increased levels of intracellular calcium are not required for the formation of attaching and
TRANSCRIPT

INFECTION AND IMMUNITY,0019-9567/98/$04.0010
Aug. 1998, p. 3900–3908 Vol. 66, No. 8
Copyright © 1998, American Society for Microbiology. All Rights Reserved.
Increased Levels of Intracellular Calcium Are Not Required forthe Formation of Attaching and Effacing Lesions by
Enteropathogenic and EnterohemorrhagicEscherichia coli
CHRISTOPHER BAIN,1 ROGERIA KELLER,2 GEORGINA K. COLLINGTON,1
LUIZ R. TRABULSI,3 AND STUART KNUTTON1*
Institute of Child Health, University of Birmingham, Birmingham B4 6NH, United Kingdom,1
and Departmento de Parasitologia, Instituto de Ciencias Biomedicas II, Universidadede Sao Paulo,2 and Laboratorio Especial de Microbiologia,
Instituto Butantan,3 Sao Paulo, Brazil
Received 9 February 1998/Returned for modification 12 March 1998/Accepted 4 May 1998
Elevated concentrations of intracellular calcium ([Ca]i) have been implicated as an important signallingevent during attaching and effacing (A/E) lesion formation by enteropathogenic Escherichia coli (EPEC). Thehighly localized nature of the cytoskeletal and cell surface alterations occurring during A/E lesion formationsuggests that there should be equally localized EPEC-induced signalling events. To analyze further the calciumresponses to infection of HEp-2 cells by EPEC, we employed calcium-imaging fluorescence microscopy, whichallows both temporal and spatial measurements of [Ca]i in live cells. Using this imaging technique, not onlywere we unable to detect any significant elevation in [Ca]i at sites of A/E EPEC adhesion, but, with several differentclassical EPEC and enterohemorrhagic E. coli (EHEC) strains and three different infection procedures, eachof which resulted in extensive A/E bacterial adhesion, we were unable to detect any significant alterations in[Ca]i in infected cells compared to uninfected cells. In addition, chelation of intracellular free calcium with bis-(aminophenoxy)-ethane-N,N,N*,N*-tetraacetic acid (BAPTA) did not, as previously reported, prevent A/E le-sion formation. We conclude that increased [Ca]i are not required for A/E lesion formation by EPEC and EHEC.
Enteropathogenic Escherichia coli (EPEC) strains remain animportant cause of severe infantile diarrheal disease in manyparts of the developing world (8). EPEC colonizes the intesti-nal mucosa and produces attaching and effacing (A/E) lesions,which are characterized by localized destruction of brush bor-der microvilli, intimate bacterial adhesion, and formation of acomplex cytoskeletal structure beneath intimately attachedbacteria (23). Accumulation of polymerized actin beneath bac-teria results in the formation of cup-like pedestal structures inthe apical enterocyte membrane (22). EPEC produces mor-phologically similar A/E lesions in a variety of cultured celllines (21). The determinants of A/E lesion formation havebeen localized to a large pathogenicity island on the EPECchromosome termed the locus of enterocyte effacement or(LEE) (27). Encoded within the LEE are a type III secretionsystem (16); an outer membrane protein adhesin, intimin (17);a translocated intimin receptor, Tir (18); and three EPECsecreted proteins (Esps), EspA, EspB, and EspD, each ofwhich has been shown to be required for signal transduction tohost cells and A/E lesion formation (12, 20, 25). Enterohem-orrhagic E. coli (EHEC) strains, which cause a hemorrhagiccolitis, possess a homologous LEE and also produce A/E le-sions (27).
In vitro studies employing defined mutants support a three-stage model of A/E lesion formation (10). Stage 1 involves aninitial nonintimate bacterial adhesion to epithelial cells, whichis thought to be promoted by a plasmid-encoded bundle-form-
ing pilus (BFP). In stage 2 signal transduction in host cells istriggered by Esps, resulting in cytoskeletal rearrangements,microvillous effacement, and tyrosine phosphorylation of Tirfollowing translocation to the host cell membrane (18). Stage 3involves intimin binding to tyrosine-phosphorylated Tir to pro-duce the characteristic intimate attachment; pedestal forma-tion results from further cytoskeletal reorganization and poly-merization beneath intimately attached bacteria.
Early studies of EPEC signal transduction examined secondmessengers and demonstrated increased concentrations of in-tracellular calcium ([Ca]i) in EPEC-infected epithelial cells(2, 11). The subsequent demonstration of inositol phosphatefluxes, including increased levels of inositol triphosphate (11,13), suggested possible stimulation of the classical phospho-lipase C pathway and release of calcium from inositol triphos-phate-sensitive intracellular stores. Activation of brush bordervillin by calcium, disruption of the microvillous actin cytoskel-eton, and vesiculation of the microvillous membrane provideda plausible model for EPEC-induced brush border effacement(4). One difficulty with such a model, however, was how such acalcium signal could give rise to the highly localized cytoskel-etal changes seen during EPEC infection: microvillous efface-ment occurs only at sites of intimate bacterial adhesion. Wehypothesized that if calcium signalling is important, calciumsignals should be equally localized within the cell to sites ofA/E lesion formation. To test this hypothesis and analyze fur-ther the calcium responses to infection of HEp-2 cells byEPEC (and EHEC), we employed calcium-imaging fluores-cence microscopy, which not only allows real-time measure-ments of [Ca]i in infected cells but also allows the spatialdistribution of [Ca]i to be determined and thus correlated withA/E lesion formation. Here we report that not only were we
* Corresponding author. Mailing address: Institute of Child Health,University of Birmingham, Clinical Research Block, Whittal St., Bir-mingham B4 6NH, United Kingdom. Phone: 44 121 333 8746. Fax: 44121 333 8701. E-mail: [email protected].
