implications of heparan sulfate and heparanase in...
TRANSCRIPT

ACTAUNIVERSITATIS
UPSALIENSISUPPSALA
2017
Digital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Medicine 1303
Implications of Heparan Sulfateand Heparanase in InflammatoryDiseases
ANDREAS DIGRE
ISSN 1651-6206ISBN 978-91-554-9828-3urn:nbn:se:uu:diva-315699

Dissertation presented at Uppsala University to be publicly examined in A1:111a, BMC,Husargatan 3, Uppsala, Friday, 7 April 2017 at 13:15 for the degree of Doctor of Philosophy(Faculty of Medicine). The examination will be conducted in English. Faculty examiner:Professor MD Liliana Schaefer (Department of General Pharmacology and Toxicology,Goethe University, Frankfurt, Germany).
AbstractDigre, A. 2017. Implications of Heparan Sulfate and Heparanase in Inflammatory Diseases.Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine1303. 68 pp. Uppsala: Acta Universitatis Upsaliensis. ISBN 978-91-554-9828-3.
Heparan sulfate (HS), an unbranched sulfated carbohydrate chain, and the HS-degradingenzyme heparanase play important roles in physiological and pathological processes during allstages of life, from early embryogenesis to ageing. Accumulated information shows that HSand heparanase are involved in inflammatory processes and associated diseases, e.g. rheumatoidarthritis (RA) and Alzheimer’s disease.
In this thesis I have investigated the role of HS and heparanase (Hpa) in inflammatory-relatedpathologies. In the first project, Hpa overexpressing mice (Hpa-tg) were induced with a murinemodel of RA. We found a pro-inflammatory role of Hpa through enhancing the activity of T-cells and innate immune cells, which contributed to an augmented severity of clinical symptomsin the Hpa-tg mice.
In my second project, we revealed co-current interaction of heparin with both ApoA1 andSAA of HDL isolated from plasma of inflamed mouse. Mass spectrometry analysis indicatedclose proximity of ApoA1 and SAA on the HDL surface, providing a molecular and structuralmechanism for the simultaneous binding of heparin to apoA1 and SAA.
In my third project, we investigated the role of Hpa in AA amyloid formation and resolutionin mice in a model of AA-amyloidosis. We found a similar degree of amyloid formation inHpa-KO mice compared to the wildtype control mice, but the resolution process was faster inHpa-KO mice. The rapid clearance of deposited SAA in Hpa-KO mice was associated withupregulated expression of matrix metalloproteases. The results suggest an associated functionof ECM proteases with heparanase in the process of AA amyloid resolution.
In my fourth project, we found that overexpression of heparanase impaired inflammationassociated beta amyloid (Aβ) clearance in the brain of an Alzheimer’s disease mouse model.Examination of the cytokine profile of brain lysates revealed an overall lower inflammatoryreaction in the double transgenic (tgHpa*Swe) mice compared to single APP-tg (tg-Swe) micein response to LPS-induced inflammation.
Keywords: Heparanase, Heparan sulfate, Inflammation, Inflammatory diseases, Autoimmunediseases, Amyloidosis, Reumathoid arthritis, SAA, APP, A beta, Neuroinflammation
Andreas Digre, Department of Medical Biochemistry and Microbiology, Box 582, UppsalaUniversity, SE-75123 Uppsala, Sweden.
© Andreas Digre 2017
ISSN 1651-6206ISBN 978-91-554-9828-3urn:nbn:se:uu:diva-315699 (http://urn.kb.se/resolve?urn=urn:nbn:se:uu:diva-315699)

Dedicated to my family
“You've got to sing like there's nobody listening, Love like you'll never be hurt, Dance like there's nobody watching, And live like it's paradise on earth.” ― Susanna Clark & Richard Leigh

Supervisors: Professor Jin-Ping Li Department of Medical Biochemistry and Microbiology, Uppsala University Professor Lena Kjellén Department of Medical Biochemistry and Microbiology, Uppsala University Faculty Opponent: Professor MD Liliana Schaefer Department of General Pharmacology and Toxicology, Goethe University, Frankfurt, Germany Examining Committee: Docent Sofia Schedin Weiss Department of Neurobiology, Care Sciences and Society, Karolinska Institute Senior Professor Anders Malmström Department of Experimental Medical Science, Lund University Docent Joakim Bergström Department of Public Health and Caring Sciences, Geriatrics, Uppsala University Cover pages: The front-page illustration shows the hands from a patient with a severe degree of rheumatoid arthritis, edited graphically with an inflamma-tory fluid surrounding the hands. Case courtesy of A.Prof. Frank Gaillard, Radiopaedia.org, rID: 7245. Backside illustration shows the amyloid-beta deposition (blue-green) in a brain from a mouse overexpressing amyloid-beta precursor protein. Courtesy of Prof. Lars Nilsson.

List of Papers
This thesis is based on the following papers, which are referred to in the text by their Roman numerals.
I. Digre, A.*, Singh, K.*, Åbrink, M., Rejimers, R.M., Sandler, S., Vlodavsky, I., and Li J.-P. (2017) Overexpression of heparanase en-hances T lymphocyte activities and intensifies the inflammatory re-sponse in a model of murine rheumatoid arthritis. Submitted manu-script
II. Digre, A., Nan, J., Frank, M., and Li J.-P. (2016) Heparin interac-
tions with apoA1 and SAA in inflammation-associated HDL. Bio-chemical and Biophysical Research Communications, 474 (2) 309-314.
III. Wang, B., Tan, Y.X., Jia, J., Digre, A., Zhang, X., Vlodavsky, I.,
and Li J.-P. (2012) Accelerated resolution of AA amyloid in hepara-nase knockout mice is associated with matrix metalloproteases. PLoS One, 7(7):e39899.
IV. Jendresen, C.B., Digre, A., Cui, H., Zhang, X., Vlodavsky, I., Nils-
son, L.N.G., and Li J.-P. (2017) Overexpression of heparanase inter-feres with inflammatory-associated amyloid-beta clearance in mice. Manuscript
* These authors contributed equally to the study.
Reprints were made with permission from the respective publishers.


Contents
Introduction ................................................................................................... 111. Heparan sulfate proteoglycans (HSPGs) .............................................. 12
1.1 Proteoglycans ................................................................................. 121.2 HS - structure & function ............................................................... 131.3 Biosynthesis ................................................................................... 14
2. Heparanase ............................................................................................ 162.1 Functions ........................................................................................ 162.2 Molecular properties ...................................................................... 172.3 Expression ...................................................................................... 172.4 Enzymatic properties ..................................................................... 192.5 Heparanase 2 .................................................................................. 20
3. HS & heparanase in inflammation ........................................................ 213.1 Inflammation .................................................................................. 213.2 HS in inflammatory reactions ........................................................ 233.3 Heparanase in inflammatory reactions ........................................... 233.4 Murine models of inflammation .................................................... 26
4. HS & heparanase in rheumatoid arthritis (RA) .................................... 274.1 Rheumatoid arthritis ....................................................................... 274.2 Heparanase & RA .......................................................................... 294.3 Collagen-induced arthritis (CIA) ................................................... 29
5. HS & heparanase in amyloidosis .......................................................... 315.1 Amyloidosis ................................................................................... 315.2 Serum amyloid A & AA amyloidosis ............................................ 315.3 Alzheimer’s disease (AD) .............................................................. 33
Present investigations .................................................................................... 35Paper I ....................................................................................................... 35Paper II ..................................................................................................... 36Paper III .................................................................................................... 39Paper IV .................................................................................................... 40
Concluding remarks and future perspectives ................................................ 44Populärvetenskaplig sammanfattning ........................................................... 47Acknowledgement ......................................................................................... 49References ..................................................................................................... 52


Abbreviations
Aβ Amyloid-β AβPP Amyloid-β precursor protein ACPA Anti-citrullinated protein antibodies AD Alzheimer´s disease AFP Amyloid fibril protein ApoA1 BACE1 BBB
Apolipoprotein A-1 Beta-secretase 1 Blood-brain barrier
CAA CFA DAMP DMARD
Cerebral amyloid angioapathy Complete Freund׳s adjuvant Damage-associated molecular pattern Disease-modifying antirheumatic drug
ECM ELISA FGF
Extracellular matrix Enzyme-linked immunosorbent assay Fibroblast growth factor
GAG Glycosaminoglycan GlcA Glucuronic acid GlcN Glucosamine GlcNAc N-acetylglucosamine GlcNS N-sulfated glucosamine GlyCAM-1 GPI
Glycosylation-dependent cell adhesion molecule-1 Glycophophatidyl inisitol
HBD HS/heparin-binding domain (on heparanase) HDL High-density lipoprotein HDL-SAA High-density lipoprotein – Serum amyloid A HS Heparan sulfate HSPG Heparan sulfate proteoglycan IAPP Islet amyloid polypeptide ICAM-1 Intercellular adhesion molecule 1 IdoA Iduronic acid IL Interleukin LFA Lymphocyte function-associated antigen 1 LPS Lipopolysaccharide Mac-1 Macrophage-1 antigen MAPτ Microtubule associated protein Tau MIP2 Macrophage inflammatory protein 2

MHC Major histocompatibility complex MMP Matrix metalloproteinase NDST N-deacetylase/N-sulfotransferase NFT NSAID
Neurofibrillary tangles Non-steroidal anti-inflammatory drug
PG PAMP PRR
Proteoglycan Pathogen-associated molecular pattern Pattern recognition receptor
RA RCT RF
Rheumatoid arthritis Reverse cholesterol transport Rheumatoid factor
SAA Serum amyloid A TNF Tumor necrosis factor VCAM-1 Vascular cell adhesion molecule 1

11
Introduction
In 1979, a breakthrough study of the anticoagulant effect of heparin was published by Ulf Lindahl and his colleagues in Uppsala, where they had found that a specific sulfation pattern of the heparin glycosaminoglycan (GAG) chain was needed for it to bind to a specific site of antithrombin (AT), alter the conformation of AT and consequently stop the coagulation process by AT-induced inhibition of thrombin (Lindahl et al., 1979). The finding was of great importance as the heparin drugs could be optimized for the treatment of thrombosis. Since the 90s, there has been an explosion of new information regarding the GAGs and their tremendously broad function as proteoglycans (PGs), covering all stages of the life cycle, all cells of the metazoan organism, and most aspects of physiology. However, the research field of GAGs have continuously been complicated by the structural com-plexity of these linear polysaccharides.
In Uppsala, in the 00s, Jin-Ping Li, a previous postdoctoral researcher in Ulf Lindahl´s group, and her colleagues, developed mice with altered ex-pression of an enzyme that had been found to degrade heparan sulfate (HS) chains, heparanase. These mice, either overexpressing or lacking heparanase, were shown to be great tools to reflect the role of both heparanase and its substrate HS in various facets of physiology and pathophysiology. Hepara-nase, previously mostly known for its involvement in cancer, have recently been implicated in several other diseases, with accumulating evidence identi-fying it as a regulator of one our most basic mechanisms of immune defence, inflammation.

12
1. Heparan sulfate proteoglycans (HSPGs)
Figure 1. Heparan sulfate and heparin proteoglycans (HSPGs). HSPGs consist of core proteins linked through a linker tetrasaccharide to one or more HS/heparin chains as well as other types of GAGs, including chondroitin sulfate (CS). Three type of HSPGs include membrane-spanning- (e.g. syndecan and glypican), ECM- (e.g. perlecan) and intracellular vesicle HSPGs (serglycin).
1.1 Proteoglycans Proteoglycans (PGs) are protein-containing glycoconjugates expressed in nearly all metazoan cells (Esko et al., 2009). The macromolecules consists of a core protein with one or more covalently attached linear glycosaminogly-can (GAG) chain(s) of repeating disaccharide units. The GAG chains at-tached to PGs are divided into five major types based on structure, glycosid-ic linkage and the degree of sulfation of the carbohydrate units: chondroitin sulfate (CS), dermatan sulfate (DS), keratan sulfate (KS), heparan sulfate (HS) and heparin (Esko et al., 2009). These chains are, in varying numbers,

13
covalently linked to core proteins found in the extracellular matrix (ECM), attached to the cell membrane or stored in secretory granules (Esko et al., 2009). All this diversity have facilitated the great span of biological func-tions exerted by PGs, ranging from structural organization of cartilage tissue and the ECM to cell-cell, cell-matrix signaling and cell adhesion (Bishop et al., 2007, Esko et al., 2009).
1.2 HS - structure & function The sulfated GAG chain HS is a negatively charged site facilitating electro-static interactions with a number of biologically active proteins, e.g. mor-phogens, chemokines, growth factors, enzymes and ECM-proteins (Kjellén and Lindahl, 1991, Lyon and Gallagher, 1998, Perrimon and Bernfield, 2000, Gallagher, 2001, Esko and Selleck, 2002). HS is mainly present in the body attached to a core protein, in the form of a PG. Three subfamilies of HS proteoglycans (HSPGs) have been described: Membrane-spanning HSPGs (e.g. syndecans, betaglycan, CD44v3 and glycophophatidyl inisitol (GPI)-linked glypicans), ECM HSPGs (e.g. perlecan, collagen XVIII and agrin) and the secretory vesicle HSPG, serglycin (Gallagher, 2001, Esko and Selleck, 2002, Song and Filmus, 2002, Bishop et al., 2007). HSPGs have essential functions in the metazoan system during both embryogenesis and adult life: (I) co-receptor for e.g. growth factors, cytokines and microbes, (II) transportation and presentation of chemokines at the cell surface, (III) cell adhesion to the ECM, (IV) “storage” of growth factors and morphogens in the ECM for future release by heparanase, (V) paracrine signaling by HSPG ectodomain shedding and (VI) internalization of ligands, e.g. enzymes (Bishop et al., 2007, Bernfield et al., 1999). Hence, it is not surprising that the field of HS has expanded enormously in the last decades and new func-tions of HS are continuously being discovered (Esko et al., 2009, Bishop et al., 2007).
Heparin, which has a molecular structure very similar to HS, has been used as an anticoagulant in the clinic to treat and prevent thromboembolism for more than half a century (Gray et al., 2008). The anticoagulant activity of heparin is owed to its specific binding to antithrombin, thus vastly increasing the ability of antithrombin to bind and inhibit coagulation proteases (Gray et al., 2008). The specific binding of heparin to antithrombin is mediated by a unique pentasaccharide sequence found on a subset of heparin chains (Lam et al., 1976, Andersson et al., 1976, Thunberg et al., 1979, Rosenberg et al., 1979, Lindahl et al., 1980). The high structural similarity between HS and heparin has caused discussions regarding their definition: Both contain the same type of monosaccharide residues with the same type of modifications and connected by the same glycosidic linkage, but the degree of sulfation and epimerization, as well as core protein, differs between them (Figure 1). Heparin has a considerable higher degree of sulfation/epimerization than HS.

14
Heparin chains are also exclusively synthesized in connective tissue mast cells in the form of serglycin proteoglycans; while HS is produced in virtual-ly all metazoan cell types (Esko et al., 2009).
Figure 2. HS/heparin biosynthesis. The elongating chain of alternating GlcA and GlcN is subjected to N- and O-sulfation as well as C5-epimerization in varying degrees. The heparin chain is modified to a higher degree. The AT-binding penta-saccharide sequence on heparin is indicated.
1.3 Biosynthesis The structure and amount of HS vary significantly between cells of different tissues. However, when comparing between individuals with the same geno-type, the same levels and structure of HS is found, indicating a very stable and regulated HS production process (Ledin et al., 2004). The diverse sul-fation patterns of HS, which allow for interactions with numerous HS bind-ing proteins, are created by an orchestra of HS biosynthesis enzymes, capa-ble of HS chain deacetylation, sulfation, and epimerization (Esko and Selleck, 2002, Habuchi et al., 2004), as well as post-biosynthesis sulfatases, capable of desulfating the chain (Lamanna et al., 2007, Dhoot et al., 2001, Morimoto-Tomita et al., 2002, Ai et al., 2006). Thus, the key to HS biology lies in the biosynthesis and modification of the molecule (Figure 1). HS bio-synthesis is accomplished mainly in the Golgi compartment and is initiated

15
by the biosynthesis of a tetrasaccharide (GlcAβ3Galβ3Galβ4Xylβ3(-Ser)) onto specific serine attachment sites, consisting of a Ser-Gly dipeptide flanked by acidic amino acids, on a newly translated HSPG core protein (Esko et al., 2009, Esko and Selleck, 2002, Esko and Zhang, 1996, Sugahara and Kitagawa, 2000). This attachment is catalyzed sequentially by xylosyl-transferase 1 or 2 (XT1 and XT2)(Ponighaus et al., 2007), galactosyltrans-ferase I and II (GalTI and GalTII) and glucuronosyltransferase I (GlcATI)(Esko and Selleck, 2002). The tetrasaccharide is not specific for HS chains, but constitutes a shared feature among HS, heparin, CS and DS (Esko et al., 2009). Following the assembly of the linker tetrasaccharide, the type of GAG chain that will be synthesized is decided by addition of the next sugar residue: While the attachment of a β4GalNAc residue by GALNACT1 and GALNACT2 results in the initiation of CS/DS biosynthesis, the addition of an α4GlcNAc results in the initiation of HS/heparin formation (Esko and Selleck, 2002, Esko and Zhang, 1996, Lindahl and Li, 2009). The addition of the initial GlcNAc residue to the proximal tetrasaccharide, so called Glc-NAcT-I activity, is believed to be performed by the evolutionary preserved EXT-like 3 (EXTL3) (Esko et al., 2009, Nadanaka and Kitagawa, 2008, Kitagawa et al., 2001, Osman et al., 2004, Busse et al., 2007). However, this is still under investigation since both EXTL2 and the EXT1/EXT2 complex also have been suggested to possess this activity (Nadanaka and Kitagawa, 2008, Kitagawa et al., 1999, Kim et al., 2003, Busse et al., 2007). Although, EXTL2 GlcNAcT-I activity was recently found to inhibit further extension of the HS chain, indicating it has a role as a suppressor of HS biosynthesis (Nadanaka et al., 2013).
The co-polymerases exostosin 1 and 2 (EXT1 and EXT2) are responsible for the extension of the HS chain by adding alternation GlcA and GlcNAc residues, forming repeating disaccharide units (-GlcAβ4-GlcNAcα4-)(Esko and Selleck, 2002, McCormick et al., 1998, Lind et al., 1998). Even though both of these polymerases comprise dual GlcA and GlcNAc transferase (GlcAT, GlcNAcT) activity, formation of the EXT1/EXT2 complex results in a synergistically increased polymerase activity (Lind et al., 1998, McCormick et al., 2000).
The elongating HS chain is a target for extensive modifications, starting with the substitution of the N-acetyl group of GlcNAc residues with an N-sulfate group, in two independent steps. Both reactions are performed by the N-deacetylase/N-sulfotransferase (NDST) family of enzymes and facilitates further modifications of the HS polymer (Esko and Selleck, 2002, Aikawa et al., 2001). GlcA residues on the reducing end of a newly sulfated GlcNS residue, are subsequently subjected to both epimerization by D-glucuronyl C5 epimerase (GLCE) into L-iduronic acid (IdoA) and 2-O-sulfation by an uronosyl 2-O-sulfotransferase (2-OST) (Esko and Selleck, 2002, Valla et al., 2001, Kobayashi et al., 1996, Rong et al., 2000, Rong et al., 2001).

