formulation and evaluation of felodipine sublingual
TRANSCRIPT

www.wjpps.com Vol 3, Issue 6, 2014.
850
Bhavik et al. World Journal of Pharmacy and Pharmaceutical Sciences
FORMULATION AND EVALUATION OF FELODIPINE SUBLINGUAL
TABLET
Rohit J. Patel, Bhavik N. Patel*, Dasharath M. Patel and Chhagan N. Patel
Department of Pharmaceutics and Pharmaceutical Technology, Shri Sarvajanik Pharmacy College,
Mehsana-384001, Gujarat, India.
ABSTRACT
Objective: Felodipine is the drug of choice in hypertension and
congestive heart failure.Its bioavailability is very low about 15%.
Present investigation was undertaken to formulate sublingual tablet of
Felodipine to overcome the first pass metabolism and provide fast
onset of action. Experimental Work: The solid dispersion of
Felodipine were prepared with β-cyclodextrin, poloxamer 407, PEG
6000 and PVP K-30 in various ratios (1:2, 1:4, 1:6 1:8) and phase
solubility study was performed to select the carrier. The selected solid
dispersion was then utilized for the preparation of sublingual tablet by
direct compression utilizing different superdisintegrant like Cross
carmelose sodium, Crosspovidone, Kyron T-314 and Sodium starch
glycolate. Prepared tablets were evaluated for weight variation, thickness, friability, content
uniformity, hardness, disintegration time, wetting time and in-vitro drug release. Stability
study of optimized formulation was performed as per ICH guideline. Result: The optimized
formulation (batch F3) containing Drug-Poloxamer-407 (1:6) complex and Kyron-T314(5%)
showed greater drug dissolution (87% in 15 min) and satisfactory in vitro disintegration time
(22 sec). Stability study of optimized formulation showed that optimized formulation was
stable at accelerated environment condition. Conclusion: Felodipine sublingual tablet were
prepared successfully by the use of solid dispersion of Felodipine-poloxamer 407 (1:6)
complex using Kyron-T314 as a superdisintegrant.Thus, sublingual tablet of Felodipine could
be an alternative route to avoid gastrointestinal side effect as well as bypass hepatic first pass
metabolism. The formulated sublingual tablets may act as a potential alternate for the
Felodipine oral tablet.
WWOORRLLDD JJOOUURRNNAALL OOFF PPHHAARRMMAACCYY AANNDD PPHHAARRMMAACCEEUUTTIICCAALL SSCCIIEENNCCEESS SSJJIIFF IImmppaacctt FFaaccttoorr 22..778866
VVoolluummee 33,, IIssssuuee 66,, 885500--886644.. RReesseeaarrcchh AArrttiiccllee IISSSSNN 2278 – 4357
Article Received on 25 March 2014, Revised on 15 April 2014, Accepted on 07May 2014
*Correspondence for Author
Bhavik N. Patel
Department of Pharmaceutics
and Pharmaceutical
Technology, Shri Sarvajanik
Pharmacy College, Mehsana-
384001, Gujarat, India.

www.wjpps.com Vol 3, Issue 6, 2014.
