formation of methane sulfinic acid in the gas-phase oh-radical initiated oxidation of dimethyl...
TRANSCRIPT

Formation of Methane Sulfinic Acidin the Gas-Phase OH-RadicalInitiated Oxidation of DimethylSulfoxideC E C I L I A A R S E N E , † , ‡ I A N B A R N E S , * , †
K A R L H . B E C K E R , †
W I L L I A M F . S C H N E I D E R , §
T I M O T H Y T . W A L L I N G T O N , §
N I K O L A O S M I H A L O P O U L O S , | A N DI U L I A V . P A T R O E S C U - K L O T Z #
Physikalische Chemie/FB9, Bergische Universitat-GHWuppertal, Gauss Strasse 20, D-42097 Wuppertal, Germany,Faculty of Chemistry, Department of Analytical Chemistry,“Al.I. Cuza” University of Iasi, Carol I - 11, Iasi 6600,Romania, Ford Motor Company, SRL-3083,Dearborn, Michigan 48121-2053, Department of Chemistry,University of Crete, 300 Leof. Knossou, GR-71409 Heraklion,Greece, and National Institute for Environmental Studies,Atmospheric Environment Division, 16-2 Onogawa,Tsukuba, Ibaraki 305-0053, Japan
Dimethyl sulfoxide (CH3S(O)CH3: DMSO) is an importantproduct of dimethyl sulfide (CH3SCH3: DMS) photooxidation.The mechanism of the OH-radical initiated oxidation ofDMSO is still highly uncertain and a major aim of recentstudies has been to establish if methane sulfinic acid (CH3S-(O)OH: MSIA) is a major reaction product. In the presentwork the products of the OH-radical gas-phase oxidationof dimethyl sulfoxide have been investigated in the absenceand presence of NOx. All experiments were performed ina 1080 L reaction chamber in 1000 mbar synthetic air at 284( 2 K using long-path FT-IR spectroscopy and ionchromatography to monitor and quantify reactants andreaction products. Formation of methane sulfinic acid inhigh yield (80-99%) was observed in both in the absenceand presence of NOx, and the results support that it isthe major primary reaction product. Other products observedincluded dimethyl sulfone (CH3S(O)2CH3: DMSO2), sulfurdioxide (SO2), methane sulfonic acid (CH3S(O)2OH: MSA),and methane sulfonyl peroxynitrate (CH3S(O)2OONO2:MSPN). The formation behavior of these products is inline with their source being mainly secondary productionvia oxidation of a primary product, i.e. MSIA.
IntroductionDimethyl sulfoxide (DMSO) is considered to be an im-portant intermediate in the atmospheric oxidation ofDMS. It has been observed in laboratory chamber studiesof the OH-radical initiated oxidation of DMS (1-5) and in
the marine boundary layer (6-8). Despite its importanceas a DMS oxidation product the chemical behavior ofDMSO in the atmosphere has received relatively littleattention.
The production of DMSO in the OH-radical initiatedphotooxidation of DMS is thought to involve addition of theOH-radical to the sulfur atom of DMS to form an adductwhich can either decompose back to reactants or can reactwith molecular oxygen to form DMSO. Turnipseed et al. (9),in a pulsed laser photolysis/pulsed laser-induced fluores-cence study on the reaction of OH + DMS, have reported abranching ratio of Φ ) 0.5 ( 0.15 for HO2 production fromthe DMS-OH + O2 reaction. They assumed that HO2 wasformed via H-atom abstraction from the hydroxyl group ofthe DMS-OH adduct and that the coproduct was DMSO. Incontrast, Arsene et al. (4) measured a near unit molarformation yield for DMSO in a smog chamber study underNOx-free conditions. However, in a later study Arsene et al.(5) showed that the DMSO yield is sensitive to the NOconcentration and in the presence of NO obtained yieldssimilar to those of Turnipseed et al. (9). Although the absoluteyield of DMSO under atmospheric conditions is still uncer-tain, all of the studies confirm that its yield will be quitesubstantial.
The rate constant for the reaction of OH with DMSO atroom temperature and atmospheric pressure is approxi-mately 15 times faster than that for OH with DMS: kDMSO+OH
) 9.4 × 10-11 cm3 molecule-1 s-1 (10) compared to kDMS+OH
) 6.5 × 10-12 cm3 molecule-1 s-1 (11). Since the reaction ofDMSO with the OH-radical is fast, this removal process isvery likely the dominant atmospheric sink. Physical removalof DMSO involving uptake by aerosol and cloud dropletsincluding heterogeneous reactions on that (7, 8, 12-14) mayalso be significant.
The products of the OH-radical initiated oxidation ofDMSO in the atmosphere are poorly characterized. Productsobserved in smog chamber studies include sulfur dioxide(SO2), dimethyl sulfone (DMSO2: CH3S(O)2CH3), methane-sulfonyl peroxynitrate (MSPN: CH3S(O)2OONO2), methanesulfonic acid (MSA: CH3S(O)2OH), and methane sulfinic acid(MSIA: CH3S(O)OH) (3, 15). However, the values of theproduct yields reported in the various studies differ signifi-cantly. Production of significant amounts of SO2 and lesseramounts of DMSO2 were observed by Barnes et al. (15) in along path FT-IR study. In 1996, Sørensen et al. (3) foundproduction of SO2 and DMSO2 in roughly equal amounts,and they reported a MSIA formation yield of e 0.3%. Thevery low yield of MSIA reported by Sørensen et al. (3) is inmarked disagreement with a recent absolute study ofUrbanski et al. (16) whose investigations imply a near unityield of MSIA. Urbanski et al. (16) investigated the mechanismand kinetics of the OH + DMSO reaction at 298 K using the248 nm laser flash photolysis of H2O2 in the presence of DMSOand time-resolved tunable diode laser spectroscopic for thedetection of CH3, CH4, and SO2. They obtained a ratecoefficient of kOH+DMSO ) (8.7 ( 1.6) × 10-11 cm3 molecule-1
s-1 for the OH + DMSO reaction and a CH3 yield of 0.98 (0.12 in the absence of O2. The observed unit yield of CH3 andthe near zero yields of CH4 and SO2 allowed them to concludethat the dominant OH + DMSO reaction channel was OH-radical addition to DMSO, followed by very rapid DMSO-OH adduct decomposition to CH3 and MSIA. Urbanski et al.(16) did not directly observe MSIA but they deduced, by aprocess of elimination based on a consideration of thefollowing reaction sequence, that MSIA was the mostprobable CH3 coproduct:
* Corresponding author phone: +49 202 439 2510; fax: +49 202439 2505; e-mail: [email protected].
† Bergische Universitat-GH Wuppertal.‡ “Al.I. Cuza” University of Iasi.§ Ford Motor Company.| University of Crete.# National Institute for Environmental Studies.
