gene regulation during development by chromatin and the ...715144/fulltext01.pdfgene regulation...
TRANSCRIPT

1
Gene regulation during development by chromatin and the Super Elongation
Complex
Olle Dahlberg
Department of Molecular Biosciences
The Wenner-Gren Institute Stockholm University 2014

2
Doctoral thesis 2014 Department of Molecular Biosciences The Wenner-Gren Institute Stockholm University Stockholm, Sweden
©Olle Dahlberg, Stockholm 2014 ISBN 978-91-7447-932-4 Printed in Sweden by Universitetsservice US-AB, Stockholm 2014 Distributor: Stockholm University

3
”Nothing in biology makes sense except in the light of evolution”
This book is dedicated to my wife Jessie and our F1 gen-eration

4

5
Abstract Developmental processes are carefully controlled at the level of tran-scription to ensure that the fertilized egg develops into an adult organ-ism. The mechanisms that controls transcription of protein-coding genes ultimately ensure that the Pol II machine synthesizes mRNA from the correct set of genes in every cell type. Transcriptional control involves Pol II recruitment as well as transcriptional elongation. Re-cent genome-wide studies shows that recruitment of Pol II is often followed by an intermediate step where Pol II is halted in a promoter-proximal paused configuration. The release of Pol II from promoter-proximal pausing is thus an additional and commonly occurring mechanism in metazoan gene regulation. The serine kinase P-TEFb is part of the Super Elongation Complex that regulates the release of paused Pol II into productive elongation. However, little is known about the role of P-TEFb mediated gene expression in development. We have investigated the function of P-TEFb in early Drosophila em-bryogenesis and find that P-TEFb and other Super Elongation Com-plex subunits are critical for activation of the most early expressed genes. We demonstrate an unexpected function for Super Elongation Complex in activation of genes with non-paused Pol II. Furthermore, the Super Elongation Complex shares phenotypes with subunits of the Mediator complex to control the activation of essential developmental genes. This raises the possibility that the Super Elongation Complex has an unappreciated role in the recruitment of Pol II to promoters. The unique chromatin landscape of each cell type is comprised of post-translational chromatin modifications such as histone methyla-tions and acetylations. To study the function of histone modifications during development, we depleted the histone demethylase KDM4A in Drosophila to evaluate the role of KDM4A and histone H3 lysine 36 trimethylation (H3K36me3) in gene regulation. We find that KDM4A has a male-specific function and regulates gene expression both by catalytic-dependent and independent mechanisms. Furthermore, we used histone replacement to investigate the direct role of H3K14 acet-ylation in a multicellular organism. We show that H3K14 acetylation is essential for development, but is not cell lethal, suggesting that H3K14 acetylation has a critical role in developmental gene regula-tion. This work expands our knowledge of the mechanisms that pre-cisely controls gene regulation and transcription, and in addition high-lights the complexity of metazoan development.

6

7
List of Publication I Olle Dahlberg, Olga Shilkova and Mattias Mannervik
P-TEFb, the Super Elongation Complex and Mediator regulate non-paused, rapidly transcribed genes during early Drosophila embryo development (Submitted)
II Filip Crona, Olle Dahlberg, Lina E Lundberg, Jan Larsson
and Mattias Mannervik (2013) Gene regulation by the lysine demethylase KDM4A in Drosophila. Dev.Biol.373(2):453-463.
III Olle Dahlberg, Roshan Vaid and Mattias Mannervik
Histone 3 lysine 14 is essential in Drosophila. (Manuscript)
Article 2 is reprinted with permission from publisher Elsevier

8
Contents
Introduction 10 Transcription 12 RNA Polymerase II 14 Pol II pausing 17 DNA pause motifs 18 Factors involved in pausing 19 P-TEFb 20 Super Elongation Complex 21 The Mediator 22 Cis-regulatory modules 24 Chromatin and epigenetics 27 Post translational modifications of histones 29 Lysine methyltransferases 29 Lysine demetylases 30 Histone acetyltransferases 33 Histone deacetylases 34 Drosophila early development 35 Cellularization 37 Future perspectives 40 Paper I 42 Paper II 45 Paper III 46 Acknowledgements 48 References 50

9
Abbreviations
Pol II RNA Polymerase II CTD Pol II C-terminal domain MBT Mid-blastula transition (MBT=MZT) MZT Maternal to zygotic transition Ser2-p Phosphorylation of CTD Serine 2 SEC the Super Elongation Complex CRM Cis-regulatory module (enhancer) H3K36me3 Histone 3 Lysine 36 trimethylation H3K14ac Histone 3 Lysine 14 acetylation KMT Lysine transferase KDM Lysine demethylase HAT Histone acetyltransferace HDAC Histone deacetylace Bp Base pairs

10
Introduction
“They are in you and in me; they created us, body and mind; and their preservation is the ultimate rationale for our existence. They have come a long way, those replicators. Now they go by the name of genes, and we are their survival machines.” -Richard Dawkins, The selfish gene. The vast numbers of species that exists today or ever lived show an endless variety of beautiful forms and shapes. Marvelous species pop-ulate every corner of our planet and have done so for billions of years. Even more amazing is that when looking at any of the species on our planet, we find that they all are related with each other. All organisms share a common ancestor. All living organisms use DNA to pass down genetic information from one generation to the next. Considering that all species share a common ancestor, our own existence is explained by an unbroken chain of generations transferring their genetic infor-mation from the first organism that ever lived, until today. Our bodies eventually become old and die, but our genes are immortal and will live on. They are passed down to our children, and continue to exist. As the genes are passed on, the transmission through generations leads to mutations and changes in the genetic code. This is a prerequisite for adaptation through evolution. Evolution works many times by chang-ing old genes to perform new functions. By altering how genes are expressed, changes in development takes place 1. One of the most astounding facts about biology is that the same genes building an in-sect body are also involved in making up the human body. The con-servation of gene function throughout the evolutionary tree is intri-guing and gives us the opportunity to learn about general biological mechanisms by studying any living organism. Developmental biology is the study of an organism going from a sin-gle fertilized egg up to the point of adulthood. The mechanisms of the development of multicellular organisms have been one of the biggest mysteries in biology, and are still a great challenge for the develop-mental biologist. It has been known for many years that genes deter-mine when and where different cell types are to be formed during em-

11
bryogenesis. Genes also determine how many cells each cell type will have, where limbs will grow out and how to shape the morphology of our bodies. Animal development is among the most fascinating events ever to take place in nature. During embryonic development, a single fertilized egg cell goes through multiple divisions to form an adult organism containing different cell types and organs, ready to transfer its genes to new generations. Some of the fundamental questions in developmental biology are how cells know what kind of cell they will form and what the fundamental differences are between different cell types. What factors determines what part of the embryo that will be-come left and right, anterior/posterior and dorsal/ventral? These questions have puzzled people, and the field of developmental biology has emerged to address them. Development is driven by a precise regulation of genes involved in forming the function, shape and size of cells and tissue. The study of gene regulation is of great importance to understand development.

12
Transcription Transcription is the mechanism where the genetic information existing in the DNA is transferred into RNA. The regulation of transcription drives development in all organisms. A fundamental knowledge about how transcription is regulated is essential when deciphering the mech-anisms of development. Messenger RNA (mRNA) can further be used to synthesize protein in the process of translation. Much of the non-coding part of the genome is transcribed to generate RNA that can function as structural units such as rRNA and tRNA. In addition, non-coding RNAs such as long non-coding RNA have in more recent years caught attention. The eukaryotic genome is transcribed by the three RNA polymerase machines Pol I, Pol II and Pol III. RNA poly-merase II (Pol II) transcribes protein-coding genes. To transcribe a certain DNA sequence, Pol II needs access to the promoter sequence upstream of the DNA to be transcribed. A major obstacle for getting access to promoters is nucleosomes that occupy the promoter. Nucleo-somes are an essential part of the eukaryotic chromatin and are im-portant transcriptional regulatory components. Depletion of nucleo-somes leads to both upregulation and downregulation of transcription 2. Prior to Pol II promoter access, nucleosomes positioned around the promoter needs to be removed to make room for the transcription ma-chinery. If any cis-regulatory modules are to be used in the gene acti-vation, they too need to be cleared from nucleosomes. DNA-binding transcription factors bind nucleosome occupied region and recruits nucleosome-remodelling factors to move the nucleosomes. This will create an open and nucleosome-free region. This provides an oppor-tunity for the transcription machinery to access the DNA (FIG 1). A very basic transcriptional model includes a core promoter plus the transcribed DNA sequence. The core promoter contains DNA ele-ments that are recognized and bound by general transcription factors as well as gene-specific transcription factors 3. When access to the core promoter is granted, the Pol II machine binds in concert with a large set of general transcription factors to form a preinitiation com-plex. Transcription can be studied out in vitro, and test tube transcrip-tion has been used extensively as a model for transcription. DNA tem-plates bearing a TATA-box DNA motif require a set of general tran-scription factors for Pol II to localize and bind the promoter in vitro 4. TATA-binding protein (TBP) is the first factor to bind the promoter. TBP is a subunit of the mega-dalton TFIID complex where TBP itself binds the TATA-box DNA motif. TFIIB and TFIIA are recruited to stabilize the interaction between TBP and the TATA-box. The TATA-box is found ~30 bp upstream the transcription start site.

13
TFIIB binds TFIIB-recognition elements (BRE) upstream and down-stream of the TATA sequence, followed by binding of Pol II together with TFIIF. The last components added is TFIIE and the TFIIH hel-icase/kinase complex 3 5.
FIGURE 1. A core promoter is cleared from nucleosomes to allow for tran-scription. An arrow marks the transcription start site. A) A pioneering DNA-binding transcription factor (PF) is recruited to the promoter. B) The PF recruits nucleosome remodeling factors that can alter the nucleosome occupancy to create an open chromatin structure at the core promoter. C) The nucleosome-free region al-lows the general transcription machinery to bind DNA at the core promoter.

14
RNA Polymerase II Pol II is a multi-subunit complex where the largest subunit Rpb1 con-tains a C-terminal domain (CTD). The CTD protrudes from the Pol II protein complex and has multiple heptad repeats. The heptad repeats sequence contains variations of the consensus sequence Y1S2P3T4S5P6S7. The length of the CTD varies in different organisms. Budding yeast contain 26-29 heptad repeats, Drosophila have 45 and humans 52 6. The serines as well as tyrosines in the CTD are subjected to phosphorylations. However, potentially all amino acids in the CTD could be subjected to some kind of modification such as glycosyla-tion, phosphorylation or ubiquitination independently of each other. This could in principle give an almost infinite number of combinations of modifications at one single CTD. The Pol II CTD is not essential for transcription in vitro. However, cells with 10-12 complete heptad repeats are conditionally viable whereas less than ten repeats is cell lethal 7. Serine phosphorylations are among the most studied post-translational CTD modifications. A hyperphosphorylated CTD is cor-related with a transcriptional engaged Pol II (chromatin associated Pol II). Phosphorylation of Pol II CTD Ser5 (Ser5-p) and Ser7 (Ser7-p) takes place at the promoter while Ser2 phosphorylation (Ser2-p) is accumulated onto the CTD during elongation and reaches high levels at the 3´ end of the gene (FIG 2). During transcriptional initiation, Cdk7 of the TFIIH complex carries out Ser5-p. Ser5-p is considered to be the key event in “promoter clearance” when Pol II is released from the pre-initiation complex at the core promoter. In addition, Ser5-p is found on transcriptionally initiated Pol II that have “paused” 20-80 base pairs downstream the transcription start site 8. Ser2-p is best known for the role in release of Pol II from pause into transcriptional elongation. Ser2-p is carried out by serine kinases such as the Cdk9/Cyclin T complex P-TEFb or Cdk12/Cyclin K. A simplified commonly accepted model for Pol II transcription can be outlined as follows: (1) recruitment of unphosphorylated Pol II to the promoter followed by (2) Pol II recieves Ser5-p for its promoter clear-ance and (3) additionally receives Ser2-p mediated by the P-TEFb kinase for productive elongation 9 (FIG 3). Considering the current view on how phosphorylations controls the release of Pol II from the preinitiation complex and from pause, changing the levels of CTD phosphorylation would have a global im-pact on transcription. However, Pol II phosphorylation is very com-plex and most likely cannot be explained by simple models. Ser7-p mutations have been demonstrated to give an effect on short-noncoding RNA genes, but seem to have a less important role in the regulation of protein-coding genes 9. Yeast mutants impaired in Ser2-p

15
are viable and show little changes in global gene expression, having defects in cytokinesis due to changes in a small set of genes with roles in meiosis 10. Furthermore, the more distally located CTD repeats of higher eukaryotes are subjected to lysine 7 acetylation by the acetyl-transferase P300/KAT3B. The creation of Pol II CTD Lysine mutants showed that P300 mediated acetylation of the CTD is gene-specific and important to activate a specific set of genes during growth-factor response 11. Multiple steps in mRNA biogenesis occur at the same time and place, and this “cotranscriptionality” is key for the crosstalk between com-ponents involved in processes such as elongation and splicing 12. It has been demonstrated that Pol II CTD is important for 3´ end processing both in cells and in test tubes. Set2 is a Histone methyltransferase that have been implicated in elongation and splicing. During Pol II elonga-tion, Set2 binds the CTD via Ser2-p and Ser5-p and distributes H3K36me3 in the gene body. H3K36me3 reaches its maximal level in the 3´-end of the transcribed gene. Depletion of Set2 results in tran-scriptional defects 13. Taken together, phosphorylation of the CTD might not be as critical for global transcription as one might think. Instead, the function of different amino acid residues of Pol II CTD could have evolved into controlling different sets of genes depending on the CTD post-translational state. One could speculate that the CTD modifications could act as a “code” where different combinations of modifications carefully and precisely control the different steps in mRNA synthesis and processing.
FIGURE 2. The Pol II CTD protrudes from the largest Pol II subunit and contains multiple heptad repeats where serine 2, 5 and 7 are subjected to phosphorylations that alter the function of the polymerase.