851
Bhavik et al. World Journal of Pharmacy and Pharmaceutical Sciences
Keywords: Felodipine, Solid Dispersion, Poloxamer-407, Sublingual Tablet, Hypertension
INTRODUCTION
The oral route of administration is considered as the most widely accepted route. The unique
environment of the oral cavity offers its potential as a site for drug delivery. Because rich
blood supply and direct access to systemic circulation, the oral mucosal route is suitable for
drugs, which are susceptible to acid hydrolysis in the stomach or which are extensively
metabolized in the liver. The continuous secretion of saliva results in rapid removal of
released drug and this may desire that the oral cavity be restricted to the delivery of drugs,
which have a short systemic circulation. Tablets that disintegrate or dissolve rapidly in the
patient’s mouth are convenient for young children, especially elderly and patients who are
unable to swallow, and in some cases where potable liquids are not available. The drug can
be easily disintegrate in the presence of small volume of saliva in oral cavity. Then the
medication can be absorbed partially or entirely into the systemic circulation from blood
vessels in the sublingual mucosa, or it can be swallowed as a solution to be absorbed from
gastrointestinal tract. The sublingual route usually produces a faster onset of action than
orally administered tablets and the amount absorbed through sublingual blood vessels bypass
the hepatic first- pass metabolic processes[1-3]
Felodipine is a calcium-channel blocker used in the treatment of hypertension and angina
pectoris. Being a dihydropyridine derivative Felodipine has the advantage of being more
selective as vasodilator and having less cardiac effects than non-dihydropyridine calcium
antagonists. This benefit is abolished by the poor bioavailability of the drug, which– although
being almost completely absorbed from the gastrointestinal tract-is only 15% bioavailable
after oral administration.[4-5]
The poor oral bioavailability of Felodipine was attributable to its extensive first-pass
metabolism and the very low water solubility of the drug. The aqueous solubility of a given
drug is a very critical factor affecting drug efficacy and safety as it affects the drug
dissolution parameters and the oral bioavailability. The efficacy of a drug can be severely
limited by its poor aqueous solubility. Moreover, for poorly soluble drugs, the dissolution
step may be the rate-limiting process for drug absorption. In addition, the poor aqueous
solubility and wettability of the drug adds to the difficulties encountered in drug formulation.
As a result, many attempts have been made to improve the aqueous solubility of insoluble
drugs in order to increase drug efficacy and/or reduce side effects.[6]

www.wjpps.com Vol 3, Issue 6, 2014.
852
Bhavik et al. World Journal of Pharmacy and Pharmaceutical Sciences
MATERIALS AND METHODS
Felodipine was purchased from Torrent Pharmaceuticals Limited, Gujarat, India.Sodium
Starch Glycolate (SSG), Crospovidone, Microcrystalline Cellulose (MCC) were procured
from Yarrow Chem. Products, Mumbai, India. Kyron-314 was received as a gift sample from
Corel pharma, Ahmedabad, India. Poloxamer 407 was purchased from BASF chemical
company, Germany. Polyethylene Glycol 6000was purchased from S. D. Fine Chemical
Limited, Mumbai, India. All other materials used were of pharmaceutical or analytical grade.
Drug-Excipients Compatibility Study
The drug-excipient compatibility study was carried out by using Fourier Transform Infrared
(FTIR) spectroscopy. FTIR study was conducted using KBr powder mixing method on FTIR
spectrophotometer (FTIR-1700, Shimadzu, Kyoto, Japan) and the spectrums were recorded in
the wavelength region of 4000 - 400 cm-1.
Preparation of solid dispersion[7]
Solid dispersions were prepared by solvent wettingmethod using different ratio (1:2, 1:4, 1:6,
and 1:8) of drug with solubility enhancing agent like poloxamer-407, PVP k-30, Beta-
cyclodextrinand PEG-6000. Felodipine, dissolved in an appropriate amount of ethanol. After
complete dissolution of Felodipine, solutions were dropped onto polymeric carriers. Solvents
were removed under vacuum at room temperature. The solid dispersions obtained were
ground in a mortar and passed through sieve no.60.
Phase solubility study[8]
The excess amount of drug or solid dispersion was added to conical flasks containing 10 ml
of phosphate buffer 6.8 pH and subjected to shaking on a rotary shaker for 48 hours at 37°C.
Then the flasks were removed and content was filtered by 0.45µm membrane filter paper and
filtrate was analyzed for the drug content after appropriate dilution with phosphate buffer 6.8
pH and compared with pure drug solubility.
Differential scanning calorimetry (DSC) of solid dispersion
The thermogram of solid dispersion was obtained by differential scanning calorimeter (DSC),
on Shimadzu TA-60 model. Sample holder: DSC aluminum cell. Amount of sample taken: 5-
8 mg. Temp. Rangestudied: 30°C to 300°C. Nitrogen flow rate: 40-50 ml /min. Reference
sample: Blank DSC aluminum cell.

www.wjpps.com Vol 3, Issue 6, 2014.
853
Bhavik et al. World Journal of Pharmacy and Pharmaceutical Sciences
Preparation of sublingual tablets by direct compression method[9]
All the ingredients were passed through # 80-mesh separately. Then the ingredients were
weighed and mixed in geometrical order and compressed into tablets of 120mg by direct
compression method using 6 mm flat punches on a Rotary Tablet Compression Machine. For
optimization of super disintegrant different types of disintegrants were selected like sodium
starch glycolate, crospovidone, crosscarmalose sodium and kyron T-314 in concentration 3%.
