ilaris, inn - canakinumab · patients with suspected and confirmed hids/mkd (van der hilst et al...
TRANSCRIPT

30 Churchill Place ● Canary Wharf ● London E14 5EU ● United Kingdom
An agency of the European Union Telephone +44 (0)20 3660 6000 Facsimile +44 (0)20 3660 5520 Send a question via our website www.ema.europa.eu/contact
© European Medicines Agency, 2017. Reproduction is authorised provided the source is acknowledged.
15 December 2016 EMA/26517/2017 Committee for Medicinal Products for Human Use (CHMP)
Assessment report
ILARIS
International non-proprietary name: canakinumab
Procedure No. EMEA/H/C/001109/X/0045/G
Note Variation assessment report as adopted by the CHMP with all information of a commercially confidential nature deleted.

Assessment report EMA/26517/2017 Page 2/60
Table of contents
1. Background information on the procedure .............................................. 51.1. Submission of the dossier ..................................................................................... 5 1.2. Steps taken for the assessment of the product ........................................................ 6
2. Scientific discussion ................................................................................ 72.1. Problem statement ............................................................................................... 7 2.1.1. Disease or condition .......................................................................................... 7 2.1.2. Epidemiology .................................................................................................... 7 2.1.3. Biologic features ............................................................................................... 8 2.1.4. Clinical presentation, diagnosis ........................................................................... 8 2.1.5. Management ..................................................................................................... 9 2.2. Quality aspects .................................................................................................. 10 2.2.1. Introduction.................................................................................................... 10 2.2.2. Active Substance ............................................................................................. 11 2.2.3. Finished Medicinal Product ................................................................................ 11 2.2.4. Discussion on chemical, pharmaceutical and biological aspects.............................. 12 2.2.5. Conclusions on the chemical, pharmaceutical and biological aspects ...................... 12 2.2.6. Recommendations for future quality development ...............................................12 2.3. Non-clinical aspects ............................................................................................ 12 2.3.1. Introduction.................................................................................................... 12 2.3.2. Pharmacology ................................................................................................. 12 2.3.3. Pharmacokinetics ............................................................................................ 12 2.3.4. Toxicology ...................................................................................................... 13 2.3.5. Ecotoxicity/environmental risk assessment ......................................................... 13 2.3.6. Discussion on non-clinical aspects ..................................................................... 13 2.3.7. Conclusion on the non-clinical aspects ............................................................... 14 2.4. Clinical aspects .................................................................................................. 14 2.4.1. Introduction.................................................................................................... 14 2.4.2. Pharmacokinetics ............................................................................................ 15 Interactions ............................................................................................................. 18 2.4.3. Pharmacodynamics .......................................................................................... 19 2.4.4. Discussion on clinical pharmacology ................................................................... 20 2.4.5. Conclusions on clinical pharmacology ................................................................. 22 2.5. Clinical efficacy .................................................................................................. 23 2.5.1. Dose response studies ..................................................................................... 23 2.5.2. Main study ..................................................................................................... 23 2.5.3. Discussion on clinical efficacy ............................................................................ 38 2.5.4. Conclusions on the clinical efficacy .................................................................... 40 2.6. Clinical safety .................................................................................................... 41 2.6.1. Discussion on clinical safety .............................................................................. 45 2.6.2. Conclusions on the clinical safety ...................................................................... 47 2.6.3. PSUR cycle ..................................................................................................... 47

Assessment report EMA/26517/2017 Page 3/60
2.7. Risk Management Plan ........................................................................................ 47 2.8. Pharmacovigilance ............................................................................................. 51 2.9. Product information ............................................................................................ 52 2.9.1. User consultation ............................................................................................ 52
3. Benefit-Risk Balance ............................................................................. 52 3.1. Therapeutic Context ........................................................................................... 52 3.1.1. Disease or condition ........................................................................................ 52 3.1.2. Available therapies and unmet medical need ....................................................... 52 3.1.3. Main clinical studies ......................................................................................... 52 3.2. Favourable effects .............................................................................................. 53 3.3. Uncertainties and limitations about favourable effects ............................................. 53 3.4. Unfavourable effects ........................................................................................... 54 3.5. Uncertainties and limitations about unfavourable effects ......................................... 54 3.6. Benefit-risk assessment and discussion ................................................................. 55 3.6.1. Importance of favourable and unfavourable effects .............................................. 55 3.6.2. Balance of benefits and risks ............................................................................ 55 3.7. Conclusions ....................................................................................................... 56
4. Recommendations ................................................................................. 56
Assessment report EMA/26517/2017 Page 4/60
List of abbreviations
ACZ / ACZ885 Canakinumab/ILARIS ADA anti-drug antibody AE adverse event AIDAI Auto-Inflammatory Disease Activity Index CAPS Cryopyrin Associated Periodic Syndrome CHQ-PF50 Child Health Questionnaire – Parent Form 50 CKD chronic kidney disease crFMF colchicine resistant/intolerant Familial Mediterranean Fever CRP C-reactive protein CTCAE Common Terminology Criteria for Adverse Events FMF Familial Mediterranean Fever HIDS Hyper Immunoglobulin D Syndrome (also known as mevalonate kinase
deficiency [MKD]) IL-1β interleukin-1 beta MCS mental-component summary score MedDRA Medical dictionary for regulatory activities MKD Mevalonate Kinase Deficiency NSAID non-steroidal anti-inflammatory drug OR odds ratio Pbo placebo PCS physical component summary score PD pharmacodynamics PFS pre-filled syringe Ph.Eur. European Pharmacopoeia PK pharmacokinetic PoC proof of concept PGA Physician Global Assessment of disease activity PT preferred term q4w every 4 weeks q8w every 8 weeks QoL quality of life SAA serum amyloid A SAE serious adverse event sc subcutaneous(ly) SF-12 Medical Outcome Short Form (12) Health Survey SBP Summary of Biopharmaceutics SCE Summary of Clinical Efficacy SCP Summary of Clinical Pharmacology SCS Summary of Clinical Safety SOC system organ class TNF tumor necrosis factor TRAPS Tumor Necrosis Factor Receptor Associated Periodic Syndrome

Assessment report EMA/26517/2017 Page 5/60
1. Background information on the procedure
1.1. Submission of the dossier
Novartis Europharm Ltd submitted on 18 April 2016 a group of variation(s) consisting of an extension of the marketing authorisation and the following variation(s):
Variation(s) requested Type C.I.6.a C.I.6.a - Change(s) to therapeutic indication(s) - Addition of a new
therapeutic indication or modification of an approved one II
The MAH applied for an additional form (150 mg/ml solution for injection) and an extension of indication based on the results of the pivotal phase 3 study CACZ885N2301 to include the treatment of adults and children of 2 years of age and older with one of the following Periodic Fever Syndromes: - Tumour Necrosis Factor Receptor Associated Periodic Syndrome (TRAPS); - Hyperimmunoglobulin D Syndrome (HIDS) / Mevalonate Kinase Deficiency (MKD); - Familial Mediterranean Fever (FMF). Ilaris should be given in combination with colchicine, if appropriate. As a consequence sections 4.1, 4.2, 4.4, 4.8, 5.1 and 5.2 of the SmPC are proposed to be updated and the Package Leaflet is proposed to be updated accordingly. In addition, the annexes have been aligned with the latest QRD template v.10. A revised RMP version 11 was provided as part of the application.
The legal basis for this application refers to:
Article 7.2 of Commission Regulation (EC) No 1234/2008 – Group of variations
Information on Paediatric requirements
Pursuant to Article 8 of Regulation (EC) No 1901/2006, the application included EMA Decisions P/208/2011, P/0141/2013, P/0238/2015 and P/0239/2015 on the agreement of paediatric investigation plan(s) (PIP) and on the granting of a class waiver.
At the time of submission of the application, the PIPs P/208/2011, P/0141/2013, P/0238/2015 and P/0239/2015 were completed.
Information relating to orphan market exclusivity
Similarity
Pursuant to Article 8 of Regulation (EC) No. 141/2000 and Article 3 of Commission Regulation (EC) No 847/2000, the MAH did not submit a critical report addressing the possible similarity with authorised orphan medicinal products because there is no authorised orphan medicinal product for a condition related to the proposed indication.

Assessment report EMA/26517/2017 Page 6/60
Scientific Advice
CHMP scientific advice of the quality and clinical issues was received on 25 July 2013.
1.2. Steps taken for the assessment of the product
The Rapporteur and Co-Rapporteur appointed by the CHMP were:
Rapporteur: Jan Mueller-Berghaus Co-Rapporteur: Outi Mäki-Ikola
• The application was received by the EMA on 18 April 2016.
• The procedure started on 19 May 2016.
• The Rapporteur's first Assessment Report was circulated to all CHMP members on 4 August 2016.
• The Co-Rapporteur's first Assessment Report was circulated to all CHMP members on 4 August 2016.
• The PRAC Rapporteur's first Assessment Report was circulated to all PRAC members on 16 August 2016.
• During the meeting on 2 September 2016, the PRAC agreed on the PRAC Assessment Overview and Advice to CHMP. The PRAC Assessment Overview and Advice was sent to the MAH on 5 September 2016.
• During the meeting on 15 September 2016, the CHMP agreed on the consolidated List of Questions to be sent to the MAH. The final consolidated List of Questions was sent to the MAH on 16 September 2016 .
• The MAH submitted the responses to the CHMP consolidated List of Questions on 13 October 2016.
• The Rapporteurs circulated the Joint Assessment Report on the responses to the List of Questions to all CHMP members on 21 November 2016. The PRAC Rapporteur's first response Assessment Report was circulated to all PRAC members on 18 November 2016 .
• During the PRAC meeting on 1 December 2016, the PRAC agreed on the PRAC Assessment Overview and Advice to CHMP. The PRAC Assessment Overview and Advice was sent to the MAH on 2 December 2016 .
• The Rapporteurs circulated the updated Joint Assessment Report on the responses to the List of Questions to all CHMP members on 8 December 2016. The PRAC Rapporteur's updated response Assessment Report was circulated to all PRAC members on 9 December 2016.
• During the meeting on 15 December 2016, the CHMP, in the light of the overall data submitted and the scientific discussion within the Committee, issued a positive opinion for an extension of the marketing authorisation for ILARIS and an extension of indication.

Assessment report EMA/26517/2017 Page 7/60
2. Scientific discussion
2.1. Problem statement
2.1.1. Disease or condition
Tumour necrosis factor receptor associated periodic syndrome (TRAPS)
Ilaris is indicated for the treatment of tumour necrosis factor (TNF) receptor associated periodic syndrome (TRAPS).
Hyperimmunoglobulin D syndrome (HIDS)/mevalonate kinase deficiency (MKD)
Ilaris is indicated for the treatment of hyperimmunoglobulin D syndrome (HIDS)/mevalonate kinase deficiency (MKD).
Familial Mediterranean fever (FMF)
Ilaris is indicated for the treatment of Familial Mediterranean Fever (FMF). Ilaris should be given in combination with colchicine, if appropriate.
2.1.2. Epidemiology
The three rare diseases TRAPS, HIDS/MKD and FMF are a group of clinically distinct autoinflammatory conditions, which, together with CAPS, have been classified under a single term of periodic fever syndromes. FMF is the most frequent periodic febrile syndrome among the autoinflammatory syndromes. It is estimated that about 100,000 subjects worldwide are affected by FMF (Fonnesu et al 2009). FMF is prevalent mainly among eastern Mediterranean people: non-Ashkenazi Jews, Armenians, Turks, and Arabs, for whom the estimated prevalence is between 1/200-1/1000 (Fonnesu et al 2009). With a population of more than 67 million inhabitants, therefore, a large proportion of all the FMF cases in the world live in Turkey (Turkish FMF Study Group 2005).
Hyperimmunoglobulin D/Mevalonate kinase deficiency (HIDS/MKD) is also a rare autoinflammatory disease, the epidemiology of which is largely unknown. A prospective active surveillance study was recently conducted in Germany during a period of 3 years, by the German Paediatric Surveillance Unit for rare paediatric diseases, yielding 16 cases from 10 families (Lainka et al 2012). Based on these patients, the prevalence of HIDS/MKD in children less than 16 years of age during the 3 years of observation was estimated at 6.2 (95% CI: 3.5, 10.2) per million in this age group (Lainka et al 2012). The international HIDS/MKD database collected data about patients with suspected and confirmed HIDS/MKD (van der Hilst et al 2008). Established in 1994 by Dutch researchers, this group had by Jan 2007 collected information (submitted online by the patients’ physicians) on 244 patients out of whom a total of 126 patients with mutation-positive HIDS/MKD were identified (van der Hilst 2008).
Publications with available information on TRAPS epidemiology are extremely scarce. Clinical study groups, mostly from patients attending in- and out-patient rheumatology centers, are active at national and international level in Europe, and have published data to characterize the frequency, clinical signs, and genotypic features of the disease. A review paper reported a prevalence rate of 1 per million in the United Kingdom (Lachmann and Hawkins 2009).

Assessment report EMA/26517/2017 Page 8/60
2.1.3. Biologic features
The 3 rare diseases, FMF, HIDS/MKD, and TRAPS are a group of clinically distinct auto inflammatory conditions, which, together with CAPS, have been classified under a single term of periodic fever syndromes.
Although the underlying genetic defects and molecular aetiology differ across the periodic fever syndromes, the disease mechanism common across these autoinflammatory conditions involves abnormal activation of the innate immune system, leading to dysregulation of cytokines and excessive inflammation (Ozen and Bilginer 2014).
The overlapping clinical manifestations across the periodic fever syndromes are the recurrent episodes of systemic inflammation accompanied by high and disabling fever and characteristic signs and symptoms in target organs and body systems (i.e., serositis, neutrophilic rash, muco-cutaneous ulcers, arthralgia/arthritis, and aseptic meningitis/headaches) (Piram et al 2011, ter Haar et al 2013).
2.1.4. Clinical presentation, diagnosis
FMF is an autosomal recessive disease that affects mainly people of Mediterranean ancestry. Approximately 90% of patients with FMF experience the onset of disease before the age of 20 years. FMF is characterized by short febrile attacks caused by neutrophil -induced serosal inflammation and a gradual accumulation of amyloid in the kidneys. These febrile attacks are associated with pain in the abdomen, chest, joints, muscles, scrotum, and/or skin (Sozeri and Kasapcopur 2015).
HIDS/MKD is an autosomal recessive disorder which is characterized by febrile attacks that last 3 to 7 days occurring every 4 to 6 weeks. These attacks are often associated with abdominal pain, vomiting, diarrhea, headache, polyarthralgia, non-destructive arthritis, and/or skin lesions (Hausmann and Dedeoglu 2013, van der Hilst et al 2005). HIDS, originally named based on the high serum levels of immunoglobulin D (IgD) in the first cases described, is caused by mutations in the gene encoding mevalonate kinase, a critical enzyme acting early in the isoprenoid pathway. Therefore, HIDS is also referred to as MKD (van der Meer et al 1984, Drenth et al 1999, Stoffels and Simon 2011) and the two terms are often used interchangeably.
TRAPS is an autosomal dominant disorder that affects mostly people of northern European descent. The median age of onset is 3 years. TRAPS is characterized by febrile attacks that tend to last longer than those of FMF or HIDS. These attacks are associated with abdominal pain, severe myalgia, and painful erythema on the trunk or extremities. An estimated 14% to 25% of TRAPS patients develop reactive amyloidosis (van der Hilst et al 2005). Treatment with non-steroidal anti-inflammatory drug (NSAIDs) or glucocorticoids can reduce the severity of symptoms but does not affect the frequency of febrile attacks (Hausmann and Dedeoglu 2013).
The recurrent episodes of fever and severe localized inflammation are not explained by usual childhood infections. The episodes are usually associated with elevated serum levels of acute-phase reactants (e.g., Creactive protein (CRP), serum amyloid A (SAA), fibrinogen), an elevated erythrocyte sedimentation rate (ESR), and leukocytosis. Febrile episodes and associated symptoms in patients with these diseases can have a notable effect on quality of life (QoL), affecting school, work and interpersonal relationships (Hoffman and Simon 2009). Although the inflammatory attacks cause much morbidity and significantly decreases the QoL of patients, the major source of mortality and most serious complication in patients with FMF, HIDS/MKD or TRAPS is represented by progressive secondary amyloidosis that may develop over several years and can progress to chronic kidney disease with subsequent renal failure. Renal amyloidosis is observed in up to 60% of untreated FMF patients, up to 25% of TRAPS patients and in <10% of HIDS/MKD patients (van der Hilst et al 2005; Hoffman and Simon 2009).

