h. pylori negative gastritis - uscap …/99th/pdf/companion19h.pdf · 1 h. pylori-negative...
TRANSCRIPT

1
H. PYLORI-NEGATIVE GASTRITIS
WHAT TO DO WHEN HELICOBACTERS AREN'T THERE
Robert M Genta
Caris Diagnostics
and
University of Texas Southwestern Medical Center
Dallas, Texas
INTRODUCTION H. pylori infection was recognized as the main cause of chronic active gastritis and peptic ulcer disease just over two decades ago 1, and shortly thereafter the first effective treatments were introduced 2, 3. Since then, and likely even before its discovery, the prevalence of H. pylori infection has been steadily declining, particularly in industrialized and emerging economy countries; this is probably a reflection of improved sanitary conditions, widespread treatment of infected patients, and the pervasive use of antibiotics 4. In parallel with this decline, an increased proportion of H. pylori-negative ulcers has been reported, both in the United States 5,
6 and elsewhere 7-9. Furthermore, although no studies documenting this phenomenon have been published to date, there is a common perception amongst pathologists practicing in the Western world that chronic active gastritis with no detectable H. pylori organisms (“H. pylori –negative chronic active gastritis”) is on the rise 10 11. The often cited 1990s axiom that even a small number of polymorphonuclear neutrophils in a gastric biopsy is an almost certain indication of current H. pylori infection is no longer applicable. While no formal hypothesis has been put forward and tested, commonly offered explanations include antibiotic therapy administered to treat other infections, the masking effect of proton-pump inhibitors (PPIs), and failure to detect organisms because of inadequate sampling or sub-optimal staining techniques.
The purpose of this presentation is to help the practicing pathologist confronted with a gastric biopsy that "looks like H. pylori should be there, but is not."
WHEN AND HOW TO SEARCH FOR H. PYLORI Just as we used to say that active gastritis means H. pylori infection, we also said that there is no H. pylori infection without active gastritis. As it turns out, we were wrong on both accounts. As any experienced gastrointestinal pathologist will know, when an anti-H. pylori immunohistochemical stain (HP/IHC) is used in all gastric biopsies, one will occasionally find organisms in unexpected backgrounds, such as a virtually normal mucosa, an antral mucosa with reactive gastropathy and no active inflammation, or a fundic mucosa with a minimal sub-epithelial rim of lymphocytes and plasma cells and no neutrophils. Having been fooled by a few such cases, I advocate the preemptive use of HP/IHC in all gastric biopsies.

2
Those who cannot or choose not to routinely use the HP/IHC or other appropriate special stains for the detection of H. pylori must decide when to request such stains. When H. pylori are not detected by whatever means one uses routinely, a more sensitive stain (ideally the HP/IHC) should be used in the following circumstances: 1 - Chronic active gastritis (CAG) 2 - Focal active gastritis ("focally enhanced") 12 3 - Chronic inactive gastritis with lymphoid follicles 13 4 - Atrophic corpus gastritis, to exclude H. pylori before suggesting autoimmune gastritis 5 - When a duodenal or gastric ulcer is described in the endoscopy report, irrespective of the appearance of the gastric mucosa in the biopsy 6 - When the biopsies are obtained to confirm the success of eradication therapy 7 - Whenever suspicious speckles that could be H. pylori are seen 8 - If a MALT lymphoma is either seen or reported to have been treated in the past 14, 15
In circumstances 2 through 7 the expected (and desirable) finding is the absence of H. pylori, as in the case of a treated MALT lymphoma. H. pylori-negative MALT lymphomas do exist, but they are a distinct minority (<10%); therefore, a diligent search for the organisms is necessary. If they are not found, a suggestion for more representative sampling is usually well received by clinicians, who also need to determine the extent of the lymphoma. The true dilemma occurs in the case of H. pylori-negative CAG. THE 3R APPROACH TO H. PYLORI-NEGATIVE CAG Reconsider. When H. pylori is not found by HP/IHC one should first reconsider the initial diagnosis of CAG. 16 After critically reviewing ~400 gastric biopsy specimens that had been diagnosed as "CAG - No H. pylori detected by the H. pylori Blue stain" we found that only approximately 120 cases represented true CAG; most of the other cases were reactive gastropathy with small erosions (where neutrophils are usually abundant) 17; chronic inactive gastritis (neutrophils may have been present in the lamina propria, but were not found in the epithelium); and specimens from the cardia (where active inflammation is usually associated with reflux and not with H. pylori infection) 18, 19. Of the real CAG cases, the HP/IHC yielded a little over another 25% new positives. If going back to one's own dubious diagnoses seems futile or painful, it may be helpful to consider that each previously undetected case is a patient who will be treated and will not develop peptic ulcers, and whose risk of gastric cancer will be reduced by more than 70%. Table 1 shows possible sources of inaccurate diagnoses of CAG. Re-stain. If one is convinced that the histologic appearance is unequivocally that of CAG, a more sensitive stain should be examined. Thus, if only H&E-stained slides were prepared, a Giemsa (modified to Blue or Yellow) or a silver stain should be done; if still negative, the HP/IHC (which ought to be done in first place) should then be ordered. If the HP/IHC is negative and the impression of H. pylori gastritis is overwhelming, a second HP/IHC may be appropriate, particularly if the control was not perfect. This sequential strategy will eventually yield cases of unequivocal CAG with no H. pylori. This is the time to

3
Retreat. After reviewing the slides, restaining, and perhaps staining once more, those who adhere to the precept "You are not obliged to finish the task, but neither are you free to neglect it" will be probably satisfied that they have not neglected the task and will feel free to desist from further action. They will issue a well-worded report stating that, in spite of the characteristic histologic appearance, no H. pylori was found after a meticulous search (and a long list of CPT codes). Such reports, however, are unlikely to gratify an inquisitive clinician, who will be left wondering what should be done. Therefore, striving to finish the task is a better alternative. AFTER A DIAGNOSIS OF H. PYLORI-NEGATIVE CAG Calling the clinician and explaining the circumstances that lead to an equivocal diagnosis is probably the best way to avoid being perceived as a timid pathologist. Before discussing a case, however, it is necessary to be maximally informed. Two common clinical circumstances may decrease the gastric H. pylori load: recent use of antibiotics and proton-pump inhibitors. Other causes for the failure to detect organisms in a gastric biopsy are listed in table 2.
Antibiotic regimens not specifically prescribed for the treatment of H. pylori can temporarily attenuate or eradicate the infection in a proportion of patients, depending on the type of antibiotic used, the length of the treatment, and whether or not the patient was coincidentally using PPIs. In most patients intentional or unintentional eradication causes the disappearance of polymorphonuclear neutrophils from the gastric mucosa within days of the start of the therapy 20, 21, leaving the histopathologic impression of a chronic inactive gastritis. Incomplete and unsuccessful eradication, however, may greatly reduce the bacterial load (thus making them undetectable in certain parts of the stomach) with little or no decrease of active inflammation. If one can elicit a history of recent antibiotic treatment, a significant step is made in finding an explanation for H. pylori CAG. Furthermore, the pathologist can predict that the infection, inadequately treated, will soon reemerge, and a new set of biopsies in a few weeks will likely show organisms.
A much more common reason for the apparent disappearance of H. pylori from some compartments of the stomach (particularly the antrum) is the use of proton pump inhibitors (PPIs). 10 Now available in more than ten different preparations, some of which can be obtained over the counter, PPIs alter the gastric acid environment and induce shifts in the H. pylori populations within the stomach, usually reducing the bacterial burden in the antrum 22 while increasing the inflammation in the corpus23-25 26-28. Not only do these changes cause false negative urea breath tests 29-31, but they have also been shown to hinder the histopathologic detection of H. pylori in gastric biopsies 28. Thus, many diagnoses of H. pylori-negative CAG in antral biopsies could be avoided if samples from the gastric coypus were also available.
These explanations may convince the clinician to perform a non-invasive test (serology, urea breath test, fecal antigen detection) or to repeat the endoscopy and take more representative biopsies. Nothing could be worse than leaving the impression that H. pylori-negative CAG is a final diagnosis and the patient does not need further workup.

4
Table 1 - Causes of Helicobacter pylori-negative chronic active gastritis
Crohn’s Disease
Focally enhanced gastritis
Lymphocytic gastritis with a strong active component
Autoimmune gastritis with a strong active component
Reflux carditis (activity limited to the cardia)
Drug-related (e.g NSAIDs)
Biopsy near an erosion or ulcer
Granulomatous gastritis
Infectious (CMV, staphylococcus, syphilis)
Table 2 - Possible reasons for failure to detect H. pylori
Diagnostic error Bacteria were missed Condition is not chronic active gastritis
Sampling error Proton pump inhibitors shift bacteria from antrum into body
Factors causing an unfavorable local environment for bacteria Intestinal metaplasia Ulcer
Suppression resulting from incidental antibiotic or proton pump inhibitors use

5
REFERENCES
1. Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1984;1:1311-1315.
2. Glupczynski Y, Burette A, Nyst JF, De PC, De KE, Deltenre M. Campylobacter pylori-associated gastritis: attempts to eradicate the bacteria by various antibiotics and anti-ulcer regimens. Acta Gastroenterol Belg 1988;51:329-337.
3. Marshall BJ. Campylobacter pylori infection: diagnosis and therapy. Med J Aust 1989;151:426-427.
4. Fennerty MB. Helicobacter pylori: why it still matters in 2005. Cleve Clin J Med 2005;72 Suppl 2:S1-7; discussion S14-21.:S1-S7.
5. Jyotheeswaran S, Shah AN, Jin HO, Potter GD, Ona FV, Chey WY. Prevalence of Helicobacter pylori in peptic ulcer patients in greater Rochester, NY: is empirical triple therapy justified? Am J Gastroenterol 1998;93:574-578.
6. Ciociola AA, McSorley DJ, Turner K, Sykes D, Palmer JB. Helicobacter pylori infection rates in duodenal ulcer patients in the United States may be lower than previously estimated. Am J Gastroenterol 1999;94:1834-1840.
7. Jang HJ, Choi MH, Shin WG, Kim KH, Chung YW, Kim KO, Park CH, Baek IH, Baik KH, Kae SH, Kim HY. Has peptic ulcer disease changed during the past ten years in Korea? A prospective multi-center study. Dig Dis Sci 2008;53:1527-1531.
8. McColl KE. Helicobacter pylori negative ulcer disease. Dig Liver Dis 2000;32:125-127.
9. Ong TZ, Hawkey CJ, Ho KY. Nonsteroidal anti-inflammatory drug use is a significant cause of peptic ulcer disease in a tertiary hospital in Singapore: a prospective study. J Clin Gastroenterol 2006;40:795-800.
10. Genta RM, Schuler CM, Lash RH. Helicobacter pylori-negative chronic active gastritis: a new entity or the result of widespread acid inhibition? 134 ed. 2008:A - 125.
11. Genta RM. Where have all the Helicobacters gone? J Clin Gastroenterol 2007;41:727-728.
12. Oberhuber G, Hirsch M, Stolte M. High incidence of upper gastrointestinal tract involvement in Crohn's disease. Virchows Arch 1998;432:49-52.
13. Genta RM, Hamner HW. The significance of lymphoid follicles in the interpretation of gastric biopsy specimens. Arch Pathol Lab Med 1994;118:740-743.

6
14. Fischbach W, Chan AO, Wong BC. Helicobacter pylori and Gastric Malignancy. Helicobacter 2005;10 Suppl 1:34-9.:34-39.
15. Fischbach W, Goebeler ME, Ruskone-Fourmestraux A, Wundisch T, Neubauer A, Raderer M, Savio A. Most patients with minimal histological residuals of gastric MALT lymphoma after successful eradication of Helicobacter pylori can be managed safely by a watch and wait strategy: experience from a large international series. Gut 2007;56:1685-1687.
16. Lash RH, Lauwers GY, Odze RD, Genta RM. Inflammatory Disorders of the Stomach. In: Odze RD and Goldblum JR, eds. Surgical Pathology of the GI TRact, Liver, Biliary Tract, and Pancreas. Second ed. Philadelphia: Sunders Elsevier, 2009:269-320.
17. Genta RM. Differential diagnosis of reactive gastropathy. Semin Diagn Pathol 2005;22:273-283.
18. Glickman JN, Fox V, Antonioli DA, Wang HH, Odze RD. Morphology of the cardia and significance of carditis in pediatric patients. Am J Surg Pathol 2002;26:1032-1039.
19. Odze RD. Pathology of the gastroesophageal junction. Semin Diagn Pathol 2005;22:256-265.
20. Go MF, Lew GM, Lichtenberger LM, Genta RM, Graham DY. Gastric mucosal hydrophobicity and Helicobacter pylori: response to antimicrobial therapy. Am J Gastroenterol 1993;88:1362-1365.
21. Franceschi F, Genta RM, Sepulveda AR. Gastric mucosa: long-term outcome after cure of Helicobacter pylori infection. J Gastroenterol 2002;37 Suppl 13:17-23.:17-23.
22. Graham DY, Genta R, Evans DG, Reddy R, Clarridge JE, Olson CA, Edmonds AL, Siepman N. Helicobacter pylori does not migrate from the antrum to the corpus in response to omeprazole. Am J Gastroenterol 1996;91:2120-2124.
23. Genta RM. Acid suppression and gastric atrophy: sifting fact from fiction. Gut 1998;43 Suppl 1:S35-8.:S35-S38.
24. Genta RM. Atrophy, acid suppression and Helicobacter pylori infection: a tale of two studies. Eur J Gastroenterol Hepatol 1999;11 Suppl 2:S29-33; discussion S43-5.:S29-S33.
25. Genta RM, Rindi G, Fiocca R, Magner DJ, D'Amico D, Levine DS. Effects of 6-12 months of esomeprazole treatment on the gastric mucosa. Am J Gastroenterol 2003;98:1257-1265.
26. Kuipers EJ, Lundell L, Klinkenberg-Knol EC, Havu N, Festen HP, Liedman B, Lamers CB, Jansen JB, Dalenback J, Snel P, Nelis GF, Meuwissen SG. Atrophic gastritis and Helicobacter pylori infection in patients with reflux esophagitis treated with omeprazole or fundoplication. N Engl J Med 1996;334:1018-1022.

7
27. Kuipers EJ, Uyterlinde AM, Pena AS, Hazenberg HJ, Bloemena E, Lindeman J, Klinkenberg-Knol EC, Meuwissen SG. Increase of Helicobacter pylori-associated corpus gastritis during acid suppressive therapy: implications for long-term safety. Am J Gastroenterol 1995;90:1401-1406.
28. Graham DY, Opekun AR, Yamaoka Y, Osato MS, El-Zimaity HM. Early events in proton pump inhibitor-associated exacerbation of corpus gastritis. Aliment Pharmacol Ther 2003;17:193-200.
29. Adachi K, Fujishiro H, Mihara T, Komazawa Y, Kinoshita Y. Influence of lansoprazole, famotidine, roxatidine and rebamipide administration on the urea breath test for the diagnosis of Helicobacter pylori infection. J Gastroenterol Hepatol 2003;18:168-171.
30. Graham DY, Opekun AR, Hammoud F, Yamaoka Y, Reddy R, Osato MS, El-Zimaity HM. Studies regarding the mechanism of false negative urea breath tests with proton pump inhibitors. Am J Gastroenterol 2003;98:1005-1009.
31. Laine L, Estrada R, Trujillo M, Knigge K, Fennerty MB. Effect of proton-pump inhibitor therapy on diagnostic testing for Helicobacter pylori. Ann Intern Med 1998;129:547-550.

