immunodeficiencies and autoimmune diseases martin liška
TRANSCRIPT

Immunodeficiencies and
autoimmune diseases
Martin Liška

Immunodeficiencies
• Humoral – innate immunity - complement, MBL acquired immunity – immunoglobulins (B lymphocytes)
• Cell mediated immunity – innate immunity – phagocytes - acquired immunity – T lymphocytes
• Primary – congenital, genetically defined, symptoms predominantly at early age
• Secondary – the onset of symptoms at any age chronic diseases effect of irradiation immunosuppression surgical intervention, injuries stress

Immunodeficiencies – critical life periods in respect to symptoms onset
• Newborn age - severe primary disorders of cell mediated immunity
• 6 mth. – 2 yrs. – severe humoral immunodeficiencies cong./transient
• 3 - 5 yrs. – transient and selective humoral immunodeficiencies, secondary immunodeficiencies
• 15 – 20 yrs. – hormonal instability, thymus involution, life-style changes, some typical infections first symptoms of CVID
• Middleage – often excessive workload, stress first symptoms of autoimmune disorders (also immunodeficiency)
• Advanced and old age – rather symptoms of severe secondary immunodeficiencies,
repercussion of functional disorders

Immunodeficiencies – major clinical features
• Antibodies - microbial infections (encapsulated bacteria) respiratory - pneumonia, sinusitis, otitis
GIT – diarrhea• Complement system – microbial infections (pyogenic), sepsis edema (HAE) – C1-INH deficiency• T lymphocytes - bacterial, fungal, viral GIT – diarrhoea respiratory – pneumonia, sinusitis• Phagocytes - abscesses, recurrent purulent skin infections granulomatous inflammation

I. Primary immunodeficiencies – phagocytic cell defects
1/ Quantitative – decreased numbers of granulocytes –neutrophil elastase mutation
Congenital chronic agranulocytosis
Cyclic agranulocytosis (neutropenia)

I. Primary immunodeficiencies – phagocytic cell defects
2/ Qualitative – phagocytes functional disorders, various enzyme deficits, inability of phagocytes to degrade the ingested material
a/ Chronic Granulomatous Disease (CGD)• Approximately in 60% X-linked • Enzymatic inability to generate toxic oxygen metabolites (H2O2) during
oxygen consumption) - result of defect in neutrophilic cytochrome b (part of complex containing NADPH oxidase)
• Inability to kill bacteria such as Staph.aureus, Pseud.aeruginosa that produce catalase
• Clinical features: granulomas of skin, organs• Treatment: long-term ATB administration

II. Primary immunodeficiencies – B cell disorders
Bruton’s X-linked hypogamaglobulinemia• Blockage of the maturation of pre-B lymphocytes into B lymphocytes (tyrosine
kinase defect)• Undetectable or very low serum levels of Ig• Pneumonia, pyogenic otitis, complicated sinusitis, increased occurrence of
pulmonary fibrosis• Treatment: life-long IVIG substitution
CVID – Common Variable ImmunoDeficiency• B cell functional disorder, mostly low levels of IgG and IgA• Symptoms’ onset between 2nd and 3rd decade• Recurrent respiratory tract infections (pneumonia)• Treatment: IVIG substitution

II. Primary immunodeficiencies – B cell disorders
Selective IgA deficiency• Disorder of B cell function • Recurrent mild/moderate infections (respiratory, GIT, urinary tract) or
asymptomatic• Risk of reaction to live attenuated vaccines or generation of anti-IgA
antibodies after a blood transfusion
Selective IgG subclasses or specific IgG deficiency • B cell function disorder• Onset of symptoms in childhood, mostly respiratory tract infections
caused by encapsulated bacteria (H.influenzae, Pneumococci) Transient hypogammaglobulinemia of infancy

III. Primary immunodeficiencies – T cell disorders
diGeorge syndrome• Disorder of prethymocytes maturation due to absence of thymus (disorder
of development of 3rd and 4th branchial pouch) • Congenital heart diseases• The onset of symptoms after the birth – hypocalcemic spasms and
manifestations of cong.heart disease• Immunodeficiency could be only mild, the numbers of T lymphocytes later
usually become normal• Treatment symptomatic

