indoles - startseite...of heterocyclic chemistry. the subdivisions have been designed to cover the...
TRANSCRIPT

INDOLES Part Four
The Monoterpenoid Indole Alkaloids
Edited by
J. Edwin Saxton Department of Organic Chemistry
The University of Leeds United Kingdom
AN INTERSCIENCE@ PUBLICATION
JOHN WILEY AND SONS
NEW YORK * CHICHESTER * BRISBANE * TORONTO * SINGAPORE


INDOLES
Part Four
The Monoterpenoid Indole Alkaloids
171is is the twenty-jifth volume in the series
THE CHEMISTRY OF HETEROCYCLIC COMPOUNDS

THE CHEMISTRY OF HETEROCYCLIC COMPOUNDS
A SERIES OF MONOGRAPHS
ARNOLD WEISSBERGER and EDWARD C. TAYLOR
Editors

INDOLES Part Four
The Monoterpenoid Indole Alkaloids
Edited by
J. Edwin Saxton Department of Organic Chemistry
The University of Leeds United Kingdom
AN INTERSCIENCE@ PUBLICATION
JOHN WILEY AND SONS
NEW YORK * CHICHESTER * BRISBANE * TORONTO * SINGAPORE

An Interscience@ hblication
Copyright 0 1983 by John Wiley & Sons, Inc.
All rights reserved. Published simultaneously in Canada.
Reproduction or translation of any part of this work beyond that permitted by Section 107 or 108 of the 1976 United States Copyright Act without the permission of the copyright owner is unlawful. Requests for permission or further information should be addressed to the Permissions Department, John Wiley & Sons, Inc.
Library of Congress Cataloging in Publication Data Main entry under title: Indoles
(The Chemistry of heterocyclic compomds; v. 35,
“An Interscience publication.” Includes indexes. 1. Indole alkaloids. I. Saxton, J. Edwin (John
pt. 4)
Edwin), 1927- . 11. Series: Chemistry of heterocyclic compounds; v. 25, pt. 4. QP801.145M66 1983 574.19’242 82-21958 ISBN 0471-89748-5

Contributors
RICHARD T. BROWN Department of Chemistry The University of Manchester Manchester MI3 9PL, United Kingdom
JUN-CHAO CAI School of Pharmacy University of Wisconsin Madison, Wisconsin Shanghai Institute of Materia Medica Chinese Academy of Sciences, Shanghai People’s Republic of China
GEOFFREY A. CORDELL Department of Pharmacognosy and
University of Illinois a t the Medical
Chicago, Illinois
Pharmacology
Center
WILLIAM A. CREASEY Department of Pharmacology School of Medicine, University of
Philadelphia, Pennsylvania Pennsylvania
GUENTER GRETHE Research Laboratories Hoffinann-La Roche Inc. Nutley, New Jersey
RICHARD B. HERBERT Department of Organic Chemistry The University of Leeds Leeds LS2 9JT, United Kingdom
HENRI-PHILIPPE HUSSON Institut de chimie des Substances
91 190 Gif;sur- Yvette, France Naturelles, CNRS
C. RICHARD HUTCHINSON School of Pharmacy University of Wisconsin Madison, Wisconsin
JOHN A. JOULE Department of Chemistry The University of Manchester Manchester MI 3 9PL. United Kingdom
J. EDWIN SAXTON Department of Organic Chemistry The University of Leeds Leeds LS2 9JT, United Kingdom
MILAN R. USKOKOVIC Research Laboratories Hoffinann-La Roche Inc. Nutley, New Jersey


The Chemistry of Heterocyclic Compounds
The chemistry of heterocyclic compounds is one of the most complex branches of organic chemistry. It is equally interesting for its theoretical implications, for the diversity of its synthetic procedures, and for the physiological and industrial significance of heterocyclic compounds.
A field of such importance and intrinsic difficulty should be made as readily accessible as possible, and the lack of a modern detailed and comprehensive presentation of heterocyclic chemistry is therefore keenly felt. It is the intention of the present series to fill this gap by expert presentations of the various branches of heterocyclic chemistry. The subdivisions have been designed to cover the field in its entirety by monographs which reflect the importance and the interrelations of the various compounds, and accommodate the specific interests of the authors.
In order to continue to make heterocyclic chemistry as readily accessible as possible new editions are planned for those areas where the respective volumes in the first edition have become obsolete by overwhelming progress. If, however, the changes are not too great so that the first editions can be brought up-to-date by supplementary volumes, supplements to the respective volumes will be published in the first edition.
Research Laboratories Eastman Kodak Company Rochester, New York
ARNOLD WEISSBERGER
EDWARD TAYLOR Princeton University Princeton, New Jersey