1
Gastric Lumps and Bumps: A tale of two polyps
Henry D. Appelman, M.D.
Department of Pathology
University of Michigan
General Comments
The stomach is far less rich in polyps than is the colon. As a result, we know much less
about gastric polyps than colonic polyps, and the literature tends to be much more recent
and sparser. About 5 to 10% of biopsies of endoscopic gastric polyps are normal
mucosa. The reasons for this include the biopsy forceps missing the lesion, an intramural
lesion that is not available to endoscopic biopsy, normal mucosa that somehow turned
into an endoscopic bump and finally the pathologist doesn’t know a polyp when he or she
sees one. There are about 10 named gastric polyps involving the mucosa including two
surface adenomas, a deep adenoma, two heterotopias, juvenile and Peutz-Jeghers polyps,
polyps with no names and no literatures and finally the two that will be discussed here,
fundic gland polyps and hyperplastic polyps. From a clinician’s standpoint, the main
issue is whether the polyp is neoplastic, and if it is, then whether it is benign or
malignant. If it is not neoplastic, then in general, there does not seem to be a great
interest in polyp type, but we type it anyway. There are a few exceptions to this. It
appears from our practice that there is so little interest on the part of the endoscopists in
the mucosa adjacent to a polyp or in the mucosa in the rest of the stomach, that it is only
biopsied infrequently.
The two common polyps, hyperplastic and fundic gland, have not been rigidly defined in
the literature or in the textbooks, and as a result, we do not know their histologic limits.
Hyperplastic polyps
Historical perspective: During the 1960’s and 1970’s, the most common gastric polyp
had three names from 3 different years, regenerative polyp in 1965 1, hyperplastic polyp
in 19712 and hyperplaseogenous in 1973
4. The hyperplastic designation won over the
other two. In the early literature, these polyps were described as mainly single, although
a few patients made multiples. Histologically, they had a complex surface architecture,
deep cysts, and excess stroma. The architecture included coarse villiform changes on the
surface, ulcers both in the flat and villiform areas, an edematous inflamed lamina propria,
striking distortion of the pits, and glands did not participate. There were a variety of
different types of epithelium including normal pit or foveolar epithelium, hypertrophic pit
epithelium with huge distended goblet cells, and regenerative epithelium with the typical
syncytial undifferentiated cytoplasm and vesicular nuclei that characterize regenerative
epithelium everywhere. In some polyps, the epithelium is more atypical with elongated
stratified nuclei, more frequent mitoses, and less cytoplasmic mucin. Intestinal
metaplasia occurs, but is not a dominant component, and in one study from Japan, it was
mentioned that the likelihood of finding intestinal metaplasia in the polyp was less than

2
finding it in the surrounding mucosa11
. Of course this comment was based on Japanese
stomachs which commonly have intestinal metaplasia. I cannot find comparable
information on intestinal metaplasia in United States stomachs with these polyps.
Hyperplastic polyps are said to occur in three settings. The first, the sporadic
polyp is the most common. The second and third settings are based on the findings of
somewhat similar changes in polypoid mucosa on the gastric side of a gastroenteric
anastomosis, and on the top of the proximal-most gastric folds, originally described in
refluxers, but also found in non-refluxers. It turns out that the anastomosis and proximal
fold polyps never quite achieve the distortion, exuberance and stroma of the sporadic
polyps, so it is questionable whether they are all the same.
Elster in 1976 stated that “hyperplastic polyps of the stomach have no counterpart
in other parts of the gastrointestinal tract and are thereby organotypical”4. This was
reemphasized by Hattori from Japan nine years later, and it is clear that this is a true
analysis11
. We see occasional polyps in other sites, especially the colon, that have
somewhat similar architecture and stroma, but they never quite look the same.
Is there a universally accepted, clear-cut definition of the hyperplastic polyp? In the
literature and in the textbooks it becomes hardly any author ever actually defines it, but
the authors do describe it, although the descriptions are not necessarily comparable. In
one description from Stolte et al from Germany in 199512
, a hyperplastic polyp was
characterized by lengthening, tortuosity, and variable cystic dilatation of foveolae,
widening of the stroma and edema, increased numbers and dilatation of the capillaries
and apical erosions. However, this is a description and not a definition, and it does not
tell us what features are required for the diagnosis of hyperplastic polyp and to what
extent they must be present. As a result, whenever a study on hyperplastic polyps was
published, it was not clear if the authors of those studies actually analyzed the same
lesions. We also have no idea how the full-blown polyp develops. Does it start from a
localized focus of pit expansion and stromal edema and inflammation or simply from a
small polyp containing only elongated foveolae, a lesion which has been designated as
focal or polypoid foveolar hyperplasia? Not only don’t we know how this lesion evolves,
but we have no clue as to what is the stimulus for its development. We don’t know if it is
inflammatory or neoplastic or a mixture. If we look at small lesions that have all of the
architectural and stromal features, they appear to arise in the pit or foveolar compartment
and look like they are tacked onto the surface. We must be aware of the possibility that
since there are no minimal requirements for the diagnosis of hyperplastic polyp, a lot of
things are thrown into that category that may not belong there, and some of these are
presumably included in published studies of hyperplastic polyps. This is a common
situation throughout not just pathology but medicine in general, namely a tendency to try
to fit everything into existing categories, rather than separating them into categories that
currently are not named.
There is a fact (or rumor) that hyperplastic polyps occur in inflamed stomachs13,
14
,
as much as 40% with Helicobacter pylori, and others with atrophic gastritis including the
autoimmune type. Reports also indicate that there may be additional chemical or reactive
gastropathy, but this is so common in stomachs these days, that its significance is
questionable. Data such as this suggests that these are inflammatory lesions, but we don’t
know that for a fact.

3
It is also stated that dysplasia occurs in anywhere from 1 to 20% of hyperplastic
polyps, and this is size related with more dysplasia in larger lesions. Cancers have even
been described in a very, very few polyps. There is clearly something wrong with data
that puts anything between 1 and 20%. The inflammatory and dysplastic data had been
based on studies of a polyp that had and still has no minimal diagnostic criteria and also
on publications some of which are based on studies including only whole intact polyps
and on other studies that included biopsies as well which potentially introduced sampling
problems.
What separates low-grade dysplasia from regenerative epithelium in these polyps
is exactly the same thing that separates low-grade dysplasia from regenerative epithelium
everywhere else in the gastrointestinal tract. It is sometimes impossible to tell which
kind of epithelium is present. These polyps are sitting in stomachs that contain a lot of
solid material and which have churning and mixing motility activities, so these polyps are
probably banged around a lot. Therefore, whatever the authors call this epithelium is
what it gets published as, and perhaps this accounts for the dysplasia rate that varies from
1 to 20%. Furthermore, is there anything that separates a hyperplastic polyp with
dysplasia from an adenoma with secondary hyperplastic polyp changes? We have seen
some adenomas in which fairly extensive hyperplastic polyp-like changes involve the
surface. Biopsies may exacerbate this distinction because of sampling.
The neoplastic associations, regardless of how we interpret the data, color
surveillance recommendations. For instance, the American Society for Gastrointestinal
Endoscopy has published guidelines in 2006 that are available on its website
(www.ASGE.org). These guidelines state that the polyps should be endoscopically
excised wherever feasible and clinically appropriate. No surveillance endoscopy is
necessary after adequate sampling (there is no definition of adequate sampling) or
removal of nondysplastic polyps. Topographical biopsy “mapping” may be useful to
detect the presence of gastritis and intestinal metaplasia.
Topographical biopsy mapping is not done at our institution. I am sure that there
are institutions where it is the rule, but I don’t even know that for a fact.
Summary: we do not know what hyperplastic polyps are, what causes them, what their
precursors are, and there are no minimal criteria for diagnosis. It is not clear that all
hyperplastic polyp studies have actually studied the same polyps, so the results of these
studied probably should not be pooled for analysis. The dysplasia/carcinoma risk is not
settled. Nevertheless, we will keep trying!
Fundic gland polyps
General Comments: Beginning in the 1970’s we started seeing another gastric polyp
which also had little published information. This is what we now call a fundic gland
polyp (FGP). These days it is the most common of all gastric polyps, probably 7 to 1 over
its closest competitor, the hyperplastic polyp. Sometimes these are part of a syndrome,
familial adenomatous polyposis, that includes cancers, but cancers for all intents and
purposes don’t occur in fundic gland polyps. They also tend to carpet the fundus in the
body, especially in the patients with FAP. As with hyperplastic polyps, they also appear
to be tacked onto the top of the normal oxyntic mucosa indicating that they have
developed within the pit compartment.

4
Historical perspective: It appears that the first recognition or name for fundic gland
polyp was by Elster in 1976. He referred to these as “cysts of gastric glands”, and then a
year later he changes the name to “fundic gland cysts”4,15
. These were described as
“fundic glandular cysts in otherwise normal gastric mucosa”. However, an analysis of
the illustrations in his papers indicates that this otherwise normal gastric mucosa is really
not normal but the superficial glands are disorganized, clustered, often budding and
branching. Watanabe from Japan in an analysis of gastric lesions in FAP may have been
the first to use the fundic gland polyp designation16
. In his paper, there was no definition
of fundic gland polyp, just many illustrations and descriptions such as “simple
hyperplasia of fundic glands”. Actually, it is difficult to recognize any hyperplasia, since
there are fewer glands per unit area in FGPs than in normal oxyntic mucosa. What was
mentioned in Watanabe’s paper was the fact that the glands were irregular, tortuous,
sometimes branching; that is, they were disorganized. Before the Elster and Watanabe
papers, we only recognized two types of gastric polyps, adenomas and hyperplastic
polyps. Fundic gland polyps were probably included in the mix. We did not recognize
them as separate, so we probably called them hyperplastic, since they did not look like
adenomas. They don’t have adenoma-like dysplasia.
Diagnostic criteria: In the twelve months from August, 2005 through July, 2006, four
pathologists in the gastrointestinal subspecialty sign-out service at the University of
Michigan made the diagnosis of fundic gland polyps in 306 patients, 16 of whom had
familial adenomatous polyposis. The age distribution of patients with these polyps
corresponds to the age of the patients who were biopsied during upper endoscopy. There
were twice as many women as men, but this has been found in other studies. Regardless,
when I asked my colleagues, and even myself how to define a fundic gland polyp and to
list the minimal diagnostic criteria, there was no consensus. Nevertheless, I showed them
several polyps which had a variety of changes including short pits, long pits, clusters of
glands beneath the surface that varied from area to area, cysts, some of which were gland
cysts, some pit cysts and some mixed cysts, and expanded lamina propria which often
had a lot of smooth muscle. Everybody diagnosed them as fundic gland polyps, and the
basic reasons for diagnosis boiled down to “because they looks like it”. I showed the
same polyps to a bunch of our house officers and they came up with the same diagnosis
for the same reason. Therefore, we seem to know what a fundic gland polyp looks like,
and we can teach other people how to recognize it, but we have a great deal of difficulty
defining what it is leads us to that diagnosis.
As was true for hyperplastic polyps, there are no published minimal criteria that
will allow a bump in the gastric oxyntic mucosa to be called a FGP. A look at the
literature and in the textbooks and even in the World Health Organization Classification
of Tumors published in 2000, the definitions are anything but uniform.
The Appelman Approach: FGPs for dummies (for what it is worth): FGPs are
architecturally complex but cytologically simple lesions. They are architecturally
complex in that the entire mucosa is structurally altered when compared to normal, and
they are cytologically simple because all cells are mature gastric epithelial cells. Perhaps
because of this combination, they have been referred to as hamartomas. There are two
sets of architectural abnormalities, epithelial and stromal. The intensity of these changes
varies greatly from one polyp to another. The epithelial architectural alterations involve
both the pits and the glands. The pit changes involve length. In most areas of these

5
polyps, the pits are shorter than normal, but in some polyps, they are longer. Pits also
extend deeply into the mucosa and form cysts. The gland changes are more complex and
include clusters of glands beneath the surface, parietal cells in the upper parts of the pits
or even on the surface, glands with irregular branches and buds, and cystic glands toward
the middle and deeper parts of the polyps. Actually, there are many cysts that have a
mixture of pit and gland epithelium. The stromal changes include an increase in the
amount of lamina propria when compared to normal oxyntic mucosa. This bonus stroma
may be edematous, may have inflammation, and often has smooth muscle bundles,
probably not extending from the muscularis mucosae, considering that these polyps are
tacked onto the surface. The bigger the polyp, the larger the number and size of the cysts.
This suggests that these polyps enlarge by an increase in number and size of cysts.
Fundic gland polyps are said to occur in three settings. First are those that are
associated with familial adenomatous polyposis, in other words in patients with a
germline mutation in the FAP gene. Second are sporadic polyps, that is, not in FAP
patients, but occurring in people who are not taking proton pump inhibitors. Third are
sporadic polyps occurring in patients who have been treated with proton pump inhibitors.
Genetic Changes: The polyps in different patients with FAP do not look the same, but
neither do the polyps from single FAP patients. However, they all have variations on the
same abnormalities including pit and gland architecture and lamina propria changes. In
one study on FAP patients with known germline APC gene mutations, APC gene
alterations were found in at least one polyp in 9 of these 11 patients, but not all polyps
from individual patients had the detectable gene alterations23
. Conceivably, this variation
may be due to the fact that this is a 9-year-old study, and there may be better detection
systems now. Furthermore, no individual patient had more than one APC gene alteration
in the polyps.
Genetic changes were also analyzed in sporadic fundic gland polyps that have no
APC gene defects. In one study from the USA, 52 of 57 polyps from 40 patients have
beta-catenin mutations24
. In this study, there was no mention if the patients were taking
proton pump inhibitors. In another study from Japan, 29 of 45 such polyps from 35
patients had the beta-catenin mutation, and none of these patients were on long term
proton pump inhibitors25
. In both these studies, the mutations were found in both the pit
epithelium and in the gland cyst epithelium in almost all polyps, and different mutations
were found in different polyps from the same patients, in contrast to the findings of the
FAP patients. In another study of patients taking proton pump inhibitors, CpG island
methylation was found to be more common in sporadic polyps, but we have not had any
data since the publication of that study 6 years ago28
.
Do proton pump inhibitors cause FGPs? Twenty years ago these polyps were
curiosities, but there has been a striking increase in incidence that seems to coincide with
the increased use of PPIs. The data regarding cause and effect are conflicting. In one
study from the USA, two groups of patients were compared, a larger group not taking
proton pump inhibitors who only had a single upper endoscopy, and a smaller group
taking PPIs who had both an initial exam and a follow-up exam if they had no polyps at
the first exam20
. Comparing these two groups, FGPs were much more common in the
PPI group, but so were other polyps, and in this study, there was no second endoscopy for
the group not taking PPIs. Also, FGPs were described simply as composed of cystic
fundic glands, nothing more and nothing less.

6
The second study from Germany compared over 28,000 patients not taking PPIs
with over 2200 patients on the drugs, and the prevalence of FGPs was identical22
. We do
know that PPIs induce hypertrophy of parietal cells with formation of apical snouts, and
they also induce fundic gland cysts. Both of these changes appear to increase with
increasing length of time the patients are taking PPIs. Both of these changes appear to be
secondary to the hypergastrinemia that results from inhibition of gastric acid production
by the drugs.
FGPs contain many parietal cells. The parietal cells in the polyps seem to
respond to PPIs as do the native parietal cells, namely they undergo hypertrophy and
develop snouts. Perhaps PPIs do not cause FGPs, but they may make tiny ones bigger
and endoscopically apparent as a result of the parietal cell hypertrophy and increase in
gland cysts21
.
Finally, there is some literature suggesting that in patients with fundic gland polyps,
there is an increased risk for colonic adenomas and carcinomas. However, the
studies do not all come to the same conclusion9, 30
. In a recent study, FGPs were
associated with increased prevalence of hyperplastic colonic polyps in men and colonic
adenomas in women mainly over 60 years of age, but there was no increased association
with adenocarcinoma.
Summary: Fundic gland polyps occur in familial adenomatous polyposis but also
spontaneously, and their incidence has increased at the same time as has the use of proton
pump inhibitors. They are architecturally complex and cytologically simple polyps.
There is no proof that PPIs induce polyps with the same architectural complexity that
occurs in FAP patients. The parietal cells in FGPs respond to PPIs exactly like parietal
cells in flat mucosa. This may make some tiny FGPs enlarge and become endoscopic
polyps. Genetic abnormalities have been found in both FAP and sporadic FGPs, and
there may be some alteration in PPI associated polyps. The association between FGPs
and colonic neoplasia is not clearly established. Finally, until we have a rigid definition
of the minimal criteria for a fundic gland polyp, we will have no clue as to whether PPIs
or anything else cause them.
References for Stomach Polyps
General Polyp References
1. Ming S-C, Goldman H. Gastric polyps. A histogenetic classification and its relation
to carcinoma. Cance. 18:721-726, 1965
2. Tomasulo J. Gastric polyps. Histologic types and their relationship to gastric
carcinoma. Vanver. 27:1346-1355, 1971
3. Goldman DS, Appelman HD: Gastric mucosal polyps. Am J Clin Pathol
1972;58:434-444
4. Elster K. Histologic classification of gastric polyps. In: Morson BC, ed. Pathology of
the Gastrointestinal Tract. Berlin: Springer-Verlag, 1976:77-93
5. Nakamura T, Nakano G-I. Histopathological classification and malignant change in
gastric polyps. J Clin Pathol. 38:754-764, 1985

7
6. Appelman HD. Localized and extensive expansions of the gastric mucosa: mucosal
polyps and giant folds. In: Appelman HD, ed. Pathology of the Esophagus, Stomach
and Duodenum. New York: Churchill Livingstone, 1984:79-104
7. Appelman HD. Chapter 9, Non-neoplastic tumor-like lesions, predominantly
epithelial, in Lewin KJ, Appelman HD. Tumors of the Esophagus and Stomach.
Fascicle 18, Third Series, Atlas of Tumor Pathology, Armed Forces Institute of
Pathology, Washington, D.C., 1996:183-232
8. Park DY, Lauwers GY. Gastric polyps: classification and management. Arch Pathol
Lab Med. 132:633-640, 2008
9. Carmack SW, Genta RM, Schuler CM, Saboorian MH. The current spectrum of
gastric polyps: a 1-year national study of over 120,000 patients. Am J Gaastroenterol.
104:1524-1532, 2009
10. Carmack SW, Genta RM, Graham DY, Lauwers GY. Management of gastric polyps: a
pathology-based guide for gastroenterologists. Nat. Rev. Gastroenterol. Hepatol 6:331-341,
2009
Hyperplastic Polyp References
11. Hattori T. Morphologic range of hyperplastic polyps and carcinomas arising in
hyperplastic polyps of the stomach. J Clin Pathol. 1985;38:622-630
12. Stolte M, Bethke B, Sticht T, Burkhard U. Differentiation of focal foveolar
hyperplasia from hyperplastic polyps in gastric biopsy material. Path Res Pract.
191:1198-1202, 1995
13. Abraham SC, Singh VK, Yardley JH, Wu T-T. Hyperplastic polyps of the stomach.
Association with histologic patterns of gastritis and gastric atrophy. Am J Surg
Pathol. 25:500-507, 2001
14. Jain R, Chetty R. Gastric hyperplastic polyps: a review. Dig Dis Sci. 54:1839-46, 2009.
Fundic Gland Polyp References
15. Elster K, Eidt H, Ottenjann R, Rosch W, Seifert E. The glandular cyst, a polypoid
lesion of the gastric mucosa. Dtsch Med Wochenschr. 1977 Feb 11;102(6):183-7.
16. Watanabe H, Enjoji M, Yao T, Ohsato K. Gastric lesions in familial adenomatosis
coli. Their incidence and histologic analysis. Hum Pathol. 9:269-283, 1978
17. Sipponen P, Laxen F, Seppala K. Cystic “hamartomatous” gastric polyps: a disorder
of oxyntic glands. Histopathol. 7:729-737, 1983
18. Iida M, Yao T, Watanabe H, Itoh H, Iwashita A. fundic gland polyposis in patients
without familial adenomatosis coli: its incidence and clinical features. Gastroenterol.
86:1437-1442, 1984
19. Lee RG, Burt RW. The histopathology of fundic gland polyps of the stomach. Am J
Clin Pathol. 86:498-503, 1986
20. Choudhry U, Boyce HW Jr, Coppola D. Proton pump inhibitor-associated gastric
polyps. A retrospective analysis of their frequency, and endoscopic, histologic and
untrastructural characteristics. Am J Clin Pathol. 110:615-621, 1998
21. Cats A, Schenk BE, Bloemena E, et al. Parietal cell trotrusions and fundic gland cysts
during omeprazole maintenance treatment. Hum Pathol. 31:684-690, 2000
22. Vieth M. Stolts M. Fundic gland polyps are not induced by proton pump inhibitor
therapy. Am J Clin Pathol. 116:716-720, 2001
23. Abraham SC, Nobukawa B, Giardiella FM, et al. Fundic gland polyps in familial
adenomatous polyposis. Neo-plasms with frequent somatic adenomatous polyposis
coli gene alterations. Am J Pathol. 157:747-754, 2000

