iezy-drug: a web server for identifying the interaction between
TRANSCRIPT

Hindawi Publishing CorporationBioMed Research InternationalVolume 2013 Article ID 701317 13 pageshttpdxdoiorg1011552013701317
Research ArticleiEzy-Drug A Web Server for Identifying the Interaction betweenEnzymes and Drugs in Cellular Networking
Jian-Liang Min1 Xuan Xiao123 and Kuo-Chen Chou34
1 Computer Department Jing-De-Zhen Ceramic Institute Jing-De-Zhen 333046 China2 Information School ZheJiang Textile amp Fashion College NingBo 315211 China3 Gordon Life Science Institute Belmont MA 02478 USA4Center of Excellence in Genomic Medicine Research (CEGMR) King Abdulaziz University Jeddah Saudi Arabia
Correspondence should be addressed to Xuan Xiao xxiaogordonlifescienceorg
Received 7 August 2013 Accepted 17 September 2013
Academic Editor Tatsuya Akutsu
Copyright copy 2013 Jian-Liang Min et alThis is an open access article distributed under the Creative Commons Attribution Licensewhich permits unrestricted use distribution and reproduction in any medium provided the original work is properly cited
With the features of extremely high selectivity and efficiency in catalyzing almost all the chemical reactions in cells enzymes playvitally important roles for the life of an organism and hence have become frequent targets for drug design An essential step indeveloping drugs by targeting enzymes is to identify drug-enzyme interactions in cells It is both time-consuming and costly todo this purely by means of experimental techniques alone Although some computational methods were developed in this regardbased on the knowledge of the three-dimensional structure of enzyme unfortunately their usage is quite limited because three-dimensional structures of many enzymes are still unknown Here we reported a sequence-based predictor called ldquoiEzy-Drugrdquoin which each drug compound was formulated by a molecular fingerprint with 258 feature components each enzyme by theChoursquos pseudo amino acid composition generated via incorporating sequential evolution information and physicochemical featuresderived from its sequence and the prediction engine was operated by the fuzzy K-nearest neighbor algorithm The overall successrate achieved by iEzy-Drug via rigorous cross-validations was about 91 Moreover to maximize the convenience for the majorityof experimental scientists a user-friendly web server was established by which users can easily obtain their desired results
1 Introduction
Enzymes are biomacromolecules that catalyze almost all thechemical reactions essential for the life of a cell [1] Mostenzymes are proteins although some RNA molecules havebeen identified to possess the function of enzyme as well Ascatalysts enzymes possess two exceptional features one is ofhigh efficiency and the other of high selectivity For instancethe second-order rate constant between some enzymes andtheir substrates [2] was surprisingly high [3] which couldalmost reach the upper limit of diffusion-controlled reactionrate according to the calculation and analysis by Chouand coworkers [4ndash6] The high selectivity or specificity ofenzymes was likened to the ldquolock-and-keyrdquo model implyingthat an accurate fit is required between the active site of anenzyme and its substrate for the catalytic reaction to occurOwing to the previous unique features enzymes play acrucial role in controlling and regulating the order of
chemical reactions in cells that is vitally important fortheir survival It is also because of this that enzymes areexcellent drug targets and actually many drugs are enzymeinhibitors For example some peptide inhibitors againstHIVAIDS [7ndash10] and SARS (severe acute respiratory syn-drome) [11ndash13] were based on the Choursquos distorted keytheory [14] as illustrated in Figure 1 where (a) shows agood fit for a cleavable octapeptide with the active siteof HIV-protease and (b) shows that the peptide hasbecome an ideal inhibitor or ldquodistorted keyrdquo after its scis-sile bond is modified For a brief introduction about theChoursquos distorted key theory and its application for design-ing peptide drugs see aWikipedia article at httpenwikipediaorgwikiChoursquos distorted key theory for peptide drugs
To develop enzyme-targeting drugs an essential step isto identify drug-enzyme interaction in cellular networking[15] The completion of the human genome project and the
2 BioMed Research International
(a) (b)
Figure 1 A schematic drawing to illustrate how to use Choursquos distorted key theory to develop peptide drugs against HIVAIDS (a) shows agood fitting and binding of a peptide to the active site of HIV protease right before it is cleaved by the enzyme (b) shows that the peptide hasbecome a noncleavable one after its scissile bond is modified although it can still tightly bind to the active site Such a modified peptide orldquodistorted keyrdquo will automatically become an inhibitor candidate against HIV protease
emergence of molecular medicine have provided excellentopportunity to discover unknown target enzymes for drugsMany efforts were made in this regard by computationallyanalyzing drug-enzyme interactions The most commonlyused approaches are docking simulations (see eg [16ndash19])and protein cleavage site analysis (see eg [8 12 13]) basedon Choursquos distorted key theory [14] However the latterapproach is mainly used to find peptide drugs Comparedwith the smaller organic compounds although peptide drugshave the advantage of low toxicity to human body theyhave the shortcoming of poor metabolic stability and lowbioavailability due to their inability to readily crossing thrumembrane barriers such as the intestinal and blood-brainbarriers [20] In contrast the molecular docking is indeed auseful vehicle for investigating the interaction of an enzymereceptor with its organic inhibitor and revealing their bindingmechanism as demonstrated by a series of studies [11 19ndash23] However to conduct molecular docking a necessaryprerequisite is the availability of the 3D (three dimensional)structure of the targeted enzyme Unfortunately the 3Dstructures of many enzymes are still unknown Although X-ray crystallography is a powerful tool in determining the 3Dstructures of enzymes it is time-consuming and expensiveParticularly not all enzymes can be successfully crystallizedFor example membrane enzymes are very difficult to crys-tallize and most of them will not dissolve in normal solventsTherefore so far very few membrane enzyme 3D structureshave been determined Although NMR is indeed a verypowerful tool in determining the 3D structures of membraneproteins as indicated by a series of recent publications (seeeg [24ndash30]) it is time-consuming and costly To acquire thestructural information in a timelymanner one has to resort tovarious structural bioinformatics tools (see eg [18 31 32])Unfortunately the number of templates for developing highquality 3D structures by structural bioinformatics is verylimited
Therefore it would save us a lot of time and moneyif we could identify the interactions between enzymes anddrugs before carrying out any intense study in this regardIn view of this the present study was initiated in an attemptto develop a computational method based on the sequence-derived features that can be used to predict the drug-enzymeinteractions in cellular networking
As summarized in a comprehensive review [33] anddemonstrated by a series of recent publications [34ndash37] tosuccessfully develop the desiredmethod we need to considerthe following procedures (i) construct or select a validbenchmark dataset to train and test the predictor (ii) denotethe drug-enzyme samples with an effective formulation thatcan truly reflect their intrinsic relation with the target to bepredicted (iii) introduce or develop a powerful algorithm(or engine) to operate the prediction (iv) conduct a rigorouscross-validation to objectively evaluate its anticipated accu-racy (v) establish a user-friendly web-server for the predictorthat is freely accessible to the public Next let us elaboratehow to deal with these procedures one by one
2 Materials and Methods
21 Benchmark Dataset The data used in this study werecollected from Kyoto Encyclopedia of Genes and Genomes(KEGG) [38] at httpwwwkeggjpkegg which is a data-base resource for understanding high-level functions andutilities of the biological system such as the cell theorganism and the ecosystem from molecular-level infor-mation especially large-scale molecular datasets generatedby genome sequencing and other high-throughput exper-imental technologies For the current study the benchmarkdataset S can be formulated as
S = S+
cup Sminus
(1)
where S+ is the positive subset that consists of the interactiveenzyme-drug pairs only while Sminus is the negative subsetthat contains of the noninteractive enzyme-drug pairs onlyand the symbol cup represents the union in the set theoryHere the ldquointeractiverdquo pair means the pair whose twocounterparts are interacted with each other in the drug-target networks as defined in the KEGG database [38]while the ldquononinteractiverdquo pair means that its two counterparts are not interacted with each other in the drug-targetnetworks The positive dataset S+ contains 2719 enzyme-drug pairs derived from Yamanishi et al [39] The negativedataset Sminus contains 5438 noninteractive enzyme-drug pairswhich were derived according to the following procedures(i) separating each of the pairs in S+ into single drug and
BioMed Research International 3
enzyme (ii) recoupling each of the single drugs with eachof the single enzymes into pairs in a way that none of themoccurred inS+ (iii) randomly picking the pairs thus formeduntil they reached the number two times as many as thepairs in S+ The 2719 interactive enzyme-drug pairs and5438 noninteractive enzyme-drug pairs are given in OnlineSupporting Information S1 (see Supplementary Materialavailable online at httpdxdoiorg1011552013701317) Allthe detailed information for the compounds or drugs listedthere can be found in the KEGG database via their codes
22 Sample Representation Since each of the samples in thecurrent network system contains an enzyme (protein) anda drug a combination of the following two approaches wasadopted to represent the enzyme-drug pair samples
221 Drug
(a) 2D Molecular Fingerprints Although the number ofdrugs is extremely large most of them are small organicmolecules and are composed of some fixed small structures[40] The identification of small molecules or structures canbe used to detect the drug-target interactions [41] Molec-ular fingerprints are bit-string representations of molecularstructure and properties [42] It should be pointed outthat there are many types of structural representations thathave been suggested for the description of drug moleculesincluding physicochemical properties [43] chemical graphs[44] topological indices [45] 3D pharmacophore patternsandmolecular fields In the current study let us use the simpleand generally adopted 2Dmolecular fingerprints to representdrug molecules as described below
First for each of the drugs concerned we can obtaina MOL file from the KEGG database [38] via its codethat contains the detailed information of chemical structureSecond we can convert the MOL file format into its 2Dmolecular fingerprint file format by using a chemical toolboxsoftware called OpenBabel [46] which can be downloadedfrom the website at httpopenbabelorg The current ver-sion of OpenBabel can generate four types of fingerprintsFP2 FP3 FP4 and MACCS In the current study we usedthe FP2 fingerprint format It is a path-based fingerprint thatidentifies small molecule fragments based on all linear andring substructures and maps them onto a bit-string using ahash function (somewhat similar to the daylight fingerprints[47 48]) It is a length of 256-bit hexadecimal string obtainedfrom theOpenBabel andwe can convert it to a 256-bit vectorThen a molecular fingerprint can be formulated as a 256-Dvector given by
MF = [1198601 sdot sdot sdot 119860119895 sdot sdot sdot 119860256]T (2)
where 119860119895(119895 = 1 2 256) is an integer between 0 and 15
and T is the matrix transpose operatorIn order to capture as much useful information from
a molecular fingerprint as possible we can also convertthe above 256-bit hexadecimal string into a 1024-bit binaryvector which is a digital sequence only including 0 and 1
and consider two different digital signal characteristics for thedigital sequence as follows
(b) Information Entropy Shannon proposed that any infor-mation is redundant and redundant size is related withthe occurrence probability or uncertainty of each symbolsuch as numbers letters or words among the informationThe information entropy for a system with a probabilitydistribution 119875(119909
119894) for two classes information entropy [49] is
defined as
119867119909= minussum
119909
119875 (119909119894) log2119875 (119909119894) (119894 = 0 1) (3)
where 119875(119909119894) represents the occurrence probability of num-
ber 119894 in the aforementioned 1024-bit binary vector andthe information entropy 119867
119909is a measure value of the
information amount For example for the digital sequence100100011010010 the value of the information entropy 119867
119909
thus obtained is
119875 (1199090) =
9
15
= 06
119875 (1199091) =
6
15
= 04
119867119909= minus (06 times log
206 + 04 times log
204) = 0971
(4)
(c) Complexity Factor The Lempel-Ziv (LZ) complexity [50]reflects the order that is retained in the sequence and hencewas adopted in this study For each step only two operationswere allowed in the process to get the LZ complexity eithercopying the longest section from the part of a nonemptysequence or generating an additional symbol mark thatensures the uniqueness of per component 119878(119894
119896minus1rarr 119894119896) Its
substring is expressed by
119878 (119894 997888rarr 119895) = 119898119894119898119894+1119898119894+2sdot sdot sdot 119898119895
(1 le 119894 le 119895 le 119871) (5)
where 1198981represents the 1st digital value 119898
2the 2nd value
and so forth A nonempty digital sequence is synthesizedaccording to the following formula
Syn (119878) = 119878 (1 997888rarr 1198941) ∙ 119878 (119894
1+ 1 997888rarr 119894
2)
∙ sdot sdot sdot ∙ 119878 (119894119898minus1
+ 1 997888rarr 119894119871)
(6)
Suppose that 119878 = 11989811198982119898311989841198985sdot sdot sdot 119898119871has been recon-
structed by the subsymbol 119898119903which is viewed as the newly
inserted symbol The substring up to 119898119903will be denoted by
119878(1 rarr 119903)∙ where the bold dot ∙ indicates that 119898119903is a newly
inserted symbol for checkingwhether the rest of the substring119878(119903+1 rarr 119871) can be reconstructed by a simple process At firstsuppose 119878(119902) = 119898
119903+ 1 and see whether 119878(119902) is the substring
for the subsequence 119878(1 rarr 119903) which means deleting thelast symbol from the substring 119878(1 rarr 119903)119878(119902) If the answeris ldquonordquo we insert 119878(119902) into the sequence followed by a dot ∙Thus it could not be obtained by the same operation If theanswer is ldquoyesrdquo no new symbol is needed and we can go on toproceed with 119878(119902) = 119898
119903+1119898119903+2
and repeat the same previousprocedure The LZ complexity is the number of dots (plus
4 BioMed Research International
one if the string is not terminated by a dot) For example forthe sequence 100100011010010 syn(119875) and the correspondingcomplexity factor CF are described as
Syn (119878) = 1 ∙ 0 ∙ 01 ∙ 000 ∙ 11 ∙ 01001 ∙ 0
CF = 7(7)
Thus by adding the information entropy 119867119909(4) and com-
plexity factor CF (7) into the molecular fingerprint MF (2)we obtained a total of (256 + 1 + 1) = 258 feature elements torepresent a drug compound that is it can now be formulatedas a 258-D vector given by
119863 = [11986011198602sdot sdot sdot 119860
256119867119909
CF]T (8)
where 119860119894has the same meaning as in (2) while 119867
119909and CF
are the information entropy and complexity factor respec-tively as described in the previous two sections
222 Enzyme The sequences of the enzymes involved inthis study are given in Online Supporting Information S2Now the problem is how to effectively represent these enzymesequences for the current study Generally speaking thereare two kinds of approaches to formulate enzyme sequencesthe sequential model and the nonsequential or discretemodel [51] The most typical sequential representation foran enzyme sample E with 119871 residues is its entire amino acidsequence that is
E = 1198771119877211987731198774119877511987761198777sdot sdot sdot 119877119871 (9)
where 1198771represents the 1st residue 119877
2the 2nd residue and
so forth An enzyme sample thus formulated can contain itsmost complete information This is an obvious advantage ofthe sequential representation To get the desired results thesequence-similarity-search-based tools such as BLAST [5253] are usually utilized to conduct the prediction Howeverthis kind of approach failed to work when the query enzymedid not have significant homology to enzyme of known char-acters Thus various nonsequential representation modelswere proposed The simplest nonsequential model for anenzyme was based on its amino acid composition (AAC) asdefined by
E = [11989111198912sdot sdot sdot 11989120]
T (10)
where 119891119906(119906 = 1 2 20) are the normalized occurrence
frequencies of the 20 native amino acids [54ndash56] in theenzyme E and T has the same meaning as in (2) and (8)The AAC-discrete model was widely used for identifyingvarious attributes of proteins (see eg [57ndash61]) Howeveras can be seen from (10) all the sequence order effectswere lost by using the AAC-discrete model This is its mainshortcoming To avoid completely losing the sequence-orderinformation the pseudo amino acid composition [62 63]or Choursquos PseAAC [3] was proposed to replace the simpleAAC model Since the concept of PseAAC was proposedin 2001 [62] it has penetrated into almost all the fields of
protein attribute predictions and computational proteomicssuch as predicting supersecondary structure [64] predictingmetalloproteinase family [65] predicting membrane pro-tein types [66 67] predicting protein structural class [68]discriminating outer membrane proteins [69] identifyingantibacterial peptides [70] identifying allergenic proteins[71] identifying bacterial virulent proteins [72] predictingprotein subcellular location [73 74] identifying GPCRs andtheir types [75] identifying protein quaternary structuralattributes [76] predicting protein submitochondria locations[77] identifying risk type of human papillomaviruses [78]identifying cyclin proteins [79] predicting GABA(A) recep-tor proteins [80] and predicting cysteine S-nitrosylation sitesin proteins [81] among many others (see a long list of paperscited in the References section of [33]) Recently the conceptof PseAAC was further extended to represent the featurevectors of DNA and nucleotides [36 82] as well as otherbiological samples (see eg [83 84]) Because it has beenwidely and increasingly used recently two powerful soft-wares called ldquoPseAAC-Builderrdquo [85] and ldquopropyrdquo [86] wereestablished for generating various special Choursquos pseudo-amino acid compositions in addition to the web-serverPseAAC [87] built in 2008 According to a recent review [33]the general formof Choursquos PseAAC for an enzyme sample canbe formulated by
E = [12059511205952sdot sdot sdot 120595119906sdot sdot sdot 120595Ω]
T (11)
where the subscript Ω is an integer and its value as wellas the components 120595
119906(119906 = 1 2 Ω) will depend on
how to extract the desired information from the amino acidsequence of E (cf (10)) Next let us describe how to extractuseful information from the benchmark datasetS andOnlineSupporting Information S2 to define the enzyme samplesconcerned via (11)
To incorporate as much useful information as possiblefrom an enzyme sample we are to approach this problemfrom three different angles followed by incorporating thefeature elements thus obtained into the general form ofPseAAC of (11)(a) Amino Acid Composition The components of amino acidcomposition have beenwidely used to predict various proteinattributes [57ndash61] In this study they were also included as thefirst 20 elements in the general Choursquos PseAAC (cf (11)) thatis
120595119906= 119891119906
(119906 = 1 2 20) (12)
where 119891119906has the same meaning as in (10)
(b) Dipeptide Composition Dipeptide composition has beenused to predict the protein secondary structural contents[88 89] as well as various protein attributes (see eg [90ndash93]) The number of different dipeptides is 20 times 20 = 400Suppose that the normalized occurrence frequencies of the400 dipeptides in an enzyme sample are given by
119891
(2)
119906(119906 = 1 2 400) (13)
BioMed Research International 5
Incorporating the above 400 dipeptide components into (11)we have
120595119906+20
= 119891
(2)
119906(119906 = 1 2 400) (14)
(c) Sequential Evolution Information Biology is a naturalscience with a historic dimension All biological specieshave developed starting out from a very limited number ofancestral species Their evolution involves changes of singleresidues insertions and deletions of several residues [94]gene doubling and gene fusion With these changes accu-mulated for a long period of time many similarities betweeninitial and resultant amino acid sequences are graduallyeliminated but the corresponding proteins may still sharemany common attributes [18] such as having basically thesame biological function and residing at a same subcellularlocation To extract the sequential evolution information anduse it to define the components of (11) the PSSM (PositionSpecific Scoring Matrix) was used as described next
According to Schaffer et al [95] the sequence evolutioninformation of enzyme E with 119871 amino acid residues can beexpressed by an 119871 times 20matrix as given by
P(0)PSSM =
[
[
[
[
[
[
[
119864
0
1rarr1119864
0
1rarr2sdot sdot sdot 119864
0
1rarr20
119864
0
2rarr1119864
0
2rarr2sdot sdot sdot 119864
0
2rarr20
119864
0
119871rarr1119864
0
119871rarr2sdot sdot sdot 119864
0
119871rarr20
]
]
]
]
]
]
]
(15)
where 1198640119894rarr 119895
represents the original score of the 119894th aminoacid residue (119894 = 1 2 119871) in the enzyme sequence changedto amino acid type 119895 (119895 = 1 2 20) in the process ofevolution Here the numerical codes 1 2 20 are used torepresent the 20 native amino acid types denoted by A CD E F G H I K L M N P Q R S T V W and Y The119871 times 20 scores in (15) were generated by using PSI-BLAST[96] to search the UniProtKBSwiss-Prot database (Release2013-05) through three iterations with 0001 as the 119864 valuecutoff for multiple sequence alignment against the sequenceof the enzyme E In order to make every element in (15) bescaled from their original score ranges into region of [0 1]we performed a conversion through the standard sigmoidfunction to make it become
P(1)PSSM =
[
[
[
[
[
[
[
119864
1
1rarr1119864
1
1rarr2sdot sdot sdot 119864
1
1rarr20
119864
1
2rarr1119864
1
2rarr2sdot sdot sdot 119864
1
2rarr20
119864
1
119871rarr1119864
1
119871rarr2sdot sdot sdot 119864
1
119871rarr20
]
]
]
]
]
]
]
(16)
where
119864
1
119894rarr 119895=
1
1 + 119890
minus1198640
119894rarr 119895
(1 le 119894 le 119871 1 le 119895 le 20) (17)
Nowwe extract the useful information from (16) to define thecomponents of (11) via the following approach
120595119906+420
= ℓ119906
(119906 = 1 2 20) (18)
where
ℓ119895=
1
119871
times
119871
sum
119896=1
119864
1
119896rarr119895(119895 = 1 2 20) (19)
(d) Grey System Model Approach The grey system theory[97] is quite useful in dealing with complicated systems thatlack sufficient information or need to process uncertaininformation According to the grey system theory we canextract the following information from the 119895th column of(16) that is
[
119886
119895
1
119886
119895
2
] = (BT119895B119895)
minus1
BT119895U119895
(119895 = 1 2 20) (20)
where
B119895=
[
[
[
[
[
[
[
[
[
[
minus119864
1
2rarr119895minus119864
1
1rarr119895minus 05119864
1
2rarr1198951
minus119864
1
3rarr119895minus
2
sum
119894=1
119864
1
119894rarr 119895minus 05119864
1
3rarr1198951
minus119864
1
119871rarr119895minus
119871minus1
sum
119894=1
119864
1
119894rarr 119895minus 05119864
1
119871rarr1198951
]
]
]
]
]
]
]
]
]
]
U119895=
[
[
[
[
[
[
119864
1
2rarr119895minus 119864
1
1rarr119895
119864
1
3rarr119895minus 119864
1
2rarr119895
119864
1
119871rarr119895minus 119864
1
119871minus1rarr119895
]
]
]
]
]
]
(21)
Therefore based on the grey system theory and (20) we canextract another 20 times 2 = 40 quantities from (16) to definethe components of (11) that is
120593119895=
1199081119886
119895
1when 119895 is an odd number
1199082119886
119895
2when 119895 is an even number
1 le 119895 le 20
(22)
where 1198861198951and 119886119895
2are given by (20) 119908
1and 119908
2are weight
factors which were all set to 1 in the current studySubstituting the elements in (12) (14) (18) and (22) we
finally obtain a total of Ω = 20 + 400 + 20 + 40 = 480
components for the PseAAC of (11) where
120595119906=
119891119906
when 1 le 119906 le 20119891
(2)
119906when 21 le 119906 le 420
ℓ119906
when 421 le 119906 le 440120593119906
when 440 le 119906 le 480
(23)
In other words in this study (11) or Choursquos PseAAC is a 480-D vector whose 480 components are given by (23) derivedfrom the amino acid composition dipeptide compositionsequential evolution information and grey system theory(e) Representing Enzyme-Drug Pairs Now the pair betweenan enzyme molecule E and a drug compound D can beformulated by combing (8) and (11) as given by
G = D oplus E
= [1198601sdot sdot sdot 119860
256119867119909
CF 1205951sdot sdot sdot 120595480]
T
(24)
6 BioMed Research International
where G represents the enzyme-drug pair oplus the orthogonalsum [51] and each of the (258 + 480) = 738 feature elementsis given in (8) and (23)
For the convenience of the later formulation let us use119909119894(119894 = 1 2 738) to represent the 738 components of (24)
that is
G = [11990911199092sdot sdot sdot 119909119894sdot sdot sdot 119909738]
T (25)
To optimize the prediction results different weights wereusually tested for each of the elements in (25) However sinceit would consume a lot of computational time for a total of 738weight factors here let us adopt the normalization approachto deal with this problem as done in [98 99] that is convert119909119894in (25) to 119910
119894according to the following equation
119910119894=
2tanminus1 (119909119894)
120587
(119894 = 1 2 738) (26)
where tanminus1 means arctangent By means of (26) everycomponent in (25) will be converted into the range of[minus1 1] that is we have minus1 le 119910
119894le 1 As demonstrated
in [98 99] the normalization approach via (26) was quiteeffective in enhancing the quality of prediction operated ina high dimension space Therefore in this study we wouldnot to take the procedure of optimizing the weight factorssignificantly reducing the computational times
23 Fuzzy 119870-Nearest Neighbour Algorithm The 119870-NN (119870-Nearest Neighbor) classifier is quite popular in patternrecognition community owing to its good performance andsimple-to-use feature According to the 119870-NN rule [100]named also as the ldquovoting 119870-NN rulerdquo the query sampleshould be assigned to the subset represented by a majorityof its 119870 nearest neighbors as illustrated in Figure 5 of [33]
Fuzzy 119870-NN classification method [101] is a specialvariation of the119870-NNclassification family Instead of roughlyassigning the label based on a voting from the 119870 nearestneighbors it attempts to estimate the membership valuesthat indicate how much degree the query sample belongsto the classes concerned Obviously it is impossible for anycharacteristic description to contain complete informationwhich would make the classification ambiguous In view ofthis the fuzzy principle is very reasonable and particularlyuseful in dealing with complicated biological systems such asidentifying nuclear receptor subfamilies [102] characterizingthe structure of fast-folding proteins [103] classifying Gprotein-coupled receptors [104] predicting protein quater-nary structural attributes [105] predicting protein structuralclasses [106 107] and so forth
Next let us give a brief introduction how to use thefuzzy119870-NN approach to identify the interactions between theenzymes and the drug compounds in the network concerned
Supposing that S(119873) = G1G2 G
119873 is a set of vec-
tors representing 119873 enzyme-drug pairs in a training set
classified into two classes 119862+ 119862minus where 119862+ denotes theinteractive pair class while 119862minus the noninteractive pair classSlowast(G) = Glowast
1Glowast2 Glowast
119870 sub S(119873) is the subset of the 119870
nearest neighbor pairs to the query pair G Thus the fuzzymembership value for the query pair G in the two classes ofS(119873) is given by
120583
+
(G) =sum
119870
119895=1120583
+(Glowast119895) 119889(GGlowast
119895)
minus2(120593minus1)
sum
119870
119895=1119889(GGlowast
119895)
minus2(120593minus1)
120583
minus
(G) =sum
119870
119895=1120583
minus(Glowast119895) 119889(GGlowast
119895)
minus2(120593minus1)
sum
119870
119895=1119889(GGlowast
119895)
minus2(120593minus1)
(27)
where 119870 is the number of the nearest neighbors counted forthe query pairG 120583+(Glowast
119895) and 120583minus(Glowast
119895) the fuzzy membership
values of the training sample Glowast119895to the class 119862+ and 119862minus
respectively as will be further defined next 119889(GGlowast119895) the
cosine distance between G and its 119895th nearest pair Glowast119895in
the training dataset S(119873) 120593(gt 1) the fuzzy coefficient fordetermining how heavily the distance is weighted whencalculating each nearest neighborrsquos contribution to the mem-bership value Note that the parameters 119870 and 120593 will affectthe computation result of (27) and they will be optimizedby a grid-search as will be described later Also variousother metrics can be chosen for 119889(GGlowast
119895) such as Euclidean
distance Hamming distance [108] andMahalanobis distance[55 109]
The quantitative definitions for the aforementioned120583
+(Glowast119895) and 120583minus(Glowast
119895) in (27) are given by
120583
+
(Glowast119895) =
1 if Glowast119895isin 119862
+
0 otherwise
120583
minus
(Glowast119895) =
1 if Glowast119895isin 119862
minus
0 otherwise
(28)
Substituting the results obtained by (27) into (28) it followsthat if 120583+(G) gt 120583
minus(G) then the query pair G is an
interactive couple otherwise noninteractive In other wordsthe outcome can be formulated as
G isin
119862
+ if 120583+ (G) gt 120583minus (G)
119862
minus otherwise
(29)
If there is a tie between 120583+(G) and 120583minus(G) the query pair Gwill be randomly assigned to one of the two classes Howevercase like that is quite rare and in this study never happened
The predictor thus established is called iEzy-Drugwhere ldquoirdquo means identify and ldquoEzy-Drugrdquo means the inter-action between enzyme and drug To provide an intuitiveoverall picture a flowchart is provided in Figure 2 to showthe process of how the classifier works in identifying enzyme-drug interactions
BioMed Research International 7
Input a queryprotein sequence and
a drug code
Create Chous PseAAC of a protein
Create feature vector of a drug
Feature fusion
engineTraining dataset
Yes No
Noninteractive enzyme drug pair
Interactive enzymedrug pair
AACDipeptideSeq Evol
Grey model
Mol FptInfo entropy
CF
FKNN operation
Figure 2 A flowchart to show the operation process of the iEzy-Drug predictor See the text for further explanation
24 Criteria for Performance Evaluation In the literaturethe following equation set is often used for examining theperformance quality of a predictor
Sn = TPTP + FN
Sp = TNTN + FP
Acc = TP + TNTP + TN + FP + FN
MCC = (TP times TN) minus (FP times FN)radic(TP + FP) (TP + FN) (TN + FP) (TN + FN)
(30)
where TP represents the true positive TN the true negativeFP the false positive FN the false negative Sn the sensitiv-ity Sp the specificity Acc the accuracy MCC the Mathewrsquoscorrelation coefficient
To most biologists however the four metrics as formu-lated in (30) are not quite intuitive and easier-to-understandparticularly for the Mathewrsquos correlation coefficient Herelet us adopt the Choursquos symbols to formulate the previousfour metrics By means of Choursquos symbols [111 112] the rates
of correct predictions for the interactive enzyme-drug pairsin dataset S+ and the noninteractive enzyme-drug pairs indataset Sminus are respectively defined by (cf (1))
Λ
+
=
119873
+minus 119873
+
minus
119873
+ for the interactive enzyme-drug pairs
Λ
minus
=
119873
minusminus 119873
minus
+
119873
minus for the noninteractive enzyme-drug pairs
(31)
where 119873+ is the total number of the interactive enzyme-drug pairs investigated while 119873+
minusis the number of the
interactive enzyme-drug pairs incorrectly predicted as thenoninteractive enzyme-drug pairs 119873minus is the total numberof the noninteractive enzyme-drug pairs investigated while119873
minus
+is the number of the noninteractive enzyme-drug pairs
incorrectly predicted as the interactive enzyme-drug pairsThe overall success prediction rate is given by [113] as follows
Λ =
Λ
+119873
++ Λ
minus119873
minus
119873
++ 119873
minus= 1 minus
119873
+
minus+ 119873
minus
+
119873
++ 119873
minus
(32)
It is obvious from (31)-(32) that if and only if none ofthe interactive enzyme-drug pairs and the noninteractiveenzyme-drug pairs are mispredicted that is 119873+
minus= 119873
minus
+= 0
and Λ+ = Λ
minus= 1 we have the overall success rate Λ = 1
Otherwise the overall success rate would be smaller than 1The relations between the symbols in (32) and those in
(30) are given by
TP = 119873+ minus 119873+minus
TN = 119873
minus
minus 119873
minus
+
FP = 119873minus+
FN = 119873
+
minus
(33)
Substituting (33) into (30) and also noting (31)-(32) we obtain
Sn = Λ+ = 1 minus119873
+
minus
119873
+
Sp = Λminus = 1 minus119873
minus
+
119873
minus
Acc = Λ = 1 minus119873
+
minus+ 119873
minus
+
119873
++ 119873
minus
MCC =1 minus ((119873
+
minus119873
+) + (119873
minus
+119873
minus))
radic(1 + (119873
minus
+minus 119873
+
minus) 119873
+) (1 + (119873
+
minusminus 119873
minus
+) 119873
minus)
(34)
Now it is obvious to see from (34) when 119873
+
minus= 0
meaning none of the interactive enzyme-drug pairs was
8 BioMed Research International
02
46
810
1
15
2089
0895
09
0905
091
0915
092
ACC
K120593
Figure 3 A 3D plot to show how the parameter in (27) wasoptimized for the iEzy-Drug predictor
mispredicted to be a noninteractive enzyme-drug pair wehave the sensitivity Sn = 1 while 119873+
minus= 119873
+ meaning thatall the interactive enzyme-drug pairs weremispredicted to bethe noninteractive enzyme-drug pairs we have the sensitivitySn = 0 Likewise when 119873minus
+= 0 meaning none of the
noninteractive enzyme-drug pairs wasmispredicted we havethe specificity Sp = 1 while 119873minus
+= 119873
minus meaning all thenoninteractive enzyme-drug pairs were incorrectly predictedas interactive enzyme-drug pairs we have the specificity Sp =0 When119873+
minus= 119873
minus
+= 0 meaning that none of the interactive
enzyme-drug pairs in the dataset S+ and none of the nonin-teractive enzyme-drug pairs in Sminus was incorrectly predictedwe have the overall accuracy Acc = Λ = 1 while 119873+
minus= 119873
+
and 119873minus+= 119873
minus meaning that all the interactive enzyme-drugpairs in the dataset S+ and all the noninteractive enzyme-drug pairs in Sminus were mispredicted we have the overallaccuracy Acc = Λ = 0 The MCC correlation coefficient isusually used for measuring the quality of binary (two-class)classifications When 119873+
minus= 119873
minus
+= 0 meaning that none
of the interactive enzyme-drug pairs in the dataset S+ andnone of the noninteractive enzyme-drug pairs in Sminus weremispredicted we have MCC = 1 when 119873+
minus= 119873
+2 and
119873
minus
+= 119873
minus2 we have MCC = 0 meaning no better than
random prediction when 119873+minus= 119873
+ and 119873minus+= 119873
minus we haveMCC = minus1 meaning total disagreement between predictionand observation As we can see from the previous discussionit is much more intuitive and easier-to-understand whenusing (34) to examine a predictor for its sensitivity specificityoverall accuracy and Mathewrsquos correlation coefficient It isinstructive to point out that themetrics as defined in (30) and(34) are valid for single label systems for multilabel systemsa set of more complicated metrics should be used as given in[114]
3 Results and Discussion
31 Cross-Validation How to properly examine the predic-tion quality is a key for developing a new predictor and
False positive rate
True
pos
itive
rate
1
01
02
03
04
05
06
07
08
09
0
101 02 03 04 05 06 07 08 090
BaselineiEzy-Drug AUC = 09377
Figure 4 A plot for the ROC curve to quantitatively show theperformance of the iEzy-Drug predictor
estimating its potential application value Generally speak-ing the following three cross-validation methods are oftenused to examine a predictor of its effectiveness in practicalapplication independent dataset test subsampling or 119870-fold(such as 5-fold 7-fold or 10-fold) test and jackknife test[108] However as elaborated by a penetrating analysis in[115] considerable arbitrariness exists in the independentdataset test Also as demonstrated by (27)ndash(29) in [33]the subsampling test (or 119870-fold cross-validation) cannotavoid arbitrariness either Only the jackknife test is the leastarbitrary that can always yield a unique result for a givenbenchmark dataset Therefore the jackknife test has beenwidely recognized and increasingly utilized by investigatorsto examine the quality of various predictors (see eg [66 7174 80]) Accordingly the success rate by the jackknife test wasalso used to optimize the two uncertain parameters 119870 and 120593in (27) The result thus obtained is shown in Figure 3 fromwhich we obtain when 119870 = 6 and 120593 = 15 the iEzy-Drugpredictor reaches its optimized status
The success rates thus obtained by the jackknife test inidentifying interactive Enzyme-drug pairs or noninteractiveenzyme-drug pairs on the benchmark dataset S (cf OnlineSupporting Information S1) are given in Table 1 where forfacilitating comparison the corresponding result by He et al[110] is also given As we can see from the table the overallaccuracy Acc achieved by iEzy-Drug was 9103 remarkablyhigher than 8548 the corresponding rate obtained byHe et al [110] on the same benchmark Furthermore listedin Table 1 are also the values obtained by iEzy-Drug for theother three metrics that is Sn = 9081 Sp = 9114 andMCC = 8039 indicating that the accuracy of iEzy-Drug isnot only very high but also quite stale
To provide a graphical illustration to show the per-formance of the current binary classifier iEzy-Drug asits discrimination threshold is varied a 2D plot calledReceiver Operating Characteristic (ROC) curve [116 117]was also given (Figure 4) In the ROC curve the verticalcoordinate 119884 is for the true positive rate or Sn (cf (34))while horizontal coordinate 119883 for the false positive rate
BioMed Research International 9
Read Me Data Citation
Enter both the protein sequence and the drug code (example) the number of query pairs is limited at 10 or less for each submission
Submit Clear
You can download the program and run it on your own local computer
iEzy-Drug identifying the interaction between enzymes anddrugs in cellular networking
Figure 5 A semiscreenshot to show the top page of the iEzy-Drugweb-server Its web-site address is at httpwwwjci-bioinfocniEzy-Drug
Table 1 The jackknife success rates obtained with iEzy-Drug in identifying interactive enzyme-drug pairs and noninteractive enzyme-drugpairs for the benchmark dataset S (cf Online Supporting Information S1)
Method Acc Sn Sp MCC
iEzy-Druga 74258157 = 9103 24692719 = 9081 49565438 = 9114 8039NN predictorb 8548 NA NA NAaSee (27) where the parameters119870 = 6 and 120593 = 15bSee [110]
or 1-Sp The best possible prediction method would yielda point with the coordinate (0 1) representing 100 truepositive rate (sensitivity Sn) and 0 false positive rate or100 specificity Therefore the (0 1) point is also calleda perfect classification A completely random guess wouldgive a point along a diagonal from the point (0 0) to (1 1)The area under the ROC curve also called Area Under theROC (AUROC) is often used to indicate the performancequality of a binary classifier the value 05 of AUROCis equivalent to random prediction while 1 of AUROCrepresents a perfect one As we can see from Figure 4the AUROC value obtained by iEzy-Drug is 09377
The reason why iEzy-Drug can remarkably improve theprediction quality is that it has introduced the 2D molecularfingerprints to represent drug samples see Online SupportingInformation S3 for the detailed fingerprint expressions for thedrugs listed in Online Supporting Information S1 and thatit has successfully used PseAAC to incorporate the featuresderived from the sequences of enzymes that are essentialfor identifying the interaction of enzymes with drugs in thecellular networking
To enhance the value of its practical applications theweb server for iEzy-Drug has been established that canbe freely accessible at httpwwwjci-bioinfocniEzy-DrugIt is anticipated that the web server will become a usefulhigh throughput tool for both basic research and drug
development in the relevant areas or at the very least playa complementary role to the existing method [39 110 118] forwhich so far noweb-serverwhatsoever has been provided yet
32 The Protocol or User Guide For the convenience of thevast majority of biologists and pharmaceutical scientists herelet us provide a step-by-step guide to show how the userscan easily get the desired result by means of the web serverwithout the need to follow the complicated mathematicalequations presented in this paper for the process of develop-ing the predictor and its integrityStep 1 Open the web server at the site httpwwwjci-bio-infocniEzy-Drug and you will see the top page of thepredictor on your computer screen as shown in Figure 5Click on the ReadMe button to see a brief introduction aboutiEzy-Drug predictor and the caveat when using itStep 2 Either type or copypaste the query pairs into theinput box at the center of Figure 5 Each query pair consistsof two parts one is for the protein sequence and the other forthe drug The enzyme sequence should be in FASTA formatwhile the drug in the KEGG code Examples for the querypairs input can be seen by clicking on the Example buttonright above the input boxStep 3 Click on the Submit button to see the predicted resultFor example if you use the four query pairs in the Example
10 BioMed Research International
window as the input after clicking the Submit button youwill see on your screen that the ldquohsa 10056rdquo enzyme andthe ldquoD0021rdquo drug are an interactive pair and that the ldquohsa100rdquo enzyme and the ldquoD0037rdquo drug are also an interactivepair but that the ldquohsa 3295rdquo enzyme and the ldquoD00889rdquo drugare not an interactive pair and that the ldquohsa 7366rdquo enzymeand the ldquoD03601rdquo drug are not an interactive pair eitherAll these results are fully consistent with the experimentalobservations It takes about 3 minutes before the results areshown on the screenStep 4 Click on the Citation button to find the relevant paperthat documents the detailed development and algorithm ofiEzy-DurgStep 5 Click on the Data button to download the benchmarkdataset used to train and test the iEzy-Durg predictorStep 6 The program code is also available by clicking thebutton download on the lower panel of Figure 5
Acknowledgments
The authors would like to thank the three anonymousreviewers whose constructive comments are very helpful forstrengthening the presentation of the paper This work wassupported by the Grants from the National Natural ScienceFoundation of China (no 31260273) the Jiangxi ProvincialForeign Scientific and Technological Cooperation Project(no 20120BDH80023) Natural Science Foundation of JiangxiProvince China (no 2010GZS0122 20122BAB201020) theDepartment of Education of Jiangxi Province (GJJ12490)the LuoDi plan of the Department of Education of JiangxiProvince (KJLD12083) and the Jiangxi Provincial Founda-tion for Leaders of Disciplines in Science (20113BCB22008)The funders had no role in study design data collection andanalysis decision to publish or preparation of the paper
References
[1] A Bairoch ldquoThe ENZYME database in 2000rdquo Nucleic AcidsResearch vol 28 no 1 pp 304ndash305 2000
[2] S H Koenig and R D Brown ldquoH2CO3as substrate for carbonic
anhydrase in the dehydration of H2CO3(minus)rdquo Proceedings of the
National Academy of Sciences of the United States of Americavol 69 no 9 pp 2422ndash2425 1972
[3] S X Lin and J Lapointe ldquoTheoretical and experimental biologyin onerdquo Journal of Biomedical Science and Engineering vol 6 pp435ndash442 2013
[4] K C Chou and S P Jiang ldquoStudies on the rate of diffusion-controlled reactions of enzymes Spatial factor and force fieldfactorrdquo Scientia Sinica vol 17 no 5 pp 664ndash680 1974
[5] K C Chou ldquoThe kinetics of the combination reaction betweenenzyme and substraterdquo Scientia Sinica vol 19 no 4 pp 505ndash528 1976
[6] K C Chou and G P Zhou ldquoRole of the protein outside activesite on the diffusion-controlled reaction of enzymerdquo Journal ofthe American Chemical Society vol 104 no 5 pp 1409ndash14131982
[7] R A Poorman A G Tomasselli R L Heinrikson and FJ Kezdy ldquoA cumulative specificity model for proteases from
human immunodeficiency virus types 1 and 2 inferred fromstatistical analysis of an extended substrate data baserdquo TheJournal of Biological Chemistry vol 266 no 22 pp 14554ndash145611991
[8] K-C Chou ldquoA vectorized sequence-coupling model for pre-dicting HIV protease cleavage sites in proteinsrdquo The Journal ofBiological Chemistry vol 268 no 23 pp 16938ndash16948 1993
[9] G Z Liang and S Z Li ldquoA new sequence representation asapplied in better specificity elucidation for human immunod-eficiency virus type 1 proteaserdquo Biopolymers vol 88 no 3 pp401ndash412 2007
[10] J J Chou ldquoPredicting cleavability of peptide sequences byHIVprotease via correlation-angle approachrdquo Journal of ProteinChemistry vol 12 no 3 pp 291ndash302 1993
[11] Q S Du S Wang D Q Wei S Sirois and K C ChouldquoMolecular modeling and chemical modification for findingpeptide inhibitor against severe acute respiratory syndromecoronavirus main proteinaserdquo Analytical Biochemistry vol 337no 2 pp 262ndash270 2005
[12] Y Gan H Huang Y Huang et al ldquoSynthesis and activity ofan octapeptide inhibitor designed for SARS coronavirus mainproteinaserdquo Peptides vol 27 no 4 pp 622ndash625 2006
[13] Q S Du H Sun and K C Chou ldquoInhibitor design for SARScoronavirus main protease based on lsquodistorted key theoryrsquordquoMedicinal Chemistry vol 3 no 1 pp 1ndash6 2007
[14] K C Chou ldquoPrediction of human immunodeficiency virusprotease cleavage sites in proteinsrdquoAnalytical Biochemistry vol233 no 1 pp 1ndash14 1996
[15] J Knowles and G Gromo ldquoTarget selection in drug discoveryrdquoNature Reviews Drug Discovery vol 2 no 1 pp 63ndash69 2003
[16] A C Cheng R G Coleman K T Smyth et al ldquoStructure-basedmaximal affinity model predicts small-molecule druggabilityrdquoNature Biotechnology vol 25 no 1 pp 71ndash75 2007
[17] M Rarey B Kramer T Lengauer and G Klebe ldquoA fast flexibledocking method using an incremental construction algorithmrdquoJournal of Molecular Biology vol 261 no 3 pp 470ndash489 1996
[18] K C Chou ldquoReview structural bioinformatics and its impactto biomedical sciencerdquo Current Medicinal Chemistry vol 11 no16 pp 2105ndash2134 2004
[19] K C Chou D Q Wei and W Z Zhong ldquoBinding mechanismof coronavirus main proteinase with ligands and its implicationto drug design against SARSrdquo Biochemical and BiophysicalResearch Communications vol 308 pp 148ndash151 2003 (Erratumin Biochemical and Biophysical Research Communications vol310 p 675 2003)
[20] K C Chou D Q Wei Q S Du S Sirois and W Z ZhongldquoProgress in computational approach to drug developmentagaint SARSrdquo Current Medicinal Chemistry vol 13 no 27 pp3263ndash3270 2006
[21] G P Zhou and F A Troy ldquoNMR studies on how the bindingcomplex of polyisoprenol recognition sequence peptides andpolyisoprenols can modulate membrane structurerdquo CurrentProtein and Peptide Science vol 6 no 5 pp 399ndash411 2005
[22] R B Huang Q S Du C H Wang and K C Chou ldquoAn in-depth analysis of the biological functional studies based on theNMR M2 channel structure of influenza A virusrdquo Biochemicaland Biophysical Research Communications vol 377 no 4 pp1243ndash1247 2008
[23] Q S Du R B Huang C H Wang X M Li and K C ChouldquoEnergetic analysis of the two controversial drug binding sitesof the M2 proton channel in influenza A virusrdquo Journal ofTheoretical Biology vol 259 no 1 pp 159ndash164 2009
BioMed Research International 11
[24] M J Berardi W M Shih S C Harrison and J J Chou ldquoMito-chondrial uncoupling protein 2 structure determined by NMRmolecular fragment searchingrdquo Nature vol 476 no 7358 pp109ndash114 2011
[25] J R Schnell and J J Chou ldquoStructure andmechanism of theM2proton channel of influenza A virusrdquo Nature vol 451 no 7178pp 591ndash595 2008
[26] B OuYang S Xie M J Berardi et al ldquoUnusual architecture ofthe p7 channel from hepatitis C virusrdquoNature vol 498 pp 521ndash525 2013
[27] K Oxenoid and J J Chou ldquoThe structure of phospholambanpentamer reveals a channel-like architecture in membranesrdquoProceedings of the National Academy of Sciences of the UnitedStates of America vol 102 no 31 pp 10870ndash10875 2005
[28] M E Call KWWucherpfennig and J J Chou ldquoThe structuralbasis for intramembrane assembly of an activating immunore-ceptor complexrdquo Nature Immunology vol 11 no 11 pp 1023ndash1029 2010
[29] R M Pielak J R Schnell and J J Chou ldquoMechanism of druginhibition and drug resistance of influenza A M2 channelrdquoProceedings of the National Academy of Sciences of the UnitedStates of America vol 106 no 18 pp 7379ndash7384 2009
[30] J Wang R M Pielak M A McClintock and J J ChouldquoSolution structure and functional analysis of the influenza Bproton channelrdquo Nature Structural and Molecular Biology vol16 no 12 pp 1267ndash1271 2009
[31] K C Chou ldquoCoupling interaction between thromboxane A2receptor and alpha-13 subunit of guanine nucleotide-bindingproteinrdquo Journal of Proteome Research vol 4 no 5 pp 1681ndash1686 2005
[32] K C Chou ldquoInsights from modeling three-dimensional struc-tures of the human potassium and sodium channelsrdquo Journal ofProteome Research vol 3 no 4 pp 856ndash861 2004
[33] K C Chou ldquoSome remarks on protein attribute prediction andpseudo amino acid compositionrdquo Journal ofTheoretical Biologyvol 273 no 1 pp 236ndash247 2011
[34] W Z Lin J A Fang X Xiao and K C Chou ldquoiLoc-animal amulti-label learning classifier for predicting subcellular local-ization of animal proteinsrdquo Molecular BioSystems vol 9 pp634ndash644 2013
[35] X Xiao PWangW Z Lin J H Jia and K C Chou ldquoiAMP-2La two-level multi-label classifier for identifying antimicrobialpeptides and their functional typesrdquo Analytical Biochemistryvol 436 pp 168ndash177 2013
[36] W Chen P M Feng H Lin and K C Chou ldquoiRSpot-PseDNCidentify recombination spots with pseudo dinucleotide compo-sitionrdquo Nucleic Acids Research vol 41 article e69 2013
[37] K C Chou Z C Wu and X Xiao ldquoILoc-Hum using theaccumulation-label scale to predict subcellular locations ofhuman proteins with both single and multiple sitesrdquoMolecularBioSystems vol 8 no 2 pp 629ndash641 2012
[38] M Kotera M Hirakawa T Tokimatsu S Goto and MKanehisa ldquoThe KEGG databases and tools facilitating omicsanalysis latest developments involving human diseases andpharmaceuticalsrdquo Methods in Molecular Biology vol 802 pp19ndash39 2012
[39] Y YamanishiM Araki A GutteridgeWHonda andM Kane-hisa ldquoPrediction of drug-target interaction networks from theintegration of chemical and genomic spacesrdquo Bioinformaticsvol 24 no 13 pp i232ndashi240 2008
[40] P Finn S Muggleton D Page and A Srinivasan ldquoPharma-cophore discovery using the Inductive Logic Programmingsystem PROGOLrdquo Machine Learning vol 30 no 2-3 pp 241ndash270 1998
[41] I Vogt D Stumpfe H E Ahmed and J Bajorath ldquoMethodsfor computer-aided chemical biology Part 2 evaluation of com-pound selectivity using 2D molecular fingerprintsrdquo ChemicalBiology and Drug Design vol 70 pp 195ndash205 2007
[42] H Eckert and J Bajorath ldquoMolecular similarity analysis in vir-tual screening foundations limitations and novel approachesrdquoDrug Discovery Today vol 12 no 5-6 pp 225ndash233 2007
[43] S Laurent L V Elst and R N Muller ldquoComparative studyof the physicochemical properties of six clinical low molecularweight gadolinium contrast agentsrdquo Contrast Media amp Molecu-lar Imaging vol 1 no 3 pp 128ndash137 2006
[44] E Gregori-Puigjane R Garriga-Sust and J Mestres ldquoIndexingmolecules with chemical graph identifiersrdquo Journal of Compu-tational Chemistry vol 32 no 12 pp 2638ndash2646 2011
[45] B Ren ldquoApplication of novel atom-type AI topological indicesto QSPR studies of alkanesrdquo Computers and Chemistry vol 26no 4 pp 357ndash369 2002
[46] N M OrsquoBoyle M Banck C A James C Morley T Vander-meersch and G R Hutchison ldquoOpen Babel an open chemicaltoolboxrdquo Journal of Cheminformatics vol 3 p 33 2011
[47] V J Gillet P Willett and J Bradshaw ldquoSimilarity searchingusing reduced graphsrdquo Journal of Chemical Information andComputer Sciences vol 43 no 2 pp 338ndash345 2003
[48] D Butina ldquoUnsupervised data base clustering based on day-lightrsquos fingerprint and Tanimoto similarity a fast and automatedway to cluster small and large data setsrdquo Journal of ChemicalInformation and Computer Sciences vol 39 no 4 pp 747ndash7501999
[49] C E Shannon ldquoA mathematical theory of communicationrdquoACM SIGMOBILE Mobile Computing and CommunicationsReview vol 5 pp 3ndash55 2001
[50] V D Gusev L A Nemytikova and N A Chuzhanova ldquoOn thecomplexity measures of genetic sequencesrdquo Bioinformatics vol15 no 12 pp 994ndash999 1999
[51] K C Chou and H B Shen ldquoReview recent progress in proteinsubcellular location predictionrdquo Analytical Biochemistry vol370 no 1 pp 1ndash16 2007
[52] S F Altschul ldquoEvaluating the statistical significance of multipledistinct local alignmentsrdquo in Theoretical and ComputationalMethods in Genome Research S Suhai Ed pp 1ndash14 PlenumNew York NY USA 1997
[53] J C Wootton and S Federhen ldquoStatistics of local complexity inamino acid sequences and sequence databasesrdquo Computers andChemistry vol 17 no 2 pp 149ndash163 1993
[54] H Nakashima K Nishikawa and T Ooi ldquoThe folding type ofa protein is relevant to the amino acid compositionrdquo Journal ofBiochemistry vol 99 no 1 pp 153ndash162 1986
[55] K C Chou and C T Zhang ldquoPredicting protein folding typesby distance functions that make allowances for amino acidinteractionsrdquo The Journal of Biological Chemistry vol 269 no35 pp 22014ndash22020 1994
[56] K-C Chou ldquoA novel approach to predicting protein structuralclasses in a (20-1)-D amino acid composition spacerdquo Proteinsvol 21 no 4 pp 319ndash344 1995
[57] H Nakashima and K Nishikawa ldquoDiscrimination of intracel-lular and extracellular proteins using amino acid compositionand residue-pair frequenciesrdquo Journal of Molecular Biology vol238 no 1 pp 54ndash61 1994
12 BioMed Research International
[58] G P Zhou ldquoAn intriguing controversy over protein structuralclass predictionrdquo Journal of Protein Chemistry vol 17 no 8 pp729ndash738 1998
[59] I Bahar A R Atilgan R L Jernigan and B Erman ldquoUnder-standing the recognition of protein structural classes by aminoacid compositionrdquo Proteins vol 29 pp 172ndash185 1997
[60] J Cedano P Aloy J A Perez-Pons and E Querol ldquoRelationbetween amino acid composition and cellular location ofproteinsrdquo Journal of Molecular Biology vol 266 no 3 pp 594ndash600 1997
[61] G P Zhou and K Doctor ldquoSubcellular location prediction ofapoptosis proteinsrdquo Proteins vol 50 no 1 pp 44ndash48 2003
[62] KChou ldquoPrediction of protein cellular attributes using pseudo-amino acid compositionrdquo Proteins vol 43 pp 246ndash255 2001(Erratum in Proteins vol 44 p 60 2001)
[63] K C Chou ldquoUsing amphiphilic pseudo amino acid composi-tion to predict enzyme subfamily classesrdquo Bioinformatics vol21 no 1 pp 10ndash19 2005
[64] D Zou Z He J He and Y Xia ldquoSupersecondary structureprediction using Choursquos pseudo amino acid compositionrdquoJournal of Computational Chemistry vol 32 no 2 pp 271ndash2782011
[65] M M Beigi M Behjati and H Mohabatkar ldquoPrediction ofmetalloproteinase family based on the concept ofChoursquos pseudoamino acid composition using a machine learning approachrdquoJournal of Structural and Functional Genomics vol 12 no 4 pp191ndash197 2011
[66] Y K Chen and K B Li ldquoPredicting membrane protein typesby incorporating protein topology domains signal peptidesand physicochemical properties into the general form of Choursquospseudo amino acid compositionrdquo Journal ofTheoretical Biologyvol 318 pp 1ndash12 2013
[67] C Huang and J Q Yuan ldquoA multilabel model based on Choursquospseudo-amino acid composition for identifying membraneproteins with both single and multiple functional typesrdquo TheJournal of Membrane Biology vol 246 pp 327ndash334 2013
[68] S S Sahu and G Panda ldquoA novel feature representationmethod based on Choursquos pseudo amino acid composition forprotein structural class predictionrdquo Computational Biology andChemistry vol 34 no 5-6 pp 320ndash327 2010
[69] M Hayat and A Khan ldquoDiscriminating outer membraneproteins with fuzzy K-nearest neighbor algorithms based on thegeneral form of Choursquos PseAACrdquo Protein and Peptide Lettersvol 19 no 4 pp 411ndash421 2012
[70] MKhosravian F K FaramarziMM BeigiM Behbahani andH Mohabatkar ldquoPredicting antibacterial peptides by the con-cept of Choursquos pseudo-amino acid composition and machinelearning methodsrdquo Protein amp Peptide Letters vol 20 pp 180ndash186 2013
[71] HMohabatkarMM Beigi K Abdolahi and SMohsenzadehldquoPrediction of allergenic proteins by means of the concept ofChoursquos pseudo amino acid composition and amachine learningapproachrdquoMedicinal Chemistry vol 9 pp 133ndash137 2013
[72] L Nanni A Lumini D Gupta and A Garg ldquoIdentifyingbacterial virulent proteins by fusing a set of classifiers basedon variants of Choursquos pseudo amino acid composition and onevolutionary informationrdquo IEEEACM Transactions on Compu-tational Biology and Bioinformatics vol 9 no 2 pp 467ndash4752012
[73] T H Chang L C Wu T Y Lee S P Chen H D Huang andJ T Horng ldquoEuLoc a web-server for accurately predict protein
subcellular localization in eukaryotes by incorporating variousfeatures of sequence segments into the general form of ChoursquosPseAACrdquo Journal of Computer-Aided Molecular Design vol 27pp 91ndash103 2013
[74] S Zhang Y Zhang H Yang C Zhao and Q Pan ldquoUsing theconcept of Choursquos pseudo amino acid composition to predictprotein subcellular localization an approach by incorporatingevolutionary information and von Neumann entropiesrdquo AminoAcids vol 34 no 4 pp 565ndash572 2008
[75] R Zia Ur and A Khan ldquoIdentifying GPCRs and their typeswith Choursquos pseudo amino acid composition an approach frommulti-scale energy representation and position specific scoringmatrixrdquo Protein amp Peptide Letters vol 19 pp 890ndash903 2012
[76] X Y Sun S P Shi J D Qiu S B Suo S Y Huang and R PLiang ldquoIdentifying protein quaternary structural attributes byincorporating physicochemical properties into the general formof Choursquos PseAAC via discrete wavelet transformrdquo MolecularBioSystems vol 8 pp 3178ndash3184 2012
[77] L Nanni and A Lumini ldquoGenetic programming for creatingChoursquos pseudo amino acid based features for submitochondrialocalizationrdquo Amino Acids vol 34 no 4 pp 653ndash660 2008
[78] M Esmaeili H Mohabatkar and S Mohsenzadeh ldquoUsing theconcept of Choursquos pseudo amino acid composition for risk typeprediction of human papillomavirusesrdquo Journal of TheoreticalBiology vol 263 no 2 pp 203ndash209 2010
[79] H Mohabatkar ldquoPrediction of cyclin proteins using Choursquospseudo amino acid compositionrdquo Protein and Peptide Lettersvol 17 no 10 pp 1207ndash1214 2010
[80] H Mohabatkar M M Beigi and A Esmaeili ldquoPredictionof GABA(A) receptor proteins using the concept of Choursquospseudo-amino acid composition and support vector machinerdquoJournal of Theoretical Biology vol 281 no 1 pp 18ndash23 2011
[81] Y Xu J Ding L Y Wu and K C Chou ldquoiSNO-PseAAC pre-dict cysteine S-nitrosylation sites in proteins by incorporatingposition specific amino acid propensity into pseudo amino acidcompositionrdquo PLoS ONE vol 8 Article ID e55844 2013
[82] W Chen H Lin PM Feng C Ding Y C Zuo andK C ChouldquoiNuc-PhysChem a sequence-based predictor for identifyingnucleosomes via physicochemical propertiesrdquo PLoS ONE vol7 Article ID e47843 2012
[83] B Li T Huang L Liu Y Cai and K C Chou ldquoIdentificationof colorectal cancer related genes with mrmr and shortest pathin protein-protein interaction networkrdquo PLoS ONE vol 7 no 4Article ID e33393 2012