Environ. Sci. Technol. 2002, 36, 5155-5163
10.1021/es020035u CCC: $22.00 2002 American Chemical Society VOL. 36, NO. 23, 2002 / ENVIRONMENTAL SCIENCE & TECHNOLOGY 9 5155Published on Web 11/02/2002

Recent observations of relatively high yields of MSIA andMSA in the oxidation of DMS using ion chromatographyfrom this laboratory in combination with the results ofUrbanski et al. (16) led us to conclude that the main sourceof these compounds was most probably the result of theOH-radical induced further oxidation of the primarilyproduced DMSO product (4). To substantiate these conclu-sions a detailed study directed at detection and quantificationof MSIA and MSA formation in the OH-radical initiatedoxidation of DMSO both in the absence and in the presenceof NOx has been performed in our laboratory. We report hereresults from a product study of the reaction of OH radicalswith DMSO in a large volume photoreactor at 284 K andatmospheric pressure using both ion chromatography andlong-path in situ FT-IR spectroscopy to monitor reactantsand products. Since an infrared spectrum of MSIA is notavailable, ion chromatography in combination with cryogenicsampling was used to monitor its formation.
The experiments have been performed at the lowesttemperature possible in the reactor. Apart from increasingthe rate of reaction of OH with DMSO which will reduce thetime constant of the experiments, reduction in temperaturereduces secondary consumption of MSIA by OH radicals.Reaction of MSIA with OH is predicted to proceed via anabstraction reaction (17) the importance of which willdecrease with decreasing temperature.
Experimental SectionThe product analyses of the OH-radical initiated oxidationof DMSO both in the absence and in the presence of NOx
were performed in a 1080 L quartz glass reaction chamberequipped with 32 super actinic fluorescent lamps (PhilipsTL 05/40 W: 320 < λ < 480, λmax ) 360 nm) and 32 low-pressure mercury lamps (Philips TUV 40 W: λmax ) 254 nm).All experiments were performed at a total pressure of 1000mbar of synthetic air at 284 ( 2 K. Photolysis of H2O2 in theabsence of NOx
and photolysis of CH3ONO in the presence of NOx
were used as OH-radical sources. Mixtures of DMSO/H2O2/synthetic air and DMSO/CH3ONO/NOx/synthetic air werephotolyzed using the germicidal lamps (λmax ) 254 nm) andthe 32 super actinic fluorescent lamps (λmax ) 360 nm),respectively. Typical initial concentrations of DMSO andH2O2, were 1.0-1.5 ppm and approximately 10 ppm, re-spectively (1 ppm ) 2.45 × 1013 molecules cm-3 at 298 K and1 atm). The initial NO concentration was varied between 1and 2.0 ppm, and the NO2 concentration was 0.06-0.16 ppm.Reactants were flushed into the reactor by injection into astream of synthetic air via various inlet ports. The inlet port
for DMSO was heated to 70-100 °C to avoid deposition ofDMSO on the wall of the inlet system.
The concentration-time behavior of reactants and prod-ucts were monitored in situ using long path (492 m) FT-IRspectroscopy (Bruker IFS 88) equipped with KBr beam splitterand HgCdTe detector. Infrared spectra were recorded in the650-4000 wavelength range with a resolution of 1 cm-1.Typically 5 or 6 spectra, derived from 32 co-added inter-ferograms, were collected over irradiation periods of ap-proximately 5 min. The concentration of commerciallyavailable reactants and products were determined by com-puter-aided spectral subtraction using calibrated referencespectra generated in this laboratory. Methanesulfonyl per-oxynitrate (MSPN) is not a commercially available compound;its IR absorption coefficients have been estimated from acalibration involving in situ production in the reactionchamber using the 254 nm photolysis of methyl thiolformate(CH3SC(O)H, MTF) in synthetic air in the presence of NO2
(18). The calibration was based on the amount of MTFconsumed and the amount of SO2 formed with the differencein the sulfur being attributed to MSPN formation.
Products such as MSIA and MSA were analyzed using anOdyssey Ion Chromatography (IC) System from Alltechconsisting of a 7725/7725i rheodyne valve, a 526 HPLC pump,an ERIS 100 HP autosuppressor, and a 550 conductivitydetector. For the anion separation an Allsep hydrophilic anionexchange column (particle size 7 µm, 100 mm length × 4.6mm id) based on methacrilate with quaternary aminefunctional groups operated at ambient temperature was used.A mobile phase consisting of a solution of bicarbonate (0.24mM) and carbonate (0.3 mM) sodium salts was chosen aseluent. During the analysis the flow of the eluent wasmaintained at a rate of 1.0 mL min-1. Working standardswere prepared using MSIA and MSA sodium salts purchasedfrom Lancaster (97% purity) and Aldrich (99.5% purity),respectively, and Milli-Q water (R ) 18.5 MR cm-1). Calibra-tions of the MSIA and MSA anions were performed weekly,and every day one or two working standards were injectedfor comparison. Compounds were identified by comparisonof the retention time, tR, of peaks of the unknown samplewith the retention times of peaks in the chromatograms ofstandard solutions. Figure 1 illustrates that the heights of theIC peaks corresponding to the CH3S(O)O- and CH3S(O)2O-
anions were proportional to the concentrations of CH3S(O)O-
and CH3S(O)2O-, respectively. In the range of 0.2-12 µg mL-1
correlation coefficients better than 0.997 (for CH3S(O)O-)and 0.992 (for CH3S(O)2O-) were obtained.
The system used for sampling of MSIA and MSA from thereaction chamber consisted of a short Teflon tube attachedto two empty glass cryogenic U-tube traps connected in series,a flow controller, and a pump as indicated in Figure 2. The
CH3S(O)CH3 + OH f CH3S(O)CH2 + H2O
CH3S(O)CH3 + OH + M f CH3S(O)(OH)CH3 + M
CH3S(O)(OH)CH3 + M f CH3S(O)OH + CH3 + M
CH3S(O)CH3 + OH f [CH3S(O)(OH)CH3]* f
CH3 + CH3S(O)OH
H2O2 + hν (λ ) 254 nm) f OH + OH
CH3ONO + hν (λmax ) 360 nm) f CH3O + NO
CH3O + O2 f HCHO + HO2
HO2 + NO f OH + NO2
FIGURE 1. Calibration curves for methane sulfinate (CH3S(O)O-)and methane sulfonate (CH3S(O)2O-).
5156 9 ENVIRONMENTAL SCIENCE & TECHNOLOGY / VOL. 36, NO. 23, 2002

glass cryogenic U-tubes were immersed in an ethanol-liquidnitrogen slush bath (-112 °C). The pump was adjusted topass air from the reactor at a rate of 1 L min-1 through thecold trap and about 5 L of air were sampled. To improve theefficiency of trapping a glass sinter was inserted into eachof the sampling tubes. Due to the low temperature, anymethane sulfinate or methane sulfonate passing through thetrap condensed on the walls of the tubes. Tests with a thirdtube showed only very minor traces of the ions indicatingthat the collection efficiency with two tubes was sufficientand that breakthrough of the sample was negligible. Thevery high yields of MSIA obtained in the experiments alsosupport that the cryogenic trapping was efficient.