8
24. Abraham SC, Nobukawa B, Giardiello FM, et al. Sporadic fundic gland polyps.
Common gastric polyps arising through activating mutations in the ß-catenin gene.
Am J Pathol. 158:1005-1010, 2001
25. Sekine S, Shibata T, Yamauchi Y, et al. ß-catenin mutations in sporadic fundic gland
polyps. Virchows Arch. 440:381-386, 2002
26. Abraham SC, Park SJ, Mugartegui L, et al. Sporadic fundic gland polyps with
epithelial dysplasia. Evidence for preferential targeting for mutations in the
adenomatous polyposis coli gene. Am J Pathol. 161:1735-1742, 2002
27. Torberson M, Le J-H, Cruz-Correa M, et al. Sporadic fundic gland polyposis: a
clinical, histological, and molecular analysis. Mod Pathol. 15:716-723, 2002
28. Abraham SC, Park SJ, Cruz-Correa M, et al. Frequent CpG island methylation in
sporadic and syndromic gastric fundic gland polyps. Am J Clin Pathol. 122:740-746,
2004
29. Burt RW. Gastric fundic gland polyps. Gastroenterol. 125:1462-1469, 2003
30. Teichmann J, Weickert U, Riemann JF. Gastric fundic gland polyps and colonic
polyps—is there a link, really? Eur J Med Res. 13:192-195, 2008

Lauwers-Early gastric neoplasms
1
EARLY GASTRIC NEOPLASMS: DIAGNOSES AND IMPLICATIONS
Gregory Y. Lauwers, M.D. Director, Division of Surgical Pathology
Director, Gastrointestinal Pathology Service Massachusetts General Hospital and
Harvard Medical School Boston, Massachusetts, USA
Despite a marked decline in incidence in the West and some decrease in the East, gastric
cancer remains a significant cause of morbidity and cancer related deaths worldwide. The
prevalence of gastric cancer is closely related to prevalence of Helicobacter pylori infection and
shows wide geographic variation. Subsequent chronic gastritis, atrophy, and intestinal metaplasia
are lesions that confer a high risk for the development of gastric cancer, while gastric dysplasia, the
penultimate stage of the carcinogenetic cascade, is a direct neoplastic precursor lesion.2, 4, 14-16, 18, 22,
36, 41, 60, 64 Evidence for gastric dysplasia as a direct precursor of adenocarcinoma stems from
observational studies reporting high-grade dysplasia (HGD) in close proximity to 40-100% of early
gastric cancers and 5-80% of advanced adenocarcinomas.48, 61, 95 Moreover, dysplasia is also a
marker of increased risk for cancer elsewhere in the gastric mucosa.
As with gastric cancer, the prevalence of dysplasia shows wide geographic variations. The
difference is likely related to variations in the genetic makeup of the population, as well as
variations in environmental factors, such as the prevalence of Helicobacter pylori infection (and
subtype) and the age at which the infection is acquired.2, 6, 11, 14-16, 18, 22, 25, 51, 60, 64, 79, 95 The frequency of
dysplasia also varies with the underlying etiology. For instance, prevalence rates of up to 40%
have been reported in patients with pernicious anemia but the disease confers only a moderate
increase in risk of developing gastric cancer.3, 6, 33, 94 In the setting of familial adenomatous polyposis
(FAP), flat or polypoid dysplasias, typically antral in location, are frequently multiple and may be
seen in 2-50% of patients.7, 8, 10, 21, 31, 38, 74, 75, 80, 82, 86, 87 Patients with a gastric remnant status post
gastrectomy, Menetrier’s disease, or Peutz-Jegher’s syndrome also are at increased risk.5, 44, 81, 89

Lauwers-Early gastric neoplasms
2
Classification of Gastric Epithelial Dysplasia
The classification of dysplastic lesions has been controversial, with various diagnostic
criteria used across the world. Japanese authors refer to these as borderline (Group 3 or 4)
lesions, while the terms gastric adenoma (for raised lesions) and gastric dysplasia (for
flat/depressed lesions) have been widely used in the Western literature.32, 67, 77
Earlier guidelines for the diagnosis and grading of gastric dysplasia embraced a three-
tiered system of mild, moderate and severe dysplasia. As in any segment of the gastrointestinal
tract, dysplasia was defined as "unequivocally neoplastic epithelium that may be associated with or
give rise to invasive adenocarcinoma."54, 57, 66 Later schemes have proposed a two-tiered system of
low- and high-grade dysplasia,48, 69, 70, 78 which has proven to be more reproducible and provides a
clinically meaningful risk stratification.32, 47 The WHO recommends the terminology of non-invasive
low-grade and high-grade intraepithelial neoplasia, and defines carcinoma as invasion into the
lamina propria or beyond.34 However, the terminology of adenoma/dysplasia is widely entrenched,
and continues to be used, particularly in North America.
A significant debate has occurred over differentiating adenocarcinoma from high-grade
dysplasia. Furthermore, the complexity of cyto-architectural features has been considered to be of
paramount importance for the diagnosis of carcinoma in Japan, while breach of the basement
membrane and invasion into the lamina propria has been considered the sine qua non of
malignancy and hence a prerequisite for the diagnosis of cancer in the West.47, 76 As an attempt to
bridge differences, the Vienna classification was developed as a consensus between Western and
Asian investigators.78 This consensus view takes into account the discrepancies in the reporting of
dysplasia between Japanese and Western pathologists. For example, non-invasive intramucosal
neoplastic lesions with high-grade cellular and/or architectural atypia are classified as
"intramucosal carcinoma" in Japan, whereas similar lesions are diagnosed as high-grade dysplasia
by most Western pathologists. In the Vienna classification, high-grade lesions without invasion of
the lamina propria and adenocarcinomas with invasion confined to the lamina propria, are now
placed into a single diagnostic category, a rationale supported by current endoscopic management.

Lauwers-Early gastric neoplasms
3
Phenotypic variants of gastric dysplasia
Most examples of dysplasia have an "intestinal" phenotype, i.e., resembling colonic
adenomas. These lesions are commonly referred to as adenomatous (or type I) dysplasia. The
histologic characteristics include crowded glands lined by tall columnar cells with pencillate,
overlapping, and hyperchromatic nuclei which show pseudostratification and inconspicuous
nucleoli.42 Other less common histologic variants of include foveolar (type II or non-adenomatous)
dysplasia and pyloric type dysplasia.42 The distinctive feature of Type II dysplasia is the presence
of glands lined by either low cuboidal or columnar epithelium with pale-clear cytoplasm, round-oval,
vesicular nuclei and variably prominent nucleoli. Although prior studies have suggested that this
form of dysplasia is almost always low-grade,1 more recent studies indicate that Type II dysplasia
may be associated with distinct clinico-pathological characteristics and is more often high-grade
when evaluated in a high-risk population.63 Some authors have indicated that Type II dysplasia is
more commonly associated with poorly-differentiated adenocarcinoma.42, 56, 58
Pyloric type dysplasia is a recently recognized type of dysplasia. Frequently observed in
the body fundus, it is commonly seen in the older population. Some series indicate that it is
commonly shows high grade dysplasia.13
Tubule neck (or globoid) dysplasia is exceedingly rare and is believed to be a precursor of
diffuse-type gastric carcinoma.30 It occurs in non-metaplastic gastric epithelium and appears as
enlarged, clear cells occupying the gland neck region and confined within the basement
membrane.
In the setting of inherited germline E-cadherin/CDH1 gene mutation, prophylactic
gastrectomies have shown examples of "signet ring cell carcinoma in situ," often with a "pagetoid"
spread between the gastric foveolar and glandular epithelium within the basement membrane.12, 50
These changes are often multifocal and have a predilection for the proximal stomach and the body-
antral transitional zone.

Lauwers-Early gastric neoplasms
4
Grading of gastric dysplasia
The two-tiered scheme of low and high grade is widely used in all classification schemes.
Practically, gastric biopsies need to be categorized into one of several categories: negative for
dysplasia, indefinite for dysplasia, low grade dysplasia/adenoma, high grade dysplasia/adenoma,
intramucosal carcinoma or an invasive adenocarcinoma.
Indefinite for dysplasia
There are cases for which one cannot establish a diagnosis with certainty. Commonly it
means differentiating between reactive epithelial changes and dysplasia. It should be seen as a
provisional designation that emphasizes the need to follow up the patient and to obtain additional
biopsies. Alternatively, it should not be used as a wastebasket term for all cases with reactive
atypia which are obviously in response to inflammatory or direct mucosal injury. Clues to the
reactive nature of the epithelial changes includes the presence of vascular congestion and a
gradual rather then abrupt transition between the atypical and adjacent normal cells.54, 66
Low-grade dysplasia (Adenoma; Non-invasive intraepithelial neoplasia-low grade)
We used the term "adenoma" for elevated mucosal lesions and low grade dysplasia for flat
lesions that show minimal architectural disarray and cytological atypia.32, 47, 54, 66, 95 As mentioned
earlier, in most cases, the morphological appearance is reminiscent of colonic adenomas and the
lesions often occur in a background of intestinal metaplasia. The criteria for separating Type II
dysplasia into low and high grade categories are not well established. The presence of gastric
foveolar type epithelium with elongated, hyperchromatic nuclei that show some degree of
pseudostratification is categorized as low-grade Type II dysplasia. Although a designation of low-
grade implies a comparatively reduced risk of malignant transformation, it must be recognized that
low-grade dysplasia occurring in a background of extensive intestinal metaplasia may be
associated with a higher risk of malignancy.71

Lauwers-Early gastric neoplasms
5
High-grade dysplasia (Adenoma with high-grade dysplasia; Non-invasive intraepithelial neoplasia-
high grade)
Marked cytological atypia or architectural complexity deserve a diagnosis of high-grade
dysplasia. High grade dysplastic glands are commonly lined by rounded, pleomorphic nuclei that
show prominent nucleoli and loss of polarity. Marked irregularities of the nuclear membrane and
clumping of chromatin are also features often associated with high grade dysplasia. However,
marked glandular crowding, budding, and intra-luminal bridges should raise the question of early
gastric cancer. Typical or atypical mitoses may be present in either low grade or high grade
dysplasia, but are more often and more easily discernible in the latter category.32, 47, 69
Intramucosal adenocarcinoma
The controversy and disparity in literature regarding separation of "dysplasia" from
"carcinoma" has already been alluded to above. The current approach to this problem is based on
two facts: 1) "invasion," particularly when limited to the lamina propria, is difficult to identify on
routine histology, and 2) intramucosal adenocarcinomas have a less than 10% risk of nodal
metastases24 and neoplastic lesions with invasion of the lamina propria but confined to the mucosa
are, therefore, amenable to a conservative approach through endoscopic resection. Currently,
lesions which show marked architectural atypia in the form of fused glandular pattern, cribriforming
or intra-luminal necrosis, as well as those that show definite evidence of invasion into the lamina
propria in the form of single cells or small clusters of cells, are categorized as intramucosal
adenocarcinoma.
Characteristics of intramucosal of adenocarcinomas
Intramucosal adenocarcinoma belong to the category of early gastric cancers (EGC),
which are defined as invasive adenocarcinomas confined to the mucosa or submucosa, whether
lymph node metastasis is present or not.29, 49 In Western series, EGCs represent between 15% and
21% of all newly diagnosed cancers, while in Japan, they account for over 50% of the cases.23, 28, 37,
83 The higher prevalence of gastric cancer, a more liberal use of upper endoscopy, perhaps a
better technique including chromoendoscopy, and a difference in diagnostic criteria may explain
the difference.

Lauwers-Early gastric neoplasms
6
Most EGCs are small, measuring between 2 cm and 5 cm and localized on the lesser
curvature and around the angulus.49, 55 Multiple tumors are seen in 3% to 13% of the patients, and
are associated with a worse prognosis.23, 53
The Paris classification divides EGCs into 3 types based on the endoscopic macroscopic
appearance (figure 2): Protruded (type I), superficial (type II), and excavated (type III).40 Type II is
further subdivided into IIa (elevated type), IIb (flat type), and IIc (depressed type). Superficial (type
II) EGCs account for about 80% of the cases, with type IIc being the most common subtype.90 Type
IIb accounts for 58% of small tumors measuring less than 5 mm.45 Notably, this endoscopic
classification has shown to be a good indicator of the risk for nodal metastasis, reportedly low in
type Ia or IIa EGC.19
The majority of EGCs are well differentiated glandular carcinomas. Tubular and papillary
variants represent 52% and 37% of cases, respectively, and can be difficult to differentiate from
dysplasia (see above). Signet ring cell carcinoma and poorly differentiated carcinoma represent
26% and 14% of the cases, and are usually depressed or ulcerated (types IIc and III).23, 49, 90 Diffuse
type EGCs tend to have a greater depth of invasion.19
Progression and outcome of early gastric neoplasms
A) Low –grade dysplasia
Although assessing regression of low-grade dysplasia is difficult because of sampling
issues and inter-observer variation in the diagnosis, it has been reported in 38-75%, while
persistence is seen in 19-50% of cases.6, 9, 46
Historical data have reported progression to adenocarcinoma in 0-23% of patients with
LGD within a span of 1-4 years, but recent studies indicated a lower risk of progression (0-9%),
while there is a significant risk of malignant transformation associated in high-grade (10-100%).68, 91

Lauwers-Early gastric neoplasms
7
B) High-grade dysplasia
This diagnosis is more ominous, since HGD has been noted to persist in 14-58% of the
cases and to progress to cancer in 60-85% of patients over a median interval of 4-48 months.20, 27,
43, 46, 70, 73, 91 Regression, though, also has been reported, and varies from 0-16%.
C) Intramucosal adenocarcinoma
In a series of patients diagnosed with EGC and followed without surgery, 63% of the
tumors progressed to advanced carcinomas over a span of 6 to 88 months.84 However, when
resected, the prognosis of EGCs is excellent, with a five-year survival rate greater than 90% in
most series.17, 23, 26, 39, 83 The size and depth of invasion are the two major prognostic indicators, with
the larger the diameter, the greater the risk of submucosal infiltration.28, 52, 93 Notably, the risk of
invasion should not be overlooked even in very small tumors. In one series, 15.5% of 3-5 mm
EGCs invaded the submucosa.62 Even for intramucosal EGCs, lymph node metastases have been
reported in up to 7% of cases. However, the five-year survival remains close to 100%.23, 52, 93 For
EGCs extending into the submucosa, the rate of lymph node metastases is between 8% and 25%,
and the five-year survival is 80% to 90%.23, 93
Management of early gastric neoplasms
Given the demonstrated low rate of malignant transformation of low grade dysplasis and
the development of newer endoscopic imaging techniques, such as chromoendoscopy, annual
endoscopic surveillance with re-biopsy is typically performed and surgical resection is not
necessary.72, 88 Similarly, a diagnosis of indefinite for dysplasia should also prompt endoscopic
surveillance and biopsy.
Although a diagnosis of high-grade dysplasis in years past was the indication for surgery,
nowdays, this diagnosis as well as a diagnosis of intramucosal adenocarcinoma (provided deep
submucosal invasion is ruled out with certainty with endoscopic ultrasound) will be managed
endoscopically since endoscopic mucosal resection and submucosal dissection offer definitive
therapy.