In the postwar years, and particularly during the 30 years since the isolation of reserpine in 1952, enormous interest has been shown in the monoterpenoid indole alkaloids, an interest which even now shows few signs of abating. The result is thaf these alkaloids now constitute probably the largest single group in the whole of the alkaloid field, with well over 1000 members, the vast majority of whose structures have been securely established. This impressive array of structures underlines the apparently limitless ingenuity of nature in the construction of alkaloids from two simple precursors, namely, tryptophan (which usually undergoes little structural modification in its incorporation into the final alkaloid) and a monoterpene unit derived in the first place from mevalonic acid, via geraniol. In parallel with this intensive activity in isolation and structure elucidation there has been a predictable surge of interest in the laboratory synthesis of these alkaloids, and in their biosynthesis, the details of which are now reasonably well understood.
The chemistry of these alkaloids has been reviewed comprehensively and periodically in the monographs edited by the late Dr. R. H. F. Manske and annually in the Royal Society of Chemistry’s Specialist Periodical Reports. Hence in order to acquire a true perspective of the area the reader is obliged to consult several volumes. In contrast, space limitations do not permit detailed treatment of this vast field in a one-volume survey of alkaloids, and the recent texts that fall into this category, superb as they are, can only provide a cursory treatment of this fascinating area.
This volume therefore constitutes an attempt to provide a one-volume summary of the chemistry, biosynthesis, and pharmacology of the monoterpenoid indole alkaloids which, although not exhaustive, nevertheless gives a reasonably complete picture of the present state of the art, and with copious references to the original literature allows the reader to trace all the recent significant work. Since emphasis has been placed on the publications of the last 30 years the older, classical work on the Strychnos and Cinchona groups has not been discussed in detail, and attention has naturally been focused on the alkaloids of the corynantheine, heteroyohimbine, yohimbine, aspidospermine, and catharanthine groups, as well as the bisindole alkaloids, which were comparatively little known in 1952 and which have yielded to structural investigation by modem methods in the intervening years.
Many of the monoterpenoid indole alkaloids exhibit a well-defined pharma- cological activity, and several of them have found clinical use. Indeed, the possibility of discovering new alkaloids with useful pharmacological activity still provides the stimulus to investigationsin this area as much as the intellectual rewards of structure elucidation and synthesis do. In the final chapter of this book the pharmacology and biochemistry of these alkaloids are discussed in some detail.
In general the literature has been surveyed to mid-1981, although a few later references have also been included.
ix

X Preface
The Editor warmly thanks Mrs. M. Romanowicz for secretarial assistance, and Mr. T. Lanigan for his original suggestion that there was a need for a volume of this kind.
J. EDWIN SAXTON
Leeds, United Kingdom August 1983

Contents
Part Four
I.
IT.
III.
rv.
V.
M.
w.
WI .
a.
X.
XI.
STRUCTUML AND BIOSYNTHETIC RELATIONSHIPS
RICHARD B. HERBERT
ALKALOIDS OF ARISTOTELZA SPECIES
J. EDWIN SAXTON
THE CORYNANTHEINE-HETEROYOHIMBINE GROUP
RICHARD T. BROWN
THE YOHIMBINE GROUP
RICHARD T. BROWN
THE SARPAGINE-AJMALINE GROUP
JOHN A. JOULE
THE ULEJNE-ELLIPTICINE-VALLESAMINE GROUP
JOHN A. JOULE
THE STR YCHNOS ALKALOIDS
HENRI-PHILPPE HUSSON
THE ASPIDOSPERMINE GROUP
J. EDWIN SAXTON
THE EBURNAMINE-VINCAMINE GROUP
J. EDWIN SAXTON
THE IBOGAMINE-CATHARANTHINE GROUP
GEOFFREY A. CORDELL
THE BISINDOLE ALKALOIDS
GEOFFREY A. CORDELL xi
1
47
63
147
201
265
293
33 1
439
467
539

xii Contents
XII. THE CZNCHONA GROUP
GUENTER GRETHE and MILAN R. U S K O K O V ~
W I . CAMPTOTHECIN
C. RICHARD HUTCHINSON and JUN-CHAO CAI
XIV. PHARMACOLOGY, BIOCHEMISTRY, AND CLINICAL APPLICATIONS OF THE MONOTERPENOID ALKALOIDS
WILLIAM A. CREASEY
AUTHOR INDEX
SUBJECT INDEX
729
753
783
83 1
865

