heme oxygenase paper
TRANSCRIPT

1
Formation and Decay of Hydroperoxo-Ferric Heme Complex in Horseradish Peroxidase Studied by Cryoradiolysis.
Ilia G. Denisov#,*,1, Thomas M. Makris+, Stephen G. Sligar#,+,*
Department of Biochemistry#, Center for Biophysics and Computational Biology+, Beckman
Institute*, University of Illinois, Urbana-Champaign, IL, 61801.
Keywords: Horseradish peroxidase; hydroperoxo-ferric intermediate; peroxo-ferric intermediate;
radiolytic reduction; Compound I; EPR spectra; UV/Vis spectra
Running Title: Hydroperoxo-ferric HRP
Correspondence should be addressed to:
Dr. Stephen G. Sligar
University of Illinois
Department of Biochemistry
505 S. Goodwin Avenue
Urbana, IL, 61801
E-mail: [email protected]
Phone: 217-244-9872
Fax: 217-265-4073
Copyright 2002 by The American Society for Biochemistry and Molecular Biology, Inc.
JBC Papers in Press. Published on September 4, 2002 as Manuscript M207949200 by guest on A
pril 5, 2018http://w
ww
.jbc.org/D
ownloaded from

2
SUMMARY
Using radiolytic reduction of the oxy-ferrous horseradish peroxidase (HRP) at 77 K, we observed
the formation and decay of the putative intermediate, the hydroperoxo-ferric heme complex, often
called “Compound 0”. This intermediate is common for several different enzyme systems as the
precursor of the “Compound I” (ferryl-oxo π-cation radical) intermediate. EPR and UV/Vis
absorption spectra show that protonation of the primary intermediate of radiolytic reduction, the
peroxo-ferric complex, to form the hydroperoxo-ferric complex is completed only after annealing
at temperatures 150 – 180 K. After further annealing at 195 - 205 K, this complex directly
transforms to ferric HRP, without any observable intervening species. The lack of Compound I
formation is explained by inability of the enzyme to deliver the second proton to the distal oxygen
atom of hydroperoxide ligand, shown to be necessary for dioxygen bond heterolysis on the
“oxidase pathway”, which is non-physiological for HRP. Alternatively, the physiological substrate
H2O2 brings both protons to the active site of HRP, and Compound I is subsequently formed via
rearrangement of the proton from the proximal to the distal oxygen atom of the bound peroxide.
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

3
INTRODUCTION
Horseradish Peroxidase (HRP)2 is a highly characterized heme enzyme, which has historically
served as a paradigm for evaluating the rules of formation of redox active heme-oxygen
intermediates, including the ferryl-oxo heme complexes commonly known as “Compound I” and
“Compound II.” A detailed mechanism for Compound I formation in the reaction of H2O2 with
peroxidases was formulated by Poulos and Kraut on the basis of structural analysis of cytochrome c
peroxidase active center (1), and has been successfully applied to other enzymes (2-5):
The essential features of this mechanism also provide a framework for the analysis of new systems,
which involve formation of redox-active heme-oxygen intermediates (6-8). A crucial point in the
formation of the ferryl-oxo species is the second protonation of the distal oxygen atom of the bound
hydroperoxo species, followed by the heterolytic scission of O-O bond. The importance of these
elementary steps has been confirmed by quantum chemical methods (9-11). A vast amount of
kinetic data obtained in mutagenesis studies of the distal pocket (5,12-15), provide an additional
!"#$%
&'(%
&'(%)*+
!"#$%
&'(%
&'(%)*+
!",$%
&'(%
&'(%)*+
-%-%
&%
&$%
-%
$&.-%
$/%
$&.-.%
!"#$%&'(#$)$"( *+,-./"-.0.12"--%3(3.4/5"0( 6.4/.7&,(8(
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

4
support for the evaluation of the second protonation of the distal oxygen as the most important, and
in some cases, the rate-limiting step in catalysis.
Despite many efforts, experimental characterization of this step, as well as the detailed properties of
the hydroperoxo-ferric heme (Fe3+P-OOH-) complex in peroxidases, is still far from complete. One
reason is due to the low stability of such complexes (16), as their transient character leads to a lack
of accumulation during the reaction course. Several studies of these intermediates in wild-type and
mutant HRP (17-19) and microperoxidase (20-22) by means of stopped-flow methods have
produced absorption spectra that could be tentatively assigned to Fe3+P-OOH- (hydroperoxo- anion)
or Fe3+P- H2O2 (neutral hydrogen peroxide) complex via comparison with theoretical estimates
(23). Even so, we are aware of yet only one successful isolation of an (Fe3+P-OOH-) intermediate in
an H2O2 driven heme protein system, which was obtained using a distal pocket mutant of Mb (24).
The difficulties in the kinetic resolution of the (Fe3+P-OOH-) intermediate on this pathway may be
caused by the fact that both protons necessary for the Compound I formation are brought into the
active site of the enzyme with the H2O2 molecule itself. Apparently, the subsequent 1-2 proton shift
is fast and not rate limiting under most conditions (5,12-15,19).
An alternative way of generating (Fe3+P-OOH-) intermediates with high yield is the cryoradiolytic
reduction of the corresponding oxy-ferrous complexes using ionizing radiation (25,26). This
method mimics the oxidase/oxygenase pathway of oxygen activation, which utilizes molecular
oxygen instead of H2O2, and includes the reduction of the bound oxygen and delivery of two
external protons to form Compound I (6,27). As a result of the one-electron reduction of oxy-
ferrous precursors at 77 K, peroxo/hydroperoxo- ferric heme enzyme complexes can be stabilized
at low temperatures (26,28-30). The (Fe3+P-OOH-) intermediates of cytochromes P450 (31-34),
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