16
FIGURE 3. A simple model of Pol II-mediated transcription. A) Pol II is recruit-ed to the core promoter together with general transcription factors. This forms the preinitiation complex. B) Pol II promoter clearance is mediated by the TFIIH com-plex that generates Ser5-p. At this stage, Pol II start transcription but can pause downstream the transcription start site (indicated by black arrow). C) Release of Pol II into transcriptional elongation by P-TEFb-mediated phosphorylation of Pol II CTD as well as negative elongation factors.

17
Pol II pausing Recruitment of Pol II to the gene is a major rate-limiting step that con-trols the transcriptional activity. Once Pol II is recruited to the pro-moter along with the general transcription factors (FIG 3A), transcrip-tional elongation can be divided into two separate stages. In the first stage (early elongating), Pol II is released from the core promoter as-sociated pre-initiation complex and initiates RNA synthesis (FIG 3 B). In the next stage, Pol II progresses throughout the gene body towards the 3´ end of the gene (productive elongation) (FIG 3 C). An interme-diate step has been described as a major role in metazoan transcrip-tion. This involves a promoter proximal pausing of Pol II immediately after the step of early elongation 14. Previous yeast studies looking at rate-limiting factors during Pol II mediated transcription showed that recruitment of the polymerase to the gene is a major rate-limiting step during transcription 15. However, other early data such as studies of the heat-shock gene hsp70 in Drosophila suggested an additional, dif-ferent mechanism where Pol II is recruited to the gene but unable to go into productive elongation before heat shock induction 16 17. In em-bryos, the gene Sloppy-paired 1 was one of the first developmental genes demonstrated to have paused Pol II 18. Thus, the polymerase is positioned downstream the transcription start site in a promoter prox-imal paused state, and is released into transcriptional elongation only when the gene is activated. The pausing behavior of Pol II was for many years seen as a less common mechanism for gene activation in metazoan organisms whereas RNA Polymerase pausing in bacteria has been generally accepted 14. Pol II can be recruited to an inactive gene and stay tethered to the promoter without being released into elongation. The half-life of gene-associated Pol II has been estimated stably pause for approximately 5-15 min at the inactive gene 19 20. However, a majority of all genes in early Drosophila embryos have paused Pol II and are being active at the same time. This means that Pol II is recruited, paused and released continuously as long as the gene is active. With new genome-wide methods introduced to the sci-entific community, it has become clear that Pol II pausing is a com-mon feature and that pausing play a major role in transcription of the eukaryotic genome. Recent genome-wide studies show that Pol II pausing occurs 20-80 bp downstream of the transcription start site at thousands of genes 21 8. Estimations have been made that at least 70% of all active genes in the Drosophila S2 cell line contain paused Pol II 22. What makes Pol II pause? Different molecular models of pausing

18
have been proposed. One model is involving a kinetic model where Pol II elongation is competing with pausing factors inhibiting the ma-chine from elongation. Another model depends on the position of the +1 nucleosome acting as a physical barrier that stops Pol II from elon-gating any further. A third model involves pausing factors that bind Pol II and tether it to DNA, and thus fixating the polymerase in a paused state 14. Taken together, Pol II pausing is accepted as a major regulatory step in gene regulation such as in the transcription of de-velopmental genes. However, the biological and molecular functions of the factors involved in pausing remain unclear, and the regulation of Pol II is an extremely complex event. There is most likely no uni-form mechanism that can fully explain how pausing occurs at different paused genes occupied with different factors and having different core promoter sequences. We can make a model where Pol II regulation is divided into two different types. The first is a regulation based solely on the rate of Pol II recruitment to the gene that is immediately fol-lowed by transcriptional elongation. The second type of regulation is controlled primary at the level of Pol II recruitment to the gene plus an additional regulation at the level of Pol II release from promoter-proximal pause. Hence, genes can have paused Pol II even though polymerase recruitment is the rate-limiting step 23. Pause DNA motifs Pol II pausing strength has been linked to the DNA sequence situated around the TSS and some DNA motifs are enriched at genes showing more pausing. One DNA-motif that is enriched at active paused Pol II genes is the GAGA motif. GAGA-motifs are generally located up-stream the core promoter at -80 to -75 bp. The GAGA-motif can re-cruit the GAGA-factor (GAF or Trl). In Drosophila early embryos, GAGA-containing genes show a high degree of pausing 24. However, over 2000 Pol II bound promoters that lack GAF instead have the core promoter element Motif 1 25. As with many GAGA-containing genes, Motif 1 genes are paused and lack a TATA consensus motif in the core promoter. Thus Motif 1, GAGA and TATA tend to be mutually exclusive. The M1BP protein binds Motif 1 and associates with a spe-cific set of genes in Drosophila. GAF genes and M1BP bound have different nucleosome patterns. The strong +1 nucleosome signal in M1BP-bound genes suggests that Pol II pausing is achieved with the assistance of a nucleosome barrier. The low nucleosome signal at GAF-bound genes suggests that Pol II utilizes a different pausing

19
mechanism at those genes 26. So far, no mammalian counterpart of GAF or M1BP has been described. Factors involved in pausing The DRB-sensitivity inducing factor (DSIF) and negative elongation factor (NELF) were originally biochemically purified and identified as factors that are responsible for the effect of DRB addition. Addition of the nucleoside analog DRB to cell cultures results in an inhibition of Pol II elongation. However, DRB does not to inhibit Pol II itself, and in vitro assays using purified Pol II and general transcription factors does not respond to DRB. DSIF is composed of the subunits Spt4 and Spt5. NELF contain the four subunits A, B or C, D and E where NELF E contains an RNA recognition motif 14. DSIF and NELF are identified as factors involved in pausing and the regulation of transcription. NELF and DSIF can interact with each other 27 and stabilize Pol II pausing 14. DSIF and NELF have been suggested to bind Pol II and also short nascent RNA as it emerges from the polymerase 28. NELF and DSIF act as transcriptional inhibi-tors in vitro, 27 and are responsible for Pol II pausing on the hsp70 gene in vivo 29. Pol II-associated DSIF and NELF are subjected to phosphorylations that mediate the release of Pol II into elongation. Spt5 of DSIF contains a Pol II CTD-like repetitive sequence in its C-terminal region (CTR), and release of paused Pol II into elongation is mediated by phosphorylation of the CTR domain by the P-TEFb com-plex. Furthermore, Ser2-p of Pol II dissociates NELF from Pol II. However, NELF does not seem to bind directly to the Pol II CTD since CTD-less Pol II is still being sensitive to DSIF and NELF in-duced pausing 27 21. In vivo data show that both DSIF and NELF are critical for correct development. NELF associates with Pol II at active genes and depletion of NELF by RNAi in Drosophila S2 cells results in a decreased Pol II occupany at many promoters 20,30. However, sev-eral studies indicate a gene-specific function rather than DSIF and NELF would be equally important for all genes. Maternal depletion of NELF in Drosophila embryos results in embryos with multiple lethal phenotypes including abnormal nuclear morphology. Interestingly, several different segmentation genes are expressed at normal levels and NELF depletion only gives specific effects on transcription of slp1 enhancer-promoter transgenes 31. In a similar fashion, the W049 allele of DSIF subunit Spt5 results in gene-specific effects on pair-rule genes in Drosophila embryos with derepression of some, but not all eve and runt stripes 32. In addition, the DSIF allele foggy isolated from zebra fish involves a point mutation in the c-terminus of Spt5, produc-

20
ing an allele very similar to the DrosophilaW049 allele. The foggy-allele also give specific developmental defects such as a reduction of dopamine-containing neurons as well as an increase in serotonin-containing neurons in the hypothalamus 33. The gene specific pheno-types of the DSIF and NELF alleles suggest that some genes depends more on NELF and DSIF than others. P-TEFb The core P-TEFb complex consists of two components, the kinase Cdk9 and Cyclin T. P-TEFb can be found as a part of the Super Elon-gation Complex (SEC) or stored inactive in a complex consisting of a 7SK RNA and HEXIM 34. In addition, recent mass-spectrometric ap-proaches have revealed that P-TEFb can be found in several other pro-tein complexes 35. P-TEFb is considered to be the main factor that ultimately releases Pol II from pausing. This is mediated via phosphorylation of Pol II CTD Ser2 (FIG 3 C) 36 37 38 39 as well as phosphorylation of NELF and DSIF 40. The P-TEFb kinase activity can be blocked by the drug fla-vopiridol, and treatment of cells with the inhibitor block most Pol II mediated transcription 36 and result in an increase of paused Pol II unable to release into productive elongation 20. In contrast, the Pol II CTD Ser2 and the kinase activity of P-TEFb is only essential for cor-rect processing but not for the transcriptional elongation of small nu-clear RNAs (snRNA) 41. These studies indicates two different roles for P-TEFb: One as having the role of being the main Pol II pause to re-lease switch, and in addition having a different role in cotranscription-al processes. Although P-TEFb has been considered to work as a Pol II pause-to-release factor, in vitro assays using phosphorylated peptides show specificity towards CTD Ser5-p. This is not considered to correlate with Pol II pause-to-release but rather with Pol II in transcriptional initiation 42. In mouse ES cells, knockdown of either of two existing Cyclin T proteins give distinctly different effects on gene regulation with different groups of genes being affected 43. This suggests that P-TEFb might have specific target genes rather than function as a gen-eral transcription factor for all genes. In Drosophila, depletion of P-TEFb subunit Cdk9 leads to lower levels of chromatin associated Ser2-phosphorylated Pol II. When RNAi was used to knock down Cdk9, CTD Ser2-p levels were reduced in an expected manner 44. In-terestingly, the level of Ser5-p was reduced to the same extent. One might speculate that the reduced Ser5-p levels could be an indirect

21
effect from a failure in TFIIH kinase activity upon Cdk9 depletion. Another explanation could be a more direct effect where P-TEFb have an additional role to mediate the phosphorylation of Ser5 in correla-tion with the in vitro findings in 42. In addition, stainings of polytene chromosomes show that ELL of the Super Elongation Complex (dis-cussed in next chapter) does no longer bind to chromatin in absence of Cdk9. This indicates that Cdk9 plays a role in the recruitment or sta-bility of the Super Elongation Complex. Depletion of Cdk9 did not have an effect of DSIF binding, suggesting that DSIF bind inde-pendently of P-TEFb 44. Co-stainings with antibodies against P-TEFb and hyperphosphorylated Pol II do not show a perfect correlation. This suggests that P-TEFb do not only work in transcriptional elonga-tion 38. In addition, the Cdk9 homolog Cdk12 have been shown to colocalize with hyperphosphorylated (elongating) Pol II, but only partly co-localize with P-TEFb. Interestingly, the Cdk12 distribution over an activated hsp70 gene is spread out throughout the gene body as if the protein is associated with Pol II during elongation rather than associate with the promoter 45. This is in contrast to the distribution of P-TEFb that mostly is associated with the transcription start site 46. Cdk12 and its partner Cyclin K are in contrast to P-TEFb both essen-tial for oogenesis. We have not been able to produce offspring mater-nally depleted from either Cdk12 or Cyclin K. This indicates that P-TEFb and the Cdk12 complex play separate roles during oogenesis. To study the maternal effects of Cdk12, other approaches have to be taken. Injections of early embryos with drugs or antibodies that targets Cdk12 could give further information about its gene-regulatory role during early embryogenesis. The Super Elongation Complex The super elongation complex (SEC) is a P-TEFb-containing complex that is involved in gene activation and can mediate rapid induction of transcription irrespectively of Pol II pausing 47. Besides P-TEFb, the mammalian Super elongation complex (SEC) consists of three types of Eleven-nineteen lys-rich leukaemia (ELL) proteins (ELL1, ELL2 and ELL3) that previously were characterized as Pol II elongation factors. Members of the AF4/FMR2 family (AFF1, AFF2, AFF3 and AFF4) 48 are essential for the stability of the complex. The gene MLL is often rearranged in acute childhood haematological diseases where MLL in-frame translocations are found in a majority of the cases. MLL is the homolog of the yeast Set1 and the Drosophila

