hdac6 inhibition restores ciliary expression and...
TRANSCRIPT

Therapeutics, Targets, and Chemical Biology
HDAC6 Inhibition Restores Ciliary Expression andDecreases Tumor Growth
Sergio A. Gradilone, Brynn N. Radtke, Pamela S. Bogert, Bing Q. Huang, Gabriella B. Gajdos, andNicholas F. LaRusso
AbstractPrimary cilia are multisensory organelles recently found to be absent in some tumor cells, but the
mechanisms of deciliation and the role of cilia in tumor biology remain unclear. Cholangiocytes, the epithelialcells lining the biliary tree, normally express primary cilia and their interaction with bile componentsregulates multiple processes, including proliferation and transport. Using cholangiocarcinoma as a model, wefound that primary cilia are reduced in cholangiocarcinoma by a mechanism involving histone deacetylase 6(HDAC6). The experimental deciliation of normal cholangiocyte cells increased the proliferation rate andinduced anchorage-independent growth. Furthermore, deciliation induced the activation of mitogen-acti-vated protein kinase and Hedgehog signaling, two important pathways involved in cholangiocarcinomadevelopment. We found that HDAC6 is overexpressed in cholangiocarcinoma and overexpression of HDAC6in normal cholangiocytes induced deciliation and increased both proliferation and anchorage-independentgrowth. To evaluate the effect of cilia restoration on tumor cells, we targeted HDAC6 by short hairpin RNA(shRNA) or by the pharmacologic inhibitor, tubastatin-A. Both approaches restored the expression of primarycilia in cholangiocarcinoma cell lines and decreased cell proliferation and anchorage-independent growth.The effects of tubastatin-A were abolished when cholangiocarcinoma cells were rendered unable toregenerate cilia by stable transfection of IFT88-shRNA. Finally, inhibition of HDAC6 by tubastatin-A alsoinduced a significant decrease in tumor growth in a cholangiocarcinoma animal model. Our data support akey role for primary cilia in malignant transformation, provide a plausible mechanism for their involvement,and suggest that restoration of primary cilia in tumor cells by HDAC6 targeting may be a potentialtherapeutic approach for cholangiocarcinoma. Cancer Res; 73(7); 2259–70. �2013 AACR.
IntroductionPrimary cilia aremicrotubule-based organelles that function
asmultisensors of the extracellular environment (1). Interest inprimary cilia has increasedmarkedly over the last 15 years, as itwas observed that mutations in genes required for the assem-bly and/or the sensory properties of cilia result in diversehuman disorders such as visceral epithelial hyperplasia, poly-cystic kidneys, pancreas, and liver among other abnormalities(2). Recent observations also suggest a relationship betweenciliary structure/function and tumorigenesis. For example,Aurora A kinase mediates ciliary disassembly and is over-
expressed in many epithelial cancers (3). Nek8, a kinaseexpressed in primary cilia that regulates ciliogenesis, is increas-ed in breast cancer (2, 4); and the loss of the von Hippel-Lindau(VHL) tumor suppressor gene inhibits ciliogenesis and isassociated with renal cancers (5, 6). Also, mutations in miceof Tg737, the mammalian homolog of Chlamydomonas IFT88,a key component for ciliary formation (7), accelerate therate at which chemical carcinogens induce liver neoplasms(8). Finally, very recent findings showed reduced expressionof cilia in pancreatic ductal adenocarcinoma (2), renal cancer(6), astrocytoma/glioblastoma (9), and breast cancer (10).While these data suggest that ciliary dysfunction may be asso-ciated with cancer development, the mechanisms leadingto ciliary reduction in tumor cells as well as the consequen-ces of such a loss remain poorly understood and are the subjectof the present manuscript.
Cholangiocarcinoma (CCA) is a malignancy thought to bederived from cholangiocytes, the epithelial cells lining thebiliary tree. Cholangiocarcinoma is a highly aggressive tumorwhose incidence has been increasingworldwide over the past 2decades, now accounting for 10% to 15% of all hepatobiliarymalignancies. Advanced cholangiocarcinoma has a devastat-ing prognosis, with a median survival of less than 24 months(11, 12).
Authors' Affiliation:Department ofMedicine, Division ofGastroenterologyandHepatology,MayoCenter forCell Signalling inGastroenterology,MayoClinic, Rochester, Minnesota
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
Corresponding Author: Sergio A. Gradilone, Department of Medicine,Division of Gastroenterology and Hepatology, Mayo Center for CellSignalling in Gastroenterology, Mayo Clinic, 200 First Street SW,Rochester, MN 55905. Phone: 507-284-1006; Fax: 507-284-0762;E-mail: [email protected]
doi: 10.1158/0008-5472.CAN-12-2938
�2013 American Association for Cancer Research.
CancerResearch
www.aacrjournals.org 2259
on July 28, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 31, 2013; DOI: 10.1158/0008-5472.CAN-12-2938

Cholangiocytes normally express primary cilia extendingfrom their apical plasma membrane into the ductal lumen.In cholangiocytes, the primary cilium functions as a multi-sensor of the extracellular milieu detecting a wide varietyof chemical and physical stimuli. Indeed, we reported thatcholangiocyte primary cilia are mechano-, chemo-, and osmo-sensory organelles (13–16).
In the present manuscript, we describe that ciliary expres-sion is decreased in cholangiocarcinoma by a mechanisminvolving overexpression of histone deacetylase 6 (HDAC6).We found that targeting HDAC6 in cholangiocarcinoma cellsdecreases the tumorigenic phenotype of the cells in a ciliary re-expression–dependentmanner in vitro and in an animalmodelof cholangiocarcinoma. The data not only shed light on themechanisms by which ciliary disassembly facilitate malignanttransformation but also identify a potential molecular targetfor cholangiocarcinoma.
Materials and MethodsCell lines and culture
The normal human cholangiocytes (H69 and NHC) and thenormal rat (NRC) cell lines were maintained as previouslydescribed (13, 17, 18). The human cholangiocarcinoma celllines [HuCCT-1 (ref. 19) and KMCH (ref. 20)] and the ratcholangiocarcinoma cell line (BDEneu; refs. 21, 22) were cul-tured in Dulbecco's Modified Eagles' Media (DMEM) supple-mented with 10% FBS, 100 units/mL penicillin, 100 mg/mLstreptomycin, and 100 mg/L insulin.
Real-time PCRTotal RNA was extracted using TRIzol reagent (Invitrogen)
and synthesized into cDNA using SuperScript III First-StrandSynthesis System for RT-PCR (Invitrogen). Quantitative PCRfor HDAC6 was carried out using 1 mL of cDNA and the LightCycler Fast Start DNA MasterPlus SYBR Green I kit (RocheDiagnostics) as previously described (23). The primers usedwere HDAC6 sense (50-AGTCTTATGGATGGCTATTGCATG-30), HDAC6 antisense (50-TGGACCAGTTAGAGGCCTTCAGG-30), PTCH1 sense (50-CGCTGTCTTCCTTCTGAACC-30), andPTCH1 antisense (50- ATCAGCACTCCCAGCAGAGT-30). IFT88expression was analyzed using the TaqMan Gene ExpressionAssay (Assay ID Hs00197926_m1) from Applied Biosystemsfollowing the manufacturer's directions. The samples werenormalized to 18S rRNA.
