gruporegionalandaluzsoc iedad# …seqa.es/graseqa2012/boletin_graseqa_6_2013.pdf ·...
TRANSCRIPT

BOLETÍN GRASEQA
XVIII Reunión de la SEQA: Mini-Symposium Aceite de Oliva
Nº 6 OCTUBRE 2013
Depósito Legal: J 1559-‐2011
ISSN: 2254-‐1241
GRUPO REGIONAL ANDALUZ SOCIEDAD
ESPAÑOLA DE QUÍMICA ANALÍTICA

CONTENIDOS
Pag.
INVESTIGACIÓN GRASEQA (XVIII Reunión de la SEQA: Mini-‐Symposium Aceite de Oliva) Estudio del proteoma de la aceituna y del aceite de oliva Clara Esteve, María Concepción García y Maria Luisa Marina, Universidad de Alcalá de Henares
3
Caracterización de compuestos volátiles en aceite de oliva virgen mediante espectrometría de movilidad iónica Rocío Garrido Delgado, María del Mar Dobao Prieto y Lourdes Arce Jiménez, Universidad de Córdoba
16
GRUPOS DE INVESTIGACIÓN GRASEQA 31 TESIS DOCTORALES GRASEQA 33 NOTICIAS Y ACTUALIDAD GRASEQA 41 AGENDA 44
COMITÉ EDITORIAL
EDITOR: Juan Francisco García Reyes, Universidad de Jaén, Departamento de Química Física y Analítica. Edificio B3. Tel.: 953-‐213040. E-‐mail: [email protected] Universidad de Almería María del Mar Aguilera Luiz ([email protected] )
José Luis Martínez Vidal ([email protected] ) Antonia Garrido Frenich ([email protected])
Universidad de Cádiz Dolores Bellido Milla ([email protected] )
Carlos Moreno ([email protected] ) Enrique Durán Guerrero ([email protected] )
Universidad de Córdoba Rafael Lucena Rodríguez ([email protected] )
Feliciano Priego Capote ([email protected] ) Loreto Lunar ([email protected] )
Universidad de Granada Luis Fermín Capitán Vallvey ([email protected] )
Luis Cuadros Rodríguez ([email protected] ) Universidad de Huelva Jose Luis Gómez Ariza ([email protected] )
Tamara García Barrera ([email protected] ) Macarena González Fernández [email protected]
Universidad de Jaén Antonio Molina Díaz ([email protected] ) Natividad Ramos Martos ([email protected] )
Universidad de Málaga María del Mar López Guerrero ([email protected] ) Miguel Hernández López ([email protected] )
Universidad de Sevilla Alfonso Guiraum ([email protected] )Mercedes Villar Navarro ([email protected]) María Ramos Payán ([email protected] )

INVESTIGACIÓN GRASEQA: XVIII Reunión de la SEQA/Mini-‐Symposium Aceite de Oliva
3
Estudio del proteoma de la aceituna y del aceite de oliva
CLARA ESTEVE*, MARÍA CONCEPCIÓN GARCÍA y MARÍA LUISA MARINA UNIVERSIDAD DE ALCALÁ DE HENARES, Facultad de Química, Departamento de Química Analítica, Ctra.
Madrid-‐Barcelona km 33.6, 28871 Alcalá de Henares. E-‐mail: [email protected]
1. Introducción El olivo es la única especie de la familia Oleaceae con fruto comestible, la oliva o aceituna. Aunque se producen aceitunas a nivel mundial, la mayor parte de la producción sigue realizándose en la región mediterránea. A partir de la aceituna se obtiene el aceite de oliva, muy apreciado por sus características organolépticas y su elevado valor nutricional. Es uno de los pocos aceites vegetales que se obtiene por extracción en frío, sin necesidad de refinado, conservando íntegro su contenido en compuestos minoritarios esenciales. La aceituna, y especialmente el aceite de oliva, son alimentos incluidos entre los más representativos de la dieta mediterránea. Numerosos estudios han puesto de manifiesto que en los países donde existe un elevado consumo de aceite de oliva se da una menor incidencia de enfermedades cardiovasculares, obesidad, síndrome metabólico, diabetes tipo 2 e hipertensión [1, 2].
Son numerosos los trabajos descritos en bibliografía en los que se ha investigado en profundidad la composición de la aceituna y del aceite de oliva, encontrándose estudios sobre ácidos grasos, polifenoles, esteroles, etc. Sin embargo, hasta el momento ha sido muy poco el interés que ha suscitado el estudio del contenido proteico de la aceituna y del aceite de oliva a pesar de que la presencia de proteínas se ha asociado a la estabilidad del aceite frente a la oxidación, a su turbidez y a su potencial alergénico [3-‐5]. De hecho, durante mucho tiempo las proteínas y péptidos presentes en el aceite de oliva se consideraron como impurezas y no fue hasta el año 2001 cuando Hidalgo y col.
[6] las describieron, por primera vez, como componentes minoritarios.
Como se ha comentado anteriormente, se han relacionado las proteínas presentes en la aceituna y el aceite de oliva con su alergenicidad. Aunque la mayor parte de los pacientes alérgicos al olivo lo son al polen, en los últimos años se han descrito de forma creciente casos de dermatitis por contacto con la aceituna o el aceite y casos de alergia asociada a la ingesta de aceituna y de aceite de oliva [7].
La mayor parte de los alérgenos alimentarios son de naturaleza proteica [8], por lo que no es descabellado proponer a las proteínas contenidas en la aceituna y el aceite de oliva como los causantes de dichas alergias. Además, cabe destacar los casos de reacciones alérgicas a la aceituna en pacientes que previamente habían recibido inmunoterapia para la alergia al polen del olivo [9, 10]. Los autores de estos trabajos sugieren la presencia de una estructura común entre alérgenos presentes en el polen y en el fruto. A este fenómeno se le conoce como reactividad cruzada, y se han descrito numerosos casos entre polen y alimentos de origen vegetal [11]. Aunque los alérgenos del polen del olivo han sido ampliamente estudiados, identificando hasta el momento un total de doce (Ole e 1-‐12), tan solo un alérgeno ha sido descrito en la pulpa del fruto, recibiendo el nombre de Ole e 13 [7].
Las proteínas en la aceituna están distribuidas en la piel, la pulpa y la semilla. El contenido proteico de la semilla es más elevado que en el resto del fruto y, por ello, ésta ha sido propuesta como una importante fuente de proteínas [12, 13]. La semilla de la aceituna contiene mayoritariamente proteínas de almacenamiento y oleosinas, llegando a

BOLETÍN GRASEQA Nº 6 OCTUBRE 2013
4
representar un 70% del total del contenido proteico [13]. En cuanto al aceite de oliva, el número de estudios que describen la presencia de proteínas en el mismo es muy limitado. Las proteínas pueden pasar del fruto al aceite durante el prensado. El proceso de extracción del aceite incluye habitualmente un proceso de filtrado para eliminar los sólidos en suspensión y la humedad [14], aunque no existen resultados concluyentes de su influencia sobre el contenido de proteínas [4, 15]. Sin embargo, sí que se han encontrado diferencias entre el contenido de proteínas de aceites refinados y no refinados [16].
2. Extracción de las proteínas de la aceituna y del aceite de oliva En comparación con otros organismos, la extracción de proteínas de la aceituna es especialmente problemática debido a la naturaleza grasa de su matriz y al elevado contenido en compuestos interferentes como lípidos, polifenoles o flavonoides [17, 18], que pueden interferir en gran medida en la extracción e identificación de las proteínas. Además, las proteínas de la aceituna se encuentran en muy baja concentración en comparación con otros componentes de la misma, habiéndose estimado unas concentraciones de 1,5-‐18 mg/g [17, 19-‐21] y de 32 mg/g [13], para la pulpa y para el hueso, respectivamente.
La extracción de las proteínas del hueso o de la semilla se ha llevado a cabo siguiendo dos estrategias generales. La extracción de las proteínas totales o de las proteínas de almacenamiento de la semilla (SSPs) se ha realizado generalmente empleando un tampón Tris(hidroximetilaminometano)-‐ácido clorhídrico (Tris) a pH 6,8-‐7,5 en presencia de diferentes aditivos que favorecen la extracción proteica como dodecilsulfato sódico (SDS), ditiotreitol (DTT), 2-‐mercaptoetanol o urea [22-‐25]. Con el fin de mejorar su rendimiento se ha incluido el uso de una sonda de ultrasonidos focalizada para facilitar la extracción [22]. A continuación, las proteínas se aislan por precipitación con acetona y centrifugación. Por otro lado, la
extracción de las oleosinas se ha realizado homogeneizando la muestra con un tampón HEPES (ácido 4-‐(2-‐Hidroxietil)-‐1-‐Pireracinil-‐Etanosulfónico) a pH 7,5 en presencia de sacarosa, sales, complejos de coordinación y ácido ascórbico [13, 26, 27]. De esta forma, se consigue extraer una capa grasa que contiene los oleosomas, los cuales son seguidamente aislados y purificados mediante la eliminación de los ácidos grasos y triacilgliceroles por adición de un disolvente orgánico como una mezcla cloroformo:metanol (MeOH) [13, 26] o dietiléter [27].
La extracción de las proteínas de la pulpa de la aceituna presenta dos dificultades adicionales: una menor concentración de proteínas en comparación con la semilla y la presencia de una elevada cantidad de compuestos interferentes. De hecho, se ha comprobado que cuando se utilizan tampones acuosos en la extracción y disolventes orgánicos en la precipitación, las proteínas aisladas vienen acompañadas de compuestos de bajo peso molecular como los polifenoles [21].
Wang y col. [21] desarrollaron un método general para la extracción de proteínas de tejidos vegetales recalcitrantes combinando lavados con ácido tricloroacético (TCA)/acetona y MeOH para eliminar los interferentes seguidos de una extracción de las proteínas con fenol. Además, en nuestro grupo de investigación se ha realizado una comparativa de diferentes métodos de extracción de las proteínas de la pulpa empleando tres procedimientos distintos: la eliminación de los compuestos interferentes previa a la extracción de las proteínas mediante la realización de lavados con acetona/agua/TCA, la eliminación de los compuestos interferentes tras la extracción de las proteínas mediante la precipitación de estas en un medio orgánico y una combinación de ambas estrategias [28]. En base a estos tres procedimientos, se diseñaron un total de siete métodos de extracción distintos. El método que permitió una extracción de las proteínas simultánea a una eliminación de compuestos indeseados constaba de un paso de pre-‐lavado con TCA/acetona seguido de una extracción con

INVESTIGACIÓN GRASEQA: XVIII Reunión de la SEQA/Mini-‐Symposium Aceite de Oliva
5
Tris/SDS/DTT y precipitación de las proteínas con acetona. Por otro lado, se ha aplicado también a la extracción de las proteínas de la pulpa el mismo método utilizado para la semilla, basado en el aislamiento de los oleosomas mediante el uso de cloroformo/MeOH [6, 20]. Además se han propuesto un método para la extracción específica de la taumatina, de gran interés por su alergenicidad [7], en el que el fruto se desgrasa con éter-‐etanol, las proteínas se extraen con tampón fosfato y después se aíslan mediante diálisis [29].
En cuanto al aceite de oliva, la ausencia de metodologías que se puedan aplicar a una matriz de naturaleza tan apolar unido a la extremadamente baja concentración de proteínas (estimada entre 0,05 y 2,40 mg/kg [4, 6, 16, 26]) en relación a la presencia de compuestos interferentes han impedido que, a día de hoy, se tenga certeza sobre la identidad de las especies proteicas que pasan de la aceituna al aceite de oliva [30].
Tradicionalmente, la extracción de las proteínas del aceite de oliva se ha llevado a cabo haciendo una primera extracción con disolventes acuosos/orgánicos seguida de un paso de aislamiento de las proteínas. En el método de extracción más ampliamente utilizado, las proteínas se precipitaban a baja temperatura con acetona [6, 26, 31] o una mezcla de acetona:hexano (1:1, v/v) [16]. Seguidamente, las proteínas precipitadas se separaban del sobrenadante mediante la utilización de un filtro Whatman o por centrifugación.
Otros autores han empleado también este método introduciendo algunas modificaciones [4, 32, 33]. Sin embargo, la utilización de un filtro Whatman como método para aislar las proteínas se ha puesto en entredicho, tras observar el mismo perfil cuando se realizaba un blanco del filtro [34]. Por otro lado, Martín-‐Hernández y col. [16] evaluaron cinco métodos diferentes de extracción de las proteínas de diversos aceites. Todos ellos estaban basados en la adición de diferentes disolventes orgánicos o mezclas de disolventes que contenían acetona, hexano, MeOH, isopropanol y/o agua, estableciéndose
como método más adecuado el basado en la extracción con acetona:hexano (1:1, v/v). Por último, en nuestro grupo de investigación se ha hecho una comparación de diferentes métodos convencionales, incluyendo la extracción líquido-‐líquido en condiciones desnaturalizantes y la precipitación con diferentes disolventes orgánicos [34]. Los extractos proteicos obtenidos mediante la utilización de estos métodos de extracción mostraron la presencia de proteínas en un intervalo de masas de hasta 30 kDa. La comparación de este perfil con los obtenidos a partir de la semilla y la pulpa de la aceituna, todas en un rango de hasta 50 kDa, permitió observar la posible correspondencia de las proteínas del aceite con las de la pulpa y la semilla.
Una herramienta adicional en la extracción de proteínas de matrices vegetales o alimentarias es el empleo de enzimas, que ha permitido una separación más eficiente de las mismas de otros compuestos como grasas, carbohidratos o aromas. Por ejemplo, la extracción asistida por la enzima alcalasa ha sido empleada en la extracción de las proteínas del orujo, uno de los residuos sólidos que quedan tras la obtención del aceite de oliva. En este estudio se observó que el empleo de alcalasa permitía aumentar el rendimiento de la extracción de las proteínas entre un 5 y un 30% [35]. Sin embargo, tanto en el caso del hueso [22] como de la pulpa [28], no se ha conseguido mejorar la extracción de las proteínas mediante el uso de enzimas, a pesar de haber utilizado enzimas de naturaleza proteasa, carbohidrasa y lipasa.
Cuando lo que se ha querido llevar a cabo ha sido un estudio proteómico, en el interés se focaliza sobre las proteínas de baja abundancia, han aparecido nuevas complicaciones. Las proteínas expresadas en una célula tienen un intervalo dinámico de concentraciones muy grande que complica el análisis proteómico [36, 37]. Por ejemplo, en las semillas, proteínas como las de almacenamiento (SSPs) pueden estar presentes en niveles de 105-‐107 moléculas por célula. Estas proteínas de alta abundancia dificultan la detección de las proteínas de baja

BOLETÍN GRASEQA Nº 6 OCTUBRE 2013
6
abundancia cuya concentración se encuentra por debajo de las 10-‐100 moléculas por célula.
Son numerosas las técnicas desarrolladas para el enriquecimiento de muestras utilizando diferentes biomoléculas como anticuerpos, proteínas, péptidos y nucleótidos [38]. Mientras que estas técnicas se basan en la substracción de las proteínas de alta abundancia, existen otras técnicas basadas en el enriquecimiento de las proteínas de baja bundancia, como en el uso de bibliotecas de péptidos [39]. Existe una biblioteca de ligandos peptídicos que se encuentra disponible comercialmente con el nombre de ProteoMiner (Bio-‐Rad) [40].
Se trata de una biblioteca combinacional de ligandos peptídicos (CPLLs) consistente en una colección de hexapéptidos sintéticos unidos covalentemente a un soporte cromatográfico esférico poroso. Cuando esta biblioteca de péptidos se incuba con una mezcla de proteínas cada hexapéptido interacciona específicamente con una proteína, en función de sus propiedades físico-‐químicas.
Las especies que se encuentran en exceso rápidamente saturan su ligando correspondiente, mientras que las especies poco o muy poco abundantes quedan absorbidas completamente. Las proteínas no enlazadas se eliminan con un paso de lavado, mientras que las proteínas unidas a las esferas son después recuperadas. Así se consigue, de forma simultánea, la eliminación de las proteínas de alta abundancia y el enriquecimiento de las proteínas de baja abundancia, disminuyendo el intervalo dinámico de concentración de las proteínas.
La extracción de las proteínas de la semilla y de la pulpa de la aceituna utilizando CPLLs se ha llevado a cabo empleando diferentes bibliotecas comerciales y fabricadas en el laboratorio del Prof. Righetti (Politécnico de Milán), a pHs 7,4 o pH 2,2 en presencia de TFA al 0,1% (v/v) [41].
Las incubaciones se realizaron durante toda la noche y seguidamente las proteínas se recuperaron empleando un tampón que contenía SDS. En comparación con el método de extracción convencional, la captura con CPLLs permitía aumentar de forma significativa el número de proteínas extraídas, especialmente en el caso de la pulpa. Tal y como se muestra en la Figura 1, mientras que para la muestra control se observa una única banda, la muestra correspondiente a la captura con CPLLs a pH 2,2 en presencia de 0,1% de TFA muestra un perfil con un mayor número de bandas comprendidas entre pesos moleculares de 10 y 100 kDa. Este enriquecimiento en el perfil proteico se verá después reflejado en el número de proteínas identificadas mediante espectrometría de masas (MS).
En el caso del aceite la extracción con CPLLs, y debido a la ausencia de información acerca de la utilización de esta metodología para la captura de proteínas en matrices tan lipídicas como el aceite de oliva, se emplearon dos estrategias distintas: extracción directa de las proteínas a partir del aceite y extracción de las proteínas en presencia de micelas agua-‐aceite [34]. Ninguna de las estrategias utilizadas resultó ser adecuada probablemente debido a la naturaleza lipídica de la matriz que impide la interacción CPLLs-‐proteína.