5
heme oxygenase (35), Mb (26,29,30), and Hb (25,28) have been isolated and characterized using
EPR and optical absorption spectroscopy. Several X-ray structures of intermediates produced by
radiolytic reduction of the crystals of corresponding oxy-ferrous precursors, including Compound
III in HRP (36), are also available (36-38), although none of these studies was able to detect the
(Fe3+P-OOH-) complex. Earlier investigations using cryoradiolysis (30), or pulse radiolysis at
ambient conditions (39) for reduction of oxy-ferrous HRP, have not provided unambiguous
assignment of observed intermediate states. Detailed studies of the reductive pathway of oxygen
activation in HRP, where the associated Compound I state is relatively stable, could provide critical
input into the mechanism for oxygen-oxygen bond scission and the requirements for specific distal
pocket residues in generating higher valent metal – oxo complexes thought to be operating in the
heme-thiolate oxygenases such as cytochrome P450. To investigate this path, we used the
cryogenic radiolytic reduction of oxy-ferrous HRP to prepare the peroxo/hydroperoxo ferric
intermediate and monitor its protonation and possible Compound I formation, using EPR and
optical absorption spectroscopy. As a reference point, we also document the cryoradiolytic
reduction of Compound I, prepared using the classic reaction of the ferric enzyme with hydrogen
peroxide.
METHODS
Ferrous HRP was prepared by anaerobic reduction of the ferric HRP (Type XII, Sigma) with
dithionite and subsequent anaerobic chromatography on a Sephadex G25 column (Pharmacia) to
remove the excess dithionite and decomposition products. The enzyme solution was mixed with
buffered deoxygenated glycerol (GC grade, Sigma) to give a final concentration of 25 - 50 µM of
HRP in 70% (v/v) glycerol and 0.1 M phosphate-citrate buffer, pH 6.2, oxygenated at 270 K by
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

6
bubbling air and stirring for 30 s, and cooled to 77 K, as described (33,34). All samples for optical
spectroscopy were prepared in UV-enhanced methacrylate semimicro spectroscopic cells (Fisher
Scientific, Pittsburgh, PA) and used with 4.3 mm path. The ferric HRP samples used for the control
were prepared in the same fashion, cooled to 77 K and irradiated together with other samples.
Compound I and Compound II were prepared according to published procedures (3, and references
therein). Compound I was obtained by manual mixing 70% glycerol/buffer solutions of the ferric
HRP and 1.2 molar excess of H2O2 at low temperature (260 - 270 K). After mixing for 20 - 30 sec,
the sample was placed into a thermostat and quickly cooled to 200 K. The sample was then slowly
cooled to 77K, utilizing UV/Vis spectroscopy to monitor the purity of the preparation. Compound
II was prepared by mixing of 75% glycerol/buffer solutions of the ferric HRP, containing fourfold
molar excess of the substrate, 3,5-dimethylphenol, with a fourfold molar excess of H2O2 in the
similar conditions. The spectra of Compound I and Compound II showed weak temperature
dependence and were similar to those reported in the literature (40,41).
UV/Vis spectra were measured on a Cary 3 spectrophotometer (Varian, Australia) in an unsilvered
dewar (33) using liquid nitrogen as a cooling agent. ESR spectra were obtained at the University of
Illinois EPR Resource Center on a Varian E-122 X-Band (9.08 GHz) spectrometer with a protein
concentration of 0.4 mM. Typical conditions include a microwave power of 0.5mW with
modulation amplitude of 12.5 Gauss at 100 kHz. An Air Products (Allentown, PA) liquid helium
flow system was used for measurements at 4 to 20K. Samples for EPR were prepared in a similar
manner as those for UV/Vis experiments, with a final glycerol concentration of 20% (v/v).
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

