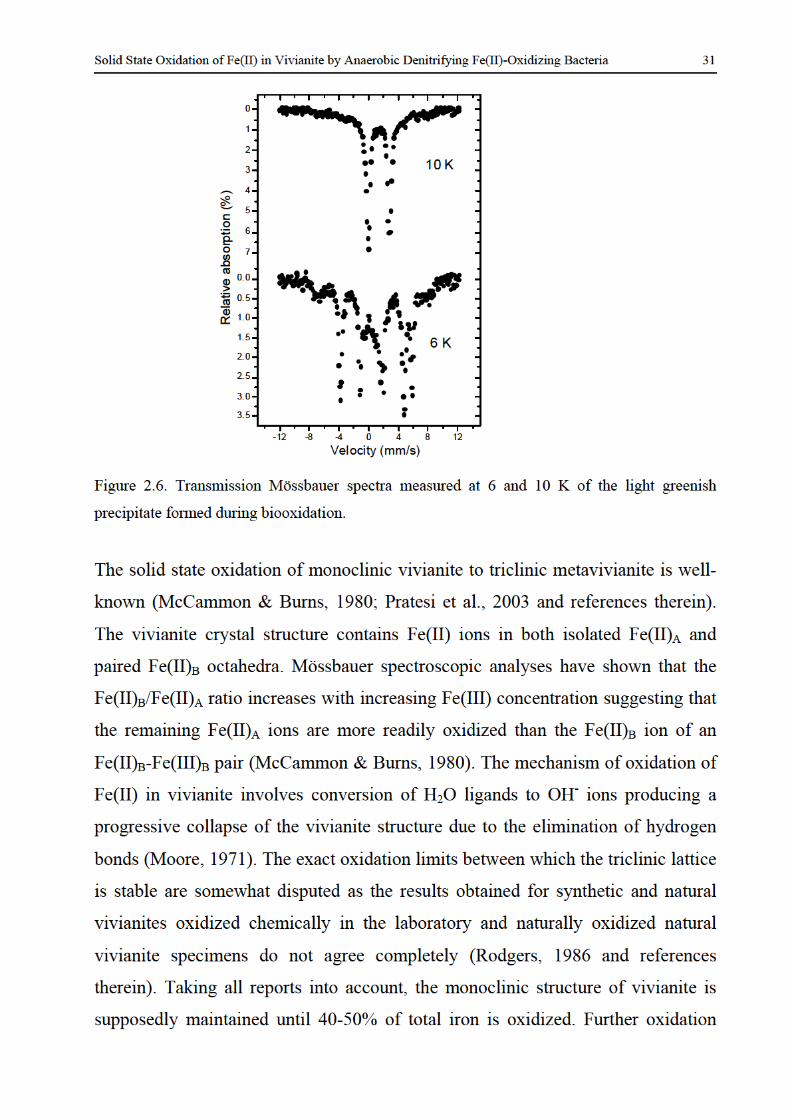
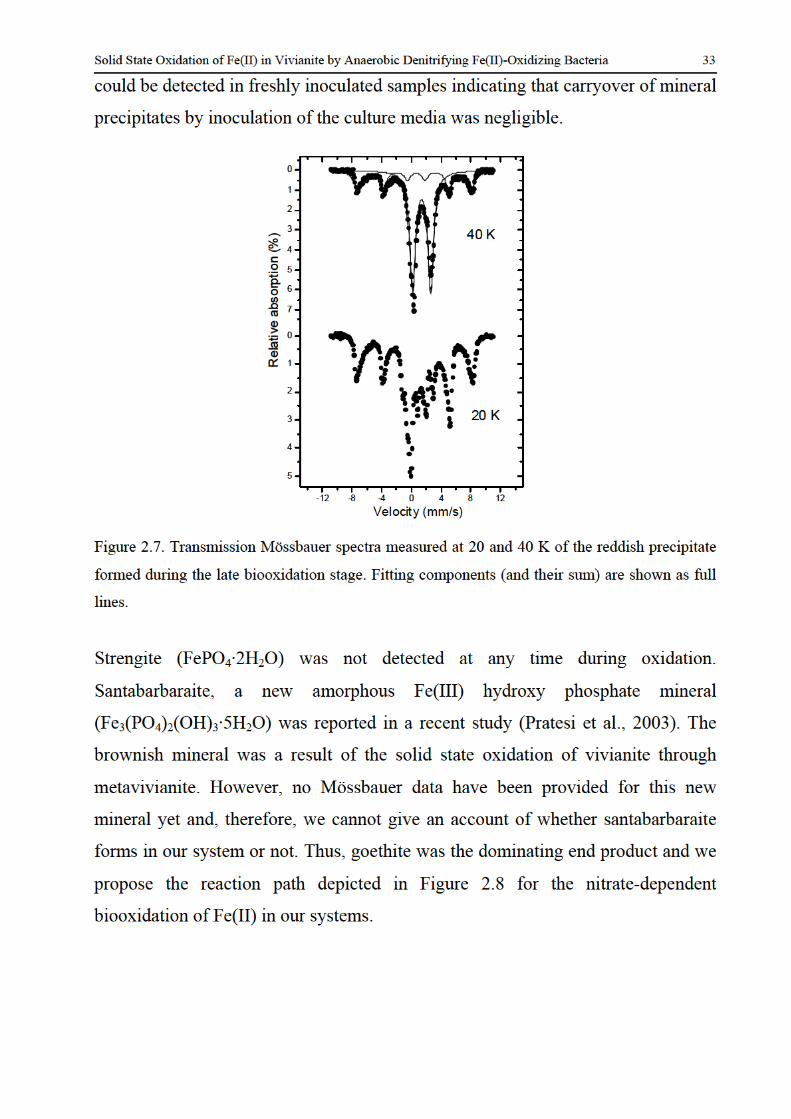
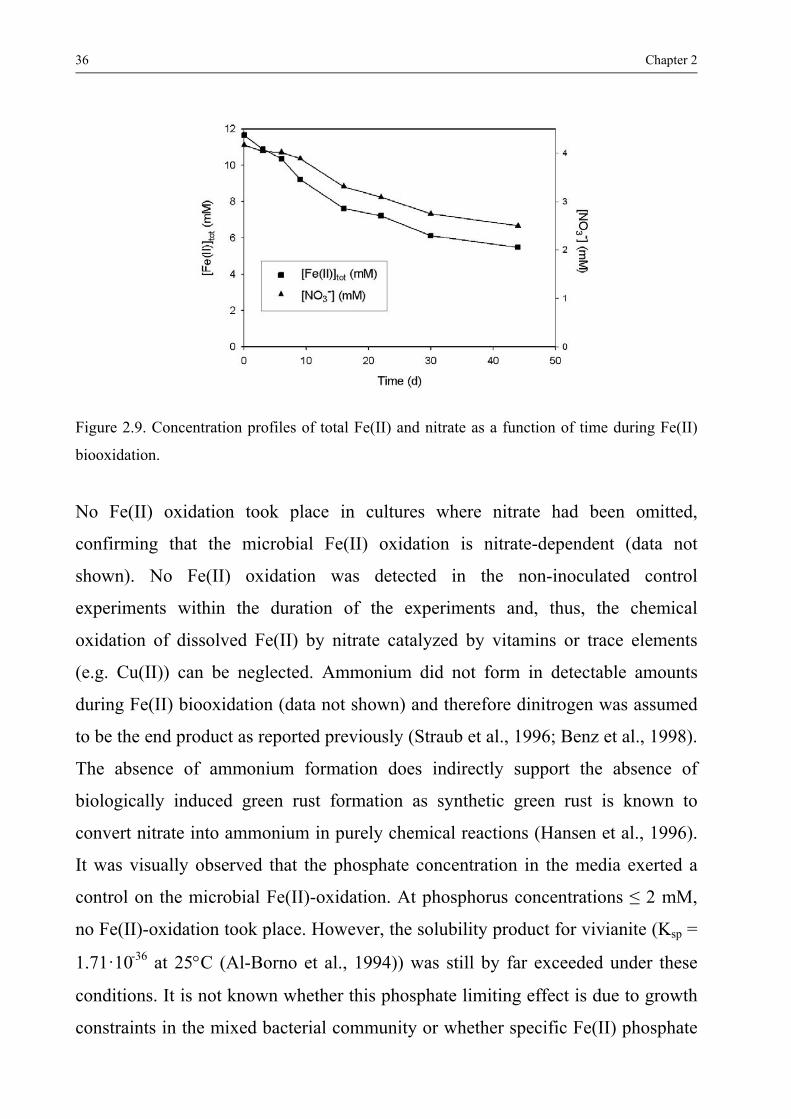
formation and redox reactions of green rusts under
TRANSCRIPT

Diss ETH No 15492
Formation and Redox Reactions of Green Rusts under Geochemical Conditions found
in Natural Soils and Sediments
A dissertation submitted to the
SWISS FEDERAL INSTITUTE OF TECHNOLOGY
for the degree of
DOCTOR OF NATURAL SCIENCES
presented by
MARIANNE ERBS
MSc in environmental chemistry
born January 13 1973
in Haderslev Denmark
Accepted on recommendation of
Prof Dr Rene P Schwarzenbach examiner
Prof Dr Stefan B Haderlein co-examiner
Prof Dr Hans CB Hansen co-examiner
Zuumlrich 2004
In fond memory of my mother
Esther Kristine Erbs (1949-2002)
who taught me how to be strong feel joy and bear compassion
I dedicate this work to her Without her support care and love
I would never have been the person I am today
To dare is to lose ones footing momentarily Not to dare is to lose oneself
Soslashren Kierkegaard
Acknowledgements
I would like to thank Stefan Haderlein Hans Christian B Hansen and Rene
Schwarzenbach for their supervision of this work Without the encouragement and
confidence of HCB Hansen and former colleagues at the Royal Veterinary and
Agricultural University in Copenhagen I would never have pursued a PhD and
without the understanding of Rene Schwarzenbach after the tragic death of my
mother I would not have had the time necessary to finish it
I thank Christian Bender Koch Hanne Nancke-Krogh Susanne Guldberg and
Henrik T Andersen (Royal Veterinary and Agricultural University Denmark) for
their valuable contribution to my work I would also like to express my gratitude to
former and present members of the Contaminant Hydrology Group from whom I
have received many benefits I mourn the loss of Denis Mavrocordatos (EAWAG)
who provided technical assistance in the electron microscopy lab and I will always
keep the sunny hours in his company in fond memory Finally I would like to
thank Kristina Straub and Bernhard Schink (University of Constance Germany)
who welcomed me in their lab for a week and taught me how to work with strict
anaerobic bacteria
I gratefully acknowledge the grant which I received from the Danish Research
Agency
Table of Contents
Table of Contents Zusammenfassung I Summary V 1 General Introduction 1 11 Iron cycling in the subsurface 1 12 Green rusts 3 13 Microbial formation of green rusts 7 14 Redox reactions of green rusts 8 15 Outline of the thesis 10 References 11 2 Solid State Oxidation of Vivianite by Anaerobic
Denitrifying Fe(II)-Oxidizing Bacteria 17 Abstract 17 21 Introduction 17 22 Materials and methods 22
221 Microorganisms and media 22 222 Characterisation of precipitates 23 223 Biooxidation experiments 24 224 Analytical methods 25
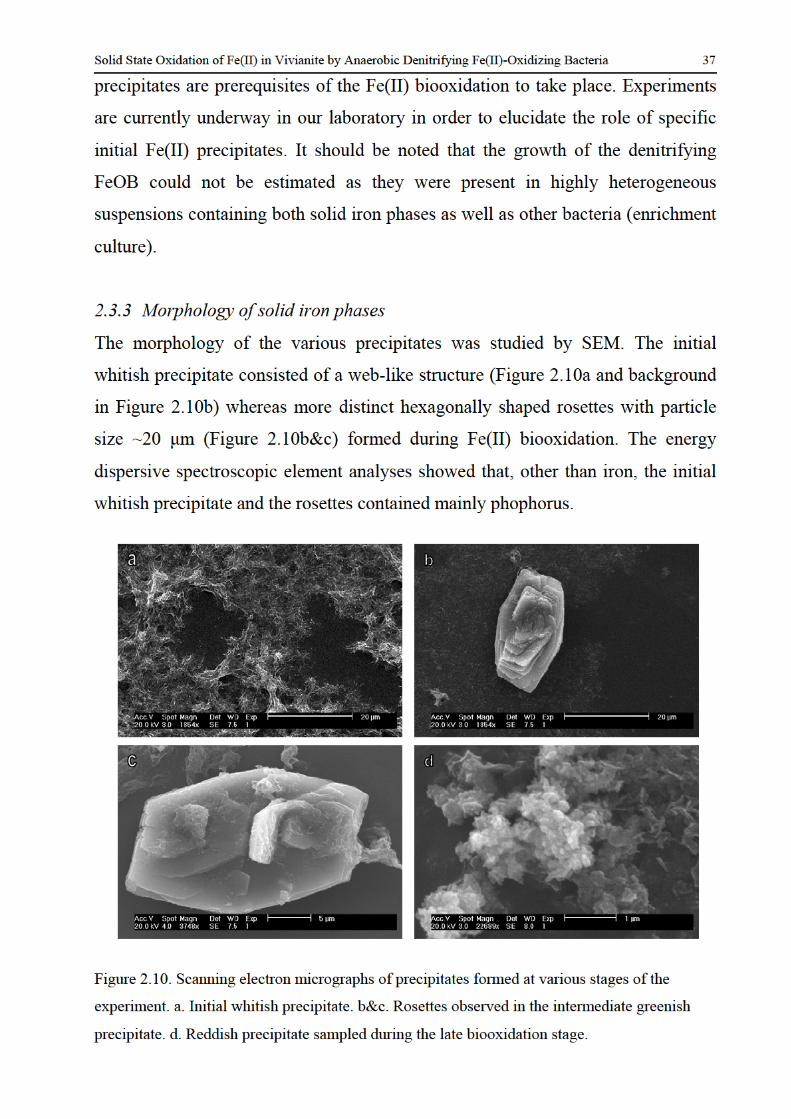
23 Results and discussion 25 231 Identification of solid iron-containing phases 25 232 Factors controlling the rate and extent of Fe(II) biooxidation 34 233 Morphology of solid iron phases 37
24 Conclusions 38 References 39 3 Formation of Layered Iron Hydroxides by
Microbial Fe(III) Reduction 43 Abstract 43 31 Introduction 44 32 Materials and methods 47
321 Preparation of iron oxide coatings 47 322 Mineral characterisation 48 323 Culture conditions and cell preparation 48 324 Bioreduction experiments 49
Table of Contents
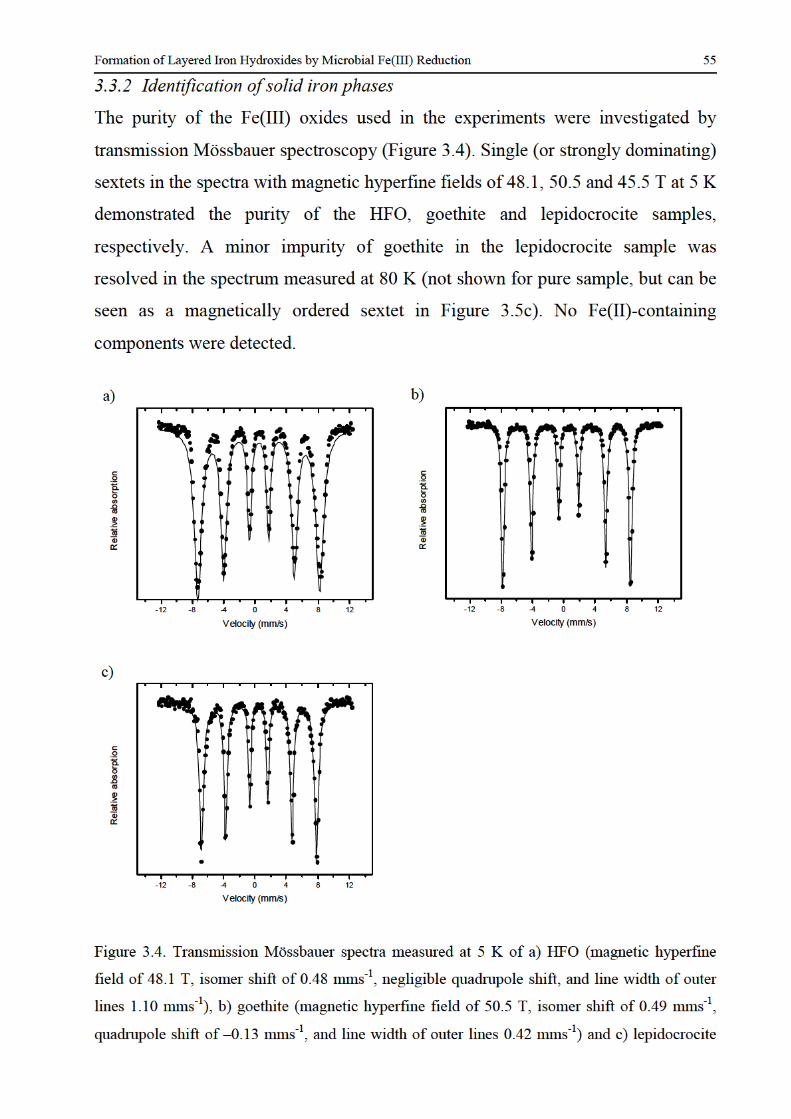
325 Analytical methods 50 33 Results and discussion 50
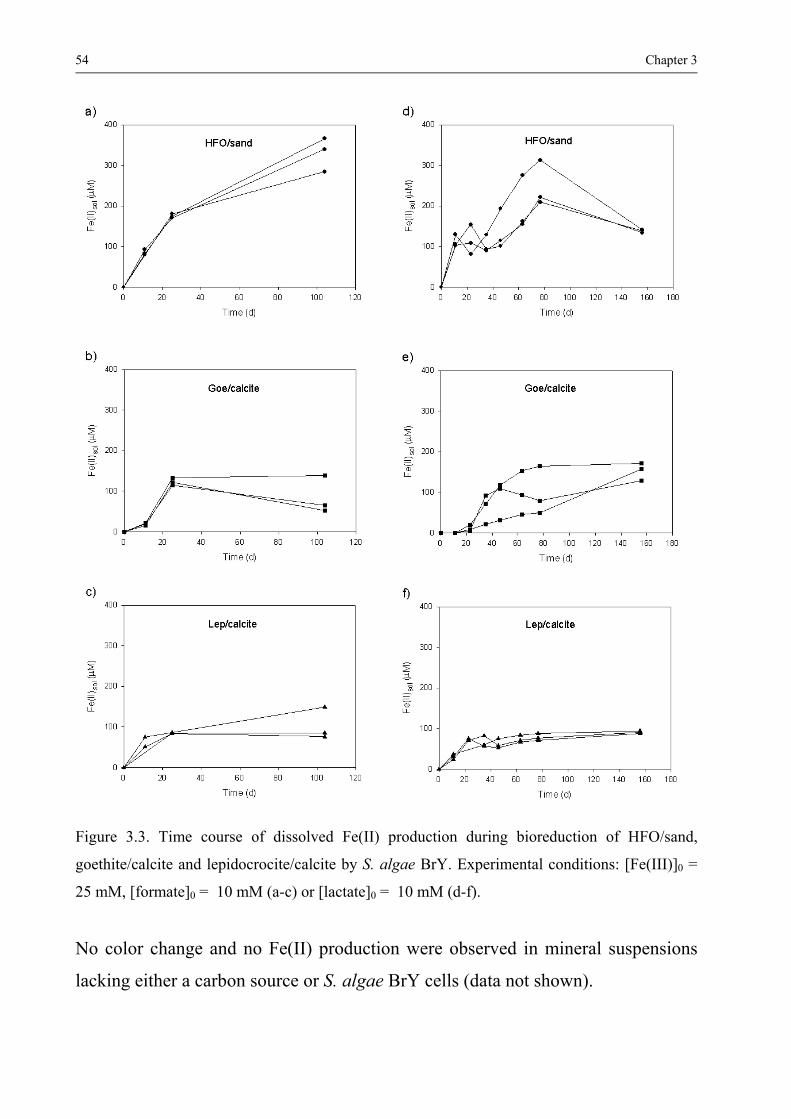
331 Fe(II) production and suspension colour changes 50 332 Identification of solid iron phases 55 333 Factors controlling the identity of the secondary iron minerals 58 334 Factors controlling the rate and extent of Fe(III) bioreduction 59
34 Conclusions 60 References 61 4 Reduction of Nitroaromatic Probe Compounds by Sulphate
Green Rust The Effect of Probe Compound Charge 65 Abstract 65 41 Introduction 66 42 Materials and methods 71
421 Synthesis of GR-SO4 71 422 Mineral characterisation 72 423 Lyophilization and determination of specific surface area 72 424 Estimation of the one-electron reduction potential for 4-NPA 73 425 Kinetic experiments 74 426 Analytical methods 74
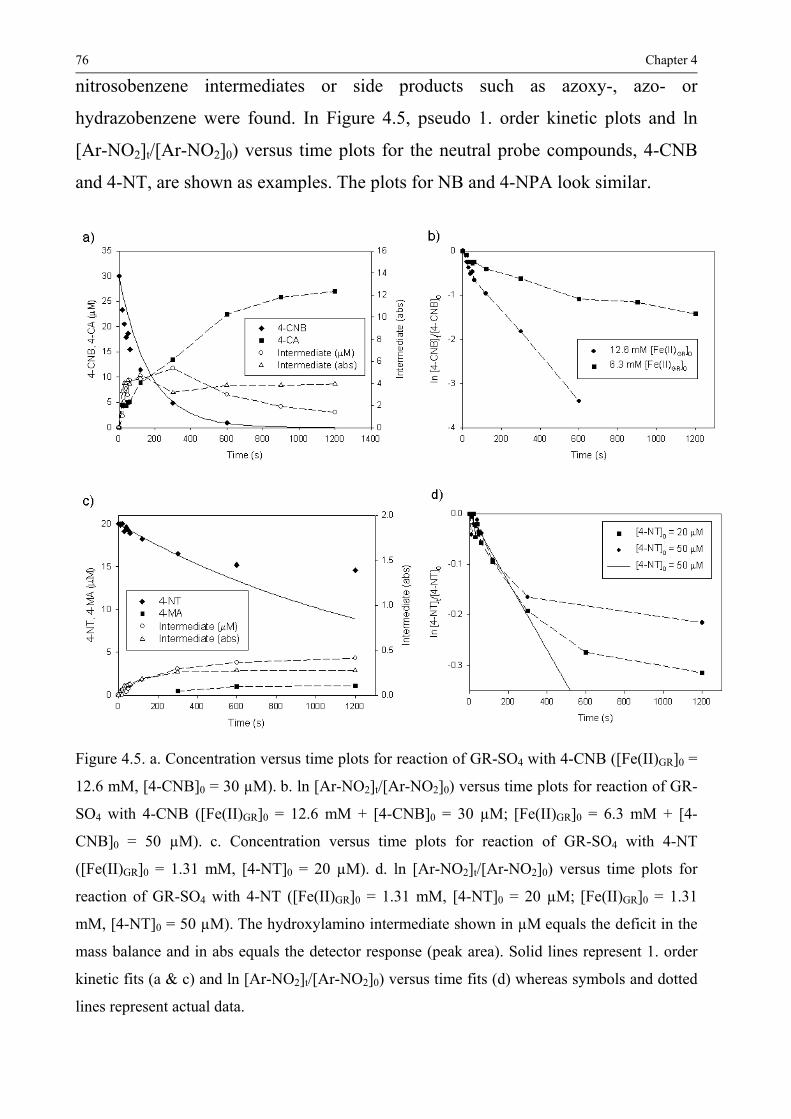
43 Results and discussion 75 431 Product formation and reaction kinetics 75 432 Comparison of rate constants for the different NACs 79 433 Factors influencing the reaction rate 82 434 Comparison with rate constants obtained for other Fe(II) containing
mineral systems 83 435 Depletion of reactive sites 85 436 The role of external and internal reactive sites 86
44 Conclusions 89 References 91 5 Reductive Transformation of Trichloroacetate in Abiotic
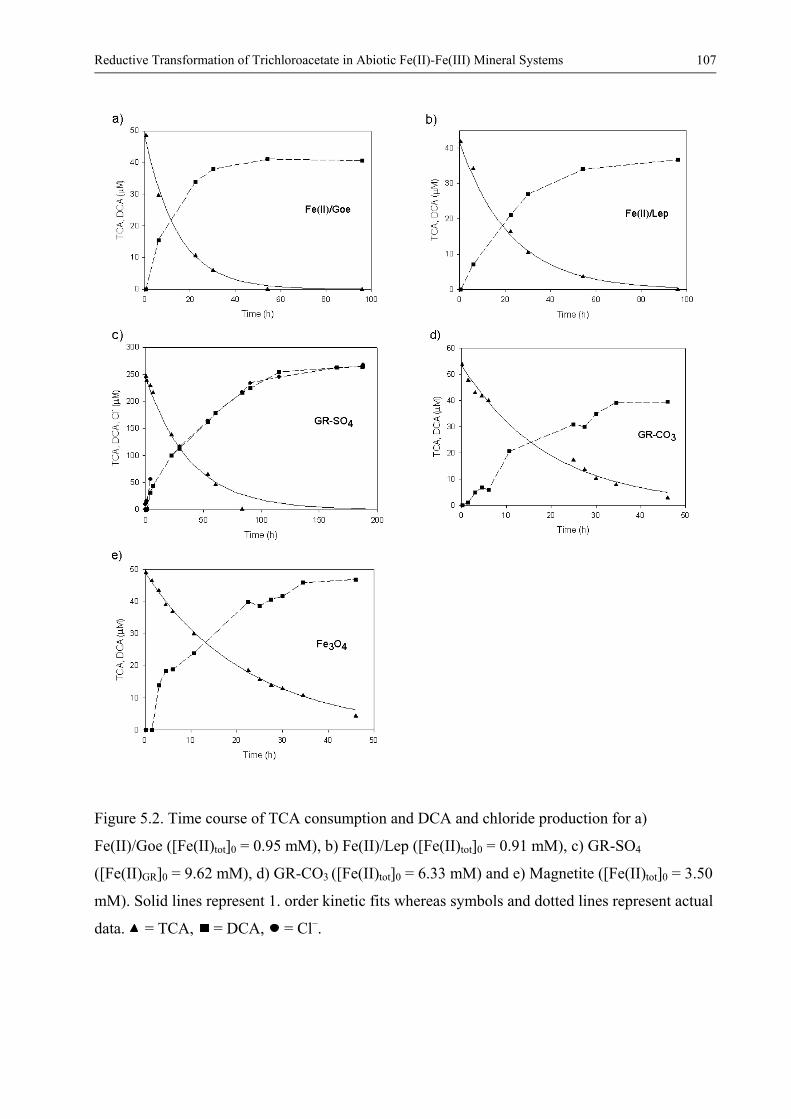
Fe(II)-Fe(III) Mineral Systems 97 Abstract 97 51 Introduction 98 52 Materials and methods 101
521 Synthesis of GRs and magnetite 102 522 Preparation of iron oxide coatings 102 523 Mineral characterisation 103 524 Kinetic experiments 103
Table of Contents
525 Analytical methods 104 53 Results and discussion 105
531 Product formation and reaction kinetics 105 532 Comparing rate constants obtained for the various Fe(II)-Fe(III)
mineral systems 109 533 Comparing with rate constants obtained for other chlorinated
aliphatic compound 112 534 Factors controlling the reactivity of surface-bound Fe(II) 114 535 Comparison with biotic and other abiotic systems 118
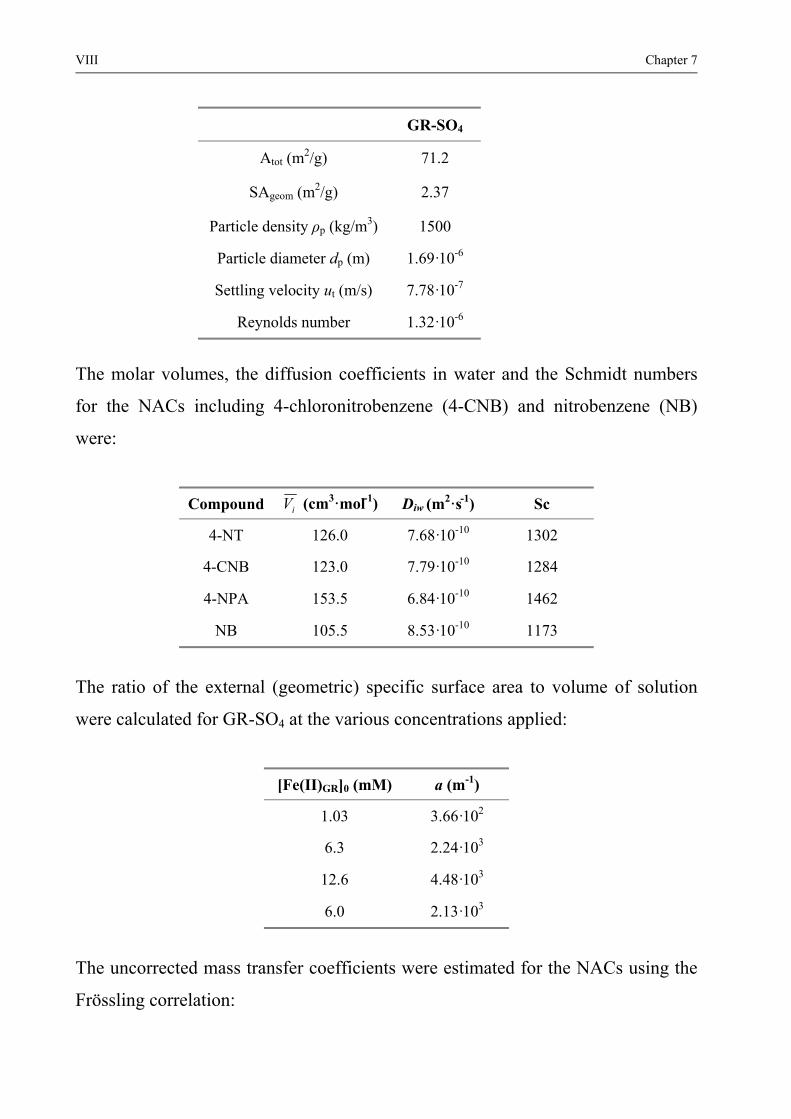
54 Conclusions 119 References 120 6 Conclusions and Outlook 125 References 128 7 Supporting Information I 71 Estimation of the one-electron reduction potential for 4-NPA I 72 The rate-limiting step IV 721 Mass transfer (diffusion) limited kinetics V
722 Surface saturation limited kinetics IX 73 External surface area of GR-SO4 and GR-CO3 XI 74 Van der Waals radii XIV 75 Adsorption of Fe(II) onto Fe(III) oxides XVI References XVIII Curriculum Vitae
Zusammenfassung I
Zusammenfassung Geschichtete Fe(II)-Fe(III)-Hydroxide (Gruumlner Rost) gehoumlren zur Gruppe der
Fe(II)-haltigen Mineralsysteme (zB Magnetit (Fe3O4) Siderit (FeCO3) Vivianit
(Fe2(PO4)2sdot8H2O) Fe(II)-Sulfide sowie an die Oberflaumlche von Fe(III)-Oxiden und
Tonmineralien gebundenes zweiwertiges Eisen) die die Aktivitaumlt von Fe(II) in
suboxischen und anoxischen Boumlden und Sedimenten kontrollieren Gruumlner Rost
Phasen (GRs) bestehen aus planaren positiv geladenen trioktaedrischen Fe(II)-
Fe(III)-Hydroxidschichten die durch hydratisierte Anionen in den
Zwischenschichten ausgeglichen werden Ihre generelle Zusammensetzung ist
[FeII(6-x)FeIII
x(OH)12]x+[(A)xnmiddotyH2O]x- wobei x = 09 - 42 ist A entspricht einem n-
valenten Anion (zB CO32- Clndash oder SO4
2-) und y repraumlsentiert die Anzahl
Wassermolekuumlle in der Zwischenschicht GRs sind wichtige intermediaumlre Phasen
die durch unvollstaumlndige Oxidation von Fe(II) oder teilweise Reduktion von Fe(III)
gebildet werden koumlnnen Sie koumlnnen in suboxischen nicht-sauren eisenhaltigen
natuumlrlichen wie auch technischen Systemen auftreten so wie in Wasser gesaumlttigten
Boumlden und interstitiellen Sedimenten Rohrleitungen in der
Trinkwasserversorgung Stahlpfosten in marinen Sedimenten Stahlbeton und in
reaktiven durchlaumlssigen Waumlnden aus nullwertigem Eisen zur in-situ Sanierung von
Altlasten und Aquiferen Aufgrund ihrer Schichtstruktur den anionischen
Zwischenschichten und der hohen spezifischen Oberflaumlchen sind GRs reaktive
Ionentauscher und Sorbentien von Anionen Des Weiteren wurde gezeigt dass
GRs eine Reihe anorganischer und organischer Schadstoffe reduzieren koumlnnen
Durch Immobilisierung und Transformation koumlnnen GRs somit eine wichtige Rolle
fuumlr das Abbauverhalten und den Transport solcher Schadstoffe in suboxischen
Boumlden und Sedimenten spielen Die Resultate dieser Dissertation tragen zum
Verstaumlndnis uumlber die Bildung und Reaktivitaumlt von Fe(II)-haltigen Mineralsystemen
wie GRs Vivianit Magnetit und an Goethit (α-FeOOH)- und Lepidokrozit (γ-
FeOOH)-Oberflaumlchen gebundenes Fe(II) in der Natur bei
II Zusammenfassung
Um die Rolle von Bakterien bei der Bildung von GRs in natuumlrlichen Boumlden und
Sedimenten aufzuklaumlren wurden Eisenminerale untersucht die als Folge der
Aktivitaumlt von eisenrespirierenden Bakterien gebildet wurden Kapitel 2 beschreibt
die Untersuchungen von eisenhaltigen Produkten die von anaeroben autotrophen
denitrifizierenden Fe(II)-oxidierenden Bakterien (FeOB) gebildet wurden Ein
Bikarbonat- und Phosphat-reiches Kulturmedium bot den nitratreduzierenden
FeOB optimale Bedingungen Fe(II) lag zu Anfang der Reaktion als weisses
Fe(II)-Hydroxyphosphat (Vivianit) und als geloumlstes Fe(II) vor Die Ergebnisse
zeigten dass die denitrifizierenden FeOB amorphen Goethit via ein gruumlnes Fe(III)-
angereichertes Vivianit-Zwischenprodukt bildeten Die Analyse mit Moumlssbauer
Spektroskopie deutet nicht auf eine Bildung von GR hin
In Kapitel 3 werden jene Eisenmineralien beschrieben die waumlhrend der Reduktion
verbreiteter Fe(III)-Oxide durch anaerobe dissimilative Fe(III)-reduzierende
Mikroorganismen Shewanella algae BrY gebildet wurden Um natuumlrliche
Zustaumlnde zu simulieren wurden Fe(III)-Oxide als Beschichtungen auf
Silikatpartikel (Modellsystem fuumlr Sandboumlden) oder Calcitpartikel (CaCO3
Modellsystem fuumlr kalkhaltige Boumlden) aufgetragen sowie synthetische
Elektronencarrier und hochkonzentrierte kuumlnstliche pH-Puffer ausgeschlossen
Die erforschten Mineralsysteme umfassten GoethitCalcit- LepidokrozitCalcit-
und FerrihydritSand-Suspensionen S algae BrY reduzierte beachtliche Mengen
des eingesetzten Fe(III) und es bildeten sich gruumlne und schwarze Festphasen
innerhalb von 1-2 Wochen nach der Animpfung Moumlssbauer Spektroskopie der
gruumlnen und schwarzen Praumlzipitate zeigte dass sich diese aus GR und Vivianit
zusammensetzen
Die Reaktivitaumlt synthetischer GRs gegenuumlber reduzierbaren organischen
Schadstoffen wurde erkundet um die potentielle Bedeutung von GR-Phasen fuumlr
das Schicksal solcher Verbindungen abzuschaumltzen Zu diesem Zweck wurden
Nitroaromaten (NACs) und Chloracetate als Modellverbindungen benutzt um
Zusammenfassung III
umweltrelevante Redoxreaktionen zu studieren In Kapitel 4 wurde die relative
Reaktivitaumlt von aumlusseren und inneren reaktiven Stellen in synthetischem Sulfat-
Gruumlnem Rost (GR-SO4) anhand von strukturaumlhnlichen ldquoreaktiven
Sondenmolekuumllenrdquo mit unterschiedlichen Ladungen untersucht Als reaktive
Sondenmolekuumlle wurden Nitrobenzen 2-Nitrophenol 4-Nitrotoluen 4-
Chlornitrobenzen und 4-Nitrophenylessigsaumlure verwendet Die Ergebnisse zeigen
dass GR-SO4 die NACs vollstaumlndig zu den entsprechenden Anilinen reduzierte
Die Reaktionen folgten einer pseudo 1 Ordnungs Kinetik bezuumlglich NAC und die
auf Oberflaumlche normalisierten pseudo 1 Ordnungs Geschwindigkeitskonstanten
(Anfangsraten) waren 016ndash465middot10-4 s-1middotm-2middotL fuumlr [Fe(II)GR]0 = 103-1260 mM
[NAC]0 = 20-102 microM und pH 84-86 Weder durch Einbezug von
Massentransferlimitierung noch von Oberflaumlchensaumlttigungskinetik war es moumlglich
die aumlhnlichen Oberflaumlchennormalisierten pseudo 1 Ordnungs
Geschwindigkeitskonstanten fuumlr die Reduktion der neutralen und anionischen
NACs durch GR-SO4 zu erklaumlren Dieser Umstand laumlsst vermuten dass die
Reaktion zwischen NAC und GR-SO4 an den externen reaktiven Fe(II)-Stellen
stattfindet Bei niedrigen Fe(II)GR-Anfangskonzentrationen wurden die externen
reaktiven Fe(II)-Stellen aufgebraucht und die Regenerierung von neuen externen
reaktiven Stellen haben schliesslich die Geschwindigkeit der Reduktion von NACs
durch GR-SO4 kontrolliert
In Kapitel 5 wurde die Reaktivitaumlt von verschiedenen umweltrelevanten Fe(II)-
Fe(III)-Mineralsystemen gegenuumlber Trichloressigsaumlure (TCA) und
Dichloressigsaumlure (DCA) in Batchexperimenten die natuumlrliche Bedingungen
imitierten untersucht Die Fe(II)-Fe(III)-Systeme umfassten Sulfat-Gruumlner Rost
Carbonat-Gruumlner Rost Magnetit Fe(II)Goethit und Fe(II)Lepidokrozit TCA
wurde von allen Fe(II)-haltigen Mineralien zu DCA reduziert Die Reaktionen
folgten einer pseudo 1 Ordnungs Kinetik bezuumlglich TCA und die auf Oberflaumlche
normalisierten pseudo 1 Ordnungs Geschwindigkeitskonstanten betrugen 033ndash
76middot10-5 min-1middotm-2middotL bei [Fe(II)]0 = 025ndash116 mM [TCA]0 = 15ndash1000 microM und pH
IV Zusammenfassung
70ndash87 Die Ergebnisse zeigen keine signifikanten Unterschiede zwischen den
verschiedenen Fe(II)-Fe(III)-Systemen bezuumlglich Produkteverteilung und
oberflaumlchen-normalisierten pseudo 1 Ordnungs Geschwindigkeits-konstanten In
keinem der Systeme wurde DCA innerhalb des experimentellen Zeitraums zu
Monochloressigsaumlure oder Essigsaumlure weiter reduziert
Die Ergebnisse die in dieser Dissertation praumlsentiert werden zeigen dass
mikrobiologische Prozesse fuumlr die Oxidation von Vivianit-Phasen im Untergrund
verantwortlich sein koumlnnen Zudem wurde nachgewiesen dass GRs bei tiefen
Kohlenstoff- und Fe(III)-Konzentrationen sowie durch Ausschluss von
kuumlnstlichen Elektronencarriern und pH-Pufferung mikrobiell gebildet werden
koumlnnen Ferner zeigten Befunde dass GRs eine bedeutende Rolle fuumlr die reduktive
Transformation von NACs und TCA in natuumlrlichen Boumlden und Sedimenten spielen
koumlnnen
Summary V
Summary
Layered iron(II)-iron(III)-hydroxides (green rusts) belong to the group of Fe(II)-
bearing mineral systems eg magnetite (Fe3O4) siderite (FeCO3) vivianite
(Fe2(PO4)2sdot8H2O) Fe(II) sulfides as well as Fe(II) associated with Fe(III) oxide
and clay mineral surfaces that control the Fe(II) activity in suboxic and anoxic
soils and sediments Green rusts (GRs) consist of plane positively charged
trioctahedral Fe(II)-Fe(III) hydroxide sheets balanced by hydrated anions in the
interlayers and hold the general formula [FeII(6-x)FeIII
x(OH)12]x+[(A)xnmiddotyH2O]x-
where x = 09 - 42 A is an n-valent anion eg CO32- Clndash or SO4
2- and y is the
number of water molecules in the interlayer GRs are important intermediate
phases formed by partial oxidation of Fe(II) or partial reduction of Fe(III) and they
have been found in suboxic non-acid iron-rich natural environments such as
hydromorphic soils and intertidal sediments and in engineering systems including
pipeline distribution systems for drinking water steel sheet piles in marine
sediments reinforced concrete and permeable reactive barriers of zero-valent iron
implemented for on-site remediation of contaminants Due to their layered
structures anionic interlayers and high specific surface areas GRs represent
reactive ion exchangers and sorbents of anions In addition GRs have been shown
to reduce a range of inorganic and organic pollutants Thus through sequestration
and reductive transformation GRs may play an important role in the fate and
transport of contaminants in suboxic soils and sediments The work presented in
this dissertation adds to the understanding of how Fe(II)-bearing minerals like
GRs vivianite magnetite and Fe(II) associated with goethite (α-FeOOH) and
lepidocrocite (γ-FeOOH) may form and react in nature
In order to elucidate the role of bacteria in the formation of GRs in natural soils
and sediments we studied the iron mineral phases forming as a result of the
activity of iron-respiring bacteria In the study described in chapter 2 the Fe-
containing products formed by anaerobic autotrophic denitrifying Fe(II)-oxidizing
VI Summary
bacteria (FeOB) were examined The culture medium applied contained high levels
of bicarbonate and phosphate and is typically used in this kind of studies as it
provides excellent conditions for the nitrate-reducing FeOB Fe(II) was present
initially as a whitish solid Fe(II) hydroxy phosphate (vivianite) and as soluble
Fe(II) The results obtained demonstrate that the denitrifying FeOB produce poorly
crystalline goethite via a greenish Fe(III)-enriched vivianite intermediate
Moumlssbauer spectroscopic analyses provided no significant evidence of green rust
formation
In chapter 3 the Fe-containing products formed during reduction of common
Fe(III) oxides by the anaerobic dissimilatory Fe(III)-reducing microorganism
Shewanella algae BrY are discussed In order to simulate natural conditions
Fe(III) oxides were applied as coatings on silica (model system for sandy soils) or
calcite (CaCO3) particles (model system for calcareous soils) and synthetic
electron shuttles as well as highly concentrated artificial pH buffers were excluded
The mineral systems studied include goethitecalcite lepidocrocitecalcite and
hydrous ferric oxidesand suspensions S algae BrY reduced substantial amounts
of the initial Fe(III) and green and blackish mineral phases were produced within
1-2 weeks after inoculation Moumlssbauer spectroscopic analyses showed that the
green and black precipitates consisted of GR and vivianite
The reactivity of synthetic GRs towards reducible organic pollutants was
investigated in order to asses the potential significance of GR phases for the fate of
such compounds To this end we used nitroaromatic compounds (NACs) and
chlorinated acetates as suitable model compounds for studying environmentally
relevant redox reactions In the work described in chapter 4 the relative reactivity
of outer and inner Fe(II) reactive sites in synthetic sulfate green rust (GR-SO4) was
studied using a series of structurally closely related compounds with different
charge properties as ldquoreactive probesrdquo The probe compounds included
nitrobenzene 2-nitrophenol 4-nitrotoluene 4-chloronitrobenzene and 4-
Summary VII
nitrophenylacetic acid The results show that NACs are completely reduced to their
corresponding anilines by GR-SO4 The reactions followed pseudo 1 order
kinetics with respect to NAC and the surface area-normalised pseudo 1 order rate
constants (initial rates) obtained were 016ndash465middot10-4 s-1middotm-2middotL at [Fe(II)GR]0 = 103-
1260 mM [NAC]0 = 20-102 microM and pH 84-86 Neither mass transfer control nor
surface saturation kinetics could explain the similarity of the surface-normalised
pseudo 1 order rate constants obtained for the reduction of the neutral and anionic
NACs by GR-SO4 These observations suggest that the reaction between NAC and
GR-SO4 takes place at the external reactive Fe(II) sites At low initial Fe(II)GR
concentrations the external reactive Fe(II) sites were depleted and the regeneration
of new external reactive sites eventually controlled the reduction of the NACs by
GR-SO4
Finally the reactivity of various Fe(II)-Fe(III) mineral systems towards
trichloroacetic acid (TCA) and dichloroacetate (DCA) has been investigated in
laboratory batch experiments imitating natural conditions (Chapter 5) The Fe(II)-
Fe(III)-systems investigated included GR-SO4 carbonate green rust magnetite
Fe(II)goethite and Fe(II)lepidocrocite TCA was readily reduced to DCA by all
Fe(II)-containing minerals The reactions followed pseudo 1 order kinetics with
respect to TCA and the surface area-normalised pseudo 1 order rate constants
obtained were 033ndash76middot10-5 min-1middotm-2middotL at [Fe(II)]0 = 025ndash116 mM [TCA]0 =
15ndash1000 microM and pH 70ndash87 Our results showed no significant differences
regarding product distribution and surface area-normalised reaction rate constants
between the Fe(II)-Fe(III)-systems DCA was not further reduced to
monochloroacetate (MCA) or acetate in any of the systems within the time frame
in our experiments
The results presented in chapter 2 indicate that microbiological processes may be
responsible for the oxidation of vivianite phases in natural subsurface
environments In chapter 3 we demonstrated that GRs may be produced
VIII Summary
microbially at conditions including low carbon and Fe(III) concentrations as well
as the exclusion of synthetic electron shuttles and pH buffers The results obtained
in chapter 4 and 5 show that GRs transform NACs and TCA readily The reductive
transformation of NACs and TCA by GRs is relevant to understanding the
processes responsible for their degradation in the subsurface and the development
of innovative technologies for their remediation
General Introduction 1
1 General Introduction
11 Iron cycling in the subsurface
Iron is the fourth most abundant element (4-5 mass) and the most abundant redox
sensitive element in the Earthrsquos crust It is found as Fe(II) and Fe(III) in a number
of minerals in rocks soils and sediments Under anoxic conditions solid Fe(III)-
containing minerals can be reduced to soluble Fe(II) once the more energetically
favoured electron donors - nitrate and manganese(IV) oxides - have been
consumed Dissolved Fe(II) can be reoxidized to insoluble Fe(III) microbially or
abiotically upon exposure to oxygen Due to this ready alternation between the
Fe(II) and Fe(III) redox states iron plays a major role in controlling the redox
potential and the carbon cycling in subsurface environments (Nealson amp Saffarini
1994)
Nonenzymatic processes were previously considered to account for most of the
Fe(III) reduction in subsurface environments The significance of bacteria in the
biogeochemical cycling of iron has been broadly recognized over the past two
decades Dissimilatory Fe(III)-reducing bacteria (DIRB) that gain energy by
coupling the oxidation of hydrogen or organic compounds to the reduction of
Fe(III) oxides have been known for many years but their biogeochemical
importance was only widely acknowledged about a decade ago (reviewed by
Lovley 1997) Fe(III) bioreduction accounts for a major fraction of the carbon
oxidation in many different anoxic environments and in the presence of sufficient
amounts of reactive Fe(III) microbial Fe(III) reduction may even inhibit sulphate
reduction and methanogenesis (King 1990 Lovley amp Phillips 1986) In fact most
of the Fe(III) reduction in the Fe(III) reduction zone of aquatic sediments and
aquifers is enzymatically catalyzed by microorganisms (Lovley et al 1991) A
wide diversity of DIRB distributed among several different phylogenetic groups
2 Chapter 1 is known today The two most studied DIRB are the obligate anaerobic Geobacter
spp and the facultatively anaerobic Shewanella spp (Figure 11)
Aerobic oxidation of Fe(II)-containing minerals by lithotrophic acidophilic and
neutrophilic bacteria has been known for many years but their broad significance
in the biogeochemical cycling of iron has only been recognized over the past two
decades Both acidophilic (Thiobacillus ferrooxidans) and neutrophilic
(Gallionella ferruginea Leptothrix ochracea Sphaerotilus natans) aerobic Fe(II)-
oxidizing bacteria (FeOB) have been isolated (Hanert 1992 Kuenen et al 1992
Mulder amp Deinema 1992)
Figure 11 The microbial iron cycle
Anaerobic Fe(II) oxidation by phototrophic purple non-sulfur bacteria utilizing
Fe(II) as an electron donor in the light was recognized only a decade ago (Widdel
et al 1993) Subsequently it was demonstrated that the biological oxidation of
Fe(II) in the absence of oxygen is possible by light-independent chemotrophic
microorganisms using nitrate as the electron acceptor (Straub et al 1996) Thus
the microbial iron cycle includes anaerobic Fe(III)-reducing microorganisms and
aerobic as well as anaerobic Fe(II)-oxidizing bacteria (Figure 11)
General Introduction 3
12 Green rusts
Iron oxides iron hydroxides and iron oxyhydroxides (collectively termed iron
oxides or Fe(III) oxides) are ubiquitous in the pedosphere where they originate
from aerobic weathering of surface magmatic rocks such as ferromagnesium
silicates and pyrite (Cornell amp Schwertmann 1996) Goethite (α-FeOOH)
lepidocrocite (γ-FeOOH) ferrihydrite (Fe5HO8sdot4H2O) hematite (α-Fe2O3)
magnetite (Fe3O4) maghemite (γ-Fe2O3) and akageneite (β-FeOOH) constitute the
most important iron oxides in soils and sediments (Schwertmann amp Cornell 1991)
The formation and transformation of iron oxides depend on pH solution
composition redox potential temperature rate of oxidationreduction and degree
and rate of hydrationdehydration Iron oxides are important to many soil
properties such as colour pH and redox buffer capacity aggregation with other
soil particles as well as retention of anions and cations (Cornell amp Schwertmann
1996) A number of Fe(II)-bearing minerals including Fe(II)-containing clays (eg
smectites vermiculites and micas) magnetite siderite (FeCO3) vivianite
(Fe2(PO4)2sdot8H2O) Fe(II) sulphides and green rusts (layered Fe(II)-Fe(III)
hydroxides) may be present in soils and sediments under suboxic and anoxic
conditions Green rusts are believed to play a central role as metastable
intermediates in the redox cycling of iron at circumneutral pH in aquatic and
terrestrial environments
Green rusts (GRs) are layered iron(II)-iron(III)-hydroxides consisting of plane
positively charged trioctahedral Fe(II)-Fe(III) hydroxide sheets balanced by
hydrated anions in the interlayers (cf Figure 41 this work) GRs belong
structually to the pyroaurite-sjoumlgrenite group of layered hydroxides and they hold
the general formula [FeII(6-x)FeIII
x(OH)12]x+[(A)xnmiddotyH2O]x- where x = 09 - 42 A is
an n-valent anion eg CO32- Clndash or SO4
2- and y is the number of water molecules
in the interlayer The three most common and investigated green rust forms include
chloride GR (GR-Cl) sulphate GR (GR-SO4) and carbonate GR (GR-CO3)
Generally GRs are crystallographically classified into the GRI (rhombohedral
4 Chapter 1 GR-Cl and GR-CO3) and GRII (hexagonal GR-SO4) crystal systems The GR
interlayer thickness is a function of both the size and the charge of the interlayer
anion Tetrahedrally coordinated anions like sulphate lead to larger interlayer
distances than smaller monoatomic anions like chloride or planar ions like
carbonate (Mendiboure amp Schoumlllhorn 1986) Not only size but also charge density
plays a role for the interlayer spacing That is for anions having the same number
of valence electrons anions with smaller ionic radii (higher electron density) are
bound more strongly and therefore result in smaller interlayer spacings The
interlayer in GR-SO4 is composed of two consecutive planes of anions and water
whereas GR-Cl and GR-CO3 interlayers consist of only one single plane (Simon et
al 2003)
GRs are important intermediate phases formed by partial oxidation of Fe(II) or
partial reduction of Fe(III) In neutral and weakly alkaline solutions the oxidation
of dissolved Fe(II) always passes through solid GR phases (Bernal et al 1959)
GRs may also form during oxidation of zero-valent iron and as a result of the
combination of Fe(II) and Fe(III) at circumneutral pH (Figure 12)
Figure 12 Formation and transformation of GRs Fe3O4 = magnetite γ-Fe2O3 = maghemite α-
FeOOH = goethite γ-FeOOH = lepidocrocite akageneite = β-FeOOH
General Introduction 5
Oxidation of GR-CO3 usually produces goethite and magnetite-maghemite
whereas GR-Cl and GR-SO4 transform into lepidocrocite and magnetite-
maghemite depending on pH and oxidation rate (Bernal et al 1959 Taylor 1980
Carlson amp Schwertmann 1990) The brown δ-FeOOH is formed by vigorous
oxidation of GR using air or a 30 aqueous solution of hydrogen peroxide (Bernal
et al 1959 Misawa et al 1974) Black ferromagnetic magnetite forms by slow
oxidation of GR whereas lepidocrocite forms at high oxidation rates (Misawa et
al 1974) The presence of chloride is a prerequisite for the formation of
akageneite (Bernal et al 1959 Refait amp Genin 1997)
A substantial amount of work has been conducted in order to estimate the free
energies of formation of green rusts The free energies of formation reported for
the carbonate and sulphate GRs fall in the range 4234ndash4384 kJsdotmol-1 as determined
from solution data monitored during anoxic alkalimetric titrations and from
reduction potential (Eh) and pH recordings monitored during oxidation of GRs in
aqueous solution (Hansen et al 1994 Drissi et al 1995 Genin et al 1996) The
free energies of formation provided allow for estimation of the stability domains of
GRs in Eh-pH phase diagrams (Drissi et al 1995 Genin et al 1996) As
evidenced from such diagrams (Figure 13) the stability domain of GR-SO4 lies
within pH 6-8 and Eh -700 ndash -400 mV depending on the activities of Fe(II) and
sulphate (compare Figures 13aampb) This agrees with the natural GR occurrences
found in suboxic non-acid iron-rich environments such as hydromorphic soils and
intertidal sediments (Al-Agha et al 1995 Trolard et al 1996 Genin et al 1998)
In addition GRs have been found as corrosion products in numerous engineering
systems including a pipeline distribution system for drinking water steel sheet
piles in marine sediments reinforced concrete (ferro-concrete) and permeable
reactive barriers of zero-valent iron implemented for on-site remediation of organic
and inorganic contaminants (Tuovinen et al 1980 Nielsen 1976 Genin et al
1991 Roh et al 2000)
6 Chapter 1
Figure 13 Eh-pH phase diagrams of GR-SO4 a) a = 10+2Fe-3 = 10minus2
4SOa -3 and b) a = 10+2Fe-2
= 10minus24SOa -1
The stability domains of GR-Cl and GR-CO3 are similar to the stability domain of
GR-SO4 At Fe(II) and sulphate activities lower than depicted in Figure 13b the
stability domain of GR-SO4 will be situated at higher pH and lower Eh Other
dissolved species present at anoxic conditions such as phosphate sulphide
carbonate and organic ligands may exert considerable effects on the availability of
Fe(II) and Fe(III) At anoxic and circumneutral conditions vivianite
(Fe2(PO4)2sdot8H2O) controls the Fe(II) activity even at very low phosphate
concentrations (Nriagu amp Dell 1974) The formation of solid Fe(II) sulphides and
siderite (FeCO3) as well as the complexation of Fe(II) and Fe(III) by organic
ligands may also control the activity of Fe(II) in the subsurface and thereby
interfere with the formation of GRs
Due to their layered structures anionic interlayers and high specific surface areas
GRs represent reactive ion exchangers and sorbents of environmentally concerning
anions eg arsenate and selenate (Myneni et al 1997 Randall et al 2001) In
addition GRs may incorporate divalent transition metal cations like Ni2+ Zn2+
Cd2+ Co2+ and Mg2+ by isomorphic substitution for Fe2+ in the hydroxide layers
General Introduction 7
(Tamaura 1985 Tamaura 1986 Refait et al 1994 Parmar et al 2001 Refait et
al 2001) Furthermore GRs have been shown to reduce a range of inorganic
contaminants such as nitrite nitrate selenate chromate uranyl pertechnetate and
the transition metals AgI AuIII CuII and HgII as well as organic pollutants
including halogenated ethanes ethenes and methanes (Hansen et al 1994 Hansen
et al 1996 Myneni et al 1997 Erbs et al 1999 Loyaux-Lawniczak et al 1999
Cui amp Spahiu 2002 Lee amp Batchelor 2002b Heasman et al 2003 OrsquoLoughlin et
al 2003aampb Pepper et al 2003 Elsner et al 2004 OrsquoLoughlin amp Burris 2004)
Thus through sequestration and reductive transformation GRs may play an
important role in the fate and transport of contaminants in suboxic soils and
sediments It should be noted that the rate constants reported for the reduction of
these inorganic and organic pollutants by GRs cannot be directly compared as the
various studies were conducted at very different experimental conditions
13 Microbial formation of green rusts
Generally one would expect that biogenic minerals have chemical compositions
and crystal habits similar to those produced by nonenzymatic processes as they are
governed by the same equilibrium principles In fact since the latter stages of
mineralization are abiotically driven and since the secondary Fe(II)-containing
minerals are formed indirectly by electron transfer outside the bacterial cell and not
directly inside the bacterial cell the type of iron mineral formed is a function of the
environmental conditions in which the bacteria live ie the same microorganism
form different minerals in different environments
The microbial formation of GRs resulting from bioreduction of various Fe(III)
oxides including ferrihydrite goethite and lepidocrocite by strains of the
anaerobic dissimilatory DIRB Shewanella putrefaciens has been reported
repeatedly over the last years (Fredrickson et al 1998 Kukkadapu et al 2001
Parmar et al 2001 Ona-Nguema et al 2002aampb Glasauer et al 2003)
However no evidence of biogenic formation of GRs at natural geochemical
8 Chapter 1 conditions have been offered and it is still unknown whether this process may take
place at natural conditions comprising low nutrient levels low iron concentrations
and the absence of synthetic electron shuttles and highly concentrated artificial pH
buffers Moreover the biotic formation of GRs by anaerobic denitrifying Fe(II)-
oxidizing bacteria has been suggested but the phases still need to be properly
identified (Chaudhuri et al 2001) In order to elucidate the role of bacteria in the
formation of GRs in natural soils and sediments we studied the iron mineral
phases forming as a result of the activity of iron-respiring bacteria (Chapters 2 and
3)
14 Redox reactions of green rusts
Fe(II) is one of the most abundant reductants present in aquatic and terrestrial
environments under suboxic and anoxic conditions (Lyngkilde amp Christensen
1992 Ruumlgge et al 1998) In these environments Fe(II) may be present as soluble
organic and inorganic complexes as surface complexes and as a host of Fe(II)-
bearing minerals Although aqueous Fe(II) complexes may reduce a number of
contaminants Fe(II) associated with mineral surfaces and structural Fe(II) present
in the mineral lattice in Fe(II)-containing minerals are often more powerful
reductants Fe(II)-bearing minerals including GRs magnetite siderite Fe(II)
sulphides as well as Fe(II)-carrying Fe(III) oxide and clay mineral surfaces have
been shown to reduce a number of organic and inorganic contaminants such as
nitroaromatic compounds chlorinated aliphatics chromate uranyl pertechnetate
nitrate monochloramine and carbamate pesticides (Klausen et al 1995 Cui amp
Eriksen 1996 Butler amp Hayes 1998amp1999 Erbs et al 1999 Liger et al 1999
Loyaux-Lawniczak et al 1999 Amonette et al 2000 Hwang amp Batchelor 2000
Hansen et al 2001 Gander et al 2002 Lee amp Batchelor 2002aampb Pecher et al
2002 Vikesland amp Valentine 2002 Hofstetter et al 2003 OrsquoLoughlin et al
2003aampb Strathmann amp Stone 2003 Elsner et al 2004 OrsquoLoughlin amp Burris
2004) However only few comparative studies on the reactivity of Fe(II)-bearing
minerals exist (Lee amp Batchelor 2002b Elsner et al 2004) When examining the
General Introduction 9
reaction rates of the reductive transformation of NACs and chlorinated aliphatics
by GRs and other Fe(II)-bearing minerals reported in these studies the rate
constants for GRs are mostly among the highest rates reported and in some cases
even higher than the rate constants for Fe(II) sulphides Thus GRs may play an
important role in the transformation of reducible contaminants in the subsurface
Nitroaromatic compounds (NACs) are widely applied as explosives herbicides
insecticides solvents and intermediates in the synthesis of dyes and pesticides
(Hartter 1985 Rosenblatt et al 1991) NACs are ubiquitous in the subsurface
environment and pose a health risk due to their toxicity (Rickert 1985) In anoxic
environments reduction of the nitro group is generally the first step during abiotic
or microbial transformation of the NACs (Macalady et al 1986) The
transformation reaction generally produces the corresponding aromatic amines and
minor amounts of intermediates (hydroxylamines and nitroso compounds) as well
as coupling products (azo and azoxy compounds) These products may be of
similar or even greater environmental concern
Trichloroacetic acid (TCA) is ubiquitous in soils and the concentrations reported
range from lt005 to 380 microgkg (Euro Chlor 2001 McCulloch 2002 Ahlers et al
2003) On account of its phytotoxicity suspected human carcinogenicity and
widespread occurrence TCA is of considerable environmental concern especially
in the terrestrial compartment (Ahlers et al 2003) Moreover the daughter
compounds of TCA - dichloroacetic acid (DCA) and monochloroacetic acid
(MCA) - are also toxins and suspected human carcinogens as well as widespread in
the environment (Reimann et al 1996 Berg et al 2000 Ahlers et al 2003 and
references therein) In this work the reactivity of synthetic green rusts towards
nitroaromatic compounds (NACs) and the reactivity of various Fe(II)-Fe(III)
mineral systems including synthetic GRs towards chlorinated acetates have been
studied (Chapters 4 and 5)
10 Chapter 1 15 Outline of the thesis
An examination of the Fe-containing products produced during solid state
oxidation of vivianite by anaerobic autotrophic denitrifying Fe(II)-oxidizing
bacteria is presented in chapter 2 The Fe(II)-oxidizing bacteria were cultured in a
mineral medium containing high levels of bicarbonate and phosphate which is
typically used in this kind of studies as it provides excellent conditions for the
nitrate-reducing FeOB The solid iron phases forming were investigated by
transmission Moumlssbauer spectroscopy infrared spectroscopy and scanning electron
microscopy
Chapter 3 includes a study on the Fe-containing products formed during reduction
of common Fe(III) oxides by the anaerobic dissimilatory Fe(III)-reducing
microorganism Shewanella algae BrY In order to simulate natural conditions
Fe(III) oxides were applied as coatings on silica (model system for sandy soils) or
calcite particles (model system for calcareous soils) and synthetic electron shuttles
as well as highly concentrated artificial pH buffers were excluded The mineral
systems studied include goethitecalcite lepidocrocitecalcite and hydrous ferric
oxidesand suspensions The solid iron phases produced were examined by
transmission Moumlssbauer spectroscopy
A study on the relative reactivity of outer and inner Fe(II) sites in synthetic GR-
SO4 by using a series of structurally closely related compounds with different
charge properties as ldquoreactive probesrdquo is presented in chapter 4 The probe
compounds included nitrobenzene 2-nitrophenol 4-nitrotoluene 4-
chloronitrobenzene and 4-nitrophenylacetic acid
In chapter 5 an investigation of the reactivity of various Fe(II)-Fe(III) mineral
systems towards TCA and DCA is presented The study included laboratory batch
experiments imitating natural conditions The Fe(II)-Fe(III)-systems investigated
included GR-SO4 carbonate green rust magnetite Fe(II)goethite and
General Introduction 11
Fe(II)lepidocrocite The reactivities of the Fe(II)-Fe(III) mineral systems were
examined by comparing their surface-normalized rate constants
The results and environmental implications of this work are summarized in chapter
6 References Ahlers J Regelmann J Riedhammer C (2003) Environmental risk assessment of airborne trichloroacetic acid - a contribution to the discussion of the significance of anthropogenic and natural sources Chemosphere 52 531-537 Al-Agha MR Burley SD Curtis CD Esson J (1995) Complex cementation textures and authigenic mineral assemblages in recent concretions from the Lincolnshire Wash (east coast UK) driven by Fe(0) to Fe(II) oxidation Journal of the Geological Society 152 157-171 Amonette JE Workman DJ Kennedy DW Fruchter JS Gorby YA (2000) Dechlorination of carbon tetrachloride by Fe(II) associated with goethite Environmental Science and Technology 34 4606-4613 Berg M Muumlller SR Muumlhlemann J Wiedmer A Schwarzenbach RP (2000) Concentrations and mass fluxes of chloroacetic acids and trifluoroacetic acid in rain and natural waters in Switzerland Environmental Science and Technology 34 2675-2683 Bernal JD Dasgupta DR Mackay AL (1959) The oxides and hydroxides of iron and their structural inter-relationships Clay Minerals Bulletin 4 15-30 Butler EC Hayes KF (1998) Effects of solution composition and pH on the reductive dechlorination of hexachloroethane by iron sulfide Environmental Science and Technology 32 1276-1284 Butler EC Hayes KF (1999) Kinetics of the transformation of trichloroethylene and tetrachloroethylene by iron sulfide Environmental Science and Technology 33 2021-2027 Carlson L Schwertmann U (1990) The effect of CO2 and oxidation rate on the formation of goethite versus lepidocrocite from an Fe(II) system at pH 6 and 7 Clay Minerals 25 65-71 Chaudhuri SK Lack JG Coates JD (2001) Biogenic magnetite formation through anaerobic biooxidation of Fe(II) Applied and Environmental Microbiology 67 2844-2848 Cornell RM Schwertmann U (1996) The iron oxides Structure properties reactions occurrence and uses VCH Verlagsgesellschaft mbH Weinheim Cui D Eriksen TE (1996) Reduction of pertechnetate by ferrous iron in solution influence of sorbed and precipitated Fe(II) Environmental Science and Technology 30 2259-2262
Cui D Spahiu K (2002) The reduction of U(VI) on corroded iron under anoxic conditions Radiochemica Acta 90 623-628
12 Chapter 1 Drissi SH Refait Ph Abdelmoula M Geacutenin JMR (1995) The preparation and thermodynamic properties of Fe(II)-Fe(III) hydroxide-carbonate (green rust I) Pourbaix diagram of iron in carbonate-containing aqueous media Corrosion Science 37 2025-2041 Elsner M Haderlein SB Schwarzenbach RP (2004) Reactivity of Fe(II)-bearing minerals towards reductive transformation of organic contaminants Environmental Science and Technology 38 799-807 Erbs M Hansen HCB Olsen CE (1999) Reductive dechlorination of carbon tetrachloride using iron(II)iron(III)-hydroxide-sulphate (green rust) Environmental Science and Technology 33 307-311 Euro Chlor (2001) Trichloroacetic acid in the environment a dossier Euro Chlor Brussels and the European Chlorinated Solvent Association Fredrickson JK Zachara JM Kennedy DW Dong H Onstott TC Hinman NW Li S (1998) Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium Geochimica et Cosmochimica Acta 62 3239-3257 Gander JW Parkin GF Scherer MM (2002) Kinetics of 111-trichloroethane transformation by iron sulfide and a methanogenic consortium Environmental Science and Technology 36 4540-4546 Geacutenin JMR Bourrieacute G Trolard F Abdelmoula M Jaffrezic A Refait Ph Maitre V Humbert B Herbillon A (1998) Thermodynamic equilibria in aqueous suspensions of synthetic and natural Fe(II)-Fe(III) green rusts Occurrences of the mineral in hydromorphic soils Environmental Science and Technology 32 1058-1068 Geacutenin JMR Olowe AA Benbouzid-Rollet ND Prieur D Confente M Resiak B (1991) The simultaneous presence of green rust 2 and sulfate reducing bacteria in the corrosion of steel sheet piles in a harbour area Hyperfine Interactions 69 875-878 Geacutenin JMR Olowe AA Refait Ph Simon L (1996) On the stoichiometry and Pourbaix diagram of Fe(II)-Fe(III) hydroxy-sulphate of sulphate-containing green rust 2 An electrochemical and Moumlssbauer spectroscopy study Corrosion Science 38 1751-1762 Glasauer S Weidler PG Langley S Beveridge TJ (2003) Controls on Fe reduction and mineral formation by a subsurface bacterium Geochimica et Cosmochimica Acta 67 1277- 1288 Hanert HH (1992) The genus Gallionella In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 4082-4088 Hansen HCB Borggaard OK Soslashrensen J (1994) Evaluation of the free energy of formation of iron(II)iron(III)-hydroxidesulphate (Green Rust) and its reduction of nitrite Geochimica et Cosmochimica Acta 58 2599-2608 Hansen HCB Guldberg S Erbs M Koch CB (2001) Kinetics of nitrate reduction by green rusts ndash effects of interlayer anion and Fe(II)Fe(III) ratio Applied Clay Science 18 81-91
General Introduction 13
Hansen HCB Koch CB Nancke-Krogh H Borggaard OK Soslashrensen J (1996) Abiotic nitrate reduction to ammonium Key role of green rust Environmental Science and Technology 30 2053-2056 Hartter DR (1985) The use and importance of nitroaromatic chemicals in the chemical industry In Toxicity of nitroaromatic compounds Rickert DE (ed) Hemisphere Publishing Corporation 1-13 Heasman DM Sherman DM Ragnarsdottir KV (2003) The reduction of aqueous Au3+ by sulfide minerals and green rust phases American Mineralogist 88 725-738 Hofstetter TB Schwarzenbach RP Haderlein SB (2003) Reactivity of Fe(II) species associated with clay minerals Environmental Science and Technology 37 519-528 Hwang I Batchelor B (2000) Reductive dechlorination of tetrachloroethylene by Fe(II) in cement slurries Environmental Science and Technology 34 5017-5022 King GM (1990) Effects of added manganic and ferric oxides on sulfate reduction and sulfide oxidation in intertidal sediments FEMS Microbiology Ecology 73 131-138 Klausen J Troumlber SP Haderlein SB Schwarzenbach RP (1995) Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions Environmental Science and Technology 29 2396-2404 Kuenen JG Robertson LA Tuovinen OH (1992) The genera Thiobacillus Thiomicrospira and Thiosphaera In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 2618-2624
Kukkadapu RK Zachara JM Smith SC Fredrickson JK Liu C (2001) Dissimilatory bacterial reduction of Al-substituted goethite in subsurface sediments Geochimica et Cosmochimica Acta 65 2913-2924 Lee W Batchelor B (2002a) Abiotic reductive dechlorination of chlorinated ethylenes by iron- bearing soil minerals 1 Pyrite and magnetite Environmental Science and Technology 36 5147- 5154 Lee W Batchelor B (2002b) Abiotic reductive dechlorination of chlorinated ethylenes by iron-bearing soil minerals 2 Green rust Environmental Science and Technology 36 5348- 5354 Liger E Charlet L Van Cappellen P (1999) Surface catalysis of uranium (VI) reduction by iron(II) Geochimica et Cosmochimica Acta 63 2939-2955 Lovley DR (1997) Microbial Fe(III) reduction in subsurface environments FEMS Microbiology Reviews 20 305-313 Lovley DR Phillips EJP (1986) Organic matter mineralization with reduction of ferric iron in anaerobic sediments Applied and Environmental Microbiology 51 683-689 Lovley DR Phillips EJP Lonergan DJ (1991) Enzymatic versus nonenzymatic mechanisms for Fe(III) reduction in aquatic sediments Environmental Science and Technology 25 1062-1067
14 Chapter 1 Loyaux-Lawniczak S Refait Ph Lecomte P Ehrhardt J Geacutenin JMR (1999) The reduction of chromate ions by Fe(II) layered hydroxides Hydrology and Earth System Sciences 3 593-599 Lyngkilde J Christensen TH (1992) Redox zones of a landfill leachate pollution plume (Vejen Denmark) Journal of Contaminant Hydrology 10 273-289 Macalady DL Tratnyek PG Grundl TJ (1986) Abiotic reduction reactions of anthropogenic organic chemicals in anaerobic systems A critical review Journal of Contaminant Hydrology 1 1-28 McCulloch A (2002) Trichloroacetic acid in the environment Chemosphere 47 667-686 Mendiboure A Schoumlllhorn A (1986) Formation and anion exchange reactions of layered transition metal hydroxides [Ni1-xMx](OH)2(CO3)x2(H2O)z (M = Fe Co) Revue de Chimie Mineacuterale 23 819-827 Misawa T Hashimoto K Shimodaira S (1974) The mechanism of formation of iron oxide and oxyhydroxides in aqueous solutions at room temperature Corrosion Science 14 131-149 Mulder EG Deinema MH (1992) The sheathed bacteria In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 2618-2624 Myneni SCB Tokunaga TK Brown Jr GE (1997) Abiotic selenium redox transformations in the presence of Fe(IIIII) oxides Science 278 1106-1109 Nealson KH Saffarini D (1994) Iron and manganese in anaerobic respiration Environmental significance physiology and regulation Annual Review of Microbiology 48 311-343 Nielsen A (1976) Hvid groslashn og sort rust Beskrivelse af en korrosionsskade paring et svoslashmmebassin Nordisk Betong 2 21-24 Nriagu JO Dell CI (1974) Diagenetic formation of iron phosphates in recent lake sediments American Mineralogist 59 934-946 OLoughlin EJ Burris DR (2004) Reduction of halogenated ethanes by green rust Environmental Toxicology and Chemistry 23 41-48 OLoughlin EJ Kelly SD Cook RE Csencsits R Kemner KM (2003a) Reduction of uranium(VI) by mixed iron(II)iron(III) hydroxide (green rust) Formation of UO2 nanoparticles Environmental Science and Technology 37 721-727 OLoughlin EJ Kelly SD Kemner KM Csencsits R Cook RE (2003b) Reduction of AgI AuIII CuII and HgII by FeIIFeIII hydroxysulfate green rust Chemosphere 53 437-446 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002a) Iron (IIIII) hydroxycarbonate green rust formation and stabilization from lepidocrocite bioreduction Environmental Science and Technology 36 16-20 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002b) Microbial reduction of lepidocrocite γ-FeOOH by Shewanella putrefaciens The
General Introduction 15
formation of green rust Hyperfine Interactions 139140 231-237 Parmar N Gorby YA Beveridge TJ Ferris FG (2001) Formation of green rust and immobilization of nickel in response to bacterial reduction of hydrous ferric oxide Geomicrobiology Journal 18 375-385 Pecher K Haderlein SB Schwarzenbach RP (2002) Reduction of polyhalogenated methanes by surface-bound Fe(II) in aqueous suspensions of iron oxides Environmental Science and Technology 36 1734-1741 Pepper SE Bunker DJ Bryan ND Livens FR Charnock JM Pattrick RAD Collison D (2003) Treatment of radioactive wastes An X-ray absorption spectroscopy study of the reaction of technetium with green rust Journal of Colloid and Interface Science 268 408- 412 Randall SR Sherman DM Ragnarsdottir KV (2001) Sorption of As(V) on green rust (Fe4(II)Fe2(III)(OH)12SO4
3H2O) and lepidocrocite (γ-FeOOH) Surface complexes from EXAFS spectroscopy Geochimica et Cosmochimica Acta 65 1015-1023 Refait Ph Abdelmoula M Trolard F Geacutenin JMR Ehrhardt JJ Bourrieacute G (2001) Moumlssbauer and XAS study of a green rust mineral the partial substitution of Fe2+ by Mg2+ American Mineralogist 86 731-739 Refait Ph Drissi SH Marie Y Geacutenin JMR (1994) The substitution of Fe2+ ions by Ni2+ ions in green rust one compounds Hyperfine Interactions 90 389-394 Refait Ph Geacutenin JMR (1997) The mechanisms of oxidation of ferrous hydroxychloride β- Fe2(OH)3Cl in aqueous solution The formation of akaganeite vs goethite Corrosion Science 39 539-553 Reimann S Grob K Frank H (1996) Chloroacetic acids in rainwater Environmental Science and Technology 30 2340-2344 Rickert DE (1985) Toxicity of nitroaromatic compounds Hemisphere Publishing Corporation 1-13 Roh Y Lee SY Elless MP (2000) Characterization of corrosion products in the permeable reactive barriers Environmental Geology 40 184-194 Rosenblatt DH Burrows EP Mitchell WR Parmer DL (1991) Organic explosives and related compounds In The Handbook of Environmental Chemistry Anthropogenic compounds Hutzinger O (Ed) Springer-Verlag 195-234 Ruumlgge K Hofstetter TB Haderlein SB Bjerg PL Knudsen S Zraurig C Mosbaeligk H Christensen TH (1998) Characterization of predominant reductants in an anaerobic leachate- affected aquifer by nitroaromatic probe compounds Environmental Science and Technology 32 23-31 Schwertmann U Cornell RM (1991) Iron oxides in the laboratory Preparation and characterization VCH Verlagsgesellschaft mbH Weinheim Simon L Francois M Refait Ph Renaudin G Lelaurain M Geacutenin JMR (2003)
16 Chapter 1 Structure of the Fe(II-III)-layered double hydroxysulphate green rust two from Rietveld analysis Solid State Sciences 5 327-334 Strathmann TJ Stone AT (2003) Mineral surface catalysis of reactions between FeII and oxime carbamate pesticides Geochimica et Cosmochimica Acta 67 2775-2791 Straub KL Benz M Schink B Widdel F (1996) Anaerobic nitrate-dependent microbial oxidation of ferrous iron Applied and Environmental Microbiology 62 1458-1460 Tamaura Y (1985) ZnII-bearing green rust II and its spontaneous transformation into ZnII- bearing ferrite in aqueous solution Bulletin of the Chemical Society of Japan 58 2951-2954 Tamaura Y (1986) Ni(II)-bearing green rust II and its spontaneous transformation into Ni(II)- bearing ferrites Bulletin of the Chemical Society of Japan 59 1829-1832 Taylor RM (1980) Formation and properties of Fe(II)Fe(III)-hydroxycarbonate and its possible significance in soil formation Clay Minerals 15 369-382 Trolard F Abdelmoula M Bourrieacute G Humbert B Geacutenin JMR (1996) Mise en eacutevidence dun constituant de type rouilles vertes dans les sols hydromorphes Proposition de lexistence dun nouveau mineacuteral la fougeacuterite Geacuteosciences de surface Comptes Rendus de LrsquoAcademie des Sciences 323 1015-1022 Tuovinen OH Button KS Vuorinen A Carlson L Mair DM Yut LA (1980) Bacterial chemical and mineralogical characteristics of tubercles in distribution pipelines Journal of the American Water Works Association 72 626-635 Vikesland PJ Valentine RL (2002) Iron oxide surface-catalyzed oxidation of ferrous iron by monochloramine implications of oxide type and carbonate on reactivity Environmental Science and Technology 36 512-519 Widdel F Schnell S Heising S Ehrenreich A Assmus B Schink B (1993) Ferrous iron oxidation by anoxygenic phototrophic bacteria Nature 362 834-836
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 17
2 Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria
Abstract
This work investigated the Fe-containing products formed by anaerobic
autotrophic denitrifying Fe(II)-oxidizing bacteria in a specific bicarbonate buffered
(30 mM HCO3- pH 70) culture media containing 10 mM Fe(II) 4 mM nitrate and
4 mM phosphate Fe(II) was present initially as a whitish vivianite-like
(Fe3(PO4)2middot8H2O) precipitate and as soluble Fe(II) The initial phase of the
oxidation produced a greenish metavivianite-like ((FeII3-xFeIII
x)(PO4)2(OH)xmiddot(8-
x)H2O x gt 12) phase In the late oxidation phase a reddish precipitate of poorly
crystalline goethite (α-FeOOH) dominated the colour of the media in coexistence
with Fe(II)-containing siderite (FeCO3) The increasing amounts of Fe(III) present
in the ldquovivianiterdquo and ldquometavivianiterdquo structures were accompanied by an
increasing intensity in the green colour as the Fe(II) biooxidation progressed This
colour development has produced the idea of biogenic green rusts (layered Fe(II)-
Fe(III) hydroxides) in several studies on nitrate-dependent Fe(II) biooxidation
However in this work no evidence of green rust formation mediated by anaerobic
denitrifying Fe(II)-oxidizing bacteria was obtained
21 Introduction
Aerobic oxidation of Fe(II)-containing minerals by lithotrophic acidophilic and
neutrophilic bacteria has been known for many years but their broad significance
in the biogeochemical cycling of iron has only been recognized over the past two
decades Aerobic Fe(II)-oxidizing bacteria (FeOB) have been isolated from acidic
ecosystems (Thiobacillus ferrooxidans) neutral eutrophic systems (Sphaerotilus
natans Leptothrix ochracea) and neutral oligotrophic systems (Gallionella
ferruginea) (Hanert 1992 Kuenen et al 1992 Mulder amp Deinema 1992) At
neutral pH Fe(II) is unstable in the presence of oxygen and is rapidly oxidized to
the insoluble Fe(III) Hence the only pH neutral environments where soluble
18 Chapter 2
Fe(II) is available for aerobic FeOB are at interfaces between oxic and anoxic
conditions The aerobic neutrophilic FeOB (Leptothrix ochracea Gallionella
ferruginea and Sphaerotilus natans) live at such interfaces and are usually
associated with the yellowishreddish ferric deposits formed there
Over the past several years there has been a growing recognition that other less
readily detectable types of bacteria are involved in Fe(II) oxidation in ecosystems
at circumneutral pH For example it has been reported that neutrophilic FeOB are
abundant at the Loihi seamount hydrothermal vents and play a major role in the
Fe(III) oxide deposition (Emerson amp Moyer 2002) Similarly unidentified
neutrophilic obligate lithotrophic FeOB have been isolated from the rhizosphere of
wetlands plants where they are closely associated with deposits of amorphous
Fe(III) oxides (Emerson et al 1999) It was previously believed that Fe(III) oxide
deposits associated with sheaths were produced biologically whereas Fe(III) oxide
deposits not associated with cells were produced abiotically Recently the
formation of amorphous Fe(III) oxide in gradient tubes has been attributed to the
action of FeOB (Sobolev amp Roden 2001) The authors attribute 90 of the
oxidation to biological processes and indicated that the organisms seem to produce
a mobile form of Fe(III) that diffuses away from the cells before being
precipitated thereby avoiding encrustation of the cells They suggest that such
soluble Fe(III) complexes might be substrates for closely associated Fe(III)-
reducing bacteria Such an arrangement might allow close coupling between
microbial Fe(II) oxidation and Fe(III) reduction within millimeters of the oxic-
anoxic interface
Anaerobic Fe(II) oxidation by phototrophic purple non-sulfur bacteria utilizing
Fe(II) as an electron donor in the light was recognized only a decade ago (Widdel
et al 1993) Subsequently it was demonstrated that the biological oxidation of
Fe(II) in the absence of oxygen is possible by light-independent chemotrophic
microbial activity using nitrate as the electron acceptor (Straub et al 1996) In
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 19
addition studies conducted in gradient cultures revealed that nitrate-reducing
strains could also oxidize Fe(II) with molecular oxygen (Benz et al 1998) Hence
these Fe(II)-oxidizing strains may use nitrate as well as oxygen as electron
acceptors The microbial oxidation of Fe(II) was coupled to stoichiometric
reduction of nitrate to N2 and only one strain produced traces of N2O as a by-
product (Straub et al 1996 Benz et al 1998) The authors proposed the formation
of 2-line ferrihydrite as the end product of Fe(II) biooxidation The chemical
reduction of nitrate by Fe(II) requires a catalyst eg at least 10 microM Cu2+ in order
to take place at significant rates and may thus be considered insignificant under the
conditions applied in our study (Moraghan amp Buresh 1976) The chemical
oxidation of Fe(II) with nitrous oxide has not been observed However nitrite can
oxidize Fe(II) chemically (Moraghan amp Buresh 1977 Straub et al 1996) but this
process is considered insignificant at the conditions applied here No denitrifying
Fe(II)-oxidizing enrichment culture has been found to produce ammonium from
nitrate
Both lithoheterotrophic (depending on organic cosubstrates such as acetate) and
strictly lithoautotrophic nitrate-reducing FeOB have been found in various marine
and freshwater sediments However most isolates depend on organic cosubstrates
for cell biosynthesis (Benz et al 1998) Most probable number estimations
showed that denitrifying FeOB accounted for 00006-08 of the acetate-oxidizing
denitrifying microbial population Lithotrophic FeOB accounted for less than
00001 of the total bacterial community Attempts to isolate CO2-fixing nitrate-
dependent FeOB from lithotrophic cultures have failed (Straub amp Buchholz-
Cleven 1998) Mixotrophic FeOB accounted for 0004-004 of the total bacterial
community In addition microbial nitrate-dependent Fe(II) oxidation was
demonstrated in a flooded paddy soil as well as in activated sludge from a
wastewater treatment plant (Nielsen amp Nielsen 1998 Ratering amp Schnell 2001)
Since the activity is not restricted to sunlight exposed habitats microbial nitrate-
dependent Fe(II) oxidation is supposedly more important on a global scale than
20 Chapter 2
anaerobic Fe(II) oxidation by phototrophic bacteria Furthermore it has been
reported that anaerobic denitrifying FeOB aptly oxidize biogenic Fe(II) minerals
formed by bioreduction of synthetic goethite and ferrihydrite and that anaerobic
Fe(III)-reducing bacteria readily reduce Fe(III) minerals formed by biooxidation of
Fe(II) (Weber et al 2001 Straub et al 1998) Hence autotrophic denitrifying
FeOB may play a significant role in the nitrogen and iron cycles in subsurface
environments where the nitrate and the Fe(II) zones overlap and organic carbon
supply is limited (Figure 21)
Figure 21 The microbial iron cycle linking the carbon and nitrogen cycles
Phosphate is released into the environment through natural processes such as rock
weathering and decomposition of dead organic material and anthropogenic
activities eg wastewater effluents and application of manure and fertilizers in
horti- and agriculture In anoxic soils and sediments phosphate may be sequestered
by sorption onto Fe(III) oxides (Williams et al 1971 Patrick amp Khalid 1974)
Phosphate strongly influences the type morphology and properties of Fe(III)
oxides formed by oxidation and hydrolysis of Fe(II) salts as well as the degree of
their transformation (Kandori et al 1992 Cumplido et al 2000 Benali et al
2001) Phosphate may also be retained by precipitation of Fe(II) phosphates such
as the monoclinic vivianite (Fe3(PO4)2middot8H2O) which is the most important stable
Fe(II) orthophosphate solid encountered in the subsurface under most conditions
(Nriagu 1972) At anoxic and circumneutral conditions the whitish vivianite
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 21
controls the Fe(II) activity even at very low phosphate concentrations (Nriagu amp
Dell 1974) Vivianite occurs as a secondary mineral in the gossans of metallic ore
deposits and as a weathering product of primary iron-manganese phosphates in
pegmatites (Gaines et al 1997) Moreover natural vivianite occurrences have
been identified in a number of lake and river sediments (Zwaan amp Kortenbout van
der Sluys 1971 Nriagu amp Dell 1974 Postma 1981 Nembrini et al 1983
Henderson et al 1984 Dodd et al 2003 House 2003 and references therein)
Vivianite is also found in sewage sludge as a result of the wastewater treatment
where iron salts are added in order to remove phosphate (Seitz et al 1973) It is
however still indefinite how ubiquitous vivianite is in nature Furthermore only
little is known about the mechanism of vivianite formation and the role played by
sedimentary Fe(III) oxides Anaerobic Fe(III)-reducing microorganisms may
reduce Fe(III) oxides thereby releasing the iron as soluble Fe(II) and mobilizing
the phosphate adsorbed to the Fe(III) oxides (Lovley 1997) It has been suggested
that vivianite is formed by precipitation following reductive dissolution of Fe(III)
oxides (Manning et al 1981 Manning amp Jones 1982) However it has also been
proposed that the transformation of Fe(III) oxides to vivianite occurs topotactically
and not via reductive dissolution (Nembrini et al 1983) Vivianite was shown to
form microbially as a result of the activity of the anaerobic Fe(III)-reducing
bacteria Shewanella putrefaciens in the presence of high Fe(III)-citrate and
phosphate concentrations (Jorand et al 2000) Moreover vivianite formation by
bioreduction of Fe(III) in hydrous ferric oxide and in smectite has been reported
(Fredrickson et al 1998 Dong et al 2003)
Only little is known about the oxidation products of vivianite Metavivianite a
greenish triclinic iron hydroxy phosphate mineral was first described by Ritz et al
(1974) and it was later found to coexist with vivianite in several natural sediment
samples (Henderson et al 1984) Once the Fe(III) content became evident the true
composition of metavivianite ((FeII3-xFeIII
x)(PO4)2(OH)x
22 Chapter 2
middot(8-x)H2O x gt 12)) was established (Rodgers amp Johnston 1985 Rodgers 1986
and references therein) The formation of intermediate greenish precipitates during
oxidation of fluffy colourless Fe(II) precipitates by anoxic phototrophic
microorganisms and nitrate-dependent FeOB have been reported (Ehrenreich amp
Widdel 1994 Chaudhuri et al 2001) Since both studies were conducted in
bicarbonate buffered mineral media (22-30 mM HCO3ndash pH 70-72) containing
37-5 mM phosphate we assume that the initial fluffy whitish precipitates
consisted mainly of vivianite Chaudhuri et al (2001) proposed that the
intermediate green phases produces by the denitrifying FeOB consist of carbonate
green rust (GR-CO3) but no convincing evidence of this biogenic GR-CO3 has been
provided yet The major objective of this work was to examine the Fe-containing
products forming during the course of biooxidation of vivianite by non-
phototrophic anaerobic denitrifying Fe(II)-oxidizing bacteria
2 2 Materials and methods
All handling and sampling of solutions and suspensions were carried out under
sterile and strict anoxic conditions All chemicals were pa quality
221 Microorganisms and media
Enrichment cultures of nitrate-reducing FeOB taken from town ditches (Bremen
Germany) were grown in anoxic bicarbonate-buffered (30 mM HCO3ndash 90
N210 CO2 pH 70) mineral media containing 4 mM phosphate as well as
essential trace elements and vitamins (Table 21 Straub amp Buchholz-Cleven
1998) Ammonium was omitted from the media in order to facilitate detection of
ammonium possibly produced by reduction of nitrate The techniques used for
preparation of media and cultivation of bacteria under anoxic conditions have been
described by Widdel amp Bak (1992) 05 M aqueous stock solutions of FeCl2 or
FeSO4 were prepared in 100 mL glass flasks by reacting 65 mmol of iron powder
(particle size 10 microm Merck) with 100 mL deoxygenated 10 M HCl or 05 M
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 23
H2SO4 respectively The solutions were magnetically stirred and heated (~80degC)
during reaction until the H2(g) production had ceased (ge 1 hour) The FeCl2 and
FeSO4 stock solutions were stored under a small Ar overpressure at 5degC
Table 21 Composition of the mineral medium (adopted from Straub amp Buchholz-Cleven
(1998))
Components Concentration (M) KH2PO4 15middot10-3
K2HPO4 25middot10-3
MgSO4middot7H2O 10middot10-3
CaCl2middot2H2O 50middot10-4
H3BO3 56middot10-5
ZnSO4middot7H2O 10middot10-6
Na2MoO4middot2H2O 40middot10-6
CuSO4middot5H2O 20middot10-7
MnSO4middotH2O 10middot10-6
Na2SeO4 12middot10-5
CoCl2middot6H2O 50middot10-6
NiCl2middot6H2O 80middot10-6
NaCl 10middot10-5
NaHCO3ndash 30middot10-2
Cyanocobalamine (vitamin B12) 37middot10-8
p-aminobenzoic acid (vitamin Hrsquo) 36middot10-7
D(+)-biotin (vitamin H) 41middot10-8
Nicotinic acid (Niacin) 81middot10-7
Ca-D(+)-pantothenate (vitamin B5) 52middot10-8
Pyridoxamine dihydrochloride 96middot10-7
Thiaminechloridehydrochloride (vitamin B1) 15middot10-7
NaNO3 40middot10-3
FeSO4 or FeCl2 0010
222 Characterisation of precipitates
In order to optimize the characterization and distinction between the spectral
components transmission Moumlssbauer spectra were obtained at temperatures
between 5 K and 250 K and in external magnetic fields of 4 T (parallel to the γ-ray
direction) using a conventional constant acceleration spectrometer and a source of 57Co in Rh The spectrometer was calibrated using a 125 microm foil of α-Fe at room
temperature and isomer shifts are given relative to the centroid of the spectrum of
this absorber The spectra were fitted using simple Lorentzian line shape Infrared
(IR) spectra were obtained using a Perkin Elmer FT-IR 2000 spectrometer and the
24 Chapter 2
KBr pellet technique Scanning electron microscopy (SEM) was carried out in
order to study the morphology and composition of the precipitates Specimens for
SEM were prepared by depositing suspended particles onto an aluminum stub
coated with a carbon sticker The stub was quickly transferred into a sputtering
chamber and coated with a thin Pt film (~20 nm) In order to avoid interfering Pt
signals in the energy dispersive spectra the stubs were in some cases not coated
with Pt but quickly transferred to the SEM chamber for evacuation Measurements
were performed using a Philips XL30 equipped with a LaB6 source and an
accelerating voltage of 20 kV and an EDAX eDXi X-ray dispersive spectrometer
223 Biooxidation experiments
The biooxidation experiments were conducted in 50-400 mL butyl rubber
stoppered bottles with a 90 N210 CO2 headspace constituting 10 of the total
volume Prior to inoculation 4 mM NaNO3 was added as the electron acceptor and
10 mM Fe2+ (as chloride or sulphate) as the electron donor to the mineral media
Control experiments were performed in the same media only they were not
inoculated Addition of ferrous iron to the media induced an immediate
precipitation of a solid whitish material The whitish precipitate was collected on
022 microm polyvinylidendifluorid (Durapore Millipore) filters and stored in an
anoxic atmosphere until further measurements Old outgrown media suspensions
that had turned reddish in color due to a precipitate produced by the denitrifying
FeOB were used as inocula Inoculum volume was kept lt1 of the culture volume
in order to prevent the reddish inoculum from dominating over the initial whitish
precipitate Cultures were incubated in the dark at room temperature and gently
agitated once every day Typical color successions for the media were initial
whitish precipitates turning first more and more greenish over time and then finally
turning reddish (see Fig 22) The color developed uniformly without any
indications of multiple phases in the precipitate At different time intervals
suspension samples were withdrawn using 90 N210 CO2-flushed polyethylene
syringes The precipitates were collected on 022 microm polyvinylidendifluorid
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 25
(Durapore Millipore) filters and analyzed by Moumlssbauer spectroscopy and SEM
Nonfiltered suspension samples were digested in 01 M HCl and chemically
analyzed for Fe(II) NO3- and NH4
+
224 Analytical methods
Fe2+ was determined using a modified phenanthroline method (Fadrus amp Maly
1975) Nitrate was quantified by ion chromatography (Morales et al 2000) and
ammonium was measured photometrically using the indophenol reaction (Rossum
amp Villarruz 1963)
23 Results and discussion
231 Identification of solid iron-containing phases
In most cases the mineral media for cultivating denitrifying FeOB contained 10
mM FeCl2 or FeSO4 4 mM NO3ndash 4 mM total phosphorus and 30 mM HCO3
- at pH
70 ([HCO3-] = 2138middot[CO3
2-] at pH 70) Whitish flocs precipitated immediately at
these initial conditions when Fe(II) was added to the media (Figure 22a) Such
colourless flocs have been reported to precipitate in similar mineral media (10 mM
Fe(II) 37-5 mM phosphate 22-30 mM HCO3ndash pH 70-72) (Ehrenreich amp
Widdel 1994 Chaudhuri et al 2001) Our Fe(II) measurements showed that 20-
50 of the total Fe(II) added was present in this initial white precipitate
26 Chapter 2
a
210- Figure 22 Colour of suspended material in the growth media during Fe(II) biooxidation a)
Initial whitish precipitate prior to inoculation b) Inte1mediate greenish phase fo1med within 2-3
days after inoculation c) reddish precipitate at late stage of biooxidation (gt5-6 days)
The precipitates were filtered and investigated by Mossbauer and IR spectroscopy
The transmission Mossbauer spectra obtained for the initial whitish precipitate at
temperatures between 20 and 250 K are shown in Figure 23 The spectrum
measured at 250 K consists of two fairly well-resolved Fe(II) doublets (see
parameters in Table 22) The change in line-overlap with decreasing temperature
is primarily ascribed to differences in the temperature dependence of the
quadrupole splitting of the two components From the spectra at 10 and 6 K
(Figure 24) it can be concluded that magnetic ordering takes place between these
two temperatures and that only one transition occurs (indicating the presence of
only one phase) The parameters of one of the Fe(II) doublets at 250 K (designated
B in Table 22) are in very good agreement with previously published values for
the vivianite Fe(Il)8 site at room temperature (eg McCammon amp Burns 1980)
and the ordering temperature also agrees well with an assignment as vivianite
(Forsyth et al 1970) However the second Fe(II) doublet in the initial colourless
precipitate (Table 22) has parameters that deviate from previously reported values
by having a smaller quadrupole splitting (indicating a less distorted coordination)
and a significantly higher relative intensity and line width These effects might be
due to the presence of numerous defects in the vivianite crystal lattice particularly
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 27
affecting the Fe(II)A sites It should be noted that further components may be added
to the fit in order to improve its statistics Nevertheless we decided to include no
further components as suggested by the finding of one magnetic ordering only
Accordingly our interpretation of the Moumlssbauer results for the initial white
precipitate suggests a highly defective vivianite having a distribution of local
coordination environments particular in the A site This assignment is further
supported by a major absorption band due to phosphate anions in the infrared
spectrum at approximately 1000 cm-1 and the absence of other complex anions
(data not shown) Thus the whitish precipitate is referred to as a vivianite-like
(ldquovivianiterdquo) precipitate
0
3 5
3 0
2 5
2 0
1 5
1 0
0 5
0 0
20 K
Velocity (mms)
Rel
ativ
e ab
sorp
tion
()
3
2
1
0
80 K
3 5
3 0
2 5
2 0
1 5
1 0
0 5
0 0
150 K
-5 -4 -3 -2 -1 0 1 2 3 4 5
2 5
2 0
1 5
1 0
0 5
0 0
250 K
Figure 23 Transmission Moumlssbauer spectra measured between 250 and 20 K of the initial
whitish precipitate prior to inoculation (see Fig 22a) Fitting components (and their sum) are
shown as full lines
28 Chapter 2
Table 22 Selected Mossbauer parameters of the doublet components in the spectra obtained for
different precipitates
Precipitates Temperature Isomer shift Quadrupole Line width Area (K) (mms-1) splitting (mms-1) (mms-1) ()
Whitish Fe(II)B 250 127 309 035 38
Fe(II)A 250 128 181 051 62
Dark greenish Fe(II)B 250 126 305 023 17
Fe(II)A 250 132 238 051 44
Fe(III) 250 036 085 040 38
Reddish-orange 40 138 244 094 67 Fe(II) Hyperfine parameters are generally given with uncertainties of 003 mms- the spectral area with an uncertainty of 3
1005
1000
0995
0990
- 0985 ~ e c 0980 0
-~ 0975 E c nl b 1000 g ~ Qi 0995 0
0990
0985
0980 -12 -8
~ -~
bullbull bull Ibull bull bullbull bull bull ~ bull bull ~
It
10 K
bull bull bull bull (J ~ i~ 6K bullmiddotf bull bull
~ bull bull bull
-4 0 4 8 12
Velocity (mmls)
Figure 24 Transmission Mossbauer spectra measured at 10 and 6 K of the initial whitish
precipitate prior to inoculation (see Fig 22a)
In general the color of the media suspension changed from whitish into light green
color within 2-3 days after inoculation (Figure 22b ) This transformation occurred
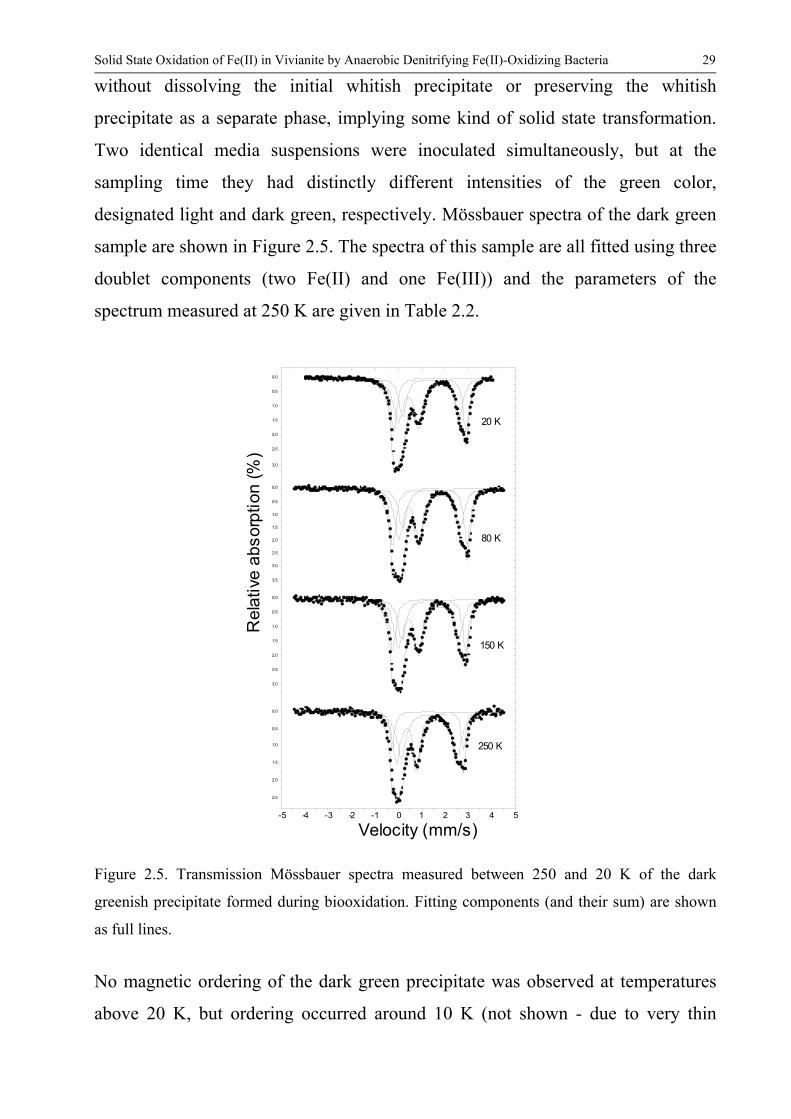
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 29
without dissolving the initial whitish precipitate or preserving the whitish
precipitate as a separate phase implying some kind of solid state transformation
Two identical media suspensions were inoculated simultaneously but at the
sampling time they had distinctly different intensities of the green color
designated light and dark green respectively Moumlssbauer spectra of the dark green
sample are shown in Figure 25 The spectra of this sample are all fitted using three
doublet components (two Fe(II) and one Fe(III)) and the parameters of the
spectrum measured at 250 K are given in Table 22
30
25
20
15
10
05
00
20 K
Velocity (mms)
Rel
ativ
e ab
sorp
tion
()
35
30
25
20
15
10
05
00
80 K
30
25
20
15
10
05
00
150 K
-5 -4 -3 -2 -1 0 1 2 3 4 5
25
20
15
10
05
00
250 K
Figure 25 Transmission Moumlssbauer spectra measured between 250 and 20 K of the dark
greenish precipitate formed during biooxidation Fitting components (and their sum) are shown
as full lines
No magnetic ordering of the dark green precipitate was observed at temperatures
above 20 K but ordering occurred around 10 K (not shown - due to very thin
30 Chapter 2
samples this was not investigated in details) The two greenish samples had very
similar parameters only differing in the relative intensity of Fe(III) 26 and 38
in the light greenish and dark greenish samples respectively Assuming the
spectral area of a component to be proportional to the abundance of the species in
the solid these results indicate a correlation between the intensity of the green
color and the content of Fe(III) in the precipitate The parameters of the Fe(II)
doublets in the dark green precipitate (Table 22) were in very good agreement
with previously published values for vivianite with a non-negligible Fe(III) content
(McCammon amp Burns 1980) whereas the Fe(III) component in particular had a
higher quadrupole splitting The observation that magnetic ordering of both Fe(II)
and Fe(III) occurred at similar temperature for the light green phase (Figure 26) is
a strong indication that they both belong to the same phase The absence of the
component with the low quadrupole splitting in the spectra of both green samples
might indicate that the initial vivianite-like phase crystallized into a more well-
defined vivianite over time However freshly prepared and long-term aged (gt1
year) suspensions of the initial vivianite-like precipitate did not differ significantly
Hence we suggest that the recrystallization of the vivianite-like precipitate can be
explained by Fe(II) biooxidation
Solid State Oxidation ofFe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria
0
2
3
- 4
~ 5 c ~ 6 e-0 7 -2 nl Q) 00 gt ~ 05 Qi 0 10
15
20
25
30
35
bullbullbull hi 6~
bullbullbullbull bull bull bullbull bull 10 K bull bull
bull bull bull bull bull bull bull
~~ ~~ lf 6K bull bull bullbull middot bull bull r bull
bull bull bull bull bull bullbull bull bull bull - 12 -a -4 0 4 8 12
Velocity (mms)
31
Figure 26 Transmission Mossbauer spectrn measured at 6 and 10 K of the light greenish
precipitate fonned during biooxidation
The solid state oxidation of monoclinic vivianite to triclinic metavivianite is well-
known (McCammon amp Bums 1980 Pratesi et al 2003 and references therein)
The vivianite crystal structure contains Fe(II) ions in both isolated Fe(II)A and
paired Fe(Il)8 octahedra Mossbauer spectroscopic analyses have shown that the
Fe(Il)8 Fe(II)A ratio increases with increasing Fe(III) concentration suggesting that
the remaining Fe(II)A ions are more readily oxidized than the Fe(II)a ion of an
Fe(Il)8 -Fe(III)8 pair (McCammon amp Bums 1980) The mechanism of oxidation of
Fe(II) in vivianite involves conversion of H20 ligands to OH- ions producing a
progressive collapse of the vivianite structure due to the elimination of hydrogen
bonds (Moore 1971) The exact oxidation limits between which the triclinic lattice
is stable are somewhat disputed as the results obtained for synthetic and natural
vivianites oxidized chemically in the laboratory and naturally oxidized natural
vivianite specimens do not agree completely (Rodgers 1986 and references
therein) Taking all reports into account the monoclinic structure of vivianite is
supposedly maintained until 40-50 of total iron is oxidized Further oxidation
32 Chapter 2
leads to the formation of the triclinic metavivianite in which the FeA site is fully
oxidized whereas the oxidation of the FeB ranges from 20 to almost 100 Thus
the triclinic metavivianite structure persists close to complete oxidation of total
iron The Moumlssbauer results obtained in this study are consistent with the vivianite
solid state oxidation mechanism reported by McCammon amp Burns (1980) Thus
we propose that the intermediate greenish precipitate is a metavivianite-like
(ldquometavivianiterdquo) phase It should be noted that a minor oxidation of dissolved
Fe(II) may have occurred even though the solid state oxidation of Fe(II) was
predominant
Within 5-6 days after inoculation the greenish intermediate was transformed into a
reddish product (Figure 22c) The magnetically ordered sextet in the spectrum of
the red phase (Figure 27) measured at 40 K was due to goethite (α-FeOOH)
(magnetic hyperfine field of 470 T and a quadrupole shift of -01 mms-1 cp
Moslashrup et al 1983) The sextet deviated from ideal goethite by its asymmetric line
shape and its low ordering temperature (around 100 K ndash data not shown) and thus
the goethite was poorly crystalline It is very likely that the presence of phosphate
in the media retarded the crystal growth of goethite The unusual reddish colour of
the goethite might also be explained by the presence of phosphate The spectrum at
40 K was however dominated by a Fe(II) doublet (Table 22) that ordered
magnetically between 40 and 20 K (Figure 27) The hyperfine parameters and the
magnetic ordering temperature indicated that this component was due to siderite
(FeCO3) having a magnetic ordering temperature of 38 K (Jacobs 1963) The
siderite component may have formed as a result of the microbial activity changing
the chemistry of the solution and precipitating a major part of the remaining
dissolved Fe(II) at this stage The characteristic vivianite Fe(II) doublet with large
quadrupole splitting was not detected in this sample The reddish precipitate
contained considerably less Fe(III) than the greenish precipitate (only 33 as
estimated from the spectral area) None of the components in the reddish sample
Solid State Oxidation ofFe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 33
could be detected in freshly inoculated samples indicating that carryover of mineral
precipitates by inoculation of the culture media was negligible
0
2
- 3
~4 c
Q 5
e 6 0
~ 7 g
0 3l Q)
a 1
2
3
4
5
-12 a
40K
bull bull (
4 0 4 8 12 Velocity (mms)
Figure 27 Transmission Mossbauer spectra measured at 20 and 40 K of the reddish precipitate
fo1med during the late biooxidation stage Fitting components (and their sum) are shown as full
lines
Strengite (FeP04middot2H20) was not detected at any time during oxidation
Santabarbaraite a new amorphous F e(III) hydroxy phosphate mineral
(Fe3(P04)i(OH)3middot5H20) was reported in a recent study (Pratesi et al 2003) The
brownish mineral was a result of the solid state oxidation of vivianite through
metavivianite However no Mossbauer data have been provided for this new
mineral yet and therefore we cannot give an account of whether santabarbaraite
forms in our system or not Thus goethite was the dominating end product and we
propose the reaction path depicted in Figure 28 for the nitrate-dependent
biooxidation of Fe(II) in our systems
34 Chapter 2
Fe3(PO4)2middot8H2O (FeII3-xFeIII
x)(PO4)2(OH)xmiddot(8-x)H2O α-FeOOH
NO3- N2 NO3
- N2
ldquoVivianiterdquo ldquoMetavivianiterdquo Goethite
Figure 28 Proposed reaction path and iron-containing minerals forming during solid state
oxidation of vivianite by denitrifying FeOB at the experimental conditions applied in this study
The biotic formation of layered Fe(II)-Fe(III) hydroxides (green rusts) by
anaerobic denitrifying Fe(II)-oxidizing bacteria has been suggested but proper
identification of these phases still lacks (Chaudhuri et al 2001) We cannot rule
out that small amounts of green rusts (GRs) perhaps a phosphate intercalated GR
(Hansen amp Poulsen 1999) might have been present here during the greenish
intermediate ldquometavivianiterdquo oxidation stage When present in low concentrations
especially in mixtures including other iron minerals it is very difficult to identify
GRs even with Moumlssbauer spectroscopy At least two complementary methods
such as X-ray diffraction (XRD) and Moumlssbauer spectroscopy are required for
proper identification and characterization of GRs However the precipitates
collected in this work were poorly crystalline and did not allow for XRD analysis
Electron micrographs including energy dispersive X-ray spectroscopy suspension
colour and mineral stability calculations do not suffice as evidence Hence no
convincing evidence of GR formation facilitated by denitrifying FeOB has been
provided so far The blue-green colours of metavivianite and green rust minerals
originate from Fe(II)-Fe(III) charge transfer between adjacent Fe(II) and Fe(III)
ions in edge-shared octahedra (Faye et al 1968) The greenish suspension colour
occurring during the intermediate phase has incited the idea of biogenic GRs in
studies on nitrate-dependent Fe(II) biooxidation (Chaudhuri et al 2001 Lack et
al 2002aampb) However our results indicate that this reasoning is misleading
232 Factors controlling the rate and extent of Fe(II) biooxidation
Generally it was found that maximally 20-64 of the initial Fe(II) amount was
oxidized to Fe(III) (Figure 29) This indicates some limitations in the accessibility
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 35
of Fe(II) in the system Based solely on stoichiometry considerations the
microorganisms are expected to oxidize 5 mol Fe(II) for every 1 mol nitrate
reduced to dinitrogen However as exemplified in Figure 28 this ratio was mostly
lt4 which can be explained by the consumption of nitrogen as a result of microbial
growth At initial [Fe(II)][NO3ndash] ratios lt5 nitrate is in excess and should not limit
the extent of the biooxidation Thus the lack of complete biooxidation could not be
due to exhaustion of nitrate Furthermore all growth essential nutrients were more
than sufficiently applied hence the incomplete Fe(II) biooxidation was not caused
by lack of nutrients The most reasonable explanation therefore seems to be that an
increasingly limited access to the electron donor over time inhibits complete long-
term Fe(II) biooxidation At least four mechanisms may cause this inhibition 1)
the Fe(II) becomes isolated within the structure of the mixed Fe(II)-Fe(III)
minerals forming during biooxidation or underneath a passive Fe(III)-bearing
surface film on the initial Fe(II) precipitates 2) the FeOB cell surface becomes
covered with a passive Fe(III)-bearing surface film 3) the Fe(II) biooxidation is
controlled by the rate of dissolution of the initial Fe(II) minerals or 4) the reaction
proceeds primarily by biooxidation of dissolved Fe(II) whose concentration
gradually decreases due to changes in solid phase composition The actual
mechanisms whereby the surface-associated Fe(III) can inhibit Fe(II) biooxidation
are unknown but they may involve both kinetic and thermodynamic constraints on
the electron transfer The Moumlssbauer results obtained in this work strongly suggest
that the Fe(II) biooxidation occurred mainly in the solid state of the initial
ldquovivianiterdquo phase However we cannot rule out that some dissolved Fe(II) was
oxidized as well
36 Chapter 2
Figure 29 Concentration profiles of total Fe(II) and nitrate as a function of time during Fe(II)
biooxidation
No Fe(II) oxidation took place in cultures where nitrate had been omitted
confirming that the microbial Fe(II) oxidation is nitrate-dependent (data not
shown) No Fe(II) oxidation was detected in the non-inoculated control
experiments within the duration of the experiments and thus the chemical
oxidation of dissolved Fe(II) by nitrate catalyzed by vitamins or trace elements
(eg Cu(II)) can be neglected Ammonium did not form in detectable amounts
during Fe(II) biooxidation (data not shown) and therefore dinitrogen was assumed
to be the end product as reported previously (Straub et al 1996 Benz et al 1998)
The absence of ammonium formation does indirectly support the absence of
biologically induced green rust formation as synthetic green rust is known to
convert nitrate into ammonium in purely chemical reactions (Hansen et al 1996)
It was visually observed that the phosphate concentration in the media exerted a
control on the microbial Fe(II)-oxidation At phosphorus concentrations le 2 mM
no Fe(II)-oxidation took place However the solubility product for vivianite (Ksp =
171middot10-36 at 25degC (Al-Borno et al 1994)) was still by far exceeded under these
conditions It is not known whether this phosphate limiting effect is due to growth
constraints in the mixed bacterial community or whether specific Fe(II) phosphate
Solid State Oxidation ofFe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 37
precipitates are prerequisites of the Fe(II) biooxidation to take place Experiments
are currently underway in our laboratory in order to elucidate the role of specific
initial Fe(II) precipitates It should be noted that the growth of the denitrifying
FeOB could not be estimated as they were present in highly heterogeneous
suspensions containing both solid iron phases as well as other bacteria (enrichment
culture)
233 Morphology of solid iron phases
The morphology of the various precipitates was studied by SEM The initial
whitish precipitate consisted of a web-like structure (Figure 21 Oa and background
in Figure 21 Ob) whereas more distinct hexagonally shaped rosettes with particle
size ~20 microm (Figure 2lObampc) formed during Fe(II) biooxidation The energy
dispersive spectroscopic element analyses showed that other than iron the initial
whitish precipitate and the rosettes contained mainly phophorus
Figure 210 Scanning electron micro graphs of precipitates fo1med at various stages of the
experiment a Initial whitish precipitate bampc Rosettes observed in the intennediate greenish
precipitate d Reddish precipitate sampled during the late biooxidation stage
38 Chapter 2
These observations are interpreted as vivianite forming a web-like morphology in
the initial whitish precipitate and partly transforming into hexagonal particles in
the greenish colored stage The interpretations are supported by similar vivianite
morphologies reported including pseudo-hexagonal vivianite crystals of low
symmetry resulting from microbial Fe(III) reduction of HFO and platy rosettes of
vivianite crystals formed during bioreduction of Fe(III) in smectite (Fredrickson et
al 1998 Dong et al 2003) It was not possible to associate the morphology
observed in the reddish precipitate with the minerals identified in this phase
(Figure 210d)
24 Conclusions
This work demonstrated that anaerobic autotrophic denitrifying Fe(II)-oxidizing
bacteria produce poorly crystalline goethite by solid state oxidation of ldquovivianiterdquo
via a ldquometavivianiterdquo intermediate The increasing amount of Fe(III) forming in the
vivianite structure was accompanied by an increasing intensity in the green colour
as the Fe(II) biooxidation progressed Moumlssbauer spectroscopic analyses provided
no significant evidence of green rust formation The finding of microbially
oxidized vivianite in this study raises the question of the oxidation state of
vivianite specimens from natural sediments Vivianite is generally believed to be
an ideal Fe(II) hydroxy phosphate mineral and the presence of Fe(III) is explained
by aerial oxidation upon sampling The results presented here indicate that
microbiological processes may be responsible for the oxidation of vivianite and
metavivianite in natural subsurface environments Acknowledgments
We would like to thank Dr K Straub for providing and advising us how to culture the nitrate-
reducing FeOB Furthermore we thank Dr C B Koch for performing the Moumlssbauer analyses
and Dr D Mavrocordatos for performing the SEM analyses
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 39
References Al-Borno A Tomson MB (1994) The temperature dependence of the solubility product constant of vivianite Geochimica et Cosmochimica Acta 58 5373-5378 Benali O Abdelmoula M Refait Ph Geacutenin JMR (2001) Effect of orthophosphate on the oxidation products of Fe(II)-Fe(III) hydroxycarbonate The transformation of green rust to ferrihydrite Geochimica et Cosmochimica Acta 65 1715-1726 Benz M Brune A Schink B (1998) Anaerobic and aerobic oxidation of ferrous iron at neutral pH by chemohetorotrophic nitrate-reducing bacteria Archives of Microbiology 169 159-165 Chaudhuri SK Lack JG Coates JD (2001) Biogenic magnetite formation through anaerobic biooxidation of Fe(II) Applied and Environmental Microbiology 67 2844-2848 Cumplido J Barron V Torrent J (2000) Effect of phosphate on the formation of nanophase lepidocrocite from Fe(II) sulfate Clays and Clay Minerals 48 503-510 Dodd J Large DJ Fortey NJ Kemp S Styles M Wetton P Milodowski A (2003) Geochemistry and petrography of phosphorus in urban canal bed sediment Applied Geochemistry 18 259-267 Dong H Kostka JE Kim J (2003) Microscopic evidence for microbial dissolution of smectite Clays and Clay Minerals 51 502-512 Ehrenreich A Widdel F (1994) Anaerobic oxidation of ferrous iron by purple bacteria a new type of phototrophic metabolism Applied and Environmental Microbiology 60 4517-4526 Emerson D Moyer CL (2002) Neutrophilic Fe-oxidizing bacteria are abundant at the Loihi seamount hydrothermal vents and play a major role in Fe oxide deposition Applied and Environmental Microbiology 68 3085-3093 Emerson D Weiss JV Megonigal JP (1999) Iron-oxidizing bacteria are associated with ferric hydroxide precipitates (Fe-plaque) on the roots of wetland plants Applied and Environmental Microbiology 65 2758-2761 Fadrus H Maly J (1975) Suppression of iron(III) interference in the determination of iron(II) in water by the 110-phenanthroline method The Analyst 100 549-554 Faye GH Manning PG Nickel EH (1968) The polarized optical absorption spectra of tourmaline cordierite chloritoid and vivianite ferrous-ferric electronic interaction as a source of pleochroism American Mineralogist 53 1174-1201 Forsyth JB Johnson CE Wilkonson C (1970) The magnetic structure of vivianite Fe3(PO4)2middot8H2O Journal of Physics Part C Solid State Physics 3 1127-1139 Fredrickson JK Zachara JM Kennedy DW Dong H Onstott TC Hinman NW Li S (1998) Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium Geochimica et Cosmochimica Acta 62 3239-3257 Gaines RV Skinner HCW Foord EE Mason B Rosenzweig A (1997) Danas new
40 Chapter 2 mineralogy 8th ed John Wiley amp Sons Inc Hanert HH (1992) The genus Gallionella In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 4082-4088 Hansen HCB Koch CB Nancke-Krogh H Borggaard OK Soerensen J (1996) Abiotic nitrate reduction to ammonium Key role of green rust Environmental Science and Technology 30 2053-2056 Hansen HCB Poulsen IF (1999) Interaction of synthetic sulphate green rust with phosphate and the crystallization of vivianite Clays and Clay Minerals 47 312-318 Henderson GS Black PM Ridgers KA Rankin PC (1984) New data on New Zealand vivianite and metavivianite New Zealand Journal of Geology and Geophysics 27 367-378 House WA (2003) Geochemical cycling of phosphorus in rivers Applied Geochemistry 18 739-748 Jacobs IS (1963) Metamagnetism of siderite (FeCO3) Journal of Applied Physics 34 1106-1107 Jorand F Appenzeller BMR Abdelmoula M Refait Ph Block J-C Geacutenin JMR (2000) Assessment of vivianite formation in Shewanella putrefaciens culture Environmental Technology 21 1001-1005 Kandori K Uchida S Kataoka S Ishikawa T (1992) Effects of silicate and phosphate ions on the formation of ferric oxide hydroxide particles Journal of Materials Science 27 719-728 Kuenen JG Robertson LA Tuovinen OH (1992) The genera Thiobacillus Thiomicrospira and Thiosphaera In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 2618-2624 Lack JG Chaudhuri SK Chakraborty R Achenbach LA Coates JD (2002a) Anaerobic biooxidation of Fe(II) by Dechlorosoma suillum Microbial Ecology 43 424-431 Lack JG Chaudhuri SK Kelly SD Kemner KM OConnor SM Coates JD (2002b) Immobilization of radionuclides and heavy metals through anaerobic bio-oxidation of Fe(II) Applied and Environmental Microbiology 68 2704-2710 Lovley DR (1997) Microbial Fe(III) reduction in subsurface environments FEMS Microbiology Reviews 20 305-313 Manning PG Birchall T Jones W (1981) Ferric hydroxides in surficial sediments of the great lakes and their role in phosphorus availability a Moumlssbauer spectral study Canadian Mineralogist 19 525-530 Manning PG Jones W (1982) The binding capacity of ferric hydroxides for non-apatite inorganic phosphorus in sediments of the depositional basins of Lakes Erie and Ontario Canadian Mineralogist 20 169-176 McCammon CA Burns RG (1980) The oxidation mechanism of vivianite as studied by Moumlssbauer spectroscopy American Mineralogist 65 361-366 Moore PB (1971) The Fe2+
3(H2O)n(PO4)2 homologous series crystal-chemical relationships
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 41
and oxidized equivalents American Mineralogist 56 1-17 Moraghan JT Buresh RJ (1976) Chemical reduction of nitrate by ferrous iron Journal of Environmental Quality 5 320-325 Moraghan JT Buresh RJ (1977) Chemical reduction of nitrite and nitrous oxide by ferrous iron Journal of American Soil Science Society 40 47-50 Morales JA de Graterol LS Mesa J (2000) Determination of chloride sulfate and nitrate in groundwater samples by ion chromatography Journal of Chromatography A 884 185-190 Mulder EG Deinema MH (1992) The sheathed bacteria In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 2618-2624 Moslashrup S Madsen MB Franck J Villadsen J Koch CJW (1983) A new interpretation of Moumlssbauer spectra of microcrystalline goethiterdquosuper-ferromagnetismrdquo of ldquosuper-spin-glassrdquo behaviour Journal of Magnetism and Magnetic Materials 40 163-174 Nembrini GP Capobianco JA Viel M Williams AF (1983) A Moumlssbauer and chemical study of the formation of vivianite in sediments of Lago Maggiore (Italy) Geochimica et Cosmochimica Acta 47 1459-1464 Nielsen JL Nielsen PH (1998) Microbial nitrate-dependent oxidation of ferrous iron in activated sludge Environmental Science and Technology 32 3556-3561 Nriagu JO (1972) Stability of vivianite and ion-pair formation in the system Fe3(PO4)2-H3PO4- H2O Geochimica et Cosmochimica Acta 36 459-470 Nriagu JO Dell CI (1974) Diagenetic formation of iron phosphates in recent lake sediments American Mineralogist 59 934-946 Patrick Jr WH Khalid RA (1974) Phosphate release and sorption by soils and sediments Effect of aerobic and anaerobic conditions Science 186 53-55 Postma D (1981) Formation of siderite and vivianite and the pore-water composition of a recent bog sediment in Denmark Chemical Geology 31 225-244 Pratesi G Cipriani C Giuli G Birch WD (2003) Santabarbaraite a new amorphous phosphate mineral European Journal of Mineralogy 15 185-192 Ratering S Schnell S (2001) Nitrate-dependent iron(II) oxidation in paddy soil Environmental Microbiology 3 100-109 Ritz C Essene EJ Peacor DR (1974) Metavivianite Fe3(PO4)2middot8H2O a new mineral American Mineralogist 59 896-899 Rodgers KA (1986) Metavivianite and kerchenite a review Mineralogical Magazine 50 687- 691 Rodgers KA Johnston JH (1985) Type metavivianite Moumlssbauer evidence for a revised composition Neues Jahrbuch fuumlr Mineralogie-Monatshefte 12 539-542
42 Chapter 2 Rossum JR Villarruz PA (1963) Determination of ammonia by the indophenol method Journal of American Water Works Association 55 657-658 Seitz MA Riedner RJ Malhotra SK Kipp RJ (1973) Iron-phosphate compound identification in sewage sludge residue Environmental Science and Technology 7 354-357 Sobolev D Roden EE (2001) Suboxic deposition of ferric iron by bacteria in opposing gradients of Fe(II) and oxygen at circumneutral pH Applied and Environmental Microbiology 67 1328-1334 Straub KL Benz M Schink B Widdel F (1996) Anaerobic nitrate-dependent microbial oxidation of ferrous iron Applied and Environmental Microbiology 62 1458-1460 Straub KL Buchholz-Cleven BEE (1998) Enumeration and detection of anaerobic ferrous iron-oxidizing nitrate-reducing bacteria from diverse European sediments Applied and Environmental Microbiology 64 4846-4856 Straub KL Hanzlik M Buchholz-Cleven BEE (1998) The use of biologically produced ferrihydrite for the isolation of novel iron-reducing bacteria Systematic and Applied Microbiology 21 442-449 Weber KA Picardal FW Roden EE (2001) Microbially catalyzed nitrate-dependent oxidation of biogenic solid-phase Fe(II) compounds Environmental Science and Technology 35 1644-1650 Widdel F Bak F (1992) Gram-negative mesophilic sulfate-reducing bacteria In The Prokaryotes (Balows A Truumlper HG Dworkin M Harder W Schleifer K-H (eds)) Springer 2nd ed 3352-3378
Widdel F Schnell S Heising S Ehrenreich A Assmus B Schink B (1993) Ferrous iron oxidation by anoxygenic phototrophic bacteria Nature 362 834-836 Williams JDH Syers JK Shukla SS Harris RF Armstrong DE (1971) Levels of inorganic and total phosphorus in lake sediments as related to other sediment parameters Environmental Science and Technology 5 1113-1120
Zwaan PC Kortenbout van der Sluys G (1971) Vivianite crystals from Hare Noord Brabant Province The Netherlands Scripta Geology 6 1-7
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 43
3 Formation of Layered Iron Hydroxides by Microbial Fe(III)
Reduction Abstract
Many inorganic and organic pollutants may be degraded by microorganisms in the
subsurface However a wide range of contaminants including chromate nitrate
radionuclides nitroaromatic compounds chlorinated aliphatics and carbamate
pesticides may also be chemically transformed by reduction reactions involving
layered iron(II)-iron(III)-hydroxides (green rusts) Hence green rusts (GRs) may
play a potentially important role in the fate and transport of pollutants in iron-rich
suboxic soils and sediments Yet only little is known about the formation of GRs
in these environments The biotic formation of GRs mediated by the anaerobic
dissimilatory Fe(III)-reducing bacteria Shewanella spp has been reported and
proposed in several studies However the experimental conditions applied were
mostly not natural and the evidence of GR formation provided may be questioned
This work investigated the Fe-containing products formed by the facultatively
anaerobic Fe(III)-reducing microorganism Shewanella algae BrY in culture
media containing 4-10 mM formate or lactate and 8-27 mM Fe(III) In order to
simulate natural conditions Fe(III) oxides were applied as coatings on silica
(model system for sandy soils) or calcite particles (model system for calcareous
soils) and synthetic electron shuttles as well as highly concentrated artificial pH
buffers were excluded S algae BrY reduced 19-72 of the initial Fe(III) when
grown in goethitecalcite lepidocrocitecalcite or hydrous ferric oxidesand mineral
systems and green or blackish mineral phases were produced within 1-2 weeks
after inoculation Moumlssbauer spectroscopic analyses indicated that the green and
blackish precipitates were dominated by vivianite (Fe3(PO4)2sdot8H2O) and green rust
44 Chapter 3
31 Introduction
The significance of bacteria in the biogeochemical cycling of iron has been broadly
recognized over the past two decades Chemical processes were previously
considered to account for most of the Fe(III) reduction in subsurface environments
Dissimilatory Fe(III)-reducing bacteria (DIRB) that gain energy by coupling the
oxidation of hydrogen or organic compounds to the reduction of Fe(III) oxides
have been known for many years but their biogeochemical importance was
acknowledged only a decade ago (reviewed by Lovley 1997) DIRB transfer
electrons to extracellular Fe(III) without assimilating the iron Fe(III) bioreduction
accounts for a major fraction of the carbon oxidation in many different
environments and in the presence of high amounts of reactive Fe(III) microbial
Fe(III) reduction may even inhibit sulfate reduction and methanogenesis (King
1990 Lovley amp Phillips 1986) In fact most of the Fe(III) reduction in the Fe(III)
reduction zone of aquatic sediments and aquifers is thought to be enzymatically
catalyzed by microorganisms (Lovley et al 1991) However the relative
importance of microbial and chemical processes involved in the Fe(III) reduction
are still somewhat disputed among microbiologists and geochemists
A wide diversity of Fe(III)-reducing bacteria which fall in a number of different
phylogenetic groups is known today Both organisms growing by respiration and
by fermentation have been isolated and identified (Lovley 1991 Nealson amp
Saffarini 1994) Hydrogen short- and long-chained fatty acids amino acids
sugars and aromatic compounds may serve as electron donors for Fe(III)
bioreduction The enzymes responsible for dissimilatory Fe(III) reduction are outer
membrane associated ferric reductases (Lower et al 2001 and references therein)
Iron reducing bacteria may utilize alternative electron acceptors such as O2 nitrate
S0 sulfate humic substances contaminant metals and metalloids as well as
chlorinated solvents The first organism shown to couple respiratory growth to
dissimilatory iron reduction was Pseudomonas ferrireductans now known as
Shewanella oneidensis but previously classified as Alteromonas putrefaciens and
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 45
Shewanella putrefaciens (Venkateswaran et al 1999) Various DIRB including
the obligate anaerobic Geobacter sp and the facultatively anaerobic Shewanella
sp have been isolated from both marine and freshwater sediments soil and
aquifers (Thamdrup 2000 and references therein)
The redox potentials of oxidized and reduced iron couples and thus the energy
yield available from Fe(III) reduction depend strongly on the specific iron phases
involved In soil and aquatic environments Fe(III) oxides mainly occur in
association with other sediment particles as aggregates or coatings Amorphous
and poorly crystalline Fe(III) oxides usually make up 20 or less of the iron
content in a sediment (Thamdrup 2000) They are the main products of abiotic and
biotic Fe(II) oxidation in sediments and they constitute the most important phases
for microbial Fe(III) reduction Until recently it was generally believed that DIRB
reduced insoluble Fe(III) oxides only by direct contact with the Fe(III) oxide
thereby allowing electron transfer from the cell to the Fe(III) oxide surface
However over the past several years there has been a growing recognition that
DIRB may use different strategies in order to access the solid Fe(III) oxides These
strategies include solubilization of Fe(III) by synthetic or natural Fe(III) chelators
and Fe(III) reduction via electron shuttling with soluble humic substances or
microbially produced electron shuttles (Nevin amp Lovley 2002 and references
therein Turick et al 2003) The Fe(III) complexing agents may also stimulate
Fe(III) oxide reduction indirectly by chelation and thus removal of Fe(II) from
the cell and the Fe(III) oxide surfaces Both chelated Fe(III) and soluble electron
shuttles are more accessible to Fe(III) reductases than solid Fe(III) oxides In
contrast to Geobacter metallireducens S algae produces and releases extracellular
electron shuttling compounds (Nevin amp Lovley 2000) However in the absence of
soluble electron shuttles reversible adhesion is required for reduction of solid
Fe(III) oxides by S algae BrY (Das amp Caccavo 2000) Shewanella algae BrY
adheres readily and preferentially to a range of solid Fe(III) oxides such as
ferrihydrite goethite and hematite (Das amp Caccavo 2001) The adhesion
46 Chapter 3
mechanisms are not completely understood but recent results suggest that the
adhesion is mediated by cell surface proteins and independent of cell motility
(Caccavo amp Das 2002)
The microbial formation of GRs resulting from bioreduction of various Fe(III)
oxides including ferrihydrite goethite and lepidocrocite by strains of the
anaerobic DIRB Shewanella putrefaciens has been reported repeatedly over the
last years (Fredrickson et al 1998 Kukkadapu et al 2001 Liu et al 2001
Parmar et al 2001 Ona-Nguema et al 2002aampb Glasauer et al 2003)
However no evidence of biogenic formation of GRs at natural geochemical
conditions have been offered and it is still unknown whether this process may take
place at natural conditions comprising low carbon and iron concentrations as well
as the absence of synthetic electron shuttles and highly concentrated artificial pH
buffers GRs are layered iron(II)-iron(III)-hydroxides with anionic interlayers and
they hold the general formula [FeII(6-x)FeIII
x(OH)12]x+[(A)xnmiddotyH2O]x- where x = 09 -
42 A is an n-valent anion eg CO32- Clndash or SO4
2- and y is the number of water
molecules in the interlayer In circumneutral solutions the oxidation of dissolved
Fe(II) always passes through solid GR phases (Bernal et al 1959) This agrees
with the natural GR occurrences found in suboxic nonacid iron-rich environments
such as hydromorphic soils and intertidal sediments (Al-Agha et al 1995 Trolard
et al 1996 Genin et al 1998) In addition GRs have been found as corrosion
products in numerous engineering systems eg in a pipeline distribution system
for drinking water steel sheet piles in marine sediments reinforced concrete
(ferro-concrete) and permeable reactive barriers of zero-valent iron implemented
for on-site remediation of organic and inorganic contaminants (Tuovinen et al
1980 Nielsen 1976 Genin et al 1991 Roh et al 2000) Through sequestration
and reductive transformation GRs may play an important role in the fate and
transport of organic and inorganic pollutants in suboxic iron-rich soils and
sediments (see Chapters 4 amp 5 in this work and references therein)
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 47
The major goal of this work was to examine the iron minerals forming during the
course of Fe(III) bioreduction of hydrous ferric oxide goethite and lepidocrocite
Two model systems simulating sandy and calcareous soils in subsurface
environments were designed in order to investigate the formation of iron minerals
at conditions including low carbon levels low Fe(III) concentrations applied as
Fe(III) oxide coatings on sand or calcite no electron shuttle and no synthetic pH
buffers
32 Materials and methods
All handling and sampling of solutions and suspensions were carried out at strict
anoxic conditions Standard sterile techniques were used throughout (Hungate
1969 Miller amp Wolin 1974) Only the iron oxide coatings were not autoclaved in
order to avoid the iron oxides from transforming Goethite (acicular particles with
size 01 times 06 microm specific surface area 16 m2g) and lepidocrocite (acicular
particles with size 005 times 03 microm specific surface area 18 m2g) were purchased as
fine powders from Bayer (Bayferrox 910 and 943) Calcite (grain size 170-350 microm
Pluumlss-Staufer AG) and sea sand (dominantly quartz grain size 01-03 mm Riedel-
de Haeumln) were used as Fe(III) oxide coating bearing minerals
321 Preparation of iron oxide coatings
Two grams of goethite (goe) or lepidocrocite (lep) and 100 g calcite were
combined with 200 mL deionized water (DIW) in a 500 mL polyethylene flask
Hydrous ferric oxide (HFO) was synthesized by dissolving 4 g Fe(NO3)3middot9H2O in
70 mL DIW followed by slow neutralization under magnetic stirring till pH 7 with
approximately 30 mL 1 M NaOH (method modified after Schwertmann amp Cornell
1991) The HFO coating was made by combining 100 mL freshly precipitated
HFO with 900 mL deionized water and 50 g sea sand in a polyethylene bottle The
suspensions containing the iron oxide coatings were gently agitated on a
reciprocating shaker for 24 h and left to stand for another 24 h Excess Fe(III)
oxides and salts were removed from the coated material by repeated decantation
48 Chapter 3
and washing with 003 M NaNO3 followed by washing with DIW until clear
runoff Finally the coatings were collected on folding filters and air dried The
amount of HFO goethite and lepidocrocite coated onto sand and calcite after
washing and drying was quantified to 7-11 mg Fe(III)g sand or calcite
322 Mineral characterisation
The identity and purity of the HFO synthesized were examined by means of X-ray
diffraction (XRD) measurements The XRD analyses were performed on a Scintag
XDS 2000 using Co Kα radiation (45 kV 40 mA) using divergence scatter and
receiving slits of 1deg 05deg and 02 mm respectively Samples were scanned
between 6 and 80 deg2θ with a scan speed of 1 deg2θmin Mineral suspension samples
for transmission Moumlssbauer spectroscopic analysis were collected on 02 microm filters
in an anoxic glove box (Coy Laboratory Products Inc) transferred to Perspex
capsules and stored in liquid nitrogen until measurement Moumlssbauer spectra were
obtained between 250 and 5 K using a conventional constant acceleration
spectrometer and a source of 57Co in Rh The spectrometer was calibrated using a
125 microm foil of α-Fe at room temperature and isomer shifts are given relative to
the centroid of this absorber The spectra were fitted using simple Lorentzian line
shape and it was assumed that all positions have identical f-factors
323 Culture conditions and cell preparation
Shewanella algae BrY is a motile gram-negative rod which was isolated first from
anoxic estuary sediments (Caccavo et al 1992) S algae BrY was grown
aerobically in tryptic soy broth (30 gL CASO-bouillon Merck) at 28degC on a rotary
shaker at 150 rpm for 16-18 h Cells were harvested by centrifugation (6000 rpm times
g 4ordmC 15 min) during the late exponential ndash early stationary growth phase at
OD660 ~ 06 Optimal Fe(III) reductase activity is expressed at this stage of growth
(Roden amp Zachara 1996) The cells were washed twice in oxic 50 mM PIPES
[piperazine-NNacute-bis(2-ethanesulfonic acid)] buffer (pH 70) and resuspended in
culture medium containing no Fe(III) and no carbon source Washed cell
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 49
suspensions were used as inocula for Fe(III) reduction experiments Oxygen was
expelled from the inoculum by extensive purging with 100 N2(g) (9999999
purity) Working stock cultures of S algae BrY were maintained aerobically on
tryptic soy agar plates at ambient temperature
324 Bioreduction experiments
All anaerobic incubations were carried out in anoxic serum vials (25 mL) or test
tubes (13 mL) sealed with thick (10-13 mm) butyl rubber stoppers and aluminum
crimp caps or plastic screw caps The basal culture medium (Table 31) was
prepared according to Kostka amp Nealson (1998) but with a phosphate
concentration of 2 mM and the exclusion of Fe(II) and EDTA
(ethylenediaminetetraacetic acid) The medium was amended with 4-10 mM
lactate or formate and 8-27 mM Fe(III) The Fe(III) was applied as Fe(III) oxide
coatings on sand or calcite The suspensions were purged extensively with 100
N2(g) (HFOsand suspensions) or 995 N205 CO2(g) (goecalcite and
lepcalcite suspensions) prior to inoculation The calcareous systems were buffered
at pH ~ 76 through a natural buffer system (CaCO3(s) + 995 N205 CO2(g))
whereas the sandy systems contained no pH buffer (100 N2(g) pH 55-60)
Inoculum size made up 5 of the total volume Cultures were incubated dark at
room temperature and gently agitated once every day At different time intervals
suspension samples for Fe(II) and Moumlssbauer analysis were withdrawn from the
reaction mixture using 100 N2(g) or 995 N205 CO2(g)-flushed sterile
disposable syringes and hypodermic needles Suspension samples for Fe(II)
analysis were digested in 01 M HCl for 30 min
50 Chapter 3
Table 31 Composition of the mineral medium (modified from Kostka amp Nealson (1998))
Components Concentration (M)
(NH4)2SO4 00143 KH2PO4 73middot10-4
K2HPO4 13middot10-3
MgSO4middot7H2O 10middot10-3
CaCl2middot2H2O 50middot10-4
H3BO3 56middot10-5
ZnSO4middot7H2O 10middot10-6
Na2MoO4middot2H2O 40middot10-6
CuSO4middot5H2O 20middot10-7
MnSO4middotH2O 10middot10-6
Na2SeO4 12middot10-5
CoCl2middot6H2O 50middot10-6
NiCl2middot6H2O 80middot10-6
NaCl 10middot10-5
L-arginine 11middot10-4
L-serine 19middot10-4
L-glutamic acid 14middot10-4
Lactate or formate 4-10middot10-3
Fe(III) 8-27middot10-3
325 Analytical methods
Fe(II) was determined using a modified phenanthroline method (Fadrus amp Maly
1975) The total amount of Fe(III) coated on calcite and sand was determined by
atomic absorption spectroscopy following dissolution in 6 M HCl(aq) for 24 h
33 Results and discussion
331 Fe(II) production and suspension colour changes
Strongly chelating agents such as EDTA were omitted from the culture medium in
order to prevent complexation of Fe(II) and Fe(III) which interferes with
precipitation of Fe(II) and Fe(II)-Fe(III) mineral phases Within 1-2 weeks after
inoculation Shewanella algae BrY produced green mineral phases in media
suspensions containing lepidocrocite and goethite as coatings on calcite and 4-10
mM formate or lactate (Figure 31) The formation of the green precipitates was
generally slower for the lepidocrocite coating than for the goethite coating The
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 51
blue-green colours of the phases produced most likely originate from Fe(II)-Fe(III)
charge transfer between adjacent Fe(II) and Fe(III) ions in edge-shared octahedra
(Faye et al 1968) Dark brown and blackish products were formed when the
bacteria were inoculated on HFO coated sand (Figure 32)
Figure 31 Culture tubes containing a) goethite and b) lepidocrocite coated calcite in culture
medium The left tubes of the pair were not inoculated whereas the right tubes were
photographed 5 months after inoculation with S algae BrY Experimental conditions [formate]0
= 4 mM [Fe(III)]0 = 8 mM 995 N205 CO2(g) pH 76
52 Chapter 3
Figure 32 Culture tubes containing HFO coated sand in culture medium Tubes 1 and 2 to the
left were not inoculated whereas tubes 3-5 to the right were photographed a) 13 days and b) 21
days after inoculation with S algae BrY Experimental conditions [lactate]0 = 10 mM [Fe(III)]0
= 25 mM 100 N2(g) pH 55-60
The green and black colours did not change to other colours (observed for gt1
year) indicating that the microbial Fe(III) reduction ceased at these mineral stages
The concentrations of dissolved ferrous iron (Fe(II)sol) estimated during Fe(III)
bioreduction were generally low (Figure 33) When comparing the final Fe(II)sol
amounts produced and the slopes of the Fe(II)sol formation curves for HFO
goethite and lepidocrocite in Figure 33 it can be seen that the final Fe(II)sol
amount and the Fe(II)sol production rate both follow the order HFO gt goethite gt
lepidocrocite at similar cell densities regardless of the carbon source applied This
suggests that bioreduction by S algae BrY is more facile for HFO than for goethite
and lepidocrocite at the experimental conditions employed here It should be noted
that the final Fe(II)sol amounts and the Fe(II)sol production rates reported in this
work have not been normalised with respect to the specific surface areas of the iron
oxides and coating-bearing solids applied The reactivity trend is consistent with
previous findings demonstrating higher reducibility of natural and poorly
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 53
crystalline Fe(III) oxides as compared to synthetic crystalline Fe(III) oxides
(Zachara et al 1998) The authors ascribed these differences in reducibility to
differences in particle size surface area and crystal defects of the Fe(III) oxides In
some cases the dissolved Fe(II) concentration decreased again with time (Figure
33 b-d) This indicates that the Fe(II) formed was incorporated into solid phases
forming andor adsorbed onto the calcite sand or Fe(III) oxide surfaces The solid
Fe(II) concentrations were not estimated spectrophotometrically The solid
material was generally low in total iron and therefore saving it for Moumlssbauer
spectroscopic analysis was given highest priority
54 Chapter 3
Figure 33 Time course of dissolved Fe(II) production during bioreduction of HFOsand
goethitecalcite and lepidocrocitecalcite by S algae BrY Experimental conditions [Fe(III)]0 =
25 mM [formate]0 = 10 mM (a-c) or [lactate]0 = 10 mM (d-f)
No color change and no Fe(II) production were observed in mineral suspensions
lacking either a carbon source or S algae BrY cells (data not shown)
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 55
332 Identification of solid iron phases
The purity of the Fe(III) oxides used in the experiments were investigated by
transmission Mossbauer spectroscopy (Figure 34) Single (or strongly dominating)
sextets in the spectra with magnetic hyperfine fields of 48 1 505 and 45 5 Tat 5 K
demonstrated the purity of the HFO goethite and lepidocrocite samples
respectively A minor impurity of goethite in the lepidocrocite sample was
resolved in the spectrum measured at 80 K (not shown for pure sample but can be
seen as a magnetically ordered sextet in Figure 35c) No Fe(II)-containing
components were detected
a)
c)
middot 12 -8 -4 4 8 12
Velocity (mmls
bull middot12 -8 -4 0 4 12
Velocity (mmls)
b)
middot12 -8 -4 4 8 12
Velocity (mmls
Figure 34 Transmission Mossbauer spectra measured at 5 K of a) HFO (magnetic hyperfine
field of 481 T isomer shift of 048 1nrns-1 negligible quadrupole shift and line width of outer
lines 110 rmns-1) b) goethite (magnetic hyperfine field of 505 T isomer shift of 049 rmns-1
quadmpole shift of -013 1nrns-1 and line width of outer lines 042 rnrns-1
) and c) lepidocrocite
56 Chapter 3
(magnetic hyperfine field of 455 T isomer shift of 050 mms-1 quadrupole shift of ndash001 mms-1
and line width of outer lines 060 mms-1) prior to inoculation Simple Lorenztian fits are shown
The oxidation state and coordination of Fe in the microbially reduced HFO
goethite and lepidocrocite samples were also examined by transmission Moumlssbauer
spectroscopy (Figure 35) The bioreduced HFO goethite and lepidocrocite samples
cultured on formate contained Fe(II) holding similar coordination as inferred from
the similarity of the hyperfine parameters (see legend in Figure 35) but different
relative intensities (72 19 and 71 respectively) The major part of the Fe(III)
remaining in the bioreduced samples were coordinated similarly to the Fe(III)
present in the initial Fe(III) oxide The coordination of Fe(II) in the bioreduced
lepidocrocite samples cultured on lactate was slightly different (a smaller
quadrupole splitting of 288 mms-1 for the ferrous component dominates ndash data not
shown) The exact mineralogy of the Fe(II) present in the green phases was not
fully resolved but its coordination is very akin to one of the Fe(II) sites in vivianite
(see Chapter 2 this work) and synthetic green rusts (Koch 1998) These findings
agree with other reports on the bioformation of vivianite and green rusts by
Shewanella putrefaciens CN32 although the evidence provided may be discussed
(Fredrickson et al 1998 Glasauer et al 2003 Parmar et al 2001) Our
Moumlssbauer data on the green phases did not allow for a detailed account of the type
of green rust produced However when considering solution composition (see
Table 31) and the high affinity of GR interlayers for carbonate it is reasonable to
assume that carbonate GR was formed (Hansen amp Taylor 1991) Due to the high
amounts of Fe(III) in the oxides present in the experiments it was difficult to probe
a possible content of Fe(III) in the vivianite with certainty The differences in the
number of Fe(II) positions in the Moumlssbauer spectra and particular the different
temperatures at which magnetic ordering takes place can be employed in order to
distinguish between green rust and vivianite Preliminary Moumlssbauer data obtained
for the blackish precipitates formed in the HFOsand suspensions indicate that they
hold no resemblance to magnetite eventhough the colour suggests so On the
contrary the black precipitates seemed to be more similar to synthetic green rusts
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 57
Mossbauer spectroscopic measurements are currently underway in order to resolve
the Fe(II) coordinations in the greenish and blackish phases
a) b)
c c g Q e- e 0 0 1l 1l
-~ bull ~
iii ~ Qi bull a bull
-12 -8 4 0 4 12
4 -3 -2 -1 0 1 2 4 Velocity (mmts) Velocity ( rmis)
c)
-12 -8 4 8 12
v elocity (m mis)
Figure 35 Transmission Mossbauer spectra of the black and green phases fo1med within 1-2
weeks after inoculation of a) HFO (measured at 130 K) b) goethitecalcite (measured at 80 K)
and c) lepidocrocitecalcite (measured at 80 K) with S algae BrY Experimental conditions
[fo1m ate]0 = 4 mM [Fe(III)]o = 8 mM 995 Ni05 C02(g) pH 76 The quadrnpole
splittings and isomer shifts for the Fe(II) components in the three systems are a) 293 nnns-1 and
126 mmsmiddot1 b) 308 rmnsmiddot1 and131 rmnsmiddot1 and c) 322 mmsmiddot1 and 132 mmsmiddot1 Simple Lorenztian
fits are shown
The evidence provided in many of the studies proposing biogenic GRs is not all
too convincing but it strongly suggests the probability of microbially produced GR
being present The challenge encountered is that when present in low
58 Chapter 3
concentrations especially in mixtures including other iron minerals it is very
difficult to identify GRs using conventional solid phase analysis methods even
with Moumlssbauer spectroscopy At least two complementary methods such as XRD
and Moumlssbauer spectroscopy are required for proper identification and
characterization of GRs However in this work the solid materials were generally
too low in total iron to allow for XRD analysis Moreover the highly
heterogeneous suspensions were dominated by the coating-bearing sand and calcite
solids Electron micrographs including energy dispersive X-ray spectroscopy
suspension colour and mineral stability calculations do not suffice as evidence The
most convincing evidence provided so far involves an atypical GR-CO3 with an
Fe(II)Fe(III) ratio of 1 (Ona-Nguema et al 2002aampb) This GR-CO3 was formed
as a result of lepidocrocite reduction by Shewanella putrefaciens CIP 8040 at
conditions comprising high nutrient levels (50-75 mM formate) high Fe(III)
concentrations (80-300 mM) and a synthetic electron shuttle (100 microM
anthraquinone-26-disulfonate (AQDS)) at initial pH 75 Hence the results
reported during recent years suggest that microbial formation of GR may be
possible The results presented here indicate that GRs may be produced
microbially at conditions including low carbon and Fe(III) concentrations as well
as the exclusion of synthetic electron shuttles and pH buffers
333 Factors controlling the identity of the secondary iron minerals
In general one would expect that biogenic minerals have chemical compositions
and crystal habits similar to those produced by nonenzymatic processes as they are
governed by the same equilibrium principles In fact since the latter stages of
mineralization are inorganically driven and the secondary Fe(II)-containing
minerals are formed indirectly by electron transfer outside the bacterial cell and not
directly inside the bacterial cell the type of iron mineral formed is a function of the
environmental conditions in which the bacteria live ie the same microorganism
form different minerals in different environments The key factors controlling the
identity of the secondary iron minerals include medium composition electron
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 59
donor and electron acceptor concentrations mineral aging as well as adsorbed ions
(Zachara et al 2002) The main factor controlling the nature of the secondary
mineral products are the respiration-driven biogenic Fe(II) supply rate and
magnitude and its surface reaction with the residual oxide and other sorbed ions
(Zachara et al 2002) Especially solution and medium composition have a strong
impact on the nature of the Fe(II)-containing biomineralization products forming
Accordingly siderite (FeCO3) and magnetite (Fe3O4) were the secondary solid
phases resulting from the bioreduction of ferrihydrite by Shewanella putrefaciens
CN32 in bicarbonate buffered medium (pH 71) containing no phosphate whereas
siderite and vivianite were the secondary iron minerals dominating in bicarbonate
buffered medium (pH 74) containing 4 mM phosphate (Zachara et al 2002) This
is explained by the inhibiting effect of phosphate on crystallization of magnetite
(Couling amp Mann 1985 Fredrickson et al 1998)
334 Factors controlling the rate and extent of Fe(III) bioreduction
In this study the extent of Fe(III) bioreduction was estimated to 19-72 by
transmission Moumlssbauer measurements In fact complete microbial reduction of
crystalline Fe(III) minerals has never been observed in laboratory batch culture
studies (Roden amp Urrutia 2002) It has been found that Fe(II) does not inhibit
Fe(III) reductase activity through an enzyme inhibition mechanism (Roden amp
Urrutia 2002) Hence other chemical andor physiologic factors control the
bioavailability of solid Fe(III) phases and thus the extent of their microbial Fe(III)
reduction The initial rate and long-term extent of microbial reduction of
amorphous and crystalline Fe(III) oxides including HFO goethite and hematite
were linearly correlated with oxide surface area (Roden amp Zachara 1996)
Association of biogenic Fe(II) with Fe(III) oxide and DIRB cell surfaces reduced
the long-term extent of crystalline Fe(III) oxide bioreduction (Roden amp Urrutia
2002) These results were explained by Fe(II) surface complexes andor
precipitates creating a passive Fe(II)-bearing surface film providing direct physical
interference with the electron transfer from the DIRB cells to Fe(III) However the
60 Chapter 3
real mechanisms whereby the surface-associated Fe(II) inhibits Fe(III) oxide
bioreduction are unclear but they most likely involve both kinetic and
thermodynamic constraints on the electron transfer Culture medium composition
in particular the presence and the concentration of phosphate as well as Fe(II)
chelating ligands also exert an influence on the extent of the microbial reduction
of Fe(III) oxides The extent of Fe(III) bioreduction was inhibited by high
phosphate concentrations which favoured surfacebulk precipitation processes
(Urrutia et al 1998) The carbon sources most frequently applied in Fe(III)
bioreduction studies include malate citrate and other di- and tricarboxylic acids
which are not only easily metabolizable carbon sources but also eminent Fe(II) and
Fe(III) chelators In this study we employed formate and lactate as carbon sources
since they are the weakest complexing agents of Fe(II) and Fe(III) among the C1-
C3 monocarboxylic acids (Martell 1964) Thus we expect less dissolution of
prevailing precipitates by complexation as compared to other studies
34 Conclusions
This work demonstrated that Shewanella algae BrY reduced 19-72 of initial
Fe(III) when grown in culture media containing 4-10 mM formate or lactate and 8-
27 mM Fe(III) applied as goethite or lepidocrocite coatings on calcite (pH 76) or
HFO coatings on sand (pH 55-60) Within 1-2 weeks after inoculation green
mineral phases were produced in the goethitecalcite and lepidocrocitecalcite
mineral systems whereas black precipitates formed in the HFOsand suspensions
Moumlssbauer spectroscopic analyses indicated that the greenish and blackish phases
most likely were mineral mixtures dominated by vivianite and green rust Thus the
results indicate that GRs may be produced microbially at conditions including low
carbon and Fe(III) concentrations as well as the exclusion of synthetic electron
shuttles and pH buffers
Acknowledgments
We would like to thank Dr R Gerlach for providing us the Shewanella algae BrY culture and
Dr C B Koch for performing the Moumlssbauer analyses
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 61
References Al-Agha MR Burley SD Curtis CD Esson J (1995) Complex cementation textures and authigenic mineral assemblages in recent concretions from the Lincolnshire Wash (east coast UK) driven by Fe(0) to Fe(II) oxidation Journal of the Geological Society 152 157-171 Bernal JD Dasgupta DR Mackay AL (1959) The oxides and hydroxides of iron and their structural inter-relationships Clay Minerals Bulletin 4 15-30 Caccavo Jr F Blakemore RP Lovley DR (1992) A hydrogen-oxidizing Fe(III)-reducing microorganism from the Great Bay Estuary New Hampshire Applied and Environmental Microbiology 58 3211-3216 Caccavo Jr F Das A (2002) Adhesion of dissimilatory Fe(III)-reducing bacteria to Fe(III) minerals Geomicrobiology Journal 19 161-177 Couling SB Mann S (1985) The influence of inorganic phosphate on the crystallization of magnetite (Fe3O4) from aqueous solution Journal of the Chemical Society Chemical Communications 1713-1715 Das A Caccavo Jr F (2000) Dissimilatory Fe(III) oxide reduction by Shewanella alga BrY requires adhesion Current Microbiology 40 344-347
Das A Caccavo Jr F (2001) Adhesion of the dissimilatory Fe(III)-reducing bacterium Shewanella alga BrY to crystalline Fe(III) oxides Current Microbiology 42 151-154 Fadrus H Maly J (1975) Suppression of iron(III) interference in the determination of iron(II) in water by the 110-phenanthroline method The Analyst 100 549-554 Faye GH Manning PG Nickel EH (1968) The polarized optical absorption spectra of tourmaline cordierite chloritoid and vivianite ferrous-ferric electronic interaction as a source of pleochroism American Mineralogist 53 1174-1201 Fredrickson JK Zachara JM Kennedy DW Dong H Onstott TC Hinman NW Li S (1998) Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium Geochimica et Cosmochimica Acta 62 3239-3257 Geacutenin JMR Olowe AA Benbouzid-Rollet ND Prieur D Confente M Resiak B (1991) The simultaneous presence of green rust 2 and sulfate reducing bacteria in the corrosion of steel sheet piles in a harbour area Hyperfine Interactions 69 875-878 Geacutenin JMR Bourrieacute G Trolard F Abdelmoula M Jaffrezic A Refait Ph Maitre V Humbert B Herbillon A (1998) Thermodynamic equilibria in aqueous suspensions of synthetic and natural Fe(II)-Fe(III) green rusts Occurrences of the mineral in hydromorphic soils Environmental Science and Technology 32 1058-1068 Glasauer S Weidler PG Langley S Beveridge TJ (2003) Controls on Fe reduction and mineral formation by a subsurface bacterium Geochimica et Cosmochimica Acta 67 1277- 1288 Hansen HCB Taylor RM (1991) The use of glycerol intercalates in the exchange of CO3
2- with SO4
2- NO3- or Cl- in pyroaurite-type compounds Clay Minerals 26 311-327
62 Chapter 3
Hungate RE (1969) A roll tube method for cultivation of strict anaerobes Methods in Microbiology 3B 117-132 King GM (1990) Effects of added manganic and ferric oxides on sulfate reduction and sulfide oxidation in intertidal sediments FEMS Microbiology Ecology 73 131-138 Koch CB (1998) Structures and properties of anionic clay minerals Hyperfine Interactions 117 131 -157 Kostka J Nealson KH (1998) Isolation cultivation and characterization of iron- and manganese reducing bacteria In Techniques in Microbial Ecology Burlage RS Atlas R Stahl D Geesey G Sayler G (eds) Oxford University Press Inc 58-78 Kukkadapu RK Zachara JM Smith SC Fredrickson JK Liu C (2001) Dissimilatory bacterial reduction of Al-substituted goethite in subsurface sediments Geochimica et Cosmochimica Acta 65 2913-2924
Liu C Zachara JM Gorby YA Szecsody JE Brown CF (2001) Microbial reduction of Fe(III) and sorptionprecipitation of Fe(II) on Shewanella putrefaciens strain CN32 Environmental Science and Technology 35 1385-1393 Lovley DR (1991) Dissimilatory Fe(III) and Mn(IV) reduction Microbiological Reviews 55 259-287 Lovley DR (1997) Microbial Fe(III) reduction in subsurface environments FEMS Microbiology Reviews 20 305-313 Lovley DR Phillips EJP (1986) Organic matter mineralization with reduction of ferric iron in anaerobic sediments Applied and Environmental Microbiology 51 683-689 Lovley DR Phillips EJP Lonergan DJ (1991) Enzymatic versus nonenzymatic mechanisms for Fe(III) reduction in aquatic sediments Environmental Science and Technology 25 1062-1067 Lower SK Hochella Jr MF Beveridge TJ (2001) Bacterial recognition of mineral surfaces Nanoscale interactions between Shewanella and α-FeOOH Science 292 1360-1363 Martell AE (1964) Stability constants of metal-ion complexes Part 2 Organic including macromolecule ligands The Chemical Society London 2 ed Miller TL Wolin MJ (1974) A serum bottle modification of the Hungate technique for cultivating obligate anaerobes Applied Microbiology 27 985-987 Nealson KH Saffarini D (1994) Iron and manganese in anaerobic respiration Environmental significance physiology and regulation Annual Review of Microbiology 48 311-343 Nevin KP Lovley DR (2000) Lack of production of electron-shuttling compounds or solubilization of Fe(III) during reduction of insoluble Fe(III) oxide by G metallireducens Applied and Environmental Microbiology 66 2248-2251 Nevin KP Lovley DR (2002) Mechanisms for Fe(III) oxide reduction in sedimentary
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 63
environments Geomicrobiology Journal 19 141-159 Nielsen A (1976) Hvid groslashn og sort rust Beskrivelse af en korrosionsskade paring et svoslashmmebassin Nordisk Betong 2 21-24 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002a) Iron (IIIII) hydroxycarbonate green rust formation and stabilization from lepidocrocite bioreduction Environmental Science and Technology 36 16-20 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002b) Microbial reduction of lepidocrocite γ-FeOOH by Shewanella putrefaciens The formation of green rust Hyperfine Interactions 139140 231-237 Parmar N Gorby YA Beveridge TJ Ferris FG (2001) Formation of green rust and immobilization of nickel in response to bacterial reduction of hydrous ferric oxide Geomicrobiology Journal 18 375-385 Roden EE Urrutia MM (2002) Influence of biogenic Fe(II) on bacterial crystalline Fe(III) oxide reduction Geomicrobiology Journal 19 209-251 Roden EE Zachara JM (1996) Microbial reduction of crystalline iron(III) oxides Influence of oxide surface area and potential for cell growth Environmental Science and Technology 30 1618-1628 Roh Y Lee SY Elless MP (2000) Characterization of corrosion products in the permeable reactive barriers Environmental Geology 40 184-194 Schwertmann U Cornell RM (1991) Iron oxides in the laboratory Preparation and characterization VCH Verlagsgesellschaft mbH Weinheim Thamdrup B (2000) Bacterial manganese and iron reduction in aquatic sediments In Advances in Microbial Ecology (Schink B ed) Kluwer AcademicPlenum Publishers New York 41-84 Trolard F Abdelmoula M Bourrieacute G Humbert B Geacutenin JMR (1996) Mise en eacutevidence dun constituant de type rouilles vertes dans les sols hydromorphes Proposition de lexistence dun nouveau mineacuteral la fougeacuterite Geacuteosciences de surface Comptes Rendus de LrsquoAcademie des Sciences 323 1015-1022 Tuovinen OH Button KS Vuorinen A Carlson L Mair DM Yut LA (1980) Bacterial chemical and mineralogical characteristics of tubercles in distribution pipelines Journal of the American Water Works Association 72 626-635 Turick CE Caccavo Jr F Tisa LS (2003) Electron transfer from Shewanella algae BrY to hydrous ferric oxide is mediated by cell-associated melanin FEMS Microbiology Letters 220 99-104 Urrutia MM Roden EE Fredrickson JK Zachara JM (1998) Microbial and surface chemistry controls on reduction of synthetic Fe(III) oxide minerals by the dissimilatory iron- reducing bacterium Shewanella alga Geomicrobiology 15 269-291 Venkateswaran K Moser DP Dollhopf ME Lies DP Saffarini DA MacGregor BJ Ringelberg DB White DC Nishijima M Sano H Burghardt J Stackebrandt E
64 Chapter 3
Nealson KH (1999) Polyphasic taxonomy of the genus Shewanella and description of Shewanella oneidensis sp nov International Journal of Systematic Bacteriology 49 705-724 Zachara JM Fredrickson JK Li S Kennedy DW Smith SC Gassman PL (1998) Bacterial reduction of crystalline Fe3+ oxides in single phase suspensions and subsurface materials American Mineralogist 83 1426-1443
Zachara JM Kukkadapu RK Fredrickson JK Gorby YA Smith SC (2002) Biomineralization of poorly crystalline Fe(III) oxides by dissimilatory metal reducing bacteria (DMRB) Geomicrobiology Journal 19 179-207
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 65
4 Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust The Effect of Probe Compound Charge
Abstract
Layered iron(II)-iron(III)-hydroxides (green rusts) may play an important role in
controlling the fate and transport of many organic and inorganic contaminants in
iron-rich suboxic soils and sediments Unlike most other iron oxides green rusts
(GRs) contain not only external Fe(II) reactive sites at the basal planes and at the
edges but also internal sites in the space between consecutive Fe(II)-Fe(III)
hydroxide layers The GR interlayer thickness is a function of both the size and the
charge of the interlayer anion Whether a given oxidant has access to the internal
sites in GRs is dependent on its charge We investigated the reductive
transformation of nitroaromatic compounds (NACs) by GR-SO4 and studied the
effect of NAC charge on the reactivity towards GR-SO4 A series of structurally
closely related compounds with different charge properties including nitrobenzene
4-nitrotoluene 4-chloronitrobenzene and 4-nitrophenylacetic acid were used as
probe compounds The NACs were completely reduced to their corresponding
anilines by GR-SO4 The reactions followed pseudo 1 order kinetics with respect
to NAC and the surface area-normalised pseudo 1 order rate constants obtained
were 016ndash465middot10-4 s-1middotm-2middotL at [Fe(II)GR]0 = 103-1260 mM [NAC]0 = 20-102
microM and pH 84-86 Neither mass transfer control nor surface saturation kinetics
could account for the rather unexpected similarity of the surface area-normalised
pseudo 1 order rate constants obtained for the reduction of the neutral and anionic
NACs by GR-SO4 These observations suggest that the anionic NACs did not have
an enhanced access to the inner or outer Fe(II)-GR reactive sites as compared to
the neutral NACs Hence the reaction between NAC and GR-SO4 primarily took
place at the edges of GR-SO4
66 Chapter 4
41 Introduction
Layered iron(II)-iron(III)-hydroxides (green rusts) are intermediate phases formed
by partial oxidation of Fe(II) or partial reduction of Fe(III) In neutral and weakly
alkaline solutions the oxidation of dissolved Fe(II) always passes through solid
green rust (GR) phases (Bernal et al 1959) This agrees with the natural GR
occurrences found in suboxic non-acid iron-rich environments such as
hydromorphic soils and intertidal sediments (Al-Agha et al 1995 Trolard et al
1996 Genin et al 1998) In addition GRs have been found as corrosion products
in numerous engineered systems ie a pipeline distribution system for drinking
water steel sheet piles in marine sediments reinforced concrete (ferro-concrete)
and permeable reactive barriers of zero-valent iron implemented for on-site
remediation of organic and inorganic contaminants (Tuovinen et al 1980 Nielsen
1976 Genin et al 1991 Roh et al 2000) Furthermore the microbial formation
of GRs resulting from bioreduction of Fe(III) oxides by strains of the anaerobic
dissimilatory Fe(III) reducing bacteria Shewanella putrefaciens has been reported
increasingly over the last 5 years (Fredrickson et al 1998 Kukkadapu et al 2001
Parmar et al 2001 Ona-Nguema et al 2002 Glasauer et al 2003) Moreover
the biotic formation of GRs by anaerobic denitrifying Fe(II) oxidizing bacteria has
been proposed but proper identification of the GR phases still lacks (Chaudhuri et
al 2001) All these indications of microbial GR formation infer the importance of
GRs as a link between geochemical and biological processes in natural systems
GRs form platy crystals with the general formula [FeII(6-
x)FeIIIx(OH)12]x+[(A)xnmiddotyH2O]x- where x = 09 - 42 A is an n-valent anion eg
CO32- Clndash or SO4
2- and y is the number of water molecules in the interlayer The
crystal structure consists of positively charged hydroxide sheets with Fe(II) and
Fe(III) cations having octahedral hydroxyl coordination The Fe(III) in the
hydroxide layers creates a net positive charge which is balanced by hydrated
anions in the interlayers (Figure 41) The interlayers have a higher affinity for
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 67
divalent anions than for monovalent anions (Miyata 1983) Among the 3 most
common GR forms the affinity follows the order CO32- gt SO4
2- gt Clndash The extreme
preference shown for carbonate hinders further access and exchange except under
certain conditions (Hansen amp Taylor 1991) Non-carbonate forms are readily
exchanged with other anions when dispersed in a solution containing the
exchanging anion (Mendiboure amp Schoumlllhorn 1986)
c
ba
Figure 41 Green rust layer structure The hydroxide layers and the interlayers are connected by
hydrogen bonds (not shown) The GR-SO4 crystal structure is characterised by the hexagonal
unit cell having a = b = 055 nm and c = 110 nm (Simon et al 2003) The unit cell consists of
one double layer (a double layer is a hydroxide layer and an interlayer) ie the hydroxide layer
constitutes 049 nm and the interlayer 061 nm in GR-SO4
The GR interlayer thickness (extending in the c axis direction Figure 41) is a
function of both the size and the charge of the interlayer anion Tetrahedrally
coordinated anions like sulphate lead to larger interlayer distances than smaller
monoatomic anions like chloride or planar ions like carbonate (Mendiboure amp
Schoumlllhorn 1986) Not only size but also charge density plays a role for the
interlayer spacing That is for anions having the same number of valence
electrons anions with smaller ionic radii (higher electron density) are bound more
strongly and therefore result in smaller interlayer spacings
Due to their layered structures anionic interlayers and high specific surface areas
GRs represent reactive ion exchangers and sorbents of anions eg arsenate
selenate and phosphate (Myneni et al 1997 Hansen amp Poulsen 1999 Randall et
al 2001) In addition GR may incorporate heavy metal cations by isomorphic
68 Chapter 4
substitution into the GR hydroxide layers (Tamaura 1985 Tamaura 1986)
Furthermore GRs have been shown to reduce a range of inorganic contaminants
such as nitrite nitrate selenate chromate uranyl pertechnetate and the transition
metals AgI AuIII CuII and HgII as well as organic pollutants including halogenated
ethanes ethenes and methanes (Hansen et al 1994 Hansen et al 1996 Myneni et
al 1997 Erbs et al 1999 Loyaux-Lawniczak et al 1999 Cui amp Spahiu 2002
Lee amp Batchelor 2002b Heasman et al 2003 OrsquoLoughlin et al 2003a amp 2003b
Pepper et al 2003 Elsner et al 2004 OrsquoLoughlin amp Burris 2004) Thus through
sequestration and reductive transformation GRs may play an important role in
controlling the fate and transport of contaminants in suboxic soils and sediments
In a previous study the effects of interlayer anion and Fe(II)Fe(III) ratio in GRs
on the reduction rate of nitrate were investigated (Hansen et al 2001) It was
found that the rate of nitrate reduction to ammonium increased with increasing
Fe(II)Fe(III) ratio and decreased when exchanging a monovalent interlayer anion
(chloride) with a divalent anion (sulphate) The results suggest that for anionic
oxidants like nitrate Fe(II) within the hydroxide layer is available from the outside
basal planes and from the edges as well as through the interlayer under certain
conditions (Figure 42) However oxidants with different charge properties
(cations neutral molecules) may exhibit different affinities for the various reactive
Fe(II) sites present in GR
As the reactive sites are located inat the Fe(II)-Fe(III) hydroxide layers the rate of
reaction depends on the hydroxide layer area which can be accessed by the
oxidant If the oxidant can exchange with the interlayer anion reaction can take
place both at outer and inner surfaces of the GR particles and in total more
reactive sites are available for the reaction However it was found that nitrate
cannot penetrate the interlayer when carbonate or sulphate constitutes the
interlayer anions (Hansen amp Koch 1998) This agrees with the fact that the
interlayers have a lower affinity for monovalent anions than for divalent anions
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 69
(Miyata 1983) However when nitrate was forced into the interlayer by extracting
the interlayer sulphate through precipitation of barium sulphate outside the GR
particles the observed 40 fold increase in rate of nitrate reduction almost equalled
the increase in exposed surface area of the Fe(II)-Fe(III) hydroxide layers (Hansen
amp Koch 1998) From these observations it is expected that the rate of reaction
depends on the particular GR form the crystallite size and the ease with which an
oxidant can exchange with An- in the GR interlayer (Figure 42) Due to
electrostatic interactions we expect anions to be attracted to the positively charged
outer and inner surfaces to a higher degree than cations and neutral compounds If
this theory holds we may expect oxidants with similar intrinsic reactivity (similar
one-electron reduction potentials) to react in the following order anionic gt non-
charged gt cationic (Figure 43) granting that we do not normalise the rate
constants with respect to the amount of oxidant sorbed
Figure 42 Reaction of a probe compound at basal planes at edges and in the interlayer of GR
The hypothesis only holds in cases where the oxidants possess the same intrinsic
reactivities If the relative reactivities of the probe compounds differ greatly from
what would be expected when considering only their reduction potentials
70 Chapter 4
compound specific effects such as charge properties might explain this and the
relative reactivities may follow a pattern like the one depicted in Figure 43
Figure 43 Hypothetical plot of observed reaction rate constants for the reactions between
cationic neutral and anionic probe compounds and GR-SO4 assuming that the oxidant charge
controls its reactivity towards GR
In this work we investigated the reductive transformation of NACs by GR-SO4
Furthermore the effect of NAC charge on the rate of reaction and the possible
access to the internal reactive sites in GR-SO4 were assessed When quantifying
Fe(II) in GRs by means of acid digestion it is not possible to distinguish between
the reactive sites accessible from the outside (at the basal planes or at the edges) or
through the interlayer However we designed an indirect method to gain insight
into the relative importance of the various reactive sites by using a series of
structurally closely related compounds with different charge properties as ldquoreactive
probesrdquo Neutral and anionic probes were needed in order to access all Fe(II)
reactive sites According to our hypothesis cationic and non-charged oxidants
should provide information about the reactivity of the outer Fe(II) reactive sites in
GR whereas the anionic oxidants should provide information about the reactivity
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 71
of both outer and inner Fe(II) reactive sites We chose five nitro aromatic
compounds (NACs) - representing an important group of reducible organic
pollutants - as probe compounds (Figure 43) This class of compounds is not only
of great environmental concern but also comprises suitable model compounds for
studying redox reactions potentially relevant in the environment Moreover they
react readily with Fe(II) surface species associated with iron oxides or clay
minerals transforming them into well-defined easily detected products allowing
mass and electron balances to be established (Hofstetter et al 2003 Klausen et al
1995 Schultz amp Grundl 2000) Our main goals were to establish the rate law and
estimate the surface area-normalised reaction rates for the reaction of the probe
compounds with GR-SO4 in order to assess the importance of the Fe(II) reactive
sites accessible through the interlayer relative to the Fe(II) reactive sites accessible
at the outer surface in GR-SO4
4 2 Materials and methods
All handling and sampling of solutions and suspensions were carried out under
strict anoxic conditions All chemicals were pa quality or better Methanolic stock
solutions (5 mM) of nitrobenzene (NB) 4-nitrotoluene (4-NT) 4-
chloronitrobenzene (4-CNB) and 4-nitrophenylacetic acid (4-NPA) were prepared
in deoxygenated methanol Several attempts to synthesize the cationic probe
compound 4-(NNN-trimethylammonium)-nitrobenzene failed and therefore the
study had to be carried out with only neutral and anionic oxidants The sulphate
GR form was chosen as it is the most stable form and thus the easiest to work
with in the lab
421 Synthesis of GR-SO4
GR-SO4 was synthesized by controlled air oxidation of an FeSO4 solution at a
constant pH of 700 according to the procedure given by Koch amp Hansen (1997)
The GR-SO4 suspension was washed with deoxygenated deionised water and
72 Chapter 4
separated on a folding filter redispersed in deoxygenated 25 mM Na2SO4(aq) in
order to stabilize the GR-SO4 and prevent it from transforming into magnetite
spontaneously Washing separation and redispersion of the GR-SO4 suspension
were conducted in an anoxic glove box (Coy Laboratory Products Inc) All
suspensions and solutions were deoxygenated by Ar-purging (999998 Ar
Carbagas)
422 Mineral characterisation
The identity and purity of the GR-SO4 suspensions were examined by means of X-
ray diffraction measurements The XRD analyses were performed on a Scintag
XDS 2000 using Cu Kα radiation (45 kV 40 mA) Glycerol smears made
according to Hansen (1989) were scanned between 6 and 80 deg2θ with a scan speed
of 1 deg2θmin
423 Lyophilization and determination of specific surface area
Simple air-drying of the GR mineral in the glove box resulted in big flakes with
very low surface areas hence a more suitable lyophilization method was adopted
from Elsner et al (2004) The GR-SO4 suspensions were lyophilised using
Schlenk-type glassware The set-up consisted of a 1 L round bottom flask and a
200 mL glass finger connected by a crescent-shaped bridge equipped with an
evacuation outlet and a stopcock All ground joints and fittings were attached using
high-vacuum grease The washed and resuspended GR-SO4 suspensions were
filled into the glass finger and the freeze-drying apparatus was assembled and
closed before taking it out of the glove box The suspension was frozen by
carefully submerging the lower part of the glass finger into liquid nitrogen for a
few hours Subsequently the evacuation outlet was connected to a vacuum pump
by a metal hose Following a short evacuation of the metal hose the lyophilization
apparatus was evacuated for several minutes by gently opening the stopcock The
evacuation was terminated by closing the stopcock and disconnecting the vacuum
pump The apparatus position was now reversed by removing the glass finger from
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 73
and immersing the round bottom flask into liquid nitrogen As any other
lyophilization method this method depends on sublimation of the ice from the
frozen sample and its recondensation on a cool surface in this case the round
bottom flask Generally it took 1-2 d for the mineral to dry The apparatus was
disassemled in the glove box and the fine powder stored under anoxic conditions
The specific surface area (SSA) of GR-SO4 was determined by the BET multi-
point method using N2 adsorption (Brunauer et al 1938) Powder samples were
filled into sample burettes in the glove box and the generously greased stopcocks
closed Samples and burettes were evacuated prior to connecting them to the BET-
instrument (Sorptomatic 1990 Fisons)
424 Estimation of the one-electron reduction potential for 4-NPA
Kinetic experiments in 100 mL Viton stoppered and alu-crimp capped serum vials
were carried out under the exclusion of oxygen as described by Hofstetter et al
(1999) The homogeneous aqueous solutions contained 50 mM KH2PO4 buffer
(pH = 660) 5 mM Na2S redox buffer and 20 microM juglone (8-hydroxy-14-
naphthoquinone) added as deoxygenated 20 mM methanolic stock solution The
solutions were equilibrated at least one day prior to 4-NPA addition To start the
reaction 50 microM 4-NPA was added as deoxygenated 20 mM methanolic stock
solution The vials were agitated on a roller apparatus in the dark at 21ordmC Control
experiments were prepared similarly except for the addition of juglone At
different time intervals aqueous samples were withdrawn with a syringe and
collected in 18 mL HPLC vials containing 100 microL 1 M HCl The sample vials
were sealed with Teflon-coated silicone septa and plastic screw caps and vortexed
for 10 s The samples were stored at -20degC and analysed without further treatment
For comparison experiments with 4-NT were also conducted See Supporting
Information 71 for more information on the one-electron reduction potentials
74 Chapter 4
425 Kinetic experiments
All reactions took place at pH 84-86 where GR-SO4 tends to stabilize and buffer
itself Samples for Fe(II) and XRD analysis were withdrawn prior to reaction Due
to the fast reactions the experiments were conducted in 10 mL single-use
polyethylene syringes (BD Plastipak) in the glove box To start reaction 40-200
microL 5 mM methanolic stock solutions of NAC were quickly added to 10 mL GR-
SO4 suspension (1-12 mM Fe(II)GR) washed and resuspended in 25 mM
Na2SO4(aq) A Teflon filter (25 mm x 02 microm BGB Analytik) was quickly
mounted on the tip of the syringe and the syringe was vigorously shaken between
sampling At different time intervals filtered suspension samples were collected in
18 mL HPLC vials The HPLC vials were sealed with Teflon-coated silicone septa
and plastic screw caps The samples were stored at -20degC and analysed without
further treatment Absorption of NAC in the syringe and in the Teflon filter
evaluated in blank experiments with NAC added to 25 mM Na2SO4(aq) was found
to be negligible
426 Analytical methods
Initial total and aqueous Fe(II) were determined using a modified phenanthroline
method (Fadrus and Maly 1975) In order to determine [Fe(II)aq] and [Fe(II)total] 1
mL filtered (022 microm) and 1 mL unfiltered GR-SO4 suspension samples were
withdrawn and each treated with 18 mL 01 M HCl for at least 30 min From these
acid digests 01 mL was added to 05 mL Fe(II)-reagent and 19 mL deionised
water (DIW) added up The Fe(II) content in GR-SO4 was estimated as the
difference [Fe(II)GR] = [Fe(II)total] - [Fe(II)aq] The NACs and their corresponding
intermediates and products formed during reduction by GR-SO4 were quantified by
reversed-phase HPLC Separation was performed on a LiChrospher 100 RP-18 (5
microm 125 times 4 mm ID) reversed-phase column coupled with a LiChroCART 100 RP-
18 (4 times 4 mm ID) precolumn Analytical conditions were isocratic and the eluent
consisted of 10 mM hydroxylammonium chloride in various DIWCH3OH
mixtures (vv 3565 and pH 70 for 4-NT and 4-CNB 955 and pH 60 for
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 75
4-NPA) The injection volume was 20 microLand the flow-rate 10 mLmin HPLC
analyses were performed using a Gynkotek High Precision Pump M480 Gynkotek
Gina 50 autosampler and a diode array UV detector (340s Gynkotek) UV-VIS
detection was carried out at the wavelengths of maximum absorption for the
various nitro aromatic and aniline analytes
43 Results and discussion
431 Productformation and reaction kinetics
The reduction of the aromatic nitro group occurs via nitroso- and hydroxylamino-
intermediates where 2 electrons are transferred in each reaction step (Figure 44)
0 --0 H OH H H --0 N N N N
2e- 2H+ H20 + 2e-~ 2e- 2H+ H20
~ ~ R R R
Nitro benzene Nitrosobenzene Hydroxylamine Aniline
Figure 44 Reductive transfonnation pathway of NA Cs
Thus in order to reduce 1 Ar-N02 completely to Ar-NH2 6 electrons
corresponding to 6 mol Fe(II) are needed As magnetite was the major iron phase
formed during reaction (XRD results not shown) we assume the following
reaction stoichiometry
The aniline product was not formed at the same rate as the nitro compound
degraded which is consistent with the detection of early eluting hydroxylamine
intermediates during the course of the reaction (Figure 45a amp 45c) No traces of
76 Chapter 4
nitrosobenzene intermediates or side products such as azoxy- azo- or
hydrazobenzene were found In Figure 45 pseudo 1 order kinetic plots and ln
[Ar-NO2]t[Ar-NO2]0) versus time plots for the neutral probe compounds 4-CNB
and 4-NT are shown as examples The plots for NB and 4-NPA look similar
Figure 45 a Concentration versus time plots for reaction of GR-SO4 with 4-CNB ([Fe(II)GR]0 =
126 mM [4-CNB]0 = 30 microM) b ln [Ar-NO2]t[Ar-NO2]0) versus time plots for reaction of GR-
SO4 with 4-CNB ([Fe(II)GR]0 = 126 mM + [4-CNB]0 = 30 microM [Fe(II)GR]0 = 63 mM + [4-
CNB]0 = 50 microM) c Concentration versus time plots for reaction of GR-SO4 with 4-NT
([Fe(II)GR]0 = 131 mM [4-NT]0 = 20 microM) d ln [Ar-NO2]t[Ar-NO2]0) versus time plots for
reaction of GR-SO4 with 4-NT ([Fe(II)GR]0 = 131 mM [4-NT]0 = 20 microM [Fe(II)GR]0 = 131
mM [4-NT]0 = 50 microM) The hydroxylamino intermediate shown in microM equals the deficit in the
mass balance and in abs equals the detector response (peak area) Solid lines represent 1 order
kinetic fits (a amp c) and ln [Ar-NO2]t[Ar-NO2]0) versus time fits (d) whereas symbols and dotted
lines represent actual data
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 77
At intial Fe(II)GR concentrations in large excess of initial Ar-NO2 concentration
we found a pseudo 1 order rate law for the degradation of Ar-NO2 by GR-SO4
[ ] [ ] [ b 2
a GR
2 ArNOFe(II) ArNOsdotsdot=minus k
dtd ]
where a = 1 b = 1 and the observed pseudo 1 order rate constant kobs = k middot
[Fe(II)GR] At high [Fe(II)GR]0[Ar-NO2]0 ratios the nitro compound was
transformed completely into the aniline product within reaction duration and the
degradation curves of the nitro compound were shaped according to pseudo 1
order kinetics (data points follow solid line in Figure 45a) In some instances ie
at low [Fe(II)GR]0[Ar-NO2]0 ratios the reactions did not follow pseudo 1 order
kinetics for the whole duration of reaction (data points deviate from solid line in
Figure 45c) Hence in order to allow comparison all the pseudo 1 order rate
constants were calculated as initial rates (ie max first two half-lives) from linear
fits of (time ln [Ar-NO2]t[Ar-NO2]0)-plots (Figure 45b amp 45d) Surface area-
normalised pseudo 1 order rate constants are shown in Table 41
Tabl
e 4
1 S
urfa
ce a
rea-
norm
alis
ed p
seud
o 1
ord
er ra
te c
onst
ants
for t
he re
duct
ive
trans
form
atio
n of
4-n
itrot
olue
ne (4
-NT)
4-
chlo
roni
trobe
nzen
e (4
-CN
B) a
nd 4
-nitr
ophe
nyla
cetic
aci
d (4
-NPA
) by
GR
-SO
4
Exp
erim
ent
Age
GR
(d
) [F
e(II
) GR] 0
(mM
) [N
AC
] 0 (micro
M)
[Fe(
II) G
R] 0
[N
AC
] 0∆[
ArN
O2]
(microM
) af b
k obs
(s-1
) ck o
bs (s
-1middotm
-2middotL
) d
GR
-SO
4 + 4
-NT
3 1
103
20
51
5
109
54
5
7
65middot1
0-46
95middot1
0-5
GR
-SO
4 + 4
-NT
3 1
103
50
20
6
174
34
8
7
41middot1
0-46
74middot1
0-5
GR
-SO
4 + 4
-NT
3 1
103
10
0 10
3
214
21
4
2
63middot1
0-42
39middot1
0-5
GR
-SO
4 + 4
-CN
B 1
1
103
55
18
7
177
32
2
4
21middot1
0-43
83middot1
0-5
GR
-SO
4 + 4
-CN
B 1
1
103
10
2 10
1
165
16
2
2
37middot1
0-42
15middot1
0-5
GR
-SO
4 + 4
-NPA
1
1 1
03
25
412
9
7 38
8
4
82middot1
0-44
38middot1
0-5
GR
-SO
4 + 4
-NPA
1
1 1
03
46
224
9
9 21
5
6
37middot1
0-45
79middot1
0-5
GR
-SO
4 + 4
-NPA
1
1 1
03
100
103
13
7
137
196
middot10-4
178
middot10-5
GR
-SO
4 + 4
-NT
2 15
1
31
20
655
5
4 27
0
6
74middot1
0-44
82middot1
0-5
GR
-SO
4 + 4
-NT
2 15
1
31
50
262
9
7 19
4
5
89middot1
0-44
21middot1
0-5
GR
-SO
4 + 4
-NT
4 2
126
0 50
25
2 49
1
982
110
middot10-2
817
middot10-5
GR
-SO
4 + 4
-NT
4 2
630
50
12
6 42
6
852
186
middot10-3
276
middot10-5
GR
-SO
4 + 4
-CN
B 2
2
126
0 30
42
0 29
0
967
925
middot10-3
687
middot10-5
GR
-SO
4 + 4
-CN
B 2
2
630
50
12
6 38
0
760
136
middot10-3
202
middot10-5
GR
-SO
4 + 4
-NPA
2
2 12
60
40
315
371
92
8
5
96middot1
0-34
43middot1
0-5
GR
-SO
4 + 4
-NPA
2
2 6
30
45
140
273
60
7
1
09middot1
0-31
62middot1
0-5
a A
mou
nt o
f NA
C re
duce
d by
GR
-SO
4 at r
eact
ion
term
inat
ion
b F
ract
ion
of in
itial
ly a
dded
NA
C tr
ansf
orm
ed b
y G
R-S
O4 a
t rea
ctio
n te
rmin
atio
n c
Pse
udo
1
orde
r rat
e co
nsta
nts c
alcu
late
d as
initi
al ra
tes
ie m
ax f
irst t
wo
half-
lives
d S
urfa
ce a
rea-
norm
alis
ed p
seud
o 1
ord
er ra
te c
onst
ants
The
are
a of
GR
-SO
4 per
L
su
spen
sion
was
cal
cula
ted
as frac14
middot[Fe
(II)
GR] 0middot
600
gmiddotm
ol-1
middot71
2 m
2 middotg-1
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 79
432 Comparison of rate constants for the different NACs
Even for NACs holding very different one-electron transfer reduction potentials
( ) their reactivities differed only little in Fe(II)-Fe(III) systems such as the
Fe(II)goethite system (slope a = 06 for linear free energy relationship (LFER)
between k
1hE
obs and Hofstetter et al 1999) and the Fe(II)magnetite system
(LFER slope a = 034 Klausen et al 1995) When considering only the for the
reductive transformation reactions of the NACs applied in this study (Table 42)
we expect the surface area-normalised pseudo 1 order rate constants for the
reduction of the NACs to follow the order 4-CNB gt NB gt 4-NT gt 4-NPA Based
on log k
1hE
1hE
obs versus correlations obtained in Fe(II)goethite systems we expect 4-
CNB to react 6 times faster than 4-NPA (Hofstetter et al 1999)
1hE
Table 42 One-electron reduction potentials and relative reactivities in Fe(II)-magnetite and GR-
SO4 systems for the nitro aromatic probe compounds
Compound pKa Eh1
acute (mV) krel (Fe3O4) cd krel (GR-SO4) ce
4-Chlornitrobenzene - -450 a 122 148
Nitrobenzene - -486 a 1 1
4-Nitrotoluene - -500 a 057 176
4-Nitrophenylacetic acid 385 -543 b - 123 a Values from references cited in Hofstetter et al 1999 b Estimated at pH 660 using a LFER (Hofstetter et al 1999 see Supporting Information 71) c Reactivity relative to NB d Values from Klausen et al 1995 e Values from this work
A comparison of the relative rate constants of the NACs obtained for their
transformation by GR-SO4 (this work) and by magnetite (Klausen et al 1995)
shows that they do not differ significantly from each other in any of the mineral
systems (Table 42) When considering charge effects we expect the anionic probe
compounds to react faster with GR-SO4 than the neutral probe compounds
provided that they sorb preferentially within the GR-SO4 interlayers and that Fe(II)
in the interlayers are equally or more reactive than external Fe(II) sites Still the
surface area-normalised kobs values obtained for NB 4-NT 4-CNB and 4-NPA
under various experimental conditions did not differ significantly from each other
80 Chapter 4
(Figure 46 Table 41) The anionic probe compound 4-NPA did not react
significantly faster with GR-SO4 than the neutral probe compounds NB 4-NT and
4-CNB This may indicate that 4-NPA does not significantly interact with reactive
Fe(II) sites in the interlayer Alternatively the negative charge carried by 4-NPA
may be compensating for the lower intrinsic reactivity as compared to the neutral
probe compounds thus explaining the similarity in rate constants for 4-NPA and
the neutral probe compounds Finally other factors than intrinsic reactivity or
charge of the probe compounds such as regeneration of reactive sites or formation
of the magnetite phases may control the overall reactivity of the system
Figure 46 Actual plot of surface area-normalised pseudo 1 order rate constants for the reactions
between neutral and anionic probe compounds and GR-SO4