CHAPTER I
Structural and Biosy nthetic Relationships RICHARD B. HERBERT
Department of Organic Chemistry, The University of Leeds, Leeds, United Kingdom
I. Introduction . . . . . . . . . . . . . 11. Biosynthesis . . . . . . . . . . . . .
A. Corynanthe-Sttychnos, Aspidosperma, and Iboga Alkaloids B. Vincamine . . . . . . . . . . . . C. Oxindole Alkaloids . . . . . . . . . . D. Apparicine and Uleine . . . . . . . . . E. Cinchona Alkaloids . . . . . . . . . . F. Camptothecin . . . . . . . . . . . G. Bisindole Alkaloids . . . . . . . . . .
111. Structural Relationships . . . . . . . . . . A. Alkaloids with a Nonrearranged Secologanin Skeleton. . B. Alkaloids with a Rearranged Secologanin Skeleton . . C. Oxindole Alkaloids . . . . . . . . . . D. Bisindole Alkaloids . . . . . . . . . .
References . . . . . . . . . . . . . .
. . . .
. . . .
. . . .
. . . .
. . . .
. . . .
. . . .
. . . .
. . . .
. . . .
. . . .
. . . .
. . . .
. . . .
. . . .
1
2 2
12 12 13 14 14 15
17 19 29 32 34
40
I. INTRODUCTION
The individual chapters in this volume attest to the rich variety of structures obtaining within the largest group of alkaloids, those that contain an indole, or related, fragment and a rearranged monoterpene unit,* and that are found most frequently in plants of the Apocynaceae, Loganiaceae, and Rubiaceae families.' The purpose of this account is to attempt a correlation of the various types within a framework of structural relationships. Any such classification must depend on a recognition of the biosynthetic pathways thus far delineated, so it is with a con- sideration of these pathways that we begin.
The subject has been reviewed extensively and the reader is referred t o some of these reviews for alternative treatment?
* The Aristotelia indole alkaloids, for example, 1, are distinct from those under discussion here. They have a regular monoterpene unit. A possible biosynthetic scheme has been proposed.'
1
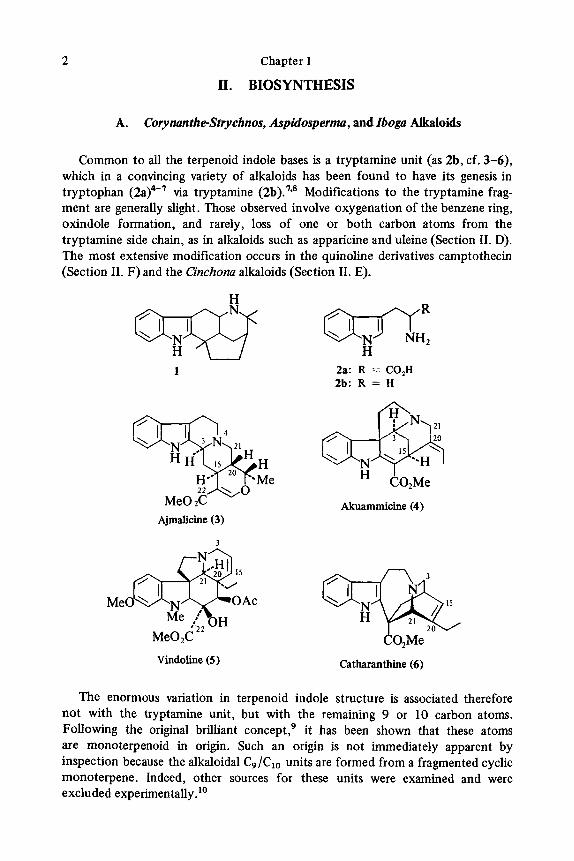
2 Chapter I
II. BIOSYNTHESIS
A. CorynantheSfrychnos, Aspidosperm, and Zboga Alkaloids
Common to all the terpenoid indole bases is a tryptamine unit (as 2b, cf. 3-6), which in a convincing variety of alkaloids has been found to have its genesis in tryptophan (2a)4-' via tryptamine (2b).'*8 Modifications to the tryptamine frag- ment are generally slight. Those observed involve oxygenation of the benzene ring, oxindole formation, and rarely, loss of one or both carbon atoms from the tryptamine side chain, as in alkaloids such as apparicine and uleine (Section 11. D). The most extensive modification occurs in the quinoline derivatives camptothecin (Section 11. F) and the Cinchona alkaloids (Section 11. E).
1 2a: R =- C0,H 2b: R = H
H
Akuammicine (4) Ajmalicine (3)
3
H
Vindoline (5 ) Catharanthine (6)
The enormous variation in terpenoid indole structure is associated therefore not with the tryptamine unit, but with the remaining 9 or 10 carbon atoms. Following the original brilliant concept: it has been shown that these atoms are monoterpenoid in origin. Such an origin is not immediately apparent by inspection because the alkaloidal C9/Clo units are formed from a fragmented cyclic monoterpene. Indeed, other sources for these units were examined and were excluded experimentally."

