guia: international conference on harmonisation...
TRANSCRIPT

GCP - ICH
GUIA: INTERNATIONAL CONFERENCE ON HARMONISATION OF
TECHNICAL REQUIREMENTS FOR REGISTRATION OF
PHARMACEUTICALS FOR HUMAN USE
ICH
CONSIDERACIONES GENERALES PARA PROTOCOLOS CLINICOS:
EUROPA, JAPON, USA
• PROTECCION DE LOS SUJETOS PARA LOS PROTOCOLOS CLINICOS
• ESTUDIOS NO CLINICOS
•CALIDAD DE LA INVESTIGACION MEDICINAL
•FASES DEL DESARROLLO CLINICO

GCP - ICH
ICH E6 Protection of clinical trial subjects
•The Declaration of Helsinki
Good Clinical Practice (GCP)
DISEÑO, MONITOREO, RECOLECCION DE DATOS DE PROTOCOLOS
CLINCOS QUE ASEGURAN LA CREDIBILIDAD DE RESULTADOS Y
PROTEGE LOS DERECHOS Y CONFIDENCIALIDAD DE LOS PACIENTES
INVOLUCRADOS
•LOS DERECHOS Y SEGURIDAD DE LOS INDIVIDUOS ES LO MAS IMPORTANTE
•LA RESPONSABILIDAD SOBRE LA SALUD DE LOS INDIVIDUOS DEPENDE DEL
MEDICO
•CADA INDIVIDUO INVOLUCRADO DEBE FIRMAR UN CONSENTIMIENTO POR
ESCRITO PREVIO AL PROTOCOLO CLINICO
•LOS RESULTADOS DEBEN SER CONFIDENCIALES, ARCHIVADOS Y ACCESIBLES
PARA ESTUDIO Y VERIFICACION

GCP - ICH
ICH E9 and E10
The protocol should specify methods of allocation to treatment groups and
blinding
a) Randomisation
In conducting a controlled trial, randomised allocation is the preferred means
of assuring comparability of test groups and minimising the possibility of
selection bias.
b) Blinding
Blinding is an important means of reducing or minimising the risk of biased
study outcomes. A trial where the treatment assignment is not known by the
study participant because of the use of placebo or other methods of masking
the intervention, is referred to as a single blind study. When the investigator
and sponsor staff who are involved in the treatment or clinical evaluation of
the subjects and analysis of data are also unaware of the treatment
assignments, the study is double blind.
c) Compliance
Methods used to evaluate patient usage of the test drug should be specified
in the protocol and the actual usage documented.

GCP - ICH
ICH – E8 Non-Clinical Studies (using animals)
a) duration and total exposure proposed
b) characteristics of the drug (e.g. long half life, biotechnology products)
c) disease or condition targeted for treatment
d) route of administration
Non-clinical information including toxicology, pharmacology and
pharmacokinetics to support clinical trials
DEFINIR:
•ESPECIES DE ANIMALES USADAS
•NUMERO Y SEXO DE LOS ANIMALES USADOS EN CADA GRUPO
•DOSIS DE LAS UNIDADES (mg/kg)
•INTERVALOS DE LAS DOSIS
•RUTA DE ADMINISTRACION
•INFORMACION SOBRE LA DISTRIBUCION SISTEMICA
•DURACION DEL SEGUIMIENTO DESPUES DE LA EXPOSICION
•ICH E6 : Before any clinical trial is carried out, results of non-clinical
investigations or previous human studies should be sufficient to indicate that
the drug is acceptably safe for the proposed investigation in humans.