16
After these modifications, a specific pattern of modified regions, do-mains, appears in the HS chain: Regions rich in GlcA and GlcNAc residues, designated N-acetylated (NA) domains, regions highly modified with IdoA and GlcNS residues, entitled N-sulfated (NS) domains and the NA/NS do-mains in between with more or less equivalent amounts of NA and NS glu-cosamine residues (Maccarana et al., 1996). The GlcNS residues of the NS domains of the HS chain are then further sulfated by the glucosaminyl 6-O-sulfotransferase (6-OST) and the glucosaminyl 3-O-sulfotransferase (3-OST) families of enzymes (Esko and Selleck, 2002, Habuchi et al., 1998, Habuchi et al., 2000, Shworak et al., 1997, Shworak et al., 1999).
Post-biosynthetically, the 6-O-sulfate groups are removed in specific re-gions of the HS chain, at the cell surface, by extracellular sulfatase 1 and 2 (SULF1 and SULF2) (Lamanna et al., 2007, Dhoot et al., 2001, Morimoto-Tomita et al., 2002, Ai et al., 2006).
2. Heparanase 2.1 Functions Heparanase is an essentially lysosomal mammalian protein with multiple functions, both intracellularly and extracellularly following excretion (Vlodavsky et al., 2013). Due to its endo-β-D-glucuronidase activity, hepa-ranase is able to cleave the glycosidic bonds of both HS and heparin between GlcA and GlcN units, releasing chain fragments of 4-7kDa (Freeman and Parish, 1998, Pikas et al., 1998). The fact that HS is involved in a vast range of biological processes makes heparanase a highly significant regulator of those processes. For example, cleavage of HS in the ECM results in release of sequestered signal molecules, including cytokines, growth factors and chemokines (Vlodavsky et al., 2013). HS fragmentation also contributes to ECM reorganization, thus permitting increased cell motility and tissue inva-sion (Dempsey et al., 2000). Heparanase also possesses non-enzymatic prop-erties such as the upregulation of tissue factor in vascular endothelial and cancer cells (Nadir et al., 2006), enhancement of Akt signalling and PI3K-dependent endothelial cell migration (Gingis-Velitski et al., 2004a), and facilitation of cell adhesion (Levy-Adam et al., 2008). Furthermore, via both enzymatic and non-enzymatic mechanisms, heparanase affects expression of essential genes, such as growth factor hepatocyte growth factor (HGF), vas-cular endothelial growth factor (VEGF), which stimulates angiogenesis, and the ECM degrading matrix metalloproteinase (MMP)-9 (Zetser et al., 2006, Purushothaman et al., 2008, Ramani et al., 2011). Altogether, heparanase`s great repertoire of functions together with the widespread abundance of its enzymatic substrates makes it an important biomolecule in numerous physio-

17
logical and pathological conditions, including embryogenesis, inflammation, autoimmune disorders, amyloidosis and cancer.
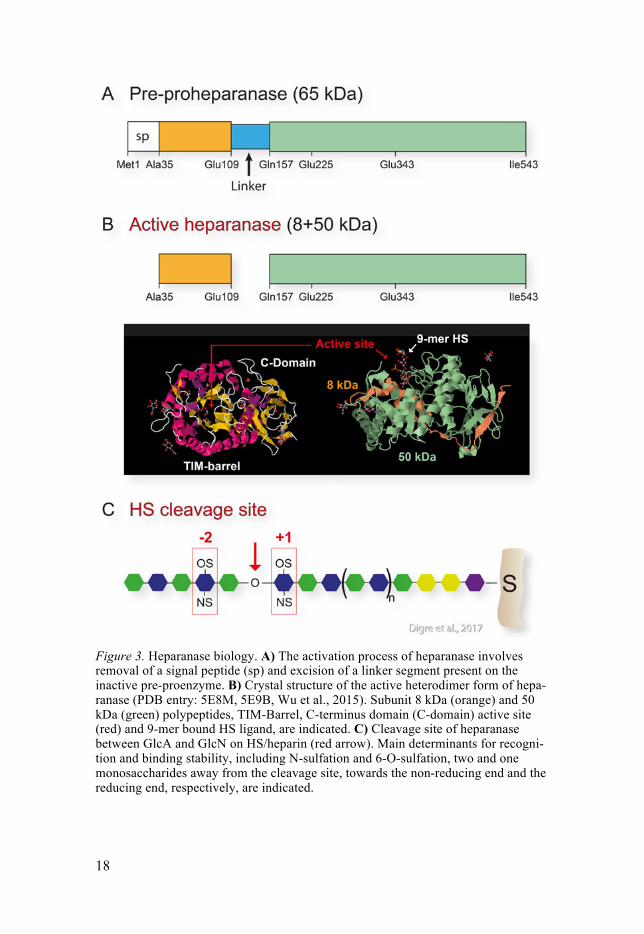
2.2 Molecular properties A predominant form of heparanase is present in mammals, encoded by the HPSE gene. The mRNA is translated into an inactive pre-proenzyme of 543 amino acid, with the molecular weight of 65kDa (Figure 3)(Hulett et al., 1999, Kussie et al., 1999, Toyoshima and Nakajima, 1999, Vlodavsky et al., 1999). After removal of the signal peptide from the pre-proheparanase in the ER, latent proheparanase is transported by vesicles through the Golgi com-partment to the cell surface. At the cell surface the latent heparanase binds membrane-bound HSPGs through HS/heparin binding domains (HBDs) and rapidly becomes internalized (Gingis-Velitski et al., 2004b, Levy-Adam et al., 2005). The internalization can also be assisted through proheparanase binding to low-density lipoprotein receptor-related proteins and mannose 6-phosphate receptors (Vreys et al., 2005). After internalization, the hepara-nase/HSPG complex is transported to lysosomes where proheparanase is processed, by the protease cathepsin-L, into the enzymatically active form by removing a 6 kDa internal linking segment, and joining the two remain-ing polypeptides of 50kDa and 8kDa into a heterodimer (Figure 2)(Zetser et al., 2004, Abboud-Jarrous et al., 2005, Abboud-Jarrous et al., 2008). The optimal pH for the catalytic activity of heparanase has been found to be mildly acidic, between 5-6 (Gilat et al., 1995). The glutamic acids at position 225 and 343 are proposed catalytic residues for the endo-β-D-glucuronidase activity of heparanase, serving as proton donor and nucleophile, respectively (Hulett et al., 2000, Wu et al., 2015). A TIM-barrel fold contains the active site together with a catalytically essential C-terminal-domain (C-domain) (Figure 2). The C-domain has also been shown to be involved in the secre-tion of heparanase and in the non-enzymatic facilitation of Akt phosphoryla-tion, cell proliferation and tumor xenograft progression (Fux et al., 2009).
2.3 Expression After the discovery of heparanase (Ogren and Lindahl, 1975), its activity quickly became associated with tumor progression and its expression has since then been found to be upregulated in essentially all tumors examined (Vlodavsky et al., 1983, Nakajima et al., 1984, Vlodavsky et al., 2008).
18
Figure 3. Heparanase biology. A) The activation process of heparanase involves removal of a signal peptide (sp) and excision of a linker segment present on the inactive pre-proenzyme. B) Crystal structure of the active heterodimer form of hepa-ranase (PDB entry: 5E8M, 5E9B, Wu et al., 2015). Subunit 8 kDa (orange) and 50 kDa (green) polypeptides, TIM-Barrel, C-terminus domain (C-domain) active site (red) and 9-mer bound HS ligand, are indicated. C) Cleavage site of heparanase between GlcA and GlcN on HS/heparin (red arrow). Main determinants for recogni-tion and binding stability, including N-sulfation and 6-O-sulfation, two and one monosaccharides away from the cleavage site, towards the non-reducing end and the reducing end, respectively, are indicated.

19
In contrast, healthy tissue, including connective tissue cells and most normal epithelia, express very low or no heparanase mRNA, with the excep-tion of the placenta, keratinocytes, platelets and activated cells of the im-mune system (Parish et al., 2001, Vlodavsky et al., 2001). The high probabil-ity of pathological development as a consequence of increased heparanase activity implies that there should exist a strict regulation of its activity in our bodies and this has indeed turned out to be the case. Heparanase regulation exists (I) on the post-transcriptional level, through cathepsin-L-dependent activation (Abboud-Jarrous et al., 2008), (II) on the gene level by early growth response transcription factors, p53, inflammatory cytokines and hy-poxia (Lerner et al., 2011, Chen et al., 2004, de Mestre et al., 2005, Baraz et al., 2006, Sandwall et al., 2009) and (III) by means of regulating the secre-tion of the lysosomally stored activated form of heparanase by physiological concentrations of ADP and ATP (Goldshmidt et al., 2002, Shafat et al., 2006).
Transgenic mice that either overexpress the human heparanase gene (Hpa-tg)(Zcharia et al., 2004) or lack the expression of the endogenous gene (Hpa-KO)(Zcharia et al., 2009) have been established. Both mouse models have mild phenotypes, as they appear normal, are fertile and have normal life spans. However, they have been proven to be excellent tools for studying HS and heparanase in numerous physiological and pathological processes (Vlodavsky et al., 2013). The HS chains from newborn and adult Hpa-tg mouse tissues are considerably shorter than those in wildtype mice. Physio-logical studies of Hpa-tg mice revealed lower food consumption and body weight, increased rate of hair growth, increased levels of urinary protein, excess branching and widening of ducts in mammary glands linked to in-creased neovascularization and disrupted basal membranes (Zcharia et al., 2004). Studies of the Hpa-KO mouse lacked an obvious phenotype, even though tissues contained longer HS chains and were completely deficient of heparanase (Zcharia et al., 2009). This phenomenon was ascribed to an up-regulated expression of MMPs, being able to take over some of the tasks normally performed by heparanase, such as ECM modulation (Wang et al., 2012).
2.4 Enzymatic properties The substrate specificity of the endo-β-D-glucuronidase activity of hepara-nase was elucidated in a step-wise manner. Dagmar Pikas and her colleagues utilized heparanase, isolated from human hepatoma and platelets, together with heparin, HS and capsular polysaccharides generated from Escherichia coli, K5, modified in varying ways to mimic the different modification steps in endogenous HS biosynthesis (Pikas et al., 1998). An 8-mer heparin, with only one cleavage site, was found to be cleavable. However, the unmodified K5 polysaccharide, with the same backbone as unmodified HS/heparin, was

20
non-degradable. Furthermore, chemical modification of the K5 backbone by N-deacetylation/N-sulfation or partial enzymatic C-5 epimerization of GlcA to IdoA, did not facilitate degradation. Instead, sole addition of O-sulfation on the K5 backbone enabled degradation. Subsequent O-desulfation analysis revealed a critical 2-O-sulfation on a hexuronic acid, two monosaccharides away from the cleavage site towards the reducing end.
In a later study a more complex view of the substrate specificity emerged, when purified recombinant heparanase and a library of diversely sulfated tetra- and hexasaccharides, isolated from porcine intestinal heparin and bo-vine kidney HS, were used (Okada et al., 2002). This study revealed that the minimum size of the backbone for heparanase recognition is a trisaccharide and that certain combinations of sulfation patterns around the cleavage site are important. However, this study could not identify any sulfated sequence specifically recognized by heparanase.
Peterson and Liu performed two studies where purified recombinant hep-aranase and enzymatically synthesized polysaccharides and structurally de-fined oligosaccharide substrates was used (Peterson and Liu, 2010, Peterson and Liu, 2012). The studies suggested plasticity in heparanase substrate recognition without any specific sulfation sequence specificity, but instead a requirement for sulfation with a bias towards various sulfated patterns at and nearby the cleavage site. Concerning the catalytic mode of action, it was proposed that two different mechanisms for substrate processing exists: con-secutive and gapped cleavage, with different distance between sites of cleav-age (Peterson and Liu, 2012). A possible ability of heparanase to bind re-leased fragments allosterically was also suggested.
Recently, crystal structures of heparanase, with or without bound specific HS-like substrates, were analyzed (Wu et al., 2015). The structure analysis was in agreement with earlier findings, recognizing the need for sulfated areas of HS/heparin substrate to enable interaction with heparanase. Fur-thermore, the recognized part of HS in the binding cleft was confirmed to be a trisaccharide. Main determinants for recognition and binding stability was suggested to be N-sulfation and 6-O-sulfation, two and one monosaccharides away from the cleavage site, towards the non-reducing end and the reducing end, respectively.
2.5 Heparanase 2 A homologous gene of heparanase has also been found, denoted heparanase 2. It has three splice variants and shares approx. 40% sequence identity with heparanase (McKenzie et al., 2000). Although it does not possess endo-β-D-glucuronidase enzymatic activity, it has been shown to be widely expressed in healthy tissue and to be able to bind strongly to HS/heparin and hepara-nase, suggesting a significant role as an inhibitor of heparanase activity (Levy-Adam et al., 2010). The inhibition is further supported by a study

21
where increased protein expression of heparanase 2 was correlated with a favorable outcome of gastric cancer (Zhang et al., 2013). Additionally, a recent in vivo experiment showed that overexpression of heparanase 2 in head and neck cancer cells reduced tumor growth and tumor vascularization in immunodeficient NOD/SCID mice (Gross-Cohen et al., 2016). This effect was shown to be HS-independent and unrelated to heparanase activity.
3. HS & heparanase in inflammation 3.1 Inflammation The healthy adult human body is in a functional state of tissue homeostasis. When the homeostasis is disturbed, the body swiftly responds with a com-plex orchestration of potent countermeasures to restore it. A central part in the course of restoration is an immunovascular response entitled inflamma-tion. Inflammation is the major response of the non-specific innate part of the immune system. The potentially harmful causative stimuli of acute in-flammatory responses can be of physical, chemical, biological and psycho-logical nature, e.g. blunt trauma, heat, cold or infectious organisms (Serhan et al., 2010). In the early stage of an acute inflammatory response, immune cells (leukocytes), mainly neutrophils, are recruited to the site of injury (so called “homing”). This process begins with the activation of the local vascu-lar endothelial cells that initiate expression of different types of adhesion molecules and chemoattractants on their cell surface (Serhan et al., 2010). This allows leukocytes in the bloodstream, expressing P-selectin glycopro-tein ligand (PSGL)-1 and L-selectin on their cell surfaces, to interact transi-ently with endothelial P-selectin, HSPGs and glycosylation-dependent cell adhesion molecule (GlyCAM)-1. The transient interactions cause the leuko-cytes to slow down and roll along the vessel wall (Gotte, 2003, Ley et al., 2007). Chemokines (e.g. macrophage inflammatory protein (MIP)-2 and interleukin (IL)-8) bound to HSPGs presented on the endothelial cell surface then activate leukocyte cell surface integrins (e.g. lymphocyte function-associated antigen (LFA)-1 and macrophage-1 antigen (Mac-1)) that interact strongly with endothelial immunoglobulin family of adhesion proteins (e.g. intercellular adhesion molecule (ICAM)-1 and vascular cell adhesion mole-cule (VCAM)-1), resulting in leukocyte arrest (Gotte, 2003). Following ar-rest, leukocytes are guided along the endothelial surface in a crawling mo-tion, via integrin interaction and chemotaxis. At a location with high amount of presented chemokines, the leukocytes transmigrate into the inflamed sub-endothelial tissue within minutes (Ley et al., 2007, Phillipson et al., 2006, Massena et al., 2010, Hepper et al., 2012, Heit et al., 2008). Other early in-flammatory events include vasodilation (“widening”) and increased permea-bility of the blood vessel, to enable a flow of fluid and plasma protein into

22
the site of injury, causing swelling (edema) (Serhan et al., 2010). Plasma proteins with important functions at the site of inflammation include (I) in-nate immunity complement proteins that combat pathogens and toxins in various ways, and (II) opsonizing antibodies and acute-phase reactants, also with diverse functions, that facilitate an efficient immune response (Abbas et al., 2012).
The entire process of inflammation is orchestrated through a diverse col-lection of inflammatory mediators, including cytokines, chemokines, com-plement proteins, acute phase reactants and resident cell-derived small-molecule mediators (Serhan et al., 2010). Secretion of cytokines by tissue cells is an early response to infection or tissue damage, and a critical event to activate an acute inflammatory response (Abbas et al., 2012). Tumor necro-sis factor (TNF), IL-1 and IL-6 are the most central pro-inflammatory cyto-kines of the innate immune system since they are involved in initiation of many essential steps of the inflammatory response. These steps include the induction of (I) adhesion molecules and chemokines instrumental in leuko-cyte-endothelial interaction during extravasation, (II) fever, (III) liver syn-thesis of acute-phase proteins, (IV) glycogen/fat catabolism and (V) apopto-sis as well as (VI) pro-inflammatory activation of numerous cell types that consequently express and secrete various inflammatory mediators on their own, thus creating a cascade of effector molecules and an easily accessible environment of apoptosis, phagocytosis and tissue generation. TNF, IL-1 and IL-6 are mainly supplied by mast cells and tissue macrophages, but also by local cells such as endothelial and epithelial cells (Abbas et al., 2012).
Following the successful destruction of harmful stimuli, and regeneration of the damaged tissue, the inflammatory reaction is resolved, usually within days. Edema fluid gets transported back to the blood via draining lymphat-ics, together with a part of the leukocytes. Other leukocytes are cleared by apoptosis and subsequent phagocytosis by local macrophages (Serhan et al., 2010).
In some cases, the acute inflammation is not properly resolved and enters a chronic state of inflammation due to incomplete elimination of an infec-tion, prolonged tissue injury or inappropriate, persisting activation of the immune system. During chronic inflammation, the dominating leukocyte recruited to the site of inflammation is changed from neutrophils to mono-cytes, activated to mature into macrophages. These macrophages commonly work together with lymphocytes of the secondary antigen-specific adaptive immune system to create continuous cascades of pro-inflammatory media-tors, so called flares, leading to tissue remodeling through angiogenesis and fibrosis (Abbas et al., 2012).

23
3.2 HS in inflammatory reactions HS is involved in numerous inflammatory events. Interactions between HS and toll-like receptor 4 (TLR-4) have been shown to trigger an inflammatory response (Johnson et al., 2002, Brunn et al., 2005, Akbarshahi et al., 2011, O'Callaghan et al., 2015). As mentioned previously, HS plays an essential role in leukocyte-endothelial cell interaction and the resulting extravasation from the blood into the site of injury, both as an adhesion molecule, interact-ing with leukocyte L-selectin, and through presentation of the large group of chemokines guiding the leukocytes to the site of transmigration (Gotte, 2003, Ley et al., 2007)(Figure 3A). Among inflammatory mediators, HS does not only bind chemokines, but also non-chemotactic cytokines. Bio-chemical in vitro studies have shown interactions between HS and MIP-2 (Massena et al., 2010), MIP-1α (Stringer et al., 2003) and “regulated on acti-vation, normal T cell expressed and secreted” (RANTES) (Vives et al., 2002) as well as the cytokines IL-2 (Najjam et al., 1998), IL-8 (Spillmann et al., 1998), and IL-10 (Salek-Ardakani et al., 2000). HS also stores these in-flammatory mediators in the ECM, making them readily available for an immediate inflammatory response (Bishop et al., 2007, Bernfield et al., 1999).
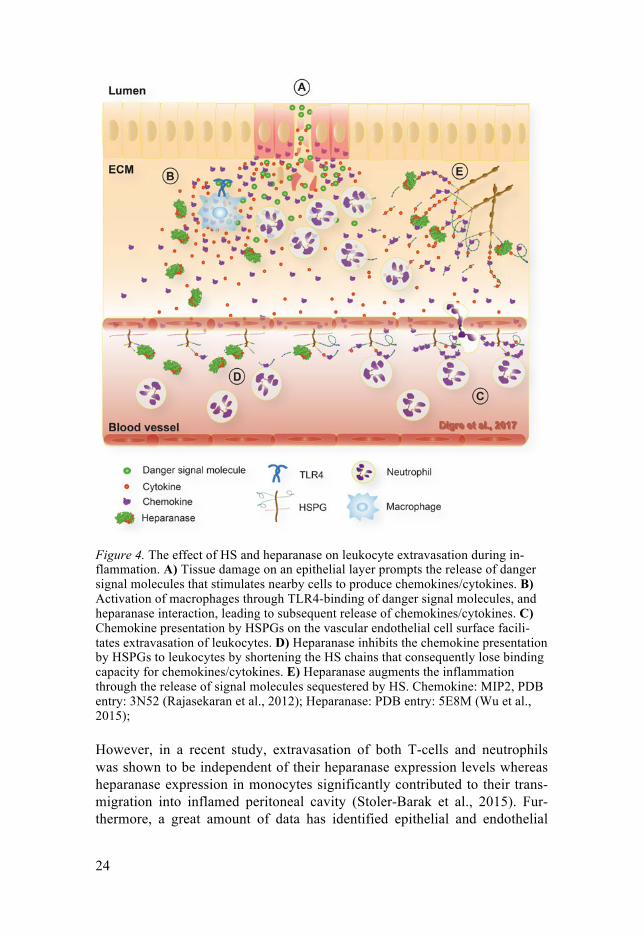
3.3 Heparanase in inflammatory reactions As an HS-fragmenting/modifying enzyme, heparanase is believed to be in-volved in all the actions of HS. This implies a diverse role of heparanase in various stages of inflammation, including the initiation process, homing and extravasation of leukocytes, regulation of the local inflammatory response at the site of injury and final resolution of inflammation. Heparanase activity was early discovered in neutrophils and activated T-cells, hypothesized to contribute to extravasation and migration to the site of injury in inflammato-ry settings (Naparstek et al., 1984, Matzner et al., 1985, Fridman et al., 1987, Lider et al., 1989, Vlodavsky et al., 1992).
24
Figure 4. The effect of HS and heparanase on leukocyte extravasation during in-flammation. A) Tissue damage on an epithelial layer prompts the release of danger signal molecules that stimulates nearby cells to produce chemokines/cytokines. B) Activation of macrophages through TLR4-binding of danger signal molecules, and heparanase interaction, leading to subsequent release of chemokines/cytokines. C) Chemokine presentation by HSPGs on the vascular endothelial cell surface facili-tates extravasation of leukocytes. D) Heparanase inhibits the chemokine presentation by HSPGs to leukocytes by shortening the HS chains that consequently lose binding capacity for chemokines/cytokines. E) Heparanase augments the inflammation through the release of signal molecules sequestered by HS. Chemokine: MIP2, PDB entry: 3N52 (Rajasekaran et al., 2012); Heparanase: PDB entry: 5E8M (Wu et al., 2015);
However, in a recent study, extravasation of both T-cells and neutrophils was shown to be independent of their heparanase expression levels whereas heparanase expression in monocytes significantly contributed to their trans-migration into inflamed peritoneal cavity (Stoler-Barak et al., 2015). Fur-thermore, a great amount of data has identified epithelial and endothelial