Composition of batches A1 to A4 shown in Table 01.
Table 01: Composition of batches A1 to A4
Ingredient Quantity per tablet (mg) A1 A2 A3 A4
Drug+ poloxamer407 35 35 35 35 Cross carmelose sodium 3.6 - - - Cross povidone - 3.6 - - Kyron-T314 - - 3.6 - Sodium starch glycolate - - - 3.6 Microcrystalline cellulose 53.8 53.8 53.8 53.8 Mannitol 24 24 24 24 Talc 2.4 2.4 2.4 2.4 Aerosil 1.2 1.2 1.2 1.2 Total weight 120
Evaluation of tablets
Hardness [10]
The test was done as per the standard methods. The hardness of three randomly
selectedtablets from each formulation (F1 to F15) was determined by placing each tablet
diagonallybetween the two plungers of tablet hardness tester (with the nozzle) and applying
pressureuntil the tablet broke down into two parts completely and the reading on the scale
was noteddown in Kg/cm2. The results are presented in Tables 04.
Thickness [10]
The thickness of three randomly selected tablets from each formulation was determined
inmm using a micrometre screw. The average values were calculated. The results
arepresented in Table 04.

www.wjpps.com Vol 3, Issue 6, 2014.
854
Bhavik et al. World Journal of Pharmacy and Pharmaceutical Sciences
Weight variation[10]
Weight variation test was done as per standard procedure. Twenty tablets from each
formulationwere weighed using an electronic balance and the average weight was
calculated.The results are shown in Table 04.
Friability [10]
The friability of tablets using 10 tablets as a sample was measured using a Roche
Friabilator.Tablets were rotated at 25 rpm for 4 minutes or up to 100 revolutions. The tablets
were takenout, deducted and reweighted. The percentage friability was calculated from the
loss inweight as given in equation below. The weight loss should not more than 1%.The
results areshown in Table 04.
%Friability = (initial weight – final weight) x 100 (initial weight)
Drug Content [10]
Ten randomly selected tablets from each formulation were finely powdered andpowder
equivalent to 5mg of Felodipine was accurately weighed andtransferred to 100 ml volumetric
flasks containing 50 ml of methanol. Theflasks were shaken to mix the contents thoroughly.
The volume was made up to the markwith methanol and filtered. Two ml of the filtrate was
suitably diluted andFelodipine content was estimated at 360 nm using a double beam UV-
visiblespectrophotometer. The results are presented in Tables 04.
Wetting Time [11]
The tablets wetting time was measured by a procedure modified from that reported by Bi etal.
The tablet was placed at the centre of two layers of absorbent paper fitted into a dish. Afterthe
paper was thoroughly wetted with distilled water, excess water was completely drainedout of
the dish. The time required for the water to diffuse from the wetted absorbent
paperthroughout the entire tablet was then recorded using a stopwatch. The results are
presented inTable 04.
In- vitro Disintegration Time [11]
In- vitro Disintegration times for sublingual tablets were determined using USP
tabletdisintegration apparatus with phosphate buffer of pH 6.8 as medium. The volume of
mediumwas 900 ml and temp was 37± 2 °C. The time in seconds taken for complete
disintegration ofthe tablets with no palatable mass remaining in the apparatus was measured.
The results arepresented in Table 04.

www.wjpps.com Vol 3, Issue 6, 2014.
855
Bhavik et al. World Journal of Pharmacy and Pharmaceutical Sciences
In- vitro drug release study [11]
In-vitro release rate of Felodipine sublingual tablets was carried out usingUnited State
Pharmacopoeia (USP) dissolution testing apparatus (Paddle method). Thedissolution test was
carried out using 900 ml of 6.8 pH phosphate buffer, at 37± 20‘C and 50rpm. A sample (5
ml) of the solution was withdrawn from the dissolution apparatus at 5, 10, 15, 20, 25 and 30
min. The samples were replaced with fresh dissolution medium of samequantity. The samples
were filtered through whatman filter paper No 40 and analysed forFelodipineafter appropriate
dilution by UV spectrophotometer at 364 nm. Thepercentage drug release was calculated
using an equation obtained from the calibration curve.The results are presented in Fig 07.