ImmunfluorescencesLiver sections were incubated with antibodies against acet-
ylated a-tubulin (1:500, Sigma-Aldrich), IFT88 (1:100, Protein-tech), CK19 (1:100, Santa Cruz Biotechnology or Abcam),g-tubulin (1:500, Sigma-Aldrich), proliferating cell nuclearantigen (PCNA; 1:1,000, Santa Cruz Biotechnology), and/orHDAC6 (1:100, Abcam) overnight at 4�C followedby incubationfor 1 hourwithfluorescent secondary antibodies (1:100). Nucleiwere stained with 40,6-diamino-2-phenylindole (DAPI; ProlongGold w/DAPI, Invitrogen). For HDAC6-flag expression analy-sis, cells were transfected with the Addgene plasmid 13823(Dr. Eric Verdin; ref. 24) using Fugene reagent (Roche). After 3
days of incubation in media without serum, cells were fixedand stained for ciliary markers acetylated a-tubulin and/orIFT88 and ciliated cells were analyzed under the confocalmicroscopy.
Scanning electron microscopyCells were processed as previously described (25).
Chemical and molecular deciliationChemical deciliation was carried out by treatment with 4
mmol/L chloral hydrate as previously described (15). Molec-ular deciliation was obtained by stably transfection with ift88,Kif3a, or Cep164 short hairpin RNA (shRNA) plasmids (Sup-plementary Table S1).
Proliferation assaysProliferation assays were conducted using the CellTiter 96
AQueous Non-Radioactive Cell Proliferation Assay (MTS; Pro-mega) and/or counting cells using the Cellometer Auto4(Nexcelom Bioscience) cell counter.
Anchorage-independent growthAnchorage-independent growth was assessed by growing
cells in soft agar. About 25,000 cells suspended in 0.4% agar inculture media were layered over a 1% agar layer in a 6-wellplate. Media were added twice a week and pictures were takenafter 14 to 21 days of incubation. The number and size ofcolonies were analyzed using the Gel-Pro software.
Invasion assaysInvasion assays were conducted using the CytoSelect 24-
Well Cell Invasion Assay Kit (Cell Biolabs, Inc.) following themanufacturer's directions.
Western blotsProtein fractions were subjected to SDS-PAGE and trans-
ferred to nitrocellulose membranes. After blocking, blotswere incubated overnight at 4�C with one of the followingantibodies: HDAC6 (1:1,000, Santa Cruz Biotechnology), Erk(1:2,000, Abcam), p-Erk (1:1,000, BD Biosciences), Gli1 (1:500,Abcam), IFT88 (1:1,000, Proteintech), Kif3a (1:500, SantaCruz Biotechnology), Cep164 (1:500, Genetex), IL-6 (1:500,Santa Cruz), bcl-2 (1:1,000, Santa Cruz), actin (1:5000, Sig-ma-Aldrich or Abcam), and acetylated-a-tubulin (1:5,000,Sigma-Aldrich); washed and incubated for 1 hour atroom temperature with horseradish peroxidase-conjugated(1:5,000, Invitrogen) or IRdye 680 or 800 (1:15,000, Odyssey)corresponding secondary antibody. For protein detection,ECL system or Odyssey Liquor Scanner was used, and theGel-Pro Analyzer 6.0 software was used for densitometricanalysis.
In vivo experimentsAll animal experimentation was carried out in accord-
ance with and approved by the Institutional Animal Careand Use Committee. In vivo cell transplantation was carriedout in adult fisher 344 male rats (Harlan) with initial meanbody weights ranging between 200 and 250 g, as previously
Gradilone et al.
Cancer Res; 73(7) April 1, 2013 Cancer Research2260
on July 28, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 31, 2013; DOI: 10.1158/0008-5472.CAN-12-2938

described (21, 26, 27). Five days after tumor implantation,animals were treated daily with tubastatin-A (10 mg/kg bodyweight intraperitoneally) or vehicle for 7 days. After treat-ment, animals were euthanized and the livers were removedfor analysis.
Statistical analysisData are expressed as mean � SE. Statistical analyses were
conducted by one-way ANOVAwith Bonferroni post hoc test tocompare more than 2 groups and by the Student t test tocompare 2 groups. Results were considered statistically differ-ent at P < 0.05.
ResultsPrimary cilia are reduced in cholangiocarcinoma in vivoand in vitroTo assess the expression of primary cilia in cholangio-
carcinoma, we stained liver samples from 21 patients with
cholangiocarcinoma and 6 normal controls with the ciliarymarkers, acetylated a-tubulin and/or IFT88, and the cho-langiocyte marker, CK19. We found that while 100% of bileducts from normal controls show primary cilia, only 20% areciliated in cholangiocarcinoma samples (Fig. 1A–E). Wefound a similar situation when we stained a commerciallyavailable cholangiocarcinoma tissue array [AccuMax ArrayA205(II); Supplementary Fig. S1]. To further explore thedifferential expression of primary cilia, we assessed theirexpression on 2 normal human cholangiocyte cell lines (H69,NHC) and the cholangiocarcinoma cell lines Hucct-1 andKMCH. Ciliary expression was induced by serum starvation(15) and assessed by staining with acetylated a-tubulin,IFT88, and the centrosome marker, g-tubulin; our resultsshowed that only 3% (Hucct-1) and 1.8% (KMCH) of thecholangiocarcinoma cell lines express cilia, whereas in thenormal cell lines, cilia were found in 67% (H69) and 61%(NHC) of the cells, respectively (Fig. 1F–H). Finally, toconfirm ciliary loss in cholangiocarcinoma cells, we
Figure 1. Primary cilia are reducedin cholangiocarcinoma. Confocalimmunofluorescence for 2 ciliarymarkers, acetylated-a-tubulin in red,and IFT88 in purple. Nuclei arestained in blue with DAPI. Cilia areeasily appreciated on the bile ductlumen of control normal humantissue and on normal cholangiocytecell lines (A, C, F). Even though thered and purple signals weresaturated, reduced amount of ciliarystructures were found oncholangiocarcinoma samples (B, D)or in the cell lines HuCCT-1 (G) andKMCH as shown in theaccompanying quantifications (E, H).In vivo, the cholangiocyte markerCK19, and in vitro, the centrosomemarker g-tubulin, were stained ingreen. �, P < 0.0001, n ¼ 21;#, P < 0.001, n ¼ 110. I–L, scanningelectron microscopy of the apicalsurface of normal rat cholangiocytesand the rat CCA cells BDEneu.
Normal CCA
A B
C D
F G
I J
K L
H
E
# #0
10203040506070
H69 NHC Hucct-1 KMCH
Cilia
ted
ce
lls
(%)
*0
25
50
75
100
Normal CCA
Cilia
ted
bile
du
cts
(%
)
The Role of Cilia in Cholangiocarcinoma
www.aacrjournals.org Cancer Res; 73(7) April 1, 2013 2261
on July 28, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 31, 2013; DOI: 10.1158/0008-5472.CAN-12-2938

conducted scanning electron microscopy of the apical sur-face of normal and cholangiocarcinoma cells (Fig. 1I–L).