25
cells as the main source of heparanase during inflammation, and the expres-sion of heparanase in these cells is induced in response to inflammatory cy-tokines (Chen et al., 2004, Edovitsky et al., 2006, Lerner et al., 2011, Schmidt et al., 2012, Goldberg et al., 2013). An HS-containing glycocalyx layer blocks leukocyte binding sites on vascular endothelial cells during non-inflammatory conditions (Constantinescu et al., 2003, Weinbaum et al., 2007). In a model of acute inflammatory lung injury, the activity of hepara-nase was increased and resulted in degradation of the pulmonary endothelial glycocalyx, leading to exposure of endothelial surface adhesion molecules and increased neutrophil recruitment (Schmidt et al., 2012). This phenome-non was attenuated in Hpa-KO mice, suggesting a pro-inflammatory role of heparanase in leukocyte homing to the site of injury. However, heparanase overexpression was shown to attenuate chemokine-guided leukocyte crawl-ing along the endothelial surface due to the reduced ability of truncated en-dothelial surface HS to bind and present chemokines, thereby limiting the inflammatory response (Massena et al., 2010)(Figure 3C). In line with this, leukocyte recruitment and activation were attenuated in models of inflamma-tory hyperalgesia and neuroinflammation in Hpa-tg mice (Li et al., 2012, Zhang et al., 2012). The anti-inflammatory effect of heparanase in those studies is proposed to be an effect of HS fragments, constantly present in Hpa-tg mice, acting as inhibitors of HS/chemokine-interactions (Young, 2008, Goldberg et al., 2013).
Several lines of evidence associate heparanase both with the induction and attenuation of inflammation due to its modulating function of the inter-play between HS and TLR-4 on leukocytes (mainly resident tissue macro-phages), and by regulating the mast cell storage of inflammatory mediators in secretory granules (Brunn et al., 2005, Lerner et al., 2011, Wang et al., 2011, Blich et al., 2013, Brennan et al., 2012, Goodall et al., 2014, O'Callaghan et al., 2015). In a model of chronic inflammatory ulcerative colitis, heparanase was further suggested to drive the mechanism behind the disorder in a viscous self-sustaining cycle (Lerner et al., 2011). In the sug-gested model, macrophages produce TNFα and cathepsin-L that leads to induction and activation of heparanase from the colon epithelial cells. This, in turn, facilitates further stimulation of macrophages because of their in-creased exposure to the intestinal microbial flora. In more recent studies, heparanase was found to activate and induce cytokine expression in macro-phages. Whether the activation is dependent on its enzymatic modulation of HS or not is unclear (Goodall et al., 2014, Gutter-Kapon et al., 2016). On the other hand, microglial response to LPS via TLR-4 and CD14 was disturbed in Hpa-tg mice, suggesting an agonist role of intact HSPGs in TLR-4 stimu-lation of an innate immune response (O'Callaghan et al., 2015).
Collectively, the role of heparanase in inflammatory settings is multifac-eted, as it is involved in both pro- and anti-inflammatory mechanisms. The most obvious explanation of this phenomenon is the broad functional range

26
of its substrate HS. Nevertheless, more studies are needed to unravel the action of heparanase in various physiological and pathological scenarios.
3.4 Murine models of inflammation The induction step of inflammation includes detection of pathogen- and damage-associated molecular patterns (PAMPs and DAMPs) by pattern recognition receptors (PRRs) (Serhan et al., 2010). These are evolutionary conserved germline-encoded receptors that recognize (I) molecular struc-tures that are characteristic of microbial pathogens but not mammalian cells (e.g. bacterial cell wall structures, fungal mannans and viral double-stranded RNA), as well as (II) endogenous molecules produced or released from damaged and dying cells (e.g. heat shock proteins and nuclear proteins) (Abbas et al., 2012). Such ligand molecules are utilized to induce and study inflammation and inflammatory-associated diseases in animal models. Com-plete Freund׳s adjuvant (CFA), lipopolysaccharide (LPS) and silver nitrate (AgNO3) were utilized in my studies.
CFA consists of a non-metabolizable oil mixture of paraffin oil and mannide monooleate along with heat-killed Mycobacterium tuberculosis. It is used extensively to potentiate the immune response towards antigens, which are mixed together with the CFA as a water-in-oil emulsion and sub-sequently injected into animals (Freund et al., 1937, Petrovsky and Aguilar, 2004). Since the bacterial component of CFA contains many PAMPs, nu-merous PRRs are stimulated, including TLR2, -4 and -9, C-type lectin recep-tor (CLR) Mincle, and NOD-like receptor (NLR) 2 (Coffman et al., 2010, Kleinnijenhuis et al., 2011). CFA stimulation creates a strong TH1/TH17 type adaptive immune response with production of cytokines, such as TNFα, IL-1, IL-6, IL-17 and interferon (IFN)-γ, as well as chemokines and adhesion molecules (Serhan et al., 2010).
LPS is a structurally heterogeneous cell wall component of Gram-negative bacteria (O'Neill et al., 2013). The lipid moiety of LPS, Lipid A, is responsible for its inflammation-stimulating activity (Westphal and Luderitz, 1954). The discovery of the LPS-receptor TLR4 is historically considered to have paved the way for our current understanding of the innate immune sys-tem, and its importance was recognized with the Nobel Prize in 2011 (O'Neill et al., 2013). LPS stimulation of TLR4 on leukocytes leads to a strong TH1/TH17 type inflammatory response with IL-1/TNFα/IFN-γ produc-tion through the nuclear factor (NF) –κB pathway (Abbas et al., 2012, Park et al., 2015).
Silver nitrate is an oxidative agent that can induce a sterile inflammation in mice caused by tissue damage and subsequent DAMP activation. Among other purposes, it is utilized for its ability to induce production of acute phase proteins, such as serum amyloid A (SAA) (Ishihara, 1973, Skinner et al., 1977). Experimental models with subcutaneous injections into mice dur-

27
ing 2-3 weeks or a single injection together with amyloid enhancing factor (AEF) are utilized for development of amyloid protein A (AA) amyloidosis (Axelrad et al., 1982).
4. HS & heparanase in rheumatoid arthritis (RA) 4.1 Rheumatoid arthritis Self-tolerance is the fundamental ability of the immune system not to re-spond to endogenous antigens. An ability acquired through previous expo-sure to these antigens. When self-tolerance fails, the immune system initiates immunologic responses towards self-antigens, a condition termed autoim-munity (Abbas et al., 2012). Rheumatoid arthritis (RA) is an autoimmune disorder causing chronic inflammation in the synovium (synovitis) of small and large joints of the extremities, such as fingers, elbows, shoulders, knees, ankles and toes (Abbas et al., 2012). It is a disease with systemic manifesta-tion of inflammation, such as pulmonary fibrosis and vasculitis, and it often co-occurs with other diseases, including atherosclerosis, amyloidosis and ischemic heart disease (NCCCC, 2009). Arthritis, meaning joint inflamma-tion, generates formation of synovial fibrous tissue, called pannus, and de-struction of the articular framework, including cartilage and bone tissues. In the most severe scenario, the joint ends become fused, known as ankylosis (Abbas et al., 2012). The disease is found in around 1% of the adult popula-tion worldwide and has been traced back thousands of years to Native Amer-ican tribes in North America (Rothschild et al., 1988, Firestein, 2003). Inter-estingly, evidence of the presence of RA in Europe first emerged in the early 17th century, suggesting a late emergence of a causative factor (Firestein, 2003).
The pathology of RA involves various cell types of the innate and adap-tive immune system, as well as local cells of the joint tissue. T-cells, includ-ing pro-inflammatory CD4+ TH1 and TH17 cells, macrophages, activated B cells, antibody-producing plasma cells and neutrophils are found in the in-flamed synovium of RA patients (Abbas et al., 2012). These cells work to-gether with the local cells in orchestrating inflammatory-mediated tissue injury through an array of cytokines, including TNFα, IL-1, IL-6, IL-8, IL-17 and interferon (IFN)-γ, as well as proteolytic enzymes such as MMPs, serine proteases and aggrecanases (Firestein, 2003, Abbas et al., 2012). Ac-tivation and resulting hyperplasia of the lining fibroblast-like cells of the local synovial membrane, leads to their invasion into the synovial space and pannus-formation on the cartilage surfaces where they start do degrade the cartilage layer, and later on the underlying bone tissue (Firestein, 2003). The process of pannus formation has been compared to the formation of a tumor, due to the characteristics of the pannus-forming hyper-proliferating and de-

28
differentiating synoviocytes, and the tissue environment within the pannus with activated angiogenesis, oxidative stress and hypoxia (Firestein, 2003).
In addition to the cell-mediated mechanisms, there is also a humoral im-mune response involved in the pathology of RA. Autoantibodies, able to bind to Fc regions of other antibodies, are frequently found in the circulation of RA patients and are used in the diagnosis of the disease. They are called Rheuma-toid factors (RFs) and form antibody-antibody immune complexes that have been proposed to be key elements in autoimmunity (Abbas et al., 2012). The enzymatic conversion of arginine to citrulline on proteins is a common modi-fication in inflammatory environments but the presence of antibodies against it is suggested to contribute to the development of RA. Autoantibody, recogniz-ing the citrullinated peptides (anti-citrullinated protein antibodies; ACPAs), has been shown to be present in more than 70% of RA patients, and has a higher specificity for RA than RF, even though both are present in numerous inflammatory conditions (Nishimura et al., 2007). In addition to the articular destruction, there are also non-articular systemic consequences of RA, includ-ing vasculitis and lung injury (Abbas et al., 2012).
The underlying causes of RA are not fully understood but include a com-bination of genetic and environmental factors that generate a tissue environ-ment in which self-tolerance is overcome. Among the genetic factors, many alleles of the HLA-DRB1 gene, coding for major histocompatibility complex (MHC) component HLA-DR4, involved in antigen presentation, are highly associated with the disease (Nepom et al., 1989). These alleles all share a five amino acid sequence motif in HLA-DR4, known as the “shared epitope” (Gregersen et al., 1987). Interestingly, HLA-DR4 and synthesized peptides containing the “shared epitope” has been shown to interact with a citrullinat-ed protein, and consequently activate an innate immune response (Ling et al., 2013). Among environmental factors, tobacco and infections have the highest association with RA (Abbas et al., 2012). Together with the finding that tobacco can be traced back to early Native American tribe members in North America, which have been found to present skeletal RA symptoms, makes it a very strong candidate for studies regarding RA induction.
The goal of current treatments of RA is to limit the disease progression or even achieve full remission of the condition. These treatments include dis-ease-modifying anti-rheumatic drugs (DMARDs) such as methotrexate and biologic agents, such as the cytokine inhibitor anti-TNF. There are also symptom-managing treatments including non-steroidal anti-inflammatory drugs (NSAIDs) and corticosteroids (Burch and Onysko, 2012, Schuna, 2011). These are broad anti-immune and anti-inflammatory drugs, having a systemic immunosuppressive effect, resulting in increased susceptibility to infections. Therefore, future treatments that are currently explored focus on antigen-specific therapies that can restore self-tolerance, mainly through the utilization of modified immunosuppressive regulatory T cells (Thomas, 2013).

29
4.2 Heparanase & RA Inflammatory-mediated leukocyte recruitment and ECM remodeling as well as immune response activation and angiogenesis are common processes in RA pathogenesis in which heparanase has been implicated (Vlodavsky et al., 2013). The involvement of heparanase in inflammatory processes and as a regulator of innate immune activation was discussed earlier (see chapter 3.3 “Heparanase in inflammation”). Angiogenesis is essential to the formation of “tumor-like” pannus during the development of RA by facilitating synovio-cyte and immune cell proliferation (Matsuno et al., 2002, Roccaro et al., 2005, Wang et al., 2005). Heparanase has many proangiogenic properties related to its ability to cleave HS in the ECM. This ECM remodeling facili-tates cell migration and proliferation of budding vascular endothelial cells along with the release of various proangiogenic mediators such as fibroblast growth factor (FGF), ECM degrading MMPs, proinflammatory cytokines, cell attracting chemokines and platelet factor 4 (PF-4)(Karateev, 2003). Tak-ing all of these potential RA-functions into account, heparanase expression is likely to be found in the joint of RA patients. Indeed, a study showed a more than 100-fold increase of heparanase activity in synovial fluid and tissue from the knee joint of patients with RA as compared to patients with osteoar-thritis and patients without arthritis. This was accompanied by upregulated heparanase mRNA expression in synovial tissue (Li et al., 2008).
4.3 Collagen-induced arthritis (CIA)
Figure 5. Collagen-induced arthritis C57Bl/6 mouse with severe swelling of hind and front paws.
Several inducible models have been developed to mimic the pathology of RA. These involve procedures of immunization with (I) joint related proteins (e.g. collagens, proteoglycans, glucose-6-phosphate isomerase), (II) antigen-specific mediators (antibodies or T-cells) and (III) adjuvant (e.g. CFA, pris-tine or mannan). The available models vary in their ability to mimic the pa-thology and their coverage of different stages of RA (Holmdahl, 2015).
Collagen-induced arthritis (CIA) is a well-established model of RA, used in both mice and rats (Holmdahl et al., 1989). The induction involves subcu-taneous injections of collagen type II (CII) emulsified in complete (and in-

30
complete) Freund׳s adjuvant (CFA) (Holmdahl et al., 2002). The CII injected into mice is of chicken, bovine or porcine origin, where C57Bl/6 and DBA/1 mice is particularly susceptible to chicken and bovine CII, respectively (Wooley et al., 1985). CFA with different amounts of heat-killed Mycobac-terium tuberculosis is mixed with the antigen as a water-in-oil emulsion and subsequently injected into an animal model (Freund et al., 1937, Petrovsky and Aguilar, 2004). Incomplete Freund׳s adjuvant (IFA), lacking the bacteri-al components, is used together with CII to induce CIA in rats and to boost the immune response in mice about 3 weeks post CFA-induction (McNamee et al., 2015). The most susceptible mouse strain for CIA is the DBA/1 mouse, because of its expression of the MHC type-II molecule haplotype H-2q, which is important for antigen recognition (Wooley et al., 1985). Howev-er, DBA/1 mice do not develop a chronic type of arthritis and since genetic models of mice most commonly are on the C57Bl/6 background strain, gen-erating DBA/1 mice with a genetic construct normally requires years of tedi-ous back-crossing work (Inglis et al., 2007). The C57Bl/6 mice carry the H-2b haplotype and were previously believed to be resistant to CIA. However, the development of alternative induction protocols with chicken CII and an increased amount of M. tuberculosis has enabled arthritis induction also in these mice, with around 50-75% of the mice developing visible arthritis (Inglis et al., 2008, Campbell et al., 2000). In contrast to DBA/1 mice, C57Bl/6 mice develop a chronic type of arthritis, which is similar to RA in humans (Inglis et al., 2007). The paw swelling is milder than in DBA/1 mice, but with a gradual increase of severity. However, the arthritogenic mechanisms driving the C57Bl/6 induction are not clear: A humoral re-sponse towards CII with anti-CII antibodies is present, but there have been conflicting results concerning the ability of T-cells to cross-react with CII (Inglis et al., 2007, Backlund et al., 2013). Recently, a rapid back-cross into an H-2q haplotype has been made possible by Holmdahl and his colleagues, with the use of C57Bl/6N.Q mice (Backlund et al., 2013). Since these mice express the H-2q haplotype on a C57Bl/6 background, only three generations of crossings with genetically modified C57Bl/6 mice are needed to produce homozygotic expression of the H-2q haplotype. Thus, providing confirmed CII-specific T-cell responsiveness in the CIA mouse model together with the genetic alteration of interest.
Additional to the adaptive response, CIA is also accompanied by a di-verse innate response, mainly brought on by the bacterial components of CFA (Billiau and Matthys, 2011). It has been proposed that CFA has a main role in the development of arthritis in the CIA model, where the initial stage of the model is characterized by an innate immune response towards CFA, which in turn facilitates the following adaptive auto-immune response to-wards articular auto-antigens (Billiau and Matthys, 2011). This assumption is based on the observed development of arthritis in rats and mice injected with CFA only, suggesting a limited importance of injected auto-antigens in

31
the CIA model (Trentham et al., 1977, Geboes et al., 2007). Instead, system-ic inflammatory induction as well as previously observed enhanced expan-sion of myeloid leukocyte precursors (myelopoiesis), is suggested to be of critical importance for subsequent leukocyte infiltration, inflammation and destruction of the joint (Matthys et al., 1999, Billiau and Matthys, 2011).
5. HS & heparanase in amyloidosis 5.1 Amyloidosis To date, 31 human amyloid fibril proteins (AFPs) are known to have the pathological ability to misfold and aggregate into large insoluble fibrillar structures denoted amyloid (Sipe et al., 2014, Harrison et al., 2007). The amyloid fibrils are deposited in specific tissues and accumulate over time. The accumulation damages the surrounding cell structures and cause in-flammation and extensive cell death that compromises organ functions. This condition is known as amyloidosis and can potentially lead to life threaten-ing failure of an entire organ (Sipe et al., 2014). Amyloid formation is a re-sult of native soluble AFPs becoming misfolded. As all other proteins, the polypeptide chains of AFPs are initially synthesized and folded into their native, biologically functional, soluble form in the ER. However, a small fraction of the soluble AFPs will at a later time point misfold and aggregate into highly-ordered β-pleated sheet structures, thus forming rigid, insoluble, linear fibrils of about 10 nm width (Sipe et al., 2014). This can occur locally or systemically in multiple organs, depending on whether the amyloid pro-tein is expressed nearby the deposition site or whether it is a plasma protein. All amyloid fibrils bind the dye Congo red and display a green, yellow or orange birefringence upon polarization microscopy (Sipe et al., 2014).
HS has been extensively associated with amyloid deposits. In fact, HS in-teracts directly with AFPs and also accelerates the misfolding of AFPs into amyloid fibrils (Zhang and Li, 2010). However, the exact function of HS in the development of different amyloidoses is still unclear.
5.2 Serum amyloid A & AA amyloidosis Serum amyloid A (SAA) is an acute phase apolipoprotein that is associated with the cholesterol-transporting high-density lipoprotein (HDL) particles in blood (Benditt et al., 1982, Gabay and Kushner, 1999). The importance of its physiological function is strongly supported by the fact that SAA has been extensively conserved throughout more than 500 million years of evolution (Santiago et al., 2000, Santiago-Cardona et al., 2003). The physiological function of SAA is related to its substantial acute phase response during inflammation. Here, the plasma concentration of SAA is upregulated by a

32
1000-fold (0.001-1mg/ml) via liver synthesis. Hence, making SAA the most abundant apolipoprotein in HDL particles (Gabay and Kushner, 1999).
The precise role of SAA during inflammation is still unknown, but sug-gestions include regulation of the immune response as well as recycling of cholesterol generated at sites of injury (Kisilevsky and Manley, 2012). The reverse cholesterol transport (RCT) from the periphery to the liver by HDL during non-inflammatory conditions is a well-studied process associated with inhibition of atherosclerotic plaque formation (Goldbourt et al., 1997). HDL targets macrophages for obtaining cholesterol, both during normal (vascular macrophages) and inflammatory (macrophages at the site of in-flammation) conditions, and this interaction can be facilitated by HS (Kinkley et al., 2006, Tam et al., 2008, Kisilevsky and Manley, 2012).
A vast range of alterations in lipid metabolism occurs during inflamma-tion, including (I) redirection of lipoproteins to the site of injury (Kisilevsky and Subrahmanyan, 1992), (II) increased delivery of lipids to host-defending leukocytes (Hardardottir et al., 1994), (III) binding-inhibition of viruses and toxic microbial products such as LPS (Sernatinger et al., 1988, Pajkrt et al., 1996), and (IV) lysis of parasites (Hajduk et al., 1989). These are efficient countermeasures taken to ensure the survival of the organism, but they are also disturbances of the normal lipid metabolism, one of them being reduced RCT (Esteve et al., 2005). If the inflammation is not resolved and becomes chronic, the beneficial properties of the lipid acute phase response becomes harmful, increasing the risk for cardiovascular diseases caused by the ham-pered RCT (Wallberg-Jonsson et al., 1996), increased lipid accumulation inside macrophages resulting in foam cell formation along vascular walls, and subsequent development of atherosclerosis (Esteve et al., 2005).
Another lipid-associated consequence of chronic inflammation is the devel-opment of Amyloid A (AA) amyloidosis caused by the aggregation of SAA fragments into amyloid fibrils (Cunnane, 2001, Merlini and Westermark, 2004). This amyloid is normally formed in kidney, spleen and liver, leading to proteinuria, subsequent progressive renal dysfunction and finally degeneration of the kidney (Lachmann and Hawkins, 2006, Lachmann et al., 2007). The cause of SAA amyloid formation is unknown and even though the disease is associated with high levels of plasma SAA it is still only a fraction of the pa-tients with chronic inflammation that develops the disease. This suggests that additional factors are important for facilitating the formation and accumulation of AA. HS has been found to be extensively associated with AA amyloid de-posits both via direct structural interaction and by observed local upregulation of HSPGs around amyloid deposits (Snow and Kisilevsky, 1985, Snow et al., 1987a). Furthermore, HS can dissociate SAA from the HDL particle, depend-ent on a minimal HS chain length of 12-14 monosaccharides units (Noborn et al., 2012). The interaction between HS and SAA has been found to be pH-sensitive, occurring only at mildly acidic conditions, around pH 5 (Elimova et al., 2009, Noborn et al., 2012).

