homogeneous catalytic hydrogenation of organic compounds...
TRANSCRIPT

Indian Journal of chemistryVol. 31A, April 1992, pp. 243-250
Homogeneous catalytic hydrogenation of organic compounds usingorthometallated schiff base complexes of palladium(ll)
Deb K Mukherjee, Biman K Pallt & Chitta R Saha"Department of Chemistry, Indian Institute of Technology, Kharagpur 721 302
Received 21 August 1991; revised and accepted 3 December 1991
The dinuclear orthometallated schiff base complexes of palladium(II) are efficient catalysts for dihy-drogen reduction of organic - N02, >C=O, >C=C<, > C=N -, - N=N - and -C=N groupsunder normal or high pressure conditions. The dependence of the catalytic activity on the nature of thebridging group and the schiffbase ligands has been established. The experimental conditions control thenature and number of the reduction products in case of nitroaromatics. Reduction mechanism has beenproposed on the basis of the nature of intermediate and final reduction products, change of physico-chemical properties of the catalyst solutions, polarographic data of tbeligands and the complexes and thekinetic data.
Numerous methods have been developed for the ca-talytic reduction of organic nitrocompounds 1-7,al-kenes8-12 and alkynes8•13.14 in homogeneous phase,but relatively few investigations have been made onthe reduction of nitriles":'? and aliphatic nitrocom-poundsI8-22. The importance of the reduction lies inthe use of the products for the manufacture of dyes,drugs, pharmaceuticals etc. The elucidation of un-ambiguous reaction mechanism for the reductionswas difficult due to several limirarions'v-". We haveinvestigated the catalytic activities of various dinuc-lear orthopalladated complexes with differentlyC,N-substituted schiff base ligands in order to findout the influence of electron density and steric envi-ronment at different points of the complex on theircatalytic efficiencies. This may help in the modifica-tion of the catalyst system. The present paper re-ports the results of such investigations.
Materials and MethodsPure and recrystallised solid reagents, predistilled
solvents and pure, dry and deoxygenated hydrogenand nitrogen gases were used for all the experi-ments. Dimethylformamide (DMF) was dried bystoring it over CaH2 under N2 for 24 h followed bydistillation under reduced pressure. H2 and N 2gaseswere deoxygenated and dried by passing them suc-cessively through alkaline pyrogallol, liquid N2 trapand silica gel tower before introduction into thereaction system.
The complexes used in the present investigationwere prepared according to the literature methods"and purified by recrystallization from dichlorome-
thane/ n-hexane and their purities were checked byphysico-chemical means before use as catalysts.
Analyses of the product mixture were done on aVarian 3700 gas chromatograph equipped with aflame ionization detector using a S.S. columnpacked with 25% SE-30 on Chromosorb W mesh.Temperature programming was done in the range180° to 300°C with the increase of temperature atthe rate of 1rC/min. Vibrational, electronic andPMR spectra were recorded in Perkin-Elmer-883,Shimadzu MPC-3100, and Varian EM-390, 90MHz spectrometers respectively. Molecular weightsof the complexes in C6H6 and DMF were deter-mined in a Knauer Dampdruck osmometer.
Preparation of the catalystsThe schiff base ligands were prepared by reflux-
ing equimolar quantities of the appropriate alde-hyde or ketone and amine in benzene or xylene24.25.The sterically hindered schiff bases required ex-tended reflux period (2-3 days) in xylene. The pre-paration of benzophenoneanil using appropriatereagents required the presence of POCl3 in the me-dium"; Reaction of the schiff base ligand and Pd(II)acetate in refluxing acetic acid resulted in the preci-pitation of the corresponding complex which wasfurther purified by repeated crystallization from sol-vent mixture+', The purities of the complexes werechecked by TLC, IR and electronic spectral data.The metathetical reaction of acetato-bridged com-plexes. with NaCl or NaBr in acetone yielded thecorresponding chloro- and bromo-analogues".
Another series of orthopalladated complexes was

244 INDIAN J CHEM, SEe. A, APRIL 1992
prepared using the secondary amine ill).The latterwere obtained by the dihydrogen reduction of thecorresponding schiff base ligand, ill in the pres-ence of the presently investigated catalysts or other-wise.". Reaction of Pd(OAch with ill) in retluxingAcOH produced Pd2(LH2}z(OAc)2' The corre-sponding chloro- and bromo-analogues were pre-pared by metathetical reactions. The catalytic acti-vities of the orthometallated complexes,Pd2(LH2)2X2 (Fig. 1a) towards the reduction pro-cesses were found to be comparable to those ofPd2LzX2(Fig. 1b).
Reduction procedureThe procedures for normal and high pressure re-
ductions were mentioned in our earlier papers's". Incase of normal pressure reduction, the yellow DMFsolution of the catalyst turned deep greenish brownwithin 10 min on stirring under hydrogen at 20°C.The substrate addition at this stage slightly faded thesolution colour and hydrogen absorption started ata maximum rate which was measured using a gradu-ated gas reservoir. The intermediate samples (0.20ml) were withdrawn from the reaction mixture at aninterval of 10 min and analysed immediately byGLC. In the case of high pressure reductions, the fi-nal reaction mixture was quenched and the compo-nents were estimated by GLC using appropriate co-lumns. The intermediate sample analysis in this casewas made at an interval of 30 min only.