3900
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/i
ai o
n 19
Nov
embe
r 20
21 b
y 20
1.18
3.96
.226
.

unable to detect any significant elevation in [Ca]i at sites ofA/E EPEC adhesion, but we were unable to detect any signif-icant alterations in [Ca]i in EPEC- and EHEC-infected HEp-2cells compared to uninfected cells.
MATERIALS AND METHODS
Bacterial strains. The EPEC and EHEC strains used in the study are listed inTable 1. Stock cultures were subcultured into Mueller-Hinton broth and incu-bated aerobically at 37°C for 18 h.
Infection of cultured HEp-2 cells. HEp-2 cells were grown on 25-mm-diameterglass coverslips in 199 medium (Sigma) supplemented with 10% fetal calf serumand 5% glutamine. Subconfluent cultures were briefly washed in phosphate-buffered saline (PBS) and transferred to six-well tissue culture plates containing4 ml of HEPES-buffered Eagle’s minimum essential medium (MEM) containing2% newborn calf serum (NCS) and 0.5% D-mannose. For calcium-imaging stud-ies three different bacterial infection procedures were used.
(i) “Spindown” infection. The spindown infection protocol used a methodrecently developed in our laboratory to achieve rapid synchronous EPEC infec-tion of a cell monolayer (6). Briefly, 30 ml of an overnight bacterial broth culturewas subcultured into 3 ml of tissue culture medium in a 30-mm-diameter petridish for 4 h at 37°C to induce expression of virulence factors, including BFP,Esps, and intimin; BFP expression resulted in spontaneous aggregation of bac-teria. A 0.5-ml volume of the aggregated bacterial suspension was placed in aLeiden coverslip chamber containing a cell monolayer preloaded with the fluo-rescent calcium-imaging dye fura-2 (see below), and the chamber was centri-fuged at 700 3 g for 2 min. The culture medium was immediately replaced with0.5 ml of fresh medium, and calcium imaging was performed immediately for 1 has described below.
(ii) “Settledown” infection. In the spindown protocol there was a short (up to5-min) delay between the centrifugation step and commencement of calciumimaging, during which time alterations in cell calcium could have occurred. Inorder to eliminate the possibility that we might miss rapid alterations in cellcalcium, we used a protocol similar to the spindown procedure except that thecentrifugation step was omitted and calcium imaging was begun immediatelyupon addition of bacteria.
(iii) Gradual infection. The gradual infection protocol essentially reproducedthe method originally used by Baldwin et al. (2), in which calcium levels wereassayed in cells following infection with EPEC for 1, 2, 3, 4, and 6 h. Coverslipsin six-well plates containing 4 ml of MEM plus 2% NCS were inoculated with 80ml of bacterial broth culture and incubated at 37°C for up to 6 h. Cells wereloaded with fura-2 during the 40 min prior to the appropriate imaging time point.Washed coverslips were mounted in the Leiden chamber and transferred to themicroscope, and calcium imaging was performed as described below.
Measurement of intracellular free calcium. Intracellular calcium concentra-tions were determined by digital fluorescence imaging with the ratiometric,dual-excitation wavelength dye fura-2. Fura-2 was loaded into cells as the ace-toxymethyl (AM) ester derivative of fura-2, fura-2 AM (Molecular Probes),which, being more hydrophobic than the dye itself, enters mammalian cells moreeasily. Once inside the cell, endogenous esterases cleave the AM group, trappingthe dye inside the cells. HEp-2 cells were loaded with fura-2 by incubating cellmonolayers in loading medium (MEM containing 10% NCS and 2 mg of fura-2AM per ml for 40 min at 32°C, with the reduced temperature being used to helpreduce compartmentalization of the dye. Monolayers were rinsed three times infresh medium to remove extracellular dye.
The dye-loaded cell monolayers were mounted in Leiden coverslip chambersand transferred to an open perfusion chamber, which was maintained at 37°Cand mounted on the stage of a Zeiss Axiovert 135TV inverted microscope, andimaged by using a 403 achrostigmat oil immersion phase-contrast lens. TheCa-free and Ca-bound forms of fura-2 fluoresce maximally at 380 and 340 nm,respectively. These excitation wavelengths were provided by a Zeiss XBO75Xenon lamp and transmitted through filters mounted in a computer-controlledfilter wheel. Emitted fluorescence was detected with a high-sensitivity ExtendedISIS Intensified charge-coupled device video camera (Photonic Science Ltd.).The camera and filter wheel were connected to an Apple Macintosh 6100/60
computer running the dedicated calcium image analysis software IonVision III(Image Processing and Vision Company Ltd., Coventry, United Kingdom),which was used to control the experiment, capture the relevant fluorescenceimages, and calculate both the ratiometric images and the [Ca]i values by usingion standards. Pseudocolor ratio images produced immediately after each imagepair was recorded allowed real-time alterations in calcium levels to be monitoreddirectly during an experiment; a more detailed statistical analysis of the data wasperformed later by using saved image pairs. Complementary phase-contrastimages of the same microscope fields used for calcium imaging were recordedwith a Hamamatsu C3077 charge-coupled device video camera.