IV. Primary immunodeficiencies – combined defects of T and B cells
SCID – Severe Combined ImmunoDeficiency• X-linked recessive or AR disease, combined disorder of humoral and cell
mediated immunity• Severe disorder (patients often die during first 2 years of life), onset of
symptoms soon after birth (severe diarrhoea, pneumonia, meningitis, BCGitis)
• Immunological features: typically lymphopenia and thymus hypoplasia• Forms: AR form – often enzymatic deficiency (ADA, PNP) that leads to
accumulation of metabolites toxic to DNA synthesis (lymphocytes) X-linked form – disorder of stem-cell Treatment: ATB, IVIG BMT is of critical significance gene therapy

V. Primary immunodeficiencies – complement system disorders
Hereditary angioedema (HAE)
• Absence or functional deficiency of C1-inhibitor• Anaphylactoid reactions with skin and/or mucosal (oral, laryngeal, gut)
edemas caused by activation of kinin system • Injuries or surgical/stomatological interventions are mostly triggering
factor • Laryngeal edemas could be life-threatening, immediate treatment is
necessary !• Treatment: preventive – androgens, EACA immediate – C1-INH concentrate or fresh frozen plasma administration - newly bradykinin receptor antagonist (icatibant)

Secondary immunodeficiencies
• Acute and chronic viral infections – infectious mononucleosis, influenza• Metabolic disorders – diabetes mellitus, uremia• Autoimmune diseases – autoantibodies against immunocompetent cells
(neutrophils, lymphocytes); autoimmune phenomena also after administration of certain drugs (e.g. oxacilin, quinidine)
• Chronic GIT diseases• Malignant diseases (leukemia)• Hypersplenism/asplenia• Burn, postoperative status, injuries• Severe nutritional disorders• Chronic infections• Ionizing radiation• Drug induced immunodeficiencies (chemotherapy)• Immunosupressissive therapyve therapy
• Chronic stress• Chronic exposure to harmful chemical substances

Secondary immunodeficiencies - A.I.D.S.
• Caused by retrovirus HIV 1 or HIV 2• Virus has a tropism for cells bearing CD4 surface marker (Th CD4+
lymphocytes); also affects macrophages and CNS cells• Viral genome transcribes into human DNA and infected cell provides viral
replication• Transmission: sexual intercourse contact with blood endouterine (mother – fetus, breast milk)• Phases: acute (flu-like sy) asymptomatic – several years, viral replication symptomatic – infections, autoimmune disorders, malignancy,
allergy final – systemic breakdown, opportune infections

A.I.D.S. - Treatment
• Reverse transcriptase inhibitors (e.g.zidovudine)• Protease inhibitors - block the viral protease enzyme• Combined drug therapy• Antimicrobial agents

AUTOIMMUNE DISEASES

Autoimmune disease
• Results from a failure of self-tolerance• Immunological tolerance is specific
unresponsiveness to an antigen• All individuals are tolerant of their own (self)
antigens

AUTOIMMUNE PATOLOGICAL RESPONSE- ETIOLOGY
• the diseases are chronic and usually irreversible• incidence: 5%-7% of population, higher frequencies in
women, increases with age• factors contribute to autoimmunity: - internal (HLA association, polymorphism of cytokine genes,
defect in genes regulating apoptosis, polymorphism in genes for TCR a H immunoglobulin chains, association with immunodeficiency, hormonal factors)
- external (infection, stress by activation of neuroendocrine axis and hormonal dysbalance, drug and ionization through modification of autoantigens)

CLINICAL CATEGORIES
• systemic - affect many organs and tissue
• organoleptic - affect predominantly one organ accompanied
by affection of other organs (inflammatory bowel diseases, celiac disease, AI hepatitis, pulmonary fibrosis)
• organ specific - affect one organ or group of organs connected
with development or function