After the collection, 5 mL Milli-Q water was added to theU-tubes to dissolve the sampled material which was thentransferred to a small storage flask. A 200 µL aliquot of thecollected sample was injected into the IC system via aninjection loop, and the samples were normally analyzed 4 hafter collection. The detection limits for the ion chromato-graphic analysis were estimated as three times the standarddeviation of duplicate analyses of low level samples (below0.2 µg mL-1) and were about 0.012 µg mL-1 for CH3S(O)O-
and 0.014 µg mL-1 for CH3S(O)2O-.
Test experiments showed that when the samples werecollected at the end of the irradiation period the measuredyields of both MSIA and MSA were much lower than thoseobtained in experiments where the samples were collectedcontinuously during irradiation. These differences in themeasured yields are attributed to reaction of MSIA with OHradicals and other loss processes such as wall uptake (17).Since the rate coefficients for these loss processes are notknown, to obtain a better semiquantitative estimate of theyield of MSIA a continuous sampling approach was employedfor the experiments. The amounts of methane sulfinate andmethane sulfonate formed were calculated as follows using1 ppb (liquid) ) 1 µg L-1 ) 1 ng mL-1 of water: the number
of moles of gas in “X” L of sampled air at 284 K and 1 atmis
therefore the gas-phase concentration of [A]gas is given by
where MWa is the molar weight of substance A, [A]liquid is themeasured liquid-phase concentration in ppb, and W(mL) isthe amount of water used for collection. The gas-phase molaryields were then calculated against the amount of DMSconsumed over the sampling period.
ResultsMethane Sulfinic Acid Formation in the Photooxidation ofDMSO. As mentioned earlier the infrared spectrum of MSIAis not presently known, and it was, therefore, not possibleto establish the formation of this compound in the gas phaseusing long path FT-IR spectroscopy. Consequently, ionchromatography was the analytical tool used to detect andsemiquantify MSIA formation in the photooxidation ofDMSO.
Figure 3a,b shows chromatograms that demonstrate thedetection and resolution of the CH3S(O)O- and CH3S(O)2O-
anions from MSIA and MSA, sampled from (a) DMSO-H2O2-synthetic air and (b) DMSO-CH3ONO-NOx-synthetic airoxidation systems, respectively. The identity of a third peakappearing in both chromatograms is not certain. Calibrationexperiments show that it contains the SO4
2- anion fromH2SO4. However, tests of the column and detector used inthe ion chromatographic analysis showed that the systemalso gives a response to a possible impurity in DMSO which
FIGURE 2. Experimental setup used for sampling MSIA and MSA from the reaction chamber.
n ) PVRT
)1(atm) × “X”(L)
0.082 × 284(K)) “n” mol
[A]gas )[A]liquid × W
MWa × n× 109 ppb
VOL. 36, NO. 23, 2002 / ENVIRONMENTAL SCIENCE & TECHNOLOGY 9 5157

appears as a peak with almost the same retention time (tR)as that for SO4
2-. Tests of the system response includedinjection of (a) the eluent (no signal), (b) a sample collectedfrom pure synthetic air (no signal), (c) a sample from a DMSO/synthetic air mixture (appearance of a peak with tR ) 9.65min), (d) a sample from a DMSO/H2O2/synthetic air mixture(appearance of a peak with tR ) 9.71 min), (e) a standardsolution of CH3S(O)O- (peak with tR ) 6.23 min), (f) a standardsolution of CH3S(O)2O- (peak with tR ) 7.05 min), (g) astandard solution of SO4
2- (peak with tR ) 9.72 min), and (h)a water solution of pure DMSO (appearance of a peak withtR ) 9.69 min). The chromatograms from these tests are showncollectively in Figure 4. Since the sulfate anion peak is overlaidwith a peak associated with DMSO, it was not possible todetermine the sulfate anion yield in the experiments.
A possible problem in the quantification of MSIA andMSA with the sampling method and ion chromatographicanalysis used here is formation of these compounds in liquidphase and/or heterogeneous reactions between H2O2 andDMSO on the surface of the sampling lines and U-tube trap.In the dark it is known that H2O2 can oxidize DMSO to DMSO2
on surfaces, and if the tubing is exposed to light OH radicalscould be formed which can oxidize DMSO to MSIA andsubsequently MSA. However, investigations of the H2O2
initiated oxidation of DMSO in the liquid phase at temper-atures between 4 and 30 °C (19, 20) suggest that ourmeasurements are not affected by contributions from liquid-phase chemistry. In these studies a slow rate constant of 5× 10-6 M-1 s-1 was measured for the reaction between DMSOand H2O2 at 4 °C. Since in the present study the sampleswere collected in an ethanol-liquid nitrogen bath (-112 °C)and the time scale for the analysis, i.e. addition of Milli-Qwater for dissolving and transferring the sampled materialto the IC, was short, oxidation of any sampled DMSO withH2O2 in liquid phase is expected to be minimal. This is furthersupported by the good agreement with the results obtainedusing the photolysis of CH3ONO as the OH radical source(see below).
The yields of CH3S(O)O- and CH3S(O)2O- anions frommeasurements performed at 284 K using the samplescollected from the irradiation of DMSO/H2O2/synthetic airand DMSO/CH3ONO/NOx/synthetic air mixtures are pre-sented in Tables 1 and 2, respectively. Since the reactionsystem was probed continuously, the values in Tables 1 and2 do not represent the true formation yields of the acidicspecies; they are integral yield values for the time period ofthe experiment incorporating both production and lossprocesses.
The results presented in both Tables 1 and 2 consist infact of two sets of experiments: (i) experiments where thequantification of methane sulfinic and methane sulfonic acidcould be affected by photooxidation of DMSO in the liquid
phase (sampling system exposed to room light) and (ii)experiments where the quantification of both acids wasperformed under dark conditions (i.e. the sampling systemwas wrapped in aluminum foil to shield it from light). Asseen in Tables 1 and 2 the measured yields of MSIA are veryhigh (84-99%) for both the unshielded and shielded samplingtechniques. Measurements made with the dark samplingsystem give MSIA yields (84-89%) which are slightly lowerthan those observed in the unshielded experiments (∼99%).Obviously exposure of the sampling system to room lightwould appear to enhance the MSIA yield probably viaphotoinduced aqueous phase OH radical oxidation of DMSOand highlights the need to protect sampling systems for MSIAagainst the effects of photo activation. Considering the
FIGURE 3. Chromatograms demonstrating the ion chromatographicmeasurements of (1) MSIA, (2) MSA, and (3) a peak comprising ofSO4
2- from H2SO4 and an unidentified contribution from DMSO fromsamples collected during irradiation of (a) a DMSO/H2O2/syntheticair mixture and (b) a DMSO/CH3ONO/NOx/synthetic air. Note thegood resolution between the MSIA and MSA peaks.
