hydrogen bonding: ab initio accuracy from fast interatomic ...€¦ · hydrogen bonding: ab initio...
TRANSCRIPT

© 2018 Optibrium Ltd.Optibrium™, StarDrop™, Auto-Modeller™, Card View™ and Glowing Molecule™ are trademarks of Optibrium Ltd.
Mario Öeren, PhD – [email protected]
Hydrogen Bonding: Ab Initio Accuracy From Fast Interatomic Gaussian Approximation Potentials22nd August 2018

© 2018 Optibrium Ltd.
Introduction
• Gaussian Approximation Potential (GAP)
− A short introduction
− Practical example
− Ab initio accuracy without electrons
• The Hydrogen Bond Model
− How GAPs are applied
• Results
− Accuracy of the model
− Speed-up
− Improvements for the model
2
© 2018 Optibrium Ltd.
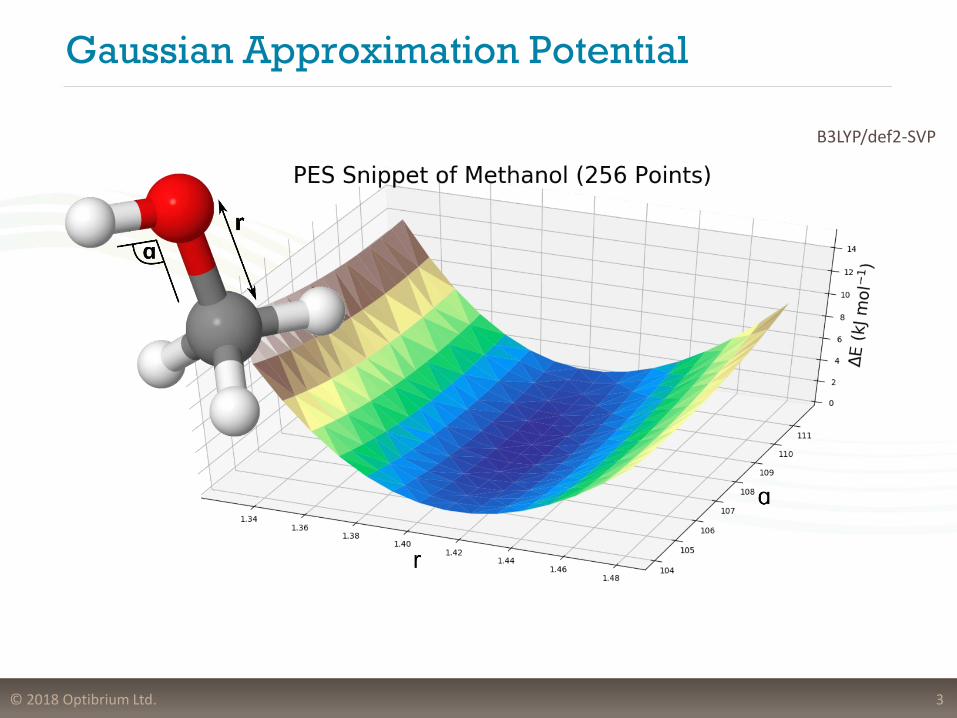
Gaussian Approximation Potential
3
B3LYP/def2-SVP