INVESTIGACIÓN GRASEQA: XVIII Reunión de la SEQA/Mini-‐Symposium Aceite de Oliva
7
Figura 1. Gel de SDS-‐PAGE correspondiente a los extractos obtenidos a partir de la pulpa de la aceituna. Comparación del extracto proteico de la pulpa utilizado como control y el extracto tratado con CPLLs a pH 2.2. La tinción se realizó con Coomassie Blue micelar. De la referencia 41, Figura 3.a.
3. Obtención de perfiles proteicos y aplicación a la diferenciación de cultivos La composición de las aceitunas y el aceite de oliva obtenidos a partir de diferentes genotipos de olivo puede variar de forma considerable afectando a sus características y calidad. La antigüedad de este tipo de cultivo, que se cree que es superior a los 6000 años, unido a su hibridación y selección ha provocado que hoy en día se conozcan más de 1500 variedades de olivo en todo el mundo [42], muchas de las cuales pueden considerarse sinónimas (misma variedad con distintos nombres) u homónimas (distintas variedades con el mismo nombre). Este hecho ha provocado gran confusión a la hora de caracterizar este cultivo y ha dificultado las labores de clasificación e identificación del mismo. Además, el elevado precio que pueden alcanzar los aceites de oliva virgen y la existencia de denominaciones de origen han provocado que en los últimos años la
comunidad científica haya realizado considerables esfuerzos por desarrollar diferentes metodologías para llevar a cabo su caracterización y clasificación [43].
Tradicionalmente, la identificación de genotipos de olivo se ha llevado a cabo en base a caracteres morfológicos de la planta (hoja, inflorescencia, fruto, endocarpio, etc.) y/o agronómicos [42]. Además, en los últimos años se han desarrollado técnicas de caracterización más novedosas basadas en el estudio de diferentes marcadores, tanto composicionales como genéticos [43]. Sin embargo, ninguna de estas estrategias ha resultado totalmente satisfactoria para la clasificación varietal del olivo, surgiendo la necesidad de encontrar nuevos marcadores. Teniendo en cuenta que a partir del ADN se expresan las proteínas contenidas en cada organismo, es posible pensar en el potencial que puede presentar el estudio de estas como estrategia alternativa en los estudios de autentificación de aceitunas y aceites de oliva.
250150
KDa
10075
50
37
2520
1510
Mr CPPLspH 2.2CTRL
4567
1
2
3
111098
1213
3
21
250150
KDa
10075
50
37
2520
1510
Mr CPPLspH 2.2CTRL
4567
1
2
3
111098
1213
3
21

BOLETÍN GRASEQA Nº 6 OCTUBRE 2013
8
En los últimos años, la comparación de perfiles, tanto aminoacídicos como proteicos, se han convertido en una herramienta analítica muy útil para la caracterización de distintas variedades de especies vegetales [44, 45]. Estos perfiles se obtienen, principalmente, por técnicas separativas electroforéticas o cromatográficas. Así, el análisis de los aminoácidos resultantes de la hidrólisis ácida de las proteínas extraídas del aceite de oliva, llevado a cabo tanto mediante cromatografía de líquidos de alta eficacia (HPLC) con detector ultravioleta (UV) [46] como con MS [31] ha sido una de las estrategias empleadas para la caracterización varietal del aceite de oliva. Sin embargo, la comparación de perfiles aminoacídicos no resultó ser una estrategia útil en la diferenciación entre variedades genéticas del aceite de oliva.
Por otro lado, las proteínas de la aceituna se han separado mediante HPLC. Las limitaciones habituales de esta técnica en la separación de proteínas, debido a la reducida difusividad de estas [47], se ha visto solventada gracias al uso de columnas de cromatografía líquida de ultra-‐alta eficacia (UPLC) [22, 28]. Estas columnas, basadas en rellenos con partículas con tamaño por debajo de los 2 µm, han permitido un aumento significativo en la eficacia en la separación cromatográfica en comparación con el uso de columnas de sílice convencional, perfusivas, monolíticas y peliculares [22]. Cuando se separaron las proteínas del hueso de la aceituna se emplearon un total de ocho columnas cromatográficas, siendo las columnas de UPLC las únicas que permitieron una buena resolución del perfil proteico [22]. Los perfiles cromatográficos obtenidos mostraban hasta 13 picos con diferentes áreas en función de la variedad.
Se analizaron un total de 91 muestras pertenecientes a 29 variedades diferentes de aceituna. La utilización de técnicas de análisis multivariante permitió encontrar las variables con mayor potencial para diferenciar variedades de aceituna. De este modo, se consiguió la correcta clasificación del 99% de las muestras de aceituna analizadas. Las variedades de aceituna que se analizaron
habían crecido bajo las mismas condiciones agronómicas, demostrándose que las proteínas presentes en el hueso de la aceituna tienen un gran potencial como marcadores de trazabilidad del origen botánico de las aceitunas constituyendo una interesante alternativa a los métodos existentes para diferenciar variedades de este cultivo. En el caso de la pulpa de la aceituna [28], las proteínas se separaban en un tiempo de análisis de 20 min observando tres picos mayoritarios en casi todas las variedades. Aunque el tamaño de estos picos variaba en función de la variedad de aceituna, los perfiles encontrados al inyectar extractos correspondientes a diferentes variedades de aceituna resultaban ser más similares y con menor capacidad de clasificación que los que se obtuvieron a partir del hueso.
La separación de las proteínas tanto del fruto de la aceituna como del aceite de oliva también se ha llevado a cabo mediante electroforesis capilar (CE) [32, 48, 49]. La separación de las proteínas del fruto se realizó, en primer lugar, empleando electroforesis capilar en zona (CZE), lo que permitió obtener un perfil con numerosos picos atribuibles a proteínas [48]. Por un lado se analizaron aceitunas crudas de las variedades Arbequina, Picual y Hojiblanca de dos orígenes geográficos diferentes (Jaén y Toledo), y se observó que, para estas aceitunas, el perfil proteico cambiaba en función del origen botánico, obteniendo también diferencias en el perfil en función del origen geográfico de la aceituna (Figura 2). Además, se utilizó la electroforesis capilar en gel (CGE) empleando un gel diseñado para separar compuestos en un rango efectivo de entre 10 y 225 kDa, permitiendo separar los polifenoles de baja masa molecular de las proteínas de mayor masa molecular [49].
El análisis de los extractos proteicos procedentes de 20 variedades de aceituna cultivadas bajo las mismas condiciones climatológicas permitió observar, en todos los casos, 7 picos electroforéticos correspondientes a proteínas. La aplicación de técnicas de análisis discriminante permitió en este caso la correcta clasificación de 16 de las 20 variedades de aceituna analizadas,

INVESTIGACIÓN GRASEQA: XVIII Reunión de la SEQA/Mini-‐Symposium Aceite de Oliva
9
agrupándose en cuatro grupos cuando se utilizó como factor de clasificación el origen geográfico. Por último, se llevó a cabo la separación de un extracto proteico de aceite de oliva por CZE [32]. El método empleado combinó una preconcentración en el capilar para aumentar la intensidad de la señal con un recubrimiento dinámico del capilar con UltraTrol para evitar las posibles interacciones entre las proteínas y la pared del capilar. Se analizaron los aceites de oliva monovarietales más ampliamente comercializados en el mercado español: Picual, Hojiblanca y Arbequina. En este trabajo se obtuvieron perfiles proteicos en los que se diferenciaban tres zonas en función de su poder discriminativo. La diferenciación de las tres variedades de aceite de oliva monovarietales también fue posible realizando un análisis discriminante. Los resultados obtenidos en este trabajo de investigación muestran, por primera vez, el potencial de las proteínas presentes en el aceite de oliva como marcadores de trazabilidad del origen botánico de los mismos. De esta forma, se obtuvieron hasta siete picos atribuibles a proteínas presentes en el aceite oliva, pudiendo ser clasificados de acuerdo a la variedad de aceituna de la que procedían.
4. Estudio de la aceituna y del aceite de oliva mediante técnicas proteómicas Con el fin de aumentar la información disponible sobre las proteínas contenidas tanto en la aceituna como en el aceite de oliva se han aplicado para su identificación estrategias proteómicas. La proteómica es un estudio a gran escala de las proteínas presentes en un organismo en un determinado momento y en unas determinadas condiciones [50]. La proteómica de plantas representa solo un pequeño porcentaje de los trabajos publicados en proteómica hasta el momento, estando limitado por dos grandes problemas: la ausencia de bases de datos de los genomas de los organismos de los que proceden [51] y la dificultad de obtener extractos proteicos de alta pureza.
Hasta el momento no se habían sido muchos los esfuerzos realizados por identificar las proteínas presentes tanto en la aceituna como en el aceite de oliva. En bibliografía tan solo existían dos ejemplos en los que se había empleado la degradación de Edman, un método de secuenciación de aminoácidos, para la identificación parcial de las SSPs de la semilla [23] y la taumatina en la pulpa de la aceituna [29]. Sin embargo, hasta la fecha no existía ningún ejemplo en el que se hubieran empleado técnicas modernas de identificación de proteínas ni en la aceituna ni en el aceite de oliva.
En los últimos años se han aplicado, por primera vez, técnicas proteómicas basadas en el uso de MS a la identificación de las proteínas contenidas en la aceituna, permitiendo estudiar en profundidad el proteoma de la misma. Debido a que solo una mínima parte de su genoma se encuentra secuenciado, la mayor parte de la identificación de proteínas debe realizarse por homología con secuencias de proteínas de organismos con genomas ya secuenciados, como arabidopsis thaliana, glycine max o vitis vinifera. En los trabajos de proteómica que se han aplicado al estudio de las proteínas de la aceituna y del aceite de oliva se ha empleado
bottom-‐upcabo, el primer paso que se debe llevar a cabo es la separación de la proteína o proteínas de interés del resto de la muestra mediante técnicas electroforéticas, como la electroforesis en gel de poliacrilamida con SDS (SDS-‐PAGE) [41, 52]. Seguidamente la proteína se digiere utilizando un agente proteolítico, que generalmente es la enzima tripsina, que produce cortes predecibles y específicos en su secuencia aminoacídica. A continuación, la mezcla de péptidos característicos producidos se analiza mediante MS. Han sido dos las estrategias de MS empleadas en la identificación de proteínas. La primera es la conocida como peptide-‐mass fingerprint [53], la cual se basa en la medida de todas las masas de los péptidos generados durante la digestión, y para la cual la técnica empleada ha sido habitualmente ionización por desorción láser

BOLETÍN GRASEQA Nº 6 OCTUBRE 2013
10
asistida por matriz-‐tiempo de vuelo (MALDI-‐TOF.
Figura 2. Perfil proteico obtenido para muestras de aceituna de las variedades Arbequina, Picual y Hojiblanca de dos orígenes geográficos diferentes (Toledo y Jaén), extraídas siguiendo una extracción con cloroformo/MeOH y precipitación de las proteínas. Los picos asignados a proteínas están marcados con (*). De la referencia 48, Figura 4.
Esta estrategia ha sido útil cuando el interés era el de identificar una sola proteína y cuando la secuencia de la misma se
encontraba en las bases de datos [52]. La segunda estrategia de MS que se ha empleado en la identificación de proteínas de
0 5 10 15 20 25 30 35 40 45
0
10
20
30
40
50
60
70
80
** ***
*** *
*
** **
*****
*
******
*
* *
Hojiblanca
Picual
mU
A
min
Arbequina
*
Geographical origin: Jaén
0 5 10 15 20 25 30 35 40 45
0
10
20
30
40
50
60
70
80
*
*****
***
* **
* * * * *
***
*
*
Hojiblanca
Picual
Arbequina
mU
A
min
Geographical origin: Toledo

INVESTIGACIÓN GRASEQA: XVIII Reunión de la SEQA/Mini-‐Symposium Aceite de Oliva
11
la aceituna y del aceite de oliva es la conocida como peptide fragment fingerprinting, para la cual el sistema más empleado es la ionización por electronebulización-‐espectrometría de masas en tándem (ESI-‐MS/MS), acoplado a un sistema de HPLC que regula la entrada de péptidos en el espectrómetro de masas. Esta técnica se basa en la fragmentación de los péptidos obteniendo información estructural de los mismos a partir de la cual es posible la identificación de la proteína por comparación con las bases de datos [54] como SwissProt o NCBInr [55] gracias al uso de motores de búsqueda como MASCOT o SEQUEST.
Así, en el primer estudio proteómico que se realizó del fruto de la aceituna, la separación mediante SDS-‐PAGE del extracto obtenido permitió observar una banda mayoritaria con una masa molecular de 24 kDa [52]. Esta banda fue cortada, digerida con tripsina y analizada mediante MALDI-‐TOF y nanoLC-‐ESI-‐MS/MS. El análisis de MALDI-‐TOF permitió determinar que la proteína mayoritaria de la pulpa de la aceituna pertenece a la familia de las taumatinas y el análisis mediante nanoLC-‐MS/MS permitió además detectar variaciones aminoacídicas de la misma en función de la variedad. Se identificaron además en esta banda otras proteínas de baja abundancia, destacando la presencia de la proteína Cu/Zn-‐superóxido dismutasa, que ha sido descrita como alérgeno en el polen del olivo, con el nombre Ole e 5 [7] estando además implicada en la defensa celular debido a su gran capacidad antioxidante [56]. Además, muchos otros péptidos mostraban homología con péptidos de proteínas de otras plantas, pudiéndose identificar hasta un total de 49 proteínas, de entre las cuales destaca la presencia de numerosas proteínas ribosomales.
Por otro lado, se llevó a cabo la captura de las proteínas de baja y muy baja abundancia procedentes de la pulpa y la semilla de aceituna mediante el empleo de CPLLs con diferentes propiedades químicas [41]. Las proteínas aisladas se separaron por SDS-‐PAGE, se digirieron con tripsina y se analizaron mediante nanoLC-‐MS/MS. En esta ocasión, se consiguió identificar un total de 231 proteínas para la pulpa, de las cuales 172
fueron identificadas gracias al empleo de las CPLLs, lo que supone un aumento del 400% respecto a las proteínas identificadas en la muestra control. De entre las proteínas identificadas 9 fueron asignadas a proteínas de la aceituna, de entre las que destacan la presencia de una Cu/Zn-‐SOD, una -‐1,3-‐glucanasa y una isoflavona reductasa, las cuales han sido identificadas como los alérgenos del polen Ole e 5, 9 y 12 [7] y podrían dar explicación al caso de alergenicidad cruzada entre polen y fruto del olivo. En el caso de la semilla fue posible identificar un total de 61 proteínas, siendo la primera ocasión en la que se realiza un estudio en profundidad de las proteínas contenidas en la semilla. De entre las proteínas identificadas destacan la presencia de un gran número de isoformas de histonas, globulinas y oleosinas. En este punto vale la pena resaltar el hecho de que aunque el contenido de proteínas en la semilla de aceituna sea superior al que presenta la pulpa, el número de proteínas diferentes que constituyen el proteoma de la pulpa es superior al que se observa en el proteoma de la semilla
De este estudio cabe destacar el hecho de que la mayor parte de la identificación de las proteínas solo pudo realizarse por homología con proteínas de otras especies vegetales como Populus trichocarpa, Zea mays o Vitis vinífera. De hecho, tan solo el 1,4% de las proteínas de la semilla y el 4% de las proteínas de la pulpa fueron asignadas a proteínas de Olea europaea, poniendo de manifiesto la dificultad de trabajar con organismos cuyos genomas no han sido totalmente secuenciados. Además, es importante resaltar el hecho de que entre todas las proteínas identificadas, diez fueron observadas, simultáneamente, en el hueso y la pulpa de la aceituna. De entre ellas destacan la variante larga de GluB-‐5, una glutenina y dos globulinas 11S.
El análisis de la funcionalidad de las proteínas encontradas en la semilla y la pulpa de aceituna se ha llevado a cabo gracias al software QuickGO disponible en la red [57]. Este análisis ha permitido observar claras diferencias entre ambos proteomas [41].