Results and DiscussionThe compounds have low solubilities in non-po-
lar solvents like benzene, cyclohexane, etc., and
moderate solubilities in mild coordinating solventslike DMF, DMSO, etc. The reduction rate of a givensubstrate was highest in DMF medium and hencemost of the reactions were carried out in this medi-um. Detailed investigations were made with theacetato-bridged complexes due to their better solu-bilities and higher catalytic activities (Table 1).
Reduction of nitroaromatics, alkenes, alkynes andother organic substrates
The catalytic hydrogenation of nitroaromaticsleads to the formation of the corresponding anilinesin almost all cases except nitrophenol and m-dinitrobenzene where the final products were onlythe corresponding hydroxylarnines (Table 2). Anal-ysis of the intermediate reacton samples indicatedthe primary formation of hydroxylamines in the caseof substrates which are finally reduced to the corre-sponding anilines. The formation of coupled pro-ducts such as azo- or. azoxybenzene was not ob-
{11 x,~ /R-C Pd'>{~rf
I 2R2
(bJVI R': CH)
(a)R': C6H5VII
VIII R', C6H5
x: OAc,
Table I-Relative catalytic activity of Pd(II) schiff base complexes in DMF medium at 25°C
Catalyst Nitrobenzene Styrene
Cat.Conc. Sub, Cone, lni tial rate of Cat. Cone, Sub.Conc. Initial rate of(mol. lit - I X 10 -4) (mol.lit - I) H2 abs. (mol.Iit "! x 10-4) (mol. lit-I) H2 abs.
(m1.min-l) (m1.min-l)
Pd2(mbih(OAch 4.8 0.32 7.90 2.4 0,86 12.90Pd2(BANh(OAch 4.8 0.32 6.30 2.4 0.86 11.95
Pd2(pTBANlz(OAch 4.8 0.32 6,05 2.4 0.86 11.70Pd2(mTBANlz(OAc}z 4.8 0.32 5.85 2.4 0.86 11.55Pd2(dmBAN)2(OAc}z 4.8 0.32 5,80 2.4 0.86 10.30Pd2(apkt)2(OAc}z 4.8 0.32 5.65 2.4 0.86 07.10Pd2(mbpkth(OAc}z 4.8 0.32 3.20 2.4 0.86 04.55Pd2(bpkth(OAc)2 4.8 0.32 2.4 0.86 01.10
mbi = N-methylbenzaldimine; BAN =N-phenylbenzaldimine; pTBAN =(N-{p-tolyl)benzaldimine); mTBAN = (N-( m-tolyl)be~za.ldi-mine); dmBAN = (N-(3,5-dimethyl)benzaldimine); apkt =N-phenylacetophenoneketimine; bpkt =N-phenylbenzophenoneketlmme;mbpkt = N-methylbenzophenoneketimine.
MUKHERJEE et al: CATALYTIC HYDROGENATION USING SCHIFF BASE COMPLEXES OF Pd(II) 245
Table 2-0ptiroum conditions and yields of main products at 1 atm pressure of Hydrogen and 25°C using (a) = Pd2(mbih(OAc h.(b) = Pd2(pTBANMOAc)2. (c)= Pd2(apkth(OAch as catalysts
Substrate Catalyst Initial Initial Product %(mollit-t x lO-4) rate of turnover
H2-abs. number(ml.min t+) (min-I)
1 Nitrobenzene a 4.8 7.90 14.60 Aniline 98 (a)b 4.6 5.50 10.60 94 (b)c 4.6 5.20 08.80 96 (c)
2 o-Chloronitro- a 4.8 2.70 05.00 o-Chloroaniline 93 (a)benzene b 4.6 2.40 04.60 88 (b)
c 4.6 2.15 04.40 88 (c)
3 p-Chloronitro- a 4.8 3.40 06.30 p-Chloroaniline 90 (a)benzene b 4.6 2.70 05.20 86 (b)
c 4.2 2.20 04.65 83 (c)
4 m-Dinitrobenzene a 4.8 7.82 12.40 m-Phenylenehydroxyl- 94 (a)b 4.6 6.30 09.80 amine 83 (b)c 4.2 5.70 07.65 80 (c)
5 o-Nitrotoluene a 4.8 2.82 05.20 o-Toluidine 96 (a)b 4.6 2.35 04.50 90 (b)c 4.2 2.20 04.05 92 (c)
6 Pent-f-ene a 4.2 5.40 31.40 Pentane 66 (a) Pent-2-ene 30 (a)b 5.6 4.15 18.80 66 (b) 30 (b)c 5.2 3.15 15.30 58 (c) 40 (c)
7 Hex-l-ene a 4.2 5.05 28.60 Hexane 64 (a) Hex-2-ene 28 (a)b 5.6 3.70 15.80 60 (b) 30 (b)c 5.2 2.70 12.80 66 (c) 30 (c)