© 2018 Optibrium Ltd.
Gaussian Approximation Potential
4
B3LYP/def2-SVP

© 2018 Optibrium Ltd.
• Input from an ab-initio calculation:
− Cartesian Coordinates
− Energy Values
− Force Values
• Representation of geometry
− Invariant with respect to permutational, rotational, reflectional and translational symmetry
• Smooth Overlap of Atomic Positions (SOAP)
− Finite cut-off
− Complicated environments
− Size of the system does not matter
• Interatomic potential fit using Gaussian Processes
− Gaussian Approximation Potential (GAP)
Gaussian Approximation Potential
5
10.1103/PhysRevB.87.18411510.1002/qua.24927
B3LYP/def2-SVP
© 2018 Optibrium Ltd.
0
4
8
12
16
0 4 8 12 16
ΔE
kJ m
ol−1
(GA
P)
ΔE kJ mol−1 (DFT)
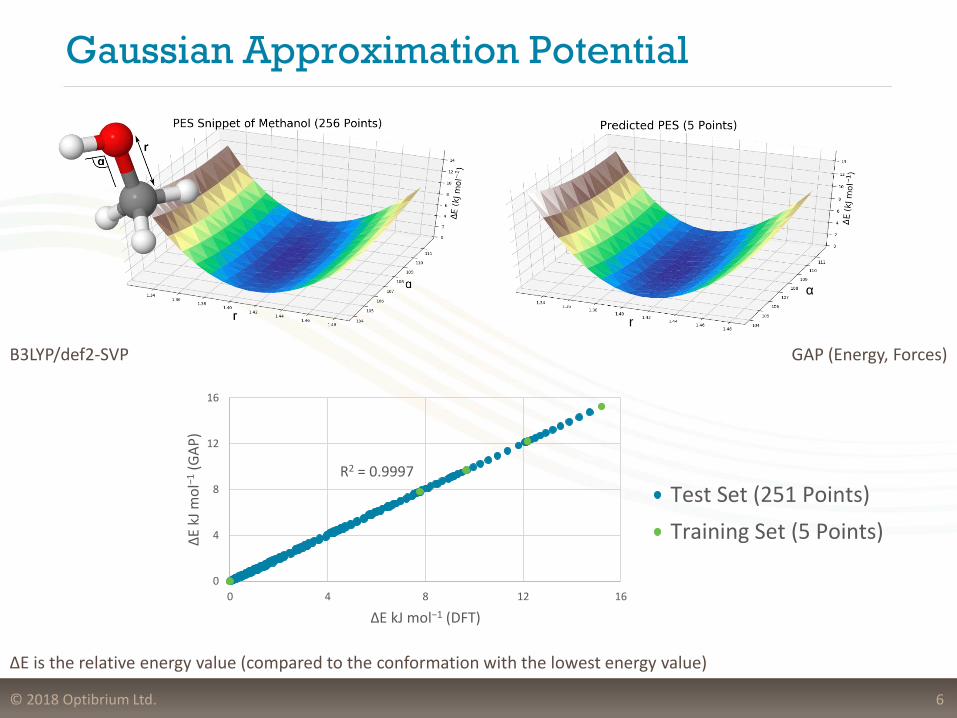
Test Set (251 Points)
Training Set (5 Points)
Gaussian Approximation Potential
6
B3LYP/def2-SVP GAP (Energy, Forces)
R2 = 0.9997
ΔE is the relative energy value (compared to the conformation with the lowest energy value)
r
α