7
The radiolytic reduction of the enzyme and reference samples was achieved by exposing to gamma-
radiation from a 60Co source (dose rate 11 kGy/hour, for 4 or 8 hours) (Notre Dame Radiation Lab,
Notre Dame University, South Bend, IN). The samples were fully immersed in liquid nitrogen
during irradiation. Background absorption due to the radiolysis products was subtracted as
described (33) using irradiated buffer/glycerol reference samples. These samples were irradiated at
77 K together with the protein samples, and 40 absorption spectra were taken at different
temperatures from 80 K – 240 K. The background spectra for any arbitrary temperature in this
range were then calculated using linear interpolation.
RESULTS AND DISCUSSION
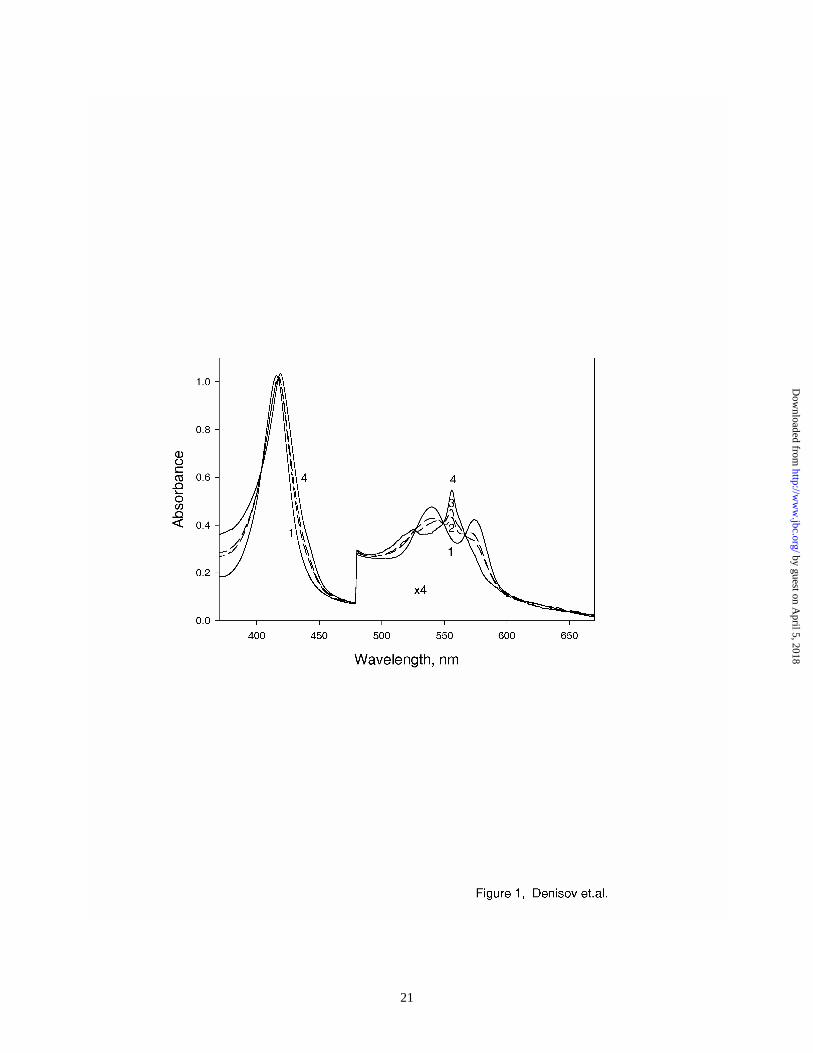
The spectra of oxy-HRP before and after irradiation at 77 K are shown in Figure 1. The radiolytic
reduction of the oxy-ferrous heme complex in HRP results in formation of a primary intermediate,
peroxo/hydroperoxo - ferric intermediate, as has been observed with other heme proteins (26,29-
35,42,43). The yield of this intermediate (35% - 50%) was estimated from the dose dependent
absorption spectra, using the spectra of oxy-HRP as the reference. The spectrum of the pure (Fe3+P-
OOH-) HRP complex, calculated from multiple experiments with different yields, is also shown in
Figure 1. The absorption spectrum of this intermediate has a red-shifted Soret band (maximum at
419 nm, compared to 416 nm for oxy-ferrous HRP) and blue-shifted Q-bands (556 and 526 nm).
This spectrum is in a very good agreement with the spectrum of the (Fe3+P-OOH-) complex of
heme oxygenase3 , and with the kinetically resolved spectrum of the same intermediate obtained in
stopped-flow studies of the reaction of Mb mutant H64Q with H2O2 (24). This intermediate of the
reaction of H2O2 with heme enzymes is very unstable at ambient conditions, and previously was
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