8 CycIohexene a 4.2 3.15 19.30 CycIohexane 88 (a)b 5.6 3.30 14.70 80 (b)c 5.2 2.40 09.80 80 (c)
9 Benzaldehyde a 5.7 2.70 14.20 Benzylalcohol 96 (a)b 4.8 1.15 13.lO 90 (b)c 4.8 1.00 04.55 90 (c)
10 Benzylidine aniline a 5.9 2.55 13.80 N-Phenylbenzylamine 96 (a)b 4.8 1.00 07.80 90 (b)c 4.8 0.70 02.60 93 (c)
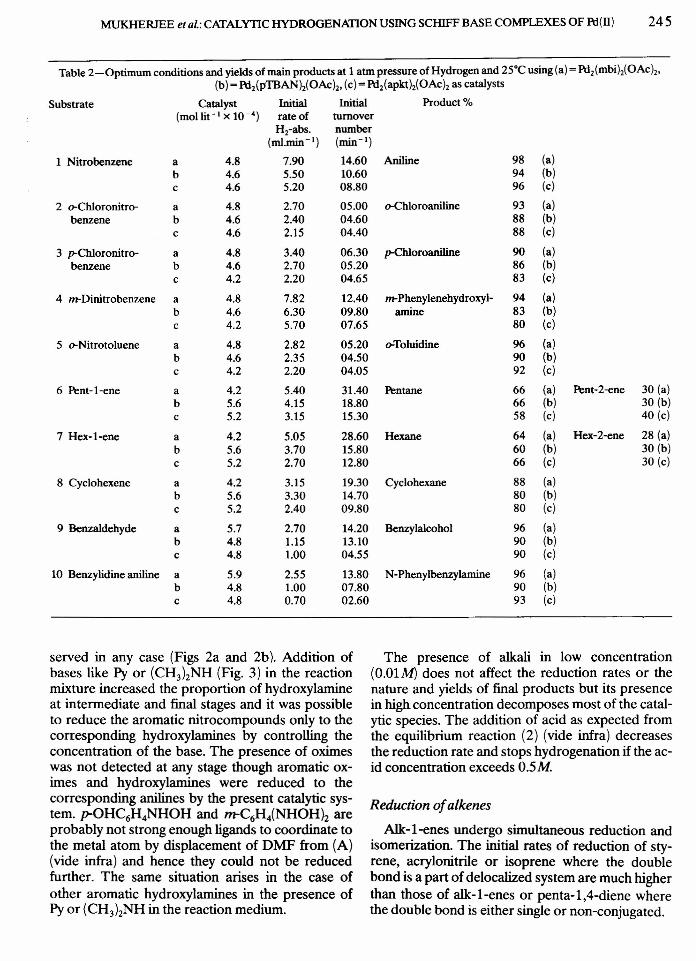
served in any case (Figs 2a and 2b). Addition ofbases like Py or (CH3}zNH (Fig. 3) in the reactionmixture increased the proportion of hydroxylamineat intermediate and final stages and it was possibleto reduce the aromatic nitrocompounds only to thecorresponding hydroxylamines by controlling theconcentration of the base. The presence of oximeswas not detected at any stage though aromatic ox-imes and hydroxylamines were reduced to thecorresponding anilines by the present catalytic sys-tem. p-OHC6H4NHOH and m-C6H4(NHOH}z areprobably not strong enough ligands to coordinate tothe metal atom by displacement of DMF from (A)(vide infra) and hence they could not be reducedfurther. The same situation arises in the case ofother aromatic hydroxylamines in the presence ofPy or (CH3)2NH in the reaction medium.
The presence of alkali in low concentration(O.OIM) does not affect the reduction rates or thenature and yields of final products but its presencein high concentration decomposes most of the catal-ytic species. The addition of acid as expected fromthe equilibrium reaction (2) (vide infra) decreasesthe reduction rate and stops hydrogenation if the ac-id concentration exceeds O.5M.
Reduction of alkenes
Alk-l-enes undergo simultaneous reduction andisomerization. The initial rates of reduction of sty-rene, acrylonitrile or isoprene where the doublebond is a part of delocalized system are much higherthan those of alk-T-enes or penta-I,4-diene wherethe double bond is either single or non-conjugated.
246 INDIAN J CHEM, SEe. A, APRIL 1992
100.
'" 80C 0'"c8.E 600u'0:' 40.,0~
20
o 10 20 30 40 50 60 70
Time in min
Fig. 2a-Reduction of nitrobenzene with Pd2(mbi)2(OAch inDMF at 25°C and under 1 atm. of hydrogen.Pd2(mbi)2(OAc)2 =4.82 x 10-4 mol dm >', [nitrobenzene) =0.32mol dm - 3. 0 = nitrobenzene; ~ = aniline; .& = phenylhydroxyl-
amine.
on 80C••c8. 60Eouo;! 40
••o:l:20
100~---------------------------------'
90o
Tim~ in min
Fig. 2b-Reduction of nitrobenzene with Pd2(apkth(OAch inDMF at 25·C and under 1 atm. of hydrogen.Pd2(apktlz(OAch=4.82 x 10-4 mol dm-J, [nitroben-zene) = 0.32 mol dm - 3; ~ = nitrobenzene; ~ = aniline;
o = phenylhydroxylamine.
Reduction of nitroalkanes, benzonitrile and benzo-phenone
The reduction of these substrates is possible onlyunder high pressure and high temperature condi-tions. The reduction rate decreases with increasingmolecular weight of the nitro alkanes and increaseswith increasing branching at the a-carbon atom(Table 3). Relatively higher hydrogen pressure andhigher temperature are necessary for the reductionof benzophenone and benzonitrile. The former pro-duced the corresponding alcohol while the latterwas reduced to a mixture of benzylamine (15-20%)and dibenzylamine (80-85%).
