects

Calculate distributions of the K-tuples. For K=1 – nucleotide/amino acid frequencies. For K=5 – 45 (205) possible 5-tuples Various methods for scoring distances Report K=5 as seemingly optimal
ACCGT
GGTAC
ATTGC
AACGG
GCTAT
ATGCG
GTTGC
Genome AGenome AACCGT
GGTAC
ATTGC
AACGG
GCTAT
ATGCG
GTTGC
Genome BGenome B
Qi et al.: Composition Vector

For every position in Genome A, find the
longest common substring in Genome B.
AGGCTTAGATCGAGGCTAGGATCCCCTTAGCGAGGCTTAGATCGAGGCTAGGATCCCCTTAGCGAGGCTTAGATCGAGGCTAGGATCCCCTTAGCGAGGCTTAGATCGAGGCTAGGATCCCCTTAGCG
AAAGCTACCTGGATGAAGGTAGGCTGCGCCCTTTAAAGCTACCTGGATGAAGGTAGGCTGCGCCCTTTAAAGCTACCTGGATGAAGGTAGGCTGCGCCCTTTAAAGCTACCTGGATGAAGGTAGGCTGCGCCCTTT
Genome A
Genome B
Our Approach: Average Common Substring (ACS)

For every position in Genome A, find the
longest common substring in Genome B.
AGGCTTAGATCGAGGCTTAGATCGAAGGCTAGGATCCCCTTAGCGGGCTAGGATCCCCTTAGCG
AAAAAAGCTGCTAACCTGGCCTGGAATGTGAAAAGGTGGTAAGGCTGGCTGGCGCCCTTTCGCCCTTT
Genome A
Genome B
Our Approach: ACS (cont.)

For every position in Genome A, find the
longest common substring in Genome B.
AGGCTTAGATCGAGGCTTAGATCGAGAGGCTAGGATCCCCTTAGCGGCTAGGATCCCCTTAGCG
AAAAAGAGCTACCTGGATGACTACCTGGATGAAGAGGTGTAGAGGCTGCGCCCTTTGCTGCGCCCTTT
Genome A
Genome B
Our Approach: ACS (cont.)

For every position in Genome A, find the
longest common substring in Genome B.
AGGCTTAGATCGAGGCTTAGATCGAGGAGGCTAGGATCCCCTTAGCGCTAGGATCCCCTTAGCG
AAAGCTACCTGGATGAAAAGCTACCTGGATGAAGGAGGTTAGGAGGCTGCGCCCTTTCTGCGCCCTTT
Genome A
Genome B
Our Approach: ACS (cont.)

For every position in Genome A, find the
longest common substring in Genome B.
AGGCTTAGATCGAGGCTTAGATCGAGGCAGGCTAGGATCCCCTTAGCGTAGGATCCCCTTAGCG
AAAGCTACCTGGATGAAGGTAAAGCTACCTGGATGAAGGTAGGCAGGCTACGCCCTTTTACGCCCTTT
Genome A
Genome B
Our Approach: ACS (cont.)

For every position in Genome A, find the length
of longest common substring in Genome B. In this case, ( )=5.
AGGCTTAGATCGAGGCTTAGATCGAGGCTAGGCTAGGATCCCCTTAGCGAGGATCCCCTTAGCG
AAAGCTACCTGGATGAAGGTAAAGCTACCTGGATGAAGGTAGGCTAGGCTGCGCCCTTTGCGCCCTTT
Genome A
Genome B
Our Approach: ACS (cont.)

For every position in Genome A, find the length
of longest common substring in Genome B. In this case, ( )=5. ACS= average ( ) = L(Genome A, Genome B)
AGGCTTAGATCGAGGCTTAGATCGAGGCTAGGCTAGGATCCCCTTAGCGAGGATCCCCTTAGCG
AAAGCTACCTGGATGAAGGTAAAGCTACCTGGATGAAGGTAGGCTAGGCTGCGCCCTTTGCGCCCTTT
Genome A
Genome B
Our Approach: ACS (cont.)