Assessment report EMA/26517/2017 Page 9/60
2.1.5. Management
To date, there are no approved therapies for HIDS/MKD or TRAPS. Certain medications have been used to treat the symptoms of these periodic fever syndromes, but these provide limited control over the relapsing febrile attacks and none treat the underlying inflammation. It has been reported that acute treatment with NSAIDs or corticosteroids can reduce the symptoms associated with febrile attacks in some patients. Literature reports have suggested that both anakinra (a human IL-1 receptor antagonist) and eternacept (a TNF-α inhibitor) might be effective for the treatment of periodic fever syndromes. However, while anakinra may have a practical limitation due to the need for daily injections and etanercept may have some limited utility, both are not approved to treat these conditions.
For patients with FMF, colchicine has shown to be effective in controlling febrile attacks and preventing secondary amyloidosis in the majority of these patients. However, colchicine is not universally approved for the treatment of FMF and is associated with the significant side effects of diarrhea and transient elevation of transaminases and the rare side effects of liver dysfunction, leukopenia, and neuromyopathy. Colchicine doses must be adjusted in patients with impaired renal or liver function, and other medications can affect the metabolism of colchicine. Patients who do not respond to or are intolerant to colchicine have very few, if any, treatment options.
Thus, there is a unmet medical need for a therapy that provide both relief of fever and symptoms as well as effective disease control in patients with FMF in whom colchicine is contraindicated, is not tolerated, or does not provide an adequate response, HIDS/MKD, and TRAPS.
About the product
Canakinumab, a human IgG1 kappa antibody directed against IL-1β, is already authorized in the EU for the treatment of Cryopyrin-Associated Periodic Syndromes (CAPS) in adults and children (≥ 2 years), for systemic juvenile idiopathic arthritis (sJIA) in children (≥ 2 years) and for gouty arthritis in adults. IL-1β is a pro-inflammatory cytokine that is elaborated by a variety of cell types, particularly mononuclear phagocytes, in response to injury, infection and cellular activation. IL-1β is known to play a dominant role in the pathophysiology of a number of inflammatory diseases including the group of auto inflammatory conditions known as periodic fever syndromes.
The claimed indications are:
Tumour necrosis factor receptor associated periodic syndrome (TRAPS)
Ilaris is indicated for the treatment of tumour necrosis factor (TNF) receptor associated periodic syndrome (TRAPS).
Hyperimmunoglobulin D syndrome (HIDS)/mevalonate kinase deficiency (MKD)
Ilaris is indicated for the treatment of hyperimmunoglobulin D syndrome (HIDS)/mevalonate kinase deficiency (MKD).
Familial Mediterranean fever (FMF)
Ilaris is indicated for the treatment of familial Mediterranean fever (FMF) in patients in whom colchicine is contraindicated, is not tolerated, or does not provide an adequate response despite the highest tolerable dose of colchicine.
The approved indications are:

Assessment report EMA/26517/2017 Page 10/60
Tumour necrosis factor receptor associated periodic syndrome (TRAPS)
Ilaris is indicated for the treatment of tumour necrosis factor (TNF) receptor associated periodic syndrome (TRAPS).
Hyperimmunoglobulin D syndrome (HIDS)/mevalonate kinase deficiency (MKD)
Ilaris is indicated for the treatment of hyperimmunoglobulin D syndrome (HIDS)/mevalonate kinase deficiency (MKD).
Familial Mediterranean fever (FMF)
Ilaris is indicated for the treatment of Familial Mediterranean Fever (FMF). Ilaris should be given in combination with colchicine, if appropriate.
The recommended starting dose of Ilaris in TRAPS, HIDS/MKD and FMF patients is 150 mg for patients with body weight > 40 kg and 2 mg/kg for patients with body weight ≥ 7.5 kg and ≤ 40 kg. This is administered every four weeks as a single dose via subcutaneous injection
Type of Application and aspects on development
The MAH applied for an extension of the indication for the treatment of Periodic Fever Syndromes. The present variation is grouped with a line extension to register a new pharmaceutical form i.e., 150 mg/ml solution for injection.
The pivotal Study N2301 was still ongoing at the time of MA variation application submission (24-week randomized withdrawal phase has been finalised and 72-week open-label treatment phase has not yet been finalised).
Consultation with PEI (CHMP Rapporteur) took place on 8 October 2015 and with Fimea (CHMP Co-Rapporteur) on 25 September 2015. In both of these pre-submission meetings, the content of the quality and clinical parts of dossier were discussed and agreed on. CHMP scientific advice of the quality and clinical issues was received on 25 July 2013.
The proposed indications of TRAPS, HIDS/MKD and FMF for canakinumab are supported by Paediatric Investigation Plans (PIP) which all have been completed and received positive opinion on compliance check from the Paediatric Committee (PDCO) on 26 February 2016
2.2. Quality aspects
2.2.1. Introduction
The current commercially available dosage forms of Ilaris are a lyophilised 150 mg powder for solution for injection and 150 mg powder and solvent for solution for injection. The present extension application is submitted to register canakinumab 150 mg/1 mL solution for injection in vial. The present and proposed dosage forms are for subcutaneous use. To support this, data for the solution for injection in vial is provided with the submission package including additional data for “solution for injection in prefilled syringe”. The solution for injection in prefilled syringe data is considered supportive only and the prefilled syringe presentation is not registered with this submission.

Assessment report EMA/26517/2017 Page 11/60
2.2.2. Active Substance
The entire Ilaris CTD 3.2.S Module related to the existing presentations (EU/1/09/564/001-003) remains unchanged.
2.2.3. Finished Medicinal Product
The currently approved Ilaris presentation is a lyophilisate which contains 150 mg of canakinumab in histidine buffer with sucrose and polysorbate 80 as additional excipients. The proposed liquid formulation in single-use vial contains 150 mg/mL canakinumab. Mannitol is used as stabiliser instead of sucrose, and the content of histidine and polysorbate 80 is lower (see Table 1). The suitability of the liquid formulation was demonstrated in formulation robustness studies. The nature and contents of the container consist of 1 mL of solution for injection in a vial (type I glass) with a (laminated chlorobutyl rubber) stopper and flip-off (aluminium) cap. The container closure system meets the Ph. Eur. requirements. The primary packaging of the liquid presentation is comparable to that of the lyophilisate; only the vial size is smaller. Packs contain one vial. Needles are not provided by the manufacturer. Process validation of the manufacture of canakinumab solution in vial was performed based on the production of consecutive validation batches at commercial manufacturing scale. The data provided indicate that the process is capable of consistently manufacturing a finished product which meets the specifications.The applicant has demonstrated the analytical comparability of the finished product for all presentations. The comparability of canakinumab solution in vial to the lyophilisate was thoroughly assessed in line with ICH Q5E. Canakinumab solution in vial and lyophilisate demonstrated comparable physico-chemical properties and potency after storage at the long term storage conditions (2 years, 2°C - 8°C). The excipients used for formulation of the finished product comply with the requirements of the Ph. Eur. None of the excipients used in the manufacture of the finished product are of human or animal origin. No novel excipients have been introduced in the manufacture of the finished product. Finished product release specifications were set based on release and stability data of canakinumab solution. Where appropriate, the acceptance criteria are identical to those for the lyophilisate. Additional tests are used to control purity of the canakinumab solution in vial and some acceptance criteria are more stringent than for control of the lyophilisate (purity by reducing SDS-PAGE, purity by SEC and charge heterogeneity by CEX). This is endorsed. The resulting specifications for release and stability testing for canakinumab solution in vial are considered acceptable. The applicant has appropriately justified the proposed limits in their response. The limits are supported by clinical studies and are identical to the limits of commercial Ilaris finished product. The test methods are described, with references to Ph. Eur where appropriate. Non-compendial methods were validated according to ICH Q2 with acceptable results. Batch analysis data have been provided for eight batches of canakinumab solution in vial (clinical, registration and process validation batches). The batch release results of the finished product lots are within the specification limits, consistent and highly similar indicating that the manufacturing process is under control. Stability studies for canakinumab solution in vial are conducted in line with ICH Q5C and Q1A. A photostability study has been performed according to ICH Q1B.
A shelf life of 24 months at 2°C-8°C is proposed for canakinumab liquid in vial. To support the claim the applicant has provided stability results of the liquid in vial registration batches that are representative of commercial scale process and manufactured during October 2014 at Novartis Stein AG (Switzerland). Data of 12 months of ongoing stability studies is presented for long term conditions (5°C) including 6 months for accelerated (25°C) and stressed (30°C/40°C) conditions. In addition 24 months supportive data for a clinical liquid in vial batch is presented.

Assessment report EMA/26517/2017 Page 12/60
Furthermore, supportive stability data (36 months) for three PFS batches are provided for long-term, accelerated and stressed conditions. The batches were manufactured during 2010. The data from the PFS batches are considered supportive since the formulation of the solution in vial and in PFS is identical and comparability of canakinumab in the PFS and the vial presentation was shown. Importantly, a comparative stability profile was demonstrated. Overall the 24 month shelf life (2°C-8°C) is acceptable. In relation to adventitious agents, none of the excipients used for the solution formulation are of human or animal origin. Thus, viral and TSE safety is still sufficiently demonstrated.
2.2.4. Discussion on chemical, pharmaceutical and biological aspects
For the most part information on development and manufacture of the finished product is presented in a satisfactory manner and no major issues were identified during the assessment of the dossier. The few points for clarification raised during the review were satisfactorily addressed by the applicant.
2.2.5. Conclusions on the chemical, pharmaceutical and biological aspects
From a quality point of view the line extension is approvable when Ilaris is used in accordance with the SmPC and package leaflet.
2.3. Non-clinical aspects
2.3.1. Introduction
Non new non-clinical studies have been performed.
However additional pharmacology information and an update of the pharmacokinetic data for the specific periodic fever syndromes indications FMF, HIDS/MKD and TRAPS have been provided. This includes comparative systemic exposure ratios in nonclinical toxicology species marmoset and patients with periodic fever to confirm the adequacy of previous toxicology data to support the clinical use of canakinumab in the new indications.
2.3.2. Pharmacology
No new non-clinical pharmacology studies have been submitted for this application.
Literature data has been provided regarding the role of IL-1β in the individual periodic fever syndromes.
Periodic fever syndromes may be caused by different genetic defects which are transmitted in either dominant or recessive Mendelian inheritance. The underlying gene defects lead directly to the activation of the inflammasome and increased secretion of IL-1β (as in CAPS) or by indirect mechanisms activating the inflammasome in an unregulated way (as in HIDS/MKD, TRAPS or FMF). IL-1β induces the fever response in humans. Therefore, idiopathic fever attacks are suggestive of a dysregulation of the pathways leading to inflammasome activation.
2.3.3. Pharmacokinetics
Given the species-specificity of canakinumab, the marmoset monkey has been used in the nonclinical setting for understanding the pharmacokinetics of canakinumab (data provided at the time of the initial marketing authorisation). Table 3 provides a comparison of PK parameters in marmoset monkeys, rhesus monkeys, healthy volunteers and periodic fever syndrome patients.

Assessment report EMA/26517/2017 Page 13/60
Table 3 - Comparative pharmacokinetics (Mean (SD)) of ACZ885
2.3.4. Toxicology
No new toxicology studies were performed.
2.3.5. Ecotoxicity/environmental risk assessment
As a protein, canakinumab is not expected to pose a risk to the environment
According to Directive 2001/83/EC and Guideline EMEA/CHMP/SWP/4447/00 corr2, medicinal products consisting of substances occurring naturally in the environment, such as electrolytes, vitamins, proteins etc. do not need to be accompanied by an environmental risk assessment because they are unlikely to result in significant risk to the environment.
2.3.6. Discussion on non-clinical aspects
No new non-clinical studies have been submitted which was considered acceptable by CHMP.

Assessment report EMA/26517/2017 Page 14/60
The applicant has sufficiently delineated the rationale for using canakinumab in patients with period fever syndromes based on literature data. The available data provided a sufficient rationale for blocking IL-1β in these indications.
A comparison of PK parameters in marmoset monkey, rhesus monkey, healthy volunteers and patients with periodic fever syndromes was provided in this application. The available data provides a sufficiently high exposure multiple to support the proposed treatment in patients with periodic fever syndromes.
No new toxicology studies were performed. However, toxicology of canakinumab in the liquid formulation was evaluated in a 13-week SC study in marmosets. This study was already reviewed at the time of MAA and supports the use of the proposed liquid formulation.
2.3.7. Conclusion on the non-clinical aspects
The available non-clinical data support the proposed line extension (canakinumab solution for injection in vial) and the variation to extend the indication.
2.4. Clinical aspects
2.4.1. Introduction
In order to obtain a marketing authorization of canakinumab for the treatment of Periodic Fever Syndromes (TRAPS, HIDS/MKD, and FMF) at a recommended dose of 150 mg (or 2 mg/kg in patient weighing ≤ 40 kg) administered subcutaneously every 4 weeks, data from four Phase II studies (D2203, D2402, D2204, DTR01) and one phase III study (N2301) in patients with the Periodic Fever Syndromes TRAPS, HIDS/MKDS, and colchicine resistant/intolerant Familial Mediterranean Fever (crFMF) were evaluated.
CHMP scientific advice on quality and clinical development was received on 25 July 2013.
The proposed indications of TRAPS, HIDS/MKD and FMF for canakinumab are supported by Paediatric Investigation Plans (PIP) which all have been completed and received positive opinion on compliance check from the Paediatric Committee (PDCO) on 26 February 2016.
GCP
The Clinical trials were performed in accordance with GCP as claimed by the MAH.
The MAH has provided a statement to the effect that clinical trials conducted outside the community were carried out in accordance with the ethical standards of Directive 2001/20/EC.
• Tabular overview of clinical studies
The clinical studies conducted in patients with TRAPS, HIDS/MKD and crFMF patients are presented in the Table 4 below. In addition, a BE study (Study A2104) of the comparability between the canakinumab solution in pre-filled syringe and the currently authorized lyophilized formulation was carried out.

Assessment report EMA/26517/2017 Page 15/60
Table 4 - Clinical studies in patients with Periodic Fever Syndromes
2.4.2. Pharmacokinetics
Bioequivalence between marketed 150 mg powder for solution for injection and 150 mg solution for injection in pre-filled syringe (Study A2104)
Study A2104 was a randomized, open-label, single-dose, parallel-group study in healthy subjects to determine the bioequivalence of a liquid presentation of ACZ885 (canakinumab) with respect to the powder for solution for injection ACZ885 following 150 mg subcutaneous administration.
The primary objective was to demonstrate the bioequivalence of a single subcutaneous dose of a 150 mg solution for injection in pre-filled syringe with respect to the marketed 150 mg powder for solution for injection. Secondary objectives were to evaluate the safety and tolerability after a single subcutaneous dose of a solution for injection presentation of ACZ885 packaged in a pre-filled syringe and also to evaluate the pharmacodynamics of ACZ885 based on total IL-1β, after a single subcutaneous dose of a solution for injection of ACZ885 packaged in a pre-filled syringe and after a single subcutaneous dose of the powder for solution for injection presentation of ACZ885.
Despite the minor difference in composition between solution for injection in pre-filled syringe (mannitol) and powder for solution for injection (sucrose), the bioequivalence study A2104 demonstrated the comparable systemic exposure of canakinumab between the two presentations.
In addition, comparability of physicochemical properties between solution for injection in vial and powder for solution for injection has been shown by release testing, characterization test and stability test.
Comparability of solutions for injection in pre-filled syringe and in vial

Assessment report EMA/26517/2017 Page 16/60
Canakinumab is available as powder for solution for injection, as a lyophilisate form that is reconstituted with Water for Injections for subcutaneous administration at 150 mg/mL. A canakinumab solution for injection in pre-filled syringe is also in clinical development (Phase 3 study CACZ885M2301). A new presentation solution for injection in vial has been developed and used in Study N2301.
The solution for injection in pre-filled syringe and in vial has the same composition. The only difference is the primary packaging. However, the solution for injection in vial has a primary packaging the same as powder for solution for injection except for the size of the type I glass vial.
The formulation for intended use and used in the pivotal phase 3 study is a solution in a vial instead of PFS, as the latter does not allow weight-based dosing required for the conduct of the study. It has the same composition as the solution in PFS. Extractable volume is considered comparable. Supportive evidence comes from the population PK model results which did not indicate a difference in bioavailability between the solution for injection in vial vs. powder for solution for injection.
Pharmacokinetic in target population of PF syndromes
Pharmacokinetic (PK) data from 20 TRAPS patients, 9 HDS/MKD patients and 16 crFMF patients are available from four phase II studies.
In the Phase III study N2301, PK pre-dose samples at week 5, 9, 13 and 17 (representing Cmin) from n=181 patients of all three cohorts were available. Only one sample (week 3) represented canakinumab concentration within a dose interval (2 weeks after administration of the first dose). In the group receiving 150 mg q4w (no flare) Cmin levels increased in all three disease cohorts from 6.5 - 10.4 mcg/mL up to 13.5 - 20 µg/mL after the 4th dose. The highest canakinumab levels were observed in the TRAPS cohort, the lowest in the HIDS cohort. In the group of patients who up-titrated from 150 mg to 300 mg q4w (≥ 1 flare) mean Ctrough canakinumab levels at week 17 were increased in all cohorts (23-30 mcg/mL).
The population PK study involved patients from studies N2301 (crFMF, HIDS/MKD, TRAPS, phase 3, up to day 113), D2203 (TRAPS, phase 2 PoC up to day 253), D2304 (CAPS, phase 3 up to day 337), D2306 (CAPS, phase 3, up to day 729), and D2308 (CAPS, phase 3, up to day 169). These data were used to describe the pharmacokinetics of ACZ885 in the new disease conditions of periodic fever syndromes (crFMF, HIDS/MKD, TRAPS) and assessing comparability with CAPS. The studies included several dosing regimens with subcutaneous administration. The population PK model was built based on 3318 ACZ885 serum concentrations from 362 patients with periodic fever syndromes and CAPS.
Based on the population PK analysis, for a typical patient with Periodic Fever Syndromes (TRAPS, HIDS/MKD, and crFMF) weighing 55 kg (70 kg), the estimated serum clearance of canakinumab was 0.14 ± 0.04 L/day (0.17 L/day). The corresponding volume of distribution was 4.96 ± 1.35 L. Serum clearance of canakinumab and its volume of distribution were dependent on bodyweight in an allometric relationship. The exponent for clearance was estimated to be 0.86 and 1.02 for the total volume of distribution. Canakinumab minimal concentration at steady state following 150 mg sc q4w dosing regimens was estimated to be 16.4 ± 7.4 mcg/mL. The estimated steady state AUCtau was 636.7 ± 260.2 μg.day/mL. The elimination half-life of canakinumab in crFMF, HIDS/MKD and TRAPS patients is about 25.7 ± 6.5 days.
Special populations
Impaired renal function
There were no severely renal impaired subjects included in the Periodic Fever Syndromes (TRAPS, HIDS/MKD, and crFMF) studies. Since canakinumab is a human IgG immunoglobulin with large molecular size (~150 kDa),