Table 3- Yields of main products of the reduction of nitroalkanes, benzophenone and benzonitrile using (a) Pd2(mbi}z(OAc lz;(b) Pd2( apkt lz(0AC)2as catalysts under high pressure and high temperature conditions
[Substrate) [Catalyst) Hydrogen Temp. Reaction Products (%)(mol.dm v x 10-4) pressure ·C time
(KNm-Z) (hr)
1 Nitromethane a 5.7 11.3 x 103 70 6.00 Methylamine (93)(0.25 M) b 5.2 11.7 x 103 85 6.00 (90)
2 Nitroethane a 5.7 11.3 x 103 70 6.00 Ethylamine (92)(0.25 M) b 5.2 11.7 x 103 90 6.50 (92)
3 I-Nitropropane a 5.7 11.3'< 103 75 5.50 1-Aminopropane (90)(0.25 M) b 5.2 11.7 x 103 80 7.00 (84)
4 2-Nitropropane a 5.7 11.3 x 103 75 5.70 2-Aminopropane (88)(0.25 M) b 5.0 11.7 X 10J 80 7.00 (80)
5 Benzophenone a 5.6 11.7 x 103 75 6.50 Diphenylmethanol (90)(0.30 M) b 4.8 11.7 x 103 80 7.00 (88)
6 Benzonitrile a 5.6 11.7 x 103 80 7.00 Dibenzylamine (80)(0.30 M) b 4.8 11.7 x 103 90 8.00 (64)
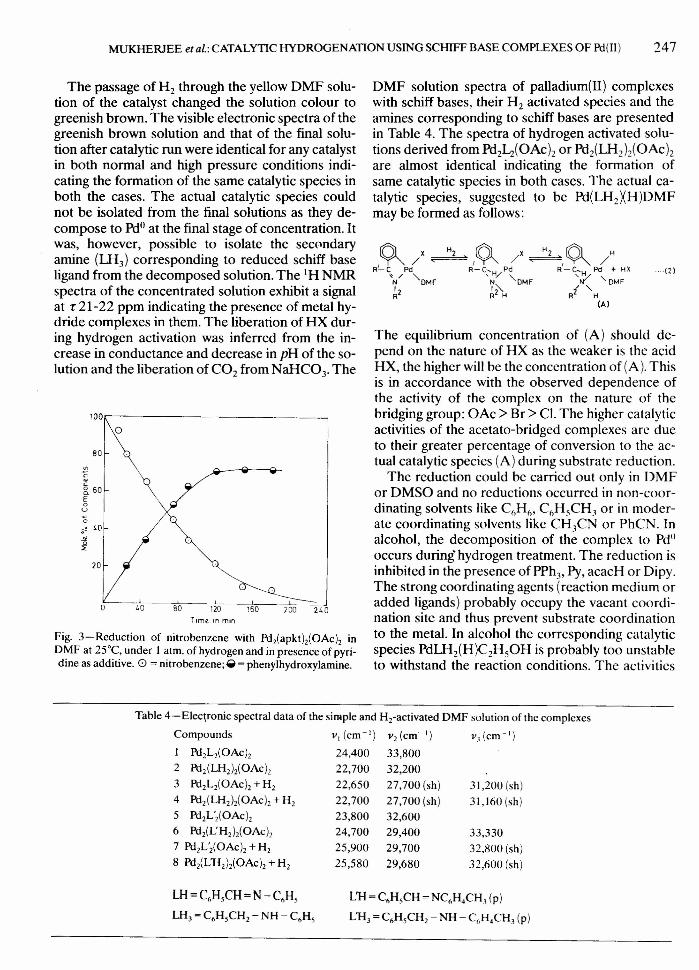
The DRS and solution spectra of any complex ca-talyst in benzene solution are almost identical butdiffer appreciably from those taken in DMF orDMSO. The molecular weight measurements of thecomplexes in dilute DMF solution indicated nearly100% dissociation. These facts together with thebridge cleavage of the dinuclear complexes by PPh3,
Py etc.23,28 suggest them to undergo the followingreaction in DMF solution.