8
characterized only via decomposition of transient absorption spectra in stopped-flow studies or via
freeze-quench techniques (12 – 21, 24). The same (Fe3+P-OOH-) complex is firmly identified as an
intermediate of the reductive activation of the bound dioxygen in heme proteins, as demonstrated
by cryoradiolytic reduction studies (26,29,30,32-35) and quantum chemical calculations (10,11,23).
Unfortunately, it was impossible to resolve the spectra of (Fe3+P-OOH-) complex at wavelengths
shorter than 360 nm due to the high absorption of the products of radiolysis of glycerol and the
protein matrix. Thus, we cannot confirm the true hyperporphyrin shape of the Soret band of
(Fe3+P-OOH-) complex, as calculated by Harris and Loew (23) and often used as a criterion to
distinguish between the neutral peroxide – ferric (Fe3+P-HOOH) and hydroperoxo-anion – ferric
(Fe3+P-OOH-) complexes based on their absorption spectra in the Soret region. (19,44).
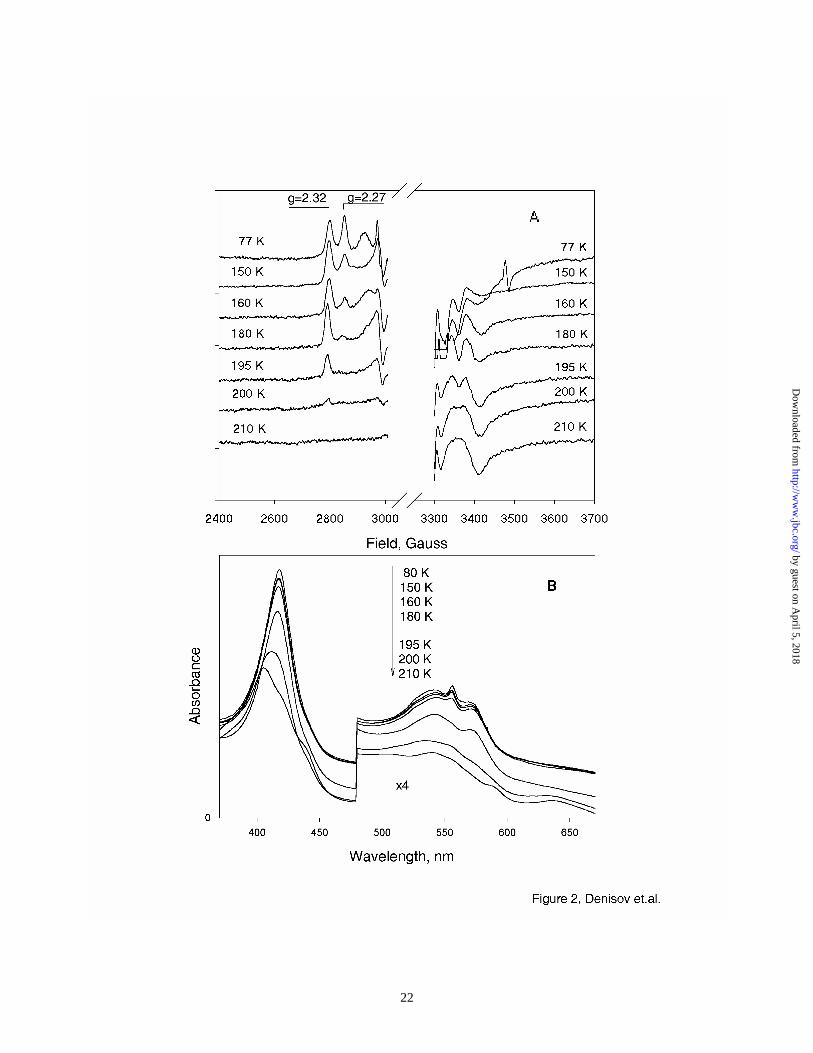
Figure 2 shows the annealing of the (Fe3+P-OOH-) complex monitored by EPR and UV/Vis spectra.
In the EPR spectra, the presence of two species in the primary intermediate is clearly resolved.
Based on the analogous studies of other heme proteins (25,29,42,43), these are assigned as the
unprotonated peroxo-ferric heme complex (Fe3+P-OO2-, g3=2.27), and the protonated, hydroperoxo-
ferric heme complex (Fe3+P-OOH-, g3=2.32). Gradual annealing at 140 – 160 K results in
disappearance of the feature at g3=2.27 and increase at g3=2.32, as the first protonation of peroxo-
ferric complex and formation of hydroperoxo- intermediate is completed. These results are in
agreement with EPR spectra obtained using cryoradiolytic reduction studies of oxy-HRP in 50%
ethylene glycol (30).
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

9
The analysis of absorption spectra of the similar samples measured at 90K – 160 K does not reveal
any significant changes that can be assigned to the protonation of peroxo-ferric intermediate. Thus,
at least under the conditions of our experiments, the optical spectra in this region are not sensitive
enough to distinguish clearly between the unprotonated and protonated peroxo/hydroperoxo ferric
heme complex of HRP. Recently we obtained EPR and UV/Vis spectra of peroxo/hydroperoxo
intermediates of the distal mutants of cytochrome P450 CYP101, in which the first protonation of a
peroxo-ferric heme complex could be detected in optical absorption spectra as a three nanometer
blue shift of Soret, and a similar blue shift of the Qα band from 561 nm to 558 nm4. However, the
relative spectral differences between oxy-ferrous and peroxo/hydroperoxo- ferric heme complexes
in the Soret region are weaker in HRP and in heme oxygenase than in P450 enzymes (33,34). This
difference, which is caused by the different proximal ligands in these enzymes,
imidazole/imidazolate in HRP and heme oxygenase versus thiolate in P450, also results in different
sensitivity of the optical absorption spectra to the protonation of the distal peroxo- ligand.
Further annealing by increasing the sample temperature from 180K to 210K shows the decay of
(Fe3+P-OOH-) complex with no evidence for the formation of a Compound I intermediate. EPR
spectra of the samples annealed at 195K, 200K, and 210K, show gradual decrease of the signal
from (Fe3+P-OOH-) complex and formation of the ferric HRP with no other observable
intermediates. This result is consistent with two scenarios. One would involve the transient
formation of Compound I, which is quickly reduced to generate the EPR-silent Compound II, and
after the second reduction returns to the ferric enzyme. Another possibility is the direct decay of
(Fe3+P-OOH-) intermediate through dissociation of the ligand and return to the ferric resting state of
HRP. Because EPR-silent species may be involved in the evolution of this system, EPR
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