Assessment report EMA/26517/2017 Page 17/60
little intact immunoglobulin can be filtered by the kidney, hence little antibody is expected to be excreted in the urine.
Impaired hepatic function
No formal study has been performed with canakinumab in patients with impaired hepatic function as it is known that the majority of IgG elimination occurs via intracellular catabolism, following fluid-phase or receptor mediated endocytosis.
Gender
PK data from 171 male and 191 female patients were included in the population PK model from Periodic Fever Syndromes (TRAPS, HIDS/MKD, and crFMF) and CAPS studies. No gender-related difference was observed in canakinumab clearance after correction for bodyweight in either patient population.
Race
The database in the population PK model included 27 Japanese patients (8 from Periodic Fever Syndromes (TRAPS, HIDS/MKD, and crFMF)). Effect of ethnicity (Japanese vs. non- Japanese) was assessed by estimating relative changes on clearance in the final population PK model. The difference of the typical clearance between Japanese vs. non-Japanese patients was minimal. The ratio of the two estimated clearances were 1.07 and the corresponding 95% CI was within 20% (0.96-1.20), meeting the standard bioequivalence criteria.
Weight
The one-compartment model selected as base model already contained bodyweight as a covariate. The canakinumab clearance versus bodyweight relationship had an estimated allometric coefficient of 0.86 (95% CI: 0.80 – 0.91).
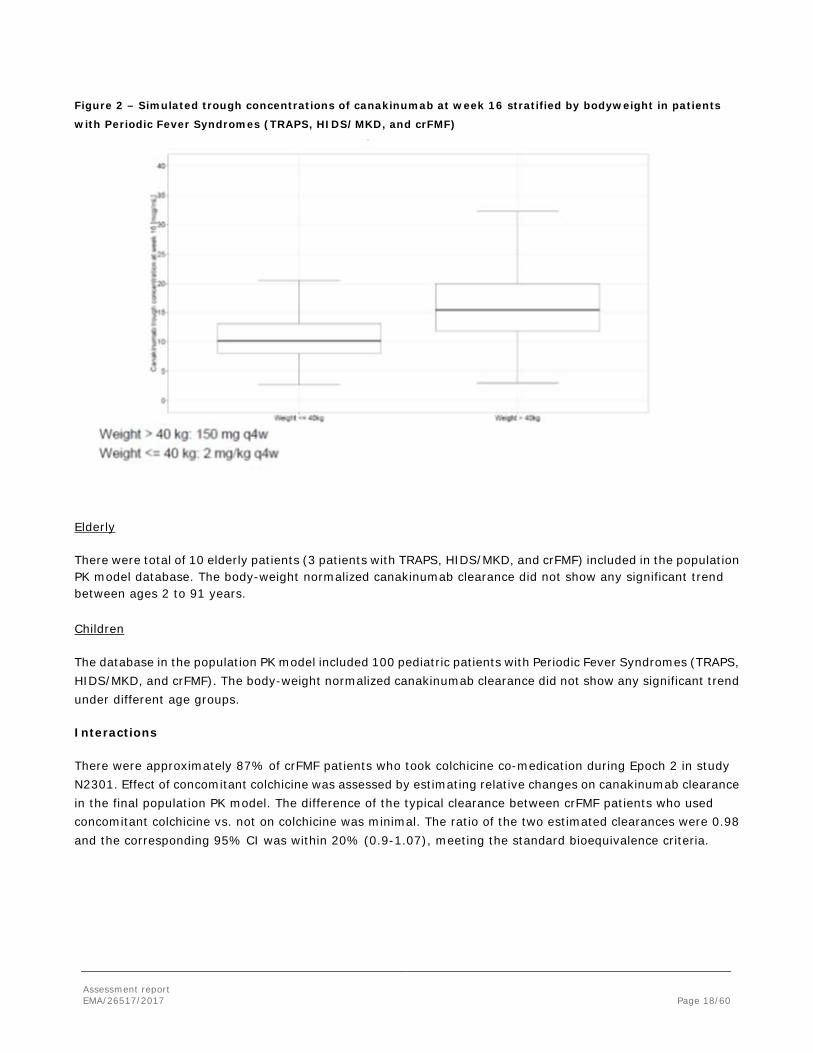
When stratified by bodyweight, approximately 20% higher exposure was predicted for trough concentrations at week 16 for the higher bodyweight group (> 40 kg) when given 150 mg q4w vs. the lower bodyweight group (≤ 40 kg) given 2 mg/kg q4w (Figure 2).
Assessment report EMA/26517/2017 Page 18/60
Figure 2 – Simulated trough concentrations of canakinumab at week 16 stratified by bodyweight in patients
with Periodic Fever Syndromes (TRAPS, HIDS/MKD, and crFMF)
Elderly
There were total of 10 elderly patients (3 patients with TRAPS, HIDS/MKD, and crFMF) included in the population PK model database. The body-weight normalized canakinumab clearance did not show any significant trend between ages 2 to 91 years.
Children
The database in the population PK model included 100 pediatric patients with Periodic Fever Syndromes (TRAPS, HIDS/MKD, and crFMF). The body-weight normalized canakinumab clearance did not show any significant trend under different age groups.
Interactions
There were approximately 87% of crFMF patients who took colchicine co-medication during Epoch 2 in study N2301. Effect of concomitant colchicine was assessed by estimating relative changes on canakinumab clearance in the final population PK model. The difference of the typical clearance between crFMF patients who used concomitant colchicine vs. not on colchicine was minimal. The ratio of the two estimated clearances were 0.98 and the corresponding 95% CI was within 20% (0.9-1.07), meeting the standard bioequivalence criteria.

Assessment report EMA/26517/2017 Page 19/60
2.4.3. Pharmacodynamics
Primary and Secondary pharmacology
Total IL-1ß
Canakinumab binding to human IL-1ß neutralizes its bioactivity and results in the formation of a canakinumab-IL-1ß complex. Since the complex is cleared slower than the free IL-1ß, an increase in total IL-1ß is usually observed after canakinumab administration.
The same kit for determination of total IL-1ß in PF syndrome studies has been used as in previous studies. In study N2301, HIDS/MKD patients showed generally higher IL-1ß baseline values (up to 10 ng/L) compared to crFMF and TRAPS patients (< 1 ng/L).
There was an observed separation between the 150 mg (2 mg/kg) and 300 mg (4 mg/kg) dose levels of median IL-1β levels.
An increase in total IL-1ß after administration of canakinumab was observed in patients with Periodic Fever Syndromes. A trend for higher total IL-1ß concentrations at week 2 in non-responders was observed in all patients with Periodic Fever Syndromes (TRAPS, HIDS/MKD, and crFMF), especially in HIDS/MKD and TRAPS patients.
Canakinumab treatment was associated with a decrease of IL-1ß-induced downstream mediators including IL-1ß pathway related genes, acute phase proteins such as serum amyloid (SAA) and C-reactive protein (CRP).
Immunogenicity
The recently developed Immunogenicity assay with improved drug tolerance has now been used in the phase 3 study N2301 after it had been firstly applied in the SJIA Study G2301E1. In contrast, in studies D2203, D2402, D2204, and DTR01 the former assay was used for immunogenicity measurements. It was confirmed in cross-comparison measurements that both MSD based assays are capable of detecting anti-drug antibodies, but the improved MSD method provided a better performance in sensitivity and drug tolerance.
In study N2301, during screening, 33 samples out of 639 were above the Screening Cut Point, but could not be confirmed in the second confirmation assay. Therefore, all samples were reported as immunogenicity negative. Overall, the incidence of treatment related anti-canakinumab antibodies was <1% in patients with Periodic Fever Syndromes (TRAPS, HIDS/MKD, and crFMF).
PKPD relationship
The exposure-efficacy and exposure-safety relationships of canakinumab in TRAPS, HIDS/MKD, and crFMF patients were investigated in the Phase III study N2301. Predicted canakinumab concentrations were approximately 30% lower in HIDS/MKD and TRAPS patients at the time of flare compared to no flare. Furthermore, in HIDS/MKD patients this trend was also observed in the subset of patients (11/15) in whom flares occurred despite up-titration to a dose of 300 mg or 4 mg/kg. In contrast, for crFMF patients there was no difference between concentrations at flare and no flare.
An exposure-response model for the probability of flare suggested a difference in drug sensitivity between disease cohorts, with HIDS/MKD and TRAPS patients requiring approximately 10-fold higher drug concentrations to reach efficacy.
While mean neutrophil levels decreased following canakinumab treatment, this did not translate into a higher occurrence of notable abnormalities (CTC grade ≥1) with increased concentrations of canakinumab. Likewise,

Assessment report EMA/26517/2017 Page 20/60
the occurrence of notable abnormalities for lymphocytes count, leukocytes and platelets count did not increase with canakinumab concentrations either.
2.4.4. Discussion on clinical pharmacology
Three PK-study reports have been submitted in order to support this application: a bioequivalence study (ACZ885A2104), popPK study of ACZ885 in patients with periodic fever syndromes and a PK/PD modelling report.
The bioequivalence study A2104 demonstrated the comparable systemic exposure of canakinumab between a solution for injection in pre-filled syringe (PFS) and the currently authorized powder for solution for injection. However, the formulation for intended use and used in the pivotal phase 3 study is a solution in a vial instead of PFS. It has the same composition as the solution in PFS and the extractable volume is considered comparable. It is noted that the BE-study ACZ885A2104 has been performed with a liquid formulation of ACZ885 in pre-filled syringe. As this application concerns only a liquid formulation in a vial, comparability has been demonstrated at quality level of 1) the powder for solution of injection and solution for injection in prefilled syringe, 2) the powder for solution of injection and solution for injection in vial, and 3) the solution for injection in prefilled syringe and solution for injection in vial (see Quality section). Thus, the quality of ACZ885 is comparable for both presentations. Supportive evidence for comparability comes from the population PK model analysis which did not indicate a difference in bioavailability between the solution for injection in vial vs. powder for solution for injection. Thus, bioequivalence can be assumed between all three formulations.
With regard to bioanalytical methods used in this application, several concerns were raised during the procedure. The MAH responded to the concerns, however, there is still an overall concern on the consistency and reliable performance of the bioanalytical methods. Especially a) it seems that the commercial kit used for determination of IL-1β in human serum has a very high rate of inter-batch kit variability, b) in determination of anti-ACZ885 antibodies new reagent batches have caused signal inconsistencies across the MSD plates, c) there are clear deficiencies in the biological characterisation of the used control antibodies, d) a very high inter-run imprecision has been observed for the determination of neutralizing antibodies, the root-cause for the imprecision is unknown, e) the use of several different control antibodies in the transfer and validation of the assays hampers the comparison of assay performance. However, as these methods and analyses are not necessary for concluding on a positive benefit/risk balance of canakinumab in the treatment of the applied indications, these deficiencies are not pursued further during this variation procedure. Nevertheless, the following recommendation is made with regards to the reliability and consistency of the bioanalytical assays for the future: For future applications the characterisation of control antibodies should include also characterisation of functionality and the use of different types of control antibodies should be clearly considered and justified to allow also comparison of assays in, e.g., method transfer situations. The Applicant is recommended to introduce measures to appropriately control and qualify new reagent batches and commercial kits as well as to control the high imprecision and variability observed for the bioanalytical assays in order to avoid inconsistencies in assay performance.
Pharmacokinetic (PK) data from 20 TRAPS patients, 9 HDS/MKD patients and 16 crFMF patients are available from four phase II studies. Descriptive PK statistics showed canakinumab levels as expected.
In the Phase III study N2301, PK pre-dose samples from n= of all three cohorts were available. The highest canakinumab levels were observed in the TRAPS cohort, the lowest in the HIDS cohort. It is unclear whether this is confounded by body weight.

Assessment report EMA/26517/2017 Page 21/60
Based on the population PK analysis, for a typical patient with Periodic Fever Syndromes (TRAPS, HIDS/MKD, and crFMF) the estimated serum clearance of canakinumab was in accordance with CL values obtained for other patient populations (e.g. CAPS, sJIA).
A previous population PK-binding model of ACZ885 in CAPS patients (ACZ885 CAPS Extension Modeling Report) demonstrated that the PK data that was available for that analysis can be well described with a linear two-compartment model, first-order absorption for the sc administration and constant rate infusion for the intravenous administration. As the present dataset lacks rich sampling with sc dosing in periodic fevers syndromes, absorption rate and bioavailability were fixed in the present analysis at the previously estimated values in the CAPS patient population (ACZ885 CAPS Extension Modeling Report). This is considered appropriate.
Overall, the PK characteristics of canakinumab observed in crFMF, HIDS/MKD and TRAPS patients are consistent with PK observed in other indications and are typical for a human IgG1 molecule following sc dose administration. However, it is noted that clearance estimates represent CL/F assuming a fixed value for Fabs = 65 % (subcutaneous bioavailability) for all patients which was estimated in the first PKPD model analysis for CAPS patients. Therefore, variability in CL is also affected by possible differences in SC bioavailability between the patients and populations. The estimated individual clearances were generally consistent across all disease conditions in Periodic Fevers Syndromes after adjusting for body weight.
Median Ctrough levels at week 16 in patients with PF syndromes were higher in the group > 40 kg (receiving 150 mg) than in the group <= 40 kg body weight (receiving 2 mg/kg) (15 vs. 10 mcg/mL). This is expected due to the dosage regimen, since patients weighing 41-75 kg receive a relatively lower dose per kg (< 2 mg/kg) when applying 150 mg flat dose. Patients between 41-50 kg body weight receive the highest dose per kg (> 3 mg/kg).
The applicant stated that clearance and volume of distribution were not impacted by age (above 2 years old) and albumin level at baseline in patients with Periodic Fever Syndromes (TRAPS, HIDS/MKD, or crFMF) after correction for the subject’s bodyweight.
The intended dosage regimen (change from 2 mg/kg to 150 mg flat at 40 kg) is the same as authorised for CAPS patients with the exception of the dose interval which is 4-weekly here compared to 8-weekly in CAPS. Therefore, as expected from the dosage regimen, steady state levels reached in PFS patients are markedly higher than in CAPS patients and are in the average range of those in sJIA patients after 4 mg/kg q4w.
The effect of colchicine co-medication in study N2301 (approximately 87% of crFMF patients took colchicine co-medication) was investigated by population PK analysis. The result support the conclusion that colchicine co-medication had no impact on pharmacokinetics of canakinumab in patients with Periodic Fever Syndromes (TRAPS, HIDS/MKD, and crFMF).
An increase in total IL-1ß after administration of canakinumab indicating successful binding over time was observed in patients with Periodic Fever Syndromes. Further discussion on the reason for the observation of about 10-fold higher IL-1ß baseline values in HIDS/MKD patients and on the impact on the exposure-response relationship in this subgroup was requested.
The MAH speculates that, differently from FMF and TRAPS, in HIDS/MKD the activation of the inflammasome, which triggers the conversion of inactive pro-IL1 into IL-1α and IL-1β may be activated through additional pathways not activated in the other 2 diseases, such as the impairment of the isoprenoid biosynthesis and provides literature references supporting this hypothesis. Furthermore, from the tendency of higher total IL-1β concentrations at day 15 in non-responders vs. responders which was particularly visible in HIDS/MKD, the MAH

Assessment report EMA/26517/2017 Page 22/60
speculates that this could be the reason why such patients were more likely to require up-titration. Although speculative the explanations are considered plausible.
The trend for higher total IL-1ß concentrations at week 2 in non-responders is in accordance with higher canakinumab exposure levels in these patients due to dose up-titration. There was a separation between 150 mg (2 mg/kg) and 300 mg (4 mg/kg) dose levels, indicating availability of IL-1β for binding with a higher dose of the drug.
Canakinumab treatment was associated with a decrease of IL-1ß-induced downstream mediators including IL-1ß pathway related genes and acute phase proteins. This adds to the evidence that canakinumab neutralizes the activity and down-regulates the production of IL-1ß in vivo.
Canakinumab showed a low immunogenicity potential in PF syndrome patients (< 1%), even lower than observed before in other populations (up to 3% in gouty arthritis). The applicant was requested to discuss the possible reasons including the impact of the change from sucrose to mannitol in the formulation.
The MAH is of the opinion that the lower incidence of immunogenicity reported in study N2301 is not related to the change in stabilizer from sucrose to mannitol, but could rather be attributed to a better specificity of the new ADA assay. The explanation that the new assay is more specific (i.e. produced less false positives) is supported by the cross-validation results. But in contrast, the gained improvement in sensitivity on the other hand favours an increase in the number of true positive samples which is in opposite to the observed lower overall ADA incidence. However, it is agreed that it is an early stage of collecting immunogenicity data with this new assay.
The ability to detect a neutralising antibody response is very low during therapy due to the low drug tolerance of the Nab assay (3.4 µg/mL). However, since no positive screening results were detected in study N2301, this is less relevant in this case.
An exposure-response relationship between the probability of flare and canakinumab serum concentrations was observed in patients with Periodic Fever syndromes. The reason for the different sensitivity in the relationships between the subpopulations is unclear. The finding that HIDS/MKD and TRAPS patients require approximately 10-fold higher drug concentrations to reach efficacy is consistent with the greater need of up-titration observed in these two disease conditions in study N2301.
No clear exposure-safety relationship was observed. This is in accordance with previous observations in other indications. While mean neutrophil levels decreased following canakinumab treatment, this did not translate into a higher occurrence of notable abnormalities (CTC grade ≥1) with increased concentrations of canakinumab.
2.4.5. Conclusions on clinical pharmacology
The data submitted was considered acceptable by CHMP.
However, with regard to bioanalytical methods used in this application, several concerns were raised during the procedure.
As such, CHMP has recommended that for future applications the characterisation of control antibodies should include also characterisation of functionality and the use of different types of control antibodies should be clearly considered and justified to allow also comparison of assays in, e.g., method transfer situations. The Applicant should introduce measures to appropriately control and qualify new reagent batches and commercial kits as well as to control the high imprecision and variability observed for the bioanalytical assays in order to avoid inconsistencies in assay performance.

Assessment report EMA/26517/2017 Page 23/60
2.5. Clinical efficacy
2.5.1. Dose response studies
The initial canakinumab dosing regimen defined for Study N2301 (150 mg sc or 2 mg/kg sc) was based on evidence in approved indications (e.g., CAPS - [Study A2102]) and from completed open-label Phase 2 studies in crFMF, HIDS/MKD and TRAPS. Because of the rarity of these 3 conditions, no formal dose finding study was conducted.
2.5.2. Main study
A randomized, double-blind, placebo controlled study of canakinumab in patients with Hereditary Periodic Fevers (TRAPS, HIDS, or crFMF), with subsequent randomized withdrawal/dosing frequency reduction and open-label long term treatment epochs
Methods
Study Participants
The study population consisted of male and female patients (≥2 years old) with clinically and genetically confirmed hereditary periodic fevers: TRAPS (cohort 1), HIDS (cohort 2), and crFMF (cohort 3).
The study population consisted of male and female patients ≥ 2 years of age at the time of the screening visit for the randomized cohorts and patients > 28 days but < 2 years old for the nonrandomized treatment arm.
For randomized crFMF patients: diagnosis of type 1 FMF disease according to Tel Hashomer criteria but no active flare at the time of screening, at least one of the known MEFV gene exon 10 mutations, and 1 of the following 2 criteria: documented active disease despite colchicine therapy or documented intolerance to effective doses of colchicine. At randomization, patients must have had acute crFMF flare characterized by inflammation and serositis, active clinical crFMF flare as evidenced by PGA ≥ 2, and CRP > 10 mg/L.
For randomized HIDS patients: clinical diagnosis of HIDS but no active flare at the time of screening, genetic/enzymatic diagnosis of HIDS, and prior documented history of ≥ 3 febrile acute HIDS flares in a 6-month period when not receiving prophylactic treatment. At randomization, patients must have had active clinical HIDS flare as evidenced by PGA ≥ 2 and CRP > 10 mg/L.
For randomized TRAPS patients: clinical diagnosis of TRAPS but no active flare at the time of screening, mutation of the TNFRSF1A gene, and chronic or recurrent disease activity periodicity. At randomization, patients must have had active clinical TRAPS flare as evidenced by PGA ≥ 2 and CRP > 10 mg/L.
Treatments
Canakinumab solution for injection was provided in vials that contained 150 mg/mL canakinumab in a 1 mL solution.
Placebo solution for injection was provided in vials and matched the canakinumab solution.
Total duration of treatment is up to 113 weeks. As of the data cut-off date for this primary analysis (end of Epoch 2), all patients completed Week 16 of treatment.