GCP - ICH
ESTUDIOS NO CLINICOS
ESTUDIOS DE SEGURIDAD:
DEBE DETERMINARSE LA DOSIS EN FUNCION DE ESTUDIOS TOXICOLOGICOS
ESTUDIAR:
•DOSIS UNICA
•DOSIS REPETIDAS
•CARCINOGENICIDAD
•ALERGIAS
•TOXICIDAD REPRODUCTIVA
•GENOTOXICIDAD

GCP - ICH
ESTUDIOS FARMACOLOGICOS Y FARMACOCINETICOS:
•MECANISMO DE ACCION Y PRINCIPALES EFECTOS PRODUCIDOS
•NATURALEZA Y FRECUENCIA DEL EFECTO
•RELACION DOSIS RESPUESTA EN RELACION CON LA DURACCION DE
LA ACCION
•ESTUDIO DE LA RUTA CLINICA DE ADMINISTRACION
•FARMACOLOGIA SISTEMICA GENERAL, OBSERVACION DE ORGANOS
AFECTADOS
•ESTUDIOS DE ABSORCION, DISTRIBUCION, METABOLISMO Y
EXCRECION
•REVERSIBILIDAD DE LOS EFECTOS
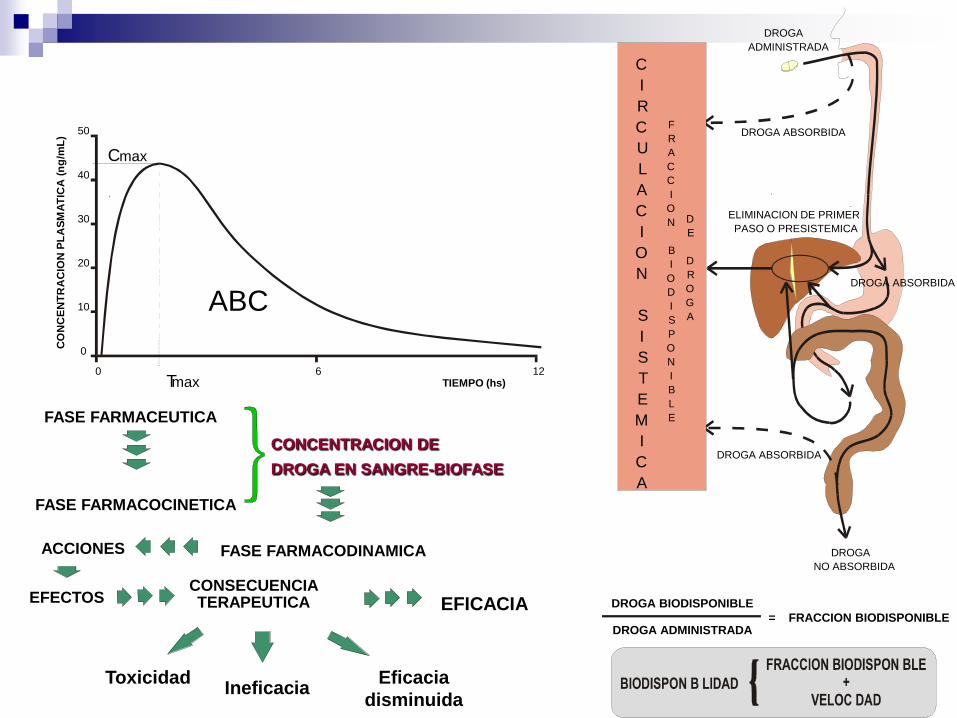
C
I
R
C
U
L
A
C
I
O
N
S
I
S
T
E
M
I
C
A
F
R
A
C
C
I
O
N
B
I
O
D
I
S
P
O
N
I
B
L
E
D
E
D
R
O
G
A
DROGA
ADMINISTRADA
DROGA ABSORBIDA
ELIMINACION DE PRIMER
PASO O PRESISTEMICA
DROGA ABSORBIDA
DROGA