Lauwers-Early gastric neoplasms
8
Endoscopic mucosal resection has rapidly become the treatment of choice in association
with endoscopic ultrasound for staging. The primary criteria of EGC amenable to EMR are elevated
lesions less than 2 cm in size, depressed lesions less than 1 cm in size without ulceration, and the
absence of lymph node metastasis. Endoscopic submucosal dissection (ESD) is a more recent
method developed in order to increase the en bloc and R0 resection rate, especially for lesions
larger than 20 mm in diameter. Drawbacks of endoscopic submucosal dissection include the fact
that it is technically a substantially more difficult procedure and that it is associated with a higher
perforation rate.35, 59, 65, 92 Finally, the eradication of H. pylori improves prognosis of patients with
early neoplasms. In a study of 132 patients with EGC who underwent EMR, no new cases of
gastric cancer were observed after resection when H. pylori was eradicated; in contrast, 13.5% of
untreated patients had new early-stage intestinal-type gastric cancer.85

Lauwers-Early gastric neoplasms
9
REFERENCES
1. Abraham SC, Montgomery EA, Singh VK, et al. Gastric adenomas: intestinal-type and gastric-type adenomas differ in the risk of adenocarcinoma and presence of background mucosal pathology. Am J Surg Pathol. 2002;26:1276-1285. 2. Asaka M, Takeda H, Sugiyama T, et al. What role does Helicobacter pylori play in gastric cancer? Gastroenterology. 1997;113:S56-60. 3. Aste H, Sciallero S, Pugliese V, et al. The clinical significance of gastric epithelial dysplasia. Endoscopy. 1986;18:174-176. 4. Baik SC, Youn HS, Chung MH, et al. Increased oxidative DNA damage in Helicobacter pylori-infected human gastric mucosa. Cancer Res. 1996;56:1279-1282. 5. Bassily R, Smallwood RA, Crotty B. Risk of gastric cancer is not increased after partial gastrectomy. J Gastroenterol Hepatol. 2000;15:762-765. 6. Bearzi I, Brancorsini D, Santinelli A, et al. Gastric dysplasia: a ten-year follow-up study. Pathol Res Pract. 1994;190:61-68. 7. Bertoni G, Sassatelli R, Nigrisoli E, et al. Dysplastic changes in gastric fundic gland polyps of patients with familial adenomatous polyposis. Ital J Gastroenterol Hepatol. 1999;31:192-197. 8. Bulow S, Lauritsen KB, Johansen A, et al. Gastroduodenal polyps in familial polyposis coli. Dis Colon Rectum. 1985;28:90-93. 9. Burke AP, Sobin LH, Shekitka KM, et al. Dysplasia of the stomach and Barrett esophagus: a follow-up study. Mod Pathol. 1991;4:336-341. 10. Burt RW, Berenson MM, Lee RG, et al. Upper gastrointestinal polyps in Gardner's syndrome. Gastroenterology. 1984;86:295-301. 11. Camilleri JP, Potet F, Amat C, et al. Gastric mucosal dysplasia: preliminary results of a prospective study of patients followed for periods of up to six years. In: Ming SC, ed. Precursors of Gastric Cancer. New York: Praeger; 1984:83-92. 12. Carneiro F, Huntsman DG, Smyrk TC, et al. Model of the early development of diffuse gastric cancer in E-cadherin mutation carriers and its implications for patient screening. J Pathol. 2004;203:681-687. 13. Chen ZM, Scudiere JR, Abraham SC, et al. Pyloric gland adenoma: an entity distinct from gastric foveolar type adenoma. Am J Surg Pathol. 2009;33:186-193. 14. Correa P. The epidemiology and pathogenesis of chronic gastritis: three etiologic entities. Front Gastrointest Res. 1980;6:98-108. 15. Correa P. Clinical implications of recent developments in gastric cancer pathology and epidemiology. Semin Oncol. 1985;12:2-10. 16. Correa P. A human model of gastric carcinogenesis. Cancer Res. 1988;48:3554-3560. 17. Correa P. Human gastric carcinogenesis: a multistep and multifactorial process-- First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992;52:6735-6740. 18. Correa P, Tahara E. Stomach. In: Henson D, Albores-Saavedra J, eds. Pathology of Incipient Neoplasia. Philadelphia: WB Saunders; 1993. 19. Craanen ME, Dekker W, Ferwerda J, et al. Early gastric cancer: a clinicopathologic study. J Clin Gastroenterol. 1991;13:274-283. 20. Di Gregorio C, Morandi P, Fante R, et al. Gastric dysplasia. A follow-up study. Am J Gastroenterol. 1993;88:1714-1719. 21. Domizio P, Talbot IC, Spigelman AD, et al. Upper gastrointestinal pathology in familial adenomatous polyposis: results from a prospective study of 102 patients. J Clin Pathol. 1990;43:738-743. 22. EUROGAST. An international association between Helicobacter pylori infection and gastric cancer. The EUROGAST Study Group. Lancet. 1993;341:1359-1362. 23. Everett SM, Axon AT. Early gastric cancer in Europe. Gut. 1997;41:142-150. 24. Everett SM, Axon AT. Early gastric cancer in Europe. Gut. 1997;41:142-150.

Lauwers-Early gastric neoplasms
10
25. Farinati F, Rugge M, Di Mario F, et al. Early and advanced gastric cancer in the follow-up of moderate and severe gastric dysplasia patients. A prospective study. I.G.G.E.D.--Interdisciplinary Group on Gastric Epithelial Dysplasia. Endoscopy. 1993;25:261-264. 26. Farley DR, Donohue JH. Early gastric cancer. Surg Clin North Am. 1992;72:401-421. 27. Fertitta AM, Comin U, Terruzzi V, et al. Clinical significance of gastric dysplasia: a multicenter follow-up study. Gastrointestinal Endoscopic Pathology Study Group. Endoscopy. 1993;25:265-268. 28. Folli S, Dente M, Dell'Amore D, et al. Early gastric cancer: prognostic factors in 223 patients. Br J Surg. 1995;82:952-956. 29. Fujita S. Biology of early gastric carcinoma. Pathol Res Pract. 1978;163:297-309. 30. Ghandur-Mnaymneh L, Paz J, Roldan E, et al. Dysplasia of nonmetaplastic gastric mucosa. A proposal for its classification and its possible relationship to diffuse-type gastric carcinoma. Am J Surg Pathol. 1988;12:96-114. 31. Goedde TA, Rodriguez-Bigas MA, Herrera L, et al. Gastroduodenal polyps in familial adenomatous polyposis. Surg Oncol. 1992;1:357-361. 32. Goldstein NS, Lewin KJ. Gastric epithelial dysplasia and adenoma: historical review and histological criteria for grading. Hum Pathol. 1997;28:127-133. 33. Graem N, Fischer AB, Beck H. Dysplasia and carcinoma in the Billroth II resected stomach 27-35 years post-operatively. Acta Pathol Microbiol Immunol Scand [A]. 1984;92:185-188. 34. Hamilton S, Aaltonen L, eds. Pathology and Genetics of Tumours of the Digestive System. Lyon, France: IARC Press; 2000. 35. Hiki Y, Shimao H, Mieno H, et al. Modified treatment of early gastric cancer: evaluation of endoscopic treatment of early gastric cancers with respect to treatment indication groups. World J Surg. 1995;19:517-522. 36. Hill M. Epidemiology and mechanism of gastric carcinogenesis. In: Reed P, Carbonia M, Johnston B, et al., eds. New trends in gastric cancer background and videoscopy. London: Kluwer Academic; 1989:3-12. 37. Hisamichi S. Screening for gastric cancer. World J Surg. 1989;13:31-37. 38. Iida M, Yao T, Itoh H, et al. Natural history of gastric adenomas in patients with familial adenomatosis coli/Gardner's syndrome. Cancer. 1988;61:605-611. 39. Itoh H, Oohata Y, Nakamura K, et al. Complete ten-year postgastrectomy follow-up of early gastric cancer. Am J Surg. 1989;158:14-16. 40. Japanese Research Society for Gastric Cancer. Group classification of gastric biopsy specimens. In: Nishi M, Omori Y, Miwa K, eds. Japanese Classification of Gastric Carcinoma. Tokyo: Kanehara; 1995:74-76. 41. Jass JR. Role of intestinal metaplasia in the histogenesis of gastric carcinoma. J Clin Pathol. 1980;33:801-810. 42. Jass JR. A classification of gastric dysplasia. Histopathology. 1983;7:181-193. 43. Kokkola A, Haapiainen R, Laxen F, et al. Risk of gastric carcinoma in patients with mucosal dysplasia associated with atrophic gastritis: a follow up study. J Clin Pathol. 1996;49:979-984. 44. Kondo K. Duodenogastric reflux and gastric stump carcinoma. Gastric Cancer. 2002;5:16-22. 45. Kurihara M, Shirakabe H, Yarita T, et al. Diagnosis of small early gastric cancer by X-ray, endoscopy, and biopsy. Cancer Detect Prev. 1981;4:377-383. 46. Lansdown M, Quirke P, Dixon MF, et al. High grade dysplasia of the gastric mucosa: a marker for gastric carcinoma. Gut. 1990;31:977-983. 47. Lauwers GY, Riddell RH. Gastric epithelial dysplasia. Gut. 1999;45:784-790. 48. Lewin K, Appleman H. Atlas of Tumor Pathology: Tumors of the esophagus and stomach. Third series. Washington, D. C.: Armed Forces Institute of Pathology; 1996. 49. Lewin KJ, Appleman HD. Carcinoma of the stomach. Tumors of the esophagus and stomach. Chapter 11. Atlas of Tumor Pathology (Third Series Fascicle 18). Washington: Armed Forces Institute of Pathology.; 1996:245-330.

Lauwers-Early gastric neoplasms
11
50. Lynch HT, Grady W, Suriano G, et al. Gastric cancer: new genetic developments. J Surg Oncol. 2005;90:114-133; discussion 133. 51. Machado JC, Pharoah P, Sousa S, et al. Interleukin 1B and interleukin 1RN polymorphisms are associated with increased risk of gastric carcinoma. Gastroenterology. 2001;121:823-829. 52. Maehara Y, Orita H, Okuyama T, et al. Predictors of lymph node metastasis in early gastric cancer. Br J Surg. 1992;79:245-247. 53. Marrano D, Viti G, Grigioni W, et al. Synchronous and metachronous cancer of the stomach. Eur J Surg Oncol. 1987;13:493-498. 54. Ming SC, Bajtai A, Correa P, et al. Gastric dysplasia. Significance and pathologic criteria. Cancer. 1984;54:1794-1801. 55. Ming SC, Hirota T. Malignant Epithelial Tumors of the Stomach. (Chapter 27). In: Ming SC, Goldman H, eds. Pathology of the Gastrointestinal Tract. Baltimore: Williams & Wilkins; 1998. 56. Morson BC, Jass JR, Sobin LH. Precancerous lesions of the gastrointestinal tract: A histological classification. London: Bailliere Tindall; 1985. 57. Morson BC, Sobin LH, Grundmann E, et al. Precancerous conditions and epithelial dysplasia in the stomach. J Clin Pathol. 1980;33:711-721. 58. Murayama H, Kikuchi M, Enjoji M, et al. Changes in gastric mucosa that antedate gastric carcinoma. Cancer. 1990;66:2017-2026. 59. Noda M, Kodama T, Atsumi M, et al. Possibilities and limitations of endoscopic resection for early gastric cancer. Endoscopy. 1997;29:361-365. 60. Nomura A, Stemmermann GN, Chyou PH, et al. Helicobacter pylori infection and gastric carcinoma among Japanese Americans in Hawaii. N Engl J Med. 1991;325:1132-1136. 61. Oehlert W, Keller P, Henke M, et al. Gastric mucosal dysplasia: what is its clinical significance? Front Gastrointest Res. 1979;4:173-182. 62. Oohara T, Tohma H, Takezoe K, et al. Minute gastric cancers less than 5 mm in diameter. Cancer. 1982;50:801-810. 63. Park DY, Srivastava A, Kim GH, et al. Adenomatous and foveolar gastric dysplasia. Disctinct patterns of mucin expression and background intestinal metaplasia. Am J Surg Pathol. in press. 64. Parsonnet J, Friedman GD, Vandersteen DP, et al. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med. 1991;325:1127-1131. 65. Probst A, Golger D, Arnholdt H, et al. Endoscopic submucosal dissection of early cancers, flat adenomas, and submucosal tumors in the gastrointestinal tract. Clin Gastroenterol Hepatol. 2009;7:149-155. 66. Riddell RH, Goldman H, Ransohoff DF, et al. Dysplasia in inflammatory bowel disease: standardized classification with provisional clinical applications. Hum Pathol. 1983;14:931-968. 67. Riddell RH, Iwafuchi M. Problems arising from eastern and western classification systems for gastrointestinal dysplasia and carcinoma: are they resolvable? Histopathology. 1998;33:197-202. 68. Rugge M, Cassaro M, Di Mario F, et al. The long term outcome of gastric non-invasive neoplasia. Gut. 2003;52:1111-1116. 69. Rugge M, Correa P, Dixon MF, et al. Gastric dysplasia: the Padova international classification. Am J Surg Pathol. 2000;24:167-176. 70. Rugge M, Farinati F, Di Mario F, et al. Gastric epithelial dysplasia: a prospective multicenter follow-up study from the Interdisciplinary Group on Gastric Epithelial Dysplasia. Hum Pathol. 1991;22:1002-1008. 71. Rugge M, Leandro G, Farinati F, et al. Gastric epithelial dysplasia. How clinicopathologic background relates to management. Cancer. 1995;76:376-382. 72. Rugge M, Nitti D, Farinati F, et al. Non-invasive neoplasia of the stomach. Eur J Gastroenterol Hepatol. 2005;17:1191-1196. 73. Saraga EP, Gardiol D, Costa J. Gastric dysplasia. A histological follow-up study. Am J Surg Pathol. 1987;11:788-796.

Lauwers-Early gastric neoplasms
12
74. Sarre RG, Frost AG, Jagelman DG, et al. Gastric and duodenal polyps in familial adenomatous polyposis: a prospective study of the nature and prevalence of upper gastrointestinal polyps. Gut. 1987;28:306-314. 75. Sawada T, Muto T. Familial adenomatous polyposis: should patients undergo surveillance of the upper gastrointestinal tract? Endoscopy. 1995;27:6-11. 76. Schlemper RJ, Itabashi M, Kato Y, et al. Differences in diagnostic criteria for gastric carcinoma between Japanese and western pathologists. Lancet. 1997;349:1725-1729. 77. Schlemper RJ, Kato Y, Stolte M. Review of histological classifications of gastrointestinal epithelial neoplasia: differences in diagnosis of early carcinomas between Japanese and Western pathologists. Journal of gastroenterology. 2001;36:445-456. 78. Schlemper RJ, Riddell RH, Kato Y, et al. The Vienna classification of gastrointestinal epithelial neoplasia. Gut. 2000;47:251-255. 79. Serck-Hanssen A. Precancerous lesions of the stomach. Scand J Gastroenterol Suppl. 1979;54:104-105. 80. Shemesh E, Bat L. A prospective evaluation of the upper gastrointestinal tract and periampullary region in patients with Gardner syndrome. Am J Gastroenterol. 1985;80:825-827. 81. Shinmura K, Goto M, Tao H, et al. A novel STK11 germline mutation in two siblings with Peutz-Jeghers syndrome complicated by primary gastric cancer. Clin Genet. 2005;67:81-86. 82. Spigelman AD, Williams CB, Talbot IC, et al. Upper gastrointestinal cancer in patients with familial adenomatous polyposis. Lancet. 1989;2:783-785. 83. Sue-Ling HM, Martin I, Griffith J, et al. Early gastric cancer: 46 cases treated in one surgical department. Gut. 1992;33:1318-1322. 84. Tsukuma H, Mishima T, Oshima A. Prospective study of "early" gastric cancer. Int J Cancer. 1983;31:421-426. 85. Uemura N, Mukai T, Okamoto S, et al. Effect of Helicobacter pylori eradication on subsequent development of cancer after endoscopic resection of early gastric cancer. Cancer Epidemiol Biomarkers Prev. 1997;6:639-642. 86. Utsunomiya J, Maki T, Iwama T, et al. Gastric lesion of familial polyposis coli. Cancer. 1974;34:745-754. 87. Watanabe H, Enjoji M, Yao T, et al. Gastric lesions in familial adenomatosis coli: their incidence and histologic analysis. Hum Pathol. 1978;9:269-283. 88. Weinstein WM, Goldstein NS. Gastric dysplasia and its management. Gastroenterology. 1994;107:1543-1545. 89. Wood MG, Bates C, Brown RC, et al. Intramucosal carcinoma of the gastric antrum complicating Menetrier's disease. J Clin Pathol. 1983;36:1071-1075. 90. Xuan ZX, Ueyama T, Yao T, et al. Time trends of early gastric carcinoma. A clinicopathologic analysis of 2846 cases. Cancer. 1993;72:2889-2894. 91. Yamada H, Ikegami M, Shimoda T, et al. Long-term follow-up study of gastric adenoma/dysplasia. Endoscopy. 2004;36:390-396. 92. Yasuda K. Endoscopic ultrasonic probes and mucosectomy for early gastric carcinoma. Gastrointest Endosc. 1996;43:S29-31. 93. Yasuda K, Shiraishi N, Suematsu T, et al. Rate of detection of lymph node metastasis is correlated with the depth of submucosal invasion in early stage gastric carcinoma. Cancer. 1999;85:2119-2123. 94. Ye W, Nyren O. Risk of cancers of the oesophagus and stomach by histology or subsite in patients hospitalised for pernicious anaemia. Gut. 2003;52:938-941. 95. Zhang Y. Typing and grading of gastric dysplasia. In: Zhang Y, Kawai K, eds. Precancerous conditions and lesions of the stomach. Berlin: Springer-Verlag; 1993:64-84.