DMFPd2L2X2 ~ 2Pd.L.X.DMFHL= schiff base ligandX = OAc, CI, Br
... (1)
MUKHERJEE et al.: CATALYTIC HYDROGENATION USING SCHIFF BASE COMPLEXES OF Pd(II) 247
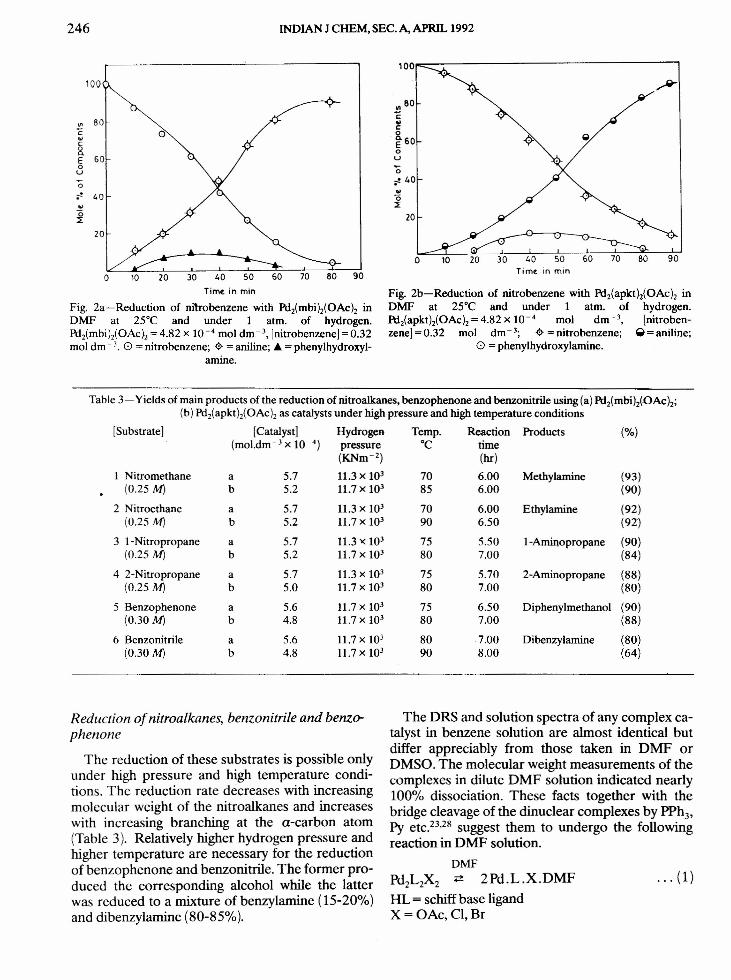
The passage of H2 through the yellow DMF solu-tion of the catalyst changed the solution colour togreenish brown. The visible electronic spectra of thegreenish brown solution and that of the final solu-tion after catalytic run were identical for any catalystin both normal and high pressure conditions indi-cating the formation of the same catalytic species inboth the cases. The actual catalytic species couldnot be isolated from the final solutions as they de-compose to PdQat the final stage of concentration. Itwas, however, possible to isolate the secondaryamine (LH3) corresponding to reduced schiff baseligand from the decomposed solution. The IH NMRspectra of the concentrated solution exhibit a signalat r 21-22 ppm indicating the presence of metal hy-dride complexes in them. The liberation of HX dur-ing hydrogen activation was inferred from the in-crease in conductance and decrease in pH of the so-lution and the liberation of CO2 from NaHC03. The
100.---------------,
o
80
'""§c::5. 60Eou"0;" 40
i
~' ,J160 200 240
20
o 40 eo 120TIme. In rmn
Fig. 3-Reduction of nitrobenzene with Pdz(apkth(OAch inDMF at 25°C, under 1 atm. of hydrogen and in presence of pyri-dine as additive. 0 = nitrobenzene; ~ = phenylhydroxylamine.
DMF solution spectra of palladium(II) complexeswith schiff bases, their H2 activated species and theamines corresponding to schiff bases are presentedin Table 4. The spectra of hydrogen activated solu-tions derived from Pdzl-2( OAC)2or Pd2(LH2lz(OAc lzare almost identical indicating the formation ofsame catalytic species in both cases. The actual ca-talytic species, suggested to be Pd(LH2)(H)DMFmay be formed as follows:
The equilibrium concentration of (A) should de-pend on the nature of HX as the weaker is the acidHX, the higher will be the concentration of (A). Thisis in accordance with the observed dependence ofthe activity of the complex on the nature of thebridging group: OAc > Br > CI. The higher catalyticactivities of the aceta to-bridged complexes are dueto their greater percentage of conversion to the ac-tual catalytic species (A) during substrate reduction.
The reduction could be carried out only in DMFor DMSO and no reductions occurred in non-coor-dinating solvents like CoHo, COH)CH-' or in moder-ate coordinating solvents like CH)CN or PhCN. Inalcohol, the decomposition of the complex to Pd"occurs during hydrogen treatment. The reduction isinhibited in the presence of Pl'h., Py, acacH or Dipy.The strong coordinating agents (reaction medium oradded ligands) probably occupy the vacant coordi-nation site and thus prevent substrate coordinationto the metal. In alcohol the corresponding catalyticspecies PdLH2(H)C2HsOH is probably too unstableto withstand the reaction conditions. The activities
Table 4-Eleqronic spectral data of the simple and H2-activated DMF solution of the complexes
Compounds VI (em -I) V1 (cm-I) v, (ern -I)
1 Pd2L2(OAc)1 24,400 33,8002 Pd2(LHzh(OAc)2 22,700 32,2003 Pd2L2(OAc)2+H2 22,650 27,700 (sh)
4 Pd2(LH2h(OAch + H2 22,700 27,700 (sh)5 Pd2L'2(OAch 23,800 32,6006 Pd2(L'H2h(OAch 24,700 29,4007 Pd2L'2(OAch + H2 25,900 29,7008Pd2(LB2h(OAch + H2 25,580 29,680
LH = C.HsCH = N - C.HsLH3 = C.HsCH2 - NH - C6HS
31,200(sh)3] ,160 (sh)
33,33032,800 (sh)32,600 (sh)
L'H =C.HsCH = NC6H4CH] (p)
LBJ =C.HsCH2 - NH -C6H4CH3 (p)

248 INDIAN J CHEM, SEe. A, APRIL 1992
of the complexes decrease in the presence of lowconcentration of added HX (X = bridging group)and are lost completely if the concentration exceeds0.1 M. All these results support the existence of eq-uilibrium (Eq. 2) in the reaction medium.