22
protein Trithorax. Set1 was identified in yeast as a H3K4 methyltrans-ferase and is a subunit in the large COMPASS complex (complex of proteins associated with Set1). Among the most occurring MLL trans-locations in disease are fusions with nuclear-localized proteins such as components of the SEC 49. MLL normally regulates many Hox, Pax and Wnt target genes, and mis-activation of the MLL-associated genes is achieved by the recruitment of the SEC complex in association with P-TEFb. The Drosophila protein Lilliputian (Lilli) is the homolog of AFF4 50. Lilli controls the activation of early zygotic genes such as the cellular-ization gene Sry-a and the pair-rule gene fushi-tarazu (ftz) as well as hkb 50 and sxl 51. The activation of ftz is mediated in concert with the transcription factor Runt 51. However, Lilli does not control expres-sion of even-skipped or tailless 50. Lilli colocalizes with elongating hyperphosphorylated Pol II as seen on polytene chromosomes, and the human AFF4 is recruited to heat-shock loci upon induction together with other components of SEC 48. Our data indicates that different subunits of the SEC are critical for early development. The Mediator complex The Mediator complex is considered a general transcription factor due to its role in global regulation of transcription of protein coding-and non-protein coding genes. Mediator is believed to regulate the for-mation and structure of the preinitiation complex, and most factors in the preinitiation complex physically or functionally interact with parts of the Mediator. The Mediator is conserved in all eukaryotic organ-isms and much of the subunit composition has been defined based on mass spectrometry studies. In yeast, some Mediator subunits are re-quired for viability and for the transcription of all genes, while other subunits are non-essential and gene-selective. The structure of Media-tor can be separated into different domains with a head, middle and tail domain as well as a Cdk8-containing kinase domain. The Media-tor has a function of transferring signals from sequence-specific tran-scription factors to the preinitiation complex 52 (FIG 4). The Mediator exists in compositionally distinct variants, and the variable composi-tion is a key factor for enabling gene and tissue specificity. Each Me-diator subunit interacts with different transcription factors suggesting a way of Mediator to act gene-specific depending on context. For ex-ample, MED1 show protein-protein interactions with nuclear receptors and specifically regulates nuclear-receptor target genes 53. Emerging evidence suggests that Mediator directly interacts with Pol II and thus

23
can function as a “molecular bridge” between upstream cis-regulatory modules and the core promoter. Crystallization of the Mediator head module demonstrates that MED6, MED8 and MED17 directly interact with the Pol II CTD. Furthermore, the kinase activity of TFIIH is stimulated by Mediator 54 leading to CTD Ser5-p that in turn dimin-ishes the contact between Mediator and CTD 55. This indicates that recruitment of the Mediator complex can mediate the release of Pol II from the preinitiation complex. Is Mediator also involved in the re-cruitment of P-TEFb? Indeed, MED23 depletion decreases CTD Ser5-p as well as Ser2-p at the target gene Egr1 in ES cells, whereas Pol II occupancy stays unaffected. The decrease in Pol II phosphorylation is explained by MED23 having a role in the recruitment of P-TEFb to Egr1, likely due to the physical interaction with P-TEFb subunit Cdk9 56. In addition, other Mediator subunits can release Pol II from pausing via recruitment of the Super elongation complex. MED26 directly recruits TFIID and later the Super Elongation Complex 57. A similar mechanism is seen in hypoxia where the transcription factor HIF1A enhances Pol II pause-release at hypoxia-induced genes by the re-cruitment of a variant of the Mediator containing CDK8, MED1 and MED26. This is again followed by the recruitment of SEC that leads to gene activation 58. Taken together, the Mediator is involved in a majority of the different stages of transcription. It interacts with the Pol II at promoters through recruitment by gene-specific transcription factors. Next, the Mediator directs factors that release Pol II into early elongation. Finally, Media-tor recruits Pol II pause-release factors such as the SEC to drive Pol II into productive elongation. Although much is known about the role of Mediator, little work has focused on the function of Mediator in early development. We mater-nally depleted a majority of the Mediator subunits and showed that the head module MED20 and MED22 functionally interacts with the Su-per Elongation Complex during the activation of early zygotic genes. To further characterize Mediator in development, our small Mediator screen demonstrates the possibility to generate maternal depletion of a number of Mediator subunits. This could in the future be a tool to fur-ther characterize the role of the different Mediator subunits in metazo-an early development.

24
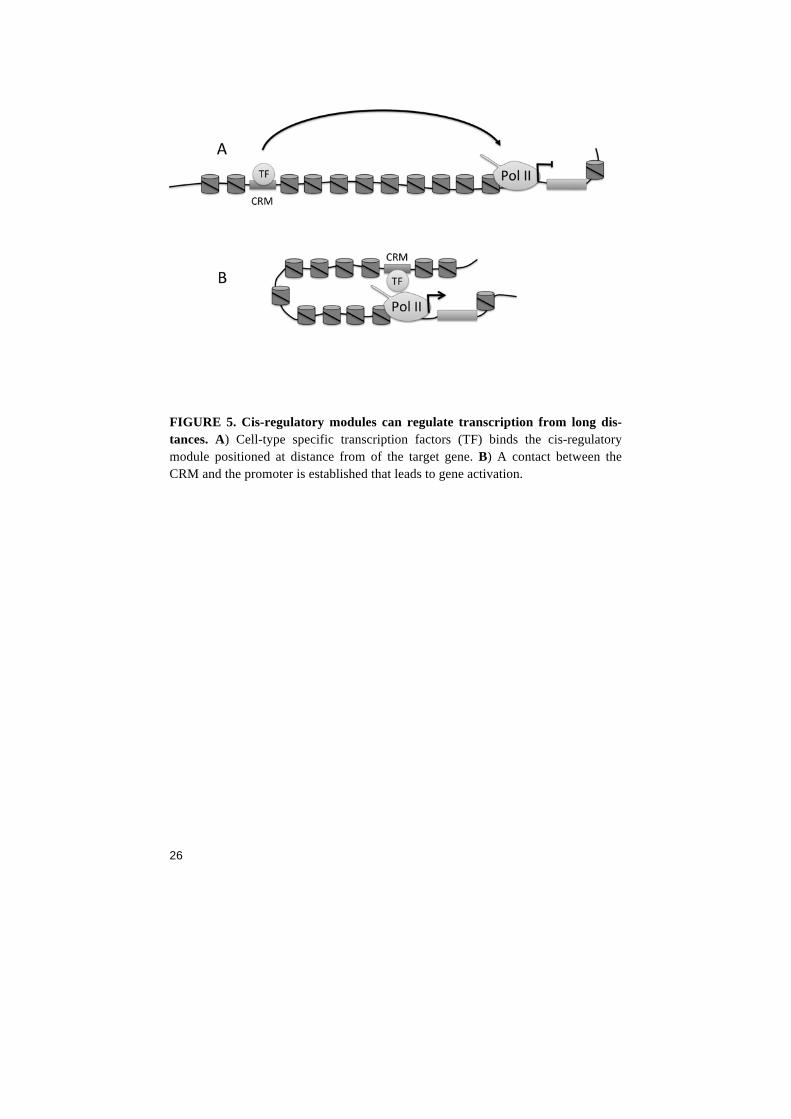
FIGURE 4. The multi-subunit Mediator complex can be divided into anatomical parts: tail, middle and head module and a kinase module. Additional DNA-binding transcription factors (TF) interact with the different subunits of Mediator to achieve gene-specificity. The head domain containing MED20 and MED22 directly interacts with the CTD of Pol II via MED6, MED8 and MED17. Cis-Regulatory Modules All multicellular organisms need to tightly regulate gene expression in a spatial and temporal manner. The metazoan genome consists of large intergenic sequences that do not contain protein-coding genes but have important information about gene regulation. During development, the organism has a need to transcribe its genes in specific cell types and in a spatial-temporal manner to form different cell types. The way a gene can be expressed in patterns is controlled by cis-regulatory modules (CRM). CRM´s are often called “enhanc-ers” due to their ability to positively affect transcription. However, CRM´s are not only upregulating gene expression but can as well work in the opposite direction. CRM´s consist of DNA sequences lo-cated upstream or downstream of the gene's transcription start site. CRM´s can be positioned close to the transcription start site, in introns or as long as several hundred kilobases from the regulated gene 59. Historically, transcriptional enhancers were originally discovered in viral genomes and metazoan enhancers were first demonstrated in mouse myeloma cells to have a function in the upregulation of beta-globin genes 60. The exact mechanism for a CRM´s ability to regulate transcription is not known, but CRM´s controls gene expression by activating or repressing actions in a context-dependent manner. Some studies suggests CRM´s to work by recruiting transcription factors and form a DNA “loop” that brings the CRM in close proximity to the

25
promoter. The gene looping is mediated by factors such as the Media-tor complex and the mechanism might explain why an activating pro-tein can function on a gene from a long distance 61 (FIG 5). Recently, genome wide data analyses have suggested that the binding of tran-scription factors can mark out regions that act as CRM´s. For exam-ple, ChIP-seq data show that the acetyltransferase P300 can predict tissue-specific active CRM´s during mouse development 62. In addi-tion, specific post-translational chromatin signatures mark out active and inactive CRM´s. H3K4me1 in combination with H3K27ac have been suggested as a main chromatin signature for active CRM´s in human embryonic stem cells whereas the combination H3K4me1/H3K27me3 mark out “poised enhancers” that will become active in later stages 63. Enhancers are bound and transcribed by Pol II that produces non-coding short RNA´s (eRNA). The function of eRNA transcription is still discussed, but might have a function in keeping an open chroma-tin at enhancers 64. In Drosophila, much effort has been spent trying to elucidate the func-tion of CRM´s. A common strategy to study CRM´s has been to create transgenic reporter constructs where the DNA sequence of the CRM is cloned in front of a core promoter containing a transcription start site. A DNA sequence whose transcription can be monitored (e.g. green fluorescent protein) is placed downstream the transcription start site. Recently, innovative large-scale assays have been designed to find all functional CRM´s in cell culture and in vivo 66,67. However, the choice of core promoter might impact the outcome of the assay. Core pro-moters differ from each other in terms of transcriptional strength and timing 65. Using a CRM plus its gene´s endogenous core promoter will most likely give the best result when studying spatial and temporal activity.
26
FIGURE 5. Cis-regulatory modules can regulate transcription from long dis-tances. A) Cell-type specific transcription factors (TF) binds the cis-regulatory module positioned at distance from of the target gene. B) A contact between the CRM and the promoter is established that leads to gene activation.

27
Chromatin and epigenetics “…It is therefore very desirable that a method should be available for obtaining many genetically identical individuals among the verte-brates. Such individuals have been produced in the case of the frog, Xenopus laevis, by making use of the technique of nuclear transplan-tation.” -J.B Gurdon, 1962 Chromatin and epigenetics are two tightly linked terms. The word epigenetics means above genetics and was originally used by Conrad Waddington (1905-1975) to describe a phenomenon that could not be explained simply by genetics 68. Today, epigenetics is defined as “the study of mitotically and/or meiotically heritable changes in gene func-tion that cannot be explained by changes in DNA sequence”. The def-inition of epigenetics has changed over the years, and will most likely continue to evolve with new discoveries. What is important to re-member is the fundamental question: How can a fertilized egg cell give rise to a whole organism 69? A mammal contains at least two hundred different cell types, which differ from each other in shape, size and function. Each cell type ex-presses defined sets of genes during its journey from a pluripotent state to fully differentiated. During this lineage commitment, new genes are constantly being activated at the same time as others are silenced. In the early Drosophila embryo, some genes such as the cel-lularization genes are only expressed during a short period in early development. This is followed by a complete inactivation of those genes that is maintained during the remaining life of the fly. Anything else could result in a complete disaster for the animal. Cell specific gene regulatory mechanisms are maintained due to an epigenetic memory. Epigenetic changes during linage commitment can be kept for the whole life of an organism. For example, brain cells can contin-ue to stay as brain cells for 100 years. However, John Gurdon showed more than 50 years ago that it is possible to reprogram a fully differ-entiated genome. By inserting a somatic nucleus into a Xenopus frog egg lacking its own nucleus, the genome of the somatic nuclei was completely overridden and reprogrammed by the egg cell proteins 70. The somatic chromatin was reprogrammed to recapitulate the expres-sion of a fertilized egg cell. In this way, the artificial zygote started dividing and developed into an adult frog. This early experiments showed that the epigenome is indeed plastic and will change under the right conditions. Every cell type is maintained by cell-specific epigenetic marks that cover its genome. Epigenetics regulate what genes that will be in a

28
silent mode, what genes that are active and what genes that will have the ability to be turned on in later stages. In addition, epigenetics is the reason why a daughter cell can express exactly the same set of genes as its mother. This is due to a transfer of epigenetic marks from one generation of cells to the next. The typical eukaryotic cell stores its DNA in the nuclei, and in contrast to prokaryotic organisms, all known eukaryotic organisms are storing their DNA within chromatin. Chromatin is the term used to describe DNA wrapped around nucleo-somes as well as other proteins in close proximity to DNA. Chromatin is used to pack, store and organize the cell´s genetic material. The core protein components of chromatin are DNA and histones. Histones are found in octameric complexes and make up the nucleosomes. Nucleo-somes are built up with two copies each of the histones H3, H4, H2A and H2B. A sequence of 147 base pairs is wrapped around each nu-cleosome. To access the information in DNA, chromatin needs to open up and nucleosomes have to move from the sequence to be used. Evolution has solved this problem by providing ATP-dependent nucleosome-remodeling enzymes that can slide or push nucleosomes and provide a piece of naked DNA 71. The position of nucleosomes is established by the combination of DNA sequences, nucleosome remodeling factors as well as transcription factors and the general transcription machin-ery. Nucleosomes are depleted from active promoters and cis-regulatory regions and correct nucleosome positioning is essential for transcription 72. The position of nucleosomes around promoters is or-ganized in a pattern of a -1 nucleosome upstream the transcription start site and +1 nucleosome downstream. The region at the transcrip-tion start site is nucleosome-free. The +1 nucleosome has been sug-gested to contribute to Pol II pausing when the transcription machine collides with the nucleosome 73. Core promoter sequence motifs corre-lated with Pol II pausing such as the GAGA motif (bound by GAF) and the Motif 1 (bound by M1BP) have distinctly different nucleo-some organization around the promoter. GAF-bound genes have al-most devoid of an organized nucleosome positioning, whereas M1BP-bound genes show highly organized nucleosomes. The GAF genes generally have more NELF-binding than M1BP-bound genes, and GAF genes show a higher degree of pausing 26.