[84] Y Jiang T Huang C Lei Y F Gao Y D Cai and K C ChouldquoSignal propagation in protein interaction network duringcolorectal cancer progressionrdquo BioMed Research Internationalvol 2013 Article ID 287019 9 pages 2013
[85] P Du X Wang C Xu and Y Gao ldquoPseAAC-Builder across-platform stand-alone program for generating variousspecial Choursquos pseudo-amino acid compositionsrdquo AnalyticalBiochemistry vol 425 no 2 pp 117ndash119 2012
[86] D S Cao Q S Xu and Y Z Liang ldquoPropy a tool to generatevarious modes of Choursquos PseAACrdquo Bioinformatics vol 29 pp960ndash962 2013
[87] H Shen and K C Chou ldquoPseAAC a flexible web serverfor generating various kinds of protein pseudo amino acidcompositionrdquo Analytical Biochemistry vol 373 no 2 pp 386ndash388 2008
[88] K C Chou ldquoUsing pair-coupled amino acid composition topredict protein secondary structure contentrdquo Journal of ProteinChemistry vol 18 no 4 pp 473ndash480 1999
BioMed Research International 13
[89] W Liu and K C Chou ldquoPrediction of protein secondarystructure contentrdquo Protein Engineering vol 12 no 12 pp 1041ndash1050 1999
[90] M Bhasin and G P S Raghava ldquoClassification of nuclearreceptors based on amino acid composition and dipeptidecompositionrdquo The Journal of Biological Chemistry vol 279 no22 pp 23262ndash23266 2004
[91] H Lin andH Ding ldquoPredicting ion channels and their types bythe dipeptidemode of pseudo amino acid compositionrdquo Journalof Theoretical Biology vol 269 no 1 pp 64ndash69 2011
[92] H Lin and Q Li ldquoUsing pseudo amino acid composition topredict protein structural class approached by incorporating400 dipeptide componentsrdquo Journal of Computational Chem-istry vol 28 no 9 pp 1463ndash1466 2007
[93] L Nanni and A Lumini ldquoCombing ontologies and dipeptidecomposition for predicting DNA-binding proteinsrdquo AminoAcids vol 34 no 4 pp 635ndash641 2008
[94] K-C Chou ldquoThe convergence-divergence duality in lectindomains of selectin family and its implicationsrdquo FEBS Lettersvol 363 no 1-2 pp 123ndash126 1995
[95] A A Schaffer L Aravind T L Madden et al ldquoImprovingthe accuracy of PSI-BLAST protein database searches withcomposition-based statistics and other refinementsrdquo NucleicAcids Research vol 29 no 14 pp 2994ndash3005 2001
[96] S F Altschul and E V Koonin ldquoIterated profile searches withPSI-BLAST a tool for discovery in protein databasesrdquo Trends inBiochemical Sciences vol 23 no 11 pp 444ndash447 1998
[97] J Deng ldquoGrey entropy and grey target decision makingrdquoJournal of Grey System vol 22 no 1 pp 1ndash24 2010
[98] B M Bolstad R A Irizarry M Astrand and T P Speed ldquoAcomparison of normalizationmethods for high density oligonu-cleotide array data based on variance and biasrdquo Bioinformaticsvol 19 no 2 pp 185ndash193 2003
[99] J-Y Shi S-W Zhang Q Pan Y-M Cheng and J XieldquoPrediction of protein subcellular localization by support vectormachines using multi-scale energy and pseudo amino acidcompositionrdquo Amino Acids vol 33 no 1 pp 69ndash74 2007
[100] T Denoeux ldquoA 120581-nearest neighbor classification rule based onDempster-Shafer theoryrdquo IEEE Transactions on Systems Manand Cybernetics vol 25 no 5 pp 804ndash813 1995
[101] J M Keller M R Gray and J A Givens ldquoA fuzzy k-nearestneighbours algorithmrdquo IEEE Transactions on Systems Man andCybernetics vol 15 no 4 pp 580ndash585 1985
[102] X Xiao P Wang and K Chou ldquoiNR-physchem a sequence-based predictor for identifying nuclear receptors and theirsubfamilies via physical-chemical property matrixrdquo PLoS ONEvol 7 no 2 Article ID e30869 2012
[103] I Roterman L Konieczny W Jurkowski K Prymula and MBanach ldquoTwo-intermediate model to characterize the structureof fast-folding proteinsrdquo Journal of Theoretical Biology vol 283no 1 pp 60ndash70 2011
[104] X Xiao P Wang and K C Chou ldquoGPCR-2L predicting Gprotein-coupled receptors and their types by hybridizing twodifferentmodes of pseudo amino acid compositionsrdquoMolecularBioSystems vol 7 no 3 pp 911ndash919 2011
[105] X Xiao P Wang and K C Chou ldquoQuat-2L a web-server forpredicting protein quaternary structural attributesrdquo MolecularDiversity vol 15 no 1 pp 149ndash155 2011
[106] X Zheng C Li and J Wang ldquoAn information-theoreticapproach to the prediction of protein structural classrdquo Journalof Computational Chemistry vol 31 no 6 pp 1201ndash1206 2010
[107] H Shen J Yang X Liu and K C Chou ldquoUsing supervisedfuzzy clustering to predict protein structural classesrdquo Biochem-ical and Biophysical Research Communications vol 334 no 2pp 577ndash581 2005
[108] K-C Chou and C-T Zhang ldquoReview prediction of proteinstructural classesrdquo Critical Reviews in Biochemistry and Molec-ular Biology vol 30 no 4 pp 275ndash349 1995
[109] P C Mahalanobis ldquoOn the generalized distance in statisticsrdquoProceedings of the National Institute of Sciences of India vol 2pp 49ndash55 1936
[110] R M Centor ldquoSignal detectability the use of ROC curves andtheir analysesrdquoMedical Decision Making vol 11 no 2 pp 102ndash106 1991
[111] K-C Chou ldquoUsing subsite coupling to predict signal peptidesrdquoProtein Engineering vol 14 no 2 pp 75ndash79 2001
[112] K C Chou ldquoPrediction of protein signal sequences and theircleavage sitesrdquo Proteins vol 42 pp 136ndash139 2001
[113] K C Chou ldquoPrediction of signal peptides using scaledwindowrdquoPeptides vol 22 no 12 pp 1973ndash1979 2001
[114] K C Chou ldquoSome remarks on predicting multi-label attributesinmolecular biosystemsrdquoMolecular Biosystems vol 9 pp 1092ndash1100 2013
[115] K C Chou and H B Shen ldquoCell-PLoc 2 0 an improvedpackage of web-servers for predicting subcellular localization ofproteins in various organismsrdquoNatural Science vol 2 pp 1090ndash1103 2010
[116] M Gribskov and N L Robinson ldquoUse of receiver operatingcharacteristic (ROC) analysis to evaluate sequence matchingrdquoComputers and Chemistry vol 20 no 1 pp 25ndash33 1996
[117] Y Yamanishi M Kotera M Kanehisa and S Goto ldquoDrug-target interaction prediction from chemical genomic and phar-macological data in an integrated frameworkrdquo Bioinformaticsvol 26 no 12 pp i246ndashi254 2010
[118] Z He J Zhang X Shi et al ldquoPredicting drug-target interactionnetworks based on functional groups and biological featuresrdquoPLoS ONE vol 5 no 3 Article ID e9603 2010
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

2 BioMed Research International
(a) (b)
Figure 1 A schematic drawing to illustrate how to use Choursquos distorted key theory to develop peptide drugs against HIVAIDS (a) shows agood fitting and binding of a peptide to the active site of HIV protease right before it is cleaved by the enzyme (b) shows that the peptide hasbecome a noncleavable one after its scissile bond is modified although it can still tightly bind to the active site Such a modified peptide orldquodistorted keyrdquo will automatically become an inhibitor candidate against HIV protease
emergence of molecular medicine have provided excellentopportunity to discover unknown target enzymes for drugsMany efforts were made in this regard by computationallyanalyzing drug-enzyme interactions The most commonlyused approaches are docking simulations (see eg [16ndash19])and protein cleavage site analysis (see eg [8 12 13]) basedon Choursquos distorted key theory [14] However the latterapproach is mainly used to find peptide drugs Comparedwith the smaller organic compounds although peptide drugshave the advantage of low toxicity to human body theyhave the shortcoming of poor metabolic stability and lowbioavailability due to their inability to readily crossing thrumembrane barriers such as the intestinal and blood-brainbarriers [20] In contrast the molecular docking is indeed auseful vehicle for investigating the interaction of an enzymereceptor with its organic inhibitor and revealing their bindingmechanism as demonstrated by a series of studies [11 19ndash23] However to conduct molecular docking a necessaryprerequisite is the availability of the 3D (three dimensional)structure of the targeted enzyme Unfortunately the 3Dstructures of many enzymes are still unknown Although X-ray crystallography is a powerful tool in determining the 3Dstructures of enzymes it is time-consuming and expensiveParticularly not all enzymes can be successfully crystallizedFor example membrane enzymes are very difficult to crys-tallize and most of them will not dissolve in normal solventsTherefore so far very few membrane enzyme 3D structureshave been determined Although NMR is indeed a verypowerful tool in determining the 3D structures of membraneproteins as indicated by a series of recent publications (seeeg [24ndash30]) it is time-consuming and costly To acquire thestructural information in a timelymanner one has to resort tovarious structural bioinformatics tools (see eg [18 31 32])Unfortunately the number of templates for developing highquality 3D structures by structural bioinformatics is verylimited
Therefore it would save us a lot of time and moneyif we could identify the interactions between enzymes anddrugs before carrying out any intense study in this regardIn view of this the present study was initiated in an attemptto develop a computational method based on the sequence-derived features that can be used to predict the drug-enzymeinteractions in cellular networking
As summarized in a comprehensive review [33] anddemonstrated by a series of recent publications [34ndash37] tosuccessfully develop the desiredmethod we need to considerthe following procedures (i) construct or select a validbenchmark dataset to train and test the predictor (ii) denotethe drug-enzyme samples with an effective formulation thatcan truly reflect their intrinsic relation with the target to bepredicted (iii) introduce or develop a powerful algorithm(or engine) to operate the prediction (iv) conduct a rigorouscross-validation to objectively evaluate its anticipated accu-racy (v) establish a user-friendly web-server for the predictorthat is freely accessible to the public Next let us elaboratehow to deal with these procedures one by one
2 Materials and Methods
21 Benchmark Dataset The data used in this study werecollected from Kyoto Encyclopedia of Genes and Genomes(KEGG) [38] at httpwwwkeggjpkegg which is a data-base resource for understanding high-level functions andutilities of the biological system such as the cell theorganism and the ecosystem from molecular-level infor-mation especially large-scale molecular datasets generatedby genome sequencing and other high-throughput exper-imental technologies For the current study the benchmarkdataset S can be formulated as
S = S+
cup Sminus
(1)
where S+ is the positive subset that consists of the interactiveenzyme-drug pairs only while Sminus is the negative subsetthat contains of the noninteractive enzyme-drug pairs onlyand the symbol cup represents the union in the set theoryHere the ldquointeractiverdquo pair means the pair whose twocounterparts are interacted with each other in the drug-target networks as defined in the KEGG database [38]while the ldquononinteractiverdquo pair means that its two counterparts are not interacted with each other in the drug-targetnetworks The positive dataset S+ contains 2719 enzyme-drug pairs derived from Yamanishi et al [39] The negativedataset Sminus contains 5438 noninteractive enzyme-drug pairswhich were derived according to the following procedures(i) separating each of the pairs in S+ into single drug and
BioMed Research International 3
enzyme (ii) recoupling each of the single drugs with eachof the single enzymes into pairs in a way that none of themoccurred inS+ (iii) randomly picking the pairs thus formeduntil they reached the number two times as many as thepairs in S+ The 2719 interactive enzyme-drug pairs and5438 noninteractive enzyme-drug pairs are given in OnlineSupporting Information S1 (see Supplementary Materialavailable online at httpdxdoiorg1011552013701317) Allthe detailed information for the compounds or drugs listedthere can be found in the KEGG database via their codes
22 Sample Representation Since each of the samples in thecurrent network system contains an enzyme (protein) anda drug a combination of the following two approaches wasadopted to represent the enzyme-drug pair samples
221 Drug
(a) 2D Molecular Fingerprints Although the number ofdrugs is extremely large most of them are small organicmolecules and are composed of some fixed small structures[40] The identification of small molecules or structures canbe used to detect the drug-target interactions [41] Molec-ular fingerprints are bit-string representations of molecularstructure and properties [42] It should be pointed outthat there are many types of structural representations thathave been suggested for the description of drug moleculesincluding physicochemical properties [43] chemical graphs[44] topological indices [45] 3D pharmacophore patternsandmolecular fields In the current study let us use the simpleand generally adopted 2Dmolecular fingerprints to representdrug molecules as described below
First for each of the drugs concerned we can obtaina MOL file from the KEGG database [38] via its codethat contains the detailed information of chemical structureSecond we can convert the MOL file format into its 2Dmolecular fingerprint file format by using a chemical toolboxsoftware called OpenBabel [46] which can be downloadedfrom the website at httpopenbabelorg The current ver-sion of OpenBabel can generate four types of fingerprintsFP2 FP3 FP4 and MACCS In the current study we usedthe FP2 fingerprint format It is a path-based fingerprint thatidentifies small molecule fragments based on all linear andring substructures and maps them onto a bit-string using ahash function (somewhat similar to the daylight fingerprints[47 48]) It is a length of 256-bit hexadecimal string obtainedfrom theOpenBabel andwe can convert it to a 256-bit vectorThen a molecular fingerprint can be formulated as a 256-Dvector given by
MF = [1198601 sdot sdot sdot 119860119895 sdot sdot sdot 119860256]T (2)
where 119860119895(119895 = 1 2 256) is an integer between 0 and 15
and T is the matrix transpose operatorIn order to capture as much useful information from
a molecular fingerprint as possible we can also convertthe above 256-bit hexadecimal string into a 1024-bit binaryvector which is a digital sequence only including 0 and 1
and consider two different digital signal characteristics for thedigital sequence as follows
(b) Information Entropy Shannon proposed that any infor-mation is redundant and redundant size is related withthe occurrence probability or uncertainty of each symbolsuch as numbers letters or words among the informationThe information entropy for a system with a probabilitydistribution 119875(119909
119894) for two classes information entropy [49] is
defined as
119867119909= minussum
119909
119875 (119909119894) log2119875 (119909119894) (119894 = 0 1) (3)
where 119875(119909119894) represents the occurrence probability of num-
ber 119894 in the aforementioned 1024-bit binary vector andthe information entropy 119867
119909is a measure value of the
information amount For example for the digital sequence100100011010010 the value of the information entropy 119867
119909
thus obtained is
119875 (1199090) =
9
15
= 06
119875 (1199091) =
6
15
= 04
119867119909= minus (06 times log
206 + 04 times log
204) = 0971
(4)
(c) Complexity Factor The Lempel-Ziv (LZ) complexity [50]reflects the order that is retained in the sequence and hencewas adopted in this study For each step only two operationswere allowed in the process to get the LZ complexity eithercopying the longest section from the part of a nonemptysequence or generating an additional symbol mark thatensures the uniqueness of per component 119878(119894
119896minus1rarr 119894119896) Its
substring is expressed by
119878 (119894 997888rarr 119895) = 119898119894119898119894+1119898119894+2sdot sdot sdot 119898119895
(1 le 119894 le 119895 le 119871) (5)
where 1198981represents the 1st digital value 119898
2the 2nd value
and so forth A nonempty digital sequence is synthesizedaccording to the following formula
Syn (119878) = 119878 (1 997888rarr 1198941) ∙ 119878 (119894
1+ 1 997888rarr 119894
2)
∙ sdot sdot sdot ∙ 119878 (119894119898minus1
+ 1 997888rarr 119894119871)
(6)
Suppose that 119878 = 11989811198982119898311989841198985sdot sdot sdot 119898119871has been recon-
structed by the subsymbol 119898119903which is viewed as the newly
inserted symbol The substring up to 119898119903will be denoted by
119878(1 rarr 119903)∙ where the bold dot ∙ indicates that 119898119903is a newly
inserted symbol for checkingwhether the rest of the substring119878(119903+1 rarr 119871) can be reconstructed by a simple process At firstsuppose 119878(119902) = 119898
119903+ 1 and see whether 119878(119902) is the substring
for the subsequence 119878(1 rarr 119903) which means deleting thelast symbol from the substring 119878(1 rarr 119903)119878(119902) If the answeris ldquonordquo we insert 119878(119902) into the sequence followed by a dot ∙Thus it could not be obtained by the same operation If theanswer is ldquoyesrdquo no new symbol is needed and we can go on toproceed with 119878(119902) = 119898
119903+1119898119903+2
and repeat the same previousprocedure The LZ complexity is the number of dots (plus
4 BioMed Research International
one if the string is not terminated by a dot) For example forthe sequence 100100011010010 syn(119875) and the correspondingcomplexity factor CF are described as
Syn (119878) = 1 ∙ 0 ∙ 01 ∙ 000 ∙ 11 ∙ 01001 ∙ 0
CF = 7(7)
Thus by adding the information entropy 119867119909(4) and com-
plexity factor CF (7) into the molecular fingerprint MF (2)we obtained a total of (256 + 1 + 1) = 258 feature elements torepresent a drug compound that is it can now be formulatedas a 258-D vector given by
119863 = [11986011198602sdot sdot sdot 119860
256119867119909
CF]T (8)
where 119860119894has the same meaning as in (2) while 119867
119909and CF
are the information entropy and complexity factor respec-tively as described in the previous two sections
222 Enzyme The sequences of the enzymes involved inthis study are given in Online Supporting Information S2Now the problem is how to effectively represent these enzymesequences for the current study Generally speaking thereare two kinds of approaches to formulate enzyme sequencesthe sequential model and the nonsequential or discretemodel [51] The most typical sequential representation foran enzyme sample E with 119871 residues is its entire amino acidsequence that is
E = 1198771119877211987731198774119877511987761198777sdot sdot sdot 119877119871 (9)
where 1198771represents the 1st residue 119877
2the 2nd residue and
so forth An enzyme sample thus formulated can contain itsmost complete information This is an obvious advantage ofthe sequential representation To get the desired results thesequence-similarity-search-based tools such as BLAST [5253] are usually utilized to conduct the prediction Howeverthis kind of approach failed to work when the query enzymedid not have significant homology to enzyme of known char-acters Thus various nonsequential representation modelswere proposed The simplest nonsequential model for anenzyme was based on its amino acid composition (AAC) asdefined by
E = [11989111198912sdot sdot sdot 11989120]
T (10)
where 119891119906(119906 = 1 2 20) are the normalized occurrence
frequencies of the 20 native amino acids [54ndash56] in theenzyme E and T has the same meaning as in (2) and (8)The AAC-discrete model was widely used for identifyingvarious attributes of proteins (see eg [57ndash61]) Howeveras can be seen from (10) all the sequence order effectswere lost by using the AAC-discrete model This is its mainshortcoming To avoid completely losing the sequence-orderinformation the pseudo amino acid composition [62 63]or Choursquos PseAAC [3] was proposed to replace the simpleAAC model Since the concept of PseAAC was proposedin 2001 [62] it has penetrated into almost all the fields of
protein attribute predictions and computational proteomicssuch as predicting supersecondary structure [64] predictingmetalloproteinase family [65] predicting membrane pro-tein types [66 67] predicting protein structural class [68]discriminating outer membrane proteins [69] identifyingantibacterial peptides [70] identifying allergenic proteins[71] identifying bacterial virulent proteins [72] predictingprotein subcellular location [73 74] identifying GPCRs andtheir types [75] identifying protein quaternary structuralattributes [76] predicting protein submitochondria locations[77] identifying risk type of human papillomaviruses [78]identifying cyclin proteins [79] predicting GABA(A) recep-tor proteins [80] and predicting cysteine S-nitrosylation sitesin proteins [81] among many others (see a long list of paperscited in the References section of [33]) Recently the conceptof PseAAC was further extended to represent the featurevectors of DNA and nucleotides [36 82] as well as otherbiological samples (see eg [83 84]) Because it has beenwidely and increasingly used recently two powerful soft-wares called ldquoPseAAC-Builderrdquo [85] and ldquopropyrdquo [86] wereestablished for generating various special Choursquos pseudo-amino acid compositions in addition to the web-serverPseAAC [87] built in 2008 According to a recent review [33]the general formof Choursquos PseAAC for an enzyme sample canbe formulated by
E = [12059511205952sdot sdot sdot 120595119906sdot sdot sdot 120595Ω]
T (11)
where the subscript Ω is an integer and its value as wellas the components 120595
119906(119906 = 1 2 Ω) will depend on
how to extract the desired information from the amino acidsequence of E (cf (10)) Next let us describe how to extractuseful information from the benchmark datasetS andOnlineSupporting Information S2 to define the enzyme samplesconcerned via (11)
To incorporate as much useful information as possiblefrom an enzyme sample we are to approach this problemfrom three different angles followed by incorporating thefeature elements thus obtained into the general form ofPseAAC of (11)(a) Amino Acid Composition The components of amino acidcomposition have beenwidely used to predict various proteinattributes [57ndash61] In this study they were also included as thefirst 20 elements in the general Choursquos PseAAC (cf (11)) thatis
120595119906= 119891119906
(119906 = 1 2 20) (12)
where 119891119906has the same meaning as in (10)
(b) Dipeptide Composition Dipeptide composition has beenused to predict the protein secondary structural contents[88 89] as well as various protein attributes (see eg [90ndash93]) The number of different dipeptides is 20 times 20 = 400Suppose that the normalized occurrence frequencies of the400 dipeptides in an enzyme sample are given by
119891
(2)
119906(119906 = 1 2 400) (13)
BioMed Research International 5
Incorporating the above 400 dipeptide components into (11)we have
120595119906+20
= 119891
(2)
119906(119906 = 1 2 400) (14)
(c) Sequential Evolution Information Biology is a naturalscience with a historic dimension All biological specieshave developed starting out from a very limited number ofancestral species Their evolution involves changes of singleresidues insertions and deletions of several residues [94]gene doubling and gene fusion With these changes accu-mulated for a long period of time many similarities betweeninitial and resultant amino acid sequences are graduallyeliminated but the corresponding proteins may still sharemany common attributes [18] such as having basically thesame biological function and residing at a same subcellularlocation To extract the sequential evolution information anduse it to define the components of (11) the PSSM (PositionSpecific Scoring Matrix) was used as described next
According to Schaffer et al [95] the sequence evolutioninformation of enzyme E with 119871 amino acid residues can beexpressed by an 119871 times 20matrix as given by
P(0)PSSM =
[
[
[
[
[
[
[
119864
0
1rarr1119864
0
1rarr2sdot sdot sdot 119864
0
1rarr20
119864
0
2rarr1119864
0
2rarr2sdot sdot sdot 119864
0
2rarr20
119864
0
119871rarr1119864
0
119871rarr2sdot sdot sdot 119864
0
119871rarr20
]
]
]
]
]
]
]
(15)
where 1198640119894rarr 119895
represents the original score of the 119894th aminoacid residue (119894 = 1 2 119871) in the enzyme sequence changedto amino acid type 119895 (119895 = 1 2 20) in the process ofevolution Here the numerical codes 1 2 20 are used torepresent the 20 native amino acid types denoted by A CD E F G H I K L M N P Q R S T V W and Y The119871 times 20 scores in (15) were generated by using PSI-BLAST[96] to search the UniProtKBSwiss-Prot database (Release2013-05) through three iterations with 0001 as the 119864 valuecutoff for multiple sequence alignment against the sequenceof the enzyme E In order to make every element in (15) bescaled from their original score ranges into region of [0 1]we performed a conversion through the standard sigmoidfunction to make it become
P(1)PSSM =
[
[
[
[
[
[
[
119864
1
1rarr1119864
1
1rarr2sdot sdot sdot 119864
1
1rarr20
119864
1
2rarr1119864
1
2rarr2sdot sdot sdot 119864
1
2rarr20
119864
1
119871rarr1119864
1
119871rarr2sdot sdot sdot 119864
1
119871rarr20
]
]
]
]
]
]
]
(16)
where
119864
1
119894rarr 119895=
1
1 + 119890
minus1198640
119894rarr 119895
(1 le 119894 le 119871 1 le 119895 le 20) (17)
Nowwe extract the useful information from (16) to define thecomponents of (11) via the following approach
120595119906+420
= ℓ119906
(119906 = 1 2 20) (18)
where
ℓ119895=
1
119871
times
119871
sum
119896=1
119864
1
119896rarr119895(119895 = 1 2 20) (19)
(d) Grey System Model Approach The grey system theory[97] is quite useful in dealing with complicated systems thatlack sufficient information or need to process uncertaininformation According to the grey system theory we canextract the following information from the 119895th column of(16) that is
[
119886
119895
1
119886
119895
2
] = (BT119895B119895)
minus1
BT119895U119895
(119895 = 1 2 20) (20)
where
B119895=
[
[
[
[
[
[
[
[
[
[
minus119864
1
2rarr119895minus119864
1
1rarr119895minus 05119864
1
2rarr1198951
minus119864
1
3rarr119895minus
2
sum
119894=1
119864
1
119894rarr 119895minus 05119864
1
3rarr1198951
minus119864
1
119871rarr119895minus
119871minus1
sum
119894=1
119864
1
119894rarr 119895minus 05119864
1
119871rarr1198951
]
]
]
]
]
]
]
]
]
]
U119895=
[
[
[
[
[
[
119864
1
2rarr119895minus 119864
1
1rarr119895
119864
1
3rarr119895minus 119864
1
2rarr119895
119864
1
119871rarr119895minus 119864
1
119871minus1rarr119895
]
]
]
]
]
]
(21)
Therefore based on the grey system theory and (20) we canextract another 20 times 2 = 40 quantities from (16) to definethe components of (11) that is
120593119895=
1199081119886
119895
1when 119895 is an odd number
1199082119886
119895
2when 119895 is an even number
1 le 119895 le 20
(22)
where 1198861198951and 119886119895
2are given by (20) 119908
1and 119908
2are weight
factors which were all set to 1 in the current studySubstituting the elements in (12) (14) (18) and (22) we
finally obtain a total of Ω = 20 + 400 + 20 + 40 = 480
components for the PseAAC of (11) where
120595119906=
119891119906
when 1 le 119906 le 20119891
(2)
119906when 21 le 119906 le 420
ℓ119906
when 421 le 119906 le 440120593119906
when 440 le 119906 le 480
(23)
In other words in this study (11) or Choursquos PseAAC is a 480-D vector whose 480 components are given by (23) derivedfrom the amino acid composition dipeptide compositionsequential evolution information and grey system theory(e) Representing Enzyme-Drug Pairs Now the pair betweenan enzyme molecule E and a drug compound D can beformulated by combing (8) and (11) as given by
G = D oplus E
= [1198601sdot sdot sdot 119860
256119867119909
CF 1205951sdot sdot sdot 120595480]
T
(24)
6 BioMed Research International
where G represents the enzyme-drug pair oplus the orthogonalsum [51] and each of the (258 + 480) = 738 feature elementsis given in (8) and (23)
For the convenience of the later formulation let us use119909119894(119894 = 1 2 738) to represent the 738 components of (24)
that is
G = [11990911199092sdot sdot sdot 119909119894sdot sdot sdot 119909738]
T (25)
To optimize the prediction results different weights wereusually tested for each of the elements in (25) However sinceit would consume a lot of computational time for a total of 738weight factors here let us adopt the normalization approachto deal with this problem as done in [98 99] that is convert119909119894in (25) to 119910
119894according to the following equation
119910119894=
2tanminus1 (119909119894)
120587
(119894 = 1 2 738) (26)
where tanminus1 means arctangent By means of (26) everycomponent in (25) will be converted into the range of[minus1 1] that is we have minus1 le 119910
119894le 1 As demonstrated
in [98 99] the normalization approach via (26) was quiteeffective in enhancing the quality of prediction operated ina high dimension space Therefore in this study we wouldnot to take the procedure of optimizing the weight factorssignificantly reducing the computational times
23 Fuzzy 119870-Nearest Neighbour Algorithm The 119870-NN (119870-Nearest Neighbor) classifier is quite popular in patternrecognition community owing to its good performance andsimple-to-use feature According to the 119870-NN rule [100]named also as the ldquovoting 119870-NN rulerdquo the query sampleshould be assigned to the subset represented by a majorityof its 119870 nearest neighbors as illustrated in Figure 5 of [33]
Fuzzy 119870-NN classification method [101] is a specialvariation of the119870-NNclassification family Instead of roughlyassigning the label based on a voting from the 119870 nearestneighbors it attempts to estimate the membership valuesthat indicate how much degree the query sample belongsto the classes concerned Obviously it is impossible for anycharacteristic description to contain complete informationwhich would make the classification ambiguous In view ofthis the fuzzy principle is very reasonable and particularlyuseful in dealing with complicated biological systems such asidentifying nuclear receptor subfamilies [102] characterizingthe structure of fast-folding proteins [103] classifying Gprotein-coupled receptors [104] predicting protein quater-nary structural attributes [105] predicting protein structuralclasses [106 107] and so forth
Next let us give a brief introduction how to use thefuzzy119870-NN approach to identify the interactions between theenzymes and the drug compounds in the network concerned
Supposing that S(119873) = G1G2 G
119873 is a set of vec-
tors representing 119873 enzyme-drug pairs in a training set
classified into two classes 119862+ 119862minus where 119862+ denotes theinteractive pair class while 119862minus the noninteractive pair classSlowast(G) = Glowast
1Glowast2 Glowast
119870 sub S(119873) is the subset of the 119870
nearest neighbor pairs to the query pair G Thus the fuzzymembership value for the query pair G in the two classes ofS(119873) is given by
120583
+
(G) =sum
119870
119895=1120583
+(Glowast119895) 119889(GGlowast
119895)
minus2(120593minus1)
sum
119870
119895=1119889(GGlowast
119895)
minus2(120593minus1)
120583
minus
(G) =sum
119870
119895=1120583
minus(Glowast119895) 119889(GGlowast
119895)
minus2(120593minus1)
sum
119870
119895=1119889(GGlowast
119895)
minus2(120593minus1)
(27)
where 119870 is the number of the nearest neighbors counted forthe query pairG 120583+(Glowast
119895) and 120583minus(Glowast
119895) the fuzzy membership
values of the training sample Glowast119895to the class 119862+ and 119862minus
respectively as will be further defined next 119889(GGlowast119895) the
cosine distance between G and its 119895th nearest pair Glowast119895in
the training dataset S(119873) 120593(gt 1) the fuzzy coefficient fordetermining how heavily the distance is weighted whencalculating each nearest neighborrsquos contribution to the mem-bership value Note that the parameters 119870 and 120593 will affectthe computation result of (27) and they will be optimizedby a grid-search as will be described later Also variousother metrics can be chosen for 119889(GGlowast
119895) such as Euclidean
distance Hamming distance [108] andMahalanobis distance[55 109]
The quantitative definitions for the aforementioned120583
+(Glowast119895) and 120583minus(Glowast
119895) in (27) are given by
120583
+
(Glowast119895) =
1 if Glowast119895isin 119862
+
0 otherwise
120583
minus
(Glowast119895) =
1 if Glowast119895isin 119862
minus
0 otherwise
(28)
Substituting the results obtained by (27) into (28) it followsthat if 120583+(G) gt 120583
minus(G) then the query pair G is an
interactive couple otherwise noninteractive In other wordsthe outcome can be formulated as
G isin
119862
+ if 120583+ (G) gt 120583minus (G)
119862
minus otherwise
(29)
If there is a tie between 120583+(G) and 120583minus(G) the query pair Gwill be randomly assigned to one of the two classes Howevercase like that is quite rare and in this study never happened
The predictor thus established is called iEzy-Drugwhere ldquoirdquo means identify and ldquoEzy-Drugrdquo means the inter-action between enzyme and drug To provide an intuitiveoverall picture a flowchart is provided in Figure 2 to showthe process of how the classifier works in identifying enzyme-drug interactions
BioMed Research International 7
Input a queryprotein sequence and
a drug code
Create Chous PseAAC of a protein
Create feature vector of a drug
Feature fusion
engineTraining dataset
Yes No
Noninteractive enzyme drug pair
Interactive enzymedrug pair
AACDipeptideSeq Evol
Grey model
Mol FptInfo entropy
CF
FKNN operation
Figure 2 A flowchart to show the operation process of the iEzy-Drug predictor See the text for further explanation
24 Criteria for Performance Evaluation In the literaturethe following equation set is often used for examining theperformance quality of a predictor
Sn = TPTP + FN
Sp = TNTN + FP
Acc = TP + TNTP + TN + FP + FN
MCC = (TP times TN) minus (FP times FN)radic(TP + FP) (TP + FN) (TN + FP) (TN + FN)
(30)
where TP represents the true positive TN the true negativeFP the false positive FN the false negative Sn the sensitiv-ity Sp the specificity Acc the accuracy MCC the Mathewrsquoscorrelation coefficient
To most biologists however the four metrics as formu-lated in (30) are not quite intuitive and easier-to-understandparticularly for the Mathewrsquos correlation coefficient Herelet us adopt the Choursquos symbols to formulate the previousfour metrics By means of Choursquos symbols [111 112] the rates
of correct predictions for the interactive enzyme-drug pairsin dataset S+ and the noninteractive enzyme-drug pairs indataset Sminus are respectively defined by (cf (1))
Λ
+
=
119873
+minus 119873
+
minus
119873
+ for the interactive enzyme-drug pairs
Λ
minus
=
119873
minusminus 119873
minus
+
119873
minus for the noninteractive enzyme-drug pairs
(31)
where 119873+ is the total number of the interactive enzyme-drug pairs investigated while 119873+
minusis the number of the
interactive enzyme-drug pairs incorrectly predicted as thenoninteractive enzyme-drug pairs 119873minus is the total numberof the noninteractive enzyme-drug pairs investigated while119873
minus
+is the number of the noninteractive enzyme-drug pairs
incorrectly predicted as the interactive enzyme-drug pairsThe overall success prediction rate is given by [113] as follows
Λ =
Λ
+119873
++ Λ
minus119873
minus
119873
++ 119873
minus= 1 minus
119873
+
minus+ 119873
minus
+
119873
++ 119873
minus
(32)
It is obvious from (31)-(32) that if and only if none ofthe interactive enzyme-drug pairs and the noninteractiveenzyme-drug pairs are mispredicted that is 119873+
minus= 119873
minus
+= 0
and Λ+ = Λ
minus= 1 we have the overall success rate Λ = 1
Otherwise the overall success rate would be smaller than 1The relations between the symbols in (32) and those in
(30) are given by
TP = 119873+ minus 119873+minus
TN = 119873
minus
minus 119873
minus
+
FP = 119873minus+
FN = 119873
+
minus
(33)
Substituting (33) into (30) and also noting (31)-(32) we obtain
Sn = Λ+ = 1 minus119873
+
minus
119873
+
Sp = Λminus = 1 minus119873
minus
+
119873
minus
Acc = Λ = 1 minus119873
+
minus+ 119873
minus
+
119873
++ 119873
minus
MCC =1 minus ((119873
+
minus119873
+) + (119873
minus
+119873
minus))
radic(1 + (119873
minus
+minus 119873
+
minus) 119873
+) (1 + (119873
+
minusminus 119873
minus
+) 119873
minus)
(34)
Now it is obvious to see from (34) when 119873
+
minus= 0
meaning none of the interactive enzyme-drug pairs was
8 BioMed Research International
02
46
810
1
15
2089
0895
09
0905
091
0915
092
ACC
K120593
Figure 3 A 3D plot to show how the parameter in (27) wasoptimized for the iEzy-Drug predictor
mispredicted to be a noninteractive enzyme-drug pair wehave the sensitivity Sn = 1 while 119873+
minus= 119873
+ meaning thatall the interactive enzyme-drug pairs weremispredicted to bethe noninteractive enzyme-drug pairs we have the sensitivitySn = 0 Likewise when 119873minus
+= 0 meaning none of the
noninteractive enzyme-drug pairs wasmispredicted we havethe specificity Sp = 1 while 119873minus
+= 119873
minus meaning all thenoninteractive enzyme-drug pairs were incorrectly predictedas interactive enzyme-drug pairs we have the specificity Sp =0 When119873+
minus= 119873
minus
+= 0 meaning that none of the interactive
enzyme-drug pairs in the dataset S+ and none of the nonin-teractive enzyme-drug pairs in Sminus was incorrectly predictedwe have the overall accuracy Acc = Λ = 1 while 119873+
minus= 119873
+
and 119873minus+= 119873
minus meaning that all the interactive enzyme-drugpairs in the dataset S+ and all the noninteractive enzyme-drug pairs in Sminus were mispredicted we have the overallaccuracy Acc = Λ = 0 The MCC correlation coefficient isusually used for measuring the quality of binary (two-class)classifications When 119873+
minus= 119873
minus
+= 0 meaning that none
of the interactive enzyme-drug pairs in the dataset S+ andnone of the noninteractive enzyme-drug pairs in Sminus weremispredicted we have MCC = 1 when 119873+
minus= 119873
+2 and
119873
minus
+= 119873
minus2 we have MCC = 0 meaning no better than
random prediction when 119873+minus= 119873
+ and 119873minus+= 119873
minus we haveMCC = minus1 meaning total disagreement between predictionand observation As we can see from the previous discussionit is much more intuitive and easier-to-understand whenusing (34) to examine a predictor for its sensitivity specificityoverall accuracy and Mathewrsquos correlation coefficient It isinstructive to point out that themetrics as defined in (30) and(34) are valid for single label systems for multilabel systemsa set of more complicated metrics should be used as given in[114]
3 Results and Discussion
31 Cross-Validation How to properly examine the predic-tion quality is a key for developing a new predictor and
False positive rate
True
pos
itive
rate
1
01
02
03
04
05
06
07
08
09
0
101 02 03 04 05 06 07 08 090
BaselineiEzy-Drug AUC = 09377
Figure 4 A plot for the ROC curve to quantitatively show theperformance of the iEzy-Drug predictor
estimating its potential application value Generally speak-ing the following three cross-validation methods are oftenused to examine a predictor of its effectiveness in practicalapplication independent dataset test subsampling or 119870-fold(such as 5-fold 7-fold or 10-fold) test and jackknife test[108] However as elaborated by a penetrating analysis in[115] considerable arbitrariness exists in the independentdataset test Also as demonstrated by (27)ndash(29) in [33]the subsampling test (or 119870-fold cross-validation) cannotavoid arbitrariness either Only the jackknife test is the leastarbitrary that can always yield a unique result for a givenbenchmark dataset Therefore the jackknife test has beenwidely recognized and increasingly utilized by investigatorsto examine the quality of various predictors (see eg [66 7174 80]) Accordingly the success rate by the jackknife test wasalso used to optimize the two uncertain parameters 119870 and 120593in (27) The result thus obtained is shown in Figure 3 fromwhich we obtain when 119870 = 6 and 120593 = 15 the iEzy-Drugpredictor reaches its optimized status
The success rates thus obtained by the jackknife test inidentifying interactive Enzyme-drug pairs or noninteractiveenzyme-drug pairs on the benchmark dataset S (cf OnlineSupporting Information S1) are given in Table 1 where forfacilitating comparison the corresponding result by He et al[110] is also given As we can see from the table the overallaccuracy Acc achieved by iEzy-Drug was 9103 remarkablyhigher than 8548 the corresponding rate obtained byHe et al [110] on the same benchmark Furthermore listedin Table 1 are also the values obtained by iEzy-Drug for theother three metrics that is Sn = 9081 Sp = 9114 andMCC = 8039 indicating that the accuracy of iEzy-Drug isnot only very high but also quite stale
To provide a graphical illustration to show the per-formance of the current binary classifier iEzy-Drug asits discrimination threshold is varied a 2D plot calledReceiver Operating Characteristic (ROC) curve [116 117]was also given (Figure 4) In the ROC curve the verticalcoordinate 119884 is for the true positive rate or Sn (cf (34))while horizontal coordinate 119883 for the false positive rate
BioMed Research International 9
Read Me Data Citation
Enter both the protein sequence and the drug code (example) the number of query pairs is limited at 10 or less for each submission
Submit Clear
You can download the program and run it on your own local computer
iEzy-Drug identifying the interaction between enzymes anddrugs in cellular networking
Figure 5 A semiscreenshot to show the top page of the iEzy-Drugweb-server Its web-site address is at httpwwwjci-bioinfocniEzy-Drug
Table 1 The jackknife success rates obtained with iEzy-Drug in identifying interactive enzyme-drug pairs and noninteractive enzyme-drugpairs for the benchmark dataset S (cf Online Supporting Information S1)
Method Acc Sn Sp MCC
iEzy-Druga 74258157 = 9103 24692719 = 9081 49565438 = 9114 8039NN predictorb 8548 NA NA NAaSee (27) where the parameters119870 = 6 and 120593 = 15bSee [110]
or 1-Sp The best possible prediction method would yielda point with the coordinate (0 1) representing 100 truepositive rate (sensitivity Sn) and 0 false positive rate or100 specificity Therefore the (0 1) point is also calleda perfect classification A completely random guess wouldgive a point along a diagonal from the point (0 0) to (1 1)The area under the ROC curve also called Area Under theROC (AUROC) is often used to indicate the performancequality of a binary classifier the value 05 of AUROCis equivalent to random prediction while 1 of AUROCrepresents a perfect one As we can see from Figure 4the AUROC value obtained by iEzy-Drug is 09377
The reason why iEzy-Drug can remarkably improve theprediction quality is that it has introduced the 2D molecularfingerprints to represent drug samples see Online SupportingInformation S3 for the detailed fingerprint expressions for thedrugs listed in Online Supporting Information S1 and thatit has successfully used PseAAC to incorporate the featuresderived from the sequences of enzymes that are essentialfor identifying the interaction of enzymes with drugs in thecellular networking
To enhance the value of its practical applications theweb server for iEzy-Drug has been established that canbe freely accessible at httpwwwjci-bioinfocniEzy-DrugIt is anticipated that the web server will become a usefulhigh throughput tool for both basic research and drug
development in the relevant areas or at the very least playa complementary role to the existing method [39 110 118] forwhich so far noweb-serverwhatsoever has been provided yet
32 The Protocol or User Guide For the convenience of thevast majority of biologists and pharmaceutical scientists herelet us provide a step-by-step guide to show how the userscan easily get the desired result by means of the web serverwithout the need to follow the complicated mathematicalequations presented in this paper for the process of develop-ing the predictor and its integrityStep 1 Open the web server at the site httpwwwjci-bio-infocniEzy-Drug and you will see the top page of thepredictor on your computer screen as shown in Figure 5Click on the ReadMe button to see a brief introduction aboutiEzy-Drug predictor and the caveat when using itStep 2 Either type or copypaste the query pairs into theinput box at the center of Figure 5 Each query pair consistsof two parts one is for the protein sequence and the other forthe drug The enzyme sequence should be in FASTA formatwhile the drug in the KEGG code Examples for the querypairs input can be seen by clicking on the Example buttonright above the input boxStep 3 Click on the Submit button to see the predicted resultFor example if you use the four query pairs in the Example
10 BioMed Research International
window as the input after clicking the Submit button youwill see on your screen that the ldquohsa 10056rdquo enzyme andthe ldquoD0021rdquo drug are an interactive pair and that the ldquohsa100rdquo enzyme and the ldquoD0037rdquo drug are also an interactivepair but that the ldquohsa 3295rdquo enzyme and the ldquoD00889rdquo drugare not an interactive pair and that the ldquohsa 7366rdquo enzymeand the ldquoD03601rdquo drug are not an interactive pair eitherAll these results are fully consistent with the experimentalobservations It takes about 3 minutes before the results areshown on the screenStep 4 Click on the Citation button to find the relevant paperthat documents the detailed development and algorithm ofiEzy-DurgStep 5 Click on the Data button to download the benchmarkdataset used to train and test the iEzy-Durg predictorStep 6 The program code is also available by clicking thebutton download on the lower panel of Figure 5
Acknowledgments
The authors would like to thank the three anonymousreviewers whose constructive comments are very helpful forstrengthening the presentation of the paper This work wassupported by the Grants from the National Natural ScienceFoundation of China (no 31260273) the Jiangxi ProvincialForeign Scientific and Technological Cooperation Project(no 20120BDH80023) Natural Science Foundation of JiangxiProvince China (no 2010GZS0122 20122BAB201020) theDepartment of Education of Jiangxi Province (GJJ12490)the LuoDi plan of the Department of Education of JiangxiProvince (KJLD12083) and the Jiangxi Provincial Founda-tion for Leaders of Disciplines in Science (20113BCB22008)The funders had no role in study design data collection andanalysis decision to publish or preparation of the paper
References
[1] A Bairoch ldquoThe ENZYME database in 2000rdquo Nucleic AcidsResearch vol 28 no 1 pp 304ndash305 2000
[2] S H Koenig and R D Brown ldquoH2CO3as substrate for carbonic
anhydrase in the dehydration of H2CO3(minus)rdquo Proceedings of the
National Academy of Sciences of the United States of Americavol 69 no 9 pp 2422ndash2425 1972
[3] S X Lin and J Lapointe ldquoTheoretical and experimental biologyin onerdquo Journal of Biomedical Science and Engineering vol 6 pp435ndash442 2013
[4] K C Chou and S P Jiang ldquoStudies on the rate of diffusion-controlled reactions of enzymes Spatial factor and force fieldfactorrdquo Scientia Sinica vol 17 no 5 pp 664ndash680 1974
[5] K C Chou ldquoThe kinetics of the combination reaction betweenenzyme and substraterdquo Scientia Sinica vol 19 no 4 pp 505ndash528 1976
[6] K C Chou and G P Zhou ldquoRole of the protein outside activesite on the diffusion-controlled reaction of enzymerdquo Journal ofthe American Chemical Society vol 104 no 5 pp 1409ndash14131982
[7] R A Poorman A G Tomasselli R L Heinrikson and FJ Kezdy ldquoA cumulative specificity model for proteases from
human immunodeficiency virus types 1 and 2 inferred fromstatistical analysis of an extended substrate data baserdquo TheJournal of Biological Chemistry vol 266 no 22 pp 14554ndash145611991
[8] K-C Chou ldquoA vectorized sequence-coupling model for pre-dicting HIV protease cleavage sites in proteinsrdquo The Journal ofBiological Chemistry vol 268 no 23 pp 16938ndash16948 1993
[9] G Z Liang and S Z Li ldquoA new sequence representation asapplied in better specificity elucidation for human immunod-eficiency virus type 1 proteaserdquo Biopolymers vol 88 no 3 pp401ndash412 2007
[10] J J Chou ldquoPredicting cleavability of peptide sequences byHIVprotease via correlation-angle approachrdquo Journal of ProteinChemistry vol 12 no 3 pp 291ndash302 1993
[11] Q S Du S Wang D Q Wei S Sirois and K C ChouldquoMolecular modeling and chemical modification for findingpeptide inhibitor against severe acute respiratory syndromecoronavirus main proteinaserdquo Analytical Biochemistry vol 337no 2 pp 262ndash270 2005
[12] Y Gan H Huang Y Huang et al ldquoSynthesis and activity ofan octapeptide inhibitor designed for SARS coronavirus mainproteinaserdquo Peptides vol 27 no 4 pp 622ndash625 2006
[13] Q S Du H Sun and K C Chou ldquoInhibitor design for SARScoronavirus main protease based on lsquodistorted key theoryrsquordquoMedicinal Chemistry vol 3 no 1 pp 1ndash6 2007
[14] K C Chou ldquoPrediction of human immunodeficiency virusprotease cleavage sites in proteinsrdquoAnalytical Biochemistry vol233 no 1 pp 1ndash14 1996
[15] J Knowles and G Gromo ldquoTarget selection in drug discoveryrdquoNature Reviews Drug Discovery vol 2 no 1 pp 63ndash69 2003
[16] A C Cheng R G Coleman K T Smyth et al ldquoStructure-basedmaximal affinity model predicts small-molecule druggabilityrdquoNature Biotechnology vol 25 no 1 pp 71ndash75 2007
[17] M Rarey B Kramer T Lengauer and G Klebe ldquoA fast flexibledocking method using an incremental construction algorithmrdquoJournal of Molecular Biology vol 261 no 3 pp 470ndash489 1996
[18] K C Chou ldquoReview structural bioinformatics and its impactto biomedical sciencerdquo Current Medicinal Chemistry vol 11 no16 pp 2105ndash2134 2004
[19] K C Chou D Q Wei and W Z Zhong ldquoBinding mechanismof coronavirus main proteinase with ligands and its implicationto drug design against SARSrdquo Biochemical and BiophysicalResearch Communications vol 308 pp 148ndash151 2003 (Erratumin Biochemical and Biophysical Research Communications vol310 p 675 2003)
[20] K C Chou D Q Wei Q S Du S Sirois and W Z ZhongldquoProgress in computational approach to drug developmentagaint SARSrdquo Current Medicinal Chemistry vol 13 no 27 pp3263ndash3270 2006
[21] G P Zhou and F A Troy ldquoNMR studies on how the bindingcomplex of polyisoprenol recognition sequence peptides andpolyisoprenols can modulate membrane structurerdquo CurrentProtein and Peptide Science vol 6 no 5 pp 399ndash411 2005
[22] R B Huang Q S Du C H Wang and K C Chou ldquoAn in-depth analysis of the biological functional studies based on theNMR M2 channel structure of influenza A virusrdquo Biochemicaland Biophysical Research Communications vol 377 no 4 pp1243ndash1247 2008
[23] Q S Du R B Huang C H Wang X M Li and K C ChouldquoEnergetic analysis of the two controversial drug binding sitesof the M2 proton channel in influenza A virusrdquo Journal ofTheoretical Biology vol 259 no 1 pp 159ndash164 2009
BioMed Research International 11
[24] M J Berardi W M Shih S C Harrison and J J Chou ldquoMito-chondrial uncoupling protein 2 structure determined by NMRmolecular fragment searchingrdquo Nature vol 476 no 7358 pp109ndash114 2011
[25] J R Schnell and J J Chou ldquoStructure andmechanism of theM2proton channel of influenza A virusrdquo Nature vol 451 no 7178pp 591ndash595 2008
[26] B OuYang S Xie M J Berardi et al ldquoUnusual architecture ofthe p7 channel from hepatitis C virusrdquoNature vol 498 pp 521ndash525 2013
[27] K Oxenoid and J J Chou ldquoThe structure of phospholambanpentamer reveals a channel-like architecture in membranesrdquoProceedings of the National Academy of Sciences of the UnitedStates of America vol 102 no 31 pp 10870ndash10875 2005
[28] M E Call KWWucherpfennig and J J Chou ldquoThe structuralbasis for intramembrane assembly of an activating immunore-ceptor complexrdquo Nature Immunology vol 11 no 11 pp 1023ndash1029 2010
[29] R M Pielak J R Schnell and J J Chou ldquoMechanism of druginhibition and drug resistance of influenza A M2 channelrdquoProceedings of the National Academy of Sciences of the UnitedStates of America vol 106 no 18 pp 7379ndash7384 2009
[30] J Wang R M Pielak M A McClintock and J J ChouldquoSolution structure and functional analysis of the influenza Bproton channelrdquo Nature Structural and Molecular Biology vol16 no 12 pp 1267ndash1271 2009
[31] K C Chou ldquoCoupling interaction between thromboxane A2receptor and alpha-13 subunit of guanine nucleotide-bindingproteinrdquo Journal of Proteome Research vol 4 no 5 pp 1681ndash1686 2005
[32] K C Chou ldquoInsights from modeling three-dimensional struc-tures of the human potassium and sodium channelsrdquo Journal ofProteome Research vol 3 no 4 pp 856ndash861 2004
[33] K C Chou ldquoSome remarks on protein attribute prediction andpseudo amino acid compositionrdquo Journal ofTheoretical Biologyvol 273 no 1 pp 236ndash247 2011
[34] W Z Lin J A Fang X Xiao and K C Chou ldquoiLoc-animal amulti-label learning classifier for predicting subcellular local-ization of animal proteinsrdquo Molecular BioSystems vol 9 pp634ndash644 2013
[35] X Xiao PWangW Z Lin J H Jia and K C Chou ldquoiAMP-2La two-level multi-label classifier for identifying antimicrobialpeptides and their functional typesrdquo Analytical Biochemistryvol 436 pp 168ndash177 2013
[36] W Chen P M Feng H Lin and K C Chou ldquoiRSpot-PseDNCidentify recombination spots with pseudo dinucleotide compo-sitionrdquo Nucleic Acids Research vol 41 article e69 2013
[37] K C Chou Z C Wu and X Xiao ldquoILoc-Hum using theaccumulation-label scale to predict subcellular locations ofhuman proteins with both single and multiple sitesrdquoMolecularBioSystems vol 8 no 2 pp 629ndash641 2012
[38] M Kotera M Hirakawa T Tokimatsu S Goto and MKanehisa ldquoThe KEGG databases and tools facilitating omicsanalysis latest developments involving human diseases andpharmaceuticalsrdquo Methods in Molecular Biology vol 802 pp19ndash39 2012
[39] Y YamanishiM Araki A GutteridgeWHonda andM Kane-hisa ldquoPrediction of drug-target interaction networks from theintegration of chemical and genomic spacesrdquo Bioinformaticsvol 24 no 13 pp i232ndashi240 2008
[40] P Finn S Muggleton D Page and A Srinivasan ldquoPharma-cophore discovery using the Inductive Logic Programmingsystem PROGOLrdquo Machine Learning vol 30 no 2-3 pp 241ndash270 1998
[41] I Vogt D Stumpfe H E Ahmed and J Bajorath ldquoMethodsfor computer-aided chemical biology Part 2 evaluation of com-pound selectivity using 2D molecular fingerprintsrdquo ChemicalBiology and Drug Design vol 70 pp 195ndash205 2007
[42] H Eckert and J Bajorath ldquoMolecular similarity analysis in vir-tual screening foundations limitations and novel approachesrdquoDrug Discovery Today vol 12 no 5-6 pp 225ndash233 2007
[43] S Laurent L V Elst and R N Muller ldquoComparative studyof the physicochemical properties of six clinical low molecularweight gadolinium contrast agentsrdquo Contrast Media amp Molecu-lar Imaging vol 1 no 3 pp 128ndash137 2006
[44] E Gregori-Puigjane R Garriga-Sust and J Mestres ldquoIndexingmolecules with chemical graph identifiersrdquo Journal of Compu-tational Chemistry vol 32 no 12 pp 2638ndash2646 2011
[45] B Ren ldquoApplication of novel atom-type AI topological indicesto QSPR studies of alkanesrdquo Computers and Chemistry vol 26no 4 pp 357ndash369 2002
[46] N M OrsquoBoyle M Banck C A James C Morley T Vander-meersch and G R Hutchison ldquoOpen Babel an open chemicaltoolboxrdquo Journal of Cheminformatics vol 3 p 33 2011
[47] V J Gillet P Willett and J Bradshaw ldquoSimilarity searchingusing reduced graphsrdquo Journal of Chemical Information andComputer Sciences vol 43 no 2 pp 338ndash345 2003
[48] D Butina ldquoUnsupervised data base clustering based on day-lightrsquos fingerprint and Tanimoto similarity a fast and automatedway to cluster small and large data setsrdquo Journal of ChemicalInformation and Computer Sciences vol 39 no 4 pp 747ndash7501999
[49] C E Shannon ldquoA mathematical theory of communicationrdquoACM SIGMOBILE Mobile Computing and CommunicationsReview vol 5 pp 3ndash55 2001
[50] V D Gusev L A Nemytikova and N A Chuzhanova ldquoOn thecomplexity measures of genetic sequencesrdquo Bioinformatics vol15 no 12 pp 994ndash999 1999
[51] K C Chou and H B Shen ldquoReview recent progress in proteinsubcellular location predictionrdquo Analytical Biochemistry vol370 no 1 pp 1ndash16 2007
[52] S F Altschul ldquoEvaluating the statistical significance of multipledistinct local alignmentsrdquo in Theoretical and ComputationalMethods in Genome Research S Suhai Ed pp 1ndash14 PlenumNew York NY USA 1997
[53] J C Wootton and S Federhen ldquoStatistics of local complexity inamino acid sequences and sequence databasesrdquo Computers andChemistry vol 17 no 2 pp 149ndash163 1993
[54] H Nakashima K Nishikawa and T Ooi ldquoThe folding type ofa protein is relevant to the amino acid compositionrdquo Journal ofBiochemistry vol 99 no 1 pp 153ndash162 1986
[55] K C Chou and C T Zhang ldquoPredicting protein folding typesby distance functions that make allowances for amino acidinteractionsrdquo The Journal of Biological Chemistry vol 269 no35 pp 22014ndash22020 1994
[56] K-C Chou ldquoA novel approach to predicting protein structuralclasses in a (20-1)-D amino acid composition spacerdquo Proteinsvol 21 no 4 pp 319ndash344 1995
[57] H Nakashima and K Nishikawa ldquoDiscrimination of intracel-lular and extracellular proteins using amino acid compositionand residue-pair frequenciesrdquo Journal of Molecular Biology vol238 no 1 pp 54ndash61 1994
12 BioMed Research International
[58] G P Zhou ldquoAn intriguing controversy over protein structuralclass predictionrdquo Journal of Protein Chemistry vol 17 no 8 pp729ndash738 1998
[59] I Bahar A R Atilgan R L Jernigan and B Erman ldquoUnder-standing the recognition of protein structural classes by aminoacid compositionrdquo Proteins vol 29 pp 172ndash185 1997
[60] J Cedano P Aloy J A Perez-Pons and E Querol ldquoRelationbetween amino acid composition and cellular location ofproteinsrdquo Journal of Molecular Biology vol 266 no 3 pp 594ndash600 1997
[61] G P Zhou and K Doctor ldquoSubcellular location prediction ofapoptosis proteinsrdquo Proteins vol 50 no 1 pp 44ndash48 2003
[62] KChou ldquoPrediction of protein cellular attributes using pseudo-amino acid compositionrdquo Proteins vol 43 pp 246ndash255 2001(Erratum in Proteins vol 44 p 60 2001)
[63] K C Chou ldquoUsing amphiphilic pseudo amino acid composi-tion to predict enzyme subfamily classesrdquo Bioinformatics vol21 no 1 pp 10ndash19 2005
[64] D Zou Z He J He and Y Xia ldquoSupersecondary structureprediction using Choursquos pseudo amino acid compositionrdquoJournal of Computational Chemistry vol 32 no 2 pp 271ndash2782011
[65] M M Beigi M Behjati and H Mohabatkar ldquoPrediction ofmetalloproteinase family based on the concept ofChoursquos pseudoamino acid composition using a machine learning approachrdquoJournal of Structural and Functional Genomics vol 12 no 4 pp191ndash197 2011
[66] Y K Chen and K B Li ldquoPredicting membrane protein typesby incorporating protein topology domains signal peptidesand physicochemical properties into the general form of Choursquospseudo amino acid compositionrdquo Journal ofTheoretical Biologyvol 318 pp 1ndash12 2013
[67] C Huang and J Q Yuan ldquoA multilabel model based on Choursquospseudo-amino acid composition for identifying membraneproteins with both single and multiple functional typesrdquo TheJournal of Membrane Biology vol 246 pp 327ndash334 2013
[68] S S Sahu and G Panda ldquoA novel feature representationmethod based on Choursquos pseudo amino acid composition forprotein structural class predictionrdquo Computational Biology andChemistry vol 34 no 5-6 pp 320ndash327 2010
[69] M Hayat and A Khan ldquoDiscriminating outer membraneproteins with fuzzy K-nearest neighbor algorithms based on thegeneral form of Choursquos PseAACrdquo Protein and Peptide Lettersvol 19 no 4 pp 411ndash421 2012
[70] MKhosravian F K FaramarziMM BeigiM Behbahani andH Mohabatkar ldquoPredicting antibacterial peptides by the con-cept of Choursquos pseudo-amino acid composition and machinelearning methodsrdquo Protein amp Peptide Letters vol 20 pp 180ndash186 2013
[71] HMohabatkarMM Beigi K Abdolahi and SMohsenzadehldquoPrediction of allergenic proteins by means of the concept ofChoursquos pseudo amino acid composition and amachine learningapproachrdquoMedicinal Chemistry vol 9 pp 133ndash137 2013
[72] L Nanni A Lumini D Gupta and A Garg ldquoIdentifyingbacterial virulent proteins by fusing a set of classifiers basedon variants of Choursquos pseudo amino acid composition and onevolutionary informationrdquo IEEEACM Transactions on Compu-tational Biology and Bioinformatics vol 9 no 2 pp 467ndash4752012
[73] T H Chang L C Wu T Y Lee S P Chen H D Huang andJ T Horng ldquoEuLoc a web-server for accurately predict protein
subcellular localization in eukaryotes by incorporating variousfeatures of sequence segments into the general form of ChoursquosPseAACrdquo Journal of Computer-Aided Molecular Design vol 27pp 91ndash103 2013
[74] S Zhang Y Zhang H Yang C Zhao and Q Pan ldquoUsing theconcept of Choursquos pseudo amino acid composition to predictprotein subcellular localization an approach by incorporatingevolutionary information and von Neumann entropiesrdquo AminoAcids vol 34 no 4 pp 565ndash572 2008