Deciliation of normal cholangiocytes inducesproliferation, anchorage-independent growth, andinvasion
To explore the potential relationship between ciliary lossand cholangiocyte phenotype, we assessed the effect ofdeciliation on normal human (i.e., H69) and rat (NRC)cholangiocyte cell lines. First, we induced chemical decilia-tion by ClHy treatment (15); deciliation by this manipulationincreased normal cholangiocyte proliferation by 2-fold(Fig. 2A). We complemented this approach of chemicaldeciliation by molecular deciliation using specific shRNAsagainst IFT88, Kif3a, or Cep164 (Supplementary Fig. S2); thisapproach caused an increase in cell proliferation by 1.89-,1.77-, and 1.63-fold, respectively (Fig. 2B). The increased
proliferation was confirmed by nucleotide incorporationassays (Supplementary Fig. S3). Furthermore, cell-cycle anal-ysis showed an increase in G2–M and a decrease in G1–G0
phases in IFT88-shRNA deciliated cells compared with nor-mal NT-shRNA controls (Supplementary Fig. S4), furthersupporting increased proliferation. To further characterizethe effect of ciliary loss, we assessed anchorage-independentgrowth and invasion and found that deciliation by IFT88shRNAs induced both parameters by 6- and 3-fold, respec-tively, and by 3-fold and 2-fold, respectively, when deciliationwas induced by Kif3a shRNAs (Fig. 2C and D). Finally,we analyzed the status of the hedgehog signaling pathwayby real-time PCR (RT-PCR) of patched mRNA and Westernblots for Gli1, interleukin (IL)-6, and Bcl2; and the mitogen-activated protein kinase (MAPK) signaling pathway byWestern blotting and found that deciliation induced acti-vation of both hedgehog and MAPK signaling pathways
0
0.5
1
0
0.5
1
ClHy [mmol/L]
0 4
ClHy [mmol/L]
0 4
NRCs H69
0
0.5
1
Pro
life
rati
on
rate
(Ab
s 4
92 n
m)
Pro
life
rati
on
rate
(Ab
s 4
92 n
m)
0
2
4
6
8
NT IFT88 Kif3a
0
2
4
6
8
NT IFT88 Kif3a
Co
lon
ies
nu
mb
er
(fo
ld in
cre
as
e)
Inv
as
ion
ra
te
(fo
ld i
nc
rea
se
)
0
1
2
3
Control ClHy IFT88
Pa
tch
ed
mR
NA
(fo
ld in
cre
as
e) p-ERK
t-ERK
Gli1
IL-6
Actin
A B
C
DE F
* **
*
*
*
*
* *
Control 4 mM ClHyH69 NT shRNA H69 IFT88 shRNA
Figure 2. Cholangiocyte deciliation induces a malignant-like phenotype. A, scanning electron microscopy (SEM) images from control NRCs showed normalcilia (left) butwhen treatedwithClHy for 24 hours, ciliawere absent (right). Proliferation ratesweremeasured byMTSassay. NRCs andH69 cellswere followedduring 3 days after deciliation. B, SEM (top) and immunofluorescence (bottom) images from scrambled nontarget (NT) control and IFT88 shRNA H69–transfected cells cultured 3 days in minimal medium. Proliferation rates for IFT88-, Kif3a-, Cep164-, or nontarget (NT) shRNA stable–transfected cells weremeasured byMTS assay. C, nontarget (NT) control and IFT88 or Kif3a shRNA–transfected cells were cultured 21 days in soft agar. Colonies quantification forIFT88 and Kif3a shRNAs cells showed a 6-fold and 3-fold increase comparedwith NT shRNA cells, respectively. D, invasion assays where cells were culturedon polycarbonate membrane inserts coated with uniform layer of basement membrane in 24-well plates (CytoSelect 24-Well Cell Invasion Assay, CellBiolabs). E, NRCs and H69 cells were deciliated with ClHy and IFT88 shRNAs, respectively, and Patched mRNA levels, a marker of the Hedgehog pathwayactivation,were quantifiedbyRT-PCR. F, normal cells stably transfectedwithKif3a,Cep164, or IFT88 shRNAs showelevated p-ERK/t-ERK ratio,Gli1, and IL6compared with NT controls.
Gradilone et al.
Cancer Res; 73(7) April 1, 2013 Cancer Research2262
on July 28, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 31, 2013; DOI: 10.1158/0008-5472.CAN-12-2938

(Fig. 2E and F, Supplementary Fig. S2). Taken together, thesedata suggest that experimentally induced deciliation stimu-lates a phenotypic transformation of normal cholangiocytesin culture to a malignant phenotype.
HDAC6 is overexpressed in cholangiocarcinoma anddecreases ciliary expressionTo explore the potential mechanisms involved in the cho-
langiocarcinoma decreased ciliary expression, we analyzedHDAC6, a tubulin deacetylase reported to induce ciliaryresorption in the immortalized retinal pigment epithelial cellline hTERT-RPE1 (3); moreover, evidence in the literaturesuggests an important role for HDAC6 overexpression intumorigenesis (28–31). We assessed the expression of HDAC6by Western blot analysis on 2 different cholangiocarcinomacell lines (Hucct-1 and KMCH) and found increased expres-sion in both (on average, 100%) of HDAC6 compared withnormal cultured cholangiocytes (H69). Overexpression ofHDAC6 correlated with the decreased amount of its target,acetylated a-tubulin (Fig. 3A), supporting the validity ofour analyses. The mRNA level was analyzed by qRT-PCR andno significant differences were found (Fig. 3B), suggesting aposttranscriptional regulatory pathway. Using confocal immu-nofluorescence microscopy, we also observed that HDAC6was overexpressed in liver specimens from 10 patients withcholangiocarcinoma compared with 11 normals (Fig. 3C).We found a similar situation when we stained the commer-cially available cholangiocarcinoma tissue array [AccuMaxArray A205(II); Supplementary Fig. S5].To assess the role of HDAC6 overexpression in cholangio-
cyte ciliary loss, we transfected NHC cells with a HDAC6-flagexpression vector. Confocal immunofluorescences using anti-
flag and HDAC6 antibodies showed the expression of theHDAC6-flag construct (Fig. 4A). As predicted, when the cellswere co-stained with the ciliary markers, acetylated-a-tubulinand IFT88, the cells that were positive for the flag epitopeshowed reduced cilia expression compared with the surround-ing nontransfected cells (Fig. 4B). After stable transfected cellswere obtained by antibiotic selection, Western blot analysisshowed the overexpression of HDAC6 in the stably transfect-ed cells using anti-flag antibodies (Fig. 4C). In addition to thedecreased ciliary expression (Fig. 4D), these cells showedan increased proliferation rate (1.7-fold; Fig. 4E), increasedanchorage-independent growth (1.5-fold; Fig. 4F), and de-creased protein levels of acetylated-a-tubulin comparedwith empty vector–transfected cells (Fig. 4G). Taken together,these results suggest that the overexpression of HDAC6in normal cholangiocytes correlates with the reduction ofprimary cilia expression and the acquisition of a malignant-like phenotype.
Targeting HDAC6 induces ciliary restoration andreverses the malignant phenotype ofcholangiocarcinoma cells
As HDAC6 overexpression seems to contribute importantlyto cholangiocarcinoma ciliary loss, we inhibited HDAC6 ex-pression by specific shRNAs (Fig. 5A) or with the HDAC6inhibitor, tubastatin-A (32). These approaches both inducedan increase in acetylated-a-tubulin levels and the restorationof primary cilia expression in the cholangiocarcinoma celllines (3.3- and 18-fold, respectively; Fig. 5A, D, and E); andthe restoration of primary cilia correlated with downregu-lated Hedgehog (Hh) and MAPK signaling pathways (Fig.5C), as well as decreased cell proliferation rates (decreased in
Figure 3. HDAC6 expression incholangiocarcinoma. A,Western blotanalysis of HDAC6 proteinexpression showed increased levelsin different tumor cell lines comparedwith control H69 cells. Theupregulation of HDAC6 correlateswith the decreased amount ofacetylated a-tubulin. B, qRT-PCRshowed no significant differences inthe messenger level of HDAC6. C,confocal immunofluorescenceimages for HDAC6 on normal andcholangiocarcinoma (CCA) humanliver samples.