Selection of concentration of super disintegrant in tablet formulation
Sublingual tablets of Felodipine were prepared by direct compression. All the ingredients
were passed through # 80-mesh separately. Then the ingredients were weighed and mixed in
geometrical order and compressed into tablets of 120mg by direct compression method using
6 mm flat punches on a Rotary Tablet Compression Machine (Rimek 10 station minipress).
For selection of concentration of super disintegrantkyron T-314 in 3%, 4% and 5% were
taken. Composition of batches F1 to F3 shown in Table 02
Table 02: Formula for different concentration of superdisintegrant
Ingredient Quantity per tablet (mg)
F1 F2 F3 Drug+ poloxamer407 35 35 35 Kyron-T314 (3%) 3.6 Kyron-T314 (4%) 4.8 Kyron-T314 (5%) 6 Microcrystalline cellulose 53.8 52.6 51.4 Mannitol 24 24 24 Talc 2.4 2.4 2.4 Aerosil 1.2 1.2 1.2 Total weight 120
Stability studies of the optimized formulation
Stability testing of drug products begins as a part of drug discovery and ends with the demise
of the compound or commercial product. To assess the drug and formulation stability,
stability studies were done according to ICH guidelines Q1C. The stability studies were
carried out on the optimized formulations as per ICH guidelines Q1C. The optimized

www.wjpps.com Vol 3, Issue 6, 2014.
856
Bhavik et al. World Journal of Pharmacy and Pharmaceutical Sciences
formulation sealed in aluminum packaging and kept in humidity chamber maintained 40 ± 2
°C / 75 ± 5 % RH for 1 month. The optimized formulation sealed in aluminum foil was also
kept at room temperature and humidity condition. At the end of studies, samples were
analyzed for the % drug release and drug content.
RESULT AND DISCUSSION
Drug-excipientscompatibility study
The FTIR spectra of Felodipine showed a characteristic peaks of Felodipine appeared at 3355
cm-1(N-H stretching), 800 cm-1 (C-Cl stretching), 1500–1415 cm-1 (skeleton vibration of
aromatic C=C ring stretching), 1000-1300 cm-1 (C-O stretching).
The incompatibility between the Drug and Excipients were studied by FTIR
spectroscopy.The spectral data of pure drug and drug-excipient mixtures are presented in
‘Fig.01-02’.The results indicate that there was no chemical incompatibility between drug and
excipients used in formulation.
Figure 01: FTIR spectra of Felodipine
Figure 02: FTIR spectra of Felodipine + Excipients
www.wjpps.com Vol 3, Issue 6, 2014.
857
Bhavik et al. World Journal of Pharmacy and Pharmaceutical Sciences
Phase solubility study
Phase solubility study of Felodipine was conducted in phosphate buffer (pH 6.8) and the
result of phase solubility study are given in Table 03. The solubility of pure drug was only
19.17µg/mL.Solubility of Felodipine in phosphate buffer (pH 6.8): 20.56 µg/mL.The
solubility of Felodipine-poloxamer 407 (1:6) and Felodipine-poloxamer 407 (1:8) complex
was 410.58µg/mL and 429.41 µg/mL respectively. Above both complex there is no much
more difference in solubility so Felodipine-poloxamer-407 (1:6) was optimized.
Table 03: Solubility study of drug-carrier complex
Drug-Carrier
Complex
Method Poloxamer 407
PVP K-30
PEG 6000 Betacyclodextrin
Solubility(µg/ml) 1:2 PM 27.05 7.05 10.58 9.41
1:4 PM 103.52 9.41 12.94 14.11
1:6 PM 212.94 12.94 11.76 16.47
1:8 PM 321.17 10.58 8.23 17.64
1:2 SD 52.94 34.11 25.88 21.17
1:4 SD 195.29 47.05 42.35 24.70
1:6 SD 410.58 32.94 63.52 25.88
1:8 SD 429.41 36.47 56.47 27.05
Figure 03: Comparison of solubility study
DSC of solid dispersion
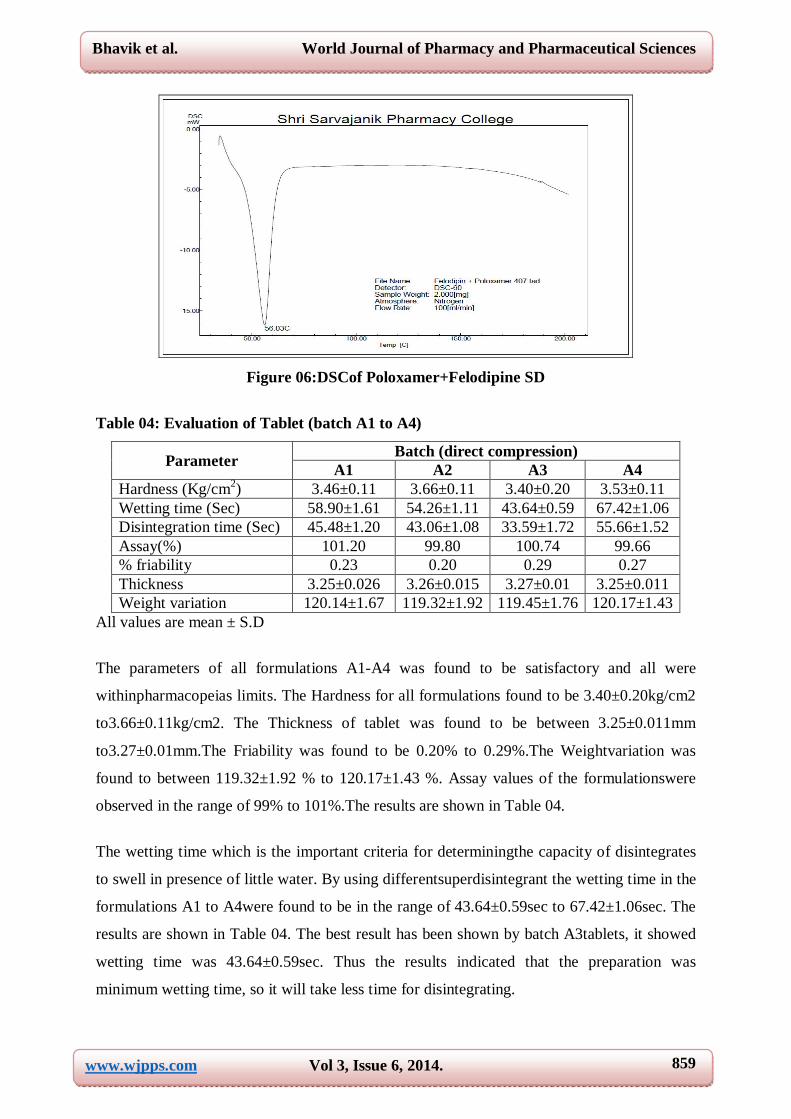
The DSC thermogram of felodipine is shown in figure 05 exhibited an sharp
endothermicpeak at 151.37°C, corresponding to its melting point, whereas nosuch peak was

www.wjpps.com Vol 3, Issue 6, 2014.
858
Bhavik et al. World Journal of Pharmacy and Pharmaceutical Sciences
observed in solid dispersion particles prepared with poloxamer-407, suggesting thatfelodipine
was molecularly dispersed and in an amorphous form. From the aboveobservation it may be
suggested that the physical state of felodipine changed from crystallineto amorphous during
the solvent evaporation process. It has been known that transforming thephysical state of the
drug to the amorphous or partially amorphous state lead to a high energystate and high
disorder, resulting in enhanced solubility and faster dissolution.
Figure 04: DSCof Poloxamer 407
Figure 05: DSCof Felodipine
www.wjpps.com Vol 3, Issue 6, 2014.
