gynaecological oncology top 2015 articles supplement … · gynaecological oncology top 2015...
TRANSCRIPT

Gynaecological Oncology TOP 2015 ARTICLES SUPPLEMENT
CONTENTSREVIEW: The next steps in improving the outcomes of advanced ovarian cancer Women’s Health Vol. 11 Issue 3
REVIEW: The next steps in cervical screening Women’s Health Vol. 11 Issue 2
DRUG EVALUATION: Olaparib: an oral PARP-1 and PARP-2 inhibitor with promising activity in ovarian cancer Future Oncology Vol. 11 Issue 5
Powered by

355 (2015) 11(3), 355–367 ISSN 1745-5057
part of
Review
10.2217/WHE.15.6 © 2015 Future Medicine Ltd
Review 2015/04/2811
3
2015
Worldwide ovarian cancer affects over 200,000 women per year. Overall survival rates are poor due to two predominate reasons. First, the majority of patients present with advanced disease creating significant difficulty with effecting disease eradication. Second, acquisition of chemotherapy resistance results in untreatable progressive disease. Advances in treatment of advanced ovarian cancer involve a spectrum of interventions including improvements in frontline debulking surgery and combination chemotherapy. Anti-angiogenic factors have been shown to have activity in frontline and recurrent disease while novel chemotherapeutic agents and targeted treatments are in development particularly for disease that is resistant to platinum-based chemotherapy. These developments aim to improve the progression-free and overall survival of women with advanced ovarian cancer
Keywords: adjuvant chemotherapy • BRCA • debulking surgery • immunotherapy • intraperitoneal chemotherapy • neoadjuvant chemotherapy • ovarian cancer • PARP inhibitors • platinum resistance • TP53
Ovarian cancer is the third most common gynecological malignancy worldwide but it is the most lethal, responsible for 160,000 deaths per year [1]. Following surgical deb-ulking, response rates to initial chemotherapy are high with up to 75% of patients having a complete clinical response [2]. However, the majority of patients will still go on to relapse. Once relapse has occurred treatment is not curative and unfortunately cure rates have changed little in the last 20 years. Ovarian cancer frequently responds to multiple lines of chemotherapy and new discoveries have increased the progression-free survival (PFS) and overall survival (OS) [3]. In this review, we discuss the recent discoveries and changes in the management of advanced ovarian can-cer that have improved survival and we shed light on novel treatments and current areas of research.
Advances in surgeryPrimary – maximal effort – cytoreductive surgery followed by chemotherapy is the
mainstay of initial treatment for advanced ovarian cancer; Féderation Internationale de Gynécologie et d’Obstétrique (FIGO) stage IC-IV. Research in the 20th century in Europe and USA demonstrated that removal of as much visible (macroscopic) tumor as is technically possible prior to systemic che-motherapy was the most important factor in improving patient outcomes [4]. Initially, optimal debulking was defined as residual disease of less than 2 cm in the lesion of greatest diameter. More recently it has been shown that those patients who have no mac-roscopic disease at the end of surgery have greatly improved survival compared with those who were debulked with residual dis-ease [5]. Data analysis has shown that patients who have no visible postoperative residual disease have a 5-year survival of 63%, but in those where the largest residual lesion is 1–10 mm this falls to 28.6%; demonstrat-ing the poor prognosis for any residual dis-ease however small [6]. Therefore complete macroscopic debulking surgery has become
The next steps in improving the outcomes of advanced ovarian cancer
Mark R Openshaw*,1, Christina Fotopoulou1, Sarah Blagden1 & Hani Gabra1
1Department of Medical Oncology,
Hammersmith Hospital, Imperial College
NHS, London, UK
*Author for correspondence:

356 (2015) 11(3)
Figure 1. Overall survival at 1–5 years is dependent upon the size of largest residual tumor deposit following debulking surgery. Data taken with permission from [6].
10090
807060
504030
2010
00 1–10 >10
Tumour residual (mm)
Ove
rall
surv
ival
(%
)
1 year2 years5 years
future science group
Review Openshaw, Fotopoulou, Blagden & Gabra
the target for primary surgery. Five-year survival falls further to 21.3% when residual disease is greater than 10 mm indicating that upfront surgery cytoreduction is of value even if total macroscopic tumor clearance is not feasible. This reduction in survival with increasing residual disease is demonstrated in Figure 1. The ben-efit of optimal surgical debulking within the peritoneal cavity is present even in patients with FIGO stage IV disease [7]. However, the reason for being classed as stage FIGO IV disease is important; stage IV due to positive pleura cytology alone (now stage IVA) is asso-ciated with a much higher OS than that due to intra-parenchymal hepatic metastases (now stage IVB) and this finding is reflected in the 2014 update in FIGO staging [8].
As surgery is operator dependent, improving surgi-cal skill and technique may improve outcomes if all patients are operated on with the aim of achieving no macroscopic residual disease. A large metanalysis of 3126 patients [6] showed that postoperative residual disease overrides the FIGO stage in terms of predict-ing prognosis, with advanced stage optimally debulked patients showing higher survival rates than subop-timally debulked patients of a lower stage. However, there is a subset of patients in whom complete mac-roscopic debulking is not technically possible due to distribution of disease and therefore these patients have poorer outcomes. It is not clear whether these patients are disadvantaged because their disease is unresectable and therefore improvement in surgical technique may improve their outlook or whether such patients have disease in which inability to carry out resection is a surrogate marker of unfavorable disease biology.
A review of the data of SCOTROC by Craw-ford et al. [7] shed light on this issue of favorable versus unfavorable disease. It first showed that patients in
the UK generally had a lower proportion of patients that were optimally debulked compared with similar patients in Australia and the USA, which is an ongo-ing area of controversy. It also showed that optimal debulking was associated with improved PFS mainly in patients with less extensive disease at diagnosis, rather than across all patients as might be expected if degree of debulking is a universally critical issue in ovarian cancer. This suggests that more extensive disease may represent a more aggressive subtype of disease and that the poor outcomes from incomplete debulking in these patients may be due to differences in tumor biology rather than lack of technical skill. This may also suggest why complete removal is asso-ciated with better PFS; because it is a less aggressive subtype of disease. Definitive evidence for this theory is still sought.
Neoadjuvant chemotherapyAlthough primary cytoreductive surgery is considered the standard of care in many centers there has been considerable interest in the use of neoadjuvant chemo-therapy followed by interval debulking surgery, due to the morbidity associated with maximal effort cytore-ductive surgery. In their 2010 paper, Vergote et al. [9] presented a randomized trial in which patients with stage IIIC and IV ovarian cancer were randomized to primary cytoreductive surgery or neoadjuvant chemo-therapy followed by interval debulking. The results showed no difference in survival between the two groups suggesting that neoadjuvant chemotherapy followed by interval debulking was not inferior. Since primary cytoreductive surgery carries greater morbid-ity and mortality risk due to the extent of surgery, it may be interpreted that neoadjuvant chemotherapy is preferable. However, in this trial surgical quality was strongly inhomogeneous with total macroscopic tumor clearance rates ranging from 3.9 to 63%. In addition OS was just 29 months in the upfront sur-gery group, compared with 49 months in a compa-rable group in the GOG172 [10] study carried out in the USA. These differences in survival are believed by many practitioners to be primarily due to a lower rate of low-volume debulking in the Vergote et al. study which contributed to the view held by the majority of US practitioners that primary debulking surgery is still the best practice for most women with ovar-ian cancer. This leaves unanswered the question of adequate timing of surgery with standardized total macroscopic debulking. The differences between US and European philosophies mean that neoadjuvant chemotherapy remains controversial and surgical management of ovarian cancer is typically much less aggressive in Europe than in the USA.

www.futuremedicine.com 357future science group
The next steps in improving the outcomes of advanced ovarian cancer Review
In Vergote et al. 80.6% of the neoadjuvant group of patients were resected to less than 1 cm with no con-current increase in OS, as compared with expectations from upfront surgery. This suggests that the survival advantage of being optimally debulked is lost if neo-adjuvant chemotherapy has been given and therefore that the definition of postoperative residual disease after chemotherapy is not the same as the definition of residual disease in chemo-naive patients. This poses the question of oncologic safety and validity of surgery at a delayed time point and the need to redefine the meaning of optimal surgery in the neoadjuvant setting.
This view is supported by the Gynecologic Oncology Group (GOG) 152 trial in which patients with advanced ovarian cancer and residual tumor exceeding 1 cm after primary surgery despite maximal surgical effort in the hands of experienced surgeons, were randomized after three cycles of adjuvant chemotherapy to interval cyto-reductive surgery plus three further cycles of chemother-apy or to chemotherapy alone [11]. In this study patients had higher optimal debulking rates after three cycles of chemotherapy compared with their initial surgery, but there was no survival advantage of reoperation after sub-optimal initial debulking. This study emphasizes the point that the impact of tumor residual disease depends on the timing of the cytoreductive procedure. In order to address the issue of survival benefit of upfront deb-ulking counterbalanced with higher surgical morbidity, a further trial is currently being developed with strictly defined qualification criteria for the participating centers to ensure surgical quality. This will answer the ques-tion of the optimal timing of chemotherapy. The study is planned as a collaboration between the GOG, AGO, GINECO, NOGGO, MITO and MANGO organiza-tions and is expected to include more than 700 primary patients with advanced stage III and IV disease
Secondary debulking surgeryDespite the strong evidence for primary cytoreduc-tive surgery in ovarian cancer, the evidence for sur-gery after disease relapse is less well defined. Surgery at relapse has two aims; cytoreduction or palliation of otherwise untreatable symptoms. Even in cases where cytoreduction is the aim, surgery is not curative.
In many centers relapses are surgically excised based on local practice before further chemotherapy is administered in the belief that this improves survival. Retrospective analysis of the DESKTOP dataset of patients who have undergone selective secondary deb-ulking surgery has shown significant improvement in survival if recurrent disease was completely resected; median survival in complete resection 45.2 versus 19.7 months if incomplete (hazard ratio [HR]: 3.71; 95% CI: 2.27–6.05; p < 0.001) [12]. A study prospec-
tively randomizing resectable patients to secondary deb-ulking surgery or chemotherapy, DESKTOP III [13], has been designed to directly compare these strategies. In this trial patients with presumed resectable disease are randomized between second-line chemotherapy or secondary debulking followed by chemotherapy. This trial, together with the equivalent American trial GOG 213 [14] will define the prospective value of surgical cytoreduction in platinum-sensitive relapse.
Of note, in all retrospective series so far, cytoreduc-tive surgery at relapse has been shown to be associated with prolongation of OS and PFS only if a total macro-scopic tumor clearance could be obtained. However, it is difficult to predict this preoperatively. Based on the DESKTOP dataset the German Arbeitsgemeinschaft Gynäkologische Onkologie (AGO) defined a clinical ‘AGO score’ which could predict total macroscopic tumor clearance in more than two of three patients if all of the following factors were met; ascites less than 500 ml, tumor-free surgery at initial presentation of disease and good performance status at relapse. The number and localization of the relapsed lesions were of no significant value if total macroscopic tumor clear-ance could be obtained. Interestingly, in the DESK-TOP series the patients with disseminated peritoneal carcinomatosis at relapse had the same survival as those without peritoneal carcinomatosis if they had no macroscopic disease postoperatively [15].
There is also evidence that the value of cytoreductive surgery holds beyond first disease recurrence, such that wherever complete surgical resection of disease is pos-sible and the disease is responsive to platinum chemo-therapy it may be recommended, since surgical effort appears to be associated with significantly prolonged OS and PFS [16].
Intraoperative mappingGiven the importance of complete macroscopic cyto-reductive therapy for prolonged survival in ovarian cancer, advances that can improve the visualization of miniscule tumor deposits during surgery would be beneficial. Ovarian cancer cells are known to over express folate receptors and intra-operative visualiza-tion of cancer cells by the development of a folate receptor-α-targeted fluorescent agent has shown exper-imental potential in these patients. By helping to guide surgeons in their debulking efforts these experiments aim to increase the degree of tumor clearance and therefore survival [17]. Other agents such as integrin fluorescent imaging are in development and may come to play an important role in fully resecting advanced ovarian cancer [18]. However, all of the agents described in this section are in the early stages of research and development.

358 (2015) 11(3) future science group
Review Openshaw, Fotopoulou, Blagden & Gabra
Adjuvant chemotherapyAdjuvant chemotherapy results in a long-term cure in 10–15% of advanced ovarian cancer patients and improves OS in the remainder [3]. For more than a decade platinum analog-based adjuvant chemo-therapy regimens have been the standard of care fol-lowing primary cytoreductive surgery. Carboplatin which has equivalent efficacy and comparatively less toxicity compared with cisplatin has become the most widely used platinum-based agent [3]. Many differ-ent platinum containing combinations have been investigated and carboplatin plus paclitaxel given on a 3 weekly basis has become the consensus frontline treatment [19]. Despite this consensus there is evidence that the benefit of adding paclitaxel to a carboplatin regimen is small and that in patients who are unable to tolerate paclitaxel, carboplatin alone is a reasonable first-line option [20]. The most important dose limiting toxicity of paclitaxel is neuropathy, the risk of which can be reduced with the substitution of docetaxel but at the cost of increased myelosuppression [21].
Dose dense regimesA study by Katsumata et al. [22] in Japan has sug-gested that dose dense paclitaxel may provide a sig-nificant survival advantage over standard treatment. In this trial women were randomized to a conven-tional regimen of 3 weekly carboplatin and pacli-taxel or a dose dense regimen of weekly paclitaxel plus 3 weekly carboplatin. Median OS was higher in the dose dense regimen group (100.5 months) than in the conventional treatment group (62.2 months; HR: 0.79; CI: 0.63–0.99; p = 0.039) [23].To confirm these findings in a wider ethnic group the Interna-tional Collaboration on Ovarian Neoplasm (ICON)8 study [24] is currently trialing conventional 3 weekly carboplatin and paclitaxel, to dose dense paclitaxel, with conventional and dose dense carboplatin. Two trials have already published further data on dose-dense paclitaxel. The completed Multicenter Italian Trials in Ovarian Cancer (MITO-7) study showed no difference between its trial arms of three weekly carboplatin (AUC 6) plus paclitaxel (175 mg/m2) versus weekly carboplatin (AUC 2) plus paclitaxel (60 mg/m2) [25]. However, the lower total dose of weekly paclitaxel used in this study may have been responsible for the negative study findings. GOG262 has published preliminary results [26]. This study compared dose-dense weekly paclitaxel (80 mg/m2) and carboplatin (AUC 6) with a conventional three weekly carboplatin and paclitaxel. Preliminary results show no difference between the two arms. However, crucially both arms provided the option of additional bevacizumab. This addition makes results difficult to
interpret as it appears that bevacizumab may reduce the advantage of dose dense paclitaxel.
Angiogenesis inhibitorsDespite the improvement in chemotherapy regimens the overall improvement in PFS since the discovery of the platinum agents has been disappointing. There-fore, recent research has been focused on understand-ing the biology of ovarian cancer in order to discover new methods of targeting the disease.
Angiogenesis is a complex process regulated by a number of endogenous pro- and anti-angiogenic fac-tors and plays a key role in the growth and metastases of solid tumors including ovarian cancer. VEGF is a key mediator of angiogenesis and expression of intra-tumoral VEGF and its receptor are associated with poor prognosis in ovarian cancer [27]. Bevacizumab is a monoclonal antibody that binds to the isoform VEGF-A and has proven efficacy in a range of cancers. The ICON7 [28] and the GOG 218 study [29] were two landmark trials that defined the role of bevacizumab in front-line treatment of advanced ovarian cancer. Patients were randomized to three weekly carbo-platin and paclitaxel with or without bevacizumab. Median PFS was improved by 1.5 months in ICON7 (HR: 0.81; 95% CI: 0.70–0.94; p = 0.004) and 3.8 months in GOG218 (HR: 0.72; 95% CI: 0.62–0.82; p < 0.001) and was restricted to those patients who received maintenance bevacizumab for a year or more. Although PFS was improved with bevacizumab in GOG218 and ICON 7, a benefit in OS has not been shown. The PFS advantage then appeared to wane once bevacizumab was discontinued and the optimal period of time to continue the drug remains unclear. Bevacizumab is usually well tolerated but common toxicities include hypertension and proteinuria. An increase in the frequency of bowel perforation has also been observed. Bevacizumab is the first anti-angio-genesis drug to show an improvement in PFS and on the basis of these trials the EMA has approved beva-cizumab for front line use. In contrast the National Institute for Clinical Excellence has not provided funding on the basis of lack of cost–effectiveness [30], as bevacizumab costs GB£128,000–£161,000 per quality-adjusted life year (QALY) gained. The appro-priate role for bevacizumab in ovarian cancer in the up-front adjuvant setting is not yet determined.
A number of VEGF pathway inhibitors have been trialed besides bevacizumab. Pazopanib is an oral tyro-sine kinase inhibitor that inhibits several kinase recep-tors including VEGF, PDGF and FGF. The PDGF and FGF pathways have been shown to be involved in angiogenesis and may be involved in the resis-tance mechanisms to VEGF receptor inhibitors [27].

www.futuremedicine.com 359future science group
The next steps in improving the outcomes of advanced ovarian cancer Review
AGO-OVAR-16 [31] is a large Phase III trial compar-ing 24 months of maintenance pazopanib vs placebo in 940 women with advanced ovarian cancer. Patients in the pazopanib arm had a PFS of 17.9 months ver-sus 12.3 months in the placebo arm (HR: 0.76; 95% CI: 0.64–0.91; p = 0.002). The most common tox-icity was hypertension (31% grade 3/4), with other toxicities including diarrhoea, hepatic impairment and neutropenia. The result of this trial suggests that maintenance pazopanib is a potential oral alternative to bevacizumab, although head to head comparison trials have not been undertaken. However, as the use of VEGF antagonists in the up-front setting remains controversial, they have not been incorporated into clinical trials as the ‘standard’ arm in the treatment of advanced disease.
Alternative angiogenic pathways are also being tar-geted as potential treatments for ovarian cancer. One of these pathways involves angiopoietins; which are cytokines involved in the regulation of angiogenesis and inflammation. Novel peptibodies which selec-tively neutralize specific angiopoietins have shown promise in Phase II clinical trials in recurrent ovarian cancer [27] and in the future may provide new adjuvant treatments.
Intraperitoneal chemotherapyAs the development of new targeted therapies for ovar-ian cancer has been limited, research has also focused on delivering existing chemotherapeutic agents in novel ways. Since ovarian cancer frequently has an intraperitoneal distribution at diagnosis, intraperi-toneal instillation of chemotherapy has been investi-gated as a way of exposing tumor tissue to extremely high levels of chemotherapy. Studies have shown that intraperitoneal concentrations of intraperitoneal cis-platin may reach 12–21-fold the concentration of the plasma [32]. Therefore the intraperitoneal route may be expected to have improved disease control in addition to reduced systemic adverse effects. The first random-ized trial of intraperitoneal chemotherapy, published in 1996 GOG 104 [33], supported both of these aspects. In this trial patients were given the same dose of cisplatin (100 mg/m2) intraperitoneal or intravenous (iv.) along with cyclophosphamide iv. for six cycles. Median sur-vival in the intraperitoneal arm was significantly longer at 49 months compared with 41 months in the iv. arm (HR: 0.76; CI: 0.61–0.96; p = 0.02). Grade III/IV neutropenia, clinical hearing loss, and neuromuscu-lar toxic effects were all reduced in the intraperito-neal group. Two subsequent trials; GOG114 [34] and GOG172 [10] confirmed the finding of increased sur-vival. However, they showed increased grade III/IV adverse effects, particularly fatigue, gastrointestinal
and hematological effects. As a result of these adverse effects only 18% and 42% of patients completed the assigned intraperitoneal chemotherapy in the respec-tive trials leading to serious concerns about intraperi-toneal toxicity. An additional problem of GOG172 and GOG114 trials lay in their design. While GOG104 was a simple substitution of iv. for intraperitoneal che-motherapy, both of the subsequent trials had unequal control and treatment arms (Figure 2) such that it is dif-ficult to be certain that the positive results of these tri-als were due to superiority of the intraperitoneal route. Consistent evidence of significantly increased survival led the National Cancer Institute (NCI) in the USA to recommend the use of frontline intraperitoneal chemo-therapy in 2006 [35]. Improvements in port placement and management may significantly reduce toxicities and there are a number of centers worldwide that use and develop this promising form of therapy. However, lack of experience with this route of chemotherapy and uncertainty over its risk of complications has meant that it has yet to be widely adopted.
Areas for further research include substituting cis-platin with carboplatin to look for reduced intraperi-toneal toxicity and a definitive trial with balanced che-motherapy doses to confirm that intraperitoneal is a superior route of administration.
In an exploratory retrospective subanalysis of GOG172 by Lesnock et al. the tumors were analyzed for expression of the Breast Cancer 1 (BRCA1) pro-tein [36]. This study showed that patients with tumors that had reduced BRCA1 expression had longer OS in response to intraperitoneal chemotherapy (84 months) than iv. (47 months; HR: 0.67; 0.67 CI: 0.47–0.97; p = 0.03). However, the benefit of intraperitoneal vs. iv. chemotherapy was lost in tumors with normal BRCA1 expression. It would therefore appear that the benefit of intraperitoneal chemotherapy may be restricted to those with reduced BRCA1 expression. Prospective trials are needed to confirm this finding and with a 36 month survival advantage it would appear prudent to design trials involving intraperitoneal treatment in those with aberrant BRCA1 expression, particularly those with inherited BRCA1 mutations.
BRCA1- & BRCA2-related diseaseBRCA1 and BRCA2 are dominantly inherited can-cer susceptibility genes that were first discovered in families with high rates of breast cancer. Mutations in these genes have been found to be common in women with high-grade serous cancer (HGSC), which is the most common histological subtype of ovarian can-cer. BRCA germline mutations are seen in 13–16% of HGSC and somatic mutations are detected in another 6% of cases. The lifetime risk of HGSC is up to 60%