[75] R Zia Ur and A Khan ldquoIdentifying GPCRs and their typeswith Choursquos pseudo amino acid composition an approach frommulti-scale energy representation and position specific scoringmatrixrdquo Protein amp Peptide Letters vol 19 pp 890ndash903 2012
[76] X Y Sun S P Shi J D Qiu S B Suo S Y Huang and R PLiang ldquoIdentifying protein quaternary structural attributes byincorporating physicochemical properties into the general formof Choursquos PseAAC via discrete wavelet transformrdquo MolecularBioSystems vol 8 pp 3178ndash3184 2012
[77] L Nanni and A Lumini ldquoGenetic programming for creatingChoursquos pseudo amino acid based features for submitochondrialocalizationrdquo Amino Acids vol 34 no 4 pp 653ndash660 2008
[78] M Esmaeili H Mohabatkar and S Mohsenzadeh ldquoUsing theconcept of Choursquos pseudo amino acid composition for risk typeprediction of human papillomavirusesrdquo Journal of TheoreticalBiology vol 263 no 2 pp 203ndash209 2010
[79] H Mohabatkar ldquoPrediction of cyclin proteins using Choursquospseudo amino acid compositionrdquo Protein and Peptide Lettersvol 17 no 10 pp 1207ndash1214 2010
[80] H Mohabatkar M M Beigi and A Esmaeili ldquoPredictionof GABA(A) receptor proteins using the concept of Choursquospseudo-amino acid composition and support vector machinerdquoJournal of Theoretical Biology vol 281 no 1 pp 18ndash23 2011
[81] Y Xu J Ding L Y Wu and K C Chou ldquoiSNO-PseAAC pre-dict cysteine S-nitrosylation sites in proteins by incorporatingposition specific amino acid propensity into pseudo amino acidcompositionrdquo PLoS ONE vol 8 Article ID e55844 2013
[82] W Chen H Lin PM Feng C Ding Y C Zuo andK C ChouldquoiNuc-PhysChem a sequence-based predictor for identifyingnucleosomes via physicochemical propertiesrdquo PLoS ONE vol7 Article ID e47843 2012
[83] B Li T Huang L Liu Y Cai and K C Chou ldquoIdentificationof colorectal cancer related genes with mrmr and shortest pathin protein-protein interaction networkrdquo PLoS ONE vol 7 no 4Article ID e33393 2012
[84] Y Jiang T Huang C Lei Y F Gao Y D Cai and K C ChouldquoSignal propagation in protein interaction network duringcolorectal cancer progressionrdquo BioMed Research Internationalvol 2013 Article ID 287019 9 pages 2013
[85] P Du X Wang C Xu and Y Gao ldquoPseAAC-Builder across-platform stand-alone program for generating variousspecial Choursquos pseudo-amino acid compositionsrdquo AnalyticalBiochemistry vol 425 no 2 pp 117ndash119 2012
[86] D S Cao Q S Xu and Y Z Liang ldquoPropy a tool to generatevarious modes of Choursquos PseAACrdquo Bioinformatics vol 29 pp960ndash962 2013
[87] H Shen and K C Chou ldquoPseAAC a flexible web serverfor generating various kinds of protein pseudo amino acidcompositionrdquo Analytical Biochemistry vol 373 no 2 pp 386ndash388 2008
[88] K C Chou ldquoUsing pair-coupled amino acid composition topredict protein secondary structure contentrdquo Journal of ProteinChemistry vol 18 no 4 pp 473ndash480 1999
BioMed Research International 13
[89] W Liu and K C Chou ldquoPrediction of protein secondarystructure contentrdquo Protein Engineering vol 12 no 12 pp 1041ndash1050 1999
[90] M Bhasin and G P S Raghava ldquoClassification of nuclearreceptors based on amino acid composition and dipeptidecompositionrdquo The Journal of Biological Chemistry vol 279 no22 pp 23262ndash23266 2004
[91] H Lin andH Ding ldquoPredicting ion channels and their types bythe dipeptidemode of pseudo amino acid compositionrdquo Journalof Theoretical Biology vol 269 no 1 pp 64ndash69 2011
[92] H Lin and Q Li ldquoUsing pseudo amino acid composition topredict protein structural class approached by incorporating400 dipeptide componentsrdquo Journal of Computational Chem-istry vol 28 no 9 pp 1463ndash1466 2007
[93] L Nanni and A Lumini ldquoCombing ontologies and dipeptidecomposition for predicting DNA-binding proteinsrdquo AminoAcids vol 34 no 4 pp 635ndash641 2008
[94] K-C Chou ldquoThe convergence-divergence duality in lectindomains of selectin family and its implicationsrdquo FEBS Lettersvol 363 no 1-2 pp 123ndash126 1995
[95] A A Schaffer L Aravind T L Madden et al ldquoImprovingthe accuracy of PSI-BLAST protein database searches withcomposition-based statistics and other refinementsrdquo NucleicAcids Research vol 29 no 14 pp 2994ndash3005 2001
[96] S F Altschul and E V Koonin ldquoIterated profile searches withPSI-BLAST a tool for discovery in protein databasesrdquo Trends inBiochemical Sciences vol 23 no 11 pp 444ndash447 1998
[97] J Deng ldquoGrey entropy and grey target decision makingrdquoJournal of Grey System vol 22 no 1 pp 1ndash24 2010
[98] B M Bolstad R A Irizarry M Astrand and T P Speed ldquoAcomparison of normalizationmethods for high density oligonu-cleotide array data based on variance and biasrdquo Bioinformaticsvol 19 no 2 pp 185ndash193 2003
[99] J-Y Shi S-W Zhang Q Pan Y-M Cheng and J XieldquoPrediction of protein subcellular localization by support vectormachines using multi-scale energy and pseudo amino acidcompositionrdquo Amino Acids vol 33 no 1 pp 69ndash74 2007
[100] T Denoeux ldquoA 120581-nearest neighbor classification rule based onDempster-Shafer theoryrdquo IEEE Transactions on Systems Manand Cybernetics vol 25 no 5 pp 804ndash813 1995
[101] J M Keller M R Gray and J A Givens ldquoA fuzzy k-nearestneighbours algorithmrdquo IEEE Transactions on Systems Man andCybernetics vol 15 no 4 pp 580ndash585 1985
[102] X Xiao P Wang and K Chou ldquoiNR-physchem a sequence-based predictor for identifying nuclear receptors and theirsubfamilies via physical-chemical property matrixrdquo PLoS ONEvol 7 no 2 Article ID e30869 2012
[103] I Roterman L Konieczny W Jurkowski K Prymula and MBanach ldquoTwo-intermediate model to characterize the structureof fast-folding proteinsrdquo Journal of Theoretical Biology vol 283no 1 pp 60ndash70 2011
[104] X Xiao P Wang and K C Chou ldquoGPCR-2L predicting Gprotein-coupled receptors and their types by hybridizing twodifferentmodes of pseudo amino acid compositionsrdquoMolecularBioSystems vol 7 no 3 pp 911ndash919 2011
[105] X Xiao P Wang and K C Chou ldquoQuat-2L a web-server forpredicting protein quaternary structural attributesrdquo MolecularDiversity vol 15 no 1 pp 149ndash155 2011
[106] X Zheng C Li and J Wang ldquoAn information-theoreticapproach to the prediction of protein structural classrdquo Journalof Computational Chemistry vol 31 no 6 pp 1201ndash1206 2010
[107] H Shen J Yang X Liu and K C Chou ldquoUsing supervisedfuzzy clustering to predict protein structural classesrdquo Biochem-ical and Biophysical Research Communications vol 334 no 2pp 577ndash581 2005
[108] K-C Chou and C-T Zhang ldquoReview prediction of proteinstructural classesrdquo Critical Reviews in Biochemistry and Molec-ular Biology vol 30 no 4 pp 275ndash349 1995
[109] P C Mahalanobis ldquoOn the generalized distance in statisticsrdquoProceedings of the National Institute of Sciences of India vol 2pp 49ndash55 1936
[110] R M Centor ldquoSignal detectability the use of ROC curves andtheir analysesrdquoMedical Decision Making vol 11 no 2 pp 102ndash106 1991
[111] K-C Chou ldquoUsing subsite coupling to predict signal peptidesrdquoProtein Engineering vol 14 no 2 pp 75ndash79 2001
[112] K C Chou ldquoPrediction of protein signal sequences and theircleavage sitesrdquo Proteins vol 42 pp 136ndash139 2001
[113] K C Chou ldquoPrediction of signal peptides using scaledwindowrdquoPeptides vol 22 no 12 pp 1973ndash1979 2001
[114] K C Chou ldquoSome remarks on predicting multi-label attributesinmolecular biosystemsrdquoMolecular Biosystems vol 9 pp 1092ndash1100 2013
[115] K C Chou and H B Shen ldquoCell-PLoc 2 0 an improvedpackage of web-servers for predicting subcellular localization ofproteins in various organismsrdquoNatural Science vol 2 pp 1090ndash1103 2010
[116] M Gribskov and N L Robinson ldquoUse of receiver operatingcharacteristic (ROC) analysis to evaluate sequence matchingrdquoComputers and Chemistry vol 20 no 1 pp 25ndash33 1996
[117] Y Yamanishi M Kotera M Kanehisa and S Goto ldquoDrug-target interaction prediction from chemical genomic and phar-macological data in an integrated frameworkrdquo Bioinformaticsvol 26 no 12 pp i246ndashi254 2010
[118] Z He J Zhang X Shi et al ldquoPredicting drug-target interactionnetworks based on functional groups and biological featuresrdquoPLoS ONE vol 5 no 3 Article ID e9603 2010
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

BioMed Research International 3
enzyme (ii) recoupling each of the single drugs with eachof the single enzymes into pairs in a way that none of themoccurred inS+ (iii) randomly picking the pairs thus formeduntil they reached the number two times as many as thepairs in S+ The 2719 interactive enzyme-drug pairs and5438 noninteractive enzyme-drug pairs are given in OnlineSupporting Information S1 (see Supplementary Materialavailable online at httpdxdoiorg1011552013701317) Allthe detailed information for the compounds or drugs listedthere can be found in the KEGG database via their codes
22 Sample Representation Since each of the samples in thecurrent network system contains an enzyme (protein) anda drug a combination of the following two approaches wasadopted to represent the enzyme-drug pair samples
221 Drug
(a) 2D Molecular Fingerprints Although the number ofdrugs is extremely large most of them are small organicmolecules and are composed of some fixed small structures[40] The identification of small molecules or structures canbe used to detect the drug-target interactions [41] Molec-ular fingerprints are bit-string representations of molecularstructure and properties [42] It should be pointed outthat there are many types of structural representations thathave been suggested for the description of drug moleculesincluding physicochemical properties [43] chemical graphs[44] topological indices [45] 3D pharmacophore patternsandmolecular fields In the current study let us use the simpleand generally adopted 2Dmolecular fingerprints to representdrug molecules as described below
First for each of the drugs concerned we can obtaina MOL file from the KEGG database [38] via its codethat contains the detailed information of chemical structureSecond we can convert the MOL file format into its 2Dmolecular fingerprint file format by using a chemical toolboxsoftware called OpenBabel [46] which can be downloadedfrom the website at httpopenbabelorg The current ver-sion of OpenBabel can generate four types of fingerprintsFP2 FP3 FP4 and MACCS In the current study we usedthe FP2 fingerprint format It is a path-based fingerprint thatidentifies small molecule fragments based on all linear andring substructures and maps them onto a bit-string using ahash function (somewhat similar to the daylight fingerprints[47 48]) It is a length of 256-bit hexadecimal string obtainedfrom theOpenBabel andwe can convert it to a 256-bit vectorThen a molecular fingerprint can be formulated as a 256-Dvector given by
MF = [1198601 sdot sdot sdot 119860119895 sdot sdot sdot 119860256]T (2)
where 119860119895(119895 = 1 2 256) is an integer between 0 and 15
and T is the matrix transpose operatorIn order to capture as much useful information from
a molecular fingerprint as possible we can also convertthe above 256-bit hexadecimal string into a 1024-bit binaryvector which is a digital sequence only including 0 and 1
and consider two different digital signal characteristics for thedigital sequence as follows
(b) Information Entropy Shannon proposed that any infor-mation is redundant and redundant size is related withthe occurrence probability or uncertainty of each symbolsuch as numbers letters or words among the informationThe information entropy for a system with a probabilitydistribution 119875(119909
119894) for two classes information entropy [49] is
defined as
119867119909= minussum
119909
119875 (119909119894) log2119875 (119909119894) (119894 = 0 1) (3)
where 119875(119909119894) represents the occurrence probability of num-
ber 119894 in the aforementioned 1024-bit binary vector andthe information entropy 119867
119909is a measure value of the
information amount For example for the digital sequence100100011010010 the value of the information entropy 119867
119909
thus obtained is
119875 (1199090) =
9
15
= 06
119875 (1199091) =
6
15
= 04
119867119909= minus (06 times log
206 + 04 times log
204) = 0971
(4)
(c) Complexity Factor The Lempel-Ziv (LZ) complexity [50]reflects the order that is retained in the sequence and hencewas adopted in this study For each step only two operationswere allowed in the process to get the LZ complexity eithercopying the longest section from the part of a nonemptysequence or generating an additional symbol mark thatensures the uniqueness of per component 119878(119894
119896minus1rarr 119894119896) Its
substring is expressed by
119878 (119894 997888rarr 119895) = 119898119894119898119894+1119898119894+2sdot sdot sdot 119898119895
(1 le 119894 le 119895 le 119871) (5)
where 1198981represents the 1st digital value 119898
2the 2nd value
and so forth A nonempty digital sequence is synthesizedaccording to the following formula
Syn (119878) = 119878 (1 997888rarr 1198941) ∙ 119878 (119894
1+ 1 997888rarr 119894
2)
∙ sdot sdot sdot ∙ 119878 (119894119898minus1
+ 1 997888rarr 119894119871)
(6)
Suppose that 119878 = 11989811198982119898311989841198985sdot sdot sdot 119898119871has been recon-
structed by the subsymbol 119898119903which is viewed as the newly
inserted symbol The substring up to 119898119903will be denoted by
119878(1 rarr 119903)∙ where the bold dot ∙ indicates that 119898119903is a newly
inserted symbol for checkingwhether the rest of the substring119878(119903+1 rarr 119871) can be reconstructed by a simple process At firstsuppose 119878(119902) = 119898
119903+ 1 and see whether 119878(119902) is the substring
for the subsequence 119878(1 rarr 119903) which means deleting thelast symbol from the substring 119878(1 rarr 119903)119878(119902) If the answeris ldquonordquo we insert 119878(119902) into the sequence followed by a dot ∙Thus it could not be obtained by the same operation If theanswer is ldquoyesrdquo no new symbol is needed and we can go on toproceed with 119878(119902) = 119898
119903+1119898119903+2
and repeat the same previousprocedure The LZ complexity is the number of dots (plus
4 BioMed Research International
one if the string is not terminated by a dot) For example forthe sequence 100100011010010 syn(119875) and the correspondingcomplexity factor CF are described as
Syn (119878) = 1 ∙ 0 ∙ 01 ∙ 000 ∙ 11 ∙ 01001 ∙ 0
CF = 7(7)
Thus by adding the information entropy 119867119909(4) and com-
plexity factor CF (7) into the molecular fingerprint MF (2)we obtained a total of (256 + 1 + 1) = 258 feature elements torepresent a drug compound that is it can now be formulatedas a 258-D vector given by
119863 = [11986011198602sdot sdot sdot 119860
256119867119909
CF]T (8)
where 119860119894has the same meaning as in (2) while 119867
119909and CF
are the information entropy and complexity factor respec-tively as described in the previous two sections
222 Enzyme The sequences of the enzymes involved inthis study are given in Online Supporting Information S2Now the problem is how to effectively represent these enzymesequences for the current study Generally speaking thereare two kinds of approaches to formulate enzyme sequencesthe sequential model and the nonsequential or discretemodel [51] The most typical sequential representation foran enzyme sample E with 119871 residues is its entire amino acidsequence that is
E = 1198771119877211987731198774119877511987761198777sdot sdot sdot 119877119871 (9)
where 1198771represents the 1st residue 119877
2the 2nd residue and
so forth An enzyme sample thus formulated can contain itsmost complete information This is an obvious advantage ofthe sequential representation To get the desired results thesequence-similarity-search-based tools such as BLAST [5253] are usually utilized to conduct the prediction Howeverthis kind of approach failed to work when the query enzymedid not have significant homology to enzyme of known char-acters Thus various nonsequential representation modelswere proposed The simplest nonsequential model for anenzyme was based on its amino acid composition (AAC) asdefined by
E = [11989111198912sdot sdot sdot 11989120]
T (10)
where 119891119906(119906 = 1 2 20) are the normalized occurrence
frequencies of the 20 native amino acids [54ndash56] in theenzyme E and T has the same meaning as in (2) and (8)The AAC-discrete model was widely used for identifyingvarious attributes of proteins (see eg [57ndash61]) Howeveras can be seen from (10) all the sequence order effectswere lost by using the AAC-discrete model This is its mainshortcoming To avoid completely losing the sequence-orderinformation the pseudo amino acid composition [62 63]or Choursquos PseAAC [3] was proposed to replace the simpleAAC model Since the concept of PseAAC was proposedin 2001 [62] it has penetrated into almost all the fields of
protein attribute predictions and computational proteomicssuch as predicting supersecondary structure [64] predictingmetalloproteinase family [65] predicting membrane pro-tein types [66 67] predicting protein structural class [68]discriminating outer membrane proteins [69] identifyingantibacterial peptides [70] identifying allergenic proteins[71] identifying bacterial virulent proteins [72] predictingprotein subcellular location [73 74] identifying GPCRs andtheir types [75] identifying protein quaternary structuralattributes [76] predicting protein submitochondria locations[77] identifying risk type of human papillomaviruses [78]identifying cyclin proteins [79] predicting GABA(A) recep-tor proteins [80] and predicting cysteine S-nitrosylation sitesin proteins [81] among many others (see a long list of paperscited in the References section of [33]) Recently the conceptof PseAAC was further extended to represent the featurevectors of DNA and nucleotides [36 82] as well as otherbiological samples (see eg [83 84]) Because it has beenwidely and increasingly used recently two powerful soft-wares called ldquoPseAAC-Builderrdquo [85] and ldquopropyrdquo [86] wereestablished for generating various special Choursquos pseudo-amino acid compositions in addition to the web-serverPseAAC [87] built in 2008 According to a recent review [33]the general formof Choursquos PseAAC for an enzyme sample canbe formulated by
E = [12059511205952sdot sdot sdot 120595119906sdot sdot sdot 120595Ω]
T (11)
where the subscript Ω is an integer and its value as wellas the components 120595
119906(119906 = 1 2 Ω) will depend on
how to extract the desired information from the amino acidsequence of E (cf (10)) Next let us describe how to extractuseful information from the benchmark datasetS andOnlineSupporting Information S2 to define the enzyme samplesconcerned via (11)
To incorporate as much useful information as possiblefrom an enzyme sample we are to approach this problemfrom three different angles followed by incorporating thefeature elements thus obtained into the general form ofPseAAC of (11)(a) Amino Acid Composition The components of amino acidcomposition have beenwidely used to predict various proteinattributes [57ndash61] In this study they were also included as thefirst 20 elements in the general Choursquos PseAAC (cf (11)) thatis
120595119906= 119891119906
(119906 = 1 2 20) (12)
where 119891119906has the same meaning as in (10)
(b) Dipeptide Composition Dipeptide composition has beenused to predict the protein secondary structural contents[88 89] as well as various protein attributes (see eg [90ndash93]) The number of different dipeptides is 20 times 20 = 400Suppose that the normalized occurrence frequencies of the400 dipeptides in an enzyme sample are given by
119891
(2)
119906(119906 = 1 2 400) (13)
BioMed Research International 5
Incorporating the above 400 dipeptide components into (11)we have
120595119906+20
= 119891
(2)
119906(119906 = 1 2 400) (14)
(c) Sequential Evolution Information Biology is a naturalscience with a historic dimension All biological specieshave developed starting out from a very limited number ofancestral species Their evolution involves changes of singleresidues insertions and deletions of several residues [94]gene doubling and gene fusion With these changes accu-mulated for a long period of time many similarities betweeninitial and resultant amino acid sequences are graduallyeliminated but the corresponding proteins may still sharemany common attributes [18] such as having basically thesame biological function and residing at a same subcellularlocation To extract the sequential evolution information anduse it to define the components of (11) the PSSM (PositionSpecific Scoring Matrix) was used as described next
According to Schaffer et al [95] the sequence evolutioninformation of enzyme E with 119871 amino acid residues can beexpressed by an 119871 times 20matrix as given by
P(0)PSSM =
[
[
[
[
[
[
[
119864
0
1rarr1119864
0
1rarr2sdot sdot sdot 119864
0
1rarr20
119864
0
2rarr1119864
0
2rarr2sdot sdot sdot 119864
0
2rarr20
119864
0
119871rarr1119864
0
119871rarr2sdot sdot sdot 119864
0
119871rarr20
]
]
]
]
]
]
]
(15)
where 1198640119894rarr 119895
represents the original score of the 119894th aminoacid residue (119894 = 1 2 119871) in the enzyme sequence changedto amino acid type 119895 (119895 = 1 2 20) in the process ofevolution Here the numerical codes 1 2 20 are used torepresent the 20 native amino acid types denoted by A CD E F G H I K L M N P Q R S T V W and Y The119871 times 20 scores in (15) were generated by using PSI-BLAST[96] to search the UniProtKBSwiss-Prot database (Release2013-05) through three iterations with 0001 as the 119864 valuecutoff for multiple sequence alignment against the sequenceof the enzyme E In order to make every element in (15) bescaled from their original score ranges into region of [0 1]we performed a conversion through the standard sigmoidfunction to make it become
P(1)PSSM =
[
[
[
[
[
[
[
119864
1
1rarr1119864
1
1rarr2sdot sdot sdot 119864
1
1rarr20
119864
1
2rarr1119864
1
2rarr2sdot sdot sdot 119864
1
2rarr20
119864
1
119871rarr1119864
1
119871rarr2sdot sdot sdot 119864
1
119871rarr20
]
]
]
]
]
]
]
(16)
where
119864
1
119894rarr 119895=
1
1 + 119890
minus1198640
119894rarr 119895
(1 le 119894 le 119871 1 le 119895 le 20) (17)
Nowwe extract the useful information from (16) to define thecomponents of (11) via the following approach
120595119906+420
= ℓ119906
(119906 = 1 2 20) (18)
where
ℓ119895=
1
119871
times
119871
sum
119896=1
119864
1
119896rarr119895(119895 = 1 2 20) (19)
(d) Grey System Model Approach The grey system theory[97] is quite useful in dealing with complicated systems thatlack sufficient information or need to process uncertaininformation According to the grey system theory we canextract the following information from the 119895th column of(16) that is
[
119886
119895
1
119886
119895
2
] = (BT119895B119895)
minus1
BT119895U119895
(119895 = 1 2 20) (20)
where
B119895=
[
[
[
[
[
[
[
[
[
[
minus119864
1
2rarr119895minus119864
1
1rarr119895minus 05119864
1
2rarr1198951
minus119864
1
3rarr119895minus
2
sum
119894=1
119864
1
119894rarr 119895minus 05119864
1
3rarr1198951
minus119864
1
119871rarr119895minus
119871minus1
sum
119894=1
119864
1
119894rarr 119895minus 05119864
1
119871rarr1198951
]
]
]
]
]
]
]
]
]
]
U119895=
[
[
[
[
[
[
119864
1
2rarr119895minus 119864
1
1rarr119895
119864
1
3rarr119895minus 119864
1
2rarr119895
119864
1
119871rarr119895minus 119864
1
119871minus1rarr119895
]
]
]
]
]
]
(21)
Therefore based on the grey system theory and (20) we canextract another 20 times 2 = 40 quantities from (16) to definethe components of (11) that is
120593119895=
1199081119886
119895
1when 119895 is an odd number
1199082119886
119895
2when 119895 is an even number
1 le 119895 le 20
(22)
where 1198861198951and 119886119895
2are given by (20) 119908
1and 119908
2are weight
factors which were all set to 1 in the current studySubstituting the elements in (12) (14) (18) and (22) we
finally obtain a total of Ω = 20 + 400 + 20 + 40 = 480
components for the PseAAC of (11) where
120595119906=
119891119906
when 1 le 119906 le 20119891
(2)
119906when 21 le 119906 le 420
ℓ119906
when 421 le 119906 le 440120593119906
when 440 le 119906 le 480
(23)
In other words in this study (11) or Choursquos PseAAC is a 480-D vector whose 480 components are given by (23) derivedfrom the amino acid composition dipeptide compositionsequential evolution information and grey system theory(e) Representing Enzyme-Drug Pairs Now the pair betweenan enzyme molecule E and a drug compound D can beformulated by combing (8) and (11) as given by
G = D oplus E
= [1198601sdot sdot sdot 119860
256119867119909
CF 1205951sdot sdot sdot 120595480]
T
(24)
6 BioMed Research International
where G represents the enzyme-drug pair oplus the orthogonalsum [51] and each of the (258 + 480) = 738 feature elementsis given in (8) and (23)
For the convenience of the later formulation let us use119909119894(119894 = 1 2 738) to represent the 738 components of (24)
that is
G = [11990911199092sdot sdot sdot 119909119894sdot sdot sdot 119909738]
T (25)
To optimize the prediction results different weights wereusually tested for each of the elements in (25) However sinceit would consume a lot of computational time for a total of 738weight factors here let us adopt the normalization approachto deal with this problem as done in [98 99] that is convert119909119894in (25) to 119910
119894according to the following equation
119910119894=
2tanminus1 (119909119894)
120587
(119894 = 1 2 738) (26)
where tanminus1 means arctangent By means of (26) everycomponent in (25) will be converted into the range of[minus1 1] that is we have minus1 le 119910
119894le 1 As demonstrated
in [98 99] the normalization approach via (26) was quiteeffective in enhancing the quality of prediction operated ina high dimension space Therefore in this study we wouldnot to take the procedure of optimizing the weight factorssignificantly reducing the computational times
23 Fuzzy 119870-Nearest Neighbour Algorithm The 119870-NN (119870-Nearest Neighbor) classifier is quite popular in patternrecognition community owing to its good performance andsimple-to-use feature According to the 119870-NN rule [100]named also as the ldquovoting 119870-NN rulerdquo the query sampleshould be assigned to the subset represented by a majorityof its 119870 nearest neighbors as illustrated in Figure 5 of [33]
Fuzzy 119870-NN classification method [101] is a specialvariation of the119870-NNclassification family Instead of roughlyassigning the label based on a voting from the 119870 nearestneighbors it attempts to estimate the membership valuesthat indicate how much degree the query sample belongsto the classes concerned Obviously it is impossible for anycharacteristic description to contain complete informationwhich would make the classification ambiguous In view ofthis the fuzzy principle is very reasonable and particularlyuseful in dealing with complicated biological systems such asidentifying nuclear receptor subfamilies [102] characterizingthe structure of fast-folding proteins [103] classifying Gprotein-coupled receptors [104] predicting protein quater-nary structural attributes [105] predicting protein structuralclasses [106 107] and so forth
Next let us give a brief introduction how to use thefuzzy119870-NN approach to identify the interactions between theenzymes and the drug compounds in the network concerned
Supposing that S(119873) = G1G2 G
119873 is a set of vec-
tors representing 119873 enzyme-drug pairs in a training set
classified into two classes 119862+ 119862minus where 119862+ denotes theinteractive pair class while 119862minus the noninteractive pair classSlowast(G) = Glowast
1Glowast2 Glowast
119870 sub S(119873) is the subset of the 119870
nearest neighbor pairs to the query pair G Thus the fuzzymembership value for the query pair G in the two classes ofS(119873) is given by
120583
+
(G) =sum
119870
119895=1120583
+(Glowast119895) 119889(GGlowast
119895)
minus2(120593minus1)
sum
119870
119895=1119889(GGlowast
119895)
minus2(120593minus1)
120583
minus
(G) =sum
119870
119895=1120583
minus(Glowast119895) 119889(GGlowast
119895)
minus2(120593minus1)
sum
119870
119895=1119889(GGlowast
119895)
minus2(120593minus1)
(27)
where 119870 is the number of the nearest neighbors counted forthe query pairG 120583+(Glowast
119895) and 120583minus(Glowast
119895) the fuzzy membership
values of the training sample Glowast119895to the class 119862+ and 119862minus
respectively as will be further defined next 119889(GGlowast119895) the
cosine distance between G and its 119895th nearest pair Glowast119895in
the training dataset S(119873) 120593(gt 1) the fuzzy coefficient fordetermining how heavily the distance is weighted whencalculating each nearest neighborrsquos contribution to the mem-bership value Note that the parameters 119870 and 120593 will affectthe computation result of (27) and they will be optimizedby a grid-search as will be described later Also variousother metrics can be chosen for 119889(GGlowast
119895) such as Euclidean
distance Hamming distance [108] andMahalanobis distance[55 109]
The quantitative definitions for the aforementioned120583
+(Glowast119895) and 120583minus(Glowast
119895) in (27) are given by
120583
+
(Glowast119895) =
1 if Glowast119895isin 119862
+
0 otherwise
120583
minus
(Glowast119895) =
1 if Glowast119895isin 119862
minus
0 otherwise
(28)
Substituting the results obtained by (27) into (28) it followsthat if 120583+(G) gt 120583
minus(G) then the query pair G is an
interactive couple otherwise noninteractive In other wordsthe outcome can be formulated as
G isin
119862
+ if 120583+ (G) gt 120583minus (G)
119862
minus otherwise
(29)
If there is a tie between 120583+(G) and 120583minus(G) the query pair Gwill be randomly assigned to one of the two classes Howevercase like that is quite rare and in this study never happened
The predictor thus established is called iEzy-Drugwhere ldquoirdquo means identify and ldquoEzy-Drugrdquo means the inter-action between enzyme and drug To provide an intuitiveoverall picture a flowchart is provided in Figure 2 to showthe process of how the classifier works in identifying enzyme-drug interactions
BioMed Research International 7
Input a queryprotein sequence and
a drug code
Create Chous PseAAC of a protein
Create feature vector of a drug
Feature fusion
engineTraining dataset
Yes No
Noninteractive enzyme drug pair
Interactive enzymedrug pair
AACDipeptideSeq Evol
Grey model
Mol FptInfo entropy
CF
FKNN operation
Figure 2 A flowchart to show the operation process of the iEzy-Drug predictor See the text for further explanation
24 Criteria for Performance Evaluation In the literaturethe following equation set is often used for examining theperformance quality of a predictor
Sn = TPTP + FN
Sp = TNTN + FP
Acc = TP + TNTP + TN + FP + FN
MCC = (TP times TN) minus (FP times FN)radic(TP + FP) (TP + FN) (TN + FP) (TN + FN)
(30)
where TP represents the true positive TN the true negativeFP the false positive FN the false negative Sn the sensitiv-ity Sp the specificity Acc the accuracy MCC the Mathewrsquoscorrelation coefficient
To most biologists however the four metrics as formu-lated in (30) are not quite intuitive and easier-to-understandparticularly for the Mathewrsquos correlation coefficient Herelet us adopt the Choursquos symbols to formulate the previousfour metrics By means of Choursquos symbols [111 112] the rates
of correct predictions for the interactive enzyme-drug pairsin dataset S+ and the noninteractive enzyme-drug pairs indataset Sminus are respectively defined by (cf (1))
Λ
+
=
119873
+minus 119873
+
minus
119873
+ for the interactive enzyme-drug pairs
Λ
minus
=
119873
minusminus 119873
minus
+
119873
minus for the noninteractive enzyme-drug pairs
(31)
where 119873+ is the total number of the interactive enzyme-drug pairs investigated while 119873+
minusis the number of the
interactive enzyme-drug pairs incorrectly predicted as thenoninteractive enzyme-drug pairs 119873minus is the total numberof the noninteractive enzyme-drug pairs investigated while119873
minus
+is the number of the noninteractive enzyme-drug pairs
incorrectly predicted as the interactive enzyme-drug pairsThe overall success prediction rate is given by [113] as follows
Λ =
Λ
+119873
++ Λ
minus119873
minus
119873
++ 119873
minus= 1 minus
119873
+
minus+ 119873
minus
+
119873
++ 119873
minus
(32)
It is obvious from (31)-(32) that if and only if none ofthe interactive enzyme-drug pairs and the noninteractiveenzyme-drug pairs are mispredicted that is 119873+
minus= 119873
minus
+= 0
and Λ+ = Λ
minus= 1 we have the overall success rate Λ = 1
Otherwise the overall success rate would be smaller than 1The relations between the symbols in (32) and those in
(30) are given by
TP = 119873+ minus 119873+minus
TN = 119873
minus
minus 119873
minus
+
FP = 119873minus+
FN = 119873
+
minus
(33)
Substituting (33) into (30) and also noting (31)-(32) we obtain
Sn = Λ+ = 1 minus119873
+
minus
119873
+
Sp = Λminus = 1 minus119873
minus
+
119873
minus
Acc = Λ = 1 minus119873
+
minus+ 119873
minus
+
119873
++ 119873
minus
MCC =1 minus ((119873
+
minus119873
+) + (119873
minus
+119873
minus))
radic(1 + (119873
minus
+minus 119873
+
minus) 119873
+) (1 + (119873
+
minusminus 119873
minus
+) 119873
minus)
(34)
Now it is obvious to see from (34) when 119873
+
minus= 0
meaning none of the interactive enzyme-drug pairs was
8 BioMed Research International
02
46
810
1
15
2089
0895
09
0905
091
0915
092
ACC
K120593
Figure 3 A 3D plot to show how the parameter in (27) wasoptimized for the iEzy-Drug predictor
mispredicted to be a noninteractive enzyme-drug pair wehave the sensitivity Sn = 1 while 119873+
minus= 119873
+ meaning thatall the interactive enzyme-drug pairs weremispredicted to bethe noninteractive enzyme-drug pairs we have the sensitivitySn = 0 Likewise when 119873minus
+= 0 meaning none of the
noninteractive enzyme-drug pairs wasmispredicted we havethe specificity Sp = 1 while 119873minus
+= 119873
minus meaning all thenoninteractive enzyme-drug pairs were incorrectly predictedas interactive enzyme-drug pairs we have the specificity Sp =0 When119873+
minus= 119873
minus
+= 0 meaning that none of the interactive
enzyme-drug pairs in the dataset S+ and none of the nonin-teractive enzyme-drug pairs in Sminus was incorrectly predictedwe have the overall accuracy Acc = Λ = 1 while 119873+
minus= 119873
+
and 119873minus+= 119873
minus meaning that all the interactive enzyme-drugpairs in the dataset S+ and all the noninteractive enzyme-drug pairs in Sminus were mispredicted we have the overallaccuracy Acc = Λ = 0 The MCC correlation coefficient isusually used for measuring the quality of binary (two-class)classifications When 119873+
minus= 119873
minus
+= 0 meaning that none
of the interactive enzyme-drug pairs in the dataset S+ andnone of the noninteractive enzyme-drug pairs in Sminus weremispredicted we have MCC = 1 when 119873+
minus= 119873
+2 and
119873
minus
+= 119873
minus2 we have MCC = 0 meaning no better than
random prediction when 119873+minus= 119873
+ and 119873minus+= 119873
minus we haveMCC = minus1 meaning total disagreement between predictionand observation As we can see from the previous discussionit is much more intuitive and easier-to-understand whenusing (34) to examine a predictor for its sensitivity specificityoverall accuracy and Mathewrsquos correlation coefficient It isinstructive to point out that themetrics as defined in (30) and(34) are valid for single label systems for multilabel systemsa set of more complicated metrics should be used as given in[114]
3 Results and Discussion
31 Cross-Validation How to properly examine the predic-tion quality is a key for developing a new predictor and
False positive rate
True
pos
itive
rate
1
01
02
03
04
05
06
07
08
09
0
101 02 03 04 05 06 07 08 090
BaselineiEzy-Drug AUC = 09377
Figure 4 A plot for the ROC curve to quantitatively show theperformance of the iEzy-Drug predictor
estimating its potential application value Generally speak-ing the following three cross-validation methods are oftenused to examine a predictor of its effectiveness in practicalapplication independent dataset test subsampling or 119870-fold(such as 5-fold 7-fold or 10-fold) test and jackknife test[108] However as elaborated by a penetrating analysis in[115] considerable arbitrariness exists in the independentdataset test Also as demonstrated by (27)ndash(29) in [33]the subsampling test (or 119870-fold cross-validation) cannotavoid arbitrariness either Only the jackknife test is the leastarbitrary that can always yield a unique result for a givenbenchmark dataset Therefore the jackknife test has beenwidely recognized and increasingly utilized by investigatorsto examine the quality of various predictors (see eg [66 7174 80]) Accordingly the success rate by the jackknife test wasalso used to optimize the two uncertain parameters 119870 and 120593in (27) The result thus obtained is shown in Figure 3 fromwhich we obtain when 119870 = 6 and 120593 = 15 the iEzy-Drugpredictor reaches its optimized status
The success rates thus obtained by the jackknife test inidentifying interactive Enzyme-drug pairs or noninteractiveenzyme-drug pairs on the benchmark dataset S (cf OnlineSupporting Information S1) are given in Table 1 where forfacilitating comparison the corresponding result by He et al[110] is also given As we can see from the table the overallaccuracy Acc achieved by iEzy-Drug was 9103 remarkablyhigher than 8548 the corresponding rate obtained byHe et al [110] on the same benchmark Furthermore listedin Table 1 are also the values obtained by iEzy-Drug for theother three metrics that is Sn = 9081 Sp = 9114 andMCC = 8039 indicating that the accuracy of iEzy-Drug isnot only very high but also quite stale
To provide a graphical illustration to show the per-formance of the current binary classifier iEzy-Drug asits discrimination threshold is varied a 2D plot calledReceiver Operating Characteristic (ROC) curve [116 117]was also given (Figure 4) In the ROC curve the verticalcoordinate 119884 is for the true positive rate or Sn (cf (34))while horizontal coordinate 119883 for the false positive rate
BioMed Research International 9
Read Me Data Citation
Enter both the protein sequence and the drug code (example) the number of query pairs is limited at 10 or less for each submission
Submit Clear
You can download the program and run it on your own local computer
iEzy-Drug identifying the interaction between enzymes anddrugs in cellular networking
Figure 5 A semiscreenshot to show the top page of the iEzy-Drugweb-server Its web-site address is at httpwwwjci-bioinfocniEzy-Drug
Table 1 The jackknife success rates obtained with iEzy-Drug in identifying interactive enzyme-drug pairs and noninteractive enzyme-drugpairs for the benchmark dataset S (cf Online Supporting Information S1)
Method Acc Sn Sp MCC
iEzy-Druga 74258157 = 9103 24692719 = 9081 49565438 = 9114 8039NN predictorb 8548 NA NA NAaSee (27) where the parameters119870 = 6 and 120593 = 15bSee [110]
or 1-Sp The best possible prediction method would yielda point with the coordinate (0 1) representing 100 truepositive rate (sensitivity Sn) and 0 false positive rate or100 specificity Therefore the (0 1) point is also calleda perfect classification A completely random guess wouldgive a point along a diagonal from the point (0 0) to (1 1)The area under the ROC curve also called Area Under theROC (AUROC) is often used to indicate the performancequality of a binary classifier the value 05 of AUROCis equivalent to random prediction while 1 of AUROCrepresents a perfect one As we can see from Figure 4the AUROC value obtained by iEzy-Drug is 09377
The reason why iEzy-Drug can remarkably improve theprediction quality is that it has introduced the 2D molecularfingerprints to represent drug samples see Online SupportingInformation S3 for the detailed fingerprint expressions for thedrugs listed in Online Supporting Information S1 and thatit has successfully used PseAAC to incorporate the featuresderived from the sequences of enzymes that are essentialfor identifying the interaction of enzymes with drugs in thecellular networking
To enhance the value of its practical applications theweb server for iEzy-Drug has been established that canbe freely accessible at httpwwwjci-bioinfocniEzy-DrugIt is anticipated that the web server will become a usefulhigh throughput tool for both basic research and drug
development in the relevant areas or at the very least playa complementary role to the existing method [39 110 118] forwhich so far noweb-serverwhatsoever has been provided yet
32 The Protocol or User Guide For the convenience of thevast majority of biologists and pharmaceutical scientists herelet us provide a step-by-step guide to show how the userscan easily get the desired result by means of the web serverwithout the need to follow the complicated mathematicalequations presented in this paper for the process of develop-ing the predictor and its integrityStep 1 Open the web server at the site httpwwwjci-bio-infocniEzy-Drug and you will see the top page of thepredictor on your computer screen as shown in Figure 5Click on the ReadMe button to see a brief introduction aboutiEzy-Drug predictor and the caveat when using itStep 2 Either type or copypaste the query pairs into theinput box at the center of Figure 5 Each query pair consistsof two parts one is for the protein sequence and the other forthe drug The enzyme sequence should be in FASTA formatwhile the drug in the KEGG code Examples for the querypairs input can be seen by clicking on the Example buttonright above the input boxStep 3 Click on the Submit button to see the predicted resultFor example if you use the four query pairs in the Example
10 BioMed Research International
window as the input after clicking the Submit button youwill see on your screen that the ldquohsa 10056rdquo enzyme andthe ldquoD0021rdquo drug are an interactive pair and that the ldquohsa100rdquo enzyme and the ldquoD0037rdquo drug are also an interactivepair but that the ldquohsa 3295rdquo enzyme and the ldquoD00889rdquo drugare not an interactive pair and that the ldquohsa 7366rdquo enzymeand the ldquoD03601rdquo drug are not an interactive pair eitherAll these results are fully consistent with the experimentalobservations It takes about 3 minutes before the results areshown on the screenStep 4 Click on the Citation button to find the relevant paperthat documents the detailed development and algorithm ofiEzy-DurgStep 5 Click on the Data button to download the benchmarkdataset used to train and test the iEzy-Durg predictorStep 6 The program code is also available by clicking thebutton download on the lower panel of Figure 5
Acknowledgments
The authors would like to thank the three anonymousreviewers whose constructive comments are very helpful forstrengthening the presentation of the paper This work wassupported by the Grants from the National Natural ScienceFoundation of China (no 31260273) the Jiangxi ProvincialForeign Scientific and Technological Cooperation Project(no 20120BDH80023) Natural Science Foundation of JiangxiProvince China (no 2010GZS0122 20122BAB201020) theDepartment of Education of Jiangxi Province (GJJ12490)the LuoDi plan of the Department of Education of JiangxiProvince (KJLD12083) and the Jiangxi Provincial Founda-tion for Leaders of Disciplines in Science (20113BCB22008)The funders had no role in study design data collection andanalysis decision to publish or preparation of the paper
References
[1] A Bairoch ldquoThe ENZYME database in 2000rdquo Nucleic AcidsResearch vol 28 no 1 pp 304ndash305 2000
[2] S H Koenig and R D Brown ldquoH2CO3as substrate for carbonic
anhydrase in the dehydration of H2CO3(minus)rdquo Proceedings of the
National Academy of Sciences of the United States of Americavol 69 no 9 pp 2422ndash2425 1972
[3] S X Lin and J Lapointe ldquoTheoretical and experimental biologyin onerdquo Journal of Biomedical Science and Engineering vol 6 pp435ndash442 2013
[4] K C Chou and S P Jiang ldquoStudies on the rate of diffusion-controlled reactions of enzymes Spatial factor and force fieldfactorrdquo Scientia Sinica vol 17 no 5 pp 664ndash680 1974
[5] K C Chou ldquoThe kinetics of the combination reaction betweenenzyme and substraterdquo Scientia Sinica vol 19 no 4 pp 505ndash528 1976
[6] K C Chou and G P Zhou ldquoRole of the protein outside activesite on the diffusion-controlled reaction of enzymerdquo Journal ofthe American Chemical Society vol 104 no 5 pp 1409ndash14131982
[7] R A Poorman A G Tomasselli R L Heinrikson and FJ Kezdy ldquoA cumulative specificity model for proteases from
human immunodeficiency virus types 1 and 2 inferred fromstatistical analysis of an extended substrate data baserdquo TheJournal of Biological Chemistry vol 266 no 22 pp 14554ndash145611991
[8] K-C Chou ldquoA vectorized sequence-coupling model for pre-dicting HIV protease cleavage sites in proteinsrdquo The Journal ofBiological Chemistry vol 268 no 23 pp 16938ndash16948 1993
[9] G Z Liang and S Z Li ldquoA new sequence representation asapplied in better specificity elucidation for human immunod-eficiency virus type 1 proteaserdquo Biopolymers vol 88 no 3 pp401ndash412 2007
[10] J J Chou ldquoPredicting cleavability of peptide sequences byHIVprotease via correlation-angle approachrdquo Journal of ProteinChemistry vol 12 no 3 pp 291ndash302 1993
[11] Q S Du S Wang D Q Wei S Sirois and K C ChouldquoMolecular modeling and chemical modification for findingpeptide inhibitor against severe acute respiratory syndromecoronavirus main proteinaserdquo Analytical Biochemistry vol 337no 2 pp 262ndash270 2005
[12] Y Gan H Huang Y Huang et al ldquoSynthesis and activity ofan octapeptide inhibitor designed for SARS coronavirus mainproteinaserdquo Peptides vol 27 no 4 pp 622ndash625 2006
[13] Q S Du H Sun and K C Chou ldquoInhibitor design for SARScoronavirus main protease based on lsquodistorted key theoryrsquordquoMedicinal Chemistry vol 3 no 1 pp 1ndash6 2007
[14] K C Chou ldquoPrediction of human immunodeficiency virusprotease cleavage sites in proteinsrdquoAnalytical Biochemistry vol233 no 1 pp 1ndash14 1996
[15] J Knowles and G Gromo ldquoTarget selection in drug discoveryrdquoNature Reviews Drug Discovery vol 2 no 1 pp 63ndash69 2003
[16] A C Cheng R G Coleman K T Smyth et al ldquoStructure-basedmaximal affinity model predicts small-molecule druggabilityrdquoNature Biotechnology vol 25 no 1 pp 71ndash75 2007
[17] M Rarey B Kramer T Lengauer and G Klebe ldquoA fast flexibledocking method using an incremental construction algorithmrdquoJournal of Molecular Biology vol 261 no 3 pp 470ndash489 1996
[18] K C Chou ldquoReview structural bioinformatics and its impactto biomedical sciencerdquo Current Medicinal Chemistry vol 11 no16 pp 2105ndash2134 2004
[19] K C Chou D Q Wei and W Z Zhong ldquoBinding mechanismof coronavirus main proteinase with ligands and its implicationto drug design against SARSrdquo Biochemical and BiophysicalResearch Communications vol 308 pp 148ndash151 2003 (Erratumin Biochemical and Biophysical Research Communications vol310 p 675 2003)
[20] K C Chou D Q Wei Q S Du S Sirois and W Z ZhongldquoProgress in computational approach to drug developmentagaint SARSrdquo Current Medicinal Chemistry vol 13 no 27 pp3263ndash3270 2006
[21] G P Zhou and F A Troy ldquoNMR studies on how the bindingcomplex of polyisoprenol recognition sequence peptides andpolyisoprenols can modulate membrane structurerdquo CurrentProtein and Peptide Science vol 6 no 5 pp 399ndash411 2005
[22] R B Huang Q S Du C H Wang and K C Chou ldquoAn in-depth analysis of the biological functional studies based on theNMR M2 channel structure of influenza A virusrdquo Biochemicaland Biophysical Research Communications vol 377 no 4 pp1243ndash1247 2008
[23] Q S Du R B Huang C H Wang X M Li and K C ChouldquoEnergetic analysis of the two controversial drug binding sitesof the M2 proton channel in influenza A virusrdquo Journal ofTheoretical Biology vol 259 no 1 pp 159ndash164 2009
BioMed Research International 11
[24] M J Berardi W M Shih S C Harrison and J J Chou ldquoMito-chondrial uncoupling protein 2 structure determined by NMRmolecular fragment searchingrdquo Nature vol 476 no 7358 pp109ndash114 2011
[25] J R Schnell and J J Chou ldquoStructure andmechanism of theM2proton channel of influenza A virusrdquo Nature vol 451 no 7178pp 591ndash595 2008
[26] B OuYang S Xie M J Berardi et al ldquoUnusual architecture ofthe p7 channel from hepatitis C virusrdquoNature vol 498 pp 521ndash525 2013
[27] K Oxenoid and J J Chou ldquoThe structure of phospholambanpentamer reveals a channel-like architecture in membranesrdquoProceedings of the National Academy of Sciences of the UnitedStates of America vol 102 no 31 pp 10870ndash10875 2005
[28] M E Call KWWucherpfennig and J J Chou ldquoThe structuralbasis for intramembrane assembly of an activating immunore-ceptor complexrdquo Nature Immunology vol 11 no 11 pp 1023ndash1029 2010
[29] R M Pielak J R Schnell and J J Chou ldquoMechanism of druginhibition and drug resistance of influenza A M2 channelrdquoProceedings of the National Academy of Sciences of the UnitedStates of America vol 106 no 18 pp 7379ndash7384 2009
[30] J Wang R M Pielak M A McClintock and J J ChouldquoSolution structure and functional analysis of the influenza Bproton channelrdquo Nature Structural and Molecular Biology vol16 no 12 pp 1267ndash1271 2009
[31] K C Chou ldquoCoupling interaction between thromboxane A2receptor and alpha-13 subunit of guanine nucleotide-bindingproteinrdquo Journal of Proteome Research vol 4 no 5 pp 1681ndash1686 2005
[32] K C Chou ldquoInsights from modeling three-dimensional struc-tures of the human potassium and sodium channelsrdquo Journal ofProteome Research vol 3 no 4 pp 856ndash861 2004
[33] K C Chou ldquoSome remarks on protein attribute prediction andpseudo amino acid compositionrdquo Journal ofTheoretical Biologyvol 273 no 1 pp 236ndash247 2011
[34] W Z Lin J A Fang X Xiao and K C Chou ldquoiLoc-animal amulti-label learning classifier for predicting subcellular local-ization of animal proteinsrdquo Molecular BioSystems vol 9 pp634ndash644 2013
[35] X Xiao PWangW Z Lin J H Jia and K C Chou ldquoiAMP-2La two-level multi-label classifier for identifying antimicrobialpeptides and their functional typesrdquo Analytical Biochemistryvol 436 pp 168ndash177 2013
[36] W Chen P M Feng H Lin and K C Chou ldquoiRSpot-PseDNCidentify recombination spots with pseudo dinucleotide compo-sitionrdquo Nucleic Acids Research vol 41 article e69 2013
[37] K C Chou Z C Wu and X Xiao ldquoILoc-Hum using theaccumulation-label scale to predict subcellular locations ofhuman proteins with both single and multiple sitesrdquoMolecularBioSystems vol 8 no 2 pp 629ndash641 2012
[38] M Kotera M Hirakawa T Tokimatsu S Goto and MKanehisa ldquoThe KEGG databases and tools facilitating omicsanalysis latest developments involving human diseases andpharmaceuticalsrdquo Methods in Molecular Biology vol 802 pp19ndash39 2012
[39] Y YamanishiM Araki A GutteridgeWHonda andM Kane-hisa ldquoPrediction of drug-target interaction networks from theintegration of chemical and genomic spacesrdquo Bioinformaticsvol 24 no 13 pp i232ndashi240 2008
[40] P Finn S Muggleton D Page and A Srinivasan ldquoPharma-cophore discovery using the Inductive Logic Programmingsystem PROGOLrdquo Machine Learning vol 30 no 2-3 pp 241ndash270 1998
[41] I Vogt D Stumpfe H E Ahmed and J Bajorath ldquoMethodsfor computer-aided chemical biology Part 2 evaluation of com-pound selectivity using 2D molecular fingerprintsrdquo ChemicalBiology and Drug Design vol 70 pp 195ndash205 2007
[42] H Eckert and J Bajorath ldquoMolecular similarity analysis in vir-tual screening foundations limitations and novel approachesrdquoDrug Discovery Today vol 12 no 5-6 pp 225ndash233 2007
[43] S Laurent L V Elst and R N Muller ldquoComparative studyof the physicochemical properties of six clinical low molecularweight gadolinium contrast agentsrdquo Contrast Media amp Molecu-lar Imaging vol 1 no 3 pp 128ndash137 2006
[44] E Gregori-Puigjane R Garriga-Sust and J Mestres ldquoIndexingmolecules with chemical graph identifiersrdquo Journal of Compu-tational Chemistry vol 32 no 12 pp 2638ndash2646 2011
[45] B Ren ldquoApplication of novel atom-type AI topological indicesto QSPR studies of alkanesrdquo Computers and Chemistry vol 26no 4 pp 357ndash369 2002
[46] N M OrsquoBoyle M Banck C A James C Morley T Vander-meersch and G R Hutchison ldquoOpen Babel an open chemicaltoolboxrdquo Journal of Cheminformatics vol 3 p 33 2011
[47] V J Gillet P Willett and J Bradshaw ldquoSimilarity searchingusing reduced graphsrdquo Journal of Chemical Information andComputer Sciences vol 43 no 2 pp 338ndash345 2003
[48] D Butina ldquoUnsupervised data base clustering based on day-lightrsquos fingerprint and Tanimoto similarity a fast and automatedway to cluster small and large data setsrdquo Journal of ChemicalInformation and Computer Sciences vol 39 no 4 pp 747ndash7501999
[49] C E Shannon ldquoA mathematical theory of communicationrdquoACM SIGMOBILE Mobile Computing and CommunicationsReview vol 5 pp 3ndash55 2001
[50] V D Gusev L A Nemytikova and N A Chuzhanova ldquoOn thecomplexity measures of genetic sequencesrdquo Bioinformatics vol15 no 12 pp 994ndash999 1999
[51] K C Chou and H B Shen ldquoReview recent progress in proteinsubcellular location predictionrdquo Analytical Biochemistry vol370 no 1 pp 1ndash16 2007
[52] S F Altschul ldquoEvaluating the statistical significance of multipledistinct local alignmentsrdquo in Theoretical and ComputationalMethods in Genome Research S Suhai Ed pp 1ndash14 PlenumNew York NY USA 1997
[53] J C Wootton and S Federhen ldquoStatistics of local complexity inamino acid sequences and sequence databasesrdquo Computers andChemistry vol 17 no 2 pp 149ndash163 1993
[54] H Nakashima K Nishikawa and T Ooi ldquoThe folding type ofa protein is relevant to the amino acid compositionrdquo Journal ofBiochemistry vol 99 no 1 pp 153ndash162 1986
[55] K C Chou and C T Zhang ldquoPredicting protein folding typesby distance functions that make allowances for amino acidinteractionsrdquo The Journal of Biological Chemistry vol 269 no35 pp 22014ndash22020 1994
[56] K-C Chou ldquoA novel approach to predicting protein structuralclasses in a (20-1)-D amino acid composition spacerdquo Proteinsvol 21 no 4 pp 319ndash344 1995
[57] H Nakashima and K Nishikawa ldquoDiscrimination of intracel-lular and extracellular proteins using amino acid compositionand residue-pair frequenciesrdquo Journal of Molecular Biology vol238 no 1 pp 54ndash61 1994
12 BioMed Research International
[58] G P Zhou ldquoAn intriguing controversy over protein structuralclass predictionrdquo Journal of Protein Chemistry vol 17 no 8 pp729ndash738 1998
[59] I Bahar A R Atilgan R L Jernigan and B Erman ldquoUnder-standing the recognition of protein structural classes by aminoacid compositionrdquo Proteins vol 29 pp 172ndash185 1997
[60] J Cedano P Aloy J A Perez-Pons and E Querol ldquoRelationbetween amino acid composition and cellular location ofproteinsrdquo Journal of Molecular Biology vol 266 no 3 pp 594ndash600 1997
[61] G P Zhou and K Doctor ldquoSubcellular location prediction ofapoptosis proteinsrdquo Proteins vol 50 no 1 pp 44ndash48 2003
[62] KChou ldquoPrediction of protein cellular attributes using pseudo-amino acid compositionrdquo Proteins vol 43 pp 246ndash255 2001(Erratum in Proteins vol 44 p 60 2001)
[63] K C Chou ldquoUsing amphiphilic pseudo amino acid composi-tion to predict enzyme subfamily classesrdquo Bioinformatics vol21 no 1 pp 10ndash19 2005
[64] D Zou Z He J He and Y Xia ldquoSupersecondary structureprediction using Choursquos pseudo amino acid compositionrdquoJournal of Computational Chemistry vol 32 no 2 pp 271ndash2782011
[65] M M Beigi M Behjati and H Mohabatkar ldquoPrediction ofmetalloproteinase family based on the concept ofChoursquos pseudoamino acid composition using a machine learning approachrdquoJournal of Structural and Functional Genomics vol 12 no 4 pp191ndash197 2011
[66] Y K Chen and K B Li ldquoPredicting membrane protein typesby incorporating protein topology domains signal peptidesand physicochemical properties into the general form of Choursquospseudo amino acid compositionrdquo Journal ofTheoretical Biologyvol 318 pp 1ndash12 2013
[67] C Huang and J Q Yuan ldquoA multilabel model based on Choursquospseudo-amino acid composition for identifying membraneproteins with both single and multiple functional typesrdquo TheJournal of Membrane Biology vol 246 pp 327ndash334 2013
[68] S S Sahu and G Panda ldquoA novel feature representationmethod based on Choursquos pseudo amino acid composition forprotein structural class predictionrdquo Computational Biology andChemistry vol 34 no 5-6 pp 320ndash327 2010
[69] M Hayat and A Khan ldquoDiscriminating outer membraneproteins with fuzzy K-nearest neighbor algorithms based on thegeneral form of Choursquos PseAACrdquo Protein and Peptide Lettersvol 19 no 4 pp 411ndash421 2012
[70] MKhosravian F K FaramarziMM BeigiM Behbahani andH Mohabatkar ldquoPredicting antibacterial peptides by the con-cept of Choursquos pseudo-amino acid composition and machinelearning methodsrdquo Protein amp Peptide Letters vol 20 pp 180ndash186 2013
[71] HMohabatkarMM Beigi K Abdolahi and SMohsenzadehldquoPrediction of allergenic proteins by means of the concept ofChoursquos pseudo amino acid composition and amachine learningapproachrdquoMedicinal Chemistry vol 9 pp 133ndash137 2013
[72] L Nanni A Lumini D Gupta and A Garg ldquoIdentifyingbacterial virulent proteins by fusing a set of classifiers basedon variants of Choursquos pseudo amino acid composition and onevolutionary informationrdquo IEEEACM Transactions on Compu-tational Biology and Bioinformatics vol 9 no 2 pp 467ndash4752012
[73] T H Chang L C Wu T Y Lee S P Chen H D Huang andJ T Horng ldquoEuLoc a web-server for accurately predict protein
subcellular localization in eukaryotes by incorporating variousfeatures of sequence segments into the general form of ChoursquosPseAACrdquo Journal of Computer-Aided Molecular Design vol 27pp 91ndash103 2013
[74] S Zhang Y Zhang H Yang C Zhao and Q Pan ldquoUsing theconcept of Choursquos pseudo amino acid composition to predictprotein subcellular localization an approach by incorporatingevolutionary information and von Neumann entropiesrdquo AminoAcids vol 34 no 4 pp 565ndash572 2008
[75] R Zia Ur and A Khan ldquoIdentifying GPCRs and their typeswith Choursquos pseudo amino acid composition an approach frommulti-scale energy representation and position specific scoringmatrixrdquo Protein amp Peptide Letters vol 19 pp 890ndash903 2012
[76] X Y Sun S P Shi J D Qiu S B Suo S Y Huang and R PLiang ldquoIdentifying protein quaternary structural attributes byincorporating physicochemical properties into the general formof Choursquos PseAAC via discrete wavelet transformrdquo MolecularBioSystems vol 8 pp 3178ndash3184 2012
[77] L Nanni and A Lumini ldquoGenetic programming for creatingChoursquos pseudo amino acid based features for submitochondrialocalizationrdquo Amino Acids vol 34 no 4 pp 653ndash660 2008
[78] M Esmaeili H Mohabatkar and S Mohsenzadeh ldquoUsing theconcept of Choursquos pseudo amino acid composition for risk typeprediction of human papillomavirusesrdquo Journal of TheoreticalBiology vol 263 no 2 pp 203ndash209 2010
[79] H Mohabatkar ldquoPrediction of cyclin proteins using Choursquospseudo amino acid compositionrdquo Protein and Peptide Lettersvol 17 no 10 pp 1207ndash1214 2010
[80] H Mohabatkar M M Beigi and A Esmaeili ldquoPredictionof GABA(A) receptor proteins using the concept of Choursquospseudo-amino acid composition and support vector machinerdquoJournal of Theoretical Biology vol 281 no 1 pp 18ndash23 2011
[81] Y Xu J Ding L Y Wu and K C Chou ldquoiSNO-PseAAC pre-dict cysteine S-nitrosylation sites in proteins by incorporatingposition specific amino acid propensity into pseudo amino acidcompositionrdquo PLoS ONE vol 8 Article ID e55844 2013
[82] W Chen H Lin PM Feng C Ding Y C Zuo andK C ChouldquoiNuc-PhysChem a sequence-based predictor for identifyingnucleosomes via physicochemical propertiesrdquo PLoS ONE vol7 Article ID e47843 2012
[83] B Li T Huang L Liu Y Cai and K C Chou ldquoIdentificationof colorectal cancer related genes with mrmr and shortest pathin protein-protein interaction networkrdquo PLoS ONE vol 7 no 4Article ID e33393 2012
[84] Y Jiang T Huang C Lei Y F Gao Y D Cai and K C ChouldquoSignal propagation in protein interaction network duringcolorectal cancer progressionrdquo BioMed Research Internationalvol 2013 Article ID 287019 9 pages 2013
[85] P Du X Wang C Xu and Y Gao ldquoPseAAC-Builder across-platform stand-alone program for generating variousspecial Choursquos pseudo-amino acid compositionsrdquo AnalyticalBiochemistry vol 425 no 2 pp 117ndash119 2012
[86] D S Cao Q S Xu and Y Z Liang ldquoPropy a tool to generatevarious modes of Choursquos PseAACrdquo Bioinformatics vol 29 pp960ndash962 2013
[87] H Shen and K C Chou ldquoPseAAC a flexible web serverfor generating various kinds of protein pseudo amino acidcompositionrdquo Analytical Biochemistry vol 373 no 2 pp 386ndash388 2008
[88] K C Chou ldquoUsing pair-coupled amino acid composition topredict protein secondary structure contentrdquo Journal of ProteinChemistry vol 18 no 4 pp 473ndash480 1999