In heterogeneous reactions mass transfer in bulk solution becomes the rate-
limiting step when the surface reaction is much faster than the diffusion of the
reacting species to the reactive surface In cases where mass transfer controls the
overall rate of reaction the observed pseudo 1 order rate constant kobs ge kLmiddota
where kL is the calculated mass transfer coefficient (mmiddots-1) and a is the ratio of the
external (geometric) specific surface area to volume of solution (m-1) (see
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 81
Supporting Information 72) Mass transfer controlled reactions between GR-SO4
particles and the NACs in bulk solution would explain the similar pseudo 1 order
rate constants obtained for the NACs in this work However when comparing our
estimates of kLmiddota with kobs (see Supporting Information 72) we found that the rates
of mass transfer for all 4 NACs exceed the observed rate constants by at least 3
orders of magnitude at every initial Fe(II)GR concentration Thus the reactions of
the given NACs with GR-SO4 are not likely to be mass transfer limited under the
experimental conditions applied here
Since mass transfer in bulk solution does not control the reaction between GR-SO4
and NACs the overall reaction rate may be surface saturation controlled During
the reductive transformation of NACs not only the parent compound but also
various intermediates forming may compete for the restricted number of reactive
sites present in GR-SO4 This competition may constitute the rate limiting step in
the overall reactivity and may even be enhanced if the number of reactive sites is
depleted during reaction However surface saturation kinetics would not explain
the unexpected similarity of the pseudo 1 order rate constants obtained for the
NACs but it could explain the bent curves observed at low initial Fe(II)GR
concentrations (Figure 45d) The kinetically deviating cases at low [Fe(II)GR]0
were evaluated according to Langmuir-Hinshelwood kinetics (see Supporting
Information 72) Our experimental data did not agree with the Langmuir-
Hinshelwood rate law for any of the NACs (regression results not shown)
Simplifying the rate law by assuming that the aniline product or the
hydroxylamino intermediate or both did not compete for the reactive sites did not
improve the regression Thus the Langmuir-Hinshelwood model cannot explain
the deviations from pseudo 1 order kinetics observed at [Fe(II)GR]0 in our GR-SO4
system and it does not suffice as the correct reaction mechanism nor as the rate-
limiting step
82 Chapter 4
If the adsorption follows a saturation-type sorption isoterm (eg Langmuir) the
sorbate (oxidant) concentration at the surface will vary non-linearily with the total
amount of oxidant added This dependence will have to be taken into account when
establishing rate laws for the heterogeneous reactions and when testing the
hypothesis that the reaction rates depend on the sorbed concentration of the
oxidants However at the high reaction rates observed here we could not quantify
sorption Since the measured initial NAC concentrations corresponded to the
nominal amount of NAC added we assume that transformation and not sorption
was responsible for the consumption of NAC
433 Factors influencing the reaction rate
In general numerous compound- and system-specific factors influence redox
reactions One very important factor is pH which influences the speciation of
dissociable compounds as well as the stability of GR and the formation of other
iron minerals in the system pH has a strong impact on the sorption and therefore
the availability of ionisable oxidants such as carboxylic acids At pH ~ 84 where
our experiments were conducted 4-NPA (pKa = 385) is completely dissociated
Our experiments conducted with GR-SO4 and NB showed that pH was constant
during reaction In addition solution pH has an effect on the surface speciation
From other Fe(II)-Fe(III) systems such as Fe(II) surface species associated with
iron oxides or clay minerals it is well-known that other reactive hydroxylated
Fe(II)-Fe(III)-hydroxo surface complexes can form at higher pH (Charlet et al
1998 Liger et al 1999) Williams amp Scherer (2001) reported a small decrease (5
fold) in the reduction rate of chromate with GR-CO3 when increasing pH from 50
to 90 This decrease may be due to the alternating speciation of the Fe(II) surface
sites on GR-CO3 and of chromate in solution (pKa (H2CrO4) = 08 pKa (HCrO4ndash) =
65) when raising pH (Williams amp Scherer 2001) In contrast other studies have
reported small increases (4 fold) in the reduction rates of nitrate and
trichloroethene with GR-SO4 when increasing pH from 71 to 84 and from 68 to
101 respectively (Koch amp Hansen 1997 Lee amp Batchelor 2002b)
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 83
In this work all experiments were carried out in the presence of 25 mM
Na2SO4(aq) in order to minimize GR-SO4 dissolution and spontaneous
transformation into magnetite Preliminary results from experiments conducted
with NB show that the bulk concentration of Na2SO4 has only a very small impact
on the rate ie increasing the concentration of Na2SO4(aq) in the GR-SO4
suspension from 5 to 25 mM reduced the observed rate constant by a factor of 2
At Na2SO4 concentrations above 25 mM the effect leveled off and therefore
[Na2SO4] = 25 mM was chosen for this work Portions of the same GR-SO4
suspension were used for kinetic experiments over a period of two weeks No
significant aging effects eg rate constants decreasing as a function of GR age
were observed within this time frame
434 Comparison with rate constants obtained for other Fe(II) containing
mineral systems
A recent study compared the reactivity of various Fe(II) containing iron mineral
systems towards organic probe compounds representing different classes of
pollutants (Elsner et al 2004) The reductive transformation of 4-CNB was
investigated for the Fe(III) minerals goethite (α-FeOOH) lepidocrocite (γ-
FeOOH) and hematite (α-Fe2O3) as well as for the Fe(II)-Fe(III) oxide magnetite
(Fe3O4) All experiments were conducted in the presence of 1 mM dissolved Fe(II)
at pH 72 As seen from the surface area-normalised pseudo 1 order rate constants
in Figure 47 the reduction of 4-CNB by the Fe(II)-amended goethite
lepidocrocite and magnetite systems was up to 40 times faster than its reduction by
GR-SO4 The reduction rate obtained for the Fe(II)hematite system was only
slightly higher than the rate for the blank containing no iron mineral but aqueous
Fe(II) solely
84 Chapter 4
Figure 47 Surface area-normalised pseudo 1 order rate constants for the degradation of 4-CNB
by GR-SO4 (open square this work) and various Fe(II) containing mineral systems (solid circles
Elsner et al 2004) Experimental conditions applied by Elsner et al 1 mM aqueous Fe(II) pH
72 25 m2 mineral surface areaL GR-SO4 = green rust sulphate α-FeOOH = goethite Fe3O4 =
magnetite γ-FeOOH = lepidocrocite α-Fe2O3 = hematite
The experiments with GR-SO4 in our study were carried out at pH 84 whereas the
experiments comprising the other systems in Figure 47 were conducted at pH 72
As the reactivity of GR is expected to increase with pH (Lee amp Batchelor 2002b
Koch amp Hansen 1997) the lower of GR-SO4 compared to other Fe(II) systems
cannot be explained by differences in pH values GR-SO4 might just contain fewer
or less reactive surface sites than Fe(II)-amended goethite lepidocrocite and
magnetite suspensions These findings contrast those of other studies which found
higher surface area-normalised pseudo 1 order rate constants for dechlorination
reactions for GR-SO4 than for magnetite (Lee amp Batchelor 2002a amp 2002b Elsner
et al 2004) The different reactivity orders of the Fe(II)-bearing minerals found
for chlorinated aliphatics and nitro aromatics suggest that effects other than pH and
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 85
the intrinsic reduction potentials of the reacting species play a role for the
reactivity of these Fe(II)-bearing minerals
435 Depletion of reactive sites
Assuming that the platy hexagonal GR-SO4 crystals hold an average width of 1 microm
and an average particle thickness of 35 nm (Hansen amp Koch 1998) the ratio of
outer surface area to total surface area AouterAtotal ~ 131 (see Supporting
Information 73) This means that only 3 of the total surface area in GR-SO4 is
available at the external surface Thus as the interlayer sulphate in GR-SO4 is not
readily exchanged with the anionic NAC applied we conclude that 4-NPA and
other NACs only react with the Fe(II) sites at the external GR-SO4 surface In
Table 43 the actual amounts of NAC reduced by GR-SO4 during reaction is
compared with the amount of NAC which theoretically can be reduced by the
initial amount of external reactive Fe(II) sites in GR-SO4 at AouterAtot ~ 131
Table 43 The actual amounts of NAC reduced by GR-SO4 during reaction ∆[NAC]act
compared with the amount of NAC which stoichiometrically should be reduced by the initial
amount of external reactive Fe(II) sites in GR-SO4 ∆[Ar-NO2]theory (calculated as
[Fe(II)GR]0(31middot6) assuming an even distribution of Fe(II) throughout the GR-SO4 structure)
Calculated for [NAC]0 ~ 50 microM
[Fe(II)GR]0 (mM)
∆[Ar-NO2]theory(microM)
∆[4-NT]act (microM)
∆[4-CNB]act (microM)
∆[4-NPA]act (microM)
103 55 174 177 99
63 339 426 380 273
As seen in Table 43 the actual amounts of NAC reduced by GR-SO4 during
reaction are in most cases higher than the amount of NAC which should be
reduced at the given [Fe(II)GR]0 according to reaction stoichiometry This indicates
that new external reactive sites were regenerated eg the Fe(III) phases produced
peel off the GR surface exposing new Fe(II) sites or that outermost internal
86 Chapter 4
reactive sites in close vicinity to the edges are available for reaction as well Lee
and Batchelor (2000b) also found the experimentally observed reduction capacity
of GR-SO4 for chlorinated ethylenes to be 2-3 orders of magnitudes lower than the
estimated reduction capacity including all Fe(II) in GR-SO4
At low initial Fe(II)GR concentrations only a fraction of NAC was reduced within
the reaction time observed (Figure 45c) even though there was stoichiometric
excess of Fe(II)-GR present The fraction of initial Ar-NO2 reduced by GR-SO4 at
reaction termination decreased as [Fe(II)GR]0 decreased (Table 41) and was
accompanied by a change in apparent rate laws with time (compare Figures 45b amp
d) In order to explain these observations we propose that the NACs react only at
external reactive Fe(II) sites and that the regeneration of new external reactive sites
is much slower than the reduction of NAC by GR-SO4 Thus the fast reduction of
NAC taking place at the external reactive sites represents the pseudo 1 order
behaviour whereas depletion of external reactive sites and their slow regeneration
are represented by the second bent part of the (time ln [Ar-NO2]t[Ar-NO2]0)-
curves deviating from pseudo 1 order kinetics Hence at low [Fe(II)GR]0 the
regeneration of reactive sites will eventually control the overall reaction rate
Depletion of available Fe(II) was also observed during the fast reduction of
chromate by GR-CO3 when the initial chromate concentration was increased or
when the GR-CO3 suspension was respiked with chromate repeatedly (Williams amp
Scherer 2001)
436 The role of external and internal reactive sites
It is reasonable to assume that GRs hold adsorption properties similar to other
layered double hydroxides such as hydrotalcites The sorption of 246-
trinitrophenol (TNP) and 245-trichlorophenol (TCP) on chloride and carbonate
intercalated hydrotalcites (HT-Cl = Mg3Al(OH)8ClmiddotyH2O HT-CO3 =
Mg6Al2(OH)16CO3middotyH2O) has been investigated (Hermosin et al 1993 Ulibarri et
al 1995 Ulibarri et al 2001) The authors found that the adsorption of TCP on
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 87
HT-CO3 was very low and that TCP adsorbs only on the external surface sites of
HT-CO3 (Hermosin et al 1993) Furthermore is was reported that the adsorption
of TNP on HT was dramatically affected by the nature of the interlayer anion ie
the adsorption of TNP was considerably higher on HT-Cl than on HT-CO3
(Ulibarri et al 2001) For HT-Cl interlayer anion exchange of chloride with TNP
was detected by XRD analysis and an expansion of the characteristic basal d003
spacing from 79 Ǻ to 132 Ǻ confirmed the presence of TNP in the HT interlayer
(Ulibarri et al 1995) Collating the results reported for HTs with GRs it is not
likely that the divalent SO42- in GR-SO4 is exchanged with the monovalent 4-NPA
Chacirctelet et al (1996) investigated the adsorption of mono- and divalent anions
onin the outer and inner adsorption sites in HT by varying the zetapotential with
pH in the presence of various electrolytes The authors found that SO42- adsorbs on
the external HT surfaces by formation of outer-sphere complexes whereas chloride
hardly adsorbed on HT Moreover it was reported that the adsorption of sulphate
onto HT was not strongly affected by the presence of chloride while sulphate on
the contrary inhibited the adsorption of chloride on HT Studies applying
spectroscopic analyses have investigated the sorption of oxyanions at external and
internal GR-SO4 surfaces (Myneni et al 1997 Randall et al 2001) Selenate was
adsorbed only on the outer GR-SO4 surface when added after GR formation
whereas it was primarily coprecipitated into the interlayer when present during GR
formation Thus for selenate its presence during GR formation is a prerequisite of
its incorporation in the GR interlayer Selenate is readily reduced by GR-SO4 and
the rates of reduction of coprecipitated selenate were very similar to the reduction
rates of selenate adsorbed at the outer GR surface (Myneni et al 1997) This
finding suggests that the outer and inner reactive Fe(II) sites in GR-SO4 hold
similar reactivities
Results based on electron microscopy reported that the reduction of uranyl took
place primarily at the edges of hexagonal GR-SO4 particles (OrsquoLoughlin et al
2003a) In another recent study XRD characterization of the GR-SO4 crystals
88 Chapter 4
during reaction with trichloroacetate (TCA) indicated that TCA did not enter the
GR-SO4 interlayer during reaction (Chapter 5 this work) The average GR-SO4
particle thickness perpendicular to the basal plane was constant during reaction
implying that TCA reacts only at the edges and not at the basal planes Assuming
that the platy hexagonal GR-SO4 crystals hold an average width of 1 microm and an
average particle thickness of 35 nm (Hansen amp Koch 1998) the ratio of edge
surface area to outer surface area is AedgeAouter ~ 130 (see Supporting Information
73) Hence only 3 of the outer surface area in GR-SO4 is available at the edges
Once more the regeneration of new external reactive sites is strongly inferred as
the actual amounts of NAC reduced by GR-SO4 during reaction are much higher
than the amount of NAC which may be reduced by the reactive edge sites present
initially Assuming that the NACs react at the edges only and if employing the
AedgeAouter in the estimation of the rate constants the surface area-normalised
pseudo 1 order rate constants for GR-SO4 would be 30 times higher than the rate
constants depicted in Figure 47 Thus the reactivity of GR-SO4 normalised to its
reactive surface area is higher than the reactivity normalised to its outer surface
area determined by the BET method (N2 adsorption)
The reduction of chromate has been examined in the presence of all the common
GR forms (Bond amp Fendorf 2003 Loyaux-Lawniczak et al 1999 Loyaux-
Lawniczak et al 2000 Williams amp Scherer 2001) The results reported by Bond
amp Fendorf (2003) confirm that not only the surface area of GR but also the
interlayer spacing (interlayer anion size) and interlayer anion charge play an
important role for the reaction rate Hence it follows that coordination (size) and
charge of the oxidant determine its access to the internal sites in GRs
The results obtained for all 4 NACs support what has been reported for nitrate and
TCA At [Fe(II)GR]0 = 2-10 mM and [NO3-]0 = 143 mM pseudo 1 order rate
constants for the reduction of nitrate by GR-SO4 were 158middot10-7 s-1middotm-2middotL (Hansen
et al 2001) This reaction rate increased 40 times by adding barium nitrate instead
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 89
of sodium nitrate thereby precipitating the interlayer sulphate as barium sulphate
and enhancing access to the interlayer Though barium addition changes the GR-
SO4 system dramatically it indicates the importance of interlayer anion exchange
(Hansen amp Koch 1998) The rate constant reported for nitrate (no barium added) is
100-1000 smaller than the rate constants obtained for the NACs in this work
Moreover the reaction kinetics for nitrate did not deviate from pseudo 1 order
kinetics At [Fe(II)GR]0 = 025-104 mM and [TCA]0 = 50 microM-1 mM pseudo 1
order rate constants for the reduction of TCA by GR-CO3 or GR-SO4 were 65middot10-7
s-1middotm-2middotL (Chapter 5 this work) The rate constant for TCA is 10-1000 smaller than
the rate constants for the NACs and the reaction kinetics for TCA did not deviate
from pseudo 1 order kinetics This suggests that the overall reductive
transformation of slowly reacting oxidants such as nitrate and TCA is not
controlled by the rate of regeneration of external Fe(II) reactive sites Altogether
the results reported for selenate chromate and nitrate clearly demonstrate that
these anionic oxidants react primarily with external reactive sites in GR-SO4 Only
under certain conditions ie adding the oxidant prior to GR-SO4 formation or
extracting the interlayer sulphate through precipitation with barium outside the
GR-SO4 particles do the oxidants have access to the interlayer Our findings
suggest that both the neutral and anionic nitro aromatic probe compounds applied
here also react exclusively with the external reactive sites in GR-SO4 Supposedly
the neutral and monovalent charge states of the NACs hinder their access to the
GR-SO4 interlayer A divalent anionic nitro aromatic probe compound might
exchange with the interlayer sulphate more readily and gain access to the inner
Fe(II) reactive sites in GR-SO4 only divalent anionic NACs are not commercially
available
44 Conclusions
This work demonstrates that NACs are completely reduced to their corresponding
anilines by GR-SO4 The surface area-normalised pseudo 1 order rate constants
obtained for the reduction of the neutral and anionic NACs by GR-SO4 under
90 Chapter 4
various experimental conditions did not differ significantly from each other despite
their different charges Neither mass transfer control nor surface saturation kinetics
could account for the similarity of the pseudo 1 order rate constants obtained for
the NACs These observations suggest that the anionic NACs do not have an
enhanced access to inner or outer Fe(II)-GR reactive sites as compared to the
neutral NACs Based on our estimations of the molecular sizes of the NACs we
propose that the charge and not the size of the NACs controls their access to the
internal reactive sites in GRs Hence the reaction between NAC and GR-SO4 takes
place primarily at the external reactive Fe(II) sites This work further demonstrated
that the reduction of the NACs by GR-SO4 only followed pseudo 1 order kinetics
throughout the whole reaction at high initial Fe(II)GR concentrations At low initial
Fe(II)GR concentrations the NACs were not reduced completely within the reaction
time observed though according to reaction stoichiometry the total Fe(II)-GR
present should be sufficient to reduce the whole amount of NAC This means that
at some point during the reaction the external reactive Fe(II) sites were depleted
and the regeneration of new external reactive sites was much slower than the
reduction of the NACs by GR-SO4 The reduction of 4-CNB by GR-SO4 reported
here was 10-100 times slower than its reduction by other Fe(II)-Fe(III) systems
such as goethite lepidocrocite and magnetite suspensions amended with Fe(II)
(Elsner et al 2004)
The results obtained in this work infer that under natural geochemical conditions
where GR-SO4 presumably forms in low concentrations the rate of regeneration of
external Fe(II) reactive sites may control the overall reductive transformation of
fast reacting pollutants by GR-SO4 Thus not only abiotic processes such as
interchanging redox conditions created by water level alterations but also the direct
microbial formation through Fe(III)-reducing bacteria may govern the formation of
GRs and the renewal of external Fe(II) reactive sites in GRs This holds both for
natural systems like iron-rich suboxic soils and sediments as well as engineered
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 91
systems like permeable reactive barriers of zero-valent iron implemented for on-
site remediation of organic and inorganic contaminants
Acknowledgments
We would like to thank Henrik T Andersen for performing the NB kinetic experiments and
Hanne Nancke-Krogh for technical assistance in the laboratory
References
Al-Agha MR Burley SD Curtis CD Esson J (1995) Complex cementation textures and authigenic mineral assemblages in recent concretions from the Lincolnshire Wash (east coast UK) driven by Fe(0) to Fe(II) oxidation Journal of the Geological Society 152 157-171 Bernal JD Dasgupta DR Mackay AL (1959) The oxides and hydroxides of iron and their structural inter-relationships Clay Minerals Bulletin 4 15-30 Bond DL Fendorf S (2003) Kinetics and structural constraints of chromate reduction of green rusts Environmental Science and Technology 37 2750-2757 Brunauer S Emmett PH Teller E (1938) Adsorption of gases in multimolecular layers Journal of American Chemical Society 60 309-319 Charlet L Silvester E Liger E (1998) N-compound reduction and actinide immobilisation in surficial fluids by Fe(II) The surface FeIIIFeIIOH0 species as major reductant Chemical Geology 151 85-93 Chacirctelet L Bottero JY Yvon J Bouchelaghem A (1996) Competition between monovalent and divalent anions for calcined and uncalcined hydrotalcite anion exchange and adsorption sites Colloids and Surfaces A Physicochemical and Engineering Aspects 111 167-175 Chaudhuri SK Lack JG Coates JD (2001) Biogenic magnetite formation through anaerobic biooxidation of Fe(II) Applied and Environmental Microbiology 67 2844-2848 Cui D Spahiu K (2002) The reduction of U(VI) on corroded iron under anoxic conditions Radiochemica Acta 90 623-628 Elsner M Haderlein SB Schwarzenbach RP (2004) Reactivity of Fe(II)-bearing minerals towards reductive transformation of organic contaminants Environmental Science and Technology 38 799-807 Erbs M Hansen HCB Olsen CE (1999) Reductive dechlorination of carbon tetrachloride using iron(II)iron(III)-hydroxide-sulphate (green rust) Environmental Science and Technology 33 307-311 Fadrus H Maly J (1975) Suppression of iron(III) interference in the determination of iron(II) in water by the 110-phenanthroline method The Analyst 100 549-554 Fredrickson JK Zachara JM Kennedy DW Dong H Onstott TC Hinman NW Li S
92 Chapter 4 (1998) Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium Geochimica et Cosmochimica Acta 62 3239-3257 Geacutenin JMR Bourrieacute G Trolard F Abdelmoula M Jaffrezic A Refait Ph Maitre V Humbert B Herbillon A (1998) Thermodynamic equilibria in aqueous suspensions of synthetic and natural Fe(II)-Fe(III) green rusts Occurrences of the mineral in hydromorphic soils Environmental Science and Technology 32 1058-1068
Geacutenin JMR Olowe AA Benbouzid-Rollet ND Prieur D Confente M Resiak B (1991) The simultaneous presence of green rust 2 and sulfate reducing bacteria in the corrosion of steel sheet piles in a harbour area Hyperfine Interactions 69 875-878 Glasauer S Weidler PG Langley S Beveridge TJ (2003) Controls on Fe reduction and mineral formation by a subsurface bacterium Geochimica et Cosmochimica Acta 67 1277- 1288 Hansen HCB (1989) Composition stabilization and light absorption of Fe(II)Fe(III) hydroxycarbonate (green rust) Clay Minerals 24 663-669 Hansen HCB Borggaard OK Soslashrensen J (1994) Evaluation of the free energy of formation of iron(II)iron(III)-hydroxidesulphate (Green Rust) and its reduction of nitrite Geochimica et Cosmochimica Acta 58 2599-2608 Hansen HCB Guldberg S Erbs M Koch CB (2001) Kinetics of nitrate reduction by green rusts ndash effects of interlayer anion and Fe(II)Fe(III) ratio Applied Clay Science 18 81-91 Hansen HCB Koch CB (1998) Reduction of nitrate to ammonium by sulphate green rust Activation energy and interlayer reaction mechanism Clay Minerals 33 87-101 Hansen HCB Koch CB Nancke-Krogh H Borggaard OK Soerensen J (1996) Abiotic nitrate reduction to ammonium Key role of green rust Environmental Science and Technology 30 2053-2056 Hansen HCB Poulsen IF (1999) Interaction of synthetic sulphate green rust with phosphate and the crystallization of vivianite Clays and Clay Minerals 47 312-318 Hansen HCB Taylor RM (1991) The use of glycerol intercalates in the exchange of CO3
2- with SO4
2- NO3- or Cl- in pyroaurite-type compounds Clay Minerals 26 311-327
Heasman DM Sherman DM Ragnarsdottir KV (2003) The reduction of aqueous Au3+ by sulfide minerals and green rust phases American Mineralogist 88 725-738 Hermosin MC Pavlovic I Ulibarri MA Cornejo J (1993) Trichlorophenol adsorption on layered double hydroxide a potential sorbent Journal of Environmental Science and Health A28 1875-1888 Hofstetter TB Heijmann CG Haderlein SB Holliger C Schwarzenbach RP (1999) Complete reduction of TNT and other (poly)nitroaromatic compounds under iron-reducing subsurface conditions Environmental Science and Technology 33 1479-1487 Hofstetter TB Schwarzenbach RP Haderlein SB (2003) Reactivity of Fe(II) species associated with clay minerals Environmental Science and Technology 37 519-528
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 93
Klausen J Troumlber SP Haderlein SB Schwarzenbach RP (1995) Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions Environmental Science and Technology 29 2396-2404 Koch CB Hansen HCB (1997) Reduction of nitrate to ammonium by sulphate green rust Advances in GeoEcology 30 373-393 Kukkadapu RK Zachara JM Smith SC Fredrickson JK Liu C (2001) Dissimilatory bacterial reduction of Al-substituted goethite in subsurface sediments Geochimica et Cosmochimica Acta 65 2913-2924 Lee W Batchelor B (2002a) Abiotic reductive dechlorination of chlorinated ethylenes by iron- bearing soil minerals 1 Pyrite and magnetite Environmental Science and Technology 36 5147- 5154 Lee W Batchelor B (2002b) Abiotic reductive dechlorination of chlorinated ethylenes by iron-bearing soil minerals 2 Green rust Environmental Science and Technology 36 5348- 5354 Liger E Charlet L Van Cappellen P (1999) Surface catalysis of uranium (VI) reduction by iron(II) Geochimica et Cosmochimica Acta 63 2939-2955 Loyaux-Lawniczak S Refait Ph Ehrhardt J Lecomte P Geacutenin JMR (2000) Trapping of Cr by formation of ferrihydrite during the reduction of chromate ions by Fe(II)-Fe(III) hydroxysalt green rusts Environmental Science and Technology 34 438-443 Loyaux-Lawniczak S Refait Ph Lecomte P Ehrhardt J Geacutenin JMR (1999) The reduction of chromate ions by Fe(II) layered hydroxides Hydrology and Earth System Sciences 3 593-599 Mendiboure A Schoumlllhorn A (1986) Formation and anion exchange reactions of layered transition metal hydroxides [Ni1-xMx](OH)2(CO3)x2(H2O)z (M = Fe Co) Revue de Chimie Mineacuterale 23 819-827 Miyata S (1983) Anion-exchange properties of hydrotalcite-like compounds Clays and Clay Minerals 31 305-311 Myneni SCB Tokunaga TK Brown Jr GE (1997) Abiotic selenium redox transformations in the presence of Fe(IIIII) oxides Science 278 1106-1109 Nielsen A (1976) Hvid groslashn og sort rust Beskrivelse af en korrosionsskade paring et svoslashmmebassin Nordisk Betong 2 21-24 OLoughlin EJ Burris DR (2004) Reduction of halogenated ethanes by green rust Environmental Toxicology and Chemistry 23 41-48 OLoughlin EJ Kelly SD Cook RE Csencsits R Kemner KM (2003a) Reduction of uranium(VI) by mixed iron(II)iron(III) hydroxide (green rust) Formation of UO2 nanoparticles Environmental Science and Technology 37 721-727
94 Chapter 4 OLoughlin EJ Kelly SD Kemner KM Csencsits R Cook RE (2003b) Reduction of AgI AuIII CuII and HgII by FeIIFeIII hydroxysulfate green rust Chemosphere 53 437-446 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002) Iron (IIIII) hydroxycarbonate green rust formation and stabilization from lepidocrocite bioreduction Environmental Science and Technology 36 16-20 Parmar N Gorby YA Beveridge TJ Ferris FG (2001) Formation of green rust and immobilization of nickel in response to bacterial reduction of hydrous ferric oxide Geomicrobiology Journal 18 375-385 Pepper SE Bunker DJ Bryan ND Livens FR Charnock JM Pattrick RAD Collison D (2003) Treatment of radioactive wastes An X-ray absorption spectroscopy study of the reaction of technetium with green rust Journal of Colloid and Interface Science 268 408- 412 Randall SR Sherman DM Ragnarsdottir KV (2001) Sorption of As(V) on green rust (Fe4(II)Fe2(III)(OH)12SO4
3H2O) and lepidocrocite (γ-FeOOH) Surface complexes from EXAFS spectroscopy Geochimica et Cosmochimica Acta 65 1015-1023 Roh Y Lee SY Elless MP (2000) Characterization of corrosion products in the permeable reactive barriers Environmental Geology 40 184-194 Schultz CA Grundl TJ (2000) pH dependence on reduction rate of 4-Cl-nitrobenzene by Fe(II)montmorillonite systems Environmental Science and Technology 34 3641-3648 Simon L Francois M Refait Ph Renaudin G Lelaurain M Geacutenin JMR (2003) Structure of the Fe(II-III)-layered double hydroxysulphate green rust two from Rietveld analysis Solid State Sciences 5 327-334 Tamaura Y (1986) Ni(II)-bearing green rust II and its spontaneous transformation into Ni(II)- bearing ferrites Bulletin of the Chemical Society of Japan 59 1829-1832 Tamaura Y (1985) ZnII-bearing green rust II and its spontaneous transformation into ZnII- bearing ferrite in aqueous solution Bulletin of the Chemical Society of Japan 58 2951-2954 Trolard F Abdelmoula M Bourrieacute G Humbert B Geacutenin JMR (1996) Mise en eacutevidence dun constituant de type rouilles vertes dans les sols hydromorphes Proposition de lexistence dun nouveau mineacuteral la fougeacuterite Geacuteosciences de surface Comptes Rendus de LrsquoAcademie des Sciences 323 1015-1022 Tuovinen OH Button KS Vuorinen A Carlson L Mair DM Yut LA (1980) Bacterial chemical and mineralogical characteristics of tubercles in distribution pipelines Journal of the American Water Works Association 72 626-635 Ulibarri MA Pavlovic I Barriga C Hermosin MC Cornejo J (2001) Adsorption of anionic species on hydrotalcite-like compounds effect of interlayer anion and crystallinity Applied Clay Science 18 17-27 Ulibarri MA Pavlovic I Hermosin MC Cornejo J (1995) Hydrotalcite-like compounds as potential sorbents of phenols from water Applied Clay Science 10 131-145
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 95
Williams AGB Scherer MM (2001) Kinetics of chromate reduction by carbonate green rust Environmental Science and Technology 35 3488-3494
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 97
5 Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems
Abstract
Trichloroacetate (TCA) is a widespread environmental contaminant with proven
phytotoxicity and suspected human carcinogenicity In order to assess the global
cycling of TCA and to predict its fate in subsurface environments information
regarding the reactivity and product distribution of TCA degradation is needed
Due to the high oxidation state of TCA conditions for oxidative transformation
pathways in soils and groundwater are unfavorable However in suboxic soils and
sediments Fe(II)-bearing minerals are potential reactants for reductive
dehalogenation reactions of TCA as has been demonstrated for other halogenated
contaminants We examined the reactivity of various Fe(II)-Fe(III) mineral
systems towards TCA and dichloroacetate (DCA) its expected transformation
product in laboratory batch experiments imitating natural conditions ie low
initial Fe(II) Fe(III) and TCADCA concentrations and no artificial buffer The
Fe(II)-Fe(III)-systems investigated included sulfate green rust (GR-SO4) carbonate
green rust (GR-CO3) magnetite Fe(II)goethite and Fe(II)lepidocrocite
Trichloroacetate was readily reduced to DCA by all Fe(II)-bearing minerals The
reactions generally followed pseudo 1 order kinetics with respect to TCA The
surface area-normalised pseudo 1 order rate constants obtained (035ndash76middot10-5 min-
1middotm-2middotL at [Fe(II)]0 = 020ndash122 mM [TCA]0 = 15ndash1000 microM and pH 70ndash87)
showed no striking differences regarding product distribution and surface area-
normalised reaction rate constants between the Fe(II)-Fe(III)-systems The
stoichiometrically formed DCA was not further reduced to monochloroacetate
(MCA) or acetate in any of the systems within the time frame in our experiments
To our knowledge this is the first published report on abiotic transformation of
TCA by Fe(II)-bearing minerals Our results imply that processes involving
reactive Fe(II)-bearing minerals may play a significant role in controlling the fate
98 Chapter 5
of TCA in natural subsurface environments and that DCA found in the subsurface
may be formed by such processes
51 Introduction
Trichloroacetic acid (TCA) has been applied as a herbicide for many years until its
use was banned in the late 1980acutes (Berg et al 2000) Today TCA is mainly used
as an etching agent in the metal industry as a swelling solvent in the plastic
production and as a bleaching agent in the paper and pulp manufacture (Muumlller et
al 1996) Other anthropogenic sources include formation of TCA as a result of the
chlorine based disinfecting process used in drinking water treatment and the
atmospheric photooxidation of chlorinated solvents including tetrachloroethene
and 111-trichloroethane (McCulloch 2002) Only very little information is
available on the TCA production volumes and even less is known about the
amount of TCA released into the environment as a result of its industrial
applications Due to its low volatility and high aqueous solubility TCA is easily
washed out of the atmosphere into the aquatic and terrestrial biospheres As TCA
is found in almost every ecosystem around the globe including non-urban and
non-industrial sites the relative contributions from anthropogenic and natural
sources are currently being debated (McCulloch 2002 Ahlers et al 2003)
Trichloroacetic acid is omnipresent in soils and the concentrations reported are
very variable ranging from lt005 microgkg to 380 microgkg (Euro Chlor 2001
McCulloch 2002 Ahlers et al 2003) Both abiotic and enzymatically catalyzed
formation of TCA from humic acids have been demonstrated in laboratory studies
(Haiber et al 1996 Hoekstra et al 1999b Fahimi et al 2003) Furthermore the
in situ natural formation of TCA from anthropogenic or natural tetrachloroethene
or 111-trichloroethane in biota has been suggested (Hoekstra et al 1999a
McCulloch 2002) Such natural sources may explain part of the TCA
concentrations found in soils but their environmental significance is still unknown
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 99
On account of its phytotoxicity suspected human carcinogenicity and widespread
occurrence TCA is of considerable environmental concern especially in the
terrestrial compartment The TCA concentrations found in soil air and water in
pre-industrial times were far below the present ones (Jordan amp Frank 1999 Ahlers
et al 2003) Based on the current TCA concentrations detected in soils the
European Commission proposed risk reduction measures concerning
tetrachloroethene - a precursor of TCA - to be taken immediately (Ahlers et al
2003 and references therein) Occurrences of monochloroacetic acid (MCA) and
dichloroacetic acid (DCA) reported include surface waters marine waters
precipitation ice (glaciers) and air (Reimann et al 1996 Berg et al 2000 Scott et
al 2000 Scott et al 2002) Based on the concentrations reported for the aquatic
environments it is reasonable to assume that MCA and DCA are omnipresent in
soils as well Sources of MCA and DCA include production in the chemical
industry photooxidation of chlorinated aliphatics in the atmosphere and reductive
transformation of TCA (Reimann et al 1996 Ahlers et al 2003 and references
therein) MCA and DCA are also toxins and suspected human carcinogens (Kuumlhn
amp Pattard 1990) hence not only TCA but also its daughter compounds are
pollutants of environmental concern
In subsurface environments TCA may be removed by sorption seepage chemical
transformation microbial degradation and plant uptake followed by metabolic
degradation andor physical removal at harvest (Foy 1975) There is little or no
evidence of abiotic transformations of TCA in the literature Only one recent study
demonstrated the reductive dechlorination of TCA to MCA by Fe(0) (Hozalski et
al 2001) It has been reported that the degradation of TCA in soil is slow and
mainly mediated by microorganisms but only little is known about the bacteria and
processes involved (Lignell et al 1984) Biodegradation of TCA has been found at
both oxic and anoxic conditions An aerobic microorganism capable of growing on
TCA as the sole carbon and energy source has been characterised (Yu amp Welander
1995) Moreover anaerobic bacteria coupling co-metabolic growth to reductive
100 Chapter 5
dechlorination of TCA have been isolated (Weightman et al 1992 De Wever et
al 2000) However more information regarding the abiotic and biotic
transformation of TCA is needed in order to assess the fate and transport of TCA in
natural subsurface environments
It is well-known that Fe(II) present in minerals or associated with mineral surfaces
is a much stronger reductant than Fe(II) in solution The enhanced reactivity of a
structural or surface-bound Fe(II) center can be rationalized by the increased
electron density donated by hydroxyl ligands and a stabilization of the Fe(III)
oxidation state by the hydroxyl ligands (Luther 1990) Fe(II)-bearing minerals
including layered Fe(II)-Fe(III) hydroxides (green rusts) magnetite (Fe3O4)
siderite (FeCO3) Fe(II) sulfides as well as Fe(II)-carrying Fe(III) oxides and clay
minerals have also been shown to reduce a range of organic and inorganic
contaminants such as nitro aromatic compounds chlorinated aliphatics chromate
uranyl pertechnetate nitrate monochloramine and carbamate pesticides (Chapter
4 this work Klausen et al 1995 Cui amp Eriksen 1996 Erbs et al 1999 Liger et
al 1999 Loyaux-Lawniczak et al 1999 Amonette et al 2000 Hansen et al
2001 Pecher et al 2002 Vikesland amp Valentine 2002 Hofstetter et al 2003
OrsquoLoughlin and Burris 2003 OrsquoLoughlin et al 2003a amp 2003b Strathmann amp
Stone 2003 Elsner et al 2004a) Laboratory and field studies showed that even in
geochemically highly heterogeneous anoxic aquifer sediments Fe(II) adsorbed to
Fe(III) (hydr)oxide surfaces was the dominant reductant of nitroaromatic and
halogenated contaminants (Ruumlgge et al 1998 Hofstetter et al 1999 Kenneke amp
Weber 2003) Only little is known about the nature of the Fe(II) species associated
with Fe(III) oxide surfaces but reactive hydroxylated Fe(II)-Fe(III)-hydroxo
surface complexes associated with hematite and magnetite above pH 65 have been
proposed (Charlet et al 1998aampb Liger et al 1999) Due to the presence of
structural Fe(II) within the mineral lattice the reactivity of Fe(II) associated with
mixed valent Fe(II)-Fe(III) minerals such as green rusts magnetite and reduced
ferruginous clay minerals may hold another reactivity than Fe(II) associated with
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 101
pure Fe(III) oxides However Fe(II) adsorbed on Fe(III) oxides such as goethite
hematite and lepidocrocite may also hold different reactivities as the Fe(III) oxides
contain different crystal and surface structures
Since chlorinated ethanes and ethenes such as hexachloroethane 111-
trichloroethane tetrachloroethene and trichloroethene are susceptible to chemical
reduction by a range of Fe(II)-bearing minerals including magnetite GR-SO4
Fe(II) sulfides and Fe(II)-carrying Fe(III) oxides (Butler amp Hayes 1998 amp 1999
Hwang amp Batchelor 2000 Gander et al 2002 Lee amp Batchelor 2002aampb Elsner
et al 2004a) we hypothesized that TCA may be transformed by Fe(II)-bearing
minerals as well The main goals of this work were to study such reactions and
establish product distribution and surface area-normalised reaction rates for the
reductive dechlorination of TCA by Fe(II)-Fe(III) mineral systems common in
nature
5 2 Materials and methods
No synthetic buffers were applied and iron concentrations were kept low The
calcareous systems were pH-controlled at 76 through a natural buffer system
(CaCO3(s) + 995 N205 CO2(g)) All handling and sampling of solutions and
suspensions were carried out under strict anoxic conditions Goethite (acicular
particles with size 01 times 06 microm specific surface area 16 m2g) and lepidocrocite
(acicular particles with size 005 times 03 microm specific surface area 18 m2g) were
purchased as fine powders from Bayer (Bayferrox 910 and 943) Calcite (grain size
170-350 microm Pluumlss-Staufer AG) was used as a buffer or as a Fe(III)-oxide-bearing
mineral In order to simulate natural conditions the iron minerals were applied as
coatings on calcite particles (model system for calcareous soils) in some
experiments Trichloroacetic acid dichloroacetic acid and monochloroacetic acid
were pa quality (Fluka)
102 Chapter 5
521 Synthesis of GRs and magnetite
GR-CO3 was synthesized by controlled air oxidation of an FeCl2 solution at a
constant pH of 700 (titrated with 1 M Na2CO3) according to the procedure given
by Hansen amp Koch (1997) 05 M aqueous stock solutions of FeCl2 were prepared
in 100 mL glass flasks by reacting 65 mmol of iron powder (particle size 10 microm
Merck) with 100 mL deoxygenated 10 M HCl The solutions were magnetically
stirred and heated (~80degC) during reaction until the H2(g) production had ceased (ge
2 hours) The FeCl2 solutions were stored in the dark under a small Ar
overpressure at 5degC The GR-CO3 suspensions were washed with deoxygenated
deionised water (DIW) separated on a folding filter (medium filtration rate cotton
linterhigh alpha pulp Schleicher amp Schuell) and redispersed in deoxygenated
DIW Washing separation and redispersion of the GR-CO3 suspension were
conducted in an anoxic glove box (92 N28 H2 Coy Laboratory Products Inc)
All suspensions and solutions were deoxygenated by Ar-purging (999998 Ar
Carbagas) Magnetite was synthesized by further aerial oxidation of GR-CO3 at pH
700 until consumption of 1 M Na2CO3 ceased GR-SO4 was synthesized by
controlled air oxidation of an FeSO4 solution at a constant pH of 70 according to
the procedure given by Koch amp Hansen (1997) The GR-SO4 suspension was
washed with deoxygenated DIW separated on a glass filter funnel (pore size 4
Duran) and redispersed in deoxygenated DIW Washing separation and
redispersion of the GR-SO4 suspension were conducted in an anoxic glove bag
(999995 Ar Aldrich)
522 Preparation of iron oxide coatings
Two grams of goethite (goe) or lepidocrocite (lep) and 100 g calcite were
combined with 200 mL DIW in a 500 mL polyethylene flask The suspension was
gently agitated on a reciprocating shaker for 24 h and left to stand for another 24 h
Excess Fe(III) oxides and salts were removed from the coated material by repeated
decantation and washing with DIW in polyethylene flasks until clear runoff
Finally the coatings were collected on folding filters and air dried The amount of
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 103
goethite and lepidocrocite coated onto calcite after washing and drying was
quantified to 10-11 mg Fe(III)g calcite
523 Mineral characterisation
The identity and purity of the GR-CO3 GR-SO4 and magnetite suspensions were
examined by means of X-ray diffraction (XRD) The XRD analyses were
performed on a Scintag XDS 2000 using Co Kα radiation (45 kV 40 mA) or a
Siemens D5000 XRD applying Co Kα radiation (40 kV 40 mA) Glycerol smears
made according to Hansen (1989) were scanned between 6 and 80 deg2θ with a scan
speed of 1 deg2θmin The specific surface area (SSA) of calcite was determined by
the BET multi-point method using N2 adsorption (Brunauer et al 1938) Powder
samples were filled into sample burettes in the glove box and the generously
lubricated stopcocks closed Samples and burettes were evacuated prior to
connecting them to the BET-instrument (Sorptomatic 1990 Fisons)
524 Kinetic experiments
All reactions were carried out in 25-100 mL serum vials sealed with stoppers
(Viton or Teflon coated rubber) and aluminum crimp caps Kinetic experiments
were conducted with GR-SO4 GR-CO3 magnetite Fe(II)goethite and
Fe(II)lepidocrocite at room temperature In most cases pH was controlled through
the carbonate-bicarbonate buffer system by adding calcite to suspensions
containing the iron minerals solely or by adding the iron minerals as coatings on
calcite Furthermore the calcite containing suspensions were deoxygenated with
05 CO2995 N2(g) thereby attaining an initial pH of 76-77 The GR-CO3 and
magnetite suspensions were deoxygenated with 100 N2(g) and no additional pH
buffer was added The goethite and lepidocrocite suspensions were amended with
300-1000 microM FeCl2(aq) and equilibrated gt 20 h prior to TCADCA addition See
Table 51 for more details on the experimental conditions To start the reaction 50
microM - 1 mM TCA or DCA was added to the mineral suspensions from aqueous
anoxic stock solutions The reaction vials were agitated gently on a roller apparatus
104 Chapter 5
or a shaking table (35 rpm) in order to minimize abrasion of the iron oxide mineral
coatings At appropriate time intervals suspension samples were withdrawn using
Ar(g)- 100 N2(g)- or 995 N205 CO2(g)-flushed sterile disposable syringes
and hypodermic needles The suspension samples were filtered (02 microm Teflon)
and collected for quantification of chloride and the chlorinated acetic acids The
samples were stored at -20degC and analysed without further treatment
525 Analytical methods
Total and aqueous Fe(II) were determined using a modified phenanthroline method
(Fadrus and Maly 1975) For determining [Fe(II)aq] and [Fe(II)total] 1 mL filtered
(02 microm Teflon) and 1 mL unfiltered mineral suspension were added to 18 mL 01
M HCl respectively and allowed to dissolve for 30 min From these acid digests
01 mL was added to 05 mL Fe(II)-phenanthroline-buffer-reagent and 19 mL
DIW added up Estimates of the structural or adsorbed Fe(II) in the Fe(II)-Fe(III)
mineral systems were estimated as the difference [Fe(II)solid] = [Fe(II)total] -
[Fe(II)aq] The total amount of Fe(III) coated on calcite was determined by atomic
absorption spectroscopy following dissolution in 6 M HCl(aq) for 24 h At low
initial TCA concentrations (le 50 microM) the chlorinated acetic acids were quantified
by means of a modified ion interaction (or paired-ion) chromatographic method
(Sarzanini et al 1999) Separation was performed on a LiChrospher 100 RP-18 (5
microm 125 times 4 mm ID) reversed-phase column coupled with a LiChroCART 100 RP-
18 (4 times 4 mm ID) precolumn Analytical conditions were isocratic and the eluent
consisted of 50 aqueous solution of 35 mM cetyltrimethylammonium chloride
(pH 50) and 50 CH3CN The injection volume was 20 microL and the flow-rate 10
mLmin HPLC analyses of the chloroacetates were performed using a Gynkotek
Pump M480 Gynkotek Gina 50 auto sampler and a diode array UV detector (340s
Gynkotek) UV-VIS detection was carried out at 200 nm At higher initial TCA
concentrations the chlorinated acetic acids were quantified by a modified HPLC
method (Husain et al 1992) Separation was performed on a ChromSphere C-18
(10 microm 250 times 46 mm ID) reversed-phase column Analytical conditions were
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 105
isocratic and the eluent consisted of 015 M (NH4)2SO4(aq) pH 55 The injection
volume was 20 microL and the flow-rate 10 mLmin HPLC analyses were performed
using a Series 10 Liquid Chromatographic Pump (Perkin-Elmer) and a SPD-10 A
VP UV-VIS detector (Shimadzu) UV-VIS detection was carried out at 210 nm
Chloride was determined in the GR-SO4 kinetic experiments using a flow injection
system with spectrophotometric detection (Cheregi amp Danet 1997)
53 Results and discussion
531 Product formation and reaction kinetics
Trichloroacetate was readily reduced to DCA by all the Fe(II)-bearing minerals
examined Only DCA was detected within the reaction time in all the Fe(II)-Fe(III)
mineral systems Experiments conducted with the various Fe(II)-Fe(III) mineral
systems and DCA confirmed that no significant reduction of DCA took place (data
not shown) Hence it is reasonable to assume that the further hydrogenolysis of
DCA to MCA is too slow to be detected within the experimental time frame here
The mass balance of TCA and DCA was almost complete in all suspensions ruling
out any alternative reaction pathways to reductive dechlorination Decarboxylation
of TCA producing chloroform and carbon dioxide requires high temperatures and
is therefore assumed not to take place at the experimental conditions applied here
(Atkins et al 1984) Based on these results we propose that the reductive
dechlorination of TCA by Fe(II)-bearing minerals proceeds via hydrogenolysis
(replacement of halogen by hydrogen) as reported for the transformation of TCA
by zero-valent iron (Hozalski et al 2001) Thus in order to reduce TCA to DCA
2 electrons corresponding to 2 Fe(II) are needed (Figure 51)
106 Chapter 5
Cl3CC
O
O- Cl2HCC
O
O-
2e- H+ Cl-
TCA DCA Figure 51 Proposed reductive transformation pathway of TCA
In the Fe(II)goe and Fe(II)lep systems we detected no TCA transformation in the
absence of either aqueous Fe(II) or pure or calcite-associated goethite and
lepidocrocite These results strongly indicate that reactive Fe(II) species associated
with the goethite and lepidocrocite surfaces are the reductants for TCA in these
systems The Fe(III) phases forming in the mineral suspensions were not
characterised and therefore the reaction stoichiometry cannot be assessed
At initial Fe(II) concentrations in large excess of initial TCA concentration we
found a pseudo 1 order rate law for the degradation of TCA by Fe(II)
[ ] [ ] [ b a TCAFe(II) TCA
sdotsdot=minus kdt
d ]
where a = 1 b = 1 and the observed pseudo 1 order rate constant kobs = k middot [Fe(II)]
At all [Fe(II)]0[TCA]0 ratios studied (6-738) TCA was transformed almost
quantitatively into DCA and the reaction kinetics followed pseudo 1 order kinetics
with respect to TCA (Figure 52) The observed pseudo 1 order rate constants for
the transformation of TCA by the various Fe(II)-Fe(III) mineral systems were
calculated as initial rates (ie max first two half-lives) from linear fits of (time ln
[TCA]t[TCA]0)-plots (Table 51) The amount of chloride produced during
reaction with GR-SO4 was always equivalent to the amount of TCA transformed
into DCA (Figure 52c) This also indicates that no significant further reduction of
DCA took place in GR-SO4 suspensions
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 107
Figure 52 Time course of TCA consumption and DCA and chloride production for a)
Fe(II)Goe ([Fe(II)tot]0 = 095 mM) b) Fe(II)Lep ([Fe(II)tot]0 = 091 mM) c) GR-SO4
([Fe(II)GR]0 = 962 mM) d) GR-CO3 ([Fe(II)tot]0 = 633 mM) and e) Magnetite ([Fe(II)tot]0 = 350
mM) Solid lines represent 1 order kinetic fits whereas symbols and dotted lines represent actual
data = TCA = DCA = Clndash
T
able
51
Exp
erim
enta
l con
ditio
ns a
nd p
seud
o 1
ord
er ra
te c
onst
ants
for t
he re
duct
ive
trans
form
atio
n of
TC
A b
y va
rious
Fe(
II)-
Fe(I
II) c
onta
inin
g m
iner
al sy
stem
s
Syst
em
Susp
ensi
on a
ge
(d)
[Fe(
II)] s
olid
a (m
M)
[Fe(
II)] a
q b
(mM
) [T
CA
] 0 (micro
M)
pHin
itcpH
endd
k obs
e (min
-1)
Surf
ace
area
(m
2 L)
k obs
f
(min
-1m
-2middotL
)
Fe(I
I)aq
1
0
030
434
nd
76
gn
dn
dn
d
Fe(I
I)G
oe
1
002
024
429
nd
78
g1
021
0-47
1 i
143
10-5
Fe(I
I)G
oe
1
013
094
543
77
70
225
10-4
71
i3
161
0-5
Fe(I
I)G
oe
coat
ing
1
023
40
066
484
765
80
g6
401
0-454
0 j
119
10-5
Fe(I
I)G
oe
coat
ing
1
0
150
8048
6n
d7
6 g12
43
10-4
540
j2
301
0-5
Fe(I
I)L
ep
1
0
020
2315
7n
d7
8 g0
751
0-48
0 i
094
10-5
Fe(I
I)L
ep
coat
ing
1
016
30
137
470
765
80
g2
821
0-454
0 j
052
10-5
Fe(I
I)L
ep
coat
ing
1
0
100
8141
7n
d7
7 g8
311
0-454
0 j
154
10-5
Fe3O
41
3
380
1251
38
107
8g
830
10-4
16
k5
311
0-4
Fe3O
477
112
56
556
70
70
153
10-4
52
k2
951
0-5
GR
-CO
31
5
940
3950
37
658
4g
761
10-4
419
l1
821
0-5
GR
-CO
32
7
60
147
88
568
2940
81
0-453
6 l
761
10-5
GR
-CO
332
73
003
563
85
80
490
10-4
515
l0
951
0-5
GR
-CO
314
2
3
530
005
629
nd
87 g
513
10-4
249
l2
061
0-5
GR
-SO
41
5
17-1
217
086
-13
910
5n
dn
d3
601
0-492
6 m
039
10-5
GR
-SO
41
6
22-1
027
093
-14
527
0n
dn
d3
761
0-488
1 m
043
10-5
GR
-SO
41
7
05-1
014
077
-17
950
0n
dn
d3
741
0-4 9
18
m0
411
0-5
GR
-SO
41
5
17-1
051
060
-16
510
00n
dn
d2
891
0-483
7 m
035
10-5
n
d =
not
det
ecte
d a
Ini
tial s
truct
ural
or a
dsor
bed
Fe(I
I) e
stim
ated
as [
Fe(I
I)to
tal]
ndash [F
e(II
) aq]
b In
itial
dis
solv
ed F
e(II
) mea
sure
d c
Sus
pens
ion
pH p
rior t
o TC
A a
dditi
on d
Sus
pens
ion
pH a
t rea
ctio
n
te
rmin
atio
n e
Pse
udo
1 o
rder
rate
con
stan
ts fo
r the
con
sum
ptio
n of
TC
A c
alcu
late
d fr
om in
itial
rate
s (m
ax f
irst t
wo
half-
lives
) f
Surf
ace
area
-nor
mal
ised
pse
udo
1 o
rder
rate
con
stan
ts g
pH
con
trol
th
roug
h pu
re C
aCO
3 and
05
C
O2(g
) h
pH
con
trol t
hrou
gh F
e(II
I) o
xide
-coa
ted
calc
ite a
nd 0
5
CO
2(g)
i Es
timat
ed u
sing
the
SSA
of t
he F
e(II
I) o
xide
app
lied
j E
stim
ated
usi
ng th
e SS
A o
f cal
cite
~1
m2 g
k E
stim
ated
ass
umin
g SS
A =
4 m
2 g (S
chw
ertm
ann
amp C
orne
ll 1
991)
frac12middot[
Fe(I
I) sol
id] 0middot
232
gmiddotm
ol-1
middot4 m
2 middotg-1
l E
stim
ated
ass
umin
g SS
A =
47
m2 g
(Will
iam
s amp S
cher
er 2
001)
frac14middot[F
e(II
) GR] 0middot
600
gmiddotm
ol-1
middot47
m2 middotg
-1 m
Est
imat
ed a
s in l
but u
sing
SSA
= 7
12
m2 middotg
-1 (C
hapt
er 4
thi
s wor
k)
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 109
532 Comparing rate constants obtained for the various Fe(II)-Fe(III) mineral
systems
Data for the systems containing iron oxide coated calcite were very similar to the
data obtained for the pure iron oxides (not shown in Figure 53) Since no SSA was
determined for magnetite in this study a SSA of 4 m2g was assumed
(Schwertmann amp Cornell 1991) However it should be noted that the magnetite
synthesized by Schwertmann and Cornell (1991) was prepared differently (ie
oxidation of Fe(II) by nitrate in a heated alkaline solution) from the magnetite
applied in this study The surface area-normalised pseudo 1 order kobs values
obtained for GR-CO3 GR-SO4 Fe(II)goethite and Fe(II)lepidocrocite were all
within the same order of magnitude (Figure 53a)
Figure 53 Average surface area-normalised pseudo 1 order rate constants for the degradation of
a) TCA (this work) b) hexachloroethane (Elsner et al 2004a) and c) carbon tetrachloride
(Amonette et al 2000 Pecher et al 2002 OrsquoLoughlin et al 2003c Elsner et al 2004b) by
GR-SO4 GR-CO3 (suspension age 1 d) Fe3O4 Fe(II)α-FeOOH and Fe(II)γ-FeOOH
Experimental conditions applied in this work [Fe(II)tot]0 = 025-107 mM in the goethite and
lepidocrocite suspensions [Fe(II)tot]0 = 025-116 mM in the GR-SO4 and GR-CO3 suspensions
pH 70-86 71-926 m2 mineral surface areaL Experimental conditions applied by Elsner et al
1 mM aqueous Fe(II) 25 m2 mineral surface areaL Experimental conditions applied in
references employed in c) [Fe(II)tot]0 = 1-83 mM 25-275 m2 mineral surface areaL GR-SO4 =
110 Chapter 5
sulfate green rust GR-CO3 = carbonate green rust Fe3O4 = magnetite α-FeOOH = goethite γ-
FeOOH = lepidocrocite
When comparing the rate constants for the Fe(II)-Fe(III) mineral systems found for
reduction of TCA in this study (Figure 2a) mixed valent Fe(II)-Fe(III) minerals
such as green rusts and magnetite containing structural Fe(II) within the mineral
lattice do not seem to be significantly more reactive than Fe(II)-Fe(III) mineral
systems containing Fe(II) associated with pure Fe(III) oxides Unlike most other
iron oxides GRs contain not only external Fe(II) reactive sites at the surface but
also internal sites in the space between consecutive Fe(II)-Fe(III) hydroxide layers
The GR interlayer thickness is a function of both the size and the charge of the
interlayer anion For solutes the Fe(II) within the GR hydroxide layer is accessible
at the outside basal planes and at the edges as well as through the interlayer under
certain conditions (see Figure 42 Chapter 4 this work) Due to electrostatic
forces oxidants holding different charge properties (anions cations neutral
molecules) may exhibit different affinities for the various reactive Fe(II) sites
present in GR As the reactive sites are located inat the Fe(II)-Fe(III) hydroxide
layers the rate of reaction depends on the hydroxide layer area which can be
accessed by the oxidant If the oxidant is able to exchange with the interlayer
anion reaction can take place both at outer and inner surfaces of the GR particles
and in total more reactive sites are available for the reaction Thus oxidant size
and charge primarily control its access to the internal sites in GRs XRD
characterization of the GR-SO4 crystals during reaction with TCA demonstrated
that the GR-SO4 interlayer spacing did not vary during reaction (Table 52) This
may indicate that TCA did not enter the GR-SO4 interlayers
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 111 Table 52 Diffraction angle d-spacing and width at half peak height (Wfrac12) for the 001 GR-SO4
diffraction peak as a function of time during reaction with TCA ([Fe(II)GR]0 = 4 mM [TCA]0 = 1
mM)
Time (min) Angle (deg2θ)
d001-spacing(nm)
Wfrac12(degθ)
0 9483 10821 0273
10 9494 10809 0287
215 9522 10777 0263
330 9550 10745 0273
510 9524 10775 0277
855 9509 10791 0253
1160 9467 10839 0268
We roughly estimated the molecular size of TCA by summing the covalent radii of
the individual atoms (see Supporting Information 74) When comparing the
molecular size of TCA with the GR-SO4 interlayer spacing of 061 nm it can be
concluded that only when the C-C bond is oriented perpendicular to the interlayer
plane does the size of TCA exceed the GR-SO4 interlayer spacing In contrast the
size of TCA exceeds the GR-CO3 interlayer spacing (026 nm) regardless of its
orientation Hence if TCA was intercalated in the GR-CO3 interlayer we would
expect the interlayer spacing to expand The same holds for intercalation of a
vertically oriented TCA in the GR-SO4 interlayer Supposedly both the low charge
and the size of TCA impeded its access to the GR-SO4 and GR-CO3 interlayers ie
the divalent sulphate and carbonate in the GR interlayers did not readily exchange
with the monovalent TCA since GR interlayers generally have a higher affinity for
divalent anions than for monovalent anions (Miyata 1983) Thus TCA did neither
access nor react with internal Fe(II) reactive sites in GR-SO4 which means that the
reaction between TCA and GR-SO4 took place at the external reactive Fe(II) sites
solely It is reasonable to assume that the same holds for the reaction between TCA
and GR-CO3 No significant aging effects eg rate constants varying as a function
of GR age were observed within 142 days (see Table 51) However the SSAs of
112 Chapter 5
the GR suspensions holding ages up to 142 days were not measured but estimated
assuming that the GR SSA did not decrease within the time frame
According to the Scherrer formula the width at half peak height (Wfrac12) of a
diffraction peak is inversely proportional to the average crystal dimension
perpendicular to the given crystal plane (Klug amp Alexander 1974) The average
GR-SO4 particle thickness perpendicular to the basal plane (Wfrac12 Table 52) was
constant during reaction implying that TCA reacts only at the edges and not at the
basal planes Assuming that the platy hexagonal GR-SO4 and GR-CO3 crystals
hold an average width of 1 microm and an average particle thickness of 35 nm (Hansen
and Koch 1998) the ratio of edge surface area to outer surface area is AedgeAouter ~
130 for GR-SO4 and 121 for GR-CO3 (see Supporting Information 73) This
means that only 3 of the outer surface area in GR-SO4 and 5 of the outer
surface area in GR-CO3 are available at the edges Assuming that TCA reacts at the
edges only and if employing the AedgeAouter in the estimation of the rate constants
the surface area-normalised pseudo 1 order rate constants for GR-SO4 and GR-
CO3 would be 20-30 times higher than the rate constants depicted in Figure 53a
Thus the reactivity of GRs normalised to their reactive surface area is much higher
than the reactivity normalised to their total surface area
533 Comparing with rate constants obtained for other chlorinated aliphatic
compounds
Though care must be taken when comparing kinetic parameters obtained at
different experimental conditions (eg pH [Fe(II)]0[TCA]0 ratios surface area to
volume ratios etc) it is interesting to compare our results to those reported for
hexachloroethane (Figure 53b data from Elsner et al 2004a) The reductive
transformation of hexachloroethane was investigated for various Fe(II)-bearing
minerals including Fe(II)goethite Fe(II)lepidocrocite and GR-SO4 in the presence
of 1 mM dissolved Fe(II) and 25 m2 mineral surface areaL at pH 72 except for the
GR-SO4 suspensions in which the dissolved Fe(II) concentrations were slightly
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 113
higher and pH = 8 The pseudo 1 order rate constants reported for
hexachloroethane are in the range 18middot10-4 ndash 75middot10-3 h-1middotm-2middotL (Elsner et al 2004a)
When comparing Figure 53a with Figure 53b it can be seen that the differences
in intrinsic reactivity of the Fe(II)-bearing mineral systems are more pronounced
for hexachloroethane than for TCA
Caution should also be advised to the different reaction mechanisms by which
hexachloroethane and TCA react The transfer of a single electron and the
formation of an alkyl radical upon removal of a chlorine atom constitute the first
and in most cases the rate-limiting step in the reduction of chlorinated aliphatic
compounds (Vogel et al 1987) Depending on the chemical structure of the
chlorinated aliphatic compound the resulting free alkyl radical may undergo
hydrogenolysis chloroelimination or dimerizationcoupling In the case of TCA
the free dichloroacetate radical most likely undergoes hydrogenolysis The almost
quantitative transformation of TCA to DCA confirms that hydrogenolysis is the
prevalent reaction mechanism in our mineral systems The pentachloroethyl radical
formed from hexachloroethane may undergo hydrogenolysis (producing
pentachloroethane) or dichloroelimination (producing tetrachloroethene) Elsner et
al (2004a) found that hexachloroethane was transformed quantitatively into
tetrachloroethylene for all minerals which strongly indicates that
dichloroelimination was the dominating reaction mechanism Another
polychlorinated aliphatic compound transformed mainly by hydrogenolysis under
reducing conditions is carbon tetrachloride Several studies have investigated the
reductive dechlorination of carbon tetrachloride by various Fe(II)-bearing minerals
including Fe(II)goethite and GR-SO4 and reported pseudo 1 order rate constants
in the order 152middot10-4 ndash 640middot10-4 h-1middotm-2middotL for Fe(II)goethite and 864middot10-4 h-1middotm-2middotL
for GR-SO4 (Amonette et al 2000 Pecher et al 2002 OLoughlin et al 2003c
Elsner et al 2004b) When comparing Figure 53a with Figure 53c it can be seen
that the range of magnitude of the rate constants and the differences in intrinsic
114 Chapter 5
reactivity of the Fe(II)-bearing mineral systems are similar for carbon tetrachloride
and TCA
534 Factors controlling the reactivity of surface-bound Fe(II)
The reactivity of an oxidant towards Fe(II) surface species cannot be predicted
from the reduction potentials of the redox couple alone In heterogeneous systems
processes such as mass transfer and adsorptiondesorption may have a rate-limiting
effect on the overall reaction rate If the adsorption follows a saturation-type
sorption isotherm (eg Langmuir) the sorbate (oxidant) concentration at the
surface will vary non-linearly with the total amount of oxidant added This
dependence will have to be taken into account when establishing rate laws for the
heterogeneous reactions and when testing the hypothesis that the reaction rates
depend on the sorbed concentration of the oxidants pH has a strong impact on the
sorption and thereby on the availability of ionizable oxidants At the pH values
applied here the chloroacetates are fully dissociated (pKa (TCA) = 066 pKa
(DCA) = 135 pKa (MCA) = 287) However we found the sorption of TCA to be
negligible in suspensions of pure calcite goethitecalcite and lepidocrocitecalcite
at pH 76-77 Moreover the mass balance of TCA and DCA was almost complete
in all suspensions and therefore loss of TCA or DCA due to adsorption at mineral
surfaces or incorporation in the GR anion interlayers can be ruled out Calcite has a
much lower adsorption capacity than most iron oxides hence we anticipate that
goethite and lepidocrocite control the adsorption of TCA and DCA in both the pure
FeOOH and the FeOOHcalcite suspensions This was supported by our
experimental results demonstrating that the presence of a calcite surface - either
pure or as a support for goethite and lepidocrocite coatings - did not exert any
noticeable effect on the reaction rates (see Table 51) In addition the surface area-
normalised rate constants for mineral systems containing goethite or lepidocrocite
in pure form and mineral systems containing goethite or lepidocrocite as coatings
on calcite were very similar In heterogeneous reactions mass transfer in bulk
solution becomes the rate-limiting step when the surface reaction is much faster
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 115
than the diffusion of the reacting species to the reactive surface However at the
low rate constants obtained here the reaction of TCA with the Fe(II)-bearing
minerals is not likely to be mass transfer limited (see Supporting Information 72)
One very important factor affecting heterogeneous redox reactions is pH which
influences the speciation of the complexes in solution and at mineral surfaces as
well as the stability of the more soluble Fe(II)-containing minerals such as GRs In
contrast to aqueous Fe(II) complexes it is not possible to predict the reactivity of
Fe(II) surface species as their reduction potentials are unknown In the absence of
specifically adsorbing solutes other than H+ the surface charge of the Fe(III)
oxides goethite and lepidocrocite is determined by the surface densities of the
charged surface species equivFeOH2+ and equivFeOndash whereas the surface charge of calcite
is determined by the density of the surface species equivCO3ndash equivCaOH2
+ and equivCaOndash
(Stumm 1992 Van Cappellen et al 1993) The point of zero charge (pHpzc) of
pure calcite is in the pH range 7-11 and depends on the partial pressure of carbon
dioxide pCO2 The higher the pCO2 the lower the pHpzc At the experimental
conditions applied here (05 = 0005 atm CO2(g)) the pHpzc = 82 for calcite
(Table 53) As only 10-11 mg Fe(III) of goethite and lepidocrocite was coated
onto calcite we assumed a pHpzc of 82 for the goethite and lepidocrocite coated
calcite particles as well The pHpzc values for green rusts are unknown
116 Chapter 5
Table 53 Specific surface areas and point of zero charge of the various iron minerals in pure form as well as goethite and lepidocrocite coated onto calcite
Mineral Structural formula SSA (m2g) pHpzc
GR-SO4 FeII4FeIII
2(OH)12SO4middot3H2O 71 a -
GR-CO3 FeII4FeIII
2(OH)12CO3middot3H2O 47 b -
Magnetite Fe3O4 - 69 e
Goethite α-FeOOH 16 c 85 f
Lepidocrocite γ-FeOOH 18 c 73 e
Calcite CaCO3 le 1 d 82 g
Goe coating - le 1 d 82 h
Lep coating - le 1 d 82 h
a Chapter 4 this work b Williams amp Scherer 2001 c Product information by Bayer d The SSA of calcite was
quantified to le 1 m2g The detection limit of our BET method was 1 m2g e Charlet et al 1998a f Liger et al
1999 g Van Cappellen et al 1993 h Same as for calcite
The surface hydroxyl groups on iron oxides may be both singly (equivFe-OH) doubly
(equivFe2-OH) triply (equivFe3-OH) and geminally (equivFe-(OH)2) coordinated (Cornell amp
Schwertmann 1996 Stumm 1992) The differently coordinated surface hydroxyl
groups are not equally reactive Adsorption reactions involve only singly
coordinated surface groups and therefore only this kind of hydroxyl groups on iron
oxides will be considered here (Cornell amp Schwertmann 1996) Hence the
predominant surface sites available for adsorption in pure suspensions of Fe(III)
oxides are equivFeOH0 equivFeOH2+ and equivFeOndash In the presence of dissolved Fe(II)
equivFeIIIOFeIIOH0 equivFeIIIOFeIIOndash and equivFeIIIOFeII+ constitute the main reactive sites at
the Fe(III) oxide surfaces (Liger et a 1999) Assuming that Fe2+ and other cationic
Fe(II) species are the dominating adsorbates on the mineral surfaces in our
experiments we expect the actual pHpzc to be higher than the pHpzc of the pure
oxides listed in Table 53 Hence at pHlt82 where most of our experiments were
conducted all the mineral surfaces presumably carry net positive charges
At pH 70 where Fe2+ is still the predominant Fe(II) species in solution (~50) we
expect that equivFeIIIOFeIIOH equivFeIIIOFeIIOndash and equivFeIIIOFeII+ constitute the main
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 117
reactive sites at the Fe(III) oxide surfaces as suggested by Liger et a 1999 As pH
increases from 70 to 87 the Fe(II) carbonate complexes become increasingly
important in solution at the expense of the Fe2+ FeCl+ FeSO40 and FeOH+ species
(King 1998) Fe(II) carbonate complexes do not bind at the oxide surface as
readily as the aquo or hydroxo complexes of Fe(II) but carbonate itself sorbs
readily to Fe(III) oxide surfaces through which the Fe(III) oxide surface is coated
by inner-sphere monodentate equivFeIIIOCOOH0 surface complexes (Villalobos amp
Leckie 2000 amp 2001) The presence of carbonate shifted the sorption edge for the
Fe(II) adsorption on goethite from pH 58 to 78 and the authors hypothesized this
to be a result of the formation of aqueous and surface Fe(II)-carbonate complexes
and to competition between carbonate and Fe(II) for Fe(III) oxide surface sites
(Vikesland amp Valentine 2002) Similarly monodentate surface complexes like
equivFeIIICl0 and equivFeIIIOSO3ndash as well as ternary monodentate surface complexes like
equivFeIIIOFeIICl0 and equivFeIIIOFeIIOSO3ndash and ternary bidentate surface complexes such
as (equivFeIIIO)2FeIIOSO3 may form at Fe(III) oxide surfaces when Fe(II) chloride and
sulfate are present in solution (Ostergren et al 2000 Kim et al 2004) However
the effects of anionic ligands such as chloride and sulfate on Fe(II) adsorption at
Fe(III) oxide surfaces and the reactivity of Fe(II) carbonate chloride and sulfate
surface sites are still unknown and need to be evaluated (see Supporting
Information 75) Thus we do not know whether chloride and sulfate decrease or
increase the Fe(II) sorption in our mineral systems We can only report that we did
not detect any significant differences in the rate of TCA transformation between
the mineral suspensions containing carbonate chloride and sulfate respectively
Hence we anticipate that equivFeIIIOFeIIOH equivFeIIIOFeIIOndash and equivFeIIIOFeII+ constitute
the main reactive sites at the Fe(III) oxide surfaces within the whole pH range 70-
87 This might also explain why we did not detect any obvious systematic pH
effect in the Fe(II)-Fe(III)-systems (see Table 51) In the case of
hexachloroethane the reactivity order GR-SO4gtgoethitegtmagnetitegtlepidocrocite
may be rationalized by the variations in surface site densities and total amount of
118 Chapter 5
Fe(II) sorbed on the iron minerals (see Supporting Information 75) as well as the
different speciations and reactivities of the Fe(II) surface sites on the iron minerals
535 Comparison with biotic and other abiotic systems
Only one report on abiotic transformation of TCA is found in the literature and the
study demonstrates the reductive dechlorination of TCA to MCA by Fe(0)
(Hozalski et al 2001) The authors reported a pseudo 1 order rate constant of
60middot10-4 min-1middotm-2middotL for the transformation of TCA to DCA and a pseudo 1 order
rate constant of 225middot10-4 min-1middotm-2middotL for the transformation of DCA to MCA at
[Fe(0)]0 = 025 M [TCA]0 = 100-200 microM and pH 36-62 The rate constant for
TCA reduction by Fe(0) is 10-300 times faster than the rate constants reported for
the Fe(II)-bearing mineral systems here
There is abundant evidence that soil microorganisms and fungi can dechlorinate
TCA but only little is known about the bacteria and processes involved in the
biodegradation of TCA Biotransformation of TCA has been found at both oxic
and anoxic conditions Most of the microorganisms isolated grow feebly on TCA
as a sole source of carbon (Foy 1975 Weightman et al 1992 De Wever et al
2000) Only one bacterium capable of growing on TCA as the sole carbon and
energy source has been characterized (Yu amp Welander 1995) In addition
anaerobic bacteria coupling co-metabolic growth to reductive dechlorination of
TCA have been isolated (Weightman et al 1992 De Wever et al 2000) The
inability to grow on the less chlorinated acids DCA and MCA is a notable feature
of both the aerobic and anaerobic bacteria Complete transformation of TCA to
methane and carbon dioxide has only been found when abiotic and biotic processes
were combined (Egli et al 1989) The abiotic transformation of TCA to DCA
occurred spontaneously in the presence of sterile activated charcoal whereas the
DCA formed was further degraded to methane and carbon dioxide by a mixed
culture of methanogenic bacteria However the abiotic reductant(s) responsible for
the transformation of TCA to DCA was not reported (Egli et al 1989)
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 119
The rate constants obtained in this work suggest that the Fe(II)-bearing mineral
systems may be important reductants of TCA in natural suboxic environments In
natural iron-rich soils holding specific surface areas of 22 m2g (Kenneke amp
Weber 2003) average bulk densities of 265 gcm3 and porosities of 25 and
containing 2 iron oxides a rough estimation of the half-life of TCA amounts to
47 minutes when applying the average surface area-normalised rate constant
obtained for all the Fe(II)-Fe(III) mineral systems in this work (1middot10-3 h-1middotm-2middotL)
This estimation is based on the assumption that enough reactive Fe(II) is available
in these soils The natural iron-reducing sediment investigated by Kenneke and
Weber (2003) contained 80 microM Fe(II) in the soil solution and 315 micromole Fe(II) per
g sediment At such low Fe(II) concentrations the overall rate of abiotic
transformation of TCA in natural soils and sediments is most likely limited by the
regeneration of reactive Fe(II) Hence the continuous regeneration of reactive
Fe(II) surface sites by adsorption of abiotically or microbially produced Fe(II) may
further the long-term abiotic transformation of TCA in such environments
54 Conclusions
This work demonstrates that various Fe(II)-Fe(III) minerals systems including GR-
SO4 GR-CO3 magnetite Fe(II)goethite and Fe(II)lepidocrocite readily transform
TCA to DCA Dichloroacetate was not further reduced to MCA or acetate by any
of the Fe(II)-bearing minerals The surface area-normalised pseudo 1 order rate
constants obtained for the reductive transformation of TCA by the various Fe(II)-
bearing minerals did not differ significantly from each other The results obtained
in this work infer that under natural geochemical conditions Fe(II)-bearing mineral
systems may play an important role in the overall transformation of TCA Thus
not only microbial degradation but also abiotic reductive transformation of TCA by
Fe(II)-bearing minerals may govern the fate of TCA in natural subsurface
environments This holds both for natural systems like iron-rich suboxic soils and
sediments as well as engineered systems like permeable reactive barriers of zero-
120 Chapter 5
valent iron implemented for on-site remediation where both Fe(0) and solid or
surface-bound Fe(II) corrosion intermediates may transform TCA
Acknowledgments
We would like to thank Susanne Guldberg for performing the experimental work comprising
GR-SO4
References
Ahlers J Regelmann J Riedhammer C (2003) Environmental risk assessment of airborne trichloroacetic acid - a contribution to the discussion of the significance of anthropogenic and natural sources Chemosphere 52 531-537 Amonette JE Workman DJ Kennedy DW Fruchter JS Gorby YA (2000) Dechlorination of carbon tetrachloride by Fe(II) associated with goethite Environmental Science and Technology 34 4606-4613 Atkins PJ Gold V Marsh R (1984) The decarboxylation of trichloroacetic acid and the reactions of the trichloromethyl anion with 135-trinitrobenzene and with hydrogen ions kinetic measurements in dimethyl sulphoxide solution Journal of the Chemical Society Perkin Transactions 2 7 1239-1245 Berg M Muumlller SR Muumlhlemann J Wiedmer A Schwarzenbach RP (2000) Concentrations and mass fluxes of chloroacetic acids and trifluoroacetic acid in rain and natural waters in Switzerland Environmental Science and Technology 34 2675-2683 Brunauer S Emmett PH Teller E (1938) Adsorption of gases in multimolecular layers Journal of American Chemical Society 60 309-319 Butler EC Hayes KF (1998) Effects of solution composition and pH on the reductive dechlorination of hexachloroethane by iron sulfide Environmental Science and Technology 32 1276-1284 Butler EC Hayes KF (1999) Kinetics of the transformation of trichloroethylene and tetrachloroethylene by iron sulfide Environmental Science and Technology 33 2021-2027 Charlet L Liger E Gerasimo P (1998a) Decontamination of TCE- and U-rich water by granular iron Role of sorbed Fe(II) Journal of Environmental Engineering 124 25-30 Charlet L Silvester E Liger E (1998b) N-compound reduction and actinide immobilisation in surficial fluids by Fe(II) the surface FeIIIFeIIOH0 species as major reductant Chemical Geology 151 85-93 Cheregi M Danet AF (1997) Flow injection determination of chloride ions with spectrophotometric detection Analytical Letters 30 2847-2858 Cornell RM Schwertmann U (1996) The iron oxides Structure properties reactions occurrence and uses VCH Verlagsgesellschaft mbH Weinheim
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 121 Cui D Eriksen TE (1996) Reduction of pertechnetate by ferrous iron in solution influence of sorbed and precipitated Fe(II) Environmental Science and Technology 30 2259-2262
Egli C Thuumler M Suter D Cook AM Leisinger T (1989) Monochloro- and dichloroacetic acids as carbon and energy sources for a stable methanogenic mixed culture Archives of Microbiology 152 218-223 Elsner M Haderlein SB Schwarzenbach RP (2004a) Reactivity of Fe(II)-bearing minerals towards reductive transformation of organic contaminants Environmental Science and Technology 38 799-807 Elsner M Haderlein SB Kellerhals T Luzi S Zwank L Angst W Schwarzenbach RP (2004b) Mechanisms and products of surface-mediated reductive dehalogenation of carbon tetrachloride by Fe(II) on goethite Environmental Science and Technology 38 2058-2066 Erbs M Hansen HCB Olsen CE (1999) Reductive dechlorination of carbon tetrachloride using iron(II)iron(III)-hydroxide-sulphate (green rust) Environmental Science and Technology 33 307-311 Euro Chlor (2001) Trichloroacetic acid in the environment a dossier Euro Chlor Brussels and the European Chlorinated Solvent Association Fadrus H Maly J (1975) Suppression of iron(III) interference in the determination of iron(II) in water by the 110-phenanthroline method The Analyst 100 549-554 Fahimi IJ Keppler F Schoumller HF (2003) Formation of chloroacetic acids from soil humic acid and phenolic moieties Chemosphere 52 513-520 Foy CL (1975) The chlorinated aliphatic acids In Herbicides Chemistry degradation and mode of action Kearney PC Kaufman DD (eds) Marcel Dekker Inc 399-452 Gander JW Parkin GF Scherer MM (2002) Kinetics of 111-trichloroethane transformation by iron sulfide and a methanogenic consortium Environmental Science and Technology 36 4540-4546 Haiber G Jacob G Niedan V Nkusi G Schoumller HF (1996) The occurrence of trichloroacetic acid (TCAA) ndash indications of a natural production Chemosphere 33 839-849
Hansen HCB (1989) Composition stabilization and light absorption of Fe(II)Fe(III) hydroxycarbonate (green rust) Clay Minerals 24 663-669 Hansen HCB Guldberg S Erbs M Koch CB (2001) Kinetics of nitrate reduction by green rusts ndash effects of interlayer anion and Fe(II)Fe(III) ratio Applied Clay Science 18 81-91 Hansen HCB Koch CB (1997) A comparison of nitrate reduction by carbonate and sulphate forms of green rust Kodama H Mermut A R Torrance J K (eds) Proceedings of the 11th International Clay Conference Ottawa Canada Clays for our future 11 295-302 Hoekstra EJ de Leer EWB Brinkman UATh (1999a) Mass balance of trichloroacetic acid in the soil top layer Chemosphere 38 551-563 Hoekstra EJ de Leer EWB Brinkman UATh (1999b) Findings supporting the natural
122 Chapter 5
formation of trichloroacetic acid in soil Chemosphere 38 2875-2883 Hofstetter TB Heijman CG Haderlein SB Holliger HC Schwarzenbach RP (1999) Complete reduction of TNT and other (poly)nitroaromatic compounds under iron-reducing subsurface conditions Environmental Science and Technology 33 1479-1487 Hofstetter TB Schwarzenbach RP Haderlein SB (2003) Reactivity of Fe(II) species associated with clay minerals Environmental Science and Technology 37 519-528 Hozalski RM Zhang L Arnold WA (2001) Reduction of haloacetic acids by Fe0 Implications for treatment and fate Environmental Science and Technology 35 2258-2263 Husain S Narsimha R Alvi SN Rao RN (1992) Monitoring the effluents of the trichloroacetic acid process by high-performance liquid chromatography Journal of Chromatography 600 316-319 Hwang I Batchelor B (2000) Reductive dechlorination of tetrachloroethylene by Fe(II) in cement slurries Environmental Science and Technology 34 5017-5022 Jordan A Frank H (1999) Trifluoroacetate in the environment Evidence for sources other than HFCHCFCs Environmental Science and Technology 33 522-527 Kenneke JF Weber EJ (2003) Reductive dehalogenation of halomethanes in iron- and sulfate-reducing sediments 1 reactivity pattern analysis Environmental Science and Technology 37 713-720 Kim CS Rytuba JJ Brown Jr GE (2004) EXAFS study of mercury(II) sorption to Fe- and Al-(hydr)oxides II Effects of chloride and sulphate Journal of Colloid and Interface Science 270 9-20 King DW (1998) Role of carbonate speciation on the oxidation rate of Fe(II) in aquatic systems Environmental Science and Technology 32 2997-3003 Klausen J Troumlber SP Haderlein SB Schwarzenbach RP (1995) Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions Environmental Science and Technology 29 2396-2404 Klug PH Alexander LE (1974) X-ray diffraction procedures John Wiley amp Sons Inc Koch CB Hansen HCB (1997) Reduction of nitrate to ammonium by sulphate green rust Advances in GeoEcology 30 373-393 Kuumlhn R Pattard M (1990) Results of the harmful effects of water pollutants to green algae (Scenedesmus subspicatus) in the cell multiplication inhibition test Water Research 24 31-38 Lee W Batchelor B (2002a) Abiotic reductive dechlorination of chlorinated ethylenes by iron- bearing soil minerals 1 Pyrite and magnetite Environmental Science and Technology 36 5147- 5154 Lee W Batchelor B (2002b) Abiotic reductive dechlorination of chlorinated ethylenes by iron-bearing soil minerals 2 Green rust Environmental Science and Technology 36 5348- 5354
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 123 Liger E Charlet L Van Cappellen P (1999) Surface catalysis of uranium (VI) reduction by iron(II) Geochimica et Cosmochimica Acta 63 2939-2955 Lignell R Heinonen-Tanski H Uusi-Rauva A (1984) Degradation of trichloroacetic acid (TCA) in soil Acta Agriculturae Scandinavia 34 3-8 Loyaux-Lawniczak S Refait Ph Lecomte P Ehrhardt J Geacutenin JMR (1999) The reduction of chromate ions by Fe(II) layered hydroxides Hydrology and Earth System Sciences 3 593-599 Luther III GW (1990) The Frontier-Molecular-Orbital theory approach in geochemical processes in W Stumm Ed Aquatic Chemical kinetics John Wiley and Sons New York pp 173-198 McCulloch A (2002) Trichloroacetic acid in the environment Chemosphere 47 667-686 Miyata S (1983) Anion-exchange properties of hydrotalcite-like compounds Clays and Clay Minerals 31 305-311 Muumlller SR Zweifel H-R Kinnison DJ Jacobsen JA Meier MA Ulrich MM Schwarzenbach RP (1996) Occurrence sources and fate of trichloroacetic acid in Swiss lakes Environmental Toxicology and Chemistry 15 1470-1478 OLoughlin EJ Burris DR (2003) Reduction of halogenated ethanes by green rust Environmental Toxicology and Chemistry 23 41-48 OLoughlin EJ Kelly SD Cook RE Csencsits R Kemner KM (2003a) Reduction of uranium(VI) by mixed iron(II)iron(III) hydroxide (green rust) Formation of UO2 nanoparticles Environmental Science and Technology 37 721-727 OLoughlin EJ Kelly SD Kemner KM Csencsits R Cook RE (2003b) Reduction of AgI AuIII CuII and HgII by FeIIFeIII hydroxysulfate green rust Chemosphere 53 437-446 OLoughlin EJ Kemner KM Burris DR (2003c) Effects of AgI AuIII and CuII on the reductive dechlorination of carbon tetrachloride by green rust Environmental Science and Technology 37 2905-2912 Ostergren JD Brown Jr GE Parks GA Persson P (2000) Inorganic ligand effects on Pb(II) sorption to goethite (α-FeOOH) II Sulfate Journal of Colloid and Interface Science 225 483-493 Pecher K Haderlein SB Schwarzenbach RP (2002) Reduction of polyhalogenated methanes by surface-bound Fe(II) in aqueous suspensions of iron oxides Environmental Science and Technology 36 1734-1741 Reimann S Grob K Frank H (1996) Chloroacetic acids in rainwater Environmental Science and Technology 30 2340-2344 Ruumlgge K Hofstetter TB Haderlein SB Bjerg PL Knudsen S Zraurig C Mosbaeligk H Christensen TH (1998) Characterization of predominant reductants in an anaerobic leachate- affected aquifer by nitroaromatic probe compounds Environmental Science and Technology 32
124 Chapter 5
23-31 Sarzanini C Bruzzoniti MC Mentasti E (1999) Preconcentration and separation of haloacetic acids by ion chromatography Journal of Chromatography A850 197-211 Schwertmann U Cornell RM (1991) Iron oxides in the laboratory Preparation and characterization VCH Verlagsgesellschaft mbH Weinheim Scott BF Mactavish DC Spencer C Strachan WMJ Muir DCG (2000) Haloacetic acids in Canadian lake waters and precipitation Environmental Science and Technology 34 4266-4272 Scott BF Spencer C Marvin CH Mactavish DC Muir DCG (2002) Distribution of haloacetic acids in the water columns of the Laurentian Great Lakes and Lake Malawi Environmental Science and Technology 36 1893-1898 Strathmann TJ Stone AT (2003) Mineral surface catalysis of reactions between FeII and oxime carbamate pesticides Geochimica et Cosmochimica Acta 67 2775-2791 Stumm W (1992) Chemistry of the solid-water interface John Wiley amp Sons Inc Van Cappellen P Charlet L Stumm W Wersin P (1993) A surface complexation model of the carbonate mineral-aqueous solution interface Geochimica et Cosmochimica Acta 57 3505- 3518 Vikesland PJ Valentine RL (2002) Iron oxide surface-catalyzed oxidation of ferrous iron by monochloramine implications of oxide type and carbonate on reactivity Environmental Science and Technology 36 512-519 Villalobos M Leckie JO (2000) Carbonate adsorption on goethite under closed and open CO2 conditions Geochimica et Cosmochimica Acta 64 3787-3802 Villalobos M Leckie JO (2001) Surface complexation modeling and FTIR study of carbonate adsorption to goethite Journal of Colloid and Interface Science 235 15-32 Vogel TM Criddle CS McCarty PL (1987) Transformations of halogenated aliphatic compounds Environmental Science and Technology 21 722-736 Weightman AL Weightman AJ Slater JH (1992) Microbial dehalogenation of trichloroacetic acid World Journal of Microbiology and Biotechnology 8 512-518 De Wever H Cole JR Fettig MR Hogan DA Tiedje JM (2000) Reductive dehalogenation of trichloroacetic acid by Trichlorobacter thiogenes gen nov spnov Applied and Environmental Microbiology 66 2297-2301 Williams AGB Scherer MM (2001) Kinetics of chromate reduction by carbonate green rust Environmental Science and Technology 35 3488-3494 Yu P Welander T (1995) Growth of an aerobic bacterium with trichloroacetic acid as the sole source of energy and carbon Applied Microbiology and Biotechnology 42 769-774
Conclusions and Outlook 125
6 Conclusions and Outlook The work presented in this dissertation adds to the understanding of how Fe(II)-
bearing minerals like green rusts (GRs) vivianite (Fe2(PO4)2sdot8H2O) magnetite
(Fe3O4) and Fe(II) associated with goethite and lepidocrocite may form and react in
nature In order to elucidate the role of bacteria in the formation of GRs in natural
soils and sediments we studied the iron mineral phases forming as a result of the
activity of iron-respiring bacteria In chapter 2 the Fe-containing products formed
by anaerobic autotrophic denitrifying Fe(II)-oxidizing bacteria (FeOB) were
examined The culture medium applied contained high levels of bicarbonate and
phosphate and is typically used in this kind of studies as it provides excellent
conditions for the nitrate-reducing FeOB Fe(II) was present initially as a whitish
solid Fe(II) hydroxy phosphate (vivianite) and as soluble Fe(II) The results
obtained demonstrate that the denitrifying FeOB produce poorly crystalline
goethite via a greenish Fe(III)-enriched vivianite intermediate Moumlssbauer
spectroscopic analyses provided no evidence of green rust formation At low
phosphate concentrations where vivianite does not control the Fe(II) activity it is
reasonable to assume that siderite (FeCO3) precipitates initially and that carbonate
GR phases may form during biooxidation At low bicarbonate concentrations we
would expect Fe(II) sulfate or chloride species to dominate initially (depending on
the Fe(II) source applied) and sulfate GR or chloride GR to form during
biooxidation In chapter 3 we investigated the Fe-containing products formed
during reduction of common Fe(III) oxides by the anaerobic dissimilatory Fe(III)-
reducing microorganism Shewanella algae BrY S algae BrY reduced substantial
amounts of the initial Fe(III) and green and blackish mineral phases were produced
within 1-2 weeks after inoculation Moumlssbauer spectroscopic analyses showed that
the green and black precipitates consisted of green rust and vivianite
We studied the reactivity of synthetic GRs towards reducible organic pollutants in
order to asses the potential significance of GR phases for the fate of such
126 Chapter 6
compounds To this end we used nitroaromatic compounds (NACs) and
chlorinated acetates as suitable model compounds for studying redox reactions
potentially relevant in the environment In chapter 4 we investigated the relative
reactivity of outer and inner Fe(II) reactive sites in synthetic sulfate green rust
(GR-SO4) by using a series of structurally closely related compounds with different
charge properties as ldquoreactive probesrdquo The probe compounds included
nitrobenzene 2-nitrophenol 4-nitrotoluene 4-chloronitrobenzene and 4-
nitrophenylacetic acid Our results demonstrated that NACs are completely
reduced to their corresponding anilines by GR-SO4 The reactions followed pseudo
1 order kinetics with respect to NAC and the surface area-normalised pseudo 1
order rate constants obtained were 016ndash465middot10-4 s-1middotm-2middotL at [Fe(II)GR]0 = 103-
1260 mM [NAC]0 = 20-102 microM and pH 84-86 Neither mass transfer control nor
surface saturation kinetics could account for the similarity of the surface-
normalised pseudo 1 order rate constants obtained for the reduction of the neutral
and anionic NACs by GR-SO4 These observations suggest that the reaction
between NAC and GR-SO4 takes place at the external reactive Fe(II) sites At low
initial Fe(II)GR concentrations the external reactive Fe(II) sites were depleted and
the regeneration of new external reactive sites eventually controlled the reduction
of the NACs by GR-SO4 In chapter 5 we examined the reactivity of various
Fe(II)-Fe(III) mineral systems towards trichloroacetic acid (TCA) and
dichloroacetate (DCA) in laboratory batch experiments imitating natural
conditions The Fe(II)-Fe(III)-systems investigated included GR-SO4 carbonate
green rust magnetite Fe(II)goethite and Fe(II)lepidocrocite TCA was readily
reduced to DCA by all Fe(II)-containing minerals The reactions followed pseudo
1 order kinetics with respect to TCA and the surface area-normalised pseudo 1
order rate constants obtained were 033ndash76middot10-5 min-1middotm-2middotL at [Fe(II)]0 = 025ndash
116 mM [TCA]0 = 15ndash1000 microM and pH 70ndash87 Our results showed no
significant differences regarding product distribution and surface area-normalised
reaction rate constants between the Fe(II)-Fe(III)-systems DCA was not further
Conclusions and Outlook 127
reduced to monochloroacetate (MCA) or acetate in any of the systems within the
time frame in our experiments
As suggested in chapters 2 and 3 sufficient evidence must be provided and caution
should be exercised when proclaiming new biogenic minerals The study of
microbially produced GRs is still in its infancy and more research is needed in
order to elucidate the role of bacteria in the formation of GRs in natural soils and
sediments The results presented in chapter 2 indicate that microbiological
processes may be responsible for the oxidation of vivianite and metavivianite
((FeII3-xFeIII
x)(PO4)2(OH)xmiddot(8-x)H2O x gt 12) in natural subsurface environments
In chapter 3 we demonstrated that GRs may be produced microbially at conditions
including low carbon and Fe(III) concentrations as well as the exclusion of
synthetic electron shuttles and pH buffers The role of microbial processes in the
redox cycling of iron in the subsurface and the ways in which these processes can
be coupled to contaminant remediation are currently active areas of research Zero-
valent iron has been the most extensively studied reductant for the treatment of
many inorganic and organic contaminants and is currently the most commonly
used material for the construction of permeable reactive barriers (PRB) but a
detailed understanding of the processes involved in the reduction of these
pollutants by Fe(0) is lacking (Scherer et al 2000) Potentially reactive Fe(II)-
bearing corrosion products identified in iron metal columns and barriers include
magnetite siderite Fe(II) sulfides green rusts as well as Fe(II) sorbed to mineral
surfaces (Gu et al 1999 Roh et al 2000) The formation of reactive Fe(II)-
bearing minerals like GRs may explain the effective long-term operation of zero-
valent iron PRBs despite the formation of thick oxide films Thus natural in situ
PRBs might be created by stimulating the activity of anaerobic dissimilatory
Fe(III)-reducing bacteria and the subsequent formation of Fe(II) species such as
GRs Furthermore suspensions of synthetic GRs which are easily prepared from
relatively inexpensive commodity chemicals may also be injected and dispersed
into the subsurface
128 Chapter 6
The reductive transformation of NACs and TCA by GRs is relevant to
understanding the processes responsible for their degradation in the subsurface and
the development of innovative technologies for their remediation The results
obtained in chapters 4 and 5 indicate that GRs may play a significant role in the
reductive transformation of NACs and TCA in natural subsurface environments
Furthermore our results suggest that mainly the outer Fe(II) sites in GRs are
utilized in the reaction with neutral and monovalent anionic compounds and that
these sites may be replenished eg by reduction of the oxidized surface sites or
adsorption of Fe(II) from solution The continuous restoration of Fe(II) surface
sites in GRs may promote their long-term reactivity towards reducible
contaminants
References
Gu B Phelps TJ Liang L Dickey MJ Roh Y Kinsall BL Palumbo AV Jacobs GK (1999) Biochemical dynamics in zero-valent iron columns Implications for permeable reactive barriers Environmental Science and Technology 33 2170-2177 Roh Y Lee SY Elless MP (2000) Characterization of corrosion products in the permeable reactive barriers Environmental Geology 40 184-194 Scherer MM Richter S Valentine RL Alvarez PJJ (2000) Chemistry and microbiology of permeable reactive barriers for In Situ groundwater clean up Critical Reviews in Environmental Science and Technology 30 363-411
Supporting Information I
7 Supporting Information
71 Estimation of the one-electron reduction potential for 4-NPA
The one-electron reduction potential of the half-reaction for a given NAC 1hE
ArNO2 + e- ArNO2
can be used for comparing reduction rates of different NACs in a given system
The formation of the nitroaryl radical is the rate-determining step in the overall rate
of the reduction of a NAC to the corresponding aniline The difference between the
of a NAC and a given reductant is proportional to the change in standard free
energy for the transfer of the first electron ∆G
1hE
1degrsquo If a linear relationship between
the free energy of activation and ∆G1degrsquo is assumed the values of various NACs
can be a measure of their relative reactivity with a given reductant
1hE
As neither the one-electron reduction potential for 4-nitrophenylacetic acid (4-
NPA) nor the Hammett constant for the acetic acid substituent could be found in
the literature the one-electron reduction potential for 4-NPA was estimated by
application of a linear free energy relationship (LFER) to experimental data
Kinetic experiments were conducted in order to obtain the pseudo 1 order rate
constant for the reduction of 4-NPA by a model hydroquinone (reduced
juglone (8-hydroxy-14-naphthoquinone) in the presence of HS
minusHJUGk
ndash) The reduction of
a NAC by juglone follows the rate law
[ ] [ ] [ ] [ ] [ ] [ ]2222 ArNOJUGfkArNOHJUGkArNOk
dtArNOd
totHJUGHJUGHJUGobs sdotsdotsdot=sdotsdot=sdot=minus minusminusminusminus
and the was deducted from a LFER 1hE
II Chapter 7
bEak hHJUG +sdot=minus 059160
log1
for which a and b values have been established for a range of NACs with known
values (Hofstetter et al 1999) An excellent correlation of and log
has been found to exist over a range of 250 mV corresponding to more than 5 order
of magnitude for This is due to the fact that the actual transfer of the first
electron is the rate-determining step under the experimental conditions chosen
1hE 1
hE minusHJUGk
minusHJUGk
For comparison experiments with 4-nitrotoluene (4-NT) were also conducted The
pseudo 1 order rate constants for the reduction of 4-NPA with juglone were
corrected for the reduction of 4-NPA with only HSndash (control experiments
containing no juglone)
[ ]minuslowast
minusminus
minus
minus=
HJUGkk
k HSHJUGHJUG
where (MminusHJUGk -1middots-1) is the rate constant for a compound in the presence of only
juglone (slowastminusHJUGk -1) is the pseudo 1 order rate constant for a compound in the
presence of both juglone and HSndash (sminusHSk -1) is the pseudo 1 order rate constant for
the control reaction in the presence of only HSndash and [HJUGndash] (M) is the
concentration of the reactive dissociated HJUG- form (nondissociated
hydroquinone species are very nonreactive as compared to the monophenolate
species)
Supporting Information III
OH 0 OH OH
+ e- + H+ = + e- + H+ =
0 0
pl(( ox) = 8 00 PK1 (red) = 6 60
JUG HJUG
OH OH
OH
pKa2(red) = 10 60
Figure 7 1 Oxidized and reduced juglone fo1m s
Kinetic experiments in homogeneous anoxic aqueous solutions contained 5 mM
HS- 20 microM total juglone 50 mM KH2P04 buffer and were conducted at pH =
660 corresponding to a concentration of the reactive dissociated juglone form
[HJUG] = 10 microM
By using the LFER
E1 logkHJUG- = 125 middot 005~16 + 923
the following values were obtained
(Hofstetter et al 1999)
NAC k (M-1 -1) HJUG- middots log kHJUG- E~ (mV)
4-NT
4-NPA
311 middot10middot7
164middot10middot7
847middot 10-8
116middot 10middot7
226middot10middot2
489middot10middot3
-165
-231
-515
-546
IV Chapter 7
lowast
minusHJUGk -values are averages of triplicates whereas -values are averages of
duplicates The determined for 4-NT in this work (-515 mV) differs 3 from
the -value of -500 mV reported in the literature (Meisel amp Neta 1975
Wardman 1989) Hence it is assumed that the -value determined for 4-NPA
also differs by 3
minusHSk
1hE
1hE
1hE
Note that even for NACs holding very different values the difference in their
reactivities are much less pronounced in Fe(II)-Fe(III) systems such as the
Fe(II)goethite system (LFER slope a = 06 Hofstetter et al 1999) and the
Fe(II)magnetite system (LFER slope a = 034 Klausen et al 1995) as compared
to the jugloneH
1hE
2S system (a = 125) Furthermore it should be noted that all
LFERs mentioned here were established for neutral NACs and in this work we
have simply assumed that the LFERs are also valid for anionic NACs
72 The rate-limiting step
The overall rate of a reaction is equal to the rate of the slowest step in the
mechanism In heterogeneous reactions eg a compound reacting at the surface of
suspended particles in bulk solution the overall process by which the
heterogeneous reactions proceed may be broken down into a sequence of
individual diffusion steps and reaction steps 1) Mass transfer (diffusion) of the
reactant from the bulk fluid to the external surface of the solid phase 2)
Adsorption of reactant onto the solid surface 3) Reaction on the solid surface 4)
Desorption of the products from the solid surface 5) Mass transfer of the products
from the external solid surface to the bulk fluid Hence the rate of reaction of a
compound reacting at the surface of suspended particles in bulk solution may be
either mass transfer adsorptiondesorption or surface reaction limited When the
diffusion steps are much faster than the reaction steps the mass transfer or
diffusion steps do not affect the overall reaction rate However if the reaction steps
Supporting Information V
are very fast compared with the diffusion steps mass transport affects the reaction
rate Here only the external mass transfer is considered ie the diffusion of
reactants or products between the bulk fluid and the external surface of the solid
phase The additional internal mass transfer resistance for particles containing
substantial internal surface area is not addressed
721 Mass transfer (diffusion) limited kinetics
The overall rate constant can be represented by a system of resistances in series
(Fogler 1999 Arnold et al 1999)
⎟⎟⎠
⎞⎜⎜⎝
⎛+=
minusgeomSALobs kkak1111
where kobs is the observed rate constant kL is the mass transfer coefficient (mmiddots-1) a
is the ratio of the external (geometric) specific surface area to volume of solution
(m-1) and kSA-geom is the intrinsic rate constant of the reaction normalized to the
external specific surface area rather than the BET specific surface area By
comparing kLmiddota with kobs one can estimate the role of mass transfer on the rate of
reaction Thus if kLmiddota gtgt kobs mass transfer is so fast that it has no impact on the
reaction rate whereas if kLmiddota le kobs mass transfer is the rate limiting step
In fluid dynamics the Reynolds number Re is used for determining whether a
flow is laminar or turbulent
νtp ud sdot
=Re
where dp is the particle diameter (m) ut is the terminal particle settling velocity
(mmiddots-1) and ν is the kinematic fluid viscosity (m2middots-1) ν = η ρ where η is the
(absolute) dynamic fluid viscosity in centipoise (1 centipoise = 1 mPamiddots = 10-3
kgmiddotm-1middots-1) and ρ is the fluid density (kgmiddotm-3)
VI Chapter 7
At Re lt 1 we can apply Stokersquos particle settling velocity Stokersquos law is an
equation relating the terminal settling velocity of a smooth rigid sphere in a
viscous fluid of known density and viscosity to the diameter of the sphere when
subjected to a known force field
( )η
ρρsdot
minussdotsdot=
18
2pp
t
dgu (mmiddots-1)
where g = 981 mmiddots-2 is the gravitational constant ρp is the particle density (kgmiddotm-3)
The Sherwood number is the main parameter for prediction of the mass transfer
process
in fluid dynamics
3121 ScRe602Sh sdotsdot+=sdot
=lowast
lowast
iw
pL
Ddk
where Diw is the diffusion coefficient of the compound i in water (m2middots-1) is the
minimum (uncorrected) value of the mass transfer coefficient and Sc is the
Schmidt number This relation is often referred to as the Froumlssling correlation The
particle diameter is a key parameter in the Froumlssling correlation and the external
mass transfer coefficient varies with square of the particle size for smaller
particles
lowastLk
The Schmidt number is the ratio of the kinematic fluid viscosity and the diffusion
coefficient of the compound i in water
iwDν
=Sc
Supporting Information VII
According to Harriott (1962) the actual mass transfer coefficient kL is 15 times
greater than the minimum value of the mass transfer coefficient The
uncertainty in k
lowastLk
Lmiddota associated with particle sphericity and roughness issues are
believed not to exceed a factor of 2
The diffusion coefficient of a compound i in water can be estimated as (Hayduk amp
Laudie 1974)
5890141
9102613
iiw
VD
sdot
sdot=
minus
η (m2middots-1)
where iV is the molar volume of the compound i (cm3middotmol-1) estimated according
to Fuller et al 1966
Assuming spherical particles the external (geometric) specific surface area and the
particle diameter are calculated from the measured BET specific surface area Atot
assuming that our GR-SO4 has a AtotAouter ~ 30 similar to the one reported by
Hansen amp Koch (1998)
( ) ( ) ( )pppp
p
pp
p
dd
dV
SAAρρπ
πρ 1000
6
100061100030
SA3
2tot
geom sdot=
sdotsdot
sdot=
sdot== (m2middotg-1)
In our aqueous GR-SO4 system the density ρ = 1000 kgmiddotm-3 the absolute dynamic
viscosity η = 10-3 Pamiddots and the kinematic viscosity ν = 10-6 m2middots-1 for water The
GR-SO4 particle specific parameters used is found below
VIII Chapter 7
GR-SO4
Atot (m2g) 712
SAgeom (m2g) 237
Particle density ρp (kgm3) 1500
Particle diameter dp (m) 169middot10-6
Settling velocity ut (ms) 778middot10-7
Reynolds number 132middot10-6
The molar volumes the diffusion coefficients in water and the Schmidt numbers
for the NACs including 4-chloronitrobenzene (4-CNB) and nitrobenzene (NB)
were
Compound iV (cm3middotmol-1) Diw (m2middots-1) Sc
4-NT 1260 768middot10-10 1302
4-CNB 1230 779middot10-10 1284
4-NPA 1535 684middot10-10 1462
NB 1055 853middot10-10 1173
The ratio of the external (geometric) specific surface area to volume of solution
were calculated for GR-SO4 at the various concentrations applied
[Fe(II)GR]0 (mM) a (m-1)
103 366middot102
63 224middot103
126 448middot103
60 213middot103
The uncorrected mass transfer coefficients were estimated for the NACs using the
Froumlssling correlation
Supporting Information IX
[Fe(II)GR]0 (mM) lowastLk (mmiddots-1)
4-NT 912middot10-4
4-CNB 925middot10-4
4-NPA 812middot10-4
NB 101middot10-3
Finally kLmiddota was calculated and compared with the experimental 1 order rate
constants kobs obtained for the NACs
Compound [Fe(II)GR]0 (mM) kLmiddota (s-1) kobs (s-1) a
4-NT 103 050 420middot10-4
63 307 140middot10-3
126 613 590middot10-3
4-CNB 103 051 740middot10-4
63 311 170middot10-3
126 622 460middot10-3
4-NPA 103 045 640middot10-4
63 273 109middot10-3
126 546 473middot10-3
NB 60 324 137middot10-3 b
a Experimental pseudo 1 order rate constant at 50 microM [Ar-NO2]0 b Experimental pseudo 1 order rate constant at 10 microM [Ar-NO2]0
When comparing kLmiddota with kobs it can be seen that the rates of mass transfer for all
3 NACs exceed the observed rate constants by at least 3 or 4 orders of magnitude
at every initial Fe(II)GR concentration Thus the reaction of the given NACs with
GR-SO4 is not subject to mass transfer limitations under the experimental
conditions applied here
722 Surface saturation limited kinetics
More than 75 of all heterogeneous reactions that are not diffusion-limited are
surface-reaction-limited rather than adsorption- or desorption-limited We now
X Chapter 7
look at the reaction A = B = C where an intermediate B is formed In our system
A = Ar-NO2 B = Ar-NHOH and C = Ar-NH2 In this case the surface reaction is
assumed to be a single-site mechanism where only the site S on which A or B is
adsorbed is involved in the reaction forming B or C
KA
Adsorption 1 A + S = AmiddotS
kS1
Surface reaction 1 AmiddotS = BmiddotS
KB-1
Desorption 1 BmiddotS = B + S
KB
Adsorption 2 B + S = BmiddotS
kS2
Surface reaction 2 BmiddotS = CmiddotS
KC-1
Desorption 2 CmiddotS = C + S
The rate law for this surface-reaction limited single-site mechanism involving an
intermediate follows Langmuir-Hinshelwood kinetics (adopted from Fogler 1999)
CCBBAA
AAsitesSA
CKCKCKCKCk
dtdC
sdot+sdot+sdot+sdotsdotsdot
=minus1
1
Supporting Information XI
where kS1 is the intrinsic rate constant of the surface reaction transforming A into
the intermediate B Csites is the concentration of reactive sites S on the solid
surface KA KB and KC are the adsorption constants for A B and C at the reactive
surface sites and CA CB and CC are the concentrations of A B and C in the bulk
fluid Two major assumptions of the Langmuir isotherm imply that there is a fixed
number of localised surface sites present on the surface and that the activity of the
surface towards adsorption desorption or surface reaction is independent of
surface coverage
Hence fitting -∆CA∆t to CA CB and CC using a nonlinear curve fitting software
such as SigmaPlot may provide one with the intrinsic rate constant and the
adsorptions constants If KB and KC gtgt KA the intermediate and the product are
strongly competing with the reactant for vacant reactive surface sites
Our data was not fitted successfully by the Langmuir-Hinshelwood rate law
(regression results not shown) Simplifying the rate law by excluding either the
term KCmiddotCC or KBmiddotCB or both (assuming that the aniline product or the
hydroxylaniline intermediate or both did not compete for the reactive sites) did not
improve the regression The Langmuir-Hinshelwood rate law for a dual-site
mechanism did not fit our data either Thus Langmuir-Hinshelwood kinetics
cannot explain the reaction mechanism of the given NACs in our GR-SO4 system
73 External surface area of GR-SO4 and GR-CO3
The GR-SO4 unit cell consists of one double layer (d001 = 11 nm) ie one
hydroxide layer (049 nm ) and one interlayer (061 nm) Hexagonal GR-SO4
particles holding an average width of 1 microm (Figure 72) an average particle
thickness of 35 nm (Hansen amp Koch 1998) and a hydroxide layer thickness of
049 nm have a surface area of the basal plane
Abasal = 1 microm middot 1 microm ndash 2 middot 05 microm middot 025 microm = 075 microm2
XII Chapter 7
and a surface area of the edges
Aedge = (2 middot 05 microm + 4 middot 056 microm) middot 000049 microm = 00016 microm2
Figure 72 The hexagonal platy morphology of GR particles holding an average width of 1 microm
The particle thickness is the mean crystal thickness perpendicular to the 003 plane
as determined from the 003 reflections in an X-ray diffractogram A GR-SO4
particle holding a thickness of 35 nm contains 35 nm11 nm = 318 double layers
The GR-CO3 unit cell consists of one double layer (d001 = 075 nm) ie one
hydroxide layer (049 nm ) and one interlayer (026 nm) Hence a GR-CO3 particle
holding a thickness of 35 nm contains 35 nm075 nm = 467 double layers
The outer surface area of a GR-SO4 particle including outer basal planes and
edges is
Aouter = 222 microm 155microm 00016318microm 0752 =sdot+sdot
and the total surface area of a GR-SO4 particle including both inner and outer
basal planes as well as edges is
Supporting Information XIII
Atot = 222 microm 478)microm 00016microm 0752(318 =+sdotsdot
Hence the ratio of outer surface area to total surface area is
131microm 478microm 155
AA
2
2
tot
outer asymp=
Furthermore the ratio of edge surface area to outer surface area is
130microm 155
microm 00016318AA
2
2
outer
edge asympsdot
=
For GR-CO3 the outer surface area including outer basal planes and edges is
Aouter = 222 microm 157microm 00016746microm 0752 =sdot+sdot
and the total surface area of a GR-CO3 particle including both inner and outer
basal planes as well as edges is
Atot = 222 microm 701)microm 00016microm 0752(467 =+sdotsdot
Hence the ratio of outer surface area to total surface area is
145microm 701microm 157
AA
2
2
tot
outer asymp=
Furthermore the ratio of edge surface area to outer surface area is
121microm 157
microm 00016467AA
2
2
outer
edge asympsdot
=
XIV Chapter 7
74 Van der Waals radii
The size of polyatomic molecules can be estimated by summing the van der Waals
radii of the
individual atoms Van der Waals radii or nonbonded radii can be pictured as the
radii of hard spherical atoms (Figure 73)
Figure 73 Schematic of neighboring nonbonded atoms with van der Waals radii rA and rB
Assuming that the spheres of neighboring nonbonded atoms just touch (Figure
73) the highest possible ion or molecule size Ms can be estimated as the sum of
the van der Waals radii
Ms = 2middotrA + 2middotrB + (1)
Taking Paulingrsquos rule for nonmetals into account we can estimate the real size of
polyatomic ions bound by covalent bonds (Pauling 1960) The van der Waals
radius is larger than the covalent radius because it involves the interposition of two
electron pairs between the atoms rather than one The rule states that the van der
Waals radius of an atom exceeds its covalent radius by ~008 nm (overlap in
Figure 74)
Figure 74 Schematic of atoms undergoing covalent bonding
Supporting Information XV
Thus the size of polyatomic ions bound by covalent bonds Ms can now be
estimated as the sum of the van der Waals radii subtracted by 008 nm
Ms = 2middot(rA - 008 nm) + 2middot(rB - 008 nm) + (2)
We estimated the molecular size of the NACs (Table 72) by means of equation (2)
and the van der Waals radii of the atoms in Table 71
Table 71 Van der Waals radii of various atoms Values from Pauling 1960
Atom vdW radii (nm)
H 0120
O 0140
N 0150
C 0170
Cl 0181
S 0185
In order to make the calculations it was assumed that all atoms were spherical and
that all bond angles were 90deg or 180deg (linear structures) In addition no distinctions
were made between single and double bonds The molecular sizes of the NACs
were estimated with the benzene ring representing the xy plane
Table 72 Molecular sizes of the NACs a Thickness z of the xy plane
Compound Ms (x) (nm) Ms (y) (nm) Ms (z) (nm)a
NB 054 080 036
4-NT 054 106 036
4-CNB 054 100 036
4-NPA 054 136 036
XVI Chapter 7
Note that the molecular sizes in Table 72 are only rough estimations
For comparison with the GR-SO4 interlayer spacing (061 nm) we consider three
possible orientations of the NACs in the GR-SO4 interlayer 1) The NAC xyz
coordination is equivalent to the crystal abc coordination (z = c = 036 nm) 2) the
NAC xy plane is parallel to the crystal bc plane (z = a = 054 nm) and 3) the NAC
xy plane is parallel to the crystal ac plane (z = b = 080-136 nm) Hence the sizes
of the NACs do not hinder their access to the GR-SO4 interlayer Only when
oriented vertically do the sizes of the NACs (z = b = 080-136 nm) exceed the GR-
SO4 interlayer spacing
The molecular size of trichloroacetate (TCA) was also estimated by means of
equation (2) and the atomic van der Waals radii in Table 71 When the TCA
aliphatic chain is assumed to represent the x direction (Ms (x) = 066 nm) the
molecular size in the y and z directions ranges from 045-053 nm depending on the
free rotation of the C-C bond Thus only if the C-C bond is oriented perpendicular
to the crystal ab plane does the size of TCA exceed the GR-SO4 interlayer spacing
(061 nm) In contrast the size of TCA exceeds the GR-CO3 interlayer spacing
(026 nm) regardless of its orientation
75 Adsorption of Fe(II) onto Fe(III) oxides
As seen from the Fe(II) sorption isotherms Fe(II) sorption varies widely between
the Fe(III) oxides as a function of solution pH (Figure 75) Average surface
densities of approximately 2 singly coordinated sitesnm2 iron oxide have been
suggested for goethite and lepidocrocite (Cornell amp Schwertmann 1996) The
similar surface site densities of goethite and lepidocrocite might explain their
similar Fe(II) adsorption isotherms (Figure 75)
Supporting Information XVII
Figure 75 Fe(II) adsorption edges for ferrihydrite goethite hematite lepidocrocite and
magnetite in the absence of other specifically adsorbing cations and anions (from Vikesland amp
Valentine 2002 and references therein) The total number of surface sites was in excess of the
total Fe(II) concentrations in all experiments
Dissolved cations or anions may specifically adsorb at the calcite and Fe(III) oxide
surfaces by exchanging for H+ or OHndash at the equivCO3H0 equivCaOH0 equivFeOH0 and
equivFeIIIOFeIIOH0 surface sites At the experimental conditions applied here within a
pH range 70-87 the dominant species of interest in solution are Fe2+ HCO3ndash
CO32ndash Clndash SO4
2ndash (only in the GR-SO4 systems) and the anionic TCA and DCA In
addition Fe2+ readily forms aqueous complexes with hydroxide carbonate
chloride and sulfate whereby the species FeOH+ FeHCO3+ Fe(OH)(CO3)ndash
FeCO30 Fe(CO3)2
2ndash FeCl+ and FeSO40 may occur (Millero amp Hawke 1992) At
pH 70-87 we expect the Fe(II) species Fe2+ FeCO30 Fe(OH)(CO3)ndash FeOH+ and
Fe(CO3)22ndash to dominate in the GR-CO3 and CaCO3(s)CO2(g) buffered magnetite
suspensions In the goecalcite and lepcalcite suspensions we expect the FeCl+
species to dominate as well whereas the Fe2+ FeSO40 and FeOH+ species most
XVIII Chapter 7
likely dominate in the GR-SO4 suspensions Anionic inorganic ligands like
carbonate chloride and sulfate can lower or enhance the adsorption of Fe(II) due to
a) formation of stable nonadsorbing Fe(II) ligand aqueous complexes b) formation
of Fe(II) ligand Fe(III) oxide surface complexes which can lead to surface
precipitation at high Fe(II) and ligand concentrations c) competitive ligand
sorption to the Fe(III) oxide surface blocking reactive sorption sites at the surface
and d) diminution of the positive charge at the Fe(III) oxide surface (at pH levels
below the point of zero charge (pHpzc) of the Fe(III) oxide) thereby decreasing the
electrostatic repulsion of cations by the Fe(III) oxide surface Specifically adsorbed
cations increase the pHpzc whereas specifically adsorbed anions decrease the pHpzc
References Arnold WA Ball WP Roberts AL (1999) Polychlorinated ethane reaction with zero-valent zinc Pathways and rate control Journal of Contaminant Hydrology 40 183-200 Cornell RM Schwertmann U (1996) The iron oxides Structure properties reactions occurrence and uses VCH Verlagsgesellschaft mbH Weinheim Fogler HS (1999) Elements of chemical reaction engineering 3rd ed Prentice Hall Fuller EN Schettler PD Giddings JC (1966) A new method for prediction of binary gas-phase diffusion coefficients Industrial and Engineering Chemistry 58 19-27 Hansen HCB Koch CB (1998) Reduction of nitrate to ammonium by sulphate green rust Activation energy and interlayer reaction mechanism Clay Minerals 33 87-101 Harriott P (1962) Mass transfer to particles Part I Suspended in agitated tanks AIChE Journal 8 93-102 Hayduk W Laudie H (1974) Prediction of diffusion coefficients for nonelectrolytes in dilute aqueous solutions AIChE Journal 20 611-615 Hofstetter TB Heijmann CG Haderlein SB Holliger C Schwarzenbach RP (1999) Complete reduction of TNT and other (poly)nitroaromatic compounds under iron-reducing subsurface conditions Environmental Science and Technology 33 1479-1487 Klausen J Troumlber SP Haderlein SB Schwarzenbach RP (1995) Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions Environmental Science and Technology 29 2396-2404 Meisel D Neta P (1975) One-electron redox potentials of nitro compounds and radiosensitizers Correlation with spin densities of their radical anions Journal of the American Chemical Society 97 5198-5203
Supporting Information XIX
Millero FJ Hawke DJ (1992) Ionic interactions of divalent metals in natural waters Marine Chemistry 40 19-48 Pauling L (1960) The nature of the chemical bond 3rd ed Cornell University Press Ithaca Vikesland PJ Valentine RL (2002) Iron oxide surface-catalyzed oxidation of ferrous iron by monochloramine implications of oxide type and carbonate on reactivity Environmental Science and Technology 36 512-519 Wardman P (1989) Reduction potentials of one-eletron couples involving free radicals in aqueous solution The Journal of Physical Chemistry Reference Data 18 1637-1755
Curriculum Vitae 13011973 Born in Haderslev Denmark 1988-1991 Mathematical high school Haderslev Katedralskole Denmark 1991-1992 Sabbatical year 1992-1995 B Sc in environmental chemistry University of Copenhagen
(KU) Denmark B Sc thesis 1995 ldquoMethane oxidizing bacteria in soilrdquo
1995-1998 M Sc in environmental chemistry University of Copenhagen
Denmark 1997-1998 diploma thesis ldquoReductive dechlorination of carbon tetrachloride and chloroform in presence of iron(II)iron(III)-hydroxides (green rust)rdquo
1998-1999 Research and teaching assistant at the Chemistry Department
The Royal Veterinary amp Agricultural University (KVL) Denmark
1999-2004 PhD in environmental sciences Swiss Federal Institute of
Technology Zuumlrich (ETHZ) and Swiss Federal Institute for Environmental Science and Technology (EAWAG) Switzerland Docoral thesis ldquoFormation and redox reactions of green rusts under geochemical conditions found in natural soils and sedimentsrdquo
2000-2002 Teaching assistent at the Swiss Federal Institute of Technology
Zuumlrich and supervision of diploma students 2002 Microbial Diversity summer course (7 weeks) at the Marine
Biological Laboratory Woods Hole Massachusetts USA

In fond memory of my mother
Esther Kristine Erbs (1949-2002)
who taught me how to be strong feel joy and bear compassion
I dedicate this work to her Without her support care and love
I would never have been the person I am today
To dare is to lose ones footing momentarily Not to dare is to lose oneself
Soslashren Kierkegaard
Acknowledgements
I would like to thank Stefan Haderlein Hans Christian B Hansen and Rene
Schwarzenbach for their supervision of this work Without the encouragement and
confidence of HCB Hansen and former colleagues at the Royal Veterinary and
Agricultural University in Copenhagen I would never have pursued a PhD and
without the understanding of Rene Schwarzenbach after the tragic death of my
mother I would not have had the time necessary to finish it
I thank Christian Bender Koch Hanne Nancke-Krogh Susanne Guldberg and
Henrik T Andersen (Royal Veterinary and Agricultural University Denmark) for
their valuable contribution to my work I would also like to express my gratitude to
former and present members of the Contaminant Hydrology Group from whom I
have received many benefits I mourn the loss of Denis Mavrocordatos (EAWAG)
who provided technical assistance in the electron microscopy lab and I will always
keep the sunny hours in his company in fond memory Finally I would like to
thank Kristina Straub and Bernhard Schink (University of Constance Germany)
who welcomed me in their lab for a week and taught me how to work with strict
anaerobic bacteria
I gratefully acknowledge the grant which I received from the Danish Research
Agency
Table of Contents
Table of Contents Zusammenfassung I Summary V 1 General Introduction 1 11 Iron cycling in the subsurface 1 12 Green rusts 3 13 Microbial formation of green rusts 7 14 Redox reactions of green rusts 8 15 Outline of the thesis 10 References 11 2 Solid State Oxidation of Vivianite by Anaerobic
Denitrifying Fe(II)-Oxidizing Bacteria 17 Abstract 17 21 Introduction 17 22 Materials and methods 22
221 Microorganisms and media 22 222 Characterisation of precipitates 23 223 Biooxidation experiments 24 224 Analytical methods 25
23 Results and discussion 25 231 Identification of solid iron-containing phases 25 232 Factors controlling the rate and extent of Fe(II) biooxidation 34 233 Morphology of solid iron phases 37
24 Conclusions 38 References 39 3 Formation of Layered Iron Hydroxides by
Microbial Fe(III) Reduction 43 Abstract 43 31 Introduction 44 32 Materials and methods 47
321 Preparation of iron oxide coatings 47 322 Mineral characterisation 48 323 Culture conditions and cell preparation 48 324 Bioreduction experiments 49
Table of Contents
325 Analytical methods 50 33 Results and discussion 50
331 Fe(II) production and suspension colour changes 50 332 Identification of solid iron phases 55 333 Factors controlling the identity of the secondary iron minerals 58 334 Factors controlling the rate and extent of Fe(III) bioreduction 59
34 Conclusions 60 References 61 4 Reduction of Nitroaromatic Probe Compounds by Sulphate
Green Rust The Effect of Probe Compound Charge 65 Abstract 65 41 Introduction 66 42 Materials and methods 71
421 Synthesis of GR-SO4 71 422 Mineral characterisation 72 423 Lyophilization and determination of specific surface area 72 424 Estimation of the one-electron reduction potential for 4-NPA 73 425 Kinetic experiments 74 426 Analytical methods 74
43 Results and discussion 75 431 Product formation and reaction kinetics 75 432 Comparison of rate constants for the different NACs 79 433 Factors influencing the reaction rate 82 434 Comparison with rate constants obtained for other Fe(II) containing
mineral systems 83 435 Depletion of reactive sites 85 436 The role of external and internal reactive sites 86
44 Conclusions 89 References 91 5 Reductive Transformation of Trichloroacetate in Abiotic
Fe(II)-Fe(III) Mineral Systems 97 Abstract 97 51 Introduction 98 52 Materials and methods 101
521 Synthesis of GRs and magnetite 102 522 Preparation of iron oxide coatings 102 523 Mineral characterisation 103 524 Kinetic experiments 103
Table of Contents
525 Analytical methods 104 53 Results and discussion 105
531 Product formation and reaction kinetics 105 532 Comparing rate constants obtained for the various Fe(II)-Fe(III)
mineral systems 109 533 Comparing with rate constants obtained for other chlorinated
aliphatic compound 112 534 Factors controlling the reactivity of surface-bound Fe(II) 114 535 Comparison with biotic and other abiotic systems 118
54 Conclusions 119 References 120 6 Conclusions and Outlook 125 References 128 7 Supporting Information I 71 Estimation of the one-electron reduction potential for 4-NPA I 72 The rate-limiting step IV 721 Mass transfer (diffusion) limited kinetics V
722 Surface saturation limited kinetics IX 73 External surface area of GR-SO4 and GR-CO3 XI 74 Van der Waals radii XIV 75 Adsorption of Fe(II) onto Fe(III) oxides XVI References XVIII Curriculum Vitae
Zusammenfassung I
Zusammenfassung Geschichtete Fe(II)-Fe(III)-Hydroxide (Gruumlner Rost) gehoumlren zur Gruppe der
Fe(II)-haltigen Mineralsysteme (zB Magnetit (Fe3O4) Siderit (FeCO3) Vivianit
(Fe2(PO4)2sdot8H2O) Fe(II)-Sulfide sowie an die Oberflaumlche von Fe(III)-Oxiden und
Tonmineralien gebundenes zweiwertiges Eisen) die die Aktivitaumlt von Fe(II) in
suboxischen und anoxischen Boumlden und Sedimenten kontrollieren Gruumlner Rost
Phasen (GRs) bestehen aus planaren positiv geladenen trioktaedrischen Fe(II)-
Fe(III)-Hydroxidschichten die durch hydratisierte Anionen in den
Zwischenschichten ausgeglichen werden Ihre generelle Zusammensetzung ist
[FeII(6-x)FeIII
x(OH)12]x+[(A)xnmiddotyH2O]x- wobei x = 09 - 42 ist A entspricht einem n-
valenten Anion (zB CO32- Clndash oder SO4
2-) und y repraumlsentiert die Anzahl
Wassermolekuumlle in der Zwischenschicht GRs sind wichtige intermediaumlre Phasen
die durch unvollstaumlndige Oxidation von Fe(II) oder teilweise Reduktion von Fe(III)
gebildet werden koumlnnen Sie koumlnnen in suboxischen nicht-sauren eisenhaltigen
natuumlrlichen wie auch technischen Systemen auftreten so wie in Wasser gesaumlttigten
Boumlden und interstitiellen Sedimenten Rohrleitungen in der
Trinkwasserversorgung Stahlpfosten in marinen Sedimenten Stahlbeton und in
reaktiven durchlaumlssigen Waumlnden aus nullwertigem Eisen zur in-situ Sanierung von
Altlasten und Aquiferen Aufgrund ihrer Schichtstruktur den anionischen
Zwischenschichten und der hohen spezifischen Oberflaumlchen sind GRs reaktive
Ionentauscher und Sorbentien von Anionen Des Weiteren wurde gezeigt dass
GRs eine Reihe anorganischer und organischer Schadstoffe reduzieren koumlnnen
Durch Immobilisierung und Transformation koumlnnen GRs somit eine wichtige Rolle
fuumlr das Abbauverhalten und den Transport solcher Schadstoffe in suboxischen
Boumlden und Sedimenten spielen Die Resultate dieser Dissertation tragen zum
Verstaumlndnis uumlber die Bildung und Reaktivitaumlt von Fe(II)-haltigen Mineralsystemen
wie GRs Vivianit Magnetit und an Goethit (α-FeOOH)- und Lepidokrozit (γ-
FeOOH)-Oberflaumlchen gebundenes Fe(II) in der Natur bei
II Zusammenfassung
Um die Rolle von Bakterien bei der Bildung von GRs in natuumlrlichen Boumlden und
Sedimenten aufzuklaumlren wurden Eisenminerale untersucht die als Folge der
Aktivitaumlt von eisenrespirierenden Bakterien gebildet wurden Kapitel 2 beschreibt
die Untersuchungen von eisenhaltigen Produkten die von anaeroben autotrophen
denitrifizierenden Fe(II)-oxidierenden Bakterien (FeOB) gebildet wurden Ein
Bikarbonat- und Phosphat-reiches Kulturmedium bot den nitratreduzierenden
FeOB optimale Bedingungen Fe(II) lag zu Anfang der Reaktion als weisses
Fe(II)-Hydroxyphosphat (Vivianit) und als geloumlstes Fe(II) vor Die Ergebnisse
zeigten dass die denitrifizierenden FeOB amorphen Goethit via ein gruumlnes Fe(III)-
angereichertes Vivianit-Zwischenprodukt bildeten Die Analyse mit Moumlssbauer
Spektroskopie deutet nicht auf eine Bildung von GR hin
In Kapitel 3 werden jene Eisenmineralien beschrieben die waumlhrend der Reduktion
verbreiteter Fe(III)-Oxide durch anaerobe dissimilative Fe(III)-reduzierende
Mikroorganismen Shewanella algae BrY gebildet wurden Um natuumlrliche
Zustaumlnde zu simulieren wurden Fe(III)-Oxide als Beschichtungen auf
Silikatpartikel (Modellsystem fuumlr Sandboumlden) oder Calcitpartikel (CaCO3
Modellsystem fuumlr kalkhaltige Boumlden) aufgetragen sowie synthetische
Elektronencarrier und hochkonzentrierte kuumlnstliche pH-Puffer ausgeschlossen
Die erforschten Mineralsysteme umfassten GoethitCalcit- LepidokrozitCalcit-
und FerrihydritSand-Suspensionen S algae BrY reduzierte beachtliche Mengen
des eingesetzten Fe(III) und es bildeten sich gruumlne und schwarze Festphasen
innerhalb von 1-2 Wochen nach der Animpfung Moumlssbauer Spektroskopie der
gruumlnen und schwarzen Praumlzipitate zeigte dass sich diese aus GR und Vivianit
zusammensetzen
Die Reaktivitaumlt synthetischer GRs gegenuumlber reduzierbaren organischen
Schadstoffen wurde erkundet um die potentielle Bedeutung von GR-Phasen fuumlr
das Schicksal solcher Verbindungen abzuschaumltzen Zu diesem Zweck wurden
Nitroaromaten (NACs) und Chloracetate als Modellverbindungen benutzt um
Zusammenfassung III
umweltrelevante Redoxreaktionen zu studieren In Kapitel 4 wurde die relative
Reaktivitaumlt von aumlusseren und inneren reaktiven Stellen in synthetischem Sulfat-
Gruumlnem Rost (GR-SO4) anhand von strukturaumlhnlichen ldquoreaktiven
Sondenmolekuumllenrdquo mit unterschiedlichen Ladungen untersucht Als reaktive
Sondenmolekuumlle wurden Nitrobenzen 2-Nitrophenol 4-Nitrotoluen 4-
Chlornitrobenzen und 4-Nitrophenylessigsaumlure verwendet Die Ergebnisse zeigen
dass GR-SO4 die NACs vollstaumlndig zu den entsprechenden Anilinen reduzierte
Die Reaktionen folgten einer pseudo 1 Ordnungs Kinetik bezuumlglich NAC und die
auf Oberflaumlche normalisierten pseudo 1 Ordnungs Geschwindigkeitskonstanten
(Anfangsraten) waren 016ndash465middot10-4 s-1middotm-2middotL fuumlr [Fe(II)GR]0 = 103-1260 mM
[NAC]0 = 20-102 microM und pH 84-86 Weder durch Einbezug von
Massentransferlimitierung noch von Oberflaumlchensaumlttigungskinetik war es moumlglich
die aumlhnlichen Oberflaumlchennormalisierten pseudo 1 Ordnungs
Geschwindigkeitskonstanten fuumlr die Reduktion der neutralen und anionischen
NACs durch GR-SO4 zu erklaumlren Dieser Umstand laumlsst vermuten dass die
Reaktion zwischen NAC und GR-SO4 an den externen reaktiven Fe(II)-Stellen
stattfindet Bei niedrigen Fe(II)GR-Anfangskonzentrationen wurden die externen
reaktiven Fe(II)-Stellen aufgebraucht und die Regenerierung von neuen externen
reaktiven Stellen haben schliesslich die Geschwindigkeit der Reduktion von NACs
durch GR-SO4 kontrolliert
In Kapitel 5 wurde die Reaktivitaumlt von verschiedenen umweltrelevanten Fe(II)-
Fe(III)-Mineralsystemen gegenuumlber Trichloressigsaumlure (TCA) und
Dichloressigsaumlure (DCA) in Batchexperimenten die natuumlrliche Bedingungen
imitierten untersucht Die Fe(II)-Fe(III)-Systeme umfassten Sulfat-Gruumlner Rost
Carbonat-Gruumlner Rost Magnetit Fe(II)Goethit und Fe(II)Lepidokrozit TCA
wurde von allen Fe(II)-haltigen Mineralien zu DCA reduziert Die Reaktionen
folgten einer pseudo 1 Ordnungs Kinetik bezuumlglich TCA und die auf Oberflaumlche
normalisierten pseudo 1 Ordnungs Geschwindigkeitskonstanten betrugen 033ndash
76middot10-5 min-1middotm-2middotL bei [Fe(II)]0 = 025ndash116 mM [TCA]0 = 15ndash1000 microM und pH
IV Zusammenfassung
70ndash87 Die Ergebnisse zeigen keine signifikanten Unterschiede zwischen den
verschiedenen Fe(II)-Fe(III)-Systemen bezuumlglich Produkteverteilung und
oberflaumlchen-normalisierten pseudo 1 Ordnungs Geschwindigkeits-konstanten In
keinem der Systeme wurde DCA innerhalb des experimentellen Zeitraums zu
Monochloressigsaumlure oder Essigsaumlure weiter reduziert
Die Ergebnisse die in dieser Dissertation praumlsentiert werden zeigen dass
mikrobiologische Prozesse fuumlr die Oxidation von Vivianit-Phasen im Untergrund
verantwortlich sein koumlnnen Zudem wurde nachgewiesen dass GRs bei tiefen
Kohlenstoff- und Fe(III)-Konzentrationen sowie durch Ausschluss von
kuumlnstlichen Elektronencarriern und pH-Pufferung mikrobiell gebildet werden
koumlnnen Ferner zeigten Befunde dass GRs eine bedeutende Rolle fuumlr die reduktive
Transformation von NACs und TCA in natuumlrlichen Boumlden und Sedimenten spielen
koumlnnen
Summary V
Summary
Layered iron(II)-iron(III)-hydroxides (green rusts) belong to the group of Fe(II)-
bearing mineral systems eg magnetite (Fe3O4) siderite (FeCO3) vivianite
(Fe2(PO4)2sdot8H2O) Fe(II) sulfides as well as Fe(II) associated with Fe(III) oxide
and clay mineral surfaces that control the Fe(II) activity in suboxic and anoxic
soils and sediments Green rusts (GRs) consist of plane positively charged
trioctahedral Fe(II)-Fe(III) hydroxide sheets balanced by hydrated anions in the
interlayers and hold the general formula [FeII(6-x)FeIII
x(OH)12]x+[(A)xnmiddotyH2O]x-
where x = 09 - 42 A is an n-valent anion eg CO32- Clndash or SO4
2- and y is the
number of water molecules in the interlayer GRs are important intermediate
phases formed by partial oxidation of Fe(II) or partial reduction of Fe(III) and they
have been found in suboxic non-acid iron-rich natural environments such as
hydromorphic soils and intertidal sediments and in engineering systems including
pipeline distribution systems for drinking water steel sheet piles in marine
sediments reinforced concrete and permeable reactive barriers of zero-valent iron
implemented for on-site remediation of contaminants Due to their layered
structures anionic interlayers and high specific surface areas GRs represent
reactive ion exchangers and sorbents of anions In addition GRs have been shown
to reduce a range of inorganic and organic pollutants Thus through sequestration
and reductive transformation GRs may play an important role in the fate and
transport of contaminants in suboxic soils and sediments The work presented in
this dissertation adds to the understanding of how Fe(II)-bearing minerals like
GRs vivianite magnetite and Fe(II) associated with goethite (α-FeOOH) and
lepidocrocite (γ-FeOOH) may form and react in nature
In order to elucidate the role of bacteria in the formation of GRs in natural soils
and sediments we studied the iron mineral phases forming as a result of the
activity of iron-respiring bacteria In the study described in chapter 2 the Fe-
containing products formed by anaerobic autotrophic denitrifying Fe(II)-oxidizing
VI Summary
bacteria (FeOB) were examined The culture medium applied contained high levels
of bicarbonate and phosphate and is typically used in this kind of studies as it
provides excellent conditions for the nitrate-reducing FeOB Fe(II) was present
initially as a whitish solid Fe(II) hydroxy phosphate (vivianite) and as soluble
Fe(II) The results obtained demonstrate that the denitrifying FeOB produce poorly
crystalline goethite via a greenish Fe(III)-enriched vivianite intermediate
Moumlssbauer spectroscopic analyses provided no significant evidence of green rust
formation
In chapter 3 the Fe-containing products formed during reduction of common
Fe(III) oxides by the anaerobic dissimilatory Fe(III)-reducing microorganism
Shewanella algae BrY are discussed In order to simulate natural conditions
Fe(III) oxides were applied as coatings on silica (model system for sandy soils) or
calcite (CaCO3) particles (model system for calcareous soils) and synthetic
electron shuttles as well as highly concentrated artificial pH buffers were excluded
The mineral systems studied include goethitecalcite lepidocrocitecalcite and
hydrous ferric oxidesand suspensions S algae BrY reduced substantial amounts
of the initial Fe(III) and green and blackish mineral phases were produced within
1-2 weeks after inoculation Moumlssbauer spectroscopic analyses showed that the
green and black precipitates consisted of GR and vivianite
The reactivity of synthetic GRs towards reducible organic pollutants was
investigated in order to asses the potential significance of GR phases for the fate of
such compounds To this end we used nitroaromatic compounds (NACs) and
chlorinated acetates as suitable model compounds for studying environmentally
relevant redox reactions In the work described in chapter 4 the relative reactivity
of outer and inner Fe(II) reactive sites in synthetic sulfate green rust (GR-SO4) was
studied using a series of structurally closely related compounds with different
charge properties as ldquoreactive probesrdquo The probe compounds included
nitrobenzene 2-nitrophenol 4-nitrotoluene 4-chloronitrobenzene and 4-
Summary VII
nitrophenylacetic acid The results show that NACs are completely reduced to their
corresponding anilines by GR-SO4 The reactions followed pseudo 1 order
kinetics with respect to NAC and the surface area-normalised pseudo 1 order rate
constants (initial rates) obtained were 016ndash465middot10-4 s-1middotm-2middotL at [Fe(II)GR]0 = 103-
1260 mM [NAC]0 = 20-102 microM and pH 84-86 Neither mass transfer control nor
surface saturation kinetics could explain the similarity of the surface-normalised
pseudo 1 order rate constants obtained for the reduction of the neutral and anionic
NACs by GR-SO4 These observations suggest that the reaction between NAC and
GR-SO4 takes place at the external reactive Fe(II) sites At low initial Fe(II)GR
concentrations the external reactive Fe(II) sites were depleted and the regeneration
of new external reactive sites eventually controlled the reduction of the NACs by
GR-SO4
Finally the reactivity of various Fe(II)-Fe(III) mineral systems towards
trichloroacetic acid (TCA) and dichloroacetate (DCA) has been investigated in
laboratory batch experiments imitating natural conditions (Chapter 5) The Fe(II)-
Fe(III)-systems investigated included GR-SO4 carbonate green rust magnetite
Fe(II)goethite and Fe(II)lepidocrocite TCA was readily reduced to DCA by all
Fe(II)-containing minerals The reactions followed pseudo 1 order kinetics with
respect to TCA and the surface area-normalised pseudo 1 order rate constants
obtained were 033ndash76middot10-5 min-1middotm-2middotL at [Fe(II)]0 = 025ndash116 mM [TCA]0 =
15ndash1000 microM and pH 70ndash87 Our results showed no significant differences
regarding product distribution and surface area-normalised reaction rate constants
between the Fe(II)-Fe(III)-systems DCA was not further reduced to
monochloroacetate (MCA) or acetate in any of the systems within the time frame
in our experiments
The results presented in chapter 2 indicate that microbiological processes may be
responsible for the oxidation of vivianite phases in natural subsurface
environments In chapter 3 we demonstrated that GRs may be produced
VIII Summary
microbially at conditions including low carbon and Fe(III) concentrations as well
as the exclusion of synthetic electron shuttles and pH buffers The results obtained
in chapter 4 and 5 show that GRs transform NACs and TCA readily The reductive
transformation of NACs and TCA by GRs is relevant to understanding the
processes responsible for their degradation in the subsurface and the development
of innovative technologies for their remediation
General Introduction 1
1 General Introduction
11 Iron cycling in the subsurface
Iron is the fourth most abundant element (4-5 mass) and the most abundant redox
sensitive element in the Earthrsquos crust It is found as Fe(II) and Fe(III) in a number
of minerals in rocks soils and sediments Under anoxic conditions solid Fe(III)-
containing minerals can be reduced to soluble Fe(II) once the more energetically
favoured electron donors - nitrate and manganese(IV) oxides - have been
consumed Dissolved Fe(II) can be reoxidized to insoluble Fe(III) microbially or
abiotically upon exposure to oxygen Due to this ready alternation between the
Fe(II) and Fe(III) redox states iron plays a major role in controlling the redox
potential and the carbon cycling in subsurface environments (Nealson amp Saffarini
1994)
Nonenzymatic processes were previously considered to account for most of the
Fe(III) reduction in subsurface environments The significance of bacteria in the
biogeochemical cycling of iron has been broadly recognized over the past two
decades Dissimilatory Fe(III)-reducing bacteria (DIRB) that gain energy by
coupling the oxidation of hydrogen or organic compounds to the reduction of
Fe(III) oxides have been known for many years but their biogeochemical
importance was only widely acknowledged about a decade ago (reviewed by
Lovley 1997) Fe(III) bioreduction accounts for a major fraction of the carbon
oxidation in many different anoxic environments and in the presence of sufficient
amounts of reactive Fe(III) microbial Fe(III) reduction may even inhibit sulphate
reduction and methanogenesis (King 1990 Lovley amp Phillips 1986) In fact most
of the Fe(III) reduction in the Fe(III) reduction zone of aquatic sediments and
aquifers is enzymatically catalyzed by microorganisms (Lovley et al 1991) A
wide diversity of DIRB distributed among several different phylogenetic groups
2 Chapter 1 is known today The two most studied DIRB are the obligate anaerobic Geobacter
spp and the facultatively anaerobic Shewanella spp (Figure 11)
Aerobic oxidation of Fe(II)-containing minerals by lithotrophic acidophilic and
neutrophilic bacteria has been known for many years but their broad significance
in the biogeochemical cycling of iron has only been recognized over the past two
decades Both acidophilic (Thiobacillus ferrooxidans) and neutrophilic
(Gallionella ferruginea Leptothrix ochracea Sphaerotilus natans) aerobic Fe(II)-
oxidizing bacteria (FeOB) have been isolated (Hanert 1992 Kuenen et al 1992
Mulder amp Deinema 1992)
Figure 11 The microbial iron cycle
Anaerobic Fe(II) oxidation by phototrophic purple non-sulfur bacteria utilizing
Fe(II) as an electron donor in the light was recognized only a decade ago (Widdel
et al 1993) Subsequently it was demonstrated that the biological oxidation of
Fe(II) in the absence of oxygen is possible by light-independent chemotrophic
microorganisms using nitrate as the electron acceptor (Straub et al 1996) Thus
the microbial iron cycle includes anaerobic Fe(III)-reducing microorganisms and
aerobic as well as anaerobic Fe(II)-oxidizing bacteria (Figure 11)
General Introduction 3
12 Green rusts
Iron oxides iron hydroxides and iron oxyhydroxides (collectively termed iron
oxides or Fe(III) oxides) are ubiquitous in the pedosphere where they originate
from aerobic weathering of surface magmatic rocks such as ferromagnesium
silicates and pyrite (Cornell amp Schwertmann 1996) Goethite (α-FeOOH)
lepidocrocite (γ-FeOOH) ferrihydrite (Fe5HO8sdot4H2O) hematite (α-Fe2O3)
magnetite (Fe3O4) maghemite (γ-Fe2O3) and akageneite (β-FeOOH) constitute the
most important iron oxides in soils and sediments (Schwertmann amp Cornell 1991)
The formation and transformation of iron oxides depend on pH solution
composition redox potential temperature rate of oxidationreduction and degree
and rate of hydrationdehydration Iron oxides are important to many soil
properties such as colour pH and redox buffer capacity aggregation with other
soil particles as well as retention of anions and cations (Cornell amp Schwertmann
1996) A number of Fe(II)-bearing minerals including Fe(II)-containing clays (eg
smectites vermiculites and micas) magnetite siderite (FeCO3) vivianite
(Fe2(PO4)2sdot8H2O) Fe(II) sulphides and green rusts (layered Fe(II)-Fe(III)
hydroxides) may be present in soils and sediments under suboxic and anoxic
conditions Green rusts are believed to play a central role as metastable
intermediates in the redox cycling of iron at circumneutral pH in aquatic and
terrestrial environments
Green rusts (GRs) are layered iron(II)-iron(III)-hydroxides consisting of plane
positively charged trioctahedral Fe(II)-Fe(III) hydroxide sheets balanced by
hydrated anions in the interlayers (cf Figure 41 this work) GRs belong
structually to the pyroaurite-sjoumlgrenite group of layered hydroxides and they hold
the general formula [FeII(6-x)FeIII
x(OH)12]x+[(A)xnmiddotyH2O]x- where x = 09 - 42 A is
an n-valent anion eg CO32- Clndash or SO4
2- and y is the number of water molecules
in the interlayer The three most common and investigated green rust forms include
chloride GR (GR-Cl) sulphate GR (GR-SO4) and carbonate GR (GR-CO3)
Generally GRs are crystallographically classified into the GRI (rhombohedral
4 Chapter 1 GR-Cl and GR-CO3) and GRII (hexagonal GR-SO4) crystal systems The GR
interlayer thickness is a function of both the size and the charge of the interlayer
anion Tetrahedrally coordinated anions like sulphate lead to larger interlayer
distances than smaller monoatomic anions like chloride or planar ions like
carbonate (Mendiboure amp Schoumlllhorn 1986) Not only size but also charge density
plays a role for the interlayer spacing That is for anions having the same number
of valence electrons anions with smaller ionic radii (higher electron density) are
bound more strongly and therefore result in smaller interlayer spacings The
interlayer in GR-SO4 is composed of two consecutive planes of anions and water
whereas GR-Cl and GR-CO3 interlayers consist of only one single plane (Simon et
al 2003)
GRs are important intermediate phases formed by partial oxidation of Fe(II) or
partial reduction of Fe(III) In neutral and weakly alkaline solutions the oxidation
of dissolved Fe(II) always passes through solid GR phases (Bernal et al 1959)
GRs may also form during oxidation of zero-valent iron and as a result of the
combination of Fe(II) and Fe(III) at circumneutral pH (Figure 12)
Figure 12 Formation and transformation of GRs Fe3O4 = magnetite γ-Fe2O3 = maghemite α-
FeOOH = goethite γ-FeOOH = lepidocrocite akageneite = β-FeOOH
General Introduction 5
Oxidation of GR-CO3 usually produces goethite and magnetite-maghemite
whereas GR-Cl and GR-SO4 transform into lepidocrocite and magnetite-
maghemite depending on pH and oxidation rate (Bernal et al 1959 Taylor 1980
Carlson amp Schwertmann 1990) The brown δ-FeOOH is formed by vigorous
oxidation of GR using air or a 30 aqueous solution of hydrogen peroxide (Bernal
et al 1959 Misawa et al 1974) Black ferromagnetic magnetite forms by slow
oxidation of GR whereas lepidocrocite forms at high oxidation rates (Misawa et
al 1974) The presence of chloride is a prerequisite for the formation of
akageneite (Bernal et al 1959 Refait amp Genin 1997)
A substantial amount of work has been conducted in order to estimate the free
energies of formation of green rusts The free energies of formation reported for
the carbonate and sulphate GRs fall in the range 4234ndash4384 kJsdotmol-1 as determined
from solution data monitored during anoxic alkalimetric titrations and from
reduction potential (Eh) and pH recordings monitored during oxidation of GRs in
aqueous solution (Hansen et al 1994 Drissi et al 1995 Genin et al 1996) The
free energies of formation provided allow for estimation of the stability domains of
GRs in Eh-pH phase diagrams (Drissi et al 1995 Genin et al 1996) As
evidenced from such diagrams (Figure 13) the stability domain of GR-SO4 lies
within pH 6-8 and Eh -700 ndash -400 mV depending on the activities of Fe(II) and
sulphate (compare Figures 13aampb) This agrees with the natural GR occurrences
found in suboxic non-acid iron-rich environments such as hydromorphic soils and
intertidal sediments (Al-Agha et al 1995 Trolard et al 1996 Genin et al 1998)
In addition GRs have been found as corrosion products in numerous engineering
systems including a pipeline distribution system for drinking water steel sheet
piles in marine sediments reinforced concrete (ferro-concrete) and permeable
reactive barriers of zero-valent iron implemented for on-site remediation of organic
and inorganic contaminants (Tuovinen et al 1980 Nielsen 1976 Genin et al
1991 Roh et al 2000)
6 Chapter 1
Figure 13 Eh-pH phase diagrams of GR-SO4 a) a = 10+2Fe-3 = 10minus2
4SOa -3 and b) a = 10+2Fe-2
= 10minus24SOa -1
The stability domains of GR-Cl and GR-CO3 are similar to the stability domain of
GR-SO4 At Fe(II) and sulphate activities lower than depicted in Figure 13b the
stability domain of GR-SO4 will be situated at higher pH and lower Eh Other
dissolved species present at anoxic conditions such as phosphate sulphide
carbonate and organic ligands may exert considerable effects on the availability of
Fe(II) and Fe(III) At anoxic and circumneutral conditions vivianite
(Fe2(PO4)2sdot8H2O) controls the Fe(II) activity even at very low phosphate
concentrations (Nriagu amp Dell 1974) The formation of solid Fe(II) sulphides and
siderite (FeCO3) as well as the complexation of Fe(II) and Fe(III) by organic
ligands may also control the activity of Fe(II) in the subsurface and thereby
interfere with the formation of GRs
Due to their layered structures anionic interlayers and high specific surface areas
GRs represent reactive ion exchangers and sorbents of environmentally concerning
anions eg arsenate and selenate (Myneni et al 1997 Randall et al 2001) In
addition GRs may incorporate divalent transition metal cations like Ni2+ Zn2+
Cd2+ Co2+ and Mg2+ by isomorphic substitution for Fe2+ in the hydroxide layers
General Introduction 7
(Tamaura 1985 Tamaura 1986 Refait et al 1994 Parmar et al 2001 Refait et
al 2001) Furthermore GRs have been shown to reduce a range of inorganic
contaminants such as nitrite nitrate selenate chromate uranyl pertechnetate and
the transition metals AgI AuIII CuII and HgII as well as organic pollutants
including halogenated ethanes ethenes and methanes (Hansen et al 1994 Hansen
et al 1996 Myneni et al 1997 Erbs et al 1999 Loyaux-Lawniczak et al 1999
Cui amp Spahiu 2002 Lee amp Batchelor 2002b Heasman et al 2003 OrsquoLoughlin et
al 2003aampb Pepper et al 2003 Elsner et al 2004 OrsquoLoughlin amp Burris 2004)
Thus through sequestration and reductive transformation GRs may play an
important role in the fate and transport of contaminants in suboxic soils and
sediments It should be noted that the rate constants reported for the reduction of
these inorganic and organic pollutants by GRs cannot be directly compared as the
various studies were conducted at very different experimental conditions
13 Microbial formation of green rusts
Generally one would expect that biogenic minerals have chemical compositions
and crystal habits similar to those produced by nonenzymatic processes as they are
governed by the same equilibrium principles In fact since the latter stages of
mineralization are abiotically driven and since the secondary Fe(II)-containing
minerals are formed indirectly by electron transfer outside the bacterial cell and not
directly inside the bacterial cell the type of iron mineral formed is a function of the
environmental conditions in which the bacteria live ie the same microorganism
form different minerals in different environments
The microbial formation of GRs resulting from bioreduction of various Fe(III)
oxides including ferrihydrite goethite and lepidocrocite by strains of the
anaerobic dissimilatory DIRB Shewanella putrefaciens has been reported
repeatedly over the last years (Fredrickson et al 1998 Kukkadapu et al 2001
Parmar et al 2001 Ona-Nguema et al 2002aampb Glasauer et al 2003)
However no evidence of biogenic formation of GRs at natural geochemical
8 Chapter 1 conditions have been offered and it is still unknown whether this process may take
place at natural conditions comprising low nutrient levels low iron concentrations
and the absence of synthetic electron shuttles and highly concentrated artificial pH
buffers Moreover the biotic formation of GRs by anaerobic denitrifying Fe(II)-
oxidizing bacteria has been suggested but the phases still need to be properly
identified (Chaudhuri et al 2001) In order to elucidate the role of bacteria in the
formation of GRs in natural soils and sediments we studied the iron mineral
phases forming as a result of the activity of iron-respiring bacteria (Chapters 2 and
3)
14 Redox reactions of green rusts
Fe(II) is one of the most abundant reductants present in aquatic and terrestrial
environments under suboxic and anoxic conditions (Lyngkilde amp Christensen
1992 Ruumlgge et al 1998) In these environments Fe(II) may be present as soluble
organic and inorganic complexes as surface complexes and as a host of Fe(II)-
bearing minerals Although aqueous Fe(II) complexes may reduce a number of
contaminants Fe(II) associated with mineral surfaces and structural Fe(II) present
in the mineral lattice in Fe(II)-containing minerals are often more powerful
reductants Fe(II)-bearing minerals including GRs magnetite siderite Fe(II)
sulphides as well as Fe(II)-carrying Fe(III) oxide and clay mineral surfaces have
been shown to reduce a number of organic and inorganic contaminants such as
nitroaromatic compounds chlorinated aliphatics chromate uranyl pertechnetate
nitrate monochloramine and carbamate pesticides (Klausen et al 1995 Cui amp
Eriksen 1996 Butler amp Hayes 1998amp1999 Erbs et al 1999 Liger et al 1999
Loyaux-Lawniczak et al 1999 Amonette et al 2000 Hwang amp Batchelor 2000
Hansen et al 2001 Gander et al 2002 Lee amp Batchelor 2002aampb Pecher et al
2002 Vikesland amp Valentine 2002 Hofstetter et al 2003 OrsquoLoughlin et al
2003aampb Strathmann amp Stone 2003 Elsner et al 2004 OrsquoLoughlin amp Burris
2004) However only few comparative studies on the reactivity of Fe(II)-bearing
minerals exist (Lee amp Batchelor 2002b Elsner et al 2004) When examining the
General Introduction 9
reaction rates of the reductive transformation of NACs and chlorinated aliphatics
by GRs and other Fe(II)-bearing minerals reported in these studies the rate
constants for GRs are mostly among the highest rates reported and in some cases
even higher than the rate constants for Fe(II) sulphides Thus GRs may play an
important role in the transformation of reducible contaminants in the subsurface
Nitroaromatic compounds (NACs) are widely applied as explosives herbicides
insecticides solvents and intermediates in the synthesis of dyes and pesticides
(Hartter 1985 Rosenblatt et al 1991) NACs are ubiquitous in the subsurface
environment and pose a health risk due to their toxicity (Rickert 1985) In anoxic
environments reduction of the nitro group is generally the first step during abiotic
or microbial transformation of the NACs (Macalady et al 1986) The
transformation reaction generally produces the corresponding aromatic amines and
minor amounts of intermediates (hydroxylamines and nitroso compounds) as well
as coupling products (azo and azoxy compounds) These products may be of
similar or even greater environmental concern
Trichloroacetic acid (TCA) is ubiquitous in soils and the concentrations reported
range from lt005 to 380 microgkg (Euro Chlor 2001 McCulloch 2002 Ahlers et al
2003) On account of its phytotoxicity suspected human carcinogenicity and
widespread occurrence TCA is of considerable environmental concern especially
in the terrestrial compartment (Ahlers et al 2003) Moreover the daughter
compounds of TCA - dichloroacetic acid (DCA) and monochloroacetic acid
(MCA) - are also toxins and suspected human carcinogens as well as widespread in
the environment (Reimann et al 1996 Berg et al 2000 Ahlers et al 2003 and
references therein) In this work the reactivity of synthetic green rusts towards
nitroaromatic compounds (NACs) and the reactivity of various Fe(II)-Fe(III)
mineral systems including synthetic GRs towards chlorinated acetates have been
studied (Chapters 4 and 5)
10 Chapter 1 15 Outline of the thesis
An examination of the Fe-containing products produced during solid state
oxidation of vivianite by anaerobic autotrophic denitrifying Fe(II)-oxidizing
bacteria is presented in chapter 2 The Fe(II)-oxidizing bacteria were cultured in a
mineral medium containing high levels of bicarbonate and phosphate which is
typically used in this kind of studies as it provides excellent conditions for the
nitrate-reducing FeOB The solid iron phases forming were investigated by
transmission Moumlssbauer spectroscopy infrared spectroscopy and scanning electron
microscopy
Chapter 3 includes a study on the Fe-containing products formed during reduction
of common Fe(III) oxides by the anaerobic dissimilatory Fe(III)-reducing
microorganism Shewanella algae BrY In order to simulate natural conditions
Fe(III) oxides were applied as coatings on silica (model system for sandy soils) or
calcite particles (model system for calcareous soils) and synthetic electron shuttles
as well as highly concentrated artificial pH buffers were excluded The mineral
systems studied include goethitecalcite lepidocrocitecalcite and hydrous ferric
oxidesand suspensions The solid iron phases produced were examined by
transmission Moumlssbauer spectroscopy
A study on the relative reactivity of outer and inner Fe(II) sites in synthetic GR-
SO4 by using a series of structurally closely related compounds with different
charge properties as ldquoreactive probesrdquo is presented in chapter 4 The probe
compounds included nitrobenzene 2-nitrophenol 4-nitrotoluene 4-
chloronitrobenzene and 4-nitrophenylacetic acid
In chapter 5 an investigation of the reactivity of various Fe(II)-Fe(III) mineral
systems towards TCA and DCA is presented The study included laboratory batch
experiments imitating natural conditions The Fe(II)-Fe(III)-systems investigated
included GR-SO4 carbonate green rust magnetite Fe(II)goethite and
General Introduction 11
Fe(II)lepidocrocite The reactivities of the Fe(II)-Fe(III) mineral systems were
examined by comparing their surface-normalized rate constants
The results and environmental implications of this work are summarized in chapter
6 References Ahlers J Regelmann J Riedhammer C (2003) Environmental risk assessment of airborne trichloroacetic acid - a contribution to the discussion of the significance of anthropogenic and natural sources Chemosphere 52 531-537 Al-Agha MR Burley SD Curtis CD Esson J (1995) Complex cementation textures and authigenic mineral assemblages in recent concretions from the Lincolnshire Wash (east coast UK) driven by Fe(0) to Fe(II) oxidation Journal of the Geological Society 152 157-171 Amonette JE Workman DJ Kennedy DW Fruchter JS Gorby YA (2000) Dechlorination of carbon tetrachloride by Fe(II) associated with goethite Environmental Science and Technology 34 4606-4613 Berg M Muumlller SR Muumlhlemann J Wiedmer A Schwarzenbach RP (2000) Concentrations and mass fluxes of chloroacetic acids and trifluoroacetic acid in rain and natural waters in Switzerland Environmental Science and Technology 34 2675-2683 Bernal JD Dasgupta DR Mackay AL (1959) The oxides and hydroxides of iron and their structural inter-relationships Clay Minerals Bulletin 4 15-30 Butler EC Hayes KF (1998) Effects of solution composition and pH on the reductive dechlorination of hexachloroethane by iron sulfide Environmental Science and Technology 32 1276-1284 Butler EC Hayes KF (1999) Kinetics of the transformation of trichloroethylene and tetrachloroethylene by iron sulfide Environmental Science and Technology 33 2021-2027 Carlson L Schwertmann U (1990) The effect of CO2 and oxidation rate on the formation of goethite versus lepidocrocite from an Fe(II) system at pH 6 and 7 Clay Minerals 25 65-71 Chaudhuri SK Lack JG Coates JD (2001) Biogenic magnetite formation through anaerobic biooxidation of Fe(II) Applied and Environmental Microbiology 67 2844-2848 Cornell RM Schwertmann U (1996) The iron oxides Structure properties reactions occurrence and uses VCH Verlagsgesellschaft mbH Weinheim Cui D Eriksen TE (1996) Reduction of pertechnetate by ferrous iron in solution influence of sorbed and precipitated Fe(II) Environmental Science and Technology 30 2259-2262
Cui D Spahiu K (2002) The reduction of U(VI) on corroded iron under anoxic conditions Radiochemica Acta 90 623-628
12 Chapter 1 Drissi SH Refait Ph Abdelmoula M Geacutenin JMR (1995) The preparation and thermodynamic properties of Fe(II)-Fe(III) hydroxide-carbonate (green rust I) Pourbaix diagram of iron in carbonate-containing aqueous media Corrosion Science 37 2025-2041 Elsner M Haderlein SB Schwarzenbach RP (2004) Reactivity of Fe(II)-bearing minerals towards reductive transformation of organic contaminants Environmental Science and Technology 38 799-807 Erbs M Hansen HCB Olsen CE (1999) Reductive dechlorination of carbon tetrachloride using iron(II)iron(III)-hydroxide-sulphate (green rust) Environmental Science and Technology 33 307-311 Euro Chlor (2001) Trichloroacetic acid in the environment a dossier Euro Chlor Brussels and the European Chlorinated Solvent Association Fredrickson JK Zachara JM Kennedy DW Dong H Onstott TC Hinman NW Li S (1998) Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium Geochimica et Cosmochimica Acta 62 3239-3257 Gander JW Parkin GF Scherer MM (2002) Kinetics of 111-trichloroethane transformation by iron sulfide and a methanogenic consortium Environmental Science and Technology 36 4540-4546 Geacutenin JMR Bourrieacute G Trolard F Abdelmoula M Jaffrezic A Refait Ph Maitre V Humbert B Herbillon A (1998) Thermodynamic equilibria in aqueous suspensions of synthetic and natural Fe(II)-Fe(III) green rusts Occurrences of the mineral in hydromorphic soils Environmental Science and Technology 32 1058-1068 Geacutenin JMR Olowe AA Benbouzid-Rollet ND Prieur D Confente M Resiak B (1991) The simultaneous presence of green rust 2 and sulfate reducing bacteria in the corrosion of steel sheet piles in a harbour area Hyperfine Interactions 69 875-878 Geacutenin JMR Olowe AA Refait Ph Simon L (1996) On the stoichiometry and Pourbaix diagram of Fe(II)-Fe(III) hydroxy-sulphate of sulphate-containing green rust 2 An electrochemical and Moumlssbauer spectroscopy study Corrosion Science 38 1751-1762 Glasauer S Weidler PG Langley S Beveridge TJ (2003) Controls on Fe reduction and mineral formation by a subsurface bacterium Geochimica et Cosmochimica Acta 67 1277- 1288 Hanert HH (1992) The genus Gallionella In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 4082-4088 Hansen HCB Borggaard OK Soslashrensen J (1994) Evaluation of the free energy of formation of iron(II)iron(III)-hydroxidesulphate (Green Rust) and its reduction of nitrite Geochimica et Cosmochimica Acta 58 2599-2608 Hansen HCB Guldberg S Erbs M Koch CB (2001) Kinetics of nitrate reduction by green rusts ndash effects of interlayer anion and Fe(II)Fe(III) ratio Applied Clay Science 18 81-91
General Introduction 13
Hansen HCB Koch CB Nancke-Krogh H Borggaard OK Soslashrensen J (1996) Abiotic nitrate reduction to ammonium Key role of green rust Environmental Science and Technology 30 2053-2056 Hartter DR (1985) The use and importance of nitroaromatic chemicals in the chemical industry In Toxicity of nitroaromatic compounds Rickert DE (ed) Hemisphere Publishing Corporation 1-13 Heasman DM Sherman DM Ragnarsdottir KV (2003) The reduction of aqueous Au3+ by sulfide minerals and green rust phases American Mineralogist 88 725-738 Hofstetter TB Schwarzenbach RP Haderlein SB (2003) Reactivity of Fe(II) species associated with clay minerals Environmental Science and Technology 37 519-528 Hwang I Batchelor B (2000) Reductive dechlorination of tetrachloroethylene by Fe(II) in cement slurries Environmental Science and Technology 34 5017-5022 King GM (1990) Effects of added manganic and ferric oxides on sulfate reduction and sulfide oxidation in intertidal sediments FEMS Microbiology Ecology 73 131-138 Klausen J Troumlber SP Haderlein SB Schwarzenbach RP (1995) Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions Environmental Science and Technology 29 2396-2404 Kuenen JG Robertson LA Tuovinen OH (1992) The genera Thiobacillus Thiomicrospira and Thiosphaera In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 2618-2624