360 (2015) 11(3)
Figure 2. Design of GOG114and GOG172. ip.: Intraperitoneal; iv..: Intravenous.
GOG114
Stage III ovarian cancerdebulked to ≤1 cm
GOG172
Stage III ovarian cancerdebulked to ≤1 cm
Arm 1Cisplatin 75 mg/m2 iv.Paclitaxel 135mg/m2 iv.21-day cycle × 6
Carboplatin AUC 9 x 2 iv.*28 day cycle x 2Cisplatin 100 mg/m2 ip.**Paclitaxel 135 mg/m2 iv.21-day cycle × 6
Paclitaxel 135 mg/m2 iv. day 1 Cisplatin 75 mg/m2 iv. day 221-day cycle × 6
Paclitaxel 135 mg/m2 iv.Cisplatin 100 mg/m2 ip. day 2†
Paclitaxel 60 mg/m2 ip. day 8‡
21-day cycle × 6
Arm 2
Arm 1
Arm 2
Control arm*Additional dose of iv. carboplatin**Increased dose ip. cisplatin
------ Trial arm†Increased dose ip. cisplatin‡Additional dose ip. paclitaxel
future science group
Review Openshaw, Fotopoulou, Blagden & Gabra
in BRCA1 mutation carriers, and up to 27% in those with BRCA2 [37]. Women who are known carriers are offered pre-emptive salipingo-oophrectomy after com-pletion of their families to substantially reduce their risk of developing ovarian cancer.
Patients with HGSC that carry BRCA muta-tions have been shown to be particularly sensitive to platinum-based chemotherapy, and frequently have sustained response to repeated treatments with these agents [38]. This increased sensitivity to platinum agents may explain the prolonged survival in the intra-peritoneal trials mentioned previously, since BRCA patients may have particular sensitivity to the higher intratumoral concentrations of platinum agents.
PARP inhibitorsThe BRCA genes encode proteins that sense DNA damage and are involved in double standed (ds)DNA repair via homologous recombination. Cell lines carrying these mutations are therefore deficient in dsDNA repair and rely upon single stranded (ss)DNA repair mechanisms to resolve DNA damage. Poly(ADP-ribose) polymerase (PARP) enzymes are a family of proteins shown to be involved in ssDNA repair. PARP inhibitors are a new class of drug that
prevent PARP enzyme activity leading to persistence of spontaneous ssDNA breaks that progress to dsDNA breaks on replication [39]. BRCA mutated cells cannot repair these breaks leading to cell death, while nor-mal cells are spared. This targeting of BRCA mutated cells by exploitation of their specific sensitivity to PARP inhibitors is called ‘synthetic lethality’ and has been the focus of a great deal of research in recent years. Olaparib is one of a number of PARP inhibi-tors that have been investigated. A Phase III study of maintenance olaparib versus placebo has already been completed in women with recurrent HGSC [40]. Women receiving olaparib had an improvement in PFS which was much more pronounced in those who harboured a BRCA mutation (11.5 vs 5.6 months; p < 0.00001) [41]. This evidence, although involving retrospective BRCA evaluation, suggests that testing for BRCA status in women with HGSC has clinical significance. A Phase III trial called SOLO (Study of OLaparib in Ovarian cancer) has been designed to determine the benefit of olaparib as a maintenance monotherapy specifically in BRCA mutated ovar-ian cancer patients. This strategy will be evaluated in both the first-line (SOLO1) [42] and second-line (SOLO2) [43] setting.

www.futuremedicine.com 361future science group
The next steps in improving the outcomes of advanced ovarian cancer Review
Predicting relapseUnfortunately the majority of patients with advanced ovarian cancer will go on to have relapse of their dis-ease after frontline surgery and chemotherapy. There are many options for surveillance to detect relapse including serial measurement of the tumor marker cancer antigen 125 (CA125), physical exam and a number of imaging modalities. Significant controversy exists about the rela-tive effectiveness of each modality and their effect on survival and whilst detection of recurrences by physical exam alone is rare (4% of relapses), the combination of examination, history and elevated CA125 will detect up to 90% of recurrences [2].
CA125 is a glycoprotein which is elevated in up to 88% of advanced ovarian cancers and has a role in both monitoring disease response and in follow-up. Rising CA125 may precede development of symptoms by a number of months and represents a significant manage-ment dilemma; whether to treat when CA125 is first elevated or on the development of symptoms? This was addressed in a study by Rustin et al. [44] in which women who were in complete remission were randomized to early treatment of relapse on asymptomatic CA125 rise or treatment only on symptomatic recurrence. There was no significant difference between the two treatment arms (median survival: 25.7 vs 27.1 months) but women in the early treatment arm received more chemotherapy overall and had an earlier deterioration in quality of life. Therefore measuring CA125 alone is not sufficient to dictate timing of further treatment but must be used in conjunction with other investigations. Continuing developments in chemotherapy treatment and increas-ing use of secondary debulking constantly change the options for disease treatment and mean that early detec-tion may in the future allow improved outcomes
Although there are many imaging modalities avail-able for ovarian cancer, there has been no OS benefit shown for serial radiological studies [2]. The most com-monly used modality to guide treatment is computer tomography (CT). However, as an elevated CA125 may precede CT findings by many months alternative imaging has been investigated to decide who requires immediate treatment. 18Fluorine deoxyglucose PET is a method of imaging based on the increased glucose metabolism of malignant tumors. It has been shown to detect ovarian relapses at an earlier stage during follow-up with a sensitivity of up to 96% [45]. This suggests that fluorine deoxyglucose PET could play a role in early detection of relapses but it has yet to been proven to be useful in clinical trials.
Platinum-sensitive relapseAt relapse the period of time that has elapsed since last treatment with platinum-based compounds is prog-
nostic [3]. Patients who have relapsed more than 1 year after their last platinum-based treatment are classified as platinum sensitive and have approximately a 60% chance of response to further platinum containing chemotherapy. If relapse occurs following a platinum-free interval (PFI) of 6–12 months patients are classi-fied as partially platinum sensitive and have a reduced response rate of 25–40% to platinum agents. Patients who relapse with a PFI of less than 6 months are clas-sified as platinum resistant and are not rechallenged with platinum-based regimes due to unacceptably low response rates.
In platinum sensitive relapse, second-line treatment will most commonly involve carboplatin. Research into combination therapy has shown that the addi-tion of paclitaxel to a platinum regime gives superior survival at relapse [46]. However, this combination is accompanied by significant toxicity including periph-eral neuropathy, neutropenia and alopecia. Carbopla-tin plus gemcitabine is one alternative regime which has also shown superiority to carboplatin alone [47]. The CALYPSO [48] study compared the combination of pegylated liposomal doxorubicin (PLD) and carbo-platin with standard carboplatin and paclitaxel at first relapse. The PLD arm had superior PFS (HR: 0.82; 95% CI: 0.72–0.94; p = 0.005), although in subse-quent analysis there was no difference in OS, poten-tially due to unequal post study cross-over [49]. Overall more patients discontinued treatment in the standard arm due to severe nonhematologic toxicity, with the PLD arm having significantly less neuropathy and alopecia, but more nausea, mucositis and hand-foot syndrome. Therefore in the relapse setting carboplatin plus PLD is less toxic and likely more effective than carboplatin plus paclitaxel.
Angiogenesis inhibitorsAnti-angiogenic agents are known to have activity in the setting of platinum sensitive recurrence. The larg-est randomized Phase III trial is the OCEANS trial [50] in which carboplatin and gemcitabine was trialled with and without the addition of concurrent bevaci-zumab, which was continued until disease progression. PFS was superior in the bevacizumab arm (HR: 0.48; 95% CI: 0.39–0.60; p < 0.0001) and has therefore been approved for use by the EMA and the cancer drugs fund (CDF) for first-line platinum-sensitive relapse in conjunction with carboplatin and gemcitabine only. There is currently no published data on the effective-ness of bevacizumab for relapse after having received the drug in the first-line setting.
Cediranib is an oral tyrosine kinase inhibitor that also has inhibitory effects on VEGF/PDGF/FGF receptors and is similar in action to pazopanib [27].

362 (2015) 11(3) future science group
Review Openshaw, Fotopoulou, Blagden & Gabra
The ICON6 study enrolled patients following their first relapse with ovarian cancer. The control arm was standard chemotherapy, with two experimental arms of concurrent cediranib plus placebo maintenance or concurrent plus 18 months maintenance cedira-nib. The trial showed the combination of cediranib and chemotherapy provided a 2.7 month advantage in OS (17.6 vs 20.3 months; HR: 0.7; p =0.04) and also extended PFS by 3.1 months (9.4 vs 12.5 months; HR: 0.68; p = 0.0022) thus showing a significant survival benefit to cediranib use [51].
The OCEANS trial which showed improvement in PFS but not OS and the ICON 6 trial which showed modest OS and PFS improvement strengthen the evi-dence for a benefit from targeting the angiogenesis pathway in recurrent ovarian cancer, in addition to the evidence for targeting it in the frontline setting. On publication of the final results of ICON6 cedira-nib will likely be an oral alternative to bevacizumab in recurrent ovarian cancer.
Partially platinum-sensitive relapseThe trials thus far described in this section primarily recruited patients with platinum-sensitive recurrence. For patients that relapse with a PFI of 6–12 months there is less consensus regarding rechallenge with plat-inum-based regimes due to decreased response rates. In addition, platinum-based toxicities may be greater with a short PFI and therefore there is need for alter-native nonplatinum agents. Trabectedin is a chemo-therapy agent originally derived from the sea squirt Ecteinascidia turbinate, that has proven efficacy in relapsed ovarian cancer [52]. In the OVA-301 trial tra-bectedin plus PLD was shown to have superior activity to PLD alone in relapsed ovarian cancer [53]. A total of 214 patients in this study had partially platinum sensi-tive disease and a subanalysis of these patients showed a median PFS of 7.4 months in the combination arm versus 5.5 months in the PLD arm (HR: 0.65; 95% CI: 0.45–0.92; p = 0.015) [54]. This was the largest dif-ference for all PFI subsets analyzed therefore showing evidence for a specific benefit of trabectedin/PLD in partially platinum-sensitive disease. In addition, clini-cal data suggest that the artificial expansion of PFI with an intervening nonplatinum therapy may be ben-eficial [55], possibly by reversing platinum resistance. Within OVA-301 partially platinum sensitive patients in the trabectedin/PLD arm survived significantly longer after subsequent platinum treatment (HR: 0.63; p = 0.0357; median: 13.3 vs 9.8 months) than those on PLD alone, thus supporting the hypothesis that artificially increasing PFI is important. The addi-tional benefit for trabectedin/PLD therapy in terms of PFS and survival after further platinum therapy
provides a new option for partially platinum-sensi-tive patients. However, because platinum containing regimes remain a reasonable treatment option in this group and the evidence is based on subset analysis only, a trial is planned comparing carboplatin/PLD using the regimen evaluated in the CALYPSO trial with trabectedin/PLD [54].
Platinum-resistant relapseWhere the PFI is less than 6 months platinum agents have low response rates and the median survival is less than 12 months [56]. The mainstay of treatment is single agent therapies which have varying levels of activity. Weekly paclitaxel appears to have response rates of up to 25% [57]. Other commonly used thera-pies such as topotecan and PLD have response rates of 10–15% [56]. Improvements on these poor response rates have been sought by adding anti-angiogenic fac-tors. The AURELIA study investigated the addition of bevacizumab to single agent therapy from any of the previous mentioned single agents. Choice of ther-apy was investigator directed depending on previous patient treatment tolerances. The addition of bevaci-zumab showed a PFS advantage with a median PFS of 6.7 months with chemotherapy plus bevacizumab ver-sus 3.4 months with chemotherapy alone (HR: 0.48; 95% CI: 0.38–0.60; p < 0.001) [58]. The improve-ment in PFS is not dissimilar to the 1.5–3.8-month improvement in PFS seen in the adjuvant use of beva-cizumab [28,29]. However, in the platinum-resistant set-ting there is considerably less exposure to bevacizumab to gain the same improvement in PFS. This shows that anti-angiogenic treatment is active in all presentations of ovarian cancer resulting in an approximate 3 month improvement in PFS independent of PFI.
Novel chemotherapeutic agentsIn the search for novel nonplatinum-based chemo-therapeutic agents various lines have been investi-gated. Over 80% of ovarian cancers overexpress the membrane bound folate receptor (FR), whereas it is poorly expressed or absent in most normal cells mak-ing it an excellent candidate for targeted therapy. Farletuzumab, an FR targeted antibody, has shown disappointing results in Phase III trials [18]. How-ever, vintafolide, a folate conjugated with the vinca alkaloid desacetylvinblastine monohydrazine, is spe-cifically designed to target the FR delivering potent chemotherapy directly to cells overexpressing FR. PRECEDENT [59] is a Phase II study which assessed vintafolide with PLD versus PLD alone in recur-rent ovarian cancer. This showed an improvement in PFS with a median survival of 5.0 months for the combination arm versus 2.7 months for the PLD

www.futuremedicine.com 363future science group
The next steps in improving the outcomes of advanced ovarian cancer Review
alone arm (HR: 0.63; 95% CI: 0.41–0.96; p = 0.031). This trial was run alongside evaluation of etarfolatide, which is a folate-technetium conjugate imaging agent that selectively binds FR-expressing cells analogous to the action of vintafolide. This allowed imaging of FR-expressing tumors via single photon emission computed tomography. Patients who had 100% of lesions positive for FR, had an improved PFS median of 5.5 months in the experimental arm compared with 1.5 months for PLD alone. PROCEED, a random-ized placebo controlled trial of vintafolide alongside etarfolatide imaging has been undertaken [60], but enrolment has been suspended due to a disappointing interim analysis. It is hoped that future developments of FR targeted drugs may be more successful.
Another area of interest has been in inhibition of the Phosphoinositide 3-kinase (PI3K)/AKT pathway. This has been shown to be overactive in ovarian can-cer and may be involved in the resistance of ovarian cancer to platinum agents [61]. Inhibition of AKT is postulated to reverse chemotherapeutic resistance and therefore a Phase I/II study is underway to investi-gate a novel AKT inhibitor GSK2110183 when given alongside carboplatin and paclitaxel [62].
ImmunotherapyThere is evidence that the human immune system generates tumor reactive immune cells that have anti-tumor potential. Therefore modulation of this response represents a rational therapeutic approach to the treatment of ovarian cancer [18]. The most well developed area of research in ovarian cancer immu-notherapy is dendritic cell (DC)-based vaccines. DCs capture, process and present antigens including tumor-associated antigens thereby inducing the gen-eration of effector and memory T cells [63]. The activ-ity of these cells is believed to be responsible for the observed antitumor effects. The design of DC therapy in ovarian cancer is in the early stages but there have been encouraging findings in the treatment of pros-tate cancer [64]. DC vaccines are usually prepared ex vivo. Autologous immune cells collected from patients are treated with a range of peptides, mRNA, proteins, tumor lysates or other agents before reintroduction to the patient [63]. The best immunizing antigens are currently unknown but the use of a cell surface associ-ated mucin 1 dendritic vaccination is the most exten-sively studied and CA125 responses have been seen in response to their use [65]. A Phase II trial (CAN-003) randomizing 56 patients in remission after first or sec-ond-line therapy to DC vaccine or standard observa-tional care has demonstrated safe use of a mucin 1 DC vaccine and a significant increase in PFS in patients after second-line therapy (median PFS for observation
4.94 months vs ‘not reached’ but greater than 12.91 months for DC vaccine [p = 0.04]) [66]. A larger ran-domized Phase II trial called CANVAS has been initi-ated to further investigate this vaccine’s effectiveness in first- and second-line remission [67].
Programed cell death 1 (PD-1) is an inhibitory surface receptor expressed by a number of immune cells including B and T cells that binds to the pro-gramed cell death ligand 1 (PD-L1). The expression of PD-L1 in tumors is inversely correlated with sur-vival of patients, as tumor cells are believed to activate the PD-1 inhibitory pathway to evade the immune response. In animal models antibody mediated PD-1 blockade enhanced tumor immunity [68], suggesting that targeted interventions against this pathway may increase antitumor immunity in ovarian cancer and may compliment developments in DC vaccination.
Future perspectiveAs we begin to understand how different ovarian can-cers respond to standard chemotherapy, novel agents and targeted agents, the tailoring of treatment to a cancer’s histological and genetic makeup will become increasingly important. Whole-exome DNA sequenc-ing of 316 ovarian cancer samples has shown that TP53 mutations occur in virtually all (96%) patients and that a number of genes are mutated in a subset of high-grade serous ovarian cancers. BRCA1 and BRCA2 had germline mutations in 9 and 8% of cases, respectively, and there were somatic mutations in a further 3% of cases. Six other genes were also shown to be statistically recurrently mutated: RB1, NF1, FAT3, CSMD3, GABRA6 and CDK12 [69]. Although the clinical significance of these mutations has yet to be fully elucidated, it lends credence to the theory that there may be other genetic mutations besides BRCA that could be targeted to improve outcomes. Spatial heterogeneity of cancer mutations within the same person suggests that single biopsies should not be used as the sole specimen when deciding on personalized medicine [70].
The TP53 tumor suppressor gene is a key regulator of cell cycle arrest or cell death upon oncogenic stress. Most TP53 mutations are missense mutations that result in dysfunctional p53 protein allowing cell survival and division. Restoration of wild type p53 function can therefore induce cell death and tumor regression. APR-246 is a compound that can restore wild type confor-mation to mutant p53. It has been tested in a Phase I/II clinical trial involving 22 patients with hematological malignancies or hormone-refractory prostate cancer. Drug safety has been confirmed and clinical effects have been observed [71]. Due to the frequency of TP53 mutations in ovarian cancer, restoring p53 activity with