In spindown and settledown infection experiments, one ratio image was takenevery minute from the same area of cells for the 60-min duration of the exper-iment; in the gradual infection experiments, five ratio images from different fieldswere taken at each time point to obtain a representative value for the wholemonolayer. At the end of some experiments, cells were exposed to the calciumionophore ionomycin (10 mM) or to the calcium agonist histamine (0.1 M) toconfirm the responsiveness of the cells and the detection system. At the end ofother experiments, cells were fixed and stained for actin to detect A/E lesionformation (22).
Chelation of intracellular calcium. To assess the ability of EPEC to produceA/E lesions in the absence of elevated intracellular calcium levels, intracellularcalcium was chelated by incubating HEp-2 cell monolayers with 10 mM AM bis-(aminophenoxy)-ethane-N,N,N9,N9-tetraacetic acid (BAPTA-AM) in MEM–2%NCS for 45 min at 37°C. The spindown protocol was used to infect washed cellmonolayers with EPEC for 1 h at 37°C, after which time the cells were washedagain and examined by the fluorescence actin-staining (FAS) test for A/E lesionformation (22). The calcium-buffering capacity of BAPTA-loaded cells was as-sessed by comparing the histamine responses of control and BAPTA-treatedHEp-2 cells by using the IonVision system.
Fluorescence staining of filamentous actin (FAS test). Formalin-fixed andwashed cells were permeabilized by treating coverslips with 0.1% Triton X-100 inPBS for 4 min. After three washes in PBS, coverslips were stained with a 5-mg/mlsolution of fluorescein isothiocyanate phalloidin (Sigma) in PBS for 20 min tospecifically stain filamentous actin (22). Coverslips were washed a further threetimes in PBS and mounted in glycerol-PBS. Cells were examined by incident-light fluorescence with a Leitz Dialux microscope. Fluorescence and phase-con-trast micrographs of the same field were made.
Live-dead assay. The viability of cells following EPEC infection was assessedby fluorescence microscopy with the commercial EukoLight Viability/Cytotoxic-ity Kit (Molecular Probes) containing calcein-AM and ethidium homodimer. Atthe end of appropriate experiments, HEp-2 cell monolayers were incubated withthe live-dead kit solution (2 mM calcein-AM and 4 mM ethidium homodimer inPBS) for 45 min at room temperature, washed, and examined by incident-lightfluorescence for viable (green) and dead (red) cells.
Statistical analyses. Results are presented as means 6 standard errors of themeans. The data were analyzed by Student’s t test, and P , 0.05 was consideredstatistically significant.
RESULTS
The resting [Ca]i in uninfected HEp-2 cells, determined byaveraging [Ca]i in cells from five different microscope fields,was 151 6 34 nM (n 5 7); this value is comparable to basallevels reported by others (2, 11). To confirm that the HEp-2cells being used were responsive to calcium agonists and thatthe imaging system was capable of detecting changes in [Ca]iunder the conditions employed, fura-2-loaded HEp-2 cells werestimulated with 1 mM histamine during imaging. This resultedin large, though variable, rises in [Ca]i of 435 6 273% (n 5 5).
Based on knowledge of BFP and Esp expression in mid-exponential-phase cultures of EPEC grown in tissue culturemedium, we developed an infection protocol (spindown infec-tion) which achieved a rapid synchronous EPEC infection ofHEp-2 cell monolayers (6). Time course experiments in whichcoverslips were fixed and examined by using the FAS testshowed that A/E lesions were produced after 10 to 20 min (6).We used this rapid infection protocol to measure [Ca]i inHEp-2 cell monolayers infected with EPEC strain E2348/69,with calcium levels being determined in a single microscopefield of cells at 1-min intervals for 60 min. In a typical exper-iment there were ;15 to 20 cells in a microscope field, and atleast 75% of cells possessed adherent microcolonies of A/Ebacteria as assessed at the end of an experiment by phase-contrast microscopy and the FAS test (Fig. 1A). Irrespective ofwhether we measured [Ca]i averaged over all cells in the field,
TABLE 1. EPEC and EHEC strains used in this study
Strain Serotype Description Origin
E2348/69 O127:H6 EPEC isolate United Kingdom20293 O111: EPEC isolate United Kingdom7958 O126: EPEC isolate France172/1 O55:H2 EPEC isolate BrazilE29962 O157:H7 EHEC isolate United Kingdom85-170 O157:H7 EHEC isolate spontaneously
cured of phages encodingShiga-like toxins
United States
VOL. 66, 1998 INTRACELLULAR CALCIUM AND A/E LESION FORMATION 3901
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/i
ai o
n 19
Nov
embe
r 20
21 b
y 20
1.18
3.96
.226
.

in individual cells, or in specific regions of cells containing at-tached bacterial microcolonies (identified by comparison withphase-contrast micrographs taken at the end of the experi-ment), we were unable to detect anything other than minorfluctuations in [Ca]i throughout the experimental period. Re-
sults of a typical experiment are illustrated in Fig. 2, whichshows pseudocolor ratio images taken at 2-min intervals. Al-though A/E lesion formation occurred between images 5 and10, no significant alterations in [Ca]i were detected during thisperiod. However, ;4- to 5-fold increases in [Ca]i were seen
FIG. 1. FAS tests performed at the end of typical calcium-imaging experiments. The micrographs show fluorescence (left panels) and corresponding phase-contrastimages (right panels) of HEp-2 cell monolayers spindown infected with EPEC strain E2348/69 for 1 h (A), gradually infected with EPEC strain E2348/69 for 6 h (B),gradually infected with EHEC strain E29962 for 6 h (C), and treated with BAPTA and then spindown infected with EPEC strain E2348/69 for 1 h (D). Each infectionprocedure resulted in good colonization of cells with A/E EPEC (A and B), although adhesion by EHEC strains was significantly less than that by EPEC strains (C);characteristic EPEC-induced actin accretion occurred in BAPTA-treated cells (D). Magnification, 3320.