Kukkadapu RK Zachara JM Smith SC Fredrickson JK Liu C (2001) Dissimilatory bacterial reduction of Al-substituted goethite in subsurface sediments Geochimica et Cosmochimica Acta 65 2913-2924 Lee W Batchelor B (2002a) Abiotic reductive dechlorination of chlorinated ethylenes by iron- bearing soil minerals 1 Pyrite and magnetite Environmental Science and Technology 36 5147- 5154 Lee W Batchelor B (2002b) Abiotic reductive dechlorination of chlorinated ethylenes by iron-bearing soil minerals 2 Green rust Environmental Science and Technology 36 5348- 5354 Liger E Charlet L Van Cappellen P (1999) Surface catalysis of uranium (VI) reduction by iron(II) Geochimica et Cosmochimica Acta 63 2939-2955 Lovley DR (1997) Microbial Fe(III) reduction in subsurface environments FEMS Microbiology Reviews 20 305-313 Lovley DR Phillips EJP (1986) Organic matter mineralization with reduction of ferric iron in anaerobic sediments Applied and Environmental Microbiology 51 683-689 Lovley DR Phillips EJP Lonergan DJ (1991) Enzymatic versus nonenzymatic mechanisms for Fe(III) reduction in aquatic sediments Environmental Science and Technology 25 1062-1067
14 Chapter 1 Loyaux-Lawniczak S Refait Ph Lecomte P Ehrhardt J Geacutenin JMR (1999) The reduction of chromate ions by Fe(II) layered hydroxides Hydrology and Earth System Sciences 3 593-599 Lyngkilde J Christensen TH (1992) Redox zones of a landfill leachate pollution plume (Vejen Denmark) Journal of Contaminant Hydrology 10 273-289 Macalady DL Tratnyek PG Grundl TJ (1986) Abiotic reduction reactions of anthropogenic organic chemicals in anaerobic systems A critical review Journal of Contaminant Hydrology 1 1-28 McCulloch A (2002) Trichloroacetic acid in the environment Chemosphere 47 667-686 Mendiboure A Schoumlllhorn A (1986) Formation and anion exchange reactions of layered transition metal hydroxides [Ni1-xMx](OH)2(CO3)x2(H2O)z (M = Fe Co) Revue de Chimie Mineacuterale 23 819-827 Misawa T Hashimoto K Shimodaira S (1974) The mechanism of formation of iron oxide and oxyhydroxides in aqueous solutions at room temperature Corrosion Science 14 131-149 Mulder EG Deinema MH (1992) The sheathed bacteria In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 2618-2624 Myneni SCB Tokunaga TK Brown Jr GE (1997) Abiotic selenium redox transformations in the presence of Fe(IIIII) oxides Science 278 1106-1109 Nealson KH Saffarini D (1994) Iron and manganese in anaerobic respiration Environmental significance physiology and regulation Annual Review of Microbiology 48 311-343 Nielsen A (1976) Hvid groslashn og sort rust Beskrivelse af en korrosionsskade paring et svoslashmmebassin Nordisk Betong 2 21-24 Nriagu JO Dell CI (1974) Diagenetic formation of iron phosphates in recent lake sediments American Mineralogist 59 934-946 OLoughlin EJ Burris DR (2004) Reduction of halogenated ethanes by green rust Environmental Toxicology and Chemistry 23 41-48 OLoughlin EJ Kelly SD Cook RE Csencsits R Kemner KM (2003a) Reduction of uranium(VI) by mixed iron(II)iron(III) hydroxide (green rust) Formation of UO2 nanoparticles Environmental Science and Technology 37 721-727 OLoughlin EJ Kelly SD Kemner KM Csencsits R Cook RE (2003b) Reduction of AgI AuIII CuII and HgII by FeIIFeIII hydroxysulfate green rust Chemosphere 53 437-446 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002a) Iron (IIIII) hydroxycarbonate green rust formation and stabilization from lepidocrocite bioreduction Environmental Science and Technology 36 16-20 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002b) Microbial reduction of lepidocrocite γ-FeOOH by Shewanella putrefaciens The
General Introduction 15
formation of green rust Hyperfine Interactions 139140 231-237 Parmar N Gorby YA Beveridge TJ Ferris FG (2001) Formation of green rust and immobilization of nickel in response to bacterial reduction of hydrous ferric oxide Geomicrobiology Journal 18 375-385 Pecher K Haderlein SB Schwarzenbach RP (2002) Reduction of polyhalogenated methanes by surface-bound Fe(II) in aqueous suspensions of iron oxides Environmental Science and Technology 36 1734-1741 Pepper SE Bunker DJ Bryan ND Livens FR Charnock JM Pattrick RAD Collison D (2003) Treatment of radioactive wastes An X-ray absorption spectroscopy study of the reaction of technetium with green rust Journal of Colloid and Interface Science 268 408- 412 Randall SR Sherman DM Ragnarsdottir KV (2001) Sorption of As(V) on green rust (Fe4(II)Fe2(III)(OH)12SO4
3H2O) and lepidocrocite (γ-FeOOH) Surface complexes from EXAFS spectroscopy Geochimica et Cosmochimica Acta 65 1015-1023 Refait Ph Abdelmoula M Trolard F Geacutenin JMR Ehrhardt JJ Bourrieacute G (2001) Moumlssbauer and XAS study of a green rust mineral the partial substitution of Fe2+ by Mg2+ American Mineralogist 86 731-739 Refait Ph Drissi SH Marie Y Geacutenin JMR (1994) The substitution of Fe2+ ions by Ni2+ ions in green rust one compounds Hyperfine Interactions 90 389-394 Refait Ph Geacutenin JMR (1997) The mechanisms of oxidation of ferrous hydroxychloride β- Fe2(OH)3Cl in aqueous solution The formation of akaganeite vs goethite Corrosion Science 39 539-553 Reimann S Grob K Frank H (1996) Chloroacetic acids in rainwater Environmental Science and Technology 30 2340-2344 Rickert DE (1985) Toxicity of nitroaromatic compounds Hemisphere Publishing Corporation 1-13 Roh Y Lee SY Elless MP (2000) Characterization of corrosion products in the permeable reactive barriers Environmental Geology 40 184-194 Rosenblatt DH Burrows EP Mitchell WR Parmer DL (1991) Organic explosives and related compounds In The Handbook of Environmental Chemistry Anthropogenic compounds Hutzinger O (Ed) Springer-Verlag 195-234 Ruumlgge K Hofstetter TB Haderlein SB Bjerg PL Knudsen S Zraurig C Mosbaeligk H Christensen TH (1998) Characterization of predominant reductants in an anaerobic leachate- affected aquifer by nitroaromatic probe compounds Environmental Science and Technology 32 23-31 Schwertmann U Cornell RM (1991) Iron oxides in the laboratory Preparation and characterization VCH Verlagsgesellschaft mbH Weinheim Simon L Francois M Refait Ph Renaudin G Lelaurain M Geacutenin JMR (2003)
16 Chapter 1 Structure of the Fe(II-III)-layered double hydroxysulphate green rust two from Rietveld analysis Solid State Sciences 5 327-334 Strathmann TJ Stone AT (2003) Mineral surface catalysis of reactions between FeII and oxime carbamate pesticides Geochimica et Cosmochimica Acta 67 2775-2791 Straub KL Benz M Schink B Widdel F (1996) Anaerobic nitrate-dependent microbial oxidation of ferrous iron Applied and Environmental Microbiology 62 1458-1460 Tamaura Y (1985) ZnII-bearing green rust II and its spontaneous transformation into ZnII- bearing ferrite in aqueous solution Bulletin of the Chemical Society of Japan 58 2951-2954 Tamaura Y (1986) Ni(II)-bearing green rust II and its spontaneous transformation into Ni(II)- bearing ferrites Bulletin of the Chemical Society of Japan 59 1829-1832 Taylor RM (1980) Formation and properties of Fe(II)Fe(III)-hydroxycarbonate and its possible significance in soil formation Clay Minerals 15 369-382 Trolard F Abdelmoula M Bourrieacute G Humbert B Geacutenin JMR (1996) Mise en eacutevidence dun constituant de type rouilles vertes dans les sols hydromorphes Proposition de lexistence dun nouveau mineacuteral la fougeacuterite Geacuteosciences de surface Comptes Rendus de LrsquoAcademie des Sciences 323 1015-1022 Tuovinen OH Button KS Vuorinen A Carlson L Mair DM Yut LA (1980) Bacterial chemical and mineralogical characteristics of tubercles in distribution pipelines Journal of the American Water Works Association 72 626-635 Vikesland PJ Valentine RL (2002) Iron oxide surface-catalyzed oxidation of ferrous iron by monochloramine implications of oxide type and carbonate on reactivity Environmental Science and Technology 36 512-519 Widdel F Schnell S Heising S Ehrenreich A Assmus B Schink B (1993) Ferrous iron oxidation by anoxygenic phototrophic bacteria Nature 362 834-836
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 17
2 Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria
Abstract
This work investigated the Fe-containing products formed by anaerobic
autotrophic denitrifying Fe(II)-oxidizing bacteria in a specific bicarbonate buffered
(30 mM HCO3- pH 70) culture media containing 10 mM Fe(II) 4 mM nitrate and
4 mM phosphate Fe(II) was present initially as a whitish vivianite-like
(Fe3(PO4)2middot8H2O) precipitate and as soluble Fe(II) The initial phase of the
oxidation produced a greenish metavivianite-like ((FeII3-xFeIII
x)(PO4)2(OH)xmiddot(8-
x)H2O x gt 12) phase In the late oxidation phase a reddish precipitate of poorly
crystalline goethite (α-FeOOH) dominated the colour of the media in coexistence
with Fe(II)-containing siderite (FeCO3) The increasing amounts of Fe(III) present
in the ldquovivianiterdquo and ldquometavivianiterdquo structures were accompanied by an
increasing intensity in the green colour as the Fe(II) biooxidation progressed This
colour development has produced the idea of biogenic green rusts (layered Fe(II)-
Fe(III) hydroxides) in several studies on nitrate-dependent Fe(II) biooxidation
However in this work no evidence of green rust formation mediated by anaerobic
denitrifying Fe(II)-oxidizing bacteria was obtained
21 Introduction
Aerobic oxidation of Fe(II)-containing minerals by lithotrophic acidophilic and
neutrophilic bacteria has been known for many years but their broad significance
in the biogeochemical cycling of iron has only been recognized over the past two
decades Aerobic Fe(II)-oxidizing bacteria (FeOB) have been isolated from acidic
ecosystems (Thiobacillus ferrooxidans) neutral eutrophic systems (Sphaerotilus
natans Leptothrix ochracea) and neutral oligotrophic systems (Gallionella
ferruginea) (Hanert 1992 Kuenen et al 1992 Mulder amp Deinema 1992) At
neutral pH Fe(II) is unstable in the presence of oxygen and is rapidly oxidized to
the insoluble Fe(III) Hence the only pH neutral environments where soluble
18 Chapter 2
Fe(II) is available for aerobic FeOB are at interfaces between oxic and anoxic
conditions The aerobic neutrophilic FeOB (Leptothrix ochracea Gallionella
ferruginea and Sphaerotilus natans) live at such interfaces and are usually
associated with the yellowishreddish ferric deposits formed there
Over the past several years there has been a growing recognition that other less
readily detectable types of bacteria are involved in Fe(II) oxidation in ecosystems
at circumneutral pH For example it has been reported that neutrophilic FeOB are
abundant at the Loihi seamount hydrothermal vents and play a major role in the
Fe(III) oxide deposition (Emerson amp Moyer 2002) Similarly unidentified
neutrophilic obligate lithotrophic FeOB have been isolated from the rhizosphere of
wetlands plants where they are closely associated with deposits of amorphous
Fe(III) oxides (Emerson et al 1999) It was previously believed that Fe(III) oxide
deposits associated with sheaths were produced biologically whereas Fe(III) oxide
deposits not associated with cells were produced abiotically Recently the
formation of amorphous Fe(III) oxide in gradient tubes has been attributed to the
action of FeOB (Sobolev amp Roden 2001) The authors attribute 90 of the
oxidation to biological processes and indicated that the organisms seem to produce
a mobile form of Fe(III) that diffuses away from the cells before being
precipitated thereby avoiding encrustation of the cells They suggest that such
soluble Fe(III) complexes might be substrates for closely associated Fe(III)-
reducing bacteria Such an arrangement might allow close coupling between
microbial Fe(II) oxidation and Fe(III) reduction within millimeters of the oxic-
anoxic interface
Anaerobic Fe(II) oxidation by phototrophic purple non-sulfur bacteria utilizing
Fe(II) as an electron donor in the light was recognized only a decade ago (Widdel
et al 1993) Subsequently it was demonstrated that the biological oxidation of
Fe(II) in the absence of oxygen is possible by light-independent chemotrophic
microbial activity using nitrate as the electron acceptor (Straub et al 1996) In
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 19
addition studies conducted in gradient cultures revealed that nitrate-reducing
strains could also oxidize Fe(II) with molecular oxygen (Benz et al 1998) Hence
these Fe(II)-oxidizing strains may use nitrate as well as oxygen as electron
acceptors The microbial oxidation of Fe(II) was coupled to stoichiometric
reduction of nitrate to N2 and only one strain produced traces of N2O as a by-
product (Straub et al 1996 Benz et al 1998) The authors proposed the formation
of 2-line ferrihydrite as the end product of Fe(II) biooxidation The chemical
reduction of nitrate by Fe(II) requires a catalyst eg at least 10 microM Cu2+ in order
to take place at significant rates and may thus be considered insignificant under the
conditions applied in our study (Moraghan amp Buresh 1976) The chemical
oxidation of Fe(II) with nitrous oxide has not been observed However nitrite can
oxidize Fe(II) chemically (Moraghan amp Buresh 1977 Straub et al 1996) but this
process is considered insignificant at the conditions applied here No denitrifying
Fe(II)-oxidizing enrichment culture has been found to produce ammonium from
nitrate
Both lithoheterotrophic (depending on organic cosubstrates such as acetate) and
strictly lithoautotrophic nitrate-reducing FeOB have been found in various marine
and freshwater sediments However most isolates depend on organic cosubstrates
for cell biosynthesis (Benz et al 1998) Most probable number estimations
showed that denitrifying FeOB accounted for 00006-08 of the acetate-oxidizing
denitrifying microbial population Lithotrophic FeOB accounted for less than
00001 of the total bacterial community Attempts to isolate CO2-fixing nitrate-
dependent FeOB from lithotrophic cultures have failed (Straub amp Buchholz-
Cleven 1998) Mixotrophic FeOB accounted for 0004-004 of the total bacterial
community In addition microbial nitrate-dependent Fe(II) oxidation was
demonstrated in a flooded paddy soil as well as in activated sludge from a
wastewater treatment plant (Nielsen amp Nielsen 1998 Ratering amp Schnell 2001)
Since the activity is not restricted to sunlight exposed habitats microbial nitrate-
dependent Fe(II) oxidation is supposedly more important on a global scale than
20 Chapter 2
anaerobic Fe(II) oxidation by phototrophic bacteria Furthermore it has been
reported that anaerobic denitrifying FeOB aptly oxidize biogenic Fe(II) minerals
formed by bioreduction of synthetic goethite and ferrihydrite and that anaerobic
Fe(III)-reducing bacteria readily reduce Fe(III) minerals formed by biooxidation of
Fe(II) (Weber et al 2001 Straub et al 1998) Hence autotrophic denitrifying
FeOB may play a significant role in the nitrogen and iron cycles in subsurface
environments where the nitrate and the Fe(II) zones overlap and organic carbon
supply is limited (Figure 21)
Figure 21 The microbial iron cycle linking the carbon and nitrogen cycles
Phosphate is released into the environment through natural processes such as rock
weathering and decomposition of dead organic material and anthropogenic
activities eg wastewater effluents and application of manure and fertilizers in
horti- and agriculture In anoxic soils and sediments phosphate may be sequestered
by sorption onto Fe(III) oxides (Williams et al 1971 Patrick amp Khalid 1974)
Phosphate strongly influences the type morphology and properties of Fe(III)
oxides formed by oxidation and hydrolysis of Fe(II) salts as well as the degree of
their transformation (Kandori et al 1992 Cumplido et al 2000 Benali et al
2001) Phosphate may also be retained by precipitation of Fe(II) phosphates such
as the monoclinic vivianite (Fe3(PO4)2middot8H2O) which is the most important stable
Fe(II) orthophosphate solid encountered in the subsurface under most conditions
(Nriagu 1972) At anoxic and circumneutral conditions the whitish vivianite
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 21
controls the Fe(II) activity even at very low phosphate concentrations (Nriagu amp
Dell 1974) Vivianite occurs as a secondary mineral in the gossans of metallic ore
deposits and as a weathering product of primary iron-manganese phosphates in
pegmatites (Gaines et al 1997) Moreover natural vivianite occurrences have
been identified in a number of lake and river sediments (Zwaan amp Kortenbout van
der Sluys 1971 Nriagu amp Dell 1974 Postma 1981 Nembrini et al 1983
Henderson et al 1984 Dodd et al 2003 House 2003 and references therein)
Vivianite is also found in sewage sludge as a result of the wastewater treatment
where iron salts are added in order to remove phosphate (Seitz et al 1973) It is
however still indefinite how ubiquitous vivianite is in nature Furthermore only
little is known about the mechanism of vivianite formation and the role played by
sedimentary Fe(III) oxides Anaerobic Fe(III)-reducing microorganisms may
reduce Fe(III) oxides thereby releasing the iron as soluble Fe(II) and mobilizing
the phosphate adsorbed to the Fe(III) oxides (Lovley 1997) It has been suggested
that vivianite is formed by precipitation following reductive dissolution of Fe(III)
oxides (Manning et al 1981 Manning amp Jones 1982) However it has also been
proposed that the transformation of Fe(III) oxides to vivianite occurs topotactically
and not via reductive dissolution (Nembrini et al 1983) Vivianite was shown to
form microbially as a result of the activity of the anaerobic Fe(III)-reducing
bacteria Shewanella putrefaciens in the presence of high Fe(III)-citrate and
phosphate concentrations (Jorand et al 2000) Moreover vivianite formation by
bioreduction of Fe(III) in hydrous ferric oxide and in smectite has been reported
(Fredrickson et al 1998 Dong et al 2003)
Only little is known about the oxidation products of vivianite Metavivianite a
greenish triclinic iron hydroxy phosphate mineral was first described by Ritz et al
(1974) and it was later found to coexist with vivianite in several natural sediment
samples (Henderson et al 1984) Once the Fe(III) content became evident the true
composition of metavivianite ((FeII3-xFeIII
x)(PO4)2(OH)x
22 Chapter 2
middot(8-x)H2O x gt 12)) was established (Rodgers amp Johnston 1985 Rodgers 1986
and references therein) The formation of intermediate greenish precipitates during
oxidation of fluffy colourless Fe(II) precipitates by anoxic phototrophic
microorganisms and nitrate-dependent FeOB have been reported (Ehrenreich amp
Widdel 1994 Chaudhuri et al 2001) Since both studies were conducted in
bicarbonate buffered mineral media (22-30 mM HCO3ndash pH 70-72) containing
37-5 mM phosphate we assume that the initial fluffy whitish precipitates
consisted mainly of vivianite Chaudhuri et al (2001) proposed that the
intermediate green phases produces by the denitrifying FeOB consist of carbonate
green rust (GR-CO3) but no convincing evidence of this biogenic GR-CO3 has been
provided yet The major objective of this work was to examine the Fe-containing
products forming during the course of biooxidation of vivianite by non-
phototrophic anaerobic denitrifying Fe(II)-oxidizing bacteria
2 2 Materials and methods
All handling and sampling of solutions and suspensions were carried out under
sterile and strict anoxic conditions All chemicals were pa quality
221 Microorganisms and media
Enrichment cultures of nitrate-reducing FeOB taken from town ditches (Bremen
Germany) were grown in anoxic bicarbonate-buffered (30 mM HCO3ndash 90
N210 CO2 pH 70) mineral media containing 4 mM phosphate as well as
essential trace elements and vitamins (Table 21 Straub amp Buchholz-Cleven
1998) Ammonium was omitted from the media in order to facilitate detection of
ammonium possibly produced by reduction of nitrate The techniques used for
preparation of media and cultivation of bacteria under anoxic conditions have been
described by Widdel amp Bak (1992) 05 M aqueous stock solutions of FeCl2 or
FeSO4 were prepared in 100 mL glass flasks by reacting 65 mmol of iron powder
(particle size 10 microm Merck) with 100 mL deoxygenated 10 M HCl or 05 M
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 23
H2SO4 respectively The solutions were magnetically stirred and heated (~80degC)
during reaction until the H2(g) production had ceased (ge 1 hour) The FeCl2 and
FeSO4 stock solutions were stored under a small Ar overpressure at 5degC
Table 21 Composition of the mineral medium (adopted from Straub amp Buchholz-Cleven
(1998))
Components Concentration (M) KH2PO4 15middot10-3
K2HPO4 25middot10-3
MgSO4middot7H2O 10middot10-3
CaCl2middot2H2O 50middot10-4
H3BO3 56middot10-5
ZnSO4middot7H2O 10middot10-6
Na2MoO4middot2H2O 40middot10-6
CuSO4middot5H2O 20middot10-7
MnSO4middotH2O 10middot10-6
Na2SeO4 12middot10-5
CoCl2middot6H2O 50middot10-6
NiCl2middot6H2O 80middot10-6
NaCl 10middot10-5
NaHCO3ndash 30middot10-2
Cyanocobalamine (vitamin B12) 37middot10-8
p-aminobenzoic acid (vitamin Hrsquo) 36middot10-7
D(+)-biotin (vitamin H) 41middot10-8
Nicotinic acid (Niacin) 81middot10-7
Ca-D(+)-pantothenate (vitamin B5) 52middot10-8
Pyridoxamine dihydrochloride 96middot10-7
Thiaminechloridehydrochloride (vitamin B1) 15middot10-7
NaNO3 40middot10-3
FeSO4 or FeCl2 0010
222 Characterisation of precipitates
In order to optimize the characterization and distinction between the spectral
components transmission Moumlssbauer spectra were obtained at temperatures
between 5 K and 250 K and in external magnetic fields of 4 T (parallel to the γ-ray
direction) using a conventional constant acceleration spectrometer and a source of 57Co in Rh The spectrometer was calibrated using a 125 microm foil of α-Fe at room
temperature and isomer shifts are given relative to the centroid of the spectrum of
this absorber The spectra were fitted using simple Lorentzian line shape Infrared
(IR) spectra were obtained using a Perkin Elmer FT-IR 2000 spectrometer and the
24 Chapter 2
KBr pellet technique Scanning electron microscopy (SEM) was carried out in
order to study the morphology and composition of the precipitates Specimens for
SEM were prepared by depositing suspended particles onto an aluminum stub
coated with a carbon sticker The stub was quickly transferred into a sputtering
chamber and coated with a thin Pt film (~20 nm) In order to avoid interfering Pt
signals in the energy dispersive spectra the stubs were in some cases not coated
with Pt but quickly transferred to the SEM chamber for evacuation Measurements
were performed using a Philips XL30 equipped with a LaB6 source and an
accelerating voltage of 20 kV and an EDAX eDXi X-ray dispersive spectrometer
223 Biooxidation experiments
The biooxidation experiments were conducted in 50-400 mL butyl rubber
stoppered bottles with a 90 N210 CO2 headspace constituting 10 of the total
volume Prior to inoculation 4 mM NaNO3 was added as the electron acceptor and
10 mM Fe2+ (as chloride or sulphate) as the electron donor to the mineral media
Control experiments were performed in the same media only they were not
inoculated Addition of ferrous iron to the media induced an immediate
precipitation of a solid whitish material The whitish precipitate was collected on
022 microm polyvinylidendifluorid (Durapore Millipore) filters and stored in an
anoxic atmosphere until further measurements Old outgrown media suspensions
that had turned reddish in color due to a precipitate produced by the denitrifying
FeOB were used as inocula Inoculum volume was kept lt1 of the culture volume
in order to prevent the reddish inoculum from dominating over the initial whitish
precipitate Cultures were incubated in the dark at room temperature and gently
agitated once every day Typical color successions for the media were initial
whitish precipitates turning first more and more greenish over time and then finally
turning reddish (see Fig 22) The color developed uniformly without any
indications of multiple phases in the precipitate At different time intervals
suspension samples were withdrawn using 90 N210 CO2-flushed polyethylene
syringes The precipitates were collected on 022 microm polyvinylidendifluorid
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 25
(Durapore Millipore) filters and analyzed by Moumlssbauer spectroscopy and SEM
Nonfiltered suspension samples were digested in 01 M HCl and chemically
analyzed for Fe(II) NO3- and NH4
+
224 Analytical methods
Fe2+ was determined using a modified phenanthroline method (Fadrus amp Maly
1975) Nitrate was quantified by ion chromatography (Morales et al 2000) and
ammonium was measured photometrically using the indophenol reaction (Rossum
amp Villarruz 1963)
23 Results and discussion
231 Identification of solid iron-containing phases
In most cases the mineral media for cultivating denitrifying FeOB contained 10
mM FeCl2 or FeSO4 4 mM NO3ndash 4 mM total phosphorus and 30 mM HCO3
- at pH
70 ([HCO3-] = 2138middot[CO3
2-] at pH 70) Whitish flocs precipitated immediately at
these initial conditions when Fe(II) was added to the media (Figure 22a) Such
colourless flocs have been reported to precipitate in similar mineral media (10 mM
Fe(II) 37-5 mM phosphate 22-30 mM HCO3ndash pH 70-72) (Ehrenreich amp
Widdel 1994 Chaudhuri et al 2001) Our Fe(II) measurements showed that 20-
50 of the total Fe(II) added was present in this initial white precipitate
26 Chapter 2
a
210- Figure 22 Colour of suspended material in the growth media during Fe(II) biooxidation a)
Initial whitish precipitate prior to inoculation b) Inte1mediate greenish phase fo1med within 2-3
days after inoculation c) reddish precipitate at late stage of biooxidation (gt5-6 days)
The precipitates were filtered and investigated by Mossbauer and IR spectroscopy
The transmission Mossbauer spectra obtained for the initial whitish precipitate at
temperatures between 20 and 250 K are shown in Figure 23 The spectrum
measured at 250 K consists of two fairly well-resolved Fe(II) doublets (see
parameters in Table 22) The change in line-overlap with decreasing temperature
is primarily ascribed to differences in the temperature dependence of the
quadrupole splitting of the two components From the spectra at 10 and 6 K
(Figure 24) it can be concluded that magnetic ordering takes place between these
two temperatures and that only one transition occurs (indicating the presence of
only one phase) The parameters of one of the Fe(II) doublets at 250 K (designated
B in Table 22) are in very good agreement with previously published values for
the vivianite Fe(Il)8 site at room temperature (eg McCammon amp Burns 1980)
and the ordering temperature also agrees well with an assignment as vivianite
(Forsyth et al 1970) However the second Fe(II) doublet in the initial colourless
precipitate (Table 22) has parameters that deviate from previously reported values
by having a smaller quadrupole splitting (indicating a less distorted coordination)
and a significantly higher relative intensity and line width These effects might be
due to the presence of numerous defects in the vivianite crystal lattice particularly
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 27
affecting the Fe(II)A sites It should be noted that further components may be added
to the fit in order to improve its statistics Nevertheless we decided to include no
further components as suggested by the finding of one magnetic ordering only
Accordingly our interpretation of the Moumlssbauer results for the initial white
precipitate suggests a highly defective vivianite having a distribution of local
coordination environments particular in the A site This assignment is further
supported by a major absorption band due to phosphate anions in the infrared
spectrum at approximately 1000 cm-1 and the absence of other complex anions
(data not shown) Thus the whitish precipitate is referred to as a vivianite-like
(ldquovivianiterdquo) precipitate
0
3 5
3 0
2 5
2 0
1 5
1 0
0 5
0 0
20 K
Velocity (mms)
Rel
ativ
e ab
sorp
tion
()
3
2
1
0
80 K
3 5
3 0
2 5
2 0
1 5
1 0
0 5
0 0
150 K
-5 -4 -3 -2 -1 0 1 2 3 4 5
2 5
2 0
1 5
1 0
0 5
0 0
250 K
Figure 23 Transmission Moumlssbauer spectra measured between 250 and 20 K of the initial
whitish precipitate prior to inoculation (see Fig 22a) Fitting components (and their sum) are
shown as full lines
28 Chapter 2
Table 22 Selected Mossbauer parameters of the doublet components in the spectra obtained for
different precipitates
Precipitates Temperature Isomer shift Quadrupole Line width Area (K) (mms-1) splitting (mms-1) (mms-1) ()
Whitish Fe(II)B 250 127 309 035 38
Fe(II)A 250 128 181 051 62
Dark greenish Fe(II)B 250 126 305 023 17
Fe(II)A 250 132 238 051 44
Fe(III) 250 036 085 040 38
Reddish-orange 40 138 244 094 67 Fe(II) Hyperfine parameters are generally given with uncertainties of 003 mms- the spectral area with an uncertainty of 3
1005
1000
0995
0990
- 0985 ~ e c 0980 0
-~ 0975 E c nl b 1000 g ~ Qi 0995 0
0990
0985
0980 -12 -8
~ -~
bullbull bull Ibull bull bullbull bull bull ~ bull bull ~
It
10 K
bull bull bull bull (J ~ i~ 6K bullmiddotf bull bull
~ bull bull bull
-4 0 4 8 12
Velocity (mmls)
Figure 24 Transmission Mossbauer spectra measured at 10 and 6 K of the initial whitish
precipitate prior to inoculation (see Fig 22a)
In general the color of the media suspension changed from whitish into light green
color within 2-3 days after inoculation (Figure 22b ) This transformation occurred
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 29
without dissolving the initial whitish precipitate or preserving the whitish
precipitate as a separate phase implying some kind of solid state transformation
Two identical media suspensions were inoculated simultaneously but at the
sampling time they had distinctly different intensities of the green color
designated light and dark green respectively Moumlssbauer spectra of the dark green
sample are shown in Figure 25 The spectra of this sample are all fitted using three
doublet components (two Fe(II) and one Fe(III)) and the parameters of the
spectrum measured at 250 K are given in Table 22
30
25
20
15
10
05
00
20 K
Velocity (mms)
Rel
ativ
e ab
sorp
tion
()
35
30
25
20
15
10
05
00
80 K
30
25
20
15
10
05
00
150 K
-5 -4 -3 -2 -1 0 1 2 3 4 5
25
20
15
10
05
00
250 K
Figure 25 Transmission Moumlssbauer spectra measured between 250 and 20 K of the dark
greenish precipitate formed during biooxidation Fitting components (and their sum) are shown
as full lines
No magnetic ordering of the dark green precipitate was observed at temperatures
above 20 K but ordering occurred around 10 K (not shown - due to very thin
30 Chapter 2
samples this was not investigated in details) The two greenish samples had very
similar parameters only differing in the relative intensity of Fe(III) 26 and 38
in the light greenish and dark greenish samples respectively Assuming the
spectral area of a component to be proportional to the abundance of the species in
the solid these results indicate a correlation between the intensity of the green
color and the content of Fe(III) in the precipitate The parameters of the Fe(II)
doublets in the dark green precipitate (Table 22) were in very good agreement
with previously published values for vivianite with a non-negligible Fe(III) content
(McCammon amp Burns 1980) whereas the Fe(III) component in particular had a
higher quadrupole splitting The observation that magnetic ordering of both Fe(II)
and Fe(III) occurred at similar temperature for the light green phase (Figure 26) is
a strong indication that they both belong to the same phase The absence of the
component with the low quadrupole splitting in the spectra of both green samples
might indicate that the initial vivianite-like phase crystallized into a more well-
defined vivianite over time However freshly prepared and long-term aged (gt1
year) suspensions of the initial vivianite-like precipitate did not differ significantly
Hence we suggest that the recrystallization of the vivianite-like precipitate can be
explained by Fe(II) biooxidation
Solid State Oxidation ofFe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria
0
2
3
- 4
~ 5 c ~ 6 e-0 7 -2 nl Q) 00 gt ~ 05 Qi 0 10
15
20
25
30
35
bullbullbull hi 6~
bullbullbullbull bull bull bullbull bull 10 K bull bull
bull bull bull bull bull bull bull
~~ ~~ lf 6K bull bull bullbull middot bull bull r bull
bull bull bull bull bull bullbull bull bull bull - 12 -a -4 0 4 8 12
Velocity (mms)
31
Figure 26 Transmission Mossbauer spectrn measured at 6 and 10 K of the light greenish
precipitate fonned during biooxidation
The solid state oxidation of monoclinic vivianite to triclinic metavivianite is well-
known (McCammon amp Bums 1980 Pratesi et al 2003 and references therein)
The vivianite crystal structure contains Fe(II) ions in both isolated Fe(II)A and
paired Fe(Il)8 octahedra Mossbauer spectroscopic analyses have shown that the
Fe(Il)8 Fe(II)A ratio increases with increasing Fe(III) concentration suggesting that
the remaining Fe(II)A ions are more readily oxidized than the Fe(II)a ion of an
Fe(Il)8 -Fe(III)8 pair (McCammon amp Bums 1980) The mechanism of oxidation of
Fe(II) in vivianite involves conversion of H20 ligands to OH- ions producing a
progressive collapse of the vivianite structure due to the elimination of hydrogen
bonds (Moore 1971) The exact oxidation limits between which the triclinic lattice
is stable are somewhat disputed as the results obtained for synthetic and natural
vivianites oxidized chemically in the laboratory and naturally oxidized natural
vivianite specimens do not agree completely (Rodgers 1986 and references
therein) Taking all reports into account the monoclinic structure of vivianite is
supposedly maintained until 40-50 of total iron is oxidized Further oxidation
32 Chapter 2
leads to the formation of the triclinic metavivianite in which the FeA site is fully
oxidized whereas the oxidation of the FeB ranges from 20 to almost 100 Thus
the triclinic metavivianite structure persists close to complete oxidation of total
iron The Moumlssbauer results obtained in this study are consistent with the vivianite
solid state oxidation mechanism reported by McCammon amp Burns (1980) Thus
we propose that the intermediate greenish precipitate is a metavivianite-like
(ldquometavivianiterdquo) phase It should be noted that a minor oxidation of dissolved
Fe(II) may have occurred even though the solid state oxidation of Fe(II) was
predominant
Within 5-6 days after inoculation the greenish intermediate was transformed into a
reddish product (Figure 22c) The magnetically ordered sextet in the spectrum of
the red phase (Figure 27) measured at 40 K was due to goethite (α-FeOOH)
(magnetic hyperfine field of 470 T and a quadrupole shift of -01 mms-1 cp
Moslashrup et al 1983) The sextet deviated from ideal goethite by its asymmetric line
shape and its low ordering temperature (around 100 K ndash data not shown) and thus
the goethite was poorly crystalline It is very likely that the presence of phosphate
in the media retarded the crystal growth of goethite The unusual reddish colour of
the goethite might also be explained by the presence of phosphate The spectrum at
40 K was however dominated by a Fe(II) doublet (Table 22) that ordered
magnetically between 40 and 20 K (Figure 27) The hyperfine parameters and the
magnetic ordering temperature indicated that this component was due to siderite
(FeCO3) having a magnetic ordering temperature of 38 K (Jacobs 1963) The
siderite component may have formed as a result of the microbial activity changing
the chemistry of the solution and precipitating a major part of the remaining
dissolved Fe(II) at this stage The characteristic vivianite Fe(II) doublet with large
quadrupole splitting was not detected in this sample The reddish precipitate
contained considerably less Fe(III) than the greenish precipitate (only 33 as
estimated from the spectral area) None of the components in the reddish sample
Solid State Oxidation ofFe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 33
could be detected in freshly inoculated samples indicating that carryover of mineral
precipitates by inoculation of the culture media was negligible
0
2
- 3
~4 c
Q 5
e 6 0
~ 7 g
0 3l Q)
a 1
2
3
4
5
-12 a
40K
bull bull (
4 0 4 8 12 Velocity (mms)
Figure 27 Transmission Mossbauer spectra measured at 20 and 40 K of the reddish precipitate
fo1med during the late biooxidation stage Fitting components (and their sum) are shown as full
lines
Strengite (FeP04middot2H20) was not detected at any time during oxidation
Santabarbaraite a new amorphous F e(III) hydroxy phosphate mineral
(Fe3(P04)i(OH)3middot5H20) was reported in a recent study (Pratesi et al 2003) The
brownish mineral was a result of the solid state oxidation of vivianite through
metavivianite However no Mossbauer data have been provided for this new
mineral yet and therefore we cannot give an account of whether santabarbaraite
forms in our system or not Thus goethite was the dominating end product and we
propose the reaction path depicted in Figure 28 for the nitrate-dependent
biooxidation of Fe(II) in our systems
34 Chapter 2
Fe3(PO4)2middot8H2O (FeII3-xFeIII
x)(PO4)2(OH)xmiddot(8-x)H2O α-FeOOH
NO3- N2 NO3
- N2
ldquoVivianiterdquo ldquoMetavivianiterdquo Goethite
Figure 28 Proposed reaction path and iron-containing minerals forming during solid state
oxidation of vivianite by denitrifying FeOB at the experimental conditions applied in this study
The biotic formation of layered Fe(II)-Fe(III) hydroxides (green rusts) by
anaerobic denitrifying Fe(II)-oxidizing bacteria has been suggested but proper
identification of these phases still lacks (Chaudhuri et al 2001) We cannot rule
out that small amounts of green rusts (GRs) perhaps a phosphate intercalated GR
(Hansen amp Poulsen 1999) might have been present here during the greenish
intermediate ldquometavivianiterdquo oxidation stage When present in low concentrations
especially in mixtures including other iron minerals it is very difficult to identify
GRs even with Moumlssbauer spectroscopy At least two complementary methods
such as X-ray diffraction (XRD) and Moumlssbauer spectroscopy are required for
proper identification and characterization of GRs However the precipitates
collected in this work were poorly crystalline and did not allow for XRD analysis
Electron micrographs including energy dispersive X-ray spectroscopy suspension
colour and mineral stability calculations do not suffice as evidence Hence no
convincing evidence of GR formation facilitated by denitrifying FeOB has been
provided so far The blue-green colours of metavivianite and green rust minerals
originate from Fe(II)-Fe(III) charge transfer between adjacent Fe(II) and Fe(III)
ions in edge-shared octahedra (Faye et al 1968) The greenish suspension colour
occurring during the intermediate phase has incited the idea of biogenic GRs in
studies on nitrate-dependent Fe(II) biooxidation (Chaudhuri et al 2001 Lack et
al 2002aampb) However our results indicate that this reasoning is misleading
232 Factors controlling the rate and extent of Fe(II) biooxidation
Generally it was found that maximally 20-64 of the initial Fe(II) amount was
oxidized to Fe(III) (Figure 29) This indicates some limitations in the accessibility
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 35
of Fe(II) in the system Based solely on stoichiometry considerations the
microorganisms are expected to oxidize 5 mol Fe(II) for every 1 mol nitrate
reduced to dinitrogen However as exemplified in Figure 28 this ratio was mostly
lt4 which can be explained by the consumption of nitrogen as a result of microbial
growth At initial [Fe(II)][NO3ndash] ratios lt5 nitrate is in excess and should not limit
the extent of the biooxidation Thus the lack of complete biooxidation could not be
due to exhaustion of nitrate Furthermore all growth essential nutrients were more
than sufficiently applied hence the incomplete Fe(II) biooxidation was not caused
by lack of nutrients The most reasonable explanation therefore seems to be that an
increasingly limited access to the electron donor over time inhibits complete long-
term Fe(II) biooxidation At least four mechanisms may cause this inhibition 1)
the Fe(II) becomes isolated within the structure of the mixed Fe(II)-Fe(III)
minerals forming during biooxidation or underneath a passive Fe(III)-bearing
surface film on the initial Fe(II) precipitates 2) the FeOB cell surface becomes
covered with a passive Fe(III)-bearing surface film 3) the Fe(II) biooxidation is
controlled by the rate of dissolution of the initial Fe(II) minerals or 4) the reaction
proceeds primarily by biooxidation of dissolved Fe(II) whose concentration
gradually decreases due to changes in solid phase composition The actual
mechanisms whereby the surface-associated Fe(III) can inhibit Fe(II) biooxidation
are unknown but they may involve both kinetic and thermodynamic constraints on
the electron transfer The Moumlssbauer results obtained in this work strongly suggest
that the Fe(II) biooxidation occurred mainly in the solid state of the initial
ldquovivianiterdquo phase However we cannot rule out that some dissolved Fe(II) was
oxidized as well
36 Chapter 2
Figure 29 Concentration profiles of total Fe(II) and nitrate as a function of time during Fe(II)
biooxidation
No Fe(II) oxidation took place in cultures where nitrate had been omitted
confirming that the microbial Fe(II) oxidation is nitrate-dependent (data not
shown) No Fe(II) oxidation was detected in the non-inoculated control
experiments within the duration of the experiments and thus the chemical
oxidation of dissolved Fe(II) by nitrate catalyzed by vitamins or trace elements
(eg Cu(II)) can be neglected Ammonium did not form in detectable amounts
during Fe(II) biooxidation (data not shown) and therefore dinitrogen was assumed
to be the end product as reported previously (Straub et al 1996 Benz et al 1998)
The absence of ammonium formation does indirectly support the absence of
biologically induced green rust formation as synthetic green rust is known to
convert nitrate into ammonium in purely chemical reactions (Hansen et al 1996)
It was visually observed that the phosphate concentration in the media exerted a
control on the microbial Fe(II)-oxidation At phosphorus concentrations le 2 mM
no Fe(II)-oxidation took place However the solubility product for vivianite (Ksp =
171middot10-36 at 25degC (Al-Borno et al 1994)) was still by far exceeded under these
conditions It is not known whether this phosphate limiting effect is due to growth
constraints in the mixed bacterial community or whether specific Fe(II) phosphate
Solid State Oxidation ofFe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 37
precipitates are prerequisites of the Fe(II) biooxidation to take place Experiments
are currently underway in our laboratory in order to elucidate the role of specific
initial Fe(II) precipitates It should be noted that the growth of the denitrifying
FeOB could not be estimated as they were present in highly heterogeneous
suspensions containing both solid iron phases as well as other bacteria (enrichment
culture)
233 Morphology of solid iron phases
The morphology of the various precipitates was studied by SEM The initial
whitish precipitate consisted of a web-like structure (Figure 21 Oa and background
in Figure 21 Ob) whereas more distinct hexagonally shaped rosettes with particle
size ~20 microm (Figure 2lObampc) formed during Fe(II) biooxidation The energy
dispersive spectroscopic element analyses showed that other than iron the initial
whitish precipitate and the rosettes contained mainly phophorus
Figure 210 Scanning electron micro graphs of precipitates fo1med at various stages of the
experiment a Initial whitish precipitate bampc Rosettes observed in the intennediate greenish
precipitate d Reddish precipitate sampled during the late biooxidation stage
38 Chapter 2
These observations are interpreted as vivianite forming a web-like morphology in
the initial whitish precipitate and partly transforming into hexagonal particles in
the greenish colored stage The interpretations are supported by similar vivianite
morphologies reported including pseudo-hexagonal vivianite crystals of low
symmetry resulting from microbial Fe(III) reduction of HFO and platy rosettes of
vivianite crystals formed during bioreduction of Fe(III) in smectite (Fredrickson et
al 1998 Dong et al 2003) It was not possible to associate the morphology
observed in the reddish precipitate with the minerals identified in this phase
(Figure 210d)
24 Conclusions
This work demonstrated that anaerobic autotrophic denitrifying Fe(II)-oxidizing
bacteria produce poorly crystalline goethite by solid state oxidation of ldquovivianiterdquo
via a ldquometavivianiterdquo intermediate The increasing amount of Fe(III) forming in the
vivianite structure was accompanied by an increasing intensity in the green colour
as the Fe(II) biooxidation progressed Moumlssbauer spectroscopic analyses provided
no significant evidence of green rust formation The finding of microbially
oxidized vivianite in this study raises the question of the oxidation state of
vivianite specimens from natural sediments Vivianite is generally believed to be
an ideal Fe(II) hydroxy phosphate mineral and the presence of Fe(III) is explained
by aerial oxidation upon sampling The results presented here indicate that
microbiological processes may be responsible for the oxidation of vivianite and
metavivianite in natural subsurface environments Acknowledgments
We would like to thank Dr K Straub for providing and advising us how to culture the nitrate-
reducing FeOB Furthermore we thank Dr C B Koch for performing the Moumlssbauer analyses
and Dr D Mavrocordatos for performing the SEM analyses
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 39
References Al-Borno A Tomson MB (1994) The temperature dependence of the solubility product constant of vivianite Geochimica et Cosmochimica Acta 58 5373-5378 Benali O Abdelmoula M Refait Ph Geacutenin JMR (2001) Effect of orthophosphate on the oxidation products of Fe(II)-Fe(III) hydroxycarbonate The transformation of green rust to ferrihydrite Geochimica et Cosmochimica Acta 65 1715-1726 Benz M Brune A Schink B (1998) Anaerobic and aerobic oxidation of ferrous iron at neutral pH by chemohetorotrophic nitrate-reducing bacteria Archives of Microbiology 169 159-165 Chaudhuri SK Lack JG Coates JD (2001) Biogenic magnetite formation through anaerobic biooxidation of Fe(II) Applied and Environmental Microbiology 67 2844-2848 Cumplido J Barron V Torrent J (2000) Effect of phosphate on the formation of nanophase lepidocrocite from Fe(II) sulfate Clays and Clay Minerals 48 503-510 Dodd J Large DJ Fortey NJ Kemp S Styles M Wetton P Milodowski A (2003) Geochemistry and petrography of phosphorus in urban canal bed sediment Applied Geochemistry 18 259-267 Dong H Kostka JE Kim J (2003) Microscopic evidence for microbial dissolution of smectite Clays and Clay Minerals 51 502-512 Ehrenreich A Widdel F (1994) Anaerobic oxidation of ferrous iron by purple bacteria a new type of phototrophic metabolism Applied and Environmental Microbiology 60 4517-4526 Emerson D Moyer CL (2002) Neutrophilic Fe-oxidizing bacteria are abundant at the Loihi seamount hydrothermal vents and play a major role in Fe oxide deposition Applied and Environmental Microbiology 68 3085-3093 Emerson D Weiss JV Megonigal JP (1999) Iron-oxidizing bacteria are associated with ferric hydroxide precipitates (Fe-plaque) on the roots of wetland plants Applied and Environmental Microbiology 65 2758-2761 Fadrus H Maly J (1975) Suppression of iron(III) interference in the determination of iron(II) in water by the 110-phenanthroline method The Analyst 100 549-554 Faye GH Manning PG Nickel EH (1968) The polarized optical absorption spectra of tourmaline cordierite chloritoid and vivianite ferrous-ferric electronic interaction as a source of pleochroism American Mineralogist 53 1174-1201 Forsyth JB Johnson CE Wilkonson C (1970) The magnetic structure of vivianite Fe3(PO4)2middot8H2O Journal of Physics Part C Solid State Physics 3 1127-1139 Fredrickson JK Zachara JM Kennedy DW Dong H Onstott TC Hinman NW Li S (1998) Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium Geochimica et Cosmochimica Acta 62 3239-3257 Gaines RV Skinner HCW Foord EE Mason B Rosenzweig A (1997) Danas new
40 Chapter 2 mineralogy 8th ed John Wiley amp Sons Inc Hanert HH (1992) The genus Gallionella In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 4082-4088 Hansen HCB Koch CB Nancke-Krogh H Borggaard OK Soerensen J (1996) Abiotic nitrate reduction to ammonium Key role of green rust Environmental Science and Technology 30 2053-2056 Hansen HCB Poulsen IF (1999) Interaction of synthetic sulphate green rust with phosphate and the crystallization of vivianite Clays and Clay Minerals 47 312-318 Henderson GS Black PM Ridgers KA Rankin PC (1984) New data on New Zealand vivianite and metavivianite New Zealand Journal of Geology and Geophysics 27 367-378 House WA (2003) Geochemical cycling of phosphorus in rivers Applied Geochemistry 18 739-748 Jacobs IS (1963) Metamagnetism of siderite (FeCO3) Journal of Applied Physics 34 1106-1107 Jorand F Appenzeller BMR Abdelmoula M Refait Ph Block J-C Geacutenin JMR (2000) Assessment of vivianite formation in Shewanella putrefaciens culture Environmental Technology 21 1001-1005 Kandori K Uchida S Kataoka S Ishikawa T (1992) Effects of silicate and phosphate ions on the formation of ferric oxide hydroxide particles Journal of Materials Science 27 719-728 Kuenen JG Robertson LA Tuovinen OH (1992) The genera Thiobacillus Thiomicrospira and Thiosphaera In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 2618-2624 Lack JG Chaudhuri SK Chakraborty R Achenbach LA Coates JD (2002a) Anaerobic biooxidation of Fe(II) by Dechlorosoma suillum Microbial Ecology 43 424-431 Lack JG Chaudhuri SK Kelly SD Kemner KM OConnor SM Coates JD (2002b) Immobilization of radionuclides and heavy metals through anaerobic bio-oxidation of Fe(II) Applied and Environmental Microbiology 68 2704-2710 Lovley DR (1997) Microbial Fe(III) reduction in subsurface environments FEMS Microbiology Reviews 20 305-313 Manning PG Birchall T Jones W (1981) Ferric hydroxides in surficial sediments of the great lakes and their role in phosphorus availability a Moumlssbauer spectral study Canadian Mineralogist 19 525-530 Manning PG Jones W (1982) The binding capacity of ferric hydroxides for non-apatite inorganic phosphorus in sediments of the depositional basins of Lakes Erie and Ontario Canadian Mineralogist 20 169-176 McCammon CA Burns RG (1980) The oxidation mechanism of vivianite as studied by Moumlssbauer spectroscopy American Mineralogist 65 361-366 Moore PB (1971) The Fe2+
3(H2O)n(PO4)2 homologous series crystal-chemical relationships
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 41
and oxidized equivalents American Mineralogist 56 1-17 Moraghan JT Buresh RJ (1976) Chemical reduction of nitrate by ferrous iron Journal of Environmental Quality 5 320-325 Moraghan JT Buresh RJ (1977) Chemical reduction of nitrite and nitrous oxide by ferrous iron Journal of American Soil Science Society 40 47-50 Morales JA de Graterol LS Mesa J (2000) Determination of chloride sulfate and nitrate in groundwater samples by ion chromatography Journal of Chromatography A 884 185-190 Mulder EG Deinema MH (1992) The sheathed bacteria In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 2618-2624 Moslashrup S Madsen MB Franck J Villadsen J Koch CJW (1983) A new interpretation of Moumlssbauer spectra of microcrystalline goethiterdquosuper-ferromagnetismrdquo of ldquosuper-spin-glassrdquo behaviour Journal of Magnetism and Magnetic Materials 40 163-174 Nembrini GP Capobianco JA Viel M Williams AF (1983) A Moumlssbauer and chemical study of the formation of vivianite in sediments of Lago Maggiore (Italy) Geochimica et Cosmochimica Acta 47 1459-1464 Nielsen JL Nielsen PH (1998) Microbial nitrate-dependent oxidation of ferrous iron in activated sludge Environmental Science and Technology 32 3556-3561 Nriagu JO (1972) Stability of vivianite and ion-pair formation in the system Fe3(PO4)2-H3PO4- H2O Geochimica et Cosmochimica Acta 36 459-470 Nriagu JO Dell CI (1974) Diagenetic formation of iron phosphates in recent lake sediments American Mineralogist 59 934-946 Patrick Jr WH Khalid RA (1974) Phosphate release and sorption by soils and sediments Effect of aerobic and anaerobic conditions Science 186 53-55 Postma D (1981) Formation of siderite and vivianite and the pore-water composition of a recent bog sediment in Denmark Chemical Geology 31 225-244 Pratesi G Cipriani C Giuli G Birch WD (2003) Santabarbaraite a new amorphous phosphate mineral European Journal of Mineralogy 15 185-192 Ratering S Schnell S (2001) Nitrate-dependent iron(II) oxidation in paddy soil Environmental Microbiology 3 100-109 Ritz C Essene EJ Peacor DR (1974) Metavivianite Fe3(PO4)2middot8H2O a new mineral American Mineralogist 59 896-899 Rodgers KA (1986) Metavivianite and kerchenite a review Mineralogical Magazine 50 687- 691 Rodgers KA Johnston JH (1985) Type metavivianite Moumlssbauer evidence for a revised composition Neues Jahrbuch fuumlr Mineralogie-Monatshefte 12 539-542
42 Chapter 2 Rossum JR Villarruz PA (1963) Determination of ammonia by the indophenol method Journal of American Water Works Association 55 657-658 Seitz MA Riedner RJ Malhotra SK Kipp RJ (1973) Iron-phosphate compound identification in sewage sludge residue Environmental Science and Technology 7 354-357 Sobolev D Roden EE (2001) Suboxic deposition of ferric iron by bacteria in opposing gradients of Fe(II) and oxygen at circumneutral pH Applied and Environmental Microbiology 67 1328-1334 Straub KL Benz M Schink B Widdel F (1996) Anaerobic nitrate-dependent microbial oxidation of ferrous iron Applied and Environmental Microbiology 62 1458-1460 Straub KL Buchholz-Cleven BEE (1998) Enumeration and detection of anaerobic ferrous iron-oxidizing nitrate-reducing bacteria from diverse European sediments Applied and Environmental Microbiology 64 4846-4856 Straub KL Hanzlik M Buchholz-Cleven BEE (1998) The use of biologically produced ferrihydrite for the isolation of novel iron-reducing bacteria Systematic and Applied Microbiology 21 442-449 Weber KA Picardal FW Roden EE (2001) Microbially catalyzed nitrate-dependent oxidation of biogenic solid-phase Fe(II) compounds Environmental Science and Technology 35 1644-1650 Widdel F Bak F (1992) Gram-negative mesophilic sulfate-reducing bacteria In The Prokaryotes (Balows A Truumlper HG Dworkin M Harder W Schleifer K-H (eds)) Springer 2nd ed 3352-3378
Widdel F Schnell S Heising S Ehrenreich A Assmus B Schink B (1993) Ferrous iron oxidation by anoxygenic phototrophic bacteria Nature 362 834-836 Williams JDH Syers JK Shukla SS Harris RF Armstrong DE (1971) Levels of inorganic and total phosphorus in lake sediments as related to other sediment parameters Environmental Science and Technology 5 1113-1120
Zwaan PC Kortenbout van der Sluys G (1971) Vivianite crystals from Hare Noord Brabant Province The Netherlands Scripta Geology 6 1-7
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 43
3 Formation of Layered Iron Hydroxides by Microbial Fe(III)
Reduction Abstract
Many inorganic and organic pollutants may be degraded by microorganisms in the
subsurface However a wide range of contaminants including chromate nitrate
radionuclides nitroaromatic compounds chlorinated aliphatics and carbamate
pesticides may also be chemically transformed by reduction reactions involving
layered iron(II)-iron(III)-hydroxides (green rusts) Hence green rusts (GRs) may
play a potentially important role in the fate and transport of pollutants in iron-rich
suboxic soils and sediments Yet only little is known about the formation of GRs
in these environments The biotic formation of GRs mediated by the anaerobic
dissimilatory Fe(III)-reducing bacteria Shewanella spp has been reported and
proposed in several studies However the experimental conditions applied were
mostly not natural and the evidence of GR formation provided may be questioned
This work investigated the Fe-containing products formed by the facultatively
anaerobic Fe(III)-reducing microorganism Shewanella algae BrY in culture
media containing 4-10 mM formate or lactate and 8-27 mM Fe(III) In order to
simulate natural conditions Fe(III) oxides were applied as coatings on silica
(model system for sandy soils) or calcite particles (model system for calcareous
soils) and synthetic electron shuttles as well as highly concentrated artificial pH
buffers were excluded S algae BrY reduced 19-72 of the initial Fe(III) when
grown in goethitecalcite lepidocrocitecalcite or hydrous ferric oxidesand mineral
systems and green or blackish mineral phases were produced within 1-2 weeks
after inoculation Moumlssbauer spectroscopic analyses indicated that the green and
blackish precipitates were dominated by vivianite (Fe3(PO4)2sdot8H2O) and green rust
44 Chapter 3
31 Introduction
The significance of bacteria in the biogeochemical cycling of iron has been broadly
recognized over the past two decades Chemical processes were previously
considered to account for most of the Fe(III) reduction in subsurface environments
Dissimilatory Fe(III)-reducing bacteria (DIRB) that gain energy by coupling the
oxidation of hydrogen or organic compounds to the reduction of Fe(III) oxides
have been known for many years but their biogeochemical importance was
acknowledged only a decade ago (reviewed by Lovley 1997) DIRB transfer
electrons to extracellular Fe(III) without assimilating the iron Fe(III) bioreduction
accounts for a major fraction of the carbon oxidation in many different
environments and in the presence of high amounts of reactive Fe(III) microbial
Fe(III) reduction may even inhibit sulfate reduction and methanogenesis (King
1990 Lovley amp Phillips 1986) In fact most of the Fe(III) reduction in the Fe(III)
reduction zone of aquatic sediments and aquifers is thought to be enzymatically
catalyzed by microorganisms (Lovley et al 1991) However the relative
importance of microbial and chemical processes involved in the Fe(III) reduction
are still somewhat disputed among microbiologists and geochemists
A wide diversity of Fe(III)-reducing bacteria which fall in a number of different
phylogenetic groups is known today Both organisms growing by respiration and
by fermentation have been isolated and identified (Lovley 1991 Nealson amp
Saffarini 1994) Hydrogen short- and long-chained fatty acids amino acids
sugars and aromatic compounds may serve as electron donors for Fe(III)
bioreduction The enzymes responsible for dissimilatory Fe(III) reduction are outer
membrane associated ferric reductases (Lower et al 2001 and references therein)
Iron reducing bacteria may utilize alternative electron acceptors such as O2 nitrate
S0 sulfate humic substances contaminant metals and metalloids as well as
chlorinated solvents The first organism shown to couple respiratory growth to
dissimilatory iron reduction was Pseudomonas ferrireductans now known as
Shewanella oneidensis but previously classified as Alteromonas putrefaciens and
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 45
Shewanella putrefaciens (Venkateswaran et al 1999) Various DIRB including
the obligate anaerobic Geobacter sp and the facultatively anaerobic Shewanella
sp have been isolated from both marine and freshwater sediments soil and
aquifers (Thamdrup 2000 and references therein)
The redox potentials of oxidized and reduced iron couples and thus the energy
yield available from Fe(III) reduction depend strongly on the specific iron phases
involved In soil and aquatic environments Fe(III) oxides mainly occur in
association with other sediment particles as aggregates or coatings Amorphous
and poorly crystalline Fe(III) oxides usually make up 20 or less of the iron
content in a sediment (Thamdrup 2000) They are the main products of abiotic and
biotic Fe(II) oxidation in sediments and they constitute the most important phases
for microbial Fe(III) reduction Until recently it was generally believed that DIRB
reduced insoluble Fe(III) oxides only by direct contact with the Fe(III) oxide
thereby allowing electron transfer from the cell to the Fe(III) oxide surface
However over the past several years there has been a growing recognition that
DIRB may use different strategies in order to access the solid Fe(III) oxides These
strategies include solubilization of Fe(III) by synthetic or natural Fe(III) chelators
and Fe(III) reduction via electron shuttling with soluble humic substances or
microbially produced electron shuttles (Nevin amp Lovley 2002 and references
therein Turick et al 2003) The Fe(III) complexing agents may also stimulate
Fe(III) oxide reduction indirectly by chelation and thus removal of Fe(II) from
the cell and the Fe(III) oxide surfaces Both chelated Fe(III) and soluble electron
shuttles are more accessible to Fe(III) reductases than solid Fe(III) oxides In
contrast to Geobacter metallireducens S algae produces and releases extracellular
electron shuttling compounds (Nevin amp Lovley 2000) However in the absence of
soluble electron shuttles reversible adhesion is required for reduction of solid
Fe(III) oxides by S algae BrY (Das amp Caccavo 2000) Shewanella algae BrY
adheres readily and preferentially to a range of solid Fe(III) oxides such as
ferrihydrite goethite and hematite (Das amp Caccavo 2001) The adhesion
46 Chapter 3
mechanisms are not completely understood but recent results suggest that the
adhesion is mediated by cell surface proteins and independent of cell motility
(Caccavo amp Das 2002)
The microbial formation of GRs resulting from bioreduction of various Fe(III)
oxides including ferrihydrite goethite and lepidocrocite by strains of the
anaerobic DIRB Shewanella putrefaciens has been reported repeatedly over the
last years (Fredrickson et al 1998 Kukkadapu et al 2001 Liu et al 2001
Parmar et al 2001 Ona-Nguema et al 2002aampb Glasauer et al 2003)
However no evidence of biogenic formation of GRs at natural geochemical
conditions have been offered and it is still unknown whether this process may take
place at natural conditions comprising low carbon and iron concentrations as well
as the absence of synthetic electron shuttles and highly concentrated artificial pH
buffers GRs are layered iron(II)-iron(III)-hydroxides with anionic interlayers and
they hold the general formula [FeII(6-x)FeIII
x(OH)12]x+[(A)xnmiddotyH2O]x- where x = 09 -
42 A is an n-valent anion eg CO32- Clndash or SO4
2- and y is the number of water
molecules in the interlayer In circumneutral solutions the oxidation of dissolved
Fe(II) always passes through solid GR phases (Bernal et al 1959) This agrees
with the natural GR occurrences found in suboxic nonacid iron-rich environments
such as hydromorphic soils and intertidal sediments (Al-Agha et al 1995 Trolard
et al 1996 Genin et al 1998) In addition GRs have been found as corrosion
products in numerous engineering systems eg in a pipeline distribution system
for drinking water steel sheet piles in marine sediments reinforced concrete
(ferro-concrete) and permeable reactive barriers of zero-valent iron implemented
for on-site remediation of organic and inorganic contaminants (Tuovinen et al
1980 Nielsen 1976 Genin et al 1991 Roh et al 2000) Through sequestration
and reductive transformation GRs may play an important role in the fate and
transport of organic and inorganic pollutants in suboxic iron-rich soils and
sediments (see Chapters 4 amp 5 in this work and references therein)
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 47
The major goal of this work was to examine the iron minerals forming during the
course of Fe(III) bioreduction of hydrous ferric oxide goethite and lepidocrocite
Two model systems simulating sandy and calcareous soils in subsurface
environments were designed in order to investigate the formation of iron minerals
at conditions including low carbon levels low Fe(III) concentrations applied as
Fe(III) oxide coatings on sand or calcite no electron shuttle and no synthetic pH
buffers
32 Materials and methods
All handling and sampling of solutions and suspensions were carried out at strict
anoxic conditions Standard sterile techniques were used throughout (Hungate
1969 Miller amp Wolin 1974) Only the iron oxide coatings were not autoclaved in
order to avoid the iron oxides from transforming Goethite (acicular particles with
size 01 times 06 microm specific surface area 16 m2g) and lepidocrocite (acicular
particles with size 005 times 03 microm specific surface area 18 m2g) were purchased as
fine powders from Bayer (Bayferrox 910 and 943) Calcite (grain size 170-350 microm
Pluumlss-Staufer AG) and sea sand (dominantly quartz grain size 01-03 mm Riedel-
de Haeumln) were used as Fe(III) oxide coating bearing minerals
321 Preparation of iron oxide coatings
Two grams of goethite (goe) or lepidocrocite (lep) and 100 g calcite were
combined with 200 mL deionized water (DIW) in a 500 mL polyethylene flask
Hydrous ferric oxide (HFO) was synthesized by dissolving 4 g Fe(NO3)3middot9H2O in
70 mL DIW followed by slow neutralization under magnetic stirring till pH 7 with
approximately 30 mL 1 M NaOH (method modified after Schwertmann amp Cornell
1991) The HFO coating was made by combining 100 mL freshly precipitated
HFO with 900 mL deionized water and 50 g sea sand in a polyethylene bottle The
suspensions containing the iron oxide coatings were gently agitated on a
reciprocating shaker for 24 h and left to stand for another 24 h Excess Fe(III)
oxides and salts were removed from the coated material by repeated decantation
48 Chapter 3
and washing with 003 M NaNO3 followed by washing with DIW until clear
runoff Finally the coatings were collected on folding filters and air dried The
amount of HFO goethite and lepidocrocite coated onto sand and calcite after
washing and drying was quantified to 7-11 mg Fe(III)g sand or calcite
322 Mineral characterisation
The identity and purity of the HFO synthesized were examined by means of X-ray
diffraction (XRD) measurements The XRD analyses were performed on a Scintag
XDS 2000 using Co Kα radiation (45 kV 40 mA) using divergence scatter and
receiving slits of 1deg 05deg and 02 mm respectively Samples were scanned
between 6 and 80 deg2θ with a scan speed of 1 deg2θmin Mineral suspension samples
for transmission Moumlssbauer spectroscopic analysis were collected on 02 microm filters
in an anoxic glove box (Coy Laboratory Products Inc) transferred to Perspex
capsules and stored in liquid nitrogen until measurement Moumlssbauer spectra were
obtained between 250 and 5 K using a conventional constant acceleration
spectrometer and a source of 57Co in Rh The spectrometer was calibrated using a
125 microm foil of α-Fe at room temperature and isomer shifts are given relative to
the centroid of this absorber The spectra were fitted using simple Lorentzian line
shape and it was assumed that all positions have identical f-factors
323 Culture conditions and cell preparation
Shewanella algae BrY is a motile gram-negative rod which was isolated first from
anoxic estuary sediments (Caccavo et al 1992) S algae BrY was grown
aerobically in tryptic soy broth (30 gL CASO-bouillon Merck) at 28degC on a rotary
shaker at 150 rpm for 16-18 h Cells were harvested by centrifugation (6000 rpm times
g 4ordmC 15 min) during the late exponential ndash early stationary growth phase at
OD660 ~ 06 Optimal Fe(III) reductase activity is expressed at this stage of growth
(Roden amp Zachara 1996) The cells were washed twice in oxic 50 mM PIPES
[piperazine-NNacute-bis(2-ethanesulfonic acid)] buffer (pH 70) and resuspended in
culture medium containing no Fe(III) and no carbon source Washed cell
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 49
suspensions were used as inocula for Fe(III) reduction experiments Oxygen was
expelled from the inoculum by extensive purging with 100 N2(g) (9999999
purity) Working stock cultures of S algae BrY were maintained aerobically on
tryptic soy agar plates at ambient temperature
324 Bioreduction experiments
All anaerobic incubations were carried out in anoxic serum vials (25 mL) or test
tubes (13 mL) sealed with thick (10-13 mm) butyl rubber stoppers and aluminum
crimp caps or plastic screw caps The basal culture medium (Table 31) was
prepared according to Kostka amp Nealson (1998) but with a phosphate
concentration of 2 mM and the exclusion of Fe(II) and EDTA
(ethylenediaminetetraacetic acid) The medium was amended with 4-10 mM
lactate or formate and 8-27 mM Fe(III) The Fe(III) was applied as Fe(III) oxide
coatings on sand or calcite The suspensions were purged extensively with 100
N2(g) (HFOsand suspensions) or 995 N205 CO2(g) (goecalcite and
lepcalcite suspensions) prior to inoculation The calcareous systems were buffered
at pH ~ 76 through a natural buffer system (CaCO3(s) + 995 N205 CO2(g))
whereas the sandy systems contained no pH buffer (100 N2(g) pH 55-60)
Inoculum size made up 5 of the total volume Cultures were incubated dark at
room temperature and gently agitated once every day At different time intervals
suspension samples for Fe(II) and Moumlssbauer analysis were withdrawn from the
reaction mixture using 100 N2(g) or 995 N205 CO2(g)-flushed sterile
disposable syringes and hypodermic needles Suspension samples for Fe(II)
analysis were digested in 01 M HCl for 30 min
50 Chapter 3
Table 31 Composition of the mineral medium (modified from Kostka amp Nealson (1998))
Components Concentration (M)
(NH4)2SO4 00143 KH2PO4 73middot10-4
K2HPO4 13middot10-3
MgSO4middot7H2O 10middot10-3
CaCl2middot2H2O 50middot10-4
H3BO3 56middot10-5
ZnSO4middot7H2O 10middot10-6
Na2MoO4middot2H2O 40middot10-6
CuSO4middot5H2O 20middot10-7
MnSO4middotH2O 10middot10-6
Na2SeO4 12middot10-5
CoCl2middot6H2O 50middot10-6
NiCl2middot6H2O 80middot10-6
NaCl 10middot10-5
L-arginine 11middot10-4
L-serine 19middot10-4
L-glutamic acid 14middot10-4
Lactate or formate 4-10middot10-3
Fe(III) 8-27middot10-3
325 Analytical methods
Fe(II) was determined using a modified phenanthroline method (Fadrus amp Maly
1975) The total amount of Fe(III) coated on calcite and sand was determined by
atomic absorption spectroscopy following dissolution in 6 M HCl(aq) for 24 h
33 Results and discussion
331 Fe(II) production and suspension colour changes
Strongly chelating agents such as EDTA were omitted from the culture medium in
order to prevent complexation of Fe(II) and Fe(III) which interferes with
precipitation of Fe(II) and Fe(II)-Fe(III) mineral phases Within 1-2 weeks after
inoculation Shewanella algae BrY produced green mineral phases in media
suspensions containing lepidocrocite and goethite as coatings on calcite and 4-10
mM formate or lactate (Figure 31) The formation of the green precipitates was
generally slower for the lepidocrocite coating than for the goethite coating The
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 51
blue-green colours of the phases produced most likely originate from Fe(II)-Fe(III)
charge transfer between adjacent Fe(II) and Fe(III) ions in edge-shared octahedra
(Faye et al 1968) Dark brown and blackish products were formed when the
bacteria were inoculated on HFO coated sand (Figure 32)
Figure 31 Culture tubes containing a) goethite and b) lepidocrocite coated calcite in culture
medium The left tubes of the pair were not inoculated whereas the right tubes were
photographed 5 months after inoculation with S algae BrY Experimental conditions [formate]0
= 4 mM [Fe(III)]0 = 8 mM 995 N205 CO2(g) pH 76
52 Chapter 3
Figure 32 Culture tubes containing HFO coated sand in culture medium Tubes 1 and 2 to the
left were not inoculated whereas tubes 3-5 to the right were photographed a) 13 days and b) 21
days after inoculation with S algae BrY Experimental conditions [lactate]0 = 10 mM [Fe(III)]0
= 25 mM 100 N2(g) pH 55-60
The green and black colours did not change to other colours (observed for gt1
year) indicating that the microbial Fe(III) reduction ceased at these mineral stages
The concentrations of dissolved ferrous iron (Fe(II)sol) estimated during Fe(III)
bioreduction were generally low (Figure 33) When comparing the final Fe(II)sol
amounts produced and the slopes of the Fe(II)sol formation curves for HFO
goethite and lepidocrocite in Figure 33 it can be seen that the final Fe(II)sol
amount and the Fe(II)sol production rate both follow the order HFO gt goethite gt
lepidocrocite at similar cell densities regardless of the carbon source applied This
suggests that bioreduction by S algae BrY is more facile for HFO than for goethite
and lepidocrocite at the experimental conditions employed here It should be noted
that the final Fe(II)sol amounts and the Fe(II)sol production rates reported in this
work have not been normalised with respect to the specific surface areas of the iron
oxides and coating-bearing solids applied The reactivity trend is consistent with
previous findings demonstrating higher reducibility of natural and poorly
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 53
crystalline Fe(III) oxides as compared to synthetic crystalline Fe(III) oxides
(Zachara et al 1998) The authors ascribed these differences in reducibility to
differences in particle size surface area and crystal defects of the Fe(III) oxides In
some cases the dissolved Fe(II) concentration decreased again with time (Figure
33 b-d) This indicates that the Fe(II) formed was incorporated into solid phases
forming andor adsorbed onto the calcite sand or Fe(III) oxide surfaces The solid
Fe(II) concentrations were not estimated spectrophotometrically The solid
material was generally low in total iron and therefore saving it for Moumlssbauer
spectroscopic analysis was given highest priority
54 Chapter 3
Figure 33 Time course of dissolved Fe(II) production during bioreduction of HFOsand
goethitecalcite and lepidocrocitecalcite by S algae BrY Experimental conditions [Fe(III)]0 =
25 mM [formate]0 = 10 mM (a-c) or [lactate]0 = 10 mM (d-f)
No color change and no Fe(II) production were observed in mineral suspensions
lacking either a carbon source or S algae BrY cells (data not shown)
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 55
332 Identification of solid iron phases
The purity of the Fe(III) oxides used in the experiments were investigated by
transmission Mossbauer spectroscopy (Figure 34) Single (or strongly dominating)
sextets in the spectra with magnetic hyperfine fields of 48 1 505 and 45 5 Tat 5 K
demonstrated the purity of the HFO goethite and lepidocrocite samples
respectively A minor impurity of goethite in the lepidocrocite sample was
resolved in the spectrum measured at 80 K (not shown for pure sample but can be
seen as a magnetically ordered sextet in Figure 35c) No Fe(II)-containing
components were detected
a)
c)
middot 12 -8 -4 4 8 12
Velocity (mmls
bull middot12 -8 -4 0 4 12
Velocity (mmls)
b)
middot12 -8 -4 4 8 12
Velocity (mmls
Figure 34 Transmission Mossbauer spectra measured at 5 K of a) HFO (magnetic hyperfine
field of 481 T isomer shift of 048 1nrns-1 negligible quadrupole shift and line width of outer
lines 110 rmns-1) b) goethite (magnetic hyperfine field of 505 T isomer shift of 049 rmns-1
quadmpole shift of -013 1nrns-1 and line width of outer lines 042 rnrns-1
) and c) lepidocrocite
56 Chapter 3
(magnetic hyperfine field of 455 T isomer shift of 050 mms-1 quadrupole shift of ndash001 mms-1
and line width of outer lines 060 mms-1) prior to inoculation Simple Lorenztian fits are shown
The oxidation state and coordination of Fe in the microbially reduced HFO
goethite and lepidocrocite samples were also examined by transmission Moumlssbauer
spectroscopy (Figure 35) The bioreduced HFO goethite and lepidocrocite samples
cultured on formate contained Fe(II) holding similar coordination as inferred from
the similarity of the hyperfine parameters (see legend in Figure 35) but different
relative intensities (72 19 and 71 respectively) The major part of the Fe(III)
remaining in the bioreduced samples were coordinated similarly to the Fe(III)
present in the initial Fe(III) oxide The coordination of Fe(II) in the bioreduced
lepidocrocite samples cultured on lactate was slightly different (a smaller
quadrupole splitting of 288 mms-1 for the ferrous component dominates ndash data not
shown) The exact mineralogy of the Fe(II) present in the green phases was not
fully resolved but its coordination is very akin to one of the Fe(II) sites in vivianite
(see Chapter 2 this work) and synthetic green rusts (Koch 1998) These findings
agree with other reports on the bioformation of vivianite and green rusts by
Shewanella putrefaciens CN32 although the evidence provided may be discussed
(Fredrickson et al 1998 Glasauer et al 2003 Parmar et al 2001) Our
Moumlssbauer data on the green phases did not allow for a detailed account of the type
of green rust produced However when considering solution composition (see
Table 31) and the high affinity of GR interlayers for carbonate it is reasonable to
assume that carbonate GR was formed (Hansen amp Taylor 1991) Due to the high
amounts of Fe(III) in the oxides present in the experiments it was difficult to probe
a possible content of Fe(III) in the vivianite with certainty The differences in the
number of Fe(II) positions in the Moumlssbauer spectra and particular the different
temperatures at which magnetic ordering takes place can be employed in order to
distinguish between green rust and vivianite Preliminary Moumlssbauer data obtained
for the blackish precipitates formed in the HFOsand suspensions indicate that they
hold no resemblance to magnetite eventhough the colour suggests so On the
contrary the black precipitates seemed to be more similar to synthetic green rusts
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 57
Mossbauer spectroscopic measurements are currently underway in order to resolve
the Fe(II) coordinations in the greenish and blackish phases
a) b)
c c g Q e- e 0 0 1l 1l
-~ bull ~
iii ~ Qi bull a bull
-12 -8 4 0 4 12
4 -3 -2 -1 0 1 2 4 Velocity (mmts) Velocity ( rmis)
c)
-12 -8 4 8 12
v elocity (m mis)
Figure 35 Transmission Mossbauer spectra of the black and green phases fo1med within 1-2
weeks after inoculation of a) HFO (measured at 130 K) b) goethitecalcite (measured at 80 K)
and c) lepidocrocitecalcite (measured at 80 K) with S algae BrY Experimental conditions
[fo1m ate]0 = 4 mM [Fe(III)]o = 8 mM 995 Ni05 C02(g) pH 76 The quadrnpole
splittings and isomer shifts for the Fe(II) components in the three systems are a) 293 nnns-1 and
126 mmsmiddot1 b) 308 rmnsmiddot1 and131 rmnsmiddot1 and c) 322 mmsmiddot1 and 132 mmsmiddot1 Simple Lorenztian
fits are shown
The evidence provided in many of the studies proposing biogenic GRs is not all
too convincing but it strongly suggests the probability of microbially produced GR
being present The challenge encountered is that when present in low
58 Chapter 3
concentrations especially in mixtures including other iron minerals it is very
difficult to identify GRs using conventional solid phase analysis methods even
with Moumlssbauer spectroscopy At least two complementary methods such as XRD
and Moumlssbauer spectroscopy are required for proper identification and
characterization of GRs However in this work the solid materials were generally
too low in total iron to allow for XRD analysis Moreover the highly
heterogeneous suspensions were dominated by the coating-bearing sand and calcite
solids Electron micrographs including energy dispersive X-ray spectroscopy
suspension colour and mineral stability calculations do not suffice as evidence The
most convincing evidence provided so far involves an atypical GR-CO3 with an
Fe(II)Fe(III) ratio of 1 (Ona-Nguema et al 2002aampb) This GR-CO3 was formed
as a result of lepidocrocite reduction by Shewanella putrefaciens CIP 8040 at
conditions comprising high nutrient levels (50-75 mM formate) high Fe(III)
concentrations (80-300 mM) and a synthetic electron shuttle (100 microM
anthraquinone-26-disulfonate (AQDS)) at initial pH 75 Hence the results
reported during recent years suggest that microbial formation of GR may be
possible The results presented here indicate that GRs may be produced
microbially at conditions including low carbon and Fe(III) concentrations as well
as the exclusion of synthetic electron shuttles and pH buffers
333 Factors controlling the identity of the secondary iron minerals
In general one would expect that biogenic minerals have chemical compositions
and crystal habits similar to those produced by nonenzymatic processes as they are
governed by the same equilibrium principles In fact since the latter stages of
mineralization are inorganically driven and the secondary Fe(II)-containing
minerals are formed indirectly by electron transfer outside the bacterial cell and not
directly inside the bacterial cell the type of iron mineral formed is a function of the
environmental conditions in which the bacteria live ie the same microorganism
form different minerals in different environments The key factors controlling the
identity of the secondary iron minerals include medium composition electron
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 59
donor and electron acceptor concentrations mineral aging as well as adsorbed ions
(Zachara et al 2002) The main factor controlling the nature of the secondary
mineral products are the respiration-driven biogenic Fe(II) supply rate and
magnitude and its surface reaction with the residual oxide and other sorbed ions
(Zachara et al 2002) Especially solution and medium composition have a strong
impact on the nature of the Fe(II)-containing biomineralization products forming
Accordingly siderite (FeCO3) and magnetite (Fe3O4) were the secondary solid
phases resulting from the bioreduction of ferrihydrite by Shewanella putrefaciens
CN32 in bicarbonate buffered medium (pH 71) containing no phosphate whereas
siderite and vivianite were the secondary iron minerals dominating in bicarbonate
buffered medium (pH 74) containing 4 mM phosphate (Zachara et al 2002) This
is explained by the inhibiting effect of phosphate on crystallization of magnetite
(Couling amp Mann 1985 Fredrickson et al 1998)
334 Factors controlling the rate and extent of Fe(III) bioreduction
In this study the extent of Fe(III) bioreduction was estimated to 19-72 by
transmission Moumlssbauer measurements In fact complete microbial reduction of
crystalline Fe(III) minerals has never been observed in laboratory batch culture
studies (Roden amp Urrutia 2002) It has been found that Fe(II) does not inhibit
Fe(III) reductase activity through an enzyme inhibition mechanism (Roden amp
Urrutia 2002) Hence other chemical andor physiologic factors control the
bioavailability of solid Fe(III) phases and thus the extent of their microbial Fe(III)
reduction The initial rate and long-term extent of microbial reduction of
amorphous and crystalline Fe(III) oxides including HFO goethite and hematite
were linearly correlated with oxide surface area (Roden amp Zachara 1996)
Association of biogenic Fe(II) with Fe(III) oxide and DIRB cell surfaces reduced
the long-term extent of crystalline Fe(III) oxide bioreduction (Roden amp Urrutia
2002) These results were explained by Fe(II) surface complexes andor
precipitates creating a passive Fe(II)-bearing surface film providing direct physical
interference with the electron transfer from the DIRB cells to Fe(III) However the
60 Chapter 3
real mechanisms whereby the surface-associated Fe(II) inhibits Fe(III) oxide
bioreduction are unclear but they most likely involve both kinetic and
thermodynamic constraints on the electron transfer Culture medium composition
in particular the presence and the concentration of phosphate as well as Fe(II)
chelating ligands also exert an influence on the extent of the microbial reduction
of Fe(III) oxides The extent of Fe(III) bioreduction was inhibited by high
phosphate concentrations which favoured surfacebulk precipitation processes
(Urrutia et al 1998) The carbon sources most frequently applied in Fe(III)
bioreduction studies include malate citrate and other di- and tricarboxylic acids
which are not only easily metabolizable carbon sources but also eminent Fe(II) and
Fe(III) chelators In this study we employed formate and lactate as carbon sources
since they are the weakest complexing agents of Fe(II) and Fe(III) among the C1-
C3 monocarboxylic acids (Martell 1964) Thus we expect less dissolution of
prevailing precipitates by complexation as compared to other studies
34 Conclusions
This work demonstrated that Shewanella algae BrY reduced 19-72 of initial
Fe(III) when grown in culture media containing 4-10 mM formate or lactate and 8-
27 mM Fe(III) applied as goethite or lepidocrocite coatings on calcite (pH 76) or
HFO coatings on sand (pH 55-60) Within 1-2 weeks after inoculation green
mineral phases were produced in the goethitecalcite and lepidocrocitecalcite
mineral systems whereas black precipitates formed in the HFOsand suspensions
Moumlssbauer spectroscopic analyses indicated that the greenish and blackish phases
most likely were mineral mixtures dominated by vivianite and green rust Thus the
results indicate that GRs may be produced microbially at conditions including low
carbon and Fe(III) concentrations as well as the exclusion of synthetic electron
shuttles and pH buffers
Acknowledgments
We would like to thank Dr R Gerlach for providing us the Shewanella algae BrY culture and
Dr C B Koch for performing the Moumlssbauer analyses
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 61
References Al-Agha MR Burley SD Curtis CD Esson J (1995) Complex cementation textures and authigenic mineral assemblages in recent concretions from the Lincolnshire Wash (east coast UK) driven by Fe(0) to Fe(II) oxidation Journal of the Geological Society 152 157-171 Bernal JD Dasgupta DR Mackay AL (1959) The oxides and hydroxides of iron and their structural inter-relationships Clay Minerals Bulletin 4 15-30 Caccavo Jr F Blakemore RP Lovley DR (1992) A hydrogen-oxidizing Fe(III)-reducing microorganism from the Great Bay Estuary New Hampshire Applied and Environmental Microbiology 58 3211-3216 Caccavo Jr F Das A (2002) Adhesion of dissimilatory Fe(III)-reducing bacteria to Fe(III) minerals Geomicrobiology Journal 19 161-177 Couling SB Mann S (1985) The influence of inorganic phosphate on the crystallization of magnetite (Fe3O4) from aqueous solution Journal of the Chemical Society Chemical Communications 1713-1715 Das A Caccavo Jr F (2000) Dissimilatory Fe(III) oxide reduction by Shewanella alga BrY requires adhesion Current Microbiology 40 344-347
Das A Caccavo Jr F (2001) Adhesion of the dissimilatory Fe(III)-reducing bacterium Shewanella alga BrY to crystalline Fe(III) oxides Current Microbiology 42 151-154 Fadrus H Maly J (1975) Suppression of iron(III) interference in the determination of iron(II) in water by the 110-phenanthroline method The Analyst 100 549-554 Faye GH Manning PG Nickel EH (1968) The polarized optical absorption spectra of tourmaline cordierite chloritoid and vivianite ferrous-ferric electronic interaction as a source of pleochroism American Mineralogist 53 1174-1201 Fredrickson JK Zachara JM Kennedy DW Dong H Onstott TC Hinman NW Li S (1998) Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium Geochimica et Cosmochimica Acta 62 3239-3257 Geacutenin JMR Olowe AA Benbouzid-Rollet ND Prieur D Confente M Resiak B (1991) The simultaneous presence of green rust 2 and sulfate reducing bacteria in the corrosion of steel sheet piles in a harbour area Hyperfine Interactions 69 875-878 Geacutenin JMR Bourrieacute G Trolard F Abdelmoula M Jaffrezic A Refait Ph Maitre V Humbert B Herbillon A (1998) Thermodynamic equilibria in aqueous suspensions of synthetic and natural Fe(II)-Fe(III) green rusts Occurrences of the mineral in hydromorphic soils Environmental Science and Technology 32 1058-1068 Glasauer S Weidler PG Langley S Beveridge TJ (2003) Controls on Fe reduction and mineral formation by a subsurface bacterium Geochimica et Cosmochimica Acta 67 1277- 1288 Hansen HCB Taylor RM (1991) The use of glycerol intercalates in the exchange of CO3
2- with SO4
2- NO3- or Cl- in pyroaurite-type compounds Clay Minerals 26 311-327
62 Chapter 3
Hungate RE (1969) A roll tube method for cultivation of strict anaerobes Methods in Microbiology 3B 117-132 King GM (1990) Effects of added manganic and ferric oxides on sulfate reduction and sulfide oxidation in intertidal sediments FEMS Microbiology Ecology 73 131-138 Koch CB (1998) Structures and properties of anionic clay minerals Hyperfine Interactions 117 131 -157 Kostka J Nealson KH (1998) Isolation cultivation and characterization of iron- and manganese reducing bacteria In Techniques in Microbial Ecology Burlage RS Atlas R Stahl D Geesey G Sayler G (eds) Oxford University Press Inc 58-78 Kukkadapu RK Zachara JM Smith SC Fredrickson JK Liu C (2001) Dissimilatory bacterial reduction of Al-substituted goethite in subsurface sediments Geochimica et Cosmochimica Acta 65 2913-2924
Liu C Zachara JM Gorby YA Szecsody JE Brown CF (2001) Microbial reduction of Fe(III) and sorptionprecipitation of Fe(II) on Shewanella putrefaciens strain CN32 Environmental Science and Technology 35 1385-1393 Lovley DR (1991) Dissimilatory Fe(III) and Mn(IV) reduction Microbiological Reviews 55 259-287 Lovley DR (1997) Microbial Fe(III) reduction in subsurface environments FEMS Microbiology Reviews 20 305-313 Lovley DR Phillips EJP (1986) Organic matter mineralization with reduction of ferric iron in anaerobic sediments Applied and Environmental Microbiology 51 683-689 Lovley DR Phillips EJP Lonergan DJ (1991) Enzymatic versus nonenzymatic mechanisms for Fe(III) reduction in aquatic sediments Environmental Science and Technology 25 1062-1067 Lower SK Hochella Jr MF Beveridge TJ (2001) Bacterial recognition of mineral surfaces Nanoscale interactions between Shewanella and α-FeOOH Science 292 1360-1363 Martell AE (1964) Stability constants of metal-ion complexes Part 2 Organic including macromolecule ligands The Chemical Society London 2 ed Miller TL Wolin MJ (1974) A serum bottle modification of the Hungate technique for cultivating obligate anaerobes Applied Microbiology 27 985-987 Nealson KH Saffarini D (1994) Iron and manganese in anaerobic respiration Environmental significance physiology and regulation Annual Review of Microbiology 48 311-343 Nevin KP Lovley DR (2000) Lack of production of electron-shuttling compounds or solubilization of Fe(III) during reduction of insoluble Fe(III) oxide by G metallireducens Applied and Environmental Microbiology 66 2248-2251 Nevin KP Lovley DR (2002) Mechanisms for Fe(III) oxide reduction in sedimentary
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 63
environments Geomicrobiology Journal 19 141-159 Nielsen A (1976) Hvid groslashn og sort rust Beskrivelse af en korrosionsskade paring et svoslashmmebassin Nordisk Betong 2 21-24 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002a) Iron (IIIII) hydroxycarbonate green rust formation and stabilization from lepidocrocite bioreduction Environmental Science and Technology 36 16-20 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002b) Microbial reduction of lepidocrocite γ-FeOOH by Shewanella putrefaciens The formation of green rust Hyperfine Interactions 139140 231-237 Parmar N Gorby YA Beveridge TJ Ferris FG (2001) Formation of green rust and immobilization of nickel in response to bacterial reduction of hydrous ferric oxide Geomicrobiology Journal 18 375-385 Roden EE Urrutia MM (2002) Influence of biogenic Fe(II) on bacterial crystalline Fe(III) oxide reduction Geomicrobiology Journal 19 209-251 Roden EE Zachara JM (1996) Microbial reduction of crystalline iron(III) oxides Influence of oxide surface area and potential for cell growth Environmental Science and Technology 30 1618-1628 Roh Y Lee SY Elless MP (2000) Characterization of corrosion products in the permeable reactive barriers Environmental Geology 40 184-194 Schwertmann U Cornell RM (1991) Iron oxides in the laboratory Preparation and characterization VCH Verlagsgesellschaft mbH Weinheim Thamdrup B (2000) Bacterial manganese and iron reduction in aquatic sediments In Advances in Microbial Ecology (Schink B ed) Kluwer AcademicPlenum Publishers New York 41-84 Trolard F Abdelmoula M Bourrieacute G Humbert B Geacutenin JMR (1996) Mise en eacutevidence dun constituant de type rouilles vertes dans les sols hydromorphes Proposition de lexistence dun nouveau mineacuteral la fougeacuterite Geacuteosciences de surface Comptes Rendus de LrsquoAcademie des Sciences 323 1015-1022 Tuovinen OH Button KS Vuorinen A Carlson L Mair DM Yut LA (1980) Bacterial chemical and mineralogical characteristics of tubercles in distribution pipelines Journal of the American Water Works Association 72 626-635 Turick CE Caccavo Jr F Tisa LS (2003) Electron transfer from Shewanella algae BrY to hydrous ferric oxide is mediated by cell-associated melanin FEMS Microbiology Letters 220 99-104 Urrutia MM Roden EE Fredrickson JK Zachara JM (1998) Microbial and surface chemistry controls on reduction of synthetic Fe(III) oxide minerals by the dissimilatory iron- reducing bacterium Shewanella alga Geomicrobiology 15 269-291 Venkateswaran K Moser DP Dollhopf ME Lies DP Saffarini DA MacGregor BJ Ringelberg DB White DC Nishijima M Sano H Burghardt J Stackebrandt E
64 Chapter 3
Nealson KH (1999) Polyphasic taxonomy of the genus Shewanella and description of Shewanella oneidensis sp nov International Journal of Systematic Bacteriology 49 705-724 Zachara JM Fredrickson JK Li S Kennedy DW Smith SC Gassman PL (1998) Bacterial reduction of crystalline Fe3+ oxides in single phase suspensions and subsurface materials American Mineralogist 83 1426-1443
Zachara JM Kukkadapu RK Fredrickson JK Gorby YA Smith SC (2002) Biomineralization of poorly crystalline Fe(III) oxides by dissimilatory metal reducing bacteria (DMRB) Geomicrobiology Journal 19 179-207
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 65
4 Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust The Effect of Probe Compound Charge
Abstract
Layered iron(II)-iron(III)-hydroxides (green rusts) may play an important role in
controlling the fate and transport of many organic and inorganic contaminants in
iron-rich suboxic soils and sediments Unlike most other iron oxides green rusts
(GRs) contain not only external Fe(II) reactive sites at the basal planes and at the
edges but also internal sites in the space between consecutive Fe(II)-Fe(III)
hydroxide layers The GR interlayer thickness is a function of both the size and the
charge of the interlayer anion Whether a given oxidant has access to the internal
sites in GRs is dependent on its charge We investigated the reductive
transformation of nitroaromatic compounds (NACs) by GR-SO4 and studied the
effect of NAC charge on the reactivity towards GR-SO4 A series of structurally
closely related compounds with different charge properties including nitrobenzene
4-nitrotoluene 4-chloronitrobenzene and 4-nitrophenylacetic acid were used as
probe compounds The NACs were completely reduced to their corresponding
anilines by GR-SO4 The reactions followed pseudo 1 order kinetics with respect
to NAC and the surface area-normalised pseudo 1 order rate constants obtained
were 016ndash465middot10-4 s-1middotm-2middotL at [Fe(II)GR]0 = 103-1260 mM [NAC]0 = 20-102
microM and pH 84-86 Neither mass transfer control nor surface saturation kinetics
could account for the rather unexpected similarity of the surface area-normalised
pseudo 1 order rate constants obtained for the reduction of the neutral and anionic
NACs by GR-SO4 These observations suggest that the anionic NACs did not have
an enhanced access to the inner or outer Fe(II)-GR reactive sites as compared to
the neutral NACs Hence the reaction between NAC and GR-SO4 primarily took
place at the edges of GR-SO4
66 Chapter 4
41 Introduction
Layered iron(II)-iron(III)-hydroxides (green rusts) are intermediate phases formed
by partial oxidation of Fe(II) or partial reduction of Fe(III) In neutral and weakly
alkaline solutions the oxidation of dissolved Fe(II) always passes through solid
green rust (GR) phases (Bernal et al 1959) This agrees with the natural GR
occurrences found in suboxic non-acid iron-rich environments such as
hydromorphic soils and intertidal sediments (Al-Agha et al 1995 Trolard et al
1996 Genin et al 1998) In addition GRs have been found as corrosion products
in numerous engineered systems ie a pipeline distribution system for drinking
water steel sheet piles in marine sediments reinforced concrete (ferro-concrete)
and permeable reactive barriers of zero-valent iron implemented for on-site
remediation of organic and inorganic contaminants (Tuovinen et al 1980 Nielsen
1976 Genin et al 1991 Roh et al 2000) Furthermore the microbial formation
of GRs resulting from bioreduction of Fe(III) oxides by strains of the anaerobic
dissimilatory Fe(III) reducing bacteria Shewanella putrefaciens has been reported
increasingly over the last 5 years (Fredrickson et al 1998 Kukkadapu et al 2001
Parmar et al 2001 Ona-Nguema et al 2002 Glasauer et al 2003) Moreover
the biotic formation of GRs by anaerobic denitrifying Fe(II) oxidizing bacteria has
been proposed but proper identification of the GR phases still lacks (Chaudhuri et
al 2001) All these indications of microbial GR formation infer the importance of
GRs as a link between geochemical and biological processes in natural systems
GRs form platy crystals with the general formula [FeII(6-
x)FeIIIx(OH)12]x+[(A)xnmiddotyH2O]x- where x = 09 - 42 A is an n-valent anion eg
CO32- Clndash or SO4
2- and y is the number of water molecules in the interlayer The
crystal structure consists of positively charged hydroxide sheets with Fe(II) and
Fe(III) cations having octahedral hydroxyl coordination The Fe(III) in the
hydroxide layers creates a net positive charge which is balanced by hydrated
anions in the interlayers (Figure 41) The interlayers have a higher affinity for
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 67
divalent anions than for monovalent anions (Miyata 1983) Among the 3 most
common GR forms the affinity follows the order CO32- gt SO4
2- gt Clndash The extreme
preference shown for carbonate hinders further access and exchange except under
certain conditions (Hansen amp Taylor 1991) Non-carbonate forms are readily
exchanged with other anions when dispersed in a solution containing the
exchanging anion (Mendiboure amp Schoumlllhorn 1986)
c
ba
Figure 41 Green rust layer structure The hydroxide layers and the interlayers are connected by
hydrogen bonds (not shown) The GR-SO4 crystal structure is characterised by the hexagonal
unit cell having a = b = 055 nm and c = 110 nm (Simon et al 2003) The unit cell consists of
one double layer (a double layer is a hydroxide layer and an interlayer) ie the hydroxide layer
constitutes 049 nm and the interlayer 061 nm in GR-SO4
The GR interlayer thickness (extending in the c axis direction Figure 41) is a
function of both the size and the charge of the interlayer anion Tetrahedrally
coordinated anions like sulphate lead to larger interlayer distances than smaller
monoatomic anions like chloride or planar ions like carbonate (Mendiboure amp
Schoumlllhorn 1986) Not only size but also charge density plays a role for the
interlayer spacing That is for anions having the same number of valence
electrons anions with smaller ionic radii (higher electron density) are bound more
strongly and therefore result in smaller interlayer spacings
Due to their layered structures anionic interlayers and high specific surface areas
GRs represent reactive ion exchangers and sorbents of anions eg arsenate
selenate and phosphate (Myneni et al 1997 Hansen amp Poulsen 1999 Randall et
al 2001) In addition GR may incorporate heavy metal cations by isomorphic
68 Chapter 4
substitution into the GR hydroxide layers (Tamaura 1985 Tamaura 1986)
Furthermore GRs have been shown to reduce a range of inorganic contaminants
such as nitrite nitrate selenate chromate uranyl pertechnetate and the transition
metals AgI AuIII CuII and HgII as well as organic pollutants including halogenated
ethanes ethenes and methanes (Hansen et al 1994 Hansen et al 1996 Myneni et
al 1997 Erbs et al 1999 Loyaux-Lawniczak et al 1999 Cui amp Spahiu 2002
Lee amp Batchelor 2002b Heasman et al 2003 OrsquoLoughlin et al 2003a amp 2003b
Pepper et al 2003 Elsner et al 2004 OrsquoLoughlin amp Burris 2004) Thus through
sequestration and reductive transformation GRs may play an important role in
controlling the fate and transport of contaminants in suboxic soils and sediments
In a previous study the effects of interlayer anion and Fe(II)Fe(III) ratio in GRs
on the reduction rate of nitrate were investigated (Hansen et al 2001) It was
found that the rate of nitrate reduction to ammonium increased with increasing
Fe(II)Fe(III) ratio and decreased when exchanging a monovalent interlayer anion
(chloride) with a divalent anion (sulphate) The results suggest that for anionic
oxidants like nitrate Fe(II) within the hydroxide layer is available from the outside
basal planes and from the edges as well as through the interlayer under certain
conditions (Figure 42) However oxidants with different charge properties
(cations neutral molecules) may exhibit different affinities for the various reactive
Fe(II) sites present in GR
As the reactive sites are located inat the Fe(II)-Fe(III) hydroxide layers the rate of
reaction depends on the hydroxide layer area which can be accessed by the
oxidant If the oxidant can exchange with the interlayer anion reaction can take
place both at outer and inner surfaces of the GR particles and in total more
reactive sites are available for the reaction However it was found that nitrate
cannot penetrate the interlayer when carbonate or sulphate constitutes the
interlayer anions (Hansen amp Koch 1998) This agrees with the fact that the
interlayers have a lower affinity for monovalent anions than for divalent anions
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 69
(Miyata 1983) However when nitrate was forced into the interlayer by extracting
the interlayer sulphate through precipitation of barium sulphate outside the GR
particles the observed 40 fold increase in rate of nitrate reduction almost equalled
the increase in exposed surface area of the Fe(II)-Fe(III) hydroxide layers (Hansen
amp Koch 1998) From these observations it is expected that the rate of reaction
depends on the particular GR form the crystallite size and the ease with which an
oxidant can exchange with An- in the GR interlayer (Figure 42) Due to
electrostatic interactions we expect anions to be attracted to the positively charged
outer and inner surfaces to a higher degree than cations and neutral compounds If
this theory holds we may expect oxidants with similar intrinsic reactivity (similar
one-electron reduction potentials) to react in the following order anionic gt non-
charged gt cationic (Figure 43) granting that we do not normalise the rate
constants with respect to the amount of oxidant sorbed
Figure 42 Reaction of a probe compound at basal planes at edges and in the interlayer of GR
The hypothesis only holds in cases where the oxidants possess the same intrinsic
reactivities If the relative reactivities of the probe compounds differ greatly from
what would be expected when considering only their reduction potentials
70 Chapter 4
compound specific effects such as charge properties might explain this and the
relative reactivities may follow a pattern like the one depicted in Figure 43
Figure 43 Hypothetical plot of observed reaction rate constants for the reactions between
cationic neutral and anionic probe compounds and GR-SO4 assuming that the oxidant charge
controls its reactivity towards GR
In this work we investigated the reductive transformation of NACs by GR-SO4
Furthermore the effect of NAC charge on the rate of reaction and the possible
access to the internal reactive sites in GR-SO4 were assessed When quantifying
Fe(II) in GRs by means of acid digestion it is not possible to distinguish between
the reactive sites accessible from the outside (at the basal planes or at the edges) or
through the interlayer However we designed an indirect method to gain insight
into the relative importance of the various reactive sites by using a series of
structurally closely related compounds with different charge properties as ldquoreactive
probesrdquo Neutral and anionic probes were needed in order to access all Fe(II)
reactive sites According to our hypothesis cationic and non-charged oxidants
should provide information about the reactivity of the outer Fe(II) reactive sites in
GR whereas the anionic oxidants should provide information about the reactivity
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 71
of both outer and inner Fe(II) reactive sites We chose five nitro aromatic
compounds (NACs) - representing an important group of reducible organic
pollutants - as probe compounds (Figure 43) This class of compounds is not only
of great environmental concern but also comprises suitable model compounds for
studying redox reactions potentially relevant in the environment Moreover they
react readily with Fe(II) surface species associated with iron oxides or clay
minerals transforming them into well-defined easily detected products allowing
mass and electron balances to be established (Hofstetter et al 2003 Klausen et al
1995 Schultz amp Grundl 2000) Our main goals were to establish the rate law and
estimate the surface area-normalised reaction rates for the reaction of the probe
compounds with GR-SO4 in order to assess the importance of the Fe(II) reactive
sites accessible through the interlayer relative to the Fe(II) reactive sites accessible
at the outer surface in GR-SO4
4 2 Materials and methods
All handling and sampling of solutions and suspensions were carried out under
strict anoxic conditions All chemicals were pa quality or better Methanolic stock
solutions (5 mM) of nitrobenzene (NB) 4-nitrotoluene (4-NT) 4-
chloronitrobenzene (4-CNB) and 4-nitrophenylacetic acid (4-NPA) were prepared
in deoxygenated methanol Several attempts to synthesize the cationic probe
compound 4-(NNN-trimethylammonium)-nitrobenzene failed and therefore the
study had to be carried out with only neutral and anionic oxidants The sulphate
GR form was chosen as it is the most stable form and thus the easiest to work
with in the lab
421 Synthesis of GR-SO4
GR-SO4 was synthesized by controlled air oxidation of an FeSO4 solution at a
constant pH of 700 according to the procedure given by Koch amp Hansen (1997)
The GR-SO4 suspension was washed with deoxygenated deionised water and
72 Chapter 4
separated on a folding filter redispersed in deoxygenated 25 mM Na2SO4(aq) in
order to stabilize the GR-SO4 and prevent it from transforming into magnetite
spontaneously Washing separation and redispersion of the GR-SO4 suspension
were conducted in an anoxic glove box (Coy Laboratory Products Inc) All
suspensions and solutions were deoxygenated by Ar-purging (999998 Ar
Carbagas)
422 Mineral characterisation
The identity and purity of the GR-SO4 suspensions were examined by means of X-
ray diffraction measurements The XRD analyses were performed on a Scintag
XDS 2000 using Cu Kα radiation (45 kV 40 mA) Glycerol smears made
according to Hansen (1989) were scanned between 6 and 80 deg2θ with a scan speed
of 1 deg2θmin
423 Lyophilization and determination of specific surface area
Simple air-drying of the GR mineral in the glove box resulted in big flakes with
very low surface areas hence a more suitable lyophilization method was adopted
from Elsner et al (2004) The GR-SO4 suspensions were lyophilised using
Schlenk-type glassware The set-up consisted of a 1 L round bottom flask and a
200 mL glass finger connected by a crescent-shaped bridge equipped with an
evacuation outlet and a stopcock All ground joints and fittings were attached using
high-vacuum grease The washed and resuspended GR-SO4 suspensions were
filled into the glass finger and the freeze-drying apparatus was assembled and
closed before taking it out of the glove box The suspension was frozen by
carefully submerging the lower part of the glass finger into liquid nitrogen for a
few hours Subsequently the evacuation outlet was connected to a vacuum pump
by a metal hose Following a short evacuation of the metal hose the lyophilization
apparatus was evacuated for several minutes by gently opening the stopcock The
evacuation was terminated by closing the stopcock and disconnecting the vacuum
pump The apparatus position was now reversed by removing the glass finger from
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 73
and immersing the round bottom flask into liquid nitrogen As any other
lyophilization method this method depends on sublimation of the ice from the
frozen sample and its recondensation on a cool surface in this case the round
bottom flask Generally it took 1-2 d for the mineral to dry The apparatus was
disassemled in the glove box and the fine powder stored under anoxic conditions
The specific surface area (SSA) of GR-SO4 was determined by the BET multi-
point method using N2 adsorption (Brunauer et al 1938) Powder samples were
filled into sample burettes in the glove box and the generously greased stopcocks
closed Samples and burettes were evacuated prior to connecting them to the BET-
instrument (Sorptomatic 1990 Fisons)
424 Estimation of the one-electron reduction potential for 4-NPA
Kinetic experiments in 100 mL Viton stoppered and alu-crimp capped serum vials
were carried out under the exclusion of oxygen as described by Hofstetter et al
(1999) The homogeneous aqueous solutions contained 50 mM KH2PO4 buffer
(pH = 660) 5 mM Na2S redox buffer and 20 microM juglone (8-hydroxy-14-
naphthoquinone) added as deoxygenated 20 mM methanolic stock solution The
solutions were equilibrated at least one day prior to 4-NPA addition To start the
reaction 50 microM 4-NPA was added as deoxygenated 20 mM methanolic stock
solution The vials were agitated on a roller apparatus in the dark at 21ordmC Control
experiments were prepared similarly except for the addition of juglone At
different time intervals aqueous samples were withdrawn with a syringe and
collected in 18 mL HPLC vials containing 100 microL 1 M HCl The sample vials
were sealed with Teflon-coated silicone septa and plastic screw caps and vortexed
for 10 s The samples were stored at -20degC and analysed without further treatment
For comparison experiments with 4-NT were also conducted See Supporting
Information 71 for more information on the one-electron reduction potentials
74 Chapter 4
425 Kinetic experiments
All reactions took place at pH 84-86 where GR-SO4 tends to stabilize and buffer
itself Samples for Fe(II) and XRD analysis were withdrawn prior to reaction Due
to the fast reactions the experiments were conducted in 10 mL single-use
polyethylene syringes (BD Plastipak) in the glove box To start reaction 40-200
microL 5 mM methanolic stock solutions of NAC were quickly added to 10 mL GR-
SO4 suspension (1-12 mM Fe(II)GR) washed and resuspended in 25 mM
Na2SO4(aq) A Teflon filter (25 mm x 02 microm BGB Analytik) was quickly
mounted on the tip of the syringe and the syringe was vigorously shaken between
sampling At different time intervals filtered suspension samples were collected in
18 mL HPLC vials The HPLC vials were sealed with Teflon-coated silicone septa
and plastic screw caps The samples were stored at -20degC and analysed without
further treatment Absorption of NAC in the syringe and in the Teflon filter
evaluated in blank experiments with NAC added to 25 mM Na2SO4(aq) was found
to be negligible
426 Analytical methods
Initial total and aqueous Fe(II) were determined using a modified phenanthroline
method (Fadrus and Maly 1975) In order to determine [Fe(II)aq] and [Fe(II)total] 1
mL filtered (022 microm) and 1 mL unfiltered GR-SO4 suspension samples were
withdrawn and each treated with 18 mL 01 M HCl for at least 30 min From these
acid digests 01 mL was added to 05 mL Fe(II)-reagent and 19 mL deionised
water (DIW) added up The Fe(II) content in GR-SO4 was estimated as the
difference [Fe(II)GR] = [Fe(II)total] - [Fe(II)aq] The NACs and their corresponding
intermediates and products formed during reduction by GR-SO4 were quantified by
reversed-phase HPLC Separation was performed on a LiChrospher 100 RP-18 (5
microm 125 times 4 mm ID) reversed-phase column coupled with a LiChroCART 100 RP-
18 (4 times 4 mm ID) precolumn Analytical conditions were isocratic and the eluent
consisted of 10 mM hydroxylammonium chloride in various DIWCH3OH
mixtures (vv 3565 and pH 70 for 4-NT and 4-CNB 955 and pH 60 for
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 75
4-NPA) The injection volume was 20 microLand the flow-rate 10 mLmin HPLC
analyses were performed using a Gynkotek High Precision Pump M480 Gynkotek
Gina 50 autosampler and a diode array UV detector (340s Gynkotek) UV-VIS
detection was carried out at the wavelengths of maximum absorption for the
various nitro aromatic and aniline analytes
43 Results and discussion
431 Productformation and reaction kinetics
The reduction of the aromatic nitro group occurs via nitroso- and hydroxylamino-
intermediates where 2 electrons are transferred in each reaction step (Figure 44)
0 --0 H OH H H --0 N N N N
2e- 2H+ H20 + 2e-~ 2e- 2H+ H20
~ ~ R R R
Nitro benzene Nitrosobenzene Hydroxylamine Aniline
Figure 44 Reductive transfonnation pathway of NA Cs
Thus in order to reduce 1 Ar-N02 completely to Ar-NH2 6 electrons
corresponding to 6 mol Fe(II) are needed As magnetite was the major iron phase
formed during reaction (XRD results not shown) we assume the following
reaction stoichiometry
The aniline product was not formed at the same rate as the nitro compound
degraded which is consistent with the detection of early eluting hydroxylamine
intermediates during the course of the reaction (Figure 45a amp 45c) No traces of
76 Chapter 4
nitrosobenzene intermediates or side products such as azoxy- azo- or
hydrazobenzene were found In Figure 45 pseudo 1 order kinetic plots and ln
[Ar-NO2]t[Ar-NO2]0) versus time plots for the neutral probe compounds 4-CNB
and 4-NT are shown as examples The plots for NB and 4-NPA look similar
Figure 45 a Concentration versus time plots for reaction of GR-SO4 with 4-CNB ([Fe(II)GR]0 =
126 mM [4-CNB]0 = 30 microM) b ln [Ar-NO2]t[Ar-NO2]0) versus time plots for reaction of GR-
SO4 with 4-CNB ([Fe(II)GR]0 = 126 mM + [4-CNB]0 = 30 microM [Fe(II)GR]0 = 63 mM + [4-
CNB]0 = 50 microM) c Concentration versus time plots for reaction of GR-SO4 with 4-NT
([Fe(II)GR]0 = 131 mM [4-NT]0 = 20 microM) d ln [Ar-NO2]t[Ar-NO2]0) versus time plots for
reaction of GR-SO4 with 4-NT ([Fe(II)GR]0 = 131 mM [4-NT]0 = 20 microM [Fe(II)GR]0 = 131
mM [4-NT]0 = 50 microM) The hydroxylamino intermediate shown in microM equals the deficit in the
mass balance and in abs equals the detector response (peak area) Solid lines represent 1 order
kinetic fits (a amp c) and ln [Ar-NO2]t[Ar-NO2]0) versus time fits (d) whereas symbols and dotted
lines represent actual data
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 77
At intial Fe(II)GR concentrations in large excess of initial Ar-NO2 concentration
we found a pseudo 1 order rate law for the degradation of Ar-NO2 by GR-SO4
[ ] [ ] [ b 2
a GR
2 ArNOFe(II) ArNOsdotsdot=minus k
dtd ]
where a = 1 b = 1 and the observed pseudo 1 order rate constant kobs = k middot
[Fe(II)GR] At high [Fe(II)GR]0[Ar-NO2]0 ratios the nitro compound was
transformed completely into the aniline product within reaction duration and the
degradation curves of the nitro compound were shaped according to pseudo 1
order kinetics (data points follow solid line in Figure 45a) In some instances ie
at low [Fe(II)GR]0[Ar-NO2]0 ratios the reactions did not follow pseudo 1 order
kinetics for the whole duration of reaction (data points deviate from solid line in
Figure 45c) Hence in order to allow comparison all the pseudo 1 order rate
constants were calculated as initial rates (ie max first two half-lives) from linear
fits of (time ln [Ar-NO2]t[Ar-NO2]0)-plots (Figure 45b amp 45d) Surface area-
normalised pseudo 1 order rate constants are shown in Table 41
Tabl
e 4
1 S
urfa
ce a
rea-
norm
alis
ed p
seud
o 1
ord
er ra
te c
onst
ants
for t
he re
duct
ive
trans
form
atio
n of
4-n
itrot
olue
ne (4
-NT)
4-
chlo
roni
trobe
nzen
e (4
-CN
B) a
nd 4
-nitr
ophe
nyla
cetic
aci
d (4
-NPA
) by
GR
-SO
4
Exp
erim
ent
Age
GR
(d
) [F
e(II
) GR] 0
(mM
) [N
AC
] 0 (micro
M)
[Fe(
II) G
R] 0
[N
AC
] 0∆[
ArN
O2]
(microM
) af b
k obs
(s-1
) ck o
bs (s
-1middotm
-2middotL
) d
GR
-SO
4 + 4
-NT
3 1
103
20
51
5
109
54
5
7
65middot1
0-46
95middot1
0-5
GR
-SO
4 + 4
-NT
3 1
103
50
20
6
174
34
8
7
41middot1
0-46
74middot1
0-5
GR
-SO
4 + 4
-NT
3 1
103
10
0 10
3
214
21
4
2
63middot1
0-42
39middot1
0-5
GR
-SO
4 + 4
-CN
B 1
1
103
55
18
7
177
32
2
4
21middot1
0-43
83middot1
0-5
GR
-SO
4 + 4
-CN
B 1
1
103
10
2 10
1
165
16
2
2
37middot1
0-42
15middot1
0-5
GR
-SO
4 + 4
-NPA
1
1 1
03
25
412
9
7 38
8
4
82middot1
0-44
38middot1
0-5
GR
-SO
4 + 4
-NPA
1
1 1
03
46
224
9
9 21
5
6
37middot1
0-45
79middot1
0-5
GR
-SO
4 + 4
-NPA
1
1 1
03
100
103
13
7
137
196
middot10-4
178
middot10-5
GR
-SO
4 + 4
-NT
2 15
1
31
20
655
5
4 27
0
6
74middot1
0-44
82middot1
0-5
GR
-SO
4 + 4
-NT
2 15
1
31
50
262
9
7 19
4
5
89middot1
0-44
21middot1
0-5
GR
-SO
4 + 4
-NT
4 2
126
0 50
25
2 49
1
982
110
middot10-2
817
middot10-5
GR
-SO
4 + 4
-NT
4 2
630
50
12
6 42
6
852
186
middot10-3
276
middot10-5
GR
-SO
4 + 4
-CN
B 2
2
126
0 30
42
0 29
0
967
925
middot10-3
687
middot10-5
GR
-SO
4 + 4
-CN
B 2
2
630
50
12
6 38
0
760
136
middot10-3
202
middot10-5
GR
-SO
4 + 4
-NPA
2
2 12
60
40
315
371
92
8
5
96middot1
0-34
43middot1
0-5
GR
-SO
4 + 4
-NPA
2
2 6
30
45
140
273
60
7
1
09middot1
0-31
62middot1
0-5
a A
mou
nt o
f NA
C re
duce
d by
GR
-SO
4 at r
eact
ion
term
inat
ion
b F
ract
ion
of in
itial
ly a
dded
NA
C tr
ansf
orm
ed b
y G
R-S
O4 a
t rea
ctio
n te
rmin
atio
n c
Pse
udo
1
orde
r rat
e co
nsta
nts c
alcu
late
d as
initi
al ra
tes
ie m
ax f
irst t
wo
half-
lives
d S
urfa
ce a
rea-
norm
alis
ed p
seud
o 1
ord
er ra
te c
onst
ants
The
are
a of
GR
-SO
4 per
L
su
spen
sion
was
cal
cula
ted
as frac14
middot[Fe
(II)
GR] 0middot
600
gmiddotm
ol-1
middot71
2 m
2 middotg-1
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 79
432 Comparison of rate constants for the different NACs
Even for NACs holding very different one-electron transfer reduction potentials
( ) their reactivities differed only little in Fe(II)-Fe(III) systems such as the
Fe(II)goethite system (slope a = 06 for linear free energy relationship (LFER)
between k
1hE
obs and Hofstetter et al 1999) and the Fe(II)magnetite system
(LFER slope a = 034 Klausen et al 1995) When considering only the for the
reductive transformation reactions of the NACs applied in this study (Table 42)
we expect the surface area-normalised pseudo 1 order rate constants for the
reduction of the NACs to follow the order 4-CNB gt NB gt 4-NT gt 4-NPA Based
on log k
1hE
1hE
obs versus correlations obtained in Fe(II)goethite systems we expect 4-
CNB to react 6 times faster than 4-NPA (Hofstetter et al 1999)
1hE
Table 42 One-electron reduction potentials and relative reactivities in Fe(II)-magnetite and GR-
SO4 systems for the nitro aromatic probe compounds
Compound pKa Eh1
acute (mV) krel (Fe3O4) cd krel (GR-SO4) ce
4-Chlornitrobenzene - -450 a 122 148
Nitrobenzene - -486 a 1 1
4-Nitrotoluene - -500 a 057 176
4-Nitrophenylacetic acid 385 -543 b - 123 a Values from references cited in Hofstetter et al 1999 b Estimated at pH 660 using a LFER (Hofstetter et al 1999 see Supporting Information 71) c Reactivity relative to NB d Values from Klausen et al 1995 e Values from this work
A comparison of the relative rate constants of the NACs obtained for their
transformation by GR-SO4 (this work) and by magnetite (Klausen et al 1995)
shows that they do not differ significantly from each other in any of the mineral
systems (Table 42) When considering charge effects we expect the anionic probe
compounds to react faster with GR-SO4 than the neutral probe compounds
provided that they sorb preferentially within the GR-SO4 interlayers and that Fe(II)
in the interlayers are equally or more reactive than external Fe(II) sites Still the
surface area-normalised kobs values obtained for NB 4-NT 4-CNB and 4-NPA
under various experimental conditions did not differ significantly from each other
80 Chapter 4
(Figure 46 Table 41) The anionic probe compound 4-NPA did not react
significantly faster with GR-SO4 than the neutral probe compounds NB 4-NT and
4-CNB This may indicate that 4-NPA does not significantly interact with reactive
Fe(II) sites in the interlayer Alternatively the negative charge carried by 4-NPA
may be compensating for the lower intrinsic reactivity as compared to the neutral
probe compounds thus explaining the similarity in rate constants for 4-NPA and
the neutral probe compounds Finally other factors than intrinsic reactivity or
charge of the probe compounds such as regeneration of reactive sites or formation
of the magnetite phases may control the overall reactivity of the system
Figure 46 Actual plot of surface area-normalised pseudo 1 order rate constants for the reactions
between neutral and anionic probe compounds and GR-SO4
In heterogeneous reactions mass transfer in bulk solution becomes the rate-
limiting step when the surface reaction is much faster than the diffusion of the
reacting species to the reactive surface In cases where mass transfer controls the
overall rate of reaction the observed pseudo 1 order rate constant kobs ge kLmiddota
where kL is the calculated mass transfer coefficient (mmiddots-1) and a is the ratio of the
external (geometric) specific surface area to volume of solution (m-1) (see
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 81
Supporting Information 72) Mass transfer controlled reactions between GR-SO4
particles and the NACs in bulk solution would explain the similar pseudo 1 order
rate constants obtained for the NACs in this work However when comparing our
estimates of kLmiddota with kobs (see Supporting Information 72) we found that the rates
of mass transfer for all 4 NACs exceed the observed rate constants by at least 3
orders of magnitude at every initial Fe(II)GR concentration Thus the reactions of
the given NACs with GR-SO4 are not likely to be mass transfer limited under the
experimental conditions applied here
Since mass transfer in bulk solution does not control the reaction between GR-SO4
and NACs the overall reaction rate may be surface saturation controlled During
the reductive transformation of NACs not only the parent compound but also
various intermediates forming may compete for the restricted number of reactive
sites present in GR-SO4 This competition may constitute the rate limiting step in
the overall reactivity and may even be enhanced if the number of reactive sites is
depleted during reaction However surface saturation kinetics would not explain
the unexpected similarity of the pseudo 1 order rate constants obtained for the
NACs but it could explain the bent curves observed at low initial Fe(II)GR
concentrations (Figure 45d) The kinetically deviating cases at low [Fe(II)GR]0
were evaluated according to Langmuir-Hinshelwood kinetics (see Supporting
Information 72) Our experimental data did not agree with the Langmuir-
Hinshelwood rate law for any of the NACs (regression results not shown)
Simplifying the rate law by assuming that the aniline product or the
hydroxylamino intermediate or both did not compete for the reactive sites did not
improve the regression Thus the Langmuir-Hinshelwood model cannot explain
the deviations from pseudo 1 order kinetics observed at [Fe(II)GR]0 in our GR-SO4
system and it does not suffice as the correct reaction mechanism nor as the rate-
limiting step
82 Chapter 4
If the adsorption follows a saturation-type sorption isoterm (eg Langmuir) the
sorbate (oxidant) concentration at the surface will vary non-linearily with the total
amount of oxidant added This dependence will have to be taken into account when
establishing rate laws for the heterogeneous reactions and when testing the
hypothesis that the reaction rates depend on the sorbed concentration of the
oxidants However at the high reaction rates observed here we could not quantify
sorption Since the measured initial NAC concentrations corresponded to the
nominal amount of NAC added we assume that transformation and not sorption
was responsible for the consumption of NAC
433 Factors influencing the reaction rate
In general numerous compound- and system-specific factors influence redox
reactions One very important factor is pH which influences the speciation of
dissociable compounds as well as the stability of GR and the formation of other
iron minerals in the system pH has a strong impact on the sorption and therefore
the availability of ionisable oxidants such as carboxylic acids At pH ~ 84 where
our experiments were conducted 4-NPA (pKa = 385) is completely dissociated
Our experiments conducted with GR-SO4 and NB showed that pH was constant
during reaction In addition solution pH has an effect on the surface speciation
From other Fe(II)-Fe(III) systems such as Fe(II) surface species associated with
iron oxides or clay minerals it is well-known that other reactive hydroxylated
Fe(II)-Fe(III)-hydroxo surface complexes can form at higher pH (Charlet et al
1998 Liger et al 1999) Williams amp Scherer (2001) reported a small decrease (5
fold) in the reduction rate of chromate with GR-CO3 when increasing pH from 50
to 90 This decrease may be due to the alternating speciation of the Fe(II) surface
sites on GR-CO3 and of chromate in solution (pKa (H2CrO4) = 08 pKa (HCrO4ndash) =
65) when raising pH (Williams amp Scherer 2001) In contrast other studies have
reported small increases (4 fold) in the reduction rates of nitrate and
trichloroethene with GR-SO4 when increasing pH from 71 to 84 and from 68 to
101 respectively (Koch amp Hansen 1997 Lee amp Batchelor 2002b)
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 83
In this work all experiments were carried out in the presence of 25 mM
Na2SO4(aq) in order to minimize GR-SO4 dissolution and spontaneous
transformation into magnetite Preliminary results from experiments conducted
with NB show that the bulk concentration of Na2SO4 has only a very small impact
on the rate ie increasing the concentration of Na2SO4(aq) in the GR-SO4
suspension from 5 to 25 mM reduced the observed rate constant by a factor of 2
At Na2SO4 concentrations above 25 mM the effect leveled off and therefore
[Na2SO4] = 25 mM was chosen for this work Portions of the same GR-SO4
suspension were used for kinetic experiments over a period of two weeks No
significant aging effects eg rate constants decreasing as a function of GR age
were observed within this time frame
434 Comparison with rate constants obtained for other Fe(II) containing
mineral systems
A recent study compared the reactivity of various Fe(II) containing iron mineral
systems towards organic probe compounds representing different classes of
pollutants (Elsner et al 2004) The reductive transformation of 4-CNB was
investigated for the Fe(III) minerals goethite (α-FeOOH) lepidocrocite (γ-
FeOOH) and hematite (α-Fe2O3) as well as for the Fe(II)-Fe(III) oxide magnetite
(Fe3O4) All experiments were conducted in the presence of 1 mM dissolved Fe(II)
at pH 72 As seen from the surface area-normalised pseudo 1 order rate constants
in Figure 47 the reduction of 4-CNB by the Fe(II)-amended goethite
lepidocrocite and magnetite systems was up to 40 times faster than its reduction by
GR-SO4 The reduction rate obtained for the Fe(II)hematite system was only
slightly higher than the rate for the blank containing no iron mineral but aqueous
Fe(II) solely
84 Chapter 4
Figure 47 Surface area-normalised pseudo 1 order rate constants for the degradation of 4-CNB
by GR-SO4 (open square this work) and various Fe(II) containing mineral systems (solid circles
Elsner et al 2004) Experimental conditions applied by Elsner et al 1 mM aqueous Fe(II) pH
72 25 m2 mineral surface areaL GR-SO4 = green rust sulphate α-FeOOH = goethite Fe3O4 =
magnetite γ-FeOOH = lepidocrocite α-Fe2O3 = hematite
The experiments with GR-SO4 in our study were carried out at pH 84 whereas the
experiments comprising the other systems in Figure 47 were conducted at pH 72
As the reactivity of GR is expected to increase with pH (Lee amp Batchelor 2002b
Koch amp Hansen 1997) the lower of GR-SO4 compared to other Fe(II) systems
cannot be explained by differences in pH values GR-SO4 might just contain fewer
or less reactive surface sites than Fe(II)-amended goethite lepidocrocite and
magnetite suspensions These findings contrast those of other studies which found
higher surface area-normalised pseudo 1 order rate constants for dechlorination
reactions for GR-SO4 than for magnetite (Lee amp Batchelor 2002a amp 2002b Elsner
et al 2004) The different reactivity orders of the Fe(II)-bearing minerals found
for chlorinated aliphatics and nitro aromatics suggest that effects other than pH and
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 85
the intrinsic reduction potentials of the reacting species play a role for the
reactivity of these Fe(II)-bearing minerals
435 Depletion of reactive sites
Assuming that the platy hexagonal GR-SO4 crystals hold an average width of 1 microm
and an average particle thickness of 35 nm (Hansen amp Koch 1998) the ratio of
outer surface area to total surface area AouterAtotal ~ 131 (see Supporting
Information 73) This means that only 3 of the total surface area in GR-SO4 is
available at the external surface Thus as the interlayer sulphate in GR-SO4 is not
readily exchanged with the anionic NAC applied we conclude that 4-NPA and
other NACs only react with the Fe(II) sites at the external GR-SO4 surface In
Table 43 the actual amounts of NAC reduced by GR-SO4 during reaction is
compared with the amount of NAC which theoretically can be reduced by the
initial amount of external reactive Fe(II) sites in GR-SO4 at AouterAtot ~ 131
Table 43 The actual amounts of NAC reduced by GR-SO4 during reaction ∆[NAC]act
compared with the amount of NAC which stoichiometrically should be reduced by the initial
amount of external reactive Fe(II) sites in GR-SO4 ∆[Ar-NO2]theory (calculated as
[Fe(II)GR]0(31middot6) assuming an even distribution of Fe(II) throughout the GR-SO4 structure)
Calculated for [NAC]0 ~ 50 microM
[Fe(II)GR]0 (mM)
∆[Ar-NO2]theory(microM)
∆[4-NT]act (microM)
∆[4-CNB]act (microM)
∆[4-NPA]act (microM)
103 55 174 177 99
63 339 426 380 273
As seen in Table 43 the actual amounts of NAC reduced by GR-SO4 during
reaction are in most cases higher than the amount of NAC which should be
reduced at the given [Fe(II)GR]0 according to reaction stoichiometry This indicates
that new external reactive sites were regenerated eg the Fe(III) phases produced
peel off the GR surface exposing new Fe(II) sites or that outermost internal
86 Chapter 4
reactive sites in close vicinity to the edges are available for reaction as well Lee
and Batchelor (2000b) also found the experimentally observed reduction capacity
of GR-SO4 for chlorinated ethylenes to be 2-3 orders of magnitudes lower than the
estimated reduction capacity including all Fe(II) in GR-SO4
At low initial Fe(II)GR concentrations only a fraction of NAC was reduced within
the reaction time observed (Figure 45c) even though there was stoichiometric
excess of Fe(II)-GR present The fraction of initial Ar-NO2 reduced by GR-SO4 at
reaction termination decreased as [Fe(II)GR]0 decreased (Table 41) and was
accompanied by a change in apparent rate laws with time (compare Figures 45b amp
d) In order to explain these observations we propose that the NACs react only at
external reactive Fe(II) sites and that the regeneration of new external reactive sites
is much slower than the reduction of NAC by GR-SO4 Thus the fast reduction of
NAC taking place at the external reactive sites represents the pseudo 1 order
behaviour whereas depletion of external reactive sites and their slow regeneration
are represented by the second bent part of the (time ln [Ar-NO2]t[Ar-NO2]0)-
curves deviating from pseudo 1 order kinetics Hence at low [Fe(II)GR]0 the
regeneration of reactive sites will eventually control the overall reaction rate
Depletion of available Fe(II) was also observed during the fast reduction of
chromate by GR-CO3 when the initial chromate concentration was increased or
when the GR-CO3 suspension was respiked with chromate repeatedly (Williams amp
Scherer 2001)
436 The role of external and internal reactive sites
It is reasonable to assume that GRs hold adsorption properties similar to other
layered double hydroxides such as hydrotalcites The sorption of 246-
trinitrophenol (TNP) and 245-trichlorophenol (TCP) on chloride and carbonate
intercalated hydrotalcites (HT-Cl = Mg3Al(OH)8ClmiddotyH2O HT-CO3 =
Mg6Al2(OH)16CO3middotyH2O) has been investigated (Hermosin et al 1993 Ulibarri et
al 1995 Ulibarri et al 2001) The authors found that the adsorption of TCP on
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 87
HT-CO3 was very low and that TCP adsorbs only on the external surface sites of
HT-CO3 (Hermosin et al 1993) Furthermore is was reported that the adsorption
of TNP on HT was dramatically affected by the nature of the interlayer anion ie
the adsorption of TNP was considerably higher on HT-Cl than on HT-CO3
(Ulibarri et al 2001) For HT-Cl interlayer anion exchange of chloride with TNP
was detected by XRD analysis and an expansion of the characteristic basal d003
spacing from 79 Ǻ to 132 Ǻ confirmed the presence of TNP in the HT interlayer
(Ulibarri et al 1995) Collating the results reported for HTs with GRs it is not
likely that the divalent SO42- in GR-SO4 is exchanged with the monovalent 4-NPA
Chacirctelet et al (1996) investigated the adsorption of mono- and divalent anions
onin the outer and inner adsorption sites in HT by varying the zetapotential with
pH in the presence of various electrolytes The authors found that SO42- adsorbs on
the external HT surfaces by formation of outer-sphere complexes whereas chloride
hardly adsorbed on HT Moreover it was reported that the adsorption of sulphate
onto HT was not strongly affected by the presence of chloride while sulphate on
the contrary inhibited the adsorption of chloride on HT Studies applying
spectroscopic analyses have investigated the sorption of oxyanions at external and
internal GR-SO4 surfaces (Myneni et al 1997 Randall et al 2001) Selenate was
adsorbed only on the outer GR-SO4 surface when added after GR formation
whereas it was primarily coprecipitated into the interlayer when present during GR
formation Thus for selenate its presence during GR formation is a prerequisite of
its incorporation in the GR interlayer Selenate is readily reduced by GR-SO4 and
the rates of reduction of coprecipitated selenate were very similar to the reduction
rates of selenate adsorbed at the outer GR surface (Myneni et al 1997) This
finding suggests that the outer and inner reactive Fe(II) sites in GR-SO4 hold
similar reactivities
Results based on electron microscopy reported that the reduction of uranyl took
place primarily at the edges of hexagonal GR-SO4 particles (OrsquoLoughlin et al
2003a) In another recent study XRD characterization of the GR-SO4 crystals
88 Chapter 4
during reaction with trichloroacetate (TCA) indicated that TCA did not enter the
GR-SO4 interlayer during reaction (Chapter 5 this work) The average GR-SO4
particle thickness perpendicular to the basal plane was constant during reaction
implying that TCA reacts only at the edges and not at the basal planes Assuming
that the platy hexagonal GR-SO4 crystals hold an average width of 1 microm and an
average particle thickness of 35 nm (Hansen amp Koch 1998) the ratio of edge
surface area to outer surface area is AedgeAouter ~ 130 (see Supporting Information
73) Hence only 3 of the outer surface area in GR-SO4 is available at the edges
Once more the regeneration of new external reactive sites is strongly inferred as
the actual amounts of NAC reduced by GR-SO4 during reaction are much higher
than the amount of NAC which may be reduced by the reactive edge sites present
initially Assuming that the NACs react at the edges only and if employing the
AedgeAouter in the estimation of the rate constants the surface area-normalised
pseudo 1 order rate constants for GR-SO4 would be 30 times higher than the rate
constants depicted in Figure 47 Thus the reactivity of GR-SO4 normalised to its
reactive surface area is higher than the reactivity normalised to its outer surface
area determined by the BET method (N2 adsorption)
The reduction of chromate has been examined in the presence of all the common
GR forms (Bond amp Fendorf 2003 Loyaux-Lawniczak et al 1999 Loyaux-
Lawniczak et al 2000 Williams amp Scherer 2001) The results reported by Bond
amp Fendorf (2003) confirm that not only the surface area of GR but also the
interlayer spacing (interlayer anion size) and interlayer anion charge play an
important role for the reaction rate Hence it follows that coordination (size) and
charge of the oxidant determine its access to the internal sites in GRs
The results obtained for all 4 NACs support what has been reported for nitrate and
TCA At [Fe(II)GR]0 = 2-10 mM and [NO3-]0 = 143 mM pseudo 1 order rate
constants for the reduction of nitrate by GR-SO4 were 158middot10-7 s-1middotm-2middotL (Hansen
et al 2001) This reaction rate increased 40 times by adding barium nitrate instead
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 89
of sodium nitrate thereby precipitating the interlayer sulphate as barium sulphate
and enhancing access to the interlayer Though barium addition changes the GR-
SO4 system dramatically it indicates the importance of interlayer anion exchange
(Hansen amp Koch 1998) The rate constant reported for nitrate (no barium added) is
100-1000 smaller than the rate constants obtained for the NACs in this work
Moreover the reaction kinetics for nitrate did not deviate from pseudo 1 order
kinetics At [Fe(II)GR]0 = 025-104 mM and [TCA]0 = 50 microM-1 mM pseudo 1
order rate constants for the reduction of TCA by GR-CO3 or GR-SO4 were 65middot10-7
s-1middotm-2middotL (Chapter 5 this work) The rate constant for TCA is 10-1000 smaller than
the rate constants for the NACs and the reaction kinetics for TCA did not deviate
from pseudo 1 order kinetics This suggests that the overall reductive
transformation of slowly reacting oxidants such as nitrate and TCA is not
controlled by the rate of regeneration of external Fe(II) reactive sites Altogether
the results reported for selenate chromate and nitrate clearly demonstrate that
these anionic oxidants react primarily with external reactive sites in GR-SO4 Only
under certain conditions ie adding the oxidant prior to GR-SO4 formation or
extracting the interlayer sulphate through precipitation with barium outside the
GR-SO4 particles do the oxidants have access to the interlayer Our findings
suggest that both the neutral and anionic nitro aromatic probe compounds applied
here also react exclusively with the external reactive sites in GR-SO4 Supposedly
the neutral and monovalent charge states of the NACs hinder their access to the
GR-SO4 interlayer A divalent anionic nitro aromatic probe compound might
exchange with the interlayer sulphate more readily and gain access to the inner
Fe(II) reactive sites in GR-SO4 only divalent anionic NACs are not commercially
available
44 Conclusions
This work demonstrates that NACs are completely reduced to their corresponding
anilines by GR-SO4 The surface area-normalised pseudo 1 order rate constants
obtained for the reduction of the neutral and anionic NACs by GR-SO4 under
90 Chapter 4
various experimental conditions did not differ significantly from each other despite
their different charges Neither mass transfer control nor surface saturation kinetics
could account for the similarity of the pseudo 1 order rate constants obtained for
the NACs These observations suggest that the anionic NACs do not have an
enhanced access to inner or outer Fe(II)-GR reactive sites as compared to the
neutral NACs Based on our estimations of the molecular sizes of the NACs we
propose that the charge and not the size of the NACs controls their access to the
internal reactive sites in GRs Hence the reaction between NAC and GR-SO4 takes
place primarily at the external reactive Fe(II) sites This work further demonstrated
that the reduction of the NACs by GR-SO4 only followed pseudo 1 order kinetics
throughout the whole reaction at high initial Fe(II)GR concentrations At low initial
Fe(II)GR concentrations the NACs were not reduced completely within the reaction
time observed though according to reaction stoichiometry the total Fe(II)-GR
present should be sufficient to reduce the whole amount of NAC This means that
at some point during the reaction the external reactive Fe(II) sites were depleted
and the regeneration of new external reactive sites was much slower than the
reduction of the NACs by GR-SO4 The reduction of 4-CNB by GR-SO4 reported
here was 10-100 times slower than its reduction by other Fe(II)-Fe(III) systems
such as goethite lepidocrocite and magnetite suspensions amended with Fe(II)
(Elsner et al 2004)
The results obtained in this work infer that under natural geochemical conditions
where GR-SO4 presumably forms in low concentrations the rate of regeneration of
external Fe(II) reactive sites may control the overall reductive transformation of
fast reacting pollutants by GR-SO4 Thus not only abiotic processes such as
interchanging redox conditions created by water level alterations but also the direct
microbial formation through Fe(III)-reducing bacteria may govern the formation of
GRs and the renewal of external Fe(II) reactive sites in GRs This holds both for
natural systems like iron-rich suboxic soils and sediments as well as engineered
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 91
systems like permeable reactive barriers of zero-valent iron implemented for on-
site remediation of organic and inorganic contaminants
Acknowledgments
We would like to thank Henrik T Andersen for performing the NB kinetic experiments and
Hanne Nancke-Krogh for technical assistance in the laboratory
References
Al-Agha MR Burley SD Curtis CD Esson J (1995) Complex cementation textures and authigenic mineral assemblages in recent concretions from the Lincolnshire Wash (east coast UK) driven by Fe(0) to Fe(II) oxidation Journal of the Geological Society 152 157-171 Bernal JD Dasgupta DR Mackay AL (1959) The oxides and hydroxides of iron and their structural inter-relationships Clay Minerals Bulletin 4 15-30 Bond DL Fendorf S (2003) Kinetics and structural constraints of chromate reduction of green rusts Environmental Science and Technology 37 2750-2757 Brunauer S Emmett PH Teller E (1938) Adsorption of gases in multimolecular layers Journal of American Chemical Society 60 309-319 Charlet L Silvester E Liger E (1998) N-compound reduction and actinide immobilisation in surficial fluids by Fe(II) The surface FeIIIFeIIOH0 species as major reductant Chemical Geology 151 85-93 Chacirctelet L Bottero JY Yvon J Bouchelaghem A (1996) Competition between monovalent and divalent anions for calcined and uncalcined hydrotalcite anion exchange and adsorption sites Colloids and Surfaces A Physicochemical and Engineering Aspects 111 167-175 Chaudhuri SK Lack JG Coates JD (2001) Biogenic magnetite formation through anaerobic biooxidation of Fe(II) Applied and Environmental Microbiology 67 2844-2848 Cui D Spahiu K (2002) The reduction of U(VI) on corroded iron under anoxic conditions Radiochemica Acta 90 623-628 Elsner M Haderlein SB Schwarzenbach RP (2004) Reactivity of Fe(II)-bearing minerals towards reductive transformation of organic contaminants Environmental Science and Technology 38 799-807 Erbs M Hansen HCB Olsen CE (1999) Reductive dechlorination of carbon tetrachloride using iron(II)iron(III)-hydroxide-sulphate (green rust) Environmental Science and Technology 33 307-311 Fadrus H Maly J (1975) Suppression of iron(III) interference in the determination of iron(II) in water by the 110-phenanthroline method The Analyst 100 549-554 Fredrickson JK Zachara JM Kennedy DW Dong H Onstott TC Hinman NW Li S
92 Chapter 4 (1998) Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium Geochimica et Cosmochimica Acta 62 3239-3257 Geacutenin JMR Bourrieacute G Trolard F Abdelmoula M Jaffrezic A Refait Ph Maitre V Humbert B Herbillon A (1998) Thermodynamic equilibria in aqueous suspensions of synthetic and natural Fe(II)-Fe(III) green rusts Occurrences of the mineral in hydromorphic soils Environmental Science and Technology 32 1058-1068
Geacutenin JMR Olowe AA Benbouzid-Rollet ND Prieur D Confente M Resiak B (1991) The simultaneous presence of green rust 2 and sulfate reducing bacteria in the corrosion of steel sheet piles in a harbour area Hyperfine Interactions 69 875-878 Glasauer S Weidler PG Langley S Beveridge TJ (2003) Controls on Fe reduction and mineral formation by a subsurface bacterium Geochimica et Cosmochimica Acta 67 1277- 1288 Hansen HCB (1989) Composition stabilization and light absorption of Fe(II)Fe(III) hydroxycarbonate (green rust) Clay Minerals 24 663-669 Hansen HCB Borggaard OK Soslashrensen J (1994) Evaluation of the free energy of formation of iron(II)iron(III)-hydroxidesulphate (Green Rust) and its reduction of nitrite Geochimica et Cosmochimica Acta 58 2599-2608 Hansen HCB Guldberg S Erbs M Koch CB (2001) Kinetics of nitrate reduction by green rusts ndash effects of interlayer anion and Fe(II)Fe(III) ratio Applied Clay Science 18 81-91 Hansen HCB Koch CB (1998) Reduction of nitrate to ammonium by sulphate green rust Activation energy and interlayer reaction mechanism Clay Minerals 33 87-101 Hansen HCB Koch CB Nancke-Krogh H Borggaard OK Soerensen J (1996) Abiotic nitrate reduction to ammonium Key role of green rust Environmental Science and Technology 30 2053-2056 Hansen HCB Poulsen IF (1999) Interaction of synthetic sulphate green rust with phosphate and the crystallization of vivianite Clays and Clay Minerals 47 312-318 Hansen HCB Taylor RM (1991) The use of glycerol intercalates in the exchange of CO3
2- with SO4
2- NO3- or Cl- in pyroaurite-type compounds Clay Minerals 26 311-327
Heasman DM Sherman DM Ragnarsdottir KV (2003) The reduction of aqueous Au3+ by sulfide minerals and green rust phases American Mineralogist 88 725-738 Hermosin MC Pavlovic I Ulibarri MA Cornejo J (1993) Trichlorophenol adsorption on layered double hydroxide a potential sorbent Journal of Environmental Science and Health A28 1875-1888 Hofstetter TB Heijmann CG Haderlein SB Holliger C Schwarzenbach RP (1999) Complete reduction of TNT and other (poly)nitroaromatic compounds under iron-reducing subsurface conditions Environmental Science and Technology 33 1479-1487 Hofstetter TB Schwarzenbach RP Haderlein SB (2003) Reactivity of Fe(II) species associated with clay minerals Environmental Science and Technology 37 519-528
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 93
Klausen J Troumlber SP Haderlein SB Schwarzenbach RP (1995) Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions Environmental Science and Technology 29 2396-2404 Koch CB Hansen HCB (1997) Reduction of nitrate to ammonium by sulphate green rust Advances in GeoEcology 30 373-393 Kukkadapu RK Zachara JM Smith SC Fredrickson JK Liu C (2001) Dissimilatory bacterial reduction of Al-substituted goethite in subsurface sediments Geochimica et Cosmochimica Acta 65 2913-2924 Lee W Batchelor B (2002a) Abiotic reductive dechlorination of chlorinated ethylenes by iron- bearing soil minerals 1 Pyrite and magnetite Environmental Science and Technology 36 5147- 5154 Lee W Batchelor B (2002b) Abiotic reductive dechlorination of chlorinated ethylenes by iron-bearing soil minerals 2 Green rust Environmental Science and Technology 36 5348- 5354 Liger E Charlet L Van Cappellen P (1999) Surface catalysis of uranium (VI) reduction by iron(II) Geochimica et Cosmochimica Acta 63 2939-2955 Loyaux-Lawniczak S Refait Ph Ehrhardt J Lecomte P Geacutenin JMR (2000) Trapping of Cr by formation of ferrihydrite during the reduction of chromate ions by Fe(II)-Fe(III) hydroxysalt green rusts Environmental Science and Technology 34 438-443 Loyaux-Lawniczak S Refait Ph Lecomte P Ehrhardt J Geacutenin JMR (1999) The reduction of chromate ions by Fe(II) layered hydroxides Hydrology and Earth System Sciences 3 593-599 Mendiboure A Schoumlllhorn A (1986) Formation and anion exchange reactions of layered transition metal hydroxides [Ni1-xMx](OH)2(CO3)x2(H2O)z (M = Fe Co) Revue de Chimie Mineacuterale 23 819-827 Miyata S (1983) Anion-exchange properties of hydrotalcite-like compounds Clays and Clay Minerals 31 305-311 Myneni SCB Tokunaga TK Brown Jr GE (1997) Abiotic selenium redox transformations in the presence of Fe(IIIII) oxides Science 278 1106-1109 Nielsen A (1976) Hvid groslashn og sort rust Beskrivelse af en korrosionsskade paring et svoslashmmebassin Nordisk Betong 2 21-24 OLoughlin EJ Burris DR (2004) Reduction of halogenated ethanes by green rust Environmental Toxicology and Chemistry 23 41-48 OLoughlin EJ Kelly SD Cook RE Csencsits R Kemner KM (2003a) Reduction of uranium(VI) by mixed iron(II)iron(III) hydroxide (green rust) Formation of UO2 nanoparticles Environmental Science and Technology 37 721-727
94 Chapter 4 OLoughlin EJ Kelly SD Kemner KM Csencsits R Cook RE (2003b) Reduction of AgI AuIII CuII and HgII by FeIIFeIII hydroxysulfate green rust Chemosphere 53 437-446 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002) Iron (IIIII) hydroxycarbonate green rust formation and stabilization from lepidocrocite bioreduction Environmental Science and Technology 36 16-20 Parmar N Gorby YA Beveridge TJ Ferris FG (2001) Formation of green rust and immobilization of nickel in response to bacterial reduction of hydrous ferric oxide Geomicrobiology Journal 18 375-385 Pepper SE Bunker DJ Bryan ND Livens FR Charnock JM Pattrick RAD Collison D (2003) Treatment of radioactive wastes An X-ray absorption spectroscopy study of the reaction of technetium with green rust Journal of Colloid and Interface Science 268 408- 412 Randall SR Sherman DM Ragnarsdottir KV (2001) Sorption of As(V) on green rust (Fe4(II)Fe2(III)(OH)12SO4
3H2O) and lepidocrocite (γ-FeOOH) Surface complexes from EXAFS spectroscopy Geochimica et Cosmochimica Acta 65 1015-1023 Roh Y Lee SY Elless MP (2000) Characterization of corrosion products in the permeable reactive barriers Environmental Geology 40 184-194 Schultz CA Grundl TJ (2000) pH dependence on reduction rate of 4-Cl-nitrobenzene by Fe(II)montmorillonite systems Environmental Science and Technology 34 3641-3648 Simon L Francois M Refait Ph Renaudin G Lelaurain M Geacutenin JMR (2003) Structure of the Fe(II-III)-layered double hydroxysulphate green rust two from Rietveld analysis Solid State Sciences 5 327-334 Tamaura Y (1986) Ni(II)-bearing green rust II and its spontaneous transformation into Ni(II)- bearing ferrites Bulletin of the Chemical Society of Japan 59 1829-1832 Tamaura Y (1985) ZnII-bearing green rust II and its spontaneous transformation into ZnII- bearing ferrite in aqueous solution Bulletin of the Chemical Society of Japan 58 2951-2954 Trolard F Abdelmoula M Bourrieacute G Humbert B Geacutenin JMR (1996) Mise en eacutevidence dun constituant de type rouilles vertes dans les sols hydromorphes Proposition de lexistence dun nouveau mineacuteral la fougeacuterite Geacuteosciences de surface Comptes Rendus de LrsquoAcademie des Sciences 323 1015-1022 Tuovinen OH Button KS Vuorinen A Carlson L Mair DM Yut LA (1980) Bacterial chemical and mineralogical characteristics of tubercles in distribution pipelines Journal of the American Water Works Association 72 626-635 Ulibarri MA Pavlovic I Barriga C Hermosin MC Cornejo J (2001) Adsorption of anionic species on hydrotalcite-like compounds effect of interlayer anion and crystallinity Applied Clay Science 18 17-27 Ulibarri MA Pavlovic I Hermosin MC Cornejo J (1995) Hydrotalcite-like compounds as potential sorbents of phenols from water Applied Clay Science 10 131-145
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 95
Williams AGB Scherer MM (2001) Kinetics of chromate reduction by carbonate green rust Environmental Science and Technology 35 3488-3494
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 97
5 Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems
Abstract
Trichloroacetate (TCA) is a widespread environmental contaminant with proven
phytotoxicity and suspected human carcinogenicity In order to assess the global
cycling of TCA and to predict its fate in subsurface environments information
regarding the reactivity and product distribution of TCA degradation is needed
Due to the high oxidation state of TCA conditions for oxidative transformation
pathways in soils and groundwater are unfavorable However in suboxic soils and
sediments Fe(II)-bearing minerals are potential reactants for reductive
dehalogenation reactions of TCA as has been demonstrated for other halogenated
contaminants We examined the reactivity of various Fe(II)-Fe(III) mineral
systems towards TCA and dichloroacetate (DCA) its expected transformation
product in laboratory batch experiments imitating natural conditions ie low
initial Fe(II) Fe(III) and TCADCA concentrations and no artificial buffer The
Fe(II)-Fe(III)-systems investigated included sulfate green rust (GR-SO4) carbonate
green rust (GR-CO3) magnetite Fe(II)goethite and Fe(II)lepidocrocite
Trichloroacetate was readily reduced to DCA by all Fe(II)-bearing minerals The
reactions generally followed pseudo 1 order kinetics with respect to TCA The
surface area-normalised pseudo 1 order rate constants obtained (035ndash76middot10-5 min-
1middotm-2middotL at [Fe(II)]0 = 020ndash122 mM [TCA]0 = 15ndash1000 microM and pH 70ndash87)
showed no striking differences regarding product distribution and surface area-
normalised reaction rate constants between the Fe(II)-Fe(III)-systems The
stoichiometrically formed DCA was not further reduced to monochloroacetate
(MCA) or acetate in any of the systems within the time frame in our experiments
To our knowledge this is the first published report on abiotic transformation of
TCA by Fe(II)-bearing minerals Our results imply that processes involving
reactive Fe(II)-bearing minerals may play a significant role in controlling the fate
98 Chapter 5
of TCA in natural subsurface environments and that DCA found in the subsurface
may be formed by such processes
51 Introduction
Trichloroacetic acid (TCA) has been applied as a herbicide for many years until its
use was banned in the late 1980acutes (Berg et al 2000) Today TCA is mainly used
as an etching agent in the metal industry as a swelling solvent in the plastic
production and as a bleaching agent in the paper and pulp manufacture (Muumlller et
al 1996) Other anthropogenic sources include formation of TCA as a result of the
chlorine based disinfecting process used in drinking water treatment and the
atmospheric photooxidation of chlorinated solvents including tetrachloroethene
and 111-trichloroethane (McCulloch 2002) Only very little information is
available on the TCA production volumes and even less is known about the
amount of TCA released into the environment as a result of its industrial
applications Due to its low volatility and high aqueous solubility TCA is easily
washed out of the atmosphere into the aquatic and terrestrial biospheres As TCA
is found in almost every ecosystem around the globe including non-urban and
non-industrial sites the relative contributions from anthropogenic and natural
sources are currently being debated (McCulloch 2002 Ahlers et al 2003)
Trichloroacetic acid is omnipresent in soils and the concentrations reported are
very variable ranging from lt005 microgkg to 380 microgkg (Euro Chlor 2001
McCulloch 2002 Ahlers et al 2003) Both abiotic and enzymatically catalyzed
formation of TCA from humic acids have been demonstrated in laboratory studies
(Haiber et al 1996 Hoekstra et al 1999b Fahimi et al 2003) Furthermore the
in situ natural formation of TCA from anthropogenic or natural tetrachloroethene
or 111-trichloroethane in biota has been suggested (Hoekstra et al 1999a
McCulloch 2002) Such natural sources may explain part of the TCA
concentrations found in soils but their environmental significance is still unknown
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 99
On account of its phytotoxicity suspected human carcinogenicity and widespread
occurrence TCA is of considerable environmental concern especially in the
terrestrial compartment The TCA concentrations found in soil air and water in
pre-industrial times were far below the present ones (Jordan amp Frank 1999 Ahlers
et al 2003) Based on the current TCA concentrations detected in soils the
European Commission proposed risk reduction measures concerning
tetrachloroethene - a precursor of TCA - to be taken immediately (Ahlers et al
2003 and references therein) Occurrences of monochloroacetic acid (MCA) and
dichloroacetic acid (DCA) reported include surface waters marine waters
precipitation ice (glaciers) and air (Reimann et al 1996 Berg et al 2000 Scott et
al 2000 Scott et al 2002) Based on the concentrations reported for the aquatic
environments it is reasonable to assume that MCA and DCA are omnipresent in
soils as well Sources of MCA and DCA include production in the chemical
industry photooxidation of chlorinated aliphatics in the atmosphere and reductive
transformation of TCA (Reimann et al 1996 Ahlers et al 2003 and references
therein) MCA and DCA are also toxins and suspected human carcinogens (Kuumlhn
amp Pattard 1990) hence not only TCA but also its daughter compounds are
pollutants of environmental concern
In subsurface environments TCA may be removed by sorption seepage chemical
transformation microbial degradation and plant uptake followed by metabolic
degradation andor physical removal at harvest (Foy 1975) There is little or no
evidence of abiotic transformations of TCA in the literature Only one recent study
demonstrated the reductive dechlorination of TCA to MCA by Fe(0) (Hozalski et
al 2001) It has been reported that the degradation of TCA in soil is slow and
mainly mediated by microorganisms but only little is known about the bacteria and
processes involved (Lignell et al 1984) Biodegradation of TCA has been found at
both oxic and anoxic conditions An aerobic microorganism capable of growing on
TCA as the sole carbon and energy source has been characterised (Yu amp Welander
1995) Moreover anaerobic bacteria coupling co-metabolic growth to reductive
100 Chapter 5
dechlorination of TCA have been isolated (Weightman et al 1992 De Wever et
al 2000) However more information regarding the abiotic and biotic
transformation of TCA is needed in order to assess the fate and transport of TCA in
natural subsurface environments
It is well-known that Fe(II) present in minerals or associated with mineral surfaces
is a much stronger reductant than Fe(II) in solution The enhanced reactivity of a
structural or surface-bound Fe(II) center can be rationalized by the increased
electron density donated by hydroxyl ligands and a stabilization of the Fe(III)
oxidation state by the hydroxyl ligands (Luther 1990) Fe(II)-bearing minerals
including layered Fe(II)-Fe(III) hydroxides (green rusts) magnetite (Fe3O4)
siderite (FeCO3) Fe(II) sulfides as well as Fe(II)-carrying Fe(III) oxides and clay
minerals have also been shown to reduce a range of organic and inorganic
contaminants such as nitro aromatic compounds chlorinated aliphatics chromate
uranyl pertechnetate nitrate monochloramine and carbamate pesticides (Chapter
4 this work Klausen et al 1995 Cui amp Eriksen 1996 Erbs et al 1999 Liger et
al 1999 Loyaux-Lawniczak et al 1999 Amonette et al 2000 Hansen et al
2001 Pecher et al 2002 Vikesland amp Valentine 2002 Hofstetter et al 2003
OrsquoLoughlin and Burris 2003 OrsquoLoughlin et al 2003a amp 2003b Strathmann amp
Stone 2003 Elsner et al 2004a) Laboratory and field studies showed that even in
geochemically highly heterogeneous anoxic aquifer sediments Fe(II) adsorbed to
Fe(III) (hydr)oxide surfaces was the dominant reductant of nitroaromatic and
halogenated contaminants (Ruumlgge et al 1998 Hofstetter et al 1999 Kenneke amp
Weber 2003) Only little is known about the nature of the Fe(II) species associated
with Fe(III) oxide surfaces but reactive hydroxylated Fe(II)-Fe(III)-hydroxo
surface complexes associated with hematite and magnetite above pH 65 have been
proposed (Charlet et al 1998aampb Liger et al 1999) Due to the presence of
structural Fe(II) within the mineral lattice the reactivity of Fe(II) associated with
mixed valent Fe(II)-Fe(III) minerals such as green rusts magnetite and reduced
ferruginous clay minerals may hold another reactivity than Fe(II) associated with
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 101
pure Fe(III) oxides However Fe(II) adsorbed on Fe(III) oxides such as goethite
hematite and lepidocrocite may also hold different reactivities as the Fe(III) oxides
contain different crystal and surface structures
Since chlorinated ethanes and ethenes such as hexachloroethane 111-
trichloroethane tetrachloroethene and trichloroethene are susceptible to chemical
reduction by a range of Fe(II)-bearing minerals including magnetite GR-SO4
Fe(II) sulfides and Fe(II)-carrying Fe(III) oxides (Butler amp Hayes 1998 amp 1999
Hwang amp Batchelor 2000 Gander et al 2002 Lee amp Batchelor 2002aampb Elsner
et al 2004a) we hypothesized that TCA may be transformed by Fe(II)-bearing
minerals as well The main goals of this work were to study such reactions and
establish product distribution and surface area-normalised reaction rates for the
reductive dechlorination of TCA by Fe(II)-Fe(III) mineral systems common in
nature
5 2 Materials and methods
No synthetic buffers were applied and iron concentrations were kept low The
calcareous systems were pH-controlled at 76 through a natural buffer system
(CaCO3(s) + 995 N205 CO2(g)) All handling and sampling of solutions and
suspensions were carried out under strict anoxic conditions Goethite (acicular
particles with size 01 times 06 microm specific surface area 16 m2g) and lepidocrocite
(acicular particles with size 005 times 03 microm specific surface area 18 m2g) were
purchased as fine powders from Bayer (Bayferrox 910 and 943) Calcite (grain size
170-350 microm Pluumlss-Staufer AG) was used as a buffer or as a Fe(III)-oxide-bearing
mineral In order to simulate natural conditions the iron minerals were applied as
coatings on calcite particles (model system for calcareous soils) in some
experiments Trichloroacetic acid dichloroacetic acid and monochloroacetic acid
were pa quality (Fluka)
102 Chapter 5
521 Synthesis of GRs and magnetite
GR-CO3 was synthesized by controlled air oxidation of an FeCl2 solution at a
constant pH of 700 (titrated with 1 M Na2CO3) according to the procedure given
by Hansen amp Koch (1997) 05 M aqueous stock solutions of FeCl2 were prepared
in 100 mL glass flasks by reacting 65 mmol of iron powder (particle size 10 microm
Merck) with 100 mL deoxygenated 10 M HCl The solutions were magnetically
stirred and heated (~80degC) during reaction until the H2(g) production had ceased (ge
2 hours) The FeCl2 solutions were stored in the dark under a small Ar
overpressure at 5degC The GR-CO3 suspensions were washed with deoxygenated
deionised water (DIW) separated on a folding filter (medium filtration rate cotton
linterhigh alpha pulp Schleicher amp Schuell) and redispersed in deoxygenated
DIW Washing separation and redispersion of the GR-CO3 suspension were
conducted in an anoxic glove box (92 N28 H2 Coy Laboratory Products Inc)
All suspensions and solutions were deoxygenated by Ar-purging (999998 Ar
Carbagas) Magnetite was synthesized by further aerial oxidation of GR-CO3 at pH
700 until consumption of 1 M Na2CO3 ceased GR-SO4 was synthesized by
controlled air oxidation of an FeSO4 solution at a constant pH of 70 according to
the procedure given by Koch amp Hansen (1997) The GR-SO4 suspension was
washed with deoxygenated DIW separated on a glass filter funnel (pore size 4
Duran) and redispersed in deoxygenated DIW Washing separation and
redispersion of the GR-SO4 suspension were conducted in an anoxic glove bag
(999995 Ar Aldrich)
522 Preparation of iron oxide coatings
Two grams of goethite (goe) or lepidocrocite (lep) and 100 g calcite were
combined with 200 mL DIW in a 500 mL polyethylene flask The suspension was
gently agitated on a reciprocating shaker for 24 h and left to stand for another 24 h
Excess Fe(III) oxides and salts were removed from the coated material by repeated
decantation and washing with DIW in polyethylene flasks until clear runoff
Finally the coatings were collected on folding filters and air dried The amount of
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 103
goethite and lepidocrocite coated onto calcite after washing and drying was
quantified to 10-11 mg Fe(III)g calcite
523 Mineral characterisation
The identity and purity of the GR-CO3 GR-SO4 and magnetite suspensions were
examined by means of X-ray diffraction (XRD) The XRD analyses were
performed on a Scintag XDS 2000 using Co Kα radiation (45 kV 40 mA) or a
Siemens D5000 XRD applying Co Kα radiation (40 kV 40 mA) Glycerol smears
made according to Hansen (1989) were scanned between 6 and 80 deg2θ with a scan
speed of 1 deg2θmin The specific surface area (SSA) of calcite was determined by
the BET multi-point method using N2 adsorption (Brunauer et al 1938) Powder
samples were filled into sample burettes in the glove box and the generously
lubricated stopcocks closed Samples and burettes were evacuated prior to
connecting them to the BET-instrument (Sorptomatic 1990 Fisons)
524 Kinetic experiments
All reactions were carried out in 25-100 mL serum vials sealed with stoppers
(Viton or Teflon coated rubber) and aluminum crimp caps Kinetic experiments