364 (2015) 11(3) future science group
Review Openshaw, Fotopoulou, Blagden & Gabra
APR-246 is an area of current research. PiSARRO, a Phase Ib/II study assessing carboplatin combination chemotherapy with or without APR-246 in relapsed platinum-sensitive disease has been designed [72] spe-cifically to investigate the effectiveness of modifying dysfunctional p53 to attain disease control.
A potential barrier to targeted therapy is that the mutations may change over time, such as when the tumor becomes resistant to chemotherapy treatment. There-fore, a method of detecting these changes is important
and in early development are techniques such as cir-culating tumor DNA analysis [73]. The ALFApump® system provides an alternative way to obtain whole cell DNA. It has been developed to continuously drain asci-tes into the bladder [74] and is currently being trialed in advanced ovarian cancer. An exciting possibility is to continuously monitor the genetic mutations of cancer cells present within urine due to the presence of the pump. As the urine may include cells from multiple tumor sites within the peritoneal cavity it may provide
Executive summary
Advances in surgery• Primary cytoreductive surgery to no macroscopic disease shows the greatest improvements in 5-year survival.• The benefit of optimal surgical debulking within the peritoneal cavity is present even in patients with stage IV
disease.• Neoadjuvant chemotherapy reduces morbidity due to surgery but there are concerns that surgery at a delayed
time point may compromise effective surgical disease clearance.• Debulking surgery at relapse may be indicated if complete macroscopic tumor clearance can be achieved.
Ongoing trial DESKTOP III.Adjuvant chemotherapy• Carboplatin and paclitaxel three weekly is the standard of care. The intraperitoneal route may be considered
in selected patients.• Dose dense paclitaxel given with carboplatin may have a survival advantage. Ongoing research is with ICON8.• Bevacizumab and pazopanib in addition to standard chemotherapy improve progression-free survival.• Intraperitoneal chemotherapy has superior efficacy at the potential cost of increased adverse effects.Breast cancer (BRCA)-related disease• BRCA-related disease is more sensitive to platinum-based chemotherapy.• Intraperitoneal platinum chemotherapy may be more effective in BRCA mutation carriers.• PARP inhibitors show an improvement in progression-free survival on initial trials. Further research will be
undertaken in the SOLO 1 & SOLO 2 trials.Predicting relapse• The majority of patients with advanced ovarian cancer will go on to have relapse of their disease.• There is evidence that CA125 monitoring is not necessary during follow-up.• An asymptomatic CA125 rise with no lesion on imaging represents a prognostic dilemma and current
information indicates they do not need treatment until symptomatic.Platinum & partially platinum-sensitive relapse• Rechallenge with a platinum-containing regime is mandatory if tolerated.• The combination of pegylated liposomal doxorubicin and carboplatin is less toxic and likely more effective
than carboplatin paclitaxel.• Use of bevacizumab or cediranib show a progression-free survival advantage when given with standard
chemotherapy.• Trabectedin is a novel chemotherapy agent with appears most effective in partially platinum-sensitive disease.Platinum-resistant relapse• Where the platinum-free interval is less than 6 months platinum agents have low response rates.• Chemo-monotherapy is the mainstay of treatment.• The addition of bevacizumab to chemo-monotherapy in the AURELIA study showed a progression-free survival
advantage similar to that seen in the adjuvant setting.• Folate-receptor targeted chemotherapy agents are an area of current interest.• Immunotherapy using dendritic cell vaccines is a promising area of future research.Future perspective• The use of targeted therapies based on the detected mutations of the tumors will be of increasing importance
in tailoring anticancer therapy.• TP53 is almost ubiquitously mutated in ovarian cancer and drugs that can modify dysfunctional p53 are an
important area of research. Ongoing trial PiSARRO.• Spatial and temporal variation in tumor characteristics and mutations is a significant barrier to effective
personalized medicine.

www.futuremedicine.com 365future science group
The next steps in improving the outcomes of advanced ovarian cancer Review
a method to monitor both spatial and temporal genetic change allowing a more comprehensive assessment for personalized medical care.
In summary the treatment of ovarian cancer draws on a wide knowledge of expertise. Advances in its treatment are diverse and include new surgical treat-ments, use of an increasing number of imagining modalities, new uses of existing therapies and the development of novel chemotherapeutic agents and targeted therapies. It is likely that each of these will have an impact on the disease such that there will be a continued improvement in 5-year survival and a gradual increase in the percentage of patients who
achieve a long-term cure. The future is promising, and with each step forward the lives of patients will be inexorably improved.
Financial & competing interests disclosureS Blagden has received an honorarium from Roche and re-
search funding from Merck. The authors have no other rele-
vant affiliations or financial involvement with any organization
or entity with a financial interest in or financial conflict with
the subject matter or materials discussed in the manuscript
apart from those disclosed.
No writing assistance was utilized in the production of this
manuscript.
ReferencesPapers of special note have been highlighted as: • of interest; •• of considerable interest
1 Lozano R, Naghavi M, Foreman K et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380(9859), 2095–2128 (2012).
2 Marcus CS, Maxwell GL, Darcy KM, Hamilton CA, Mcguire WP. Current approaches and challenges in managing and monitoring treatment response in ovarian cancer. J. Cancer 5(1), 25–30 (2014).
3 Cannistra SA. Cancer of the ovary. N. Engl. J. Med. 351(24), 2519–2529 (2004).
4 Hacker NF, Berek JS, Lagasse LD, Nieberg RK, Elashoff RM. Primary cytoreductive surgery for epithelial ovarian cancer. Obstet. Gynecol. 61(4), 413–420 (1983).
5 Eisenkop SM, Nalick RH, Wang HJ, Teng NN. Peritoneal implant elimination during cytoreductive surgery for ovarian cancer: impact on survival. Gynecol. Oncol. 51(2), 224–229 (1993).
6 Du Bois A, Reuss A, Pujade-Lauraine E, Harter P, Ray-Coquard I, Pfisterer J. Role of surgical outcome as prognostic factor in advanced epithelial ovarian cancer: a combined exploratory analysis of 3 prospectively randomized Phase 3 multicenter trials: by the Arbeitsgemeinschaft Gynaekologische Onkologie Studiengruppe Ovarialkarzinom (AGO-OVAR) and the Groupe d’Investigateurs Nationaux Pour les Etudes des Cancers de l’Ovaire (GINECO). Cancer 115(6), 1234–1244 (2009).
•• Evidence that the goal of primary surgery should be complete macroscopic clearance.
7 Crawford SC, Vasey PA, Paul J, Hay A, Davis JA, Kaye SB. Does aggressive surgery only benefit patients with less advanced ovarian cancer? Results from an international comparison within the SCOTROC-1 Trial. J. Clin. Oncol. 23(34), 8802–8811 (2005).
8 Mutch DG, Prat J. 2014 FIGO staging for ovarian, fallopian tube and peritoneal cancer. Gynecol. Oncol. 133(3), 401–404 (2014).
9 Vergote I, Trope CG, Amant F et al. Neoadjuvant chemotherapy or primary surgery in stage IIIC or IV ovarian cancer. N. Engl. J. Med. 363(10), 943–953 (2010).
10 Armstrong DK, Bundy B, Wenzel L et al. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N. Engl. J. Med. 354(1), 34–43 (2006).
11 Rose PG, Nerenstone S, Brady MF et al. Secondary surgical cytoreduction for advanced ovarian carcinoma. N. Engl. J. Med. 351(24), 2489–2497 (2004).
12 Harter P, Du Bois A, Hahmann M et al. Surgery in recurrent ovarian cancer: the Arbeitsgemeinschaft Gynaekologische Onkologie (AGO) DESKTOP OVAR trial. Ann. Surg. Oncol. 13(12), 1702–1710 (2006).
13 ClinicalTrials Database: NCT01166737. https://clinicaltrials.gov/ct2/show/NCT01166737
14 ClinicalTrials Database: NCT00565851. https://clinicaltrials.gov/ct2/show/NCT00565851
15 Harter P, Hahmann M, Lueck HJ et al. Surgery for recurrent ovarian cancer: role of peritoneal carcinomatosis: exploratory analysis of the DESKTOP I Trial about risk factors, surgical implications, and prognostic value of peritoneal carcinomatosis. Ann. Surg. Oncol. 16(5), 1324–1330 (2009).
16 Fotopoulou C, Zang R, Gultekin M et al. Value of tertiary cytoreductive surgery in epithelial ovarian cancer: an international multicenter evaluation. Ann. Surg. Oncol. 20(4), 1348–1354 (2013).
17 Van Dam GM, Themelis G, Crane LM et al. Intraoperative tumor-specific fluorescence imaging in ovarian cancer by folate receptor-alpha targeting: first in-human results. Nat. Med. 17(10), 1315–1319 (2011).
• Early indication that the development of intraoperative tumour imaging may revolutionize debulking surgery.
18 Schmid BC, Oehler MK. New perspectives in ovarian cancer treatment. Maturitas 77(2), 128–136 (2014).
19 Du Bois A, Quinn M, Thigpen T et al. 2004 consensus statements on the management of ovarian cancer: final document of the 3rd International Gynecologic Cancer Intergroup Ovarian Cancer Consensus Conference (GCIG OCCC 2004). Ann. Oncol. 16(Suppl. 8), viii7–viii12 (2005).
20 Group TICONI. Paclitaxel plus carboplatin versus standard chemotherapy with either single-agent carboplatin or

366 (2015) 11(3) future science group
Review Openshaw, Fotopoulou, Blagden & Gabra
cyclophosphamide, doxorubicin, and cisplatin in women with ovarian cancer: the ICON3 randomised trial. Lancet 360(9332), 505–515 (2002).
21 Vasey PA, Jayson GC, Gordon A et al. Phase III randomized trial of docetaxel-carboplatin versus paclitaxel-carboplatin as first-line chemotherapy for ovarian carcinoma. J. Natl. Cancer Inst. 96(22), 1682–1691 (2004).
22 Katsumata N, Yasuda M, Takahashi F et al. Dose-dense paclitaxel once a week in combination with carboplatin every 3 weeks for advanced ovarian cancer: a Phase 3, open-label, randomised controlled trial. Lancet 374(9698), 1331–1338 (2009).
• Initial evidence for frontline dose dense paclitaxel.
23 Katsumata N, Yasuda M, Isonishi S et al. Long-term results of dose-dense paclitaxel and carboplatin versus conventional paclitaxel and carboplatin for treatment of advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer (JGOG 3016): a randomised, controlled, open-label trial. Lancet Oncol. 14(10), 1020–1026 (2013).
24 ClinicalTrials Database: NCT01654146. https://clinicaltrials.gov/ct2/show/NCT01654146
25 Pignata S, Scambia G, Katsaros D et al. Carboplatin plus paclitaxel once a week versus every 3 weeks in patients with advanced ovarian cancer (MITO-7): a randomised, multicentre, open-label, Phase 3 trial. Lancet Oncol. 15(4), 396–405 (2014).
26 Chan J, Brady MF, Penson R et al. Phase III trial of every-3-weeks paclitaxel vs. dose dense weekly paclitaxel with carboplatin +/- bevacizumab in epithelial ovarian, peritoneal, fallopian tube cancer: GOG 262 (NCT01167712). Int. J. Gynecol. Cancer 23(Suppl), 9–10 (2013).
27 Burger RA. Overview of anti-angiogenic agents in development for ovarian cancer. Gynecol. Oncol. 121(1), 230–238 (2011).
28 Perren TJ, Swart AM, Pfisterer J et al. A Phase 3 trial of bevacizumab in ovarian cancer. N. Engl. J. Med. 365(26), 2484–2496 (2011).
29 Burger RA, Brady MF, Bookman MA et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N. Engl. J. Med. 365(26), 2473–2483 (2011).
30 Dyer M, Richardson J, Robertson J, Adam J. NICE Bevacizumab in combination with paclitaxel and carboplatin for first-line treatment of advanced ovarian cancer. Lancet Oncol. 14(8), 689–690 (2013).
31 Du Bois A, Floquet A, Kim JW et al. Randomized, double-blind, Phase III trial of pazopanib versus placebo in women who have not progressed after first-line chemotherapy for advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer (AEOC): Results of an international Intergroup trial (AGO-OVAR16). Program and Abstracts of the 49th Annual Meeting of the American Society of Clinical Oncology. Chicago, IL, USA, 2013 (Abstract LBA5503).
32 Howell SB, Pfeifle CL, Wung WE et al. Intraperitoneal cisplatin with systemic thiosulfate protection. Ann. Intern. Med. 97(6), 845–851 (1982).
33 Alberts DS, Liu PY, Hannigan EV et al. Intraperitoneal cisplatin plus intravenous cyclophosphamide versus
intravenous cisplatin plus intravenous cyclophosphamide for stage III ovarian cancer. N. Engl. J. Med. 335(26), 1950–1955 (1996).
•• Initial evidence of the improved efficacy of chemotherapy via the intraperitoneal route.
34 Markman M, Bundy BN, Alberts DS et al. Phase III trial of standard-dose intravenous cisplatin plus paclitaxel versus moderately high-dose carboplatin followed by intravenous paclitaxel and intraperitoneal cisplatin in small-volume stage III ovarian carcinoma: an intergroup study of the Gynecologic Oncology Group, Southwestern Oncology Group, and Eastern Cooperative Oncology Group. J. Clin. Oncol. 19(4), 1001–1007 (2001).
35 Institute NC. NCI clinical announcement: intraperitoneal chemotherapy for ovarian cancer. (2006).
36 Lesnock JL, Darcy KM, Tian C et al. BRCA1 expression and improved survival in ovarian cancer patients treated with intraperitoneal cisplatin and paclitaxel: a Gynecologic Oncology Group Study. Br. J. Cancer 108(6), 1231–1237 (2013).
37 George SH, Shaw P. BRCA and early events in the development of serous ovarian cancer. Front. Oncol. 4, 5 (2014).
38 Foulkes WD. BRCA1 and BRCA2: chemosensitivity, treatment outcomes and prognosis. Fam. Cancer 5(2), 135–142 (2006).
39 Liu JF, Konstantinopoulos PA, Matulonis UA. PARP inhibitors in ovarian cancer: current status and future promise. Gynecol. Oncol. 133(2), 362–369 (2014).
40 Ledermann J, Harter P, Gourley C et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N. Engl. J. Med. 366(15), 1382–1392 (2012).
41 Ledermann J, Harter P, Gourley C et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised Phase 2 trial. Lancet Oncol. 15(8), 852–861 (2014).
• Evidence for the increased efficacy of poly(ADP-ribose) polymerase inhibitors in breast cancer (BRCA)-mutated patients.
42 ClinicalTrials Database: NCT01844986. https://clinicaltrials.gov/ct2/show/NCT01844986
43 ClinicalTrials Database: NCT01874353. https://clinicaltrials.gov/ct2/show/NCT01874353
44 Rustin GJ, Van Der Burg ME, Griffin CL et al. Early versus delayed treatment of relapsed ovarian cancer (MRC OV05/EORTC 55955): a randomised trial. Lancet 376(9747), 1155–1163 (2010).
45 Zimny M, Siggelkow W, Schroder W et al. 2-[Fluorine-18]-fluoro-2-deoxy-d-glucose positron emission tomography in the diagnosis of recurrent ovarian cancer. Gynecol. Oncol. 83(2), 310–315 (2001).
46 Parmar MK, Ledermann JA, Colombo N et al. Paclitaxel plus platinum-based chemotherapy versus conventional platinum-based chemotherapy in women with relapsed ovarian cancer: the ICON4/AGO-OVAR-2.2 trial. Lancet 361(9375), 2099–2106 (2003).

www.futuremedicine.com 367future science group
The next steps in improving the outcomes of advanced ovarian cancer Review
47 Pfisterer J, Plante M, Vergote I et al. Gemcitabine plus carboplatin compared with carboplatin in patients with platinum-sensitive recurrent ovarian cancer: an intergroup trial of the AGO-OVAR, the NCIC CTG, and the EORTC GCG. J. Clin. Oncol. 24(29), 4699–4707 (2006).
48 Pujade-Lauraine E, Wagner U, Aavall-Lundqvist E et al. Pegylated liposomal doxorubicin and carboplatin compared with paclitaxel and carboplatin for patients with platinum-sensitive ovarian cancer in late relapse. J. Clin. Oncol. 28(20), 3323–3329 (2010).
49 Wagner U, Marth C, Largillier R et al. Final overall survival results of Phase III GCIG CALYPSO trial of pegylated liposomal doxorubicin and carboplatin vs paclitaxel and carboplatin in platinum-sensitive ovarian cancer patients. Br. J. Cancer 107(4), 588–591 (2012).
50 Aghajanian C, Blank SV, Goff BA et al. OCEANS: a randomized, double-blind, placebo-controlled Phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J. Clin. Oncol. 30(17), 2039–2045 (2012).
51 Ledermann JA, Perren TJ, Raja FA. Randomised double-blind Phase III trial of cediranib (AZD 2171) in relapsed platinum sensitive ovarian cancer: Results of the ICON6 trial. Presented at: The 17th European Cancer Congress (ECC2013). Amsterdam, Netherlands, 27 September–1 October 2013.
52 Ferrandina G, Salutari V, Vincenzi B et al. Trabectedin as single agent in the salvage treatment of heavily treated ovarian cancer patients: a retrospective, multicenter study. Gynecol. Oncol. 130(3), 505–510 (2013).
53 Monk BJ, Herzog TJ, Kaye SB et al. Trabectedin plus pegylated liposomal doxorubicin (PLD) versus PLD in recurrent ovarian cancer: overall survival analysis. Eur. J. Cancer 48(15), 2361–2368 (2012).
54 Poveda A, Vergote I, Tjulandin S et al. Trabectedin plus pegylated liposomal doxorubicin in relapsed ovarian cancer: outcomes in the partially platinum-sensitive (platinum-free interval 6–12 months) subpopulation of OVA-301 Phase III randomized trial. Ann. Oncol. 22(1), 39–48 (2011).
55 Kavanagh J, Tresukosol D, Edwards C et al. Carboplatin reinduction after taxane in patients with platinum-refractory epithelial ovarian cancer. J. Clin. Oncol. 13(7), 1584–1588 (1995).
56 Davis A, Tinker AV, Friedlander M. “Platinum resistant” ovarian cancer: what is it, who to treat and how to measure benefit? Gynecol. Oncol. 133(3), 624–631 (2014).
57 Markman M, Hall J, Spitz D et al. Phase II trial of weekly single-agent paclitaxel in platinum/paclitaxel-refractory ovarian cancer. J. Clin. Oncol. 20(9), 2365–2369 (2002).
58 Pujade-Lauraine E, Hilpert F, Weber B et al. Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: the AURELIA Open-Label Randomized Phase III Trial. J. Clin. Oncol. 32(13), 1302–1308 (2014).
59 Naumann RW, Coleman RL, Burger RA et al. PRECEDENT: a randomized Phase II trial comparing vintafolide (EC145) and pegylated liposomal doxorubicin
(PLD) in combination versus PLD alone in patients with platinum-resistant ovarian cancer. J. Clin. Oncol. 31(35), 4400–4406 (2013).
60 ClinicalTrials Database: NCT01170650. https://clinicaltrials.gov/ct2/show/NCT01170650
61 Yuan ZQ, Feldman RI, Sussman GE, Coppola D, Nicosia SV, Cheng JQ. AKT2 inhibition of cisplatin-induced JNK/p38 and Bax activation by phosphorylation of ASK1: implication of AKT2 in chemoresistance. J. Biol. Chem. 278(26), 23432–23440 (2003).
62 Clinicaltrials.Gov. Dose-finding Study in platinum-resistant ovarian cancer NCT01653912. https://clinicaltrials.gov/ct2/show/NCT01653912
63 Stiff PJ, Czerlanis C, Drakes ML. Dendritic cell immunotherapy in ovarian cancer. Expert Rev. Anticancer Ther. 13(1), 43–53 (2013).
• Important reading on the devlopment of immunotherapy in ovarian cancer.
64 Kantoff PW, Higano CS, Shore ND et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 363(5), 411–422 (2010).
65 Mitchell PL, Quinn MA, Grant PT et al. A Phase 2, single-arm study of an autologous dendritic cell treatment against mucin 1 in patients with advanced epithelial ovarian cancer. J. Immunother. Cancer 2, 16 (2014).
66 Gray JH, Gargosky SE. Progression-free survival in ovarian cancer patients in second remission with mucin-1 autologous dendritic cell therapy. Presented at: The 50th Annual Meeting of the American Society of Clinical Oncology. Chicago, IL, USA, 31 May 2014 (Abstract 5504).
67 ClinicalTrials Database: NCT01521143. https://clinicaltrials.gov/ct2/show/NCT01521143
68 Duraiswamy J, Freeman GJ, Coukos G. Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res. 73(23), 6900–6912 (2013).
69 Network. CGaR. Integrated genomic analyses of ovarian carcinoma. Nature 474(7353), 609–615 (2011).
•• Significant advances in the understanding of genetic mutations in ovarian cancer.
70 Gerlinger M, Rowan AJ, Horswell S et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 366(10), 883–892 (2012).
71 Bykov VJ, Wiman KG. Mutant p53 reactivation by small molecules makes its way to the clinic. FEBS Lett. 588(16), 2622–2627 (2014).
72 ClinicalTrials Database: NCT02098343. https://clinicaltrials.gov/ct2/show/NCT02098343
73 Dawson SJ, Rosenfeld N, Caldas C. Circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 369(1), 93–94 (2013).
74 Bellot P, Welker MW, Soriano G et al. Automated low flow pump system for the treatment of refractory ascites: a multi-center safety and efficacy study. J. Hepatol. 58(5), 922–927 (2013).