BOLETÍN GRASEQA Nº 6 OCTUBRE 2013
12
Aunque existen algunas proteínas presentes en el hueso y en la pulpa con las mismas funcionalidades, también se observan otras proteínas con funcionalidades que se dan de forma exclusiva en uno de ellos. Así, por ejemplo, solo en la semilla se encontraron proteínas de unión a ADN, de regulación y transcripción del ADN y proteínas de membrana y solo en la pulpa de aceituna se encontraron proteínas ribosomales, proteínas implicadas en procesos metabólicos y proteínas del cloroplasto.
En relación al aceite de oliva, la utilización de aceites de oliva virgen extra sin filtrar permitió observar algunas bandas poco intensas por SDS-‐PAGE [34]. La digestión tríptica de estas bandas y su posterior análisis mediante nanoLC-‐LTQ-‐Orbitrap-‐XL ha permitido, por primera vez, identificar un total de diez proteínas en el aceite de oliva. La asignación de los espectros se realizó por homología con otras especies. Al igual que había ocurrido anteriormente en los casos de la pulpa y la semilla de la aceituna [41], hubo una gran cantidad de espectros que no pudieron ser asignados a ninguna proteína debido a que el genoma del olivo no ha sido aún secuenciado. De estas diez proteínas, tres eran proteínas de plantas y una de ellas se correspondía con una proteína identificada previamente en el hueso, lo que apoya la idea de que las proteínas del fruto pueden pasar al aceite de oliva durante su elaboración y sobrevivir en el mismo. Las otras siete proteínas identificadas fueron asignadas a bacterias. La presencia de estas proteínas puede explicarse teniendo en cuenta que es muy común encontrar bacterias en alimentos obtenidos mediante procedimientos como los utilizados en la producción de aceite o vino. Aunque este trabajo es estudio preliminar, se trata de un hallazgo único teniendo en cuenta que, hasta la fecha, nunca se había conseguido identificar mediante MS proteínas presentes en un aceite vegetal.
5. Conclusiones A pesar de la escasa atención que se había prestado hasta el momento, ha sido en los últimos años cuando la comunidad científica
se ha centrado en el estudio de las proteínas de la aceituna y del aceite de oliva. La dificultad de extraer estas proteínas, debido a la complejidad de la matriz con un elevado contenido de lípidos y compuestos interferentes y una concentración baja de proteínas, ha hecho que una gran parte del esfuerzo de estos estudios se haya puesto en el desarrollo de nuevas metodologías de extracción más sencillas y eficientes en cada uno de los nuevos trabajos. Además, la aplicación la aplicación al estudio de dichas proteínas de técnicas de aislamiento novedosas como son las CPLLs ha permitido profundizar en el conocimiento de las mismas. El uso de estas proteínas como marcadores genéticos, mediante la obtención de perfiles proteicos mediante técnicas electroforéticas y cromatográficas, ha demostrado ser una herramienta muy útil para la caracterización varietal y botánica tanto de la aceituna como del aceite de oliva. Finalmente, la aplicación de técnicas proteómicas ha permitido dar un gran paso adelante en el conocimiento de estas proteínas, permitiendo, por primera, identificar un gran número de las mismas, y obteniendo así una visión más amplia del proteoma de estas matrices. Agradecimientos M. C. García agradece al Ministerio de Ciencia e Innovación y al Ministerio de Economía y Competitividad, la concesión de los proyectos CTQ2009-‐11252 y AGL2012-‐36362, respectivamente. M. L. Marina y M. C. García agradecen a la comunidad de Madrid y al programa europeo FEDER la concesión del proyecto S-‐2009/AGR-‐1464. C. Esteve agradece a la Universidad de Alcalá y al Ministerio de Educación, Cultura y Deporte (España) la concesión de dos contratos pre-‐doctorales.
Referencias [1] H.M. Roche, M.J. Gibney, A. Kafatos, A. Zampelas, C.M. Williams, Beneficial properties of olive oil, Food Res. Int. 33 (2000) 227. [2] J. López-‐Miranda, F. Pérez-‐Jiménez, E. Ros, R. De Caterina, L. Badimon, M.I. Covas, E. Escrich, J.M. Ordovás, F. Soriguer, R. Abia, C. Alarcon de la Lastra, M. Battino, D. Corella, J. Chamorro-‐Quirós, J. Delgado-‐Lista, D. Giugliano, K. Esposito, R. Estruch, J.M. Fernández-‐Real, J.J. Gaforio, C. La Vecchia, D. Lairon, F. López-‐Segura, P. Mata, J.A. Menendez, F.J. Muriana, J. Osada, D.B. Panagiotakos, J.A. Paniagua, P. Pérez-‐Martínez, J.

INVESTIGACIÓN GRASEQA: XVIII Reunión de la SEQA/Mini-‐Symposium Aceite de Oliva
13
Perona, M.A. Peinado, M. Pineda-‐Priego, H.E. Poulsen, J.L. Quiles, M.C. Ramírez-‐Tortosa, J. Ruano, L. Serra-‐Majem, R. Sola, M. Solanas, V. Solfrizzi, R. De la Torre-‐Fornell, A. Trichopoulou, M. Uceda, J.M. Villalba-‐Montoro, J.R. Villar-‐Ortiz, F. Visioli, N. Yiannakouris, Olive oil and health: Summary of the II international conference on olive oil and health consensus report, Jaen and Cordoba (Spain) 2008, Nutr. Metab. Cardiovasc. Dis. 20 (2008) 284. [3] M.D. Georgalaki, T.G. Sotiroudis, A. Xenakis, The presence of oxidizing enzyme act in virgin olive oil. JAOCS 75 (1998) 155. [4] A. Koidis, D. Boskou, The contents of proteins and phospholipids in cloudy (veiled) virgin olive oils, Eur. J. Lipid Sci. Technol. 108 (2006) 323. [5] F.J. Hidalgo, R. Zamora, Peptides and proteins in edible oils: Stability, allergenicity, and new processing trends, Trends Food Sci. Technol. 17 (2006) 56. [6] F.J. Hidalgo, M. Alaiz, R. Zamora, Determination of peptides and proteins in fats and oils, Anal. Chem. 73 (2001) 698. [7] C. Esteve, C. Montealegre, M.L. Marina, M.C. García, Analysis of olive allergens, Talanta. 92 (2012) 1. [8] D.G. Ebo, W.J. Stevens, Ige-‐mediated food allergy -‐ Extensive review of the literature, Acta Clin. Belg. 56 (2001) 234. [9] M. Unsel, O. Ardeniz, N. Mete, R. Ersoy, A.Z. Sin, O. Gulbahar, A. Kokuludag, Food Allergy due to Olive, J. Investig. Allergol. Clin. Immunol. 19 (2009) 497. [10] F. Feo Brito, P. Mur Gimeno, B. Bartolomé, A. Castro, F. Guerra, Anaphylaxis Due to Olive Fruit After Pollen Immunotherapy, J. Investig. Allergol. Clin. Immunol. 21 (2001) 160. [11] S. Vieths, S. Scheurer, B. Ballmer-‐Weber, Current understanding of cross-‐reactivity of food allergens and pollen, Ann. N. Y. Acad. Sci. 964 (2002) 47. [12] S. Miladi, D.M. Hegsted, Olive Kernels as a Source of Protein, Nutr. Rep. Int. 9 (1974) 117. [13] G. Rodríguez, A. Lama, R. Rodríguez, A. Jiménez, R. Guillen, J. Fernández-‐Bolanos, Olive stone an attractive source of bioactive and valuable compounds, Bioresour. Technol. 99 (2008) 5261. [14] J. Lozano-‐Sánchez, L. Cerretani, A. Bendini, A. Segura-‐Carretero, A. Fernández-‐Gutiérrez, Filtration process of extra virgin olive oil: effect on minor components, oxidative stability and sensorial and physicochemical characteristics, Trends Food. Sci. Technol. 21 (2010) 201. [15] G.A.E. Wong, C.M. King, Occupational allergic contact dermatitis from olive oil in pizza making, Contact Derm. 50 (2004) 102. [16] C. Martín-‐Hernández, S. Benet, L. Obert, Determination of proteins in refined and nonrefined oils, J. Agric. Food Chem. 56 (2008) 4348. [17] C.D. Stalikas, Extraction, separation, and detection methods for phenolic acids and flavonoids, J. Sep. Sci. 30 (2007) 3268. [18] M. Savarese, E. De Marco, R. Sacchi, Characterization of phenolic extracts from olives (Olea europaea cv. Pisciottana) by electrospray ionization mass spectrometry, Food Chem. 105 (2007) 761. [19] R. Briante, M. Patumi, S. Limongelli, F. Febbraio, C. Vaccaro, A. Di Salle, F. La Cara, R. Nucci, Changes in
phenolic and enzymatic activities content during fruit ripening in two Italian cultivars of Olea europaea L, Plant Sci. 162 (2002) 791. [20] R. Zamora, M. Alaiz, F.J. Hidalgo, Influence of cultivar and fruit ripening on olive (Olea europaea) fruit protein content, composition, and antioxidant activity, J. Agric. Food Chem. 49 (2001) 4267. [21] W. Wang, R. Vignani, M. Scali, M. Cresti, A universal and rapid protocol for protein extraction from recalcitrant plant tissues for proteomic analysis, Electrophoresis 27 (2006) 2782. [22] C. Esteve, C. Del Río, M.L. Marina, M.C. García, First ultraperformance liquid chromatography based strategy for profiling intact proteins in complex matrices: application to the evaluation of the performance of olive (Olea europaea L.) stone proteins for cultivar fingerprinting, J. Agric. Food Chem. 58 (2010) 8176. [23] W. Wang, J. De Dios Alché, M.I. Rodríguez-‐García. Characterization of olive seed storage proteins, Acta Physiol. Plant 29 (2007) 439. [24] J. De Dios Alché, J.C. Jiménez-‐López, W. Wang, A.J. Castro-‐López, M.I, Rodríguez-‐García, Biochemical characterization and cellular localization of 11S type storage proteins in olive (Olea europaea L.) seeds, J. Agric. Food Chem. 54 (2006) 5562. [25] W. Wang, J. De Dios Alché, A.J. Castro, M.I. Rodríguez-‐García, Characterization of seed storage proteins and their synthesis during seed development in Olea europaea, Int. J. Dev. Biol. 45 (2001) S63. [26] F.J. Hidalgo, M. Alaiz, R. Zamora, Low molecular weight polypeptides in virgin and refined olive oils, JAOCS 79 (2002) 685. [27] J.H.E. Ross, J. Sánchez, F. Millán, D.J. Murphy, Differential Presence of Oleosins in Oleogenic Seed and Mesocarp Tissues in Olive (Olea-‐Europaea) and Avocado (Persea-‐Americana), Plant Sci. 93 (1993) 203. [28] C. Esteve, C. Del Río, M.L. Marina, M.C. García, Development of an ultra-‐high performance liquid chromatography analytical methodology for the profiling of olive (Olea europaea L.) pulp proteins, Anal. Chim. Acta 690 (2011) 129. [29] O. Palomares, M. Alcantara, J. Quiralte, M. Villalba, F. Garzon, R. Rodríguez, Airway disease and thaumatin-‐like protein in an olive-‐oil mill worker, N. Engl. J. Med. 358 (2008) 1306. [30] C. Montealegre, C. Esteve, M.C. García, M.L. Marina, Proteins in olive fruit and oil, Crit. Rev. Food Sci. Nutr. doi:10.1080/10408398.2011.598639. [31] M.J. Lerma-‐García, G. Ramis-‐Ramos, J.M. Herrero-‐Martínez, E.F. Simó-‐Alfonso, Classification of vegetable oils according to their botanical origin using amino acid profiles established by direct infusion mass spectrometry, Rapid Commun. Mass Spectrom. 21 (2007) 3751. [32] C. Montealegre, M.L. Marina, C. García-‐Ruiz, Separation of proteins from olive oil by CE: An approximation to the differentiation of monovarietal olive oils, Electrophoresis 31 (2010) 2218. [33] V. Concha-‐Herrera, M.J. Lerma-‐García, J.M. Herrero-‐Martínez, E.F. Simó-‐Alfonso, Classification of vegetable oils according to their botanical origin using amino acid profiles established by High Performance Liquid

BOLETÍN GRASEQA Nº 6 OCTUBRE 2013
14
Chromatography with UV-‐vis detection: A first approach, Food Chem. 120 (2010) 1149.
P.G. Righetti, Analytical approaches for the characterization and identification of olive (Olea europaea) oil proteins, J. Agric. Food Chem, 2013,. DOI: 10.1021/jf4028359. [35] J. Vioque, A. Clemente, R. Sánchez-‐Vioque, J. Pedroche, F. Millán, Effect of Alcalase (TM) on olive pomace protein extraction, JAOCS 77 (2000) 181. [36] G.L. Corthals, V.C. Wasinger, D.F. Hochstrasser, J.C. Sánchez, The dynamic range of protein expression: A challenge for proteomic research, Electrophoresis 21 (2000) 1104. [37] S. Surinova, R. Schiess, R. Huettenhain, F. Cerciello, B. Wollscheid, R. Aebersold, On the Development of Plasma Protein Biomarkers, J. Proteome Res. 10 (2011) 5. [38] X. Fang, W. Zhang, Affinity separation and enrichment methods in proteomic analysis, J. Proteomics 71 (2008) 284. [39] V. Thulasiraman, S.H. Lin, L. Gheorghiu, J. Lathrop, L. Lomas, D. Hammond, E. Boschetti, Reduction of the concentration difference of proteins in biological liquids using a library of combinatorial ligands, Electrophoresis 26 (2005) 3561. [40] E. Boschetti, P.G. Righetti, The ProteoMiner in the proteomic arena: A non-‐depleting tool for discovering low-‐abundance species, J. Proteomics 71 (2008) 255.
Citterio, P.G. Righetti, Identification of olive (Olea europaea) seed and pulp proteins by nLC-‐MS/MS via combinational peptide ligand libraries, J. Proteomics 75 (2012) 2396. [42] L. Rallo, D. Barranco, J.M. Caballero, C. del Río, A. Martín, J. Tous, I. Trujillo, Variedades de Olivo en España. Madrid: Mundi-‐Prensa; 2005. [43] C. Montealegre, M.L. Marina, C. García-‐Ruiz, Traceability Markers to the Botanical Origin in Olive Oils, J. Agric. Food Chem. 58 (2010) 28. [44] J.M. Rodríguez-‐Nogales, M.C. García, M.L. Marina, Analysis of European and North American maize inbred and hybrid lines by monolithic and perfusion reversed-‐phase high-‐performance chromatography and multivariate analysis, J. Agric. Food Chem. 54 (2006) 8702. [45] W. Seilmeier, I. Valdez, E. Mendez, H. Wieser, Comparative investigations of gluten proteins from different wheat species -‐ II. Characterization of omega-‐gliadins, Eur. Food Res. Technol. 212 (2001) 355.
[46] F.J. Hidalgo, M. Alaiz, R. Zamora, Low molecular weight prolypeptides inn virgin and refined olive oils, JAOCS 79 (2002) 685. [47] A.L. Capriotti, C. Cavaliere, P. Foglia, R. Samperi, A. Lagana, Intact protein separation by chromatographic and/or electrophoretic techniques for top-‐down proteomics, J. Chromatogr. A 1218 (2011) 8760. [48] C. Montealegre, M.L. Marina, C. García-‐Ruiz, Separation of Olive Proteins Combining a Simple Extraction Method and a Selective Capillary Electrophoresis (CE) Approach: Application to Raw and Table Olive Samples, J. Agric. Food Chem. 58 (2010) 11808. [49] C. Montealegre, M.C. García, C. del Río, M.L. Marina ML, C. García-‐Ruiz, Separation of olive proteins by capillary gel electrophoresis, Talanta 97 (2012) 420. [50] V.C. Wasinger, S.J. Cordwell, A. Cerpapoljak, J.X. Yan, A.A. Gooley, M.R. Wilkins, M.W. Duncan, R Harris, K.L. Williams, I. Humpherysmith, Progress with Gene-‐Product Mapping of the Mollicutes -‐ Mycoplasma-‐Genitalium, Electrophoresis 16 (1995) 1090. [51] G.K. Agrawal, D. Job, M. Zivy, V.P. Agrawal, R.A. Bradshaw, M.J. Dunn, P.A. Haynes, K.J. van Wijk, S. Kikuchi, J. Renaut, W. Weckwerth, R. Rakwal, Time to articulate a vision for the future of plant proteomics -‐ A global perspective: An initiative for establishing the International Plant Proteomics Organization (INPPO), Proteomics 11 (2011) 1559. [52] C. Esteve, B. Cañas, E. Moreno-‐Gordaliza, C. Del Río, M.C. García, M.L. Marina, Identification of olive (Olea europaea) pulp proteins by matrix-‐assisted laser-‐desorption/ionization time-‐of-‐flight mass spectrometry and nano-‐liquid chromatography tandem mass spectrometry, J. Agric. Food Chem. 59 (2011) 12093. [53] W.J. Henzel, C. Watanabe, J.T. Stults, Protein identification: The origins of peptide mass fingerprinting, J. Am. Soc. Mass Spectrom. 14 (2003) 931. [54] M. Pischetsrieder, R. Baeuerlein, Proteome research in food science, Chem. Soc. Rev. 38 (2009) 2600. [55] A.I. Nesvizhskii, O. Vitek, R. Aebersold, Analysis and validation of proteomic data generated by tandem mass spectrometry, Nat. Meth. 4 (2007) 787. [56] D.D. Mruk, B. Silvestrini, M.Y. Mo, C.Y. Cheng, Antioxidant superoxide dismutase a review: its function, regulation in the tesis, and role in male fertility. Contraception 65 (2002) 305. [57] www.ebi.ac.uk/QuickGO

INVESTIGACIÓN GRASEQA: XVIII Reunión de la SEQA/Mini-‐Symposium Aceite de Oliva
15
Clara Esteve Gil es Doctora en Ciencias Químicas por la Universidad de Alcalá desde marzo de 2013. Se licenció en Química por la Universidad de Valencia en 2008, habiendo disfrutado de una beca Sicue-‐Séneca para cursar un año en la Universidad de Alcalá, donde se especializó en Química Médica. Su tesis doctoral, dirigida por las Dras. Mª Luisa Marina y Mª Concepción García, estuvo centrada en el estudio de las proteínas contenidas en la aceituna y el aceite de oliva, la aplicación de perfiles proteicos a la caracterización varietal del olivo, y la evaluación de las propiedades bioactivas de los hidrolizados de la semilla de la aceituna. Durante la realización de su tesis doctoral tuvo la oportunidad de realizar dos estancias pre-‐doctorales en la Universidad Complutense de Madrid, bajo la dirección del Dr. Benito Cañas, y en el Politécnico de Milán, bajo la dirección del Dr. Pier Giorgio Righetti. Actualmente se encuentra trabajando como investigador post-‐doctoral en el Departamento de Análisis Instrumental y Química Ambiental del CSIC bajo la dirección de las Dras. Belén Gómara y María José González, centrando su trabajo en el desarrollo de métodos para la determinación de contaminantes emergentes relacionados con el envasado y conservación de los alimentos en productos lácteos.
María Concepción García López es Profesora Titular de Química Analítica en la Universidad de Alcalá desde el 2003. En el año 1999 se doctoró en Ciencias Químicas consiguiendo el Premio Extraordinario de Doctorado. Posteriormente, consiguió un contrato Marie Curie para realizar una estancia de un año en el Departamento de Química Analítica y Espectroscopía Aplicada de la Vrije Universiteit Amsterdam. En el año 2002 recibió el Premio para Jóvenes Investigadores de la Universidad de Alcalá. Además de su labor docente, ha dirigido numerosos trabajos de investigación y tesis doctorales y ha participado o sido investigadora principal en diferentes proyectos de investigación financiados con fondos europeos, nacionales o regionales. Es co-‐autora de más de 60 artículos científicos publicados en revistas de prestigio internacional y de 9 capítulos de libro. La investigación realizada durante toda su carrera profesional se ha centrado en el desarrollo de metodologías para la separación, caracterización, determinación e identificación de proteínas y péptidos en alimentos utilizando técnicas cromatográficas y espectrometría de masas.
María Luisa Marina es Catedrática de Química Analítica en la Universidad de Alcalá desde 2004. Se doctoró en Ciencias Químicas en esta Universidad en 1985 obteniendo la máxima calificación para su Tesis Doctoral y el Premio Extraordinario de Doctorado. Fue investigadora posdoctoral en 1986 en el INSTN, CEA, Saclay (Francia). Ha desarrollado una intensa actividad docente e investigadora en el Área de Química Analítica durante los últimos 30 años. Es responsable del grupo de investigación de la
de (micro)-‐de numerosos proyectos de investigación financiados con fondos europeos, nacionales y regionales. Es co-‐ Analysis and Detection in Capillary
l Chemistry Series (Elsevier) y co-‐autora de 20 capítulos de libros publicados por editoriales internacionales de prestigio. El número de artículos científicos publicados en revistas alto prestigio internacional (JCR) del que es co-‐autora supera los 200 y ha participado en numerosos congresos y reuniones científicas tanto de índole nacional como internacional. Su actividad investigadora se enmarca en el desarrollo de estrategias analíticas innovadoras mediante técnicas de separación y micro-‐ y nano-‐separación tanto electroforéticas como cromatográficas acopladas a espectrometría de masas aplicadas a la proteómica, peptidómica y metabolómica y a las separaciones quirales para su implementación en los campos farmacéutico, medioambiental y alimentario.