33
Taken together, these studies suggests HS as an essential factor in the de-velopment of AA amyloidosis as well as a potential receptor for HDL-SAA during inflammatory-associated cholesterol transport.
5.3 Alzheimer’s disease (AD) Age-related diseases have increased in frequency as a result of the increased human life expectancy. Among these, dementia is the biggest threat to a contended life in old age and an economical burden on society (Suzman and Beard, 2011). The most common form of dementia is Alzheimer´s disease (AD), a progressive neurodegenerative type of disease of the central nervous system that leads to impaired cognitive function of the memory, orientation and rational thinking, hence accompanied with drastic changes in behaviour (van Horssen et al., 2003, Skaper, 2012). The underlying pathology of AD includes (I) extracellular amyloid senile plaque deposition, (II) neurofibrilla-ry tangles (NFTs) of hyperphosphorylated microtubule associated protein Tau (MAPτ) in nerve cell soma and dendrites, and (III) cerebral amyloid angioapathy (CAA) (Zhang and Li, 2010).
The main component of both senile plaque and CAA is Aβ peptide, gen-erated from the AFP Aβ precursor peptide (AβPP), a large transmembrane glycoprotein. AβPP is processed in two steps to generate Aβ peptide. Initial-ly, the enzyme β-secretase (BACE1) cleaves off a soluble extracellular fragment of AβPP. The remaining membrane-anchored Aβ domain is subse-quently released into the ECM by γ-secretase, mainly as peptides of 40 or 42 amino acid residues (Aβ40, Aβ42)(Skaper, 2012).
MAPτ is normally involved in stabilizing the microtubule structures in-side neurons. However, following abnormal phosphorylation, MAPτ dissoci-ates from the microtubule structure and aggregates into paired helical fila-ments leading to neuronal inactivity and cell death (Johnson and Bailey, 2002).
Neuroinflammation is a common feature of the neurodegenerative disease Alzheimer’s disease (AD)(Akiyama et al., 2000). Because of the disastrous consequences of severe inflammation in the brain, with neuron death and functional derangement, the brain holds several anatomic features to impair initiation of adaptive immunity towards antigens, including: (I) reduced dia-pedesis and delivery of inflammatory mediators via the brain microvascula-ture (the so called blood-brain barrier (BBB)), (II) absence of conventional lymphatic drainage and a low level of antigen-presentation, and (III) a high expression of anti-inflammatory mediators and receptors (Abbas et al., 2012). The local leukocytes of the brain includes microglia and astrocytes that are activated during inflammation and work together with recruited vas-cular monocytes that migrate across the BBB and mature into activated mac-rophages (Nakanishi, 2003, Town et al., 2005, Gate et al., 2010). The acti-vated leukocytes secrete various inflammatory mediators, in the form of

34
cytokines, chemokines and proteolytic enzymes. However, the underlying mechanisms of neuroinflammation and their implications on AD are not fully understood. Accumulated data indicate a crucial role of neuroinflam-mation in AD as a disease-promoting factor (Heneka et al., 2015). Aβ amy-loid deposition causes chronic activation of the innate immune system and impairment of microglial clearance functions. In contrast, there is also evi-dence showing that activation of microglia and infiltration of vascular mon-ocytes contributes to Aβ amyloid clearance through phagocytosis (Zuroff et al., 2017). Additionally, inhibition of anti-inflammatory pathways in mouse models has been shown to induce Aβ amyloid resolution (Guillot-Sestier et al., 2015).
Similar to other amyloid diseases, HS has been shown to be highly asso-ciated with Aβ amyloid deposits in AD, including senile plaques, vasculature deposits and NFTs (Zhang and Li, 2010). The role of HS during AD devel-opment is not clear, but studies have implicated HSPGs in facilitating amy-loid plaque formation (Castillo et al., 1997, Castillo et al., 1998, Cotman et al., 2000), and inhibiting Aβ degradation (Biroc et al., 1993, Gupta-Bansal et al., 1995). HS/HSPG and heparin have also been shown to be involved in the processing of AβPP into Aβ, by affecting the action of BACE1 (Leveugle et al., 1997, Scholefield et al., 2003, Cui et al., 2011). A recent study of our group using human AβPP (with a Swedish mutation) overexpressing mice (tgSwe) that also overexpress heparanase (tgHpa*Swe) supported the role of HS as a promoter of Aβ plaque formation, as the increased heparanase-mediated fragmentation of HS resulted in inhibition of plaque formation compared to the tgSwe mice not overexpressing heparanase (Jendresen et al., 2015).

35
Present investigations
Paper I Aim The aim of the study was to examine the effect of heparanase overexpression in a collagen-induced murine model for RA.
Background The participation of HS in inflammatory processes has been extensively demonstrated, ranging from the initiation process, homing and extravasation of leukocytes, to regulation of the local inflammatory response at the site of injury and final resolution of inflammation (Goldberg et al., 2013, Kumar et al., 2015). Structural alteration of HS by heparanase overexpression is known to affect the interaction of HS with its ligands, thus our heparanase overexpressing (Hpa-tg) mice are excellent tools for studying the various inflammatory-related functions of HS.
When the Hpa-tg mouse was used in an acute inflammatory model by in-jection of bacterial cytotoxic LPS, leukocyte transmigration was shown to be impaired compared to control mice (Massena et al., 2010). A similar out-come was observed when we applied the same model to study neuroinflam-mation in Hpa-tg mice, displaying a decreased recruitment of leukocytes to the site of inflammation (Zhang et al., 2012). In contrast, a chronic inflam-matory ulcerative colitis model and a lung sepsis model demonstrated a pro-inflammatory effect of heparanase linked to the pro-inflammatory mediator TNFα (Lerner et al., 2011, Schmidt et al., 2012). Our data also indicate a pro-inflammatory interactive relationship between TNFα and heparanase overexpressing synovial fibroblasts. As TNFα-mediated inflammatory mechanisms play an essential role in RA pathology, these data are of great interest.
Results and discussion The demonstrated increase of heparanase activity in the synovium of RA patients prompted us to investigate the role of heparanase in RA pathology (Li et al., 2008). We utilized an established inducible model of RA, modified

36
for C57Bl/6 mice, through injections of chicken collagen together with com-plete Freund´s adjuvant (CIA), using our Hpa-tg mice together with wildtype (WT) control mice. C57Bl/6 mice that were previously believed to be re-sistant to CIA, gave an incidence around 50% with the modified CIA proto-col. Clinical data demonstrated no significant difference in incidence be-tween the two groups. However, the severity of observed RA symptoms was higher and seemed to appear earlier in the Hpa-tg mice. The average clinical score was 32 among the 6 Hpa-tg mice that developed symptoms, compared to an average score of 12 in the WT mice. The clinical scores correlated well with the severity of inflammatory processes on the cellular level, where the mouse with highest score displayed the highest degree of cell infiltration and bone destruction in afflicted joints.
Examination of cells from thymus, spleen and lymph nodes revealed in-creased innate and adaptive immune responses of the Hpa-tg mice, reflected by increased proportions of macrophages, antigen presenting cells and plasmacytoid dendritic cells as well as Helios-positive CD4+ and CD8+ T cells. Extensive studies have pointed out T-cells as being central in RA pa-thology. T-cells have been found to comprise 30-50% of the cells present in the synovium of RA patients (Bartok and Firestein, 2010). We observed an essentially equal proliferation of splenic lymphocytes from Hpa-tg and WT mice, with or without T-cell-stimulating ConA. However, splenic lympho-cytes isolated from CIA-induced mice revealed a higher proliferation poten-tial in Hpa-tg mice, indicating a role of T-cells in generating the more severe symptoms observed in Hpa-tg CIA mice as well as supporting a pathological role of T-cells in RA.
In conclusion, our results confirm a disease-contributing role of hepara-nase expression in a murine model of RA providing novel insights into the mechanisms of the human variant of the disease. The heparanase-dependent upregulation of symptoms is, at least to a degree, brought on by an enhanced TNFα and T-cell activity. Consequently, these results indicate the possible therapeutic effect of using heparanase-inhibiting agents to reduce the de-structive inflammatory processes raging in RA.
Paper II Aim The aim of the study was to investigate the mechanism responsible for HS-mediated dissociation of acute phase HDL-SAA particles as well as to iden-tify the apolipoproteins involved.

37
Background HS has been found to play a critical role in the formation of many different forms of amyloid. Experimental models have helped to uncover an extensive association between HS and the systemic amyloidogenic plasma protein serum amyloid A (SAA) (Zhang and Li, 2010). SAA is an acute-phase pro-tein, normally associated with cholesterol transporting high-density lipopro-tein (HDL) particles, that can aggregate into amyloid fibrils that is deposited mainly in kidney, liver and spleen giving rise to the progressive disorder AA amyloidosis (Westermark et al., 2015). The deposition of SAA is partly ex-plained by its dramatic upregulation in response to pro-inflammatory cyto-kines, increasing its concentration in blood by a 1000-fold (Gabay and Kushner, 1999). AA amyloidosis is highly associated with chronic inflam-matory conditions where the concentration of SAA remains high. However, the large majority of people with chronic inflammation does not develop the disease, prompting the question as to what other factors are needed for dis-ease progression.
Early studies revealed an HS/heparin-binding motif in SAA. The interac-tion of SAA and HS/heparin was later found to result in AA aggregation at mildly acidic pH (Elimova et al., 2009). Recent in vitro studies have further investigated the efficiency of HS to facilitate AA fibril formation from SAA1.1 and different regions of the SAA peptide (Egashira et al., 2011, Aguilera et al., 2014). Our recent investigation uncovered a size-dependable ability of HS/heparin to dissociate SAA from the HDL-SAA particle (Noborn et al., 2012). This study theorized that dissociation of HDL-SAA can only be catalyzed by a simultaneous binding between HS/heparin and two other apolipoproteins.
Except for SAA, another possible candidate for being involved in the dis-sociation process is apolipoprotein A1 (ApoA1). ApoA1 is a plasma apolipoprotein composing the major protein part of HDL, which is involved in reverse cholesterol transport from the periphery to the liver. The level of apoA1 in plasma is inversely associated with the occurrence of atherosclero-sis, and this function is impaired in carriers of mutations in apoA1 (Holleboom et al., 2013). Several mutations in the apoA1 gene have been identified, and the corresponding mutant proteins are associated with abnor-mal aggregation of the protein and amyloidosis (Mangione et al., 2001). Recent studies have provided evidence that mutant apoA1 interacts with cell surface HS (Kuwabara et al., 2015).
Results and discussion To find out whether heparin, apart from SAA, also interacts with apoA1, samples of HDL-SAA were incubated with heparin for 0-6 hours followed by a centrifugation step. Separation with SDS-Urea-PAGE followed by

38
Coomassie blue staining revealed that incubation of HDL-SAA in the pres-ence of heparin at a mild acidic condition (pH5.0) resulted in dissociation of both apoA1 and SAA. The dissociation already occurred at the 0 incubation time point, indicating a rapid interaction of the polysaccharide with the apolipoproteins. However, apoA1 and SAA displayed a different time-dependent pattern. Since SAA was completely precipitated after incubation of 1 h, in subsequent experiments, all incubations were carried out for 1 h.
To determine whether heparin was co-precipitated with the apolipopro-teins, the SDS-PAGE gels were stained with Alcian blue (staining of sulfat-ed glycosaminoglycans, e.g. heparin) followed by silver staining to increase the intensity. Indeed, positive staining for heparin was seen in the pellet frac-tions indicating co-precipitation of heparin with the lipoproteins. Due to observed excess of unbound heparin we performed a dose-dependent incuba-tion where both 5 and 50 µg/ml heparin was shown to precipitate both SAA and ApoA1, where a major portion of SAA was lost from the supernatant fraction in 50 µg/ml heparin incubations, but only a minor part of ApoA1. Additionally, a significant portion of SAA was unaccounted for in the 50 µg/ml heparin incubations, indicating formation of insoluble SAA aggre-gates.
Next, we examined the effect of heparin chain length on the dissociation of the apoA1 and SAA from the lipoprotein particles. SDS-PAGE analysis of the incubation products revealed a pattern that displayed an increased dissociation of the apolipoproteins with an increase in heparin chain length. Notably, though the apoA1 dissociation was increased with the increase in heparin chain length, it seems that the oligomers of 12-sugar units had a capacity comparable to native heparin (average 50 sugar units). In contrast, dissociation of SAA was 40% in samples incubated with the 12-mers, and increased further to 60% when incubated with the native heparin.
The simultaneous and almost proportional dissociation of apoA1 and SAA from HDL suggest that the two proteins might be located closely to one another in the lipoprotein particles. This was examined by MS analysis of the digested peptides following chemical crosslinking. Multiple crosslinks between apoA1 and SAA were detected, suggesting that domains in SAA and apoA proteins are located very close to each other (within 25 Å) in the HDL particles and possibly interact as well.
In summary, our data provides experimental evidence for the molecular interaction of apoA1 and SAA with heparin and reveals close proximity of apoA1 and SAA in the HDL-SAA particle. Though both apoA1 and SAA interact with heparin, they also appear to have distinctive individual features in this interaction.

39
Paper III Aim The aim of the study was to investigate how the absence of heparanase af-fected amyloid resolution in a murine model of AA amyloidosis.
Background Extracts of amyloid-containing tissue, denoted amyloid enhancing factor, (AEF), can be used to induce AA amyloidosis in mice (Kisilevsky et al., 1995). An intravenous injection of the AEF in combination with inflammato-ry stimulation, e.g. subcutaneous injection of silver nitrate, prompts a rapid process of amyloid deposition in the spleen and liver within 36-48 hours (Axelrad et al., 1982). The amyloid deposits subsequently gradually regress to a point where they can no longer be detected, through a process of amy-loid clearance (Hawkins and Pepys, 1990). This process was shown to in-volve formation of anti-AA autoantibodies and Fc-mediated phagocytosis (Nystrom and Westermark, 2012).
Accumulated data have shown that the development of AA-amyloidosis is dependent on cellular HS (Snow et al., 1991, Ancsin and Kisilevsky, 2004, Li et al., 2005). Our group previously demonstrated that overexpression of heparanase attenuated the development of AEF-induced AA-amyloidosis(Li et al., 2005), validating the essential role of HS in SAA deposition. The ra-tionale of the phenomenon is that an increased concentration of heparanase leads to production of shorter fragments of HS, which are unable to form complex with and induce aggregation of SAA.
Results and discussion With this in mind, we anticipated that our mice lacking heparanase (Hpa-KO), would facilitate an increased rate and degree of amyloid deposition, as well as hampered amyloid clearance, as a result of non-degraded longer HS chains (Zcharia et al., 2009). To investigate this, Hpa-KO and C57Bl/6 (WT) mice were subjected to the inducible model of AA-amyloidosis, by intrave-nous injection with 100 µg of AEF followed by subcutaneous injection of 0.2 ml 1% silver nitrate. Spleen, kidney and liver were collected for exami-nation of amyloid deposits 7, 10, 21, 42 and 72 days post induction (DPI). Unexpectedly, Congo red staining of the organs collected 7 DPI displayed similar amounts of amyloid when compared between Hpa-KO and WT mice, in all three organs. The spleen contained substantial amounts of amyloid compared to moderate amounts in the liver and essentially no amyloid in the kidney. The organs collected 10-72 DPI were utilized for evaluation of amy-loid resolution. Surprisingly, Hpa-KO mice exhibited increased rate of amy-

40
loid clearance compared to WT mice. Spleens from both groups, collected 42 and 72 DPI, lacked Congo red positive signals. However, Hpa-KO spleens collected 10 and especially 21 DPI, displayed substantial decrease of Congo red positive staining compared to WT. Immunostaining of SAA in spleens collected 14 DPI, in comparison with Congo red staining, revealed, in addition to decreased amount of staining in Hpa-KO, a differential locali-zation of non-fibril SAA and fibril SAA. The Congo red positive SAA was localized to the perifollicular areas of the spleen while anti-SAA staining was more diffusely distributed. Immunoblotting of the splenic amyloid, ex-tracted overnight in 5% SDS at 60°C, revealed, again, seemingly weaker staining of SAA in Hpa-KO. However, comparable intensity of staining was found in the soluble samples of the splenic lysates, indicating rapid metabo-lism of SAA following fibril degradation.
To explain the difference in amyloid resolution between the two groups, the spleens and livers were analyzed by immunostaining as well as im-munoblotting for expression of matrix metalloproteinases (MMP) 2 and 9. Indeed, increased expression of MMP-2 and -9 were detected in Hpa-KO mouse spleen and liver, respectively. Finally, AEF degradation by MMP-9 was confirmed by SDS-PAGE analysis of incubated samples, to validate role of MMPs in the process of amyloid clearance.
Together, the results suggest there exists a co-regulating mechanism be-tween the proteases and heparanase in the ECM to ensure continuous home-ostasis during various challenges. Such a dynamic mechanism could explain the clinically observed ability of organs to evade build up of damaging levels of SAA spawned amyloid deposits during periods of persistently high plas-ma levels of SAA found in patients with chronic inflammation. Hence, MMPs and heparanase could potentially serve as prognostic markers in pa-tients suffering from AA amyloidosis.
Paper IV Aim The aim of the study is to examine the role of heparanase expression in re-solving/clearance of deposited Aβ in a mouse model of AD combined with an inflammation model induced by LPS-stimulation.
Background Examination of brains from patients with Alzheimer’s disease (AD) and mice overexpressing amyloid β precursor protein (AβPP-tg), has shown col-ocalization of HS and HS proteoglycans (HSPGs) with Aβ amyloid deposits (Snow et al., 1987b, O'Callaghan et al., 2008, Lord et al., 2011). HS/HSPGs

41
have additionally been found to promote formation of Aβ plaques (Castillo et al., 1997, Cotman et al., 2000) and inhibit its degradation (Biroc et al., 1993, Gupta-Bansal et al., 1995). In a previous study of our group, a double transgenic mouse was generated by crossing the Hpa-tg mouse with an AD mouse model, overexpressing a human AβPP gene with a Swedish mutation (tgSwe)(Citron et al., 1992). The results showed that overexpression of hepa-ranase in this mouse significantly lowered Aβ burden in the tgSwe mice. The suggested explanation of the phenomenon is that an enhanced fragmentation of HS by heparanase inhibits HS-promoted Aβ aggregation (Jendresen et al., 2015).
Neuroinflammation is a common feature of the neurodegenerative disease Alzheimer’s disease (AD)(Akiyama et al., 2000). Local leukocytes include microglia and astrocytes that are activated during inflammation and work together with recruited vascular monocytes that migrate across the BBB and mature into activated macrophages (Nakanishi, 2003, Town et al., 2005, Gate et al., 2010). The activated leukocytes secrete various inflammatory mediators, in the form of cytokines, chemokines and proteolytic enzymes. However, the underlying mechanisms of neuroinflammation and their impli-cations on AD are not fully understood. A paradox seems to exist in neuroin-flammation. The amyloid deposits in AD seem to activate the inflammatory response, resulting in activation of immune cells that have the ability of clearing away the amyloid, thus amyloid deposits facilitate their own clear-ance (Bates et al., 2009, Solito and Sastre, 2012). A recent study of our group employed Hpa-tg mice to study the role of HS in neuroinflammation using an acute inflammatory model where mice were injected with bacterial cytotoxic lipopolysaccharide (LPS)(Zhang et al., 2012). Heparanase overex-pression was shown to reduce the neuroinflammatory response and inhibit the recruitment of monocytes by an impaired ability of the cells to transmi-grate through the BBB. Furthermore, clearance of Aβ-amyloid injected into the brain was attenuated in Hpa-tg mice.
Together, these data suggests both pro-AD and anti-AD functions of HS and heparanase, where disease-promoting functions of HS seems more prominent, and vice versa for its degrading enzyme.
Results and discussion In light of the hampered clearance of Aβ in Hpa-tg mice when using an arti-ficial model of Aβ deposition, we wanted to further investigate this phenom-enon in a mouse model with endogenous deposition of Aβ. The transgenic mouse model generated in our earlier study, overexpressing both Hpa and AβPP (tgHpa*Swe), was therefore utilized to study the role of heparan sul-fate in neuroinflammation, and its implication in Aβ plaque formation and inflammatory-mediated clearance. The double-transgenic tgHpa*Swe mice and mice expressing only AβPP (tgSwe) were subjected to intra-peritoneal

42
injections of LPS at the age of 13.5 months; an age when the mice have a considerate amount of deposited amyloid. The mice were then sacrificed 1.5 months post-injection to collect the brains. One half of the brain was fixed and used for histology staining of amyloid deposits, and the other half was homogenized for biochemical analysis of Aβ and inflammatory markers.
Quantification of cerebral cortex immunohistochemical staining of the main component of Aβ-burden: Aβ1-40 (Philipson et al., 2009), revealed a trend towards decreased Aβ-burden in LPS-treated tgSwe mice compared to vehicle-treated tgSwe mice. This trend was not observed in tgHpa*Swe mice that are known from our previous study to develop less amyloid than the tgSwe mice already under naïve conditions (Jendresen et al., 2015). Further analysis of total amyloid burden by Congo red staining revealed a significant LPS-induced decrease in the tgSwe mice, and again, this was not seen in the tgHpa*Swe mice. Quantification of cerebral amyloid angioapathy (CAA) did however not show any significant differences due to LPS-treatment, in either group.
ELISA quantification of different types of amyloid, based on solubility, revealed a LPS-induced decrease of formic acid (FA) and SDS-soluble Aβ in tgSwe mice. In addition, the tween-soluble Aβ of LPS-treated tgSwe mice increased. Together, indicating a LPS-stimulated resolution of the deposited Aβ plaque. A similar pattern was seen when investigating the effect of hepa-ranase on Aβ-solubility, with less formic acid (FA) and SDS-soluble Aβ and a higher ratio of tween-soluble Aβ to total Aβ. Thus, indicating a hampered ability of HS to facilitate Aβ plaque formation, leading to an accumulated amount of non-fibrillar soluble Aβ.
As a mean to understand the observed pattern of amyloid deposition in the different groups of mice, we examined the levels of inflammatory cyto-kines in the brain lysates. Even though the levels of most cytokines were increased in the LPS-stimulated tgHpa*Swe mice compared to vehicle-treated tgHpa*Swe mice, overall lower levels of cytokines were found in both groups of tgHpa*Swe mice. This impaired inflammatory response is in agreement with previous observations of impaired microglial response in tgHpa mouse brain (Zhang et al., 2012, O'Callaghan et al., 2015).
A model of “macrophage colony-stimulating factor (M-CSF) resistance” has been proposed, where sufficient production of pro-inflammatory cyto-kine granulocyte macrophage colony-stimulating factor (GM-CSF), in com-bination with IFN-gamma or LPS stimulation, will interfere with the consti-tutively expressed non-inflammatory signals by M-CSF and result in pro-inflammatory M1-phenotypic polarization of macrophages. Hence, a ratio between GM-CSF and M-CSF can be used to evaluate the inflammatory status of the brain, where an increased ratio correlates with increased severi-ty of inflammation. Here, tgSwe mice exhibited a two-fold higher ratio of GM-CSF/M-CSF than WT and tgHpa*Swe mice in the vehicle-treated groups. This indicates that the increased amyloid burden of the tgSwe mice

43
fueled a chronic type neuroinflammation. This is in agreement with the low-ered ratio in the LPS-treated tgSwe mice, where the amyloid burden was reduced.
All together, this study illustrates the implications of heparanase in in-flammation-associated Aβ clearance. Together with our previous studies within this line, we are able to conclude that heparan sulfate and heparanase play pivotal roles in the Aβ pathology of Alzheimer’s disease.

