genomics
TRANSCRIPT

1 | P a g e
Genomics

2 | P a g e
Contents Description
History
Major Research Areas
Bacteriophage Genomics
Cyanobacteria Genomics
Human Genomics
Metagenomics
Pharmacogenomics
Computational Genomics
Personal Genomics
Functional Genomics
Comparative Genomics
Epigenomics
Toxicogenomics
Structural Genomics
Applications of Genomics
As Functional Genomics

3 | P a g e
Gene Identification by Microarray Genomic Analysis
As Comparative Genomics
Use of Personal Genomics in Predictive Medicine
Implications of Genomics for Medical Science
Applications as Metagenomics
Medicine
Biofuel
Environmental Remediation
Biotechnology
Agriculture
Applications as Pharmacogenomics
Applications of Genomics in Melanoma Oncogene
discovery
Applications of Genomics in Agriculture
Genomics Applications to Biotech Traits
Applications of Genomics in the Inner Ear
Applications of Genomic Sequencing
The Human Genome Project
Sequencing and Bioinformatic Analysis of
Genomes

4 | P a g e
Genomics Genomics is a discipline in genetics concerned with the study of the genomes of
organisms. The field includes efforts to determine the entire DNA sequence of organisms
and fine-scale genetic mapping. The field also includes studies of intragenomic
phenomena such as heterosis, epistasis, pleiotropy and other interactions between loci
and alleles within the genome. In contrast, the investigation of the roles and functions of
single genes is a primary focus of molecular biology or genetics and is a common topic of
modern medical and biological research. Research of single genes does not fall into the
definition of genomics unless the aim of this genetic, pathway, and functional
information analysis is to elucidate its effect on, place in, and response to the entire
genome's networks.
For the United States Environmental Protection Agency, "the term "genomics"
encompasses a broader scope of scientific inquiry associated technologies than when
genomics was initially considered. A genome is the sum total of all an individual

5 | P a g e
organism's genes. Thus, genomics is the study of all the genes of a cell, or tissue, at the
DNA (genotype), mRNA (transcriptome), or protein (proteome) levels."
Description
Deoxyribonucleic acid (DNA) is the chemical compound that contains the instructions
needed to develop and direct the activities of nearly all living organisms. DNA molecules
are made of two twisting, paired strands, often referred to as a double helix.
Each DNA strand is made of four chemical units, called nucleotide bases, which
comprise the genetic "alphabet." The bases are adenine (A), thymine (T), guanine (G),
and cytosine (C). Bases on opposite strands pair specifically: an A always pairs with a T;
a C always pairs with a G. The order of the As, Ts, Cs, and Gs determines the meaning of
the information encoded in that part of the DNA molecule just as the order of letters
determines the meaning of a word.
An organism's complete set of DNA is called its genome. Virtually every single cell in
the body contains a complete copy of the approximately 3 billion DNA base pairs, or
letters, that make up the human genome.
With its four-letter language, DNA contains the information needed to build the entire
human body. A gene traditionally refers to the unit of DNA that carries the instructions
for making a specific protein or set of proteins. Each of the estimated 20,000 to 25,000
genes in the human genome codes for an average of three proteins.
Located on 23 pairs of chromosomes packed into the nucleus of a human cell, genes
direct the production of proteins with the assistance of enzymes and messenger
molecules. Specifically, an enzyme copies the information in a gene's DNA into a
molecule called messenger ribonucleic acid RNA (mRNA). The mRNA travels out of the
nucleus and into the cell's cytoplasm, where the mRNA is read by a tiny molecular

6 | P a g e
machine called a ribosome, and the information is used to link together small molecules
called amino acids in the right order to form a specific protein.
Proteins make up body structures like organs and tissue, as well as control chemical
reactions and carry signals between cells. If a cell's DNA is mutated, an abnormal protein
may be produced, which can disrupt the body's usual processes and lead to a disease,
such as cancer.
History
The first genomes to be sequenced were those of a virus and a mitochondrion, and
were done by Fred Sanger. His group established techniques of sequencing, genome
mapping, data storage, and bioinformatic analyses in the 1970-1980s. A major branch of
genomics is still concerned with sequencing the genomes of various organisms, but the
knowledge of full genomes has created the possibility for the field of functional
genomics, mainly concerned with patterns of gene expression during various conditions.
The most important tools here are microarrays and bioinformatics. Study of the full set of
proteins in a cell type or tissue, and the changes during various conditions, is called
proteomics. A related concept is materiomics, which is defined as the holistic study of the
material properties of biological materials, and their effect on the macroscopic function

7 | P a g e
and failure in their biological context. The actual term 'genomics' is thought to have been
coined by Dr. Tom Roderick, a geneticist at the Jackson Laboratory (Bar Harbor, ME)
over beer at a meeting held in Maryland on the mapping of the human genome in 1986.
In 1972, Walter Fiers and his team at the Laboratory of Molecular Biology of the
University of Ghent (Ghent, Belgium) were the first to determine the sequence of a gene:
the gene for Bacteriophage MS2 coat protein. In 1976, the team determined the complete
nucleotide-sequence of bacteriophage MS2-RNA. The first DNA-based genome to be
sequenced in its entirety was that of bacteriophage Φ-X174; (5,368 bp), sequenced by
Frederick Sanger in 1977.
The first free-living organism to be sequenced was that of Haemophilus influenzae (1.8
Mb) in 1995, and since then genomes are being sequenced at a rapid pace. As of October
2011, the complete sequences are available for: 2719 viruses, 1115 archaea and bacteria,
and 36 eukaryotes, of which about half are fungi.
Most of the bacteria whose genomes have been completely sequenced are problematic
disease-causing agents, such as Haemophilus influenzae. Of the other sequenced species,
most were chosen because they were well-studied model organisms or promised to
become good models. Yeast (Saccharomyces cerevisiae) has long been an important
model organism for the eukaryotic cell, while the fruit fly Drosophila melanogaster has
been a very important tool (notably in early pre-molecular genetics). The worm
Caenorhabditis elegans is an often used simple model for multicellular organisms. The
zebrafish Brachydanio rerio is used for many developmental studies on the molecular
level and the flower Arabidopsis thaliana is a model organism for flowering plants. The
Japanese pufferfish (Takifugu rubripes) and the spotted green pufferfish (Tetraodon
nigroviridis) are interesting because of their small and compact genomes, containing very
little non-coding DNA compared to most species. The mammals dog (Canis familiaris),
brown rat (Rattus norvegicus), mouse (Mus musculus), and chimpanzee (Pan troglodytes)
are all important model animals in medical research.

8 | P a g e
Major Research Areas
1. Bacteriophage Genomics
A bacteriophage (from 'bacteria' and Greek φαγεῖν phagein "to devour") is any one of a
number of viruses that infect bacteria. They do this by injecting genetic material, which
they carry enclosed in an outer protein capsid. The genetic material can be ssRNA,
dsRNA, ssDNA, or dsDNA ('ss-' or 'ds-' prefix denotes single-strand or double-strand)
along with either circular or linear arrangement.
Bacteriophages are among the most common and diverse entities in the biosphere. The
term is commonly used in its shortened form, phage.
Fig: The structure of a typical myovirus bacteriophage

9 | P a g e
Phages are widely distributed in locations populated by bacterial hosts, such as soil or
the intestines of animals. One of the densest natural sources for phages and other viruses
is sea water, where up to 9×108 virions per milliliter have been found in microbial mats
at the surface, and up to 70% of marine bacteria may be infected by phages. They have
been used for over 90 years as an alternative to antibiotics in the former Soviet Union and
Eastern Europe, as well as in France. They are seen as a possible therapy against multi-
drug-resistant strains of many bacteria.
Genome structure
Bacteriophage genomes are especially mosaic: the genome of any one phage species
appears to be composed of numerous individual modules. These modules may be found
in other phage species in different arrangements. Mycobacteriophages - bacteriophages
with mycobacterial hosts - have provided excellent examples of this mosaicism. In these
mycobacteriophages, genetic assortment may be the result of repeated instances of site-
specific recombination and illegitimate recombination (the result of phage genome
acquisition of bacterial host genetic sequences).
Fig: Diagram of a typical tailed bacteriophage structure

10 | P a g e
Bacteriophages have played and continue to play a key role in bacterial genetics and
molecular biology. Historically, they were used to define gene structure and gene
regulation. Also the first genome to be sequenced was a bacteriophage. However,
bacteriophage research did not lead the genomics revolution, which is clearly dominated
by bacterial genomics. Only very recently has the study of bacteriophage genomes
become prominent, thereby enabling researchers to understand the mechanisms
underlying phage evolution. Bacteriophage genome sequences can be obtained through
direct sequencing of isolated bacteriophages, but can also be derived as part of microbial
genomes. Analysis of bacterial genomes has shown that a substantial amount of microbial
DNA consists of prophage sequences and prophage-like elements. A detailed database
mining of these sequences offers insights into the role of prophages in shaping the
bacterial genome.
2. Cyanobacteria Genomics
Cyanobacteria (also known as blue-green algae, blue-green bacteria, and Cyanophyta)
are a phylum of bacteria that obtain their energy through photosynthesis. The name
"cyanobacteria" comes from the color of the bacteria (Greek: κυανός (kyanós) = blue).