201 (2015) 11(2), 201–212 ISSN 1745-5057
part of
Review
10.2217/WHE.14.70 © 2015 Future Medicine Ltd
Review11
2
2015
Cervical cancer is fourth most common cancer among women with four-fifths of the global burden in low- and middle-income countries (LMICs). Persistent infection with one of the high-risk types of human papillomaviruses (HPV), particularly HPV 16/18, is the central cause of cervical neoplasia. Progress in developing feasible, alternative screening methods in LMICs and HPV vaccines have further improved cervical cancer prevention prospects. While existing screening programs in high-income countries should be re-organized, in view of the downstream effects of national HPV vaccination programs, LMICs should introduce national programs to vaccinate single year cohorts of girls aged 9–13 years with two or three doses and screen 30–35-year-old women with HPV testing to pragmatically decrease their high disease burden.
Keywords: cervix cancer • cytology • HPV testing • HPV vaccination • natural history • precancerous lesions • prevention • screening • VIA
Cervical cancer is the fourth most common cancer in the world with a high incidence and mortality in low- and middle-income countries (LMIC) in sub-Saharan Africa, Central and South America, Asia and Ocea-nia. Of the estimated 528,000 new cases and 267,000 cervical cancer deaths in the world, LMICs account for 445,000 new cases and 230,000 deaths [1]. Lack of effective pre-vention and screening programs or existing suboptimally performing cytology screening programs, high prevalence of HPV infection in the general populations and of HIV infec-tion in certain populations have been respon-sible for the high burden of cervical cancer in LMICs. The knowledge that persistent infection with one of the oncogenic human papillomaviruses (HPV) is the necessary cause for cervical cancer has led to innova-tive disease control strategies for prevention and early detection that may lead to potential elimination of cervical cancer.
HPV vaccination to prevent HPV 16 and 18 infections has been introduced as part of national immunization programs (NIPs) in more than 60 countries including some
LMICs at national or subnational levels, and eventually HPV vaccination will be implemented in more countries. New HPV vaccines that will target a wide spectrum of cancer-causing HPV infections will have even more impact on preventing HPV infec-tions than the currently available vaccines. In due course, widespread HPV vaccination will have a telling impact on the incidence and prevalence of cancer causing HPV infections leading to lower frequency of precancerous lesions. Cervical screening programs will need to adapt in the post HPV vaccination era to maintain their efficiency and effec-tiveness to further reduce cervical cancer risk and their eventual elimination in a cost-effective manner. This task is now becoming more urgent given the relatively rapid impact of the HPV vaccination in real-world settings as seen in Australia, Denmark and other countries [2–9]. The observed stability on the HPV type distribution in cervical cancer specimens over 70 years from 1940 to 2007 predicts a high and stable impact of HPV vaccination in reducing the cervical cancer burden in future vaccinated generations [10].
The next steps in cervical screening
Rengaswamy Sankaranarayanan*,1, You-lin Qiao2 & Namory Keita3
1Section of Early Detection & Prevention,
International Agency for Research on
Cancer, Lyon, France 2Department of Cancer Epidemiology,
National Cancer Center, Cancer Hospital,
Chinese Academy of Medical Sciences
& Peking Union Medical College, Beijing
100021, China 3Department of Gynecology & Obstetrics,
University Hospital Conakry, Donka,
Guinée Conakry, Guinea
*Author for correspondence:

202 (2015) 11(2) future science group
Review Sankaranarayanan, Qiao & Keita
Australia was the first country to introduce HPV vaccination as part of its NIP in April 2007, the first country to offer it to boys in 2014 and the first country to conduct a comprehensive review of cervical screening to adapt to the post HPV vaccination scenario [2,6,9,11]. In this chapter, we briefly review how cervical cancer screening programs need to cost-effectively adapt over the next few years both in LMICs as well as in high-income countries in the context of increasing primary prevention initiatives and new developments in the way that one can screen for cervical cancer.
HPV infection & cervical cancerCervical cancer is a rare long-term outcome of a com-mon persistent infection with one of the high-risk HPV types such as HPV 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59 and 68. Persistent high-risk HPV infection is a necessary cause for cervical precancer-ous lesions and cancer and HPV 16 and 18 are associ-ated with 70–75% of the cervical cancer cases across the world [12]. HPV viral DNA integration in cervi-cal cancer cells and the reduction in high-grade cer-vical intraepithelial neoplasia (CIN3), following HPV vaccination, support the causal role of HPV infection.
Early age at first sexual intercourse and at first child birth and more lifetime sexual partners are the main risk factors for genital HPV infection in women [13]. The life-time probability of ever being infected with HPV is as high as 80–90% and peak HPV acquisition occurs in adolescence and early adulthood. However, 90% of the infections become undetectable within 2 years of acquisition. In 5–10% of women the infec-tion may persist leading to a high risk for cervical can-cer. Since only a small proportion of HPV infected women ever progress to invasive cancer, other fac-tors such as the type and duration of HPV infection, immunocompromised states such as HIV infection, poor nutritional status and smoking may be involved in cervical carcinogenesis. HPV 16 and 18 are more likely to persist than other high-risk HPV infections, which often resolve spontaneously. HIV-infected women suffer from a high frequency of incident, per-sistent and progressive HPV infection. Antiretroviral therapy (ART) has no impact on the high risk of HPV infection and the cumulative incidence of cervical cancer among HIV-infected women. Increased sur-vival of HIV-infected women on ART in a moderately immunocompromised state increases the risk and the development of cervical neoplasia.
Factors such as multiparity, early age at first full-term pregnancy, long term use of oral contraceptives, poor sanitation and hygiene, co-infection with other agents (e.g., herpes simplex virus 2, chlamidia trocho-matis) and smoking may modulate the progression of
HPV infection to cervical neoplasia and are associated with an increased risk of cervical cancer [14–18]. Multi-parity explains the highest proportion of cervical can-cer among HPV-infected women. A general decline in parity to some extent accounts for the declining trends in cervical cancer incidence seen in countries with no screening programs. Male circumcision and barrier protection during sexual intercourse may reduce the risk of HPV infection and cervical cancer [19].
Natural history of cervical neoplasiaThe natural history of cervical cancer involves four dis-tinct stages namely HPV infection of the metaplastic epithelium of the transformation zone (TZ), long-term HPV infection persistence, clonal progression of HPV infected epithelium to high-grade cervical cancer pre-cursor lesions (CIN3) and progression of CIN3 to inva-sive cancer [20]. The fact that it takes over 2–3 decades from the time of HPV infection to cancer occurrence facilitates decisions on appropriate age-groups to screen and optimal frequency of screening.
HPV infection is ubiquitous among women of repro-ductive age and the peak of HPV infection is seen in women below 25 years of age followed by a decline that plateaus around 30–35 years and in some developing countries a second peak is observed in women between 45 and 50 years. Since the rate of incident infections decline steadily with age, infections acquired at a young age contribute to most cervical cancers. Among HPV-infected women, the most important determinant of cancer risk is persistence, particularly HPV 16 persis-tence. A minority of women may demonstrate minor cellular abnormalities, such atypical squamous cells of undetermined significance (ASCUS), low-grade squa-mous intraepithelial lesions (LSIL) or atypical glan-dular cells of undetermined significance (AGUS), on cytology or CIN1 on histology within months or years following incident and transient HPV infections. In a great majority (>80%), infection clears within 2 years and the low-grade lesions resolve [20].
Persistent HPV infections may progress to high-grade squamous intraepithelial lesions (HSIL) or CIN2 and 3 or adenocarcinoma in situ (AIS). Around 70% of CIN2 lesions in women under 25 years, and 40–50% in older women, regress. CIN3 is considered as the real precursor of cervical cancer, although 20–30% of these regress. The peak of CIN3 is observed between 25 and 35 years of age. The time between getting infected with HPV and developing CIN3 is shorter than the time between CIN3 developing into invasive cancer. Repeated HPV positivity conveys substantially more risk of CIN3 than a single HPV-positive test. Among women testing positive at two HPV tests 2-years apart, 19.3% had an absolute risk of CIN3 or worse lesions

www.futuremedicine.com 203future science group
The next steps in cervical screening Review
(CIN 3+) at 12 years; for HPV 16, the risk of CIN 3 + at 3, 5 and 12 years of follow-up were 8.9, 23.8 and 47.4%, respectively [20]. Around 10–20% of untreated CIN3/AIS lesions may progress to invasive cancer in 5 years and 40–50% of untreated CIN3 progress within 30 years, whereas less than 1% of adequately treated CIN3/AIS progress to cancer [21–24].
The risk of CIN3 or worse lesions and cervical cancer is extremely low for several years following one or two negative HPV tests [25,26]. The group of HPV-negative women included those once positive, but whose infec-tions cleared, and those who never acquired HPV infec-tion, however, it is not possible to distinguish between the above groups due to lack of accurate serology. The low risk of CIN3 or worse lesions in HPV-negative women indicates that infections, once cleared, rarely reappear and do not cause substantial CIN3 lesions.
Histologically, the most frequent type of cervical cancer is squamous cell carcinoma arising from squa-mous intraepithelial precursor lesions accounting for 80–90% of cases in different regions of the world; adenocarcinoma (AC) and its subtypes developing from glandular precursor lesions account for 10–20% of cases [27]. Rare types of cervical cancers include adenoid cystic carcinoma, adenoid basal and small cell carcinoma and carcinosarcoma, which do not have any known precursor lesions; other rare cervical cancers include sarcoma, melanoma and primary lymphoma.
PreventionThe fact that high-risk HPV infections cause almost all cervical cancers have led to two new approaches for cervical cancer prevention: HPV vaccination to prevent infections in younger women (≤18 years old) and HPV testing in older women (≥30 years old). Pri-mary prevention of cervical cancer is based on healthy life styles, improved socio-economic status, awareness, empowerment of women with education and better social status, male circumcision, improved hygiene and HPV vaccination. The slow decline in cervical cancer incidence in many LMICs without screening programs is due to socio-economic development, improvements in education and awareness, better sanitation and increasing family planning practices.
Prophylactic HPV vaccination with the currently available vaccines is a major strategy for cervical cancer prevention. The currently available quadrivalent vac-cine targets oncogenic HPV 16 and 18 types as well as 6 and 11; the bivalent vaccine targets HPV 16 and 18. Efficacy against vaccine HPV-type related CIN3 in HPV-naïve populations in Phase III clinical trials exceeded 99% [28].
HPV vaccination has the potential to prevent 70% of cervical cancers in adequately vaccinated populations
and 75–80% cases if the cross-protection is taken into account. The evidence on safety and efficacy strongly supports the introduction of HPV vaccination in NIPs. More than 200 million vaccine doses have been used so far with excellent safety and tolerability pro-files. Mild-to-moderate injection-site symptoms, head-ache and fatigue were the most common adverse events following HPV vaccination.
HPV vaccination is currently part of the NIP in 62 countries targeting pre-adolescent and adolescent girls and ‘catch up’ immunization of older cohorts with variable upper age limits (Figure 1). Decisions by governments to implement HPV vaccination in NIPs, which involved substantial investment, have in part been based on findings from cost–effectiveness stud-ies. The major barriers for HPV vaccination include high costs of the vaccine, significant antivaccine pro-paganda, political and media frenzies spreading misin-formation and exaggerated adverse event coverage, and the logistics of finding suitable health service platforms and vaccinating pre-adolescent and adolescent girls. Government commitment, procuring HPV vaccines at affordable prices through tiered pricing, Global Alli-ance for Vaccines and Immunization (GAVI) Alliance and negotiated pricing, education of the community at large, awareness about the safety and efficacy are criti-cal to introduce HPV vaccination in NIPs in LMICs. Among the LMICs, Bhutan, Malaysia, Uzbekistan, Fiji, Rwanda, South Africa, Zambia, Uganda, Pan-ama, Mexico, Brazil, Chile, Argentina, Colombia, Peru, Uruguay and Paraguay have implemented HPV vaccination as part of NIP with very high coverage of the target populations (>90% third dose coverage) and excellent safety profile [29–31]. In fact, ten LMICs (nine in sub-Saharan Africa, i.e., Ghana, Kenya, Madagas-car, Malawi, Mozambique, Niger, Sierra Leone, Tan-zania and Zimbabwe as well as Laos in Asia) have introduced HPV vaccination demonstration projects supported by GAVI, as a prelude to future national programs (Figure 1). Uganda, Rwanda and Uzbekistan are currently receiving support from GAVI alliance for HPV vaccination in their wider NIP. The introduction of HPV vaccination as part of NIPs in some LMICs has happened in spite of antivaccination campaigns in different countries [32–34].
Emerging evidence from evaluation of HPV vacci-nation programs around the world has shown that the number of young people with vaccine targeted HPV infections and the frequency of precancerous lesions and genital warts among vaccinated cohorts is falling and protection is expected to be long term [7,8,35–37]. In April 2007, Australia introduced quadrivalent HPV vaccination for 12–13-year-old girls through schools in their NIP followed by a catch-up vaccination for

204 (2015) 11(2)
Figure 1. World map showing countries with HPV vaccination implemented as part of the national immunization programs or as GAVI-assisted demonstration projects. HPV: Human papillomaviruses; GAVI: Global Alliance for Vaccines and Immunization.
National program
GAVI-supported HPV vaccination demonstration projects 2014
National program supported by GAVI
future science group
Review Sankaranarayanan, Qiao & Keita
13–26-year-olds via schools, community-based pro-grams and general practices during July–December 2009. Third dose coverage among the 12–13-year-old primary target group exceeded a modest 70% reflecting a real world setting. A significant decline in the preva-lence of HPV types 16/18/6/11 after introduction of a national HPV vaccination program in Australia [36]. Further evidence of vaccine effectiveness have emerged since then as indicated by the 77% fall in prevalence of HPV vaccine-related infections, 90% reduction in genital warts and 48% reduction in CIN3/AIS lesions in the vaccine targeted age-group [6,35]. The rapid drop in HPV prevalence in Australia corresponds to the previous model-based projections on the effects of vaccination [38].
A statistically significant decline in the risk for HPV16/18 associated CIN2 and 3 lesions among vac-cinated women has been reported in a preliminary study in the USA [39]. Long-term, population-based cancer registry follow-up of 874 vaccinated, 875 pla-cebo-vaccinated and an unvaccinated cohort of 15,719 women in Finland found no cases of CIN3 or cervi-cal cancer among the vaccinees, three cases of CIN3 among the placebo vaccinated women (incidence rate = 87.1 per 100,000), and 59 cases of CIN3 and three can-cer cases among the unvaccinated women (incidence rate = 93.8 per 100,000) [37]. In a program of rou-tine national surveillance following HPV vaccination in Scotland, a significant decline in HPV 16 and 18 prevalence was demonstrated among HPV vaccinated
women (prevalence = 150/1100 [13.6%]) compared with unvaccinated women (prevalence = 1018/3418 [29.5%]) was found in a cross-sectional study involv-ing 1000 women aged 20–21 years recruited annually during 2009–2012 [8].
Six years from the introduction of HPV vaccination in Denmark in 2006, the risk of atypia or worse lesions and CIN 2 and 3 lesions have significantly declined among vaccinated women; there is a 44% reduction in CIN2–3 lesions among vaccinated cohorts of women born during 1991–1992 and a 73% reduction in CIN 2–3 lesions in the 1993–94 birth cohorts [7]. Although there was no information on the HPV types responsible for the detected cervical lesions, the lower risk among vaccinated women seems to be due to a reduced risk for HPV16/18 lesions. These early findings are instruc-tive to encourage LMICs to roll out HPV vaccination programs to prevent cervical cancer.
Reduced number of doses from the current three-dose schedule will lower costs, ease logistics of vaccine delivery, increase accessibility and improve compli-ance with vaccine schedules. The two doses may be administered over a 2-year period, with the interval between the first and second doses being of at least 6 months. Whereas most countries use the three-dose schedule, a two-dose regimen is being used in some countries such as Canada, Chile, Colombia, Mexico, South Africa and Switzerland. England will switch over to a two-dose schedule from Sep-tember 2014 onwards; the guidelines in the UK for

www.futuremedicine.com 205future science group
The next steps in cervical screening Review
switching over to two-dose regimen suggest that the first dose can be given at any time during school year 8 (12–13-year-old girls) followed by a minimum of 6 months and a maximum of 24 months between doses; for operational purposes a 12-month gap between the first and second doses has been recom-mended. The use of the two-dose regime in the above countries is based on the recent research evidence that immunogenicity following two doses in adoles-cent girls is noninferior to that following a three-dose course in the age-group where efficacy against persis-tent infection and precancerous lesions has been dem-onstrated [40–45]. Based on findings that immunoge-nicity of two doses is comparable to three doses in 9–4-year-old girls, the European Medical Agency and ten countries in Central and North America, Africa and Asia have licensed the use of two-dose regimes.
Future perspective for screening in LMICsIt is well established that screening asymptomatic, apparently healthy women with cervical screening tests such as conventional cytology (Pap smear), liquid-based cytology, HPV testing and visual inspection with acetic acid (VIA) and treating detected precancerous lesions can effectively prevent cervical cancer. High-income countries have integrated large-scale cytology screening in public health services and have achieved high cumulative coverage rates by screening women at frequent intervals of 2–5 years, investigating screen-positive women with colposcopy, directed biopsies and treating those detected with CIN2–3 and AIS leading to substantial declines in cervical cancer incidence and mortality [46]. However, most LMICs do not have the capacity to initiate and sustain quality-assured cytol-ogy screening programs in view of the underdeveloped health services, several competing priorities, lack of resources and variable commitment to provide pre-ventive health care; they are largely unsuccessful in reducing cervical cancer mortality in some LMICs where they exist [47,48]. The high risk of cervical can-cer, the inability to introduce quality-assured cytology screening and the need for affordable and simple tests have led to the evaluation of innovative approaches for cervical cancer prevention in LMICs.
VIA is the most widely evaluated alternative test in LMICs which allows a single visit ‘see and treat’ approach. It is a point of care screening test in low-resource settings given the limited consumable and infrastructure requirements, immediate results allowing further investigations and treatment in the same sitting and the ease with which the providers can be trained, despite the variation in accuracy and reproducibility due to the subjective nature of the test; realistic sen-sitivity and specificity of a single, quality-assured VIA
in detecting CIN2–3 lesions is around 50 and 85%, respectively [48]. VIA involves naked eye visualization of cervix under bright light 1 min after the application of 5% acetic acid; detection of dense acetowhitening in the cervix abutting the squamocolumnar junction constitutes a positive test outcome [49]. Visual inspec-tion with Lugol’s iodine may be used as an adjunctive test following VIA to assess equivocal VIA results or to assess if the acetowhite lesions turn yellow after iodine application. The accuracy of VIA in detecting CIN2–3 lesions in postmenopausal women is not satisfactory due to the inability to visualize the TZ adequately. However, VIA performs better in HIV-infected women than in the general population due to the increased prevalence of large high-grade lesions [50,51].
In two randomized trials in India, VIA screening was associated with 30–35% reduction in cervical can-cer mortality [52,53]. The safety, acceptability and effec-tiveness of a single visit ‘screen and treat’ approach in which VIA-positive women who eligible for cryother-apy are treated in the same sitting has been well estab-lished [54,55]. Women with cryotherapy ineligible pre-cancerous lesions may be referred for excisional methods such as loop electrosurgical excision procedure for diag-nosis and treatment. The single-visit approach repre-sents a major innovation for scaling-up cervical cancer screening in LMICs and has been adopted by many countries in sub-Saharan Africa and Asia [48]. Despite all its limitations, implementation of VIA screening in LMICs will facilitate the building up of infrastructure and human resources that may facilitate the introduc-tion of affordable HPV tests in future [48]. VIA-based ‘screen-and-treat’ has been implemented through HIV care services in sub-Saharan African countries [53] and in more than 20 provinces in Thailand.
HPV testing, the most reproducible of all cervi-cal screening tests, involves detecting HPV DNA or mRNA in cervical cells collected by pelvic examination or by self-sampling. Since only a negligible minority of women have ever been screened in LMICs, collecting cervical samples by pelvic examination is preferable as it provides an opportunity to visually inspect cervix for cancer. HPV testing is more sensitive, but less specific than cytology for the detection of precancerous lesions and cancer. The accuracy of HPV testing in detect-ing CIN2+ lesions exceeds 90% and CIN3+ exceeds 95%; although the specificity was lower than cytology, the difference was not always significant [56]. Lower cumulative incidence of CIN3+ lesions or cancer has been reported in women aged 30 years or above who were HPV-negative compared with cytology-negative women [26,56,57]. A pooled 53% reduction in CIN3+ lesions was found in HPV-negative women compared with cytology-negative women at enrollment in four