Structural and Biosynthetic Relationships 3
Close examination of the various alkaloidal C9/Clo terpenoid units reveals broadly three major types: first, that of the Corynanthe-Strychnos group, for example, ajmalicine (3) and akuammicine (4) where the unit is simplified as in 10; second, that of the Aspidospem group, for example, vindoline (5 ) with the C9/C10 unit represented by 11; and third, that of the Iboga group, for example, catharanthine (6), which is a third variant (9). The relationship of these skeletal types to a com- mon cyclopentane monoterpene skeleton is illustrated in Scheme 1, in which the tenth atom (C-22), which is sometimes missing, is depicted as a carbomethoxy group.
H F
C02H 9 10
HO
7 8 (3R)-Mevalonic acid
0
Iboga (9)
b - 0 ____,
22 C02Me 0 0
Corynunfhe-Strychnos (10) Aspidosperma (1 1)
Scheme 1
Essential proof that these C9/C10 units are terpenoid in origin and related in the way shown has come with extensive and rigorous experimentation. It was established that the C9/Clo units of representatives of the three groups of alkaloids are each derived from two molecules of mevalonic a ~ i d ~ ~ " - ' ~ linked initially in the normal head-to-tail fashion, elaborated along a pathway which includes geraniol (7)/nerol (14)12-" and the cyclopentane monoterpene, loganin (13)15J6 (cf. Scheme 1). Not only was loganin (13), in contrast to three other cyclopentanoid monoterpenes, a specific precursor, but also its biosynthesis from geraniol and its presence in Cbrharunthus roseus G. Don (syn. Unca roseu L.), the plant used for most of the experiments, could be demonstrated (Scheme 2; 12 has, like 3, the skeleton of type 10). These results secure loganin as an intermediate in alkaloid biosynthesis; it stands as a key compound along the biosynthetic pathway.
As observed in other systems, the biosynthesis of the alkaloidal monoterpene unit is from (3R)-mevalonic acid, not the (3s) i~omer. '~ The transformation of C-2 or C-3' of mevalonate through C-9 and (2-10 of the intermediate 8 into alkaloids

4 Chapter I
Geraniol
Perivine (12) Ajmalicine
OAc
Me0,C
Vindoline
Scheme 2
o&+/ . I
CO,Me A
Catharanthine
was observed to occur with loss of identity between these termini, as observed in the biosynthesis of cyclopentanoid m~noterpenes.'~
Further experimental results established18 that deoxyloganin (17) is to be sited as an intermediate in biosynthesis before loganin (13), and the hydroxy derivatives 15 and 16, of geraniol(7) and nerol(14), respectively, must also be included in the
(Scheme 3). The failure of various other derivatives of geraniol and nerol to act as precursors17419 restricts the range of possible intermediates beyond 15 and 16, and leads to a plausible mechanism for cyclization via a trialdehyde (Scheme 4), which accounts for the observation that label passing from mevalonate through C-9 and C-10 of the acyclic terpenes becomes equally distributed between the corresponding positions in loganin and the alkaloid^.'^^"^
Alkaloid derivation from loganin (13) must involve cyclopentane ring cleavage (cf. 8 + 10 in Scheme l), which may be rationalized in terms of either of the two mechanisms shown in Scheme 5, to give secologanin (18). Actual involvement of this fragmented terpene in alkaloid biosynthesis is proved by the observations that it is a natural constituent of C. roseus, is derived from loganin (13) in this plant, and is a precursor for representative terpenoid indole alkaloids.2' From secologanin the pathway leads logically to the Rhuzyu sfrictu Decaisne base, strictosidine (19),

Structural and Biosynthetic Relationships 5
f Mevalonate
+CH20H
Geraniol
t l
14 I I
Nerol(14)
Loganin (13) .+ Alkaloids
t
1 CH,OH - L
.* OGlU
15
t l
14 I I
C02Me
CH,OH t *CH,OH
16
17
Scheme 3
CHO CHO CHO
@:: __* V c O H - Loganin(13)
OH Me CHO Me
Scheme 4
Me 7 - O H
as the first alkaloid; it is one of two epimeric bases obtained by chemically con- densing secologanin (18) with tryptamine (2b) (Scheme 6).422 Both this base and its C-3 epimer, vincoside (20), were shown to be natural constituents of C roseus and to be derived from loganin (13).a23 The earliest results indicated that vincoside (20), not strictosidine (19), was the terpenoid indole alkaloid precursor,8 though this involved C-3 epimerization (cf. 3 and 4). However, recent results obtained with enzyme preparations from plant tissue cultures,24-30 as well as with whole plants, have shown unambiguously that strictosidine (19) is the true intermediate in the formation of the Corynanthe-Strychnos type with both the 3 a (e.g., 3, 25, and 26) and 3 0 configuration (e.g., 27 and 28), as well as vindoline (9, catharanthine (6), and a variety of other alkaloid^.^^-^ In the case of the 30 alkaloids the incorpo- ration of strictosidine occurs, not surprisingly, with loss of the C-3 proton; this proton is retained through the formation of alkaloids with 3 a configuration.3l (The ipecac alkaloids include a C 9 / C l o terpenoid unit derived along a pathway related to that described Here the alkaloids, with opposite stereo- chemistry at positions equivalent to those at C-3 in terpenoid indole alkaloids, are derived by contrast from precursors of the same .)