were conducted with GR-SO4 GR-CO3 magnetite Fe(II)goethite and
Fe(II)lepidocrocite at room temperature In most cases pH was controlled through
the carbonate-bicarbonate buffer system by adding calcite to suspensions
containing the iron minerals solely or by adding the iron minerals as coatings on
calcite Furthermore the calcite containing suspensions were deoxygenated with
05 CO2995 N2(g) thereby attaining an initial pH of 76-77 The GR-CO3 and
magnetite suspensions were deoxygenated with 100 N2(g) and no additional pH
buffer was added The goethite and lepidocrocite suspensions were amended with
300-1000 microM FeCl2(aq) and equilibrated gt 20 h prior to TCADCA addition See
Table 51 for more details on the experimental conditions To start the reaction 50
microM - 1 mM TCA or DCA was added to the mineral suspensions from aqueous
anoxic stock solutions The reaction vials were agitated gently on a roller apparatus
104 Chapter 5
or a shaking table (35 rpm) in order to minimize abrasion of the iron oxide mineral
coatings At appropriate time intervals suspension samples were withdrawn using
Ar(g)- 100 N2(g)- or 995 N205 CO2(g)-flushed sterile disposable syringes
and hypodermic needles The suspension samples were filtered (02 microm Teflon)
and collected for quantification of chloride and the chlorinated acetic acids The
samples were stored at -20degC and analysed without further treatment
525 Analytical methods
Total and aqueous Fe(II) were determined using a modified phenanthroline method
(Fadrus and Maly 1975) For determining [Fe(II)aq] and [Fe(II)total] 1 mL filtered
(02 microm Teflon) and 1 mL unfiltered mineral suspension were added to 18 mL 01
M HCl respectively and allowed to dissolve for 30 min From these acid digests
01 mL was added to 05 mL Fe(II)-phenanthroline-buffer-reagent and 19 mL
DIW added up Estimates of the structural or adsorbed Fe(II) in the Fe(II)-Fe(III)
mineral systems were estimated as the difference [Fe(II)solid] = [Fe(II)total] -
[Fe(II)aq] The total amount of Fe(III) coated on calcite was determined by atomic
absorption spectroscopy following dissolution in 6 M HCl(aq) for 24 h At low
initial TCA concentrations (le 50 microM) the chlorinated acetic acids were quantified
by means of a modified ion interaction (or paired-ion) chromatographic method
(Sarzanini et al 1999) Separation was performed on a LiChrospher 100 RP-18 (5
microm 125 times 4 mm ID) reversed-phase column coupled with a LiChroCART 100 RP-
18 (4 times 4 mm ID) precolumn Analytical conditions were isocratic and the eluent
consisted of 50 aqueous solution of 35 mM cetyltrimethylammonium chloride
(pH 50) and 50 CH3CN The injection volume was 20 microL and the flow-rate 10
mLmin HPLC analyses of the chloroacetates were performed using a Gynkotek
Pump M480 Gynkotek Gina 50 auto sampler and a diode array UV detector (340s
Gynkotek) UV-VIS detection was carried out at 200 nm At higher initial TCA
concentrations the chlorinated acetic acids were quantified by a modified HPLC
method (Husain et al 1992) Separation was performed on a ChromSphere C-18
(10 microm 250 times 46 mm ID) reversed-phase column Analytical conditions were
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 105
isocratic and the eluent consisted of 015 M (NH4)2SO4(aq) pH 55 The injection
volume was 20 microL and the flow-rate 10 mLmin HPLC analyses were performed
using a Series 10 Liquid Chromatographic Pump (Perkin-Elmer) and a SPD-10 A
VP UV-VIS detector (Shimadzu) UV-VIS detection was carried out at 210 nm
Chloride was determined in the GR-SO4 kinetic experiments using a flow injection
system with spectrophotometric detection (Cheregi amp Danet 1997)
53 Results and discussion
531 Product formation and reaction kinetics
Trichloroacetate was readily reduced to DCA by all the Fe(II)-bearing minerals
examined Only DCA was detected within the reaction time in all the Fe(II)-Fe(III)
mineral systems Experiments conducted with the various Fe(II)-Fe(III) mineral
systems and DCA confirmed that no significant reduction of DCA took place (data
not shown) Hence it is reasonable to assume that the further hydrogenolysis of
DCA to MCA is too slow to be detected within the experimental time frame here
The mass balance of TCA and DCA was almost complete in all suspensions ruling
out any alternative reaction pathways to reductive dechlorination Decarboxylation
of TCA producing chloroform and carbon dioxide requires high temperatures and
is therefore assumed not to take place at the experimental conditions applied here
(Atkins et al 1984) Based on these results we propose that the reductive
dechlorination of TCA by Fe(II)-bearing minerals proceeds via hydrogenolysis
(replacement of halogen by hydrogen) as reported for the transformation of TCA
by zero-valent iron (Hozalski et al 2001) Thus in order to reduce TCA to DCA
2 electrons corresponding to 2 Fe(II) are needed (Figure 51)
106 Chapter 5
Cl3CC
O
O- Cl2HCC
O
O-
2e- H+ Cl-
TCA DCA Figure 51 Proposed reductive transformation pathway of TCA
In the Fe(II)goe and Fe(II)lep systems we detected no TCA transformation in the
absence of either aqueous Fe(II) or pure or calcite-associated goethite and
lepidocrocite These results strongly indicate that reactive Fe(II) species associated
with the goethite and lepidocrocite surfaces are the reductants for TCA in these
systems The Fe(III) phases forming in the mineral suspensions were not
characterised and therefore the reaction stoichiometry cannot be assessed
At initial Fe(II) concentrations in large excess of initial TCA concentration we
found a pseudo 1 order rate law for the degradation of TCA by Fe(II)
[ ] [ ] [ b a TCAFe(II) TCA
sdotsdot=minus kdt
d ]
where a = 1 b = 1 and the observed pseudo 1 order rate constant kobs = k middot [Fe(II)]
At all [Fe(II)]0[TCA]0 ratios studied (6-738) TCA was transformed almost
quantitatively into DCA and the reaction kinetics followed pseudo 1 order kinetics
with respect to TCA (Figure 52) The observed pseudo 1 order rate constants for
the transformation of TCA by the various Fe(II)-Fe(III) mineral systems were
calculated as initial rates (ie max first two half-lives) from linear fits of (time ln
[TCA]t[TCA]0)-plots (Table 51) The amount of chloride produced during
reaction with GR-SO4 was always equivalent to the amount of TCA transformed
into DCA (Figure 52c) This also indicates that no significant further reduction of
DCA took place in GR-SO4 suspensions
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 107
Figure 52 Time course of TCA consumption and DCA and chloride production for a)
Fe(II)Goe ([Fe(II)tot]0 = 095 mM) b) Fe(II)Lep ([Fe(II)tot]0 = 091 mM) c) GR-SO4
([Fe(II)GR]0 = 962 mM) d) GR-CO3 ([Fe(II)tot]0 = 633 mM) and e) Magnetite ([Fe(II)tot]0 = 350
mM) Solid lines represent 1 order kinetic fits whereas symbols and dotted lines represent actual
data = TCA = DCA = Clndash
T
able
51
Exp
erim
enta
l con
ditio
ns a
nd p
seud
o 1
ord
er ra
te c
onst
ants
for t
he re
duct
ive
trans
form
atio
n of
TC
A b
y va
rious
Fe(
II)-
Fe(I
II) c
onta
inin
g m
iner
al sy
stem
s
Syst
em
Susp
ensi
on a
ge
(d)
[Fe(
II)] s
olid
a (m
M)
[Fe(
II)] a
q b
(mM
) [T
CA
] 0 (micro
M)
pHin
itcpH
endd
k obs
e (min
-1)
Surf
ace
area
(m
2 L)
k obs
f
(min
-1m
-2middotL
)
Fe(I
I)aq
1
0
030
434
nd
76
gn
dn
dn
d
Fe(I
I)G
oe
1
002
024
429
nd
78
g1
021
0-47
1 i
143
10-5
Fe(I
I)G
oe
1
013
094
543
77
70
225
10-4
71
i3
161
0-5
Fe(I
I)G
oe
coat
ing
1
023
40
066
484
765
80
g6
401
0-454
0 j
119
10-5
Fe(I
I)G
oe
coat
ing
1
0
150
8048
6n
d7
6 g12
43
10-4
540
j2
301
0-5
Fe(I
I)L
ep
1
0
020
2315
7n
d7
8 g0
751
0-48
0 i
094
10-5
Fe(I
I)L
ep
coat
ing
1
016
30
137
470
765
80
g2
821
0-454
0 j
052
10-5
Fe(I
I)L
ep
coat
ing
1
0
100
8141
7n
d7
7 g8
311
0-454
0 j
154
10-5
Fe3O
41
3
380
1251
38
107
8g
830
10-4
16
k5
311
0-4
Fe3O
477
112
56
556
70
70
153
10-4
52
k2
951
0-5
GR
-CO
31
5
940
3950
37
658
4g
761
10-4
419
l1
821
0-5
GR
-CO
32
7
60
147
88
568
2940
81
0-453
6 l
761
10-5
GR
-CO
332
73
003
563
85
80
490
10-4
515
l0
951
0-5
GR
-CO
314
2
3
530
005
629
nd
87 g
513
10-4
249
l2
061
0-5
GR
-SO
41
5
17-1
217
086
-13
910
5n
dn
d3
601
0-492
6 m
039
10-5
GR
-SO
41
6
22-1
027
093
-14
527
0n
dn
d3
761
0-488
1 m
043
10-5
GR
-SO
41
7
05-1
014
077
-17
950
0n
dn
d3
741
0-4 9
18
m0
411
0-5
GR
-SO
41
5
17-1
051
060
-16
510
00n
dn
d2
891
0-483
7 m
035
10-5
n
d =
not
det
ecte
d a
Ini
tial s
truct
ural
or a
dsor
bed
Fe(I
I) e
stim
ated
as [
Fe(I
I)to
tal]
ndash [F
e(II
) aq]
b In
itial
dis
solv
ed F
e(II
) mea
sure
d c
Sus
pens
ion
pH p
rior t
o TC
A a
dditi
on d
Sus
pens
ion
pH a
t rea
ctio
n
te
rmin
atio
n e
Pse
udo
1 o
rder
rate
con
stan
ts fo
r the
con
sum
ptio
n of
TC
A c
alcu
late
d fr
om in
itial
rate
s (m
ax f
irst t
wo
half-
lives
) f
Surf
ace
area
-nor
mal
ised
pse
udo
1 o
rder
rate
con
stan
ts g
pH
con
trol
th
roug
h pu
re C
aCO
3 and
05
C
O2(g
) h
pH
con
trol t
hrou
gh F
e(II
I) o
xide
-coa
ted
calc
ite a
nd 0
5
CO
2(g)
i Es
timat
ed u
sing
the
SSA
of t
he F
e(II
I) o
xide
app
lied
j E
stim
ated
usi
ng th
e SS
A o
f cal
cite
~1
m2 g
k E
stim
ated
ass
umin
g SS
A =
4 m
2 g (S
chw
ertm
ann
amp C
orne
ll 1
991)
frac12middot[
Fe(I
I) sol
id] 0middot
232
gmiddotm
ol-1
middot4 m
2 middotg-1
l E
stim
ated
ass
umin
g SS
A =
47
m2 g
(Will
iam
s amp S
cher
er 2
001)
frac14middot[F
e(II
) GR] 0middot
600
gmiddotm
ol-1
middot47
m2 middotg
-1 m
Est
imat
ed a
s in l
but u
sing
SSA
= 7
12
m2 middotg
-1 (C
hapt
er 4
thi
s wor
k)
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 109
532 Comparing rate constants obtained for the various Fe(II)-Fe(III) mineral
systems
Data for the systems containing iron oxide coated calcite were very similar to the
data obtained for the pure iron oxides (not shown in Figure 53) Since no SSA was
determined for magnetite in this study a SSA of 4 m2g was assumed
(Schwertmann amp Cornell 1991) However it should be noted that the magnetite
synthesized by Schwertmann and Cornell (1991) was prepared differently (ie
oxidation of Fe(II) by nitrate in a heated alkaline solution) from the magnetite
applied in this study The surface area-normalised pseudo 1 order kobs values
obtained for GR-CO3 GR-SO4 Fe(II)goethite and Fe(II)lepidocrocite were all
within the same order of magnitude (Figure 53a)
Figure 53 Average surface area-normalised pseudo 1 order rate constants for the degradation of
a) TCA (this work) b) hexachloroethane (Elsner et al 2004a) and c) carbon tetrachloride
(Amonette et al 2000 Pecher et al 2002 OrsquoLoughlin et al 2003c Elsner et al 2004b) by
GR-SO4 GR-CO3 (suspension age 1 d) Fe3O4 Fe(II)α-FeOOH and Fe(II)γ-FeOOH
Experimental conditions applied in this work [Fe(II)tot]0 = 025-107 mM in the goethite and
lepidocrocite suspensions [Fe(II)tot]0 = 025-116 mM in the GR-SO4 and GR-CO3 suspensions
pH 70-86 71-926 m2 mineral surface areaL Experimental conditions applied by Elsner et al
1 mM aqueous Fe(II) 25 m2 mineral surface areaL Experimental conditions applied in
references employed in c) [Fe(II)tot]0 = 1-83 mM 25-275 m2 mineral surface areaL GR-SO4 =
110 Chapter 5
sulfate green rust GR-CO3 = carbonate green rust Fe3O4 = magnetite α-FeOOH = goethite γ-
FeOOH = lepidocrocite
When comparing the rate constants for the Fe(II)-Fe(III) mineral systems found for
reduction of TCA in this study (Figure 2a) mixed valent Fe(II)-Fe(III) minerals
such as green rusts and magnetite containing structural Fe(II) within the mineral
lattice do not seem to be significantly more reactive than Fe(II)-Fe(III) mineral
systems containing Fe(II) associated with pure Fe(III) oxides Unlike most other
iron oxides GRs contain not only external Fe(II) reactive sites at the surface but
also internal sites in the space between consecutive Fe(II)-Fe(III) hydroxide layers
The GR interlayer thickness is a function of both the size and the charge of the
interlayer anion For solutes the Fe(II) within the GR hydroxide layer is accessible
at the outside basal planes and at the edges as well as through the interlayer under
certain conditions (see Figure 42 Chapter 4 this work) Due to electrostatic
forces oxidants holding different charge properties (anions cations neutral
molecules) may exhibit different affinities for the various reactive Fe(II) sites
present in GR As the reactive sites are located inat the Fe(II)-Fe(III) hydroxide
layers the rate of reaction depends on the hydroxide layer area which can be
accessed by the oxidant If the oxidant is able to exchange with the interlayer
anion reaction can take place both at outer and inner surfaces of the GR particles
and in total more reactive sites are available for the reaction Thus oxidant size
and charge primarily control its access to the internal sites in GRs XRD
characterization of the GR-SO4 crystals during reaction with TCA demonstrated
that the GR-SO4 interlayer spacing did not vary during reaction (Table 52) This
may indicate that TCA did not enter the GR-SO4 interlayers
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 111 Table 52 Diffraction angle d-spacing and width at half peak height (Wfrac12) for the 001 GR-SO4
diffraction peak as a function of time during reaction with TCA ([Fe(II)GR]0 = 4 mM [TCA]0 = 1
mM)
Time (min) Angle (deg2θ)
d001-spacing(nm)
Wfrac12(degθ)
0 9483 10821 0273
10 9494 10809 0287
215 9522 10777 0263
330 9550 10745 0273
510 9524 10775 0277
855 9509 10791 0253
1160 9467 10839 0268
We roughly estimated the molecular size of TCA by summing the covalent radii of
the individual atoms (see Supporting Information 74) When comparing the
molecular size of TCA with the GR-SO4 interlayer spacing of 061 nm it can be
concluded that only when the C-C bond is oriented perpendicular to the interlayer
plane does the size of TCA exceed the GR-SO4 interlayer spacing In contrast the
size of TCA exceeds the GR-CO3 interlayer spacing (026 nm) regardless of its
orientation Hence if TCA was intercalated in the GR-CO3 interlayer we would
expect the interlayer spacing to expand The same holds for intercalation of a
vertically oriented TCA in the GR-SO4 interlayer Supposedly both the low charge
and the size of TCA impeded its access to the GR-SO4 and GR-CO3 interlayers ie
the divalent sulphate and carbonate in the GR interlayers did not readily exchange
with the monovalent TCA since GR interlayers generally have a higher affinity for
divalent anions than for monovalent anions (Miyata 1983) Thus TCA did neither
access nor react with internal Fe(II) reactive sites in GR-SO4 which means that the
reaction between TCA and GR-SO4 took place at the external reactive Fe(II) sites
solely It is reasonable to assume that the same holds for the reaction between TCA
and GR-CO3 No significant aging effects eg rate constants varying as a function
of GR age were observed within 142 days (see Table 51) However the SSAs of
112 Chapter 5
the GR suspensions holding ages up to 142 days were not measured but estimated
assuming that the GR SSA did not decrease within the time frame
According to the Scherrer formula the width at half peak height (Wfrac12) of a
diffraction peak is inversely proportional to the average crystal dimension
perpendicular to the given crystal plane (Klug amp Alexander 1974) The average
GR-SO4 particle thickness perpendicular to the basal plane (Wfrac12 Table 52) was
constant during reaction implying that TCA reacts only at the edges and not at the
basal planes Assuming that the platy hexagonal GR-SO4 and GR-CO3 crystals
hold an average width of 1 microm and an average particle thickness of 35 nm (Hansen
and Koch 1998) the ratio of edge surface area to outer surface area is AedgeAouter ~
130 for GR-SO4 and 121 for GR-CO3 (see Supporting Information 73) This
means that only 3 of the outer surface area in GR-SO4 and 5 of the outer
surface area in GR-CO3 are available at the edges Assuming that TCA reacts at the
edges only and if employing the AedgeAouter in the estimation of the rate constants
the surface area-normalised pseudo 1 order rate constants for GR-SO4 and GR-
CO3 would be 20-30 times higher than the rate constants depicted in Figure 53a
Thus the reactivity of GRs normalised to their reactive surface area is much higher
than the reactivity normalised to their total surface area
533 Comparing with rate constants obtained for other chlorinated aliphatic
compounds
Though care must be taken when comparing kinetic parameters obtained at
different experimental conditions (eg pH [Fe(II)]0[TCA]0 ratios surface area to
volume ratios etc) it is interesting to compare our results to those reported for
hexachloroethane (Figure 53b data from Elsner et al 2004a) The reductive
transformation of hexachloroethane was investigated for various Fe(II)-bearing
minerals including Fe(II)goethite Fe(II)lepidocrocite and GR-SO4 in the presence
of 1 mM dissolved Fe(II) and 25 m2 mineral surface areaL at pH 72 except for the
GR-SO4 suspensions in which the dissolved Fe(II) concentrations were slightly
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 113
higher and pH = 8 The pseudo 1 order rate constants reported for
hexachloroethane are in the range 18middot10-4 ndash 75middot10-3 h-1middotm-2middotL (Elsner et al 2004a)
When comparing Figure 53a with Figure 53b it can be seen that the differences
in intrinsic reactivity of the Fe(II)-bearing mineral systems are more pronounced
for hexachloroethane than for TCA
Caution should also be advised to the different reaction mechanisms by which
hexachloroethane and TCA react The transfer of a single electron and the
formation of an alkyl radical upon removal of a chlorine atom constitute the first
and in most cases the rate-limiting step in the reduction of chlorinated aliphatic
compounds (Vogel et al 1987) Depending on the chemical structure of the
chlorinated aliphatic compound the resulting free alkyl radical may undergo
hydrogenolysis chloroelimination or dimerizationcoupling In the case of TCA
the free dichloroacetate radical most likely undergoes hydrogenolysis The almost
quantitative transformation of TCA to DCA confirms that hydrogenolysis is the
prevalent reaction mechanism in our mineral systems The pentachloroethyl radical
formed from hexachloroethane may undergo hydrogenolysis (producing
pentachloroethane) or dichloroelimination (producing tetrachloroethene) Elsner et
al (2004a) found that hexachloroethane was transformed quantitatively into
tetrachloroethylene for all minerals which strongly indicates that
dichloroelimination was the dominating reaction mechanism Another
polychlorinated aliphatic compound transformed mainly by hydrogenolysis under
reducing conditions is carbon tetrachloride Several studies have investigated the
reductive dechlorination of carbon tetrachloride by various Fe(II)-bearing minerals
including Fe(II)goethite and GR-SO4 and reported pseudo 1 order rate constants
in the order 152middot10-4 ndash 640middot10-4 h-1middotm-2middotL for Fe(II)goethite and 864middot10-4 h-1middotm-2middotL
for GR-SO4 (Amonette et al 2000 Pecher et al 2002 OLoughlin et al 2003c
Elsner et al 2004b) When comparing Figure 53a with Figure 53c it can be seen
that the range of magnitude of the rate constants and the differences in intrinsic
114 Chapter 5
reactivity of the Fe(II)-bearing mineral systems are similar for carbon tetrachloride
and TCA
534 Factors controlling the reactivity of surface-bound Fe(II)
The reactivity of an oxidant towards Fe(II) surface species cannot be predicted
from the reduction potentials of the redox couple alone In heterogeneous systems
processes such as mass transfer and adsorptiondesorption may have a rate-limiting
effect on the overall reaction rate If the adsorption follows a saturation-type
sorption isotherm (eg Langmuir) the sorbate (oxidant) concentration at the
surface will vary non-linearly with the total amount of oxidant added This
dependence will have to be taken into account when establishing rate laws for the
heterogeneous reactions and when testing the hypothesis that the reaction rates
depend on the sorbed concentration of the oxidants pH has a strong impact on the
sorption and thereby on the availability of ionizable oxidants At the pH values
applied here the chloroacetates are fully dissociated (pKa (TCA) = 066 pKa
(DCA) = 135 pKa (MCA) = 287) However we found the sorption of TCA to be
negligible in suspensions of pure calcite goethitecalcite and lepidocrocitecalcite
at pH 76-77 Moreover the mass balance of TCA and DCA was almost complete
in all suspensions and therefore loss of TCA or DCA due to adsorption at mineral
surfaces or incorporation in the GR anion interlayers can be ruled out Calcite has a
much lower adsorption capacity than most iron oxides hence we anticipate that
goethite and lepidocrocite control the adsorption of TCA and DCA in both the pure
FeOOH and the FeOOHcalcite suspensions This was supported by our
experimental results demonstrating that the presence of a calcite surface - either
pure or as a support for goethite and lepidocrocite coatings - did not exert any
noticeable effect on the reaction rates (see Table 51) In addition the surface area-
normalised rate constants for mineral systems containing goethite or lepidocrocite
in pure form and mineral systems containing goethite or lepidocrocite as coatings
on calcite were very similar In heterogeneous reactions mass transfer in bulk
solution becomes the rate-limiting step when the surface reaction is much faster
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 115
than the diffusion of the reacting species to the reactive surface However at the
low rate constants obtained here the reaction of TCA with the Fe(II)-bearing
minerals is not likely to be mass transfer limited (see Supporting Information 72)
One very important factor affecting heterogeneous redox reactions is pH which
influences the speciation of the complexes in solution and at mineral surfaces as
well as the stability of the more soluble Fe(II)-containing minerals such as GRs In
contrast to aqueous Fe(II) complexes it is not possible to predict the reactivity of
Fe(II) surface species as their reduction potentials are unknown In the absence of
specifically adsorbing solutes other than H+ the surface charge of the Fe(III)
oxides goethite and lepidocrocite is determined by the surface densities of the
charged surface species equivFeOH2+ and equivFeOndash whereas the surface charge of calcite
is determined by the density of the surface species equivCO3ndash equivCaOH2
+ and equivCaOndash
(Stumm 1992 Van Cappellen et al 1993) The point of zero charge (pHpzc) of
pure calcite is in the pH range 7-11 and depends on the partial pressure of carbon
dioxide pCO2 The higher the pCO2 the lower the pHpzc At the experimental
conditions applied here (05 = 0005 atm CO2(g)) the pHpzc = 82 for calcite
(Table 53) As only 10-11 mg Fe(III) of goethite and lepidocrocite was coated
onto calcite we assumed a pHpzc of 82 for the goethite and lepidocrocite coated
calcite particles as well The pHpzc values for green rusts are unknown
116 Chapter 5
Table 53 Specific surface areas and point of zero charge of the various iron minerals in pure form as well as goethite and lepidocrocite coated onto calcite
Mineral Structural formula SSA (m2g) pHpzc
GR-SO4 FeII4FeIII
2(OH)12SO4middot3H2O 71 a -
GR-CO3 FeII4FeIII
2(OH)12CO3middot3H2O 47 b -
Magnetite Fe3O4 - 69 e
Goethite α-FeOOH 16 c 85 f
Lepidocrocite γ-FeOOH 18 c 73 e
Calcite CaCO3 le 1 d 82 g
Goe coating - le 1 d 82 h
Lep coating - le 1 d 82 h
a Chapter 4 this work b Williams amp Scherer 2001 c Product information by Bayer d The SSA of calcite was
quantified to le 1 m2g The detection limit of our BET method was 1 m2g e Charlet et al 1998a f Liger et al
1999 g Van Cappellen et al 1993 h Same as for calcite
The surface hydroxyl groups on iron oxides may be both singly (equivFe-OH) doubly
(equivFe2-OH) triply (equivFe3-OH) and geminally (equivFe-(OH)2) coordinated (Cornell amp
Schwertmann 1996 Stumm 1992) The differently coordinated surface hydroxyl
groups are not equally reactive Adsorption reactions involve only singly
coordinated surface groups and therefore only this kind of hydroxyl groups on iron
oxides will be considered here (Cornell amp Schwertmann 1996) Hence the
predominant surface sites available for adsorption in pure suspensions of Fe(III)
oxides are equivFeOH0 equivFeOH2+ and equivFeOndash In the presence of dissolved Fe(II)
equivFeIIIOFeIIOH0 equivFeIIIOFeIIOndash and equivFeIIIOFeII+ constitute the main reactive sites at
the Fe(III) oxide surfaces (Liger et a 1999) Assuming that Fe2+ and other cationic
Fe(II) species are the dominating adsorbates on the mineral surfaces in our
experiments we expect the actual pHpzc to be higher than the pHpzc of the pure
oxides listed in Table 53 Hence at pHlt82 where most of our experiments were
conducted all the mineral surfaces presumably carry net positive charges
At pH 70 where Fe2+ is still the predominant Fe(II) species in solution (~50) we
expect that equivFeIIIOFeIIOH equivFeIIIOFeIIOndash and equivFeIIIOFeII+ constitute the main
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 117
reactive sites at the Fe(III) oxide surfaces as suggested by Liger et a 1999 As pH
increases from 70 to 87 the Fe(II) carbonate complexes become increasingly
important in solution at the expense of the Fe2+ FeCl+ FeSO40 and FeOH+ species
(King 1998) Fe(II) carbonate complexes do not bind at the oxide surface as
readily as the aquo or hydroxo complexes of Fe(II) but carbonate itself sorbs
readily to Fe(III) oxide surfaces through which the Fe(III) oxide surface is coated
by inner-sphere monodentate equivFeIIIOCOOH0 surface complexes (Villalobos amp
Leckie 2000 amp 2001) The presence of carbonate shifted the sorption edge for the
Fe(II) adsorption on goethite from pH 58 to 78 and the authors hypothesized this
to be a result of the formation of aqueous and surface Fe(II)-carbonate complexes
and to competition between carbonate and Fe(II) for Fe(III) oxide surface sites
(Vikesland amp Valentine 2002) Similarly monodentate surface complexes like
equivFeIIICl0 and equivFeIIIOSO3ndash as well as ternary monodentate surface complexes like
equivFeIIIOFeIICl0 and equivFeIIIOFeIIOSO3ndash and ternary bidentate surface complexes such
as (equivFeIIIO)2FeIIOSO3 may form at Fe(III) oxide surfaces when Fe(II) chloride and
sulfate are present in solution (Ostergren et al 2000 Kim et al 2004) However
the effects of anionic ligands such as chloride and sulfate on Fe(II) adsorption at
Fe(III) oxide surfaces and the reactivity of Fe(II) carbonate chloride and sulfate
surface sites are still unknown and need to be evaluated (see Supporting
Information 75) Thus we do not know whether chloride and sulfate decrease or
increase the Fe(II) sorption in our mineral systems We can only report that we did
not detect any significant differences in the rate of TCA transformation between
the mineral suspensions containing carbonate chloride and sulfate respectively
Hence we anticipate that equivFeIIIOFeIIOH equivFeIIIOFeIIOndash and equivFeIIIOFeII+ constitute
the main reactive sites at the Fe(III) oxide surfaces within the whole pH range 70-
87 This might also explain why we did not detect any obvious systematic pH
effect in the Fe(II)-Fe(III)-systems (see Table 51) In the case of
hexachloroethane the reactivity order GR-SO4gtgoethitegtmagnetitegtlepidocrocite
may be rationalized by the variations in surface site densities and total amount of
118 Chapter 5
Fe(II) sorbed on the iron minerals (see Supporting Information 75) as well as the
different speciations and reactivities of the Fe(II) surface sites on the iron minerals
535 Comparison with biotic and other abiotic systems
Only one report on abiotic transformation of TCA is found in the literature and the
study demonstrates the reductive dechlorination of TCA to MCA by Fe(0)
(Hozalski et al 2001) The authors reported a pseudo 1 order rate constant of
60middot10-4 min-1middotm-2middotL for the transformation of TCA to DCA and a pseudo 1 order
rate constant of 225middot10-4 min-1middotm-2middotL for the transformation of DCA to MCA at
[Fe(0)]0 = 025 M [TCA]0 = 100-200 microM and pH 36-62 The rate constant for
TCA reduction by Fe(0) is 10-300 times faster than the rate constants reported for
the Fe(II)-bearing mineral systems here
There is abundant evidence that soil microorganisms and fungi can dechlorinate
TCA but only little is known about the bacteria and processes involved in the
biodegradation of TCA Biotransformation of TCA has been found at both oxic
and anoxic conditions Most of the microorganisms isolated grow feebly on TCA
as a sole source of carbon (Foy 1975 Weightman et al 1992 De Wever et al
2000) Only one bacterium capable of growing on TCA as the sole carbon and
energy source has been characterized (Yu amp Welander 1995) In addition
anaerobic bacteria coupling co-metabolic growth to reductive dechlorination of
TCA have been isolated (Weightman et al 1992 De Wever et al 2000) The
inability to grow on the less chlorinated acids DCA and MCA is a notable feature
of both the aerobic and anaerobic bacteria Complete transformation of TCA to
methane and carbon dioxide has only been found when abiotic and biotic processes
were combined (Egli et al 1989) The abiotic transformation of TCA to DCA
occurred spontaneously in the presence of sterile activated charcoal whereas the
DCA formed was further degraded to methane and carbon dioxide by a mixed
culture of methanogenic bacteria However the abiotic reductant(s) responsible for
the transformation of TCA to DCA was not reported (Egli et al 1989)
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 119
The rate constants obtained in this work suggest that the Fe(II)-bearing mineral
systems may be important reductants of TCA in natural suboxic environments In
natural iron-rich soils holding specific surface areas of 22 m2g (Kenneke amp
Weber 2003) average bulk densities of 265 gcm3 and porosities of 25 and
containing 2 iron oxides a rough estimation of the half-life of TCA amounts to
47 minutes when applying the average surface area-normalised rate constant
obtained for all the Fe(II)-Fe(III) mineral systems in this work (1middot10-3 h-1middotm-2middotL)
This estimation is based on the assumption that enough reactive Fe(II) is available
in these soils The natural iron-reducing sediment investigated by Kenneke and
Weber (2003) contained 80 microM Fe(II) in the soil solution and 315 micromole Fe(II) per
g sediment At such low Fe(II) concentrations the overall rate of abiotic
transformation of TCA in natural soils and sediments is most likely limited by the
regeneration of reactive Fe(II) Hence the continuous regeneration of reactive
Fe(II) surface sites by adsorption of abiotically or microbially produced Fe(II) may
further the long-term abiotic transformation of TCA in such environments
54 Conclusions
This work demonstrates that various Fe(II)-Fe(III) minerals systems including GR-
SO4 GR-CO3 magnetite Fe(II)goethite and Fe(II)lepidocrocite readily transform
TCA to DCA Dichloroacetate was not further reduced to MCA or acetate by any
of the Fe(II)-bearing minerals The surface area-normalised pseudo 1 order rate
constants obtained for the reductive transformation of TCA by the various Fe(II)-
bearing minerals did not differ significantly from each other The results obtained
in this work infer that under natural geochemical conditions Fe(II)-bearing mineral
systems may play an important role in the overall transformation of TCA Thus
not only microbial degradation but also abiotic reductive transformation of TCA by
Fe(II)-bearing minerals may govern the fate of TCA in natural subsurface
environments This holds both for natural systems like iron-rich suboxic soils and
sediments as well as engineered systems like permeable reactive barriers of zero-
120 Chapter 5
valent iron implemented for on-site remediation where both Fe(0) and solid or
surface-bound Fe(II) corrosion intermediates may transform TCA
Acknowledgments
We would like to thank Susanne Guldberg for performing the experimental work comprising
GR-SO4
References
Ahlers J Regelmann J Riedhammer C (2003) Environmental risk assessment of airborne trichloroacetic acid - a contribution to the discussion of the significance of anthropogenic and natural sources Chemosphere 52 531-537 Amonette JE Workman DJ Kennedy DW Fruchter JS Gorby YA (2000) Dechlorination of carbon tetrachloride by Fe(II) associated with goethite Environmental Science and Technology 34 4606-4613 Atkins PJ Gold V Marsh R (1984) The decarboxylation of trichloroacetic acid and the reactions of the trichloromethyl anion with 135-trinitrobenzene and with hydrogen ions kinetic measurements in dimethyl sulphoxide solution Journal of the Chemical Society Perkin Transactions 2 7 1239-1245 Berg M Muumlller SR Muumlhlemann J Wiedmer A Schwarzenbach RP (2000) Concentrations and mass fluxes of chloroacetic acids and trifluoroacetic acid in rain and natural waters in Switzerland Environmental Science and Technology 34 2675-2683 Brunauer S Emmett PH Teller E (1938) Adsorption of gases in multimolecular layers Journal of American Chemical Society 60 309-319 Butler EC Hayes KF (1998) Effects of solution composition and pH on the reductive dechlorination of hexachloroethane by iron sulfide Environmental Science and Technology 32 1276-1284 Butler EC Hayes KF (1999) Kinetics of the transformation of trichloroethylene and tetrachloroethylene by iron sulfide Environmental Science and Technology 33 2021-2027 Charlet L Liger E Gerasimo P (1998a) Decontamination of TCE- and U-rich water by granular iron Role of sorbed Fe(II) Journal of Environmental Engineering 124 25-30 Charlet L Silvester E Liger E (1998b) N-compound reduction and actinide immobilisation in surficial fluids by Fe(II) the surface FeIIIFeIIOH0 species as major reductant Chemical Geology 151 85-93 Cheregi M Danet AF (1997) Flow injection determination of chloride ions with spectrophotometric detection Analytical Letters 30 2847-2858 Cornell RM Schwertmann U (1996) The iron oxides Structure properties reactions occurrence and uses VCH Verlagsgesellschaft mbH Weinheim
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 121 Cui D Eriksen TE (1996) Reduction of pertechnetate by ferrous iron in solution influence of sorbed and precipitated Fe(II) Environmental Science and Technology 30 2259-2262
Egli C Thuumler M Suter D Cook AM Leisinger T (1989) Monochloro- and dichloroacetic acids as carbon and energy sources for a stable methanogenic mixed culture Archives of Microbiology 152 218-223 Elsner M Haderlein SB Schwarzenbach RP (2004a) Reactivity of Fe(II)-bearing minerals towards reductive transformation of organic contaminants Environmental Science and Technology 38 799-807 Elsner M Haderlein SB Kellerhals T Luzi S Zwank L Angst W Schwarzenbach RP (2004b) Mechanisms and products of surface-mediated reductive dehalogenation of carbon tetrachloride by Fe(II) on goethite Environmental Science and Technology 38 2058-2066 Erbs M Hansen HCB Olsen CE (1999) Reductive dechlorination of carbon tetrachloride using iron(II)iron(III)-hydroxide-sulphate (green rust) Environmental Science and Technology 33 307-311 Euro Chlor (2001) Trichloroacetic acid in the environment a dossier Euro Chlor Brussels and the European Chlorinated Solvent Association Fadrus H Maly J (1975) Suppression of iron(III) interference in the determination of iron(II) in water by the 110-phenanthroline method The Analyst 100 549-554 Fahimi IJ Keppler F Schoumller HF (2003) Formation of chloroacetic acids from soil humic acid and phenolic moieties Chemosphere 52 513-520 Foy CL (1975) The chlorinated aliphatic acids In Herbicides Chemistry degradation and mode of action Kearney PC Kaufman DD (eds) Marcel Dekker Inc 399-452 Gander JW Parkin GF Scherer MM (2002) Kinetics of 111-trichloroethane transformation by iron sulfide and a methanogenic consortium Environmental Science and Technology 36 4540-4546 Haiber G Jacob G Niedan V Nkusi G Schoumller HF (1996) The occurrence of trichloroacetic acid (TCAA) ndash indications of a natural production Chemosphere 33 839-849
Hansen HCB (1989) Composition stabilization and light absorption of Fe(II)Fe(III) hydroxycarbonate (green rust) Clay Minerals 24 663-669 Hansen HCB Guldberg S Erbs M Koch CB (2001) Kinetics of nitrate reduction by green rusts ndash effects of interlayer anion and Fe(II)Fe(III) ratio Applied Clay Science 18 81-91 Hansen HCB Koch CB (1997) A comparison of nitrate reduction by carbonate and sulphate forms of green rust Kodama H Mermut A R Torrance J K (eds) Proceedings of the 11th International Clay Conference Ottawa Canada Clays for our future 11 295-302 Hoekstra EJ de Leer EWB Brinkman UATh (1999a) Mass balance of trichloroacetic acid in the soil top layer Chemosphere 38 551-563 Hoekstra EJ de Leer EWB Brinkman UATh (1999b) Findings supporting the natural
122 Chapter 5
formation of trichloroacetic acid in soil Chemosphere 38 2875-2883 Hofstetter TB Heijman CG Haderlein SB Holliger HC Schwarzenbach RP (1999) Complete reduction of TNT and other (poly)nitroaromatic compounds under iron-reducing subsurface conditions Environmental Science and Technology 33 1479-1487 Hofstetter TB Schwarzenbach RP Haderlein SB (2003) Reactivity of Fe(II) species associated with clay minerals Environmental Science and Technology 37 519-528 Hozalski RM Zhang L Arnold WA (2001) Reduction of haloacetic acids by Fe0 Implications for treatment and fate Environmental Science and Technology 35 2258-2263 Husain S Narsimha R Alvi SN Rao RN (1992) Monitoring the effluents of the trichloroacetic acid process by high-performance liquid chromatography Journal of Chromatography 600 316-319 Hwang I Batchelor B (2000) Reductive dechlorination of tetrachloroethylene by Fe(II) in cement slurries Environmental Science and Technology 34 5017-5022 Jordan A Frank H (1999) Trifluoroacetate in the environment Evidence for sources other than HFCHCFCs Environmental Science and Technology 33 522-527 Kenneke JF Weber EJ (2003) Reductive dehalogenation of halomethanes in iron- and sulfate-reducing sediments 1 reactivity pattern analysis Environmental Science and Technology 37 713-720 Kim CS Rytuba JJ Brown Jr GE (2004) EXAFS study of mercury(II) sorption to Fe- and Al-(hydr)oxides II Effects of chloride and sulphate Journal of Colloid and Interface Science 270 9-20 King DW (1998) Role of carbonate speciation on the oxidation rate of Fe(II) in aquatic systems Environmental Science and Technology 32 2997-3003 Klausen J Troumlber SP Haderlein SB Schwarzenbach RP (1995) Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions Environmental Science and Technology 29 2396-2404 Klug PH Alexander LE (1974) X-ray diffraction procedures John Wiley amp Sons Inc Koch CB Hansen HCB (1997) Reduction of nitrate to ammonium by sulphate green rust Advances in GeoEcology 30 373-393 Kuumlhn R Pattard M (1990) Results of the harmful effects of water pollutants to green algae (Scenedesmus subspicatus) in the cell multiplication inhibition test Water Research 24 31-38 Lee W Batchelor B (2002a) Abiotic reductive dechlorination of chlorinated ethylenes by iron- bearing soil minerals 1 Pyrite and magnetite Environmental Science and Technology 36 5147- 5154 Lee W Batchelor B (2002b) Abiotic reductive dechlorination of chlorinated ethylenes by iron-bearing soil minerals 2 Green rust Environmental Science and Technology 36 5348- 5354
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 123 Liger E Charlet L Van Cappellen P (1999) Surface catalysis of uranium (VI) reduction by iron(II) Geochimica et Cosmochimica Acta 63 2939-2955 Lignell R Heinonen-Tanski H Uusi-Rauva A (1984) Degradation of trichloroacetic acid (TCA) in soil Acta Agriculturae Scandinavia 34 3-8 Loyaux-Lawniczak S Refait Ph Lecomte P Ehrhardt J Geacutenin JMR (1999) The reduction of chromate ions by Fe(II) layered hydroxides Hydrology and Earth System Sciences 3 593-599 Luther III GW (1990) The Frontier-Molecular-Orbital theory approach in geochemical processes in W Stumm Ed Aquatic Chemical kinetics John Wiley and Sons New York pp 173-198 McCulloch A (2002) Trichloroacetic acid in the environment Chemosphere 47 667-686 Miyata S (1983) Anion-exchange properties of hydrotalcite-like compounds Clays and Clay Minerals 31 305-311 Muumlller SR Zweifel H-R Kinnison DJ Jacobsen JA Meier MA Ulrich MM Schwarzenbach RP (1996) Occurrence sources and fate of trichloroacetic acid in Swiss lakes Environmental Toxicology and Chemistry 15 1470-1478 OLoughlin EJ Burris DR (2003) Reduction of halogenated ethanes by green rust Environmental Toxicology and Chemistry 23 41-48 OLoughlin EJ Kelly SD Cook RE Csencsits R Kemner KM (2003a) Reduction of uranium(VI) by mixed iron(II)iron(III) hydroxide (green rust) Formation of UO2 nanoparticles Environmental Science and Technology 37 721-727 OLoughlin EJ Kelly SD Kemner KM Csencsits R Cook RE (2003b) Reduction of AgI AuIII CuII and HgII by FeIIFeIII hydroxysulfate green rust Chemosphere 53 437-446 OLoughlin EJ Kemner KM Burris DR (2003c) Effects of AgI AuIII and CuII on the reductive dechlorination of carbon tetrachloride by green rust Environmental Science and Technology 37 2905-2912 Ostergren JD Brown Jr GE Parks GA Persson P (2000) Inorganic ligand effects on Pb(II) sorption to goethite (α-FeOOH) II Sulfate Journal of Colloid and Interface Science 225 483-493 Pecher K Haderlein SB Schwarzenbach RP (2002) Reduction of polyhalogenated methanes by surface-bound Fe(II) in aqueous suspensions of iron oxides Environmental Science and Technology 36 1734-1741 Reimann S Grob K Frank H (1996) Chloroacetic acids in rainwater Environmental Science and Technology 30 2340-2344 Ruumlgge K Hofstetter TB Haderlein SB Bjerg PL Knudsen S Zraurig C Mosbaeligk H Christensen TH (1998) Characterization of predominant reductants in an anaerobic leachate- affected aquifer by nitroaromatic probe compounds Environmental Science and Technology 32
124 Chapter 5
23-31 Sarzanini C Bruzzoniti MC Mentasti E (1999) Preconcentration and separation of haloacetic acids by ion chromatography Journal of Chromatography A850 197-211 Schwertmann U Cornell RM (1991) Iron oxides in the laboratory Preparation and characterization VCH Verlagsgesellschaft mbH Weinheim Scott BF Mactavish DC Spencer C Strachan WMJ Muir DCG (2000) Haloacetic acids in Canadian lake waters and precipitation Environmental Science and Technology 34 4266-4272 Scott BF Spencer C Marvin CH Mactavish DC Muir DCG (2002) Distribution of haloacetic acids in the water columns of the Laurentian Great Lakes and Lake Malawi Environmental Science and Technology 36 1893-1898 Strathmann TJ Stone AT (2003) Mineral surface catalysis of reactions between FeII and oxime carbamate pesticides Geochimica et Cosmochimica Acta 67 2775-2791 Stumm W (1992) Chemistry of the solid-water interface John Wiley amp Sons Inc Van Cappellen P Charlet L Stumm W Wersin P (1993) A surface complexation model of the carbonate mineral-aqueous solution interface Geochimica et Cosmochimica Acta 57 3505- 3518 Vikesland PJ Valentine RL (2002) Iron oxide surface-catalyzed oxidation of ferrous iron by monochloramine implications of oxide type and carbonate on reactivity Environmental Science and Technology 36 512-519 Villalobos M Leckie JO (2000) Carbonate adsorption on goethite under closed and open CO2 conditions Geochimica et Cosmochimica Acta 64 3787-3802 Villalobos M Leckie JO (2001) Surface complexation modeling and FTIR study of carbonate adsorption to goethite Journal of Colloid and Interface Science 235 15-32 Vogel TM Criddle CS McCarty PL (1987) Transformations of halogenated aliphatic compounds Environmental Science and Technology 21 722-736 Weightman AL Weightman AJ Slater JH (1992) Microbial dehalogenation of trichloroacetic acid World Journal of Microbiology and Biotechnology 8 512-518 De Wever H Cole JR Fettig MR Hogan DA Tiedje JM (2000) Reductive dehalogenation of trichloroacetic acid by Trichlorobacter thiogenes gen nov spnov Applied and Environmental Microbiology 66 2297-2301 Williams AGB Scherer MM (2001) Kinetics of chromate reduction by carbonate green rust Environmental Science and Technology 35 3488-3494 Yu P Welander T (1995) Growth of an aerobic bacterium with trichloroacetic acid as the sole source of energy and carbon Applied Microbiology and Biotechnology 42 769-774
Conclusions and Outlook 125
6 Conclusions and Outlook The work presented in this dissertation adds to the understanding of how Fe(II)-
bearing minerals like green rusts (GRs) vivianite (Fe2(PO4)2sdot8H2O) magnetite
(Fe3O4) and Fe(II) associated with goethite and lepidocrocite may form and react in
nature In order to elucidate the role of bacteria in the formation of GRs in natural
soils and sediments we studied the iron mineral phases forming as a result of the
activity of iron-respiring bacteria In chapter 2 the Fe-containing products formed
by anaerobic autotrophic denitrifying Fe(II)-oxidizing bacteria (FeOB) were
examined The culture medium applied contained high levels of bicarbonate and
phosphate and is typically used in this kind of studies as it provides excellent
conditions for the nitrate-reducing FeOB Fe(II) was present initially as a whitish
solid Fe(II) hydroxy phosphate (vivianite) and as soluble Fe(II) The results
obtained demonstrate that the denitrifying FeOB produce poorly crystalline
goethite via a greenish Fe(III)-enriched vivianite intermediate Moumlssbauer
spectroscopic analyses provided no evidence of green rust formation At low
phosphate concentrations where vivianite does not control the Fe(II) activity it is
reasonable to assume that siderite (FeCO3) precipitates initially and that carbonate
GR phases may form during biooxidation At low bicarbonate concentrations we
would expect Fe(II) sulfate or chloride species to dominate initially (depending on
the Fe(II) source applied) and sulfate GR or chloride GR to form during
biooxidation In chapter 3 we investigated the Fe-containing products formed
during reduction of common Fe(III) oxides by the anaerobic dissimilatory Fe(III)-
reducing microorganism Shewanella algae BrY S algae BrY reduced substantial
amounts of the initial Fe(III) and green and blackish mineral phases were produced
within 1-2 weeks after inoculation Moumlssbauer spectroscopic analyses showed that
the green and black precipitates consisted of green rust and vivianite
We studied the reactivity of synthetic GRs towards reducible organic pollutants in
order to asses the potential significance of GR phases for the fate of such
126 Chapter 6
compounds To this end we used nitroaromatic compounds (NACs) and
chlorinated acetates as suitable model compounds for studying redox reactions
potentially relevant in the environment In chapter 4 we investigated the relative
reactivity of outer and inner Fe(II) reactive sites in synthetic sulfate green rust
(GR-SO4) by using a series of structurally closely related compounds with different
charge properties as ldquoreactive probesrdquo The probe compounds included
nitrobenzene 2-nitrophenol 4-nitrotoluene 4-chloronitrobenzene and 4-
nitrophenylacetic acid Our results demonstrated that NACs are completely
reduced to their corresponding anilines by GR-SO4 The reactions followed pseudo
1 order kinetics with respect to NAC and the surface area-normalised pseudo 1
order rate constants obtained were 016ndash465middot10-4 s-1middotm-2middotL at [Fe(II)GR]0 = 103-
1260 mM [NAC]0 = 20-102 microM and pH 84-86 Neither mass transfer control nor
surface saturation kinetics could account for the similarity of the surface-
normalised pseudo 1 order rate constants obtained for the reduction of the neutral
and anionic NACs by GR-SO4 These observations suggest that the reaction
between NAC and GR-SO4 takes place at the external reactive Fe(II) sites At low
initial Fe(II)GR concentrations the external reactive Fe(II) sites were depleted and
the regeneration of new external reactive sites eventually controlled the reduction
of the NACs by GR-SO4 In chapter 5 we examined the reactivity of various
Fe(II)-Fe(III) mineral systems towards trichloroacetic acid (TCA) and
dichloroacetate (DCA) in laboratory batch experiments imitating natural
conditions The Fe(II)-Fe(III)-systems investigated included GR-SO4 carbonate
green rust magnetite Fe(II)goethite and Fe(II)lepidocrocite TCA was readily
reduced to DCA by all Fe(II)-containing minerals The reactions followed pseudo
1 order kinetics with respect to TCA and the surface area-normalised pseudo 1
order rate constants obtained were 033ndash76middot10-5 min-1middotm-2middotL at [Fe(II)]0 = 025ndash
116 mM [TCA]0 = 15ndash1000 microM and pH 70ndash87 Our results showed no
significant differences regarding product distribution and surface area-normalised
reaction rate constants between the Fe(II)-Fe(III)-systems DCA was not further
Conclusions and Outlook 127
reduced to monochloroacetate (MCA) or acetate in any of the systems within the
time frame in our experiments
As suggested in chapters 2 and 3 sufficient evidence must be provided and caution
should be exercised when proclaiming new biogenic minerals The study of
microbially produced GRs is still in its infancy and more research is needed in
order to elucidate the role of bacteria in the formation of GRs in natural soils and
sediments The results presented in chapter 2 indicate that microbiological
processes may be responsible for the oxidation of vivianite and metavivianite
((FeII3-xFeIII
x)(PO4)2(OH)xmiddot(8-x)H2O x gt 12) in natural subsurface environments
In chapter 3 we demonstrated that GRs may be produced microbially at conditions
including low carbon and Fe(III) concentrations as well as the exclusion of
synthetic electron shuttles and pH buffers The role of microbial processes in the
redox cycling of iron in the subsurface and the ways in which these processes can
be coupled to contaminant remediation are currently active areas of research Zero-
valent iron has been the most extensively studied reductant for the treatment of
many inorganic and organic contaminants and is currently the most commonly
used material for the construction of permeable reactive barriers (PRB) but a
detailed understanding of the processes involved in the reduction of these
pollutants by Fe(0) is lacking (Scherer et al 2000) Potentially reactive Fe(II)-
bearing corrosion products identified in iron metal columns and barriers include
magnetite siderite Fe(II) sulfides green rusts as well as Fe(II) sorbed to mineral
surfaces (Gu et al 1999 Roh et al 2000) The formation of reactive Fe(II)-
bearing minerals like GRs may explain the effective long-term operation of zero-
valent iron PRBs despite the formation of thick oxide films Thus natural in situ
PRBs might be created by stimulating the activity of anaerobic dissimilatory
Fe(III)-reducing bacteria and the subsequent formation of Fe(II) species such as
GRs Furthermore suspensions of synthetic GRs which are easily prepared from
relatively inexpensive commodity chemicals may also be injected and dispersed
into the subsurface
128 Chapter 6
The reductive transformation of NACs and TCA by GRs is relevant to
understanding the processes responsible for their degradation in the subsurface and
the development of innovative technologies for their remediation The results
obtained in chapters 4 and 5 indicate that GRs may play a significant role in the
reductive transformation of NACs and TCA in natural subsurface environments
Furthermore our results suggest that mainly the outer Fe(II) sites in GRs are
utilized in the reaction with neutral and monovalent anionic compounds and that
these sites may be replenished eg by reduction of the oxidized surface sites or
adsorption of Fe(II) from solution The continuous restoration of Fe(II) surface
sites in GRs may promote their long-term reactivity towards reducible
contaminants
References
Gu B Phelps TJ Liang L Dickey MJ Roh Y Kinsall BL Palumbo AV Jacobs GK (1999) Biochemical dynamics in zero-valent iron columns Implications for permeable reactive barriers Environmental Science and Technology 33 2170-2177 Roh Y Lee SY Elless MP (2000) Characterization of corrosion products in the permeable reactive barriers Environmental Geology 40 184-194 Scherer MM Richter S Valentine RL Alvarez PJJ (2000) Chemistry and microbiology of permeable reactive barriers for In Situ groundwater clean up Critical Reviews in Environmental Science and Technology 30 363-411
Supporting Information I
7 Supporting Information
71 Estimation of the one-electron reduction potential for 4-NPA
The one-electron reduction potential of the half-reaction for a given NAC 1hE
ArNO2 + e- ArNO2
can be used for comparing reduction rates of different NACs in a given system
The formation of the nitroaryl radical is the rate-determining step in the overall rate
of the reduction of a NAC to the corresponding aniline The difference between the
of a NAC and a given reductant is proportional to the change in standard free
energy for the transfer of the first electron ∆G
1hE
1degrsquo If a linear relationship between
the free energy of activation and ∆G1degrsquo is assumed the values of various NACs
can be a measure of their relative reactivity with a given reductant
1hE
As neither the one-electron reduction potential for 4-nitrophenylacetic acid (4-
NPA) nor the Hammett constant for the acetic acid substituent could be found in
the literature the one-electron reduction potential for 4-NPA was estimated by
application of a linear free energy relationship (LFER) to experimental data
Kinetic experiments were conducted in order to obtain the pseudo 1 order rate
constant for the reduction of 4-NPA by a model hydroquinone (reduced
juglone (8-hydroxy-14-naphthoquinone) in the presence of HS
minusHJUGk
ndash) The reduction of
a NAC by juglone follows the rate law
[ ] [ ] [ ] [ ] [ ] [ ]2222 ArNOJUGfkArNOHJUGkArNOk
dtArNOd
totHJUGHJUGHJUGobs sdotsdotsdot=sdotsdot=sdot=minus minusminusminusminus
and the was deducted from a LFER 1hE
II Chapter 7
bEak hHJUG +sdot=minus 059160
log1
for which a and b values have been established for a range of NACs with known
values (Hofstetter et al 1999) An excellent correlation of and log
has been found to exist over a range of 250 mV corresponding to more than 5 order
of magnitude for This is due to the fact that the actual transfer of the first
electron is the rate-determining step under the experimental conditions chosen
1hE 1
hE minusHJUGk
minusHJUGk
For comparison experiments with 4-nitrotoluene (4-NT) were also conducted The
pseudo 1 order rate constants for the reduction of 4-NPA with juglone were
corrected for the reduction of 4-NPA with only HSndash (control experiments
containing no juglone)
[ ]minuslowast
minusminus
minus
minus=
HJUGkk
k HSHJUGHJUG
where (MminusHJUGk -1middots-1) is the rate constant for a compound in the presence of only
juglone (slowastminusHJUGk -1) is the pseudo 1 order rate constant for a compound in the
presence of both juglone and HSndash (sminusHSk -1) is the pseudo 1 order rate constant for
the control reaction in the presence of only HSndash and [HJUGndash] (M) is the
concentration of the reactive dissociated HJUG- form (nondissociated
hydroquinone species are very nonreactive as compared to the monophenolate
species)
Supporting Information III
OH 0 OH OH
+ e- + H+ = + e- + H+ =
0 0
pl(( ox) = 8 00 PK1 (red) = 6 60
JUG HJUG
OH OH
OH
pKa2(red) = 10 60
Figure 7 1 Oxidized and reduced juglone fo1m s
Kinetic experiments in homogeneous anoxic aqueous solutions contained 5 mM
HS- 20 microM total juglone 50 mM KH2P04 buffer and were conducted at pH =
660 corresponding to a concentration of the reactive dissociated juglone form
[HJUG] = 10 microM
By using the LFER
E1 logkHJUG- = 125 middot 005~16 + 923
the following values were obtained
(Hofstetter et al 1999)
NAC k (M-1 -1) HJUG- middots log kHJUG- E~ (mV)
4-NT
4-NPA
311 middot10middot7
164middot10middot7
847middot 10-8
116middot 10middot7
226middot10middot2
489middot10middot3
-165
-231
-515
-546
IV Chapter 7
lowast
minusHJUGk -values are averages of triplicates whereas -values are averages of
duplicates The determined for 4-NT in this work (-515 mV) differs 3 from
the -value of -500 mV reported in the literature (Meisel amp Neta 1975
Wardman 1989) Hence it is assumed that the -value determined for 4-NPA
also differs by 3
minusHSk
1hE
1hE
1hE
Note that even for NACs holding very different values the difference in their
reactivities are much less pronounced in Fe(II)-Fe(III) systems such as the
Fe(II)goethite system (LFER slope a = 06 Hofstetter et al 1999) and the
Fe(II)magnetite system (LFER slope a = 034 Klausen et al 1995) as compared
to the jugloneH
1hE
2S system (a = 125) Furthermore it should be noted that all
LFERs mentioned here were established for neutral NACs and in this work we
have simply assumed that the LFERs are also valid for anionic NACs
72 The rate-limiting step
The overall rate of a reaction is equal to the rate of the slowest step in the
mechanism In heterogeneous reactions eg a compound reacting at the surface of
suspended particles in bulk solution the overall process by which the
heterogeneous reactions proceed may be broken down into a sequence of
individual diffusion steps and reaction steps 1) Mass transfer (diffusion) of the
reactant from the bulk fluid to the external surface of the solid phase 2)
Adsorption of reactant onto the solid surface 3) Reaction on the solid surface 4)
Desorption of the products from the solid surface 5) Mass transfer of the products
from the external solid surface to the bulk fluid Hence the rate of reaction of a
compound reacting at the surface of suspended particles in bulk solution may be
either mass transfer adsorptiondesorption or surface reaction limited When the
diffusion steps are much faster than the reaction steps the mass transfer or
diffusion steps do not affect the overall reaction rate However if the reaction steps
Supporting Information V
are very fast compared with the diffusion steps mass transport affects the reaction
rate Here only the external mass transfer is considered ie the diffusion of
reactants or products between the bulk fluid and the external surface of the solid
phase The additional internal mass transfer resistance for particles containing
substantial internal surface area is not addressed
721 Mass transfer (diffusion) limited kinetics
The overall rate constant can be represented by a system of resistances in series
(Fogler 1999 Arnold et al 1999)
⎟⎟⎠
⎞⎜⎜⎝
⎛+=
minusgeomSALobs kkak1111
where kobs is the observed rate constant kL is the mass transfer coefficient (mmiddots-1) a
is the ratio of the external (geometric) specific surface area to volume of solution
(m-1) and kSA-geom is the intrinsic rate constant of the reaction normalized to the
external specific surface area rather than the BET specific surface area By
comparing kLmiddota with kobs one can estimate the role of mass transfer on the rate of
reaction Thus if kLmiddota gtgt kobs mass transfer is so fast that it has no impact on the
reaction rate whereas if kLmiddota le kobs mass transfer is the rate limiting step
In fluid dynamics the Reynolds number Re is used for determining whether a
flow is laminar or turbulent
νtp ud sdot
=Re
where dp is the particle diameter (m) ut is the terminal particle settling velocity
(mmiddots-1) and ν is the kinematic fluid viscosity (m2middots-1) ν = η ρ where η is the
(absolute) dynamic fluid viscosity in centipoise (1 centipoise = 1 mPamiddots = 10-3
kgmiddotm-1middots-1) and ρ is the fluid density (kgmiddotm-3)
VI Chapter 7
At Re lt 1 we can apply Stokersquos particle settling velocity Stokersquos law is an
equation relating the terminal settling velocity of a smooth rigid sphere in a
viscous fluid of known density and viscosity to the diameter of the sphere when
subjected to a known force field
( )η
ρρsdot
minussdotsdot=
18
2pp
t
dgu (mmiddots-1)
where g = 981 mmiddots-2 is the gravitational constant ρp is the particle density (kgmiddotm-3)
The Sherwood number is the main parameter for prediction of the mass transfer
process
in fluid dynamics
3121 ScRe602Sh sdotsdot+=sdot
=lowast
lowast
iw
pL
Ddk
where Diw is the diffusion coefficient of the compound i in water (m2middots-1) is the
minimum (uncorrected) value of the mass transfer coefficient and Sc is the
Schmidt number This relation is often referred to as the Froumlssling correlation The
particle diameter is a key parameter in the Froumlssling correlation and the external
mass transfer coefficient varies with square of the particle size for smaller
particles
lowastLk
The Schmidt number is the ratio of the kinematic fluid viscosity and the diffusion
coefficient of the compound i in water
iwDν
=Sc
Supporting Information VII
According to Harriott (1962) the actual mass transfer coefficient kL is 15 times
greater than the minimum value of the mass transfer coefficient The
uncertainty in k
lowastLk
Lmiddota associated with particle sphericity and roughness issues are
believed not to exceed a factor of 2
The diffusion coefficient of a compound i in water can be estimated as (Hayduk amp
Laudie 1974)
5890141
9102613
iiw
VD
sdot
sdot=
minus
η (m2middots-1)
where iV is the molar volume of the compound i (cm3middotmol-1) estimated according
to Fuller et al 1966
Assuming spherical particles the external (geometric) specific surface area and the
particle diameter are calculated from the measured BET specific surface area Atot
assuming that our GR-SO4 has a AtotAouter ~ 30 similar to the one reported by
Hansen amp Koch (1998)
( ) ( ) ( )pppp
p
pp
p
dd
dV
SAAρρπ
πρ 1000
6
100061100030
SA3
2tot
geom sdot=
sdotsdot
sdot=
sdot== (m2middotg-1)
In our aqueous GR-SO4 system the density ρ = 1000 kgmiddotm-3 the absolute dynamic
viscosity η = 10-3 Pamiddots and the kinematic viscosity ν = 10-6 m2middots-1 for water The
GR-SO4 particle specific parameters used is found below
VIII Chapter 7
GR-SO4
Atot (m2g) 712
SAgeom (m2g) 237
Particle density ρp (kgm3) 1500
Particle diameter dp (m) 169middot10-6
Settling velocity ut (ms) 778middot10-7
Reynolds number 132middot10-6
The molar volumes the diffusion coefficients in water and the Schmidt numbers
for the NACs including 4-chloronitrobenzene (4-CNB) and nitrobenzene (NB)
were
Compound iV (cm3middotmol-1) Diw (m2middots-1) Sc
4-NT 1260 768middot10-10 1302
4-CNB 1230 779middot10-10 1284
4-NPA 1535 684middot10-10 1462
NB 1055 853middot10-10 1173
The ratio of the external (geometric) specific surface area to volume of solution
were calculated for GR-SO4 at the various concentrations applied
[Fe(II)GR]0 (mM) a (m-1)
103 366middot102
63 224middot103
126 448middot103
60 213middot103
The uncorrected mass transfer coefficients were estimated for the NACs using the
Froumlssling correlation
Supporting Information IX
[Fe(II)GR]0 (mM) lowastLk (mmiddots-1)
4-NT 912middot10-4
4-CNB 925middot10-4
4-NPA 812middot10-4
NB 101middot10-3
Finally kLmiddota was calculated and compared with the experimental 1 order rate
constants kobs obtained for the NACs
Compound [Fe(II)GR]0 (mM) kLmiddota (s-1) kobs (s-1) a
4-NT 103 050 420middot10-4
63 307 140middot10-3
126 613 590middot10-3
4-CNB 103 051 740middot10-4
63 311 170middot10-3
126 622 460middot10-3
4-NPA 103 045 640middot10-4
63 273 109middot10-3
126 546 473middot10-3
NB 60 324 137middot10-3 b
a Experimental pseudo 1 order rate constant at 50 microM [Ar-NO2]0 b Experimental pseudo 1 order rate constant at 10 microM [Ar-NO2]0
When comparing kLmiddota with kobs it can be seen that the rates of mass transfer for all
3 NACs exceed the observed rate constants by at least 3 or 4 orders of magnitude
at every initial Fe(II)GR concentration Thus the reaction of the given NACs with
GR-SO4 is not subject to mass transfer limitations under the experimental
conditions applied here
722 Surface saturation limited kinetics
More than 75 of all heterogeneous reactions that are not diffusion-limited are
surface-reaction-limited rather than adsorption- or desorption-limited We now
X Chapter 7
look at the reaction A = B = C where an intermediate B is formed In our system
A = Ar-NO2 B = Ar-NHOH and C = Ar-NH2 In this case the surface reaction is
assumed to be a single-site mechanism where only the site S on which A or B is
adsorbed is involved in the reaction forming B or C
KA
Adsorption 1 A + S = AmiddotS
kS1
Surface reaction 1 AmiddotS = BmiddotS
KB-1
Desorption 1 BmiddotS = B + S
KB
Adsorption 2 B + S = BmiddotS
kS2
Surface reaction 2 BmiddotS = CmiddotS
KC-1
Desorption 2 CmiddotS = C + S
The rate law for this surface-reaction limited single-site mechanism involving an
intermediate follows Langmuir-Hinshelwood kinetics (adopted from Fogler 1999)
CCBBAA
AAsitesSA
CKCKCKCKCk
dtdC
sdot+sdot+sdot+sdotsdotsdot
=minus1
1
Supporting Information XI
where kS1 is the intrinsic rate constant of the surface reaction transforming A into
the intermediate B Csites is the concentration of reactive sites S on the solid
surface KA KB and KC are the adsorption constants for A B and C at the reactive
surface sites and CA CB and CC are the concentrations of A B and C in the bulk
fluid Two major assumptions of the Langmuir isotherm imply that there is a fixed
number of localised surface sites present on the surface and that the activity of the
surface towards adsorption desorption or surface reaction is independent of
surface coverage
Hence fitting -∆CA∆t to CA CB and CC using a nonlinear curve fitting software
such as SigmaPlot may provide one with the intrinsic rate constant and the
adsorptions constants If KB and KC gtgt KA the intermediate and the product are
strongly competing with the reactant for vacant reactive surface sites
Our data was not fitted successfully by the Langmuir-Hinshelwood rate law
(regression results not shown) Simplifying the rate law by excluding either the
term KCmiddotCC or KBmiddotCB or both (assuming that the aniline product or the
hydroxylaniline intermediate or both did not compete for the reactive sites) did not
improve the regression The Langmuir-Hinshelwood rate law for a dual-site
mechanism did not fit our data either Thus Langmuir-Hinshelwood kinetics
cannot explain the reaction mechanism of the given NACs in our GR-SO4 system
73 External surface area of GR-SO4 and GR-CO3
The GR-SO4 unit cell consists of one double layer (d001 = 11 nm) ie one
hydroxide layer (049 nm ) and one interlayer (061 nm) Hexagonal GR-SO4
particles holding an average width of 1 microm (Figure 72) an average particle
thickness of 35 nm (Hansen amp Koch 1998) and a hydroxide layer thickness of
049 nm have a surface area of the basal plane
Abasal = 1 microm middot 1 microm ndash 2 middot 05 microm middot 025 microm = 075 microm2
XII Chapter 7
and a surface area of the edges
Aedge = (2 middot 05 microm + 4 middot 056 microm) middot 000049 microm = 00016 microm2
Figure 72 The hexagonal platy morphology of GR particles holding an average width of 1 microm
The particle thickness is the mean crystal thickness perpendicular to the 003 plane
as determined from the 003 reflections in an X-ray diffractogram A GR-SO4
particle holding a thickness of 35 nm contains 35 nm11 nm = 318 double layers
The GR-CO3 unit cell consists of one double layer (d001 = 075 nm) ie one
hydroxide layer (049 nm ) and one interlayer (026 nm) Hence a GR-CO3 particle
holding a thickness of 35 nm contains 35 nm075 nm = 467 double layers
The outer surface area of a GR-SO4 particle including outer basal planes and
edges is
Aouter = 222 microm 155microm 00016318microm 0752 =sdot+sdot
and the total surface area of a GR-SO4 particle including both inner and outer
basal planes as well as edges is
Supporting Information XIII
Atot = 222 microm 478)microm 00016microm 0752(318 =+sdotsdot
Hence the ratio of outer surface area to total surface area is
131microm 478microm 155
AA
2
2
tot
outer asymp=
Furthermore the ratio of edge surface area to outer surface area is
130microm 155
microm 00016318AA
2
2
outer
edge asympsdot
=
For GR-CO3 the outer surface area including outer basal planes and edges is
Aouter = 222 microm 157microm 00016746microm 0752 =sdot+sdot
and the total surface area of a GR-CO3 particle including both inner and outer
basal planes as well as edges is
Atot = 222 microm 701)microm 00016microm 0752(467 =+sdotsdot
Hence the ratio of outer surface area to total surface area is
145microm 701microm 157
AA
2
2
tot
outer asymp=
Furthermore the ratio of edge surface area to outer surface area is
121microm 157
microm 00016467AA
2
2
outer
edge asympsdot
=
XIV Chapter 7
74 Van der Waals radii
The size of polyatomic molecules can be estimated by summing the van der Waals
radii of the
individual atoms Van der Waals radii or nonbonded radii can be pictured as the
radii of hard spherical atoms (Figure 73)
Figure 73 Schematic of neighboring nonbonded atoms with van der Waals radii rA and rB
Assuming that the spheres of neighboring nonbonded atoms just touch (Figure
73) the highest possible ion or molecule size Ms can be estimated as the sum of
the van der Waals radii
Ms = 2middotrA + 2middotrB + (1)
Taking Paulingrsquos rule for nonmetals into account we can estimate the real size of
polyatomic ions bound by covalent bonds (Pauling 1960) The van der Waals
radius is larger than the covalent radius because it involves the interposition of two
electron pairs between the atoms rather than one The rule states that the van der
Waals radius of an atom exceeds its covalent radius by ~008 nm (overlap in
Figure 74)
Figure 74 Schematic of atoms undergoing covalent bonding
Supporting Information XV
Thus the size of polyatomic ions bound by covalent bonds Ms can now be
estimated as the sum of the van der Waals radii subtracted by 008 nm
Ms = 2middot(rA - 008 nm) + 2middot(rB - 008 nm) + (2)
We estimated the molecular size of the NACs (Table 72) by means of equation (2)
and the van der Waals radii of the atoms in Table 71
Table 71 Van der Waals radii of various atoms Values from Pauling 1960
Atom vdW radii (nm)
H 0120
O 0140
N 0150
C 0170
Cl 0181
S 0185
In order to make the calculations it was assumed that all atoms were spherical and
that all bond angles were 90deg or 180deg (linear structures) In addition no distinctions
were made between single and double bonds The molecular sizes of the NACs
were estimated with the benzene ring representing the xy plane
Table 72 Molecular sizes of the NACs a Thickness z of the xy plane
Compound Ms (x) (nm) Ms (y) (nm) Ms (z) (nm)a
NB 054 080 036
4-NT 054 106 036
4-CNB 054 100 036
4-NPA 054 136 036
XVI Chapter 7
Note that the molecular sizes in Table 72 are only rough estimations
For comparison with the GR-SO4 interlayer spacing (061 nm) we consider three
possible orientations of the NACs in the GR-SO4 interlayer 1) The NAC xyz
coordination is equivalent to the crystal abc coordination (z = c = 036 nm) 2) the
NAC xy plane is parallel to the crystal bc plane (z = a = 054 nm) and 3) the NAC
xy plane is parallel to the crystal ac plane (z = b = 080-136 nm) Hence the sizes
of the NACs do not hinder their access to the GR-SO4 interlayer Only when
oriented vertically do the sizes of the NACs (z = b = 080-136 nm) exceed the GR-
SO4 interlayer spacing
The molecular size of trichloroacetate (TCA) was also estimated by means of
equation (2) and the atomic van der Waals radii in Table 71 When the TCA
aliphatic chain is assumed to represent the x direction (Ms (x) = 066 nm) the
molecular size in the y and z directions ranges from 045-053 nm depending on the
free rotation of the C-C bond Thus only if the C-C bond is oriented perpendicular
to the crystal ab plane does the size of TCA exceed the GR-SO4 interlayer spacing
(061 nm) In contrast the size of TCA exceeds the GR-CO3 interlayer spacing
(026 nm) regardless of its orientation
75 Adsorption of Fe(II) onto Fe(III) oxides
As seen from the Fe(II) sorption isotherms Fe(II) sorption varies widely between
the Fe(III) oxides as a function of solution pH (Figure 75) Average surface
densities of approximately 2 singly coordinated sitesnm2 iron oxide have been
suggested for goethite and lepidocrocite (Cornell amp Schwertmann 1996) The
similar surface site densities of goethite and lepidocrocite might explain their
similar Fe(II) adsorption isotherms (Figure 75)
Supporting Information XVII
Figure 75 Fe(II) adsorption edges for ferrihydrite goethite hematite lepidocrocite and
magnetite in the absence of other specifically adsorbing cations and anions (from Vikesland amp
Valentine 2002 and references therein) The total number of surface sites was in excess of the
total Fe(II) concentrations in all experiments
Dissolved cations or anions may specifically adsorb at the calcite and Fe(III) oxide
surfaces by exchanging for H+ or OHndash at the equivCO3H0 equivCaOH0 equivFeOH0 and
equivFeIIIOFeIIOH0 surface sites At the experimental conditions applied here within a
pH range 70-87 the dominant species of interest in solution are Fe2+ HCO3ndash
CO32ndash Clndash SO4
2ndash (only in the GR-SO4 systems) and the anionic TCA and DCA In
addition Fe2+ readily forms aqueous complexes with hydroxide carbonate
chloride and sulfate whereby the species FeOH+ FeHCO3+ Fe(OH)(CO3)ndash
FeCO30 Fe(CO3)2
2ndash FeCl+ and FeSO40 may occur (Millero amp Hawke 1992) At
pH 70-87 we expect the Fe(II) species Fe2+ FeCO30 Fe(OH)(CO3)ndash FeOH+ and
Fe(CO3)22ndash to dominate in the GR-CO3 and CaCO3(s)CO2(g) buffered magnetite
suspensions In the goecalcite and lepcalcite suspensions we expect the FeCl+
species to dominate as well whereas the Fe2+ FeSO40 and FeOH+ species most
XVIII Chapter 7
likely dominate in the GR-SO4 suspensions Anionic inorganic ligands like
carbonate chloride and sulfate can lower or enhance the adsorption of Fe(II) due to
a) formation of stable nonadsorbing Fe(II) ligand aqueous complexes b) formation
of Fe(II) ligand Fe(III) oxide surface complexes which can lead to surface
precipitation at high Fe(II) and ligand concentrations c) competitive ligand
sorption to the Fe(III) oxide surface blocking reactive sorption sites at the surface
and d) diminution of the positive charge at the Fe(III) oxide surface (at pH levels
below the point of zero charge (pHpzc) of the Fe(III) oxide) thereby decreasing the
electrostatic repulsion of cations by the Fe(III) oxide surface Specifically adsorbed
cations increase the pHpzc whereas specifically adsorbed anions decrease the pHpzc
References Arnold WA Ball WP Roberts AL (1999) Polychlorinated ethane reaction with zero-valent zinc Pathways and rate control Journal of Contaminant Hydrology 40 183-200 Cornell RM Schwertmann U (1996) The iron oxides Structure properties reactions occurrence and uses VCH Verlagsgesellschaft mbH Weinheim Fogler HS (1999) Elements of chemical reaction engineering 3rd ed Prentice Hall Fuller EN Schettler PD Giddings JC (1966) A new method for prediction of binary gas-phase diffusion coefficients Industrial and Engineering Chemistry 58 19-27 Hansen HCB Koch CB (1998) Reduction of nitrate to ammonium by sulphate green rust Activation energy and interlayer reaction mechanism Clay Minerals 33 87-101 Harriott P (1962) Mass transfer to particles Part I Suspended in agitated tanks AIChE Journal 8 93-102 Hayduk W Laudie H (1974) Prediction of diffusion coefficients for nonelectrolytes in dilute aqueous solutions AIChE Journal 20 611-615 Hofstetter TB Heijmann CG Haderlein SB Holliger C Schwarzenbach RP (1999) Complete reduction of TNT and other (poly)nitroaromatic compounds under iron-reducing subsurface conditions Environmental Science and Technology 33 1479-1487 Klausen J Troumlber SP Haderlein SB Schwarzenbach RP (1995) Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions Environmental Science and Technology 29 2396-2404 Meisel D Neta P (1975) One-electron redox potentials of nitro compounds and radiosensitizers Correlation with spin densities of their radical anions Journal of the American Chemical Society 97 5198-5203
Supporting Information XIX
Millero FJ Hawke DJ (1992) Ionic interactions of divalent metals in natural waters Marine Chemistry 40 19-48 Pauling L (1960) The nature of the chemical bond 3rd ed Cornell University Press Ithaca Vikesland PJ Valentine RL (2002) Iron oxide surface-catalyzed oxidation of ferrous iron by monochloramine implications of oxide type and carbonate on reactivity Environmental Science and Technology 36 512-519 Wardman P (1989) Reduction potentials of one-eletron couples involving free radicals in aqueous solution The Journal of Physical Chemistry Reference Data 18 1637-1755
Curriculum Vitae 13011973 Born in Haderslev Denmark 1988-1991 Mathematical high school Haderslev Katedralskole Denmark 1991-1992 Sabbatical year 1992-1995 B Sc in environmental chemistry University of Copenhagen
(KU) Denmark B Sc thesis 1995 ldquoMethane oxidizing bacteria in soilrdquo
1995-1998 M Sc in environmental chemistry University of Copenhagen
Denmark 1997-1998 diploma thesis ldquoReductive dechlorination of carbon tetrachloride and chloroform in presence of iron(II)iron(III)-hydroxides (green rust)rdquo
1998-1999 Research and teaching assistant at the Chemistry Department
The Royal Veterinary amp Agricultural University (KVL) Denmark
1999-2004 PhD in environmental sciences Swiss Federal Institute of
Technology Zuumlrich (ETHZ) and Swiss Federal Institute for Environmental Science and Technology (EAWAG) Switzerland Docoral thesis ldquoFormation and redox reactions of green rusts under geochemical conditions found in natural soils and sedimentsrdquo
2000-2002 Teaching assistent at the Swiss Federal Institute of Technology
Zuumlrich and supervision of diploma students 2002 Microbial Diversity summer course (7 weeks) at the Marine
Biological Laboratory Woods Hole Massachusetts USA

Acknowledgements
I would like to thank Stefan Haderlein Hans Christian B Hansen and Rene
Schwarzenbach for their supervision of this work Without the encouragement and
confidence of HCB Hansen and former colleagues at the Royal Veterinary and
Agricultural University in Copenhagen I would never have pursued a PhD and
without the understanding of Rene Schwarzenbach after the tragic death of my
mother I would not have had the time necessary to finish it
I thank Christian Bender Koch Hanne Nancke-Krogh Susanne Guldberg and
Henrik T Andersen (Royal Veterinary and Agricultural University Denmark) for
their valuable contribution to my work I would also like to express my gratitude to
former and present members of the Contaminant Hydrology Group from whom I
have received many benefits I mourn the loss of Denis Mavrocordatos (EAWAG)
who provided technical assistance in the electron microscopy lab and I will always
keep the sunny hours in his company in fond memory Finally I would like to
thank Kristina Straub and Bernhard Schink (University of Constance Germany)
who welcomed me in their lab for a week and taught me how to work with strict
anaerobic bacteria
I gratefully acknowledge the grant which I received from the Danish Research
Agency
Table of Contents
Table of Contents Zusammenfassung I Summary V 1 General Introduction 1 11 Iron cycling in the subsurface 1 12 Green rusts 3 13 Microbial formation of green rusts 7 14 Redox reactions of green rusts 8 15 Outline of the thesis 10 References 11 2 Solid State Oxidation of Vivianite by Anaerobic
Denitrifying Fe(II)-Oxidizing Bacteria 17 Abstract 17 21 Introduction 17 22 Materials and methods 22
221 Microorganisms and media 22 222 Characterisation of precipitates 23 223 Biooxidation experiments 24 224 Analytical methods 25
23 Results and discussion 25 231 Identification of solid iron-containing phases 25 232 Factors controlling the rate and extent of Fe(II) biooxidation 34 233 Morphology of solid iron phases 37
24 Conclusions 38 References 39 3 Formation of Layered Iron Hydroxides by
Microbial Fe(III) Reduction 43 Abstract 43 31 Introduction 44 32 Materials and methods 47
321 Preparation of iron oxide coatings 47 322 Mineral characterisation 48 323 Culture conditions and cell preparation 48 324 Bioreduction experiments 49
Table of Contents
325 Analytical methods 50 33 Results and discussion 50
331 Fe(II) production and suspension colour changes 50 332 Identification of solid iron phases 55 333 Factors controlling the identity of the secondary iron minerals 58 334 Factors controlling the rate and extent of Fe(III) bioreduction 59
34 Conclusions 60 References 61 4 Reduction of Nitroaromatic Probe Compounds by Sulphate
Green Rust The Effect of Probe Compound Charge 65 Abstract 65 41 Introduction 66 42 Materials and methods 71
421 Synthesis of GR-SO4 71 422 Mineral characterisation 72 423 Lyophilization and determination of specific surface area 72 424 Estimation of the one-electron reduction potential for 4-NPA 73 425 Kinetic experiments 74 426 Analytical methods 74
43 Results and discussion 75 431 Product formation and reaction kinetics 75 432 Comparison of rate constants for the different NACs 79 433 Factors influencing the reaction rate 82 434 Comparison with rate constants obtained for other Fe(II) containing
mineral systems 83 435 Depletion of reactive sites 85 436 The role of external and internal reactive sites 86
44 Conclusions 89 References 91 5 Reductive Transformation of Trichloroacetate in Abiotic
Fe(II)-Fe(III) Mineral Systems 97 Abstract 97 51 Introduction 98 52 Materials and methods 101
521 Synthesis of GRs and magnetite 102 522 Preparation of iron oxide coatings 102 523 Mineral characterisation 103 524 Kinetic experiments 103
Table of Contents
525 Analytical methods 104 53 Results and discussion 105
531 Product formation and reaction kinetics 105 532 Comparing rate constants obtained for the various Fe(II)-Fe(III)
mineral systems 109 533 Comparing with rate constants obtained for other chlorinated
aliphatic compound 112 534 Factors controlling the reactivity of surface-bound Fe(II) 114 535 Comparison with biotic and other abiotic systems 118
54 Conclusions 119 References 120 6 Conclusions and Outlook 125 References 128 7 Supporting Information I 71 Estimation of the one-electron reduction potential for 4-NPA I 72 The rate-limiting step IV 721 Mass transfer (diffusion) limited kinetics V
722 Surface saturation limited kinetics IX 73 External surface area of GR-SO4 and GR-CO3 XI 74 Van der Waals radii XIV 75 Adsorption of Fe(II) onto Fe(III) oxides XVI References XVIII Curriculum Vitae
Zusammenfassung I
Zusammenfassung Geschichtete Fe(II)-Fe(III)-Hydroxide (Gruumlner Rost) gehoumlren zur Gruppe der
Fe(II)-haltigen Mineralsysteme (zB Magnetit (Fe3O4) Siderit (FeCO3) Vivianit
(Fe2(PO4)2sdot8H2O) Fe(II)-Sulfide sowie an die Oberflaumlche von Fe(III)-Oxiden und
Tonmineralien gebundenes zweiwertiges Eisen) die die Aktivitaumlt von Fe(II) in
suboxischen und anoxischen Boumlden und Sedimenten kontrollieren Gruumlner Rost
Phasen (GRs) bestehen aus planaren positiv geladenen trioktaedrischen Fe(II)-
Fe(III)-Hydroxidschichten die durch hydratisierte Anionen in den
Zwischenschichten ausgeglichen werden Ihre generelle Zusammensetzung ist
[FeII(6-x)FeIII
x(OH)12]x+[(A)xnmiddotyH2O]x- wobei x = 09 - 42 ist A entspricht einem n-
valenten Anion (zB CO32- Clndash oder SO4
2-) und y repraumlsentiert die Anzahl
Wassermolekuumlle in der Zwischenschicht GRs sind wichtige intermediaumlre Phasen
die durch unvollstaumlndige Oxidation von Fe(II) oder teilweise Reduktion von Fe(III)
gebildet werden koumlnnen Sie koumlnnen in suboxischen nicht-sauren eisenhaltigen
natuumlrlichen wie auch technischen Systemen auftreten so wie in Wasser gesaumlttigten
Boumlden und interstitiellen Sedimenten Rohrleitungen in der
Trinkwasserversorgung Stahlpfosten in marinen Sedimenten Stahlbeton und in
reaktiven durchlaumlssigen Waumlnden aus nullwertigem Eisen zur in-situ Sanierung von
Altlasten und Aquiferen Aufgrund ihrer Schichtstruktur den anionischen
Zwischenschichten und der hohen spezifischen Oberflaumlchen sind GRs reaktive
Ionentauscher und Sorbentien von Anionen Des Weiteren wurde gezeigt dass
GRs eine Reihe anorganischer und organischer Schadstoffe reduzieren koumlnnen
Durch Immobilisierung und Transformation koumlnnen GRs somit eine wichtige Rolle
fuumlr das Abbauverhalten und den Transport solcher Schadstoffe in suboxischen
Boumlden und Sedimenten spielen Die Resultate dieser Dissertation tragen zum
Verstaumlndnis uumlber die Bildung und Reaktivitaumlt von Fe(II)-haltigen Mineralsystemen
wie GRs Vivianit Magnetit und an Goethit (α-FeOOH)- und Lepidokrozit (γ-
FeOOH)-Oberflaumlchen gebundenes Fe(II) in der Natur bei
II Zusammenfassung
Um die Rolle von Bakterien bei der Bildung von GRs in natuumlrlichen Boumlden und
Sedimenten aufzuklaumlren wurden Eisenminerale untersucht die als Folge der
Aktivitaumlt von eisenrespirierenden Bakterien gebildet wurden Kapitel 2 beschreibt
die Untersuchungen von eisenhaltigen Produkten die von anaeroben autotrophen
denitrifizierenden Fe(II)-oxidierenden Bakterien (FeOB) gebildet wurden Ein
Bikarbonat- und Phosphat-reiches Kulturmedium bot den nitratreduzierenden
FeOB optimale Bedingungen Fe(II) lag zu Anfang der Reaktion als weisses
Fe(II)-Hydroxyphosphat (Vivianit) und als geloumlstes Fe(II) vor Die Ergebnisse
zeigten dass die denitrifizierenden FeOB amorphen Goethit via ein gruumlnes Fe(III)-
angereichertes Vivianit-Zwischenprodukt bildeten Die Analyse mit Moumlssbauer
Spektroskopie deutet nicht auf eine Bildung von GR hin
In Kapitel 3 werden jene Eisenmineralien beschrieben die waumlhrend der Reduktion
verbreiteter Fe(III)-Oxide durch anaerobe dissimilative Fe(III)-reduzierende
Mikroorganismen Shewanella algae BrY gebildet wurden Um natuumlrliche
Zustaumlnde zu simulieren wurden Fe(III)-Oxide als Beschichtungen auf
Silikatpartikel (Modellsystem fuumlr Sandboumlden) oder Calcitpartikel (CaCO3
Modellsystem fuumlr kalkhaltige Boumlden) aufgetragen sowie synthetische
Elektronencarrier und hochkonzentrierte kuumlnstliche pH-Puffer ausgeschlossen
Die erforschten Mineralsysteme umfassten GoethitCalcit- LepidokrozitCalcit-
und FerrihydritSand-Suspensionen S algae BrY reduzierte beachtliche Mengen
des eingesetzten Fe(III) und es bildeten sich gruumlne und schwarze Festphasen
innerhalb von 1-2 Wochen nach der Animpfung Moumlssbauer Spektroskopie der
gruumlnen und schwarzen Praumlzipitate zeigte dass sich diese aus GR und Vivianit
zusammensetzen
Die Reaktivitaumlt synthetischer GRs gegenuumlber reduzierbaren organischen
Schadstoffen wurde erkundet um die potentielle Bedeutung von GR-Phasen fuumlr
das Schicksal solcher Verbindungen abzuschaumltzen Zu diesem Zweck wurden
Nitroaromaten (NACs) und Chloracetate als Modellverbindungen benutzt um
Zusammenfassung III
umweltrelevante Redoxreaktionen zu studieren In Kapitel 4 wurde die relative
Reaktivitaumlt von aumlusseren und inneren reaktiven Stellen in synthetischem Sulfat-
Gruumlnem Rost (GR-SO4) anhand von strukturaumlhnlichen ldquoreaktiven
Sondenmolekuumllenrdquo mit unterschiedlichen Ladungen untersucht Als reaktive
Sondenmolekuumlle wurden Nitrobenzen 2-Nitrophenol 4-Nitrotoluen 4-
Chlornitrobenzen und 4-Nitrophenylessigsaumlure verwendet Die Ergebnisse zeigen
dass GR-SO4 die NACs vollstaumlndig zu den entsprechenden Anilinen reduzierte
Die Reaktionen folgten einer pseudo 1 Ordnungs Kinetik bezuumlglich NAC und die
auf Oberflaumlche normalisierten pseudo 1 Ordnungs Geschwindigkeitskonstanten
(Anfangsraten) waren 016ndash465middot10-4 s-1middotm-2middotL fuumlr [Fe(II)GR]0 = 103-1260 mM
[NAC]0 = 20-102 microM und pH 84-86 Weder durch Einbezug von
Massentransferlimitierung noch von Oberflaumlchensaumlttigungskinetik war es moumlglich
die aumlhnlichen Oberflaumlchennormalisierten pseudo 1 Ordnungs
Geschwindigkeitskonstanten fuumlr die Reduktion der neutralen und anionischen
NACs durch GR-SO4 zu erklaumlren Dieser Umstand laumlsst vermuten dass die
Reaktion zwischen NAC und GR-SO4 an den externen reaktiven Fe(II)-Stellen
stattfindet Bei niedrigen Fe(II)GR-Anfangskonzentrationen wurden die externen
reaktiven Fe(II)-Stellen aufgebraucht und die Regenerierung von neuen externen
reaktiven Stellen haben schliesslich die Geschwindigkeit der Reduktion von NACs
durch GR-SO4 kontrolliert
In Kapitel 5 wurde die Reaktivitaumlt von verschiedenen umweltrelevanten Fe(II)-
Fe(III)-Mineralsystemen gegenuumlber Trichloressigsaumlure (TCA) und
Dichloressigsaumlure (DCA) in Batchexperimenten die natuumlrliche Bedingungen
imitierten untersucht Die Fe(II)-Fe(III)-Systeme umfassten Sulfat-Gruumlner Rost
Carbonat-Gruumlner Rost Magnetit Fe(II)Goethit und Fe(II)Lepidokrozit TCA
wurde von allen Fe(II)-haltigen Mineralien zu DCA reduziert Die Reaktionen
folgten einer pseudo 1 Ordnungs Kinetik bezuumlglich TCA und die auf Oberflaumlche
normalisierten pseudo 1 Ordnungs Geschwindigkeitskonstanten betrugen 033ndash
76middot10-5 min-1middotm-2middotL bei [Fe(II)]0 = 025ndash116 mM [TCA]0 = 15ndash1000 microM und pH
IV Zusammenfassung
70ndash87 Die Ergebnisse zeigen keine signifikanten Unterschiede zwischen den
verschiedenen Fe(II)-Fe(III)-Systemen bezuumlglich Produkteverteilung und
oberflaumlchen-normalisierten pseudo 1 Ordnungs Geschwindigkeits-konstanten In
keinem der Systeme wurde DCA innerhalb des experimentellen Zeitraums zu
Monochloressigsaumlure oder Essigsaumlure weiter reduziert
Die Ergebnisse die in dieser Dissertation praumlsentiert werden zeigen dass
mikrobiologische Prozesse fuumlr die Oxidation von Vivianit-Phasen im Untergrund
verantwortlich sein koumlnnen Zudem wurde nachgewiesen dass GRs bei tiefen
Kohlenstoff- und Fe(III)-Konzentrationen sowie durch Ausschluss von
kuumlnstlichen Elektronencarriern und pH-Pufferung mikrobiell gebildet werden
koumlnnen Ferner zeigten Befunde dass GRs eine bedeutende Rolle fuumlr die reduktive
Transformation von NACs und TCA in natuumlrlichen Boumlden und Sedimenten spielen
koumlnnen
Summary V
Summary
Layered iron(II)-iron(III)-hydroxides (green rusts) belong to the group of Fe(II)-
bearing mineral systems eg magnetite (Fe3O4) siderite (FeCO3) vivianite
(Fe2(PO4)2sdot8H2O) Fe(II) sulfides as well as Fe(II) associated with Fe(III) oxide
and clay mineral surfaces that control the Fe(II) activity in suboxic and anoxic
soils and sediments Green rusts (GRs) consist of plane positively charged
trioctahedral Fe(II)-Fe(III) hydroxide sheets balanced by hydrated anions in the
interlayers and hold the general formula [FeII(6-x)FeIII
x(OH)12]x+[(A)xnmiddotyH2O]x-
where x = 09 - 42 A is an n-valent anion eg CO32- Clndash or SO4
2- and y is the
number of water molecules in the interlayer GRs are important intermediate
phases formed by partial oxidation of Fe(II) or partial reduction of Fe(III) and they
have been found in suboxic non-acid iron-rich natural environments such as
hydromorphic soils and intertidal sediments and in engineering systems including
pipeline distribution systems for drinking water steel sheet piles in marine
sediments reinforced concrete and permeable reactive barriers of zero-valent iron
implemented for on-site remediation of contaminants Due to their layered
structures anionic interlayers and high specific surface areas GRs represent
reactive ion exchangers and sorbents of anions In addition GRs have been shown
to reduce a range of inorganic and organic pollutants Thus through sequestration
and reductive transformation GRs may play an important role in the fate and
transport of contaminants in suboxic soils and sediments The work presented in
this dissertation adds to the understanding of how Fe(II)-bearing minerals like
GRs vivianite magnetite and Fe(II) associated with goethite (α-FeOOH) and
lepidocrocite (γ-FeOOH) may form and react in nature
In order to elucidate the role of bacteria in the formation of GRs in natural soils
and sediments we studied the iron mineral phases forming as a result of the
activity of iron-respiring bacteria In the study described in chapter 2 the Fe-
containing products formed by anaerobic autotrophic denitrifying Fe(II)-oxidizing
VI Summary
bacteria (FeOB) were examined The culture medium applied contained high levels
of bicarbonate and phosphate and is typically used in this kind of studies as it
provides excellent conditions for the nitrate-reducing FeOB Fe(II) was present
initially as a whitish solid Fe(II) hydroxy phosphate (vivianite) and as soluble
Fe(II) The results obtained demonstrate that the denitrifying FeOB produce poorly
crystalline goethite via a greenish Fe(III)-enriched vivianite intermediate
Moumlssbauer spectroscopic analyses provided no significant evidence of green rust
formation
In chapter 3 the Fe-containing products formed during reduction of common
Fe(III) oxides by the anaerobic dissimilatory Fe(III)-reducing microorganism
Shewanella algae BrY are discussed In order to simulate natural conditions
Fe(III) oxides were applied as coatings on silica (model system for sandy soils) or
calcite (CaCO3) particles (model system for calcareous soils) and synthetic
electron shuttles as well as highly concentrated artificial pH buffers were excluded
The mineral systems studied include goethitecalcite lepidocrocitecalcite and
hydrous ferric oxidesand suspensions S algae BrY reduced substantial amounts
of the initial Fe(III) and green and blackish mineral phases were produced within
1-2 weeks after inoculation Moumlssbauer spectroscopic analyses showed that the
green and black precipitates consisted of GR and vivianite
The reactivity of synthetic GRs towards reducible organic pollutants was
investigated in order to asses the potential significance of GR phases for the fate of
such compounds To this end we used nitroaromatic compounds (NACs) and
chlorinated acetates as suitable model compounds for studying environmentally
relevant redox reactions In the work described in chapter 4 the relative reactivity
of outer and inner Fe(II) reactive sites in synthetic sulfate green rust (GR-SO4) was
studied using a series of structurally closely related compounds with different
charge properties as ldquoreactive probesrdquo The probe compounds included
nitrobenzene 2-nitrophenol 4-nitrotoluene 4-chloronitrobenzene and 4-
Summary VII
nitrophenylacetic acid The results show that NACs are completely reduced to their
corresponding anilines by GR-SO4 The reactions followed pseudo 1 order
kinetics with respect to NAC and the surface area-normalised pseudo 1 order rate
constants (initial rates) obtained were 016ndash465middot10-4 s-1middotm-2middotL at [Fe(II)GR]0 = 103-
1260 mM [NAC]0 = 20-102 microM and pH 84-86 Neither mass transfer control nor
surface saturation kinetics could explain the similarity of the surface-normalised
pseudo 1 order rate constants obtained for the reduction of the neutral and anionic
NACs by GR-SO4 These observations suggest that the reaction between NAC and
GR-SO4 takes place at the external reactive Fe(II) sites At low initial Fe(II)GR
concentrations the external reactive Fe(II) sites were depleted and the regeneration
of new external reactive sites eventually controlled the reduction of the NACs by
GR-SO4
Finally the reactivity of various Fe(II)-Fe(III) mineral systems towards
trichloroacetic acid (TCA) and dichloroacetate (DCA) has been investigated in
laboratory batch experiments imitating natural conditions (Chapter 5) The Fe(II)-
Fe(III)-systems investigated included GR-SO4 carbonate green rust magnetite
Fe(II)goethite and Fe(II)lepidocrocite TCA was readily reduced to DCA by all
Fe(II)-containing minerals The reactions followed pseudo 1 order kinetics with
respect to TCA and the surface area-normalised pseudo 1 order rate constants
obtained were 033ndash76middot10-5 min-1middotm-2middotL at [Fe(II)]0 = 025ndash116 mM [TCA]0 =
15ndash1000 microM and pH 70ndash87 Our results showed no significant differences
regarding product distribution and surface area-normalised reaction rate constants
between the Fe(II)-Fe(III)-systems DCA was not further reduced to
monochloroacetate (MCA) or acetate in any of the systems within the time frame
in our experiments
The results presented in chapter 2 indicate that microbiological processes may be
responsible for the oxidation of vivianite phases in natural subsurface
environments In chapter 3 we demonstrated that GRs may be produced
VIII Summary
microbially at conditions including low carbon and Fe(III) concentrations as well
as the exclusion of synthetic electron shuttles and pH buffers The results obtained
in chapter 4 and 5 show that GRs transform NACs and TCA readily The reductive
transformation of NACs and TCA by GRs is relevant to understanding the
processes responsible for their degradation in the subsurface and the development
of innovative technologies for their remediation
General Introduction 1
1 General Introduction
11 Iron cycling in the subsurface
Iron is the fourth most abundant element (4-5 mass) and the most abundant redox
sensitive element in the Earthrsquos crust It is found as Fe(II) and Fe(III) in a number
of minerals in rocks soils and sediments Under anoxic conditions solid Fe(III)-
containing minerals can be reduced to soluble Fe(II) once the more energetically
favoured electron donors - nitrate and manganese(IV) oxides - have been
consumed Dissolved Fe(II) can be reoxidized to insoluble Fe(III) microbially or
abiotically upon exposure to oxygen Due to this ready alternation between the
Fe(II) and Fe(III) redox states iron plays a major role in controlling the redox
potential and the carbon cycling in subsurface environments (Nealson amp Saffarini
1994)
Nonenzymatic processes were previously considered to account for most of the
Fe(III) reduction in subsurface environments The significance of bacteria in the
biogeochemical cycling of iron has been broadly recognized over the past two
decades Dissimilatory Fe(III)-reducing bacteria (DIRB) that gain energy by
coupling the oxidation of hydrogen or organic compounds to the reduction of
Fe(III) oxides have been known for many years but their biogeochemical
importance was only widely acknowledged about a decade ago (reviewed by
Lovley 1997) Fe(III) bioreduction accounts for a major fraction of the carbon
oxidation in many different anoxic environments and in the presence of sufficient
amounts of reactive Fe(III) microbial Fe(III) reduction may even inhibit sulphate
reduction and methanogenesis (King 1990 Lovley amp Phillips 1986) In fact most
of the Fe(III) reduction in the Fe(III) reduction zone of aquatic sediments and
aquifers is enzymatically catalyzed by microorganisms (Lovley et al 1991) A
wide diversity of DIRB distributed among several different phylogenetic groups
2 Chapter 1 is known today The two most studied DIRB are the obligate anaerobic Geobacter
spp and the facultatively anaerobic Shewanella spp (Figure 11)
Aerobic oxidation of Fe(II)-containing minerals by lithotrophic acidophilic and
neutrophilic bacteria has been known for many years but their broad significance
in the biogeochemical cycling of iron has only been recognized over the past two
decades Both acidophilic (Thiobacillus ferrooxidans) and neutrophilic
(Gallionella ferruginea Leptothrix ochracea Sphaerotilus natans) aerobic Fe(II)-
oxidizing bacteria (FeOB) have been isolated (Hanert 1992 Kuenen et al 1992
Mulder amp Deinema 1992)
Figure 11 The microbial iron cycle
Anaerobic Fe(II) oxidation by phototrophic purple non-sulfur bacteria utilizing
Fe(II) as an electron donor in the light was recognized only a decade ago (Widdel
et al 1993) Subsequently it was demonstrated that the biological oxidation of
Fe(II) in the absence of oxygen is possible by light-independent chemotrophic
microorganisms using nitrate as the electron acceptor (Straub et al 1996) Thus
the microbial iron cycle includes anaerobic Fe(III)-reducing microorganisms and
aerobic as well as anaerobic Fe(II)-oxidizing bacteria (Figure 11)
General Introduction 3
12 Green rusts
Iron oxides iron hydroxides and iron oxyhydroxides (collectively termed iron
oxides or Fe(III) oxides) are ubiquitous in the pedosphere where they originate
from aerobic weathering of surface magmatic rocks such as ferromagnesium
silicates and pyrite (Cornell amp Schwertmann 1996) Goethite (α-FeOOH)
lepidocrocite (γ-FeOOH) ferrihydrite (Fe5HO8sdot4H2O) hematite (α-Fe2O3)
magnetite (Fe3O4) maghemite (γ-Fe2O3) and akageneite (β-FeOOH) constitute the
most important iron oxides in soils and sediments (Schwertmann amp Cornell 1991)
The formation and transformation of iron oxides depend on pH solution
composition redox potential temperature rate of oxidationreduction and degree
and rate of hydrationdehydration Iron oxides are important to many soil
properties such as colour pH and redox buffer capacity aggregation with other
soil particles as well as retention of anions and cations (Cornell amp Schwertmann
1996) A number of Fe(II)-bearing minerals including Fe(II)-containing clays (eg
smectites vermiculites and micas) magnetite siderite (FeCO3) vivianite
(Fe2(PO4)2sdot8H2O) Fe(II) sulphides and green rusts (layered Fe(II)-Fe(III)
hydroxides) may be present in soils and sediments under suboxic and anoxic
conditions Green rusts are believed to play a central role as metastable
intermediates in the redox cycling of iron at circumneutral pH in aquatic and
terrestrial environments
Green rusts (GRs) are layered iron(II)-iron(III)-hydroxides consisting of plane
positively charged trioctahedral Fe(II)-Fe(III) hydroxide sheets balanced by
hydrated anions in the interlayers (cf Figure 41 this work) GRs belong
structually to the pyroaurite-sjoumlgrenite group of layered hydroxides and they hold
the general formula [FeII(6-x)FeIII
x(OH)12]x+[(A)xnmiddotyH2O]x- where x = 09 - 42 A is
an n-valent anion eg CO32- Clndash or SO4
2- and y is the number of water molecules
in the interlayer The three most common and investigated green rust forms include
chloride GR (GR-Cl) sulphate GR (GR-SO4) and carbonate GR (GR-CO3)
Generally GRs are crystallographically classified into the GRI (rhombohedral
4 Chapter 1 GR-Cl and GR-CO3) and GRII (hexagonal GR-SO4) crystal systems The GR
interlayer thickness is a function of both the size and the charge of the interlayer
anion Tetrahedrally coordinated anions like sulphate lead to larger interlayer
distances than smaller monoatomic anions like chloride or planar ions like
carbonate (Mendiboure amp Schoumlllhorn 1986) Not only size but also charge density
plays a role for the interlayer spacing That is for anions having the same number
of valence electrons anions with smaller ionic radii (higher electron density) are
bound more strongly and therefore result in smaller interlayer spacings The
interlayer in GR-SO4 is composed of two consecutive planes of anions and water
whereas GR-Cl and GR-CO3 interlayers consist of only one single plane (Simon et
al 2003)
GRs are important intermediate phases formed by partial oxidation of Fe(II) or
partial reduction of Fe(III) In neutral and weakly alkaline solutions the oxidation
of dissolved Fe(II) always passes through solid GR phases (Bernal et al 1959)
GRs may also form during oxidation of zero-valent iron and as a result of the
combination of Fe(II) and Fe(III) at circumneutral pH (Figure 12)
Figure 12 Formation and transformation of GRs Fe3O4 = magnetite γ-Fe2O3 = maghemite α-
FeOOH = goethite γ-FeOOH = lepidocrocite akageneite = β-FeOOH
General Introduction 5
Oxidation of GR-CO3 usually produces goethite and magnetite-maghemite
whereas GR-Cl and GR-SO4 transform into lepidocrocite and magnetite-
maghemite depending on pH and oxidation rate (Bernal et al 1959 Taylor 1980
Carlson amp Schwertmann 1990) The brown δ-FeOOH is formed by vigorous
oxidation of GR using air or a 30 aqueous solution of hydrogen peroxide (Bernal
et al 1959 Misawa et al 1974) Black ferromagnetic magnetite forms by slow
oxidation of GR whereas lepidocrocite forms at high oxidation rates (Misawa et
al 1974) The presence of chloride is a prerequisite for the formation of
akageneite (Bernal et al 1959 Refait amp Genin 1997)
A substantial amount of work has been conducted in order to estimate the free
energies of formation of green rusts The free energies of formation reported for
the carbonate and sulphate GRs fall in the range 4234ndash4384 kJsdotmol-1 as determined
from solution data monitored during anoxic alkalimetric titrations and from
reduction potential (Eh) and pH recordings monitored during oxidation of GRs in
aqueous solution (Hansen et al 1994 Drissi et al 1995 Genin et al 1996) The
free energies of formation provided allow for estimation of the stability domains of
GRs in Eh-pH phase diagrams (Drissi et al 1995 Genin et al 1996) As
evidenced from such diagrams (Figure 13) the stability domain of GR-SO4 lies
within pH 6-8 and Eh -700 ndash -400 mV depending on the activities of Fe(II) and
sulphate (compare Figures 13aampb) This agrees with the natural GR occurrences
found in suboxic non-acid iron-rich environments such as hydromorphic soils and
intertidal sediments (Al-Agha et al 1995 Trolard et al 1996 Genin et al 1998)
In addition GRs have been found as corrosion products in numerous engineering
systems including a pipeline distribution system for drinking water steel sheet
piles in marine sediments reinforced concrete (ferro-concrete) and permeable
reactive barriers of zero-valent iron implemented for on-site remediation of organic
and inorganic contaminants (Tuovinen et al 1980 Nielsen 1976 Genin et al
1991 Roh et al 2000)
6 Chapter 1
Figure 13 Eh-pH phase diagrams of GR-SO4 a) a = 10+2Fe-3 = 10minus2
4SOa -3 and b) a = 10+2Fe-2
= 10minus24SOa -1
The stability domains of GR-Cl and GR-CO3 are similar to the stability domain of
GR-SO4 At Fe(II) and sulphate activities lower than depicted in Figure 13b the
stability domain of GR-SO4 will be situated at higher pH and lower Eh Other
dissolved species present at anoxic conditions such as phosphate sulphide
carbonate and organic ligands may exert considerable effects on the availability of
Fe(II) and Fe(III) At anoxic and circumneutral conditions vivianite
(Fe2(PO4)2sdot8H2O) controls the Fe(II) activity even at very low phosphate
concentrations (Nriagu amp Dell 1974) The formation of solid Fe(II) sulphides and
siderite (FeCO3) as well as the complexation of Fe(II) and Fe(III) by organic
ligands may also control the activity of Fe(II) in the subsurface and thereby
interfere with the formation of GRs
Due to their layered structures anionic interlayers and high specific surface areas
GRs represent reactive ion exchangers and sorbents of environmentally concerning
anions eg arsenate and selenate (Myneni et al 1997 Randall et al 2001) In
addition GRs may incorporate divalent transition metal cations like Ni2+ Zn2+
Cd2+ Co2+ and Mg2+ by isomorphic substitution for Fe2+ in the hydroxide layers
General Introduction 7
(Tamaura 1985 Tamaura 1986 Refait et al 1994 Parmar et al 2001 Refait et
al 2001) Furthermore GRs have been shown to reduce a range of inorganic
contaminants such as nitrite nitrate selenate chromate uranyl pertechnetate and
the transition metals AgI AuIII CuII and HgII as well as organic pollutants
including halogenated ethanes ethenes and methanes (Hansen et al 1994 Hansen
et al 1996 Myneni et al 1997 Erbs et al 1999 Loyaux-Lawniczak et al 1999
Cui amp Spahiu 2002 Lee amp Batchelor 2002b Heasman et al 2003 OrsquoLoughlin et
al 2003aampb Pepper et al 2003 Elsner et al 2004 OrsquoLoughlin amp Burris 2004)
Thus through sequestration and reductive transformation GRs may play an
important role in the fate and transport of contaminants in suboxic soils and
sediments It should be noted that the rate constants reported for the reduction of
these inorganic and organic pollutants by GRs cannot be directly compared as the
various studies were conducted at very different experimental conditions
13 Microbial formation of green rusts
Generally one would expect that biogenic minerals have chemical compositions
and crystal habits similar to those produced by nonenzymatic processes as they are
governed by the same equilibrium principles In fact since the latter stages of
mineralization are abiotically driven and since the secondary Fe(II)-containing
minerals are formed indirectly by electron transfer outside the bacterial cell and not
directly inside the bacterial cell the type of iron mineral formed is a function of the
environmental conditions in which the bacteria live ie the same microorganism
form different minerals in different environments
The microbial formation of GRs resulting from bioreduction of various Fe(III)
oxides including ferrihydrite goethite and lepidocrocite by strains of the
anaerobic dissimilatory DIRB Shewanella putrefaciens has been reported
repeatedly over the last years (Fredrickson et al 1998 Kukkadapu et al 2001
Parmar et al 2001 Ona-Nguema et al 2002aampb Glasauer et al 2003)
However no evidence of biogenic formation of GRs at natural geochemical
8 Chapter 1 conditions have been offered and it is still unknown whether this process may take
place at natural conditions comprising low nutrient levels low iron concentrations
and the absence of synthetic electron shuttles and highly concentrated artificial pH
buffers Moreover the biotic formation of GRs by anaerobic denitrifying Fe(II)-
oxidizing bacteria has been suggested but the phases still need to be properly
identified (Chaudhuri et al 2001) In order to elucidate the role of bacteria in the
formation of GRs in natural soils and sediments we studied the iron mineral
phases forming as a result of the activity of iron-respiring bacteria (Chapters 2 and
3)
14 Redox reactions of green rusts
Fe(II) is one of the most abundant reductants present in aquatic and terrestrial
environments under suboxic and anoxic conditions (Lyngkilde amp Christensen
1992 Ruumlgge et al 1998) In these environments Fe(II) may be present as soluble
organic and inorganic complexes as surface complexes and as a host of Fe(II)-
bearing minerals Although aqueous Fe(II) complexes may reduce a number of
contaminants Fe(II) associated with mineral surfaces and structural Fe(II) present
in the mineral lattice in Fe(II)-containing minerals are often more powerful
reductants Fe(II)-bearing minerals including GRs magnetite siderite Fe(II)
sulphides as well as Fe(II)-carrying Fe(III) oxide and clay mineral surfaces have
been shown to reduce a number of organic and inorganic contaminants such as
nitroaromatic compounds chlorinated aliphatics chromate uranyl pertechnetate
nitrate monochloramine and carbamate pesticides (Klausen et al 1995 Cui amp
Eriksen 1996 Butler amp Hayes 1998amp1999 Erbs et al 1999 Liger et al 1999
Loyaux-Lawniczak et al 1999 Amonette et al 2000 Hwang amp Batchelor 2000
Hansen et al 2001 Gander et al 2002 Lee amp Batchelor 2002aampb Pecher et al
2002 Vikesland amp Valentine 2002 Hofstetter et al 2003 OrsquoLoughlin et al
2003aampb Strathmann amp Stone 2003 Elsner et al 2004 OrsquoLoughlin amp Burris
2004) However only few comparative studies on the reactivity of Fe(II)-bearing
minerals exist (Lee amp Batchelor 2002b Elsner et al 2004) When examining the
General Introduction 9
reaction rates of the reductive transformation of NACs and chlorinated aliphatics
by GRs and other Fe(II)-bearing minerals reported in these studies the rate
constants for GRs are mostly among the highest rates reported and in some cases
even higher than the rate constants for Fe(II) sulphides Thus GRs may play an
important role in the transformation of reducible contaminants in the subsurface
Nitroaromatic compounds (NACs) are widely applied as explosives herbicides
insecticides solvents and intermediates in the synthesis of dyes and pesticides
(Hartter 1985 Rosenblatt et al 1991) NACs are ubiquitous in the subsurface
environment and pose a health risk due to their toxicity (Rickert 1985) In anoxic
environments reduction of the nitro group is generally the first step during abiotic
or microbial transformation of the NACs (Macalady et al 1986) The
transformation reaction generally produces the corresponding aromatic amines and
minor amounts of intermediates (hydroxylamines and nitroso compounds) as well
as coupling products (azo and azoxy compounds) These products may be of
similar or even greater environmental concern
Trichloroacetic acid (TCA) is ubiquitous in soils and the concentrations reported
range from lt005 to 380 microgkg (Euro Chlor 2001 McCulloch 2002 Ahlers et al
2003) On account of its phytotoxicity suspected human carcinogenicity and
widespread occurrence TCA is of considerable environmental concern especially
in the terrestrial compartment (Ahlers et al 2003) Moreover the daughter
compounds of TCA - dichloroacetic acid (DCA) and monochloroacetic acid
(MCA) - are also toxins and suspected human carcinogens as well as widespread in
the environment (Reimann et al 1996 Berg et al 2000 Ahlers et al 2003 and
references therein) In this work the reactivity of synthetic green rusts towards
nitroaromatic compounds (NACs) and the reactivity of various Fe(II)-Fe(III)
mineral systems including synthetic GRs towards chlorinated acetates have been
studied (Chapters 4 and 5)
10 Chapter 1 15 Outline of the thesis
An examination of the Fe-containing products produced during solid state
oxidation of vivianite by anaerobic autotrophic denitrifying Fe(II)-oxidizing
bacteria is presented in chapter 2 The Fe(II)-oxidizing bacteria were cultured in a
mineral medium containing high levels of bicarbonate and phosphate which is
typically used in this kind of studies as it provides excellent conditions for the
nitrate-reducing FeOB The solid iron phases forming were investigated by
transmission Moumlssbauer spectroscopy infrared spectroscopy and scanning electron
microscopy
Chapter 3 includes a study on the Fe-containing products formed during reduction
of common Fe(III) oxides by the anaerobic dissimilatory Fe(III)-reducing
microorganism Shewanella algae BrY In order to simulate natural conditions
Fe(III) oxides were applied as coatings on silica (model system for sandy soils) or
calcite particles (model system for calcareous soils) and synthetic electron shuttles
as well as highly concentrated artificial pH buffers were excluded The mineral
systems studied include goethitecalcite lepidocrocitecalcite and hydrous ferric
oxidesand suspensions The solid iron phases produced were examined by
transmission Moumlssbauer spectroscopy
A study on the relative reactivity of outer and inner Fe(II) sites in synthetic GR-
SO4 by using a series of structurally closely related compounds with different
charge properties as ldquoreactive probesrdquo is presented in chapter 4 The probe
compounds included nitrobenzene 2-nitrophenol 4-nitrotoluene 4-
chloronitrobenzene and 4-nitrophenylacetic acid
In chapter 5 an investigation of the reactivity of various Fe(II)-Fe(III) mineral
systems towards TCA and DCA is presented The study included laboratory batch
experiments imitating natural conditions The Fe(II)-Fe(III)-systems investigated
included GR-SO4 carbonate green rust magnetite Fe(II)goethite and
General Introduction 11
Fe(II)lepidocrocite The reactivities of the Fe(II)-Fe(III) mineral systems were
examined by comparing their surface-normalized rate constants
The results and environmental implications of this work are summarized in chapter
6 References Ahlers J Regelmann J Riedhammer C (2003) Environmental risk assessment of airborne trichloroacetic acid - a contribution to the discussion of the significance of anthropogenic and natural sources Chemosphere 52 531-537 Al-Agha MR Burley SD Curtis CD Esson J (1995) Complex cementation textures and authigenic mineral assemblages in recent concretions from the Lincolnshire Wash (east coast UK) driven by Fe(0) to Fe(II) oxidation Journal of the Geological Society 152 157-171 Amonette JE Workman DJ Kennedy DW Fruchter JS Gorby YA (2000) Dechlorination of carbon tetrachloride by Fe(II) associated with goethite Environmental Science and Technology 34 4606-4613 Berg M Muumlller SR Muumlhlemann J Wiedmer A Schwarzenbach RP (2000) Concentrations and mass fluxes of chloroacetic acids and trifluoroacetic acid in rain and natural waters in Switzerland Environmental Science and Technology 34 2675-2683 Bernal JD Dasgupta DR Mackay AL (1959) The oxides and hydroxides of iron and their structural inter-relationships Clay Minerals Bulletin 4 15-30 Butler EC Hayes KF (1998) Effects of solution composition and pH on the reductive dechlorination of hexachloroethane by iron sulfide Environmental Science and Technology 32 1276-1284 Butler EC Hayes KF (1999) Kinetics of the transformation of trichloroethylene and tetrachloroethylene by iron sulfide Environmental Science and Technology 33 2021-2027 Carlson L Schwertmann U (1990) The effect of CO2 and oxidation rate on the formation of goethite versus lepidocrocite from an Fe(II) system at pH 6 and 7 Clay Minerals 25 65-71 Chaudhuri SK Lack JG Coates JD (2001) Biogenic magnetite formation through anaerobic biooxidation of Fe(II) Applied and Environmental Microbiology 67 2844-2848 Cornell RM Schwertmann U (1996) The iron oxides Structure properties reactions occurrence and uses VCH Verlagsgesellschaft mbH Weinheim Cui D Eriksen TE (1996) Reduction of pertechnetate by ferrous iron in solution influence of sorbed and precipitated Fe(II) Environmental Science and Technology 30 2259-2262
Cui D Spahiu K (2002) The reduction of U(VI) on corroded iron under anoxic conditions Radiochemica Acta 90 623-628
12 Chapter 1 Drissi SH Refait Ph Abdelmoula M Geacutenin JMR (1995) The preparation and thermodynamic properties of Fe(II)-Fe(III) hydroxide-carbonate (green rust I) Pourbaix diagram of iron in carbonate-containing aqueous media Corrosion Science 37 2025-2041 Elsner M Haderlein SB Schwarzenbach RP (2004) Reactivity of Fe(II)-bearing minerals towards reductive transformation of organic contaminants Environmental Science and Technology 38 799-807 Erbs M Hansen HCB Olsen CE (1999) Reductive dechlorination of carbon tetrachloride using iron(II)iron(III)-hydroxide-sulphate (green rust) Environmental Science and Technology 33 307-311 Euro Chlor (2001) Trichloroacetic acid in the environment a dossier Euro Chlor Brussels and the European Chlorinated Solvent Association Fredrickson JK Zachara JM Kennedy DW Dong H Onstott TC Hinman NW Li S (1998) Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium Geochimica et Cosmochimica Acta 62 3239-3257 Gander JW Parkin GF Scherer MM (2002) Kinetics of 111-trichloroethane transformation by iron sulfide and a methanogenic consortium Environmental Science and Technology 36 4540-4546 Geacutenin JMR Bourrieacute G Trolard F Abdelmoula M Jaffrezic A Refait Ph Maitre V Humbert B Herbillon A (1998) Thermodynamic equilibria in aqueous suspensions of synthetic and natural Fe(II)-Fe(III) green rusts Occurrences of the mineral in hydromorphic soils Environmental Science and Technology 32 1058-1068 Geacutenin JMR Olowe AA Benbouzid-Rollet ND Prieur D Confente M Resiak B (1991) The simultaneous presence of green rust 2 and sulfate reducing bacteria in the corrosion of steel sheet piles in a harbour area Hyperfine Interactions 69 875-878 Geacutenin JMR Olowe AA Refait Ph Simon L (1996) On the stoichiometry and Pourbaix diagram of Fe(II)-Fe(III) hydroxy-sulphate of sulphate-containing green rust 2 An electrochemical and Moumlssbauer spectroscopy study Corrosion Science 38 1751-1762 Glasauer S Weidler PG Langley S Beveridge TJ (2003) Controls on Fe reduction and mineral formation by a subsurface bacterium Geochimica et Cosmochimica Acta 67 1277- 1288 Hanert HH (1992) The genus Gallionella In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 4082-4088 Hansen HCB Borggaard OK Soslashrensen J (1994) Evaluation of the free energy of formation of iron(II)iron(III)-hydroxidesulphate (Green Rust) and its reduction of nitrite Geochimica et Cosmochimica Acta 58 2599-2608 Hansen HCB Guldberg S Erbs M Koch CB (2001) Kinetics of nitrate reduction by green rusts ndash effects of interlayer anion and Fe(II)Fe(III) ratio Applied Clay Science 18 81-91
General Introduction 13
Hansen HCB Koch CB Nancke-Krogh H Borggaard OK Soslashrensen J (1996) Abiotic nitrate reduction to ammonium Key role of green rust Environmental Science and Technology 30 2053-2056 Hartter DR (1985) The use and importance of nitroaromatic chemicals in the chemical industry In Toxicity of nitroaromatic compounds Rickert DE (ed) Hemisphere Publishing Corporation 1-13 Heasman DM Sherman DM Ragnarsdottir KV (2003) The reduction of aqueous Au3+ by sulfide minerals and green rust phases American Mineralogist 88 725-738 Hofstetter TB Schwarzenbach RP Haderlein SB (2003) Reactivity of Fe(II) species associated with clay minerals Environmental Science and Technology 37 519-528 Hwang I Batchelor B (2000) Reductive dechlorination of tetrachloroethylene by Fe(II) in cement slurries Environmental Science and Technology 34 5017-5022 King GM (1990) Effects of added manganic and ferric oxides on sulfate reduction and sulfide oxidation in intertidal sediments FEMS Microbiology Ecology 73 131-138 Klausen J Troumlber SP Haderlein SB Schwarzenbach RP (1995) Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions Environmental Science and Technology 29 2396-2404 Kuenen JG Robertson LA Tuovinen OH (1992) The genera Thiobacillus Thiomicrospira and Thiosphaera In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 2618-2624
Kukkadapu RK Zachara JM Smith SC Fredrickson JK Liu C (2001) Dissimilatory bacterial reduction of Al-substituted goethite in subsurface sediments Geochimica et Cosmochimica Acta 65 2913-2924 Lee W Batchelor B (2002a) Abiotic reductive dechlorination of chlorinated ethylenes by iron- bearing soil minerals 1 Pyrite and magnetite Environmental Science and Technology 36 5147- 5154 Lee W Batchelor B (2002b) Abiotic reductive dechlorination of chlorinated ethylenes by iron-bearing soil minerals 2 Green rust Environmental Science and Technology 36 5348- 5354 Liger E Charlet L Van Cappellen P (1999) Surface catalysis of uranium (VI) reduction by iron(II) Geochimica et Cosmochimica Acta 63 2939-2955 Lovley DR (1997) Microbial Fe(III) reduction in subsurface environments FEMS Microbiology Reviews 20 305-313 Lovley DR Phillips EJP (1986) Organic matter mineralization with reduction of ferric iron in anaerobic sediments Applied and Environmental Microbiology 51 683-689 Lovley DR Phillips EJP Lonergan DJ (1991) Enzymatic versus nonenzymatic mechanisms for Fe(III) reduction in aquatic sediments Environmental Science and Technology 25 1062-1067
14 Chapter 1 Loyaux-Lawniczak S Refait Ph Lecomte P Ehrhardt J Geacutenin JMR (1999) The reduction of chromate ions by Fe(II) layered hydroxides Hydrology and Earth System Sciences 3 593-599 Lyngkilde J Christensen TH (1992) Redox zones of a landfill leachate pollution plume (Vejen Denmark) Journal of Contaminant Hydrology 10 273-289 Macalady DL Tratnyek PG Grundl TJ (1986) Abiotic reduction reactions of anthropogenic organic chemicals in anaerobic systems A critical review Journal of Contaminant Hydrology 1 1-28 McCulloch A (2002) Trichloroacetic acid in the environment Chemosphere 47 667-686 Mendiboure A Schoumlllhorn A (1986) Formation and anion exchange reactions of layered transition metal hydroxides [Ni1-xMx](OH)2(CO3)x2(H2O)z (M = Fe Co) Revue de Chimie Mineacuterale 23 819-827 Misawa T Hashimoto K Shimodaira S (1974) The mechanism of formation of iron oxide and oxyhydroxides in aqueous solutions at room temperature Corrosion Science 14 131-149 Mulder EG Deinema MH (1992) The sheathed bacteria In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 2618-2624 Myneni SCB Tokunaga TK Brown Jr GE (1997) Abiotic selenium redox transformations in the presence of Fe(IIIII) oxides Science 278 1106-1109 Nealson KH Saffarini D (1994) Iron and manganese in anaerobic respiration Environmental significance physiology and regulation Annual Review of Microbiology 48 311-343 Nielsen A (1976) Hvid groslashn og sort rust Beskrivelse af en korrosionsskade paring et svoslashmmebassin Nordisk Betong 2 21-24 Nriagu JO Dell CI (1974) Diagenetic formation of iron phosphates in recent lake sediments American Mineralogist 59 934-946 OLoughlin EJ Burris DR (2004) Reduction of halogenated ethanes by green rust Environmental Toxicology and Chemistry 23 41-48 OLoughlin EJ Kelly SD Cook RE Csencsits R Kemner KM (2003a) Reduction of uranium(VI) by mixed iron(II)iron(III) hydroxide (green rust) Formation of UO2 nanoparticles Environmental Science and Technology 37 721-727 OLoughlin EJ Kelly SD Kemner KM Csencsits R Cook RE (2003b) Reduction of AgI AuIII CuII and HgII by FeIIFeIII hydroxysulfate green rust Chemosphere 53 437-446 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002a) Iron (IIIII) hydroxycarbonate green rust formation and stabilization from lepidocrocite bioreduction Environmental Science and Technology 36 16-20 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002b) Microbial reduction of lepidocrocite γ-FeOOH by Shewanella putrefaciens The
General Introduction 15
formation of green rust Hyperfine Interactions 139140 231-237 Parmar N Gorby YA Beveridge TJ Ferris FG (2001) Formation of green rust and immobilization of nickel in response to bacterial reduction of hydrous ferric oxide Geomicrobiology Journal 18 375-385 Pecher K Haderlein SB Schwarzenbach RP (2002) Reduction of polyhalogenated methanes by surface-bound Fe(II) in aqueous suspensions of iron oxides Environmental Science and Technology 36 1734-1741 Pepper SE Bunker DJ Bryan ND Livens FR Charnock JM Pattrick RAD Collison D (2003) Treatment of radioactive wastes An X-ray absorption spectroscopy study of the reaction of technetium with green rust Journal of Colloid and Interface Science 268 408- 412 Randall SR Sherman DM Ragnarsdottir KV (2001) Sorption of As(V) on green rust (Fe4(II)Fe2(III)(OH)12SO4
3H2O) and lepidocrocite (γ-FeOOH) Surface complexes from EXAFS spectroscopy Geochimica et Cosmochimica Acta 65 1015-1023 Refait Ph Abdelmoula M Trolard F Geacutenin JMR Ehrhardt JJ Bourrieacute G (2001) Moumlssbauer and XAS study of a green rust mineral the partial substitution of Fe2+ by Mg2+ American Mineralogist 86 731-739 Refait Ph Drissi SH Marie Y Geacutenin JMR (1994) The substitution of Fe2+ ions by Ni2+ ions in green rust one compounds Hyperfine Interactions 90 389-394 Refait Ph Geacutenin JMR (1997) The mechanisms of oxidation of ferrous hydroxychloride β- Fe2(OH)3Cl in aqueous solution The formation of akaganeite vs goethite Corrosion Science 39 539-553 Reimann S Grob K Frank H (1996) Chloroacetic acids in rainwater Environmental Science and Technology 30 2340-2344 Rickert DE (1985) Toxicity of nitroaromatic compounds Hemisphere Publishing Corporation 1-13 Roh Y Lee SY Elless MP (2000) Characterization of corrosion products in the permeable reactive barriers Environmental Geology 40 184-194 Rosenblatt DH Burrows EP Mitchell WR Parmer DL (1991) Organic explosives and related compounds In The Handbook of Environmental Chemistry Anthropogenic compounds Hutzinger O (Ed) Springer-Verlag 195-234 Ruumlgge K Hofstetter TB Haderlein SB Bjerg PL Knudsen S Zraurig C Mosbaeligk H Christensen TH (1998) Characterization of predominant reductants in an anaerobic leachate- affected aquifer by nitroaromatic probe compounds Environmental Science and Technology 32 23-31 Schwertmann U Cornell RM (1991) Iron oxides in the laboratory Preparation and characterization VCH Verlagsgesellschaft mbH Weinheim Simon L Francois M Refait Ph Renaudin G Lelaurain M Geacutenin JMR (2003)
16 Chapter 1 Structure of the Fe(II-III)-layered double hydroxysulphate green rust two from Rietveld analysis Solid State Sciences 5 327-334 Strathmann TJ Stone AT (2003) Mineral surface catalysis of reactions between FeII and oxime carbamate pesticides Geochimica et Cosmochimica Acta 67 2775-2791 Straub KL Benz M Schink B Widdel F (1996) Anaerobic nitrate-dependent microbial oxidation of ferrous iron Applied and Environmental Microbiology 62 1458-1460 Tamaura Y (1985) ZnII-bearing green rust II and its spontaneous transformation into ZnII- bearing ferrite in aqueous solution Bulletin of the Chemical Society of Japan 58 2951-2954 Tamaura Y (1986) Ni(II)-bearing green rust II and its spontaneous transformation into Ni(II)- bearing ferrites Bulletin of the Chemical Society of Japan 59 1829-1832 Taylor RM (1980) Formation and properties of Fe(II)Fe(III)-hydroxycarbonate and its possible significance in soil formation Clay Minerals 15 369-382 Trolard F Abdelmoula M Bourrieacute G Humbert B Geacutenin JMR (1996) Mise en eacutevidence dun constituant de type rouilles vertes dans les sols hydromorphes Proposition de lexistence dun nouveau mineacuteral la fougeacuterite Geacuteosciences de surface Comptes Rendus de LrsquoAcademie des Sciences 323 1015-1022 Tuovinen OH Button KS Vuorinen A Carlson L Mair DM Yut LA (1980) Bacterial chemical and mineralogical characteristics of tubercles in distribution pipelines Journal of the American Water Works Association 72 626-635 Vikesland PJ Valentine RL (2002) Iron oxide surface-catalyzed oxidation of ferrous iron by monochloramine implications of oxide type and carbonate on reactivity Environmental Science and Technology 36 512-519 Widdel F Schnell S Heising S Ehrenreich A Assmus B Schink B (1993) Ferrous iron oxidation by anoxygenic phototrophic bacteria Nature 362 834-836
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 17
2 Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria
Abstract
This work investigated the Fe-containing products formed by anaerobic
autotrophic denitrifying Fe(II)-oxidizing bacteria in a specific bicarbonate buffered
(30 mM HCO3- pH 70) culture media containing 10 mM Fe(II) 4 mM nitrate and
4 mM phosphate Fe(II) was present initially as a whitish vivianite-like
(Fe3(PO4)2middot8H2O) precipitate and as soluble Fe(II) The initial phase of the
oxidation produced a greenish metavivianite-like ((FeII3-xFeIII
x)(PO4)2(OH)xmiddot(8-
x)H2O x gt 12) phase In the late oxidation phase a reddish precipitate of poorly
crystalline goethite (α-FeOOH) dominated the colour of the media in coexistence
with Fe(II)-containing siderite (FeCO3) The increasing amounts of Fe(III) present
in the ldquovivianiterdquo and ldquometavivianiterdquo structures were accompanied by an
increasing intensity in the green colour as the Fe(II) biooxidation progressed This
colour development has produced the idea of biogenic green rusts (layered Fe(II)-
Fe(III) hydroxides) in several studies on nitrate-dependent Fe(II) biooxidation
However in this work no evidence of green rust formation mediated by anaerobic
denitrifying Fe(II)-oxidizing bacteria was obtained
21 Introduction
Aerobic oxidation of Fe(II)-containing minerals by lithotrophic acidophilic and
neutrophilic bacteria has been known for many years but their broad significance
in the biogeochemical cycling of iron has only been recognized over the past two
decades Aerobic Fe(II)-oxidizing bacteria (FeOB) have been isolated from acidic
ecosystems (Thiobacillus ferrooxidans) neutral eutrophic systems (Sphaerotilus
natans Leptothrix ochracea) and neutral oligotrophic systems (Gallionella
ferruginea) (Hanert 1992 Kuenen et al 1992 Mulder amp Deinema 1992) At
neutral pH Fe(II) is unstable in the presence of oxygen and is rapidly oxidized to
the insoluble Fe(III) Hence the only pH neutral environments where soluble
18 Chapter 2
Fe(II) is available for aerobic FeOB are at interfaces between oxic and anoxic
conditions The aerobic neutrophilic FeOB (Leptothrix ochracea Gallionella
ferruginea and Sphaerotilus natans) live at such interfaces and are usually
associated with the yellowishreddish ferric deposits formed there
Over the past several years there has been a growing recognition that other less
readily detectable types of bacteria are involved in Fe(II) oxidation in ecosystems
at circumneutral pH For example it has been reported that neutrophilic FeOB are
abundant at the Loihi seamount hydrothermal vents and play a major role in the
Fe(III) oxide deposition (Emerson amp Moyer 2002) Similarly unidentified
neutrophilic obligate lithotrophic FeOB have been isolated from the rhizosphere of
wetlands plants where they are closely associated with deposits of amorphous
Fe(III) oxides (Emerson et al 1999) It was previously believed that Fe(III) oxide
deposits associated with sheaths were produced biologically whereas Fe(III) oxide
deposits not associated with cells were produced abiotically Recently the
formation of amorphous Fe(III) oxide in gradient tubes has been attributed to the
action of FeOB (Sobolev amp Roden 2001) The authors attribute 90 of the
oxidation to biological processes and indicated that the organisms seem to produce
a mobile form of Fe(III) that diffuses away from the cells before being
precipitated thereby avoiding encrustation of the cells They suggest that such
soluble Fe(III) complexes might be substrates for closely associated Fe(III)-
reducing bacteria Such an arrangement might allow close coupling between
microbial Fe(II) oxidation and Fe(III) reduction within millimeters of the oxic-
anoxic interface
Anaerobic Fe(II) oxidation by phototrophic purple non-sulfur bacteria utilizing
Fe(II) as an electron donor in the light was recognized only a decade ago (Widdel
et al 1993) Subsequently it was demonstrated that the biological oxidation of
Fe(II) in the absence of oxygen is possible by light-independent chemotrophic
microbial activity using nitrate as the electron acceptor (Straub et al 1996) In
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 19
addition studies conducted in gradient cultures revealed that nitrate-reducing
strains could also oxidize Fe(II) with molecular oxygen (Benz et al 1998) Hence
these Fe(II)-oxidizing strains may use nitrate as well as oxygen as electron
acceptors The microbial oxidation of Fe(II) was coupled to stoichiometric
reduction of nitrate to N2 and only one strain produced traces of N2O as a by-
product (Straub et al 1996 Benz et al 1998) The authors proposed the formation
of 2-line ferrihydrite as the end product of Fe(II) biooxidation The chemical
reduction of nitrate by Fe(II) requires a catalyst eg at least 10 microM Cu2+ in order
to take place at significant rates and may thus be considered insignificant under the
conditions applied in our study (Moraghan amp Buresh 1976) The chemical
oxidation of Fe(II) with nitrous oxide has not been observed However nitrite can
oxidize Fe(II) chemically (Moraghan amp Buresh 1977 Straub et al 1996) but this
process is considered insignificant at the conditions applied here No denitrifying
Fe(II)-oxidizing enrichment culture has been found to produce ammonium from
nitrate
Both lithoheterotrophic (depending on organic cosubstrates such as acetate) and
strictly lithoautotrophic nitrate-reducing FeOB have been found in various marine
and freshwater sediments However most isolates depend on organic cosubstrates
for cell biosynthesis (Benz et al 1998) Most probable number estimations
showed that denitrifying FeOB accounted for 00006-08 of the acetate-oxidizing
denitrifying microbial population Lithotrophic FeOB accounted for less than
00001 of the total bacterial community Attempts to isolate CO2-fixing nitrate-
dependent FeOB from lithotrophic cultures have failed (Straub amp Buchholz-
Cleven 1998) Mixotrophic FeOB accounted for 0004-004 of the total bacterial
community In addition microbial nitrate-dependent Fe(II) oxidation was
demonstrated in a flooded paddy soil as well as in activated sludge from a
wastewater treatment plant (Nielsen amp Nielsen 1998 Ratering amp Schnell 2001)
Since the activity is not restricted to sunlight exposed habitats microbial nitrate-
dependent Fe(II) oxidation is supposedly more important on a global scale than
20 Chapter 2
anaerobic Fe(II) oxidation by phototrophic bacteria Furthermore it has been
reported that anaerobic denitrifying FeOB aptly oxidize biogenic Fe(II) minerals
formed by bioreduction of synthetic goethite and ferrihydrite and that anaerobic
Fe(III)-reducing bacteria readily reduce Fe(III) minerals formed by biooxidation of
Fe(II) (Weber et al 2001 Straub et al 1998) Hence autotrophic denitrifying
FeOB may play a significant role in the nitrogen and iron cycles in subsurface
environments where the nitrate and the Fe(II) zones overlap and organic carbon
supply is limited (Figure 21)
Figure 21 The microbial iron cycle linking the carbon and nitrogen cycles
Phosphate is released into the environment through natural processes such as rock
weathering and decomposition of dead organic material and anthropogenic
activities eg wastewater effluents and application of manure and fertilizers in
horti- and agriculture In anoxic soils and sediments phosphate may be sequestered
by sorption onto Fe(III) oxides (Williams et al 1971 Patrick amp Khalid 1974)
Phosphate strongly influences the type morphology and properties of Fe(III)
oxides formed by oxidation and hydrolysis of Fe(II) salts as well as the degree of
their transformation (Kandori et al 1992 Cumplido et al 2000 Benali et al
2001) Phosphate may also be retained by precipitation of Fe(II) phosphates such
as the monoclinic vivianite (Fe3(PO4)2middot8H2O) which is the most important stable
Fe(II) orthophosphate solid encountered in the subsurface under most conditions
(Nriagu 1972) At anoxic and circumneutral conditions the whitish vivianite
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 21
controls the Fe(II) activity even at very low phosphate concentrations (Nriagu amp
Dell 1974) Vivianite occurs as a secondary mineral in the gossans of metallic ore
deposits and as a weathering product of primary iron-manganese phosphates in
pegmatites (Gaines et al 1997) Moreover natural vivianite occurrences have
been identified in a number of lake and river sediments (Zwaan amp Kortenbout van
der Sluys 1971 Nriagu amp Dell 1974 Postma 1981 Nembrini et al 1983
Henderson et al 1984 Dodd et al 2003 House 2003 and references therein)
Vivianite is also found in sewage sludge as a result of the wastewater treatment
where iron salts are added in order to remove phosphate (Seitz et al 1973) It is
however still indefinite how ubiquitous vivianite is in nature Furthermore only
little is known about the mechanism of vivianite formation and the role played by
sedimentary Fe(III) oxides Anaerobic Fe(III)-reducing microorganisms may
reduce Fe(III) oxides thereby releasing the iron as soluble Fe(II) and mobilizing
the phosphate adsorbed to the Fe(III) oxides (Lovley 1997) It has been suggested
that vivianite is formed by precipitation following reductive dissolution of Fe(III)
oxides (Manning et al 1981 Manning amp Jones 1982) However it has also been
proposed that the transformation of Fe(III) oxides to vivianite occurs topotactically
and not via reductive dissolution (Nembrini et al 1983) Vivianite was shown to
form microbially as a result of the activity of the anaerobic Fe(III)-reducing
bacteria Shewanella putrefaciens in the presence of high Fe(III)-citrate and
phosphate concentrations (Jorand et al 2000) Moreover vivianite formation by
bioreduction of Fe(III) in hydrous ferric oxide and in smectite has been reported
(Fredrickson et al 1998 Dong et al 2003)
Only little is known about the oxidation products of vivianite Metavivianite a
greenish triclinic iron hydroxy phosphate mineral was first described by Ritz et al
(1974) and it was later found to coexist with vivianite in several natural sediment
samples (Henderson et al 1984) Once the Fe(III) content became evident the true
composition of metavivianite ((FeII3-xFeIII
x)(PO4)2(OH)x
22 Chapter 2
middot(8-x)H2O x gt 12)) was established (Rodgers amp Johnston 1985 Rodgers 1986
and references therein) The formation of intermediate greenish precipitates during
oxidation of fluffy colourless Fe(II) precipitates by anoxic phototrophic
microorganisms and nitrate-dependent FeOB have been reported (Ehrenreich amp
Widdel 1994 Chaudhuri et al 2001) Since both studies were conducted in
bicarbonate buffered mineral media (22-30 mM HCO3ndash pH 70-72) containing
37-5 mM phosphate we assume that the initial fluffy whitish precipitates
consisted mainly of vivianite Chaudhuri et al (2001) proposed that the
intermediate green phases produces by the denitrifying FeOB consist of carbonate
green rust (GR-CO3) but no convincing evidence of this biogenic GR-CO3 has been
provided yet The major objective of this work was to examine the Fe-containing
products forming during the course of biooxidation of vivianite by non-
phototrophic anaerobic denitrifying Fe(II)-oxidizing bacteria
2 2 Materials and methods
All handling and sampling of solutions and suspensions were carried out under
sterile and strict anoxic conditions All chemicals were pa quality
221 Microorganisms and media
Enrichment cultures of nitrate-reducing FeOB taken from town ditches (Bremen
Germany) were grown in anoxic bicarbonate-buffered (30 mM HCO3ndash 90
N210 CO2 pH 70) mineral media containing 4 mM phosphate as well as
essential trace elements and vitamins (Table 21 Straub amp Buchholz-Cleven
1998) Ammonium was omitted from the media in order to facilitate detection of
ammonium possibly produced by reduction of nitrate The techniques used for
preparation of media and cultivation of bacteria under anoxic conditions have been
described by Widdel amp Bak (1992) 05 M aqueous stock solutions of FeCl2 or
FeSO4 were prepared in 100 mL glass flasks by reacting 65 mmol of iron powder
(particle size 10 microm Merck) with 100 mL deoxygenated 10 M HCl or 05 M
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 23
H2SO4 respectively The solutions were magnetically stirred and heated (~80degC)
during reaction until the H2(g) production had ceased (ge 1 hour) The FeCl2 and
FeSO4 stock solutions were stored under a small Ar overpressure at 5degC
Table 21 Composition of the mineral medium (adopted from Straub amp Buchholz-Cleven
(1998))
Components Concentration (M) KH2PO4 15middot10-3
K2HPO4 25middot10-3
MgSO4middot7H2O 10middot10-3
CaCl2middot2H2O 50middot10-4
H3BO3 56middot10-5
ZnSO4middot7H2O 10middot10-6
Na2MoO4middot2H2O 40middot10-6
CuSO4middot5H2O 20middot10-7
MnSO4middotH2O 10middot10-6
Na2SeO4 12middot10-5
CoCl2middot6H2O 50middot10-6
NiCl2middot6H2O 80middot10-6
NaCl 10middot10-5
NaHCO3ndash 30middot10-2
Cyanocobalamine (vitamin B12) 37middot10-8
p-aminobenzoic acid (vitamin Hrsquo) 36middot10-7
D(+)-biotin (vitamin H) 41middot10-8
Nicotinic acid (Niacin) 81middot10-7
Ca-D(+)-pantothenate (vitamin B5) 52middot10-8
Pyridoxamine dihydrochloride 96middot10-7
Thiaminechloridehydrochloride (vitamin B1) 15middot10-7
NaNO3 40middot10-3
FeSO4 or FeCl2 0010
222 Characterisation of precipitates
In order to optimize the characterization and distinction between the spectral
components transmission Moumlssbauer spectra were obtained at temperatures
between 5 K and 250 K and in external magnetic fields of 4 T (parallel to the γ-ray
direction) using a conventional constant acceleration spectrometer and a source of 57Co in Rh The spectrometer was calibrated using a 125 microm foil of α-Fe at room
temperature and isomer shifts are given relative to the centroid of the spectrum of
this absorber The spectra were fitted using simple Lorentzian line shape Infrared
(IR) spectra were obtained using a Perkin Elmer FT-IR 2000 spectrometer and the
24 Chapter 2
KBr pellet technique Scanning electron microscopy (SEM) was carried out in
order to study the morphology and composition of the precipitates Specimens for
SEM were prepared by depositing suspended particles onto an aluminum stub
coated with a carbon sticker The stub was quickly transferred into a sputtering
chamber and coated with a thin Pt film (~20 nm) In order to avoid interfering Pt
signals in the energy dispersive spectra the stubs were in some cases not coated
with Pt but quickly transferred to the SEM chamber for evacuation Measurements
were performed using a Philips XL30 equipped with a LaB6 source and an
accelerating voltage of 20 kV and an EDAX eDXi X-ray dispersive spectrometer
223 Biooxidation experiments
The biooxidation experiments were conducted in 50-400 mL butyl rubber
stoppered bottles with a 90 N210 CO2 headspace constituting 10 of the total
volume Prior to inoculation 4 mM NaNO3 was added as the electron acceptor and
10 mM Fe2+ (as chloride or sulphate) as the electron donor to the mineral media
Control experiments were performed in the same media only they were not
inoculated Addition of ferrous iron to the media induced an immediate
precipitation of a solid whitish material The whitish precipitate was collected on
022 microm polyvinylidendifluorid (Durapore Millipore) filters and stored in an
anoxic atmosphere until further measurements Old outgrown media suspensions
that had turned reddish in color due to a precipitate produced by the denitrifying
FeOB were used as inocula Inoculum volume was kept lt1 of the culture volume
in order to prevent the reddish inoculum from dominating over the initial whitish
precipitate Cultures were incubated in the dark at room temperature and gently
agitated once every day Typical color successions for the media were initial
whitish precipitates turning first more and more greenish over time and then finally
turning reddish (see Fig 22) The color developed uniformly without any
indications of multiple phases in the precipitate At different time intervals
suspension samples were withdrawn using 90 N210 CO2-flushed polyethylene
syringes The precipitates were collected on 022 microm polyvinylidendifluorid
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 25
(Durapore Millipore) filters and analyzed by Moumlssbauer spectroscopy and SEM
Nonfiltered suspension samples were digested in 01 M HCl and chemically
analyzed for Fe(II) NO3- and NH4
+
224 Analytical methods
Fe2+ was determined using a modified phenanthroline method (Fadrus amp Maly
1975) Nitrate was quantified by ion chromatography (Morales et al 2000) and
ammonium was measured photometrically using the indophenol reaction (Rossum
amp Villarruz 1963)
23 Results and discussion
231 Identification of solid iron-containing phases
In most cases the mineral media for cultivating denitrifying FeOB contained 10
mM FeCl2 or FeSO4 4 mM NO3ndash 4 mM total phosphorus and 30 mM HCO3
- at pH
70 ([HCO3-] = 2138middot[CO3
2-] at pH 70) Whitish flocs precipitated immediately at
these initial conditions when Fe(II) was added to the media (Figure 22a) Such
colourless flocs have been reported to precipitate in similar mineral media (10 mM
Fe(II) 37-5 mM phosphate 22-30 mM HCO3ndash pH 70-72) (Ehrenreich amp
Widdel 1994 Chaudhuri et al 2001) Our Fe(II) measurements showed that 20-
50 of the total Fe(II) added was present in this initial white precipitate
26 Chapter 2
a
210- Figure 22 Colour of suspended material in the growth media during Fe(II) biooxidation a)
Initial whitish precipitate prior to inoculation b) Inte1mediate greenish phase fo1med within 2-3
days after inoculation c) reddish precipitate at late stage of biooxidation (gt5-6 days)
The precipitates were filtered and investigated by Mossbauer and IR spectroscopy
The transmission Mossbauer spectra obtained for the initial whitish precipitate at
temperatures between 20 and 250 K are shown in Figure 23 The spectrum
measured at 250 K consists of two fairly well-resolved Fe(II) doublets (see
parameters in Table 22) The change in line-overlap with decreasing temperature
is primarily ascribed to differences in the temperature dependence of the
quadrupole splitting of the two components From the spectra at 10 and 6 K
(Figure 24) it can be concluded that magnetic ordering takes place between these
two temperatures and that only one transition occurs (indicating the presence of
only one phase) The parameters of one of the Fe(II) doublets at 250 K (designated
B in Table 22) are in very good agreement with previously published values for
the vivianite Fe(Il)8 site at room temperature (eg McCammon amp Burns 1980)
and the ordering temperature also agrees well with an assignment as vivianite
(Forsyth et al 1970) However the second Fe(II) doublet in the initial colourless
precipitate (Table 22) has parameters that deviate from previously reported values
by having a smaller quadrupole splitting (indicating a less distorted coordination)
and a significantly higher relative intensity and line width These effects might be
due to the presence of numerous defects in the vivianite crystal lattice particularly
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 27
affecting the Fe(II)A sites It should be noted that further components may be added
to the fit in order to improve its statistics Nevertheless we decided to include no
further components as suggested by the finding of one magnetic ordering only
Accordingly our interpretation of the Moumlssbauer results for the initial white
precipitate suggests a highly defective vivianite having a distribution of local
coordination environments particular in the A site This assignment is further
supported by a major absorption band due to phosphate anions in the infrared
spectrum at approximately 1000 cm-1 and the absence of other complex anions
(data not shown) Thus the whitish precipitate is referred to as a vivianite-like
(ldquovivianiterdquo) precipitate
0
3 5
3 0
2 5
2 0
1 5
1 0
0 5
0 0
20 K
Velocity (mms)
Rel
ativ
e ab
sorp
tion
()
3
2
1
0
80 K
3 5
3 0
2 5
2 0
1 5
1 0
0 5
0 0
150 K
-5 -4 -3 -2 -1 0 1 2 3 4 5
2 5
2 0
1 5
1 0
0 5
0 0
250 K
Figure 23 Transmission Moumlssbauer spectra measured between 250 and 20 K of the initial
whitish precipitate prior to inoculation (see Fig 22a) Fitting components (and their sum) are
shown as full lines
28 Chapter 2
Table 22 Selected Mossbauer parameters of the doublet components in the spectra obtained for
different precipitates
Precipitates Temperature Isomer shift Quadrupole Line width Area (K) (mms-1) splitting (mms-1) (mms-1) ()
Whitish Fe(II)B 250 127 309 035 38
Fe(II)A 250 128 181 051 62
Dark greenish Fe(II)B 250 126 305 023 17
Fe(II)A 250 132 238 051 44
Fe(III) 250 036 085 040 38
Reddish-orange 40 138 244 094 67 Fe(II) Hyperfine parameters are generally given with uncertainties of 003 mms- the spectral area with an uncertainty of 3
1005
1000
0995
0990
- 0985 ~ e c 0980 0
-~ 0975 E c nl b 1000 g ~ Qi 0995 0
0990
0985
0980 -12 -8
~ -~
bullbull bull Ibull bull bullbull bull bull ~ bull bull ~
It
10 K
bull bull bull bull (J ~ i~ 6K bullmiddotf bull bull
~ bull bull bull
-4 0 4 8 12
Velocity (mmls)
Figure 24 Transmission Mossbauer spectra measured at 10 and 6 K of the initial whitish
precipitate prior to inoculation (see Fig 22a)
In general the color of the media suspension changed from whitish into light green
color within 2-3 days after inoculation (Figure 22b ) This transformation occurred
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 29
without dissolving the initial whitish precipitate or preserving the whitish
precipitate as a separate phase implying some kind of solid state transformation
Two identical media suspensions were inoculated simultaneously but at the
sampling time they had distinctly different intensities of the green color
designated light and dark green respectively Moumlssbauer spectra of the dark green
sample are shown in Figure 25 The spectra of this sample are all fitted using three
doublet components (two Fe(II) and one Fe(III)) and the parameters of the
spectrum measured at 250 K are given in Table 22
30
25
20
15
10
05
00
20 K
Velocity (mms)
Rel
ativ
e ab
sorp
tion
()
35
30
25
20
15
10
05
00
80 K
30
25
20
15
10
05
00
150 K
-5 -4 -3 -2 -1 0 1 2 3 4 5
25
20
15
10
05
00
250 K
Figure 25 Transmission Moumlssbauer spectra measured between 250 and 20 K of the dark
greenish precipitate formed during biooxidation Fitting components (and their sum) are shown
as full lines
No magnetic ordering of the dark green precipitate was observed at temperatures
above 20 K but ordering occurred around 10 K (not shown - due to very thin
30 Chapter 2
samples this was not investigated in details) The two greenish samples had very
similar parameters only differing in the relative intensity of Fe(III) 26 and 38
in the light greenish and dark greenish samples respectively Assuming the
spectral area of a component to be proportional to the abundance of the species in
the solid these results indicate a correlation between the intensity of the green
color and the content of Fe(III) in the precipitate The parameters of the Fe(II)
doublets in the dark green precipitate (Table 22) were in very good agreement
with previously published values for vivianite with a non-negligible Fe(III) content
(McCammon amp Burns 1980) whereas the Fe(III) component in particular had a
higher quadrupole splitting The observation that magnetic ordering of both Fe(II)
and Fe(III) occurred at similar temperature for the light green phase (Figure 26) is
a strong indication that they both belong to the same phase The absence of the
component with the low quadrupole splitting in the spectra of both green samples
might indicate that the initial vivianite-like phase crystallized into a more well-
defined vivianite over time However freshly prepared and long-term aged (gt1
year) suspensions of the initial vivianite-like precipitate did not differ significantly
Hence we suggest that the recrystallization of the vivianite-like precipitate can be
explained by Fe(II) biooxidation
Solid State Oxidation ofFe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria
0
2
3
- 4
~ 5 c ~ 6 e-0 7 -2 nl Q) 00 gt ~ 05 Qi 0 10
15
20
25
30
35
bullbullbull hi 6~
bullbullbullbull bull bull bullbull bull 10 K bull bull
bull bull bull bull bull bull bull
~~ ~~ lf 6K bull bull bullbull middot bull bull r bull
bull bull bull bull bull bullbull bull bull bull - 12 -a -4 0 4 8 12
Velocity (mms)
31
Figure 26 Transmission Mossbauer spectrn measured at 6 and 10 K of the light greenish
precipitate fonned during biooxidation
The solid state oxidation of monoclinic vivianite to triclinic metavivianite is well-
known (McCammon amp Bums 1980 Pratesi et al 2003 and references therein)
The vivianite crystal structure contains Fe(II) ions in both isolated Fe(II)A and
paired Fe(Il)8 octahedra Mossbauer spectroscopic analyses have shown that the
Fe(Il)8 Fe(II)A ratio increases with increasing Fe(III) concentration suggesting that
the remaining Fe(II)A ions are more readily oxidized than the Fe(II)a ion of an
Fe(Il)8 -Fe(III)8 pair (McCammon amp Bums 1980) The mechanism of oxidation of
Fe(II) in vivianite involves conversion of H20 ligands to OH- ions producing a
progressive collapse of the vivianite structure due to the elimination of hydrogen
bonds (Moore 1971) The exact oxidation limits between which the triclinic lattice
is stable are somewhat disputed as the results obtained for synthetic and natural
vivianites oxidized chemically in the laboratory and naturally oxidized natural
vivianite specimens do not agree completely (Rodgers 1986 and references
therein) Taking all reports into account the monoclinic structure of vivianite is
supposedly maintained until 40-50 of total iron is oxidized Further oxidation
32 Chapter 2
leads to the formation of the triclinic metavivianite in which the FeA site is fully
oxidized whereas the oxidation of the FeB ranges from 20 to almost 100 Thus
the triclinic metavivianite structure persists close to complete oxidation of total
iron The Moumlssbauer results obtained in this study are consistent with the vivianite
solid state oxidation mechanism reported by McCammon amp Burns (1980) Thus
we propose that the intermediate greenish precipitate is a metavivianite-like
(ldquometavivianiterdquo) phase It should be noted that a minor oxidation of dissolved
Fe(II) may have occurred even though the solid state oxidation of Fe(II) was
predominant
Within 5-6 days after inoculation the greenish intermediate was transformed into a
reddish product (Figure 22c) The magnetically ordered sextet in the spectrum of
the red phase (Figure 27) measured at 40 K was due to goethite (α-FeOOH)
(magnetic hyperfine field of 470 T and a quadrupole shift of -01 mms-1 cp
Moslashrup et al 1983) The sextet deviated from ideal goethite by its asymmetric line
shape and its low ordering temperature (around 100 K ndash data not shown) and thus
the goethite was poorly crystalline It is very likely that the presence of phosphate
in the media retarded the crystal growth of goethite The unusual reddish colour of
the goethite might also be explained by the presence of phosphate The spectrum at
40 K was however dominated by a Fe(II) doublet (Table 22) that ordered
magnetically between 40 and 20 K (Figure 27) The hyperfine parameters and the
magnetic ordering temperature indicated that this component was due to siderite
(FeCO3) having a magnetic ordering temperature of 38 K (Jacobs 1963) The
siderite component may have formed as a result of the microbial activity changing
the chemistry of the solution and precipitating a major part of the remaining
dissolved Fe(II) at this stage The characteristic vivianite Fe(II) doublet with large
quadrupole splitting was not detected in this sample The reddish precipitate
contained considerably less Fe(III) than the greenish precipitate (only 33 as
estimated from the spectral area) None of the components in the reddish sample
Solid State Oxidation ofFe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 33
could be detected in freshly inoculated samples indicating that carryover of mineral
precipitates by inoculation of the culture media was negligible
0
2
- 3
~4 c
Q 5
e 6 0
~ 7 g
0 3l Q)
a 1
2
3
4
5
-12 a
40K
bull bull (
4 0 4 8 12 Velocity (mms)
Figure 27 Transmission Mossbauer spectra measured at 20 and 40 K of the reddish precipitate
fo1med during the late biooxidation stage Fitting components (and their sum) are shown as full
lines
Strengite (FeP04middot2H20) was not detected at any time during oxidation
Santabarbaraite a new amorphous F e(III) hydroxy phosphate mineral
(Fe3(P04)i(OH)3middot5H20) was reported in a recent study (Pratesi et al 2003) The
brownish mineral was a result of the solid state oxidation of vivianite through
metavivianite However no Mossbauer data have been provided for this new
mineral yet and therefore we cannot give an account of whether santabarbaraite
forms in our system or not Thus goethite was the dominating end product and we
propose the reaction path depicted in Figure 28 for the nitrate-dependent
biooxidation of Fe(II) in our systems
34 Chapter 2
Fe3(PO4)2middot8H2O (FeII3-xFeIII
x)(PO4)2(OH)xmiddot(8-x)H2O α-FeOOH
NO3- N2 NO3
- N2
ldquoVivianiterdquo ldquoMetavivianiterdquo Goethite
Figure 28 Proposed reaction path and iron-containing minerals forming during solid state
oxidation of vivianite by denitrifying FeOB at the experimental conditions applied in this study
The biotic formation of layered Fe(II)-Fe(III) hydroxides (green rusts) by
anaerobic denitrifying Fe(II)-oxidizing bacteria has been suggested but proper
identification of these phases still lacks (Chaudhuri et al 2001) We cannot rule
out that small amounts of green rusts (GRs) perhaps a phosphate intercalated GR
(Hansen amp Poulsen 1999) might have been present here during the greenish
intermediate ldquometavivianiterdquo oxidation stage When present in low concentrations
especially in mixtures including other iron minerals it is very difficult to identify
GRs even with Moumlssbauer spectroscopy At least two complementary methods
such as X-ray diffraction (XRD) and Moumlssbauer spectroscopy are required for
proper identification and characterization of GRs However the precipitates
collected in this work were poorly crystalline and did not allow for XRD analysis
Electron micrographs including energy dispersive X-ray spectroscopy suspension
colour and mineral stability calculations do not suffice as evidence Hence no
convincing evidence of GR formation facilitated by denitrifying FeOB has been
provided so far The blue-green colours of metavivianite and green rust minerals
originate from Fe(II)-Fe(III) charge transfer between adjacent Fe(II) and Fe(III)
ions in edge-shared octahedra (Faye et al 1968) The greenish suspension colour
occurring during the intermediate phase has incited the idea of biogenic GRs in
studies on nitrate-dependent Fe(II) biooxidation (Chaudhuri et al 2001 Lack et
al 2002aampb) However our results indicate that this reasoning is misleading
232 Factors controlling the rate and extent of Fe(II) biooxidation
Generally it was found that maximally 20-64 of the initial Fe(II) amount was
oxidized to Fe(III) (Figure 29) This indicates some limitations in the accessibility
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 35
of Fe(II) in the system Based solely on stoichiometry considerations the
microorganisms are expected to oxidize 5 mol Fe(II) for every 1 mol nitrate
reduced to dinitrogen However as exemplified in Figure 28 this ratio was mostly
lt4 which can be explained by the consumption of nitrogen as a result of microbial
growth At initial [Fe(II)][NO3ndash] ratios lt5 nitrate is in excess and should not limit
the extent of the biooxidation Thus the lack of complete biooxidation could not be
due to exhaustion of nitrate Furthermore all growth essential nutrients were more
than sufficiently applied hence the incomplete Fe(II) biooxidation was not caused
by lack of nutrients The most reasonable explanation therefore seems to be that an
increasingly limited access to the electron donor over time inhibits complete long-
term Fe(II) biooxidation At least four mechanisms may cause this inhibition 1)
the Fe(II) becomes isolated within the structure of the mixed Fe(II)-Fe(III)
minerals forming during biooxidation or underneath a passive Fe(III)-bearing
surface film on the initial Fe(II) precipitates 2) the FeOB cell surface becomes
covered with a passive Fe(III)-bearing surface film 3) the Fe(II) biooxidation is
controlled by the rate of dissolution of the initial Fe(II) minerals or 4) the reaction
proceeds primarily by biooxidation of dissolved Fe(II) whose concentration
gradually decreases due to changes in solid phase composition The actual
mechanisms whereby the surface-associated Fe(III) can inhibit Fe(II) biooxidation
are unknown but they may involve both kinetic and thermodynamic constraints on
the electron transfer The Moumlssbauer results obtained in this work strongly suggest
that the Fe(II) biooxidation occurred mainly in the solid state of the initial
ldquovivianiterdquo phase However we cannot rule out that some dissolved Fe(II) was
oxidized as well
36 Chapter 2
Figure 29 Concentration profiles of total Fe(II) and nitrate as a function of time during Fe(II)
biooxidation
No Fe(II) oxidation took place in cultures where nitrate had been omitted
confirming that the microbial Fe(II) oxidation is nitrate-dependent (data not
shown) No Fe(II) oxidation was detected in the non-inoculated control
experiments within the duration of the experiments and thus the chemical
oxidation of dissolved Fe(II) by nitrate catalyzed by vitamins or trace elements
(eg Cu(II)) can be neglected Ammonium did not form in detectable amounts
during Fe(II) biooxidation (data not shown) and therefore dinitrogen was assumed
to be the end product as reported previously (Straub et al 1996 Benz et al 1998)
The absence of ammonium formation does indirectly support the absence of
biologically induced green rust formation as synthetic green rust is known to
convert nitrate into ammonium in purely chemical reactions (Hansen et al 1996)
It was visually observed that the phosphate concentration in the media exerted a
control on the microbial Fe(II)-oxidation At phosphorus concentrations le 2 mM
no Fe(II)-oxidation took place However the solubility product for vivianite (Ksp =
171middot10-36 at 25degC (Al-Borno et al 1994)) was still by far exceeded under these
conditions It is not known whether this phosphate limiting effect is due to growth
constraints in the mixed bacterial community or whether specific Fe(II) phosphate
Solid State Oxidation ofFe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 37
precipitates are prerequisites of the Fe(II) biooxidation to take place Experiments
are currently underway in our laboratory in order to elucidate the role of specific
initial Fe(II) precipitates It should be noted that the growth of the denitrifying
FeOB could not be estimated as they were present in highly heterogeneous
suspensions containing both solid iron phases as well as other bacteria (enrichment
culture)
233 Morphology of solid iron phases
The morphology of the various precipitates was studied by SEM The initial
whitish precipitate consisted of a web-like structure (Figure 21 Oa and background
in Figure 21 Ob) whereas more distinct hexagonally shaped rosettes with particle
size ~20 microm (Figure 2lObampc) formed during Fe(II) biooxidation The energy
dispersive spectroscopic element analyses showed that other than iron the initial
whitish precipitate and the rosettes contained mainly phophorus
Figure 210 Scanning electron micro graphs of precipitates fo1med at various stages of the
experiment a Initial whitish precipitate bampc Rosettes observed in the intennediate greenish
precipitate d Reddish precipitate sampled during the late biooxidation stage
38 Chapter 2
These observations are interpreted as vivianite forming a web-like morphology in
the initial whitish precipitate and partly transforming into hexagonal particles in
the greenish colored stage The interpretations are supported by similar vivianite
morphologies reported including pseudo-hexagonal vivianite crystals of low
symmetry resulting from microbial Fe(III) reduction of HFO and platy rosettes of
vivianite crystals formed during bioreduction of Fe(III) in smectite (Fredrickson et
al 1998 Dong et al 2003) It was not possible to associate the morphology
observed in the reddish precipitate with the minerals identified in this phase
(Figure 210d)
24 Conclusions
This work demonstrated that anaerobic autotrophic denitrifying Fe(II)-oxidizing
bacteria produce poorly crystalline goethite by solid state oxidation of ldquovivianiterdquo
via a ldquometavivianiterdquo intermediate The increasing amount of Fe(III) forming in the
vivianite structure was accompanied by an increasing intensity in the green colour
as the Fe(II) biooxidation progressed Moumlssbauer spectroscopic analyses provided
no significant evidence of green rust formation The finding of microbially
oxidized vivianite in this study raises the question of the oxidation state of
vivianite specimens from natural sediments Vivianite is generally believed to be
an ideal Fe(II) hydroxy phosphate mineral and the presence of Fe(III) is explained
by aerial oxidation upon sampling The results presented here indicate that
microbiological processes may be responsible for the oxidation of vivianite and
metavivianite in natural subsurface environments Acknowledgments
We would like to thank Dr K Straub for providing and advising us how to culture the nitrate-
reducing FeOB Furthermore we thank Dr C B Koch for performing the Moumlssbauer analyses
and Dr D Mavrocordatos for performing the SEM analyses
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 39
References Al-Borno A Tomson MB (1994) The temperature dependence of the solubility product constant of vivianite Geochimica et Cosmochimica Acta 58 5373-5378 Benali O Abdelmoula M Refait Ph Geacutenin JMR (2001) Effect of orthophosphate on the oxidation products of Fe(II)-Fe(III) hydroxycarbonate The transformation of green rust to ferrihydrite Geochimica et Cosmochimica Acta 65 1715-1726 Benz M Brune A Schink B (1998) Anaerobic and aerobic oxidation of ferrous iron at neutral pH by chemohetorotrophic nitrate-reducing bacteria Archives of Microbiology 169 159-165 Chaudhuri SK Lack JG Coates JD (2001) Biogenic magnetite formation through anaerobic biooxidation of Fe(II) Applied and Environmental Microbiology 67 2844-2848 Cumplido J Barron V Torrent J (2000) Effect of phosphate on the formation of nanophase lepidocrocite from Fe(II) sulfate Clays and Clay Minerals 48 503-510 Dodd J Large DJ Fortey NJ Kemp S Styles M Wetton P Milodowski A (2003) Geochemistry and petrography of phosphorus in urban canal bed sediment Applied Geochemistry 18 259-267 Dong H Kostka JE Kim J (2003) Microscopic evidence for microbial dissolution of smectite Clays and Clay Minerals 51 502-512 Ehrenreich A Widdel F (1994) Anaerobic oxidation of ferrous iron by purple bacteria a new type of phototrophic metabolism Applied and Environmental Microbiology 60 4517-4526 Emerson D Moyer CL (2002) Neutrophilic Fe-oxidizing bacteria are abundant at the Loihi seamount hydrothermal vents and play a major role in Fe oxide deposition Applied and Environmental Microbiology 68 3085-3093 Emerson D Weiss JV Megonigal JP (1999) Iron-oxidizing bacteria are associated with ferric hydroxide precipitates (Fe-plaque) on the roots of wetland plants Applied and Environmental Microbiology 65 2758-2761 Fadrus H Maly J (1975) Suppression of iron(III) interference in the determination of iron(II) in water by the 110-phenanthroline method The Analyst 100 549-554 Faye GH Manning PG Nickel EH (1968) The polarized optical absorption spectra of tourmaline cordierite chloritoid and vivianite ferrous-ferric electronic interaction as a source of pleochroism American Mineralogist 53 1174-1201 Forsyth JB Johnson CE Wilkonson C (1970) The magnetic structure of vivianite Fe3(PO4)2middot8H2O Journal of Physics Part C Solid State Physics 3 1127-1139 Fredrickson JK Zachara JM Kennedy DW Dong H Onstott TC Hinman NW Li S (1998) Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium Geochimica et Cosmochimica Acta 62 3239-3257 Gaines RV Skinner HCW Foord EE Mason B Rosenzweig A (1997) Danas new
40 Chapter 2 mineralogy 8th ed John Wiley amp Sons Inc Hanert HH (1992) The genus Gallionella In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 4082-4088 Hansen HCB Koch CB Nancke-Krogh H Borggaard OK Soerensen J (1996) Abiotic nitrate reduction to ammonium Key role of green rust Environmental Science and Technology 30 2053-2056 Hansen HCB Poulsen IF (1999) Interaction of synthetic sulphate green rust with phosphate and the crystallization of vivianite Clays and Clay Minerals 47 312-318 Henderson GS Black PM Ridgers KA Rankin PC (1984) New data on New Zealand vivianite and metavivianite New Zealand Journal of Geology and Geophysics 27 367-378 House WA (2003) Geochemical cycling of phosphorus in rivers Applied Geochemistry 18 739-748 Jacobs IS (1963) Metamagnetism of siderite (FeCO3) Journal of Applied Physics 34 1106-1107 Jorand F Appenzeller BMR Abdelmoula M Refait Ph Block J-C Geacutenin JMR (2000) Assessment of vivianite formation in Shewanella putrefaciens culture Environmental Technology 21 1001-1005 Kandori K Uchida S Kataoka S Ishikawa T (1992) Effects of silicate and phosphate ions on the formation of ferric oxide hydroxide particles Journal of Materials Science 27 719-728 Kuenen JG Robertson LA Tuovinen OH (1992) The genera Thiobacillus Thiomicrospira and Thiosphaera In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 2618-2624 Lack JG Chaudhuri SK Chakraborty R Achenbach LA Coates JD (2002a) Anaerobic biooxidation of Fe(II) by Dechlorosoma suillum Microbial Ecology 43 424-431 Lack JG Chaudhuri SK Kelly SD Kemner KM OConnor SM Coates JD (2002b) Immobilization of radionuclides and heavy metals through anaerobic bio-oxidation of Fe(II) Applied and Environmental Microbiology 68 2704-2710 Lovley DR (1997) Microbial Fe(III) reduction in subsurface environments FEMS Microbiology Reviews 20 305-313 Manning PG Birchall T Jones W (1981) Ferric hydroxides in surficial sediments of the great lakes and their role in phosphorus availability a Moumlssbauer spectral study Canadian Mineralogist 19 525-530 Manning PG Jones W (1982) The binding capacity of ferric hydroxides for non-apatite inorganic phosphorus in sediments of the depositional basins of Lakes Erie and Ontario Canadian Mineralogist 20 169-176 McCammon CA Burns RG (1980) The oxidation mechanism of vivianite as studied by Moumlssbauer spectroscopy American Mineralogist 65 361-366 Moore PB (1971) The Fe2+
3(H2O)n(PO4)2 homologous series crystal-chemical relationships
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 41
and oxidized equivalents American Mineralogist 56 1-17 Moraghan JT Buresh RJ (1976) Chemical reduction of nitrate by ferrous iron Journal of Environmental Quality 5 320-325 Moraghan JT Buresh RJ (1977) Chemical reduction of nitrite and nitrous oxide by ferrous iron Journal of American Soil Science Society 40 47-50 Morales JA de Graterol LS Mesa J (2000) Determination of chloride sulfate and nitrate in groundwater samples by ion chromatography Journal of Chromatography A 884 185-190 Mulder EG Deinema MH (1992) The sheathed bacteria In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 2618-2624 Moslashrup S Madsen MB Franck J Villadsen J Koch CJW (1983) A new interpretation of Moumlssbauer spectra of microcrystalline goethiterdquosuper-ferromagnetismrdquo of ldquosuper-spin-glassrdquo behaviour Journal of Magnetism and Magnetic Materials 40 163-174 Nembrini GP Capobianco JA Viel M Williams AF (1983) A Moumlssbauer and chemical study of the formation of vivianite in sediments of Lago Maggiore (Italy) Geochimica et Cosmochimica Acta 47 1459-1464 Nielsen JL Nielsen PH (1998) Microbial nitrate-dependent oxidation of ferrous iron in activated sludge Environmental Science and Technology 32 3556-3561 Nriagu JO (1972) Stability of vivianite and ion-pair formation in the system Fe3(PO4)2-H3PO4- H2O Geochimica et Cosmochimica Acta 36 459-470 Nriagu JO Dell CI (1974) Diagenetic formation of iron phosphates in recent lake sediments American Mineralogist 59 934-946 Patrick Jr WH Khalid RA (1974) Phosphate release and sorption by soils and sediments Effect of aerobic and anaerobic conditions Science 186 53-55 Postma D (1981) Formation of siderite and vivianite and the pore-water composition of a recent bog sediment in Denmark Chemical Geology 31 225-244 Pratesi G Cipriani C Giuli G Birch WD (2003) Santabarbaraite a new amorphous phosphate mineral European Journal of Mineralogy 15 185-192 Ratering S Schnell S (2001) Nitrate-dependent iron(II) oxidation in paddy soil Environmental Microbiology 3 100-109 Ritz C Essene EJ Peacor DR (1974) Metavivianite Fe3(PO4)2middot8H2O a new mineral American Mineralogist 59 896-899 Rodgers KA (1986) Metavivianite and kerchenite a review Mineralogical Magazine 50 687- 691 Rodgers KA Johnston JH (1985) Type metavivianite Moumlssbauer evidence for a revised composition Neues Jahrbuch fuumlr Mineralogie-Monatshefte 12 539-542
42 Chapter 2 Rossum JR Villarruz PA (1963) Determination of ammonia by the indophenol method Journal of American Water Works Association 55 657-658 Seitz MA Riedner RJ Malhotra SK Kipp RJ (1973) Iron-phosphate compound identification in sewage sludge residue Environmental Science and Technology 7 354-357 Sobolev D Roden EE (2001) Suboxic deposition of ferric iron by bacteria in opposing gradients of Fe(II) and oxygen at circumneutral pH Applied and Environmental Microbiology 67 1328-1334 Straub KL Benz M Schink B Widdel F (1996) Anaerobic nitrate-dependent microbial oxidation of ferrous iron Applied and Environmental Microbiology 62 1458-1460 Straub KL Buchholz-Cleven BEE (1998) Enumeration and detection of anaerobic ferrous iron-oxidizing nitrate-reducing bacteria from diverse European sediments Applied and Environmental Microbiology 64 4846-4856 Straub KL Hanzlik M Buchholz-Cleven BEE (1998) The use of biologically produced ferrihydrite for the isolation of novel iron-reducing bacteria Systematic and Applied Microbiology 21 442-449 Weber KA Picardal FW Roden EE (2001) Microbially catalyzed nitrate-dependent oxidation of biogenic solid-phase Fe(II) compounds Environmental Science and Technology 35 1644-1650 Widdel F Bak F (1992) Gram-negative mesophilic sulfate-reducing bacteria In The Prokaryotes (Balows A Truumlper HG Dworkin M Harder W Schleifer K-H (eds)) Springer 2nd ed 3352-3378
Widdel F Schnell S Heising S Ehrenreich A Assmus B Schink B (1993) Ferrous iron oxidation by anoxygenic phototrophic bacteria Nature 362 834-836 Williams JDH Syers JK Shukla SS Harris RF Armstrong DE (1971) Levels of inorganic and total phosphorus in lake sediments as related to other sediment parameters Environmental Science and Technology 5 1113-1120
Zwaan PC Kortenbout van der Sluys G (1971) Vivianite crystals from Hare Noord Brabant Province The Netherlands Scripta Geology 6 1-7
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 43
3 Formation of Layered Iron Hydroxides by Microbial Fe(III)
Reduction Abstract
Many inorganic and organic pollutants may be degraded by microorganisms in the
subsurface However a wide range of contaminants including chromate nitrate
radionuclides nitroaromatic compounds chlorinated aliphatics and carbamate
pesticides may also be chemically transformed by reduction reactions involving
layered iron(II)-iron(III)-hydroxides (green rusts) Hence green rusts (GRs) may
play a potentially important role in the fate and transport of pollutants in iron-rich
suboxic soils and sediments Yet only little is known about the formation of GRs
in these environments The biotic formation of GRs mediated by the anaerobic
dissimilatory Fe(III)-reducing bacteria Shewanella spp has been reported and
proposed in several studies However the experimental conditions applied were
mostly not natural and the evidence of GR formation provided may be questioned
This work investigated the Fe-containing products formed by the facultatively
anaerobic Fe(III)-reducing microorganism Shewanella algae BrY in culture
media containing 4-10 mM formate or lactate and 8-27 mM Fe(III) In order to
simulate natural conditions Fe(III) oxides were applied as coatings on silica
(model system for sandy soils) or calcite particles (model system for calcareous
soils) and synthetic electron shuttles as well as highly concentrated artificial pH
buffers were excluded S algae BrY reduced 19-72 of the initial Fe(III) when
grown in goethitecalcite lepidocrocitecalcite or hydrous ferric oxidesand mineral
systems and green or blackish mineral phases were produced within 1-2 weeks
after inoculation Moumlssbauer spectroscopic analyses indicated that the green and
blackish precipitates were dominated by vivianite (Fe3(PO4)2sdot8H2O) and green rust
44 Chapter 3
31 Introduction
The significance of bacteria in the biogeochemical cycling of iron has been broadly
recognized over the past two decades Chemical processes were previously
considered to account for most of the Fe(III) reduction in subsurface environments
Dissimilatory Fe(III)-reducing bacteria (DIRB) that gain energy by coupling the
oxidation of hydrogen or organic compounds to the reduction of Fe(III) oxides
have been known for many years but their biogeochemical importance was
acknowledged only a decade ago (reviewed by Lovley 1997) DIRB transfer
electrons to extracellular Fe(III) without assimilating the iron Fe(III) bioreduction
accounts for a major fraction of the carbon oxidation in many different
environments and in the presence of high amounts of reactive Fe(III) microbial
Fe(III) reduction may even inhibit sulfate reduction and methanogenesis (King
1990 Lovley amp Phillips 1986) In fact most of the Fe(III) reduction in the Fe(III)
reduction zone of aquatic sediments and aquifers is thought to be enzymatically
catalyzed by microorganisms (Lovley et al 1991) However the relative
importance of microbial and chemical processes involved in the Fe(III) reduction
are still somewhat disputed among microbiologists and geochemists
A wide diversity of Fe(III)-reducing bacteria which fall in a number of different
phylogenetic groups is known today Both organisms growing by respiration and
by fermentation have been isolated and identified (Lovley 1991 Nealson amp
Saffarini 1994) Hydrogen short- and long-chained fatty acids amino acids
sugars and aromatic compounds may serve as electron donors for Fe(III)
bioreduction The enzymes responsible for dissimilatory Fe(III) reduction are outer
membrane associated ferric reductases (Lower et al 2001 and references therein)
Iron reducing bacteria may utilize alternative electron acceptors such as O2 nitrate
S0 sulfate humic substances contaminant metals and metalloids as well as
chlorinated solvents The first organism shown to couple respiratory growth to
dissimilatory iron reduction was Pseudomonas ferrireductans now known as
Shewanella oneidensis but previously classified as Alteromonas putrefaciens and
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 45
Shewanella putrefaciens (Venkateswaran et al 1999) Various DIRB including
the obligate anaerobic Geobacter sp and the facultatively anaerobic Shewanella
sp have been isolated from both marine and freshwater sediments soil and
aquifers (Thamdrup 2000 and references therein)
The redox potentials of oxidized and reduced iron couples and thus the energy
yield available from Fe(III) reduction depend strongly on the specific iron phases
involved In soil and aquatic environments Fe(III) oxides mainly occur in
association with other sediment particles as aggregates or coatings Amorphous
and poorly crystalline Fe(III) oxides usually make up 20 or less of the iron
content in a sediment (Thamdrup 2000) They are the main products of abiotic and
biotic Fe(II) oxidation in sediments and they constitute the most important phases
for microbial Fe(III) reduction Until recently it was generally believed that DIRB
reduced insoluble Fe(III) oxides only by direct contact with the Fe(III) oxide
thereby allowing electron transfer from the cell to the Fe(III) oxide surface
However over the past several years there has been a growing recognition that
DIRB may use different strategies in order to access the solid Fe(III) oxides These
strategies include solubilization of Fe(III) by synthetic or natural Fe(III) chelators
and Fe(III) reduction via electron shuttling with soluble humic substances or
microbially produced electron shuttles (Nevin amp Lovley 2002 and references
therein Turick et al 2003) The Fe(III) complexing agents may also stimulate
Fe(III) oxide reduction indirectly by chelation and thus removal of Fe(II) from
the cell and the Fe(III) oxide surfaces Both chelated Fe(III) and soluble electron
shuttles are more accessible to Fe(III) reductases than solid Fe(III) oxides In
contrast to Geobacter metallireducens S algae produces and releases extracellular
electron shuttling compounds (Nevin amp Lovley 2000) However in the absence of
soluble electron shuttles reversible adhesion is required for reduction of solid
Fe(III) oxides by S algae BrY (Das amp Caccavo 2000) Shewanella algae BrY
adheres readily and preferentially to a range of solid Fe(III) oxides such as
ferrihydrite goethite and hematite (Das amp Caccavo 2001) The adhesion
46 Chapter 3
mechanisms are not completely understood but recent results suggest that the
adhesion is mediated by cell surface proteins and independent of cell motility
(Caccavo amp Das 2002)
The microbial formation of GRs resulting from bioreduction of various Fe(III)
oxides including ferrihydrite goethite and lepidocrocite by strains of the
anaerobic DIRB Shewanella putrefaciens has been reported repeatedly over the
last years (Fredrickson et al 1998 Kukkadapu et al 2001 Liu et al 2001
Parmar et al 2001 Ona-Nguema et al 2002aampb Glasauer et al 2003)
However no evidence of biogenic formation of GRs at natural geochemical
conditions have been offered and it is still unknown whether this process may take
place at natural conditions comprising low carbon and iron concentrations as well
as the absence of synthetic electron shuttles and highly concentrated artificial pH
buffers GRs are layered iron(II)-iron(III)-hydroxides with anionic interlayers and
they hold the general formula [FeII(6-x)FeIII
x(OH)12]x+[(A)xnmiddotyH2O]x- where x = 09 -
42 A is an n-valent anion eg CO32- Clndash or SO4
2- and y is the number of water
molecules in the interlayer In circumneutral solutions the oxidation of dissolved
Fe(II) always passes through solid GR phases (Bernal et al 1959) This agrees
with the natural GR occurrences found in suboxic nonacid iron-rich environments
such as hydromorphic soils and intertidal sediments (Al-Agha et al 1995 Trolard
et al 1996 Genin et al 1998) In addition GRs have been found as corrosion
products in numerous engineering systems eg in a pipeline distribution system
for drinking water steel sheet piles in marine sediments reinforced concrete
(ferro-concrete) and permeable reactive barriers of zero-valent iron implemented
for on-site remediation of organic and inorganic contaminants (Tuovinen et al
1980 Nielsen 1976 Genin et al 1991 Roh et al 2000) Through sequestration
and reductive transformation GRs may play an important role in the fate and
transport of organic and inorganic pollutants in suboxic iron-rich soils and
sediments (see Chapters 4 amp 5 in this work and references therein)
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 47
The major goal of this work was to examine the iron minerals forming during the
course of Fe(III) bioreduction of hydrous ferric oxide goethite and lepidocrocite
Two model systems simulating sandy and calcareous soils in subsurface
environments were designed in order to investigate the formation of iron minerals
at conditions including low carbon levels low Fe(III) concentrations applied as
Fe(III) oxide coatings on sand or calcite no electron shuttle and no synthetic pH
buffers
32 Materials and methods
All handling and sampling of solutions and suspensions were carried out at strict
anoxic conditions Standard sterile techniques were used throughout (Hungate
1969 Miller amp Wolin 1974) Only the iron oxide coatings were not autoclaved in
order to avoid the iron oxides from transforming Goethite (acicular particles with
size 01 times 06 microm specific surface area 16 m2g) and lepidocrocite (acicular
particles with size 005 times 03 microm specific surface area 18 m2g) were purchased as
fine powders from Bayer (Bayferrox 910 and 943) Calcite (grain size 170-350 microm
Pluumlss-Staufer AG) and sea sand (dominantly quartz grain size 01-03 mm Riedel-
de Haeumln) were used as Fe(III) oxide coating bearing minerals
321 Preparation of iron oxide coatings
Two grams of goethite (goe) or lepidocrocite (lep) and 100 g calcite were
combined with 200 mL deionized water (DIW) in a 500 mL polyethylene flask
Hydrous ferric oxide (HFO) was synthesized by dissolving 4 g Fe(NO3)3middot9H2O in
70 mL DIW followed by slow neutralization under magnetic stirring till pH 7 with
approximately 30 mL 1 M NaOH (method modified after Schwertmann amp Cornell
1991) The HFO coating was made by combining 100 mL freshly precipitated
HFO with 900 mL deionized water and 50 g sea sand in a polyethylene bottle The
suspensions containing the iron oxide coatings were gently agitated on a
reciprocating shaker for 24 h and left to stand for another 24 h Excess Fe(III)
oxides and salts were removed from the coated material by repeated decantation
48 Chapter 3
and washing with 003 M NaNO3 followed by washing with DIW until clear
runoff Finally the coatings were collected on folding filters and air dried The
amount of HFO goethite and lepidocrocite coated onto sand and calcite after
washing and drying was quantified to 7-11 mg Fe(III)g sand or calcite
322 Mineral characterisation
The identity and purity of the HFO synthesized were examined by means of X-ray
diffraction (XRD) measurements The XRD analyses were performed on a Scintag
XDS 2000 using Co Kα radiation (45 kV 40 mA) using divergence scatter and
receiving slits of 1deg 05deg and 02 mm respectively Samples were scanned
between 6 and 80 deg2θ with a scan speed of 1 deg2θmin Mineral suspension samples
for transmission Moumlssbauer spectroscopic analysis were collected on 02 microm filters
in an anoxic glove box (Coy Laboratory Products Inc) transferred to Perspex
capsules and stored in liquid nitrogen until measurement Moumlssbauer spectra were
obtained between 250 and 5 K using a conventional constant acceleration
spectrometer and a source of 57Co in Rh The spectrometer was calibrated using a
125 microm foil of α-Fe at room temperature and isomer shifts are given relative to
the centroid of this absorber The spectra were fitted using simple Lorentzian line
shape and it was assumed that all positions have identical f-factors
323 Culture conditions and cell preparation
Shewanella algae BrY is a motile gram-negative rod which was isolated first from
anoxic estuary sediments (Caccavo et al 1992) S algae BrY was grown
aerobically in tryptic soy broth (30 gL CASO-bouillon Merck) at 28degC on a rotary
shaker at 150 rpm for 16-18 h Cells were harvested by centrifugation (6000 rpm times
g 4ordmC 15 min) during the late exponential ndash early stationary growth phase at
OD660 ~ 06 Optimal Fe(III) reductase activity is expressed at this stage of growth
(Roden amp Zachara 1996) The cells were washed twice in oxic 50 mM PIPES
[piperazine-NNacute-bis(2-ethanesulfonic acid)] buffer (pH 70) and resuspended in
culture medium containing no Fe(III) and no carbon source Washed cell
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 49
suspensions were used as inocula for Fe(III) reduction experiments Oxygen was
expelled from the inoculum by extensive purging with 100 N2(g) (9999999
purity) Working stock cultures of S algae BrY were maintained aerobically on
tryptic soy agar plates at ambient temperature
324 Bioreduction experiments
All anaerobic incubations were carried out in anoxic serum vials (25 mL) or test
tubes (13 mL) sealed with thick (10-13 mm) butyl rubber stoppers and aluminum
crimp caps or plastic screw caps The basal culture medium (Table 31) was
prepared according to Kostka amp Nealson (1998) but with a phosphate
concentration of 2 mM and the exclusion of Fe(II) and EDTA
(ethylenediaminetetraacetic acid) The medium was amended with 4-10 mM
lactate or formate and 8-27 mM Fe(III) The Fe(III) was applied as Fe(III) oxide
coatings on sand or calcite The suspensions were purged extensively with 100
N2(g) (HFOsand suspensions) or 995 N205 CO2(g) (goecalcite and
lepcalcite suspensions) prior to inoculation The calcareous systems were buffered
at pH ~ 76 through a natural buffer system (CaCO3(s) + 995 N205 CO2(g))
whereas the sandy systems contained no pH buffer (100 N2(g) pH 55-60)
Inoculum size made up 5 of the total volume Cultures were incubated dark at
room temperature and gently agitated once every day At different time intervals
suspension samples for Fe(II) and Moumlssbauer analysis were withdrawn from the
reaction mixture using 100 N2(g) or 995 N205 CO2(g)-flushed sterile
disposable syringes and hypodermic needles Suspension samples for Fe(II)
analysis were digested in 01 M HCl for 30 min
50 Chapter 3
Table 31 Composition of the mineral medium (modified from Kostka amp Nealson (1998))
Components Concentration (M)
(NH4)2SO4 00143 KH2PO4 73middot10-4
K2HPO4 13middot10-3
MgSO4middot7H2O 10middot10-3
CaCl2middot2H2O 50middot10-4
H3BO3 56middot10-5
ZnSO4middot7H2O 10middot10-6
Na2MoO4middot2H2O 40middot10-6
CuSO4middot5H2O 20middot10-7
MnSO4middotH2O 10middot10-6
Na2SeO4 12middot10-5
CoCl2middot6H2O 50middot10-6
NiCl2middot6H2O 80middot10-6
NaCl 10middot10-5
L-arginine 11middot10-4
L-serine 19middot10-4
L-glutamic acid 14middot10-4
Lactate or formate 4-10middot10-3
Fe(III) 8-27middot10-3
325 Analytical methods
Fe(II) was determined using a modified phenanthroline method (Fadrus amp Maly
1975) The total amount of Fe(III) coated on calcite and sand was determined by
atomic absorption spectroscopy following dissolution in 6 M HCl(aq) for 24 h
33 Results and discussion
331 Fe(II) production and suspension colour changes
Strongly chelating agents such as EDTA were omitted from the culture medium in
order to prevent complexation of Fe(II) and Fe(III) which interferes with
precipitation of Fe(II) and Fe(II)-Fe(III) mineral phases Within 1-2 weeks after
inoculation Shewanella algae BrY produced green mineral phases in media
suspensions containing lepidocrocite and goethite as coatings on calcite and 4-10
mM formate or lactate (Figure 31) The formation of the green precipitates was
generally slower for the lepidocrocite coating than for the goethite coating The
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 51
blue-green colours of the phases produced most likely originate from Fe(II)-Fe(III)
charge transfer between adjacent Fe(II) and Fe(III) ions in edge-shared octahedra
(Faye et al 1968) Dark brown and blackish products were formed when the
bacteria were inoculated on HFO coated sand (Figure 32)
Figure 31 Culture tubes containing a) goethite and b) lepidocrocite coated calcite in culture
medium The left tubes of the pair were not inoculated whereas the right tubes were
photographed 5 months after inoculation with S algae BrY Experimental conditions [formate]0
= 4 mM [Fe(III)]0 = 8 mM 995 N205 CO2(g) pH 76
52 Chapter 3
Figure 32 Culture tubes containing HFO coated sand in culture medium Tubes 1 and 2 to the
left were not inoculated whereas tubes 3-5 to the right were photographed a) 13 days and b) 21
days after inoculation with S algae BrY Experimental conditions [lactate]0 = 10 mM [Fe(III)]0
= 25 mM 100 N2(g) pH 55-60
The green and black colours did not change to other colours (observed for gt1
year) indicating that the microbial Fe(III) reduction ceased at these mineral stages
The concentrations of dissolved ferrous iron (Fe(II)sol) estimated during Fe(III)
bioreduction were generally low (Figure 33) When comparing the final Fe(II)sol
amounts produced and the slopes of the Fe(II)sol formation curves for HFO
goethite and lepidocrocite in Figure 33 it can be seen that the final Fe(II)sol
amount and the Fe(II)sol production rate both follow the order HFO gt goethite gt
lepidocrocite at similar cell densities regardless of the carbon source applied This
suggests that bioreduction by S algae BrY is more facile for HFO than for goethite
and lepidocrocite at the experimental conditions employed here It should be noted
that the final Fe(II)sol amounts and the Fe(II)sol production rates reported in this
work have not been normalised with respect to the specific surface areas of the iron
oxides and coating-bearing solids applied The reactivity trend is consistent with
previous findings demonstrating higher reducibility of natural and poorly
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 53
crystalline Fe(III) oxides as compared to synthetic crystalline Fe(III) oxides
(Zachara et al 1998) The authors ascribed these differences in reducibility to
differences in particle size surface area and crystal defects of the Fe(III) oxides In
some cases the dissolved Fe(II) concentration decreased again with time (Figure
33 b-d) This indicates that the Fe(II) formed was incorporated into solid phases
forming andor adsorbed onto the calcite sand or Fe(III) oxide surfaces The solid
Fe(II) concentrations were not estimated spectrophotometrically The solid
material was generally low in total iron and therefore saving it for Moumlssbauer
spectroscopic analysis was given highest priority
54 Chapter 3
Figure 33 Time course of dissolved Fe(II) production during bioreduction of HFOsand
goethitecalcite and lepidocrocitecalcite by S algae BrY Experimental conditions [Fe(III)]0 =
25 mM [formate]0 = 10 mM (a-c) or [lactate]0 = 10 mM (d-f)
No color change and no Fe(II) production were observed in mineral suspensions
lacking either a carbon source or S algae BrY cells (data not shown)
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 55
332 Identification of solid iron phases
The purity of the Fe(III) oxides used in the experiments were investigated by
transmission Mossbauer spectroscopy (Figure 34) Single (or strongly dominating)
sextets in the spectra with magnetic hyperfine fields of 48 1 505 and 45 5 Tat 5 K
demonstrated the purity of the HFO goethite and lepidocrocite samples
respectively A minor impurity of goethite in the lepidocrocite sample was
resolved in the spectrum measured at 80 K (not shown for pure sample but can be
seen as a magnetically ordered sextet in Figure 35c) No Fe(II)-containing
components were detected
a)
c)
middot 12 -8 -4 4 8 12
Velocity (mmls
bull middot12 -8 -4 0 4 12
Velocity (mmls)
b)
middot12 -8 -4 4 8 12
Velocity (mmls
Figure 34 Transmission Mossbauer spectra measured at 5 K of a) HFO (magnetic hyperfine
field of 481 T isomer shift of 048 1nrns-1 negligible quadrupole shift and line width of outer
lines 110 rmns-1) b) goethite (magnetic hyperfine field of 505 T isomer shift of 049 rmns-1
quadmpole shift of -013 1nrns-1 and line width of outer lines 042 rnrns-1
) and c) lepidocrocite
56 Chapter 3
(magnetic hyperfine field of 455 T isomer shift of 050 mms-1 quadrupole shift of ndash001 mms-1
and line width of outer lines 060 mms-1) prior to inoculation Simple Lorenztian fits are shown
The oxidation state and coordination of Fe in the microbially reduced HFO
goethite and lepidocrocite samples were also examined by transmission Moumlssbauer
spectroscopy (Figure 35) The bioreduced HFO goethite and lepidocrocite samples
cultured on formate contained Fe(II) holding similar coordination as inferred from
the similarity of the hyperfine parameters (see legend in Figure 35) but different
relative intensities (72 19 and 71 respectively) The major part of the Fe(III)
remaining in the bioreduced samples were coordinated similarly to the Fe(III)
present in the initial Fe(III) oxide The coordination of Fe(II) in the bioreduced
lepidocrocite samples cultured on lactate was slightly different (a smaller
quadrupole splitting of 288 mms-1 for the ferrous component dominates ndash data not
shown) The exact mineralogy of the Fe(II) present in the green phases was not
fully resolved but its coordination is very akin to one of the Fe(II) sites in vivianite
(see Chapter 2 this work) and synthetic green rusts (Koch 1998) These findings
agree with other reports on the bioformation of vivianite and green rusts by
Shewanella putrefaciens CN32 although the evidence provided may be discussed
(Fredrickson et al 1998 Glasauer et al 2003 Parmar et al 2001) Our
Moumlssbauer data on the green phases did not allow for a detailed account of the type
of green rust produced However when considering solution composition (see
Table 31) and the high affinity of GR interlayers for carbonate it is reasonable to
assume that carbonate GR was formed (Hansen amp Taylor 1991) Due to the high
amounts of Fe(III) in the oxides present in the experiments it was difficult to probe
a possible content of Fe(III) in the vivianite with certainty The differences in the
number of Fe(II) positions in the Moumlssbauer spectra and particular the different
temperatures at which magnetic ordering takes place can be employed in order to
distinguish between green rust and vivianite Preliminary Moumlssbauer data obtained
for the blackish precipitates formed in the HFOsand suspensions indicate that they
hold no resemblance to magnetite eventhough the colour suggests so On the
contrary the black precipitates seemed to be more similar to synthetic green rusts
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 57
Mossbauer spectroscopic measurements are currently underway in order to resolve
the Fe(II) coordinations in the greenish and blackish phases
a) b)
c c g Q e- e 0 0 1l 1l
-~ bull ~
iii ~ Qi bull a bull
-12 -8 4 0 4 12
4 -3 -2 -1 0 1 2 4 Velocity (mmts) Velocity ( rmis)
c)
-12 -8 4 8 12
v elocity (m mis)
Figure 35 Transmission Mossbauer spectra of the black and green phases fo1med within 1-2
weeks after inoculation of a) HFO (measured at 130 K) b) goethitecalcite (measured at 80 K)
and c) lepidocrocitecalcite (measured at 80 K) with S algae BrY Experimental conditions
[fo1m ate]0 = 4 mM [Fe(III)]o = 8 mM 995 Ni05 C02(g) pH 76 The quadrnpole
splittings and isomer shifts for the Fe(II) components in the three systems are a) 293 nnns-1 and
126 mmsmiddot1 b) 308 rmnsmiddot1 and131 rmnsmiddot1 and c) 322 mmsmiddot1 and 132 mmsmiddot1 Simple Lorenztian
fits are shown
The evidence provided in many of the studies proposing biogenic GRs is not all
too convincing but it strongly suggests the probability of microbially produced GR
being present The challenge encountered is that when present in low
58 Chapter 3
concentrations especially in mixtures including other iron minerals it is very
difficult to identify GRs using conventional solid phase analysis methods even
with Moumlssbauer spectroscopy At least two complementary methods such as XRD
and Moumlssbauer spectroscopy are required for proper identification and
characterization of GRs However in this work the solid materials were generally
too low in total iron to allow for XRD analysis Moreover the highly
heterogeneous suspensions were dominated by the coating-bearing sand and calcite
solids Electron micrographs including energy dispersive X-ray spectroscopy
suspension colour and mineral stability calculations do not suffice as evidence The
most convincing evidence provided so far involves an atypical GR-CO3 with an
Fe(II)Fe(III) ratio of 1 (Ona-Nguema et al 2002aampb) This GR-CO3 was formed
as a result of lepidocrocite reduction by Shewanella putrefaciens CIP 8040 at
conditions comprising high nutrient levels (50-75 mM formate) high Fe(III)
concentrations (80-300 mM) and a synthetic electron shuttle (100 microM
anthraquinone-26-disulfonate (AQDS)) at initial pH 75 Hence the results
reported during recent years suggest that microbial formation of GR may be
possible The results presented here indicate that GRs may be produced
microbially at conditions including low carbon and Fe(III) concentrations as well
as the exclusion of synthetic electron shuttles and pH buffers
333 Factors controlling the identity of the secondary iron minerals
In general one would expect that biogenic minerals have chemical compositions
and crystal habits similar to those produced by nonenzymatic processes as they are
governed by the same equilibrium principles In fact since the latter stages of
mineralization are inorganically driven and the secondary Fe(II)-containing
minerals are formed indirectly by electron transfer outside the bacterial cell and not
directly inside the bacterial cell the type of iron mineral formed is a function of the
environmental conditions in which the bacteria live ie the same microorganism
form different minerals in different environments The key factors controlling the
identity of the secondary iron minerals include medium composition electron
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 59
donor and electron acceptor concentrations mineral aging as well as adsorbed ions
(Zachara et al 2002) The main factor controlling the nature of the secondary
mineral products are the respiration-driven biogenic Fe(II) supply rate and
magnitude and its surface reaction with the residual oxide and other sorbed ions
(Zachara et al 2002) Especially solution and medium composition have a strong
impact on the nature of the Fe(II)-containing biomineralization products forming
Accordingly siderite (FeCO3) and magnetite (Fe3O4) were the secondary solid
phases resulting from the bioreduction of ferrihydrite by Shewanella putrefaciens
CN32 in bicarbonate buffered medium (pH 71) containing no phosphate whereas
siderite and vivianite were the secondary iron minerals dominating in bicarbonate
buffered medium (pH 74) containing 4 mM phosphate (Zachara et al 2002) This
is explained by the inhibiting effect of phosphate on crystallization of magnetite
(Couling amp Mann 1985 Fredrickson et al 1998)
334 Factors controlling the rate and extent of Fe(III) bioreduction
In this study the extent of Fe(III) bioreduction was estimated to 19-72 by
transmission Moumlssbauer measurements In fact complete microbial reduction of
crystalline Fe(III) minerals has never been observed in laboratory batch culture
studies (Roden amp Urrutia 2002) It has been found that Fe(II) does not inhibit
Fe(III) reductase activity through an enzyme inhibition mechanism (Roden amp
Urrutia 2002) Hence other chemical andor physiologic factors control the
bioavailability of solid Fe(III) phases and thus the extent of their microbial Fe(III)
reduction The initial rate and long-term extent of microbial reduction of
amorphous and crystalline Fe(III) oxides including HFO goethite and hematite
were linearly correlated with oxide surface area (Roden amp Zachara 1996)
Association of biogenic Fe(II) with Fe(III) oxide and DIRB cell surfaces reduced
the long-term extent of crystalline Fe(III) oxide bioreduction (Roden amp Urrutia
2002) These results were explained by Fe(II) surface complexes andor
precipitates creating a passive Fe(II)-bearing surface film providing direct physical
interference with the electron transfer from the DIRB cells to Fe(III) However the
60 Chapter 3
real mechanisms whereby the surface-associated Fe(II) inhibits Fe(III) oxide
bioreduction are unclear but they most likely involve both kinetic and
thermodynamic constraints on the electron transfer Culture medium composition
in particular the presence and the concentration of phosphate as well as Fe(II)
chelating ligands also exert an influence on the extent of the microbial reduction
of Fe(III) oxides The extent of Fe(III) bioreduction was inhibited by high
phosphate concentrations which favoured surfacebulk precipitation processes
(Urrutia et al 1998) The carbon sources most frequently applied in Fe(III)
bioreduction studies include malate citrate and other di- and tricarboxylic acids
which are not only easily metabolizable carbon sources but also eminent Fe(II) and
Fe(III) chelators In this study we employed formate and lactate as carbon sources
since they are the weakest complexing agents of Fe(II) and Fe(III) among the C1-
C3 monocarboxylic acids (Martell 1964) Thus we expect less dissolution of
prevailing precipitates by complexation as compared to other studies
34 Conclusions
This work demonstrated that Shewanella algae BrY reduced 19-72 of initial
Fe(III) when grown in culture media containing 4-10 mM formate or lactate and 8-
27 mM Fe(III) applied as goethite or lepidocrocite coatings on calcite (pH 76) or
HFO coatings on sand (pH 55-60) Within 1-2 weeks after inoculation green
mineral phases were produced in the goethitecalcite and lepidocrocitecalcite
mineral systems whereas black precipitates formed in the HFOsand suspensions
Moumlssbauer spectroscopic analyses indicated that the greenish and blackish phases
most likely were mineral mixtures dominated by vivianite and green rust Thus the
results indicate that GRs may be produced microbially at conditions including low
carbon and Fe(III) concentrations as well as the exclusion of synthetic electron
shuttles and pH buffers
Acknowledgments
We would like to thank Dr R Gerlach for providing us the Shewanella algae BrY culture and
Dr C B Koch for performing the Moumlssbauer analyses
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 61
References Al-Agha MR Burley SD Curtis CD Esson J (1995) Complex cementation textures and authigenic mineral assemblages in recent concretions from the Lincolnshire Wash (east coast UK) driven by Fe(0) to Fe(II) oxidation Journal of the Geological Society 152 157-171 Bernal JD Dasgupta DR Mackay AL (1959) The oxides and hydroxides of iron and their structural inter-relationships Clay Minerals Bulletin 4 15-30 Caccavo Jr F Blakemore RP Lovley DR (1992) A hydrogen-oxidizing Fe(III)-reducing microorganism from the Great Bay Estuary New Hampshire Applied and Environmental Microbiology 58 3211-3216 Caccavo Jr F Das A (2002) Adhesion of dissimilatory Fe(III)-reducing bacteria to Fe(III) minerals Geomicrobiology Journal 19 161-177 Couling SB Mann S (1985) The influence of inorganic phosphate on the crystallization of magnetite (Fe3O4) from aqueous solution Journal of the Chemical Society Chemical Communications 1713-1715 Das A Caccavo Jr F (2000) Dissimilatory Fe(III) oxide reduction by Shewanella alga BrY requires adhesion Current Microbiology 40 344-347
Das A Caccavo Jr F (2001) Adhesion of the dissimilatory Fe(III)-reducing bacterium Shewanella alga BrY to crystalline Fe(III) oxides Current Microbiology 42 151-154 Fadrus H Maly J (1975) Suppression of iron(III) interference in the determination of iron(II) in water by the 110-phenanthroline method The Analyst 100 549-554 Faye GH Manning PG Nickel EH (1968) The polarized optical absorption spectra of tourmaline cordierite chloritoid and vivianite ferrous-ferric electronic interaction as a source of pleochroism American Mineralogist 53 1174-1201 Fredrickson JK Zachara JM Kennedy DW Dong H Onstott TC Hinman NW Li S (1998) Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium Geochimica et Cosmochimica Acta 62 3239-3257 Geacutenin JMR Olowe AA Benbouzid-Rollet ND Prieur D Confente M Resiak B (1991) The simultaneous presence of green rust 2 and sulfate reducing bacteria in the corrosion of steel sheet piles in a harbour area Hyperfine Interactions 69 875-878 Geacutenin JMR Bourrieacute G Trolard F Abdelmoula M Jaffrezic A Refait Ph Maitre V Humbert B Herbillon A (1998) Thermodynamic equilibria in aqueous suspensions of synthetic and natural Fe(II)-Fe(III) green rusts Occurrences of the mineral in hydromorphic soils Environmental Science and Technology 32 1058-1068 Glasauer S Weidler PG Langley S Beveridge TJ (2003) Controls on Fe reduction and mineral formation by a subsurface bacterium Geochimica et Cosmochimica Acta 67 1277- 1288 Hansen HCB Taylor RM (1991) The use of glycerol intercalates in the exchange of CO3
2- with SO4
2- NO3- or Cl- in pyroaurite-type compounds Clay Minerals 26 311-327
62 Chapter 3
Hungate RE (1969) A roll tube method for cultivation of strict anaerobes Methods in Microbiology 3B 117-132 King GM (1990) Effects of added manganic and ferric oxides on sulfate reduction and sulfide oxidation in intertidal sediments FEMS Microbiology Ecology 73 131-138 Koch CB (1998) Structures and properties of anionic clay minerals Hyperfine Interactions 117 131 -157 Kostka J Nealson KH (1998) Isolation cultivation and characterization of iron- and manganese reducing bacteria In Techniques in Microbial Ecology Burlage RS Atlas R Stahl D Geesey G Sayler G (eds) Oxford University Press Inc 58-78 Kukkadapu RK Zachara JM Smith SC Fredrickson JK Liu C (2001) Dissimilatory bacterial reduction of Al-substituted goethite in subsurface sediments Geochimica et Cosmochimica Acta 65 2913-2924
Liu C Zachara JM Gorby YA Szecsody JE Brown CF (2001) Microbial reduction of Fe(III) and sorptionprecipitation of Fe(II) on Shewanella putrefaciens strain CN32 Environmental Science and Technology 35 1385-1393 Lovley DR (1991) Dissimilatory Fe(III) and Mn(IV) reduction Microbiological Reviews 55 259-287 Lovley DR (1997) Microbial Fe(III) reduction in subsurface environments FEMS Microbiology Reviews 20 305-313 Lovley DR Phillips EJP (1986) Organic matter mineralization with reduction of ferric iron in anaerobic sediments Applied and Environmental Microbiology 51 683-689 Lovley DR Phillips EJP Lonergan DJ (1991) Enzymatic versus nonenzymatic mechanisms for Fe(III) reduction in aquatic sediments Environmental Science and Technology 25 1062-1067 Lower SK Hochella Jr MF Beveridge TJ (2001) Bacterial recognition of mineral surfaces Nanoscale interactions between Shewanella and α-FeOOH Science 292 1360-1363 Martell AE (1964) Stability constants of metal-ion complexes Part 2 Organic including macromolecule ligands The Chemical Society London 2 ed Miller TL Wolin MJ (1974) A serum bottle modification of the Hungate technique for cultivating obligate anaerobes Applied Microbiology 27 985-987 Nealson KH Saffarini D (1994) Iron and manganese in anaerobic respiration Environmental significance physiology and regulation Annual Review of Microbiology 48 311-343 Nevin KP Lovley DR (2000) Lack of production of electron-shuttling compounds or solubilization of Fe(III) during reduction of insoluble Fe(III) oxide by G metallireducens Applied and Environmental Microbiology 66 2248-2251 Nevin KP Lovley DR (2002) Mechanisms for Fe(III) oxide reduction in sedimentary
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 63
environments Geomicrobiology Journal 19 141-159 Nielsen A (1976) Hvid groslashn og sort rust Beskrivelse af en korrosionsskade paring et svoslashmmebassin Nordisk Betong 2 21-24 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002a) Iron (IIIII) hydroxycarbonate green rust formation and stabilization from lepidocrocite bioreduction Environmental Science and Technology 36 16-20 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002b) Microbial reduction of lepidocrocite γ-FeOOH by Shewanella putrefaciens The formation of green rust Hyperfine Interactions 139140 231-237 Parmar N Gorby YA Beveridge TJ Ferris FG (2001) Formation of green rust and immobilization of nickel in response to bacterial reduction of hydrous ferric oxide Geomicrobiology Journal 18 375-385 Roden EE Urrutia MM (2002) Influence of biogenic Fe(II) on bacterial crystalline Fe(III) oxide reduction Geomicrobiology Journal 19 209-251 Roden EE Zachara JM (1996) Microbial reduction of crystalline iron(III) oxides Influence of oxide surface area and potential for cell growth Environmental Science and Technology 30 1618-1628 Roh Y Lee SY Elless MP (2000) Characterization of corrosion products in the permeable reactive barriers Environmental Geology 40 184-194 Schwertmann U Cornell RM (1991) Iron oxides in the laboratory Preparation and characterization VCH Verlagsgesellschaft mbH Weinheim Thamdrup B (2000) Bacterial manganese and iron reduction in aquatic sediments In Advances in Microbial Ecology (Schink B ed) Kluwer AcademicPlenum Publishers New York 41-84 Trolard F Abdelmoula M Bourrieacute G Humbert B Geacutenin JMR (1996) Mise en eacutevidence dun constituant de type rouilles vertes dans les sols hydromorphes Proposition de lexistence dun nouveau mineacuteral la fougeacuterite Geacuteosciences de surface Comptes Rendus de LrsquoAcademie des Sciences 323 1015-1022 Tuovinen OH Button KS Vuorinen A Carlson L Mair DM Yut LA (1980) Bacterial chemical and mineralogical characteristics of tubercles in distribution pipelines Journal of the American Water Works Association 72 626-635 Turick CE Caccavo Jr F Tisa LS (2003) Electron transfer from Shewanella algae BrY to hydrous ferric oxide is mediated by cell-associated melanin FEMS Microbiology Letters 220 99-104 Urrutia MM Roden EE Fredrickson JK Zachara JM (1998) Microbial and surface chemistry controls on reduction of synthetic Fe(III) oxide minerals by the dissimilatory iron- reducing bacterium Shewanella alga Geomicrobiology 15 269-291 Venkateswaran K Moser DP Dollhopf ME Lies DP Saffarini DA MacGregor BJ Ringelberg DB White DC Nishijima M Sano H Burghardt J Stackebrandt E
64 Chapter 3
Nealson KH (1999) Polyphasic taxonomy of the genus Shewanella and description of Shewanella oneidensis sp nov International Journal of Systematic Bacteriology 49 705-724 Zachara JM Fredrickson JK Li S Kennedy DW Smith SC Gassman PL (1998) Bacterial reduction of crystalline Fe3+ oxides in single phase suspensions and subsurface materials American Mineralogist 83 1426-1443
Zachara JM Kukkadapu RK Fredrickson JK Gorby YA Smith SC (2002) Biomineralization of poorly crystalline Fe(III) oxides by dissimilatory metal reducing bacteria (DMRB) Geomicrobiology Journal 19 179-207
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 65
4 Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust The Effect of Probe Compound Charge
Abstract
Layered iron(II)-iron(III)-hydroxides (green rusts) may play an important role in
controlling the fate and transport of many organic and inorganic contaminants in
iron-rich suboxic soils and sediments Unlike most other iron oxides green rusts
(GRs) contain not only external Fe(II) reactive sites at the basal planes and at the
edges but also internal sites in the space between consecutive Fe(II)-Fe(III)
hydroxide layers The GR interlayer thickness is a function of both the size and the
charge of the interlayer anion Whether a given oxidant has access to the internal
sites in GRs is dependent on its charge We investigated the reductive
transformation of nitroaromatic compounds (NACs) by GR-SO4 and studied the
effect of NAC charge on the reactivity towards GR-SO4 A series of structurally
closely related compounds with different charge properties including nitrobenzene
4-nitrotoluene 4-chloronitrobenzene and 4-nitrophenylacetic acid were used as
probe compounds The NACs were completely reduced to their corresponding
anilines by GR-SO4 The reactions followed pseudo 1 order kinetics with respect
to NAC and the surface area-normalised pseudo 1 order rate constants obtained
were 016ndash465middot10-4 s-1middotm-2middotL at [Fe(II)GR]0 = 103-1260 mM [NAC]0 = 20-102
microM and pH 84-86 Neither mass transfer control nor surface saturation kinetics
could account for the rather unexpected similarity of the surface area-normalised
pseudo 1 order rate constants obtained for the reduction of the neutral and anionic
NACs by GR-SO4 These observations suggest that the anionic NACs did not have
an enhanced access to the inner or outer Fe(II)-GR reactive sites as compared to
the neutral NACs Hence the reaction between NAC and GR-SO4 primarily took
place at the edges of GR-SO4
66 Chapter 4
41 Introduction
Layered iron(II)-iron(III)-hydroxides (green rusts) are intermediate phases formed
by partial oxidation of Fe(II) or partial reduction of Fe(III) In neutral and weakly
alkaline solutions the oxidation of dissolved Fe(II) always passes through solid
green rust (GR) phases (Bernal et al 1959) This agrees with the natural GR
occurrences found in suboxic non-acid iron-rich environments such as
hydromorphic soils and intertidal sediments (Al-Agha et al 1995 Trolard et al
1996 Genin et al 1998) In addition GRs have been found as corrosion products
in numerous engineered systems ie a pipeline distribution system for drinking
water steel sheet piles in marine sediments reinforced concrete (ferro-concrete)
and permeable reactive barriers of zero-valent iron implemented for on-site
remediation of organic and inorganic contaminants (Tuovinen et al 1980 Nielsen
1976 Genin et al 1991 Roh et al 2000) Furthermore the microbial formation
of GRs resulting from bioreduction of Fe(III) oxides by strains of the anaerobic
dissimilatory Fe(III) reducing bacteria Shewanella putrefaciens has been reported
increasingly over the last 5 years (Fredrickson et al 1998 Kukkadapu et al 2001
Parmar et al 2001 Ona-Nguema et al 2002 Glasauer et al 2003) Moreover
the biotic formation of GRs by anaerobic denitrifying Fe(II) oxidizing bacteria has
been proposed but proper identification of the GR phases still lacks (Chaudhuri et
al 2001) All these indications of microbial GR formation infer the importance of
GRs as a link between geochemical and biological processes in natural systems
GRs form platy crystals with the general formula [FeII(6-
x)FeIIIx(OH)12]x+[(A)xnmiddotyH2O]x- where x = 09 - 42 A is an n-valent anion eg
CO32- Clndash or SO4
2- and y is the number of water molecules in the interlayer The
crystal structure consists of positively charged hydroxide sheets with Fe(II) and
Fe(III) cations having octahedral hydroxyl coordination The Fe(III) in the
hydroxide layers creates a net positive charge which is balanced by hydrated
anions in the interlayers (Figure 41) The interlayers have a higher affinity for
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 67
divalent anions than for monovalent anions (Miyata 1983) Among the 3 most
common GR forms the affinity follows the order CO32- gt SO4
2- gt Clndash The extreme
preference shown for carbonate hinders further access and exchange except under
certain conditions (Hansen amp Taylor 1991) Non-carbonate forms are readily
exchanged with other anions when dispersed in a solution containing the
exchanging anion (Mendiboure amp Schoumlllhorn 1986)
c
ba
Figure 41 Green rust layer structure The hydroxide layers and the interlayers are connected by
hydrogen bonds (not shown) The GR-SO4 crystal structure is characterised by the hexagonal
unit cell having a = b = 055 nm and c = 110 nm (Simon et al 2003) The unit cell consists of
one double layer (a double layer is a hydroxide layer and an interlayer) ie the hydroxide layer
constitutes 049 nm and the interlayer 061 nm in GR-SO4
The GR interlayer thickness (extending in the c axis direction Figure 41) is a
function of both the size and the charge of the interlayer anion Tetrahedrally
coordinated anions like sulphate lead to larger interlayer distances than smaller
monoatomic anions like chloride or planar ions like carbonate (Mendiboure amp
Schoumlllhorn 1986) Not only size but also charge density plays a role for the
interlayer spacing That is for anions having the same number of valence
electrons anions with smaller ionic radii (higher electron density) are bound more
strongly and therefore result in smaller interlayer spacings
Due to their layered structures anionic interlayers and high specific surface areas
GRs represent reactive ion exchangers and sorbents of anions eg arsenate
selenate and phosphate (Myneni et al 1997 Hansen amp Poulsen 1999 Randall et
al 2001) In addition GR may incorporate heavy metal cations by isomorphic
68 Chapter 4
substitution into the GR hydroxide layers (Tamaura 1985 Tamaura 1986)
Furthermore GRs have been shown to reduce a range of inorganic contaminants
such as nitrite nitrate selenate chromate uranyl pertechnetate and the transition
metals AgI AuIII CuII and HgII as well as organic pollutants including halogenated
ethanes ethenes and methanes (Hansen et al 1994 Hansen et al 1996 Myneni et
al 1997 Erbs et al 1999 Loyaux-Lawniczak et al 1999 Cui amp Spahiu 2002
Lee amp Batchelor 2002b Heasman et al 2003 OrsquoLoughlin et al 2003a amp 2003b
Pepper et al 2003 Elsner et al 2004 OrsquoLoughlin amp Burris 2004) Thus through
sequestration and reductive transformation GRs may play an important role in
controlling the fate and transport of contaminants in suboxic soils and sediments
In a previous study the effects of interlayer anion and Fe(II)Fe(III) ratio in GRs
on the reduction rate of nitrate were investigated (Hansen et al 2001) It was
found that the rate of nitrate reduction to ammonium increased with increasing
Fe(II)Fe(III) ratio and decreased when exchanging a monovalent interlayer anion
(chloride) with a divalent anion (sulphate) The results suggest that for anionic
oxidants like nitrate Fe(II) within the hydroxide layer is available from the outside
basal planes and from the edges as well as through the interlayer under certain
conditions (Figure 42) However oxidants with different charge properties
(cations neutral molecules) may exhibit different affinities for the various reactive
Fe(II) sites present in GR
As the reactive sites are located inat the Fe(II)-Fe(III) hydroxide layers the rate of
reaction depends on the hydroxide layer area which can be accessed by the
oxidant If the oxidant can exchange with the interlayer anion reaction can take
place both at outer and inner surfaces of the GR particles and in total more
reactive sites are available for the reaction However it was found that nitrate
cannot penetrate the interlayer when carbonate or sulphate constitutes the
interlayer anions (Hansen amp Koch 1998) This agrees with the fact that the
interlayers have a lower affinity for monovalent anions than for divalent anions
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 69
(Miyata 1983) However when nitrate was forced into the interlayer by extracting
the interlayer sulphate through precipitation of barium sulphate outside the GR
particles the observed 40 fold increase in rate of nitrate reduction almost equalled
the increase in exposed surface area of the Fe(II)-Fe(III) hydroxide layers (Hansen
amp Koch 1998) From these observations it is expected that the rate of reaction
depends on the particular GR form the crystallite size and the ease with which an
oxidant can exchange with An- in the GR interlayer (Figure 42) Due to
electrostatic interactions we expect anions to be attracted to the positively charged
outer and inner surfaces to a higher degree than cations and neutral compounds If
this theory holds we may expect oxidants with similar intrinsic reactivity (similar
one-electron reduction potentials) to react in the following order anionic gt non-
charged gt cationic (Figure 43) granting that we do not normalise the rate
constants with respect to the amount of oxidant sorbed
Figure 42 Reaction of a probe compound at basal planes at edges and in the interlayer of GR
The hypothesis only holds in cases where the oxidants possess the same intrinsic
reactivities If the relative reactivities of the probe compounds differ greatly from
what would be expected when considering only their reduction potentials
70 Chapter 4
compound specific effects such as charge properties might explain this and the
relative reactivities may follow a pattern like the one depicted in Figure 43
Figure 43 Hypothetical plot of observed reaction rate constants for the reactions between
cationic neutral and anionic probe compounds and GR-SO4 assuming that the oxidant charge
controls its reactivity towards GR
In this work we investigated the reductive transformation of NACs by GR-SO4
Furthermore the effect of NAC charge on the rate of reaction and the possible
access to the internal reactive sites in GR-SO4 were assessed When quantifying
Fe(II) in GRs by means of acid digestion it is not possible to distinguish between
the reactive sites accessible from the outside (at the basal planes or at the edges) or
through the interlayer However we designed an indirect method to gain insight
into the relative importance of the various reactive sites by using a series of
structurally closely related compounds with different charge properties as ldquoreactive
probesrdquo Neutral and anionic probes were needed in order to access all Fe(II)
reactive sites According to our hypothesis cationic and non-charged oxidants
should provide information about the reactivity of the outer Fe(II) reactive sites in
GR whereas the anionic oxidants should provide information about the reactivity
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 71
of both outer and inner Fe(II) reactive sites We chose five nitro aromatic
compounds (NACs) - representing an important group of reducible organic
pollutants - as probe compounds (Figure 43) This class of compounds is not only
of great environmental concern but also comprises suitable model compounds for
studying redox reactions potentially relevant in the environment Moreover they
react readily with Fe(II) surface species associated with iron oxides or clay
minerals transforming them into well-defined easily detected products allowing
mass and electron balances to be established (Hofstetter et al 2003 Klausen et al
1995 Schultz amp Grundl 2000) Our main goals were to establish the rate law and
estimate the surface area-normalised reaction rates for the reaction of the probe
compounds with GR-SO4 in order to assess the importance of the Fe(II) reactive
sites accessible through the interlayer relative to the Fe(II) reactive sites accessible
at the outer surface in GR-SO4
4 2 Materials and methods
All handling and sampling of solutions and suspensions were carried out under
strict anoxic conditions All chemicals were pa quality or better Methanolic stock
solutions (5 mM) of nitrobenzene (NB) 4-nitrotoluene (4-NT) 4-
chloronitrobenzene (4-CNB) and 4-nitrophenylacetic acid (4-NPA) were prepared
in deoxygenated methanol Several attempts to synthesize the cationic probe
compound 4-(NNN-trimethylammonium)-nitrobenzene failed and therefore the
study had to be carried out with only neutral and anionic oxidants The sulphate
GR form was chosen as it is the most stable form and thus the easiest to work
with in the lab
421 Synthesis of GR-SO4
GR-SO4 was synthesized by controlled air oxidation of an FeSO4 solution at a
constant pH of 700 according to the procedure given by Koch amp Hansen (1997)
The GR-SO4 suspension was washed with deoxygenated deionised water and
72 Chapter 4
separated on a folding filter redispersed in deoxygenated 25 mM Na2SO4(aq) in
order to stabilize the GR-SO4 and prevent it from transforming into magnetite
spontaneously Washing separation and redispersion of the GR-SO4 suspension
were conducted in an anoxic glove box (Coy Laboratory Products Inc) All
suspensions and solutions were deoxygenated by Ar-purging (999998 Ar
Carbagas)
422 Mineral characterisation
The identity and purity of the GR-SO4 suspensions were examined by means of X-
ray diffraction measurements The XRD analyses were performed on a Scintag
XDS 2000 using Cu Kα radiation (45 kV 40 mA) Glycerol smears made
according to Hansen (1989) were scanned between 6 and 80 deg2θ with a scan speed
of 1 deg2θmin
423 Lyophilization and determination of specific surface area
Simple air-drying of the GR mineral in the glove box resulted in big flakes with
very low surface areas hence a more suitable lyophilization method was adopted
from Elsner et al (2004) The GR-SO4 suspensions were lyophilised using
Schlenk-type glassware The set-up consisted of a 1 L round bottom flask and a
200 mL glass finger connected by a crescent-shaped bridge equipped with an
evacuation outlet and a stopcock All ground joints and fittings were attached using
high-vacuum grease The washed and resuspended GR-SO4 suspensions were
filled into the glass finger and the freeze-drying apparatus was assembled and
closed before taking it out of the glove box The suspension was frozen by
carefully submerging the lower part of the glass finger into liquid nitrogen for a
few hours Subsequently the evacuation outlet was connected to a vacuum pump
by a metal hose Following a short evacuation of the metal hose the lyophilization
apparatus was evacuated for several minutes by gently opening the stopcock The
evacuation was terminated by closing the stopcock and disconnecting the vacuum
pump The apparatus position was now reversed by removing the glass finger from
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 73
and immersing the round bottom flask into liquid nitrogen As any other
lyophilization method this method depends on sublimation of the ice from the
frozen sample and its recondensation on a cool surface in this case the round
bottom flask Generally it took 1-2 d for the mineral to dry The apparatus was
disassemled in the glove box and the fine powder stored under anoxic conditions
The specific surface area (SSA) of GR-SO4 was determined by the BET multi-
point method using N2 adsorption (Brunauer et al 1938) Powder samples were
filled into sample burettes in the glove box and the generously greased stopcocks
closed Samples and burettes were evacuated prior to connecting them to the BET-
instrument (Sorptomatic 1990 Fisons)
424 Estimation of the one-electron reduction potential for 4-NPA
Kinetic experiments in 100 mL Viton stoppered and alu-crimp capped serum vials
were carried out under the exclusion of oxygen as described by Hofstetter et al
(1999) The homogeneous aqueous solutions contained 50 mM KH2PO4 buffer
(pH = 660) 5 mM Na2S redox buffer and 20 microM juglone (8-hydroxy-14-
naphthoquinone) added as deoxygenated 20 mM methanolic stock solution The
solutions were equilibrated at least one day prior to 4-NPA addition To start the
reaction 50 microM 4-NPA was added as deoxygenated 20 mM methanolic stock
solution The vials were agitated on a roller apparatus in the dark at 21ordmC Control
experiments were prepared similarly except for the addition of juglone At
different time intervals aqueous samples were withdrawn with a syringe and
collected in 18 mL HPLC vials containing 100 microL 1 M HCl The sample vials
were sealed with Teflon-coated silicone septa and plastic screw caps and vortexed
for 10 s The samples were stored at -20degC and analysed without further treatment
For comparison experiments with 4-NT were also conducted See Supporting
Information 71 for more information on the one-electron reduction potentials
74 Chapter 4
425 Kinetic experiments
All reactions took place at pH 84-86 where GR-SO4 tends to stabilize and buffer
itself Samples for Fe(II) and XRD analysis were withdrawn prior to reaction Due
to the fast reactions the experiments were conducted in 10 mL single-use
polyethylene syringes (BD Plastipak) in the glove box To start reaction 40-200
microL 5 mM methanolic stock solutions of NAC were quickly added to 10 mL GR-
SO4 suspension (1-12 mM Fe(II)GR) washed and resuspended in 25 mM
Na2SO4(aq) A Teflon filter (25 mm x 02 microm BGB Analytik) was quickly
mounted on the tip of the syringe and the syringe was vigorously shaken between
sampling At different time intervals filtered suspension samples were collected in
18 mL HPLC vials The HPLC vials were sealed with Teflon-coated silicone septa
and plastic screw caps The samples were stored at -20degC and analysed without
further treatment Absorption of NAC in the syringe and in the Teflon filter
evaluated in blank experiments with NAC added to 25 mM Na2SO4(aq) was found
to be negligible
426 Analytical methods
Initial total and aqueous Fe(II) were determined using a modified phenanthroline
method (Fadrus and Maly 1975) In order to determine [Fe(II)aq] and [Fe(II)total] 1
mL filtered (022 microm) and 1 mL unfiltered GR-SO4 suspension samples were
withdrawn and each treated with 18 mL 01 M HCl for at least 30 min From these
acid digests 01 mL was added to 05 mL Fe(II)-reagent and 19 mL deionised
water (DIW) added up The Fe(II) content in GR-SO4 was estimated as the
difference [Fe(II)GR] = [Fe(II)total] - [Fe(II)aq] The NACs and their corresponding
intermediates and products formed during reduction by GR-SO4 were quantified by
reversed-phase HPLC Separation was performed on a LiChrospher 100 RP-18 (5
microm 125 times 4 mm ID) reversed-phase column coupled with a LiChroCART 100 RP-
18 (4 times 4 mm ID) precolumn Analytical conditions were isocratic and the eluent
consisted of 10 mM hydroxylammonium chloride in various DIWCH3OH
mixtures (vv 3565 and pH 70 for 4-NT and 4-CNB 955 and pH 60 for
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 75
4-NPA) The injection volume was 20 microLand the flow-rate 10 mLmin HPLC
analyses were performed using a Gynkotek High Precision Pump M480 Gynkotek
Gina 50 autosampler and a diode array UV detector (340s Gynkotek) UV-VIS
detection was carried out at the wavelengths of maximum absorption for the
various nitro aromatic and aniline analytes
43 Results and discussion
431 Productformation and reaction kinetics
The reduction of the aromatic nitro group occurs via nitroso- and hydroxylamino-
intermediates where 2 electrons are transferred in each reaction step (Figure 44)
0 --0 H OH H H --0 N N N N
2e- 2H+ H20 + 2e-~ 2e- 2H+ H20
~ ~ R R R
Nitro benzene Nitrosobenzene Hydroxylamine Aniline
Figure 44 Reductive transfonnation pathway of NA Cs
Thus in order to reduce 1 Ar-N02 completely to Ar-NH2 6 electrons
corresponding to 6 mol Fe(II) are needed As magnetite was the major iron phase
formed during reaction (XRD results not shown) we assume the following
reaction stoichiometry
The aniline product was not formed at the same rate as the nitro compound
degraded which is consistent with the detection of early eluting hydroxylamine
intermediates during the course of the reaction (Figure 45a amp 45c) No traces of
76 Chapter 4
nitrosobenzene intermediates or side products such as azoxy- azo- or
hydrazobenzene were found In Figure 45 pseudo 1 order kinetic plots and ln
[Ar-NO2]t[Ar-NO2]0) versus time plots for the neutral probe compounds 4-CNB
and 4-NT are shown as examples The plots for NB and 4-NPA look similar
Figure 45 a Concentration versus time plots for reaction of GR-SO4 with 4-CNB ([Fe(II)GR]0 =
126 mM [4-CNB]0 = 30 microM) b ln [Ar-NO2]t[Ar-NO2]0) versus time plots for reaction of GR-
SO4 with 4-CNB ([Fe(II)GR]0 = 126 mM + [4-CNB]0 = 30 microM [Fe(II)GR]0 = 63 mM + [4-
CNB]0 = 50 microM) c Concentration versus time plots for reaction of GR-SO4 with 4-NT
([Fe(II)GR]0 = 131 mM [4-NT]0 = 20 microM) d ln [Ar-NO2]t[Ar-NO2]0) versus time plots for
reaction of GR-SO4 with 4-NT ([Fe(II)GR]0 = 131 mM [4-NT]0 = 20 microM [Fe(II)GR]0 = 131
mM [4-NT]0 = 50 microM) The hydroxylamino intermediate shown in microM equals the deficit in the
mass balance and in abs equals the detector response (peak area) Solid lines represent 1 order
kinetic fits (a amp c) and ln [Ar-NO2]t[Ar-NO2]0) versus time fits (d) whereas symbols and dotted
lines represent actual data
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 77
At intial Fe(II)GR concentrations in large excess of initial Ar-NO2 concentration
we found a pseudo 1 order rate law for the degradation of Ar-NO2 by GR-SO4
[ ] [ ] [ b 2
a GR
2 ArNOFe(II) ArNOsdotsdot=minus k
dtd ]
where a = 1 b = 1 and the observed pseudo 1 order rate constant kobs = k middot
[Fe(II)GR] At high [Fe(II)GR]0[Ar-NO2]0 ratios the nitro compound was
transformed completely into the aniline product within reaction duration and the
degradation curves of the nitro compound were shaped according to pseudo 1
order kinetics (data points follow solid line in Figure 45a) In some instances ie
at low [Fe(II)GR]0[Ar-NO2]0 ratios the reactions did not follow pseudo 1 order
kinetics for the whole duration of reaction (data points deviate from solid line in
Figure 45c) Hence in order to allow comparison all the pseudo 1 order rate
constants were calculated as initial rates (ie max first two half-lives) from linear
fits of (time ln [Ar-NO2]t[Ar-NO2]0)-plots (Figure 45b amp 45d) Surface area-
normalised pseudo 1 order rate constants are shown in Table 41
Tabl
e 4
1 S
urfa
ce a
rea-
norm
alis
ed p
seud
o 1
ord
er ra
te c
onst
ants
for t
he re
duct
ive
trans
form
atio
n of
4-n
itrot
olue
ne (4
-NT)
4-
chlo
roni
trobe
nzen
e (4
-CN
B) a
nd 4
-nitr
ophe
nyla
cetic
aci
d (4
-NPA
) by
GR
-SO
4
Exp
erim
ent
Age
GR
(d
) [F
e(II
) GR] 0
(mM
) [N
AC
] 0 (micro
M)
[Fe(
II) G
R] 0
[N
AC
] 0∆[
ArN
O2]
(microM
) af b
k obs
(s-1
) ck o
bs (s
-1middotm
-2middotL
) d
GR
-SO
4 + 4
-NT
3 1
103
20
51
5
109
54
5
7
65middot1
0-46
95middot1
0-5
GR
-SO
4 + 4
-NT
3 1
103
50
20
6
174
34
8
7
41middot1
0-46
74middot1
0-5
GR
-SO
4 + 4
-NT
3 1
103
10
0 10
3
214
21
4
2
63middot1
0-42
39middot1
0-5
GR
-SO
4 + 4
-CN
B 1
1
103
55
18
7
177
32
2
4
21middot1
0-43
83middot1
0-5
GR
-SO
4 + 4
-CN
B 1
1
103
10
2 10
1
165
16
2
2
37middot1
0-42
15middot1
0-5
GR
-SO
4 + 4
-NPA
1
1 1
03
25
412
9
7 38
8
4
82middot1
0-44
38middot1
0-5
GR
-SO
4 + 4
-NPA
1
1 1
03
46
224
9
9 21
5
6
37middot1
0-45
79middot1
0-5
GR
-SO
4 + 4
-NPA
1
1 1
03
100
103
13
7
137
196
middot10-4
178
middot10-5
GR
-SO
4 + 4
-NT
2 15
1
31
20
655
5
4 27
0
6
74middot1
0-44
82middot1
0-5
GR
-SO
4 + 4
-NT
2 15
1
31
50
262
9
7 19
4
5
89middot1
0-44
21middot1
0-5
GR
-SO
4 + 4
-NT
4 2
126
0 50
25
2 49
1
982
110
middot10-2
817
middot10-5
GR
-SO
4 + 4
-NT
4 2
630
50
12
6 42
6
852
186
middot10-3
276
middot10-5
GR
-SO
4 + 4
-CN
B 2
2
126
0 30
42
0 29
0
967
925
middot10-3
687
middot10-5
GR
-SO
4 + 4
-CN
B 2
2
630
50
12
6 38
0
760
136
middot10-3
202
middot10-5
GR
-SO
4 + 4
-NPA
2
2 12
60
40
315
371
92
8
5
96middot1
0-34
43middot1
0-5
GR
-SO
4 + 4
-NPA
2
2 6
30
45
140
273
60
7
1
09middot1
0-31
62middot1
0-5
a A
mou
nt o
f NA
C re
duce
d by
GR
-SO
4 at r
eact
ion
term
inat
ion
b F
ract
ion
of in
itial
ly a
dded
NA
C tr
ansf
orm
ed b
y G
R-S
O4 a
t rea
ctio
n te
rmin
atio
n c
Pse
udo
1
orde
r rat
e co
nsta
nts c
alcu
late
d as
initi
al ra
tes
ie m
ax f
irst t
wo
half-
lives
d S
urfa
ce a
rea-
norm
alis
ed p
seud
o 1
ord
er ra
te c
onst
ants
The
are
a of
GR
-SO
4 per
L
su
spen
sion
was
cal
cula
ted
as frac14
middot[Fe
(II)
GR] 0middot
600
gmiddotm
ol-1
middot71
2 m
2 middotg-1
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 79
432 Comparison of rate constants for the different NACs
Even for NACs holding very different one-electron transfer reduction potentials
( ) their reactivities differed only little in Fe(II)-Fe(III) systems such as the
Fe(II)goethite system (slope a = 06 for linear free energy relationship (LFER)
between k
1hE
obs and Hofstetter et al 1999) and the Fe(II)magnetite system
(LFER slope a = 034 Klausen et al 1995) When considering only the for the
reductive transformation reactions of the NACs applied in this study (Table 42)
we expect the surface area-normalised pseudo 1 order rate constants for the
reduction of the NACs to follow the order 4-CNB gt NB gt 4-NT gt 4-NPA Based
on log k
1hE
1hE
obs versus correlations obtained in Fe(II)goethite systems we expect 4-
CNB to react 6 times faster than 4-NPA (Hofstetter et al 1999)
1hE
Table 42 One-electron reduction potentials and relative reactivities in Fe(II)-magnetite and GR-
SO4 systems for the nitro aromatic probe compounds
Compound pKa Eh1
acute (mV) krel (Fe3O4) cd krel (GR-SO4) ce
4-Chlornitrobenzene - -450 a 122 148
Nitrobenzene - -486 a 1 1
4-Nitrotoluene - -500 a 057 176
4-Nitrophenylacetic acid 385 -543 b - 123 a Values from references cited in Hofstetter et al 1999 b Estimated at pH 660 using a LFER (Hofstetter et al 1999 see Supporting Information 71) c Reactivity relative to NB d Values from Klausen et al 1995 e Values from this work
A comparison of the relative rate constants of the NACs obtained for their
transformation by GR-SO4 (this work) and by magnetite (Klausen et al 1995)
shows that they do not differ significantly from each other in any of the mineral
systems (Table 42) When considering charge effects we expect the anionic probe
compounds to react faster with GR-SO4 than the neutral probe compounds
provided that they sorb preferentially within the GR-SO4 interlayers and that Fe(II)
in the interlayers are equally or more reactive than external Fe(II) sites Still the
surface area-normalised kobs values obtained for NB 4-NT 4-CNB and 4-NPA
under various experimental conditions did not differ significantly from each other
80 Chapter 4
(Figure 46 Table 41) The anionic probe compound 4-NPA did not react
significantly faster with GR-SO4 than the neutral probe compounds NB 4-NT and
4-CNB This may indicate that 4-NPA does not significantly interact with reactive
Fe(II) sites in the interlayer Alternatively the negative charge carried by 4-NPA
may be compensating for the lower intrinsic reactivity as compared to the neutral
probe compounds thus explaining the similarity in rate constants for 4-NPA and
the neutral probe compounds Finally other factors than intrinsic reactivity or
charge of the probe compounds such as regeneration of reactive sites or formation
of the magnetite phases may control the overall reactivity of the system
Figure 46 Actual plot of surface area-normalised pseudo 1 order rate constants for the reactions
between neutral and anionic probe compounds and GR-SO4
In heterogeneous reactions mass transfer in bulk solution becomes the rate-
limiting step when the surface reaction is much faster than the diffusion of the
reacting species to the reactive surface In cases where mass transfer controls the
overall rate of reaction the observed pseudo 1 order rate constant kobs ge kLmiddota
where kL is the calculated mass transfer coefficient (mmiddots-1) and a is the ratio of the
external (geometric) specific surface area to volume of solution (m-1) (see
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 81
Supporting Information 72) Mass transfer controlled reactions between GR-SO4
particles and the NACs in bulk solution would explain the similar pseudo 1 order
rate constants obtained for the NACs in this work However when comparing our
estimates of kLmiddota with kobs (see Supporting Information 72) we found that the rates
of mass transfer for all 4 NACs exceed the observed rate constants by at least 3
orders of magnitude at every initial Fe(II)GR concentration Thus the reactions of
the given NACs with GR-SO4 are not likely to be mass transfer limited under the
experimental conditions applied here
Since mass transfer in bulk solution does not control the reaction between GR-SO4
and NACs the overall reaction rate may be surface saturation controlled During
the reductive transformation of NACs not only the parent compound but also
various intermediates forming may compete for the restricted number of reactive
sites present in GR-SO4 This competition may constitute the rate limiting step in
the overall reactivity and may even be enhanced if the number of reactive sites is
depleted during reaction However surface saturation kinetics would not explain
the unexpected similarity of the pseudo 1 order rate constants obtained for the
NACs but it could explain the bent curves observed at low initial Fe(II)GR
concentrations (Figure 45d) The kinetically deviating cases at low [Fe(II)GR]0
were evaluated according to Langmuir-Hinshelwood kinetics (see Supporting
Information 72) Our experimental data did not agree with the Langmuir-
Hinshelwood rate law for any of the NACs (regression results not shown)
Simplifying the rate law by assuming that the aniline product or the
hydroxylamino intermediate or both did not compete for the reactive sites did not
improve the regression Thus the Langmuir-Hinshelwood model cannot explain
the deviations from pseudo 1 order kinetics observed at [Fe(II)GR]0 in our GR-SO4
system and it does not suffice as the correct reaction mechanism nor as the rate-
limiting step
82 Chapter 4
If the adsorption follows a saturation-type sorption isoterm (eg Langmuir) the
sorbate (oxidant) concentration at the surface will vary non-linearily with the total
amount of oxidant added This dependence will have to be taken into account when
establishing rate laws for the heterogeneous reactions and when testing the
hypothesis that the reaction rates depend on the sorbed concentration of the
oxidants However at the high reaction rates observed here we could not quantify
sorption Since the measured initial NAC concentrations corresponded to the
nominal amount of NAC added we assume that transformation and not sorption
was responsible for the consumption of NAC
433 Factors influencing the reaction rate
In general numerous compound- and system-specific factors influence redox
reactions One very important factor is pH which influences the speciation of
dissociable compounds as well as the stability of GR and the formation of other
iron minerals in the system pH has a strong impact on the sorption and therefore
the availability of ionisable oxidants such as carboxylic acids At pH ~ 84 where
our experiments were conducted 4-NPA (pKa = 385) is completely dissociated
Our experiments conducted with GR-SO4 and NB showed that pH was constant
during reaction In addition solution pH has an effect on the surface speciation
From other Fe(II)-Fe(III) systems such as Fe(II) surface species associated with
iron oxides or clay minerals it is well-known that other reactive hydroxylated
Fe(II)-Fe(III)-hydroxo surface complexes can form at higher pH (Charlet et al
1998 Liger et al 1999) Williams amp Scherer (2001) reported a small decrease (5
fold) in the reduction rate of chromate with GR-CO3 when increasing pH from 50
to 90 This decrease may be due to the alternating speciation of the Fe(II) surface
sites on GR-CO3 and of chromate in solution (pKa (H2CrO4) = 08 pKa (HCrO4ndash) =
65) when raising pH (Williams amp Scherer 2001) In contrast other studies have
reported small increases (4 fold) in the reduction rates of nitrate and
trichloroethene with GR-SO4 when increasing pH from 71 to 84 and from 68 to
101 respectively (Koch amp Hansen 1997 Lee amp Batchelor 2002b)
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 83
In this work all experiments were carried out in the presence of 25 mM
Na2SO4(aq) in order to minimize GR-SO4 dissolution and spontaneous
transformation into magnetite Preliminary results from experiments conducted
with NB show that the bulk concentration of Na2SO4 has only a very small impact
on the rate ie increasing the concentration of Na2SO4(aq) in the GR-SO4
suspension from 5 to 25 mM reduced the observed rate constant by a factor of 2
At Na2SO4 concentrations above 25 mM the effect leveled off and therefore
[Na2SO4] = 25 mM was chosen for this work Portions of the same GR-SO4
suspension were used for kinetic experiments over a period of two weeks No
significant aging effects eg rate constants decreasing as a function of GR age
were observed within this time frame
434 Comparison with rate constants obtained for other Fe(II) containing
mineral systems
A recent study compared the reactivity of various Fe(II) containing iron mineral
systems towards organic probe compounds representing different classes of
pollutants (Elsner et al 2004) The reductive transformation of 4-CNB was
investigated for the Fe(III) minerals goethite (α-FeOOH) lepidocrocite (γ-
FeOOH) and hematite (α-Fe2O3) as well as for the Fe(II)-Fe(III) oxide magnetite
(Fe3O4) All experiments were conducted in the presence of 1 mM dissolved Fe(II)
at pH 72 As seen from the surface area-normalised pseudo 1 order rate constants
in Figure 47 the reduction of 4-CNB by the Fe(II)-amended goethite
lepidocrocite and magnetite systems was up to 40 times faster than its reduction by
GR-SO4 The reduction rate obtained for the Fe(II)hematite system was only
slightly higher than the rate for the blank containing no iron mineral but aqueous
Fe(II) solely
84 Chapter 4
Figure 47 Surface area-normalised pseudo 1 order rate constants for the degradation of 4-CNB
by GR-SO4 (open square this work) and various Fe(II) containing mineral systems (solid circles
Elsner et al 2004) Experimental conditions applied by Elsner et al 1 mM aqueous Fe(II) pH
72 25 m2 mineral surface areaL GR-SO4 = green rust sulphate α-FeOOH = goethite Fe3O4 =
magnetite γ-FeOOH = lepidocrocite α-Fe2O3 = hematite
The experiments with GR-SO4 in our study were carried out at pH 84 whereas the
experiments comprising the other systems in Figure 47 were conducted at pH 72
As the reactivity of GR is expected to increase with pH (Lee amp Batchelor 2002b
Koch amp Hansen 1997) the lower of GR-SO4 compared to other Fe(II) systems
cannot be explained by differences in pH values GR-SO4 might just contain fewer
or less reactive surface sites than Fe(II)-amended goethite lepidocrocite and
magnetite suspensions These findings contrast those of other studies which found
higher surface area-normalised pseudo 1 order rate constants for dechlorination
reactions for GR-SO4 than for magnetite (Lee amp Batchelor 2002a amp 2002b Elsner
et al 2004) The different reactivity orders of the Fe(II)-bearing minerals found
for chlorinated aliphatics and nitro aromatics suggest that effects other than pH and
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 85
the intrinsic reduction potentials of the reacting species play a role for the
reactivity of these Fe(II)-bearing minerals
435 Depletion of reactive sites
Assuming that the platy hexagonal GR-SO4 crystals hold an average width of 1 microm
and an average particle thickness of 35 nm (Hansen amp Koch 1998) the ratio of
outer surface area to total surface area AouterAtotal ~ 131 (see Supporting
Information 73) This means that only 3 of the total surface area in GR-SO4 is
available at the external surface Thus as the interlayer sulphate in GR-SO4 is not
readily exchanged with the anionic NAC applied we conclude that 4-NPA and
other NACs only react with the Fe(II) sites at the external GR-SO4 surface In
Table 43 the actual amounts of NAC reduced by GR-SO4 during reaction is
compared with the amount of NAC which theoretically can be reduced by the
initial amount of external reactive Fe(II) sites in GR-SO4 at AouterAtot ~ 131
Table 43 The actual amounts of NAC reduced by GR-SO4 during reaction ∆[NAC]act
compared with the amount of NAC which stoichiometrically should be reduced by the initial
amount of external reactive Fe(II) sites in GR-SO4 ∆[Ar-NO2]theory (calculated as
[Fe(II)GR]0(31middot6) assuming an even distribution of Fe(II) throughout the GR-SO4 structure)
Calculated for [NAC]0 ~ 50 microM
[Fe(II)GR]0 (mM)
∆[Ar-NO2]theory(microM)
∆[4-NT]act (microM)
∆[4-CNB]act (microM)
∆[4-NPA]act (microM)
103 55 174 177 99
63 339 426 380 273
As seen in Table 43 the actual amounts of NAC reduced by GR-SO4 during
reaction are in most cases higher than the amount of NAC which should be
reduced at the given [Fe(II)GR]0 according to reaction stoichiometry This indicates
that new external reactive sites were regenerated eg the Fe(III) phases produced
peel off the GR surface exposing new Fe(II) sites or that outermost internal
86 Chapter 4
reactive sites in close vicinity to the edges are available for reaction as well Lee
and Batchelor (2000b) also found the experimentally observed reduction capacity
of GR-SO4 for chlorinated ethylenes to be 2-3 orders of magnitudes lower than the
estimated reduction capacity including all Fe(II) in GR-SO4
At low initial Fe(II)GR concentrations only a fraction of NAC was reduced within
the reaction time observed (Figure 45c) even though there was stoichiometric
excess of Fe(II)-GR present The fraction of initial Ar-NO2 reduced by GR-SO4 at
reaction termination decreased as [Fe(II)GR]0 decreased (Table 41) and was
accompanied by a change in apparent rate laws with time (compare Figures 45b amp
d) In order to explain these observations we propose that the NACs react only at
external reactive Fe(II) sites and that the regeneration of new external reactive sites
is much slower than the reduction of NAC by GR-SO4 Thus the fast reduction of
NAC taking place at the external reactive sites represents the pseudo 1 order
behaviour whereas depletion of external reactive sites and their slow regeneration
are represented by the second bent part of the (time ln [Ar-NO2]t[Ar-NO2]0)-
curves deviating from pseudo 1 order kinetics Hence at low [Fe(II)GR]0 the
regeneration of reactive sites will eventually control the overall reaction rate
Depletion of available Fe(II) was also observed during the fast reduction of
chromate by GR-CO3 when the initial chromate concentration was increased or
when the GR-CO3 suspension was respiked with chromate repeatedly (Williams amp
Scherer 2001)
436 The role of external and internal reactive sites
It is reasonable to assume that GRs hold adsorption properties similar to other
layered double hydroxides such as hydrotalcites The sorption of 246-
trinitrophenol (TNP) and 245-trichlorophenol (TCP) on chloride and carbonate
intercalated hydrotalcites (HT-Cl = Mg3Al(OH)8ClmiddotyH2O HT-CO3 =
Mg6Al2(OH)16CO3middotyH2O) has been investigated (Hermosin et al 1993 Ulibarri et
al 1995 Ulibarri et al 2001) The authors found that the adsorption of TCP on
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 87
HT-CO3 was very low and that TCP adsorbs only on the external surface sites of
HT-CO3 (Hermosin et al 1993) Furthermore is was reported that the adsorption
of TNP on HT was dramatically affected by the nature of the interlayer anion ie
the adsorption of TNP was considerably higher on HT-Cl than on HT-CO3
(Ulibarri et al 2001) For HT-Cl interlayer anion exchange of chloride with TNP
was detected by XRD analysis and an expansion of the characteristic basal d003
spacing from 79 Ǻ to 132 Ǻ confirmed the presence of TNP in the HT interlayer
(Ulibarri et al 1995) Collating the results reported for HTs with GRs it is not
likely that the divalent SO42- in GR-SO4 is exchanged with the monovalent 4-NPA
Chacirctelet et al (1996) investigated the adsorption of mono- and divalent anions
onin the outer and inner adsorption sites in HT by varying the zetapotential with
pH in the presence of various electrolytes The authors found that SO42- adsorbs on
the external HT surfaces by formation of outer-sphere complexes whereas chloride
hardly adsorbed on HT Moreover it was reported that the adsorption of sulphate
onto HT was not strongly affected by the presence of chloride while sulphate on
the contrary inhibited the adsorption of chloride on HT Studies applying
spectroscopic analyses have investigated the sorption of oxyanions at external and
internal GR-SO4 surfaces (Myneni et al 1997 Randall et al 2001) Selenate was
adsorbed only on the outer GR-SO4 surface when added after GR formation
whereas it was primarily coprecipitated into the interlayer when present during GR
formation Thus for selenate its presence during GR formation is a prerequisite of
its incorporation in the GR interlayer Selenate is readily reduced by GR-SO4 and
the rates of reduction of coprecipitated selenate were very similar to the reduction
rates of selenate adsorbed at the outer GR surface (Myneni et al 1997) This
finding suggests that the outer and inner reactive Fe(II) sites in GR-SO4 hold
similar reactivities
Results based on electron microscopy reported that the reduction of uranyl took
place primarily at the edges of hexagonal GR-SO4 particles (OrsquoLoughlin et al
2003a) In another recent study XRD characterization of the GR-SO4 crystals
88 Chapter 4
during reaction with trichloroacetate (TCA) indicated that TCA did not enter the
GR-SO4 interlayer during reaction (Chapter 5 this work) The average GR-SO4
particle thickness perpendicular to the basal plane was constant during reaction
implying that TCA reacts only at the edges and not at the basal planes Assuming
that the platy hexagonal GR-SO4 crystals hold an average width of 1 microm and an
average particle thickness of 35 nm (Hansen amp Koch 1998) the ratio of edge
surface area to outer surface area is AedgeAouter ~ 130 (see Supporting Information
73) Hence only 3 of the outer surface area in GR-SO4 is available at the edges
Once more the regeneration of new external reactive sites is strongly inferred as
the actual amounts of NAC reduced by GR-SO4 during reaction are much higher
than the amount of NAC which may be reduced by the reactive edge sites present
initially Assuming that the NACs react at the edges only and if employing the
AedgeAouter in the estimation of the rate constants the surface area-normalised
pseudo 1 order rate constants for GR-SO4 would be 30 times higher than the rate
constants depicted in Figure 47 Thus the reactivity of GR-SO4 normalised to its
reactive surface area is higher than the reactivity normalised to its outer surface
area determined by the BET method (N2 adsorption)
The reduction of chromate has been examined in the presence of all the common
GR forms (Bond amp Fendorf 2003 Loyaux-Lawniczak et al 1999 Loyaux-
Lawniczak et al 2000 Williams amp Scherer 2001) The results reported by Bond
amp Fendorf (2003) confirm that not only the surface area of GR but also the
interlayer spacing (interlayer anion size) and interlayer anion charge play an
important role for the reaction rate Hence it follows that coordination (size) and
charge of the oxidant determine its access to the internal sites in GRs
The results obtained for all 4 NACs support what has been reported for nitrate and
TCA At [Fe(II)GR]0 = 2-10 mM and [NO3-]0 = 143 mM pseudo 1 order rate
constants for the reduction of nitrate by GR-SO4 were 158middot10-7 s-1middotm-2middotL (Hansen
et al 2001) This reaction rate increased 40 times by adding barium nitrate instead
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 89
of sodium nitrate thereby precipitating the interlayer sulphate as barium sulphate
and enhancing access to the interlayer Though barium addition changes the GR-
SO4 system dramatically it indicates the importance of interlayer anion exchange
(Hansen amp Koch 1998) The rate constant reported for nitrate (no barium added) is
100-1000 smaller than the rate constants obtained for the NACs in this work
Moreover the reaction kinetics for nitrate did not deviate from pseudo 1 order
kinetics At [Fe(II)GR]0 = 025-104 mM and [TCA]0 = 50 microM-1 mM pseudo 1
order rate constants for the reduction of TCA by GR-CO3 or GR-SO4 were 65middot10-7
s-1middotm-2middotL (Chapter 5 this work) The rate constant for TCA is 10-1000 smaller than
the rate constants for the NACs and the reaction kinetics for TCA did not deviate
from pseudo 1 order kinetics This suggests that the overall reductive
transformation of slowly reacting oxidants such as nitrate and TCA is not
controlled by the rate of regeneration of external Fe(II) reactive sites Altogether
the results reported for selenate chromate and nitrate clearly demonstrate that
these anionic oxidants react primarily with external reactive sites in GR-SO4 Only
under certain conditions ie adding the oxidant prior to GR-SO4 formation or
extracting the interlayer sulphate through precipitation with barium outside the
GR-SO4 particles do the oxidants have access to the interlayer Our findings
suggest that both the neutral and anionic nitro aromatic probe compounds applied
here also react exclusively with the external reactive sites in GR-SO4 Supposedly
the neutral and monovalent charge states of the NACs hinder their access to the
GR-SO4 interlayer A divalent anionic nitro aromatic probe compound might
exchange with the interlayer sulphate more readily and gain access to the inner
Fe(II) reactive sites in GR-SO4 only divalent anionic NACs are not commercially
available
44 Conclusions
This work demonstrates that NACs are completely reduced to their corresponding
anilines by GR-SO4 The surface area-normalised pseudo 1 order rate constants
obtained for the reduction of the neutral and anionic NACs by GR-SO4 under
90 Chapter 4
various experimental conditions did not differ significantly from each other despite
their different charges Neither mass transfer control nor surface saturation kinetics
could account for the similarity of the pseudo 1 order rate constants obtained for
the NACs These observations suggest that the anionic NACs do not have an
enhanced access to inner or outer Fe(II)-GR reactive sites as compared to the
neutral NACs Based on our estimations of the molecular sizes of the NACs we
propose that the charge and not the size of the NACs controls their access to the
internal reactive sites in GRs Hence the reaction between NAC and GR-SO4 takes
place primarily at the external reactive Fe(II) sites This work further demonstrated
that the reduction of the NACs by GR-SO4 only followed pseudo 1 order kinetics
throughout the whole reaction at high initial Fe(II)GR concentrations At low initial
Fe(II)GR concentrations the NACs were not reduced completely within the reaction
time observed though according to reaction stoichiometry the total Fe(II)-GR
present should be sufficient to reduce the whole amount of NAC This means that
at some point during the reaction the external reactive Fe(II) sites were depleted
and the regeneration of new external reactive sites was much slower than the
reduction of the NACs by GR-SO4 The reduction of 4-CNB by GR-SO4 reported
here was 10-100 times slower than its reduction by other Fe(II)-Fe(III) systems
such as goethite lepidocrocite and magnetite suspensions amended with Fe(II)
(Elsner et al 2004)
The results obtained in this work infer that under natural geochemical conditions
where GR-SO4 presumably forms in low concentrations the rate of regeneration of
external Fe(II) reactive sites may control the overall reductive transformation of
fast reacting pollutants by GR-SO4 Thus not only abiotic processes such as
interchanging redox conditions created by water level alterations but also the direct
microbial formation through Fe(III)-reducing bacteria may govern the formation of
GRs and the renewal of external Fe(II) reactive sites in GRs This holds both for
natural systems like iron-rich suboxic soils and sediments as well as engineered
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 91
systems like permeable reactive barriers of zero-valent iron implemented for on-
site remediation of organic and inorganic contaminants
Acknowledgments
We would like to thank Henrik T Andersen for performing the NB kinetic experiments and
Hanne Nancke-Krogh for technical assistance in the laboratory
References
Al-Agha MR Burley SD Curtis CD Esson J (1995) Complex cementation textures and authigenic mineral assemblages in recent concretions from the Lincolnshire Wash (east coast UK) driven by Fe(0) to Fe(II) oxidation Journal of the Geological Society 152 157-171 Bernal JD Dasgupta DR Mackay AL (1959) The oxides and hydroxides of iron and their structural inter-relationships Clay Minerals Bulletin 4 15-30 Bond DL Fendorf S (2003) Kinetics and structural constraints of chromate reduction of green rusts Environmental Science and Technology 37 2750-2757 Brunauer S Emmett PH Teller E (1938) Adsorption of gases in multimolecular layers Journal of American Chemical Society 60 309-319 Charlet L Silvester E Liger E (1998) N-compound reduction and actinide immobilisation in surficial fluids by Fe(II) The surface FeIIIFeIIOH0 species as major reductant Chemical Geology 151 85-93 Chacirctelet L Bottero JY Yvon J Bouchelaghem A (1996) Competition between monovalent and divalent anions for calcined and uncalcined hydrotalcite anion exchange and adsorption sites Colloids and Surfaces A Physicochemical and Engineering Aspects 111 167-175 Chaudhuri SK Lack JG Coates JD (2001) Biogenic magnetite formation through anaerobic biooxidation of Fe(II) Applied and Environmental Microbiology 67 2844-2848 Cui D Spahiu K (2002) The reduction of U(VI) on corroded iron under anoxic conditions Radiochemica Acta 90 623-628 Elsner M Haderlein SB Schwarzenbach RP (2004) Reactivity of Fe(II)-bearing minerals towards reductive transformation of organic contaminants Environmental Science and Technology 38 799-807 Erbs M Hansen HCB Olsen CE (1999) Reductive dechlorination of carbon tetrachloride using iron(II)iron(III)-hydroxide-sulphate (green rust) Environmental Science and Technology 33 307-311 Fadrus H Maly J (1975) Suppression of iron(III) interference in the determination of iron(II) in water by the 110-phenanthroline method The Analyst 100 549-554 Fredrickson JK Zachara JM Kennedy DW Dong H Onstott TC Hinman NW Li S
92 Chapter 4 (1998) Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium Geochimica et Cosmochimica Acta 62 3239-3257 Geacutenin JMR Bourrieacute G Trolard F Abdelmoula M Jaffrezic A Refait Ph Maitre V Humbert B Herbillon A (1998) Thermodynamic equilibria in aqueous suspensions of synthetic and natural Fe(II)-Fe(III) green rusts Occurrences of the mineral in hydromorphic soils Environmental Science and Technology 32 1058-1068
Geacutenin JMR Olowe AA Benbouzid-Rollet ND Prieur D Confente M Resiak B (1991) The simultaneous presence of green rust 2 and sulfate reducing bacteria in the corrosion of steel sheet piles in a harbour area Hyperfine Interactions 69 875-878 Glasauer S Weidler PG Langley S Beveridge TJ (2003) Controls on Fe reduction and mineral formation by a subsurface bacterium Geochimica et Cosmochimica Acta 67 1277- 1288 Hansen HCB (1989) Composition stabilization and light absorption of Fe(II)Fe(III) hydroxycarbonate (green rust) Clay Minerals 24 663-669 Hansen HCB Borggaard OK Soslashrensen J (1994) Evaluation of the free energy of formation of iron(II)iron(III)-hydroxidesulphate (Green Rust) and its reduction of nitrite Geochimica et Cosmochimica Acta 58 2599-2608 Hansen HCB Guldberg S Erbs M Koch CB (2001) Kinetics of nitrate reduction by green rusts ndash effects of interlayer anion and Fe(II)Fe(III) ratio Applied Clay Science 18 81-91 Hansen HCB Koch CB (1998) Reduction of nitrate to ammonium by sulphate green rust Activation energy and interlayer reaction mechanism Clay Minerals 33 87-101 Hansen HCB Koch CB Nancke-Krogh H Borggaard OK Soerensen J (1996) Abiotic nitrate reduction to ammonium Key role of green rust Environmental Science and Technology 30 2053-2056 Hansen HCB Poulsen IF (1999) Interaction of synthetic sulphate green rust with phosphate and the crystallization of vivianite Clays and Clay Minerals 47 312-318 Hansen HCB Taylor RM (1991) The use of glycerol intercalates in the exchange of CO3
2- with SO4
2- NO3- or Cl- in pyroaurite-type compounds Clay Minerals 26 311-327
Heasman DM Sherman DM Ragnarsdottir KV (2003) The reduction of aqueous Au3+ by sulfide minerals and green rust phases American Mineralogist 88 725-738 Hermosin MC Pavlovic I Ulibarri MA Cornejo J (1993) Trichlorophenol adsorption on layered double hydroxide a potential sorbent Journal of Environmental Science and Health A28 1875-1888 Hofstetter TB Heijmann CG Haderlein SB Holliger C Schwarzenbach RP (1999) Complete reduction of TNT and other (poly)nitroaromatic compounds under iron-reducing subsurface conditions Environmental Science and Technology 33 1479-1487 Hofstetter TB Schwarzenbach RP Haderlein SB (2003) Reactivity of Fe(II) species associated with clay minerals Environmental Science and Technology 37 519-528
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 93
Klausen J Troumlber SP Haderlein SB Schwarzenbach RP (1995) Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions Environmental Science and Technology 29 2396-2404 Koch CB Hansen HCB (1997) Reduction of nitrate to ammonium by sulphate green rust Advances in GeoEcology 30 373-393 Kukkadapu RK Zachara JM Smith SC Fredrickson JK Liu C (2001) Dissimilatory bacterial reduction of Al-substituted goethite in subsurface sediments Geochimica et Cosmochimica Acta 65 2913-2924 Lee W Batchelor B (2002a) Abiotic reductive dechlorination of chlorinated ethylenes by iron- bearing soil minerals 1 Pyrite and magnetite Environmental Science and Technology 36 5147- 5154 Lee W Batchelor B (2002b) Abiotic reductive dechlorination of chlorinated ethylenes by iron-bearing soil minerals 2 Green rust Environmental Science and Technology 36 5348- 5354 Liger E Charlet L Van Cappellen P (1999) Surface catalysis of uranium (VI) reduction by iron(II) Geochimica et Cosmochimica Acta 63 2939-2955 Loyaux-Lawniczak S Refait Ph Ehrhardt J Lecomte P Geacutenin JMR (2000) Trapping of Cr by formation of ferrihydrite during the reduction of chromate ions by Fe(II)-Fe(III) hydroxysalt green rusts Environmental Science and Technology 34 438-443 Loyaux-Lawniczak S Refait Ph Lecomte P Ehrhardt J Geacutenin JMR (1999) The reduction of chromate ions by Fe(II) layered hydroxides Hydrology and Earth System Sciences 3 593-599 Mendiboure A Schoumlllhorn A (1986) Formation and anion exchange reactions of layered transition metal hydroxides [Ni1-xMx](OH)2(CO3)x2(H2O)z (M = Fe Co) Revue de Chimie Mineacuterale 23 819-827 Miyata S (1983) Anion-exchange properties of hydrotalcite-like compounds Clays and Clay Minerals 31 305-311 Myneni SCB Tokunaga TK Brown Jr GE (1997) Abiotic selenium redox transformations in the presence of Fe(IIIII) oxides Science 278 1106-1109 Nielsen A (1976) Hvid groslashn og sort rust Beskrivelse af en korrosionsskade paring et svoslashmmebassin Nordisk Betong 2 21-24 OLoughlin EJ Burris DR (2004) Reduction of halogenated ethanes by green rust Environmental Toxicology and Chemistry 23 41-48 OLoughlin EJ Kelly SD Cook RE Csencsits R Kemner KM (2003a) Reduction of uranium(VI) by mixed iron(II)iron(III) hydroxide (green rust) Formation of UO2 nanoparticles Environmental Science and Technology 37 721-727
94 Chapter 4 OLoughlin EJ Kelly SD Kemner KM Csencsits R Cook RE (2003b) Reduction of AgI AuIII CuII and HgII by FeIIFeIII hydroxysulfate green rust Chemosphere 53 437-446 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002) Iron (IIIII) hydroxycarbonate green rust formation and stabilization from lepidocrocite bioreduction Environmental Science and Technology 36 16-20 Parmar N Gorby YA Beveridge TJ Ferris FG (2001) Formation of green rust and immobilization of nickel in response to bacterial reduction of hydrous ferric oxide Geomicrobiology Journal 18 375-385 Pepper SE Bunker DJ Bryan ND Livens FR Charnock JM Pattrick RAD Collison D (2003) Treatment of radioactive wastes An X-ray absorption spectroscopy study of the reaction of technetium with green rust Journal of Colloid and Interface Science 268 408- 412 Randall SR Sherman DM Ragnarsdottir KV (2001) Sorption of As(V) on green rust (Fe4(II)Fe2(III)(OH)12SO4
3H2O) and lepidocrocite (γ-FeOOH) Surface complexes from EXAFS spectroscopy Geochimica et Cosmochimica Acta 65 1015-1023 Roh Y Lee SY Elless MP (2000) Characterization of corrosion products in the permeable reactive barriers Environmental Geology 40 184-194 Schultz CA Grundl TJ (2000) pH dependence on reduction rate of 4-Cl-nitrobenzene by Fe(II)montmorillonite systems Environmental Science and Technology 34 3641-3648 Simon L Francois M Refait Ph Renaudin G Lelaurain M Geacutenin JMR (2003) Structure of the Fe(II-III)-layered double hydroxysulphate green rust two from Rietveld analysis Solid State Sciences 5 327-334 Tamaura Y (1986) Ni(II)-bearing green rust II and its spontaneous transformation into Ni(II)- bearing ferrites Bulletin of the Chemical Society of Japan 59 1829-1832 Tamaura Y (1985) ZnII-bearing green rust II and its spontaneous transformation into ZnII- bearing ferrite in aqueous solution Bulletin of the Chemical Society of Japan 58 2951-2954 Trolard F Abdelmoula M Bourrieacute G Humbert B Geacutenin JMR (1996) Mise en eacutevidence dun constituant de type rouilles vertes dans les sols hydromorphes Proposition de lexistence dun nouveau mineacuteral la fougeacuterite Geacuteosciences de surface Comptes Rendus de LrsquoAcademie des Sciences 323 1015-1022 Tuovinen OH Button KS Vuorinen A Carlson L Mair DM Yut LA (1980) Bacterial chemical and mineralogical characteristics of tubercles in distribution pipelines Journal of the American Water Works Association 72 626-635 Ulibarri MA Pavlovic I Barriga C Hermosin MC Cornejo J (2001) Adsorption of anionic species on hydrotalcite-like compounds effect of interlayer anion and crystallinity Applied Clay Science 18 17-27 Ulibarri MA Pavlovic I Hermosin MC Cornejo J (1995) Hydrotalcite-like compounds as potential sorbents of phenols from water Applied Clay Science 10 131-145
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 95
Williams AGB Scherer MM (2001) Kinetics of chromate reduction by carbonate green rust Environmental Science and Technology 35 3488-3494
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 97
5 Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems
Abstract
Trichloroacetate (TCA) is a widespread environmental contaminant with proven
phytotoxicity and suspected human carcinogenicity In order to assess the global
cycling of TCA and to predict its fate in subsurface environments information
regarding the reactivity and product distribution of TCA degradation is needed
Due to the high oxidation state of TCA conditions for oxidative transformation
pathways in soils and groundwater are unfavorable However in suboxic soils and
sediments Fe(II)-bearing minerals are potential reactants for reductive
dehalogenation reactions of TCA as has been demonstrated for other halogenated
contaminants We examined the reactivity of various Fe(II)-Fe(III) mineral
systems towards TCA and dichloroacetate (DCA) its expected transformation
product in laboratory batch experiments imitating natural conditions ie low
initial Fe(II) Fe(III) and TCADCA concentrations and no artificial buffer The
Fe(II)-Fe(III)-systems investigated included sulfate green rust (GR-SO4) carbonate
green rust (GR-CO3) magnetite Fe(II)goethite and Fe(II)lepidocrocite
Trichloroacetate was readily reduced to DCA by all Fe(II)-bearing minerals The
reactions generally followed pseudo 1 order kinetics with respect to TCA The
surface area-normalised pseudo 1 order rate constants obtained (035ndash76middot10-5 min-
1middotm-2middotL at [Fe(II)]0 = 020ndash122 mM [TCA]0 = 15ndash1000 microM and pH 70ndash87)
showed no striking differences regarding product distribution and surface area-
normalised reaction rate constants between the Fe(II)-Fe(III)-systems The
stoichiometrically formed DCA was not further reduced to monochloroacetate
(MCA) or acetate in any of the systems within the time frame in our experiments
To our knowledge this is the first published report on abiotic transformation of
TCA by Fe(II)-bearing minerals Our results imply that processes involving
reactive Fe(II)-bearing minerals may play a significant role in controlling the fate
98 Chapter 5
of TCA in natural subsurface environments and that DCA found in the subsurface
may be formed by such processes
51 Introduction
Trichloroacetic acid (TCA) has been applied as a herbicide for many years until its
use was banned in the late 1980acutes (Berg et al 2000) Today TCA is mainly used
as an etching agent in the metal industry as a swelling solvent in the plastic
production and as a bleaching agent in the paper and pulp manufacture (Muumlller et
al 1996) Other anthropogenic sources include formation of TCA as a result of the
chlorine based disinfecting process used in drinking water treatment and the
atmospheric photooxidation of chlorinated solvents including tetrachloroethene
and 111-trichloroethane (McCulloch 2002) Only very little information is
available on the TCA production volumes and even less is known about the
amount of TCA released into the environment as a result of its industrial
applications Due to its low volatility and high aqueous solubility TCA is easily
washed out of the atmosphere into the aquatic and terrestrial biospheres As TCA
is found in almost every ecosystem around the globe including non-urban and
non-industrial sites the relative contributions from anthropogenic and natural
sources are currently being debated (McCulloch 2002 Ahlers et al 2003)
Trichloroacetic acid is omnipresent in soils and the concentrations reported are
very variable ranging from lt005 microgkg to 380 microgkg (Euro Chlor 2001
McCulloch 2002 Ahlers et al 2003) Both abiotic and enzymatically catalyzed
formation of TCA from humic acids have been demonstrated in laboratory studies
(Haiber et al 1996 Hoekstra et al 1999b Fahimi et al 2003) Furthermore the
in situ natural formation of TCA from anthropogenic or natural tetrachloroethene
or 111-trichloroethane in biota has been suggested (Hoekstra et al 1999a
McCulloch 2002) Such natural sources may explain part of the TCA
concentrations found in soils but their environmental significance is still unknown
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 99
On account of its phytotoxicity suspected human carcinogenicity and widespread
occurrence TCA is of considerable environmental concern especially in the
terrestrial compartment The TCA concentrations found in soil air and water in
pre-industrial times were far below the present ones (Jordan amp Frank 1999 Ahlers
et al 2003) Based on the current TCA concentrations detected in soils the
European Commission proposed risk reduction measures concerning
tetrachloroethene - a precursor of TCA - to be taken immediately (Ahlers et al
2003 and references therein) Occurrences of monochloroacetic acid (MCA) and
dichloroacetic acid (DCA) reported include surface waters marine waters
precipitation ice (glaciers) and air (Reimann et al 1996 Berg et al 2000 Scott et
al 2000 Scott et al 2002) Based on the concentrations reported for the aquatic
environments it is reasonable to assume that MCA and DCA are omnipresent in
soils as well Sources of MCA and DCA include production in the chemical
industry photooxidation of chlorinated aliphatics in the atmosphere and reductive
transformation of TCA (Reimann et al 1996 Ahlers et al 2003 and references
therein) MCA and DCA are also toxins and suspected human carcinogens (Kuumlhn
amp Pattard 1990) hence not only TCA but also its daughter compounds are
pollutants of environmental concern
In subsurface environments TCA may be removed by sorption seepage chemical
transformation microbial degradation and plant uptake followed by metabolic
degradation andor physical removal at harvest (Foy 1975) There is little or no
evidence of abiotic transformations of TCA in the literature Only one recent study
demonstrated the reductive dechlorination of TCA to MCA by Fe(0) (Hozalski et
al 2001) It has been reported that the degradation of TCA in soil is slow and
mainly mediated by microorganisms but only little is known about the bacteria and
processes involved (Lignell et al 1984) Biodegradation of TCA has been found at
both oxic and anoxic conditions An aerobic microorganism capable of growing on
TCA as the sole carbon and energy source has been characterised (Yu amp Welander
1995) Moreover anaerobic bacteria coupling co-metabolic growth to reductive
100 Chapter 5
dechlorination of TCA have been isolated (Weightman et al 1992 De Wever et
al 2000) However more information regarding the abiotic and biotic
transformation of TCA is needed in order to assess the fate and transport of TCA in
natural subsurface environments
It is well-known that Fe(II) present in minerals or associated with mineral surfaces
is a much stronger reductant than Fe(II) in solution The enhanced reactivity of a
structural or surface-bound Fe(II) center can be rationalized by the increased
electron density donated by hydroxyl ligands and a stabilization of the Fe(III)
oxidation state by the hydroxyl ligands (Luther 1990) Fe(II)-bearing minerals
including layered Fe(II)-Fe(III) hydroxides (green rusts) magnetite (Fe3O4)
siderite (FeCO3) Fe(II) sulfides as well as Fe(II)-carrying Fe(III) oxides and clay
minerals have also been shown to reduce a range of organic and inorganic
contaminants such as nitro aromatic compounds chlorinated aliphatics chromate
uranyl pertechnetate nitrate monochloramine and carbamate pesticides (Chapter
4 this work Klausen et al 1995 Cui amp Eriksen 1996 Erbs et al 1999 Liger et
al 1999 Loyaux-Lawniczak et al 1999 Amonette et al 2000 Hansen et al
2001 Pecher et al 2002 Vikesland amp Valentine 2002 Hofstetter et al 2003
OrsquoLoughlin and Burris 2003 OrsquoLoughlin et al 2003a amp 2003b Strathmann amp
Stone 2003 Elsner et al 2004a) Laboratory and field studies showed that even in
geochemically highly heterogeneous anoxic aquifer sediments Fe(II) adsorbed to
Fe(III) (hydr)oxide surfaces was the dominant reductant of nitroaromatic and
halogenated contaminants (Ruumlgge et al 1998 Hofstetter et al 1999 Kenneke amp
Weber 2003) Only little is known about the nature of the Fe(II) species associated
with Fe(III) oxide surfaces but reactive hydroxylated Fe(II)-Fe(III)-hydroxo
surface complexes associated with hematite and magnetite above pH 65 have been
proposed (Charlet et al 1998aampb Liger et al 1999) Due to the presence of
structural Fe(II) within the mineral lattice the reactivity of Fe(II) associated with
mixed valent Fe(II)-Fe(III) minerals such as green rusts magnetite and reduced
ferruginous clay minerals may hold another reactivity than Fe(II) associated with
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 101
pure Fe(III) oxides However Fe(II) adsorbed on Fe(III) oxides such as goethite
hematite and lepidocrocite may also hold different reactivities as the Fe(III) oxides
contain different crystal and surface structures
Since chlorinated ethanes and ethenes such as hexachloroethane 111-
trichloroethane tetrachloroethene and trichloroethene are susceptible to chemical
reduction by a range of Fe(II)-bearing minerals including magnetite GR-SO4
Fe(II) sulfides and Fe(II)-carrying Fe(III) oxides (Butler amp Hayes 1998 amp 1999
Hwang amp Batchelor 2000 Gander et al 2002 Lee amp Batchelor 2002aampb Elsner
et al 2004a) we hypothesized that TCA may be transformed by Fe(II)-bearing
minerals as well The main goals of this work were to study such reactions and
establish product distribution and surface area-normalised reaction rates for the
reductive dechlorination of TCA by Fe(II)-Fe(III) mineral systems common in
nature
5 2 Materials and methods
No synthetic buffers were applied and iron concentrations were kept low The
calcareous systems were pH-controlled at 76 through a natural buffer system
(CaCO3(s) + 995 N205 CO2(g)) All handling and sampling of solutions and
suspensions were carried out under strict anoxic conditions Goethite (acicular
particles with size 01 times 06 microm specific surface area 16 m2g) and lepidocrocite
(acicular particles with size 005 times 03 microm specific surface area 18 m2g) were
purchased as fine powders from Bayer (Bayferrox 910 and 943) Calcite (grain size
170-350 microm Pluumlss-Staufer AG) was used as a buffer or as a Fe(III)-oxide-bearing
mineral In order to simulate natural conditions the iron minerals were applied as
coatings on calcite particles (model system for calcareous soils) in some
experiments Trichloroacetic acid dichloroacetic acid and monochloroacetic acid
were pa quality (Fluka)
102 Chapter 5
521 Synthesis of GRs and magnetite
GR-CO3 was synthesized by controlled air oxidation of an FeCl2 solution at a
constant pH of 700 (titrated with 1 M Na2CO3) according to the procedure given
by Hansen amp Koch (1997) 05 M aqueous stock solutions of FeCl2 were prepared
in 100 mL glass flasks by reacting 65 mmol of iron powder (particle size 10 microm
Merck) with 100 mL deoxygenated 10 M HCl The solutions were magnetically
stirred and heated (~80degC) during reaction until the H2(g) production had ceased (ge
2 hours) The FeCl2 solutions were stored in the dark under a small Ar
overpressure at 5degC The GR-CO3 suspensions were washed with deoxygenated
deionised water (DIW) separated on a folding filter (medium filtration rate cotton
linterhigh alpha pulp Schleicher amp Schuell) and redispersed in deoxygenated
DIW Washing separation and redispersion of the GR-CO3 suspension were
conducted in an anoxic glove box (92 N28 H2 Coy Laboratory Products Inc)
All suspensions and solutions were deoxygenated by Ar-purging (999998 Ar
Carbagas) Magnetite was synthesized by further aerial oxidation of GR-CO3 at pH
700 until consumption of 1 M Na2CO3 ceased GR-SO4 was synthesized by
controlled air oxidation of an FeSO4 solution at a constant pH of 70 according to
the procedure given by Koch amp Hansen (1997) The GR-SO4 suspension was
washed with deoxygenated DIW separated on a glass filter funnel (pore size 4
Duran) and redispersed in deoxygenated DIW Washing separation and
redispersion of the GR-SO4 suspension were conducted in an anoxic glove bag
(999995 Ar Aldrich)
522 Preparation of iron oxide coatings
Two grams of goethite (goe) or lepidocrocite (lep) and 100 g calcite were
combined with 200 mL DIW in a 500 mL polyethylene flask The suspension was
gently agitated on a reciprocating shaker for 24 h and left to stand for another 24 h
Excess Fe(III) oxides and salts were removed from the coated material by repeated
decantation and washing with DIW in polyethylene flasks until clear runoff
Finally the coatings were collected on folding filters and air dried The amount of
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 103
goethite and lepidocrocite coated onto calcite after washing and drying was
quantified to 10-11 mg Fe(III)g calcite
523 Mineral characterisation
The identity and purity of the GR-CO3 GR-SO4 and magnetite suspensions were
examined by means of X-ray diffraction (XRD) The XRD analyses were
performed on a Scintag XDS 2000 using Co Kα radiation (45 kV 40 mA) or a
Siemens D5000 XRD applying Co Kα radiation (40 kV 40 mA) Glycerol smears
made according to Hansen (1989) were scanned between 6 and 80 deg2θ with a scan
speed of 1 deg2θmin The specific surface area (SSA) of calcite was determined by
the BET multi-point method using N2 adsorption (Brunauer et al 1938) Powder
samples were filled into sample burettes in the glove box and the generously
lubricated stopcocks closed Samples and burettes were evacuated prior to
connecting them to the BET-instrument (Sorptomatic 1990 Fisons)
524 Kinetic experiments
All reactions were carried out in 25-100 mL serum vials sealed with stoppers
(Viton or Teflon coated rubber) and aluminum crimp caps Kinetic experiments
were conducted with GR-SO4 GR-CO3 magnetite Fe(II)goethite and
Fe(II)lepidocrocite at room temperature In most cases pH was controlled through
the carbonate-bicarbonate buffer system by adding calcite to suspensions
containing the iron minerals solely or by adding the iron minerals as coatings on
calcite Furthermore the calcite containing suspensions were deoxygenated with
05 CO2995 N2(g) thereby attaining an initial pH of 76-77 The GR-CO3 and
magnetite suspensions were deoxygenated with 100 N2(g) and no additional pH
buffer was added The goethite and lepidocrocite suspensions were amended with
300-1000 microM FeCl2(aq) and equilibrated gt 20 h prior to TCADCA addition See
Table 51 for more details on the experimental conditions To start the reaction 50
microM - 1 mM TCA or DCA was added to the mineral suspensions from aqueous
anoxic stock solutions The reaction vials were agitated gently on a roller apparatus
104 Chapter 5
or a shaking table (35 rpm) in order to minimize abrasion of the iron oxide mineral
coatings At appropriate time intervals suspension samples were withdrawn using
Ar(g)- 100 N2(g)- or 995 N205 CO2(g)-flushed sterile disposable syringes
and hypodermic needles The suspension samples were filtered (02 microm Teflon)
and collected for quantification of chloride and the chlorinated acetic acids The
samples were stored at -20degC and analysed without further treatment
525 Analytical methods
Total and aqueous Fe(II) were determined using a modified phenanthroline method
(Fadrus and Maly 1975) For determining [Fe(II)aq] and [Fe(II)total] 1 mL filtered
(02 microm Teflon) and 1 mL unfiltered mineral suspension were added to 18 mL 01
M HCl respectively and allowed to dissolve for 30 min From these acid digests
01 mL was added to 05 mL Fe(II)-phenanthroline-buffer-reagent and 19 mL
DIW added up Estimates of the structural or adsorbed Fe(II) in the Fe(II)-Fe(III)
mineral systems were estimated as the difference [Fe(II)solid] = [Fe(II)total] -
[Fe(II)aq] The total amount of Fe(III) coated on calcite was determined by atomic
absorption spectroscopy following dissolution in 6 M HCl(aq) for 24 h At low
initial TCA concentrations (le 50 microM) the chlorinated acetic acids were quantified
by means of a modified ion interaction (or paired-ion) chromatographic method
(Sarzanini et al 1999) Separation was performed on a LiChrospher 100 RP-18 (5
microm 125 times 4 mm ID) reversed-phase column coupled with a LiChroCART 100 RP-
18 (4 times 4 mm ID) precolumn Analytical conditions were isocratic and the eluent
consisted of 50 aqueous solution of 35 mM cetyltrimethylammonium chloride
(pH 50) and 50 CH3CN The injection volume was 20 microL and the flow-rate 10
mLmin HPLC analyses of the chloroacetates were performed using a Gynkotek
Pump M480 Gynkotek Gina 50 auto sampler and a diode array UV detector (340s
Gynkotek) UV-VIS detection was carried out at 200 nm At higher initial TCA
concentrations the chlorinated acetic acids were quantified by a modified HPLC
method (Husain et al 1992) Separation was performed on a ChromSphere C-18
(10 microm 250 times 46 mm ID) reversed-phase column Analytical conditions were
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 105
isocratic and the eluent consisted of 015 M (NH4)2SO4(aq) pH 55 The injection
volume was 20 microL and the flow-rate 10 mLmin HPLC analyses were performed
using a Series 10 Liquid Chromatographic Pump (Perkin-Elmer) and a SPD-10 A
VP UV-VIS detector (Shimadzu) UV-VIS detection was carried out at 210 nm
Chloride was determined in the GR-SO4 kinetic experiments using a flow injection
system with spectrophotometric detection (Cheregi amp Danet 1997)
53 Results and discussion
531 Product formation and reaction kinetics
Trichloroacetate was readily reduced to DCA by all the Fe(II)-bearing minerals
examined Only DCA was detected within the reaction time in all the Fe(II)-Fe(III)
mineral systems Experiments conducted with the various Fe(II)-Fe(III) mineral
systems and DCA confirmed that no significant reduction of DCA took place (data
not shown) Hence it is reasonable to assume that the further hydrogenolysis of
DCA to MCA is too slow to be detected within the experimental time frame here
The mass balance of TCA and DCA was almost complete in all suspensions ruling
out any alternative reaction pathways to reductive dechlorination Decarboxylation
of TCA producing chloroform and carbon dioxide requires high temperatures and
is therefore assumed not to take place at the experimental conditions applied here
(Atkins et al 1984) Based on these results we propose that the reductive
dechlorination of TCA by Fe(II)-bearing minerals proceeds via hydrogenolysis
(replacement of halogen by hydrogen) as reported for the transformation of TCA
by zero-valent iron (Hozalski et al 2001) Thus in order to reduce TCA to DCA
2 electrons corresponding to 2 Fe(II) are needed (Figure 51)
106 Chapter 5
Cl3CC
O
O- Cl2HCC
O
O-
2e- H+ Cl-
TCA DCA Figure 51 Proposed reductive transformation pathway of TCA
In the Fe(II)goe and Fe(II)lep systems we detected no TCA transformation in the
absence of either aqueous Fe(II) or pure or calcite-associated goethite and
lepidocrocite These results strongly indicate that reactive Fe(II) species associated
with the goethite and lepidocrocite surfaces are the reductants for TCA in these
systems The Fe(III) phases forming in the mineral suspensions were not
characterised and therefore the reaction stoichiometry cannot be assessed
At initial Fe(II) concentrations in large excess of initial TCA concentration we
found a pseudo 1 order rate law for the degradation of TCA by Fe(II)
[ ] [ ] [ b a TCAFe(II) TCA
sdotsdot=minus kdt
d ]
where a = 1 b = 1 and the observed pseudo 1 order rate constant kobs = k middot [Fe(II)]
At all [Fe(II)]0[TCA]0 ratios studied (6-738) TCA was transformed almost
quantitatively into DCA and the reaction kinetics followed pseudo 1 order kinetics
with respect to TCA (Figure 52) The observed pseudo 1 order rate constants for
the transformation of TCA by the various Fe(II)-Fe(III) mineral systems were
calculated as initial rates (ie max first two half-lives) from linear fits of (time ln
[TCA]t[TCA]0)-plots (Table 51) The amount of chloride produced during
reaction with GR-SO4 was always equivalent to the amount of TCA transformed
into DCA (Figure 52c) This also indicates that no significant further reduction of
DCA took place in GR-SO4 suspensions
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 107
Figure 52 Time course of TCA consumption and DCA and chloride production for a)
Fe(II)Goe ([Fe(II)tot]0 = 095 mM) b) Fe(II)Lep ([Fe(II)tot]0 = 091 mM) c) GR-SO4
([Fe(II)GR]0 = 962 mM) d) GR-CO3 ([Fe(II)tot]0 = 633 mM) and e) Magnetite ([Fe(II)tot]0 = 350
mM) Solid lines represent 1 order kinetic fits whereas symbols and dotted lines represent actual
data = TCA = DCA = Clndash
T
able
51
Exp
erim
enta
l con
ditio
ns a
nd p
seud
o 1
ord
er ra
te c
onst
ants
for t
he re
duct
ive
trans
form
atio
n of
TC
A b
y va
rious
Fe(
II)-
Fe(I
II) c
onta
inin
g m
iner
al sy
stem
s
Syst
em
Susp
ensi
on a
ge
(d)
[Fe(
II)] s
olid
a (m
M)
[Fe(
II)] a
q b
(mM
) [T
CA
] 0 (micro
M)
pHin
itcpH
endd
k obs
e (min
-1)
Surf
ace
area
(m
2 L)
k obs
f
(min
-1m
-2middotL
)
Fe(I
I)aq
1
0
030
434
nd
76
gn
dn
dn
d
Fe(I
I)G
oe
1
002
024
429
nd
78
g1
021
0-47
1 i
143
10-5
Fe(I
I)G
oe
1
013
094
543
77
70
225
10-4
71
i3
161
0-5
Fe(I
I)G
oe
coat
ing
1
023
40
066
484
765
80
g6
401
0-454
0 j
119
10-5
Fe(I
I)G
oe
coat
ing
1
0
150
8048
6n
d7
6 g12
43
10-4
540
j2
301
0-5
Fe(I
I)L
ep
1
0
020
2315
7n
d7
8 g0
751
0-48
0 i
094
10-5
Fe(I
I)L
ep
coat
ing
1
016
30
137
470
765
80
g2
821
0-454
0 j
052
10-5
Fe(I
I)L
ep
coat
ing
1
0
100
8141
7n
d7
7 g8
311
0-454
0 j
154
10-5
Fe3O
41
3
380
1251
38
107
8g
830
10-4
16
k5
311
0-4
Fe3O
477
112
56
556
70
70
153
10-4
52
k2
951
0-5
GR
-CO
31
5
940
3950
37
658
4g
761
10-4
419
l1
821
0-5
GR
-CO
32
7
60
147
88
568
2940
81
0-453
6 l
761
10-5
GR
-CO
332
73
003
563
85
80
490
10-4
515
l0
951
0-5
GR
-CO
314
2
3
530
005
629
nd
87 g
513
10-4
249
l2
061
0-5
GR
-SO
41
5
17-1
217
086
-13
910
5n
dn
d3
601
0-492
6 m
039
10-5
GR
-SO
41
6
22-1
027
093
-14
527
0n
dn
d3
761
0-488
1 m
043
10-5
GR
-SO
41
7
05-1
014
077
-17
950
0n
dn
d3
741
0-4 9
18
m0
411
0-5
GR
-SO
41
5
17-1
051
060
-16
510
00n
dn
d2
891
0-483
7 m
035
10-5
n
d =
not
det
ecte
d a
Ini
tial s
truct
ural
or a
dsor
bed
Fe(I
I) e
stim
ated
as [
Fe(I
I)to
tal]
ndash [F
e(II
) aq]
b In
itial
dis
solv
ed F
e(II
) mea
sure
d c
Sus
pens
ion
pH p
rior t
o TC
A a
dditi
on d
Sus
pens
ion
pH a
t rea
ctio
n
te
rmin
atio
n e
Pse
udo
1 o
rder
rate
con
stan
ts fo
r the
con
sum
ptio
n of
TC
A c
alcu
late
d fr
om in
itial
rate
s (m
ax f
irst t
wo
half-
lives
) f
Surf
ace
area
-nor
mal
ised
pse
udo
1 o
rder
rate
con
stan
ts g
pH
con
trol
th
roug
h pu
re C
aCO
3 and
05
C
O2(g
) h
pH
con
trol t
hrou
gh F
e(II
I) o
xide
-coa
ted
calc
ite a
nd 0
5
CO
2(g)
i Es
timat
ed u
sing
the
SSA
of t
he F
e(II
I) o
xide
app
lied
j E
stim
ated
usi
ng th
e SS
A o
f cal
cite
~1
m2 g
k E
stim
ated
ass
umin
g SS
A =
4 m
2 g (S
chw
ertm
ann
amp C
orne
ll 1
991)
frac12middot[
Fe(I
I) sol
id] 0middot
232
gmiddotm
ol-1
middot4 m
2 middotg-1
l E
stim
ated
ass
umin
g SS
A =
47
m2 g
(Will
iam
s amp S
cher
er 2
001)
frac14middot[F
e(II
) GR] 0middot
600
gmiddotm
ol-1
middot47
m2 middotg
-1 m
Est
imat
ed a
s in l
but u
sing
SSA
= 7
12
m2 middotg
-1 (C
hapt
er 4
thi
s wor
k)
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 109
532 Comparing rate constants obtained for the various Fe(II)-Fe(III) mineral
systems
Data for the systems containing iron oxide coated calcite were very similar to the
data obtained for the pure iron oxides (not shown in Figure 53) Since no SSA was
determined for magnetite in this study a SSA of 4 m2g was assumed
(Schwertmann amp Cornell 1991) However it should be noted that the magnetite
synthesized by Schwertmann and Cornell (1991) was prepared differently (ie
oxidation of Fe(II) by nitrate in a heated alkaline solution) from the magnetite
applied in this study The surface area-normalised pseudo 1 order kobs values
obtained for GR-CO3 GR-SO4 Fe(II)goethite and Fe(II)lepidocrocite were all
within the same order of magnitude (Figure 53a)
Figure 53 Average surface area-normalised pseudo 1 order rate constants for the degradation of
a) TCA (this work) b) hexachloroethane (Elsner et al 2004a) and c) carbon tetrachloride
(Amonette et al 2000 Pecher et al 2002 OrsquoLoughlin et al 2003c Elsner et al 2004b) by
GR-SO4 GR-CO3 (suspension age 1 d) Fe3O4 Fe(II)α-FeOOH and Fe(II)γ-FeOOH
Experimental conditions applied in this work [Fe(II)tot]0 = 025-107 mM in the goethite and
lepidocrocite suspensions [Fe(II)tot]0 = 025-116 mM in the GR-SO4 and GR-CO3 suspensions
pH 70-86 71-926 m2 mineral surface areaL Experimental conditions applied by Elsner et al
1 mM aqueous Fe(II) 25 m2 mineral surface areaL Experimental conditions applied in
references employed in c) [Fe(II)tot]0 = 1-83 mM 25-275 m2 mineral surface areaL GR-SO4 =
110 Chapter 5
sulfate green rust GR-CO3 = carbonate green rust Fe3O4 = magnetite α-FeOOH = goethite γ-
FeOOH = lepidocrocite
When comparing the rate constants for the Fe(II)-Fe(III) mineral systems found for
reduction of TCA in this study (Figure 2a) mixed valent Fe(II)-Fe(III) minerals
such as green rusts and magnetite containing structural Fe(II) within the mineral
lattice do not seem to be significantly more reactive than Fe(II)-Fe(III) mineral
systems containing Fe(II) associated with pure Fe(III) oxides Unlike most other
iron oxides GRs contain not only external Fe(II) reactive sites at the surface but
also internal sites in the space between consecutive Fe(II)-Fe(III) hydroxide layers
The GR interlayer thickness is a function of both the size and the charge of the
interlayer anion For solutes the Fe(II) within the GR hydroxide layer is accessible
at the outside basal planes and at the edges as well as through the interlayer under
certain conditions (see Figure 42 Chapter 4 this work) Due to electrostatic
forces oxidants holding different charge properties (anions cations neutral
molecules) may exhibit different affinities for the various reactive Fe(II) sites
present in GR As the reactive sites are located inat the Fe(II)-Fe(III) hydroxide
layers the rate of reaction depends on the hydroxide layer area which can be
accessed by the oxidant If the oxidant is able to exchange with the interlayer
anion reaction can take place both at outer and inner surfaces of the GR particles
and in total more reactive sites are available for the reaction Thus oxidant size
and charge primarily control its access to the internal sites in GRs XRD
characterization of the GR-SO4 crystals during reaction with TCA demonstrated
that the GR-SO4 interlayer spacing did not vary during reaction (Table 52) This
may indicate that TCA did not enter the GR-SO4 interlayers
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 111 Table 52 Diffraction angle d-spacing and width at half peak height (Wfrac12) for the 001 GR-SO4
diffraction peak as a function of time during reaction with TCA ([Fe(II)GR]0 = 4 mM [TCA]0 = 1
mM)
Time (min) Angle (deg2θ)
d001-spacing(nm)
Wfrac12(degθ)
0 9483 10821 0273
10 9494 10809 0287
215 9522 10777 0263
330 9550 10745 0273
510 9524 10775 0277
855 9509 10791 0253
1160 9467 10839 0268
We roughly estimated the molecular size of TCA by summing the covalent radii of
the individual atoms (see Supporting Information 74) When comparing the
molecular size of TCA with the GR-SO4 interlayer spacing of 061 nm it can be
concluded that only when the C-C bond is oriented perpendicular to the interlayer
plane does the size of TCA exceed the GR-SO4 interlayer spacing In contrast the
size of TCA exceeds the GR-CO3 interlayer spacing (026 nm) regardless of its
orientation Hence if TCA was intercalated in the GR-CO3 interlayer we would
expect the interlayer spacing to expand The same holds for intercalation of a
vertically oriented TCA in the GR-SO4 interlayer Supposedly both the low charge
and the size of TCA impeded its access to the GR-SO4 and GR-CO3 interlayers ie
the divalent sulphate and carbonate in the GR interlayers did not readily exchange
with the monovalent TCA since GR interlayers generally have a higher affinity for
divalent anions than for monovalent anions (Miyata 1983) Thus TCA did neither
access nor react with internal Fe(II) reactive sites in GR-SO4 which means that the
reaction between TCA and GR-SO4 took place at the external reactive Fe(II) sites
solely It is reasonable to assume that the same holds for the reaction between TCA
and GR-CO3 No significant aging effects eg rate constants varying as a function
of GR age were observed within 142 days (see Table 51) However the SSAs of
112 Chapter 5
the GR suspensions holding ages up to 142 days were not measured but estimated
assuming that the GR SSA did not decrease within the time frame
According to the Scherrer formula the width at half peak height (Wfrac12) of a
diffraction peak is inversely proportional to the average crystal dimension
perpendicular to the given crystal plane (Klug amp Alexander 1974) The average
GR-SO4 particle thickness perpendicular to the basal plane (Wfrac12 Table 52) was
constant during reaction implying that TCA reacts only at the edges and not at the
basal planes Assuming that the platy hexagonal GR-SO4 and GR-CO3 crystals
hold an average width of 1 microm and an average particle thickness of 35 nm (Hansen
and Koch 1998) the ratio of edge surface area to outer surface area is AedgeAouter ~
130 for GR-SO4 and 121 for GR-CO3 (see Supporting Information 73) This
means that only 3 of the outer surface area in GR-SO4 and 5 of the outer
surface area in GR-CO3 are available at the edges Assuming that TCA reacts at the
edges only and if employing the AedgeAouter in the estimation of the rate constants
the surface area-normalised pseudo 1 order rate constants for GR-SO4 and GR-
CO3 would be 20-30 times higher than the rate constants depicted in Figure 53a
Thus the reactivity of GRs normalised to their reactive surface area is much higher
than the reactivity normalised to their total surface area
533 Comparing with rate constants obtained for other chlorinated aliphatic
compounds
Though care must be taken when comparing kinetic parameters obtained at
different experimental conditions (eg pH [Fe(II)]0[TCA]0 ratios surface area to
volume ratios etc) it is interesting to compare our results to those reported for
hexachloroethane (Figure 53b data from Elsner et al 2004a) The reductive
transformation of hexachloroethane was investigated for various Fe(II)-bearing
minerals including Fe(II)goethite Fe(II)lepidocrocite and GR-SO4 in the presence
of 1 mM dissolved Fe(II) and 25 m2 mineral surface areaL at pH 72 except for the
GR-SO4 suspensions in which the dissolved Fe(II) concentrations were slightly
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 113
higher and pH = 8 The pseudo 1 order rate constants reported for
hexachloroethane are in the range 18middot10-4 ndash 75middot10-3 h-1middotm-2middotL (Elsner et al 2004a)
When comparing Figure 53a with Figure 53b it can be seen that the differences
in intrinsic reactivity of the Fe(II)-bearing mineral systems are more pronounced
for hexachloroethane than for TCA
Caution should also be advised to the different reaction mechanisms by which
hexachloroethane and TCA react The transfer of a single electron and the
formation of an alkyl radical upon removal of a chlorine atom constitute the first
and in most cases the rate-limiting step in the reduction of chlorinated aliphatic
compounds (Vogel et al 1987) Depending on the chemical structure of the
chlorinated aliphatic compound the resulting free alkyl radical may undergo
hydrogenolysis chloroelimination or dimerizationcoupling In the case of TCA
the free dichloroacetate radical most likely undergoes hydrogenolysis The almost
quantitative transformation of TCA to DCA confirms that hydrogenolysis is the
prevalent reaction mechanism in our mineral systems The pentachloroethyl radical
formed from hexachloroethane may undergo hydrogenolysis (producing
pentachloroethane) or dichloroelimination (producing tetrachloroethene) Elsner et
al (2004a) found that hexachloroethane was transformed quantitatively into
tetrachloroethylene for all minerals which strongly indicates that
dichloroelimination was the dominating reaction mechanism Another
polychlorinated aliphatic compound transformed mainly by hydrogenolysis under
reducing conditions is carbon tetrachloride Several studies have investigated the
reductive dechlorination of carbon tetrachloride by various Fe(II)-bearing minerals
including Fe(II)goethite and GR-SO4 and reported pseudo 1 order rate constants
in the order 152middot10-4 ndash 640middot10-4 h-1middotm-2middotL for Fe(II)goethite and 864middot10-4 h-1middotm-2middotL
for GR-SO4 (Amonette et al 2000 Pecher et al 2002 OLoughlin et al 2003c
Elsner et al 2004b) When comparing Figure 53a with Figure 53c it can be seen
that the range of magnitude of the rate constants and the differences in intrinsic
114 Chapter 5
reactivity of the Fe(II)-bearing mineral systems are similar for carbon tetrachloride
and TCA
534 Factors controlling the reactivity of surface-bound Fe(II)
The reactivity of an oxidant towards Fe(II) surface species cannot be predicted
from the reduction potentials of the redox couple alone In heterogeneous systems
processes such as mass transfer and adsorptiondesorption may have a rate-limiting
effect on the overall reaction rate If the adsorption follows a saturation-type
sorption isotherm (eg Langmuir) the sorbate (oxidant) concentration at the
surface will vary non-linearly with the total amount of oxidant added This
dependence will have to be taken into account when establishing rate laws for the
heterogeneous reactions and when testing the hypothesis that the reaction rates
depend on the sorbed concentration of the oxidants pH has a strong impact on the
sorption and thereby on the availability of ionizable oxidants At the pH values
applied here the chloroacetates are fully dissociated (pKa (TCA) = 066 pKa
(DCA) = 135 pKa (MCA) = 287) However we found the sorption of TCA to be
negligible in suspensions of pure calcite goethitecalcite and lepidocrocitecalcite
at pH 76-77 Moreover the mass balance of TCA and DCA was almost complete
in all suspensions and therefore loss of TCA or DCA due to adsorption at mineral
surfaces or incorporation in the GR anion interlayers can be ruled out Calcite has a
much lower adsorption capacity than most iron oxides hence we anticipate that
goethite and lepidocrocite control the adsorption of TCA and DCA in both the pure
FeOOH and the FeOOHcalcite suspensions This was supported by our
experimental results demonstrating that the presence of a calcite surface - either
pure or as a support for goethite and lepidocrocite coatings - did not exert any
noticeable effect on the reaction rates (see Table 51) In addition the surface area-
normalised rate constants for mineral systems containing goethite or lepidocrocite
in pure form and mineral systems containing goethite or lepidocrocite as coatings
on calcite were very similar In heterogeneous reactions mass transfer in bulk
solution becomes the rate-limiting step when the surface reaction is much faster
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 115
than the diffusion of the reacting species to the reactive surface However at the
low rate constants obtained here the reaction of TCA with the Fe(II)-bearing
minerals is not likely to be mass transfer limited (see Supporting Information 72)
One very important factor affecting heterogeneous redox reactions is pH which
influences the speciation of the complexes in solution and at mineral surfaces as
well as the stability of the more soluble Fe(II)-containing minerals such as GRs In
contrast to aqueous Fe(II) complexes it is not possible to predict the reactivity of
Fe(II) surface species as their reduction potentials are unknown In the absence of
specifically adsorbing solutes other than H+ the surface charge of the Fe(III)
oxides goethite and lepidocrocite is determined by the surface densities of the
charged surface species equivFeOH2+ and equivFeOndash whereas the surface charge of calcite
is determined by the density of the surface species equivCO3ndash equivCaOH2
+ and equivCaOndash
(Stumm 1992 Van Cappellen et al 1993) The point of zero charge (pHpzc) of
pure calcite is in the pH range 7-11 and depends on the partial pressure of carbon
dioxide pCO2 The higher the pCO2 the lower the pHpzc At the experimental
conditions applied here (05 = 0005 atm CO2(g)) the pHpzc = 82 for calcite
(Table 53) As only 10-11 mg Fe(III) of goethite and lepidocrocite was coated
onto calcite we assumed a pHpzc of 82 for the goethite and lepidocrocite coated
calcite particles as well The pHpzc values for green rusts are unknown
116 Chapter 5
Table 53 Specific surface areas and point of zero charge of the various iron minerals in pure form as well as goethite and lepidocrocite coated onto calcite
Mineral Structural formula SSA (m2g) pHpzc
GR-SO4 FeII4FeIII
2(OH)12SO4middot3H2O 71 a -
GR-CO3 FeII4FeIII
2(OH)12CO3middot3H2O 47 b -
Magnetite Fe3O4 - 69 e
Goethite α-FeOOH 16 c 85 f
Lepidocrocite γ-FeOOH 18 c 73 e
Calcite CaCO3 le 1 d 82 g
Goe coating - le 1 d 82 h
Lep coating - le 1 d 82 h
a Chapter 4 this work b Williams amp Scherer 2001 c Product information by Bayer d The SSA of calcite was
quantified to le 1 m2g The detection limit of our BET method was 1 m2g e Charlet et al 1998a f Liger et al
1999 g Van Cappellen et al 1993 h Same as for calcite
The surface hydroxyl groups on iron oxides may be both singly (equivFe-OH) doubly
(equivFe2-OH) triply (equivFe3-OH) and geminally (equivFe-(OH)2) coordinated (Cornell amp
Schwertmann 1996 Stumm 1992) The differently coordinated surface hydroxyl
groups are not equally reactive Adsorption reactions involve only singly
coordinated surface groups and therefore only this kind of hydroxyl groups on iron
oxides will be considered here (Cornell amp Schwertmann 1996) Hence the
predominant surface sites available for adsorption in pure suspensions of Fe(III)
oxides are equivFeOH0 equivFeOH2+ and equivFeOndash In the presence of dissolved Fe(II)
equivFeIIIOFeIIOH0 equivFeIIIOFeIIOndash and equivFeIIIOFeII+ constitute the main reactive sites at
the Fe(III) oxide surfaces (Liger et a 1999) Assuming that Fe2+ and other cationic
Fe(II) species are the dominating adsorbates on the mineral surfaces in our
experiments we expect the actual pHpzc to be higher than the pHpzc of the pure
oxides listed in Table 53 Hence at pHlt82 where most of our experiments were
conducted all the mineral surfaces presumably carry net positive charges
At pH 70 where Fe2+ is still the predominant Fe(II) species in solution (~50) we
expect that equivFeIIIOFeIIOH equivFeIIIOFeIIOndash and equivFeIIIOFeII+ constitute the main
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 117
reactive sites at the Fe(III) oxide surfaces as suggested by Liger et a 1999 As pH
increases from 70 to 87 the Fe(II) carbonate complexes become increasingly
important in solution at the expense of the Fe2+ FeCl+ FeSO40 and FeOH+ species
(King 1998) Fe(II) carbonate complexes do not bind at the oxide surface as
readily as the aquo or hydroxo complexes of Fe(II) but carbonate itself sorbs
readily to Fe(III) oxide surfaces through which the Fe(III) oxide surface is coated
by inner-sphere monodentate equivFeIIIOCOOH0 surface complexes (Villalobos amp
Leckie 2000 amp 2001) The presence of carbonate shifted the sorption edge for the
Fe(II) adsorption on goethite from pH 58 to 78 and the authors hypothesized this
to be a result of the formation of aqueous and surface Fe(II)-carbonate complexes
and to competition between carbonate and Fe(II) for Fe(III) oxide surface sites
(Vikesland amp Valentine 2002) Similarly monodentate surface complexes like
equivFeIIICl0 and equivFeIIIOSO3ndash as well as ternary monodentate surface complexes like
equivFeIIIOFeIICl0 and equivFeIIIOFeIIOSO3ndash and ternary bidentate surface complexes such
as (equivFeIIIO)2FeIIOSO3 may form at Fe(III) oxide surfaces when Fe(II) chloride and
sulfate are present in solution (Ostergren et al 2000 Kim et al 2004) However
the effects of anionic ligands such as chloride and sulfate on Fe(II) adsorption at
Fe(III) oxide surfaces and the reactivity of Fe(II) carbonate chloride and sulfate
surface sites are still unknown and need to be evaluated (see Supporting
Information 75) Thus we do not know whether chloride and sulfate decrease or
increase the Fe(II) sorption in our mineral systems We can only report that we did
not detect any significant differences in the rate of TCA transformation between
the mineral suspensions containing carbonate chloride and sulfate respectively
Hence we anticipate that equivFeIIIOFeIIOH equivFeIIIOFeIIOndash and equivFeIIIOFeII+ constitute
the main reactive sites at the Fe(III) oxide surfaces within the whole pH range 70-
87 This might also explain why we did not detect any obvious systematic pH
effect in the Fe(II)-Fe(III)-systems (see Table 51) In the case of
hexachloroethane the reactivity order GR-SO4gtgoethitegtmagnetitegtlepidocrocite
may be rationalized by the variations in surface site densities and total amount of
118 Chapter 5
Fe(II) sorbed on the iron minerals (see Supporting Information 75) as well as the
different speciations and reactivities of the Fe(II) surface sites on the iron minerals
535 Comparison with biotic and other abiotic systems
Only one report on abiotic transformation of TCA is found in the literature and the
study demonstrates the reductive dechlorination of TCA to MCA by Fe(0)
(Hozalski et al 2001) The authors reported a pseudo 1 order rate constant of
60middot10-4 min-1middotm-2middotL for the transformation of TCA to DCA and a pseudo 1 order
rate constant of 225middot10-4 min-1middotm-2middotL for the transformation of DCA to MCA at
[Fe(0)]0 = 025 M [TCA]0 = 100-200 microM and pH 36-62 The rate constant for
TCA reduction by Fe(0) is 10-300 times faster than the rate constants reported for
the Fe(II)-bearing mineral systems here
There is abundant evidence that soil microorganisms and fungi can dechlorinate
TCA but only little is known about the bacteria and processes involved in the
biodegradation of TCA Biotransformation of TCA has been found at both oxic
and anoxic conditions Most of the microorganisms isolated grow feebly on TCA
as a sole source of carbon (Foy 1975 Weightman et al 1992 De Wever et al
2000) Only one bacterium capable of growing on TCA as the sole carbon and
energy source has been characterized (Yu amp Welander 1995) In addition
anaerobic bacteria coupling co-metabolic growth to reductive dechlorination of
TCA have been isolated (Weightman et al 1992 De Wever et al 2000) The
inability to grow on the less chlorinated acids DCA and MCA is a notable feature
of both the aerobic and anaerobic bacteria Complete transformation of TCA to
methane and carbon dioxide has only been found when abiotic and biotic processes
were combined (Egli et al 1989) The abiotic transformation of TCA to DCA
occurred spontaneously in the presence of sterile activated charcoal whereas the
DCA formed was further degraded to methane and carbon dioxide by a mixed
culture of methanogenic bacteria However the abiotic reductant(s) responsible for
the transformation of TCA to DCA was not reported (Egli et al 1989)
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 119
The rate constants obtained in this work suggest that the Fe(II)-bearing mineral
systems may be important reductants of TCA in natural suboxic environments In
natural iron-rich soils holding specific surface areas of 22 m2g (Kenneke amp
Weber 2003) average bulk densities of 265 gcm3 and porosities of 25 and
containing 2 iron oxides a rough estimation of the half-life of TCA amounts to
47 minutes when applying the average surface area-normalised rate constant
obtained for all the Fe(II)-Fe(III) mineral systems in this work (1middot10-3 h-1middotm-2middotL)
This estimation is based on the assumption that enough reactive Fe(II) is available
in these soils The natural iron-reducing sediment investigated by Kenneke and
Weber (2003) contained 80 microM Fe(II) in the soil solution and 315 micromole Fe(II) per
g sediment At such low Fe(II) concentrations the overall rate of abiotic
transformation of TCA in natural soils and sediments is most likely limited by the
regeneration of reactive Fe(II) Hence the continuous regeneration of reactive
Fe(II) surface sites by adsorption of abiotically or microbially produced Fe(II) may
further the long-term abiotic transformation of TCA in such environments
54 Conclusions
This work demonstrates that various Fe(II)-Fe(III) minerals systems including GR-
SO4 GR-CO3 magnetite Fe(II)goethite and Fe(II)lepidocrocite readily transform
TCA to DCA Dichloroacetate was not further reduced to MCA or acetate by any
of the Fe(II)-bearing minerals The surface area-normalised pseudo 1 order rate
constants obtained for the reductive transformation of TCA by the various Fe(II)-
bearing minerals did not differ significantly from each other The results obtained
in this work infer that under natural geochemical conditions Fe(II)-bearing mineral
systems may play an important role in the overall transformation of TCA Thus
not only microbial degradation but also abiotic reductive transformation of TCA by
Fe(II)-bearing minerals may govern the fate of TCA in natural subsurface
environments This holds both for natural systems like iron-rich suboxic soils and
sediments as well as engineered systems like permeable reactive barriers of zero-
120 Chapter 5
valent iron implemented for on-site remediation where both Fe(0) and solid or
surface-bound Fe(II) corrosion intermediates may transform TCA
Acknowledgments
We would like to thank Susanne Guldberg for performing the experimental work comprising
GR-SO4
References
Ahlers J Regelmann J Riedhammer C (2003) Environmental risk assessment of airborne trichloroacetic acid - a contribution to the discussion of the significance of anthropogenic and natural sources Chemosphere 52 531-537 Amonette JE Workman DJ Kennedy DW Fruchter JS Gorby YA (2000) Dechlorination of carbon tetrachloride by Fe(II) associated with goethite Environmental Science and Technology 34 4606-4613 Atkins PJ Gold V Marsh R (1984) The decarboxylation of trichloroacetic acid and the reactions of the trichloromethyl anion with 135-trinitrobenzene and with hydrogen ions kinetic measurements in dimethyl sulphoxide solution Journal of the Chemical Society Perkin Transactions 2 7 1239-1245 Berg M Muumlller SR Muumlhlemann J Wiedmer A Schwarzenbach RP (2000) Concentrations and mass fluxes of chloroacetic acids and trifluoroacetic acid in rain and natural waters in Switzerland Environmental Science and Technology 34 2675-2683 Brunauer S Emmett PH Teller E (1938) Adsorption of gases in multimolecular layers Journal of American Chemical Society 60 309-319 Butler EC Hayes KF (1998) Effects of solution composition and pH on the reductive dechlorination of hexachloroethane by iron sulfide Environmental Science and Technology 32 1276-1284 Butler EC Hayes KF (1999) Kinetics of the transformation of trichloroethylene and tetrachloroethylene by iron sulfide Environmental Science and Technology 33 2021-2027 Charlet L Liger E Gerasimo P (1998a) Decontamination of TCE- and U-rich water by granular iron Role of sorbed Fe(II) Journal of Environmental Engineering 124 25-30 Charlet L Silvester E Liger E (1998b) N-compound reduction and actinide immobilisation in surficial fluids by Fe(II) the surface FeIIIFeIIOH0 species as major reductant Chemical Geology 151 85-93 Cheregi M Danet AF (1997) Flow injection determination of chloride ions with spectrophotometric detection Analytical Letters 30 2847-2858 Cornell RM Schwertmann U (1996) The iron oxides Structure properties reactions occurrence and uses VCH Verlagsgesellschaft mbH Weinheim
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 121 Cui D Eriksen TE (1996) Reduction of pertechnetate by ferrous iron in solution influence of sorbed and precipitated Fe(II) Environmental Science and Technology 30 2259-2262
Egli C Thuumler M Suter D Cook AM Leisinger T (1989) Monochloro- and dichloroacetic acids as carbon and energy sources for a stable methanogenic mixed culture Archives of Microbiology 152 218-223 Elsner M Haderlein SB Schwarzenbach RP (2004a) Reactivity of Fe(II)-bearing minerals towards reductive transformation of organic contaminants Environmental Science and Technology 38 799-807 Elsner M Haderlein SB Kellerhals T Luzi S Zwank L Angst W Schwarzenbach RP (2004b) Mechanisms and products of surface-mediated reductive dehalogenation of carbon tetrachloride by Fe(II) on goethite Environmental Science and Technology 38 2058-2066 Erbs M Hansen HCB Olsen CE (1999) Reductive dechlorination of carbon tetrachloride using iron(II)iron(III)-hydroxide-sulphate (green rust) Environmental Science and Technology 33 307-311 Euro Chlor (2001) Trichloroacetic acid in the environment a dossier Euro Chlor Brussels and the European Chlorinated Solvent Association Fadrus H Maly J (1975) Suppression of iron(III) interference in the determination of iron(II) in water by the 110-phenanthroline method The Analyst 100 549-554 Fahimi IJ Keppler F Schoumller HF (2003) Formation of chloroacetic acids from soil humic acid and phenolic moieties Chemosphere 52 513-520 Foy CL (1975) The chlorinated aliphatic acids In Herbicides Chemistry degradation and mode of action Kearney PC Kaufman DD (eds) Marcel Dekker Inc 399-452 Gander JW Parkin GF Scherer MM (2002) Kinetics of 111-trichloroethane transformation by iron sulfide and a methanogenic consortium Environmental Science and Technology 36 4540-4546 Haiber G Jacob G Niedan V Nkusi G Schoumller HF (1996) The occurrence of trichloroacetic acid (TCAA) ndash indications of a natural production Chemosphere 33 839-849
Hansen HCB (1989) Composition stabilization and light absorption of Fe(II)Fe(III) hydroxycarbonate (green rust) Clay Minerals 24 663-669 Hansen HCB Guldberg S Erbs M Koch CB (2001) Kinetics of nitrate reduction by green rusts ndash effects of interlayer anion and Fe(II)Fe(III) ratio Applied Clay Science 18 81-91 Hansen HCB Koch CB (1997) A comparison of nitrate reduction by carbonate and sulphate forms of green rust Kodama H Mermut A R Torrance J K (eds) Proceedings of the 11th International Clay Conference Ottawa Canada Clays for our future 11 295-302 Hoekstra EJ de Leer EWB Brinkman UATh (1999a) Mass balance of trichloroacetic acid in the soil top layer Chemosphere 38 551-563 Hoekstra EJ de Leer EWB Brinkman UATh (1999b) Findings supporting the natural
122 Chapter 5
formation of trichloroacetic acid in soil Chemosphere 38 2875-2883 Hofstetter TB Heijman CG Haderlein SB Holliger HC Schwarzenbach RP (1999) Complete reduction of TNT and other (poly)nitroaromatic compounds under iron-reducing subsurface conditions Environmental Science and Technology 33 1479-1487 Hofstetter TB Schwarzenbach RP Haderlein SB (2003) Reactivity of Fe(II) species associated with clay minerals Environmental Science and Technology 37 519-528 Hozalski RM Zhang L Arnold WA (2001) Reduction of haloacetic acids by Fe0 Implications for treatment and fate Environmental Science and Technology 35 2258-2263 Husain S Narsimha R Alvi SN Rao RN (1992) Monitoring the effluents of the trichloroacetic acid process by high-performance liquid chromatography Journal of Chromatography 600 316-319 Hwang I Batchelor B (2000) Reductive dechlorination of tetrachloroethylene by Fe(II) in cement slurries Environmental Science and Technology 34 5017-5022 Jordan A Frank H (1999) Trifluoroacetate in the environment Evidence for sources other than HFCHCFCs Environmental Science and Technology 33 522-527 Kenneke JF Weber EJ (2003) Reductive dehalogenation of halomethanes in iron- and sulfate-reducing sediments 1 reactivity pattern analysis Environmental Science and Technology 37 713-720 Kim CS Rytuba JJ Brown Jr GE (2004) EXAFS study of mercury(II) sorption to Fe- and Al-(hydr)oxides II Effects of chloride and sulphate Journal of Colloid and Interface Science 270 9-20 King DW (1998) Role of carbonate speciation on the oxidation rate of Fe(II) in aquatic systems Environmental Science and Technology 32 2997-3003 Klausen J Troumlber SP Haderlein SB Schwarzenbach RP (1995) Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions Environmental Science and Technology 29 2396-2404 Klug PH Alexander LE (1974) X-ray diffraction procedures John Wiley amp Sons Inc Koch CB Hansen HCB (1997) Reduction of nitrate to ammonium by sulphate green rust Advances in GeoEcology 30 373-393 Kuumlhn R Pattard M (1990) Results of the harmful effects of water pollutants to green algae (Scenedesmus subspicatus) in the cell multiplication inhibition test Water Research 24 31-38 Lee W Batchelor B (2002a) Abiotic reductive dechlorination of chlorinated ethylenes by iron- bearing soil minerals 1 Pyrite and magnetite Environmental Science and Technology 36 5147- 5154 Lee W Batchelor B (2002b) Abiotic reductive dechlorination of chlorinated ethylenes by iron-bearing soil minerals 2 Green rust Environmental Science and Technology 36 5348- 5354
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 123 Liger E Charlet L Van Cappellen P (1999) Surface catalysis of uranium (VI) reduction by iron(II) Geochimica et Cosmochimica Acta 63 2939-2955 Lignell R Heinonen-Tanski H Uusi-Rauva A (1984) Degradation of trichloroacetic acid (TCA) in soil Acta Agriculturae Scandinavia 34 3-8 Loyaux-Lawniczak S Refait Ph Lecomte P Ehrhardt J Geacutenin JMR (1999) The reduction of chromate ions by Fe(II) layered hydroxides Hydrology and Earth System Sciences 3 593-599 Luther III GW (1990) The Frontier-Molecular-Orbital theory approach in geochemical processes in W Stumm Ed Aquatic Chemical kinetics John Wiley and Sons New York pp 173-198 McCulloch A (2002) Trichloroacetic acid in the environment Chemosphere 47 667-686 Miyata S (1983) Anion-exchange properties of hydrotalcite-like compounds Clays and Clay Minerals 31 305-311 Muumlller SR Zweifel H-R Kinnison DJ Jacobsen JA Meier MA Ulrich MM Schwarzenbach RP (1996) Occurrence sources and fate of trichloroacetic acid in Swiss lakes Environmental Toxicology and Chemistry 15 1470-1478 OLoughlin EJ Burris DR (2003) Reduction of halogenated ethanes by green rust Environmental Toxicology and Chemistry 23 41-48 OLoughlin EJ Kelly SD Cook RE Csencsits R Kemner KM (2003a) Reduction of uranium(VI) by mixed iron(II)iron(III) hydroxide (green rust) Formation of UO2 nanoparticles Environmental Science and Technology 37 721-727 OLoughlin EJ Kelly SD Kemner KM Csencsits R Cook RE (2003b) Reduction of AgI AuIII CuII and HgII by FeIIFeIII hydroxysulfate green rust Chemosphere 53 437-446 OLoughlin EJ Kemner KM Burris DR (2003c) Effects of AgI AuIII and CuII on the reductive dechlorination of carbon tetrachloride by green rust Environmental Science and Technology 37 2905-2912 Ostergren JD Brown Jr GE Parks GA Persson P (2000) Inorganic ligand effects on Pb(II) sorption to goethite (α-FeOOH) II Sulfate Journal of Colloid and Interface Science 225 483-493 Pecher K Haderlein SB Schwarzenbach RP (2002) Reduction of polyhalogenated methanes by surface-bound Fe(II) in aqueous suspensions of iron oxides Environmental Science and Technology 36 1734-1741 Reimann S Grob K Frank H (1996) Chloroacetic acids in rainwater Environmental Science and Technology 30 2340-2344 Ruumlgge K Hofstetter TB Haderlein SB Bjerg PL Knudsen S Zraurig C Mosbaeligk H Christensen TH (1998) Characterization of predominant reductants in an anaerobic leachate- affected aquifer by nitroaromatic probe compounds Environmental Science and Technology 32
124 Chapter 5
23-31 Sarzanini C Bruzzoniti MC Mentasti E (1999) Preconcentration and separation of haloacetic acids by ion chromatography Journal of Chromatography A850 197-211 Schwertmann U Cornell RM (1991) Iron oxides in the laboratory Preparation and characterization VCH Verlagsgesellschaft mbH Weinheim Scott BF Mactavish DC Spencer C Strachan WMJ Muir DCG (2000) Haloacetic acids in Canadian lake waters and precipitation Environmental Science and Technology 34 4266-4272 Scott BF Spencer C Marvin CH Mactavish DC Muir DCG (2002) Distribution of haloacetic acids in the water columns of the Laurentian Great Lakes and Lake Malawi Environmental Science and Technology 36 1893-1898 Strathmann TJ Stone AT (2003) Mineral surface catalysis of reactions between FeII and oxime carbamate pesticides Geochimica et Cosmochimica Acta 67 2775-2791 Stumm W (1992) Chemistry of the solid-water interface John Wiley amp Sons Inc Van Cappellen P Charlet L Stumm W Wersin P (1993) A surface complexation model of the carbonate mineral-aqueous solution interface Geochimica et Cosmochimica Acta 57 3505- 3518 Vikesland PJ Valentine RL (2002) Iron oxide surface-catalyzed oxidation of ferrous iron by monochloramine implications of oxide type and carbonate on reactivity Environmental Science and Technology 36 512-519 Villalobos M Leckie JO (2000) Carbonate adsorption on goethite under closed and open CO2 conditions Geochimica et Cosmochimica Acta 64 3787-3802 Villalobos M Leckie JO (2001) Surface complexation modeling and FTIR study of carbonate adsorption to goethite Journal of Colloid and Interface Science 235 15-32 Vogel TM Criddle CS McCarty PL (1987) Transformations of halogenated aliphatic compounds Environmental Science and Technology 21 722-736 Weightman AL Weightman AJ Slater JH (1992) Microbial dehalogenation of trichloroacetic acid World Journal of Microbiology and Biotechnology 8 512-518 De Wever H Cole JR Fettig MR Hogan DA Tiedje JM (2000) Reductive dehalogenation of trichloroacetic acid by Trichlorobacter thiogenes gen nov spnov Applied and Environmental Microbiology 66 2297-2301 Williams AGB Scherer MM (2001) Kinetics of chromate reduction by carbonate green rust Environmental Science and Technology 35 3488-3494 Yu P Welander T (1995) Growth of an aerobic bacterium with trichloroacetic acid as the sole source of energy and carbon Applied Microbiology and Biotechnology 42 769-774
Conclusions and Outlook 125
6 Conclusions and Outlook The work presented in this dissertation adds to the understanding of how Fe(II)-
bearing minerals like green rusts (GRs) vivianite (Fe2(PO4)2sdot8H2O) magnetite
(Fe3O4) and Fe(II) associated with goethite and lepidocrocite may form and react in
nature In order to elucidate the role of bacteria in the formation of GRs in natural
soils and sediments we studied the iron mineral phases forming as a result of the
activity of iron-respiring bacteria In chapter 2 the Fe-containing products formed
by anaerobic autotrophic denitrifying Fe(II)-oxidizing bacteria (FeOB) were
examined The culture medium applied contained high levels of bicarbonate and
phosphate and is typically used in this kind of studies as it provides excellent
conditions for the nitrate-reducing FeOB Fe(II) was present initially as a whitish
solid Fe(II) hydroxy phosphate (vivianite) and as soluble Fe(II) The results
obtained demonstrate that the denitrifying FeOB produce poorly crystalline
goethite via a greenish Fe(III)-enriched vivianite intermediate Moumlssbauer
spectroscopic analyses provided no evidence of green rust formation At low
phosphate concentrations where vivianite does not control the Fe(II) activity it is
reasonable to assume that siderite (FeCO3) precipitates initially and that carbonate
GR phases may form during biooxidation At low bicarbonate concentrations we
would expect Fe(II) sulfate or chloride species to dominate initially (depending on
the Fe(II) source applied) and sulfate GR or chloride GR to form during
biooxidation In chapter 3 we investigated the Fe-containing products formed
during reduction of common Fe(III) oxides by the anaerobic dissimilatory Fe(III)-
reducing microorganism Shewanella algae BrY S algae BrY reduced substantial
amounts of the initial Fe(III) and green and blackish mineral phases were produced
within 1-2 weeks after inoculation Moumlssbauer spectroscopic analyses showed that
the green and black precipitates consisted of green rust and vivianite
We studied the reactivity of synthetic GRs towards reducible organic pollutants in
order to asses the potential significance of GR phases for the fate of such
126 Chapter 6
compounds To this end we used nitroaromatic compounds (NACs) and
chlorinated acetates as suitable model compounds for studying redox reactions
potentially relevant in the environment In chapter 4 we investigated the relative
reactivity of outer and inner Fe(II) reactive sites in synthetic sulfate green rust
(GR-SO4) by using a series of structurally closely related compounds with different
charge properties as ldquoreactive probesrdquo The probe compounds included
nitrobenzene 2-nitrophenol 4-nitrotoluene 4-chloronitrobenzene and 4-
nitrophenylacetic acid Our results demonstrated that NACs are completely
reduced to their corresponding anilines by GR-SO4 The reactions followed pseudo
1 order kinetics with respect to NAC and the surface area-normalised pseudo 1
order rate constants obtained were 016ndash465middot10-4 s-1middotm-2middotL at [Fe(II)GR]0 = 103-
1260 mM [NAC]0 = 20-102 microM and pH 84-86 Neither mass transfer control nor
surface saturation kinetics could account for the similarity of the surface-
normalised pseudo 1 order rate constants obtained for the reduction of the neutral
and anionic NACs by GR-SO4 These observations suggest that the reaction
between NAC and GR-SO4 takes place at the external reactive Fe(II) sites At low
initial Fe(II)GR concentrations the external reactive Fe(II) sites were depleted and
the regeneration of new external reactive sites eventually controlled the reduction
of the NACs by GR-SO4 In chapter 5 we examined the reactivity of various
Fe(II)-Fe(III) mineral systems towards trichloroacetic acid (TCA) and
dichloroacetate (DCA) in laboratory batch experiments imitating natural
conditions The Fe(II)-Fe(III)-systems investigated included GR-SO4 carbonate
green rust magnetite Fe(II)goethite and Fe(II)lepidocrocite TCA was readily
reduced to DCA by all Fe(II)-containing minerals The reactions followed pseudo
1 order kinetics with respect to TCA and the surface area-normalised pseudo 1
order rate constants obtained were 033ndash76middot10-5 min-1middotm-2middotL at [Fe(II)]0 = 025ndash
116 mM [TCA]0 = 15ndash1000 microM and pH 70ndash87 Our results showed no
significant differences regarding product distribution and surface area-normalised
reaction rate constants between the Fe(II)-Fe(III)-systems DCA was not further
Conclusions and Outlook 127
reduced to monochloroacetate (MCA) or acetate in any of the systems within the
time frame in our experiments
As suggested in chapters 2 and 3 sufficient evidence must be provided and caution
should be exercised when proclaiming new biogenic minerals The study of
microbially produced GRs is still in its infancy and more research is needed in
order to elucidate the role of bacteria in the formation of GRs in natural soils and
sediments The results presented in chapter 2 indicate that microbiological
processes may be responsible for the oxidation of vivianite and metavivianite
((FeII3-xFeIII
x)(PO4)2(OH)xmiddot(8-x)H2O x gt 12) in natural subsurface environments
In chapter 3 we demonstrated that GRs may be produced microbially at conditions
including low carbon and Fe(III) concentrations as well as the exclusion of
synthetic electron shuttles and pH buffers The role of microbial processes in the
redox cycling of iron in the subsurface and the ways in which these processes can
be coupled to contaminant remediation are currently active areas of research Zero-
valent iron has been the most extensively studied reductant for the treatment of
many inorganic and organic contaminants and is currently the most commonly
used material for the construction of permeable reactive barriers (PRB) but a
detailed understanding of the processes involved in the reduction of these
pollutants by Fe(0) is lacking (Scherer et al 2000) Potentially reactive Fe(II)-
bearing corrosion products identified in iron metal columns and barriers include
magnetite siderite Fe(II) sulfides green rusts as well as Fe(II) sorbed to mineral
surfaces (Gu et al 1999 Roh et al 2000) The formation of reactive Fe(II)-
bearing minerals like GRs may explain the effective long-term operation of zero-
valent iron PRBs despite the formation of thick oxide films Thus natural in situ
PRBs might be created by stimulating the activity of anaerobic dissimilatory
Fe(III)-reducing bacteria and the subsequent formation of Fe(II) species such as
GRs Furthermore suspensions of synthetic GRs which are easily prepared from
relatively inexpensive commodity chemicals may also be injected and dispersed
into the subsurface
128 Chapter 6
The reductive transformation of NACs and TCA by GRs is relevant to
understanding the processes responsible for their degradation in the subsurface and
the development of innovative technologies for their remediation The results
obtained in chapters 4 and 5 indicate that GRs may play a significant role in the
reductive transformation of NACs and TCA in natural subsurface environments
Furthermore our results suggest that mainly the outer Fe(II) sites in GRs are
utilized in the reaction with neutral and monovalent anionic compounds and that
these sites may be replenished eg by reduction of the oxidized surface sites or
adsorption of Fe(II) from solution The continuous restoration of Fe(II) surface
sites in GRs may promote their long-term reactivity towards reducible
contaminants
References
Gu B Phelps TJ Liang L Dickey MJ Roh Y Kinsall BL Palumbo AV Jacobs GK (1999) Biochemical dynamics in zero-valent iron columns Implications for permeable reactive barriers Environmental Science and Technology 33 2170-2177 Roh Y Lee SY Elless MP (2000) Characterization of corrosion products in the permeable reactive barriers Environmental Geology 40 184-194 Scherer MM Richter S Valentine RL Alvarez PJJ (2000) Chemistry and microbiology of permeable reactive barriers for In Situ groundwater clean up Critical Reviews in Environmental Science and Technology 30 363-411
Supporting Information I
7 Supporting Information
71 Estimation of the one-electron reduction potential for 4-NPA
The one-electron reduction potential of the half-reaction for a given NAC 1hE
ArNO2 + e- ArNO2
can be used for comparing reduction rates of different NACs in a given system
The formation of the nitroaryl radical is the rate-determining step in the overall rate
of the reduction of a NAC to the corresponding aniline The difference between the
of a NAC and a given reductant is proportional to the change in standard free
energy for the transfer of the first electron ∆G
1hE
1degrsquo If a linear relationship between
the free energy of activation and ∆G1degrsquo is assumed the values of various NACs
can be a measure of their relative reactivity with a given reductant
1hE
As neither the one-electron reduction potential for 4-nitrophenylacetic acid (4-
NPA) nor the Hammett constant for the acetic acid substituent could be found in
the literature the one-electron reduction potential for 4-NPA was estimated by
application of a linear free energy relationship (LFER) to experimental data
Kinetic experiments were conducted in order to obtain the pseudo 1 order rate
constant for the reduction of 4-NPA by a model hydroquinone (reduced
juglone (8-hydroxy-14-naphthoquinone) in the presence of HS
minusHJUGk
ndash) The reduction of
a NAC by juglone follows the rate law
[ ] [ ] [ ] [ ] [ ] [ ]2222 ArNOJUGfkArNOHJUGkArNOk
dtArNOd
totHJUGHJUGHJUGobs sdotsdotsdot=sdotsdot=sdot=minus minusminusminusminus
and the was deducted from a LFER 1hE
II Chapter 7
bEak hHJUG +sdot=minus 059160
log1
for which a and b values have been established for a range of NACs with known
values (Hofstetter et al 1999) An excellent correlation of and log
has been found to exist over a range of 250 mV corresponding to more than 5 order
of magnitude for This is due to the fact that the actual transfer of the first
electron is the rate-determining step under the experimental conditions chosen
1hE 1
hE minusHJUGk
minusHJUGk
For comparison experiments with 4-nitrotoluene (4-NT) were also conducted The
pseudo 1 order rate constants for the reduction of 4-NPA with juglone were
corrected for the reduction of 4-NPA with only HSndash (control experiments
containing no juglone)
[ ]minuslowast
minusminus
minus
minus=
HJUGkk
k HSHJUGHJUG
where (MminusHJUGk -1middots-1) is the rate constant for a compound in the presence of only
juglone (slowastminusHJUGk -1) is the pseudo 1 order rate constant for a compound in the
presence of both juglone and HSndash (sminusHSk -1) is the pseudo 1 order rate constant for
the control reaction in the presence of only HSndash and [HJUGndash] (M) is the
concentration of the reactive dissociated HJUG- form (nondissociated
hydroquinone species are very nonreactive as compared to the monophenolate
species)
Supporting Information III
OH 0 OH OH
+ e- + H+ = + e- + H+ =
0 0
pl(( ox) = 8 00 PK1 (red) = 6 60
JUG HJUG
OH OH
OH
pKa2(red) = 10 60
Figure 7 1 Oxidized and reduced juglone fo1m s
Kinetic experiments in homogeneous anoxic aqueous solutions contained 5 mM
HS- 20 microM total juglone 50 mM KH2P04 buffer and were conducted at pH =
660 corresponding to a concentration of the reactive dissociated juglone form
[HJUG] = 10 microM
By using the LFER
E1 logkHJUG- = 125 middot 005~16 + 923
the following values were obtained
(Hofstetter et al 1999)
NAC k (M-1 -1) HJUG- middots log kHJUG- E~ (mV)
4-NT
4-NPA
311 middot10middot7
164middot10middot7
847middot 10-8
116middot 10middot7
226middot10middot2
489middot10middot3
-165
-231
-515
-546
IV Chapter 7
lowast
minusHJUGk -values are averages of triplicates whereas -values are averages of
duplicates The determined for 4-NT in this work (-515 mV) differs 3 from
the -value of -500 mV reported in the literature (Meisel amp Neta 1975
Wardman 1989) Hence it is assumed that the -value determined for 4-NPA
also differs by 3
minusHSk
1hE
1hE
1hE
Note that even for NACs holding very different values the difference in their
reactivities are much less pronounced in Fe(II)-Fe(III) systems such as the
Fe(II)goethite system (LFER slope a = 06 Hofstetter et al 1999) and the
Fe(II)magnetite system (LFER slope a = 034 Klausen et al 1995) as compared
to the jugloneH
1hE
2S system (a = 125) Furthermore it should be noted that all
LFERs mentioned here were established for neutral NACs and in this work we
have simply assumed that the LFERs are also valid for anionic NACs
72 The rate-limiting step
The overall rate of a reaction is equal to the rate of the slowest step in the
mechanism In heterogeneous reactions eg a compound reacting at the surface of
suspended particles in bulk solution the overall process by which the
heterogeneous reactions proceed may be broken down into a sequence of
individual diffusion steps and reaction steps 1) Mass transfer (diffusion) of the
reactant from the bulk fluid to the external surface of the solid phase 2)
Adsorption of reactant onto the solid surface 3) Reaction on the solid surface 4)
Desorption of the products from the solid surface 5) Mass transfer of the products
from the external solid surface to the bulk fluid Hence the rate of reaction of a
compound reacting at the surface of suspended particles in bulk solution may be
either mass transfer adsorptiondesorption or surface reaction limited When the
diffusion steps are much faster than the reaction steps the mass transfer or
diffusion steps do not affect the overall reaction rate However if the reaction steps
Supporting Information V
are very fast compared with the diffusion steps mass transport affects the reaction
rate Here only the external mass transfer is considered ie the diffusion of
reactants or products between the bulk fluid and the external surface of the solid
phase The additional internal mass transfer resistance for particles containing
substantial internal surface area is not addressed
721 Mass transfer (diffusion) limited kinetics
The overall rate constant can be represented by a system of resistances in series
(Fogler 1999 Arnold et al 1999)
⎟⎟⎠
⎞⎜⎜⎝
⎛+=
minusgeomSALobs kkak1111
where kobs is the observed rate constant kL is the mass transfer coefficient (mmiddots-1) a
is the ratio of the external (geometric) specific surface area to volume of solution
(m-1) and kSA-geom is the intrinsic rate constant of the reaction normalized to the
external specific surface area rather than the BET specific surface area By
comparing kLmiddota with kobs one can estimate the role of mass transfer on the rate of
reaction Thus if kLmiddota gtgt kobs mass transfer is so fast that it has no impact on the
reaction rate whereas if kLmiddota le kobs mass transfer is the rate limiting step
In fluid dynamics the Reynolds number Re is used for determining whether a
flow is laminar or turbulent
νtp ud sdot
=Re
where dp is the particle diameter (m) ut is the terminal particle settling velocity
(mmiddots-1) and ν is the kinematic fluid viscosity (m2middots-1) ν = η ρ where η is the
(absolute) dynamic fluid viscosity in centipoise (1 centipoise = 1 mPamiddots = 10-3
kgmiddotm-1middots-1) and ρ is the fluid density (kgmiddotm-3)
VI Chapter 7
At Re lt 1 we can apply Stokersquos particle settling velocity Stokersquos law is an
equation relating the terminal settling velocity of a smooth rigid sphere in a
viscous fluid of known density and viscosity to the diameter of the sphere when
subjected to a known force field
( )η
ρρsdot
minussdotsdot=
18
2pp
t
dgu (mmiddots-1)
where g = 981 mmiddots-2 is the gravitational constant ρp is the particle density (kgmiddotm-3)
The Sherwood number is the main parameter for prediction of the mass transfer
process
in fluid dynamics
3121 ScRe602Sh sdotsdot+=sdot
=lowast
lowast
iw
pL
Ddk
where Diw is the diffusion coefficient of the compound i in water (m2middots-1) is the
minimum (uncorrected) value of the mass transfer coefficient and Sc is the
Schmidt number This relation is often referred to as the Froumlssling correlation The
particle diameter is a key parameter in the Froumlssling correlation and the external
mass transfer coefficient varies with square of the particle size for smaller
particles
lowastLk
The Schmidt number is the ratio of the kinematic fluid viscosity and the diffusion
coefficient of the compound i in water
iwDν
=Sc
Supporting Information VII
According to Harriott (1962) the actual mass transfer coefficient kL is 15 times
greater than the minimum value of the mass transfer coefficient The
uncertainty in k
lowastLk
Lmiddota associated with particle sphericity and roughness issues are
believed not to exceed a factor of 2
The diffusion coefficient of a compound i in water can be estimated as (Hayduk amp
Laudie 1974)
5890141
9102613
iiw
VD
sdot
sdot=
minus
η (m2middots-1)
where iV is the molar volume of the compound i (cm3middotmol-1) estimated according
to Fuller et al 1966
Assuming spherical particles the external (geometric) specific surface area and the
particle diameter are calculated from the measured BET specific surface area Atot
assuming that our GR-SO4 has a AtotAouter ~ 30 similar to the one reported by
Hansen amp Koch (1998)
( ) ( ) ( )pppp
p
pp
p
dd
dV
SAAρρπ
πρ 1000
6
100061100030
SA3
2tot
geom sdot=
sdotsdot
sdot=
sdot== (m2middotg-1)
In our aqueous GR-SO4 system the density ρ = 1000 kgmiddotm-3 the absolute dynamic
viscosity η = 10-3 Pamiddots and the kinematic viscosity ν = 10-6 m2middots-1 for water The
GR-SO4 particle specific parameters used is found below
VIII Chapter 7
GR-SO4
Atot (m2g) 712
SAgeom (m2g) 237
Particle density ρp (kgm3) 1500
Particle diameter dp (m) 169middot10-6
Settling velocity ut (ms) 778middot10-7
Reynolds number 132middot10-6
The molar volumes the diffusion coefficients in water and the Schmidt numbers
for the NACs including 4-chloronitrobenzene (4-CNB) and nitrobenzene (NB)
were
Compound iV (cm3middotmol-1) Diw (m2middots-1) Sc
4-NT 1260 768middot10-10 1302
4-CNB 1230 779middot10-10 1284
4-NPA 1535 684middot10-10 1462
NB 1055 853middot10-10 1173
The ratio of the external (geometric) specific surface area to volume of solution
were calculated for GR-SO4 at the various concentrations applied
[Fe(II)GR]0 (mM) a (m-1)
103 366middot102
63 224middot103
126 448middot103
60 213middot103
The uncorrected mass transfer coefficients were estimated for the NACs using the
Froumlssling correlation
Supporting Information IX
[Fe(II)GR]0 (mM) lowastLk (mmiddots-1)
4-NT 912middot10-4
4-CNB 925middot10-4
4-NPA 812middot10-4
NB 101middot10-3
Finally kLmiddota was calculated and compared with the experimental 1 order rate
constants kobs obtained for the NACs
Compound [Fe(II)GR]0 (mM) kLmiddota (s-1) kobs (s-1) a
4-NT 103 050 420middot10-4
63 307 140middot10-3
126 613 590middot10-3
4-CNB 103 051 740middot10-4
63 311 170middot10-3
126 622 460middot10-3
4-NPA 103 045 640middot10-4
63 273 109middot10-3
126 546 473middot10-3
NB 60 324 137middot10-3 b
a Experimental pseudo 1 order rate constant at 50 microM [Ar-NO2]0 b Experimental pseudo 1 order rate constant at 10 microM [Ar-NO2]0
When comparing kLmiddota with kobs it can be seen that the rates of mass transfer for all
3 NACs exceed the observed rate constants by at least 3 or 4 orders of magnitude
at every initial Fe(II)GR concentration Thus the reaction of the given NACs with
GR-SO4 is not subject to mass transfer limitations under the experimental
conditions applied here
722 Surface saturation limited kinetics
More than 75 of all heterogeneous reactions that are not diffusion-limited are
surface-reaction-limited rather than adsorption- or desorption-limited We now
X Chapter 7
look at the reaction A = B = C where an intermediate B is formed In our system
A = Ar-NO2 B = Ar-NHOH and C = Ar-NH2 In this case the surface reaction is
assumed to be a single-site mechanism where only the site S on which A or B is
adsorbed is involved in the reaction forming B or C
KA
Adsorption 1 A + S = AmiddotS
kS1
Surface reaction 1 AmiddotS = BmiddotS
KB-1
Desorption 1 BmiddotS = B + S
KB
Adsorption 2 B + S = BmiddotS
kS2
Surface reaction 2 BmiddotS = CmiddotS
KC-1
Desorption 2 CmiddotS = C + S
The rate law for this surface-reaction limited single-site mechanism involving an
intermediate follows Langmuir-Hinshelwood kinetics (adopted from Fogler 1999)
CCBBAA
AAsitesSA
CKCKCKCKCk
dtdC
sdot+sdot+sdot+sdotsdotsdot
=minus1
1
Supporting Information XI
where kS1 is the intrinsic rate constant of the surface reaction transforming A into
the intermediate B Csites is the concentration of reactive sites S on the solid
surface KA KB and KC are the adsorption constants for A B and C at the reactive
surface sites and CA CB and CC are the concentrations of A B and C in the bulk
fluid Two major assumptions of the Langmuir isotherm imply that there is a fixed
number of localised surface sites present on the surface and that the activity of the
surface towards adsorption desorption or surface reaction is independent of
surface coverage
Hence fitting -∆CA∆t to CA CB and CC using a nonlinear curve fitting software
such as SigmaPlot may provide one with the intrinsic rate constant and the
adsorptions constants If KB and KC gtgt KA the intermediate and the product are
strongly competing with the reactant for vacant reactive surface sites
Our data was not fitted successfully by the Langmuir-Hinshelwood rate law
(regression results not shown) Simplifying the rate law by excluding either the
term KCmiddotCC or KBmiddotCB or both (assuming that the aniline product or the
hydroxylaniline intermediate or both did not compete for the reactive sites) did not
improve the regression The Langmuir-Hinshelwood rate law for a dual-site
mechanism did not fit our data either Thus Langmuir-Hinshelwood kinetics
cannot explain the reaction mechanism of the given NACs in our GR-SO4 system
73 External surface area of GR-SO4 and GR-CO3
The GR-SO4 unit cell consists of one double layer (d001 = 11 nm) ie one
hydroxide layer (049 nm ) and one interlayer (061 nm) Hexagonal GR-SO4
particles holding an average width of 1 microm (Figure 72) an average particle
thickness of 35 nm (Hansen amp Koch 1998) and a hydroxide layer thickness of
049 nm have a surface area of the basal plane
Abasal = 1 microm middot 1 microm ndash 2 middot 05 microm middot 025 microm = 075 microm2
XII Chapter 7
and a surface area of the edges
Aedge = (2 middot 05 microm + 4 middot 056 microm) middot 000049 microm = 00016 microm2
Figure 72 The hexagonal platy morphology of GR particles holding an average width of 1 microm
The particle thickness is the mean crystal thickness perpendicular to the 003 plane
as determined from the 003 reflections in an X-ray diffractogram A GR-SO4
particle holding a thickness of 35 nm contains 35 nm11 nm = 318 double layers
The GR-CO3 unit cell consists of one double layer (d001 = 075 nm) ie one
hydroxide layer (049 nm ) and one interlayer (026 nm) Hence a GR-CO3 particle
holding a thickness of 35 nm contains 35 nm075 nm = 467 double layers
The outer surface area of a GR-SO4 particle including outer basal planes and
edges is
Aouter = 222 microm 155microm 00016318microm 0752 =sdot+sdot
and the total surface area of a GR-SO4 particle including both inner and outer
basal planes as well as edges is
Supporting Information XIII
Atot = 222 microm 478)microm 00016microm 0752(318 =+sdotsdot
Hence the ratio of outer surface area to total surface area is
131microm 478microm 155
AA
2
2
tot
outer asymp=
Furthermore the ratio of edge surface area to outer surface area is
130microm 155
microm 00016318AA
2
2
outer
edge asympsdot
=
For GR-CO3 the outer surface area including outer basal planes and edges is
Aouter = 222 microm 157microm 00016746microm 0752 =sdot+sdot
and the total surface area of a GR-CO3 particle including both inner and outer
basal planes as well as edges is
Atot = 222 microm 701)microm 00016microm 0752(467 =+sdotsdot
Hence the ratio of outer surface area to total surface area is
145microm 701microm 157
AA
2
2
tot
outer asymp=
Furthermore the ratio of edge surface area to outer surface area is
121microm 157
microm 00016467AA
2
2
outer
edge asympsdot
=
XIV Chapter 7
74 Van der Waals radii
The size of polyatomic molecules can be estimated by summing the van der Waals
radii of the
individual atoms Van der Waals radii or nonbonded radii can be pictured as the
radii of hard spherical atoms (Figure 73)
Figure 73 Schematic of neighboring nonbonded atoms with van der Waals radii rA and rB
Assuming that the spheres of neighboring nonbonded atoms just touch (Figure
73) the highest possible ion or molecule size Ms can be estimated as the sum of
the van der Waals radii
Ms = 2middotrA + 2middotrB + (1)
Taking Paulingrsquos rule for nonmetals into account we can estimate the real size of
polyatomic ions bound by covalent bonds (Pauling 1960) The van der Waals
radius is larger than the covalent radius because it involves the interposition of two
electron pairs between the atoms rather than one The rule states that the van der
Waals radius of an atom exceeds its covalent radius by ~008 nm (overlap in
Figure 74)
Figure 74 Schematic of atoms undergoing covalent bonding
Supporting Information XV
Thus the size of polyatomic ions bound by covalent bonds Ms can now be
estimated as the sum of the van der Waals radii subtracted by 008 nm
Ms = 2middot(rA - 008 nm) + 2middot(rB - 008 nm) + (2)
We estimated the molecular size of the NACs (Table 72) by means of equation (2)
and the van der Waals radii of the atoms in Table 71
Table 71 Van der Waals radii of various atoms Values from Pauling 1960
Atom vdW radii (nm)
H 0120
O 0140
N 0150
C 0170
Cl 0181
S 0185
In order to make the calculations it was assumed that all atoms were spherical and
that all bond angles were 90deg or 180deg (linear structures) In addition no distinctions
were made between single and double bonds The molecular sizes of the NACs
were estimated with the benzene ring representing the xy plane
Table 72 Molecular sizes of the NACs a Thickness z of the xy plane
Compound Ms (x) (nm) Ms (y) (nm) Ms (z) (nm)a
NB 054 080 036
4-NT 054 106 036
4-CNB 054 100 036
4-NPA 054 136 036
XVI Chapter 7
Note that the molecular sizes in Table 72 are only rough estimations
For comparison with the GR-SO4 interlayer spacing (061 nm) we consider three
possible orientations of the NACs in the GR-SO4 interlayer 1) The NAC xyz
coordination is equivalent to the crystal abc coordination (z = c = 036 nm) 2) the
NAC xy plane is parallel to the crystal bc plane (z = a = 054 nm) and 3) the NAC
xy plane is parallel to the crystal ac plane (z = b = 080-136 nm) Hence the sizes
of the NACs do not hinder their access to the GR-SO4 interlayer Only when
oriented vertically do the sizes of the NACs (z = b = 080-136 nm) exceed the GR-
SO4 interlayer spacing
The molecular size of trichloroacetate (TCA) was also estimated by means of
equation (2) and the atomic van der Waals radii in Table 71 When the TCA
aliphatic chain is assumed to represent the x direction (Ms (x) = 066 nm) the
molecular size in the y and z directions ranges from 045-053 nm depending on the
free rotation of the C-C bond Thus only if the C-C bond is oriented perpendicular
to the crystal ab plane does the size of TCA exceed the GR-SO4 interlayer spacing
(061 nm) In contrast the size of TCA exceeds the GR-CO3 interlayer spacing
(026 nm) regardless of its orientation
75 Adsorption of Fe(II) onto Fe(III) oxides
As seen from the Fe(II) sorption isotherms Fe(II) sorption varies widely between
the Fe(III) oxides as a function of solution pH (Figure 75) Average surface
densities of approximately 2 singly coordinated sitesnm2 iron oxide have been
suggested for goethite and lepidocrocite (Cornell amp Schwertmann 1996) The
similar surface site densities of goethite and lepidocrocite might explain their
similar Fe(II) adsorption isotherms (Figure 75)
Supporting Information XVII
Figure 75 Fe(II) adsorption edges for ferrihydrite goethite hematite lepidocrocite and
magnetite in the absence of other specifically adsorbing cations and anions (from Vikesland amp
Valentine 2002 and references therein) The total number of surface sites was in excess of the
total Fe(II) concentrations in all experiments
Dissolved cations or anions may specifically adsorb at the calcite and Fe(III) oxide
surfaces by exchanging for H+ or OHndash at the equivCO3H0 equivCaOH0 equivFeOH0 and
equivFeIIIOFeIIOH0 surface sites At the experimental conditions applied here within a
pH range 70-87 the dominant species of interest in solution are Fe2+ HCO3ndash
CO32ndash Clndash SO4
2ndash (only in the GR-SO4 systems) and the anionic TCA and DCA In
addition Fe2+ readily forms aqueous complexes with hydroxide carbonate
chloride and sulfate whereby the species FeOH+ FeHCO3+ Fe(OH)(CO3)ndash
FeCO30 Fe(CO3)2
2ndash FeCl+ and FeSO40 may occur (Millero amp Hawke 1992) At
pH 70-87 we expect the Fe(II) species Fe2+ FeCO30 Fe(OH)(CO3)ndash FeOH+ and
Fe(CO3)22ndash to dominate in the GR-CO3 and CaCO3(s)CO2(g) buffered magnetite
suspensions In the goecalcite and lepcalcite suspensions we expect the FeCl+
species to dominate as well whereas the Fe2+ FeSO40 and FeOH+ species most
XVIII Chapter 7
likely dominate in the GR-SO4 suspensions Anionic inorganic ligands like
carbonate chloride and sulfate can lower or enhance the adsorption of Fe(II) due to
a) formation of stable nonadsorbing Fe(II) ligand aqueous complexes b) formation
of Fe(II) ligand Fe(III) oxide surface complexes which can lead to surface
precipitation at high Fe(II) and ligand concentrations c) competitive ligand
sorption to the Fe(III) oxide surface blocking reactive sorption sites at the surface
and d) diminution of the positive charge at the Fe(III) oxide surface (at pH levels
below the point of zero charge (pHpzc) of the Fe(III) oxide) thereby decreasing the
electrostatic repulsion of cations by the Fe(III) oxide surface Specifically adsorbed
cations increase the pHpzc whereas specifically adsorbed anions decrease the pHpzc
References Arnold WA Ball WP Roberts AL (1999) Polychlorinated ethane reaction with zero-valent zinc Pathways and rate control Journal of Contaminant Hydrology 40 183-200 Cornell RM Schwertmann U (1996) The iron oxides Structure properties reactions occurrence and uses VCH Verlagsgesellschaft mbH Weinheim Fogler HS (1999) Elements of chemical reaction engineering 3rd ed Prentice Hall Fuller EN Schettler PD Giddings JC (1966) A new method for prediction of binary gas-phase diffusion coefficients Industrial and Engineering Chemistry 58 19-27 Hansen HCB Koch CB (1998) Reduction of nitrate to ammonium by sulphate green rust Activation energy and interlayer reaction mechanism Clay Minerals 33 87-101 Harriott P (1962) Mass transfer to particles Part I Suspended in agitated tanks AIChE Journal 8 93-102 Hayduk W Laudie H (1974) Prediction of diffusion coefficients for nonelectrolytes in dilute aqueous solutions AIChE Journal 20 611-615 Hofstetter TB Heijmann CG Haderlein SB Holliger C Schwarzenbach RP (1999) Complete reduction of TNT and other (poly)nitroaromatic compounds under iron-reducing subsurface conditions Environmental Science and Technology 33 1479-1487 Klausen J Troumlber SP Haderlein SB Schwarzenbach RP (1995) Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions Environmental Science and Technology 29 2396-2404 Meisel D Neta P (1975) One-electron redox potentials of nitro compounds and radiosensitizers Correlation with spin densities of their radical anions Journal of the American Chemical Society 97 5198-5203
Supporting Information XIX
Millero FJ Hawke DJ (1992) Ionic interactions of divalent metals in natural waters Marine Chemistry 40 19-48 Pauling L (1960) The nature of the chemical bond 3rd ed Cornell University Press Ithaca Vikesland PJ Valentine RL (2002) Iron oxide surface-catalyzed oxidation of ferrous iron by monochloramine implications of oxide type and carbonate on reactivity Environmental Science and Technology 36 512-519 Wardman P (1989) Reduction potentials of one-eletron couples involving free radicals in aqueous solution The Journal of Physical Chemistry Reference Data 18 1637-1755
Curriculum Vitae 13011973 Born in Haderslev Denmark 1988-1991 Mathematical high school Haderslev Katedralskole Denmark 1991-1992 Sabbatical year 1992-1995 B Sc in environmental chemistry University of Copenhagen
(KU) Denmark B Sc thesis 1995 ldquoMethane oxidizing bacteria in soilrdquo
1995-1998 M Sc in environmental chemistry University of Copenhagen
Denmark 1997-1998 diploma thesis ldquoReductive dechlorination of carbon tetrachloride and chloroform in presence of iron(II)iron(III)-hydroxides (green rust)rdquo
1998-1999 Research and teaching assistant at the Chemistry Department
The Royal Veterinary amp Agricultural University (KVL) Denmark
1999-2004 PhD in environmental sciences Swiss Federal Institute of
Technology Zuumlrich (ETHZ) and Swiss Federal Institute for Environmental Science and Technology (EAWAG) Switzerland Docoral thesis ldquoFormation and redox reactions of green rusts under geochemical conditions found in natural soils and sedimentsrdquo
2000-2002 Teaching assistent at the Swiss Federal Institute of Technology
Zuumlrich and supervision of diploma students 2002 Microbial Diversity summer course (7 weeks) at the Marine
Biological Laboratory Woods Hole Massachusetts USA

Table of Contents
Table of Contents Zusammenfassung I Summary V 1 General Introduction 1 11 Iron cycling in the subsurface 1 12 Green rusts 3 13 Microbial formation of green rusts 7 14 Redox reactions of green rusts 8 15 Outline of the thesis 10 References 11 2 Solid State Oxidation of Vivianite by Anaerobic
Denitrifying Fe(II)-Oxidizing Bacteria 17 Abstract 17 21 Introduction 17 22 Materials and methods 22
221 Microorganisms and media 22 222 Characterisation of precipitates 23 223 Biooxidation experiments 24 224 Analytical methods 25
23 Results and discussion 25 231 Identification of solid iron-containing phases 25 232 Factors controlling the rate and extent of Fe(II) biooxidation 34 233 Morphology of solid iron phases 37
24 Conclusions 38 References 39 3 Formation of Layered Iron Hydroxides by
Microbial Fe(III) Reduction 43 Abstract 43 31 Introduction 44 32 Materials and methods 47
321 Preparation of iron oxide coatings 47 322 Mineral characterisation 48 323 Culture conditions and cell preparation 48 324 Bioreduction experiments 49
Table of Contents
325 Analytical methods 50 33 Results and discussion 50
331 Fe(II) production and suspension colour changes 50 332 Identification of solid iron phases 55 333 Factors controlling the identity of the secondary iron minerals 58 334 Factors controlling the rate and extent of Fe(III) bioreduction 59
34 Conclusions 60 References 61 4 Reduction of Nitroaromatic Probe Compounds by Sulphate
Green Rust The Effect of Probe Compound Charge 65 Abstract 65 41 Introduction 66 42 Materials and methods 71
421 Synthesis of GR-SO4 71 422 Mineral characterisation 72 423 Lyophilization and determination of specific surface area 72 424 Estimation of the one-electron reduction potential for 4-NPA 73 425 Kinetic experiments 74 426 Analytical methods 74
43 Results and discussion 75 431 Product formation and reaction kinetics 75 432 Comparison of rate constants for the different NACs 79 433 Factors influencing the reaction rate 82 434 Comparison with rate constants obtained for other Fe(II) containing
mineral systems 83 435 Depletion of reactive sites 85 436 The role of external and internal reactive sites 86
44 Conclusions 89 References 91 5 Reductive Transformation of Trichloroacetate in Abiotic
Fe(II)-Fe(III) Mineral Systems 97 Abstract 97 51 Introduction 98 52 Materials and methods 101
521 Synthesis of GRs and magnetite 102 522 Preparation of iron oxide coatings 102 523 Mineral characterisation 103 524 Kinetic experiments 103
Table of Contents
525 Analytical methods 104 53 Results and discussion 105
531 Product formation and reaction kinetics 105 532 Comparing rate constants obtained for the various Fe(II)-Fe(III)
mineral systems 109 533 Comparing with rate constants obtained for other chlorinated
aliphatic compound 112 534 Factors controlling the reactivity of surface-bound Fe(II) 114 535 Comparison with biotic and other abiotic systems 118
54 Conclusions 119 References 120 6 Conclusions and Outlook 125 References 128 7 Supporting Information I 71 Estimation of the one-electron reduction potential for 4-NPA I 72 The rate-limiting step IV 721 Mass transfer (diffusion) limited kinetics V
722 Surface saturation limited kinetics IX 73 External surface area of GR-SO4 and GR-CO3 XI 74 Van der Waals radii XIV 75 Adsorption of Fe(II) onto Fe(III) oxides XVI References XVIII Curriculum Vitae
Zusammenfassung I
Zusammenfassung Geschichtete Fe(II)-Fe(III)-Hydroxide (Gruumlner Rost) gehoumlren zur Gruppe der
Fe(II)-haltigen Mineralsysteme (zB Magnetit (Fe3O4) Siderit (FeCO3) Vivianit
(Fe2(PO4)2sdot8H2O) Fe(II)-Sulfide sowie an die Oberflaumlche von Fe(III)-Oxiden und
Tonmineralien gebundenes zweiwertiges Eisen) die die Aktivitaumlt von Fe(II) in
suboxischen und anoxischen Boumlden und Sedimenten kontrollieren Gruumlner Rost
Phasen (GRs) bestehen aus planaren positiv geladenen trioktaedrischen Fe(II)-
Fe(III)-Hydroxidschichten die durch hydratisierte Anionen in den
Zwischenschichten ausgeglichen werden Ihre generelle Zusammensetzung ist
[FeII(6-x)FeIII
x(OH)12]x+[(A)xnmiddotyH2O]x- wobei x = 09 - 42 ist A entspricht einem n-
valenten Anion (zB CO32- Clndash oder SO4
2-) und y repraumlsentiert die Anzahl
Wassermolekuumlle in der Zwischenschicht GRs sind wichtige intermediaumlre Phasen
die durch unvollstaumlndige Oxidation von Fe(II) oder teilweise Reduktion von Fe(III)
gebildet werden koumlnnen Sie koumlnnen in suboxischen nicht-sauren eisenhaltigen
natuumlrlichen wie auch technischen Systemen auftreten so wie in Wasser gesaumlttigten
Boumlden und interstitiellen Sedimenten Rohrleitungen in der
Trinkwasserversorgung Stahlpfosten in marinen Sedimenten Stahlbeton und in
reaktiven durchlaumlssigen Waumlnden aus nullwertigem Eisen zur in-situ Sanierung von
Altlasten und Aquiferen Aufgrund ihrer Schichtstruktur den anionischen
Zwischenschichten und der hohen spezifischen Oberflaumlchen sind GRs reaktive
Ionentauscher und Sorbentien von Anionen Des Weiteren wurde gezeigt dass
GRs eine Reihe anorganischer und organischer Schadstoffe reduzieren koumlnnen
Durch Immobilisierung und Transformation koumlnnen GRs somit eine wichtige Rolle
fuumlr das Abbauverhalten und den Transport solcher Schadstoffe in suboxischen
Boumlden und Sedimenten spielen Die Resultate dieser Dissertation tragen zum
Verstaumlndnis uumlber die Bildung und Reaktivitaumlt von Fe(II)-haltigen Mineralsystemen
wie GRs Vivianit Magnetit und an Goethit (α-FeOOH)- und Lepidokrozit (γ-
FeOOH)-Oberflaumlchen gebundenes Fe(II) in der Natur bei
II Zusammenfassung
Um die Rolle von Bakterien bei der Bildung von GRs in natuumlrlichen Boumlden und
Sedimenten aufzuklaumlren wurden Eisenminerale untersucht die als Folge der
Aktivitaumlt von eisenrespirierenden Bakterien gebildet wurden Kapitel 2 beschreibt
die Untersuchungen von eisenhaltigen Produkten die von anaeroben autotrophen
denitrifizierenden Fe(II)-oxidierenden Bakterien (FeOB) gebildet wurden Ein
Bikarbonat- und Phosphat-reiches Kulturmedium bot den nitratreduzierenden
FeOB optimale Bedingungen Fe(II) lag zu Anfang der Reaktion als weisses
Fe(II)-Hydroxyphosphat (Vivianit) und als geloumlstes Fe(II) vor Die Ergebnisse
zeigten dass die denitrifizierenden FeOB amorphen Goethit via ein gruumlnes Fe(III)-
angereichertes Vivianit-Zwischenprodukt bildeten Die Analyse mit Moumlssbauer
Spektroskopie deutet nicht auf eine Bildung von GR hin
In Kapitel 3 werden jene Eisenmineralien beschrieben die waumlhrend der Reduktion
verbreiteter Fe(III)-Oxide durch anaerobe dissimilative Fe(III)-reduzierende
Mikroorganismen Shewanella algae BrY gebildet wurden Um natuumlrliche
Zustaumlnde zu simulieren wurden Fe(III)-Oxide als Beschichtungen auf
Silikatpartikel (Modellsystem fuumlr Sandboumlden) oder Calcitpartikel (CaCO3
Modellsystem fuumlr kalkhaltige Boumlden) aufgetragen sowie synthetische
Elektronencarrier und hochkonzentrierte kuumlnstliche pH-Puffer ausgeschlossen
Die erforschten Mineralsysteme umfassten GoethitCalcit- LepidokrozitCalcit-
und FerrihydritSand-Suspensionen S algae BrY reduzierte beachtliche Mengen
des eingesetzten Fe(III) und es bildeten sich gruumlne und schwarze Festphasen
innerhalb von 1-2 Wochen nach der Animpfung Moumlssbauer Spektroskopie der
gruumlnen und schwarzen Praumlzipitate zeigte dass sich diese aus GR und Vivianit
zusammensetzen
Die Reaktivitaumlt synthetischer GRs gegenuumlber reduzierbaren organischen
Schadstoffen wurde erkundet um die potentielle Bedeutung von GR-Phasen fuumlr
das Schicksal solcher Verbindungen abzuschaumltzen Zu diesem Zweck wurden
Nitroaromaten (NACs) und Chloracetate als Modellverbindungen benutzt um
Zusammenfassung III
umweltrelevante Redoxreaktionen zu studieren In Kapitel 4 wurde die relative
Reaktivitaumlt von aumlusseren und inneren reaktiven Stellen in synthetischem Sulfat-
Gruumlnem Rost (GR-SO4) anhand von strukturaumlhnlichen ldquoreaktiven
Sondenmolekuumllenrdquo mit unterschiedlichen Ladungen untersucht Als reaktive
Sondenmolekuumlle wurden Nitrobenzen 2-Nitrophenol 4-Nitrotoluen 4-
Chlornitrobenzen und 4-Nitrophenylessigsaumlure verwendet Die Ergebnisse zeigen
dass GR-SO4 die NACs vollstaumlndig zu den entsprechenden Anilinen reduzierte
Die Reaktionen folgten einer pseudo 1 Ordnungs Kinetik bezuumlglich NAC und die
auf Oberflaumlche normalisierten pseudo 1 Ordnungs Geschwindigkeitskonstanten
(Anfangsraten) waren 016ndash465middot10-4 s-1middotm-2middotL fuumlr [Fe(II)GR]0 = 103-1260 mM
[NAC]0 = 20-102 microM und pH 84-86 Weder durch Einbezug von
Massentransferlimitierung noch von Oberflaumlchensaumlttigungskinetik war es moumlglich
die aumlhnlichen Oberflaumlchennormalisierten pseudo 1 Ordnungs
Geschwindigkeitskonstanten fuumlr die Reduktion der neutralen und anionischen
NACs durch GR-SO4 zu erklaumlren Dieser Umstand laumlsst vermuten dass die
Reaktion zwischen NAC und GR-SO4 an den externen reaktiven Fe(II)-Stellen
stattfindet Bei niedrigen Fe(II)GR-Anfangskonzentrationen wurden die externen
reaktiven Fe(II)-Stellen aufgebraucht und die Regenerierung von neuen externen
reaktiven Stellen haben schliesslich die Geschwindigkeit der Reduktion von NACs
durch GR-SO4 kontrolliert
In Kapitel 5 wurde die Reaktivitaumlt von verschiedenen umweltrelevanten Fe(II)-
Fe(III)-Mineralsystemen gegenuumlber Trichloressigsaumlure (TCA) und
Dichloressigsaumlure (DCA) in Batchexperimenten die natuumlrliche Bedingungen
imitierten untersucht Die Fe(II)-Fe(III)-Systeme umfassten Sulfat-Gruumlner Rost
Carbonat-Gruumlner Rost Magnetit Fe(II)Goethit und Fe(II)Lepidokrozit TCA
wurde von allen Fe(II)-haltigen Mineralien zu DCA reduziert Die Reaktionen
folgten einer pseudo 1 Ordnungs Kinetik bezuumlglich TCA und die auf Oberflaumlche
normalisierten pseudo 1 Ordnungs Geschwindigkeitskonstanten betrugen 033ndash
76middot10-5 min-1middotm-2middotL bei [Fe(II)]0 = 025ndash116 mM [TCA]0 = 15ndash1000 microM und pH
IV Zusammenfassung
70ndash87 Die Ergebnisse zeigen keine signifikanten Unterschiede zwischen den
verschiedenen Fe(II)-Fe(III)-Systemen bezuumlglich Produkteverteilung und
oberflaumlchen-normalisierten pseudo 1 Ordnungs Geschwindigkeits-konstanten In
keinem der Systeme wurde DCA innerhalb des experimentellen Zeitraums zu
Monochloressigsaumlure oder Essigsaumlure weiter reduziert
Die Ergebnisse die in dieser Dissertation praumlsentiert werden zeigen dass
mikrobiologische Prozesse fuumlr die Oxidation von Vivianit-Phasen im Untergrund
verantwortlich sein koumlnnen Zudem wurde nachgewiesen dass GRs bei tiefen
Kohlenstoff- und Fe(III)-Konzentrationen sowie durch Ausschluss von
kuumlnstlichen Elektronencarriern und pH-Pufferung mikrobiell gebildet werden
koumlnnen Ferner zeigten Befunde dass GRs eine bedeutende Rolle fuumlr die reduktive
Transformation von NACs und TCA in natuumlrlichen Boumlden und Sedimenten spielen
koumlnnen
Summary V
Summary
Layered iron(II)-iron(III)-hydroxides (green rusts) belong to the group of Fe(II)-
bearing mineral systems eg magnetite (Fe3O4) siderite (FeCO3) vivianite
(Fe2(PO4)2sdot8H2O) Fe(II) sulfides as well as Fe(II) associated with Fe(III) oxide
and clay mineral surfaces that control the Fe(II) activity in suboxic and anoxic
soils and sediments Green rusts (GRs) consist of plane positively charged
trioctahedral Fe(II)-Fe(III) hydroxide sheets balanced by hydrated anions in the
interlayers and hold the general formula [FeII(6-x)FeIII
x(OH)12]x+[(A)xnmiddotyH2O]x-
where x = 09 - 42 A is an n-valent anion eg CO32- Clndash or SO4
2- and y is the
number of water molecules in the interlayer GRs are important intermediate
phases formed by partial oxidation of Fe(II) or partial reduction of Fe(III) and they
have been found in suboxic non-acid iron-rich natural environments such as
hydromorphic soils and intertidal sediments and in engineering systems including
pipeline distribution systems for drinking water steel sheet piles in marine
sediments reinforced concrete and permeable reactive barriers of zero-valent iron
implemented for on-site remediation of contaminants Due to their layered
structures anionic interlayers and high specific surface areas GRs represent
reactive ion exchangers and sorbents of anions In addition GRs have been shown
to reduce a range of inorganic and organic pollutants Thus through sequestration
and reductive transformation GRs may play an important role in the fate and
transport of contaminants in suboxic soils and sediments The work presented in
this dissertation adds to the understanding of how Fe(II)-bearing minerals like
GRs vivianite magnetite and Fe(II) associated with goethite (α-FeOOH) and
lepidocrocite (γ-FeOOH) may form and react in nature
In order to elucidate the role of bacteria in the formation of GRs in natural soils
and sediments we studied the iron mineral phases forming as a result of the
activity of iron-respiring bacteria In the study described in chapter 2 the Fe-
containing products formed by anaerobic autotrophic denitrifying Fe(II)-oxidizing
VI Summary
bacteria (FeOB) were examined The culture medium applied contained high levels
of bicarbonate and phosphate and is typically used in this kind of studies as it
provides excellent conditions for the nitrate-reducing FeOB Fe(II) was present
initially as a whitish solid Fe(II) hydroxy phosphate (vivianite) and as soluble
Fe(II) The results obtained demonstrate that the denitrifying FeOB produce poorly
crystalline goethite via a greenish Fe(III)-enriched vivianite intermediate
Moumlssbauer spectroscopic analyses provided no significant evidence of green rust
formation
In chapter 3 the Fe-containing products formed during reduction of common
Fe(III) oxides by the anaerobic dissimilatory Fe(III)-reducing microorganism
Shewanella algae BrY are discussed In order to simulate natural conditions
Fe(III) oxides were applied as coatings on silica (model system for sandy soils) or
calcite (CaCO3) particles (model system for calcareous soils) and synthetic
electron shuttles as well as highly concentrated artificial pH buffers were excluded
The mineral systems studied include goethitecalcite lepidocrocitecalcite and
hydrous ferric oxidesand suspensions S algae BrY reduced substantial amounts
of the initial Fe(III) and green and blackish mineral phases were produced within
1-2 weeks after inoculation Moumlssbauer spectroscopic analyses showed that the
green and black precipitates consisted of GR and vivianite
The reactivity of synthetic GRs towards reducible organic pollutants was
investigated in order to asses the potential significance of GR phases for the fate of
such compounds To this end we used nitroaromatic compounds (NACs) and
chlorinated acetates as suitable model compounds for studying environmentally
relevant redox reactions In the work described in chapter 4 the relative reactivity
of outer and inner Fe(II) reactive sites in synthetic sulfate green rust (GR-SO4) was
studied using a series of structurally closely related compounds with different
charge properties as ldquoreactive probesrdquo The probe compounds included
nitrobenzene 2-nitrophenol 4-nitrotoluene 4-chloronitrobenzene and 4-
Summary VII
nitrophenylacetic acid The results show that NACs are completely reduced to their
corresponding anilines by GR-SO4 The reactions followed pseudo 1 order
kinetics with respect to NAC and the surface area-normalised pseudo 1 order rate
constants (initial rates) obtained were 016ndash465middot10-4 s-1middotm-2middotL at [Fe(II)GR]0 = 103-
1260 mM [NAC]0 = 20-102 microM and pH 84-86 Neither mass transfer control nor
surface saturation kinetics could explain the similarity of the surface-normalised
pseudo 1 order rate constants obtained for the reduction of the neutral and anionic
NACs by GR-SO4 These observations suggest that the reaction between NAC and
GR-SO4 takes place at the external reactive Fe(II) sites At low initial Fe(II)GR
concentrations the external reactive Fe(II) sites were depleted and the regeneration
of new external reactive sites eventually controlled the reduction of the NACs by
GR-SO4
Finally the reactivity of various Fe(II)-Fe(III) mineral systems towards
trichloroacetic acid (TCA) and dichloroacetate (DCA) has been investigated in
laboratory batch experiments imitating natural conditions (Chapter 5) The Fe(II)-
Fe(III)-systems investigated included GR-SO4 carbonate green rust magnetite
Fe(II)goethite and Fe(II)lepidocrocite TCA was readily reduced to DCA by all
Fe(II)-containing minerals The reactions followed pseudo 1 order kinetics with
respect to TCA and the surface area-normalised pseudo 1 order rate constants
obtained were 033ndash76middot10-5 min-1middotm-2middotL at [Fe(II)]0 = 025ndash116 mM [TCA]0 =
15ndash1000 microM and pH 70ndash87 Our results showed no significant differences
regarding product distribution and surface area-normalised reaction rate constants
between the Fe(II)-Fe(III)-systems DCA was not further reduced to
monochloroacetate (MCA) or acetate in any of the systems within the time frame
in our experiments
The results presented in chapter 2 indicate that microbiological processes may be
responsible for the oxidation of vivianite phases in natural subsurface
environments In chapter 3 we demonstrated that GRs may be produced
VIII Summary
microbially at conditions including low carbon and Fe(III) concentrations as well
as the exclusion of synthetic electron shuttles and pH buffers The results obtained
in chapter 4 and 5 show that GRs transform NACs and TCA readily The reductive
transformation of NACs and TCA by GRs is relevant to understanding the
processes responsible for their degradation in the subsurface and the development
of innovative technologies for their remediation
General Introduction 1
1 General Introduction
11 Iron cycling in the subsurface
Iron is the fourth most abundant element (4-5 mass) and the most abundant redox
sensitive element in the Earthrsquos crust It is found as Fe(II) and Fe(III) in a number
of minerals in rocks soils and sediments Under anoxic conditions solid Fe(III)-
containing minerals can be reduced to soluble Fe(II) once the more energetically
favoured electron donors - nitrate and manganese(IV) oxides - have been
consumed Dissolved Fe(II) can be reoxidized to insoluble Fe(III) microbially or
abiotically upon exposure to oxygen Due to this ready alternation between the
Fe(II) and Fe(III) redox states iron plays a major role in controlling the redox
potential and the carbon cycling in subsurface environments (Nealson amp Saffarini
1994)
Nonenzymatic processes were previously considered to account for most of the
Fe(III) reduction in subsurface environments The significance of bacteria in the
biogeochemical cycling of iron has been broadly recognized over the past two
decades Dissimilatory Fe(III)-reducing bacteria (DIRB) that gain energy by
coupling the oxidation of hydrogen or organic compounds to the reduction of
Fe(III) oxides have been known for many years but their biogeochemical
importance was only widely acknowledged about a decade ago (reviewed by
Lovley 1997) Fe(III) bioreduction accounts for a major fraction of the carbon
oxidation in many different anoxic environments and in the presence of sufficient
amounts of reactive Fe(III) microbial Fe(III) reduction may even inhibit sulphate
reduction and methanogenesis (King 1990 Lovley amp Phillips 1986) In fact most
of the Fe(III) reduction in the Fe(III) reduction zone of aquatic sediments and
aquifers is enzymatically catalyzed by microorganisms (Lovley et al 1991) A
wide diversity of DIRB distributed among several different phylogenetic groups
2 Chapter 1 is known today The two most studied DIRB are the obligate anaerobic Geobacter
spp and the facultatively anaerobic Shewanella spp (Figure 11)
Aerobic oxidation of Fe(II)-containing minerals by lithotrophic acidophilic and
neutrophilic bacteria has been known for many years but their broad significance
in the biogeochemical cycling of iron has only been recognized over the past two
decades Both acidophilic (Thiobacillus ferrooxidans) and neutrophilic
(Gallionella ferruginea Leptothrix ochracea Sphaerotilus natans) aerobic Fe(II)-
oxidizing bacteria (FeOB) have been isolated (Hanert 1992 Kuenen et al 1992
Mulder amp Deinema 1992)
Figure 11 The microbial iron cycle
Anaerobic Fe(II) oxidation by phototrophic purple non-sulfur bacteria utilizing
Fe(II) as an electron donor in the light was recognized only a decade ago (Widdel
et al 1993) Subsequently it was demonstrated that the biological oxidation of
Fe(II) in the absence of oxygen is possible by light-independent chemotrophic
microorganisms using nitrate as the electron acceptor (Straub et al 1996) Thus
the microbial iron cycle includes anaerobic Fe(III)-reducing microorganisms and
aerobic as well as anaerobic Fe(II)-oxidizing bacteria (Figure 11)
General Introduction 3
12 Green rusts
Iron oxides iron hydroxides and iron oxyhydroxides (collectively termed iron
oxides or Fe(III) oxides) are ubiquitous in the pedosphere where they originate
from aerobic weathering of surface magmatic rocks such as ferromagnesium
silicates and pyrite (Cornell amp Schwertmann 1996) Goethite (α-FeOOH)
lepidocrocite (γ-FeOOH) ferrihydrite (Fe5HO8sdot4H2O) hematite (α-Fe2O3)
magnetite (Fe3O4) maghemite (γ-Fe2O3) and akageneite (β-FeOOH) constitute the
most important iron oxides in soils and sediments (Schwertmann amp Cornell 1991)
The formation and transformation of iron oxides depend on pH solution
composition redox potential temperature rate of oxidationreduction and degree
and rate of hydrationdehydration Iron oxides are important to many soil
properties such as colour pH and redox buffer capacity aggregation with other
soil particles as well as retention of anions and cations (Cornell amp Schwertmann
1996) A number of Fe(II)-bearing minerals including Fe(II)-containing clays (eg
smectites vermiculites and micas) magnetite siderite (FeCO3) vivianite
(Fe2(PO4)2sdot8H2O) Fe(II) sulphides and green rusts (layered Fe(II)-Fe(III)
hydroxides) may be present in soils and sediments under suboxic and anoxic
conditions Green rusts are believed to play a central role as metastable
intermediates in the redox cycling of iron at circumneutral pH in aquatic and
terrestrial environments
Green rusts (GRs) are layered iron(II)-iron(III)-hydroxides consisting of plane
positively charged trioctahedral Fe(II)-Fe(III) hydroxide sheets balanced by
hydrated anions in the interlayers (cf Figure 41 this work) GRs belong
structually to the pyroaurite-sjoumlgrenite group of layered hydroxides and they hold
the general formula [FeII(6-x)FeIII
x(OH)12]x+[(A)xnmiddotyH2O]x- where x = 09 - 42 A is
an n-valent anion eg CO32- Clndash or SO4
2- and y is the number of water molecules
in the interlayer The three most common and investigated green rust forms include
chloride GR (GR-Cl) sulphate GR (GR-SO4) and carbonate GR (GR-CO3)
Generally GRs are crystallographically classified into the GRI (rhombohedral
4 Chapter 1 GR-Cl and GR-CO3) and GRII (hexagonal GR-SO4) crystal systems The GR
interlayer thickness is a function of both the size and the charge of the interlayer
anion Tetrahedrally coordinated anions like sulphate lead to larger interlayer
distances than smaller monoatomic anions like chloride or planar ions like
carbonate (Mendiboure amp Schoumlllhorn 1986) Not only size but also charge density
plays a role for the interlayer spacing That is for anions having the same number
of valence electrons anions with smaller ionic radii (higher electron density) are
bound more strongly and therefore result in smaller interlayer spacings The
interlayer in GR-SO4 is composed of two consecutive planes of anions and water
whereas GR-Cl and GR-CO3 interlayers consist of only one single plane (Simon et
al 2003)
GRs are important intermediate phases formed by partial oxidation of Fe(II) or
partial reduction of Fe(III) In neutral and weakly alkaline solutions the oxidation
of dissolved Fe(II) always passes through solid GR phases (Bernal et al 1959)
GRs may also form during oxidation of zero-valent iron and as a result of the
combination of Fe(II) and Fe(III) at circumneutral pH (Figure 12)
Figure 12 Formation and transformation of GRs Fe3O4 = magnetite γ-Fe2O3 = maghemite α-
FeOOH = goethite γ-FeOOH = lepidocrocite akageneite = β-FeOOH
General Introduction 5
Oxidation of GR-CO3 usually produces goethite and magnetite-maghemite
whereas GR-Cl and GR-SO4 transform into lepidocrocite and magnetite-
maghemite depending on pH and oxidation rate (Bernal et al 1959 Taylor 1980
Carlson amp Schwertmann 1990) The brown δ-FeOOH is formed by vigorous
oxidation of GR using air or a 30 aqueous solution of hydrogen peroxide (Bernal
et al 1959 Misawa et al 1974) Black ferromagnetic magnetite forms by slow
oxidation of GR whereas lepidocrocite forms at high oxidation rates (Misawa et
al 1974) The presence of chloride is a prerequisite for the formation of
akageneite (Bernal et al 1959 Refait amp Genin 1997)
A substantial amount of work has been conducted in order to estimate the free
energies of formation of green rusts The free energies of formation reported for
the carbonate and sulphate GRs fall in the range 4234ndash4384 kJsdotmol-1 as determined
from solution data monitored during anoxic alkalimetric titrations and from
reduction potential (Eh) and pH recordings monitored during oxidation of GRs in
aqueous solution (Hansen et al 1994 Drissi et al 1995 Genin et al 1996) The
free energies of formation provided allow for estimation of the stability domains of
GRs in Eh-pH phase diagrams (Drissi et al 1995 Genin et al 1996) As
evidenced from such diagrams (Figure 13) the stability domain of GR-SO4 lies
within pH 6-8 and Eh -700 ndash -400 mV depending on the activities of Fe(II) and
sulphate (compare Figures 13aampb) This agrees with the natural GR occurrences
found in suboxic non-acid iron-rich environments such as hydromorphic soils and
intertidal sediments (Al-Agha et al 1995 Trolard et al 1996 Genin et al 1998)
In addition GRs have been found as corrosion products in numerous engineering
systems including a pipeline distribution system for drinking water steel sheet
piles in marine sediments reinforced concrete (ferro-concrete) and permeable
reactive barriers of zero-valent iron implemented for on-site remediation of organic
and inorganic contaminants (Tuovinen et al 1980 Nielsen 1976 Genin et al
1991 Roh et al 2000)
6 Chapter 1
Figure 13 Eh-pH phase diagrams of GR-SO4 a) a = 10+2Fe-3 = 10minus2
4SOa -3 and b) a = 10+2Fe-2
= 10minus24SOa -1
The stability domains of GR-Cl and GR-CO3 are similar to the stability domain of
GR-SO4 At Fe(II) and sulphate activities lower than depicted in Figure 13b the
stability domain of GR-SO4 will be situated at higher pH and lower Eh Other
dissolved species present at anoxic conditions such as phosphate sulphide
carbonate and organic ligands may exert considerable effects on the availability of
Fe(II) and Fe(III) At anoxic and circumneutral conditions vivianite
(Fe2(PO4)2sdot8H2O) controls the Fe(II) activity even at very low phosphate
concentrations (Nriagu amp Dell 1974) The formation of solid Fe(II) sulphides and
siderite (FeCO3) as well as the complexation of Fe(II) and Fe(III) by organic
ligands may also control the activity of Fe(II) in the subsurface and thereby
interfere with the formation of GRs
Due to their layered structures anionic interlayers and high specific surface areas
GRs represent reactive ion exchangers and sorbents of environmentally concerning
anions eg arsenate and selenate (Myneni et al 1997 Randall et al 2001) In
addition GRs may incorporate divalent transition metal cations like Ni2+ Zn2+
Cd2+ Co2+ and Mg2+ by isomorphic substitution for Fe2+ in the hydroxide layers
General Introduction 7
(Tamaura 1985 Tamaura 1986 Refait et al 1994 Parmar et al 2001 Refait et
al 2001) Furthermore GRs have been shown to reduce a range of inorganic
contaminants such as nitrite nitrate selenate chromate uranyl pertechnetate and
the transition metals AgI AuIII CuII and HgII as well as organic pollutants
including halogenated ethanes ethenes and methanes (Hansen et al 1994 Hansen
et al 1996 Myneni et al 1997 Erbs et al 1999 Loyaux-Lawniczak et al 1999
Cui amp Spahiu 2002 Lee amp Batchelor 2002b Heasman et al 2003 OrsquoLoughlin et
al 2003aampb Pepper et al 2003 Elsner et al 2004 OrsquoLoughlin amp Burris 2004)
Thus through sequestration and reductive transformation GRs may play an
important role in the fate and transport of contaminants in suboxic soils and
sediments It should be noted that the rate constants reported for the reduction of
these inorganic and organic pollutants by GRs cannot be directly compared as the
various studies were conducted at very different experimental conditions
13 Microbial formation of green rusts
Generally one would expect that biogenic minerals have chemical compositions
and crystal habits similar to those produced by nonenzymatic processes as they are
governed by the same equilibrium principles In fact since the latter stages of
mineralization are abiotically driven and since the secondary Fe(II)-containing
minerals are formed indirectly by electron transfer outside the bacterial cell and not
directly inside the bacterial cell the type of iron mineral formed is a function of the
environmental conditions in which the bacteria live ie the same microorganism
form different minerals in different environments
The microbial formation of GRs resulting from bioreduction of various Fe(III)
oxides including ferrihydrite goethite and lepidocrocite by strains of the
anaerobic dissimilatory DIRB Shewanella putrefaciens has been reported
repeatedly over the last years (Fredrickson et al 1998 Kukkadapu et al 2001
Parmar et al 2001 Ona-Nguema et al 2002aampb Glasauer et al 2003)
However no evidence of biogenic formation of GRs at natural geochemical
8 Chapter 1 conditions have been offered and it is still unknown whether this process may take
place at natural conditions comprising low nutrient levels low iron concentrations
and the absence of synthetic electron shuttles and highly concentrated artificial pH
buffers Moreover the biotic formation of GRs by anaerobic denitrifying Fe(II)-
oxidizing bacteria has been suggested but the phases still need to be properly
identified (Chaudhuri et al 2001) In order to elucidate the role of bacteria in the
formation of GRs in natural soils and sediments we studied the iron mineral
phases forming as a result of the activity of iron-respiring bacteria (Chapters 2 and
3)
14 Redox reactions of green rusts
Fe(II) is one of the most abundant reductants present in aquatic and terrestrial
environments under suboxic and anoxic conditions (Lyngkilde amp Christensen
1992 Ruumlgge et al 1998) In these environments Fe(II) may be present as soluble
organic and inorganic complexes as surface complexes and as a host of Fe(II)-
bearing minerals Although aqueous Fe(II) complexes may reduce a number of
contaminants Fe(II) associated with mineral surfaces and structural Fe(II) present
in the mineral lattice in Fe(II)-containing minerals are often more powerful
reductants Fe(II)-bearing minerals including GRs magnetite siderite Fe(II)
sulphides as well as Fe(II)-carrying Fe(III) oxide and clay mineral surfaces have
been shown to reduce a number of organic and inorganic contaminants such as
nitroaromatic compounds chlorinated aliphatics chromate uranyl pertechnetate
nitrate monochloramine and carbamate pesticides (Klausen et al 1995 Cui amp
Eriksen 1996 Butler amp Hayes 1998amp1999 Erbs et al 1999 Liger et al 1999
Loyaux-Lawniczak et al 1999 Amonette et al 2000 Hwang amp Batchelor 2000
Hansen et al 2001 Gander et al 2002 Lee amp Batchelor 2002aampb Pecher et al
2002 Vikesland amp Valentine 2002 Hofstetter et al 2003 OrsquoLoughlin et al
2003aampb Strathmann amp Stone 2003 Elsner et al 2004 OrsquoLoughlin amp Burris
2004) However only few comparative studies on the reactivity of Fe(II)-bearing
minerals exist (Lee amp Batchelor 2002b Elsner et al 2004) When examining the
General Introduction 9
reaction rates of the reductive transformation of NACs and chlorinated aliphatics
by GRs and other Fe(II)-bearing minerals reported in these studies the rate
constants for GRs are mostly among the highest rates reported and in some cases
even higher than the rate constants for Fe(II) sulphides Thus GRs may play an
important role in the transformation of reducible contaminants in the subsurface
Nitroaromatic compounds (NACs) are widely applied as explosives herbicides
insecticides solvents and intermediates in the synthesis of dyes and pesticides
(Hartter 1985 Rosenblatt et al 1991) NACs are ubiquitous in the subsurface
environment and pose a health risk due to their toxicity (Rickert 1985) In anoxic
environments reduction of the nitro group is generally the first step during abiotic
or microbial transformation of the NACs (Macalady et al 1986) The
transformation reaction generally produces the corresponding aromatic amines and
minor amounts of intermediates (hydroxylamines and nitroso compounds) as well
as coupling products (azo and azoxy compounds) These products may be of
similar or even greater environmental concern
Trichloroacetic acid (TCA) is ubiquitous in soils and the concentrations reported
range from lt005 to 380 microgkg (Euro Chlor 2001 McCulloch 2002 Ahlers et al
2003) On account of its phytotoxicity suspected human carcinogenicity and
widespread occurrence TCA is of considerable environmental concern especially
in the terrestrial compartment (Ahlers et al 2003) Moreover the daughter
compounds of TCA - dichloroacetic acid (DCA) and monochloroacetic acid
(MCA) - are also toxins and suspected human carcinogens as well as widespread in
the environment (Reimann et al 1996 Berg et al 2000 Ahlers et al 2003 and
references therein) In this work the reactivity of synthetic green rusts towards
nitroaromatic compounds (NACs) and the reactivity of various Fe(II)-Fe(III)
mineral systems including synthetic GRs towards chlorinated acetates have been
studied (Chapters 4 and 5)
10 Chapter 1 15 Outline of the thesis
An examination of the Fe-containing products produced during solid state
oxidation of vivianite by anaerobic autotrophic denitrifying Fe(II)-oxidizing
bacteria is presented in chapter 2 The Fe(II)-oxidizing bacteria were cultured in a
mineral medium containing high levels of bicarbonate and phosphate which is
typically used in this kind of studies as it provides excellent conditions for the
nitrate-reducing FeOB The solid iron phases forming were investigated by
transmission Moumlssbauer spectroscopy infrared spectroscopy and scanning electron
microscopy
Chapter 3 includes a study on the Fe-containing products formed during reduction
of common Fe(III) oxides by the anaerobic dissimilatory Fe(III)-reducing
microorganism Shewanella algae BrY In order to simulate natural conditions
Fe(III) oxides were applied as coatings on silica (model system for sandy soils) or
calcite particles (model system for calcareous soils) and synthetic electron shuttles
as well as highly concentrated artificial pH buffers were excluded The mineral
systems studied include goethitecalcite lepidocrocitecalcite and hydrous ferric
oxidesand suspensions The solid iron phases produced were examined by
transmission Moumlssbauer spectroscopy
A study on the relative reactivity of outer and inner Fe(II) sites in synthetic GR-
SO4 by using a series of structurally closely related compounds with different
charge properties as ldquoreactive probesrdquo is presented in chapter 4 The probe
compounds included nitrobenzene 2-nitrophenol 4-nitrotoluene 4-
chloronitrobenzene and 4-nitrophenylacetic acid
In chapter 5 an investigation of the reactivity of various Fe(II)-Fe(III) mineral
systems towards TCA and DCA is presented The study included laboratory batch
experiments imitating natural conditions The Fe(II)-Fe(III)-systems investigated
included GR-SO4 carbonate green rust magnetite Fe(II)goethite and
General Introduction 11
Fe(II)lepidocrocite The reactivities of the Fe(II)-Fe(III) mineral systems were
examined by comparing their surface-normalized rate constants
The results and environmental implications of this work are summarized in chapter
6 References Ahlers J Regelmann J Riedhammer C (2003) Environmental risk assessment of airborne trichloroacetic acid - a contribution to the discussion of the significance of anthropogenic and natural sources Chemosphere 52 531-537 Al-Agha MR Burley SD Curtis CD Esson J (1995) Complex cementation textures and authigenic mineral assemblages in recent concretions from the Lincolnshire Wash (east coast UK) driven by Fe(0) to Fe(II) oxidation Journal of the Geological Society 152 157-171 Amonette JE Workman DJ Kennedy DW Fruchter JS Gorby YA (2000) Dechlorination of carbon tetrachloride by Fe(II) associated with goethite Environmental Science and Technology 34 4606-4613 Berg M Muumlller SR Muumlhlemann J Wiedmer A Schwarzenbach RP (2000) Concentrations and mass fluxes of chloroacetic acids and trifluoroacetic acid in rain and natural waters in Switzerland Environmental Science and Technology 34 2675-2683 Bernal JD Dasgupta DR Mackay AL (1959) The oxides and hydroxides of iron and their structural inter-relationships Clay Minerals Bulletin 4 15-30 Butler EC Hayes KF (1998) Effects of solution composition and pH on the reductive dechlorination of hexachloroethane by iron sulfide Environmental Science and Technology 32 1276-1284 Butler EC Hayes KF (1999) Kinetics of the transformation of trichloroethylene and tetrachloroethylene by iron sulfide Environmental Science and Technology 33 2021-2027 Carlson L Schwertmann U (1990) The effect of CO2 and oxidation rate on the formation of goethite versus lepidocrocite from an Fe(II) system at pH 6 and 7 Clay Minerals 25 65-71 Chaudhuri SK Lack JG Coates JD (2001) Biogenic magnetite formation through anaerobic biooxidation of Fe(II) Applied and Environmental Microbiology 67 2844-2848 Cornell RM Schwertmann U (1996) The iron oxides Structure properties reactions occurrence and uses VCH Verlagsgesellschaft mbH Weinheim Cui D Eriksen TE (1996) Reduction of pertechnetate by ferrous iron in solution influence of sorbed and precipitated Fe(II) Environmental Science and Technology 30 2259-2262
Cui D Spahiu K (2002) The reduction of U(VI) on corroded iron under anoxic conditions Radiochemica Acta 90 623-628
12 Chapter 1 Drissi SH Refait Ph Abdelmoula M Geacutenin JMR (1995) The preparation and thermodynamic properties of Fe(II)-Fe(III) hydroxide-carbonate (green rust I) Pourbaix diagram of iron in carbonate-containing aqueous media Corrosion Science 37 2025-2041 Elsner M Haderlein SB Schwarzenbach RP (2004) Reactivity of Fe(II)-bearing minerals towards reductive transformation of organic contaminants Environmental Science and Technology 38 799-807 Erbs M Hansen HCB Olsen CE (1999) Reductive dechlorination of carbon tetrachloride using iron(II)iron(III)-hydroxide-sulphate (green rust) Environmental Science and Technology 33 307-311 Euro Chlor (2001) Trichloroacetic acid in the environment a dossier Euro Chlor Brussels and the European Chlorinated Solvent Association Fredrickson JK Zachara JM Kennedy DW Dong H Onstott TC Hinman NW Li S (1998) Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium Geochimica et Cosmochimica Acta 62 3239-3257 Gander JW Parkin GF Scherer MM (2002) Kinetics of 111-trichloroethane transformation by iron sulfide and a methanogenic consortium Environmental Science and Technology 36 4540-4546 Geacutenin JMR Bourrieacute G Trolard F Abdelmoula M Jaffrezic A Refait Ph Maitre V Humbert B Herbillon A (1998) Thermodynamic equilibria in aqueous suspensions of synthetic and natural Fe(II)-Fe(III) green rusts Occurrences of the mineral in hydromorphic soils Environmental Science and Technology 32 1058-1068 Geacutenin JMR Olowe AA Benbouzid-Rollet ND Prieur D Confente M Resiak B (1991) The simultaneous presence of green rust 2 and sulfate reducing bacteria in the corrosion of steel sheet piles in a harbour area Hyperfine Interactions 69 875-878 Geacutenin JMR Olowe AA Refait Ph Simon L (1996) On the stoichiometry and Pourbaix diagram of Fe(II)-Fe(III) hydroxy-sulphate of sulphate-containing green rust 2 An electrochemical and Moumlssbauer spectroscopy study Corrosion Science 38 1751-1762 Glasauer S Weidler PG Langley S Beveridge TJ (2003) Controls on Fe reduction and mineral formation by a subsurface bacterium Geochimica et Cosmochimica Acta 67 1277- 1288 Hanert HH (1992) The genus Gallionella In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 4082-4088 Hansen HCB Borggaard OK Soslashrensen J (1994) Evaluation of the free energy of formation of iron(II)iron(III)-hydroxidesulphate (Green Rust) and its reduction of nitrite Geochimica et Cosmochimica Acta 58 2599-2608 Hansen HCB Guldberg S Erbs M Koch CB (2001) Kinetics of nitrate reduction by green rusts ndash effects of interlayer anion and Fe(II)Fe(III) ratio Applied Clay Science 18 81-91
General Introduction 13
Hansen HCB Koch CB Nancke-Krogh H Borggaard OK Soslashrensen J (1996) Abiotic nitrate reduction to ammonium Key role of green rust Environmental Science and Technology 30 2053-2056 Hartter DR (1985) The use and importance of nitroaromatic chemicals in the chemical industry In Toxicity of nitroaromatic compounds Rickert DE (ed) Hemisphere Publishing Corporation 1-13 Heasman DM Sherman DM Ragnarsdottir KV (2003) The reduction of aqueous Au3+ by sulfide minerals and green rust phases American Mineralogist 88 725-738 Hofstetter TB Schwarzenbach RP Haderlein SB (2003) Reactivity of Fe(II) species associated with clay minerals Environmental Science and Technology 37 519-528 Hwang I Batchelor B (2000) Reductive dechlorination of tetrachloroethylene by Fe(II) in cement slurries Environmental Science and Technology 34 5017-5022 King GM (1990) Effects of added manganic and ferric oxides on sulfate reduction and sulfide oxidation in intertidal sediments FEMS Microbiology Ecology 73 131-138 Klausen J Troumlber SP Haderlein SB Schwarzenbach RP (1995) Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions Environmental Science and Technology 29 2396-2404 Kuenen JG Robertson LA Tuovinen OH (1992) The genera Thiobacillus Thiomicrospira and Thiosphaera In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 2618-2624
Kukkadapu RK Zachara JM Smith SC Fredrickson JK Liu C (2001) Dissimilatory bacterial reduction of Al-substituted goethite in subsurface sediments Geochimica et Cosmochimica Acta 65 2913-2924 Lee W Batchelor B (2002a) Abiotic reductive dechlorination of chlorinated ethylenes by iron- bearing soil minerals 1 Pyrite and magnetite Environmental Science and Technology 36 5147- 5154 Lee W Batchelor B (2002b) Abiotic reductive dechlorination of chlorinated ethylenes by iron-bearing soil minerals 2 Green rust Environmental Science and Technology 36 5348- 5354 Liger E Charlet L Van Cappellen P (1999) Surface catalysis of uranium (VI) reduction by iron(II) Geochimica et Cosmochimica Acta 63 2939-2955 Lovley DR (1997) Microbial Fe(III) reduction in subsurface environments FEMS Microbiology Reviews 20 305-313 Lovley DR Phillips EJP (1986) Organic matter mineralization with reduction of ferric iron in anaerobic sediments Applied and Environmental Microbiology 51 683-689 Lovley DR Phillips EJP Lonergan DJ (1991) Enzymatic versus nonenzymatic mechanisms for Fe(III) reduction in aquatic sediments Environmental Science and Technology 25 1062-1067
14 Chapter 1 Loyaux-Lawniczak S Refait Ph Lecomte P Ehrhardt J Geacutenin JMR (1999) The reduction of chromate ions by Fe(II) layered hydroxides Hydrology and Earth System Sciences 3 593-599 Lyngkilde J Christensen TH (1992) Redox zones of a landfill leachate pollution plume (Vejen Denmark) Journal of Contaminant Hydrology 10 273-289 Macalady DL Tratnyek PG Grundl TJ (1986) Abiotic reduction reactions of anthropogenic organic chemicals in anaerobic systems A critical review Journal of Contaminant Hydrology 1 1-28 McCulloch A (2002) Trichloroacetic acid in the environment Chemosphere 47 667-686 Mendiboure A Schoumlllhorn A (1986) Formation and anion exchange reactions of layered transition metal hydroxides [Ni1-xMx](OH)2(CO3)x2(H2O)z (M = Fe Co) Revue de Chimie Mineacuterale 23 819-827 Misawa T Hashimoto K Shimodaira S (1974) The mechanism of formation of iron oxide and oxyhydroxides in aqueous solutions at room temperature Corrosion Science 14 131-149 Mulder EG Deinema MH (1992) The sheathed bacteria In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 2618-2624 Myneni SCB Tokunaga TK Brown Jr GE (1997) Abiotic selenium redox transformations in the presence of Fe(IIIII) oxides Science 278 1106-1109 Nealson KH Saffarini D (1994) Iron and manganese in anaerobic respiration Environmental significance physiology and regulation Annual Review of Microbiology 48 311-343 Nielsen A (1976) Hvid groslashn og sort rust Beskrivelse af en korrosionsskade paring et svoslashmmebassin Nordisk Betong 2 21-24 Nriagu JO Dell CI (1974) Diagenetic formation of iron phosphates in recent lake sediments American Mineralogist 59 934-946 OLoughlin EJ Burris DR (2004) Reduction of halogenated ethanes by green rust Environmental Toxicology and Chemistry 23 41-48 OLoughlin EJ Kelly SD Cook RE Csencsits R Kemner KM (2003a) Reduction of uranium(VI) by mixed iron(II)iron(III) hydroxide (green rust) Formation of UO2 nanoparticles Environmental Science and Technology 37 721-727 OLoughlin EJ Kelly SD Kemner KM Csencsits R Cook RE (2003b) Reduction of AgI AuIII CuII and HgII by FeIIFeIII hydroxysulfate green rust Chemosphere 53 437-446 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002a) Iron (IIIII) hydroxycarbonate green rust formation and stabilization from lepidocrocite bioreduction Environmental Science and Technology 36 16-20 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002b) Microbial reduction of lepidocrocite γ-FeOOH by Shewanella putrefaciens The
General Introduction 15
formation of green rust Hyperfine Interactions 139140 231-237 Parmar N Gorby YA Beveridge TJ Ferris FG (2001) Formation of green rust and immobilization of nickel in response to bacterial reduction of hydrous ferric oxide Geomicrobiology Journal 18 375-385 Pecher K Haderlein SB Schwarzenbach RP (2002) Reduction of polyhalogenated methanes by surface-bound Fe(II) in aqueous suspensions of iron oxides Environmental Science and Technology 36 1734-1741 Pepper SE Bunker DJ Bryan ND Livens FR Charnock JM Pattrick RAD Collison D (2003) Treatment of radioactive wastes An X-ray absorption spectroscopy study of the reaction of technetium with green rust Journal of Colloid and Interface Science 268 408- 412 Randall SR Sherman DM Ragnarsdottir KV (2001) Sorption of As(V) on green rust (Fe4(II)Fe2(III)(OH)12SO4
3H2O) and lepidocrocite (γ-FeOOH) Surface complexes from EXAFS spectroscopy Geochimica et Cosmochimica Acta 65 1015-1023 Refait Ph Abdelmoula M Trolard F Geacutenin JMR Ehrhardt JJ Bourrieacute G (2001) Moumlssbauer and XAS study of a green rust mineral the partial substitution of Fe2+ by Mg2+ American Mineralogist 86 731-739 Refait Ph Drissi SH Marie Y Geacutenin JMR (1994) The substitution of Fe2+ ions by Ni2+ ions in green rust one compounds Hyperfine Interactions 90 389-394 Refait Ph Geacutenin JMR (1997) The mechanisms of oxidation of ferrous hydroxychloride β- Fe2(OH)3Cl in aqueous solution The formation of akaganeite vs goethite Corrosion Science 39 539-553 Reimann S Grob K Frank H (1996) Chloroacetic acids in rainwater Environmental Science and Technology 30 2340-2344 Rickert DE (1985) Toxicity of nitroaromatic compounds Hemisphere Publishing Corporation 1-13 Roh Y Lee SY Elless MP (2000) Characterization of corrosion products in the permeable reactive barriers Environmental Geology 40 184-194 Rosenblatt DH Burrows EP Mitchell WR Parmer DL (1991) Organic explosives and related compounds In The Handbook of Environmental Chemistry Anthropogenic compounds Hutzinger O (Ed) Springer-Verlag 195-234 Ruumlgge K Hofstetter TB Haderlein SB Bjerg PL Knudsen S Zraurig C Mosbaeligk H Christensen TH (1998) Characterization of predominant reductants in an anaerobic leachate- affected aquifer by nitroaromatic probe compounds Environmental Science and Technology 32 23-31 Schwertmann U Cornell RM (1991) Iron oxides in the laboratory Preparation and characterization VCH Verlagsgesellschaft mbH Weinheim Simon L Francois M Refait Ph Renaudin G Lelaurain M Geacutenin JMR (2003)
16 Chapter 1 Structure of the Fe(II-III)-layered double hydroxysulphate green rust two from Rietveld analysis Solid State Sciences 5 327-334 Strathmann TJ Stone AT (2003) Mineral surface catalysis of reactions between FeII and oxime carbamate pesticides Geochimica et Cosmochimica Acta 67 2775-2791 Straub KL Benz M Schink B Widdel F (1996) Anaerobic nitrate-dependent microbial oxidation of ferrous iron Applied and Environmental Microbiology 62 1458-1460 Tamaura Y (1985) ZnII-bearing green rust II and its spontaneous transformation into ZnII- bearing ferrite in aqueous solution Bulletin of the Chemical Society of Japan 58 2951-2954 Tamaura Y (1986) Ni(II)-bearing green rust II and its spontaneous transformation into Ni(II)- bearing ferrites Bulletin of the Chemical Society of Japan 59 1829-1832 Taylor RM (1980) Formation and properties of Fe(II)Fe(III)-hydroxycarbonate and its possible significance in soil formation Clay Minerals 15 369-382 Trolard F Abdelmoula M Bourrieacute G Humbert B Geacutenin JMR (1996) Mise en eacutevidence dun constituant de type rouilles vertes dans les sols hydromorphes Proposition de lexistence dun nouveau mineacuteral la fougeacuterite Geacuteosciences de surface Comptes Rendus de LrsquoAcademie des Sciences 323 1015-1022 Tuovinen OH Button KS Vuorinen A Carlson L Mair DM Yut LA (1980) Bacterial chemical and mineralogical characteristics of tubercles in distribution pipelines Journal of the American Water Works Association 72 626-635 Vikesland PJ Valentine RL (2002) Iron oxide surface-catalyzed oxidation of ferrous iron by monochloramine implications of oxide type and carbonate on reactivity Environmental Science and Technology 36 512-519 Widdel F Schnell S Heising S Ehrenreich A Assmus B Schink B (1993) Ferrous iron oxidation by anoxygenic phototrophic bacteria Nature 362 834-836
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 17
2 Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria
Abstract
This work investigated the Fe-containing products formed by anaerobic
autotrophic denitrifying Fe(II)-oxidizing bacteria in a specific bicarbonate buffered
(30 mM HCO3- pH 70) culture media containing 10 mM Fe(II) 4 mM nitrate and
4 mM phosphate Fe(II) was present initially as a whitish vivianite-like
(Fe3(PO4)2middot8H2O) precipitate and as soluble Fe(II) The initial phase of the
oxidation produced a greenish metavivianite-like ((FeII3-xFeIII
x)(PO4)2(OH)xmiddot(8-
x)H2O x gt 12) phase In the late oxidation phase a reddish precipitate of poorly
crystalline goethite (α-FeOOH) dominated the colour of the media in coexistence
with Fe(II)-containing siderite (FeCO3) The increasing amounts of Fe(III) present
in the ldquovivianiterdquo and ldquometavivianiterdquo structures were accompanied by an
increasing intensity in the green colour as the Fe(II) biooxidation progressed This
colour development has produced the idea of biogenic green rusts (layered Fe(II)-
Fe(III) hydroxides) in several studies on nitrate-dependent Fe(II) biooxidation
However in this work no evidence of green rust formation mediated by anaerobic
denitrifying Fe(II)-oxidizing bacteria was obtained
21 Introduction
Aerobic oxidation of Fe(II)-containing minerals by lithotrophic acidophilic and
neutrophilic bacteria has been known for many years but their broad significance
in the biogeochemical cycling of iron has only been recognized over the past two
decades Aerobic Fe(II)-oxidizing bacteria (FeOB) have been isolated from acidic
ecosystems (Thiobacillus ferrooxidans) neutral eutrophic systems (Sphaerotilus
natans Leptothrix ochracea) and neutral oligotrophic systems (Gallionella
ferruginea) (Hanert 1992 Kuenen et al 1992 Mulder amp Deinema 1992) At
neutral pH Fe(II) is unstable in the presence of oxygen and is rapidly oxidized to
the insoluble Fe(III) Hence the only pH neutral environments where soluble
18 Chapter 2
Fe(II) is available for aerobic FeOB are at interfaces between oxic and anoxic
conditions The aerobic neutrophilic FeOB (Leptothrix ochracea Gallionella
ferruginea and Sphaerotilus natans) live at such interfaces and are usually
associated with the yellowishreddish ferric deposits formed there
Over the past several years there has been a growing recognition that other less
readily detectable types of bacteria are involved in Fe(II) oxidation in ecosystems
at circumneutral pH For example it has been reported that neutrophilic FeOB are
abundant at the Loihi seamount hydrothermal vents and play a major role in the
Fe(III) oxide deposition (Emerson amp Moyer 2002) Similarly unidentified
neutrophilic obligate lithotrophic FeOB have been isolated from the rhizosphere of
wetlands plants where they are closely associated with deposits of amorphous
Fe(III) oxides (Emerson et al 1999) It was previously believed that Fe(III) oxide
deposits associated with sheaths were produced biologically whereas Fe(III) oxide
deposits not associated with cells were produced abiotically Recently the
formation of amorphous Fe(III) oxide in gradient tubes has been attributed to the
action of FeOB (Sobolev amp Roden 2001) The authors attribute 90 of the
oxidation to biological processes and indicated that the organisms seem to produce
a mobile form of Fe(III) that diffuses away from the cells before being
precipitated thereby avoiding encrustation of the cells They suggest that such
soluble Fe(III) complexes might be substrates for closely associated Fe(III)-
reducing bacteria Such an arrangement might allow close coupling between
microbial Fe(II) oxidation and Fe(III) reduction within millimeters of the oxic-
anoxic interface
Anaerobic Fe(II) oxidation by phototrophic purple non-sulfur bacteria utilizing
Fe(II) as an electron donor in the light was recognized only a decade ago (Widdel
et al 1993) Subsequently it was demonstrated that the biological oxidation of
Fe(II) in the absence of oxygen is possible by light-independent chemotrophic
microbial activity using nitrate as the electron acceptor (Straub et al 1996) In
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 19
addition studies conducted in gradient cultures revealed that nitrate-reducing
strains could also oxidize Fe(II) with molecular oxygen (Benz et al 1998) Hence
these Fe(II)-oxidizing strains may use nitrate as well as oxygen as electron
acceptors The microbial oxidation of Fe(II) was coupled to stoichiometric
reduction of nitrate to N2 and only one strain produced traces of N2O as a by-
product (Straub et al 1996 Benz et al 1998) The authors proposed the formation
of 2-line ferrihydrite as the end product of Fe(II) biooxidation The chemical
reduction of nitrate by Fe(II) requires a catalyst eg at least 10 microM Cu2+ in order
to take place at significant rates and may thus be considered insignificant under the
conditions applied in our study (Moraghan amp Buresh 1976) The chemical
oxidation of Fe(II) with nitrous oxide has not been observed However nitrite can
oxidize Fe(II) chemically (Moraghan amp Buresh 1977 Straub et al 1996) but this
process is considered insignificant at the conditions applied here No denitrifying
Fe(II)-oxidizing enrichment culture has been found to produce ammonium from
nitrate
Both lithoheterotrophic (depending on organic cosubstrates such as acetate) and
strictly lithoautotrophic nitrate-reducing FeOB have been found in various marine
and freshwater sediments However most isolates depend on organic cosubstrates
for cell biosynthesis (Benz et al 1998) Most probable number estimations
showed that denitrifying FeOB accounted for 00006-08 of the acetate-oxidizing
denitrifying microbial population Lithotrophic FeOB accounted for less than
00001 of the total bacterial community Attempts to isolate CO2-fixing nitrate-
dependent FeOB from lithotrophic cultures have failed (Straub amp Buchholz-
Cleven 1998) Mixotrophic FeOB accounted for 0004-004 of the total bacterial
community In addition microbial nitrate-dependent Fe(II) oxidation was
demonstrated in a flooded paddy soil as well as in activated sludge from a
wastewater treatment plant (Nielsen amp Nielsen 1998 Ratering amp Schnell 2001)
Since the activity is not restricted to sunlight exposed habitats microbial nitrate-
dependent Fe(II) oxidation is supposedly more important on a global scale than
20 Chapter 2
anaerobic Fe(II) oxidation by phototrophic bacteria Furthermore it has been
reported that anaerobic denitrifying FeOB aptly oxidize biogenic Fe(II) minerals
formed by bioreduction of synthetic goethite and ferrihydrite and that anaerobic
Fe(III)-reducing bacteria readily reduce Fe(III) minerals formed by biooxidation of
Fe(II) (Weber et al 2001 Straub et al 1998) Hence autotrophic denitrifying
FeOB may play a significant role in the nitrogen and iron cycles in subsurface
environments where the nitrate and the Fe(II) zones overlap and organic carbon
supply is limited (Figure 21)
Figure 21 The microbial iron cycle linking the carbon and nitrogen cycles
Phosphate is released into the environment through natural processes such as rock
weathering and decomposition of dead organic material and anthropogenic
activities eg wastewater effluents and application of manure and fertilizers in
horti- and agriculture In anoxic soils and sediments phosphate may be sequestered
by sorption onto Fe(III) oxides (Williams et al 1971 Patrick amp Khalid 1974)
Phosphate strongly influences the type morphology and properties of Fe(III)
oxides formed by oxidation and hydrolysis of Fe(II) salts as well as the degree of
their transformation (Kandori et al 1992 Cumplido et al 2000 Benali et al
2001) Phosphate may also be retained by precipitation of Fe(II) phosphates such
as the monoclinic vivianite (Fe3(PO4)2middot8H2O) which is the most important stable
Fe(II) orthophosphate solid encountered in the subsurface under most conditions
(Nriagu 1972) At anoxic and circumneutral conditions the whitish vivianite
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 21
controls the Fe(II) activity even at very low phosphate concentrations (Nriagu amp
Dell 1974) Vivianite occurs as a secondary mineral in the gossans of metallic ore
deposits and as a weathering product of primary iron-manganese phosphates in
pegmatites (Gaines et al 1997) Moreover natural vivianite occurrences have
been identified in a number of lake and river sediments (Zwaan amp Kortenbout van
der Sluys 1971 Nriagu amp Dell 1974 Postma 1981 Nembrini et al 1983
Henderson et al 1984 Dodd et al 2003 House 2003 and references therein)
Vivianite is also found in sewage sludge as a result of the wastewater treatment
where iron salts are added in order to remove phosphate (Seitz et al 1973) It is
however still indefinite how ubiquitous vivianite is in nature Furthermore only
little is known about the mechanism of vivianite formation and the role played by
sedimentary Fe(III) oxides Anaerobic Fe(III)-reducing microorganisms may
reduce Fe(III) oxides thereby releasing the iron as soluble Fe(II) and mobilizing
the phosphate adsorbed to the Fe(III) oxides (Lovley 1997) It has been suggested
that vivianite is formed by precipitation following reductive dissolution of Fe(III)
oxides (Manning et al 1981 Manning amp Jones 1982) However it has also been
proposed that the transformation of Fe(III) oxides to vivianite occurs topotactically
and not via reductive dissolution (Nembrini et al 1983) Vivianite was shown to
form microbially as a result of the activity of the anaerobic Fe(III)-reducing
bacteria Shewanella putrefaciens in the presence of high Fe(III)-citrate and
phosphate concentrations (Jorand et al 2000) Moreover vivianite formation by
bioreduction of Fe(III) in hydrous ferric oxide and in smectite has been reported
(Fredrickson et al 1998 Dong et al 2003)
Only little is known about the oxidation products of vivianite Metavivianite a
greenish triclinic iron hydroxy phosphate mineral was first described by Ritz et al
(1974) and it was later found to coexist with vivianite in several natural sediment
samples (Henderson et al 1984) Once the Fe(III) content became evident the true
composition of metavivianite ((FeII3-xFeIII
x)(PO4)2(OH)x
22 Chapter 2
middot(8-x)H2O x gt 12)) was established (Rodgers amp Johnston 1985 Rodgers 1986
and references therein) The formation of intermediate greenish precipitates during
oxidation of fluffy colourless Fe(II) precipitates by anoxic phototrophic
microorganisms and nitrate-dependent FeOB have been reported (Ehrenreich amp
Widdel 1994 Chaudhuri et al 2001) Since both studies were conducted in
bicarbonate buffered mineral media (22-30 mM HCO3ndash pH 70-72) containing
37-5 mM phosphate we assume that the initial fluffy whitish precipitates
consisted mainly of vivianite Chaudhuri et al (2001) proposed that the
intermediate green phases produces by the denitrifying FeOB consist of carbonate
green rust (GR-CO3) but no convincing evidence of this biogenic GR-CO3 has been
provided yet The major objective of this work was to examine the Fe-containing
products forming during the course of biooxidation of vivianite by non-
phototrophic anaerobic denitrifying Fe(II)-oxidizing bacteria
2 2 Materials and methods
All handling and sampling of solutions and suspensions were carried out under
sterile and strict anoxic conditions All chemicals were pa quality
221 Microorganisms and media
Enrichment cultures of nitrate-reducing FeOB taken from town ditches (Bremen
Germany) were grown in anoxic bicarbonate-buffered (30 mM HCO3ndash 90
N210 CO2 pH 70) mineral media containing 4 mM phosphate as well as
essential trace elements and vitamins (Table 21 Straub amp Buchholz-Cleven
1998) Ammonium was omitted from the media in order to facilitate detection of
ammonium possibly produced by reduction of nitrate The techniques used for
preparation of media and cultivation of bacteria under anoxic conditions have been
described by Widdel amp Bak (1992) 05 M aqueous stock solutions of FeCl2 or
FeSO4 were prepared in 100 mL glass flasks by reacting 65 mmol of iron powder
(particle size 10 microm Merck) with 100 mL deoxygenated 10 M HCl or 05 M
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 23
H2SO4 respectively The solutions were magnetically stirred and heated (~80degC)
during reaction until the H2(g) production had ceased (ge 1 hour) The FeCl2 and
FeSO4 stock solutions were stored under a small Ar overpressure at 5degC
Table 21 Composition of the mineral medium (adopted from Straub amp Buchholz-Cleven
(1998))
Components Concentration (M) KH2PO4 15middot10-3
K2HPO4 25middot10-3
MgSO4middot7H2O 10middot10-3
CaCl2middot2H2O 50middot10-4
H3BO3 56middot10-5
ZnSO4middot7H2O 10middot10-6
Na2MoO4middot2H2O 40middot10-6
CuSO4middot5H2O 20middot10-7
MnSO4middotH2O 10middot10-6
Na2SeO4 12middot10-5
CoCl2middot6H2O 50middot10-6
NiCl2middot6H2O 80middot10-6
NaCl 10middot10-5
NaHCO3ndash 30middot10-2
Cyanocobalamine (vitamin B12) 37middot10-8
p-aminobenzoic acid (vitamin Hrsquo) 36middot10-7
D(+)-biotin (vitamin H) 41middot10-8
Nicotinic acid (Niacin) 81middot10-7
Ca-D(+)-pantothenate (vitamin B5) 52middot10-8
Pyridoxamine dihydrochloride 96middot10-7
Thiaminechloridehydrochloride (vitamin B1) 15middot10-7
NaNO3 40middot10-3
FeSO4 or FeCl2 0010
222 Characterisation of precipitates
In order to optimize the characterization and distinction between the spectral
components transmission Moumlssbauer spectra were obtained at temperatures
between 5 K and 250 K and in external magnetic fields of 4 T (parallel to the γ-ray
direction) using a conventional constant acceleration spectrometer and a source of 57Co in Rh The spectrometer was calibrated using a 125 microm foil of α-Fe at room
temperature and isomer shifts are given relative to the centroid of the spectrum of
this absorber The spectra were fitted using simple Lorentzian line shape Infrared
(IR) spectra were obtained using a Perkin Elmer FT-IR 2000 spectrometer and the
24 Chapter 2
KBr pellet technique Scanning electron microscopy (SEM) was carried out in
order to study the morphology and composition of the precipitates Specimens for
SEM were prepared by depositing suspended particles onto an aluminum stub
coated with a carbon sticker The stub was quickly transferred into a sputtering
chamber and coated with a thin Pt film (~20 nm) In order to avoid interfering Pt
signals in the energy dispersive spectra the stubs were in some cases not coated
with Pt but quickly transferred to the SEM chamber for evacuation Measurements
were performed using a Philips XL30 equipped with a LaB6 source and an
accelerating voltage of 20 kV and an EDAX eDXi X-ray dispersive spectrometer
223 Biooxidation experiments
The biooxidation experiments were conducted in 50-400 mL butyl rubber
stoppered bottles with a 90 N210 CO2 headspace constituting 10 of the total
volume Prior to inoculation 4 mM NaNO3 was added as the electron acceptor and
10 mM Fe2+ (as chloride or sulphate) as the electron donor to the mineral media
Control experiments were performed in the same media only they were not
inoculated Addition of ferrous iron to the media induced an immediate
precipitation of a solid whitish material The whitish precipitate was collected on
022 microm polyvinylidendifluorid (Durapore Millipore) filters and stored in an
anoxic atmosphere until further measurements Old outgrown media suspensions
that had turned reddish in color due to a precipitate produced by the denitrifying
FeOB were used as inocula Inoculum volume was kept lt1 of the culture volume
in order to prevent the reddish inoculum from dominating over the initial whitish
precipitate Cultures were incubated in the dark at room temperature and gently
agitated once every day Typical color successions for the media were initial
whitish precipitates turning first more and more greenish over time and then finally
turning reddish (see Fig 22) The color developed uniformly without any
indications of multiple phases in the precipitate At different time intervals
suspension samples were withdrawn using 90 N210 CO2-flushed polyethylene
syringes The precipitates were collected on 022 microm polyvinylidendifluorid
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 25
(Durapore Millipore) filters and analyzed by Moumlssbauer spectroscopy and SEM
Nonfiltered suspension samples were digested in 01 M HCl and chemically
analyzed for Fe(II) NO3- and NH4
+
224 Analytical methods
Fe2+ was determined using a modified phenanthroline method (Fadrus amp Maly
1975) Nitrate was quantified by ion chromatography (Morales et al 2000) and
ammonium was measured photometrically using the indophenol reaction (Rossum
amp Villarruz 1963)
23 Results and discussion
231 Identification of solid iron-containing phases
In most cases the mineral media for cultivating denitrifying FeOB contained 10
mM FeCl2 or FeSO4 4 mM NO3ndash 4 mM total phosphorus and 30 mM HCO3
- at pH
70 ([HCO3-] = 2138middot[CO3
2-] at pH 70) Whitish flocs precipitated immediately at
these initial conditions when Fe(II) was added to the media (Figure 22a) Such
colourless flocs have been reported to precipitate in similar mineral media (10 mM
Fe(II) 37-5 mM phosphate 22-30 mM HCO3ndash pH 70-72) (Ehrenreich amp
Widdel 1994 Chaudhuri et al 2001) Our Fe(II) measurements showed that 20-
50 of the total Fe(II) added was present in this initial white precipitate
26 Chapter 2
a
210- Figure 22 Colour of suspended material in the growth media during Fe(II) biooxidation a)
Initial whitish precipitate prior to inoculation b) Inte1mediate greenish phase fo1med within 2-3
days after inoculation c) reddish precipitate at late stage of biooxidation (gt5-6 days)
The precipitates were filtered and investigated by Mossbauer and IR spectroscopy
The transmission Mossbauer spectra obtained for the initial whitish precipitate at
temperatures between 20 and 250 K are shown in Figure 23 The spectrum
measured at 250 K consists of two fairly well-resolved Fe(II) doublets (see
parameters in Table 22) The change in line-overlap with decreasing temperature
is primarily ascribed to differences in the temperature dependence of the
quadrupole splitting of the two components From the spectra at 10 and 6 K
(Figure 24) it can be concluded that magnetic ordering takes place between these
two temperatures and that only one transition occurs (indicating the presence of
only one phase) The parameters of one of the Fe(II) doublets at 250 K (designated
B in Table 22) are in very good agreement with previously published values for
the vivianite Fe(Il)8 site at room temperature (eg McCammon amp Burns 1980)
and the ordering temperature also agrees well with an assignment as vivianite
(Forsyth et al 1970) However the second Fe(II) doublet in the initial colourless
precipitate (Table 22) has parameters that deviate from previously reported values
by having a smaller quadrupole splitting (indicating a less distorted coordination)
and a significantly higher relative intensity and line width These effects might be
due to the presence of numerous defects in the vivianite crystal lattice particularly
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 27
affecting the Fe(II)A sites It should be noted that further components may be added
to the fit in order to improve its statistics Nevertheless we decided to include no
further components as suggested by the finding of one magnetic ordering only
Accordingly our interpretation of the Moumlssbauer results for the initial white
precipitate suggests a highly defective vivianite having a distribution of local
coordination environments particular in the A site This assignment is further
supported by a major absorption band due to phosphate anions in the infrared
spectrum at approximately 1000 cm-1 and the absence of other complex anions
(data not shown) Thus the whitish precipitate is referred to as a vivianite-like
(ldquovivianiterdquo) precipitate
0
3 5
3 0
2 5
2 0
1 5
1 0
0 5
0 0
20 K
Velocity (mms)
Rel
ativ
e ab
sorp
tion
()
3
2
1
0
80 K
3 5
3 0
2 5
2 0
1 5
1 0
0 5
0 0
150 K
-5 -4 -3 -2 -1 0 1 2 3 4 5
2 5
2 0
1 5
1 0
0 5
0 0
250 K
Figure 23 Transmission Moumlssbauer spectra measured between 250 and 20 K of the initial
whitish precipitate prior to inoculation (see Fig 22a) Fitting components (and their sum) are
shown as full lines
28 Chapter 2
Table 22 Selected Mossbauer parameters of the doublet components in the spectra obtained for
different precipitates
Precipitates Temperature Isomer shift Quadrupole Line width Area (K) (mms-1) splitting (mms-1) (mms-1) ()
Whitish Fe(II)B 250 127 309 035 38
Fe(II)A 250 128 181 051 62
Dark greenish Fe(II)B 250 126 305 023 17
Fe(II)A 250 132 238 051 44
Fe(III) 250 036 085 040 38
Reddish-orange 40 138 244 094 67 Fe(II) Hyperfine parameters are generally given with uncertainties of 003 mms- the spectral area with an uncertainty of 3
1005
1000
0995
0990
- 0985 ~ e c 0980 0
-~ 0975 E c nl b 1000 g ~ Qi 0995 0
0990
0985
0980 -12 -8
~ -~
bullbull bull Ibull bull bullbull bull bull ~ bull bull ~
It
10 K
bull bull bull bull (J ~ i~ 6K bullmiddotf bull bull
~ bull bull bull
-4 0 4 8 12
Velocity (mmls)
Figure 24 Transmission Mossbauer spectra measured at 10 and 6 K of the initial whitish
precipitate prior to inoculation (see Fig 22a)
In general the color of the media suspension changed from whitish into light green
color within 2-3 days after inoculation (Figure 22b ) This transformation occurred
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 29
without dissolving the initial whitish precipitate or preserving the whitish
precipitate as a separate phase implying some kind of solid state transformation
Two identical media suspensions were inoculated simultaneously but at the
sampling time they had distinctly different intensities of the green color
designated light and dark green respectively Moumlssbauer spectra of the dark green
sample are shown in Figure 25 The spectra of this sample are all fitted using three
doublet components (two Fe(II) and one Fe(III)) and the parameters of the
spectrum measured at 250 K are given in Table 22
30
25
20
15
10
05
00
20 K
Velocity (mms)
Rel
ativ
e ab
sorp
tion
()
35
30
25
20
15
10
05
00
80 K
30
25
20
15
10
05
00
150 K
-5 -4 -3 -2 -1 0 1 2 3 4 5
25
20
15
10
05
00
250 K
Figure 25 Transmission Moumlssbauer spectra measured between 250 and 20 K of the dark
greenish precipitate formed during biooxidation Fitting components (and their sum) are shown
as full lines
No magnetic ordering of the dark green precipitate was observed at temperatures
above 20 K but ordering occurred around 10 K (not shown - due to very thin
30 Chapter 2
samples this was not investigated in details) The two greenish samples had very
similar parameters only differing in the relative intensity of Fe(III) 26 and 38
in the light greenish and dark greenish samples respectively Assuming the
spectral area of a component to be proportional to the abundance of the species in
the solid these results indicate a correlation between the intensity of the green
color and the content of Fe(III) in the precipitate The parameters of the Fe(II)
doublets in the dark green precipitate (Table 22) were in very good agreement
with previously published values for vivianite with a non-negligible Fe(III) content
(McCammon amp Burns 1980) whereas the Fe(III) component in particular had a
higher quadrupole splitting The observation that magnetic ordering of both Fe(II)
and Fe(III) occurred at similar temperature for the light green phase (Figure 26) is
a strong indication that they both belong to the same phase The absence of the
component with the low quadrupole splitting in the spectra of both green samples
might indicate that the initial vivianite-like phase crystallized into a more well-
defined vivianite over time However freshly prepared and long-term aged (gt1
year) suspensions of the initial vivianite-like precipitate did not differ significantly
Hence we suggest that the recrystallization of the vivianite-like precipitate can be
explained by Fe(II) biooxidation
Solid State Oxidation ofFe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria
0
2
3
- 4
~ 5 c ~ 6 e-0 7 -2 nl Q) 00 gt ~ 05 Qi 0 10
15
20
25
30
35
bullbullbull hi 6~
bullbullbullbull bull bull bullbull bull 10 K bull bull
bull bull bull bull bull bull bull
~~ ~~ lf 6K bull bull bullbull middot bull bull r bull
bull bull bull bull bull bullbull bull bull bull - 12 -a -4 0 4 8 12
Velocity (mms)
31
Figure 26 Transmission Mossbauer spectrn measured at 6 and 10 K of the light greenish
precipitate fonned during biooxidation
The solid state oxidation of monoclinic vivianite to triclinic metavivianite is well-
known (McCammon amp Bums 1980 Pratesi et al 2003 and references therein)
The vivianite crystal structure contains Fe(II) ions in both isolated Fe(II)A and
paired Fe(Il)8 octahedra Mossbauer spectroscopic analyses have shown that the
Fe(Il)8 Fe(II)A ratio increases with increasing Fe(III) concentration suggesting that
the remaining Fe(II)A ions are more readily oxidized than the Fe(II)a ion of an
Fe(Il)8 -Fe(III)8 pair (McCammon amp Bums 1980) The mechanism of oxidation of
Fe(II) in vivianite involves conversion of H20 ligands to OH- ions producing a
progressive collapse of the vivianite structure due to the elimination of hydrogen
bonds (Moore 1971) The exact oxidation limits between which the triclinic lattice
is stable are somewhat disputed as the results obtained for synthetic and natural
vivianites oxidized chemically in the laboratory and naturally oxidized natural
vivianite specimens do not agree completely (Rodgers 1986 and references
therein) Taking all reports into account the monoclinic structure of vivianite is
supposedly maintained until 40-50 of total iron is oxidized Further oxidation
32 Chapter 2
leads to the formation of the triclinic metavivianite in which the FeA site is fully
oxidized whereas the oxidation of the FeB ranges from 20 to almost 100 Thus
the triclinic metavivianite structure persists close to complete oxidation of total
iron The Moumlssbauer results obtained in this study are consistent with the vivianite
solid state oxidation mechanism reported by McCammon amp Burns (1980) Thus
we propose that the intermediate greenish precipitate is a metavivianite-like
(ldquometavivianiterdquo) phase It should be noted that a minor oxidation of dissolved
Fe(II) may have occurred even though the solid state oxidation of Fe(II) was
predominant
Within 5-6 days after inoculation the greenish intermediate was transformed into a
reddish product (Figure 22c) The magnetically ordered sextet in the spectrum of
the red phase (Figure 27) measured at 40 K was due to goethite (α-FeOOH)
(magnetic hyperfine field of 470 T and a quadrupole shift of -01 mms-1 cp
Moslashrup et al 1983) The sextet deviated from ideal goethite by its asymmetric line
shape and its low ordering temperature (around 100 K ndash data not shown) and thus
the goethite was poorly crystalline It is very likely that the presence of phosphate
in the media retarded the crystal growth of goethite The unusual reddish colour of
the goethite might also be explained by the presence of phosphate The spectrum at
40 K was however dominated by a Fe(II) doublet (Table 22) that ordered
magnetically between 40 and 20 K (Figure 27) The hyperfine parameters and the
magnetic ordering temperature indicated that this component was due to siderite
(FeCO3) having a magnetic ordering temperature of 38 K (Jacobs 1963) The
siderite component may have formed as a result of the microbial activity changing
the chemistry of the solution and precipitating a major part of the remaining
dissolved Fe(II) at this stage The characteristic vivianite Fe(II) doublet with large
quadrupole splitting was not detected in this sample The reddish precipitate
contained considerably less Fe(III) than the greenish precipitate (only 33 as
estimated from the spectral area) None of the components in the reddish sample
Solid State Oxidation ofFe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 33
could be detected in freshly inoculated samples indicating that carryover of mineral
precipitates by inoculation of the culture media was negligible
0
2
- 3
~4 c
Q 5
e 6 0
~ 7 g
0 3l Q)
a 1
2
3
4
5
-12 a
40K
bull bull (
4 0 4 8 12 Velocity (mms)
Figure 27 Transmission Mossbauer spectra measured at 20 and 40 K of the reddish precipitate
fo1med during the late biooxidation stage Fitting components (and their sum) are shown as full
lines
Strengite (FeP04middot2H20) was not detected at any time during oxidation
Santabarbaraite a new amorphous F e(III) hydroxy phosphate mineral
(Fe3(P04)i(OH)3middot5H20) was reported in a recent study (Pratesi et al 2003) The
brownish mineral was a result of the solid state oxidation of vivianite through
metavivianite However no Mossbauer data have been provided for this new
mineral yet and therefore we cannot give an account of whether santabarbaraite
forms in our system or not Thus goethite was the dominating end product and we
propose the reaction path depicted in Figure 28 for the nitrate-dependent
biooxidation of Fe(II) in our systems
34 Chapter 2
Fe3(PO4)2middot8H2O (FeII3-xFeIII
x)(PO4)2(OH)xmiddot(8-x)H2O α-FeOOH
NO3- N2 NO3
- N2
ldquoVivianiterdquo ldquoMetavivianiterdquo Goethite
Figure 28 Proposed reaction path and iron-containing minerals forming during solid state
oxidation of vivianite by denitrifying FeOB at the experimental conditions applied in this study
The biotic formation of layered Fe(II)-Fe(III) hydroxides (green rusts) by
anaerobic denitrifying Fe(II)-oxidizing bacteria has been suggested but proper
identification of these phases still lacks (Chaudhuri et al 2001) We cannot rule
out that small amounts of green rusts (GRs) perhaps a phosphate intercalated GR
(Hansen amp Poulsen 1999) might have been present here during the greenish
intermediate ldquometavivianiterdquo oxidation stage When present in low concentrations
especially in mixtures including other iron minerals it is very difficult to identify
GRs even with Moumlssbauer spectroscopy At least two complementary methods
such as X-ray diffraction (XRD) and Moumlssbauer spectroscopy are required for
proper identification and characterization of GRs However the precipitates
collected in this work were poorly crystalline and did not allow for XRD analysis
Electron micrographs including energy dispersive X-ray spectroscopy suspension
colour and mineral stability calculations do not suffice as evidence Hence no
convincing evidence of GR formation facilitated by denitrifying FeOB has been
provided so far The blue-green colours of metavivianite and green rust minerals
originate from Fe(II)-Fe(III) charge transfer between adjacent Fe(II) and Fe(III)
ions in edge-shared octahedra (Faye et al 1968) The greenish suspension colour
occurring during the intermediate phase has incited the idea of biogenic GRs in
studies on nitrate-dependent Fe(II) biooxidation (Chaudhuri et al 2001 Lack et
al 2002aampb) However our results indicate that this reasoning is misleading
232 Factors controlling the rate and extent of Fe(II) biooxidation
Generally it was found that maximally 20-64 of the initial Fe(II) amount was
oxidized to Fe(III) (Figure 29) This indicates some limitations in the accessibility
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 35
of Fe(II) in the system Based solely on stoichiometry considerations the
microorganisms are expected to oxidize 5 mol Fe(II) for every 1 mol nitrate
reduced to dinitrogen However as exemplified in Figure 28 this ratio was mostly
lt4 which can be explained by the consumption of nitrogen as a result of microbial
growth At initial [Fe(II)][NO3ndash] ratios lt5 nitrate is in excess and should not limit
the extent of the biooxidation Thus the lack of complete biooxidation could not be
due to exhaustion of nitrate Furthermore all growth essential nutrients were more
than sufficiently applied hence the incomplete Fe(II) biooxidation was not caused
by lack of nutrients The most reasonable explanation therefore seems to be that an
increasingly limited access to the electron donor over time inhibits complete long-
term Fe(II) biooxidation At least four mechanisms may cause this inhibition 1)
the Fe(II) becomes isolated within the structure of the mixed Fe(II)-Fe(III)
minerals forming during biooxidation or underneath a passive Fe(III)-bearing
surface film on the initial Fe(II) precipitates 2) the FeOB cell surface becomes
covered with a passive Fe(III)-bearing surface film 3) the Fe(II) biooxidation is
controlled by the rate of dissolution of the initial Fe(II) minerals or 4) the reaction
proceeds primarily by biooxidation of dissolved Fe(II) whose concentration
gradually decreases due to changes in solid phase composition The actual
mechanisms whereby the surface-associated Fe(III) can inhibit Fe(II) biooxidation
are unknown but they may involve both kinetic and thermodynamic constraints on
the electron transfer The Moumlssbauer results obtained in this work strongly suggest
that the Fe(II) biooxidation occurred mainly in the solid state of the initial
ldquovivianiterdquo phase However we cannot rule out that some dissolved Fe(II) was
oxidized as well
36 Chapter 2
Figure 29 Concentration profiles of total Fe(II) and nitrate as a function of time during Fe(II)
biooxidation
No Fe(II) oxidation took place in cultures where nitrate had been omitted
confirming that the microbial Fe(II) oxidation is nitrate-dependent (data not
shown) No Fe(II) oxidation was detected in the non-inoculated control
experiments within the duration of the experiments and thus the chemical
oxidation of dissolved Fe(II) by nitrate catalyzed by vitamins or trace elements
(eg Cu(II)) can be neglected Ammonium did not form in detectable amounts
during Fe(II) biooxidation (data not shown) and therefore dinitrogen was assumed
to be the end product as reported previously (Straub et al 1996 Benz et al 1998)
The absence of ammonium formation does indirectly support the absence of
biologically induced green rust formation as synthetic green rust is known to
convert nitrate into ammonium in purely chemical reactions (Hansen et al 1996)
It was visually observed that the phosphate concentration in the media exerted a
control on the microbial Fe(II)-oxidation At phosphorus concentrations le 2 mM
no Fe(II)-oxidation took place However the solubility product for vivianite (Ksp =
171middot10-36 at 25degC (Al-Borno et al 1994)) was still by far exceeded under these
conditions It is not known whether this phosphate limiting effect is due to growth
constraints in the mixed bacterial community or whether specific Fe(II) phosphate
Solid State Oxidation ofFe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 37
precipitates are prerequisites of the Fe(II) biooxidation to take place Experiments
are currently underway in our laboratory in order to elucidate the role of specific
initial Fe(II) precipitates It should be noted that the growth of the denitrifying
FeOB could not be estimated as they were present in highly heterogeneous
suspensions containing both solid iron phases as well as other bacteria (enrichment
culture)
233 Morphology of solid iron phases
The morphology of the various precipitates was studied by SEM The initial
whitish precipitate consisted of a web-like structure (Figure 21 Oa and background
in Figure 21 Ob) whereas more distinct hexagonally shaped rosettes with particle
size ~20 microm (Figure 2lObampc) formed during Fe(II) biooxidation The energy
dispersive spectroscopic element analyses showed that other than iron the initial
whitish precipitate and the rosettes contained mainly phophorus
Figure 210 Scanning electron micro graphs of precipitates fo1med at various stages of the
experiment a Initial whitish precipitate bampc Rosettes observed in the intennediate greenish
precipitate d Reddish precipitate sampled during the late biooxidation stage
38 Chapter 2
These observations are interpreted as vivianite forming a web-like morphology in
the initial whitish precipitate and partly transforming into hexagonal particles in
the greenish colored stage The interpretations are supported by similar vivianite
morphologies reported including pseudo-hexagonal vivianite crystals of low
symmetry resulting from microbial Fe(III) reduction of HFO and platy rosettes of
vivianite crystals formed during bioreduction of Fe(III) in smectite (Fredrickson et
al 1998 Dong et al 2003) It was not possible to associate the morphology
observed in the reddish precipitate with the minerals identified in this phase
(Figure 210d)
24 Conclusions
This work demonstrated that anaerobic autotrophic denitrifying Fe(II)-oxidizing
bacteria produce poorly crystalline goethite by solid state oxidation of ldquovivianiterdquo
via a ldquometavivianiterdquo intermediate The increasing amount of Fe(III) forming in the
vivianite structure was accompanied by an increasing intensity in the green colour
as the Fe(II) biooxidation progressed Moumlssbauer spectroscopic analyses provided
no significant evidence of green rust formation The finding of microbially
oxidized vivianite in this study raises the question of the oxidation state of
vivianite specimens from natural sediments Vivianite is generally believed to be
an ideal Fe(II) hydroxy phosphate mineral and the presence of Fe(III) is explained
by aerial oxidation upon sampling The results presented here indicate that
microbiological processes may be responsible for the oxidation of vivianite and
metavivianite in natural subsurface environments Acknowledgments
We would like to thank Dr K Straub for providing and advising us how to culture the nitrate-
reducing FeOB Furthermore we thank Dr C B Koch for performing the Moumlssbauer analyses
and Dr D Mavrocordatos for performing the SEM analyses
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 39
References Al-Borno A Tomson MB (1994) The temperature dependence of the solubility product constant of vivianite Geochimica et Cosmochimica Acta 58 5373-5378 Benali O Abdelmoula M Refait Ph Geacutenin JMR (2001) Effect of orthophosphate on the oxidation products of Fe(II)-Fe(III) hydroxycarbonate The transformation of green rust to ferrihydrite Geochimica et Cosmochimica Acta 65 1715-1726 Benz M Brune A Schink B (1998) Anaerobic and aerobic oxidation of ferrous iron at neutral pH by chemohetorotrophic nitrate-reducing bacteria Archives of Microbiology 169 159-165 Chaudhuri SK Lack JG Coates JD (2001) Biogenic magnetite formation through anaerobic biooxidation of Fe(II) Applied and Environmental Microbiology 67 2844-2848 Cumplido J Barron V Torrent J (2000) Effect of phosphate on the formation of nanophase lepidocrocite from Fe(II) sulfate Clays and Clay Minerals 48 503-510 Dodd J Large DJ Fortey NJ Kemp S Styles M Wetton P Milodowski A (2003) Geochemistry and petrography of phosphorus in urban canal bed sediment Applied Geochemistry 18 259-267 Dong H Kostka JE Kim J (2003) Microscopic evidence for microbial dissolution of smectite Clays and Clay Minerals 51 502-512 Ehrenreich A Widdel F (1994) Anaerobic oxidation of ferrous iron by purple bacteria a new type of phototrophic metabolism Applied and Environmental Microbiology 60 4517-4526 Emerson D Moyer CL (2002) Neutrophilic Fe-oxidizing bacteria are abundant at the Loihi seamount hydrothermal vents and play a major role in Fe oxide deposition Applied and Environmental Microbiology 68 3085-3093 Emerson D Weiss JV Megonigal JP (1999) Iron-oxidizing bacteria are associated with ferric hydroxide precipitates (Fe-plaque) on the roots of wetland plants Applied and Environmental Microbiology 65 2758-2761 Fadrus H Maly J (1975) Suppression of iron(III) interference in the determination of iron(II) in water by the 110-phenanthroline method The Analyst 100 549-554 Faye GH Manning PG Nickel EH (1968) The polarized optical absorption spectra of tourmaline cordierite chloritoid and vivianite ferrous-ferric electronic interaction as a source of pleochroism American Mineralogist 53 1174-1201 Forsyth JB Johnson CE Wilkonson C (1970) The magnetic structure of vivianite Fe3(PO4)2middot8H2O Journal of Physics Part C Solid State Physics 3 1127-1139 Fredrickson JK Zachara JM Kennedy DW Dong H Onstott TC Hinman NW Li S (1998) Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium Geochimica et Cosmochimica Acta 62 3239-3257 Gaines RV Skinner HCW Foord EE Mason B Rosenzweig A (1997) Danas new
40 Chapter 2 mineralogy 8th ed John Wiley amp Sons Inc Hanert HH (1992) The genus Gallionella In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 4082-4088 Hansen HCB Koch CB Nancke-Krogh H Borggaard OK Soerensen J (1996) Abiotic nitrate reduction to ammonium Key role of green rust Environmental Science and Technology 30 2053-2056 Hansen HCB Poulsen IF (1999) Interaction of synthetic sulphate green rust with phosphate and the crystallization of vivianite Clays and Clay Minerals 47 312-318 Henderson GS Black PM Ridgers KA Rankin PC (1984) New data on New Zealand vivianite and metavivianite New Zealand Journal of Geology and Geophysics 27 367-378 House WA (2003) Geochemical cycling of phosphorus in rivers Applied Geochemistry 18 739-748 Jacobs IS (1963) Metamagnetism of siderite (FeCO3) Journal of Applied Physics 34 1106-1107 Jorand F Appenzeller BMR Abdelmoula M Refait Ph Block J-C Geacutenin JMR (2000) Assessment of vivianite formation in Shewanella putrefaciens culture Environmental Technology 21 1001-1005 Kandori K Uchida S Kataoka S Ishikawa T (1992) Effects of silicate and phosphate ions on the formation of ferric oxide hydroxide particles Journal of Materials Science 27 719-728 Kuenen JG Robertson LA Tuovinen OH (1992) The genera Thiobacillus Thiomicrospira and Thiosphaera In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 2618-2624 Lack JG Chaudhuri SK Chakraborty R Achenbach LA Coates JD (2002a) Anaerobic biooxidation of Fe(II) by Dechlorosoma suillum Microbial Ecology 43 424-431 Lack JG Chaudhuri SK Kelly SD Kemner KM OConnor SM Coates JD (2002b) Immobilization of radionuclides and heavy metals through anaerobic bio-oxidation of Fe(II) Applied and Environmental Microbiology 68 2704-2710 Lovley DR (1997) Microbial Fe(III) reduction in subsurface environments FEMS Microbiology Reviews 20 305-313 Manning PG Birchall T Jones W (1981) Ferric hydroxides in surficial sediments of the great lakes and their role in phosphorus availability a Moumlssbauer spectral study Canadian Mineralogist 19 525-530 Manning PG Jones W (1982) The binding capacity of ferric hydroxides for non-apatite inorganic phosphorus in sediments of the depositional basins of Lakes Erie and Ontario Canadian Mineralogist 20 169-176 McCammon CA Burns RG (1980) The oxidation mechanism of vivianite as studied by Moumlssbauer spectroscopy American Mineralogist 65 361-366 Moore PB (1971) The Fe2+
3(H2O)n(PO4)2 homologous series crystal-chemical relationships
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 41
and oxidized equivalents American Mineralogist 56 1-17 Moraghan JT Buresh RJ (1976) Chemical reduction of nitrate by ferrous iron Journal of Environmental Quality 5 320-325 Moraghan JT Buresh RJ (1977) Chemical reduction of nitrite and nitrous oxide by ferrous iron Journal of American Soil Science Society 40 47-50 Morales JA de Graterol LS Mesa J (2000) Determination of chloride sulfate and nitrate in groundwater samples by ion chromatography Journal of Chromatography A 884 185-190 Mulder EG Deinema MH (1992) The sheathed bacteria In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 2618-2624 Moslashrup S Madsen MB Franck J Villadsen J Koch CJW (1983) A new interpretation of Moumlssbauer spectra of microcrystalline goethiterdquosuper-ferromagnetismrdquo of ldquosuper-spin-glassrdquo behaviour Journal of Magnetism and Magnetic Materials 40 163-174 Nembrini GP Capobianco JA Viel M Williams AF (1983) A Moumlssbauer and chemical study of the formation of vivianite in sediments of Lago Maggiore (Italy) Geochimica et Cosmochimica Acta 47 1459-1464 Nielsen JL Nielsen PH (1998) Microbial nitrate-dependent oxidation of ferrous iron in activated sludge Environmental Science and Technology 32 3556-3561 Nriagu JO (1972) Stability of vivianite and ion-pair formation in the system Fe3(PO4)2-H3PO4- H2O Geochimica et Cosmochimica Acta 36 459-470 Nriagu JO Dell CI (1974) Diagenetic formation of iron phosphates in recent lake sediments American Mineralogist 59 934-946 Patrick Jr WH Khalid RA (1974) Phosphate release and sorption by soils and sediments Effect of aerobic and anaerobic conditions Science 186 53-55 Postma D (1981) Formation of siderite and vivianite and the pore-water composition of a recent bog sediment in Denmark Chemical Geology 31 225-244 Pratesi G Cipriani C Giuli G Birch WD (2003) Santabarbaraite a new amorphous phosphate mineral European Journal of Mineralogy 15 185-192 Ratering S Schnell S (2001) Nitrate-dependent iron(II) oxidation in paddy soil Environmental Microbiology 3 100-109 Ritz C Essene EJ Peacor DR (1974) Metavivianite Fe3(PO4)2middot8H2O a new mineral American Mineralogist 59 896-899 Rodgers KA (1986) Metavivianite and kerchenite a review Mineralogical Magazine 50 687- 691 Rodgers KA Johnston JH (1985) Type metavivianite Moumlssbauer evidence for a revised composition Neues Jahrbuch fuumlr Mineralogie-Monatshefte 12 539-542
42 Chapter 2 Rossum JR Villarruz PA (1963) Determination of ammonia by the indophenol method Journal of American Water Works Association 55 657-658 Seitz MA Riedner RJ Malhotra SK Kipp RJ (1973) Iron-phosphate compound identification in sewage sludge residue Environmental Science and Technology 7 354-357 Sobolev D Roden EE (2001) Suboxic deposition of ferric iron by bacteria in opposing gradients of Fe(II) and oxygen at circumneutral pH Applied and Environmental Microbiology 67 1328-1334 Straub KL Benz M Schink B Widdel F (1996) Anaerobic nitrate-dependent microbial oxidation of ferrous iron Applied and Environmental Microbiology 62 1458-1460 Straub KL Buchholz-Cleven BEE (1998) Enumeration and detection of anaerobic ferrous iron-oxidizing nitrate-reducing bacteria from diverse European sediments Applied and Environmental Microbiology 64 4846-4856 Straub KL Hanzlik M Buchholz-Cleven BEE (1998) The use of biologically produced ferrihydrite for the isolation of novel iron-reducing bacteria Systematic and Applied Microbiology 21 442-449 Weber KA Picardal FW Roden EE (2001) Microbially catalyzed nitrate-dependent oxidation of biogenic solid-phase Fe(II) compounds Environmental Science and Technology 35 1644-1650 Widdel F Bak F (1992) Gram-negative mesophilic sulfate-reducing bacteria In The Prokaryotes (Balows A Truumlper HG Dworkin M Harder W Schleifer K-H (eds)) Springer 2nd ed 3352-3378
Widdel F Schnell S Heising S Ehrenreich A Assmus B Schink B (1993) Ferrous iron oxidation by anoxygenic phototrophic bacteria Nature 362 834-836 Williams JDH Syers JK Shukla SS Harris RF Armstrong DE (1971) Levels of inorganic and total phosphorus in lake sediments as related to other sediment parameters Environmental Science and Technology 5 1113-1120
Zwaan PC Kortenbout van der Sluys G (1971) Vivianite crystals from Hare Noord Brabant Province The Netherlands Scripta Geology 6 1-7
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 43
3 Formation of Layered Iron Hydroxides by Microbial Fe(III)
Reduction Abstract
Many inorganic and organic pollutants may be degraded by microorganisms in the
subsurface However a wide range of contaminants including chromate nitrate
radionuclides nitroaromatic compounds chlorinated aliphatics and carbamate
pesticides may also be chemically transformed by reduction reactions involving
layered iron(II)-iron(III)-hydroxides (green rusts) Hence green rusts (GRs) may
play a potentially important role in the fate and transport of pollutants in iron-rich
suboxic soils and sediments Yet only little is known about the formation of GRs
in these environments The biotic formation of GRs mediated by the anaerobic
dissimilatory Fe(III)-reducing bacteria Shewanella spp has been reported and
proposed in several studies However the experimental conditions applied were
mostly not natural and the evidence of GR formation provided may be questioned
This work investigated the Fe-containing products formed by the facultatively
anaerobic Fe(III)-reducing microorganism Shewanella algae BrY in culture
media containing 4-10 mM formate or lactate and 8-27 mM Fe(III) In order to
simulate natural conditions Fe(III) oxides were applied as coatings on silica
(model system for sandy soils) or calcite particles (model system for calcareous
soils) and synthetic electron shuttles as well as highly concentrated artificial pH
buffers were excluded S algae BrY reduced 19-72 of the initial Fe(III) when
grown in goethitecalcite lepidocrocitecalcite or hydrous ferric oxidesand mineral
systems and green or blackish mineral phases were produced within 1-2 weeks
after inoculation Moumlssbauer spectroscopic analyses indicated that the green and
blackish precipitates were dominated by vivianite (Fe3(PO4)2sdot8H2O) and green rust
44 Chapter 3
31 Introduction
The significance of bacteria in the biogeochemical cycling of iron has been broadly
recognized over the past two decades Chemical processes were previously
considered to account for most of the Fe(III) reduction in subsurface environments
Dissimilatory Fe(III)-reducing bacteria (DIRB) that gain energy by coupling the
oxidation of hydrogen or organic compounds to the reduction of Fe(III) oxides
have been known for many years but their biogeochemical importance was
acknowledged only a decade ago (reviewed by Lovley 1997) DIRB transfer
electrons to extracellular Fe(III) without assimilating the iron Fe(III) bioreduction
accounts for a major fraction of the carbon oxidation in many different
environments and in the presence of high amounts of reactive Fe(III) microbial
Fe(III) reduction may even inhibit sulfate reduction and methanogenesis (King
1990 Lovley amp Phillips 1986) In fact most of the Fe(III) reduction in the Fe(III)
reduction zone of aquatic sediments and aquifers is thought to be enzymatically
catalyzed by microorganisms (Lovley et al 1991) However the relative
importance of microbial and chemical processes involved in the Fe(III) reduction
are still somewhat disputed among microbiologists and geochemists
A wide diversity of Fe(III)-reducing bacteria which fall in a number of different
phylogenetic groups is known today Both organisms growing by respiration and
by fermentation have been isolated and identified (Lovley 1991 Nealson amp
Saffarini 1994) Hydrogen short- and long-chained fatty acids amino acids
sugars and aromatic compounds may serve as electron donors for Fe(III)
bioreduction The enzymes responsible for dissimilatory Fe(III) reduction are outer
membrane associated ferric reductases (Lower et al 2001 and references therein)
Iron reducing bacteria may utilize alternative electron acceptors such as O2 nitrate
S0 sulfate humic substances contaminant metals and metalloids as well as
chlorinated solvents The first organism shown to couple respiratory growth to
dissimilatory iron reduction was Pseudomonas ferrireductans now known as
Shewanella oneidensis but previously classified as Alteromonas putrefaciens and
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 45
Shewanella putrefaciens (Venkateswaran et al 1999) Various DIRB including
the obligate anaerobic Geobacter sp and the facultatively anaerobic Shewanella
sp have been isolated from both marine and freshwater sediments soil and
aquifers (Thamdrup 2000 and references therein)
The redox potentials of oxidized and reduced iron couples and thus the energy
yield available from Fe(III) reduction depend strongly on the specific iron phases
involved In soil and aquatic environments Fe(III) oxides mainly occur in
association with other sediment particles as aggregates or coatings Amorphous
and poorly crystalline Fe(III) oxides usually make up 20 or less of the iron
content in a sediment (Thamdrup 2000) They are the main products of abiotic and
biotic Fe(II) oxidation in sediments and they constitute the most important phases
for microbial Fe(III) reduction Until recently it was generally believed that DIRB
reduced insoluble Fe(III) oxides only by direct contact with the Fe(III) oxide
thereby allowing electron transfer from the cell to the Fe(III) oxide surface
However over the past several years there has been a growing recognition that
DIRB may use different strategies in order to access the solid Fe(III) oxides These
strategies include solubilization of Fe(III) by synthetic or natural Fe(III) chelators
and Fe(III) reduction via electron shuttling with soluble humic substances or
microbially produced electron shuttles (Nevin amp Lovley 2002 and references
therein Turick et al 2003) The Fe(III) complexing agents may also stimulate
Fe(III) oxide reduction indirectly by chelation and thus removal of Fe(II) from
the cell and the Fe(III) oxide surfaces Both chelated Fe(III) and soluble electron
shuttles are more accessible to Fe(III) reductases than solid Fe(III) oxides In
contrast to Geobacter metallireducens S algae produces and releases extracellular
electron shuttling compounds (Nevin amp Lovley 2000) However in the absence of
soluble electron shuttles reversible adhesion is required for reduction of solid
Fe(III) oxides by S algae BrY (Das amp Caccavo 2000) Shewanella algae BrY
adheres readily and preferentially to a range of solid Fe(III) oxides such as
ferrihydrite goethite and hematite (Das amp Caccavo 2001) The adhesion
46 Chapter 3
mechanisms are not completely understood but recent results suggest that the
adhesion is mediated by cell surface proteins and independent of cell motility
(Caccavo amp Das 2002)
The microbial formation of GRs resulting from bioreduction of various Fe(III)
oxides including ferrihydrite goethite and lepidocrocite by strains of the
anaerobic DIRB Shewanella putrefaciens has been reported repeatedly over the
last years (Fredrickson et al 1998 Kukkadapu et al 2001 Liu et al 2001
Parmar et al 2001 Ona-Nguema et al 2002aampb Glasauer et al 2003)
However no evidence of biogenic formation of GRs at natural geochemical
conditions have been offered and it is still unknown whether this process may take
place at natural conditions comprising low carbon and iron concentrations as well
as the absence of synthetic electron shuttles and highly concentrated artificial pH
buffers GRs are layered iron(II)-iron(III)-hydroxides with anionic interlayers and
they hold the general formula [FeII(6-x)FeIII
x(OH)12]x+[(A)xnmiddotyH2O]x- where x = 09 -
42 A is an n-valent anion eg CO32- Clndash or SO4
2- and y is the number of water
molecules in the interlayer In circumneutral solutions the oxidation of dissolved
Fe(II) always passes through solid GR phases (Bernal et al 1959) This agrees
with the natural GR occurrences found in suboxic nonacid iron-rich environments
such as hydromorphic soils and intertidal sediments (Al-Agha et al 1995 Trolard
et al 1996 Genin et al 1998) In addition GRs have been found as corrosion
products in numerous engineering systems eg in a pipeline distribution system
for drinking water steel sheet piles in marine sediments reinforced concrete
(ferro-concrete) and permeable reactive barriers of zero-valent iron implemented
for on-site remediation of organic and inorganic contaminants (Tuovinen et al
1980 Nielsen 1976 Genin et al 1991 Roh et al 2000) Through sequestration
and reductive transformation GRs may play an important role in the fate and
transport of organic and inorganic pollutants in suboxic iron-rich soils and
sediments (see Chapters 4 amp 5 in this work and references therein)
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 47
The major goal of this work was to examine the iron minerals forming during the
course of Fe(III) bioreduction of hydrous ferric oxide goethite and lepidocrocite
Two model systems simulating sandy and calcareous soils in subsurface
environments were designed in order to investigate the formation of iron minerals
at conditions including low carbon levels low Fe(III) concentrations applied as
Fe(III) oxide coatings on sand or calcite no electron shuttle and no synthetic pH
buffers
32 Materials and methods
All handling and sampling of solutions and suspensions were carried out at strict
anoxic conditions Standard sterile techniques were used throughout (Hungate
1969 Miller amp Wolin 1974) Only the iron oxide coatings were not autoclaved in
order to avoid the iron oxides from transforming Goethite (acicular particles with
size 01 times 06 microm specific surface area 16 m2g) and lepidocrocite (acicular
particles with size 005 times 03 microm specific surface area 18 m2g) were purchased as
fine powders from Bayer (Bayferrox 910 and 943) Calcite (grain size 170-350 microm
Pluumlss-Staufer AG) and sea sand (dominantly quartz grain size 01-03 mm Riedel-
de Haeumln) were used as Fe(III) oxide coating bearing minerals
321 Preparation of iron oxide coatings
Two grams of goethite (goe) or lepidocrocite (lep) and 100 g calcite were
combined with 200 mL deionized water (DIW) in a 500 mL polyethylene flask
Hydrous ferric oxide (HFO) was synthesized by dissolving 4 g Fe(NO3)3middot9H2O in
70 mL DIW followed by slow neutralization under magnetic stirring till pH 7 with
approximately 30 mL 1 M NaOH (method modified after Schwertmann amp Cornell
1991) The HFO coating was made by combining 100 mL freshly precipitated
HFO with 900 mL deionized water and 50 g sea sand in a polyethylene bottle The
suspensions containing the iron oxide coatings were gently agitated on a
reciprocating shaker for 24 h and left to stand for another 24 h Excess Fe(III)
oxides and salts were removed from the coated material by repeated decantation
48 Chapter 3
and washing with 003 M NaNO3 followed by washing with DIW until clear
runoff Finally the coatings were collected on folding filters and air dried The
amount of HFO goethite and lepidocrocite coated onto sand and calcite after
washing and drying was quantified to 7-11 mg Fe(III)g sand or calcite
322 Mineral characterisation
The identity and purity of the HFO synthesized were examined by means of X-ray
diffraction (XRD) measurements The XRD analyses were performed on a Scintag
XDS 2000 using Co Kα radiation (45 kV 40 mA) using divergence scatter and
receiving slits of 1deg 05deg and 02 mm respectively Samples were scanned
between 6 and 80 deg2θ with a scan speed of 1 deg2θmin Mineral suspension samples
for transmission Moumlssbauer spectroscopic analysis were collected on 02 microm filters
in an anoxic glove box (Coy Laboratory Products Inc) transferred to Perspex
capsules and stored in liquid nitrogen until measurement Moumlssbauer spectra were
obtained between 250 and 5 K using a conventional constant acceleration
spectrometer and a source of 57Co in Rh The spectrometer was calibrated using a
125 microm foil of α-Fe at room temperature and isomer shifts are given relative to
the centroid of this absorber The spectra were fitted using simple Lorentzian line
shape and it was assumed that all positions have identical f-factors
323 Culture conditions and cell preparation
Shewanella algae BrY is a motile gram-negative rod which was isolated first from
anoxic estuary sediments (Caccavo et al 1992) S algae BrY was grown
aerobically in tryptic soy broth (30 gL CASO-bouillon Merck) at 28degC on a rotary
shaker at 150 rpm for 16-18 h Cells were harvested by centrifugation (6000 rpm times
g 4ordmC 15 min) during the late exponential ndash early stationary growth phase at
OD660 ~ 06 Optimal Fe(III) reductase activity is expressed at this stage of growth
(Roden amp Zachara 1996) The cells were washed twice in oxic 50 mM PIPES
[piperazine-NNacute-bis(2-ethanesulfonic acid)] buffer (pH 70) and resuspended in
culture medium containing no Fe(III) and no carbon source Washed cell
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 49
suspensions were used as inocula for Fe(III) reduction experiments Oxygen was
expelled from the inoculum by extensive purging with 100 N2(g) (9999999
purity) Working stock cultures of S algae BrY were maintained aerobically on
tryptic soy agar plates at ambient temperature
324 Bioreduction experiments
All anaerobic incubations were carried out in anoxic serum vials (25 mL) or test
tubes (13 mL) sealed with thick (10-13 mm) butyl rubber stoppers and aluminum
crimp caps or plastic screw caps The basal culture medium (Table 31) was
prepared according to Kostka amp Nealson (1998) but with a phosphate
concentration of 2 mM and the exclusion of Fe(II) and EDTA
(ethylenediaminetetraacetic acid) The medium was amended with 4-10 mM
lactate or formate and 8-27 mM Fe(III) The Fe(III) was applied as Fe(III) oxide
coatings on sand or calcite The suspensions were purged extensively with 100
N2(g) (HFOsand suspensions) or 995 N205 CO2(g) (goecalcite and
lepcalcite suspensions) prior to inoculation The calcareous systems were buffered
at pH ~ 76 through a natural buffer system (CaCO3(s) + 995 N205 CO2(g))
whereas the sandy systems contained no pH buffer (100 N2(g) pH 55-60)
Inoculum size made up 5 of the total volume Cultures were incubated dark at
room temperature and gently agitated once every day At different time intervals
suspension samples for Fe(II) and Moumlssbauer analysis were withdrawn from the
reaction mixture using 100 N2(g) or 995 N205 CO2(g)-flushed sterile
disposable syringes and hypodermic needles Suspension samples for Fe(II)
analysis were digested in 01 M HCl for 30 min
50 Chapter 3
Table 31 Composition of the mineral medium (modified from Kostka amp Nealson (1998))
Components Concentration (M)
(NH4)2SO4 00143 KH2PO4 73middot10-4
K2HPO4 13middot10-3
MgSO4middot7H2O 10middot10-3
CaCl2middot2H2O 50middot10-4
H3BO3 56middot10-5
ZnSO4middot7H2O 10middot10-6
Na2MoO4middot2H2O 40middot10-6
CuSO4middot5H2O 20middot10-7
MnSO4middotH2O 10middot10-6
Na2SeO4 12middot10-5
CoCl2middot6H2O 50middot10-6
NiCl2middot6H2O 80middot10-6
NaCl 10middot10-5
L-arginine 11middot10-4
L-serine 19middot10-4
L-glutamic acid 14middot10-4
Lactate or formate 4-10middot10-3
Fe(III) 8-27middot10-3
325 Analytical methods
Fe(II) was determined using a modified phenanthroline method (Fadrus amp Maly
1975) The total amount of Fe(III) coated on calcite and sand was determined by
atomic absorption spectroscopy following dissolution in 6 M HCl(aq) for 24 h
33 Results and discussion
331 Fe(II) production and suspension colour changes
Strongly chelating agents such as EDTA were omitted from the culture medium in
order to prevent complexation of Fe(II) and Fe(III) which interferes with
precipitation of Fe(II) and Fe(II)-Fe(III) mineral phases Within 1-2 weeks after
inoculation Shewanella algae BrY produced green mineral phases in media
suspensions containing lepidocrocite and goethite as coatings on calcite and 4-10
mM formate or lactate (Figure 31) The formation of the green precipitates was
generally slower for the lepidocrocite coating than for the goethite coating The
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 51
blue-green colours of the phases produced most likely originate from Fe(II)-Fe(III)
charge transfer between adjacent Fe(II) and Fe(III) ions in edge-shared octahedra
(Faye et al 1968) Dark brown and blackish products were formed when the
bacteria were inoculated on HFO coated sand (Figure 32)
Figure 31 Culture tubes containing a) goethite and b) lepidocrocite coated calcite in culture
medium The left tubes of the pair were not inoculated whereas the right tubes were
photographed 5 months after inoculation with S algae BrY Experimental conditions [formate]0
= 4 mM [Fe(III)]0 = 8 mM 995 N205 CO2(g) pH 76
52 Chapter 3
Figure 32 Culture tubes containing HFO coated sand in culture medium Tubes 1 and 2 to the
left were not inoculated whereas tubes 3-5 to the right were photographed a) 13 days and b) 21
days after inoculation with S algae BrY Experimental conditions [lactate]0 = 10 mM [Fe(III)]0
= 25 mM 100 N2(g) pH 55-60
The green and black colours did not change to other colours (observed for gt1
year) indicating that the microbial Fe(III) reduction ceased at these mineral stages
The concentrations of dissolved ferrous iron (Fe(II)sol) estimated during Fe(III)
bioreduction were generally low (Figure 33) When comparing the final Fe(II)sol
amounts produced and the slopes of the Fe(II)sol formation curves for HFO
goethite and lepidocrocite in Figure 33 it can be seen that the final Fe(II)sol
amount and the Fe(II)sol production rate both follow the order HFO gt goethite gt
lepidocrocite at similar cell densities regardless of the carbon source applied This
suggests that bioreduction by S algae BrY is more facile for HFO than for goethite
and lepidocrocite at the experimental conditions employed here It should be noted
that the final Fe(II)sol amounts and the Fe(II)sol production rates reported in this
work have not been normalised with respect to the specific surface areas of the iron
oxides and coating-bearing solids applied The reactivity trend is consistent with
previous findings demonstrating higher reducibility of natural and poorly
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 53
crystalline Fe(III) oxides as compared to synthetic crystalline Fe(III) oxides
(Zachara et al 1998) The authors ascribed these differences in reducibility to
differences in particle size surface area and crystal defects of the Fe(III) oxides In
some cases the dissolved Fe(II) concentration decreased again with time (Figure
33 b-d) This indicates that the Fe(II) formed was incorporated into solid phases
forming andor adsorbed onto the calcite sand or Fe(III) oxide surfaces The solid
Fe(II) concentrations were not estimated spectrophotometrically The solid
material was generally low in total iron and therefore saving it for Moumlssbauer
spectroscopic analysis was given highest priority
54 Chapter 3
Figure 33 Time course of dissolved Fe(II) production during bioreduction of HFOsand
goethitecalcite and lepidocrocitecalcite by S algae BrY Experimental conditions [Fe(III)]0 =
25 mM [formate]0 = 10 mM (a-c) or [lactate]0 = 10 mM (d-f)
No color change and no Fe(II) production were observed in mineral suspensions
lacking either a carbon source or S algae BrY cells (data not shown)
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 55
332 Identification of solid iron phases
The purity of the Fe(III) oxides used in the experiments were investigated by
transmission Mossbauer spectroscopy (Figure 34) Single (or strongly dominating)
sextets in the spectra with magnetic hyperfine fields of 48 1 505 and 45 5 Tat 5 K
demonstrated the purity of the HFO goethite and lepidocrocite samples
respectively A minor impurity of goethite in the lepidocrocite sample was
resolved in the spectrum measured at 80 K (not shown for pure sample but can be
seen as a magnetically ordered sextet in Figure 35c) No Fe(II)-containing
components were detected
a)
c)
middot 12 -8 -4 4 8 12
Velocity (mmls
bull middot12 -8 -4 0 4 12
Velocity (mmls)
b)
middot12 -8 -4 4 8 12
Velocity (mmls
Figure 34 Transmission Mossbauer spectra measured at 5 K of a) HFO (magnetic hyperfine
field of 481 T isomer shift of 048 1nrns-1 negligible quadrupole shift and line width of outer
lines 110 rmns-1) b) goethite (magnetic hyperfine field of 505 T isomer shift of 049 rmns-1
quadmpole shift of -013 1nrns-1 and line width of outer lines 042 rnrns-1
) and c) lepidocrocite
56 Chapter 3
(magnetic hyperfine field of 455 T isomer shift of 050 mms-1 quadrupole shift of ndash001 mms-1
and line width of outer lines 060 mms-1) prior to inoculation Simple Lorenztian fits are shown
The oxidation state and coordination of Fe in the microbially reduced HFO
goethite and lepidocrocite samples were also examined by transmission Moumlssbauer
spectroscopy (Figure 35) The bioreduced HFO goethite and lepidocrocite samples
cultured on formate contained Fe(II) holding similar coordination as inferred from
the similarity of the hyperfine parameters (see legend in Figure 35) but different
relative intensities (72 19 and 71 respectively) The major part of the Fe(III)
remaining in the bioreduced samples were coordinated similarly to the Fe(III)
present in the initial Fe(III) oxide The coordination of Fe(II) in the bioreduced
lepidocrocite samples cultured on lactate was slightly different (a smaller
quadrupole splitting of 288 mms-1 for the ferrous component dominates ndash data not
shown) The exact mineralogy of the Fe(II) present in the green phases was not
fully resolved but its coordination is very akin to one of the Fe(II) sites in vivianite
(see Chapter 2 this work) and synthetic green rusts (Koch 1998) These findings
agree with other reports on the bioformation of vivianite and green rusts by
Shewanella putrefaciens CN32 although the evidence provided may be discussed
(Fredrickson et al 1998 Glasauer et al 2003 Parmar et al 2001) Our
Moumlssbauer data on the green phases did not allow for a detailed account of the type
of green rust produced However when considering solution composition (see
Table 31) and the high affinity of GR interlayers for carbonate it is reasonable to
assume that carbonate GR was formed (Hansen amp Taylor 1991) Due to the high
amounts of Fe(III) in the oxides present in the experiments it was difficult to probe
a possible content of Fe(III) in the vivianite with certainty The differences in the
number of Fe(II) positions in the Moumlssbauer spectra and particular the different
temperatures at which magnetic ordering takes place can be employed in order to
distinguish between green rust and vivianite Preliminary Moumlssbauer data obtained
for the blackish precipitates formed in the HFOsand suspensions indicate that they
hold no resemblance to magnetite eventhough the colour suggests so On the
contrary the black precipitates seemed to be more similar to synthetic green rusts
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 57
Mossbauer spectroscopic measurements are currently underway in order to resolve
the Fe(II) coordinations in the greenish and blackish phases
a) b)
c c g Q e- e 0 0 1l 1l
-~ bull ~
iii ~ Qi bull a bull
-12 -8 4 0 4 12
4 -3 -2 -1 0 1 2 4 Velocity (mmts) Velocity ( rmis)
c)
-12 -8 4 8 12
v elocity (m mis)
Figure 35 Transmission Mossbauer spectra of the black and green phases fo1med within 1-2
weeks after inoculation of a) HFO (measured at 130 K) b) goethitecalcite (measured at 80 K)
and c) lepidocrocitecalcite (measured at 80 K) with S algae BrY Experimental conditions
[fo1m ate]0 = 4 mM [Fe(III)]o = 8 mM 995 Ni05 C02(g) pH 76 The quadrnpole
splittings and isomer shifts for the Fe(II) components in the three systems are a) 293 nnns-1 and
126 mmsmiddot1 b) 308 rmnsmiddot1 and131 rmnsmiddot1 and c) 322 mmsmiddot1 and 132 mmsmiddot1 Simple Lorenztian
fits are shown
The evidence provided in many of the studies proposing biogenic GRs is not all
too convincing but it strongly suggests the probability of microbially produced GR
being present The challenge encountered is that when present in low
58 Chapter 3
concentrations especially in mixtures including other iron minerals it is very
difficult to identify GRs using conventional solid phase analysis methods even
with Moumlssbauer spectroscopy At least two complementary methods such as XRD
and Moumlssbauer spectroscopy are required for proper identification and
characterization of GRs However in this work the solid materials were generally
too low in total iron to allow for XRD analysis Moreover the highly
heterogeneous suspensions were dominated by the coating-bearing sand and calcite
solids Electron micrographs including energy dispersive X-ray spectroscopy
suspension colour and mineral stability calculations do not suffice as evidence The
most convincing evidence provided so far involves an atypical GR-CO3 with an
Fe(II)Fe(III) ratio of 1 (Ona-Nguema et al 2002aampb) This GR-CO3 was formed
as a result of lepidocrocite reduction by Shewanella putrefaciens CIP 8040 at
conditions comprising high nutrient levels (50-75 mM formate) high Fe(III)
concentrations (80-300 mM) and a synthetic electron shuttle (100 microM
anthraquinone-26-disulfonate (AQDS)) at initial pH 75 Hence the results
reported during recent years suggest that microbial formation of GR may be
possible The results presented here indicate that GRs may be produced
microbially at conditions including low carbon and Fe(III) concentrations as well
as the exclusion of synthetic electron shuttles and pH buffers
333 Factors controlling the identity of the secondary iron minerals
In general one would expect that biogenic minerals have chemical compositions
and crystal habits similar to those produced by nonenzymatic processes as they are
governed by the same equilibrium principles In fact since the latter stages of
mineralization are inorganically driven and the secondary Fe(II)-containing
minerals are formed indirectly by electron transfer outside the bacterial cell and not
directly inside the bacterial cell the type of iron mineral formed is a function of the
environmental conditions in which the bacteria live ie the same microorganism
form different minerals in different environments The key factors controlling the
identity of the secondary iron minerals include medium composition electron
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 59
donor and electron acceptor concentrations mineral aging as well as adsorbed ions
(Zachara et al 2002) The main factor controlling the nature of the secondary
mineral products are the respiration-driven biogenic Fe(II) supply rate and
magnitude and its surface reaction with the residual oxide and other sorbed ions
(Zachara et al 2002) Especially solution and medium composition have a strong
impact on the nature of the Fe(II)-containing biomineralization products forming
Accordingly siderite (FeCO3) and magnetite (Fe3O4) were the secondary solid
phases resulting from the bioreduction of ferrihydrite by Shewanella putrefaciens
CN32 in bicarbonate buffered medium (pH 71) containing no phosphate whereas
siderite and vivianite were the secondary iron minerals dominating in bicarbonate
buffered medium (pH 74) containing 4 mM phosphate (Zachara et al 2002) This
is explained by the inhibiting effect of phosphate on crystallization of magnetite
(Couling amp Mann 1985 Fredrickson et al 1998)
334 Factors controlling the rate and extent of Fe(III) bioreduction
In this study the extent of Fe(III) bioreduction was estimated to 19-72 by
transmission Moumlssbauer measurements In fact complete microbial reduction of
crystalline Fe(III) minerals has never been observed in laboratory batch culture
studies (Roden amp Urrutia 2002) It has been found that Fe(II) does not inhibit
Fe(III) reductase activity through an enzyme inhibition mechanism (Roden amp
Urrutia 2002) Hence other chemical andor physiologic factors control the
bioavailability of solid Fe(III) phases and thus the extent of their microbial Fe(III)
reduction The initial rate and long-term extent of microbial reduction of
amorphous and crystalline Fe(III) oxides including HFO goethite and hematite
were linearly correlated with oxide surface area (Roden amp Zachara 1996)
Association of biogenic Fe(II) with Fe(III) oxide and DIRB cell surfaces reduced
the long-term extent of crystalline Fe(III) oxide bioreduction (Roden amp Urrutia
2002) These results were explained by Fe(II) surface complexes andor
precipitates creating a passive Fe(II)-bearing surface film providing direct physical
interference with the electron transfer from the DIRB cells to Fe(III) However the
60 Chapter 3
real mechanisms whereby the surface-associated Fe(II) inhibits Fe(III) oxide
bioreduction are unclear but they most likely involve both kinetic and
thermodynamic constraints on the electron transfer Culture medium composition
in particular the presence and the concentration of phosphate as well as Fe(II)
chelating ligands also exert an influence on the extent of the microbial reduction
of Fe(III) oxides The extent of Fe(III) bioreduction was inhibited by high
phosphate concentrations which favoured surfacebulk precipitation processes
(Urrutia et al 1998) The carbon sources most frequently applied in Fe(III)
bioreduction studies include malate citrate and other di- and tricarboxylic acids
which are not only easily metabolizable carbon sources but also eminent Fe(II) and
Fe(III) chelators In this study we employed formate and lactate as carbon sources
since they are the weakest complexing agents of Fe(II) and Fe(III) among the C1-
C3 monocarboxylic acids (Martell 1964) Thus we expect less dissolution of
prevailing precipitates by complexation as compared to other studies
34 Conclusions
This work demonstrated that Shewanella algae BrY reduced 19-72 of initial
Fe(III) when grown in culture media containing 4-10 mM formate or lactate and 8-
27 mM Fe(III) applied as goethite or lepidocrocite coatings on calcite (pH 76) or
HFO coatings on sand (pH 55-60) Within 1-2 weeks after inoculation green
mineral phases were produced in the goethitecalcite and lepidocrocitecalcite
mineral systems whereas black precipitates formed in the HFOsand suspensions
Moumlssbauer spectroscopic analyses indicated that the greenish and blackish phases
most likely were mineral mixtures dominated by vivianite and green rust Thus the
results indicate that GRs may be produced microbially at conditions including low
carbon and Fe(III) concentrations as well as the exclusion of synthetic electron
shuttles and pH buffers
Acknowledgments
We would like to thank Dr R Gerlach for providing us the Shewanella algae BrY culture and
Dr C B Koch for performing the Moumlssbauer analyses
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 61
References Al-Agha MR Burley SD Curtis CD Esson J (1995) Complex cementation textures and authigenic mineral assemblages in recent concretions from the Lincolnshire Wash (east coast UK) driven by Fe(0) to Fe(II) oxidation Journal of the Geological Society 152 157-171 Bernal JD Dasgupta DR Mackay AL (1959) The oxides and hydroxides of iron and their structural inter-relationships Clay Minerals Bulletin 4 15-30 Caccavo Jr F Blakemore RP Lovley DR (1992) A hydrogen-oxidizing Fe(III)-reducing microorganism from the Great Bay Estuary New Hampshire Applied and Environmental Microbiology 58 3211-3216 Caccavo Jr F Das A (2002) Adhesion of dissimilatory Fe(III)-reducing bacteria to Fe(III) minerals Geomicrobiology Journal 19 161-177 Couling SB Mann S (1985) The influence of inorganic phosphate on the crystallization of magnetite (Fe3O4) from aqueous solution Journal of the Chemical Society Chemical Communications 1713-1715 Das A Caccavo Jr F (2000) Dissimilatory Fe(III) oxide reduction by Shewanella alga BrY requires adhesion Current Microbiology 40 344-347
Das A Caccavo Jr F (2001) Adhesion of the dissimilatory Fe(III)-reducing bacterium Shewanella alga BrY to crystalline Fe(III) oxides Current Microbiology 42 151-154 Fadrus H Maly J (1975) Suppression of iron(III) interference in the determination of iron(II) in water by the 110-phenanthroline method The Analyst 100 549-554 Faye GH Manning PG Nickel EH (1968) The polarized optical absorption spectra of tourmaline cordierite chloritoid and vivianite ferrous-ferric electronic interaction as a source of pleochroism American Mineralogist 53 1174-1201 Fredrickson JK Zachara JM Kennedy DW Dong H Onstott TC Hinman NW Li S (1998) Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium Geochimica et Cosmochimica Acta 62 3239-3257 Geacutenin JMR Olowe AA Benbouzid-Rollet ND Prieur D Confente M Resiak B (1991) The simultaneous presence of green rust 2 and sulfate reducing bacteria in the corrosion of steel sheet piles in a harbour area Hyperfine Interactions 69 875-878 Geacutenin JMR Bourrieacute G Trolard F Abdelmoula M Jaffrezic A Refait Ph Maitre V Humbert B Herbillon A (1998) Thermodynamic equilibria in aqueous suspensions of synthetic and natural Fe(II)-Fe(III) green rusts Occurrences of the mineral in hydromorphic soils Environmental Science and Technology 32 1058-1068 Glasauer S Weidler PG Langley S Beveridge TJ (2003) Controls on Fe reduction and mineral formation by a subsurface bacterium Geochimica et Cosmochimica Acta 67 1277- 1288 Hansen HCB Taylor RM (1991) The use of glycerol intercalates in the exchange of CO3
2- with SO4
2- NO3- or Cl- in pyroaurite-type compounds Clay Minerals 26 311-327
62 Chapter 3
Hungate RE (1969) A roll tube method for cultivation of strict anaerobes Methods in Microbiology 3B 117-132 King GM (1990) Effects of added manganic and ferric oxides on sulfate reduction and sulfide oxidation in intertidal sediments FEMS Microbiology Ecology 73 131-138 Koch CB (1998) Structures and properties of anionic clay minerals Hyperfine Interactions 117 131 -157 Kostka J Nealson KH (1998) Isolation cultivation and characterization of iron- and manganese reducing bacteria In Techniques in Microbial Ecology Burlage RS Atlas R Stahl D Geesey G Sayler G (eds) Oxford University Press Inc 58-78 Kukkadapu RK Zachara JM Smith SC Fredrickson JK Liu C (2001) Dissimilatory bacterial reduction of Al-substituted goethite in subsurface sediments Geochimica et Cosmochimica Acta 65 2913-2924
Liu C Zachara JM Gorby YA Szecsody JE Brown CF (2001) Microbial reduction of Fe(III) and sorptionprecipitation of Fe(II) on Shewanella putrefaciens strain CN32 Environmental Science and Technology 35 1385-1393 Lovley DR (1991) Dissimilatory Fe(III) and Mn(IV) reduction Microbiological Reviews 55 259-287 Lovley DR (1997) Microbial Fe(III) reduction in subsurface environments FEMS Microbiology Reviews 20 305-313 Lovley DR Phillips EJP (1986) Organic matter mineralization with reduction of ferric iron in anaerobic sediments Applied and Environmental Microbiology 51 683-689 Lovley DR Phillips EJP Lonergan DJ (1991) Enzymatic versus nonenzymatic mechanisms for Fe(III) reduction in aquatic sediments Environmental Science and Technology 25 1062-1067 Lower SK Hochella Jr MF Beveridge TJ (2001) Bacterial recognition of mineral surfaces Nanoscale interactions between Shewanella and α-FeOOH Science 292 1360-1363 Martell AE (1964) Stability constants of metal-ion complexes Part 2 Organic including macromolecule ligands The Chemical Society London 2 ed Miller TL Wolin MJ (1974) A serum bottle modification of the Hungate technique for cultivating obligate anaerobes Applied Microbiology 27 985-987 Nealson KH Saffarini D (1994) Iron and manganese in anaerobic respiration Environmental significance physiology and regulation Annual Review of Microbiology 48 311-343 Nevin KP Lovley DR (2000) Lack of production of electron-shuttling compounds or solubilization of Fe(III) during reduction of insoluble Fe(III) oxide by G metallireducens Applied and Environmental Microbiology 66 2248-2251 Nevin KP Lovley DR (2002) Mechanisms for Fe(III) oxide reduction in sedimentary
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 63
environments Geomicrobiology Journal 19 141-159 Nielsen A (1976) Hvid groslashn og sort rust Beskrivelse af en korrosionsskade paring et svoslashmmebassin Nordisk Betong 2 21-24 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002a) Iron (IIIII) hydroxycarbonate green rust formation and stabilization from lepidocrocite bioreduction Environmental Science and Technology 36 16-20 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002b) Microbial reduction of lepidocrocite γ-FeOOH by Shewanella putrefaciens The formation of green rust Hyperfine Interactions 139140 231-237 Parmar N Gorby YA Beveridge TJ Ferris FG (2001) Formation of green rust and immobilization of nickel in response to bacterial reduction of hydrous ferric oxide Geomicrobiology Journal 18 375-385 Roden EE Urrutia MM (2002) Influence of biogenic Fe(II) on bacterial crystalline Fe(III) oxide reduction Geomicrobiology Journal 19 209-251 Roden EE Zachara JM (1996) Microbial reduction of crystalline iron(III) oxides Influence of oxide surface area and potential for cell growth Environmental Science and Technology 30 1618-1628 Roh Y Lee SY Elless MP (2000) Characterization of corrosion products in the permeable reactive barriers Environmental Geology 40 184-194 Schwertmann U Cornell RM (1991) Iron oxides in the laboratory Preparation and characterization VCH Verlagsgesellschaft mbH Weinheim Thamdrup B (2000) Bacterial manganese and iron reduction in aquatic sediments In Advances in Microbial Ecology (Schink B ed) Kluwer AcademicPlenum Publishers New York 41-84 Trolard F Abdelmoula M Bourrieacute G Humbert B Geacutenin JMR (1996) Mise en eacutevidence dun constituant de type rouilles vertes dans les sols hydromorphes Proposition de lexistence dun nouveau mineacuteral la fougeacuterite Geacuteosciences de surface Comptes Rendus de LrsquoAcademie des Sciences 323 1015-1022 Tuovinen OH Button KS Vuorinen A Carlson L Mair DM Yut LA (1980) Bacterial chemical and mineralogical characteristics of tubercles in distribution pipelines Journal of the American Water Works Association 72 626-635 Turick CE Caccavo Jr F Tisa LS (2003) Electron transfer from Shewanella algae BrY to hydrous ferric oxide is mediated by cell-associated melanin FEMS Microbiology Letters 220 99-104 Urrutia MM Roden EE Fredrickson JK Zachara JM (1998) Microbial and surface chemistry controls on reduction of synthetic Fe(III) oxide minerals by the dissimilatory iron- reducing bacterium Shewanella alga Geomicrobiology 15 269-291 Venkateswaran K Moser DP Dollhopf ME Lies DP Saffarini DA MacGregor BJ Ringelberg DB White DC Nishijima M Sano H Burghardt J Stackebrandt E
64 Chapter 3
Nealson KH (1999) Polyphasic taxonomy of the genus Shewanella and description of Shewanella oneidensis sp nov International Journal of Systematic Bacteriology 49 705-724 Zachara JM Fredrickson JK Li S Kennedy DW Smith SC Gassman PL (1998) Bacterial reduction of crystalline Fe3+ oxides in single phase suspensions and subsurface materials American Mineralogist 83 1426-1443
Zachara JM Kukkadapu RK Fredrickson JK Gorby YA Smith SC (2002) Biomineralization of poorly crystalline Fe(III) oxides by dissimilatory metal reducing bacteria (DMRB) Geomicrobiology Journal 19 179-207
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 65
4 Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust The Effect of Probe Compound Charge
Abstract
Layered iron(II)-iron(III)-hydroxides (green rusts) may play an important role in
controlling the fate and transport of many organic and inorganic contaminants in
iron-rich suboxic soils and sediments Unlike most other iron oxides green rusts
(GRs) contain not only external Fe(II) reactive sites at the basal planes and at the
edges but also internal sites in the space between consecutive Fe(II)-Fe(III)
hydroxide layers The GR interlayer thickness is a function of both the size and the
charge of the interlayer anion Whether a given oxidant has access to the internal
sites in GRs is dependent on its charge We investigated the reductive
transformation of nitroaromatic compounds (NACs) by GR-SO4 and studied the
effect of NAC charge on the reactivity towards GR-SO4 A series of structurally
closely related compounds with different charge properties including nitrobenzene
4-nitrotoluene 4-chloronitrobenzene and 4-nitrophenylacetic acid were used as
probe compounds The NACs were completely reduced to their corresponding
anilines by GR-SO4 The reactions followed pseudo 1 order kinetics with respect
to NAC and the surface area-normalised pseudo 1 order rate constants obtained
were 016ndash465middot10-4 s-1middotm-2middotL at [Fe(II)GR]0 = 103-1260 mM [NAC]0 = 20-102
microM and pH 84-86 Neither mass transfer control nor surface saturation kinetics
could account for the rather unexpected similarity of the surface area-normalised
pseudo 1 order rate constants obtained for the reduction of the neutral and anionic
NACs by GR-SO4 These observations suggest that the anionic NACs did not have
an enhanced access to the inner or outer Fe(II)-GR reactive sites as compared to
the neutral NACs Hence the reaction between NAC and GR-SO4 primarily took
place at the edges of GR-SO4
66 Chapter 4
41 Introduction
Layered iron(II)-iron(III)-hydroxides (green rusts) are intermediate phases formed
by partial oxidation of Fe(II) or partial reduction of Fe(III) In neutral and weakly
alkaline solutions the oxidation of dissolved Fe(II) always passes through solid
green rust (GR) phases (Bernal et al 1959) This agrees with the natural GR
occurrences found in suboxic non-acid iron-rich environments such as
hydromorphic soils and intertidal sediments (Al-Agha et al 1995 Trolard et al
1996 Genin et al 1998) In addition GRs have been found as corrosion products
in numerous engineered systems ie a pipeline distribution system for drinking
water steel sheet piles in marine sediments reinforced concrete (ferro-concrete)
and permeable reactive barriers of zero-valent iron implemented for on-site
remediation of organic and inorganic contaminants (Tuovinen et al 1980 Nielsen
1976 Genin et al 1991 Roh et al 2000) Furthermore the microbial formation
of GRs resulting from bioreduction of Fe(III) oxides by strains of the anaerobic
dissimilatory Fe(III) reducing bacteria Shewanella putrefaciens has been reported
increasingly over the last 5 years (Fredrickson et al 1998 Kukkadapu et al 2001
Parmar et al 2001 Ona-Nguema et al 2002 Glasauer et al 2003) Moreover
the biotic formation of GRs by anaerobic denitrifying Fe(II) oxidizing bacteria has
been proposed but proper identification of the GR phases still lacks (Chaudhuri et
al 2001) All these indications of microbial GR formation infer the importance of
GRs as a link between geochemical and biological processes in natural systems
GRs form platy crystals with the general formula [FeII(6-
x)FeIIIx(OH)12]x+[(A)xnmiddotyH2O]x- where x = 09 - 42 A is an n-valent anion eg
CO32- Clndash or SO4
2- and y is the number of water molecules in the interlayer The
crystal structure consists of positively charged hydroxide sheets with Fe(II) and
Fe(III) cations having octahedral hydroxyl coordination The Fe(III) in the
hydroxide layers creates a net positive charge which is balanced by hydrated
anions in the interlayers (Figure 41) The interlayers have a higher affinity for
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 67
divalent anions than for monovalent anions (Miyata 1983) Among the 3 most
common GR forms the affinity follows the order CO32- gt SO4
2- gt Clndash The extreme
preference shown for carbonate hinders further access and exchange except under
certain conditions (Hansen amp Taylor 1991) Non-carbonate forms are readily
exchanged with other anions when dispersed in a solution containing the
exchanging anion (Mendiboure amp Schoumlllhorn 1986)
c
ba
Figure 41 Green rust layer structure The hydroxide layers and the interlayers are connected by
hydrogen bonds (not shown) The GR-SO4 crystal structure is characterised by the hexagonal
unit cell having a = b = 055 nm and c = 110 nm (Simon et al 2003) The unit cell consists of
one double layer (a double layer is a hydroxide layer and an interlayer) ie the hydroxide layer
constitutes 049 nm and the interlayer 061 nm in GR-SO4
The GR interlayer thickness (extending in the c axis direction Figure 41) is a
function of both the size and the charge of the interlayer anion Tetrahedrally
coordinated anions like sulphate lead to larger interlayer distances than smaller
monoatomic anions like chloride or planar ions like carbonate (Mendiboure amp
Schoumlllhorn 1986) Not only size but also charge density plays a role for the
interlayer spacing That is for anions having the same number of valence
electrons anions with smaller ionic radii (higher electron density) are bound more
strongly and therefore result in smaller interlayer spacings
Due to their layered structures anionic interlayers and high specific surface areas
GRs represent reactive ion exchangers and sorbents of anions eg arsenate
selenate and phosphate (Myneni et al 1997 Hansen amp Poulsen 1999 Randall et
al 2001) In addition GR may incorporate heavy metal cations by isomorphic
68 Chapter 4
substitution into the GR hydroxide layers (Tamaura 1985 Tamaura 1986)
Furthermore GRs have been shown to reduce a range of inorganic contaminants
such as nitrite nitrate selenate chromate uranyl pertechnetate and the transition
metals AgI AuIII CuII and HgII as well as organic pollutants including halogenated
ethanes ethenes and methanes (Hansen et al 1994 Hansen et al 1996 Myneni et
al 1997 Erbs et al 1999 Loyaux-Lawniczak et al 1999 Cui amp Spahiu 2002
Lee amp Batchelor 2002b Heasman et al 2003 OrsquoLoughlin et al 2003a amp 2003b
Pepper et al 2003 Elsner et al 2004 OrsquoLoughlin amp Burris 2004) Thus through
sequestration and reductive transformation GRs may play an important role in
controlling the fate and transport of contaminants in suboxic soils and sediments
In a previous study the effects of interlayer anion and Fe(II)Fe(III) ratio in GRs
on the reduction rate of nitrate were investigated (Hansen et al 2001) It was
found that the rate of nitrate reduction to ammonium increased with increasing
Fe(II)Fe(III) ratio and decreased when exchanging a monovalent interlayer anion
(chloride) with a divalent anion (sulphate) The results suggest that for anionic
oxidants like nitrate Fe(II) within the hydroxide layer is available from the outside
basal planes and from the edges as well as through the interlayer under certain
conditions (Figure 42) However oxidants with different charge properties
(cations neutral molecules) may exhibit different affinities for the various reactive
Fe(II) sites present in GR
As the reactive sites are located inat the Fe(II)-Fe(III) hydroxide layers the rate of
reaction depends on the hydroxide layer area which can be accessed by the
oxidant If the oxidant can exchange with the interlayer anion reaction can take
place both at outer and inner surfaces of the GR particles and in total more
reactive sites are available for the reaction However it was found that nitrate
cannot penetrate the interlayer when carbonate or sulphate constitutes the
interlayer anions (Hansen amp Koch 1998) This agrees with the fact that the
interlayers have a lower affinity for monovalent anions than for divalent anions
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 69
(Miyata 1983) However when nitrate was forced into the interlayer by extracting
the interlayer sulphate through precipitation of barium sulphate outside the GR
particles the observed 40 fold increase in rate of nitrate reduction almost equalled
the increase in exposed surface area of the Fe(II)-Fe(III) hydroxide layers (Hansen
amp Koch 1998) From these observations it is expected that the rate of reaction
depends on the particular GR form the crystallite size and the ease with which an
oxidant can exchange with An- in the GR interlayer (Figure 42) Due to
electrostatic interactions we expect anions to be attracted to the positively charged
outer and inner surfaces to a higher degree than cations and neutral compounds If
this theory holds we may expect oxidants with similar intrinsic reactivity (similar
one-electron reduction potentials) to react in the following order anionic gt non-
charged gt cationic (Figure 43) granting that we do not normalise the rate
constants with respect to the amount of oxidant sorbed
Figure 42 Reaction of a probe compound at basal planes at edges and in the interlayer of GR
The hypothesis only holds in cases where the oxidants possess the same intrinsic
reactivities If the relative reactivities of the probe compounds differ greatly from
what would be expected when considering only their reduction potentials
70 Chapter 4
compound specific effects such as charge properties might explain this and the
relative reactivities may follow a pattern like the one depicted in Figure 43
Figure 43 Hypothetical plot of observed reaction rate constants for the reactions between
cationic neutral and anionic probe compounds and GR-SO4 assuming that the oxidant charge
controls its reactivity towards GR
In this work we investigated the reductive transformation of NACs by GR-SO4
Furthermore the effect of NAC charge on the rate of reaction and the possible
access to the internal reactive sites in GR-SO4 were assessed When quantifying
Fe(II) in GRs by means of acid digestion it is not possible to distinguish between
the reactive sites accessible from the outside (at the basal planes or at the edges) or
through the interlayer However we designed an indirect method to gain insight
into the relative importance of the various reactive sites by using a series of
structurally closely related compounds with different charge properties as ldquoreactive
probesrdquo Neutral and anionic probes were needed in order to access all Fe(II)
reactive sites According to our hypothesis cationic and non-charged oxidants
should provide information about the reactivity of the outer Fe(II) reactive sites in
GR whereas the anionic oxidants should provide information about the reactivity
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 71
of both outer and inner Fe(II) reactive sites We chose five nitro aromatic
compounds (NACs) - representing an important group of reducible organic
pollutants - as probe compounds (Figure 43) This class of compounds is not only
of great environmental concern but also comprises suitable model compounds for
studying redox reactions potentially relevant in the environment Moreover they
react readily with Fe(II) surface species associated with iron oxides or clay
minerals transforming them into well-defined easily detected products allowing
mass and electron balances to be established (Hofstetter et al 2003 Klausen et al
1995 Schultz amp Grundl 2000) Our main goals were to establish the rate law and
estimate the surface area-normalised reaction rates for the reaction of the probe
compounds with GR-SO4 in order to assess the importance of the Fe(II) reactive
sites accessible through the interlayer relative to the Fe(II) reactive sites accessible
at the outer surface in GR-SO4
4 2 Materials and methods
All handling and sampling of solutions and suspensions were carried out under
strict anoxic conditions All chemicals were pa quality or better Methanolic stock
solutions (5 mM) of nitrobenzene (NB) 4-nitrotoluene (4-NT) 4-
chloronitrobenzene (4-CNB) and 4-nitrophenylacetic acid (4-NPA) were prepared
in deoxygenated methanol Several attempts to synthesize the cationic probe
compound 4-(NNN-trimethylammonium)-nitrobenzene failed and therefore the
study had to be carried out with only neutral and anionic oxidants The sulphate
GR form was chosen as it is the most stable form and thus the easiest to work
with in the lab
421 Synthesis of GR-SO4
GR-SO4 was synthesized by controlled air oxidation of an FeSO4 solution at a
constant pH of 700 according to the procedure given by Koch amp Hansen (1997)
The GR-SO4 suspension was washed with deoxygenated deionised water and
72 Chapter 4
separated on a folding filter redispersed in deoxygenated 25 mM Na2SO4(aq) in
order to stabilize the GR-SO4 and prevent it from transforming into magnetite
spontaneously Washing separation and redispersion of the GR-SO4 suspension
were conducted in an anoxic glove box (Coy Laboratory Products Inc) All
suspensions and solutions were deoxygenated by Ar-purging (999998 Ar
Carbagas)
422 Mineral characterisation
The identity and purity of the GR-SO4 suspensions were examined by means of X-
ray diffraction measurements The XRD analyses were performed on a Scintag
XDS 2000 using Cu Kα radiation (45 kV 40 mA) Glycerol smears made
according to Hansen (1989) were scanned between 6 and 80 deg2θ with a scan speed
of 1 deg2θmin
423 Lyophilization and determination of specific surface area
Simple air-drying of the GR mineral in the glove box resulted in big flakes with
very low surface areas hence a more suitable lyophilization method was adopted
from Elsner et al (2004) The GR-SO4 suspensions were lyophilised using
Schlenk-type glassware The set-up consisted of a 1 L round bottom flask and a
200 mL glass finger connected by a crescent-shaped bridge equipped with an
evacuation outlet and a stopcock All ground joints and fittings were attached using
high-vacuum grease The washed and resuspended GR-SO4 suspensions were
filled into the glass finger and the freeze-drying apparatus was assembled and
closed before taking it out of the glove box The suspension was frozen by
carefully submerging the lower part of the glass finger into liquid nitrogen for a
few hours Subsequently the evacuation outlet was connected to a vacuum pump
by a metal hose Following a short evacuation of the metal hose the lyophilization
apparatus was evacuated for several minutes by gently opening the stopcock The
evacuation was terminated by closing the stopcock and disconnecting the vacuum
pump The apparatus position was now reversed by removing the glass finger from
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 73
and immersing the round bottom flask into liquid nitrogen As any other
lyophilization method this method depends on sublimation of the ice from the
frozen sample and its recondensation on a cool surface in this case the round
bottom flask Generally it took 1-2 d for the mineral to dry The apparatus was
disassemled in the glove box and the fine powder stored under anoxic conditions
The specific surface area (SSA) of GR-SO4 was determined by the BET multi-
point method using N2 adsorption (Brunauer et al 1938) Powder samples were
filled into sample burettes in the glove box and the generously greased stopcocks
closed Samples and burettes were evacuated prior to connecting them to the BET-
instrument (Sorptomatic 1990 Fisons)
424 Estimation of the one-electron reduction potential for 4-NPA
Kinetic experiments in 100 mL Viton stoppered and alu-crimp capped serum vials
were carried out under the exclusion of oxygen as described by Hofstetter et al
(1999) The homogeneous aqueous solutions contained 50 mM KH2PO4 buffer
(pH = 660) 5 mM Na2S redox buffer and 20 microM juglone (8-hydroxy-14-
naphthoquinone) added as deoxygenated 20 mM methanolic stock solution The
solutions were equilibrated at least one day prior to 4-NPA addition To start the
reaction 50 microM 4-NPA was added as deoxygenated 20 mM methanolic stock
solution The vials were agitated on a roller apparatus in the dark at 21ordmC Control
experiments were prepared similarly except for the addition of juglone At
different time intervals aqueous samples were withdrawn with a syringe and
collected in 18 mL HPLC vials containing 100 microL 1 M HCl The sample vials
were sealed with Teflon-coated silicone septa and plastic screw caps and vortexed
for 10 s The samples were stored at -20degC and analysed without further treatment
For comparison experiments with 4-NT were also conducted See Supporting
Information 71 for more information on the one-electron reduction potentials
74 Chapter 4
425 Kinetic experiments
All reactions took place at pH 84-86 where GR-SO4 tends to stabilize and buffer
itself Samples for Fe(II) and XRD analysis were withdrawn prior to reaction Due
to the fast reactions the experiments were conducted in 10 mL single-use
polyethylene syringes (BD Plastipak) in the glove box To start reaction 40-200
microL 5 mM methanolic stock solutions of NAC were quickly added to 10 mL GR-
SO4 suspension (1-12 mM Fe(II)GR) washed and resuspended in 25 mM
Na2SO4(aq) A Teflon filter (25 mm x 02 microm BGB Analytik) was quickly
mounted on the tip of the syringe and the syringe was vigorously shaken between
sampling At different time intervals filtered suspension samples were collected in
18 mL HPLC vials The HPLC vials were sealed with Teflon-coated silicone septa
and plastic screw caps The samples were stored at -20degC and analysed without
further treatment Absorption of NAC in the syringe and in the Teflon filter
evaluated in blank experiments with NAC added to 25 mM Na2SO4(aq) was found
to be negligible
426 Analytical methods
Initial total and aqueous Fe(II) were determined using a modified phenanthroline
method (Fadrus and Maly 1975) In order to determine [Fe(II)aq] and [Fe(II)total] 1
mL filtered (022 microm) and 1 mL unfiltered GR-SO4 suspension samples were
withdrawn and each treated with 18 mL 01 M HCl for at least 30 min From these
acid digests 01 mL was added to 05 mL Fe(II)-reagent and 19 mL deionised
water (DIW) added up The Fe(II) content in GR-SO4 was estimated as the
difference [Fe(II)GR] = [Fe(II)total] - [Fe(II)aq] The NACs and their corresponding
intermediates and products formed during reduction by GR-SO4 were quantified by
reversed-phase HPLC Separation was performed on a LiChrospher 100 RP-18 (5
microm 125 times 4 mm ID) reversed-phase column coupled with a LiChroCART 100 RP-
18 (4 times 4 mm ID) precolumn Analytical conditions were isocratic and the eluent
consisted of 10 mM hydroxylammonium chloride in various DIWCH3OH
mixtures (vv 3565 and pH 70 for 4-NT and 4-CNB 955 and pH 60 for
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 75
4-NPA) The injection volume was 20 microLand the flow-rate 10 mLmin HPLC
analyses were performed using a Gynkotek High Precision Pump M480 Gynkotek
Gina 50 autosampler and a diode array UV detector (340s Gynkotek) UV-VIS
detection was carried out at the wavelengths of maximum absorption for the
various nitro aromatic and aniline analytes
43 Results and discussion
431 Productformation and reaction kinetics
The reduction of the aromatic nitro group occurs via nitroso- and hydroxylamino-
intermediates where 2 electrons are transferred in each reaction step (Figure 44)
0 --0 H OH H H --0 N N N N
2e- 2H+ H20 + 2e-~ 2e- 2H+ H20
~ ~ R R R
Nitro benzene Nitrosobenzene Hydroxylamine Aniline
Figure 44 Reductive transfonnation pathway of NA Cs
Thus in order to reduce 1 Ar-N02 completely to Ar-NH2 6 electrons
corresponding to 6 mol Fe(II) are needed As magnetite was the major iron phase
formed during reaction (XRD results not shown) we assume the following
reaction stoichiometry
The aniline product was not formed at the same rate as the nitro compound
degraded which is consistent with the detection of early eluting hydroxylamine
intermediates during the course of the reaction (Figure 45a amp 45c) No traces of
76 Chapter 4
nitrosobenzene intermediates or side products such as azoxy- azo- or
hydrazobenzene were found In Figure 45 pseudo 1 order kinetic plots and ln
[Ar-NO2]t[Ar-NO2]0) versus time plots for the neutral probe compounds 4-CNB
and 4-NT are shown as examples The plots for NB and 4-NPA look similar
Figure 45 a Concentration versus time plots for reaction of GR-SO4 with 4-CNB ([Fe(II)GR]0 =
126 mM [4-CNB]0 = 30 microM) b ln [Ar-NO2]t[Ar-NO2]0) versus time plots for reaction of GR-
SO4 with 4-CNB ([Fe(II)GR]0 = 126 mM + [4-CNB]0 = 30 microM [Fe(II)GR]0 = 63 mM + [4-
CNB]0 = 50 microM) c Concentration versus time plots for reaction of GR-SO4 with 4-NT
([Fe(II)GR]0 = 131 mM [4-NT]0 = 20 microM) d ln [Ar-NO2]t[Ar-NO2]0) versus time plots for
reaction of GR-SO4 with 4-NT ([Fe(II)GR]0 = 131 mM [4-NT]0 = 20 microM [Fe(II)GR]0 = 131
mM [4-NT]0 = 50 microM) The hydroxylamino intermediate shown in microM equals the deficit in the
mass balance and in abs equals the detector response (peak area) Solid lines represent 1 order
kinetic fits (a amp c) and ln [Ar-NO2]t[Ar-NO2]0) versus time fits (d) whereas symbols and dotted
lines represent actual data
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 77
At intial Fe(II)GR concentrations in large excess of initial Ar-NO2 concentration
we found a pseudo 1 order rate law for the degradation of Ar-NO2 by GR-SO4
[ ] [ ] [ b 2
a GR
2 ArNOFe(II) ArNOsdotsdot=minus k
dtd ]
where a = 1 b = 1 and the observed pseudo 1 order rate constant kobs = k middot
[Fe(II)GR] At high [Fe(II)GR]0[Ar-NO2]0 ratios the nitro compound was
transformed completely into the aniline product within reaction duration and the
degradation curves of the nitro compound were shaped according to pseudo 1
order kinetics (data points follow solid line in Figure 45a) In some instances ie
at low [Fe(II)GR]0[Ar-NO2]0 ratios the reactions did not follow pseudo 1 order
kinetics for the whole duration of reaction (data points deviate from solid line in
Figure 45c) Hence in order to allow comparison all the pseudo 1 order rate
constants were calculated as initial rates (ie max first two half-lives) from linear
fits of (time ln [Ar-NO2]t[Ar-NO2]0)-plots (Figure 45b amp 45d) Surface area-
normalised pseudo 1 order rate constants are shown in Table 41
Tabl
e 4
1 S
urfa
ce a
rea-
norm
alis
ed p
seud
o 1
ord
er ra
te c
onst
ants
for t
he re
duct
ive
trans
form
atio
n of
4-n
itrot
olue
ne (4
-NT)
4-
chlo
roni
trobe
nzen
e (4
-CN
B) a
nd 4
-nitr
ophe
nyla
cetic
aci
d (4
-NPA
) by
GR
-SO
4
Exp
erim
ent
Age
GR
(d
) [F
e(II
) GR] 0
(mM
) [N
AC
] 0 (micro
M)
[Fe(
II) G
R] 0
[N
AC
] 0∆[
ArN
O2]
(microM
) af b
k obs
(s-1
) ck o
bs (s
-1middotm
-2middotL
) d
GR
-SO
4 + 4
-NT
3 1
103
20
51
5
109
54
5
7
65middot1
0-46
95middot1
0-5
GR
-SO
4 + 4
-NT
3 1
103
50
20
6
174
34
8
7
41middot1
0-46
74middot1
0-5
GR
-SO
4 + 4
-NT
3 1
103
10
0 10
3
214
21
4
2
63middot1
0-42
39middot1
0-5
GR
-SO
4 + 4
-CN
B 1
1
103
55
18
7
177
32
2
4
21middot1
0-43
83middot1
0-5
GR
-SO
4 + 4
-CN
B 1
1
103
10
2 10
1
165
16
2
2
37middot1
0-42
15middot1
0-5
GR
-SO
4 + 4
-NPA
1
1 1
03
25
412
9
7 38
8
4
82middot1
0-44
38middot1
0-5
GR
-SO
4 + 4
-NPA
1
1 1
03
46
224
9
9 21
5
6
37middot1
0-45
79middot1
0-5
GR
-SO
4 + 4
-NPA
1
1 1
03
100
103
13
7
137
196
middot10-4
178
middot10-5
GR
-SO
4 + 4
-NT
2 15
1
31
20
655
5
4 27
0
6
74middot1
0-44
82middot1
0-5
GR
-SO
4 + 4
-NT
2 15
1
31
50
262
9
7 19
4
5
89middot1
0-44
21middot1
0-5
GR
-SO
4 + 4
-NT
4 2
126
0 50
25
2 49
1
982
110
middot10-2
817
middot10-5
GR
-SO
4 + 4
-NT
4 2
630
50
12
6 42
6
852
186
middot10-3
276
middot10-5
GR
-SO
4 + 4
-CN
B 2
2
126
0 30
42
0 29
0
967
925
middot10-3
687
middot10-5
GR
-SO
4 + 4
-CN
B 2
2
630
50
12
6 38
0
760
136
middot10-3
202
middot10-5
GR
-SO
4 + 4
-NPA
2
2 12
60
40
315
371
92
8
5
96middot1
0-34
43middot1
0-5
GR
-SO
4 + 4
-NPA
2
2 6
30
45
140
273
60
7
1
09middot1
0-31
62middot1
0-5
a A
mou
nt o
f NA
C re
duce
d by
GR
-SO
4 at r
eact
ion
term
inat
ion
b F
ract
ion
of in
itial
ly a
dded
NA
C tr
ansf
orm
ed b
y G
R-S
O4 a
t rea
ctio
n te
rmin
atio
n c
Pse
udo
1
orde
r rat
e co
nsta
nts c
alcu
late
d as
initi
al ra
tes
ie m
ax f
irst t
wo
half-
lives
d S
urfa
ce a
rea-
norm
alis
ed p
seud
o 1
ord
er ra
te c
onst
ants
The
are
a of
GR
-SO
4 per
L
su
spen
sion
was
cal
cula
ted
as frac14
middot[Fe
(II)
GR] 0middot
600
gmiddotm
ol-1
middot71
2 m
2 middotg-1
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 79
432 Comparison of rate constants for the different NACs
Even for NACs holding very different one-electron transfer reduction potentials
( ) their reactivities differed only little in Fe(II)-Fe(III) systems such as the
Fe(II)goethite system (slope a = 06 for linear free energy relationship (LFER)
between k
1hE
obs and Hofstetter et al 1999) and the Fe(II)magnetite system
(LFER slope a = 034 Klausen et al 1995) When considering only the for the
reductive transformation reactions of the NACs applied in this study (Table 42)
we expect the surface area-normalised pseudo 1 order rate constants for the
reduction of the NACs to follow the order 4-CNB gt NB gt 4-NT gt 4-NPA Based
on log k
1hE
1hE
obs versus correlations obtained in Fe(II)goethite systems we expect 4-
CNB to react 6 times faster than 4-NPA (Hofstetter et al 1999)
1hE
Table 42 One-electron reduction potentials and relative reactivities in Fe(II)-magnetite and GR-
SO4 systems for the nitro aromatic probe compounds
Compound pKa Eh1
acute (mV) krel (Fe3O4) cd krel (GR-SO4) ce
4-Chlornitrobenzene - -450 a 122 148
Nitrobenzene - -486 a 1 1
4-Nitrotoluene - -500 a 057 176
4-Nitrophenylacetic acid 385 -543 b - 123 a Values from references cited in Hofstetter et al 1999 b Estimated at pH 660 using a LFER (Hofstetter et al 1999 see Supporting Information 71) c Reactivity relative to NB d Values from Klausen et al 1995 e Values from this work
A comparison of the relative rate constants of the NACs obtained for their
transformation by GR-SO4 (this work) and by magnetite (Klausen et al 1995)
shows that they do not differ significantly from each other in any of the mineral
systems (Table 42) When considering charge effects we expect the anionic probe
compounds to react faster with GR-SO4 than the neutral probe compounds
provided that they sorb preferentially within the GR-SO4 interlayers and that Fe(II)
in the interlayers are equally or more reactive than external Fe(II) sites Still the
surface area-normalised kobs values obtained for NB 4-NT 4-CNB and 4-NPA
under various experimental conditions did not differ significantly from each other
80 Chapter 4
(Figure 46 Table 41) The anionic probe compound 4-NPA did not react
significantly faster with GR-SO4 than the neutral probe compounds NB 4-NT and
4-CNB This may indicate that 4-NPA does not significantly interact with reactive
Fe(II) sites in the interlayer Alternatively the negative charge carried by 4-NPA
may be compensating for the lower intrinsic reactivity as compared to the neutral
probe compounds thus explaining the similarity in rate constants for 4-NPA and
the neutral probe compounds Finally other factors than intrinsic reactivity or
charge of the probe compounds such as regeneration of reactive sites or formation
of the magnetite phases may control the overall reactivity of the system
Figure 46 Actual plot of surface area-normalised pseudo 1 order rate constants for the reactions
between neutral and anionic probe compounds and GR-SO4
In heterogeneous reactions mass transfer in bulk solution becomes the rate-
limiting step when the surface reaction is much faster than the diffusion of the
reacting species to the reactive surface In cases where mass transfer controls the
overall rate of reaction the observed pseudo 1 order rate constant kobs ge kLmiddota
where kL is the calculated mass transfer coefficient (mmiddots-1) and a is the ratio of the
external (geometric) specific surface area to volume of solution (m-1) (see
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 81
Supporting Information 72) Mass transfer controlled reactions between GR-SO4
particles and the NACs in bulk solution would explain the similar pseudo 1 order
rate constants obtained for the NACs in this work However when comparing our
estimates of kLmiddota with kobs (see Supporting Information 72) we found that the rates
of mass transfer for all 4 NACs exceed the observed rate constants by at least 3
orders of magnitude at every initial Fe(II)GR concentration Thus the reactions of
the given NACs with GR-SO4 are not likely to be mass transfer limited under the
experimental conditions applied here
Since mass transfer in bulk solution does not control the reaction between GR-SO4
and NACs the overall reaction rate may be surface saturation controlled During
the reductive transformation of NACs not only the parent compound but also
various intermediates forming may compete for the restricted number of reactive
sites present in GR-SO4 This competition may constitute the rate limiting step in
the overall reactivity and may even be enhanced if the number of reactive sites is
depleted during reaction However surface saturation kinetics would not explain
the unexpected similarity of the pseudo 1 order rate constants obtained for the
NACs but it could explain the bent curves observed at low initial Fe(II)GR
concentrations (Figure 45d) The kinetically deviating cases at low [Fe(II)GR]0
were evaluated according to Langmuir-Hinshelwood kinetics (see Supporting
Information 72) Our experimental data did not agree with the Langmuir-
Hinshelwood rate law for any of the NACs (regression results not shown)
Simplifying the rate law by assuming that the aniline product or the
hydroxylamino intermediate or both did not compete for the reactive sites did not
improve the regression Thus the Langmuir-Hinshelwood model cannot explain
the deviations from pseudo 1 order kinetics observed at [Fe(II)GR]0 in our GR-SO4
system and it does not suffice as the correct reaction mechanism nor as the rate-
limiting step
82 Chapter 4
If the adsorption follows a saturation-type sorption isoterm (eg Langmuir) the
sorbate (oxidant) concentration at the surface will vary non-linearily with the total
amount of oxidant added This dependence will have to be taken into account when
establishing rate laws for the heterogeneous reactions and when testing the
hypothesis that the reaction rates depend on the sorbed concentration of the
oxidants However at the high reaction rates observed here we could not quantify
sorption Since the measured initial NAC concentrations corresponded to the
nominal amount of NAC added we assume that transformation and not sorption
was responsible for the consumption of NAC
433 Factors influencing the reaction rate
In general numerous compound- and system-specific factors influence redox
reactions One very important factor is pH which influences the speciation of
dissociable compounds as well as the stability of GR and the formation of other
iron minerals in the system pH has a strong impact on the sorption and therefore
the availability of ionisable oxidants such as carboxylic acids At pH ~ 84 where
our experiments were conducted 4-NPA (pKa = 385) is completely dissociated
Our experiments conducted with GR-SO4 and NB showed that pH was constant
during reaction In addition solution pH has an effect on the surface speciation
From other Fe(II)-Fe(III) systems such as Fe(II) surface species associated with
iron oxides or clay minerals it is well-known that other reactive hydroxylated
Fe(II)-Fe(III)-hydroxo surface complexes can form at higher pH (Charlet et al
1998 Liger et al 1999) Williams amp Scherer (2001) reported a small decrease (5
fold) in the reduction rate of chromate with GR-CO3 when increasing pH from 50
to 90 This decrease may be due to the alternating speciation of the Fe(II) surface
sites on GR-CO3 and of chromate in solution (pKa (H2CrO4) = 08 pKa (HCrO4ndash) =
65) when raising pH (Williams amp Scherer 2001) In contrast other studies have
reported small increases (4 fold) in the reduction rates of nitrate and
trichloroethene with GR-SO4 when increasing pH from 71 to 84 and from 68 to
101 respectively (Koch amp Hansen 1997 Lee amp Batchelor 2002b)
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 83
In this work all experiments were carried out in the presence of 25 mM
Na2SO4(aq) in order to minimize GR-SO4 dissolution and spontaneous
transformation into magnetite Preliminary results from experiments conducted
with NB show that the bulk concentration of Na2SO4 has only a very small impact
on the rate ie increasing the concentration of Na2SO4(aq) in the GR-SO4
suspension from 5 to 25 mM reduced the observed rate constant by a factor of 2
At Na2SO4 concentrations above 25 mM the effect leveled off and therefore
[Na2SO4] = 25 mM was chosen for this work Portions of the same GR-SO4
suspension were used for kinetic experiments over a period of two weeks No
significant aging effects eg rate constants decreasing as a function of GR age
were observed within this time frame
434 Comparison with rate constants obtained for other Fe(II) containing
mineral systems
A recent study compared the reactivity of various Fe(II) containing iron mineral
systems towards organic probe compounds representing different classes of
pollutants (Elsner et al 2004) The reductive transformation of 4-CNB was
investigated for the Fe(III) minerals goethite (α-FeOOH) lepidocrocite (γ-
FeOOH) and hematite (α-Fe2O3) as well as for the Fe(II)-Fe(III) oxide magnetite
(Fe3O4) All experiments were conducted in the presence of 1 mM dissolved Fe(II)
at pH 72 As seen from the surface area-normalised pseudo 1 order rate constants
in Figure 47 the reduction of 4-CNB by the Fe(II)-amended goethite
lepidocrocite and magnetite systems was up to 40 times faster than its reduction by
GR-SO4 The reduction rate obtained for the Fe(II)hematite system was only
slightly higher than the rate for the blank containing no iron mineral but aqueous
Fe(II) solely
84 Chapter 4
Figure 47 Surface area-normalised pseudo 1 order rate constants for the degradation of 4-CNB
by GR-SO4 (open square this work) and various Fe(II) containing mineral systems (solid circles
Elsner et al 2004) Experimental conditions applied by Elsner et al 1 mM aqueous Fe(II) pH
72 25 m2 mineral surface areaL GR-SO4 = green rust sulphate α-FeOOH = goethite Fe3O4 =
magnetite γ-FeOOH = lepidocrocite α-Fe2O3 = hematite
The experiments with GR-SO4 in our study were carried out at pH 84 whereas the
experiments comprising the other systems in Figure 47 were conducted at pH 72
As the reactivity of GR is expected to increase with pH (Lee amp Batchelor 2002b
Koch amp Hansen 1997) the lower of GR-SO4 compared to other Fe(II) systems
cannot be explained by differences in pH values GR-SO4 might just contain fewer
or less reactive surface sites than Fe(II)-amended goethite lepidocrocite and
magnetite suspensions These findings contrast those of other studies which found
higher surface area-normalised pseudo 1 order rate constants for dechlorination
reactions for GR-SO4 than for magnetite (Lee amp Batchelor 2002a amp 2002b Elsner
et al 2004) The different reactivity orders of the Fe(II)-bearing minerals found
for chlorinated aliphatics and nitro aromatics suggest that effects other than pH and
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 85
the intrinsic reduction potentials of the reacting species play a role for the
reactivity of these Fe(II)-bearing minerals
435 Depletion of reactive sites
Assuming that the platy hexagonal GR-SO4 crystals hold an average width of 1 microm
and an average particle thickness of 35 nm (Hansen amp Koch 1998) the ratio of
outer surface area to total surface area AouterAtotal ~ 131 (see Supporting
Information 73) This means that only 3 of the total surface area in GR-SO4 is
available at the external surface Thus as the interlayer sulphate in GR-SO4 is not
readily exchanged with the anionic NAC applied we conclude that 4-NPA and
other NACs only react with the Fe(II) sites at the external GR-SO4 surface In
Table 43 the actual amounts of NAC reduced by GR-SO4 during reaction is
compared with the amount of NAC which theoretically can be reduced by the
initial amount of external reactive Fe(II) sites in GR-SO4 at AouterAtot ~ 131
Table 43 The actual amounts of NAC reduced by GR-SO4 during reaction ∆[NAC]act
compared with the amount of NAC which stoichiometrically should be reduced by the initial
amount of external reactive Fe(II) sites in GR-SO4 ∆[Ar-NO2]theory (calculated as
[Fe(II)GR]0(31middot6) assuming an even distribution of Fe(II) throughout the GR-SO4 structure)
Calculated for [NAC]0 ~ 50 microM
[Fe(II)GR]0 (mM)
∆[Ar-NO2]theory(microM)
∆[4-NT]act (microM)
∆[4-CNB]act (microM)
∆[4-NPA]act (microM)
103 55 174 177 99
63 339 426 380 273
As seen in Table 43 the actual amounts of NAC reduced by GR-SO4 during
reaction are in most cases higher than the amount of NAC which should be
reduced at the given [Fe(II)GR]0 according to reaction stoichiometry This indicates
that new external reactive sites were regenerated eg the Fe(III) phases produced
peel off the GR surface exposing new Fe(II) sites or that outermost internal
86 Chapter 4
reactive sites in close vicinity to the edges are available for reaction as well Lee
and Batchelor (2000b) also found the experimentally observed reduction capacity
of GR-SO4 for chlorinated ethylenes to be 2-3 orders of magnitudes lower than the
estimated reduction capacity including all Fe(II) in GR-SO4
At low initial Fe(II)GR concentrations only a fraction of NAC was reduced within
the reaction time observed (Figure 45c) even though there was stoichiometric
excess of Fe(II)-GR present The fraction of initial Ar-NO2 reduced by GR-SO4 at
reaction termination decreased as [Fe(II)GR]0 decreased (Table 41) and was
accompanied by a change in apparent rate laws with time (compare Figures 45b amp
d) In order to explain these observations we propose that the NACs react only at
external reactive Fe(II) sites and that the regeneration of new external reactive sites
is much slower than the reduction of NAC by GR-SO4 Thus the fast reduction of
NAC taking place at the external reactive sites represents the pseudo 1 order
behaviour whereas depletion of external reactive sites and their slow regeneration
are represented by the second bent part of the (time ln [Ar-NO2]t[Ar-NO2]0)-
curves deviating from pseudo 1 order kinetics Hence at low [Fe(II)GR]0 the
regeneration of reactive sites will eventually control the overall reaction rate
Depletion of available Fe(II) was also observed during the fast reduction of
chromate by GR-CO3 when the initial chromate concentration was increased or
when the GR-CO3 suspension was respiked with chromate repeatedly (Williams amp
Scherer 2001)
436 The role of external and internal reactive sites
It is reasonable to assume that GRs hold adsorption properties similar to other
layered double hydroxides such as hydrotalcites The sorption of 246-
trinitrophenol (TNP) and 245-trichlorophenol (TCP) on chloride and carbonate
intercalated hydrotalcites (HT-Cl = Mg3Al(OH)8ClmiddotyH2O HT-CO3 =
Mg6Al2(OH)16CO3middotyH2O) has been investigated (Hermosin et al 1993 Ulibarri et
al 1995 Ulibarri et al 2001) The authors found that the adsorption of TCP on
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 87
HT-CO3 was very low and that TCP adsorbs only on the external surface sites of
HT-CO3 (Hermosin et al 1993) Furthermore is was reported that the adsorption
of TNP on HT was dramatically affected by the nature of the interlayer anion ie
the adsorption of TNP was considerably higher on HT-Cl than on HT-CO3
(Ulibarri et al 2001) For HT-Cl interlayer anion exchange of chloride with TNP
was detected by XRD analysis and an expansion of the characteristic basal d003
spacing from 79 Ǻ to 132 Ǻ confirmed the presence of TNP in the HT interlayer
(Ulibarri et al 1995) Collating the results reported for HTs with GRs it is not
likely that the divalent SO42- in GR-SO4 is exchanged with the monovalent 4-NPA
Chacirctelet et al (1996) investigated the adsorption of mono- and divalent anions
onin the outer and inner adsorption sites in HT by varying the zetapotential with
pH in the presence of various electrolytes The authors found that SO42- adsorbs on
the external HT surfaces by formation of outer-sphere complexes whereas chloride
hardly adsorbed on HT Moreover it was reported that the adsorption of sulphate
onto HT was not strongly affected by the presence of chloride while sulphate on
the contrary inhibited the adsorption of chloride on HT Studies applying
spectroscopic analyses have investigated the sorption of oxyanions at external and
internal GR-SO4 surfaces (Myneni et al 1997 Randall et al 2001) Selenate was
adsorbed only on the outer GR-SO4 surface when added after GR formation
whereas it was primarily coprecipitated into the interlayer when present during GR
formation Thus for selenate its presence during GR formation is a prerequisite of
its incorporation in the GR interlayer Selenate is readily reduced by GR-SO4 and
the rates of reduction of coprecipitated selenate were very similar to the reduction
rates of selenate adsorbed at the outer GR surface (Myneni et al 1997) This
finding suggests that the outer and inner reactive Fe(II) sites in GR-SO4 hold
similar reactivities
Results based on electron microscopy reported that the reduction of uranyl took
place primarily at the edges of hexagonal GR-SO4 particles (OrsquoLoughlin et al
2003a) In another recent study XRD characterization of the GR-SO4 crystals
88 Chapter 4
during reaction with trichloroacetate (TCA) indicated that TCA did not enter the
GR-SO4 interlayer during reaction (Chapter 5 this work) The average GR-SO4
particle thickness perpendicular to the basal plane was constant during reaction
implying that TCA reacts only at the edges and not at the basal planes Assuming
that the platy hexagonal GR-SO4 crystals hold an average width of 1 microm and an
average particle thickness of 35 nm (Hansen amp Koch 1998) the ratio of edge
surface area to outer surface area is AedgeAouter ~ 130 (see Supporting Information
73) Hence only 3 of the outer surface area in GR-SO4 is available at the edges
Once more the regeneration of new external reactive sites is strongly inferred as
the actual amounts of NAC reduced by GR-SO4 during reaction are much higher
than the amount of NAC which may be reduced by the reactive edge sites present
initially Assuming that the NACs react at the edges only and if employing the
AedgeAouter in the estimation of the rate constants the surface area-normalised
pseudo 1 order rate constants for GR-SO4 would be 30 times higher than the rate
constants depicted in Figure 47 Thus the reactivity of GR-SO4 normalised to its
reactive surface area is higher than the reactivity normalised to its outer surface
area determined by the BET method (N2 adsorption)
The reduction of chromate has been examined in the presence of all the common
GR forms (Bond amp Fendorf 2003 Loyaux-Lawniczak et al 1999 Loyaux-
Lawniczak et al 2000 Williams amp Scherer 2001) The results reported by Bond
amp Fendorf (2003) confirm that not only the surface area of GR but also the
interlayer spacing (interlayer anion size) and interlayer anion charge play an
important role for the reaction rate Hence it follows that coordination (size) and
charge of the oxidant determine its access to the internal sites in GRs
The results obtained for all 4 NACs support what has been reported for nitrate and
TCA At [Fe(II)GR]0 = 2-10 mM and [NO3-]0 = 143 mM pseudo 1 order rate
constants for the reduction of nitrate by GR-SO4 were 158middot10-7 s-1middotm-2middotL (Hansen
et al 2001) This reaction rate increased 40 times by adding barium nitrate instead
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 89
of sodium nitrate thereby precipitating the interlayer sulphate as barium sulphate
and enhancing access to the interlayer Though barium addition changes the GR-
SO4 system dramatically it indicates the importance of interlayer anion exchange
(Hansen amp Koch 1998) The rate constant reported for nitrate (no barium added) is
100-1000 smaller than the rate constants obtained for the NACs in this work
Moreover the reaction kinetics for nitrate did not deviate from pseudo 1 order
kinetics At [Fe(II)GR]0 = 025-104 mM and [TCA]0 = 50 microM-1 mM pseudo 1
order rate constants for the reduction of TCA by GR-CO3 or GR-SO4 were 65middot10-7
s-1middotm-2middotL (Chapter 5 this work) The rate constant for TCA is 10-1000 smaller than
the rate constants for the NACs and the reaction kinetics for TCA did not deviate
from pseudo 1 order kinetics This suggests that the overall reductive
transformation of slowly reacting oxidants such as nitrate and TCA is not
controlled by the rate of regeneration of external Fe(II) reactive sites Altogether
the results reported for selenate chromate and nitrate clearly demonstrate that
these anionic oxidants react primarily with external reactive sites in GR-SO4 Only
under certain conditions ie adding the oxidant prior to GR-SO4 formation or
extracting the interlayer sulphate through precipitation with barium outside the
GR-SO4 particles do the oxidants have access to the interlayer Our findings
suggest that both the neutral and anionic nitro aromatic probe compounds applied
here also react exclusively with the external reactive sites in GR-SO4 Supposedly
the neutral and monovalent charge states of the NACs hinder their access to the
GR-SO4 interlayer A divalent anionic nitro aromatic probe compound might
exchange with the interlayer sulphate more readily and gain access to the inner
Fe(II) reactive sites in GR-SO4 only divalent anionic NACs are not commercially
available
44 Conclusions
This work demonstrates that NACs are completely reduced to their corresponding
anilines by GR-SO4 The surface area-normalised pseudo 1 order rate constants
obtained for the reduction of the neutral and anionic NACs by GR-SO4 under
90 Chapter 4
various experimental conditions did not differ significantly from each other despite
their different charges Neither mass transfer control nor surface saturation kinetics
could account for the similarity of the pseudo 1 order rate constants obtained for
the NACs These observations suggest that the anionic NACs do not have an
enhanced access to inner or outer Fe(II)-GR reactive sites as compared to the
neutral NACs Based on our estimations of the molecular sizes of the NACs we
propose that the charge and not the size of the NACs controls their access to the
internal reactive sites in GRs Hence the reaction between NAC and GR-SO4 takes
place primarily at the external reactive Fe(II) sites This work further demonstrated
that the reduction of the NACs by GR-SO4 only followed pseudo 1 order kinetics
throughout the whole reaction at high initial Fe(II)GR concentrations At low initial
Fe(II)GR concentrations the NACs were not reduced completely within the reaction
time observed though according to reaction stoichiometry the total Fe(II)-GR
present should be sufficient to reduce the whole amount of NAC This means that
at some point during the reaction the external reactive Fe(II) sites were depleted
and the regeneration of new external reactive sites was much slower than the
reduction of the NACs by GR-SO4 The reduction of 4-CNB by GR-SO4 reported
here was 10-100 times slower than its reduction by other Fe(II)-Fe(III) systems
such as goethite lepidocrocite and magnetite suspensions amended with Fe(II)
(Elsner et al 2004)
The results obtained in this work infer that under natural geochemical conditions
where GR-SO4 presumably forms in low concentrations the rate of regeneration of
external Fe(II) reactive sites may control the overall reductive transformation of
fast reacting pollutants by GR-SO4 Thus not only abiotic processes such as
interchanging redox conditions created by water level alterations but also the direct
microbial formation through Fe(III)-reducing bacteria may govern the formation of
GRs and the renewal of external Fe(II) reactive sites in GRs This holds both for
natural systems like iron-rich suboxic soils and sediments as well as engineered
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 91
systems like permeable reactive barriers of zero-valent iron implemented for on-
site remediation of organic and inorganic contaminants
Acknowledgments
We would like to thank Henrik T Andersen for performing the NB kinetic experiments and
Hanne Nancke-Krogh for technical assistance in the laboratory
References
Al-Agha MR Burley SD Curtis CD Esson J (1995) Complex cementation textures and authigenic mineral assemblages in recent concretions from the Lincolnshire Wash (east coast UK) driven by Fe(0) to Fe(II) oxidation Journal of the Geological Society 152 157-171 Bernal JD Dasgupta DR Mackay AL (1959) The oxides and hydroxides of iron and their structural inter-relationships Clay Minerals Bulletin 4 15-30 Bond DL Fendorf S (2003) Kinetics and structural constraints of chromate reduction of green rusts Environmental Science and Technology 37 2750-2757 Brunauer S Emmett PH Teller E (1938) Adsorption of gases in multimolecular layers Journal of American Chemical Society 60 309-319 Charlet L Silvester E Liger E (1998) N-compound reduction and actinide immobilisation in surficial fluids by Fe(II) The surface FeIIIFeIIOH0 species as major reductant Chemical Geology 151 85-93 Chacirctelet L Bottero JY Yvon J Bouchelaghem A (1996) Competition between monovalent and divalent anions for calcined and uncalcined hydrotalcite anion exchange and adsorption sites Colloids and Surfaces A Physicochemical and Engineering Aspects 111 167-175 Chaudhuri SK Lack JG Coates JD (2001) Biogenic magnetite formation through anaerobic biooxidation of Fe(II) Applied and Environmental Microbiology 67 2844-2848 Cui D Spahiu K (2002) The reduction of U(VI) on corroded iron under anoxic conditions Radiochemica Acta 90 623-628 Elsner M Haderlein SB Schwarzenbach RP (2004) Reactivity of Fe(II)-bearing minerals towards reductive transformation of organic contaminants Environmental Science and Technology 38 799-807 Erbs M Hansen HCB Olsen CE (1999) Reductive dechlorination of carbon tetrachloride using iron(II)iron(III)-hydroxide-sulphate (green rust) Environmental Science and Technology 33 307-311 Fadrus H Maly J (1975) Suppression of iron(III) interference in the determination of iron(II) in water by the 110-phenanthroline method The Analyst 100 549-554 Fredrickson JK Zachara JM Kennedy DW Dong H Onstott TC Hinman NW Li S
92 Chapter 4 (1998) Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium Geochimica et Cosmochimica Acta 62 3239-3257 Geacutenin JMR Bourrieacute G Trolard F Abdelmoula M Jaffrezic A Refait Ph Maitre V Humbert B Herbillon A (1998) Thermodynamic equilibria in aqueous suspensions of synthetic and natural Fe(II)-Fe(III) green rusts Occurrences of the mineral in hydromorphic soils Environmental Science and Technology 32 1058-1068
Geacutenin JMR Olowe AA Benbouzid-Rollet ND Prieur D Confente M Resiak B (1991) The simultaneous presence of green rust 2 and sulfate reducing bacteria in the corrosion of steel sheet piles in a harbour area Hyperfine Interactions 69 875-878 Glasauer S Weidler PG Langley S Beveridge TJ (2003) Controls on Fe reduction and mineral formation by a subsurface bacterium Geochimica et Cosmochimica Acta 67 1277- 1288 Hansen HCB (1989) Composition stabilization and light absorption of Fe(II)Fe(III) hydroxycarbonate (green rust) Clay Minerals 24 663-669 Hansen HCB Borggaard OK Soslashrensen J (1994) Evaluation of the free energy of formation of iron(II)iron(III)-hydroxidesulphate (Green Rust) and its reduction of nitrite Geochimica et Cosmochimica Acta 58 2599-2608 Hansen HCB Guldberg S Erbs M Koch CB (2001) Kinetics of nitrate reduction by green rusts ndash effects of interlayer anion and Fe(II)Fe(III) ratio Applied Clay Science 18 81-91 Hansen HCB Koch CB (1998) Reduction of nitrate to ammonium by sulphate green rust Activation energy and interlayer reaction mechanism Clay Minerals 33 87-101 Hansen HCB Koch CB Nancke-Krogh H Borggaard OK Soerensen J (1996) Abiotic nitrate reduction to ammonium Key role of green rust Environmental Science and Technology 30 2053-2056 Hansen HCB Poulsen IF (1999) Interaction of synthetic sulphate green rust with phosphate and the crystallization of vivianite Clays and Clay Minerals 47 312-318 Hansen HCB Taylor RM (1991) The use of glycerol intercalates in the exchange of CO3
2- with SO4
2- NO3- or Cl- in pyroaurite-type compounds Clay Minerals 26 311-327
Heasman DM Sherman DM Ragnarsdottir KV (2003) The reduction of aqueous Au3+ by sulfide minerals and green rust phases American Mineralogist 88 725-738 Hermosin MC Pavlovic I Ulibarri MA Cornejo J (1993) Trichlorophenol adsorption on layered double hydroxide a potential sorbent Journal of Environmental Science and Health A28 1875-1888 Hofstetter TB Heijmann CG Haderlein SB Holliger C Schwarzenbach RP (1999) Complete reduction of TNT and other (poly)nitroaromatic compounds under iron-reducing subsurface conditions Environmental Science and Technology 33 1479-1487 Hofstetter TB Schwarzenbach RP Haderlein SB (2003) Reactivity of Fe(II) species associated with clay minerals Environmental Science and Technology 37 519-528
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 93
Klausen J Troumlber SP Haderlein SB Schwarzenbach RP (1995) Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions Environmental Science and Technology 29 2396-2404 Koch CB Hansen HCB (1997) Reduction of nitrate to ammonium by sulphate green rust Advances in GeoEcology 30 373-393 Kukkadapu RK Zachara JM Smith SC Fredrickson JK Liu C (2001) Dissimilatory bacterial reduction of Al-substituted goethite in subsurface sediments Geochimica et Cosmochimica Acta 65 2913-2924 Lee W Batchelor B (2002a) Abiotic reductive dechlorination of chlorinated ethylenes by iron- bearing soil minerals 1 Pyrite and magnetite Environmental Science and Technology 36 5147- 5154 Lee W Batchelor B (2002b) Abiotic reductive dechlorination of chlorinated ethylenes by iron-bearing soil minerals 2 Green rust Environmental Science and Technology 36 5348- 5354 Liger E Charlet L Van Cappellen P (1999) Surface catalysis of uranium (VI) reduction by iron(II) Geochimica et Cosmochimica Acta 63 2939-2955 Loyaux-Lawniczak S Refait Ph Ehrhardt J Lecomte P Geacutenin JMR (2000) Trapping of Cr by formation of ferrihydrite during the reduction of chromate ions by Fe(II)-Fe(III) hydroxysalt green rusts Environmental Science and Technology 34 438-443 Loyaux-Lawniczak S Refait Ph Lecomte P Ehrhardt J Geacutenin JMR (1999) The reduction of chromate ions by Fe(II) layered hydroxides Hydrology and Earth System Sciences 3 593-599 Mendiboure A Schoumlllhorn A (1986) Formation and anion exchange reactions of layered transition metal hydroxides [Ni1-xMx](OH)2(CO3)x2(H2O)z (M = Fe Co) Revue de Chimie Mineacuterale 23 819-827 Miyata S (1983) Anion-exchange properties of hydrotalcite-like compounds Clays and Clay Minerals 31 305-311 Myneni SCB Tokunaga TK Brown Jr GE (1997) Abiotic selenium redox transformations in the presence of Fe(IIIII) oxides Science 278 1106-1109 Nielsen A (1976) Hvid groslashn og sort rust Beskrivelse af en korrosionsskade paring et svoslashmmebassin Nordisk Betong 2 21-24 OLoughlin EJ Burris DR (2004) Reduction of halogenated ethanes by green rust Environmental Toxicology and Chemistry 23 41-48 OLoughlin EJ Kelly SD Cook RE Csencsits R Kemner KM (2003a) Reduction of uranium(VI) by mixed iron(II)iron(III) hydroxide (green rust) Formation of UO2 nanoparticles Environmental Science and Technology 37 721-727
94 Chapter 4 OLoughlin EJ Kelly SD Kemner KM Csencsits R Cook RE (2003b) Reduction of AgI AuIII CuII and HgII by FeIIFeIII hydroxysulfate green rust Chemosphere 53 437-446 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002) Iron (IIIII) hydroxycarbonate green rust formation and stabilization from lepidocrocite bioreduction Environmental Science and Technology 36 16-20 Parmar N Gorby YA Beveridge TJ Ferris FG (2001) Formation of green rust and immobilization of nickel in response to bacterial reduction of hydrous ferric oxide Geomicrobiology Journal 18 375-385 Pepper SE Bunker DJ Bryan ND Livens FR Charnock JM Pattrick RAD Collison D (2003) Treatment of radioactive wastes An X-ray absorption spectroscopy study of the reaction of technetium with green rust Journal of Colloid and Interface Science 268 408- 412 Randall SR Sherman DM Ragnarsdottir KV (2001) Sorption of As(V) on green rust (Fe4(II)Fe2(III)(OH)12SO4
3H2O) and lepidocrocite (γ-FeOOH) Surface complexes from EXAFS spectroscopy Geochimica et Cosmochimica Acta 65 1015-1023 Roh Y Lee SY Elless MP (2000) Characterization of corrosion products in the permeable reactive barriers Environmental Geology 40 184-194 Schultz CA Grundl TJ (2000) pH dependence on reduction rate of 4-Cl-nitrobenzene by Fe(II)montmorillonite systems Environmental Science and Technology 34 3641-3648 Simon L Francois M Refait Ph Renaudin G Lelaurain M Geacutenin JMR (2003) Structure of the Fe(II-III)-layered double hydroxysulphate green rust two from Rietveld analysis Solid State Sciences 5 327-334 Tamaura Y (1986) Ni(II)-bearing green rust II and its spontaneous transformation into Ni(II)- bearing ferrites Bulletin of the Chemical Society of Japan 59 1829-1832 Tamaura Y (1985) ZnII-bearing green rust II and its spontaneous transformation into ZnII- bearing ferrite in aqueous solution Bulletin of the Chemical Society of Japan 58 2951-2954 Trolard F Abdelmoula M Bourrieacute G Humbert B Geacutenin JMR (1996) Mise en eacutevidence dun constituant de type rouilles vertes dans les sols hydromorphes Proposition de lexistence dun nouveau mineacuteral la fougeacuterite Geacuteosciences de surface Comptes Rendus de LrsquoAcademie des Sciences 323 1015-1022 Tuovinen OH Button KS Vuorinen A Carlson L Mair DM Yut LA (1980) Bacterial chemical and mineralogical characteristics of tubercles in distribution pipelines Journal of the American Water Works Association 72 626-635 Ulibarri MA Pavlovic I Barriga C Hermosin MC Cornejo J (2001) Adsorption of anionic species on hydrotalcite-like compounds effect of interlayer anion and crystallinity Applied Clay Science 18 17-27 Ulibarri MA Pavlovic I Hermosin MC Cornejo J (1995) Hydrotalcite-like compounds as potential sorbents of phenols from water Applied Clay Science 10 131-145
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 95
Williams AGB Scherer MM (2001) Kinetics of chromate reduction by carbonate green rust Environmental Science and Technology 35 3488-3494
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 97
5 Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems
Abstract
Trichloroacetate (TCA) is a widespread environmental contaminant with proven
phytotoxicity and suspected human carcinogenicity In order to assess the global
cycling of TCA and to predict its fate in subsurface environments information
regarding the reactivity and product distribution of TCA degradation is needed
Due to the high oxidation state of TCA conditions for oxidative transformation
pathways in soils and groundwater are unfavorable However in suboxic soils and
sediments Fe(II)-bearing minerals are potential reactants for reductive
dehalogenation reactions of TCA as has been demonstrated for other halogenated
contaminants We examined the reactivity of various Fe(II)-Fe(III) mineral
systems towards TCA and dichloroacetate (DCA) its expected transformation
product in laboratory batch experiments imitating natural conditions ie low
initial Fe(II) Fe(III) and TCADCA concentrations and no artificial buffer The
Fe(II)-Fe(III)-systems investigated included sulfate green rust (GR-SO4) carbonate
green rust (GR-CO3) magnetite Fe(II)goethite and Fe(II)lepidocrocite
Trichloroacetate was readily reduced to DCA by all Fe(II)-bearing minerals The
reactions generally followed pseudo 1 order kinetics with respect to TCA The
surface area-normalised pseudo 1 order rate constants obtained (035ndash76middot10-5 min-
1middotm-2middotL at [Fe(II)]0 = 020ndash122 mM [TCA]0 = 15ndash1000 microM and pH 70ndash87)
showed no striking differences regarding product distribution and surface area-
normalised reaction rate constants between the Fe(II)-Fe(III)-systems The
stoichiometrically formed DCA was not further reduced to monochloroacetate
(MCA) or acetate in any of the systems within the time frame in our experiments
To our knowledge this is the first published report on abiotic transformation of
TCA by Fe(II)-bearing minerals Our results imply that processes involving
reactive Fe(II)-bearing minerals may play a significant role in controlling the fate
98 Chapter 5
of TCA in natural subsurface environments and that DCA found in the subsurface
may be formed by such processes
51 Introduction
Trichloroacetic acid (TCA) has been applied as a herbicide for many years until its
use was banned in the late 1980acutes (Berg et al 2000) Today TCA is mainly used
as an etching agent in the metal industry as a swelling solvent in the plastic
production and as a bleaching agent in the paper and pulp manufacture (Muumlller et
al 1996) Other anthropogenic sources include formation of TCA as a result of the
chlorine based disinfecting process used in drinking water treatment and the
atmospheric photooxidation of chlorinated solvents including tetrachloroethene
and 111-trichloroethane (McCulloch 2002) Only very little information is
available on the TCA production volumes and even less is known about the
amount of TCA released into the environment as a result of its industrial
applications Due to its low volatility and high aqueous solubility TCA is easily
washed out of the atmosphere into the aquatic and terrestrial biospheres As TCA
is found in almost every ecosystem around the globe including non-urban and
non-industrial sites the relative contributions from anthropogenic and natural
sources are currently being debated (McCulloch 2002 Ahlers et al 2003)
Trichloroacetic acid is omnipresent in soils and the concentrations reported are
very variable ranging from lt005 microgkg to 380 microgkg (Euro Chlor 2001
McCulloch 2002 Ahlers et al 2003) Both abiotic and enzymatically catalyzed
formation of TCA from humic acids have been demonstrated in laboratory studies
(Haiber et al 1996 Hoekstra et al 1999b Fahimi et al 2003) Furthermore the
in situ natural formation of TCA from anthropogenic or natural tetrachloroethene
or 111-trichloroethane in biota has been suggested (Hoekstra et al 1999a
McCulloch 2002) Such natural sources may explain part of the TCA
concentrations found in soils but their environmental significance is still unknown
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 99
On account of its phytotoxicity suspected human carcinogenicity and widespread
occurrence TCA is of considerable environmental concern especially in the
terrestrial compartment The TCA concentrations found in soil air and water in
pre-industrial times were far below the present ones (Jordan amp Frank 1999 Ahlers
et al 2003) Based on the current TCA concentrations detected in soils the
European Commission proposed risk reduction measures concerning
tetrachloroethene - a precursor of TCA - to be taken immediately (Ahlers et al
2003 and references therein) Occurrences of monochloroacetic acid (MCA) and
dichloroacetic acid (DCA) reported include surface waters marine waters
precipitation ice (glaciers) and air (Reimann et al 1996 Berg et al 2000 Scott et
al 2000 Scott et al 2002) Based on the concentrations reported for the aquatic
environments it is reasonable to assume that MCA and DCA are omnipresent in
soils as well Sources of MCA and DCA include production in the chemical
industry photooxidation of chlorinated aliphatics in the atmosphere and reductive
transformation of TCA (Reimann et al 1996 Ahlers et al 2003 and references
therein) MCA and DCA are also toxins and suspected human carcinogens (Kuumlhn
amp Pattard 1990) hence not only TCA but also its daughter compounds are
pollutants of environmental concern
In subsurface environments TCA may be removed by sorption seepage chemical
transformation microbial degradation and plant uptake followed by metabolic
degradation andor physical removal at harvest (Foy 1975) There is little or no
evidence of abiotic transformations of TCA in the literature Only one recent study
demonstrated the reductive dechlorination of TCA to MCA by Fe(0) (Hozalski et
al 2001) It has been reported that the degradation of TCA in soil is slow and
mainly mediated by microorganisms but only little is known about the bacteria and
processes involved (Lignell et al 1984) Biodegradation of TCA has been found at
both oxic and anoxic conditions An aerobic microorganism capable of growing on
TCA as the sole carbon and energy source has been characterised (Yu amp Welander
1995) Moreover anaerobic bacteria coupling co-metabolic growth to reductive
100 Chapter 5
dechlorination of TCA have been isolated (Weightman et al 1992 De Wever et
al 2000) However more information regarding the abiotic and biotic
transformation of TCA is needed in order to assess the fate and transport of TCA in
natural subsurface environments
It is well-known that Fe(II) present in minerals or associated with mineral surfaces
is a much stronger reductant than Fe(II) in solution The enhanced reactivity of a
structural or surface-bound Fe(II) center can be rationalized by the increased
electron density donated by hydroxyl ligands and a stabilization of the Fe(III)
oxidation state by the hydroxyl ligands (Luther 1990) Fe(II)-bearing minerals
including layered Fe(II)-Fe(III) hydroxides (green rusts) magnetite (Fe3O4)
siderite (FeCO3) Fe(II) sulfides as well as Fe(II)-carrying Fe(III) oxides and clay
minerals have also been shown to reduce a range of organic and inorganic
contaminants such as nitro aromatic compounds chlorinated aliphatics chromate
uranyl pertechnetate nitrate monochloramine and carbamate pesticides (Chapter
4 this work Klausen et al 1995 Cui amp Eriksen 1996 Erbs et al 1999 Liger et
al 1999 Loyaux-Lawniczak et al 1999 Amonette et al 2000 Hansen et al
2001 Pecher et al 2002 Vikesland amp Valentine 2002 Hofstetter et al 2003
OrsquoLoughlin and Burris 2003 OrsquoLoughlin et al 2003a amp 2003b Strathmann amp
Stone 2003 Elsner et al 2004a) Laboratory and field studies showed that even in
geochemically highly heterogeneous anoxic aquifer sediments Fe(II) adsorbed to
Fe(III) (hydr)oxide surfaces was the dominant reductant of nitroaromatic and
halogenated contaminants (Ruumlgge et al 1998 Hofstetter et al 1999 Kenneke amp
Weber 2003) Only little is known about the nature of the Fe(II) species associated
with Fe(III) oxide surfaces but reactive hydroxylated Fe(II)-Fe(III)-hydroxo
surface complexes associated with hematite and magnetite above pH 65 have been
proposed (Charlet et al 1998aampb Liger et al 1999) Due to the presence of
structural Fe(II) within the mineral lattice the reactivity of Fe(II) associated with
mixed valent Fe(II)-Fe(III) minerals such as green rusts magnetite and reduced
ferruginous clay minerals may hold another reactivity than Fe(II) associated with
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 101
pure Fe(III) oxides However Fe(II) adsorbed on Fe(III) oxides such as goethite
hematite and lepidocrocite may also hold different reactivities as the Fe(III) oxides
contain different crystal and surface structures
Since chlorinated ethanes and ethenes such as hexachloroethane 111-
trichloroethane tetrachloroethene and trichloroethene are susceptible to chemical
reduction by a range of Fe(II)-bearing minerals including magnetite GR-SO4
Fe(II) sulfides and Fe(II)-carrying Fe(III) oxides (Butler amp Hayes 1998 amp 1999
Hwang amp Batchelor 2000 Gander et al 2002 Lee amp Batchelor 2002aampb Elsner
et al 2004a) we hypothesized that TCA may be transformed by Fe(II)-bearing
minerals as well The main goals of this work were to study such reactions and
establish product distribution and surface area-normalised reaction rates for the
reductive dechlorination of TCA by Fe(II)-Fe(III) mineral systems common in
nature
5 2 Materials and methods
No synthetic buffers were applied and iron concentrations were kept low The
calcareous systems were pH-controlled at 76 through a natural buffer system
(CaCO3(s) + 995 N205 CO2(g)) All handling and sampling of solutions and
suspensions were carried out under strict anoxic conditions Goethite (acicular
particles with size 01 times 06 microm specific surface area 16 m2g) and lepidocrocite
(acicular particles with size 005 times 03 microm specific surface area 18 m2g) were
purchased as fine powders from Bayer (Bayferrox 910 and 943) Calcite (grain size
170-350 microm Pluumlss-Staufer AG) was used as a buffer or as a Fe(III)-oxide-bearing
mineral In order to simulate natural conditions the iron minerals were applied as
coatings on calcite particles (model system for calcareous soils) in some
experiments Trichloroacetic acid dichloroacetic acid and monochloroacetic acid
were pa quality (Fluka)
102 Chapter 5
521 Synthesis of GRs and magnetite
GR-CO3 was synthesized by controlled air oxidation of an FeCl2 solution at a
constant pH of 700 (titrated with 1 M Na2CO3) according to the procedure given
by Hansen amp Koch (1997) 05 M aqueous stock solutions of FeCl2 were prepared
in 100 mL glass flasks by reacting 65 mmol of iron powder (particle size 10 microm
Merck) with 100 mL deoxygenated 10 M HCl The solutions were magnetically
stirred and heated (~80degC) during reaction until the H2(g) production had ceased (ge
2 hours) The FeCl2 solutions were stored in the dark under a small Ar
overpressure at 5degC The GR-CO3 suspensions were washed with deoxygenated
deionised water (DIW) separated on a folding filter (medium filtration rate cotton
linterhigh alpha pulp Schleicher amp Schuell) and redispersed in deoxygenated
DIW Washing separation and redispersion of the GR-CO3 suspension were
conducted in an anoxic glove box (92 N28 H2 Coy Laboratory Products Inc)
All suspensions and solutions were deoxygenated by Ar-purging (999998 Ar
Carbagas) Magnetite was synthesized by further aerial oxidation of GR-CO3 at pH
700 until consumption of 1 M Na2CO3 ceased GR-SO4 was synthesized by
controlled air oxidation of an FeSO4 solution at a constant pH of 70 according to
the procedure given by Koch amp Hansen (1997) The GR-SO4 suspension was
washed with deoxygenated DIW separated on a glass filter funnel (pore size 4
Duran) and redispersed in deoxygenated DIW Washing separation and
redispersion of the GR-SO4 suspension were conducted in an anoxic glove bag
(999995 Ar Aldrich)
522 Preparation of iron oxide coatings
Two grams of goethite (goe) or lepidocrocite (lep) and 100 g calcite were
combined with 200 mL DIW in a 500 mL polyethylene flask The suspension was
gently agitated on a reciprocating shaker for 24 h and left to stand for another 24 h
Excess Fe(III) oxides and salts were removed from the coated material by repeated
decantation and washing with DIW in polyethylene flasks until clear runoff
Finally the coatings were collected on folding filters and air dried The amount of
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 103
goethite and lepidocrocite coated onto calcite after washing and drying was
quantified to 10-11 mg Fe(III)g calcite
523 Mineral characterisation
The identity and purity of the GR-CO3 GR-SO4 and magnetite suspensions were
examined by means of X-ray diffraction (XRD) The XRD analyses were
performed on a Scintag XDS 2000 using Co Kα radiation (45 kV 40 mA) or a
Siemens D5000 XRD applying Co Kα radiation (40 kV 40 mA) Glycerol smears
made according to Hansen (1989) were scanned between 6 and 80 deg2θ with a scan
speed of 1 deg2θmin The specific surface area (SSA) of calcite was determined by
the BET multi-point method using N2 adsorption (Brunauer et al 1938) Powder
samples were filled into sample burettes in the glove box and the generously
lubricated stopcocks closed Samples and burettes were evacuated prior to
connecting them to the BET-instrument (Sorptomatic 1990 Fisons)
524 Kinetic experiments
All reactions were carried out in 25-100 mL serum vials sealed with stoppers
(Viton or Teflon coated rubber) and aluminum crimp caps Kinetic experiments
were conducted with GR-SO4 GR-CO3 magnetite Fe(II)goethite and
Fe(II)lepidocrocite at room temperature In most cases pH was controlled through
the carbonate-bicarbonate buffer system by adding calcite to suspensions
containing the iron minerals solely or by adding the iron minerals as coatings on
calcite Furthermore the calcite containing suspensions were deoxygenated with
05 CO2995 N2(g) thereby attaining an initial pH of 76-77 The GR-CO3 and
magnetite suspensions were deoxygenated with 100 N2(g) and no additional pH
buffer was added The goethite and lepidocrocite suspensions were amended with
300-1000 microM FeCl2(aq) and equilibrated gt 20 h prior to TCADCA addition See
Table 51 for more details on the experimental conditions To start the reaction 50
microM - 1 mM TCA or DCA was added to the mineral suspensions from aqueous
anoxic stock solutions The reaction vials were agitated gently on a roller apparatus
104 Chapter 5
or a shaking table (35 rpm) in order to minimize abrasion of the iron oxide mineral
coatings At appropriate time intervals suspension samples were withdrawn using
Ar(g)- 100 N2(g)- or 995 N205 CO2(g)-flushed sterile disposable syringes
and hypodermic needles The suspension samples were filtered (02 microm Teflon)
and collected for quantification of chloride and the chlorinated acetic acids The
samples were stored at -20degC and analysed without further treatment
525 Analytical methods
Total and aqueous Fe(II) were determined using a modified phenanthroline method
(Fadrus and Maly 1975) For determining [Fe(II)aq] and [Fe(II)total] 1 mL filtered
(02 microm Teflon) and 1 mL unfiltered mineral suspension were added to 18 mL 01
M HCl respectively and allowed to dissolve for 30 min From these acid digests
01 mL was added to 05 mL Fe(II)-phenanthroline-buffer-reagent and 19 mL
DIW added up Estimates of the structural or adsorbed Fe(II) in the Fe(II)-Fe(III)
mineral systems were estimated as the difference [Fe(II)solid] = [Fe(II)total] -
[Fe(II)aq] The total amount of Fe(III) coated on calcite was determined by atomic
absorption spectroscopy following dissolution in 6 M HCl(aq) for 24 h At low
initial TCA concentrations (le 50 microM) the chlorinated acetic acids were quantified
by means of a modified ion interaction (or paired-ion) chromatographic method
(Sarzanini et al 1999) Separation was performed on a LiChrospher 100 RP-18 (5
microm 125 times 4 mm ID) reversed-phase column coupled with a LiChroCART 100 RP-
18 (4 times 4 mm ID) precolumn Analytical conditions were isocratic and the eluent
consisted of 50 aqueous solution of 35 mM cetyltrimethylammonium chloride
(pH 50) and 50 CH3CN The injection volume was 20 microL and the flow-rate 10
mLmin HPLC analyses of the chloroacetates were performed using a Gynkotek
Pump M480 Gynkotek Gina 50 auto sampler and a diode array UV detector (340s
Gynkotek) UV-VIS detection was carried out at 200 nm At higher initial TCA
concentrations the chlorinated acetic acids were quantified by a modified HPLC
method (Husain et al 1992) Separation was performed on a ChromSphere C-18
(10 microm 250 times 46 mm ID) reversed-phase column Analytical conditions were
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 105
isocratic and the eluent consisted of 015 M (NH4)2SO4(aq) pH 55 The injection
volume was 20 microL and the flow-rate 10 mLmin HPLC analyses were performed
using a Series 10 Liquid Chromatographic Pump (Perkin-Elmer) and a SPD-10 A
VP UV-VIS detector (Shimadzu) UV-VIS detection was carried out at 210 nm
Chloride was determined in the GR-SO4 kinetic experiments using a flow injection
system with spectrophotometric detection (Cheregi amp Danet 1997)
53 Results and discussion
531 Product formation and reaction kinetics
Trichloroacetate was readily reduced to DCA by all the Fe(II)-bearing minerals
examined Only DCA was detected within the reaction time in all the Fe(II)-Fe(III)
mineral systems Experiments conducted with the various Fe(II)-Fe(III) mineral
systems and DCA confirmed that no significant reduction of DCA took place (data
not shown) Hence it is reasonable to assume that the further hydrogenolysis of
DCA to MCA is too slow to be detected within the experimental time frame here
The mass balance of TCA and DCA was almost complete in all suspensions ruling
out any alternative reaction pathways to reductive dechlorination Decarboxylation
of TCA producing chloroform and carbon dioxide requires high temperatures and
is therefore assumed not to take place at the experimental conditions applied here
(Atkins et al 1984) Based on these results we propose that the reductive
dechlorination of TCA by Fe(II)-bearing minerals proceeds via hydrogenolysis
(replacement of halogen by hydrogen) as reported for the transformation of TCA
by zero-valent iron (Hozalski et al 2001) Thus in order to reduce TCA to DCA
2 electrons corresponding to 2 Fe(II) are needed (Figure 51)
106 Chapter 5
Cl3CC
O
O- Cl2HCC
O
O-
2e- H+ Cl-
TCA DCA Figure 51 Proposed reductive transformation pathway of TCA
In the Fe(II)goe and Fe(II)lep systems we detected no TCA transformation in the
absence of either aqueous Fe(II) or pure or calcite-associated goethite and
lepidocrocite These results strongly indicate that reactive Fe(II) species associated
with the goethite and lepidocrocite surfaces are the reductants for TCA in these
systems The Fe(III) phases forming in the mineral suspensions were not
characterised and therefore the reaction stoichiometry cannot be assessed
At initial Fe(II) concentrations in large excess of initial TCA concentration we
found a pseudo 1 order rate law for the degradation of TCA by Fe(II)
[ ] [ ] [ b a TCAFe(II) TCA
sdotsdot=minus kdt
d ]
where a = 1 b = 1 and the observed pseudo 1 order rate constant kobs = k middot [Fe(II)]
At all [Fe(II)]0[TCA]0 ratios studied (6-738) TCA was transformed almost
quantitatively into DCA and the reaction kinetics followed pseudo 1 order kinetics
with respect to TCA (Figure 52) The observed pseudo 1 order rate constants for
the transformation of TCA by the various Fe(II)-Fe(III) mineral systems were
calculated as initial rates (ie max first two half-lives) from linear fits of (time ln
[TCA]t[TCA]0)-plots (Table 51) The amount of chloride produced during
reaction with GR-SO4 was always equivalent to the amount of TCA transformed
into DCA (Figure 52c) This also indicates that no significant further reduction of
DCA took place in GR-SO4 suspensions
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 107
Figure 52 Time course of TCA consumption and DCA and chloride production for a)
Fe(II)Goe ([Fe(II)tot]0 = 095 mM) b) Fe(II)Lep ([Fe(II)tot]0 = 091 mM) c) GR-SO4
([Fe(II)GR]0 = 962 mM) d) GR-CO3 ([Fe(II)tot]0 = 633 mM) and e) Magnetite ([Fe(II)tot]0 = 350
mM) Solid lines represent 1 order kinetic fits whereas symbols and dotted lines represent actual
data = TCA = DCA = Clndash
T
able
51
Exp
erim
enta
l con
ditio
ns a
nd p
seud
o 1
ord
er ra
te c
onst
ants
for t
he re
duct
ive
trans
form
atio
n of
TC
A b
y va
rious
Fe(
II)-
Fe(I
II) c
onta
inin
g m
iner
al sy
stem
s
Syst
em
Susp
ensi
on a
ge
(d)
[Fe(
II)] s
olid
a (m
M)
[Fe(
II)] a
q b
(mM
) [T
CA
] 0 (micro
M)
pHin
itcpH
endd
k obs
e (min
-1)
Surf
ace
area
(m
2 L)
k obs
f
(min
-1m
-2middotL
)
Fe(I
I)aq
1
0
030
434
nd
76
gn
dn
dn
d
Fe(I
I)G
oe
1
002
024
429
nd
78
g1
021
0-47
1 i
143
10-5
Fe(I
I)G
oe
1
013
094
543
77
70
225
10-4
71
i3
161
0-5
Fe(I
I)G
oe
coat
ing
1
023
40
066
484
765
80
g6
401
0-454
0 j
119
10-5
Fe(I
I)G
oe
coat
ing
1
0
150
8048
6n
d7
6 g12
43
10-4
540
j2
301
0-5
Fe(I
I)L
ep
1
0
020
2315
7n
d7
8 g0
751
0-48
0 i
094
10-5
Fe(I
I)L
ep
coat
ing
1
016
30
137
470
765
80
g2
821
0-454
0 j
052
10-5
Fe(I
I)L
ep
coat
ing
1
0
100
8141
7n
d7
7 g8
311
0-454
0 j
154
10-5
Fe3O
41
3
380
1251
38
107
8g
830
10-4
16
k5
311
0-4
Fe3O
477
112
56
556
70
70
153
10-4
52
k2
951
0-5
GR
-CO
31
5
940
3950
37
658
4g
761
10-4
419
l1
821
0-5
GR
-CO
32
7
60
147
88
568
2940
81
0-453
6 l
761
10-5
GR
-CO
332
73
003
563
85
80
490
10-4
515
l0
951
0-5
GR
-CO
314
2
3
530
005
629
nd
87 g
513
10-4
249
l2
061
0-5
GR
-SO
41
5
17-1
217
086
-13
910
5n
dn
d3
601
0-492
6 m
039
10-5
GR
-SO
41
6
22-1
027
093
-14
527
0n
dn
d3
761
0-488
1 m
043
10-5
GR
-SO
41
7
05-1
014
077
-17
950
0n
dn
d3
741
0-4 9
18
m0
411
0-5
GR
-SO
41
5
17-1
051
060
-16
510
00n
dn
d2
891
0-483
7 m
035
10-5
n
d =
not
det
ecte
d a
Ini
tial s
truct
ural
or a
dsor
bed
Fe(I
I) e
stim
ated
as [
Fe(I
I)to
tal]
ndash [F
e(II
) aq]
b In
itial
dis
solv
ed F
e(II
) mea
sure
d c
Sus
pens
ion
pH p
rior t
o TC
A a
dditi
on d
Sus
pens
ion
pH a
t rea
ctio
n
te
rmin
atio
n e
Pse
udo
1 o
rder
rate
con
stan
ts fo
r the
con
sum
ptio
n of
TC
A c
alcu
late
d fr
om in
itial
rate
s (m
ax f
irst t
wo
half-
lives
) f
Surf
ace
area
-nor
mal
ised
pse
udo
1 o
rder
rate
con
stan
ts g
pH
con
trol
th
roug
h pu
re C
aCO
3 and
05
C
O2(g
) h
pH
con
trol t
hrou
gh F
e(II
I) o
xide
-coa
ted
calc
ite a
nd 0
5
CO
2(g)
i Es
timat
ed u
sing
the
SSA
of t
he F
e(II
I) o
xide
app
lied
j E
stim
ated
usi
ng th
e SS
A o
f cal
cite
~1
m2 g
k E
stim
ated
ass
umin
g SS
A =
4 m
2 g (S
chw
ertm
ann
amp C
orne
ll 1
991)
frac12middot[
Fe(I
I) sol
id] 0middot
232
gmiddotm
ol-1
middot4 m
2 middotg-1
l E
stim
ated
ass
umin
g SS
A =
47
m2 g
(Will
iam
s amp S
cher
er 2
001)
frac14middot[F
e(II
) GR] 0middot
600
gmiddotm
ol-1
middot47
m2 middotg
-1 m
Est
imat
ed a
s in l
but u
sing
SSA
= 7
12
m2 middotg
-1 (C
hapt
er 4
thi
s wor
k)
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 109
532 Comparing rate constants obtained for the various Fe(II)-Fe(III) mineral
systems
Data for the systems containing iron oxide coated calcite were very similar to the
data obtained for the pure iron oxides (not shown in Figure 53) Since no SSA was
determined for magnetite in this study a SSA of 4 m2g was assumed
(Schwertmann amp Cornell 1991) However it should be noted that the magnetite
synthesized by Schwertmann and Cornell (1991) was prepared differently (ie
oxidation of Fe(II) by nitrate in a heated alkaline solution) from the magnetite
applied in this study The surface area-normalised pseudo 1 order kobs values
obtained for GR-CO3 GR-SO4 Fe(II)goethite and Fe(II)lepidocrocite were all
within the same order of magnitude (Figure 53a)
Figure 53 Average surface area-normalised pseudo 1 order rate constants for the degradation of
a) TCA (this work) b) hexachloroethane (Elsner et al 2004a) and c) carbon tetrachloride
(Amonette et al 2000 Pecher et al 2002 OrsquoLoughlin et al 2003c Elsner et al 2004b) by
GR-SO4 GR-CO3 (suspension age 1 d) Fe3O4 Fe(II)α-FeOOH and Fe(II)γ-FeOOH
Experimental conditions applied in this work [Fe(II)tot]0 = 025-107 mM in the goethite and
lepidocrocite suspensions [Fe(II)tot]0 = 025-116 mM in the GR-SO4 and GR-CO3 suspensions
pH 70-86 71-926 m2 mineral surface areaL Experimental conditions applied by Elsner et al
1 mM aqueous Fe(II) 25 m2 mineral surface areaL Experimental conditions applied in
references employed in c) [Fe(II)tot]0 = 1-83 mM 25-275 m2 mineral surface areaL GR-SO4 =
110 Chapter 5
sulfate green rust GR-CO3 = carbonate green rust Fe3O4 = magnetite α-FeOOH = goethite γ-
FeOOH = lepidocrocite
When comparing the rate constants for the Fe(II)-Fe(III) mineral systems found for
reduction of TCA in this study (Figure 2a) mixed valent Fe(II)-Fe(III) minerals
such as green rusts and magnetite containing structural Fe(II) within the mineral
lattice do not seem to be significantly more reactive than Fe(II)-Fe(III) mineral
systems containing Fe(II) associated with pure Fe(III) oxides Unlike most other
iron oxides GRs contain not only external Fe(II) reactive sites at the surface but
also internal sites in the space between consecutive Fe(II)-Fe(III) hydroxide layers
The GR interlayer thickness is a function of both the size and the charge of the
interlayer anion For solutes the Fe(II) within the GR hydroxide layer is accessible
at the outside basal planes and at the edges as well as through the interlayer under
certain conditions (see Figure 42 Chapter 4 this work) Due to electrostatic
forces oxidants holding different charge properties (anions cations neutral
molecules) may exhibit different affinities for the various reactive Fe(II) sites
present in GR As the reactive sites are located inat the Fe(II)-Fe(III) hydroxide
layers the rate of reaction depends on the hydroxide layer area which can be
accessed by the oxidant If the oxidant is able to exchange with the interlayer
anion reaction can take place both at outer and inner surfaces of the GR particles
and in total more reactive sites are available for the reaction Thus oxidant size
and charge primarily control its access to the internal sites in GRs XRD
characterization of the GR-SO4 crystals during reaction with TCA demonstrated
that the GR-SO4 interlayer spacing did not vary during reaction (Table 52) This
may indicate that TCA did not enter the GR-SO4 interlayers
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 111 Table 52 Diffraction angle d-spacing and width at half peak height (Wfrac12) for the 001 GR-SO4
diffraction peak as a function of time during reaction with TCA ([Fe(II)GR]0 = 4 mM [TCA]0 = 1
mM)
Time (min) Angle (deg2θ)
d001-spacing(nm)
Wfrac12(degθ)
0 9483 10821 0273
10 9494 10809 0287
215 9522 10777 0263
330 9550 10745 0273
510 9524 10775 0277
855 9509 10791 0253
1160 9467 10839 0268
We roughly estimated the molecular size of TCA by summing the covalent radii of
the individual atoms (see Supporting Information 74) When comparing the
molecular size of TCA with the GR-SO4 interlayer spacing of 061 nm it can be
concluded that only when the C-C bond is oriented perpendicular to the interlayer
plane does the size of TCA exceed the GR-SO4 interlayer spacing In contrast the
size of TCA exceeds the GR-CO3 interlayer spacing (026 nm) regardless of its
orientation Hence if TCA was intercalated in the GR-CO3 interlayer we would
expect the interlayer spacing to expand The same holds for intercalation of a
vertically oriented TCA in the GR-SO4 interlayer Supposedly both the low charge
and the size of TCA impeded its access to the GR-SO4 and GR-CO3 interlayers ie
the divalent sulphate and carbonate in the GR interlayers did not readily exchange
with the monovalent TCA since GR interlayers generally have a higher affinity for
divalent anions than for monovalent anions (Miyata 1983) Thus TCA did neither
access nor react with internal Fe(II) reactive sites in GR-SO4 which means that the
reaction between TCA and GR-SO4 took place at the external reactive Fe(II) sites
solely It is reasonable to assume that the same holds for the reaction between TCA
and GR-CO3 No significant aging effects eg rate constants varying as a function
of GR age were observed within 142 days (see Table 51) However the SSAs of
112 Chapter 5
the GR suspensions holding ages up to 142 days were not measured but estimated
assuming that the GR SSA did not decrease within the time frame
According to the Scherrer formula the width at half peak height (Wfrac12) of a
diffraction peak is inversely proportional to the average crystal dimension
perpendicular to the given crystal plane (Klug amp Alexander 1974) The average
GR-SO4 particle thickness perpendicular to the basal plane (Wfrac12 Table 52) was
constant during reaction implying that TCA reacts only at the edges and not at the
basal planes Assuming that the platy hexagonal GR-SO4 and GR-CO3 crystals
hold an average width of 1 microm and an average particle thickness of 35 nm (Hansen
and Koch 1998) the ratio of edge surface area to outer surface area is AedgeAouter ~
130 for GR-SO4 and 121 for GR-CO3 (see Supporting Information 73) This
means that only 3 of the outer surface area in GR-SO4 and 5 of the outer
surface area in GR-CO3 are available at the edges Assuming that TCA reacts at the
edges only and if employing the AedgeAouter in the estimation of the rate constants
the surface area-normalised pseudo 1 order rate constants for GR-SO4 and GR-
CO3 would be 20-30 times higher than the rate constants depicted in Figure 53a
Thus the reactivity of GRs normalised to their reactive surface area is much higher
than the reactivity normalised to their total surface area
533 Comparing with rate constants obtained for other chlorinated aliphatic
compounds
Though care must be taken when comparing kinetic parameters obtained at
different experimental conditions (eg pH [Fe(II)]0[TCA]0 ratios surface area to
volume ratios etc) it is interesting to compare our results to those reported for
hexachloroethane (Figure 53b data from Elsner et al 2004a) The reductive
transformation of hexachloroethane was investigated for various Fe(II)-bearing
minerals including Fe(II)goethite Fe(II)lepidocrocite and GR-SO4 in the presence
of 1 mM dissolved Fe(II) and 25 m2 mineral surface areaL at pH 72 except for the
GR-SO4 suspensions in which the dissolved Fe(II) concentrations were slightly
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 113
higher and pH = 8 The pseudo 1 order rate constants reported for
hexachloroethane are in the range 18middot10-4 ndash 75middot10-3 h-1middotm-2middotL (Elsner et al 2004a)
When comparing Figure 53a with Figure 53b it can be seen that the differences
in intrinsic reactivity of the Fe(II)-bearing mineral systems are more pronounced
for hexachloroethane than for TCA
Caution should also be advised to the different reaction mechanisms by which
hexachloroethane and TCA react The transfer of a single electron and the
formation of an alkyl radical upon removal of a chlorine atom constitute the first
and in most cases the rate-limiting step in the reduction of chlorinated aliphatic
compounds (Vogel et al 1987) Depending on the chemical structure of the
chlorinated aliphatic compound the resulting free alkyl radical may undergo
hydrogenolysis chloroelimination or dimerizationcoupling In the case of TCA
the free dichloroacetate radical most likely undergoes hydrogenolysis The almost
quantitative transformation of TCA to DCA confirms that hydrogenolysis is the
prevalent reaction mechanism in our mineral systems The pentachloroethyl radical
formed from hexachloroethane may undergo hydrogenolysis (producing
pentachloroethane) or dichloroelimination (producing tetrachloroethene) Elsner et
al (2004a) found that hexachloroethane was transformed quantitatively into
tetrachloroethylene for all minerals which strongly indicates that
dichloroelimination was the dominating reaction mechanism Another
polychlorinated aliphatic compound transformed mainly by hydrogenolysis under
reducing conditions is carbon tetrachloride Several studies have investigated the
reductive dechlorination of carbon tetrachloride by various Fe(II)-bearing minerals
including Fe(II)goethite and GR-SO4 and reported pseudo 1 order rate constants
in the order 152middot10-4 ndash 640middot10-4 h-1middotm-2middotL for Fe(II)goethite and 864middot10-4 h-1middotm-2middotL
for GR-SO4 (Amonette et al 2000 Pecher et al 2002 OLoughlin et al 2003c
Elsner et al 2004b) When comparing Figure 53a with Figure 53c it can be seen
that the range of magnitude of the rate constants and the differences in intrinsic
114 Chapter 5
reactivity of the Fe(II)-bearing mineral systems are similar for carbon tetrachloride
and TCA
534 Factors controlling the reactivity of surface-bound Fe(II)
The reactivity of an oxidant towards Fe(II) surface species cannot be predicted
from the reduction potentials of the redox couple alone In heterogeneous systems
processes such as mass transfer and adsorptiondesorption may have a rate-limiting
effect on the overall reaction rate If the adsorption follows a saturation-type
sorption isotherm (eg Langmuir) the sorbate (oxidant) concentration at the
surface will vary non-linearly with the total amount of oxidant added This
dependence will have to be taken into account when establishing rate laws for the
heterogeneous reactions and when testing the hypothesis that the reaction rates
depend on the sorbed concentration of the oxidants pH has a strong impact on the
sorption and thereby on the availability of ionizable oxidants At the pH values
applied here the chloroacetates are fully dissociated (pKa (TCA) = 066 pKa
(DCA) = 135 pKa (MCA) = 287) However we found the sorption of TCA to be
negligible in suspensions of pure calcite goethitecalcite and lepidocrocitecalcite
at pH 76-77 Moreover the mass balance of TCA and DCA was almost complete
in all suspensions and therefore loss of TCA or DCA due to adsorption at mineral
surfaces or incorporation in the GR anion interlayers can be ruled out Calcite has a
much lower adsorption capacity than most iron oxides hence we anticipate that
goethite and lepidocrocite control the adsorption of TCA and DCA in both the pure
FeOOH and the FeOOHcalcite suspensions This was supported by our
experimental results demonstrating that the presence of a calcite surface - either
pure or as a support for goethite and lepidocrocite coatings - did not exert any
noticeable effect on the reaction rates (see Table 51) In addition the surface area-
normalised rate constants for mineral systems containing goethite or lepidocrocite
in pure form and mineral systems containing goethite or lepidocrocite as coatings
on calcite were very similar In heterogeneous reactions mass transfer in bulk
solution becomes the rate-limiting step when the surface reaction is much faster
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 115
than the diffusion of the reacting species to the reactive surface However at the
low rate constants obtained here the reaction of TCA with the Fe(II)-bearing
minerals is not likely to be mass transfer limited (see Supporting Information 72)
One very important factor affecting heterogeneous redox reactions is pH which
influences the speciation of the complexes in solution and at mineral surfaces as
well as the stability of the more soluble Fe(II)-containing minerals such as GRs In
contrast to aqueous Fe(II) complexes it is not possible to predict the reactivity of
Fe(II) surface species as their reduction potentials are unknown In the absence of
specifically adsorbing solutes other than H+ the surface charge of the Fe(III)
oxides goethite and lepidocrocite is determined by the surface densities of the
charged surface species equivFeOH2+ and equivFeOndash whereas the surface charge of calcite
is determined by the density of the surface species equivCO3ndash equivCaOH2
+ and equivCaOndash
(Stumm 1992 Van Cappellen et al 1993) The point of zero charge (pHpzc) of
pure calcite is in the pH range 7-11 and depends on the partial pressure of carbon
dioxide pCO2 The higher the pCO2 the lower the pHpzc At the experimental
conditions applied here (05 = 0005 atm CO2(g)) the pHpzc = 82 for calcite
(Table 53) As only 10-11 mg Fe(III) of goethite and lepidocrocite was coated
onto calcite we assumed a pHpzc of 82 for the goethite and lepidocrocite coated
calcite particles as well The pHpzc values for green rusts are unknown
116 Chapter 5
Table 53 Specific surface areas and point of zero charge of the various iron minerals in pure form as well as goethite and lepidocrocite coated onto calcite
Mineral Structural formula SSA (m2g) pHpzc
GR-SO4 FeII4FeIII
2(OH)12SO4middot3H2O 71 a -
GR-CO3 FeII4FeIII
2(OH)12CO3middot3H2O 47 b -
Magnetite Fe3O4 - 69 e
Goethite α-FeOOH 16 c 85 f
Lepidocrocite γ-FeOOH 18 c 73 e
Calcite CaCO3 le 1 d 82 g
Goe coating - le 1 d 82 h
Lep coating - le 1 d 82 h
a Chapter 4 this work b Williams amp Scherer 2001 c Product information by Bayer d The SSA of calcite was
quantified to le 1 m2g The detection limit of our BET method was 1 m2g e Charlet et al 1998a f Liger et al
1999 g Van Cappellen et al 1993 h Same as for calcite
The surface hydroxyl groups on iron oxides may be both singly (equivFe-OH) doubly
(equivFe2-OH) triply (equivFe3-OH) and geminally (equivFe-(OH)2) coordinated (Cornell amp
Schwertmann 1996 Stumm 1992) The differently coordinated surface hydroxyl
groups are not equally reactive Adsorption reactions involve only singly
coordinated surface groups and therefore only this kind of hydroxyl groups on iron
oxides will be considered here (Cornell amp Schwertmann 1996) Hence the
predominant surface sites available for adsorption in pure suspensions of Fe(III)
oxides are equivFeOH0 equivFeOH2+ and equivFeOndash In the presence of dissolved Fe(II)
equivFeIIIOFeIIOH0 equivFeIIIOFeIIOndash and equivFeIIIOFeII+ constitute the main reactive sites at
the Fe(III) oxide surfaces (Liger et a 1999) Assuming that Fe2+ and other cationic
Fe(II) species are the dominating adsorbates on the mineral surfaces in our
experiments we expect the actual pHpzc to be higher than the pHpzc of the pure
oxides listed in Table 53 Hence at pHlt82 where most of our experiments were
conducted all the mineral surfaces presumably carry net positive charges
At pH 70 where Fe2+ is still the predominant Fe(II) species in solution (~50) we
expect that equivFeIIIOFeIIOH equivFeIIIOFeIIOndash and equivFeIIIOFeII+ constitute the main
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 117
reactive sites at the Fe(III) oxide surfaces as suggested by Liger et a 1999 As pH
increases from 70 to 87 the Fe(II) carbonate complexes become increasingly
important in solution at the expense of the Fe2+ FeCl+ FeSO40 and FeOH+ species
(King 1998) Fe(II) carbonate complexes do not bind at the oxide surface as
readily as the aquo or hydroxo complexes of Fe(II) but carbonate itself sorbs
readily to Fe(III) oxide surfaces through which the Fe(III) oxide surface is coated
by inner-sphere monodentate equivFeIIIOCOOH0 surface complexes (Villalobos amp
Leckie 2000 amp 2001) The presence of carbonate shifted the sorption edge for the
Fe(II) adsorption on goethite from pH 58 to 78 and the authors hypothesized this
to be a result of the formation of aqueous and surface Fe(II)-carbonate complexes
and to competition between carbonate and Fe(II) for Fe(III) oxide surface sites
(Vikesland amp Valentine 2002) Similarly monodentate surface complexes like
equivFeIIICl0 and equivFeIIIOSO3ndash as well as ternary monodentate surface complexes like
equivFeIIIOFeIICl0 and equivFeIIIOFeIIOSO3ndash and ternary bidentate surface complexes such
as (equivFeIIIO)2FeIIOSO3 may form at Fe(III) oxide surfaces when Fe(II) chloride and
sulfate are present in solution (Ostergren et al 2000 Kim et al 2004) However
the effects of anionic ligands such as chloride and sulfate on Fe(II) adsorption at
Fe(III) oxide surfaces and the reactivity of Fe(II) carbonate chloride and sulfate
surface sites are still unknown and need to be evaluated (see Supporting
Information 75) Thus we do not know whether chloride and sulfate decrease or
increase the Fe(II) sorption in our mineral systems We can only report that we did
not detect any significant differences in the rate of TCA transformation between
the mineral suspensions containing carbonate chloride and sulfate respectively
Hence we anticipate that equivFeIIIOFeIIOH equivFeIIIOFeIIOndash and equivFeIIIOFeII+ constitute
the main reactive sites at the Fe(III) oxide surfaces within the whole pH range 70-
87 This might also explain why we did not detect any obvious systematic pH
effect in the Fe(II)-Fe(III)-systems (see Table 51) In the case of
hexachloroethane the reactivity order GR-SO4gtgoethitegtmagnetitegtlepidocrocite
may be rationalized by the variations in surface site densities and total amount of
118 Chapter 5
Fe(II) sorbed on the iron minerals (see Supporting Information 75) as well as the
different speciations and reactivities of the Fe(II) surface sites on the iron minerals
535 Comparison with biotic and other abiotic systems
Only one report on abiotic transformation of TCA is found in the literature and the
study demonstrates the reductive dechlorination of TCA to MCA by Fe(0)
(Hozalski et al 2001) The authors reported a pseudo 1 order rate constant of
60middot10-4 min-1middotm-2middotL for the transformation of TCA to DCA and a pseudo 1 order
rate constant of 225middot10-4 min-1middotm-2middotL for the transformation of DCA to MCA at
[Fe(0)]0 = 025 M [TCA]0 = 100-200 microM and pH 36-62 The rate constant for
TCA reduction by Fe(0) is 10-300 times faster than the rate constants reported for
the Fe(II)-bearing mineral systems here
There is abundant evidence that soil microorganisms and fungi can dechlorinate
TCA but only little is known about the bacteria and processes involved in the
biodegradation of TCA Biotransformation of TCA has been found at both oxic
and anoxic conditions Most of the microorganisms isolated grow feebly on TCA
as a sole source of carbon (Foy 1975 Weightman et al 1992 De Wever et al
2000) Only one bacterium capable of growing on TCA as the sole carbon and
energy source has been characterized (Yu amp Welander 1995) In addition
anaerobic bacteria coupling co-metabolic growth to reductive dechlorination of
TCA have been isolated (Weightman et al 1992 De Wever et al 2000) The
inability to grow on the less chlorinated acids DCA and MCA is a notable feature
of both the aerobic and anaerobic bacteria Complete transformation of TCA to
methane and carbon dioxide has only been found when abiotic and biotic processes
were combined (Egli et al 1989) The abiotic transformation of TCA to DCA
occurred spontaneously in the presence of sterile activated charcoal whereas the
DCA formed was further degraded to methane and carbon dioxide by a mixed
culture of methanogenic bacteria However the abiotic reductant(s) responsible for
the transformation of TCA to DCA was not reported (Egli et al 1989)
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 119
The rate constants obtained in this work suggest that the Fe(II)-bearing mineral
systems may be important reductants of TCA in natural suboxic environments In
natural iron-rich soils holding specific surface areas of 22 m2g (Kenneke amp
Weber 2003) average bulk densities of 265 gcm3 and porosities of 25 and
containing 2 iron oxides a rough estimation of the half-life of TCA amounts to
47 minutes when applying the average surface area-normalised rate constant
obtained for all the Fe(II)-Fe(III) mineral systems in this work (1middot10-3 h-1middotm-2middotL)
This estimation is based on the assumption that enough reactive Fe(II) is available
in these soils The natural iron-reducing sediment investigated by Kenneke and
Weber (2003) contained 80 microM Fe(II) in the soil solution and 315 micromole Fe(II) per
g sediment At such low Fe(II) concentrations the overall rate of abiotic
transformation of TCA in natural soils and sediments is most likely limited by the
regeneration of reactive Fe(II) Hence the continuous regeneration of reactive
Fe(II) surface sites by adsorption of abiotically or microbially produced Fe(II) may
further the long-term abiotic transformation of TCA in such environments
54 Conclusions
This work demonstrates that various Fe(II)-Fe(III) minerals systems including GR-
SO4 GR-CO3 magnetite Fe(II)goethite and Fe(II)lepidocrocite readily transform
TCA to DCA Dichloroacetate was not further reduced to MCA or acetate by any
of the Fe(II)-bearing minerals The surface area-normalised pseudo 1 order rate
constants obtained for the reductive transformation of TCA by the various Fe(II)-
bearing minerals did not differ significantly from each other The results obtained
in this work infer that under natural geochemical conditions Fe(II)-bearing mineral
systems may play an important role in the overall transformation of TCA Thus
not only microbial degradation but also abiotic reductive transformation of TCA by
Fe(II)-bearing minerals may govern the fate of TCA in natural subsurface
environments This holds both for natural systems like iron-rich suboxic soils and
sediments as well as engineered systems like permeable reactive barriers of zero-
120 Chapter 5
valent iron implemented for on-site remediation where both Fe(0) and solid or
surface-bound Fe(II) corrosion intermediates may transform TCA
Acknowledgments
We would like to thank Susanne Guldberg for performing the experimental work comprising
GR-SO4
References
Ahlers J Regelmann J Riedhammer C (2003) Environmental risk assessment of airborne trichloroacetic acid - a contribution to the discussion of the significance of anthropogenic and natural sources Chemosphere 52 531-537 Amonette JE Workman DJ Kennedy DW Fruchter JS Gorby YA (2000) Dechlorination of carbon tetrachloride by Fe(II) associated with goethite Environmental Science and Technology 34 4606-4613 Atkins PJ Gold V Marsh R (1984) The decarboxylation of trichloroacetic acid and the reactions of the trichloromethyl anion with 135-trinitrobenzene and with hydrogen ions kinetic measurements in dimethyl sulphoxide solution Journal of the Chemical Society Perkin Transactions 2 7 1239-1245 Berg M Muumlller SR Muumlhlemann J Wiedmer A Schwarzenbach RP (2000) Concentrations and mass fluxes of chloroacetic acids and trifluoroacetic acid in rain and natural waters in Switzerland Environmental Science and Technology 34 2675-2683 Brunauer S Emmett PH Teller E (1938) Adsorption of gases in multimolecular layers Journal of American Chemical Society 60 309-319 Butler EC Hayes KF (1998) Effects of solution composition and pH on the reductive dechlorination of hexachloroethane by iron sulfide Environmental Science and Technology 32 1276-1284 Butler EC Hayes KF (1999) Kinetics of the transformation of trichloroethylene and tetrachloroethylene by iron sulfide Environmental Science and Technology 33 2021-2027 Charlet L Liger E Gerasimo P (1998a) Decontamination of TCE- and U-rich water by granular iron Role of sorbed Fe(II) Journal of Environmental Engineering 124 25-30 Charlet L Silvester E Liger E (1998b) N-compound reduction and actinide immobilisation in surficial fluids by Fe(II) the surface FeIIIFeIIOH0 species as major reductant Chemical Geology 151 85-93 Cheregi M Danet AF (1997) Flow injection determination of chloride ions with spectrophotometric detection Analytical Letters 30 2847-2858 Cornell RM Schwertmann U (1996) The iron oxides Structure properties reactions occurrence and uses VCH Verlagsgesellschaft mbH Weinheim
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 121 Cui D Eriksen TE (1996) Reduction of pertechnetate by ferrous iron in solution influence of sorbed and precipitated Fe(II) Environmental Science and Technology 30 2259-2262
Egli C Thuumler M Suter D Cook AM Leisinger T (1989) Monochloro- and dichloroacetic acids as carbon and energy sources for a stable methanogenic mixed culture Archives of Microbiology 152 218-223 Elsner M Haderlein SB Schwarzenbach RP (2004a) Reactivity of Fe(II)-bearing minerals towards reductive transformation of organic contaminants Environmental Science and Technology 38 799-807 Elsner M Haderlein SB Kellerhals T Luzi S Zwank L Angst W Schwarzenbach RP (2004b) Mechanisms and products of surface-mediated reductive dehalogenation of carbon tetrachloride by Fe(II) on goethite Environmental Science and Technology 38 2058-2066 Erbs M Hansen HCB Olsen CE (1999) Reductive dechlorination of carbon tetrachloride using iron(II)iron(III)-hydroxide-sulphate (green rust) Environmental Science and Technology 33 307-311 Euro Chlor (2001) Trichloroacetic acid in the environment a dossier Euro Chlor Brussels and the European Chlorinated Solvent Association Fadrus H Maly J (1975) Suppression of iron(III) interference in the determination of iron(II) in water by the 110-phenanthroline method The Analyst 100 549-554 Fahimi IJ Keppler F Schoumller HF (2003) Formation of chloroacetic acids from soil humic acid and phenolic moieties Chemosphere 52 513-520 Foy CL (1975) The chlorinated aliphatic acids In Herbicides Chemistry degradation and mode of action Kearney PC Kaufman DD (eds) Marcel Dekker Inc 399-452 Gander JW Parkin GF Scherer MM (2002) Kinetics of 111-trichloroethane transformation by iron sulfide and a methanogenic consortium Environmental Science and Technology 36 4540-4546 Haiber G Jacob G Niedan V Nkusi G Schoumller HF (1996) The occurrence of trichloroacetic acid (TCAA) ndash indications of a natural production Chemosphere 33 839-849
Hansen HCB (1989) Composition stabilization and light absorption of Fe(II)Fe(III) hydroxycarbonate (green rust) Clay Minerals 24 663-669 Hansen HCB Guldberg S Erbs M Koch CB (2001) Kinetics of nitrate reduction by green rusts ndash effects of interlayer anion and Fe(II)Fe(III) ratio Applied Clay Science 18 81-91 Hansen HCB Koch CB (1997) A comparison of nitrate reduction by carbonate and sulphate forms of green rust Kodama H Mermut A R Torrance J K (eds) Proceedings of the 11th International Clay Conference Ottawa Canada Clays for our future 11 295-302 Hoekstra EJ de Leer EWB Brinkman UATh (1999a) Mass balance of trichloroacetic acid in the soil top layer Chemosphere 38 551-563 Hoekstra EJ de Leer EWB Brinkman UATh (1999b) Findings supporting the natural
122 Chapter 5
formation of trichloroacetic acid in soil Chemosphere 38 2875-2883 Hofstetter TB Heijman CG Haderlein SB Holliger HC Schwarzenbach RP (1999) Complete reduction of TNT and other (poly)nitroaromatic compounds under iron-reducing subsurface conditions Environmental Science and Technology 33 1479-1487 Hofstetter TB Schwarzenbach RP Haderlein SB (2003) Reactivity of Fe(II) species associated with clay minerals Environmental Science and Technology 37 519-528 Hozalski RM Zhang L Arnold WA (2001) Reduction of haloacetic acids by Fe0 Implications for treatment and fate Environmental Science and Technology 35 2258-2263 Husain S Narsimha R Alvi SN Rao RN (1992) Monitoring the effluents of the trichloroacetic acid process by high-performance liquid chromatography Journal of Chromatography 600 316-319 Hwang I Batchelor B (2000) Reductive dechlorination of tetrachloroethylene by Fe(II) in cement slurries Environmental Science and Technology 34 5017-5022 Jordan A Frank H (1999) Trifluoroacetate in the environment Evidence for sources other than HFCHCFCs Environmental Science and Technology 33 522-527 Kenneke JF Weber EJ (2003) Reductive dehalogenation of halomethanes in iron- and sulfate-reducing sediments 1 reactivity pattern analysis Environmental Science and Technology 37 713-720 Kim CS Rytuba JJ Brown Jr GE (2004) EXAFS study of mercury(II) sorption to Fe- and Al-(hydr)oxides II Effects of chloride and sulphate Journal of Colloid and Interface Science 270 9-20 King DW (1998) Role of carbonate speciation on the oxidation rate of Fe(II) in aquatic systems Environmental Science and Technology 32 2997-3003 Klausen J Troumlber SP Haderlein SB Schwarzenbach RP (1995) Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions Environmental Science and Technology 29 2396-2404 Klug PH Alexander LE (1974) X-ray diffraction procedures John Wiley amp Sons Inc Koch CB Hansen HCB (1997) Reduction of nitrate to ammonium by sulphate green rust Advances in GeoEcology 30 373-393 Kuumlhn R Pattard M (1990) Results of the harmful effects of water pollutants to green algae (Scenedesmus subspicatus) in the cell multiplication inhibition test Water Research 24 31-38 Lee W Batchelor B (2002a) Abiotic reductive dechlorination of chlorinated ethylenes by iron- bearing soil minerals 1 Pyrite and magnetite Environmental Science and Technology 36 5147- 5154 Lee W Batchelor B (2002b) Abiotic reductive dechlorination of chlorinated ethylenes by iron-bearing soil minerals 2 Green rust Environmental Science and Technology 36 5348- 5354
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 123 Liger E Charlet L Van Cappellen P (1999) Surface catalysis of uranium (VI) reduction by iron(II) Geochimica et Cosmochimica Acta 63 2939-2955 Lignell R Heinonen-Tanski H Uusi-Rauva A (1984) Degradation of trichloroacetic acid (TCA) in soil Acta Agriculturae Scandinavia 34 3-8 Loyaux-Lawniczak S Refait Ph Lecomte P Ehrhardt J Geacutenin JMR (1999) The reduction of chromate ions by Fe(II) layered hydroxides Hydrology and Earth System Sciences 3 593-599 Luther III GW (1990) The Frontier-Molecular-Orbital theory approach in geochemical processes in W Stumm Ed Aquatic Chemical kinetics John Wiley and Sons New York pp 173-198 McCulloch A (2002) Trichloroacetic acid in the environment Chemosphere 47 667-686 Miyata S (1983) Anion-exchange properties of hydrotalcite-like compounds Clays and Clay Minerals 31 305-311 Muumlller SR Zweifel H-R Kinnison DJ Jacobsen JA Meier MA Ulrich MM Schwarzenbach RP (1996) Occurrence sources and fate of trichloroacetic acid in Swiss lakes Environmental Toxicology and Chemistry 15 1470-1478 OLoughlin EJ Burris DR (2003) Reduction of halogenated ethanes by green rust Environmental Toxicology and Chemistry 23 41-48 OLoughlin EJ Kelly SD Cook RE Csencsits R Kemner KM (2003a) Reduction of uranium(VI) by mixed iron(II)iron(III) hydroxide (green rust) Formation of UO2 nanoparticles Environmental Science and Technology 37 721-727 OLoughlin EJ Kelly SD Kemner KM Csencsits R Cook RE (2003b) Reduction of AgI AuIII CuII and HgII by FeIIFeIII hydroxysulfate green rust Chemosphere 53 437-446 OLoughlin EJ Kemner KM Burris DR (2003c) Effects of AgI AuIII and CuII on the reductive dechlorination of carbon tetrachloride by green rust Environmental Science and Technology 37 2905-2912 Ostergren JD Brown Jr GE Parks GA Persson P (2000) Inorganic ligand effects on Pb(II) sorption to goethite (α-FeOOH) II Sulfate Journal of Colloid and Interface Science 225 483-493 Pecher K Haderlein SB Schwarzenbach RP (2002) Reduction of polyhalogenated methanes by surface-bound Fe(II) in aqueous suspensions of iron oxides Environmental Science and Technology 36 1734-1741 Reimann S Grob K Frank H (1996) Chloroacetic acids in rainwater Environmental Science and Technology 30 2340-2344 Ruumlgge K Hofstetter TB Haderlein SB Bjerg PL Knudsen S Zraurig C Mosbaeligk H Christensen TH (1998) Characterization of predominant reductants in an anaerobic leachate- affected aquifer by nitroaromatic probe compounds Environmental Science and Technology 32
124 Chapter 5
23-31 Sarzanini C Bruzzoniti MC Mentasti E (1999) Preconcentration and separation of haloacetic acids by ion chromatography Journal of Chromatography A850 197-211 Schwertmann U Cornell RM (1991) Iron oxides in the laboratory Preparation and characterization VCH Verlagsgesellschaft mbH Weinheim Scott BF Mactavish DC Spencer C Strachan WMJ Muir DCG (2000) Haloacetic acids in Canadian lake waters and precipitation Environmental Science and Technology 34 4266-4272 Scott BF Spencer C Marvin CH Mactavish DC Muir DCG (2002) Distribution of haloacetic acids in the water columns of the Laurentian Great Lakes and Lake Malawi Environmental Science and Technology 36 1893-1898 Strathmann TJ Stone AT (2003) Mineral surface catalysis of reactions between FeII and oxime carbamate pesticides Geochimica et Cosmochimica Acta 67 2775-2791 Stumm W (1992) Chemistry of the solid-water interface John Wiley amp Sons Inc Van Cappellen P Charlet L Stumm W Wersin P (1993) A surface complexation model of the carbonate mineral-aqueous solution interface Geochimica et Cosmochimica Acta 57 3505- 3518 Vikesland PJ Valentine RL (2002) Iron oxide surface-catalyzed oxidation of ferrous iron by monochloramine implications of oxide type and carbonate on reactivity Environmental Science and Technology 36 512-519 Villalobos M Leckie JO (2000) Carbonate adsorption on goethite under closed and open CO2 conditions Geochimica et Cosmochimica Acta 64 3787-3802 Villalobos M Leckie JO (2001) Surface complexation modeling and FTIR study of carbonate adsorption to goethite Journal of Colloid and Interface Science 235 15-32 Vogel TM Criddle CS McCarty PL (1987) Transformations of halogenated aliphatic compounds Environmental Science and Technology 21 722-736 Weightman AL Weightman AJ Slater JH (1992) Microbial dehalogenation of trichloroacetic acid World Journal of Microbiology and Biotechnology 8 512-518 De Wever H Cole JR Fettig MR Hogan DA Tiedje JM (2000) Reductive dehalogenation of trichloroacetic acid by Trichlorobacter thiogenes gen nov spnov Applied and Environmental Microbiology 66 2297-2301 Williams AGB Scherer MM (2001) Kinetics of chromate reduction by carbonate green rust Environmental Science and Technology 35 3488-3494 Yu P Welander T (1995) Growth of an aerobic bacterium with trichloroacetic acid as the sole source of energy and carbon Applied Microbiology and Biotechnology 42 769-774
Conclusions and Outlook 125
6 Conclusions and Outlook The work presented in this dissertation adds to the understanding of how Fe(II)-
bearing minerals like green rusts (GRs) vivianite (Fe2(PO4)2sdot8H2O) magnetite
(Fe3O4) and Fe(II) associated with goethite and lepidocrocite may form and react in
nature In order to elucidate the role of bacteria in the formation of GRs in natural
soils and sediments we studied the iron mineral phases forming as a result of the
activity of iron-respiring bacteria In chapter 2 the Fe-containing products formed
by anaerobic autotrophic denitrifying Fe(II)-oxidizing bacteria (FeOB) were
examined The culture medium applied contained high levels of bicarbonate and
phosphate and is typically used in this kind of studies as it provides excellent
conditions for the nitrate-reducing FeOB Fe(II) was present initially as a whitish
solid Fe(II) hydroxy phosphate (vivianite) and as soluble Fe(II) The results
obtained demonstrate that the denitrifying FeOB produce poorly crystalline
goethite via a greenish Fe(III)-enriched vivianite intermediate Moumlssbauer
spectroscopic analyses provided no evidence of green rust formation At low
phosphate concentrations where vivianite does not control the Fe(II) activity it is
reasonable to assume that siderite (FeCO3) precipitates initially and that carbonate
GR phases may form during biooxidation At low bicarbonate concentrations we
would expect Fe(II) sulfate or chloride species to dominate initially (depending on
the Fe(II) source applied) and sulfate GR or chloride GR to form during
biooxidation In chapter 3 we investigated the Fe-containing products formed
during reduction of common Fe(III) oxides by the anaerobic dissimilatory Fe(III)-
reducing microorganism Shewanella algae BrY S algae BrY reduced substantial
amounts of the initial Fe(III) and green and blackish mineral phases were produced
within 1-2 weeks after inoculation Moumlssbauer spectroscopic analyses showed that
the green and black precipitates consisted of green rust and vivianite
We studied the reactivity of synthetic GRs towards reducible organic pollutants in
order to asses the potential significance of GR phases for the fate of such
126 Chapter 6
compounds To this end we used nitroaromatic compounds (NACs) and
chlorinated acetates as suitable model compounds for studying redox reactions
potentially relevant in the environment In chapter 4 we investigated the relative
reactivity of outer and inner Fe(II) reactive sites in synthetic sulfate green rust
(GR-SO4) by using a series of structurally closely related compounds with different
charge properties as ldquoreactive probesrdquo The probe compounds included
nitrobenzene 2-nitrophenol 4-nitrotoluene 4-chloronitrobenzene and 4-
nitrophenylacetic acid Our results demonstrated that NACs are completely
reduced to their corresponding anilines by GR-SO4 The reactions followed pseudo
1 order kinetics with respect to NAC and the surface area-normalised pseudo 1
order rate constants obtained were 016ndash465middot10-4 s-1middotm-2middotL at [Fe(II)GR]0 = 103-
1260 mM [NAC]0 = 20-102 microM and pH 84-86 Neither mass transfer control nor
surface saturation kinetics could account for the similarity of the surface-
normalised pseudo 1 order rate constants obtained for the reduction of the neutral
and anionic NACs by GR-SO4 These observations suggest that the reaction
between NAC and GR-SO4 takes place at the external reactive Fe(II) sites At low
initial Fe(II)GR concentrations the external reactive Fe(II) sites were depleted and
the regeneration of new external reactive sites eventually controlled the reduction
of the NACs by GR-SO4 In chapter 5 we examined the reactivity of various
Fe(II)-Fe(III) mineral systems towards trichloroacetic acid (TCA) and
dichloroacetate (DCA) in laboratory batch experiments imitating natural
conditions The Fe(II)-Fe(III)-systems investigated included GR-SO4 carbonate
green rust magnetite Fe(II)goethite and Fe(II)lepidocrocite TCA was readily
reduced to DCA by all Fe(II)-containing minerals The reactions followed pseudo
1 order kinetics with respect to TCA and the surface area-normalised pseudo 1
order rate constants obtained were 033ndash76middot10-5 min-1middotm-2middotL at [Fe(II)]0 = 025ndash
116 mM [TCA]0 = 15ndash1000 microM and pH 70ndash87 Our results showed no
significant differences regarding product distribution and surface area-normalised
reaction rate constants between the Fe(II)-Fe(III)-systems DCA was not further
Conclusions and Outlook 127
reduced to monochloroacetate (MCA) or acetate in any of the systems within the
time frame in our experiments
As suggested in chapters 2 and 3 sufficient evidence must be provided and caution
should be exercised when proclaiming new biogenic minerals The study of
microbially produced GRs is still in its infancy and more research is needed in
order to elucidate the role of bacteria in the formation of GRs in natural soils and
sediments The results presented in chapter 2 indicate that microbiological
processes may be responsible for the oxidation of vivianite and metavivianite
((FeII3-xFeIII
x)(PO4)2(OH)xmiddot(8-x)H2O x gt 12) in natural subsurface environments
In chapter 3 we demonstrated that GRs may be produced microbially at conditions
including low carbon and Fe(III) concentrations as well as the exclusion of
synthetic electron shuttles and pH buffers The role of microbial processes in the
redox cycling of iron in the subsurface and the ways in which these processes can
be coupled to contaminant remediation are currently active areas of research Zero-
valent iron has been the most extensively studied reductant for the treatment of
many inorganic and organic contaminants and is currently the most commonly
used material for the construction of permeable reactive barriers (PRB) but a
detailed understanding of the processes involved in the reduction of these
pollutants by Fe(0) is lacking (Scherer et al 2000) Potentially reactive Fe(II)-
bearing corrosion products identified in iron metal columns and barriers include
magnetite siderite Fe(II) sulfides green rusts as well as Fe(II) sorbed to mineral
surfaces (Gu et al 1999 Roh et al 2000) The formation of reactive Fe(II)-
bearing minerals like GRs may explain the effective long-term operation of zero-
valent iron PRBs despite the formation of thick oxide films Thus natural in situ
PRBs might be created by stimulating the activity of anaerobic dissimilatory
Fe(III)-reducing bacteria and the subsequent formation of Fe(II) species such as
GRs Furthermore suspensions of synthetic GRs which are easily prepared from
relatively inexpensive commodity chemicals may also be injected and dispersed
into the subsurface
128 Chapter 6
The reductive transformation of NACs and TCA by GRs is relevant to
understanding the processes responsible for their degradation in the subsurface and
the development of innovative technologies for their remediation The results
obtained in chapters 4 and 5 indicate that GRs may play a significant role in the
reductive transformation of NACs and TCA in natural subsurface environments
Furthermore our results suggest that mainly the outer Fe(II) sites in GRs are
utilized in the reaction with neutral and monovalent anionic compounds and that
these sites may be replenished eg by reduction of the oxidized surface sites or
adsorption of Fe(II) from solution The continuous restoration of Fe(II) surface
sites in GRs may promote their long-term reactivity towards reducible
contaminants
References
Gu B Phelps TJ Liang L Dickey MJ Roh Y Kinsall BL Palumbo AV Jacobs GK (1999) Biochemical dynamics in zero-valent iron columns Implications for permeable reactive barriers Environmental Science and Technology 33 2170-2177 Roh Y Lee SY Elless MP (2000) Characterization of corrosion products in the permeable reactive barriers Environmental Geology 40 184-194 Scherer MM Richter S Valentine RL Alvarez PJJ (2000) Chemistry and microbiology of permeable reactive barriers for In Situ groundwater clean up Critical Reviews in Environmental Science and Technology 30 363-411
Supporting Information I
7 Supporting Information
71 Estimation of the one-electron reduction potential for 4-NPA
The one-electron reduction potential of the half-reaction for a given NAC 1hE
ArNO2 + e- ArNO2
can be used for comparing reduction rates of different NACs in a given system
The formation of the nitroaryl radical is the rate-determining step in the overall rate
of the reduction of a NAC to the corresponding aniline The difference between the
of a NAC and a given reductant is proportional to the change in standard free
energy for the transfer of the first electron ∆G
1hE
1degrsquo If a linear relationship between
the free energy of activation and ∆G1degrsquo is assumed the values of various NACs
can be a measure of their relative reactivity with a given reductant
1hE
As neither the one-electron reduction potential for 4-nitrophenylacetic acid (4-
NPA) nor the Hammett constant for the acetic acid substituent could be found in
the literature the one-electron reduction potential for 4-NPA was estimated by
application of a linear free energy relationship (LFER) to experimental data
Kinetic experiments were conducted in order to obtain the pseudo 1 order rate
constant for the reduction of 4-NPA by a model hydroquinone (reduced
juglone (8-hydroxy-14-naphthoquinone) in the presence of HS
minusHJUGk
ndash) The reduction of
a NAC by juglone follows the rate law
[ ] [ ] [ ] [ ] [ ] [ ]2222 ArNOJUGfkArNOHJUGkArNOk
dtArNOd
totHJUGHJUGHJUGobs sdotsdotsdot=sdotsdot=sdot=minus minusminusminusminus
and the was deducted from a LFER 1hE
II Chapter 7
bEak hHJUG +sdot=minus 059160
log1
for which a and b values have been established for a range of NACs with known
values (Hofstetter et al 1999) An excellent correlation of and log
has been found to exist over a range of 250 mV corresponding to more than 5 order
of magnitude for This is due to the fact that the actual transfer of the first
electron is the rate-determining step under the experimental conditions chosen
1hE 1
hE minusHJUGk
minusHJUGk
For comparison experiments with 4-nitrotoluene (4-NT) were also conducted The
pseudo 1 order rate constants for the reduction of 4-NPA with juglone were
corrected for the reduction of 4-NPA with only HSndash (control experiments
containing no juglone)
[ ]minuslowast
minusminus
minus
minus=
HJUGkk
k HSHJUGHJUG
where (MminusHJUGk -1middots-1) is the rate constant for a compound in the presence of only
juglone (slowastminusHJUGk -1) is the pseudo 1 order rate constant for a compound in the
presence of both juglone and HSndash (sminusHSk -1) is the pseudo 1 order rate constant for
the control reaction in the presence of only HSndash and [HJUGndash] (M) is the
concentration of the reactive dissociated HJUG- form (nondissociated
hydroquinone species are very nonreactive as compared to the monophenolate
species)
Supporting Information III
OH 0 OH OH
+ e- + H+ = + e- + H+ =
0 0
pl(( ox) = 8 00 PK1 (red) = 6 60
JUG HJUG
OH OH
OH
pKa2(red) = 10 60
Figure 7 1 Oxidized and reduced juglone fo1m s
Kinetic experiments in homogeneous anoxic aqueous solutions contained 5 mM
HS- 20 microM total juglone 50 mM KH2P04 buffer and were conducted at pH =
660 corresponding to a concentration of the reactive dissociated juglone form
[HJUG] = 10 microM
By using the LFER
E1 logkHJUG- = 125 middot 005~16 + 923
the following values were obtained
(Hofstetter et al 1999)
NAC k (M-1 -1) HJUG- middots log kHJUG- E~ (mV)
4-NT
4-NPA
311 middot10middot7
164middot10middot7
847middot 10-8
116middot 10middot7
226middot10middot2
489middot10middot3
-165
-231
-515
-546
IV Chapter 7
lowast
minusHJUGk -values are averages of triplicates whereas -values are averages of
duplicates The determined for 4-NT in this work (-515 mV) differs 3 from
the -value of -500 mV reported in the literature (Meisel amp Neta 1975
Wardman 1989) Hence it is assumed that the -value determined for 4-NPA
also differs by 3
minusHSk
1hE
1hE
1hE
Note that even for NACs holding very different values the difference in their
reactivities are much less pronounced in Fe(II)-Fe(III) systems such as the
Fe(II)goethite system (LFER slope a = 06 Hofstetter et al 1999) and the
Fe(II)magnetite system (LFER slope a = 034 Klausen et al 1995) as compared
to the jugloneH
1hE
2S system (a = 125) Furthermore it should be noted that all
LFERs mentioned here were established for neutral NACs and in this work we
have simply assumed that the LFERs are also valid for anionic NACs
72 The rate-limiting step
The overall rate of a reaction is equal to the rate of the slowest step in the
mechanism In heterogeneous reactions eg a compound reacting at the surface of
suspended particles in bulk solution the overall process by which the
heterogeneous reactions proceed may be broken down into a sequence of
individual diffusion steps and reaction steps 1) Mass transfer (diffusion) of the
reactant from the bulk fluid to the external surface of the solid phase 2)
Adsorption of reactant onto the solid surface 3) Reaction on the solid surface 4)
Desorption of the products from the solid surface 5) Mass transfer of the products
from the external solid surface to the bulk fluid Hence the rate of reaction of a
compound reacting at the surface of suspended particles in bulk solution may be
either mass transfer adsorptiondesorption or surface reaction limited When the
diffusion steps are much faster than the reaction steps the mass transfer or
diffusion steps do not affect the overall reaction rate However if the reaction steps
Supporting Information V
are very fast compared with the diffusion steps mass transport affects the reaction
rate Here only the external mass transfer is considered ie the diffusion of
reactants or products between the bulk fluid and the external surface of the solid
phase The additional internal mass transfer resistance for particles containing
substantial internal surface area is not addressed
721 Mass transfer (diffusion) limited kinetics
The overall rate constant can be represented by a system of resistances in series
(Fogler 1999 Arnold et al 1999)
⎟⎟⎠
⎞⎜⎜⎝
⎛+=
minusgeomSALobs kkak1111
where kobs is the observed rate constant kL is the mass transfer coefficient (mmiddots-1) a
is the ratio of the external (geometric) specific surface area to volume of solution
(m-1) and kSA-geom is the intrinsic rate constant of the reaction normalized to the
external specific surface area rather than the BET specific surface area By
comparing kLmiddota with kobs one can estimate the role of mass transfer on the rate of
reaction Thus if kLmiddota gtgt kobs mass transfer is so fast that it has no impact on the
reaction rate whereas if kLmiddota le kobs mass transfer is the rate limiting step
In fluid dynamics the Reynolds number Re is used for determining whether a
flow is laminar or turbulent
νtp ud sdot
=Re
where dp is the particle diameter (m) ut is the terminal particle settling velocity
(mmiddots-1) and ν is the kinematic fluid viscosity (m2middots-1) ν = η ρ where η is the
(absolute) dynamic fluid viscosity in centipoise (1 centipoise = 1 mPamiddots = 10-3
kgmiddotm-1middots-1) and ρ is the fluid density (kgmiddotm-3)
VI Chapter 7
At Re lt 1 we can apply Stokersquos particle settling velocity Stokersquos law is an
equation relating the terminal settling velocity of a smooth rigid sphere in a
viscous fluid of known density and viscosity to the diameter of the sphere when
subjected to a known force field
( )η
ρρsdot
minussdotsdot=
18
2pp
t
dgu (mmiddots-1)
where g = 981 mmiddots-2 is the gravitational constant ρp is the particle density (kgmiddotm-3)
The Sherwood number is the main parameter for prediction of the mass transfer
process
in fluid dynamics
3121 ScRe602Sh sdotsdot+=sdot
=lowast
lowast
iw
pL
Ddk
where Diw is the diffusion coefficient of the compound i in water (m2middots-1) is the
minimum (uncorrected) value of the mass transfer coefficient and Sc is the
Schmidt number This relation is often referred to as the Froumlssling correlation The
particle diameter is a key parameter in the Froumlssling correlation and the external
mass transfer coefficient varies with square of the particle size for smaller
particles
lowastLk
The Schmidt number is the ratio of the kinematic fluid viscosity and the diffusion
coefficient of the compound i in water
iwDν
=Sc
Supporting Information VII
According to Harriott (1962) the actual mass transfer coefficient kL is 15 times
greater than the minimum value of the mass transfer coefficient The
uncertainty in k
lowastLk
Lmiddota associated with particle sphericity and roughness issues are
believed not to exceed a factor of 2
The diffusion coefficient of a compound i in water can be estimated as (Hayduk amp
Laudie 1974)
5890141
9102613
iiw
VD
sdot
sdot=
minus
η (m2middots-1)
where iV is the molar volume of the compound i (cm3middotmol-1) estimated according
to Fuller et al 1966
Assuming spherical particles the external (geometric) specific surface area and the
particle diameter are calculated from the measured BET specific surface area Atot
assuming that our GR-SO4 has a AtotAouter ~ 30 similar to the one reported by
Hansen amp Koch (1998)
( ) ( ) ( )pppp
p
pp
p
dd
dV
SAAρρπ
πρ 1000
6
100061100030
SA3
2tot
geom sdot=
sdotsdot
sdot=
sdot== (m2middotg-1)
In our aqueous GR-SO4 system the density ρ = 1000 kgmiddotm-3 the absolute dynamic
viscosity η = 10-3 Pamiddots and the kinematic viscosity ν = 10-6 m2middots-1 for water The
GR-SO4 particle specific parameters used is found below
VIII Chapter 7
GR-SO4
Atot (m2g) 712
SAgeom (m2g) 237
Particle density ρp (kgm3) 1500
Particle diameter dp (m) 169middot10-6
Settling velocity ut (ms) 778middot10-7
Reynolds number 132middot10-6
The molar volumes the diffusion coefficients in water and the Schmidt numbers
for the NACs including 4-chloronitrobenzene (4-CNB) and nitrobenzene (NB)
were
Compound iV (cm3middotmol-1) Diw (m2middots-1) Sc
4-NT 1260 768middot10-10 1302
4-CNB 1230 779middot10-10 1284
4-NPA 1535 684middot10-10 1462
NB 1055 853middot10-10 1173
The ratio of the external (geometric) specific surface area to volume of solution
were calculated for GR-SO4 at the various concentrations applied
[Fe(II)GR]0 (mM) a (m-1)
103 366middot102
63 224middot103
126 448middot103
60 213middot103
The uncorrected mass transfer coefficients were estimated for the NACs using the
Froumlssling correlation
Supporting Information IX
[Fe(II)GR]0 (mM) lowastLk (mmiddots-1)
4-NT 912middot10-4
4-CNB 925middot10-4
4-NPA 812middot10-4
NB 101middot10-3
Finally kLmiddota was calculated and compared with the experimental 1 order rate
constants kobs obtained for the NACs
Compound [Fe(II)GR]0 (mM) kLmiddota (s-1) kobs (s-1) a
4-NT 103 050 420middot10-4
63 307 140middot10-3
126 613 590middot10-3
4-CNB 103 051 740middot10-4
63 311 170middot10-3
126 622 460middot10-3
4-NPA 103 045 640middot10-4
63 273 109middot10-3
126 546 473middot10-3
NB 60 324 137middot10-3 b
a Experimental pseudo 1 order rate constant at 50 microM [Ar-NO2]0 b Experimental pseudo 1 order rate constant at 10 microM [Ar-NO2]0
When comparing kLmiddota with kobs it can be seen that the rates of mass transfer for all
3 NACs exceed the observed rate constants by at least 3 or 4 orders of magnitude
at every initial Fe(II)GR concentration Thus the reaction of the given NACs with
GR-SO4 is not subject to mass transfer limitations under the experimental
conditions applied here
722 Surface saturation limited kinetics
More than 75 of all heterogeneous reactions that are not diffusion-limited are
surface-reaction-limited rather than adsorption- or desorption-limited We now
X Chapter 7
look at the reaction A = B = C where an intermediate B is formed In our system
A = Ar-NO2 B = Ar-NHOH and C = Ar-NH2 In this case the surface reaction is
assumed to be a single-site mechanism where only the site S on which A or B is
adsorbed is involved in the reaction forming B or C
KA
Adsorption 1 A + S = AmiddotS
kS1
Surface reaction 1 AmiddotS = BmiddotS
KB-1
Desorption 1 BmiddotS = B + S
KB
Adsorption 2 B + S = BmiddotS
kS2
Surface reaction 2 BmiddotS = CmiddotS
KC-1
Desorption 2 CmiddotS = C + S
The rate law for this surface-reaction limited single-site mechanism involving an
intermediate follows Langmuir-Hinshelwood kinetics (adopted from Fogler 1999)
CCBBAA
AAsitesSA
CKCKCKCKCk
dtdC
sdot+sdot+sdot+sdotsdotsdot
=minus1
1
Supporting Information XI
where kS1 is the intrinsic rate constant of the surface reaction transforming A into
the intermediate B Csites is the concentration of reactive sites S on the solid
surface KA KB and KC are the adsorption constants for A B and C at the reactive
surface sites and CA CB and CC are the concentrations of A B and C in the bulk
fluid Two major assumptions of the Langmuir isotherm imply that there is a fixed
number of localised surface sites present on the surface and that the activity of the
surface towards adsorption desorption or surface reaction is independent of
surface coverage
Hence fitting -∆CA∆t to CA CB and CC using a nonlinear curve fitting software
such as SigmaPlot may provide one with the intrinsic rate constant and the
adsorptions constants If KB and KC gtgt KA the intermediate and the product are
strongly competing with the reactant for vacant reactive surface sites
Our data was not fitted successfully by the Langmuir-Hinshelwood rate law
(regression results not shown) Simplifying the rate law by excluding either the
term KCmiddotCC or KBmiddotCB or both (assuming that the aniline product or the
hydroxylaniline intermediate or both did not compete for the reactive sites) did not
improve the regression The Langmuir-Hinshelwood rate law for a dual-site
mechanism did not fit our data either Thus Langmuir-Hinshelwood kinetics
cannot explain the reaction mechanism of the given NACs in our GR-SO4 system
73 External surface area of GR-SO4 and GR-CO3
The GR-SO4 unit cell consists of one double layer (d001 = 11 nm) ie one
hydroxide layer (049 nm ) and one interlayer (061 nm) Hexagonal GR-SO4
particles holding an average width of 1 microm (Figure 72) an average particle
thickness of 35 nm (Hansen amp Koch 1998) and a hydroxide layer thickness of
049 nm have a surface area of the basal plane
Abasal = 1 microm middot 1 microm ndash 2 middot 05 microm middot 025 microm = 075 microm2
XII Chapter 7
and a surface area of the edges
Aedge = (2 middot 05 microm + 4 middot 056 microm) middot 000049 microm = 00016 microm2
Figure 72 The hexagonal platy morphology of GR particles holding an average width of 1 microm
The particle thickness is the mean crystal thickness perpendicular to the 003 plane
as determined from the 003 reflections in an X-ray diffractogram A GR-SO4
particle holding a thickness of 35 nm contains 35 nm11 nm = 318 double layers
The GR-CO3 unit cell consists of one double layer (d001 = 075 nm) ie one
hydroxide layer (049 nm ) and one interlayer (026 nm) Hence a GR-CO3 particle
holding a thickness of 35 nm contains 35 nm075 nm = 467 double layers
The outer surface area of a GR-SO4 particle including outer basal planes and
edges is
Aouter = 222 microm 155microm 00016318microm 0752 =sdot+sdot
and the total surface area of a GR-SO4 particle including both inner and outer
basal planes as well as edges is
Supporting Information XIII
Atot = 222 microm 478)microm 00016microm 0752(318 =+sdotsdot
Hence the ratio of outer surface area to total surface area is
131microm 478microm 155
AA
2
2
tot
outer asymp=
Furthermore the ratio of edge surface area to outer surface area is
130microm 155
microm 00016318AA
2
2
outer
edge asympsdot
=
For GR-CO3 the outer surface area including outer basal planes and edges is
Aouter = 222 microm 157microm 00016746microm 0752 =sdot+sdot
and the total surface area of a GR-CO3 particle including both inner and outer
basal planes as well as edges is
Atot = 222 microm 701)microm 00016microm 0752(467 =+sdotsdot
Hence the ratio of outer surface area to total surface area is
145microm 701microm 157
AA
2
2
tot
outer asymp=
Furthermore the ratio of edge surface area to outer surface area is
121microm 157
microm 00016467AA
2
2
outer
edge asympsdot
=
XIV Chapter 7
74 Van der Waals radii
The size of polyatomic molecules can be estimated by summing the van der Waals
radii of the
individual atoms Van der Waals radii or nonbonded radii can be pictured as the
radii of hard spherical atoms (Figure 73)
Figure 73 Schematic of neighboring nonbonded atoms with van der Waals radii rA and rB
Assuming that the spheres of neighboring nonbonded atoms just touch (Figure
73) the highest possible ion or molecule size Ms can be estimated as the sum of
the van der Waals radii
Ms = 2middotrA + 2middotrB + (1)
Taking Paulingrsquos rule for nonmetals into account we can estimate the real size of
polyatomic ions bound by covalent bonds (Pauling 1960) The van der Waals
radius is larger than the covalent radius because it involves the interposition of two
electron pairs between the atoms rather than one The rule states that the van der
Waals radius of an atom exceeds its covalent radius by ~008 nm (overlap in
Figure 74)
Figure 74 Schematic of atoms undergoing covalent bonding
Supporting Information XV
Thus the size of polyatomic ions bound by covalent bonds Ms can now be
estimated as the sum of the van der Waals radii subtracted by 008 nm
Ms = 2middot(rA - 008 nm) + 2middot(rB - 008 nm) + (2)
We estimated the molecular size of the NACs (Table 72) by means of equation (2)
and the van der Waals radii of the atoms in Table 71
Table 71 Van der Waals radii of various atoms Values from Pauling 1960
Atom vdW radii (nm)
H 0120
O 0140
N 0150
C 0170
Cl 0181
S 0185
In order to make the calculations it was assumed that all atoms were spherical and
that all bond angles were 90deg or 180deg (linear structures) In addition no distinctions
were made between single and double bonds The molecular sizes of the NACs
were estimated with the benzene ring representing the xy plane
Table 72 Molecular sizes of the NACs a Thickness z of the xy plane
Compound Ms (x) (nm) Ms (y) (nm) Ms (z) (nm)a
NB 054 080 036
4-NT 054 106 036
4-CNB 054 100 036
4-NPA 054 136 036
XVI Chapter 7
Note that the molecular sizes in Table 72 are only rough estimations
For comparison with the GR-SO4 interlayer spacing (061 nm) we consider three
possible orientations of the NACs in the GR-SO4 interlayer 1) The NAC xyz
coordination is equivalent to the crystal abc coordination (z = c = 036 nm) 2) the
NAC xy plane is parallel to the crystal bc plane (z = a = 054 nm) and 3) the NAC
xy plane is parallel to the crystal ac plane (z = b = 080-136 nm) Hence the sizes
of the NACs do not hinder their access to the GR-SO4 interlayer Only when
oriented vertically do the sizes of the NACs (z = b = 080-136 nm) exceed the GR-
SO4 interlayer spacing
The molecular size of trichloroacetate (TCA) was also estimated by means of
equation (2) and the atomic van der Waals radii in Table 71 When the TCA
aliphatic chain is assumed to represent the x direction (Ms (x) = 066 nm) the
molecular size in the y and z directions ranges from 045-053 nm depending on the
free rotation of the C-C bond Thus only if the C-C bond is oriented perpendicular
to the crystal ab plane does the size of TCA exceed the GR-SO4 interlayer spacing
(061 nm) In contrast the size of TCA exceeds the GR-CO3 interlayer spacing
(026 nm) regardless of its orientation
75 Adsorption of Fe(II) onto Fe(III) oxides
As seen from the Fe(II) sorption isotherms Fe(II) sorption varies widely between
the Fe(III) oxides as a function of solution pH (Figure 75) Average surface
densities of approximately 2 singly coordinated sitesnm2 iron oxide have been
suggested for goethite and lepidocrocite (Cornell amp Schwertmann 1996) The
similar surface site densities of goethite and lepidocrocite might explain their
similar Fe(II) adsorption isotherms (Figure 75)
Supporting Information XVII
Figure 75 Fe(II) adsorption edges for ferrihydrite goethite hematite lepidocrocite and
magnetite in the absence of other specifically adsorbing cations and anions (from Vikesland amp
Valentine 2002 and references therein) The total number of surface sites was in excess of the
total Fe(II) concentrations in all experiments
Dissolved cations or anions may specifically adsorb at the calcite and Fe(III) oxide
surfaces by exchanging for H+ or OHndash at the equivCO3H0 equivCaOH0 equivFeOH0 and
equivFeIIIOFeIIOH0 surface sites At the experimental conditions applied here within a
pH range 70-87 the dominant species of interest in solution are Fe2+ HCO3ndash
CO32ndash Clndash SO4
2ndash (only in the GR-SO4 systems) and the anionic TCA and DCA In
addition Fe2+ readily forms aqueous complexes with hydroxide carbonate
chloride and sulfate whereby the species FeOH+ FeHCO3+ Fe(OH)(CO3)ndash
FeCO30 Fe(CO3)2
2ndash FeCl+ and FeSO40 may occur (Millero amp Hawke 1992) At
pH 70-87 we expect the Fe(II) species Fe2+ FeCO30 Fe(OH)(CO3)ndash FeOH+ and
Fe(CO3)22ndash to dominate in the GR-CO3 and CaCO3(s)CO2(g) buffered magnetite
suspensions In the goecalcite and lepcalcite suspensions we expect the FeCl+
species to dominate as well whereas the Fe2+ FeSO40 and FeOH+ species most
XVIII Chapter 7
likely dominate in the GR-SO4 suspensions Anionic inorganic ligands like
carbonate chloride and sulfate can lower or enhance the adsorption of Fe(II) due to
a) formation of stable nonadsorbing Fe(II) ligand aqueous complexes b) formation
of Fe(II) ligand Fe(III) oxide surface complexes which can lead to surface
precipitation at high Fe(II) and ligand concentrations c) competitive ligand
sorption to the Fe(III) oxide surface blocking reactive sorption sites at the surface
and d) diminution of the positive charge at the Fe(III) oxide surface (at pH levels
below the point of zero charge (pHpzc) of the Fe(III) oxide) thereby decreasing the
electrostatic repulsion of cations by the Fe(III) oxide surface Specifically adsorbed
cations increase the pHpzc whereas specifically adsorbed anions decrease the pHpzc
References Arnold WA Ball WP Roberts AL (1999) Polychlorinated ethane reaction with zero-valent zinc Pathways and rate control Journal of Contaminant Hydrology 40 183-200 Cornell RM Schwertmann U (1996) The iron oxides Structure properties reactions occurrence and uses VCH Verlagsgesellschaft mbH Weinheim Fogler HS (1999) Elements of chemical reaction engineering 3rd ed Prentice Hall Fuller EN Schettler PD Giddings JC (1966) A new method for prediction of binary gas-phase diffusion coefficients Industrial and Engineering Chemistry 58 19-27 Hansen HCB Koch CB (1998) Reduction of nitrate to ammonium by sulphate green rust Activation energy and interlayer reaction mechanism Clay Minerals 33 87-101 Harriott P (1962) Mass transfer to particles Part I Suspended in agitated tanks AIChE Journal 8 93-102 Hayduk W Laudie H (1974) Prediction of diffusion coefficients for nonelectrolytes in dilute aqueous solutions AIChE Journal 20 611-615 Hofstetter TB Heijmann CG Haderlein SB Holliger C Schwarzenbach RP (1999) Complete reduction of TNT and other (poly)nitroaromatic compounds under iron-reducing subsurface conditions Environmental Science and Technology 33 1479-1487 Klausen J Troumlber SP Haderlein SB Schwarzenbach RP (1995) Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions Environmental Science and Technology 29 2396-2404 Meisel D Neta P (1975) One-electron redox potentials of nitro compounds and radiosensitizers Correlation with spin densities of their radical anions Journal of the American Chemical Society 97 5198-5203
Supporting Information XIX
Millero FJ Hawke DJ (1992) Ionic interactions of divalent metals in natural waters Marine Chemistry 40 19-48 Pauling L (1960) The nature of the chemical bond 3rd ed Cornell University Press Ithaca Vikesland PJ Valentine RL (2002) Iron oxide surface-catalyzed oxidation of ferrous iron by monochloramine implications of oxide type and carbonate on reactivity Environmental Science and Technology 36 512-519 Wardman P (1989) Reduction potentials of one-eletron couples involving free radicals in aqueous solution The Journal of Physical Chemistry Reference Data 18 1637-1755
Curriculum Vitae 13011973 Born in Haderslev Denmark 1988-1991 Mathematical high school Haderslev Katedralskole Denmark 1991-1992 Sabbatical year 1992-1995 B Sc in environmental chemistry University of Copenhagen
(KU) Denmark B Sc thesis 1995 ldquoMethane oxidizing bacteria in soilrdquo
1995-1998 M Sc in environmental chemistry University of Copenhagen
Denmark 1997-1998 diploma thesis ldquoReductive dechlorination of carbon tetrachloride and chloroform in presence of iron(II)iron(III)-hydroxides (green rust)rdquo
1998-1999 Research and teaching assistant at the Chemistry Department
The Royal Veterinary amp Agricultural University (KVL) Denmark
1999-2004 PhD in environmental sciences Swiss Federal Institute of
Technology Zuumlrich (ETHZ) and Swiss Federal Institute for Environmental Science and Technology (EAWAG) Switzerland Docoral thesis ldquoFormation and redox reactions of green rusts under geochemical conditions found in natural soils and sedimentsrdquo
2000-2002 Teaching assistent at the Swiss Federal Institute of Technology
Zuumlrich and supervision of diploma students 2002 Microbial Diversity summer course (7 weeks) at the Marine
Biological Laboratory Woods Hole Massachusetts USA

Table of Contents
325 Analytical methods 50 33 Results and discussion 50
331 Fe(II) production and suspension colour changes 50 332 Identification of solid iron phases 55 333 Factors controlling the identity of the secondary iron minerals 58 334 Factors controlling the rate and extent of Fe(III) bioreduction 59
34 Conclusions 60 References 61 4 Reduction of Nitroaromatic Probe Compounds by Sulphate
Green Rust The Effect of Probe Compound Charge 65 Abstract 65 41 Introduction 66 42 Materials and methods 71
421 Synthesis of GR-SO4 71 422 Mineral characterisation 72 423 Lyophilization and determination of specific surface area 72 424 Estimation of the one-electron reduction potential for 4-NPA 73 425 Kinetic experiments 74 426 Analytical methods 74
43 Results and discussion 75 431 Product formation and reaction kinetics 75 432 Comparison of rate constants for the different NACs 79 433 Factors influencing the reaction rate 82 434 Comparison with rate constants obtained for other Fe(II) containing
mineral systems 83 435 Depletion of reactive sites 85 436 The role of external and internal reactive sites 86
44 Conclusions 89 References 91 5 Reductive Transformation of Trichloroacetate in Abiotic
Fe(II)-Fe(III) Mineral Systems 97 Abstract 97 51 Introduction 98 52 Materials and methods 101
521 Synthesis of GRs and magnetite 102 522 Preparation of iron oxide coatings 102 523 Mineral characterisation 103 524 Kinetic experiments 103
Table of Contents
525 Analytical methods 104 53 Results and discussion 105
531 Product formation and reaction kinetics 105 532 Comparing rate constants obtained for the various Fe(II)-Fe(III)
mineral systems 109 533 Comparing with rate constants obtained for other chlorinated
aliphatic compound 112 534 Factors controlling the reactivity of surface-bound Fe(II) 114 535 Comparison with biotic and other abiotic systems 118
54 Conclusions 119 References 120 6 Conclusions and Outlook 125 References 128 7 Supporting Information I 71 Estimation of the one-electron reduction potential for 4-NPA I 72 The rate-limiting step IV 721 Mass transfer (diffusion) limited kinetics V
722 Surface saturation limited kinetics IX 73 External surface area of GR-SO4 and GR-CO3 XI 74 Van der Waals radii XIV 75 Adsorption of Fe(II) onto Fe(III) oxides XVI References XVIII Curriculum Vitae
Zusammenfassung I
Zusammenfassung Geschichtete Fe(II)-Fe(III)-Hydroxide (Gruumlner Rost) gehoumlren zur Gruppe der
Fe(II)-haltigen Mineralsysteme (zB Magnetit (Fe3O4) Siderit (FeCO3) Vivianit
(Fe2(PO4)2sdot8H2O) Fe(II)-Sulfide sowie an die Oberflaumlche von Fe(III)-Oxiden und
Tonmineralien gebundenes zweiwertiges Eisen) die die Aktivitaumlt von Fe(II) in
suboxischen und anoxischen Boumlden und Sedimenten kontrollieren Gruumlner Rost
Phasen (GRs) bestehen aus planaren positiv geladenen trioktaedrischen Fe(II)-
Fe(III)-Hydroxidschichten die durch hydratisierte Anionen in den
Zwischenschichten ausgeglichen werden Ihre generelle Zusammensetzung ist
[FeII(6-x)FeIII
x(OH)12]x+[(A)xnmiddotyH2O]x- wobei x = 09 - 42 ist A entspricht einem n-
valenten Anion (zB CO32- Clndash oder SO4
2-) und y repraumlsentiert die Anzahl
Wassermolekuumlle in der Zwischenschicht GRs sind wichtige intermediaumlre Phasen
die durch unvollstaumlndige Oxidation von Fe(II) oder teilweise Reduktion von Fe(III)
gebildet werden koumlnnen Sie koumlnnen in suboxischen nicht-sauren eisenhaltigen
natuumlrlichen wie auch technischen Systemen auftreten so wie in Wasser gesaumlttigten
Boumlden und interstitiellen Sedimenten Rohrleitungen in der
Trinkwasserversorgung Stahlpfosten in marinen Sedimenten Stahlbeton und in
reaktiven durchlaumlssigen Waumlnden aus nullwertigem Eisen zur in-situ Sanierung von
Altlasten und Aquiferen Aufgrund ihrer Schichtstruktur den anionischen
Zwischenschichten und der hohen spezifischen Oberflaumlchen sind GRs reaktive
Ionentauscher und Sorbentien von Anionen Des Weiteren wurde gezeigt dass
GRs eine Reihe anorganischer und organischer Schadstoffe reduzieren koumlnnen
Durch Immobilisierung und Transformation koumlnnen GRs somit eine wichtige Rolle
fuumlr das Abbauverhalten und den Transport solcher Schadstoffe in suboxischen
Boumlden und Sedimenten spielen Die Resultate dieser Dissertation tragen zum
Verstaumlndnis uumlber die Bildung und Reaktivitaumlt von Fe(II)-haltigen Mineralsystemen
wie GRs Vivianit Magnetit und an Goethit (α-FeOOH)- und Lepidokrozit (γ-
FeOOH)-Oberflaumlchen gebundenes Fe(II) in der Natur bei
II Zusammenfassung
Um die Rolle von Bakterien bei der Bildung von GRs in natuumlrlichen Boumlden und
Sedimenten aufzuklaumlren wurden Eisenminerale untersucht die als Folge der
Aktivitaumlt von eisenrespirierenden Bakterien gebildet wurden Kapitel 2 beschreibt
die Untersuchungen von eisenhaltigen Produkten die von anaeroben autotrophen
denitrifizierenden Fe(II)-oxidierenden Bakterien (FeOB) gebildet wurden Ein
Bikarbonat- und Phosphat-reiches Kulturmedium bot den nitratreduzierenden
FeOB optimale Bedingungen Fe(II) lag zu Anfang der Reaktion als weisses
Fe(II)-Hydroxyphosphat (Vivianit) und als geloumlstes Fe(II) vor Die Ergebnisse
zeigten dass die denitrifizierenden FeOB amorphen Goethit via ein gruumlnes Fe(III)-
angereichertes Vivianit-Zwischenprodukt bildeten Die Analyse mit Moumlssbauer
Spektroskopie deutet nicht auf eine Bildung von GR hin
In Kapitel 3 werden jene Eisenmineralien beschrieben die waumlhrend der Reduktion
verbreiteter Fe(III)-Oxide durch anaerobe dissimilative Fe(III)-reduzierende
Mikroorganismen Shewanella algae BrY gebildet wurden Um natuumlrliche
Zustaumlnde zu simulieren wurden Fe(III)-Oxide als Beschichtungen auf
Silikatpartikel (Modellsystem fuumlr Sandboumlden) oder Calcitpartikel (CaCO3
Modellsystem fuumlr kalkhaltige Boumlden) aufgetragen sowie synthetische
Elektronencarrier und hochkonzentrierte kuumlnstliche pH-Puffer ausgeschlossen
Die erforschten Mineralsysteme umfassten GoethitCalcit- LepidokrozitCalcit-
und FerrihydritSand-Suspensionen S algae BrY reduzierte beachtliche Mengen
des eingesetzten Fe(III) und es bildeten sich gruumlne und schwarze Festphasen
innerhalb von 1-2 Wochen nach der Animpfung Moumlssbauer Spektroskopie der
gruumlnen und schwarzen Praumlzipitate zeigte dass sich diese aus GR und Vivianit
zusammensetzen
Die Reaktivitaumlt synthetischer GRs gegenuumlber reduzierbaren organischen
Schadstoffen wurde erkundet um die potentielle Bedeutung von GR-Phasen fuumlr
das Schicksal solcher Verbindungen abzuschaumltzen Zu diesem Zweck wurden
Nitroaromaten (NACs) und Chloracetate als Modellverbindungen benutzt um
Zusammenfassung III
umweltrelevante Redoxreaktionen zu studieren In Kapitel 4 wurde die relative
Reaktivitaumlt von aumlusseren und inneren reaktiven Stellen in synthetischem Sulfat-
Gruumlnem Rost (GR-SO4) anhand von strukturaumlhnlichen ldquoreaktiven
Sondenmolekuumllenrdquo mit unterschiedlichen Ladungen untersucht Als reaktive
Sondenmolekuumlle wurden Nitrobenzen 2-Nitrophenol 4-Nitrotoluen 4-
Chlornitrobenzen und 4-Nitrophenylessigsaumlure verwendet Die Ergebnisse zeigen
dass GR-SO4 die NACs vollstaumlndig zu den entsprechenden Anilinen reduzierte
Die Reaktionen folgten einer pseudo 1 Ordnungs Kinetik bezuumlglich NAC und die
auf Oberflaumlche normalisierten pseudo 1 Ordnungs Geschwindigkeitskonstanten
(Anfangsraten) waren 016ndash465middot10-4 s-1middotm-2middotL fuumlr [Fe(II)GR]0 = 103-1260 mM
[NAC]0 = 20-102 microM und pH 84-86 Weder durch Einbezug von
Massentransferlimitierung noch von Oberflaumlchensaumlttigungskinetik war es moumlglich
die aumlhnlichen Oberflaumlchennormalisierten pseudo 1 Ordnungs
Geschwindigkeitskonstanten fuumlr die Reduktion der neutralen und anionischen
NACs durch GR-SO4 zu erklaumlren Dieser Umstand laumlsst vermuten dass die
Reaktion zwischen NAC und GR-SO4 an den externen reaktiven Fe(II)-Stellen
stattfindet Bei niedrigen Fe(II)GR-Anfangskonzentrationen wurden die externen
reaktiven Fe(II)-Stellen aufgebraucht und die Regenerierung von neuen externen
reaktiven Stellen haben schliesslich die Geschwindigkeit der Reduktion von NACs
durch GR-SO4 kontrolliert
In Kapitel 5 wurde die Reaktivitaumlt von verschiedenen umweltrelevanten Fe(II)-
Fe(III)-Mineralsystemen gegenuumlber Trichloressigsaumlure (TCA) und
Dichloressigsaumlure (DCA) in Batchexperimenten die natuumlrliche Bedingungen
imitierten untersucht Die Fe(II)-Fe(III)-Systeme umfassten Sulfat-Gruumlner Rost
Carbonat-Gruumlner Rost Magnetit Fe(II)Goethit und Fe(II)Lepidokrozit TCA
wurde von allen Fe(II)-haltigen Mineralien zu DCA reduziert Die Reaktionen
folgten einer pseudo 1 Ordnungs Kinetik bezuumlglich TCA und die auf Oberflaumlche
normalisierten pseudo 1 Ordnungs Geschwindigkeitskonstanten betrugen 033ndash
76middot10-5 min-1middotm-2middotL bei [Fe(II)]0 = 025ndash116 mM [TCA]0 = 15ndash1000 microM und pH
IV Zusammenfassung
70ndash87 Die Ergebnisse zeigen keine signifikanten Unterschiede zwischen den
verschiedenen Fe(II)-Fe(III)-Systemen bezuumlglich Produkteverteilung und
oberflaumlchen-normalisierten pseudo 1 Ordnungs Geschwindigkeits-konstanten In
keinem der Systeme wurde DCA innerhalb des experimentellen Zeitraums zu
Monochloressigsaumlure oder Essigsaumlure weiter reduziert
Die Ergebnisse die in dieser Dissertation praumlsentiert werden zeigen dass
mikrobiologische Prozesse fuumlr die Oxidation von Vivianit-Phasen im Untergrund
verantwortlich sein koumlnnen Zudem wurde nachgewiesen dass GRs bei tiefen
Kohlenstoff- und Fe(III)-Konzentrationen sowie durch Ausschluss von
kuumlnstlichen Elektronencarriern und pH-Pufferung mikrobiell gebildet werden
koumlnnen Ferner zeigten Befunde dass GRs eine bedeutende Rolle fuumlr die reduktive
Transformation von NACs und TCA in natuumlrlichen Boumlden und Sedimenten spielen
koumlnnen
Summary V
Summary
Layered iron(II)-iron(III)-hydroxides (green rusts) belong to the group of Fe(II)-
bearing mineral systems eg magnetite (Fe3O4) siderite (FeCO3) vivianite
(Fe2(PO4)2sdot8H2O) Fe(II) sulfides as well as Fe(II) associated with Fe(III) oxide
and clay mineral surfaces that control the Fe(II) activity in suboxic and anoxic
soils and sediments Green rusts (GRs) consist of plane positively charged
trioctahedral Fe(II)-Fe(III) hydroxide sheets balanced by hydrated anions in the
interlayers and hold the general formula [FeII(6-x)FeIII
x(OH)12]x+[(A)xnmiddotyH2O]x-
where x = 09 - 42 A is an n-valent anion eg CO32- Clndash or SO4
2- and y is the
number of water molecules in the interlayer GRs are important intermediate
phases formed by partial oxidation of Fe(II) or partial reduction of Fe(III) and they
have been found in suboxic non-acid iron-rich natural environments such as
hydromorphic soils and intertidal sediments and in engineering systems including
pipeline distribution systems for drinking water steel sheet piles in marine
sediments reinforced concrete and permeable reactive barriers of zero-valent iron
implemented for on-site remediation of contaminants Due to their layered
structures anionic interlayers and high specific surface areas GRs represent
reactive ion exchangers and sorbents of anions In addition GRs have been shown
to reduce a range of inorganic and organic pollutants Thus through sequestration
and reductive transformation GRs may play an important role in the fate and
transport of contaminants in suboxic soils and sediments The work presented in
this dissertation adds to the understanding of how Fe(II)-bearing minerals like
GRs vivianite magnetite and Fe(II) associated with goethite (α-FeOOH) and
lepidocrocite (γ-FeOOH) may form and react in nature
In order to elucidate the role of bacteria in the formation of GRs in natural soils
and sediments we studied the iron mineral phases forming as a result of the
activity of iron-respiring bacteria In the study described in chapter 2 the Fe-
containing products formed by anaerobic autotrophic denitrifying Fe(II)-oxidizing
VI Summary
bacteria (FeOB) were examined The culture medium applied contained high levels
of bicarbonate and phosphate and is typically used in this kind of studies as it
provides excellent conditions for the nitrate-reducing FeOB Fe(II) was present
initially as a whitish solid Fe(II) hydroxy phosphate (vivianite) and as soluble
Fe(II) The results obtained demonstrate that the denitrifying FeOB produce poorly
crystalline goethite via a greenish Fe(III)-enriched vivianite intermediate
Moumlssbauer spectroscopic analyses provided no significant evidence of green rust
formation
In chapter 3 the Fe-containing products formed during reduction of common
Fe(III) oxides by the anaerobic dissimilatory Fe(III)-reducing microorganism
Shewanella algae BrY are discussed In order to simulate natural conditions
Fe(III) oxides were applied as coatings on silica (model system for sandy soils) or
calcite (CaCO3) particles (model system for calcareous soils) and synthetic
electron shuttles as well as highly concentrated artificial pH buffers were excluded
The mineral systems studied include goethitecalcite lepidocrocitecalcite and
hydrous ferric oxidesand suspensions S algae BrY reduced substantial amounts
of the initial Fe(III) and green and blackish mineral phases were produced within
1-2 weeks after inoculation Moumlssbauer spectroscopic analyses showed that the
green and black precipitates consisted of GR and vivianite
The reactivity of synthetic GRs towards reducible organic pollutants was
investigated in order to asses the potential significance of GR phases for the fate of
such compounds To this end we used nitroaromatic compounds (NACs) and
chlorinated acetates as suitable model compounds for studying environmentally
relevant redox reactions In the work described in chapter 4 the relative reactivity
of outer and inner Fe(II) reactive sites in synthetic sulfate green rust (GR-SO4) was
studied using a series of structurally closely related compounds with different
charge properties as ldquoreactive probesrdquo The probe compounds included
nitrobenzene 2-nitrophenol 4-nitrotoluene 4-chloronitrobenzene and 4-
Summary VII
nitrophenylacetic acid The results show that NACs are completely reduced to their
corresponding anilines by GR-SO4 The reactions followed pseudo 1 order
kinetics with respect to NAC and the surface area-normalised pseudo 1 order rate