BioMed Research International 13
[89] W Liu and K C Chou ldquoPrediction of protein secondarystructure contentrdquo Protein Engineering vol 12 no 12 pp 1041ndash1050 1999
[90] M Bhasin and G P S Raghava ldquoClassification of nuclearreceptors based on amino acid composition and dipeptidecompositionrdquo The Journal of Biological Chemistry vol 279 no22 pp 23262ndash23266 2004
[91] H Lin andH Ding ldquoPredicting ion channels and their types bythe dipeptidemode of pseudo amino acid compositionrdquo Journalof Theoretical Biology vol 269 no 1 pp 64ndash69 2011
[92] H Lin and Q Li ldquoUsing pseudo amino acid composition topredict protein structural class approached by incorporating400 dipeptide componentsrdquo Journal of Computational Chem-istry vol 28 no 9 pp 1463ndash1466 2007
[93] L Nanni and A Lumini ldquoCombing ontologies and dipeptidecomposition for predicting DNA-binding proteinsrdquo AminoAcids vol 34 no 4 pp 635ndash641 2008
[94] K-C Chou ldquoThe convergence-divergence duality in lectindomains of selectin family and its implicationsrdquo FEBS Lettersvol 363 no 1-2 pp 123ndash126 1995
[95] A A Schaffer L Aravind T L Madden et al ldquoImprovingthe accuracy of PSI-BLAST protein database searches withcomposition-based statistics and other refinementsrdquo NucleicAcids Research vol 29 no 14 pp 2994ndash3005 2001
[96] S F Altschul and E V Koonin ldquoIterated profile searches withPSI-BLAST a tool for discovery in protein databasesrdquo Trends inBiochemical Sciences vol 23 no 11 pp 444ndash447 1998
[97] J Deng ldquoGrey entropy and grey target decision makingrdquoJournal of Grey System vol 22 no 1 pp 1ndash24 2010
[98] B M Bolstad R A Irizarry M Astrand and T P Speed ldquoAcomparison of normalizationmethods for high density oligonu-cleotide array data based on variance and biasrdquo Bioinformaticsvol 19 no 2 pp 185ndash193 2003
[99] J-Y Shi S-W Zhang Q Pan Y-M Cheng and J XieldquoPrediction of protein subcellular localization by support vectormachines using multi-scale energy and pseudo amino acidcompositionrdquo Amino Acids vol 33 no 1 pp 69ndash74 2007
[100] T Denoeux ldquoA 120581-nearest neighbor classification rule based onDempster-Shafer theoryrdquo IEEE Transactions on Systems Manand Cybernetics vol 25 no 5 pp 804ndash813 1995
[101] J M Keller M R Gray and J A Givens ldquoA fuzzy k-nearestneighbours algorithmrdquo IEEE Transactions on Systems Man andCybernetics vol 15 no 4 pp 580ndash585 1985
[102] X Xiao P Wang and K Chou ldquoiNR-physchem a sequence-based predictor for identifying nuclear receptors and theirsubfamilies via physical-chemical property matrixrdquo PLoS ONEvol 7 no 2 Article ID e30869 2012
[103] I Roterman L Konieczny W Jurkowski K Prymula and MBanach ldquoTwo-intermediate model to characterize the structureof fast-folding proteinsrdquo Journal of Theoretical Biology vol 283no 1 pp 60ndash70 2011
[104] X Xiao P Wang and K C Chou ldquoGPCR-2L predicting Gprotein-coupled receptors and their types by hybridizing twodifferentmodes of pseudo amino acid compositionsrdquoMolecularBioSystems vol 7 no 3 pp 911ndash919 2011
[105] X Xiao P Wang and K C Chou ldquoQuat-2L a web-server forpredicting protein quaternary structural attributesrdquo MolecularDiversity vol 15 no 1 pp 149ndash155 2011
[106] X Zheng C Li and J Wang ldquoAn information-theoreticapproach to the prediction of protein structural classrdquo Journalof Computational Chemistry vol 31 no 6 pp 1201ndash1206 2010
[107] H Shen J Yang X Liu and K C Chou ldquoUsing supervisedfuzzy clustering to predict protein structural classesrdquo Biochem-ical and Biophysical Research Communications vol 334 no 2pp 577ndash581 2005
[108] K-C Chou and C-T Zhang ldquoReview prediction of proteinstructural classesrdquo Critical Reviews in Biochemistry and Molec-ular Biology vol 30 no 4 pp 275ndash349 1995
[109] P C Mahalanobis ldquoOn the generalized distance in statisticsrdquoProceedings of the National Institute of Sciences of India vol 2pp 49ndash55 1936
[110] R M Centor ldquoSignal detectability the use of ROC curves andtheir analysesrdquoMedical Decision Making vol 11 no 2 pp 102ndash106 1991
[111] K-C Chou ldquoUsing subsite coupling to predict signal peptidesrdquoProtein Engineering vol 14 no 2 pp 75ndash79 2001
[112] K C Chou ldquoPrediction of protein signal sequences and theircleavage sitesrdquo Proteins vol 42 pp 136ndash139 2001
[113] K C Chou ldquoPrediction of signal peptides using scaledwindowrdquoPeptides vol 22 no 12 pp 1973ndash1979 2001
[114] K C Chou ldquoSome remarks on predicting multi-label attributesinmolecular biosystemsrdquoMolecular Biosystems vol 9 pp 1092ndash1100 2013
[115] K C Chou and H B Shen ldquoCell-PLoc 2 0 an improvedpackage of web-servers for predicting subcellular localization ofproteins in various organismsrdquoNatural Science vol 2 pp 1090ndash1103 2010
[116] M Gribskov and N L Robinson ldquoUse of receiver operatingcharacteristic (ROC) analysis to evaluate sequence matchingrdquoComputers and Chemistry vol 20 no 1 pp 25ndash33 1996
[117] Y Yamanishi M Kotera M Kanehisa and S Goto ldquoDrug-target interaction prediction from chemical genomic and phar-macological data in an integrated frameworkrdquo Bioinformaticsvol 26 no 12 pp i246ndashi254 2010
[118] Z He J Zhang X Shi et al ldquoPredicting drug-target interactionnetworks based on functional groups and biological featuresrdquoPLoS ONE vol 5 no 3 Article ID e9603 2010
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

4 BioMed Research International
one if the string is not terminated by a dot) For example forthe sequence 100100011010010 syn(119875) and the correspondingcomplexity factor CF are described as
Syn (119878) = 1 ∙ 0 ∙ 01 ∙ 000 ∙ 11 ∙ 01001 ∙ 0
CF = 7(7)
Thus by adding the information entropy 119867119909(4) and com-
plexity factor CF (7) into the molecular fingerprint MF (2)we obtained a total of (256 + 1 + 1) = 258 feature elements torepresent a drug compound that is it can now be formulatedas a 258-D vector given by
119863 = [11986011198602sdot sdot sdot 119860
256119867119909
CF]T (8)
where 119860119894has the same meaning as in (2) while 119867
119909and CF
are the information entropy and complexity factor respec-tively as described in the previous two sections
222 Enzyme The sequences of the enzymes involved inthis study are given in Online Supporting Information S2Now the problem is how to effectively represent these enzymesequences for the current study Generally speaking thereare two kinds of approaches to formulate enzyme sequencesthe sequential model and the nonsequential or discretemodel [51] The most typical sequential representation foran enzyme sample E with 119871 residues is its entire amino acidsequence that is
E = 1198771119877211987731198774119877511987761198777sdot sdot sdot 119877119871 (9)
where 1198771represents the 1st residue 119877
2the 2nd residue and
so forth An enzyme sample thus formulated can contain itsmost complete information This is an obvious advantage ofthe sequential representation To get the desired results thesequence-similarity-search-based tools such as BLAST [5253] are usually utilized to conduct the prediction Howeverthis kind of approach failed to work when the query enzymedid not have significant homology to enzyme of known char-acters Thus various nonsequential representation modelswere proposed The simplest nonsequential model for anenzyme was based on its amino acid composition (AAC) asdefined by
E = [11989111198912sdot sdot sdot 11989120]
T (10)
where 119891119906(119906 = 1 2 20) are the normalized occurrence
frequencies of the 20 native amino acids [54ndash56] in theenzyme E and T has the same meaning as in (2) and (8)The AAC-discrete model was widely used for identifyingvarious attributes of proteins (see eg [57ndash61]) Howeveras can be seen from (10) all the sequence order effectswere lost by using the AAC-discrete model This is its mainshortcoming To avoid completely losing the sequence-orderinformation the pseudo amino acid composition [62 63]or Choursquos PseAAC [3] was proposed to replace the simpleAAC model Since the concept of PseAAC was proposedin 2001 [62] it has penetrated into almost all the fields of
protein attribute predictions and computational proteomicssuch as predicting supersecondary structure [64] predictingmetalloproteinase family [65] predicting membrane pro-tein types [66 67] predicting protein structural class [68]discriminating outer membrane proteins [69] identifyingantibacterial peptides [70] identifying allergenic proteins[71] identifying bacterial virulent proteins [72] predictingprotein subcellular location [73 74] identifying GPCRs andtheir types [75] identifying protein quaternary structuralattributes [76] predicting protein submitochondria locations[77] identifying risk type of human papillomaviruses [78]identifying cyclin proteins [79] predicting GABA(A) recep-tor proteins [80] and predicting cysteine S-nitrosylation sitesin proteins [81] among many others (see a long list of paperscited in the References section of [33]) Recently the conceptof PseAAC was further extended to represent the featurevectors of DNA and nucleotides [36 82] as well as otherbiological samples (see eg [83 84]) Because it has beenwidely and increasingly used recently two powerful soft-wares called ldquoPseAAC-Builderrdquo [85] and ldquopropyrdquo [86] wereestablished for generating various special Choursquos pseudo-amino acid compositions in addition to the web-serverPseAAC [87] built in 2008 According to a recent review [33]the general formof Choursquos PseAAC for an enzyme sample canbe formulated by
E = [12059511205952sdot sdot sdot 120595119906sdot sdot sdot 120595Ω]
T (11)
where the subscript Ω is an integer and its value as wellas the components 120595
119906(119906 = 1 2 Ω) will depend on
how to extract the desired information from the amino acidsequence of E (cf (10)) Next let us describe how to extractuseful information from the benchmark datasetS andOnlineSupporting Information S2 to define the enzyme samplesconcerned via (11)
To incorporate as much useful information as possiblefrom an enzyme sample we are to approach this problemfrom three different angles followed by incorporating thefeature elements thus obtained into the general form ofPseAAC of (11)(a) Amino Acid Composition The components of amino acidcomposition have beenwidely used to predict various proteinattributes [57ndash61] In this study they were also included as thefirst 20 elements in the general Choursquos PseAAC (cf (11)) thatis
120595119906= 119891119906
(119906 = 1 2 20) (12)
where 119891119906has the same meaning as in (10)
(b) Dipeptide Composition Dipeptide composition has beenused to predict the protein secondary structural contents[88 89] as well as various protein attributes (see eg [90ndash93]) The number of different dipeptides is 20 times 20 = 400Suppose that the normalized occurrence frequencies of the400 dipeptides in an enzyme sample are given by
119891
(2)
119906(119906 = 1 2 400) (13)
BioMed Research International 5
Incorporating the above 400 dipeptide components into (11)we have
120595119906+20
= 119891
(2)
119906(119906 = 1 2 400) (14)
(c) Sequential Evolution Information Biology is a naturalscience with a historic dimension All biological specieshave developed starting out from a very limited number ofancestral species Their evolution involves changes of singleresidues insertions and deletions of several residues [94]gene doubling and gene fusion With these changes accu-mulated for a long period of time many similarities betweeninitial and resultant amino acid sequences are graduallyeliminated but the corresponding proteins may still sharemany common attributes [18] such as having basically thesame biological function and residing at a same subcellularlocation To extract the sequential evolution information anduse it to define the components of (11) the PSSM (PositionSpecific Scoring Matrix) was used as described next
According to Schaffer et al [95] the sequence evolutioninformation of enzyme E with 119871 amino acid residues can beexpressed by an 119871 times 20matrix as given by
P(0)PSSM =
[
[
[
[
[
[
[
119864
0
1rarr1119864
0
1rarr2sdot sdot sdot 119864
0
1rarr20
119864
0
2rarr1119864
0
2rarr2sdot sdot sdot 119864
0
2rarr20
119864
0
119871rarr1119864
0
119871rarr2sdot sdot sdot 119864
0
119871rarr20
]
]
]
]
]
]
]
(15)
where 1198640119894rarr 119895
represents the original score of the 119894th aminoacid residue (119894 = 1 2 119871) in the enzyme sequence changedto amino acid type 119895 (119895 = 1 2 20) in the process ofevolution Here the numerical codes 1 2 20 are used torepresent the 20 native amino acid types denoted by A CD E F G H I K L M N P Q R S T V W and Y The119871 times 20 scores in (15) were generated by using PSI-BLAST[96] to search the UniProtKBSwiss-Prot database (Release2013-05) through three iterations with 0001 as the 119864 valuecutoff for multiple sequence alignment against the sequenceof the enzyme E In order to make every element in (15) bescaled from their original score ranges into region of [0 1]we performed a conversion through the standard sigmoidfunction to make it become
P(1)PSSM =
[
[
[
[
[
[
[
119864
1
1rarr1119864
1
1rarr2sdot sdot sdot 119864
1
1rarr20
119864
1
2rarr1119864
1
2rarr2sdot sdot sdot 119864
1
2rarr20
119864
1
119871rarr1119864
1
119871rarr2sdot sdot sdot 119864
1
119871rarr20
]
]
]
]
]
]
]
(16)
where
119864
1
119894rarr 119895=
1
1 + 119890
minus1198640
119894rarr 119895
(1 le 119894 le 119871 1 le 119895 le 20) (17)
Nowwe extract the useful information from (16) to define thecomponents of (11) via the following approach
120595119906+420
= ℓ119906
(119906 = 1 2 20) (18)
where
ℓ119895=
1
119871
times
119871
sum
119896=1
119864
1
119896rarr119895(119895 = 1 2 20) (19)
(d) Grey System Model Approach The grey system theory[97] is quite useful in dealing with complicated systems thatlack sufficient information or need to process uncertaininformation According to the grey system theory we canextract the following information from the 119895th column of(16) that is
[
119886
119895
1
119886
119895
2
] = (BT119895B119895)
minus1
BT119895U119895
(119895 = 1 2 20) (20)
where
B119895=
[
[
[
[
[
[
[
[
[
[
minus119864
1
2rarr119895minus119864
1
1rarr119895minus 05119864
1
2rarr1198951
minus119864
1
3rarr119895minus
2
sum
119894=1
119864
1
119894rarr 119895minus 05119864
1
3rarr1198951
minus119864
1
119871rarr119895minus
119871minus1
sum
119894=1
119864
1
119894rarr 119895minus 05119864
1
119871rarr1198951
]
]
]
]
]
]
]
]
]
]
U119895=
[
[
[
[
[
[
119864
1
2rarr119895minus 119864
1
1rarr119895
119864
1
3rarr119895minus 119864
1
2rarr119895
119864
1
119871rarr119895minus 119864
1
119871minus1rarr119895
]
]
]
]
]
]
(21)
Therefore based on the grey system theory and (20) we canextract another 20 times 2 = 40 quantities from (16) to definethe components of (11) that is
120593119895=
1199081119886
119895
1when 119895 is an odd number
1199082119886
119895
2when 119895 is an even number
1 le 119895 le 20
(22)
where 1198861198951and 119886119895
2are given by (20) 119908
1and 119908
2are weight
factors which were all set to 1 in the current studySubstituting the elements in (12) (14) (18) and (22) we
finally obtain a total of Ω = 20 + 400 + 20 + 40 = 480
components for the PseAAC of (11) where
120595119906=
119891119906
when 1 le 119906 le 20119891
(2)
119906when 21 le 119906 le 420
ℓ119906
when 421 le 119906 le 440120593119906
when 440 le 119906 le 480
(23)
In other words in this study (11) or Choursquos PseAAC is a 480-D vector whose 480 components are given by (23) derivedfrom the amino acid composition dipeptide compositionsequential evolution information and grey system theory(e) Representing Enzyme-Drug Pairs Now the pair betweenan enzyme molecule E and a drug compound D can beformulated by combing (8) and (11) as given by
G = D oplus E
= [1198601sdot sdot sdot 119860
256119867119909
CF 1205951sdot sdot sdot 120595480]
T
(24)
6 BioMed Research International
where G represents the enzyme-drug pair oplus the orthogonalsum [51] and each of the (258 + 480) = 738 feature elementsis given in (8) and (23)
For the convenience of the later formulation let us use119909119894(119894 = 1 2 738) to represent the 738 components of (24)
that is
G = [11990911199092sdot sdot sdot 119909119894sdot sdot sdot 119909738]
T (25)
To optimize the prediction results different weights wereusually tested for each of the elements in (25) However sinceit would consume a lot of computational time for a total of 738weight factors here let us adopt the normalization approachto deal with this problem as done in [98 99] that is convert119909119894in (25) to 119910
119894according to the following equation
119910119894=
2tanminus1 (119909119894)
120587
(119894 = 1 2 738) (26)
where tanminus1 means arctangent By means of (26) everycomponent in (25) will be converted into the range of[minus1 1] that is we have minus1 le 119910
119894le 1 As demonstrated
in [98 99] the normalization approach via (26) was quiteeffective in enhancing the quality of prediction operated ina high dimension space Therefore in this study we wouldnot to take the procedure of optimizing the weight factorssignificantly reducing the computational times
23 Fuzzy 119870-Nearest Neighbour Algorithm The 119870-NN (119870-Nearest Neighbor) classifier is quite popular in patternrecognition community owing to its good performance andsimple-to-use feature According to the 119870-NN rule [100]named also as the ldquovoting 119870-NN rulerdquo the query sampleshould be assigned to the subset represented by a majorityof its 119870 nearest neighbors as illustrated in Figure 5 of [33]
Fuzzy 119870-NN classification method [101] is a specialvariation of the119870-NNclassification family Instead of roughlyassigning the label based on a voting from the 119870 nearestneighbors it attempts to estimate the membership valuesthat indicate how much degree the query sample belongsto the classes concerned Obviously it is impossible for anycharacteristic description to contain complete informationwhich would make the classification ambiguous In view ofthis the fuzzy principle is very reasonable and particularlyuseful in dealing with complicated biological systems such asidentifying nuclear receptor subfamilies [102] characterizingthe structure of fast-folding proteins [103] classifying Gprotein-coupled receptors [104] predicting protein quater-nary structural attributes [105] predicting protein structuralclasses [106 107] and so forth
Next let us give a brief introduction how to use thefuzzy119870-NN approach to identify the interactions between theenzymes and the drug compounds in the network concerned
Supposing that S(119873) = G1G2 G
119873 is a set of vec-
tors representing 119873 enzyme-drug pairs in a training set
classified into two classes 119862+ 119862minus where 119862+ denotes theinteractive pair class while 119862minus the noninteractive pair classSlowast(G) = Glowast
1Glowast2 Glowast
119870 sub S(119873) is the subset of the 119870
nearest neighbor pairs to the query pair G Thus the fuzzymembership value for the query pair G in the two classes ofS(119873) is given by
120583
+
(G) =sum
119870
119895=1120583
+(Glowast119895) 119889(GGlowast
119895)
minus2(120593minus1)
sum
119870
119895=1119889(GGlowast
119895)
minus2(120593minus1)
120583
minus
(G) =sum
119870
119895=1120583
minus(Glowast119895) 119889(GGlowast
119895)
minus2(120593minus1)
sum
119870
119895=1119889(GGlowast
119895)
minus2(120593minus1)
(27)
where 119870 is the number of the nearest neighbors counted forthe query pairG 120583+(Glowast
119895) and 120583minus(Glowast
119895) the fuzzy membership
values of the training sample Glowast119895to the class 119862+ and 119862minus
respectively as will be further defined next 119889(GGlowast119895) the
cosine distance between G and its 119895th nearest pair Glowast119895in
the training dataset S(119873) 120593(gt 1) the fuzzy coefficient fordetermining how heavily the distance is weighted whencalculating each nearest neighborrsquos contribution to the mem-bership value Note that the parameters 119870 and 120593 will affectthe computation result of (27) and they will be optimizedby a grid-search as will be described later Also variousother metrics can be chosen for 119889(GGlowast
119895) such as Euclidean
distance Hamming distance [108] andMahalanobis distance[55 109]
The quantitative definitions for the aforementioned120583
+(Glowast119895) and 120583minus(Glowast
119895) in (27) are given by
120583
+
(Glowast119895) =
1 if Glowast119895isin 119862
+
0 otherwise
120583
minus
(Glowast119895) =
1 if Glowast119895isin 119862
minus
0 otherwise
(28)
Substituting the results obtained by (27) into (28) it followsthat if 120583+(G) gt 120583
minus(G) then the query pair G is an
interactive couple otherwise noninteractive In other wordsthe outcome can be formulated as
G isin
119862
+ if 120583+ (G) gt 120583minus (G)
119862
minus otherwise
(29)
If there is a tie between 120583+(G) and 120583minus(G) the query pair Gwill be randomly assigned to one of the two classes Howevercase like that is quite rare and in this study never happened
The predictor thus established is called iEzy-Drugwhere ldquoirdquo means identify and ldquoEzy-Drugrdquo means the inter-action between enzyme and drug To provide an intuitiveoverall picture a flowchart is provided in Figure 2 to showthe process of how the classifier works in identifying enzyme-drug interactions
BioMed Research International 7
Input a queryprotein sequence and
a drug code
Create Chous PseAAC of a protein
Create feature vector of a drug
Feature fusion
engineTraining dataset
Yes No
Noninteractive enzyme drug pair
Interactive enzymedrug pair
AACDipeptideSeq Evol
Grey model
Mol FptInfo entropy
CF
FKNN operation
Figure 2 A flowchart to show the operation process of the iEzy-Drug predictor See the text for further explanation
24 Criteria for Performance Evaluation In the literaturethe following equation set is often used for examining theperformance quality of a predictor
Sn = TPTP + FN
Sp = TNTN + FP
Acc = TP + TNTP + TN + FP + FN
MCC = (TP times TN) minus (FP times FN)radic(TP + FP) (TP + FN) (TN + FP) (TN + FN)
(30)
where TP represents the true positive TN the true negativeFP the false positive FN the false negative Sn the sensitiv-ity Sp the specificity Acc the accuracy MCC the Mathewrsquoscorrelation coefficient
To most biologists however the four metrics as formu-lated in (30) are not quite intuitive and easier-to-understandparticularly for the Mathewrsquos correlation coefficient Herelet us adopt the Choursquos symbols to formulate the previousfour metrics By means of Choursquos symbols [111 112] the rates
of correct predictions for the interactive enzyme-drug pairsin dataset S+ and the noninteractive enzyme-drug pairs indataset Sminus are respectively defined by (cf (1))
Λ
+
=
119873
+minus 119873
+
minus
119873
+ for the interactive enzyme-drug pairs
Λ
minus
=
119873
minusminus 119873
minus
+
119873
minus for the noninteractive enzyme-drug pairs
(31)
where 119873+ is the total number of the interactive enzyme-drug pairs investigated while 119873+
minusis the number of the
interactive enzyme-drug pairs incorrectly predicted as thenoninteractive enzyme-drug pairs 119873minus is the total numberof the noninteractive enzyme-drug pairs investigated while119873
minus
+is the number of the noninteractive enzyme-drug pairs
incorrectly predicted as the interactive enzyme-drug pairsThe overall success prediction rate is given by [113] as follows
Λ =
Λ
+119873
++ Λ
minus119873
minus
119873
++ 119873
minus= 1 minus
119873
+
minus+ 119873
minus
+
119873
++ 119873
minus
(32)
It is obvious from (31)-(32) that if and only if none ofthe interactive enzyme-drug pairs and the noninteractiveenzyme-drug pairs are mispredicted that is 119873+
minus= 119873
minus
+= 0
and Λ+ = Λ
minus= 1 we have the overall success rate Λ = 1
Otherwise the overall success rate would be smaller than 1The relations between the symbols in (32) and those in
(30) are given by
TP = 119873+ minus 119873+minus
TN = 119873
minus
minus 119873
minus
+
FP = 119873minus+
FN = 119873
+
minus
(33)
Substituting (33) into (30) and also noting (31)-(32) we obtain
Sn = Λ+ = 1 minus119873
+
minus
119873
+
Sp = Λminus = 1 minus119873
minus
+
119873
minus
Acc = Λ = 1 minus119873
+
minus+ 119873
minus
+
119873
++ 119873
minus
MCC =1 minus ((119873
+
minus119873
+) + (119873
minus
+119873
minus))
radic(1 + (119873
minus
+minus 119873
+
minus) 119873
+) (1 + (119873
+
minusminus 119873
minus
+) 119873
minus)
(34)
Now it is obvious to see from (34) when 119873
+
minus= 0
meaning none of the interactive enzyme-drug pairs was
8 BioMed Research International
02
46
810
1
15
2089
0895
09
0905
091
0915
092
ACC
K120593
Figure 3 A 3D plot to show how the parameter in (27) wasoptimized for the iEzy-Drug predictor
mispredicted to be a noninteractive enzyme-drug pair wehave the sensitivity Sn = 1 while 119873+
minus= 119873
+ meaning thatall the interactive enzyme-drug pairs weremispredicted to bethe noninteractive enzyme-drug pairs we have the sensitivitySn = 0 Likewise when 119873minus
+= 0 meaning none of the
noninteractive enzyme-drug pairs wasmispredicted we havethe specificity Sp = 1 while 119873minus
+= 119873
minus meaning all thenoninteractive enzyme-drug pairs were incorrectly predictedas interactive enzyme-drug pairs we have the specificity Sp =0 When119873+
minus= 119873
minus
+= 0 meaning that none of the interactive
enzyme-drug pairs in the dataset S+ and none of the nonin-teractive enzyme-drug pairs in Sminus was incorrectly predictedwe have the overall accuracy Acc = Λ = 1 while 119873+
minus= 119873
+
and 119873minus+= 119873
minus meaning that all the interactive enzyme-drugpairs in the dataset S+ and all the noninteractive enzyme-drug pairs in Sminus were mispredicted we have the overallaccuracy Acc = Λ = 0 The MCC correlation coefficient isusually used for measuring the quality of binary (two-class)classifications When 119873+
minus= 119873
minus
+= 0 meaning that none
of the interactive enzyme-drug pairs in the dataset S+ andnone of the noninteractive enzyme-drug pairs in Sminus weremispredicted we have MCC = 1 when 119873+
minus= 119873
+2 and
119873
minus
+= 119873
minus2 we have MCC = 0 meaning no better than
random prediction when 119873+minus= 119873
+ and 119873minus+= 119873
minus we haveMCC = minus1 meaning total disagreement between predictionand observation As we can see from the previous discussionit is much more intuitive and easier-to-understand whenusing (34) to examine a predictor for its sensitivity specificityoverall accuracy and Mathewrsquos correlation coefficient It isinstructive to point out that themetrics as defined in (30) and(34) are valid for single label systems for multilabel systemsa set of more complicated metrics should be used as given in[114]
3 Results and Discussion
31 Cross-Validation How to properly examine the predic-tion quality is a key for developing a new predictor and
False positive rate
True
pos
itive
rate
1
01
02
03
04
05
06
07
08
09
0
101 02 03 04 05 06 07 08 090
BaselineiEzy-Drug AUC = 09377
Figure 4 A plot for the ROC curve to quantitatively show theperformance of the iEzy-Drug predictor
estimating its potential application value Generally speak-ing the following three cross-validation methods are oftenused to examine a predictor of its effectiveness in practicalapplication independent dataset test subsampling or 119870-fold(such as 5-fold 7-fold or 10-fold) test and jackknife test[108] However as elaborated by a penetrating analysis in[115] considerable arbitrariness exists in the independentdataset test Also as demonstrated by (27)ndash(29) in [33]the subsampling test (or 119870-fold cross-validation) cannotavoid arbitrariness either Only the jackknife test is the leastarbitrary that can always yield a unique result for a givenbenchmark dataset Therefore the jackknife test has beenwidely recognized and increasingly utilized by investigatorsto examine the quality of various predictors (see eg [66 7174 80]) Accordingly the success rate by the jackknife test wasalso used to optimize the two uncertain parameters 119870 and 120593in (27) The result thus obtained is shown in Figure 3 fromwhich we obtain when 119870 = 6 and 120593 = 15 the iEzy-Drugpredictor reaches its optimized status
The success rates thus obtained by the jackknife test inidentifying interactive Enzyme-drug pairs or noninteractiveenzyme-drug pairs on the benchmark dataset S (cf OnlineSupporting Information S1) are given in Table 1 where forfacilitating comparison the corresponding result by He et al[110] is also given As we can see from the table the overallaccuracy Acc achieved by iEzy-Drug was 9103 remarkablyhigher than 8548 the corresponding rate obtained byHe et al [110] on the same benchmark Furthermore listedin Table 1 are also the values obtained by iEzy-Drug for theother three metrics that is Sn = 9081 Sp = 9114 andMCC = 8039 indicating that the accuracy of iEzy-Drug isnot only very high but also quite stale
To provide a graphical illustration to show the per-formance of the current binary classifier iEzy-Drug asits discrimination threshold is varied a 2D plot calledReceiver Operating Characteristic (ROC) curve [116 117]was also given (Figure 4) In the ROC curve the verticalcoordinate 119884 is for the true positive rate or Sn (cf (34))while horizontal coordinate 119883 for the false positive rate
BioMed Research International 9
Read Me Data Citation
Enter both the protein sequence and the drug code (example) the number of query pairs is limited at 10 or less for each submission
Submit Clear
You can download the program and run it on your own local computer
iEzy-Drug identifying the interaction between enzymes anddrugs in cellular networking
Figure 5 A semiscreenshot to show the top page of the iEzy-Drugweb-server Its web-site address is at httpwwwjci-bioinfocniEzy-Drug
Table 1 The jackknife success rates obtained with iEzy-Drug in identifying interactive enzyme-drug pairs and noninteractive enzyme-drugpairs for the benchmark dataset S (cf Online Supporting Information S1)
Method Acc Sn Sp MCC
iEzy-Druga 74258157 = 9103 24692719 = 9081 49565438 = 9114 8039NN predictorb 8548 NA NA NAaSee (27) where the parameters119870 = 6 and 120593 = 15bSee [110]
or 1-Sp The best possible prediction method would yielda point with the coordinate (0 1) representing 100 truepositive rate (sensitivity Sn) and 0 false positive rate or100 specificity Therefore the (0 1) point is also calleda perfect classification A completely random guess wouldgive a point along a diagonal from the point (0 0) to (1 1)The area under the ROC curve also called Area Under theROC (AUROC) is often used to indicate the performancequality of a binary classifier the value 05 of AUROCis equivalent to random prediction while 1 of AUROCrepresents a perfect one As we can see from Figure 4the AUROC value obtained by iEzy-Drug is 09377
The reason why iEzy-Drug can remarkably improve theprediction quality is that it has introduced the 2D molecularfingerprints to represent drug samples see Online SupportingInformation S3 for the detailed fingerprint expressions for thedrugs listed in Online Supporting Information S1 and thatit has successfully used PseAAC to incorporate the featuresderived from the sequences of enzymes that are essentialfor identifying the interaction of enzymes with drugs in thecellular networking
To enhance the value of its practical applications theweb server for iEzy-Drug has been established that canbe freely accessible at httpwwwjci-bioinfocniEzy-DrugIt is anticipated that the web server will become a usefulhigh throughput tool for both basic research and drug
development in the relevant areas or at the very least playa complementary role to the existing method [39 110 118] forwhich so far noweb-serverwhatsoever has been provided yet
32 The Protocol or User Guide For the convenience of thevast majority of biologists and pharmaceutical scientists herelet us provide a step-by-step guide to show how the userscan easily get the desired result by means of the web serverwithout the need to follow the complicated mathematicalequations presented in this paper for the process of develop-ing the predictor and its integrityStep 1 Open the web server at the site httpwwwjci-bio-infocniEzy-Drug and you will see the top page of thepredictor on your computer screen as shown in Figure 5Click on the ReadMe button to see a brief introduction aboutiEzy-Drug predictor and the caveat when using itStep 2 Either type or copypaste the query pairs into theinput box at the center of Figure 5 Each query pair consistsof two parts one is for the protein sequence and the other forthe drug The enzyme sequence should be in FASTA formatwhile the drug in the KEGG code Examples for the querypairs input can be seen by clicking on the Example buttonright above the input boxStep 3 Click on the Submit button to see the predicted resultFor example if you use the four query pairs in the Example
10 BioMed Research International
window as the input after clicking the Submit button youwill see on your screen that the ldquohsa 10056rdquo enzyme andthe ldquoD0021rdquo drug are an interactive pair and that the ldquohsa100rdquo enzyme and the ldquoD0037rdquo drug are also an interactivepair but that the ldquohsa 3295rdquo enzyme and the ldquoD00889rdquo drugare not an interactive pair and that the ldquohsa 7366rdquo enzymeand the ldquoD03601rdquo drug are not an interactive pair eitherAll these results are fully consistent with the experimentalobservations It takes about 3 minutes before the results areshown on the screenStep 4 Click on the Citation button to find the relevant paperthat documents the detailed development and algorithm ofiEzy-DurgStep 5 Click on the Data button to download the benchmarkdataset used to train and test the iEzy-Durg predictorStep 6 The program code is also available by clicking thebutton download on the lower panel of Figure 5
Acknowledgments
The authors would like to thank the three anonymousreviewers whose constructive comments are very helpful forstrengthening the presentation of the paper This work wassupported by the Grants from the National Natural ScienceFoundation of China (no 31260273) the Jiangxi ProvincialForeign Scientific and Technological Cooperation Project(no 20120BDH80023) Natural Science Foundation of JiangxiProvince China (no 2010GZS0122 20122BAB201020) theDepartment of Education of Jiangxi Province (GJJ12490)the LuoDi plan of the Department of Education of JiangxiProvince (KJLD12083) and the Jiangxi Provincial Founda-tion for Leaders of Disciplines in Science (20113BCB22008)The funders had no role in study design data collection andanalysis decision to publish or preparation of the paper
References
[1] A Bairoch ldquoThe ENZYME database in 2000rdquo Nucleic AcidsResearch vol 28 no 1 pp 304ndash305 2000
[2] S H Koenig and R D Brown ldquoH2CO3as substrate for carbonic
anhydrase in the dehydration of H2CO3(minus)rdquo Proceedings of the
National Academy of Sciences of the United States of Americavol 69 no 9 pp 2422ndash2425 1972
[3] S X Lin and J Lapointe ldquoTheoretical and experimental biologyin onerdquo Journal of Biomedical Science and Engineering vol 6 pp435ndash442 2013
[4] K C Chou and S P Jiang ldquoStudies on the rate of diffusion-controlled reactions of enzymes Spatial factor and force fieldfactorrdquo Scientia Sinica vol 17 no 5 pp 664ndash680 1974
[5] K C Chou ldquoThe kinetics of the combination reaction betweenenzyme and substraterdquo Scientia Sinica vol 19 no 4 pp 505ndash528 1976
[6] K C Chou and G P Zhou ldquoRole of the protein outside activesite on the diffusion-controlled reaction of enzymerdquo Journal ofthe American Chemical Society vol 104 no 5 pp 1409ndash14131982
[7] R A Poorman A G Tomasselli R L Heinrikson and FJ Kezdy ldquoA cumulative specificity model for proteases from
human immunodeficiency virus types 1 and 2 inferred fromstatistical analysis of an extended substrate data baserdquo TheJournal of Biological Chemistry vol 266 no 22 pp 14554ndash145611991
[8] K-C Chou ldquoA vectorized sequence-coupling model for pre-dicting HIV protease cleavage sites in proteinsrdquo The Journal ofBiological Chemistry vol 268 no 23 pp 16938ndash16948 1993
[9] G Z Liang and S Z Li ldquoA new sequence representation asapplied in better specificity elucidation for human immunod-eficiency virus type 1 proteaserdquo Biopolymers vol 88 no 3 pp401ndash412 2007
[10] J J Chou ldquoPredicting cleavability of peptide sequences byHIVprotease via correlation-angle approachrdquo Journal of ProteinChemistry vol 12 no 3 pp 291ndash302 1993
[11] Q S Du S Wang D Q Wei S Sirois and K C ChouldquoMolecular modeling and chemical modification for findingpeptide inhibitor against severe acute respiratory syndromecoronavirus main proteinaserdquo Analytical Biochemistry vol 337no 2 pp 262ndash270 2005
[12] Y Gan H Huang Y Huang et al ldquoSynthesis and activity ofan octapeptide inhibitor designed for SARS coronavirus mainproteinaserdquo Peptides vol 27 no 4 pp 622ndash625 2006
[13] Q S Du H Sun and K C Chou ldquoInhibitor design for SARScoronavirus main protease based on lsquodistorted key theoryrsquordquoMedicinal Chemistry vol 3 no 1 pp 1ndash6 2007
[14] K C Chou ldquoPrediction of human immunodeficiency virusprotease cleavage sites in proteinsrdquoAnalytical Biochemistry vol233 no 1 pp 1ndash14 1996
[15] J Knowles and G Gromo ldquoTarget selection in drug discoveryrdquoNature Reviews Drug Discovery vol 2 no 1 pp 63ndash69 2003
[16] A C Cheng R G Coleman K T Smyth et al ldquoStructure-basedmaximal affinity model predicts small-molecule druggabilityrdquoNature Biotechnology vol 25 no 1 pp 71ndash75 2007
[17] M Rarey B Kramer T Lengauer and G Klebe ldquoA fast flexibledocking method using an incremental construction algorithmrdquoJournal of Molecular Biology vol 261 no 3 pp 470ndash489 1996
[18] K C Chou ldquoReview structural bioinformatics and its impactto biomedical sciencerdquo Current Medicinal Chemistry vol 11 no16 pp 2105ndash2134 2004
[19] K C Chou D Q Wei and W Z Zhong ldquoBinding mechanismof coronavirus main proteinase with ligands and its implicationto drug design against SARSrdquo Biochemical and BiophysicalResearch Communications vol 308 pp 148ndash151 2003 (Erratumin Biochemical and Biophysical Research Communications vol310 p 675 2003)
[20] K C Chou D Q Wei Q S Du S Sirois and W Z ZhongldquoProgress in computational approach to drug developmentagaint SARSrdquo Current Medicinal Chemistry vol 13 no 27 pp3263ndash3270 2006
[21] G P Zhou and F A Troy ldquoNMR studies on how the bindingcomplex of polyisoprenol recognition sequence peptides andpolyisoprenols can modulate membrane structurerdquo CurrentProtein and Peptide Science vol 6 no 5 pp 399ndash411 2005
[22] R B Huang Q S Du C H Wang and K C Chou ldquoAn in-depth analysis of the biological functional studies based on theNMR M2 channel structure of influenza A virusrdquo Biochemicaland Biophysical Research Communications vol 377 no 4 pp1243ndash1247 2008
[23] Q S Du R B Huang C H Wang X M Li and K C ChouldquoEnergetic analysis of the two controversial drug binding sitesof the M2 proton channel in influenza A virusrdquo Journal ofTheoretical Biology vol 259 no 1 pp 159ndash164 2009
BioMed Research International 11
[24] M J Berardi W M Shih S C Harrison and J J Chou ldquoMito-chondrial uncoupling protein 2 structure determined by NMRmolecular fragment searchingrdquo Nature vol 476 no 7358 pp109ndash114 2011
[25] J R Schnell and J J Chou ldquoStructure andmechanism of theM2proton channel of influenza A virusrdquo Nature vol 451 no 7178pp 591ndash595 2008
[26] B OuYang S Xie M J Berardi et al ldquoUnusual architecture ofthe p7 channel from hepatitis C virusrdquoNature vol 498 pp 521ndash525 2013
[27] K Oxenoid and J J Chou ldquoThe structure of phospholambanpentamer reveals a channel-like architecture in membranesrdquoProceedings of the National Academy of Sciences of the UnitedStates of America vol 102 no 31 pp 10870ndash10875 2005
[28] M E Call KWWucherpfennig and J J Chou ldquoThe structuralbasis for intramembrane assembly of an activating immunore-ceptor complexrdquo Nature Immunology vol 11 no 11 pp 1023ndash1029 2010
[29] R M Pielak J R Schnell and J J Chou ldquoMechanism of druginhibition and drug resistance of influenza A M2 channelrdquoProceedings of the National Academy of Sciences of the UnitedStates of America vol 106 no 18 pp 7379ndash7384 2009
[30] J Wang R M Pielak M A McClintock and J J ChouldquoSolution structure and functional analysis of the influenza Bproton channelrdquo Nature Structural and Molecular Biology vol16 no 12 pp 1267ndash1271 2009
[31] K C Chou ldquoCoupling interaction between thromboxane A2receptor and alpha-13 subunit of guanine nucleotide-bindingproteinrdquo Journal of Proteome Research vol 4 no 5 pp 1681ndash1686 2005
[32] K C Chou ldquoInsights from modeling three-dimensional struc-tures of the human potassium and sodium channelsrdquo Journal ofProteome Research vol 3 no 4 pp 856ndash861 2004
[33] K C Chou ldquoSome remarks on protein attribute prediction andpseudo amino acid compositionrdquo Journal ofTheoretical Biologyvol 273 no 1 pp 236ndash247 2011
[34] W Z Lin J A Fang X Xiao and K C Chou ldquoiLoc-animal amulti-label learning classifier for predicting subcellular local-ization of animal proteinsrdquo Molecular BioSystems vol 9 pp634ndash644 2013
[35] X Xiao PWangW Z Lin J H Jia and K C Chou ldquoiAMP-2La two-level multi-label classifier for identifying antimicrobialpeptides and their functional typesrdquo Analytical Biochemistryvol 436 pp 168ndash177 2013
[36] W Chen P M Feng H Lin and K C Chou ldquoiRSpot-PseDNCidentify recombination spots with pseudo dinucleotide compo-sitionrdquo Nucleic Acids Research vol 41 article e69 2013
[37] K C Chou Z C Wu and X Xiao ldquoILoc-Hum using theaccumulation-label scale to predict subcellular locations ofhuman proteins with both single and multiple sitesrdquoMolecularBioSystems vol 8 no 2 pp 629ndash641 2012
[38] M Kotera M Hirakawa T Tokimatsu S Goto and MKanehisa ldquoThe KEGG databases and tools facilitating omicsanalysis latest developments involving human diseases andpharmaceuticalsrdquo Methods in Molecular Biology vol 802 pp19ndash39 2012
[39] Y YamanishiM Araki A GutteridgeWHonda andM Kane-hisa ldquoPrediction of drug-target interaction networks from theintegration of chemical and genomic spacesrdquo Bioinformaticsvol 24 no 13 pp i232ndashi240 2008
[40] P Finn S Muggleton D Page and A Srinivasan ldquoPharma-cophore discovery using the Inductive Logic Programmingsystem PROGOLrdquo Machine Learning vol 30 no 2-3 pp 241ndash270 1998
[41] I Vogt D Stumpfe H E Ahmed and J Bajorath ldquoMethodsfor computer-aided chemical biology Part 2 evaluation of com-pound selectivity using 2D molecular fingerprintsrdquo ChemicalBiology and Drug Design vol 70 pp 195ndash205 2007
[42] H Eckert and J Bajorath ldquoMolecular similarity analysis in vir-tual screening foundations limitations and novel approachesrdquoDrug Discovery Today vol 12 no 5-6 pp 225ndash233 2007
[43] S Laurent L V Elst and R N Muller ldquoComparative studyof the physicochemical properties of six clinical low molecularweight gadolinium contrast agentsrdquo Contrast Media amp Molecu-lar Imaging vol 1 no 3 pp 128ndash137 2006
[44] E Gregori-Puigjane R Garriga-Sust and J Mestres ldquoIndexingmolecules with chemical graph identifiersrdquo Journal of Compu-tational Chemistry vol 32 no 12 pp 2638ndash2646 2011
[45] B Ren ldquoApplication of novel atom-type AI topological indicesto QSPR studies of alkanesrdquo Computers and Chemistry vol 26no 4 pp 357ndash369 2002
[46] N M OrsquoBoyle M Banck C A James C Morley T Vander-meersch and G R Hutchison ldquoOpen Babel an open chemicaltoolboxrdquo Journal of Cheminformatics vol 3 p 33 2011
[47] V J Gillet P Willett and J Bradshaw ldquoSimilarity searchingusing reduced graphsrdquo Journal of Chemical Information andComputer Sciences vol 43 no 2 pp 338ndash345 2003
[48] D Butina ldquoUnsupervised data base clustering based on day-lightrsquos fingerprint and Tanimoto similarity a fast and automatedway to cluster small and large data setsrdquo Journal of ChemicalInformation and Computer Sciences vol 39 no 4 pp 747ndash7501999
[49] C E Shannon ldquoA mathematical theory of communicationrdquoACM SIGMOBILE Mobile Computing and CommunicationsReview vol 5 pp 3ndash55 2001
[50] V D Gusev L A Nemytikova and N A Chuzhanova ldquoOn thecomplexity measures of genetic sequencesrdquo Bioinformatics vol15 no 12 pp 994ndash999 1999
[51] K C Chou and H B Shen ldquoReview recent progress in proteinsubcellular location predictionrdquo Analytical Biochemistry vol370 no 1 pp 1ndash16 2007
[52] S F Altschul ldquoEvaluating the statistical significance of multipledistinct local alignmentsrdquo in Theoretical and ComputationalMethods in Genome Research S Suhai Ed pp 1ndash14 PlenumNew York NY USA 1997
[53] J C Wootton and S Federhen ldquoStatistics of local complexity inamino acid sequences and sequence databasesrdquo Computers andChemistry vol 17 no 2 pp 149ndash163 1993
[54] H Nakashima K Nishikawa and T Ooi ldquoThe folding type ofa protein is relevant to the amino acid compositionrdquo Journal ofBiochemistry vol 99 no 1 pp 153ndash162 1986
[55] K C Chou and C T Zhang ldquoPredicting protein folding typesby distance functions that make allowances for amino acidinteractionsrdquo The Journal of Biological Chemistry vol 269 no35 pp 22014ndash22020 1994
[56] K-C Chou ldquoA novel approach to predicting protein structuralclasses in a (20-1)-D amino acid composition spacerdquo Proteinsvol 21 no 4 pp 319ndash344 1995
[57] H Nakashima and K Nishikawa ldquoDiscrimination of intracel-lular and extracellular proteins using amino acid compositionand residue-pair frequenciesrdquo Journal of Molecular Biology vol238 no 1 pp 54ndash61 1994
12 BioMed Research International
[58] G P Zhou ldquoAn intriguing controversy over protein structuralclass predictionrdquo Journal of Protein Chemistry vol 17 no 8 pp729ndash738 1998
[59] I Bahar A R Atilgan R L Jernigan and B Erman ldquoUnder-standing the recognition of protein structural classes by aminoacid compositionrdquo Proteins vol 29 pp 172ndash185 1997
[60] J Cedano P Aloy J A Perez-Pons and E Querol ldquoRelationbetween amino acid composition and cellular location ofproteinsrdquo Journal of Molecular Biology vol 266 no 3 pp 594ndash600 1997
[61] G P Zhou and K Doctor ldquoSubcellular location prediction ofapoptosis proteinsrdquo Proteins vol 50 no 1 pp 44ndash48 2003
[62] KChou ldquoPrediction of protein cellular attributes using pseudo-amino acid compositionrdquo Proteins vol 43 pp 246ndash255 2001(Erratum in Proteins vol 44 p 60 2001)
[63] K C Chou ldquoUsing amphiphilic pseudo amino acid composi-tion to predict enzyme subfamily classesrdquo Bioinformatics vol21 no 1 pp 10ndash19 2005
[64] D Zou Z He J He and Y Xia ldquoSupersecondary structureprediction using Choursquos pseudo amino acid compositionrdquoJournal of Computational Chemistry vol 32 no 2 pp 271ndash2782011
[65] M M Beigi M Behjati and H Mohabatkar ldquoPrediction ofmetalloproteinase family based on the concept ofChoursquos pseudoamino acid composition using a machine learning approachrdquoJournal of Structural and Functional Genomics vol 12 no 4 pp191ndash197 2011
[66] Y K Chen and K B Li ldquoPredicting membrane protein typesby incorporating protein topology domains signal peptidesand physicochemical properties into the general form of Choursquospseudo amino acid compositionrdquo Journal ofTheoretical Biologyvol 318 pp 1ndash12 2013
[67] C Huang and J Q Yuan ldquoA multilabel model based on Choursquospseudo-amino acid composition for identifying membraneproteins with both single and multiple functional typesrdquo TheJournal of Membrane Biology vol 246 pp 327ndash334 2013
[68] S S Sahu and G Panda ldquoA novel feature representationmethod based on Choursquos pseudo amino acid composition forprotein structural class predictionrdquo Computational Biology andChemistry vol 34 no 5-6 pp 320ndash327 2010
[69] M Hayat and A Khan ldquoDiscriminating outer membraneproteins with fuzzy K-nearest neighbor algorithms based on thegeneral form of Choursquos PseAACrdquo Protein and Peptide Lettersvol 19 no 4 pp 411ndash421 2012
[70] MKhosravian F K FaramarziMM BeigiM Behbahani andH Mohabatkar ldquoPredicting antibacterial peptides by the con-cept of Choursquos pseudo-amino acid composition and machinelearning methodsrdquo Protein amp Peptide Letters vol 20 pp 180ndash186 2013
[71] HMohabatkarMM Beigi K Abdolahi and SMohsenzadehldquoPrediction of allergenic proteins by means of the concept ofChoursquos pseudo amino acid composition and amachine learningapproachrdquoMedicinal Chemistry vol 9 pp 133ndash137 2013
[72] L Nanni A Lumini D Gupta and A Garg ldquoIdentifyingbacterial virulent proteins by fusing a set of classifiers basedon variants of Choursquos pseudo amino acid composition and onevolutionary informationrdquo IEEEACM Transactions on Compu-tational Biology and Bioinformatics vol 9 no 2 pp 467ndash4752012
[73] T H Chang L C Wu T Y Lee S P Chen H D Huang andJ T Horng ldquoEuLoc a web-server for accurately predict protein
subcellular localization in eukaryotes by incorporating variousfeatures of sequence segments into the general form of ChoursquosPseAACrdquo Journal of Computer-Aided Molecular Design vol 27pp 91ndash103 2013
[74] S Zhang Y Zhang H Yang C Zhao and Q Pan ldquoUsing theconcept of Choursquos pseudo amino acid composition to predictprotein subcellular localization an approach by incorporatingevolutionary information and von Neumann entropiesrdquo AminoAcids vol 34 no 4 pp 565ndash572 2008
[75] R Zia Ur and A Khan ldquoIdentifying GPCRs and their typeswith Choursquos pseudo amino acid composition an approach frommulti-scale energy representation and position specific scoringmatrixrdquo Protein amp Peptide Letters vol 19 pp 890ndash903 2012
[76] X Y Sun S P Shi J D Qiu S B Suo S Y Huang and R PLiang ldquoIdentifying protein quaternary structural attributes byincorporating physicochemical properties into the general formof Choursquos PseAAC via discrete wavelet transformrdquo MolecularBioSystems vol 8 pp 3178ndash3184 2012
[77] L Nanni and A Lumini ldquoGenetic programming for creatingChoursquos pseudo amino acid based features for submitochondrialocalizationrdquo Amino Acids vol 34 no 4 pp 653ndash660 2008
[78] M Esmaeili H Mohabatkar and S Mohsenzadeh ldquoUsing theconcept of Choursquos pseudo amino acid composition for risk typeprediction of human papillomavirusesrdquo Journal of TheoreticalBiology vol 263 no 2 pp 203ndash209 2010
[79] H Mohabatkar ldquoPrediction of cyclin proteins using Choursquospseudo amino acid compositionrdquo Protein and Peptide Lettersvol 17 no 10 pp 1207ndash1214 2010
[80] H Mohabatkar M M Beigi and A Esmaeili ldquoPredictionof GABA(A) receptor proteins using the concept of Choursquospseudo-amino acid composition and support vector machinerdquoJournal of Theoretical Biology vol 281 no 1 pp 18ndash23 2011
[81] Y Xu J Ding L Y Wu and K C Chou ldquoiSNO-PseAAC pre-dict cysteine S-nitrosylation sites in proteins by incorporatingposition specific amino acid propensity into pseudo amino acidcompositionrdquo PLoS ONE vol 8 Article ID e55844 2013
[82] W Chen H Lin PM Feng C Ding Y C Zuo andK C ChouldquoiNuc-PhysChem a sequence-based predictor for identifyingnucleosomes via physicochemical propertiesrdquo PLoS ONE vol7 Article ID e47843 2012
[83] B Li T Huang L Liu Y Cai and K C Chou ldquoIdentificationof colorectal cancer related genes with mrmr and shortest pathin protein-protein interaction networkrdquo PLoS ONE vol 7 no 4Article ID e33393 2012
[84] Y Jiang T Huang C Lei Y F Gao Y D Cai and K C ChouldquoSignal propagation in protein interaction network duringcolorectal cancer progressionrdquo BioMed Research Internationalvol 2013 Article ID 287019 9 pages 2013
[85] P Du X Wang C Xu and Y Gao ldquoPseAAC-Builder across-platform stand-alone program for generating variousspecial Choursquos pseudo-amino acid compositionsrdquo AnalyticalBiochemistry vol 425 no 2 pp 117ndash119 2012
[86] D S Cao Q S Xu and Y Z Liang ldquoPropy a tool to generatevarious modes of Choursquos PseAACrdquo Bioinformatics vol 29 pp960ndash962 2013
[87] H Shen and K C Chou ldquoPseAAC a flexible web serverfor generating various kinds of protein pseudo amino acidcompositionrdquo Analytical Biochemistry vol 373 no 2 pp 386ndash388 2008
[88] K C Chou ldquoUsing pair-coupled amino acid composition topredict protein secondary structure contentrdquo Journal of ProteinChemistry vol 18 no 4 pp 473ndash480 1999
BioMed Research International 13
[89] W Liu and K C Chou ldquoPrediction of protein secondarystructure contentrdquo Protein Engineering vol 12 no 12 pp 1041ndash1050 1999
[90] M Bhasin and G P S Raghava ldquoClassification of nuclearreceptors based on amino acid composition and dipeptidecompositionrdquo The Journal of Biological Chemistry vol 279 no22 pp 23262ndash23266 2004
[91] H Lin andH Ding ldquoPredicting ion channels and their types bythe dipeptidemode of pseudo amino acid compositionrdquo Journalof Theoretical Biology vol 269 no 1 pp 64ndash69 2011
[92] H Lin and Q Li ldquoUsing pseudo amino acid composition topredict protein structural class approached by incorporating400 dipeptide componentsrdquo Journal of Computational Chem-istry vol 28 no 9 pp 1463ndash1466 2007
[93] L Nanni and A Lumini ldquoCombing ontologies and dipeptidecomposition for predicting DNA-binding proteinsrdquo AminoAcids vol 34 no 4 pp 635ndash641 2008
[94] K-C Chou ldquoThe convergence-divergence duality in lectindomains of selectin family and its implicationsrdquo FEBS Lettersvol 363 no 1-2 pp 123ndash126 1995
[95] A A Schaffer L Aravind T L Madden et al ldquoImprovingthe accuracy of PSI-BLAST protein database searches withcomposition-based statistics and other refinementsrdquo NucleicAcids Research vol 29 no 14 pp 2994ndash3005 2001
[96] S F Altschul and E V Koonin ldquoIterated profile searches withPSI-BLAST a tool for discovery in protein databasesrdquo Trends inBiochemical Sciences vol 23 no 11 pp 444ndash447 1998
[97] J Deng ldquoGrey entropy and grey target decision makingrdquoJournal of Grey System vol 22 no 1 pp 1ndash24 2010
[98] B M Bolstad R A Irizarry M Astrand and T P Speed ldquoAcomparison of normalizationmethods for high density oligonu-cleotide array data based on variance and biasrdquo Bioinformaticsvol 19 no 2 pp 185ndash193 2003
[99] J-Y Shi S-W Zhang Q Pan Y-M Cheng and J XieldquoPrediction of protein subcellular localization by support vectormachines using multi-scale energy and pseudo amino acidcompositionrdquo Amino Acids vol 33 no 1 pp 69ndash74 2007
[100] T Denoeux ldquoA 120581-nearest neighbor classification rule based onDempster-Shafer theoryrdquo IEEE Transactions on Systems Manand Cybernetics vol 25 no 5 pp 804ndash813 1995
[101] J M Keller M R Gray and J A Givens ldquoA fuzzy k-nearestneighbours algorithmrdquo IEEE Transactions on Systems Man andCybernetics vol 15 no 4 pp 580ndash585 1985
[102] X Xiao P Wang and K Chou ldquoiNR-physchem a sequence-based predictor for identifying nuclear receptors and theirsubfamilies via physical-chemical property matrixrdquo PLoS ONEvol 7 no 2 Article ID e30869 2012
[103] I Roterman L Konieczny W Jurkowski K Prymula and MBanach ldquoTwo-intermediate model to characterize the structureof fast-folding proteinsrdquo Journal of Theoretical Biology vol 283no 1 pp 60ndash70 2011
[104] X Xiao P Wang and K C Chou ldquoGPCR-2L predicting Gprotein-coupled receptors and their types by hybridizing twodifferentmodes of pseudo amino acid compositionsrdquoMolecularBioSystems vol 7 no 3 pp 911ndash919 2011
[105] X Xiao P Wang and K C Chou ldquoQuat-2L a web-server forpredicting protein quaternary structural attributesrdquo MolecularDiversity vol 15 no 1 pp 149ndash155 2011
[106] X Zheng C Li and J Wang ldquoAn information-theoreticapproach to the prediction of protein structural classrdquo Journalof Computational Chemistry vol 31 no 6 pp 1201ndash1206 2010
[107] H Shen J Yang X Liu and K C Chou ldquoUsing supervisedfuzzy clustering to predict protein structural classesrdquo Biochem-ical and Biophysical Research Communications vol 334 no 2pp 577ndash581 2005
[108] K-C Chou and C-T Zhang ldquoReview prediction of proteinstructural classesrdquo Critical Reviews in Biochemistry and Molec-ular Biology vol 30 no 4 pp 275ndash349 1995
[109] P C Mahalanobis ldquoOn the generalized distance in statisticsrdquoProceedings of the National Institute of Sciences of India vol 2pp 49ndash55 1936
[110] R M Centor ldquoSignal detectability the use of ROC curves andtheir analysesrdquoMedical Decision Making vol 11 no 2 pp 102ndash106 1991
[111] K-C Chou ldquoUsing subsite coupling to predict signal peptidesrdquoProtein Engineering vol 14 no 2 pp 75ndash79 2001
[112] K C Chou ldquoPrediction of protein signal sequences and theircleavage sitesrdquo Proteins vol 42 pp 136ndash139 2001
[113] K C Chou ldquoPrediction of signal peptides using scaledwindowrdquoPeptides vol 22 no 12 pp 1973ndash1979 2001
[114] K C Chou ldquoSome remarks on predicting multi-label attributesinmolecular biosystemsrdquoMolecular Biosystems vol 9 pp 1092ndash1100 2013
[115] K C Chou and H B Shen ldquoCell-PLoc 2 0 an improvedpackage of web-servers for predicting subcellular localization ofproteins in various organismsrdquoNatural Science vol 2 pp 1090ndash1103 2010
[116] M Gribskov and N L Robinson ldquoUse of receiver operatingcharacteristic (ROC) analysis to evaluate sequence matchingrdquoComputers and Chemistry vol 20 no 1 pp 25ndash33 1996
[117] Y Yamanishi M Kotera M Kanehisa and S Goto ldquoDrug-target interaction prediction from chemical genomic and phar-macological data in an integrated frameworkrdquo Bioinformaticsvol 26 no 12 pp i246ndashi254 2010
[118] Z He J Zhang X Shi et al ldquoPredicting drug-target interactionnetworks based on functional groups and biological featuresrdquoPLoS ONE vol 5 no 3 Article ID e9603 2010
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

BioMed Research International 5
Incorporating the above 400 dipeptide components into (11)we have
120595119906+20
= 119891
(2)
119906(119906 = 1 2 400) (14)
(c) Sequential Evolution Information Biology is a naturalscience with a historic dimension All biological specieshave developed starting out from a very limited number ofancestral species Their evolution involves changes of singleresidues insertions and deletions of several residues [94]gene doubling and gene fusion With these changes accu-mulated for a long period of time many similarities betweeninitial and resultant amino acid sequences are graduallyeliminated but the corresponding proteins may still sharemany common attributes [18] such as having basically thesame biological function and residing at a same subcellularlocation To extract the sequential evolution information anduse it to define the components of (11) the PSSM (PositionSpecific Scoring Matrix) was used as described next
According to Schaffer et al [95] the sequence evolutioninformation of enzyme E with 119871 amino acid residues can beexpressed by an 119871 times 20matrix as given by
P(0)PSSM =
[
[
[
[
[
[
[
119864
0
1rarr1119864
0
1rarr2sdot sdot sdot 119864
0
1rarr20
119864
0
2rarr1119864
0
2rarr2sdot sdot sdot 119864
0
2rarr20
119864
0
119871rarr1119864
0
119871rarr2sdot sdot sdot 119864
0
119871rarr20
]
]
]
]
]
]
]
(15)
where 1198640119894rarr 119895
represents the original score of the 119894th aminoacid residue (119894 = 1 2 119871) in the enzyme sequence changedto amino acid type 119895 (119895 = 1 2 20) in the process ofevolution Here the numerical codes 1 2 20 are used torepresent the 20 native amino acid types denoted by A CD E F G H I K L M N P Q R S T V W and Y The119871 times 20 scores in (15) were generated by using PSI-BLAST[96] to search the UniProtKBSwiss-Prot database (Release2013-05) through three iterations with 0001 as the 119864 valuecutoff for multiple sequence alignment against the sequenceof the enzyme E In order to make every element in (15) bescaled from their original score ranges into region of [0 1]we performed a conversion through the standard sigmoidfunction to make it become
P(1)PSSM =
[
[
[
[
[
[
[
119864
1
1rarr1119864
1
1rarr2sdot sdot sdot 119864
1
1rarr20
119864
1
2rarr1119864
1
2rarr2sdot sdot sdot 119864
1
2rarr20
119864
1
119871rarr1119864
1
119871rarr2sdot sdot sdot 119864
1
119871rarr20
]
]
]
]
]
]
]
(16)
where
119864
1
119894rarr 119895=
1
1 + 119890
minus1198640
119894rarr 119895
(1 le 119894 le 119871 1 le 119895 le 20) (17)
Nowwe extract the useful information from (16) to define thecomponents of (11) via the following approach
120595119906+420
= ℓ119906
(119906 = 1 2 20) (18)
where
ℓ119895=
1
119871
times
119871
sum
119896=1
119864
1
119896rarr119895(119895 = 1 2 20) (19)
(d) Grey System Model Approach The grey system theory[97] is quite useful in dealing with complicated systems thatlack sufficient information or need to process uncertaininformation According to the grey system theory we canextract the following information from the 119895th column of(16) that is
[
119886
119895
1
119886
119895
2
] = (BT119895B119895)
minus1
BT119895U119895
(119895 = 1 2 20) (20)
where
B119895=
[
[
[
[
[
[
[
[
[
[
minus119864
1
2rarr119895minus119864
1
1rarr119895minus 05119864
1
2rarr1198951
minus119864
1
3rarr119895minus
2
sum
119894=1
119864
1
119894rarr 119895minus 05119864
1
3rarr1198951
minus119864
1
119871rarr119895minus
119871minus1
sum