44
Concluding remarks and future perspectives
Collectively, HS and heparanase play a significant role in all facets of in-flammation, from the initiation process, homing and extravasation of leuko-cytes, to the regulation of the local inflammatory response at the site of inju-ry and the final resolution of inflammation. These processes are of great importance in the disorders that are the subject of this thesis (RA, AA-amyloidosis and AD). After a century of substantial advancement in medical science and modernized living conditions, these disorders remain incurable, and are continuously becoming more prevalent in an ageing world popula-tion. This section aims to highlight potentially causative factors of these modern-day inflammatory diseases, as to identify key inflammatory mecha-nisms to target for future HS/heparanase research.
The modern-day inflammatory diseases Inflammation occurs frequently throughout our body in a highly controlled manner to fend of environmental challenges threatening our existence. It is an essential mechanism of our body. The consequence of potentially patho-logical inflammatory reactions is therefore an acceptable risk from an evolu-tionary perspective. In contrast, it can often be unacceptable and life threat-ening, from an individual’s perspective, when inflammatory processes be-come pathological. The list of inflammation-related pathological conse-quences is long, including well known disorders such as cancer, Alzheimer’s disease, cardiovascular disease, diabetes, rheumatoid arthritis, sepsis and amyloidosis. Many of the inflammation-associated diseases are also inter-connected, as demonstrated by a tendency of patients to develop multiple diseases simultaneously (Fabbri et al., 2015). The logical question is thus: How is inflammation regulated?
The inflammatory response is controlled by leukocytes of the immune system and the local tissular cells, through their production of inflammatory mediators and expression of inflammation-associated receptors. However, under most circumstances, these cells only respond as long as they are stimu-lated by danger molecules: PAMPs and DAMPs. Such stimulation is a re-quirement for an inflammatory response, since the body is always trying to maintain a state of homeostasis, which promotes survival, and even though inflammation is beneficial in response to infections and tissue damage, it is also harmful to our body. Hence, the body will only sustain inflammatory processes as long as it is necessary for survival. Consequently, when the

45
danger molecules are removed, the inflammation will succumb to the anti-inflammatory pressure and the tissue will return to homeostasis. Hence, ex-cept for cases with genetic defects in proteins of the inflammatory signaling pathways (Rigante et al., 2016), persistent (or chronic) inflammation is a result of the body’s inability to remove the underlying cause of danger mole-cule production.
Since the industrial revolution, humans have developed a lifestyle very different from the one that were instrumental in our evolutionary develop-ment. We are now exposed to a large number of new causative agents for injuries and danger molecule production. Genes that are evolutionary con-served to protect us from starvation, infection, injury and predation may now, in a lifestyle void of these challenges, contribute to the development of low-grade chronic inflammation-related diseases (Kotas and Medzhitov, 2015). The modern lifestyle includes changes in (I) diet, (II) hygiene, (III) chemical exposure, and an (IV) ever-increasing life expectancy brought on by an advanced healthcare and improved living conditions.
(I) Our diet has since before the agricultural age changed from wild plants and animal foods into a diet where 72% of the total energy consumed comes from refined sugars, refined vegetable oils, highly processed cereals, dairy products and a high consumption of alcohol (Ilich et al., 2014). The modern diet in combination with our gut microbes, contributes to constant low-grade chronic inflammation associated with the development of numerous disor-ders, such as obesity, diabetes, cardiovascular disease, autoimmune disorders as well as inflammatory intestinal diseases such as inflammatory bowels disease and colon cancer (Ilich et al., 2014, Breithaupt, 2014).
(II) Exposure to microbes early on in life is important to acquire an opti-mized balance between immune host defence and tolerance. Dysregulation of this balance, due to improved hygiene, has been linked to increased sus-ceptibility to allergies and inflammatory diseases (Stefka et al., 2014).
(III) Following the industrial revolution, humans were introduced to ex-posure by many new, potentially harmful, chemicals. However, an agent of exposure, introduced to humans far earlier, has through its addictive nature today become the greatest contributor to the development of inflammatory-associated diseases: tobacco. Tobacco smoking is the leading cause of pre-ventable death worldwide with an estimated 6 million annual deaths (Warren et al., 2014).
(IV) Advancement in medicine and improved living conditions has re-sulted in an ageing world population (Niccoli and Partridge, 2012). Func-tionality of many bodily processes decreases with age, thus causing homeo-static imbalance leading to the development of diseases, such as cancer, car-diovascular disease and neurodegeneration. The loss of functions is believed to be a result of an evolutionary trade-off between reproducibility and life expectancy (Williams, 1957). George C. Williams theorized that organisms with shorter live spans had a higher probability for increased numbers of

46
offspring. Neurodegenerative diseases such as Alzheimer’s disease is be-coming increasingly common in the ageing world population and represent a dreaded situation where death is preceded by a gradual loss of the essence of being (Wyss-Coray, 2016). To date, there are no drugs available to halt the disease progression and only a few drugs available for treating the symptoms of the disease (Islam and Tabrez, 2017). Hence, there is a great demand for an increased understanding of the disease progression, especially regarding the age-induced loss of functions, and development of novel therapeutics.
The modern lifestyle has indeed replaced previous detrimental diseases with new modern inflammatory-related pathologies, associated with the im-mune system, gut microbes, chemical exposure and pathways of cellular maintenance. A multifaceted molecule like HS, highly involved in all those aspects, undoubtedly makes an interesting target for future research, together with its degrading enzyme heparanase.

47
Populärvetenskaplig sammanfattning
Heparansulfat (HS) är en ogrenad sulfaterad kolhydratkedja, som tillsam-mans med det HS-nedbrytande enzymet heparanas, har en viktig roll i fysio-logiska och sjukdomsrelaterade processer under alla stadier i livet. Från ägg-cell till ålderns höst. Under en livstid genomgår kroppen prövningar från både omgivning och eventuella brister i arvsmassan. Vårt första försvar mot skada består av en immunovaskulär process: inflammation. När denna pro-cess inte kan stängas av och fortgår i en kronisk inflammation, tar kroppen skada, vilket leder till utveckling av olika sjukdomar. Kumulativ data visar att HS och heparanas är inblandade i olika aspekter av sådana sjukdomar, t.ex. vid ledgångsreumatism.
I detta arbete har jag undersökt vilken roll HS och heparanas spelar i olika inflammationsrelaterade sjukdomar med hjälp av möss som antingen över-producerar (Hpa-tg) eller saknar produktion (Hpa-KO) av heparanas.
I mitt första projekt använde vi en musmodell för att skildra sjukdomsför-loppet i reumatisk artrit (RA) i Hpa-tg möss. Vi fann en pro-inflammatorisk roll för heparanas som ledde till ökad aktivitet hos olika immunceller, bl.a. T celler, neutrofiler och antigen-presenterande celler. Detta i sin tur gjorde att de kliniska symptomen av RA blev värre bland Hpa-tg-mössen.
Serum amyloid A (SAA) är ett protein som finns i högdensitetslipoprotein (HDL)-partiklar och deltar i kolesteroltransport, både under inflammation och vid sjukdomen AA-amyloidos. Vid AA-amyloidos tappar SAA sin na-turliga form, klumpar ihop sig till olösligt plack, s.k. amyloid, upplagras i mjälte, njure och lever och leder i värsta fall till organsvikt och döden. I mitt andra arbete fann vi att heparin kunde binda de två HDL-proteinerna SAA och ApoA1, samtidigt, och våra resultat tydde på att SAA och ApoA1 befin-ner sig nära varandra i HDL-partikeln. I mitt tredje arbete observerade vi att ansamlingen av amyloid i mjälte hos möss med AA-amyloidos snabbare löstes upp hos möss som saknade heparanas. Detta sammanföll med ökad mängd matrix-metalloproteinaser (MMP), vilket indikerade att heparanas och MMP arbetar tillsammans för att lösa upp amyloid.
I mitt fjärde arbete undersökte vi hur Hpa och HS påverkar beta amyloid (Aβ) i en musmodell av Alzheimer’s sjukdom (AD) med överproduktion av en sjuklig variant av proteinet (AβPP), den så kallade ”svenska mutationen” (tgSwe). Vi fann att kronisk inflammation gjorde att ansamlat Aβ amyloid i hjärnan hos tgSwe-mössen löstes upp snabbare. Detta gällde inte möss som överproducerade både AβPP och Hpa (tgHpa*Swe). Efterföljande analys av

48
inflammatoriska cytokiner visade att tgHpa*Swe mössen generellt sett hade mindre inflammation. Sammanfattningsvis ledde alltså Hpa-överproduktion i en musmodell av Alzheimer’s sjukdom till minskad inflammation och att mer Aβ amyloid stannade kvar i hjärnan.

49
Acknowledgement
Present work has been carried out at the Department of Medical Biochemis-try and Microbiology at Uppsala University, Sweden. Financial support was provided by grants from the Swedish Research Council.
I would like to express my sincere gratitude to all the people who con-tributed to my work as well as helped and encouraged me throughout this adventure as a PhD student. My warmest thanks goes out to:
My supervisor Jin-Ping Li, thanks for greeting me with a warmth since the first day, believing in me, giving me the privileged and prestigious posi-tion as a doctoral student under your excellent supervision, being there for me when I needed consultation and guiding me at every turn. I have felt like a prince, and been very happy under your wings. You are truly a great scien-tist, always curious of new findings and enthusiastic about exploring new fields of science. I really enjoyed our venture into the field of autoimmune diseases. You have always given all your heart to ensure the continued suc-cess of the group and its members. Thank you!
My co-supervisor Lena Kjellén, thank you a lot! For reading my email, taking me in as your master student and introducing me to this amazing lab full of amazing people of present and past. I would be somewhere complete-ly different doing something completely different without you. Thanks for the guidance during both the master work and the following years. And you are just wonderful.
Magnus Åbrink, thank you for being my guide in both the jungle of Rheumathoid arthritis and leading me to the path of comfortable ways of doing animal experiments. You are such a pleasant man to be around.
Kailash Singh, thanks for sharing your expertise regarding both FACS analysis but also your vast knowledge in the field of immunology. You real-ly helped me along the way towards understanding how our immune system works, and you are such a humble person and a good friend.
Rogier Reijmers, thanks for your impressive efforts of contribution with those delicate lymph nodes and your constructive comments throughout the RA project, you are truly a great researcher.
Jie Nan and Martin Frank, thank you for your hard work in analysing the HDL-SAA structure. I am very fascinated with the ability of both of your techniques to enable visualization of structures too small for the eye to per-ceive.

50
Thank you, thank you, thank you, to all co-authors and collaborators for taking some of the mountain of work of my shoulders, contributing with excellent data, sharing your great research skills and making my work possi-ble.
Dr. Ramesh Babu Namburi, thank you for being there for me since I came to the lab, being an awesome friend, helping me with all my problems, sharing countless discussions, trips and working out HARD with me! You are truly an achiever, a doer, and nothing can stop you. Love you man.
Future Dr. Tahira Batool, thank you for being you. You are a good per-son, who is fighting your way through life. Thank you for letting me be me and bombard you with more or less good lessons and advice. It has helped me a lot into becoming a humble and wise person. Good luck in your life in Sweden! ☺
Huuuge immense vast thanks to present and former group members Ta-hira Batool, Hao “Eric” Cui, Jian-Ping Fang, Gan-Lin Zhang, Shilpa Mallesh, Tian-Yi Song, Xiao Zhang, Yin-Xia Tan, Fredrik Noborn, Xin Liu, Hiroshi Nakato, Fabian Baltes, Sher, Erika, Elin, for contributions in work as well as in turning me into a clever(ish) happy guy!
Anna Eriksson, you definitely deserve a huge thank you for how you not only helped me but all the people in the corridor. You deserve an additional PhD degree in lab teaching ☺.
I would like to mention my previous supervisor, Audrey Deligny, thank you for teaching me (as a clueless master student) a great conduct of labora-tory practice. It has helped me a lot!
Emma and Bene, thanks for being awesome friends here in Uppsala. You have filled my life with joy and endured many hours of listening to my many thoughts flying around in my head about the research life but mostly life itself. Love you guys!
Tabea Dierker, yes you cleaned up my thesis kappa with incredible thor-oughness, very thankful for that, but mostly I want to thank you for being an otherwise great human being who always helps others, come with great ad-vice and find humor in the little things. You are also an exemplary research-er.
Thanks to Dorothe, Inger, Andrew, Sara, Rashmi, Xiao-Qun, Beata, Paul, Johan, Peder, Anh-Tri, Maria, Wahida and all you other D9:4 peo-ple as well as to some great helpful and nice IMBIM people that ended up in a not so great corridor (everyone knows D9:4 is the best☺), we had so much fun and you have helped me in countless ways along the way! I´m deeply sorry if I forgot anyone, but you have to excuse my Alzheimery brain after all this thesis writing about Alzheimer’s disease. ☺ I really cherish all my encounters in life!
Grzegorz Furman and Roland Bergdahl (and Armin ☺), thank you for being great friends. We have had non-stop fun in the ARG-group around Ultuna and BMC during our studies here in Uppsala. We are for sure

51
bomberman experts after all those recesses together. Love you guys! Group hug! ☺
I would like to express my great appreciation and love for my dear goofy family and my large group of relatives (tjocka släkten). Without you I would be nothing:
My parents, Hanno Höjlind and Evy-Ann Digre, have given me uncon-ditional love and support throughout my life. Words cannot express my love and gratitude to you.
My siblings, Robert, Veronica, Pernilla and Victoria Digre, have filled my entire life with love and laughter (and battles ☺). You are the second, third, fourth, fifth halves of me.
To my nephew, Emanuel, who loved my magnifying glass I got him for Christmas: science is waiting for you.☺ I Love you, whatever you choose to do with your life. (..But do science. ☺)
To all my aunts, uncles, grandfathers, grandmothers, cousins and bo-nus relatives, I would like to send a lot of love and gratitude for support and wonderful times together. You are such great and warm people.
Last but not least, I would like to thank all my dear friends outside the sphere of Uppsala University, including Dick Hedberg, Robin Eriksson, Christoffer Kauppi, Kim Larsson and Kim Persson. Thanks for the sup-port and glorious times together in tents, at festivals, parties, LANs, movie nights, hockey games in “ladan”. Love you all! ☺

52
References
ABBAS, A. K., LICHTMAN, A. H. & PILLAI, S. 2012. Cellular and molecular immunology, Philadelphia, Elsevier/Saunders, c2012.
ABBOUD-JARROUS, G., ATZMON, R., PERETZ, T., PALERMO, C., GADEA, B. B., JOYCE, J. A. & VLODAVSKY, I. 2008. Cathepsin L is responsible for processing and activation of proheparanase through multiple cleavages of a linker segment. The Journal of Biological Chemistry, 283, 18167-76.
ABBOUD-JARROUS, G., RANGINI-GUETTA, Z., AINGORN, H., ATZMON, R., ELGAVISH, S., PERETZ, T. & VLODAVSKY, I. 2005. Site-directed mutagenesis, proteolytic cleavage, and activation of human proheparanase. The Journal of Biological Chemistry, 280, 13568-75.
AGUILERA, J. J., ZHANG, F., BEAUDET, J. M., LINHARDT, R. J. & COLON, W. 2014. Divergent effect of glycosaminoglycans on the in vitro aggregation of serum amyloid A. Biochimie, 104, 70-80.
AI, X., DO, A. T., KUSCHE-GULLBERG, M., LINDAHL, U., LU, K. & EMERSON, C. P., JR. 2006. Substrate specificity and domain functions of extracellular heparan sulfate 6-O-endosulfatases, QSulf1 and QSulf2. The Journal of Biological Chemistry, 281, 4969-76.
AIKAWA, J., GROBE, K., TSUJIMOTO, M. & ESKO, J. D. 2001. Multiple Isozymes of Heparan Sulfate/Heparin GlcNAc N-Deacetylase/N-Sulfotransferase: Structure and Activity of the Fourth Member, NDST4. J. Biol. Chem., 276, 5876-5882.
AKBARSHAHI, H., AXELSSON, J. B., SAID, K., MALMSTROM, A., FISCHER, H. & ANDERSSON, R. 2011. TLR4 dependent heparan sulphate-induced pancreatic inflammatory response is IRF3-mediated. Journal of translational medicine, 9, 219.
AKIYAMA, H., BARGER, S., BARNUM, S., BRADT, B., BAUER, J., COLE, G. M., COOPER, N. R., EIKELENBOOM, P., EMMERLING, M., FIEBICH, B. L., FINCH, C. E., FRAUTSCHY, S., GRIFFIN, W. S., HAMPEL, H., HULL, M., LANDRETH, G., LUE, L., MRAK, R., MACKENZIE, I. R., MCGEER, P. L., O'BANION, M. K., PACHTER, J., PASINETTI, G., PLATA-SALAMAN, C., ROGERS, J., RYDEL, R., SHEN, Y., STREIT, W., STROHMEYER, R., TOOYOMA, I., VAN MUISWINKEL, F. L., VEERHUIS, R., WALKER, D., WEBSTER, S., WEGRZYNIAK, B., WENK, G. & WYSS-CORAY, T. 2000. Inflammation and Alzheimer's disease. Neurobiology of aging, 21, 383-421.
ANCSIN, J. B. & KISILEVSKY, R. 2004. A binding site for highly sulfated heparan sulfate is identified in the N terminus of the circumsporozoite protein: significance for malarial sporozoite attachment to hepatocytes. The Journal of biological chemistry, 279, 21824-32.
ANDERSSON, L. O., BARROWCLIFFE, T. W., HOLMER, E., JOHNSON, E. A. & SIMS, G. E. 1976. Anticoagulant properties of heparin fractionated by affinity chromatography on matrix-bound antithrombin iii and by gel filtration. Thrombosis Research, 9, 575-83.