HDAC6
Acet-αα-tubulin
Actin
0
0.2
0.4
0.6
0.8
1
H69 Hucct-1 KMCH
H69
Huc
ct-1
KM
CH
HD
AC
6/A
cti
n r
ati
o
0
0.5
1
1.5
2
2.5
H69 Hucct-1 KMCH
Ac
et-
α-tu
bu
lin
/Acti
n r
ati
o
0
10
20
30
40
50
60
70
H69 Hucct-1 KMCH
Co
py
nu
mb
er/
1E
+06
18
S
A
B C
Normal CCA
The Role of Cilia in Cholangiocarcinoma
www.aacrjournals.org Cancer Res; 73(7) April 1, 2013 2263
on July 28, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 31, 2013; DOI: 10.1158/0008-5472.CAN-12-2938

average by 50%; Fig. 5B and F) and invasion (decreased by40%; Fig. 5G). To analyze whether the restoration of cilia is amajor reason for these phenotypic changes, we repeated the
experiments in KMCH cells stably transfected with IFT88-shRNA to prevent ciliogenesis. In the experiments in whichcholangiocarcinoma cells were prevented from developing
HDAC6
Flag DAPI ac-a-tub Flag
ift88 MergeMerge
A B
C
MW EV HDAC6-
flag
Flag
Actin
D
E F
HDAC6
Acet-α-tubulin
Actin
G
0
20
40
60
80
100
EV HDAC6-flag
Cilia
ted
ce
lls (
%)
*
*
0
0.2
0.4
0.6
0.8
1
EV HDAC6-Flag
Pro
life
rati
on
rate
(Ab
s 4
92
nm
)
0
100
200
300
400
500
600
EV HDAC6-Flag
Co
lon
y n
um
be
r
*
Figure4. Effect ofHDAC6overexpression in normal cholangiocytes. A,NHCcellswere transfectedwithHDAC6-flagexpression vector, andprotein expressionwas analyzed by confocal immunofluorescence using anti-flag (green) and anti-HDAC6 antibodies (red); nuclei were stained in blue with DAPI. B, confocalimmunofluorescence using anti-flag (purple), anti-acetylated-a-tubulin (red), and anti-ift88 (green) showed that cells overexpressing HDAC6 do not growcilia comparedwith nontransfected surrounding cells. C, NHCcellswere stably transfectedwith empty vector orwith HDAC6-flag expression vector.Westernblot analysis showed the expression of HDAC6-flag. D, confocal immunofluorescence on cells cultured 2 days in cilia-promoting media showed ciliaryexpression by acetylated-a-tubulin staining (red) and centrioles (purple). Note that cilia are easily detected by the basal bodies in empty vector–transfectedcholangiocytes (EV), whereas they are mainly absent on the HDAC6-flag–transfected cells. Nuclei are stained in blue with DAPI. E, MTS proliferation assaycomparing empty vector (EV) and HDAC6-flag stably transfected cells (P < 0.05). F, colony number after 14 days of growth in soft agar (P < 0.05). G, Westernblot analysis of acetylated-a-tubulin on NHC cells stable transfected with EV and HDAC6-flag.
Gradilone et al.
Cancer Res; 73(7) April 1, 2013 Cancer Research2264
on July 28, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 31, 2013; DOI: 10.1158/0008-5472.CAN-12-2938
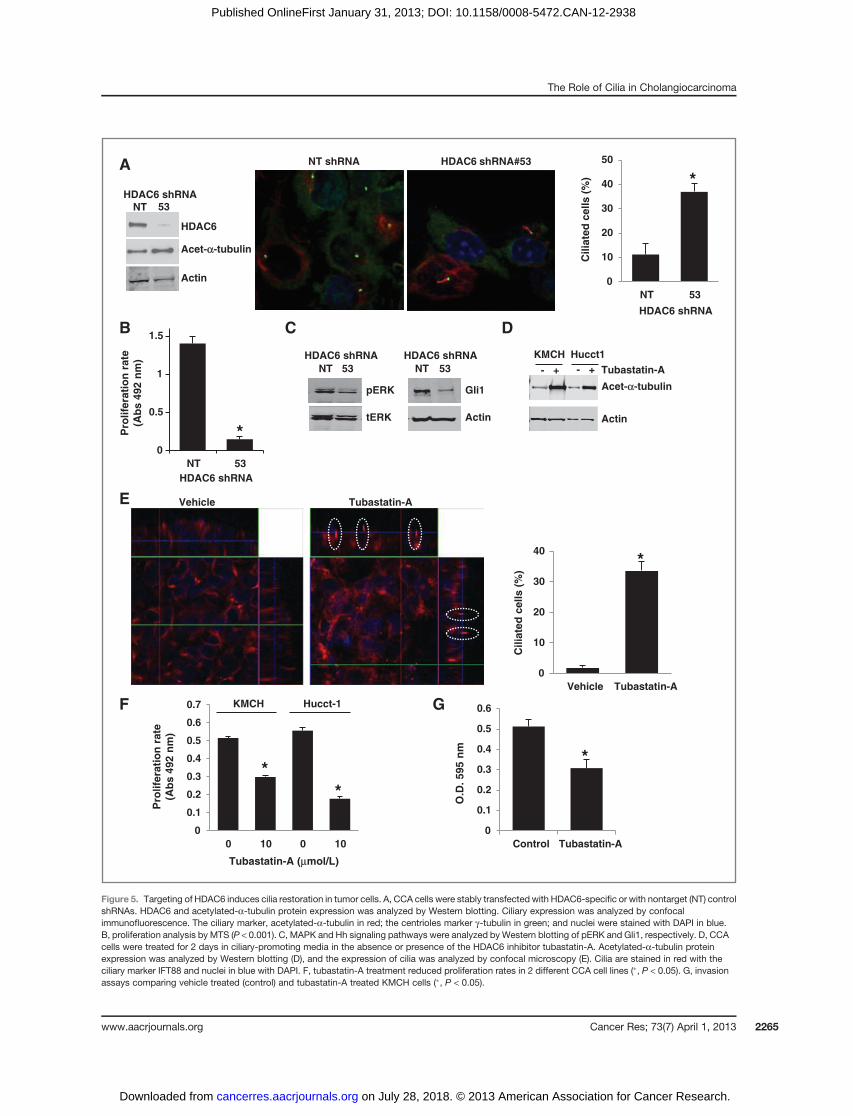
A
B
pERK
tERK Actin
Gli1
NT 53
HDAC6 shRNA
NT 53
HDAC6 shRNA
C
E
F G
HDAC6
Acet-αα-tubulin
Acet-α-tubulin
Actin
NT 53HDAC6 shRNA
0
10
20
30
40
50
NT 53
HDAC6 shRNA
Cilia
ted
cells (
%) *
Pro
life
rati
on
ra
te
(Ab
s 4
92
nm
)
HDAC6 shRNA
*
- + Tubastatin-A
KMCH Hucct1
Actin
- +
0
10
20
30
40
Vehicle Tubastatin-A
Cilia
ted
cells (
%)
*
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0 10 0 10
Tubastatin-A (μmol/L)
KMCH Hucct-1
*
*
0
0.1
0.2
0.3
0.4
0.5
0.6
Control Tubastatin-A
O.D
. 5
95
nm
*
D
0
0.5
1
1.5
NT 53
Pro
life
rati
on
ra
te
(Ab
s 4
92
nm
)NT shRNA HDAC6 shRNA#53
Vehicle Tubastatin-A
Figure 5. Targeting of HDAC6 induces cilia restoration in tumor cells. A, CCA cells were stably transfected with HDAC6-specific or with nontarget (NT) controlshRNAs. HDAC6 and acetylated-a-tubulin protein expression was analyzed by Western blotting. Ciliary expression was analyzed by confocalimmunofluorescence. The ciliary marker, acetylated-a-tubulin in red; the centrioles marker g-tubulin in green; and nuclei were stained with DAPI in blue.B, proliferation analysis by MTS (P < 0.001). C, MAPK and Hh signaling pathways were analyzed by Western blotting of pERK and Gli1, respectively. D, CCAcells were treated for 2 days in ciliary-promoting media in the absence or presence of the HDAC6 inhibitor tubastatin-A. Acetylated-a-tubulin proteinexpression was analyzed by Western blotting (D), and the expression of cilia was analyzed by confocal microscopy (E). Cilia are stained in red with theciliary marker IFT88 and nuclei in blue with DAPI. F, tubastatin-A treatment reduced proliferation rates in 2 different CCA cell lines (�, P < 0.05). G, invasionassays comparing vehicle treated (control) and tubastatin-A treated KMCH cells (�, P < 0.05).