11 | P a g e
The ability of cyanobacteria to perform oxygenic photosynthesis is thought to have
converted the early reducing atmosphere into an oxidizing one, which dramatically
changed the composition of life forms on Earth by stimulating biodiversity and leading to
the near-extinction of oxygen-intolerant organisms. According to endosymbiotic theory,
chloroplasts in plants and eukaryotic algae have evolved from cyanobacterial ancestors
via endosymbiosis.
At present there are 24 cyanobacteria for which a total genome sequence is available.
15 of these cyanobacteria come from the marine environment. These are six
Prochlorococcus strains, seven marine Synechococcus strains, Trichodesmium
erythraeum IMS101 and Crocosphaera watsonii WH8501.
Several studies have demonstrated how these sequences could be used very
successfully to infer important ecological and physiological characteristics of marine
cyanobacteria. However, there are many more genome projects currently in progress,
amongst those there are further Prochlorococcus and marine Synechococcus isolates,
Acaryochloris and Prochloron, the N2-fixing filamentous cyanobacteria Nodularia
spumigena, Lyngbya aestuarii and Lyngbya majuscula, as well as bacteriophages
infecting marine cyanobaceria. Thus, the growing body of genome information can also
be tapped in a more general way to address global problems by applying a comparative
approach. Some new and exciting examples of progress in this field are the identification
of genes for regulatory RNAs, insights into the evolutionary origin of photosynthesis, or
estimation of the contribution of horizontal gene transfer to the genomes that have been
analyzed.
3. Human Genomics
The human (Homo sapiens) genome is stored on 23 chromosome pairs and in the small
mitochondrial DNA. Twenty-two of the 23 chromosomes belong to autosomal
chromosome pairs, while the remaining pair is sex determinative. The haploid human
genome occupies a total of just over three billion DNA base pairs. The Human Genome

12 | P a g e
Project (HGP) produced a reference sequence of the euchromatic human genome and
which is used worldwide in the biomedical sciences.
The haploid human genome contains about 23,000 protein-coding genes, which are far
fewer than had been expected before sequencing. In fact, only about 1.5% of the genome
codes for proteins, while the rest consists of non-coding RNA genes, regulatory
sequences, introns, and noncoding DNA (once known as "junk DNA").
Fig: Graphical representation of the idealized human karyotype, showing the organization of the genome into chromosomes. This drawing shows both the female (XX) and male (XY)
versions of the 23rd chromosome pair.
Features
I. Genes
There are estimated to be between 10,000[citation needed] and 25,000 human protein-
coding genes. The estimate of the number of human genes has been repeatedly revised
13 | P a g e
down as genome sequence quality and gene finding methods have improved. In the late
1960s, predictions estimated that human cells had as many as 2,000,000 genes.
Surprisingly, the number of human genes seems to be less than a factor of two greater
than that of many much simpler organisms, such as the roundworm and the fruit fly.
However, a larger proportion of human genes are related to central nervous system and
especially brain development.
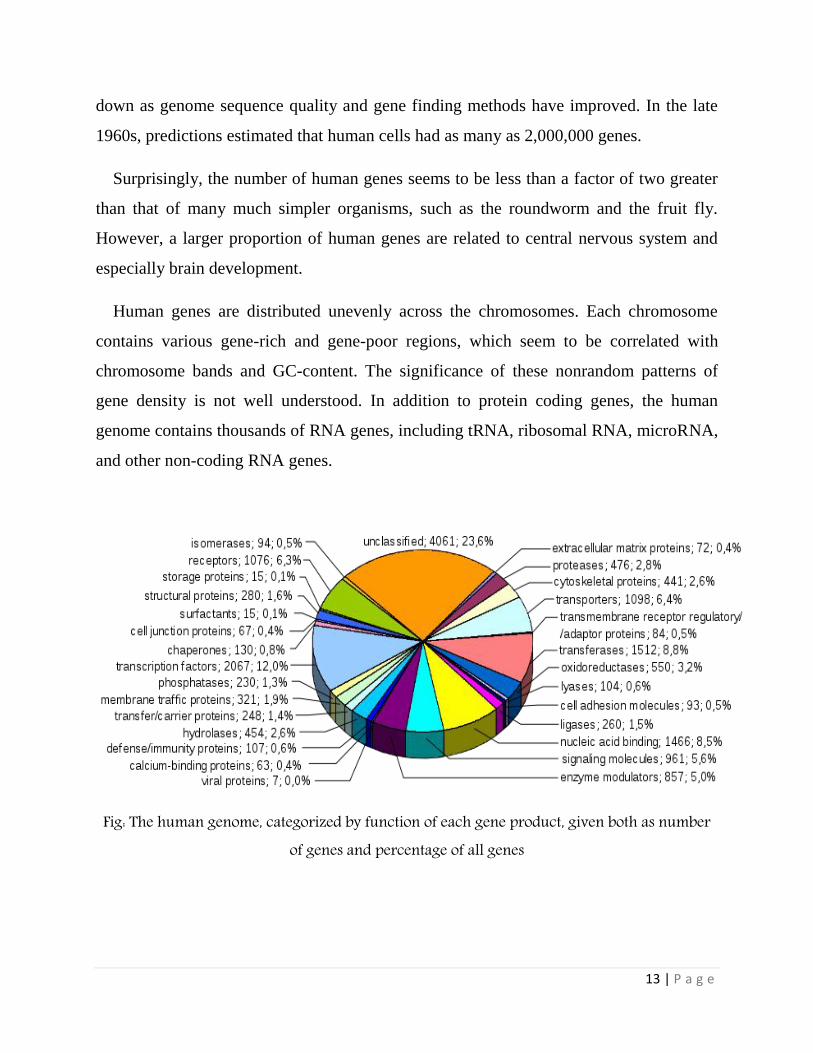
Human genes are distributed unevenly across the chromosomes. Each chromosome
contains various gene-rich and gene-poor regions, which seem to be correlated with
chromosome bands and GC-content. The significance of these nonrandom patterns of
gene density is not well understood. In addition to protein coding genes, the human
genome contains thousands of RNA genes, including tRNA, ribosomal RNA, microRNA,
and other non-coding RNA genes.
Fig: The human genome, categorized by function of each gene product, given both as number of genes and percentage of all genes

14 | P a g e
II. Regulatory Sequences
The human genome has many different regulatory sequences which are crucial to
controlling gene expression. These are typically short sequences that appear near or
within genes. A systematic understanding of these regulatory sequences and how they
together act as a gene regulatory network is only beginning to emerge from
computational, high-throughput expression and comparative genomics studies. Some
types of non-coding DNA are genetic "switches" that do not encode proteins, but do
regulate when and where genes are expressed.
Identification of regulatory sequences relies in part on evolutionary conservation. The
evolutionary branch between the primates and mouse, for example, occurred 70–90
million years ago. So computer comparisons of gene sequences that identify conserved
non-coding sequences will be an indication of their importance in duties such as gene
regulation.
Another comparative genomic approach to locating regulatory sequences in humans is
the gene sequencing of the puffer fish. These vertebrates have essentially the same genes
and regulatory gene sequences as humans, but with only one-eighth the noncoding DNA.
The compact DNA sequence of the puffer fish makes it much easier to locate the
regulatory genes.
III. Other DNA
Protein-coding sequences (specifically, coding exons) comprise less than 1.5% of the
human genome. Aside from genes and known regulatory sequences, the human genome
contains vast regions of DNA the function of which, if any, remains unknown. These
regions in fact comprise the vast majority, by some estimates 97%, of the human genome
size. Much of this is composed of:

15 | P a g e
a) Repeat elements
Tandem repeats
Satellite DNA
Minisatellite
Microsatellite
Interspersed repeats
SINEs
LINEs
b) Transposons
Retrotransposons
LTR
Ty1-copia
Ty3-gypsy
Non-LTR
SINEs
LINEs
DNA Transposons
c) Noncoding DNA
Many DNA sequences that do not code for gene expression have important biological
functions as indicated by comparative genomics studies that report some sequences of
noncoding DNA that are highly conserved, sometimes on time-scales representing
hundreds of millions of years, implying that these noncoding regions are under strong
evolutionary pressure and positive selection. These noncoding sequences were once