The complex Pd2L2( OAc h (ill = C6HsCH = NH)could not be synthesized and hence its catalytic acti-vities could not be studied. The catalytic activities ofPd2L2(OAch and their corresponding Pd2(ill2)2
(GAc], are almost identical and their yellow DMFsolutions undergo the same type of transformationsduring hydrogen activation and substrate reduction.The complexes Pd2L2(OAch (Fig. l b) with varyingL can be arranged in the following order accordingto their catalytic activities:
I> II - III - IV - V> VI - VII > VIIIThe complex with N-methylbenzaldimine (com-plex-I) required low induction period and showedhighest catalytic activity both under normal andhigh pressure conditions. The activities of (II to V)though considerably less than that of I, are almostequal. The solutions derived from ketirnine com-plexes (complex VI to VITI) are highly sensitive toair and decompose to PdQin the presence of tracesof air. The complexes may be divided into fourgroups depending on their activities. The first group(compound-I) having only alkyl substituent on N-at-om of the > C = N - group has the highest activitywhile the second group (compound II-Y) having on-ly aryl substituents on the N atom is far less reactive.The activities, however, do not change appreciablywith the number of alkyl substituents on the phenylring. The third group (compound VI and VII) withalkyl and aryl substituents on C and N atom respect-ively or vice-versa have far less activities while thefourth group (compound VIII) having aryl substitu-ents on both the atoms of the azomethine group isleast reactive.
The above results show that the substituents oneither C or N atoms of > C = N - group decreasethe catalytic activities of the complexes and that thearyl substituents are far more effective in this re-spect. The structures of the complexes Pd2L2X2 sug-gest planarity of the chelate ring with the conse-quent electron delocalization on it. Any change inelectron density caused by the nature of the substi-tuent at the C or N atom of the > C = N - group willbe partially distributed in the chelate ring. The effectof substitutions on the electron density and stericcrowding around the metal atom appears inappreci-able and only its involvement in the rate determin-ing step should not influence the catalytic activitiesof the complexes to such a great extent as observedexperimentally. This is, however, possible if the
> C = N - group itself is directly involved in therate determining step. The substitutions on C and Natoms will stabilize the > C = N - group by sterical-ly hindering the attack of H2 on it and the aryl sub-stituents will further stabilize the azomethine groupby delocalizing the latter's rr-electron density ontheir own rings. Thus the complex catalysts contain-ing more stable > C = N - group are expected to beless efficient as found experimentally.
The polarographic reductions of the schiff base li-gands ill, the' corresponding secondary aminesill3, and their corresponding orthopalladated com-plexes were studied in DMF solution in the range of- 0.1 V to - 1.7 V. The studies were made using te-traethyl ammonium iodide as the supporting elec-trolyte and the potentials are referred to the usualsaturated colomel electrode. Each of the schiff baseligands and its corresponding complex showed twowell defined reduction steps each involving oneelectron. The linearity of the plot of E, versus log( if(id - i) in each case indicated the reversibility of theprocess. The E 1/2 and n values and the shape of thepolarograrns of the ligand and the correspondingcomplex are almost identical. (E 1(2)1 value of the li-gands and the complexes lie within the range - 0.49V to - 0.54 V while the corresponding (E 1d~va-lues fall in the range -0.715 V to -0.73 V. Thecorresponding reduced ligands Ll-I, and their com-plexes do not exhibit any reduction wave in the giv-en range. The results suggest that the E 1/2 and n va-lues obtained both for the schiff base ligand and itscomplex are actually the reduction values of the li-gands only. The schiff base ligand in the complex,therefore, undergoes an overall 2-electron reversi-ble reduction which may be represented as follows(Eq.3):
Kinetic studies and mechanismKinetic studies using the two most active com-
plexes as catalytic system and nitrobenzene and sty-rene as substrates have been made under normalpressure at 25°C. Studies under high pressure con-ditions were carried out using the same two catalyticsystems and CH3N02 as substrate at 110°C. Therate was determined by analysis of the product com-position at different time intervals and the initialrates were determined from graphical extrapolationof the rate curve to zero time.
MUKHERJEE et al.: CATALYTIC HYDROGENATION USING SCHIFF BASE COMPLEXES OF Pd(II) 249
Variation of catalyst concentrationKinetic studies revealed that the initial rates of hy-
drogenation of nitrobenzene and styrene were firstorder dependent on [catalyst].