The Role of Cilia in Cholangiocarcinoma
www.aacrjournals.org Cancer Res; 73(7) April 1, 2013 2265
on July 28, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 31, 2013; DOI: 10.1158/0008-5472.CAN-12-2938
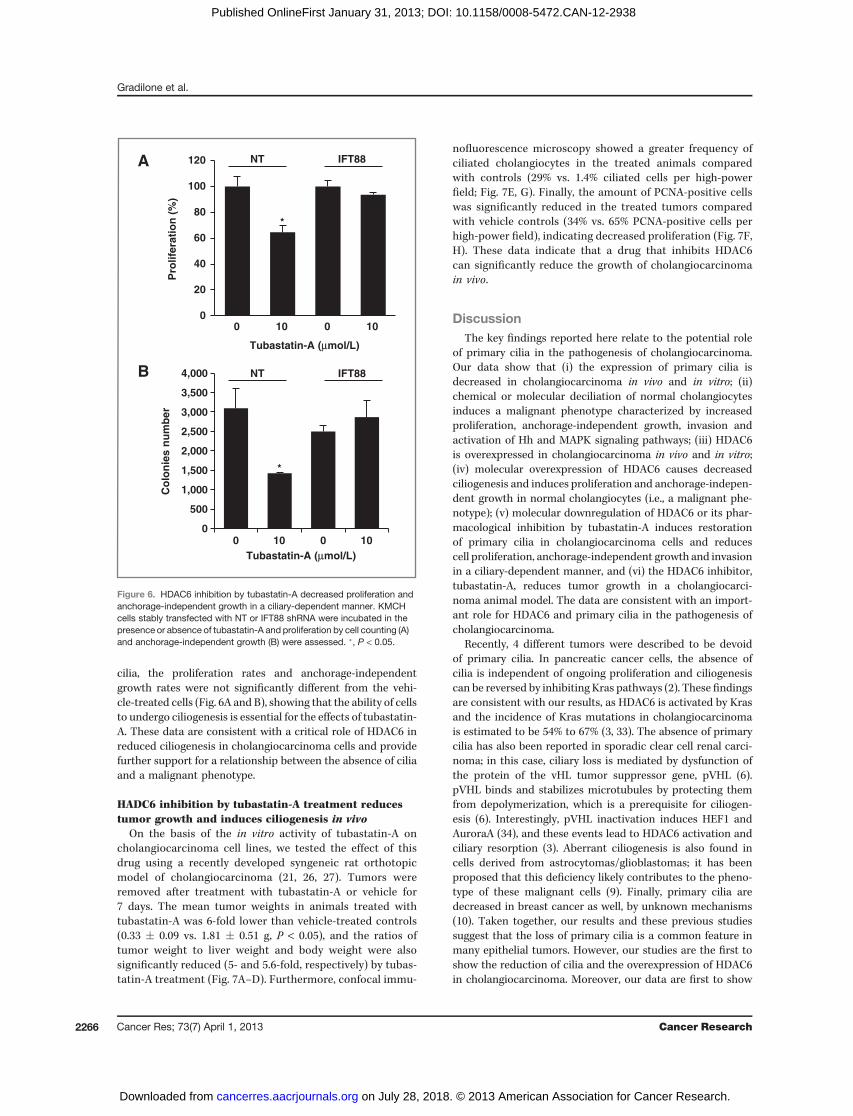
cilia, the proliferation rates and anchorage-independentgrowth rates were not significantly different from the vehi-cle-treated cells (Fig. 6A and B), showing that the ability of cellsto undergo ciliogenesis is essential for the effects of tubastatin-A. These data are consistent with a critical role of HDAC6 inreduced ciliogenesis in cholangiocarcinoma cells and providefurther support for a relationship between the absence of ciliaand a malignant phenotype.
HADC6 inhibition by tubastatin-A treatment reducestumor growth and induces ciliogenesis in vivo
On the basis of the in vitro activity of tubastatin-A oncholangiocarcinoma cell lines, we tested the effect of thisdrug using a recently developed syngeneic rat orthotopicmodel of cholangiocarcinoma (21, 26, 27). Tumors wereremoved after treatment with tubastatin-A or vehicle for7 days. The mean tumor weights in animals treated withtubastatin-A was 6-fold lower than vehicle-treated controls(0.33 � 0.09 vs. 1.81 � 0.51 g, P < 0.05), and the ratios oftumor weight to liver weight and body weight were alsosignificantly reduced (5- and 5.6-fold, respectively) by tubas-tatin-A treatment (Fig. 7A–D). Furthermore, confocal immu-
nofluorescence microscopy showed a greater frequency ofciliated cholangiocytes in the treated animals comparedwith controls (29% vs. 1.4% ciliated cells per high-powerfield; Fig. 7E, G). Finally, the amount of PCNA-positive cellswas significantly reduced in the treated tumors comparedwith vehicle controls (34% vs. 65% PCNA-positive cells perhigh-power field), indicating decreased proliferation (Fig. 7F,H). These data indicate that a drug that inhibits HDAC6can significantly reduce the growth of cholangiocarcinomain vivo.
DiscussionThe key findings reported here relate to the potential role
of primary cilia in the pathogenesis of cholangiocarcinoma.Our data show that (i) the expression of primary cilia isdecreased in cholangiocarcinoma in vivo and in vitro; (ii)chemical or molecular deciliation of normal cholangiocytesinduces a malignant phenotype characterized by increasedproliferation, anchorage-independent growth, invasion andactivation of Hh and MAPK signaling pathways; (iii) HDAC6is overexpressed in cholangiocarcinoma in vivo and in vitro;(iv) molecular overexpression of HDAC6 causes decreasedciliogenesis and induces proliferation and anchorage-indepen-dent growth in normal cholangiocytes (i.e., a malignant phe-notype); (v) molecular downregulation of HDAC6 or its phar-macological inhibition by tubastatin-A induces restorationof primary cilia in cholangiocarcinoma cells and reducescell proliferation, anchorage-independent growth and invasionin a ciliary-dependent manner, and (vi) the HDAC6 inhibitor,tubastatin-A, reduces tumor growth in a cholangiocarci-noma animal model. The data are consistent with an import-ant role for HDAC6 and primary cilia in the pathogenesis ofcholangiocarcinoma.