FIGURE 4. Example of chromatograms obtained by injection of (a)the eluent, (b) a sample collected with only synthetic air in thereaction chamber, (c) a sample collected with a DMSO/syntheticair mixture in the chamber, (d) a sample collected with a DMSO/H2O2/synthetic air mixture in the chamber, (e) a standard solutionof CH3S(O)O-, (f) a standard solution of CH3S(O)2O-, (g) a standardsolution of SO4
2-, and (h) a sample of pure DMSO.
TABLE 1: Ion Chromatographic Analysis of CH3S(O)O- andCH3S(O)2O- Anions from Samples Collected during theOxidation of DMSO in 1000 mbar Synthetic Air at 284 K Usingthe Photolysis of H2O2 as the OH Radical Source
CH3S(O)O- CH3S(O)2O-
sampling system expt no. vol (L) (ppm) (% S) (ppm) (% S)
exposed to hν 1 4.52 0.63 99.9 0.14 22.52 4.71 0.53 98.7 0.12 22.23 4.95 0.58 99.5 0.14 23.8
dark 4 4.05 0.41 86.9 0.05 10.45 4.81 0.45 84.5 0.08 14.26 4.82 0.46 89.2 0.04 7.8
5158 9 ENVIRONMENTAL SCIENCE & TECHNOLOGY / VOL. 36, NO. 23, 2002

semiquantitative nature of the MSIA determination thisartifact in the yield is relatively small and does not affect themajor conclusions from the study.
Gas-Phase DMSO Oxidation Products. The sulfur-containing products observed and quantified in the gas phasein the irradiation of DMSO/H2O2/synthetic air reactionmixtures, by long path FT-IR spectroscopy, included SO2,DMSO2, and MSA. In the irradiation of DMSO/CH3ONO/NOx/synthetic air reaction mixtures, besides the sulfur-containing products just listed, production of MSPN wasalso observed.
Formation yields of SO2, DMSO2, MSA, and MSPN havebeen obtained from plots of their concentration versus theamount of DMSO consumed. Since the irradiation periodsemployed in the experiments were short, corrections forsecondary loss processes of the products, i.e. loss to the wallsor reaction with OH-radicals, were insignificant and wereneglected in the correction of the product concentrations.Correction was, however, made for the wall loss of DMSOusing a first-order loss rate of kwall ) 7 × 10-5 s-1 for 284 K,obtained in dark experiments. This correction only accountsfor approximately 5% of the DMSO decay.
Figure 5a-d shows examples of plots of the DMSO2 (a),SO2 (b), MSA (c), and MSPN (d) concentrations versus theconsumption of DMSO at 284 K in the absence and in thepresence of NOx. After an induction period these plots weregenerally fairly linear, and the yields listed in Tables 3 and4 for experiments performed with and without NOx, respec-tively, have been derived from the slopes of the linear sections.As will be discussed below, since we believe that thecompounds are formed in secondary reactions the yields ascalculated relative to the decay of DMSO do not have muchphysical significant. However, previous product yield de-terminations have also been based on DMSO decay, and thepresent data set helps to highlight the reason for the largedisparity between reported literature yields.
DiscussionYields of Gas-Phase Products. The shapes of the yield-timeprofiles for SO2, DMSO2, and MSA in all of these experimentsshow that these compounds are not main primary productsof the oxidation of DMSO. There are pronounced delaysobserved in the formation of these species in the gas phase,which are not in line with primary production pathways.This behavior is clearly visible in the plots of the productconcentrations versus the consumption of DMSO shown inFigure 5a-d. The delay in production is much morepronounced for the NOx-free experiments. This is probablyreasonable since with NO in the system any peroxide radicalsformed will be rapidly converted to alkoxy radicals (RO2 +NO f RO + NO2) which will react further to form the observedproducts. In the absence of NO the products will be formedby the cross reactions of peroxy radicals which are generallymuch slower than reactions of peroxy radicals with NO.
The yields of the stable secondary products in the systemas calculated relative to the decay of DMSO are very low at
284 K with SO2, DMSO2, MSA, and MSPN accounting for only∼15% S in the presence of NOx and SO2, DMSO2 and MSAaccounting for ∼12% S in the absence of NOx. The resultsreported here represent part of a more extensive series ofexperiments on the oxidation products from the OH radicalinitiated oxidation of DMSO at different temperatures andNOx concentrations (21). In the other studies the yields ofthese compounds were also low.
For methane sulfonic acid yield information was obtainedboth from long-path FT-IR spectroscopy and IC. The yieldsof MSA obtained using IC are much higher than thoseobtained using long-path FT-IR spectroscopy (Tables 1-4).This is not surprising since the yields of MSA obtained usingIC are composite values representing the sum of contributionsfrom gas-phase MSA, particulate phase MSA and possiblecontributions from the oxidation of MSIA to MSA, whereasthe FT-IR values represent pure gas-phase MSA yields. Theformation of MSA in the gas-phase oxidation of DMSOobserved in the present study is a clear indication that thiscompound, in contrast to previous mechanistic thinking thatthe abstraction channel is the principal formation channel(17, 22), can also be formed via the addition channel of theOH-radical initiated oxidation of DMS.
DMSO2 could be formed from oxidation of DMSO throughaddition of OH to DMSO to form an adduct which subse-quently reacts with O2:
However, the delay in the observation of the formationof DMSO2 suggests this pathway is not operative. Theformation of DMSO2 was found to be sensitive to the amountof NO, and in experiments with high NO (21), which are notpresented here, the formation of DMSO2 could be nearlysuppressed. We suspect that reactions of peroxy radicals withDMSO are the probable source of DMSO2 in the presentexperiments. Finally, since DMSO2, SO2, and MSA are formedin secondary reactions, it is not surprising that there arelarge differences in the reported literature yields.
Yield of MSIA. In the present study high and similar yieldsof MSIA have been observed in the gas-phase OH-radicalinitiated oxidation of DMSO using two very different OH-radical sources. It was also observed that the measured MSIAyields did not depend strongly on whether the Teflonsampling line was exposed to room light. The present resultsstrongly support the suggestion from the absolute study ofUrbanski et al. (16) that MSIA is the major primary productof the oxidation of DMSO by OH-radicals, through theaddition channel:
This mechanism is in contradiction with the experimentalobservations of Sørensen et al. (3). In the study performedby Sørensen et al. (3) a MSIA yield of 1.5 ( 1.1% was measuredfrom the oxidation of DMS by OH radicals, while in the OH-radical initiated oxidation of DMSO they obtained a MSIAyield smaller than 0.3%. Urbanski et al. (16) concluded intheir study that observation of a relatively high yield of MSIAin the oxidation of DMS in the study of Sørensen et al. (3),but not in the oxidation of DMSO, was incompatible withtheir observations. We believe that the explanation for theapparent discrepancy between the results of Urbanski et al.(16) and our work with those of Sørensen et al. (3) lies in theexperimental conditions and sampling method employedby these authors. In the study of Sørensen et al. (3) the
TABLE 2: Ion Chromatographic Analysis of CH3S(O)O- andCH3S(O)2O- Anions from Samples Collected during theOxidation of DMSO in 1000 mbar Synthetic Air at 284 K Usingthe Photolysis of CH3ONO in the Presence of Different InitialConcentrations of NOx as the OH Radical Source
CH3S(O)O- CH3S(O)2O-
sampling systemexptno.