),( BAL
From ACS to Our Distance: Intuition High L( A , B ) indicates higher similarity.
Should normalize to account for length of B.

)log(
),(
B
BAL
From ACS to Our Distance: Intuition High L( A , B ) indicates higher similarity.
Should normalize to account for length of B. Still, we want distance rather than similarity.

From ACS to Our Distance: Intuition High L( A , B ) indicates higher similarity.
Should normalize to account for length of B. Still, we want distance rather than similarity.
)||(~
)||(~
),(
),(
)log(
),(
)log()||(
~
ABDBADBAD
AAL
A
BAL
BBAD
s

)||(~
)||(~
),(
),(
)log(
),(
)log()||(
~
ABDBADBAD
AAL
A
BAL
BBAD
s
High L( A , B ) indicates higher similarity.
Should normalize to account for length of B. Still, we want distance rather than similarity. And want to have D( A , A ) = 0 .
From ACS to Our Distance: Intuition

log( ) log( )( || )
( , ) (
( , ) ( || ) ( ||
,
)
)
sD A B
B A
D A B
D A BL A B L A A
D B A
High L( A , B ) indicates higher similarity.
Should normalize to account for length of B. Still, we want distance rather than similarity. And want to have D( A , A ) = 0 .
Finally, we want to ensure symmetry.
From ACS to Our Distance: Intuition

Comparison to Human (H)
9.134.570.9x106E. coli
8.974.822x106S. Cerevisiae (yeast)
5.565.2911x106Arabidopsis Thaliana
2.1122.9712x106Mus Musculus (mouse)
Ds(H,*)L(H,*)Proteome
sizeSpecies

What Good is this Weird Measure?
1) Our “ACS distance” is related to an information theoretic measure thatis close to Kullback Leibler relative entropy between two distributions.
2) The proof of the pudding is in the eating: Will show this “weird measure” is empirically good.

Define = number of bits required to describe distribution p, given q.
is closely related to Kullback Leibler
relative entropy
An Info Theoretic Measure( || )D p q
1 1lim ( ) log
( )
1log log
(
( )
|| )
( || )( )
l l
ll l
x X
P p
p xl q x
E E q x
p
p qq
q
X
D
D
( )log| )
( )( | P
pp q
XE
q XD
( || )D p q

Both and are common “distance measures” between two probability distributions p and q.
In general, the two “distances” are neither symmetric, nor satisfy triangle inequality.
An Info Theoretic Measure( || )D p q ( || )p qD

Suppose p and q are Markovian probabilitydistributions on strings, and A, B are generated by them. Abraham Wyner (1993) showed that w.h.p
Relations Between ACS and
,
,
log( )( || )
( , )
( , ) ( ||
( || )
(
li
|| ) ( |
) ( ||
m
)
| )
A B
A B
s
BD A B
L A B
D A B
D p q
D p
D A B D
q
B A
D q p
( || )D p q

Computation distance of two k long genomes: Naïve implementation requires O(k2)
(disaster on billion letters long genomes) With suffix trees/arrays: Total time for
computing is O(k) (much nicer).
ACS Implementation and Complexity
1 2( , )sD g g

Results and Comparisons Many genomes and proteomes Small ribosomal subunit ML tree Compare to other whole-genome methods Quantitative and qualitative evaluation

Benchmark dataset – 75 species 191 species (all non-viral proteomes in NCBI) 1,865 viral genomes 34 mitochondrial DNA of mammals (same as Li et al.)
Four Datasets Used

Benchmark Dataset – 75 Species Genomes and proteomes of archaea,
bacteria and eukarya Tree topologies reconstructed from
distance matrix using Neighbor Joining (Saitou and Nei 1987)
Reference tree and distance matrix obtained from the RDP (ribosomal database)

Benchmark dataset Genomes/Proteomes of 75 species from archaea, bacteria and
eukarya with known genomes, proteomes, and with RDP entries.
Methods implemented and tested : ACS (Ours) “Lempel Ziv complexity” (Otu and Sayhood) K-mers composition vectors (Qi et al.).
Results: Quantitative Evaluations
Tree Evaluation
04.05.35.33.5E
4.0
5.3
5.3
3.5
E
03.42.44.6D
3.403.42.3C
2.43.401.2B
4.62.31.20A
DCBA
A
B
E
DC
Tested Methods

