glycoprotein 130 receptor signaling mediates a-cell
TRANSCRIPT

Samuel Z. Chow,1 Madeleine Speck,1 Piriya Yoganathan,1 Dominika Nackiewicz,1 Ann Maria Hansen,2
Mette Ladefoged,2 Björn Rabe,3 Stefan Rose-John,3 Peter J. Voshol,4 Francis C. Lynn,1
Pedro L. Herrera,5 Werner Müller,6 Helga Ellingsgaard,7 and Jan A. Ehses1
Glycoprotein 130 ReceptorSignaling Mediates a-CellDysfunction in a Rodent Modelof Type 2 DiabetesDiabetes 2014;63:2984–2995 | DOI: 10.2337/db13-1121
Dysregulated glucagon secretion accompanies islet in-flammation in type 2 diabetes. We recently discoveredthat interleukin (IL)-6 stimulates glucagon secretionfrom human and rodent islets. IL-6 family cytokinesrequire the glycoprotein 130 (gp130) receptor to signal.In this study, we elucidated the effects of a-cell gp130receptor signaling on glycemic control in type 2 diabe-tes. IL-6 family cytokines were elevated in islets in ro-dent models of this disease. gp130 receptor activationincreased STAT3 phosphorylation in primary a-cellsand stimulated glucagon secretion. Pancreatic a-cellgp130 knockout (agp130KO) mice showed no differ-ences in glycemic control, a-cell function, or a-cellmass. However, when subjected to streptozotocin plushigh-fat diet to induce islet inflammation and pathophys-iology modeling type 2 diabetes, agp130KO mice hadreduced fasting glycemia, improved glucose tolerance,reduced fasting insulin, and improved a-cell function.Hyperinsulinemic-euglycemic clamps revealed no dif-ferences in insulin sensitivity. We conclude that in a set-ting of islet inflammation and pathophysiology modelingtype 2 diabetes, activation of a-cell gp130 receptor sig-naling has deleterious effects on a-cell function, pro-moting hyperglycemia. Antagonism of a-cell gp130
receptor signaling may be useful for the treatmentof type 2 diabetes.
Islet inflammation (1,2) and pancreatic a-cell dysfunction(3–6) contribute to hyperglycemia in patients with type 2diabetes. Pancreatic islets from patients with type 2 diabe-tes are infiltrated with macrophages (7,8), express elevatedproinflammatory cytokines (9,10), and express features offibrosis (11), consistent with reports from animals andprimates with this disease (7,12–18). The detrimentaleffects of inflammation on islet b-cell function were re-cently confirmed, when the interleukin (IL)-1 receptor an-tagonist reduced hyperglycemia and improved b-cellinsulin secretion in patients with type 2 diabetes (1).
Pancreatic a-cell dysfunction is detectable in the earlystages of type 2 diabetes, where the inverse relationshipbetween pulsatile insulin and glucagon secretion is lost(19). This dysregulation is associated with relative hyper-glucagonemia, which contributes to the development offasting hyperglycemia associated with frank type 2 diabe-tes (6). Indeed, inhibition of glucagon action by variousmeans reduces hyperglycemia in rodent models of thisdisease (5,6).
1Department of Surgery, Faculty of Medicine, University of British Columbia, Childand Family Research Institute, Vancouver, British Columbia, Canada2Diabetes Research Unit, Novo Nordisk A/S, Måløv, Denmark3Institute of Biochemistry, Medical Faculty, Christian Albrechts University of Kiel,Kiel, Germany4Institute of Metabolic Science, University of Cambridge Metabolic ResearchLaboratories and National Institute for Health Research Biomedical ResearchCentre, Addenbrooke’s Hospital, Cambridge, U.K.5Department of Genetic Medicine and Development, Faculty of Medicine, Univer-sity of Geneva, Geneva, Switzerland6Faculty of Life Sciences, University of Manchester, Manchester, U.K.7The Centre of Inflammation and Metabolism and the Centre for Physical ActivityResearch, Department of Infectious Diseases, Rigshospitalet, University ofCopenhagen, Copenhagen, Denmark
Corresponding author: Jan A. Ehses, [email protected].
Received 19 July 2013 and accepted 7 April 2014.
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db13-1121/-/DC1.
S.Z.C. and M.S. contributed equally to this work.
© 2014 by the American Diabetes Association. Readers may use this article aslong as the work is properly cited, the use is educational and not for profit, andthe work is not altered.
2984 Diabetes Volume 63, September 2014
ISLETSTUDIES

We recently identified IL-6 as a key cytokine elevatedduring islet inflammation in rodents with type 2 diabetes(7,20), with effects on a-cell glucagon secretion, GLP-1secretion, and survival (21,22). IL-6 is part of a familyof IL-6–type cytokines that all share the same commonsignal transducer for signaling, the glycoprotein 130(gp130) (23). In support of a role for IL-6 cytokines inhuman disease, a recent study of global gene expression inhuman islets identified a group of coexpressed genes as-sociated with type 2 diabetes. Within this group of genes,expression of two IL-6 family cytokines, IL-6 and IL-11,were both increased in islets from patients with type 2diabetes: IL-6 was increased 2.75-fold, while IL-11 mRNAwas increased 1.61-fold (24).
Despite these advances, there remain significant gapsin our knowledge regarding how the gp130 receptorregulates a-cell function. In our previous reports, we wereunable to distinguish systemic IL-6 effects from tissue-specific IL-6 cytokine-mediated effects on the a-cell. Inaddition, we did not evaluate how IL-6 family cytokinesregulate glycemia via actions on glucagon or GLP-1 secre-tion in a setting of islet inflammation and decreased b-cellmass modeling type 2 diabetes (21,22). Thus we generateda-cell gp130 receptor knockout (agp130KO) mice and in-vestigated the role of the a-cell in a nongenetic rodentmodel of type 2 diabetes.
RESEARCH DESIGN AND METHODS
AnimalsMale rodents were used for all experiments. Goto-Kakizaki (GK) rats (Taconic), Wistar rats (Taconic),BKS.Cg-m+/+Leprdb/BomTac (db/db) mice (Taconic), C57BL/6Jmice (CDM), and Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J(mT/mG) mice (The Jackson Laboratory) were from com-mercial sources. Gp130fl/fl and Gcg-Cre mice were providedby W. Müller (25) and P. Herrera (26), respectively. Gcg-Cre mice were bred with mT/mG mice to produce Gcg-CremT/mG mice. All rodents were housed at the Child andFamily Research Institute (CFRI) animal facility, main-tained on a 12-h light/dark cycle, and fed a normal chowdiet (13 kcal% fat). All procedures were approved by theUniversity of British Columbia Committee on Animal Careor the Principles of Laboratory Care (Denmark).
