human resistin promotes neutrophil proinflammatory ... journal of immunology human resistin...
TRANSCRIPT

of May 13, 2018.This information is current as
Severity of Acute Lung InjuryExtracellular Trap Formation and IncreasesProinflammatory Activation and Neutrophil Human Resistin Promotes Neutrophil
Abraham and Jaroslaw W. ZmijewskiGregoire, Jessy Deshane, Jean Francois Pittet, Edward Shaoning Jiang, Dae Won Park, Jean-Marc Tadie, Murielle
ol.1302764http://www.jimmunol.org/content/early/2014/04/08/jimmun
published online 9 April 2014J Immunol
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2014 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on May 13, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on May 13, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
Human Resistin Promotes Neutrophil ProinflammatoryActivation and Neutrophil Extracellular Trap Formation andIncreases Severity of Acute Lung Injury
Shaoning Jiang,* Dae Won Park,*,† Jean-Marc Tadie,*,‡ Murielle Gregoire,x
Jessy Deshane,* Jean Francois Pittet,{ Edward Abraham,‖ and Jaroslaw W. Zmijewski*
Although resistin was recently found to modulate insulin resistance in preclinical models of type II diabetes and obesity, recent
studies also suggested that resistin has proinflammatory properties. We examined whether the human-specific variant of resistin
affects neutrophil activation and the severity of LPS-induced acute lung injury. Because human and mouse resistin have distinct
patterns of tissue distribution, experiments were performed using humanized resistin mice that exclusively express human resistin
(hRTN+/2/2) but are deficient in mouse resistin. Enhanced production of TNF-a or MIP-2 was found in LPS-treated hRtn+/2/2
neutrophils compared with control Rtn2/2/2 neutrophils. Expression of human resistin inhibited the activation of AMP-
activated protein kinase, a major sensor and regulator of cellular bioenergetics that also is implicated in inhibiting inflammatory
activity of neutrophils and macrophages. In addition to the ability of resistin to sensitize neutrophils to LPS stimulation, human
resistin enhanced neutrophil extracellular trap formation. In LPS-induced acute lung injury, humanized resistin mice demon-
strated enhanced production of proinflammatory cytokines, more severe pulmonary edema, increased neutrophil extracellular
trap formation, and elevated concentration of the alarmins HMGB1 and histone 3 in the lungs. Our results suggest that human
resistin may play an important contributory role in enhancing TLR4-induced inflammatory responses, and it may be a target for
future therapies aimed at reducing the severity of acute lung injury and other inflammatory situations in which neutrophils play
a major role. The Journal of Immunology, 2014, 192: 000–000.
Resistin is a secretory cysteine-rich protein that belongs tothe FIZZ protein family and is characterized as an insulinresistance factor found in a mice model of type II diabetes
and obesity (1–4). Recent studies showed that resistin plays animportant role in regulating glucose homeostasis, as well as thepathophysiology of insulin resistance, in rodents (5). In particular,loss of resistin was shown to improve insulin sensitivity (6), andhyperresistinemia results in insulin resistance that predisposes totype II diabetes mellitus (5). In addition to regulation of glucosehomeostasis, including enhancing insulin sensitivity, resistin wasrecently implicated in the development of cardiovascular dis-orders. For example, cardiac hypertrophy resulted from expression
of resistin in diabetic rat hearts (7). The proposed mechanism ofaction for resistin’s effects on inducing hypertrophy of ventricularmyocytes relates to inhibition of AMP-activated protein kinase(AMPK), a major sensor and regulator of bioenergetics at cellularand organism levels (8, 9). Although mouse resistins RELMa andRELMb and human resistin have been implicated in inflammation(10–12), variant resistins have distinct patterns of tissue distribu-tion and, therefore, appear to have compartment-specific effects.Unlike the expression of rodent resistin, which is limited to adi-pocytes, human resistin is primarily produced by macrophages andneutrophils, and significant amounts of resistin are found in thelungs (3, 13, 14). Increased expression of human resistin occurs inimmune disorders, including dysregulated inflammatory con-ditions (3, 13, 15). For example, systemic amounts of humanresistin are elevated for prolonged periods in septic patients (16,17). However, the contributory role of resistin in inflammation andorgan injury has not been well characterized. Only high concen-trations of human resistin were reported to directly stimulate cy-tokine production, such as TNF-a, by RAW 264.7 cells (18).Although i.p. administration of purified murine resistin has modestinflammatory effects in mice, a marked increase in the severity ofliver injury only occurred when resistin was combined with en-dotoxin challenge (19).Neutrophils play an essential role in innate immune and in-
flammatory responses directed toward eradication of microbialinfection (20, 21). However, exaggerated proinflammatory acti-vation, often accompanied by the release of neutrophil extracel-lular traps (NETs), is frequently associated with collateral tissuedamage and organ dysfunction, including development of acutelung injury (ALI) (22–26). Recent studies, including results ob-tained in our laboratory, established the important link amongmetabolism, neutrophil activation, and inflammation (27–29).Although AMPK is a major metabolic sensor and regulator of
*Department of Medicine, University of Alabama at Birmingham, Birmingham, AL35294; †Division of Infectious Diseases, Korea University Ansan Hospital, Ansan425-707, Korea; ‡Service des Maladies Infectieuses et Reanimation Medicale, CentreHospitalier Universitaire; xINSERM, Unite Mixte de Recherche U917, Universite deRennes 1, Rennes 35033, France; {Department of Anesthesiology, University ofAlabama at Birmingham, Birmingham, AL 35294-0012; and ‖Office of the Dean,Wake Forest University School of Medicine, Winston-Salem, NC 27157
Received for publication October 15, 2013. Accepted for publication March 10,2014.
This work was supported in part by National Institutes of Health Grants GM87748and HL107585 (to J.W.Z.). hRTN+/2/2 and RTN2/2/2 mice were generated withfunding from National Institutes of Health Grant DK49210 and the TransgenicMouse Core of the Penn Diabetes Research Center (Philadephia, PA; National Insti-tutes of Health Grant DK1952).
Address correspondence and reprint requests to Dr. Jaroslaw W. Zmijewski, Divisionof Pulmonary, Allergy, and Critical Care Medicine, Department of Medicine, Uni-versity of Alabama at Birmingham, 901 19th Street South, BMRII 304, Birmingham,AL 35294. E-mail address: [email protected]
Abbreviations used in this article: AICAR, 5-aminoimidazole-4-carboxamide-1-b-D-ribofuranoside; ALI, acute lung injury; AMPK, AMP-activated protein kinase;ARDS, acute respiratory distress syndrome; BAL, bronchoalveolar lavage; i.t., intra-tracheal; NET, neutrophil extracellular trap.
Copyright� 2014 by The American Association of Immunologists, Inc. 0022-1767/14/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1302764
Published April 9, 2014, doi:10.4049/jimmunol.1302764 by guest on M
ay 13, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

energy production (30, 31), AMPK also can inhibit NF-kB–as-sociated signaling in TLR2- or TLR4-stimulated neutrophils andmacrophages (27, 32). Moreover, mice that received the AMPKactivators metformin or 5-aminoimidazole-4-carboxamide-1-b-D-ribofuranoside (AICAR) were partially protected from endotoxin-induced ALI (27). Although activation of AMPK before LPSchallenge can suppress the inflammatory response in many celltypes, little is known about mechanisms responsible for the ob-served inhibition of AMPK activity during inflammatory responses(27, 33, 34).Recent studies suggest that resistin is able to diminish AMPK
activity (7, 35, 36). Because activation of AMPK has anti-inflammatory functions, we examined the hypothesis that inter-actions between human resistin and AMPK result in increasedneutrophil proinflammatory activation and enhance the develop-ment and severity of LPS-induced ALI. Given the well-characterizeddifferences in structure and localization of human and murineresistin, our experiments were performed using mice deficient inrodent resistin (RTN2/2) as well as humanized resistin mice(hRTN+/2/-), which exclusively expressed the human variant ofresistin.