10
spectroscopy does not provide complete characterization of the (Fe3+P-OOH-) fate in HRP. Thus,
the optical absorption spectra of the similar samples were studied, and the reference experiments
using cryoradiolytic reduction of Compound I were performed to assign the annealing pathway of
(Fe3+P-OOH-) complex.
Figure 2B shows the absorption spectra of the radiolytically reduced oxy-ferrous HRP measured at
190 – 210 K. At temperatures above the glass transition of the solvent matrix, the hydroperoxo-
ferric complex gradually disappears, as clearly seen by simultaneous decrease of the sharp
maximum at 557 nm and of the Soret band at 420 nm. No increase of absorption at 650 nm
characteristic for Compound I porphyrin π-cation radical was detected in annealing experiments.
On the contrary, the bands at 407 nm and 635 nm are observed, corresponding to the spectrum of
the resting form of HRP, possibly perturbed by the presence of the dissociated hydroperoxo-anion/
hydrogen peroxide at the distal pocket of the enzyme. At higher temperatures this spectrum, as well
as the spectrum of the remaining oxy-HRP disappears, and the characteristic spectrum of the
carbonmonoxy-ferrous complex of HRP emerges with Soret band at 421 nm and Q bands at 540
nm and 572 nm (not shown). The CO adduct is the result of the nonspecific reduction of the heme
enzyme by the products of radiolysis, and binding of CO, which is one of the products of glycerol
radiolysis (45), and is always observed after thawing of irradiated frozen solutions of the heme
enzymes (33,34,46-48). In a control experiment, the dissolution of nonirradiated metMb into a
glycerol/buffer reference sample, which was irradiated at 77 K and thawed, resulted in formation of
fully reduced Mb-CO.
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

11
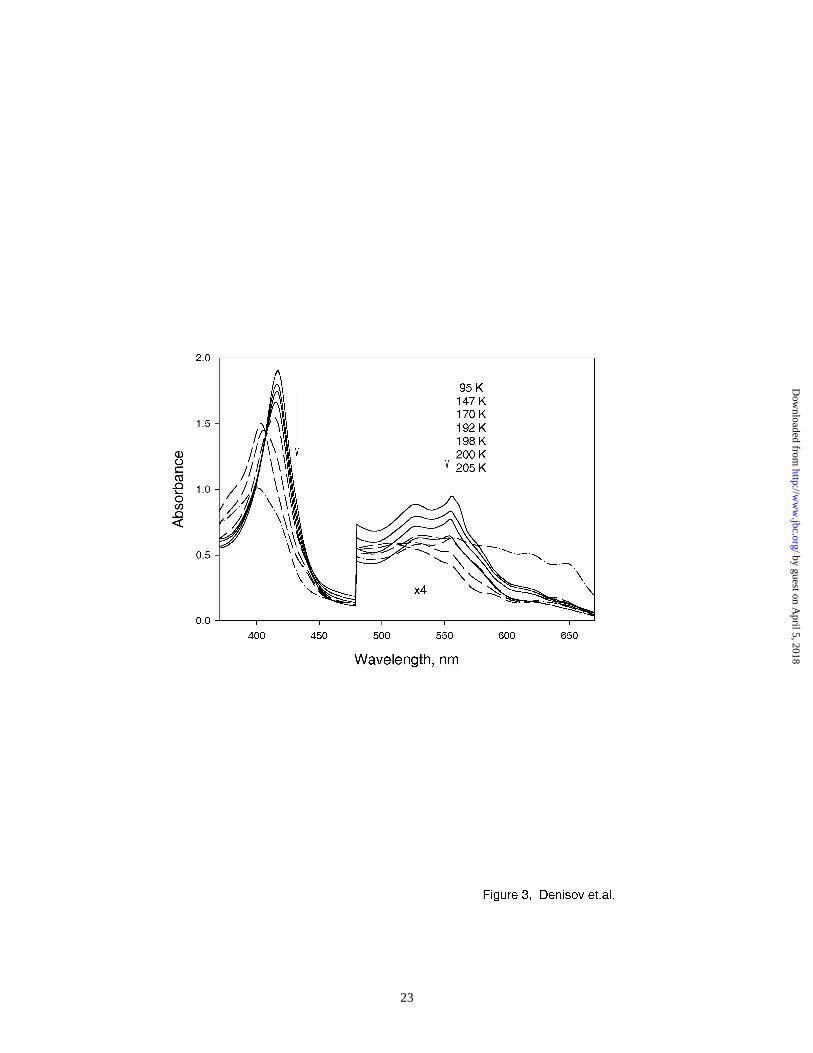
In order to probe the reactivity of Compound I under radiolytic conditions we separately generated
this intermediate using H2O2, as described in Methods. Figure 3 shows the spectra of Compound I
before and after radiolytic reduction. The spectrum of Compound I obtained at low temperature is
in a good agreement with earlier reports (40,41). Radiolytic reduction of Compound I at 77 K
results in formation of a new species with the absorption spectra similar to those reported for
Compound II. Direct formation of Compound II confirms the chemical competence of
cryoradiolysis on HRP intermediates, and the true one-electron reduction as a result of cryogenic
radiolytic treatment of heme enzyme intermediates. At temperatures below 190 K, the shape of the
spectrum of Compound II does not change significantly, which implies that it is relatively stable
and does not undergo further reduction by products of radiolysis until annealed above the glass
transition temperature. At 198 K, this intermediate begins to disappear gradually, with concomitant
formation of ferric HRP (Fig. 3, spectra at 198 K – 205 K). The latter process is consistent with
one-electron reduction of Compound II, as occurs in the peroxidase enzymatic cycle under ambient
conditions.
Results shown in Fig. 3 suggest that the annealing of (Fe3+P-OOH-) complex in HRP (Fig. 2B) to
form ferric HRP does not follow a path through Compound I and Compound II. Comparison of Fig.
2B and Fig. 3 reveals substantial differences in observed spectra and annealing behavior of reduced
oxy-HRP and Compound II. Although the spectrum of Compound II has almost the same peak
positions (418 nm, 526 nm and 558 nm, Fig. 3) as the (Fe3+P-OOH-) intermediate (419 nm, 526 nm
and 556 nm), the spectra of these two species are not identical. The Qα band at 556 nm has
substantially higher amplitude than the Qβ band at 526 nm in the spectrum of (Fe3+P-OOH-)
complex, while in the spectrum of Compound II these two bands are almost of the same intensity.
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