Assessment report EMA/26517/2017 Page 24/60
Objectives
This clinical study report presents primary, secondary and exploratory endpoint efficacy and safety results up to the end of Epoch 2. All results for objectives pertaining to Epochs 3 and 4 will be reported separately.
The primary hypothesis tested was the superiority of canakinumab (150 mg or 2mg/kg q4w) relative to placebo with respect to the proportion of responders in the randomized treatment epoch (Epoch 2).
The primary objective of the randomized treatment epoch (Epoch 2) and of the overall study is to demonstrate that canakinumab treatment at a dose of 150 mg (or 2 mg/kg in patient weighing ≤ 40 kg) sc every 4 weeks is superior to placebo in achieving a clinically meaningful reduction of disease activity defined as resolution of the index flare at Day 15 and no new disease flares over 16 weeks of treatment (up to the end of Epoch 2).
The secondary objectives of the study (Epoch 2 only) are:
• To evaluate the percentage of patients who achieve a Physician Global Assessment of disease activity (PGA) < 2 (“minimal” or “none”) at the end of Week 16
• To evaluate the percentage of patients with the serologic remission at the end of Week 16 (defined as C-reactive protein (CRP) ≤ 10 mg/L)
• To evaluate the percentage of patients with normalized serum amyloid A (SAA) level at the end of Week 16 (defined as SAA ≤ 10 mg/L)
• To evaluate the long-term safety and tolerability and immunogenicity of canakinumab • To evaluate the pharmacokinetics/ pharmacodynamics of canakinumab
Outcomes/endpoints
Efficacy assessments consisted of the following: resolution of the index flare, new disease flare, Physician Global Assessment of Disease Activity (PGA), inflammatory markers (CRP and SAA), Patient/Parent’s Global Assessment (PPGA), physician’s severity assessment of key disease-specific signs and symptoms, number of fever episodes, use of rescue medication, and improvements in health related-quality of life measurements as assessed with the Auto-Inflammatory Disease Activity Index (AIDAI), Child Health Questionnaire – Parent Form 50 (CHQ-PF50), Medical Outcome Short Form (12) Health Survey (SF-12) and Sheehan Disability Scale (SDS).
The primary efficacy variable of the randomized treatment epoch (Epoch 2) and for the overall study was the proportion of responders within each cohort.
A responder was defined as a patient who had resolution of his/her index disease flare at Day 15 (PGA < 2, and CRP within normal range (≤ 10 mg/L) or reduction ≥ 70% from baseline) and did not experience a new flare (PGA ≥ 2 and CRP ≥ 30 mg/L) from the time of the resolution of the index flare until the end of Epoch 2.
Sample size
It was calculated that with a sample size of 28 subjects per treatment group Fisher's exact test with a 2.5% one-sided significance level would have 90% power to detect a treatment difference of 45% assuming a responder rate of 65% for the canakinumab treatment group and of 20% in the placebo group within each of the 3 disease cohorts (crFMF, HIDS/MKD, TRAPS). Rounded to 30 subjects per treatment arm and disease cohort, a total of 180 patients were to be enrolled.

Assessment report EMA/26517/2017 Page 25/60
Due to recruitment difficulties only 46 TRAPS patients were randomized, providing about 83% power to detect a treatment difference of 45%.
Randomisation
At baseline of epoch 2 subjects were randomized in a 1:1 ratio to receive either Canakinumab 150 mg or placebo. Randomization was stratified for disease cohort.
All patients randomized to canakinumab in epoch 2 and completing epoch 2 were to then be re-randomized (in epoch 3) in a ratio of 1:1 to receive either Canakinumab or placebo.
Randomisation was performed via IRT.
Blinding (masking)
Patients, investigator staff, persons performing the assessments, and data analysts remained blind to the identity of the randomized treatment assignment until week 16 database lock for the primary endpoint analysis, performed once all patients had completed epoch 2.
Statistical methods
The primary efficacy parameter, proportion of responders at week 16, was analysed separately for each disease cohort (crFMF, HIDS/MKD, TRAPS) based on the FAS (all patients randomized to epoch 2 who received at least 1 dose of study drug). Patients who needed dose escalation in the canakinumab arms, who crossed-over from placebo to canakinumab, or who discontinued from the study due to any reason prior to evaluating the primary endpoint were considered as non-responders in the analysis.
The primary hypothesis tested was the superiority of canakinumab relative to placebo with respect to the proportion of responders. Within each disease cohort this hypothesis was tested at a one-sided 2.5% level by means of Fisher’s exact test. The proportion of responders (including its 95%-CI according Clopper-Pearson) as well as the odds ratio and risk difference between treatments with the corresponding 95% CI was presented for each disease cohort.
For sensitivity purposes the primary analysis was repeated using the PPS and using a definition of the resolution of the index flare and new flare based on centralized SAA values. Further sensitivity analyses of the primary efficacy parameter were performed to assess the robustness of study results.
The secondary efficacy parameters at week 16 (frequency of patients a) with PGA < 2, b) in serologic remission (defined as CRP ≤ 10 mg/L), c) with a normalized SAA level at week 16 (defined as SAA ≤ 10 mg/L)) were analyzed using a logistic regression model with treatment group and baseline PGA, CRP or SAA as explanatory variables.
The primary and secondary efficacy variables were analyzed within the following subgroups: prior use of any biologics (no, yes), age groups (< 18 years, ≥ 18 years), and colchicine status (negative, positive; performed for the crFMF cohort only).
In order to control the type I error (within each disease cohort) a hierarchical testing procedure was applied separately for each disease cohort (testing was continued as long as each test showed statistical significance at the 2.5% level (1-sided)):

Assessment report EMA/26517/2017 Page 26/60
• Primary objective: superiority of Canakinumab vs placebo with respect to the percentage of responders at week 16
• Secondary objectives:
o superiority of Canakinumab vs placebo with respect to the percentage of patients with PGA < 2 at week 16.
o superiority of Canakinumab vs placebo with respect the percentage of patients with CRP ≤ 10 mg/L at week 16.
o superiority of Canakinumab vs placebo with respect the percentage of patients with SAA ≤ 10 mg/L at week 16.
Results
Conduct of the study
The study protocol was amended twice. The key features of each amendment are given below:
Amendment 1 (15-Jul-2014) introduced the following changes:
- changed Section 6.5.6 Pregnancy and assessments of fertility to reference the use of effective contraception in accordance with locally approved prescribing information
Amendment 2 (22-Oct-2014) introduced the following changes:
- updated exploratory objectives to reflect the request from the Paediatric Committee (PDCO) at the European Medicines Agency to include patients > 28 days but < 2 years of age in addition to patients ≥ 2 years of age
- clarified how patients in the randomized and non-randomized cohorts were managed:
- study entry time and treatment was harmonized between the patients who were > 28 days but < 2 years of age and Japanese crFMF patients with non-exon 10 mutations
- clarified the definition of the resolution of index flare
- clarified the blinded escape criteria
Both amendments were not considered to have affected the interpretation of study results as they occurred prior to study unblinding for the Week 16 primary endpoint analysis.
Other changes in study conduct
Study recruitment was stopped when 2/3 of patients were enrolled in the slowest enrolling cohort (TRAPS). The aim to randomize at least 60 patients per cohort was achieved for crFMF and HIDS/MKD cohorts (63 and 72 patients randomized, respectively). As recruitment in the TRAPS cohort was difficult due to the extreme rarity of the disease, overall study recruitment was stopped when 46 TRAPS patients were randomized.

Assessment report EMA/26517/2017 Page 27/60
Baseline data
Demographic and baseline disease characteristics were generally comparable between the randomized canakinumab and placebo groups in all 3 disease cohorts. The studied population of crFMF, HIDS/MKD and TRAPS patients all had genetic and past medical history of the respective disease, as per inclusion criteria, and also had active disease as defined by clinical and serological criteria at the time of randomization.
- For crFMF cohort, the mean age was 22 years and 46% of the cohort was < 18 years old, with 3 patients < 6 years old. The proportion of males and females was well balanced. Most crFMF patients were Caucasian and all had confirmed mutation in the MEFV gene. The median duration of disease was 14.7 years, with a median 18 flares per year, and most patients had PGA moderate or severe disease activity.
- For HIDS/MKD cohort, the mean age was 13.5 years and 75% of the cohort was < 18 years old, with 17 patients < 6 years old. There were slightly more females (59.7%) than males (40.3%). Most HIDS/MKD patients were Caucasian and all had confirmed mutation in the MVK (mevalonate kinase) gene. The median duration of disease was 9.8 years, with a median 12 flares per year, and the majority of patients had PGA moderate or severe disease activity.
- For TRAPS cohort, the mean age was 22 years and 59% of the cohort was < 18 years old, with 7 patients < 6 years old. There was an equal number of males and females. Most TRAPS patients were Caucasian and all had confirmed mutation in the TNFRSF1A gene. The median duration of disease was 8.2 years, with a median 9 flares per year, and over half of patients had PGA moderate (47.8%) or severe (8.7%) disease.
Numbers analysed
A total of 180 patients (60 per cohort, 30 per treatment arm) were planned to be randomized. A total of 181 patients were randomized and 4 patients entered the non-randomized open-label treatment arm.
In the crFMF cohort, 100 patients were screened and 63 patients were randomized (31 to 150 mg q4w canakinumab and 32 to placebo). In the HIDS/MKD cohort, 98 patients were screened and 72 patients were randomized (37 to 150 mg q4w canakinumab and 35 to placebo). In the TRAPS cohort, 82 patients were screened and 46 patients were randomized (22 to 150 mg q4w canakinumab and 24 to placebo). All randomized patients were included in the Full Analysis Set (for efficacy analysis) and the Safety set (for safety analysis). Four non-randomized patients (2 crFMF patients with non-exon 10 mutations and 2 HIDS/MKD patients > 28 days but < 2 years old) received open-label treatment.
Outcomes and estimation
The primary endpoint was met for all three cohorts and canakinumab was statistically superior to placebo in resolving the index disease flare at day 15 and in preventing new flares before the end of week 16. The best results can be seen for the crFMF cohort (one-sided p-value: < 0.0001) and slightly weaker response, although still statistically significant, in the TRAPS cohort (one-sided p-value: 0.0050).
More patients achieved a complete response with canakinumab than with placebo, regardless of age, prior use of biologics and concomitant use of colchicine (for crFMF). Although the majority of patients were well-controlled with canakinumab 150 mg q4w, 16% of crFMF patients, 32% of HIDS/MKD patients and 36% of TRAPS patients required dose escalation to 300 mg q4w.

Assessment report EMA/26517/2017 Page 28/60
Table 5 – Primary analysis: comparison between treatment groups for the proportion of responders after 16
weeks by cohort (Full analysis set)
The study included the option for dose escalation in case the index flare did not resolve by day 15. This escape option had to be used by 16% of crFMF patients, 32% of HIDS/MKD patients, and 36%of TRAPS patients. However, this option enabled especially crFMF and TRAPS patients to reach resolution of index flare by day 29.
In the HIDS cohort, only 4 of 15 patients (26.7%) achieved the resolution of index flare after dose escalation at day 29. This corresponds to the primary endpoint results in this cohort with 35.14% responders in comparison to 61.29% in crFMF patients and 45.45% in TRAPS patients. Though, as the primary endpoint is met in all three cohorts this is not of major relevance.
The majority of crFMF, HIDS/MKD and TRAPS patients did not re-flare over 16 weeks of treatment in Epoch 2. Among canakinumab-treated patients (150 mg or 300 mg q4w), 90.3%, 64.9% and 77.3% of crFMF, HIDS/MKD and TRAPS patients, respectively, had no new flare (i.e., PGA ≥ 2 and CRP ≥ 30 mg/L) after resolution of the index flare up to the end of Week 16.
Secondary efficacy results
Canakinumab was superior to placebo in the secondary endpoints of PGA < 2 and CRP ≤ 10 mg/L at Week 16 in crFMF, HIDS/MKD and TRAPS cohorts based on results of the testing hierarchy. For SAA ≤ 10 mg/L at Week 16, canakinumab showed greater improvements in all disease cohorts, with a statistically significant difference for TRAPS patients.
- In each randomized cohort, a significantly higher proportion of patients in the 150 mg q4w canakinumab group achieved a PGA score < 2 (no to minimal disease activity) at the end of Epoch 2 compared with placebo. Canakinumab was superior to placebo for PGA < 2 after 16 weeks for crFMF (OR=16.96, p<0.0001), HIDS/MKD (OR=13.63, p=0.0006), and TRAPS (OR=23.79, p=0.0028)
- In each randomized cohort, a significantly higher proportion of patients in the canakinumab 150 mg q4w group achieved serological remission defined as CRP ≤ 10 mg/L at the end of Epoch 2 compared with placebo. Canakinumab was superior to placebo for CRP ≤ 10 mg/L after 16 weeks for crFMF (OR=29.78, p<0.0001), HIDS/MKD (OR=12.71, p=0.0010) and TRAPS (OR=6.64, p=0.0149).
- Canakinumab normalized SAA to ≤ 10 mg/L for a significantly higher proportion of patients in the TRAPS cohort compared with placebo after 16 weeks (OR=16.69, p=0.0235). There were also greater improvements in the normalization of SAA at Week 16 with canakinumab treatment in crFMF patients (25.81% vs. 0% for placebo) and in HIDS/MKD patients (13.51% vs. 2.86% for placebo).

Assessment report EMA/26517/2017 Page 29/60
Exploratory analyses of the secondary endpoints showed that dose escalation further improved response rates for PGA < 2, CRP ≤ 10 mg/L and SAA ≤ 10 mg/L after 16 weeks. The proportion of patients with PGA < 2 (10%, 22% and 23% higher, respectively, for crFMF, HIDS/MKD and TRAPS cohorts), CRP ≤ 10 mg/L (7%, 8% and 27% higher, respectively) or with SAA ≤ 10 mg/L (5% higher for both HIDS/MKD and TRAPS; no change for crFMF) increased with up-titration to 300 mg q4w canakinumab before Day 29.
Table 6 – Results for the primary and secondary efficacy variables after 16 weeks by cohort (Full analysis set)
Physician Global Assessment (PGA) improvement over time
Rapid and sustained improvements in PGA disease activity scores were observed in patients randomised to
receive treatment with Ilaris 150 mg every 4 weeks across all three disease cohorts. A high proportion of
patients shifted from “moderate” or “severe” (PGA > 2) to “no” or “minimal” disease activity (PGA < 2) as early
as day 15, and this response was maintained up to the end of Part II.
Auto-inflammatory disease activity index score (AIDAI)
The AIDAI score is a daily patient diary made up of the following 12 components: fever, ≥ 38°C, overall
symptoms, abdominal pain; nausea/vomiting, diarrhoea, headaches, chest pain, painful nodes, arthralgia or
myalgia, swelling of the joints, eye manifestations, and skin rash. Overall, reductions in the median AIDAI score
were observed by week 2 for patients randomised to Ilaris 150 mg every 4 weeks and continued up to the end
of week 16 in all three disease cohorts.
Time to fever resolution
The median time to first resolution of fever was 1 to 1.5 days shorter for patients randomised to Ilaris 150 mg
every 4 weeks compared with placebo in all three disease cohorts.