Tree evaluation Reference tree: “Accepted” tree obtained
from ribosomal database project (Cole et al. 2003)
Tree Distance: Robinson-Foulds (1981)
Results: Quantitative Evaluations
Tree Evaluation
04.05.35.33.5E
4.0
5.3
5.3
3.5
E
03.42.44.6D
3.403.42.3C
2.43.401.2B
4.62.31.20A
DCBA
A
B
E
DC
Tested Methods

Robinson-Foulds Distance Each tree edge partitions species into 2
sets. Search which partitions exist only in one of
the trees.
AA
BB
CC
DD EE
AA
BB
EE
DD CC
Tree ATree A Tree BTree B
A,B C,D,E A,B C,D,ECommon Common PartitionPartition
xx
yy

AA
BB
CC
DD EE
AA
BB
EE
DD CC
Tree ATree A Tree BTree B
D,E
A,B,C
Robinson-Foulds Distance
xx
yyPartitionPartition
Not in BNot in B
Each tree edge partitions species into 2 sets.
Search which partitions exist only in one of the trees.

Distance = number of edges inducing partitions existing only in one of the trees.
For n leaves, distance ranges from 0 through 2n-6.
Robinson-Foulds Distance
AA
BB
CC
DD EE
AA
BB
EE
DD CC
Tree ATree A Tree BTree B
D,E
A,B,Cxx
yyPartitionPartition
Not in BNot in B
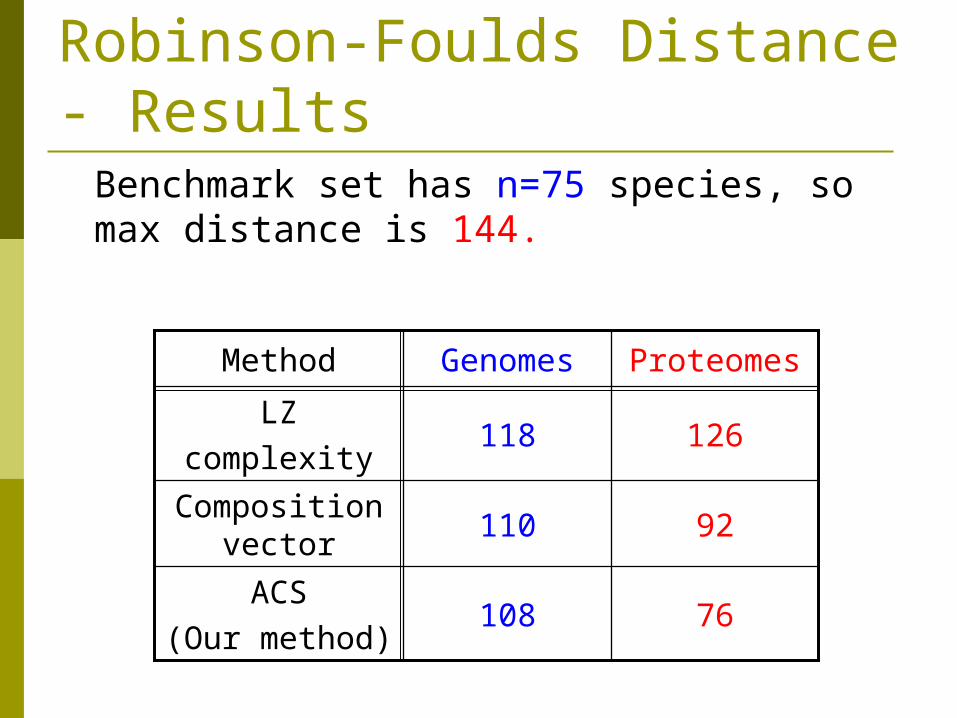
Robinson-Foulds Distance - Results
Benchmark set has n=75 species, so max distance is 144.
76108ACS
(Our method)
92110Composition
vector
126118LZ
complexity
ProteomesGenomesMethod