Recently, 4 different tumors were described to be devoidof primary cilia. In pancreatic cancer cells, the absence ofcilia is independent of ongoing proliferation and ciliogenesiscan be reversed by inhibiting Kras pathways (2). These findingsare consistent with our results, as HDAC6 is activated by Krasand the incidence of Kras mutations in cholangiocarcinomais estimated to be 54% to 67% (3, 33). The absence of primarycilia has also been reported in sporadic clear cell renal carci-noma; in this case, ciliary loss is mediated by dysfunction ofthe protein of the vHL tumor suppressor gene, pVHL (6).pVHL binds and stabilizes microtubules by protecting themfrom depolymerization, which is a prerequisite for ciliogen-esis (6). Interestingly, pVHL inactivation induces HEF1 andAuroraA (34), and these events lead to HDAC6 activation andciliary resorption (3). Aberrant ciliogenesis is also found incells derived from astrocytomas/glioblastomas; it has beenproposed that this deficiency likely contributes to the pheno-type of these malignant cells (9). Finally, primary cilia aredecreased in breast cancer as well, by unknown mechanisms(10). Taken together, our results and these previous studiessuggest that the loss of primary cilia is a common feature inmany epithelial tumors. However, our studies are the first toshow the reduction of cilia and the overexpression of HDAC6in cholangiocarcinoma. Moreover, our data are first to show
0
20
40
60
80
100
120
0 10 0 10
Pro
life
rati
on
(%
)NT IFT88
*
*
Tubastatin-A (μmol/L)
A
0
500
1,000
1,500
2,000
2,500
3,000
3,500
4,000
0 10 0 10
Tubastatin-A (μmol/L)
NT IFT88
Co
lon
ies
nu
mb
er
B
Figure 6. HDAC6 inhibition by tubastatin-A decreased proliferation andanchorage-independent growth in a ciliary-dependent manner. KMCHcells stably transfected with NT or IFT88 shRNA were incubated in thepresence or absence of tubastatin-A and proliferation by cell counting (A)and anchorage-independent growth (B) were assessed. �, P < 0.05.
Gradilone et al.
Cancer Res; 73(7) April 1, 2013 Cancer Research2266
on July 28, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 31, 2013; DOI: 10.1158/0008-5472.CAN-12-2938

that restoration of primary cilia by targeting HDAC6 is apotential therapeutic approach for cholangiocarcinoma andperhaps other tumors characterized by defective ciliogenesis.On the other hand, the situation is different in medullo-
blastoma and basal cell carcinoma; these tumors are mainlyciliated and cilia are required for the growth of tumors bearingan activation mutation at the level of smoothened (ciliary-dependent activator of the Hh signaling pathway). In contrast,
if the tumors have an activation mutation of the downstreameffector of the pathway, the transcription factor Gli2, primarycilia play a similar role as described in the present work, thatis, inhibition of tumor development (35, 36). In cholangiocar-cinoma, consistent with our results, the aberrant activationof Gli transcription factors has been described (37, 38).
HDAC6 is a unique member of the histone deacetylasefamily because, unlike other histone deacetylases, it does not
A B C D
0.0
0.5
1.0
1.5
2.0
2.5
Control Tubastatin-A
Tu
mo
r w
eig
ht
(g)
*
0
5
10
15
20
25
Control Tubastatin-A
Tu
mo
r/liv
er
we
igh
t ra
tio
(%
)
*
0.0
0.5
1.0
1.5
Control Tubastatin-A
Tu
mo
r/b
od
y w
eig
ht
rati
o (
%)
*
E
F
0
20
40
60
80
100
120
Vehicle Tubastatin-A
Noncilia Cilia
%
0
10
20
30
40
50
60
70
Vehicle Tubastatin-A
PC
NA
/DA
PI
% *
Control Tubastatin-A
Co
ntr
ol
Tu
ba
sta
tin
-AC
on
tro
lT
ub
as
tati
n-A
Figure 7. Effect of tubastatin-A on cholangiocarcinoma growth in vivo. The effect of tubastatin-A was tested on an orthotopic, syngeneic CCA model in rats.A, livers were removed after 7 days of treatment and tumors were dissected. Tumor weights, tumor/liver weight, and tumor/body weight ratios werecalculated and compared (B–D). Tumor sections were stained with the ciliary marker acetylated-a-tubulin in red and the cholangiocyte marker CK-7 in green(E) or with the proliferation marker PCNA in green (F); nuclei were stained in blue with DAPI. The amount of ciliated and PCNA-positive cells per field werequantified (G, H). �, P < 0.05, n ¼ 5 for controls and n ¼ 6 for tubastatin-A.
The Role of Cilia in Cholangiocarcinoma
www.aacrjournals.org Cancer Res; 73(7) April 1, 2013 2267
on July 28, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 31, 2013; DOI: 10.1158/0008-5472.CAN-12-2938

interact with histones (i.e., inhibition of HDAC6 inhibits dea-cetylation of a-tubulin without affecting histone acetylation;ref. 39). HDAC6 not only deacetylates a-tubulin but alsocortactin, Hsp90, and the redox regulatory proteins, PrxI andII (40). Moreover, HDAC6 has been identified as a key regulatorof many processes that are linked to cancer (e.g., cell survival,motility, and metastasis), making it an attractive therapeutictarget (39). On the basis of our results, it appears that theantitumorigenic effect of HDAC6 inhibition in cholangiocar-cinoma mainly depends on ciliary restoration, as KMCH cellsstably transfected with ift88 shRNA did not respond to tubas-tatin-A treatment. We acknowledge that the involvement ofthe other targets of HDAC6 cannot be confidently excluded.
Previous work with embryonic kidney cells, mammary epi-thelial cells, mouse embryonic fibroblasts, and ovarian andcancer cell lines showed that HDAC6 is not only important forRas- or ErbB2-dependent oncogenic transformation of primarycells but also is required for maintaining the anchorage-inde-pendent growth of established cancer cell lines (28). The exactmolecular mechanisms mediating such a tumor-promotingeffect remains unknown. Our results in cholangiocarcinomacells suggest that HDAC6 mediates the oncogenic-inducedloss of primary cilia and the subsequent derepression oftumorigenic signaling pathways such as Hh and MAPK. Themechanisms by which HDAC6 is overexpressed in cholangio-carcinoma remain to be elucidated. As we found alterationsin protein but not inmRNA levels for HDAC6, posttranslationalregulations (e.g., HCAC6 turnover rates, the downregulationof microRNAs potentially targeting HDAC6 mRNA, etc.), arepossibilities.
Hedgehog and extracellular signal–regulated kinase (ERK)1/2 pathways are both activated in cholangiocarcinoma (37,38, 41), and the dual targeting of these pathways coordinatelydecrease proliferation and survival of cholangiocarcinomacells (41). IL6 and bcl-2, both targets of hedgehog signaling(42–44), are also activated in cholangiocarcinoma (45, 46).Our results show that the experimental deciliation of normalcholangiocytes induced the activation of both hedgehogand MAPK pathways, consistent with the concept that cilianormally act as a negative regulator of these pathways incholangiocytes. Importantly, our results also show that therestoration of primary cilia, by targeting HDAC6 in cholangio-carcinoma cells, decreases Hh and MAPK signaling pathways.
