high-performance computing for reconstructing phylogenies from gene-order data david a. bader...
Post on 21-Dec-2015
216 views
TRANSCRIPT

High-Performance Computing forReconstructing Phylogenies from
Gene-Order Data
David A. BaderElectrical & Computer Engineering
University of New Mexico
http://hpc.eece.unm.edu/

11 November 2001High-Performance for Phylogeny Reconstruction, David A. Bader2
Computational Biology Special Interest Group, HPC 2001
Acknowledgment of Support
• National Science Foundation • CAREER: High-Performance Algorithms for Scientific
Applications (00-93039)• ITR: Algorithms for Irregular Discrete Computations on
SMPs (00-81404)• DEB: Ecosystem Studies: Self-Organization of Semi-Arid
Landscapes: Test of Optimality Principles (99-10123)• ITR/AP: Reconstructing Complex Evolutionary Histories
(01-21377)• DEB Comparative Chloroplast Genomics: Integrating
Computational Methods, Molecular Evolution, and Phylogeny (01-20709)
• ITR/AP(DEB): Computing Optimal Phylogenetic Trees under Genome Rearrangement Metrics (01-13095)
• PACI: NCSA/Alliance, NPACI/SDSC, PSC• Sun Microsystems

11 November 2001High-Performance for Phylogeny Reconstruction, David A. Bader3
Computational Biology Special Interest Group, HPC 2001
Algorithms that Scale from the Blade to the Fire

11 November 2001High-Performance for Phylogeny Reconstruction, David A. Bader4
Computational Biology Special Interest Group, HPC 2001
Commercial Aspects of Phylogeny Reconstruction
• Identification of microorganisms• public health entomology• sequence motifs for groups are patented• example: differentiating tuberculosis strains
• Dynamics of microbial communities• pesticide exposure: identify and quantify microbes in soil
• Vaccine development• variants of a cell wall or protein coat component• porcine reproductive and respiratory syndrome virus isolates from US and Europe were separate
populations• HIV studied through DNA markers
• Biochemical pathways• antibacterials and herbicides
Glyphosate (Roundup, Rodeo , and Pondmaster ): first herbicide targeted at a pathway not present in mammals• phylogenetic distribution of a pathway is studied by the pharmaceutical industry before a drug is
developed
• Pharmaceutical industry• predicting the natural ligands for cell surface receptors which are potential drug targets• a single family, G protein coupled receptors (GPCRs), contains 40% of the targets of most pharm.
companies

11 November 2001High-Performance for Phylogeny Reconstruction, David A. Bader5
Computational Biology Special Interest Group, HPC 2001
GRAPPA: Genome Rearrangements Analysis
• Genome Rearrangements Analysis under Parsimony and other Phylogenetic Algorithms
• http://www.cs.unm.edu/~moret/GRAPPA/• Open-source• already used by other computational phylogeny groups,
Caprara, Pevzner, LANL, FBI, PharmCos.
• Gene-order Phylogeny Reconstruction• Breakpoint Median• Inversion Median
• over one-million fold speedup from previous codes• Parallelism
• Scales linearly with the number of processors
• Developed using Sun Forte C

11 November 2001High-Performance for Phylogeny Reconstruction, David A. Bader6
Computational Biology Special Interest Group, HPC 2001
Molecular Data for Phylogeny
• simple DNA sequence: nucleotides• low-level functionality: amino acids, etc.• genomic level: genes• (next is functional level: proteomics, etc.)
Biologists now have full gene sequences for many single-chromosome organisms and organelles (e.g., mitochondria, chloroplasts) and for more and more larger organisms
11 November 2001High-Performance for Phylogeny Reconstruction, David A. Bader7
Computational Biology Special Interest Group, HPC 2001
Gene Order Phylogeny
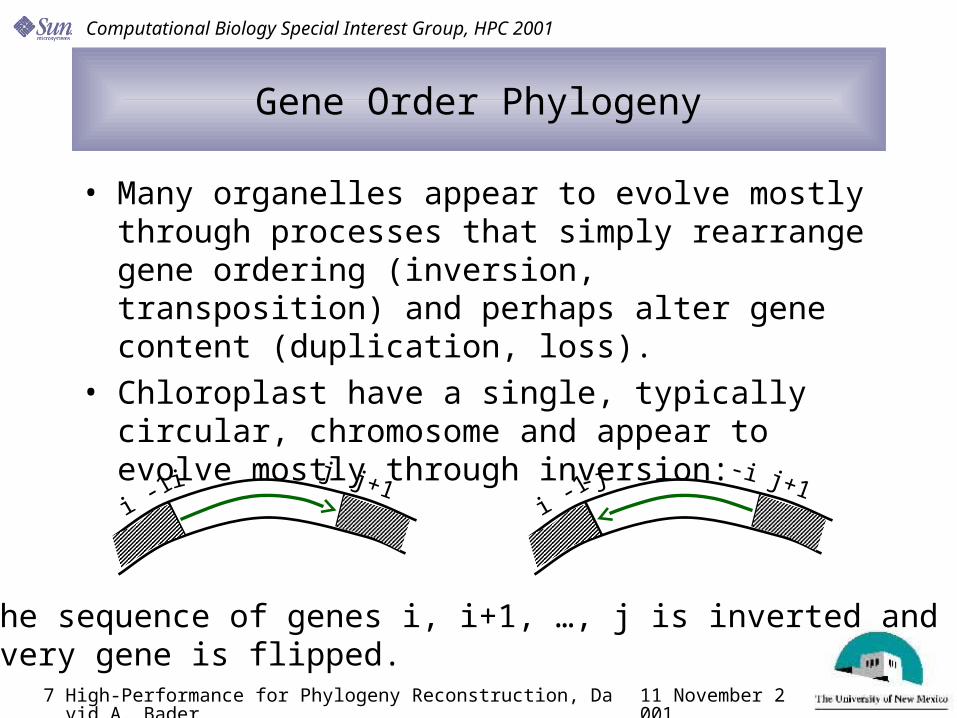
• Many organelles appear to evolve mostly through processes that simply rearrange gene ordering (inversion, transposition) and perhaps alter gene content (duplication, loss).
• Chloroplast have a single, typically circular, chromosome and appear to evolve mostly through inversion:
i -1i j j+1
i -1-j -i j+1
The sequence of genes i, i+1, …, j is inverted and every gene is flipped.