All Proteomes Dataset 191 proteomes from NCBI Genome 11 Eukarya, 19 Archaea, 161 Bacteria Compared to NCBI Taxonomy

191 proteomes from NCBI Genome 11 Eukarya, 19 Archaea, 161 Bacteria Compared to NCBI Taxonomy
All Proteomes Dataset
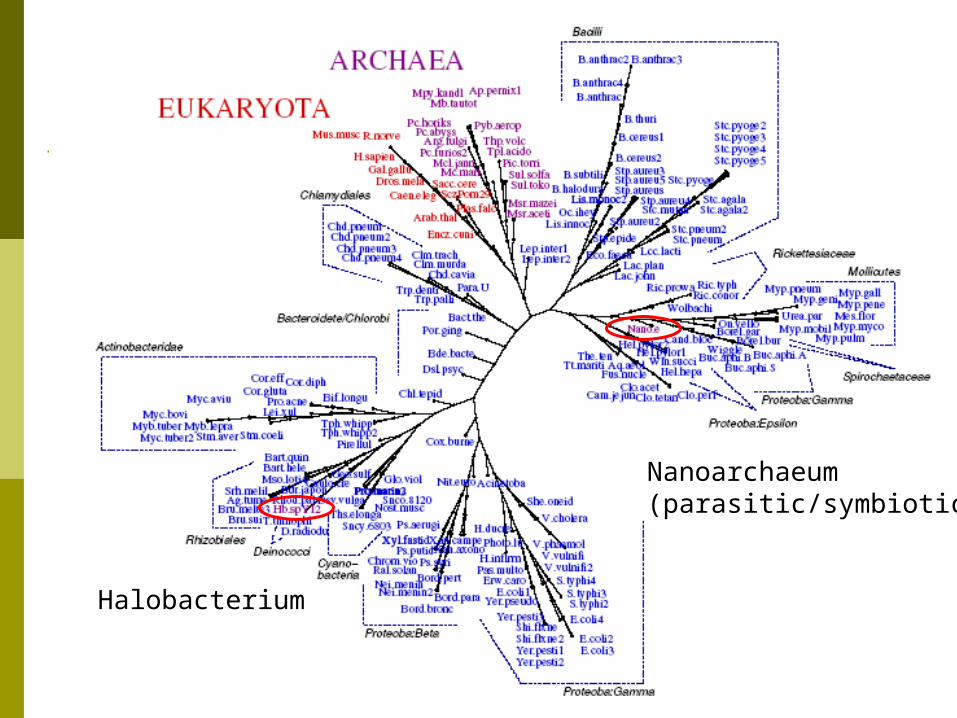
191 proteomes from NCBI Genome 11 Eukarya, 19 Archaea, 161 Bacteria Compared to NCBI Taxonomy
All Proteomes Dataset
Halobacterium
Nanoarchaeum(parasitic/symbiotic)

Viral Forest 1865 viral genomes from EBI Split into super-families:
dsDNA ssDNA dsRNA ssRNA positive ssRNA negative Retroids Satellite nucleic acid

83 Reverse-transcriptases: Hepatitis B viruses Circular dsDNA ssRNA
Retroid TreeAvian
Mammalian

Each segment treated separately 174 segments of 74 viruses.
ssRNA Negative Tree

Mammalian mtDNA Tree
Avian
Mammalian

Throwing Branch Lengths In
Intelligent Design ?

Additional Directions attempted
Naïve introduction of mismatches Division into segments Weighted combinations of genome and
proteome data Bottom line (subject to change):
Simple is beautiful.

Summary Whole genome/proteome phylogeny based
on ACS method Effective algorithm Information theoretic justification Successful reconstruction of known
phylogenies.

Future work
Statistical significance Improved branch lengths estimation Handle large eukaryotic genomes via
improved suffix array routines (e.g. by Stephan Kurtz enhanced suffix arrays - smaller memory requirements)
This should enable to have a full comparison of proteome vs. genome trees.
Not there yet.

Thank you !
Questions ?