119894=1
119864
1
119894rarr 119895minus 05119864
1
119871rarr1198951
]
]
]
]
]
]
]
]
]
]
U119895=
[
[
[
[
[
[
119864
1
2rarr119895minus 119864
1
1rarr119895
119864
1
3rarr119895minus 119864
1
2rarr119895
119864
1
119871rarr119895minus 119864
1
119871minus1rarr119895
]
]
]
]
]
]
(21)
Therefore based on the grey system theory and (20) we canextract another 20 times 2 = 40 quantities from (16) to definethe components of (11) that is
120593119895=
1199081119886
119895
1when 119895 is an odd number
1199082119886
119895
2when 119895 is an even number
1 le 119895 le 20
(22)
where 1198861198951and 119886119895
2are given by (20) 119908
1and 119908
2are weight
factors which were all set to 1 in the current studySubstituting the elements in (12) (14) (18) and (22) we
finally obtain a total of Ω = 20 + 400 + 20 + 40 = 480
components for the PseAAC of (11) where
120595119906=
119891119906
when 1 le 119906 le 20119891
(2)
119906when 21 le 119906 le 420
ℓ119906
when 421 le 119906 le 440120593119906
when 440 le 119906 le 480
(23)
In other words in this study (11) or Choursquos PseAAC is a 480-D vector whose 480 components are given by (23) derivedfrom the amino acid composition dipeptide compositionsequential evolution information and grey system theory(e) Representing Enzyme-Drug Pairs Now the pair betweenan enzyme molecule E and a drug compound D can beformulated by combing (8) and (11) as given by
G = D oplus E
= [1198601sdot sdot sdot 119860
256119867119909
CF 1205951sdot sdot sdot 120595480]
T
(24)
6 BioMed Research International
where G represents the enzyme-drug pair oplus the orthogonalsum [51] and each of the (258 + 480) = 738 feature elementsis given in (8) and (23)
For the convenience of the later formulation let us use119909119894(119894 = 1 2 738) to represent the 738 components of (24)
that is
G = [11990911199092sdot sdot sdot 119909119894sdot sdot sdot 119909738]
T (25)
To optimize the prediction results different weights wereusually tested for each of the elements in (25) However sinceit would consume a lot of computational time for a total of 738weight factors here let us adopt the normalization approachto deal with this problem as done in [98 99] that is convert119909119894in (25) to 119910
119894according to the following equation
119910119894=
2tanminus1 (119909119894)
120587
(119894 = 1 2 738) (26)
where tanminus1 means arctangent By means of (26) everycomponent in (25) will be converted into the range of[minus1 1] that is we have minus1 le 119910
119894le 1 As demonstrated
in [98 99] the normalization approach via (26) was quiteeffective in enhancing the quality of prediction operated ina high dimension space Therefore in this study we wouldnot to take the procedure of optimizing the weight factorssignificantly reducing the computational times
23 Fuzzy 119870-Nearest Neighbour Algorithm The 119870-NN (119870-Nearest Neighbor) classifier is quite popular in patternrecognition community owing to its good performance andsimple-to-use feature According to the 119870-NN rule [100]named also as the ldquovoting 119870-NN rulerdquo the query sampleshould be assigned to the subset represented by a majorityof its 119870 nearest neighbors as illustrated in Figure 5 of [33]
Fuzzy 119870-NN classification method [101] is a specialvariation of the119870-NNclassification family Instead of roughlyassigning the label based on a voting from the 119870 nearestneighbors it attempts to estimate the membership valuesthat indicate how much degree the query sample belongsto the classes concerned Obviously it is impossible for anycharacteristic description to contain complete informationwhich would make the classification ambiguous In view ofthis the fuzzy principle is very reasonable and particularlyuseful in dealing with complicated biological systems such asidentifying nuclear receptor subfamilies [102] characterizingthe structure of fast-folding proteins [103] classifying Gprotein-coupled receptors [104] predicting protein quater-nary structural attributes [105] predicting protein structuralclasses [106 107] and so forth
Next let us give a brief introduction how to use thefuzzy119870-NN approach to identify the interactions between theenzymes and the drug compounds in the network concerned
Supposing that S(119873) = G1G2 G
119873 is a set of vec-
tors representing 119873 enzyme-drug pairs in a training set
classified into two classes 119862+ 119862minus where 119862+ denotes theinteractive pair class while 119862minus the noninteractive pair classSlowast(G) = Glowast
1Glowast2 Glowast
119870 sub S(119873) is the subset of the 119870
nearest neighbor pairs to the query pair G Thus the fuzzymembership value for the query pair G in the two classes ofS(119873) is given by
120583
+
(G) =sum
119870
119895=1120583
+(Glowast119895) 119889(GGlowast
119895)
minus2(120593minus1)
sum
119870
119895=1119889(GGlowast
119895)
minus2(120593minus1)
120583
minus
(G) =sum
119870
119895=1120583
minus(Glowast119895) 119889(GGlowast
119895)
minus2(120593minus1)
sum
119870
119895=1119889(GGlowast
119895)
minus2(120593minus1)
(27)
where 119870 is the number of the nearest neighbors counted forthe query pairG 120583+(Glowast
119895) and 120583minus(Glowast
119895) the fuzzy membership
values of the training sample Glowast119895to the class 119862+ and 119862minus
respectively as will be further defined next 119889(GGlowast119895) the
cosine distance between G and its 119895th nearest pair Glowast119895in
the training dataset S(119873) 120593(gt 1) the fuzzy coefficient fordetermining how heavily the distance is weighted whencalculating each nearest neighborrsquos contribution to the mem-bership value Note that the parameters 119870 and 120593 will affectthe computation result of (27) and they will be optimizedby a grid-search as will be described later Also variousother metrics can be chosen for 119889(GGlowast
119895) such as Euclidean
distance Hamming distance [108] andMahalanobis distance[55 109]
The quantitative definitions for the aforementioned120583
+(Glowast119895) and 120583minus(Glowast
119895) in (27) are given by
120583
+
(Glowast119895) =
1 if Glowast119895isin 119862
+
0 otherwise
120583
minus
(Glowast119895) =
1 if Glowast119895isin 119862
minus
0 otherwise
(28)
Substituting the results obtained by (27) into (28) it followsthat if 120583+(G) gt 120583
minus(G) then the query pair G is an
interactive couple otherwise noninteractive In other wordsthe outcome can be formulated as
G isin
119862
+ if 120583+ (G) gt 120583minus (G)
119862
minus otherwise
(29)
If there is a tie between 120583+(G) and 120583minus(G) the query pair Gwill be randomly assigned to one of the two classes Howevercase like that is quite rare and in this study never happened
The predictor thus established is called iEzy-Drugwhere ldquoirdquo means identify and ldquoEzy-Drugrdquo means the inter-action between enzyme and drug To provide an intuitiveoverall picture a flowchart is provided in Figure 2 to showthe process of how the classifier works in identifying enzyme-drug interactions
BioMed Research International 7
Input a queryprotein sequence and
a drug code
Create Chous PseAAC of a protein
Create feature vector of a drug
Feature fusion
engineTraining dataset
Yes No
Noninteractive enzyme drug pair
Interactive enzymedrug pair
AACDipeptideSeq Evol
Grey model
Mol FptInfo entropy
CF
FKNN operation
Figure 2 A flowchart to show the operation process of the iEzy-Drug predictor See the text for further explanation
24 Criteria for Performance Evaluation In the literaturethe following equation set is often used for examining theperformance quality of a predictor
Sn = TPTP + FN
Sp = TNTN + FP
Acc = TP + TNTP + TN + FP + FN
MCC = (TP times TN) minus (FP times FN)radic(TP + FP) (TP + FN) (TN + FP) (TN + FN)
(30)
where TP represents the true positive TN the true negativeFP the false positive FN the false negative Sn the sensitiv-ity Sp the specificity Acc the accuracy MCC the Mathewrsquoscorrelation coefficient
To most biologists however the four metrics as formu-lated in (30) are not quite intuitive and easier-to-understandparticularly for the Mathewrsquos correlation coefficient Herelet us adopt the Choursquos symbols to formulate the previousfour metrics By means of Choursquos symbols [111 112] the rates
of correct predictions for the interactive enzyme-drug pairsin dataset S+ and the noninteractive enzyme-drug pairs indataset Sminus are respectively defined by (cf (1))
Λ
+
=
119873
+minus 119873
+
minus
119873
+ for the interactive enzyme-drug pairs
Λ
minus
=
119873
minusminus 119873
minus
+
119873
minus for the noninteractive enzyme-drug pairs
(31)
where 119873+ is the total number of the interactive enzyme-drug pairs investigated while 119873+
minusis the number of the
interactive enzyme-drug pairs incorrectly predicted as thenoninteractive enzyme-drug pairs 119873minus is the total numberof the noninteractive enzyme-drug pairs investigated while119873
minus
+is the number of the noninteractive enzyme-drug pairs
incorrectly predicted as the interactive enzyme-drug pairsThe overall success prediction rate is given by [113] as follows
Λ =
Λ
+119873
++ Λ
minus119873
minus
119873
++ 119873
minus= 1 minus
119873
+
minus+ 119873
minus
+
119873
++ 119873
minus
(32)
It is obvious from (31)-(32) that if and only if none ofthe interactive enzyme-drug pairs and the noninteractiveenzyme-drug pairs are mispredicted that is 119873+
minus= 119873
minus
+= 0
and Λ+ = Λ
minus= 1 we have the overall success rate Λ = 1
Otherwise the overall success rate would be smaller than 1The relations between the symbols in (32) and those in
(30) are given by
TP = 119873+ minus 119873+minus
TN = 119873
minus
minus 119873
minus
+
FP = 119873minus+
FN = 119873
+
minus
(33)
Substituting (33) into (30) and also noting (31)-(32) we obtain
Sn = Λ+ = 1 minus119873
+
minus
119873
+
Sp = Λminus = 1 minus119873
minus
+
119873
minus
Acc = Λ = 1 minus119873
+
minus+ 119873
minus
+
119873
++ 119873
minus
MCC =1 minus ((119873
+
minus119873
+) + (119873
minus
+119873
minus))
radic(1 + (119873
minus
+minus 119873
+
minus) 119873
+) (1 + (119873
+
minusminus 119873
minus
+) 119873
minus)
(34)
Now it is obvious to see from (34) when 119873
+
minus= 0
meaning none of the interactive enzyme-drug pairs was
8 BioMed Research International
02
46
810
1
15
2089
0895
09
0905
091
0915
092
ACC
K120593
Figure 3 A 3D plot to show how the parameter in (27) wasoptimized for the iEzy-Drug predictor
mispredicted to be a noninteractive enzyme-drug pair wehave the sensitivity Sn = 1 while 119873+
minus= 119873
+ meaning thatall the interactive enzyme-drug pairs weremispredicted to bethe noninteractive enzyme-drug pairs we have the sensitivitySn = 0 Likewise when 119873minus
+= 0 meaning none of the
noninteractive enzyme-drug pairs wasmispredicted we havethe specificity Sp = 1 while 119873minus
+= 119873
minus meaning all thenoninteractive enzyme-drug pairs were incorrectly predictedas interactive enzyme-drug pairs we have the specificity Sp =0 When119873+
minus= 119873
minus
+= 0 meaning that none of the interactive
enzyme-drug pairs in the dataset S+ and none of the nonin-teractive enzyme-drug pairs in Sminus was incorrectly predictedwe have the overall accuracy Acc = Λ = 1 while 119873+
minus= 119873
+
and 119873minus+= 119873
minus meaning that all the interactive enzyme-drugpairs in the dataset S+ and all the noninteractive enzyme-drug pairs in Sminus were mispredicted we have the overallaccuracy Acc = Λ = 0 The MCC correlation coefficient isusually used for measuring the quality of binary (two-class)classifications When 119873+
minus= 119873
minus
+= 0 meaning that none
of the interactive enzyme-drug pairs in the dataset S+ andnone of the noninteractive enzyme-drug pairs in Sminus weremispredicted we have MCC = 1 when 119873+
minus= 119873
+2 and
119873
minus
+= 119873
minus2 we have MCC = 0 meaning no better than
random prediction when 119873+minus= 119873
+ and 119873minus+= 119873
minus we haveMCC = minus1 meaning total disagreement between predictionand observation As we can see from the previous discussionit is much more intuitive and easier-to-understand whenusing (34) to examine a predictor for its sensitivity specificityoverall accuracy and Mathewrsquos correlation coefficient It isinstructive to point out that themetrics as defined in (30) and(34) are valid for single label systems for multilabel systemsa set of more complicated metrics should be used as given in[114]
3 Results and Discussion
31 Cross-Validation How to properly examine the predic-tion quality is a key for developing a new predictor and
False positive rate
True
pos
itive
rate
1
01
02
03
04
05
06
07
08
09
0
101 02 03 04 05 06 07 08 090
BaselineiEzy-Drug AUC = 09377
Figure 4 A plot for the ROC curve to quantitatively show theperformance of the iEzy-Drug predictor
estimating its potential application value Generally speak-ing the following three cross-validation methods are oftenused to examine a predictor of its effectiveness in practicalapplication independent dataset test subsampling or 119870-fold(such as 5-fold 7-fold or 10-fold) test and jackknife test[108] However as elaborated by a penetrating analysis in[115] considerable arbitrariness exists in the independentdataset test Also as demonstrated by (27)ndash(29) in [33]the subsampling test (or 119870-fold cross-validation) cannotavoid arbitrariness either Only the jackknife test is the leastarbitrary that can always yield a unique result for a givenbenchmark dataset Therefore the jackknife test has beenwidely recognized and increasingly utilized by investigatorsto examine the quality of various predictors (see eg [66 7174 80]) Accordingly the success rate by the jackknife test wasalso used to optimize the two uncertain parameters 119870 and 120593in (27) The result thus obtained is shown in Figure 3 fromwhich we obtain when 119870 = 6 and 120593 = 15 the iEzy-Drugpredictor reaches its optimized status
The success rates thus obtained by the jackknife test inidentifying interactive Enzyme-drug pairs or noninteractiveenzyme-drug pairs on the benchmark dataset S (cf OnlineSupporting Information S1) are given in Table 1 where forfacilitating comparison the corresponding result by He et al[110] is also given As we can see from the table the overallaccuracy Acc achieved by iEzy-Drug was 9103 remarkablyhigher than 8548 the corresponding rate obtained byHe et al [110] on the same benchmark Furthermore listedin Table 1 are also the values obtained by iEzy-Drug for theother three metrics that is Sn = 9081 Sp = 9114 andMCC = 8039 indicating that the accuracy of iEzy-Drug isnot only very high but also quite stale
To provide a graphical illustration to show the per-formance of the current binary classifier iEzy-Drug asits discrimination threshold is varied a 2D plot calledReceiver Operating Characteristic (ROC) curve [116 117]was also given (Figure 4) In the ROC curve the verticalcoordinate 119884 is for the true positive rate or Sn (cf (34))while horizontal coordinate 119883 for the false positive rate
BioMed Research International 9
Read Me Data Citation
Enter both the protein sequence and the drug code (example) the number of query pairs is limited at 10 or less for each submission
Submit Clear
You can download the program and run it on your own local computer
iEzy-Drug identifying the interaction between enzymes anddrugs in cellular networking
Figure 5 A semiscreenshot to show the top page of the iEzy-Drugweb-server Its web-site address is at httpwwwjci-bioinfocniEzy-Drug
Table 1 The jackknife success rates obtained with iEzy-Drug in identifying interactive enzyme-drug pairs and noninteractive enzyme-drugpairs for the benchmark dataset S (cf Online Supporting Information S1)
Method Acc Sn Sp MCC
iEzy-Druga 74258157 = 9103 24692719 = 9081 49565438 = 9114 8039NN predictorb 8548 NA NA NAaSee (27) where the parameters119870 = 6 and 120593 = 15bSee [110]
or 1-Sp The best possible prediction method would yielda point with the coordinate (0 1) representing 100 truepositive rate (sensitivity Sn) and 0 false positive rate or100 specificity Therefore the (0 1) point is also calleda perfect classification A completely random guess wouldgive a point along a diagonal from the point (0 0) to (1 1)The area under the ROC curve also called Area Under theROC (AUROC) is often used to indicate the performancequality of a binary classifier the value 05 of AUROCis equivalent to random prediction while 1 of AUROCrepresents a perfect one As we can see from Figure 4the AUROC value obtained by iEzy-Drug is 09377
The reason why iEzy-Drug can remarkably improve theprediction quality is that it has introduced the 2D molecularfingerprints to represent drug samples see Online SupportingInformation S3 for the detailed fingerprint expressions for thedrugs listed in Online Supporting Information S1 and thatit has successfully used PseAAC to incorporate the featuresderived from the sequences of enzymes that are essentialfor identifying the interaction of enzymes with drugs in thecellular networking
To enhance the value of its practical applications theweb server for iEzy-Drug has been established that canbe freely accessible at httpwwwjci-bioinfocniEzy-DrugIt is anticipated that the web server will become a usefulhigh throughput tool for both basic research and drug
development in the relevant areas or at the very least playa complementary role to the existing method [39 110 118] forwhich so far noweb-serverwhatsoever has been provided yet
32 The Protocol or User Guide For the convenience of thevast majority of biologists and pharmaceutical scientists herelet us provide a step-by-step guide to show how the userscan easily get the desired result by means of the web serverwithout the need to follow the complicated mathematicalequations presented in this paper for the process of develop-ing the predictor and its integrityStep 1 Open the web server at the site httpwwwjci-bio-infocniEzy-Drug and you will see the top page of thepredictor on your computer screen as shown in Figure 5Click on the ReadMe button to see a brief introduction aboutiEzy-Drug predictor and the caveat when using itStep 2 Either type or copypaste the query pairs into theinput box at the center of Figure 5 Each query pair consistsof two parts one is for the protein sequence and the other forthe drug The enzyme sequence should be in FASTA formatwhile the drug in the KEGG code Examples for the querypairs input can be seen by clicking on the Example buttonright above the input boxStep 3 Click on the Submit button to see the predicted resultFor example if you use the four query pairs in the Example
10 BioMed Research International
window as the input after clicking the Submit button youwill see on your screen that the ldquohsa 10056rdquo enzyme andthe ldquoD0021rdquo drug are an interactive pair and that the ldquohsa100rdquo enzyme and the ldquoD0037rdquo drug are also an interactivepair but that the ldquohsa 3295rdquo enzyme and the ldquoD00889rdquo drugare not an interactive pair and that the ldquohsa 7366rdquo enzymeand the ldquoD03601rdquo drug are not an interactive pair eitherAll these results are fully consistent with the experimentalobservations It takes about 3 minutes before the results areshown on the screenStep 4 Click on the Citation button to find the relevant paperthat documents the detailed development and algorithm ofiEzy-DurgStep 5 Click on the Data button to download the benchmarkdataset used to train and test the iEzy-Durg predictorStep 6 The program code is also available by clicking thebutton download on the lower panel of Figure 5
Acknowledgments
The authors would like to thank the three anonymousreviewers whose constructive comments are very helpful forstrengthening the presentation of the paper This work wassupported by the Grants from the National Natural ScienceFoundation of China (no 31260273) the Jiangxi ProvincialForeign Scientific and Technological Cooperation Project(no 20120BDH80023) Natural Science Foundation of JiangxiProvince China (no 2010GZS0122 20122BAB201020) theDepartment of Education of Jiangxi Province (GJJ12490)the LuoDi plan of the Department of Education of JiangxiProvince (KJLD12083) and the Jiangxi Provincial Founda-tion for Leaders of Disciplines in Science (20113BCB22008)The funders had no role in study design data collection andanalysis decision to publish or preparation of the paper
References
[1] A Bairoch ldquoThe ENZYME database in 2000rdquo Nucleic AcidsResearch vol 28 no 1 pp 304ndash305 2000
[2] S H Koenig and R D Brown ldquoH2CO3as substrate for carbonic
anhydrase in the dehydration of H2CO3(minus)rdquo Proceedings of the
National Academy of Sciences of the United States of Americavol 69 no 9 pp 2422ndash2425 1972
[3] S X Lin and J Lapointe ldquoTheoretical and experimental biologyin onerdquo Journal of Biomedical Science and Engineering vol 6 pp435ndash442 2013
[4] K C Chou and S P Jiang ldquoStudies on the rate of diffusion-controlled reactions of enzymes Spatial factor and force fieldfactorrdquo Scientia Sinica vol 17 no 5 pp 664ndash680 1974
[5] K C Chou ldquoThe kinetics of the combination reaction betweenenzyme and substraterdquo Scientia Sinica vol 19 no 4 pp 505ndash528 1976
[6] K C Chou and G P Zhou ldquoRole of the protein outside activesite on the diffusion-controlled reaction of enzymerdquo Journal ofthe American Chemical Society vol 104 no 5 pp 1409ndash14131982
[7] R A Poorman A G Tomasselli R L Heinrikson and FJ Kezdy ldquoA cumulative specificity model for proteases from
human immunodeficiency virus types 1 and 2 inferred fromstatistical analysis of an extended substrate data baserdquo TheJournal of Biological Chemistry vol 266 no 22 pp 14554ndash145611991
[8] K-C Chou ldquoA vectorized sequence-coupling model for pre-dicting HIV protease cleavage sites in proteinsrdquo The Journal ofBiological Chemistry vol 268 no 23 pp 16938ndash16948 1993
[9] G Z Liang and S Z Li ldquoA new sequence representation asapplied in better specificity elucidation for human immunod-eficiency virus type 1 proteaserdquo Biopolymers vol 88 no 3 pp401ndash412 2007
[10] J J Chou ldquoPredicting cleavability of peptide sequences byHIVprotease via correlation-angle approachrdquo Journal of ProteinChemistry vol 12 no 3 pp 291ndash302 1993
[11] Q S Du S Wang D Q Wei S Sirois and K C ChouldquoMolecular modeling and chemical modification for findingpeptide inhibitor against severe acute respiratory syndromecoronavirus main proteinaserdquo Analytical Biochemistry vol 337no 2 pp 262ndash270 2005
[12] Y Gan H Huang Y Huang et al ldquoSynthesis and activity ofan octapeptide inhibitor designed for SARS coronavirus mainproteinaserdquo Peptides vol 27 no 4 pp 622ndash625 2006
[13] Q S Du H Sun and K C Chou ldquoInhibitor design for SARScoronavirus main protease based on lsquodistorted key theoryrsquordquoMedicinal Chemistry vol 3 no 1 pp 1ndash6 2007
[14] K C Chou ldquoPrediction of human immunodeficiency virusprotease cleavage sites in proteinsrdquoAnalytical Biochemistry vol233 no 1 pp 1ndash14 1996
[15] J Knowles and G Gromo ldquoTarget selection in drug discoveryrdquoNature Reviews Drug Discovery vol 2 no 1 pp 63ndash69 2003
[16] A C Cheng R G Coleman K T Smyth et al ldquoStructure-basedmaximal affinity model predicts small-molecule druggabilityrdquoNature Biotechnology vol 25 no 1 pp 71ndash75 2007
[17] M Rarey B Kramer T Lengauer and G Klebe ldquoA fast flexibledocking method using an incremental construction algorithmrdquoJournal of Molecular Biology vol 261 no 3 pp 470ndash489 1996
[18] K C Chou ldquoReview structural bioinformatics and its impactto biomedical sciencerdquo Current Medicinal Chemistry vol 11 no16 pp 2105ndash2134 2004
[19] K C Chou D Q Wei and W Z Zhong ldquoBinding mechanismof coronavirus main proteinase with ligands and its implicationto drug design against SARSrdquo Biochemical and BiophysicalResearch Communications vol 308 pp 148ndash151 2003 (Erratumin Biochemical and Biophysical Research Communications vol310 p 675 2003)
[20] K C Chou D Q Wei Q S Du S Sirois and W Z ZhongldquoProgress in computational approach to drug developmentagaint SARSrdquo Current Medicinal Chemistry vol 13 no 27 pp3263ndash3270 2006
[21] G P Zhou and F A Troy ldquoNMR studies on how the bindingcomplex of polyisoprenol recognition sequence peptides andpolyisoprenols can modulate membrane structurerdquo CurrentProtein and Peptide Science vol 6 no 5 pp 399ndash411 2005
[22] R B Huang Q S Du C H Wang and K C Chou ldquoAn in-depth analysis of the biological functional studies based on theNMR M2 channel structure of influenza A virusrdquo Biochemicaland Biophysical Research Communications vol 377 no 4 pp1243ndash1247 2008
[23] Q S Du R B Huang C H Wang X M Li and K C ChouldquoEnergetic analysis of the two controversial drug binding sitesof the M2 proton channel in influenza A virusrdquo Journal ofTheoretical Biology vol 259 no 1 pp 159ndash164 2009
BioMed Research International 11
[24] M J Berardi W M Shih S C Harrison and J J Chou ldquoMito-chondrial uncoupling protein 2 structure determined by NMRmolecular fragment searchingrdquo Nature vol 476 no 7358 pp109ndash114 2011
[25] J R Schnell and J J Chou ldquoStructure andmechanism of theM2proton channel of influenza A virusrdquo Nature vol 451 no 7178pp 591ndash595 2008
[26] B OuYang S Xie M J Berardi et al ldquoUnusual architecture ofthe p7 channel from hepatitis C virusrdquoNature vol 498 pp 521ndash525 2013
[27] K Oxenoid and J J Chou ldquoThe structure of phospholambanpentamer reveals a channel-like architecture in membranesrdquoProceedings of the National Academy of Sciences of the UnitedStates of America vol 102 no 31 pp 10870ndash10875 2005
[28] M E Call KWWucherpfennig and J J Chou ldquoThe structuralbasis for intramembrane assembly of an activating immunore-ceptor complexrdquo Nature Immunology vol 11 no 11 pp 1023ndash1029 2010
[29] R M Pielak J R Schnell and J J Chou ldquoMechanism of druginhibition and drug resistance of influenza A M2 channelrdquoProceedings of the National Academy of Sciences of the UnitedStates of America vol 106 no 18 pp 7379ndash7384 2009
[30] J Wang R M Pielak M A McClintock and J J ChouldquoSolution structure and functional analysis of the influenza Bproton channelrdquo Nature Structural and Molecular Biology vol16 no 12 pp 1267ndash1271 2009
[31] K C Chou ldquoCoupling interaction between thromboxane A2receptor and alpha-13 subunit of guanine nucleotide-bindingproteinrdquo Journal of Proteome Research vol 4 no 5 pp 1681ndash1686 2005
[32] K C Chou ldquoInsights from modeling three-dimensional struc-tures of the human potassium and sodium channelsrdquo Journal ofProteome Research vol 3 no 4 pp 856ndash861 2004
[33] K C Chou ldquoSome remarks on protein attribute prediction andpseudo amino acid compositionrdquo Journal ofTheoretical Biologyvol 273 no 1 pp 236ndash247 2011
[34] W Z Lin J A Fang X Xiao and K C Chou ldquoiLoc-animal amulti-label learning classifier for predicting subcellular local-ization of animal proteinsrdquo Molecular BioSystems vol 9 pp634ndash644 2013
[35] X Xiao PWangW Z Lin J H Jia and K C Chou ldquoiAMP-2La two-level multi-label classifier for identifying antimicrobialpeptides and their functional typesrdquo Analytical Biochemistryvol 436 pp 168ndash177 2013
[36] W Chen P M Feng H Lin and K C Chou ldquoiRSpot-PseDNCidentify recombination spots with pseudo dinucleotide compo-sitionrdquo Nucleic Acids Research vol 41 article e69 2013
[37] K C Chou Z C Wu and X Xiao ldquoILoc-Hum using theaccumulation-label scale to predict subcellular locations ofhuman proteins with both single and multiple sitesrdquoMolecularBioSystems vol 8 no 2 pp 629ndash641 2012
[38] M Kotera M Hirakawa T Tokimatsu S Goto and MKanehisa ldquoThe KEGG databases and tools facilitating omicsanalysis latest developments involving human diseases andpharmaceuticalsrdquo Methods in Molecular Biology vol 802 pp19ndash39 2012
[39] Y YamanishiM Araki A GutteridgeWHonda andM Kane-hisa ldquoPrediction of drug-target interaction networks from theintegration of chemical and genomic spacesrdquo Bioinformaticsvol 24 no 13 pp i232ndashi240 2008
[40] P Finn S Muggleton D Page and A Srinivasan ldquoPharma-cophore discovery using the Inductive Logic Programmingsystem PROGOLrdquo Machine Learning vol 30 no 2-3 pp 241ndash270 1998
[41] I Vogt D Stumpfe H E Ahmed and J Bajorath ldquoMethodsfor computer-aided chemical biology Part 2 evaluation of com-pound selectivity using 2D molecular fingerprintsrdquo ChemicalBiology and Drug Design vol 70 pp 195ndash205 2007
[42] H Eckert and J Bajorath ldquoMolecular similarity analysis in vir-tual screening foundations limitations and novel approachesrdquoDrug Discovery Today vol 12 no 5-6 pp 225ndash233 2007
[43] S Laurent L V Elst and R N Muller ldquoComparative studyof the physicochemical properties of six clinical low molecularweight gadolinium contrast agentsrdquo Contrast Media amp Molecu-lar Imaging vol 1 no 3 pp 128ndash137 2006
[44] E Gregori-Puigjane R Garriga-Sust and J Mestres ldquoIndexingmolecules with chemical graph identifiersrdquo Journal of Compu-tational Chemistry vol 32 no 12 pp 2638ndash2646 2011
[45] B Ren ldquoApplication of novel atom-type AI topological indicesto QSPR studies of alkanesrdquo Computers and Chemistry vol 26no 4 pp 357ndash369 2002
[46] N M OrsquoBoyle M Banck C A James C Morley T Vander-meersch and G R Hutchison ldquoOpen Babel an open chemicaltoolboxrdquo Journal of Cheminformatics vol 3 p 33 2011
[47] V J Gillet P Willett and J Bradshaw ldquoSimilarity searchingusing reduced graphsrdquo Journal of Chemical Information andComputer Sciences vol 43 no 2 pp 338ndash345 2003
[48] D Butina ldquoUnsupervised data base clustering based on day-lightrsquos fingerprint and Tanimoto similarity a fast and automatedway to cluster small and large data setsrdquo Journal of ChemicalInformation and Computer Sciences vol 39 no 4 pp 747ndash7501999
[49] C E Shannon ldquoA mathematical theory of communicationrdquoACM SIGMOBILE Mobile Computing and CommunicationsReview vol 5 pp 3ndash55 2001
[50] V D Gusev L A Nemytikova and N A Chuzhanova ldquoOn thecomplexity measures of genetic sequencesrdquo Bioinformatics vol15 no 12 pp 994ndash999 1999
[51] K C Chou and H B Shen ldquoReview recent progress in proteinsubcellular location predictionrdquo Analytical Biochemistry vol370 no 1 pp 1ndash16 2007
[52] S F Altschul ldquoEvaluating the statistical significance of multipledistinct local alignmentsrdquo in Theoretical and ComputationalMethods in Genome Research S Suhai Ed pp 1ndash14 PlenumNew York NY USA 1997
[53] J C Wootton and S Federhen ldquoStatistics of local complexity inamino acid sequences and sequence databasesrdquo Computers andChemistry vol 17 no 2 pp 149ndash163 1993
[54] H Nakashima K Nishikawa and T Ooi ldquoThe folding type ofa protein is relevant to the amino acid compositionrdquo Journal ofBiochemistry vol 99 no 1 pp 153ndash162 1986
[55] K C Chou and C T Zhang ldquoPredicting protein folding typesby distance functions that make allowances for amino acidinteractionsrdquo The Journal of Biological Chemistry vol 269 no35 pp 22014ndash22020 1994
[56] K-C Chou ldquoA novel approach to predicting protein structuralclasses in a (20-1)-D amino acid composition spacerdquo Proteinsvol 21 no 4 pp 319ndash344 1995
[57] H Nakashima and K Nishikawa ldquoDiscrimination of intracel-lular and extracellular proteins using amino acid compositionand residue-pair frequenciesrdquo Journal of Molecular Biology vol238 no 1 pp 54ndash61 1994
12 BioMed Research International
[58] G P Zhou ldquoAn intriguing controversy over protein structuralclass predictionrdquo Journal of Protein Chemistry vol 17 no 8 pp729ndash738 1998
[59] I Bahar A R Atilgan R L Jernigan and B Erman ldquoUnder-standing the recognition of protein structural classes by aminoacid compositionrdquo Proteins vol 29 pp 172ndash185 1997
[60] J Cedano P Aloy J A Perez-Pons and E Querol ldquoRelationbetween amino acid composition and cellular location ofproteinsrdquo Journal of Molecular Biology vol 266 no 3 pp 594ndash600 1997
[61] G P Zhou and K Doctor ldquoSubcellular location prediction ofapoptosis proteinsrdquo Proteins vol 50 no 1 pp 44ndash48 2003
[62] KChou ldquoPrediction of protein cellular attributes using pseudo-amino acid compositionrdquo Proteins vol 43 pp 246ndash255 2001(Erratum in Proteins vol 44 p 60 2001)
[63] K C Chou ldquoUsing amphiphilic pseudo amino acid composi-tion to predict enzyme subfamily classesrdquo Bioinformatics vol21 no 1 pp 10ndash19 2005
[64] D Zou Z He J He and Y Xia ldquoSupersecondary structureprediction using Choursquos pseudo amino acid compositionrdquoJournal of Computational Chemistry vol 32 no 2 pp 271ndash2782011
[65] M M Beigi M Behjati and H Mohabatkar ldquoPrediction ofmetalloproteinase family based on the concept ofChoursquos pseudoamino acid composition using a machine learning approachrdquoJournal of Structural and Functional Genomics vol 12 no 4 pp191ndash197 2011
[66] Y K Chen and K B Li ldquoPredicting membrane protein typesby incorporating protein topology domains signal peptidesand physicochemical properties into the general form of Choursquospseudo amino acid compositionrdquo Journal ofTheoretical Biologyvol 318 pp 1ndash12 2013
[67] C Huang and J Q Yuan ldquoA multilabel model based on Choursquospseudo-amino acid composition for identifying membraneproteins with both single and multiple functional typesrdquo TheJournal of Membrane Biology vol 246 pp 327ndash334 2013
[68] S S Sahu and G Panda ldquoA novel feature representationmethod based on Choursquos pseudo amino acid composition forprotein structural class predictionrdquo Computational Biology andChemistry vol 34 no 5-6 pp 320ndash327 2010
[69] M Hayat and A Khan ldquoDiscriminating outer membraneproteins with fuzzy K-nearest neighbor algorithms based on thegeneral form of Choursquos PseAACrdquo Protein and Peptide Lettersvol 19 no 4 pp 411ndash421 2012
[70] MKhosravian F K FaramarziMM BeigiM Behbahani andH Mohabatkar ldquoPredicting antibacterial peptides by the con-cept of Choursquos pseudo-amino acid composition and machinelearning methodsrdquo Protein amp Peptide Letters vol 20 pp 180ndash186 2013
[71] HMohabatkarMM Beigi K Abdolahi and SMohsenzadehldquoPrediction of allergenic proteins by means of the concept ofChoursquos pseudo amino acid composition and amachine learningapproachrdquoMedicinal Chemistry vol 9 pp 133ndash137 2013
[72] L Nanni A Lumini D Gupta and A Garg ldquoIdentifyingbacterial virulent proteins by fusing a set of classifiers basedon variants of Choursquos pseudo amino acid composition and onevolutionary informationrdquo IEEEACM Transactions on Compu-tational Biology and Bioinformatics vol 9 no 2 pp 467ndash4752012
[73] T H Chang L C Wu T Y Lee S P Chen H D Huang andJ T Horng ldquoEuLoc a web-server for accurately predict protein
subcellular localization in eukaryotes by incorporating variousfeatures of sequence segments into the general form of ChoursquosPseAACrdquo Journal of Computer-Aided Molecular Design vol 27pp 91ndash103 2013
[74] S Zhang Y Zhang H Yang C Zhao and Q Pan ldquoUsing theconcept of Choursquos pseudo amino acid composition to predictprotein subcellular localization an approach by incorporatingevolutionary information and von Neumann entropiesrdquo AminoAcids vol 34 no 4 pp 565ndash572 2008
[75] R Zia Ur and A Khan ldquoIdentifying GPCRs and their typeswith Choursquos pseudo amino acid composition an approach frommulti-scale energy representation and position specific scoringmatrixrdquo Protein amp Peptide Letters vol 19 pp 890ndash903 2012
[76] X Y Sun S P Shi J D Qiu S B Suo S Y Huang and R PLiang ldquoIdentifying protein quaternary structural attributes byincorporating physicochemical properties into the general formof Choursquos PseAAC via discrete wavelet transformrdquo MolecularBioSystems vol 8 pp 3178ndash3184 2012
[77] L Nanni and A Lumini ldquoGenetic programming for creatingChoursquos pseudo amino acid based features for submitochondrialocalizationrdquo Amino Acids vol 34 no 4 pp 653ndash660 2008
[78] M Esmaeili H Mohabatkar and S Mohsenzadeh ldquoUsing theconcept of Choursquos pseudo amino acid composition for risk typeprediction of human papillomavirusesrdquo Journal of TheoreticalBiology vol 263 no 2 pp 203ndash209 2010
[79] H Mohabatkar ldquoPrediction of cyclin proteins using Choursquospseudo amino acid compositionrdquo Protein and Peptide Lettersvol 17 no 10 pp 1207ndash1214 2010
[80] H Mohabatkar M M Beigi and A Esmaeili ldquoPredictionof GABA(A) receptor proteins using the concept of Choursquospseudo-amino acid composition and support vector machinerdquoJournal of Theoretical Biology vol 281 no 1 pp 18ndash23 2011
[81] Y Xu J Ding L Y Wu and K C Chou ldquoiSNO-PseAAC pre-dict cysteine S-nitrosylation sites in proteins by incorporatingposition specific amino acid propensity into pseudo amino acidcompositionrdquo PLoS ONE vol 8 Article ID e55844 2013
[82] W Chen H Lin PM Feng C Ding Y C Zuo andK C ChouldquoiNuc-PhysChem a sequence-based predictor for identifyingnucleosomes via physicochemical propertiesrdquo PLoS ONE vol7 Article ID e47843 2012
[83] B Li T Huang L Liu Y Cai and K C Chou ldquoIdentificationof colorectal cancer related genes with mrmr and shortest pathin protein-protein interaction networkrdquo PLoS ONE vol 7 no 4Article ID e33393 2012
[84] Y Jiang T Huang C Lei Y F Gao Y D Cai and K C ChouldquoSignal propagation in protein interaction network duringcolorectal cancer progressionrdquo BioMed Research Internationalvol 2013 Article ID 287019 9 pages 2013
[85] P Du X Wang C Xu and Y Gao ldquoPseAAC-Builder across-platform stand-alone program for generating variousspecial Choursquos pseudo-amino acid compositionsrdquo AnalyticalBiochemistry vol 425 no 2 pp 117ndash119 2012
[86] D S Cao Q S Xu and Y Z Liang ldquoPropy a tool to generatevarious modes of Choursquos PseAACrdquo Bioinformatics vol 29 pp960ndash962 2013
[87] H Shen and K C Chou ldquoPseAAC a flexible web serverfor generating various kinds of protein pseudo amino acidcompositionrdquo Analytical Biochemistry vol 373 no 2 pp 386ndash388 2008
[88] K C Chou ldquoUsing pair-coupled amino acid composition topredict protein secondary structure contentrdquo Journal of ProteinChemistry vol 18 no 4 pp 473ndash480 1999
BioMed Research International 13
[89] W Liu and K C Chou ldquoPrediction of protein secondarystructure contentrdquo Protein Engineering vol 12 no 12 pp 1041ndash1050 1999
[90] M Bhasin and G P S Raghava ldquoClassification of nuclearreceptors based on amino acid composition and dipeptidecompositionrdquo The Journal of Biological Chemistry vol 279 no22 pp 23262ndash23266 2004
[91] H Lin andH Ding ldquoPredicting ion channels and their types bythe dipeptidemode of pseudo amino acid compositionrdquo Journalof Theoretical Biology vol 269 no 1 pp 64ndash69 2011
[92] H Lin and Q Li ldquoUsing pseudo amino acid composition topredict protein structural class approached by incorporating400 dipeptide componentsrdquo Journal of Computational Chem-istry vol 28 no 9 pp 1463ndash1466 2007
[93] L Nanni and A Lumini ldquoCombing ontologies and dipeptidecomposition for predicting DNA-binding proteinsrdquo AminoAcids vol 34 no 4 pp 635ndash641 2008
[94] K-C Chou ldquoThe convergence-divergence duality in lectindomains of selectin family and its implicationsrdquo FEBS Lettersvol 363 no 1-2 pp 123ndash126 1995
[95] A A Schaffer L Aravind T L Madden et al ldquoImprovingthe accuracy of PSI-BLAST protein database searches withcomposition-based statistics and other refinementsrdquo NucleicAcids Research vol 29 no 14 pp 2994ndash3005 2001
[96] S F Altschul and E V Koonin ldquoIterated profile searches withPSI-BLAST a tool for discovery in protein databasesrdquo Trends inBiochemical Sciences vol 23 no 11 pp 444ndash447 1998
[97] J Deng ldquoGrey entropy and grey target decision makingrdquoJournal of Grey System vol 22 no 1 pp 1ndash24 2010
[98] B M Bolstad R A Irizarry M Astrand and T P Speed ldquoAcomparison of normalizationmethods for high density oligonu-cleotide array data based on variance and biasrdquo Bioinformaticsvol 19 no 2 pp 185ndash193 2003
[99] J-Y Shi S-W Zhang Q Pan Y-M Cheng and J XieldquoPrediction of protein subcellular localization by support vectormachines using multi-scale energy and pseudo amino acidcompositionrdquo Amino Acids vol 33 no 1 pp 69ndash74 2007
[100] T Denoeux ldquoA 120581-nearest neighbor classification rule based onDempster-Shafer theoryrdquo IEEE Transactions on Systems Manand Cybernetics vol 25 no 5 pp 804ndash813 1995
[101] J M Keller M R Gray and J A Givens ldquoA fuzzy k-nearestneighbours algorithmrdquo IEEE Transactions on Systems Man andCybernetics vol 15 no 4 pp 580ndash585 1985
[102] X Xiao P Wang and K Chou ldquoiNR-physchem a sequence-based predictor for identifying nuclear receptors and theirsubfamilies via physical-chemical property matrixrdquo PLoS ONEvol 7 no 2 Article ID e30869 2012
[103] I Roterman L Konieczny W Jurkowski K Prymula and MBanach ldquoTwo-intermediate model to characterize the structureof fast-folding proteinsrdquo Journal of Theoretical Biology vol 283no 1 pp 60ndash70 2011
[104] X Xiao P Wang and K C Chou ldquoGPCR-2L predicting Gprotein-coupled receptors and their types by hybridizing twodifferentmodes of pseudo amino acid compositionsrdquoMolecularBioSystems vol 7 no 3 pp 911ndash919 2011
[105] X Xiao P Wang and K C Chou ldquoQuat-2L a web-server forpredicting protein quaternary structural attributesrdquo MolecularDiversity vol 15 no 1 pp 149ndash155 2011
[106] X Zheng C Li and J Wang ldquoAn information-theoreticapproach to the prediction of protein structural classrdquo Journalof Computational Chemistry vol 31 no 6 pp 1201ndash1206 2010
[107] H Shen J Yang X Liu and K C Chou ldquoUsing supervisedfuzzy clustering to predict protein structural classesrdquo Biochem-ical and Biophysical Research Communications vol 334 no 2pp 577ndash581 2005
[108] K-C Chou and C-T Zhang ldquoReview prediction of proteinstructural classesrdquo Critical Reviews in Biochemistry and Molec-ular Biology vol 30 no 4 pp 275ndash349 1995
[109] P C Mahalanobis ldquoOn the generalized distance in statisticsrdquoProceedings of the National Institute of Sciences of India vol 2pp 49ndash55 1936
[110] R M Centor ldquoSignal detectability the use of ROC curves andtheir analysesrdquoMedical Decision Making vol 11 no 2 pp 102ndash106 1991
[111] K-C Chou ldquoUsing subsite coupling to predict signal peptidesrdquoProtein Engineering vol 14 no 2 pp 75ndash79 2001
[112] K C Chou ldquoPrediction of protein signal sequences and theircleavage sitesrdquo Proteins vol 42 pp 136ndash139 2001
[113] K C Chou ldquoPrediction of signal peptides using scaledwindowrdquoPeptides vol 22 no 12 pp 1973ndash1979 2001
[114] K C Chou ldquoSome remarks on predicting multi-label attributesinmolecular biosystemsrdquoMolecular Biosystems vol 9 pp 1092ndash1100 2013
[115] K C Chou and H B Shen ldquoCell-PLoc 2 0 an improvedpackage of web-servers for predicting subcellular localization ofproteins in various organismsrdquoNatural Science vol 2 pp 1090ndash1103 2010
[116] M Gribskov and N L Robinson ldquoUse of receiver operatingcharacteristic (ROC) analysis to evaluate sequence matchingrdquoComputers and Chemistry vol 20 no 1 pp 25ndash33 1996
[117] Y Yamanishi M Kotera M Kanehisa and S Goto ldquoDrug-target interaction prediction from chemical genomic and phar-macological data in an integrated frameworkrdquo Bioinformaticsvol 26 no 12 pp i246ndashi254 2010
[118] Z He J Zhang X Shi et al ldquoPredicting drug-target interactionnetworks based on functional groups and biological featuresrdquoPLoS ONE vol 5 no 3 Article ID e9603 2010
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

6 BioMed Research International
where G represents the enzyme-drug pair oplus the orthogonalsum [51] and each of the (258 + 480) = 738 feature elementsis given in (8) and (23)
For the convenience of the later formulation let us use119909119894(119894 = 1 2 738) to represent the 738 components of (24)
that is
G = [11990911199092sdot sdot sdot 119909119894sdot sdot sdot 119909738]
T (25)
To optimize the prediction results different weights wereusually tested for each of the elements in (25) However sinceit would consume a lot of computational time for a total of 738weight factors here let us adopt the normalization approachto deal with this problem as done in [98 99] that is convert119909119894in (25) to 119910
119894according to the following equation
119910119894=
2tanminus1 (119909119894)
120587
(119894 = 1 2 738) (26)
where tanminus1 means arctangent By means of (26) everycomponent in (25) will be converted into the range of[minus1 1] that is we have minus1 le 119910
119894le 1 As demonstrated
in [98 99] the normalization approach via (26) was quiteeffective in enhancing the quality of prediction operated ina high dimension space Therefore in this study we wouldnot to take the procedure of optimizing the weight factorssignificantly reducing the computational times
23 Fuzzy 119870-Nearest Neighbour Algorithm The 119870-NN (119870-Nearest Neighbor) classifier is quite popular in patternrecognition community owing to its good performance andsimple-to-use feature According to the 119870-NN rule [100]named also as the ldquovoting 119870-NN rulerdquo the query sampleshould be assigned to the subset represented by a majorityof its 119870 nearest neighbors as illustrated in Figure 5 of [33]
Fuzzy 119870-NN classification method [101] is a specialvariation of the119870-NNclassification family Instead of roughlyassigning the label based on a voting from the 119870 nearestneighbors it attempts to estimate the membership valuesthat indicate how much degree the query sample belongsto the classes concerned Obviously it is impossible for anycharacteristic description to contain complete informationwhich would make the classification ambiguous In view ofthis the fuzzy principle is very reasonable and particularlyuseful in dealing with complicated biological systems such asidentifying nuclear receptor subfamilies [102] characterizingthe structure of fast-folding proteins [103] classifying Gprotein-coupled receptors [104] predicting protein quater-nary structural attributes [105] predicting protein structuralclasses [106 107] and so forth
Next let us give a brief introduction how to use thefuzzy119870-NN approach to identify the interactions between theenzymes and the drug compounds in the network concerned
Supposing that S(119873) = G1G2 G
119873 is a set of vec-
tors representing 119873 enzyme-drug pairs in a training set
classified into two classes 119862+ 119862minus where 119862+ denotes theinteractive pair class while 119862minus the noninteractive pair classSlowast(G) = Glowast
1Glowast2 Glowast
119870 sub S(119873) is the subset of the 119870
nearest neighbor pairs to the query pair G Thus the fuzzymembership value for the query pair G in the two classes ofS(119873) is given by
120583
+
(G) =sum
119870
119895=1120583
+(Glowast119895) 119889(GGlowast
119895)
minus2(120593minus1)
sum
119870
119895=1119889(GGlowast
119895)
minus2(120593minus1)
120583
minus
(G) =sum
119870
119895=1120583
minus(Glowast119895) 119889(GGlowast
119895)
minus2(120593minus1)
sum
119870
119895=1119889(GGlowast
119895)
minus2(120593minus1)
(27)
where 119870 is the number of the nearest neighbors counted forthe query pairG 120583+(Glowast
119895) and 120583minus(Glowast
119895) the fuzzy membership
values of the training sample Glowast119895to the class 119862+ and 119862minus
respectively as will be further defined next 119889(GGlowast119895) the
cosine distance between G and its 119895th nearest pair Glowast119895in
the training dataset S(119873) 120593(gt 1) the fuzzy coefficient fordetermining how heavily the distance is weighted whencalculating each nearest neighborrsquos contribution to the mem-bership value Note that the parameters 119870 and 120593 will affectthe computation result of (27) and they will be optimizedby a grid-search as will be described later Also variousother metrics can be chosen for 119889(GGlowast
119895) such as Euclidean
distance Hamming distance [108] andMahalanobis distance[55 109]
The quantitative definitions for the aforementioned120583
+(Glowast119895) and 120583minus(Glowast
119895) in (27) are given by
120583
+
(Glowast119895) =
1 if Glowast119895isin 119862
+
0 otherwise
120583
minus
(Glowast119895) =
1 if Glowast119895isin 119862
minus
0 otherwise
(28)
Substituting the results obtained by (27) into (28) it followsthat if 120583+(G) gt 120583
minus(G) then the query pair G is an
interactive couple otherwise noninteractive In other wordsthe outcome can be formulated as
G isin
119862
+ if 120583+ (G) gt 120583minus (G)
119862
minus otherwise
(29)
If there is a tie between 120583+(G) and 120583minus(G) the query pair Gwill be randomly assigned to one of the two classes Howevercase like that is quite rare and in this study never happened
The predictor thus established is called iEzy-Drugwhere ldquoirdquo means identify and ldquoEzy-Drugrdquo means the inter-action between enzyme and drug To provide an intuitiveoverall picture a flowchart is provided in Figure 2 to showthe process of how the classifier works in identifying enzyme-drug interactions
BioMed Research International 7
Input a queryprotein sequence and
a drug code
Create Chous PseAAC of a protein
Create feature vector of a drug
Feature fusion
engineTraining dataset
Yes No
Noninteractive enzyme drug pair
Interactive enzymedrug pair
AACDipeptideSeq Evol
Grey model
Mol FptInfo entropy
CF
FKNN operation
Figure 2 A flowchart to show the operation process of the iEzy-Drug predictor See the text for further explanation
24 Criteria for Performance Evaluation In the literaturethe following equation set is often used for examining theperformance quality of a predictor
Sn = TPTP + FN
Sp = TNTN + FP
Acc = TP + TNTP + TN + FP + FN
MCC = (TP times TN) minus (FP times FN)radic(TP + FP) (TP + FN) (TN + FP) (TN + FN)
(30)
where TP represents the true positive TN the true negativeFP the false positive FN the false negative Sn the sensitiv-ity Sp the specificity Acc the accuracy MCC the Mathewrsquoscorrelation coefficient
To most biologists however the four metrics as formu-lated in (30) are not quite intuitive and easier-to-understandparticularly for the Mathewrsquos correlation coefficient Herelet us adopt the Choursquos symbols to formulate the previousfour metrics By means of Choursquos symbols [111 112] the rates
of correct predictions for the interactive enzyme-drug pairsin dataset S+ and the noninteractive enzyme-drug pairs indataset Sminus are respectively defined by (cf (1))
Λ
+
=
119873
+minus 119873
+
minus
119873
+ for the interactive enzyme-drug pairs
Λ
minus
=
119873
minusminus 119873
minus
+
119873
minus for the noninteractive enzyme-drug pairs
(31)
where 119873+ is the total number of the interactive enzyme-drug pairs investigated while 119873+
minusis the number of the
interactive enzyme-drug pairs incorrectly predicted as thenoninteractive enzyme-drug pairs 119873minus is the total numberof the noninteractive enzyme-drug pairs investigated while119873
minus
+is the number of the noninteractive enzyme-drug pairs
incorrectly predicted as the interactive enzyme-drug pairsThe overall success prediction rate is given by [113] as follows
Λ =
Λ
+119873
++ Λ
minus119873
minus
119873
++ 119873
minus= 1 minus
119873
+
minus+ 119873
minus
+
119873
++ 119873
minus
(32)
It is obvious from (31)-(32) that if and only if none ofthe interactive enzyme-drug pairs and the noninteractiveenzyme-drug pairs are mispredicted that is 119873+
minus= 119873
minus
+= 0
and Λ+ = Λ
minus= 1 we have the overall success rate Λ = 1
Otherwise the overall success rate would be smaller than 1The relations between the symbols in (32) and those in
(30) are given by
TP = 119873+ minus 119873+minus
TN = 119873
minus
minus 119873
minus
+
FP = 119873minus+
FN = 119873
+
minus
(33)
Substituting (33) into (30) and also noting (31)-(32) we obtain
Sn = Λ+ = 1 minus119873
+
minus
119873
+
Sp = Λminus = 1 minus119873
minus
+
119873
minus
Acc = Λ = 1 minus119873
+
minus+ 119873
minus
+
119873
++ 119873
minus
MCC =1 minus ((119873
+
minus119873
+) + (119873
minus
+119873
minus))
radic(1 + (119873
minus
+minus 119873
+
minus) 119873
+) (1 + (119873
+
minusminus 119873
minus
+) 119873
minus)
(34)
Now it is obvious to see from (34) when 119873
+
minus= 0
meaning none of the interactive enzyme-drug pairs was
8 BioMed Research International
02
46
810
1
15
2089
0895
09
0905
091
0915
092
ACC
K120593
Figure 3 A 3D plot to show how the parameter in (27) wasoptimized for the iEzy-Drug predictor
mispredicted to be a noninteractive enzyme-drug pair wehave the sensitivity Sn = 1 while 119873+
minus= 119873
+ meaning thatall the interactive enzyme-drug pairs weremispredicted to bethe noninteractive enzyme-drug pairs we have the sensitivitySn = 0 Likewise when 119873minus
+= 0 meaning none of the
noninteractive enzyme-drug pairs wasmispredicted we havethe specificity Sp = 1 while 119873minus
+= 119873
minus meaning all thenoninteractive enzyme-drug pairs were incorrectly predictedas interactive enzyme-drug pairs we have the specificity Sp =0 When119873+
minus= 119873
minus
+= 0 meaning that none of the interactive
enzyme-drug pairs in the dataset S+ and none of the nonin-teractive enzyme-drug pairs in Sminus was incorrectly predictedwe have the overall accuracy Acc = Λ = 1 while 119873+
minus= 119873
+
and 119873minus+= 119873
minus meaning that all the interactive enzyme-drugpairs in the dataset S+ and all the noninteractive enzyme-drug pairs in Sminus were mispredicted we have the overallaccuracy Acc = Λ = 0 The MCC correlation coefficient isusually used for measuring the quality of binary (two-class)classifications When 119873+
minus= 119873
minus
+= 0 meaning that none
of the interactive enzyme-drug pairs in the dataset S+ andnone of the noninteractive enzyme-drug pairs in Sminus weremispredicted we have MCC = 1 when 119873+
minus= 119873
+2 and
119873
minus
+= 119873
minus2 we have MCC = 0 meaning no better than
random prediction when 119873+minus= 119873
+ and 119873minus+= 119873
minus we haveMCC = minus1 meaning total disagreement between predictionand observation As we can see from the previous discussionit is much more intuitive and easier-to-understand whenusing (34) to examine a predictor for its sensitivity specificityoverall accuracy and Mathewrsquos correlation coefficient It isinstructive to point out that themetrics as defined in (30) and(34) are valid for single label systems for multilabel systemsa set of more complicated metrics should be used as given in[114]
3 Results and Discussion
31 Cross-Validation How to properly examine the predic-tion quality is a key for developing a new predictor and
False positive rate
True
pos
itive
rate
1
01
02
03
04
05
06
07
08
09
0
101 02 03 04 05 06 07 08 090
BaselineiEzy-Drug AUC = 09377
Figure 4 A plot for the ROC curve to quantitatively show theperformance of the iEzy-Drug predictor
estimating its potential application value Generally speak-ing the following three cross-validation methods are oftenused to examine a predictor of its effectiveness in practicalapplication independent dataset test subsampling or 119870-fold(such as 5-fold 7-fold or 10-fold) test and jackknife test[108] However as elaborated by a penetrating analysis in[115] considerable arbitrariness exists in the independentdataset test Also as demonstrated by (27)ndash(29) in [33]the subsampling test (or 119870-fold cross-validation) cannotavoid arbitrariness either Only the jackknife test is the leastarbitrary that can always yield a unique result for a givenbenchmark dataset Therefore the jackknife test has beenwidely recognized and increasingly utilized by investigatorsto examine the quality of various predictors (see eg [66 7174 80]) Accordingly the success rate by the jackknife test wasalso used to optimize the two uncertain parameters 119870 and 120593in (27) The result thus obtained is shown in Figure 3 fromwhich we obtain when 119870 = 6 and 120593 = 15 the iEzy-Drugpredictor reaches its optimized status
The success rates thus obtained by the jackknife test inidentifying interactive Enzyme-drug pairs or noninteractiveenzyme-drug pairs on the benchmark dataset S (cf OnlineSupporting Information S1) are given in Table 1 where forfacilitating comparison the corresponding result by He et al[110] is also given As we can see from the table the overallaccuracy Acc achieved by iEzy-Drug was 9103 remarkablyhigher than 8548 the corresponding rate obtained byHe et al [110] on the same benchmark Furthermore listedin Table 1 are also the values obtained by iEzy-Drug for theother three metrics that is Sn = 9081 Sp = 9114 andMCC = 8039 indicating that the accuracy of iEzy-Drug isnot only very high but also quite stale
To provide a graphical illustration to show the per-formance of the current binary classifier iEzy-Drug asits discrimination threshold is varied a 2D plot calledReceiver Operating Characteristic (ROC) curve [116 117]was also given (Figure 4) In the ROC curve the verticalcoordinate 119884 is for the true positive rate or Sn (cf (34))while horizontal coordinate 119883 for the false positive rate
BioMed Research International 9
Read Me Data Citation
Enter both the protein sequence and the drug code (example) the number of query pairs is limited at 10 or less for each submission
Submit Clear
You can download the program and run it on your own local computer
iEzy-Drug identifying the interaction between enzymes anddrugs in cellular networking
Figure 5 A semiscreenshot to show the top page of the iEzy-Drugweb-server Its web-site address is at httpwwwjci-bioinfocniEzy-Drug
Table 1 The jackknife success rates obtained with iEzy-Drug in identifying interactive enzyme-drug pairs and noninteractive enzyme-drugpairs for the benchmark dataset S (cf Online Supporting Information S1)
Method Acc Sn Sp MCC
iEzy-Druga 74258157 = 9103 24692719 = 9081 49565438 = 9114 8039NN predictorb 8548 NA NA NAaSee (27) where the parameters119870 = 6 and 120593 = 15bSee [110]
or 1-Sp The best possible prediction method would yielda point with the coordinate (0 1) representing 100 truepositive rate (sensitivity Sn) and 0 false positive rate or100 specificity Therefore the (0 1) point is also calleda perfect classification A completely random guess wouldgive a point along a diagonal from the point (0 0) to (1 1)The area under the ROC curve also called Area Under theROC (AUROC) is often used to indicate the performancequality of a binary classifier the value 05 of AUROCis equivalent to random prediction while 1 of AUROCrepresents a perfect one As we can see from Figure 4the AUROC value obtained by iEzy-Drug is 09377
The reason why iEzy-Drug can remarkably improve theprediction quality is that it has introduced the 2D molecularfingerprints to represent drug samples see Online SupportingInformation S3 for the detailed fingerprint expressions for thedrugs listed in Online Supporting Information S1 and thatit has successfully used PseAAC to incorporate the featuresderived from the sequences of enzymes that are essentialfor identifying the interaction of enzymes with drugs in thecellular networking
To enhance the value of its practical applications theweb server for iEzy-Drug has been established that canbe freely accessible at httpwwwjci-bioinfocniEzy-DrugIt is anticipated that the web server will become a usefulhigh throughput tool for both basic research and drug
development in the relevant areas or at the very least playa complementary role to the existing method [39 110 118] forwhich so far noweb-serverwhatsoever has been provided yet
32 The Protocol or User Guide For the convenience of thevast majority of biologists and pharmaceutical scientists herelet us provide a step-by-step guide to show how the userscan easily get the desired result by means of the web serverwithout the need to follow the complicated mathematicalequations presented in this paper for the process of develop-ing the predictor and its integrityStep 1 Open the web server at the site httpwwwjci-bio-infocniEzy-Drug and you will see the top page of thepredictor on your computer screen as shown in Figure 5Click on the ReadMe button to see a brief introduction aboutiEzy-Drug predictor and the caveat when using itStep 2 Either type or copypaste the query pairs into theinput box at the center of Figure 5 Each query pair consistsof two parts one is for the protein sequence and the other forthe drug The enzyme sequence should be in FASTA formatwhile the drug in the KEGG code Examples for the querypairs input can be seen by clicking on the Example buttonright above the input boxStep 3 Click on the Submit button to see the predicted resultFor example if you use the four query pairs in the Example
10 BioMed Research International
window as the input after clicking the Submit button youwill see on your screen that the ldquohsa 10056rdquo enzyme andthe ldquoD0021rdquo drug are an interactive pair and that the ldquohsa100rdquo enzyme and the ldquoD0037rdquo drug are also an interactivepair but that the ldquohsa 3295rdquo enzyme and the ldquoD00889rdquo drugare not an interactive pair and that the ldquohsa 7366rdquo enzymeand the ldquoD03601rdquo drug are not an interactive pair eitherAll these results are fully consistent with the experimentalobservations It takes about 3 minutes before the results areshown on the screenStep 4 Click on the Citation button to find the relevant paperthat documents the detailed development and algorithm ofiEzy-DurgStep 5 Click on the Data button to download the benchmarkdataset used to train and test the iEzy-Durg predictorStep 6 The program code is also available by clicking thebutton download on the lower panel of Figure 5