1
Update On Gastric Cancer: Molecular Pathology and Targeted Therapies
Antonia R. Sepulveda MD., PhD,
Department of Pathology and Laboratory Medicine,
University of Pennsylvania, Philadelphia, PA
Gastric carcinoma is the fourth most frequent cancer worldwide, representing the second most
common cause of death from cancer (approximately 700,000/year)1. In the United States 21,259
new cases of stomach cancer were estimated in 2007, remaining stable at 21,130 new cases in
2009 2. The incidence of gastric cancers (GC) involving the distal stomach, body and fundus,
which have been associated with Helicobacter infection have declined over past decades, while
adenocarcinomas of the cardia and gastroesophageal GE-junction (GEJ) is increasing. Combined
figures for GE junction and gastric cancer indicate 1.4 million new cases diagnosed annually
with 1.1 million attributed deaths 3.
Clinical trials of targeted therapies for advanced gastric cancer have generally included gastric
and gastroesophageal junction adenocarcinomas. Additionally, some trials used the classification
of GE junction adenocarcinomas as described by Siewert, classifying GE junction carcinomas
into types I, II and III depending on the relative extent of involvement of the esophagus and
stomach 4.
Histopathologically and genetically, gastric and gastro-esophageal junction cancers are
heterogeneous and are influenced by gene-environment interactions resulting in activation of
multiple molecular pathways. The molecular subtypes of gastric cancer include three main
groups of tumors characterized by either the chromosomal instability pathway (CIN), the
microsatellite instability pathway (MSI), and the CpG island methylator phenotype pathway
(reviewed in 5). Currently, it is not clear whether and how these subtypes of gastric and GEJ
carcinomas can be useful in clinical practice to predict specific pathways with mutational and
regulatory alterations that may interfere with targeted therapies.
Surgical resection is the only potentially curative option for gastric cancer and is recommended
for stages Tis-T3N0-N2M0 or T4N0M0 6. For tumors not amenable to surgical curative
resection, including locally advanced, recurrent, or metastatic cancers, a number of
chemotherapy regimens can be used, albeit with limited success, such that 5 year survival rates
for advanced gastric and GE junction cancers remain extremely poor at 20-50% for stages II-III
and 5-10% for stage IV tumors. Recently, a number of agents that target specific molecules in
cancer related pathways have become available and are being tested in patients with gastric and
GE junction carcinomas. Here we will review the targeted therapies that have advanced to phase
II or III clinical trials and offer promise in the treatment of these cancers. In addition, the specific
roles of pathology and molecular testing as it relates to specific targeted therapies will be
discussed.
Cell surface receptor inhibitors
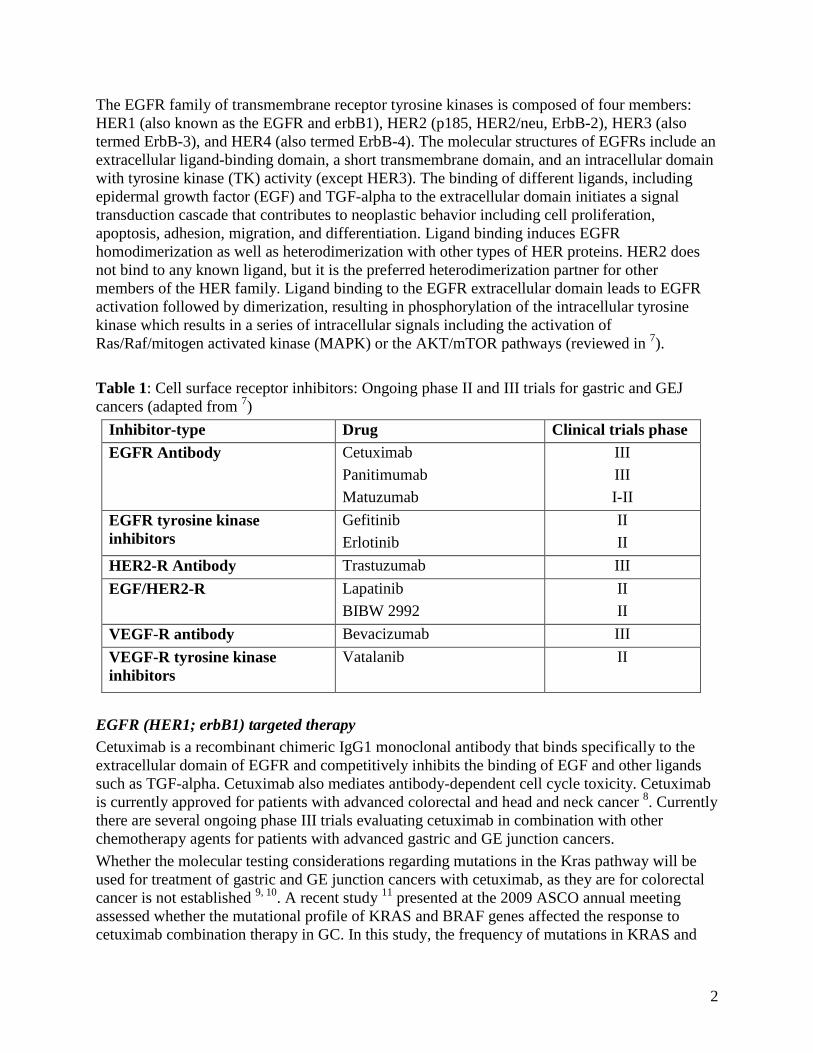
EGFR Family Inhibitors
Current available therapies target the EGFR pathways through inhibition of the EGFR using two
different mechanisms:1) inhibition of the EGFR via monoclonal antibodies (i.e. cetuximab,
matuzamab, panitumumab, trastuzumab) or 2) tyrosine kinase inhibitors (i.e. gefitinib, erlotinib).
2
The EGFR family of transmembrane receptor tyrosine kinases is composed of four members:
HER1 (also known as the EGFR and erbB1), HER2 (p185, HER2/neu, ErbB-2), HER3 (also
termed ErbB-3), and HER4 (also termed ErbB-4). The molecular structures of EGFRs include an
extracellular ligand-binding domain, a short transmembrane domain, and an intracellular domain
with tyrosine kinase (TK) activity (except HER3). The binding of different ligands, including
epidermal growth factor (EGF) and TGF-alpha to the extracellular domain initiates a signal
transduction cascade that contributes to neoplastic behavior including cell proliferation,
apoptosis, adhesion, migration, and differentiation. Ligand binding induces EGFR
homodimerization as well as heterodimerization with other types of HER proteins. HER2 does
not bind to any known ligand, but it is the preferred heterodimerization partner for other
members of the HER family. Ligand binding to the EGFR extracellular domain leads to EGFR
activation followed by dimerization, resulting in phosphorylation of the intracellular tyrosine
kinase which results in a series of intracellular signals including the activation of
Ras/Raf/mitogen activated kinase (MAPK) or the AKT/mTOR pathways (reviewed in 7).
Table 1: Cell surface receptor inhibitors: Ongoing phase II and III trials for gastric and GEJ
cancers (adapted from 7)
Inhibitor-type Drug Clinical trials phase
EGFR Antibody Cetuximab
Panitimumab
Matuzumab
III
III
I-II
EGFR tyrosine kinase
inhibitors
Gefitinib
Erlotinib
II
II
HER2-R Antibody Trastuzumab III
EGF/HER2-R Lapatinib
BIBW 2992
II
II
VEGF-R antibody Bevacizumab III
VEGF-R tyrosine kinase
inhibitors
Vatalanib
II
EGFR (HER1; erbB1) targeted therapy
Cetuximab is a recombinant chimeric IgG1 monoclonal antibody that binds specifically to the
extracellular domain of EGFR and competitively inhibits the binding of EGF and other ligands
such as TGF-alpha. Cetuximab also mediates antibody-dependent cell cycle toxicity. Cetuximab
is currently approved for patients with advanced colorectal and head and neck cancer 8. Currently
there are several ongoing phase III trials evaluating cetuximab in combination with other
chemotherapy agents for patients with advanced gastric and GE junction cancers.
Whether the molecular testing considerations regarding mutations in the Kras pathway will be
used for treatment of gastric and GE junction cancers with cetuximab, as they are for colorectal
cancer is not established 9, 10
. A recent study 11
presented at the 2009 ASCO annual meeting
assessed whether the mutational profile of KRAS and BRAF genes affected the response to
cetuximab combination therapy in GC. In this study, the frequency of mutations in KRAS and

3
BRAF was lower as compared to colorectal cancer, and the mutational status of KRAS and
BRAF genes did not correlate with the response to cetuximab-based therapy in advanced gastric
cancer patients. Forty four tumor samples were collected from patients with locally advanced or
metastatic GC undergoing cetuximab combination therapy as first-line treatment in two
consecutive phase II studies, FOLCETUX Study and DOCETUX Study. The mutational status of
KRAS (exon 2) and BRAF (exon 15) was detected by PCR amplification followed by direct
sequencing. KRAS and BRAF mutations were detected in 5 (11.4%) and 1 (2.3%), respectively,
of the 44 tumors analyzed. These frequencies are consistent with those previously reported in
GC. The only BRAF mutation found in 1 sample was the classic V600E substitution. As a
whole, 13.6 % of the analyzed tumors carried a mutation in either KRAS or BRAF genes. K-
RAS and BRAF mutations were, as expected, mutually exclusive. As this study examined a
small cohort, additional studies are warranted before clinical guidelines can be established.
Panitumumab is the first fully human monoclonal antibody (IgG2) specific to EGFR. It has been
used successfully in patients with advanced colorectal cancer who failed standard therapies 12
. A
phase III trial (REAL III) is due to start investigating the role of panitumumab in combination
therapy for locally advanced or metastatic gastric or GEJ cancer.
The EGFR tyrosine kinase inhibitors have proved minimal evidence of efficacy in gastric
carcinomas, but studies are ongoing 7.
HER2 (HER2/neu, ErbB-2) targeted therapies
HER2 has no known ligand (orphan receptor), and preferentially heterodimerizes with HER3
which lacks intrinsic tyrosine kinase activity. The HER2, and the HER2/HER3 heterodimer is
likely to be the most effective complex for activating pathways downstream of EGF receptors 13,
14.
Overexpression and amplification of HER2 has been described in 6-35% of gastric and GEJ
adenocarcinomas 15;16
(Table 2).
Trastuzumab (Herceptin) is a monoclonal antibody which specifically targets HER2 protein by
directly binding the extracellular domain of the receptor. Trastuzumab enhances survival rates in
both primary and metastatic HER2-positive breast cancer patients. The efficacy of trastuzumab
in breast cancer patients has led to investigate its antitumor activity in patients with HER2-
positive cancers, including gastric adenocarcinomas.
HER2/neu positivity rates have been reported to be more frequent in intestinal type gastric
cancer (21.5%) than in diffuse gastric cancer (2%) or mixed types (5%) 15
; these findings were
confirmed in the ToGA trial with HER2 positivity being more frequent in intestinal than
diffuse/mixed cancer (32.2% vs 6.1%/20.4%), respectively 17
. In addition, HER-2/neu
amplification in gastric carcinoma is associated with poor outcome 15
and has been shown to be
an independent prognostic factor 18
.
Results from the largest study to date (ToGA trial) evaluating the addition of trastuzumab
(Herceptin) to chemotherapy in HER2-positive advanced gastric cancer were reported at the
2009 ASCO meeting 17, 19
. The ToGA trial is the first randomized Phase III trial providing
prospective information on HER2-positivity rates in GC (Table 2). The trial enrolled 3,883
patients from 24 countries. A HER2-scoring system modified from the protocol in breast cancer
was used: a score of IHC 3+ and/or FISH positive was defined as HER2 positive. The modified
HER2-scoring system showed concordance between IHC and FISH results of 87.5%. In breast
cancer most IHC 0/1 samples are FISH negative but, in the ToGA cohort, the frequency of IHC
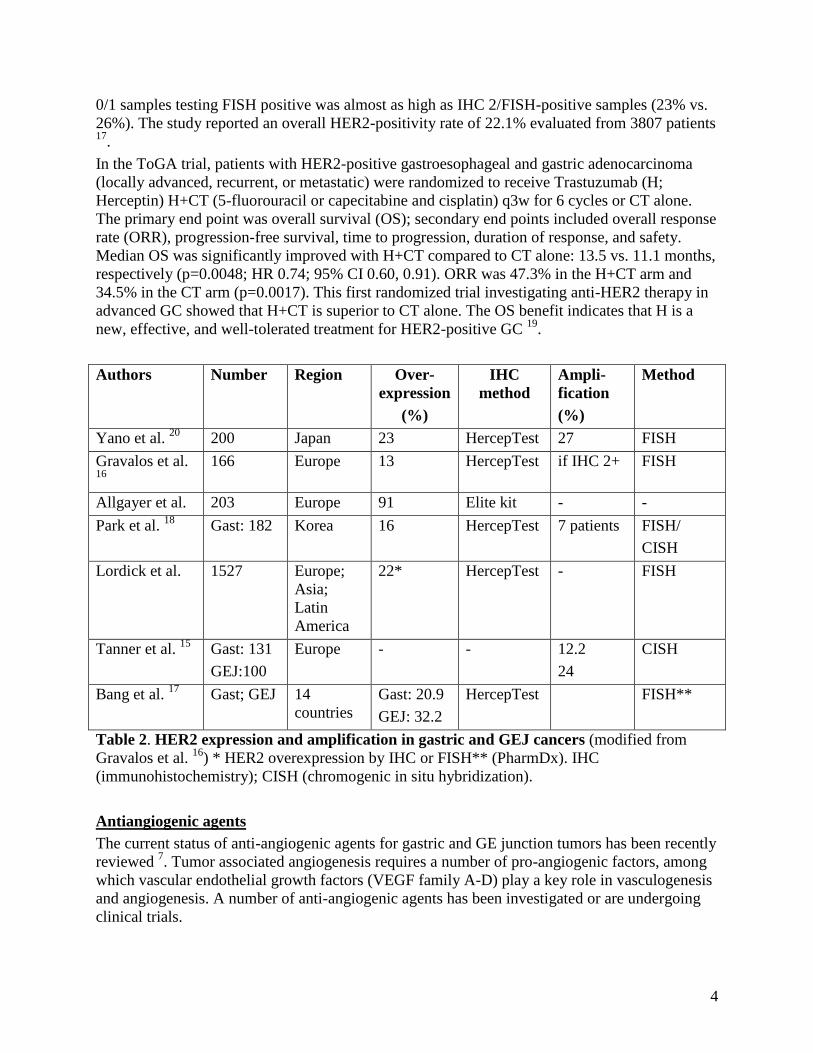
4
0/1 samples testing FISH positive was almost as high as IHC 2/FISH-positive samples (23% vs.
26%). The study reported an overall HER2-positivity rate of 22.1% evaluated from 3807 patients 17
.
In the ToGA trial, patients with HER2-positive gastroesophageal and gastric adenocarcinoma
(locally advanced, recurrent, or metastatic) were randomized to receive Trastuzumab (H;
Herceptin) H+CT (5-fluorouracil or capecitabine and cisplatin) q3w for 6 cycles or CT alone.
The primary end point was overall survival (OS); secondary end points included overall response
rate (ORR), progression-free survival, time to progression, duration of response, and safety.
Median OS was significantly improved with H+CT compared to CT alone: 13.5 vs. 11.1 months,
respectively (p=0.0048; HR 0.74; 95% CI 0.60, 0.91). ORR was 47.3% in the H+CT arm and
34.5% in the CT arm (p=0.0017). This first randomized trial investigating anti-HER2 therapy in
advanced GC showed that H+CT is superior to CT alone. The OS benefit indicates that H is a
new, effective, and well-tolerated treatment for HER2-positive GC 19
.
Authors Number
Region Over-
expression
(%)
IHC
method
Ampli-
fication
(%)
Method
Yano et al. 20
200 Japan 23 HercepTest 27 FISH
Gravalos et al. 16
166 Europe 13 HercepTest if IHC 2+ FISH
Allgayer et al. 203 Europe 91 Elite kit - -
Park et al. 18
Gast: 182 Korea 16 HercepTest 7 patients FISH/
CISH
Lordick et al. 1527 Europe;
Asia;
Latin
America
22* HercepTest - FISH
Tanner et al. 15
Gast: 131
GEJ:100
Europe - - 12.2
24
CISH
Bang et al. 17
Gast; GEJ 14
countries
Gast: 20.9
GEJ: 32.2
HercepTest FISH**
Table 2. HER2 expression and amplification in gastric and GEJ cancers (modified from
Gravalos et al. 16
) * HER2 overexpression by IHC or FISH** (PharmDx). IHC
(immunohistochemistry); CISH (chromogenic in situ hybridization).
Antiangiogenic agents
The current status of anti-angiogenic agents for gastric and GE junction tumors has been recently
reviewed 7. Tumor associated angiogenesis requires a number of pro-angiogenic factors, among
which vascular endothelial growth factors (VEGF family A-D) play a key role in vasculogenesis
and angiogenesis. A number of anti-angiogenic agents has been investigated or are undergoing
clinical trials.