12
Thus, the appearance of Compound II spectra in the optical annealing studies of (Fe3+P-OOH-)
species would have been detected, if the pathway through Compound I and Compound II was the
operative one. Instead, in all such experiments, the spectrum of ferric HRP emerged at the expense
of (Fe3+P-OOH-) spectrum, and the direct decay of hydroperoxo-ferric complex, or the so-called
peroxide shunt, was observed.
The lack of Compound I formation from its immediate precursor, (Fe3+P-OOH-) complex, may be
considered as an unexpected result, because HRP is known for its facile formation of the stable
Compound I species. However, there are important mechanistic differences between the enzymatic
cycle of this enzyme, which uses H2O2 as the oxygen donor, and the oxidase/oxygenase path
explored in the current studies. The (Fe3+P-OOH-) complex is formed as the precursor of
Compound I in both systems, and the second protonation of the β oxygen atom in this complex is
the decisive step to form Compound I. The specific delivery of this second proton is crucial for
Compound I formation from (Fe3+P-OOH-) complex (10,11,23,52), since the latter is very unstable
and quickly disappears even at low temperatures (16-18). The perturbation of this proton delivery
system caused by mutations at the active site was shown to result in nonproductive reduction of
oxygen and formation of hydrogen peroxide, or “uncoupling” in the cytochrome P450 catalytic
cycle (49,50). In HRP, the (Fe3+P-OOH-) complex is formed from H2O2 via deprotonation of the
neutral hydrogen peroxide at the distal pocket of the enzyme, and the same proton can be used for
the second protonation of the β oxygen via the rearrangement catalyzed by the distal His42 and
Arg38 side chains (1,51,52). If dioxygen is used instead of H2O2, both protonation steps have to be
provided by the enzyme, as occurs in the P450 monooxygenases (32,53,54). Since this path is non-
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

13
physiological for HRP, the enzyme fails to deliver the second proton required for Compound I
formation, and an uncoupling is observed.
In conclusion, we prepared the main redox intermediates of the HRP enzymatic cycle and
performed the radiolytic reduction of these species. The (Fe3+P-OOH-) intermediate, the precursor
of Compound I formation, was isolated via radiolytic reduction of oxy-HRP. Absorption and EPR
spectra of this intermediate are reported with much higher resolution and precision than in previous
studies. The first protonation of the peroxo-ferric heme complex, the primary product of the
reduction of oxy-ferrous HRP, is completed only at the temperatures higher than 150 K, as was
observed in D251N mutant of P450 CYP101 (32,43), where the proton delivery mechanism is
impaired (50,53,55). In wild-type P450 CYP101, the primary intermediate of radiolytic reduction
of oxy-ferrous enzyme at 77 K is the fully protonated hydroperoxo-ferric complex (32,43).
Comparison with similar studies on other heme proteins (25,26,28-30,35,42) shows that the ability
to deliver a proton to the peroxo-ferric heme complex correlates with the function of the protein, as
this protonation is most effective in P450s and least effective in Mb, which does not carry out
dioxygen bond scission during its normal function.
Annealing of the reduced oxy-ferrous complex of HRP results in completion of the first protonation
of the peroxide ligand and formation of the ferric hydroperoxo intermediate. Formally, this
intermediate is the same as the one formed in the reaction of H2O2 with heme proteins, which is a
precursor for the formation of Compound I in HRP and other peroxidases. However, when obtained
by reduction of the oxy-ferrous heme complex, the peroxo-ferric intermediate via this path still
needs two protons provided by the enzyme to affect O-O bond scission. This difference between
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

14
peroxidase (using H2O2) and oxygenase/oxidase (using O2) pathways is essential in the subsequent
evolution of this intermediate. Annealing at higher temperatures of (Fe3+P-OOH-) intermediate in
HRP did not result in Compound I formation. We conclude, that Compound I is not formed on this
pathway, because the delivery of the second proton to the distal (β) oxygen atom of the
hydroperoxo-anion ligand is not optimized in HRP. These results confirm the importance of
protonation mechanisms in heme enzymes, which use dioxygen for the formation of the redox
active heme oxygen intermediates.
ACKNOWLEDGMENTS
We gratefully appreciate the help provided by Dr. John Bentley while using the 60Co source in the
Notre Dame Radiation Laboratory (Notre Dame University, IN). Irradiations were conducted at the
Notre Dame Radiation Laboratory, which is a facility of the U.S. Department of Energy, Office of
Basic Energy Sciences. Useful discussions with Dr. S.-C. Hung are gratefully acknowledged. We
thank Ms. Aretta Weber for the expert editorial assistance. NIH Research Resources Grant P-41-
RR01811 is acknowledged for the use of IERC resources. The work was supported by a merit
award from the National Institutes of Health R37GM31756.
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