53
AXELRAD, M. A., KISILEVSKY, R., WILLMER, J., CHEN, S. J. & SKINNER, M. 1982. Further characterization of amyloid-enhancing factor. Laboratory investigation; a journal of technical methods and pathology, 47, 139-46.
BACKLUND, J., LI, C., JANSSON, E., CARLSEN, S., MERKY, P., NANDAKUMAR, K. S., HAAG, S., YTTERBERG, J., ZUBAREV, R. A. & HOLMDAHL, R. 2013. C57BL/6 mice need MHC class II Aq to develop collagen-induced arthritis dependent on autoreactive T cells. Annals of the rheumatic diseases, 72, 1225-32.
BARAZ, L., HAUPT, Y., ELKIN, M., PERETZ, T. & VLODAVSKY, I. 2006. Tumor suppressor p53 regulates heparanase gene expression. Oncogene, 25, 3939-47.
BARTOK, B. & FIRESTEIN, G. S. 2010. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunological reviews, 233, 233-55.
BATES, K. A., VERDILE, G., LI, Q. X., AMES, D., HUDSON, P., MASTERS, C. L. & MARTINS, R. N. 2009. Clearance mechanisms of Alzheimer's amyloid-beta peptide: implications for therapeutic design and diagnostic tests. Molecular psychiatry, 14, 469-86.
BENDITT, E. P., HOFFMAN, J. S., ERIKSEN, N., PARMELEE, D. C. & WALSH, K. A. 1982. SAA, an apoprotein of HDL: its structure and function. Annals of the New York Academy of Sciences, 389, 183-9.
BERNFIELD, M., GOTTE, M., PARK, P. W., REIZES, O., FITZGERALD, M. L., LINCECUM, J. & ZAKO, M. 1999. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem, 68, 729-77.
BILLIAU, A. & MATTHYS, P. 2011. Collagen-induced arthritis and related animal models: how much of their pathogenesis is auto-immune, how much is auto-inflammatory? Cytokine & growth factor reviews, 22, 339-44.
BIROC, S. L., PAYAN, D. G. & FISHER, J. M. 1993. Isoforms of Agrin Are Widely Expressed in the Developing Rat and May Function as Protease Inhibitors. Developmental Brain Research, 75, 119-129.
BISHOP, J. R., SCHUKSZ, M. & ESKO, J. D. 2007. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature, 446, 1030-7.
BLICH, M., GOLAN, A., ARVATZ, G., SEBBAG, A., SHAFAT, I., SABO, E., COHEN-KAPLAN, V., PETCHERSKI, S., AVNIEL-POLAK, S., EITAN, A., HAMMERMAN, H., ARONSON, D., AXELMAN, E., ILAN, N., NUSSBAUM, G. & VLODAVSKY, I. 2013. Macrophage activation by heparanase is mediated by TLR-2 and TLR-4 and associates with plaque progression. Arteriosclerosis, thrombosis, and vascular biology, 33, e56-65.
BREITHAUPT, H. Immune Homeostasis and Inflammatory Disease. Immune Homeostasis and Inflammatory Disease: A Herrenhausen Symposium, November 6-8, 2014 2014 Herrenhausen Palace Conference Center, Hanover, Germany. Nature Medicine.
BRENNAN, T. V., LIN, L., HUANG, X., CARDONA, D. M., LI, Z., DREDGE, K., CHAO, N. J. & YANG, Y. 2012. Heparan sulfate, an endogenous TLR4 agonist, promotes acute GVHD after allogeneic stem cell transplantation. Blood, 120, 2899-908.
BRUNN, G. J., BUNGUM, M. K., JOHNSON, G. B. & PLATT, J. L. 2005. Conditional signaling by Toll-like receptor 4. FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 19, 872-4.
BURCH, J. & ONYSKO, M. 2012. Current standards and future treatments of rheumatoid arthritis. Formulary Journal, 47, 359-368.

54
BUSSE, M., FETA, A., PRESTO, J., WILEN, M., GRONNING, M., KJELLEN, L. & KUSCHE-GULLBERG, M. 2007. Contribution of EXT1, EXT2, and EXTL3 to heparan sulfate chain elongation. The Journal of Biological Chemistry, 282, 32802-10.
CAMPBELL, I. K., HAMILTON, J. A. & WICKS, I. P. 2000. Collagen-induced arthritis in C57BL/6 (H-2b) mice: new insights into an important disease model of rheumatoid arthritis. European journal of immunology, 30, 1568-75.
CASTILLO, G. M., CUMMINGS, J. A., YANG, W., JUDGE, M. E., SHEARDOWN, M. J., RIMVALL, K., HANSEN, J. B. & SNOW, A. D. 1998. Sulfate content and specific glycosaminoglycan backbone of perlecan are critical for perlecan's enhancement of islet amyloid polypeptide (amylin) fibril formation. Diabetes, 47, 612-20.
CASTILLO, G. M., NGO, C., CUMMINGS, J., WIGHT, T. N. & SNOW, A. D. 1997. Perlecan binds to the beta-amyloid proteins (A beta) of Alzheimer's disease, accelerates A beta fibril formation, and maintains A beta fibril stability. Journal of neurochemistry, 69, 2452-65.
CHEN, G., WANG, D., VIKRAMADITHYAN, R., YAGYU, H., SAXENA, U., PILLARISETTI, S. & GOLDBERG, I. J. 2004. Inflammatory cytokines and fatty acids regulate endothelial cell heparanase expression. Biochemistry, 43, 4971-7.
CITRON, M., OLTERSDORF, T., HAASS, C., MCCONLOGUE, L., HUNG, A. Y., SEUBERT, P., VIGO-PELFREY, C., LIEBERBURG, I. & SELKOE, D. J. 1992. Mutation of the beta-amyloid precursor protein in familial Alzheimer's disease increases beta-protein production. Nature, 360, 672-4.
COFFMAN, R. L., SHER, A. & SEDER, R. A. 2010. Vaccine adjuvants: putting innate immunity to work. Immunity, 33, 492-503.
CONSTANTINESCU, A. A., VINK, H. & SPAAN, J. A. 2003. Endothelial cell glycocalyx modulates immobilization of leukocytes at the endothelial surface. Arteriosclerosis, thrombosis, and vascular biology, 23, 1541-7.
COTMAN, S. L., HALFTER, W. & COLE, G. J. 2000. Agrin binds to beta-amyloid (Abeta), accelerates abeta fibril formation, and is localized to Abeta deposits in Alzheimer's disease brain. Molecular and cellular neurosciences, 15, 183-98.
CUI, H., HUNG, A. C., KLAVER, D. W., SUZUKI, T., FREEMAN, C., NARKOWICZ, C., JACOBSON, G. A. & SMALL, D. H. 2011. Effects of heparin and enoxaparin on APP processing and Abeta production in primary cortical neurons from Tg2576 mice. PLoS ONE, 6, e23007.
CUNNANE, G. 2001. Amyloid precursors and amyloidosis in inflammatory arthritis. Current opinion in rheumatology, 13, 67-73.
DE MESTRE, A. M., RAO, S., HORNBY, J. R., SOE-HTWE, T., KHACHIGIAN, L. M. & HULETT, M. D. 2005. Early growth response gene 1 (EGR1) regulates heparanase gene transcription in tumor cells. J Biol Chem, 280, 35136-47.
DEMPSEY, L. A., BRUNN, G. J. & PLATT, J. L. 2000. Heparanase, a potential regulator of cell-matrix interactions. Trends Biochem Sci, 25, 349-51.
DHOOT, G. K., GUSTAFSSON, M. K., AI, X., SUN, W., STANDIFORD, D. M. & EMERSON, C. P., JR. 2001. Regulation of Wnt signaling and embryo patterning by an extracellular sulfatase. Science, 293, 1663-6.
EDOVITSKY, E., LERNER, I., ZCHARIA, E., PERETZ, T., VLODAVSKY, I. & ELKIN, M. 2006. Role of endothelial heparanase in delayed-type hypersensitivity. Blood, 107, 3609-16.
EGASHIRA, M., TAKASE, H., YAMAMOTO, I., TANAKA, M. & SAITO, H. 2011. Identification of regions responsible for heparin-induced amyloidogenesis

55
of human serum amyloid A using its fragment peptides. Arch Biochem Biophys, 511, 101-6.
ELIMOVA, E., KISILEVSKY, R. & ANCSIN, J. B. 2009. Heparan sulfate promotes the aggregation of HDL-associated serum amyloid A: evidence for a proamyloidogenic histidine molecular switch. FASEB J, 23, 3436-48.
ESKO, J. D., KIMATA, K. & LINDAHL, U. 2009. Proteoglycans and Sulfated Glycosaminoglycans. In: VARKI, A., CUMMINGS, R. D., ESKO, J. D., FREEZE, H. H., STANLEY, P., BERTOZZI, C. R., HART, G. W. & ETZLER, M. E. (eds.) Essentials of Glycobiology. 2nd ed. Cold Spring Harbor (NY).
ESKO, J. D. & SELLECK, S. B. 2002. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem, 71, 435-71.
ESKO, J. D. & ZHANG, L. 1996. Influence of core protein sequence on glycosaminoglycan. Curr Opin Struct Biol, 6, 663-670.
ESTEVE, E., RICART, W. & FERNANDEZ-REAL, J. M. 2005. Dyslipidemia and inflammation: an evolutionary conserved mechanism. Clin Nutr, 24, 16-31.
FABBRI, E., ZOLI, M., GONZALEZ-FREIRE, M., SALIVE, M. E., STUDENSKI, S. A. & FERRUCCI, L. 2015. Aging and Multimorbidity: New Tasks, Priorities, and Frontiers for Integrated Gerontological and Clinical Research. Journal of the American Medical Directors Association, 16, 640-7.
FIRESTEIN, G. S. 2003. Evolving concepts of rheumatoid arthritis. Nature, 423, 356-61.
FREEMAN, C. & PARISH, C. R. 1998. Human platelet heparanase: purification, characterization and catalytic activity. Biochem J, 330, 1341-50.
FREUND, J., CASALS, J. & HOSMER, E. P. 1937. Sensitization and antibody formation after injection of tubercle bacilli and paraffin oil. Proceedings of the Society for Experimental Biology and Medicine, 37, 509-513.
FRIDMAN, R., LIDER, O., NAPARSTEK, Y., FUKS, Z., VLODAVSKY, I. & COHEN, I. R. 1987. Soluble antigen induces T lymphocytes to secrete an endoglycosidase that degrades the heparan sulfate moiety of subendothelial extracellular matrix. Journal of cellular physiology, 130, 85-92.
FUX, L., FEIBISH, N., COHEN-KAPLAN, V., GINGIS-VELITSKI, S., FELD, S., GEFFEN, C., VLODAVSKY, I. & ILAN, N. 2009. Structure-function approach identifies a COOH-terminal domain that mediates heparanase signaling. Cancer research, 69, 1758-67.
GABAY, C. & KUSHNER, I. 1999. Acute-phase proteins and other systemic responses to inflammation. The New England journal of medicine, 340, 448-54.
GALLAGHER, J. T. 2001. Heparan sulfate: growth control with a restricted sequence menu. J Clin Invest, 108, 357-61.
GATE, D., REZAI-ZADEH, K., JODRY, D., RENTSENDORJ, A. & TOWN, T. 2010. Macrophages in Alzheimer's disease: the blood-borne identity. Journal of neural transmission, 117, 961-70.
GEBOES, L., DE KLERCK, B., VAN BALEN, M., KELCHTERMANS, H., MITERA, T., BOON, L., DE WOLF-PEETERS, C. & MATTHYS, P. 2007. Freund's complete adjuvant induces arthritis in mice lacking a functional interferon-gamma receptor by triggering tumor necrosis factor alpha-driven osteoclastogenesis. Arthritis and rheumatism, 56, 2595-607.
GILAT, D., HERSHKOVIZ, R., GOLDKORN, I., CAHALON, L., KORNER, G., VLODAVSKY, I. & LIDER, O. 1995. Molecular behavior adapts to context: heparanase functions as an extracellular matrix-degrading enzyme or as a T cell adhesion molecule, depending on the local pH. J Exp Med, 181, 1929-34.

56
GINGIS-VELITSKI, S., ZETSER, A., FLUGELMAN, M. Y., VLODAVSKY, I. & ILAN, N. 2004a. Heparanase induces endothelial cell migration via protein kinase B/Akt activation. J Biol Chem, 279, 23536-41.
GINGIS-VELITSKI, S., ZETSER, A., KAPLAN, V., BEN-ZAKEN, O., COHEN, E., LEVY-ADAM, F., BASHENKO, Y., FLUGELMAN, M. Y., VLODAVSKY, I. & ILAN, N. 2004b. Heparanase uptake is mediated by cell membrane heparan sulfate proteoglycans. The Journal of Biological Chemistry, 279, 44084-92.
GOLDBERG, R., MEIROVITZ, A., HIRSHOREN, N., BULVIK, R., BINDER, A., RUBINSTEIN, A. M. & ELKIN, M. 2013. Versatile role of heparanase in inflammation. Matrix biology : journal of the International Society for Matrix Biology, 32, 234-40.
GOLDBOURT, U., YAARI, S. & MEDALIE, J. H. 1997. Isolated low HDL cholesterol as a risk factor for coronary heart disease mortality. A 21-year follow-up of 8000 men. Arteriosclerosis, thrombosis, and vascular biology, 17, 107-13.
GOLDSHMIDT, O., ZCHARIA, E., ABRAMOVITCH, R., METZGER, S., AINGORN, H., FRIEDMANN, Y., SCHIRRMACHER, V., MITRANI, E. & VLODAVSKY, I. 2002. Cell surface expression and secretion of heparanase markedly promote tumor angiogenesis and metastasis. Proc Natl Acad Sci U S A, 99, 10031-6.
GOODALL, K. J., POON, I. K., PHIPPS, S. & HULETT, M. D. 2014. Soluble heparan sulfate fragments generated by heparanase trigger the release of pro-inflammatory cytokines through TLR-4. PLoS ONE, 9, e109596.
GOTTE, M. 2003. Syndecans in inflammation. FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 17, 575-91.
GRAY, E., MULLOY, B. & BARROWCLIFFE, T. W. 2008. Heparin and low-molecular-weight heparin. Thromb Haemost, 99, 807-18.
GREGERSEN, P. K., SILVER, J. & WINCHESTER, R. J. 1987. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis and rheumatism, 30, 1205-13.
GROSS-COHEN, M., FELD, S., DOWECK, I., NEUFELD, G., HASSON, P., ARVATZ, G., BARASH, U., NARODITSKY, I., ILAN, N. & VLODAVSKY, I. 2016. Heparanase 2 Attenuates Head and Neck Tumor Vascularity and Growth. Cancer research, 76, 2791-801.
GUILLOT-SESTIER, M. V., DOTY, K. R. & TOWN, T. 2015. Innate Immunity Fights Alzheimer's Disease. Trends in neurosciences, 38, 674-81.
GUPTA-BANSAL, R., FREDERICKSON, R. C. & BRUNDEN, K. R. 1995. Proteoglycan-mediated inhibition of A beta proteolysis. A potential cause of senile plaque accumulation. The Journal of Biological Chemistry, 270, 18666-71.
GUTTER-KAPON, L., ALISHEKEVITZ, D., SHAKED, Y., LI, J. P., ARONHEIM, A., ILAN, N. & VLODAVSKI, I. 2016. Heparanase is required for activation and function of macrophages. PNAS.
HABUCHI, H., HABUCHI, O. & KIMATA, K. 2004. Sulfation pattern in glycosaminoglycan: does it have a code? Glycoconjugate journal, 21, 47-52.
HABUCHI, H., KOYAYASHI, M. & KIMATA, K. 1998. Molecular Characterization and Expression of Heparan-sulfate-6-Sulfotransferase.
Complete cDNA cloning in human and partial cloning in Chinese hamster ovary cells. J. Biol. Chem., 273, 9208-9213.
HABUCHI, H., TANAKA, M., HABUCHI, O., YOSHIDA, K., SUZUKI, H., BAN, K. & KIMATA, K. 2000. The occurrence of three isoforms of heparan sulfate 6-

57
O- sulfotransferase having different specificities for hexuronic acid adjacent to the targeted N-sulfoglucosamine. J Biol Chem, 275, 2859-68.
HAJDUK, S. L., MOORE, D. R., VASUDEVACHARYA, J., SIQUEIRA, H., TORRI, A. F., TYTLER, E. M. & ESKO, J. D. 1989. Lysis of Trypanosoma brucei by a toxic subspecies of human high density lipoprotein. The Journal of Biological Chemistry, 264, 5210-7.
HARDARDOTTIR, I., GRUNFELD, C. & FEINGOLD, K. R. 1994. Effects of endotoxin and cytokines on lipid metabolism. Current opinion in lipidology, 5, 207-15.
HARRISON, R. S., SHARPE, P. C., SINGH, Y. & FAIRLIE, D. P. 2007. Amyloid peptides and proteins in review. Reviews of physiology, biochemistry and pharmacology, 159, 1-77.
HAWKINS, P. N. & PEPYS, M. B. 1990. A primed state exists in vivo following histological regression of amyloidosis. Clinical and experimental immunology, 81, 325-8.
HEIT, B., ROBBINS, S. M., DOWNEY, C. M., GUAN, Z., COLARUSSO, P., MILLER, B. J., JIRIK, F. R. & KUBES, P. 2008. PTEN functions to 'prioritize' chemotactic cues and prevent 'distraction' in migrating neutrophils. Nature immunology, 9, 743-52.
HENEKA, M. T., GOLENBOCK, D. T. & LATZ, E. 2015. Innate immunity in Alzheimer's disease. Nature immunology, 16, 229-36.
HEPPER, I., SCHYMEINSKY, J., WECKBACH, L. T., JAKOB, S. M., FROMMHOLD, D., SIXT, M., LASCHINGER, M., SPERANDIO, M. & WALZOG, B. 2012. The mammalian actin-binding protein 1 is critical for spreading and intraluminal crawling of neutrophils under flow conditions. Journal of immunology, 188, 4590-601.
HOLLEBOOM, A. G., JAKULJ, L., FRANSSEN, R., DECARIS, J., VERGEER, M., KOETSVELD, J., LUCHOOMUN, J., GLASS, A., HELLERSTEIN, M. K., KASTELEIN, J. J., HOVINGH, G. K., KUIVENHOVEN, J. A., GROEN, A. K., TURNER, S. M. & STROES, E. S. 2013. In vivo tissue cholesterol efflux is reduced in carriers of a mutation in APOA1. Journal of lipid research, 54, 1964-71.
HOLMDAHL, R. 2015. Animal models for rheumatoid arthritis [Online]. London: Henry Stewart Talks Ltd. Available: https://hstalks.com/bs/3076/ [Accessed July 30, 2015.
HOLMDAHL, R., ANDERSSON, M. E., GOLDSCHMIDT, T. J., JANSSON, L., KARLSSON, M., MALMSTROM, V. & MO, J. 1989. Collagen induced arthritis as an experimental model for rheumatoid arthritis. Immunogenetics, pathogenesis and autoimmunity. APMIS : acta pathologica, microbiologica, et immunologica Scandinavica, 97, 575-84.
HOLMDAHL, R., BOCKERMANN, R., BACKLUND, J. & YAMADA, H. 2002. The molecular pathogenesis of collagen-induced arthritis in mice--a model for rheumatoid arthritis. Ageing research reviews, 1, 135-47.
HULETT, M. D., FREEMAN, C., HAMDORF, B. J., BAKER, R. T., HARRIS, M. J. & PARISH, C. R. 1999. Cloning of mammalian heparanase, an important enzyme in tumor invasion and metastasis. Nat Med, 5, 803-9.
HULETT, M. D., HORNBY, J. R., OHMS, S. J., ZUEGG, J., FREEMAN, C., GREADY, J. E. & PARISH, C. R. 2000. Identification of active-site residues of the pro-metastatic endoglycosidase heparanase. Biochemistry, 39, 15659-67.
ILICH, J. Z., KELLY, O. J., KIM, Y. & SPICER, M. T. 2014. Low-grade chronic inflammation perpetuated by modern diet as a promoter of obesity and osteoporosis. Arhiv za higijenu rada i toksikologiju, 65, 139-48.