SYSTEMIC AUTOIMMUNE DISEASES
• Systemic lupus erythematosus• Rheumathoid arthritis• Sjögren‘s syndrome• Dermatopolymyositis• Systemic sclerosis• Mixed connective tissue disease • Vasculitis

SYSTEMIC LUPUS ERYTHEMATOSUS
• chronic, inflammatory, multiorgan disorder
• autoantibodies react with nuclear material and attack cell function, immune complexes with dsDNA deposit in the tissue
• general symptoms: include malaise, fever, weight loss• multiple tissue are involved including the skin, mucosa,
kidney, joints, brain and cardiovascular system
• characteristic features: butterfly rash, renal involvement, CNS manifestation, pulmonary fibrosis

DIAGNOSTIC TESTS
• a elevated ESR (erythrocyte sedimentation rate), low CRP, trombocytopenia, leucopenia, hemolytic anemia, decreased levels of complement compounds (C4, C3), elevated serum Ig levels, immune complexes in serum

AUTOANTIBODIES
• Autoantibodies: ANA, dsDNA (double-stranded), ENA (SS-A/Ro, SS-A/La), Sm, against histones, phospholipids

RHEUMATOID ARTHRITIS
• chronic, inflammatory disease with systemic involvement• characterized by an inflammatory joint lesion in the synovial membrane,
destruction of the cartilage and bone, results in the joint deformation• clinical features: arthritis, fever, fatigue, weakness, weight loss• systemic features: vasculitis, pericarditis, uveitis, nodules under skin,
intersticial pulmonary fibrosis• diagnostic tests: elevated C- reactive protein and ESR, elevated serum gammaglobulin levels - autoantibodies against IgG = rheumatoid factor (RF), a-CCP (cyclic citrulline peptid), ANA - X-rays of hands and legs- show a periarticular porosis, marginal erosion

SJÖGREN‘S SYNDROME
• chronic inflammatory disease affecting exocrine glands• the primary targets are the lacrimal and salivary gland duct epithelium• general features: malaise, weakness, fever• primary syndrome - features: dry eyes and dry mouth, swollen salivary
glands, dryness of the nose, larynx, bronchi and vaginal mucosa, involvement kidney, central and periferal nervous system, arthritis
• secondary syndrome – is associated with others AI diseases (SLE, RA, sclerodermia, polymyositis, primary biliary cirhosis,AI thyroiditis)
• autoantibodies against ENA (SS-A, SS-B), ANA, RF• The Schirmer test - measures the production of tears

Heliotrope rash is a violaceous eruption on the upper eyelids, often with swelling
• a connective-tissue disease related to polymyositis (PM) that is characterized by inflammation of the muscles and the skin.
Gottron's sign is an erythematous, scaly eruption occurring in symmetric fashion over the MCP and interphalangeal joints
Dermatopolymyositis

Dermatopolymyositis
• Elevated creatine phosphokinase (CPK)
• muscle biopsy (a mixed B- and T-cell perivascular inflammatory infiltrate, perifascicular muscle fiber atrophy)
• EMG (electromyogram)• autoantibodies - ENA (Jo-1)