6 Chapter I
H
- b
Me0,C L O e ( 1 3 ) , /
\ / \ /
4
Secologanin (18)
Scheme 5
Recent work with enzyme preparations from plant tissue cultures (notably C roseus) has provided exciting results which precisely define the early steps up to and beyond strictosidine. It has been found that tryptamine (2b) and secologanin (18) condense together under enzyme catalysis to give strictosidine (19) stereo- specificaiiy. The enzyme that carries out the condensation has been isolated, purified, and characterized.J6 Subsequent modification of strictosidine (19) first requires loss of the glucose moiety; two strictosidinespecific glucosidases have been isolated and ~haracterized.~’ Biosynthesis with the enzyme preparations stops with the formation of strictosidine (19) in the presence of a 0-glucosidase
In the absence of inhibitor biosynthesis can proceed normally and the alkaloids ajmalicine (3), its C-19 epimer (29), and tetrahydroalstonine (30) are f ~ r m e d . ~ ’ - ~ ~ The enzyme preparation requires reduced pyridine nucleotide (NADPH or NADH); in the absence of coenzyme, 20,21 -dehydroajmalicine (23) (cathenamine) is found to a c c u m ~ l a t e ~ * ~ ~ and has been identified as an alkaloid of Guettarda eximia plants.38 Moreover, cathenamine is enzymatically converted into 3, 29, and 30 in the presence of NADPH.26327 Cathenamine is thus very probably a normal intermediate in alkaloid biosynthesis in intact plants.
Two further compounds, 21 and 22, have been identified as biosynthetic inter- mediates. The latter, 4,21 -dehydrogeissoschizine (22), has, like cathenamine (23), been isolated from G. eximia plants.39 On incubation with an enzyme preparation in the presence of NADPH, 22 was converted into 3,29, and 30. In the absence of NADPH, cathenamine (23) was formed; under NADPH-regenerating conditions, geissoschizine (24) was formed. From this evidence and the observation that unlabeled 22 diluted the incorporation of radioactivity from [ 1-14C] -tryptamine into 3, 29, and 30 and that 22 was formed from radioactive tryptamine plus secologanin,it is clear that 22 is a normal biosynthetic intermediate?’ The evidence implicating 21 as an intermediate is more circumstantial and comes from the
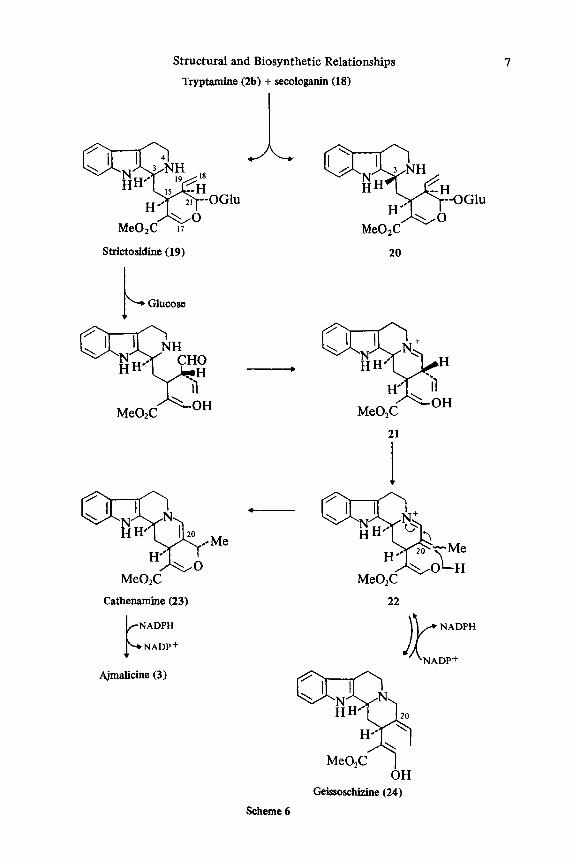
Structural and Biosynthetic Relationships
Tryptamine (2b) + secologanin (18)
--OGlu HH” l 9 /
H ” ‘0
MeO,C 17
-- H 21 --OGlu
Strictosidine (19) 20
Glucose
7
H He’ H ____, CHO
HI’ \ OH MeOzC
21
.-Me
MeOzC
Cathenamhe (23) 22
NADPH
NADPi
Ajmalicine (3)
NADPH
NADP+
Me0,C 1 OH
Geissoschizine (24)
Scheme 6