vol(L)
NO(ppb)
NO2(ppb) (ppm) (% S) (ppm) (% S)
exposed to hν 7 4.56 961 61 0.43 92.2 0.08 16.18 4.82 2046 156 0.88 99.2 0.19 22.1
dark 9 5.36 932 85 0.32 79.9 0.05 11.610 4.81 1771 123 0.33 84.4 0.06 15.5
OH + CH3S(O)CH3 f (CH3)2S(O)(OH)
(CH3)2S(O)(OH) + O2 f CH3S(O)2CH3 + HO2
CH3S(O)CH3 + OH f (CH3)2S(O)(OH)98thermal decomposition
CH3S(O)OH + CH3
VOL. 36, NO. 23, 2002 / ENVIRONMENTAL SCIENCE & TECHNOLOGY 9 5159

experiments were performed at a temperature of 295 ( 3 Kcompared with 284 ( 2 K employed during our measurementsand the samples were collected at the end of the irradiationperiod and not during the irradiation as in the present study.It is expected that MSIA will be very susceptible to bothphysical removal, i.e. wall/aerosol loss processes, and fastfurther oxidation with OH-radicals via H-atom abstraction(17). Therefore, continuous cryogenic sampling at lowreaction chamber temperatures will be much more efficientin trapping the highly reactive MSIA than measurementsperformed at room temperature and long irradiation times
with filter sample collection at the end of the experiment.Although quantification of the MSIA yield in the reaction
is difficult and is only semiquantitative in this study, theresults suggest a near unit yield. If an abstraction channelwas operative in the reaction of OH with DMSO, it wouldlead to production of CH3S(O)CH2OO radicals:
The further reaction of the CH3S(O)CH2OO radicals wouldbe expected to lead to CH3SO radicals and eventually SO2
which would appear as a primary product:
Since the experimental observations give no indicationfor primary formation of SO2, this channel is considered tobe of minor significance.
The coproduct of MSIA is the CH3 radical. In the systemusing methyl nitrite (CH3ONO) as the OH radical source CH3
radicals are also formed so no useful mechanistic informa-tion can be obtained from this system on the fate of CH3.However, in the experiments using the photolysis of H2O2 asthe OH source consideration of the fate of these radicalscan give some useful mechanistic information. In the absenceof NO the CH3 radicals will initially add O2 to form methylperoxy radicals. In the initial stages of the reaction theseradicals will essentially undergo self-reaction with a minorcontribution from reaction with HO2 radicals formed fromOH + H2O2 and OH + HCHO. The self-reaction of CH3OOproceeds via two channels (23, 24):
FIGURE 5. Plots of the DMSO2, SO2, MSA, MSPN concentration versus the consumption of DMSO at 284 K and 1000 mbar of syntheticair in the absence and presence of NOx.
TABLE 3: Yields of the Gas-Phase Products Observed in theOH-Radical Initiated Oxidation of DMSO in 1000 mbarSynthetic Air at 284 K in the Absence of NOx
a
product yields (% molar)
expt no. DMSO2 SO2 MSA total sulfur
1 3.35 4.55 4.78 12.72 2.95 4.01 4.95 11.93 3.19 3.89 4.01 11.14 3.65 4.13 4.78 12.35 3.51 3.95 4.95 11.76 3.23 4.25 4.01 11.2
a The yields are calculated relative to the decay of DMSO.
TABLE 4: Molar Yields of the Gas-Phase Products Observed inthe OH-Radical Initiated Oxidation of DMSO in 1000 mbarSynthetic Air at 284 K in the Presence of NOx
a
NOx (ppb) product yields (% molar)
expt no. NO NO2 DMSO2 SO2 MSA MSPN total sulfur
7 961 61 3.36 3.67 5.21 2.25 14.88 2046 156 3.15 3.48 4.75 2.36 14.19 932 85 3.04 4.12 5.36 2.17 15.8
10 1771 123 2.86 3.87 5.11 1.99 14.8a The yields are calculated relative to the decay of DMSO.
CH3S(O)CH3 + OH f CH3S(O)CH2 + H2O
CH3S(O)CH2 + O2 + M f CH3S(O)CH2OO + M
CH3S(O)CH2OO + NO f CH3S(O)CH2O + NO2
CH3S(O)CH2OO + RO2 f CH3S(O)CH2O + RO + O2
CH3S(O)CH2O + ∆ f CH3S(O) + HCHO
CH3S(O) + O2/NO f f SO2
5160 9 ENVIRONMENTAL SCIENCE & TECHNOLOGY / VOL. 36, NO. 23, 2002
Both HCHO and CH3OH are observed as primary productsin experiments on OH + DMSO using H2O2 as the OH-radicalsource. The molar yield of CH3OH is 5.39% which is close tothat expected from the self-reaction of CH3OO radicals. Theobserved formation of HCHO and CH3OH is consistent withthe reaction of OH with DMSO leading to MSIA and CH3
radicals.Residual Infrared Spectrum. Although MSIA is probably
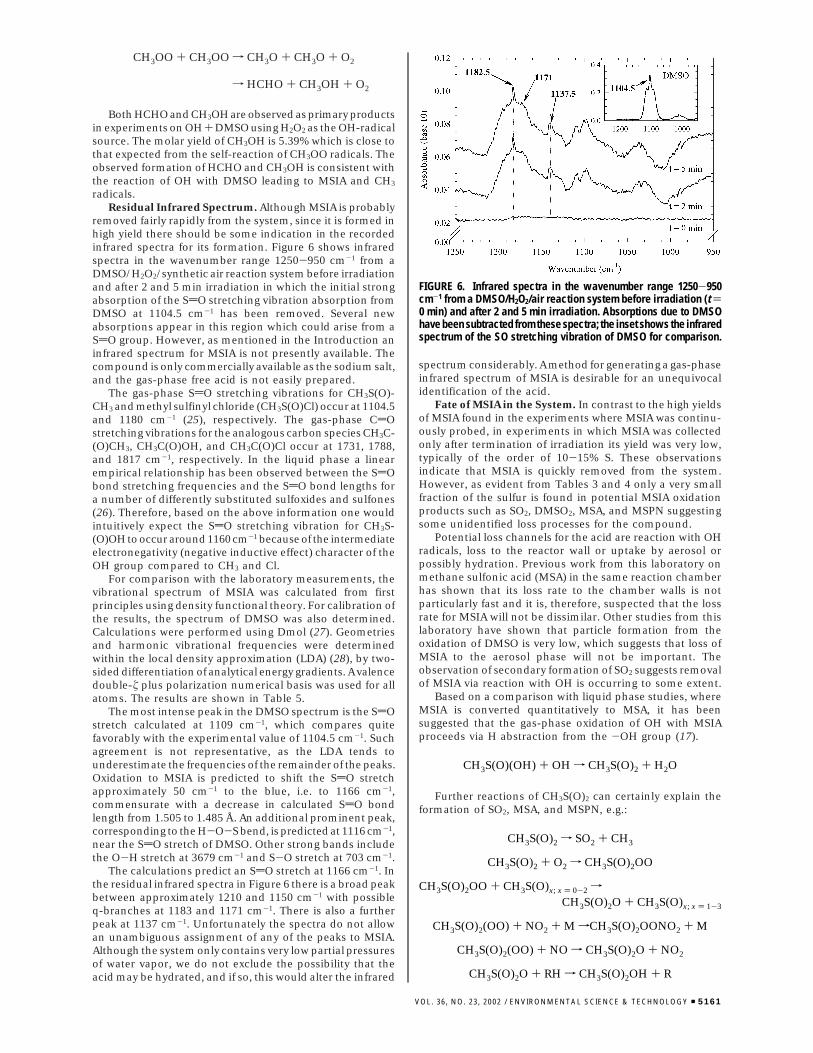
removed fairly rapidly from the system, since it is formed inhigh yield there should be some indication in the recordedinfrared spectra for its formation. Figure 6 shows infraredspectra in the wavenumber range 1250-950 cm-1 from aDMSO/H2O2/synthetic air reaction system before irradiationand after 2 and 5 min irradiation in which the initial strongabsorption of the SdO stretching vibration absorption fromDMSO at 1104.5 cm-1 has been removed. Several newabsorptions appear in this region which could arise from aSdO group. However, as mentioned in the Introduction aninfrared spectrum for MSIA is not presently available. Thecompound is only commercially available as the sodium salt,and the gas-phase free acid is not easily prepared.
