gper functions as a tumor suppressor in triple-negative breast cancer cells
TRANSCRIPT

1 3
J Cancer Res Clin OncolDOI 10.1007/s00432-014-1620-8
ORIgInal aRtICle - CanCeR ReseaRCh
GPER functions as a tumor suppressor in triple‑negative breast cancer cells
Christine Weißenborn · Tanja Ignatov · Hans‑Joachim Ochel · Serban Dan Costa · Ana Claudia Zenclussen · Zoya Ignatova · Atanas Ignatov
Received: 5 February 2014 / accepted: 10 February 2014 © springer-Verlag Berlin heidelberg 2014
factors, such as radiation, and gPeR amount inversely cor-related with the p53 expression level.Conclusions Overall, our results establish the protective role in breast cancer tumorigenesis, and the cell surface expression of gPeR makes it an excellent potential thera-peutic target for triple-negative breast cancer.
Keywords gPeR · gPR30 · Breast cancer · tumor suppression · tnBC
Introduction
several distinct subtypes of invasive breast cancers, the most common cancer types in women, have been identified using microarray-based approach (Perou et al. 2000). One of them, the triple-negative breast cancer (tnBC), is nega-tive for estrogen, progesterone, and heR2 receptors, which limits the options for optimal adjuvant therapy. the iden-tification of potential therapeutic targets for this subtype remains a clinical challenge (Perou et al. 2000; schneider et al. 2008).
More than 50 % of breast cancer patients express high levels of the orphan g-protein-coupled receptor, gPR30, a membrane-bound estrogen receptor (gPeR) (arias-Pulido et al. 2010; Filardo et al. 2006; Ignatov et al. 2011; Kuo et al. 2007; liu et al. 2009; tu et al. 2009), whose expression is favorable for patients’ survival (arias-Pulido et al. 2010; Ignatov et al. 2011). similar results were also obtained for ovarian cancer patients (Ignatov et al. 2013a). the function of gPeR, however, in the disease pathology is controversial and still a subject of intense debate. the identification and first functional studies on gPeR pro-posed rapid nongenomic effects of estrogen (Prossnitz et al. 2008). studies with high-affinity non-steroidal receptor
Abstract Background the orphan, membrane-bound estrogen receptor (gPeR) is expressed at high levels in a large frac-tion of breast cancer patients and its expression is favorable for patients’ survival.Methods We investigated the role of gPeR as a poten-tial tumor suppressor in triple-negative breast cancer cells MDa-MB-231 and MDa-MB-468 using cell cycle analy-sis and apoptosis assay. the constitutive activity of gPeR was investigated.Results gPeR-specific activation with g-1 agonist inhib-ited breast cancer cell growth in concentration-dependent manner via induction of the cell cycle arrest in g2/M phase, enhanced phosphorylation of histone h3 and caspase-3-me-diated apoptosis. analysis of the methylation status of the gPeR promoter in the triple-negative breast cancer cells and in tissues derived from breast cancer patients revealed that gPeR amount is regulated by epigenetic mechanisms and gPeR expression is inactivated by promoter methyla-tion. Furthermore, gPeR expression was induced by stress
C. Weißenborn · t. Ignatov · s. D. Costa · a. Ignatov (*) Department of Obstetrics and gynecology, University of Magdeburg, gerhart-hauptmann str 35, Magdeburg, germanye-mail: [email protected]
C. Weißenborn · a. C. Zenclussen Department of experimental Obstetrics and gynaecology, University of Magdeburg, Magdeburg, germany
h.-J. Ochel Department of Radiotherapy, University of Magdeburg, Magdeburg, germany
Z. Ignatova Department of Biochemistry and Biology, University of Potsdam, Potsdam, germany

J Cancer Res Clin Oncol
1 3
agonist g-1 and antagonist g-15 (Bologa et al. 2006; Den-nis et al. 2009) suggest that gPeR mediates the prolifera-tive effects of estrogen in many estrogen-related cancers (albanito et al. 2007; Filardo et al. 2000; Ignatov et al. 2010b, c; Vivacqua et al. 2006). Conversely, studies in vari-ous cell cultures show that gPeR can act as an inhibitor on cell growth and proliferation (ariazi et al. 2010; Chan et al. 2010; gao et al. 2011; holm et al. 2011; Wang et al. 2012). thereby, gPeR blocks the cell cycle progression in g1 or g2/M phase of estrogen receptor-positive breast cancer cells without any apoptotic effect (ahola et al. 2002; ariazi et al. 2010). In vivo, the gPeR expression is down-regulated during breast cancer progression, suggest-ing the potential role of gPeR in tumor suppression (Igna-tov et al. 2013b). the influence of various environmental and milieu-specific factors, including the presence or the absence of estrogen (Fan et al. 2009; Ignatov et al. 2010c; leblanc et al. 2007), may explain in part those polarized observations. an alternative plausible explanation might be the fact that gPeR-induced stimulation of cell prolif-eration is mainly observed after receptor stimulation with non-specific gPeR agonists such as estrogen and tamox-ifen (albanito et al. 2008; Bologa et al. 2006; Filardo et al. 2000, 2002; girgert et al. 2012; Ignatov et al. 2010c; Mag-giolini et al. 2004). Importantly, the tnBC is negative for estrogen receptor implying that underlying mechanisms that control the gPeR expression may significantly differ than in other type of breast cancer.
here, we demonstrate that gPeR mediates the inhibi-tory effect of g-1 on triple-negative breast cancer cells via induction of the cell cycle arrest and cell apoptosis. gPeR expression is regulated by epigenetic mechanisms both in vitro and in vivo. Moreover, gPeR expression is stimulated via radiation which acts as Dna-damaging agent. Our results propose that gPeR may act as a potential therapeu-tic target in tnBC.
Materials and methods
Cell culture and treatment
MDa-MB-231, MDa-MB-468, MCF-7, and heK-293 cells were obtained from Cell lines services (germany) and routinely cultured in DMeM/F12 (Pan Biotech) supplemented with 10 % FBs, 100 U/ml penicillin, and 100 μg/μl streptomycin at 37 °C in a humidified 5 % CO2 atmosphere. If necessary, the cells were synchronized by estrogen withdrawal for 24 h using phenol red-free medium supplemented with 10 % charcoal-stripped steroid-depleted FBs. thereafter, the cells were treated as indicated in the figure legends, and the cell count was measured with a Coulter counter as described (Filardo et al. 2006).