8 Chapter I
OMe
16H 20H 3H __
a Mitragynine (26) a a-Yohimbine (25) a Yohimbine (34) P P Speciociliatine (28) P Corynanthine (35) a 0
MeOzC Reserpiline (27)
RI+R~ H
19-H 20-H __- 29 a P
Tetrahydroalstonine (30) 5 a
Me
OH Sitsirkine (31): R' = CO,Me, Rz = CH,OH 33
Isositsirikine (32): R' = CH,OH, R* = C0,Me
isolation of sitsirikine (31) and isositsirikine (32) as new and exclusive products from a C. roseus enzyme preparation incubated with strictosidine (19) and potassium borohydride!l Also potentially involved in this area of biosynthesis is 33, isolated from G. eximia, and convertible chemically into cathenamine (23)?2 It is a reasonable substrate biochemically for obtaining different stereochemistries at C-19 and C-20. Within the enzyme system ajmalicine (3) is not converted into the epimers 29 or 30, and so the differing stereochemistries at these centers must be irreversibly set up at an earlier stage, that is, possibly through 33.
The Coyrunthe-type alkaloids, represented by ajmalicine (3) and mitragynine

Structural and Biosynthetic Relationships 9
36
(26), are the simplest variation of the strictosidine skeleton. Only slightly more complex are alkaloids of the yohimbine (25, 34) type which have a further carbocyclic ring. a-Yohimbine (25) and its stereoisomers yohimbine (34) and corynanthine (35) have been shown to have a genesis along the strictosidine path- way, as e~pec ted .~ ' "~ Plausibly an intermediate of type 36 is involved.
Simple rearrangement of strictosidine (19) affords the Strychnos skeleton, exemplified by akuammicine (4), the known biosynthetic steps to which have already been referred to; further results are given below. Strychnine (37) is slightly more complex in that it has an additional Cz unit ((2-22 and C-23). It has, how- ever, the expected origins in tryptophan (h), and in mevalonate via geraniol (7); the Cz unit arises from acetate.44 A search45 for later intermediates in strychnine biosynthesis has revealed that geissoschizine (24; but see discussion below) and the Wieland-Gumlich aldehyde (40) are precursors, and negative results with geissoschizal (41) and diaboline (42) have suggested, respectively, that 39 (i.e., skeletal rearrange- ment before loss of C1 unit) and 38 may be intermediates. The deduced pathway is illustrated in Scheme 7.
OH
38 Strychnine (37)
18
Corynanthe skeleton (as 24)
39 Wieland-Gumlich
aldehyde (40): R = H 42: R = COMe
Scheme 7

10 Chapter I
43
All the alkaloids so far mentioned have a biogenesis via strictosidine (19). It is implicit in their close structural relationship to 19 and clear from the foregoing experimental results that alkaloids with the Corynanthe skeleton are the first to be formed, followed by those of the Strychnos type. It is a reasonable hypothesis that the other skeletons are elaborations of the Corynanthe skeleton, and this is sup- ported by the results of biosynthetic experiments. First, alkaloids with Corynanthe- type structures appear in C roseus seedlings before bases of the Iboga and Aspidosperma type.& Second, it has been found that the CoFyynanthe base, geissoschizine (24) (but not its double-bond isomer, 43),14 is an intact precursor for representatives of the Aspidospemta and Iboga groups: and also those with the Strychnos skeleton, represented by akuammicine (4)47 and strychnine (37).45
Although the above results with geissoschizine (24) in whole plants indicate that it is an intermediate in the elaboration of more complex alkaloids, experiments with a cellfree preparation from a C roseus tissue culture which synthesizes Corymntize bases indicate that herc at least 24 is on a shulit fiom the main path- way, entering this pathway by an NADP+-dependent reaction just prior to cathenamine (23)48 (see Scheme 6). The deduced relationship, however, between the Corynanthe skeleton and those of the Iboga and Aspidospemta alkaloids still stands; one may conclude either that a compound related to geissoschizine (24) is directly involved or that, for plants producing more complex alkaloids, geissoschizine (24) is not at a terminus of biosynthesis but is an intermediate. The observedI4 loss of tritium from C-2 of loganin (13) (= C-20 in 24) in the formation of the three alkaloid groups is accommodated through the intermediacy of 22, where the equivalent site, C-20, like that of geissoschizine (24), is devoid of a hydrogen atom.
The results of orthodox feeding experiments and those in which the sequence of alkaloid formation is obtained by noting the appearance of precursor label in individual alkaloids in relation to time have led to the c o n c l ~ s i o n ~ " ~ that preakuammicine (44), stemmadenine (45), and tabersonine (48) are further important intermediates in the biosynthesis of both catharanthine (6) (Zboga type) and vindoline ( 5 ) (Aspidosperma type). The formation of tabersonine (48) may be rationalized in terms of the pathway illustrated in Scheme 8. Central to this hypo- thetical route is the enamine (47) arrived at by fragmentation of stemmadenine (45). Although alternative cyclization paths from this enamine could give both the lboga type (as 6 ) and the Aspidosperma type (as 5) , such a simple idea is argued against by the incorporation of tabersonine (48) into catharanthine (6),& unless the conversion of 47 into 48 is reversible (Scheme 8). The validity of 47 as a biosynthetic intermediate is supported by the isolation of simple derivatives of