Acknowledgments
The authors would like to thank the three anonymousreviewers whose constructive comments are very helpful forstrengthening the presentation of the paper This work wassupported by the Grants from the National Natural ScienceFoundation of China (no 31260273) the Jiangxi ProvincialForeign Scientific and Technological Cooperation Project(no 20120BDH80023) Natural Science Foundation of JiangxiProvince China (no 2010GZS0122 20122BAB201020) theDepartment of Education of Jiangxi Province (GJJ12490)the LuoDi plan of the Department of Education of JiangxiProvince (KJLD12083) and the Jiangxi Provincial Founda-tion for Leaders of Disciplines in Science (20113BCB22008)The funders had no role in study design data collection andanalysis decision to publish or preparation of the paper
References
[1] A Bairoch ldquoThe ENZYME database in 2000rdquo Nucleic AcidsResearch vol 28 no 1 pp 304ndash305 2000
[2] S H Koenig and R D Brown ldquoH2CO3as substrate for carbonic
anhydrase in the dehydration of H2CO3(minus)rdquo Proceedings of the
National Academy of Sciences of the United States of Americavol 69 no 9 pp 2422ndash2425 1972
[3] S X Lin and J Lapointe ldquoTheoretical and experimental biologyin onerdquo Journal of Biomedical Science and Engineering vol 6 pp435ndash442 2013
[4] K C Chou and S P Jiang ldquoStudies on the rate of diffusion-controlled reactions of enzymes Spatial factor and force fieldfactorrdquo Scientia Sinica vol 17 no 5 pp 664ndash680 1974
[5] K C Chou ldquoThe kinetics of the combination reaction betweenenzyme and substraterdquo Scientia Sinica vol 19 no 4 pp 505ndash528 1976
[6] K C Chou and G P Zhou ldquoRole of the protein outside activesite on the diffusion-controlled reaction of enzymerdquo Journal ofthe American Chemical Society vol 104 no 5 pp 1409ndash14131982
[7] R A Poorman A G Tomasselli R L Heinrikson and FJ Kezdy ldquoA cumulative specificity model for proteases from
human immunodeficiency virus types 1 and 2 inferred fromstatistical analysis of an extended substrate data baserdquo TheJournal of Biological Chemistry vol 266 no 22 pp 14554ndash145611991
[8] K-C Chou ldquoA vectorized sequence-coupling model for pre-dicting HIV protease cleavage sites in proteinsrdquo The Journal ofBiological Chemistry vol 268 no 23 pp 16938ndash16948 1993
[9] G Z Liang and S Z Li ldquoA new sequence representation asapplied in better specificity elucidation for human immunod-eficiency virus type 1 proteaserdquo Biopolymers vol 88 no 3 pp401ndash412 2007
[10] J J Chou ldquoPredicting cleavability of peptide sequences byHIVprotease via correlation-angle approachrdquo Journal of ProteinChemistry vol 12 no 3 pp 291ndash302 1993
[11] Q S Du S Wang D Q Wei S Sirois and K C ChouldquoMolecular modeling and chemical modification for findingpeptide inhibitor against severe acute respiratory syndromecoronavirus main proteinaserdquo Analytical Biochemistry vol 337no 2 pp 262ndash270 2005
[12] Y Gan H Huang Y Huang et al ldquoSynthesis and activity ofan octapeptide inhibitor designed for SARS coronavirus mainproteinaserdquo Peptides vol 27 no 4 pp 622ndash625 2006
[13] Q S Du H Sun and K C Chou ldquoInhibitor design for SARScoronavirus main protease based on lsquodistorted key theoryrsquordquoMedicinal Chemistry vol 3 no 1 pp 1ndash6 2007
[14] K C Chou ldquoPrediction of human immunodeficiency virusprotease cleavage sites in proteinsrdquoAnalytical Biochemistry vol233 no 1 pp 1ndash14 1996
[15] J Knowles and G Gromo ldquoTarget selection in drug discoveryrdquoNature Reviews Drug Discovery vol 2 no 1 pp 63ndash69 2003
[16] A C Cheng R G Coleman K T Smyth et al ldquoStructure-basedmaximal affinity model predicts small-molecule druggabilityrdquoNature Biotechnology vol 25 no 1 pp 71ndash75 2007
[17] M Rarey B Kramer T Lengauer and G Klebe ldquoA fast flexibledocking method using an incremental construction algorithmrdquoJournal of Molecular Biology vol 261 no 3 pp 470ndash489 1996
[18] K C Chou ldquoReview structural bioinformatics and its impactto biomedical sciencerdquo Current Medicinal Chemistry vol 11 no16 pp 2105ndash2134 2004
[19] K C Chou D Q Wei and W Z Zhong ldquoBinding mechanismof coronavirus main proteinase with ligands and its implicationto drug design against SARSrdquo Biochemical and BiophysicalResearch Communications vol 308 pp 148ndash151 2003 (Erratumin Biochemical and Biophysical Research Communications vol310 p 675 2003)
[20] K C Chou D Q Wei Q S Du S Sirois and W Z ZhongldquoProgress in computational approach to drug developmentagaint SARSrdquo Current Medicinal Chemistry vol 13 no 27 pp3263ndash3270 2006
[21] G P Zhou and F A Troy ldquoNMR studies on how the bindingcomplex of polyisoprenol recognition sequence peptides andpolyisoprenols can modulate membrane structurerdquo CurrentProtein and Peptide Science vol 6 no 5 pp 399ndash411 2005
[22] R B Huang Q S Du C H Wang and K C Chou ldquoAn in-depth analysis of the biological functional studies based on theNMR M2 channel structure of influenza A virusrdquo Biochemicaland Biophysical Research Communications vol 377 no 4 pp1243ndash1247 2008
[23] Q S Du R B Huang C H Wang X M Li and K C ChouldquoEnergetic analysis of the two controversial drug binding sitesof the M2 proton channel in influenza A virusrdquo Journal ofTheoretical Biology vol 259 no 1 pp 159ndash164 2009
BioMed Research International 11
[24] M J Berardi W M Shih S C Harrison and J J Chou ldquoMito-chondrial uncoupling protein 2 structure determined by NMRmolecular fragment searchingrdquo Nature vol 476 no 7358 pp109ndash114 2011
[25] J R Schnell and J J Chou ldquoStructure andmechanism of theM2proton channel of influenza A virusrdquo Nature vol 451 no 7178pp 591ndash595 2008
[26] B OuYang S Xie M J Berardi et al ldquoUnusual architecture ofthe p7 channel from hepatitis C virusrdquoNature vol 498 pp 521ndash525 2013
[27] K Oxenoid and J J Chou ldquoThe structure of phospholambanpentamer reveals a channel-like architecture in membranesrdquoProceedings of the National Academy of Sciences of the UnitedStates of America vol 102 no 31 pp 10870ndash10875 2005
[28] M E Call KWWucherpfennig and J J Chou ldquoThe structuralbasis for intramembrane assembly of an activating immunore-ceptor complexrdquo Nature Immunology vol 11 no 11 pp 1023ndash1029 2010
[29] R M Pielak J R Schnell and J J Chou ldquoMechanism of druginhibition and drug resistance of influenza A M2 channelrdquoProceedings of the National Academy of Sciences of the UnitedStates of America vol 106 no 18 pp 7379ndash7384 2009
[30] J Wang R M Pielak M A McClintock and J J ChouldquoSolution structure and functional analysis of the influenza Bproton channelrdquo Nature Structural and Molecular Biology vol16 no 12 pp 1267ndash1271 2009
[31] K C Chou ldquoCoupling interaction between thromboxane A2receptor and alpha-13 subunit of guanine nucleotide-bindingproteinrdquo Journal of Proteome Research vol 4 no 5 pp 1681ndash1686 2005
[32] K C Chou ldquoInsights from modeling three-dimensional struc-tures of the human potassium and sodium channelsrdquo Journal ofProteome Research vol 3 no 4 pp 856ndash861 2004
[33] K C Chou ldquoSome remarks on protein attribute prediction andpseudo amino acid compositionrdquo Journal ofTheoretical Biologyvol 273 no 1 pp 236ndash247 2011
[34] W Z Lin J A Fang X Xiao and K C Chou ldquoiLoc-animal amulti-label learning classifier for predicting subcellular local-ization of animal proteinsrdquo Molecular BioSystems vol 9 pp634ndash644 2013
[35] X Xiao PWangW Z Lin J H Jia and K C Chou ldquoiAMP-2La two-level multi-label classifier for identifying antimicrobialpeptides and their functional typesrdquo Analytical Biochemistryvol 436 pp 168ndash177 2013
[36] W Chen P M Feng H Lin and K C Chou ldquoiRSpot-PseDNCidentify recombination spots with pseudo dinucleotide compo-sitionrdquo Nucleic Acids Research vol 41 article e69 2013
[37] K C Chou Z C Wu and X Xiao ldquoILoc-Hum using theaccumulation-label scale to predict subcellular locations ofhuman proteins with both single and multiple sitesrdquoMolecularBioSystems vol 8 no 2 pp 629ndash641 2012
[38] M Kotera M Hirakawa T Tokimatsu S Goto and MKanehisa ldquoThe KEGG databases and tools facilitating omicsanalysis latest developments involving human diseases andpharmaceuticalsrdquo Methods in Molecular Biology vol 802 pp19ndash39 2012
[39] Y YamanishiM Araki A GutteridgeWHonda andM Kane-hisa ldquoPrediction of drug-target interaction networks from theintegration of chemical and genomic spacesrdquo Bioinformaticsvol 24 no 13 pp i232ndashi240 2008
[40] P Finn S Muggleton D Page and A Srinivasan ldquoPharma-cophore discovery using the Inductive Logic Programmingsystem PROGOLrdquo Machine Learning vol 30 no 2-3 pp 241ndash270 1998
[41] I Vogt D Stumpfe H E Ahmed and J Bajorath ldquoMethodsfor computer-aided chemical biology Part 2 evaluation of com-pound selectivity using 2D molecular fingerprintsrdquo ChemicalBiology and Drug Design vol 70 pp 195ndash205 2007
[42] H Eckert and J Bajorath ldquoMolecular similarity analysis in vir-tual screening foundations limitations and novel approachesrdquoDrug Discovery Today vol 12 no 5-6 pp 225ndash233 2007
[43] S Laurent L V Elst and R N Muller ldquoComparative studyof the physicochemical properties of six clinical low molecularweight gadolinium contrast agentsrdquo Contrast Media amp Molecu-lar Imaging vol 1 no 3 pp 128ndash137 2006
[44] E Gregori-Puigjane R Garriga-Sust and J Mestres ldquoIndexingmolecules with chemical graph identifiersrdquo Journal of Compu-tational Chemistry vol 32 no 12 pp 2638ndash2646 2011
[45] B Ren ldquoApplication of novel atom-type AI topological indicesto QSPR studies of alkanesrdquo Computers and Chemistry vol 26no 4 pp 357ndash369 2002
[46] N M OrsquoBoyle M Banck C A James C Morley T Vander-meersch and G R Hutchison ldquoOpen Babel an open chemicaltoolboxrdquo Journal of Cheminformatics vol 3 p 33 2011
[47] V J Gillet P Willett and J Bradshaw ldquoSimilarity searchingusing reduced graphsrdquo Journal of Chemical Information andComputer Sciences vol 43 no 2 pp 338ndash345 2003
[48] D Butina ldquoUnsupervised data base clustering based on day-lightrsquos fingerprint and Tanimoto similarity a fast and automatedway to cluster small and large data setsrdquo Journal of ChemicalInformation and Computer Sciences vol 39 no 4 pp 747ndash7501999
[49] C E Shannon ldquoA mathematical theory of communicationrdquoACM SIGMOBILE Mobile Computing and CommunicationsReview vol 5 pp 3ndash55 2001
[50] V D Gusev L A Nemytikova and N A Chuzhanova ldquoOn thecomplexity measures of genetic sequencesrdquo Bioinformatics vol15 no 12 pp 994ndash999 1999
[51] K C Chou and H B Shen ldquoReview recent progress in proteinsubcellular location predictionrdquo Analytical Biochemistry vol370 no 1 pp 1ndash16 2007
[52] S F Altschul ldquoEvaluating the statistical significance of multipledistinct local alignmentsrdquo in Theoretical and ComputationalMethods in Genome Research S Suhai Ed pp 1ndash14 PlenumNew York NY USA 1997
[53] J C Wootton and S Federhen ldquoStatistics of local complexity inamino acid sequences and sequence databasesrdquo Computers andChemistry vol 17 no 2 pp 149ndash163 1993
[54] H Nakashima K Nishikawa and T Ooi ldquoThe folding type ofa protein is relevant to the amino acid compositionrdquo Journal ofBiochemistry vol 99 no 1 pp 153ndash162 1986
[55] K C Chou and C T Zhang ldquoPredicting protein folding typesby distance functions that make allowances for amino acidinteractionsrdquo The Journal of Biological Chemistry vol 269 no35 pp 22014ndash22020 1994
[56] K-C Chou ldquoA novel approach to predicting protein structuralclasses in a (20-1)-D amino acid composition spacerdquo Proteinsvol 21 no 4 pp 319ndash344 1995
[57] H Nakashima and K Nishikawa ldquoDiscrimination of intracel-lular and extracellular proteins using amino acid compositionand residue-pair frequenciesrdquo Journal of Molecular Biology vol238 no 1 pp 54ndash61 1994
12 BioMed Research International
[58] G P Zhou ldquoAn intriguing controversy over protein structuralclass predictionrdquo Journal of Protein Chemistry vol 17 no 8 pp729ndash738 1998
[59] I Bahar A R Atilgan R L Jernigan and B Erman ldquoUnder-standing the recognition of protein structural classes by aminoacid compositionrdquo Proteins vol 29 pp 172ndash185 1997
[60] J Cedano P Aloy J A Perez-Pons and E Querol ldquoRelationbetween amino acid composition and cellular location ofproteinsrdquo Journal of Molecular Biology vol 266 no 3 pp 594ndash600 1997
[61] G P Zhou and K Doctor ldquoSubcellular location prediction ofapoptosis proteinsrdquo Proteins vol 50 no 1 pp 44ndash48 2003
[62] KChou ldquoPrediction of protein cellular attributes using pseudo-amino acid compositionrdquo Proteins vol 43 pp 246ndash255 2001(Erratum in Proteins vol 44 p 60 2001)
[63] K C Chou ldquoUsing amphiphilic pseudo amino acid composi-tion to predict enzyme subfamily classesrdquo Bioinformatics vol21 no 1 pp 10ndash19 2005
[64] D Zou Z He J He and Y Xia ldquoSupersecondary structureprediction using Choursquos pseudo amino acid compositionrdquoJournal of Computational Chemistry vol 32 no 2 pp 271ndash2782011
[65] M M Beigi M Behjati and H Mohabatkar ldquoPrediction ofmetalloproteinase family based on the concept ofChoursquos pseudoamino acid composition using a machine learning approachrdquoJournal of Structural and Functional Genomics vol 12 no 4 pp191ndash197 2011
[66] Y K Chen and K B Li ldquoPredicting membrane protein typesby incorporating protein topology domains signal peptidesand physicochemical properties into the general form of Choursquospseudo amino acid compositionrdquo Journal ofTheoretical Biologyvol 318 pp 1ndash12 2013
[67] C Huang and J Q Yuan ldquoA multilabel model based on Choursquospseudo-amino acid composition for identifying membraneproteins with both single and multiple functional typesrdquo TheJournal of Membrane Biology vol 246 pp 327ndash334 2013
[68] S S Sahu and G Panda ldquoA novel feature representationmethod based on Choursquos pseudo amino acid composition forprotein structural class predictionrdquo Computational Biology andChemistry vol 34 no 5-6 pp 320ndash327 2010
[69] M Hayat and A Khan ldquoDiscriminating outer membraneproteins with fuzzy K-nearest neighbor algorithms based on thegeneral form of Choursquos PseAACrdquo Protein and Peptide Lettersvol 19 no 4 pp 411ndash421 2012
[70] MKhosravian F K FaramarziMM BeigiM Behbahani andH Mohabatkar ldquoPredicting antibacterial peptides by the con-cept of Choursquos pseudo-amino acid composition and machinelearning methodsrdquo Protein amp Peptide Letters vol 20 pp 180ndash186 2013
[71] HMohabatkarMM Beigi K Abdolahi and SMohsenzadehldquoPrediction of allergenic proteins by means of the concept ofChoursquos pseudo amino acid composition and amachine learningapproachrdquoMedicinal Chemistry vol 9 pp 133ndash137 2013
[72] L Nanni A Lumini D Gupta and A Garg ldquoIdentifyingbacterial virulent proteins by fusing a set of classifiers basedon variants of Choursquos pseudo amino acid composition and onevolutionary informationrdquo IEEEACM Transactions on Compu-tational Biology and Bioinformatics vol 9 no 2 pp 467ndash4752012
[73] T H Chang L C Wu T Y Lee S P Chen H D Huang andJ T Horng ldquoEuLoc a web-server for accurately predict protein
subcellular localization in eukaryotes by incorporating variousfeatures of sequence segments into the general form of ChoursquosPseAACrdquo Journal of Computer-Aided Molecular Design vol 27pp 91ndash103 2013
[74] S Zhang Y Zhang H Yang C Zhao and Q Pan ldquoUsing theconcept of Choursquos pseudo amino acid composition to predictprotein subcellular localization an approach by incorporatingevolutionary information and von Neumann entropiesrdquo AminoAcids vol 34 no 4 pp 565ndash572 2008
[75] R Zia Ur and A Khan ldquoIdentifying GPCRs and their typeswith Choursquos pseudo amino acid composition an approach frommulti-scale energy representation and position specific scoringmatrixrdquo Protein amp Peptide Letters vol 19 pp 890ndash903 2012
[76] X Y Sun S P Shi J D Qiu S B Suo S Y Huang and R PLiang ldquoIdentifying protein quaternary structural attributes byincorporating physicochemical properties into the general formof Choursquos PseAAC via discrete wavelet transformrdquo MolecularBioSystems vol 8 pp 3178ndash3184 2012
[77] L Nanni and A Lumini ldquoGenetic programming for creatingChoursquos pseudo amino acid based features for submitochondrialocalizationrdquo Amino Acids vol 34 no 4 pp 653ndash660 2008
[78] M Esmaeili H Mohabatkar and S Mohsenzadeh ldquoUsing theconcept of Choursquos pseudo amino acid composition for risk typeprediction of human papillomavirusesrdquo Journal of TheoreticalBiology vol 263 no 2 pp 203ndash209 2010
[79] H Mohabatkar ldquoPrediction of cyclin proteins using Choursquospseudo amino acid compositionrdquo Protein and Peptide Lettersvol 17 no 10 pp 1207ndash1214 2010
[80] H Mohabatkar M M Beigi and A Esmaeili ldquoPredictionof GABA(A) receptor proteins using the concept of Choursquospseudo-amino acid composition and support vector machinerdquoJournal of Theoretical Biology vol 281 no 1 pp 18ndash23 2011
[81] Y Xu J Ding L Y Wu and K C Chou ldquoiSNO-PseAAC pre-dict cysteine S-nitrosylation sites in proteins by incorporatingposition specific amino acid propensity into pseudo amino acidcompositionrdquo PLoS ONE vol 8 Article ID e55844 2013
[82] W Chen H Lin PM Feng C Ding Y C Zuo andK C ChouldquoiNuc-PhysChem a sequence-based predictor for identifyingnucleosomes via physicochemical propertiesrdquo PLoS ONE vol7 Article ID e47843 2012
[83] B Li T Huang L Liu Y Cai and K C Chou ldquoIdentificationof colorectal cancer related genes with mrmr and shortest pathin protein-protein interaction networkrdquo PLoS ONE vol 7 no 4Article ID e33393 2012
[84] Y Jiang T Huang C Lei Y F Gao Y D Cai and K C ChouldquoSignal propagation in protein interaction network duringcolorectal cancer progressionrdquo BioMed Research Internationalvol 2013 Article ID 287019 9 pages 2013
[85] P Du X Wang C Xu and Y Gao ldquoPseAAC-Builder across-platform stand-alone program for generating variousspecial Choursquos pseudo-amino acid compositionsrdquo AnalyticalBiochemistry vol 425 no 2 pp 117ndash119 2012
[86] D S Cao Q S Xu and Y Z Liang ldquoPropy a tool to generatevarious modes of Choursquos PseAACrdquo Bioinformatics vol 29 pp960ndash962 2013
[87] H Shen and K C Chou ldquoPseAAC a flexible web serverfor generating various kinds of protein pseudo amino acidcompositionrdquo Analytical Biochemistry vol 373 no 2 pp 386ndash388 2008
[88] K C Chou ldquoUsing pair-coupled amino acid composition topredict protein secondary structure contentrdquo Journal of ProteinChemistry vol 18 no 4 pp 473ndash480 1999
BioMed Research International 13
[89] W Liu and K C Chou ldquoPrediction of protein secondarystructure contentrdquo Protein Engineering vol 12 no 12 pp 1041ndash1050 1999
[90] M Bhasin and G P S Raghava ldquoClassification of nuclearreceptors based on amino acid composition and dipeptidecompositionrdquo The Journal of Biological Chemistry vol 279 no22 pp 23262ndash23266 2004
[91] H Lin andH Ding ldquoPredicting ion channels and their types bythe dipeptidemode of pseudo amino acid compositionrdquo Journalof Theoretical Biology vol 269 no 1 pp 64ndash69 2011
[92] H Lin and Q Li ldquoUsing pseudo amino acid composition topredict protein structural class approached by incorporating400 dipeptide componentsrdquo Journal of Computational Chem-istry vol 28 no 9 pp 1463ndash1466 2007
[93] L Nanni and A Lumini ldquoCombing ontologies and dipeptidecomposition for predicting DNA-binding proteinsrdquo AminoAcids vol 34 no 4 pp 635ndash641 2008
[94] K-C Chou ldquoThe convergence-divergence duality in lectindomains of selectin family and its implicationsrdquo FEBS Lettersvol 363 no 1-2 pp 123ndash126 1995
[95] A A Schaffer L Aravind T L Madden et al ldquoImprovingthe accuracy of PSI-BLAST protein database searches withcomposition-based statistics and other refinementsrdquo NucleicAcids Research vol 29 no 14 pp 2994ndash3005 2001
[96] S F Altschul and E V Koonin ldquoIterated profile searches withPSI-BLAST a tool for discovery in protein databasesrdquo Trends inBiochemical Sciences vol 23 no 11 pp 444ndash447 1998
[97] J Deng ldquoGrey entropy and grey target decision makingrdquoJournal of Grey System vol 22 no 1 pp 1ndash24 2010
[98] B M Bolstad R A Irizarry M Astrand and T P Speed ldquoAcomparison of normalizationmethods for high density oligonu-cleotide array data based on variance and biasrdquo Bioinformaticsvol 19 no 2 pp 185ndash193 2003
[99] J-Y Shi S-W Zhang Q Pan Y-M Cheng and J XieldquoPrediction of protein subcellular localization by support vectormachines using multi-scale energy and pseudo amino acidcompositionrdquo Amino Acids vol 33 no 1 pp 69ndash74 2007
[100] T Denoeux ldquoA 120581-nearest neighbor classification rule based onDempster-Shafer theoryrdquo IEEE Transactions on Systems Manand Cybernetics vol 25 no 5 pp 804ndash813 1995
[101] J M Keller M R Gray and J A Givens ldquoA fuzzy k-nearestneighbours algorithmrdquo IEEE Transactions on Systems Man andCybernetics vol 15 no 4 pp 580ndash585 1985
[102] X Xiao P Wang and K Chou ldquoiNR-physchem a sequence-based predictor for identifying nuclear receptors and theirsubfamilies via physical-chemical property matrixrdquo PLoS ONEvol 7 no 2 Article ID e30869 2012
[103] I Roterman L Konieczny W Jurkowski K Prymula and MBanach ldquoTwo-intermediate model to characterize the structureof fast-folding proteinsrdquo Journal of Theoretical Biology vol 283no 1 pp 60ndash70 2011
[104] X Xiao P Wang and K C Chou ldquoGPCR-2L predicting Gprotein-coupled receptors and their types by hybridizing twodifferentmodes of pseudo amino acid compositionsrdquoMolecularBioSystems vol 7 no 3 pp 911ndash919 2011
[105] X Xiao P Wang and K C Chou ldquoQuat-2L a web-server forpredicting protein quaternary structural attributesrdquo MolecularDiversity vol 15 no 1 pp 149ndash155 2011
[106] X Zheng C Li and J Wang ldquoAn information-theoreticapproach to the prediction of protein structural classrdquo Journalof Computational Chemistry vol 31 no 6 pp 1201ndash1206 2010
[107] H Shen J Yang X Liu and K C Chou ldquoUsing supervisedfuzzy clustering to predict protein structural classesrdquo Biochem-ical and Biophysical Research Communications vol 334 no 2pp 577ndash581 2005
[108] K-C Chou and C-T Zhang ldquoReview prediction of proteinstructural classesrdquo Critical Reviews in Biochemistry and Molec-ular Biology vol 30 no 4 pp 275ndash349 1995
[109] P C Mahalanobis ldquoOn the generalized distance in statisticsrdquoProceedings of the National Institute of Sciences of India vol 2pp 49ndash55 1936
[110] R M Centor ldquoSignal detectability the use of ROC curves andtheir analysesrdquoMedical Decision Making vol 11 no 2 pp 102ndash106 1991
[111] K-C Chou ldquoUsing subsite coupling to predict signal peptidesrdquoProtein Engineering vol 14 no 2 pp 75ndash79 2001
[112] K C Chou ldquoPrediction of protein signal sequences and theircleavage sitesrdquo Proteins vol 42 pp 136ndash139 2001
[113] K C Chou ldquoPrediction of signal peptides using scaledwindowrdquoPeptides vol 22 no 12 pp 1973ndash1979 2001
[114] K C Chou ldquoSome remarks on predicting multi-label attributesinmolecular biosystemsrdquoMolecular Biosystems vol 9 pp 1092ndash1100 2013
[115] K C Chou and H B Shen ldquoCell-PLoc 2 0 an improvedpackage of web-servers for predicting subcellular localization ofproteins in various organismsrdquoNatural Science vol 2 pp 1090ndash1103 2010
[116] M Gribskov and N L Robinson ldquoUse of receiver operatingcharacteristic (ROC) analysis to evaluate sequence matchingrdquoComputers and Chemistry vol 20 no 1 pp 25ndash33 1996
[117] Y Yamanishi M Kotera M Kanehisa and S Goto ldquoDrug-target interaction prediction from chemical genomic and phar-macological data in an integrated frameworkrdquo Bioinformaticsvol 26 no 12 pp i246ndashi254 2010
[118] Z He J Zhang X Shi et al ldquoPredicting drug-target interactionnetworks based on functional groups and biological featuresrdquoPLoS ONE vol 5 no 3 Article ID e9603 2010
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology
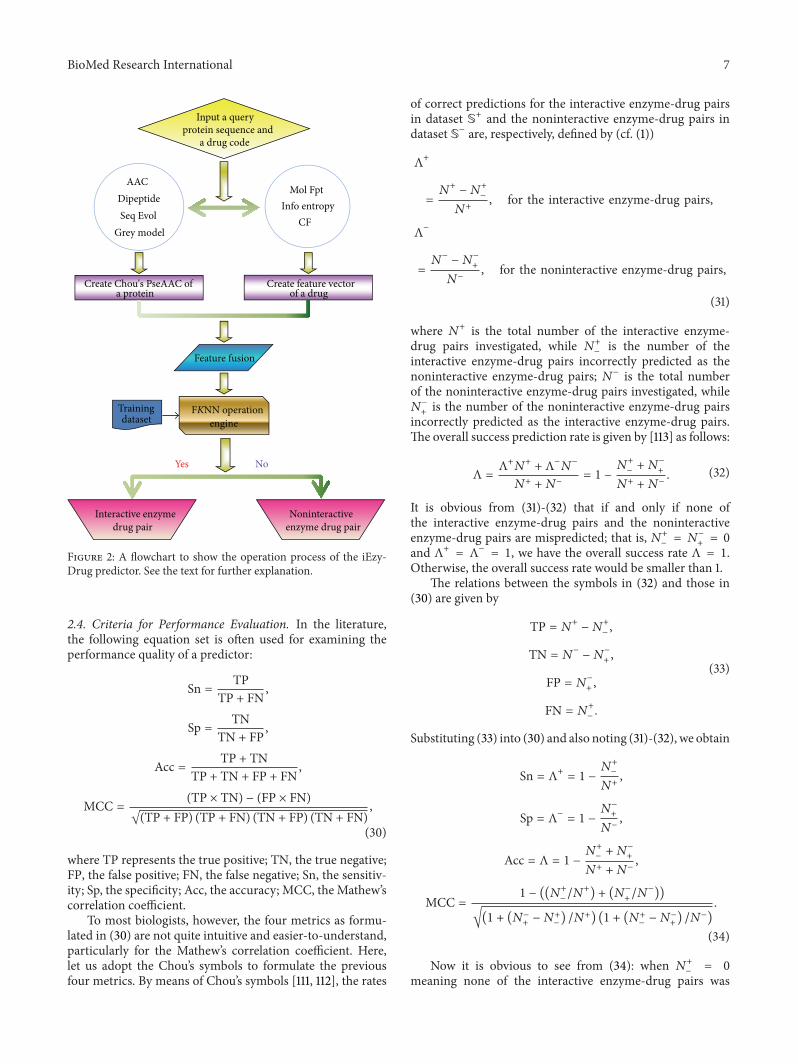
BioMed Research International 7
Input a queryprotein sequence and
a drug code
Create Chous PseAAC of a protein
Create feature vector of a drug
Feature fusion
engineTraining dataset
Yes No
Noninteractive enzyme drug pair
Interactive enzymedrug pair
AACDipeptideSeq Evol
Grey model
Mol FptInfo entropy
CF
FKNN operation
Figure 2 A flowchart to show the operation process of the iEzy-Drug predictor See the text for further explanation
24 Criteria for Performance Evaluation In the literaturethe following equation set is often used for examining theperformance quality of a predictor
Sn = TPTP + FN
Sp = TNTN + FP
Acc = TP + TNTP + TN + FP + FN
MCC = (TP times TN) minus (FP times FN)radic(TP + FP) (TP + FN) (TN + FP) (TN + FN)
(30)
where TP represents the true positive TN the true negativeFP the false positive FN the false negative Sn the sensitiv-ity Sp the specificity Acc the accuracy MCC the Mathewrsquoscorrelation coefficient
To most biologists however the four metrics as formu-lated in (30) are not quite intuitive and easier-to-understandparticularly for the Mathewrsquos correlation coefficient Herelet us adopt the Choursquos symbols to formulate the previousfour metrics By means of Choursquos symbols [111 112] the rates
of correct predictions for the interactive enzyme-drug pairsin dataset S+ and the noninteractive enzyme-drug pairs indataset Sminus are respectively defined by (cf (1))
Λ
+
=
119873
+minus 119873
+
minus
119873
+ for the interactive enzyme-drug pairs
Λ
minus
=
119873
minusminus 119873
minus
+
119873
minus for the noninteractive enzyme-drug pairs
(31)
where 119873+ is the total number of the interactive enzyme-drug pairs investigated while 119873+
minusis the number of the
interactive enzyme-drug pairs incorrectly predicted as thenoninteractive enzyme-drug pairs 119873minus is the total numberof the noninteractive enzyme-drug pairs investigated while119873
minus
+is the number of the noninteractive enzyme-drug pairs
incorrectly predicted as the interactive enzyme-drug pairsThe overall success prediction rate is given by [113] as follows
Λ =
Λ
+119873
++ Λ
minus119873
minus
119873
++ 119873
minus= 1 minus
119873
+
minus+ 119873
minus
+
119873
++ 119873
minus
(32)
It is obvious from (31)-(32) that if and only if none ofthe interactive enzyme-drug pairs and the noninteractiveenzyme-drug pairs are mispredicted that is 119873+
minus= 119873
minus
+= 0
and Λ+ = Λ
minus= 1 we have the overall success rate Λ = 1
Otherwise the overall success rate would be smaller than 1The relations between the symbols in (32) and those in
(30) are given by
TP = 119873+ minus 119873+minus
TN = 119873
minus
minus 119873
minus
+
FP = 119873minus+
FN = 119873
+
minus
(33)
Substituting (33) into (30) and also noting (31)-(32) we obtain
Sn = Λ+ = 1 minus119873
+
minus
119873
+
Sp = Λminus = 1 minus119873
minus
+
119873
minus
Acc = Λ = 1 minus119873
+
minus+ 119873
minus
+
119873
++ 119873
minus
MCC =1 minus ((119873
+
minus119873
+) + (119873
minus
+119873
minus))
radic(1 + (119873
minus
+minus 119873
+
minus) 119873
+) (1 + (119873
+
minusminus 119873
minus
+) 119873
minus)
(34)
Now it is obvious to see from (34) when 119873
+
minus= 0
meaning none of the interactive enzyme-drug pairs was
8 BioMed Research International
02
46
810
1
15
2089
0895
09
0905
091
0915
092
ACC
K120593
Figure 3 A 3D plot to show how the parameter in (27) wasoptimized for the iEzy-Drug predictor
mispredicted to be a noninteractive enzyme-drug pair wehave the sensitivity Sn = 1 while 119873+
minus= 119873
+ meaning thatall the interactive enzyme-drug pairs weremispredicted to bethe noninteractive enzyme-drug pairs we have the sensitivitySn = 0 Likewise when 119873minus
+= 0 meaning none of the
noninteractive enzyme-drug pairs wasmispredicted we havethe specificity Sp = 1 while 119873minus
+= 119873
minus meaning all thenoninteractive enzyme-drug pairs were incorrectly predictedas interactive enzyme-drug pairs we have the specificity Sp =0 When119873+
minus= 119873
minus
+= 0 meaning that none of the interactive
enzyme-drug pairs in the dataset S+ and none of the nonin-teractive enzyme-drug pairs in Sminus was incorrectly predictedwe have the overall accuracy Acc = Λ = 1 while 119873+
minus= 119873
+
and 119873minus+= 119873
minus meaning that all the interactive enzyme-drugpairs in the dataset S+ and all the noninteractive enzyme-drug pairs in Sminus were mispredicted we have the overallaccuracy Acc = Λ = 0 The MCC correlation coefficient isusually used for measuring the quality of binary (two-class)classifications When 119873+
minus= 119873
minus
+= 0 meaning that none
of the interactive enzyme-drug pairs in the dataset S+ andnone of the noninteractive enzyme-drug pairs in Sminus weremispredicted we have MCC = 1 when 119873+
minus= 119873
+2 and
119873
minus
+= 119873
minus2 we have MCC = 0 meaning no better than
random prediction when 119873+minus= 119873
+ and 119873minus+= 119873
minus we haveMCC = minus1 meaning total disagreement between predictionand observation As we can see from the previous discussionit is much more intuitive and easier-to-understand whenusing (34) to examine a predictor for its sensitivity specificityoverall accuracy and Mathewrsquos correlation coefficient It isinstructive to point out that themetrics as defined in (30) and(34) are valid for single label systems for multilabel systemsa set of more complicated metrics should be used as given in[114]
3 Results and Discussion
31 Cross-Validation How to properly examine the predic-tion quality is a key for developing a new predictor and
False positive rate
True
pos
itive
rate
1
01
02
03
04
05
06
07
08
09
0
101 02 03 04 05 06 07 08 090
BaselineiEzy-Drug AUC = 09377
Figure 4 A plot for the ROC curve to quantitatively show theperformance of the iEzy-Drug predictor
estimating its potential application value Generally speak-ing the following three cross-validation methods are oftenused to examine a predictor of its effectiveness in practicalapplication independent dataset test subsampling or 119870-fold(such as 5-fold 7-fold or 10-fold) test and jackknife test[108] However as elaborated by a penetrating analysis in[115] considerable arbitrariness exists in the independentdataset test Also as demonstrated by (27)ndash(29) in [33]the subsampling test (or 119870-fold cross-validation) cannotavoid arbitrariness either Only the jackknife test is the leastarbitrary that can always yield a unique result for a givenbenchmark dataset Therefore the jackknife test has beenwidely recognized and increasingly utilized by investigatorsto examine the quality of various predictors (see eg [66 7174 80]) Accordingly the success rate by the jackknife test wasalso used to optimize the two uncertain parameters 119870 and 120593in (27) The result thus obtained is shown in Figure 3 fromwhich we obtain when 119870 = 6 and 120593 = 15 the iEzy-Drugpredictor reaches its optimized status
The success rates thus obtained by the jackknife test inidentifying interactive Enzyme-drug pairs or noninteractiveenzyme-drug pairs on the benchmark dataset S (cf OnlineSupporting Information S1) are given in Table 1 where forfacilitating comparison the corresponding result by He et al[110] is also given As we can see from the table the overallaccuracy Acc achieved by iEzy-Drug was 9103 remarkablyhigher than 8548 the corresponding rate obtained byHe et al [110] on the same benchmark Furthermore listedin Table 1 are also the values obtained by iEzy-Drug for theother three metrics that is Sn = 9081 Sp = 9114 andMCC = 8039 indicating that the accuracy of iEzy-Drug isnot only very high but also quite stale
To provide a graphical illustration to show the per-formance of the current binary classifier iEzy-Drug asits discrimination threshold is varied a 2D plot calledReceiver Operating Characteristic (ROC) curve [116 117]was also given (Figure 4) In the ROC curve the verticalcoordinate 119884 is for the true positive rate or Sn (cf (34))while horizontal coordinate 119883 for the false positive rate
BioMed Research International 9
Read Me Data Citation
Enter both the protein sequence and the drug code (example) the number of query pairs is limited at 10 or less for each submission
Submit Clear
You can download the program and run it on your own local computer
iEzy-Drug identifying the interaction between enzymes anddrugs in cellular networking
Figure 5 A semiscreenshot to show the top page of the iEzy-Drugweb-server Its web-site address is at httpwwwjci-bioinfocniEzy-Drug
Table 1 The jackknife success rates obtained with iEzy-Drug in identifying interactive enzyme-drug pairs and noninteractive enzyme-drugpairs for the benchmark dataset S (cf Online Supporting Information S1)
Method Acc Sn Sp MCC
iEzy-Druga 74258157 = 9103 24692719 = 9081 49565438 = 9114 8039NN predictorb 8548 NA NA NAaSee (27) where the parameters119870 = 6 and 120593 = 15bSee [110]
or 1-Sp The best possible prediction method would yielda point with the coordinate (0 1) representing 100 truepositive rate (sensitivity Sn) and 0 false positive rate or100 specificity Therefore the (0 1) point is also calleda perfect classification A completely random guess wouldgive a point along a diagonal from the point (0 0) to (1 1)The area under the ROC curve also called Area Under theROC (AUROC) is often used to indicate the performancequality of a binary classifier the value 05 of AUROCis equivalent to random prediction while 1 of AUROCrepresents a perfect one As we can see from Figure 4the AUROC value obtained by iEzy-Drug is 09377
The reason why iEzy-Drug can remarkably improve theprediction quality is that it has introduced the 2D molecularfingerprints to represent drug samples see Online SupportingInformation S3 for the detailed fingerprint expressions for thedrugs listed in Online Supporting Information S1 and thatit has successfully used PseAAC to incorporate the featuresderived from the sequences of enzymes that are essentialfor identifying the interaction of enzymes with drugs in thecellular networking
To enhance the value of its practical applications theweb server for iEzy-Drug has been established that canbe freely accessible at httpwwwjci-bioinfocniEzy-DrugIt is anticipated that the web server will become a usefulhigh throughput tool for both basic research and drug
development in the relevant areas or at the very least playa complementary role to the existing method [39 110 118] forwhich so far noweb-serverwhatsoever has been provided yet
32 The Protocol or User Guide For the convenience of thevast majority of biologists and pharmaceutical scientists herelet us provide a step-by-step guide to show how the userscan easily get the desired result by means of the web serverwithout the need to follow the complicated mathematicalequations presented in this paper for the process of develop-ing the predictor and its integrityStep 1 Open the web server at the site httpwwwjci-bio-infocniEzy-Drug and you will see the top page of thepredictor on your computer screen as shown in Figure 5Click on the ReadMe button to see a brief introduction aboutiEzy-Drug predictor and the caveat when using itStep 2 Either type or copypaste the query pairs into theinput box at the center of Figure 5 Each query pair consistsof two parts one is for the protein sequence and the other forthe drug The enzyme sequence should be in FASTA formatwhile the drug in the KEGG code Examples for the querypairs input can be seen by clicking on the Example buttonright above the input boxStep 3 Click on the Submit button to see the predicted resultFor example if you use the four query pairs in the Example
10 BioMed Research International
window as the input after clicking the Submit button youwill see on your screen that the ldquohsa 10056rdquo enzyme andthe ldquoD0021rdquo drug are an interactive pair and that the ldquohsa100rdquo enzyme and the ldquoD0037rdquo drug are also an interactivepair but that the ldquohsa 3295rdquo enzyme and the ldquoD00889rdquo drugare not an interactive pair and that the ldquohsa 7366rdquo enzymeand the ldquoD03601rdquo drug are not an interactive pair eitherAll these results are fully consistent with the experimentalobservations It takes about 3 minutes before the results areshown on the screenStep 4 Click on the Citation button to find the relevant paperthat documents the detailed development and algorithm ofiEzy-DurgStep 5 Click on the Data button to download the benchmarkdataset used to train and test the iEzy-Durg predictorStep 6 The program code is also available by clicking thebutton download on the lower panel of Figure 5
Acknowledgments
The authors would like to thank the three anonymousreviewers whose constructive comments are very helpful forstrengthening the presentation of the paper This work wassupported by the Grants from the National Natural ScienceFoundation of China (no 31260273) the Jiangxi ProvincialForeign Scientific and Technological Cooperation Project(no 20120BDH80023) Natural Science Foundation of JiangxiProvince China (no 2010GZS0122 20122BAB201020) theDepartment of Education of Jiangxi Province (GJJ12490)the LuoDi plan of the Department of Education of JiangxiProvince (KJLD12083) and the Jiangxi Provincial Founda-tion for Leaders of Disciplines in Science (20113BCB22008)The funders had no role in study design data collection andanalysis decision to publish or preparation of the paper
References
[1] A Bairoch ldquoThe ENZYME database in 2000rdquo Nucleic AcidsResearch vol 28 no 1 pp 304ndash305 2000
[2] S H Koenig and R D Brown ldquoH2CO3as substrate for carbonic
anhydrase in the dehydration of H2CO3(minus)rdquo Proceedings of the
National Academy of Sciences of the United States of Americavol 69 no 9 pp 2422ndash2425 1972
[3] S X Lin and J Lapointe ldquoTheoretical and experimental biologyin onerdquo Journal of Biomedical Science and Engineering vol 6 pp435ndash442 2013
[4] K C Chou and S P Jiang ldquoStudies on the rate of diffusion-controlled reactions of enzymes Spatial factor and force fieldfactorrdquo Scientia Sinica vol 17 no 5 pp 664ndash680 1974
[5] K C Chou ldquoThe kinetics of the combination reaction betweenenzyme and substraterdquo Scientia Sinica vol 19 no 4 pp 505ndash528 1976
[6] K C Chou and G P Zhou ldquoRole of the protein outside activesite on the diffusion-controlled reaction of enzymerdquo Journal ofthe American Chemical Society vol 104 no 5 pp 1409ndash14131982
[7] R A Poorman A G Tomasselli R L Heinrikson and FJ Kezdy ldquoA cumulative specificity model for proteases from
human immunodeficiency virus types 1 and 2 inferred fromstatistical analysis of an extended substrate data baserdquo TheJournal of Biological Chemistry vol 266 no 22 pp 14554ndash145611991
[8] K-C Chou ldquoA vectorized sequence-coupling model for pre-dicting HIV protease cleavage sites in proteinsrdquo The Journal ofBiological Chemistry vol 268 no 23 pp 16938ndash16948 1993
[9] G Z Liang and S Z Li ldquoA new sequence representation asapplied in better specificity elucidation for human immunod-eficiency virus type 1 proteaserdquo Biopolymers vol 88 no 3 pp401ndash412 2007
[10] J J Chou ldquoPredicting cleavability of peptide sequences byHIVprotease via correlation-angle approachrdquo Journal of ProteinChemistry vol 12 no 3 pp 291ndash302 1993
[11] Q S Du S Wang D Q Wei S Sirois and K C ChouldquoMolecular modeling and chemical modification for findingpeptide inhibitor against severe acute respiratory syndromecoronavirus main proteinaserdquo Analytical Biochemistry vol 337no 2 pp 262ndash270 2005
[12] Y Gan H Huang Y Huang et al ldquoSynthesis and activity ofan octapeptide inhibitor designed for SARS coronavirus mainproteinaserdquo Peptides vol 27 no 4 pp 622ndash625 2006
[13] Q S Du H Sun and K C Chou ldquoInhibitor design for SARScoronavirus main protease based on lsquodistorted key theoryrsquordquoMedicinal Chemistry vol 3 no 1 pp 1ndash6 2007
[14] K C Chou ldquoPrediction of human immunodeficiency virusprotease cleavage sites in proteinsrdquoAnalytical Biochemistry vol233 no 1 pp 1ndash14 1996
[15] J Knowles and G Gromo ldquoTarget selection in drug discoveryrdquoNature Reviews Drug Discovery vol 2 no 1 pp 63ndash69 2003
[16] A C Cheng R G Coleman K T Smyth et al ldquoStructure-basedmaximal affinity model predicts small-molecule druggabilityrdquoNature Biotechnology vol 25 no 1 pp 71ndash75 2007
[17] M Rarey B Kramer T Lengauer and G Klebe ldquoA fast flexibledocking method using an incremental construction algorithmrdquoJournal of Molecular Biology vol 261 no 3 pp 470ndash489 1996
[18] K C Chou ldquoReview structural bioinformatics and its impactto biomedical sciencerdquo Current Medicinal Chemistry vol 11 no16 pp 2105ndash2134 2004
[19] K C Chou D Q Wei and W Z Zhong ldquoBinding mechanismof coronavirus main proteinase with ligands and its implicationto drug design against SARSrdquo Biochemical and BiophysicalResearch Communications vol 308 pp 148ndash151 2003 (Erratumin Biochemical and Biophysical Research Communications vol310 p 675 2003)
[20] K C Chou D Q Wei Q S Du S Sirois and W Z ZhongldquoProgress in computational approach to drug developmentagaint SARSrdquo Current Medicinal Chemistry vol 13 no 27 pp3263ndash3270 2006
[21] G P Zhou and F A Troy ldquoNMR studies on how the bindingcomplex of polyisoprenol recognition sequence peptides andpolyisoprenols can modulate membrane structurerdquo CurrentProtein and Peptide Science vol 6 no 5 pp 399ndash411 2005
[22] R B Huang Q S Du C H Wang and K C Chou ldquoAn in-depth analysis of the biological functional studies based on theNMR M2 channel structure of influenza A virusrdquo Biochemicaland Biophysical Research Communications vol 377 no 4 pp1243ndash1247 2008
[23] Q S Du R B Huang C H Wang X M Li and K C ChouldquoEnergetic analysis of the two controversial drug binding sitesof the M2 proton channel in influenza A virusrdquo Journal ofTheoretical Biology vol 259 no 1 pp 159ndash164 2009
BioMed Research International 11
[24] M J Berardi W M Shih S C Harrison and J J Chou ldquoMito-chondrial uncoupling protein 2 structure determined by NMRmolecular fragment searchingrdquo Nature vol 476 no 7358 pp109ndash114 2011
[25] J R Schnell and J J Chou ldquoStructure andmechanism of theM2proton channel of influenza A virusrdquo Nature vol 451 no 7178pp 591ndash595 2008
[26] B OuYang S Xie M J Berardi et al ldquoUnusual architecture ofthe p7 channel from hepatitis C virusrdquoNature vol 498 pp 521ndash525 2013
[27] K Oxenoid and J J Chou ldquoThe structure of phospholambanpentamer reveals a channel-like architecture in membranesrdquoProceedings of the National Academy of Sciences of the UnitedStates of America vol 102 no 31 pp 10870ndash10875 2005
[28] M E Call KWWucherpfennig and J J Chou ldquoThe structuralbasis for intramembrane assembly of an activating immunore-ceptor complexrdquo Nature Immunology vol 11 no 11 pp 1023ndash1029 2010
[29] R M Pielak J R Schnell and J J Chou ldquoMechanism of druginhibition and drug resistance of influenza A M2 channelrdquoProceedings of the National Academy of Sciences of the UnitedStates of America vol 106 no 18 pp 7379ndash7384 2009
[30] J Wang R M Pielak M A McClintock and J J ChouldquoSolution structure and functional analysis of the influenza Bproton channelrdquo Nature Structural and Molecular Biology vol16 no 12 pp 1267ndash1271 2009
[31] K C Chou ldquoCoupling interaction between thromboxane A2receptor and alpha-13 subunit of guanine nucleotide-bindingproteinrdquo Journal of Proteome Research vol 4 no 5 pp 1681ndash1686 2005
[32] K C Chou ldquoInsights from modeling three-dimensional struc-tures of the human potassium and sodium channelsrdquo Journal ofProteome Research vol 3 no 4 pp 856ndash861 2004
[33] K C Chou ldquoSome remarks on protein attribute prediction andpseudo amino acid compositionrdquo Journal ofTheoretical Biologyvol 273 no 1 pp 236ndash247 2011
[34] W Z Lin J A Fang X Xiao and K C Chou ldquoiLoc-animal amulti-label learning classifier for predicting subcellular local-ization of animal proteinsrdquo Molecular BioSystems vol 9 pp634ndash644 2013
[35] X Xiao PWangW Z Lin J H Jia and K C Chou ldquoiAMP-2La two-level multi-label classifier for identifying antimicrobialpeptides and their functional typesrdquo Analytical Biochemistryvol 436 pp 168ndash177 2013
[36] W Chen P M Feng H Lin and K C Chou ldquoiRSpot-PseDNCidentify recombination spots with pseudo dinucleotide compo-sitionrdquo Nucleic Acids Research vol 41 article e69 2013
[37] K C Chou Z C Wu and X Xiao ldquoILoc-Hum using theaccumulation-label scale to predict subcellular locations ofhuman proteins with both single and multiple sitesrdquoMolecularBioSystems vol 8 no 2 pp 629ndash641 2012
[38] M Kotera M Hirakawa T Tokimatsu S Goto and MKanehisa ldquoThe KEGG databases and tools facilitating omicsanalysis latest developments involving human diseases andpharmaceuticalsrdquo Methods in Molecular Biology vol 802 pp19ndash39 2012
[39] Y YamanishiM Araki A GutteridgeWHonda andM Kane-hisa ldquoPrediction of drug-target interaction networks from theintegration of chemical and genomic spacesrdquo Bioinformaticsvol 24 no 13 pp i232ndashi240 2008
[40] P Finn S Muggleton D Page and A Srinivasan ldquoPharma-cophore discovery using the Inductive Logic Programmingsystem PROGOLrdquo Machine Learning vol 30 no 2-3 pp 241ndash270 1998
[41] I Vogt D Stumpfe H E Ahmed and J Bajorath ldquoMethodsfor computer-aided chemical biology Part 2 evaluation of com-pound selectivity using 2D molecular fingerprintsrdquo ChemicalBiology and Drug Design vol 70 pp 195ndash205 2007
[42] H Eckert and J Bajorath ldquoMolecular similarity analysis in vir-tual screening foundations limitations and novel approachesrdquoDrug Discovery Today vol 12 no 5-6 pp 225ndash233 2007
[43] S Laurent L V Elst and R N Muller ldquoComparative studyof the physicochemical properties of six clinical low molecularweight gadolinium contrast agentsrdquo Contrast Media amp Molecu-lar Imaging vol 1 no 3 pp 128ndash137 2006
[44] E Gregori-Puigjane R Garriga-Sust and J Mestres ldquoIndexingmolecules with chemical graph identifiersrdquo Journal of Compu-tational Chemistry vol 32 no 12 pp 2638ndash2646 2011
[45] B Ren ldquoApplication of novel atom-type AI topological indicesto QSPR studies of alkanesrdquo Computers and Chemistry vol 26no 4 pp 357ndash369 2002
[46] N M OrsquoBoyle M Banck C A James C Morley T Vander-meersch and G R Hutchison ldquoOpen Babel an open chemicaltoolboxrdquo Journal of Cheminformatics vol 3 p 33 2011
[47] V J Gillet P Willett and J Bradshaw ldquoSimilarity searchingusing reduced graphsrdquo Journal of Chemical Information andComputer Sciences vol 43 no 2 pp 338ndash345 2003
[48] D Butina ldquoUnsupervised data base clustering based on day-lightrsquos fingerprint and Tanimoto similarity a fast and automatedway to cluster small and large data setsrdquo Journal of ChemicalInformation and Computer Sciences vol 39 no 4 pp 747ndash7501999
[49] C E Shannon ldquoA mathematical theory of communicationrdquoACM SIGMOBILE Mobile Computing and CommunicationsReview vol 5 pp 3ndash55 2001
[50] V D Gusev L A Nemytikova and N A Chuzhanova ldquoOn thecomplexity measures of genetic sequencesrdquo Bioinformatics vol15 no 12 pp 994ndash999 1999
[51] K C Chou and H B Shen ldquoReview recent progress in proteinsubcellular location predictionrdquo Analytical Biochemistry vol370 no 1 pp 1ndash16 2007
[52] S F Altschul ldquoEvaluating the statistical significance of multipledistinct local alignmentsrdquo in Theoretical and ComputationalMethods in Genome Research S Suhai Ed pp 1ndash14 PlenumNew York NY USA 1997
[53] J C Wootton and S Federhen ldquoStatistics of local complexity inamino acid sequences and sequence databasesrdquo Computers andChemistry vol 17 no 2 pp 149ndash163 1993
[54] H Nakashima K Nishikawa and T Ooi ldquoThe folding type ofa protein is relevant to the amino acid compositionrdquo Journal ofBiochemistry vol 99 no 1 pp 153ndash162 1986
[55] K C Chou and C T Zhang ldquoPredicting protein folding typesby distance functions that make allowances for amino acidinteractionsrdquo The Journal of Biological Chemistry vol 269 no35 pp 22014ndash22020 1994
[56] K-C Chou ldquoA novel approach to predicting protein structuralclasses in a (20-1)-D amino acid composition spacerdquo Proteinsvol 21 no 4 pp 319ndash344 1995
[57] H Nakashima and K Nishikawa ldquoDiscrimination of intracel-lular and extracellular proteins using amino acid compositionand residue-pair frequenciesrdquo Journal of Molecular Biology vol238 no 1 pp 54ndash61 1994
12 BioMed Research International
[58] G P Zhou ldquoAn intriguing controversy over protein structuralclass predictionrdquo Journal of Protein Chemistry vol 17 no 8 pp729ndash738 1998
[59] I Bahar A R Atilgan R L Jernigan and B Erman ldquoUnder-standing the recognition of protein structural classes by aminoacid compositionrdquo Proteins vol 29 pp 172ndash185 1997
[60] J Cedano P Aloy J A Perez-Pons and E Querol ldquoRelationbetween amino acid composition and cellular location ofproteinsrdquo Journal of Molecular Biology vol 266 no 3 pp 594ndash600 1997
[61] G P Zhou and K Doctor ldquoSubcellular location prediction ofapoptosis proteinsrdquo Proteins vol 50 no 1 pp 44ndash48 2003
[62] KChou ldquoPrediction of protein cellular attributes using pseudo-amino acid compositionrdquo Proteins vol 43 pp 246ndash255 2001(Erratum in Proteins vol 44 p 60 2001)
[63] K C Chou ldquoUsing amphiphilic pseudo amino acid composi-tion to predict enzyme subfamily classesrdquo Bioinformatics vol21 no 1 pp 10ndash19 2005
[64] D Zou Z He J He and Y Xia ldquoSupersecondary structureprediction using Choursquos pseudo amino acid compositionrdquoJournal of Computational Chemistry vol 32 no 2 pp 271ndash2782011
[65] M M Beigi M Behjati and H Mohabatkar ldquoPrediction ofmetalloproteinase family based on the concept ofChoursquos pseudoamino acid composition using a machine learning approachrdquoJournal of Structural and Functional Genomics vol 12 no 4 pp191ndash197 2011
[66] Y K Chen and K B Li ldquoPredicting membrane protein typesby incorporating protein topology domains signal peptidesand physicochemical properties into the general form of Choursquospseudo amino acid compositionrdquo Journal ofTheoretical Biologyvol 318 pp 1ndash12 2013
[67] C Huang and J Q Yuan ldquoA multilabel model based on Choursquospseudo-amino acid composition for identifying membraneproteins with both single and multiple functional typesrdquo TheJournal of Membrane Biology vol 246 pp 327ndash334 2013
[68] S S Sahu and G Panda ldquoA novel feature representationmethod based on Choursquos pseudo amino acid composition forprotein structural class predictionrdquo Computational Biology andChemistry vol 34 no 5-6 pp 320ndash327 2010
[69] M Hayat and A Khan ldquoDiscriminating outer membraneproteins with fuzzy K-nearest neighbor algorithms based on thegeneral form of Choursquos PseAACrdquo Protein and Peptide Lettersvol 19 no 4 pp 411ndash421 2012
[70] MKhosravian F K FaramarziMM BeigiM Behbahani andH Mohabatkar ldquoPredicting antibacterial peptides by the con-cept of Choursquos pseudo-amino acid composition and machinelearning methodsrdquo Protein amp Peptide Letters vol 20 pp 180ndash186 2013
[71] HMohabatkarMM Beigi K Abdolahi and SMohsenzadehldquoPrediction of allergenic proteins by means of the concept ofChoursquos pseudo amino acid composition and amachine learningapproachrdquoMedicinal Chemistry vol 9 pp 133ndash137 2013
[72] L Nanni A Lumini D Gupta and A Garg ldquoIdentifyingbacterial virulent proteins by fusing a set of classifiers basedon variants of Choursquos pseudo amino acid composition and onevolutionary informationrdquo IEEEACM Transactions on Compu-tational Biology and Bioinformatics vol 9 no 2 pp 467ndash4752012
[73] T H Chang L C Wu T Y Lee S P Chen H D Huang andJ T Horng ldquoEuLoc a web-server for accurately predict protein
subcellular localization in eukaryotes by incorporating variousfeatures of sequence segments into the general form of ChoursquosPseAACrdquo Journal of Computer-Aided Molecular Design vol 27pp 91ndash103 2013
[74] S Zhang Y Zhang H Yang C Zhao and Q Pan ldquoUsing theconcept of Choursquos pseudo amino acid composition to predictprotein subcellular localization an approach by incorporatingevolutionary information and von Neumann entropiesrdquo AminoAcids vol 34 no 4 pp 565ndash572 2008
[75] R Zia Ur and A Khan ldquoIdentifying GPCRs and their typeswith Choursquos pseudo amino acid composition an approach frommulti-scale energy representation and position specific scoringmatrixrdquo Protein amp Peptide Letters vol 19 pp 890ndash903 2012
[76] X Y Sun S P Shi J D Qiu S B Suo S Y Huang and R PLiang ldquoIdentifying protein quaternary structural attributes byincorporating physicochemical properties into the general formof Choursquos PseAAC via discrete wavelet transformrdquo MolecularBioSystems vol 8 pp 3178ndash3184 2012
[77] L Nanni and A Lumini ldquoGenetic programming for creatingChoursquos pseudo amino acid based features for submitochondrialocalizationrdquo Amino Acids vol 34 no 4 pp 653ndash660 2008
[78] M Esmaeili H Mohabatkar and S Mohsenzadeh ldquoUsing theconcept of Choursquos pseudo amino acid composition for risk typeprediction of human papillomavirusesrdquo Journal of TheoreticalBiology vol 263 no 2 pp 203ndash209 2010
[79] H Mohabatkar ldquoPrediction of cyclin proteins using Choursquospseudo amino acid compositionrdquo Protein and Peptide Lettersvol 17 no 10 pp 1207ndash1214 2010
[80] H Mohabatkar M M Beigi and A Esmaeili ldquoPredictionof GABA(A) receptor proteins using the concept of Choursquospseudo-amino acid composition and support vector machinerdquoJournal of Theoretical Biology vol 281 no 1 pp 18ndash23 2011
[81] Y Xu J Ding L Y Wu and K C Chou ldquoiSNO-PseAAC pre-dict cysteine S-nitrosylation sites in proteins by incorporatingposition specific amino acid propensity into pseudo amino acidcompositionrdquo PLoS ONE vol 8 Article ID e55844 2013
[82] W Chen H Lin PM Feng C Ding Y C Zuo andK C ChouldquoiNuc-PhysChem a sequence-based predictor for identifyingnucleosomes via physicochemical propertiesrdquo PLoS ONE vol7 Article ID e47843 2012
[83] B Li T Huang L Liu Y Cai and K C Chou ldquoIdentificationof colorectal cancer related genes with mrmr and shortest pathin protein-protein interaction networkrdquo PLoS ONE vol 7 no 4Article ID e33393 2012
[84] Y Jiang T Huang C Lei Y F Gao Y D Cai and K C ChouldquoSignal propagation in protein interaction network duringcolorectal cancer progressionrdquo BioMed Research Internationalvol 2013 Article ID 287019 9 pages 2013
[85] P Du X Wang C Xu and Y Gao ldquoPseAAC-Builder across-platform stand-alone program for generating variousspecial Choursquos pseudo-amino acid compositionsrdquo AnalyticalBiochemistry vol 425 no 2 pp 117ndash119 2012
[86] D S Cao Q S Xu and Y Z Liang ldquoPropy a tool to generatevarious modes of Choursquos PseAACrdquo Bioinformatics vol 29 pp960ndash962 2013
[87] H Shen and K C Chou ldquoPseAAC a flexible web serverfor generating various kinds of protein pseudo amino acidcompositionrdquo Analytical Biochemistry vol 373 no 2 pp 386ndash388 2008
[88] K C Chou ldquoUsing pair-coupled amino acid composition topredict protein secondary structure contentrdquo Journal of ProteinChemistry vol 18 no 4 pp 473ndash480 1999
BioMed Research International 13
[89] W Liu and K C Chou ldquoPrediction of protein secondarystructure contentrdquo Protein Engineering vol 12 no 12 pp 1041ndash1050 1999
[90] M Bhasin and G P S Raghava ldquoClassification of nuclearreceptors based on amino acid composition and dipeptidecompositionrdquo The Journal of Biological Chemistry vol 279 no22 pp 23262ndash23266 2004
[91] H Lin andH Ding ldquoPredicting ion channels and their types bythe dipeptidemode of pseudo amino acid compositionrdquo Journalof Theoretical Biology vol 269 no 1 pp 64ndash69 2011
[92] H Lin and Q Li ldquoUsing pseudo amino acid composition topredict protein structural class approached by incorporating400 dipeptide componentsrdquo Journal of Computational Chem-istry vol 28 no 9 pp 1463ndash1466 2007
[93] L Nanni and A Lumini ldquoCombing ontologies and dipeptidecomposition for predicting DNA-binding proteinsrdquo AminoAcids vol 34 no 4 pp 635ndash641 2008
[94] K-C Chou ldquoThe convergence-divergence duality in lectindomains of selectin family and its implicationsrdquo FEBS Lettersvol 363 no 1-2 pp 123ndash126 1995
[95] A A Schaffer L Aravind T L Madden et al ldquoImprovingthe accuracy of PSI-BLAST protein database searches withcomposition-based statistics and other refinementsrdquo NucleicAcids Research vol 29 no 14 pp 2994ndash3005 2001
[96] S F Altschul and E V Koonin ldquoIterated profile searches withPSI-BLAST a tool for discovery in protein databasesrdquo Trends inBiochemical Sciences vol 23 no 11 pp 444ndash447 1998
[97] J Deng ldquoGrey entropy and grey target decision makingrdquoJournal of Grey System vol 22 no 1 pp 1ndash24 2010
[98] B M Bolstad R A Irizarry M Astrand and T P Speed ldquoAcomparison of normalizationmethods for high density oligonu-cleotide array data based on variance and biasrdquo Bioinformaticsvol 19 no 2 pp 185ndash193 2003
[99] J-Y Shi S-W Zhang Q Pan Y-M Cheng and J XieldquoPrediction of protein subcellular localization by support vectormachines using multi-scale energy and pseudo amino acidcompositionrdquo Amino Acids vol 33 no 1 pp 69ndash74 2007
[100] T Denoeux ldquoA 120581-nearest neighbor classification rule based onDempster-Shafer theoryrdquo IEEE Transactions on Systems Manand Cybernetics vol 25 no 5 pp 804ndash813 1995
[101] J M Keller M R Gray and J A Givens ldquoA fuzzy k-nearestneighbours algorithmrdquo IEEE Transactions on Systems Man andCybernetics vol 15 no 4 pp 580ndash585 1985
[102] X Xiao P Wang and K Chou ldquoiNR-physchem a sequence-based predictor for identifying nuclear receptors and theirsubfamilies via physical-chemical property matrixrdquo PLoS ONEvol 7 no 2 Article ID e30869 2012
[103] I Roterman L Konieczny W Jurkowski K Prymula and MBanach ldquoTwo-intermediate model to characterize the structureof fast-folding proteinsrdquo Journal of Theoretical Biology vol 283no 1 pp 60ndash70 2011
[104] X Xiao P Wang and K C Chou ldquoGPCR-2L predicting Gprotein-coupled receptors and their types by hybridizing twodifferentmodes of pseudo amino acid compositionsrdquoMolecularBioSystems vol 7 no 3 pp 911ndash919 2011
[105] X Xiao P Wang and K C Chou ldquoQuat-2L a web-server forpredicting protein quaternary structural attributesrdquo MolecularDiversity vol 15 no 1 pp 149ndash155 2011
[106] X Zheng C Li and J Wang ldquoAn information-theoreticapproach to the prediction of protein structural classrdquo Journalof Computational Chemistry vol 31 no 6 pp 1201ndash1206 2010
[107] H Shen J Yang X Liu and K C Chou ldquoUsing supervisedfuzzy clustering to predict protein structural classesrdquo Biochem-ical and Biophysical Research Communications vol 334 no 2pp 577ndash581 2005
[108] K-C Chou and C-T Zhang ldquoReview prediction of proteinstructural classesrdquo Critical Reviews in Biochemistry and Molec-ular Biology vol 30 no 4 pp 275ndash349 1995
[109] P C Mahalanobis ldquoOn the generalized distance in statisticsrdquoProceedings of the National Institute of Sciences of India vol 2pp 49ndash55 1936
[110] R M Centor ldquoSignal detectability the use of ROC curves andtheir analysesrdquoMedical Decision Making vol 11 no 2 pp 102ndash106 1991
[111] K-C Chou ldquoUsing subsite coupling to predict signal peptidesrdquoProtein Engineering vol 14 no 2 pp 75ndash79 2001
[112] K C Chou ldquoPrediction of protein signal sequences and theircleavage sitesrdquo Proteins vol 42 pp 136ndash139 2001
[113] K C Chou ldquoPrediction of signal peptides using scaledwindowrdquoPeptides vol 22 no 12 pp 1973ndash1979 2001
[114] K C Chou ldquoSome remarks on predicting multi-label attributesinmolecular biosystemsrdquoMolecular Biosystems vol 9 pp 1092ndash1100 2013
[115] K C Chou and H B Shen ldquoCell-PLoc 2 0 an improvedpackage of web-servers for predicting subcellular localization ofproteins in various organismsrdquoNatural Science vol 2 pp 1090ndash1103 2010
[116] M Gribskov and N L Robinson ldquoUse of receiver operatingcharacteristic (ROC) analysis to evaluate sequence matchingrdquoComputers and Chemistry vol 20 no 1 pp 25ndash33 1996
[117] Y Yamanishi M Kotera M Kanehisa and S Goto ldquoDrug-target interaction prediction from chemical genomic and phar-macological data in an integrated frameworkrdquo Bioinformaticsvol 26 no 12 pp i246ndashi254 2010
[118] Z He J Zhang X Shi et al ldquoPredicting drug-target interactionnetworks based on functional groups and biological featuresrdquoPLoS ONE vol 5 no 3 Article ID e9603 2010
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

8 BioMed Research International
02
46
810
1
15
2089
0895
09
0905
091
0915
092
ACC
K120593
Figure 3 A 3D plot to show how the parameter in (27) wasoptimized for the iEzy-Drug predictor
mispredicted to be a noninteractive enzyme-drug pair wehave the sensitivity Sn = 1 while 119873+
minus= 119873
+ meaning thatall the interactive enzyme-drug pairs weremispredicted to bethe noninteractive enzyme-drug pairs we have the sensitivitySn = 0 Likewise when 119873minus
+= 0 meaning none of the
noninteractive enzyme-drug pairs wasmispredicted we havethe specificity Sp = 1 while 119873minus