11 November 2001High-Performance for Phylogeny Reconstruction, David A. Bader8
Computational Biology Special Interest Group, HPC 2001
Gene Order Phylogeny (cont’d)
• The real problem• Reconstruct the “true” tree, identify the “true”
ancestral genomes, and recover on each edge the “true” sequence of evolutionary changes
• The optimization problem (parsimony)• Reconstruct a tree and ancestral genomes so as
to minimize the sum, over all tree edges, of the inferred evolutionary distance along each edge
• The surrogate problem• Do the optimization problem with a measure of
inferred evolutionary distance that lends itself to analysis

11 November 2001High-Performance for Phylogeny Reconstruction, David A. Bader9
Computational Biology Special Interest Group, HPC 2001
Breakpoint Analysis:A Surrogate for Gene Order
• Breakpoint:• an adjacent pair of genes present in one genome, but
absent in the other
• Breakpoint distance:• the total number of breakpoints between two genomes
(a true metric, similar to Hamming distance)
• Breakpoint phylogeny:• the tree and ancestral genomes that minimize the sum,
over all edges of the tree, of the breakpoint distances
Naturally, it is an NP-hard problem, even with just 3 leaves.

11 November 2001High-Performance for Phylogeny Reconstruction, David A. Bader10
Computational Biology Special Interest Group, HPC 2001
Breakpoint Analysis(Sankoff & Blanchette 1998)
• For each tree topology do• somehow assign initial genomes to the internal nodes• repeat
• for each internal node do
– compute a new genome that minimizes the distances to its three neighbors
– replace old genome by new if distance is reduced
• until no change
Sankoff & Blanchette implemented this in a C++ package
(2n-
5)!!
= (
2n-5
) (2
n-7)
…
5
3 t
rees
unkn
own
itera
tive
heur
istic
NP
-har
d

11 November 2001High-Performance for Phylogeny Reconstruction, David A. Bader11
Computational Biology Special Interest Group, HPC 2001
Algorithm Engineering Works!
• We reimplemented everything – • the original code is too slow and not as flexible as we
wanted.
• Our main dataset is a collection of chloroplast data from the flowering plant family Campanulaceae (bluebells):
• 13 genomes of 105 gene segments each
• On our old workstation:• BPAnalysis processes 10-12 trees/minute• Our implementation processes over 50,000 trees/minute
• Speedup ratio is over 5,000!!• On synthetic datasets, we see speedups from 300 to
over 50,000…

11 November 2001High-Performance for Phylogeny Reconstruction, David A. Bader12
Computational Biology Special Interest Group, HPC 2001
So… What did we do?!
• Absolutely no high-level algorithmic changes• Three low-level algorithmic changes:
• better bounding• strong upper bound initialization• “condensing”
• Completely different data representation• Two low-level algorithmic changes
• all memory is pre-allocated• some loops are hand-unrolled
• Written in C instead of C++
~10x
~10x
~ 6x
?(convenience)
*(~ 10x)*
* Well, so I lied just a little bit…