206 (2015) 11(2) future science group
Review Sankaranarayanan, Qiao & Keita
randomized trials; moreover, in three trials, the detec-tion rate of cervical cancer at round 2 was 87% lower in HPV-negative women at recruitment [57]. Following a single round of HPV testing, a significant 53% reduc-tion in the incidence of advanced cancer (stage II+) and a 48% reduction in cervical cancer mortality was observed compared with the control arm with routine care [26]. It is evident that HPV testing is more effec-tive than cytology screening as a primary screening test for women aged 30 years and above and the screening intervals for HPV-negative women can be extended to 5 years or more; a recent WHO Guidance note even recommends a 10-year interval (WHO 2013). Man-agement of women testing positive for HPV poses com-plexities, but VIA may be used for triage for treatment or referral in a two-visit ‘screen and treat’ approach [48]. Reflex cytology testing or genotyping for HPV 16 or 18 can be used to triage HPV-positive women where such capacity exists.
Cost–effectiveness studies in settings such as rural China suggest that primary careHPV screening com-pares favorably to VIA screening [58]. However, there are still considerable challenges in introducing HPV-based screening programs and in fulfilling the promise of a feasible point-of-care HPV screening test in many LMICs unless more affordable and rapid HPV tests become widely available. Even the currently available careHPV test has limitations in these contexts in view of the costs and the need for high-volume screening. In spite of these limitations, HPV testing is the promise of the future in all settings given the fact that wide-spread HPV vaccination will significantly reduce HPV infections and CIN lesions and HPV testing will be the most suitable screening test in such a scenario. A pragmatic way of introducing HPV testing in such a scenario will be to offer it once to women aged 30 or 35 years in LMICs and, if resources permit, repeating it at 10-year intervals, with the eventual wider avail-ability of affordable and rapid HPV tests. Primary care practitioners in LMICs may prescribe HPV testing for women aged 30 years and above if such tests are afford-ably available in their setting; women with a negative test may be advised a repeat test after 5–10 years and those with a positive test may be triaged with VIA in a screen and treat approach or cytology or colposcopy.
It is important to ensure that screen-positive women be followed up with diagnosis and treatment, irrespec-tive of the screening test used. The most clinically effective and cost-effective methods for reducing cervi-cal cancer incidence are those that limit the number of visits required for screening and treatment implying a single visit approach is preferred in LMICs. Recently it was estimated that US$59 million would be required to purchase treatment if cryotherapy is to be placed at
every screening facility in 23 high-risk sub-Saharan African countries and the cost per women screened ranged from US$3 to $15 and the cost per woman treated varied between $28 to $71 [59,60]. The aim of treatment of screen-positive women (in a ‘screen- and- treat’ setting) or of women with confirmed CIN is to prevent their progression to cancer by destroying or removing the entire TZ. Women with confirmed CIN1 may be followed over a 2-year period and treated if it persists or progresses; alternatively women aged 30 years and above with CIN 1 in LMICs may be treated if there is a high probability of losing them for follow-up. Treatment of young women and adolescents with any grade of CIN should be discouraged to avoid increased pregnancy-related risks. Therefore, to avoid detection and treatment of CIN in young women, screening should only commence in women over 30 in LMICs. In settings with capacity for colposcopy and histology, a ‘see-and-treat’ approach may be used where cryotherapy or cold coagulation may be offered during a colposcopy visit after taking biopsies, with histology available aposteriori, providing histological assessment of the lesion(s) treated [54].
While HPV vaccination trials have not been done widely in high-risk Asian and African populations the role of alternative tests in cervical cancer screen-ing has been more widely addressed in these popula-tions. A number of evaluations of HPV vaccination of pre-adolescent girls in developed and developing coun-tries, especially low-income countries, have universally found this intervention to be cost-effective, even in the context of existing screening programs in developed countries, whereas the cost–effectiveness in LMICs is heavily influenced by the unit cost of the vaccine [61,62]. Cost–effectiveness studies also indicate that intro-duction of vaccination in countries without national HPV vaccination at present would prevent substan-tially more cases of cervical cancer than in countries with such programs [62]. Although studies differ in their conclusions about the optimal age for catch-up vaccination, a catch-up round for young adolescent girls (12–15 years) whose future access to screening is uncertain in LMICs is worth considering and 16–20% of HPV 16/18 protection from vaccination is attributed to catch up vaccination strategy [63,64].
Although the best blend of vaccination and screen-ing is not obvious, a pragmatic way of integrating HPV vaccination and screening in LMICs is to provide HPV vaccination to a single year cohort of girls between 9–13 years of age (e.g., 11-year-old girls) with two or three doses and to provide at least a single round of cervical screening with HPV testing for women aged 30–35 years; if resources permit, catch up vaccination of 13–15-year-old girls or 13–18-year-old girls may

www.futuremedicine.com 207future science group
The next steps in cervical screening Review
be carried out and screening may be repeated after 10 years. While HPV vaccination will result in genera-tion of successive cohorts of women at low risk of HPV infection and cervical cancer as vaccination proceeds over time, the single round of screening at 30–35 years will reduce the risk of cervical cancer deaths among targeted women not yet protected by HPV vaccina-tion. As both the programs progress over two decades and more, the target population for screening will eventually comprise of increasing proportions of vacci-nated women and the role of screening will essentially be to reduce the risk among vaccine nonparticipants and those who have failed vaccination either through breakthrough infections or because of lesions caused by vaccine nonincluded types (most likely in those who were vaccinated by current vaccines rather than the next generation vaccines aiming to protect against 80–90% of cervical cancers). The timing of the effect of vaccination on cervical screening will be country-specific and will depend on any catch-up vaccination, variation in coverage, the impact of herd immunity (often not considered and not always evaluated in most cost–effectiveness studies) and the age at which screening starts. The above strategy ensures at least a low intensity screening program that is commensu-rate with the health services resources is introduced to begin with, which will eventually turn out to be a more cost-effective way of screening in the context of a long-standing HPV vaccination program, taking into account its probable downstream impact.
Conclusion & future perspectiveCervical cancer screening in high-income developed countries mostly rely on conventional cytology or liq-uid-based cytology although recently at least six coun-tries (Finland, France, Italy, Sweden, Spain, Nether-lands) in the EU have begun to implement primary HPV testing in cervical cancer screening though most of these are still in planning or piloting stages. The screening programs are organized or opportunistic, but more commonly are a mix of both types of programs. Whereas organized programs, as in Nordic countries, rely on active invitation of the target population with systematic monitoring and quality assurance, oppor-tunistic programs lack monitoring and quality control. Even today, in many countries in the EU, where the European Commission recommends organized pro-grams and has formulated guideline, there are large variations in screening policies and opportunistic screening is the only or main way to access screening; coverage of the screening tests taken within organized screening in EU ranges between 10 and 79% [65]. In Italy, for instance, almost half of the target women aged 25–64 years are screened opportunistically rather
than in organized programs [66]. The targeted age-range and the frequency at which it is repeated vary between countries. For instance, the target age-range and screening intervals are respectively 30–60 years and 5 years in The Netherlands, whereas it is 15 years and above and annual screening in Luxembourg [65]. In the Australian National Cervical Screening Pro-gram, these are currently 18–69 years and 2 years [34]. It is estimated that almost 65 million smears are taken and US$6 billion is spent in the USA, whereas Aus-tralia spends around AU$220 million annually for cervical screening [67,68]. However, the current model of cervical cancer screening in high-income countries cannot continue for long in the light of implementa-tion of HPV vaccination programs, costs incurred and logistics involved. Studies indicate that if screen-ing remains unchanged in developed countries, it will be less cost-effective in vaccinated compared with unvaccinated women [69].
As described earlier, HPV vaccination of pre-adoles-cent girls and catch-up vaccination of adolescents and young women has been introduced in developed coun-tries since 2007. HPV vaccination has been found to be cost-effective even in the context of existing screening programs; however, catch-up vaccination over the age of 18 years has been found to be less cost-effective and stud-ies differ in their conclusions about the optimal age for catch-up vaccination [69]. It may take approximately two decades for these programs to have an impact on the fre-quency of cervical precancerous lesions and at least four decades for the downstream impact on cervical cancer [70]. Since peak incidence of cervical cancer is observed in the fifth and sixth decades, health gains in terms of cer-vical cancer prevention are likely to be observed several decades from now. On the other hand, cervical screening programs deal with detecting and treating cervical pre-cancerous lesions by frequently repeated screening tests beginning in the third decade of life in most developed countries, with the high frequency of low-grade lesions in cytology (due to high frequency of HPV infections in young women) driving triaging and treatment demands of a screening program.
The existing screening programs will need to take into account the population-level downstream effects of HPV vaccination in terms of reduced HPV infec-tion frequency in future. A number of new consid-erations should be taken into account as new vacci-nated cohorts reach screening age and the changing dynamics of the cost–effectiveness equation. These include new screening strategies in populations of mixed vaccination status, delaying the starting age of screening to 25 or 30 years, increasing the screening intervals to 5 or 7 or 10 years, using HPV testing as primary screening test and new methods of triaging

208 (2015) 11(2) future science group
Review Sankaranarayanan, Qiao & Keita
Executive summary
Global burden of cervical cancer • Cervical cancer is the fourth most common cancer globally with estimated 528,000 new cases and 267,000
deaths.• Four-fifths of the global burden occur in low- and middle-income countries (LMICs) where effective prevention
and early detection programs are lacking.HPV infection & cervical carcinogenesis• Cervical cancer is a rare long-term outcome of a common infection with one of the high-risk human
papillomaviruses (HPV) types such as HPV 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59 and 68, with HPV 16 and 18 being responsible for 70–75% of the cervical cancer cases globally.
• Peak HPV acquisition occurs in adolescence and early adulthood with a life-time probability of ever being infected as high as 80–90%.
• Approximately 90% of the incident HPV infections become undetectable within 2 years of acquisition, with infection persisting in 5–10% of women conferring a high risk for cervical cancer.
• Prevention of HPV infection will prevent cervical precancerous lesions and cancer and other HPV-related cancers.
Natural history of cervical neoplasia• The natural history of cervical cancer involves HPV infection of cervical cells, HPV infection persistence, clonal
progression of HPV infected cells to high-grade cervical cancer precursor lesions (CIN3) or adenocarcinoma in situ (AIS) and their progression to invasive cancer over 2–3 decades.
• Around 10–50% of untreated CIN3/AIS lesions may progress to invasive cancer over 5–30 years, whereas only less than 1% of adequately treated CIN3/AIS progress to cancer.
Prevention of cervical cancer• Efficacy of current vaccines against HPV 16/18 related CIN3 in HPV-naïve populations in Phase III clinical trials
exceeded 99%.• The evidence on safety and efficacy strongly supports the introduction of HPV vaccination in national
immunization programs (NIPs).• HPV vaccination has been introduced in NIPs in at least 62 countries and GAVI assisted HPV vaccination
demonstration projects have been introduced in ten LMICs.• Based on research findings that immunogenicity of two doses of current HPV vaccines is comparable to three
doses in 9–14-year-old girls, two-dose vaccination has been implemented in NIPs in Canada, Colombia, Chile, Switzerland, Mexico and South Africa and England.
Future perspectives for screening in LMICs• Most LMICs do not have the capacity to initiate and sustain quality-assured cytology screening programs.• Screening tests with visual inspection with acetic acid (VIA) and single visit ‘screen-and-treat’ approach have
proved feasible, safe and effective in reducing cervical cancer burden in research settings.• HPV testing has proved as accurate, effective and highly reproducible alternative screening method with a high
negative predictive value in women above the age of 30 years.• A pragmatic way of integrating HPV vaccination and screening in LMICs is to provide vaccination to a single
year cohort of girls between 9 and 13 years of age and to provide at least a single round of HPV screening for women aged 30–35 years.
• While HPV vaccination will result in cohorts of women at low risk of HPV infection and cervical cancer, the low-intensity screening will constitute a cost-effective way of screening when downstream effects vaccination occur, in addition to reducing the risk of cervical cancer deaths among a proportion of women not yet protected by HPV vaccination.
Future perspective for screening in high-income countries• The existing cytology screening programs should be reorganized as population-level downstream effects
of HPV vaccination become evident and the target population for screening will increasingly comprise of vaccinated women.
• Screening programs should switch over from cytology to HPV testing as primary screening test and consider new methods to triage HPV-positive women using cytology or HPV genotyping, increasing the starting age of screening to 25 or 30 years and increasing screening intervals to 5, 7 or even 10 years.
• The reorganization should carefully consider any disparities in vaccination and screening uptakes, introduction of new generation of HPV vaccines such as the nonavalent vaccine, potential effects of vaccination on trends in screening participation and compliance to post screening management algorithms.
• Just as Australia was the first country to introduce HPV vaccination in NIP in 2007, it is also the first high-income country to consider cervical screening policy changes as above in the post HPV vaccination era.

www.futuremedicine.com 209future science group
The next steps in cervical screening Review
HPV-positive women such as reflex liquid-based cytol-ogy or HPV genotyping for HPV types 16 and 18. In addition, careful consideration should be given to any disparities in vaccination and screening uptakes, intro-duction of new generation of HPV vaccines such as the nonavalent vaccine, potential effects of vaccination on trends in screening participation and compliance to post screening management algorithms.
Since a large proportion of high-grade CIN are caused by HPV 16 and 18, preventing these by HPV vaccina-tion will lead to significant reduction in the frequency of high-grade lesions. There will be fewer abnormal findings on cytology leading to a diagnosis high-grade CIN and the positive predictive value of an abnormal cytology for predicting CIN 3 or worse lesions will sub-stantially decline, leaving behind at most some insig-nificant cytological abnormalities in this scenario [71]. Currently, colposcopy is the most widely used triaging investigation for cytology-positive women in developed countries. The above scenario following long-standing HPV vaccination will further challenge colposcopic interpretation of cervical findings.
As the HPV vaccination programs gain momentum and coverage, one needs to consider to raise the age of starting screening, the use of more objective primary screening tests such as HPV testing, increase the screen-ing intervals and adapt suitable management algorithms. Cost–effectiveness analyses support raising the age of initiating cervical screening to 24 years [72]. Recently the medical services advisory committee (MSAC) of Aus-tralia recommended the Australian Government that a
‘new’ cervical cancer screening test, namely HPV test-ing should replace the current Pap smear in the Austra-lian National Cervical Cancer Screening program [11]. It has recommended 5-yearly cervical screening using a primary HPV test with partial HPV genotyping and reflex liquid-based cytology triage, for HPV vaccinated and unvaccinated women 25–69 years of age, with exit testing of women 70–74 years of age, in the context of revisiting how cervical screening might be reorganized in the aftermath of HPV vaccination introduction. It is anticipated the above changes may be implemented after 2016. In the postvaccination scenario, HPV test-ing based, low-intensity screening with raised age at initiating screening and few screening rounds at wide intervals such as 7 or 10 years seems to be the most suitable option for screening in all countries.
AcknowledgementsThe authors gratefully acknowledge E Bayle, K Guinot and
S Montigny for their assistance in the preparation of this
manuscript.
Financial & competing interests disclosureThe authors have no relevant affiliations or financial involve-
ment with any organization or entity with a financial inter-
est in or financial conflict with the subject matter or mate-
rials discussed in the manuscript. This includes employment,
consultancies, honoraria, stock ownership or options, expert
testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this
manuscript.
ReferencesPapers of special note have been highlighted as: • of interest; •• of considerable interest.
1 Ferlay J, Soerjomataram I, Ervik M et al. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11. International Agency for Research on Cancer, Lyon, France. http://globocan.iarc.fr
2 Gertig DM, Brotherton JM, Budd AC, Drennan K, Chappell G, Saville AM. Impact of a population-based HPV vaccination program on cervical abnormalities: a data linkage study. BMC Med. 11, 227 (2013).
•• Describes the impact of human papillomaviruses (HPV) vaccination on cervical lesions.
3 Korostil IA, Ali H, Guy RJ, Donovan B, Law MG, Regan DG. Near elimination of genital warts in Australia predicted with extension of human papillomavirus vaccination to males. Sex. Transm. Dis. 40(11), 833–835 (2013).
4 Hariri S, Markowitz LE, Dunne EF, Unger ER. Population impact of HPV vaccines: summary of early evidence. J. Adolesc. Health 53(6), 679–682 (2013).
5 Markowitz LE, Hariri S, Lin C et al. Reduction in human papillomavirus (HPV) prevalence among young women
following HPV vaccine introduction in the United States, National Health and Nutrition Examination Surveys, 2003–2010. J. Infect. Dis. 208(3), 385–393 (2013).
6 Garland SM. The Australian experience with the human papillomavirus vaccine. Clin. Ther. 36(1), 17–23 (2014).
•• Provides a good review of the impact of HPV vaccination in a national immunization program.
7 Baldur-Felskov B, Dehlendorff C, Munk C, Kjaer SK. Early impact of human papillomavirus vaccination on cervical neoplasia – nationwide follow-up of young Danish women. J. Natl Cancer Inst. 106(3), djt460 (2014).
•• Provides a good review of the impact of HPV vaccination in a national immunization program.
8 Kavanagh K, Pollock KG, Potts A et al. Introduction and sustained high coverage of the HPV bivalent vaccine leads to a reduction in prevalence of HPV 16/18 and closely related HPV types. Br. J. Cancer 110(11), 2804–2811 (2014).
9 Brotherton JM. Human papillomavirus vaccination: where are we now? J. Paediatr. Child Health doi:10.1111/jpc.12627 (2014) (Epub ahead of print).
10 Alemany L, de Sanjosé S, Tous S et al. Time trends of human papillomavirus types in invasive cervical cancer, from 1940 to 2007. Int. J. Cancer 135(1), 88–95 (2014).

210 (2015) 11(2) future science group
Review Sankaranarayanan, Qiao & Keita
11 Australian Government. Department of Health. National Cervical Screening Program Renewal. www.cancerscreening.gov.au
12 International Agency for Research on Cancer. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Volume 90. Human Papillomavirus. IARC, Lyon, France (2007).
13 Chelimo C, Wouldes TA, Cameron LD, Elwood JM. Risk factors for and prevention of human papillomaviruses (HPV), genital warts and cervical cancer. J. Infect. 66(3), 207–217 (2013).
14 Castellsague X, Bosch FX, Munoz N. Environmental co-factors in HPV carcinogenesis. Virus Res. 89(2), 191–199 (2002).
15 Castellsague X, Munoz N. Chapter 3: Cofactors in human papillomavirus carcinogenesis – role of parity, oral contraceptives, and tobacco smoking. J. Natl Cancer Inst. Monogr. (31), 20–28 (2003).
16 Hammouda D, Munoz N, Herrero R et al. Cervical carcinoma in Algiers, Algeria: human papillomavirus and lifestyle risk factors. Int. J. Cancer 113(3), 483–489 (2005).
17 Franceschi S, Rajkumar T, Vaccarella S et al. Human papillomavirus and risk factors for cervical cancer in Chennai, India: a case-control study. Int. J. Cancer 107(1), 127–133 (2003).
18 nternational Collaboration of Epidemiological Studies of Cervical Cancer. Cervical carcinoma and reproductive factors: collaborative reanalysis of individual data on 16,563 women with cervical carcinoma and 33,542 women without cervical carcinoma from 25 epidemiological studies. Int. J. Cancer 119(5), 1108–1124 (2006).
19 Albero G, Castellsague X, Giuliano AR, Bosch FX. Male circumcision and genital human papillomavirus: a systematic review and meta-analysis. Sex. Transm. Dis. 39(2), 104–113 (2012).
20 Moscicki AB, Schiffman M, Burchell A et al. Updating the natural history of human papillomavirus and anogenital cancers. Vaccine 30(Suppl.), F24–F33 (2012).
• Provides a good review of natural history of HPV infection.
21 Ostor AG. Natural history of cervical intraepithelial neoplasia: a critical review. Int. J. Gynecol. Pathol. 12(2), 186–192 (1993).
22 McCredie MR, Sharples KJ, Paul C et al. Natural history of cervical neoplasia and risk of invasive cancer in women with cervical intraepithelial neoplasia 3: a retrospective cohort study. Lancet Oncol. 9(5), 425–434 (2008).
23 Lee KR, Flynn CE. Early invasive adenocarcinoma of the cervix. Cancer 89(5), 1048–1055 (2000).
24 Salani R, Puri I, Bristow RE. Adenocarcinoma in situ of the uterine cervix: a metaanalysis of 1278 patients evaluating the predictive value of conization margin status. Am. J. Obstet. Gynecol. 200(2), 182–185 (2009).
25 Schiffman M, Glass AG, Wentzensen N et al. A long-term prospective study of type-specific human papillomavirus infection and risk of cervical neoplasia among 20,000 women in the Portland Kaiser Cohort Study. Cancer Epidemiol. Biomarkers Prev. 20(7), 1398–1409 (2011).
26 Sankaranarayanan R, Nene BM, Shastri SS et al. HPV screening for cervical cancer in rural India. N. Engl. J. Med. 360(14), 1385–1394 (2009).
•• Provides a good insight in conducting screening trials in developing countries.
27 Forman D, Bray F, Brewster DH et al. Cancer Incidence in Five Continent. Vol. X (electronic version). IARC, Lyon, France. http://ci5.iarc.fr
28 Schiller JT, Castellsague X, Garland SM. A review of clinical trials of human papillomavirus prophylactic vaccines. Vaccine 30(Suppl.), F123–F138 (2012).
•• Provides a good overview on the efficacy of HPV vaccines.
29 Binagwaho A, Ngabo F, Wagner CM et al. Integration of comprehensive women’s health programmes into health systems: cervical cancer prevention, care and control in Rwanda. Bull. World Health Organ. 91(9), 697–703 (2013).
30 Markowitz LE, Tsu V, Deeks SL et al. Human papillomavirus vaccine introduction – the first five years. Vaccine 30(Suppl.), F139–F148 (2012).
31 Binagwaho A, Wagner CM, Gatera M, Karema C, Nutt CT, Ngabo F. Achieving high coverage in Rwanda’s national human papillomavirus vaccination programme. Bull. World Health Organ. 90(8), 623–628 (2012).
•• Describes important national experience in introducing HPV vaccination in a subsaharian African country.
32 Binagwaho A, Wagner CM, Nutt CT. HPV vaccine in Rwanda: different disease, same double standard. Lancet 378(9807), 1916 (2011).
33 van Bogaert L. Are the currently existing anti-human papillomavirus vaccines appropriate for the developing world? Ann. Med. Health Sci. Res. 3(3), 306–312 (2013).
34 Guardian Global Development. Why anti-HPV vaccine campaigners need a shot of good sense. The Guardian (2013). www.theguardian.com
35 Brotherton JM, Fridman M, May CL, Chappell G, Saville AM, Gertig DM. Early effect of the HPV vaccination programme on cervical abnormalities in Victoria, Australia: an ecological study. Lancet 377(9783), 2085–2092 (2011).
36 Tabrizi SN, Brotherton JM, Kaldor JM et al. Fall in human papillomavirus prevalence following a national vaccination program. J. Infect. Dis. 206(11), 1645–1651 (2012).
37 Rana MM, Huhtala H, Apter D et al. Understanding long-term protection of human papillomavirus vaccination against cervical carcinoma: Cancer registry-based follow-up. Int. J. Cancer 132(12), 2833–2838 (2013).
38 Smith MA, Canfell K. Testing previous model predictions against new data on human papillomavirus vaccination program outcomes. BMC Res. Notes 7, 109 (2014).
39 Powell SE, Hariri S, Steinau M et al. Impact of human papillomavirus (HPV) vaccination on HPV 16/18-related prevalence in precancerous cervical lesions. Vaccine 31(1), 109–113 (2012).
40 Kreimer AR, Rodriguez AC, Hildesheim A et al. Proof-of-principle evaluation of the efficacy of fewer than three doses