© 2018 Optibrium Ltd.
DFT
Train GAP
MD
New Data
GAP for Methanol
• How to explore the PES (obtain data points)
− Molecular dynamics simulation
− Single point calculations of MD snapshots
7
ΔE is the relative energy value
12 Degrees of Freedom!
© 2018 Optibrium Ltd.
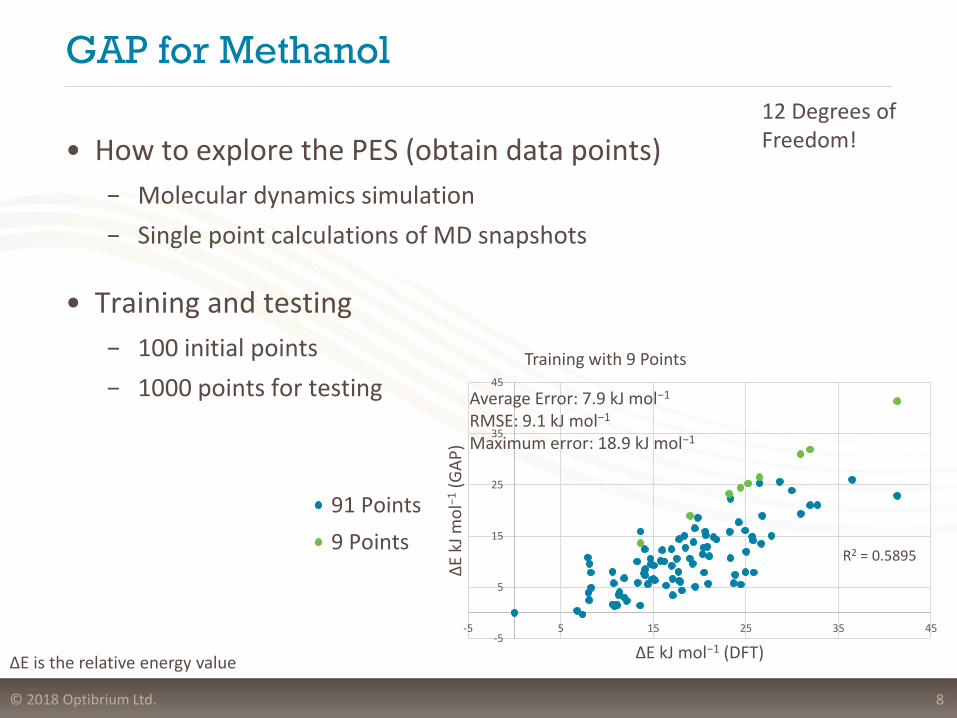
GAP for Methanol
• How to explore the PES (obtain data points)
− Molecular dynamics simulation
− Single point calculations of MD snapshots
• Training and testing
− 100 initial points
− 1000 points for testing
8
-5
5
15
25
35
45
-5 5 15 25 35 45
ΔE
kJ m
ol−1
(GA
P)
ΔE kJ mol−1 (DFT)
Training with 9 Points
91 Points
9 Points
Average Error: 7.9 kJ mol−1
RMSE: 9.1 kJ mol−1
Maximum error: 18.9 kJ mol−1
R2 = 0.5895
ΔE is the relative energy value
12 Degrees of Freedom!
© 2018 Optibrium Ltd.
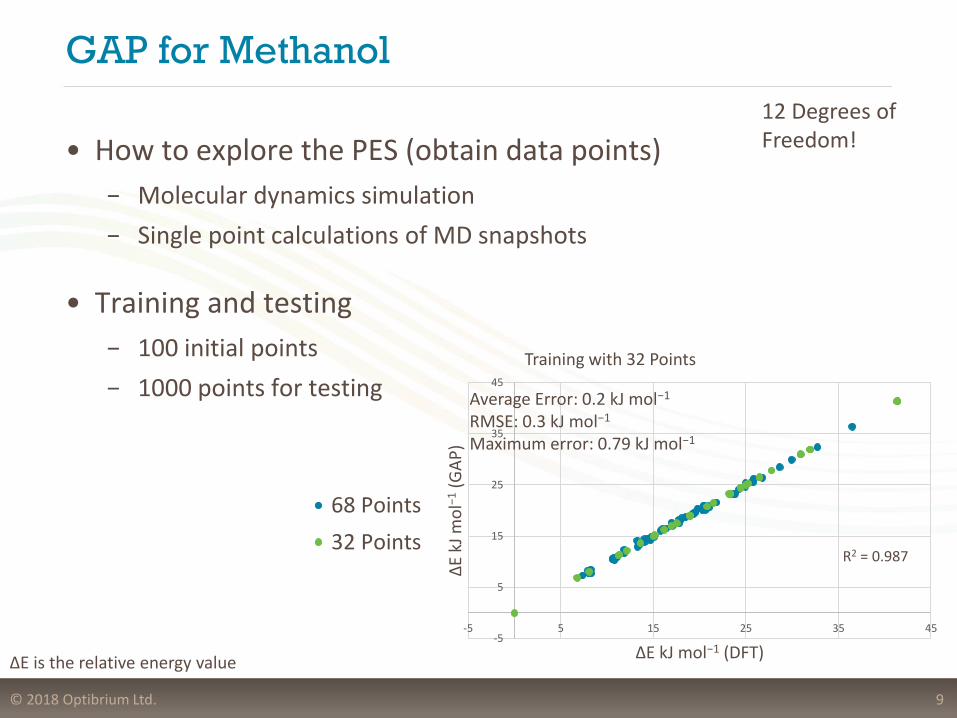
GAP for Methanol
• How to explore the PES (obtain data points)
− Molecular dynamics simulation
− Single point calculations of MD snapshots
• Training and testing
− 100 initial points
− 1000 points for testing
9
-5
5
15
25
35
45
-5 5 15 25 35 45
ΔE
kJ m
ol−1
(GA
P)
ΔE kJ mol−1 (DFT)
Training with 32 Points
68 Points
32 Points
Average Error: 0.2 kJ mol−1
RMSE: 0.3 kJ mol−1
Maximum error: 0.79 kJ mol−1
R2 = 0.987
ΔE is the relative energy value
12 Degrees of Freedom!