+= 119873
minus meaning all thenoninteractive enzyme-drug pairs were incorrectly predictedas interactive enzyme-drug pairs we have the specificity Sp =0 When119873+
minus= 119873
minus
+= 0 meaning that none of the interactive
enzyme-drug pairs in the dataset S+ and none of the nonin-teractive enzyme-drug pairs in Sminus was incorrectly predictedwe have the overall accuracy Acc = Λ = 1 while 119873+
minus= 119873
+
and 119873minus+= 119873
minus meaning that all the interactive enzyme-drugpairs in the dataset S+ and all the noninteractive enzyme-drug pairs in Sminus were mispredicted we have the overallaccuracy Acc = Λ = 0 The MCC correlation coefficient isusually used for measuring the quality of binary (two-class)classifications When 119873+
minus= 119873
minus
+= 0 meaning that none
of the interactive enzyme-drug pairs in the dataset S+ andnone of the noninteractive enzyme-drug pairs in Sminus weremispredicted we have MCC = 1 when 119873+
minus= 119873
+2 and
119873
minus
+= 119873
minus2 we have MCC = 0 meaning no better than
random prediction when 119873+minus= 119873
+ and 119873minus+= 119873
minus we haveMCC = minus1 meaning total disagreement between predictionand observation As we can see from the previous discussionit is much more intuitive and easier-to-understand whenusing (34) to examine a predictor for its sensitivity specificityoverall accuracy and Mathewrsquos correlation coefficient It isinstructive to point out that themetrics as defined in (30) and(34) are valid for single label systems for multilabel systemsa set of more complicated metrics should be used as given in[114]
3 Results and Discussion
31 Cross-Validation How to properly examine the predic-tion quality is a key for developing a new predictor and
False positive rate
True
pos
itive
rate
1
01
02
03
04
05
06
07
08
09
0
101 02 03 04 05 06 07 08 090
BaselineiEzy-Drug AUC = 09377
Figure 4 A plot for the ROC curve to quantitatively show theperformance of the iEzy-Drug predictor
estimating its potential application value Generally speak-ing the following three cross-validation methods are oftenused to examine a predictor of its effectiveness in practicalapplication independent dataset test subsampling or 119870-fold(such as 5-fold 7-fold or 10-fold) test and jackknife test[108] However as elaborated by a penetrating analysis in[115] considerable arbitrariness exists in the independentdataset test Also as demonstrated by (27)ndash(29) in [33]the subsampling test (or 119870-fold cross-validation) cannotavoid arbitrariness either Only the jackknife test is the leastarbitrary that can always yield a unique result for a givenbenchmark dataset Therefore the jackknife test has beenwidely recognized and increasingly utilized by investigatorsto examine the quality of various predictors (see eg [66 7174 80]) Accordingly the success rate by the jackknife test wasalso used to optimize the two uncertain parameters 119870 and 120593in (27) The result thus obtained is shown in Figure 3 fromwhich we obtain when 119870 = 6 and 120593 = 15 the iEzy-Drugpredictor reaches its optimized status
The success rates thus obtained by the jackknife test inidentifying interactive Enzyme-drug pairs or noninteractiveenzyme-drug pairs on the benchmark dataset S (cf OnlineSupporting Information S1) are given in Table 1 where forfacilitating comparison the corresponding result by He et al[110] is also given As we can see from the table the overallaccuracy Acc achieved by iEzy-Drug was 9103 remarkablyhigher than 8548 the corresponding rate obtained byHe et al [110] on the same benchmark Furthermore listedin Table 1 are also the values obtained by iEzy-Drug for theother three metrics that is Sn = 9081 Sp = 9114 andMCC = 8039 indicating that the accuracy of iEzy-Drug isnot only very high but also quite stale
To provide a graphical illustration to show the per-formance of the current binary classifier iEzy-Drug asits discrimination threshold is varied a 2D plot calledReceiver Operating Characteristic (ROC) curve [116 117]was also given (Figure 4) In the ROC curve the verticalcoordinate 119884 is for the true positive rate or Sn (cf (34))while horizontal coordinate 119883 for the false positive rate
BioMed Research International 9
Read Me Data Citation
Enter both the protein sequence and the drug code (example) the number of query pairs is limited at 10 or less for each submission
Submit Clear
You can download the program and run it on your own local computer
iEzy-Drug identifying the interaction between enzymes anddrugs in cellular networking
Figure 5 A semiscreenshot to show the top page of the iEzy-Drugweb-server Its web-site address is at httpwwwjci-bioinfocniEzy-Drug
Table 1 The jackknife success rates obtained with iEzy-Drug in identifying interactive enzyme-drug pairs and noninteractive enzyme-drugpairs for the benchmark dataset S (cf Online Supporting Information S1)
Method Acc Sn Sp MCC
iEzy-Druga 74258157 = 9103 24692719 = 9081 49565438 = 9114 8039NN predictorb 8548 NA NA NAaSee (27) where the parameters119870 = 6 and 120593 = 15bSee [110]
or 1-Sp The best possible prediction method would yielda point with the coordinate (0 1) representing 100 truepositive rate (sensitivity Sn) and 0 false positive rate or100 specificity Therefore the (0 1) point is also calleda perfect classification A completely random guess wouldgive a point along a diagonal from the point (0 0) to (1 1)The area under the ROC curve also called Area Under theROC (AUROC) is often used to indicate the performancequality of a binary classifier the value 05 of AUROCis equivalent to random prediction while 1 of AUROCrepresents a perfect one As we can see from Figure 4the AUROC value obtained by iEzy-Drug is 09377
The reason why iEzy-Drug can remarkably improve theprediction quality is that it has introduced the 2D molecularfingerprints to represent drug samples see Online SupportingInformation S3 for the detailed fingerprint expressions for thedrugs listed in Online Supporting Information S1 and thatit has successfully used PseAAC to incorporate the featuresderived from the sequences of enzymes that are essentialfor identifying the interaction of enzymes with drugs in thecellular networking
To enhance the value of its practical applications theweb server for iEzy-Drug has been established that canbe freely accessible at httpwwwjci-bioinfocniEzy-DrugIt is anticipated that the web server will become a usefulhigh throughput tool for both basic research and drug
development in the relevant areas or at the very least playa complementary role to the existing method [39 110 118] forwhich so far noweb-serverwhatsoever has been provided yet
32 The Protocol or User Guide For the convenience of thevast majority of biologists and pharmaceutical scientists herelet us provide a step-by-step guide to show how the userscan easily get the desired result by means of the web serverwithout the need to follow the complicated mathematicalequations presented in this paper for the process of develop-ing the predictor and its integrityStep 1 Open the web server at the site httpwwwjci-bio-infocniEzy-Drug and you will see the top page of thepredictor on your computer screen as shown in Figure 5Click on the ReadMe button to see a brief introduction aboutiEzy-Drug predictor and the caveat when using itStep 2 Either type or copypaste the query pairs into theinput box at the center of Figure 5 Each query pair consistsof two parts one is for the protein sequence and the other forthe drug The enzyme sequence should be in FASTA formatwhile the drug in the KEGG code Examples for the querypairs input can be seen by clicking on the Example buttonright above the input boxStep 3 Click on the Submit button to see the predicted resultFor example if you use the four query pairs in the Example
10 BioMed Research International
window as the input after clicking the Submit button youwill see on your screen that the ldquohsa 10056rdquo enzyme andthe ldquoD0021rdquo drug are an interactive pair and that the ldquohsa100rdquo enzyme and the ldquoD0037rdquo drug are also an interactivepair but that the ldquohsa 3295rdquo enzyme and the ldquoD00889rdquo drugare not an interactive pair and that the ldquohsa 7366rdquo enzymeand the ldquoD03601rdquo drug are not an interactive pair eitherAll these results are fully consistent with the experimentalobservations It takes about 3 minutes before the results areshown on the screenStep 4 Click on the Citation button to find the relevant paperthat documents the detailed development and algorithm ofiEzy-DurgStep 5 Click on the Data button to download the benchmarkdataset used to train and test the iEzy-Durg predictorStep 6 The program code is also available by clicking thebutton download on the lower panel of Figure 5
Acknowledgments
The authors would like to thank the three anonymousreviewers whose constructive comments are very helpful forstrengthening the presentation of the paper This work wassupported by the Grants from the National Natural ScienceFoundation of China (no 31260273) the Jiangxi ProvincialForeign Scientific and Technological Cooperation Project(no 20120BDH80023) Natural Science Foundation of JiangxiProvince China (no 2010GZS0122 20122BAB201020) theDepartment of Education of Jiangxi Province (GJJ12490)the LuoDi plan of the Department of Education of JiangxiProvince (KJLD12083) and the Jiangxi Provincial Founda-tion for Leaders of Disciplines in Science (20113BCB22008)The funders had no role in study design data collection andanalysis decision to publish or preparation of the paper
References
[1] A Bairoch ldquoThe ENZYME database in 2000rdquo Nucleic AcidsResearch vol 28 no 1 pp 304ndash305 2000
[2] S H Koenig and R D Brown ldquoH2CO3as substrate for carbonic
anhydrase in the dehydration of H2CO3(minus)rdquo Proceedings of the
National Academy of Sciences of the United States of Americavol 69 no 9 pp 2422ndash2425 1972
[3] S X Lin and J Lapointe ldquoTheoretical and experimental biologyin onerdquo Journal of Biomedical Science and Engineering vol 6 pp435ndash442 2013
[4] K C Chou and S P Jiang ldquoStudies on the rate of diffusion-controlled reactions of enzymes Spatial factor and force fieldfactorrdquo Scientia Sinica vol 17 no 5 pp 664ndash680 1974
[5] K C Chou ldquoThe kinetics of the combination reaction betweenenzyme and substraterdquo Scientia Sinica vol 19 no 4 pp 505ndash528 1976
[6] K C Chou and G P Zhou ldquoRole of the protein outside activesite on the diffusion-controlled reaction of enzymerdquo Journal ofthe American Chemical Society vol 104 no 5 pp 1409ndash14131982
[7] R A Poorman A G Tomasselli R L Heinrikson and FJ Kezdy ldquoA cumulative specificity model for proteases from
human immunodeficiency virus types 1 and 2 inferred fromstatistical analysis of an extended substrate data baserdquo TheJournal of Biological Chemistry vol 266 no 22 pp 14554ndash145611991
[8] K-C Chou ldquoA vectorized sequence-coupling model for pre-dicting HIV protease cleavage sites in proteinsrdquo The Journal ofBiological Chemistry vol 268 no 23 pp 16938ndash16948 1993
[9] G Z Liang and S Z Li ldquoA new sequence representation asapplied in better specificity elucidation for human immunod-eficiency virus type 1 proteaserdquo Biopolymers vol 88 no 3 pp401ndash412 2007
[10] J J Chou ldquoPredicting cleavability of peptide sequences byHIVprotease via correlation-angle approachrdquo Journal of ProteinChemistry vol 12 no 3 pp 291ndash302 1993
[11] Q S Du S Wang D Q Wei S Sirois and K C ChouldquoMolecular modeling and chemical modification for findingpeptide inhibitor against severe acute respiratory syndromecoronavirus main proteinaserdquo Analytical Biochemistry vol 337no 2 pp 262ndash270 2005
[12] Y Gan H Huang Y Huang et al ldquoSynthesis and activity ofan octapeptide inhibitor designed for SARS coronavirus mainproteinaserdquo Peptides vol 27 no 4 pp 622ndash625 2006
[13] Q S Du H Sun and K C Chou ldquoInhibitor design for SARScoronavirus main protease based on lsquodistorted key theoryrsquordquoMedicinal Chemistry vol 3 no 1 pp 1ndash6 2007
[14] K C Chou ldquoPrediction of human immunodeficiency virusprotease cleavage sites in proteinsrdquoAnalytical Biochemistry vol233 no 1 pp 1ndash14 1996
[15] J Knowles and G Gromo ldquoTarget selection in drug discoveryrdquoNature Reviews Drug Discovery vol 2 no 1 pp 63ndash69 2003
[16] A C Cheng R G Coleman K T Smyth et al ldquoStructure-basedmaximal affinity model predicts small-molecule druggabilityrdquoNature Biotechnology vol 25 no 1 pp 71ndash75 2007
[17] M Rarey B Kramer T Lengauer and G Klebe ldquoA fast flexibledocking method using an incremental construction algorithmrdquoJournal of Molecular Biology vol 261 no 3 pp 470ndash489 1996
[18] K C Chou ldquoReview structural bioinformatics and its impactto biomedical sciencerdquo Current Medicinal Chemistry vol 11 no16 pp 2105ndash2134 2004
[19] K C Chou D Q Wei and W Z Zhong ldquoBinding mechanismof coronavirus main proteinase with ligands and its implicationto drug design against SARSrdquo Biochemical and BiophysicalResearch Communications vol 308 pp 148ndash151 2003 (Erratumin Biochemical and Biophysical Research Communications vol310 p 675 2003)
[20] K C Chou D Q Wei Q S Du S Sirois and W Z ZhongldquoProgress in computational approach to drug developmentagaint SARSrdquo Current Medicinal Chemistry vol 13 no 27 pp3263ndash3270 2006
[21] G P Zhou and F A Troy ldquoNMR studies on how the bindingcomplex of polyisoprenol recognition sequence peptides andpolyisoprenols can modulate membrane structurerdquo CurrentProtein and Peptide Science vol 6 no 5 pp 399ndash411 2005
[22] R B Huang Q S Du C H Wang and K C Chou ldquoAn in-depth analysis of the biological functional studies based on theNMR M2 channel structure of influenza A virusrdquo Biochemicaland Biophysical Research Communications vol 377 no 4 pp1243ndash1247 2008
[23] Q S Du R B Huang C H Wang X M Li and K C ChouldquoEnergetic analysis of the two controversial drug binding sitesof the M2 proton channel in influenza A virusrdquo Journal ofTheoretical Biology vol 259 no 1 pp 159ndash164 2009
BioMed Research International 11
[24] M J Berardi W M Shih S C Harrison and J J Chou ldquoMito-chondrial uncoupling protein 2 structure determined by NMRmolecular fragment searchingrdquo Nature vol 476 no 7358 pp109ndash114 2011
[25] J R Schnell and J J Chou ldquoStructure andmechanism of theM2proton channel of influenza A virusrdquo Nature vol 451 no 7178pp 591ndash595 2008
[26] B OuYang S Xie M J Berardi et al ldquoUnusual architecture ofthe p7 channel from hepatitis C virusrdquoNature vol 498 pp 521ndash525 2013
[27] K Oxenoid and J J Chou ldquoThe structure of phospholambanpentamer reveals a channel-like architecture in membranesrdquoProceedings of the National Academy of Sciences of the UnitedStates of America vol 102 no 31 pp 10870ndash10875 2005
[28] M E Call KWWucherpfennig and J J Chou ldquoThe structuralbasis for intramembrane assembly of an activating immunore-ceptor complexrdquo Nature Immunology vol 11 no 11 pp 1023ndash1029 2010
[29] R M Pielak J R Schnell and J J Chou ldquoMechanism of druginhibition and drug resistance of influenza A M2 channelrdquoProceedings of the National Academy of Sciences of the UnitedStates of America vol 106 no 18 pp 7379ndash7384 2009
[30] J Wang R M Pielak M A McClintock and J J ChouldquoSolution structure and functional analysis of the influenza Bproton channelrdquo Nature Structural and Molecular Biology vol16 no 12 pp 1267ndash1271 2009
[31] K C Chou ldquoCoupling interaction between thromboxane A2receptor and alpha-13 subunit of guanine nucleotide-bindingproteinrdquo Journal of Proteome Research vol 4 no 5 pp 1681ndash1686 2005
[32] K C Chou ldquoInsights from modeling three-dimensional struc-tures of the human potassium and sodium channelsrdquo Journal ofProteome Research vol 3 no 4 pp 856ndash861 2004
[33] K C Chou ldquoSome remarks on protein attribute prediction andpseudo amino acid compositionrdquo Journal ofTheoretical Biologyvol 273 no 1 pp 236ndash247 2011
[34] W Z Lin J A Fang X Xiao and K C Chou ldquoiLoc-animal amulti-label learning classifier for predicting subcellular local-ization of animal proteinsrdquo Molecular BioSystems vol 9 pp634ndash644 2013
[35] X Xiao PWangW Z Lin J H Jia and K C Chou ldquoiAMP-2La two-level multi-label classifier for identifying antimicrobialpeptides and their functional typesrdquo Analytical Biochemistryvol 436 pp 168ndash177 2013
[36] W Chen P M Feng H Lin and K C Chou ldquoiRSpot-PseDNCidentify recombination spots with pseudo dinucleotide compo-sitionrdquo Nucleic Acids Research vol 41 article e69 2013
[37] K C Chou Z C Wu and X Xiao ldquoILoc-Hum using theaccumulation-label scale to predict subcellular locations ofhuman proteins with both single and multiple sitesrdquoMolecularBioSystems vol 8 no 2 pp 629ndash641 2012
[38] M Kotera M Hirakawa T Tokimatsu S Goto and MKanehisa ldquoThe KEGG databases and tools facilitating omicsanalysis latest developments involving human diseases andpharmaceuticalsrdquo Methods in Molecular Biology vol 802 pp19ndash39 2012
[39] Y YamanishiM Araki A GutteridgeWHonda andM Kane-hisa ldquoPrediction of drug-target interaction networks from theintegration of chemical and genomic spacesrdquo Bioinformaticsvol 24 no 13 pp i232ndashi240 2008
[40] P Finn S Muggleton D Page and A Srinivasan ldquoPharma-cophore discovery using the Inductive Logic Programmingsystem PROGOLrdquo Machine Learning vol 30 no 2-3 pp 241ndash270 1998
[41] I Vogt D Stumpfe H E Ahmed and J Bajorath ldquoMethodsfor computer-aided chemical biology Part 2 evaluation of com-pound selectivity using 2D molecular fingerprintsrdquo ChemicalBiology and Drug Design vol 70 pp 195ndash205 2007
[42] H Eckert and J Bajorath ldquoMolecular similarity analysis in vir-tual screening foundations limitations and novel approachesrdquoDrug Discovery Today vol 12 no 5-6 pp 225ndash233 2007
[43] S Laurent L V Elst and R N Muller ldquoComparative studyof the physicochemical properties of six clinical low molecularweight gadolinium contrast agentsrdquo Contrast Media amp Molecu-lar Imaging vol 1 no 3 pp 128ndash137 2006
[44] E Gregori-Puigjane R Garriga-Sust and J Mestres ldquoIndexingmolecules with chemical graph identifiersrdquo Journal of Compu-tational Chemistry vol 32 no 12 pp 2638ndash2646 2011
[45] B Ren ldquoApplication of novel atom-type AI topological indicesto QSPR studies of alkanesrdquo Computers and Chemistry vol 26no 4 pp 357ndash369 2002
[46] N M OrsquoBoyle M Banck C A James C Morley T Vander-meersch and G R Hutchison ldquoOpen Babel an open chemicaltoolboxrdquo Journal of Cheminformatics vol 3 p 33 2011
[47] V J Gillet P Willett and J Bradshaw ldquoSimilarity searchingusing reduced graphsrdquo Journal of Chemical Information andComputer Sciences vol 43 no 2 pp 338ndash345 2003
[48] D Butina ldquoUnsupervised data base clustering based on day-lightrsquos fingerprint and Tanimoto similarity a fast and automatedway to cluster small and large data setsrdquo Journal of ChemicalInformation and Computer Sciences vol 39 no 4 pp 747ndash7501999
[49] C E Shannon ldquoA mathematical theory of communicationrdquoACM SIGMOBILE Mobile Computing and CommunicationsReview vol 5 pp 3ndash55 2001
[50] V D Gusev L A Nemytikova and N A Chuzhanova ldquoOn thecomplexity measures of genetic sequencesrdquo Bioinformatics vol15 no 12 pp 994ndash999 1999
[51] K C Chou and H B Shen ldquoReview recent progress in proteinsubcellular location predictionrdquo Analytical Biochemistry vol370 no 1 pp 1ndash16 2007
[52] S F Altschul ldquoEvaluating the statistical significance of multipledistinct local alignmentsrdquo in Theoretical and ComputationalMethods in Genome Research S Suhai Ed pp 1ndash14 PlenumNew York NY USA 1997
[53] J C Wootton and S Federhen ldquoStatistics of local complexity inamino acid sequences and sequence databasesrdquo Computers andChemistry vol 17 no 2 pp 149ndash163 1993
[54] H Nakashima K Nishikawa and T Ooi ldquoThe folding type ofa protein is relevant to the amino acid compositionrdquo Journal ofBiochemistry vol 99 no 1 pp 153ndash162 1986
[55] K C Chou and C T Zhang ldquoPredicting protein folding typesby distance functions that make allowances for amino acidinteractionsrdquo The Journal of Biological Chemistry vol 269 no35 pp 22014ndash22020 1994
[56] K-C Chou ldquoA novel approach to predicting protein structuralclasses in a (20-1)-D amino acid composition spacerdquo Proteinsvol 21 no 4 pp 319ndash344 1995
[57] H Nakashima and K Nishikawa ldquoDiscrimination of intracel-lular and extracellular proteins using amino acid compositionand residue-pair frequenciesrdquo Journal of Molecular Biology vol238 no 1 pp 54ndash61 1994
12 BioMed Research International
[58] G P Zhou ldquoAn intriguing controversy over protein structuralclass predictionrdquo Journal of Protein Chemistry vol 17 no 8 pp729ndash738 1998
[59] I Bahar A R Atilgan R L Jernigan and B Erman ldquoUnder-standing the recognition of protein structural classes by aminoacid compositionrdquo Proteins vol 29 pp 172ndash185 1997
[60] J Cedano P Aloy J A Perez-Pons and E Querol ldquoRelationbetween amino acid composition and cellular location ofproteinsrdquo Journal of Molecular Biology vol 266 no 3 pp 594ndash600 1997
[61] G P Zhou and K Doctor ldquoSubcellular location prediction ofapoptosis proteinsrdquo Proteins vol 50 no 1 pp 44ndash48 2003
[62] KChou ldquoPrediction of protein cellular attributes using pseudo-amino acid compositionrdquo Proteins vol 43 pp 246ndash255 2001(Erratum in Proteins vol 44 p 60 2001)
[63] K C Chou ldquoUsing amphiphilic pseudo amino acid composi-tion to predict enzyme subfamily classesrdquo Bioinformatics vol21 no 1 pp 10ndash19 2005
[64] D Zou Z He J He and Y Xia ldquoSupersecondary structureprediction using Choursquos pseudo amino acid compositionrdquoJournal of Computational Chemistry vol 32 no 2 pp 271ndash2782011
[65] M M Beigi M Behjati and H Mohabatkar ldquoPrediction ofmetalloproteinase family based on the concept ofChoursquos pseudoamino acid composition using a machine learning approachrdquoJournal of Structural and Functional Genomics vol 12 no 4 pp191ndash197 2011
[66] Y K Chen and K B Li ldquoPredicting membrane protein typesby incorporating protein topology domains signal peptidesand physicochemical properties into the general form of Choursquospseudo amino acid compositionrdquo Journal ofTheoretical Biologyvol 318 pp 1ndash12 2013
[67] C Huang and J Q Yuan ldquoA multilabel model based on Choursquospseudo-amino acid composition for identifying membraneproteins with both single and multiple functional typesrdquo TheJournal of Membrane Biology vol 246 pp 327ndash334 2013
[68] S S Sahu and G Panda ldquoA novel feature representationmethod based on Choursquos pseudo amino acid composition forprotein structural class predictionrdquo Computational Biology andChemistry vol 34 no 5-6 pp 320ndash327 2010
[69] M Hayat and A Khan ldquoDiscriminating outer membraneproteins with fuzzy K-nearest neighbor algorithms based on thegeneral form of Choursquos PseAACrdquo Protein and Peptide Lettersvol 19 no 4 pp 411ndash421 2012
[70] MKhosravian F K FaramarziMM BeigiM Behbahani andH Mohabatkar ldquoPredicting antibacterial peptides by the con-cept of Choursquos pseudo-amino acid composition and machinelearning methodsrdquo Protein amp Peptide Letters vol 20 pp 180ndash186 2013
[71] HMohabatkarMM Beigi K Abdolahi and SMohsenzadehldquoPrediction of allergenic proteins by means of the concept ofChoursquos pseudo amino acid composition and amachine learningapproachrdquoMedicinal Chemistry vol 9 pp 133ndash137 2013
[72] L Nanni A Lumini D Gupta and A Garg ldquoIdentifyingbacterial virulent proteins by fusing a set of classifiers basedon variants of Choursquos pseudo amino acid composition and onevolutionary informationrdquo IEEEACM Transactions on Compu-tational Biology and Bioinformatics vol 9 no 2 pp 467ndash4752012
[73] T H Chang L C Wu T Y Lee S P Chen H D Huang andJ T Horng ldquoEuLoc a web-server for accurately predict protein
subcellular localization in eukaryotes by incorporating variousfeatures of sequence segments into the general form of ChoursquosPseAACrdquo Journal of Computer-Aided Molecular Design vol 27pp 91ndash103 2013
[74] S Zhang Y Zhang H Yang C Zhao and Q Pan ldquoUsing theconcept of Choursquos pseudo amino acid composition to predictprotein subcellular localization an approach by incorporatingevolutionary information and von Neumann entropiesrdquo AminoAcids vol 34 no 4 pp 565ndash572 2008
[75] R Zia Ur and A Khan ldquoIdentifying GPCRs and their typeswith Choursquos pseudo amino acid composition an approach frommulti-scale energy representation and position specific scoringmatrixrdquo Protein amp Peptide Letters vol 19 pp 890ndash903 2012
[76] X Y Sun S P Shi J D Qiu S B Suo S Y Huang and R PLiang ldquoIdentifying protein quaternary structural attributes byincorporating physicochemical properties into the general formof Choursquos PseAAC via discrete wavelet transformrdquo MolecularBioSystems vol 8 pp 3178ndash3184 2012
[77] L Nanni and A Lumini ldquoGenetic programming for creatingChoursquos pseudo amino acid based features for submitochondrialocalizationrdquo Amino Acids vol 34 no 4 pp 653ndash660 2008
[78] M Esmaeili H Mohabatkar and S Mohsenzadeh ldquoUsing theconcept of Choursquos pseudo amino acid composition for risk typeprediction of human papillomavirusesrdquo Journal of TheoreticalBiology vol 263 no 2 pp 203ndash209 2010
[79] H Mohabatkar ldquoPrediction of cyclin proteins using Choursquospseudo amino acid compositionrdquo Protein and Peptide Lettersvol 17 no 10 pp 1207ndash1214 2010
[80] H Mohabatkar M M Beigi and A Esmaeili ldquoPredictionof GABA(A) receptor proteins using the concept of Choursquospseudo-amino acid composition and support vector machinerdquoJournal of Theoretical Biology vol 281 no 1 pp 18ndash23 2011
[81] Y Xu J Ding L Y Wu and K C Chou ldquoiSNO-PseAAC pre-dict cysteine S-nitrosylation sites in proteins by incorporatingposition specific amino acid propensity into pseudo amino acidcompositionrdquo PLoS ONE vol 8 Article ID e55844 2013
[82] W Chen H Lin PM Feng C Ding Y C Zuo andK C ChouldquoiNuc-PhysChem a sequence-based predictor for identifyingnucleosomes via physicochemical propertiesrdquo PLoS ONE vol7 Article ID e47843 2012
[83] B Li T Huang L Liu Y Cai and K C Chou ldquoIdentificationof colorectal cancer related genes with mrmr and shortest pathin protein-protein interaction networkrdquo PLoS ONE vol 7 no 4Article ID e33393 2012
[84] Y Jiang T Huang C Lei Y F Gao Y D Cai and K C ChouldquoSignal propagation in protein interaction network duringcolorectal cancer progressionrdquo BioMed Research Internationalvol 2013 Article ID 287019 9 pages 2013
[85] P Du X Wang C Xu and Y Gao ldquoPseAAC-Builder across-platform stand-alone program for generating variousspecial Choursquos pseudo-amino acid compositionsrdquo AnalyticalBiochemistry vol 425 no 2 pp 117ndash119 2012
[86] D S Cao Q S Xu and Y Z Liang ldquoPropy a tool to generatevarious modes of Choursquos PseAACrdquo Bioinformatics vol 29 pp960ndash962 2013
[87] H Shen and K C Chou ldquoPseAAC a flexible web serverfor generating various kinds of protein pseudo amino acidcompositionrdquo Analytical Biochemistry vol 373 no 2 pp 386ndash388 2008
[88] K C Chou ldquoUsing pair-coupled amino acid composition topredict protein secondary structure contentrdquo Journal of ProteinChemistry vol 18 no 4 pp 473ndash480 1999
BioMed Research International 13
[89] W Liu and K C Chou ldquoPrediction of protein secondarystructure contentrdquo Protein Engineering vol 12 no 12 pp 1041ndash1050 1999
[90] M Bhasin and G P S Raghava ldquoClassification of nuclearreceptors based on amino acid composition and dipeptidecompositionrdquo The Journal of Biological Chemistry vol 279 no22 pp 23262ndash23266 2004
[91] H Lin andH Ding ldquoPredicting ion channels and their types bythe dipeptidemode of pseudo amino acid compositionrdquo Journalof Theoretical Biology vol 269 no 1 pp 64ndash69 2011
[92] H Lin and Q Li ldquoUsing pseudo amino acid composition topredict protein structural class approached by incorporating400 dipeptide componentsrdquo Journal of Computational Chem-istry vol 28 no 9 pp 1463ndash1466 2007
[93] L Nanni and A Lumini ldquoCombing ontologies and dipeptidecomposition for predicting DNA-binding proteinsrdquo AminoAcids vol 34 no 4 pp 635ndash641 2008
[94] K-C Chou ldquoThe convergence-divergence duality in lectindomains of selectin family and its implicationsrdquo FEBS Lettersvol 363 no 1-2 pp 123ndash126 1995
[95] A A Schaffer L Aravind T L Madden et al ldquoImprovingthe accuracy of PSI-BLAST protein database searches withcomposition-based statistics and other refinementsrdquo NucleicAcids Research vol 29 no 14 pp 2994ndash3005 2001
[96] S F Altschul and E V Koonin ldquoIterated profile searches withPSI-BLAST a tool for discovery in protein databasesrdquo Trends inBiochemical Sciences vol 23 no 11 pp 444ndash447 1998
[97] J Deng ldquoGrey entropy and grey target decision makingrdquoJournal of Grey System vol 22 no 1 pp 1ndash24 2010
[98] B M Bolstad R A Irizarry M Astrand and T P Speed ldquoAcomparison of normalizationmethods for high density oligonu-cleotide array data based on variance and biasrdquo Bioinformaticsvol 19 no 2 pp 185ndash193 2003
[99] J-Y Shi S-W Zhang Q Pan Y-M Cheng and J XieldquoPrediction of protein subcellular localization by support vectormachines using multi-scale energy and pseudo amino acidcompositionrdquo Amino Acids vol 33 no 1 pp 69ndash74 2007
[100] T Denoeux ldquoA 120581-nearest neighbor classification rule based onDempster-Shafer theoryrdquo IEEE Transactions on Systems Manand Cybernetics vol 25 no 5 pp 804ndash813 1995
[101] J M Keller M R Gray and J A Givens ldquoA fuzzy k-nearestneighbours algorithmrdquo IEEE Transactions on Systems Man andCybernetics vol 15 no 4 pp 580ndash585 1985
[102] X Xiao P Wang and K Chou ldquoiNR-physchem a sequence-based predictor for identifying nuclear receptors and theirsubfamilies via physical-chemical property matrixrdquo PLoS ONEvol 7 no 2 Article ID e30869 2012
[103] I Roterman L Konieczny W Jurkowski K Prymula and MBanach ldquoTwo-intermediate model to characterize the structureof fast-folding proteinsrdquo Journal of Theoretical Biology vol 283no 1 pp 60ndash70 2011
[104] X Xiao P Wang and K C Chou ldquoGPCR-2L predicting Gprotein-coupled receptors and their types by hybridizing twodifferentmodes of pseudo amino acid compositionsrdquoMolecularBioSystems vol 7 no 3 pp 911ndash919 2011
[105] X Xiao P Wang and K C Chou ldquoQuat-2L a web-server forpredicting protein quaternary structural attributesrdquo MolecularDiversity vol 15 no 1 pp 149ndash155 2011
[106] X Zheng C Li and J Wang ldquoAn information-theoreticapproach to the prediction of protein structural classrdquo Journalof Computational Chemistry vol 31 no 6 pp 1201ndash1206 2010
[107] H Shen J Yang X Liu and K C Chou ldquoUsing supervisedfuzzy clustering to predict protein structural classesrdquo Biochem-ical and Biophysical Research Communications vol 334 no 2pp 577ndash581 2005
[108] K-C Chou and C-T Zhang ldquoReview prediction of proteinstructural classesrdquo Critical Reviews in Biochemistry and Molec-ular Biology vol 30 no 4 pp 275ndash349 1995
[109] P C Mahalanobis ldquoOn the generalized distance in statisticsrdquoProceedings of the National Institute of Sciences of India vol 2pp 49ndash55 1936
[110] R M Centor ldquoSignal detectability the use of ROC curves andtheir analysesrdquoMedical Decision Making vol 11 no 2 pp 102ndash106 1991
[111] K-C Chou ldquoUsing subsite coupling to predict signal peptidesrdquoProtein Engineering vol 14 no 2 pp 75ndash79 2001
[112] K C Chou ldquoPrediction of protein signal sequences and theircleavage sitesrdquo Proteins vol 42 pp 136ndash139 2001
[113] K C Chou ldquoPrediction of signal peptides using scaledwindowrdquoPeptides vol 22 no 12 pp 1973ndash1979 2001
[114] K C Chou ldquoSome remarks on predicting multi-label attributesinmolecular biosystemsrdquoMolecular Biosystems vol 9 pp 1092ndash1100 2013
[115] K C Chou and H B Shen ldquoCell-PLoc 2 0 an improvedpackage of web-servers for predicting subcellular localization ofproteins in various organismsrdquoNatural Science vol 2 pp 1090ndash1103 2010
[116] M Gribskov and N L Robinson ldquoUse of receiver operatingcharacteristic (ROC) analysis to evaluate sequence matchingrdquoComputers and Chemistry vol 20 no 1 pp 25ndash33 1996
[117] Y Yamanishi M Kotera M Kanehisa and S Goto ldquoDrug-target interaction prediction from chemical genomic and phar-macological data in an integrated frameworkrdquo Bioinformaticsvol 26 no 12 pp i246ndashi254 2010
[118] Z He J Zhang X Shi et al ldquoPredicting drug-target interactionnetworks based on functional groups and biological featuresrdquoPLoS ONE vol 5 no 3 Article ID e9603 2010
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

BioMed Research International 9
Read Me Data Citation
Enter both the protein sequence and the drug code (example) the number of query pairs is limited at 10 or less for each submission
Submit Clear
You can download the program and run it on your own local computer
iEzy-Drug identifying the interaction between enzymes anddrugs in cellular networking
Figure 5 A semiscreenshot to show the top page of the iEzy-Drugweb-server Its web-site address is at httpwwwjci-bioinfocniEzy-Drug
Table 1 The jackknife success rates obtained with iEzy-Drug in identifying interactive enzyme-drug pairs and noninteractive enzyme-drugpairs for the benchmark dataset S (cf Online Supporting Information S1)
Method Acc Sn Sp MCC
iEzy-Druga 74258157 = 9103 24692719 = 9081 49565438 = 9114 8039NN predictorb 8548 NA NA NAaSee (27) where the parameters119870 = 6 and 120593 = 15bSee [110]
or 1-Sp The best possible prediction method would yielda point with the coordinate (0 1) representing 100 truepositive rate (sensitivity Sn) and 0 false positive rate or100 specificity Therefore the (0 1) point is also calleda perfect classification A completely random guess wouldgive a point along a diagonal from the point (0 0) to (1 1)The area under the ROC curve also called Area Under theROC (AUROC) is often used to indicate the performancequality of a binary classifier the value 05 of AUROCis equivalent to random prediction while 1 of AUROCrepresents a perfect one As we can see from Figure 4the AUROC value obtained by iEzy-Drug is 09377
The reason why iEzy-Drug can remarkably improve theprediction quality is that it has introduced the 2D molecularfingerprints to represent drug samples see Online SupportingInformation S3 for the detailed fingerprint expressions for thedrugs listed in Online Supporting Information S1 and thatit has successfully used PseAAC to incorporate the featuresderived from the sequences of enzymes that are essentialfor identifying the interaction of enzymes with drugs in thecellular networking
To enhance the value of its practical applications theweb server for iEzy-Drug has been established that canbe freely accessible at httpwwwjci-bioinfocniEzy-DrugIt is anticipated that the web server will become a usefulhigh throughput tool for both basic research and drug
development in the relevant areas or at the very least playa complementary role to the existing method [39 110 118] forwhich so far noweb-serverwhatsoever has been provided yet
32 The Protocol or User Guide For the convenience of thevast majority of biologists and pharmaceutical scientists herelet us provide a step-by-step guide to show how the userscan easily get the desired result by means of the web serverwithout the need to follow the complicated mathematicalequations presented in this paper for the process of develop-ing the predictor and its integrityStep 1 Open the web server at the site httpwwwjci-bio-infocniEzy-Drug and you will see the top page of thepredictor on your computer screen as shown in Figure 5Click on the ReadMe button to see a brief introduction aboutiEzy-Drug predictor and the caveat when using itStep 2 Either type or copypaste the query pairs into theinput box at the center of Figure 5 Each query pair consistsof two parts one is for the protein sequence and the other forthe drug The enzyme sequence should be in FASTA formatwhile the drug in the KEGG code Examples for the querypairs input can be seen by clicking on the Example buttonright above the input boxStep 3 Click on the Submit button to see the predicted resultFor example if you use the four query pairs in the Example
10 BioMed Research International
window as the input after clicking the Submit button youwill see on your screen that the ldquohsa 10056rdquo enzyme andthe ldquoD0021rdquo drug are an interactive pair and that the ldquohsa100rdquo enzyme and the ldquoD0037rdquo drug are also an interactivepair but that the ldquohsa 3295rdquo enzyme and the ldquoD00889rdquo drugare not an interactive pair and that the ldquohsa 7366rdquo enzymeand the ldquoD03601rdquo drug are not an interactive pair eitherAll these results are fully consistent with the experimentalobservations It takes about 3 minutes before the results areshown on the screenStep 4 Click on the Citation button to find the relevant paperthat documents the detailed development and algorithm ofiEzy-DurgStep 5 Click on the Data button to download the benchmarkdataset used to train and test the iEzy-Durg predictorStep 6 The program code is also available by clicking thebutton download on the lower panel of Figure 5
Acknowledgments
The authors would like to thank the three anonymousreviewers whose constructive comments are very helpful forstrengthening the presentation of the paper This work wassupported by the Grants from the National Natural ScienceFoundation of China (no 31260273) the Jiangxi ProvincialForeign Scientific and Technological Cooperation Project(no 20120BDH80023) Natural Science Foundation of JiangxiProvince China (no 2010GZS0122 20122BAB201020) theDepartment of Education of Jiangxi Province (GJJ12490)the LuoDi plan of the Department of Education of JiangxiProvince (KJLD12083) and the Jiangxi Provincial Founda-tion for Leaders of Disciplines in Science (20113BCB22008)The funders had no role in study design data collection andanalysis decision to publish or preparation of the paper
References
[1] A Bairoch ldquoThe ENZYME database in 2000rdquo Nucleic AcidsResearch vol 28 no 1 pp 304ndash305 2000
[2] S H Koenig and R D Brown ldquoH2CO3as substrate for carbonic
anhydrase in the dehydration of H2CO3(minus)rdquo Proceedings of the
National Academy of Sciences of the United States of Americavol 69 no 9 pp 2422ndash2425 1972
[3] S X Lin and J Lapointe ldquoTheoretical and experimental biologyin onerdquo Journal of Biomedical Science and Engineering vol 6 pp435ndash442 2013
[4] K C Chou and S P Jiang ldquoStudies on the rate of diffusion-controlled reactions of enzymes Spatial factor and force fieldfactorrdquo Scientia Sinica vol 17 no 5 pp 664ndash680 1974
[5] K C Chou ldquoThe kinetics of the combination reaction betweenenzyme and substraterdquo Scientia Sinica vol 19 no 4 pp 505ndash528 1976
[6] K C Chou and G P Zhou ldquoRole of the protein outside activesite on the diffusion-controlled reaction of enzymerdquo Journal ofthe American Chemical Society vol 104 no 5 pp 1409ndash14131982
[7] R A Poorman A G Tomasselli R L Heinrikson and FJ Kezdy ldquoA cumulative specificity model for proteases from
human immunodeficiency virus types 1 and 2 inferred fromstatistical analysis of an extended substrate data baserdquo TheJournal of Biological Chemistry vol 266 no 22 pp 14554ndash145611991
[8] K-C Chou ldquoA vectorized sequence-coupling model for pre-dicting HIV protease cleavage sites in proteinsrdquo The Journal ofBiological Chemistry vol 268 no 23 pp 16938ndash16948 1993
[9] G Z Liang and S Z Li ldquoA new sequence representation asapplied in better specificity elucidation for human immunod-eficiency virus type 1 proteaserdquo Biopolymers vol 88 no 3 pp401ndash412 2007
[10] J J Chou ldquoPredicting cleavability of peptide sequences byHIVprotease via correlation-angle approachrdquo Journal of ProteinChemistry vol 12 no 3 pp 291ndash302 1993
[11] Q S Du S Wang D Q Wei S Sirois and K C ChouldquoMolecular modeling and chemical modification for findingpeptide inhibitor against severe acute respiratory syndromecoronavirus main proteinaserdquo Analytical Biochemistry vol 337no 2 pp 262ndash270 2005
[12] Y Gan H Huang Y Huang et al ldquoSynthesis and activity ofan octapeptide inhibitor designed for SARS coronavirus mainproteinaserdquo Peptides vol 27 no 4 pp 622ndash625 2006
[13] Q S Du H Sun and K C Chou ldquoInhibitor design for SARScoronavirus main protease based on lsquodistorted key theoryrsquordquoMedicinal Chemistry vol 3 no 1 pp 1ndash6 2007
[14] K C Chou ldquoPrediction of human immunodeficiency virusprotease cleavage sites in proteinsrdquoAnalytical Biochemistry vol233 no 1 pp 1ndash14 1996
[15] J Knowles and G Gromo ldquoTarget selection in drug discoveryrdquoNature Reviews Drug Discovery vol 2 no 1 pp 63ndash69 2003
[16] A C Cheng R G Coleman K T Smyth et al ldquoStructure-basedmaximal affinity model predicts small-molecule druggabilityrdquoNature Biotechnology vol 25 no 1 pp 71ndash75 2007
[17] M Rarey B Kramer T Lengauer and G Klebe ldquoA fast flexibledocking method using an incremental construction algorithmrdquoJournal of Molecular Biology vol 261 no 3 pp 470ndash489 1996
[18] K C Chou ldquoReview structural bioinformatics and its impactto biomedical sciencerdquo Current Medicinal Chemistry vol 11 no16 pp 2105ndash2134 2004
[19] K C Chou D Q Wei and W Z Zhong ldquoBinding mechanismof coronavirus main proteinase with ligands and its implicationto drug design against SARSrdquo Biochemical and BiophysicalResearch Communications vol 308 pp 148ndash151 2003 (Erratumin Biochemical and Biophysical Research Communications vol310 p 675 2003)
[20] K C Chou D Q Wei Q S Du S Sirois and W Z ZhongldquoProgress in computational approach to drug developmentagaint SARSrdquo Current Medicinal Chemistry vol 13 no 27 pp3263ndash3270 2006
[21] G P Zhou and F A Troy ldquoNMR studies on how the bindingcomplex of polyisoprenol recognition sequence peptides andpolyisoprenols can modulate membrane structurerdquo CurrentProtein and Peptide Science vol 6 no 5 pp 399ndash411 2005
[22] R B Huang Q S Du C H Wang and K C Chou ldquoAn in-depth analysis of the biological functional studies based on theNMR M2 channel structure of influenza A virusrdquo Biochemicaland Biophysical Research Communications vol 377 no 4 pp1243ndash1247 2008
[23] Q S Du R B Huang C H Wang X M Li and K C ChouldquoEnergetic analysis of the two controversial drug binding sitesof the M2 proton channel in influenza A virusrdquo Journal ofTheoretical Biology vol 259 no 1 pp 159ndash164 2009
BioMed Research International 11
[24] M J Berardi W M Shih S C Harrison and J J Chou ldquoMito-chondrial uncoupling protein 2 structure determined by NMRmolecular fragment searchingrdquo Nature vol 476 no 7358 pp109ndash114 2011
[25] J R Schnell and J J Chou ldquoStructure andmechanism of theM2proton channel of influenza A virusrdquo Nature vol 451 no 7178pp 591ndash595 2008
[26] B OuYang S Xie M J Berardi et al ldquoUnusual architecture ofthe p7 channel from hepatitis C virusrdquoNature vol 498 pp 521ndash525 2013
[27] K Oxenoid and J J Chou ldquoThe structure of phospholambanpentamer reveals a channel-like architecture in membranesrdquoProceedings of the National Academy of Sciences of the UnitedStates of America vol 102 no 31 pp 10870ndash10875 2005
[28] M E Call KWWucherpfennig and J J Chou ldquoThe structuralbasis for intramembrane assembly of an activating immunore-ceptor complexrdquo Nature Immunology vol 11 no 11 pp 1023ndash1029 2010
[29] R M Pielak J R Schnell and J J Chou ldquoMechanism of druginhibition and drug resistance of influenza A M2 channelrdquoProceedings of the National Academy of Sciences of the UnitedStates of America vol 106 no 18 pp 7379ndash7384 2009
[30] J Wang R M Pielak M A McClintock and J J ChouldquoSolution structure and functional analysis of the influenza Bproton channelrdquo Nature Structural and Molecular Biology vol16 no 12 pp 1267ndash1271 2009
[31] K C Chou ldquoCoupling interaction between thromboxane A2receptor and alpha-13 subunit of guanine nucleotide-bindingproteinrdquo Journal of Proteome Research vol 4 no 5 pp 1681ndash1686 2005
[32] K C Chou ldquoInsights from modeling three-dimensional struc-tures of the human potassium and sodium channelsrdquo Journal ofProteome Research vol 3 no 4 pp 856ndash861 2004
[33] K C Chou ldquoSome remarks on protein attribute prediction andpseudo amino acid compositionrdquo Journal ofTheoretical Biologyvol 273 no 1 pp 236ndash247 2011
[34] W Z Lin J A Fang X Xiao and K C Chou ldquoiLoc-animal amulti-label learning classifier for predicting subcellular local-ization of animal proteinsrdquo Molecular BioSystems vol 9 pp634ndash644 2013
[35] X Xiao PWangW Z Lin J H Jia and K C Chou ldquoiAMP-2La two-level multi-label classifier for identifying antimicrobialpeptides and their functional typesrdquo Analytical Biochemistryvol 436 pp 168ndash177 2013
[36] W Chen P M Feng H Lin and K C Chou ldquoiRSpot-PseDNCidentify recombination spots with pseudo dinucleotide compo-sitionrdquo Nucleic Acids Research vol 41 article e69 2013
[37] K C Chou Z C Wu and X Xiao ldquoILoc-Hum using theaccumulation-label scale to predict subcellular locations ofhuman proteins with both single and multiple sitesrdquoMolecularBioSystems vol 8 no 2 pp 629ndash641 2012
[38] M Kotera M Hirakawa T Tokimatsu S Goto and MKanehisa ldquoThe KEGG databases and tools facilitating omicsanalysis latest developments involving human diseases andpharmaceuticalsrdquo Methods in Molecular Biology vol 802 pp19ndash39 2012
[39] Y YamanishiM Araki A GutteridgeWHonda andM Kane-hisa ldquoPrediction of drug-target interaction networks from theintegration of chemical and genomic spacesrdquo Bioinformaticsvol 24 no 13 pp i232ndashi240 2008
[40] P Finn S Muggleton D Page and A Srinivasan ldquoPharma-cophore discovery using the Inductive Logic Programmingsystem PROGOLrdquo Machine Learning vol 30 no 2-3 pp 241ndash270 1998
[41] I Vogt D Stumpfe H E Ahmed and J Bajorath ldquoMethodsfor computer-aided chemical biology Part 2 evaluation of com-pound selectivity using 2D molecular fingerprintsrdquo ChemicalBiology and Drug Design vol 70 pp 195ndash205 2007
[42] H Eckert and J Bajorath ldquoMolecular similarity analysis in vir-tual screening foundations limitations and novel approachesrdquoDrug Discovery Today vol 12 no 5-6 pp 225ndash233 2007
[43] S Laurent L V Elst and R N Muller ldquoComparative studyof the physicochemical properties of six clinical low molecularweight gadolinium contrast agentsrdquo Contrast Media amp Molecu-lar Imaging vol 1 no 3 pp 128ndash137 2006
[44] E Gregori-Puigjane R Garriga-Sust and J Mestres ldquoIndexingmolecules with chemical graph identifiersrdquo Journal of Compu-tational Chemistry vol 32 no 12 pp 2638ndash2646 2011
[45] B Ren ldquoApplication of novel atom-type AI topological indicesto QSPR studies of alkanesrdquo Computers and Chemistry vol 26no 4 pp 357ndash369 2002
[46] N M OrsquoBoyle M Banck C A James C Morley T Vander-meersch and G R Hutchison ldquoOpen Babel an open chemicaltoolboxrdquo Journal of Cheminformatics vol 3 p 33 2011
[47] V J Gillet P Willett and J Bradshaw ldquoSimilarity searchingusing reduced graphsrdquo Journal of Chemical Information andComputer Sciences vol 43 no 2 pp 338ndash345 2003
[48] D Butina ldquoUnsupervised data base clustering based on day-lightrsquos fingerprint and Tanimoto similarity a fast and automatedway to cluster small and large data setsrdquo Journal of ChemicalInformation and Computer Sciences vol 39 no 4 pp 747ndash7501999
[49] C E Shannon ldquoA mathematical theory of communicationrdquoACM SIGMOBILE Mobile Computing and CommunicationsReview vol 5 pp 3ndash55 2001
[50] V D Gusev L A Nemytikova and N A Chuzhanova ldquoOn thecomplexity measures of genetic sequencesrdquo Bioinformatics vol15 no 12 pp 994ndash999 1999
[51] K C Chou and H B Shen ldquoReview recent progress in proteinsubcellular location predictionrdquo Analytical Biochemistry vol370 no 1 pp 1ndash16 2007
[52] S F Altschul ldquoEvaluating the statistical significance of multipledistinct local alignmentsrdquo in Theoretical and ComputationalMethods in Genome Research S Suhai Ed pp 1ndash14 PlenumNew York NY USA 1997
[53] J C Wootton and S Federhen ldquoStatistics of local complexity inamino acid sequences and sequence databasesrdquo Computers andChemistry vol 17 no 2 pp 149ndash163 1993
[54] H Nakashima K Nishikawa and T Ooi ldquoThe folding type ofa protein is relevant to the amino acid compositionrdquo Journal ofBiochemistry vol 99 no 1 pp 153ndash162 1986
[55] K C Chou and C T Zhang ldquoPredicting protein folding typesby distance functions that make allowances for amino acidinteractionsrdquo The Journal of Biological Chemistry vol 269 no35 pp 22014ndash22020 1994
[56] K-C Chou ldquoA novel approach to predicting protein structuralclasses in a (20-1)-D amino acid composition spacerdquo Proteinsvol 21 no 4 pp 319ndash344 1995
[57] H Nakashima and K Nishikawa ldquoDiscrimination of intracel-lular and extracellular proteins using amino acid compositionand residue-pair frequenciesrdquo Journal of Molecular Biology vol238 no 1 pp 54ndash61 1994
12 BioMed Research International
[58] G P Zhou ldquoAn intriguing controversy over protein structuralclass predictionrdquo Journal of Protein Chemistry vol 17 no 8 pp729ndash738 1998
[59] I Bahar A R Atilgan R L Jernigan and B Erman ldquoUnder-standing the recognition of protein structural classes by aminoacid compositionrdquo Proteins vol 29 pp 172ndash185 1997
[60] J Cedano P Aloy J A Perez-Pons and E Querol ldquoRelationbetween amino acid composition and cellular location ofproteinsrdquo Journal of Molecular Biology vol 266 no 3 pp 594ndash600 1997
[61] G P Zhou and K Doctor ldquoSubcellular location prediction ofapoptosis proteinsrdquo Proteins vol 50 no 1 pp 44ndash48 2003
[62] KChou ldquoPrediction of protein cellular attributes using pseudo-amino acid compositionrdquo Proteins vol 43 pp 246ndash255 2001(Erratum in Proteins vol 44 p 60 2001)
[63] K C Chou ldquoUsing amphiphilic pseudo amino acid composi-tion to predict enzyme subfamily classesrdquo Bioinformatics vol21 no 1 pp 10ndash19 2005
[64] D Zou Z He J He and Y Xia ldquoSupersecondary structureprediction using Choursquos pseudo amino acid compositionrdquoJournal of Computational Chemistry vol 32 no 2 pp 271ndash2782011
[65] M M Beigi M Behjati and H Mohabatkar ldquoPrediction ofmetalloproteinase family based on the concept ofChoursquos pseudoamino acid composition using a machine learning approachrdquoJournal of Structural and Functional Genomics vol 12 no 4 pp191ndash197 2011
[66] Y K Chen and K B Li ldquoPredicting membrane protein typesby incorporating protein topology domains signal peptidesand physicochemical properties into the general form of Choursquospseudo amino acid compositionrdquo Journal ofTheoretical Biologyvol 318 pp 1ndash12 2013
[67] C Huang and J Q Yuan ldquoA multilabel model based on Choursquospseudo-amino acid composition for identifying membraneproteins with both single and multiple functional typesrdquo TheJournal of Membrane Biology vol 246 pp 327ndash334 2013
[68] S S Sahu and G Panda ldquoA novel feature representationmethod based on Choursquos pseudo amino acid composition forprotein structural class predictionrdquo Computational Biology andChemistry vol 34 no 5-6 pp 320ndash327 2010
[69] M Hayat and A Khan ldquoDiscriminating outer membraneproteins with fuzzy K-nearest neighbor algorithms based on thegeneral form of Choursquos PseAACrdquo Protein and Peptide Lettersvol 19 no 4 pp 411ndash421 2012
[70] MKhosravian F K FaramarziMM BeigiM Behbahani andH Mohabatkar ldquoPredicting antibacterial peptides by the con-cept of Choursquos pseudo-amino acid composition and machinelearning methodsrdquo Protein amp Peptide Letters vol 20 pp 180ndash186 2013
[71] HMohabatkarMM Beigi K Abdolahi and SMohsenzadehldquoPrediction of allergenic proteins by means of the concept ofChoursquos pseudo amino acid composition and amachine learningapproachrdquoMedicinal Chemistry vol 9 pp 133ndash137 2013
[72] L Nanni A Lumini D Gupta and A Garg ldquoIdentifyingbacterial virulent proteins by fusing a set of classifiers basedon variants of Choursquos pseudo amino acid composition and onevolutionary informationrdquo IEEEACM Transactions on Compu-tational Biology and Bioinformatics vol 9 no 2 pp 467ndash4752012
[73] T H Chang L C Wu T Y Lee S P Chen H D Huang andJ T Horng ldquoEuLoc a web-server for accurately predict protein
subcellular localization in eukaryotes by incorporating variousfeatures of sequence segments into the general form of ChoursquosPseAACrdquo Journal of Computer-Aided Molecular Design vol 27pp 91ndash103 2013
[74] S Zhang Y Zhang H Yang C Zhao and Q Pan ldquoUsing theconcept of Choursquos pseudo amino acid composition to predictprotein subcellular localization an approach by incorporatingevolutionary information and von Neumann entropiesrdquo AminoAcids vol 34 no 4 pp 565ndash572 2008
[75] R Zia Ur and A Khan ldquoIdentifying GPCRs and their typeswith Choursquos pseudo amino acid composition an approach frommulti-scale energy representation and position specific scoringmatrixrdquo Protein amp Peptide Letters vol 19 pp 890ndash903 2012
[76] X Y Sun S P Shi J D Qiu S B Suo S Y Huang and R PLiang ldquoIdentifying protein quaternary structural attributes byincorporating physicochemical properties into the general formof Choursquos PseAAC via discrete wavelet transformrdquo MolecularBioSystems vol 8 pp 3178ndash3184 2012
[77] L Nanni and A Lumini ldquoGenetic programming for creatingChoursquos pseudo amino acid based features for submitochondrialocalizationrdquo Amino Acids vol 34 no 4 pp 653ndash660 2008
[78] M Esmaeili H Mohabatkar and S Mohsenzadeh ldquoUsing theconcept of Choursquos pseudo amino acid composition for risk typeprediction of human papillomavirusesrdquo Journal of TheoreticalBiology vol 263 no 2 pp 203ndash209 2010
[79] H Mohabatkar ldquoPrediction of cyclin proteins using Choursquospseudo amino acid compositionrdquo Protein and Peptide Lettersvol 17 no 10 pp 1207ndash1214 2010
[80] H Mohabatkar M M Beigi and A Esmaeili ldquoPredictionof GABA(A) receptor proteins using the concept of Choursquospseudo-amino acid composition and support vector machinerdquoJournal of Theoretical Biology vol 281 no 1 pp 18ndash23 2011
[81] Y Xu J Ding L Y Wu and K C Chou ldquoiSNO-PseAAC pre-dict cysteine S-nitrosylation sites in proteins by incorporatingposition specific amino acid propensity into pseudo amino acidcompositionrdquo PLoS ONE vol 8 Article ID e55844 2013
[82] W Chen H Lin PM Feng C Ding Y C Zuo andK C ChouldquoiNuc-PhysChem a sequence-based predictor for identifyingnucleosomes via physicochemical propertiesrdquo PLoS ONE vol7 Article ID e47843 2012
[83] B Li T Huang L Liu Y Cai and K C Chou ldquoIdentificationof colorectal cancer related genes with mrmr and shortest pathin protein-protein interaction networkrdquo PLoS ONE vol 7 no 4Article ID e33393 2012
[84] Y Jiang T Huang C Lei Y F Gao Y D Cai and K C ChouldquoSignal propagation in protein interaction network duringcolorectal cancer progressionrdquo BioMed Research Internationalvol 2013 Article ID 287019 9 pages 2013
[85] P Du X Wang C Xu and Y Gao ldquoPseAAC-Builder across-platform stand-alone program for generating variousspecial Choursquos pseudo-amino acid compositionsrdquo AnalyticalBiochemistry vol 425 no 2 pp 117ndash119 2012
[86] D S Cao Q S Xu and Y Z Liang ldquoPropy a tool to generatevarious modes of Choursquos PseAACrdquo Bioinformatics vol 29 pp960ndash962 2013
[87] H Shen and K C Chou ldquoPseAAC a flexible web serverfor generating various kinds of protein pseudo amino acidcompositionrdquo Analytical Biochemistry vol 373 no 2 pp 386ndash388 2008
[88] K C Chou ldquoUsing pair-coupled amino acid composition topredict protein secondary structure contentrdquo Journal of ProteinChemistry vol 18 no 4 pp 473ndash480 1999
BioMed Research International 13
[89] W Liu and K C Chou ldquoPrediction of protein secondarystructure contentrdquo Protein Engineering vol 12 no 12 pp 1041ndash1050 1999
[90] M Bhasin and G P S Raghava ldquoClassification of nuclearreceptors based on amino acid composition and dipeptidecompositionrdquo The Journal of Biological Chemistry vol 279 no22 pp 23262ndash23266 2004
[91] H Lin andH Ding ldquoPredicting ion channels and their types bythe dipeptidemode of pseudo amino acid compositionrdquo Journalof Theoretical Biology vol 269 no 1 pp 64ndash69 2011
[92] H Lin and Q Li ldquoUsing pseudo amino acid composition topredict protein structural class approached by incorporating400 dipeptide componentsrdquo Journal of Computational Chem-istry vol 28 no 9 pp 1463ndash1466 2007
[93] L Nanni and A Lumini ldquoCombing ontologies and dipeptidecomposition for predicting DNA-binding proteinsrdquo AminoAcids vol 34 no 4 pp 635ndash641 2008
[94] K-C Chou ldquoThe convergence-divergence duality in lectindomains of selectin family and its implicationsrdquo FEBS Lettersvol 363 no 1-2 pp 123ndash126 1995
[95] A A Schaffer L Aravind T L Madden et al ldquoImprovingthe accuracy of PSI-BLAST protein database searches withcomposition-based statistics and other refinementsrdquo NucleicAcids Research vol 29 no 14 pp 2994ndash3005 2001
[96] S F Altschul and E V Koonin ldquoIterated profile searches withPSI-BLAST a tool for discovery in protein databasesrdquo Trends inBiochemical Sciences vol 23 no 11 pp 444ndash447 1998
[97] J Deng ldquoGrey entropy and grey target decision makingrdquoJournal of Grey System vol 22 no 1 pp 1ndash24 2010
[98] B M Bolstad R A Irizarry M Astrand and T P Speed ldquoAcomparison of normalizationmethods for high density oligonu-cleotide array data based on variance and biasrdquo Bioinformaticsvol 19 no 2 pp 185ndash193 2003
[99] J-Y Shi S-W Zhang Q Pan Y-M Cheng and J XieldquoPrediction of protein subcellular localization by support vectormachines using multi-scale energy and pseudo amino acidcompositionrdquo Amino Acids vol 33 no 1 pp 69ndash74 2007
[100] T Denoeux ldquoA 120581-nearest neighbor classification rule based onDempster-Shafer theoryrdquo IEEE Transactions on Systems Manand Cybernetics vol 25 no 5 pp 804ndash813 1995
[101] J M Keller M R Gray and J A Givens ldquoA fuzzy k-nearestneighbours algorithmrdquo IEEE Transactions on Systems Man andCybernetics vol 15 no 4 pp 580ndash585 1985
[102] X Xiao P Wang and K Chou ldquoiNR-physchem a sequence-based predictor for identifying nuclear receptors and theirsubfamilies via physical-chemical property matrixrdquo PLoS ONEvol 7 no 2 Article ID e30869 2012
[103] I Roterman L Konieczny W Jurkowski K Prymula and MBanach ldquoTwo-intermediate model to characterize the structureof fast-folding proteinsrdquo Journal of Theoretical Biology vol 283no 1 pp 60ndash70 2011
[104] X Xiao P Wang and K C Chou ldquoGPCR-2L predicting Gprotein-coupled receptors and their types by hybridizing twodifferentmodes of pseudo amino acid compositionsrdquoMolecularBioSystems vol 7 no 3 pp 911ndash919 2011
[105] X Xiao P Wang and K C Chou ldquoQuat-2L a web-server forpredicting protein quaternary structural attributesrdquo MolecularDiversity vol 15 no 1 pp 149ndash155 2011
[106] X Zheng C Li and J Wang ldquoAn information-theoreticapproach to the prediction of protein structural classrdquo Journalof Computational Chemistry vol 31 no 6 pp 1201ndash1206 2010
[107] H Shen J Yang X Liu and K C Chou ldquoUsing supervisedfuzzy clustering to predict protein structural classesrdquo Biochem-ical and Biophysical Research Communications vol 334 no 2pp 577ndash581 2005
[108] K-C Chou and C-T Zhang ldquoReview prediction of proteinstructural classesrdquo Critical Reviews in Biochemistry and Molec-ular Biology vol 30 no 4 pp 275ndash349 1995
[109] P C Mahalanobis ldquoOn the generalized distance in statisticsrdquoProceedings of the National Institute of Sciences of India vol 2pp 49ndash55 1936
[110] R M Centor ldquoSignal detectability the use of ROC curves andtheir analysesrdquoMedical Decision Making vol 11 no 2 pp 102ndash106 1991
[111] K-C Chou ldquoUsing subsite coupling to predict signal peptidesrdquoProtein Engineering vol 14 no 2 pp 75ndash79 2001
[112] K C Chou ldquoPrediction of protein signal sequences and theircleavage sitesrdquo Proteins vol 42 pp 136ndash139 2001
[113] K C Chou ldquoPrediction of signal peptides using scaledwindowrdquoPeptides vol 22 no 12 pp 1973ndash1979 2001
[114] K C Chou ldquoSome remarks on predicting multi-label attributesinmolecular biosystemsrdquoMolecular Biosystems vol 9 pp 1092ndash1100 2013
[115] K C Chou and H B Shen ldquoCell-PLoc 2 0 an improvedpackage of web-servers for predicting subcellular localization ofproteins in various organismsrdquoNatural Science vol 2 pp 1090ndash1103 2010
[116] M Gribskov and N L Robinson ldquoUse of receiver operatingcharacteristic (ROC) analysis to evaluate sequence matchingrdquoComputers and Chemistry vol 20 no 1 pp 25ndash33 1996
[117] Y Yamanishi M Kotera M Kanehisa and S Goto ldquoDrug-target interaction prediction from chemical genomic and phar-macological data in an integrated frameworkrdquo Bioinformaticsvol 26 no 12 pp i246ndashi254 2010
[118] Z He J Zhang X Shi et al ldquoPredicting drug-target interactionnetworks based on functional groups and biological featuresrdquoPLoS ONE vol 5 no 3 Article ID e9603 2010
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