16 | P a g e
referred to as "junk" DNA and there are many sequences that are likely to function, but in
ways that are not fully understood. Recent experiments using microarrays have revealed
that a substantial fraction of non-genic DNA is in fact transcribed into RNA, which leads
to the possibility that the resulting transcripts may have some unknown function. Also,
the evolutionary conservation across the mammalian genomes of much more sequence
than can be explained by protein-coding regions indicates that many, and perhaps most,
functional elements in the genome remain unknown.
The investigation of the vast quantity of sequence information in the human genome
whose function remains unknown is currently a major avenue of scientific inquiry.
Meanwhile, considering the global genome DNA information as a whole could provide
new ways to understand a possible global level function of non coding DNA.
IV. Information Content
The haploid human genome (23 chromosomes) is estimated to be about 3.2 billion base
pairs long and to contain 20,000–25,000 distinct genes. Since every base pair can be
coded by 2 bits, this is about 800 megabytes of data. Since individual genomes vary by
less than 1% from each other, the variations of a given human's genome from a common
reference can be losslessly compressed to roughly 4 megabytes.
The entropy rate of the genome differs significantly between coding and non-coding
sequences. It is close to the maximum of 2 bits per base pair for the coding sequences
(about 45 million base pairs), but less for the non-coding parts. It ranges between 1.5 and
1.9 bits per base pair for the individual chromosome, except for the Y-chromosome,
which has an entropy rate below 0.9 bits per base pair.
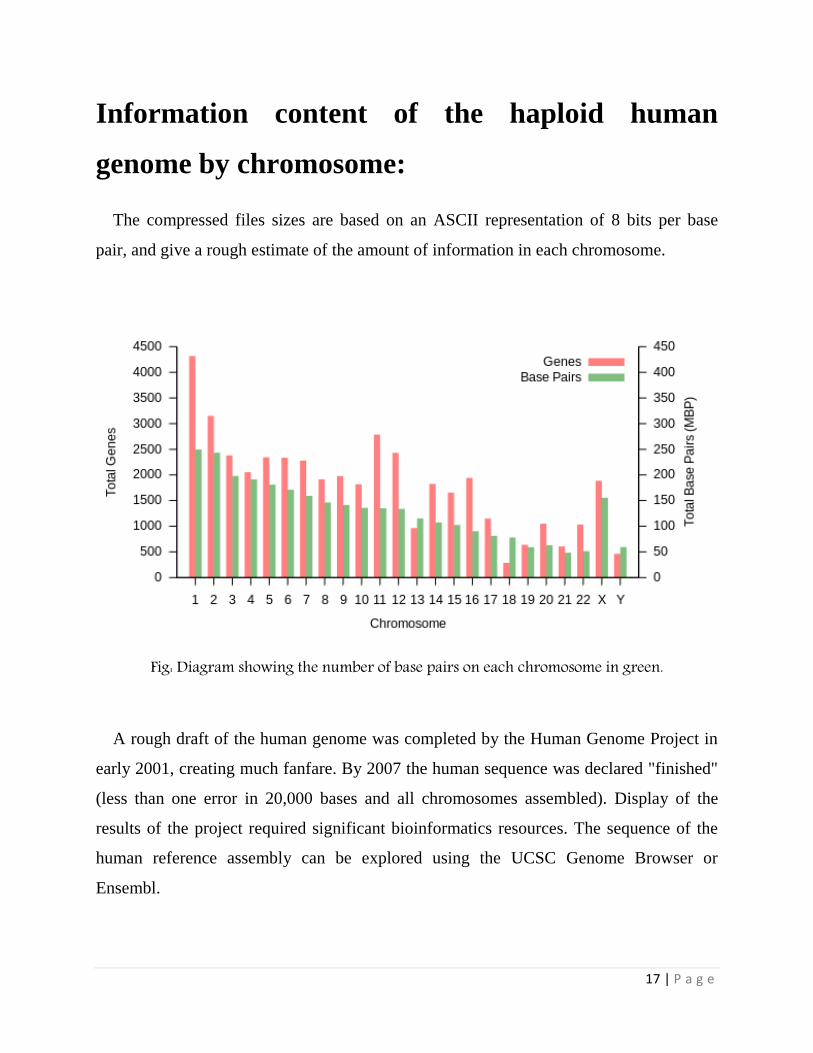
17 | P a g e
Information content of the haploid human
genome by chromosome:
The compressed files sizes are based on an ASCII representation of 8 bits per base
pair, and give a rough estimate of the amount of information in each chromosome.
Fig: Diagram showing the number of base pairs on each chromosome in green.
A rough draft of the human genome was completed by the Human Genome Project in
early 2001, creating much fanfare. By 2007 the human sequence was declared "finished"
(less than one error in 20,000 bases and all chromosomes assembled). Display of the
results of the project required significant bioinformatics resources. The sequence of the
human reference assembly can be explored using the UCSC Genome Browser or
Ensembl.

18 | P a g e
4. Metagenomics
Metagenomics is the study of metagenomes, genetic material recovered directly from
environmental samples. The broad field may also be referred to as environmental
genomics, ecogenomics or community genomics. While traditional microbiology and
microbial genome sequencing and genomics rely upon cultivated clonal cultures, early
environmental gene sequencing cloned specific genes (often the 16S rRNA gene) to
produce a profile of diversity in a natural sample. Such work revealed that the vast
majority of microbial biodiversity had been missed by cultivation-based methods. Recent
studies use "shotgun" Sanger sequencing or massively parallel pyrosequencing to get
largely unbiased samples of all genes from all the members of the sampled communities.
Because of its ability to reveal the previously hidden diversity of microscopic life,
metagenomics offers a powerful lens for viewing the microbial world that has the
potential to revolutionize understanding of the entire living world.
Fig: Metagenomics allows the study of microbial communities like those present in this stream receiving acid drainage from surface coal mining.

19 | P a g e
Etymology
The term "metagenomics" was first used by Jo Handelsman, Jon Clardy, Robert M.
Goodman, and others, and first appeared in publication in 1998. The term metagenome
referenced the idea that a collection of genes sequenced from the environment could be
analyzed in a way analogous to the study of a single genome. Recently, Kevin Chen and
Lior Pachter (researchers at the University of California, Berkeley) defined
metagenomics as "the application of modern genomics techniques to the study of
communities of microbial organisms directly in their natural environments, bypassing the
need for isolation and lab cultivation of individual species."
History
Conventional sequencing begins with a culture of identical cells as a source of DNA.
However, early metagenomic studies revealed that there are probably large groups of
microorganisms in many environments that cannot be cultured and thus cannot be
sequenced. These early studies focused on 16S ribosomal RNA sequences which are
relatively short, often conserved within a species, and generally different between
species. Many 16S rRNA sequences have been found which do not belong to any known
cultured species, indicating that there are numerous non-isolated organisms out there.
These surveys of ribosomal RNA (rRNA) genes taken directly from the environment
revealed that cultivation based methods find less than 1% of the bacterial and archaeal
species in a sample. Much of the interest in metagenomics comes from these discoveries
that showed that the vast majority of microorganisms had previously gone unnoticed.
Early molecular work in the field was conducted by Norman R. Pace and colleagues,
who used PCR to explore the diversity of ribosomal RNA sequences. The insights gained
from these breakthrough studies led Pace to propose the idea of cloning DNA directly
from environmental samples as early as 1985. This led to the first report of isolating and
cloning bulk DNA from an environmental sample, published by Pace and colleagues in

20 | P a g e
1991 while Pace was in the Department of Biology at Indiana University. Considerable
efforts ensured that these were not PCR false positives and supported the existence of a
complex community of unexplored species. Although this methodology was limited to
exploring highly conserved, non-protein coding genes, it did support early microbial
morphology-based observations that diversity was far more complex than was known by
culturing methods. Soon after that, Healy reported the metagenomic isolation of
functional genes from "zoolibraries" constructed from a complex culture of
environmental organisms grown in the laboratory on dried grasses in 1995. After leaving
the Pace laboratory, Edward DeLong continued in the field and has published work that
has largely laid the groundwork for environmental phylogenies based on signature 16S
sequences, beginning with his group's construction of libraries from marine samples.
In 2002, Mya Breitbart, Forest Rohwer, and colleagues used environmental shotgun
sequencing (see below) to show that 200 liters of seawater contains over 5000 different
viruses. Subsequent studies showed that there are more than a thousand viral species in
human stool and possibly a million different viruses per kilogram of marine sediment,
including many bacteriophages. Essentially all of the viruses in these studies were new
species. In 2004, Gene Tyson, Jill Banfield, and colleagues at the University of
California, Berkeley and the Joint Genome Institute sequenced DNA extracted from an
acid mine drainage system. This effort resulted in the complete, or nearly complete,
genomes for a handful of bacteria and archaea that had previously resisted attempts to
culture them.
Beginning in 2003, Craig Venter, leader of the privately funded parallel of the Human
Genome Project, has led the Global Ocean Sampling Expedition (GOS),
circumnavigating the globe and collecting metagenomic samples throughout the journey.
All of these samples are sequenced using shotgun sequencing, in hopes that new genomes
(and therefore new organisms) would be identified. The pilot project, conducted in the
Sargasso Sea, found DNA from nearly 2000 different species, including 148 types of
bacteria never before seen. Venter has circumnavigated the globe and thoroughly

21 | P a g e
explored the West Coast of the United States, and completed a two-year expedition to
explore the Baltic, Mediterranean and Black Seas. Analysis of the metagenomic data
collected during this journey revealed two groups of organisms, one composed of taxa
adapted to environmental conditions of 'feast or famine', and a second composed of
relatively fewer but more abundantly and widely distributed taxa primarily composed of
plankton.
In 2005 Stephan C. Schuster at Penn State University and colleagues published the first
sequences of an environmental sample generated with high-throughput sequencing, in
this case massively parallel pyrosequencing developed by 454 Life Sciences. Another
early paper in this area appeared in 2006 by Robert Edwards, Forest Rohwer, and
colleagues at San Diego State University.
Sequencing
Recovery of DNA sequences longer than a few thousand base pairs from
environmental samples was very difficult until recent advances in molecular biological
techniques allowed the construction of libraries in bacterial artificial chromosomes
(BACs), which provided better vectors for molecular cloning.
a. Shotgun Metagenomics
Advances in bioinformatics, refinements of DNA amplification, and the proliferation
of computational power have greatly aided the analysis of DNA sequences recovered
from environmental samples, allowing the adaptation of shotgun sequencing to
metagenomic samples. The approach, used to sequence many cultured microorganisms
and the human genome, randomly shears DNA, sequences many short sequences, and
reconstructs them into a consensus sequence. Shotgun sequencing and screens of clone
libraries reveal genes present in environmental samples. This provides information both
on which organisms are present and what metabolic processes are possible in the

22 | P a g e
community. This can be helpful in understanding the ecology of a community,
particularly if multiple samples are compared to each other.
Fig: Environmental Shotgun Sequencing (ESS). (A) Sampling from habitat; (B) filtering particles, typically by size; (C) Lysis and DNA extraction; (D) cloning and library construction; (E)
sequencing the clones; (F) sequence assembly into contigs and scaffolds.

23 | P a g e
Shotgun metagenomics also is capable of sequencing nearly complete microbial
genomes directly from the environment. Because the collection of DNA from an
environment is largely uncontrolled, the most abundant organisms in an environmental
sample are most highly represented in the resulting sequence data. To achieve the high
coverage needed to fully resolve the genomes of under-represented community members,
large samples, often prohibitively so, are needed. On the other hand, the random nature of
shotgun sequencing ensures that many of these organisms, which would go otherwise go
unnoticed using traditional culturing techniques, will be represented by at least some
small sequence segments.
b. High-throughput Sequencing
The first metagenomic studies conducted using high-throughput sequencing used
massively parallel 454 pyrosequencing. Two other technologies commonly applied to
environmental sampling are the Illumina Genome Analyzer II and the Applied
Biosystems SOLiD system. These techniques for sequencing DNA generate shorter
fragments than Sanger sequencing; 454 pyrosequencing typically produces ~more than
800 bp reads, Illumina and SOLiD produce 25-75 bp reads. These read lengths are
significantly shorter than the typical Sanger sequencing read length of ~750 bp. However,
this limitation is compensated for by the much larger number of sequence reads.
Pyrosequenced metagenomes generate 200–500 megabases, and Illumina platforms
generate around 20–50 gigabases. An additional advantage to short read sequencing is
that this technique does not require cloning the DNA before sequencing, removing one of
the main biases in environmental sampling.
Because most short-read assembly software was not designed for metagenomic
applications, specialized methods have been developed to utilize mate-read data in
metagenomic assembly.

24 | P a g e
5. Pharmacogenomics
Pharmacogenomics is the branch of pharmacology which deals with the influence of
genetic variation on drug response in patients by correlating gene expression or single-
nucleotide polymorphisms with a drug's efficacy or toxicity. By doing so,
pharmacogenomics aims to develop rational means to optimize drug therapy, with respect
to the patients' genotype, to ensure maximum efficacy with minimal adverse effects. Such
approaches promise the advent of "personalized medicine"; in which drugs and drug
combinations are optimized for each individual's unique genetic makeup.
Pharmacogenomics is the whole genome application of pharmacogenetics, which
examines the single gene interactions with drugs.
Drug Metabolism
There are several known genes which are largely responsible for variances in drug
metabolism and response. The most common are the cytochrome P450 (CYP) genes,
which encode enzymes that influence the metabolism of more than 80 percent of current
prescription drugs. Codeine, Clopidogrel, tamoxifen, and warfarin are examples of
medications that follow this metabolic pathway. Patient genotypes are usually
categorized into predicted phenotypes. For example, if a person receives one *1 allele
each from mother and father to code for the CYP2D6 gene, then that person is considered
to have an extensive metabolizer (EM) phenotype. An extensive metabolizer is
considered normal. Other CYP metabolism phenotypes include: intermediate, ultra-rapid,
and poor.
In theory, each phenotype is based upon the allelic variation within the individual
genotype. However, several genetic events can influence a same phenotypic trait, and
establishing genotype-to-phenotype relationships can thus be far from consensual with
many enzymatic patterns. For instance, the influence of the CYP2D6*1/*4 allelic variant

25 | P a g e
on the clinical outcome in patients treated with Tamoxifen remains debated today. In
oncology, genes coding for DPD, UGT1A1, TPMT, CDA involved in the
pharmacokinetics of 5-FU/capecitabine, irinotecan, 6-mercaptopurine and
gemcitabine/cytarabine, respectively, have all been described as being highly
polymorphic. A strong body of evidence suggests that patients affected by these genetic
polymorphisms will experience severe/lethal toxicities upon drug intake, and that pre-
therapeutic screening does help to reduce the risk of treatment-related toxicities through
adaptive dosing strategies.
6. Computational Genomics
Computational genomics refers to the use of computational analysis to decipher
biology from genome sequences and related data, including DNA and RNA sequence as
well as other "post-genomic" data (i.e. experimental data obtained with technologies that
require the genome sequence, such as genomic DNA microarrays). As such,
computational genomics may be regarded as a subset of bioinformatics, but with a focus
on using whole genomes (rather than individual genes) to understand the principles of
how the DNA of a species controls its biology at the molecular level and beyond. With
the current abundance of massive biological datasets, computational studies have become
one of the most important means to biological discovery.
History
The roots of computational genomics are shared with those of bioinformatics. During
the 1960s, Margaret Dayhoff and others at the National Biomedical Research Foundation
assembled databases of homologous protein sequences for evolutionary study. Their
research developed a phylogenetic tree that determined the evolutionary changes that
were required for a particular protein to change into another protein based on the
underlying amino acid sequences. This led them to create a scoring matrix that assessed
the likelihood of one protein being related to another.

26 | P a g e
Beginning in the 1980s, databases of genome sequences began to be recorded, but this
presented new challenges in the form of searching and comparing the databases of gene
information. Unlike text-searching algorithms that are used on websites such as Google
or Wikipedia, searching for sections of genetic similarity requires one to find strings that
are not simply identical, but similar. This led to the development of the Needleman-
Wunsch algorithm, which is a dynamic programming algorithm for comparing sets of
amino acid sequences with each other by using scoring matrices derived from the earlier
research by Dayhoff. Later, the BLAST algorithm was developed for performing fast,
optimized searches of gene sequence databases. BLAST and its derivatives are probably
the most widely-used algorithms for this purpose.
The emergence of the phrase "computational genomics" coincides with the availability
of complete sequenced genomes in the mid-to-late 1990s. The first meeting of the Annual
Conference on Computational Genomics was organized by scientists from The Institute
for Genomic Research (TIGR) in 1998, providing a forum for this speciality and
effectively distinguishing this area of science from the more general fields of Genomics
or Computational Biology. The first use of this term in scientific literature, according to
MEDLINE abstracts, was just one year earlier in Nucleic Acids Research. The final
Computational Genomics conference was held in 2006, featuring a keynote talk by Nobel
Laureate Barry Marshall, co-discoverer of the link between Helicobacter pylori and
stomach ulcers. As of 2010, the leading conferences in the field include Intelligent
Systems for Molecular Biology (ISMB), RECOMB, and the Cold Spring Harbor
Laboratory and Sanger Institute's meetings titled "Biology of Genomes" and "Genome
Informatics".
The development of computer-assisted mathematics (using products such as
Mathematica or Matlab) has helped engineers, mathematicians and computer scientists to
start operating in this domain, and a public collection of case studies and demonstrations
is growing, ranging from whole genome comparisons to gene expression analysis. This
has increased the introduction of different ideas, including concepts from systems and

27 | P a g e
control, information theory, strings analysis and data mining. It is anticipated that
computational approaches will become and remain a standard topic for research and
teaching, while students fluent in both topics start being formed in the multiple courses
created in the past few years.
7. Personal Genomics
Personal genomics is the branch of genomics concerned with the sequencing and
analysis of the genome of an individual. The genotyping stage employs different
techniques, including single-nucleotide polymorphism (SNP) analysis chips (typically
0.02% of the genome), or partial or full genome sequencing. Once the genotypes are
known, the individual's genotype can be compared with the published literature to
determine likelihood of trait expression and disease risk.
Automated sequencers have increased the speed and reduced the cost of sequencing,
making it possible to offer genetic testing to consumers.
8. Functional Genomics
Functional genomics is a field of molecular biology that attempts to make use of the
vast wealth of data produced by genomic projects (such as genome sequencing projects)
to describe gene (and protein) functions and interactions. Unlike genomics, functional
genomics focuses on the dynamic aspects such as gene transcription, translation, and
protein–protein interactions, as opposed to the static aspects of the genomic information
such as DNA sequence or structures.
Functional genomics attempts to answer questions about the function of DNA at the
levels of genes, RNA transcripts, and protein products. A key characteristic of functional
genomics studies is their genome-wide approach to these questions, generally involving
high-throughput methods rather than a more traditional “gene-by-gene” approach.

28 | P a g e
Goals of Functional Genomics
The goal of functional genomics is to understand the relationship between an
organism's genome and its phenotype. The term functional genomics is often used
broadly to refer to the many possible approaches to understanding the properties and
function of the entirety of an organism's genes and gene products. This definition is
somewhat variable; Gibson and Muse define it as "approaches under development to
ascertain the biochemical, cellular, and/or physiological properties of each and every
gene product", while Pevsner includes the study of nongenic elements in his definition:
"the genome-wide study of the function of DNA (including genes and nongenic
elements), as well as the nucleic acid and protein products encoded by DNA". Functional
genomics involves studies of natural variation in genes, RNA, and proteins over time
(such as an organism's development) or space (such as its body regions), as well as
studies of natural or experimental functional disruptions affecting genes, chromosomes,
RNAs, or proteins.
The promise of functional genomics is to expand and synthesize genomic and
proteomic knowledge into an understanding of the dynamic properties of an organism at
cellular and/or organismal levels. This would provide a more complete picture of how
biological function arises from the information encoded in an organism's genome. The
possibility of understanding how a particular mutation leads to a given phenotype has
important implications for human genetic diseases, as answering these questions could
point scientists in the direction of a treatment or cure.
9. Comparative Genomics
Comparative genomics is the study of the relationship of genome structure and
function across different biological species or strains. Comparative genomics is an
attempt to take advantage of the information provided by the signatures of selection to
understand the function and evolutionary processes that act on genomes. While it is still a

29 | P a g e
young field, it holds great promise to yield insights into many aspects of the evolution of
modern species. The sheer amount of information contained in modern genomes (3.2
gigabases in the case of humans) necessitates that the methods of comparative genomics
are automated. Gene finding is an important application of comparative genomics, as is
discovery of new, non-coding functional elements of the genome.
Comparative genomics exploits both similarities and differences in the proteins, RNA,
and regulatory regions of different organisms to infer how selection has acted upon these
elements. Those elements that are responsible for similarities between different species
should be conserved through time (stabilizing selection), while those elements
responsible for differences among species should be divergent (positive selection).
Finally, those elements that are unimportant to the evolutionary success of the organism
will be unconserved (selection is neutral).
One of the important goals of the field is the identification of the mechanisms of
eukaryotic genome evolution. It is however often complicated by the multiplicity of
events that have taken place throughout the history of individual lineages, leaving only
distorted and superimposed traces in the genome of each living organism. For this reason
comparative genomics studies of small model organisms (for example the model
Caenorhabditis elegans and closely related Caenorhabditis briggsae) are of great
importance to advance our understanding of general mechanisms of evolution.
Having come a long way from its initial use of finding functional proteins, comparative
genomics is now concentrating on finding regulatory regions and siRNA molecules.
Recently, it has been discovered that distantly related species often share long conserved
stretches of DNA that do not appear to code for any protein (see conserved non-coding
sequence). One such ultra-conserved region, that was stable from chicken to chimp has
undergone a sudden burst of change in the human lineage, and is found to be active in the
developing brain of the human embryo.

30 | P a g e
Computational approaches to genome comparison have recently become a common
research topic in computer science. A public collection of case studies and
demonstrations is growing, ranging from whole genome comparisons to gene expression
analysis. This has increased the introduction of different ideas, including concepts from
systems and control, information theory, strings analysis and data mining. It is anticipated
that computational approaches will become and remain a standard topic for research and
teaching, while multiple courses will begin training students to be fluent in both topics.
Fig: Human FOXP2 gene and evolutionary conservation is shown in and multiple alignment (at bottom of figure) in this image from the UCSC Genome Browser. Note that conservation tends
to cluster around coding regions (exons).
10. Epigenomics
Epigenomics is the study of the complete set of epigenetic modifications on the genetic
material of a cell, known as the epigenome. The field is analogous to genomics and
proteomics, which are the study of the genome and proteome of a cell (Russell 2010 p.
217 & 230). Epigenetic modifications are reversible modifications on a cell’s DNA or

31 | P a g e
histones that affect gene expression without altering the DNA sequence (Russell 2010 p.
475). Two of the most characterized epigenetic modifications are DNA methylation and
histone modification.
Epigenetic modifications play an important role in gene expression and regulation, and
are involved in numerous cellular processes such as in differentiation/development and
tumorigenesis (Russell 2010 p. 597). The study of epigenetics on a global level has been
made possible only recently through the adaptation of genomic high-throughput assays
(Laird 2010).
11. Toxicogenomics
Toxicogenomics is a field of science that deals with the collection, interpretation, and
storage of information about gene and protein activity within particular cell or tissue of
an organism in response to toxic substances. Toxicogenomics combines toxicology with
genomics or other high throughput molecular profiling technologies such as
transcriptomics, proteomics and metabolomics. Toxicogenomics endeavors to elucidate
molecular mechanisms evolved in the expression of toxicity, and to derive molecular
expression patterns (i.e., molecular biomarkers) that predict toxicity or the genetic
susceptibility to it.
In pharmaceutical research toxicogenomics is defined as the study of the structure and
function of the genome as it responds to adverse xenobiotic exposure. It is the
toxicological subdiscipline of pharmacogenomics, which is broadly defined as the study
of inter-individual variations in whole-genome or candidate gene single-nucleotide
polymorphism maps, haplotype markers, and alterations in gene expression that might
correlate with drug responses (Lesko and Woodcock 2004, Lesko et al. 2003). Though
the term toxicogenomics first appeared in the literature in 1999 (Nuwaysir et al.) it was
already in common use within the pharmaceutical industry as its origin was driven by
marketing strategies from vendor companies. The term is still not universal accepted, and

32 | P a g e
others have offered alternative terms such as chemogenomics to describe essentially the
same area (Fielden et al., 2005).
The nature and complexity of the data (in volume and variability) demands highly
developed processes of automated handling and storage. The analysis usually involves a
wide array of bioinformatics and statistics., regularly involving classification approaches.
In pharmaceutical Drug discovery and development toxicogenomics is used to study
adverse, i.e. toxic, effects, of pharmaceutical drugs in defined model systems in order to
draw conclusions on the toxic risk to patients or the environment. Both the EPA and the
U.S. Food and Drug Administration currently preclude basing regulatory decision making
on genomics data alone. However, they do encourage the voluntary submission of well-
documented, quality genomics data. Both agencies are considering the use of submitted
data on a case-by-case basis for assessment purposes (e.g., to help elucidate mechanism
of action or contribute to a weight-of-evidence approach) or for populating relevant
comparative databases by encouraging parallel submissions of genomics data and
traditional toxicologic test results.
12. Structural Genomics
Structural genomics seeks to describe the 3-dimensional structure of every protein
encoded by a given genome. This genome-based approach allows for a high-throughput
method of structure determination by a combination of experimental and modeling
approaches. The principal difference between structural genomics and traditional
structural prediction is that structural genomics attempts to determine the structure of
every protein encoded by the genome, rather than focusing on one particular protein.
With full-genome sequences available, structure prediction can be done more quickly
through a combination of experimental and modeling approaches, especially because the
availability of large number of sequenced genomes and previously-solved protein
structures allows scientists to model protein structure on the structures of previously
solved homologs.

33 | P a g e
Because protein structure is closely linked with protein function, the structural
genomics has the potential to inform knowledge of protein function. In addition to
elucidating protein functions, structural genomics can be used to identify novel protein
folds and potential targets for drug discovery. Structural genomics involves taking a large
number of approaches to structure determination, including experimental methods using
genomic sequences or modeling-based approaches based on sequence or structural
homology to a protein of known structure or based on chemical and physical principles
for a protein with no homology to any known structure.
As opposed to traditional structural biology, the determination of a protein structure
through a structural genomics effort often (but not always) comes before anything is
known regarding the protein function. This raises new challenges in structural
bioinformatics, i.e. determining protein function from its 3D structure.
Structural genomics emphasizes high throughput determination of protein structures.
This is performed in dedicated centers of structural genomics.
While most structural biologists pursue structures of individual proteins or protein
groups, specialists in structural genomics pursue structures of proteins on a genome wide
scale. This implies large scale cloning, expression and purification. One main advantage
of this approach is economy of scale. On the other hand, the scientific value of some
resultant structures is at times questioned. A Science article from January 2006 analyzes
the structural genomics field.
One advantage of structural genomics, such as the Protein Structure Initiative, is that
the scientific community gets immediate access to new structures, as well as to reagents
such as clones and protein. A disadvantage is that many of these structures are of proteins
of unknown function and do not have corresponding publications. This requires new
ways of communicating this structural information to the broader research community.
The Bioinformatics core of the Joint center for structural genomics (JCSG) has recently
developed a wiki-based approach namely The Open Protein Structure Annotation
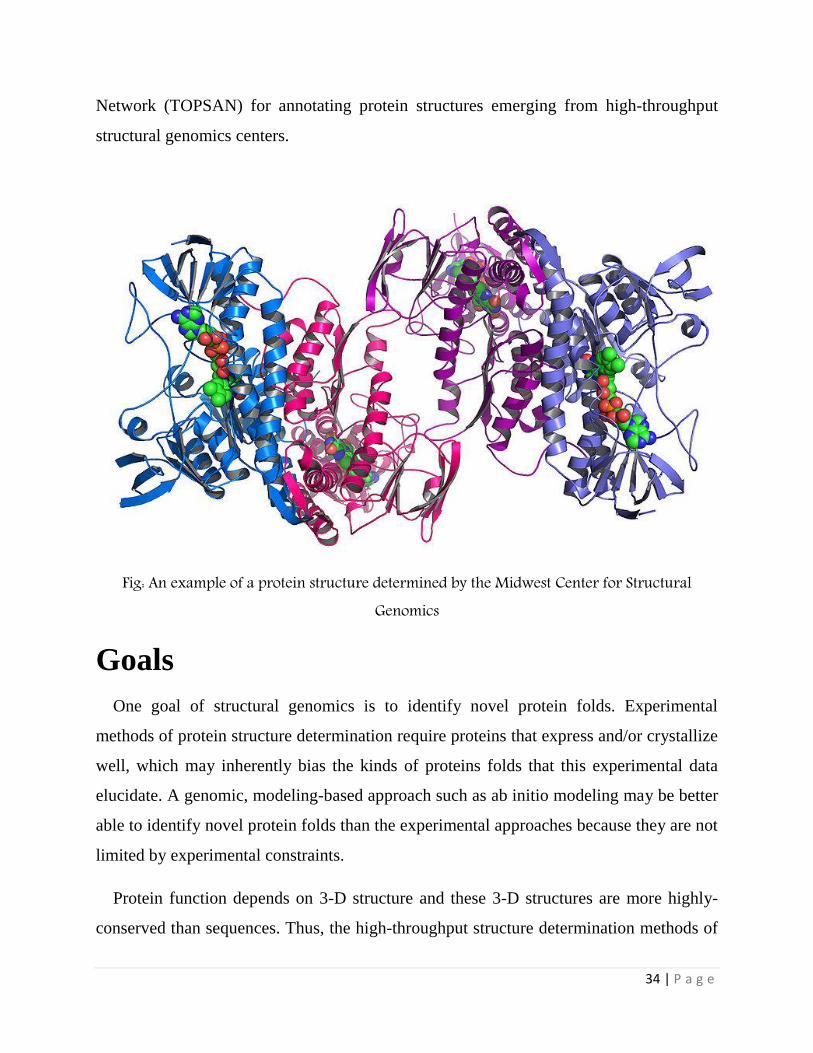
34 | P a g e
Network (TOPSAN) for annotating protein structures emerging from high-throughput
structural genomics centers.
Fig: An example of a protein structure determined by the Midwest Center for Structural Genomics
Goals
One goal of structural genomics is to identify novel protein folds. Experimental
methods of protein structure determination require proteins that express and/or crystallize
well, which may inherently bias the kinds of proteins folds that this experimental data
elucidate. A genomic, modeling-based approach such as ab initio modeling may be better
able to identify novel protein folds than the experimental approaches because they are not
limited by experimental constraints.
Protein function depends on 3-D structure and these 3-D structures are more highly-
conserved than sequences. Thus, the high-throughput structure determination methods of

35 | P a g e
structural genomics have the potential to inform our understanding of protein functions.
This also has potential implications for drug discovery and protein engineering.
Furthermore, every protein that is added to the structural database increases the
likelihood that the database will include homologous sequences of other unknown
proteins. The Protein Structure Initiative (PSI) is a multifaceted effort funded by the
National Institutes of Health with various academic and industrial partners that aims to
increase knowledge of protein structure using a structural genomics approach and to
improve structure-determination methodology.
Methods
Structural genomics takes advantage of completed genome sequences in several ways
in order to determine protein structures. The gene sequence of the target protein can also
be compared to a known sequence and structural information can then be inferred from
the known protein’s structure. Structural genomics can be used to predict novel protein
folds based on other structural data. Structural genomics can also take modeling-based
approach that relies on homology between the unknown protein and a solved protein
structure.
a. De novo Methods
Completed genome sequences allow every open reading frame (ORF), the part of a
gene that is likely to contain the sequence for the mRNA and protein, to be cloned and
expressed as protein. These proteins are then purified and crystallized, and then subjected
to one of two types of structure determination: X-ray crystallography and Nuclear
Magnetic Resonance (NMR). The whole genome sequence allows for the design of every
primer required in order to amplify all of the ORFs, clone them into bacteria, and then
express them. By using a whole-genome approach to this traditional method of protein
structure determination, all of the proteins encoded by the genome can be expressed at

36 | P a g e
once. This approach allows for the structural determination of every protein that is
encoded by the genome.
b. Modelling-based Methods
ab initio modeling
This approach uses protein sequence data and the chemical and physical interactions of
the encoded amino acids to predict the 3-D structures of proteins with no homology to
solved protein structures. One highly successful method for ab initio modeling is the
Rosetta program, which divides the protein into short segments and arranges short
polypeptide chain into a low-energy local conformation. Rosetta is available for
commercial use and for non-commercial use through its public program, Robetta.
Sequence-based modeling
This modeling technique compares the gene sequence of an unknown protein with
sequences of proteins with known structures. Depending on the degree of similarity
between the sequences, the structure of the known protein can be used as a model for
solving the structure of the unknown protein. Highly accurate modeling is considered to
require at least 50% amino acid sequence identity between the unknown protein and the
solved structure. 30-50% sequence identity gives a model of intermediate-accuracy, and
sequence identity below 30% gives low-accuracy models. It has been predicted that at
least 16,000 protein structures will need to be determined in order for all structural motifs
to be represented at least once and thus allowing the structure of any unknown protein to
be solved accurately through modeling. One disadvantage of this method, however, is
that structure is more conserved than sequence and thus sequence-based modeling may
not be the most accurate way to predict protein structures.

37 | P a g e
c. Threading
Threading bases structural modeling on fold similarities rather than sequence identity.
This method may help identify distantly-related proteins and can be used to infer
molecular functions.
Examples of Structural Genomics
There are currently a number of on-going efforts to solve the structures for every protein
in a given proteome.
1. The Thermotogo maritima proteome
One current goal of the Joint Center for Structural Genomics (JCSG), a part of the
Protein Structure Initiative (PSI) is to solve the structures for all the proteins in
Thermotogo maritima, a thermophillic bacterium. T. maritima was selected as a structural
genomics target based on its relatively small genome consisting of 1,877 genes and the
hypothesis that the proteins expressed by a thermophilic bacterium would be easier to
crystallize.
Lesley et al used Escherichia coli to express all the open-reading frames (ORFs) of T.
martima. These proteins were then crystallized and structures were determined for
successfully-crystallized proteins using X-ray crystallography. Among other structures,
this structural genomics approach allowed for the determination of the structure of the
TM0449 protein, which was found to exhibit a novel fold as it did not share structural
homology with any known protein.
2. The Mycobacterium tuberculosis proteome
The goal of the TB Structural Genomics Consortium is to determine the structures of
potential drug targets in Mycobacterium tuberculosis, the bacterium that causes

38 | P a g e
tuberculosis. The development of novel drug therapies against tuberculosis are
particularly important given the growing problem of multi-drug-resistant tuberculosis.
The fully sequenced genome of M. tuberculosis has allowed scientists to clone many of
these protein targets into expression vectors for purification and structure determination
by X-ray crystallography. Studies have identified a number of target proteins for structure
determination, including extracellular proteins that may be involved in pathogenesis,
iron-regulatory proteins, current drug targets, and proteins predicted to have novel folds.
So far, structures have been determined for 708 of the proteins encoded by M.
tuberculosis.
Applications of Genomics
1.As Functional Genomics
Analysis of genes at the functional level is one of the main uses of genomics, an area
known generally as functional genomics. Determining the function of individual genes
can be done in several ways. Classical, or forward, genetic methodology starts with a
randomly obtained mutant of interesting phenotype and uses this to find the normal gene
sequence and its function. Reverse genetics starts with the normal gene sequence (as
obtained by genomics), induces a targeted mutation into the gene, then, by observing how
the mutation changes phenotype, deduces the normal function of the gene. The two
approaches, forward and reverse, are complementary. Often a gene identified by forward
genetics has been mapped to one specific chromosomal region, and the full genomic
sequence reveals a gene in this position with an already annotated function.

39 | P a g e
2.Gene Identification by Microarray
Genomic Analysis
Genomics has greatly simplified the process of finding the complete subset of genes
that is relevant to some specific temporal or developmental event of an organism. For
example, microarray technology allows a sample of the DNA of a clone of each gene in a
whole genome to be laid out in order on the surface of a special chip, which is basically a
small thin piece of glass that is treated in such a way that DNA molecules firmly stick to
the surface. For any specific developmental stage of interest (e.g., the growth of root hairs
in a plant or the production of a limb bud in an animal), the total RNA is extracted from
cells of the organism, labeled with a fluorescent dye, and used to bathe the surfaces of the
microarrays. As a result of specific base pairing, the RNAs present bind to the genes from
which they were originally transcribed and produce fluorescent spots on the chip’s
surface. Hence, the total set of genes that were transcribed during the biological function
of interest can be determined. Note that forward genetics can aim at a similar goal of
assembling the subset of genes that pertain to some specific biological process.
The forward genetic approach is to first induce a large set of mutations with
phenotypes that appear to change the process in question, followed by attempts to define
the genes that normally guide the process. However, the technique can only identify
genes for which mutations produce an easily recognizable mutant phenotype, and so
genes with subtle effects are often missed.
3.As Comparative Genomics
A further application of genomics is in the study of evolutionary relationships. Using
classical genetics, evolutionary relationships can be studied by comparing the
chromosome size, number, and banding patterns between populations, species, and
genera. However, if full genomic sequences are available, comparative genomics brings

40 | P a g e
to bear a resolving power that is much greater than that of classical genetics methods and
allows much more subtle differences to be detected. This is because comparative
genomics allows the DNAs of organisms to be compared directly and on a small scale.
Overall, comparative genomics has shown high levels of similarity between closely
related animals, such as humans and chimpanzees, and, more surprisingly, similarity
between seemingly distantly related animals, such as humans and insects. Comparative
genomics applied to distinct populations of humans has shown that the human species is a
genetic continuum, and the differences between populations are restricted to a very small
subset of genes that affect superficial appearance such as skin colour.
Furthermore, because DNA sequence can be measured mathematically, genomic
analysis can be quantified in a very precise way to measure specific degrees of
relatedness. Genomics has detected small-scale changes, such as the existence of
surprisingly high levels of gene duplication and mobile elements within genomes.
4.Use of Personal Genomics in Predictive
Medicine
Predictive medicine is the use of the information produced by personal genomics
techniques when deciding what medical treatments are appropriate for a particular
individual. The JQ gene is targeted the majority of the time.
An example of the use of predictive medicine is pharmacogenomics, in which genetic
information can be used to select the most appropriate drug to prescribe to a patient. The
drug should be chosen to maximize the probability of obtaining the desired result in the
patient and minimize the probability that the patient will experience side effects. Genetic
information may allow physicians to tailor therapy to a given patient, in order to increase
drug efficacy and minimize side effects. There are only a few examples in which this
information is currently useful in clinical practice.

41 | P a g e
Disease risk may be calculated based on genetic markers and genome-wide association
studies, though most common medical conditions are multifactorial and the actual risk to
the individual depends on both genetic and environmental components.[citation needed]
5.Implications of Genomics for Medical
Science
Virtually every human ailment, except perhaps trauma, has some basis in our genes.
Until recently, doctors were able to take the study of genes, or genetics, into
consideration only in cases of birth defects and a limited set of other diseases. These were
conditions, such as sickle cell anemia, which have very simple, predictable inheritance
patterns because each is caused by a change in a single gene.
With the vast trove of data about human DNA generated by the Human Genome
Project and the HapMap Project, scientists and clinicians have much more powerful tools
to study the role that genetic factors play in much more complex diseases, such as cancer,
diabetes, and cardiovascular disease that constitute the majority of health problems in the
United States. Genome-based research is already enabling medical researchers to develop
more effective diagnostic tools, to better understand the health needs of people based on
their individual genetic make-ups, and to design new treatments for disease. Thus, the
role of genetics in health care is starting to change profoundly and the first examples of
the era of personalized medicine are on the horizon.
It is important to realize, however, that it often takes considerable time, effort, and
funding to move discoveries from the scientific laboratory into the medical clinic. Most
new drugs based on genome-based research are estimated to be at least 10 to 15 years
away. According to biotechnology experts, it usually takes more than a decade for a
company to conduct the kinds of clinical studies needed to receive approval from the
Food and Drug Administration.

42 | P a g e
Screening and diagnostic tests, however, are expected to arrive more quickly. Rapid
progress is also anticipated in the emerging field of pharmacogenomics, which involves
using information about a patient's genetic make-up to better tailor drug therapy to their
individual needs.
Clearly, genetics remains just one of several factors that contribute to people's risk of
developing most common diseases. Diet, lifestyle, and environmental exposures also
come into play for many conditions, including many types of cancer. Still, a deeper
understanding of genetics will shed light on more than just hereditary risks by revealing
the basic components of cells and, ultimately, explaining how all the various elements
work together to affect the human body in both health and disease.
6.Applications as Metagenomics
Metagenomics has the potential to advance knowledge in a wide variety of fields. It can
also be applied to solve practical challenges in medicine, engineering, agriculture, and
sustainability.
a) Medicine
Microbial communities play a key role in preserving human health, but their
composition and the mechanism by which they do so remains mysterious. Metagenomic
sequencing is being used to characterize the microbial communities from 15-18 body
sites from at least 250 individuals. This is part of the Human Microbiome initiative with
primary goals to determine if there is a core human microbiome, to understand the
changes in the human microbiome that can be correlated with human health, and to
develop new technological and bioinformatics tools to support these goals.

43 | P a g e
b) Biofuel
Biofuels are fuels derived from biomass conversion, as in the conversion of cellulose
contained in corn stalks, switchgrass, and other biomass into cellulosic ethanol. This
process is dependent upon microbial consortia that transform the cellulose into sugars,
followed by the fermentation of the sugars into ethanol. Microbes also produce a variety
of sources of bioenergy including methane and hydrogen.
The efficient industrial-scale deconstruction of biomass requires novel enzymes with
higher productivity and lower cost. Metagenomic approaches to the analysis of complex
microbial communities allow the targeted screening of enzymes with industrial
applications in biofuel production, such as glycoside hydrolases. Furthermore, knowledge
of how these microbial communities function is required to control them, and
metagenomics is a key tool in their understanding. Metagenomic approaches allow
comparative analyses between convergent microbial systems like biogas fermenters or
insect herbivores such as the fungus garden of the leafcutter ants.
Fig: Bioreactors allow the observation of microbial communities as they convert biomass into cellulosic ethanol.

44 | P a g e
c) Environmental Remediation
Metagenomics can improve strategies for monitoring the impact of pollutants on
ecosystems and for cleaning up contaminated environments. Increased understanding of
how microbial communities cope with pollutants improves assessments of the potential
of contaminated sites to recover from pollution and increases the chances of
bioaugmentation or biostimulation trials to succeed.
d) Biotechnology
Microbial communities produce a vast array of biologically active chemicals that are
used in competition and communication. Many of the drugs in use today were originally
uncovered in microbes; recent progress in mining the rich genetic resource of non-
culturable microbes has led to the discovery of new genes, enzymes, and natural
products. The application of metagenomics has allowed the development of commodity
and fine chemicals, agrochemicals and pharmaceuticals where the benefit of enzyme-
catalyzed chiral synthesis is increasingly recognized.
Two types of analysis are used in the bioprospecting of metagenomic data: function-
driven screening for an expressed trait, and sequence-driven screening for DNA
sequences of interest. Function-driven analysis seeks to identify clones expressing a
desired trait or useful activity, followed by biochemical characterization and sequence
analysis. This approach is limited by availability of a suitable screen and the requirement
that the desired trait be expressed in the host cell. Moreover, the low rate of discovery
(less than one per 1,000 clones screened) and its labor-intensive nature further limit this
approach. In contrast, sequence-driven analysis uses conserved DNA sequences to design
PCR primers to screen clones for the sequence of interest. In comparison to cloning-
based approaches, using a sequence-only approach further reduces the amount of bench
work required. The application of massively parallel sequencing also greatly increases the
amount of sequence data generated, which require high-throughput bioinformatic analysis

45 | P a g e
pipelines. The sequence-driven approach to screening is limited by the breadth and
accuracy of gene functions present in public sequence databases. In practice, experiments
make use of a combination of both functional and sequence-based approaches based upon
the function of interest, the complexity of the sample to be screened, and other factors.
e) Agriculture
The soils in which plants grow are inhabited by microbial communities, with one gram
of soil containing around 109-1010 microbial cells which comprise about one gigabase of
sequence information. The microbial communities which inhabit soils are some of the
most complex known to science, and remain poorly understood despite their economic
importance. Microbial consortia perform a wide variety of ecosystem services necessary
for plant growth, including fixing atmospheric nitrogen, nutrient cycling, disease
suppression, and sequester iron and other metals. Functional metagenomics strategies are
being used to explore the interactions between plants and microbes through cultivation-
independent study of these microbial communities.
By allowing insights into the role of previously uncultivated or rare community
members in nutrient cycling and the promotion of plant growth, metagenomic approaches
can contribute to improved disease detection in crops and livestock and the adaptation of
enhanced farming practices which improve crop health by harnessing the relationship
between microbes and plants.
7.Applications as Pharmacogenomics
Pharmacogenomics has applications in illnesses like cancer, cardiovascular disorders,
depression, bipolar disorder, attention deficit disorders, HIV, tuberculosis, asthma, and
diabetes.
In cancer treatment, pharmacogenomics tests are used to identify which patients are
most likely to respond to certain cancer drugs. In behavioral health, pharmacogenomic

46 | P a g e
tests provide tools for physicians and care givers to better manage medication selection
and side effect amelioration. Pharmacogenomics is also known as companion diagnostics,
meaning tests being bundled with drugs. Examples include KRAS test with cetuximab
and EGFR test with gefitinib. Beside efficacy, germline pharmacogenetics can help to
identify patients likely to undergo severe toxicities when given cytotoxics showing
impaired detoxification in relation with genetic polymorphism, such as canonical 5-FU.
In cardio vascular disorders, the main concern is response to drugs including warfarin,
clopidogrel, beta blockers, and statins.
Many people take medications called SSRIs, or selective serotonin reuptake inhibitors,
for different psychiatric disorders. Many of the medications are metabolized by CYP450
enzymes, including fluoxetine, paroxetine, and citalopram.
8.Applications of Genomics in Melanoma
Oncogene discovery
The identification of recurrent alterations in the melanoma genome has provided key
insights into the biology of melanoma genesis and progression. These discoveries have
come about as a result of the systematic deployment and integration of diverse genomic
technologies, including DNA sequencing, chromosomal copy number analysis, and gene
expression profiling.
9.Applications of Genomics in Agriculture
Animal and plant genomics and genetics play a significant role in vaccine &
therapeutics development, breeding and selection for meat quality, milk production and
pest resistance. Exactly the same principles and methods for identifying SNPs and
biomarkers in human data can be applied to livestock (sheep, pig, cow and poultry) and
plant data. The benefits for the agricultural industry are enormous.