The gas-phase SdO stretching vibrations for CH3S(O)-CH3 and methyl sulfinyl chloride (CH3S(O)Cl) occur at 1104.5and 1180 cm-1 (25), respectively. The gas-phase CdOstretching vibrations for the analogous carbon species CH3C-(O)CH3, CH3C(O)OH, and CH3C(O)Cl occur at 1731, 1788,and 1817 cm-1, respectively. In the liquid phase a linearempirical relationship has been observed between the SdObond stretching frequencies and the SdO bond lengths fora number of differently substituted sulfoxides and sulfones(26). Therefore, based on the above information one wouldintuitively expect the SdO stretching vibration for CH3S-(O)OH to occur around 1160 cm-1 because of the intermediateelectronegativity (negative inductive effect) character of theOH group compared to CH3 and Cl.
For comparison with the laboratory measurements, thevibrational spectrum of MSIA was calculated from firstprinciples using density functional theory. For calibration ofthe results, the spectrum of DMSO was also determined.Calculations were performed using Dmol (27). Geometriesand harmonic vibrational frequencies were determinedwithin the local density approximation (LDA) (28), by two-sided differentiation of analytical energy gradients. A valencedouble-ú plus polarization numerical basis was used for allatoms. The results are shown in Table 5.
The most intense peak in the DMSO spectrum is the SdOstretch calculated at 1109 cm-1, which compares quitefavorably with the experimental value of 1104.5 cm-1. Suchagreement is not representative, as the LDA tends tounderestimate the frequencies of the remainder of the peaks.Oxidation to MSIA is predicted to shift the SdO stretchapproximately 50 cm-1 to the blue, i.e. to 1166 cm-1,commensurate with a decrease in calculated SdO bondlength from 1.505 to 1.485 Å. An additional prominent peak,corresponding to the H-O-S bend, is predicted at 1116 cm-1,near the SdO stretch of DMSO. Other strong bands includethe O-H stretch at 3679 cm-1 and S-O stretch at 703 cm-1.
The calculations predict an SdO stretch at 1166 cm-1. Inthe residual infrared spectra in Figure 6 there is a broad peakbetween approximately 1210 and 1150 cm-1 with possibleq-branches at 1183 and 1171 cm-1. There is also a furtherpeak at 1137 cm-1. Unfortunately the spectra do not allowan unambiguous assignment of any of the peaks to MSIA.Although the system only contains very low partial pressuresof water vapor, we do not exclude the possibility that theacid may be hydrated, and if so, this would alter the infrared
spectrum considerably. A method for generating a gas-phaseinfrared spectrum of MSIA is desirable for an unequivocalidentification of the acid.
Fate of MSIA in the System. In contrast to the high yieldsof MSIA found in the experiments where MSIA was continu-ously probed, in experiments in which MSIA was collectedonly after termination of irradiation its yield was very low,typically of the order of 10-15% S. These observationsindicate that MSIA is quickly removed from the system.However, as evident from Tables 3 and 4 only a very smallfraction of the sulfur is found in potential MSIA oxidationproducts such as SO2, DMSO2, MSA, and MSPN suggestingsome unidentified loss processes for the compound.
Potential loss channels for the acid are reaction with OHradicals, loss to the reactor wall or uptake by aerosol orpossibly hydration. Previous work from this laboratory onmethane sulfonic acid (MSA) in the same reaction chamberhas shown that its loss rate to the chamber walls is notparticularly fast and it is, therefore, suspected that the lossrate for MSIA will not be dissimilar. Other studies from thislaboratory have shown that particle formation from theoxidation of DMSO is very low, which suggests that loss ofMSIA to the aerosol phase will not be important. Theobservation of secondary formation of SO2 suggests removalof MSIA via reaction with OH is occurring to some extent.
Based on a comparison with liquid phase studies, whereMSIA is converted quantitatively to MSA, it has beensuggested that the gas-phase oxidation of OH with MSIAproceeds via H abstraction from the -OH group (17).
Further reactions of CH3S(O)2 can certainly explain theformation of SO2, MSA, and MSPN, e.g.:
CH3OO + CH3OO f CH3O + CH3O + O2
f HCHO + CH3OH + O2
FIGURE 6. Infrared spectra in the wavenumber range 1250-950cm-1 from a DMSO/H2O2/air reaction system before irradiation (t )0 min) and after 2 and 5 min irradiation. Absorptions due to DMSOhave been subtracted from these spectra; the inset shows the infraredspectrum of the SO stretching vibration of DMSO for comparison.