Streptozotocin, High-Fat Diet, and Streptozotocin/High-Fat Diet Mouse ModelsStreptozotocin (STZ; Sigma-Aldrich) was prepared in ace-tate buffer and administered (25 mg/kg) via intraperitonealinjections for 5 consecutive days at 7–9 weeks of age. Con-trol mice were administered an intraperitoneal injection ofacetate buffer. Some mice were put on high-fat diet (HFD;58 kcal% fat with sucrose; Research Diets) 3 weeks follow-ing intraperitoneal injection of acetate buffer or STZ.
Islet Isolation and ImmunocytochemistryMouse and rat islets were isolated and cultured asdescribed (7,27,28). For pSTAT3 experiments, islets weredispersed and plated on 804G extracellular matrix–coated
8-chamber slides (Nunc) (29). Islet cells were serumstarved followed by treatment with IL-6, hyper-IL (HIL)-6 (30), leukemia inhibitory factor (LIF), or IL-27 (100 ng/mLwas used for all cytokine treatments unless otherwisestated). Cells were paraformaldehyde fixed, permeabilizedwith methanol, blocked with 5% goat serum (Invitrogen),and incubated with rabbit anti-pSTAT3 (1:100; Cell Signal-ing Technology) and mouse anti-glucagon (1:1,000; Sigma-Aldrich). Slides were incubated with Alexa 488 donkeyanti-rabbit (1:250; Jackson ImmunoResearch) and Alexa594 donkey anti-mouse (1:450; Jackson ImmunoResearch)and mounted using Vectashield with DAPI (Vector Labora-tories). Imaging was acquired with a BX61 microscope andLeica SP5 II confocal imaging system and quantified usingImage-Pro Analyzer. An average of 1,130 6 261 glucagon-positive a-cells were counted per experimental condition.
Pancreatic a-cell apoptosis was assessed by TUNELstaining as described (21) and imaged using a BX61 mi-croscope. An average of 3,164 6 722 glucagon-positivea-cells were counted per experimental condition.
Flow CytometryIsolated islets were dispersed and incubated with Fc block(1:100; eBioscience). Islet cells were stained with CD45-eFluor 450 (1:250; clone 30-F11; eBioscience), CD11b-PE(1:1,200; clone M1/70; eBioscience), Ly-6c allophycocya-nin (1:1,200; clone HK1.4; eBioscience), and the viabilitydye eFluor 506 (1:1,000; eBioscience). Unstained, singlestains, and fluorescence minus one controls were used forsetting gates and compensation with the help of the CFRIFACS core facility using a BD LSR II instrument (BDBiosciences). Cells were gated on live cells and CD45+ cellsfollowed by CD11b+Ly6c+ cells. Islets were pooled fromtwo mice per treatment per time point.
Secretion AssaysIslets were plated on 804G extracellular matrix–coatedplates for secretion assays. Aprotinin (250 kallikrein in-hibitor units/mL; Sigma-Aldrich) and dipeptidyl peptidase-4 inhibitor (50 mmol/L; Millipore) were added to each well,and islets were treated with IL-6 or HIL-6. Conditionedmedia were collected, and 70% acid ethanol was addedto islets for total hormone content. Glucagon and GLP-1were measured by radioimmunoassay (Millipore). Glucose-stimulated insulin secretion from islets was performed aspreviously published (7). For total pancreatic insulin, glu-cagon, and GLP-1 content, hormones were extracted using70% acid ethanol and determined using Luminex technol-ogy (Millipore). Total pancreatic protein concentration wasdetermined using the Pierce BCA Protein Assay kit (ThermoScientific). Ex vivo secretion of IL-6 and LIF was deter-mined by Luminex, and soluble IL-6 receptor (sIL-6R) se-cretion was assayed by ELISA (31).
Physiological MeasurementsFor blood sampling, aprotinin (250 kallikrein inhibitorunits/mL plasma; Sigma-Aldrich) and dipeptidyl peptidase-4inhibitor (50 mmol/L; Millipore) were added to the
diabetes.diabetesjournals.org Chow and Associates 2985

collection tubes. Mice were fasted overnight and injectedintraperitoneally with 1.5 g/kg body weight glucose forglucose tolerance tests (intraperitoneal glucose tolerancetest [IPGTT]). For insulin tolerance tests (ITTs), micewere fasted 2 h and injected intraperitoneally with1 unit/kg insulin (Novolin GE, Novo Nordisk). Plasma in-sulin and glucagon were measured using ELISA (ALPCOand Mercodia, respectively), total GLP-1 was measuredusing Meso Scale Discovery technology, and plasma IL-6was measured using Luminex technology (Millipore).
Hyperinsulinemic-Euglycemic ClampsHyperinsulinemic-euglycemic clamps were performed aspreviously described (32–34). Mice were fasted overnightand anesthetized with acepromazine, midazolam, and fen-tanyl, with an initial dose by intraperitoneal injection andtop-up doses by subcutaneous injection. Once fully immo-bilized from anesthesia, the tail vein was cannulated, anda 1-h basal infusion of 3H-D-glucose (1.2 mCi/h) was startedto determine steady-state tracer. Duplicate blood sampleswere taken from the saphenous vein at the end of the basalperiod to measure fasting blood glucose levels, for deter-mination of endogenous glucose production, and for insu-lin measurements. Hyperinsulinemia was induced witha constant infusion of insulin (3.5 mU/kg/min). To main-tain euglycemia (;5.0 mmol/L), a variable infusion of12.5% D-glucose was started simultaneously. Once euglyce-mia had been achieved, steady state was maintained for 60min, after which triplicate blood samples were taken forfurther analysis. Briefly, insulin concentrations were mea-sured by ELISA, and plasma samples were counted usinga 1450 MicroBeta TriLux LSC and Luminescence Counter(PerkinElmer) after extraction by trichloroacetic acid pre-cipitation. Whole-body glucose use (mmol kg21 min21) wasdetermined as the ratio of the specific activity of glucose tothe rate of 3H-D-glucose appearance. Subsequently, endog-enous glucose production (mmol kg21 min21) could becalculated as the difference between whole-body glucoseuptake and exogenous glucose infusion.
ImmunohistochemistryAntigen retrieval was performed on sectioned tissuesusing 10 mmol/L citrate buffer (Fisher Scientific) followedby blocking with 1% goat serum and donkey/goat anti-mouse IgG (1:30; eBioscience). Sections were incubatedwith guinea pig anti-insulin (1:1,000; Millipore) andmouse anti-glucagon (1:2,000) followed by incubationwith Alexa 488 donkey anti–guinea pig (1:250; JacksonImmunoResearch) and Alexa 594 donkey anti-mouse(1:450). Sections were mounted using Vectashield withDAPI and imaged on a BX61 microscope.
Pancreatic Gcg-Cre X mT/mG and mT/mG cryosectionswere paraformaldehyde fixed and equilibrated througha sucrose gradient. Tissue was frozen in optimum cuttingtemperature compound (Tissue-Tek). Sections were per-meabilized with 0.1% Triton X-100 for 30 min and thenblocked in 5% goat serum and 1% BSA (Fisher Scientific).Sections were incubated in 1% BSA and 0.1% Triton
X-100 with mouse anti-glucagon (1:2,000) followed byincubation with Alexa 647 goat anti-mouse (1:250;Abcam) and mounted with Vectashield. Images weretaken on a Leica SP5 II confocal imaging system.