Assessment report EMA/26517/2017 Page 30/60
Health-related quality of life measurements
Short-Form Health Survey (SF-12): SF-12 covers domains of both physical and mental health and was
administered in patients 18 years of age and older. Increases in the median physical component and mental
component summary scores (PCS and MCS) were observed by day 29, with PCS further improving at week 16.
The PCS was close to that of healthy population norms at week 16.
Child Health Questionnaire-Parent Form 50 (CHQ-PF50): The CHQ-PF50 covers domains of physical and
psychosocial health, assessing a child’s quality of life from the parent’s perspective. Increases in median
physical summary scores were observed by day 29 and maintained to week 16 across cohorts, likewise for
psychosocial summary scores in the FMF and HIDS/MKD cohorts.
The results of the secondary and exploratory efficacy analysis support the results of the primary endpoint. It is shown that treatment with canakinumab is able to normalize the CRP levels in all three disease cohorts in a statistically significant way in comparison to placebo. Furthermore, evaluation of PGA shows a reduction to <2 also in a statistically significant way for 64.52% of crFMF, 45.95% of HIDS and 45.45% of TRAPS patients.
These results are regarded as clinically meaningful improvement of disease status after 3 months of treatment.
Ancillary analyses
Age
Canakinumab showed greater efficacy compared to placebo with the respect to the proportion of responders after 16 weeks in both age subgroups across all 3 disease cohorts (Table 7). Similar results were also observed for the secondary efficacy endpoints of PGA < 2 and CRP ≤ 10 mg/L in crFMF, HIDS/MKD and TRAPS patients. For SAA ≤ 10 mg/L, canakinumab was more efficacious than placebo in both age subgroups in crFMF patients, while in HIDS/MKD and TRAPS patients this effect was apparent in patients < 18 years old but not in the very small number of HIDS/MKD and TRAPS patients ≥ 18 years old.
Table 7 – Comparison between treatment groups for the porportion of responders after 16 weeks by age group
(Full analysis set)

Assessment report EMA/26517/2017 Page 31/60
Concomitant use of colchicine (crFMF patients)
Most (87.3%) crFMF patients were taking concomitant colchicine during Epoch 2. In this subgroup, canakinumab showed greater efficacy than placebo with respect to the proportion of responders after 16 weeks (Table 8). The risk difference in favor of canakinumab vs. placebo (62%) in patients taking concomitant colchicine was of similar magnitude as that in the overall population (55%; Table 8), indicating the added benefit of canakinumab beyond that provided by colchicine therapy. These treatment differences were demonstrated on top of the maximal tolerated colchicine dose. Both the canakinumab and placebo group received comparable daily colchicine doses (1.6±0.89 and 1.7±0.63 mg/day, respectively), and treatment responders (1.5±0.86 mg/day) did not receive higher doses of colchicine vs. non-responders (1.7±0.71 mg/day).
Table 8 – Comparison between treatment groups for primary and secondary efficacy endpoints in crFMF
patients after 16 weks by concomitant colchicine use (Full analysis set)
Prior use of biologics
A small proportion of patients had prior exposure to biologics (23.8% of crFMF patients; 18.1% of HIDS/MKD patients; and 34.8% of TRAPS patients). In this subgroup of patients with prior exposure to biologics, there were more responders in Epoch 2 in the canakinumab group than in the placebo group for the crFMF cohort and TRAPS cohort (Table 9). In the HIDS/MKD cohort, 1 patient in the canakinumab group was a responder compared with 0 patients in the placebo group. The responder profile among patients without prior exposure to biologics was similar to the overall population, with a higher proportion of patients on canakinumab compared to placebo achieving a complete response in all 3 disease cohorts (Table 9). Similarly, there were slightly more patients with PGA < 2, CRP ≤ 10 mg/L or SAA ≤ 10 mg/L in the canakinumab group compared with the placebo group in crFMF, HIDS/MKD and TRAPS patients, although comparisons are limited by the very small number of patients with prior use of biologics. Table 9 – Comparison between treatment groups for the proportion of responders by prior use of biologics (Full analysis set)

Assessment report EMA/26517/2017 Page 32/60
Summary of main study
The following tables summarise the efficacy results from the main studies supporting the present application. These summaries should be read in conjunction with the discussion on clinical efficacy as well as the benefit risk assessment (see later sections).
Table 10 - Summary of Efficacy for trial N2301
Title:
A randomized, double-blind, placebo controlled study of canakinumab in patients with Hereditary Periodic Fevers (TRAPS, HIDS, or crFMF), with subsequent randomized withdrawal/dosing frequency reduction and open-label long term treatment epochs
Study identifier ACZ885N2301
Design Randomized, double-blind, placebo controlled
Duration of main phase: 16 weeks
Duration of Run-in phase: not applicable
Duration of Extension phase: not applicable
Hypothesis Superiority of Canakinumab in patients within each of 3 disease cohorts (TRAPS, HIDS, or crFMF),
Treatments groups
Canakinumab
Canakinumab 150 mg (or 2 mg/kg for patients weighing ≤ 40 kg) q4w for 24 weeks, n = 31 (crFMF), n = 36 (HIDS/MKD), n = 22 (TRAPS)
Placebo Placebo q4w for 24 weeks, n = 31 (crFMF), n = 33 (HIDS/MKD), n = 24 (TRAPS)

Assessment report EMA/26517/2017 Page 33/60
Endpoints and definitions
Primary endpoint
Response
resolution of the index disease flare at day 15 without experience a new flare from the time of the resolution of the index flare until the end of epoch 2
Secondary endpoint
PGA response
PGA < 2 at week 16
Secondary endpoint
CRP response
CRP ≤ 10 mg/L at week 16
Secondary endpoint
SAA response
SAA ≤ 10 mg/L at week 16
Database lock Date not provided
Results and Analysis
Analysis description Primary Analysis
Analysis population and time point description
FAS (all patients randomized to epoch 2 who received at least 1 dose of study drug during epoch 2).
Descriptive statistics and estimate variability
Disease cohort crFMF HIDS/MKD TRAPS
Treatment group Canakinumab
Placebo
Canakinumab
Placebo
Canakinumab
Placebo
Number of subject
31 32 37 35 22 24
Response
n (%)
19 (61.3%)
2 (6.3%)
13 (35.1%)
2 (5.7%)
10 (45.5%)
2 (8.3%)
95%-CI
(42.2, 78.2)
(0.8, 20.6)
(20.2, 52.5)
(0.7, 19.2)
(24.4, 67.8)
(1.0, 27.0)
Effect estimate per comparison
crFMF cohort
Comparison groups Canakinumab vs. placebo
Odds ratio 23.75
95%-CI (4.38, 227.53)
P-value < 0.0001
HIDS/MKD cohort
Odds ratio 8.94

Assessment report EMA/26517/2017 Page 34/60
95%-CI (1.72, 86.41)
P-value 0.002
TRAPS cohort
Odds ratio 9.17
95%-CI (1.51, 94.61)
P-value 0.005
Notes Within each of the 3 disease cohorts the primary endpoint was met.
Analysis description Secondary analysis (PGA response)
Descriptive statistics and estimate variability
Disease cohort crFMF HIDS/MKD TRAPS
Treatment group Canakinumab
Placebo
Canakinumab
Placebo
Canakinumab
Placebo
Number of subject
31 32 37 35 22 24
Response
n (%)
23 (74.2%)
3 (9.4%)
25 (67.6%)
2 (5.7%)
15 (68.2%)
1 (4.2%)
95%-CI
(55.4, 88.1)
(2.0, 25.0)
(50.2, 82.0)
(0.7, 14.9)
(45.1, 86.1)
(0.1, 21.1)
Effect estimate per comparison
crFMF cohort
Comparison groups Canakinumab vs. placebo
Odds ratio 26.55
95%-CI (6.23, 113.03)
P-value < 0.0001
HIDS/MKD cohort
Odds ratio 33.85
95%-CI (6.93, 99.47)
P-value < 0.0001
TRAPS cohort
Odds ratio 61.08
95%-CI (6.17, 604.74)
P-value 0.0002
Analysis description Secondary analysis (CRP response)
Descriptive statistics Disease cohort crFMF HIDS/MKD TRAPS
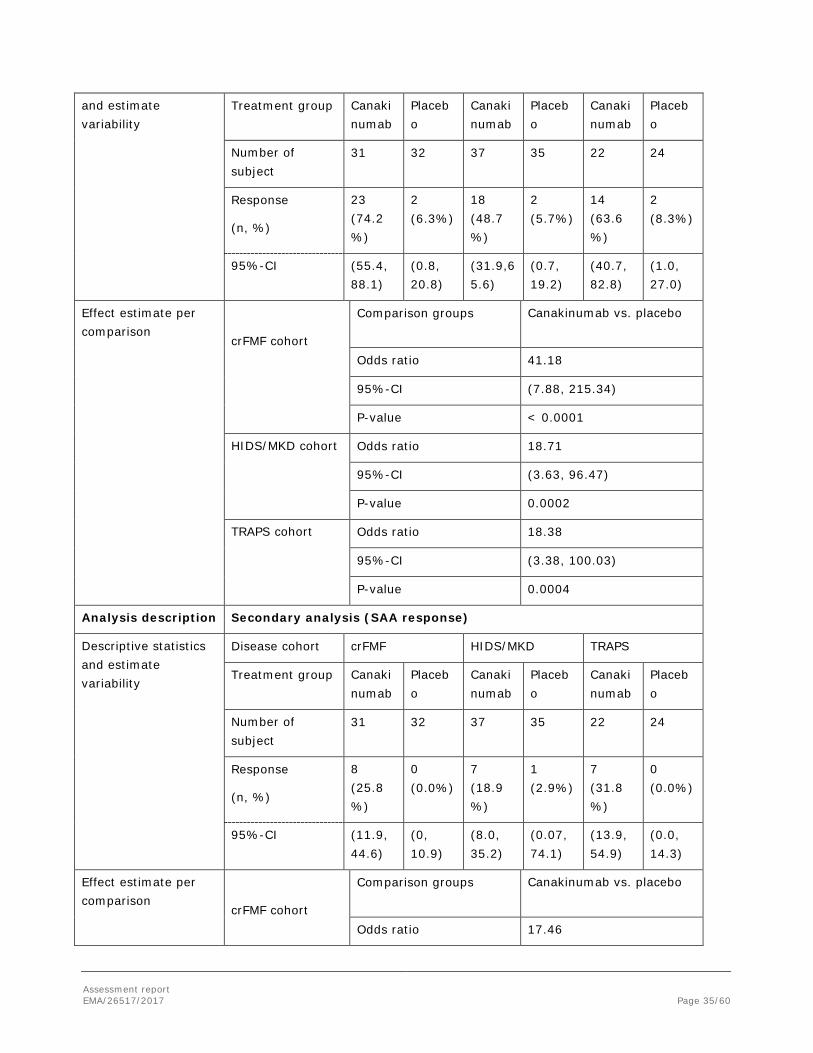
Assessment report EMA/26517/2017 Page 35/60
and estimate variability
Treatment group Canakinumab
Placebo
Canakinumab
Placebo
Canakinumab
Placebo
Number of subject
31 32 37 35 22 24
Response
(n, %)
23 (74.2%)
2 (6.3%)
18 (48.7%)
2 (5.7%)
14 (63.6%)
2 (8.3%)
95%-CI
(55.4, 88.1)
(0.8, 20.8)
(31.9,65.6)
(0.7, 19.2)
(40.7, 82.8)
(1.0, 27.0)
Effect estimate per comparison
crFMF cohort
Comparison groups Canakinumab vs. placebo
Odds ratio 41.18
95%-CI (7.88, 215.34)
P-value < 0.0001
HIDS/MKD cohort
Odds ratio 18.71
95%-CI (3.63, 96.47)
P-value 0.0002
TRAPS cohort
Odds ratio 18.38
95%-CI (3.38, 100.03)
P-value 0.0004
Analysis description Secondary analysis (SAA response)
Descriptive statistics and estimate variability
Disease cohort crFMF HIDS/MKD TRAPS
Treatment group Canakinumab
Placebo
Canakinumab
Placebo
Canakinumab
Placebo
Number of subject
31 32 37 35 22 24
Response
(n, %)
8 (25.8%)
0 (0.0%)
7 (18.9%)
1 (2.9%)
7 (31.8%)
0 (0.0%)
95%-CI
(11.9, 44.6)
(0, 10.9)
(8.0, 35.2)
(0.07, 74.1)
(13.9, 54.9)
(0.0, 14.3)
Effect estimate per comparison
crFMF cohort
Comparison groups Canakinumab vs. placebo
Odds ratio 17.46

Assessment report EMA/26517/2017 Page 36/60
95%-CI (0.92, 332.92)
P-value 0.0286
HIDS/MKD cohort
Odds ratio 8.24
95%-CI (0.92, 74.07)
P-value 0.0299
TRAPS cohort
Odds ratio 20.87
95%-CI (1.26, 344.67)
P-value 0.0169
Notes With the exception of SAA response in the HIDS/MKD cohort, the secondary efficacy endpoints indicate a statistically significant superiority of Canakinumab when compared to placebo in all 3 disease cohorts.
Analysis performed across trials (pooled analyses and meta-analysis)
There was no pooling of efficacy data from the randomized, placebo-controlled pivotal Phase 3 studies and the 4 open-label, uncontrolled Phase 2 studies, as the study design, efficacy variables and dosing regimens differ across the studies.
Supportive studies
Four uncontrolled studies contributed for further efficacy data in the target population of periodic fever syndromes. Efficacy measurements were made relative to baseline values. The design of the earlier uncontrolled Phase 2 POC trials in crFMF, HIDS/MKD and TRAPS is briefly summarized in Table 11.

Assessment report EMA/26517/2017 Page 37/60
Table 11 – Summary of uncontrolled phase 2 studies in crFMF, HIDS/MKD and TRAPS
The results of 9 patients in study DTR01 can be regarded as supportive and provide some evidence that treatment with canakinumab in patients with crFMF is able to reduce the number of attacks and at least prolongs the time to the next attack. During the follow up period without active treatment, median time to subsequent disease attack was 71 days. This is a clinically important finding although the number of treated patients is rather too small to draw final conclusions.
Study D2204 included 7 children with crFMF. Patients aged between 4 and 20 years were treated every 4 weeks with the now proposed dosing regimen of canakinumab for crFMF. The presented results are very promising although the patient numbers are small. Canakinumab treatment led to a pronounced reduction in disease flare/attack rate which is medically very important. Also the improvement of laboratory parameters shows that the underlying inflammatory process is markedly reduced.
Study D2402 in HIDS/MKD patients used a slightly different dosing regimen of 300mg every 6 weeks with options for additional dosing. Study D2402 enrolled 9 patients with a median time since diagnosis of 4.1 years, age range of 5-29 years and with genetically or enzymatically approved HIDS. The included patients can be regarded as representative for the periodic fever syndrome HIDS/MKD. The primary endpoint of study D2402 in patients with HIDS/MKD was met as the median number of flares per patient decreased from 5 (range: 3-12) in

Assessment report EMA/26517/2017 Page 38/60
the 6-month run-in period to 0 (range: 0-2) during the 6-month treatment period. The assessment of HIDS/MKD control by patient and physician improved from no disease control or poor control at baseline to good or excellent control at Day 4 until the end of the study. Laboratory parameters (CRP, SAA) normalized starting at Day 15 and this was sustained through the end of study. Canakinumab treatment therefore reduced the HIDS flare rate and reduced the flare severity. The results of this pilot study are supportive for the overall efficacy of canakinumab treatment in periodic fever syndromes and are clinically highly relevant for the affected patients.
Study D2203 was a pilot study for TRAPS in 20 patients. Study design and enrolment criteria are appropriate to allow assessment of efficacy, though in an open label setting, for this rare disease in a well-defined patient population. To be included into the study, the patients had to have a mutation of TNFRSF1A gen. Dosing scheme in study D2203 corresponds to the proposed dosing for periodic fever syndromes.
The primary objective of Study D2203 in patients with TRAPS was met. 19/20 (95.0%) patients achieved a complete or almost complete response at Day 15. Of the 19 patients with a complete/almost complete response at Day 15, 16 patients achieved this outcome already by day 8 and the remaining 3 patients did so by Day 15. Furthermore, CRP and SAA levels decreased qickly until day 8 and stayed in the normal range for four month. With respect to physician`s global assessment of TRAPS, there was a shift to less disease activity for all patients during the treatment period and all patients had no or minimal disease activity at Day 113. These results are clinically relevant in the target population and are supportive for the overall efficacy of canakinumab in the proposed new indication TRAPS.
Data on sustained efficacy in long term use is still very limited (29 patients) and so far can be regarded as supportive only.
2.5.3. Discussion on clinical efficacy
Design and conduct of clinical studies
A single pivotal Phase 3 study, N2301, which includes 3 randomized cohorts (one cohort per condition: crFMF, HIDS/MKD and TRAPS), was submitted in support of this application.
The study design followed the scientific advices received and is regarded as appropriate to investigate the efficacy in the proposed indications. No major protocol violations were recorded.
All patients in the crFMF, HIDS/MKD and TRAPS cohorts of study N2301 had a medical history of the respective condition as well as a confirmed genetic mutation. All patients had active disease as defined by clinical and serological criteria at the time of randomization. crFMF patients were either experiencing active disease despite colchicine therapy or were intolerant to effective doses of colchicine.
The chosen primary endpoint and the time point of assessment allowed appropriate evaluation of efficacy in comparison to placebo control.
Four uncontrolled studies contributed further efficacy data in the target population of periodic fever syndromes. Efficacy measurements were made relative to baseline values. The methods used for the efficacy evaluation were comparable across the studies. The primary and secondary endpoints of the pivotal study were more robust than the ones used in the phase 2 studies and adequate to demonstrate a clinically relevant treatment response. The methods for blinding in the pivotal study were acceptable. The reported protocol deviations during the pivotal study seem not to have confounded the interpretation of the study results, as verified by Per Protoxol analysis.

Assessment report EMA/26517/2017 Page 39/60
Efficacy data and additional analyses
In study N2301, 181 patients were randomized and 4 patients entered the non-randomized open label period. 63 crFMF patients were randomized, 72 with HIDS/MKD, and 46 patients with TRAPS. In view of the rarity of the three periodic fever syndromes, the numbers that were recruited and randomized are sufficient to evaluate efficacy.
The data shows that the primary endpoint was met for all three cohorts and that canakinumab was statistically superior to placebo in resolving the index disease flare at day 15 and in preventing new flares before the end of week 16. The best results can be seen for the crFMF cohort and slightly weaker response, although still statistically significant, in the TRAPS cohort. The study included the option for dose escalation in case the index flare did not resolve by day 15. This escape option had to be used by 16% of crFMF patients, 32% of HIDS/MKD patients, and 36% of TRAPS patients. However, this option enabled especially crFMF and TRAPS patients to reach resolution of index flare by day 29.
The results of the secondary and exploratory efficacy analysis support the results of the primary endpoint. It is shown that treatment with canakinumab is able to normalize the CRP levels in all three disease cohorts in a statistically significant way in comparison to placebo. Furthermore, evaluation of PGA shows a reduction to <2 also in a statistically significant way. These results are regarded as clinically meaningful improvement of disease status after 3 month of treatment.
In the exploratory analysis of the primary efficacy endpoint where the responder rate was expanded to include all the patients who achieved resolution of the index flare at Day 29 and no new flare after Day 29 up to the end of the 16-week treatment period with either the randomized treatment or with an up-titration to 300 mg q4w before Day 29, the proportion of responders increased to 71%, 72% and 57% in the crFMF, TRAPS and HIDS cohorts, respectively. The results of this analysis are clinically very relevant. As suggested by this analysis and also by the exploratory analysis of the need for blinded escape medication, it can be suggested that the patients with HIDS or TRAPS may need somewhat higher doses of canakinumab for full treatment response compared to the crFMF patients. The proportion of the patients with need for up-titration of canakinumab from Days 8 to 28 was somewhat higher in the HIDS and TRAPS cohorts compared to the crFMF cohort. An initial dose of canakinumab 150 mg given every 4 weeks and with possibility for up-titration to 300 mg every 4 weeks is justified from efficacy point of view in all applied indications. The PK/PD-modelling suggests that some HIDS-patients who were uncontrolled at 300 mg could benefit from a higher dose to reduce new flares. The MAH was requested to present and discuss all the available data in this context and it is agreed by CHMP that the present limited clinical data do not justify the recommendation for increasing the dose in some HIDS-patients >300mg.
All presented subgroup analyses are supportive to the primary and secondary analysis. Due to the partly very small cohorts of patients, the possibility to draw conclusions is limited. Subgroup analyses with regard to age, prior use of biological therapy and concomitant use of colchicine do not show any clear signals of reduced or improved efficacy.
Only two patients in the canakinumab group and 6 patients in the placebo group were not taking concomitant colchicine. Therefore, the proposed indication for the patients with contraindication or intolerance to colchicine was questioned. As colchicine is available as a licenced medicinal product for the treatment of FMF only in some Member States of the EU (Netherlands, Spain, Portugal, Slovakia and United Kingdom), the MAH was requested to discuss the possibility of extrapolating the results of concomitant use of colchicine and canakinumab in the colchicine resistant FMF to the use of canakinumab as a single agent in the treatment of crFMF and colchicine naïve FMF, i.e. regardless of the prior/ concomitant use of colchicine, based on all available data and literature.

Assessment report EMA/26517/2017 Page 40/60
According to the MAH, the data of the patients without concomitant or prior use of colchicine is too limited for reliable extrapolation of the effect of canakinumab in this FMF sub-population. It is acknowledged that there is a clinical need for the treatment of FMF in these relatively rare patients with a contraindication, intolerance or insufficient response to colchicine, or in colchicine naïve patients who do not have access to colchicine therapy (as colchicine is not licenced for the treatment of FMF in many Member States). On the other hand, it is recommended in the recent EULAR recommendation for the treatment of FMF that "colchicine should be co-administered with alternative biological therapies given that it may reduce the risk of amyloidosis despite persistence of attacks". Further, there is biological plausibility that canakinumab would have similar effect in the treatment of FMF without colchicine.
Taking into account all the above-mentioned aspects, the indication was changed as follows to cover all FMF patients with a potential to benefit from canakinumab treatment:
Ilaris is indicated for the treatment of Familial Mediterranean Fever (FMF). Ilaris should be given in combination with colchicine, if appropriate.
In the relatively small subgroup of patients (44 out of 181 participants) with prior use of biologics, the proportion of responders in the canakinumab group was of the same magnitude as in the overall crFMF and TRAPS cohorts. In contrast, the corresponding proportion was unexpectedly low (1 out of 9 patients) in the HIDS cohort. The MAH adequately discussed the possible reasons of this finding in the HIDS population.
Data on sustained efficacy in long term use is still very limited and needs further evaluation. Data presented so far can be regarded as supportive only. However, the MAH presented with the response to questions an interim study report presenting the results of Epoch 3 (week 40). After reviewing these results it can be concluded that treatment with canakinumab 150 mg given at the extended q8w dosing interval provided sufficient disease control for more than half of crFMF and TRAPS patients, while the majority of HIDS/MKD patients required a higher exposure (canakinumab 150 mg q4w, 300 mg q8w, or 300 mg q4w) to maintain disease control. Improvements in CRP, SAA, and PGA from study baseline were sustained throughout the duration of Epoch 3.
Four uncontrolled open label studies (DTR01and D2204 in crFMF, D2402 in HIDS/MKD, D2203 in TRAPS) contributed with further supportive efficacy data in the target population of periodic fever syndromes. Efficacy measurements were made relative to baseline values.
The presented results of study DRT01 in 9 crFMF patients are supportive to the efficacy of canakinumab treatment in this periodic fever syndrome. The presented results of study D2204 are also positively supportive of the main efficacy results although the patient numbers are small (7 children). The results of study D2402 are supportive for the overall efficacy of canakinumab treatment in periodic fever syndromes and are clinically highly relevant for the affected patients. 20 patients with confirmed gene mutation indicative for TRAPS were enrolled into study D2203 and are regarded as representative for the indication. The primary objective was met. These results are clinically relevant in the target population and are supportive for the overall efficacy of canakinumab in the proposed new indication TRAPS.
2.5.4. Conclusions on the clinical efficacy
Results from study N2301 show the superiority of canakinumab treatment compared to placebo treatment in the proposed indications crFMF, HIDS/MKD, and TRAPS. Laboratory parameters improved to normal levels, demonstrating reduction of inflammatory reaction. The efficacy of treatment sustained over a prolonged time even after withdrawal of study drug. Patients and physicians voted the disease activity as markedly reduced and health related quality of live parameters mostly also showed improvement.

Assessment report EMA/26517/2017 Page 41/60
Results from the four supportive phase II pilot studies were consistent with the results from phase III study N2301. All four studies reached the primary endpoints and therefore demonstrated that canakinumab was able to reduce the disease attack rate, flare severity and normalized laboratory parameters of interest (CRP, SAA).
The pivotal phase 3 study is still ongoing and will provide important information on long-term efficacy, and also importantly on the dosing in the long-term use. The so far limited long term data from the 2-year treatment in studies D2402 and D2203 is promising (majority of HIDS/MKD and TRAPS patients without re-flare) but needs further enrichment from upcoming data of period 4 of study N2301. The CHMP recommended that this data should be submitted post-approval.
2.6. Clinical safety
Patient exposure
The strategy for evaluation of safety in periodic fever syndromes is focussing on four different patient sets. Patients from study N2301 epoch 2 were pooled and include 181 patients in the three different diseases (63 crFMF patients, 72 HIDS/MKD patients, and 46 TRAPS patients) with a median duration of exposure of 113 days and a cumulative exposure of 47.6 patient-years. This safety population reflects the safety results for each patient up to Week 16 (end of Epoch 2).
The second, supportive pool presents data from study N2301 epochs 2-4 with a data cut off 25th August 2015 with a duration of exposure to placebo (9.39 patient-years) much shorter than the duration of exposure to canakinumab (114.85 patient-years) since all data collected in Epochs 3 and 4 are counted within the canakinumab treatment group.
The third pooled set is the CAPS data, already reflected in the SmPC.
Finally the fourth group of additional descriptive safety data results from the four open label pilot studies that were not pooled.
Overall, the presented patient numbers in this group of orphan indications are seen as representative although long term safety data are still lacking from study N2301 and will be presented after the end of epoch 4/study.
Adverse events
The adverse event profile of canakinumab treatment is overall mostly comparable in the new proposed indication with the approved CAPS indication.
The most prominent adverse events were infections and infestations, pyrexia and headache. The most common infections are those of the upper respiratory tract. All infections were mild or moderate in severity.
Injection site reactions seemed to occur more frequently in study N2301 groups compared to CAPS data. Apart from these injection site reactions, all other AEs were reported more frequently in the placebo population based on exposure adjusted event rates. It is noted that all patients in the pivotal phase 3 study N2301, comprising majority of the crFMF/ HIDS/ TRAPS patients, were treated with the new liquid in vial formulation of canakinumab while the approved lyophilized product was used in the CAPS-studies. Further analyses demonstrated that injection site reactions did not occur more in patients receiving liquid in vial than in those receiving lyophilized preparation, 12.4% vs. 15.9%, respectively.
Infections and infestation, injection site reactions with the most reported AES possibly related to study treatment.

Assessment report EMA/26517/2017 Page 42/60
The AE profiles observed in Study DTR01 and Study D2204 in crFMF patients, in Study D2402 in HIDS/MKD patients and in Study D2203 in TRAPS patients were consistent with the AE profile of the N2301 Phase 3 population. AEs in the infections and infestations SOC were observed more commonly, mainly due to upper respiratory tract-related infections.
Serious adverse event/deaths/other significant events
No deaths were reported in Study N2301 as of the data cut-off date (25-Aug-2015). No deaths were reported in the Phase 2 studies or in the CAPS pooled group.
In the N2301 Epoch 2 crFMF cohort, the incidence of SAEs was low in the Any ACZ group (5 patients, 8.6%). No SAE was reported in more than 1 patient. The SAEs considered by the investigator to be possibly related to study drug were granulomatous liver disease, pharyngo-tonsillitis, and atypical pneumonia. In the N2301 Epoch 2 HIDS/MKD cohort, the incidence of SAEs was 11.8% (8 patients) in the Any ACZ group. With the exception of pneumonia, reported in 2 patients in the placebo (Pbo) to 150 mg group, no SAE was reported in more than 1 patient. One SAE, neutropenia, which occurred in a patient in the Pbo group, was considered by the investigator to be related to study drug. In the N2301 Epoch 2 TRAPS cohort, the incidence of SAEs was low, affecting 4.7% (2 patients) in the Any ACZ group. No SAE was reported in more than 1 patient. No SAE was considered by the investigator to be related to study drug.
In Epochs 2-4, the incidence of SAEs in the Total ACZ group was 10.7% (18 patients). No SAE was reported in more than 1 patient in any particular cohort except umbilical hernia (2 patients (3.4%) in crFMF ACZ group) and pneumonia (3 patients (4.4%) in HIDS/MKD ACZ group). The SOC with the highest frequency of SAEs was infections and infestations (4.1% in the Total ACZ group). The exposure-adjusted SAE rate per 100 patient-years was 30.5 in the Total ACZ group and 85.2 in the Total placebo group.
No SAEs were reported during the treatment phase of crFMF Studies DTR01 or D2204. In HIDS/MKD Study D2402, a total of 14 SAEs were reported in 4 patients, none of which were considered by the investigator to be related to study treatment. In TRAPS Study D2203, 7 patients experienced an SAE during the treatment and follow-up periods, the most common of which was abdominal pain (2 patients). The profile of SAEs in the Phase 2 studies in was similar to that observed in Study N2301.
Laboratory findings
Haematology
Hematology data from patients in Study N2301 was in line with the known safety profile of canakinumab.
Most of the newly occurring or worsening hematology abnormalities in Epoch 2 in the crFMF, HIDS/MKD, and TRAPS cohorts were CTCAE grade 1 or 2. Most of these hematology abnormalities were decreases in neutrophils, leukocytes and platelets in patients receiving canakinumab. One HIDS/MKD patient and 1 TRAPS patient had a grade 3 neutropenia, and 1 HIDS/MKD patient had a grade 3 thrombocytopenia. No patient had a grade 4 hematology abnormality. The hematological profile in Epochs 2-4 was comparable to that of Epoch 2, with no additional grade 3 or 4 events in Epochs 3 or 4.
Similar to what was observed in Study N2301 Epochs 2-4, most of the newly occurring or worsening hematology abnormalities in the CAPS pooled population were grade 1 or 2. However, the Total ACZ group showed a higher incidence of grade 1 reductions in platelets (Total ACZ: 27.8%, CAPS: 0.5%), grade 1 reductions in leukocytes (Total ACZ: 21.4%, CAPS: 4.2%), grade 1 reductions in neutrophils (Total ACZ: 13.7%, CAPS: 3.7%), and grade 2 reductions in neutrophils (Total ACZ: 7.1%, CAPS: 1.6%).

Assessment report EMA/26517/2017 Page 43/60
Clinical chemistry and urinalysis
Patient clinical chemistry and urinalysis data from Study N2301 was in line with the known safety profile of canakinumab. No safety concerns were noted in any parameter. The clinical chemistry and urinalysis profile seen in Epochs 2-4 was comparable to that of Epoch 2. In all N2301 cohorts in Epoch 2 and in Epochs 2-4, all newly occurring or worsening notable post baseline abnormalities in biochemistry and urinalysis parameters were mild or CTCAE grade 1. In Epochs 2-4 in the Total ACZ group, 0 patients showed changes in creatinine clearance, gamma glutamyl transferase, cholesterol, or triglycerides. The most commonly observed change was a creatinine increase ≥ 25% from baseline, which occurred in 28.4% of patients. No patients in the Total ACZ group had a creatinine increase ≥ 3 times the ULN. Overall, clinical chemistry and urinalysis changes were comparable across the crFMF, HIDS/MKD, and TRAPS cohorts. Patients in the N2301 Epoch 2-4 group showed either a comparable number or fewer newly occurring or worsening notable post-baseline abnormalities in biochemistry and urinalysis parameters vs. the CAPS group.
Abnormalities in biochemistry and urinalysis parameters are reflecting the currently known profile of canakinumab treatment with an even lower rate compared to the CAPS patients.
Data on renal function were collected in the crFMF cohort. There were a small number of patients with improvement in kidney function.
Results of vital signs observation are mostly unremarkable apart from25 canakinumab treated patients presenting with a decrease in pulse rate >20 compared to baseline.
Safety in special populations
Table 12 – Safety in special populations

Assessment report EMA/26517/2017 Page 44/60
Safety data (AEs, notable laboratory data (by weight and dose level only), and notable vital sign data (by age only) for the N2301 patient population (Epochs 2-4) and for the CAPS pooled population were evaluated according to the following subgroups based on intrinsic demographic factors and cumulative canakinumab dose level:
- Age: ≥ 2 to < 4 years, ≥ 4 to < 6 years, ≥ 6 to < 12 years, ≥ 12 to < 18 years, ≥ 18 years to < 65 years, and ≥ 65 years
- Gender: male, female
- Race: Caucasian, Asian, Other

Assessment report EMA/26517/2017 Page 45/60
- Region: North America (US, Canada), Japan, Other
- Body weight: ≤ 40 kg, > 40 kg
- Cumulative N2301 dose level over 16 weeks: ≤ 600 mg, > 600 mg
There seem to be also a slight tendency for increased rates of haematological changes and changes in creatinine levels with a higher cumulative dose. Adverse events with a substantially higher incidence in patients in the Total ACZ group with cumulative doses >600 mg compared with patients on doses ≤600 mg included: abdominal pain, arthralgia, cough, diarrhea, headache, injection site reaction, nasopharyngitis, oropharyngeal pain, pyrexia, rhinitis, and vomiting.
The profile of AEs in each of these subgroups of intrinsic and extrinsic factors was consistent with that observed for the overall crFMF, HIDS/MKD, TRAPS, and CAPS populations
Safety related to drug-drug interactions and other interactions
No specific drug interaction studies were performed with canakinumab. No Canakinumab-immunosuppressants combination therapy toxicities were reported in Study N2301
Discontinuation due to adverse events
No AEs leading to discontinuation were reported in Epoch 2 in the crFMF or TRAPS cohort.
In Epoch 2, the incidence of AEs leading to discontinuation in the HIDS/MKD cohort was low, affecting 2.9% (2 patients) in the Any ACZ group. The 2 AEs that led to discontinuation were HIDS flare (1 patient in the Pbo to 150 mg q4w to 300 mg q4w group) and pericarditis (1 patient in the 150 mg q4w to 300 mg q4w group), which was not considered by the investigator to be related to study drug.
In Epochs 2-4, 2 additional patients experienced an AE that led to discontinuation. These patients, one in the TRAPS ACZ group and one in the HIDS/MKD placebo group, had an AE of neutropenia.
The rate of study discontinuations due to AEs is below 3%.
Post marketing experience
As reported in PSUR 12 (cut-off date 30 Jun 2015), the estimated cumulative exposure from marketing experience for canakinumab was approximately 7000 patient years.
In terms of complete exposure, and taking into account subjects treated for all indications under all dose regimens, cumulatively >3400 subjects have been exposed to canakinumab in completed and reported clinical trials. This total represents a cumulative clinical trial exposure of just over 3140 patient-years for which safety data has been collected.
This is a representative safety data base supporting the safe use of canakinumab in the different indications
2.6.1. Discussion on clinical safety
The assessment of the safety data primarily focuses on the data from study N2301 interim report of week 16 primary endpoint analysis. Safety data of the CAPS studies were already assessed in various procedures and are well reflected in the SmPC.

Assessment report EMA/26517/2017 Page 46/60
Patient population was presented in four patient sets. Overall, the presented patient numbers in this group of rare indications are seen as representative although long term safety data are still lacking from study N2301 and will be presented after the end of epoch 4/study.
The adverse event profile of canakinumab treatment is overall mostly comparable in the new proposed indication with the approved CAPS indication.
The most prominent adverse events were infections and infestations, pyrexia and headache. The most common infections are those of the upper respiratory tract. All infections were mild or moderate in severity. Other observed adverse events that occur more frequently are diarrhoea and abdominal pain. Injection site reactions occur more frequently in study N2301 groups compared to CAPS data. Apart from these injection site reactions, all other AEs were reported more frequently in the placebo population based on exposure adjusted event rates.
Infections and infestation, injection site reactions, and gastrointestinal disorders were the SOCs with the most reported AES possibly related to study treatment.
It is noted that all patients in the pivotal phase 3 study N2301, comprising majority of the crFMF/ HIDS/ TRAPS patients, were treated with the new liquid in vial formulation of canakinumab while the approved lyophilized product was used in the CAPS-studies. With further analyses the MAH was able to prove, that injection site reactions did not occur more in patients receiving liquid in vial than in those receiving lyophilized preparation, 12.4% vs. 15.9%, respectively.
No death occurred during the phase II and III studies.
The rate of SAEs does not differ markedly from the already known safety profile of canakinumab. Observations from the phase II open label studies also support this.
Results of the evaluation of haematology data shows that more patients have abnormalities in their platelet counts, neutrophil counts and leukocyte counts compared to placebo and also compared to the CAPS population. Most of the haematology abnormalities were CTC grade 1 or 2, 2 patients had a grade 3 neutropenia. Grade 1 thrombocytopenia occurred in 27.8% of canakinumab treated patients compared to 4.1% in placebo patients. This is properly reflected in the SmPC (see section 4.8). Neutropenia is a known adverse drug reaction of canakinumab and should be monitored as described in the SmPC.
According to PK/PD-modelling report, the occurrences leukopenia, neutropenia, or thrombopenia were not dependent on the serum ACZ885 concentration. The MAH has provided an analysis of the cases of thrombocytopenia, leukopenia, and neutropenia – with the related AEs – in the crFMF/HIDS/TRAPS, and CAPS populations, as well as in the placebo group. There were no significant safety issues as the observed abnormalities were reversible and did not lead to dose reductions or withdrawals. This is reflected appropriately in the SmPC (see section 4.8).
Abnormalities in biochemistry and urinalysis parameters are reflecting the currently known profile of canakinumab treatment with an even lower rate compared to the CAPS patients.
Data on renal function were collected in the crFMF cohort. There were a small number of patients with improvement in kidney function. The patient numbers are however too small to draw definite conclusions.
The presented subgroup analysis is not promoting any concerns regarding safety in special populations. The finding that female patients might have more adverse events when treated with canakinumab compared to males was already described in the original assessment for marketing authorisation and was also found in the gout population.