© 2018 Optibrium Ltd.
GAP for Methanol
• How to explore the PES (obtain data points)
− Molecular dynamics simulation
− Single point calculations of MD snapshots
• Training and testing
− 100 initial points
− 1000 points for testing
• 46 training points
10
-5
5
15
25
35
45
55
-5 5 15 25 35 45 55
ΔE
kJ m
ol−1
(GA
P)
ΔE kJ mol−1 (DFT)
Testing the GAP
1000 Points
Average Error: 0.2 kJ mol−1
RMSE: 0.2 kJ mol−1
Maximum error: 0.88 kJ mol−1
R2 = 0.987
ΔE is the relative energy value
12 Degrees of Freedom!
© 2018 Optibrium Ltd.
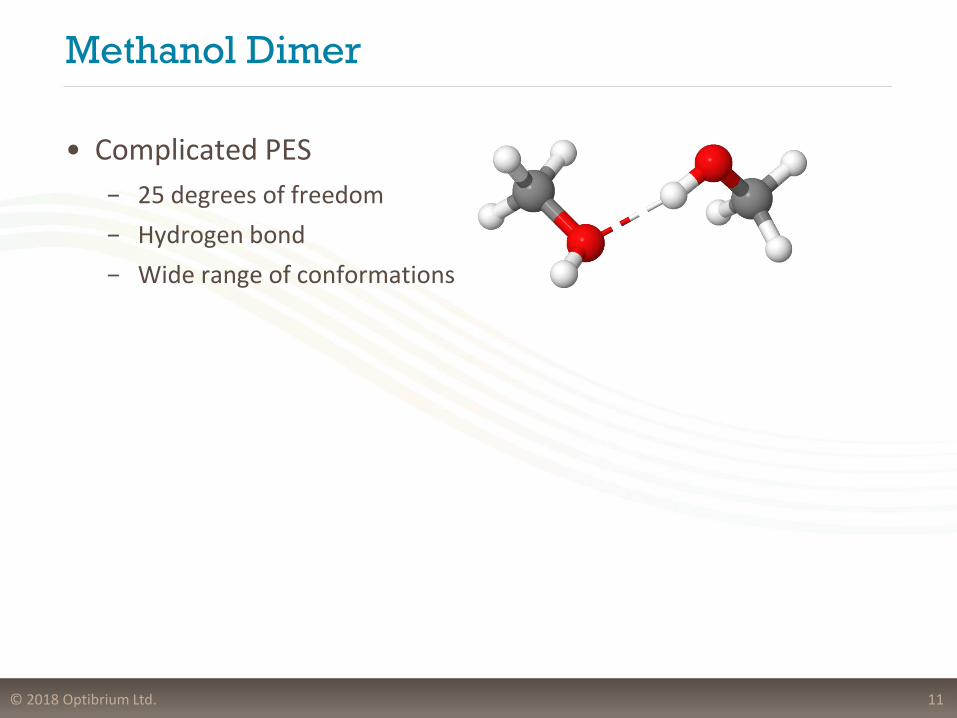
Methanol Dimer
• Complicated PES
− 25 degrees of freedom
− Hydrogen bond
− Wide range of conformations
11

© 2018 Optibrium Ltd.
Methanol Dimer
• Complicated PES
− 25 degrees of freedom
− Hydrogen bond
− Wide range of conformations
o 3.5 × 1092
o 2.4 × 105
12
R2 = 0.9682
ΔE is the relative energy value

© 2018 Optibrium Ltd.
Methanol Dimer
• Complicated PES
− 25 degrees of freedom
− Hydrogen bond
− Wide range of conformations
13
• Simplifications