Materials and MethodsMice
Humanized resistin mice (hRTN+/2/2) and resistin-knockout mice (RTN2/2/2)were generated by Dr. Mitchell A. Lazar (University of Pennsylvania, Phil-adelphia, PA), as previously described (5). Briefly, the transgenic mice on theC57BL/6 background, with expression of human resistin under the control ofthe CD68 promoter, were bred to C57BL6 resistin-knockout mice (RTN2/2/2)to generate mice that express human resistin but lack murine resistin (5).Wild-type C57BL/6 mice were purchased from the National Cancer Institute-Frederick (Frederick, MD). Male mice, 10–12 wk of age, were used forexperiments. The mice were kept under 12-h light–dark cycle conditions, withfree access to food and water. All experiments were conducted in accordancewith protocols approved by the University of Alabama at Birmingham Ani-mal Care and Use Committee.
Measurement of resistin in serum of acute respiratory distresssyndrome and septic patients
The study protocol was approved by the local ethics committee (CentreHospitalier Universitaire Rennes), and written informed consent was ob-tained from the patient or their closest relative. We studied 27 patients (13male, 14 female) who were admitted to the Medical ICU at Rennes Uni-versity Hospital; they were compared with healthy volunteers. Acute re-spiratory distress syndrome (ARDS; n = 8) was defined using the Berlindefinition (37). Septic shock (n = 13) was defined according to interna-tionally accepted criteria (38).
Materials
Recombinant human resistin expressed in HEK293 cells was purchased fromAdipoGene (San Diego, CA). Compound C was obtained from Millipore(Billerica, MA). AICAR was from Enzo Life Sciences (Plymouth Meeting,PA). PMA and Escherichia coli 0111:B4 endotoxin (LPS) were purchasedfrom Sigma-Aldrich (St. Louis, MO). Custom Ab mixtures (Abs) andnegative-selection columns for neutrophil isolation were from STEMCELLTechnologies (Vancouver, BC, Canada), whereas Abs to phospho–Thr172-AMPKa and total AMPKa, as well as NADPH oxidase subunit p-p40phox,were obtained from Cell Signaling Technology (Danvers, MA). Histone 3and b-actin Abs were from Santa Cruz Biotechnology (Santa Cruz, CA).Anti–citrulline–histone 3 Ab and mouse mAb to histone 3–FITC wereobtained from Abcam (Cambridge, MA). Emulsion oil solution containingDAPI was from Vector Laboratories (Burlingame, CA). SYTOX Greenprobe was purchased from Invitrogen (Carlsbad, CA).
Isolation of neutrophils
Bone marrow neutrophils were purified using a negative-selection columnpurification system, as previously described (27, 39). Briefly, bone marrowcell suspensions were isolated from the femur and tibia of a mouse byflushing with RPMI 1640 medium. Negative selection to purify neutrophilswas performed by incubation of the cell suspension with biotinylated
primary Abs specific for the cell surface markers F4/80, CD4, CD45R,CD5, and TER119 (STEMCELL Technologies) for 15 min at 4˚C, fol-lowed by incubation with anti-biotin tetrameric Abs (STEMCELL Tech-nologies) for 15 min. The complex of anti-tetrameric Abs and cells wasincubated with colloidal magnetic dextran iron particles (STEMCELLTechnologies) for an additional 15 min at 4˚C. The T cells, B cells, RBCs,monocytes, and macrophages were captured in a column surrounded bya magnet, allowing the neutrophils to pass through. Neutrophil purity, asdetermined by Wright–Giemsa–stained cytospin preparations, was con-sistently .98%. Viability of purified bone marrow neutrophils was de-termined after trypan blue staining and was consistently .95%. Humanneutrophils were isolated from the peripheral blood of healthy donorsusing a CD16 MicroBeads Magnetic Cell Sorting Kit (MACS; MiltenyiBiotec, San Diego, CA), according to the manufacturer’s instructions. TheCD16+ cells (neutrophils) were collected and suspended in RPMI 1640medium.
Purification and culture of peritoneal macrophages
Peritoneal macrophages were elicited in 8–10-wk-old mice using Brewerthioglycollate injected i.p. Cells were collected 4 d after injection ofBrewer thioglycollate and plated in 48-well plates (2.53 105 cells/well) inRPMI 1640 medium.
ELISA
Human resistin, TNF-a, and MIP-2 were measured in serum, neutrophilculture media, or bronchoalveolar lavage (BAL) using ELISA kits (R&DSystems, Minneapolis, MN), according to the manufacturer’s instructionsand as previously described (40, 41).
Western blot analysis
Western blot analysis was performed as described previously (27, 42, 43).Briefly, cell lysates were mixed with Laemmli sample buffer and boiled for15 min. Equal amounts of proteins were resolved by NaDodSO4-PAGEand transferred onto polyvinylidene fluoride membranes (Immobilon P;Millipore). The membranes were probed with specific Abs, as described inthe figure legends, followed by detection with HRP-conjugated goat anti-rabbit IgG or anti-mouse IgG. Bands were visualized by ECL (SuperSignal; Pierce Biotechnology, Rockford, IL) and quantified by AlphaEaseFCsoftware (Alpha Innotech, San Leandro, CA).
Measurement of NET-derived DNA
Bone marrow neutrophils (2 3 105 cells) were seeded in Costar 96-wellblack plates (Costar, Corning, NY) in the presence of 0.5% FBS andSYTOX Green (5 mM), a cell-impermeable DNA-binding dye. Plateletswere purified from whole blood collected in sodium citrate anticoagulanttubes, and platelet-rich plasma was obtained by centrifugation. The cellswere incubated with human resistin or PMA in the presence of platelets(106 cells) for the indicated time at 37˚C, and free DNA in culture mediumwas measured using time-dependent fluorescence of SYTOX Green probe(FLUOstar OPTIMA spectrophotometer microplate reader; BMG LAB-TECH, Alexandria, VA), at an excitation wavelength of 492 nm and anemission wavelength of 530 nm. To measure release of DNA in the lung ofmice, BAL fluid (50 ml) was incubated with 50 ml SYTOX Green (5 mM)for 10 min, followed by reading SYTOX Green fluorescence.
Imaging NETs and extracellular histone
Neutrophils were cultured on poly-L-lysine–coated glass coverslips andtreated as described in the figure legends. Next, cells were gently washedwith PBS and incubated with paraformaldehyde (4%) for 30 min at roomtemperature. Cells were subsequently incubated with PBS/BSA (3%) for30 min at room temperature and anti–histone 3–FITC–labeled Ab for anadditional 30 min. Neutrophils were washed with PBS, and samples weremounted with emulsion oil solution containing DAPI to visualize nuclearand released DNA. Confocal microscopy was performed, as previouslydescribed, using a confocal laser scanning microscope (LSM 710 confocalmicroscope; Carl Zeiss MicroImaging, Jena, Germany) provided by theHigh Resolution Imaging Facility at the University of Alabama at Bir-mingham (33).
ALI model
ALI was induced by intratracheal (i.t.) administration of 2 mg/kg LPS in75 ml PBS, as previously described (27, 34, 44–46). With this model, ALIis characterized by neutrophil infiltration into the lung interstitium and air-ways, development of interstitial edema, and increased pulmonary proin-flammatory cytokine production, with the greatest degree of injury being
2 HUMAN RESISTIN AND ACUTE LUNG INJURY
by guest on May 13, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

present 24 h after LPS exposure (27, 47). ALI was induced in hRTN2/2/2
or hRTN+/2/2 mice by i.t. instillation of LPS. At 24 h after LPS admin-istration, BALs were obtained by cannulating the trachea with a blunt 20-gauge needle and then lavaging the lungs three times with 1 ml iced PBS.Samples were subjected to ELISA or Western blot analysis. In additionalexperiments, lungs were processed to paraffin sections followed by H&Estaining, as previously described (45).