Assessment report EMA/26517/2017 Page 47/60
There seem to be also a slight tendency for increased rates of haematological changes and changes in creatinine levels with a higher cumulative dose of canakinumab. Adverse events with a substantially higher incidence in patients in the Total ACZ group with cumulative doses >600 mg compared with patients on doses ≤600 mg included abdominal pain, arthralgia, cough, diarrhoea, headache, injection site reaction, nasopharyngitis, oropharyngeal pain, pyrexia, rhinitis, and vomiting. Patterns were similar to the overall population. However, these findings are all limited by the relatively lower patient numbers in the different cohorts and the fact that the patient characteristics were presumably different in the two dose groups, and are therefore no final conclusion can be made. Sub-grouping the patients by age, race, and weight did not display clinically significant differences on the types or incidences of the AEs.
Immunogenicity data generated from study N2301 shows that no patient had neutralizing antibodies. Two patients in the phase II studies had treatment emergent ADA with emerging AE or loss of efficacy or changes in PK. Therefore, immunogenicity is not a concern of canakinumab treatment in the proposed indications.
The rate of study discontinuations due to AEs is below 3%.
As already mentioned data on long term use of canakinumab in the respective proposed indications is still rather limited and needs to be increased. Further data will be available after finishing of study N2301.
From the safety database all the adverse reactions reported in clinical trials and post-marketing have been included in the Summary of Product Characteristics.
2.6.2. Conclusions on the clinical safety
Treatment with canakinumab in the three proposed new indications of crFMF, HIDS/MKD, and TRAPS was associated with the occurrence of a wide variety of adverse events and serious adverse events. The most common were infections and infestations; neutropenia and events associated with the underlying disease as well as injections site reactions. Most of the events were of mild to moderate severity. No death was reported. The safety profile of canakinumab did not differ markedly from the already known and described profile in the already approved indications.
Long term safety data is still very limited in these indications but more data will become available with completion of study N2301.
In conclusion, the safety profile of canakinumab in the treatment of crFMF, HIDS/MKD, and TRAPS does not preclude the authorization of Ilaris in these indications. The clinical safety data submitted in support of this application was considerd acceptable.
2.6.3. PSUR cycle
The annex II related to the PSUR refers to the EURD list which remains unchanged.
2.7. Risk Management Plan
Safety concerns
Important identified risks
Infections Opportunistic infections Neutropenia Drug induced liver injury (DILI, hepatic transaminase and bilirubin
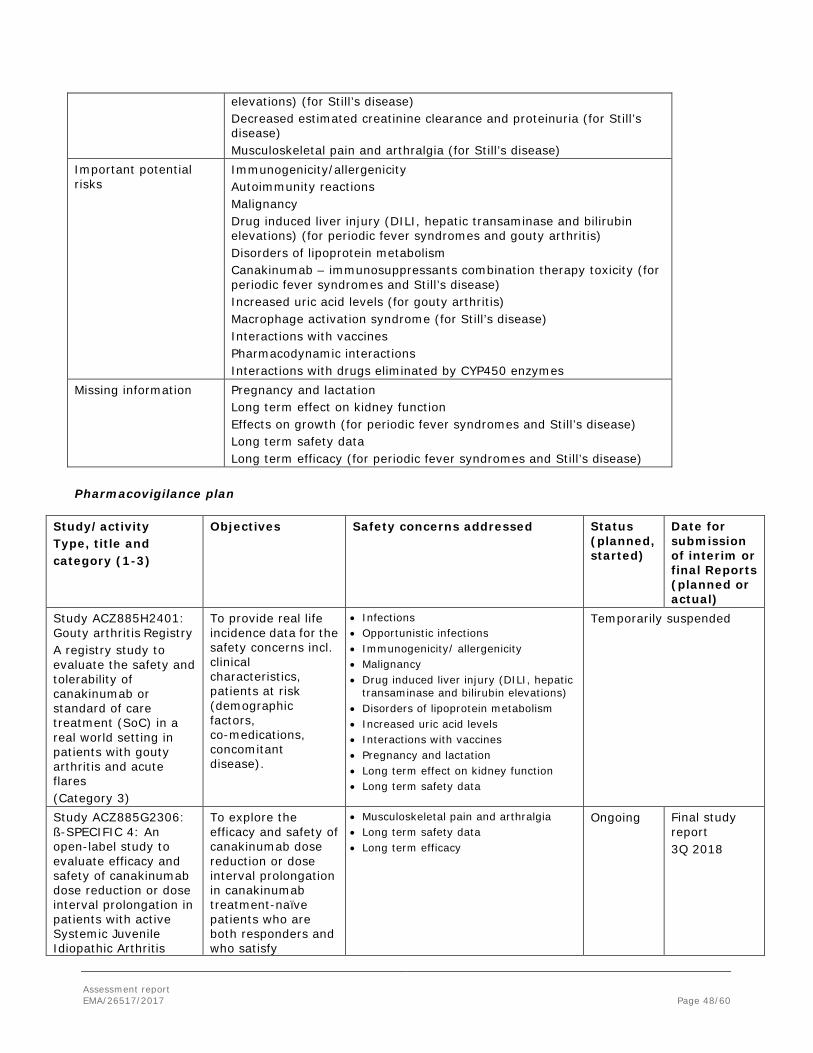
Assessment report EMA/26517/2017 Page 48/60
elevations) (for Still’s disease) Decreased estimated creatinine clearance and proteinuria (for Still’s disease) Musculoskeletal pain and arthralgia (for Still’s disease)
Important potential risks
Immunogenicity/allergenicity Autoimmunity reactions Malignancy Drug induced liver injury (DILI, hepatic transaminase and bilirubin elevations) (for periodic fever syndromes and gouty arthritis) Disorders of lipoprotein metabolism Canakinumab – immunosuppressants combination therapy toxicity (for periodic fever syndromes and Still’s disease) Increased uric acid levels (for gouty arthritis) Macrophage activation syndrome (for Still’s disease) Interactions with vaccines Pharmacodynamic interactions Interactions with drugs eliminated by CYP450 enzymes
Missing information Pregnancy and lactation Long term effect on kidney function Effects on growth (for periodic fever syndromes and Still’s disease) Long term safety data Long term efficacy (for periodic fever syndromes and Still’s disease)
Pharmacovigilance plan
Study/activity Type, title and category (1-3)
Objectives Safety concerns addressed Status (planned, started)
Date for submission of interim or final Reports (planned or actual)
Study ACZ885H2401: Gouty arthritis Registry A registry study to evaluate the safety and tolerability of canakinumab or standard of care treatment (SoC) in a real world setting in patients with gouty arthritis and acute flares (Category 3)
To provide real life incidence data for the safety concerns incl. clinical characteristics, patients at risk (demographic factors, co-medications, concomitant disease).
• Infections • Opportunistic infections • Immunogenicity/ allergenicity • Malignancy • Drug induced liver injury (DILI, hepatic
transaminase and bilirubin elevations) • Disorders of lipoprotein metabolism • Increased uric acid levels • Interactions with vaccines • Pregnancy and lactation • Long term effect on kidney function • Long term safety data
Temporarily suspended
Study ACZ885G2306: ß-SPECIFIC 4: An open-label study to evaluate efficacy and safety of canakinumab dose reduction or dose interval prolongation in patients with active Systemic Juvenile Idiopathic Arthritis
To explore the efficacy and safety of canakinumab dose reduction or dose interval prolongation in canakinumab treatment-naïve patients who are both responders and who satisfy
• Musculoskeletal pain and arthralgia • Long term safety data • Long term efficacy
Ongoing Final study report 3Q 2018

Assessment report EMA/26517/2017 Page 49/60
Study/activity Type, title and category (1-3)
Objectives Safety concerns addressed Status (planned, started)
Date for submission of interim or final Reports (planned or actual)
(SJIA) (Category 3)
pre-defined criteria for inclusion
Study ACZ885G2403: SJIA Registry
(Category 3)
To collect prospective safety, tolerability, efficacy, and treatment adherence information on juvenile idiopathic arthritis (JIA) subjects
It is expected that aspects of the following risks will be addressed: • Infections (serious infections and those
treated with IV anti-infective) • Opportunistic infections • Neutropenia • Drug-induced liver injury (DILI, hepatic
transaminase and bilirubin elevations) (routine ALT and AST levels; hepatic adverse events)
• Musculoskeletal pain and arthralgia – SAE only
• Immunogenicity/allergenicity - anaphylaxis/ hypersensitivity events
• Autoimmunity reactions - new autoimmunity disease events
• Malignancy • Disorders of lipoprotein metabolism -
routine total cholesterol and triglyerceride levels; hypercholesterolemia adverse events
• Canakinumab – immunosuppressants combination therapy toxicity – SAEs only
• Macrophage activation syndrome • Pregnancy and lactation • Long-term effects on kidney function –
SAE only • Long-term effects on growth - routine
height and weight measurements • Long-term efficacy
Planned Final study report 1Q 2024
Risk minimisation measures
Safety concern Routine risk minimization measures Additional risk minimization measures
Important identified risks Infections Labeling:
Addressed in SmPC in: Section 4.3 (Contraindication), Section 4.4 (Special warnings and precautions for use), Section 4.5 (Interaction with other medicinal products and other forms of interaction) and Section 4.8 (Undesirable effects)
Patient reminder card HCP information (as per local legislation)
Opportunistic infections Addressed in SmPC in: Section 4.4 (Special warnings and precautions for use) Section 4.8 (Undesirable effects)
Patient reminder card HCP information (as per local legislation )

Assessment report EMA/26517/2017 Page 50/60
Safety concern Routine risk minimization measures Additional risk minimization measures
Neutropenia Addressed in SmPC in: Section 4.4 (Special warnings and precautions for use) and Section 4.8 (Undesirable effects)
HCP information (as per local legislation):
Drug induced liver injury (DILI, hepatic transaminase and bilirubin elevations (for Still’s disease)
Addressed in SmPC in: Section 4.4 (Special warnings and precautions for use), and Section 4.8 (Undesirable effects)
None
Decreased estimated creatinine clearance and proteinuria (for Still’s disease)
Addressed in SmPC in: Section 4.8 (Undesirable effects)
None
Musculoskeletal pain and arthralgia (for Still’s disease)
Addressed in SmPC in: Section 4.8 (Undesirable effects)
None.
Important potential risks Immunogenicity/ allergenicity
Addressed in SmPC in: Section 4.3 (Contraindications), Section 4.4 (Special warnings and precautions for use) Section 4.8 (Undesirable effects), Section 5.1 (Pharmacodynamic properties)
HCP information (as per local legislation)
Autoimmunity reactions No risk minimization measure is considered necessary at this time. Upon the emergence of new safety findings related to autoimmunity reactions, as reviewed regularly in the PSUR, appropriate updated risk management activities will be considered.
None
Malignancy Addressed in SmPC in: Section 4.4 (Special warnings and precautions for use)
HCP information (as per local legislation)
Drug induced liver injury (DILI, hepatic transaminase and bilirubin elevations (for periodic fever syndromes and gouty arthritis)
Addressed in SmPC in: Section 4.4 (Special warnings and precautions for use), and Section 4.8 (Undesirable effects)
None
Disorders of lipoprotein metabolism
Addressed in SmPC in: Section 4.8 (Undesirable effects)
HCP information (as per local legislation)
Canakinumab – immunosuppressants combination therapy toxicity (for periodic fever syndromes and Still’s disease)
Addressed in SmPC in: Section 4.4 (Special warning and precautions for use) and Section 4.5 (Interaction with other medicinal products and other forms of interaction)
None

Assessment report EMA/26517/2017 Page 51/60
Safety concern Routine risk minimization measures Additional risk minimization measures
Increased uric acid levels (for gouty arthritis)
Addressed in SmPC in: Section 4.8 Undesirable effects
None
Macrophage activation syndrome (for Still’s disease)
Addressed in SmPC in: Section 4.4 (Special warnings and precautions for use)
Patient reminder card HCP information
Interactions with vaccines
Addressed in SmPC in: Section 4.4 (Special warnings and precautions for use) and Section 4.5 (interaction with other medicinal products and other forms of interaction)
HCP information (as per local legislation)
Pharmacodynamic interactions
Addressed in SmPC in: Section 4.4 (Special warnings and precautions for use) and Section 4.5 (interaction with other medicinal products and other forms of interaction)
None
Interactions with drugs eliminated by CYP450 enzymes
Addressed in SmPC in: Section 4.5 (Interaction with other medicinal products and other forms of interaction)
None
Missing information Pregnancy and lactation Addressed in SmPC in:
Section 4.6 (Fertility, pregnancy and lactation) HCP information (as per local legislation) Patient reminder card
Long term effect on kidney function
No risk minimization measure is considered necessary at this time
None
Effects on growth (for periodic fever syndromes and Still’s disease)
No risk minimization measure is considered necessary at this time
None
Long term safety data No risk minimization measure is considered necessary at this time
None
Long term efficacy (for periodic fever syndromes and Still’s disease)
No risk minimization measure is considered necessary at this time
None
Conclusion
The CHMP and PRAC considered that the risk management plan version 1.2 is acceptable.
2.8. Pharmacovigilance
Pharmacovigilance system
The CHMP considered that the pharmacovigilance system summary submitted by the MAH fulfils the requirements of Article 8(3) of Directive 2001/83/EC.

Assessment report EMA/26517/2017 Page 52/60
2.9. Product information
2.9.1. User consultation
The results of the user consultation with target patient groups on the package leaflet submitted by the MAH show that the package leaflet meets the criteria for readability as set out in the Guideline on the readability of the label and package leaflet of medicinal products for human use.
3. Benefit-Risk Balance
3.1. Therapeutic Context
3.1.1. Disease or condition
Tumour necrosis factor receptor associated periodic syndrome (TRAPS)
Ilaris is indicated for the treatment of tumour necrosis factor (TNF) receptor associated periodic syndrome (TRAPS).
Hyperimmunoglobulin D syndrome (HIDS)/mevalonate kinase deficiency (MKD)
Ilaris is indicated for the treatment of hyperimmunoglobulin D syndrome (HIDS)/mevalonate kinase deficiency (MKD).
Familial Mediterranean fever (FMF)
Ilaris is indicated for the treatment of Familial Mediterranean Fever (FMF). Ilaris should be given in combination with colchicine, if appropriate.
3.1.2. Available therapies and unmet medical need
To date, there are no approved therapies for HIDS/MKD or TRAPS. Certain medications have been used to treat the symptoms of these periodic fever syndromes, but these provide limited control over the relapsing febrile attacks and none treat the underlying inflammation.
For patients with FMF, colchicine has shown to be effective in controlling febrile attacks and preventing secondary amyloidosis in the majority of these patients. However, colchicine is not universally approved for the treatment of FMF and is associated with the significant side effects and patients who do not respond to or are intolerant to colchicine have very few, if any, treatment options.
Thus, there is an unmet medical need for a therapy that provides both relief of fever and symptoms as well as effective disease control.
3.1.3. Main clinical studies
Canakinumab treatment was evaluated in the described periodic fever syndromes in four uncontrolled phase II pilot studies and one Phase 3 study CACZ885N2301 in a total of 226 patients, which evaluated the efficacy and safety of canakinumab at a starting dose of 150 mg sc or 2 mg/kg for patients weighing ≤ 40 kg administered every 4 weeks (q4w) in patients with FMF, HIDS/MKD, or TRAPS compared to placebo.

Assessment report EMA/26517/2017 Page 53/60
Study N2301 is a randomized, double-blind, placebo controlled study of canakinumab in patients with Hereditary Periodic Fevers (TRAPS, HIDS, or crFMF), with subsequent randomized withdrawal/dosing frequency reduction and open-label long term treatment epochs.
3.2. Favourable effects
Study N2301 achieved the primary objective and showed that Canakinumab was superior to placebo in all 3 disease cohorts with respect to the proportion of patients who had resolution of their index flare by Day 15 and did not experience a new flare during the remainder of Epoch 2. The proportion of patients who achieved a complete response in Epoch 2 was:
- 61.3% for canakinumab vs. 6.3% for placebo in crFMF patients (OR=23.75, p<0.0001),
- 35.1% vs. 5.7% in HIDS/MKD patients (OR=8.94, p=0.002), and
- 45.5% vs. 8.3% (OR=9.17, p=0.005) in TRAPS patients.
More patients achieved a complete response with canakinumab than with placebo, regardless of age, prior use of biologics and concomitant use of colchicine (for crFMF).
Furthermore, sustained flare control was seen over 16 weeks of treatment. Among canakinumab-treated patients (150 mg or 300 mg q4w), 90.3%, 64.9% and 77.3% of crFMF, HIDS/MKD and TRAPS patients, respectively, had no new flare (i.e., PGA ≥ 2 and CRP ≥ 30 mg/L) after resolution of the index flare up to the end of Week 16.
Secondary efficacy results show that canakinumab was superior to placebo in the secondary endpoints of PGA < 2 and CRP ≤ 10 mg/L at Week 16 in crFMF, HIDS/MKD and TRAPS cohorts. For the secondary endpoint SAA ≤ 10 mg/L at Week 16, canakinumab showed greater improvements in all disease cohorts but only with a statistically significant difference for TRAPS patients.
The results of the secondary and exploratory efficacy analysis support the results of the primary endpoint. It is shown that treatment with canakinumab is able to normalize the CRP levels in all three disease cohorts in a statistically significant way in comparison to placebo. These results are regarded as clinically meaningful improvement of disease status after 3 month of treatment.
All presented subgroup analyses are supportive to the primary and secondary analysis. Due to the partly very small cohorts of patients, the possibility to draw conclusions is limited. Subgroup analyses with regard to age, prior use of biological therapy and concomitant use of colchicine do not show any clear signals of reduced or improved efficacy.
Four uncontrolled open label studies (DTR01and D2204 in crFMF, D2402 in HIDS/MKD, D2203 in TRAPS) contributed further supportive efficacy data in the target population of periodic fever syndromes (45 patients). Efficacy measurements were made relative to baseline values. The results of these pilot studies fully support the efficacy seen in the phase III study.
3.3. Uncertainties and limitations about favourable effects
Due to the rarity of the condition, the data of patients not using prior/ concomitant colchicine in the treatment of FMF are too limited to draw any firm conclusions on the efficacy of canakinumab in this FMF subpopulation.
The data of long-term efficacy of canakinumab in treatment of crFMF, HIDS and TRAPS are rather limited, and therefore the results of the 24-week withdrawal and the 72-week long-term treatment periods of the currently

Assessment report EMA/26517/2017 Page 54/60
ongoing pivotal study N2301 should be provided as a post-approval measure as soon as the results are available.
No formal dose-finding studies were conducted due to rarity of the applied indications. Further data on maintenance dosing will become available from the study N2301.
3.4. Unfavourable effects
The adverse event profile of canakinumab treatment is overall mostly comparable in the new proposed indication with the approved CAPS indication.
The most prominent adverse events were infections and infestations, pyrexia and headache. The most common infections are those of the upper respiratory tract. Infections and infestation, and injection site reactions, were the SOCs with the most reported AEs possibly related to study treatment.
The rate of SAEs does not differ markedly from the already known safety profile of canakinumab. Observations from the phase II open label studies also support this. Results of the evaluation of haematology data shows that more patients have abnormalities in their platelet counts, neutrophil counts and leukocyte counts compared to placebo and also compared to the CAPS population. Most of the haematology abnormalities were CTC grade 1 or 2, 2 patients had a grade 3 neutropenia. Grade 1 thrombocytopenia occurred in 15.9% of canakinumab treated patients (compared to 4.1% in placebo patients).
3.5. Uncertainties and limitations about unfavourable effects
According to PK/PD modeling report, the occurrences of leukopenia, neutropenia, and lymphopenia were not dependent on the serum ACZ885 concentration. The study included the option for dose escalation in case the index flare did not resolve by day 15. This escape option had to be used by 16% of crFMF patients, 32% of HIDS/MKD patients, and 36% of TRAPS patients treated with canakinumab compared to 62.5% of crFMF patients, 63% of HIDS/MKD patients, and 75% of TRAPS patients treated with placebo. However, this option enabled especially crFMF and TRAPS patients to reach resolution of index flare by day 29. One third of the patients (n= 25) had to escalate the dose to 300mg or 4mg/kg in patients >40kg bw due to not optimal efficacy. This higher cumulative dose might lead to an increased amount of adverse events. There seems to be also a slight tendency for increased rates of haematological changes and changes in creatinine levels with a higher cumulative dose of canakinumab. A higher percentage of Total ACZ patients in the >600 mg group showed a ≥25% increase from baseline in creatinine (36.4%) compared with the ≤ 600 mg group (24.6%). However, the serum creatinine values remained within the normal range. Adverse events with a substantially higher incidence in patients in the Total ACZ group with cumulative doses >600 mg compared with patients on doses ≤600 mg until 16 weeks included abdominal pain, arthralgia, cough, diarrhoea, headache, injection site reaction, nasopharyngitis, oropharyngeal pain, pyrexia, rhinitis, and vomiting. Dose dependence on these adverse events cannot be ruled out completely.
Subgroup analysis showed that female patients might have more adverse events (85.0% in females vs. 73.9% in males) when treated with canakinumab compared to males. This was already described in the original assessment for marketing authorisation and was also found in the gout population.
Data on long term use of canakinumab in the respective proposed indications is still rather limited and needs to be increased. Further data will be available after finishing of study N2301.

Assessment report EMA/26517/2017 Page 55/60
3.6. Benefit-risk assessment and discussion
3.6.1. Importance of favourable and unfavourable effects
Canakinumab treatment is superior compared to placebo treatment in the proposed indications crFMF, HIDS/MKD, and TRAPS. The onset of response in all three cohorts was within a few days. Laboratory parameters improved to normal levels, demonstrating reduction of inflammatory reaction. The efficacy of treatment sustained over a prolonged time even after withdrawal of study drug. Patients and physicians assessed the disease activity as markedly reduced and health related quality of life parameters were also mostly improved.
16-30% of patients in the different cohorts required up-titration of the dose. With the higher dose regimen, further efficacy was attained. This flexible dosing option provides a useful tool for treating physicians. However, this might result in a higher cumulative dose which could possibly be associated with an increase in adverse events and serious adverse events.
Canakinumab’s ability to reduce the disease attack rate, flare severity and normalized laboratory parameters of interest (CRP, SAA) is regarded as medically highly important. The currently limited long term data from the 2-year treatment in studies D2402 and D2203 is promising (majority of HIDS/MKD and TRAPS patients without re-flare) but not yet conclusive.
The most common adverse events were infections and infestations, neutropenia and events associated with the underlying disease as well as injections site reactions.
No deaths were reported. The safety profile of canakinumab did not differ markedly from the already known and described profile in the approved indications.
An increase in frequency can be seen for Grad 1 neutropenia and thrombocytopenia. However, this is properly addressed in the SmPC.
Long term safety data is still very limited in these indications but more data will become available with completion of study N2301.
3.6.2. Balance of benefits and risks
Canakinumab was able to demonstrate a clinically meaningful benefit in the proposed three indications in a double-blind, randomized, placebo-controlled Phase 3 confirmatory study as well as in four supportive phase II pilot studies. There were no new safety findings in the canakinumab –treated crFMF, HIDS/MKD and TRAPS patients, and the safety profile in the 3 indications were mostly comparable to the approved indication of CAPS.
Canakinumab at the proposed dosing regimen is regarded as an effective and safe treatment for the intended population who is in need for further treatment options and also for patients with an inadequate response to current therapies.
With regard to one of the proposed three new indications, colchicine resistant FMF, there is data only on a few patients without concomitant colchicine use, i.e. patients with contraindication or intolerance to colchicine. Further, colchicine has a marketing authorization for the treatment of FMF only in 5 member States of the EU (Netherlands, Spain, Portugal, Slovakia, and United Kingdom). Prevalence of this disease is highest among subjects originating from Eastern Mediterranean area, but today these patients may appear in other areas.
It is noteworthy, however, that canakinumab has been already licensed for the “symptomatic treatment of adult patients with frequent gouty arthritis attacks (at least 3 attacks in the previous 12 months) in whom

Assessment report EMA/26517/2017 Page 56/60
non-steroidal anti-inflammatory drugs (NSAIDs) and colchicine are contraindicated, are not tolerated, or do not provide an adequate response, and in whom repeated courses of corticosteroids are not appropriate (see section 5.1).”, even though colchicine has not been licensed for the treatment of gout in all MSs either (colchicine has been licensed for gout-indication in 19 MSs, according to the Art 57 database).
Overall, the MAH was requested to explore the possibility of extrapolating the crFMF-indication to all FMF-patients regardless of the prior/ concomitant use of colchicine. According to the MAH, the data of the patients without concomitant or prior use of colchicine is too limited for reliable extrapolation of the effect of canakinumab in this FMF sub-population. It is however acknowledged that there is clinical need for the treatment of FMF in these relatively rare patients with contraindication, intolerance or insufficient response to colchicine, or in colchicine naïve patients who do not have access to colchicine therapy (as colchicine is not licenced for the treatment of FMF in many Member States). The only uncertainty related to the use of canakinumab alone/ at first line treatment is related to the fact that colchicine has been suggested to prevent secondary amyloidosis in these patients “by suppressing chronic inflammatory activity” (EULAR recommendations for the management of familial Mediterranean fever Ann Rheum Dis 2016;0:1-8: "Colchicine should be co-administered with alternative biological therapies given that it may reduce the risk of amyloidosis despite persistence of attacks".) and therefore these patients should be treated with colchicine, if available. Also, there is strong biological plausibility that canakinumab would have similar effect in the treatment of FMF without colchicine. Finally, the treatment of FMF will be usually carried out by specialists, often in special centers, and therefore it is also feasible and appropriate to leave some flexibility to the indication, i.e. for the treating physician to decide.
Taking into account all the above-mentioned aspects, the indication is changed as follows to cover all FMF patients with a potential to benefit from canakinumab treatment:
Ilaris is indicated for the treatment of Familial Mediterranean Fever (FMF). Ilaris should be given in combination with colchicine, if appropriate.
The benefit-risk balance for Canakinumab in the treatment of crFMF, HIDS/MKD, and TRAPS is positive, provided that sufficient long term efficacy and safety data will be collected and the final study report of study N2301 will be submitted.
3.7. Conclusions
The overall benefit-risk of Ilaris is positive.
4. Recommendations
Outcome
Based on the CHMP review of data on quality and safety and efficacy, the CHMP considers that the risk-benefit balance of the line extension for ILARIS, to add the new pharmaceutical form 150 mg/ml solution for injection, is favourable in the following indication:
Periodic fever syndromes Ilaris is indicated for the treatment of the following autoinflammatory periodic fever syndromes in adults, adolescents and children aged 2 years and older:

Assessment report EMA/26517/2017 Page 57/60
Cryopyrin-associated periodic syndromes Ilaris is indicated for the treatment of cryopyrin-associated periodic syndromes (CAPS) including: • Muckle-Wells syndrome (MWS), • Neonatal-onset multisystem inflammatory disease (NOMID) / chronic infantile neurological, cutaneous,
articular syndrome (CINCA), • Severe forms of familial cold autoinflammatory syndrome (FCAS) / familial cold urticaria (FCU) presenting
with signs and symptoms beyond cold-induced urticarial skin rash. Tumour necrosis factor receptor associated periodic syndrome (TRAPS) Ilaris is indicated for the treatment of tumour necrosis factor (TNF) receptor associated periodic syndrome (TRAPS). Hyperimmunoglobulin D syndrome (HIDS)/mevalonate kinase deficiency (MKD) Ilaris is indicated for the treatment of hyperimmunoglobulin D syndrome (HIDS)/mevalonate kinase deficiency (MKD). Familial Mediterranean fever (FMF) Ilaris is indicated for the treatment of Familial Mediterranean Fever (FMF). Ilaris should be given in combination with colchicine, if appropriate. Ilaris is also indicated for the treatment of: Still’s disease Ilaris is indicated for the treatment of active Still’s disease including adult-onset Still’s disease (AOSD) and systemic juvenile idiopathic arthritis (SJIA) in patients aged 2 years and older who have responded inadequately to previous therapy with non-steroidal anti-inflammatory drugs (NSAIDs) and systemic corticosteroids. Ilaris can be given as monotherapy or in combination with methotrexate. Gouty arthritis Ilaris is indicated for the symptomatic treatment of adult patients with frequent gouty arthritis attacks (at least 3 attacks in the previous 12 months) in whom non-steroidal anti-inflammatory drugs (NSAIDs) and colchicine are contraindicated, are not tolerated, or do not provide an adequate response, and in whom repeated courses of corticosteroids are not appropriate. The CHMP therefore recommends the extension of the marketing authorisation for ILARIS subject to the following conditions:
Conditions or restrictions regarding supply and use
Medicinal product subject to restricted medical prescription (see Annex I: Summary of Product Characteristics, section 4.2).
Conditions and requirements of the marketing authorisation
Periodic Safety Update Reports
The requirements for submission of periodic safety update reports for this medicinal product are set out in the list of Union reference dates (EURD list) provided for under Article 107c(7) of Directive 2001/83/EC and any subsequent updates published on the European medicines web-portal.
Conditions or restrictions with regard to the safe and effective use of the medicinal product
Risk Management Plan (RMP)
The MAH shall perform the required pharmacovigilance activities and interventions detailed in the agreed RMP

Assessment report EMA/26517/2017 Page 58/60
presented in Module 1.8.2 of the marketing authorisation and any agreed subsequent updates of the RMP.
An updated RMP should be submitted:
• At the request of the European Medicines Agency;
• Whenever the risk management system is modified, especially as the result of new information being received that may lead to a significant change to the benefit/risk profile or as the result of an important (pharmacovigilance or risk minimisation) milestone being reached.
Additional risk minimisation measures
The Marketing Authorisation Holder (MAH) shall ensure that, prior to launch, all physicians who are expected to prescribe/use Ilaris are provided with a physician information pack containing the following:
• The Summary of Product Characteristics • Healthcare professional information • Patient Reminder Card
The Healthcare Professional information should contain the following key messages:
• The risk of serious infections, including opportunistic bacterial, viral and fungal infections in patients treated with Ilaris;
• For CAPS, TRAPS, HIDS/MKD, FMF and SJIA patients: the need to instruct patients on proper techniques for self-administration when the patient is willing and capable to do so, and guidance for Health Care Professionals on how to report administration errors;
• The identified or potential risk of immunogenicity that might lead to immune-mediated symptoms. For gouty arthritis patients: highlighting that intermittent therapy or re-exposure after a long treatment-free interval may be associated with an enhanced immune response (or loss of immune tolerance) to Ilaris and thus re-treated patients must be considered at risk of hypersensitivity reactions;
• For chronic therapy in CAPS: the need for Health Care Professionals to perform an annual clinical assessment of patients regarding a potential increased risk for the development of malignancies;
• As treatment with Ilaris should not be initiated in patients with neutropenia, the need to measure neutrophil counts prior to initiating treatment and again after 1 to 2 months. For chronic therapy in CAPS patients or repeated therapy in gouty arthritis patients, it is recommended to assess neutrophil counts periodically during treatment;
• For SJIA patients, the need for Health Care Professionals to be attentive to symptoms of infection or worsening of SJIA, as these are known triggers for macrophage activation syndrome (MAS) which is a known, life-threatening disorder that may develop in patients with rheumatic conditions, in particular SJIA patients. If MAS occurs, or is suspected, evaluation and treatment should be started as early as possible;
• The need to monitor patients for changes in their lipid profiles; • The unknown safety of Ilaris in pregnant and lactating women, thus the need for physicians to discuss this
risk with patients if they become or plan to become pregnant; • The proper patient management as regards the interaction with vaccination; • Where relevant, the possibility to include patients in the registry study to facilitate the collection of long
term efficacy and safety data; • The role and use of Patient Reminder Card.
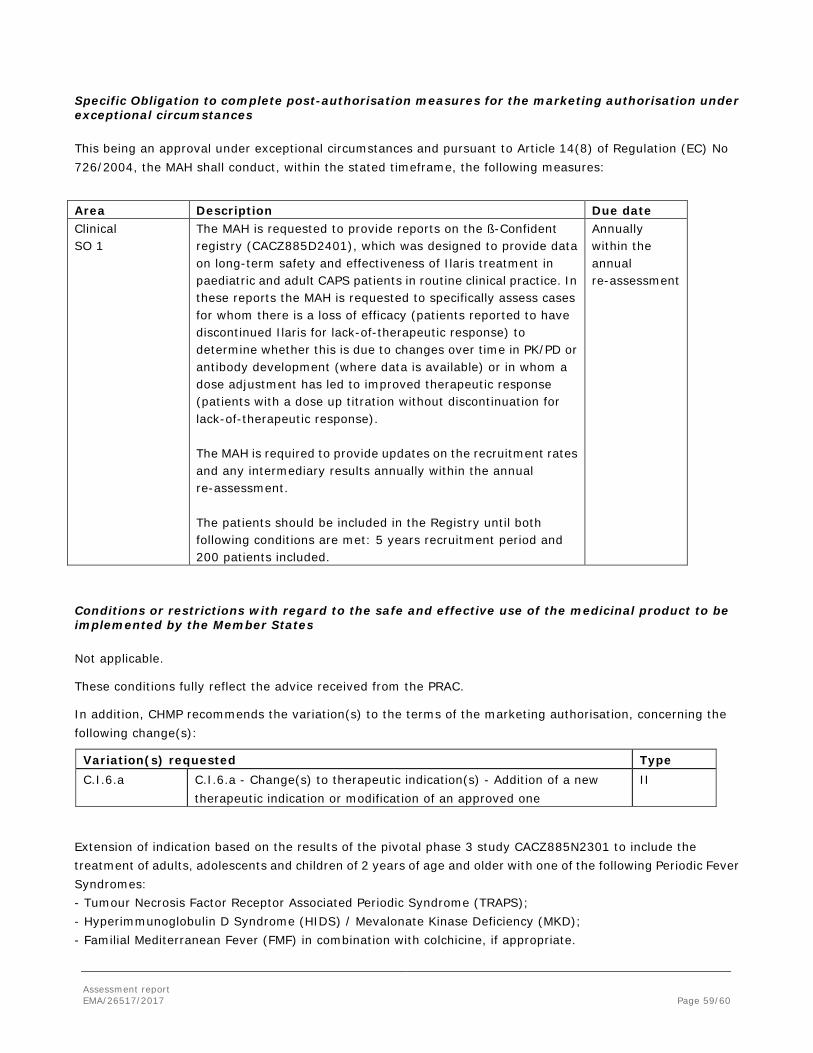
Assessment report EMA/26517/2017 Page 59/60
Specific Obligation to complete post-authorisation measures for the marketing authorisation under exceptional circumstances
This being an approval under exceptional circumstances and pursuant to Article 14(8) of Regulation (EC) No 726/2004, the MAH shall conduct, within the stated timeframe, the following measures:
Area Description Due date
Clinical SO 1
The MAH is requested to provide reports on the ß-Confident registry (CACZ885D2401), which was designed to provide data on long-term safety and effectiveness of Ilaris treatment in paediatric and adult CAPS patients in routine clinical practice. In these reports the MAH is requested to specifically assess cases for whom there is a loss of efficacy (patients reported to have discontinued Ilaris for lack-of-therapeutic response) to determine whether this is due to changes over time in PK/PD or antibody development (where data is available) or in whom a dose adjustment has led to improved therapeutic response (patients with a dose up titration without discontinuation for lack-of-therapeutic response). The MAH is required to provide updates on the recruitment rates and any intermediary results annually within the annual re-assessment. The patients should be included in the Registry until both following conditions are met: 5 years recruitment period and 200 patients included.
Annually within the annual re-assessment
Conditions or restrictions with regard to the safe and effective use of the medicinal product to be implemented by the Member States
Not applicable.
These conditions fully reflect the advice received from the PRAC.
In addition, CHMP recommends the variation(s) to the terms of the marketing authorisation, concerning the following change(s):
Variation(s) requested Type C.I.6.a C.I.6.a - Change(s) to therapeutic indication(s) - Addition of a new
therapeutic indication or modification of an approved one II
Extension of indication based on the results of the pivotal phase 3 study CACZ885N2301 to include the treatment of adults, adolescents and children of 2 years of age and older with one of the following Periodic Fever Syndromes: - Tumour Necrosis Factor Receptor Associated Periodic Syndrome (TRAPS); - Hyperimmunoglobulin D Syndrome (HIDS) / Mevalonate Kinase Deficiency (MKD); - Familial Mediterranean Fever (FMF) in combination with colchicine, if appropriate.

Assessment report EMA/26517/2017 Page 60/60
As a consequence sections 4.1, 4.2, 4.4, 4.8, 5.1 and 5.2 of the SmPC are proposed to be updated and the Annex II and the Package Leaflet is proposed to be updated accordingly. In addition, the annexes have been aligned with the latest QRD template v.10. A revised RMP version 11.2 was agreed during the procedure.