NO ABSORBIDA
DROGA ABSORBIDA
DROGA BIODISPONIBLE
DROGA ADMINISTRADA = FRACCION BIODISPONIBLE
C O
N C
E N
T R
A C
I O N
P L
A S
M A
T I C
A (
n g
/ m L
)
TIEMPO (hs)
0
0 6 12
10
20
30
40
50
T max
C max
ABC
FASE FARMACEUTICA
FASE FARMACOCINETICA
ACCIONES
EFECTOS
CONCENTRACION DE
DROGA EN SANGRE-BIOFASE
FASE FARMACODINAMICA
CONSECUENCIA TERAPEUTICA EFICACIA
Ineficacia Toxicidad Eficacia
disminuida
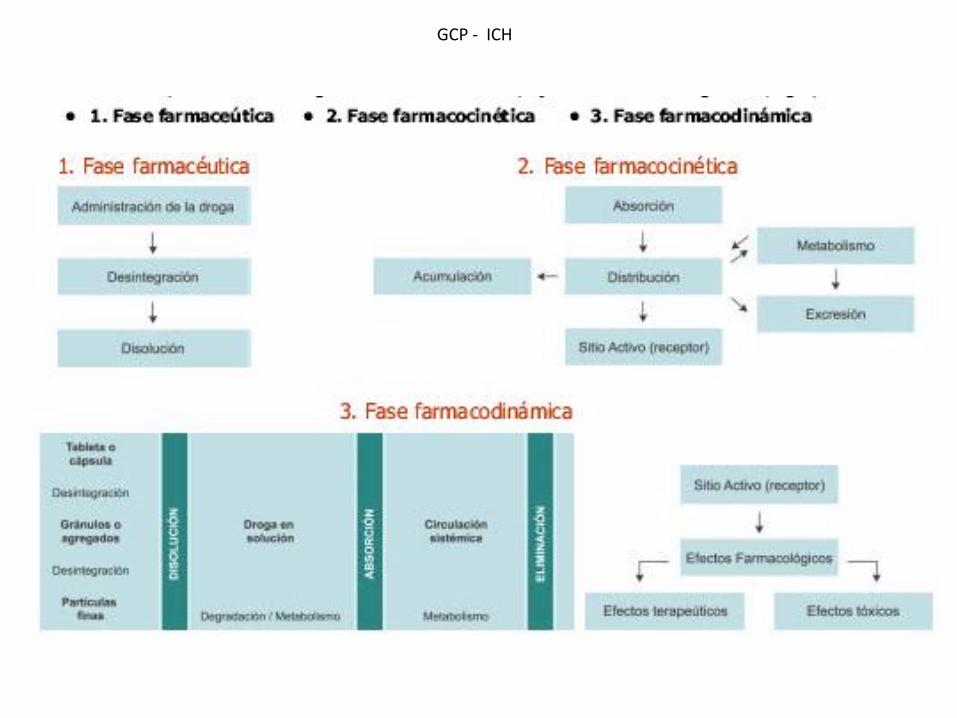
GCP - ICH
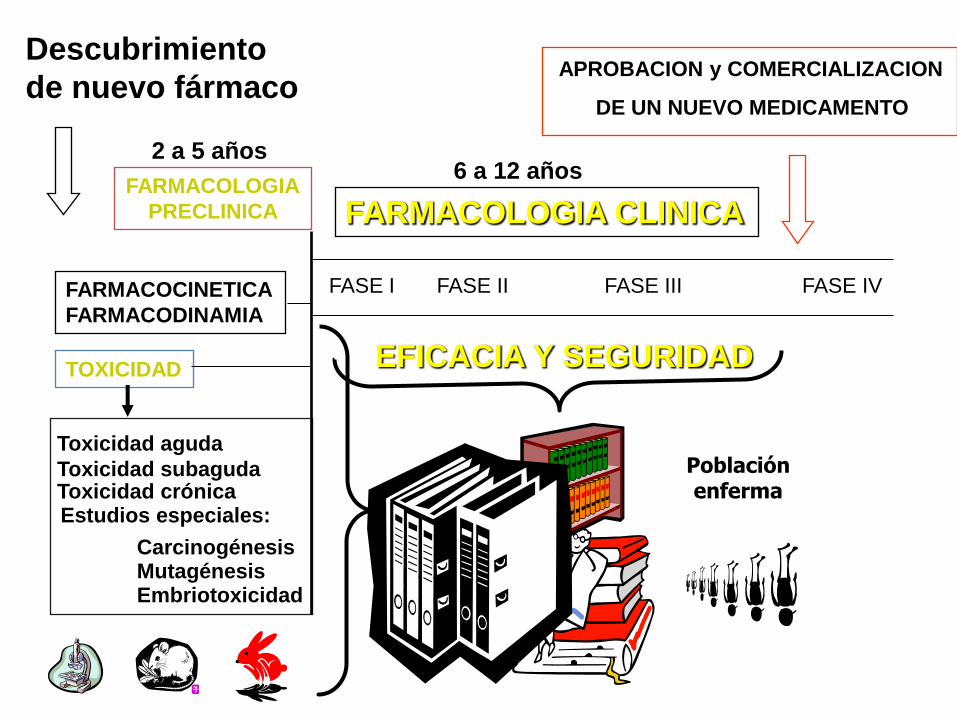
Descubrimiento
de nuevo fármaco
Toxicidad aguda
Toxicidad subaguda Toxicidad crónica Estudios especiales:
Mutagénesis Carcinogénesis
Embriotoxicidad
FARMACOCINETICA
FARMACODINAMIA
2 a 5 años
FARMACOLOGIA
PRECLINICA
TOXICIDAD
FASE I FASE II FASE III FASE IV
Población enferma
APROBACION y COMERCIALIZACION
DE UN NUEVO MEDICAMENTO
6 a 12 años
FARMACOLOGIA CLINICA
EFICACIA Y SEGURIDAD