3902 BAIN ET AL. INFECT. IMMUN.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/i
ai o
n 19
Nov
embe
r 20
21 b
y 20
1.18
3.96
.226
.

FIG. 2. Montage of pseudocolor ratio images taken at 2-min intervals, showing [Ca]i in HEp-2 cells spindown infected with E2348/69 for 1 h. The images show nosignificant alterations in [Ca]i during the period of A/E lesion formation (images 5 to 10). After 40 min (image 21), 100 mM histamine was added to the cells, resultingin a large increase in [Ca]i.
VOL. 66, 1998 INTRACELLULAR CALCIUM AND A/E LESION FORMATION 3903
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/i
ai o
n 19
Nov
embe
r 20
21 b
y 20
1.18
3.96
.226
.

immediately following the addition of 100 mM histamine after40 min, confirming the responsiveness of the cells and thedetection system.
The spindown infection protocol resulted in rapid EPECadhesion and A/E lesion formation. However, there was ashort delay between the centrifugation step and commence-ment of calcium imaging, during which time rapid calciumresponses could have been missed. We therefore used a settle-down procedure in which calcium imaging was commencedimmediately following the addition of an aggregated bacterialsuspension. Although adhesion and A/E lesion formation werenot as rapid as with the spindown procedure, by 1 h there wasequally good bacterial attachment and A/E lesion formation(data not shown). Using this settledown procedure, we werestill unable to detect any significant alteration in [Ca]i through-out the 60-min experimental period. As with the spindownexperiments, corresponding phase-contrast and FAS tests con-firmed that imaged fields possessed infected cells producingA/E lesions, and addition of histamine or ionomycin confirmedthe responsiveness of the cells and the detection system.A graphical representation of [Ca]i in typical spindown andsettledown infection experiments in comparison with thosein uninfected cells is shown in Fig. 3.
Since we were unable to detect [Ca]i in cells infected byusing rapid infection procedures, we used a gradual infectionwhich essentially reproduced the method originally used todemonstrate increased [Ca]i in EPEC-infected HEp-2 cells (2).Cells were infected with an overnight broth culture of EPEC,and [Ca]i was measured after 1, 2, 3, 4, and 6 h. Little bacterialadhesion occurred during the first hour of infection, but adhe-sion and A/E lesion formation were clearly seen at 2 h, afterwhich adhesion increased rapidly, and by 6 h all cells possessedmicrocolonies of A/E bacteria (Fig. 1B). Average [Ca]i fromfive randomly selected microscope fields were determined ateach time point, but again we were unable to detect any sig-nificant alterations in [Ca]i in cell monolayers infected withEPEC strain E2348/69, with three other classical EPEC strainsbelonging to serogroups O55, O111, and O126, or with twoO157:H7 EHEC strains (Fig. 4). Although the EHEC strainsdid not adhere to HEp-2 cells as efficiently as EPEC strains,significant levels of A/E bacterial adhesion were apparentafter a 6-h incubation (Fig. 1C). The responsiveness of thecells and detection system was again confirmed at the endof the experimental period by using histamine (data notshown).
Additional evidence for a role of cell calcium in A/E lesionformation has been the reported inhibition of lesion forma-tion following the buffering of intracellular free calcium withBAPTA. The ability of 10 mM BAPTA to buffer intracellularcalcium was confirmed by demonstrating the abrogation of thehistamine response in BAPTA-treated cells. Typical calciumresponses were seen following sequential additions of 1 mMhistamine to control HEp-2 cells monolayers, but such re-sponses were completely inhibited in BAPTA-treated cells(Fig. 5). Comparable A/E lesion formation assessed by FASoccurred in BAPTA-treated (Fig. 1D) compared to untreated(Fig. 1A) HEp-2 cells.