Systemic sclerosis
• sclerosis in the skin or other organs• Diffuse scleroderma (progressive systemic
sclerosis) is the most severe form, involves skin, will generally cause internal organ
damage (specifically the lungs and gastrointestinal tract)
• The limited form is much milder• The limited form is often referred to as CREST
syndrome (CREST is an acronym for the five main features: Calcinosis, Raynaud's syndrome, Esophageal dysmotility, Sclerodactyly, Telangiectasia
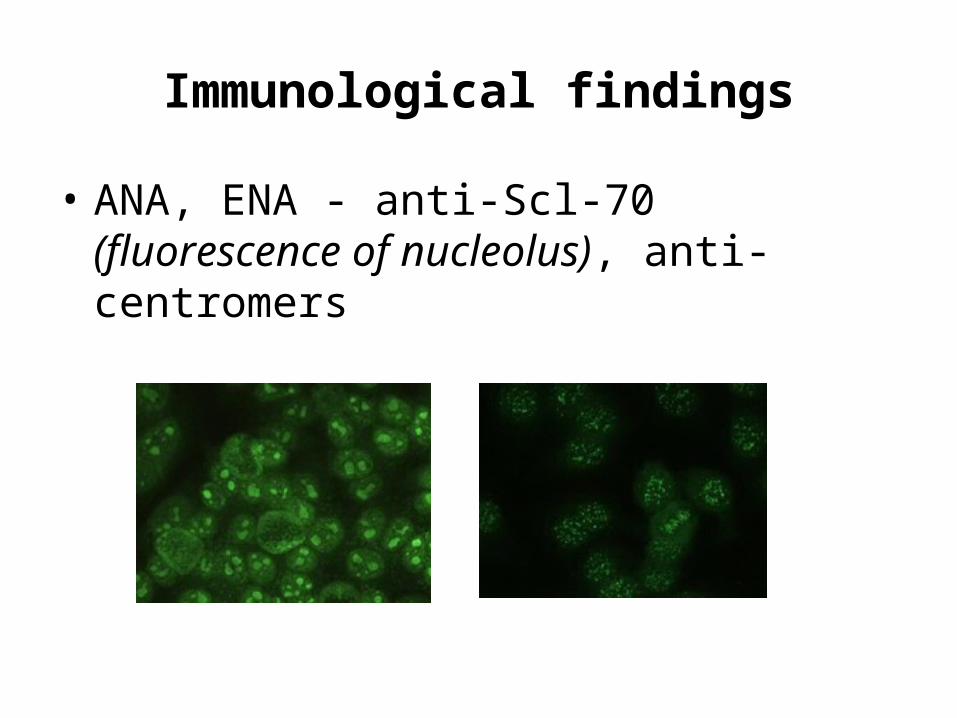
Immunological findings
• ANA, ENA - anti-Scl-70 (fluorescence of nucleolus), anti-centromers

Mixed connective tissue disease
• combines features of polymyositis, systemic lupus erythematosus, scleroderma, and dermatomyositis (overlap syndrome)
• Symptoms : joint pain/swelling, malaise, Raynaud phenomenon, muscle inflammation and sclerodactyly (thickening of the skin of the pads of the fingers)
• Distinguishing laboratory characteristics: a positive, speckled anti-nuclear antibody (ANA)
and anti-U1-RNP antibody (ENA)

Vasculitis
• characterized by inflammatory destruction of vessels leading to thrombosis and aneurysms• proliferation of the intimal part of blood-vessel wall
and fibrinoid necrosis• affect mostly lung, kidneys, skin
• diagnostic tests: elevated ESR, CRP, leucocytosis, biopsy of affected organ (necrosis, granulomas), angiography
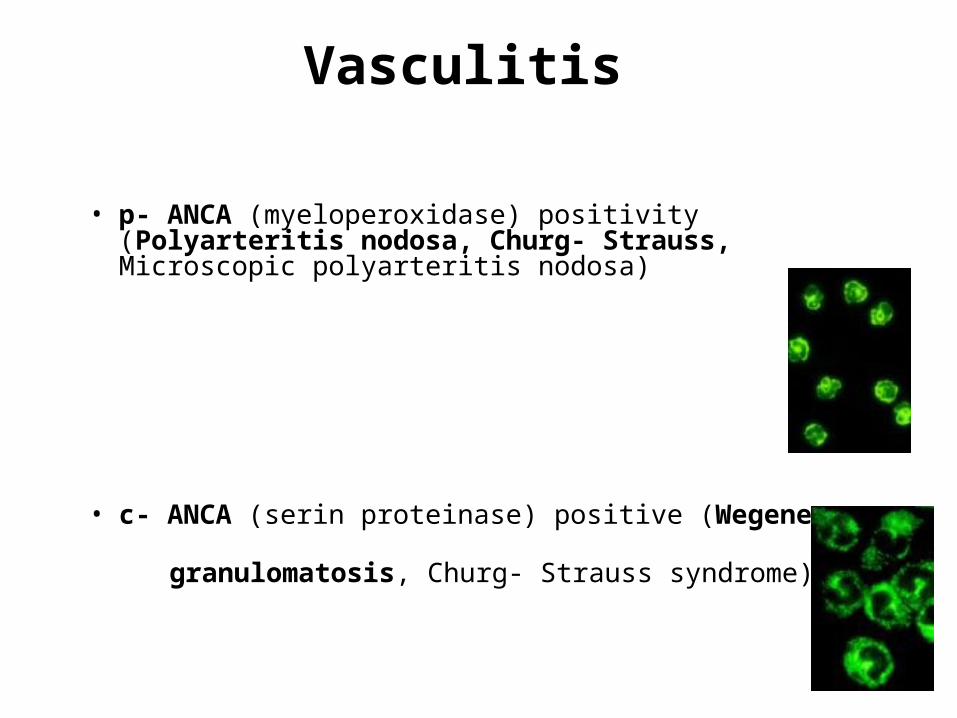
Vasculitis
• p- ANCA (myeloperoxidase) positivity (Polyarteritis nodosa, Churg- Strauss, Microscopic polyarteritis nodosa)
• c- ANCA (serin proteinase) positive (Wegener granulomatosis, Churg- Strauss syndrome)