15
REFERENCES
1. Poulos, T. L., and Kraut, J. (1980) J.Biol.Chem. 255, 8199-8205
2. English, A. M., and Tsaprailis, G. (1995) Adv.Inorg.Chem. 43, 79-125
3. Dunford, H. B. (1999) Heme Peroxidases, Wiley, New York, 507 pp.
4. Nicholls, P., Fita, I., and Loewen, P.C. (2001) Adv.Inorg.Chem. 51, 51-106
5. Veitch, N. C., and Smith, A.T. (2001) Adv.Inorg.Chem. 51, 107-162
6. Watanabe, Y., and Groves, J.T. (1992) in: The Enzymes (Sigman, D.S., ed.), 3rd. Ed., v. 20,
pp. 405-452, Academic Press, San Diego
7. Dawson, J.H. (1996) Chem.Rev. 96, 2841-2888
8. Groves, J.T., and Wang, C. C.-Y. (2000) Curr.Opin.Chem.Biol. 4, 687-695
9. Loew, G.H., and Dupuis, M. (1996) J.Am.Chem.Soc. 118, 10584-10587
10. Woon, D.E., and Loew, G.H. (1998) J.Phys.Chem. A 102, 10380-10384
11. Wirstam, M., Blomberg, M.R.A., and Siegbahn, P.E.M. (1999) J.Am.Chem.Soc. 121,
10178-10185
12. Rodrigues-Lopez, J.N., Smith, A.T., and Thorneley, R.N.F. (1996) J.Biol.Chem. 271, 4023-
4030
13. Rodrigues-Lopez, J.N., Smith, A.T., and Thorneley, R.N.F. (1997) J.Biol.Chem. 272, 389-
395
14. Tanaka, M., Ishimori, K., Mukai, M., Kitagawa, T., and Morishima, I. (1997) Biochemistry
36, 9889-9898
15. Tanaka, M., Ishimori, K., and Morishima, I. (1998) Biochemistry 37, 2629-2638
16. Tajima, K. (1989) Inorg.Chim.Acta 163, 115-122
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

16
17. Baek, H.K., and van Wart, H.E. (1989) Biochemistry 28, 5714-5719
18. Baek, H.K., and van Wart, H.E. (1992) J.Am.Chem.Soc. 114, 718-725
19. Rodrigues-Lopez, J.N., Lowe, D.J., Hernandez-Ruiz, J., Hiner, A.N.P., Garcia-Canovas, F.,
and Thorneley, R.N.F. (2001) J.Am.Chem.Soc. 123, 11838-11847
20. Primus, J.-L., Grunenwald, S., Hagedoorn, P.-L., Albrecht-Gay, A.-M., Mandon, D., and
Veeger, C. (2002) J.Am.Chem.Soc. 124, 1214-1221
21. Veeger, C. (2002) J.Inorg.Biochem. 91, 35-45
22. Yeh, H.-C., Wang, J.-S., Su, Y.O., and Lin, W.-Y. (2001) J.Biol.Inorg.Chem. 6, 770-777
23. Harris, D.L., and Loew, G.H. (1996) J.Am.Chem.Soc. 118, 10588-10594
24. Brittain, T., Baker, A.R., Butler, C.S., Little, R.H., Lowe, D.J., Greenwood, C., and
Watmough, N.J. (1997) Biochem.J. 326, 109-115
25. Symons, M.C.R., and Petersen, R.L. (1978) Proc.R.Soc.Lond. B 201, 285-300
26. Symons, M.C.R., and Petersen, R.L. (1978) Biochim.Biophys.Acta 535, 241-247
27. Groves, J.T., and Han, Y. (1995) in Cytochrome P450. Structure, Mechanism, and
Biochemistry (Ortiz de Montellano, P. R., ed), pp. 3-48., Plenum press, New York
28. Davydov, R.M. (1980) Biofizika 25, 203-207
29. Kappl, R., Hohn-Berlage, M., Huttermann, J., Bartlett, N., and Symons, M.C.R., (1985)
Biochim.Biophys.Acta 827, 327-343
30. Gasyna, Z. (1979) FEBS Lett. 106, 213-218
31. Davydov, R., Kappl, R., Hutterman, R., and Peterson, J. (1991) FEBS Lett. 295, 113-115
32. Davydov, R., Makris, T.M., Kofman, V., Werst, D.E., Sligar, S.G., and Hoffman, B.M.
(2001) J.Am.Chem.Soc. 123, 1403-1415
33. Denisov, I.G., Makris, T.M., and Sligar, S.G. (2001) J.Biol.Chem. 276, 11648-11652.
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

17
34. Denisov, I.G., Hung, S.-C., Weiss, K.E., McLean, M.A., Shiro, Y., Park., S.-Y., Champion,
P.M., and Sligar, S.G. (2001) J.Inorg.Biochem. 87, 215-226
35. Davydov, R., Kofman, V., Fujii, H., Yoshida, T., Ikeda-Saito, M., and Hoffman, B.M.
(2002) J. Am.Chem.Soc. 124, 1798-1808
36. Berglund, G.I., Carlsson, G.H., Smith, A.T., Szoke, H., Henriksen, A., and Hajdu, J. (2002)
Nature 417, 463-468.
37. Schlichting, I., Berendzen, J., Chu, K., Stock, A.M., Maves, S.A., Benson, D.E., Sweet,
R.M., Ringe, D., Petsko, G.A., and Sligar, S.G.,. (2000) Science 287, 1615-1622
38. Sjogren, T., and Hajdu, J. (2001) J.Biol.Chem. 276, 13072-13076.
39. Kobayashi, K., and Hayashi, K. (1981) J.Biol.Chem., 256, 12350-12354.
40. Blumberg, W.E., Peisach, J., Wittenberg, B.A., and Wittenberg, J.B. (1968) J.Biol.Chem.
243, 1854-1862
41. Gasyna, Z., Browett, W.R., and Stillman, M.J. (1988) Biochemistry 27, 2502-2509
42. Davydov, R.M., Yoshida, T., Ikeda-Saito, M., and Hoffman, B.M. (1999) J.Am.Chem.Soc.
121, 10656-10657.
43. Davydov, R., Macdonald, I.D.G., Makris, T.M., Sligar, S.G., and Hoffman, B.M. (1999)
J.Am.Chem.Soc. 121, 10654-10655.
44. Hiner, A.N.P., Raven, E.L., Thorneley, R.N.F., Garcia-Canovas, F., and Rodrigues-Lopez,
J.N. (2002) J.Inorg.Biochem. 91, 27-34
45. Woods, R.J., and Pikaev, A.K. (1994) Applied Radiation Chemistry: Radiation Processing,
Wiley Interscience, New York, 535 pp.
46. Magonov, S.N., Davydov, R.M., Blyumenfeld, L.A., Vilu, R.O., Arutyunyan, A.M., and
Sharonov, Yu.A. (1978) Molek.Biol. (Eng.Transl.) 12, 725-733.
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

18
47. Magonov, S.N., Davydov, R.M., Blyumenfeld, L.A., Arutyunyan, A.M., and Sharonov,
Yu.A. (1978) Molek.Biol. (Eng.Transl.) 12, 919-924.
48. Magonov, S.N., Davydov, R.M., Blyumenfeld, L.A., Vilu, R.O., Arutyunyan, A.M., and
Sharonov, Yu.A. (1978) Molek.Biol. (Eng.Transl.) 12, 913-919.
49. Imai, M., Shimada, H., Watanabe, Y., Matsushima-Hibiya, Y., Makino, R., Koga, H.,
Horiuchi, T., and Ishimura, Y. (1989) Proc.Natl.Acad.Sci. U.S.A. 86, 7823-7827
50. Martinis, S.A., Atkins, W.M., Stayton, P.S., and Sligar, S.G. (1989) J.Am.Chem.Soc. 111,
9252-9253
51. Dunford, H. B. (1991) in Peroxidases in Chemistry and Biology (Everse, J., Everse, K.E.,
Grisham, M.B., eds.) v. 2, pp. 2-24., CRC Press, Boca Raton, FL
52. Filizola, M., and Loew, G.H. (2000) J. Am.Chem.Soc. 122, 18-25
53. Vidakovic, M., Sligar, S.G., Li, H., and Poulos, T.L. (1998) Biochemistry 37, 9211-9219
54. Mueller, E.J., Loida, P.J., and Sligar, S.G. (1995) in Cytochrome P450; Structure,
Mechanism, and Biochemistry. (Ortiz de Montellano, P. R., ed), pp. 83-124, Plenum Press,
New York,
55. Gerber, N.C., and Sligar, S.G. (1994) J.Biol.Chem. 269, 4260-4266
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

19
FOOTNOTES
1 On leave from the Institute of Macromolecular Compounds, Russian Academy of Sciences,
St.Petersburg, Russia.
2 The abbreviations used are: HRP, horseradish peroxidase; (Fe3+P-OOH-), ferric
protoporphyrin IX, complex with hydroperoxo-anion; Mb, myoglobin; Hb, hemoglobin;
3 Denisov, I.G., Ikeda-Saito, M., Yoshida, T., and Sligar, S.G., submitted
4 Makris, T.M., et.al., in preparation
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

20
LEGENDS
Figure 1 Absorption spectra of oxy-HRP before irradiation (1), and after irradiation at 77 K,
with doses 44 kGy (2) and 88 kGy (3). Spectrum of pure hydroperoxo-ferric
intermediate, calculated and averaged from multiple experiments with yields 35% -
50% (4). Spectra were measured at 80 K – 140 K.
Figure 2. (A) EPR spectra of oxy-HRP irradiated at 77 K and annealed at the indicated
temperatures. All spectra were measured at 77 K. (B) Absorption spectra of oxy-
HRP irradiated at 77 K and annealed at the same temperatures as the EPR samples.
The arrow shows the direction of spectral changes with increasing temperatures.
Spectra are separated for clarity.
Figure 3. Absorption spectra of Compound II, formed by γ-irradiation of Compound I at 77 K,
and measured at the indicated temperatures (see text). Arrows show the directions
of spectral changes with increasing temperatures. Full lines, spectra of radiolytically
reduced at 77 K Compound I, measured below 195 K. Dashed lines, spectra of the
same samples, measured at 198 K – 205 K indicating gradual transition to ferric
HRP. (– . – .) spectrum of Compount I before irradiation, measured at 100 K.
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Ilia G. Denisov, Thomas M. Makris and Stephen G. Sligarstudied by cryoradiolysis
Formation and decay of hydroperoxo-ferric heme complex in horseradish peroxidase
published online September 4, 2002J. Biol. Chem.
10.1074/jbc.M207949200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on April 5, 2018
http://ww
w.jbc.org/
Dow
nloaded from