Wet-to-dry lung weight ratios
Separate groups of mice were used to measure wet-to-dry ratios and forBAL fluid acquisition. The wet-to-dry ratio was determined as reportedpreviously (45, 47). All mice used for lung wet-to-dry weight ratioswere of identical ages. Lungs were excised, blotted, and then weighed toobtain the “wet” weight. Lungs were dried in an oven at 80˚C for 7 d toobtain the “dry” weight.
Statistical analysis
Statistical significance was determined by the Wilcoxon rank-sum test(independent two-group Mann–Whitney U test), as well as the Student t testfor comparisons between two groups. Multigroup comparisons were per-formed using one-way ANOVA with the Tukey post hoc test. A p value ,0.05 was considered significant.
ResultsResistin is increased in the circulation of patients with ARDSor sepsis
As shown in Fig. 1A and B, resistin was dose dependently releasedfrom LPS-treated human neutrophils and from dHL-60 cells,a human cell line that, upon differentiation, resembles primaryneutrophils. Elevated amounts of resistin in the peripheral circu-lation were found in critically ill patients, including patients withARDS or sepsis (Fig. 1C). In particular, the highest levels ofresistin were present in plasma of septic patients.
Expression of human resistin by neutrophils and macrophagesisolated from hRTN2/2/2 and hRTN+/2/2 mice
Both human and murine resistin are implicated in the develop-ment of insulin resistance (4, 6, 48). However, murine resistin is ex-clusively expressed in adipocytes, whereas human resistin is primarily
expressed in leukocytes (1, 3, 49). To delineate the effects ofhuman resistin on neutrophil proinflammatory activation anddevelopment of ALI, we used humanized resistin mice (hRTN+/2/2)(i.e., mice that express human resistin but are deficient in murineresistin) (5). As shown in Fig. 1D, whereas modest amounts ofresistin were produced by hRTN+/2/2 bone marrow neutrophils,much greater amounts of human resistin were present after cultureof hRTN+/2/2 peritoneal macrophages. Human resistin also wasdetected in the serum of unmanipulated hRTN+/2/2 mice (Fig. 1D,right panel). Of note, the levels of human resistin in the serum ofhRTN+/2/2 mice were similar to those found in the circulation ofseptic patients (Fig. 1C).
Human resistin inhibits AMPK activation and enhancesproinflammatory activity of LPS-stimulated neutrophils
To determine the effects of human resistin on neutrophil activa-tion, hRTN2/2/2 or hRTN+/2/2 neutrophils were incubated or notwith LPS, followed by measurement of cytokines in culture media.As shown in Fig. 2A, expression of human resistin significantlyincreased TNF-a and MIP-2 production by LPS-treated neutro-phils. Of note, despite the production of human resistin, little or norelease of TNF-a or MIP-2 was found in unstimulated RTN+/2/2
neutrophils. Such results suggest that rather than directly induc-ing proinflammatory activation of neutrophils, human resistin“primed” neutrophils for a more robust response upon LPS/TLR4engagement.Previous studies suggested that resistin-mediated development
of insulin resistance was associated with inhibition of AMPKactivation (7, 35). The activation status of AMPK plays an im-portant role in regulating the proinflammatory responses of manycell types, including neutrophils and macrophages (27). As shownin Fig. 2B, exposure to the AMPK activator AICAR dose de-pendently diminished TNF-a production after LPS stimulation inhTRN2/2/2 neutrophils. However, the inhibitory effects of AICARwere diminished in neutrophils that expressed human resistin.As shown in Fig. 2C and 2D, treatment with AICAR increased
FIGURE 1. Resistin is expressed by LPS-stimu-
lated neutrophils and humanized resistin mice, as
well as in critically ill patients. Human resistin was
measured in the culture medium of LPS-treated
peripheral human neutrophils (A) or differentiated
HL-60 cells (B). (A) Neutrophils were incubated
with the indicated concentrations of LPS for 4 h
Data are mean 6 SD (n = 3). (B) HL-60 leukocytes
were differentiated into the surrogate PMNs using
1.3% DMSO treatment over 5 d. Resistin in the
culture medium of HL-60 cells was determined after
exposure to LPS for 24 h. Data are mean 6 SEM
(n = 4). (C) Amount of human resistin in plasma of
normal (control), ARDS, and septic patients. Data
are means (n = 7–13). (D) Human resistin was
measured in the media of bone marrow neutrophils
and peritoneal macrophages cultured in serum-free
conditions for 4 h. Resistin also was measured in the
serum of murine resistin-deficient mice (hRTN2/2/2)
and humanized resistin mice (hRTN+/2/2). Data are
mean 6 SD (n = 3). *p , 0.05, **p , 0.01, ***p ,0.001.
The Journal of Immunology 3
by guest on May 13, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

phosphorylation of AMPK to a greater extent in hTRN2/2/2
neutrophils compared with hTRN+/2/2 neutrophils. These datasuggest that human resistin increases neutrophil sensitivity toLPS-induced cytokine production through inhibiting AMPK ac-tivation.
Human resistin stimulates NET formation
Extensive NET formation was shown to perpetuate inflammationand cardiovascular complications (25, 50). Although NETs alsowere found in the lungs of mice with LPS-induced ALI (23), therelationship between resistin and NET formation has not beendescribed. To address this issue, bone marrow neutrophils isolatedfrom wild-type mice were cultured on poly-D-lysine–coated glasscoverslips and treated or not with recombinant human resistin.Experiments also were performed in the presence of isolatedplatelets, because platelets were shown to stimulate NETosis (22,51). Neutrophils also were incubated with PMA, an effective ac-tivator of NET formation (22, 23, 52). Fig. 3A shows that expo-sure of neutrophils to human resistin resulted in NET formation,as evidenced by DNA staining with DAPI and direct immunode-tection of histone 3 with FITC-labeled anti-H3 Abs. In particular,the appearance of extracellular histone 3 colocalized with stainingfor chromatin DNA, indicative of NET formation. NETosis wasfurther confirmed by measuring the concentrations of free DNA inculture medium using SYTOX Green fluorogenic probe, whichbecomes fluorescent upon binding to DNA (Fig. 3B) (53). Westernblotting also revealed significant increases in extracellular histone3 in culture media that were collected from resistin-treated neu-trophils (Fig. 3C). Of note, after 18 h of culture, the amount of freeDNA in the media was significantly increased in hRTN+/2/2
neutrophils compared with hRTN2/2/2 neutrophils (Fig. 3D).
AMPK inhibition promotes resistin-induced NET formation
Recent studies showed that NADPH oxidase and reactive oxygenspecies were implicated in the formation of NETs (52, 54). Asshown in Fig. 3E, exposure to human resistin increased the cit-
rullination of histone 3, a process known to stimulate chromatindecondensation prior to deployment of NETs. In addition to chro-matin relaxation, Western blotting revealed that human resistin dosedependently increased phosphorylation of the NADPH oxidasesubunit p40phox (Fig. 3F). These results suggest that human resistininduces NET formation through mechanisms that are likely to in-volve assembly of NADPH components that accompany neutrophilpriming.Because resistin inhibited AMPK activation, we next examined
whether compound C, an AMPK inhibitor, could affect NETosis.We found that inclusion of compound C in neutrophil culturesenhanced release of extracellular histone 3 to a similar extent as thatfound after PMA treatment; additive effects were observed whenneutrophils were treated with both PMA and compound C(Fig. 3G). Of note, inclusion of the AMPK activator AICAR di-minished resistin-induced increases in histone 3 citrullination(Fig. 3H). These results suggest that cross-talk between resistinand AMPK signaling is an important regulatory mechanism inneutrophil NETosis.