BOLETÍN GRASEQA Nº 6 OCTUBRE 2013
16
Caracterización de compuestos volátiles en aceite de oliva virgen mediante espectrometría
de movilidad iónica ROCÍO GARRIDO DELGADO, MARÍA DEL MAR DOBAO PRIETO y LOURDES ARCE JIMÉNEZ
UNIVERSIDAD DE CÓRDOBA, Departamento de Química Analítica, Instituto Universitario de Investigación en Química Fina y Nanoquímica (IUIQFN), Campus de Excelencia Internacional
Agroalimentario (ceiA3), Campus de Rabanales, 14071, Córdoba, España.
E-‐mail: Lourde-‐[email protected]
1. Introducción En los últimos años, los consumidores están mostrando un gran interés por conocer la calidad de distintos productos alimenticios de alto valor añadido; entre éstos, el aceite de oliva es un alimento muy apreciado por los consumidores debido a sus beneficios para la salud. El Reglamento Europeo Nº 61/2011 establece los criterios para asegurar las características organolépticas en los aceites de oliva vírgenes [1] definidos en el Anexo I del Reglamento (UE) Nº 61/2011 [2]. El método oficial que define los parámetros de calidad que deben tener las distintas categorías de aceite de oliva implica el uso de diferentes métodos de análisis físico-‐químicos además de un análisis sensorial mediante un panel de cata (Figura 1). Entre las características de los paneles de cata para evaluar la calidad del aceite de oliva hay que mencionar que hoy en día, sólo los catadores son capaces de describir el perfil sensorial de una muestra de aceite de oliva [3]. La clasificación que proporcionan los paneles de cata se establece en base a dos criterios como son la presencia o ausencia del atributo frutado y la intensidad total de defectos. Dicha clasificación establece tres categorías distintas conocidas como virgen extra, AOVE (frutado positivo, mediana del defecto nulo), virgen, AOV (frutado positivo, mediana del defecto entre 0 y 3.5) y lampante, AOL (frutado nulo, mediana del defecto mayor a
3.5). La necesidad actual de llevar a cabo este análisis sensorial para conocer finalmente la calidad de una muestra de aceite de oliva repercute en el coste por análisis y en el tiempo necesario para conocer la categoría de una muestra, lo que se traduce en un problema para la comercialización de una forma rápida, de las muestras envasadas. Así mismo, la evaluación de la calidad del aceite de oliva por paneles tiene algunos inconvenientes añadidos como son la subjetividad del análisis y la falta de paneles acreditados.
Figura 1. Parámetros que hay que analizar por el método official para determinar la calidad del aceite de oliva.
Esta situación se refleja en estos momentos en un gran interés del sector oleico por apoyar el desarrollo de métodos analíticos rápidos, simples y fiables capaces de

INVESTIGACIÓN GRASEQA: XVIII Reunión de la SEQA/Mini-‐Symposium Aceite de Oliva
17
diferenciar los tipos de aceites de oliva vírgenes en función de su calidad. Actualmente las investigaciones apuntan a que una posible vía de estudio sería la caracterización de los compuestos presentes en la fracción volátil del aceite de oliva por ser éstos los responsables de sus características organolépticas. Para alcanzar los receptores olfativos humanos las moléculas olorantes necesitan ser volátiles, así, los compuestos responsables del olor se caracterizan por tener una presión de vapor relativamente baja y sus pesos moleculares no suelen exceder de 300 Da.
El aceite de oliva está formado por unos 100 compuestos volátiles conteniendo aldehídos, hidrocarburos alifáticos y aromáticos, alcoholes, cetonas, ácidos, ésteres, éteres y derivados del tiofeno y furano. La presencia de estos compuestos depende de factores medioambientales, agronómicos, variedad de la aceituna, grado de maduración y factores tecnológicos ligados a los procesos de extracción y de envasado del aceite de oliva [4-‐8]. No siempre los compuestos volátiles a altas concentraciones son los principales responsables del aroma ya que factores químicos como la volatilidad, carácter hidrofóbico, tamaño, forma, estructura conformacional de las moléculas, tipo y posición de los grupos funcionales parecen afectar a la intensidad del olor más que su concentración [9,10]. Cada compuesto volátil contribuye al aroma total según sus niveles de umbral sensorial [11] y hay que tener en cuenta que la detección simultánea de varios defectos en un análisis sensorial se produce como la suma de los efectos de los marcadores específicos y un efecto de sinergia entre ellos [12]. Existe también relación entre el aroma y el sabor del aceite de oliva y su contenido en polifenoles, destacando la presencia de hidrotirosol, tirosol, ácido cafeico, ácido cumárico y ácido p-‐hidroxibenzoico [13,14].
Los principales responsables de los atributos positivos del aroma del aceite de oliva virgen son fundamentalmente compuestos orgánicos de cinco y seis átomos de carbonos del grupo de los aldehídos, alcoholes, ésteres y cetonas [2,5]. La mayoría de esos volátiles son
sintetizados enzimáticamente a partir de los ácidos grasos poliinsaturados, principalmente linoleico y linolénico, a través de la ruta de la lipoxigenasa. Entre los compuestos volátiles que contribuyen mayoritariamente a los atributos positivos del aroma del aceite de oliva se pueden destacar el cis-‐3-‐hexenal, hexanal y trans-‐2-‐hexenal. Otros compuestos que contribuyen a notas sensoriales positivas son el acetato de etilo, 6-‐metil-‐5-‐hepten-‐2-‐ona, acetato de 3-‐hexenilo, 1-‐hexanol, 3-‐hexen-‐1-‐ol y 2-‐hexen-‐1-‐ol [15]. Estos atributos positivos se corresponden con la percepción de aromas relacionados con hojas verdes, frutas, almendra, plátano, tomate, plantas aromáticas, dulce, picante y amargo.
Las condiciones de temperatura, alta humedad y la pérdida del epicarpio, lleva a la aceleración de procesos de autolisis de compuestos orgánicos y de las aceitunas degradadas favoreciendo la actividad microbiana en los tejidos de las aceitunas por todos los microorganismos presentes en el medio ambiente [3-‐16]. Estos procesos citados dan lugar a compuestos volátiles, responsables de los atributos negativos del aceite de oliva virgen, asociados con actividad oxidativa microbiana como las descritas para Aspergillus o Penicillum (defecto moho-‐humedad), fermentaciones aeróbicas por Acetobacterias (defecto avinado-‐avinagrado), fermentaciones anaeróbicas (defecto atrojado) y autooxidaciones (defecto rancio). De esta forma, la actual normativa europea sobre el aceite de oliva [17,18] clasifica los defectos más frecuentes en cuatro grupos: atrojado, moho-‐humedad, avinado-‐avinagrado y rancio. Los principales compuestos volátiles relacionados con esos defectos son: 1-‐octen-‐3-‐ol para moho-‐humedad; butanoato de etilo, ácidos propanoico y ácido butanoico para el defecto atrojado; ácido acético, 3-‐metil-‐butanol y acetato de etilo para el defecto avinado-‐avinagrado y algunos aldehídos saturados e insaturados y ácidos para el defecto rancio [19]. Asimismo reacciones asociadas a la manipulación, procesos tecnológicos y almacenamiento producen defectos en el aceite de oliva.

BOLETÍN GRASEQA Nº 6 OCTUBRE 2013
18
Las nuevas tendencias de parte del sector oleico están basadas en encontrar métodos analíticos complementarios a los paneles de cata. Estos métodos analíticos deben responder a dos enfoques, por un lado a la determinación de los compuestos químicos responsables del aroma del aceite de oliva y, por otro, explicar con detalle cuáles son los compuestos químicos relacionados con los atributos sensoriales. Esos enfoques tienden a combinar el uso de nuevas técnicas analíticas con herramientas estadísticas adecuadas [20]. La Cromatografía de Gases es la técnica más usada para demostrar la calidad del aceite de oliva a lo largo de los años ya que permite determinar de manera específica la composición del aroma del aceite de oliva [21-‐25]. En cambio, los Sensores Químicos o Narices Electrónicas [26,27] intentan asemejarse a la percepción sensorial que realiza el consumidor ya que ésta no se basa exclusivamente en la medida de un compuesto específico (objetivo principal de la separación cromatográfica) sino de un conjunto de ellos con intensidades variables entre los tipos de aceites.
Desde hace unos años, nuestro grupo de investigación está estudiando el potencial de la Espectrometría de Movilidad Iónica (IMS, siglas en inglés) para resolver distintos problemas de la industria agroalimentaria [28-‐30]. Así se ha usado por primera vez la IMS para demostrar la calidad del aceite de oliva [31-‐33] y lograr su clasificación. Con esta técnica, se pretende encontrar un perfil de
característica de cada categoría de aceite de oliva virgen. La IMS se caracteriza por una buena sensibilidad (límites de detección del orden de las ppb), no requiere tratamiento de muestra previo, además de la rapidez y el bajo coste por análisis comparada con otras técnicas analíticas. Sin embargo presenta algunos inconvenientes como, por ejemplo, la limitada selectividad debido a las reacciones ión-‐molécula que se producen al operar a presión atmosférica. A continuación se presenta una breve descripción de la técnica IMS.
2. Espectrometría de movilidad iónica La IMS es una técnica analítica desarrollada para la determinación de compuestos volátiles y semivolátiles que se basa en la separación en fase gaseosa de los iones en un tubo de deriva bajo la influencia de un campo eléctrico constante a presión atmosférica. La IMS se podría incluir dentro de las técnicas analíticas de vanguardia ya que puede proporcionar información analítica de un gran número de muestras de forma rápida y simple. Asimismo se puede enmarcar dentro de las técnicas de cribado ya que puede proporcionar una respuesta rápida a la presencia o ausencia de algún tipo de compuesto presente en una muestra.
El fenómeno de movilidad iónica parte de una primera etapa de ionización de la muestra mediante el empleo de una fuente de ionización, generalmente una fuente radiactiva o ultravioleta. Los iones formados se separan en el tubo de deriva dependiendo generalmente de su estructura conformacional (masa, tamaño, carga y forma). La velocidad que adquiere el ión (velocidad de deriva, vd) característica de cada ión, es proporcional al campo eléctrico (E) aplicado según la ecuación (1):
vd = K E (1)
La constante de proporcionalidad K, recibe el nombre de constante de movilidad iónica, y viene dada por la ecuación de Revercomb y Mason (2):
(2)
donde q es la carga del ión, N la densidad en número del gas amortiguador, k la constante de Boltzmann, T la temperatura absoluta, m la masa del gas amortiguador, M la masa del ión, y la sección media de colisión del ión. La constante de movilidad se puede normalizar a valores bajos de campo eléctrico y condiciones estándares de presión (P) y temperatura (T) obteniéndose así la constante de movilidad reducida (Ko) que se calcula según la ecuación (3):

INVESTIGACIÓN GRASEQA: XVIII Reunión de la SEQA/Mini-‐Symposium Aceite de Oliva
19
(cm2V-‐1s-‐1 ) (3)
En un espectrómetro de movilidad iónica (ver Figura 2) podemos distinguir cuatro zonas: región de ionización, rendija de entrada o shutter grid, región de deriva y detector. Las moléculas gaseosas entran al interior del equipo arrastradas por un gas y llegan a la cámara de ionización donde existe una fuente capaz de ionizar dichas moléculas. La etapa de ionización depende del tipo de fuente usada.
En este caso se va a describir la ionización radiactiva que es la que se ha usado en este trabajo. La fuente radiactiva (Tritio, 3H) ioniza la atmósfera circundante la cual reacciona rápidamente con las moléculas de agua que existen formando protones hidratados o iones reactantes (H2O)nH+. Estos iones reactantes reaccionan a su vez con las moléculas de la muestra que tengan una afinidad protónica mayor a la del agua (692 kJ/mol) para formar los iones productos M(H2O)H+. Dependiendo de las propiedades físico-‐químicas de las sustancias, de su concentración e incluso de las condiciones experimentales se pueden formar iones dímeros (M2(H2O)H+) e incluso trímeros (M3(H2O)H+).
Los iones formados entran a través de la rendija a la región de deriva. En esta región los iones vuelan bajo un campo eléctrico constante produciéndose la colisión con el gas
de deriva el cual fluye en contracorriente colisionando finalmente los iones formados con el detector que es una placa de Faraday. El cambio en la velocidad de los iones debido a estas colisiones está influenciado por la sección transversal de los iones. Así la IMS separa los iones en base a su tamaño, forma, masa y carga. El tiempo de tránsito de los iones a través de la región de deriva se conoce como tiempo de deriva (td) y es del orden de milisegundos.
Uno de los inconvenientes que presenta la IMS es su escaso poder de resolución. De esta forma, para aumentar la selectividad, especialmente para la determinación de compuestos individuales en una muestra compleja, a menudo es necesario acoplar un paso previo de separación mediante el empleo de distintas columnas de cromatografía de gases como son una columna multicapilar (MCC-‐IMS) o bien la columna clásica de cromatografía gaseosa de 30 m de longitud (GC-‐IMS).
El campo de aplicación de la IMS es muy variado tanto como técnica independiente como acoplada. Conviene distinguir las aplicaciones en el campo de la detección de armas químicas, drogas, y de los análisis industrial, medioambiental, clínico, forense y alimentario.
.
Figura 2. Esquema general del espectrómetro de movilidad iónica.

BOLETÍN GRASEQA Nº 6 OCTUBRE 2013
20
3. Análisis de las muestras de aceite de oliva por IMS La IMS, por sus características analíticas, es muy adecuada para el análisis de control de calidad de alimentos, control de procesos y caracterización de alimentos mediante la determinación de un compuesto específico o de una familia de ellos [28-‐34]. Las muestras de alimentos se caracterizan por una amplia variedad de matrices que pueden ser sólidas o líquidas. En IMS, sólo las muestras gaseosas se pueden introducir directamente en el espectrómetro de movilidad iónica. Por tanto, las muestras líquidas o sólidas requieren una etapa previa de preparación de la muestra. Existen diferentes formas de determinar por IMS los compuestos volátiles presentes en los alimentos [35]. La más simple es mediante la creación de un espacio de cabeza gaseoso en el vial de muestra. Por otro lado, la determinación de los compuestos volátiles se puede hacer directamente si se obtiene una respuesta selectiva o bien usando alguna técnica de separación previa al análisis por IMS, como sería el uso de columnas cromatográficas. De esta forma, la IMS es una técnica que posibilita bien el estudio global de la información que contiene el espectro o bien el estudio y caracterización de los compuestos concretos presentes en la muestra objeto de estudio.
3.1. Análisis de la información global del espectro de movilidad iónica
Los análisis de las muestras de aceite de oliva virgen mediante IMS se realizaron con el equipo FlavourSpec® de G.A.S. (Gesellschaftfür Analytische Sensorsysteme mbH, Dortmund, Alemania). En la Figura 3 se muestra un esquema del equipo usado así como las condiciones experimentales empleadas. Dicho equipo consta de automuestreador que permite el muestreo directo del espacio de cabeza de las muestras de aceite dentro del inyector del equipo (CTC-‐PAL, CTC Analytics AG, Zwingen, Suiza) para una mejor reproducibilidad de las medidas. Para el análisis, se colocó 1 g de muestra en
un vial de espacio de cabeza de 20 mL encapsulado con un septum magnético. Después de una incubación de 10 minutos a 60°C, 100 µL del espacio de cabeza se inyectó automáticamente por una jeringa calentada (80°C) dentro del inyector (80°C) del equipo IMS. Después de la inyección, el gas portador pasa a través del inyector y conduce la muestra a una columna de gases multicapilar (MCC) o de gases clásica (GC) que se encuentra a 40°C para la separación. En estas condiciones, los analitos se eluyen, en modo isotérmico, y son conducidos a la cámara de ionización del espectrómetro de movilidad iónica con una fuente de tritio (6.5 keV). Posteriormente, los iones se introducen en la región deriva. La longitud del tubo de deriva es de 6 cm donde se aplica un voltaje constante de 350 V cm-‐1 y una temperatura de 45°C. Los datos se adquirieron en el modo de polaridad positiva por el ordenador integrado en el equipo. Cada espectro se formó con la media de 32 medidas, el ancho de pulso de red de la rendija de 100 ms y la frecuencia de muestreo de 150 kHz. Esta tecnología permite el análisis totalmente automático y directo (sin tratamiento de muestra previo al análisis) de compuestos volátiles (principalmente aldehídos, ésteres, alcoholes y cetonas) presentes en las muestras de aceite de oliva.
El método inicialmente optimizado usó una MCC apolar (OV-‐5, 20 cm de largo, rellena de 5 % de difenilo y 95 % dimetilpolisiloxano), mostrando una óptima precisión obteniéndose valores de repetitividad y reproducibilidad más bajos del 8 % en todos los casos expresado como desviación estándar relativa. Bajo estas condiciones se analizaron 98 muestras de aceite (27 muestras de AOL, 28 muestras de AOV y 43 muestras de AOVE) por duplicado [33]. Estas muestras se obtuvieron del Laboratorio Oficial Agroalimentario de la Junta de Andalucía (Córdoba, España) y de una almazara de Córdoba (España) de las cosechas 2009-‐2010 y 2010-‐2011. Antes de analizar estas muestras por IMS se analizaron según el Reglamento de la Unión Europea 1989/2003 para fijar sus categorías y así tener datos de referencia fiables para poder compararlos con los datos obtenidos del análisis mediante MCC-‐IMS. En

INVESTIGACIÓN GRASEQA: XVIII Reunión de la SEQA/Mini-‐Symposium Aceite de Oliva
21
la Figura 4 se muestran los perfiles de movilidad iónica característicos de las tres categorías de aceite de oliva: AOVE, AOV, AOL donde se aprecian las distintas señales características para cada tipo de aceite.
Una vez analizadas todas las muestras de aceite de oliva virgen, los datos se trataron quimiométricamente. En primer lugar se creó el modelo quimiométrico usando un grupo de muestras para la calibración en el que el 97 % de las muestras se clasificaron correctamente en sus respectivas categorías. Con posterioridad, el modelo calibrado se validó con un conjunto de muestras excluidas de las de calibración y se obtuvo un 87 % de muestras correctamente clasificadas [33].
El uso de la información global obtenida del espectro de movilidad iónica ha resultado una aproximación interesante para la caracterización del aceite de oliva en función de su calidad. El potencial de este método se está confirmando con un estudio más amplio englobando un mayor grupo de muestras y los resultados se compararán con los obtenidos mediante el análisis de las mismas muestras de aceites por otras técnicas analíticas.
Figura 3. Esquema del Espectrómetro de Movilidad Iónica (FlavourSpec®) y condiciones experimentales empleadas en este trabajo.