859
Bhavik et al. World Journal of Pharmacy and Pharmaceutical Sciences
Figure 06:DSCof Poloxamer+Felodipine SD
Table 04: Evaluation of Tablet (batch A1 to A4)
Parameter Batch (direct compression) A1 A2 A3 A4
Hardness (Kg/cm2) 3.46±0.11 3.66±0.11 3.40±0.20 3.53±0.11 Wetting time (Sec) 58.90±1.61 54.26±1.11 43.64±0.59 67.42±1.06 Disintegration time (Sec) 45.48±1.20 43.06±1.08 33.59±1.72 55.66±1.52 Assay(%) 101.20 99.80 100.74 99.66 % friability 0.23 0.20 0.29 0.27 Thickness 3.25±0.026 3.26±0.015 3.27±0.01 3.25±0.011 Weight variation 120.14±1.67 119.32±1.92 119.45±1.76 120.17±1.43
All values are mean ± S.D
The parameters of all formulations A1-A4 was found to be satisfactory and all were
withinpharmacopeias limits. The Hardness for all formulations found to be 3.40±0.20kg/cm2
to3.66±0.11kg/cm2. The Thickness of tablet was found to be between 3.25±0.011mm
to3.27±0.01mm.The Friability was found to be 0.20% to 0.29%.The Weightvariation was
found to between 119.32±1.92 % to 120.17±1.43 %. Assay values of the formulationswere
observed in the range of 99% to 101%.The results are shown in Table 04.
The wetting time which is the important criteria for determiningthe capacity of disintegrates
to swell in presence of little water. By using differentsuperdisintegrant the wetting time in the
formulations A1 to A4were found to be in the range of 43.64±0.59sec to 67.42±1.06sec. The
results are shown in Table 04. The best result has been shown by batch A3tablets, it showed
wetting time was 43.64±0.59sec. Thus the results indicated that the preparation was
minimum wetting time, so it will take less time for disintegrating.

www.wjpps.com Vol 3, Issue 6, 2014.
860
Bhavik et al. World Journal of Pharmacy and Pharmaceutical Sciences
The disintegration time of sublingual tablets should be less because in a very short time
itshould be totally disintegrates. By using different superdisintegrant, disintegration time in
theformulations A1 to A4 were found to be in the range of 33.59±1.72sec to 55.66±1.52sec.
Theresults are shown in Table 04.The best result has been shown by batch A3 tablets, it
showedthe disintegration time was 33.59±1.72 seconds.So, kyron T-314 selected for further
study.
Table05: Cumulative percentage release(A1 to A4)
Time (min) A1 A2 A3 A4
0 0 0 0 0
5 38.1 39.63 42.37 35.42
10 55.19 58.91 61.18 51.82
15 71.23 77.64 80.64 69.09
20 85.74 89.32 94.75 81.48
25 92.82 96.31 98.82 90.33
30 97.78 98.04 99.64 95.98
Figure 07: Cumulative percentage release
All the batches contain 3% super disintegrant. Among all batches, batch A3 showed highest
drug release as compared to other batches. Batch A3 showed above 90% drug release in 20
min. So, kyron T-314 selected for further study.

www.wjpps.com Vol 3, Issue 6, 2014.
861
Bhavik et al. World Journal of Pharmacy and Pharmaceutical Sciences
Selection of concentration of super disintegrant
Table 06: Evaluation of batch F1 to F3
Parameter F1 F2 F3 Hardness(Kg/cm2) 3.60±0.20 3.87±0.23 3.93±0.115 Wetting time(Sec) 40.56±1.08 34.79±0.52 32.43±0.67 Disintegration time (Sec) 32.62±0.43 26.94±0.59 22.86±0.79 Assay (%) 100.22 99.56 99.87 % friability 0.31 0.29 0.25 Thickness 3.26±0.015 3.27±0.020 3.25±0.011 Weight variation 120.27±1.89 119.33±1.75 120.54±1.57
All values are mean ± S.D The optimization of super disintegrant was done based on the evaluation parameterslike
hardness, disintegration time, wetting time, % friability and % assay. Batch F3 contain kyron
T-314 (5%) shown minimum disintegration time and wetting timeamong all
tabletformulation.
Table 07: Cumulative percentage release for batch F1 to F3
Time(min) F1 F2 F3 0 0 0 0 5 41.12 43.12 46.59
10 61.64 64.75 67.32 15 79.88 83.47 87.48 20 93.56 94.53 96.83 25 97.91 98.19 99.14 30 99.45 99.89 99.91
Figure 08: Cumulative percentage release for batch F1 to F3
www.wjpps.com Vol 3, Issue 6, 2014.