Structural and Biosynthetic Relationships 11
Preakuammiche (44)
Tabersonine (48)
Vindoline (5)
Stemmadenine (45)
COzMe
46
1 +
47
Catharanthine (6) +--- G N k C02Me Scheme 8
secodine (49) from plants anil by the specific incorporation of labeled secodine (49) into vindoline (5); the tritium loss from secodine, labeled in the dihydro- pyridine ring, on translation into vindoline (9, is consistent with involvement of secodine (49) via a more highly oxidized intermediate (as 47).” It is to be noted that, since tritium from C-1 of loganin (equivalent t o C-21 in 45) is retained on formation of 5 and 6 , the double shift in the conversion of 45 into 46 leaves that particular proton ~naffected.’~ Necessary further clarification of the biosynthesis of Iboga and Aspidospema alkaloids probably depends now on work with isolated enzymes leading to the sort of clarity obtaining to date for the
Secodine (49)

12 Chapter I
biosynthesis of the Corynanthe alkaloids. Exciting prospects are thus opened up by the development of strains of C. roseus cultures which will indeed synthesize Strychnos, Iboga, and Aspidosperma alkaloids.”
It is a puzzling feature, particularly of the Aspidosperma alkaloid^,'^ but also of bases of the Ibogas3 and vincamine (52) series, that both enantiomeric series occur naturally, sometimes in the same plant. The chirality from earlier biosynthetic intermediates is lost on reaching secodine (49), which is achiral, but this does not explain the subsequent biological stereospecificity in both stereochemical senses. The natural occurrence of enantiomeric forms of akuammicine (4) has been given a chemical rationale.54
B. Vincamine
The terpenoid skeleton (1 1) in vincamine (52) is the same as in the Aspidospemza bases, for example, 5 and 50, suggesting that vincamine is related biosynthetically to this group of alkaloids [see Scheme 955156; for the laboratory conversion of vincadifformine (50) into vincamine see Chapter VIII] . This has been supported in a provisional way with the reported incorporations of tryptophan, stemmadenine (49, and tabersonine (48).57 It is additionally to be noted that both vincatine (51)” and vincadifformine occur with vincamine (52) in Vinca minor L.
Vincadifformine (50) Vincatine (5 1): R = Me \ / \ / \ /
(R-W \ /
‘4
Vincamine (52) Scheme 9
C. Oxindole Alkaloids
Derivation of the oxindole alkaloids mitraphylline (53) and isomitraphylline (54) from the corresponding indole derivatives has been indicated by the apparent

Structural and Biosynthetic Relationships 13
Mitraphylline (53) Isomitraphylline (54)
transformation of ajmalicine (3)and 3isoajmalicine into these alkaloids in Mitragyna purvifoliu Korth.60 Some conclusions have also been reached about interrelation- ships of oxindole alkaloids in Mitragyna species.6l
D. Apparicine and Uleine
Apparicine (59, vallesamine (59), and uleine (56) are exceptional terpenoid indole alkaloids in that their biosynthesis must involve modification to the normal tryptamine side chain with loss of C-1 . Definitive evidence on the course of uleine biosynthesis is lacking,6' but it appears that apparicine (5.5) does arise from tryptophan (2a) with the expected loss of C-2 (=C-1 in tryptamine).a Stemmadenine (45) and secodine (49) are precursors, which means that rearrange- ment to the apparicine skeleton is a late step in biosynthesis.@ Secure details on the biosynthesis of these intriguing metabolites must await further work, but a key step probably involves a cleavage of the type shown in Scheme with subse- quent bonding of C-6 to N-4 (for 55) or C-7 to (2-21 (for 56). Such a sequence has been given chemical validity by the laboratory conversion [H'O,; (CF,CO),O; hydrolysis] of stemmadenine (45) into vallesamine (59)."6 Vallesamine, preliminary
Uleine (56) A
I I
57 Scheme 10
58