29
Post-translational modifications of Histones Much focus has been put into studying the function of the histone tails that protrudes from the globular histone domain. The tails are subject-ed to a variety of post-translational modifications. Modifications of the histone tails are correlative with the epigenetic state of genes. Sev-eral types of modifications are found on histones and the combinatori-al nature of the modifications has been suggested to act as a “histone code” 74. The epigenetic state of each gene is controlled by “writer”, “reader” and “eraser” factors that are recruited to chromatin and modi-fies the histone tails. Histone modifications are well conserved from Yeast to mammals. Histone tail substitutions and histone mutants have been created in Yeast to elucidate the direct role of each modification. However, the difficulties creating metazoan histone mutants have giv-en rise to a “chicken or the egg” situation. A challenge now lies in demonstrating whether certain modifications are the outcome of e.g. gene activation, or if the modification itself is responsible for a certain transcriptional state. In the following chapters, I will focus on the modifications that are most relevant to my own work. This involves methylations and acetylations of lysines on the Histone 3 tails (FIG 6). Lysine methyltransferases Histone 3 lysines are subjected to methylations that have negative or positive effects on gene expression. Methylations are carried out by Lysine Methyl Transferases (KMT) that distributes mono-di and tri-methylation. The first KMT enzyme to be discovered that had the abil-ity to methylate histones was the KMT1A. This class of proteins con-tains a SET domain that function as the catalytic unit. The discovery of KMT1A made it possible to find other methyl transferases by ho-mology searches. A different class of enzymes only consists of the Dot1 protein 75. KMT´s have a high degree of substrate specificity as well as a precise enzymatic function such as KMT1A and B that cata-lyze the H3K9me1 to H3K9me3 reaction, while KMT1C prefers to catalyze H3K9me1 to H3K9me2. The Mixed lineage leukemia (MLL) proteins contain the SET1 do-main and belong to the KMT2 group that in humans have five mem-bers (MLL, MLL2, MLL3, MLL4, MLL5). The MLL proteins exists in COMPASS complexes and can function as gene activators mediat-ed via their function as H3K4 methyl transferases 76. MLL-mediated H3K4me3 is often found at expressed genes, making this an active mark. One function for H3K4me2/3 lies in the ability to directly re-cruit the basal transcription factor complex TFIID 77,78.

30
The recruitment of TFIID is mediated via binding of the TFIID subu-nit TAF3 to H3K4me2 or me3 79. Other histone modifications are correlated with the elongating Pol II, such as H3K36me3. During transcriptional elongation, the Pol II CTD is associated with the KMT enzyme Set2 80 81 82. Set2 deposits H3K36me1/me2/me3 on nucleosomes in the body of transcriptionally active genes 83. Considering that it specifically cover exons of tran-scribed genes, H3K36me3 has been suggested to regulate splicing 84. An additional mechanism have been proposed where Pol II CTD and H3K36me3 recruits the Histone deacetylase complex Rpd3S that de-pletes acetylation in coding sequences. Furthermore, incorporation of H3K36me3 inhibits insertion of H3K56ac in coding sequences. This maintenance of hypoacetylation is essential to inhibit spurious tran-scription from intergenic positions 85 86 87. Set2 is essential in development and RNAi-mediated Set2 depletion in Drosophila results in a global decrease in H3K36me3 levels and le-thality during pupal stage. In addition, Set2 genetically interacts with the nuclear receptor Ecdysone to regulate Ecdysone target genes 82. Lysine demethylases Histone demethylases are enzymes that catalyse removal of histone methylations. As with the KMT´s, there are two classes of Lysine de-methylases (KDM´s) where the first class to be discovered was the FAD-dependant enzyme KDM1A. The second class contains a JmjD-domain that uses alpha-ketoglutarate and iron for their catalytical function. The KDM4A family contains the JmjD domain. KDM4A catalyzes H3K9me2/3 and H3K36me2/3 demethylation. However, KDM4A are unable to remove H3K9me1/H3K36me1 75,88 89. In Dro-sophila, the KDM4A family is represented by the two proteins KDM4A and KDM4B. KDM4A mutant flies have elevated levels of H3K36me3 and a reduced lifespan 90, although the mutation is not lethal. This indicates that the precise level of H3K36me3 is not critical for viability. On the other hand, overexpression of KDM4A results in diminished H3K36me3 levels and a male-specific lethality. The sex specific effect suggests impaired H3K36me3-mediated dosage com-pensation or mis-regulation of male-specific isoforms.

31
In contrast to KDM4A, the homolog KDM4B is recessive lethal. KDM4B expression is upregulated by p53 during UV-induced DNA-damage and required for H3K9me3 demethylation 91. Recently, it was shown that KDM4B levels are upregulated in the Kdm4a mutant, and that at least one copy of either Kdm4a or Kdm4b is necessary for sur-vival. This suggests a biological redundancy between KDM4A and KDM4B 92. Interestingly, KDM4A and B homozygous double mu-tants showed a phenotype that resembled phenotypes resulting in the disruption of ecdysteroid pathways. Indeed, KDM4A physically inter-acts with the Ecdysone receptor and removes H3K9me3 at target gene promoters prior to transcription 92. The functional interactions be-tween either Set2 or KDM4A with Ecdysone indicates that H3K9 de-methylation works in concert with H3K36 methylation to achieve a specific histone code that promotes expression of Ecdysone target genes.
FIGURE 6. Modification of histone lysine residues. An empty lysine (K) can be acetylated by histone acetyltransferases or methylated by histone methyltransferases. The acetylation is removed by histone deacetylases and methylations are removed by histone demethylases.

32
FIGURE 7. The function of Histone 3 modifying enzymes during transcription-al elongation. A DNA-binding transcription factor (TF) binds a motif in an up-stream regulatory sequence and in turn recruits a demethylase (KDM4A) to remove the repressive H3K9me3 marks. MLL in the COMPASS complex distributes H3K4me3 that recruits Sgf29. This is followed by recruitment of SAGA subunits that acetylates H3K14ac. This allow for the recruitment of general transcription factors such as the TFIID complex. Simultaneously, HDAC´s are recruited to partic-ipate in the turnover of Histone acetylation that further promotes gene activation and transcription. The turnover of H3K4me3 by Lid might even more increase the re-cruitment and binding of the general transcription machinery. During transcriptional elongation, Set2 associates with the Pol II CTD and distributes H3K36me in the gene body to prohibit insertion of acetylated Histones.

33
Histone acetyltransferases One general function for DNA-binding transcription factors is to bind specific DNA motifs and recruit co-activators that further mediates gene activation. Histone acetyltransferases (HAT´s) are co-activators and have mainly been correlated with gene activation and maintenance of active transcription. Acetylation of histone tails neutralizes their positive charge that leads to a weaker interaction between nucleo-somes and DNA. This promotes nucleosome displacement and open up the chromatin for transcription. There are four mammalian HAT families: GCN5/PCAF, MYST, Nuclear receptor coactivator and CBP. Additional non-histone substrates have lead to a reclassification where HAT´s now in addition can be referred to as KAT´s (Lysine acetyltransferases). For example, the CREB-binding protein (CBP) can in addition to histones also acetylate a number of non-histone tar-gets including the RNA Pol II CTD. CPB/P300 (Drosophila Nejire) are large proteins that contain several conserved protein-binding do-mains. This allow for the response and participation in a number of different pathways 93 via interaction with a variety of transcription factors. The large SAGA coactivator complex (Spt-Ada2b-Gcn5 acetyltrans-ferase) is well conserved and can tweak chromatin to promote tran-scription. Some of the known catalytic functions of the SAGA com-plex involves binding to acetylated histones, H2B deubiquitination and histone acetylation 94. The modular nature of SAGA indicates a way of acting gene specific through different transcription factors that interacts various modules depending on cell type, similar to CBP. One mechanism that recruits SAGA to chromatin is through its subunit Sgf29 that bind H3K4me2/3 nucleosomes 95. Upon SAGA recruit-ment, the HAT subunit Gcn5 acetylates H3K9 and H3K14 residues 95. Finally, the combination of H3K4me3/ H3K14ac potentiates binding of TFIID binding via the TBP-associated factor TAF1 79. With TFIID bound to the promoter, transcription is imminent. In Drosophila, SAGA is responsible for H3K9ac and H3K14ac. Indi-vidual loss of various SAGA components results in developmental defects and lethality 96. Ada2b mutants die as early pupae and are ex-tra sensitive to irradiation-induced DNA-damage, suggesting a role for H3K9ac/H3K14ac in DNA repair 97. Similarly, both loss of the yeast H3K14 acetyltransferase Mst2 or mutations of H3K14 is critical for DNA damage response 98. Mutational analyses of Lysine residues have shown that the levels of H3K4me3 decrease when H3K14 is substitut-ed 99. One explanation to this phenomenon was recently demonstrated in Yeast where H3K14ac directly inhibits the enzymatic activity of the H3K4 demethylase Jhd2 (Drosophila gene lid) 100. In this way, each

34
gene can build an extremely versatile and unique mechanism of pre-cise gene activation by the combination of writers, readers and activa-tors. There are additional complexes that specifically acetylate H3K9 and H3K14. One being the MYST family protein Sas5 that is a component of the NuA3 complex. The NuA3 complex binds H3K4me3 and me-diate H3K14ac, very similar to the function of SAGA 101. Histone deacetylases Histone deacetylases (HDAC´s) modifies histones by the removal of acetylations from lysines. Like the KAT enzymes, HDAC´s can act on non-histone proteins and are partly localized to the cytoplasm. Since HDAC proteins are found in histone-less organisms such as bacteria, it becomes clear that HDAC´s are essential for other mechanisms than chromatin modification as well. The HDAC family is divided into three different classes: class I, class II and class III. Class I contains HDAC1/2 and HDAC3. Class II contain two sub-classes: HAC4/5/7/9 and HDAC6/10. Class III contains the sirtuin-family of HDAC proteins that has been implicated in prolonging lifespan in Drosophila. Sir2 was originally characterized as required for transcriptional and telomeric silencing and have later been impli-cated in a variety of cellular processes 102. In Yeast, Sir2 deacetylate H3K9ac and H3K14ac to promote Clr4-mediated H3K9 trimethyla-tion 103. Together with DNA binding factors, HDAC´s are recruited to chroma-tin where they deacetylate histones and thus can function as co-repressors. However, the story of biology is always more than meets the eye, and deacetylation of histones per se might not be the repres-sive mechanism. For example, inhibition of HDAC3´s catalytic activi-ty or deacetylase dead HDAC3 increases global histone acetylation as expected. Nonetheless, the protein still function as a repressor even without its enzymatic function 104. This indicates that the repressive function does not only come from the deacetylation mechanism but could rather be a result of the combined action of the whole repressive protein complex. In addition, genome-wide data show that most HDAC´s are associated with the transcription start site of active genes and positively correlates with transcription 105 106. This further chal-lenge the traditional view on HDAC´s as repressors. Recent studies have even shown that HDACs might be important for gene activation. For example, a screen in Drosophila identified HDAC3 as an activator of the hsp70 gene by mediating Pol II pause-release 107.

35
The combination of HAT´s/HDAC´s at active genes might function in acetylation turnover, possibly to “reset” the chromatin after each round of transcription. The reset function might allow the “old” preinitiation complex to release from its position to make room for a new Pol II-containing preinitiation complex at the core promoter. Drosophila early development "Two years work wasted. I have been breeding those flies for all that time and I've got nothing out of it." -Thomas Hunt Morgan The fruit fly, Drosophila melanogaster have a long history as a model organism. Drosophila used in research originates in the early 1900´s. Thomas Hunt Morgan started to use flies in his genetic research. The knowledge about the function of different biological systems has been constantly increasing ever since, much due many years of extensive fly work. Fruit fly embryos are excellent when studying developmen-tal processes. The whole developmental process after fertilization happens outside the female. Fly embryos are easy to collect in large quantities and it is possible to study developing cells and organs from the outside without the need for dissection. The study of early development is to a great degree the study of ma-ternal factors and how they control the formation of the embryo´s morphology and body plan. The mechanisms of embryogenesis differ between multicellular organisms but starts with one single cell, the fertilized egg. A fertilized egg contains information how to form the entire organism. The information exists both in the DNA of the organ-ism's genome, in the messenger RNA (mRNA), proteins and other molecules in the egg cell. In the fruit fly Drosophila melanogaster, the female fly produces her eggs in the ovaries, where she deposits RNA and proteins into the egg. This maternal contribution is used by her offspring during the first period of development before the embryo´s own zygotic transcription starts. When the egg is fertilized, the female will deposit the egg and leave it to develop. During the first hours of development, the embryo uses the maternally provided components for all of its cellullar functions. The time window when the embryo starts producing mRNA from its own genome is usually termed ma-ternal to zygotic transition (MZT) or midblastula transition (MBT) 108. The maternal factors are rapidly degraded during the MZT to initiate cellularization and to control nuclear divisions. The timing of maternal