CH3S(O)(OH) + OH f CH3S(O)2 + H2O
CH3S(O)2 f SO2 + CH3
CH3S(O)2 + O2 f CH3S(O)2OO
CH3S(O)2OO + CH3S(O)x; x ) 0-2 f
CH3S(O)2O + CH3S(O)x; x ) 1-3
CH3S(O)2(OO) + NO2 + M fCH3S(O)2OONO2 + M
CH3S(O)2(OO) + NO f CH3S(O)2O + NO2
CH3S(O)2O + RH f CH3S(O)2OH + R
VOL. 36, NO. 23, 2002 / ENVIRONMENTAL SCIENCE & TECHNOLOGY 9 5161

However, based on the large loss of MSIA and the knownchemistry of the CH3S(O)2 radical very high yields of SO2
would be expected (9, 17). Therefore, if the OH-radical is themajor oxidant the low observed SO2 yields suggest that analternative reaction channel leading to products other thanthose observed must be operative. Other possible reactionchannels include the following:
An addition channel opens up another possible route toformation of MSA. The reaction of CH3 would lead toformation of more formaldehyde in the system and in theabsence of NO also CH3OH. The further reactions of‚S(O)(OH)2 and ‚SO2OH would probably lead eventually toformation of H2SO4 or other sulfur oxy acids. A reaction ofMSIA with other radicals in the photooxidative system isanother possibility which cannot be presently excluded.
The results from the present study have firmly establishedMSIA as an important DMS secondary oxidation product viathe further oxidation of DMSO the chemistry of which hasseveral interesting implications for the atmospheric sulfurcycle. The question whether the gas-phase oxidation of DMSOin the atmosphere produces MSIA or DMSO2 is importantsince MSIA can readily oxidize to MSA, whereas DMSO2
oxidation is not expected to produce MSA. DMSO2 has oftenbeen assumed to be the main product in atmospheric models.This study shows quite convincingly that MSIA is thedominant product. There are several possible fates for MSIAin the atmosphere. MSIA is a low volatility acidic compoundand can be readily taken up by existing aerosols and dropletswhere it is now known that it will be quickly oxidized to MSA
(20). On the other hand, the gas-phase reaction of OH radicalswith MSIA has been predicted to be rapid (17), and this hasnow been confirmed experimentally (29). The experimentsindicate that SO2, a precursor of cloud condensation nuclei(CCN), is the major product. Therefore, the atmospheric fateof MSIA may well be a balance between the relativecompetitiveness between uptake and gas-phase oxidationby OH. Since in the marine atmosphere where DMS occursthe OH levels are generally low uptake will probably oftendominate. The fate of MSIA from the gas-phase oxidation ofDMSO will influence the atmospheric methanesulfonate(MS-)/nss-SO4
2- ratio and may partly explain the largevariation observed in this ratio in field experiments (22, 30).Obviously only further investigations on the chemistry ofMSIA will be able to unambiguously establish its majoratmospheric fate.
AcknowledgmentsFinancial support for this work by the European Commissionwithin the EL CID project and the BMBF within the AFSproject is gratefully acknowledged.
Literature Cited(1) Barnes, I.; Becker, K. H.; Patroescu, I. The tropospheric oxidation
of dimethyl sulfide. A new source of carbonyl sulfide. Geophys.Res. Lett. 1994, 21, 2389-2392.
(2) Barnes, I.; Becker, K. H.; Patroescu, I. FTIR products study ofthe initiated oxidation of dimethyl sulfide. Observation ofcarbonyl sulfide and dimethyl sulfoxide. Atmos. Environ. 1996,30, 1805-1814.
(3) Sørensen, S.; Falbe-Hansen, H.; Mangoni, M.; Hjorth, J.; JensenN. R. Observation of DMSO and CH3S(O)OH from the gas-phasereaction between DMS and OH. J. Atmos. Chem. 1996, 24, 299-315.
(4) Arsene, C.; Barnes, I.; Becker, K. H. FT-IR product study of thephotooxidation of dimethyl sulfide: Temperature and O2 partialpressure dependence. Phys. Chem. Chem. Phys. 1999, 1, 5463-5470.
(5) Arsene, C.; Barnes, I.; Becker, K. H.; Mocanu; R. FT-IR productstudy in the photooxidation of dimethyl sulfide in the presenceof NOx - temperature dependence. Atmos. Environ. 2001, 35,3769-3780.
TABLE 5: Calculated LDA Vibrational Spectra (cm-1) of DMSO and MSIA, Relative Intensities (Scaled to 100 Maximum), andApproximate Assignments of Selected Bands
DMSO MSIA
νobserved νcalculated intensities assignment νcalculated intensities assignment
183 0 CH3 twist 172 1 CH3 twist233 0 CH3 twist 274 38 HOS twist281 0 CSC bend 323 16301 5 336 20354 3 423 11650 7 C-S sym str 685 12 C-S str672 14 C-S asym str 703 84 S-O str844 5 904 2 CH3 rock
927 879 9 909 7 CH3 rock908 3 CH3 rock 1116 73 HOS bend
1008 982 13 CH3 rock 1166 100 SdO str1104 1109 100 SdO str 1248 20 CH3 s def
1233 9 CH3 s def 1365 6 CH3 d def1314 1256 24 CH3 s def 1374 8 CH3 d def1419 1354 11 CH3 d def 2981 1 CH3 sym str
1374 4 CH3 d def 3105 0 CH3 asym str1377 0 CH3 d def 3112 0 CH3 asym str
1444 1400 23 CH3 d def 3679 80 O-H str2933 2965 4 CH3 sym str2933 2985 4 CH3 sym str3001 3082 1 CH3 asym str3001 3083 2 CH3 asym str
3093 0 CH3 asym str3093 0 CH3 asym str
CH3S(O)(OH) + OH + M f {CH3S(O)(OH)2} +
M 98O2
CH2S(O)2(OH) + HO2 + M
CH3S(O)(OH) + OH + M f {CH3S(O)(OH)2} +M f CH3 + ‚S(O)(OH)2 + M
f ‚CH2S(O)(OH) + H2O + M
f CH4 + ‚SO2OH
5162 9 ENVIRONMENTAL SCIENCE & TECHNOLOGY / VOL. 36, NO. 23, 2002

(6) Bandy, A. R.; Thornton, D. C.; Blomquist, B. W.; Chen, S.; Wade,T. P.; Ianni, J. C.; Mitchell, G. M.; Nadler, W. Chemistry ofdimethyl sulfide in the equatorial Pacific atmosphere. Geophys.Res. Lett. 1996, 23, 741-744.
(7) Berresheim, H.; Huey, J. W.; Thorn, R. P.; Eisele, F. L.; Tanner,D. J.; Jefferson, A. Measurements of dimethyl sulfide, dimethylsulfoxide, dimethyl sulfone, and aerosol ions at Palmer Station,Antarctica. J. Geophys. Res. 1998, 103, 1629-1637.
(8) Sciare, J.; Baboukas, E.; Hancy, R.; Mihalopoulos, N.; Nguyen,B. C. Seasonal variation of dimethyl sulfoxide in rainwater atAmsterdam Island at the Southern Indian Ocean: Implicationson the biogenic sulfur cycle. J. Atmos. Chem. 1998, 30, 229-240.