www.futuremedicine.com 211future science group
The next steps in cervical screening Review
of a bivalent HPV16/18 vaccine. J. Natl Cancer Inst. 103(19), 1444–1451 (2011).
41 Romanowski B, Schwarz TF, Ferguson LM et al. Immunogenicity and safety of the HPV-16/18 AS04-adjuvanted vaccine administered as a 2-dose schedule compared with the licensed 3-dose schedule: results from a randomized study. Hum. Vaccin. 7(12), 1374–1386 (2011).
42 Safaeian M, Porras C, Pan Y et al. Durable antibody responses following one dose of the bivalent human papillomavirus L1 virus-like particle vaccine in the Costa Rica Vaccine Trial. Cancer Prev. Res. (Phila). 6(11), 1242–1250 (2013).
43 Dobson SR, McNeil S, Dionne M et al. Immunogenicity of 2 doses of HPV vaccine in younger adolescents vs 3 doses in young women: a randomized clinical trial. JAMA 309(17), 1793–1802 (2013).
44 Romanowski B, Schwarz TF, Ferguson LM et al. Immune response to the HPV-16/18 AS04-adjuvanted vaccine administered as a 2-dose or 3-dose schedule up to 4 years after vaccination: Results from a randomized study. Hum. Vaccin. Immunother. doi:10.4161/hv.28022 (2014) (Epub ahead of print).
45 Lazcano-Ponce E, Stanley M, Munoz N et al. Overcoming barriers to HPV vaccination: non-inferiority of antibody response to human papillomavirus 16/18 vaccine in adolescents vaccinated with a two-dose vs. a three-dose schedule at 21 months. Vaccine 32(6), 725–732 (2014).
46 International Agency forResearch on Cancer. IARC Handbooks of Cancer Prevention. (Volume 10). Cervix Cancer Screening. IARC, Lyon, France (2005).
47 Murillo R, Almonte M, Pereira A et al. Cervical cancer screening programs in Latin America and the Caribbean. Vaccine 26(Suppl.), L37–L48 (2008).
48 Sankaranarayanan R, Nessa A, Esmy PO, Dangou JM. Visual inspection methods for cervical cancer prevention. Best Pract. Res. Clin. Obstet. Gynaecol. 26(2), 221–232 (2012).
49 Sankaranarayanan R, Wesley R. A Practical Manual on Visual Screening for Cervical Neoplasia. IARC, Lyon, France (2003).
50 Joshi S, Sankaranarayanan R, Muwonge R, Kulkarni V, Somanathan T, Divate U. Screening of cervical neoplasia in HIV-infected women in India. AIDS 27(4), 607–615 (2013).
•• Describes the efficacy of screening tests in HIV-infected women.
51 Sahasrabuddhe VV, Bhosale RA, Kavatkar AN et al. Comparison of visual inspection with acetic acid and cervical cytology to detect high-grade cervical neoplasia among HIV-infected women in India. Int. J. Cancer 130(1), 234–240 (2012).
52 Sankaranarayanan R, Esmy PO, Rajkumar R et al. Effect of visual screening on cervical cancer incidence and mortality in Tamil Nadu, India: a cluster-randomised trial. Lancet 370(9585), 398–406 (2007).
53 Shastri SS, Mittra I, Mishra GA et al. Effect of VIA screening by primary health workers: randomized controlled study in Mumbai, India. J. Natl Cancer Inst. 106(3), dju009 (2014).
54 Sankaranarayanan R, Rajkumar R, Esmy PO et al. Effectiveness, safety and acceptability of ‘see and treat’ with cryotherapy by nurses in a cervical screening study in India. Br. J. Cancer 96(5), 738–743 (2007).
55 Mwanahamuntu MH, Sahasrabuddhe VV, Kapambwe S et al. Advancing cervical cancer prevention initiatives in resource-constrained settings: insights from the Cervical Cancer Prevention Program in Zambia. PLoS Med. 8(5), e1001032 (2011).
56 Arbyn M, Ronco G, Anttila A et al. Evidence regarding human papillomavirus testing in secondary prevention of cervical cancer. Vaccine 30(Suppl.), F88–F99 (2012).
57 Ronco G, Dillner J, Elfstrom KM et al. Efficacy of HPV-based screening for prevention of invasive cervical cancer: follow-up of four European randomised controlled trials. Lancet 383(9916), 524–532 (2014).
58 Shi JF, Canfell K, Lew JB et al. Evaluation of primary HPV-DNA testing in relation to visual inspection methods for cervical cancer screening in rural China: an epidemiologic and cost–effectiveness modelling study. BMC Cancer 11239 (2011).
59 Quentin W, Adu-Sarkodie Y, Terris-Prestholt F, Legood R, Opoku BK, Mayaud P. Costs of cervical cancer screening and treatment using visual inspection with acetic acid (VIA) and cryotherapy in Ghana: the importance of scale. Trop. Med. Int. Health 16(3), 379–389 (2011).
60 Mvundura M, Tsu V. Estimating the costs of cervical cancer screening in high-burden Sub-Saharan African countries. Int. J. Gynaecol. Obstet. 126(2), 151–155 (2014).
61 Natunen K, Lehtinen TA, Torvinen S, Lehtinen M. Cost-effectiveness of HPV-vaccination in medium or low income countries with high cervical incidence – a systematic review. J. Vaccines Vaccin. 4, 172 (2013).
62 Jit M, Brisson M, Portnoy A, Hutubessy R. Cost-effectiveness of female human papillomavirus vaccination in 179 countries: a PRIME modelling study. Lancet Glob. Health 2(7), e406–e414 (2014).
63 Baussano I, Lazzarato F, Ronco G, Dillner J, Franceschi S. Benefits of catch-up in vaccination against human papillomavirus in medium- and low-income countries. Int. J. Cancer 133(8), 1876–1881 (2013).
64 Baussano I, Dillner J, Lazzarato F, Ronco G, Franceschi S. Upscaling human papillomavirus vaccination in high-income countries: impact assessment based on transmission model. Infect. Agent Cancer 9(1), 4 (2014).
65 Anttila A, von KL, Aasmaa A et al. Cervical cancer screening policies and coverage in Europe. Eur. J. Cancer 45(15), 2649–2658 (2009).
66 Giorgi RP, Federici A, Zappa M. The cancer screening monitoring system: indicators for organised programmes and possible extension to spontaneous screening. Pathologica 105(3), 83–85 (2013).
67 Eltoum IA, Roberson J. Impact of HPV testing, HPV vaccine development, and changing screening frequency on national Pap test volume: projections from the National Health Interview Survey (NHIS). Cancer 111(1), 34–40 (2007).

212 (2015) 11(2) future science group
Review Sankaranarayanan, Qiao & Keita
68 Lew JB, Howard K, Gertig D et al. Expenditure and resource utilisation for cervical screening in Australia. BMC Health Serv. Res. 12, 446 (2012).
69 Canfell K, Chesson H, Kulasingam SL, Berkhof J, Diaz M, Kim JJ. Modeling preventative strategies against human papillomavirus-related disease in developed countries. Vaccine. 30(Suppl.), F157–F167 (2012).
70 Van de Velde, N, Boily MC, Drolet M et al. Population-level impact of the bivalent, quadrivalent, and nonavalent human
papillomavirus vaccines: a model-based analysis. J. Natl Cancer Inst. 104(22), 1712–1723 (2012).
71 Schiffman M. Integration of human papillomavirus vaccination, cytology, and human papillomavirus testing. Cancer 111(3), 145–153 (2007).
72 Kulasingam SL, Myers ER. Potential health and economic impact of adding a human papillomavirus vaccine to screening programs. JAMA 290(6), 781–789 (2003).

747ISSN 1479-6694Future Oncol. (2015) 11(5), 747–757
part of
10.2217/FON.14.313 © 2015 Future Medicine Ltd
DRUG EVALUATION
Olaparib: an oral PARP-1 and PARP-2 inhibitor with promising activity in ovarian cancer
Camille C Gunderson1 & Kathleen N Moore*,1
1University of Oklahoma Health Sciences Center, Stephenson Oklahoma Cancer Center, 800 NE 10th Street, Suite 5040, Oklahoma City,
OK 73104, USA
*Author for correspondence: Tel.: +1 405 271 7770; Fax: +1 405 271 1006; [email protected]
ABSTRACT Olaparib (Lynparza™; AZD2281) is a potent PARP-1 and PARP-2 inhibitor with biologic activity in ovarian cancer as well as other solid tumors. It has been tested in Phase I and II trials and has single-agent activity in both germline BRCA mutated and sporadic ovarian cancer. Phase III trials assessing the efficacy of olaparib in the maintenance setting following first line and platinum-sensitive recurrence are underway for patients with a germline BRCA mutation, given the inherent molecular compatibility with the drug’s mechanism of action.
KEYWORDS • BRCA mutation • homologous recombination pathway • PARP inhibitor • olaparib • ovarian cancer
Approximately 22,000 cases of ovarian cancer are diagnosed each year in the USA. Approximately 75% of cases are detected at an advanced stage and thus require adjuvant chemotherapy. Despite improvements in chemotherapy regimens and modes of delivery, ovarian cancer remains the most lethal gynecologic malignancy with approximately 14,000 deaths each year [1]. The stand-ard chemotherapy backbone for first-line adjuvant treatment after surgery has since 1996 been a platinum/taxane-based systemic regimen, when this was demonstrated to prolong progression-free survival (PFS) and overall survival (OS) in comparison with cisplatin and cyclophosphamide [2]. Six cycles are recommended, as multiple studies have demonstrated that the use of more than six cycles as adjuvant therapy does not offer a survival advantage but increases treatment-related toxic-ity [3–5]. Unfortunately, despite high initial response rates, the majority of patients recur and require additional therapies. Once patients have progressed through their third regimen of chemotherapy, the PFS expected following chemotherapy drops markedly (5.6 months), leaving patients with little to no time off chemotherapy for the remainder of their lives [6].
The quest for improved outcomes without extending the duration of conventional cytotoxic chemotherapy regimens has prompted the incorporation of biologic agents along with traditional platinum-based chemotherapy. Such agents include angiogenesis inhibitors, poly(ADP-ribose) polymerase (PARP) inhibitors, and use of various cytotoxic and biologic agents as maintenance therapy after completion of adjuvant therapy.
PARP inhibitors are a new class of anticancer drugs that have evolved rapidly since they were first developed in 2005. They target tumors that have deficits in homologous recombination repair (such as BRCA mutations) by a process known as synthetic lethality; therein, neither the deficiency in homologous recombination repair nor PARP inhibition alone is cytotoxic, but the combination of the two leads to cell death [7]. As double stranded DNA breaks accumulate, genomic stability is undermined. The initial reports detail single-agent in vitro anticancer activity in BRCA-deficient cells [8,9]. Since then, several studies have noted that PARP inhibitors are active in women with both BRCA-related and unrelated ovarian cancer in the recurrent and maintenance settings, both as single agents and in combination with chemotherapy [10]. PARP inhibitors can be synergistic

Future Oncol. (2015) 11(5)748
DRug EvAluATiOn Gunderson & Moore
future science group
with chemotherapy due to processes such as mitotic catastrophe, where unrepaired DNA damage leads to cell death [11]. PARP inhibitors have distinct potential to change the paradigm of ovarian cancer treatment in comparison to other biologic agents for several reasons: ease of administration due to oral formulation (tablet and capsule); generally acceptable toxicity and tolerability; frequency of deficits in homologous recombination in high grade serous ovarian can-cer; and synergy with both conventional chemo-therapy agents that induce DNA strand breaks and other targeted agents such as antiangiogenic drugs and PI3K inhibitors; however, the hema-tologic toxicity in combination with cytotoxic agents is considerable [12].
Overview of the marketSince the initial in vitro reports [8,9], various PARP inhibitors have been studied in women with ovarian cancer. Olaparib (Lynparza™; AZD2281) has been evaluated most extensively and has compelling data from Phase I and II tri-als; other drugs in the same class include Veliparib (ABT-888), Niraparib (MK4827), Rucaparib (CO338, AGO14699 and PF01367388) and BMN 673. Several other PARP inhibitors have been evaluated in clinical testing but do not have any reported patient data related to ovarian can-cer; these include AZD2461 (NCT01247168), CEP9722 (NCT00920595), E7449, E7016 and INO-1001.
Olaparib (Lynparza; AZD2281)Olaparib (Lynparza; formerly known as AZD2281) is an oral drug initially developed by Kudos Pharmaceuticals (in conjunction with the UK Institute of Cancer Research) and later acquired by AstraZeneca (Macclesfield, UK). It has activity against PARP-1, PARP-2 and PARP-3, although only the PARP-1 and PARP-2 p roteins repair DNA single strand breaks [13].
Olaparib was the first PARP inhibitor to be investigated and has undergone the most com-prehensive evaluation thus far. In December 2014, the European Commission granted mar-keting authorization for Lynparza (olaparib) capsules (400 mg twice daily [b.i.d.]) as the first therapy for the maintenance treatment of adult patients with platinum-sensitive relapsed BRCA-mutated (germline and/or somatic) high-grade serous epithelial ovarian, fallopian tube or primary peritoneal cancer who are in complete response or partial response to
platinum-based chemotherapy (AstraZeneca press release 18 December 2014). In addition, on 19 December 2014, the US FDA approved Lynparza (olaparib) capsules (400 mg b.i.d.) as the first monotherapy for patients with delete-rious or suspected deleterious germline BRCA-mutated advanced ovarian cancer as detected by an FDA-approved companion diagnostic test (BRACAnalysis CDx™), who have been treated with three or more prior lines of chemotherapy (AstraZeneca press release 19 December 2014).
ChemistryOlaparib (4-[(3-[4-cyclopropylcabonyl)piperazin-4-ul] carbonyl1)-4 f luroophenyl] methyl(2H) phthalazin-1-one) is an oral PARP-1 and PARP-2 inhibitor with a molecular weight of 434.47 Da and an IC
50 of 0.005 μM for PARP1
(Figure 1) [10]. The drug was developed with the aim of enhancing the cytotoxicity of certain existing cancer chemotherapeutic agents which target DNA repair.
Preclinical pharmacodynamicsIn preclinical studies, a range of tumor cell lines were analyzed for sensitivity to PARP inhibition using colony formation assays [10,14]. Enhanced olaparib sensitivity was noted (IC
50 <1 μM)
in cell lines with known BRCA1 mutations, BRCA2 mutations, or those with low expres-sion of homologous recombination genes. These results correspond with the proposed mechanism of PARP inhibitors, where deficiencies in homol-ogous recombination lead to inability to repair double strand breaks in DNA occurring due to treatment with a PARP inhibitor (synthetic lethality) [14].
The antitumor effect of olaparib has been demonstrated in BRCA-deficient mouse tumors. Specifically, a BRCA1-/- orthotopic mouse mam-mary tumor model and a BRCA2-/- orthotopic mouse model had significant antitumor activity when treated with single-agent olaparib 50 mg/kg for 28 days [15,16]. Additionally, studies have dem-onstrated that olaparib potentiates the efficacy of platinum agents in vitro and in vivo using BRCA mouse mammary models. Evers et al. reported that in BRCA2-deficient mouse mammary cell lines, olaparib induced a synergistic potentiation of cisplatin in comparison with only an additive effect with a BRCA-proficient control line [17]. Additionally, Hay et al. showed single-agent olaparib and carboplatin combination efficacy in conditionally deleted BRCA2 orthotopic in vivo