5
Bevacizumab is a chimeric monoclonal antibody that binds the VEGF-receptor and prevents the
interaction of VEGF to its receptors (Flt1 and KDR) on the surface of endothelial cells. Clinical
data of bevacizumab therapy in patients with advanced gastric or GEJ cancer is promising but is
limited to phase II trials.
Another approach to inhibit the VEGF pathway uses tyrosine kinase inhibitors directed against
the receptors of VEGF (Flt1 and KDR). There are several compounds available, some
specifically targeting VEGF receptors, such as PTK787/ZK222584 (Vatalanib) and others that
inhibit both the VEGF receptors and other tyrosine kinase receptors such as sunitinib and
sorafenib. Phase II results are available for sorafenib, in which 44 patients with advanced gastric
or GE junction cancers were treated with a combination of docetaxel, cisplatin and sorafenib,
with an objective response rate of 38.6%. The median PFS was 5.8 months and the median OS
was 14.9 months.
Other targeted therapies for gastric and GEJ cancers
A large number of specific inhibitors of molecular targets in gastric and GE junction cancer
pathways are in the early stages of clinical trials, while supporting data for others is limited to
pre-clinical studies. Among these agents are the insulin-like growth factor-I (IGF-IR) receptor
inhibitors, fibroblast growth factor (FGF) receptor inhibitors and c-Met signaling pathway
inhibitors. A number of cell cycle associated drug targets including Aurora kinase inhibitors,
polo-like kinase inhibitors and cyclin dependent kinase (cdk) inhibitors are being tested.
Another approach has involved the use of inhibitors of cancer associated epigenetic changes.
Histone deacetylase (HDAC) inhibitors are under considerable research given the potential to re-
express tumor suppressor genes silenced by hypermethylation in cancer cells. A phase II clinical
trial is ongoing.
Other agents under current trials include heat shock protein 90 (HS90) inhibitors, ubiquitin-
proteasome pathway inhibitors, PI3K/Akt/mTOR pathway inhibitors and matrix
metalloproteinase (MMP) inhibitors.

6
Bibliography
1. Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin
2005;55:74-108.
2. Pickle LW, Hao Y, Jemal A, Zou Z, Tiwari RC, Ward E, Hackey M, Holly L, Feuer EJ.
A New Method of Estimating United States and State-level Cancer Incidence Counts for
the Current Calendar Year. CA Cancer J Clin 2007;57:30-42.
3. Kamangar F, Dores GM, Anderson WF. Patterns of cancer incidence, mortality, and
prevalence across five continents: defining priorities to reduce cancer disparities in
different geographic regions of the world. J Clin Oncol 2006;24:2137-50.
4. Stein HJ, Feith M, Siewert JR. Individualized surgical strategies for cancer of the
esophagogastric junction. Ann Chir Gynaecol 2000;89:191-8.
5. Sepulveda AR, Aisner D. Molecular Basis of Diseases of the Gastrointestinal Tract In:
Coleman WB, Tsongalis GJ, eds. Molecular Pathology: Elsevier Inc, 2008.
6. Van Cutsem E, Van de Velde C, Roth A, Lordick F, Kohne CH, Cascinu S, Aapro M.
Expert opinion on management of gastric and gastro-oesophageal junction
adenocarcinoma on behalf of the European Organisation for Research and Treatment of
Cancer (EORTC)-gastrointestinal cancer group. Eur J Cancer 2008;44:182-94.
7. Arkenau HT. Gastric cancer in the era of molecularly targeted agents: current drug
development strategies. J Cancer Res Clin Oncol 2009;135:855-66.
8. Saltz LB, Meropol NJ, Loehrer PJ, Sr., Needle MN, Kopit J, Mayer RJ. Phase II trial of
cetuximab in patients with refractory colorectal cancer that expresses the epidermal
growth factor receptor. J Clin Oncol 2004;22:1201-8.
9. De Roock W, Piessevaux H, De Schutter J, Janssens M, De Hertogh G, Personeni N,
Biesmans B, Van Laethem JL, Peeters M, Humblet Y, Van Cutsem E, Tejpar S. KRAS
wild-type state predicts survival and is associated to early radiological response in
metastatic colorectal cancer treated with cetuximab. Ann Oncol 2008;19:508-15.
10. Allegra CJ, Jessup JM, Somerfield MR, Hamilton SR, Hammond EH, Hayes DF,
McAllister PK, Morton RF, Schilsky RL. American Society of Clinical Oncology
provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic
colorectal carcinoma to predict response to anti-epidermal growth factor receptor
monoclonal antibody therapy. J Clin Oncol 2009;27:2091-6.
11. Stella G, Llimpe FR, Barone C, Falcone A, Fabio FD, Martoni A, Lamba S, Ceccarelli C,
Siena S, Bardelli A, Pinto C. KRAS and BRAF mutational status as response biomarkers
to cetuximab combination therapy in advanced gastric cancer patients. J Clin Oncol
2009;27: (suppl; abstr e15503).
12. Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B, Canon JL, Van
Laethem JL, Maurel J, Richardson G, Wolf M, Amado RG. Open-label phase III trial of
panitumumab plus best supportive care compared with best supportive care alone in
patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol
2007;25:1658-64.
13. Srinivasan R, Leverton KE, Sheldon H, Hurst HC, Sarraf C, Gullick WJ. Intracellular
expression of the truncated extracellular domain of c-erbB-3/HER3. Cell Signal
2001;13:321-30.
14. Hirata A, Hosoi F, Miyagawa M, Ueda S, Naito S, Fujii T, Kuwano M, Ono M. HER2
overexpression increases sensitivity to gefitinib, an epidermal growth factor receptor

7
tyrosine kinase inhibitor, through inhibition of HER2/HER3 heterodimer formation in
lung cancer cells. Cancer Res 2005;65:4253-60.
15. Tanner M, Hollmen M, Junttila TT, Kapanen AI, Tommola S, Soini Y, Helin H, Salo J,
Joensuu H, Sihvo E, Elenius K, Isola J. Amplification of HER-2 in gastric carcinoma:
association with Topoisomerase IIalpha gene amplification, intestinal type, poor
prognosis and sensitivity to trastuzumab. Ann Oncol 2005;16:273-8.
16. Gravalos C, Jimeno A. HER2 in gastric cancer: a new prognostic factor and a novel
therapeutic target. Ann Oncol 2008;19:1523-9.
17. Bang Y, Chung H, Xu J, Lordick F, Sawaki A, Lipatov O, Al-Sakaff N, See C, Rueschoff
J, Cutsem EV. Pathological features of advanced gastric cancer (GC): Relationship to
human epidermal growth factor receptor 2 (HER2) positivity in the global screening
programme of the ToGA trial. J Clin Oncol 2009;27:(suppl; abstr 4556).
18. Park DI, Yun JW, Park JH, Oh SJ, Kim HJ, Cho YK, Sohn CI, Jeon WK, Kim BI, Yoo
CH, Son BH, Cho EY, Chae SW, Kim EJ, Sohn JH, Ryu SH, Sepulveda AR. HER-2/neu
amplification is an independent prognostic factor in gastric cancer. Dig Dis Sci
2006;51:1371-9.
19. Cutsem EV, Kang Y, Chung H, Shen L, Sawaki A, Lordick F, Hill J, Lehle M,
Feyereislova A, Bang Y. Efficacy results from the ToGA trial: A phase III study of
trastuzumab added to standard chemotherapy (CT) in first-line human epidermal growth
factor receptor 2 (HER2)-positive advanced gastric cancer (GC). J Clin Oncol 2009;
27:(suppl; abstr LBA4509)
20. Yano T, Doi T, Ohtsu A, Boku N, Hashizume K, Nakanishi M, Ochiai A. Comparison of
HER2 gene amplification assessed by fluorescence in situ hybridization and HER2
protein expression assessed by immunohistochemistry in gastric cancer. Oncol Rep
2006;15:65-71.

GASTRIC LYMPHOMAS:
25+ Years of Revolution/Evolution
Jerome S. Burke, MD
Department of Pathology Alta Bates Summit Medical Center
Berkeley, California
The gastrointestinal tract is the most common location of extranodal lymphomas and the stomach in particular serves as an excellent model to illustrate many of the general issues concerning extranodal lymphomas. The stomach is the most frequent site of involvement of extranodal gastrointestinal lymphomas with small intestine second in frequency. Large intestinal and rectal lymphomas are less common, but are found in patients with AIDS and occasionally as a complication of ulcerative colitis and Crohn disease. In a study of 371 patients with primary gastrointestinal lymphomas registered in a German Multicenter Study, stomach accounted for 277 of cases ((74.8%). Curiously, the incidence of primary gastric lymphomas in the United States is increasing, specifically in patients over 60 years of age; this increase appears independent of the AIDS epidemic and the now common diagnosis of extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue (MALT) type. The concept of extranodal marginal zone lymphoma of MALT type has revolutionized the criteria for the morphologic diagnosis of an extranodal lymphoma, specifically those lymphomas dominated by small lymphocytes. Moreover, since the initial descriptions of lymphomas of MALT in 1983 and 1984 by Drs. Peter Isaacson and Dennis Wright, the criteria have evolved and expanded to encompass not only morphologic standards, specifically a more precise definition of various cell types in MALT lymphoma, the proposals for ―lymphoepithelial lesions‖ and ―follicular colonization,‖ the relationship between MALT lymphomas and marginal zone B cells, and a scoring system for diagnosis of gastric MALT lymphoma, but also to embrace immunophenotypic, biologic, molecular genetic and clinical discoveries. Such developments include refinement of the immunophenotype, the association of gastric MALT lymphoma and Helicobacter pylori infection, the notion of acquired MALT, the biology of auto-antigen activation and continuous somatic mutations, genetic aberrations, as for example the discovery of the t(11;18)(q21;q21) chromosomal translocation in lymphomas of MALT, and the clinical implications of specific cytogenetic alterations. Revolution – Florid Gastric Lymphoid Hyperplasia (“Pseudolymphoma”) is Likely
Malignant Lymphoma!

Gastric Lymphomas; Burke - 2
In establishing a histologic diagnosis of an extranodal lymphoma, pathologists are aware that various reactive lymphoid hyperplasias exist that mimic extranodal lymphomas clinically and pathologically, such as gastric lymphoid hyperplasia and chronic lymphocytic gastritis. To complicate matters, malignant lymphoma of extranodal marginal zone (MALT) type may develop in association with these reactive lymphoid infiltrates in the stomach. In the 1960’s and 1970’s, the morphologic criteria to distinguish extranodal lymphoma from extranodal lymphoid hyperplasia were extrapolated from those used traditionally to distinguish malignant lymphoma from lymphoid hyperplasia in lymph nodes. The major criteria for this distinction included monomorphic lymphocytic infiltrates, cellular atypia, germinal centers and architectural disruption (Table 1).
Table 1 Traditional Histologic Criteria for Distinguishing Extranodal Lymphomas from
Lymphoid Hyperplasias
Extranodal Lymphomas Lymphoid Hyperplasias
Infiltrate monomorphous Infiltrate polymorphous (lymphocytes in stages of transformation)
Cytologic atypia Cytologic maturity
(lymphocytes, plasma cells, and immunoblasts)
Germinal centers uncommon Germinal centers common
(usually in center of infiltrate)
Massive infiltration with Random infiltration with architectural destruction architectural retention
Because more than 50% of gastric lymphomas are large B cell lymphomas with associated monomorphism, cellular atypia, and architectural destruction (e.g., obliteration of glands), these traditional criteria generally have proven to be reliable and applicable Reactive lymphoid conditions that are at the opposite end of the spectrum equally do not pose diagnostic problems. Lymphocytic infiltrates that exhibit polymorphism, that display a range of mature lymphocytes (including plasma cells and immunoblasts), that are associated with well-defined germinal centers and that do not destroy completely the architectural landmarks of the stomach usually can be diagnosed confidently as benign and reactive. The main difficulty in the separation of gastric extranodal lymphomas from

Gastric Lymphomas; Burke - 3
lymphoid hyperplasias concerns MALT lymphomas that are composed of small lymphocytes and that frequently are associated with germinal center formation. For these cases, the traditional histological criteria are not applicable. Multiparameter studies have revealed that many histologically ambiguous extranodal small lymphocytic proliferations in stomach are, in fact, monoclonal and, therefore, are presumed to be malignant lymphomas especially of MALT type. The application of immunologic and molecular genetic analyses to gastric small lymphocytic proliferations has served to vividly alter the traditional histologic criteria and has revealed myriad inconsistencies in these criteria (Table 2). For example, in a review of 97 cases originally diagnosed as gastric pseudolymphoma between 1970 and 1985 at the AFIP, 79% were reclassified as malignant lymphoma, with fully two-thirds of the newly classified lymphomatous cases interpreted as lymphomas of MALT type. The remaining cases were regarded as examples of lymphoid hyperplasia or atypical lymphocytic infiltrates. Consequently, the term ―pseudolymphoma‖ is regarded as imprecise and anachronistic and no longer is acceptable as a diagnostic category.
Table 2 Modifications of the Traditional Histologic Criteria for Distinguishing Extranodal
Lymphomas From Lymphoid Hyperplasia
Extranodal lymphomas may be polymorphous (e.g., peripheral T cell lymphomas)
Extranodal lymphomas may be composed of cytologically mature-appearing lymphocytes (marginal zone lymphoma of MALT type and other low-grade lymphomas with or without plasma cell differentiation)
Germinal centers may be observed at the periphery of extranodal lymphomas and in the centers of many low-grade extranodal lymphomas, especially those of MALT type
Degree of infiltration, architectural and epithelial destruction is highly variable in benign and malignant extranodal lymphocytic infiltrates
The conventional view was that extranodal lymphomas are characterized by nuclear membrane irregularities or atypia. Marginal zone lymphomas of MALT type, however, are cytologically mutable. Although MALT lymphomas typically are composed of marginal zone or centrocyte-like cells in the marginal zones, they also may have a monocytoid appearance, or they be composed of plasma cells, or they may have no, or only subtle, nuclear membrane irregularities similar to the cells of chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL); the latter variant often is readily confused with extranodal lymphoid hyperplasia. A small lymphocytic proliferation that appears mature is the histologic norm of most cases of gastric lymphoid hyperplasia, yet cytologic maturity or minimal atypia is equally the histologic hallmark of most low-grade malignant lymphomas including those of MALT. How then

Gastric Lymphomas; Burke - 4
can these two groups be distinguished in a small biopsy specimen? In many cases, it is impossible to do when applying only histological criteria. This histologic dilemma prompted the use of the noncommittal term ―extranodal small lymphocytic proliferation‖ to describe histologically ambiguous or indeterminate extranodal lymphocytic infiltrates. Current morphologic criteria used to separate a benign small lymphocytic infiltrate from a malignant small lymphocytic infiltrate includes the designations of whether the infiltrate is monomorphous, dense, or exhibits cytologic atypia or Dutcher bodies and whether this results in destruction of glands, follicles, or other structures indigenous to the stomach. If these morphologic features are unequivocally present, then the lymphocytic infiltrate likely is malignant lymphoma (see Table 3). Our perspective of the small extranodal lymphocytic proliferations that appear mature has revised dramatically with the application of immunologic markers to analyze the clonality of these lesions. Correlative immunopathologic studies have now reduced the number of cases regarded as atypical or indeterminate. Naturally, cases remain, especially small biopsy specimens, in which the histologic features or immunologic findings remain equivocal; these cases should receive not only a descriptive diagnosis but also a request for a repeat biopsy with reservation of fresh tissues to determine clonality, whether by immunohistochemistry, flow cytometry, molecular techniques, or a combination thereof. When only fixed, paraffin-embedded tissues are available, a consistent determination of clonality in a biopsy specimen dominated by small lymphocytes usually is not possible in most laboratories. In some cases, however, immunologic studies of paraffin-embedded tissues can reveal an aberrant phenotype in the suspicious small lymphocytic population, such as the coexpression of B cell antigen CD20 and T cell antigen CD43. In most instances, the aberrant immunophenotype supports the diagnosis of an extranodal gastric B cell lymphoma. Prudence is necessary in the evaluation of immunoglobulin gene rearrangement studies employing polymerase chain reaction (PCR) techniques in fixed paraffin-embedded tissues; small monoclonal bands may occur in extranodal reactive lymphoid hyperplasias, as for example in chronic active gastritis associated with Helicobacter pylori. As well as diagnostic problems encountered with the gastric lesions dominated by small lymphocytes, benign reactive germinal centers (lymphoid follicles) pose another risk in diagnosis. The presence of germinal centers is a commonly accepted histologic attribute of extranodal lymphoid hyperplasias. Germinal centers, however, are regular constituents of most gastric lymphomas of MALT. The essential morphologic characteristic of MALT lymphomas is their emulation of normal MALT as typified by Peyer patches found in the terminal ileum. The neoplastic B cells of MALT lymphomas are found in marginal-type zones surrounding reactive follicles and frequently attenuated rims of mantle zone lymphocytes. The germinal centers vary in appearance but commonly appear atrophic as a result of impingement by the

Gastric Lymphomas; Burke - 5
surrounding small lymphocytes. In MALT lymphomas of the stomach associated with peptic ulceration, germinal centers occur at the base of the ulcer and in the adjacent mucosa where they seem encroached upon and entrapped by the monotonous marginal zone lymphocytes. In some cases, the neoplastic B cells in the marginal zone invade the germinal centers in a process referred to as ―follicular colonization.‖ Follicular colonization may simulate follicular lymphoma in cases where there are numerous follicles. At times, the lymphomatous proliferation may be so extensive as to result in architectural obliteration with masking of any residual germinal centers; however, the presence of former germinal centers can be highlighted by the immunohistochemical demonstration of follicular dendritic cells employing an antibody against CD21 or CD23.
Evolution – Refinement of Morphologic Criteria, Immunophenotype, Biology, Molecular Genetics and Clinical Therapy of Gastric MALT Lymphomas
The morphologic features of extranodal marginal zone lymphoma of MALT type have witnessed gradual changes since the initial descriptions by Isaacason and Wright. Although originally thought to be of follicular center cell origin, the lymphoma cells of MALT were later designated as ―centrocyte-like‖ and currently are termed as ―marginal zone cells.‖ Gastric lymphomas of MALT type typically are characterized by an expansion of the marginal-like zones surrounding benign germinal centers. As described, marginal zone cells constitute a variety of cell types ranging from small, to intermediate-sized lymphocytes, to large cells resembling immunoblasts. The marginal zone or centrocyte-like cells are small to medium-sized lymphocytes with variable nuclear membrane irregularities resembling a centrocyte or small cleaved cell. The most common form has abundant pale-staining cytoplasm and is referred to as monocytoid. Others forms of gastric MALT exhibit plasmacytoid features or resemble small lymphocytes. Occasionally, signet ring-type cells are observed in gastric MALT lymphomas and appear to represent a peculiar type of lymphoepithelial lesion in which the foveolar cells, disaggregated by the lymphomatous infiltrate, acquire a globoid, signet ring-type appearance. Regardless of cytologic composition, the marginal zone cells of MALT cells share immunophenotypic characteristics. Gastric MALT lymphomas are B cell derived with CD20 expression and frequently containing numerous admixed CD3-positive reactive T cells. Up to 50% of cases exhibit aberrant coexpression of CD43 which can prove helpful in diagnosis. Unlike CLL/SLL and mantle cell lymphomas, the lymphomas of MALT origin lack CD5 and are without bcl-1 gene rearrangements.
MALT lymphomas differ from follicular lymphomas in that they are CD10 negative and do not exhibit rearrangements of the bcl-2 proto-oncogene. The absence of CD10 and bcl-2 positivity in gastric lymphoma with an apparent follicular architecture has been rationalized by the concept of ―follicular colonization‖. As discussed above, follicular colonization refers to the simulation of a follicular lymphoma as a result of invasion by the marginal zone cells of MALT into pre-existing reactive follicles.