36
RNA degradation is critical and degradation is mediated by elements in the 3´ UTR of the maternal mRNA´s. The zygotic gene expression is critical for degradation of maternal products. Experiments using Pol II mutants show that the onset of cellularization is dependent on zy-gotic expression 109. In pre-MBT embryos, a small set of genes is expressed as early as during nuclear cycle 8. This is the first time TATA-binding protein is seen in the nuclei. Pol II occupies only about hundred genes in pre-MBT embryos, compared to 4000 genes occupied by TBP and Pol II during MBT transition. In pre-MBT embryos, many genes are activat-ed solely by Pol II recruitment whereas Pol II pausing only exists at ten genes 110. Several anterior-posterior patterning genes as well as the P-TEFb target genes Sry-a, term and CG7271 are among the non-paused genes. Non-paused genes often have multiple Zelda binding motifs upstream the core promoter in combination with a strong TA-TA-box. An advantage with the non-pausing mechanism might be to achieve multiple rounds of transcription between each short nuclear cycle. In contrast, MBT activated genes are paused and often have the GAGA-motifs as well as DPE, MTE and PB motifs that correlates with paused Pol II 30 110,111. Interestingly, the patterning genes even-skipped, sloppy-paired 1 and runt has a ”dual” mode of activation, meaning that they are activated in pre-MBT by a non-pausing mecha-nism and later in MBT becomes paused. What are the factors that are involved in switching Pol II from non-pause to a paused state during the MBT? The non-pause to pause switch might be mediated by known factors such as NELF together with one or more of the pre-MBT activated genes. However, the few genes that show Pol II paus-ing already in pre-MBT stage suggests that pausing at these specific genes only depend on maternal factors. An interesting finding is that depletion of NELF in Drosophila em-bryos does not result in a clear effect on the endogenous expression levels of several patterning genes including slp1. Instead, maternal NELF depletion shows a differential requirement for NELF in activa-tion of slp1 and eve-reporter constructs. ChIP-data show that NELF bind the dual genes slp1 and eve and the non-paused genes Sry-a and ftz in 2h old embryos 31. The authors suggest that the NELF regulation is dictated from CRM´s rather than from the core promoter. Is it pos-sible that Pol II pausing is achieved via the activity of cis-regulatory sequences? CRM´s might not function as early as stage 8, and genes that are expressed that early will have a dispersed expression pattern irrespective of the core promoter sequence. Only later, the dual genes might come under the control of their CRM´s to achieve Pol II paus-

37
ing, possibly mediated via looping pausing factors to the promoters to mediate pausing. Zygotically expressed genes that are only active during the pre-MBT/MBT stage such as the non-paused Sry-a, term and CG7172 might be controlled by a much more ”simple” on/off mechanism that lacks the precise control that can be achieved from paused genes. Ex-periments where CRM´s from paused genes are placed in the vicinity of a gene such as the non-paused term would be very interesting and give valuable information about Pol II behavior. In addition, small screens that focus on the pre-MBT genes might re-veal novel proteins involved in the non-pause to pause switch. Other alternatives could involve protein-protein interaction assays involving the NELF subunits in pre-MBT embryos. Cellularization After oviposition, the fertilized egg contains one single diploid nucle-us. During the first hours, the nuclei rapidly undergo 13 nuclear divi-sions. The nuclei divides every 8 minutes until the embryo is filled up with ~6000 nuclei. The 6000 nuclei are a part of a syncytial blasto-derm. The syncytium is made up of multiple nuclei sharing a common cytoplasm. The shared cytoplasm promotes the diffusion of proteins and mRNA throughout the embryo. During this early time, a single plasma membrane surrounds the whole egg cell and the nuclei. The nine first nuclear divisions take place in the interior of the embryo. In nuclear cycle 9-10, the majority of nuclei reaches the embryo´s cortex and undergoes four more nuclear divisions. In the posterior end of the embryo, a small subset of nuclei is enclosed by cell membranes al-ready at stage 9. This is followed by one or two cell divisions before they arrest in G2. This group of nuclei forms the pole cells. The pole cells are primordial germ cells that stay separated from the other cells at the posterior end and later follow the germ band extension towards the dorsal side. The pole cells will later become the germ cells that give rise to the gametes. The pole cells follow a unique transcriptional programme to maintain their germ-cell identity. While somatic nuclei activate RNA Polymerase II (Pol II), the pole cells express factors such as the peptide Polar granule component (pgc) to inactivate P-TEFb. This has been suggested to keep the cells in a quiescent state 112 113 (FIG 8).

38
FIGURE 8. Rapid nuclear division during the first hours of embryogenesis. A) An egg containing a diploid nucleus. B) A subset of nuclei reaches the posterior and becomes pole cells. C) The nuclei migrate to the embryo´s cortex. Pole cells are visible at the posterior tip of the embryo. D) Cellularization starts at nuclear cycle 13. E) Cellularization is complete. F) Gastrulation initiates. At stage 13, a process starts that turn all into individual cells in a pro-cess termed cellularization. Cellularization is a mechanism found in the development of most insects and the process can be seen as a form of cytokinesis taking place on the basal side of the cells. Cellulariza-tion involves processes such as the coordination of a dynamic cyto-skeleton and membrane transport and is divided in four steps. When cycle 13 is complete, cellularization begin the first phase by an invag-ination of the plasma membrane between each nuclei. In the initial steps one and two, each nuclei elongates. Step two and three involves a continuous invagination of the plasma membrane until the leading edge (the furrow canal) of the membrane reaches the basal end of the elongated nuclei. During the process of cellularization, basal and api-cal adherens junctions appear in the cleavage furrow. In step 4, new lateral membranes are inserted. Step four ends when an actomyosin contractile ring consisting of myosin II and actin at the basal side of each cell pinches off the membrane. Thus, each somatic nucleus is turned into a monolayer of individual cells forming the cellular blas-toderm embryo (FIG 9). Both maternal and zygotic factors are known to regulate cellularization. Only a handful of factors have been shown to specifically be involved in the process. All cellularization genes

39
show a rapid induction of expression prior to cellularization, mainly with expression throughout the whole embryo. The zygotic cellulari-zation genes are among the small subset of genes that are expressed in pre-MBT embryos, in contrast to the 4000 genes that are activated during the massive maternal to zygotic transition 110. Due to short nu-clear cycles in the syncytium, early zygotically transcribed genes are much shorter that genes expressed later in development. This allows multiple cycles of transcription between each round of mitosis that normally shut down transcription 114.
FIGURE 9. Different steps in the cellularization process. A i) Cellularization starts with an invagination of the plasma membrane and an elongation of nuclei. Actin is concentrated below the plasma membrane. A ii-A iii) Actin and myosin follow the furrow canal forming the lateral membranes. Nuclei continue to elongate. A iv) The actomyosin machine pinch off and thereby creates the basal membrane. A v) A monolayer of cells is formed at the embryo´s cortex. B) Apical view of the cell monolayer after cellularization show membranes organized in a honeycomb pattern around each nucleus.

40
Future perspectives Initially, Drosophila research focused of pure genetics, but has for the last three decades much relied on forward genetic screens trying to elucidate which genes are involved in a certain process 115. Later ad-vantages made it possible to use reverse genetics based upon the emerging genome sequence data 116. The existing techniques and methods for the Drosophila system have grown in number over the years. From relying on blunt and painstaking X-ray and chemically induced mutagenesis that creates randomly generated mutations to advanced molecular methods. The detection of mRNA´s with RNA in situ hybridization is commonly used in embryos and fluorescently labeled probes has opened up for the possibilities of the generation of three-dimensional confocal imaging in multiple colors. Recently, the use of locked nucleic acid probes have proven to work for the detec-tion of miRNA´s 117 118. This might be used in future assays to label different parts of a transcript such as the 5´ and 3´ of the genes with two colors, or used to detect two or more transcripts that can not be distinguished from each other with conventional RNA hybridization methods. Using fluorescently tagged proteins, it is possible to follow cellular processes with live imaging from the early blastoderm stage all the way up to time point of the larvae hatching 24 h after egg lay-ing. Future fluorescent live imaging assays might include embryos that are carrying 10 or more cellular components tagged with different fluorescent markers. The possibilities today let us perform more or less any kind of targeted DNA changes in the fly genome. Recent dis-coveries such as the CRISPR/Cas9 system 119 120 121 122 have opened up a new world of possibilities where the limitations only lies in the time and cost. In addition, CRISPR/Cas9 open up for novel types of experiments such as targeted chromatin modification or immunopre-cipitation of specific DNA elements. The advantage of being able to deplete proteins or disrupt genes in vivo and follow the phenotypes that occur is an important a very powerful tool for the fly researcher. One of the most frequently used molecular methods is the creation of transgenic animals. Traditional methods for making transgenic flies have relied on P-element based insertion of DNA sequences, which is efficient but have the disadvantage of being random in the insertion into the genome. P-element based insertions have been used to create large collections of RNAi constructs, gene and enhancer traps as well as huge fly libraries of gene disruptions. Newer methods making use of phiC31-mediated transgenesis circumvent positional effect varia-tions among differently inserted transgenic constructs since constructs can be inserted at the same location with base pair faithfulness 123. In addition, phiC31-mediated transgenesis gives the advantage of inser-

41
tion of larger pieces of DNA e.g. bacterial artificial chromosomes into the genome. The transgenic flies described in the work in this book were made both with P-element based random insertion as well as with phiC31-mediated insertions. Future methods for creating trans-genic flies might involve negative selection markers such as pro-apoptotic alleles or the use of drug-resistant constructs to terminate all non-transgenic flies. Automated cloning/DNA synthesis in combina-tion with recent large-scale injection methods 124 125 could in addition speed up the rate of transgene generation and scientific discovery. One of the big challenges is to overcome problems with the heteroge-neous nature of the embryo. Any assay that uses whole-embryo ex-tracts from differentiated embryos deals with the issue of having a sample that contains multiple tissues and cell-types. This can be over-come by using methods that drive the whole embryo to differentiate into only one tissue. However, such methods are generally painstaking and the generation of enough material can limit the project. To over-come these problems, the establishment of cell-lines from the different cell types is a potential solution. A second option is having robust methods to sort out cells using flow-cytometry based methods. How-ever, this might not be an optimal method if the cell number per em-bryo is low. Antibodies with high affinity are essential for all molecular labs, and many studies rely on the quality of antibodies. One challenge for the future will be to efficiently produce small monoclonal antibodies such as nanobodies or peptides that are as effective as polyclonal antibod-ies. High affinity peptides that can be cloned and expressed in cultures such as bacteria will overcome the issues that generally occur when in-house produced, unique and well-working antibodies runs out. Clonable nanobodies can additionally be tagged with fluorescent markers and used to track dynamic mechanisms such as phosphoryla-tions or histone modifications in live conditions 126, or used to deplete protein function 127. The biggest challenge (and opportunities) for fu-ture science lies in the enormous amount of data that exists today and are constantly increasing. The human mind will not able to store and recall all relevant information that has ever been produced. Therefore, there will be a demand for software that automatically analyze new data and puts it in the context of all previously generated results. One of the next big steps in science could be the introduction of robot sci-entists to automate more or less all steps of the scientific discovery 128. So far, the new genome-wide datasets are at best a complement to the essential need to carefully and painstakingly dissect each gene by more traditional molecular methods.

42
Perspectives on Paper I P-TEFb was initially found to have an in vitro activity in transcrip-tional elongation in vitro. It overcomes the repressive effects of the negative elongation factors DSIF and NELF. The role of P-TEFb in early embryogenesis is poorly understood. Therefore, we investigated the function of P-TEFb by miRNA-mediated knockdown of Cdk9 and Cyclin T in Drosophila to deplete maternal proteins. Our aim was initially to characterize P-TEFb´s function in Pol II pause release. We found effects in early embryos such as a posterior lack of cells and cellularization defects. The previously identified cellularization gene Sry-a was downregulated in P-TEFb embryos. We further identified two more P-TEFb target genes, terminus and CG7271. Some of the defects could be due to a failure of Pol II recruitment to the genes Sry-a, terminus and CG7271. In addition, we found components of the Super elongation complex and two Mediator subunits to phenocopy the P-TEFb depleted embryos. We could see decreased Pol II occu-pancy at P-TEFb target gene 5´ ends as well as in the 3´ ends. Howev-er, we did not find and evidence for P-TEFb having a role in Pol II pause-release at target genes. Furthermore, the levels of Ser2 phos-phorylated Pol II was decreased at all tested genes, suggesting a global role for P-TEFb in Pol II CTD Ser2-p during early development. Ser2-p correlates with elongating Pol II. However, little is known about the direct effect of Ser2-p and its role in transcription. The near to normal expressed cellularization genes bnk, nullo and slam had decreased Ser2-p yet showed a modest decrease in Pol II occupancy. This indi-cates that decreased Pol II Ser2-p is neither regulating Pol II pause-release nor Pol II elongation efficiency at these genes. Ser2-p might play a different function such as priming Pol II for transcriptional ter-mination or mRNA processing. Furthermore, P-TEFb target genes in early embryos are activated solely by Pol II recruitment indicating that a non-paused gene cannot simply be turned into a Pol II paused gene by depletion of P-TEFb. What is the role of Mediator in early embry-onic development? In our study, depletion of either of the Mediator head domain subunits MED20 or MED22 resulted in a P-TEFb pheno-type. This suggests that the head module of Mediator regulates a spe-cific set of genes in early embryos via MED20 and MED22. Smaller variants of Mediator complexes comprised of a few Mediator subunits have been suggested to regulate specific sets of genes, and it is plausi-ble to speculate that MED20 and MED22 might

43
FIGURE 10. Activation of P-TEFb-dependent genes versus P-TEFb independ-ent genes in early embryogenesis. A) An early zygotic P-TEFb dependent gene such as terminus is bound by the pioneering factor Zelda together with a second unknown DNA-binding factor (X) that might recruit Med20, Med22, and SEC/P-TEFb. The specific combination of factors mediates recruitment of the preinitiation complex. B) Activation of a P-TEFb independent gene such as slam. Zelda allow for binding of the factor Y that recruits the preinitiation complex independently of P-TEFb/SEC or Med20 and Med22. P-TEFb regulates Pol II phosphorylation at both terminus and slam, possibly in a mechanism separate from Pol II recruitment. belong to a Mediator variant that activates some of the most early ex-pressed genes in pre-MBT embryos (FIG 10). We found a function for term in early development where overexpres-sion as well as miRNA mediated depletion of Terminus protein lead to cellular defects in the blastocyst stage. However, the function of ter-minus remains unknown. The highly similar DNA and protein se-quence (~99%) between term and its homolog CG7271 suggest that a recent gene duplication event took place in the split between the two groups D.obscura and D.melanogaster. This has resulted in two gene copies in melanogaster and one copy in obscura. In contrast, D. simu-lans only have one copy, suggesting that it have lost one copy in the split with D. sechellia. The amino acid sequence of Terminus is con-taining a predicted C2H2 zinc-finger, and protein-protein interaction data (The Drosophila Interactions Database, DroID) suggests Term to bind proteins involved in transcription and mRNA processing. The protein Elongin A (EloA) is among the protein-protein interaction partners (DroID database data). EloA have been suggested to be in-volved in suppressing transient pausing of Pol II in vitro together with the Super elongation complex member ELL 129. Interestingly, Term shows a protein-protein interaction with its homolog CG6885 (FIG 11). CG6885 have a 28% amino acid homology to Term and CG7171. CG6885 is only expressed at the same developmental stage as term and CG7271.

44
A small but potentially interesting finding is that some maternally contributed transcripts such as pgc and nanos showed a slightly in-creased RNA in situ signal. The increased levels might indicate a function for P-TEFb or any P-TEFb target genes to control the regula-tion of maternal transcript levels or maternal transcript destabilization. This study demonstrates the complexity of gene regulation and indi-cates a novel role for P-TEFb/SEC together with Mediator in early development. A critical next step will be to investigate and find how gene-specificity is achieved and what DNA-binding factors that regu-lates Pol II recruitment to genes in pre-MBT embryos.
FIGURE 11. Protein-protein interaction data from the DroID database. Terminus protein-protein interaction partners. The strength of each interactions was calculated using weighted correlation.

45
Perspectives on Paper II KDM4A can act as a demethylase that target H3K36me3 and H3K9me3. To assess to function of the JmjC protein KDM4A, we made a mutant fly based on a P-element jump out strategy. The KDM4A mutant flies were used in combination with a second KDM4A allele to avoid background effects. The mutant flies were viable and did not show any obvious phenotype although the levels of H3K36me3 were higher than normal. This is consistent with recent studies that show that KDM4A and KDM4A are biologically redun-dant. To investigate if overexpressed KDM4A resulted in an opposite effect, we used an Actin Gal4 driver in combination with a UAS-KDM4A transgene. Indeed, we could see a decrease in H3K36me3 levels in larvae overexpressing KDM4A. This further demonstrated that KDM4A regulates the levels of histone methylation. To find po-tential KDM4A targets, we prepared RNA from KDM4A mutant lar-vae. Microarray analyses of the mutant larvae indicated that about 100 genes had more than a 1.5 fold change in gene expression. This was validated by measuring the levels of cDNA from a subset of the target genes with quantitative RT-PCR. In addition, we analyzed KDM4A target genes in the larvae overexpressing KDM4A and found an oppo-site effect on gene expression compared to the KDM4A mutants. This showed that KDM4A regulate gene expression of these genes. To investigate how the catalytic function of KDM4A influence its function in regulating gene expression, we created transgenic flies that expressed a catalytic dead KDM4A (H195A) cloned together with upstream and downstream sequences. The inactive form of KDM4A was combined with the KDM4A mutant allele and the expression of KDM4A target genes was again investigated by RT-qPCR. Genes that were upregulated in the KDM4A mutant showed restored expression levels by the H195A transgene, indicating that the enzymatic function of KDM4A is not necessary for correct gene expression of those genes. In contrast, H195A larvae could not restore the expression lev-els of the down regulated target genes suggesting that the demethylase function of KDM4A is needed for their correct expression levels. Next, we studied the levels of methylated nucleosomes positioned at the KDM4A target genes by performing chromatin immune precipita-tion. We speculated that genes with higher levels of KDM4A sub-strates H3K9me3 and H3K36me3 would be more sensitive to the lev-els of KDM4A protein. Surprisingly, all of the tested genes had a min-imal amount of H3K9me3 and H3K36me3 in wild type and in KDM4A mutants. This was not due to the genes being inactive since the expression levels of some of the genes are high. One explanation for this could be that KDM4A regulates expression of these genes by

46
proxy, or via a so far unappreciated mechanism that does not involve H3K36me3 or H3K9me3. HP1a was previously demonstrated to interact with KDM4A and to stimulate its enzymatic function in vitro. In contrast to this synergistic effect, we found that the expression levels of some KDM4A target genes were restored in KDM4A/HP1 double mutant animals. This points to HP1A and KD4A having antagonizing functions. However, the potential of HP1a to stimulate KDM4A might not function at genes having very low levels of H3K36me3. At genes having high H3K36me3 levels, HP1a might function differently. It was previously reported that male KDM4A mutant flies have a re-duced lifespan suggesting that KDM4A might preferentially target male-specific gene expression. Therefore, we overexpressed KDM4A and investigated the effects on adult flies. We found a sex-specific effect where male flies were sensitive to increased levels of KDM4A to a much higher degree that females. In addition, we found that KDM4A regulates a subset of highly expressed genes on the X-chromosome. Other studies have shown that H3K36me3 affect bind-ing of the dosage-compensating MSL complex that might explain some of the male lethal effects. Together, this strengthens the evidence that KDM4A is involved in a male specific gene regulation, possibly through the dosage-compensation mechanism. The effects could also be a result of KDM4A working in male-specific splicing. To further characterize the function of KDM4A, there might be a need for sex-selection of embryos/larvae to specifically investigate gene regulation by KDM4A in males. Perspectives on Paper III Studying the direct biological outcome of histone modifications have until recently only been possible in Yeast systems. Much of the obsta-cles have come from scattered locations of histone genes in multicel-lular organisms as well as large copy numbers of each histone gene. Lately, advantages in Drosophila genetics have opened up the possi-bilities for flies as a useful model to study histone modifications. In Drosophila, the canonical histone genes are clustered within one re-gion. Recently, flies devoid of the histone cluster were made by the creation of a histone deficient chromosome. Furthermore, the histone deficiency flies were rescued by the addition of twelve transgenic his-

47
tone copies positioned in trans 130. This histone replacement method is carried out by the introduction of multiple vectors where each is con-taining a histone gene unit (HisGU). Each HisGU contains one copy of histone 1, histone 2A, histone 2B, histone 3 and histone 4. In addi-tion to this, histone replacement with mutant histones was previously proven to work 131. We have used the histone replacement approach in order to study the direct effects of several histone post-translational modifications in a multi-cellular model system. We mutated the previ-ously described HisGU plasmids at the different residues H3K14, H3K18, H3K36 and H3K79 in order to generate four different histone mutant fly strains. The transgenic flies carrying twelve copies of His-GUH3K14R were used to study the effects of an organism devoid of H3K14 acetylation. Furthermore, we used the H3K14R flies to gener-ate mitotic clones in imaginal wing discs. We show that H3K14ac is essential for viability and that H3K14 replacement leads to lethality in the embryo and larvae stage. Additionally, we conclude that H3K14ac-mediated inhibition of H3K4 demethylation is an evolution-ary conserved feature and that depletion of H3K14ac is not cell-lethal. We will further investigate the role of H3K14 acetylation and explore the three additional created histone mutations. One possibility to ex-pand the usefulness of this system could involve temporally controlled onset of the histone depletion. This could be achieved by having a wild-type histone 3 transgene under the control of e.g. a heat-shock promoter could be introduced in the histone replaced flies. This would allow for expression of wild-type histone 3 until a certain stage. To investigate the role of histone modifications in early embryos, there might be an idea to explore the possibilities of maternally deplete wild-type histones using miRNA-mediated knock down while at the same time simply overexpressing miRNA resistant mutant histones using the same maternal Gal4 driver.

48
Acknowledgements Usually when getting someone’s thesis, the acknowledgement section is the first and sometimes the only part in the book that people read. So I am taking a risk by not listing all people I have ever worked with or met during my PhD studies. Instead, I will try to mention as few people as possible by name and just say that I am very happy for the friends I have made during my time here. Especially I would like to acknowledge the colleagues I have worked together with in my lab through days, nights and weekends. I am very grateful that I had the opportunity to get to know all of you and that I got the chance to work with you. I am also very grateful for my family that for some mysteri-ous reason put up with my bad mood and grumpiness during the times of hard work in the lab. How did I end up as a PhD student in Stockholm? I´ve always had an interest in how things around me are constructed, and the passion for inventing small gadgets or picking apart broken electronics have followed me since I was young. After finishing school, I wrote an application to study ”green biology” at Umeå Uni-versity. However, I was drafted by the army and went on to do my service as a fire fighter. After this non-military service, I spent a year working in Stockholm and one year studying Swedish folk music in Dalarna. Next time I sent in my application to Umeå University, I aimed for molecular biology (white biology), even if I didn’t really knew what it was. It turned out that the first year was dedicated to chemistry courses. It was hard work but lots of fun and I remember that I was among the few students that seemed to like organic chemis-try. My time in Umeå was great, and I met many people that still are some of my closest friends. Umeå have a long history of fly genetics and even used to have a Drosophila stock center, and it was hard to avoid being taught the basics of fly genetics as a molecular biology student. I also had the chance to do work in research groups that used flies, such as Dan Hultmark´s lab. During my last months as a student, I did my exam work in Erik Johansson´s lab at the department of med-ical chemistry where I purified proteins. That was the time when I met Jessie. Since Jessie lived in Växjö, far away from Umeå, I decided to move to Växjö after finishing my studies. Living in Växjö was not very inspiring for either of us, and when I came in contact with my supervisor Mattias, we decided to move to Stockholm. Instead of taking apart radios to see how they work, I got the oppor-tunity to do similar things but on a molecular level. The time as a PhD student has been a great experience in many ways. I have made tons of friends, played lots of sports and had thousands of nice lunches at Lantis. I have learned a lot about so many cool things like how one

49
can take apart and put together pieces of DNA, and how to put the new homemade DNA back into an organism. Mattias and my other colleagues have really helped me to develop my skills in critical and scientific thinking. Much of the training has come from the hundreds of journal clubs, data clubs and group meetings where we spent end-less hours discussing figures and data. Mattias has always been sup-portive and allowed me do things in my own way, and at the same time tried hard to turn me into a thinking, independent researcher. I am very grateful that I got the chance of doing a PhD, and I have en-joyed every second of it. “Somewhere, something incredible is waiting to be known” -Carl Sagan

50
1 Carroll, S. B. Evo-devo and an expanding evolutionary synthesis: A genetic theory of morphological evolution. Cell 134, 25-36, doi:Doi 10.1016/J.Cell.2008.06.030 (2008).
2 Iyer, V. R. Nucleosome positioning: bringing order to the eukaryotic genome. Trends Cell Biol 22, 250-256, doi:Doi 10.1016/J.Tcb.2012.02.004 (2012).
3 Fuda, N. J., Ardehali, M. B. & Lis, J. T. Defining mechanisms that regulate RNA polymerase II transcription in vivo. Nature 461, 186-192, doi:Doi 10.1038/Nature08449 (2009).
4 Luse, D. S. Promoter clearance by RNA polymerase II. Bba-Gene Regul Mech 1829, 63-68, doi:Doi 10.1016/J.Bbagrm.2012.08.010 (2013).
5 He, Y., Fang, J., Taatjes, D. J. & Nogales, E. Structural visualization of key steps in human transcription initiation. Nature 495, 481-+, doi:Doi 10.1038/Nature11991 (2013).
6 Chapman, R. D., Haidemann, M., Hintermair, C. & Eick, D. Molecular evolution of the RNA polymerase IICTD. Trends in Genetics 24, 289-296, doi:Doi 10.1016/J.Tig.2008.03.010 (2008).
7 Nonet, M., Sweetser, D. & Young, R. A. Functional Redundancy and Structural Polymorphism in the Large Subunit of Rna Polymerase-Ii. Cell 50, 909-915, doi:Doi 10.1016/0092-8674(87)90517-4 (1987).
8 Adelman, K. & Lis, J. T. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet 13, 720-731, doi:Doi 10.1038/Nrg3293 (2012).
9 Napolitano, G., Lania, L. & Majello, B. RNA polymerase II CTD modifications: how many tales from a single tail. J Cell Physiol 229, 538-544, doi:10.1002/jcp.24483 (2014).
10 Saberianfar, R., Cunningham-Dunlop, S. & Karagiannis, J. Global Gene Expression Analysis of Fission Yeast Mutants Impaired in Ser-2 Phosphorylation of the RNA Pol II Carboxy Terminal Domain. Plos One 6, doi:ARTN e24694 DOI 10.1371/journal.pone.0024694 (2011).
11 Schroder, S. et al. Acetylation of RNA Polymerase II Regulates Growth-Factor-Induced Gene Transcription in Mammalian Cells. Mol Cell 52, 314-324, doi:Doi 10.1016/J.Molcel.2013.10.009 (2013).
12 Perales, R. & Bentley, D. "Cotranscriptionality": The Transcription Elongation Complex as a Nexus for Nuclear Transactions. Mol Cell 36, 178-191, doi:Doi 10.1016/J.Molcel.2009.09.018 (2009).
13 Richard, P. & Manley, J. L. Transcription termination by nuclear RNA polymerases. Gene Dev 23, 1247-1269, doi:10.1101/gad.1792809 (2009).
14 Kwak, H. & Lis, J. T. Control of Transcriptional Elongation. Annu Rev Genet 47, 483-508, doi:Doi 10.1146/Annurev-Genet-110711-155440 (2013).
15 Ptashne, M. & Gann, A. Transcriptional activation by recruitment. Nature 386, 569-577, doi:Doi 10.1038/386569a0 (1997).
16 Gilmour, D. S. & Lis, J. T. Rna Polymerase-Ii Interacts with the Promoter Region of the Noninduced Hsp70 Gene in Drosophil-Melanogaster Cells. Mol Cell Biol 6, 3984-3989 (1986).