Increased severity of LPS-induced ALI in humanized resistinmice
To explore the effects of human resistin on ALI, hRTN+/2/2 andcontrol RTN2/2/2 mice were subjected to i.t. instillation of saline(control) or LPS. Despite increased concentrations of resistin inserum and BAL of humanized resistin mice, expression of in-flammatory mediators or evidence of lung injury was negligible insaline-treated hRTN+/2/2 or resistin-deficient mice. In contrast,administration of LPS significantly increased lung injury inhRTN+/2/2 mice compared with RTN2/2/2 mice. In particular,increased wet-to-dry ratios, indicative of more severe interstitialpulmonary edema, were present in LPS-treated hRTN+/2/2 mice(Fig. 4A). Compared with resistin-deficient mice (RTN2/2/2),exposure to LPS increased the numbers of total white cells andneutrophils in BALs isolated from LPS-treated hRTN+/2/2 micecompared with control mice (Fig. 4B). Histological analysis of the
A B
C D
FIGURE 2. Human resistin diminished AMPK
activity and increased neutrophil sensitivity to LPS
challenge. (A) Levels of TNF-a and MIP-2 were
determined in the culture media of hRTN2/2/2 and
hRTN+/2/2 neutrophils. Cells were incubated with
LPS (30 ng/ml) for 4 h, and media were subjected to
ELISA. Data are mean6 SD (n = 3). (B) Amount of
TNF-a in culture media obtained from hRTN+/2/2
or RTN2/2/2 neutrophils that were treated with
AICAR for 60 min, followed by exposure to LPS (0
or 30 ng/ml) for 4.5 h. Data are mean 6 SD (n = 4).
Representative Western blots (C) and optical densi-
tometry (D) show phospho–Thr172-AMPK, AMPK,
and b-actin obtained from hRTN+/2/2 or RTN2/2/2
neutrophils treated with AICAR (0, 0, 0.1 or 0.3
mM) for 60 min. Data are mean 6 SEM (n = 3–4).
*p , 0.05, ***p , 0.001.
4 HUMAN RESISTIN AND ACUTE LUNG INJURY
by guest on May 13, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

lungs showed increased tissue damage and enhanced neutrophilinfiltration in hRTN+/2/2 mice compared with hRTN2/2/2 miceafter LPS exposure (Fig. 4D). Although modest amounts ofresistin were present at baseline in BALs from untreated hRTN+/2/2
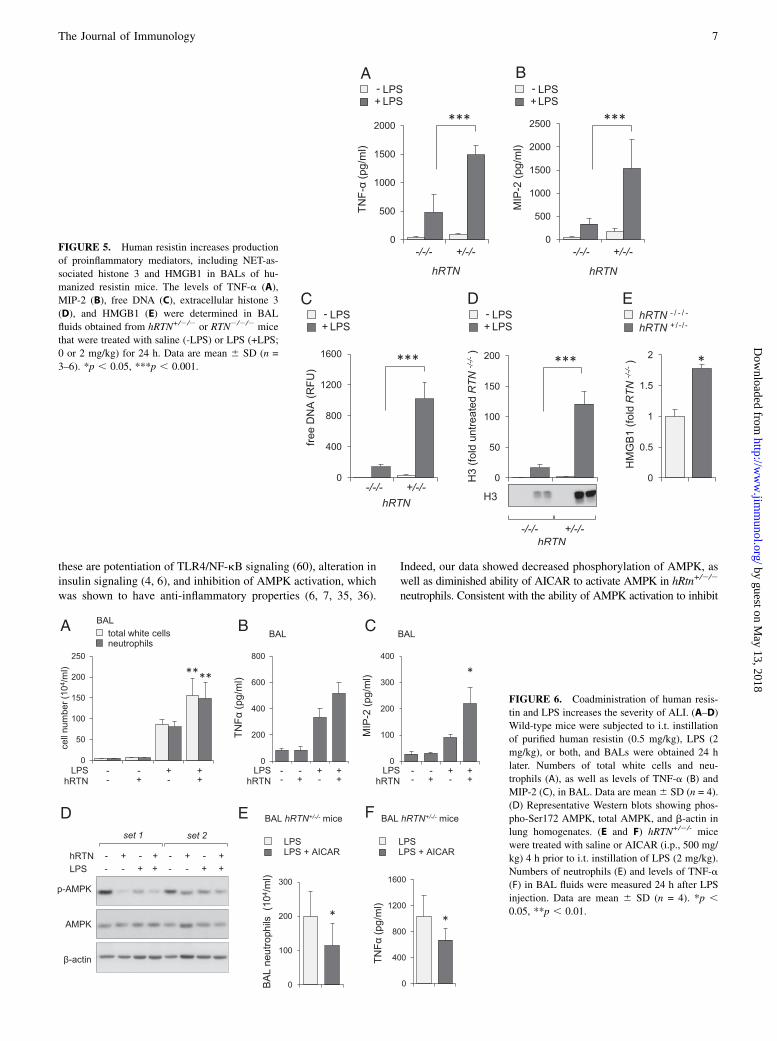
mice, significant increases in human resistin concentrations werepresent in BALs of LPS-treated hRTN+/2/2 mice (Fig. 4C). Com-pared with TNF-a and MIP-2 concentrations found in BALs fromLPS-treated hRTN2/2/2 mice, even higher concentrations of thesecytokines were present in LPS-treated hRTN+/2/2 mice (Fig. 5A,5B). hRTN+/2/2 mice also showed increased markers of NET for-mation in the lungs after LPS exposure, including free DNA(Fig. 5C) and enrichment of HMGB1 (Fig. 5E) and histone 3(Fig. 5D) in BALs. These results indicate that human resistin con-tributes to a more robust proinflammatory pulmonary response inLPS-induced ALI.
Although exposure of humanized resistin mice to LPS wasassociated with more severe ALI than in wild-type mice, suchresults may reflect the absence of mouse resistin in hRTN+/2/2
mice. Therefore, to more directly determine the contribution ofhuman resistin to the severity of ALI, wild-type mice were giveni.t. instillation of purified human resistin (0.5 mg/kg), LPS (2 mg/kg), or the combination of human resistin and LPS. As shown inFig. 6A and 6C, significantly higher numbers of total white cellsand neutrophils, as well as greater increases in TNF-a and MIP-2,were found in the BALs of mice given both human resistin andLPS compared with LPS alone. Of note, i.t. administration ofhuman resistin alone had negligible effects on BAL white cells orcytokine levels (Fig. 6A–C). In additional experiments, we ex-amined the effects of human resistin on AMPK phosphorylation inthe lung. As shown in Fig. 6D, i.t. administration of human resistin
FIGURE 3. Human resistin induces formation of NETs. Representative images (A) and quantitative data show amount of DNA (B) and histone 3
associated with NET formation. Bone marrow neutrophils isolated from wild-type C57BL/6 mice were incubated with purified human resistin (hRTN;
1 mg/ml) or PMA (100 nM) for 3 h in the presence of platelets. DNA, blue; histone 3, green. Original magnification 320. Data are mean 6 SD (n = 3).