Michaud and Yoder speculated that genes and proteinsinvolved in the structure or function of primary cilia mayrepresent new targets for small-molecule inhibitors, siRNAs,
or antibody therapeutics (47). On the basis of our data, wewould extend this concept by suggesting that restorationof primary cilia and their complex multisensory signals byHDAC6 targeting could act as a tumor suppressor mecha-nism. Indeed, the rapidly evolving field of HDAC inhibitorspromises to generate very potent and specific HDAC6 inhibi-tors, like tubastatin-A (32) and the more recently developedACY-1215 (48). The fact that mice lacking HDAC6 are viableand develop normally (49) suggests that HDAC6-specifictargeting may have minimal adverse effects. Furthermore,our in vivo experiments on a rat cholangiocarcinoma modelusing tubastatin-A showed a significant decrease in tumorgrowth associated with an increased ciliary expression, sug-gesting that the restoration of primary cilia in tumor cells bymeans of HDAC6 inhibitors may be a potential therapeuticapproach for cholangiocarcinoma.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: S.A. Gradilone, P.S. Bogert, G.B. Gajdos, N.F. LaRussoDevelopment of methodology: P.S. Bogert, N.F. LaRussoAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): S.A. Gradilone, B.N. Radtke, P.S. Bogert, B.Q. Huang, N.F. LaRussoAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): S.A. Gradilone, G.B. Gajdos, N.F. LaRussoWriting, review, and/or revision of the manuscript: S.A. Gradilone, B.N.Radtke, N.F. LaRussoAdministrative, technical, or material support (i.e., reporting or orga-nizing data, constructing databases): B.N. Radtke
AcknowledgmentsThe authors thank Drs. Tetyana Masyuk, Anatoliy Masyuk, Steven O'Hara,
and Patrick Splinter for insightful discussions; Drs. Gregory Gores and MartinFernandez-Zapico for carefully reviewing and editing the manuscript; and Dr.Alphonse Sirica for generously providing BDEneu cells for the in vivoexperiments.
Grant SupportThis work was supported by a Pilot and Feasibility Award to S.A. Gradilone
from the Mayo Clinic Center for Cell Signaling in Gastroenterology(P30DK084567), a Wendy Will Case Cancer Fund Award (S.A. Gradilone), aMayo Clinic Cancer Center Eagle Fellowship (S.A. Gradilone), a R01 DK24031 (N.F. LaRusso), and the Mayo Foundation.
The costs of publication of this article were defrayed in part by the payment ofpage charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received July 25, 2012; revised December 18, 2012; accepted January 3, 2013;published OnlineFirst January 31, 2013.
References1. Satir P, Pedersen LB, Christensen ST. The primary cilium at a glance.
J Cell Sci 2010;123:499–503.2. Seeley ES, Carriere C, Goetze T, Longnecker DS, Korc M. Pancreatic
cancer and precursor pancreatic intraepithelial neoplasia lesions aredevoid of primary cilia. Cancer Res 2009;69:422–30.
3. Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA.HEF1-dependent Aurora A activation induces disassembly of theprimary cilium. Cell 2007;129:1351–63.
4. Bowers AJ, Boylan JF. Nek8, a NIMA family kinase member, isoverexpressed in primary human breast tumors. Gene 2004;328:135–42.
5. Lutz MS, Burk RD. Primary cilium formation requires von hippel-lindau gene function in renal-derived cells. Cancer Res 2006;66:6903–7.
6. SchramlP, Frew IJ, ThomaCR,BoysenG,StruckmannK,KrekW, et al.Sporadic clear cell renal cell carcinoma but not the papillary type ischaracterized by severely reduced frequency of primary cilia. ModPathol 2009;22:31–6.
7. Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, WitmanGB, et al. Chlamydomonas IFT88 and its mouse homologue, poly-cystic kidneydiseasegene tg737, are required for assembly of cilia andflagella. J Cell Biol 2000;151:709–18.
Gradilone et al.
Cancer Res; 73(7) April 1, 2013 Cancer Research2268
on July 28, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 31, 2013; DOI: 10.1158/0008-5472.CAN-12-2938

8. Isfort RJ, Cody DB, Doersen CJ, Richards WG, Yoder BK, WilkinsonJE, et al. The tetratricopeptide repeat containing Tg737 gene isa liver neoplasia tumor suppressor gene. Oncogene 1997;15:1797–803.
9. Moser JJ, Fritzler MJ, Rattner JB. Primary ciliogenesis defects areassociated with human astrocytoma/glioblastoma cells. BMC Cancer2009;9:448.
10. Yuan K, Frolova N, Xie Y,Wang D, Cook L, Kwon YJ, et al. Primary ciliaare decreased in breast cancer: analysis of a collection of humanbreast cancer cell lines and tissues. J Histochem Cytochem 2010;58:857–70.
11. Marzioni M, Invernizzi P, Candelaresi C, Maggioni M, Saccomanno S,Selmi C, et al. Human cholangiocarcinoma development is associatedwith dysregulation of opioidergic modulation of cholangiocyte growth.Dig Liver Dis 2009;41:523–33.
12. Francis H, Alpini G, DeMorrow S. Recent advances in the regulation ofcholangiocarcinoma growth. Am J Physiol Gastrointest Liver Physiol2010;299:G1–9.
13. Masyuk AI, Huang BQ, Ward CJ, Gradilone SA, Banales JM, MasyukTV, et al. Biliary exosomes influencecholangiocyte regulatorymechan-isms and proliferation through interaction with primary cilia. AmJ Physiol Gastrointest Liver Physiol 2010;299:G990–9.
14. Masyuk AI, GradiloneSA, Banales JM,HuangBQ,Masyuk TV, LeeSO,et al. Cholangiocyte primary cilia are chemosensory organelles thatdetect biliary nucleotides via P2Y12 purinergic receptors. AmJPhysiolGastrointest Liver Physiol 2008;295:G725–34.
15. Gradilone SA, Masyuk AI, Splinter P, Banales JM, Huang B, Tietz P,et al. Cholangiocyte cilia express TRPV4 anddetect changes in luminaltonicity inducing bicarbonate secretion. Proc Natl Acad Sci U S A2007;104:19138–43.
16. Masyuk AI, Masyuk TV, Splinter PL, Huang BQ, Stroope AJ, LaRussoNF. Cholangiocyte cilia detect changes in luminal fluid flow andtransmit them into intracellular Ca2þ and cAMP signaling. Gastroen-terology 2006;131:911–20.
17. O'Hara SP, Splinter PL, Trussoni CE, Gajdos GB, Lineswala PN,LaRusso NF. Cholangiocyte N-Ras protein mediates lipopolysaccha-ride-induced interleukin 6 secretion and proliferation. J Biol Chem2011;286:30352–60.
18. Banales JM, Saez E, Uriz M, Sarvide S, Urribarri AD, Splinter P,et al. Up-regulation of microRNA 506 leads to decreased Cl(-)/HCO(3) (-) anion exchanger 2 expression in biliary epitheliumof patients with primary biliary cirrhosis. Hepatology 2012;56:687–97.
19. Miyagiwa M, Ichida T, Tokiwa T, Sato J, Sasaki H. A new humancholangiocellular carcinoma cell line (HuCC-T1) producing carbohy-drate antigen 19/9 in serum-free medium. In Vitro Cell Dev Biol1989;25:503–10.
20. Murakami T, Yano H, MaruiwaM, Sugihara S, Kojiro M. Establishmentand characterization of a human combined hepatocholangiocarci-noma cell line and its heterologous transplantation in nude mice.Hepatology 1987;7:551–6.
21. Sirica AE, Zhang Z, Lai GH, Asano T, Shen XN, Ward DJ, et al. A novel"patient-like" model of cholangiocarcinoma progression based on bileduct inoculation of tumorigenic rat cholangiocyte cell lines. Hepatol-ogy 2008;47:1178–90.
22. Lai GH, Zhang Z, Shen XN, Ward DJ, Dewitt JL, Holt SE, et al. erbB-2/neu transformed rat cholangiocytes recapitulate key cellular andmolecular features of human bile duct cancer. Gastroenterology2005;129:2047–57.
23. O'Hara SP, Splinter PL, Gajdos GB, Trussoni CE, Fernandez-ZapicoME, Chen XM, et al. NFkappaB p50-CCAAT/enhancer-bindingprotein beta (C/EBPbeta)-mediated transcriptional repression ofmicroRNA let-7i following microbial infection. J Biol Chem 2010;285:216–25.