For pancreatic b- and a-cell mass analysis, all islets inthree sections (spaced 200 mm apart) were analyzed usingImage-Pro Analyzer. Insulin and glucagon-positive areawas quantified using the area/count function. Endocrinecell mass was calculated as previously described (21).
Gene Expression AnalysisTotal RNA was extracted from islets using the MNNucleoSpin RNA II kit (Macherey-Nagel), RNA was re-versed transcribed and quantitative PCR was performedusing PrimeTime primers/probes (IDT) in a ViiA 7 real-time PCR system (ABI) as previously described (35). Dif-ferential expression was determined by the 22DDCT
method (36) with Rplp0 and 18S as reference genes.
Statistical AnalysisData are expressed as means6 SEM/SD with the number ofindividual experiments presented in the figure legends. Alldata were analyzed using the nonlinear regression analysisprogram Prism (GraphPad), and significance was tested us-ing Student t test or ANOVA with post hoc tests for multiplecomparison analysis. Significance was set at P , 0.05.
RESULTS
IL-6 Family Cytokines Are Elevated in Pancreatic IsletsFrom Rodent Models of Type 2 DiabetesWe analyzed islet IL-6 family cytokine expression in tworodent models, the GK rat and db/db mouse. Both the GKrat and the db/db mouse displayed elevated blood glucoseand declining insulin levels at 12 and 15 weeks of age,respectively, suggestive of b-cell failure (Fig. 1A, B, D, andE). Isolated islets from 8–12-week-old GK rats had signif-icantly increased levels of Il6, Lif, Clcf1, and Osm mRNArelative to Wistar controls (Fig. 1C); similar effects wereobserved in islets from 15-week-old db/db mice (comparedwith Fig. 1C and F). Interestingly, both IL-6 family cyto-kines and glycemia were more elevated in db/db micerelative to GK rats. Thus elevated IL-6 family cytokineexpression accompanies hyperglycemia and declining in-sulin levels in rodent models of type 2 diabetes, and themagnitude of islet IL-6 cytokine expression may contrib-ute to the prevailing hyperglycemia in these models.
gp130 Receptor Activation Stimulates STAT3Phosphorylation and Glucagon Secretion From a-CellsTo determine if gp130 receptor signaling is activated ina-cells following IL-6 treatment, dispersed islets werestimulated with IL-6 or HIL-6 (IL-6 fused to the sIL-6R[37]; HIL-6 can stimulate all gp130 receptors in the ab-sence of an IL-6R) and immunostained for pSTAT3. IL-6induced phosphorylation of STAT3 in a-cells within 30min to a similar degree as HIL-6 (Fig. 2A). Next weassessed the effect of different IL-6 doses and time of ex-posure on glucagon secretion in islets. IL-6 increased glu-cagon secretion in a concentration- and time-dependent
2986 a-Cell gp130 Receptor Signaling in Diabetes Diabetes Volume 63, September 2014

manner, with no significant differences in glucagon con-tent (Fig. 2B and C). Consistent with our pSTAT3 data,IL-6 stimulated glucagon secretion to a similar degree asHIL-6 (Fig. 2D and E), suggesting that a-cell gp130 re-ceptor signaling is maximally activated by IL-6 and sup-porting our previous observation that pancreatic a-cellsexpress the IL-6 transmembrane receptor (21). To ensurethat IL-6 did not induce a-cell death, causing glucagonrelease into the incubation medium, islets were treatedwith IL-6 and apoptosis visualized by TUNEL staining. IL-6protected from cell death induced by a mixture of cyto-kines (IL-1b, TNFa, and IFNg) (Fig. 2F), consistent withour previous observations under nutrient stress (21). Col-lectively, these data indicate that gp130 signaling withina-cells leads to phosphorylation of STAT3 and stimula-tion of glucagon secretion.
Generation and Validation of agp130KO MiceTo assess Cre recombinase activity within a-cells, Gcg-Cremice were crossed with mT/mG reporter mice. Pancreatic
sections revealed expression of EGFP in a proportion ofa-cells stained with glucagon, demonstrating active Crerecombinase in 44.1 6 1.5% of a-cells (Fig. 3A and B),similar to previous reports using Gcg-Cre mice (38).
Next we assessed pSTAT3 levels in response to gp130receptor activation in fl/fl controls and agp130KO islets.Stimulation of pSTAT3 with IL-6 and LIF was significantlydecreased within a-cells of agp130KO islets (Fig. 3C). IL-27failed to induce pSTAT3 within control islets, suggesting lackof IL-27 receptors on a-cells. Finally, we assessed glucagonsecretion in response to gp130 receptor activation inagp130KO islets. IL-6 stimulated glucagon secretion wasreduced by ;50% in agp130KO islets (Fig. 3D). There wasno effect of IL-6 on GLP-1 secretion following 48-h exposureof mouse islets (Fig. 3E). Thus agp130KO islets have Cre-mediated recombination occurring in a proportion of a-cellsand reduced gp130 receptor signaling, coinciding with a 50%reduction in gp130 receptor–mediated glucagon secretion.
To rule out any a-cell or intestinal L-cell developmentalabnormalities in agp130KO mice, we assessed several
Figure 1—IL-6 family cytokine expression is increased in rodents with type 2 diabetes. Nonfasted glycemia (A) and insulin levels (B) inWistar and GK rats. IL-6 family cytokine expression levels in islets from 8–12-week-old Wistar and GK rats (C). Expression levels wereassessed by quantitative real-time PCR, normalized to the housekeeping gene Rplp0, and expressed as fold control (Wistar). Nonfastedglycemia (D) and insulin levels (E) in 4–15-week-old db/dbmice. IL-6 family cytokine expression levels in islets from 4-, 8-, and 15-week-olddb/db mice (F). Expression levels were assessed by quantitative real-time PCR, normalized to the housekeeping gene Rplp0, andexpressed as fold control (4 weeks db/db). Data represent mean 6 SEM from 3–4 Wistar and 3–4 GK rats (A–C). Data representmean 6 SEM from 5–20 db/db mice (D–E) and mean 6 SD from islets pooled from $3 mice/age (F ). White bars represent Wistar/4-week db/db, gray bars represent 8-week db/db, and black bars represent GK or 15-week db/db. ★P < 0.05, ★★P < 0.01,★★★P < 0.001 as tested by Student t test. nd, not detected.
diabetes.diabetesjournals.org Chow and Associates 2987

physiological parameters in agp130KO mice. agp130KOmice displayed no difference in body weight, glucose tol-erance, glucose-stimulated insulin secretion, or insulinsensitivity at 16–18 weeks of age (Fig. 3F–I). FastingGLP-1 levels were unchanged, and pancreatic a-cell func-tion was normal, as assessed by glucose-mediated glucagonsuppression during a GTT and hypoglycemia-mediated glu-cagon secretion during an ITT (Fig. 3J and K). Lastly, b-celland a-cell mass were similar between genotypes (Fig. 3LandM). Thus agp130 receptor signaling does not appear toaffect a-cell development or function under normal chow-fed conditions.