Mtt viability assay
Mtt (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazo-lium bromide)-viability assay was performed as already described (Ignatov et al. 2003). Briefly, 2,000 cells per well were seeded and cultured in a 96-well plate in the growth medium. after 24 h, the cells were stimulated for 3 days with g-1 or DMsO as a control. thereafter, Mtt was added for 3 h in the dark. the supernatants were removed, cells were lysed in 150 μl lysis buffer (isopropanol con-taining 4 mM hCl and 0.1 % nP-40), and the absorbance at 570 nm was recorded.
Cell cycle analysis and apoptosis assay
Cells were treated with 1 μM g-1 or medium for 1–3 days. Cell cycle distribution was analyzed by propidium iodide (PI) staining using flow cytometry. apoptosis was deter-mined with FItC annexin V apoptosis Detection Kit (BD, heidelberg) following the manufacturer’s protocol.
Real-time Rt-PCR
Rna isolation and quantitative Rt-PCR were performed as previously described (schumacher et al. 2009). We used the following primers for gPeR identification: forward, 5′-agtCggatgtgaggttCag-3′ and reverse, 5′-tCt-gtgtgaggagtaCaag-3′. gPeR mRna levels were normalized to the β-actin transcript levels. all reactions were carried out on an iCycler (BioRad, germany).
Western blot analysis
the following primary antibodies were used: sc-48524-R for gPeR (1:500 dilution; santa Cruz), ab13847 for cas-pase-3 (1:1,000 dilution; abcam), OP43 for p53 (1:2,000 dilution; Calbiochem), #4138 for cyclin B1 (dilution 1:1,000; Cell signalling), #9112 for Cdc2 (dilution 1:2,000; Cell signalling); #9701 phosphoh3 (ser10) (dilution 1:2,000; Cell signalling), and a5441 for β-actin (dilution 1:10,000; sigma-aldrich). Peroxidase-conjugated anti-rabbit or anti-mouse antibodies (thermo scientific), diluted 1:2,000 or 1:5,000, respectively, were used as secondary antibodies.
siRna treatment
gPeR knockout experiments were performed as already described (Ignatov et al. 2010c) after transfection of the cells with gPeR-specific siRna (sc-60743, santa Cruz), p53-specific siRna (sc-29435, santa Cruz), or scram-bled control siRna (sc-37007, santa Cruz). the trans-fection was performed with lipofectamine 2000 (life

J Cancer Res Clin Oncol
1 3
technologies) according to the manufacturer’s protocol. Cells were analyzed 48 h after transfection.
Methylation analysis of gPeR promoter region in breast cancer cells and tissue
the methylation status of the gPeR promoter region was determined by methylation-specific PCR (MsP) as previ-ously described (Ignatov et al. 2008, 2010a). the following primers, designed with the MethPrimer software (li and Dahiya 2002), were used:
me thylated forward 5′-ggttagtagggggCgtat tC-3′;
me thylated reverse 5′-taattaCgaatttCaCaatCt Cgta-3′;
un methylated forward 5′-taaatggttagtagggggt gtattt-3′;
un methylated reverse 5′-CataattaCaaatttCaCaa tCtCata-3′.
Breast cancer cells, breast cancer tissues, and normal breast specimens were analyzed. Dna was isolated with nucleospin® Dna extraction kit (Macherey & nagel). the initial bisulfite reaction that converts unmethylated cytosines to uracils was performed with the Cpgenome Dna modification Kit (s7820, Chemicon). the MsP con-sists of two individual PCRs with distinct primers specific for methylated and unmethylated Dna sequences. We used 100 nmol/l specific forward and reverse primers, 2 μl of 2 mmol/l dntPs, 5 μl PCR buffer, 0.75 U of gotaq poly-merase (Promega), and 100 ng of template Dna in a final volume of 25 μl. the PCR conditions were as follows: 95 °C for 10 min; followed by 40 cycles of denaturation at 94 °C for 1 min, annealing at 55 °C for 1 min, and amplifi-cation at 72 °C for 1 min. the PCR products were analyzed on 3 % agarose gel stained with ethidium bromide.
Methylated and unmethylated Dna sequences were always included in the analysis. gels were interpreted only in case of working positive control. the methylation of gPeR promoter was made using yes/no decision.
Radiation treatment of breast cancer cells
MDa-MB-231, MDa-MB-468, and MCF-7 cells were grown on culture plates to 70–80 % confluence and irra-diated with 0, 2, 5, and 10 gy at room temperature. the experiments were performed after 48-h incubation.
Bioinformatic tools and statistical analysis
the following software tools were used to predict methyla-tion sites in the 5′-regions upstream of the transcription start:
http://www.ebi.ac.uk/tools/emboss/cpgplot/index.html; http://bio.dfci.harvard.edu/Methylator/.
the curve fit and nonlinear regression analysis of the dose–response curves were performed with the graphPad Prism software. Data are present as mean ± sD. In all sta-tistical analyses, two-sided tests were applied. statistical significance was determined by student’s t-test, and p val-ues <0.05 were considered as statistically significant.
Results
gPeR stimulation with g-1 inhibited the growth of triple-negative breast cancer cells via cell cycle arrest in the M-phase and caspase-dependent cell apoptosis
triple-negative MDa-MB-231 and MDa-MB-468 breast cancer cell lines were stimulated with increasing concen-trations of gPeR-specific agonist g-1 for 3 days. g-1 caused a concentration-dependent inhibition of the cell growth with an IC50 value of 0.1 μM for MDa-MB-231 and 0.3 μM for MDa-MB-468 cells (Fig. 1a). notably, MDa-MB-231 cells expressed lower levels of gPeR mRna than the MDa-MB-468 cells (Fig. 1b). to con-firm the specificity of gPeR agonist g-1, we next down-regulated the gPeR expression with siRna. Intriguingly, the down-regulation of gPeR in the absence of g-1 was associated with increased cell growth in MDa-MB-231 and MDa-MB-468 cells. these data demonstrated the constitutive activity of gPeR. In both MDa-MB-231 and MDa-MB-468 cells, the specific knockdown of the gPeR expression abrogated the inhibitory effect of g-1 (Fig. 1c). the effect was specific to the triple-negative MDa-MB-231 and MDa-MB-468 breast cancer cells; no effect was observed in the control heK-293 cells lacking gPeR (Fig. 1c).