*p , 0.05, **p , 0.01, versus untreated. (C) Representative Western blot and optical bend densitometry show the amount of histone 3 in the culture media
obtained from neutrophils treated with purified hRTN (1 mg/ml) for 3 h. Data are mean 6 SD (n = 3). **p , 0.01. (D) Amounts of free DNA in culture
media of neutrophils obtained from hRTN2/2/2 or hRTN+/2/2 mice. hRTN2/2/2 or hRTN+/2/2 neutrophils were cultured for 16 h in RPMI 1640 media and
serum (10%) from hRTN2/2/2 or hRTN+/2/2 mice. Representative Western blots show levels of citrullinated histone 3, histone 3, and b-actin in neutrophils
after treatment with purified hRTN (1 mg/ml) (E) or levels of p-p40phox in neutrophils treated with hRTN for 3 h (F). (G) Western blot analysis of ex-
tracellular histone 3 in culture media of neutrophils treated with compound C (com. C; 10 mM) for 30 min, followed by incubation with PMA (100 nM)
for 3 h. Data are mean 6 SD (n = 3–5). *p , 0.05. (H) Amount of citrullinated histone 3, histone 3, and b-actin in neutrophils treated with AICAR
(0 or 0.3 mM) for 60 min and then inclusion of purified hRTN (1 mg/ml) for the indicated times.
The Journal of Immunology 5
by guest on May 13, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

or LPS alone resulted in a modest decrease in AMPK phosphor-ylation, and exposure to LPS and human resistin produced an evengreater decrease. Of note, previous studies showed that inhibitionof AMPK activation potentiated the proinflammatory effects ofLPS (55).
Activation of AMPK in vivo partially attenuates LPS-inducedALI in humanized resistin mice
Because previous studies showed that pharmacologically inducedactivation of AMPK with AICAR diminished LPS-induced ALI(27), we examined whether a similar approach affects the severityof pulmonary injury in humanized resistin mice. In these experi-ments, hRTN+/2/2 mice were treated with saline (control, i.p.) orAICAR (i.p., 500 mg/kg) for 4 h, followed by i.t. injection of LPS(2 mg/kg), and were sacrificed 24 h later to determine the severityof lung injury. As shown in Fig. 6E and 6F, administration ofAICAR resulted in decreased neutrophil accumulation in thelungs, as well as diminished levels of BALTNF-a, compared withsaline-treated mice.
DiscussionIn these studies, we showed that human resistin enhances neutro-phil proinflammatory responses to LPS stimulation. Similar to theability of resistin to enhance TLR4-induced neutrophil activation
in vitro, more severe LPS-induced ALI was present in mice thatexpress human resistin (hRtn+/2/2). Although previous studiessuggested that resistin had proinflammatory action, our results in-dicate that human resistin itself has only modest effects on TNF-aor MIP-2 production by neutrophils. However, exposure of neu-trophils to resistin appears to prime them for enhanced activationwhen subsequently stimulated by LPS. Similarly, despite thepresence of considerable amounts of human resistin in the circu-lation under basal conditions in hRtn+/2/2 mice, only a minimalincrease in cytokines was found in BALss. In contrast, the severityof lung injury was significantly increased in hRtn+/2/2 mice afterpulmonary instillation of LPS. Of note, similar to our results, ex-cessive inflammatory responses in liver and skeletal muscle werereported in mice given resistin and LPS (19). Overall, these resultssuggest that resistin contributes to proinflammatory responses andthe development of more severe organ injury through mechanismsthat appear to involve either resistin-mediated priming effects orsynergistic interactions with TLR4 and a putative resistin receptor.Of note, previous studies showed that cross-talk between TLR4-induced cellular activation and other TLR-independent signalingcascades is an important mechanism for enhancement of proin-flammatory responses in neutrophils (56–59).Several possible mechanisms may be involved in the ability of
human resistin to increase neutrophil sensitivity to LPS. Among
FIGURE 4. Human resistin increases severity of
LPS-induced ALI. hRTN+/2/2 or control RTN2/2/2
mice were subjected to i.t. application of LPS (0 or
2 mg/kg) for 24 h. (A) Lung wet-to-dry ratios were
measured 24 h after LPS administration. Fold in-
crease above values present in mice receiving saline
alone are shown. Data are mean 6 SEM (n = 4–5).
(B) Numbers of total white cells and neutrophils in
BAL fluid from saline-treated (-LPS) or LPS-treated
(+LPS) mice. Data are mean 6 SD (n = 3–6). (C)
Human resistin was measured in BALs of hRTN+/2/2
or hRTN+/2/2 mice treated with saline (-LPS) or LPS
(+LPS) for 24 h. Data are mean 6 SD (n = 5). (D)
Representative H&E staining of lung sections ob-
tained from control (saline) or LPS-treated hRTN+/2/-
or RTN2/2/- mice. Original magnification 320. *p ,0.05, **p , 0.01, ***p , 0.001.
6 HUMAN RESISTIN AND ACUTE LUNG INJURY
by guest on May 13, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
these are potentiation of TLR4/NF-kB signaling (60), alteration ininsulin signaling (4, 6), and inhibition of AMPK activation, whichwas shown to have anti-inflammatory properties (6, 7, 35, 36).
Indeed, our data showed decreased phosphorylation of AMPK, aswell as diminished ability of AICAR to activate AMPK in hRtn+/2/2
neutrophils. Consistent with the ability of AMPK activation to inhibit
FIGURE 5. Human resistin increases production
of proinflammatory mediators, including NET-as-
sociated histone 3 and HMGB1 in BALs of hu-
manized resistin mice. The levels of TNF-a (A),
MIP-2 (B), free DNA (C), extracellular histone 3
(D), and HMGB1 (E) were determined in BAL
fluids obtained from hRTN+/2/2 or RTN2/2/2 mice
that were treated with saline (-LPS) or LPS (+LPS;
0 or 2 mg/kg) for 24 h. Data are mean 6 SD (n =
3–6). *p , 0.05, ***p , 0.001.
FIGURE 6. Coadministration of human resis-
tin and LPS increases the severity of ALI. (A–D)
Wild-type mice were subjected to i.t. instillation
of purified human resistin (0.5 mg/kg), LPS (2
mg/kg), or both, and BALs were obtained 24 h
later. Numbers of total white cells and neu-
trophils (A), as well as levels of TNF-a (B) and
MIP-2 (C), in BAL. Data are mean 6 SD (n = 4).
(D) Representative Western blots showing phos-
pho-Ser172 AMPK, total AMPK, and b-actin in
lung homogenates. (E and F) hRTN+/2/- mice
were treated with saline or AICAR (i.p., 500 mg/
kg) 4 h prior to i.t. instillation of LPS (2 mg/kg).
Numbers of neutrophils (E) and levels of TNF-a
(F) in BAL fluids were measured 24 h after LPS
injection. Data are mean 6 SD (n = 4). *p ,0.05, **p , 0.01.