10 BioMed Research International
window as the input after clicking the Submit button youwill see on your screen that the ldquohsa 10056rdquo enzyme andthe ldquoD0021rdquo drug are an interactive pair and that the ldquohsa100rdquo enzyme and the ldquoD0037rdquo drug are also an interactivepair but that the ldquohsa 3295rdquo enzyme and the ldquoD00889rdquo drugare not an interactive pair and that the ldquohsa 7366rdquo enzymeand the ldquoD03601rdquo drug are not an interactive pair eitherAll these results are fully consistent with the experimentalobservations It takes about 3 minutes before the results areshown on the screenStep 4 Click on the Citation button to find the relevant paperthat documents the detailed development and algorithm ofiEzy-DurgStep 5 Click on the Data button to download the benchmarkdataset used to train and test the iEzy-Durg predictorStep 6 The program code is also available by clicking thebutton download on the lower panel of Figure 5
Acknowledgments
The authors would like to thank the three anonymousreviewers whose constructive comments are very helpful forstrengthening the presentation of the paper This work wassupported by the Grants from the National Natural ScienceFoundation of China (no 31260273) the Jiangxi ProvincialForeign Scientific and Technological Cooperation Project(no 20120BDH80023) Natural Science Foundation of JiangxiProvince China (no 2010GZS0122 20122BAB201020) theDepartment of Education of Jiangxi Province (GJJ12490)the LuoDi plan of the Department of Education of JiangxiProvince (KJLD12083) and the Jiangxi Provincial Founda-tion for Leaders of Disciplines in Science (20113BCB22008)The funders had no role in study design data collection andanalysis decision to publish or preparation of the paper
References
[1] A Bairoch ldquoThe ENZYME database in 2000rdquo Nucleic AcidsResearch vol 28 no 1 pp 304ndash305 2000
[2] S H Koenig and R D Brown ldquoH2CO3as substrate for carbonic
anhydrase in the dehydration of H2CO3(minus)rdquo Proceedings of the
National Academy of Sciences of the United States of Americavol 69 no 9 pp 2422ndash2425 1972
[3] S X Lin and J Lapointe ldquoTheoretical and experimental biologyin onerdquo Journal of Biomedical Science and Engineering vol 6 pp435ndash442 2013
[4] K C Chou and S P Jiang ldquoStudies on the rate of diffusion-controlled reactions of enzymes Spatial factor and force fieldfactorrdquo Scientia Sinica vol 17 no 5 pp 664ndash680 1974
[5] K C Chou ldquoThe kinetics of the combination reaction betweenenzyme and substraterdquo Scientia Sinica vol 19 no 4 pp 505ndash528 1976
[6] K C Chou and G P Zhou ldquoRole of the protein outside activesite on the diffusion-controlled reaction of enzymerdquo Journal ofthe American Chemical Society vol 104 no 5 pp 1409ndash14131982
[7] R A Poorman A G Tomasselli R L Heinrikson and FJ Kezdy ldquoA cumulative specificity model for proteases from
human immunodeficiency virus types 1 and 2 inferred fromstatistical analysis of an extended substrate data baserdquo TheJournal of Biological Chemistry vol 266 no 22 pp 14554ndash145611991
[8] K-C Chou ldquoA vectorized sequence-coupling model for pre-dicting HIV protease cleavage sites in proteinsrdquo The Journal ofBiological Chemistry vol 268 no 23 pp 16938ndash16948 1993
[9] G Z Liang and S Z Li ldquoA new sequence representation asapplied in better specificity elucidation for human immunod-eficiency virus type 1 proteaserdquo Biopolymers vol 88 no 3 pp401ndash412 2007
[10] J J Chou ldquoPredicting cleavability of peptide sequences byHIVprotease via correlation-angle approachrdquo Journal of ProteinChemistry vol 12 no 3 pp 291ndash302 1993
[11] Q S Du S Wang D Q Wei S Sirois and K C ChouldquoMolecular modeling and chemical modification for findingpeptide inhibitor against severe acute respiratory syndromecoronavirus main proteinaserdquo Analytical Biochemistry vol 337no 2 pp 262ndash270 2005
[12] Y Gan H Huang Y Huang et al ldquoSynthesis and activity ofan octapeptide inhibitor designed for SARS coronavirus mainproteinaserdquo Peptides vol 27 no 4 pp 622ndash625 2006
[13] Q S Du H Sun and K C Chou ldquoInhibitor design for SARScoronavirus main protease based on lsquodistorted key theoryrsquordquoMedicinal Chemistry vol 3 no 1 pp 1ndash6 2007
[14] K C Chou ldquoPrediction of human immunodeficiency virusprotease cleavage sites in proteinsrdquoAnalytical Biochemistry vol233 no 1 pp 1ndash14 1996
[15] J Knowles and G Gromo ldquoTarget selection in drug discoveryrdquoNature Reviews Drug Discovery vol 2 no 1 pp 63ndash69 2003
[16] A C Cheng R G Coleman K T Smyth et al ldquoStructure-basedmaximal affinity model predicts small-molecule druggabilityrdquoNature Biotechnology vol 25 no 1 pp 71ndash75 2007
[17] M Rarey B Kramer T Lengauer and G Klebe ldquoA fast flexibledocking method using an incremental construction algorithmrdquoJournal of Molecular Biology vol 261 no 3 pp 470ndash489 1996
[18] K C Chou ldquoReview structural bioinformatics and its impactto biomedical sciencerdquo Current Medicinal Chemistry vol 11 no16 pp 2105ndash2134 2004
[19] K C Chou D Q Wei and W Z Zhong ldquoBinding mechanismof coronavirus main proteinase with ligands and its implicationto drug design against SARSrdquo Biochemical and BiophysicalResearch Communications vol 308 pp 148ndash151 2003 (Erratumin Biochemical and Biophysical Research Communications vol310 p 675 2003)
[20] K C Chou D Q Wei Q S Du S Sirois and W Z ZhongldquoProgress in computational approach to drug developmentagaint SARSrdquo Current Medicinal Chemistry vol 13 no 27 pp3263ndash3270 2006
[21] G P Zhou and F A Troy ldquoNMR studies on how the bindingcomplex of polyisoprenol recognition sequence peptides andpolyisoprenols can modulate membrane structurerdquo CurrentProtein and Peptide Science vol 6 no 5 pp 399ndash411 2005
[22] R B Huang Q S Du C H Wang and K C Chou ldquoAn in-depth analysis of the biological functional studies based on theNMR M2 channel structure of influenza A virusrdquo Biochemicaland Biophysical Research Communications vol 377 no 4 pp1243ndash1247 2008
[23] Q S Du R B Huang C H Wang X M Li and K C ChouldquoEnergetic analysis of the two controversial drug binding sitesof the M2 proton channel in influenza A virusrdquo Journal ofTheoretical Biology vol 259 no 1 pp 159ndash164 2009
BioMed Research International 11
[24] M J Berardi W M Shih S C Harrison and J J Chou ldquoMito-chondrial uncoupling protein 2 structure determined by NMRmolecular fragment searchingrdquo Nature vol 476 no 7358 pp109ndash114 2011
[25] J R Schnell and J J Chou ldquoStructure andmechanism of theM2proton channel of influenza A virusrdquo Nature vol 451 no 7178pp 591ndash595 2008
[26] B OuYang S Xie M J Berardi et al ldquoUnusual architecture ofthe p7 channel from hepatitis C virusrdquoNature vol 498 pp 521ndash525 2013
[27] K Oxenoid and J J Chou ldquoThe structure of phospholambanpentamer reveals a channel-like architecture in membranesrdquoProceedings of the National Academy of Sciences of the UnitedStates of America vol 102 no 31 pp 10870ndash10875 2005
[28] M E Call KWWucherpfennig and J J Chou ldquoThe structuralbasis for intramembrane assembly of an activating immunore-ceptor complexrdquo Nature Immunology vol 11 no 11 pp 1023ndash1029 2010
[29] R M Pielak J R Schnell and J J Chou ldquoMechanism of druginhibition and drug resistance of influenza A M2 channelrdquoProceedings of the National Academy of Sciences of the UnitedStates of America vol 106 no 18 pp 7379ndash7384 2009
[30] J Wang R M Pielak M A McClintock and J J ChouldquoSolution structure and functional analysis of the influenza Bproton channelrdquo Nature Structural and Molecular Biology vol16 no 12 pp 1267ndash1271 2009
[31] K C Chou ldquoCoupling interaction between thromboxane A2receptor and alpha-13 subunit of guanine nucleotide-bindingproteinrdquo Journal of Proteome Research vol 4 no 5 pp 1681ndash1686 2005
[32] K C Chou ldquoInsights from modeling three-dimensional struc-tures of the human potassium and sodium channelsrdquo Journal ofProteome Research vol 3 no 4 pp 856ndash861 2004
[33] K C Chou ldquoSome remarks on protein attribute prediction andpseudo amino acid compositionrdquo Journal ofTheoretical Biologyvol 273 no 1 pp 236ndash247 2011
[34] W Z Lin J A Fang X Xiao and K C Chou ldquoiLoc-animal amulti-label learning classifier for predicting subcellular local-ization of animal proteinsrdquo Molecular BioSystems vol 9 pp634ndash644 2013
[35] X Xiao PWangW Z Lin J H Jia and K C Chou ldquoiAMP-2La two-level multi-label classifier for identifying antimicrobialpeptides and their functional typesrdquo Analytical Biochemistryvol 436 pp 168ndash177 2013
[36] W Chen P M Feng H Lin and K C Chou ldquoiRSpot-PseDNCidentify recombination spots with pseudo dinucleotide compo-sitionrdquo Nucleic Acids Research vol 41 article e69 2013
[37] K C Chou Z C Wu and X Xiao ldquoILoc-Hum using theaccumulation-label scale to predict subcellular locations ofhuman proteins with both single and multiple sitesrdquoMolecularBioSystems vol 8 no 2 pp 629ndash641 2012
[38] M Kotera M Hirakawa T Tokimatsu S Goto and MKanehisa ldquoThe KEGG databases and tools facilitating omicsanalysis latest developments involving human diseases andpharmaceuticalsrdquo Methods in Molecular Biology vol 802 pp19ndash39 2012
[39] Y YamanishiM Araki A GutteridgeWHonda andM Kane-hisa ldquoPrediction of drug-target interaction networks from theintegration of chemical and genomic spacesrdquo Bioinformaticsvol 24 no 13 pp i232ndashi240 2008
[40] P Finn S Muggleton D Page and A Srinivasan ldquoPharma-cophore discovery using the Inductive Logic Programmingsystem PROGOLrdquo Machine Learning vol 30 no 2-3 pp 241ndash270 1998
[41] I Vogt D Stumpfe H E Ahmed and J Bajorath ldquoMethodsfor computer-aided chemical biology Part 2 evaluation of com-pound selectivity using 2D molecular fingerprintsrdquo ChemicalBiology and Drug Design vol 70 pp 195ndash205 2007
[42] H Eckert and J Bajorath ldquoMolecular similarity analysis in vir-tual screening foundations limitations and novel approachesrdquoDrug Discovery Today vol 12 no 5-6 pp 225ndash233 2007
[43] S Laurent L V Elst and R N Muller ldquoComparative studyof the physicochemical properties of six clinical low molecularweight gadolinium contrast agentsrdquo Contrast Media amp Molecu-lar Imaging vol 1 no 3 pp 128ndash137 2006
[44] E Gregori-Puigjane R Garriga-Sust and J Mestres ldquoIndexingmolecules with chemical graph identifiersrdquo Journal of Compu-tational Chemistry vol 32 no 12 pp 2638ndash2646 2011
[45] B Ren ldquoApplication of novel atom-type AI topological indicesto QSPR studies of alkanesrdquo Computers and Chemistry vol 26no 4 pp 357ndash369 2002
[46] N M OrsquoBoyle M Banck C A James C Morley T Vander-meersch and G R Hutchison ldquoOpen Babel an open chemicaltoolboxrdquo Journal of Cheminformatics vol 3 p 33 2011
[47] V J Gillet P Willett and J Bradshaw ldquoSimilarity searchingusing reduced graphsrdquo Journal of Chemical Information andComputer Sciences vol 43 no 2 pp 338ndash345 2003
[48] D Butina ldquoUnsupervised data base clustering based on day-lightrsquos fingerprint and Tanimoto similarity a fast and automatedway to cluster small and large data setsrdquo Journal of ChemicalInformation and Computer Sciences vol 39 no 4 pp 747ndash7501999
[49] C E Shannon ldquoA mathematical theory of communicationrdquoACM SIGMOBILE Mobile Computing and CommunicationsReview vol 5 pp 3ndash55 2001
[50] V D Gusev L A Nemytikova and N A Chuzhanova ldquoOn thecomplexity measures of genetic sequencesrdquo Bioinformatics vol15 no 12 pp 994ndash999 1999
[51] K C Chou and H B Shen ldquoReview recent progress in proteinsubcellular location predictionrdquo Analytical Biochemistry vol370 no 1 pp 1ndash16 2007
[52] S F Altschul ldquoEvaluating the statistical significance of multipledistinct local alignmentsrdquo in Theoretical and ComputationalMethods in Genome Research S Suhai Ed pp 1ndash14 PlenumNew York NY USA 1997
[53] J C Wootton and S Federhen ldquoStatistics of local complexity inamino acid sequences and sequence databasesrdquo Computers andChemistry vol 17 no 2 pp 149ndash163 1993
[54] H Nakashima K Nishikawa and T Ooi ldquoThe folding type ofa protein is relevant to the amino acid compositionrdquo Journal ofBiochemistry vol 99 no 1 pp 153ndash162 1986
[55] K C Chou and C T Zhang ldquoPredicting protein folding typesby distance functions that make allowances for amino acidinteractionsrdquo The Journal of Biological Chemistry vol 269 no35 pp 22014ndash22020 1994
[56] K-C Chou ldquoA novel approach to predicting protein structuralclasses in a (20-1)-D amino acid composition spacerdquo Proteinsvol 21 no 4 pp 319ndash344 1995
[57] H Nakashima and K Nishikawa ldquoDiscrimination of intracel-lular and extracellular proteins using amino acid compositionand residue-pair frequenciesrdquo Journal of Molecular Biology vol238 no 1 pp 54ndash61 1994
12 BioMed Research International
[58] G P Zhou ldquoAn intriguing controversy over protein structuralclass predictionrdquo Journal of Protein Chemistry vol 17 no 8 pp729ndash738 1998
[59] I Bahar A R Atilgan R L Jernigan and B Erman ldquoUnder-standing the recognition of protein structural classes by aminoacid compositionrdquo Proteins vol 29 pp 172ndash185 1997
[60] J Cedano P Aloy J A Perez-Pons and E Querol ldquoRelationbetween amino acid composition and cellular location ofproteinsrdquo Journal of Molecular Biology vol 266 no 3 pp 594ndash600 1997
[61] G P Zhou and K Doctor ldquoSubcellular location prediction ofapoptosis proteinsrdquo Proteins vol 50 no 1 pp 44ndash48 2003
[62] KChou ldquoPrediction of protein cellular attributes using pseudo-amino acid compositionrdquo Proteins vol 43 pp 246ndash255 2001(Erratum in Proteins vol 44 p 60 2001)
[63] K C Chou ldquoUsing amphiphilic pseudo amino acid composi-tion to predict enzyme subfamily classesrdquo Bioinformatics vol21 no 1 pp 10ndash19 2005
[64] D Zou Z He J He and Y Xia ldquoSupersecondary structureprediction using Choursquos pseudo amino acid compositionrdquoJournal of Computational Chemistry vol 32 no 2 pp 271ndash2782011
[65] M M Beigi M Behjati and H Mohabatkar ldquoPrediction ofmetalloproteinase family based on the concept ofChoursquos pseudoamino acid composition using a machine learning approachrdquoJournal of Structural and Functional Genomics vol 12 no 4 pp191ndash197 2011
[66] Y K Chen and K B Li ldquoPredicting membrane protein typesby incorporating protein topology domains signal peptidesand physicochemical properties into the general form of Choursquospseudo amino acid compositionrdquo Journal ofTheoretical Biologyvol 318 pp 1ndash12 2013
[67] C Huang and J Q Yuan ldquoA multilabel model based on Choursquospseudo-amino acid composition for identifying membraneproteins with both single and multiple functional typesrdquo TheJournal of Membrane Biology vol 246 pp 327ndash334 2013
[68] S S Sahu and G Panda ldquoA novel feature representationmethod based on Choursquos pseudo amino acid composition forprotein structural class predictionrdquo Computational Biology andChemistry vol 34 no 5-6 pp 320ndash327 2010
[69] M Hayat and A Khan ldquoDiscriminating outer membraneproteins with fuzzy K-nearest neighbor algorithms based on thegeneral form of Choursquos PseAACrdquo Protein and Peptide Lettersvol 19 no 4 pp 411ndash421 2012
[70] MKhosravian F K FaramarziMM BeigiM Behbahani andH Mohabatkar ldquoPredicting antibacterial peptides by the con-cept of Choursquos pseudo-amino acid composition and machinelearning methodsrdquo Protein amp Peptide Letters vol 20 pp 180ndash186 2013
[71] HMohabatkarMM Beigi K Abdolahi and SMohsenzadehldquoPrediction of allergenic proteins by means of the concept ofChoursquos pseudo amino acid composition and amachine learningapproachrdquoMedicinal Chemistry vol 9 pp 133ndash137 2013
[72] L Nanni A Lumini D Gupta and A Garg ldquoIdentifyingbacterial virulent proteins by fusing a set of classifiers basedon variants of Choursquos pseudo amino acid composition and onevolutionary informationrdquo IEEEACM Transactions on Compu-tational Biology and Bioinformatics vol 9 no 2 pp 467ndash4752012
[73] T H Chang L C Wu T Y Lee S P Chen H D Huang andJ T Horng ldquoEuLoc a web-server for accurately predict protein
subcellular localization in eukaryotes by incorporating variousfeatures of sequence segments into the general form of ChoursquosPseAACrdquo Journal of Computer-Aided Molecular Design vol 27pp 91ndash103 2013
[74] S Zhang Y Zhang H Yang C Zhao and Q Pan ldquoUsing theconcept of Choursquos pseudo amino acid composition to predictprotein subcellular localization an approach by incorporatingevolutionary information and von Neumann entropiesrdquo AminoAcids vol 34 no 4 pp 565ndash572 2008
[75] R Zia Ur and A Khan ldquoIdentifying GPCRs and their typeswith Choursquos pseudo amino acid composition an approach frommulti-scale energy representation and position specific scoringmatrixrdquo Protein amp Peptide Letters vol 19 pp 890ndash903 2012
[76] X Y Sun S P Shi J D Qiu S B Suo S Y Huang and R PLiang ldquoIdentifying protein quaternary structural attributes byincorporating physicochemical properties into the general formof Choursquos PseAAC via discrete wavelet transformrdquo MolecularBioSystems vol 8 pp 3178ndash3184 2012
[77] L Nanni and A Lumini ldquoGenetic programming for creatingChoursquos pseudo amino acid based features for submitochondrialocalizationrdquo Amino Acids vol 34 no 4 pp 653ndash660 2008
[78] M Esmaeili H Mohabatkar and S Mohsenzadeh ldquoUsing theconcept of Choursquos pseudo amino acid composition for risk typeprediction of human papillomavirusesrdquo Journal of TheoreticalBiology vol 263 no 2 pp 203ndash209 2010
[79] H Mohabatkar ldquoPrediction of cyclin proteins using Choursquospseudo amino acid compositionrdquo Protein and Peptide Lettersvol 17 no 10 pp 1207ndash1214 2010
[80] H Mohabatkar M M Beigi and A Esmaeili ldquoPredictionof GABA(A) receptor proteins using the concept of Choursquospseudo-amino acid composition and support vector machinerdquoJournal of Theoretical Biology vol 281 no 1 pp 18ndash23 2011
[81] Y Xu J Ding L Y Wu and K C Chou ldquoiSNO-PseAAC pre-dict cysteine S-nitrosylation sites in proteins by incorporatingposition specific amino acid propensity into pseudo amino acidcompositionrdquo PLoS ONE vol 8 Article ID e55844 2013
[82] W Chen H Lin PM Feng C Ding Y C Zuo andK C ChouldquoiNuc-PhysChem a sequence-based predictor for identifyingnucleosomes via physicochemical propertiesrdquo PLoS ONE vol7 Article ID e47843 2012
[83] B Li T Huang L Liu Y Cai and K C Chou ldquoIdentificationof colorectal cancer related genes with mrmr and shortest pathin protein-protein interaction networkrdquo PLoS ONE vol 7 no 4Article ID e33393 2012
[84] Y Jiang T Huang C Lei Y F Gao Y D Cai and K C ChouldquoSignal propagation in protein interaction network duringcolorectal cancer progressionrdquo BioMed Research Internationalvol 2013 Article ID 287019 9 pages 2013
[85] P Du X Wang C Xu and Y Gao ldquoPseAAC-Builder across-platform stand-alone program for generating variousspecial Choursquos pseudo-amino acid compositionsrdquo AnalyticalBiochemistry vol 425 no 2 pp 117ndash119 2012
[86] D S Cao Q S Xu and Y Z Liang ldquoPropy a tool to generatevarious modes of Choursquos PseAACrdquo Bioinformatics vol 29 pp960ndash962 2013
[87] H Shen and K C Chou ldquoPseAAC a flexible web serverfor generating various kinds of protein pseudo amino acidcompositionrdquo Analytical Biochemistry vol 373 no 2 pp 386ndash388 2008
[88] K C Chou ldquoUsing pair-coupled amino acid composition topredict protein secondary structure contentrdquo Journal of ProteinChemistry vol 18 no 4 pp 473ndash480 1999
BioMed Research International 13
[89] W Liu and K C Chou ldquoPrediction of protein secondarystructure contentrdquo Protein Engineering vol 12 no 12 pp 1041ndash1050 1999
[90] M Bhasin and G P S Raghava ldquoClassification of nuclearreceptors based on amino acid composition and dipeptidecompositionrdquo The Journal of Biological Chemistry vol 279 no22 pp 23262ndash23266 2004
[91] H Lin andH Ding ldquoPredicting ion channels and their types bythe dipeptidemode of pseudo amino acid compositionrdquo Journalof Theoretical Biology vol 269 no 1 pp 64ndash69 2011
[92] H Lin and Q Li ldquoUsing pseudo amino acid composition topredict protein structural class approached by incorporating400 dipeptide componentsrdquo Journal of Computational Chem-istry vol 28 no 9 pp 1463ndash1466 2007
[93] L Nanni and A Lumini ldquoCombing ontologies and dipeptidecomposition for predicting DNA-binding proteinsrdquo AminoAcids vol 34 no 4 pp 635ndash641 2008
[94] K-C Chou ldquoThe convergence-divergence duality in lectindomains of selectin family and its implicationsrdquo FEBS Lettersvol 363 no 1-2 pp 123ndash126 1995
[95] A A Schaffer L Aravind T L Madden et al ldquoImprovingthe accuracy of PSI-BLAST protein database searches withcomposition-based statistics and other refinementsrdquo NucleicAcids Research vol 29 no 14 pp 2994ndash3005 2001
[96] S F Altschul and E V Koonin ldquoIterated profile searches withPSI-BLAST a tool for discovery in protein databasesrdquo Trends inBiochemical Sciences vol 23 no 11 pp 444ndash447 1998
[97] J Deng ldquoGrey entropy and grey target decision makingrdquoJournal of Grey System vol 22 no 1 pp 1ndash24 2010
[98] B M Bolstad R A Irizarry M Astrand and T P Speed ldquoAcomparison of normalizationmethods for high density oligonu-cleotide array data based on variance and biasrdquo Bioinformaticsvol 19 no 2 pp 185ndash193 2003
[99] J-Y Shi S-W Zhang Q Pan Y-M Cheng and J XieldquoPrediction of protein subcellular localization by support vectormachines using multi-scale energy and pseudo amino acidcompositionrdquo Amino Acids vol 33 no 1 pp 69ndash74 2007
[100] T Denoeux ldquoA 120581-nearest neighbor classification rule based onDempster-Shafer theoryrdquo IEEE Transactions on Systems Manand Cybernetics vol 25 no 5 pp 804ndash813 1995
[101] J M Keller M R Gray and J A Givens ldquoA fuzzy k-nearestneighbours algorithmrdquo IEEE Transactions on Systems Man andCybernetics vol 15 no 4 pp 580ndash585 1985
[102] X Xiao P Wang and K Chou ldquoiNR-physchem a sequence-based predictor for identifying nuclear receptors and theirsubfamilies via physical-chemical property matrixrdquo PLoS ONEvol 7 no 2 Article ID e30869 2012
[103] I Roterman L Konieczny W Jurkowski K Prymula and MBanach ldquoTwo-intermediate model to characterize the structureof fast-folding proteinsrdquo Journal of Theoretical Biology vol 283no 1 pp 60ndash70 2011
[104] X Xiao P Wang and K C Chou ldquoGPCR-2L predicting Gprotein-coupled receptors and their types by hybridizing twodifferentmodes of pseudo amino acid compositionsrdquoMolecularBioSystems vol 7 no 3 pp 911ndash919 2011
[105] X Xiao P Wang and K C Chou ldquoQuat-2L a web-server forpredicting protein quaternary structural attributesrdquo MolecularDiversity vol 15 no 1 pp 149ndash155 2011
[106] X Zheng C Li and J Wang ldquoAn information-theoreticapproach to the prediction of protein structural classrdquo Journalof Computational Chemistry vol 31 no 6 pp 1201ndash1206 2010
[107] H Shen J Yang X Liu and K C Chou ldquoUsing supervisedfuzzy clustering to predict protein structural classesrdquo Biochem-ical and Biophysical Research Communications vol 334 no 2pp 577ndash581 2005
[108] K-C Chou and C-T Zhang ldquoReview prediction of proteinstructural classesrdquo Critical Reviews in Biochemistry and Molec-ular Biology vol 30 no 4 pp 275ndash349 1995
[109] P C Mahalanobis ldquoOn the generalized distance in statisticsrdquoProceedings of the National Institute of Sciences of India vol 2pp 49ndash55 1936
[110] R M Centor ldquoSignal detectability the use of ROC curves andtheir analysesrdquoMedical Decision Making vol 11 no 2 pp 102ndash106 1991
[111] K-C Chou ldquoUsing subsite coupling to predict signal peptidesrdquoProtein Engineering vol 14 no 2 pp 75ndash79 2001
[112] K C Chou ldquoPrediction of protein signal sequences and theircleavage sitesrdquo Proteins vol 42 pp 136ndash139 2001
[113] K C Chou ldquoPrediction of signal peptides using scaledwindowrdquoPeptides vol 22 no 12 pp 1973ndash1979 2001
[114] K C Chou ldquoSome remarks on predicting multi-label attributesinmolecular biosystemsrdquoMolecular Biosystems vol 9 pp 1092ndash1100 2013
[115] K C Chou and H B Shen ldquoCell-PLoc 2 0 an improvedpackage of web-servers for predicting subcellular localization ofproteins in various organismsrdquoNatural Science vol 2 pp 1090ndash1103 2010
[116] M Gribskov and N L Robinson ldquoUse of receiver operatingcharacteristic (ROC) analysis to evaluate sequence matchingrdquoComputers and Chemistry vol 20 no 1 pp 25ndash33 1996
[117] Y Yamanishi M Kotera M Kanehisa and S Goto ldquoDrug-target interaction prediction from chemical genomic and phar-macological data in an integrated frameworkrdquo Bioinformaticsvol 26 no 12 pp i246ndashi254 2010
[118] Z He J Zhang X Shi et al ldquoPredicting drug-target interactionnetworks based on functional groups and biological featuresrdquoPLoS ONE vol 5 no 3 Article ID e9603 2010
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

BioMed Research International 11
[24] M J Berardi W M Shih S C Harrison and J J Chou ldquoMito-chondrial uncoupling protein 2 structure determined by NMRmolecular fragment searchingrdquo Nature vol 476 no 7358 pp109ndash114 2011
[25] J R Schnell and J J Chou ldquoStructure andmechanism of theM2proton channel of influenza A virusrdquo Nature vol 451 no 7178pp 591ndash595 2008
[26] B OuYang S Xie M J Berardi et al ldquoUnusual architecture ofthe p7 channel from hepatitis C virusrdquoNature vol 498 pp 521ndash525 2013
[27] K Oxenoid and J J Chou ldquoThe structure of phospholambanpentamer reveals a channel-like architecture in membranesrdquoProceedings of the National Academy of Sciences of the UnitedStates of America vol 102 no 31 pp 10870ndash10875 2005
[28] M E Call KWWucherpfennig and J J Chou ldquoThe structuralbasis for intramembrane assembly of an activating immunore-ceptor complexrdquo Nature Immunology vol 11 no 11 pp 1023ndash1029 2010
[29] R M Pielak J R Schnell and J J Chou ldquoMechanism of druginhibition and drug resistance of influenza A M2 channelrdquoProceedings of the National Academy of Sciences of the UnitedStates of America vol 106 no 18 pp 7379ndash7384 2009
[30] J Wang R M Pielak M A McClintock and J J ChouldquoSolution structure and functional analysis of the influenza Bproton channelrdquo Nature Structural and Molecular Biology vol16 no 12 pp 1267ndash1271 2009
[31] K C Chou ldquoCoupling interaction between thromboxane A2receptor and alpha-13 subunit of guanine nucleotide-bindingproteinrdquo Journal of Proteome Research vol 4 no 5 pp 1681ndash1686 2005
[32] K C Chou ldquoInsights from modeling three-dimensional struc-tures of the human potassium and sodium channelsrdquo Journal ofProteome Research vol 3 no 4 pp 856ndash861 2004
[33] K C Chou ldquoSome remarks on protein attribute prediction andpseudo amino acid compositionrdquo Journal ofTheoretical Biologyvol 273 no 1 pp 236ndash247 2011
[34] W Z Lin J A Fang X Xiao and K C Chou ldquoiLoc-animal amulti-label learning classifier for predicting subcellular local-ization of animal proteinsrdquo Molecular BioSystems vol 9 pp634ndash644 2013
[35] X Xiao PWangW Z Lin J H Jia and K C Chou ldquoiAMP-2La two-level multi-label classifier for identifying antimicrobialpeptides and their functional typesrdquo Analytical Biochemistryvol 436 pp 168ndash177 2013
[36] W Chen P M Feng H Lin and K C Chou ldquoiRSpot-PseDNCidentify recombination spots with pseudo dinucleotide compo-sitionrdquo Nucleic Acids Research vol 41 article e69 2013
[37] K C Chou Z C Wu and X Xiao ldquoILoc-Hum using theaccumulation-label scale to predict subcellular locations ofhuman proteins with both single and multiple sitesrdquoMolecularBioSystems vol 8 no 2 pp 629ndash641 2012
[38] M Kotera M Hirakawa T Tokimatsu S Goto and MKanehisa ldquoThe KEGG databases and tools facilitating omicsanalysis latest developments involving human diseases andpharmaceuticalsrdquo Methods in Molecular Biology vol 802 pp19ndash39 2012
[39] Y YamanishiM Araki A GutteridgeWHonda andM Kane-hisa ldquoPrediction of drug-target interaction networks from theintegration of chemical and genomic spacesrdquo Bioinformaticsvol 24 no 13 pp i232ndashi240 2008
[40] P Finn S Muggleton D Page and A Srinivasan ldquoPharma-cophore discovery using the Inductive Logic Programmingsystem PROGOLrdquo Machine Learning vol 30 no 2-3 pp 241ndash270 1998
[41] I Vogt D Stumpfe H E Ahmed and J Bajorath ldquoMethodsfor computer-aided chemical biology Part 2 evaluation of com-pound selectivity using 2D molecular fingerprintsrdquo ChemicalBiology and Drug Design vol 70 pp 195ndash205 2007
[42] H Eckert and J Bajorath ldquoMolecular similarity analysis in vir-tual screening foundations limitations and novel approachesrdquoDrug Discovery Today vol 12 no 5-6 pp 225ndash233 2007
[43] S Laurent L V Elst and R N Muller ldquoComparative studyof the physicochemical properties of six clinical low molecularweight gadolinium contrast agentsrdquo Contrast Media amp Molecu-lar Imaging vol 1 no 3 pp 128ndash137 2006
[44] E Gregori-Puigjane R Garriga-Sust and J Mestres ldquoIndexingmolecules with chemical graph identifiersrdquo Journal of Compu-tational Chemistry vol 32 no 12 pp 2638ndash2646 2011
[45] B Ren ldquoApplication of novel atom-type AI topological indicesto QSPR studies of alkanesrdquo Computers and Chemistry vol 26no 4 pp 357ndash369 2002
[46] N M OrsquoBoyle M Banck C A James C Morley T Vander-meersch and G R Hutchison ldquoOpen Babel an open chemicaltoolboxrdquo Journal of Cheminformatics vol 3 p 33 2011
[47] V J Gillet P Willett and J Bradshaw ldquoSimilarity searchingusing reduced graphsrdquo Journal of Chemical Information andComputer Sciences vol 43 no 2 pp 338ndash345 2003
[48] D Butina ldquoUnsupervised data base clustering based on day-lightrsquos fingerprint and Tanimoto similarity a fast and automatedway to cluster small and large data setsrdquo Journal of ChemicalInformation and Computer Sciences vol 39 no 4 pp 747ndash7501999
[49] C E Shannon ldquoA mathematical theory of communicationrdquoACM SIGMOBILE Mobile Computing and CommunicationsReview vol 5 pp 3ndash55 2001
[50] V D Gusev L A Nemytikova and N A Chuzhanova ldquoOn thecomplexity measures of genetic sequencesrdquo Bioinformatics vol15 no 12 pp 994ndash999 1999
[51] K C Chou and H B Shen ldquoReview recent progress in proteinsubcellular location predictionrdquo Analytical Biochemistry vol370 no 1 pp 1ndash16 2007
[52] S F Altschul ldquoEvaluating the statistical significance of multipledistinct local alignmentsrdquo in Theoretical and ComputationalMethods in Genome Research S Suhai Ed pp 1ndash14 PlenumNew York NY USA 1997
[53] J C Wootton and S Federhen ldquoStatistics of local complexity inamino acid sequences and sequence databasesrdquo Computers andChemistry vol 17 no 2 pp 149ndash163 1993
[54] H Nakashima K Nishikawa and T Ooi ldquoThe folding type ofa protein is relevant to the amino acid compositionrdquo Journal ofBiochemistry vol 99 no 1 pp 153ndash162 1986
[55] K C Chou and C T Zhang ldquoPredicting protein folding typesby distance functions that make allowances for amino acidinteractionsrdquo The Journal of Biological Chemistry vol 269 no35 pp 22014ndash22020 1994
[56] K-C Chou ldquoA novel approach to predicting protein structuralclasses in a (20-1)-D amino acid composition spacerdquo Proteinsvol 21 no 4 pp 319ndash344 1995
[57] H Nakashima and K Nishikawa ldquoDiscrimination of intracel-lular and extracellular proteins using amino acid compositionand residue-pair frequenciesrdquo Journal of Molecular Biology vol238 no 1 pp 54ndash61 1994
12 BioMed Research International
[58] G P Zhou ldquoAn intriguing controversy over protein structuralclass predictionrdquo Journal of Protein Chemistry vol 17 no 8 pp729ndash738 1998
[59] I Bahar A R Atilgan R L Jernigan and B Erman ldquoUnder-standing the recognition of protein structural classes by aminoacid compositionrdquo Proteins vol 29 pp 172ndash185 1997
[60] J Cedano P Aloy J A Perez-Pons and E Querol ldquoRelationbetween amino acid composition and cellular location ofproteinsrdquo Journal of Molecular Biology vol 266 no 3 pp 594ndash600 1997
[61] G P Zhou and K Doctor ldquoSubcellular location prediction ofapoptosis proteinsrdquo Proteins vol 50 no 1 pp 44ndash48 2003
[62] KChou ldquoPrediction of protein cellular attributes using pseudo-amino acid compositionrdquo Proteins vol 43 pp 246ndash255 2001(Erratum in Proteins vol 44 p 60 2001)
[63] K C Chou ldquoUsing amphiphilic pseudo amino acid composi-tion to predict enzyme subfamily classesrdquo Bioinformatics vol21 no 1 pp 10ndash19 2005
[64] D Zou Z He J He and Y Xia ldquoSupersecondary structureprediction using Choursquos pseudo amino acid compositionrdquoJournal of Computational Chemistry vol 32 no 2 pp 271ndash2782011
[65] M M Beigi M Behjati and H Mohabatkar ldquoPrediction ofmetalloproteinase family based on the concept ofChoursquos pseudoamino acid composition using a machine learning approachrdquoJournal of Structural and Functional Genomics vol 12 no 4 pp191ndash197 2011
[66] Y K Chen and K B Li ldquoPredicting membrane protein typesby incorporating protein topology domains signal peptidesand physicochemical properties into the general form of Choursquospseudo amino acid compositionrdquo Journal ofTheoretical Biologyvol 318 pp 1ndash12 2013
[67] C Huang and J Q Yuan ldquoA multilabel model based on Choursquospseudo-amino acid composition for identifying membraneproteins with both single and multiple functional typesrdquo TheJournal of Membrane Biology vol 246 pp 327ndash334 2013
[68] S S Sahu and G Panda ldquoA novel feature representationmethod based on Choursquos pseudo amino acid composition forprotein structural class predictionrdquo Computational Biology andChemistry vol 34 no 5-6 pp 320ndash327 2010
[69] M Hayat and A Khan ldquoDiscriminating outer membraneproteins with fuzzy K-nearest neighbor algorithms based on thegeneral form of Choursquos PseAACrdquo Protein and Peptide Lettersvol 19 no 4 pp 411ndash421 2012
[70] MKhosravian F K FaramarziMM BeigiM Behbahani andH Mohabatkar ldquoPredicting antibacterial peptides by the con-cept of Choursquos pseudo-amino acid composition and machinelearning methodsrdquo Protein amp Peptide Letters vol 20 pp 180ndash186 2013
[71] HMohabatkarMM Beigi K Abdolahi and SMohsenzadehldquoPrediction of allergenic proteins by means of the concept ofChoursquos pseudo amino acid composition and amachine learningapproachrdquoMedicinal Chemistry vol 9 pp 133ndash137 2013
[72] L Nanni A Lumini D Gupta and A Garg ldquoIdentifyingbacterial virulent proteins by fusing a set of classifiers basedon variants of Choursquos pseudo amino acid composition and onevolutionary informationrdquo IEEEACM Transactions on Compu-tational Biology and Bioinformatics vol 9 no 2 pp 467ndash4752012
[73] T H Chang L C Wu T Y Lee S P Chen H D Huang andJ T Horng ldquoEuLoc a web-server for accurately predict protein
subcellular localization in eukaryotes by incorporating variousfeatures of sequence segments into the general form of ChoursquosPseAACrdquo Journal of Computer-Aided Molecular Design vol 27pp 91ndash103 2013
[74] S Zhang Y Zhang H Yang C Zhao and Q Pan ldquoUsing theconcept of Choursquos pseudo amino acid composition to predictprotein subcellular localization an approach by incorporatingevolutionary information and von Neumann entropiesrdquo AminoAcids vol 34 no 4 pp 565ndash572 2008
[75] R Zia Ur and A Khan ldquoIdentifying GPCRs and their typeswith Choursquos pseudo amino acid composition an approach frommulti-scale energy representation and position specific scoringmatrixrdquo Protein amp Peptide Letters vol 19 pp 890ndash903 2012
[76] X Y Sun S P Shi J D Qiu S B Suo S Y Huang and R PLiang ldquoIdentifying protein quaternary structural attributes byincorporating physicochemical properties into the general formof Choursquos PseAAC via discrete wavelet transformrdquo MolecularBioSystems vol 8 pp 3178ndash3184 2012
[77] L Nanni and A Lumini ldquoGenetic programming for creatingChoursquos pseudo amino acid based features for submitochondrialocalizationrdquo Amino Acids vol 34 no 4 pp 653ndash660 2008
[78] M Esmaeili H Mohabatkar and S Mohsenzadeh ldquoUsing theconcept of Choursquos pseudo amino acid composition for risk typeprediction of human papillomavirusesrdquo Journal of TheoreticalBiology vol 263 no 2 pp 203ndash209 2010
[79] H Mohabatkar ldquoPrediction of cyclin proteins using Choursquospseudo amino acid compositionrdquo Protein and Peptide Lettersvol 17 no 10 pp 1207ndash1214 2010
[80] H Mohabatkar M M Beigi and A Esmaeili ldquoPredictionof GABA(A) receptor proteins using the concept of Choursquospseudo-amino acid composition and support vector machinerdquoJournal of Theoretical Biology vol 281 no 1 pp 18ndash23 2011
[81] Y Xu J Ding L Y Wu and K C Chou ldquoiSNO-PseAAC pre-dict cysteine S-nitrosylation sites in proteins by incorporatingposition specific amino acid propensity into pseudo amino acidcompositionrdquo PLoS ONE vol 8 Article ID e55844 2013
[82] W Chen H Lin PM Feng C Ding Y C Zuo andK C ChouldquoiNuc-PhysChem a sequence-based predictor for identifyingnucleosomes via physicochemical propertiesrdquo PLoS ONE vol7 Article ID e47843 2012
[83] B Li T Huang L Liu Y Cai and K C Chou ldquoIdentificationof colorectal cancer related genes with mrmr and shortest pathin protein-protein interaction networkrdquo PLoS ONE vol 7 no 4Article ID e33393 2012
[84] Y Jiang T Huang C Lei Y F Gao Y D Cai and K C ChouldquoSignal propagation in protein interaction network duringcolorectal cancer progressionrdquo BioMed Research Internationalvol 2013 Article ID 287019 9 pages 2013
[85] P Du X Wang C Xu and Y Gao ldquoPseAAC-Builder across-platform stand-alone program for generating variousspecial Choursquos pseudo-amino acid compositionsrdquo AnalyticalBiochemistry vol 425 no 2 pp 117ndash119 2012
[86] D S Cao Q S Xu and Y Z Liang ldquoPropy a tool to generatevarious modes of Choursquos PseAACrdquo Bioinformatics vol 29 pp960ndash962 2013
[87] H Shen and K C Chou ldquoPseAAC a flexible web serverfor generating various kinds of protein pseudo amino acidcompositionrdquo Analytical Biochemistry vol 373 no 2 pp 386ndash388 2008
[88] K C Chou ldquoUsing pair-coupled amino acid composition topredict protein secondary structure contentrdquo Journal of ProteinChemistry vol 18 no 4 pp 473ndash480 1999
BioMed Research International 13
[89] W Liu and K C Chou ldquoPrediction of protein secondarystructure contentrdquo Protein Engineering vol 12 no 12 pp 1041ndash1050 1999
[90] M Bhasin and G P S Raghava ldquoClassification of nuclearreceptors based on amino acid composition and dipeptidecompositionrdquo The Journal of Biological Chemistry vol 279 no22 pp 23262ndash23266 2004
[91] H Lin andH Ding ldquoPredicting ion channels and their types bythe dipeptidemode of pseudo amino acid compositionrdquo Journalof Theoretical Biology vol 269 no 1 pp 64ndash69 2011
[92] H Lin and Q Li ldquoUsing pseudo amino acid composition topredict protein structural class approached by incorporating400 dipeptide componentsrdquo Journal of Computational Chem-istry vol 28 no 9 pp 1463ndash1466 2007
[93] L Nanni and A Lumini ldquoCombing ontologies and dipeptidecomposition for predicting DNA-binding proteinsrdquo AminoAcids vol 34 no 4 pp 635ndash641 2008
[94] K-C Chou ldquoThe convergence-divergence duality in lectindomains of selectin family and its implicationsrdquo FEBS Lettersvol 363 no 1-2 pp 123ndash126 1995
[95] A A Schaffer L Aravind T L Madden et al ldquoImprovingthe accuracy of PSI-BLAST protein database searches withcomposition-based statistics and other refinementsrdquo NucleicAcids Research vol 29 no 14 pp 2994ndash3005 2001
[96] S F Altschul and E V Koonin ldquoIterated profile searches withPSI-BLAST a tool for discovery in protein databasesrdquo Trends inBiochemical Sciences vol 23 no 11 pp 444ndash447 1998
[97] J Deng ldquoGrey entropy and grey target decision makingrdquoJournal of Grey System vol 22 no 1 pp 1ndash24 2010
[98] B M Bolstad R A Irizarry M Astrand and T P Speed ldquoAcomparison of normalizationmethods for high density oligonu-cleotide array data based on variance and biasrdquo Bioinformaticsvol 19 no 2 pp 185ndash193 2003
[99] J-Y Shi S-W Zhang Q Pan Y-M Cheng and J XieldquoPrediction of protein subcellular localization by support vectormachines using multi-scale energy and pseudo amino acidcompositionrdquo Amino Acids vol 33 no 1 pp 69ndash74 2007
[100] T Denoeux ldquoA 120581-nearest neighbor classification rule based onDempster-Shafer theoryrdquo IEEE Transactions on Systems Manand Cybernetics vol 25 no 5 pp 804ndash813 1995
[101] J M Keller M R Gray and J A Givens ldquoA fuzzy k-nearestneighbours algorithmrdquo IEEE Transactions on Systems Man andCybernetics vol 15 no 4 pp 580ndash585 1985
[102] X Xiao P Wang and K Chou ldquoiNR-physchem a sequence-based predictor for identifying nuclear receptors and theirsubfamilies via physical-chemical property matrixrdquo PLoS ONEvol 7 no 2 Article ID e30869 2012
[103] I Roterman L Konieczny W Jurkowski K Prymula and MBanach ldquoTwo-intermediate model to characterize the structureof fast-folding proteinsrdquo Journal of Theoretical Biology vol 283no 1 pp 60ndash70 2011
[104] X Xiao P Wang and K C Chou ldquoGPCR-2L predicting Gprotein-coupled receptors and their types by hybridizing twodifferentmodes of pseudo amino acid compositionsrdquoMolecularBioSystems vol 7 no 3 pp 911ndash919 2011
[105] X Xiao P Wang and K C Chou ldquoQuat-2L a web-server forpredicting protein quaternary structural attributesrdquo MolecularDiversity vol 15 no 1 pp 149ndash155 2011
[106] X Zheng C Li and J Wang ldquoAn information-theoreticapproach to the prediction of protein structural classrdquo Journalof Computational Chemistry vol 31 no 6 pp 1201ndash1206 2010
[107] H Shen J Yang X Liu and K C Chou ldquoUsing supervisedfuzzy clustering to predict protein structural classesrdquo Biochem-ical and Biophysical Research Communications vol 334 no 2pp 577ndash581 2005
[108] K-C Chou and C-T Zhang ldquoReview prediction of proteinstructural classesrdquo Critical Reviews in Biochemistry and Molec-ular Biology vol 30 no 4 pp 275ndash349 1995
[109] P C Mahalanobis ldquoOn the generalized distance in statisticsrdquoProceedings of the National Institute of Sciences of India vol 2pp 49ndash55 1936
[110] R M Centor ldquoSignal detectability the use of ROC curves andtheir analysesrdquoMedical Decision Making vol 11 no 2 pp 102ndash106 1991
[111] K-C Chou ldquoUsing subsite coupling to predict signal peptidesrdquoProtein Engineering vol 14 no 2 pp 75ndash79 2001
[112] K C Chou ldquoPrediction of protein signal sequences and theircleavage sitesrdquo Proteins vol 42 pp 136ndash139 2001
[113] K C Chou ldquoPrediction of signal peptides using scaledwindowrdquoPeptides vol 22 no 12 pp 1973ndash1979 2001
[114] K C Chou ldquoSome remarks on predicting multi-label attributesinmolecular biosystemsrdquoMolecular Biosystems vol 9 pp 1092ndash1100 2013
[115] K C Chou and H B Shen ldquoCell-PLoc 2 0 an improvedpackage of web-servers for predicting subcellular localization ofproteins in various organismsrdquoNatural Science vol 2 pp 1090ndash1103 2010
[116] M Gribskov and N L Robinson ldquoUse of receiver operatingcharacteristic (ROC) analysis to evaluate sequence matchingrdquoComputers and Chemistry vol 20 no 1 pp 25ndash33 1996
[117] Y Yamanishi M Kotera M Kanehisa and S Goto ldquoDrug-target interaction prediction from chemical genomic and phar-macological data in an integrated frameworkrdquo Bioinformaticsvol 26 no 12 pp i246ndashi254 2010
[118] Z He J Zhang X Shi et al ldquoPredicting drug-target interactionnetworks based on functional groups and biological featuresrdquoPLoS ONE vol 5 no 3 Article ID e9603 2010
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

12 BioMed Research International
[58] G P Zhou ldquoAn intriguing controversy over protein structuralclass predictionrdquo Journal of Protein Chemistry vol 17 no 8 pp729ndash738 1998
[59] I Bahar A R Atilgan R L Jernigan and B Erman ldquoUnder-standing the recognition of protein structural classes by aminoacid compositionrdquo Proteins vol 29 pp 172ndash185 1997
[60] J Cedano P Aloy J A Perez-Pons and E Querol ldquoRelationbetween amino acid composition and cellular location ofproteinsrdquo Journal of Molecular Biology vol 266 no 3 pp 594ndash600 1997
[61] G P Zhou and K Doctor ldquoSubcellular location prediction ofapoptosis proteinsrdquo Proteins vol 50 no 1 pp 44ndash48 2003
[62] KChou ldquoPrediction of protein cellular attributes using pseudo-amino acid compositionrdquo Proteins vol 43 pp 246ndash255 2001(Erratum in Proteins vol 44 p 60 2001)
[63] K C Chou ldquoUsing amphiphilic pseudo amino acid composi-tion to predict enzyme subfamily classesrdquo Bioinformatics vol21 no 1 pp 10ndash19 2005
[64] D Zou Z He J He and Y Xia ldquoSupersecondary structureprediction using Choursquos pseudo amino acid compositionrdquoJournal of Computational Chemistry vol 32 no 2 pp 271ndash2782011
[65] M M Beigi M Behjati and H Mohabatkar ldquoPrediction ofmetalloproteinase family based on the concept ofChoursquos pseudoamino acid composition using a machine learning approachrdquoJournal of Structural and Functional Genomics vol 12 no 4 pp191ndash197 2011
[66] Y K Chen and K B Li ldquoPredicting membrane protein typesby incorporating protein topology domains signal peptidesand physicochemical properties into the general form of Choursquospseudo amino acid compositionrdquo Journal ofTheoretical Biologyvol 318 pp 1ndash12 2013
[67] C Huang and J Q Yuan ldquoA multilabel model based on Choursquospseudo-amino acid composition for identifying membraneproteins with both single and multiple functional typesrdquo TheJournal of Membrane Biology vol 246 pp 327ndash334 2013
[68] S S Sahu and G Panda ldquoA novel feature representationmethod based on Choursquos pseudo amino acid composition forprotein structural class predictionrdquo Computational Biology andChemistry vol 34 no 5-6 pp 320ndash327 2010
[69] M Hayat and A Khan ldquoDiscriminating outer membraneproteins with fuzzy K-nearest neighbor algorithms based on thegeneral form of Choursquos PseAACrdquo Protein and Peptide Lettersvol 19 no 4 pp 411ndash421 2012
[70] MKhosravian F K FaramarziMM BeigiM Behbahani andH Mohabatkar ldquoPredicting antibacterial peptides by the con-cept of Choursquos pseudo-amino acid composition and machinelearning methodsrdquo Protein amp Peptide Letters vol 20 pp 180ndash186 2013
[71] HMohabatkarMM Beigi K Abdolahi and SMohsenzadehldquoPrediction of allergenic proteins by means of the concept ofChoursquos pseudo amino acid composition and amachine learningapproachrdquoMedicinal Chemistry vol 9 pp 133ndash137 2013
[72] L Nanni A Lumini D Gupta and A Garg ldquoIdentifyingbacterial virulent proteins by fusing a set of classifiers basedon variants of Choursquos pseudo amino acid composition and onevolutionary informationrdquo IEEEACM Transactions on Compu-tational Biology and Bioinformatics vol 9 no 2 pp 467ndash4752012
[73] T H Chang L C Wu T Y Lee S P Chen H D Huang andJ T Horng ldquoEuLoc a web-server for accurately predict protein
subcellular localization in eukaryotes by incorporating variousfeatures of sequence segments into the general form of ChoursquosPseAACrdquo Journal of Computer-Aided Molecular Design vol 27pp 91ndash103 2013
[74] S Zhang Y Zhang H Yang C Zhao and Q Pan ldquoUsing theconcept of Choursquos pseudo amino acid composition to predictprotein subcellular localization an approach by incorporatingevolutionary information and von Neumann entropiesrdquo AminoAcids vol 34 no 4 pp 565ndash572 2008
[75] R Zia Ur and A Khan ldquoIdentifying GPCRs and their typeswith Choursquos pseudo amino acid composition an approach frommulti-scale energy representation and position specific scoringmatrixrdquo Protein amp Peptide Letters vol 19 pp 890ndash903 2012
[76] X Y Sun S P Shi J D Qiu S B Suo S Y Huang and R PLiang ldquoIdentifying protein quaternary structural attributes byincorporating physicochemical properties into the general formof Choursquos PseAAC via discrete wavelet transformrdquo MolecularBioSystems vol 8 pp 3178ndash3184 2012
[77] L Nanni and A Lumini ldquoGenetic programming for creatingChoursquos pseudo amino acid based features for submitochondrialocalizationrdquo Amino Acids vol 34 no 4 pp 653ndash660 2008
[78] M Esmaeili H Mohabatkar and S Mohsenzadeh ldquoUsing theconcept of Choursquos pseudo amino acid composition for risk typeprediction of human papillomavirusesrdquo Journal of TheoreticalBiology vol 263 no 2 pp 203ndash209 2010
[79] H Mohabatkar ldquoPrediction of cyclin proteins using Choursquospseudo amino acid compositionrdquo Protein and Peptide Lettersvol 17 no 10 pp 1207ndash1214 2010
[80] H Mohabatkar M M Beigi and A Esmaeili ldquoPredictionof GABA(A) receptor proteins using the concept of Choursquospseudo-amino acid composition and support vector machinerdquoJournal of Theoretical Biology vol 281 no 1 pp 18ndash23 2011
[81] Y Xu J Ding L Y Wu and K C Chou ldquoiSNO-PseAAC pre-dict cysteine S-nitrosylation sites in proteins by incorporatingposition specific amino acid propensity into pseudo amino acidcompositionrdquo PLoS ONE vol 8 Article ID e55844 2013
[82] W Chen H Lin PM Feng C Ding Y C Zuo andK C ChouldquoiNuc-PhysChem a sequence-based predictor for identifyingnucleosomes via physicochemical propertiesrdquo PLoS ONE vol7 Article ID e47843 2012
[83] B Li T Huang L Liu Y Cai and K C Chou ldquoIdentificationof colorectal cancer related genes with mrmr and shortest pathin protein-protein interaction networkrdquo PLoS ONE vol 7 no 4Article ID e33393 2012
[84] Y Jiang T Huang C Lei Y F Gao Y D Cai and K C ChouldquoSignal propagation in protein interaction network duringcolorectal cancer progressionrdquo BioMed Research Internationalvol 2013 Article ID 287019 9 pages 2013
[85] P Du X Wang C Xu and Y Gao ldquoPseAAC-Builder across-platform stand-alone program for generating variousspecial Choursquos pseudo-amino acid compositionsrdquo AnalyticalBiochemistry vol 425 no 2 pp 117ndash119 2012
[86] D S Cao Q S Xu and Y Z Liang ldquoPropy a tool to generatevarious modes of Choursquos PseAACrdquo Bioinformatics vol 29 pp960ndash962 2013
[87] H Shen and K C Chou ldquoPseAAC a flexible web serverfor generating various kinds of protein pseudo amino acidcompositionrdquo Analytical Biochemistry vol 373 no 2 pp 386ndash388 2008
[88] K C Chou ldquoUsing pair-coupled amino acid composition topredict protein secondary structure contentrdquo Journal of ProteinChemistry vol 18 no 4 pp 473ndash480 1999
BioMed Research International 13
[89] W Liu and K C Chou ldquoPrediction of protein secondarystructure contentrdquo Protein Engineering vol 12 no 12 pp 1041ndash1050 1999
[90] M Bhasin and G P S Raghava ldquoClassification of nuclearreceptors based on amino acid composition and dipeptidecompositionrdquo The Journal of Biological Chemistry vol 279 no22 pp 23262ndash23266 2004
[91] H Lin andH Ding ldquoPredicting ion channels and their types bythe dipeptidemode of pseudo amino acid compositionrdquo Journalof Theoretical Biology vol 269 no 1 pp 64ndash69 2011
[92] H Lin and Q Li ldquoUsing pseudo amino acid composition topredict protein structural class approached by incorporating400 dipeptide componentsrdquo Journal of Computational Chem-istry vol 28 no 9 pp 1463ndash1466 2007
[93] L Nanni and A Lumini ldquoCombing ontologies and dipeptidecomposition for predicting DNA-binding proteinsrdquo AminoAcids vol 34 no 4 pp 635ndash641 2008
[94] K-C Chou ldquoThe convergence-divergence duality in lectindomains of selectin family and its implicationsrdquo FEBS Lettersvol 363 no 1-2 pp 123ndash126 1995
[95] A A Schaffer L Aravind T L Madden et al ldquoImprovingthe accuracy of PSI-BLAST protein database searches withcomposition-based statistics and other refinementsrdquo NucleicAcids Research vol 29 no 14 pp 2994ndash3005 2001
[96] S F Altschul and E V Koonin ldquoIterated profile searches withPSI-BLAST a tool for discovery in protein databasesrdquo Trends inBiochemical Sciences vol 23 no 11 pp 444ndash447 1998
[97] J Deng ldquoGrey entropy and grey target decision makingrdquoJournal of Grey System vol 22 no 1 pp 1ndash24 2010
[98] B M Bolstad R A Irizarry M Astrand and T P Speed ldquoAcomparison of normalizationmethods for high density oligonu-cleotide array data based on variance and biasrdquo Bioinformaticsvol 19 no 2 pp 185ndash193 2003
[99] J-Y Shi S-W Zhang Q Pan Y-M Cheng and J XieldquoPrediction of protein subcellular localization by support vectormachines using multi-scale energy and pseudo amino acidcompositionrdquo Amino Acids vol 33 no 1 pp 69ndash74 2007
[100] T Denoeux ldquoA 120581-nearest neighbor classification rule based onDempster-Shafer theoryrdquo IEEE Transactions on Systems Manand Cybernetics vol 25 no 5 pp 804ndash813 1995
[101] J M Keller M R Gray and J A Givens ldquoA fuzzy k-nearestneighbours algorithmrdquo IEEE Transactions on Systems Man andCybernetics vol 15 no 4 pp 580ndash585 1985
[102] X Xiao P Wang and K Chou ldquoiNR-physchem a sequence-based predictor for identifying nuclear receptors and theirsubfamilies via physical-chemical property matrixrdquo PLoS ONEvol 7 no 2 Article ID e30869 2012
[103] I Roterman L Konieczny W Jurkowski K Prymula and MBanach ldquoTwo-intermediate model to characterize the structureof fast-folding proteinsrdquo Journal of Theoretical Biology vol 283no 1 pp 60ndash70 2011
[104] X Xiao P Wang and K C Chou ldquoGPCR-2L predicting Gprotein-coupled receptors and their types by hybridizing twodifferentmodes of pseudo amino acid compositionsrdquoMolecularBioSystems vol 7 no 3 pp 911ndash919 2011
[105] X Xiao P Wang and K C Chou ldquoQuat-2L a web-server forpredicting protein quaternary structural attributesrdquo MolecularDiversity vol 15 no 1 pp 149ndash155 2011
[106] X Zheng C Li and J Wang ldquoAn information-theoreticapproach to the prediction of protein structural classrdquo Journalof Computational Chemistry vol 31 no 6 pp 1201ndash1206 2010
[107] H Shen J Yang X Liu and K C Chou ldquoUsing supervisedfuzzy clustering to predict protein structural classesrdquo Biochem-ical and Biophysical Research Communications vol 334 no 2pp 577ndash581 2005
[108] K-C Chou and C-T Zhang ldquoReview prediction of proteinstructural classesrdquo Critical Reviews in Biochemistry and Molec-ular Biology vol 30 no 4 pp 275ndash349 1995
[109] P C Mahalanobis ldquoOn the generalized distance in statisticsrdquoProceedings of the National Institute of Sciences of India vol 2pp 49ndash55 1936
[110] R M Centor ldquoSignal detectability the use of ROC curves andtheir analysesrdquoMedical Decision Making vol 11 no 2 pp 102ndash106 1991
[111] K-C Chou ldquoUsing subsite coupling to predict signal peptidesrdquoProtein Engineering vol 14 no 2 pp 75ndash79 2001
[112] K C Chou ldquoPrediction of protein signal sequences and theircleavage sitesrdquo Proteins vol 42 pp 136ndash139 2001
[113] K C Chou ldquoPrediction of signal peptides using scaledwindowrdquoPeptides vol 22 no 12 pp 1973ndash1979 2001
[114] K C Chou ldquoSome remarks on predicting multi-label attributesinmolecular biosystemsrdquoMolecular Biosystems vol 9 pp 1092ndash1100 2013
[115] K C Chou and H B Shen ldquoCell-PLoc 2 0 an improvedpackage of web-servers for predicting subcellular localization ofproteins in various organismsrdquoNatural Science vol 2 pp 1090ndash1103 2010
[116] M Gribskov and N L Robinson ldquoUse of receiver operatingcharacteristic (ROC) analysis to evaluate sequence matchingrdquoComputers and Chemistry vol 20 no 1 pp 25ndash33 1996
[117] Y Yamanishi M Kotera M Kanehisa and S Goto ldquoDrug-target interaction prediction from chemical genomic and phar-macological data in an integrated frameworkrdquo Bioinformaticsvol 26 no 12 pp i246ndashi254 2010
[118] Z He J Zhang X Shi et al ldquoPredicting drug-target interactionnetworks based on functional groups and biological featuresrdquoPLoS ONE vol 5 no 3 Article ID e9603 2010
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

BioMed Research International 13
[89] W Liu and K C Chou ldquoPrediction of protein secondarystructure contentrdquo Protein Engineering vol 12 no 12 pp 1041ndash1050 1999
[90] M Bhasin and G P S Raghava ldquoClassification of nuclearreceptors based on amino acid composition and dipeptidecompositionrdquo The Journal of Biological Chemistry vol 279 no22 pp 23262ndash23266 2004
[91] H Lin andH Ding ldquoPredicting ion channels and their types bythe dipeptidemode of pseudo amino acid compositionrdquo Journalof Theoretical Biology vol 269 no 1 pp 64ndash69 2011
[92] H Lin and Q Li ldquoUsing pseudo amino acid composition topredict protein structural class approached by incorporating400 dipeptide componentsrdquo Journal of Computational Chem-istry vol 28 no 9 pp 1463ndash1466 2007
[93] L Nanni and A Lumini ldquoCombing ontologies and dipeptidecomposition for predicting DNA-binding proteinsrdquo AminoAcids vol 34 no 4 pp 635ndash641 2008
[94] K-C Chou ldquoThe convergence-divergence duality in lectindomains of selectin family and its implicationsrdquo FEBS Lettersvol 363 no 1-2 pp 123ndash126 1995
[95] A A Schaffer L Aravind T L Madden et al ldquoImprovingthe accuracy of PSI-BLAST protein database searches withcomposition-based statistics and other refinementsrdquo NucleicAcids Research vol 29 no 14 pp 2994ndash3005 2001
[96] S F Altschul and E V Koonin ldquoIterated profile searches withPSI-BLAST a tool for discovery in protein databasesrdquo Trends inBiochemical Sciences vol 23 no 11 pp 444ndash447 1998
[97] J Deng ldquoGrey entropy and grey target decision makingrdquoJournal of Grey System vol 22 no 1 pp 1ndash24 2010
[98] B M Bolstad R A Irizarry M Astrand and T P Speed ldquoAcomparison of normalizationmethods for high density oligonu-cleotide array data based on variance and biasrdquo Bioinformaticsvol 19 no 2 pp 185ndash193 2003
[99] J-Y Shi S-W Zhang Q Pan Y-M Cheng and J XieldquoPrediction of protein subcellular localization by support vectormachines using multi-scale energy and pseudo amino acidcompositionrdquo Amino Acids vol 33 no 1 pp 69ndash74 2007
[100] T Denoeux ldquoA 120581-nearest neighbor classification rule based onDempster-Shafer theoryrdquo IEEE Transactions on Systems Manand Cybernetics vol 25 no 5 pp 804ndash813 1995
[101] J M Keller M R Gray and J A Givens ldquoA fuzzy k-nearestneighbours algorithmrdquo IEEE Transactions on Systems Man andCybernetics vol 15 no 4 pp 580ndash585 1985
[102] X Xiao P Wang and K Chou ldquoiNR-physchem a sequence-based predictor for identifying nuclear receptors and theirsubfamilies via physical-chemical property matrixrdquo PLoS ONEvol 7 no 2 Article ID e30869 2012
[103] I Roterman L Konieczny W Jurkowski K Prymula and MBanach ldquoTwo-intermediate model to characterize the structureof fast-folding proteinsrdquo Journal of Theoretical Biology vol 283no 1 pp 60ndash70 2011
[104] X Xiao P Wang and K C Chou ldquoGPCR-2L predicting Gprotein-coupled receptors and their types by hybridizing twodifferentmodes of pseudo amino acid compositionsrdquoMolecularBioSystems vol 7 no 3 pp 911ndash919 2011
[105] X Xiao P Wang and K C Chou ldquoQuat-2L a web-server forpredicting protein quaternary structural attributesrdquo MolecularDiversity vol 15 no 1 pp 149ndash155 2011
[106] X Zheng C Li and J Wang ldquoAn information-theoreticapproach to the prediction of protein structural classrdquo Journalof Computational Chemistry vol 31 no 6 pp 1201ndash1206 2010
[107] H Shen J Yang X Liu and K C Chou ldquoUsing supervisedfuzzy clustering to predict protein structural classesrdquo Biochem-ical and Biophysical Research Communications vol 334 no 2pp 577ndash581 2005
[108] K-C Chou and C-T Zhang ldquoReview prediction of proteinstructural classesrdquo Critical Reviews in Biochemistry and Molec-ular Biology vol 30 no 4 pp 275ndash349 1995
[109] P C Mahalanobis ldquoOn the generalized distance in statisticsrdquoProceedings of the National Institute of Sciences of India vol 2pp 49ndash55 1936
[110] R M Centor ldquoSignal detectability the use of ROC curves andtheir analysesrdquoMedical Decision Making vol 11 no 2 pp 102ndash106 1991
[111] K-C Chou ldquoUsing subsite coupling to predict signal peptidesrdquoProtein Engineering vol 14 no 2 pp 75ndash79 2001
[112] K C Chou ldquoPrediction of protein signal sequences and theircleavage sitesrdquo Proteins vol 42 pp 136ndash139 2001
[113] K C Chou ldquoPrediction of signal peptides using scaledwindowrdquoPeptides vol 22 no 12 pp 1973ndash1979 2001
[114] K C Chou ldquoSome remarks on predicting multi-label attributesinmolecular biosystemsrdquoMolecular Biosystems vol 9 pp 1092ndash1100 2013
[115] K C Chou and H B Shen ldquoCell-PLoc 2 0 an improvedpackage of web-servers for predicting subcellular localization ofproteins in various organismsrdquoNatural Science vol 2 pp 1090ndash1103 2010
[116] M Gribskov and N L Robinson ldquoUse of receiver operatingcharacteristic (ROC) analysis to evaluate sequence matchingrdquoComputers and Chemistry vol 20 no 1 pp 25ndash33 1996
[117] Y Yamanishi M Kotera M Kanehisa and S Goto ldquoDrug-target interaction prediction from chemical genomic and phar-macological data in an integrated frameworkrdquo Bioinformaticsvol 26 no 12 pp i246ndashi254 2010
[118] Z He J Zhang X Shi et al ldquoPredicting drug-target interactionnetworks based on functional groups and biological featuresrdquoPLoS ONE vol 5 no 3 Article ID e9603 2010
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology