il bambino con immunodeficienza presunta e vera: … · discromie sindrome di chediak-higashi...
TRANSCRIPT

Lucia Leonardi
IL BAMBINO CON
IMMUNODEFICIENZA PRESUNTA E
VERA: REVISIONE DELLE LINEE
GUIDA

LG e IDP?
ampia eterogeneità
‘singolarmente’ rare
fenotipi non ‘classici’
classificazione IDP
sempre più
complessa: >300 IDP
molecolarmente
caratterizzate
quadri clinici
sia di esordio che
evolutivi
estremamente
eterogenei

DIAGNOSI DIFFICILE CONSIDEREVOLE
RITARDO (anche per le IDP più comuni) e CORRELA
CON PROGNOSI (spesso quoad vitam
SOSPETTO DIAGNOSTICO PRECOCE (pediatra libera
scelta) E’ PERO’ POSSIBILE)
Classificazione fenotipica IDP

Il puzzle delle malattie rare
sintomi e segni che indirizzano
• il sospetto di IDP
• la natura del difetto immunologico
• la richiesta e l’interpretazione degli esami
anamnesi
Es. obiettivo

Classificazione Semplificata
IDP combinate (deficit immunità umorale + cellulare): precoci,
linfopenia, infezioni spesso fatali da virus batteri e funghi
Deficit anticorpali: 75% IDP, infezioni da p. extracellulari
IDP associate a manifestazioni sindromiche: prevale difetto dei
linfociti T infezioni da p.intracellulari, autoimmunità, neoplasie
Malattie da disregolazione immunologica: infiammazione
persistente per massiva attivazione mastocitaria febbre
Difetti congeniti dei fagociti (numero e/o funzione): infezioni
da gram- e funghi
Difetti nell’immunità intrinseca e innata : suscettibilità selettiva a
un solo patogeno
Malattie autoinfiammatorie
Deficit del complemento: infezioni da Neisseria
Al-Herz W et al.Frontiers in immunology.2014;5:162

L’anamnesi nel sospetto di IDP: infezioni
ricorrenti… ma non solo
atopia
manifestazioni cutanee

Anamnesi
DD: ID secondarie
•Prematurità, età <4 anni (THI)
•Infezioni (HIV, EBV, CMV)
•Nutrizione: deficienza vitamine, zinco
•Protidodispersione: enteropatie, sdr
nefrosica
•Iatrogene: splenectomia, post TCSE,
CTS
•Neoplasie : CLL, linfoma non Hodgkin
Anamnesi Familiare
• IDP
• Decessi per infezioni
età pediatrica
• Poliabortività
materna

IRR
Più comune presentazione di esordio delle immunodeficienze
primitive (soprattutto umorali)
Significativa causa di morbilità/mortalità tra i pazienti con IDP
Età prescolare IRR di modesta gravità e a localizzazione
prevalentemente alta: «fisiologiche»
DD: asma, ipertrofia adenoidea, MRGE, fibrosi cistica

IRR: red Flags
più di 4 episodi di otite in 12 mesi
più di 2 episodi di sinusite batterica in un anno
più di 2 episodi di polmonite in un anno
infezioni inusuali per severità
infezioni invasive da germi poco virulenti o opportunisti
infezioni ricorrenti sostenute da uno stesso patogeno (anche germi
comuni)
danno d’organo da infezioni ricorrenti/persistenti: bronchiettasie o
altre complicanze
trattamenti antibiotici protratti per inadeguata risposta alla terapia
Gray PA et al. J Paed Child Health 2012;48:202. Jesenak M et al. Front Pediatr 2014;2:77.
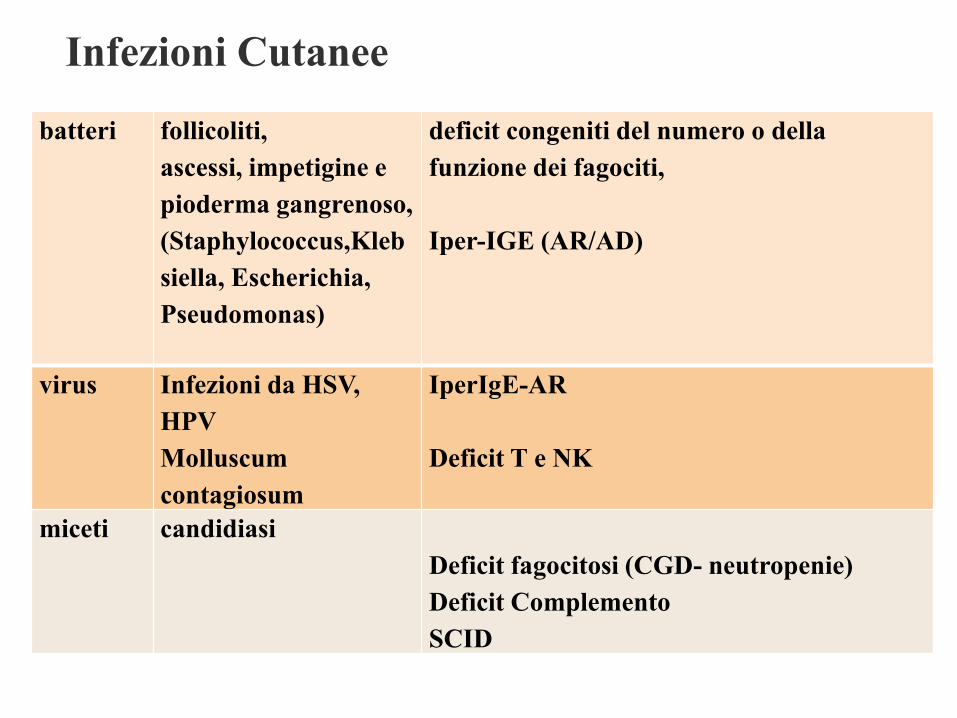
Infezioni Cutanee
batteri follicoliti,
ascessi, impetigine e
pioderma gangrenoso,
(Staphylococcus,Kleb
siella, Escherichia,
Pseudomonas)
deficit congeniti del numero o della
funzione dei fagociti,
Iper-IGE (AR/AD)
virus Infezioni da HSV,
HPV
Molluscum
contagiosum
IperIgE-AR
Deficit T e NK
miceti candidiasi
Deficit fagocitosi (CGD- neutropenie)
Deficit Complemento
SCID

Manifestazioni cutanee non infettive
Manifestazione cutanea Sospetta IDP
Eczema Sdr da Iper-IgE (AD/AR)
Sdr di Wiskott-Aldrich (eczema, microtrombocitemia, IR)
IPEX (Disordine immune, poliendocrinopatia, enteropatia
X-linked): precoce eczema +IDDM + diarrea intrattabile
SCID
Eritrodermia Sindrome di Netherton (eritroderma ittiosiforme congenito,
tricoressi invaginata, atopia)
Sindrome di Omenn (eritrodermia neonatale, alopecia,
linfoadenopatia, epatosplenomegalia)
Granulomi Malattia granulomatosa cronica (asettici)
CVID (arti e volto- aspetto similsarcoidosico)
Displasia di cute, capelli,
unghie o denti
APECED: Poliendocrinopatia autoimmune, candidiasi,
displasia ectodermica
Discromie Sindrome di Chediak-Higashi
Angioedema senza orticaria Angioedema ereditario tipi 1-3
Relan M, Lehman HK. Curr Allergy Asthma Rep 2014;14:480 (mod.)

Manifestazioni cutanee nelle IDP
ECZEMA manifestazione cutanea più comune età pediatrica
precoce e severo (spesso esordio neonatale
IDP volto e superfici estensorie
lesioni nettamente demarcate o non eritematose
non familiarità per atopia
ASCESSI CUTANEI RICORRENTI
ricircolo nell’ambiente domestico di ceppi
di stafilococco virulenti (colonizzazione nasale di portatori sani)

CGD Sdr di Omenn
HIES

Diarrea cronica
durata e severità maggiore
richiedono un regime terapeutico più intensivo e prolungato
causate da organismi atipici e opportunistici (umorali:
Giardia, Campylobacter, SCID rotavirus )
causa malassorbimento e scarso accrescimento
può associarsi a malattie infiammatorie croniche o
autoimmuni dell’intestino e ad allergia alimentare
(HIES/WAS/IgAd)
diarrea acquosa profusa accompagnata da malassorbimento
grave che non risponde alle modificazioni dietetiche e al
digiuno può essere il quadro di esordio, età precoce, di
IPEX e SCID

Ritardo crescita
quasi sempre presente (> IDP associate a sindromi)
segno precoce nella SCID: nei primi giorni di vita ritardo di
crescita+ diarrea cronica (Rotavirus, Adenovirus,
Coxsackievirus) ed infezioni opportunistiche (candidiasi orale,
infezioni respiratorie)
infezioni ricorrenti e severe ridotta
alimentazionemalassorbimento
obiettività: addome globoso, diastasi dei muscoli retti
addominali, epatosplenomegalia, disidratazione, pallore,
ittero, linfoadenomegalia
DD: cardiopatie, FC, IVU, MRGE, infezioni (TBC, HIV)

autoimmunità
fenomeni autoimmuni nelle IDP:
-come corredo sintomatologico specifico (spesso già
all’esordio) fenomeni autoimmuni poliorgano
[APECED, IPEX ]
-come complicanza citopenie, artriti, vasculiti
[soprattutto in deficit imm.umorale: CVID, IgAd*]
-quanto maggiore è la suscettibilità alle infezioni tanto minore -
o più tardiva - è la componente autoimmune e viceversa
Bacchetta R, Notarangelo LD. Front Immunol 2013; 4: 77

Autoimmunità nelle IDP
Pensare sempre a una IDP quando sintomi di autoimmunità
precoci, poliorgano o associati a IR
Follow up attento in pz con CVID/IgAD
ANApositività isolata non ha significato clinico:
l’esame non va mai richiesto in assenza di sintomatologia
specifica!