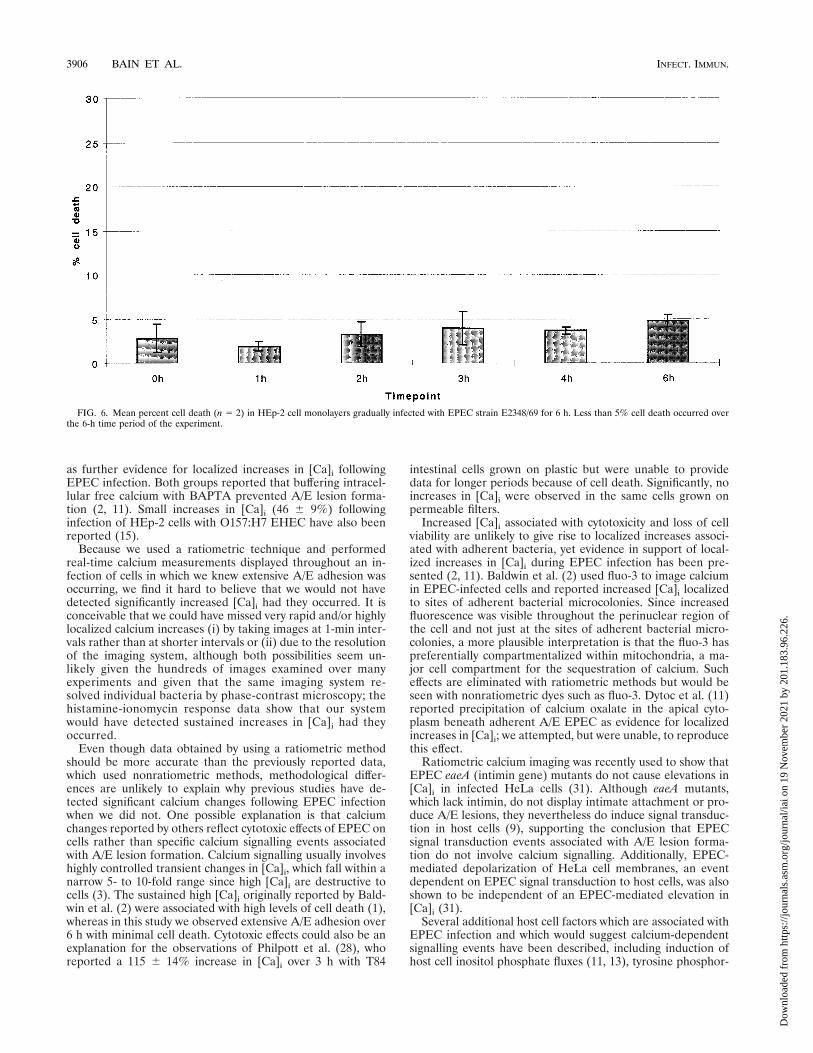
Substantial cell death following EPEC infection of HEp-2cells has previously been reported (1). Since cell viability is afactor which would affect cell calcium measurements, we as-sessed HEp-2 cell viability. In our experiments, ,5% HEp-2cell nonviability throughout the 1-h spindown or settledownexperiments (data not shown) and throughout the 6-h gradualinfection experiments with EPEC strain E2348/69 (Fig. 6) wasobserved.
DISCUSSION
In this study we attempted to measure temporal and spatialalterations in [Ca]i in EPEC- and EHEC-infected HEp-2 cellsby using fura-2 ratiometric digital fluorescence microscopy todetermine if previously reported increases in [Ca]i were local-ized to sites of bacterial adhesion and A/E lesion formation.Not only were we unable to detect any significant elevation in[Ca]i at sites of A/E EPEC adhesion, we were unable to detectany significant alterations in [Ca]i in infected cells compared touninfected cells. In addition, buffering intracellular calciumwith BAPTA did not, as previously reported, prevent A/E le-sion formation. We therefore conclude that specific calciumsignalling and increased [Ca]i are not required for the forma-tion of A/E lesions by EPEC and EHEC.
These findings contradict those of several previous studieswhich have demonstrated elevated [Ca]i in EPEC- and EHEC-infected cells. Baldwin et al. (2) first described alterations in[Ca]i in EPEC-infected epithelial cells and reported a sus-tained increase in [Ca]i following EPEC infection of HEp-2cell monolayers compared to that for control cells or cellsinfected with pathogenic E. coli strains that did not produceA/E lesions; EPEC infection resulted in a doubling of [Ca]iafter 1 h and in an ;6-fold-higher [Ca]i after a 4-h incubation.
FIG. 3. Mean [Ca]i (n 5 4) in control HEp-2 cell monolayers and HEp-2 cell monolayers infected for 1 h with EPEC strain E2348/69 by the spindown andsettledown procedures. No significant alteration in [Ca]i was seen throughout the 1-h infection period, during which extensive A/E adhesion occurred. An ;10-foldincrease in [Ca]i occurred in control cells following the addition of 1 mM ionomycin, confirming the responsiveness of the cells and the detection system.
3904 BAIN ET AL. INFECT. IMMUN.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/i
ai o
n 19
Nov
embe
r 20
21 b
y 20
1.18
3.96
.226
.

Evidence supporting release of calcium from intracellular storeswas also presented, and fluorescence micrographs of EPEC-infected cells loaded with fluo-3 were interpreted as showingincreased [Ca]i in regions of the cell corresponding to areas ofbacterial attachment (2). Dytoc et al. (11), also reported sig-
nificant increases in [Ca]i in EPEC-infected HEp-2 cells, al-though their reported increase of 125 6 40% over 6 h wassignificantly less than that reported by Baldwin et al. (2). Elec-tron microscopy studies showing precipitation of calcium ox-alate subjacent to areas of EPEC attachment were presented
FIG. 4. Mean [Ca]i (n 5 2) in control HEp-2 cell monolayers and HEp-2 cell monolayers infected for up to 6 h with four different EPEC strains and two differentEHEC strains by using the gradual infection procedure. Values are presented as percentages of the mean [Ca]i in the cells at time zero. No significant change in [Ca]iwas seen with any of the six strains over the 6-h time course. Error bars are illustrated for strain E2348/69 and are typical of those for the remainder of the strains.
FIG. 5. Effect of BAPTA on histamine-induced rises in HEp-2 cell [Ca]i. Histamine (H) (1 mM) was added to both control and BAPTA-treated cells after 7 and18 min (arrows). Histamine-stimulated rises in [Ca]i were completely abrogated in the BAPTA-treated cells, whereas considerable increases in [Ca]i were seen in theuntreated cells.