24. Fischle W, Emiliani S, Hendzel MJ, Nagase T, Nomura N, Voelter W,et al. A new family of human histone deacetylases relatedto Saccharomyces cerevisiae HDA1p. J Biol Chem 1999;274:11713–20.
25. Gradilone SA, Masyuk TV, Huang BQ, Banales JM, Lehmann GL,Radtke BN, et al. Activation of Trpv4 reduces the hyperproliferative
phenotype of cystic cholangiocytes from an animal model of ARPKD.Gastroenterology 2010;139:304–14.e2.
26. Smoot RL, Blechacz BR, Werneburg NW, Bronk SF, Sinicrope FA,Sirica AE, et al. A Bax-mediated mechanism for obatoclax-inducedapoptosis of cholangiocarcinoma cells. Cancer Res 2010;70:1960–9.
27. Fingas CD, Blechacz BR, Smoot RL, Guicciardi ME, Mott J, Bronk SF,et al. A smac mimetic reduces TNF related apoptosis inducing ligand(TRAIL)-induced invasion andmetastasis of cholangiocarcinoma cells.Hepatology 2010;52:550–61.
28. Lee YS, Lim KH, Guo X, Kawaguchi Y, Gao Y, Barrientos T, et al. Thecytoplasmic deacetylase HDAC6 is required for efficient oncogenictumorigenesis. Cancer Res 2008;68:7561–9.
29. Sakuma T, Uzawa K, Onda T, Shiiba M, Yokoe H, Shibahara T, et al.Aberrant expression of histone deacetylase 6 in oral squamous cellcarcinoma. Int J Oncol 2006;29:117–24.
30. Marcus AI, Zhou J, O'Brate A, Hamel E, Wong J, Nivens M, et al. Thesynergistic combination of the farnesyl transferase inhibitor lonafarniband paclitaxel enhances tubulin acetylation and requires a functionaltubulin deacetylase. Cancer Res 2005;65:3883–93.
31. Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL.Domain-selective small-molecule inhibitor of histone deacetylase 6(HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci U S A2003;100:4389–94.
32. Butler KV, Kalin J, Brochier C, Vistoli G, Langley B, Kozikowski AP.Rational design and simple chemistry yield a superior, neuropro-tective HDAC6 inhibitor, tubastatin A. J Am Chem Soc 2010;132:10842–6.
33. YoshikawaD,OjimaH, Kokubu A, Ochiya T, Kasai S, Hirohashi S, et al.Vandetanib (ZD6474), an inhibitor of VEGFR and EGFR signalling, as anovel molecular-targeted therapy against cholangiocarcinoma. BrJ Cancer 2009;100:1257–66.
34. Xu J, Li H, Wang B, Xu Y, Yang J, Zhang X, et al. VHL inactiva-tion induces HEF1 and Aurora kinase A. J Am Soc Nephrol 2010;21:2041–6.
35. Han YG, Kim HJ, Dlugosz AA, Ellison DW, Gilbertson RJ, Alvarez-Buylla A. Dual and opposing roles of primary cilia in medulloblastomadevelopment. Nat Med 2009;15:1062–5.
36. Wong SY, Seol AD, So PL, Ermilov AN, Bichakjian CK, Epstein EH Jr,et al. Primary cilia can bothmediate and suppressHedgehog pathway-dependent tumorigenesis. Nat Med 2009;15:1055–61.
37. Kurita S, Mott JL, Cazanave SC, Fingas CD, Guicciardi ME, BronkSF, et al. Hedgehog inhibition promotes a switch from Type II toType I cell death receptor signaling in cancer cells. PLoS One2011;6:e18330.
38. Kurita S, Mott JL, Almada LL, Bronk SF,Werneburg NW, Sun SY, et al.GLI3-dependent repressionofDR4mediateshedgehogantagonismofTRAIL-induced apoptosis. Oncogene 2010;29:4848–58.
39. Aldana-Masangkay GI, Sakamoto KM. The role of HDAC6 in cancer.J Biomed Biotechnol 2011;2011:875824.
40. Parmigiani RB, XuWS, Venta-PerezG, Erdjument-BromageH, YanevaM, Tempst P, et al. HDAC6 is a specific deacetylase of peroxiredoxinsand is involved in redox regulation. Proc Natl Acad Sci U S A 2008;105:9633–8.
41. Jinawath A, Akiyama Y, Sripa B, Yuasa Y. Dual blockade of theHedgehog and ERK1/2 pathways coordinately decreases proliferationand survival of cholangiocarcinoma cells. J Cancer Res Clin Oncol2007;133:271–8.
42. Elsawa SF, Almada LL, Ziesmer SC, Novak AJ, Witzig TE, AnsellSM, et al. GLI2 transcription factor mediates cytokine cross-talk in the tumor microenvironment. J Biol Chem 2011;286:21524–34.
43. Bigelow RL, Chari NS, Unden AB, Spurgers KB, Lee S, Roop DR,et al. Transcriptional regulation of bcl-2 mediated by the sonichedgehog signaling pathway through gli-1. J Biol Chem 2004;279:1197–205.
44. Xu XF, Guo CY, Liu J, YangWJ, Xia YJ, Xu L, et al. Gli1 maintains cellsurvival by up-regulating IGFBP6 and Bcl-2 through promoterregions in parallel manner in pancreatic cancer cells. J Carcinog2009;8:13.
The Role of Cilia in Cholangiocarcinoma
www.aacrjournals.org Cancer Res; 73(7) April 1, 2013 2269
on July 28, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 31, 2013; DOI: 10.1158/0008-5472.CAN-12-2938

45. Wehbe H, Henson R, Meng F, Mize-Berge J, Patel T. Interleukin-6contributes to growth in cholangiocarcinoma cells by aberrantpromoter methylation and gene expression. Cancer Res 2006;66:10517–24.
46. Harnois DM, Que FG, Celli A, LaRusso NF, Gores GJ. Bcl-2 is over-expressed and alters the threshold for apoptosis in a cholangiocarci-noma cell line. Hepatology 1997;26:884–90.
47. Michaud EJ, Yoder BK. The primary cilium in cell signaling and cancer.Cancer Res 2006;66:6463–7.
48. Santo L, Hideshima T, Kung AL, Tseng JC, Tamang D, Yang M,et al. Preclinical activity, pharmacodynamic, and pharmacokineticproperties of a selective HDAC6 inhibitor, ACY-1215, in combi-nation with bortezomib in multiple myeloma. Blood 2012;119:2579–89.
49. Zhang Y, Kwon S, Yamaguchi T, Cubizolles F, Rousseaux S, KneisselM, et al. Mice lacking histone deacetylase 6 have hyperacetylatedtubulin but are viable and develop normally. Mol Cell Biol 2008;28:1688–701.
Gradilone et al.
Cancer Res; 73(7) April 1, 2013 Cancer Research2270
on July 28, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 31, 2013; DOI: 10.1158/0008-5472.CAN-12-2938

2013;73:2259-2270. Published OnlineFirst January 31, 2013.Cancer Res Sergio A. Gradilone, Brynn N. Radtke, Pamela S. Bogert, et al. Tumor GrowthHDAC6 Inhibition Restores Ciliary Expression and Decreases
Updated version
10.1158/0008-5472.CAN-12-2938doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2013/01/31/0008-5472.CAN-12-2938.DC1
Access the most recent supplemental material at:
Cited articles
http://cancerres.aacrjournals.org/content/73/7/2259.full#ref-list-1
This article cites 48 articles, 20 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/73/7/2259.full#related-urls
This article has been cited by 10 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/73/7/2259To request permission to re-use all or part of this article, use this link
on July 28, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 31, 2013; DOI: 10.1158/0008-5472.CAN-12-2938