749
Figure 1. Olaparib molecule.
N
NH
O
N
O
N
O
F
Olaparib: an oral PARP-1 & PARP-2 inhibitor with promising activity in ovarian cancer DRug EvAluATiOn
future science group www.futuremedicine.com
mouse models with selective antitumor activity, and Rottenberg et al. have demonstrated that in BRCA1-deficient orthotopically transplanted in vivo mouse models, combination of platinum agents with olaparib increased both the progres-sion free and overall survival [15,16].
Additional preclinical data support the use of PARP inhibitors in setting of homolo-gous recombination deficiency other than that induced by germline BRCA1 or BRCA2 muta-tions. These conditions, often referred to as ‘BRCA-ness’, include inactivation of the Fanconi anemia pathway, reduced expression of BRCA1, hypermethylation of BRCA1 promoter, ampli-fication of the EMSY gene and inactivation of BRCA2, as well as decreased expression of pro-teins involved in homologous recombination such as RAD51 and ATM. [18,19] Further, recent preclinical data point to PTEN deficiency as a possible marker of sensitivity to PARP inhibi-tors. Mendes et al. reported on a series of tumor cell lines with PTEN deficiency where sensitiv-ity to cisplatin and PARP inhibition was much higher than in wild-type cell lines due to a defect in homologous recombination. The same effect was not seen in cell lines treated with paclitaxel and PARP inhibition. These results were con-firmed in mouse xenografts and suggest an addi-tional role for PARP inhibitors in patients who harbor PTEN mutations [18,19].
Pharmacokinetics & human metabolismFollowing the first dose of an olaparib capsule, the absorption was rapid with peak concentra-tions achieved within 1–3 h after dosing. Drug exposure increased proportionately with increas-ing dose up to 100 mg b.i.d., but beyond this, it increased less than proportionately. At the current recommended capsule dose of 400 mg b.i.d., the population estimated maximum plasma concentration at steady state (C
max ss)
ranged from 1.18 to 14.2 μg/ml, and the steady state area under the plasma concentration time curve (AUC) from 0 to 12 h ranged from 6.48 to 154 μg h/ml. Olaparib is eliminated in the urine (35–50%) and feces (12–60%) [20,21].
Clinical efficacy●● Phase I studies
In a single-agent dose-escalation Phase I study of olaparib capsules dosed continuously, the maximum tolerated dose was 400 mg b.i.d. This resulted in a partial response in nine of 19 patients with BRCA-associated breast, ovarian
or prostate cancers; furthermore, 63% (12 of 19) derived clinical benefit (Table 1) [22]. Next, an expansion cohort of 39 ovarian cancer patients with germline BRCA mutations were treated with olaparib monotherapy at a dose of 200 mg b.i.d. and 11 patients were treated with this dose during dose escalation; 40% demonstrated either RECIST or CA125 response for a median duration of response of 28 weeks. Notably, this study revealed an important association between platinum sensitivity and the clinical benefit gained from olaparib. Specifically, the more platinum-sensitive a patient, the greater response rate to olaparib (23% in platinum-refractory, 45% in platinum-resistant, and 69% in platinum- sensitive patients), likely due to defective homologous recombination pathways that may improve activity of both platinum agents and PARP inhibitors [21].
●● Phase II studiesNonrandomized studiesAudeh and colleagues evaluated germline BRCA mutated patients with recurrent ovarian cancer and demonstrated a dose–response relationship with olaparib. They recruited patients to two sequential cohorts (400 and 100 mg) and found that patients receiving 400 mg b.i.d. olaparib capsules had a response rate of 33% in com-parison to 13% in patients who received olapa-rib capsules 100 mg b.i.d. Those receiving the 100 mg b.i.d. dose also had greater frequency of progression at 16 weeks (65 vs 33%). Although this study suggests a dose-dependent response, the results should be interpreted with caution as this was not a randomized study, and addi-tionally, the patients receiving the lower dose of olaparib had poorer prognostic features than those receiving the 400 mg dose. Additionally, relatively comparable response rates were noted for both platinum-sensitive (38%) and plati-num-resistant (30%) disease [24]. In another
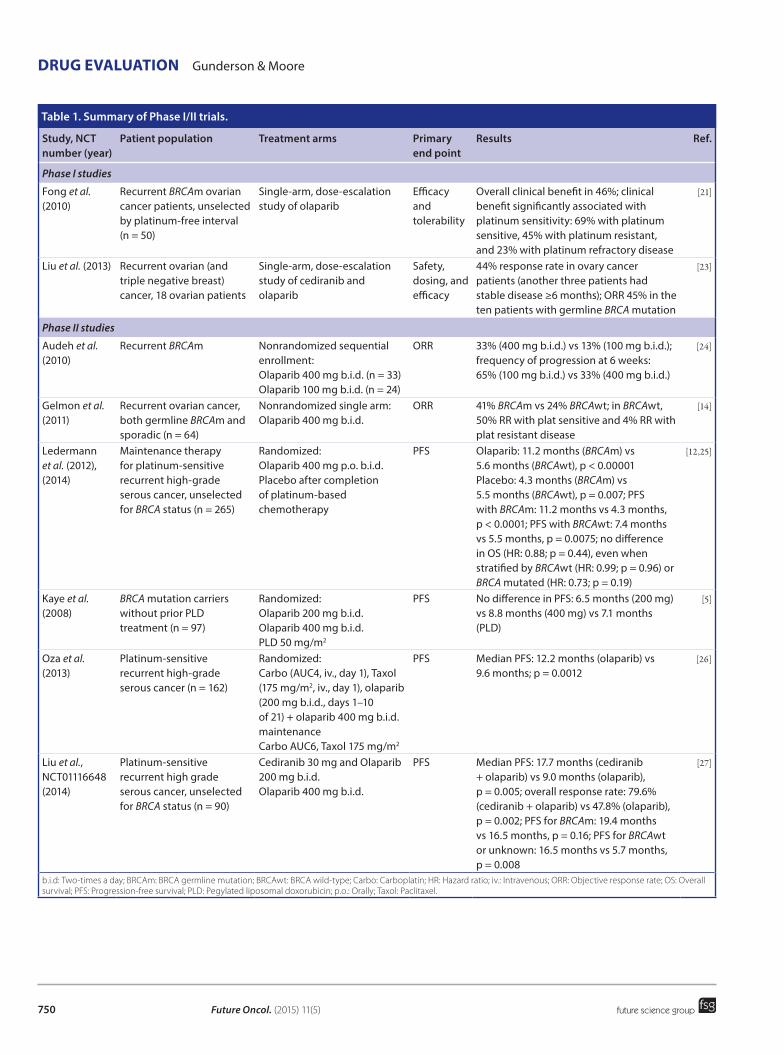
Future Oncol. (2015) 11(5)750
DRug EvAluATiOn Gunderson & Moore
future science group
Table 1. Summary of Phase I/II trials.
Study, NCT number (year)
Patient population Treatment arms Primary end point
Results Ref.
Phase I studies
Fong et al. (2010)
Recurrent BRCAm ovarian cancer patients, unselected by platinum-free interval (n = 50)
Single-arm, dose-escalation study of olaparib
Efficacy and tolerability
Overall clinical benefit in 46%; clinical benefit significantly associated with platinum sensitivity: 69% with platinum sensitive, 45% with platinum resistant, and 23% with platinum refractory disease
[21]
Liu et al. (2013) Recurrent ovarian (and triple negative breast) cancer, 18 ovarian patients
Single-arm, dose-escalation study of cediranib and olaparib
Safety, dosing, and efficacy
44% response rate in ovary cancer patients (another three patients had stable disease ≥6 months); ORR 45% in the ten patients with germline BRCA mutation
[23]
Phase II studies
Audeh et al. (2010)
Recurrent BRCAm Nonrandomized sequential enrollment: Olaparib 400 mg b.i.d. (n = 33) Olaparib 100 mg b.i.d. (n = 24)
ORR 33% (400 mg b.i.d.) vs 13% (100 mg b.i.d.); frequency of progression at 6 weeks: 65% (100 mg b.i.d.) vs 33% (400 mg b.i.d.)
[24]
Gelmon et al. (2011)
Recurrent ovarian cancer, both germline BRCAm and sporadic (n = 64)
Nonrandomized single arm: Olaparib 400 mg b.i.d.
ORR 41% BRCAm vs 24% BRCAwt; in BRCAwt, 50% RR with plat sensitive and 4% RR with plat resistant disease
[14]
Ledermann et al. (2012), (2014)
Maintenance therapy for platinum-sensitive recurrent high-grade serous cancer, unselected for BRCA status (n = 265)
Randomized: Olaparib 400 mg p.o. b.i.d. Placebo after completion of platinum-based chemotherapy
PFS Olaparib: 11.2 months (BRCAm) vs 5.6 months (BRCAwt), p < 0.00001 Placebo: 4.3 months (BRCAm) vs 5.5 months (BRCAwt), p = 0.007; PFS with BRCAm: 11.2 months vs 4.3 months, p < 0.0001; PFS with BRCAwt: 7.4 months vs 5.5 months, p = 0.0075; no difference in OS (HR: 0.88; p = 0.44), even when stratified by BRCAwt (HR: 0.99; p = 0.96) or BRCA mutated (HR: 0.73; p = 0.19)
[12,25]
Kaye et al. (2008)
BRCA mutation carriers without prior PLD treatment (n = 97)
Randomized: Olaparib 200 mg b.i.d. Olaparib 400 mg b.i.d. PLD 50 mg/m2
PFS No difference in PFS: 6.5 months (200 mg) vs 8.8 months (400 mg) vs 7.1 months (PLD)
[5]
Oza et al. (2013)
Platinum-sensitive recurrent high-grade serous cancer (n = 162)
Randomized: Carbo (AUC4, iv., day 1), Taxol (175 mg/m2, iv., day 1), olaparib (200 mg b.i.d., days 1–10 of 21) + olaparib 400 mg b.i.d. maintenance Carbo AUC6, Taxol 175 mg/m2
PFS Median PFS: 12.2 months (olaparib) vs 9.6 months; p = 0.0012
[26]
Liu et al., NCT01116648 (2014)
Platinum-sensitive recurrent high grade serous cancer, unselected for BRCA status (n = 90)
Cediranib 30 mg and Olaparib 200 mg b.i.d. Olaparib 400 mg b.i.d.
PFS Median PFS: 17.7 months (cediranib + olaparib) vs 9.0 months (olaparib), p = 0.005; overall response rate: 79.6% (cediranib + olaparib) vs 47.8% (olaparib), p = 0.002; PFS for BRCAm: 19.4 months vs 16.5 months, p = 0.16; PFS for BRCAwt or unknown: 16.5 months vs 5.7 months, p = 0.008
[27]
b.i.d: Two-times a day; BRCAm: BRCA germline mutation; BRCAwt: BRCA wild-type; Carbo: Carboplatin; HR: Hazard ratio; iv.: Intravenous; ORR: Objective response rate; OS: Overall survival; PFS: Progression-free survival; PLD: Pegylated liposomal doxorubicin; p.o.: Orally; Taxol: Paclitaxel.
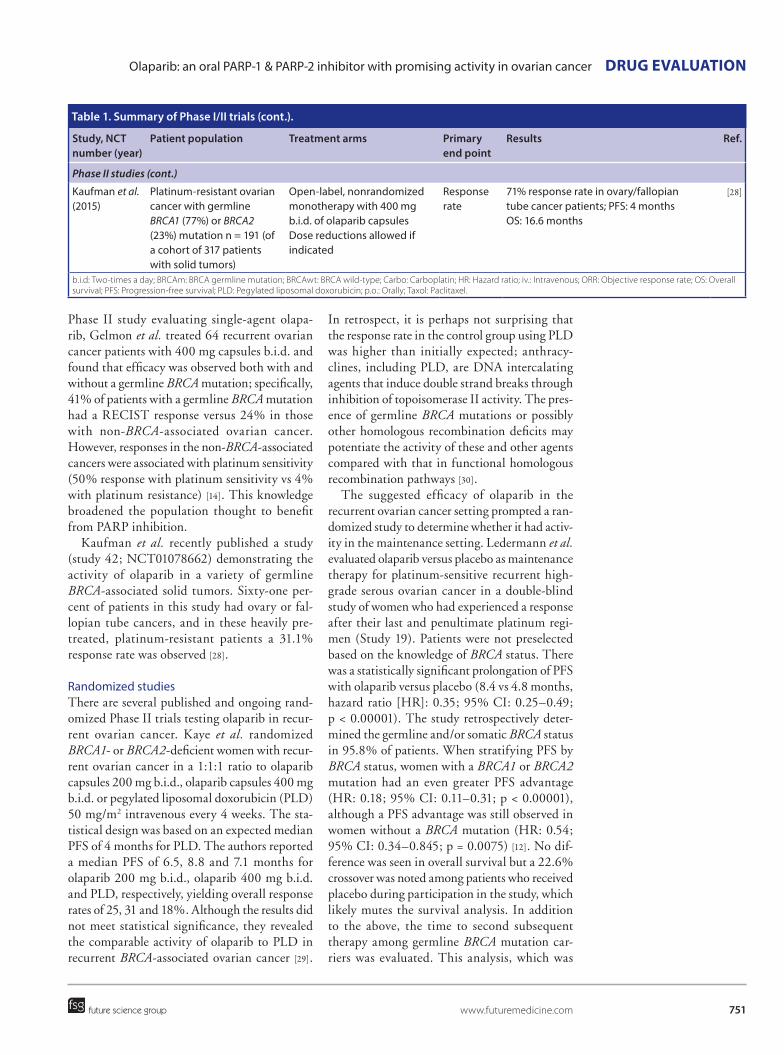
751
Olaparib: an oral PARP-1 & PARP-2 inhibitor with promising activity in ovarian cancer DRug EvAluATiOn
future science group www.futuremedicine.com
Phase II study evaluating single-agent olapa-rib, Gelmon et al. treated 64 recurrent ovarian cancer patients with 400 mg capsules b.i.d. and found that efficacy was observed both with and without a germline BRCA mutation; specifically, 41% of patients with a germline BRCA mutation had a RECIST response versus 24% in those with non-BRCA-associated ovarian cancer. However, responses in the non-BRCA-associated cancers were associated with platinum sensitivity (50% response with platinum sensitivity vs 4% with platinum resistance) [14]. This knowledge broadened the population thought to benefit from PARP inhibition.
Kaufman et al. recently published a study (study 42; NCT01078662) demonstrating the activity of olaparib in a variety of germline BRCA-associated solid tumors. Sixty-one per-cent of patients in this study had ovary or fal-lopian tube cancers, and in these heavily pre-treated, platinum-resistant patients a 31.1% response rate was observed [28].
Randomized studiesThere are several published and ongoing rand-omized Phase II trials testing olaparib in recur-rent ovarian cancer. Kaye et al. randomized BRCA1- or BRCA2-deficient women with recur-rent ovarian cancer in a 1:1:1 ratio to olaparib capsules 200 mg b.i.d., olaparib capsules 400 mg b.i.d. or pegylated liposomal doxorubicin (PLD) 50 mg/m2 intravenous every 4 weeks. The sta-tistical design was based on an expected median PFS of 4 months for PLD. The authors reported a median PFS of 6.5, 8.8 and 7.1 months for olaparib 200 mg b.i.d., olaparib 400 mg b.i.d. and PLD, respectively, yielding overall response rates of 25, 31 and 18%. Although the results did not meet statistical significance, they revealed the comparable activity of olaparib to PLD in recurrent BRCA-associated ovarian cancer [29].
In retrospect, it is perhaps not surprising that the response rate in the control group using PLD was higher than initially expected; anthracy-clines, including PLD, are DNA intercalating agents that induce double strand breaks through inhibition of topoisomerase II activity. The pres-ence of germline BRCA mutations or possibly other homologous recombination deficits may potentiate the activity of these and other agents compared with that in functional homologous recombination pathways [30].
The suggested efficacy of olaparib in the recurrent ovarian cancer setting prompted a ran-domized study to determine whether it had activ-ity in the maintenance setting. Ledermann et al. evaluated olaparib versus placebo as maintenance therapy for platinum-sensitive recurrent high-grade serous ovarian cancer in a double-blind study of women who had experienced a response after their last and penultimate platinum regi-men (Study 19). Patients were not preselected based on the knowledge of BRCA status. There was a statistically significant prolongation of PFS with olaparib versus placebo (8.4 vs 4.8 months, hazard ratio [HR]: 0.35; 95% CI: 0.25–0.49; p < 0.00001). The study retrospectively deter-mined the germline and/or somatic BRCA status in 95.8% of patients. When stratifying PFS by BRCA status, women with a BRCA1 or BRCA2 mutation had an even greater PFS advantage (HR: 0.18; 95% CI: 0.11–0.31; p < 0.00001), although a PFS advantage was still observed in women without a BRCA mutation (HR: 0.54; 95% CI: 0.34–0.845; p = 0.0075) [12]. No dif-ference was seen in overall survival but a 22.6% crossover was noted among patients who received placebo during participation in the study, which likely mutes the survival analysis. In addition to the above, the time to second subsequent therapy among germline BRCA mutation car-riers was evaluated. This analysis, which was
Study, NCT number (year)
Patient population Treatment arms Primary end point
Results Ref.
Phase II studies (cont.)
Kaufman et al. (2015)
Platinum-resistant ovarian cancer with germline BRCA1 (77%) or BRCA2 (23%) mutation n = 191 (of a cohort of 317 patients with solid tumors)
Open-label, nonrandomized monotherapy with 400 mg b.i.d. of olaparib capsules Dose reductions allowed if indicated
Response rate
71% response rate in ovary/fallopian tube cancer patients; PFS: 4 months OS: 16.6 months
[28]
b.i.d: Two-times a day; BRCAm: BRCA germline mutation; BRCAwt: BRCA wild-type; Carbo: Carboplatin; HR: Hazard ratio; iv.: Intravenous; ORR: Objective response rate; OS: Overall survival; PFS: Progression-free survival; PLD: Pegylated liposomal doxorubicin; p.o.: Orally; Taxol: Paclitaxel.
Table 1. Summary of Phase I/II trials (cont.).

Future Oncol. (2015) 11(5)752
DRug EvAluATiOn Gunderson & Moore
future science group
not pre-specified, sought to evaluate whether treatment with olaparib changed sensitivity to subsequent therapy started following olaparib therapy. The surrogate measure used for this analysis was the time interval between initiation of the first therapy following randomized treat-ment with olaparib and initiation of the second subsequent treatment in both BRCA mutation carriers and wild-type. In both groups, rand-omization to olaparib did not negatively affect the time spent on the next therapy and, in fact, patients appeared to maintain benefit (BRCA mutation: HR: 0.44; 95% CI: 0.29–0.67; p = 0.00013 vs BRCA wild-type: HR: 0.64; 95% CI: 0.42–0.96; p = 0.033) [25].
In a separate study of platinum-sensitive ovar-ian cancer (Study 41 [ClinicalTrials.gov identi-fier: NCT01081951]), Oza et al. randomized 162 women with platinum-sensitive ovarian cancer, and who had received ≤3 prior platinum-con-taining regimens in a 1:1 fashion to six cycles of carboplatin AUC 4, paclitaxel 175 mg/m2, and olaparib 200 mg b.i.d., followed by maintenance olaparib (400 mg b.i.d. continuously) or six cycles of carboplatin AUC 6 and paclitaxel 175 mg/m2 with no maintenance component. Again, patients were not selected onto this study based on known BRCA status. The authors found that median PFS was significantly higher with com-bination chemotherapy + olaparib and mainte-nance olaparib versus chemotherapy alone (12.2 vs 9.6 months, HR: 0.51; 95% CI: 0.34–0.77; p = 0.0012) [31], and similar to Ledermann et al., the PFS benefit with olaparib was greater in BRCA mutated patients (HR: 0.21; 95% CI: 0.08–0.55; p = 0.0015) with the greatest separa-tion of the Kaplan–Meier curves occurring dur-ing the maintenance phase of the study [26,32]. The benefit during the maintenance phase is of great importance in future trial design, consid-ering the particular myelotoxicity noted with concomitant administration of c onventional chemotherapy and PARP inhibitors.
Liu et al. recently published a Phase II rand-omized trial of olaparib 400 mg capsules b.i.d. versus cediranib 30 mg every day plus olapa-rib capsules 200 mg b.i.d. [27]. Cediranib is an oral tyrosine kinase inhibitor directed against VEGF receptor (VEGFR) 1, 2 and 3. In recur-rent ovarian cancer, single-agent cediranib has a response rate of 17% and a median PFS of 5.2 months [33]. Preclinical data exist to suggest synergy between olaparib and cediranib [34,35]. Phase I data from recurrent ovarian and triple
negative breast cancer using this combination led to the above dosing levels and reported a response rate of 44% [23]. In the randomized Phase II study, patients were not selected based on BRCA status, were platinum sensitive, and had high-grade serous or endometrioid histol-ogy. Among the entire study population, patients who received the combination had superior PFS of 17.7 versus 9 months (HR: 0.42; 95% CI: 0.23–0.76; p = 0.005). The overall response rate was similarly superior for the combination arm at 79.6 versus 47.8% (p = 0.002). Of the enrolled population, 52% had a BRCA germline muta-tion. Interestingly, when the cohort of patients who were BRCA wild-type or unknown were evaluated post hoc, a more significant benefit from the combination was seen in the BRCA wild-type group (PFS 16.5 vs 5.7 months; HR: 0.32; 95% CI: 0.14–0.74; p = 0.008) as com-pared with the germline BRCA group (PFS: 19.4 vs 16.5 months; HR: 0.55; 95% CI: 0.24–1.27; p = 0.16). This difference is attributed to greater synergism between the two agents in the set-ting of homologous recombination-proficient tumors or hypoxia [27]. A planned Phase III trial comparing combination olaparib and cediranib versus platinum-based chemotherapy in platinum-sensitive relapsed ovarian cancer will inform the benefit of combination biologic therapy versus cytotoxic chemotherapy in high-grade serous ovarian cancer; these results will be eagerly awaited given the impressive benefit noted with combination biologic therapy in the recently published Phase II trial [27].
●● Phase III trialsTo date, there are no published Phase III tri-als evaluating olaparib in ovarian cancer. The remarkable activity of olaparib in BRCA-associated ovarian cancer noted in the afore-mentioned studies has inspired two currently enrolling Phase III studies evaluating olapa-rib versus placebo as maintenance therapy in BRCA-associated newly diagnosed (GOG 3004, NCT01844986, SOLO-1) and platinum-sen-sitive recurrent ovarian cancer (AstraZeneca, NCT01874353, SOLO-2) after completion of platinum-based chemotherapy (see Table 2). Phase III trials of rucaparib and niraparib in the platinum-sensitive, relapsed maintenance setting are also ongoing. They are similar in design to SOLO-2 with the notable exception of allowing all patients with high-grade serous or endome-trioid cancer as well as those with known BRCA