BOLETÍN GRASEQA Nº 6 OCTUBRE 2013
22
Figura 4. Huella espectral de tres muestras de aceite correspondientes a cada categoría obtenidas mediante el equipo MCC-‐IMS
3.2. Análisis de la información específica del espectro de IMS. Estudio de compuestos volátiles del aroma del aceite de oliva
Además del estudio de la información global del espectro de IMS, el método optimizado permite obtener información específica de cada espectro de movilidad iónica con el objeto de identificar mediante MCC-‐IMS algunos de los compuestos volátiles que son los responsables de los atributos positivos y negativos del aceite de oliva. En la Tabla 1 se detallan las familias de compuestos estudiados mediante MCC-‐IMS presentes en el aroma del aceite de oliva donde se resumen las principales propiedades físico-‐químicas de los mismos. La mayoría de estos compuestos se han identificado en el aceite de oliva usando la Cromatografía de Gases-‐Espectrometría de Masas (GC-‐MS), pero es la primera vez que se están estudiando estos compuestos característicos del aceite de oliva mediante MCC-‐IMS o GC-‐IMS. Con los datos obtenidos se creará una base de datos para caracterizar mediante esta técnica el perfil aromático del aceite de oliva según su calidad, pudiendo así evaluar la posibilidad de clasificar un aceite de oliva en su
correspondiente categoría usando la información obtenida mediante el análisis de la información del espectro de movilidad iónica.
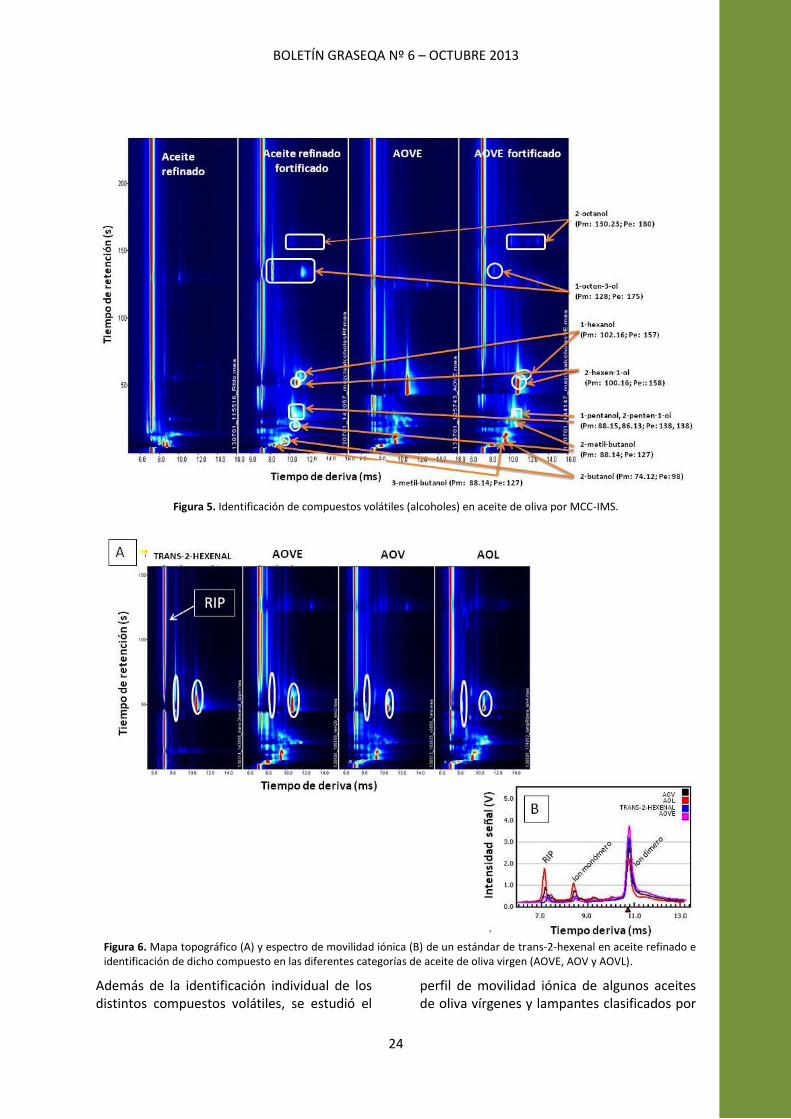
Para la identificación de compuestos volátiles en muestras de aceite de oliva, en primer lugar, se han analizado individualmente por MCC-‐IMS cada uno de los compuestos disueltos en aceite refinado para así localizar cada señal que aparece en el mapa topográfico. La identificación se hace en base a los diferentes tiempos de retención (tr) -‐ en la columna cromatográfica de gases empleada y el tiempo de deriva (td) -‐ distinta velocidad que alcanzan los iones en el tubo de deriva-‐ que presentan cada uno de los compuestos volátiles analizados. Se seleccionó aceite refinado como matriz para disolver los compuestos volátiles por semejanza con la matriz donde se están estudiando (aceite de oliva) después de comprobar que no interferían las señales del aceite refinado con las que presentaban los analitos objeto de estudio. Posteriormente, los compuestos volátiles se adicionaron, a distintas concentraciones, a las muestras de aceite de oliva y las señales correspondientes a cada analito se localizaron en el mapa topográfico. Los distintos compuestos volátiles identificados pueden presentar una o dos

INVESTIGACIÓN GRASEQA: XVIII Reunión de la SEQA/Mini-‐Symposium Aceite de Oliva
23
señales características según se forme el monómero o el dímero en función de la concentración en la que esté presente el compuesto en los aceites. En la Figura 5 se muestran, los mapas topográficos de un aceite refinado y de un AOVE al que se le han añadido 9 alcoholes para su identificación de las señales por comparación. Como se puede observar en la Figura 5, los compuestos con bajo peso molecular y punto de ebullición eluyen primero de la MCC que los de compuestos más pesados.
Entre los 10 aldehídos identificados se muestra en la Figura 6, a modo de ejemplo, el trans-‐2-‐hexenal compuesto mayoritario de la
fracción volátil del aceite de oliva y que está relacionado con el aroma de frutado que presentan los aceites de oliva virgen. En la Figura 6 A se muestra el mapa topográfico de una muestra de cada categoría de aceite de oliva (AOVE, AOV y AOL) y la señal correspondiente al estándar de trans-‐2-‐hexenal. La Figura 6 B muestra el espectro IMS característico de dicho compuesto para cada uno de los tipos de aceites donde se aprecian dos picos característicos del trans-‐2-‐hexenal correspondientes al ion monómero (td de 8.5 ms) y al ion dímero (td 10.8 ms) apareciendo el compuesto a un tiempo de retención de 46 s.
Tabla 1. Características físico-‐químicas de los compuestos volátiles identificados en el aceite de oliva mediante MCC-‐IMS. Grupo funcional Analito
Masa molecular (g/mol)
Temperatura ebullición (ºC) Presión de vapor
Afinidad Protónica (kJ/mol)
Umbral olor en aceite (mg/kg)
Aldehidos Acetaldehído 44.05 20.2 -‐ 770.9 -‐
trans-‐2-‐pentenal 84.1164 80-‐81 -‐ 839 0.30
Hexanal 100.16 130-‐131 10 mmHg (20ºC) 794.4 0.08
trans-‐2-‐hexenal 98.14 146 10 mmHg (20ºC) -‐ 0.42
trans-‐2-‐heptenal 112.17 166 1.83 mmHg (25ºC) -‐ 0.001
Octanal 128.2 171 2 mmHg (20ºC) -‐ 0.32
Decanal 156.3 207-‐209 0.15 mmHg (20ºC) -‐ -‐
Benzaldehído 106.12 178.1 834 -‐
trans-‐2-‐octenal 126.2 188-‐190 0.552 mmHg (25ºC) -‐ 0.004
trans-‐2-‐decenal 154.25 230 0.07 mmHg (25ºC) -‐ 0.01 Cetonas 2-‐butanona 72.11 79.64 70 mmHg (20ºC) 827.3 40
2-‐nonanona 142.24 192-‐195 0.645 mmHg (25ºC) -‐ <0.1
1-‐penten-‐3-‐ona 84.12 111-‐114 31.09 mmHg (25ºC) -‐ 0.7 Alcoholes 2-‐butanol 74.122 98-‐100 1.67 kPa (20ºC) 815 0.10
2-‐metil-‐butanol 88.148 127.5 3 mmHg -‐ 0.48
3-‐metil-‐butanol 88.148 131.1 3 hPa (20ºC) -‐ 0.10
1-‐pentanol 88.15 138 0.6 kPa (20ºC) 795 3.00
1-‐hexanol 102.162 157 0.124 kPa (25ºC) 799 0.4
2-‐hexen-‐1-‐ol 100.16 158-‐160 6.3 hPa (20ºC) -‐ 5.00
2-‐octanol 130.23 177-‐180 0.25 hPa (20ºC) -‐ 0.1
1-‐octen-‐3-‐ol 128.21 175 0.531 mmHg (25ºC) -‐ 0.001
cis-‐2-‐penten-‐1-‐ol 86.13 138 2.41 mmHg (25ºC) -‐ 0.25
cis-‐1-‐penten-‐3-‐ol 86.13 114-‐115 11.18 mmHg (25ºC) -‐ 0.4 Ácidos Ácido acético 60.05 117.9 11.4 mmHg (20ºC) 783.7 0.50
Ácido propionico 74.05 141 -‐ 797.2 0.72
Ácido butanoico 88.11 163.5 57 Pa (20ºC) -‐ 0.65 Ésteres Butanoato de etilo 116.16 121.3 1885 -‐ 0.03
Acetato de etilo 88.1 77 100 mmHg (27ºC) 835.5 0.94
Butanoato de propilo 130.19 143 -‐ -‐ 0.15
Acetato de hexilo 144.2 171.5 1.32 mmHg (25ºC) -‐ 0.0014
BOLETÍN GRASEQA Nº 6 OCTUBRE 2013
24
Figura 5. Identificación de compuestos volátiles (alcoholes) en aceite de oliva por MCC-‐IMS.
Figura 6. Mapa topográfico (A) y espectro de movilidad iónica (B) de un estándar de trans-‐2-‐hexenal en aceite refinado e identificación de dicho compuesto en las diferentes categorías de aceite de oliva virgen (AOVE, AOV y AOVL).
Además de la identificación individual de los distintos compuestos volátiles, se estudió el
perfil de movilidad iónica de algunos aceites de oliva vírgenes y lampantes clasificados por

INVESTIGACIÓN GRASEQA: XVIII Reunión de la SEQA/Mini-‐Symposium Aceite de Oliva
25
algunos defectos concretos como eran el avinado, atrojado-‐borras, moho-‐humedad, atrojado-‐rancio, madera húmeda-‐aceituna helada. En la Figura 7 se muestran los espectros de movilidad iónica de las muestras anteriormente citadas donde se aprecia un perfil de volátiles distinto y característico de cada uno de los defectos asociado a determinados volátiles. Se aprecia como la señal del trans-‐2-‐hexenal está presente en
algunas muestras aunque tengan defectos, lo que indica que serían muestras correspondientes a aceite de oliva virgen que aunque tengan defectos conservan cierto atributo frutado. Por el contrario, las muestras de aceite lampante no presentan apenas señal correspondiente al atributo frutado, éstas serían las muestras clasificadas con el defecto avinado y el atrojado-‐rancio.
Figura 7. Perfiles de movilidad iónica correspondientes a muestras de aceite de oliva caracterizadas con diferentes defectos sensoriales.
Para la caracterización de los demás compuestos volátiles característicos de los aceites de oliva se procedió, de igual forma que para la determinación del trans-‐2-‐hexenal, midiendo el tiempo de retención y de deriva para cada uno de los compuestos analizados. A continuación a modo de ejemplo se presentan la determinación de algunos compuestos volátiles relacionados con atributos negativos característicos del aceite de oliva virgen. En la Figura 8 se muestra la determinación del octanal, uno de los compuestos volátiles relacionados con el defecto rancio. En esta figura se muestra el espectro de movilidad iónica de una muestra de AOL que presenta como defecto mayoritario el defecto de rancio frente al espectro de movilidad iónica de un estándar de octanal preparado en una muestra de aceite de refinado. Este compuesto presentó
un pico principal (monómero) a un tiempo de deriva de 10.1 ms. A los 13 ms se observa la aparición de un pequeño pico que podría corresponder al ión dímero del octanal el cual podría incrementar su intensidad con la concentración de este compuesto en la muestra de aceite. Este compuesto aparece a un tiempo de retención de 130.6 s.
Figura 8. Espectro de movilidad iónica del octanal y de una muestra de AOL con defecto rancio.

BOLETÍN GRASEQA Nº 6 OCTUBRE 2013
26
Finalmente se muestra la identificación del propilbutanoato (Figura 9). Este compuesto está relacionado con el defecto moho en el aceite de oliva. En la Figura 9 A se muestra el mapa topográfico de una muestra de AOL que presenta principalmente este defecto y el mapa topográfico de un patrón de butanoato de propilo. Este compuesto aparece al tiempo de retención de 21 s con dos picos característicos. En la Figura 9 B se muestran los valores medios para la señal del monómero y el dímero de 5 muestras de AOL, AOV y AOVE, respectivamente. El ión dímero del butanoato de propilo es mayor en las
muestras de AOL y AOV. En el caso de este compuesto está más favorecida la formación de ión dímero (12.2 ms) el cual presenta una mayor intensidad que el ión monómero (9.3 ms) como se puede observar en la Figura 9 B. Las muestras de AOVE no deberían presentar ningún tipo de defecto, sin embargo, como se observa en la figura, las muestras de AOVE presentan señal para ese compuesto aunque inferior a las muestras de AOV y AOL. Esta señal podría estar por debajo del umbral de percepción de este compuesto, el cual tiene un valor muy bajo (0.03 mg/kg) o bien podría ser un interferente para el propilbutanoato.
Figura 9. Cuantificación de la señal del propilbutanoato en muestras de aceite de oliva.
En la Tabla 2, se detallan todos los compuestos volátiles que se han identificado en los aceites de oliva por MCC-‐IMS. En ella se correlacionan los compuestos volátiles con los defectos descritos, sus valores de movilidad iónica (tiempo deriva) y de tiempo de retención. De los 30 compuestos inicialmente seleccionados se identificaron 27 en el aceite de oliva. Sin embargo, sólo 10 se separaron de manera selectiva al añadirlos simultáneamente (trans-‐2-‐heptenal, 1-‐octen-‐
3-‐ol, trans-‐2-‐octenal, benzaldehído, hexilacetato, 2-‐butanona, propilbutanoato, trans-‐2-‐pentenal, octanal y decanal). Los ácidos acético, propiónico y butanoico característicos de los aceites de oliva no se han podido identificar en las condiciones experimentales detalladas anteriormente.
Con el objeto de mejorar la resolución de la IMS, la MCC se ha sustituido por una columna capilar de cromatografía de gases de 30 m de

INVESTIGACIÓN GRASEQA: XVIII Reunión de la SEQA/Mini-‐Symposium Aceite de Oliva
27
longitud apolar. Con el nuevo acoplamiento (GC-‐IMS) se ha mejorado la selectividad de la técnica pasando a detectar 17 compuestos al ser añadidos de manera simultánea. Los nuevos compuestos detectados son: cis-‐2-‐hexen-‐1-‐ol, trans-‐2-‐hexenal, 1-‐penten-‐3-‐ol,
acetaldehído, 1-‐penten-‐3-‐ona, 2-‐nonanona, 1-‐hexanol y 2-‐decenal. Sin embargo, el tiempo de análisis se duplica respecto al de la columna multicapilar pasando a ser de 30 min frente a los 15 min de análisis por muestra que emplea la MCC-‐IMS.
Tabla 2. Parámetros de movilidad iónica de los compuestos identificados por MCC-‐IMS en las muestras de aceite de oliva.