VOL. 66, 1998 INTRACELLULAR CALCIUM AND A/E LESION FORMATION 3905
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/i
ai o
n 19
Nov
embe
r 20
21 b
y 20
1.18
3.96
.226
.
as further evidence for localized increases in [Ca]i followingEPEC infection. Both groups reported that buffering intracel-lular free calcium with BAPTA prevented A/E lesion forma-tion (2, 11). Small increases in [Ca]i (46 6 9%) followinginfection of HEp-2 cells with O157:H7 EHEC have also beenreported (15).
Because we used a ratiometric technique and performedreal-time calcium measurements displayed throughout an in-fection of cells in which we knew extensive A/E adhesion wasoccurring, we find it hard to believe that we would not havedetected significantly increased [Ca]i had they occurred. It isconceivable that we could have missed very rapid and/or highlylocalized calcium increases (i) by taking images at 1-min inter-vals rather than at shorter intervals or (ii) due to the resolutionof the imaging system, although both possibilities seem un-likely given the hundreds of images examined over manyexperiments and given that the same imaging system re-solved individual bacteria by phase-contrast microscopy; thehistamine-ionomycin response data show that our systemwould have detected sustained increases in [Ca]i had theyoccurred.
Even though data obtained by using a ratiometric methodshould be more accurate than the previously reported data,which used nonratiometric methods, methodological differ-ences are unlikely to explain why previous studies have de-tected significant calcium changes following EPEC infectionwhen we did not. One possible explanation is that calciumchanges reported by others reflect cytotoxic effects of EPEC oncells rather than specific calcium signalling events associatedwith A/E lesion formation. Calcium signalling usually involveshighly controlled transient changes in [Ca]i, which fall within anarrow 5- to 10-fold range since high [Ca]i are destructive tocells (3). The sustained high [Ca]i originally reported by Bald-win et al. (2) were associated with high levels of cell death (1),whereas in this study we observed extensive A/E adhesion over6 h with minimal cell death. Cytotoxic effects could also be anexplanation for the observations of Philpott et al. (28), whoreported a 115 6 14% increase in [Ca]i over 3 h with T84
intestinal cells grown on plastic but were unable to providedata for longer periods because of cell death. Significantly, noincreases in [Ca]i were observed in the same cells grown onpermeable filters.
Increased [Ca]i associated with cytotoxicity and loss of cellviability are unlikely to give rise to localized increases associ-ated with adherent bacteria, yet evidence in support of local-ized increases in [Ca]i during EPEC infection has been pre-sented (2, 11). Baldwin et al. (2) used fluo-3 to image calciumin EPEC-infected cells and reported increased [Ca]i localizedto sites of adherent bacterial microcolonies. Since increasedfluorescence was visible throughout the perinuclear region ofthe cell and not just at the sites of adherent bacterial micro-colonies, a more plausible interpretation is that the fluo-3 haspreferentially compartmentalized within mitochondria, a ma-jor cell compartment for the sequestration of calcium. Sucheffects are eliminated with ratiometric methods but would beseen with nonratiometric dyes such as fluo-3. Dytoc et al. (11)reported precipitation of calcium oxalate in the apical cyto-plasm beneath adherent A/E EPEC as evidence for localizedincreases in [Ca]i; we attempted, but were unable, to reproducethis effect.
Ratiometric calcium imaging was recently used to show thatEPEC eaeA (intimin gene) mutants do not cause elevations in[Ca]i in infected HeLa cells (31). Although eaeA mutants,which lack intimin, do not display intimate attachment or pro-duce A/E lesions, they nevertheless do induce signal transduc-tion in host cells (9), supporting the conclusion that EPECsignal transduction events associated with A/E lesion forma-tion do not involve calcium signalling. Additionally, EPEC-mediated depolarization of HeLa cell membranes, an eventdependent on EPEC signal transduction to host cells, was alsoshown to be independent of an EPEC-mediated elevation in[Ca]i (31).
Several additional host cell factors which are associated withEPEC infection and which would suggest calcium-dependentsignalling events have been described, including induction ofhost cell inositol phosphate fluxes (11, 13), tyrosine phosphor-
FIG. 6. Mean percent cell death (n 5 2) in HEp-2 cell monolayers gradually infected with EPEC strain E2348/69 for 6 h. Less than 5% cell death occurred overthe 6-h time period of the experiment.
3906 BAIN ET AL. INFECT. IMMUN.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/i
ai o
n 19
Nov
embe
r 20
21 b
y 20
1.18
3.96
.226
.

ylation of phospholipase C-g1 (19), and activation of proteinkinase C (7, 26). Our data indicate that there is no significantalteration in cell calcium during A/E lesion formation but thatalterations in cell calcium may be related to cytotoxic effects ofEPEC. If this is the case, then perhaps other reported signal-ling events (e.g., increased inositol triphosphate levels) may besimilarly associated with cytotoxic effects of EPEC and shouldnow be reevaluated in assays in which minimal cell damageoccurs.
Based on morphological similarities between the effects of cal-cium and the effects of EPEC on brush border microvilli, it wasoriginally hypothesized that EPEC stimulates a localized increasein [Ca]i leading to activation of villin, disruption of the microvil-lous actin core, and vesiculation of the microvillous membrane(4). Neither we nor anybody else has demonstrated the micromo-lar calcium levels required for villin-mediated microvillous disas-sembly (4). Thus, if increased calcium is not the mechanismwhereby EPEC induces brush border effacement, then what is? Itis now clear that a number of enteric pathogens, including Yer-sinia, Salmonella, and Shigella spp., translocate into host cells viatype III secretion system virulence proteins which induce grosscytoskeletal rearrangements. Yersinia spp. translocate the YopEcytotoxin (30) and Salmonella spp. translocate a tyrosine phos-phatase, SptP (14), which cause disruption of the host cell actinmicrofilament structure. In a calcium-independent manner (5),Shigella spp. translocate the invasin IpaA, which rapidly associateswith vinculin, a protein linking actin filaments to the plasmamembrane to induce highly localized cytoskeletal rearrangementswhich result in membrane ruffling and bacterial uptake (32). In asimilar calcium-independent manner, it seems likely that EPECand EHEC, which also possess type III secretion systems, trans-locate into host cells distinct effector proteins which induce thelocalized cytoskeletal rearrangements involved in A/E lesion for-mation. Indeed, two type III secreted EPEC proteins, EspB(24, 33) and Tir (18), have recently been shown to be trans-located into host cells. Three functions have been ascribedto Tir. Tir functions as the intimate receptor for EPEC onmammalian cell surfaces, Tir nucleates actin following in-timin binding, and Tir stimulates signal transduction eventsfollowing intimin binding (18). EspB or another, as-yet-to-be-identified effector protein could be responsible for local-ized disruption of the microvillus actin cytoskeleton andmicrvillous effacement.
ACKNOWLEDGMENTS
This work was supported by grants from the Wellcome Trust (toC.B. and S.K.) and by a fellowship to R.K. from Fundacao de Amparoa Pesquisa do Estado de Sao Paulo (FAPESP) (Proc. FAPESP no.95/07033-1).
We are grateful to Henry Smith, Central Public Health Laboratory,London, United Kingdom; Jim Kaper, Center for Vaccine Develop-ment, University of Maryland, College Park; and Bernard Joly,Bacteriological Laboratory, University of Clermont-Ferrand, Cler-mont-Ferrand, France for providing strains used in the study.
REFERENCES
1. Baldwin, T. J., M. B. Lee-Delaunay, S. Knutton, and P. H. Williams. 1993.Calcium-calmodulin dependence of actin accretion and lethality in culturedHEp-2 cells infected with enteropathogenic Escherichia coli. Infect. Immun.61:760–763.
2. Baldwin, T. J., W. Ward, A. Aitken, S. Knutton, and P. H. Williams. 1991.Elevation of intracellular free calcium levels in HEp-2 cells infected withenteropathogenic Escherichia coli. Infect. Immun. 59:1599–1604.
3. Berridge, M. J. 1997. Elementary and global aspects of calcium signalling. J.Exp. Med. 200:315–319.
4. Burgess, D. R., and B. E. Prum. 1982. Reevaluation of brush border motility:
calcium induces core filament solation and microvillar vesiculation. J. CellBiol. 94:97–107.
5. Clerc, P. L., B. Berthon, M. Claret, and P. J. Sansonetti. 1989. Internaliza-tion of Shigella flexneri into HeLa cells occurs without an increase in thecytosolic Ca21 concentration. Infect. Immun. 57:2919–2922.
6. Collington, G. K., I. W. Booth, and S. Knutton. 1998. EnteropathogenicEscherichia coli (EPEC) infection rapidly modulates electrolyte transport inCaco-2 cell monolayers. Gut 42:200–207.
7. Crane, J. K., and J. S. Oh. 1997. Activation of host cell protein kinase C byenteropathogenic Escherichia coli. Infect. Immun. 65:3277–3285.
8. Donnenberg, M. S. 1995. Enteropathogenic Escherichia coli, p. 709–726. InM. J. Blaser et al. (ed.), Infections of the gastrointestinal tract. Raven Press,New York, N.Y.
9. Donnenberg, M. S., and J. B. Kaper. 1991. Construction of an eae deletionmutant of enteropathogenic Escherichia coli by using a positive-selectionsuicide vector. Infect. Immun. 59:4310–4317.
10. Donnenberg, M. S., J. B. Kaper, and B. B. Finlay. 1997. Interactions betweenenteropathogenic Escherichia coli and host epithelial cells. Trends Microbiol.5:109–114.
11. Dytoc, M., L. Fedorko, and P. M. Sherman. 1994. Signal transduction inhuman epithelial cells infected with attaching and effacing Escherichia coli invitro. Gastroenterology 106:1150–1161.
12. Foubister, V., I. Rosenshine, M. S. Donnenberg, and B. B. Finlay. 1994. TheeaeB gene of enteropathogenic Escherichia coli is necessary for signal trans-duction in epithelial cells. Infect. Immun. 62:3038–3040.
13. Foubister, V., I. Rosenshine, and B. B. Finlay. 1994. A diarrheal pathogen,enteropathogenic Escherichia coli (EPEC) triggers a flux of inositol phos-phates in infected epithelial cells. J. Exp. Med. 179:993–998.
14. Fu, Y., and J. E. Galan. 1998. The Salmonella typhimurium tyrosine phos-phatase SptP is translocated into host cells and disrupts the actin cytoskel-eton. Mol. Microbiol. 27:359–368.
15. Ismaili, A., D. J. Philpott, M. T. Dytoc, and P. M. Sherman. 1995. Signaltransduction responses following adhesion of verocytotoxin-producing Esch-erichia coli. Infect. Immun. 63:3316–3326.
16. Jarvis, K. G., J. A. Giron, A. E. Jerse, T. K. McDaniel, M. S. Donnenberg,and J. B. Kaper. 1995. Enteropathogenic Escherichia coli contains a spe-cialised secretion system necessary for the export of proteins involved inattaching and effacing lesion formation. Proc. Natl. Acad. Sci. USA 92:7996–8000.
17. Jerse, A. E., J. Yu, B. D. Tall, and J. B. Kaper. 1990. A genetic locus ofenteropathogenic Escherichia coli necessary for the production of attachingand effacing lesions on tissue culture cells. Proc. Natl. Acad. Sci. USA 87:7839–7843.
18. Kenny, B., R. DeVinney, M. Stein, D. J. Reinsheid, E. A. Frey, and B. B.Finlay. 1997. Enteropathogenic E. coli (EPEC) transfers its receptor forintimate adhesion into mammalian cells. Cell 91:511–529.
19. Kenny, B., and B. B. Finlay. 1997. Intimin-dependent binding of entero-pathogenic Escherichia coli to host cells triggers novel signaling events,including tyrosine phosphorylation of phospholipase C-gamma 1. Infect.Immun. 65:2528–2536.
20. Kenny, B., L. C. Lai, B. B. Finlay, and M. S. Donnenberg. 1996. EspA,a protein secreted by enteropathogenic Escherichia coli (EPEC) isrequired to induce signals in epithelial cells. Mol. Microbiol. 20:313–323.
21. Knutton, S., M. M. Baldini, J. B. Kaper, and A. S. McNeish. 1987. Role ofplasmid-encoded adherence factors in adhesion of enteropathogenic Esche-richia coli to HEp-2 cells. Infect. Immun. 55:78–85.
22. Knutton, S., T. J. Baldwin, P. H. Williams, and A. S. McNeish. 1989. Actinaccumulation at sites of bacterial adhesion to tissue culture cells: basis of anew diagnostic test for enteropathogenic and enterohemorrhagic Escherichiacoli. Infect. Immun. 57:1290–1298.
23. Knutton, S., D. R. Lloyd, and A. S. McNeish. 1987. Adhesion of entero-pathogenic Escherichia coli to human intestinal enterocytes and culturedhuman intestinal mucosa. Infect. Immun. 55:69–77.
24. Knutton, S., I. Rosenshine, M. J. Pallen, I. Nisan, B. C. Neves, C. Bain, C.Wolff, G. Dougan, and G. Frankel. 1998. A novel EspA-associated surfaceorganelle of enteropathogenic Escherichia coli involved in protein translo-cation into epithelial cells. EMBO J. 17:2166–2176.
25. Lai, L.-C., L. A. Wainwright, K. D. Stone, and M. S. Donnenberg. 1997.A third secreted protein that is encoded by the enteropathogenic Esch-erichia coli pathogenicity island is required for transduction of signals andfor attaching and effacing activities in host cells. Infect. Immun. 65:2211–2217.
26. Manjarrez-Hernandez, H. A., T. J. Baldin, P. H. Williams, R. Haigh, S.Knutton, and A. Aitken. 1996. Phosphorylation of myosin light chain atdistinct sites and its association with the cytoskeleton during enteropatho-genic Escherichia coli infection. Infect. Immun. 64:2368–2370.
27. McDaniel, T. K., K. G. Jarvis, M. S. Donnenberg, and J. B. Kaper. 1995. Agenetic locus of enterocyte effacement conserved among diverse enterobac-terial pathogens. Proc. Natl. Acad. Sci. USA 92:1664–1668.
28. Philpott, D. J., D. M. McKay, P. M. Sherman, and M. H. Perdue. 1996.Infection of T84 cells with enteropathogenic Escherichia coli alters barrier
VOL. 66, 1998 INTRACELLULAR CALCIUM AND A/E LESION FORMATION 3907
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/i
ai o
n 19
Nov
embe
r 20
21 b
y 20
1.18
3.96
.226
.

and transport functions. Am. J. Physiol. 270:G634–G645.29. Rosenshine, I., S. Ruschkowski, M. Stein, D. Reinscheid, S. D. Mills, and
B. B. Finlay. 1996. A pathogenic bacterium triggers epithelial signals to forma functional bacterial receptor that mediates actin pseudopod formation.EMBO J. 15:2613–2624.
30. Rosqvist, R., K.-E. Magnusson, and H. Wolf-Watz. 1994. Target cell contacttriggers expression and polarized transfer of Yersinia YopE cytotoxin intomammalian cells. EMBO J. 13:964–972.
31. Stein, M. A., D. A. Mathers, H. Yan, K. G. Baimbridge, and B. B. Finlay.
1996. Enteropathogenic Escherichia coli markedly decreases resting mem-brane potential of Caco-2 and HeLa human epithelial cells. Infect. Immun.64:4820–4825.
32. Trau van Nhieu, G., A. Ben-Ze’ev, and P. J. Sansonetti. 1997. Modulation ofbacterial entry into epithelial cells by association between vinculin and theShigella IpaA invasin. EMBO J. 16:2717–2729.
33. Wolff, C., I. Nisan, E. Hanski, G. Frankel, and I. Rosenshine. 1998. Proteintranslocation into HeLa cells by infecting enteropathogenic Escherichia coli.Mol. Microbiol. 28:143–155.
Editor: P. E. Orndorff
3908 BAIN ET AL. INFECT. IMMUN.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/i
ai o
n 19
Nov
embe
r 20
21 b
y 20
1.18
3.96
.226
.