753
Olaparib: an oral PARP-1 & PARP-2 inhibitor with promising activity in ovarian cancer DRug EvAluATiOn
future science group www.futuremedicine.com
mutations. These important trials will add to our knowledge and ability to interpret the use of PARP inhibitors in a maintenance setting.
Safety & tolerabilityThe most frequent adverse events noted with PARP inhibitors are gastrointestinal toxicity and myelosuppression. These toxicities could be problematic in ovarian cancer patients who may have overlapping myelosuppression from conven-tional chemotherapy agents or pretreated bone marrow and gastrointestinal symptoms from dis-ease burden. In the largest randomized study to date (n = 265), Ledermann et al. noted that dose interruptions (27.9 vs 8.6%) and dose reductions (22.8 vs 4.7%) were more frequent in the olapa-rib-treated patients compared with those receiv-ing placebo. Vomiting, nausea and fatigue were the most common causes of dose interruptions or reductions in the olaparib group. Incidences of grade 3–4 adverse events were 35.3 and 20.3% in the olaparib and placebo groups, respectively [12]. Study 42, which involved platinum-resistant ovarian cancer patients, many of whom had been treated with multiple prior therapies, reported anemia of any grade in 32.1% of patients (≥grade 3 in 18.7%); fatigue in 60% of patients (≥grade 3 in 6.2%); and nausea and vomiting in 61.7 and 38.9% of patients, r espectively (≥grade 3 in 0.5 and 2.6%) [28].
Future regulatory activityAs previously stated, the European Commission recently granted marketing authorization for Lynparza (olaparib) capsules (400 mg b.i.d.) as maintenance treatment of adult patients with platinum-sensitive relapsed BRCA-mutated (ger-mline and/or somatic) high-grade serous epithe-lial ovarian, fallopian tube or primary peritoneal cancer who are in complete response or partial response to platinum-based chemotherapy. Also, the FDA approved Lynparza (olaparib) cap-sules for monotherapy in the BRCA mutated,
recurrent setting after three or more lines of previous chemotherapy.
Notably, initial studies were performed uti-lizing a capsule formulation, which introduces a large pill burden for patients (16 capsules per day for a 400 mg b.i.d. dose). A tablet formula-tion has therefore been developed to facilitate delivery of olaparib doses in fewer units, and a randomized Phase I study was undertaken to select the optimal tablet dose from a safety and efficacy standpoint. Patients were randomized 1:1:1:1 to continuous dosing olaparib tablets at 200 mg three-times a day and 400 mg every day or intermittent dosing olaparib tablets of 250 mg three-times a day: 2 weeks on/1 week off, and 400 mg b.i.d.: 1 week on and 1 week off. The results of this four-arm randomization were compared with historical cohorts who had received 400 mg capsules b.i.d. or 300 mg tablets b.i.d. [36] and confirmed that 300 mg tablets b.i.d., continuous dosing maintained efficacy previously reported with the 400 mg b.i.d. capsule dose, and had an acceptable safety profile [37].
Phase III registration trials of the tablet for-mulation of olaparib as a maintenance mono-therapy in patients with BRCA mutated ovar-ian cancer following first-line platinum-based chemotherapy (SOLO-1) and as a maintenance monotherapy in patients with BRCA mutated platinum-sensitive relapsed ovarian cancer (SOLO-2) are ongoing and are nearly fully enrolled (NCT01844986 and NCT 01874353).
Conclusion●● Established data
Although several different PARP inhibitors are currently in various phases of development, olaparib was the first to be studied in ovarian cancer patients and has undergone the most extensive clinical evaluation in both germline BRCA mutated and sporadic ovarian cancer. These agents are generally well tolerated in
Table 2. Summary of ongoing Phase III trials.
NCI trial number Study arms Trial population Primary end point
Anticipated accrual
NCT01844986 (SOLO-1) Olaparib vs placebo Newly diagnosed germline BRCAm carriers, HGSC or endometrioid histology, stage III–IV after completion of platinum-based chemotherapy
PFS 344
NCT01874353 (SOLO-2) Olaparib vs placebo Recurrent platinum-sensitive ovarian cancer in germline BRCAm carriers, HGSC or endometrioid after completion of platinum-based chemotherapy
PFS 264
HGSC: High-grade serious cancer; PFS: Progression-free survival.

Future Oncol. (2015) 11(5)754
DRug EvAluATiOn Gunderson & Moore
future science group
this largely pretreated population, with a toxic-ity profile largely consisting of gastrointestinal adverse events and myelosuppression. PARP inhibitors offer a novel approach to the treatment of ovarian cancer due to their oral formulation, ease of administration, and relatively acceptable and addressable toxicities. At this point, the available data suggest appreciable prolongation of PFS. Undoubtedly, further studies are needed to delineate long-term outcomes after PARP inhibitor use; however, the duo of promising outcomes and relatively limited toxicity is excit-ing and lends support to the potential approval of PARP inhibitors as the second biologic agent for ovarian cancer, following bevacizumab.
●● Patient SelectionWhile exciting, additional questions must be answered regarding the appropriate use of olapa-rib or any other PARP inhibitor in ovarian can-cer. It will be important, for example, to under-stand whether, apart from germline BRCA1 or BRCA2 mutation carriers, there are other iden-tifiable populations that would gain meaningful clinical benefit from this class of agents.
Patients with BRCA1 or BRCA2 germline or somatic mutations are the obvious beneficiaries of PARP inhibition, given the loss of homolo-gous recombination. Women with a germline BRCA mutation represent approximately 15% of the overall ovarian cancer population, and sev-eral commercially available tests exist that sug-gests widespread testing should be feasible [38]. In fact, current clinical practice guidelines from the Society of Gynecologic Oncologists recommend germline BRCA testing for all patients with a diagnosis of high-grade serous or endometrioid ovarian cancer as standard of care, regardless of age at onset or family history. As alluded to pre-viously in the pharmacology section, there are other mutations that impact homologous recom-bination function, and patients who harbor these mutations may benefit from PARP inhibition as much as those with BRCA mutations. With further study, some of these mutations will likely also be considered as predictive biomarkers for PARP inhibitor response. The issue thus far has been that these mutations are low in frequency and were felt to be too numerous to recommend testing. Two reanalyses of data from Study 19 and Study 41 aim to identify specific popula-tions that may or may not benefit from olaparib.
Presented as an abstract at the American Society of Clinical Oncology in 2014, Lheureux et al.
(abstract 5534, full manuscript not yet published) evaluated patients on both Study 19 and 41 and focused only on those patients who received active drug and either received <6 months of olaparib or >2 years of olaparib. Genomic characteriza-tion of this entire cohort is ongoing; however, the American Society of Clinical Oncology presenta-tion focused on the BRCA status of these outliers. The authors found that across the two studies, among the 68 patients who stayed on treatment <6 months, 69% were either BRCA negative or of unknown BRCA status [39]. Given the knowl-edge we have to date, this is not surprising. What is interesting is that approximately 30% of the patients who received <6 months of olaparib had germline BRCA mutations. Why these patients received lesser benefit from olaparib given at therapeutic doses requires further elucidation. Among the 49 patients who stayed on olaparib >2 years (recall that these studies both involved maintenance olaparib), 65% were germline BRCA patients, which is logical; however, the remaining 35% were BRCA wild-type or unknown. Why these patients gained prolonged benefit from olaparib requires further molecular characteri-zation but points to a role for PARP inhibition beyond BRCA [39].
Dougherty et al. performed a retrospective analysis of tumor samples from Study 19 in which samples were sequenced for homologous recom-bination repair genes. This analysis focused on an outlier sample of long-term responders (>2 years). Among the entire Study 19 cohort, the frequency of BRCA1 or BRCA2 mutations was 53%. In addition, another 10% of patients had homolo-gous recombination gene mutations identified including BRIP1, CDK12, and RAD52 among others. If non-core homologous recombination genes such as PTEN and EMSY were included, mutations in addition to BRCA totaled 14%. Among patients who were long-term respond-ers (16% of the Study 19 cohort), 76% had a mutation in BRCA1 or 2 with enrichment in the proportion of BRCA2 patients from 17 to 39%. Nine percent of the long-term responders had homologous recombination repair mutations in core genes such as BRIP1, RAD51B, and FANCL, and 15% of the long-term group were either wild-type or had mutations in noncore homologous recombination repair genes such as PTEN and BRCA2 [40].
This important work adds to the literature and will help identify patients at highest likeli-hood to benefit from olaparib or other PARP

755
Olaparib: an oral PARP-1 & PARP-2 inhibitor with promising activity in ovarian cancer DRug EvAluATiOn
future science group www.futuremedicine.com
inhibitors. The best responders appear to be BRCA patients, but there are others who require identification.
●● Long-term effects of PARP inhibitionWhether the use and timing of PARP inhibi-tors in the treatment of ovarian cancer changes the course and behavior of the disease will likely have to await post-approval study. There is, however, some preliminary data, which is reassuring, but still incomplete. As in Study 19, Study 41 included an analysis of time to second subsequent therapy in germline BRCA mutation carriers. This analysis showed that time to sec-ond subsequent therapy was significantly longer in the olaparib group of patients compared with placebo (HR: 0.26; 95% CI: 0.11–0.59; p = 0.0013) [32]. These pre-planned retrospective analyses from Studies 41 and 19 are reassuring in that patients who received olaparib, whether they be BRCA mutation carriers or not, appeared
to maintain the benefit of olaparib rather than having a less robust response to subsequent ther-apy. Indeed, Ang et al. have shown that heavily pretreated patients who progress on PARP inhib-itor therapy retain the potential to respond to further cytotoxic chemotherapy, with an objec-tive response rate of 40% by RECIST, and no secondary BRCA mutations detected in tumor samples [41]. Additional long-term follow-up for survival is necessary.
Additionally, there is a theoretical risk of mye-lodysplastic syndrome and leukemia with the use of PARP inhibitors. This is due to the aberrant repair of double stranded DNA breaks, which accumulate with spontaneously occurring single stranded breaks [42]. Although the mechanistic rationale is supportive, to date there has not been any published data regarding the risk or inci-dence of myelodysplastic syndrome, leukemia, or other induced hematopoietic defects. Post-marketing studies will be crucial for evaluating
EXECuTivE SuMMARYMechanism of action
● Inhibits PARP enzyme from repairing single strand breaks in DNA, leading to subsequent formation of double-stranded breaks, which results in cell death.
● Stimulation of the error-prone nonhomologous end joining DNA repair pathway via phosphorylation of DNA-dependent protein kinase substrates.
● Trapping the PARP-1 and PARP-2 enzymes onto DNA, thus interfering with DNA replication.
Pharmacokinetic properties
● After oral administration, olaparib is rapidly absorbed and declines in a biphasic manner.
● Olaparib is excreted equally between urine and feces.
Clinical efficacy
● Olaparib has been shown to offer substantial response rates and significant progression-free survival prolongation in women with newly diagnosed and recurrent ovarian cancer.
Safety & tolerability
● The dose-limiting effect is myelosuppression; gastrointestinal toxicity is also a common adverse effect.
Dose & administration
● 400 mg two-times a day is the recommended capsule dose.
● Oral drugs: both capsule and tablet formulations have been evaluated, although most of the currently published data have utilized capsules.
Regulatory affairs
● Lynparza (olaparib) capsules were approved by the European Commission in December 2014 for maintenance treatment of platinum-sensitive high-grade serous ovarian cancer in BRCA-mutated women.
● Lynparza (olaparib) capsules were approved by the US FDA for treatment of recurrent ovarian cancer in women with a deleterious BRCA mutation in December 2014.

Future Oncol. (2015) 11(5)756
DRug EvAluATiOn Gunderson & Moore
future science group
and quantifying the incidence of this potential problem.
DisclaimerIn addition to the peer-review process, with the author(s) consent, the manufacturer of the product(s) discussed in this article was given the opportunity to review the manuscript for factual accuracy. Changes were made at the discretion of the author(s) and based on scientific or editorial merit only.
Financial & competing interests disclosureK Moore has been a paid consultant on advisory boards for AstraZeneca. The authors have no other relevant affilia-tions or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
ReferencesPapers of special note have been highlighted as: • of interest; •• of considerable interest
1 Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J. Clin. 64(1), 9–29 (2014).
2 McGuire WP, Hoskins WJ, Brady MF et al. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. N. Engl. J. Med. 334(1), 1–6 (1996).
3 Bookman MA. The addition of new drugs to standard therapy in the first-line treatment of ovarian cancer. Ann. Oncol. 21(Suppl. 7), vii211–vii217 (2010).
4 Dizon DS, Weitzen S, Rojan A et al. Two for good measure: six versus eight cycles of carboplatin and paclitaxel as adjuvant treatment for epithelial ovarian cancer. Gynecol. Oncol. 100(2), 417–421 (2006).
5 Kim HS, Park NH, Chung HH, Kim JW, Song YS, Kang SB. Are three additional cycles of chemotherapy useful in patients with advanced-stage epithelial ovarian cancer after a complete response to six cycles of intravenous adjuvant paclitaxel and carboplatin? Jpn. J. Clin. Oncol. 38(6), 445–450 (2008).
6 Hanker LC, Loibl S, Burchardi N et al. The impact of second to sixth line therapy on survival of relapsed ovarian cancer after primary taxane/platinum-based therapy. Ann. Oncol. 23(10), 2605–2612 (2012).
7 Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 108(2), 171–182 (2002).
8 Bryant HE, Schultz N, Thomas HD et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434(7035), 913–917 (2005).
9 Farmer H, McCabe N, Lord CJ et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434(7035), 917–921 (2005).
10 Menear KA, Adcock C, Boulter R et al. 4-[3-(4-cyclopropanecarbonylpiperazine-1-
carbonyl)-4-fluorobenzyl]-2H-phthalazin- 1-one: a novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1. J. Med. Chem. 51(20), 6581–6591 (2008).
11 Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene 23(16), 2825–2837 (2004).
•• This is the RPh2 study (Study 19) that compared maintenance olaparib versus placebo following complete or partial response to chemotherapy in platinum-sensitive disease. The addition of olaparib markedly improved progression-free survival among all patients but especially among those with a germline BRCA mutation (hazard ratio: 18).
12 Ledermann J, Harter P, Gourley C et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N. Engl. J. Med. 366(15), 1382–1392 (2012).
13 Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol. Oncol. 5(4), 387–393 (2011).
14 Gelmon KA, Tischkowitz M, Mackay H et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a Phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 12(9), 852–861 (2011).
• This was one of the earlier Phase II trials to demonstrate significant activity as a single agent for olaparib in relapsed ovarian cancer patients who carry a germline BRCA mutation.
15 Hay T, Matthews JR, Pietzka L et al. Poly(ADP-ribose) polymerase-1 inhibitor treatment regresses autochthonous Brca2/p53-mutant mammary tumors in vivo and delays tumor relapse in combination with carboplatin. Cancer Res. 69(9), 3850–3855 (2009).
16 Rottenberg S, Jaspers JE, Kersbergen A et al. High sensitivity of BRCA1-deficient
mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl Acad. Sci. USA 105(44), 17079–17084 (2008).
17 Evers B, Drost R, Schut E et al. Selective inhibition of BRCA2-deficient mammary tumor cell growth by AZD2281 and cisplatin. Clin. Cancer Res. 14(12), 3916–3925 (2008).
18 Sessa C. Update on PARP1 inhibitors in ovarian cancer. Ann. Oncol. 22(Suppl. 8), viii72–viii76 (2011).
19 Mendes-Pereira AM, Martin SA, Brough R et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol. Med. 1(6–7), 315–322 (2009).
20 Chen Y, Zhang L, Hao Q. Olaparib: a promising PARP inhibitor in ovarian cancer therapy. Arch. Gynecol. Obstet. 288(2), 367–374 (2013).
21 Fong PC, Yap TA, Boss DS et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J. Clin. Oncol. 28(15), 2512–2519 (2010).
22 Fong PC, Boss DS, Yap TA et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 361(2), 123–134 (2009).
23 Liu JF, Tolaney SM, Birrer M et al. A Phase 1 trial of the poly(ADP-ribose) polymerase inhibitor olaparib (AZD2281) in combination with the anti-angiogenic cediranib (AZD2171) in recurrent epithelial ovarian or triple-negative breast cancer. Eur. J. Cancer 49(14), 2972–2978 (2013).
24 Audeh MW, Carmichael J, Penson RT et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet 376(9737), 245–251 (2010).
25 Ledermann J, Harter P, Gourley C et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a

757future science group www.futuremedicine.com
Olaparib: an oral PARP-1 & PARP-2 inhibitor with promising activity in ovarian cancer DRug EvAluATiOn
randomised Phase 2 trial. Lancet Oncol. 15(8), 852–861 (2014).
• This post hoc analysis is from Study 19.
26 Oza A, Cibula D, Benzaquen AO et al. Olaparib plus chemotherapy, followed by maintenance monotherapy, in women with platinum sensitive recurrent serous ovarian cancer (PSR SOC): BRCA 1/2 mutation (BRCAm) and interim overall survival analyses. Eur. J. Cancer 49(Suppl. 2) (2013).
27 Liu JF, Barry WT, Birrer M et al. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: a randomised Phase 2 study. Lancet Oncol. 15(11), 1207–1214 (2014).
•• This randomized Phase II trial in platinum-sensitive patients with and without BRCA mutations demonstrated the activity of single-agent olaparib but more importantly, demonstrated the synergistic effect of combination cediranib and olaparib among patients who were BRCA unknown or wild-type.
28 Kaufman B, Shapira-Frommer R, Schmutzler RK et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 33(3), 244–250 (2015).
•• This is the largest, Phase II, single-agent study performed in multiple tumors with germline BRCA mutations. It included a large number of platinum-resistant ovarian cancer patients and demonstrated a remarkably high response rate.
29 Kaye SB, Lubinski J, Matulonis U et al. Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and
recurrent ovarian cancer. J. Clin. Oncol. 30(4), 372–379 (2012).
• This was an important trial randomizing patients with a germline BRCA mutation to one of two doses of olaparib versus pegylated liposomal doxorubicin. It not only demonstrated the similar activity of olaparib to a cytotoxic in this population but also demonstrated the chemosensitivity of BRCA patients in that pegylated liposomal doxorubicin outperformed as compared with historical data in non-BRCA patients.
30 Safra T, Rogowski O, Muggia FM. The effect of germ-line BRCA mutations on response to chemotherapy and outcome of recurrent ovarian cancer. Int. J. Gynecol. Cancer 24(3), 488–495 (2014).
31 Oza AM, Cibula D, Benzaquen AO et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: a randomised Phase 2 trial. Lancet Oncol. 16(1), 87–97 (2015).
32 Oza A, Cibula D, Benzaquen A, Poole C, Mathijssen R, Sonke G. Olaparib plus chemotherapy followed by maintenance monotherapy inpatients with platinum sensitive recurrent serous ovarian cancer (abstract). J. Clin. Oncol. 30, Abstract 28 (2012).
33 Matulonis UA, Berlin S, Ivy P et al. Cediranib, an oral inhibitor of vascular endothelial growth factor receptor kinases, is an active drug in recurrent epithelial ovarian, fallopian tube, and peritoneal cancer. J. Clin. Oncol. 27(33), 5601–5606 (2009).
34 Hegan DC, Lu Y, Stachelek GC, Crosby ME, Bindra RS, Glazer PM. Inhibition of poly(ADP-ribose) polymerase down-regulates BRCA1 and RAD51 in a pathway mediated by E2F4 and p130. Proc. Natl Acad. Sci. USA 107(5), 2201–2206 (2010).
35 Tentori L, Lacal PM, Muzi A et al. Poly(ADP-ribose) polymerase (PARP) inhibition or PARP-1 gene deletion reduces angiogenesis. Eur. J. Cancer 43(14), 2124–2133 (2007).
36 Molife L, Mateo J, McGoldrick T et al. Safety and efficacy results from two randomized expansions of a Phase I study of a tablet formulation or the PARP inhibitor, olaparib, in ovarian and breast cancer patients with BRCA1/2 mutations. J. Clin. Oncol. 30(Suppl.), Abstract 3048 (2012).
37 Mateo J, Friedlander M, Sessa C. Administration of continuous/intermittent olaparib in ovarian cancer patients with a germline BRCA1/2 mutation to determine an optimal dosing schedule for the tablet formation. Eur. J. Cancer 49(Suppl. 2), Abstract 801 (2013).
38 Zhang S, Royer R, Li S et al. Frequencies of BRCA1 and BRCA2 mutations among 1,342 unselected patients with invasive ovarian cancer. Gynecol. Oncol. 121(2), 353–357 (2011).
39 Lheureux S, Ledermann J, Kaye S et al. Characterization of ovarian cancer long-term responders on olaparib (abstract). J. Clin. Oncol. 32(5s), Abstract 5534 (2014).
40 Dougherty B, Ledermann J, Lai Z et al. Analysis of candidate homologous repair deficiency genes in a clinical trial of olaparib in patients with platinum-sensitive, relapsed serous ovarian cancer (abstract). J. Clin. Oncol. 32(5s), Abstract 5536 (2014).
41 Ang JE, Gourley C, Powell CB et al. Efficacy of chemotherapy in BRCA1/2 mutation carrier ovarian cancer in the setting of PARP inhibitor resistance: a multi-institutional study. Clin. Cancer Res. 19(19), 5485–5493 (2013).
42 Fenaux P, Morel P, Lai JL. Cytogenetics of myelodysplastic syndromes. Semin. Hematol. 33(2), 127–138 (1996).