The Journal of Immunology 7
by guest on May 13, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

inflammatory responses, decreased activation of AMPK in hRtn+/2/2
neutrophils compared with hRtn2/2/2 neutrophils was associated withmore robust TNF-a production after LPS treatment. In previousstudies, pharmacologic inhibition of AMPK or genetic deficiencyresulted in more robust activation of the TLR4/NF-kB–signalingcascade and production of inflammatory mediators in LPS-stimulatedneutrophils or IFN-g–stimulated astrocytes and microglia (61).Similarly, exposure of cells to the AMPK activators metformin,AICAR, or berberine diminished TLR4-induced activation ofneutrophils, macrophages, and endothelial cells (27, 33, 62). Theseverity of endotoxin-induced ALI was decreased in mice treatedwith either metformin or AICAR (27, 45, 63). Of note, AICARpartially attenuates LPS-induced ALI in humanized resistin mice.AMPK activation also was shown to be associated with improve-ment of vascular integrity in murine models of ALI and of airwayremodeling in preclinical models of asthma (55, 64).In addition to the ability of human resistin to increase proin-
flammatory cytokine production by neutrophils cultured with LPS,exposure of neutrophils to resistin resulted in enhanced phosphory-lation of the NADPH oxidase subunit p-p40phox and NET formation,as well as increased extracellular concentrations of the alarminsHMGB1 and histone 3 in association with NETs. Similarly, in-creased NET formation was found in lungs of hRtn+/2/2 micesubjected to LPS instillation. Although the precise mechanism re-sponsible for the ability of resistin to induce NETs has not beendetermined, NET formation, as well as release of the intranuclearproteins HMGB1 and histone 3, was shown to be coupled with morerobust inflammatory responses (22, 50, 65). For example, HMGB1was shown to contribute to the development of more severe ALI orsepsis (65). Similar to resistin, HMGB1 itself has modest proin-flammatory effects that are potentiated by combination with otherinflammatory insults, including TLR2 or TLR4 agonists or IL-1 (66).Our results also suggest that AMPK activation contributes to NETformation. In particular, activated AMPK appears to modulate NETformation through mechanisms that involved inhibition of chromatindecondensation, an essential step that precedes DNA deployment inNETosis (52, 67). Previous studies showed that NADPH oxidase islinked to activation of granular proteases and generation of NETs,independently of TLR4 engagement (68). Because AMPK also wasshown to affect neutrophil NOX2 activity (69), the ability of resistinto diminish AMPK phosphorylation provides a plausible regulatorymechanism for NOX2-induced NET formation.Our results indicate that human resistin primes neutrophils to
release greater amounts of proinflammatory mediators, such asTNF-a and MIP-2, increases NET formation, and enhances theseverity of ALI. Concentrations of resistin similar to those presentin the plasma of hRtn+/2/2 mice have been found in the circula-tion of critically ill patients, with the highest concentrations ofresistin present in patients with sepsis, an important predisposingcondition for the development of ALI (70, 71). Previous preclin-ical studies showed that increased expression of human resistincorrelated with the appearance of other inflammatory biomarkers,including IL-6, IL-12, CRP, or SOCS3 (16, 18, 72). Althoughextrapolation of potential mechanisms of organ dysfunction fromanimal models to life-threatening human conditions, such assepsis or ALI, need to be confirmed in clinical trials, our resultsobtained using humanized resistin mice not only indicate thatresistin has an important contributory role in neutrophil activationand ALI, they also suggest that resistin may be a potential ther-apeutic target in patients with organ system failures, such as ALI.
AcknowledgmentsWe thank Dr. Mitchell Lazar for providing the humanized resistin mice
(hRTN+/2/2) and the resistin-knockout mice (RTN2/2/2).
DisclosuresThe authors have no financial conflicts of interest.
References1. Schwartz, D. R., and M. A. Lazar. 2011. Human resistin: found in translation
from mouse to man. Trends Endocrinol. Metab. 22: 259–265.2. Lazar, M. A. 2007. Resistin- and Obesity-associated metabolic diseases. Horm.
Metab. Res. 39: 710–716.3. Steppan, C. M., and M. A. Lazar. 2004. The current biology of resistin. J. Intern.
Med. 255: 439–447.4. Kusminski, C. M., P. G. McTernan, and S. Kumar. 2005. Role of resistin in
obesity, insulin resistance and Type II diabetes. Clin. Sci. 109: 243–256.5. Qatanani, M., N. R. Szwergold, D. R. Greaves, R. S. Ahima, and M. A. Lazar.
2009. Macrophage-derived human resistin exacerbates adipose tissue inflam-mation and insulin resistance in mice. J. Clin. Invest. 119: 531–539.
6. Muse, E. D., S. Obici, S. Bhanot, B. P. Monia, R. A. McKay, M. W. Rajala,P. E. Scherer, and L. Rossetti. 2004. Role of resistin in diet-induced hepaticinsulin resistance. J. Clin. Invest. 114: 232–239.
7. Kang, S., E. R. Chemaly, R. J. Hajjar, and D. Lebeche. 2011. Resistin promotescardiac hypertrophy via the AMP-activated protein kinase/mammalian target ofrapamycin (AMPK/mTOR) and c-Jun N-terminal kinase/insulin receptor sub-strate 1 (JNK/IRS1) pathways. J. Biol. Chem. 286: 18465–18473.
8. Hardie, D. G., F. A. Ross, and S. A. Hawley. 2012. AMPK: a nutrient and energysensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13: 251–262.
9. Hardie, D. G., and K. Sakamoto. 2006. AMPK: a key sensor of fuel and energystatus in skeletal muscle. Physiology (Bethesda) 21: 48–60.
10. McVay, L. D., S. A. Keilbaugh, T. M. Wong, S. Kierstein, M. E. Shin, M. Lehrke,M. I. Lefterova, D. E. Shifflett, S. L. Barnes, F. Cominelli, et al. 2006. Absenceof bacterially induced RELMbeta reduces injury in the dextran sodium sulfatemodel of colitis. J. Clin. Invest. 116: 2914–2923.
11. Munitz, A., A. Waddell, L. Seidu, E. T. Cole, R. Ahrens, S. P. Hogan, andM. E. Rothenberg. 2008. Resistin-like molecule alpha enhances myeloid cellactivation and promotes colitis. J. Allergy Clin. Immunol. 122: 1200–1207.e1.
12. Osborne, L. C., K. L. Joyce, T. Alenghat, G. F. Sonnenberg, P. R. Giacomin,Y. Du, K. S. Bergstrom, B. A. Vallance, and M. G. Nair. 2013. Resistin-likemolecule a promotes pathogenic Th17 cell responses and bacterial-induced in-testinal inflammation. J. Immunol. 190: 2292–2300.
13. Pang, S. S., and Y. Y. Le. 2006. Role of resistin in inflammation andinflammation-related diseases. Cell. Mol. Immunol. 3: 29–34.
14. Patel, L., A. C. Buckels, I. J. Kinghorn, P. R. Murdock, J. D. Holbrook,C. Plumpton, C. H. Macphee, and S. A. Smith. 2003. Resistin is expressed inhuman macrophages and directly regulated by PPAR gamma activators. Bio-chem. Biophys. Res. Commun. 300: 472–476.
15. Bokarewa, M., I. Nagaev, L. Dahlberg, U. Smith, and A. Tarkowski. 2005.Resistin, an adipokine with potent proinflammatory properties. J. Immunol. 174:5789–5795.
16. Sunden-Cullberg, J., T. Nystrom, M. L. Lee, G. E. Mullins, L. Tokics,J. Andersson, A. Norrby-Teglund, and C. J. Treutiger. 2007. Pronounced ele-vation of resistin correlates with severity of disease in severe sepsis and septicshock. Crit. Care Med. 35: 1536–1542.
17. Koch, A., O. A. Gressner, E. Sanson, F. Tacke, and C. Trautwein. 2009. Serumresistin levels in critically ill patients are associated with inflammation, organdysfunction and metabolism and may predict survival of non-septic patients.Crit. Care 13: R95.
18. Silswal, N., A. K. Singh, B. Aruna, S. Mukhopadhyay, S. Ghosh, andN. Z. Ehtesham. 2005. Human resistin stimulates the pro-inflammatory cytokinesTNF-alpha and IL-12 in macrophages by NF-kappaB-dependent pathway. Bio-chem. Biophys. Res. Commun. 334: 1092–1101.
19. Beier, J. I., L. Guo, C. von Montfort, J. P. Kaiser, S. Joshi-Barve, andG. E. Arteel. 2008. New role of resistin in lipopolysaccharide-induced liverdamage in mice. J. Pharmacol. Exp. Ther. 325: 801–808.