GCP - ICH
FASE 1: FARMACOLOGIA HUMANA
•EN VOLUNTARIOS SANOS O PACIENTES CON DETERMINADA
ENFERMEDAD
•ESTIMACION DE SEGURIDAD Y TOLERABILIDAD
•ESTUDIOS FARMACOCINETICOS: ABSORCION, DISTRIBUCION,
METABOLISMO, EXCRECION
•ESTUDIOS FARMACODINAMICOS: PARA DETERMINAR DOSIS POSIBLES
DE USO

GCP - ICH
FASE 2: EXPLORACION TERAPEUTICA
•OBJETIVO: ESTUDIO DE EFECTO TERAPEUTICO
•DETERMINA DOSIS Y FRECUENCIA DE USO DEL MEDICAMENTO
•CONFIRMA DOSIS DE USO
•EVALUACION DE PUNTO FINAL DE LA TERAPIA PARA ALGUNOS
MEDICAMENTOS
•DEFINIR POBLACIONES BLANCO
•PROTOCOLOS DE CORTA DURANCION EN POBLACION DE
PACIENTES DEFINIDA USANDO MEDICIONES CLINICAS

GCP - ICH
FASE 3: CONFIRMACION TERAPEUTICA
• DEMOSTRACION O CONFIRMACION DEL BENEFICIO TERAPEUTICO
• BASES PARA APROBACION DE LICENCIA
• ESTUDIOS SOBRE RELACION DOSIS – RESPUESTA :
1. EFECTO EN POBLACIONES CON DIFERENTES ESTADIOS DE LA
ENFERMEDAD
2. EFECTOS EN COMBINACION CON OTRAS DROGAS
3. USO DE DROGAS POR LARGOS PERIODOS

GCP - ICH
FASE 4: USO TERAPEUTICO
COMIENZA DESPUES QUE LA DROGA FUE APROBADA
(DROGA SEGURA, EFICAZ Y CON DOSIS DEFINIDA)
•REDEFINE BENEFICIO/RIESGO DE LA DROGA
•INTERACCION ENTRE DROGAS
•ESTUDIOS DOSIS RESPUESTA
•ESTUDIOS EPIDEMIOLOGICOS