© 2018 Optibrium Ltd.
Methanol Dimer
• Complicated PES
− 25 degrees of freedom
− Hydrogen bond
− Wide range of conformations
14
0
5
10
15
20
25
30
0 5 10 15 20 25 30
ΔE
kJ m
ol−1
(GA
P)
ΔE kJ mol−1 (DFT)
10 000 Points For Testing
Average Error: 0.5 kJ mol−1
RMSE: 0.7 kJ mol−1
Maximum error: 3.0 kJ mol−1
• Simplifications
• 1500 TrainingPoints
R2 = 0.9682
ΔE is the relative energy value

© 2018 Optibrium Ltd.
Interaction Potential
15
EHB = 34.65 kJ mol−1

© 2018 Optibrium Ltd.
Interaction Potential
16
• Form a fragment based system
EHB = 33.85 kJ mol−1

© 2018 Optibrium Ltd.
Interaction Potential
17
• Form a fragment based system
• Hydrogen bond energy
− Methanol molecules
− Methanol dimer
E1
E2
ETotal
EHB = ETotal – E1 – E2

© 2018 Optibrium Ltd.
Interaction Potential
18
• Form a fragment based system
• Hydrogen bond energy
− Methanol molecules
− Methanol dimer
• Interaction potential
− Train potential with EHB
• Advantages
− More accurate
− Reduced time
• Atom types
EHB

© 2018 Optibrium Ltd.
Interaction Potential
19
• Form a fragment based system
• Hydrogen bond energy
− Methanol molecules
− Methanol dimer
• Interaction potential
− Train potential with EHB
• Advantages
− More accurate
− Reduced time
• Atom types
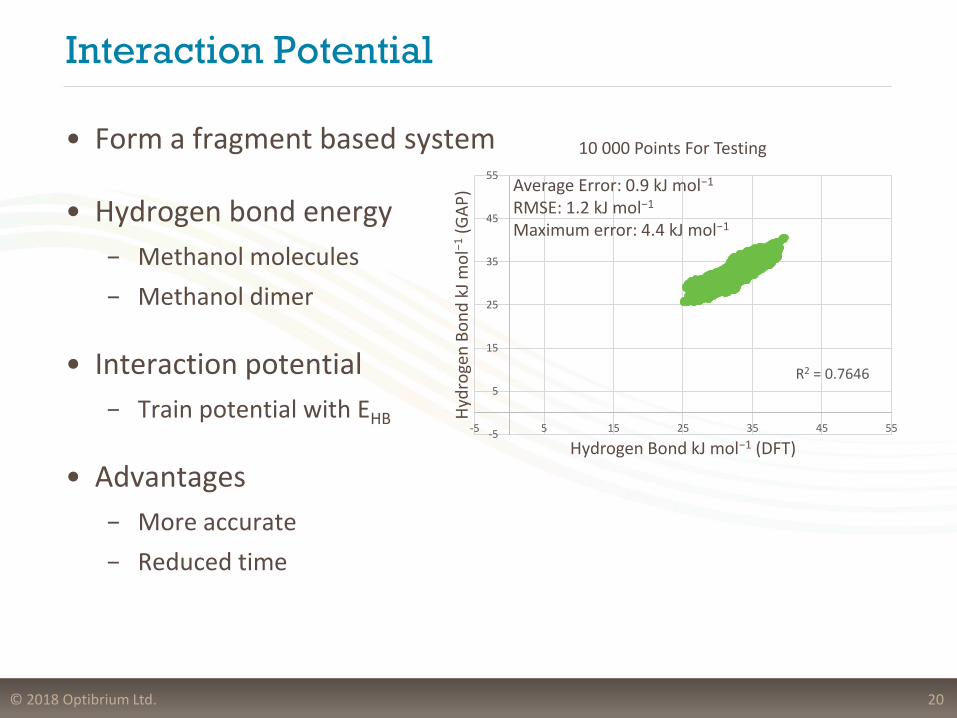
© 2018 Optibrium Ltd.
Interaction Potential
20
• Form a fragment based system
• Hydrogen bond energy
− Methanol molecules
− Methanol dimer
• Interaction potential
− Train potential with EHB
• Advantages
− More accurate
− Reduced time
• Atom types
-5
5
15
25
35
45
55
-5 5 15 25 35 45 55
Hyd
roge
n B
on
d k
J m
ol−1
(GA
P)
Hydrogen Bond kJ mol−1 (DFT)
10 000 Points For Testing
Average Error: 0.9 kJ mol−1
RMSE: 1.2 kJ mol−1
Maximum error: 4.4 kJ mol−1
R2 = 0.7646