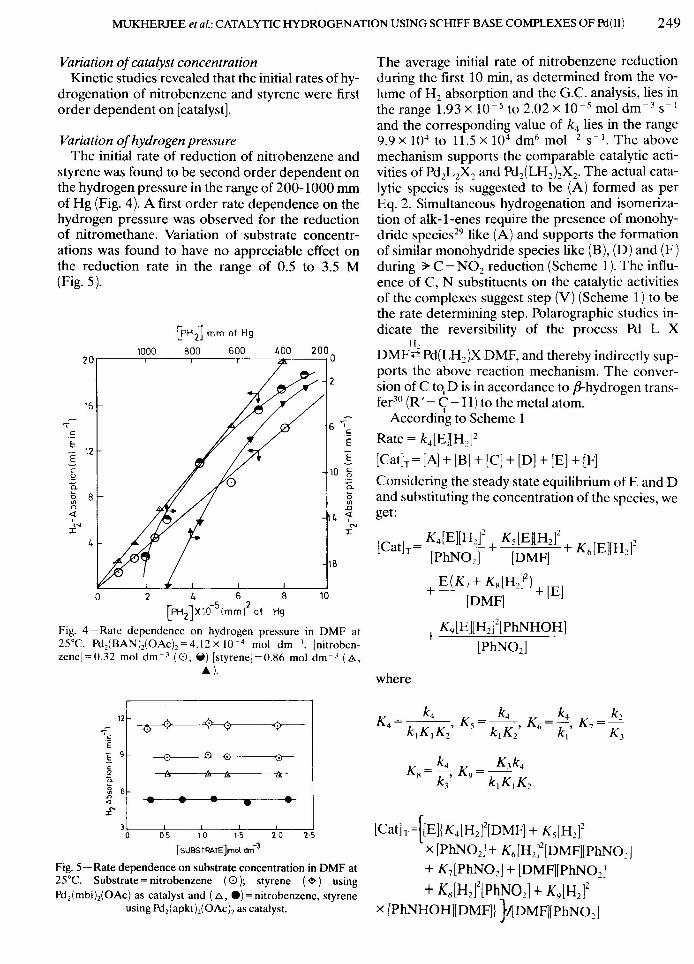
Variation of hydrogen pressureThe initial rate of reduction of nitrobenzene and
styrene was found to be second order dependent onthe hydrogen pressure in the range of 200-1000 mmof Hg (Fig. 4). A first order rate dependence on thehydrogen pressure was observed for the reductionof nitro methane. Variation of substrate concentr-ations was found to have no appreciable effect onthe reduction rate in the range of 0.5 to 3.5 M(Fig. 5).
l£'H21mm of Hg1000 800 600 GOO 200020r-----,-----,-----,---~~--~
15
cEE 12
6 .-sEE
1090.o<Il.0
4 ~N
I
4
18
a 2 4 6 8 10~ J -5 2LPH2 Xl0 Imml of Hg
Fig. 4-Rate dependence on hydrogen pressure in DMF at25°C. Pd2(BAN}z(OAc)2=4.12x 10-4 mol dm-J• [nitroben-zeneJ=O.32 mol dm-J (O, il) [styreneJ=O.H6 mol dm-J (A,
A).
12
~ 6~ ~..---.----..--~.~--~.~£'
30L--~OL5----1~O--~15~--~2~O--~25
[SUBSTRATE]\mol dm-3
Fig. 5 - Rate dependence on substrate concentration in DMF at25°C. Substrate = nitrobenzene (0); styrene ( ~ ) usingPd2(mbi}z(OAc) as catalyst and (8, e) = nitrobenzene, styrene
using Pd2(apkt}z(OAc)2 as catalyst.
The average initial rate of nitrobenzene reductionduring the first 10 min, as determined from the vo-lume of H2 absorption and the G.c. analysis, lies inthe range 1.93 x 10-5 to 2.02 X 10-5 mol dm -3 S-I
and the corresponding value of k4 lies in the range9.9x104 to 11.5x 104 dm" mol? S-I. The abovemechanism supports the comparable catalytic acti-vities of Pd2L2X2and Pd2(LH2lzX2.The actual cata-lytic species is suggested to be (A) formed as perEq. 2. Simultaneous hydrogenation and isomeriza-tion of alk-J-enes require the presence of monohy-dride species-? like (A) and supports the formationof similar monohydride species like (B), (D) and (F)during ;;..C - N02 reduction (Scheme 1). The influ-ence of C, N substituents on the catalytic activitiesof the complexes suggest step (V) (Scheme 1) to bethe rate determining step. Polarographic studies in-dicate the reversibility of the process Pd L X
H,
DMF~Pd(LH2)X DMF, and thereby indirectly sup-ports the above reaction mechanism. The conver-sion of C to,D is in accordance to p-hydrogen trans-fer30 (R' - C - H) to the metal atom.
According to Scheme 1Rate = k4[E][H2F[Catj-> [A]+ [B]+ [C] + [D] + [E] + [F]Considering the steady state equilibrium of E and Dand substituting the concentration of the species, weget:
K [E][H]2 K [E][H ]2[Cat] = 4 2 + 5 2 + K [E][H f
T [PhN02] [DMF] (, :;
E(K7 + KH[H2f) [ ]+ +E[DMF]
K9[E][H2]2[PhNHOH]+~-'------'~~'--------=-~[PhN02]
where