GCP - ICH
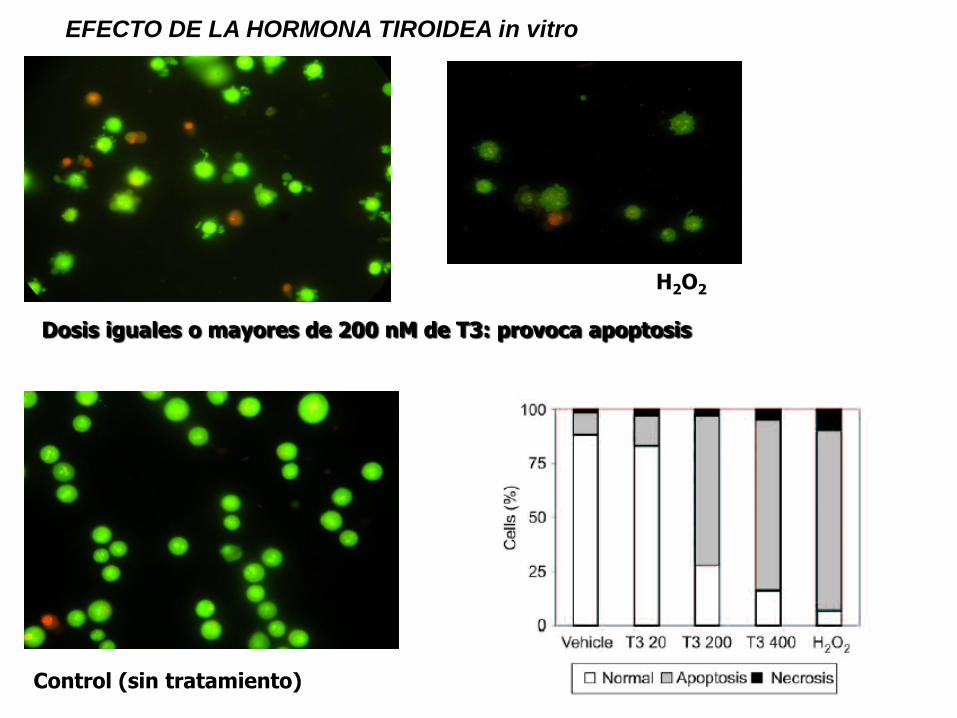
EFECTO DE LA HORMONA TIROIDEA in vitro
H2O2
Control (sin tratamiento)
Dosis iguales o mayores de 200 nM de T3: provoca apoptosis

EFECTO NO GENOMICO DE LA HORMONA TIROIDEA: ROS in vitro

ESTRÉS OXIDATIVO Y ANTIOXIDANTES
ROS AO

EFECTO NO GENOMICO DE LA HORMONA TIROIDEA in vitro

Estrategia de trabajo
Se estableció puntaje clínico de acuerdo a los síntomas y signos más comunes de hipertiroidismo: a) nerviosismo o insomnio; b) calor y/o sudoración; c) pérdida de peso o diarrea; d) taquicardia y e) temblor.
Se seleccionaron pacientes con puntaje 5. Su inclusión en los distintos grupos fue al azar. El estudio fue realizado doble ciego.

EMPLEO DE ANTIOXIDANTES EN EL HIPERTIROIDISMO
. El estudio se realizó sobre N pacientes
hipertiroideos, divididos en tres grupos:
el grupo MMI recibió Methimazol
el grupo AO (antioxidantes) recibió vit. E 200 mg,
caroteno 3 mg, vit. C 250 mg, Cu 1mg, Zn 7,5
mg, Mn 1,5 mg, Se 15 mcg. (Marca conocida, Se
definieron segun protocolo previo)
el grupo (MMI + AO) recibió los 2 tratamientos
juntos.