51
17 Rasmussen, E. B. & Lis, J. T. In vivo transcriptional pausing and cap formation on three Drosophila heat shock genes. Proc Natl Acad Sci U S A 90, 7923-7927 (1993).
18 Wang, X. L., Lee, C., Gilmour, D. S. & Gergen, J. P. Transcription elongation controls cell fate specification in the Drosophila embryo. Gene Dev 21, 1031-1036, doi:Doi 10.1101/Gad.1521207 (2007).
19 Buckley, M. S., Kwak, H., Zipfel, W. R. & Lis, J. T. Kinetics of promoter Pol II on Hsp70 reveal stable pausing and key insights into its regulation. Gene Dev 28, 14-19, doi:Doi 10.1101/Gad.231886.113 (2014).
20 Henriques, T. et al. Stable Pausing by RNA Polymerase II Provides an Opportunity to Target and Integrate Regulatory Signals. Mol Cell 52, 517-528, doi:Doi 10.1016/J.Molcel.2013.10.001 (2013).
21 Yamaguchi, Y., Shibata, H. & Handa, H. Transcription elongation factors DSIF and NELF: Promoter-proximal pausing and beyond. Bba-Gene Regul Mech 1829, 98-104, doi:Doi 10.1016/J.Bbagrm.2012.11.007 (2013).
22 Core, L. J. et al. Defining the Status of RNA Polymerase at Promoters. Cell Rep 2, 1025-1035, doi:Doi 10.1016/J.Celrep.2012.08.034 (2012).
23 Saunders, A., Core, L. J., Sutcliffe, C., Lis, J. T. & Ashe, H. L. Extensive polymerase pausing during Drosophila axis patterning enables high-level and pliable transcription. Gene Dev 27, 1146-1158, doi:Doi 10.1101/Gad.215459.113 (2013).
24 Chen, K. et al. A global change in RNA polymerase II pausing during the Drosophila midblastula transition. Elife 2, doi:ARTN e00861 DOI 10.7554/eLife.00861 (2013).
25 Ohler, U., Liao, G. C., Niemann, H. & Rubin, G. M. Computational analysis of core promoters in the Drosophila genome. Genome Biol 3, RESEARCH0087 (2002).
26 Li, J. & Gilmour, D. S. Distinct mechanisms of transcriptional pausing orchestrated by GAGA factor and M1BP, a novel transcription factor. Embo Journal 32, 1829-1841, doi:Doi 10.1038/Emboj.2013.111 (2013).
27 Yamaguchi, Y. et al. NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell 97, 41-51, doi:Doi 10.1016/S0092-8674(00)80713-8 (1999).
28 Missra, A. & Gilmour, D. S. Interactions between DSIF (DRB sensitivity inducing factor), NELF (negative elongation factor), and the Drosophila RNA polymerase II transcription elongation complex. Proc Natl Acad Sci U S A 107, 11301-11306, doi:Doi 10.1073/Pnas.1000681107 (2010).
29 Wu, C. H. et al. NELF and DSIF cause promoter proximal pausing on the hsp70 promoter in Drosophila. Gene Dev 17, 1402-1414, doi:Doi 10.1101/Gad.1091403 (2003).
30 Gilchrist, D. A. et al. Pausing of RNA polymerase II disrupts DNA-specified nucleosome organization to enable precise gene regulation. Cell 143, 540-551, doi:10.1016/j.cell.2010.10.004 (2010).
31 Wang, X. L., Hang, S. Y., Prazak, L. & Gergen, J. P. NELF Potentiates Gene Transcription in the Drosophila Embryo. Plos One 5, doi:ARTN e11498 DOI 10.1371/journal.pone.0011498 (2010).

52
32 Jennings, B. H. et al. Locus-specific requirements for Spt5 in transcriptional activation and repression in Drosophila. Current Biology 14, 1680-1684, doi:Doi 10.1016/J.Cub.2004.08.066 (2004).
33 Guo, S. et al. A regulator of transcriptional elongation controls vertebrate neuronal development. Nature 408, 366-369 (2000).
34 Li, Q. T. et al. Analysis of the large inactive P-TEFb complex indicates that it contains one 7SK molecule, a dimer of HEXIM1 or HEXIM2, and two P-TEFb molecules containing Cdk9 phosphorylated at threonine 186. Journal of Biological Chemistry 280, 28819-28826, doi:Doi 10.1074/Jbc.M502712200 (2005).
35 Ramakrishnan, R. et al. Identification of novel CDK9 and Cyclin T1-associated protein complexes (CCAPs) whose siRNA depletion enhances HIV-1 Tat function. Retrovirology 9, doi:Artn 90 Doi 10.1186/1742-4690-9-90 (2012).
36 Chao, S. H. & Price, D. H. Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. Journal of Biological Chemistry 276, 31793-31799, doi:Doi 10.1074/Jbc.M102306200 (2001).
37 Ni, Z. Y. et al. P-TEFb is critical for the maturation of RNA polymerase II into productive elongation in vivo. Mol Cell Biol 28, 1161-1170, doi:Doi 10.1128/Mcb.01859-07 (2008).
38 Lis, J. T., Mason, P., Peng, J., Price, D. H. & Werner, J. P-TEFb kinase recruitment and function at heat shock loci. Gene Dev 14, 792-803 (2000).
39 Price, D. H. P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol Cell Biol 20, 2629-2634, doi:Doi 10.1128/Mcb.20.8.2629-2634.2000 (2000).
40 Peterlin, B. M. & Price, D. H. Controlling the elongation phase of transcription with P-TEFb. Mol Cell 23, 297-305, doi:Doi 10.1016/J.Molcel.2006.06.014 (2006).
41 Medlin, J. et al. P-TEFb is not an essential elongation factor for the intronless human U2 snRNA and histone H2b genes. Embo Journal 24, 4154-4165, doi:Doi 10.1038/Sj.Emboj.7600876 (2005).
42 Czudnochowski, N., Bosken, C. A. & Geyer, M. Serine-7 but not serine-5 phosphorylation primes RNA polymerase II CTD for P-TEFb recognition. Nat Commun 3, 842, doi:10.1038/ncomms1846 (2012).
43 Kohoutek, J. et al. Cyclin T2 Is Essential for Mouse Embryogenesis. Mol Cell Biol 29, 3280-3285, doi:Doi 10.1128/Mcb.00172-09 (2009).
44 Eissenberg, J. C., Shilatifard, A., Dorokhov, N. & Michener, D. E. Cdk9 is an essential kinase in Drosophila that is required for heat shock gene expression, histone methylation and elongation factor recruitment. Molecular Genetics and Genomics 277, 101-114, doi:Doi 10.1007/S00438-006-0164-2 (2007).
45 Bartkowiak, B. et al. CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Gene Dev 24, 2303-2316, doi:Doi 10.1101/Gad.1968210 (2010).
46 Boehm, A. K., Saunders, A., Werner, J. & Lis, J. T. Transcription factor and polymerase recruitment, modification, and movement on dhsp70 in

53
vivo in the minutes following heat shock. Mol Cell Biol 23, 7628-7637, doi:Doi 10.1128/Mcb.23.21.7628-7637.2003 (2003).
47 Luo, Z., Lin, C. & Shilatifard, A. The super elongation complex (SEC) family in transcriptional control. Nat Rev Mol Cell Biol 13, 543-547, doi:10.1038/nrm3417 (2012).
48 Lin, C. et al. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell 37, 429-437, doi:10.1016/j.molcel.2010.01.026 (2010).
49 Mohan, M., Lin, C., Guest, E. & Shilatifard, A. Licensed to elongate: a molecular mechanism for MLL-based leukaemogenesis. Nat Rev Cancer 10, 721-728, doi:10.1038/nrc2915 (2010).
50 Tang, A. H., Neufeld, T. P., Rubin, G. M. & Muller, H. A. J. Transcriptional regulation of cytoskeletal functions and segmentation by a novel maternal pair-rule gene, lilliputian. Development 128, 801-813 (2001).
51 VanderZwan-Butler, C. J., Prazak, L. M. & Gergen, J. P. The HMG-box protein Lilliputian is required for Runt-dependent activation of the pair-rule gene fushi-tarazu. Dev Biol 301, 350-360, doi:Doi 10.1016/J.Ydbio.2006.10.027 (2007).
52 Yin, J. W. & Wang, G. The Mediator complex: a master coordinator of transcription and cell lineage development. Development 141, 977-987, doi:10.1242/dev.098392 (2014).
53 Poss, Z. C., Ebmeier, C. C. & Taatjes, D. J. The Mediator complex and transcription regulation. Crit Rev Biochem Mol 48, 575-608, doi:Doi 10.3109/10409238.2013.840259 (2013).
54 Kim, Y. J., Bjorklund, S., Li, Y., Sayre, M. H. & Kornberg, R. D. A Multiprotein Mediator of Transcriptional Activation and Its Interaction with the C-Terminal Repeat Domain of Rna-Polymerase-Ii. Cell 77, 599-608, doi:Doi 10.1016/0092-8674(94)90221-6 (1994).
55 Robinson, P. J. J., Bushnell, D. A., Trnka, M. J., Burlingame, A. L. & Kornberg, R. D. Structure of the Mediator Head module bound to the carboxy-terminal domain of RNA polymerase II. Proc Natl Acad Sci U S A 109, 17931-17935, doi:Doi 10.1073/Pnas.1215241109 (2012).
56 Wang, W. et al. Mediator MED23 regulates basal transcription in vivo via an interaction with P-TEFb. Transcription 4, 39-51, doi:10.4161/trns.22874 (2013).
57 Takahashi, H. et al. Human Mediator Subunit MED26 Functions as a Docking Site for Transcription Elongation Factors. Cell 146, 92-104, doi:Doi 10.1016/J.Cell.2011.06.005 (2011).
58 Galbraith, M. D. et al. HIF1A Employs CDK8-Mediator to Stimulate RNAPII Elongation in Response to Hypoxia. Cell 153, 1327-1339, doi:Doi 10.1016/J.Cell.2013.04.048 (2013).
59 Shlyueva, D., Stampfel, G. & Stark, A. Transcriptional enhancers: from properties to genome-wide predictions. Nature reviews. Genetics 15, 272-286, doi:10.1038/nrg3682 (2014).
60 Banerji, J., Olson, L. & Schaffner, W. A Lymphocyte-Specific Cellular Enhancer Is Located Downstream of the Joining Region in

54
Immunoglobulin Heavy-Chain Genes. Cell 33, 729-740, doi:Doi 10.1016/0092-8674(83)90015-6 (1983).
61 Carlsten, J. O. P., Zhu, X. F. & Gustafsson, C. M. The multitalented Mediator complex. Trends Biochem Sci 38, 531-537, doi:Doi 10.1016/J.Tibs.2013.08.007 (2013).
62 Visel, A. et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457, 854-858, doi:Doi 10.1038/Nature07730 (2009).
63 Rada-Iglesias, A. et al. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279-+, doi:Doi 10.1038/Nature09692 (2011).
64 Kim, T. K. et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 465, 182-U165, doi:Doi 10.1038/Nature09033 (2010).
65 Lagha, M. et al. Paused Pol II Coordinates Tissue Morphogenesis in the Drosophila Embryo. Cell 153, 976-987, doi:Doi 10.1016/J.Cell.2013.04.045 (2013).
66 Arnold, C. D. et al. Genome-Wide Quantitative Enhancer Activity Maps Identified by STARR-seq. Science 339, 1074-1077, doi:Doi 10.1126/Science.1232542 (2013).
67 Pfeiffer, B. D. et al. Tools for neuroanatomy and neurogenetics in Drosophila. Proc Natl Acad Sci U S A 105, 9715-9720, doi:Doi 10.1073/Pnas.0803697105 (2008).
68 Goldberg, A. D., Allis, C. D. & Bernstein, E. Epigenetics: A landscape takes shape. Cell 128, 635-638, doi:Doi 10.1016/J.Cell.2007.02.006 (2007).
69 Felsenfeld, G. A brief history of epigenetics. Cold Spring Harb Perspect Biol 6, doi:10.1101/cshperspect.a018200 (2014).
70 Gurdon, J. B. Multiple Genetically Identical Frogs. J Hered 53, 5-& (1962). 71 Mueller-Planitz, F., Klinker, H. & Becker, P. B. Nucleosome sliding
mechanisms: new twists in a looped history. Nat Struct Mol Biol 20, 1026-1032, doi:Doi 10.1038/Nsmb.2648 (2013).
72 Struhl, K. & Segal, E. Determinants of nucleosome positioning. Nat Struct Mol Biol 20, 267-273, doi:Doi 10.1038/Nsmb.2506 (2013).
73 Mavrich, T. N. et al. Nucleosome organization in the Drosophila genome. Nature 453, 358-U327, doi:Doi 10.1038/Nature06929 (2008).
74 Jenuwein, T. & Allis, C. D. Translating the histone code. Science 293, 1074-1080, doi:Doi 10.1126/Science.1063127 (2001).
75 Black, J. C., Van Rechem, C. & Whetstine, J. R. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell 48, 491-507, doi:10.1016/j.molcel.2012.11.006 (2012).
76 Shilatifard, A. The COMPASS Family of Histone H3K4 Methylases: Mechanisms of Regulation in Development and Disease Pathogenesis. Annual Review of Biochemistry, Vol 81 81, 65-95, doi:Doi 10.1146/Annurev-Biochem-051710-134100 (2012).
77 Smith, E., Lin, C. & Shilatifard, A. The super elongation complex (SEC) and MLL in development and disease. Gene Dev 25, 661-672, doi:10.1101/gad.2015411 (2011).
78 Jakovcevski, M. & Akbarian, S. Epigenetic mechanisms in neurological disease. Nat Med 18, 1194-1204, doi:Doi 10.1038/Nm.2828 (2012).

55
79 Vermeulen, M. et al. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell 131, 58-69, doi:Doi 10.1016/J.Cell.2007.08.016 (2007).
80 Li, J. X., Moazed, D. & Gygi, S. P. Association of the histone methyltransferase Set2 with RNA polymerase II plays a role in transcription elongation. Journal of Biological Chemistry 277, 49383-49388, doi:Doi 10.1074/Jbc.M209294200 (2002).
81 Li, B., Howe, L., Anderson, S., Yates, J. R. & Workman, J. L. The Set2 histone methyltransferase functions through the phosphorylated carboxyl-terminal domain of RNA polymerase II. Journal of Biological Chemistry 278, 8897-8903, doi:Doi 10.1074/Jbc.M212134200 (2003).
82 Stabell, M., Larsson, J., Aalen, R. B. & Lambertsson, A. Drosophila dSet2 functions in H3-K36 methylation and is required for development. Biochem Biophys Res Commun 359, 784-789, doi:Doi 10.1016/J.Bbrc.2007.05.189 (2007).
83 Kizer, K. O. et al. A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol Cell Biol 25, 3305-3316, doi:10.1128/MCB.25.8.3305-3316.2005 (2005).
84 Luco, R. F., Allo, M., Schor, I. E., Kornblihtt, A. R. & Misteli, T. Epigenetics in Alternative Pre-mRNA Splicing. Cell 144, 16-26, doi:Doi 10.1016/J.Cell.2010.11.056 (2011).
85 Li, B. et al. Histone H3 Lysine 36 Dimethylation (H3K36me2) Is Sufficient to Recruit the Rpd3s Histone Deacetylase Complex and to Repress Spurious Transcription. Journal of Biological Chemistry 284, 7970-7976, doi:Doi 10.1074/Jbc.M808220200 (2009).
86 Carrozza, M. J. et al. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123, 581-592, doi:Doi 10.1016/J.Cell.2005.10.023 (2005).
87 Venkatesh, S. et al. Set2 methylation of histone H3 lysine 36 suppresses histone exchange on transcribed genes. Nature 489, 452-U145, doi:Doi 10.1038/Nature11326 (2012).
88 Klose, R. J. et al. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature 442, 312-316, doi:10.1038/nature04853 (2006).
89 Whetstine, J. R. et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 125, 467-481, doi:10.1016/j.cell.2006.03.028 (2006).
90 Lorbeck, M. T. et al. The histone demethylase Dmel\Kdm4A controls genes required for life span and male-specific sex determination in Drosophila. Gene 450, 8-17, doi:10.1016/j.gene.2009.09.007 (2010).
91 Palomera-Sanchez, Z., Bucio-Mendez, A., Valadez-Graham, V., Reynaud, E. & Zurita, M. Drosophila p53 Is Required to Increase the Levels of the dKDM4B Demethylase after UV-induced DNA Damage to Demethylate Histone H3 Lysine 9. Journal of Biological Chemistry 285, 31370-31379, doi:Doi 10.1074/Jbc.M110.128462 (2010).

56
92 Tsurumi, A., Dutta, P., Yan, S. J., Sheng, R. & Li, W. X. Drosophila Kdm4 demethylases in histone H3 lysine 9 demethylation and ecdysteroid signaling. Sci Rep-Uk 3, doi:Artn 2894 Doi 10.1038/Srep02894 (2013).
93 Bedford, D. C., Kasper, L. H., Fukuyama, T. & Brindle, P. K. Target gene context influences the transcriptional requirement for the KAT3 family of CBP and p300 histone acetyltransferases. Epigenetics-Us 5, 9-15 (2010).
94 Samara, N. L. & Wolberger, C. A new chapter in the transcription SAGA. Curr Opin Struc Biol 21, 767-774, doi:10.1016/j.sbi.2011.09.004 (2011).
95 Bian, C. B. et al. Sgf29 binds histone H3K4me2/3 and is required for SAGA complex recruitment and histone H3 acetylation. Embo Journal 30, 2829-2842, doi:Doi 10.1038/Emboj.2011.193 (2011).
96 Weake, V. M. et al. Post-transcription initiation function of the ubiquitous SAGA complex in tissue-specific gene activation. Gene Dev 25, 1499-1509, doi:Doi 10.1101/Gad.2046211 (2011).
97 Qi, D., Larsson, J. & Mannervik, M. Drosophila Ada2b is required for viability and normal histone H3 acetylation. Mol Cell Biol 24, 8080-8089, doi:Doi 10.1128/Mcb.24.18.8080-8089.2004 (2004).
98 Wang, Y. et al. Histone H3 Lysine 14 Acetylation Is Required for Activation of a DNA Damage Checkpoint in Fission Yeast. Journal of Biological Chemistry 287, 4386-4393, doi:Doi 10.1074/Jbc.M111.329417 (2012).
99 Nakanishi, S. et al. A comprehensive library of histone mutants identifies nucleosomal residues required for H3K4 methylation. Nat Struct Mol Biol 15, 881-888, doi:Doi 10.1038/Nsmb.1454 (2008).
100 Maltby, V. E. et al. Histone H3K4 demethylation is negatively regulated by histone H3 acetylation in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 109, 18505-18510, doi:Doi 10.1073/Pnas.1202070109 (2012).
101 Taverna, S. D. et al. Yng1 PHD finger binding to H3 trimethylated at K4 promotes NuA3 HAT activity at K14 of H3 and transcription at a subset of targeted ORFs. Mol Cell 24, 785-796, doi:Doi 10.1016/J.Molcel.2006.10.026 (2006).
102 Park, S., Mori, R. & Shimokawa, I. Do sirtuins promote mammalian longevity?: A Critical review on its relevance to the longevity effect induced by calorie restriction. Mol Cells 35, 474-480, doi:Doi 10.1007/S10059-013-0130-X (2013).
103 Alper, B. J. et al. Sir2 is required for Clr4 to initiate centromeric heterochromatin assembly in fission yeast. Embo Journal 32, 2321-2335, doi:Doi 10.1038/Emboj.2013.143 (2013).
104 Sun, Z. et al. Deacetylase-Independent Function of HDAC3 in Transcription and Metabolism Requires Nuclear Receptor Corepressor. Mol Cell 52, 769-782, doi:Doi 10.1016/J.Molcel.2013.10.022 (2013).
105 Wang, Z. B. et al. Genome-wide Mapping of HATs and HDACs Reveals Distinct Functions in Active and Inactive Genes. Cell 138, 1019-1031, doi:Doi 10.1016/J.Cell.2009.06.049 (2009).
106 Negre, N. et al. A cis-regulatory map of the Drosophila genome. Nature 471, 527-531, doi:10.1038/nature09990 (2011).

57
107 Achary, B. G., Campbell, K. M., Co, I. S. & Gilmour, D. S. RNAi screen in Drosophila larvae identifies histone deacetylase 3 as a positive regulator of the hsp70 heat shock gene expression during heat shock. Biochim Biophys Acta, doi:10.1016/j.bbagrm.2014.02.018 (2014).
108 Tadros, W. & Lipshitz, H. D. The maternal-to-zygotic transition: a play in two acts. Development 136, 3033-3042, doi:Doi 10.1242/Dev.033183 (2009).
109 Sung, H. W., Spangenberg, S., Vogt, N. & Grosshans, J. Number of Nuclear Divisions in the Drosophila Blastoderm Controlled by Onset of Zygotic Transcription. Current Biology 23, 133-138, doi:Doi 10.1016/J.Cub.2012.12.013 (2013).
110 Chen, K. et al. A global change in RNA polymerase II pausing during the Drosophila midblastula transition. Elife 2, e00861, doi:10.7554/eLife.00861 (2013).
111 Hendrix, D. A., Hong, J. W., Zeitlinger, J., Rokhsar, D. S. & Levine, M. S. Promoter elements associated with RNA Pol II stalling in the Drosophila embryo. Proc Natl Acad Sci U S A 105, 7762-7767, doi:10.1073/pnas.0802406105 (2008).
112 Hanyu-Nakamura, K., Sonobe-Nojima, H., Tanigawa, A., Lasko, P. & Nakamura, A. Drosophila Pgc protein inhibits P-TEFb recruitment to chromatin in primordial germ cells. Nature 451, 730-U737, doi:Doi 10.1038/Nature06498 (2008).
113 Lesch, B. J. & Page, D. C. Genetics of germ cell development. Nat Rev Genet 13, 781-794, doi:Doi 10.1038/Nrg3294 (2012).
114 Heyn, P. et al. The Earliest Transcribed Zygotic Genes Are Short, Newly Evolved, and Different across Species. Cell Rep 6, 285-292, doi:Doi 10.1016/J.Celrep.2013.12.030 (2014).
115 Venken, K. J. & Bellen, H. J. Chemical mutagens, transposons, and transgenes to interrogate gene function in Drosophila melanogaster. Methods, doi:10.1016/j.ymeth.2014.02.025 (2014).
116 St Johnston, D. Using mutants, knockdowns, and transgenesis to investigate gene function in Drosophila. Wiley Interdiscip Rev Dev Biol 2, 587-613, doi:10.1002/wdev.101 (2013).
117 Kloosterman, W. P., Wienholds, E., de Bruijn, E., Kauppinen, S. & Plasterk, R. H. A. In situ detection of miRNAs in animal embryos using LNA-modified oligonucteotide probes. Nat Methods 3, 27-29, doi:Doi 10.1038/Nmeth843 (2006).
118 Lagendijk, A. K., Moulton, J. D. & Bakkers, J. Revealing details: whole mount microRNA in situ hybridization protocol for zebrafish embryos and adult tissues. Biol Open 1, 566-569, doi:10.1242/bio.2012810 (2012).
119 Bassett, A. & Liu, J. L. CRISPR/Cas9 mediated genome engineering in Drosophila. Methods, doi:10.1016/j.ymeth.2014.02.019 (2014).
120 Gratz, S. J. et al. Highly Specific and Efficient CRISPR/Cas9-Catalyzed Homology-Directed Repair in Drosophila. Genetics, doi:10.1534/genetics.113.160713 (2014).

58
121 Bassett, A. R., Tibbit, C., Ponting, C. P. & Liu, J. L. Mutagenesis and homologous recombination in Drosophila cell lines using CRISPR/Cas9. Biol Open 3, 42-49, doi:10.1242/bio.20137120 (2014).
122 Gratz, S. J. et al. Genome Engineering of Drosophila with the CRISPR RNA-Guided Cas9 Nuclease. Genetics 194, 1029-+, doi:Doi 10.1534/Genetics.113.152710 (2013).
123 Bateman, J. R., Lee, A. M. & Wu, C. T. Site-specific transformation of Drosophila via phiC31 integrase-mediated cassette exchange. Genetics 173, 769-777, doi:10.1534/genetics.106.056945 (2006).
124 Bischof, J. et al. A versatile platform for creating a comprehensive UAS-ORFeome library in Drosophila. Development 140, 2434-2442, doi:Doi 10.1242/Dev.088757 (2013).
125 Delubac, D. et al. Microfluidic system with integrated microinjector for automated Drosophila embryo injection. Lab Chip 12, 4911-4919, doi:Doi 10.1039/C2lc40104e (2012).
126 Rothbauer, U. et al. Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat Methods 3, 887-889, doi:Doi 10.1038/Nmeth953 (2006).
127 Caussinus, E., Kanca, O. & Affolter, M. Fluorescent fusion protein knockout mediated by anti-GFP nanobody. Nat Struct Mol Biol 19, 117-U142, doi:Doi 10.1038/Nsmb.2180 (2012).
128 Sparkes, A. et al. Towards Robot Scientists for autonomous scientific discovery. Autom Exp 2, 1, doi:10.1186/1759-4499-2-1 (2010).
129 Shilatifard, A., Conaway, R. C. & Conaway, J. W. The RNA polymerase II elongation complex. Annu Rev Biochem 72, 693-715, doi:Doi 10.1146/Annurev.Biochem.72.121801.161551 (2003).
130 Gunesdogan, U., Jackle, H. & Herzig, A. A genetic system to assess in vivo the functions of histones and histone modifications in higher eukaryotes. EMBO Rep 11, 772-776, doi:10.1038/embor.2010.124 (2010).
131 Pengelly, A. R., Copur, O., Jackle, H., Herzig, A. & Muller, J. A histone mutant reproduces the phenotype caused by loss of histone-modifying factor Polycomb. Science 339, 698-699, doi:10.1126/science.1231382 (2013).