20. Mantovani, A., M. A. Cassatella, C. Costantini, and S. Jaillon. 2011. Neutrophilsin the activation and regulation of innate and adaptive immunity. Nat. Rev.Immunol. 11: 519–531.
21. Grommes, J., and O. Soehnlein. 2011. Contribution of neutrophils to acute lunginjury. Mol. Med. 17: 293–307.
22. Caudrillier, A., K. Kessenbrock, B. M. Gilliss, J. X. Nguyen, M. B. Marques,M. Monestier, P. Toy, Z. Werb, and M. R. Looney. 2012. Platelets induce neu-trophil extracellular traps in transfusion-related acute lung injury. J. Clin. Invest.122: 2661–2671.
23. Tadie, J. M., H. B. Bae, S. Jiang, D. W. Park, C. P. Bell, H. Yang, J. F. Pittet,K. Tracey, V. J. Thannickal, E. Abraham, and J. W. Zmijewski. 2013. HMGB1promotes neutrophil extracellular trap formation through interactions with Toll-like receptor 4. Am. J. Physiol. Lung Cell. Mol. Physiol. 304: L342–L349.
24. Zawrotniak, M., and M. Rapala-Kozik. 2013. Neutrophil extracellular traps(NETs) - formation and implications. Acta Biochim. Pol. 60: 277–284.
25. Villanueva, E., S. Yalavarthi, C. C. Berthier, J. B. Hodgin, R. Khandpur,A. M. Lin, C. J. Rubin, W. Zhao, S. H. Olsen, M. Klinker, et al. 2011. Nettingneutrophils induce endothelial damage, infiltrate tissues, and expose immunos-timulatory molecules in systemic lupus erythematosus. J. Immunol. 187: 538–552.
26. Abraham, E. 2003. Neutrophils and acute lung injury. Crit. Care Med. 31(4, Suppl.)S195–S199.
27. Zhao, X., J. W. Zmijewski, E. Lorne, G. Liu, Y. J. Park, Y. Tsuruta, andE. Abraham. 2008. Activation of AMPK attenuates neutrophil proinflammatory
8 HUMAN RESISTIN AND ACUTE LUNG INJURY
by guest on May 13, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

activity and decreases the severity of acute lung injury. Am. J. Physiol. LungCell. Mol. Physiol. 295: L497–L504.
28. Jiang, S., D. W. Park, W. S. Stigler, J. Creighton, S. Ravi, V. Darley-Usmar, andJ. W. Zmijewski. 2013. Mitochondria and AMP-activated protein kinase-dependent mechanism of efferocytosis. J. Biol. Chem. 288: 26013–26026.
29. O’Neill, L. A., and D. G. Hardie. 2013. Metabolism of inflammation limited byAMPK and pseudo-starvation. Nature 493: 346–355.
30. Long, Y. C., and J. R. Zierath. 2006. AMP-activated protein kinase signaling inmetabolic regulation. J. Clin. Invest. 116: 1776–1783.
31. Hardie, D. G. 2007. AMP-activated protein kinase as a drug target. Annu. Rev.Pharmacol. Toxicol. 47: 185–210.
32. Sag, D., D. Carling, R. D. Stout, and J. Suttles. 2008. Adenosine 59-monophosphate-activated protein kinase promotes macrophage polarization toan anti-inflammatory functional phenotype. J. Immunol. 181: 8633–8641.
33. Bae, H. B., J. W. Zmijewski, J. S. Deshane, J. M. Tadie, D. D. Chaplin,S. Takashima, and E. Abraham. 2011. AMP-activated protein kinase enhancesthe phagocytic ability of macrophages and neutrophils. FASEB J. 25: 4358–4368.
34. Tadie, J. M., H. B. Bae, J. S. Deshane, C. P. Bell, E. R. Lazarowski,D. D. Chaplin, V. J. Thannickal, E. Abraham, and J. W. Zmijewski. 2012. Toll-like receptor 4 engagement inhibits adenosine 59-monophosphate-activatedprotein kinase activation through a high mobility group box 1 protein-dependent mechanism. Mol. Med. 18: 659–668.
35. Luo, Z., Y. Zhang, F. Li, J. He, H. Ding, L. Yan, and H. Cheng. 2009. Resistininduces insulin resistance by both AMPK-dependent and AMPK-independentmechanisms in HepG2 cells. Endocrine 36: 60–69.
36. Ou, H. C., W. J. Lee, C. M. Wu, J. F. Chen, and W. H. Sheu. 2012. Aspirinprevents resistin-induced endothelial dysfunction by modulating AMPK, ROS,and Akt/eNOS signaling. J. Vasc. Surg. 55: 1104–1115.
37. Ranieri, V. M., G. D. Rubenfeld, B. T. Thompson, N. D. Ferguson, E. Caldwell,E. Fan, L. Camporota, and A. S. Slutsky; ARDS Definition Task Force. 2012.Acute respiratory distress syndrome: the Berlin Definition. JAMA 307: 2526–2533.
38. Levy, M. M., M. P. Fink, J. C. Marshall, E. Abraham, D. Angus, D. Cook,J. Cohen, S. M. Opal, J. L. Vincent, and G. Ramsay; International Sepsis Def-initions Conference. 2003. 2001 SCCM/ESICM/ACCP/ATS/SIS InternationalSepsis Definitions Conference. Intensive Care Med. 29: 530–538.
39. Tadie, J. M., H. B. Bae, S. Banerjee, J. W. Zmijewski, and E. Abraham. 2012.Differential activation of RAGE by HMGB1 modulates neutrophil-associatedNADPH oxidase activity and bacterial killing. Am. J. Physiol. Cell Physiol.302: C249–C256.
40. Zmijewski, J. W., E. Lorne, S. Banerjee, and E. Abraham. 2009. Participation ofmitochondrial respiratory complex III in neutrophil activation and lung injury.Am. J. Physiol. Lung Cell. Mol. Physiol. 296: L624–L634.
41. Zmijewski, J. W., X. Zhao, Z. Xu, and E. Abraham. 2007. Exposure to hydrogenperoxide diminishes NF-kappaB activation, IkappaB-alpha degradation, andproteasome activity in neutrophils. Am. J. Physiol. Cell Physiol. 293: C255–C266.
42. Zmijewski, J. W., S. Banerjee, H. Bae, A. Friggeri, E. R. Lazarowski, andE. Abraham. 2010. Exposure to hydrogen peroxide induces oxidation and acti-vation of AMP-activated protein kinase. J. Biol. Chem. 285: 33154–33164.
43. Zmijewski, J. W., S. Banerjee, and E. Abraham. 2009. S-glutathionylation of theRpn2 regulatory subunit inhibits 26 S proteasomal function. J. Biol. Chem. 284:22213–22221.
44. Foster, W. M., D. M. Walters, M. Longphre, K. Macri, and L. M. Miller. 2001.Methodology for the measurement of mucociliary function in the mouse byscintigraphy. J. Appl. Physiol. 90: 1111–1117.
45. Zmijewski, J. W., E. Lorne, X. Zhao, Y. Tsuruta, Y. Sha, G. Liu, G. P. Siegal, andE. Abraham. 2008. Mitochondrial respiratory complex I regulates neutrophilactivation and severity of lung injury. Am. J. Respir. Crit. Care Med. 178: 168–179.
46. Brass, D. M., J. W. Hollingsworth, E. McElvania-Tekippe, S. Garantziotis,I. Hossain, and D. A. Schwartz. 2007. CD14 is an essential mediator of LPS-induced airway disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 293: L77–L83.
47. Zmijewski, J. W., E. Lorne, X. Zhao, Y. Tsuruta, Y. Sha, G. Liu, andE. Abraham. 2009. Antiinflammatory effects of hydrogen peroxide in neutrophilactivation and acute lung injury. Am. J. Respir. Crit. Care Med. 179: 694–704.