Pancreatic a-Cell Dysfunction and Islet Inflammationin the STZ/HFD Mouse Model of Type 2 DiabetesTo investigate the role of the a-cell gp130 receptor ina mouse model of human type 2 diabetes, we generateda nongenetic model displaying features of type 2 diabetes.Mice were administered STZ, HFD, or STZ in combinationwith HFD as depicted in Fig. 4A. STZ alone caused glucoseintolerance, tended to reduce insulin secretion in responseto glucose in vivo, had minimal effects on glucose-mediated glucagon suppression and hypoglycemia-induced glucagon secretion in vivo, and tended to reduceb-cell mass and increase a-cell mass (Fig. 4B–L). HFD
Figure 2—IL-6 and HIL-6 activate a-cell STAT3 signaling and stimulate glucagon secretion from islets. STAT3 activation in primary mousea-cells following 30-min cytokine treatment (A). Representative image indicates staining for glucagon (red), pSTAT3 (green), and DAPI(blue). Glucagon secretion (B) and glucagon content (C) following IL-6 treatment of mouse islets at the indicated doses and times.Glucagon secretion (D) and glucagon content (E ) from IL-6– and HIL-6–treated mouse islets following 48 h. TUNEL-positive a-cellswere analyzed following 24-h exposure to IL-6, cytokine mix (200 pg/mL IL-1b, 1 ng/mL TNFa, 5 ng/mL IFNg), or cytokine mix plusIL-6 (F ). Data represent mean 6 SEM from n = 3 mice performed in three independent experiments (A), n = 3–6 mice performed in twoindependent experiments (B and C), n = 6 mice performed in three independent experiments (D and E), and n = 4 mice in two independentexperiments (F ). ★P < 0.05; ★★P < 0.01; ★★★P < 0.001 vs. untreated control; #P < 0.05 vs. cytokine mix as tested by ANOVA withDunnett (A–E) or Newman-Kuels posttest (F ). Ctrl, control; cyt, cytokine; CM, cytokine mix.
2988 a-Cell gp130 Receptor Signaling in Diabetes Diabetes Volume 63, September 2014

alone increased body weight, caused glucose intolerance,increased insulin secretion in response to glucose in vivo,impaired glucose-mediated glucagon suppression, had noeffect on hypoglycemia-induced glucagon secretion in vivo,and had minimal effects on b- and a-cell mass (Fig. 4B–L).
STZ in combination with HFD caused increased bodyweight, fed hyperglycemia, glucose intolerance, and markeda-cell dysfunction (Fig. 4B–H). In addition, b-cell mass wasreduced by ;50%, in parallel with impaired glucose-stimulated insulin secretion from islets ex vivo and reduced
Figure 3—agp130KO mice exhibit partial loss of a-cell gp130 receptor function and normal glucose homeostasis and a-cell function underchow-fed conditions. Gcg-Cre mice were bred with mT/mG mice to assess activity of a-cell Cre recombinase (A and B). Images indicatenonrecombinant cells (tdTomato), recombinant cells (EGFP), and glucagon-positive cells (purple). EGFP was expressed in 44.1 6 1.5% ofa-cells (B). STAT3 activation in primary mouse a-cells from fl/fl and agp130KO mice following 30-min cytokine treatment (C). Glucagon (D)and GLP-1 (E) secretion from fl/fl and agp130KO mouse islets following IL-6 treatment. Basal glucagon secretion was 2.26 6 0.54 for fl/fland 2.35 6 0.53 for agp130KO as percentage glucagon content. Basal GLP-1 secretion was 1.97 6 0.39 for fl/fl and 1.31 6 0.17 foragp130KO as percentage GLP-1 content. Body weight of fl/fl and agp130KO mice at 16–18 weeks of age (F). IPGTT (G) (1.5 g/kg), insulinlevels during IPGTT (H), and ITT ( I) (1 unit/kg). Fasting GLP-1 levels (J) and glucagon levels during IPGTT and ITT (K). Pancreatic b- anda-cell mass (L andM). Gray bars represent fl/fl control, and light red bars represent agp130KO. Scale bars represent 50 mm. Data representn = 3–5 mice per genotype (A and B), n = 3–4 mice per genotype performed in three independent experiments (C ), n = 6 mice per genotypeperformed in two independent experiments (D and E), and 3–12 mice per genotype (F–M). ★P < 0.05; ★★P < 0.01 as tested by Student ttest. Ctrl, control; fl/fl, gp130fl/fl; KO, agp130KO.
diabetes.diabetesjournals.org Chow and Associates 2989

Figure 4—Pancreatic a-cell dysfunction and islet inflammation in STZ-, HFD-, and STZ/HFD-treated mice. Schematic of chow, STZ (25mg/kg), HFD, and STZ/HFD treatment groups (A) with weekly fed blood glucose monitoring (B). IPGTT (C) (1.5 g/kg glucose), ITT (D) (1 unit/kginsulin), and body weight (E) of chow, STZ, HFD, and STZ/HFD mice. Plasma insulin (F) and glucagon (G and H) during IPGTT and ITT,and fed insulin ( I) after 9 weeks. Pancreatic b- and a-cell mass (J and K) in treated mice with representative images (L). Glucose-stimulatedinsulin secretion (M) and insulin content (N) of islets ex vivo after 9 weeks. Pancreatic islet infiltrating monocytes (CD11b+Ly6c+ cells) inchow and STZ-treated mice (O). Expression levels of IL-6 family cytokines post-STZ injection and during HFD relative to chow mice (P). IL-6,sIL-6R, and LIF protein secretion from 100 islets/well isolated 3 weeks post-STZ (Q). Islet-derived IL-6 (R) from 100 islets/well and
2990 a-Cell gp130 Receptor Signaling in Diabetes Diabetes Volume 63, September 2014

islet insulin content (Fig. 4J–N). a-Cell mass was also sig-nificantly increased in STZ/HFD mice (Fig. 4K). Thus treat-ment with STZ plus HFD resulted in a mouse withimpaired a- and b-cell function and hyperglycemia.
To characterize islet inflammation post-STZ, isletinfiltrating monocytes (CD11b+Ly6c+ cells) were analyzedby flow cytometry. The number of islet monocytes tendedto increase at weeks 0.5 and 1 post-STZ and were signifi-cantly increased at week 2 post-STZ (Fig. 4O). To deter-mine if islet IL-6 family cytokine expression wasincreased, we analyzed islet gene expression at multipletimes post-STZ and during subsequent HFD feeding (Fig.4P). IL-27 mRNA was increased ;1 week post-STZ, OsmmRNA was increased 2 weeks post-STZ, and IL-6 and LifmRNA were increased 3 weeks post-STZ. Other IL-6 fam-ily cytokines were unchanged (Fig. 4P and not shown).Consistent with these mRNA data, IL-6 and LIF proteinsecretion were increased in islets isolated 3 weeks post-STZ, with no change in sIL-6R secretion (Fig. 4Q). IsletIL-6 secretion was not significantly increased at week 9in our treatment groups (Fig. 4R), indicating that IL-6cytokines are only transiently increased following STZand not further increased by HFD. However, plasma IL-6levels were significantly increased in our STZ/HFD groupat the end of our study (Fig. 4S). Taken together, thesedata show that STZ/HFD-treated mice have many hall-mark characteristics of type 2 diabetes, including pan-creatic a-cell dysfunction and increased expression ofpancreatic islet IL-6 family cytokines.
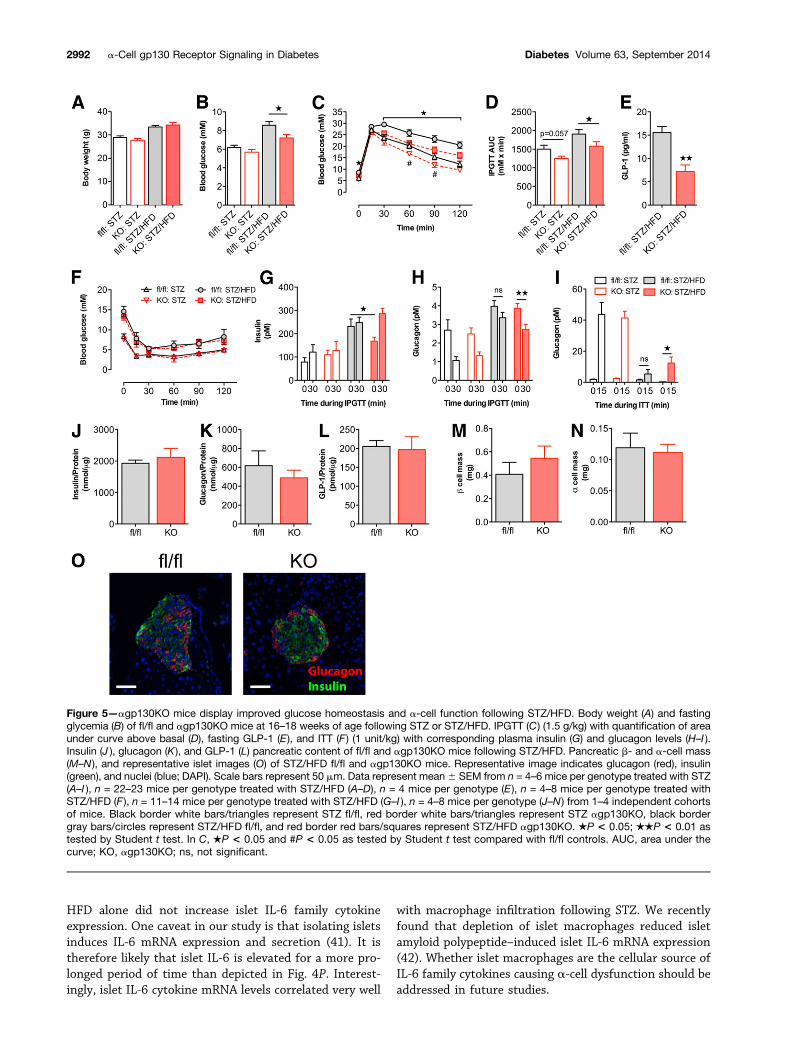
Partial Knockout of the a-Cell gp130 ReceptorProtects From Hyperglycemia Following STZ/HFDTo determine the consequence of a-cell gp130 signalingduring islet inflammation and in a pathophysiological set-ting modeling type 2 diabetes, we subjected agp130KOmice to STZ alone or STZ followed by HFD feeding.agp130KO mice displayed no difference in body weightfollowing STZ or STZ/HFD (Fig. 5A). When subjected toSTZ alone, agp130KO mice showed mildly improved glu-cose tolerance, with no differences in insulin sensitivity,insulin secretion, or a-cell function in vivo (Fig. 5C–I).However, following STZ/HFD, agp130KO mice had de-creased fasting glycemia, improved glucose tolerance,and decreased fasting insulin and showed improvementsin glucose modulated glucagon secretion in vivo (Fig. 5B–I). This was despite decreased fasting GLP-1 levels (Fig.5E). ITTs indicated no difference in insulin sensitivity inagp130KO mice (Fig. 5F). No differences in pancreaticglucagon, insulin, or GLP-1 content (Fig. 5J–L) were
observed between genotypes, along with no differencesin b- and a-cell mass (Fig. 5M–O). STZ/HFD-treatedGcg-Cre mice did not show any glucose intolerance com-pared with wild-type mice excluding any phenotypic effectof Cre recombinase expression in a-cells (SupplementaryFig. 1). Further, agp130KO mice on HFD alone showedno phenotypic differences in glucose tolerance or a- andb-cell function (Supplementary Fig. 2).
Finally, to determine if changes in insulin sensitivitycontributed to the protective phenotype in STZ/HFD-treated mice, we performed hyperinsulinemic-euglycemicclamps. There were no differences in glucose infusionrate, whole-body insulin-stimulated glucose uptake, orhepatic insulin sensitivity between genotypes (Fig. 6). In-terestingly, hepatic glucose production under fasting con-ditions tended to be reduced in agp130KO mice (Fig. 6D),consistent with reduced fasting glycemia in these mice.Taken together, these data show that gp130 receptorsignaling contributes to a-cell dysfunction in a nonge-netic model of type 2 diabetes, and partial inhibitionof gp130 receptor signaling improves glucose toleranceand protects from hyperglycemia through improveda-cell function.
DISCUSSION
Normally, glucagon secretion is increased during fastingand suppressed as blood glucose rises following a meal ina counterregulatory fashion to insulin. However, duringtype 2 diabetes, glucagon secretion is inappropriatelyelevated, contributing to hyperglycemia (5,6). Pioneeringstudies by Müller et al. (39) and elegant clamp studies byShah et al. (3) have demonstrated that lack of glucagonsuppression contributes to postprandial hyperglycemia inpeople with type 2 diabetes. The main mechanism under-lying this hyperglucagonemia is thought to be b-cell dys-function and a decrease in b-cell–derived secretoryproducts that are known to have inhibitory effects ona-cell secretion, including insulin, g-aminobutyric acid,and zinc ions (40). All of these factors inhibit glucagonsecretion, and their secretion is likely decreased in franktype 2 diabetes. Our data demonstrate that islet inflam-mation also contributes to a-cell dysfunction, with a centralrole for gp130 receptor signaling in this process and withimplications for type 2 diabetes.
agp130KO mice were protected from glucose intoler-ance following STZ alone, but not following HFD alone.This was consistent with STZ treatment causing a tran-sient increase in IL-6 and LIF secretion from islets, while
systemic IL-6 (S) during fed after 9 weeks. White bars/circles represent chow control, horizontally striped bars/white squares represent STZ,vertically striped bars/white triangles represent HFD, and black bars/triangles represent STZ/HFD. Scale bars represent 50 mm. Data rep-resent mean 6 SEM from n = 5 mice (B–I), n = 3–11 mice (J–L) per experimental group, n = 3–5 mice per experimental group (M and N), n =4–5 mice per time point (O), n = 5–6 mice per time point (P), n = 4–5 mice for each experimental group (Q–S). ★P < 0.05; ★★P < 0.01;★★★P < 0.001; ★★★★P < 0.0001 as tested by two-way ANOVA with Bonferroni post test (B–D), ANOVA with Dunnett post test (E–H),or Student t test (M–Q) compared with chow controls. Ctrl, control; ns, not significant.
diabetes.diabetesjournals.org Chow and Associates 2991
HFD alone did not increase islet IL-6 family cytokineexpression. One caveat in our study is that isolating isletsinduces IL-6 mRNA expression and secretion (41). It istherefore likely that islet IL-6 is elevated for a more pro-longed period of time than depicted in Fig. 4P. Interest-ingly, islet IL-6 cytokine mRNA levels correlated very well
with macrophage infiltration following STZ. We recentlyfound that depletion of islet macrophages reduced isletamyloid polypeptide–induced islet IL-6 mRNA expression(42). Whether islet macrophages are the cellular source ofIL-6 family cytokines causing a-cell dysfunction should beaddressed in future studies.
Figure 5—agp130KO mice display improved glucose homeostasis and a-cell function following STZ/HFD. Body weight (A) and fastingglycemia (B) of fl/fl and agp130KO mice at 16–18 weeks of age following STZ or STZ/HFD. IPGTT (C) (1.5 g/kg) with quantification of areaunder curve above basal (D), fasting GLP-1 (E), and ITT (F ) (1 unit/kg) with corresponding plasma insulin (G) and glucagon levels (H–I ).Insulin (J ), glucagon (K), and GLP-1 (L) pancreatic content of fl/fl and agp130KO mice following STZ/HFD. Pancreatic b- and a-cell mass(M–N ), and representative islet images (O) of STZ/HFD fl/fl and agp130KO mice. Representative image indicates glucagon (red), insulin(green), and nuclei (blue; DAPI). Scale bars represent 50 mm. Data represent mean6 SEM from n = 4–6 mice per genotype treated with STZ(A–I ), n = 22–23 mice per genotype treated with STZ/HFD (A–D), n = 4 mice per genotype (E ), n = 4–8 mice per genotype treated withSTZ/HFD (F ), n = 11–14 mice per genotype treated with STZ/HFD (G–I ), n = 4–8 mice per genotype (J–N ) from 1–4 independent cohortsof mice. Black border white bars/triangles represent STZ fl/fl, red border white bars/triangles represent STZ agp130KO, black bordergray bars/circles represent STZ/HFD fl/fl, and red border red bars/squares represent STZ/HFD agp130KO. ★P < 0.05; ★★P < 0.01 astested by Student t test. In C, ★P < 0.05 and #P < 0.05 as tested by Student t test compared with fl/fl controls. AUC, area under thecurve; KO, agp130KO; ns, not significant.
2992 a-Cell gp130 Receptor Signaling in Diabetes Diabetes Volume 63, September 2014

Interestingly, the combination of STZ plus HFD didnot prolong or increase islet IL-6 family cytokine expres-sion, but it did result in further impairments in a-cellfunction compared with STZ or HFD treatments alone.One possible explanation for this synergism is that HFDmediates a-cell dysfunction by causing a-cell insulin re-sistance and that STZ exacerbates these effects by de-creasing insulin and inducing inflammation. This mightexplain the synergistic effect of STZ plus HFD on glucose-mediated glucagon suppression in vivo during an IPGTT.With respect to ITT responses, hypoglycemia-induced glu-cagon secretion is strongly driven by the parasympatheticnervous system (43,44). Thus STZ acts synergisticallywith HFD to impair this response. Regardless of themechanism of action, these hypotheses are consistentwith effects of gp130 receptor signaling on a-cell func-tion and glycemic control being more pronounced in theSTZ/HFD model, compared with STZ or HFD treatmentsalone. Thus gp130 receptor signaling acts as a modifier ofa-cell function in a synergistic manner with HFD-inducedeffects on a-cell function.
Improved a-cell function in agp130KO mice treatedwith STZ/HFD resulted in reduced fasting hyperglycemia,likely due to reduced hepatic glucose output. Reducedglycemia was likely responsible for the decreased fastinginsulin observed in agp130KO mice. We ruled out anycontribution of increased insulin sensitivity in agp130KOmice treated with STZ/HFD by performing hyperinsulinemic-euglycemic clamps. However, reduced fasting glycemia mayhave improved b-cell function in agp130KO mice, likelyby reducing the effects of glucotoxicity on the b-cell. This
sequence of events may also help explain how a transientincrease in islet IL-6 cytokines can have long-lastingeffects on glycemic control.
Two independent groups recently investigated theeffect of gp130 receptor cytokines on the a-cell. McGuinnessand colleagues found that IL-62/2 mice had a blunted glu-cagon response to endotoxin that was restored by replace-ment of IL-6 (45). They went on to show that IL-6 amplifiesadrenergic-dependent glucagon secretion from mouseislets (46). In a separate study, Fernández-Millán et al.showed that a-cell mass expansion and hyperglucagone-mia during suckling in rats is partly IL-6–dependent,contributing to hyperglycemia postweaning (47). Thesestudies, our previous work (21), and data presented heresupport the notion that IL-6 cytokines are modulators ofa-cell glucagon secretion. Taking these findings into con-sideration, we propose that gp130 receptor actions arecontext-dependent and that IL-6 cytokines modulatea-cell glucagon secretion to allow for increased glucoseoutput during normal physiology (e.g., during acute in-fection or postexercise), while also driving excessivea-cell glucagon secretion and dysfunction during type 2diabetes.
Exposure of human islets to IL-6 for 4 days or enrichedhuman pancreatic a-cells to IL-6 enhances GLP-1 secre-tion in addition to glucagon secretion, perhaps via upreg-ulation of pro(hormone)-convertase 1/3 (22). Mouseislets exposed to IL-6 for 2 days did not have elevatedGLP-1 secretion. Dedifferentiation of a-cells to a pre-a-cell state has been shown to induce GLP-1 secretion(48). In addition, prolonged culture of human islets in
Figure 6—No difference in insulin sensitivity in agp130KO mice following STZ/HFD. Hyperinsulinemic-euglycemic clamps were performedafter STZ plus 6 weeks of HFD feeding in fl/fl and agp130KO littermates. Blood glucose during clamping (A) and glucose infusion rate (B).Glucose turnover (C), suppression of hepatic glucose production (D), and insulin levels (E) during clamp. Data represent mean6 SEM fromn = 7 fl/fl and n = 6 agp130KO mice from two independent cohorts. P value was determined using Student t test. GIR, glucose infusion rate;KO, agp130KO.
diabetes.diabetesjournals.org Chow and Associates 2993

vitro results in increased numbers of glucagon-positivecells due to conversion of b-cells to a-cells (49). We sur-mise that signals causing a-cell dedifferentiation or con-version of b-cells to a-cells act together with IL-6cytokines to stimulate GLP-1 secretion from a-cells. Inthe future, the mechanisms underlying IL-6–stimulateda-cell GLP-1 secretion will need to be determined andtheir role tested in vivo.
Despite decreased systemic GLP-1 levels in agp130KOmice (likely due to receptor deletion in L cells), activationof a-cell gp130 receptor signaling in a setting of reducedb-cell mass and HFD-induced insulin resistance had det-rimental effects on normal a-cell function and glycemiccontrol. This may suggest that effects of gp130 receptorcytokines on a-cell function are more important for gly-cemic control than effects on L-cell GLP-1 secretion intype 2 diabetes. While it remains to be determined whatthe mechanisms are that facilitate IL-6 cytokine actionson glucagon secretion, insight into how IL-6/gp130 influ-ences a-cell secretory processes and/or a-cell differentia-tion may allow us to identify druggable targets thatmodify the actions of the a-cell and could thereby beused for the treatment of type 2 diabetes.
Acknowledgments. The authors thank G. Soukhatcheva, M. Komba, andL. Xu (CFRI) for technical assistance provided by the CFRI islet isolation corefacility and CFRI flow cytometry core and Bruce Verchere (University of BritishColumbia) for critical review of the manuscript.Funding. This work was supported by funding from the CFRI (J.A.E.), theUniversity of British Columbia (J.A.E.), the Canadian Institutes of Health Research(PCN-110793 and PNI-120292 to J.A.E.), the Canadian Diabetes Association(OG-3-13-4097-JE), the Deutsche Forschungsgemeinschaft (Bonn, Germany;SFB877; project A1 to S.R.-J.), and the Cluster of Excellence “Inflammation atInterfaces” (to S.R.-J.). S.Z.C. is supported by a Canadian Institutes of HealthResearch Transplantation Training Program Scholarship. H.E. is supported by theDanish Council for Independent Research and the Novo Nordisk Foundation. D.N.is supported by a University of British Columbia Transplantation Training Programand Vanier Canada Graduate Scholarship. J.A.E. has salary support from a CFRIscientist award and a Canadian Diabetes Association scholar award.Duality of Interest. No potential conflicts of interest relevant to this articlewere reported.Author Contributions. S.Z.C. designed and performed experiments, an-alyzed data, and wrote the manuscript. M.S. designed and performed experimentsand analyzed data. P.Y., D.N., A.M.H., and M.L. designed and performed experi-ments. B.R. and S.R.-J. performed experiments and provided reagents. P.J.V. andH.E. designed experiments. F.C.L., P.L.H., and W.M. provided reagents and mice.J.A.E. supervised the study, designed and performed experiments, analyzed data,and wrote the manuscript. All authors edited the manuscript. J.A.E. is the guarantorof this work and, as such, had full access to all the data in the study and takesresponsibility for the integrity of the data and the accuracy of the data analysis.
References1. Larsen CM, Faulenbach M, Vaag A, et al. Interleukin-1-receptor antagonistin type 2 diabetes mellitus. N Engl J Med 2007;356:1517–15262. Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. NatRev Immunol 2011;11:98–1073. Shah P, Vella A, Basu A, Basu R, Schwenk WF, Rizza RA. Lack of sup-pression of glucagon contributes to postprandial hyperglycemia in subjects withtype 2 diabetes mellitus. J Clin Endocrinol Metab 2000;85:4053–4059
4. Baron AD, Schaeffer L, Shragg P, Kolterman OG. Role of hyperglucagonemiain maintenance of increased rates of hepatic glucose output in type II diabetics.Diabetes 1987;36:274–2835. Dunning BE, Gerich JE. The role of alpha-cell dysregulation in fasting andpostprandial hyperglycemia in type 2 diabetes and therapeutic implications.Endocr Rev 2007;28:253–2836. Unger RH, Cherrington AD. Glucagonocentric restructuring of diabetes:a pathophysiologic and therapeutic makeover. J Clin Invest 2012;122:4–127. Ehses JA, Perren A, Eppler E, et al. Increased number of islet-associatedmacrophages in type 2 diabetes. Diabetes 2007;56:2356–23708. Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG. Islet-associatedmacrophages in type 2 diabetes. Diabetologia 2009;52:1686–1688
9. Böni-Schnetzler M, Thorne J, Parnaud G, et al. Increased interleukin (IL)-1beta messenger ribonucleic acid expression in beta -cells of individuals withtype 2 diabetes and regulation of IL-1beta in human islets by glucose and au-tostimulation. J Clin Endocrinol Metab 2008;93:4065–407410. Igoillo-Esteve M, Marselli L, Cunha DA, et al. Palmitate induces a pro-inflammatory response in human pancreatic islets that mimics CCL2 ex-pression by beta cells in type 2 diabetes. Diabetologia 2010;53:1395–140511. Lee E, Ryu GR, Ko SH, et al. Antioxidant treatment may protect pancreaticbeta cells through the attenuation of islet fibrosis in an animal model of type 2diabetes. Biochem Biophys Res Commun 2011;414:397–40212. Homo-Delarche F, Calderari S, Irminger JC, et al. Islet inflammation andfibrosis in a spontaneous model of type 2 diabetes, the GK rat. Diabetes 2006;55:1625–163313. Eguchi K, Manabe I, Oishi-Tanaka Y, et al. Saturated fatty acid and TLRsignaling link b cell dysfunction and islet inflammation. Cell Metab 2012;15:518–53314. Jones HB, Nugent D, Jenkins R. Variation in characteristics of islets ofLangerhans in insulin-resistant, diabetic and non-diabetic-rat strains. Int J ExpPathol 2010;91:288–30115. Masters SL, Dunne A, Subramanian SL, et al. Activation of the NLRP3 in-flammasome by islet amyloid polypeptide provides a mechanism for enhancedIL-1b in type 2 diabetes. Nat Immunol 2010;11:897–904
16. Youm YH, Adijiang A, Vandanmagsar B, Burk D, Ravussin A, Dixit VD.Elimination of the NLRP3-ASC inflammasome protects against chronic obesity-induced pancreatic damage. Endocrinology 2011;152:4039–404517. Chentouf M, Dubois G, Jahannaut C, et al. Excessive food intake, obesityand inflammation process in Zucker fa/fa rat pancreatic islets. PLoS ONE 2011;6:e22954
18. Nicol LE, Grant WF, Comstock SM, et al. Pancreatic inflammation and in-creased islet macrophages in insulin-resistant juvenile primates [publishedcorrection appears in J Endocrinol 2013;218:X1]. J Endocrinol 2013;217:207–21319. Rohrer S, Menge BA, Grüber L, et al. Impaired crosstalk between pulsatileinsulin and glucagon secretion in prediabetic individuals. J Clin Endocrinol Metab2012;97:E791–E79520. Ehses JA, Lacraz G, Giroix MH, et al. IL-1 antagonism reduces hypergly-cemia and tissue inflammation in the type 2 diabetic GK rat. Proc Natl Acad SciU S A 2009;106:13998–1400321. Ellingsgaard H, Ehses JA, Hammar EB, et al. Interleukin-6 regulates pan-creatic alpha-cell mass expansion. Proc Natl Acad Sci U S A 2008;105:13163–1316822. Ellingsgaard H, Hauselmann I, Schuler B, et al. Interleukin-6 enhances in-sulin secretion by increasing glucagon-like peptide-1 secretion from L cells andalpha cells. Nat Med 2011;17:1481–148923. Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta2011;1813:878–88824. Mahdi T, Hänzelmann S, Salehi A, et al. Secreted frizzled-related protein 4reduces insulin secretion and is overexpressed in type 2 diabetes. Cell Metab2012;16:625–633
2994 a-Cell gp130 Receptor Signaling in Diabetes Diabetes Volume 63, September 2014

25. Betz UA, Bloch W, van den Broek M, et al. Postnatally induced inactivation ofgp130 in mice results in neurological, cardiac, hematopoietic, immunological,hepatic, and pulmonary defects. J Exp Med 1998;188:1955–196526. Herrera PL. Adult insulin- and glucagon-producing cells differentiate fromtwo independent cell lineages. Development 2000;127:2317–232227. Westwell-Roper C, Dai DL, Soukhatcheva G, et al. IL-1 blockade attenuatesislet amyloid polypeptide-induced proinflammatory cytokine release and pan-creatic islet graft dysfunction. J Immunol 2011;187:2755–276528. Ladefoged M, Buschard K, Hansen AM. Increased expression of toll-likereceptor 4 and inflammatory cytokines, interleukin-6 in particular, in islets froma mouse model of obesity and type 2 diabetes. APMIS 2013;121:531–53829. Ribaux P, Ehses JA, Lin-Marq N, et al. Induction of CXCL1 by extracellularmatrix and autocrine enhancement by interleukin-1 in rat pancreatic beta-cells.Endocrinology 2007;148:5582–559030. Peters M, Blinn G, Solem F, Fischer M, Meyer zum Büschenfelde KH, Rose-John S. In vivo and in vitro activities of the gp130-stimulating designer cytokineHyper-IL-6. J Immunol 1998;161:3575–358131. Atreya R, Mudter J, Finotto S, et al. Blockade of interleukin 6 trans signalingsuppresses T-cell resistance against apoptosis in chronic intestinal inflammation:evidence in crohn disease and experimental colitis in vivo. Nat Med 2000;6:583–58832. Voshol PJ, Jong MC, Dahlmans VE, et al. In muscle-specific lipoproteinlipase-overexpressing mice, muscle triglyceride content is increased withoutinhibition of insulin-stimulated whole-body and muscle-specific glucose uptake.Diabetes 2001;50:2585–259033. Huynh FK, Levi J, Denroche HC, et al. Disruption of hepatic leptin signalingprotects mice from age- and diet-related glucose intolerance. Diabetes 2010;59:3032–304034. Levi J, Gray SL, Speck M, et al. Acute disruption of leptin signaling in vivoleads to increased insulin levels and insulin resistance. Endocrinology 2011;152:3385–339535. Ehses JA, Meier DT, Wueest S, et al. Toll-like receptor 2-deficient mice areprotected from insulin resistance and beta cell dysfunction induced by a high-fatdiet. Diabetologia 2010;53:1795–180636. Livak KJ, Schmittgen TD. Analysis of relative gene expression data usingreal-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001;25:402–408
37. Jones SA, Scheller J, Rose-John S. Therapeutic strategies for the clinicalblockade of IL-6/gp130 signaling. J Clin Invest 2011;121:3375–338338. Lu J, Herrera PL, Carreira C, et al. Alpha cell-specific Men1 ablation triggersthe transdifferentiation of glucagon-expressing cells and insulinoma de-velopment. Gastroenterology 2010;138:1954–196539. Müller WA, Faloona GR, Aguilar-Parada E, Unger RH. Abnormal alpha-cellfunction in diabetes. Response to carbohydrate and protein ingestion. N Engl JMed 1970;283:109–11540. Kawamori D, Welters HJ, Kulkarni RN. Molecular pathways underlying thepathogenesis of pancreatic alpha-cell dysfunction. Adv Exp Med Biol 2010;654:421–44541. Johansson U, Olsson A, Gabrielsson S, Nilsson B, Korsgren O. Inflammatorymediators expressed in human islets of Langerhans: implications for islettransplantation. Biochem Biophys Res Commun 2003;308:474–47942. Westwell-Roper CY, Ehses JA, Verchere CB. Resident macrophages mediateislet amyloid polypeptide-induced islet IL-1b production and beta cell dysfunc-tion. Diabetes. 12 November 2013 [Epub ahead of print]43. Patel DG. Role of parasympathetic nervous system in glucagon response toinsulin-induced hypoglycemia in normal and diabetic rats. Metabolism 1984;33:1123–112744. Lamy CM, Sanno H, Labouèbe G, et al. Hypoglycemia-Activated GLUT2Neurons of the Nucleus Tractus Solitarius Stimulate Vagal Activity and GlucagonSecretion. Cell Metab 2014;19:527–53845. Tweedell A, Mulligan KX, Martel JE, Chueh FY, Santomango T, McGuinnessOP. Metabolic response to endotoxin in vivo in the conscious mouse: role ofinterleukin-6. Metabolism 2011;60:92–9846. Lundblad TM, Coldren A, Brissova M, Chukwuma V, Ansari T, McGuinnessOP. Interleukin-6 amplifies adrenergic-dependent glucagon secretion. Diabetes2012;61:A47747. Fernández-Millán E, de Toro-Martín J, Lizárraga-Mollinedo E, Escrivá F,Álvarez C. Role of endogenous IL-6 in the neonatal expansion and functionality ofWistar rat pancreatic alpha cells. Diabetologia 2013;56:1098–110748. Habener JF, Stanojevic V. Alpha cells come of age. Trends Endocrinol Metab2013;24:153–16349. Spijker HS, Ravelli RB, Mommaas-Kienhuis AM, et al. Conversion of maturehuman b-cells into glucagon-producing a-cells. Diabetes 2013;62:2471–2480
diabetes.diabetesjournals.org Chow and Associates 2995