Classification
• Large vessel vasculitis (Takayasu arteritis, Giant cell (temporal) arteritis)
• Medium vessel vasculitis (Polyarteritis nodosa, Wegener's granulomatosis, Kawasaki disease)
• Small vessel vasculitis (Churg-Strauss arteritis, Microscopic polyarteritis, Henoch-Schönlein purpura)
• Symptoms: fatigue, weakness, fever, arthralgias, abdominal pain, hypertension, renal insufficiency, and neurologic dysfunction

ORGANOLEPTIC AUTOIMMUNE DISEASES
• Ulcerative colitis• Crohn‘s disease• Autoimmune hepatitis• Primary biliary cirhosis• Pulmonary fibrosis

Ulcerative colitis
• chronic inflammation of the large intestine mucosa and submucosa
• features: diarrhea, bloody and mucus stools• extraintestinal features (arthritis, uveitis)• Autoantibodies: pANCA, a- large intestine

Crohn‘s disease
• the granulomatous inflammation of whole intestinal wall with ulceration and scarring that can result in abscess and fistula formation
• the inflammation of Crohn's disease the most commonly affects the terminal ileum, presents with diarrhea and is accompanied by extraintestinal features - iridocyclitis, uveitis, artritis, spondylitis
• antibodies against Saccharomyces cerevisiae (ASCA), a- pancreas
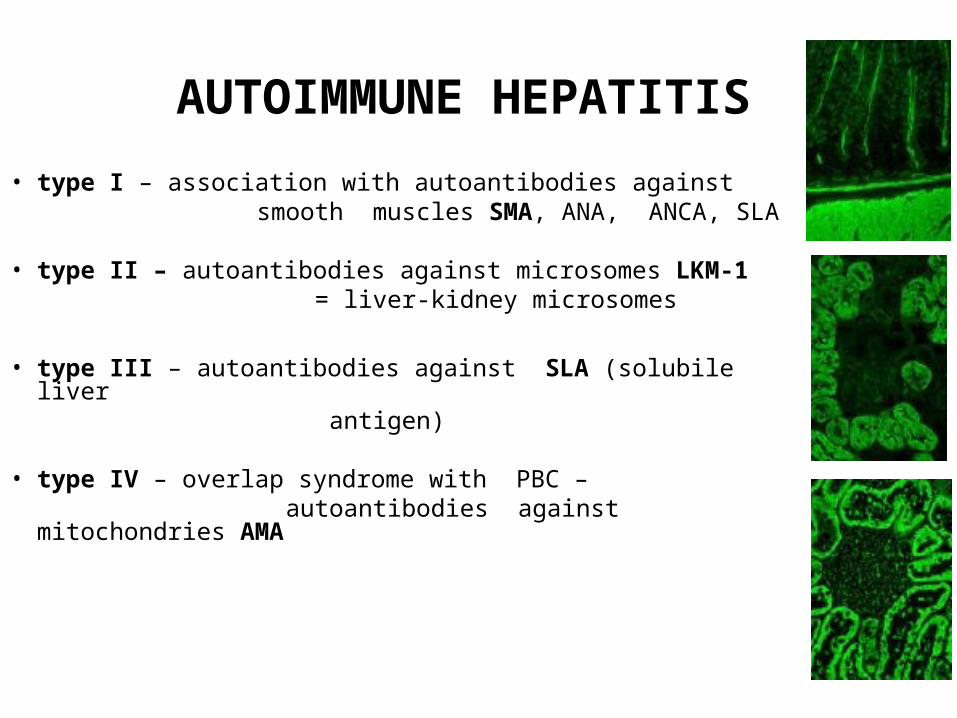
AUTOIMMUNE HEPATITIS
• type I – association with autoantibodies against smooth muscles SMA, ANA, ANCA, SLA
• type II – autoantibodies against microsomes LKM-1 = liver-kidney microsomes
• type III – autoantibodies against SLA (solubile liver
antigen)
• type IV – overlap syndrome with PBC – autoantibodies against mitochondries AMA