EFECTO DE LA HORMONA TIROIDEA: PROTOCOLO CLINICO
PACIENTES HIPERTIROIDEOS
0
20
40
60
Control Antes de
tratamiento
MMI AO
mm
ol
MD
A/m
g p
rote
ina

EFECTO DE LA HORMONA TIROIDEA: PROTOCOLO CLINICO
0
20
40
60
80
100
120
MMI 4 sem MMI 8 sem AO doble
Fre
cu
en
cia
ca
rdía
ca
TRATAMIENTO CON MMI/AO
AO puede ser utilizado como adyuvante en terapia convencional

EFECTO DE LA HORMONA TIROIDEA: PROTOCOLO CLINICO
A= MM I B= MMI+AO

GCP - ICH
CRO: CLINICAL RESEARCH ORGANIZATION
ESTUDIOS DE INVESTIGACION CLINICA

Biológicos: principios activos proteicos
extraídos de fuentes biológicas no siempre
totalmente caracterizados por medios físico-
químicos o bioquímicos. La potencia y calidad
dependen de la fuente y de los procesos.
Biotecnológicos: principios activos proteicos
producidos mediante ingeniería genética con
fuentes estables y mejor caracterizados que los
anteriores desde el punto de vista físico-
químico.

Roger SD, Mikhail A. Biosimilars: opportunity or cause for
concern? J Pharm Pharm Sci. 2007;10(3):405-10.
Biopharmaceuticals are drug products containing biotechnology-
derived proteins as active substances, and have revolutionised the
treatment of many diseases. A number of biopharmaceutical patents
are due to expire in the next few years, or have already expired. The
subsequent production of follow-on products, or 'biosimilars' has
aroused interest within the pharmaceutical industry as biosimilar
manufacturers strive to obtain part of an already large and rapidly-
growing market. The potential opportunity for price reductions
versus the originator biopharmaceuticals remains to be
determined, as the advantage of a slightly cheaper price
may be outweighed by the hypothetical increased risk of
side-effects from biosimilar molecules that are not exact
copies of their originators.

Medicamento similar: Mismo fármaco, misma concentración, forma
farmacéutica, vía de administración, posología e indicación
terapéutica del medicamento de referencia, pero no tiene
bioequivalencia comprobada con el de referencia.
Medicamento original:
El de referencia.
Eficacia y seguridad comprobada por ensayos clínicos.
Marca comercial conocida.
Marco Regulatorio en la Argentina
Disposición 5040/2006 de la ANMAT
Régimen de buenas prácticas para la realización de estudios de
biodisponibilidad/bioequivalencia.

GCP - ICH

Dos medicamentos, químicamente equiva-
lentes (principio activo y concentración
iguales), son bioequivalentes, si tienen la
misma biodisponibilidad
esto es: velocidad y magnitud de absorción sin diferencias significativas de biodisponibilidad cuando se estudian en condiciones experimentales similares y en dosis únicas o repetidas.