862
Bhavik et al. World Journal of Pharmacy and Pharmaceutical Sciences
Batch F1, F2 and F3 contain 3%, 4% and 5% super disintegrant respectively. Among all
batches, batch F3 showed highest drug release as compared to other batches. Batch F3
showed above 87.48% drug release in 15 min. So, formulation F3 was optimized.
STABILITY STUDIES OF THE OPTIMIZED FORMULATION
The stability studies were carried out on the most satisfactory formulations (Batch F3) as per
ICH guidelines Q1C. At the end of studies, samples were analyzed for the weight variation,
thickness, friability, hardness, wetting time, Disintegration time, drug content and in vitro
dissolution. The optimized formulations stored at 40 ± 2 °C / 75 ± 5 % were found stable.
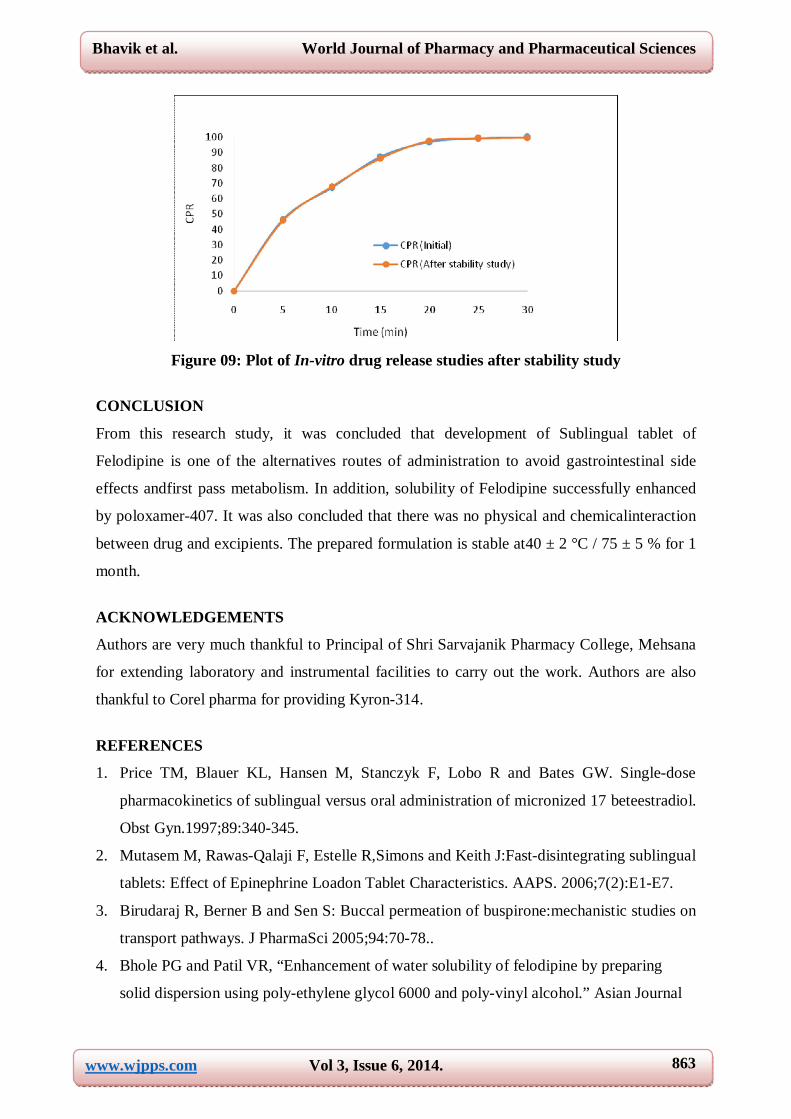
After storage at 40 ± 2 °C / 75 ± 5 %, no shape deformation in the tablets was found. Assay
of drug as well as cumulative percentage drug release was nearly similar before and after
storage. (Figure 09) So, it was clear that drug was thermally stable as well as not affected by
high humidity at 40 ± 2 °C / 75 ± 5 %.