Gastric Lymphomas; Burke - 6
The marginal zone cells of MALT not only are thought to invade residual reactive follicles, but also epithelium. As mentioned previously, epithelial invasion by the centrocyte-like cells of MALT has been referred to as a ―lymphoepithelial lesion‖ and is an important morphologic feature in the diagnosis of gastric MALT-derived lymphomas. Such lesions may be accentuated by an immunohistochemical stain for cytokeratin. Lymphoepithelial lesions are most significant as a diagnostic characteristic in the stomach, but are less relevant in other extranodal sites since they may be observed in non-lymphomatous extranodal lymphocytic infiltrates. In stomach, lymphoepithelial lesions usually are defined as invasion of gastric epithelium by three or more lymphomatous cells. A decade following the primary proposal for a lymphoma of MALT, a histologic scoring system for gastric MALT lymphoma was proposed in which invasion of epithelial structures or lymphoepithelial lesions is considered paramount to the diagnosis (Table 3).
Table 3 Gastric Marginal Zone (MALT) Lymphomas: Histologic Scoring*
Grade Description Histologic Features
0 Normal Plasma cells in lamina propria No lymphoid follicles
1 Chronic active gastritis Lymphocyte clusters in lamina propria No follicles, lymphoepithelial lesions
2 Chronic active gastritis with Prominent follicles with surrounding florid lymphoid follicle mantle zone & plasma cells formation No LELs 3 Suspicious lymphoid infiltrate Follicles surrounded by lymphocytes in LP, probably reactive that infiltrate diffusely in LP and +/-
epithelium
4 Suspicious lymphoid infiltrate Follicles surrounded by CCL cells (MZC) in LP, probably lymphoma that infiltrate diffusely in LP & epithelium
5 MZ (MALT) lymphoma Dense diffuse infiltrate of CCL cells in LP with prominent LELs *Lancet 1993:342:575-577
Employing the scoring system, a definite diagnosis of low-grade B cell lymphoma of MALT is based on the presence of a dense diffuse infiltrate of centrocyte-like cells/marginal zone cells in the lamina propria with prominent lymphoepithelial lesions. Cases regarded as suspicious lymphocytic infiltrates are those in which reactive follicles are surrounded by marginal zone cells that diffusely infiltrate into the lamina propria and into epithelium in small groups. However, caution is required as there may

Gastric Lymphomas; Burke - 7
be dense infiltrates, slight cytologic atypia, and also lymphoepithelial-like lesions in cases of lymphoid hyperplasia in the stomach. As well, the criteria may be difficult to apply. In an interobservor study, 41 H&E sections of stomach that ranged from simple gastritis to lymphoma were reviewed by 17 pathologists, including hematopathologists, gi pathologists and general pathologists. Interobservor reproducibility was suboptimal and the degree of disagreement was directly related to the pathologist’s experience in evaluating gastric biopsies for MALT lesions. The study recommended that diagnostic accuracy would be enhanced by clinical information, extensive sampling, recognition of lymphoepithelial lesions, immunophenotypic information and cytogenetic results. This recommendation was supported by another report stating that a combination of the morphologic scoring system and B cell clonality analysis by an advanced PCR method accurately discriminated chronic gastritis from covert gastric marginal zone lymphoma. PCR was particularly valuable in the interpretation of cases that exhibited an ambiguous score of grade 3 or 4. Paradoxically, most MALT-type lymphomas arise in extranodal sites without normal MALT, such as the stomach, while non-MALT lymphomas, for example Burkitt lymphoma, arise in MALT sites. In order to resolve this apparent paradox, it was proposed that lymphomas in non-MALT sites arise in a setting of ―acquired‖ MALT. This non-indigenous extranodal lymphoid tissue is thought to be acquired secondary to an infection, such as Helicobacter pylori in the stomach, or an autoimmune disease. The lymphoid infiltrates in stomach associated with Helicobacter pylori seemingly predispose patients to malignant lymphoma. There currently is sufficient histological, clinical, and epidemiologic evidence for the virtually fixed association of gastric MALT lymphomas with Helicobacter pylori infection, particularly the CagA strain. For example, in a collaborative study of more than 230,000 patients whose serum had been stored, 33 cases of gastric lymphoma developed a median of 14 years after serum collection. The patients with gastric lymphoma were significantly more likely than matched controls to have evidence of previous Helicobactor pylori infection. In contrast, no association was discovered among 31 patients who had developed nongastric non-Hodgkin lymphoma and previous Helicobactor pylori infection. In addition, molecular analysis by PCR has documented the clonal progression from Helicobacter pylori-associated chronic gastritis to MALT lymphoma of the stomach and Helicobactor pylori provides the antigenic stimulus for prolonging the clonal expansion of gastric MALT lymphomas. This view has been supported by Isaacson’s group who have exhibited evidence of ongoing Ig gene mutations in most case of MALT-type lymphoma. The discovery of ongoing mutations reinforces the perception that direct antigen stimulation is paramount in the clonal expansion of gastric MALT lymphomas. Clinically, gastric MALT lymphomas arise in adults with a peak in the seventh decade and with a male:female ratio of approximately 1.5:1. Patients commonly present with nonspecific gastritis and/or a peptic ulcer and at endoscopy, there often are reddened and slightly thickened rugae with superficial spreading of lesions without

Gastric Lymphomas; Burke - 8
formation of a tumor mass. The gastric lesions commonly are multifocal and most patients have stage IE disease. The linkage of Helicobacter pylori with gastric MALT lymphoma led to antibiotic therapy for treatment of low-grade gastric lymphomas of MALT and this therapy may induce sustained remissions in about 75% of patients. There are no apparent differences in survival and relapse-free survival between patients treated with antibiotics and with other modalities, such as local treatment, combined treatment, or chemotherapy; in fact, no consensus exists for the optimal treatment of primary gastric lymphoma. In one study of gastric MALT lymphomas treated by various modalities, the five year projected overall survival was 82%. One significant issue for pathologists is the interpretation of stomach biopsy specimens following antibiotic therapy for gastric MALT lymphoma. Clearly, no diagnostic problem exists if the biopsy reveals regression of the lymphoma with loss of lymphocytic aggregates in the lamina propria or if the biopsy exhibits a total absence of histological regression with persistence of lymphoma. Parallel to the histological scoring system employed for the initial diagnosis of gastric marginal zone lymphoma, a histological grading system has been proposed for treated patients. (Table 4).
Table 4
Gastric MZ (MALT) Lymphoma: Post-Therapy Grading*
Score Lymphoid Infiltrate LEL Stromal Changes
CR Absent or scattered plasma cells – Normal or empty LP
& small lymphoid cells in the LP &/or fibrosis
pMRD Aggregates of lymphoid cells or – Empty LP &/or
lymphoid nodules in the LP/MM fibrosis
&/or SM
rRD Dense, diffuse, or nodular +/- Focal empty LP &/or
extending around glands in the LP fibrosis
NC Dense, diffuse, or nodular +/- No changes
CR, complete histologic remission; pMRD, probable minimal residual disease;
rRD, responding residual disease; NC, no change;
LEL, lymphoepithelial lesions; LP, lamina propria; MM, muscularis mucosa; SM, submucosa *Gut 2003;52:1656
The probable minimal disease category (pMRD) does not signify a requirement for further therapy and patients are managed with followup as though they were in remission. Of interest, approximately 50% of patients with histologically negative post-antibiotic therapy gastric biopsies (CR/pMRD) exhibit persistent monoclonality, although in some patients the clone disappears with prolonged followup. In other patients, continued monoclonality may result in a delay in realizing remission.

Gastric Lymphomas; Burke - 9
However, following antibiotic therapy for gastric MALT lymphoma, the association between ongoing monoclonality and risk of relapse is tenuous. One bone of contention is that that serial gastric biopsy specimens frequently exhibit an oscillating clonal status, due perhaps to sampling or recurrent Helicobacter pylori infection. Therefore, except for clinical investigations, the determination of clonality employing PCR currently is not considered pragmatic or recommended in the evaluation of post-therapy gastric MALT lymphoma biopsy specimens. The establishment of extranodal gastric marginal zone lymphoma of MALT type as a recognized clinicopathologic entity has progressed to incorporate molecular genetics and specific chromosomal translocations. For MALT lymphomas in general, the genetic abnormalities encompass trisomies 3, 12 and 18, as well as balanced translocations, specifically t(11;18)(q21;q21), t(14;18)(q32;q21), t(1;14)(p22;q32) and t(3;14)(p14;q32). The most common translocation in gastric MALT lymphoma arising in approximately 20-30% of cases (although lower in North America) is t(11;18)(q21;q21) in which the t(11;18) fuses with amino terminal of the inhibitor of apoptosis API2 at 11q21 to the carboxyl terminal of MALT1 at 18q21 leading to a chimeric fusion product.
MALT1 is involved in antigen receptor-mediated NFB activation. The API2-MALT1 fusion product is detectable by FISH. The t(11;18)(q21;q21) is restricted to extranodal MALT lymphomas and has not been reported in other forms of marginal zone lymphoma, such as splenic or nodal, or in chronic gastritis associated with Helicobacter pylori. Gastric MALT lymphomas without a t(11;18)(q21;q21) often exhibit aneuploidy, as for example trisomy 3 or 18. In this decade, the discovery of t(11;18)(q21;q21) in some patients with gastric MALT lymphoma has led to exciting clinical prognostic correlations. Specifically, patients with gastric MALT lymphoma who prove positive for t(11;18)(q21;q21) often fail to respond to Helicobacter pylori therapy and this translocation frequently arises in patients who are Helicobacter pylori negative. Employing endosonographic staging, such t(11;18)(q21;q21) positive patients who do not respond to Helicobacter pylori eradication with antibiotics are found to have disease that has spread beyond the gastric submucosa into muscularis and/or serosa in contrast to patients with lymphoma limited to the mucosa and submucosa who generally are t(11;18)(q21;q21) negative. Moreover, t(11;18)(q21;q21) is uncommon in extranodal diffuse large B cell lymphoma and patients with this translocation rarely metamorphose to diffuse large B cell lymphoma. In contrast, aneuploid t(11;18)(q21;q21) negative patients who are unresponsive to Helicobacter pylori treatment are at risk to evolve to diffuse large B cell lymphoma. For example, microsatellite screening of gastric MALT and large B cell lymphomas display alleic imbalances limited to t(11;18)(q21;q21) negative patients that are shared by both MALT and diffuse large B cell lymphomas; this observation indicates that t(11;18)(q21;q21) negative patients are the genesis of MALT lymphomas than convert to one of large B cell type. It is therefore paramount to ascertain as to whether or not the gastric marginal zone lymphoma of MALT type is t(11;18)(q21;q21) positive since this information has prognostic and therapeutic repercussions.

Gastric Lymphomas; Burke - 10
In the 1990’s, many diffuse large B cell lymphoma cases of stomach were designated as ―high-grade MALT lymphoma‖ based on the belief that these cases had transformed from ―low-grade MALT lymphoma‖ especially when both components were present in a single specimen. Despite the likelihood that such transformations occur, the term ―high-grade MALT lymphoma‖ presently is discouraged and all such cases are interpreted as large B cell lymphoma. By current definition, marginal zone lymphoma of MALT type is strictly an indolent or low-grade extranodal lymphoma. Despite the considerable recent emphasis on MALT-type lymphomas, in fact, greater than 50% of gastric lymphomas are high-grade, diffuse large B cell lymphomas. Except in small gastroscopic biopsy specimens, differentiation of large cell lymphoma from carcinoma usually is not a problem in view of the tendency of the lymphomas to be diffuse, massive, and lacking in cohesive cell aggregates. Nonetheless, immunohistochemical verification of diagnosis is recommended and is particularly valuable in delineating cases of large cell lymphoma from gastric adenocarcinoma. The infiltration by lymphoma around, or into, partially intact gastric glands, negative mucin and keratin stains, positive reactivity for CD20, and the lack of syncytial cell aggregates or malignant acinar formation aid in the distinction of large cell lymphoma from poorly differentiated adenocarcinoma, even in small biopsy specimens. Small gastric biopsy specimens, however, are subject to sampling errors and artifactual distortion, and on occasion it may be necessary to request a second biopsy, in order to render a more complete pathologic examination. One consequential diagnostic issue is the observation of large cells in a background of a marginal zone lymphoma of MALT type in a gastric biopsy specimen. No current consensus exists as to how many large cells are required to establish the evolution from MALT lymphoma to one of diffuse large B cell type. Clearly, the presence of large cells in discrete nodular aggregates or sheets is likely an indication of transformation; however, diagnostic difficulties remain for cases in which the large cells are numerous and diffusely admixed with small marginal zone lymphocytes. In one study of 106 patients with gastric MALT lymphomas, the prognostic impact of a large cell component was assessed by semiquantitative analysis of clusters and diffusely intermingled malignant large cells; in MALT lymphomas, the observation of a diffuse large cell component in the range of 1-10%, with and without non-confluent clusters of large cells, predicted a significantly worse prognosis. In a report from Italy, the presence of scattered large cells that comprised 5–10% of the MALT lymphoma cell population was regarded as prognostically irrelevant, whereas compact clustered large cells that represented more than 10% of the MALT lymphoma proved significant, as they were associated with a worse survival.
References Abbondanzo SL, Sobin LH. Gastric ―pseudolymphoma‖: a retrospective morphologic and immunophenotypic study of 97 cases. Cancer 1997;79:1656-1663.

Gastric Lymphomas; Burke - 11
Algara P, Martinez P, Sanchez L, et al. The detection of B-cell monoclonal populations by polymerase chain reaction: accuracy of approach and application in gastric endoscopic biopsy specimens. Hum Pathol 1993;24:1184-1188. Almasri NM, Zaer FS, Iturraspe JA, et al. Contribution of flow cytometry to the diagnosis of gastric lymphomas in endoscopic biopsy specimens. Mod Pathol 1997;10:650-656. Auer IA, Gascoyne RD, Connors JM, et al. t(11;18)(q21;q21) is the most common translocation in MALT lymphomas. Ann Oncol 1997;8:979-985. Baens M, Maes B, Steyls A, et al. The product of the t(11;18), an API2-MLT fusion, marks nearly half of gastric MALT type lymphomas without large cell proliferation. Am J Pathol 2000;156:1433-1439. Bacon CM, Du MQ, Dogan A. Mucosa-associated lymphoid tissue (MALT) lymphoma: a practical guide for pathologists. J Clin Pathol 2007;60:361-372. Banks PM. Gastrointestinal lymphoproliferative disorders. Histopathology 2007;50:42-54. Barth TFE, Barth CA, Kestler HA, et al. Transcriptional profiling suggests that secondary and primary large B-cell lymphomas of the gastrointestinal (GI) tract are blastic variants of GI marginal zone lymphoma. J Pathol 2007;211:305-313. Bertoni F, Conconi A, Capella C, et al. Molecular follow-up in gastric mucosa-associated lymphoid tissue lymphomas: early analysis of the LY03 cooperative trial. Blood 2002;99:2541-2544. Burke JS. Histologic criteria for distinguishing between benign and malignant extranodal lymphoid infiltrates. Semin Diagn Pathol 1985;2:152-162. Burke JS: Extranodal hematopoietic/lymphoid disorders: an introduction. Am J Clin Pathol 1999;111(Suppl 1):S40-S45. Burke JS: Are there site-specific differences among the MALT lymphomas—morphologic, clinical? Am J Clin Pathol 1999;111(Suppl 1):S133-S143. Burke JS, Sheibani K, Nathwani, BN, et al. Monoclonal small (well-differentiated) lymphocytic proliferations of the gastrointestinal tract resembling lymphoid hyperplasia: a neoplasm of uncertain malignant potential. Hum Pathol 1987;18:1238-1245. Campo E, Chott A, Kinney MC, et al. Update on extranodal lymphomas. Conclusions of the Workshop held by the EAHP and the SH in Thessaloniki, Greece. Histopathology 2006;48:481-504. Chan JKC., Ng CS, Isaacson PG. Relationship between high-grade lymphoma and low-grade B-cell mucosa-associated lymphoid tissue lymphoma (MALToma) of the stomach. Am J Pathol 1990;136:1153-1164. Cogliatti SB, Schmid U, Schumacher U, et al. Primary B-cell gastric lymphoma: a clinicopathological study of 145 patients. Gastroenterology 1991;101:1159-1170. Collins RD. Is clonality equivalent to malignancy: specifically, is immunoglobulin gene rearrangement diagnostic of malignant lymphoma? Hum Pathol 1997;28:757-759.