58
INGLIS, J. J., CRIADO, G., MEDGHALCHI, M., ANDREWS, M., SANDISON, A., FELDMANN, M. & WILLIAMS, R. O. 2007. Collagen-induced arthritis in C57BL/6 mice is associated with a robust and sustained T-cell response to type II collagen. Arthritis research & therapy, 9, R113.
INGLIS, J. J., SIMELYTE, E., MCCANN, F. E., CRIADO, G. & WILLIAMS, R. O. 2008. Protocol for the induction of arthritis in C57BL/6 mice. Nature protocols, 3, 612-8.
ISHIHARA, T. 1973. Experimental amyloidosis using silver nitrate--electron microscopic study on the relationship between silver granules, amyloid fibrils and reticuloendothelial system. Acta pathologica japonica, 23, 439-64.
ISLAM, B. U. & TABREZ, S. 2017. Management of Alzheimer's disease-An insight of the enzymatic and other novel potential targets. International journal of biological macromolecules, 97, 700-709.
JENDRESEN, C. B., CUI, H., ZHANG, X., VLODAVSKY, I., NILSSON, L. N. & LI, J. P. 2015. Overexpression of Heparanase Lowers the Amyloid Burden in Amyloid-beta Precursor Protein Transgenic Mice. J Biol Chem, 290, 5053-64.
JOHNSON, G. B., BRUNN, G. J., KODAIRA, Y. & PLATT, J. L. 2002. Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4. Journal of immunology, 168, 5233-9.
JOHNSON, G. V. & BAILEY, C. D. 2002. Tau, where are we now? Journal of Alzheimer's disease : JAD, 4, 375-98.
KARATEEV, D. E. 2003. [Angiogenesis in rheumatoid arthritis]. Vestnik Rossiiskoi akademii meditsinskikh nauk / Rossiiskaia akademiia meditsinskikh nauk, 47-51.
KIM, B. T., KITAGAWA, H., TANAKA, J., TAMURA, J. & SUGAHARA, K. 2003. In vitro heparan sulfate polymerization: crucial roles of core protein moieties of primer substrates in addition to the EXT1-EXT2 interaction. The Journal of Biological Chemistry, 278, 41618-23.
KINKLEY, S. M., BAGSHAW, W. L., TAM, S. P. & KISILEVSKY, R. 2006. The path of murine serum amyloid A through peritoneal macrophages. Amyloid : the international journal of experimental and clinical investigation : the official journal of the International Society of Amyloidosis, 13, 123-34.
KISILEVSKY, R., LEMIEUX, L. J., FRASER, P. E., KONG, X., HULTIN, P. G. & SZAREK, W. A. 1995. Arresting amyloidosis in vivo using small-molecule anionic sulphonates or sulphates: implications for Alzheimer's disease. Nature medicine, 1, 143-8.
KISILEVSKY, R. & MANLEY, P. N. 2012. Acute-phase serum amyloid A: perspectives on its physiological and pathological roles. Amyloid : the international journal of experimental and clinical investigation : the official journal of the International Society of Amyloidosis, 19, 5-14.
KISILEVSKY, R. & SUBRAHMANYAN, L. 1992. Serum amyloid A changes high density lipoprotein's cellular affinity. A clue to serum amyloid A's principal function. Laboratory investigation; a journal of technical methods and pathology, 66, 778-85.
KITAGAWA, H., EGUSA, N., TAMURA, J. I., KUSCHE-GULLBERG, M., LINDAHL, U. & SUGAHARA, K. 2001. rib-2, a Caenorhabditis elegans homolog of the human tumor suppressor EXT genes encodes a novel alpha1,4-N-acetylglucosaminyltransferase involved in the biosynthetic initiation and elongation of heparan sulfate. The Journal of Biological Chemistry, 276, 4834-8.
KITAGAWA, H., SHIMAKAWA, H. & SUGAHARA, K. 1999. The tumor suppressor EXT-like gene EXTL2 encodes an alpha1, 4-N-acetylhexosaminyltransferase that transfers N-acetylgalactosamine and N-

59
acetylglucosamine to the common glycosaminoglycan-protein linkage region. The key enzyme for the chain initiation of heparan sulfate. The Journal of Biological Chemistry, 274, 13933-7.
KJELLÉN, L. & LINDAHL, U. 1991. Proteoglycans: Structures and interactions. Annual Review of Biochemistry, 60, 443-75.
KLEINNIJENHUIS, J., OOSTING, M., JOOSTEN, L. A., NETEA, M. G. & VAN CREVEL, R. 2011. Innate immune recognition of Mycobacterium tuberculosis. Clinical & developmental immunology, 2011, 405310.
KOBAYASHI, M., HABUCHI, H., HABUCHI, O., SAITO, M. & KIMATA, K. 1996. Purification and Characterization of Heparan Sulfate 2-Sulfotransferase from Cultured Chinese Hamster Ovary Cells. J. Biol. Chem., 271, 7645-7653.
KOTAS, M. E. & MEDZHITOV, R. 2015. Homeostasis, inflammation, and disease susceptibility. Cell, 160, 816-27.
KUMAR, A. V., KATAKAM, S. K., URBANOWITZ, A. K. & GOTTE, M. 2015. Heparan sulphate as a regulator of leukocyte recruitment in inflammation. Current protein & peptide science, 16, 77-86.
KUSSIE, P. H., HULMES, J. D., LUDWIG, D. L., PATEL, S., NAVARRO, E. C., SEDDON, A. P., GIORGIO, N. A. & BOHLEN, P. 1999. Cloning and functional expression of a human heparanase gene. Biochem Biophys Res Commun, 261, 183-7.
KUWABARA, K., NISHITSUJI, K., UCHIMURA, K., HUNG, S. C., MIZUGUCHI, M., NAKAJIMA, H., MIKAWA, S., KOBAYASHI, N., SAITO, H. & SAKASHITA, N. 2015. Cellular interaction and cytotoxicity of the iowa mutation of apolipoprotein A-I (ApoA-IIowa) amyloid mediated by sulfate moieties of heparan sulfate. The Journal of Biological Chemistry, 290, 24210-21.
LACHMANN, H. J., GOODMAN, H. J., GILBERTSON, J. A., GALLIMORE, J. R., SABIN, C. A., GILLMORE, J. D. & HAWKINS, P. N. 2007. Natural history and outcome in systemic AA amyloidosis. The New England journal of medicine, 356, 2361-71.
LACHMANN, H. J. & HAWKINS, P. N. 2006. Systemic amyloidosis. Current opinion in pharmacology, 6, 214-20.
LAM, L. H., SILBERT, J. E. & ROSENBERG, R. D. 1976. The separation of active and inactive forms of heparin. Biochemical and Biophysical Research Communications, 69, 570-7.
LAMANNA, W. C., KALUS, I., PADVA, M., BALDWIN, R. J., MERRY, C. L. & DIERKS, T. 2007. The heparanome--the enigma of encoding and decoding heparan sulfate sulfation. Journal of biotechnology, 129, 290-307.
LEDIN, J., STAATZ, W., LI, J. P., GOTTE, M., SELLECK, S., KJELLEN, L. & SPILLMANN, D. 2004. Heparan sulfate structure in mice with genetically modified heparan sulfate production. J Biol Chem, 279, 42732-42741.
LERNER, I., HERMANO, E., ZCHARIA, E., RODKIN, D., BULVIK, R., DOVINER, V., RUBINSTEIN, A. M., ISHAI-MICHAELI, R., ATZMON, R., SHERMAN, Y., MEIROVITZ, A., PERETZ, T., VLODAVSKY, I. & ELKIN, M. 2011. Heparanase powers a chronic inflammatory circuit that promotes colitis-associated tumorigenesis in mice. J Clin Invest, 121, 1709-21.
LEVEUGLE, B., DING, W., DURKIN, J. T., MISTRETTA, S., EISLE, J., MATIC, M., SIMAN, R., GREENBERG, B. D. & FILLIT, H. M. 1997. Heparin promotes beta-secretase cleavage of the Alzheimer's amyloid precursor protein. Neurochem Int, 30, 543-8.
LEVY-ADAM, F., ABBOUD-JARROUS, G., GUERRINI, M., BECCATI, D., VLODAVSKY, I. & ILAN, N. 2005. Identification and characterization of

60
heparin/heparan sulfate binding domains of the endoglycosidase heparanase. The Journal of Biological Chemistry, 280, 20457-66.
LEVY-ADAM, F., FELD, S., SUSS-TOBY, E., VLODAVSKY, I. & ILAN, N. 2008. Heparanase facilitates cell adhesion and spreading by clustering of cell surface heparan sulfate proteoglycans. PLoS ONE, 3, e2319.
LEVY-ADAM, F., ILAN, N. & VLODAVSKY, I. 2010. Tumorigenic and adhesive properties of heparanase. Seminars in cancer biology, 20, 153-60.
LEY, K., LAUDANNA, C., CYBULSKY, M. I. & NOURSHARGH, S. 2007. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nature reviews. Immunology, 7, 678-89.
LI, J. P., GALVIS, M. L., GONG, F., ZHANG, X., ZCHARIA, E., METZGER, S., VLODAVSKY, I., KISILEVSKY, R. & LINDAHL, U. 2005. In vivo fragmentation of heparan sulfate by heparanase overexpression renders mice resistant to amyloid protein A amyloidosis. Proc Natl Acad Sci U S A, 102, 6473-7.
LI, L., WANG, B., GAO, T., ZHANG, X., HAO, J. X., VLODAVSKY, I., WIESENFELD-HALLIN, Z., XU, X. J. & LI, J. P. 2012. Heparanase overexpression reduces carrageenan-induced mechanical and cold hypersensitivity in mice. Neuroscience letters, 511, 4-7.
LI, R. W., FREEMAN, C., YU, D., HINDMARSH, E. J., TYMMS, K. E., PARISH, C. R. & SMITH, P. N. 2008. Dramatic regulation of heparanase activity and angiogenesis gene expression in synovium from patients with rheumatoid arthritis. Arthritis Rheum, 58, 1590-600.
LIDER, O., BAHARAV, E., MEKORI, Y. A., MILLER, T., NAPARSTEK, Y., VLODAVSKY, I. & COHEN, I. R. 1989. Suppression of experimental autoimmune diseases and prolongation of allograft survival by treatment of animals with low doses of heparins. The Journal of clinical investigation, 83, 752-6.
LIND, T., TUFARO, F., MCCORMICK, C., LINDAHL, U. & LIDHOLT, K. 1998. The putative tumor suppressors EXT1 and EXT2 are glycosyltransferases required for the biosynthesis of heparan sulfate. The Journal of Biological Chemistry, 273, 26265-8.
LINDAHL, U., BACKSTROM, G., HOOK, M., THUNBERG, L., FRANSSON, L. A. & LINKER, A. 1979. Structure of the Antithrombin-Binding Site in Heparin. Proceedings of the National Academy of Sciences of the United States of America, 76, 3198-3202.
LINDAHL, U., BÄCKSTRÖM, G., THUNBERG, L. & LEDER, I. G. 1980. Evidence for a 3-O-sulfated D-glucosamine residue in the antithrombin-binding sequence of heparin. Proceedings of the National Academy of Sciences, 77, 6551-6555.
LINDAHL, U. & LI, J. P. 2009. Interactions between heparan sulfate and proteins-design and functional implications. Int Rev Cell Mol Biol, 276, 105-59.
LING, S., CLINE, E. N., HAUG, T. S., FOX, D. A. & HOLOSHITZ, J. 2013. Citrullinated calreticulin potentiates rheumatoid arthritis shared epitope signaling. Arthritis and rheumatism, 65, 618-26.
LORD, A., PHILIPSON, O., KLINGSTEDT, T., WESTERMARK, G., HAMMARSTROM, P., NILSSON, K. P. & NILSSON, L. N. 2011. Observations in APP bitransgenic mice suggest that diffuse and compact plaques form via independent processes in Alzheimer's disease. The American journal of pathology, 178, 2286-98.

61
LYON, M. & GALLAGHER, J. T. 1998. Bio-specific sequences and domains in heparan sulphate and the regulation of cell growth and adhesion. Matrix Biol., 17, 485-493.
MACCARANA, M., SAKURA, Y., TAWADA, A., YOSHIDA, K. & LINDAHL, U. 1996. Domain structure of heparan sulfates from bovine organs. J Biol Chem, 271, 17804-10.
MANGIONE, P., SUNDE, M., GIORGETTI, S., STOPPINI, M., ESPOSITO, G., GIANELLI, L., OBICI, L., ASTI, L., ANDREOLA, A., VIGLINO, P., MERLINI, G. & BELLOTTI, V. 2001. Amyloid fibrils derived from the apolipoprotein A1 Leu174Ser variant contain elements of ordered helical structure. Protein science : a publication of the Protein Society, 10, 187-99.
MASSENA, S., CHRISTOFFERSSON, G., HJERTSTROM, E., ZCHARIA, E., VLODAVSKY, I., AUSMEES, N., ROLNY, C., LI, J. P. & PHILLIPSON, M. 2010. A chemotactic gradient sequestered on endothelial heparan sulfate induces directional intraluminal crawling of neutrophils. Blood, 116 1924-1931.
MATSUNO, H., YUDOH, K., UZUKI, M., NAKAZAWA, F., SAWAI, T., YAMAGUCHI, N., OLSEN, B. R. & KIMURA, T. 2002. Treatment with the angiogenesis inhibitor endostatin: a novel therapy in rheumatoid arthritis. The Journal of rheumatology, 29, 890-5.
MATTHYS, P., VERMEIRE, K., MITERA, T., HEREMANS, H., HUANG, S., SCHOLS, D., DE WOLF-PEETERS, C. & BILLIAU, A. 1999. Enhanced autoimmune arthritis in IFN-gamma receptor-deficient mice is conditioned by mycobacteria in Freund's adjuvant and by increased expansion of Mac-1+ myeloid cells. Journal of immunology, 163, 3503-10.
MATZNER, Y., BAR-NER, M., YAHALOM, J., ISHAI-MICHAELI, R., FUKS, Z. & VLODAVSKY, I. 1985. Degradation of heparan sulfate in the subendothelial extracellular matrix by a readily released heparanase from human neutrophils. Possible role in invasion through basement membranes. J Clin Invest, 76, 1306-13.
MCCORMICK, C., DUNCAN, G., GOUTSOS, K. T. & TUFARO, F. 2000. The putative tumor suppressors EXT1 and EXT2 form a stable complex that accumulates in the Golgi apparatus and catalyzes the synthesis of heparan sulfate. Proceedings of the National Academy of Sciences of the United States of America, 97, 668-73.
MCCORMICK, C., LEDUC, Y., MARTINDALE, D., MATTISON, K., ESFORD, L. E., DYER, A. P. & TUFARO, F. 1998. The putative tumour suppressor EXT1 alters the expression of cell-surface heparan sulfate. Nature genetics, 19, 158-61.
MCKENZIE, E., TYSON, K., STAMPS, A., SMITH, P., TURNER, P., BARRY, R., HIRCOCK, M., PATEL, S., BARRY, E., STUBBERFIELD, C., TERRETT, J. & PAGE, M. 2000. Cloning and expression profiling of Hpa2, a novel mammalian heparanase family member. Biochem Biophys Res Commun, 276, 1170-7.
MCNAMEE, K., WILLIAMS, R. & SEED, M. 2015. Animal models of rheumatoid arthritis: How informative are they? European journal of pharmacology, 759, 278-86.
MERLINI, G. & WESTERMARK, P. 2004. The systemic amyloidoses: clearer understanding of the molecular mechanisms offers hope for more effective therapies. Journal of internal medicine, 255, 159-78.
MORIMOTO-TOMITA, M., UCHIMURA, K., WERB, Z., HEMMERICH, S. & ROSEN, S. D. 2002. Cloning and characterization of two extracellular heparin-

62
degrading endosulfatases in mice and humans. The Journal of Biological Chemistry, 277, 49175-85.
NADANAKA, S. & KITAGAWA, H. 2008. Heparan sulphate biosynthesis and disease. Journal of biochemistry, 144, 7-14.
NADANAKA, S., ZHOU, S., KAGIYAMA, S., SHOJI, N., SUGAHARA, K., SUGIHARA, K., ASANO, M. & KITAGAWA, H. 2013. EXTL2, a member of the EXT family of tumor suppressors, controls glycosaminoglycan biosynthesis in a xylose kinase-dependent manner. The Journal of Biological Chemistry, 288, 9321-33.
NADIR, Y., BRENNER, B., ZETSER, A., ILAN, N., SHAFAT, I., ZCHARIA, E., GOLDSHMIDT, O. & VLODAVSKY, I. 2006. Heparanase induces tissue factor expression in vascular endothelial and cancer cells. Journal of thrombosis and haemostasis : JTH, 4, 2443-51.
NAJJAM, S., MULLOY, B., THEZE, J., GORDON, M., GIBBS, R. & RIDER, C. C. 1998. Further characterization of the binding of human recombinant interleukin 2 to heparin and identification of putative binding sites. Glycobiology, 8, 509-16.
NAKAJIMA, M., IRIMURA, T., DI FERRANTE, N. & NICOLSON, G. L. 1984. Metastatic melanoma cell heparanase. Characterization of heparan sulfate degradation fragments produced by B16 melanoma endoglucuronidase. J Biol Chem, 259, 2283-90.
NAKANISHI, H. 2003. Microglial functions and proteases. Mol Neurobiol, 27, 163-76.
NAPARSTEK, Y., COHEN, I. R., FUKS, Z. & VLODAVSKY, I. 1984. Activated T lymphocytes produce a matrix-degrading heparan sulphate endoglycosidase. Nature, 310, 241-4.
NATIONAL COLLABORATING CENTRE FOR CHRONIC CONDITIONS (UK) 2009. Rheumatoid Arthritis: National Clinical Guideline for Management and Treatment in Adults. Rheumatoid Arthritis: National Clinical Guideline for Management and Treatment in Adults. London: Royal College of Physicians (UK)
NEPOM, G. T., BYERS, P., SEYFRIED, C., HEALEY, L. A., WILSKE, K. R., STAGE, D. & NEPOM, B. S. 1989. HLA genes associated with rheumatoid arthritis. Identification of susceptibility alleles using specific oligonucleotide probes. Arthritis and rheumatism, 32, 15-21.
NICCOLI, T. & PARTRIDGE, L. 2012. Ageing as a Risk Factor for Disease. Current Biology, 22, R741-R752.
NISHIMURA, K., SUGIYAMA, D., KOGATA, Y., TSUJI, G., NAKAZAWA, T., KAWANO, S., SAIGO, K., MORINOBU, A., KOSHIBA, M., KUNTZ, K. M., KAMAE, I. & KUMAGAI, S. 2007. Meta-analysis: diagnostic accuracy of anti-cyclic citrullinated peptide antibody and rheumatoid factor for rheumatoid arthritis. Annals of internal medicine, 146, 797-808.
NOBORN, F., ANCSIN, J. B., UBHAYASEKERA, W., KISILEVSKY, R. & LI, J. P. 2012. Heparan sulfate dissociates serum amyloid A (SAA) from acute-phase high-density lipoprotein, promoting SAA aggregation. The Journal of biological chemistry, 287, 25669-77.
NYSTROM, S. N. & WESTERMARK, G. T. 2012. AA-Amyloid is cleared by endogenous immunological mechanisms. Amyloid : the international journal of experimental and clinical investigation : the official journal of the International Society of Amyloidosis, 19, 138-45.
O'CALLAGHAN, P., LI, J. P., LANNFELT, L., LINDAHL, U. & ZHANG, X. 2015. Microglial Heparan Sulfate Proteoglycans Facilitate the Cluster-of-

63
Differentiation 14 (CD14)/Toll-like Receptor 4 (TLR4)-Dependent Inflammatory Response. The Journal of Biological Chemistry, 290, 14904-14.
O'CALLAGHAN, P., SANDWALL, E., LI, J. P., YU, H., RAVID, R., GUAN, Z. Z., VAN KUPPEVELT, T. H., NILSSON, L. N., INGELSSON, M., HYMAN, B. T., KALIMO, H., LINDAHL, U., LANNFELT, L. & ZHANG, X. 2008. Heparan Sulfate Accumulation with Abeta Deposits in Alzheimer's Disease and Tg2576 Mice is Contributed by Glial Cells. Brain Pathol, 18, 548-61.
O'NEILL, L. A., GOLENBOCK, D. & BOWIE, A. G. 2013. The history of Toll-like receptors - redefining innate immunity. Nature reviews. Immunology, 13, 453-60.
OGREN, S. & LINDAHL, U. 1975. Cleavage of macromolecular heparin by an enzyme from mouse mastocytoma. J Biol Chem, 250, 2690-7.
OKADA, Y., YAMADA, S., TOYOSHIMA, M., DONG, J., NAKAJIMA, M. & SUGAHARA, K. 2002. Structural recognition by recombinant human heparanase that plays critical roles in tumor metastasis:Hierarchical sulfate groups with differential effects and the essential target disulfated trisaccharide sequence. J Biol Chem, 3, 3.
OSMAN, N. M., NAORA, H. & OTANI, H. 2004. Glycosyltransferase encoding gene EXTL3 is differentially expressed in the developing and adult mouse cerebral cortex. Brain research. Developmental brain research, 151, 111-7.
PAJKRT, D., DORAN, J. E., KOSTER, F., LERCH, P. G., ARNET, B., VAN DER POLL, T., TEN CATE, J. W. & VAN DEVENTER, S. J. 1996. Antiinflammatory effects of reconstituted high-density lipoprotein during human endotoxemia. The Journal of experimental medicine, 184, 1601-8.
PARISH, C. R., FREEMAN, C. & HULETT, M. D. 2001. Heparanase: a key enzyme involved in cell invasion. Biochim Biophys Acta, 1471, M99-M108.
PARK, J. H., JEONG, S. Y., CHOI, A. J. & KIM, S. J. 2015. Lipopolysaccharide directly stimulates Th17 differentiation in vitro modulating phosphorylation of RelB and NF-kappaB1. Immunology letters, 165, 10-9.
PERRIMON, N. & BERNFIELD, M. 2000. Specificities of heparan sulphate proteoglycans in developmental processes. Nature, 404, 725-8.
PETERSON, S. & LIU, J. 2012. Deciphering mode of action of heparanase using structurally defined oligosaccharides. The Journal of Biological Chemistry, 287, 34836-43.
PETERSON, S. B. & LIU, J. 2010. Unraveling the specificity of heparanase utilizing synthetic substrates. J Biol Chem, 285, 14504-13.
PETROVSKY, N. & AGUILAR, J. C. 2004. Vaccine adjuvants: current state and future trends. Immunology and cell biology, 82, 488-96.
PHILIPSON, O., HAMMARSTROM, P., NILSSON, K. P., PORTELIUS, E., OLOFSSON, T., INGELSSON, M., HYMAN, B. T., BLENNOW, K., LANNFELT, L., KALIMO, H. & NILSSON, L. N. 2009. A highly insoluble state of Abeta similar to that of Alzheimer's disease brain is found in Arctic APP transgenic mice. Neurobiology of aging, 30, 1393-405.
PHILLIPSON, M., HEIT, B., COLARUSSO, P., LIU, L., BALLANTYNE, C. M. & KUBES, P. 2006. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med, 203, 2569-75.
PIKAS, D. S., LI, J. P., VLODAVSKY, I. & LINDAHL, U. 1998. Substrate specificity of heparanases from human hepatoma and platelets. J Biol Chem, 273, 18770-7.
PONIGHAUS, C., AMBROSIUS, M., CASANOVA, J. C., PRANTE, C., KUHN, J., ESKO, J. D., KLEESIEK, K. & GOTTING, C. 2007. Human