to assess the effect of g-1-stimulated gPeR on the cell cycle, we next analyzed the distribution of the cells in dif-ferent phases of the cell cycle using propidium iodide (PI) staining. the stimulation of MDa-MB-231 and MDa-MB-468 cells with 1 μM g-1 led to a significant accumula-tion of cells in g2/M phase compared to the control cells (Fig. 2a and b). the cell cycle arrest in the g2/M phase was already detectable after 24 h of g-1 stimulation and slightly decreased after 48 h (Fig. 2a and b). notably, a subpopulation of cells accumulated in the apoptotic sub-g1 phase upon g-1 treatment; the effect was stronger in MDa-MB-468 than in MDa-MB-231 cells (Fig. 2a and b).
to disentangle the nature of gPeR-induced cell cycle arrest in more details, we next investigated the expression of g2/M-phase-specific regulatory proteins cyclin B1 and Cdc2 and the phosphorylation of histone h3. g-1-induced cell cycle arrest enhanced the phosphorylation of histone

J Cancer Res Clin Oncol
1 3
h3, which is indicative of cells in mitotic phase. the effect was detectable even 6 h after g-1 stimulation of the cells and reached the highest value between 12 and 24 h (Fig. 2c and d). however, the levels of cyclin B1 and Cdc2 protein remained unchanged (Fig. 2c and d). gPeR knock down by siRna abrogated the g-1-induced cell cycle arrest and apoptosis (data not shown), suggesting again that g-1 effects are mediated by gPeR.
the observed accumulation of a sizeable fraction of cells in the apoptotic sub-g1 phase (Fig. 2a and b) led us determining the effect of gPeR on cell apoptosis. the number of apoptotic cells significantly increased in both MDa-MB-231 and MDa-MB-468 cells upon gPeR stimulation with g-1 compared to the background of
apoptotic cells among the non-stimulated cells (Fig. 3a and b): the number of apoptotic cells increased to 13.95 % in MDa-MB-231 cells (Fig. 3a) and to 24.34 % in MDa-MB-468 cells (Fig. 3b). In addition, pro-cas-pase-3 and caspase-3 increased in both MDa-MB-231 and MDa-MB-468 cells (Fig. 3c and d). taken together, these results suggest that g-1-induced gPeR activation inhibits the proliferation of triple-negative breast cancer cells via cell cycle arrest in the M-phase and caspase-dependent cell apoptosis. these analyses were performed in four additional cell lines: MCF-7, sK-BR-3, Bt-2, and MDa-MB-453. the g-1 inhibitory effect with cell cycle arrest and stimulation of cell apoptosis was observed in all cell lines investigated.
Cel
l gro
wth
(%
of c
ontr
ol)
0
50
100
150
-4-6-8-10
MDA-MB-231
MDA-MB-468
A
0
1
2
Rel
ativ
e m
RN
A e
xpre
ssio
nle
vels
of G
PE
R
MDA-MB-231 MDA-MB-468
B
C
Cel
lnum
ber
(x10
5 l)
0
100
300
400
+1µM G-1
HEK-293 MDA-MB-231
p = 0.0013 p = 0.0087
MDA-MB-468
+1µM G-1 +1µM G-1
G-1 concetration, log [M]
MDA-MB-468
MDA-MB-231
GPER
β-actin
GPER
β-actin
200
p = 0.0413p = 0.0140
Fig. 1 g-1 inhibits the growth of the triple-negative breast cancer cells. a Mtt viability assay of MDa-MB-231 and MDa-MB-468 cells treated with different concentrations of g-1 for 3 days. the number of proliferating cells is normalized to the untreated (control) cells. b qRt/PCR of the mRna expression level of gPeR presented as means of three independent replicates ±sD. gPeR signal was nor-malized to β-actin transcript levels. c silencing of the gPeR expres-sion abrogate the inhibitory effect of g-1. MDa-MB-231 and MDa-MB-486 cells were transiently transfected with gPeR-specific siRna
and followed by treatment with 1 μM g-1 for 72 h. thereafter, cell number was counted by cell counter. the expression level was nor-malized to cells transfected with scrambled siRna (control) and is presented as means of three experiments ±sD. heK-293 cells lack-ing gPeR were treated in the same way and served as control. Inset cells transfected with scrambled or gPeR-specific siRna. β-actin was used as a loading control. each experiment was repeated at least three times

J Cancer Res Clin Oncol
1 3
Promoter methylation controls gPeR expression
the observed inhibition of cell proliferation of the triple-negative breast cancer cells raised the intriguing question as to whether gPeR can act as a potential tumor suppressor in breast cancer. the clear dose-dependent inhibitory effect of the gPeR on the cell growth suggested to us that tuning the promoter by methylation may determine the gPeR expres-sion levels in the cell. We used 5-aza-2′-deoxycytidine (5-aza) to inhibit the Dna-methyltransferase, which is involved in the methylation of the promoter. Intriguingly, the treatment with 5-aza enhanced the gPeR mRna level in both MDa-MB-231 and MDa-MB-468 cells in a time-dependent manner (Fig. 4a and b).
to address whether methylation-dependent increase in the gPeR expression has the same effect on cell proliferation as the g-1-induced gPeR expression, we pre-treated the two cell lines with 5 μM 5-aza for 24 h and then incubated them with 1 μM g-1 for 72 h. Pre-treatment with 5-aza showed synergistic inhibitory effect to g-1 abrogation of the cell growth: the growth of MDa-MB-231 and MDa-MB-468 cells was reduced down to ~5 % by combined treatment with 5-aza and g-1 compared to the treatment with a single drug. these data clearly suggest that the inhibitory effect of the gPeR is proportional to its expression level and significantly decreases upon promoter methylation.
next, we analyzed the 5′-region upstream of the transla-tion start site of gPeR using MethPrimer software (li and
B
A
0
20
40
60
80
Sub G1
G-1 stimulation (h)0 24 48 72 0 24 48 72 0 24 48 72 0 24 48 72
G0/G1 S G2/M
p= 0.0032
p = 0.0002
p = 0.0084
p = 0.0404p = 0.0032
p = 0.0239
p = 0.0129
p = 0.0287
p = 0.0424
p = 0.0369
p = 0.0071
0
20
40
60
80
p = 0.0258
p = 0.0171
CC
cyclin B1
cdc2
G-1 C G-1 C G-1
0h 24h 48h 72h
β-actin
C G-1 C G-1 C G-1
0h 24h 48h 72h
C
phospho-H3
β -actin
G-1 C G-1 C G-10h 1h 6h 24h
C G-1 C G-1 C G-10h 1h 6h 24h
Per
cent
age
of c
ells
Per
cent
age
of c
ells
Sub G1
G-1 stimulation (h)0 24 48 72 0 24 48 72 0 24 48 72 0 24 48 72
G0/G1 S G2/M
D
cyclin B1
cdc2
β -actin
phospho-H3
β -actin
p = 0.0009
1 1.06 0.94 0.98 2.73 1.42 6.72 1 0.90 1.85 0.72 4.16 0.41 2.10
1 1.11 1.44 0.97 1.17 1.04 1.13
1 1.16 1.52 0.78 1.13 0.59 0.52
1 1.11 1.26 0.97 1.17 1.04 1.08
1 1.09 1.14 0.96 0.94 0.75 0.79
Fig. 2 g-1-stimulated gPeR inhibits the cell cycle in triple-negative breast cancer cells. Flow cytometry analysis of the distribution of MDa-MB-231 (a) and MDa-MB-468 (b) cells in different phases of the cell cycle after treatment with 1 μM g-1 or control for dif-ferent times. the results are presented as means of three independ-ent experiments ±sD. Western blot analysis of expression of cyclin
B1 and Cdc2 and phosphorylation of histone 3 in MDa-MB-231 (c) and MDa-MB-468 (d) cell stimulated with 1 μM g-1 or control for different times. β-actin was used as a loading control. Each Western blot is a representative example of three independent experiments. the numbers above the blots represent the relative expression units compared to the control. the control was set as 1

J Cancer Res Clin Oncol
1 3
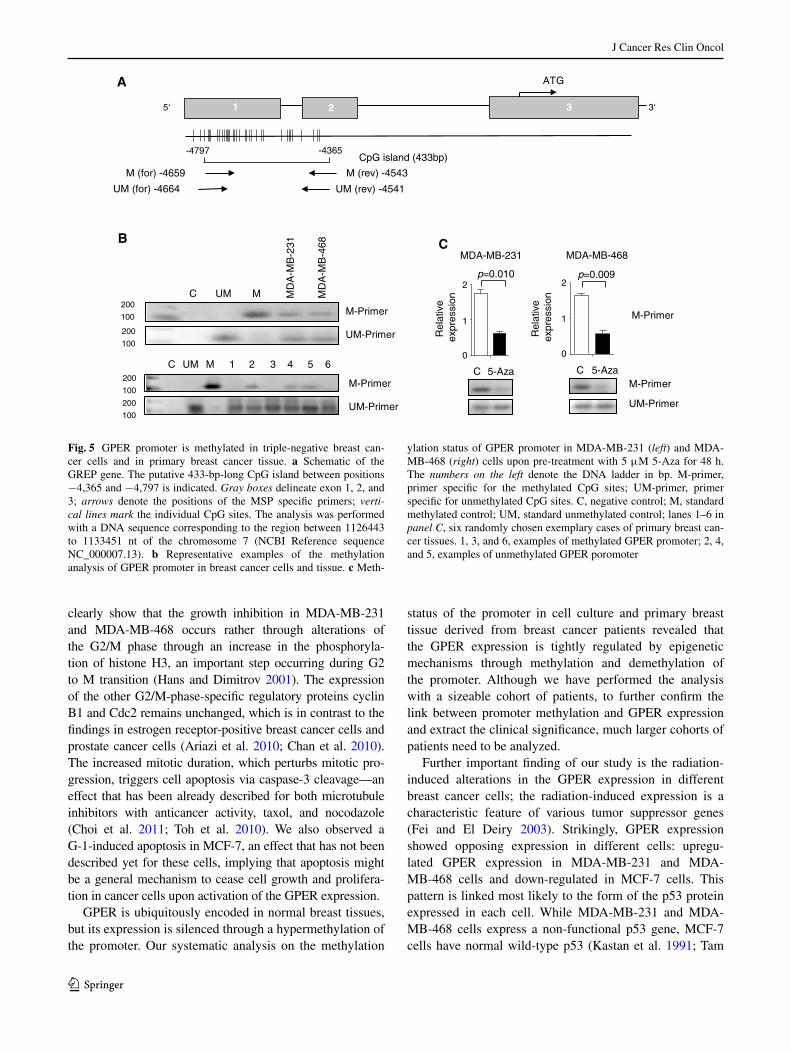
Dahiya 2002) to find any potential Cpg island(s). the hyper-methylation of Cpg islands in promoter region of tumor sup-pressor genes is associated with their inactivation (esteller et al. 2001). In the region between −4,365 and −4,797 nt upstream of the transcription start site, we identified a single 433-bp-long Cpg-reach island (Fig. 5a). Methylation-specific PCR approach confirmed experimentally the methylation within the gPeR promoter region in both breast cancer cell lines and primary breast cancer tissue (Fig. 5b). We analyzed the breast tissue of 30 breast cancer patients and compared it to a cohort of healthy individuals of the same size. In 17 out of 30 patients (56.7 %), the gPeR promoter was methylated, while the normal breast tissues contained only non-methyl-ated gPeR promoters. Furthermore, we also tested whether 5-aza alters the expression of gPeR because of changing the methylation status of the gPeR promoter. notably, pre-treat-ment with 5-aza led to a significant decrease in gPeR meth-ylation in MDa-MB-231 (p = 0.010) and MDa-MB-468 cells (p = 0.009) (Fig. 5c), thus corroborating the enhanced gPeR expression we observed in both triple-negative breast cancer cells (Fig. 4b and c).
stress-induced expression of gPeR in breast cancer cells
Important function of the tumor suppressor genes is to induce cell apoptosis and/or to arrest cell cycle progression
in response to Dna-damaging agents, such as radia-tion (sun and Yang 2010). We therefore hypothesized that gPeR as a potential tumor suppressor may also play a role in this processes. We next exposed the MDa-MB-231 and MDa-MB-468 cells to various doses of γ-radiation for 48 h and measured the levels of gPeR mRna. In both cell lines, we observed a clear dose-dependent increase in the gPeR mRna levels (Fig. 6a); the effect was much stronger in MDa-MB-231 cells. In addition, the expres-sion of the tumor suppressor p53 protein was also elevated upon radiation in both cell lines (Fig. 6b). note that MDa-MB-231 and MDa-MB-468 cells express a non-functional mutant of p53 (Kastan et al. 1991; lim et al. 2009). thus, we included in the analysis MCF-7 breast cancer cells that express wild-type p53 protein (lim et al. 2009). Intrigu-ingly, in MCF-7 cells, we observed the opposite effect: Upon radiation the, gPeR expression decreased progres-sively with the increasing dosage of radiation (Fig. 6a). thus, we hypothesized that p53 may modulate the expres-sion pattern of gPeR in an opposite direction. to test this hypothesis, p53 gene was silenced with a specific siRna in MCF-7 cells, and the expression of gPeR upon exposure of the cells to γ-radiation was monitored. p53 knockdown led to a significant increase in gPeR mRna expression after radiation (Fig. 6a), implying the direct role of wild-type p53 in gPeR expression. In addition, incubation with
0 72
Cel
lapo
ptos
is(%
)
p = 0.0007
p = 0.0064p = 0.0264
0
15
30
45A
C
B
Pro-caspase-3
Caspase-3
β-actin
24 48 0 72
Cel
l apo
ptos
is (
%)
p = 0.0026
p = 0.0015 p = 0.0005
0
15
30
45
24 48
Time (h) Time (h)
C G-1 C G-1
0h 24h 48h
MDA-MB-231 MDA-MB-468
D
Pro-caspase-3
Caspase-3
β-actin
C G-1 C G-1
0h 24h 48h
Control
G-1
Control
G-1
1 0.98 1.2 0.93 2.3 1 1 1.7 1.3 3.6
Fig. 3 g-1 induces caspase-dependent apoptosis in triple-negative breast cancer cells. the fraction of apoptotic cells increases in MDa-MB-231 (a) and MDa-MB-468 (b) cells upon treatment with 1 μM g-1 for different times. the apoptotic cells were stained with annexin V/propidium iodide, counted by flow cytometry, and normalized to the number of untreated cells. the results are presented as means of three independent replicates ±sD. the level of pro-caspase-3 and caspase-3 was enhanced in MDa-MB-231 (c) and MDa-MB-468 (d)
cells upon treatment with 1 μM g-1 for different times as indicated by Western blot. time zero represents the basal expression of cas-pase-3 before the addition of medium (c) and g-1 to the cells. β-actin was used as loading control. Each Western blot is a representative example of three independent experiments. the numbers above the blots represent the relative expression units compared to the control. the control was set as 1

J Cancer Res Clin Oncol
1 3
g-1 raised the p53 expression level in all three cell lines (Fig. 6c), corroborating the functional association between p53 and gPeR.
Discussion
here, we present an analysis on the effect of gPeR on tri-ple-negative breast cancer cells. Our observations clearly suggest that (1) gPeR-specific agonist inhibited breast can-cer cell proliferation in concentration-dependent manner, (2) gPeR activation leads to a cell cycle arrest and apop-tosis in cell-dependent fashion, (3) gPeR expression is down-regulated in breast cancer tumorigenesis, (4) aberrant gPeR expression is caused by a reversible promoter meth-ylation both in vivo and in vitro, and (5) gPeR expression is induced by stress factors such as radiation.
gPeR, a member of the gPCR family, is implicated in breast, endometrial, and ovarian cancers (Filardo et al. 2006; Fujiwara et al. 2012; smith et al. 2007, 2009). a large number of gPCR receptors, which are involved in various types of human cancers, are associated with an increased cell proliferation and tumor progression (Dorsam and
gutkind 2007). Our data clearly suggest that gPeR acti-vation in triple-negative breast cancer cells resembles the growth-specific alterations described for gPeR in other cancer types (Chan et al. 2010; Chimento et al. 2013; gao et al. 2011; holm et al. 2011; Ignatov et al. 2013a; Wang et al. 2012). the cell growth suppression through enhanced gPeR expression occurs via specific cell cycle arrest in the M-phase and caspase-3-mediated apoptosis. the effect is potentiated by the g-1 agonist, which is highly specific for the triple-negative breast cancer cells and is not a result of general cytotoxic effect as no growth defect was observed in gPeR-1-negative heK293 cells. although Wang et al. have very recently demonstrated that g-1-induced ovarian cancer cell inhibition is gPeR-independent (Wang et al. 2013), our observation suggested a gPeR-dependent activity of g-1, and these results have been confirmed by various resent observations (ariazi et al. 2010; Chan et al. 2010; Chimento et al. 2013; holm et al. 2011; Ignatov et al. 2013a). Unlike the estrogen receptor-positive MCF-7 breast cancer cells, for which g-1-induced growth inhibition through g1-phase block has been described (Fujiwara et al. 2012; gao et al. 2011), we did not observe any indications of g1-phase alter-ations in the triple-negative breast cancer cells. Our results
0
2
4
6
Rel
ativ
e m
RN
A e
xpre
ssio
nle
vels
of G
PR
30
8B
p = 0.0322
p = 0.0163
p = 0.0116
A
0
2
4
6
Rel
ativ
e m
RN
A e
xpre
ssio
nle
vels
of G
PR
30
0 24 48 72
Time (h)
p = 0.0297 p = 0.0313
0
50
100
150
Cel
l pro
lifer
atio
n (%
of c
ontr
ol)
p = 0.0338
p = 0.0489
5-Aza - + - +G-1 - - + +
D
p = 0.0016
p = 0.0157
C
Control
5-Aza
Control
5-Aza
0 24 48 72Time (h)
0
50
100
150
Cel
l pro
lifer
atio
n (%
of c
ontr
ol)
5-Aza - + - +G-1 - - + +
Fig. 4 Inhibition of promoter methylation enhances gPeR level. qRt-PCR quantification of the mRna expression of gPeR in MDa-MB-231 (a) and MDa-MB-468 (b) cells upon treatment with 5 μM 5-aza for different times. Control cells were treated the same way in a medium without 5-aza. Mtt-viability assay of MDa-MB-231
(c) and MDa-MB-468 (d) cells treated with 5 μM 5-aza for 48 h, followed by stimulation with 1 μM g-1 for 72 h. non-treated cells served as a control, and their viability was arbitrarily set as 100 %. the results are presented as means of three independent replicates ±sD
J Cancer Res Clin Oncol
1 3
clearly show that the growth inhibition in MDa-MB-231 and MDa-MB-468 occurs rather through alterations of the g2/M phase through an increase in the phosphoryla-tion of histone h3, an important step occurring during g2 to M transition (hans and Dimitrov 2001). the expression of the other g2/M-phase-specific regulatory proteins cyclin B1 and Cdc2 remains unchanged, which is in contrast to the findings in estrogen receptor-positive breast cancer cells and prostate cancer cells (ariazi et al. 2010; Chan et al. 2010). the increased mitotic duration, which perturbs mitotic pro-gression, triggers cell apoptosis via caspase-3 cleavage—an effect that has been already described for both microtubule inhibitors with anticancer activity, taxol, and nocodazole (Choi et al. 2011; toh et al. 2010). We also observed a g-1-induced apoptosis in MCF-7, an effect that has not been described yet for these cells, implying that apoptosis might be a general mechanism to cease cell growth and prolifera-tion in cancer cells upon activation of the gPeR expression.
gPeR is ubiquitously encoded in normal breast tissues, but its expression is silenced through a hypermethylation of the promoter. Our systematic analysis on the methylation
status of the promoter in cell culture and primary breast tissue derived from breast cancer patients revealed that the gPeR expression is tightly regulated by epigenetic mechanisms through methylation and demethylation of the promoter. although we have performed the analysis with a sizeable cohort of patients, to further confirm the link between promoter methylation and gPeR expression and extract the clinical significance, much larger cohorts of patients need to be analyzed.
Further important finding of our study is the radiation-induced alterations in the gPeR expression in different breast cancer cells; the radiation-induced expression is a characteristic feature of various tumor suppressor genes (Fei and el Deiry 2003). strikingly, gPeR expression showed opposing expression in different cells: upregu-lated gPeR expression in MDa-MB-231 and MDa-MB-468 cells and down-regulated in MCF-7 cells. this pattern is linked most likely to the form of the p53 protein expressed in each cell. While MDa-MB-231 and MDa-MB-468 cells express a non-functional p53 gene, MCF-7 cells have normal wild-type p53 (Kastan et al. 1991; tam
B
UM-Primer
A ATG
-4797
5‘
CpG island (433bp)
3‘1 3
-4365
25‘
M (for) -4659
UM (for) -4664
M (rev) -4543
UM (rev) -4541
C 1UM M 2 3 4 5 6
M-Primer
M-Primer
UM-Primer
C UM M MD
A-M
B-2
31
MD
A-M
B-4
68 CMDA-MB-468
M-Primer
UM-Primer
C 5-Aza
M-Primer
MDA-MB-231
C 5-Aza
0
1
2p=0.010
0
1
2p=0.009
Rel
ativ
e ex
pres
sion
Rel
ativ
e ex
pres
sion200
100
200
100
200
100
200
100
Fig. 5 gPeR promoter is methylated in triple-negative breast can-cer cells and in primary breast cancer tissue. a schematic of the gReP gene. the putative 433-bp-long Cpg island between positions −4,365 and −4,797 is indicated. Gray boxes delineate exon 1, 2, and 3; arrows denote the positions of the MsP specific primers; verti-cal lines mark the individual Cpg sites. the analysis was performed with a Dna sequence corresponding to the region between 1126443 to 1133451 nt of the chromosome 7 (nCBI Reference sequence nC_000007.13). b Representative examples of the methylation analysis of gPeR promoter in breast cancer cells and tissue. c Meth-
ylation status of gPeR promoter in MDa-MB-231 (left) and MDa-MB-468 (right) cells upon pre-treatment with 5 μM 5-aza for 48 h. the numbers on the left denote the Dna ladder in bp. M-primer, primer specific for the methylated Cpg sites; UM-primer, primer specific for unmethylated Cpg sites. C, negative control; M, standard methylated control; UM, standard unmethylated control; lanes 1–6 in panel C, six randomly chosen exemplary cases of primary breast can-cer tissues. 1, 3, and 6, examples of methylated gPeR promoter; 2, 4, and 5, examples of unmethylated gPeR poromoter

J Cancer Res Clin Oncol
1 3
et al. 1994). siRna-induced knockdown of the wild-type p53 in MCF-7 cells reverses the gPeR expression phe-notype to those of the MDa-MB-231 and MDa-MB-468 cells. thus, intact wild-type p53 exerts a protective role: It counteracts the gPeR-induced expression upon radiation. Moreover, g-1-induced activation of the breast cancer cell lines enhances the expression of p53. therefore, we sug-gest a feedback mechanism between the expression of p53 and gPeR: gPeR activation increases the p53 expression, which in turn down-regulates the expression of gPeR. In case of a non-functional p53, gPeR expression remains high and an alternative emergency mechanism is activated, which induces cell apoptosis. the cell surface expression of gPeR makes it an excellent target for tnBC.
Acknowledgments this work was supported by Deutsche Krebshilfe.
Conflict of interest this work was supported by a grant from Deutsche Krebshilfe to a.I.
References
ahola tM, Manninen t, alkio n, Ylikomi t (2002) g protein-cou-pled receptor 30 is critical for a progestin-induced growth inhibi-tion in MCF-7 breast cancer cells. endocrinology 143:3376–3384
albanito l, Madeo a, lappano R, Vivacqua a, Rago V, Carpino a, Oprea tI, Prossnitz eR, Musti aM, ando s, Maggiolini M (2007) g protein-coupled receptor 30 (gPR30) mediates gene expression changes and growth response to 17beta-estradiol and selective gPR30 ligand g-1 in ovarian cancer cells. Cancer Res 67:1859–1866
albanito l, sisci D, aquila s, Brunelli e, Vivacqua a, Madeo a, lap-pano R, Pandey DP, Picard D, Mauro l, ando s, Maggiolini M (2008) epidermal growth factor induces g protein-coupled recep-tor 30 expression in estrogen receptor-negative breast cancer cells. endocrinology 149:3799–3808
arias-Pulido h, Royce M, gong Y, Joste n, lomo l, lee sJ, Chaher n, Verschraegen C, lara J, Prossnitz eR, Cristofanilli M (2010) gPR30 and estrogen receptor expression: new insights into hor-mone dependence of inflammatory breast cancer. Breast Cancer Res treat 123:51–58
ariazi ea, Brailoiu e, Yerrum s, shupp ha, slifker MJ, Cunliffe he, Black Ma, Donato al, arterburn JB, Oprea tI, Prossnitz eR, Dun nJ, Jordan VC (2010) the g protein-coupled receptor gPR30 inhibits proliferation of estrogen receptor-positive breast cancer cells. Cancer Res 70:1184–1194
Bologa Cg, Revankar CM, Young sM, edwards Bs, arterburn JB, Kise-lyov as, Parker Ma, tkachenko se, savchuck nP, sklar la, Oprea tI, Prossnitz eR (2006) Virtual and biomolecular screening con-verge on a selective agonist for gPR30. nat Chem Biol 2:207–212
Chan QK, lam hM, ng CF, lee aY, Chan es, ng hK, ho sM, lau KM (2010) activation of gPR30 inhibits the growth of prostate cancer cells through sustained activation of erk1/2, c-jun/c-fos-dependent upregulation of p21, and induction of g(2) cell-cycle arrest. Cell Death Differ 17:1511–1523
Chimento a, Casaburi I, Bartucci M, Patrizii M, Dattilo R, avena P, ando s, Pezzi V, sirianni R (2013) selective gPeR activation
Fig. 6 γ-Radiation influences the expression of gPeR mRna. a qRt-PCR determination of the expression levels of gPeR mRna in MDa-MB-231, MDa-MB-468, MCF-7, and MCF-7 transfected with p53-specific siRna (MCF-7-p53 neg) cells after different doses of γ-radiation. b p53 expression in various breast cancer cells after different doses of γ-radiation monitored by Western blot. β-actin was used as a loading control. c p53 expression in MDa-MB-231, MDa-MB-468, and MCF-7 cells is altered upon exposure to 1 μM g-1 as compared to cells incubated in medium only (con-trol). the results are presented as means of three independent experiments ±sD. the numbers above the blots represent the relative expression units com-pared to the control. the control was set as 1
C
A
0
2.5
Rel
ativ
e ex
pres
sion
of
GP
ER
mR
NA
5
p = 0.032
p < 0.0001
p = 0.003
p = 0.005
p = 0.047
p = 0.034
p = 0.003
MCF-7-p53 negMDA-MB-468MDA-MB-231
0 2 5 10 0 2 5 10 0 2 5 10 Gy
p = 0.0023
p = 0.0097
MCF-7
0 2 5 10
B0 Gy 2 Gy 5 Gy 10 Gy
MDA-MB-468
MCF-7
MDA-MB-231
1.00 0.89 1.83 2.91
1.00 1.79 3.17 3.05
1.00 2.55 4.60 4.46
0h C G-1 C G-1
24h 48h
p53
β -actin
p53
β -actin
p53
β-actin
1.00 0.79 0.60 0.88 3.14
1.00 1.18 2.70 0.65 3.65
1.00 1.09 1.24 1.73 3.58

J Cancer Res Clin Oncol
1 3
decreases proliferation and activates apoptosis in tumor leydig cells. Cell Death Dis 4:e747
Choi hJ, Fukui M, Zhu Bt (2011) Role of cyclin B1/Cdc2 up-regula-tion in the development of mitotic prometaphase arrest in human breast cancer cells treated with nocodazole. Plos One 6:e24312
Dennis MK, Burai R, Ramesh C, Petrie WK, alcon sn, nayak tK, Bologa Cg, leitao a, Brailoiu e, Deliu e, Dun nJ, sklar la, hathaway hJ, arterburn JB, Oprea tI, Prossnitz eR (2009) In vivo effects of a gPR30 antagonist. nat Chem Biol 5:421–427
Dorsam Rt, gutkind Js (2007) g-protein-coupled receptors and can-cer. nat Rev Cancer 7:79–94
esteller M, Corn Pg, Baylin sB, herman Jg (2001) a gene hyper-methylation profile of human cancer. Cancer Res 61:3225–3229
Fan P, Yue W, Wang JP, aiyar s, li Y, Kim th, santen RJ (2009) Mecha-nisms of resistance to structurally diverse antiestrogens differ under premenopausal and postmenopausal conditions: evidence from in vitro breast cancer cell models. endocrinology 150:2036–2045
Fei P, el Deiry Ws (2003) P53 and radiation responses. Oncogene 22:5774–5783
Filardo eJ, Quinn Ja, Bland KI, Frackelton aR Jr (2000) estrogen-induced activation of erk-1 and erk-2 requires the g protein-cou-pled receptor homolog, gPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of hB-egF. Mol endocrinol 14:1649–1660
Filardo eJ, Quinn Ja, Frackelton aR Jr, Bland KI (2002) estrogen action via the g protein-coupled receptor, gPR30: stimulation of adenylyl cyclase and caMP-mediated attenuation of the epider-mal growth factor receptor-to-MaPK signaling axis. Mol endo-crinol 16:70–84
Filardo eJ, graeber Ct, Quinn Ja, Resnick MB, giri D, Delellis Ra, steinhoff MM, sabo e (2006) Distribution of gPR30, a seven membrane-spanning estrogen receptor, in primary breast cancer and its association with clinicopathologic determinants of tumor progression. Clin Cancer Res 12:6359–6366
Fujiwara s, terai Y, Kawaguchi h, takai M, Yoo s, tanaka Y, tanaka t, tsunetoh s, sasaki h, Kanemura M, tanabe a, Yamashita Y, Ohmichi M (2012) gPR30 regulates the egFR-akt cascade and predicts lower survival in patients with ovarian cancer. J Ovarian Res 5:35
gao F, Ma X, Ostmann aB, Das sK (2011) gPR30 activation opposes estrogen-dependent uterine growth via inhibition of stro-mal eRK1/2 and estrogen receptor alpha (eRalpha) phosphoryla-tion signals. endocrinology 152:1434–1447
girgert R, emons g, grundker C (2012) Inactivation of gPR30 reduces growth of triple-negative breast cancer cells: possi-ble application in targeted therapy. Breast Cancer Res treat 134:199–205
hans F, Dimitrov s (2001) histone h3 phosphorylation and cell divi-sion. Oncogene 20:3021–3027
holm a, Baldetorp B, Olde B, leeb-lundberg lM, nilsson BO (2011) the gPeR1 agonist g-1 attenuates endothelial cell pro-liferation by inhibiting Dna synthesis and accumulating cells in the s and g2 phases of the cell cycle. J Vasc Res 48:327–335
Ignatov a, lintzel J, Kreienkamp hJ, schaller hC (2003) sphingo-sine-1-phosphate is a high-affinity ligand for the g protein-cou-pled receptor gPR6 from mouse and induces intracellular Ca2+ release by activating the sphingosine-kinase pathway. Biochem Biophys Res Commun 311:329–336
Ignatov a, Bischoff J, schwarzenau C, Krebs t, Kuester D, herrmann K, Costa sD, Roessner a, semczuk a, schneider-stock R (2008) P16 alterations increase the metastatic potential of endometrial carcinoma. gynecol Oncol 111:365–371
Ignatov a, Bischoff J, Ignatov t, schwarzenau C, Krebs t, Kuester D, Costa sD, Roessner a, semczuk a, schneider-stock R (2010a) aPC promoter hypermethylation is an early event in endometrial tumorigenesis. Cancer sci 101(2):321–327
Ignatov t, eggemann h, semczuk a, smith B, Bischoff J, Roessner a, Costa sD, Kalinski t, Ignatov a (2010b) Role of gPR30 in endometrial pathology after tamoxifen for breast cancer. am J Obstet gynecol 203:595-16
Ignatov a, Ignatov t, Roessner a, Costa sD, Kalinski t (2010c) Role of gPR30 in the mechanisms of tamoxifen resistance in breast cancer MCF-7 cells. Breast Cancer Res treat 123:87–96
Ignatov a, Ignatov t, Weissenborn C, eggemann h, Bischoff J, sem-czuk a, Roessner a, Costa sD, Kalinski t (2011) g-protein-cou-pled estrogen receptor gPR30 and tamoxifen resistance in breast cancer. Breast Cancer Res treat 128:457–466
Ignatov t, Modl s, thulig M, Weissenborn C, treeck O, Ortmann O, Zenclussen a, Costa sD, Kalinski t, Ignatov a (2013a) gPeR-1 acts as a tumor suppressor in ovarian cancer. J Ovarian Res 6:51
Ignatov t, Weissenborn C, Poehlmann a, lemke a, semczuk a, Roessner a, Costa sD, Kalinski t, Ignatov a (2013b) gPeR-1 expression decreases during breast cancer tumorigenesis. Cancer Invest 31:309–315
Kastan MB, Onyekwere O, sidransky D, Vogelstein B, Craig RW (1991) Participation of p53 protein in the cellular response to Dna damage. Cancer Res 51:6304–6311
Kuo Wh, Chang lY, liu Dl, hwa hl, lin JJ, lee Ph, Chen Cn, lien hC, Yuan Rh, shun Ct, Chang KJ, hsieh FJ (2007) the interactions between gPR30 and the major biomarkers in infil-trating ductal carcinoma of the breast in an asian population. tai-wan J Obstet gynecol 46:135–145
leblanc K, sexton e, Parent s, Belanger g, Dery MC, Boucher V, asselin e (2007) effects of 4-hydroxytamoxifen, raloxifene and ICI 182 780 on survival of uterine cancer cell lines in the presence and absence of exogenous estrogens. Int J Oncol 30:477–487
li lC, Dahiya R (2002) MethPrimer: designing primers for methyla-tion PCRs. Bioinformatics 18:1427–1431
lim lY, Vidnovic n, ellisen lW, leong CO (2009) Mutant p53 medi-ates survival of breast cancer cells. Br J Cancer 101:1606–1612
liu Q, li Jg, Zheng XY, Jin F, Dong ht (2009) expression of CD133, PaX2, esa, and gPR30 in invasive ductal breast carci-nomas. Chin Med J (engl) 122:2763–2769
Maggiolini M, Vivacqua a, Fasanella g, Recchia ag, sisci D, Pezzi V, Montanaro D, Musti aM, Picard D, ando s (2004) the g protein-coupled receptor gPR30 mediates c-fos up-regulation by 17beta-estradiol and phytoestrogens in breast cancer cells. J Biol Chem 279:27008–27016
Perou CM, sorlie t, eisen MB, van de Rijn M, Jeffrey ss, Rees Ca, Pollack JR, Ross Dt, Johnsen h, akslen la, Fluge O, Perga-menschikov a, Williams C, Zhu sX, lonning Pe, Borresen-Dale al, Brown PO, Botstein D (2000) Molecular portraits of human breast tumours. nature 406:747–752
Prossnitz eR, arterburn JB, smith hO, Oprea tI, sklar la, hatha-way hJ (2008) estrogen signaling through the transmembrane g protein-coupled receptor gPR30. annu Rev Physiol 70:165–190
schneider BP, Winer eP, Foulkes WD, garber J, Perou CM, Rich-ardson a, sledge gW, Carey la (2008) triple-negative breast cancer: risk factors to potential targets. Clin Cancer Res 14:8010–8018
schumacher a, Brachwitz n, sohr s, engeland K, langwisch s, Dolaptchieva M, alexander t, taran a, Malfertheiner sF, Costa sD, Zimmermann g, nitschke C, Volk hD, alexander h, gunzer M, Zenclussen aC (2009) human chorionic gonadotropin attracts regulatory t cells into the fetal-maternal interface during early human pregnancy. J Immunol 182:5488–5497
smith hO, leslie KK, singh M, Qualls CR, Revankar CM, Joste ne, Prossnitz eR (2007) gPR30: a novel indicator of poor survival for endometrial carcinoma. am J Obstet gynecol 196:386–389
smith hO, arias-Pulido h, Kuo DY, howard t, Qualls CR, lee sJ, Verschraegen CF, hathaway hJ, Joste ne, Prossnitz eR (2009)

J Cancer Res Clin Oncol
1 3
gPR30 predicts poor survival for ovarian cancer. gynecol Oncol 114:465–471
sun W, Yang J (2010) Functional mechanisms for human tumor sup-pressors. J Cancer 1:136–140
tam sW, shay JW, Pagano M (1994) Differential expression and cell cycle regulation of the cyclin-dependent kinase 4 inhibitor p16Ink4. Cancer Res 54:5816–5820
toh Wh, nam sY, sabapathy K (2010) an essential role for p73 in regulating mitotic cell death. Cell Death Differ 17:787–800
tu g, hu D, Yang g, Yu t (2009) the correlation between gPR30 and clinicopathologic variables in breast carcinomas. technol Cancer Res treat 8:231–234
Vivacqua a, Bonofiglio D, Recchia ag, Musti aM, Picard D, ando s, Maggiolini M (2006) the g protein-coupled receptor gPR30 medi-ates the proliferative effects induced by 17beta-estradiol and hydrox-ytamoxifen in endometrial cancer cells. Mol endocrinol 20:631–646
Wang C, lv X, Jiang C, Davis Js (2012) the putative g-protein cou-pled estrogen receptor agonist g-1 suppresses proliferation of ovarian and breast cancer cells in a gPeR-independent manner. am J transl Res 4:390–402
Wang C, lv X, he C, hua g, tsai MY, Davis Js (2013) the g-pro-tein-coupled estrogen receptor agonist g-1 suppresses prolifera-tion of ovarian cancer cells by blocking tubulin polymerization. Cell Death Dis 4:e869