BOLETÍN GRASEQA Nº 6 OCTUBRE 2013
28
4. Conclusiones Los resultados obtenidos al analizar muestras de aceite de oliva de distintas categorías con IMS son válidos para considerarla como una posible técnica analítica que pudiera resolver un problema que hasta la fecha solo se puede abordar usando el análisis sensorial. Esta técnica se presenta como un posible sistema de cribado que puede ser usada por el sector oleico ya que permite realizar de forma rápida y fácil el análisis de muestras de aceite con el fin de conocer su categoría.
Observando los porcentajes obtenidos, tanto de clasificación (97 %) como de predicción (87 %) podemos afirmar que la IMS podría ser una técnica con buenas aptitudes para el estudio de la calidad de muestras de aceite de oliva y evitar las posibles pérdidas económicas que una mala clasificación puede generar en el sector oleico. Debido a la complejidad que desde un punto de vista de su composición química presentan las distintas muestras de aceite de oliva, todavía se debe invertir tiempo y esfuerzo en seguir investigando el potencial de esta técnica para poder confirmar con seguridad si su uso podría ser
viable en los laboratorios agroalimentarios de rutina.
Por otra parte, la IMS es una técnica que permite identificar algunos de los compuestos volátiles responsables de los atributos positivos y negativos asociados a los aceites de oliva vírgenes. Esta identificación también puede ayudar a mejorar los porcentajes de clasificación y predicción cuando se usa toda la información global del espectro.
Un aspecto importante a destacar de la metodología propuesta es el elevado número de muestras de aceite de oliva que se pueden analizar en una jornada de trabajo con un bajo coste además del nulo tratamiento de las muestras fuera del sistema usado. En cambio, para demostrar la calidad de un aceite de oliva usando el método oficial se necesita realizar numerosos análisis además del análisis sensorial, implicando largos tiempos de análisis para confirmar la calidad de un aceite de oliva. Estas y otras ventajas de la metodología propuesta corroboran las posibilidades que podría tener la IMS en los laboratorios de análisis de rutina para conocer la calidad de muestras de aceite de oliva.
Referencias [1] EC: Regulation No 61/2011, Off. J. Eur. Communities L232/1 (2011).
[2] EC: Regulation No. 61/2011 Annex I, Off. J. Eur. Communities L23/3 (2011).
[3] F. Angerosa, M. Servili, R. Selvaggini, A. Taticchi, S. Esposto, G. Montedoro, Volatile compounds in virgin olive oil: occurrence and their relationship with the quality, J. Chromatogr. A., 1054 (2004) 17-‐31.
[4] F. Angerosa, B. Lanza, V. Marsilio, Biogenesis of
(1996) 142-‐150.
[5] F. Angerosa, N. D' Alessandro, C. Basti, R. Vito, Biogeneration of volatile compounds in virgin olive oil: Their evolution in relation to malaxation time, J. Agric. Food Chem., 46 (1998) 2940-‐2944.
[6] F. Angerosa, R. Mostallino, C. Basti, R. Vito, Influence of malaxation temperature and time on the quality of virgin olive oils, Food Chem., 72 (2001) 19-‐28.
[7] R. Aparicio, in R. Aparicio, J. Harwood (Editors), Handbook of Olive Oil. Mundi-‐Prensa, Madrid, Spain, (2003) p. 281.
[8] M.T. Morales, F. Angerosa, R. Aparicio, Effect of the extraction conditions of virgin olive oil on the lipoxygenase cascade: chemical and sensory implications, Grasas y Aceites, 50 (1999) 114-‐121.
[9] P. Pelosi, Odorant-‐Binding proteins, Crit. Rev. Biochem. Mol. Biol., 29 (1994) 199-‐228.
[10] K.J. Rossiter, Structure-‐odor relationships, Chem. Rev., 96 (1996) 3201-‐ 3240.
[11] F. Ullrich, W. Grosch, Identification of the most intense odor compounds formed during autoxidation of methyl linolenate at room-‐temperature, J. Amer. Oil Chem. Soc., 65 (1988) 1313-‐1317.
[12] R. Aparicio, M.T. Morales, D.L. García-‐González, Towards new analyses of aroma and volatiles to understand sensory perception of olive oil, Eur. J. Lipid. Sci. Technol., 114 (2012), 1114-‐1125.
[13] F. Angerosa, R. Mostallino, C. Basti, R. Vito, Virgin olive oil odour notes: their relationships with volatile compounds from the lipoxygenase pathway and secoiridoid compounds, Food Chem., 68 (2000) 283-‐287.
[14] A. K. Kiristasis, Flavor Components of Olive Oil A review, J. Amer. Oil Chem Soc., 75, (1998) 673-‐681.
[15] R. Aparicio, M.T. Morales, M.V. Alonso, Relationship between volatile compounds and sensory attributes of

INVESTIGACIÓN GRASEQA: XVIII Reunión de la SEQA/Mini-‐Symposium Aceite de Oliva
29
olive oils by the sensory wheel, J. Amer. Oil Chem. Soc., 73 (1996) 1253-‐1264.
[16] F. Angerosa, Influence of volatile compounds on virgin olive oil quality evaluated by analytical approaches and sensor panels, Eur. J. Lipid Sci. Technol., 104 (2002) 639-‐660.
[17] Organoleptic Assessmetn of Virgin Olive Oil COI/T.20/Doc. No. 15 (1996).
[18] Official Journal of the Commission of the European Communities Regulation No. 2473/97 (1997) L341.
[19] M.T. Morales, G. Luna, R. Aparicio, Comparative study of virgin olive oil sensory defects, Food Chem., 91 (2005) 293-‐ 301.
[20] D. García-‐González, R. Aparicio, Research in Olive Oil: Challenges for Near Future, J. Agric. Food Chem., 58 (2010) 12569-‐12577.
[21] M.T. Morales, M. Tsimidou, in J. Harwood, R. Aparicio (Editors), Handbook of Olive Oil: Analysis and Properties. Aspen Publishers, Gaithersburg, MD (USA), 2000, p. 393.
[22] J.F. Cavalli, X. Fernández, L. Lizzani-‐Cuveiller, A.M. Loiseau, Comparison of static headspace, headspace solid phase microextraction, headspace sorptive extraction, and direct thermal desorption techniques on chemical composition of french olive oils, J. Agric. Food Chem., 51 (2003) 7709-‐ 7716.
[23] M. Contini, M. Esti, Effect of the matrix volatile composition in the headspace solid-‐phase microextraction analysis of extra virgin olive oil, Food Chem., 94 (2005) 143-‐150.
[24] J.F. Cavalli, X. Fernández, L. Lizzani-‐Cuvelier, A.M. Loiseau, Characterization of volatile compounds of French and Spanish virgin olive oils by HS-‐SPME: Identification of quality-‐freshness markers, Food Chem., 88 (2004) 151-‐157.
[25] C.M. Kalua, D.R. Bedgood, P.D. Prenzler, Development of a headspace solid phase microextraction-‐gas chromatography method for monitoring volatile compounds in extended time-‐course experiments of olive oil, Anal. Chim. Acta, 556 (2006) 407-‐ 414.
[26] D.L. García-‐González, R. Aparicio, Virgin olive oil quality classification combining neural network and MOS sensors, Grasas y Aceites, 51 (2003) 3515-‐3519.
[27] S. Ampuero, J.O. Bossel, The electronic nose applied to dairy products: a review,Sens. Act. B-‐ Chem, 94 (2003) 1-‐12.
[28] R. Garrido-‐Delgado, L. Arce, A. V. Guamán, A. Pardo, S. Marco, M. Valcárcel, Direct coupling of a gas liquid separator to an ion mobility spectrometer for theclassification of different white wines using chemometrics tools, Talanta, 84 (2011) 471-‐479.
[39] R. Alonso, V Rodríguez-‐Estevez, A. Dominguez-‐Vidal, M. J. Ayora-‐Cañada, L. Arce, M. Valcárcel, Ion mobility spectrometry of volatile compounds from Iberian pig fat for fast feeding regime authentication, Talanta, 76 (2008) 591-‐596.
[30] M. Menéndez, R. Garrido-‐Delgado, L. Arce, M. Valcárcel, Direct determination of volatile analytes from solid samples by UV-‐ion mobility spectrometry, J. Chromatogr. A, 1215 (2008) 8-‐14.
[31] R. Garrido-‐Delgado, F. Mercader-‐Trejo, S. Sielemann, W. de Bruyn, L. Arce, M. Valcárcel, Direct classification of olive oils by using two types of ion mobility spectrometers, Anal. Chim. Acta, 696 (2011) 108-‐115.
[32] R. Garrido-‐Delgado, F. Mercader-‐Trejo, L. Arce, M. Valcárcel, Enhancing sensitivity and selectivity in the determination of aldehydes in olive oil by use of a Tenax TA trap coupled to a UV-‐ion mobility spectrometer,J. Chromatogr. A, 1218 (2011) 7543-‐7547.
[33] R. Garrido-‐Delgado, L. Arce, M. Valcárcel, Multi-‐capillary column-‐ion mobility spectrometry: a potential screening system to differentiate virgin olive oils, Anal. Bioanl. Chem., 402 (2012) 489-‐498.
[34] W. Vautz, D. Zimmermann, M. Hartmann, J.I. Baumbach, J. Nolte, J. Jung, Ion mobility for food quality and safety, Food Add. Conta., 23 (2006) 1064-‐1073.
[35] L. Arce, M. Menéndez, R. Garrido-‐Delgado, M. Valcárcel, Sample introduction systems coupled to ion mobility spectrometry equipment for determining compounds present in gaseous, liquid and solid samples, TRAC-‐Trends Anal. Chem., 27 (2008) 139-‐150.
.

BOLETÍN GRASEQA Nº 6 OCTUBRE 2013
30
Rocío Garrido Delgado es Doctora en Ciencias Químicas por la Universidad de Córdoba (UCO) desde 2011. Su formación doctoral la realizó en el departamento de Química Analítica de la UCO bajo la dirección de la profesora Lourdes Arce y del profesor Miguel Valcárcel. Su trabajo de investigación se centró en el desarrollo de métodos analíticos de vanguardia usando la Espectrometría de Movilidad Iónica (IMS) dentro, principalmente, del campo agroalimentario. Durante sus estudios de doctorado realizó una estancia en el grupo de Santiago Marco en el Departamento de Electrónica de la Universidad de Barcelona para mejorar sus conocimientos sobre tratamiento de datos. Actualmente tiene un contrato del campus de excelencia agroalimentario (ceiA3) y trabaja en el departamento de Química Analítica de la UCO. Está participando en varios proyectos de investigación en colaboración con empresas del sector oleico y otras relacionadas con las energías renovables. Su principal actividad investigadora se centra en el desarrollo de una metodología analítica para conocer de forma rápida la calidad del aceite de oliva virgen a través de su composición volátil usando la IMS.
Mª del Mar Dobao Prieto, Dra. en Ciencias por la Universidad de Córdoba (UCO) y Licenciada en Ciencia y Tecnología de los Alimentos. Realizó su Tesis Doctoral en el Dpto. de Bioquímica y Biología Molecular bajo la dirección de los doctores Castillo Rodríguez y Martínez Luque-‐Romero. Posteriormente se incorporó a un Grupo de Trabajo del Dpto. de Química Agrícola y Edafología de la UCO, donde trabajó en aspectos relacionados con la caracterización y estudio de diversos tipos de compostaje de residuos agrícolas y urbanos y sus usos potenciales en la agricultura, dirigidos por el Dr. González Fernández. Además ha trabajado en el Laboratorio Oficial Agroalimentario de Córdoba en el Dpto. de Grasas y Aceites en la realización del análisis oficial de calidad de aceites donde perteneció al Panel Oficial de Análisis Sensorial de aceite de oliva virgen. Actualmente tiene un contrato con el Campus de Excelencia Agroalimentario (ceiA3) y trabaja en el grupo de investigación del Prof. Valcárcel (Departamento de Química Analítica de la UCO) colaborando en distintos proyectos relacionados con la determinación de compuestos volátiles por IMS en aceite de oliva.
Lourdes Arce Jiménez es Profesora Titular del Departamento de Química Analítica de la Universidad de Córdoba (UCO) desde 2007. Su vocación docente surgió siendo muy joven y la investigadora comenzó de la mano de los profesores Angel Ríos y Miguel Valcárcel; que le dirigieron su tesis doctoral (1999) centrada en el estudio de distintos sistemas de extracción de analitos de muestras agroalimentarias, usando principalmente sistemas de flujo acoplados a un equipo de Electroforesis Capilar (CE), técnica que en aquellos años era una gran desconocida en los laboratorios analíticos. En la actualidad sigue estudiando el potencial de la CE en distintas aplicaciones agroalimentarios. Al terminar su tesis doctoral tuvo la oportunidad de trabajar en distintas universidades, como profesora en la Universidad Pablo de Olavide y, como investigadora con un contrato Ramón y Cajal tanto en la UCO como en varias universidades europeas. En 2006 el Profesor Valcárcel introdujo la Espectrometría de Movilidad Iónica (IMS) como nueva técnica analítica de vanguardia en su grupo de investigación. Fruto del trabajo realizado en su equipo, se ha demostrado el potencial que tiene esta técnica para la determinación de compuestos volátiles presentes en distintos alimentos, destacando los trabajos realizados para evitar problemas en la determinación de la calidad del aceite de oliva. Los trabajos publicados con IMS han generado una gran expectación en distintas empresas del sector del aceite de oliva. La investigación realizada ha dado como fruto publicaciones científicas, capítulos de libros, codirección de tesis doctorales y trabajos fin de máster, así como la participación y/o dirección de varios proyectos de investigación nacionales e internacionales.

GRUPOS DE INVESTIGACIÓN GRASEQA
31
GRUPOS DE INVESTIGACIÓN GRASEQA
ANÁLISIS EN ALIMENTACIÓN Y MEDIOAMBIENTE (ANAMA)
UNIVERSIDAD DE GRANADA UNIVERSIDAD DE GRANADA, Departamento de Química Analítica, Facultad de Ciencias, Avda.
Fuentenueva, s/n., 18071 Granada. Tel.: 958-‐243327. E-‐mail: [email protected] Grupo de Investigación: Análisi en Alimentación y Medioambiente (FQM-‐232) Web: http://fqm232.ugr.es/ Investigador principal: Mª Gracia Bagur González. Integrantes:
DESCRIPCIÓN El grupo de investigación se creó en 1995, siendo su responsable la Doctora Mercedes Sánchez Viñas que ha mantenido esa responsabilidad hasta su reciente jubilación en 2011. Centra su actividad investigadora actual en torno a tres ejes de actuación: (a) aplicación de tratamientos estadísticos a la optimización de procesos analíticos y a la extracción de información relevante a partir de datos experimentales multivariantes; (b) puesta a punto de estrategias de separación, fraccionamiento y cuantificación de compuestos químicos presentes en alimentos (fundamentalmente de naturaleza lipídica), y aplicación de dichas estrategias para la autentificación de alimentos; y (c) aplicación de técnicas mineralógicas, químicas, etc. a estudios ambientales (en colaboración con investigadores del Dpto. de Mineralogía). En este último año se ha iniciado una nueva colaboración con el recientemente creado Instituto de Investigación Biomédica de Granada, en el ámbito de la caracterización analítica de fármacos biotecnológicos de naturaleza proteica.

BOLETÍN GRASEQA Nº 6 OCTUBRE 2013
32
LÍNEAS DE INVESTIGACIÓN ACTUALES
1.-‐ METROLOGÍA QUÍMICA, CUALIMETRÍA y QUIMIOMETRÍA APLICADA Desarrollo de protocolos para la validación de procesos analíticos y la estimación de la incertidumbre de los resultados obtenidos
Desarrollo de estrategias formales de multioptimización de procesos analíticos basadas en técnicas de experimentos estadísticamente diseñados.
Aplicación de las técnicas de tratamiento de datos multivariantes (de dos y tres vías) para la obtención de información analítica relevante, y mejora de la calidad de dicha información, presente en conjuntos de datos experimentales multivariantes, fundamentalmente obtenidos a partir de "huellas dactilares" espectrométricas y cromatográficas.
2.-‐ CALIDAD ALIMENTARIA Autentificación de aceites vegetales en relación al origen botánico, origen geográfico, presencia de aceites extraños, etc.
Desarrollo de métodos analíticos para la caracterización, y cuantificación de la proporción de aceite de oliva, de las diferentes categorías, en alimentos que lo contengan como ingrediente, incluidos las mezclas con otros aceites vegetales.
3.-‐ DETERMINACIÓN DE COMPUESTOS LIPÍDICOS Desarrollo de metodologías analíticas alternativas a las convencionales para la separación, fraccionamiento y/o aislamiento de compuestos lipídicos minoritarios (p.e.: fosfolípidos, esteroles) y su cuantificación en alimentos grasos.
4.-‐ ANÁLISIS AMBIENTAL Estudio del impacto ambiental derivado de la actividad minera mediante la combinación de técnicas mineralógicas, químicas, y de ecotoxicidad.
5.-‐ ANÁLISIS FARMACÉUTICO Desarrollo y validación de métodos analíticos de control de la calidad de fármacos biotecnológicos de naturaleza proteica.
Caracterización analítica de proteínas terapéuticas.
TRANSFERENCIA DEL CONOCIMIENTO
Línea Nº 1: ASESORAMIENTO TÉCNICO Y FORMACIÓN A PERSONAL DE LABORATORIOS ANALÍTICOS
Elaboración de procedimientos operativos de: gestión de equipos de medida e instrumentos analíticos, validación de métodos de análisis, cálculo de incertidumbres.
Cursos de formación "a la carta".
Línea Nº 2: ELABORACIÓN DE MATERIALES DE REFERENCIA CERTIFICADOS DE ACEITE DE OLIVA
Desarrollo de Materiales de Referencia Certificados de aceite de oliva, tanto de parámetros fisicoquímicos como de propiedades organolépticas, para su utilización en la validación y/o como muestras de control en la aplicación de los métodos analíticos para el control de la calidad del aceite de oliva.
Línea Nº 4: ESTUDIOS DE IMPACTO, RIESGO Y PROPUESTA DE BIORREMEDIACIÓN
Realización de estudios con objeto de proponer medidas de intervención a partir de parámetros mineralógicos, edafológicos, biológicos y analíticos.