11 November 2001High-Performance for Phylogeny Reconstruction, David A. Bader13
Computational Biology Special Interest Group, HPC 2001
One high-level algorithmic change
• (Ok, so I lied a little…)• Avoid labeling the tree if possible
• Use current best score as an upper bound.• Compute lower bound & prune tree away if lower
bound > upper bound
• Lower bound:• Get circular ordering of leaves, x1 x2 … xn
• Compute = d(x1,x2) + d(x2,x3) + … + d(xn,x1)• Then ½ is a lower bound because
• d(.) obeys the triangle inequality• every tree edge is used twice in a tree-based version of

11 November 2001High-Performance for Phylogeny Reconstruction, David A. Bader14
Computational Biology Special Interest Group, HPC 2001
Tree
a
b c
d
e
= d(a,b) + d(b,c) + d(c,d) + d(d,e) + d(e,a)
a
b c
d
e
d(a,b)
d(b,c)d(c,d)
Tree Tree version (paths)
d(d,e)d(e,a)
(Same trick as in the “twice around the tree” approximation for the TSP with triangle inequality.)

11 November 2001High-Performance for Phylogeny Reconstruction, David A. Bader15
Computational Biology Special Interest Group, HPC 2001
Algorithmic Changes (~ 10x)
1. Better bounding: skip edges that would cause degree 3 or premature cycle
2. “Condensing”: whenever the same• gene subsequence appears in all genomes, it can be
condensed into a single “superfragment”• done as static processing and on the fly before each TSP
3. Initializing the new median with the best of the old one and its three neighbors.
• Condensing is very effective on real data within families, but easily defeated by large evolutionary distances.
• (1) and (3) cause over half of the TSP instances (for finding computing “median-of-three” updated internal nodes) to be pruned away instantly.

11 November 2001High-Performance for Phylogeny Reconstruction, David A. Bader16
Computational Biology Special Interest Group, HPC 2001
Data Representation (~ 10x)
• No distance matrix for reduction of “median-of-three” to Traveling Salesperson Problem (TSP): at most 4n edges can be of interest – the others are treated as an undifferentiated pool. The adjacency lists have length 4.
• thus, linear time at each step and reduced storage.• Backtracking search has a small list of edges and only
searches among edges of cost 1 and 2 (– and 0 are always included) – still NP-hard, but often easy
• When search runs out of edges, tour is completed in linear time from the pool of edges of cost 3
• Many auxiliary arrays (á la Fortran!) to carry information on flags, degrees, other end of chains, …

11 November 2001High-Performance for Phylogeny Reconstruction, David A. Bader17
Computational Biology Special Interest Group, HPC 2001
Low-Level Coding Changes (~ 6x)
1. All storage allocated at start, with large #s of pointers passed to subroutines (no globals, to allow parallel execution).
• Avoids malloc/free overhead; Improves cache locality
2. Avoid recomputations.• Use local variables for intermediate pointers• Hand unroll loops on adjacencies to preserve locality (and to
avoid “mod” operations with circular genomes)• Speeds up addressing – never deference! & Improves cache locality
BPAnalysis uses 65MB and has a real memory footprint of ~ 12MB on our real data
Our reimplementation uses 1.6MB with a footprint of 0.6MB
11 November 2001High-Performance for Phylogeny Reconstruction, David A. Bader18
Computational Biology Special Interest Group, HPC 2001
And… How did we do it?
• 3 strategies: Profile, Profile, Profile(and use your engineering sense/nose/… )
Sun Forte 6 Analyzer
• We began with 4 main culprits:• preparing adjacency lists for the TSP• computing breakpoint distances• computing lower bounds in TSP• backtracking in TSP
• Over 10 – 12 major iterations, each of which yielded a 1.5 – 2 fold speed-up, these four switched places over and over.
11 November 2001High-Performance for Phylogeny Reconstruction, David A. Bader19
Computational Biology Special Interest Group, HPC 2001
Profiling
• And our final tally (still on the Campanulaceae dataset) is:30% backtracking (excl. LB)
20% preparing adjacency lists
20% condensing & expanding
15% computing LB
8% computing distances
7% miscellaneous overhead
• (no obvious culprits left)

11 November 2001High-Performance for Phylogeny Reconstruction, David A. Bader20
Computational Biology Special Interest Group, HPC 2001
High-Performance Computing Techniques
• Availability of hundreds of powerful processors
• Standard parallel programming interfaces (Sun HPC) • Message passing interface (MPI)• OpenMP or POSIX threads
• Algorithmic libraries for SMP clusters• SIMPLE
• Goal: make efficient use of parallelism for• exploring candidate tree topologies• sharing of improved bounds

11 November 2001High-Performance for Phylogeny Reconstruction, David A. Bader21
Computational Biology Special Interest Group, HPC 2001
Parallelization of the Phylogeny Algorithm
• Enumerating tree topologies is pleasantly parallel and allows multiple processors to independently search the tree space with little or no overhead
• Improved bounds can be broadcast to other processors without interrupting work
• Load is evenly balanced when trees are cyclically assigned (e.g. in a round-robin fashion) to the processors
• Linear speedup