Table 08: Evaluation data after stability study of optimized batch (F3)
Parameter F3 weight variation (mg) 119.62±1.46 Thickness 3.25±0.015 Diameter 6.14±0.010 Friability 0.27 Hardness 4.20±0.43 Wetting time (Sec) 35.56±1.52 Disintegration time (Sec) 24.66±0.57 Assay 99.83
All values are mean ± S.D
Table 09: In-vitro drug release studies after stability study
Time (min)
CPR (Initial)
CPR (After storage at
40 ± 2 °C / 75 ± 5 %) 0 0 0 5 46.59 45.66
10 67.32 68.12 15 87.48 86.30 20 96.83 97.74 25 99.14 99.06 30 99.91 99.67
www.wjpps.com Vol 3, Issue 6, 2014.
863
Bhavik et al. World Journal of Pharmacy and Pharmaceutical Sciences
Figure 09: Plot of In-vitro drug release studies after stability study
CONCLUSION
From this research study, it was concluded that development of Sublingual tablet of
Felodipine is one of the alternatives routes of administration to avoid gastrointestinal side
effects andfirst pass metabolism. In addition, solubility of Felodipine successfully enhanced
by poloxamer-407. It was also concluded that there was no physical and chemicalinteraction
between drug and excipients. The prepared formulation is stable at40 ± 2 °C / 75 ± 5 % for 1
month.
ACKNOWLEDGEMENTS
Authors are very much thankful to Principal of Shri Sarvajanik Pharmacy College, Mehsana
for extending laboratory and instrumental facilities to carry out the work. Authors are also
thankful to Corel pharma for providing Kyron-314.
REFERENCES
1. Price TM, Blauer KL, Hansen M, Stanczyk F, Lobo R and Bates GW. Single-dose
pharmacokinetics of sublingual versus oral administration of micronized 17 beteestradiol.
Obst Gyn.1997;89:340-345.
2. Mutasem M, Rawas-Qalaji F, Estelle R,Simons and Keith J:Fast-disintegrating sublingual
tablets: Effect of Epinephrine Loadon Tablet Characteristics. AAPS. 2006;7(2):E1-E7.
3. Birudaraj R, Berner B and Sen S: Buccal permeation of buspirone:mechanistic studies on
transport pathways. J PharmaSci 2005;94:70-78..
4. Bhole PG and Patil VR, “Enhancement of water solubility of felodipine by preparing
solid dispersion using poly-ethylene glycol 6000 and poly-vinyl alcohol.” Asian Journal

www.wjpps.com Vol 3, Issue 6, 2014.
864
Bhavik et al. World Journal of Pharmacy and Pharmaceutical Sciences
of Pharmaceutics 2009, 3(3), 240—244.
5. Drug Bank, “Drug card for Felodipine(DB01023)”, July 2013,
http://www.drugbank.ca/drugs/DB01023
6. Ladani AA, Dr Mandev and Patel B, “Enhancemnet of solubility and dissolution
attributes of Felodipine by complexation using Beta cyclodextrin and its derivative.”
IJPAN, 2011, 1(2), 64-70.
7. Kim EJ, Chun MK, Jang JS, Lee IH, Lee KR and Choi HK, “Preparation of a solid
dispersion of felodipine using a solvent wetting method.” European Journal of
Pharmaceutics and Biopharmaceutics.” 2006, 64, 200–205.
8. Krishnamoorthya V, Nagalingama A, Verma P and Parameshwarana S, “Characterization
of Olanzapine-Solid Dispersions.” Ira. J. of Pharma. Res. 2011, 10 (1), 13-23.
9. Bayrak Z, Tas C, Tasdemir U, Erol H, Ozkan C, Savaser A and Ozkan Y, “Formulation
of zolmitriptan sublingual tablets prepared by direct compression with different polymers:
In vitro and in vivo evaluation.” Eur. J. Pharm. Biopharm. 2011, 78(3), 499-505.
10. Sanada H, Yonezawa Y and Danjo K: Preparation and evaluation of compressed
tabletrapidly disintegrating in the oral cavity. Chem Pharm Bull. 1996; 44:2121-2127.
11. Edmund J: Preparation, characterization and scale of ketoconazole with
enhanceddissolution and bioavailability. Drug DevInd Pharm. 2007; 33:755-765.