TESIS DOCTORALES GRASEQA
33
TESIS DOCTORALES GRASEQA
MICOTOXINAS: APROXIMACIONES ANALÍTICAS Y METABOLÓMICAS NATALIA ARROYO MANZANARES (Universidad de Granada)
DIRECTORES: LAURA GÁMIZ GRACIA y ANA MARÍA GARCÍA CAMPAÑA GRUPO: FQM-‐302 FECHA: 26/07/2013 La problemática principal abordada en esta Tesis ha sido el desarrollo de diferentes métodos para la determinación de micotoxinas en alimentos. Para ello se han evaluado diferentes técnicas instrumentales separativas, tanto miniaturizadas como de ultra resolución acopladas a diferentes sistemas de detección de gran sensibilidad y selectividad como la fluorescencia inducida por laser (LIF) y la espectrometría de masas (MS/MS). Además se han estudiado y propuesto metodologías de tratamientos de muestra alternativas a las ya existentes que permiten aumentar la eficacia y el rendimiento de los análisis, permitiendo la determinación simultánea de micotoxinas de diversas familias. A continuación se describen brevemente los métodos analíticos desarrollados: Se ha desarrollado un método para la determinación de las aflatoxinas en arroz utilizando la cromatografía capilar electrocinética micelar (MEKC) con detección LIF y preconcentración online.
Además se ha llevado a cabo una comparación de diversos tratamientos de muestra para la determinación de ocratoxina A en diferentes tipos de vino, como son la microextracción líquido-‐líquido dispersiva (DLLME) empleando disolventes orgánicos y líquidos iónicos como extractantes, y la metodología QuEChERS. La técnica instrumental empleada ha sido la HPLC capilar-‐LIF.
Igualmente se estudió el potencial de la UHPLC-‐MS/MS junto con los tratamientos de muestra QuEChERS y DLLME en la determinación multiclase de micotoxinas en cardo mariano, frutos secos, cereales, pseudocereales y melazas de cereal.
Por otro lado, durante la realización de la estancia predoctoral en la Universidad de Gante, se ha empleado la espectrometría de masas de alta resolución (HRMS) y de múltiples etapas (MSn) para el estudio de la metabolómica de algunos de los hongos implicados en la producción de micotoxinas y otros metabolitos secundarios: Así, se desarrolló un nuevo enfoque basado en LC-‐HRMS y LC-‐MSn, para la identificación de alcaloides ergóticos nuevos o poco explorados en muestras de cereales.
Finalmente, se han determinaron los metabolitos producidos por el cluster 27 pks en el Aspergillus flavus usando LC-‐HRMS y LC-‐MSn. Asimismo, se ha estudiado por primera vez el patrón de fragmentación de estos compuestos usando ionización por electrospray en modo negativo.
REFERENCIAS N. Arroyo-‐Manzanares, L. Gámiz-‐Gracia, A.M. García-‐Campaña, J.J. Soto-‐Chinchilla, L.E. García-‐Ayuso. On-‐line preconcentration for the determination of aflatoxins in rice samples by micellar electrokinetic capillary chromatography with laser induced fluorescence detection. Electrophoresis 31 (2010) 2180 2185. N. Arroyo-‐Manzanares, L. Gámiz-‐Gracia, A.M. García-‐Campaña. Determination of Ocratoxin A by capillary HPLC with laser induced fluorescence detection. Luminescence 25 (2010) 271-‐272. N. Arroyo-‐Manzanares, A. M. García-‐Campaña, L. Gámiz-‐Gracia. Comparison of different sample treatments for the analysis of ochratoxin A in wine by capillary HPLC with laser-‐induced fluorescence detection. Analytical and Bioanalytical Chemistry 401 (2011) 2987-‐2994. N. Arroyo-‐Manzanares, L. Gámiz-‐Gracia, A.M. García-‐Campaña. Determination of ochratoxin A in wines by

BOLETÍN GRASEQA Nº 6 OCTUBRE 2013
34
capillary liquid chromatography with laser induced fluorescence detection using dispersive liquid-‐liquid microextraction. Food Chemistry 135 (2012) 368-‐372. N. Arroyo-‐Manzanares, A.M. García-‐Campaña, L. Gámiz-‐Gracia. Multiclass mycotoxin analysis in Silybum Marianum by ultra high performance liquid chromatography tandem mass spectrometry using a procedure based on QuEChERS and dispersive liquid-‐liquid microextraction. Journal of Chromatography A 1282 (2013) 11-‐19. N. Arroyo-‐Manzanares, J.F. Huertas-‐Pérez, L. Gámiz-‐Gracia, A.M. García-‐Campaña. A new approach in sample treatment combined with UHPLC-‐MS/MS for the determination of multiclass mycotoxins in edible nuts and seeds. Talanta 115 (2013) 61-‐67. N. Arroyo-‐Manzanares, J.F. Huertas-‐Pérez, A.M. García-‐Campaña, L. Gámiz-‐Gracia. Simple methodology for the determination of mycotoxins in pseudocereals, spelt and rice. Food Control 36 (2014) 94-‐101. N. Arroyo-‐Manzanares, S. Malysheva, J. Vanden Bussche, L. Vanhaecke, J. D. Di Mavungu, S. De Saeger. Holistic approach based on high resolution and multi-‐stage mass spectrometry to investigate ergot alkaloids in cereals. Talanta (en prensa). N. Arroyo-‐Manzanares, J. F. Huertas-‐Pérez, L. Gámiz-‐Gracia, A. M. García-‐Campaña. Simple and efficient methodology to determine mycotoxins in cereal syrups. Food Chemistry (enviado para su publicación)
S. V. Malysheva, N. Arroyo-‐Manzanares, J. W. Cary, K. C. Ehrlich, J. Vanden Bussche, L. Vanhaecke, D. Bhatnagar, J. D. Di Mavungu, S. De Saeger. Metabolic profiling of Aspergillus flavus: Identification and fragmentation study of the cluster 27 polyketide synthase metabolites by high resolution and multi-‐stage mass spectrometry. World Mycotoxin Journal (enviado para su publicación).
CURRICULUM VITAE
Natalia Arroyo Manzanares (Orcera, Jaén, 1984) obtuvo su grado en Ingeniería Química en 2008 por la Universidad de Granada, y durante su último curso comenzó su labor investigadora en el grupo FQM-‐302 (Departamento de Química Analítica, UGR), con una beca de colaboración. En 2009 comenzó sus estudios de Doctorado gracias a la obtención de una beca predoctoral de la Junta de Andalucía, y un año más tarde finalizó el Máster Oficial en Tecnología y Calidad de los Alimentos (UGR). Recientemente ha defendido la tesis doctoral internacional titulada "Micotoxinas: Aproximaciones analíticas y metabolómicas" obteniendo una calificación de Apto Cum Laude. Ha realizado una estancia predoctoral en la facultad de Ciencias Farmacéuticas en la Universidad de Gante (Bélgica). Actualmente disfruta de un contrato para doctores de la Universidad de Granada, continuando su investigación en el análisis de micotoxinas en alimentos.

TESIS DOCTORALES GRASEQA
35
LA ESPECTROSCOPÍA VIBRACIONAL COMO HERRAMIENTA ANALÍTICA. APLICACIONES EN ÁREAS DE INTERÉS CIENTÍFICO, AGRÍCOLA E INDUSTRIAL
MACARENA LÓPEZ SÁNCHEZ (Universidad de Jaén) DIRECTORES: MARÍA JOSÉ AYORA CAÑADA y ANTONIO MOLINA DÍAZ GRUPO: FQM-‐363/FQM-‐323 FECHA: 14/02/2013 Esta Tesis pretende contribuir a la consolidación de la espectroscopia vibracional como respuesta a necesidades analíticas actuales diversas. Se han dividido los métodos desarrollados en dos enfoques: análisis directo de muestras sólidas o semi-‐sólidas y análisis en flujo. Entre los métodos de análisis directo, se ha descrito un procedimiento para analizar muestras de productos farmacéuticos sólidos en polvo usando espectroscopía FT-‐Raman, con especial atención en el desarrollo de una metodología para la obtención de espectros de forma muy reproducible. Se ha descrito también la determinación rápida de dietilenglicol (DEG) en productos dentales adulterados combinando la espectroscopia infrarroja con reflexión total atenuada (ATR-‐FTIR) con regresión por mínimos cuadrados parciales (PLS). También se ha evaluado el potencial de las espectroscopias ATR-‐FTIR y FT-‐Raman para investigar los cambios acaecidos durante el desarrollo y maduración de la aceituna. Se relacionaron los cambios en una serie de bandas características de los espectros con la evolución del contenido de algunos constituyentes de la aceituna: aceite, agua, carotenoides y polifenoles. En la segunda parte de esta Tesis, se contemplan las ventajas ofrecidas por los métodos de análisis en flujo en el procesamiento de muestra automatizado. Se han desarrollado métodos clásicos de análisis por inyección en flujo (FIA) así como los más actuales de análisis por inyección secuencial (SIA) con detección por espectroscopía vibracional. Se ha presentado un ensayo enzimático para la monitorización de la reacción de la fructosa-‐1,6-‐bisfosfatasa con fructosa 1,6-‐bisfosfato de forma directa sin empleo de marcadores mediante espectroscopia de infrarrojo medio. Finalmente, se ha descrito un sensor en flujo con detección por espectroscopia Raman por transformada de Fourier para sulfonamidas. Dicho sensor permite la determinación rápida de sulfatiazol y sulfametoxazol en presencia de diferentes especies, principios activos y excipientes y puede aplicarse a preparaciones farmacéuticas en distintas formas de presentación. Todas las aplicaciones han demostrado ser modos rápidos y fiables de resolver problemas reales haciendo uso de la espectroscopia vibracional. REFERENCIAS
Macarena López-‐Sánchez, María José Ruedas-‐Rama, Antonio Ruiz-‐Medina, Antonio Molina-‐Díaz, María José Ayora-‐Cañada. Pharmaceutical powders analysis using FT-‐Raman spectrometry: Simultaneous determination of sulfathiazole and sulfanilamide. Talanta 74 (2008) 1603-‐1607.
María José Ruedas Rama, Macarena López-‐Sánchez, Antonio Ruiz-‐Medina, Antonio Molina-‐Díaz and María José Ayora-‐Cañada. Flow-‐through sensor with Fourier transform Raman detection for determination of sulfonamides. The Analyst 130 (2005) 1617-‐1623.
M. López-‐Sánchez, A. Domínguez-‐Vidal, M.J. Ayora-‐Cañada, A. Molina-‐Díaz. Assessment of dentifrice adulteration with diethylene glycol by means of ATR-‐FTIR spectroscopy and chemometrics. Analytica Chimica Acta 620 (2008) 113-‐119.
Macarena López-‐Sánchez, María José Ayora-‐Cañada and Antonio Molina-‐Díaz Olive Fruit Growth and Ripening as Seen by Vibrational Spectroscopy. Journal of Agricultural and Food Chemistry 58

BOLETÍN GRASEQA Nº 6 OCTUBRE 2013
36
(2010) 82-‐87. M. López-‐Sánchez, M. J. Ayora-‐Cañada, A. Molina-‐Díaz, M. Siam, W. Huber, G. Quintás, S.
Armenta, B. Lendl. Determination of enzyme activity inhibition by FTIR spectroscopy on the example of fructose bisphosphatase. Analytical and Bioanalytical Chemistry 394 (2009) 21372144.
CURRICULUM VITAE
Macarena López Sánchez (Jaén, 1980). Licenciada en Química (Universidad de Jaén, 2002). En 2003 comenzó los estudios de doctorado. En 2004 comienza su labor investigadora en el área de Química Analítica de la Universidad de Jaén, consiguiendo una Beca para formación de doctores de la Junta de Andalucía en diciembre de 2004 de cuatro años de duración. El trabajo de su Tesis doctoral se ha centrado en el empleo de la Espectroscopia Vibracional aplicada a muestras diversas utilizando métodos de análisis directo de muestras sólidas o semisólidas y métodos de análisis en flujo.
agosto de 2008), completando su formación en Espectroscopia Vibracional y sistemas de análisis en flujo. Desde febrero de 2009, trabaja como técnico de apoyo a la docencia e investigación en el área de determinación estructural del Centro de Instrumentación Científico-‐técnica de la Universidad de Jaén.

TESIS DOCTORALES GRASEQA
37
DESARROLLO DE METODOLOGÍAS ANALÍTICAS PARA LA DETERMINACIÓN DE
DAVID MORENO GONZÁLEZ (Universidad de Granada) DIRECTORES: LAURA GÁMIZ GRACIA, JUAN MANUEL BOSQUE SENDRA y ANA MARÍA GARCÍA CAMPAÑA GRUPO: FQM-‐302 FECHA: 07/05/2013 En esta Tesis se han desarrollado diferentes métodos para la determinación de residuos de carbamatos en alimentos y muestras medioambientales, considerando las recientes e importantes mejoras de las técnicas separativas en cuanto a miniaturización y aumento de la eficacia, y con objeto de explorar sus indudables ventajas. Estas técnicas, acopladas a sistemas de detección como la espectrofometría UV-‐Vis y la MS/MS permiten la cuantificación e identificación inequívoca de estos compuestos a las bajas concentraciones esperadas en estas matrices. A continuación se describe brevemente el trabajo desarrollado: o En el primer capítulo se describe el método desarrollado para la determinación de seis
carbamatos en muestras vegetales y aguas de diversa procedencia mediante HPLC capilar-‐UV como técnica instrumental y extracción en fase sólida (SPE) como técnica de tratamiento de muestra, con diferentes tipos de sorbentes dependiendo de la matriz.
o En el segundo capítulo se propone la determinación de doce carbamatos en diferentes zumos de frutas empleando CE-‐UV, aplicando la microextracción líquido-‐líquido dispersiva (DLLME) como tratamiento de muestra.
o En el tercer capítulo se empleó el acoplamiento CE-‐MS empleando una interfase de flujo coaxial o sheath-‐flow y un surfactante volátil como electrolito de fondo, para la determinación de diecisiete carbamatos en aguas de diversa procedencia y la DLLME como tratamiento de muestra.
o El siguiente capítulo consistió en la evaluación de un prototipo de interfase sin flujo adicional (sheathless) para el acoplamiento CE-‐MS. El método desarrollado se aplicó a los mismos analitos considerados en el capítulo anterior y se validó para aguas destinadas al consumo humano.
o El quinto capítulo describe un método de análisis para la determinación de veinticinco carbamatos en muestras de vino. Mediante UHPLC-‐MS/MS se consiguió la separación cromatográfica de estos plaguicidas en menos de cinco minutos. Además, se propuso un nuevo tratamiento de muestra llamado microextracción asistida por ultrasonidos con emulsificación mejorada con surfactante (UASEME).
Finalmente, en el último capítulo se evaluó la metodología QuEChERS para el análisis de treinta y tres carbamatos en productos botánicos (tés, manzanilla, tila, valeriana y tomillo) empleando de nuevo UHPLC-‐MS/MS. REFERENCIAS
D. Moreno-‐González, J.F. Huertas-‐Pérez. L. Gámiz-‐Gracia, A.M. García-‐Campaña. Determination of carbamates at trace levels in water and cucumber by capillary liquid chromatography. Int. J. Environ. Anal. Chem. 91 (2011) 1329-‐1340.
D. Moreno-‐González, L. Gámiz-‐Gracia, A.M. García-‐Campaña, J.M. Bosque-‐Sendra. Use of dispersive liquid-‐liquid microextraction for the determination of carbamates in juice samples by sweeping-‐micellar electrokinetic chromatography. Analytical and Bioanalytical Chemistry 400 (2011) 1329-‐1338.
D. Moreno-‐González, L. Gámiz-‐Gracia, J.M. Bosque-‐Sendra, A.M. García-‐Campaña. Dispersive liquid-‐liquid microextraction using a low density extraction solvent for the determination of 17 n-‐methylcarbamates by micellar electrokinetic chromatography-‐electrospray-‐mass spectrometry

BOLETÍN GRASEQA Nº 6 OCTUBRE 2013
38
employing a volatile surfactant. Journal of Chromatography A 1247 (2012) 26-‐34. D. Moreno-‐González, J.F Huertas-‐Pérez, A.M. García-‐Campaña, J.M Bosque-‐Sendra, L. Gámiz-‐Gracia.
Ultrasound-‐assisted surfactant-‐enhanced emulsification microextraction for the determination of carbamates in wines by ultra-‐high performance liquid chromatography tandem mass spectrometry. Journal of Chromatography A (1315 (2013) 1-‐7.
D. Moreno-‐González, J.F Huertas-‐Pérez, L. Gámiz-‐Gracia, J.M Bosque-‐Sendra, A.M. García-‐Campaña. Simultaneous analysis of thirty-‐three carbamates in herbal products by UHPLC-‐MS/MS using QuEChERS methodology. Food Chemistry (enviado para su publicación).
D. Moreno-‐González, R. Haselberg, L. Gámiz-‐Gracia, J.M. Bosque-‐Sendra, A. M. García-‐Campaña, G.J. de Jong, J.Sastre Toraño, G. W. Somsen. Highly sensitive micellar electrokinetic chromatography-‐mass spectrometry employing a volatile surfactant and sheathless porous tip interface. Analytical Chemistry (enviado para su publicación).
CURRICULUM VITAE
David Moreno González (Granada, 1981) estudió Química en la Universidad de Granada obteniendo su título de Licenciado en 2006, y comenzó a investigar en el grupo FQM-‐302 (Departamento de Química Analítica, UGR). En 2008 comenzó sus estudios de Doctorado, finalizando en 2013. El trabajo llevado a cabo en su tesis doctoral se ha centrado en el desarrollo de diferentes métodos para la determinación de contaminantes, fundamentalmente carbamatos, en alimentos y muestras medioambientales mediante HPLC capilar, UHPLC y CE con diversos sistemas de detección. Ha realizado una estancia predoctoral en el Departamento de Análisis Biomolecular de la Universidad de Utrecht (Holanda), que consistió en la evaluación de un prototipo de interfase sin flujo adicional (sheathless) para el acoplamiento CE-‐MS. Actualmente disfruta de un contrato de la Universidad de Granada, continuando su investigación en el análisis de residuos de contaminantes en alimentos.