Eritropoyetina
Debido a la complejidad de la molécula y
del proceso de manufactura es difícil
obtener productos idénticos al original.
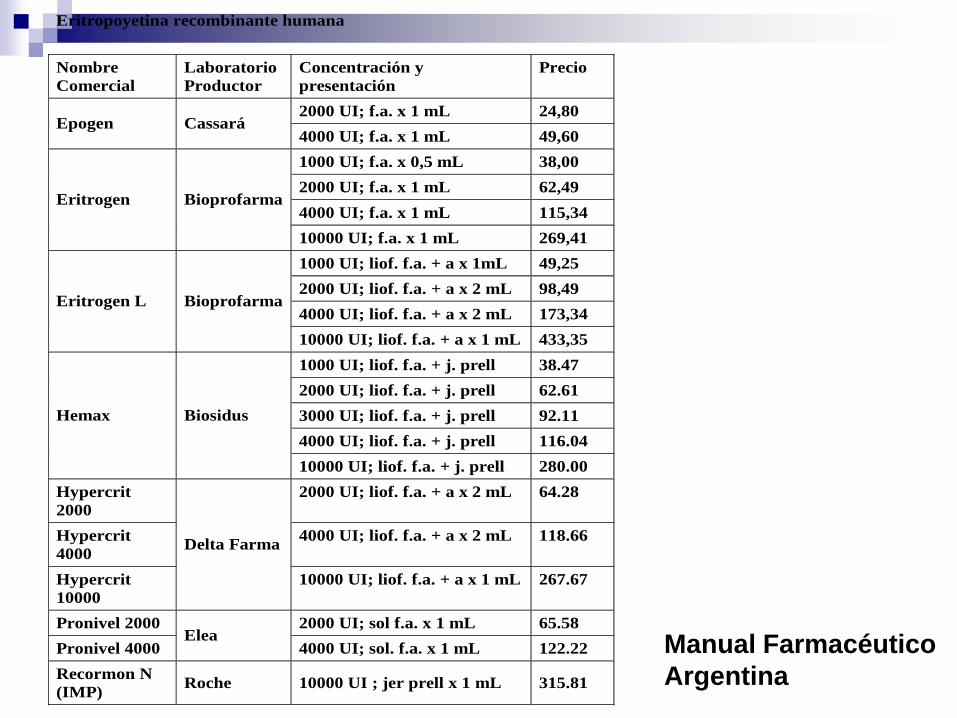
Eritropoyetina recombinante humana
Nombre
Comercial
Laboratorio
Productor
Concentración y
presentación
Precio
2000 UI; f.a. x 1 mL 24,80 Epogen Cassará
4000 UI; f.a. x 1 mL 49,60
1000 UI; f.a. x 0,5 mL 38,00
2000 UI; f.a. x 1 mL 62,49
4000 UI; f.a. x 1 mL 115,34 Eritrogen Bioprofarma
10000 UI; f.a. x 1 mL 269,41
1000 UI; liof. f.a. + a x 1mL 49,25
2000 UI; liof. f.a. + a x 2 mL 98,49
4000 UI; liof. f.a. + a x 2 mL 173,34 Eritrogen L Bioprofarma
10000 UI; liof. f.a. + a x 1 mL 433,35
1000 UI; liof. f.a. + j. prell 38.47
2000 UI; liof. f.a. + j. prell 62.61
3000 UI; liof. f.a. + j. prell 92.11
4000 UI; liof. f.a. + j. prell 116.04
Hemax Biosidus
10000 UI; liof. f.a. + j. prell 280.00
Hypercrit
2000
2000 UI; liof. f.a. + a x 2 mL 64.28
Hypercrit
4000
4000 UI; liof. f.a. + a x 2 mL 118.66
Hypercrit
10000
Delta Farma
10000 UI; liof. f.a. + a x 1 mL 267.67
Pronivel 2000 2000 UI; sol f.a. x 1 mL 65.58
Pronivel 4000 Elea
4000 UI; sol. f.a. x 1 mL 122.22
Recormon N
(IMP) Roche 10000 UI ; jer prell x 1 mL 315.81
Manual Farmacéutico
Argentina

Actualmente, los medicamentos biotecnológicos aprobados para
su comercialización ya tratan o ayudan a prevenir: ataques
cardíacos, infarto cerebral, esclerosis múltiples, leucemia,
hepatitis, artritis reumatoide, cáncer mamario, diabetes,
insuficiencia cardiaca, linfoma, cáncer renal, fibrosis quística y
otras enfermedades.
En el 2003 existían 324 medicamentos biotecnológicos bajo
desarrollo, que incluyen 154 medicinas para el cáncer, 43 para las
enfermedades infecciosas, 26 para las enfermedades autoinmunes
y 17 para AIDS/HIV y condiciones relacionadas, reafirmando este
concepto .
Se estima que el año 2012 los medicamentos biotecnológicos
representarán el 12% del total de las ventas mundiales de
medicamentos de prescripción.