[cath=[[EHK4[H2]2[DMF] + Ks[Hlx [PhN02] + K6[H2]2[DMF][PhNO:;]+ K7[PhN02] + [DMF][PhN02]
+ KH[HJ[PhN02J + K9[H2J2
x [PhNHOH][DMFll }I[DMF][PhN02]
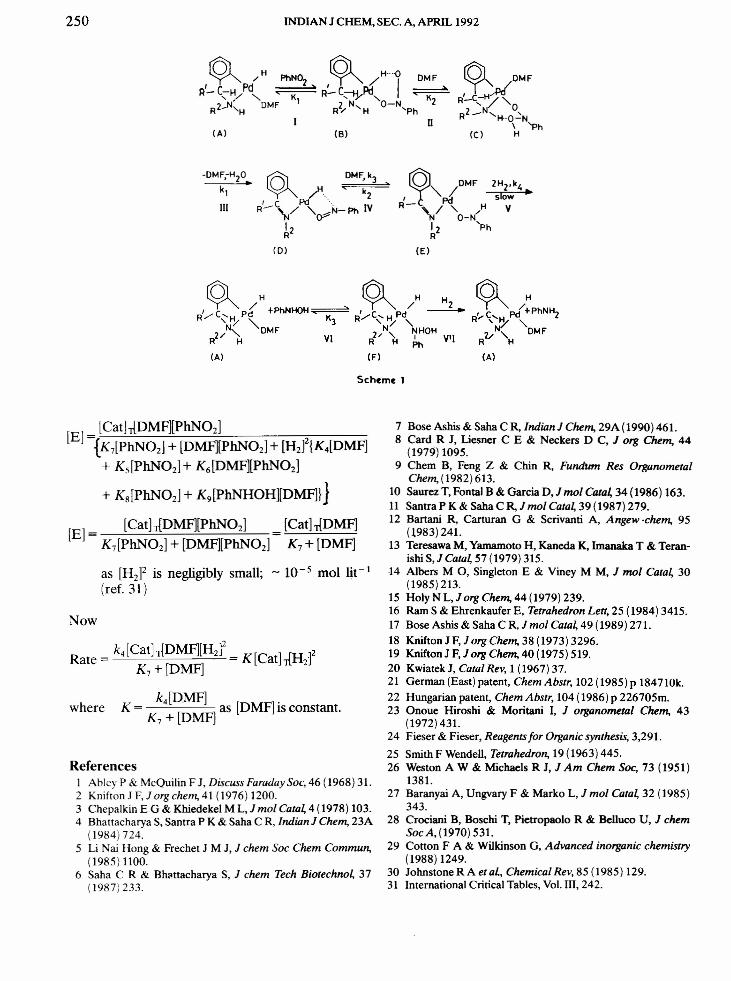
250 INDIAN J CHEM, SEe. A, APRIL 1992
~ /H PhN02
~ /H"-o DMF
R'-C-HyPd < > R~C--t..I_Pd I ~
...• "K ,'r <, K2 N' DMF 1 2 N O-N 2
R - 'H R" <, H 'Ph
(A) (6)
-DMF,-H20 •.
kl
III
u
[Cat].JDMF][PhNOz][E] ={K7[PhN02] + [DMF][PhN02] + [Hi{K4[DMF]
+ KS[PhN02] + K6[DMF][PhN02]
+ KS[PhN02] + K9[PhNHOH][DMF]}j
[E] = [Cat].JDMF][PhN02] [Cat).JDMF]K7[PhNOZ] + [DMF)[PhN02) K7 + [DMF]
as [H2F is negligibly small; - 10 - 5 mol lit - I
(ref. 31)
Now
Rate = k4[Cat].JDMF][H2]2 = K[Cat].JH fK7+[DMF) 2
k4[DMF]K = [ ] as [DMF) is constant.
K7+ DMFwhere
ReferencesI Abley P & McQuilin F J, Discuss Faraday Soc, 46 (1968) 31.2 Knifton J F, ] org chem; 41 (1976) 1200.3 Chepalkin E G & Khiedekel M L,] mol Catal; 4 (1978) 103.4 Bhattacharya S, Santra P K & Saba C R, Indian] Chern, 23A
(1984) 724.5 Li Nai Hong & Frechet J M J,] chem Soc Chem Commun,
(1985) llOO.6 Saba C R & Bhattacharya S, ] chem Tech Biotechnol; 37
(1987) 233.
Scheme 1
7 Bose Ashis & Saba C R, Indian] Chern, 29A(1990) 461.8 Card R J, Liesner C E & Neckers D C, J org Chern, 44
(1979) 1095.9 Chern B, Feng Z & Chin R, Fundum Res Organometal
Chern, (1982) 613.10 Saurez T, Fonta! B & Garcia D, J mol Casal; 34 (1986) 163.11 SantraP K & Saba C R, J mol Catal; 39 (1987) 279.12 Bartani R, Carturan G & Scrivanti A, Angew -chem; 95
(l983) 241.13 Teresawa M, Yamamoto H, Kaneda K, Imanaka T & Teran-
ishi S, J Catal; 57 (1979) 315.-l4 Albers M 0, Singleton E & Viney M M, J mol Catal; 30
(1985)213.15 Holy N L, J org Chern, 44 (1979) 239.16 Ram S & Ehrenkaufer E, Tetrahedron Lett, 25 (1984) 3415.17 Bose Ashis & Saba C R, J mol Cata~ 49 (1989) 271.18 KniftonJ F, J org Chern, 38 (1973) 3296.19 KniftonJ F,J org Chern, 40 (1975) 519.20 KwiatekJ, CatalRev, 1 (1967) 37.21 German (East) patent, Chem Abso; 102 (1985) P 18471Ok.22 Hungarian patent, ChemAbstr, 104 (1986) p 226705rn.23 Onoue Hiroshi & Moritani I, J organometal Chern, 43
(1972)431.24 Fieser & Fieser, Reagents for Organic synthesis, 3,291.25 Smith F Wendell, Tetrahedron, 19 (1963) 445.26 Weston A W & Michaels R J, JAm Chem Soc, 73 (1951)
1381.27 Baranyai A; Ungvary F & Marko L, J mol Catal; 32 (1985)
343.28 Crociani B, Boschi T, Pietropaolo R & Belluco U, J chem
Sac A, (1970) 531.29 Cotton F A & Wilkinson G, Advanced inorganic chemistry
(1988) 1249.30 lohnstoneRA et al; Chemical Rev, 85 (1985) 129.31 International Critical Tables, Vol. III, 242.