1 4 Chapter I
in vivo evidence indicates, is a precursor for apparicine. If the chemical sequence is an accurate reflection of the biosynthesis of 55 and 59, then the reported@ incorpo- ration of secodine (49) is puzzling.
E. Cinchona Alkaloids
The structure 60 for the Cinchona alkaloid cinchonamine suggested at an early stage that there was a biogenetic relationship between the terpenoid indole bases and the Cinchona alkaloids with a quinoline moiety, for example, quinine (66).6768 Indeed it was likely that alkaloids of the former type would prove to be precursors for the quinoline bases. These ideas, the results of tracer experiments show, are correct. The incorporation of trypt0phan,6’*~ geraniol (7),’2m.71 loganin ( 13),n and strictosidine (19);l establishes an early biosynthetic pathway in common with the Corynunthe alkaloids (Section 11. A). The labeling was, moreover, in accord with hypothesis (Scheme 11). Corynantheal (61), but not 43, was found to be a precursor, and is likely to be the last of the Corynanthe-type intermediatesm The next intermediate is probably the aldehyde 62, since the corresponding alcohol, cinchonamine (a), was not incorporated and half the label from [ 1-3H2] -tryptamine (labeling site corresponds to C-5 of 61) was retained. This also excludes the corre- sponding carboxylic acid as an intermediate. No information is available on the rearrangement steps that follow 62, but biosynthesis proceeds plausibly along the pathway shown in Scheme 11 . The pathway leads to the ketone, cinchonidinone (63), the intermediacy of which in biosynthesis is indicated by its natural occurrence in Cinchona plants, and its conversion into Cinchona alkaloids74; 68 is also naturally occurring. It appears that the conversion of 63 into 65 is reversible, that in vivo epimerization occurs via 63 2 67, and that 63 is a substrate for aromatic hydroxylation leading to different quinoline alkaloids.
F. Camptothecin
Camptothecin (72) represents a course of in vivo modification to the terpenoid indole skeleton leading to a quinoline alkaloid different from that of the Cinchona bases (Section 11. E). The biosynthetic pathway to camptothecin (72) follows the route to Corynanthe-type alkaloids with strictosidine (19) as the last common inte~mediate.~’?~ Biosynthesis proceeds then via the lactam 71, derived from

Structural and Biosynthetic Relationships 15
H
strictosidine (19)
H H'*
H" CHO H
corynantheal(6 1 ) 62
H
63: R = H
0 0 X,
QKf& H" *.p H
64: R=OMe H
HO
Cinchonidine (65): R = H Quinine (66): R=OMe
Cinchonine (69): R = H Quinidine (70): R = OMe
67: R = H 68: R=OMe
Scheme 11
strictosidine (19), with plausible intermediates as shown in Scheme 12, to camptothecin (72).75 Desaturation of ring D is necessarily involved in this sequence and a proton is lost from (2-14 (see 71). The results of further experiments show, not without analogy, that it is lost nonstereospecifically , indicating that removal of this particular proton is probably not enzyme mediated.77
G. Bisindole Alkaloids
By inspection it seems manifest that bisindole alkaloids, for example, vinblastine (74), have a biogenesis from monomeric terpenoid indole alkaloids, such as vindoline (5) and catharanthine (6). Investigations have been dogged, however, by poor incorporations.'* Recently more careful work and the use of cellfree prepa- rations have given positive results. Anhydrovinblastine (73), which was shown to

16 Chapter I
71 9- 0
Camptothecin (72) Scheme 12
be a natural product of Catharanthus roseus, has been found to be specifically and efficiently labeled by radioactive vindoline (5) and catharanthine (6 ) in whole plants79 and in a cellfree preparation.m In cellfree preparations vinblastine (74) was formed from ?38i*8'; 20'iieoxyleurosidine (as 73, but saturated between C-15' and C-20') also acts as a precursor for 74.83 Leurosine (75) was also generated from 5,6,and 73,but the ready chemical conversion of 73 into 7584 leads to the question whether this is a real biosynthesis. Both anhydrovinblastine (73) and leurosine (75) were found to be efficient precursors for Catharine (76)."
The chemical fusion of vindoline (5) and catharanthine (6) to give 73 occurs on treating catharanthine Nb-oxide with trifluoroacetic anhydride in the presence of vindoline (S), followed by reduction (NaBH4).= The probable mechanism for the chemical reaction is shown in Scheme 13, and it is likely that the in vivo coupling resembles this route.
Me02C Me026
Anhydrovinblastine (73) Vinblastine (74)