TESIS DOCTORALES GRASEQA
39
AVANCES EN LA DETERMINACIÓN DE RESIDUOS DE HERBICIDAS Y CEFALOSPORINAS EN MUESTRAS AMBIENTALES Y ALIMENTARIAS, MEDIANTE
TÉCNICAS MINIATURIZADAS CAROLINA QUESADA MOLINA (Universidad de Granada)
DIRECTORES: ANA MARÍA GARCÍA CAMPAÑA y M. MONSALUD del OLMO-‐IRUELA GRUPO: FQM-‐302 FECHA: 14/05/2013 En esta Tesis se han desarrollado diferentes métodos analíticos para la determinación de residuos de herbicidas y cefalosporinas, en alimentos y muestras ambientales. Se han evaluado técnicas separativas miniaturizadas, como la electroforesis capilar (CE) y cromatografía líquida capilar (capilar HPLC). Además se han propuesto tratamientos de muestra alternativos a los existentes en bibliografía, obteniéndose un incremento en la eficacia y una disminución del tiempo invertido en esta etapa. El primer capítulo de la Tesis, describe un método para la determinación de los principales productos de degradación del herbicida metribuzin en muestras de suelo y agua, utilizando CZE-‐UV. Aplica un método de preconcentración en línea, denominado apilamiento de gran volumen de muestra (LVSS). El tratamiento de muestra en suelos se basa en la extracción líquida presurizada (PLE), seguida de extracción en fase sólida (SPE). En las muestras de agua fue suficiente la SPE. Además se han calculado los valores de las constantes de disociación (pKa) de estos compuestos utilizando CZE. En el segundo capítulo se han determinado cinco sulfonilureas en muestras de uva y aguas de distinta procedencia empleando LVSS-‐CZE-‐UV. Se utilizó la SPE con relleno de HLB para las muestras de agua y C18 para las muestras de uva. El tercer capítulo presenta un método para la determinación de cinco cefalosporinas pertenecientes a la familia de -‐lactamas en aguas. En este caso se ha utilizado la SPE, combinada con LVSS como procedimiento de preconcentración en línea acoplado a CZE-‐UV. Se consiguen así límites de detección suficientemente bajos, lo que permite su aplicación a matrices donde estos residuos presentan niveles de concentración muy bajos. La determinación de cefalosporinas en carne y agua se ha desarrollado en el capítulo cuarto. Se utilizó la HPLC capilar con detección UV y extracción líquido-‐líquido asistida por sales (SALLE) para el tratamiento de la muestra. La formación previa de un par iónico entre las cefalosporinas y el CTAB mejora la eficacia de la extracción. El quinto capítulo, describe la síntesis y eficacia de un polímero impreso molecularmente (MIP) para la extracción específica de tres cefalosporinas en muestras de leche, utilizando HPLC-‐UV para las medidas analíticas. REFERENCIAS
C. Quesada-‐Molina, A.M. García-‐Campaña, M. del Olmo-‐Iruela. -‐paired extraction of cephalosporins in acetone prior to their analysis by capillary liquid chromatography in
. Talanta 115 (2013) 943-‐949.
C. Quesada-‐Molina, M. del Olmo-‐Iruela, A.M. García-‐Campaña. "Analysis of cephalosporin residues in environmental waters by capillary zone electrophoresis with off-‐line and on-‐line preconcentration". Analytical Methods 4 (2012) 2341-‐2347.

BOLETÍN GRASEQA Nº 6 OCTUBRE 2013
40
F.J. Lara, M. del Olmo-‐Iruela, C. Cruces-‐Blanco, C. Quesada-‐Molina, A.M. García-‐Campaña. -‐lactam antibiotics by liquid chromatography". Trends in
Analytical Chemistry 38 (2012) 52-‐66.
C. Quesada-‐Molina, B. Claude, A.M. García-‐Campaña, M. del Olmo-‐Iruela, P. Morin. "Convenient solid phase extraction of cephalosporins in milk using a molecularly imprinted polymer. Food Chemistry 135 (2012) 775-‐779.
C. Quesada-‐Molina, M. del Olmo-‐Iruela, A.M. García-‐Campaña. ermination of sulfonylurea herbicides in water and grape samples by capillary zone electrophoresis using large
Analytical and Bioanalytical Chemistry, 397 (2010) 2593-‐2601. C. Quesada-‐Molina, A.M. García-‐Campaña, M. del Olmo-‐Iruela. Large volume sample stacking
in capillary zone electrophoresis for the monitoring of the degradation products of metribuzin J. Chromatography A, 1164 (2007) 320-‐328.
CURRICULUM VITAE
Carolina Quesada Molina (Granada, 1979). Licenciada en Química (Universidad de Granada, 2004). En 2004 comienza su colaboración en el grupo FQM-‐302 (Departamento de Química Analítica, UGR). En 2005 comenzó los estudios de doctorado. En Octubre de 2008 consigue una Beca predoctoral FPI, dentro del programa de Formación de Personal Investigador (MICINN). El trabajo llevado a cabo en su Tesis doctoral se ha centrado en el desarrollo de diferentes métodos para la determinación de herbicidas y antibióticos en muestras ambientales y alimentarias. Ha realizado una estancia predoctoral en el Instituto de Química Orgánica y Analítica de la Universidad de Orleans (Francia), centrado en la síntesis y evaluación de polímeros impresos molecularmente (MIP) para el análisis de cefalosporinas en leche. Actualmente disfruta de un contrato puente de la Universidad de Granada, continuando su investigación en el análisis de residuos en alimentos.

ACTUALIDAD GRASEQA
41
ACTUALIDAD GRASEQA
Mercedes Gallego recibe la Medalla de Investigación de la Junta de Andalucía
El pasado 28 de Febrero de 2013 la Catedrática Mercedes Gallego Fernández del Departamento de Química Analítica de la Universidad de Córdoba fue galardonada con la medalla de investigación de la Junta de Andalucía. Mercedes Gallego, natural de Jerez de la Frontera, obtuvo la Licenciatura en Química en la Universidad Hispalense en el año 1977 y el grado de Doctor en la Universidad de Córdoba en 1980 para obtener posteriormente la Cátedra que actualmente ocupa. Su labor investigadora se ha centrado principalmente en el desarrollo de metodologías para el análisis de aguas, alimentos y fluidos biológicos, uso de técnicas cromatográficas con detección por espectrometría de masas, miniaturización y automatización. Fruto de esta intensa labor es la publicación de más de 200 artículos científicos en revistas internacionales de alto índice de impacto. Además es autora de diversas patentes y ha participado en diferentes Proyectos de Investigación I+D+i del Plan nacional, de Excelencia de la Junta de Andalucía y de la Unión Europea.
En la última década la Dra. Gallego ha realizado estudios muy novedosos sobre los compuestos emergentes cancerígenos originados en piscinas como sub-‐productos de desinfección del agua que han tenido una gran repercusión tanto a nivel internacional como nacional, siendo reconocidos estos estudios como muy importantes por la Agencia de Protección del Medio Ambiente (EPA) y por muchos organismos internacionales dedicados a la protección medioambiental. Mercedes Gallego ha sido merecedora de este premio por su meritoria labor investigadora y docente y por su carácter abierto y compresivo que le ha permitido el rodearse de personas que se han impregnado de su espíritu voluntarioso para conseguir unos excelentes resultados.
(E. Ballesteros (UJAEN) y L. Lunar (UCO))

BOLETÍN GRASEQA Nº 6 OCTUBRE 2013
42
ACTUALIDAD GRASEQA
Durante los días 17 a 19 del pasado mes de Junio se celebraron en el Palacio de Congresos Hospital de Santiago, Úbeda, la XVIII Reunión de la Sociedad Española de Química Analítica y VI Reunión de la Sociedad Española de Espectrometría de Masas (http://www.ujaen.es/eventos/seqa2013/ y http://www.ujaen.es/eventos/seem2013/), con un total de más de 320 asistentes y con una muy elevada participación de jóvenes investigadores. El programa, de elevado nivel científico, incluyó 8 conferencias plenarias, 8 sesiones de comunicaciones, una sesión de docencia y la de jóvenes investigadores, además de las sesiones de posters y de comunicaciones flash orales. Se presentaron unas 300 comunicaciones (de ellas, 40 orales) y contó con la participación de destacados científicos nacionales y extranjeros.
El programa incluía también dos minisimposios: uno sobre Arqueometría en el que los investigadores presentaron el papel de la Química Analítica puesta al servicio del estudio del patrimonio cultural, y otro sobre la Calidad del aceite de Oliva, en el que se planteó y discutió el papel que nuestra disciplina juega en este campo y los últimos avances y técnicas analíticas usadas para este fin. En el seno de las Reuniones se celebraron también la reunión del grupo de especiación, la Asamblea SEQA y la Asamblea SEEM. Además, el día 16, previo al comienzo de la Reuniones, la SEQA organizó el Workshop que tuvo una excelente acogida entre nuestros jóvenes investigadores.

ACTUALIDAD GRASEQA
43
El programa ha cubierto prácticamente todos los campos de la Química Analítica/Espectrometría de Masas, incluyendo Arqueometría, Bioanálisis y análisis forense, Medio ambiente, Calidad y seguridad de los alimentos, Nanoanálisis y miniaturización, Quimiometría y Cualimetría, Desarrollo instrumental y metodológico, Química Analítica Verde, Análisis de superficies y análisis por imagen, Especiación y trazas, Electroanálisis, Docencia…
Se otorgaron más de 100 becas a jóvenes investigadores, para la participación bien en una de las dos reuniones o, en su caso, a las dos. Además, la SEQA otorgó otras 50 ayudas adicionales para los asistentes al Workshop. Se otorgaron también 8 premios a las mejores contribuciones en forma de poster, así como el premio SEQA 2013 para jóvenes investigadores y el premio SEQA 2013 a la mejor Tesis Doctoral.
El Palacio de Congresos Hospital de Santiago resultó un marco ideal y acogedor para la celebración de las reuniones, que se desarrolló, como es habitual, en un ambiente distendido y de cordialidad, cumplió su objetivo, que era presentar y discutir los avances científicos y técnicos y los desarrollos e innovaciones en el área de la Química Analítica y la Espectrometría de Masas, así como debatir sobre los aspectos docentes de la Química Analítica, lo que permitió compartir e intercambiar experiencias y favorecer el contacto personal y, además, la incorporación y participación de los jóvenes en este foro profesional. Los actos sociales incluyeron un cóctel de recepción, una visita guiada nocturna a Úbeda y la cena. (A. Molina, UJAEN)

BOLETÍN GRASEQA Nº 6 OCTUBRE 2013
AGENDA
CONGRESO: 6th International Symposium on Recent Adavances in Food Analysis FECHAS: 5-‐8 de Noviembre de 2013 SEDE: Praga (República Checa). INFORMACIÓN: www.secyta2013.ull.es
CONGRESO: 65th Pittsburg Conference on Analytical Chemistry and Applied Spectroscopy (PITTCON 2014) FECHAS: 2-‐6 de Marzo de 2014 SEDE: Chicago (EE.UU.). INFORMACIÓN: www.pittcon.org
CONGRESO: 30th International Symposium on Microscale Bioseparations FECHAS: 27 Abril-‐1 de Mayo de 2014. SEDE: Pecs, Hungría. INFORMACIÓN: www.msb2014.org
CONGRESO: 24th Anniversary World Congress on Biosensors (Biosensors 2014) FECHAS: 27-‐30 de Mayo de 2014 SEDE: Melbourne, Australia INFORMACIÓN: www.biosensors-‐congress.elsevier.com
CONGRESO: 62nd ASMS Conference on Mass Spectrometryand Allied Topics FECHAS: 15-‐19 de Junio de 2014 SEDE: Baltimore, EE.UU. INFORMACIÓN: www.asms.org
CONGRESO: The 38th International Symposium on Environmental Analytical Chemistry-‐ISEAC38 (including Conference on Food Safety (food analytics and authencity control) FECHAS: 17-‐20 de Junio de 2014 SEDE: Lausana, Suiza INFORMACIÓN: http://www.iseac38.ch
CONGRESO: XIV Reunión del Grupo Regional Andaluz de la Sociedad Española de Química Analítica (XIV GRASEQA 2014) FECHAS: Junio 2014 SEDE: Universidad Internacional de Andalucía, Sede Antonio Machado, Baeza (Jaén). INFORMACIÓN: [email protected]
CONGRESO: 10th European Pesticide Residue Workshop (EPRW 2014) FECHAS: 30 Junio al 3 de Julio 2014 SEDE: Dublín (Irlanda). INFORMACIÓN: www.eprw2014.com
CONGRESO: 10th Annual LC/MS/MS Workshop on Environmental Applications and Food Safety FECHAS: 1-‐3 de Julio de 2014 SEDE: Barcelona. INFORMACIÓN: [email protected]
CONGRESO: DIOXIN 2014. 34th International Symposium on Halogenated Persistent Organic Pollutants FECHAS: 31 de Agosto a 5 de Septiembre de 2014. SEDE: Madrid INFORMACIÓN: www.dioxin2014.org
CONGRESO: ISC 2014 (30th International Symposium on Chromatography) FECHAS: 14 al 18 de Septiembre de 2014. SEDE: Salburgo, Austria. INFORMACIÓN: www.isc2014.at
CONGRESO: XIV JAI 2014 (14º Jornadas de Análisis Instrumental 2014 FECHAS: 29 de Septiembre al 3 de Octubre de 2014. SEDE: Barcelona. INFORMACIÓN: www.barter.es/JAI

BOLETÍN DE INSCRIPCIÓN
GRUPO REGIONAL ANDALUZ
D. / Dª :
Departamento / Sección :
Institución / Empresa :
Cargo :
DIRECCIÓN PROFESIONAL
Avda. / Calle / Plaza : Nº :
Localidad :
C.P. : Provincia :
Teléfono : Fax :
e-mail :
DIRECCIÓN PARTICULAR
Avda. / Calle / Plaza :
Nº : Portal : Esc. : Piso : Puerta : Letra :
Localidad :
C.P. : Provincia :
Teléfono : Móvil :
Fdo. :
ENVIAR A Juan Francisco García Reyes (Tesorería GRASEQA )
.
( por e-mail a: [email protected])

BOLETÍN DE INSCRIPCIÓN
GRUPO REGIONAL ANDALUZ
DOMICILIACIÓN BANCARIA(*)
Caja Ahorros / Banco:
Oficina / Sucursal :
Dirección :
Localidad :
Provincia :
Código Cuenta Cliente ( 20 dígitos ) _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _
ENVIAR A Juan Francisco García Reyes (Tesorería GRASEQA )
. ( por e-mail a: [email protected])
* Si ya se ha realizado el pago domiciliado de la cuota correspondiente al año en curso, debe ingresar directamente la cuota en: BANCO POPULAR ESPAÑOL JAÉN Urb. 20075 3272 61 0600048022
Caja Ahorros / Banco :
Oficina / Sucursal :
Dirección :
Localidad :
Provincia :
Muy Sres. míos:
A partir de la presente, agradecería a Vds. se sirvan cargar en mi Cuenta Corriente / Libreta de Ahorros Nº _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ abierta en esta entidad, los recibos de las cuotas anuales extendidas por el Grupo Regional Andaluz de la Sociedad Española de Química Analítica (GRASEQA)
Atentamente les saluda,
Fdo. :
ENVIAR A LA ENTIDAD BANCARIA

SOCIEDAD ESPAÑOLA DE QUÍMICA ANALITICA
Boletín de Inscripción Apellidos
Nombre
Titulación
Dirección Profesional
Universidad
C.P. Ciudad
Teléfono
Fax
Correo electrónico
Domicilio particular
C.P.y Ciudad
Teléfono
Nº de cuenta (20 dígitos)
Tipo de socio Numerario (Cuota 60,10 Euros)
Adherido Cuota: 30,05 Euros
* Jóvenes investigadores (menores de 35 años) / sin contrato de trabajo Enviar por e-‐mail a : [email protected] AVISO LEGAL Conforme a la Ley Orgánica, 15/1999 sobre Protección de datos de carácter personal, el firmante autoriza a SOCIEDAD ESPAÑOLA DE QUÍMICA ANALÍTICA al tratamiento de los datos personales incluidos en este impreso. Estos datos serán incluidos en el fichero (ASOCIADOS) bajo la responsabilidad de SOCIEDAD ESPAÑOLA DE QUÍMICA ANALÍTICA con la finalidad de llevar a cabo la gestión de marketing, comercial y administrativa.
Usted podrá ejercitar los derechos de acceso, rectificación, cancelación u oposición, dirigiéndose mediante carta o correo electrónico al Responsable del Fichero: [email protected]

Nº 6 – OCTUBRE 2013
Depósito Legal: J 1559-‐2011
ISSN: 2254-‐1241
GRUPO REGIONAL ANDALUZ SOCIEDAD
ESPAÑOLA DE QUÍMICA ANALÍTICA