Febbre e IDP
Sindromi Autoinfiammatorie:
ereditarie, rare, genetica nota: febbre mediterranea familiare (FMF), sdr
da difetto di mevalonato-chinasi (o sdr da Iper-IgD), sdr periodica
associata al recettore del TNF (TRAPS), sdr periodiche associate alle
criopirine (CAPS)
sindromi non ereditarie: PFAPA (febbre periodica, stomatite aftosa,
faringite e adenite cervicale)
Lachmann HJ. Clin Exp Immunol 2011; 165: 301-9
febbre persistente o ricorrente (cadenza periodica: intervalli +/- regolari)
+
sintomi infiammatori di natura articolare, gastrointestinale e/o muco-cutanea

Febbre e IDP
• episodi febbrili/infiammatori tutti uguali fra di loro, ricorrenza, s. infiammatori
• esordio clinico spesso precoce (entro i 10 anni di età)
• complicanze a lungo termine se non adeguatamente trattate (amiloidosi)
FPAPA: più comune età pediatrica (esordio sotto i 5 anni)
Diagnosi clinica, leucocitosi ed aumento IF solo durante la febbre
febbre per 3-6 gg che ricorre regolarmente
risposta al corticosteroide monodose se non risposta alla terapia corticosteroidea
bisogna considerare altre cause di febbre periodica centro 3 livello
QC:
- faringite essudativa (74%)
- aumento di volume dei linfonodi laterocervicali (70%)
- aftosi orale (68%)
- intensa iperemia faringea (64%)
- dolore addominale (36%)
- buona salute nei periodi intercritici

Fenotipo clinico orienta verso il difetto
immunologico Quadro cinico
difetto esempio
Infezione batteriche,
politopiche, dopo i 6 mesi di
vita
Immunità umorale THI, CVID, IgAD
Infezione severe batteriche,
virali, fungine e da
opportunisti dai primi mesi di
vita. Candidiasi mucocutanea.
Diarrea cronica. Arresto
crescita. Rash cutaneo
IDP combinate (cellulare-
umorale)
SCID
Infezioni precoci cutanee,
polmonari, linfonodali da
batteri e funghi
Difetti dei fagociti e
neutropenia
Neutropenia ciclica, CGD,
LAD
Meningiti batteriche, malattie
autoimmuni, orticaria -
angioedema
Difetti de complemento Deficit C1inibitore

Algoritmo Diagnostico
Semplificato IDP
Oxford Handbook of Clinical Immunology

Laboratorio
I livello può confermare il sospetto
clinico e indirizzare vs diagnosi
definitiva
• Sempre valori in assoluto e non %
• Sempre range per l’età
I livello normale, ma QC
suggestivo centro
riferimento per esami II livello

Emocromo
riduzione del valore assoluto
dei neutrofili
N<500/mm3 + >> Eos e Mono, infezioni nei primi mesi di
vitaneutropenia congenita severa
N<1000/ mm3:
transitorie (virus/farmaci) autoimmuni, cicliche, altre IDP piu
complesse
sempre striscio periferico
aumento valore assoluto dei
neutrofili
livelli 5-10 volte >normale + infezioni
batteriche ricorrenti senza produzione di pus/ ritardato distacco del
cordone ombelicale LAD
Linfocitopenia
(L<3.000/mm3 nei bambini*)
Marcata linfopenia+diarrea cronica,distrofia, IRR severe(specie se da
patogeni opportunisti),candidosi orale persistente/ sistemica, dermatite
severa o eritrodermicaSCID
sempre immunofenotipo linfocitario
trombocitopenia con
piastrine piccole (<5 μ3)
+- eczema/deficit anticorpale/neutropeniaWAS

Livelli Ig sieriche
Riduzione IgG+- riduzione di IgA e /o IgM
IgG IgM IgA IgE
XLA
CVID /N /N N
IgAd N N N
THI ?
SCID /
Iper IgM / N ?
Iper IgE N N N

Quando vaccinare? Chi?
Quando antibiotico-profilassi? Quando
antibiotico-profilassi/ terapie antibiotiche?
Follow up IDP più comuni: IgAD assoluto isolato,
CVID

Vaccinazioni- Antibioticoprofilassi
Raje N et al. Immunol Allergy Clin N Am 2015;35:599-623

Follow up IDP: I tools da consultare
http://www.aieop.org/web//?q=node/477
centro per IDP

nonostante assenza di LG
esaustive
diagnosi precoce, già sul
territorio, non solo è
auspicabile, ma possibile
attraverso il corretto approccio:
ricerca segni e sintomi e attenzione
ai fenotipi ‘non classici’
invio tempestivo centro di riferimento
diagnostica/terapia specifica
miglioramento prognosi