Gastric Lymphomas; Burke - 12
Copie_Begman C, Gaulard P, Lavergne-Slove A, et al. Proposal for a new histological grading system for post-treatment evaluation of gastric MALT lymphoma. Gut 2003;52:1656. Crump M, Gospodarowicz M, Shepherd FA. Lymphoma of the gastrointestinal tract. Semin Oncol 1999;26:324-337. De Jong D, Boot H, van Heerde P, et al. Histological grading in gastric lymphoma: pretreatment criteria and clincal relevance. Gastroenterology 1997;112:1466-1474. De Wolf-Peeters C, Achten R. The histogenesis of large-cell gastric lymphomas. Histopathology 1999;34:71-75. Deutsch AJA, Aigelsreiter A, Staber PB, et al. MALT lymphoma and extranodal diffuse large B-cell lymphoma are targeted by aberrant somatic hypermutation. Blood 2007;109:3500-3504. Dierlamm J, Baens M, Wlodarska I, et al. The apoptosis gene AP12 and a novel 18q gene, MLT, are recurrently rearranged in the t(11;18)(q21;q21) associated with mucosa-associated lymphoid tissue lymphomas. Blood 1999;93:3601-3609. Du M, Diss TC, Xu C, Peng H, et al. Ongoing mutation in MALT lymphoma immunoglobulin gene suggests that antigen stimulation plays a role in the clonal expansion. Leukemia 1996;10:1190-1197. Du MQ, Peng H-Z, Dogan A, et al: Preferential dissemination of B-cell gastric mucosa-associated lymphoid tissue (MALT) lymphoma to the splenic marginal zone. Blood 1997;90:4071-4077. Du MQ, Peng H, Liu H, et al. BCL10 gene mutation in lymphoma. Blood 2000;95:3385-3890. Du M, Peng H, Singh N, et al. The accumulation of p53 abnormalities is associated with progression of mucosa-associated lymphoid tissue lymphoma. Blood 1995;86:4587-4593. Du MQ, Xu C-F, Diss TC, et al. Intestinal dissemination of gastric mucosa-associated lymphoid tissue lymphoma. Blood 1996;88:4445-4451. Eck M, Greiner A, Schmauber B, et al. Evaluation of Helicobacter pylori in gastic MALT-type lymphoma: differences between histologic and serologic diagnosis. Mod Pathol 1999;12:1148-1151. El-Zimaity HMT, Wotherspoon A, de Jong D. Interobserver variation in the histopathological assessment of malt/malt lymphoma: towards a consensus. Blood Cells Mol Disease 2005;34:6-16. Ferreri AJM, Freschi M, Dell’Oro S, et al. Prognostic significance of the histopathologic recognition of low- and high-grade components in stage I-II B-cell gastric lymphomas. Am J Surg Pathol 2001;25:95-102. Ferrucci PF, Zucca E. Primary gastric lymphoma pathogenesis and treatment: what has changed over the past 10 years? Br J Haematol 2006;136:521-538. Ferry JA. Extranodal lymphoma. Arch Pathol Lab Med 2008;132:565-578. Genta RM, Hamner HW, Graham, DY. Gastric lymphoid follicles in helicobacter pylori infection: frequency, distribution, and response to triple therapy. Hum Pathol 1993;24:577-583. Graves FD, Linet MS, Travis LB, et al. Cancer surveillance series: non-Hodgkin’s lymphoma incidence by histologic subtype in the United States from 1978 through 1995. J Natl Cancer Inst 2000;92:1240-1251.

Gastric Lymphomas; Burke - 13
Harris NL, Isaacson PG. What are the criteria for distinguishing MALT from non-MALT lymphoma at extranodal sites? Am J Clin Pathol 1999;111(Suppl.1):S126-S132. Hsi ED, Eisbruch A, Greenson JK, et al. Classification of primary gastric lymphomas according to histologic features. Am J Surg Pathol 1998:22:17-27. Hsi ED, Greenson JK, Singleton TP, et al. Detection of immunoglobulin heavy chain gene rearrangement by polymerase chain reaction in chronic active gastritis associated with Helicobacter pylori. Hum Pathol 1996;27:290-296. Hummel M, Oeschger S, Barth TFE, et al. Wotherspoon criteria combined with B cell clonality analysis by advanced polymerase chain reaction technology discriminates covert gastric marginal zone lymphoma from chronic gastritis. Gut 2006;55:782-787. Hussell T, Isaacson PG, Crabtree JE, et al. The response of cells from low-grade B-cell gastric lymphomas of mucosa-associated lymphoid tissue to Helicobacter pylori. Lancet 1993;342:571-574. Hussell T, Isaacson, PG, Crabtree JE, et al. Immunoglobulin specificity of low grade B cell gastrointestinal lymphoma of mucosa-associated lymphoid tissue (MALT) type. Am J Pathol 1993;142:285-292. Inagaki H, Oakbe M, Seto M, et al. API2-MALT1 fusion transcripts involved in mucosa-associated lymphoid tissue lymphoma: multiplex RT-PCR detection using formalin-fixed paraffin-embedded specimens. Am J Pathol 2001;158:699-706. Isaacson PG. Lymphomas of mucosa-associated lymphoid tissue (MALT). Histopathology 1990;16:617-619. Isaacson PG, Du MQ. MALT lymphoma: from morphology to molecules. Nature Rev Cancer 2004;4:644-653. Isaacson PG, Du MQ. Gastrointestinal lymphoma; where morphology meets molecular biology. J Pathol 2005;205;255-274. Isaacson, PG, Spencer J. Malignant lymphoma of mucosa-associated lymphoid tissue. Histopathology 1987;11:445-462. Isaacson PG, Spencer J. Malignant lymphoma and autoimmune disease. Histopathology 1993;22:509-510. Isaacson PG, Spencer J, Finn T. Primary B-cell gastric lymphoma. Hum Pathol 1986;17:72-82. Isaacson PG, Spencer J, Wright DH. Classifying primary gut lymphomas. Lancet 1988;2:1148-1149. Isaacson P, Wright DH. Malignant lymphoma of mucosa-associated lymphoid tissue: a distinctive type of B-cell lymphoma. Cancer 1983;52:1410-1416. Isaacson, P, Wright DH. Extranodal malignant lymphoma arising from mucosa-associated lymphoid tissue. Cancer 1984;53:2515-2524. Isaacson PG, Wotherspoon AC, Pan, L. Follicular colonization in B-cell lymphoma of mucosa-associated lymphoid tissue. Am J Surg Pathol 1991;15:819-828.

Gastric Lymphomas; Burke - 14
Knowles DM, Jakobiec FA. Cell marker analysis of extranodal lymphoid infiltrates: to what extent does the determination of mono- or polyclonality resolve the diagnostic dilemma of malignant lymphoma v pseudolymphoma in an extranodal site? Semin Diagn Pathol 1985;2:163-168. Koch P, del Valle F, Berdel W, et al. Primary gastrointestinal non-Hodgkin’s lymphoma: I. anatomic and histologic distribution, clinical features, and survival data of 371 patients registered in the German multicenter study GIT NHL 01/92. J Clin Oncol 2001;19:3861-3873. Koch P, del Valle F, Berdel W, et al. Primary gastrointestinal non-Hodgkin’s lymphoma: II. combined surgical and conservative or conservative mangement only in localized gastric lymphoma–results of the prospective German multicenter study GIT NHL 01/92. J Clin Oncol 2001;19:3874-3883. Kurtin PJ. How do you distinguish benign from malignant extranodal small B-cell proliferations? Am J Clin Pathol 1999;111(Suppl.1):S119-S126. Lévy M, Copie-Bergman C, Gameiro C, et al. Prognostic value of translocation t(11;18) in tumoral response of low-grade gastric lymphoma of mucosa-associated lymphoid tissue type to oral chemotherapy. J Clin Oncol 2005;23:5061-5066. Liu H, Ye H, Dogan A, et al. t(11;18)(q21;q21) is associated with advanced mucosa-associated lymphoid tissue lymphoma that expresses nuclear BCL10. Blood 2001;98:1182-1187. Maes B, Demunter A, Peeters B, et al. BCL10 mutation does not represent an important pathogenic mechanism in gastric MALT-type lymphoma, and the presence of the API2-MLT fusion is associated with aberrant BCL10 expression. Blood 2002;99:1398-1404. Mitchell KA, Finn WG, Owens SR. Differences in germinal centre and non-germinal centre phenotype in gastric and intestinal diffuse large B-cell lymphomas. Leuk Lymphoma 2008;49:1717-1723. Moore I, Wright DH. Primary gastric lymphoma – a tumour of mucosa-associated lymphoid tissue; a histological and immunohistochemical study of 36 cases. Histopathology 1984;8:1025-1039. Morgner A, Miehlke S, Fischbach W, et al. Complete remission of primary high-grade B-cell gastric lymphoma after cure of Helicobacter pylori infection. J Clin Oncol 2001;19:2041-2048. Morse HC, Kearney JF, Isaacson PG, et al. Cells of the marginal zone—origins, function and neoplasia. Leuk Res 2001;25:169-178. Nakamura S, Aoyagi K, Furuse M, et al: B-cell monoclonality precedes the development of gastric MALT lymphoma in Helicobacter pylori-associated chronic gastritis. Am J Pathol 1998;152:1271-1279. Nakamura S, Yao T, Aoyagi K, et al. Helicobacter pylori and primary gastric lymphoma: a histopathologic and immunohistochemical analysis of 327 patients. Cancer 1997;79:3-11. Omonishi K, Yoshino T, Sakuma I, et al. bcl-6 protein is identified in high-grade but not low-grade mucosa-associated lymphoid tissue lymphomas of the stomach. Mod Pathol 1998;11:181-185. Osborne BM, Pugh WC. Practicality of molecular studies to evaluate small lymphocytic proliferations in endoscopic gastric biopsies. Am J Surg Pathol 1992;16:838-844. Pan L, Diss TC, Cunningham D, et al. The bcl-2 gene in primary B cell lymphoma of mucosa-associated
lymphoid tissue (MALT). Am J Pathol 1989;135:7-11.

Gastric Lymphomas; Burke - 15
Peng H, Du M, Diss TC, et al. Genetic evidence for a clonal link between low and high-grade components in gastric MALT B-cell lymphoma. Histopathology 1997;30:425-429. Radaszkiewicz T, Dragosics B, Bauer P. Gastrointestinal malignant lymphomas of the mucosa-associated lymphoid tissue: factors relevant to prognosis. Gastroenterology 1992;102:1628-1638. Ranchod M, Lewin KJ, Dorfman RF. Lymphoid hyperplasia of the gastrointestinal tract: a study of 26 cases and a review of the literature. Am J Surg. Pathol 1978;2:383-400. Rao D, Said JW. Small lymphoid proliferations in extranodal locations. Arch Pathol Lab Med 2007;131:383-396. Remstein ED, Dogan A, Einerson RR, et al. The incidence and anatomic site specificity of chromosomal translocations in primary extranodal marginal zone B-cell lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma) in North America. Am J Surg Pathol 2006;30:1546-1553. Remstein ED, James CD, Kurtin PJ. Incidence and subtype specificity of API2-MALT1 fusion transcripts in extranodal, nodal, and splenic marginal zone lymphomas. Am J Pathol 2000;156:1183-1188. Remstein ED, Kurtin PJ, James CD, et al. Mucosa-associated lymphoid tissue lymphomas with t(11;18)(q21;q21) and mucosa-associated lymphoid tissue lymphomas with aneuploidy develop along different pathogenetic pathways. Am J Pathol 2002;161:63-71. Rosenwald A, Ott G, Stilgebauer S, et al. Exclusive detection of the t(11;18)(q21;21) in extranodal marginal zone B cell lymphomas (MZBL) of MALT type in contrast to other MZBL and extranodal large B cell lymphomas. Am J Pathol 1999;155:1817-1821. Sagaert X, De Wolf-Peeters C, Noels H, et al. The pathogenesis of MALT lymphomas: where do we stand? Leukemia 2007;21:389-396. Savio A, Franzin G, Wotherspoon AC, et al. Diagnosis and posttreatment follow-up of Helicobacter pylori-positive gastric lymphoma of mucosa-associated lymphoid tissue: histology, polymerase chain reaction, or both? Blood 1996;87:1255-1260. Severson RK, Davis S. Increasing incidence of primary gastric lymphoma. Cancer 1990;66:1283-1287. Spencer J, Diss TC, Isaacson PG. Primary B cell gastric lymphoma: a genotypic analysis. Am J Pathol 1989;135:557-564. Starostik P, Greiner A, Schultz A, et al. Genetic aberrations common in gastric high-grade large B-cell lymphoma. Blood 2000;95:1180-1187. Starostik P, Patzner J, Greiner A, et al. Gastric marginal zone b-cell lymphomas of MALT type develop along 2 distinct pathogenetic pathways. Blood 2002;99:3-9. Streubel B, Lamprecht A, Dierlamm J, et al. t(14;18)(q32;q21) involving IGH and MALT1 is a frequent chromosomal aberration in MALT lymphoma. Blood 2003;101:2335-2339. Takeshita M, Iwashita A, Kurihara K, et al. Histologic and immunohistologic findings of 40 cases of gastric large B-cell lymphoma. Am J Surg Pathol 2000;24:1641-1649.

Gastric Lymphomas; Burke - 16
Thieblemont C, Bastion Y, Berger F, et al. Mucosa-associated lymphoid tissue gastrointestinal and nongastrointestinal lymphoma behavior: analysis of 108 patients. J Clin Oncol 1997;151624-1630. Thiede C, Wündisch T, Alpen B, et al. Long-term persistence of monoclonal B cells after cure of Helicobacter pylori infection and complete histologic remission in gastric mucosa-associated lymphoid tissue B-cell lymphoma. J Clin Oncol 2001;19:1600-1609. van Krieken JHJM, Raffeld M, Raghoebier S, et al. Molecular genetics of gastrointestinal non-Hodgkin's lymphomas: unusual prevalence and pattern of c-myc rearrangements in aggressive lymphomas. Blood 1990;76:797-800. Wang G, Auerbach A, Wei M, et al. t(11;18)(q21;q21) in extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue in stomach: a study of 48 cases. Mod Pathol 2009;22:79-86. Wolber RA, Owen DA, Anderson FH, et al. Lymphocytic gastritis and giant gastric folds associated with gastrointestinal protein loss. Mod Pathol 1991;4:13-15. Wotherspoon AC, Doglioni C, Diss, TC, et al. Regression of primary low-grade B-cell gastric lymphoma of mucosa-associated lymphoid tissue type after eradication of Helicobacter pylori. Lancet 1993;1342:575-577. Wotherspoon AC, Doglioni C, Isaacson PG. Low grade gastric B-cell lymphoma of mucosa-associated lymphoid tissue (MALT): a multifocal disease. Histopathology 1992;20:29-34. Wotherspoon AC, Ortiz-Hildago C, Falzon MR, et al. Helicobacter pylori-associated gastritis and primary B-cell gastric lymphoma. Lancet 1991;338:1175-1176. Wu TT, Hamilton SR. Lymphocytic gastritis: association with etiology and topology. Am J Surg Pathol 1999;23:153-158. Wündisch T, Neubauer A, Stolte M, et al. B-cell monoclonality is associated with lymphoid follicles in gastritis. Am J Surg Pathol 2003;27:882-887. Wündisch T, Thiede C, Morgner A, et al. Long-term follow-up of gastric MALT lymphoma after Helicobacter pylori eradication. J Clin Oncol 2005;23:8018-8024. Yamashita H, Watanabe H, Ajioka K, et al. When can complete regression of low-grade gastric lymphoma of mucosa-associated lymphoid tissue be predicted after Helicobacter pylori eradication? Histopathology 2000;37:131-140. Ye H, Liu H, Attygalle A, et al. Variable frequencies of t(11;18)(q21;q21) in MALT lymphomas of different sites: significant association with CagA strains of H pylori in gastric MALT lymphoma. Blood 2003;102:1012-1018. Ye H, Liu H, Raderer M, et al. High incidence of t(11;18)(q21;q21) in Helicobacter pylori–negative gastric MALT lymphoma. Blood 2003;101:2547-2550. Zucca E, Bertoni F, Roggero E, et al. Molecular analysis of the progression from Helicobacter pylori-associated chronic gastritis to mucosa-associated lymphoid-tissue lymphoma of the stomach. N Engl J Med 1998;338:804-810.

Gastric Lymphomas; Burke - 17
Zucca E, Bertoni F, Roggero E, et al. The gastric marginal zone B-cell lymphoma of MALT type. Blood 2000;96:410-419. Zukerberg LR, Ferry JA, Southern JF, et al. Lymphoid infiltrates of the stomach: evaluation of histologic criteria for the diagnosis of low-grade gastric lymphoma on endoscopic biopsy specimens. Am J Surg Pathol 1990;14:1087-1099.