AUTOIMMUNE ENDOCRINOPATHY
• Hashimoto‘s thyroiditis • Graves-Basedow disease • Diabetes mellitus I. type • Addison‘s disease • Autoimmune polyglandular syndrome

Hashimoto‘s thyroiditis
• thyroid disease result to hypothyroidism on the base of lymphocytes and plasma cells infiltrate
• autoantibodies against thyroidal peroxidase (a-TPO)
and/or against thyroglobulinu (a-TG)

Grave‘s disease
• thyrotoxicosis from overproduction of thyroid hormone (patient exhibit fatigue, nervousness, increased sweating, palpitations, weight loss,
exophtalmus)
• autoantibodies against thyrotropin receptor, autoantibodies cause thyroid cells proliferation

Diabetes mellitus (insulin- dependent)
• characterized by an inability to process sugars in the diet, due to a decrease in or total absence of insulin production
• results from immunologic destruction of the insuline- producing β-cells of the islets of Langerhans in the pancreas
• autoantibodies against GAD (glutamic acid decarboxylase = primary antigen), autoantibodies anti- islet cell, anti- insulin
• islets are infiltrated with B and T cells

AUTOIMMUNE NEUROPATHY
• Guillain-Barré syndrome (acute idiopathic polyneuritis)
• Myasthenia gravis
• Multiple sclerosis

Multiple sclerosis
• chronic demyelinizing disease with abnormal reaction T cells to myeline protein on the base of mimicry between a virus and myeline protein
• features: weakness, ataxia, impaired vision, urinary bladder dysfunction, paresthesias, mental abberations
• autoantibodies against MOG (myelin-oligodendrocyte glycoprotein)
• Magnetic resonance imaging of the brain and spine shows areas of demyelination
• The cerebrospinal fluid is tested for oligoclonal bands, can provide evidence of chronic inflammation of the central nervous system

AUTOIMMUNE CYTOPENIA
• AI hemolytic disease- autoantibodies against membrane erythrocyte antigens
• AI trombocytopenia - autoantibodies against trombocyte antigens (GPIIb/IIIa)
• AI neutropenia - autoantibodies against membrane neutrofil antigens

Treatment of autoimmune diseasesSystemic AI – non-specific immunosuppression: glucocorticoids
cytostatics:• alkylating agents - cyclophosphamide • purine analogs - azathioprine, mycophenolate,• antimetabolites - metotrexate • antibiotics - cyclosporin A, tacrolimus• monoclonal antibodies
Organ specific AI - non-specific immunosuppression
Endocrinopathies - Substitution of lacking product of endocrinal gland destroyed by AI process-insulin, thyroid gland hormones