(9) Turnipseed, A. A.; Barone, S. B.; Ravishankara, A. R. Reactionof OH with dimethyl sulfide. 2. Products and mechanisms. J.Phys. Chem. 1996, 100, 14703-14713.
(10) Hynes, A. J. and Wine, P. H. The atmospheric chemistry ofdimethyl sulfoxide (DMSO). Kinetics and mechanism of theOH + DMSO reaction. J. Atmos. Chem. 1996, 24, 23-37.
(11) Hynes, A. J.; Wine, P. H.; Semmes, D. H.Kinetics and mechanismof OH reactions with organic sulfides. J. Phys. Chem. 1986, 90,4148-4156.
(12) De Bruyn, W. J.; Shorter, J. A.; Davidovits, P.; Worsnop, D. R.;Zahniser, M. S.; Kolb, C. E. Uptake of gas-phase sulfur speciesmethane sulfonic acid, dimethyl sulfoxide and dimethyl sulfoneby aqueous surfaces. J. Geophys. Res. 1994, 99, 16927-16932.
(13) Davis, D.; Chen, G.; Kasibhatla, P.; Jefferson, A.; Tanner, D.;Eisele, F.; Lenschow, D.; Neff, W.; Berresheim, H. DMS oxidationin the Antarctic marine boundary layer: Comparison of modelsimulations and field observations of DMS, DMSO, DMSO2,H2SO4(g), MSA(g), and MSA(p). J. Geophys. Res. 1998, 103, 1657-1678.
(14) Davis, D.; Chen, G.; Bandy, A.; Thornton, D.; Eisele, F.; Mauldin,L.; Tanner, D.; Lenschow, D.; Fuelberg, H.; Huebert, B.; Heath,J.; Clarke, A.; Blake, D. Dimethyl sulfide oxidation in theEquatorial Pacific: Comparison of model simulations with fieldobservations for DMS, SO2, H2SO4(g), MSA(g), MS and NSS. J.Geophys. Res. 1999, 104, 5765-5784.
(15) Barnes, I.; Bastian, V.; Becker, K. H.; Martin, D. Fourier TransformIR studies of the reactions of dimethyl sulfoxide with OH, NO3,and Cl radicals. In Biogenic sulfur in the environment; Saltzman,E. S., Cooper, W. J., Eds.; ACS Symposium Series 393, Wash-ington, DC, 1989; pp 476-488.
(16) Urbanski, S. P.; Stickel, R. E.; Wine, P. H.. Mechanistic and kineticstudy of the gas-phase reaction of hydroxyl radical with dimethylsulfoxide. J. Phys. Chem. A 1998, 102, 10522-10529.
(17) Yin, F.; Grosjean, D.; Seinfeld, J. H. Photooxidation of dimethylsulfide and dimethyl disulfide. I. Mechanism development. J.Atmos. Chem. 1990, 11, 309-364.
(18) Patroescu, I. Reactions of Organic Sulphur Compounds in theAtmosphere, Ph.D. Thesis, University of Bucharest, Departmentof Analytical Chemistry, Romania. Becker, K. H.; Patroescu, I.Reaktionen von organischen Schwefelverbindungen in der At-mospare; Report No. 35; Bergische Universitat Wuppertal, FB9,Department of Physical Chemistry: Wuppertal, Germany, 1996.
(19) DOMAC. Dimethyl Sulfide (DMS): Oxidation Mechanism inRelation to Aerosols and Climate; DOMAC final SCA projectreport; European Commission Joint Research Center, Environ-ment Institute, EUR 19569 EN, 2000.
(20) Bardouki, H.; Barcellos da Rosa, M.; Mihalopoulos, N.; Palm,W.-U.; Zetzsch, C. Kinetics and mechanism of the oxidation ifdimethyl sulfoxide (DMSO) and methanesulphinate (MSI-) byOH radicals in aqueous medium. Atmos. Environ. 2002, 36,4627-4634.
(21) Arsene, C.; Barnes, I.; Becker, K. H.; Mocanu, R. Product Studieson the photooxidation of dimethyl sulfoxide (DMSO: CH3S(O)-CH3) in CMD; Report Lausanne; 2000; pp 17-20.
(22) Berresheim, H.; Wine, P. H.; Davis, D. D. Sulfur in theatmosphere. In Composition, Chemistry, and Climate of theAtmosphere; Singh, H. B., Ed., Van Nostrand Reinhold: ThomsonPublishing Inc.: 1995; pp 251-307.
(23) Atkinson, R.; Baulch, D. L.; Cox, R. A.; Hampson, R. F., Jr.; Kerr,J. A.; Rossi, M. J.; Troe, J. Evaluated kinetic, photochemical andheterogeneous data for atmospheric chemistry: supplementV, IUPAC subcommittee on gas kinetic data evaluation foratmospheric chemistry. J. Phys. Chem. Ref. Data 1997, 26, 521-1011.
(24) DeMore, W. B.; Sander, S. P.; Golden, D. M.; Hampson, R. F.;Kurylo, M. J.; Howard, C. J.; Ravishankara, A. R.; Kolb, C. E.;Molina, M. J. Chemical kinetics and photochemical data for usein stratospheric modeling; Evaluation number 12; JPL Publication97-4; 1997.
(25) Patroescu, I. V.; Barnes, I.; Becker, K. H. FTIR Kinetic andmechanistic study of the atmospheric chemistry of methylthiolformate. J. Phys. Chem. 1996, 100, 17207-17217.
(26) Hargittai, I. The Structure of Volatile Sulphur Compounds;Akademiai Kiado: Budapest, 1985.
(27) Delley, B. An all-electronic numerical method for solving thelocal density functional for polyatomic molecules. J. Chem. Phys.1990, 92 508-517.
(28) Vosko, S. H.; Wilk, L.; Nusair, M. Accurate spin-dependent liquidcorrelation energies for local spin density calculations: a criticalanalysis. Can J. Phys. 1980, 58, 1200-1211.
(29) EL CID, Evaluation of the Climatic Impact of Dimethyl Sulphide,Contract number: EVK2-CT-1999-00033, 2nd Year Report to theEuropean Commission, Brussels, 2002 (http://www.physchem.uni-wuppertal.de).
(30) Davis, D.; Chen, G.; Bandy, A.; Thornton, D..; Eisele, F.; Mauldin,L.; Tanner, D.; Lenschow, D.; Fuelberg, H.; Huebert, B.; Heath,J.; Clarke, A.; Blake, D. Dimethyl sulfide oxidation in theequatorial Pacific: Comparison of model simulations with fieldobservations for DMS, SO2, H2SO4(g), MSA(g), MS, and NSS. J.Geophys. Res. 1999, 105, 5765-5784.
Received for review February 18, 2002. Revised manuscriptreceived July 31, 2002. Accepted August 5, 2002.
ES020035U
VOL. 36, NO. 23, 2002 / ENVIRONMENTAL SCIENCE & TECHNOLOGY 9 5163