48. Park, H. K., M. Qatanani, E. R. Briggs, R. S. Ahima, and M. A. Lazar. 2011.Inflammatory induction of human resistin causes insulin resistance in endo-toxemic mice. Diabetes 60: 775–783.
49. Johansson, L., A. Linner, J. Sunden-Cullberg, A. Haggar, H. Herwald, K. Lore,C. J. Treutiger, and A. Norrby-Teglund. 2009. Neutrophil-derived hyper-resistinemia in severe acute streptococcal infections. J. Immunol. 183: 4047–4054.
50. Xu, J., X. Zhang, R. Pelayo, M. Monestier, C. T. Ammollo, F. Semeraro,F. B. Taylor, N. L. Esmon, F. Lupu, and C. T. Esmon. 2009. Extracellular his-tones are major mediators of death in sepsis. Nat. Med. 15: 1318–1321.
51. Clark, S. R., A. C. Ma, S. A. Tavener, B. McDonald, Z. Goodarzi, M. M. Kelly,K. D. Patel, S. Chakrabarti, E. McAvoy, G. D. Sinclair, et al. 2007. Platelet TLR4
activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat.Med. 13: 463–469.
52. Remijsen, Q., T. W. Kuijpers, E. Wirawan, S. Lippens, P. Vandenabeele, andT. Vanden Berghe. 2011. Dying for a cause: NETosis, mechanisms behind anantimicrobial cell death modality. Cell Death Differ. 18: 581–588.
53. Yipp, B. G., B. Petri, D. Salina, C. N. Jenne, B. N. Scott, L. D. Zbytnuik,K. Pittman, M. Asaduzzaman, K. Wu, H. C. Meijndert, et al. 2012. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo.Nat. Med. 18: 1386–1393.
54. Fuchs, T. A., U. Abed, C. Goosmann, R. Hurwitz, I. Schulze, V. Wahn,Y. Weinrauch, V. Brinkmann, and A. Zychlinsky. 2007. Novel cell death programleads to neutrophil extracellular traps. J. Cell Biol. 176: 231–241.
55. Xing, J., Q. Wang, K. Coughlan, B. Viollet, C. Moriasi, and M. H. Zou. 2013.Inhibition of AMP-activated protein kinase accentuates lipopolysaccharide-induced lung endothelial barrier dysfunction and lung injury in vivo. Am. J.Pathol. 182: 1021–1030.
56. Rittirsch, D., M. A. Flierl, D. E. Day, B. A. Nadeau, F. S. Zetoune, J. V. Sarma,C. M. Werner, G. A. Wanner, H. P. Simmen, M. S. Huber-Lang, and P. A. Ward.2009. Cross-talk between TLR4 and FcgammaReceptorIII (CD16) pathways.PLoS Pathog. 5: e1000464.
57. Lorne, E., X. Zhao, J. W. Zmijewski, G. Liu, Y. J. Park, Y. Tsuruta, andE. Abraham. 2009. Participation of mammalian target of rapamycin complex 1 inToll-like receptor 2- and 4-induced neutrophil activation and acute lung injury.Am. J. Respir. Cell. Mol. Biol. 41: 237–245.
58. Strassheim, D., K. Asehnoune, J. S. Park, J. Y. Kim, Q. He, D. Richter, K. Kuhn,S. Mitra, and E. Abraham. 2004. Phosphoinositide 3-kinase and Akt occupycentral roles in inflammatory responses of Toll-like receptor 2-stimulated neu-trophils. J. Immunol. 172: 5727–5733.
59. Wang, X. Q., K. Bdeir, S. Yarovoi, D. B. Cines, W. Fang, and E. Abraham. 2006.Involvement of the urokinase kringle domain in lipopolysaccharide-inducedacute lung injury. J. Immunol. 177: 5550–5557.
60. Benomar, Y., A. Gertler, P. De Lacy, D. Crepin, H. Ould Hamouda, L. Riffault,and M. Taouis. 2013. Central resistin overexposure induces insulin resistancethrough Toll-like receptor 4. Diabetes 62: 102–114.
61. Meares, G. P., H. Qin, Y. Liu, A. T. Holdbrooks, and E. N. Benveniste. 2013.AMP-activated protein kinase restricts IFN-g signaling. J. Immunol. 190: 372–380.
62. Dong, Y., M. Zhang, S. Wang, B. Liang, Z. Zhao, C. Liu, M. Wu, H. C. Choi,T. J. Lyons, and M. H. Zou. 2010. Activation of AMP-activated protein kinaseinhibits oxidized LDL-triggered endoplasmic reticulum stress in vivo. Diabetes59: 1386–1396.
63. Tsaknis, G., I. I. Siempos, P. Kopterides, N. A. Maniatis, C. Magkou,M. Kardara, S. Panoutsou, A. Kotanidou, C. Roussos, and A. Armaganidis. 2012.Metformin attenuates ventilator-induced lung injury. Crit. Care 16: R134.
64. Park, C. S., B. R. Bang, H. S. Kwon, K. A. Moon, T. B. Kim, K. Y. Lee,H. B. Moon, and Y. S. Cho. 2012. Metformin reduces airway inflammation andremodeling via activation of AMP-activated protein kinase. Biochem. Pharma-col. 84: 1660–1670.
65. Deng, Y., Z. Yang, Y. Gao, H. Xu, B. Zheng, M. Jiang, J. Xu, Z. He, andX. Wang. 2013. Toll-like receptor 4 mediates acute lung injury induced by highmobility group box-1. PLoS ONE 8: e64375.
66. Sha, Y., J. Zmijewski, Z. Xu, and E. Abraham. 2008. HMGB1 develops en-hanced proinflammatory activity by binding to cytokines. J. Immunol. 180:2531–2537.
67. Wang, Y., M. Li, S. Stadler, S. Correll, P. Li, D. Wang, R. Hayama, L. Leonelli,H. Han, S. A. Grigoryev, et al. 2009. Histone hypercitrullination mediateschromatin decondensation and neutrophil extracellular trap formation. J. CellBiol. 184: 205–213.
68. Parker, H., M. Dragunow, M. B. Hampton, A. J. Kettle, and C. C. Winterbourn.2012. Requirements for NADPH oxidase and myeloperoxidase in neutrophilextracellular trap formation differ depending on the stimulus. J. Leukoc. Biol. 92:841–849.
69. Alba, G., R. El Bekay, M. Alvarez-Maqueda, P. Chacon, A. Vega, J. Monteseirın,C. Santa Marıa, E. Pintado, F. J. Bedoya, R. Bartrons, and F. Sobrino. 2004.Stimulators of AMP-activated protein kinase inhibit the respiratory burst inhuman neutrophils. FEBS Lett. 573: 219–225.
70. Rubenfeld, G. D., E. Caldwell, E. Peabody, J. Weaver, D. P. Martin, M. Neff,E. J. Stern, and L. D. Hudson. 2005. Incidence and outcomes of acute lunginjury. N. Engl. J. Med. 353: 1685–1693.
71. Sevransky, J. E., G. S. Martin, C. Shanholtz, P. A. Mendez-Tellez, P. Pronovost,R. Brower, and D. M. Needham. 2009. Mortality in sepsis versus non-sepsisinduced acute lung injury. Crit. Care 13: R150.
72. Pirvulescu, M., I. Manduteanu, A. M. Gan, D. Stan, V. Simion, E. Butoi,M. Calin, and M. Simionescu. 2012. A novel pro-inflammatory mechanism ofaction of resistin in human endothelial cells: up-regulation of SOCS3 expressionthrough STAT3 activation. Biochem. Biophys. Res. Commun. 422: 321–326.
The Journal of Immunology 9
by guest on May 13, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from