El tema de los biosimilares presentes en los
mercados latinoamericanos ya aprobados utilizando
regulaciones que se aplican para los medicamentos
de síntesis se ve agravado en países que no tiene un
sistema de farmacovigilancia y, en los que sí lo
tienen, la actividad no forma parte de la cultura de
sus médicos.
En este contexto es riesgoso promover la
intercambiabilidad por carencia de soporte técnico.

Los productos biotecnológicos son muy sensibles a
cambios en los procesos de manufactura por lo que
se requiere el mantenimiento de condiciones de
producción:
Estables;
Reproducibles;
Consistentes y validados para el efectivo Control y
Aseguramiento de la Calidad

Estudios de bioequivalencia
Ensayo clínico
Voluntario sano
Diseño cruzado
Aleatorizado
Abierto o Ciego

La inmunogenicidad es una de las limitantes más
significativas de los productos biotecnológicos:
El sistema inmune puede detectar diferencias estructurales menores
entre proteínas que pueden ser indetectables a través de
evaluaciones analíticas pero que pueden ser biológicamente
significativas.
La producción de anticuerpos contra el producto biotecnológico
puede eventualmente tener reacciones cruzadas con proteínas
endógenas.
La presencia de anticuerpos en la mayoría de los casos no
conlleva consecuencias clínicas pero en otros puede:
Incrementar o disminuir su biodisponibilidad;
Impactar negativamente sobre la efectividad del medicamento
Tener consecuencias serias con riesgo de vida.

La inmunogenicidad pueden estar relacionada con:
Secuencia de aminoácidos; glicosilación; pureza (material celular patrón)
del biofármaco
Formulación, estabilidad y almacenamiento del producto
Dosis, intervalo de dosis y vía de administración
Sistema Inmune del Huésped
En la evaluación de la inmunogenicidad hay que considerar:
Ensayos
Sensibilidad
Detección de anticuerpos neutralizadores
Correlación de resultados clínicos
Caracterización de anticuerpos
Considerar la condición inmunológica de los pacientes
Relación de los resultados clínicos

La inmunogenicidad no se puede predecir a
partir de consideraciones de composición,
manufactura y control.
Los problemas de inmunogenicidad son quizás las
razones más convincentes para la imposición de
ensayos clínicos a los biosimilares, la única forma de
evaluarla adecuadamente.

“Biosimilares”
Productos Medicinales Biológicos Similares
Es el término oficial en Europa
A menudo se abrevia como “biosimilares”
Biológicos de continuación (follow-on)
Es el término usado actualmente en EE.UU.
En ocasiones se denominan “proteínas de continuación”
¿Cuál sería el producto de referencia?
¿Cuán similar debe ser un biosimilar?

El “Manual para las Autoridades Reguladoras de Medicamentos” de
la OMS, en la parte de la evaluación de biosimilares, excluye a
los Medicamentos Biotecnológicos por la complejidad
para aplicar el concepto de intercambiabilidad a estos
productos.
“Con algunas clases de producto, incluyendo - más evidentemente - las
formulaciones parenterales de compuestos muy solubles en agua, la
intercambiabilidad está adecuadamente segura por la aplicación de las GMP y
...”
Para otras clases de producto, incluyendo muchos biológicos como las
vacunas, el suero animal, los productos derivados de sangre humana y
plasma y productos fabricados por biotecnología, el concepto de la
intercambiabilidad tiene consideraciones complejas que no se registran en
este documento y estos productos se excluyen por consiguiente de esta
consideración.

Los problemas de inequivalencia terapéutica de los
biosimilares no sólo pueden afectar la salud de los
pacientes sino también, ...
...tener un impacto económico adverso tanto en el
paciente y su entorno, como en el sistema de salud
en general.