© 2018 Optibrium Ltd.
Interaction Potential
21
• Form a fragment based system
• Hydrogen bond energy
− Methanol molecules
− Methanol dimer
• Interaction potential
− Train potential with EHB
• Advantages
− More accurate
− Reduced time
• Atom typesImage created with the SeeSAR™ module of StarDrop™.
PDB ID: 5FDQ
StarDrop™ is a trademark of Optibrium Ltd. SeeSAR™ is a trademark of BioSolveIT GmbH.

© 2018 Optibrium Ltd.
Summary
• Hydrogen bonding model – ab initio accuracy using GAPs
− Error (RMSE): 1.2 kJ mol−1
− Time gain: 120 seconds (DFT), 0.02 seconds (GAP)
22

© 2018 Optibrium Ltd.
Summary
• Hydrogen bonding model – ab initio accuracy using GAPs
− Error (RMSE): 1.2 kJ mol−1
− Time gain: 120 seconds (DFT), 0.02 seconds (GAP)
− Big molecules (12)
Average Error: 0.54 kJ mol−1, RMSE: 0.66 kJ mol−1, Maximum Error: 1.28 kJ mol−1
23

© 2018 Optibrium Ltd.
Summary
• Hydrogen bonding model – ab initio accuracy using GAPs
− Error (RMSE): 1.2 kJ mol−1
− Time gain: 120 seconds (DFT), 0.02 seconds (GAP)
− Big molecules (12)
Average Error: 0.54 kJ mol−1, RMSE: 0.66 kJ mol−1, Maximum Error: 1.28 kJ mol−1
• Applicability
− Other dimers
E.g.methanol : formaldehyde methanol : formic acid, methanol : formic acid anion formic acid : formic acid, methanol : primary amine primary amine : primary amine
− GAPs have a wide range of applications
− Any level of theory can be used with GAPs
24

© 2018 Optibrium Ltd.
Future Work
• Über potential
− Additional dimers found in and between drug molecules and proteins
− More charged species
− Combine atom types
− Reduce the amount of dimers
• Additional interactions
− Complex dimers
− Third monomer
• Predict errors of the model
25
© 2018 Optibrium Ltd.
Future Work
• Über potential
− Additional dimers found in and between drug molecules and proteins
− More charged species
− Combine atom types
− Reduce the amount of dimers
• Additional interactions
− Complex dimers
− Third monomer
• Predict errors of the model
26

© 2018 Optibrium Ltd.
Future Work
• Über potential
− Additional dimers found in and between drug molecules and proteins
− More charged species
− Combine atom types
− Reduce the amount of dimers
• Additional interactions
− Complex dimers
− Third monomer
• Predict errors of the model
27

© 2018 Optibrium Ltd.
Future Work
• Über potential
− Additional dimers found in and between drug molecules and proteins
− More charged species
− Combine atom types
− Reduce the amount of dimers
• Additional interactions
− Complex dimers
− Third monomer
• Predict errors of the model
28

© 2018 Optibrium Ltd.
Future Work
• Über potential
− Additional dimers found in and between drug molecules and proteins
− More charged species
− Combine atom types
− Reduce the amount of dimers
• Additional interactions
− Complex dimers
− Third monomer
• Predict errors of the model
29

© 2018 Optibrium Ltd.
Future Work
• Über potential
− Additional dimers found in and between drug molecules and proteins
− More charged species
− Combine atom types
− Reduce the amount of dimers
• Additional interactions
− Complex dimers
− Third monomer
• Predict errors of the model
30

© 2018 Optibrium Ltd.
Thank You
− Peter Hunt
− Gábor Csányi
− David Ponting
− Matthew Segall
31
GAPSOAP
10.1103/PhysRevB.87.18411510.1002/qua.24927