64
xylosyltransferase II is involved in the biosynthesis of the uniform tetrasaccharide linkage region in chondroitin sulfate and heparan sulfate proteoglycans. The Journal of Biological Chemistry, 282, 5201-6.
PURUSHOTHAMAN, A., CHEN, L., YANG, Y. & SANDERSON, R. D. 2008. Heparanase stimulation of protease expression implicates it as a master regulator of the aggressive tumor phenotype in myeloma. J Biol Chem, 283, 32628-36.
RAJASEKARAN, D., KEELER, C., SYED, M. A., JONES, M. C., HARRISON, J. K., WU, D., BHANDARI, V., HODSDON, M. E. & LOLIS, E. J. 2012. A model of GAG/MIP-2/CXCR2 interfaces and its functional effects. Biochemistry, 51, 5642-54.
RAMANI, V. C., YANG, Y., REN, Y., NAN, L. & SANDERSON, R. D. 2011. Heparanase plays a dual role in driving hepatocyte growth factor (HGF) signaling by enhancing HGF expression and activity. The Journal of Biological Chemistry, 286, 6490-9.
RIGANTE, D., FREDIANI, B. & CANTARINI, L. 2016. A Comprehensive Overview of the Hereditary Periodic Fever Syndromes. Clinical reviews in allergy & immunology.
ROCCARO, A. M., RUSSO, F., CIRULLI, T., DI PIETRO, G., VACCA, A. & DAMMACCO, F. 2005. Antiangiogenesis for rheumatoid arthritis. Current drug targets. Inflammation and allergy, 4, 27-30.
RONG, J., HABUCHI, H., KIMATA, K., LINDAHL, U. & KUSCHE-GULLBERG, M. 2000. Expression of heparan sulphate L-iduronyl 2-O-sulphotransferase in human kidney 293 cells results in increased D-glucuronyl 2-O-sulphation [In Process Citation]. Biochem J, 346 Pt 2, 463-8.
RONG, J., HABUCHI, H., KIMATA, K., LINDAHL, U. & KUSCHE-GULLBERG, M. 2001. Substrate specificity of the heparan sulfate hexuronic acid 2-O-sulfotransferase. Biochemistry, 40, 5548-55.
ROSENBERG, R. D., JORDAN, R. E., FAVREAU, L. V. & LAM, L. H. 1979. Highly active heparin species with multiple binding sites for antithrombin. Biochemical and Biophysical Research Communications, 86, 1319-24.
ROTHSCHILD, B. M., TURNER, K. R. & DELUCA, M. A. 1988. Symmetrical erosive peripheral polyarthritis in the Late Archaic Period of Alabama. Science, 241, 1498-501.
SALEK-ARDAKANI, S., ARRAND, J. R., SHAW, D. & MACKETT, M. 2000. Heparin and heparan sulfate bind interleukin-10 and modulate its activity. Blood, 96, 1879-88.
SANDWALL, E., BODEVIN, S., NASSER, N. J., NEVO, E., AVIVI, A., VLODAVSKY, I. & LI, J. P. 2009. Molecular Structure of Heparan Sulfate from Spalax: IMPLICATIONS OF HEPARANASE AND HYPOXIA. J Biol Chem, 284, 3814-22.
SANTIAGO, P., ROIG-LOPEZ, J. L., SANTIAGO, C. & GARCIA-ARRARAS, J. E. 2000. Serum amyloid A protein in an echinoderm: its primary structure and expression during intestinal regeneration in the sea cucumber Holothuria glaberrima. The Journal of experimental zoology, 288, 335-44.
SANTIAGO-CARDONA, P. G., BERRIOS, C. A., RAMIREZ, F. & GARCIA-ARRARAS, J. E. 2003. Lipopolysaccharides induce intestinal serum amyloid A expression in the sea cucumber Holothuria glaberrima. Developmental and comparative immunology, 27, 105-10.
SCHMIDT, E. P., YANG, Y., JANSSEN, W. J., GANDJEVA, A., PEREZ, M. J., BARTHEL, L., ZEMANS, R. L., BOWMAN, J. C., KOYANAGI, D. E., YUNT, Z. X., SMITH, L. P., CHENG, S. S., OVERDIER, K. H., THOMPSON,

65
K. R., GERACI, M. W., DOUGLAS, I. S., PEARSE, D. B. & TUDER, R. M. 2012. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nature medicine, 18.
SCHOLEFIELD, Z., YATES, E. A., WAYNE, G., AMOUR, A., MCDOWELL, W. & TURNBULL, J. E. 2003. Heparan sulfate regulates amyloid precursor protein processing by BACE1, the Alzheimer's beta-secretase. J Cell Biol, 163, 97-107.
SCHUNA, A. A. 2011. Rheumatoid arthritis. Pharmacotherapy: A Pathophysiologic Approach. 8th ed. New York, NY: McGraw-Hill.
SERHAN, C. N., WARD, P. A. & GILROY, D. W. 2010. Fundamentals of Inflammation, New York, Cambridge University Press.
SERNATINGER, J., HOFFMAN, A., HARDMAN, D., KANE, J. P. & LEVY, J. A. 1988. Neutralization of mouse xenotropic virus by lipoproteins involves binding to the virions. The Journal of general virology, 69 ( Pt 10), 2657-61.
SHAFAT, I., VLODAVSKY, I. & ILAN, N. 2006. Characterization of mechanisms involved in secretion of active heparanase. J Biol Chem, 281, 23804-11.
SHWORAK, N. W., LIU, J., FRITZE, L. M. S., SCHWARTZ, J. J., ZHANG, L., LOGEART, D. & ROSENBERG, R. D. 1997. Molecular Cloning and Expression of Mouse and Human cDNAs Encoding Heparan Sulfate D-Glucosaminyl 3-O-Sulfotransferase. J. Biol. Chem., 272, 28008-28019.
SHWORAK, N. W., LIU, J., PETROS, L. M., ZHANG, L., KOBAYASHI, M., COPELAND, N. G., JENKINS, N. A. & ROSENBERG, R. D. 1999. Multiple isoforms of heparan sulfate D-glucosaminyl 3-O-sulfotransferase. Isolation, characterization, and expression of human cDNAs and identification of distinct genomic loci. J. Biol. Chem., 274, 5170-5184.
SIPE, J. D., BENSON, M. D., BUXBAUM, J. N., IKEDA, S., MERLINI, G., SARAIVA, M. J. & WESTERMARK, P. 2014. Nomenclature 2014: Amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid : the international journal of experimental and clinical investigation : the official journal of the International Society of Amyloidosis, 21, 221-4.
SKAPER, S. D. 2012. Alzheimer's disease and amyloid: culprit or coincidence? International review of neurobiology, 102, 277-316.
SKINNER, M., SHIRAHAMA, T., BENSON, M. D. & COHEN, A. S. 1977. Murine amyloid protein AA in casein-induced experimental amyloidosis. Laboratory investigation; a journal of technical methods and pathology, 36, 420-7.
SNOW, A. D., BRAMSON, R., MAR, H., WIGHT, T. N. & KISILEVSKY, R. 1991. A temporal and ultrastructural relationship between heparan sulfate proteoglycans and AA amyloid in experimental amyloidosis. J Histochem Cytochem, 39, 1321-30.
SNOW, A. D. & KISILEVSKY, R. 1985. Temporal relationship between glycosaminoglycan accumulation and amyloid deposition during experimental amyloidosis. A histochemical study. Laboratory investigation; a journal of technical methods and pathology, 53, 37-44.
SNOW, A. D., WILLMER, J. & KISILEVSKY, R. 1987a. A close ultrastructural relationship between sulfated proteoglycans and AA amyloid fibrils. Lab Invest, 57, 687-98.
SNOW, A. D., WILLMER, J. P. & KISILEVSKY, R. 1987b. Sulfated glycosaminoglycans in Alzheimer's disease. Human pathology, 18, 506-10.
SOLITO, E. & SASTRE, M. 2012. Microglia function in Alzheimer's disease. Frontiers in pharmacology, 3, 14.
SONG, H. H. & FILMUS, J. 2002. The role of glypicans in mammalian development. Biochimica et Biophysica Acta, 1573, 241-6.

66
SPILLMANN, D., WITT, D. & LINDAHL, U. 1998. Defining the Interleukin-8-binding Domain of Heparan Sulfate. J. Biol. Chem., 273, 15487-15493.
STEFKA, A. T., FEEHLEY, T., TRIPATHI, P., QIU, J., MCCOY, K., MAZMANIAN, S. K., TJOTA, M. Y., SEO, G. Y., CAO, S., THERIAULT, B. R., ANTONOPOULOS, D. A., ZHOU, L., CHANG, E. B., FU, Y. X. & NAGLER, C. R. 2014. Commensal bacteria protect against food allergen sensitization. Proceedings of the National Academy of Sciences of the United States of America, 111, 13145-50.
STOLER-BARAK, L., PETROVICH, E., AYCHEK, T., GUREVICH, I., TAL, O., HATZAV, M., ILAN, N., FEIGELSON, S. W., SHAKHAR, G., VLODAVSKY, I. & ALON, R. 2015. Heparanase of murine effector lymphocytes and neutrophils is not required for their diapedesis into sites of inflammation. FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 29, 2010-21.
STRINGER, S. E., NELSON, M. S. & GUPTA, P. 2003. Identification of an MIP-1alpha -binding heparan sulfate oligosaccharide that supports long-term in vitro maintenance of human LTC-ICs. Blood, 101, 2243-5.
SUGAHARA, K. & KITAGAWA, H. 2000. Recent advances in the study of the biosynthesis and functions of sulfated glycosaminoglycans. Curr Opin Struct Biol, 10, 518-27.
SUZMAN, R. & BEARD, J. 2011. Global health and ageing. 11. Available: http://www.who.int/ageing/publications/global_health.pdf.
TAM, S. P., KISILEVSKY, R. & ANCSIN, J. B. 2008. Acute-phase-HDL remodeling by heparan sulfate generates a novel lipoprotein with exceptional cholesterol efflux activity from macrophages. PLoS One, 3, e3867.
THOMAS, R. 2013. Dendritic cells and the promise of antigen-specific therapy in rheumatoid arthritis. Arthritis research & therapy, 15, 204.
THUNBERG, L., LINDAHL, U., TENGBLAD, A., LAURENT, T. C. & JACKSON, C. M. 1979. On the molecular-weight-dependence of the anticoagulant activity of heparin. Biochem J, 181, 241-43.
TOWN, T., NIKOLIC, V. & TAN, J. 2005. The microglial "activation" continuum: from innate to adaptive responses. J Neuroinflammation, 2, 24.
TOYOSHIMA, M. & NAKAJIMA, M. 1999. Human heparanase. Purification, characterization, cloning, and expression. J Biol Chem, 274, 24153-60.
TRENTHAM, D. E., TOWNES, A. S. & KANG, A. H. 1977. Autoimmunity to type II collagen an experimental model of arthritis. The Journal of experimental medicine, 146, 857-68.
VALLA, S., LI, J., ERTESVAG, H., BARBEYRON, T. & LINDAHL, U. 2001. Hexuronyl C5-epimerases in alginate and glycosaminoglycan biosynthesis. Biochimie, 83, 819-30.
VAN HORSSEN, J., WESSELING, P., VAN DEN HEUVEL, L. P., DE WAAL, R. M. & VERBEEK, M. M. 2003. Heparan sulphate proteoglycans in Alzheimer's disease and amyloid-related disorders. Lancet Neurol, 2, 482-92.
VIVES, R. R., SADIR, R., IMBERTY, A., RENCUROSI, A. & LORTAT-JACOB, H. 2002. A kinetics and modeling study of RANTES(9-68) binding to heparin reveals a mechanism of cooperative oligomerization. Biochemistry, 41, 14779-89.
VLODAVSKY, I., ELDOR, A., HAIMOVITZ-FRIEDMAN, A., MATZNER, Y., ISHAI-MICHAELI, R., LIDER, O., NAPARSTEK, Y., COHEN, I. R. & FUKS, Z. 1992. Expression of heparanase by platelets and circulating cells of the immune system: possible involvement in diapedesis and extravasation. Invasion Metastasis, 12, 112-27.

67
VLODAVSKY, I., ELKIN, M., ABBOUD-JARROUS, G., LEVI-ADAM, F., FUKS, L., SHAFAT, I. & ILAN, N. 2008. Heparanase: one molecule with multiple functions in cancer progression. Connect Tissue Res, 49, 207-10.
VLODAVSKY, I., FRIEDMANN, Y., ELKIN, M., AINGORN, H., ATZMON, R., ISHAI-MICHAELI, R., BITAN, M., PAPPO, O., PERETZ, T., MICHAL, I., SPECTOR, L. & PECKER, I. 1999. Mammalian heparanase: gene cloning, expression and function in tumor progression and metastasis [see comments]. Nat Med, 5, 793-802.
VLODAVSKY, I., FUKS, Z., BAR-NER, M., ARIAV, Y. & SCHIRRMACHER, V. 1983. Lymphoma cell-mediated degradation of sulfated proteoglycans in the subendothelial extracellular matrix: relationship to tumor cell metastasis. Cancer Res, 43, 2704-11.
VLODAVSKY, I., GOLDSHMIDT, O., ZCHARIA, E., METZGER, S., CHAJEK-SHAUL, T., ATZMON, R., GUATTA-RANGINI, Z. & FRIEDMANN, Y. 2001. Molecular properties and involvement of heparanase in cancer progression and normal development. Biochimie, 83, 831-9.
VLODAVSKY, I., IOZZO, R. V. & SANDERSON, R. D. 2013. Heparanase: multiple functions in inflammation, diabetes and atherosclerosis. Matrix biology : journal of the International Society for Matrix Biology, 32, 220-2.
VREYS, V., DELANDE, N., ZHANG, Z., COOMANS, C., ROEBROEK, A., DURR, J. & DAVID, G. 2005. Cellular uptake of mammalian heparanase precursor involves low density lipoprotein receptor-related proteins, mannose 6-phosphate receptors, and heparan sulfate proteoglycans. The Journal of Biological Chemistry, 280, 33141-8.
WALLBERG-JONSSON, S., DAHLEN, G., JOHNSON, O., OLIVECRONA, G. & RANTAPAA-DAHLQVIST, S. 1996. Lipoprotein lipase in relation to inflammatory activity in rheumatoid arthritis. Journal of internal medicine, 240, 373-80.
WANG, B., JIA, J., ZHANG, X., ZCHARIA, E., VLODAVSKY, I., PEJLER, G. & LI, J. P. 2011. Heparanase affects secretory granule homeostasis of murine mast cells through degrading heparin. The Journal of allergy and clinical immunology.
WANG, B., TAN, Y. X., JIA, J., DIGRE, A., ZHANG, X., VLODAVSKY, I. & LI, J. P. 2012. Accelerated Resolution of AA Amyloid in Heparanase Knockout Mice Is Associated with Matrix Metalloproteases. PLoS One, 7, e39899.
WANG, C. R., CHEN, S. Y., WU, C. L., LIU, M. F., JIN, Y. T., CHAO, L. & CHAO, J. 2005. Prophylactic adenovirus-mediated human kallistatin gene therapy suppresses rat arthritis by inhibiting angiogenesis and inflammation. Arthritis and rheumatism, 52, 1319-24.
WARREN, G. W., ALBERG, A. J., KRAFT, A. S. & CUMMINGS, K. M. 2014. The 2014 Surgeon General's report: "The health consequences of smoking--50 years of progress": a paradigm shift in cancer care. Cancer, 120, 1914-6.
WEINBAUM, S., TARBELL, J. M. & DAMIANO, E. R. 2007. The structure and function of the endothelial glycocalyx layer. Annual review of biomedical engineering, 9, 121-67.
WESTERMARK, G. T., FANDRICH, M. & WESTERMARK, P. 2015. AA amyloidosis: pathogenesis and targeted therapy. Annu Rev Pathol, 10, 321-44.
WESTPHAL, O. & LUDERITZ, O. 1954. Chemische Erforschung Von Lipopolysacchariden Gramnegativer Bakterien. Angewandte Chemie, 66, 407-417.
WILLIAMS, G. C. 1957. Pleiotropy, Natural-Selection, and the Evolution of Senescence. Evolution, 11, 398-411.

68
WOOLEY, P. H., LUTHRA, H. S., GRIFFITHS, M. M., STUART, J. M., HUSE, A. & DAVID, C. S. 1985. Type II collagen-induced arthritis in mice. IV. Variations in immunogenetic regulation provide evidence for multiple arthritogenic epitopes on the collagen molecule. Journal of immunology, 135, 2443-51.
WU, L., VIOLA, C. M., BRZOZOWSKI, A. M. & DAVIES, G. J. 2015. Structural characterization of human heparanase reveals insights into substrate recognition. Nature structural & molecular biology, 22, 1016-22.
WYSS-CORAY, T. 2016. Ageing, neurodegeneration and brain rejuvenation. Nature, 539, 180-186.
YOUNG, E. 2008. The anti-inflammatory effects of heparin and related compounds. Thrombosis Research, 122, 743-52.
ZCHARIA, E., JIA, J., ZHANG, X., BARAZ, L., LINDAHL, U., PERETZ, T., VLODAVSKY, I. & LI, J. P. 2009. Newly generated heparanase knock-out mice unravel co-regulation of heparanase and matrix metalloproteinases. PLoS One, 4, e5181.
ZCHARIA, E., METZGER, S., CHAJEK-SHAUL, T., AINGORN, H., ELKIN, M., FRIEDMANN, Y., WEINSTEIN, T., LI, J. P., LINDAHL, U. & VLODAVSKY, I. 2004. Transgenic expression of mammalian heparanase uncovers physiological functions of heparan sulfate in tissue morphogenesis, vascularization, and feeding behavior. Faseb J, 18, 252-63.
ZETSER, A., BASHENKO, Y., EDOVITSKY, E., LEVY-ADAM, F., VLODAVSKY, I. & ILAN, N. 2006. Heparanase induces vascular endothelial growth factor expression: correlation with p38 phosphorylation levels and Src activation. Cancer research, 66, 1455-63.
ZETSER, A., LEVY-ADAM, F., KAPLAN, V., GINGIS-VELITSKI, S., BASHENKO, Y., SCHUBERT, S., FLUGELMAN, M. Y., VLODAVSKY, I. & ILAN, N. 2004. Processing and activation of latent heparanase occurs in lysosomes. J Cell Sci, 117, 2249-58.
ZHANG, X. & LI, J. P. 2010. Heparan sulfate proteoglycans in amyloidosis. Prog Mol Biol Transl Sci, 93, 309-34.
ZHANG, X., WANG, B., O'CALLAGHAN, P., HJERTSTROM, E., JIA, J., GONG, F., ZCHARIA, E., NILSSON, L. N., LANNFELT, L., VLODAVSKY, I., LINDAHL, U. & LI, J. P. 2012. Heparanase overexpression impairs inflammatory response and macrophage-mediated clearance of amyloid-beta in murine brain. Acta neuropathologica, 124, 465-78.
ZHANG, X., XU, S., TAN, Q. & LIU, L. 2013. High expression of heparanase-2 is an independent prognostic parameter for favorable survival in gastric cancer patients. Cancer epidemiology, 37, 1010-3.
ZUROFF, L., DALEY, D., BLACK, K. L. & KORONYO-HAMAOUI, M. 2017. Clearance of cerebral Abeta in Alzheimer's disease: reassessing the role of microglia and monocytes. Cellular and molecular life sciences : CMLS.


Acta Universitatis UpsaliensisDigital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Medicine 1303
Editor: The Dean of the Faculty of Medicine
A doctoral dissertation from the Faculty of Medicine, UppsalaUniversity, is usually a summary of a number of papers. A fewcopies of the complete dissertation are kept at major Swedishresearch libraries, while the summary alone is distributedinternationally through the series Digital ComprehensiveSummaries of Uppsala Dissertations from the Faculty ofMedicine. (Prior to January, 2005, the series was publishedunder the title “Comprehensive Summaries of UppsalaDissertations from the Faculty of Medicine”.)
Distribution: publications.uu.seurn:nbn:se:uu:diva-315699
ACTAUNIVERSITATIS
UPSALIENSISUPPSALA
2017