11 November 2001High-Performance for Phylogeny Reconstruction, David A. Bader22
Computational Biology Special Interest Group, HPC 2001
Final Remarks
• Our reimplementation led to numerous extensions as well as to new theoretical results
• GRAPPA has been extended to inversion phylogeny, with linear-time algorithms for inversion distance and a new approach to exact inversion median-of-three.
• Better bounding in the next version of GRAPPA yields two more orders of magnitude speedup.
• These insights and improvements are made possible by mature development tools (Forte)
• Algorithmic engineering techniques are widely applicable
• We may not always get 6 orders of magnitude, but 3 – 4 orders should be nearly routine with most codes. (We are starting work on TBR and exact parsimony solvers.)

11 November 2001High-Performance for Phylogeny Reconstruction, David A. Bader23
Computational Biology Special Interest Group, HPC 2001
Final Remarks (cont’d)
• High-performance implementations enable:• better approximations for difficult problems (MP,
ML)• true optimization for larger instances• realistic data exploration (e.g., testing evolutionary
scenarios, assessing answers obtained through other means, etc.)
• Our analysis of the Campanulaceae dataset confirmed the conjecture of Robert Jansen et al. – that inversion is the principal process of genome evolution in cpDNA for this group.

11 November 2001High-Performance for Phylogeny Reconstruction, David A. Bader24
Computational Biology Special Interest Group, HPC 2001
Work-In-Progress and Future Work
• Tree enumeration using circular ordering• Handle unequal gene content and duplicate
genes using exemplars• Parallel branch and bound techniques
(optimized for Sun HPC Servers) for searching tree space
• Improved SPR and TBR techniques (local searches around good trees)
• Exact Algorithm for Maximum Parsimony

11 November 2001High-Performance for Phylogeny Reconstruction, David A. Bader25
Computational Biology Special Interest Group, HPC 2001
Recent publications (2001)
• A New Implementation and Detailed Study of Breakpoint Analysis, B.M.E. Moret, S. Wyman, D.A. Bader, T. Warnow, M. Yan, Sixth Pacific Symposium on Biocomputing 2001, pp. 583-594, Hawaii, January 2001.
• High-Performance Algorithm Engineering for Gene-Order Phylogenies, D.A. Bader, B. M.E. Moret, T. Warnow, S.K. Wyman, and M. Yan, DIMACS Workshop on Whole Genome Comparison, DIMACS Center, Rutgers University, Piscataway, NJ, March 2001.
• Variation in vegetation growth rates: Implications for the evolution of semi-arid landscapes , C. Restrepo, B.T. Milne, D. Bader, W. Pockman, and A. Kerkhoff, 16th Annual Symposium of the US-International Association of Landscape Ecology, Arizona State University, Tempe, April 2001.
• High-Performance Algorithm Engineering for Computational Phylogeny, B. M.E. Moret, D.A. Bader, and T. Warnow, 2001 International Conference on Computational Science, San Francisco, CA, May 2001.
• Cluster Computing: Applications, David A. Bader and Robert Pennington, The International Journal of High Performance Computing, 15(2):181-185, May 2001.
• New approaches for using gene order data in phylogeny reconstruction, R.K. Jansen, D.A. Bader, B. M. E. Moret, L.A. Raubeson, L.-S. Wang, T. Warnow, and S. Wyman. Botany 2001, Albuquerque, NM, August 2001.
• GRAPPA: a high-performance computational tool for phylogeny reconstruction from gene-order data , B. M.E. Moret, D.A. Bader, T. Warnow, S.K. Wyman, and M. Yan. Botany 2001, Albuquerque, NM, August 2001.
• Inferring phylogenies of photosynthetic organisms from chloroplast gene orders , L.A. Raubeson, D.A. Bader, B. M.E. Moret, L.-S. Wang, T. Warnow, and S.K. Wyman. Botany 2001, Albuquerque, NM, August 2001.
• Industrial Applications of High-Performance Computing for Phylogeny Reconstruction, D.A. Bader, B. M.E. Moret, and L. Vawter, SPIE ITCom: Commercial Applications for High-Performance Computing, Denver, CO, SPIE Vol. 4528, pp. 159-168, August 2001.
• Using PRAM Algorithms on a Uniform-Memory-Access Shared-Memory Architecture, D.A. Bader, A. Illendula, B. M.E. Moret, and N.R. Weisse-Bernstein, Fifth Workshop on Algorithm Engineering, Springer-Verlag LNCS 2141, 129-144, Aarhus, Denmark, August 2001.
• A Linear-Time Algorithm for Computing Inversion Distance Between Two Signed Permutations with an Experimental Study, D.A. Bader, B. M.E. Moret, and M. Yan, Journal of Computational Biology, 8(5):483-491, October 2001.