47 | P a g e
10. Genomics Applications to Biotech
Traits
Twenty years since the inception of the agricultural biotechnology era, only two
products have had a significant impact in the market place: herbicide-resistant and insect-
resistant crops. Additional products have been pursued but little success has been
achieved, principally because of limited understanding of key genetic intervention points.
Genomics tools have fueled a new strategy for identifying candidate genes. Primarily
thanks to the application of functional genomics in Arabidopsis and other plants, the
industry is now overwhelmed with candidate genes for transgenic intervention points.
This success necessitates the application of genomics to the rapid validation of gene
function and mode of action. As one example, the development of C-box binding factors

48 | P a g e
(CBFs) for enhanced freezing and drought tolerance has been rapidly advanced because
of the improved understanding generated by genomics technologies.
11. Applications of Genomics in the Inner
Ear
Understanding the development and function of the inner ear requires knowledge of
the genes expressed and the pathways involved. Such knowledge is also essential for the
development of therapeutic approaches for a wide range of inner ear diseases affecting
millions of people. The completion of the Human Genome Project and emergence of
genomics-based technologies have made it possible to analyze the expression patterns of
the inner ear genes at the whole genome level, generating an unprecedented amount of
information on gene expression patterns.
12. Applications of Genomic Sequencing
Genome sequence data now provide tools for the development of practical uses for
genetic information. DNA is an invaluable tool in forensics because - aside from identical
twins - every individual has a uniquely different DNA sequence. Repeated DNA
sequences in the human genome are sufficiently variable among individuals that they can
be used in human identity testing. The FBI uses a set of thirteen short tandem repeat
(STR) DNA sequences for the Combined DNA Index System (CODIS) database, which
contains the DNA fingerprint or profile of convicted criminals. Investigators of a crime
scene can use this information in an attempt to match the DNA profile of an unknown
sample to a convicted criminal. DNA fingerprinting can also identify victims of crime or
catastrophes, as well as many family relationships, such as paternity. While we think of
forensics in terms of identifying people, it can also be used to match donors and
recipients for organ transplants, identify species, establish pedigree, or even detect
organisms in water or food.

49 | P a g e
An unusual application of DNA fingerprinting technology is a project of Mary-Claire
King's at the University of Washington. Although her research is primarily concerned
with the identification of genetic markers for breast cancer, she also has a project to help
the "Abuelas," or grandmothers, in Argentina. In Buenos Aires in the 1970s and 1980s,
children of activists "disappeared" during the military dictatorship. The children were
placed in orphanages or illegally adopted when their parents were killed. Now King is
using mitochondrial DNA, which is inherited only maternally, to reunite the children with
their grandmothers.
The basis of many diseases is the alteration of one or more genes. Testing for such
diseases requires the examination of DNA from an individual for some change that is
known to be associated with the disease. Sometimes the change is easy to detect, such as
a large addition or deletion of DNA, or even a whole chromosome. Many changes are
very small, such as those caused by SNPs. Other changes can affect the regulation of a
gene and result in too much or too little of the gene product. In most cases if a person
inherits only one mutant copy of a gene from a parent, then the normal copy is dominant
and the person does not have the disease; however, that person is a carrier and can pass
the disease on to offspring. If two carriers produce a child and each passes the mutant
allele to the child (a one-in-four probability), that individual will have the disease.
Several different mutations in a gene often lead to a particular disease. Many diseases
result from complex interactions of multiple gene mutations, with the added effect of
environmental factors. Heart disease, type-2 diabetes and asthma are examples of such
diseases. (See the Human Evolution unit.) Many diseases do not show simple patterns of
inheritance. For example, the BRCA1 mutation is a dominant mutant allele that leads to
an increased risk for breast and ovarian cancer. (See the Cell Biology and Cancer unit.)
Although not everyone with the mutation develops the disease, the risk is much higher
than for individuals without the mutation.

50 | P a g e
Newborns commonly receive genetic testing. The tests detect genetic defects that can
be treated to prevent death or disease in the future. Apparently normal adults may also be
tested to determine whether they are carriers of alleles for cystic fibrosis, Tay-Sachs
disease (a fatal disease resulting from the improper metabolism of fat), or sickle cell
anemia. This can help them determine their risk of transmitting the disease to children.
These tests as well as others (such as for Down's syndrome) are also available for
prenatal diagnosis of diseases. As new genes are discovered that are associated with
disease, they can be used for the early detection or diagnosis of diseases such as familial
adenomatous polyposis (associated with colon cancer) or p53 tumor-suppressor gene
(associated with aggressive cancers). The ultimate value of gene testing will come with
the ability to predict more diseases, especially if such knowledge can lead to the disease's
prevention.
Gene therapy is a more ambitious endeavor: its goal is to treat or cure a disease by
providing a normal copy of the individual's mutated gene. (See the Genetically Modified
Organisms unit.) The first step in gene therapy is the introduction of the new gene into
the cells of the individual. This must be done using a vector (a gene carrier molecule),
which can be engineered in a test tube to contain the gene of interest. Viruses are the
most common vectors because they are naturally able to invade the human host cells.
These viral vectors are modified so that they can no longer cause a viral disease.
Gene therapy using viral vectors does have a few drawbacks. Patients often experience
negative side effects and expression of the desired gene introduced by viral vectors is not
always sufficiently effective. To counter these limitations, researchers are developing
new methods for the introduction of genes. One novel idea is the development of a new
artificial human chromosome that could carry large amounts of new genetic information.
This artificial chromosome would eliminate the need for recombination of the introduced
genes into an existing chromosome. Gene therapy is the long-term goal for the treatment
of genetic diseases for which there is currently no treatment or cure.

51 | P a g e
The Human Genome Project
The Human Genome Project, which was led at the National Institutes of Health (NIH)
by the National Human Genome Research Institute, produced a very high-quality version
of the human genome sequence that is freely available in public databases. That
international project was successfully completed in April 2003, under budget and more
than two years ahead of schedule.
The sequence is not that of one person, but is a composite derived from several
individuals. Therefore, it is a "representative" or generic sequence. To ensure anonymity
of the DNA donors, more blood samples (nearly 100) were collected from volunteers
than were used, and no names were attached to the samples that were analyzed. Thus, not
even the donors knew whether their samples were actually used.
The Human Genome Project was designed to generate a resource that could be used for
a broad range of biomedical studies. One such use is to look for the genetic variations
that increase risk of specific diseases, such as cancer, or to look for the type of genetic
mutations frequently seen in cancerous cells. More research can then be done to fully
understand how the genome functions and to discover the genetic basis for health and
disease.
The International HapMap Project, in which NIH also played a leading role, represents
a major step in that direction. In October 2005, the project published a comprehensive
map of human genetic variation that is already speeding the search for genes involved in
common, complex diseases, such as heart disease, diabetes, blindness, and cancer.
Another initiative that builds upon the tools and technologies created by the Human
Genome Project is The Cancer Genome Atlas pilot project. This three-year pilot, which

52 | P a g e
was launched in December 2005, will develop and test strategies for a comprehensive
exploration of the universe of genetic factors involved in cancer.
Sequencing and Bioinformatic
Analysis of Genomes
Genomic sequences are usually determined using automatic sequencing machines. In a
typical experiment to determine a genomic sequence, genomic DNA first is extracted
from a sample of cells of an organism and then is broken into many random fragments.
These fragments are cloned in a DNA vector (carrier) that is capable of carrying large
DNA inserts. Because the total amount of DNA that is required for sequencing and
additional experimental analysis is several times the total amount of DNA in an
organism’s genome, each of the cloned fragments is amplified individually by replication
inside a living bacterial cell, which reproduces rapidly and in great quantity to generate
many bacterial clones.
The cloned DNA is then extracted from the bacterial clones and is fed into the
sequencing machine. The resulting sequence data are stored in a computer. When a large
enough number of sequences from many different clones is obtained, the computer ties
them together using sequence overlaps. The result is the genomic sequence, which is then
deposited in a publicly accessible database.
A complete genomic sequence in itself is of limited use; the data must be processed to
find the genes and, if possible, their associated regulatory sequences. The need for these
detailed analyses has given rise to the field of bioinformatics, in which computer
programs scan DNA sequences looking for genes, using algorithms based on the known
features of genes, such as unique triplet sequences of nucleotides known as start and stop

53 | P a g e
codons that span a gene-sized segment of DNA or sequences of DNA that are known to
be important in regulating adjacent genes. Once candidate genes are identified, they must
be annotated to ascribe potential functions. Such annotation is generally based on known
functions of similar gene sequences in other organisms, a type of analysis made possible
by evolutionary conservation of gene sequence and function across organisms as a result
of their common ancestry. However, after annotation there is still a subset of genes for
which functions cannot be deduced; these functions gradually become revealed with
further research.
Fig: In genomics research, fragments of genomic DNA are inserted into a vector and amplified by replication in bacterial cells. In this way, large amounts of DNA can be cloned and extracted from the bacterial cells. The DNA is then sequenced and further analyzed using bioinformatics
techniques.