constants (initial rates) obtained were 016ndash465middot10-4 s-1middotm-2middotL at [Fe(II)GR]0 = 103-
1260 mM [NAC]0 = 20-102 microM and pH 84-86 Neither mass transfer control nor
surface saturation kinetics could explain the similarity of the surface-normalised
pseudo 1 order rate constants obtained for the reduction of the neutral and anionic
NACs by GR-SO4 These observations suggest that the reaction between NAC and
GR-SO4 takes place at the external reactive Fe(II) sites At low initial Fe(II)GR
concentrations the external reactive Fe(II) sites were depleted and the regeneration
of new external reactive sites eventually controlled the reduction of the NACs by
GR-SO4
Finally the reactivity of various Fe(II)-Fe(III) mineral systems towards
trichloroacetic acid (TCA) and dichloroacetate (DCA) has been investigated in
laboratory batch experiments imitating natural conditions (Chapter 5) The Fe(II)-
Fe(III)-systems investigated included GR-SO4 carbonate green rust magnetite
Fe(II)goethite and Fe(II)lepidocrocite TCA was readily reduced to DCA by all
Fe(II)-containing minerals The reactions followed pseudo 1 order kinetics with
respect to TCA and the surface area-normalised pseudo 1 order rate constants
obtained were 033ndash76middot10-5 min-1middotm-2middotL at [Fe(II)]0 = 025ndash116 mM [TCA]0 =
15ndash1000 microM and pH 70ndash87 Our results showed no significant differences
regarding product distribution and surface area-normalised reaction rate constants
between the Fe(II)-Fe(III)-systems DCA was not further reduced to
monochloroacetate (MCA) or acetate in any of the systems within the time frame
in our experiments
The results presented in chapter 2 indicate that microbiological processes may be
responsible for the oxidation of vivianite phases in natural subsurface
environments In chapter 3 we demonstrated that GRs may be produced
VIII Summary
microbially at conditions including low carbon and Fe(III) concentrations as well
as the exclusion of synthetic electron shuttles and pH buffers The results obtained
in chapter 4 and 5 show that GRs transform NACs and TCA readily The reductive
transformation of NACs and TCA by GRs is relevant to understanding the
processes responsible for their degradation in the subsurface and the development
of innovative technologies for their remediation
General Introduction 1
1 General Introduction
11 Iron cycling in the subsurface
Iron is the fourth most abundant element (4-5 mass) and the most abundant redox
sensitive element in the Earthrsquos crust It is found as Fe(II) and Fe(III) in a number
of minerals in rocks soils and sediments Under anoxic conditions solid Fe(III)-
containing minerals can be reduced to soluble Fe(II) once the more energetically
favoured electron donors - nitrate and manganese(IV) oxides - have been
consumed Dissolved Fe(II) can be reoxidized to insoluble Fe(III) microbially or
abiotically upon exposure to oxygen Due to this ready alternation between the
Fe(II) and Fe(III) redox states iron plays a major role in controlling the redox
potential and the carbon cycling in subsurface environments (Nealson amp Saffarini
1994)
Nonenzymatic processes were previously considered to account for most of the
Fe(III) reduction in subsurface environments The significance of bacteria in the
biogeochemical cycling of iron has been broadly recognized over the past two
decades Dissimilatory Fe(III)-reducing bacteria (DIRB) that gain energy by
coupling the oxidation of hydrogen or organic compounds to the reduction of
Fe(III) oxides have been known for many years but their biogeochemical
importance was only widely acknowledged about a decade ago (reviewed by
Lovley 1997) Fe(III) bioreduction accounts for a major fraction of the carbon
oxidation in many different anoxic environments and in the presence of sufficient
amounts of reactive Fe(III) microbial Fe(III) reduction may even inhibit sulphate
reduction and methanogenesis (King 1990 Lovley amp Phillips 1986) In fact most
of the Fe(III) reduction in the Fe(III) reduction zone of aquatic sediments and
aquifers is enzymatically catalyzed by microorganisms (Lovley et al 1991) A
wide diversity of DIRB distributed among several different phylogenetic groups
2 Chapter 1 is known today The two most studied DIRB are the obligate anaerobic Geobacter
spp and the facultatively anaerobic Shewanella spp (Figure 11)
Aerobic oxidation of Fe(II)-containing minerals by lithotrophic acidophilic and
neutrophilic bacteria has been known for many years but their broad significance
in the biogeochemical cycling of iron has only been recognized over the past two
decades Both acidophilic (Thiobacillus ferrooxidans) and neutrophilic
(Gallionella ferruginea Leptothrix ochracea Sphaerotilus natans) aerobic Fe(II)-
oxidizing bacteria (FeOB) have been isolated (Hanert 1992 Kuenen et al 1992
Mulder amp Deinema 1992)
Figure 11 The microbial iron cycle
Anaerobic Fe(II) oxidation by phototrophic purple non-sulfur bacteria utilizing
Fe(II) as an electron donor in the light was recognized only a decade ago (Widdel
et al 1993) Subsequently it was demonstrated that the biological oxidation of
Fe(II) in the absence of oxygen is possible by light-independent chemotrophic
microorganisms using nitrate as the electron acceptor (Straub et al 1996) Thus
the microbial iron cycle includes anaerobic Fe(III)-reducing microorganisms and
aerobic as well as anaerobic Fe(II)-oxidizing bacteria (Figure 11)
General Introduction 3
12 Green rusts
Iron oxides iron hydroxides and iron oxyhydroxides (collectively termed iron
oxides or Fe(III) oxides) are ubiquitous in the pedosphere where they originate
from aerobic weathering of surface magmatic rocks such as ferromagnesium
silicates and pyrite (Cornell amp Schwertmann 1996) Goethite (α-FeOOH)
lepidocrocite (γ-FeOOH) ferrihydrite (Fe5HO8sdot4H2O) hematite (α-Fe2O3)
magnetite (Fe3O4) maghemite (γ-Fe2O3) and akageneite (β-FeOOH) constitute the
most important iron oxides in soils and sediments (Schwertmann amp Cornell 1991)
The formation and transformation of iron oxides depend on pH solution
composition redox potential temperature rate of oxidationreduction and degree
and rate of hydrationdehydration Iron oxides are important to many soil
properties such as colour pH and redox buffer capacity aggregation with other
soil particles as well as retention of anions and cations (Cornell amp Schwertmann
1996) A number of Fe(II)-bearing minerals including Fe(II)-containing clays (eg
smectites vermiculites and micas) magnetite siderite (FeCO3) vivianite
(Fe2(PO4)2sdot8H2O) Fe(II) sulphides and green rusts (layered Fe(II)-Fe(III)
hydroxides) may be present in soils and sediments under suboxic and anoxic
conditions Green rusts are believed to play a central role as metastable
intermediates in the redox cycling of iron at circumneutral pH in aquatic and
terrestrial environments
Green rusts (GRs) are layered iron(II)-iron(III)-hydroxides consisting of plane
positively charged trioctahedral Fe(II)-Fe(III) hydroxide sheets balanced by
hydrated anions in the interlayers (cf Figure 41 this work) GRs belong
structually to the pyroaurite-sjoumlgrenite group of layered hydroxides and they hold
the general formula [FeII(6-x)FeIII
x(OH)12]x+[(A)xnmiddotyH2O]x- where x = 09 - 42 A is
an n-valent anion eg CO32- Clndash or SO4
2- and y is the number of water molecules
in the interlayer The three most common and investigated green rust forms include
chloride GR (GR-Cl) sulphate GR (GR-SO4) and carbonate GR (GR-CO3)
Generally GRs are crystallographically classified into the GRI (rhombohedral
4 Chapter 1 GR-Cl and GR-CO3) and GRII (hexagonal GR-SO4) crystal systems The GR
interlayer thickness is a function of both the size and the charge of the interlayer
anion Tetrahedrally coordinated anions like sulphate lead to larger interlayer
distances than smaller monoatomic anions like chloride or planar ions like
carbonate (Mendiboure amp Schoumlllhorn 1986) Not only size but also charge density
plays a role for the interlayer spacing That is for anions having the same number
of valence electrons anions with smaller ionic radii (higher electron density) are
bound more strongly and therefore result in smaller interlayer spacings The
interlayer in GR-SO4 is composed of two consecutive planes of anions and water
whereas GR-Cl and GR-CO3 interlayers consist of only one single plane (Simon et
al 2003)
GRs are important intermediate phases formed by partial oxidation of Fe(II) or
partial reduction of Fe(III) In neutral and weakly alkaline solutions the oxidation
of dissolved Fe(II) always passes through solid GR phases (Bernal et al 1959)
GRs may also form during oxidation of zero-valent iron and as a result of the
combination of Fe(II) and Fe(III) at circumneutral pH (Figure 12)
Figure 12 Formation and transformation of GRs Fe3O4 = magnetite γ-Fe2O3 = maghemite α-
FeOOH = goethite γ-FeOOH = lepidocrocite akageneite = β-FeOOH
General Introduction 5
Oxidation of GR-CO3 usually produces goethite and magnetite-maghemite
whereas GR-Cl and GR-SO4 transform into lepidocrocite and magnetite-
maghemite depending on pH and oxidation rate (Bernal et al 1959 Taylor 1980
Carlson amp Schwertmann 1990) The brown δ-FeOOH is formed by vigorous
oxidation of GR using air or a 30 aqueous solution of hydrogen peroxide (Bernal
et al 1959 Misawa et al 1974) Black ferromagnetic magnetite forms by slow
oxidation of GR whereas lepidocrocite forms at high oxidation rates (Misawa et
al 1974) The presence of chloride is a prerequisite for the formation of
akageneite (Bernal et al 1959 Refait amp Genin 1997)
A substantial amount of work has been conducted in order to estimate the free
energies of formation of green rusts The free energies of formation reported for
the carbonate and sulphate GRs fall in the range 4234ndash4384 kJsdotmol-1 as determined
from solution data monitored during anoxic alkalimetric titrations and from
reduction potential (Eh) and pH recordings monitored during oxidation of GRs in
aqueous solution (Hansen et al 1994 Drissi et al 1995 Genin et al 1996) The
free energies of formation provided allow for estimation of the stability domains of
GRs in Eh-pH phase diagrams (Drissi et al 1995 Genin et al 1996) As
evidenced from such diagrams (Figure 13) the stability domain of GR-SO4 lies
within pH 6-8 and Eh -700 ndash -400 mV depending on the activities of Fe(II) and
sulphate (compare Figures 13aampb) This agrees with the natural GR occurrences
found in suboxic non-acid iron-rich environments such as hydromorphic soils and
intertidal sediments (Al-Agha et al 1995 Trolard et al 1996 Genin et al 1998)
In addition GRs have been found as corrosion products in numerous engineering
systems including a pipeline distribution system for drinking water steel sheet
piles in marine sediments reinforced concrete (ferro-concrete) and permeable
reactive barriers of zero-valent iron implemented for on-site remediation of organic
and inorganic contaminants (Tuovinen et al 1980 Nielsen 1976 Genin et al
1991 Roh et al 2000)
6 Chapter 1
Figure 13 Eh-pH phase diagrams of GR-SO4 a) a = 10+2Fe-3 = 10minus2
4SOa -3 and b) a = 10+2Fe-2
= 10minus24SOa -1
The stability domains of GR-Cl and GR-CO3 are similar to the stability domain of
GR-SO4 At Fe(II) and sulphate activities lower than depicted in Figure 13b the
stability domain of GR-SO4 will be situated at higher pH and lower Eh Other
dissolved species present at anoxic conditions such as phosphate sulphide
carbonate and organic ligands may exert considerable effects on the availability of
Fe(II) and Fe(III) At anoxic and circumneutral conditions vivianite
(Fe2(PO4)2sdot8H2O) controls the Fe(II) activity even at very low phosphate
concentrations (Nriagu amp Dell 1974) The formation of solid Fe(II) sulphides and
siderite (FeCO3) as well as the complexation of Fe(II) and Fe(III) by organic
ligands may also control the activity of Fe(II) in the subsurface and thereby
interfere with the formation of GRs
Due to their layered structures anionic interlayers and high specific surface areas
GRs represent reactive ion exchangers and sorbents of environmentally concerning
anions eg arsenate and selenate (Myneni et al 1997 Randall et al 2001) In
addition GRs may incorporate divalent transition metal cations like Ni2+ Zn2+
Cd2+ Co2+ and Mg2+ by isomorphic substitution for Fe2+ in the hydroxide layers
General Introduction 7
(Tamaura 1985 Tamaura 1986 Refait et al 1994 Parmar et al 2001 Refait et
al 2001) Furthermore GRs have been shown to reduce a range of inorganic
contaminants such as nitrite nitrate selenate chromate uranyl pertechnetate and
the transition metals AgI AuIII CuII and HgII as well as organic pollutants
including halogenated ethanes ethenes and methanes (Hansen et al 1994 Hansen
et al 1996 Myneni et al 1997 Erbs et al 1999 Loyaux-Lawniczak et al 1999
Cui amp Spahiu 2002 Lee amp Batchelor 2002b Heasman et al 2003 OrsquoLoughlin et
al 2003aampb Pepper et al 2003 Elsner et al 2004 OrsquoLoughlin amp Burris 2004)
Thus through sequestration and reductive transformation GRs may play an
important role in the fate and transport of contaminants in suboxic soils and
sediments It should be noted that the rate constants reported for the reduction of
these inorganic and organic pollutants by GRs cannot be directly compared as the
various studies were conducted at very different experimental conditions
13 Microbial formation of green rusts
Generally one would expect that biogenic minerals have chemical compositions
and crystal habits similar to those produced by nonenzymatic processes as they are
governed by the same equilibrium principles In fact since the latter stages of
mineralization are abiotically driven and since the secondary Fe(II)-containing
minerals are formed indirectly by electron transfer outside the bacterial cell and not
directly inside the bacterial cell the type of iron mineral formed is a function of the
environmental conditions in which the bacteria live ie the same microorganism
form different minerals in different environments
The microbial formation of GRs resulting from bioreduction of various Fe(III)
oxides including ferrihydrite goethite and lepidocrocite by strains of the
anaerobic dissimilatory DIRB Shewanella putrefaciens has been reported
repeatedly over the last years (Fredrickson et al 1998 Kukkadapu et al 2001
Parmar et al 2001 Ona-Nguema et al 2002aampb Glasauer et al 2003)
However no evidence of biogenic formation of GRs at natural geochemical
8 Chapter 1 conditions have been offered and it is still unknown whether this process may take
place at natural conditions comprising low nutrient levels low iron concentrations
and the absence of synthetic electron shuttles and highly concentrated artificial pH
buffers Moreover the biotic formation of GRs by anaerobic denitrifying Fe(II)-
oxidizing bacteria has been suggested but the phases still need to be properly
identified (Chaudhuri et al 2001) In order to elucidate the role of bacteria in the
formation of GRs in natural soils and sediments we studied the iron mineral
phases forming as a result of the activity of iron-respiring bacteria (Chapters 2 and
3)
14 Redox reactions of green rusts
Fe(II) is one of the most abundant reductants present in aquatic and terrestrial
environments under suboxic and anoxic conditions (Lyngkilde amp Christensen
1992 Ruumlgge et al 1998) In these environments Fe(II) may be present as soluble
organic and inorganic complexes as surface complexes and as a host of Fe(II)-
bearing minerals Although aqueous Fe(II) complexes may reduce a number of
contaminants Fe(II) associated with mineral surfaces and structural Fe(II) present
in the mineral lattice in Fe(II)-containing minerals are often more powerful
reductants Fe(II)-bearing minerals including GRs magnetite siderite Fe(II)
sulphides as well as Fe(II)-carrying Fe(III) oxide and clay mineral surfaces have
been shown to reduce a number of organic and inorganic contaminants such as
nitroaromatic compounds chlorinated aliphatics chromate uranyl pertechnetate
nitrate monochloramine and carbamate pesticides (Klausen et al 1995 Cui amp
Eriksen 1996 Butler amp Hayes 1998amp1999 Erbs et al 1999 Liger et al 1999
Loyaux-Lawniczak et al 1999 Amonette et al 2000 Hwang amp Batchelor 2000
Hansen et al 2001 Gander et al 2002 Lee amp Batchelor 2002aampb Pecher et al
2002 Vikesland amp Valentine 2002 Hofstetter et al 2003 OrsquoLoughlin et al
2003aampb Strathmann amp Stone 2003 Elsner et al 2004 OrsquoLoughlin amp Burris
2004) However only few comparative studies on the reactivity of Fe(II)-bearing
minerals exist (Lee amp Batchelor 2002b Elsner et al 2004) When examining the
General Introduction 9
reaction rates of the reductive transformation of NACs and chlorinated aliphatics
by GRs and other Fe(II)-bearing minerals reported in these studies the rate
constants for GRs are mostly among the highest rates reported and in some cases
even higher than the rate constants for Fe(II) sulphides Thus GRs may play an
important role in the transformation of reducible contaminants in the subsurface
Nitroaromatic compounds (NACs) are widely applied as explosives herbicides
insecticides solvents and intermediates in the synthesis of dyes and pesticides
(Hartter 1985 Rosenblatt et al 1991) NACs are ubiquitous in the subsurface
environment and pose a health risk due to their toxicity (Rickert 1985) In anoxic
environments reduction of the nitro group is generally the first step during abiotic
or microbial transformation of the NACs (Macalady et al 1986) The
transformation reaction generally produces the corresponding aromatic amines and
minor amounts of intermediates (hydroxylamines and nitroso compounds) as well
as coupling products (azo and azoxy compounds) These products may be of
similar or even greater environmental concern
Trichloroacetic acid (TCA) is ubiquitous in soils and the concentrations reported
range from lt005 to 380 microgkg (Euro Chlor 2001 McCulloch 2002 Ahlers et al
2003) On account of its phytotoxicity suspected human carcinogenicity and
widespread occurrence TCA is of considerable environmental concern especially
in the terrestrial compartment (Ahlers et al 2003) Moreover the daughter
compounds of TCA - dichloroacetic acid (DCA) and monochloroacetic acid
(MCA) - are also toxins and suspected human carcinogens as well as widespread in
the environment (Reimann et al 1996 Berg et al 2000 Ahlers et al 2003 and
references therein) In this work the reactivity of synthetic green rusts towards
nitroaromatic compounds (NACs) and the reactivity of various Fe(II)-Fe(III)
mineral systems including synthetic GRs towards chlorinated acetates have been
studied (Chapters 4 and 5)
10 Chapter 1 15 Outline of the thesis
An examination of the Fe-containing products produced during solid state
oxidation of vivianite by anaerobic autotrophic denitrifying Fe(II)-oxidizing
bacteria is presented in chapter 2 The Fe(II)-oxidizing bacteria were cultured in a
mineral medium containing high levels of bicarbonate and phosphate which is
typically used in this kind of studies as it provides excellent conditions for the
nitrate-reducing FeOB The solid iron phases forming were investigated by
transmission Moumlssbauer spectroscopy infrared spectroscopy and scanning electron
microscopy
Chapter 3 includes a study on the Fe-containing products formed during reduction
of common Fe(III) oxides by the anaerobic dissimilatory Fe(III)-reducing
microorganism Shewanella algae BrY In order to simulate natural conditions
Fe(III) oxides were applied as coatings on silica (model system for sandy soils) or
calcite particles (model system for calcareous soils) and synthetic electron shuttles
as well as highly concentrated artificial pH buffers were excluded The mineral
systems studied include goethitecalcite lepidocrocitecalcite and hydrous ferric
oxidesand suspensions The solid iron phases produced were examined by
transmission Moumlssbauer spectroscopy
A study on the relative reactivity of outer and inner Fe(II) sites in synthetic GR-
SO4 by using a series of structurally closely related compounds with different
charge properties as ldquoreactive probesrdquo is presented in chapter 4 The probe
compounds included nitrobenzene 2-nitrophenol 4-nitrotoluene 4-
chloronitrobenzene and 4-nitrophenylacetic acid
In chapter 5 an investigation of the reactivity of various Fe(II)-Fe(III) mineral
systems towards TCA and DCA is presented The study included laboratory batch
experiments imitating natural conditions The Fe(II)-Fe(III)-systems investigated
included GR-SO4 carbonate green rust magnetite Fe(II)goethite and
General Introduction 11
Fe(II)lepidocrocite The reactivities of the Fe(II)-Fe(III) mineral systems were
examined by comparing their surface-normalized rate constants
The results and environmental implications of this work are summarized in chapter
6 References Ahlers J Regelmann J Riedhammer C (2003) Environmental risk assessment of airborne trichloroacetic acid - a contribution to the discussion of the significance of anthropogenic and natural sources Chemosphere 52 531-537 Al-Agha MR Burley SD Curtis CD Esson J (1995) Complex cementation textures and authigenic mineral assemblages in recent concretions from the Lincolnshire Wash (east coast UK) driven by Fe(0) to Fe(II) oxidation Journal of the Geological Society 152 157-171 Amonette JE Workman DJ Kennedy DW Fruchter JS Gorby YA (2000) Dechlorination of carbon tetrachloride by Fe(II) associated with goethite Environmental Science and Technology 34 4606-4613 Berg M Muumlller SR Muumlhlemann J Wiedmer A Schwarzenbach RP (2000) Concentrations and mass fluxes of chloroacetic acids and trifluoroacetic acid in rain and natural waters in Switzerland Environmental Science and Technology 34 2675-2683 Bernal JD Dasgupta DR Mackay AL (1959) The oxides and hydroxides of iron and their structural inter-relationships Clay Minerals Bulletin 4 15-30 Butler EC Hayes KF (1998) Effects of solution composition and pH on the reductive dechlorination of hexachloroethane by iron sulfide Environmental Science and Technology 32 1276-1284 Butler EC Hayes KF (1999) Kinetics of the transformation of trichloroethylene and tetrachloroethylene by iron sulfide Environmental Science and Technology 33 2021-2027 Carlson L Schwertmann U (1990) The effect of CO2 and oxidation rate on the formation of goethite versus lepidocrocite from an Fe(II) system at pH 6 and 7 Clay Minerals 25 65-71 Chaudhuri SK Lack JG Coates JD (2001) Biogenic magnetite formation through anaerobic biooxidation of Fe(II) Applied and Environmental Microbiology 67 2844-2848 Cornell RM Schwertmann U (1996) The iron oxides Structure properties reactions occurrence and uses VCH Verlagsgesellschaft mbH Weinheim Cui D Eriksen TE (1996) Reduction of pertechnetate by ferrous iron in solution influence of sorbed and precipitated Fe(II) Environmental Science and Technology 30 2259-2262
Cui D Spahiu K (2002) The reduction of U(VI) on corroded iron under anoxic conditions Radiochemica Acta 90 623-628
12 Chapter 1 Drissi SH Refait Ph Abdelmoula M Geacutenin JMR (1995) The preparation and thermodynamic properties of Fe(II)-Fe(III) hydroxide-carbonate (green rust I) Pourbaix diagram of iron in carbonate-containing aqueous media Corrosion Science 37 2025-2041 Elsner M Haderlein SB Schwarzenbach RP (2004) Reactivity of Fe(II)-bearing minerals towards reductive transformation of organic contaminants Environmental Science and Technology 38 799-807 Erbs M Hansen HCB Olsen CE (1999) Reductive dechlorination of carbon tetrachloride using iron(II)iron(III)-hydroxide-sulphate (green rust) Environmental Science and Technology 33 307-311 Euro Chlor (2001) Trichloroacetic acid in the environment a dossier Euro Chlor Brussels and the European Chlorinated Solvent Association Fredrickson JK Zachara JM Kennedy DW Dong H Onstott TC Hinman NW Li S (1998) Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium Geochimica et Cosmochimica Acta 62 3239-3257 Gander JW Parkin GF Scherer MM (2002) Kinetics of 111-trichloroethane transformation by iron sulfide and a methanogenic consortium Environmental Science and Technology 36 4540-4546 Geacutenin JMR Bourrieacute G Trolard F Abdelmoula M Jaffrezic A Refait Ph Maitre V Humbert B Herbillon A (1998) Thermodynamic equilibria in aqueous suspensions of synthetic and natural Fe(II)-Fe(III) green rusts Occurrences of the mineral in hydromorphic soils Environmental Science and Technology 32 1058-1068 Geacutenin JMR Olowe AA Benbouzid-Rollet ND Prieur D Confente M Resiak B (1991) The simultaneous presence of green rust 2 and sulfate reducing bacteria in the corrosion of steel sheet piles in a harbour area Hyperfine Interactions 69 875-878 Geacutenin JMR Olowe AA Refait Ph Simon L (1996) On the stoichiometry and Pourbaix diagram of Fe(II)-Fe(III) hydroxy-sulphate of sulphate-containing green rust 2 An electrochemical and Moumlssbauer spectroscopy study Corrosion Science 38 1751-1762 Glasauer S Weidler PG Langley S Beveridge TJ (2003) Controls on Fe reduction and mineral formation by a subsurface bacterium Geochimica et Cosmochimica Acta 67 1277- 1288 Hanert HH (1992) The genus Gallionella In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 4082-4088 Hansen HCB Borggaard OK Soslashrensen J (1994) Evaluation of the free energy of formation of iron(II)iron(III)-hydroxidesulphate (Green Rust) and its reduction of nitrite Geochimica et Cosmochimica Acta 58 2599-2608 Hansen HCB Guldberg S Erbs M Koch CB (2001) Kinetics of nitrate reduction by green rusts ndash effects of interlayer anion and Fe(II)Fe(III) ratio Applied Clay Science 18 81-91
General Introduction 13
Hansen HCB Koch CB Nancke-Krogh H Borggaard OK Soslashrensen J (1996) Abiotic nitrate reduction to ammonium Key role of green rust Environmental Science and Technology 30 2053-2056 Hartter DR (1985) The use and importance of nitroaromatic chemicals in the chemical industry In Toxicity of nitroaromatic compounds Rickert DE (ed) Hemisphere Publishing Corporation 1-13 Heasman DM Sherman DM Ragnarsdottir KV (2003) The reduction of aqueous Au3+ by sulfide minerals and green rust phases American Mineralogist 88 725-738 Hofstetter TB Schwarzenbach RP Haderlein SB (2003) Reactivity of Fe(II) species associated with clay minerals Environmental Science and Technology 37 519-528 Hwang I Batchelor B (2000) Reductive dechlorination of tetrachloroethylene by Fe(II) in cement slurries Environmental Science and Technology 34 5017-5022 King GM (1990) Effects of added manganic and ferric oxides on sulfate reduction and sulfide oxidation in intertidal sediments FEMS Microbiology Ecology 73 131-138 Klausen J Troumlber SP Haderlein SB Schwarzenbach RP (1995) Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions Environmental Science and Technology 29 2396-2404 Kuenen JG Robertson LA Tuovinen OH (1992) The genera Thiobacillus Thiomicrospira and Thiosphaera In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 2618-2624
Kukkadapu RK Zachara JM Smith SC Fredrickson JK Liu C (2001) Dissimilatory bacterial reduction of Al-substituted goethite in subsurface sediments Geochimica et Cosmochimica Acta 65 2913-2924 Lee W Batchelor B (2002a) Abiotic reductive dechlorination of chlorinated ethylenes by iron- bearing soil minerals 1 Pyrite and magnetite Environmental Science and Technology 36 5147- 5154 Lee W Batchelor B (2002b) Abiotic reductive dechlorination of chlorinated ethylenes by iron-bearing soil minerals 2 Green rust Environmental Science and Technology 36 5348- 5354 Liger E Charlet L Van Cappellen P (1999) Surface catalysis of uranium (VI) reduction by iron(II) Geochimica et Cosmochimica Acta 63 2939-2955 Lovley DR (1997) Microbial Fe(III) reduction in subsurface environments FEMS Microbiology Reviews 20 305-313 Lovley DR Phillips EJP (1986) Organic matter mineralization with reduction of ferric iron in anaerobic sediments Applied and Environmental Microbiology 51 683-689 Lovley DR Phillips EJP Lonergan DJ (1991) Enzymatic versus nonenzymatic mechanisms for Fe(III) reduction in aquatic sediments Environmental Science and Technology 25 1062-1067
14 Chapter 1 Loyaux-Lawniczak S Refait Ph Lecomte P Ehrhardt J Geacutenin JMR (1999) The reduction of chromate ions by Fe(II) layered hydroxides Hydrology and Earth System Sciences 3 593-599 Lyngkilde J Christensen TH (1992) Redox zones of a landfill leachate pollution plume (Vejen Denmark) Journal of Contaminant Hydrology 10 273-289 Macalady DL Tratnyek PG Grundl TJ (1986) Abiotic reduction reactions of anthropogenic organic chemicals in anaerobic systems A critical review Journal of Contaminant Hydrology 1 1-28 McCulloch A (2002) Trichloroacetic acid in the environment Chemosphere 47 667-686 Mendiboure A Schoumlllhorn A (1986) Formation and anion exchange reactions of layered transition metal hydroxides [Ni1-xMx](OH)2(CO3)x2(H2O)z (M = Fe Co) Revue de Chimie Mineacuterale 23 819-827 Misawa T Hashimoto K Shimodaira S (1974) The mechanism of formation of iron oxide and oxyhydroxides in aqueous solutions at room temperature Corrosion Science 14 131-149 Mulder EG Deinema MH (1992) The sheathed bacteria In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 2618-2624 Myneni SCB Tokunaga TK Brown Jr GE (1997) Abiotic selenium redox transformations in the presence of Fe(IIIII) oxides Science 278 1106-1109 Nealson KH Saffarini D (1994) Iron and manganese in anaerobic respiration Environmental significance physiology and regulation Annual Review of Microbiology 48 311-343 Nielsen A (1976) Hvid groslashn og sort rust Beskrivelse af en korrosionsskade paring et svoslashmmebassin Nordisk Betong 2 21-24 Nriagu JO Dell CI (1974) Diagenetic formation of iron phosphates in recent lake sediments American Mineralogist 59 934-946 OLoughlin EJ Burris DR (2004) Reduction of halogenated ethanes by green rust Environmental Toxicology and Chemistry 23 41-48 OLoughlin EJ Kelly SD Cook RE Csencsits R Kemner KM (2003a) Reduction of uranium(VI) by mixed iron(II)iron(III) hydroxide (green rust) Formation of UO2 nanoparticles Environmental Science and Technology 37 721-727 OLoughlin EJ Kelly SD Kemner KM Csencsits R Cook RE (2003b) Reduction of AgI AuIII CuII and HgII by FeIIFeIII hydroxysulfate green rust Chemosphere 53 437-446 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002a) Iron (IIIII) hydroxycarbonate green rust formation and stabilization from lepidocrocite bioreduction Environmental Science and Technology 36 16-20 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002b) Microbial reduction of lepidocrocite γ-FeOOH by Shewanella putrefaciens The
General Introduction 15
formation of green rust Hyperfine Interactions 139140 231-237 Parmar N Gorby YA Beveridge TJ Ferris FG (2001) Formation of green rust and immobilization of nickel in response to bacterial reduction of hydrous ferric oxide Geomicrobiology Journal 18 375-385 Pecher K Haderlein SB Schwarzenbach RP (2002) Reduction of polyhalogenated methanes by surface-bound Fe(II) in aqueous suspensions of iron oxides Environmental Science and Technology 36 1734-1741 Pepper SE Bunker DJ Bryan ND Livens FR Charnock JM Pattrick RAD Collison D (2003) Treatment of radioactive wastes An X-ray absorption spectroscopy study of the reaction of technetium with green rust Journal of Colloid and Interface Science 268 408- 412 Randall SR Sherman DM Ragnarsdottir KV (2001) Sorption of As(V) on green rust (Fe4(II)Fe2(III)(OH)12SO4
3H2O) and lepidocrocite (γ-FeOOH) Surface complexes from EXAFS spectroscopy Geochimica et Cosmochimica Acta 65 1015-1023 Refait Ph Abdelmoula M Trolard F Geacutenin JMR Ehrhardt JJ Bourrieacute G (2001) Moumlssbauer and XAS study of a green rust mineral the partial substitution of Fe2+ by Mg2+ American Mineralogist 86 731-739 Refait Ph Drissi SH Marie Y Geacutenin JMR (1994) The substitution of Fe2+ ions by Ni2+ ions in green rust one compounds Hyperfine Interactions 90 389-394 Refait Ph Geacutenin JMR (1997) The mechanisms of oxidation of ferrous hydroxychloride β- Fe2(OH)3Cl in aqueous solution The formation of akaganeite vs goethite Corrosion Science 39 539-553 Reimann S Grob K Frank H (1996) Chloroacetic acids in rainwater Environmental Science and Technology 30 2340-2344 Rickert DE (1985) Toxicity of nitroaromatic compounds Hemisphere Publishing Corporation 1-13 Roh Y Lee SY Elless MP (2000) Characterization of corrosion products in the permeable reactive barriers Environmental Geology 40 184-194 Rosenblatt DH Burrows EP Mitchell WR Parmer DL (1991) Organic explosives and related compounds In The Handbook of Environmental Chemistry Anthropogenic compounds Hutzinger O (Ed) Springer-Verlag 195-234 Ruumlgge K Hofstetter TB Haderlein SB Bjerg PL Knudsen S Zraurig C Mosbaeligk H Christensen TH (1998) Characterization of predominant reductants in an anaerobic leachate- affected aquifer by nitroaromatic probe compounds Environmental Science and Technology 32 23-31 Schwertmann U Cornell RM (1991) Iron oxides in the laboratory Preparation and characterization VCH Verlagsgesellschaft mbH Weinheim Simon L Francois M Refait Ph Renaudin G Lelaurain M Geacutenin JMR (2003)
16 Chapter 1 Structure of the Fe(II-III)-layered double hydroxysulphate green rust two from Rietveld analysis Solid State Sciences 5 327-334 Strathmann TJ Stone AT (2003) Mineral surface catalysis of reactions between FeII and oxime carbamate pesticides Geochimica et Cosmochimica Acta 67 2775-2791 Straub KL Benz M Schink B Widdel F (1996) Anaerobic nitrate-dependent microbial oxidation of ferrous iron Applied and Environmental Microbiology 62 1458-1460 Tamaura Y (1985) ZnII-bearing green rust II and its spontaneous transformation into ZnII- bearing ferrite in aqueous solution Bulletin of the Chemical Society of Japan 58 2951-2954 Tamaura Y (1986) Ni(II)-bearing green rust II and its spontaneous transformation into Ni(II)- bearing ferrites Bulletin of the Chemical Society of Japan 59 1829-1832 Taylor RM (1980) Formation and properties of Fe(II)Fe(III)-hydroxycarbonate and its possible significance in soil formation Clay Minerals 15 369-382 Trolard F Abdelmoula M Bourrieacute G Humbert B Geacutenin JMR (1996) Mise en eacutevidence dun constituant de type rouilles vertes dans les sols hydromorphes Proposition de lexistence dun nouveau mineacuteral la fougeacuterite Geacuteosciences de surface Comptes Rendus de LrsquoAcademie des Sciences 323 1015-1022 Tuovinen OH Button KS Vuorinen A Carlson L Mair DM Yut LA (1980) Bacterial chemical and mineralogical characteristics of tubercles in distribution pipelines Journal of the American Water Works Association 72 626-635 Vikesland PJ Valentine RL (2002) Iron oxide surface-catalyzed oxidation of ferrous iron by monochloramine implications of oxide type and carbonate on reactivity Environmental Science and Technology 36 512-519 Widdel F Schnell S Heising S Ehrenreich A Assmus B Schink B (1993) Ferrous iron oxidation by anoxygenic phototrophic bacteria Nature 362 834-836
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 17
2 Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria
Abstract
This work investigated the Fe-containing products formed by anaerobic
autotrophic denitrifying Fe(II)-oxidizing bacteria in a specific bicarbonate buffered
(30 mM HCO3- pH 70) culture media containing 10 mM Fe(II) 4 mM nitrate and
4 mM phosphate Fe(II) was present initially as a whitish vivianite-like
(Fe3(PO4)2middot8H2O) precipitate and as soluble Fe(II) The initial phase of the
oxidation produced a greenish metavivianite-like ((FeII3-xFeIII
x)(PO4)2(OH)xmiddot(8-
x)H2O x gt 12) phase In the late oxidation phase a reddish precipitate of poorly
crystalline goethite (α-FeOOH) dominated the colour of the media in coexistence
with Fe(II)-containing siderite (FeCO3) The increasing amounts of Fe(III) present
in the ldquovivianiterdquo and ldquometavivianiterdquo structures were accompanied by an
increasing intensity in the green colour as the Fe(II) biooxidation progressed This
colour development has produced the idea of biogenic green rusts (layered Fe(II)-
Fe(III) hydroxides) in several studies on nitrate-dependent Fe(II) biooxidation
However in this work no evidence of green rust formation mediated by anaerobic
denitrifying Fe(II)-oxidizing bacteria was obtained
21 Introduction
Aerobic oxidation of Fe(II)-containing minerals by lithotrophic acidophilic and
neutrophilic bacteria has been known for many years but their broad significance
in the biogeochemical cycling of iron has only been recognized over the past two
decades Aerobic Fe(II)-oxidizing bacteria (FeOB) have been isolated from acidic
ecosystems (Thiobacillus ferrooxidans) neutral eutrophic systems (Sphaerotilus
natans Leptothrix ochracea) and neutral oligotrophic systems (Gallionella
ferruginea) (Hanert 1992 Kuenen et al 1992 Mulder amp Deinema 1992) At
neutral pH Fe(II) is unstable in the presence of oxygen and is rapidly oxidized to
the insoluble Fe(III) Hence the only pH neutral environments where soluble
18 Chapter 2
Fe(II) is available for aerobic FeOB are at interfaces between oxic and anoxic
conditions The aerobic neutrophilic FeOB (Leptothrix ochracea Gallionella
ferruginea and Sphaerotilus natans) live at such interfaces and are usually
associated with the yellowishreddish ferric deposits formed there
Over the past several years there has been a growing recognition that other less
readily detectable types of bacteria are involved in Fe(II) oxidation in ecosystems
at circumneutral pH For example it has been reported that neutrophilic FeOB are
abundant at the Loihi seamount hydrothermal vents and play a major role in the
Fe(III) oxide deposition (Emerson amp Moyer 2002) Similarly unidentified
neutrophilic obligate lithotrophic FeOB have been isolated from the rhizosphere of
wetlands plants where they are closely associated with deposits of amorphous
Fe(III) oxides (Emerson et al 1999) It was previously believed that Fe(III) oxide
deposits associated with sheaths were produced biologically whereas Fe(III) oxide
deposits not associated with cells were produced abiotically Recently the
formation of amorphous Fe(III) oxide in gradient tubes has been attributed to the
action of FeOB (Sobolev amp Roden 2001) The authors attribute 90 of the
oxidation to biological processes and indicated that the organisms seem to produce
a mobile form of Fe(III) that diffuses away from the cells before being
precipitated thereby avoiding encrustation of the cells They suggest that such
soluble Fe(III) complexes might be substrates for closely associated Fe(III)-
reducing bacteria Such an arrangement might allow close coupling between
microbial Fe(II) oxidation and Fe(III) reduction within millimeters of the oxic-
anoxic interface
Anaerobic Fe(II) oxidation by phototrophic purple non-sulfur bacteria utilizing
Fe(II) as an electron donor in the light was recognized only a decade ago (Widdel
et al 1993) Subsequently it was demonstrated that the biological oxidation of
Fe(II) in the absence of oxygen is possible by light-independent chemotrophic
microbial activity using nitrate as the electron acceptor (Straub et al 1996) In
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 19
addition studies conducted in gradient cultures revealed that nitrate-reducing
strains could also oxidize Fe(II) with molecular oxygen (Benz et al 1998) Hence
these Fe(II)-oxidizing strains may use nitrate as well as oxygen as electron
acceptors The microbial oxidation of Fe(II) was coupled to stoichiometric
reduction of nitrate to N2 and only one strain produced traces of N2O as a by-
product (Straub et al 1996 Benz et al 1998) The authors proposed the formation
of 2-line ferrihydrite as the end product of Fe(II) biooxidation The chemical
reduction of nitrate by Fe(II) requires a catalyst eg at least 10 microM Cu2+ in order
to take place at significant rates and may thus be considered insignificant under the
conditions applied in our study (Moraghan amp Buresh 1976) The chemical
oxidation of Fe(II) with nitrous oxide has not been observed However nitrite can
oxidize Fe(II) chemically (Moraghan amp Buresh 1977 Straub et al 1996) but this
process is considered insignificant at the conditions applied here No denitrifying
Fe(II)-oxidizing enrichment culture has been found to produce ammonium from
nitrate
Both lithoheterotrophic (depending on organic cosubstrates such as acetate) and
strictly lithoautotrophic nitrate-reducing FeOB have been found in various marine
and freshwater sediments However most isolates depend on organic cosubstrates
for cell biosynthesis (Benz et al 1998) Most probable number estimations
showed that denitrifying FeOB accounted for 00006-08 of the acetate-oxidizing
denitrifying microbial population Lithotrophic FeOB accounted for less than
00001 of the total bacterial community Attempts to isolate CO2-fixing nitrate-
dependent FeOB from lithotrophic cultures have failed (Straub amp Buchholz-
Cleven 1998) Mixotrophic FeOB accounted for 0004-004 of the total bacterial
community In addition microbial nitrate-dependent Fe(II) oxidation was
demonstrated in a flooded paddy soil as well as in activated sludge from a
wastewater treatment plant (Nielsen amp Nielsen 1998 Ratering amp Schnell 2001)
Since the activity is not restricted to sunlight exposed habitats microbial nitrate-
dependent Fe(II) oxidation is supposedly more important on a global scale than
20 Chapter 2
anaerobic Fe(II) oxidation by phototrophic bacteria Furthermore it has been
reported that anaerobic denitrifying FeOB aptly oxidize biogenic Fe(II) minerals
formed by bioreduction of synthetic goethite and ferrihydrite and that anaerobic
Fe(III)-reducing bacteria readily reduce Fe(III) minerals formed by biooxidation of
Fe(II) (Weber et al 2001 Straub et al 1998) Hence autotrophic denitrifying
FeOB may play a significant role in the nitrogen and iron cycles in subsurface
environments where the nitrate and the Fe(II) zones overlap and organic carbon
supply is limited (Figure 21)
Figure 21 The microbial iron cycle linking the carbon and nitrogen cycles
Phosphate is released into the environment through natural processes such as rock
weathering and decomposition of dead organic material and anthropogenic
activities eg wastewater effluents and application of manure and fertilizers in
horti- and agriculture In anoxic soils and sediments phosphate may be sequestered
by sorption onto Fe(III) oxides (Williams et al 1971 Patrick amp Khalid 1974)
Phosphate strongly influences the type morphology and properties of Fe(III)
oxides formed by oxidation and hydrolysis of Fe(II) salts as well as the degree of
their transformation (Kandori et al 1992 Cumplido et al 2000 Benali et al
2001) Phosphate may also be retained by precipitation of Fe(II) phosphates such
as the monoclinic vivianite (Fe3(PO4)2middot8H2O) which is the most important stable
Fe(II) orthophosphate solid encountered in the subsurface under most conditions
(Nriagu 1972) At anoxic and circumneutral conditions the whitish vivianite
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 21
controls the Fe(II) activity even at very low phosphate concentrations (Nriagu amp
Dell 1974) Vivianite occurs as a secondary mineral in the gossans of metallic ore
deposits and as a weathering product of primary iron-manganese phosphates in
pegmatites (Gaines et al 1997) Moreover natural vivianite occurrences have
been identified in a number of lake and river sediments (Zwaan amp Kortenbout van
der Sluys 1971 Nriagu amp Dell 1974 Postma 1981 Nembrini et al 1983
Henderson et al 1984 Dodd et al 2003 House 2003 and references therein)
Vivianite is also found in sewage sludge as a result of the wastewater treatment
where iron salts are added in order to remove phosphate (Seitz et al 1973) It is
however still indefinite how ubiquitous vivianite is in nature Furthermore only
little is known about the mechanism of vivianite formation and the role played by
sedimentary Fe(III) oxides Anaerobic Fe(III)-reducing microorganisms may
reduce Fe(III) oxides thereby releasing the iron as soluble Fe(II) and mobilizing
the phosphate adsorbed to the Fe(III) oxides (Lovley 1997) It has been suggested
that vivianite is formed by precipitation following reductive dissolution of Fe(III)
oxides (Manning et al 1981 Manning amp Jones 1982) However it has also been
proposed that the transformation of Fe(III) oxides to vivianite occurs topotactically
and not via reductive dissolution (Nembrini et al 1983) Vivianite was shown to
form microbially as a result of the activity of the anaerobic Fe(III)-reducing
bacteria Shewanella putrefaciens in the presence of high Fe(III)-citrate and
phosphate concentrations (Jorand et al 2000) Moreover vivianite formation by
bioreduction of Fe(III) in hydrous ferric oxide and in smectite has been reported
(Fredrickson et al 1998 Dong et al 2003)
Only little is known about the oxidation products of vivianite Metavivianite a
greenish triclinic iron hydroxy phosphate mineral was first described by Ritz et al
(1974) and it was later found to coexist with vivianite in several natural sediment
samples (Henderson et al 1984) Once the Fe(III) content became evident the true
composition of metavivianite ((FeII3-xFeIII
x)(PO4)2(OH)x
22 Chapter 2
middot(8-x)H2O x gt 12)) was established (Rodgers amp Johnston 1985 Rodgers 1986
and references therein) The formation of intermediate greenish precipitates during
oxidation of fluffy colourless Fe(II) precipitates by anoxic phototrophic
microorganisms and nitrate-dependent FeOB have been reported (Ehrenreich amp
Widdel 1994 Chaudhuri et al 2001) Since both studies were conducted in
bicarbonate buffered mineral media (22-30 mM HCO3ndash pH 70-72) containing
37-5 mM phosphate we assume that the initial fluffy whitish precipitates
consisted mainly of vivianite Chaudhuri et al (2001) proposed that the
intermediate green phases produces by the denitrifying FeOB consist of carbonate
green rust (GR-CO3) but no convincing evidence of this biogenic GR-CO3 has been
provided yet The major objective of this work was to examine the Fe-containing
products forming during the course of biooxidation of vivianite by non-
phototrophic anaerobic denitrifying Fe(II)-oxidizing bacteria
2 2 Materials and methods
All handling and sampling of solutions and suspensions were carried out under
sterile and strict anoxic conditions All chemicals were pa quality
221 Microorganisms and media
Enrichment cultures of nitrate-reducing FeOB taken from town ditches (Bremen
Germany) were grown in anoxic bicarbonate-buffered (30 mM HCO3ndash 90
N210 CO2 pH 70) mineral media containing 4 mM phosphate as well as
essential trace elements and vitamins (Table 21 Straub amp Buchholz-Cleven
1998) Ammonium was omitted from the media in order to facilitate detection of
ammonium possibly produced by reduction of nitrate The techniques used for
preparation of media and cultivation of bacteria under anoxic conditions have been
described by Widdel amp Bak (1992) 05 M aqueous stock solutions of FeCl2 or
FeSO4 were prepared in 100 mL glass flasks by reacting 65 mmol of iron powder
(particle size 10 microm Merck) with 100 mL deoxygenated 10 M HCl or 05 M
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 23
H2SO4 respectively The solutions were magnetically stirred and heated (~80degC)
during reaction until the H2(g) production had ceased (ge 1 hour) The FeCl2 and
FeSO4 stock solutions were stored under a small Ar overpressure at 5degC
Table 21 Composition of the mineral medium (adopted from Straub amp Buchholz-Cleven
(1998))
Components Concentration (M) KH2PO4 15middot10-3
K2HPO4 25middot10-3
MgSO4middot7H2O 10middot10-3
CaCl2middot2H2O 50middot10-4
H3BO3 56middot10-5
ZnSO4middot7H2O 10middot10-6
Na2MoO4middot2H2O 40middot10-6
CuSO4middot5H2O 20middot10-7
MnSO4middotH2O 10middot10-6
Na2SeO4 12middot10-5
CoCl2middot6H2O 50middot10-6
NiCl2middot6H2O 80middot10-6
NaCl 10middot10-5
NaHCO3ndash 30middot10-2
Cyanocobalamine (vitamin B12) 37middot10-8
p-aminobenzoic acid (vitamin Hrsquo) 36middot10-7
D(+)-biotin (vitamin H) 41middot10-8
Nicotinic acid (Niacin) 81middot10-7
Ca-D(+)-pantothenate (vitamin B5) 52middot10-8
Pyridoxamine dihydrochloride 96middot10-7
Thiaminechloridehydrochloride (vitamin B1) 15middot10-7
NaNO3 40middot10-3
FeSO4 or FeCl2 0010
222 Characterisation of precipitates
In order to optimize the characterization and distinction between the spectral
components transmission Moumlssbauer spectra were obtained at temperatures
between 5 K and 250 K and in external magnetic fields of 4 T (parallel to the γ-ray
direction) using a conventional constant acceleration spectrometer and a source of 57Co in Rh The spectrometer was calibrated using a 125 microm foil of α-Fe at room
temperature and isomer shifts are given relative to the centroid of the spectrum of
this absorber The spectra were fitted using simple Lorentzian line shape Infrared
(IR) spectra were obtained using a Perkin Elmer FT-IR 2000 spectrometer and the
24 Chapter 2
KBr pellet technique Scanning electron microscopy (SEM) was carried out in
order to study the morphology and composition of the precipitates Specimens for
SEM were prepared by depositing suspended particles onto an aluminum stub
coated with a carbon sticker The stub was quickly transferred into a sputtering
chamber and coated with a thin Pt film (~20 nm) In order to avoid interfering Pt
signals in the energy dispersive spectra the stubs were in some cases not coated
with Pt but quickly transferred to the SEM chamber for evacuation Measurements
were performed using a Philips XL30 equipped with a LaB6 source and an
accelerating voltage of 20 kV and an EDAX eDXi X-ray dispersive spectrometer
223 Biooxidation experiments
The biooxidation experiments were conducted in 50-400 mL butyl rubber
stoppered bottles with a 90 N210 CO2 headspace constituting 10 of the total
volume Prior to inoculation 4 mM NaNO3 was added as the electron acceptor and
10 mM Fe2+ (as chloride or sulphate) as the electron donor to the mineral media
Control experiments were performed in the same media only they were not
inoculated Addition of ferrous iron to the media induced an immediate
precipitation of a solid whitish material The whitish precipitate was collected on
022 microm polyvinylidendifluorid (Durapore Millipore) filters and stored in an
anoxic atmosphere until further measurements Old outgrown media suspensions
that had turned reddish in color due to a precipitate produced by the denitrifying
FeOB were used as inocula Inoculum volume was kept lt1 of the culture volume
in order to prevent the reddish inoculum from dominating over the initial whitish
precipitate Cultures were incubated in the dark at room temperature and gently
agitated once every day Typical color successions for the media were initial
whitish precipitates turning first more and more greenish over time and then finally
turning reddish (see Fig 22) The color developed uniformly without any
indications of multiple phases in the precipitate At different time intervals
suspension samples were withdrawn using 90 N210 CO2-flushed polyethylene
syringes The precipitates were collected on 022 microm polyvinylidendifluorid
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 25
(Durapore Millipore) filters and analyzed by Moumlssbauer spectroscopy and SEM
Nonfiltered suspension samples were digested in 01 M HCl and chemically
analyzed for Fe(II) NO3- and NH4
+
224 Analytical methods
Fe2+ was determined using a modified phenanthroline method (Fadrus amp Maly
1975) Nitrate was quantified by ion chromatography (Morales et al 2000) and
ammonium was measured photometrically using the indophenol reaction (Rossum
amp Villarruz 1963)
23 Results and discussion
231 Identification of solid iron-containing phases
In most cases the mineral media for cultivating denitrifying FeOB contained 10
mM FeCl2 or FeSO4 4 mM NO3ndash 4 mM total phosphorus and 30 mM HCO3
- at pH
70 ([HCO3-] = 2138middot[CO3
2-] at pH 70) Whitish flocs precipitated immediately at
these initial conditions when Fe(II) was added to the media (Figure 22a) Such
colourless flocs have been reported to precipitate in similar mineral media (10 mM
Fe(II) 37-5 mM phosphate 22-30 mM HCO3ndash pH 70-72) (Ehrenreich amp
Widdel 1994 Chaudhuri et al 2001) Our Fe(II) measurements showed that 20-
50 of the total Fe(II) added was present in this initial white precipitate
26 Chapter 2
a
210- Figure 22 Colour of suspended material in the growth media during Fe(II) biooxidation a)
Initial whitish precipitate prior to inoculation b) Inte1mediate greenish phase fo1med within 2-3
days after inoculation c) reddish precipitate at late stage of biooxidation (gt5-6 days)
The precipitates were filtered and investigated by Mossbauer and IR spectroscopy
The transmission Mossbauer spectra obtained for the initial whitish precipitate at
temperatures between 20 and 250 K are shown in Figure 23 The spectrum
measured at 250 K consists of two fairly well-resolved Fe(II) doublets (see
parameters in Table 22) The change in line-overlap with decreasing temperature
is primarily ascribed to differences in the temperature dependence of the
quadrupole splitting of the two components From the spectra at 10 and 6 K
(Figure 24) it can be concluded that magnetic ordering takes place between these
two temperatures and that only one transition occurs (indicating the presence of
only one phase) The parameters of one of the Fe(II) doublets at 250 K (designated
B in Table 22) are in very good agreement with previously published values for
the vivianite Fe(Il)8 site at room temperature (eg McCammon amp Burns 1980)
and the ordering temperature also agrees well with an assignment as vivianite
(Forsyth et al 1970) However the second Fe(II) doublet in the initial colourless
precipitate (Table 22) has parameters that deviate from previously reported values
by having a smaller quadrupole splitting (indicating a less distorted coordination)
and a significantly higher relative intensity and line width These effects might be
due to the presence of numerous defects in the vivianite crystal lattice particularly
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 27
affecting the Fe(II)A sites It should be noted that further components may be added
to the fit in order to improve its statistics Nevertheless we decided to include no
further components as suggested by the finding of one magnetic ordering only
Accordingly our interpretation of the Moumlssbauer results for the initial white
precipitate suggests a highly defective vivianite having a distribution of local
coordination environments particular in the A site This assignment is further
supported by a major absorption band due to phosphate anions in the infrared
spectrum at approximately 1000 cm-1 and the absence of other complex anions
(data not shown) Thus the whitish precipitate is referred to as a vivianite-like
(ldquovivianiterdquo) precipitate
0
3 5
3 0
2 5
2 0
1 5
1 0
0 5
0 0
20 K
Velocity (mms)
Rel
ativ
e ab
sorp
tion
()
3
2
1
0
80 K
3 5
3 0
2 5
2 0
1 5
1 0
0 5
0 0
150 K
-5 -4 -3 -2 -1 0 1 2 3 4 5
2 5
2 0
1 5
1 0
0 5
0 0
250 K
Figure 23 Transmission Moumlssbauer spectra measured between 250 and 20 K of the initial
whitish precipitate prior to inoculation (see Fig 22a) Fitting components (and their sum) are
shown as full lines
28 Chapter 2
Table 22 Selected Mossbauer parameters of the doublet components in the spectra obtained for
different precipitates
Precipitates Temperature Isomer shift Quadrupole Line width Area (K) (mms-1) splitting (mms-1) (mms-1) ()
Whitish Fe(II)B 250 127 309 035 38
Fe(II)A 250 128 181 051 62
Dark greenish Fe(II)B 250 126 305 023 17
Fe(II)A 250 132 238 051 44
Fe(III) 250 036 085 040 38
Reddish-orange 40 138 244 094 67 Fe(II) Hyperfine parameters are generally given with uncertainties of 003 mms- the spectral area with an uncertainty of 3
1005
1000
0995
0990
- 0985 ~ e c 0980 0
-~ 0975 E c nl b 1000 g ~ Qi 0995 0
0990
0985
0980 -12 -8
~ -~
bullbull bull Ibull bull bullbull bull bull ~ bull bull ~
It
10 K
bull bull bull bull (J ~ i~ 6K bullmiddotf bull bull
~ bull bull bull
-4 0 4 8 12
Velocity (mmls)
Figure 24 Transmission Mossbauer spectra measured at 10 and 6 K of the initial whitish
precipitate prior to inoculation (see Fig 22a)
In general the color of the media suspension changed from whitish into light green
color within 2-3 days after inoculation (Figure 22b ) This transformation occurred
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 29
without dissolving the initial whitish precipitate or preserving the whitish
precipitate as a separate phase implying some kind of solid state transformation
Two identical media suspensions were inoculated simultaneously but at the
sampling time they had distinctly different intensities of the green color
designated light and dark green respectively Moumlssbauer spectra of the dark green
sample are shown in Figure 25 The spectra of this sample are all fitted using three
doublet components (two Fe(II) and one Fe(III)) and the parameters of the
spectrum measured at 250 K are given in Table 22
30
25
20
15
10
05
00
20 K
Velocity (mms)
Rel
ativ
e ab
sorp
tion
()
35
30
25
20
15
10
05
00
80 K
30
25
20
15
10
05
00
150 K
-5 -4 -3 -2 -1 0 1 2 3 4 5
25
20
15
10
05
00
250 K
Figure 25 Transmission Moumlssbauer spectra measured between 250 and 20 K of the dark
greenish precipitate formed during biooxidation Fitting components (and their sum) are shown
as full lines
No magnetic ordering of the dark green precipitate was observed at temperatures
above 20 K but ordering occurred around 10 K (not shown - due to very thin
30 Chapter 2
samples this was not investigated in details) The two greenish samples had very
similar parameters only differing in the relative intensity of Fe(III) 26 and 38
in the light greenish and dark greenish samples respectively Assuming the
spectral area of a component to be proportional to the abundance of the species in
the solid these results indicate a correlation between the intensity of the green
color and the content of Fe(III) in the precipitate The parameters of the Fe(II)
doublets in the dark green precipitate (Table 22) were in very good agreement
with previously published values for vivianite with a non-negligible Fe(III) content
(McCammon amp Burns 1980) whereas the Fe(III) component in particular had a
higher quadrupole splitting The observation that magnetic ordering of both Fe(II)
and Fe(III) occurred at similar temperature for the light green phase (Figure 26) is
a strong indication that they both belong to the same phase The absence of the
component with the low quadrupole splitting in the spectra of both green samples
might indicate that the initial vivianite-like phase crystallized into a more well-
defined vivianite over time However freshly prepared and long-term aged (gt1
year) suspensions of the initial vivianite-like precipitate did not differ significantly
Hence we suggest that the recrystallization of the vivianite-like precipitate can be
explained by Fe(II) biooxidation
Solid State Oxidation ofFe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria
0
2
3
- 4
~ 5 c ~ 6 e-0 7 -2 nl Q) 00 gt ~ 05 Qi 0 10
15
20
25
30
35
bullbullbull hi 6~
bullbullbullbull bull bull bullbull bull 10 K bull bull
bull bull bull bull bull bull bull
~~ ~~ lf 6K bull bull bullbull middot bull bull r bull
bull bull bull bull bull bullbull bull bull bull - 12 -a -4 0 4 8 12
Velocity (mms)
31
Figure 26 Transmission Mossbauer spectrn measured at 6 and 10 K of the light greenish
precipitate fonned during biooxidation
The solid state oxidation of monoclinic vivianite to triclinic metavivianite is well-
known (McCammon amp Bums 1980 Pratesi et al 2003 and references therein)
The vivianite crystal structure contains Fe(II) ions in both isolated Fe(II)A and
paired Fe(Il)8 octahedra Mossbauer spectroscopic analyses have shown that the
Fe(Il)8 Fe(II)A ratio increases with increasing Fe(III) concentration suggesting that
the remaining Fe(II)A ions are more readily oxidized than the Fe(II)a ion of an
Fe(Il)8 -Fe(III)8 pair (McCammon amp Bums 1980) The mechanism of oxidation of
Fe(II) in vivianite involves conversion of H20 ligands to OH- ions producing a
progressive collapse of the vivianite structure due to the elimination of hydrogen
bonds (Moore 1971) The exact oxidation limits between which the triclinic lattice
is stable are somewhat disputed as the results obtained for synthetic and natural
vivianites oxidized chemically in the laboratory and naturally oxidized natural
vivianite specimens do not agree completely (Rodgers 1986 and references
therein) Taking all reports into account the monoclinic structure of vivianite is
supposedly maintained until 40-50 of total iron is oxidized Further oxidation
32 Chapter 2
leads to the formation of the triclinic metavivianite in which the FeA site is fully
oxidized whereas the oxidation of the FeB ranges from 20 to almost 100 Thus
the triclinic metavivianite structure persists close to complete oxidation of total
iron The Moumlssbauer results obtained in this study are consistent with the vivianite
solid state oxidation mechanism reported by McCammon amp Burns (1980) Thus
we propose that the intermediate greenish precipitate is a metavivianite-like
(ldquometavivianiterdquo) phase It should be noted that a minor oxidation of dissolved
Fe(II) may have occurred even though the solid state oxidation of Fe(II) was
predominant
Within 5-6 days after inoculation the greenish intermediate was transformed into a
reddish product (Figure 22c) The magnetically ordered sextet in the spectrum of
the red phase (Figure 27) measured at 40 K was due to goethite (α-FeOOH)
(magnetic hyperfine field of 470 T and a quadrupole shift of -01 mms-1 cp
Moslashrup et al 1983) The sextet deviated from ideal goethite by its asymmetric line
shape and its low ordering temperature (around 100 K ndash data not shown) and thus
the goethite was poorly crystalline It is very likely that the presence of phosphate
in the media retarded the crystal growth of goethite The unusual reddish colour of
the goethite might also be explained by the presence of phosphate The spectrum at
40 K was however dominated by a Fe(II) doublet (Table 22) that ordered
magnetically between 40 and 20 K (Figure 27) The hyperfine parameters and the
magnetic ordering temperature indicated that this component was due to siderite
(FeCO3) having a magnetic ordering temperature of 38 K (Jacobs 1963) The
siderite component may have formed as a result of the microbial activity changing
the chemistry of the solution and precipitating a major part of the remaining
dissolved Fe(II) at this stage The characteristic vivianite Fe(II) doublet with large
quadrupole splitting was not detected in this sample The reddish precipitate
contained considerably less Fe(III) than the greenish precipitate (only 33 as
estimated from the spectral area) None of the components in the reddish sample
Solid State Oxidation ofFe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 33
could be detected in freshly inoculated samples indicating that carryover of mineral
precipitates by inoculation of the culture media was negligible
0
2
- 3
~4 c
Q 5
e 6 0
~ 7 g
0 3l Q)
a 1
2
3
4
5
-12 a
40K
bull bull (
4 0 4 8 12 Velocity (mms)
Figure 27 Transmission Mossbauer spectra measured at 20 and 40 K of the reddish precipitate
fo1med during the late biooxidation stage Fitting components (and their sum) are shown as full
lines
Strengite (FeP04middot2H20) was not detected at any time during oxidation
Santabarbaraite a new amorphous F e(III) hydroxy phosphate mineral
(Fe3(P04)i(OH)3middot5H20) was reported in a recent study (Pratesi et al 2003) The
brownish mineral was a result of the solid state oxidation of vivianite through
metavivianite However no Mossbauer data have been provided for this new
mineral yet and therefore we cannot give an account of whether santabarbaraite
forms in our system or not Thus goethite was the dominating end product and we
propose the reaction path depicted in Figure 28 for the nitrate-dependent
biooxidation of Fe(II) in our systems
34 Chapter 2
Fe3(PO4)2middot8H2O (FeII3-xFeIII
x)(PO4)2(OH)xmiddot(8-x)H2O α-FeOOH
NO3- N2 NO3
- N2
ldquoVivianiterdquo ldquoMetavivianiterdquo Goethite
Figure 28 Proposed reaction path and iron-containing minerals forming during solid state
oxidation of vivianite by denitrifying FeOB at the experimental conditions applied in this study
The biotic formation of layered Fe(II)-Fe(III) hydroxides (green rusts) by
anaerobic denitrifying Fe(II)-oxidizing bacteria has been suggested but proper
identification of these phases still lacks (Chaudhuri et al 2001) We cannot rule
out that small amounts of green rusts (GRs) perhaps a phosphate intercalated GR
(Hansen amp Poulsen 1999) might have been present here during the greenish
intermediate ldquometavivianiterdquo oxidation stage When present in low concentrations
especially in mixtures including other iron minerals it is very difficult to identify
GRs even with Moumlssbauer spectroscopy At least two complementary methods
such as X-ray diffraction (XRD) and Moumlssbauer spectroscopy are required for
proper identification and characterization of GRs However the precipitates
collected in this work were poorly crystalline and did not allow for XRD analysis
Electron micrographs including energy dispersive X-ray spectroscopy suspension
colour and mineral stability calculations do not suffice as evidence Hence no
convincing evidence of GR formation facilitated by denitrifying FeOB has been
provided so far The blue-green colours of metavivianite and green rust minerals
originate from Fe(II)-Fe(III) charge transfer between adjacent Fe(II) and Fe(III)
ions in edge-shared octahedra (Faye et al 1968) The greenish suspension colour
occurring during the intermediate phase has incited the idea of biogenic GRs in
studies on nitrate-dependent Fe(II) biooxidation (Chaudhuri et al 2001 Lack et
al 2002aampb) However our results indicate that this reasoning is misleading
232 Factors controlling the rate and extent of Fe(II) biooxidation
Generally it was found that maximally 20-64 of the initial Fe(II) amount was
oxidized to Fe(III) (Figure 29) This indicates some limitations in the accessibility
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 35
of Fe(II) in the system Based solely on stoichiometry considerations the
microorganisms are expected to oxidize 5 mol Fe(II) for every 1 mol nitrate
reduced to dinitrogen However as exemplified in Figure 28 this ratio was mostly
lt4 which can be explained by the consumption of nitrogen as a result of microbial
growth At initial [Fe(II)][NO3ndash] ratios lt5 nitrate is in excess and should not limit
the extent of the biooxidation Thus the lack of complete biooxidation could not be
due to exhaustion of nitrate Furthermore all growth essential nutrients were more
than sufficiently applied hence the incomplete Fe(II) biooxidation was not caused
by lack of nutrients The most reasonable explanation therefore seems to be that an
increasingly limited access to the electron donor over time inhibits complete long-
term Fe(II) biooxidation At least four mechanisms may cause this inhibition 1)
the Fe(II) becomes isolated within the structure of the mixed Fe(II)-Fe(III)
minerals forming during biooxidation or underneath a passive Fe(III)-bearing
surface film on the initial Fe(II) precipitates 2) the FeOB cell surface becomes
covered with a passive Fe(III)-bearing surface film 3) the Fe(II) biooxidation is
controlled by the rate of dissolution of the initial Fe(II) minerals or 4) the reaction
proceeds primarily by biooxidation of dissolved Fe(II) whose concentration
gradually decreases due to changes in solid phase composition The actual
mechanisms whereby the surface-associated Fe(III) can inhibit Fe(II) biooxidation
are unknown but they may involve both kinetic and thermodynamic constraints on
the electron transfer The Moumlssbauer results obtained in this work strongly suggest
that the Fe(II) biooxidation occurred mainly in the solid state of the initial
ldquovivianiterdquo phase However we cannot rule out that some dissolved Fe(II) was
oxidized as well
36 Chapter 2
Figure 29 Concentration profiles of total Fe(II) and nitrate as a function of time during Fe(II)
biooxidation
No Fe(II) oxidation took place in cultures where nitrate had been omitted
confirming that the microbial Fe(II) oxidation is nitrate-dependent (data not
shown) No Fe(II) oxidation was detected in the non-inoculated control
experiments within the duration of the experiments and thus the chemical
oxidation of dissolved Fe(II) by nitrate catalyzed by vitamins or trace elements
(eg Cu(II)) can be neglected Ammonium did not form in detectable amounts
during Fe(II) biooxidation (data not shown) and therefore dinitrogen was assumed
to be the end product as reported previously (Straub et al 1996 Benz et al 1998)
The absence of ammonium formation does indirectly support the absence of
biologically induced green rust formation as synthetic green rust is known to
convert nitrate into ammonium in purely chemical reactions (Hansen et al 1996)
It was visually observed that the phosphate concentration in the media exerted a
control on the microbial Fe(II)-oxidation At phosphorus concentrations le 2 mM
no Fe(II)-oxidation took place However the solubility product for vivianite (Ksp =
171middot10-36 at 25degC (Al-Borno et al 1994)) was still by far exceeded under these
conditions It is not known whether this phosphate limiting effect is due to growth
constraints in the mixed bacterial community or whether specific Fe(II) phosphate
Solid State Oxidation ofFe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 37
precipitates are prerequisites of the Fe(II) biooxidation to take place Experiments
are currently underway in our laboratory in order to elucidate the role of specific
initial Fe(II) precipitates It should be noted that the growth of the denitrifying
FeOB could not be estimated as they were present in highly heterogeneous
suspensions containing both solid iron phases as well as other bacteria (enrichment
culture)
233 Morphology of solid iron phases
The morphology of the various precipitates was studied by SEM The initial
whitish precipitate consisted of a web-like structure (Figure 21 Oa and background
in Figure 21 Ob) whereas more distinct hexagonally shaped rosettes with particle
size ~20 microm (Figure 2lObampc) formed during Fe(II) biooxidation The energy
dispersive spectroscopic element analyses showed that other than iron the initial
whitish precipitate and the rosettes contained mainly phophorus
Figure 210 Scanning electron micro graphs of precipitates fo1med at various stages of the
experiment a Initial whitish precipitate bampc Rosettes observed in the intennediate greenish
precipitate d Reddish precipitate sampled during the late biooxidation stage
38 Chapter 2
These observations are interpreted as vivianite forming a web-like morphology in
the initial whitish precipitate and partly transforming into hexagonal particles in
the greenish colored stage The interpretations are supported by similar vivianite
morphologies reported including pseudo-hexagonal vivianite crystals of low
symmetry resulting from microbial Fe(III) reduction of HFO and platy rosettes of
vivianite crystals formed during bioreduction of Fe(III) in smectite (Fredrickson et
al 1998 Dong et al 2003) It was not possible to associate the morphology
observed in the reddish precipitate with the minerals identified in this phase
(Figure 210d)
24 Conclusions
This work demonstrated that anaerobic autotrophic denitrifying Fe(II)-oxidizing
bacteria produce poorly crystalline goethite by solid state oxidation of ldquovivianiterdquo
via a ldquometavivianiterdquo intermediate The increasing amount of Fe(III) forming in the
vivianite structure was accompanied by an increasing intensity in the green colour
as the Fe(II) biooxidation progressed Moumlssbauer spectroscopic analyses provided
no significant evidence of green rust formation The finding of microbially
oxidized vivianite in this study raises the question of the oxidation state of
vivianite specimens from natural sediments Vivianite is generally believed to be
an ideal Fe(II) hydroxy phosphate mineral and the presence of Fe(III) is explained
by aerial oxidation upon sampling The results presented here indicate that
microbiological processes may be responsible for the oxidation of vivianite and
metavivianite in natural subsurface environments Acknowledgments
We would like to thank Dr K Straub for providing and advising us how to culture the nitrate-
reducing FeOB Furthermore we thank Dr C B Koch for performing the Moumlssbauer analyses
and Dr D Mavrocordatos for performing the SEM analyses
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 39
References Al-Borno A Tomson MB (1994) The temperature dependence of the solubility product constant of vivianite Geochimica et Cosmochimica Acta 58 5373-5378 Benali O Abdelmoula M Refait Ph Geacutenin JMR (2001) Effect of orthophosphate on the oxidation products of Fe(II)-Fe(III) hydroxycarbonate The transformation of green rust to ferrihydrite Geochimica et Cosmochimica Acta 65 1715-1726 Benz M Brune A Schink B (1998) Anaerobic and aerobic oxidation of ferrous iron at neutral pH by chemohetorotrophic nitrate-reducing bacteria Archives of Microbiology 169 159-165 Chaudhuri SK Lack JG Coates JD (2001) Biogenic magnetite formation through anaerobic biooxidation of Fe(II) Applied and Environmental Microbiology 67 2844-2848 Cumplido J Barron V Torrent J (2000) Effect of phosphate on the formation of nanophase lepidocrocite from Fe(II) sulfate Clays and Clay Minerals 48 503-510 Dodd J Large DJ Fortey NJ Kemp S Styles M Wetton P Milodowski A (2003) Geochemistry and petrography of phosphorus in urban canal bed sediment Applied Geochemistry 18 259-267 Dong H Kostka JE Kim J (2003) Microscopic evidence for microbial dissolution of smectite Clays and Clay Minerals 51 502-512 Ehrenreich A Widdel F (1994) Anaerobic oxidation of ferrous iron by purple bacteria a new type of phototrophic metabolism Applied and Environmental Microbiology 60 4517-4526 Emerson D Moyer CL (2002) Neutrophilic Fe-oxidizing bacteria are abundant at the Loihi seamount hydrothermal vents and play a major role in Fe oxide deposition Applied and Environmental Microbiology 68 3085-3093 Emerson D Weiss JV Megonigal JP (1999) Iron-oxidizing bacteria are associated with ferric hydroxide precipitates (Fe-plaque) on the roots of wetland plants Applied and Environmental Microbiology 65 2758-2761 Fadrus H Maly J (1975) Suppression of iron(III) interference in the determination of iron(II) in water by the 110-phenanthroline method The Analyst 100 549-554 Faye GH Manning PG Nickel EH (1968) The polarized optical absorption spectra of tourmaline cordierite chloritoid and vivianite ferrous-ferric electronic interaction as a source of pleochroism American Mineralogist 53 1174-1201 Forsyth JB Johnson CE Wilkonson C (1970) The magnetic structure of vivianite Fe3(PO4)2middot8H2O Journal of Physics Part C Solid State Physics 3 1127-1139 Fredrickson JK Zachara JM Kennedy DW Dong H Onstott TC Hinman NW Li S (1998) Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium Geochimica et Cosmochimica Acta 62 3239-3257 Gaines RV Skinner HCW Foord EE Mason B Rosenzweig A (1997) Danas new
40 Chapter 2 mineralogy 8th ed John Wiley amp Sons Inc Hanert HH (1992) The genus Gallionella In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 4082-4088 Hansen HCB Koch CB Nancke-Krogh H Borggaard OK Soerensen J (1996) Abiotic nitrate reduction to ammonium Key role of green rust Environmental Science and Technology 30 2053-2056 Hansen HCB Poulsen IF (1999) Interaction of synthetic sulphate green rust with phosphate and the crystallization of vivianite Clays and Clay Minerals 47 312-318 Henderson GS Black PM Ridgers KA Rankin PC (1984) New data on New Zealand vivianite and metavivianite New Zealand Journal of Geology and Geophysics 27 367-378 House WA (2003) Geochemical cycling of phosphorus in rivers Applied Geochemistry 18 739-748 Jacobs IS (1963) Metamagnetism of siderite (FeCO3) Journal of Applied Physics 34 1106-1107 Jorand F Appenzeller BMR Abdelmoula M Refait Ph Block J-C Geacutenin JMR (2000) Assessment of vivianite formation in Shewanella putrefaciens culture Environmental Technology 21 1001-1005 Kandori K Uchida S Kataoka S Ishikawa T (1992) Effects of silicate and phosphate ions on the formation of ferric oxide hydroxide particles Journal of Materials Science 27 719-728 Kuenen JG Robertson LA Tuovinen OH (1992) The genera Thiobacillus Thiomicrospira and Thiosphaera In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 2618-2624 Lack JG Chaudhuri SK Chakraborty R Achenbach LA Coates JD (2002a) Anaerobic biooxidation of Fe(II) by Dechlorosoma suillum Microbial Ecology 43 424-431 Lack JG Chaudhuri SK Kelly SD Kemner KM OConnor SM Coates JD (2002b) Immobilization of radionuclides and heavy metals through anaerobic bio-oxidation of Fe(II) Applied and Environmental Microbiology 68 2704-2710 Lovley DR (1997) Microbial Fe(III) reduction in subsurface environments FEMS Microbiology Reviews 20 305-313 Manning PG Birchall T Jones W (1981) Ferric hydroxides in surficial sediments of the great lakes and their role in phosphorus availability a Moumlssbauer spectral study Canadian Mineralogist 19 525-530 Manning PG Jones W (1982) The binding capacity of ferric hydroxides for non-apatite inorganic phosphorus in sediments of the depositional basins of Lakes Erie and Ontario Canadian Mineralogist 20 169-176 McCammon CA Burns RG (1980) The oxidation mechanism of vivianite as studied by Moumlssbauer spectroscopy American Mineralogist 65 361-366 Moore PB (1971) The Fe2+
3(H2O)n(PO4)2 homologous series crystal-chemical relationships
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 41
and oxidized equivalents American Mineralogist 56 1-17 Moraghan JT Buresh RJ (1976) Chemical reduction of nitrate by ferrous iron Journal of Environmental Quality 5 320-325 Moraghan JT Buresh RJ (1977) Chemical reduction of nitrite and nitrous oxide by ferrous iron Journal of American Soil Science Society 40 47-50 Morales JA de Graterol LS Mesa J (2000) Determination of chloride sulfate and nitrate in groundwater samples by ion chromatography Journal of Chromatography A 884 185-190 Mulder EG Deinema MH (1992) The sheathed bacteria In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 2618-2624 Moslashrup S Madsen MB Franck J Villadsen J Koch CJW (1983) A new interpretation of Moumlssbauer spectra of microcrystalline goethiterdquosuper-ferromagnetismrdquo of ldquosuper-spin-glassrdquo behaviour Journal of Magnetism and Magnetic Materials 40 163-174 Nembrini GP Capobianco JA Viel M Williams AF (1983) A Moumlssbauer and chemical study of the formation of vivianite in sediments of Lago Maggiore (Italy) Geochimica et Cosmochimica Acta 47 1459-1464 Nielsen JL Nielsen PH (1998) Microbial nitrate-dependent oxidation of ferrous iron in activated sludge Environmental Science and Technology 32 3556-3561 Nriagu JO (1972) Stability of vivianite and ion-pair formation in the system Fe3(PO4)2-H3PO4- H2O Geochimica et Cosmochimica Acta 36 459-470 Nriagu JO Dell CI (1974) Diagenetic formation of iron phosphates in recent lake sediments American Mineralogist 59 934-946 Patrick Jr WH Khalid RA (1974) Phosphate release and sorption by soils and sediments Effect of aerobic and anaerobic conditions Science 186 53-55 Postma D (1981) Formation of siderite and vivianite and the pore-water composition of a recent bog sediment in Denmark Chemical Geology 31 225-244 Pratesi G Cipriani C Giuli G Birch WD (2003) Santabarbaraite a new amorphous phosphate mineral European Journal of Mineralogy 15 185-192 Ratering S Schnell S (2001) Nitrate-dependent iron(II) oxidation in paddy soil Environmental Microbiology 3 100-109 Ritz C Essene EJ Peacor DR (1974) Metavivianite Fe3(PO4)2middot8H2O a new mineral American Mineralogist 59 896-899 Rodgers KA (1986) Metavivianite and kerchenite a review Mineralogical Magazine 50 687- 691 Rodgers KA Johnston JH (1985) Type metavivianite Moumlssbauer evidence for a revised composition Neues Jahrbuch fuumlr Mineralogie-Monatshefte 12 539-542
42 Chapter 2 Rossum JR Villarruz PA (1963) Determination of ammonia by the indophenol method Journal of American Water Works Association 55 657-658 Seitz MA Riedner RJ Malhotra SK Kipp RJ (1973) Iron-phosphate compound identification in sewage sludge residue Environmental Science and Technology 7 354-357 Sobolev D Roden EE (2001) Suboxic deposition of ferric iron by bacteria in opposing gradients of Fe(II) and oxygen at circumneutral pH Applied and Environmental Microbiology 67 1328-1334 Straub KL Benz M Schink B Widdel F (1996) Anaerobic nitrate-dependent microbial oxidation of ferrous iron Applied and Environmental Microbiology 62 1458-1460 Straub KL Buchholz-Cleven BEE (1998) Enumeration and detection of anaerobic ferrous iron-oxidizing nitrate-reducing bacteria from diverse European sediments Applied and Environmental Microbiology 64 4846-4856 Straub KL Hanzlik M Buchholz-Cleven BEE (1998) The use of biologically produced ferrihydrite for the isolation of novel iron-reducing bacteria Systematic and Applied Microbiology 21 442-449 Weber KA Picardal FW Roden EE (2001) Microbially catalyzed nitrate-dependent oxidation of biogenic solid-phase Fe(II) compounds Environmental Science and Technology 35 1644-1650 Widdel F Bak F (1992) Gram-negative mesophilic sulfate-reducing bacteria In The Prokaryotes (Balows A Truumlper HG Dworkin M Harder W Schleifer K-H (eds)) Springer 2nd ed 3352-3378
Widdel F Schnell S Heising S Ehrenreich A Assmus B Schink B (1993) Ferrous iron oxidation by anoxygenic phototrophic bacteria Nature 362 834-836 Williams JDH Syers JK Shukla SS Harris RF Armstrong DE (1971) Levels of inorganic and total phosphorus in lake sediments as related to other sediment parameters Environmental Science and Technology 5 1113-1120
Zwaan PC Kortenbout van der Sluys G (1971) Vivianite crystals from Hare Noord Brabant Province The Netherlands Scripta Geology 6 1-7
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 43
3 Formation of Layered Iron Hydroxides by Microbial Fe(III)
Reduction Abstract
Many inorganic and organic pollutants may be degraded by microorganisms in the
subsurface However a wide range of contaminants including chromate nitrate
radionuclides nitroaromatic compounds chlorinated aliphatics and carbamate
pesticides may also be chemically transformed by reduction reactions involving
layered iron(II)-iron(III)-hydroxides (green rusts) Hence green rusts (GRs) may
play a potentially important role in the fate and transport of pollutants in iron-rich
suboxic soils and sediments Yet only little is known about the formation of GRs
in these environments The biotic formation of GRs mediated by the anaerobic
dissimilatory Fe(III)-reducing bacteria Shewanella spp has been reported and
proposed in several studies However the experimental conditions applied were
mostly not natural and the evidence of GR formation provided may be questioned
This work investigated the Fe-containing products formed by the facultatively
anaerobic Fe(III)-reducing microorganism Shewanella algae BrY in culture
media containing 4-10 mM formate or lactate and 8-27 mM Fe(III) In order to
simulate natural conditions Fe(III) oxides were applied as coatings on silica
(model system for sandy soils) or calcite particles (model system for calcareous
soils) and synthetic electron shuttles as well as highly concentrated artificial pH
buffers were excluded S algae BrY reduced 19-72 of the initial Fe(III) when
grown in goethitecalcite lepidocrocitecalcite or hydrous ferric oxidesand mineral
systems and green or blackish mineral phases were produced within 1-2 weeks
after inoculation Moumlssbauer spectroscopic analyses indicated that the green and
blackish precipitates were dominated by vivianite (Fe3(PO4)2sdot8H2O) and green rust
44 Chapter 3
31 Introduction
The significance of bacteria in the biogeochemical cycling of iron has been broadly
recognized over the past two decades Chemical processes were previously
considered to account for most of the Fe(III) reduction in subsurface environments
Dissimilatory Fe(III)-reducing bacteria (DIRB) that gain energy by coupling the
oxidation of hydrogen or organic compounds to the reduction of Fe(III) oxides
have been known for many years but their biogeochemical importance was
acknowledged only a decade ago (reviewed by Lovley 1997) DIRB transfer
electrons to extracellular Fe(III) without assimilating the iron Fe(III) bioreduction
accounts for a major fraction of the carbon oxidation in many different
environments and in the presence of high amounts of reactive Fe(III) microbial
Fe(III) reduction may even inhibit sulfate reduction and methanogenesis (King
1990 Lovley amp Phillips 1986) In fact most of the Fe(III) reduction in the Fe(III)
reduction zone of aquatic sediments and aquifers is thought to be enzymatically
catalyzed by microorganisms (Lovley et al 1991) However the relative
importance of microbial and chemical processes involved in the Fe(III) reduction
are still somewhat disputed among microbiologists and geochemists
A wide diversity of Fe(III)-reducing bacteria which fall in a number of different
phylogenetic groups is known today Both organisms growing by respiration and
by fermentation have been isolated and identified (Lovley 1991 Nealson amp
Saffarini 1994) Hydrogen short- and long-chained fatty acids amino acids
sugars and aromatic compounds may serve as electron donors for Fe(III)
bioreduction The enzymes responsible for dissimilatory Fe(III) reduction are outer
membrane associated ferric reductases (Lower et al 2001 and references therein)
Iron reducing bacteria may utilize alternative electron acceptors such as O2 nitrate
S0 sulfate humic substances contaminant metals and metalloids as well as
chlorinated solvents The first organism shown to couple respiratory growth to
dissimilatory iron reduction was Pseudomonas ferrireductans now known as
Shewanella oneidensis but previously classified as Alteromonas putrefaciens and
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 45
Shewanella putrefaciens (Venkateswaran et al 1999) Various DIRB including
the obligate anaerobic Geobacter sp and the facultatively anaerobic Shewanella
sp have been isolated from both marine and freshwater sediments soil and
aquifers (Thamdrup 2000 and references therein)
The redox potentials of oxidized and reduced iron couples and thus the energy
yield available from Fe(III) reduction depend strongly on the specific iron phases
involved In soil and aquatic environments Fe(III) oxides mainly occur in
association with other sediment particles as aggregates or coatings Amorphous
and poorly crystalline Fe(III) oxides usually make up 20 or less of the iron
content in a sediment (Thamdrup 2000) They are the main products of abiotic and
biotic Fe(II) oxidation in sediments and they constitute the most important phases
for microbial Fe(III) reduction Until recently it was generally believed that DIRB
reduced insoluble Fe(III) oxides only by direct contact with the Fe(III) oxide
thereby allowing electron transfer from the cell to the Fe(III) oxide surface
However over the past several years there has been a growing recognition that
DIRB may use different strategies in order to access the solid Fe(III) oxides These
strategies include solubilization of Fe(III) by synthetic or natural Fe(III) chelators
and Fe(III) reduction via electron shuttling with soluble humic substances or
microbially produced electron shuttles (Nevin amp Lovley 2002 and references
therein Turick et al 2003) The Fe(III) complexing agents may also stimulate
Fe(III) oxide reduction indirectly by chelation and thus removal of Fe(II) from
the cell and the Fe(III) oxide surfaces Both chelated Fe(III) and soluble electron
shuttles are more accessible to Fe(III) reductases than solid Fe(III) oxides In
contrast to Geobacter metallireducens S algae produces and releases extracellular
electron shuttling compounds (Nevin amp Lovley 2000) However in the absence of
soluble electron shuttles reversible adhesion is required for reduction of solid
Fe(III) oxides by S algae BrY (Das amp Caccavo 2000) Shewanella algae BrY
adheres readily and preferentially to a range of solid Fe(III) oxides such as
ferrihydrite goethite and hematite (Das amp Caccavo 2001) The adhesion
46 Chapter 3
mechanisms are not completely understood but recent results suggest that the
adhesion is mediated by cell surface proteins and independent of cell motility
(Caccavo amp Das 2002)
The microbial formation of GRs resulting from bioreduction of various Fe(III)
oxides including ferrihydrite goethite and lepidocrocite by strains of the
anaerobic DIRB Shewanella putrefaciens has been reported repeatedly over the
last years (Fredrickson et al 1998 Kukkadapu et al 2001 Liu et al 2001
Parmar et al 2001 Ona-Nguema et al 2002aampb Glasauer et al 2003)
However no evidence of biogenic formation of GRs at natural geochemical
conditions have been offered and it is still unknown whether this process may take
place at natural conditions comprising low carbon and iron concentrations as well
as the absence of synthetic electron shuttles and highly concentrated artificial pH
buffers GRs are layered iron(II)-iron(III)-hydroxides with anionic interlayers and
they hold the general formula [FeII(6-x)FeIII
x(OH)12]x+[(A)xnmiddotyH2O]x- where x = 09 -
42 A is an n-valent anion eg CO32- Clndash or SO4
2- and y is the number of water
molecules in the interlayer In circumneutral solutions the oxidation of dissolved
Fe(II) always passes through solid GR phases (Bernal et al 1959) This agrees
with the natural GR occurrences found in suboxic nonacid iron-rich environments
such as hydromorphic soils and intertidal sediments (Al-Agha et al 1995 Trolard
et al 1996 Genin et al 1998) In addition GRs have been found as corrosion
products in numerous engineering systems eg in a pipeline distribution system
for drinking water steel sheet piles in marine sediments reinforced concrete
(ferro-concrete) and permeable reactive barriers of zero-valent iron implemented
for on-site remediation of organic and inorganic contaminants (Tuovinen et al
1980 Nielsen 1976 Genin et al 1991 Roh et al 2000) Through sequestration
and reductive transformation GRs may play an important role in the fate and
transport of organic and inorganic pollutants in suboxic iron-rich soils and
sediments (see Chapters 4 amp 5 in this work and references therein)
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 47
The major goal of this work was to examine the iron minerals forming during the
course of Fe(III) bioreduction of hydrous ferric oxide goethite and lepidocrocite
Two model systems simulating sandy and calcareous soils in subsurface
environments were designed in order to investigate the formation of iron minerals
at conditions including low carbon levels low Fe(III) concentrations applied as
Fe(III) oxide coatings on sand or calcite no electron shuttle and no synthetic pH
buffers
32 Materials and methods
All handling and sampling of solutions and suspensions were carried out at strict
anoxic conditions Standard sterile techniques were used throughout (Hungate
1969 Miller amp Wolin 1974) Only the iron oxide coatings were not autoclaved in
order to avoid the iron oxides from transforming Goethite (acicular particles with
size 01 times 06 microm specific surface area 16 m2g) and lepidocrocite (acicular
particles with size 005 times 03 microm specific surface area 18 m2g) were purchased as
fine powders from Bayer (Bayferrox 910 and 943) Calcite (grain size 170-350 microm
Pluumlss-Staufer AG) and sea sand (dominantly quartz grain size 01-03 mm Riedel-
de Haeumln) were used as Fe(III) oxide coating bearing minerals
321 Preparation of iron oxide coatings
Two grams of goethite (goe) or lepidocrocite (lep) and 100 g calcite were
combined with 200 mL deionized water (DIW) in a 500 mL polyethylene flask
Hydrous ferric oxide (HFO) was synthesized by dissolving 4 g Fe(NO3)3middot9H2O in
70 mL DIW followed by slow neutralization under magnetic stirring till pH 7 with
approximately 30 mL 1 M NaOH (method modified after Schwertmann amp Cornell
1991) The HFO coating was made by combining 100 mL freshly precipitated
HFO with 900 mL deionized water and 50 g sea sand in a polyethylene bottle The
suspensions containing the iron oxide coatings were gently agitated on a
reciprocating shaker for 24 h and left to stand for another 24 h Excess Fe(III)
oxides and salts were removed from the coated material by repeated decantation
48 Chapter 3
and washing with 003 M NaNO3 followed by washing with DIW until clear
runoff Finally the coatings were collected on folding filters and air dried The
amount of HFO goethite and lepidocrocite coated onto sand and calcite after
washing and drying was quantified to 7-11 mg Fe(III)g sand or calcite
322 Mineral characterisation
The identity and purity of the HFO synthesized were examined by means of X-ray
diffraction (XRD) measurements The XRD analyses were performed on a Scintag
XDS 2000 using Co Kα radiation (45 kV 40 mA) using divergence scatter and
receiving slits of 1deg 05deg and 02 mm respectively Samples were scanned
between 6 and 80 deg2θ with a scan speed of 1 deg2θmin Mineral suspension samples
for transmission Moumlssbauer spectroscopic analysis were collected on 02 microm filters
in an anoxic glove box (Coy Laboratory Products Inc) transferred to Perspex
capsules and stored in liquid nitrogen until measurement Moumlssbauer spectra were
obtained between 250 and 5 K using a conventional constant acceleration
spectrometer and a source of 57Co in Rh The spectrometer was calibrated using a
125 microm foil of α-Fe at room temperature and isomer shifts are given relative to
the centroid of this absorber The spectra were fitted using simple Lorentzian line
shape and it was assumed that all positions have identical f-factors
323 Culture conditions and cell preparation
Shewanella algae BrY is a motile gram-negative rod which was isolated first from
anoxic estuary sediments (Caccavo et al 1992) S algae BrY was grown
aerobically in tryptic soy broth (30 gL CASO-bouillon Merck) at 28degC on a rotary
shaker at 150 rpm for 16-18 h Cells were harvested by centrifugation (6000 rpm times
g 4ordmC 15 min) during the late exponential ndash early stationary growth phase at
OD660 ~ 06 Optimal Fe(III) reductase activity is expressed at this stage of growth
(Roden amp Zachara 1996) The cells were washed twice in oxic 50 mM PIPES
[piperazine-NNacute-bis(2-ethanesulfonic acid)] buffer (pH 70) and resuspended in
culture medium containing no Fe(III) and no carbon source Washed cell
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 49
suspensions were used as inocula for Fe(III) reduction experiments Oxygen was
expelled from the inoculum by extensive purging with 100 N2(g) (9999999
purity) Working stock cultures of S algae BrY were maintained aerobically on
tryptic soy agar plates at ambient temperature
324 Bioreduction experiments
All anaerobic incubations were carried out in anoxic serum vials (25 mL) or test
tubes (13 mL) sealed with thick (10-13 mm) butyl rubber stoppers and aluminum
crimp caps or plastic screw caps The basal culture medium (Table 31) was
prepared according to Kostka amp Nealson (1998) but with a phosphate
concentration of 2 mM and the exclusion of Fe(II) and EDTA
(ethylenediaminetetraacetic acid) The medium was amended with 4-10 mM
lactate or formate and 8-27 mM Fe(III) The Fe(III) was applied as Fe(III) oxide
coatings on sand or calcite The suspensions were purged extensively with 100
N2(g) (HFOsand suspensions) or 995 N205 CO2(g) (goecalcite and
lepcalcite suspensions) prior to inoculation The calcareous systems were buffered
at pH ~ 76 through a natural buffer system (CaCO3(s) + 995 N205 CO2(g))
whereas the sandy systems contained no pH buffer (100 N2(g) pH 55-60)
Inoculum size made up 5 of the total volume Cultures were incubated dark at
room temperature and gently agitated once every day At different time intervals
suspension samples for Fe(II) and Moumlssbauer analysis were withdrawn from the
reaction mixture using 100 N2(g) or 995 N205 CO2(g)-flushed sterile
disposable syringes and hypodermic needles Suspension samples for Fe(II)
analysis were digested in 01 M HCl for 30 min
50 Chapter 3
Table 31 Composition of the mineral medium (modified from Kostka amp Nealson (1998))
Components Concentration (M)
(NH4)2SO4 00143 KH2PO4 73middot10-4
K2HPO4 13middot10-3
MgSO4middot7H2O 10middot10-3
CaCl2middot2H2O 50middot10-4
H3BO3 56middot10-5
ZnSO4middot7H2O 10middot10-6
Na2MoO4middot2H2O 40middot10-6
CuSO4middot5H2O 20middot10-7
MnSO4middotH2O 10middot10-6
Na2SeO4 12middot10-5
CoCl2middot6H2O 50middot10-6
NiCl2middot6H2O 80middot10-6
NaCl 10middot10-5
L-arginine 11middot10-4
L-serine 19middot10-4
L-glutamic acid 14middot10-4
Lactate or formate 4-10middot10-3
Fe(III) 8-27middot10-3
325 Analytical methods
Fe(II) was determined using a modified phenanthroline method (Fadrus amp Maly
1975) The total amount of Fe(III) coated on calcite and sand was determined by
atomic absorption spectroscopy following dissolution in 6 M HCl(aq) for 24 h
33 Results and discussion
331 Fe(II) production and suspension colour changes
Strongly chelating agents such as EDTA were omitted from the culture medium in
order to prevent complexation of Fe(II) and Fe(III) which interferes with
precipitation of Fe(II) and Fe(II)-Fe(III) mineral phases Within 1-2 weeks after
inoculation Shewanella algae BrY produced green mineral phases in media
suspensions containing lepidocrocite and goethite as coatings on calcite and 4-10
mM formate or lactate (Figure 31) The formation of the green precipitates was
generally slower for the lepidocrocite coating than for the goethite coating The
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 51
blue-green colours of the phases produced most likely originate from Fe(II)-Fe(III)
charge transfer between adjacent Fe(II) and Fe(III) ions in edge-shared octahedra
(Faye et al 1968) Dark brown and blackish products were formed when the
bacteria were inoculated on HFO coated sand (Figure 32)
Figure 31 Culture tubes containing a) goethite and b) lepidocrocite coated calcite in culture
medium The left tubes of the pair were not inoculated whereas the right tubes were
photographed 5 months after inoculation with S algae BrY Experimental conditions [formate]0
= 4 mM [Fe(III)]0 = 8 mM 995 N205 CO2(g) pH 76
52 Chapter 3
Figure 32 Culture tubes containing HFO coated sand in culture medium Tubes 1 and 2 to the
left were not inoculated whereas tubes 3-5 to the right were photographed a) 13 days and b) 21
days after inoculation with S algae BrY Experimental conditions [lactate]0 = 10 mM [Fe(III)]0
= 25 mM 100 N2(g) pH 55-60
The green and black colours did not change to other colours (observed for gt1
year) indicating that the microbial Fe(III) reduction ceased at these mineral stages
The concentrations of dissolved ferrous iron (Fe(II)sol) estimated during Fe(III)
bioreduction were generally low (Figure 33) When comparing the final Fe(II)sol
amounts produced and the slopes of the Fe(II)sol formation curves for HFO
goethite and lepidocrocite in Figure 33 it can be seen that the final Fe(II)sol
amount and the Fe(II)sol production rate both follow the order HFO gt goethite gt
lepidocrocite at similar cell densities regardless of the carbon source applied This
suggests that bioreduction by S algae BrY is more facile for HFO than for goethite
and lepidocrocite at the experimental conditions employed here It should be noted
that the final Fe(II)sol amounts and the Fe(II)sol production rates reported in this
work have not been normalised with respect to the specific surface areas of the iron
oxides and coating-bearing solids applied The reactivity trend is consistent with
previous findings demonstrating higher reducibility of natural and poorly
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 53
crystalline Fe(III) oxides as compared to synthetic crystalline Fe(III) oxides
(Zachara et al 1998) The authors ascribed these differences in reducibility to
differences in particle size surface area and crystal defects of the Fe(III) oxides In
some cases the dissolved Fe(II) concentration decreased again with time (Figure
33 b-d) This indicates that the Fe(II) formed was incorporated into solid phases
forming andor adsorbed onto the calcite sand or Fe(III) oxide surfaces The solid
Fe(II) concentrations were not estimated spectrophotometrically The solid
material was generally low in total iron and therefore saving it for Moumlssbauer
spectroscopic analysis was given highest priority
54 Chapter 3
Figure 33 Time course of dissolved Fe(II) production during bioreduction of HFOsand
goethitecalcite and lepidocrocitecalcite by S algae BrY Experimental conditions [Fe(III)]0 =
25 mM [formate]0 = 10 mM (a-c) or [lactate]0 = 10 mM (d-f)
No color change and no Fe(II) production were observed in mineral suspensions
lacking either a carbon source or S algae BrY cells (data not shown)
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 55
332 Identification of solid iron phases
The purity of the Fe(III) oxides used in the experiments were investigated by
transmission Mossbauer spectroscopy (Figure 34) Single (or strongly dominating)
sextets in the spectra with magnetic hyperfine fields of 48 1 505 and 45 5 Tat 5 K
demonstrated the purity of the HFO goethite and lepidocrocite samples
respectively A minor impurity of goethite in the lepidocrocite sample was
resolved in the spectrum measured at 80 K (not shown for pure sample but can be
seen as a magnetically ordered sextet in Figure 35c) No Fe(II)-containing
components were detected
a)
c)
middot 12 -8 -4 4 8 12
Velocity (mmls
bull middot12 -8 -4 0 4 12
Velocity (mmls)
b)
middot12 -8 -4 4 8 12
Velocity (mmls
Figure 34 Transmission Mossbauer spectra measured at 5 K of a) HFO (magnetic hyperfine
field of 481 T isomer shift of 048 1nrns-1 negligible quadrupole shift and line width of outer
lines 110 rmns-1) b) goethite (magnetic hyperfine field of 505 T isomer shift of 049 rmns-1
quadmpole shift of -013 1nrns-1 and line width of outer lines 042 rnrns-1
) and c) lepidocrocite
56 Chapter 3
(magnetic hyperfine field of 455 T isomer shift of 050 mms-1 quadrupole shift of ndash001 mms-1
and line width of outer lines 060 mms-1) prior to inoculation Simple Lorenztian fits are shown
The oxidation state and coordination of Fe in the microbially reduced HFO
goethite and lepidocrocite samples were also examined by transmission Moumlssbauer
spectroscopy (Figure 35) The bioreduced HFO goethite and lepidocrocite samples
cultured on formate contained Fe(II) holding similar coordination as inferred from
the similarity of the hyperfine parameters (see legend in Figure 35) but different
relative intensities (72 19 and 71 respectively) The major part of the Fe(III)
remaining in the bioreduced samples were coordinated similarly to the Fe(III)
present in the initial Fe(III) oxide The coordination of Fe(II) in the bioreduced
lepidocrocite samples cultured on lactate was slightly different (a smaller
quadrupole splitting of 288 mms-1 for the ferrous component dominates ndash data not
shown) The exact mineralogy of the Fe(II) present in the green phases was not
fully resolved but its coordination is very akin to one of the Fe(II) sites in vivianite
(see Chapter 2 this work) and synthetic green rusts (Koch 1998) These findings
agree with other reports on the bioformation of vivianite and green rusts by
Shewanella putrefaciens CN32 although the evidence provided may be discussed
(Fredrickson et al 1998 Glasauer et al 2003 Parmar et al 2001) Our
Moumlssbauer data on the green phases did not allow for a detailed account of the type
of green rust produced However when considering solution composition (see
Table 31) and the high affinity of GR interlayers for carbonate it is reasonable to
assume that carbonate GR was formed (Hansen amp Taylor 1991) Due to the high
amounts of Fe(III) in the oxides present in the experiments it was difficult to probe
a possible content of Fe(III) in the vivianite with certainty The differences in the
number of Fe(II) positions in the Moumlssbauer spectra and particular the different
temperatures at which magnetic ordering takes place can be employed in order to
distinguish between green rust and vivianite Preliminary Moumlssbauer data obtained
for the blackish precipitates formed in the HFOsand suspensions indicate that they
hold no resemblance to magnetite eventhough the colour suggests so On the
contrary the black precipitates seemed to be more similar to synthetic green rusts
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 57
Mossbauer spectroscopic measurements are currently underway in order to resolve
the Fe(II) coordinations in the greenish and blackish phases
a) b)
c c g Q e- e 0 0 1l 1l
-~ bull ~
iii ~ Qi bull a bull
-12 -8 4 0 4 12
4 -3 -2 -1 0 1 2 4 Velocity (mmts) Velocity ( rmis)
c)
-12 -8 4 8 12
v elocity (m mis)
Figure 35 Transmission Mossbauer spectra of the black and green phases fo1med within 1-2
weeks after inoculation of a) HFO (measured at 130 K) b) goethitecalcite (measured at 80 K)
and c) lepidocrocitecalcite (measured at 80 K) with S algae BrY Experimental conditions
[fo1m ate]0 = 4 mM [Fe(III)]o = 8 mM 995 Ni05 C02(g) pH 76 The quadrnpole
splittings and isomer shifts for the Fe(II) components in the three systems are a) 293 nnns-1 and
126 mmsmiddot1 b) 308 rmnsmiddot1 and131 rmnsmiddot1 and c) 322 mmsmiddot1 and 132 mmsmiddot1 Simple Lorenztian
fits are shown
The evidence provided in many of the studies proposing biogenic GRs is not all
too convincing but it strongly suggests the probability of microbially produced GR
being present The challenge encountered is that when present in low
58 Chapter 3
concentrations especially in mixtures including other iron minerals it is very
difficult to identify GRs using conventional solid phase analysis methods even
with Moumlssbauer spectroscopy At least two complementary methods such as XRD
and Moumlssbauer spectroscopy are required for proper identification and
characterization of GRs However in this work the solid materials were generally
too low in total iron to allow for XRD analysis Moreover the highly
heterogeneous suspensions were dominated by the coating-bearing sand and calcite
solids Electron micrographs including energy dispersive X-ray spectroscopy
suspension colour and mineral stability calculations do not suffice as evidence The
most convincing evidence provided so far involves an atypical GR-CO3 with an
Fe(II)Fe(III) ratio of 1 (Ona-Nguema et al 2002aampb) This GR-CO3 was formed
as a result of lepidocrocite reduction by Shewanella putrefaciens CIP 8040 at
conditions comprising high nutrient levels (50-75 mM formate) high Fe(III)
concentrations (80-300 mM) and a synthetic electron shuttle (100 microM
anthraquinone-26-disulfonate (AQDS)) at initial pH 75 Hence the results
reported during recent years suggest that microbial formation of GR may be
possible The results presented here indicate that GRs may be produced
microbially at conditions including low carbon and Fe(III) concentrations as well
as the exclusion of synthetic electron shuttles and pH buffers
333 Factors controlling the identity of the secondary iron minerals
In general one would expect that biogenic minerals have chemical compositions
and crystal habits similar to those produced by nonenzymatic processes as they are
governed by the same equilibrium principles In fact since the latter stages of
mineralization are inorganically driven and the secondary Fe(II)-containing
minerals are formed indirectly by electron transfer outside the bacterial cell and not
directly inside the bacterial cell the type of iron mineral formed is a function of the
environmental conditions in which the bacteria live ie the same microorganism
form different minerals in different environments The key factors controlling the
identity of the secondary iron minerals include medium composition electron
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 59
donor and electron acceptor concentrations mineral aging as well as adsorbed ions
(Zachara et al 2002) The main factor controlling the nature of the secondary
mineral products are the respiration-driven biogenic Fe(II) supply rate and
magnitude and its surface reaction with the residual oxide and other sorbed ions
(Zachara et al 2002) Especially solution and medium composition have a strong
impact on the nature of the Fe(II)-containing biomineralization products forming
Accordingly siderite (FeCO3) and magnetite (Fe3O4) were the secondary solid
phases resulting from the bioreduction of ferrihydrite by Shewanella putrefaciens
CN32 in bicarbonate buffered medium (pH 71) containing no phosphate whereas
siderite and vivianite were the secondary iron minerals dominating in bicarbonate
buffered medium (pH 74) containing 4 mM phosphate (Zachara et al 2002) This
is explained by the inhibiting effect of phosphate on crystallization of magnetite
(Couling amp Mann 1985 Fredrickson et al 1998)
334 Factors controlling the rate and extent of Fe(III) bioreduction
In this study the extent of Fe(III) bioreduction was estimated to 19-72 by
transmission Moumlssbauer measurements In fact complete microbial reduction of
crystalline Fe(III) minerals has never been observed in laboratory batch culture
studies (Roden amp Urrutia 2002) It has been found that Fe(II) does not inhibit
Fe(III) reductase activity through an enzyme inhibition mechanism (Roden amp
Urrutia 2002) Hence other chemical andor physiologic factors control the
bioavailability of solid Fe(III) phases and thus the extent of their microbial Fe(III)
reduction The initial rate and long-term extent of microbial reduction of
amorphous and crystalline Fe(III) oxides including HFO goethite and hematite
were linearly correlated with oxide surface area (Roden amp Zachara 1996)
Association of biogenic Fe(II) with Fe(III) oxide and DIRB cell surfaces reduced
the long-term extent of crystalline Fe(III) oxide bioreduction (Roden amp Urrutia
2002) These results were explained by Fe(II) surface complexes andor
precipitates creating a passive Fe(II)-bearing surface film providing direct physical
interference with the electron transfer from the DIRB cells to Fe(III) However the
60 Chapter 3
real mechanisms whereby the surface-associated Fe(II) inhibits Fe(III) oxide
bioreduction are unclear but they most likely involve both kinetic and
thermodynamic constraints on the electron transfer Culture medium composition
in particular the presence and the concentration of phosphate as well as Fe(II)
chelating ligands also exert an influence on the extent of the microbial reduction
of Fe(III) oxides The extent of Fe(III) bioreduction was inhibited by high
phosphate concentrations which favoured surfacebulk precipitation processes
(Urrutia et al 1998) The carbon sources most frequently applied in Fe(III)
bioreduction studies include malate citrate and other di- and tricarboxylic acids
which are not only easily metabolizable carbon sources but also eminent Fe(II) and
Fe(III) chelators In this study we employed formate and lactate as carbon sources
since they are the weakest complexing agents of Fe(II) and Fe(III) among the C1-
C3 monocarboxylic acids (Martell 1964) Thus we expect less dissolution of
prevailing precipitates by complexation as compared to other studies
34 Conclusions
This work demonstrated that Shewanella algae BrY reduced 19-72 of initial
Fe(III) when grown in culture media containing 4-10 mM formate or lactate and 8-
27 mM Fe(III) applied as goethite or lepidocrocite coatings on calcite (pH 76) or
HFO coatings on sand (pH 55-60) Within 1-2 weeks after inoculation green
mineral phases were produced in the goethitecalcite and lepidocrocitecalcite
mineral systems whereas black precipitates formed in the HFOsand suspensions
Moumlssbauer spectroscopic analyses indicated that the greenish and blackish phases
most likely were mineral mixtures dominated by vivianite and green rust Thus the
results indicate that GRs may be produced microbially at conditions including low
carbon and Fe(III) concentrations as well as the exclusion of synthetic electron
shuttles and pH buffers
Acknowledgments
We would like to thank Dr R Gerlach for providing us the Shewanella algae BrY culture and
Dr C B Koch for performing the Moumlssbauer analyses
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 61
References Al-Agha MR Burley SD Curtis CD Esson J (1995) Complex cementation textures and authigenic mineral assemblages in recent concretions from the Lincolnshire Wash (east coast UK) driven by Fe(0) to Fe(II) oxidation Journal of the Geological Society 152 157-171 Bernal JD Dasgupta DR Mackay AL (1959) The oxides and hydroxides of iron and their structural inter-relationships Clay Minerals Bulletin 4 15-30 Caccavo Jr F Blakemore RP Lovley DR (1992) A hydrogen-oxidizing Fe(III)-reducing microorganism from the Great Bay Estuary New Hampshire Applied and Environmental Microbiology 58 3211-3216 Caccavo Jr F Das A (2002) Adhesion of dissimilatory Fe(III)-reducing bacteria to Fe(III) minerals Geomicrobiology Journal 19 161-177 Couling SB Mann S (1985) The influence of inorganic phosphate on the crystallization of magnetite (Fe3O4) from aqueous solution Journal of the Chemical Society Chemical Communications 1713-1715 Das A Caccavo Jr F (2000) Dissimilatory Fe(III) oxide reduction by Shewanella alga BrY requires adhesion Current Microbiology 40 344-347
Das A Caccavo Jr F (2001) Adhesion of the dissimilatory Fe(III)-reducing bacterium Shewanella alga BrY to crystalline Fe(III) oxides Current Microbiology 42 151-154 Fadrus H Maly J (1975) Suppression of iron(III) interference in the determination of iron(II) in water by the 110-phenanthroline method The Analyst 100 549-554 Faye GH Manning PG Nickel EH (1968) The polarized optical absorption spectra of tourmaline cordierite chloritoid and vivianite ferrous-ferric electronic interaction as a source of pleochroism American Mineralogist 53 1174-1201 Fredrickson JK Zachara JM Kennedy DW Dong H Onstott TC Hinman NW Li S (1998) Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium Geochimica et Cosmochimica Acta 62 3239-3257 Geacutenin JMR Olowe AA Benbouzid-Rollet ND Prieur D Confente M Resiak B (1991) The simultaneous presence of green rust 2 and sulfate reducing bacteria in the corrosion of steel sheet piles in a harbour area Hyperfine Interactions 69 875-878 Geacutenin JMR Bourrieacute G Trolard F Abdelmoula M Jaffrezic A Refait Ph Maitre V Humbert B Herbillon A (1998) Thermodynamic equilibria in aqueous suspensions of synthetic and natural Fe(II)-Fe(III) green rusts Occurrences of the mineral in hydromorphic soils Environmental Science and Technology 32 1058-1068 Glasauer S Weidler PG Langley S Beveridge TJ (2003) Controls on Fe reduction and mineral formation by a subsurface bacterium Geochimica et Cosmochimica Acta 67 1277- 1288 Hansen HCB Taylor RM (1991) The use of glycerol intercalates in the exchange of CO3
2- with SO4
2- NO3- or Cl- in pyroaurite-type compounds Clay Minerals 26 311-327
62 Chapter 3
Hungate RE (1969) A roll tube method for cultivation of strict anaerobes Methods in Microbiology 3B 117-132 King GM (1990) Effects of added manganic and ferric oxides on sulfate reduction and sulfide oxidation in intertidal sediments FEMS Microbiology Ecology 73 131-138 Koch CB (1998) Structures and properties of anionic clay minerals Hyperfine Interactions 117 131 -157 Kostka J Nealson KH (1998) Isolation cultivation and characterization of iron- and manganese reducing bacteria In Techniques in Microbial Ecology Burlage RS Atlas R Stahl D Geesey G Sayler G (eds) Oxford University Press Inc 58-78 Kukkadapu RK Zachara JM Smith SC Fredrickson JK Liu C (2001) Dissimilatory bacterial reduction of Al-substituted goethite in subsurface sediments Geochimica et Cosmochimica Acta 65 2913-2924
Liu C Zachara JM Gorby YA Szecsody JE Brown CF (2001) Microbial reduction of Fe(III) and sorptionprecipitation of Fe(II) on Shewanella putrefaciens strain CN32 Environmental Science and Technology 35 1385-1393 Lovley DR (1991) Dissimilatory Fe(III) and Mn(IV) reduction Microbiological Reviews 55 259-287 Lovley DR (1997) Microbial Fe(III) reduction in subsurface environments FEMS Microbiology Reviews 20 305-313 Lovley DR Phillips EJP (1986) Organic matter mineralization with reduction of ferric iron in anaerobic sediments Applied and Environmental Microbiology 51 683-689 Lovley DR Phillips EJP Lonergan DJ (1991) Enzymatic versus nonenzymatic mechanisms for Fe(III) reduction in aquatic sediments Environmental Science and Technology 25 1062-1067 Lower SK Hochella Jr MF Beveridge TJ (2001) Bacterial recognition of mineral surfaces Nanoscale interactions between Shewanella and α-FeOOH Science 292 1360-1363 Martell AE (1964) Stability constants of metal-ion complexes Part 2 Organic including macromolecule ligands The Chemical Society London 2 ed Miller TL Wolin MJ (1974) A serum bottle modification of the Hungate technique for cultivating obligate anaerobes Applied Microbiology 27 985-987 Nealson KH Saffarini D (1994) Iron and manganese in anaerobic respiration Environmental significance physiology and regulation Annual Review of Microbiology 48 311-343 Nevin KP Lovley DR (2000) Lack of production of electron-shuttling compounds or solubilization of Fe(III) during reduction of insoluble Fe(III) oxide by G metallireducens Applied and Environmental Microbiology 66 2248-2251 Nevin KP Lovley DR (2002) Mechanisms for Fe(III) oxide reduction in sedimentary
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 63
environments Geomicrobiology Journal 19 141-159 Nielsen A (1976) Hvid groslashn og sort rust Beskrivelse af en korrosionsskade paring et svoslashmmebassin Nordisk Betong 2 21-24 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002a) Iron (IIIII) hydroxycarbonate green rust formation and stabilization from lepidocrocite bioreduction Environmental Science and Technology 36 16-20 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002b) Microbial reduction of lepidocrocite γ-FeOOH by Shewanella putrefaciens The formation of green rust Hyperfine Interactions 139140 231-237 Parmar N Gorby YA Beveridge TJ Ferris FG (2001) Formation of green rust and immobilization of nickel in response to bacterial reduction of hydrous ferric oxide Geomicrobiology Journal 18 375-385 Roden EE Urrutia MM (2002) Influence of biogenic Fe(II) on bacterial crystalline Fe(III) oxide reduction Geomicrobiology Journal 19 209-251 Roden EE Zachara JM (1996) Microbial reduction of crystalline iron(III) oxides Influence of oxide surface area and potential for cell growth Environmental Science and Technology 30 1618-1628 Roh Y Lee SY Elless MP (2000) Characterization of corrosion products in the permeable reactive barriers Environmental Geology 40 184-194 Schwertmann U Cornell RM (1991) Iron oxides in the laboratory Preparation and characterization VCH Verlagsgesellschaft mbH Weinheim Thamdrup B (2000) Bacterial manganese and iron reduction in aquatic sediments In Advances in Microbial Ecology (Schink B ed) Kluwer AcademicPlenum Publishers New York 41-84 Trolard F Abdelmoula M Bourrieacute G Humbert B Geacutenin JMR (1996) Mise en eacutevidence dun constituant de type rouilles vertes dans les sols hydromorphes Proposition de lexistence dun nouveau mineacuteral la fougeacuterite Geacuteosciences de surface Comptes Rendus de LrsquoAcademie des Sciences 323 1015-1022 Tuovinen OH Button KS Vuorinen A Carlson L Mair DM Yut LA (1980) Bacterial chemical and mineralogical characteristics of tubercles in distribution pipelines Journal of the American Water Works Association 72 626-635 Turick CE Caccavo Jr F Tisa LS (2003) Electron transfer from Shewanella algae BrY to hydrous ferric oxide is mediated by cell-associated melanin FEMS Microbiology Letters 220 99-104 Urrutia MM Roden EE Fredrickson JK Zachara JM (1998) Microbial and surface chemistry controls on reduction of synthetic Fe(III) oxide minerals by the dissimilatory iron- reducing bacterium Shewanella alga Geomicrobiology 15 269-291 Venkateswaran K Moser DP Dollhopf ME Lies DP Saffarini DA MacGregor BJ Ringelberg DB White DC Nishijima M Sano H Burghardt J Stackebrandt E
64 Chapter 3
Nealson KH (1999) Polyphasic taxonomy of the genus Shewanella and description of Shewanella oneidensis sp nov International Journal of Systematic Bacteriology 49 705-724 Zachara JM Fredrickson JK Li S Kennedy DW Smith SC Gassman PL (1998) Bacterial reduction of crystalline Fe3+ oxides in single phase suspensions and subsurface materials American Mineralogist 83 1426-1443
Zachara JM Kukkadapu RK Fredrickson JK Gorby YA Smith SC (2002) Biomineralization of poorly crystalline Fe(III) oxides by dissimilatory metal reducing bacteria (DMRB) Geomicrobiology Journal 19 179-207
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 65
4 Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust The Effect of Probe Compound Charge
Abstract
Layered iron(II)-iron(III)-hydroxides (green rusts) may play an important role in
controlling the fate and transport of many organic and inorganic contaminants in
iron-rich suboxic soils and sediments Unlike most other iron oxides green rusts
(GRs) contain not only external Fe(II) reactive sites at the basal planes and at the
edges but also internal sites in the space between consecutive Fe(II)-Fe(III)
hydroxide layers The GR interlayer thickness is a function of both the size and the
charge of the interlayer anion Whether a given oxidant has access to the internal
sites in GRs is dependent on its charge We investigated the reductive
transformation of nitroaromatic compounds (NACs) by GR-SO4 and studied the
effect of NAC charge on the reactivity towards GR-SO4 A series of structurally
closely related compounds with different charge properties including nitrobenzene
4-nitrotoluene 4-chloronitrobenzene and 4-nitrophenylacetic acid were used as
probe compounds The NACs were completely reduced to their corresponding
anilines by GR-SO4 The reactions followed pseudo 1 order kinetics with respect
to NAC and the surface area-normalised pseudo 1 order rate constants obtained
were 016ndash465middot10-4 s-1middotm-2middotL at [Fe(II)GR]0 = 103-1260 mM [NAC]0 = 20-102
microM and pH 84-86 Neither mass transfer control nor surface saturation kinetics
could account for the rather unexpected similarity of the surface area-normalised
pseudo 1 order rate constants obtained for the reduction of the neutral and anionic
NACs by GR-SO4 These observations suggest that the anionic NACs did not have
an enhanced access to the inner or outer Fe(II)-GR reactive sites as compared to
the neutral NACs Hence the reaction between NAC and GR-SO4 primarily took
place at the edges of GR-SO4
66 Chapter 4
41 Introduction
Layered iron(II)-iron(III)-hydroxides (green rusts) are intermediate phases formed
by partial oxidation of Fe(II) or partial reduction of Fe(III) In neutral and weakly
alkaline solutions the oxidation of dissolved Fe(II) always passes through solid
green rust (GR) phases (Bernal et al 1959) This agrees with the natural GR
occurrences found in suboxic non-acid iron-rich environments such as
hydromorphic soils and intertidal sediments (Al-Agha et al 1995 Trolard et al
1996 Genin et al 1998) In addition GRs have been found as corrosion products
in numerous engineered systems ie a pipeline distribution system for drinking
water steel sheet piles in marine sediments reinforced concrete (ferro-concrete)
and permeable reactive barriers of zero-valent iron implemented for on-site
remediation of organic and inorganic contaminants (Tuovinen et al 1980 Nielsen
1976 Genin et al 1991 Roh et al 2000) Furthermore the microbial formation
of GRs resulting from bioreduction of Fe(III) oxides by strains of the anaerobic
dissimilatory Fe(III) reducing bacteria Shewanella putrefaciens has been reported
increasingly over the last 5 years (Fredrickson et al 1998 Kukkadapu et al 2001
Parmar et al 2001 Ona-Nguema et al 2002 Glasauer et al 2003) Moreover
the biotic formation of GRs by anaerobic denitrifying Fe(II) oxidizing bacteria has
been proposed but proper identification of the GR phases still lacks (Chaudhuri et
al 2001) All these indications of microbial GR formation infer the importance of
GRs as a link between geochemical and biological processes in natural systems
GRs form platy crystals with the general formula [FeII(6-
x)FeIIIx(OH)12]x+[(A)xnmiddotyH2O]x- where x = 09 - 42 A is an n-valent anion eg
CO32- Clndash or SO4
2- and y is the number of water molecules in the interlayer The
crystal structure consists of positively charged hydroxide sheets with Fe(II) and
Fe(III) cations having octahedral hydroxyl coordination The Fe(III) in the
hydroxide layers creates a net positive charge which is balanced by hydrated
anions in the interlayers (Figure 41) The interlayers have a higher affinity for
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 67
divalent anions than for monovalent anions (Miyata 1983) Among the 3 most
common GR forms the affinity follows the order CO32- gt SO4
2- gt Clndash The extreme
preference shown for carbonate hinders further access and exchange except under
certain conditions (Hansen amp Taylor 1991) Non-carbonate forms are readily
exchanged with other anions when dispersed in a solution containing the
exchanging anion (Mendiboure amp Schoumlllhorn 1986)
c
ba
Figure 41 Green rust layer structure The hydroxide layers and the interlayers are connected by
hydrogen bonds (not shown) The GR-SO4 crystal structure is characterised by the hexagonal
unit cell having a = b = 055 nm and c = 110 nm (Simon et al 2003) The unit cell consists of
one double layer (a double layer is a hydroxide layer and an interlayer) ie the hydroxide layer
constitutes 049 nm and the interlayer 061 nm in GR-SO4
The GR interlayer thickness (extending in the c axis direction Figure 41) is a
function of both the size and the charge of the interlayer anion Tetrahedrally
coordinated anions like sulphate lead to larger interlayer distances than smaller
monoatomic anions like chloride or planar ions like carbonate (Mendiboure amp
Schoumlllhorn 1986) Not only size but also charge density plays a role for the
interlayer spacing That is for anions having the same number of valence
electrons anions with smaller ionic radii (higher electron density) are bound more
strongly and therefore result in smaller interlayer spacings
Due to their layered structures anionic interlayers and high specific surface areas
GRs represent reactive ion exchangers and sorbents of anions eg arsenate
selenate and phosphate (Myneni et al 1997 Hansen amp Poulsen 1999 Randall et
al 2001) In addition GR may incorporate heavy metal cations by isomorphic
68 Chapter 4
substitution into the GR hydroxide layers (Tamaura 1985 Tamaura 1986)
Furthermore GRs have been shown to reduce a range of inorganic contaminants
such as nitrite nitrate selenate chromate uranyl pertechnetate and the transition
metals AgI AuIII CuII and HgII as well as organic pollutants including halogenated
ethanes ethenes and methanes (Hansen et al 1994 Hansen et al 1996 Myneni et
al 1997 Erbs et al 1999 Loyaux-Lawniczak et al 1999 Cui amp Spahiu 2002
Lee amp Batchelor 2002b Heasman et al 2003 OrsquoLoughlin et al 2003a amp 2003b
Pepper et al 2003 Elsner et al 2004 OrsquoLoughlin amp Burris 2004) Thus through
sequestration and reductive transformation GRs may play an important role in
controlling the fate and transport of contaminants in suboxic soils and sediments
In a previous study the effects of interlayer anion and Fe(II)Fe(III) ratio in GRs
on the reduction rate of nitrate were investigated (Hansen et al 2001) It was
found that the rate of nitrate reduction to ammonium increased with increasing
Fe(II)Fe(III) ratio and decreased when exchanging a monovalent interlayer anion
(chloride) with a divalent anion (sulphate) The results suggest that for anionic
oxidants like nitrate Fe(II) within the hydroxide layer is available from the outside
basal planes and from the edges as well as through the interlayer under certain
conditions (Figure 42) However oxidants with different charge properties
(cations neutral molecules) may exhibit different affinities for the various reactive
Fe(II) sites present in GR
As the reactive sites are located inat the Fe(II)-Fe(III) hydroxide layers the rate of
reaction depends on the hydroxide layer area which can be accessed by the
oxidant If the oxidant can exchange with the interlayer anion reaction can take
place both at outer and inner surfaces of the GR particles and in total more
reactive sites are available for the reaction However it was found that nitrate
cannot penetrate the interlayer when carbonate or sulphate constitutes the
interlayer anions (Hansen amp Koch 1998) This agrees with the fact that the
interlayers have a lower affinity for monovalent anions than for divalent anions
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 69
(Miyata 1983) However when nitrate was forced into the interlayer by extracting
the interlayer sulphate through precipitation of barium sulphate outside the GR
particles the observed 40 fold increase in rate of nitrate reduction almost equalled
the increase in exposed surface area of the Fe(II)-Fe(III) hydroxide layers (Hansen
amp Koch 1998) From these observations it is expected that the rate of reaction
depends on the particular GR form the crystallite size and the ease with which an
oxidant can exchange with An- in the GR interlayer (Figure 42) Due to
electrostatic interactions we expect anions to be attracted to the positively charged
outer and inner surfaces to a higher degree than cations and neutral compounds If
this theory holds we may expect oxidants with similar intrinsic reactivity (similar
one-electron reduction potentials) to react in the following order anionic gt non-
charged gt cationic (Figure 43) granting that we do not normalise the rate
constants with respect to the amount of oxidant sorbed
Figure 42 Reaction of a probe compound at basal planes at edges and in the interlayer of GR
The hypothesis only holds in cases where the oxidants possess the same intrinsic
reactivities If the relative reactivities of the probe compounds differ greatly from
what would be expected when considering only their reduction potentials
70 Chapter 4
compound specific effects such as charge properties might explain this and the
relative reactivities may follow a pattern like the one depicted in Figure 43
Figure 43 Hypothetical plot of observed reaction rate constants for the reactions between
cationic neutral and anionic probe compounds and GR-SO4 assuming that the oxidant charge
controls its reactivity towards GR
In this work we investigated the reductive transformation of NACs by GR-SO4
Furthermore the effect of NAC charge on the rate of reaction and the possible
access to the internal reactive sites in GR-SO4 were assessed When quantifying
Fe(II) in GRs by means of acid digestion it is not possible to distinguish between
the reactive sites accessible from the outside (at the basal planes or at the edges) or
through the interlayer However we designed an indirect method to gain insight
into the relative importance of the various reactive sites by using a series of
structurally closely related compounds with different charge properties as ldquoreactive
probesrdquo Neutral and anionic probes were needed in order to access all Fe(II)
reactive sites According to our hypothesis cationic and non-charged oxidants
should provide information about the reactivity of the outer Fe(II) reactive sites in
GR whereas the anionic oxidants should provide information about the reactivity
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 71
of both outer and inner Fe(II) reactive sites We chose five nitro aromatic
compounds (NACs) - representing an important group of reducible organic
pollutants - as probe compounds (Figure 43) This class of compounds is not only
of great environmental concern but also comprises suitable model compounds for
studying redox reactions potentially relevant in the environment Moreover they
react readily with Fe(II) surface species associated with iron oxides or clay
minerals transforming them into well-defined easily detected products allowing
mass and electron balances to be established (Hofstetter et al 2003 Klausen et al
1995 Schultz amp Grundl 2000) Our main goals were to establish the rate law and
estimate the surface area-normalised reaction rates for the reaction of the probe
compounds with GR-SO4 in order to assess the importance of the Fe(II) reactive
sites accessible through the interlayer relative to the Fe(II) reactive sites accessible
at the outer surface in GR-SO4
4 2 Materials and methods
All handling and sampling of solutions and suspensions were carried out under
strict anoxic conditions All chemicals were pa quality or better Methanolic stock
solutions (5 mM) of nitrobenzene (NB) 4-nitrotoluene (4-NT) 4-
chloronitrobenzene (4-CNB) and 4-nitrophenylacetic acid (4-NPA) were prepared
in deoxygenated methanol Several attempts to synthesize the cationic probe
compound 4-(NNN-trimethylammonium)-nitrobenzene failed and therefore the
study had to be carried out with only neutral and anionic oxidants The sulphate
GR form was chosen as it is the most stable form and thus the easiest to work
with in the lab
421 Synthesis of GR-SO4
GR-SO4 was synthesized by controlled air oxidation of an FeSO4 solution at a
constant pH of 700 according to the procedure given by Koch amp Hansen (1997)
The GR-SO4 suspension was washed with deoxygenated deionised water and
72 Chapter 4
separated on a folding filter redispersed in deoxygenated 25 mM Na2SO4(aq) in
order to stabilize the GR-SO4 and prevent it from transforming into magnetite
spontaneously Washing separation and redispersion of the GR-SO4 suspension
were conducted in an anoxic glove box (Coy Laboratory Products Inc) All
suspensions and solutions were deoxygenated by Ar-purging (999998 Ar
Carbagas)
422 Mineral characterisation
The identity and purity of the GR-SO4 suspensions were examined by means of X-
ray diffraction measurements The XRD analyses were performed on a Scintag
XDS 2000 using Cu Kα radiation (45 kV 40 mA) Glycerol smears made
according to Hansen (1989) were scanned between 6 and 80 deg2θ with a scan speed
of 1 deg2θmin
423 Lyophilization and determination of specific surface area
Simple air-drying of the GR mineral in the glove box resulted in big flakes with
very low surface areas hence a more suitable lyophilization method was adopted
from Elsner et al (2004) The GR-SO4 suspensions were lyophilised using
Schlenk-type glassware The set-up consisted of a 1 L round bottom flask and a
200 mL glass finger connected by a crescent-shaped bridge equipped with an
evacuation outlet and a stopcock All ground joints and fittings were attached using
high-vacuum grease The washed and resuspended GR-SO4 suspensions were
filled into the glass finger and the freeze-drying apparatus was assembled and
closed before taking it out of the glove box The suspension was frozen by
carefully submerging the lower part of the glass finger into liquid nitrogen for a
few hours Subsequently the evacuation outlet was connected to a vacuum pump
by a metal hose Following a short evacuation of the metal hose the lyophilization
apparatus was evacuated for several minutes by gently opening the stopcock The
evacuation was terminated by closing the stopcock and disconnecting the vacuum
pump The apparatus position was now reversed by removing the glass finger from
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 73
and immersing the round bottom flask into liquid nitrogen As any other
lyophilization method this method depends on sublimation of the ice from the
frozen sample and its recondensation on a cool surface in this case the round
bottom flask Generally it took 1-2 d for the mineral to dry The apparatus was
disassemled in the glove box and the fine powder stored under anoxic conditions
The specific surface area (SSA) of GR-SO4 was determined by the BET multi-
point method using N2 adsorption (Brunauer et al 1938) Powder samples were
filled into sample burettes in the glove box and the generously greased stopcocks
closed Samples and burettes were evacuated prior to connecting them to the BET-
instrument (Sorptomatic 1990 Fisons)
424 Estimation of the one-electron reduction potential for 4-NPA
Kinetic experiments in 100 mL Viton stoppered and alu-crimp capped serum vials
were carried out under the exclusion of oxygen as described by Hofstetter et al
(1999) The homogeneous aqueous solutions contained 50 mM KH2PO4 buffer
(pH = 660) 5 mM Na2S redox buffer and 20 microM juglone (8-hydroxy-14-
naphthoquinone) added as deoxygenated 20 mM methanolic stock solution The
solutions were equilibrated at least one day prior to 4-NPA addition To start the
reaction 50 microM 4-NPA was added as deoxygenated 20 mM methanolic stock
solution The vials were agitated on a roller apparatus in the dark at 21ordmC Control
experiments were prepared similarly except for the addition of juglone At
different time intervals aqueous samples were withdrawn with a syringe and
collected in 18 mL HPLC vials containing 100 microL 1 M HCl The sample vials
were sealed with Teflon-coated silicone septa and plastic screw caps and vortexed
for 10 s The samples were stored at -20degC and analysed without further treatment
For comparison experiments with 4-NT were also conducted See Supporting
Information 71 for more information on the one-electron reduction potentials
74 Chapter 4
425 Kinetic experiments
All reactions took place at pH 84-86 where GR-SO4 tends to stabilize and buffer
itself Samples for Fe(II) and XRD analysis were withdrawn prior to reaction Due
to the fast reactions the experiments were conducted in 10 mL single-use
polyethylene syringes (BD Plastipak) in the glove box To start reaction 40-200
microL 5 mM methanolic stock solutions of NAC were quickly added to 10 mL GR-
SO4 suspension (1-12 mM Fe(II)GR) washed and resuspended in 25 mM
Na2SO4(aq) A Teflon filter (25 mm x 02 microm BGB Analytik) was quickly
mounted on the tip of the syringe and the syringe was vigorously shaken between
sampling At different time intervals filtered suspension samples were collected in
18 mL HPLC vials The HPLC vials were sealed with Teflon-coated silicone septa
and plastic screw caps The samples were stored at -20degC and analysed without
further treatment Absorption of NAC in the syringe and in the Teflon filter
evaluated in blank experiments with NAC added to 25 mM Na2SO4(aq) was found
to be negligible
426 Analytical methods
Initial total and aqueous Fe(II) were determined using a modified phenanthroline
method (Fadrus and Maly 1975) In order to determine [Fe(II)aq] and [Fe(II)total] 1
mL filtered (022 microm) and 1 mL unfiltered GR-SO4 suspension samples were
withdrawn and each treated with 18 mL 01 M HCl for at least 30 min From these
acid digests 01 mL was added to 05 mL Fe(II)-reagent and 19 mL deionised
water (DIW) added up The Fe(II) content in GR-SO4 was estimated as the
difference [Fe(II)GR] = [Fe(II)total] - [Fe(II)aq] The NACs and their corresponding
intermediates and products formed during reduction by GR-SO4 were quantified by
reversed-phase HPLC Separation was performed on a LiChrospher 100 RP-18 (5
microm 125 times 4 mm ID) reversed-phase column coupled with a LiChroCART 100 RP-
18 (4 times 4 mm ID) precolumn Analytical conditions were isocratic and the eluent
consisted of 10 mM hydroxylammonium chloride in various DIWCH3OH
mixtures (vv 3565 and pH 70 for 4-NT and 4-CNB 955 and pH 60 for
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 75
4-NPA) The injection volume was 20 microLand the flow-rate 10 mLmin HPLC
analyses were performed using a Gynkotek High Precision Pump M480 Gynkotek
Gina 50 autosampler and a diode array UV detector (340s Gynkotek) UV-VIS
detection was carried out at the wavelengths of maximum absorption for the
various nitro aromatic and aniline analytes
43 Results and discussion
431 Productformation and reaction kinetics
The reduction of the aromatic nitro group occurs via nitroso- and hydroxylamino-
intermediates where 2 electrons are transferred in each reaction step (Figure 44)
0 --0 H OH H H --0 N N N N
2e- 2H+ H20 + 2e-~ 2e- 2H+ H20
~ ~ R R R
Nitro benzene Nitrosobenzene Hydroxylamine Aniline
Figure 44 Reductive transfonnation pathway of NA Cs
Thus in order to reduce 1 Ar-N02 completely to Ar-NH2 6 electrons
corresponding to 6 mol Fe(II) are needed As magnetite was the major iron phase
formed during reaction (XRD results not shown) we assume the following
reaction stoichiometry
The aniline product was not formed at the same rate as the nitro compound
degraded which is consistent with the detection of early eluting hydroxylamine
intermediates during the course of the reaction (Figure 45a amp 45c) No traces of
76 Chapter 4
nitrosobenzene intermediates or side products such as azoxy- azo- or
hydrazobenzene were found In Figure 45 pseudo 1 order kinetic plots and ln
[Ar-NO2]t[Ar-NO2]0) versus time plots for the neutral probe compounds 4-CNB
and 4-NT are shown as examples The plots for NB and 4-NPA look similar
Figure 45 a Concentration versus time plots for reaction of GR-SO4 with 4-CNB ([Fe(II)GR]0 =
126 mM [4-CNB]0 = 30 microM) b ln [Ar-NO2]t[Ar-NO2]0) versus time plots for reaction of GR-
SO4 with 4-CNB ([Fe(II)GR]0 = 126 mM + [4-CNB]0 = 30 microM [Fe(II)GR]0 = 63 mM + [4-
CNB]0 = 50 microM) c Concentration versus time plots for reaction of GR-SO4 with 4-NT
([Fe(II)GR]0 = 131 mM [4-NT]0 = 20 microM) d ln [Ar-NO2]t[Ar-NO2]0) versus time plots for
reaction of GR-SO4 with 4-NT ([Fe(II)GR]0 = 131 mM [4-NT]0 = 20 microM [Fe(II)GR]0 = 131
mM [4-NT]0 = 50 microM) The hydroxylamino intermediate shown in microM equals the deficit in the
mass balance and in abs equals the detector response (peak area) Solid lines represent 1 order
kinetic fits (a amp c) and ln [Ar-NO2]t[Ar-NO2]0) versus time fits (d) whereas symbols and dotted
lines represent actual data
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 77
At intial Fe(II)GR concentrations in large excess of initial Ar-NO2 concentration
we found a pseudo 1 order rate law for the degradation of Ar-NO2 by GR-SO4
[ ] [ ] [ b 2
a GR
2 ArNOFe(II) ArNOsdotsdot=minus k
dtd ]
where a = 1 b = 1 and the observed pseudo 1 order rate constant kobs = k middot
[Fe(II)GR] At high [Fe(II)GR]0[Ar-NO2]0 ratios the nitro compound was
transformed completely into the aniline product within reaction duration and the
degradation curves of the nitro compound were shaped according to pseudo 1
order kinetics (data points follow solid line in Figure 45a) In some instances ie
at low [Fe(II)GR]0[Ar-NO2]0 ratios the reactions did not follow pseudo 1 order
kinetics for the whole duration of reaction (data points deviate from solid line in
Figure 45c) Hence in order to allow comparison all the pseudo 1 order rate
constants were calculated as initial rates (ie max first two half-lives) from linear
fits of (time ln [Ar-NO2]t[Ar-NO2]0)-plots (Figure 45b amp 45d) Surface area-
normalised pseudo 1 order rate constants are shown in Table 41
Tabl
e 4
1 S
urfa
ce a
rea-
norm
alis
ed p
seud
o 1
ord
er ra
te c
onst
ants
for t
he re
duct
ive
trans
form
atio
n of
4-n
itrot
olue
ne (4
-NT)
4-
chlo
roni
trobe
nzen
e (4
-CN
B) a
nd 4
-nitr
ophe
nyla
cetic
aci
d (4
-NPA
) by
GR
-SO
4
Exp
erim
ent
Age
GR
(d
) [F
e(II
) GR] 0
(mM
) [N
AC
] 0 (micro
M)
[Fe(
II) G
R] 0
[N
AC
] 0∆[
ArN
O2]
(microM
) af b
k obs
(s-1
) ck o
bs (s
-1middotm
-2middotL
) d
GR
-SO
4 + 4
-NT
3 1
103
20
51
5
109
54
5
7
65middot1
0-46
95middot1
0-5
GR
-SO
4 + 4
-NT
3 1
103
50
20
6
174
34
8
7
41middot1
0-46
74middot1
0-5
GR
-SO
4 + 4
-NT
3 1
103
10
0 10
3
214
21
4
2
63middot1
0-42
39middot1
0-5
GR
-SO
4 + 4
-CN
B 1
1
103
55
18
7
177
32
2
4
21middot1
0-43
83middot1
0-5
GR
-SO
4 + 4
-CN
B 1
1
103
10
2 10
1
165
16
2
2
37middot1
0-42
15middot1
0-5
GR
-SO
4 + 4
-NPA
1
1 1
03
25
412
9
7 38
8
4
82middot1
0-44
38middot1
0-5
GR
-SO
4 + 4
-NPA
1
1 1
03
46
224
9
9 21
5
6
37middot1
0-45
79middot1
0-5
GR
-SO
4 + 4
-NPA
1
1 1
03
100
103
13
7
137
196
middot10-4
178
middot10-5
GR
-SO
4 + 4
-NT
2 15
1
31
20
655
5
4 27
0
6
74middot1
0-44
82middot1
0-5
GR
-SO
4 + 4
-NT
2 15
1
31
50
262
9
7 19
4
5
89middot1
0-44
21middot1
0-5
GR
-SO
4 + 4
-NT
4 2
126
0 50
25
2 49
1
982
110
middot10-2
817
middot10-5
GR
-SO
4 + 4
-NT
4 2
630
50
12
6 42
6
852
186
middot10-3
276
middot10-5
GR
-SO
4 + 4
-CN
B 2
2
126
0 30
42
0 29
0
967
925
middot10-3
687
middot10-5
GR
-SO
4 + 4
-CN
B 2
2
630
50
12
6 38
0
760
136
middot10-3
202
middot10-5
GR
-SO
4 + 4
-NPA
2
2 12
60
40
315
371
92
8
5
96middot1
0-34
43middot1
0-5
GR
-SO
4 + 4
-NPA
2
2 6
30
45
140
273
60
7
1
09middot1
0-31
62middot1
0-5
a A
mou
nt o
f NA
C re
duce
d by
GR
-SO
4 at r
eact
ion
term
inat
ion
b F
ract
ion
of in
itial
ly a
dded
NA
C tr
ansf
orm
ed b
y G
R-S
O4 a
t rea
ctio
n te
rmin
atio
n c
Pse
udo
1
orde
r rat
e co
nsta
nts c
alcu
late
d as
initi
al ra
tes
ie m
ax f
irst t
wo
half-
lives
d S
urfa
ce a
rea-
norm
alis
ed p
seud
o 1
ord
er ra
te c
onst
ants
The
are
a of
GR
-SO
4 per
L
su
spen
sion
was
cal
cula
ted
as frac14
middot[Fe
(II)
GR] 0middot
600
gmiddotm
ol-1
middot71
2 m
2 middotg-1
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 79
432 Comparison of rate constants for the different NACs
Even for NACs holding very different one-electron transfer reduction potentials
( ) their reactivities differed only little in Fe(II)-Fe(III) systems such as the
Fe(II)goethite system (slope a = 06 for linear free energy relationship (LFER)
between k
1hE
obs and Hofstetter et al 1999) and the Fe(II)magnetite system
(LFER slope a = 034 Klausen et al 1995) When considering only the for the
reductive transformation reactions of the NACs applied in this study (Table 42)
we expect the surface area-normalised pseudo 1 order rate constants for the
reduction of the NACs to follow the order 4-CNB gt NB gt 4-NT gt 4-NPA Based
on log k
1hE
1hE
obs versus correlations obtained in Fe(II)goethite systems we expect 4-
CNB to react 6 times faster than 4-NPA (Hofstetter et al 1999)
1hE
Table 42 One-electron reduction potentials and relative reactivities in Fe(II)-magnetite and GR-
SO4 systems for the nitro aromatic probe compounds
Compound pKa Eh1
acute (mV) krel (Fe3O4) cd krel (GR-SO4) ce
4-Chlornitrobenzene - -450 a 122 148
Nitrobenzene - -486 a 1 1
4-Nitrotoluene - -500 a 057 176
4-Nitrophenylacetic acid 385 -543 b - 123 a Values from references cited in Hofstetter et al 1999 b Estimated at pH 660 using a LFER (Hofstetter et al 1999 see Supporting Information 71) c Reactivity relative to NB d Values from Klausen et al 1995 e Values from this work
A comparison of the relative rate constants of the NACs obtained for their
transformation by GR-SO4 (this work) and by magnetite (Klausen et al 1995)
shows that they do not differ significantly from each other in any of the mineral
systems (Table 42) When considering charge effects we expect the anionic probe
compounds to react faster with GR-SO4 than the neutral probe compounds
provided that they sorb preferentially within the GR-SO4 interlayers and that Fe(II)
in the interlayers are equally or more reactive than external Fe(II) sites Still the
surface area-normalised kobs values obtained for NB 4-NT 4-CNB and 4-NPA
under various experimental conditions did not differ significantly from each other
80 Chapter 4
(Figure 46 Table 41) The anionic probe compound 4-NPA did not react
significantly faster with GR-SO4 than the neutral probe compounds NB 4-NT and
4-CNB This may indicate that 4-NPA does not significantly interact with reactive
Fe(II) sites in the interlayer Alternatively the negative charge carried by 4-NPA
may be compensating for the lower intrinsic reactivity as compared to the neutral
probe compounds thus explaining the similarity in rate constants for 4-NPA and
the neutral probe compounds Finally other factors than intrinsic reactivity or
charge of the probe compounds such as regeneration of reactive sites or formation
of the magnetite phases may control the overall reactivity of the system
Figure 46 Actual plot of surface area-normalised pseudo 1 order rate constants for the reactions
between neutral and anionic probe compounds and GR-SO4
In heterogeneous reactions mass transfer in bulk solution becomes the rate-
limiting step when the surface reaction is much faster than the diffusion of the
reacting species to the reactive surface In cases where mass transfer controls the
overall rate of reaction the observed pseudo 1 order rate constant kobs ge kLmiddota
where kL is the calculated mass transfer coefficient (mmiddots-1) and a is the ratio of the
external (geometric) specific surface area to volume of solution (m-1) (see
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 81
Supporting Information 72) Mass transfer controlled reactions between GR-SO4
particles and the NACs in bulk solution would explain the similar pseudo 1 order
rate constants obtained for the NACs in this work However when comparing our
estimates of kLmiddota with kobs (see Supporting Information 72) we found that the rates
of mass transfer for all 4 NACs exceed the observed rate constants by at least 3
orders of magnitude at every initial Fe(II)GR concentration Thus the reactions of
the given NACs with GR-SO4 are not likely to be mass transfer limited under the
experimental conditions applied here
Since mass transfer in bulk solution does not control the reaction between GR-SO4
and NACs the overall reaction rate may be surface saturation controlled During
the reductive transformation of NACs not only the parent compound but also
various intermediates forming may compete for the restricted number of reactive
sites present in GR-SO4 This competition may constitute the rate limiting step in
the overall reactivity and may even be enhanced if the number of reactive sites is
depleted during reaction However surface saturation kinetics would not explain
the unexpected similarity of the pseudo 1 order rate constants obtained for the
NACs but it could explain the bent curves observed at low initial Fe(II)GR
concentrations (Figure 45d) The kinetically deviating cases at low [Fe(II)GR]0
were evaluated according to Langmuir-Hinshelwood kinetics (see Supporting
Information 72) Our experimental data did not agree with the Langmuir-
Hinshelwood rate law for any of the NACs (regression results not shown)
Simplifying the rate law by assuming that the aniline product or the
hydroxylamino intermediate or both did not compete for the reactive sites did not
improve the regression Thus the Langmuir-Hinshelwood model cannot explain
the deviations from pseudo 1 order kinetics observed at [Fe(II)GR]0 in our GR-SO4
system and it does not suffice as the correct reaction mechanism nor as the rate-
limiting step
82 Chapter 4
If the adsorption follows a saturation-type sorption isoterm (eg Langmuir) the
sorbate (oxidant) concentration at the surface will vary non-linearily with the total
amount of oxidant added This dependence will have to be taken into account when
establishing rate laws for the heterogeneous reactions and when testing the
hypothesis that the reaction rates depend on the sorbed concentration of the
oxidants However at the high reaction rates observed here we could not quantify
sorption Since the measured initial NAC concentrations corresponded to the
nominal amount of NAC added we assume that transformation and not sorption
was responsible for the consumption of NAC
433 Factors influencing the reaction rate
In general numerous compound- and system-specific factors influence redox
reactions One very important factor is pH which influences the speciation of
dissociable compounds as well as the stability of GR and the formation of other
iron minerals in the system pH has a strong impact on the sorption and therefore
the availability of ionisable oxidants such as carboxylic acids At pH ~ 84 where
our experiments were conducted 4-NPA (pKa = 385) is completely dissociated
Our experiments conducted with GR-SO4 and NB showed that pH was constant
during reaction In addition solution pH has an effect on the surface speciation
From other Fe(II)-Fe(III) systems such as Fe(II) surface species associated with
iron oxides or clay minerals it is well-known that other reactive hydroxylated
Fe(II)-Fe(III)-hydroxo surface complexes can form at higher pH (Charlet et al
1998 Liger et al 1999) Williams amp Scherer (2001) reported a small decrease (5
fold) in the reduction rate of chromate with GR-CO3 when increasing pH from 50
to 90 This decrease may be due to the alternating speciation of the Fe(II) surface
sites on GR-CO3 and of chromate in solution (pKa (H2CrO4) = 08 pKa (HCrO4ndash) =
65) when raising pH (Williams amp Scherer 2001) In contrast other studies have
reported small increases (4 fold) in the reduction rates of nitrate and
trichloroethene with GR-SO4 when increasing pH from 71 to 84 and from 68 to
101 respectively (Koch amp Hansen 1997 Lee amp Batchelor 2002b)
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 83
In this work all experiments were carried out in the presence of 25 mM
Na2SO4(aq) in order to minimize GR-SO4 dissolution and spontaneous
transformation into magnetite Preliminary results from experiments conducted
with NB show that the bulk concentration of Na2SO4 has only a very small impact
on the rate ie increasing the concentration of Na2SO4(aq) in the GR-SO4
suspension from 5 to 25 mM reduced the observed rate constant by a factor of 2
At Na2SO4 concentrations above 25 mM the effect leveled off and therefore
[Na2SO4] = 25 mM was chosen for this work Portions of the same GR-SO4
suspension were used for kinetic experiments over a period of two weeks No
significant aging effects eg rate constants decreasing as a function of GR age
were observed within this time frame
434 Comparison with rate constants obtained for other Fe(II) containing
mineral systems
A recent study compared the reactivity of various Fe(II) containing iron mineral
systems towards organic probe compounds representing different classes of
pollutants (Elsner et al 2004) The reductive transformation of 4-CNB was
investigated for the Fe(III) minerals goethite (α-FeOOH) lepidocrocite (γ-
FeOOH) and hematite (α-Fe2O3) as well as for the Fe(II)-Fe(III) oxide magnetite
(Fe3O4) All experiments were conducted in the presence of 1 mM dissolved Fe(II)
at pH 72 As seen from the surface area-normalised pseudo 1 order rate constants
in Figure 47 the reduction of 4-CNB by the Fe(II)-amended goethite
lepidocrocite and magnetite systems was up to 40 times faster than its reduction by
GR-SO4 The reduction rate obtained for the Fe(II)hematite system was only
slightly higher than the rate for the blank containing no iron mineral but aqueous
Fe(II) solely
84 Chapter 4
Figure 47 Surface area-normalised pseudo 1 order rate constants for the degradation of 4-CNB
by GR-SO4 (open square this work) and various Fe(II) containing mineral systems (solid circles
Elsner et al 2004) Experimental conditions applied by Elsner et al 1 mM aqueous Fe(II) pH
72 25 m2 mineral surface areaL GR-SO4 = green rust sulphate α-FeOOH = goethite Fe3O4 =
magnetite γ-FeOOH = lepidocrocite α-Fe2O3 = hematite
The experiments with GR-SO4 in our study were carried out at pH 84 whereas the
experiments comprising the other systems in Figure 47 were conducted at pH 72
As the reactivity of GR is expected to increase with pH (Lee amp Batchelor 2002b
Koch amp Hansen 1997) the lower of GR-SO4 compared to other Fe(II) systems
cannot be explained by differences in pH values GR-SO4 might just contain fewer
or less reactive surface sites than Fe(II)-amended goethite lepidocrocite and
magnetite suspensions These findings contrast those of other studies which found
higher surface area-normalised pseudo 1 order rate constants for dechlorination
reactions for GR-SO4 than for magnetite (Lee amp Batchelor 2002a amp 2002b Elsner
et al 2004) The different reactivity orders of the Fe(II)-bearing minerals found
for chlorinated aliphatics and nitro aromatics suggest that effects other than pH and
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 85
the intrinsic reduction potentials of the reacting species play a role for the
reactivity of these Fe(II)-bearing minerals
435 Depletion of reactive sites
Assuming that the platy hexagonal GR-SO4 crystals hold an average width of 1 microm
and an average particle thickness of 35 nm (Hansen amp Koch 1998) the ratio of
outer surface area to total surface area AouterAtotal ~ 131 (see Supporting
Information 73) This means that only 3 of the total surface area in GR-SO4 is
available at the external surface Thus as the interlayer sulphate in GR-SO4 is not
readily exchanged with the anionic NAC applied we conclude that 4-NPA and
other NACs only react with the Fe(II) sites at the external GR-SO4 surface In
Table 43 the actual amounts of NAC reduced by GR-SO4 during reaction is
compared with the amount of NAC which theoretically can be reduced by the
initial amount of external reactive Fe(II) sites in GR-SO4 at AouterAtot ~ 131
Table 43 The actual amounts of NAC reduced by GR-SO4 during reaction ∆[NAC]act
compared with the amount of NAC which stoichiometrically should be reduced by the initial
amount of external reactive Fe(II) sites in GR-SO4 ∆[Ar-NO2]theory (calculated as
[Fe(II)GR]0(31middot6) assuming an even distribution of Fe(II) throughout the GR-SO4 structure)
Calculated for [NAC]0 ~ 50 microM
[Fe(II)GR]0 (mM)
∆[Ar-NO2]theory(microM)
∆[4-NT]act (microM)
∆[4-CNB]act (microM)
∆[4-NPA]act (microM)
103 55 174 177 99
63 339 426 380 273
As seen in Table 43 the actual amounts of NAC reduced by GR-SO4 during
reaction are in most cases higher than the amount of NAC which should be
reduced at the given [Fe(II)GR]0 according to reaction stoichiometry This indicates
that new external reactive sites were regenerated eg the Fe(III) phases produced
peel off the GR surface exposing new Fe(II) sites or that outermost internal
86 Chapter 4
reactive sites in close vicinity to the edges are available for reaction as well Lee
and Batchelor (2000b) also found the experimentally observed reduction capacity
of GR-SO4 for chlorinated ethylenes to be 2-3 orders of magnitudes lower than the
estimated reduction capacity including all Fe(II) in GR-SO4
At low initial Fe(II)GR concentrations only a fraction of NAC was reduced within
the reaction time observed (Figure 45c) even though there was stoichiometric
excess of Fe(II)-GR present The fraction of initial Ar-NO2 reduced by GR-SO4 at
reaction termination decreased as [Fe(II)GR]0 decreased (Table 41) and was
accompanied by a change in apparent rate laws with time (compare Figures 45b amp
d) In order to explain these observations we propose that the NACs react only at
external reactive Fe(II) sites and that the regeneration of new external reactive sites
is much slower than the reduction of NAC by GR-SO4 Thus the fast reduction of
NAC taking place at the external reactive sites represents the pseudo 1 order
behaviour whereas depletion of external reactive sites and their slow regeneration
are represented by the second bent part of the (time ln [Ar-NO2]t[Ar-NO2]0)-
curves deviating from pseudo 1 order kinetics Hence at low [Fe(II)GR]0 the
regeneration of reactive sites will eventually control the overall reaction rate
Depletion of available Fe(II) was also observed during the fast reduction of
chromate by GR-CO3 when the initial chromate concentration was increased or
when the GR-CO3 suspension was respiked with chromate repeatedly (Williams amp
Scherer 2001)
436 The role of external and internal reactive sites
It is reasonable to assume that GRs hold adsorption properties similar to other
layered double hydroxides such as hydrotalcites The sorption of 246-
trinitrophenol (TNP) and 245-trichlorophenol (TCP) on chloride and carbonate
intercalated hydrotalcites (HT-Cl = Mg3Al(OH)8ClmiddotyH2O HT-CO3 =
Mg6Al2(OH)16CO3middotyH2O) has been investigated (Hermosin et al 1993 Ulibarri et
al 1995 Ulibarri et al 2001) The authors found that the adsorption of TCP on
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 87
HT-CO3 was very low and that TCP adsorbs only on the external surface sites of
HT-CO3 (Hermosin et al 1993) Furthermore is was reported that the adsorption
of TNP on HT was dramatically affected by the nature of the interlayer anion ie
the adsorption of TNP was considerably higher on HT-Cl than on HT-CO3
(Ulibarri et al 2001) For HT-Cl interlayer anion exchange of chloride with TNP
was detected by XRD analysis and an expansion of the characteristic basal d003
spacing from 79 Ǻ to 132 Ǻ confirmed the presence of TNP in the HT interlayer
(Ulibarri et al 1995) Collating the results reported for HTs with GRs it is not
likely that the divalent SO42- in GR-SO4 is exchanged with the monovalent 4-NPA
Chacirctelet et al (1996) investigated the adsorption of mono- and divalent anions
onin the outer and inner adsorption sites in HT by varying the zetapotential with
pH in the presence of various electrolytes The authors found that SO42- adsorbs on
the external HT surfaces by formation of outer-sphere complexes whereas chloride
hardly adsorbed on HT Moreover it was reported that the adsorption of sulphate
onto HT was not strongly affected by the presence of chloride while sulphate on
the contrary inhibited the adsorption of chloride on HT Studies applying
spectroscopic analyses have investigated the sorption of oxyanions at external and
internal GR-SO4 surfaces (Myneni et al 1997 Randall et al 2001) Selenate was
adsorbed only on the outer GR-SO4 surface when added after GR formation
whereas it was primarily coprecipitated into the interlayer when present during GR
formation Thus for selenate its presence during GR formation is a prerequisite of
its incorporation in the GR interlayer Selenate is readily reduced by GR-SO4 and
the rates of reduction of coprecipitated selenate were very similar to the reduction
rates of selenate adsorbed at the outer GR surface (Myneni et al 1997) This
finding suggests that the outer and inner reactive Fe(II) sites in GR-SO4 hold
similar reactivities
Results based on electron microscopy reported that the reduction of uranyl took
place primarily at the edges of hexagonal GR-SO4 particles (OrsquoLoughlin et al
2003a) In another recent study XRD characterization of the GR-SO4 crystals
88 Chapter 4
during reaction with trichloroacetate (TCA) indicated that TCA did not enter the
GR-SO4 interlayer during reaction (Chapter 5 this work) The average GR-SO4
particle thickness perpendicular to the basal plane was constant during reaction
implying that TCA reacts only at the edges and not at the basal planes Assuming
that the platy hexagonal GR-SO4 crystals hold an average width of 1 microm and an
average particle thickness of 35 nm (Hansen amp Koch 1998) the ratio of edge
surface area to outer surface area is AedgeAouter ~ 130 (see Supporting Information
73) Hence only 3 of the outer surface area in GR-SO4 is available at the edges
Once more the regeneration of new external reactive sites is strongly inferred as
the actual amounts of NAC reduced by GR-SO4 during reaction are much higher
than the amount of NAC which may be reduced by the reactive edge sites present
initially Assuming that the NACs react at the edges only and if employing the
AedgeAouter in the estimation of the rate constants the surface area-normalised
pseudo 1 order rate constants for GR-SO4 would be 30 times higher than the rate
constants depicted in Figure 47 Thus the reactivity of GR-SO4 normalised to its
reactive surface area is higher than the reactivity normalised to its outer surface
area determined by the BET method (N2 adsorption)
The reduction of chromate has been examined in the presence of all the common
GR forms (Bond amp Fendorf 2003 Loyaux-Lawniczak et al 1999 Loyaux-
Lawniczak et al 2000 Williams amp Scherer 2001) The results reported by Bond
amp Fendorf (2003) confirm that not only the surface area of GR but also the
interlayer spacing (interlayer anion size) and interlayer anion charge play an
important role for the reaction rate Hence it follows that coordination (size) and
charge of the oxidant determine its access to the internal sites in GRs
The results obtained for all 4 NACs support what has been reported for nitrate and
TCA At [Fe(II)GR]0 = 2-10 mM and [NO3-]0 = 143 mM pseudo 1 order rate
constants for the reduction of nitrate by GR-SO4 were 158middot10-7 s-1middotm-2middotL (Hansen
et al 2001) This reaction rate increased 40 times by adding barium nitrate instead
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 89
of sodium nitrate thereby precipitating the interlayer sulphate as barium sulphate
and enhancing access to the interlayer Though barium addition changes the GR-
SO4 system dramatically it indicates the importance of interlayer anion exchange
(Hansen amp Koch 1998) The rate constant reported for nitrate (no barium added) is
100-1000 smaller than the rate constants obtained for the NACs in this work
Moreover the reaction kinetics for nitrate did not deviate from pseudo 1 order
kinetics At [Fe(II)GR]0 = 025-104 mM and [TCA]0 = 50 microM-1 mM pseudo 1
order rate constants for the reduction of TCA by GR-CO3 or GR-SO4 were 65middot10-7
s-1middotm-2middotL (Chapter 5 this work) The rate constant for TCA is 10-1000 smaller than
the rate constants for the NACs and the reaction kinetics for TCA did not deviate
from pseudo 1 order kinetics This suggests that the overall reductive
transformation of slowly reacting oxidants such as nitrate and TCA is not
controlled by the rate of regeneration of external Fe(II) reactive sites Altogether
the results reported for selenate chromate and nitrate clearly demonstrate that
these anionic oxidants react primarily with external reactive sites in GR-SO4 Only
under certain conditions ie adding the oxidant prior to GR-SO4 formation or
extracting the interlayer sulphate through precipitation with barium outside the
GR-SO4 particles do the oxidants have access to the interlayer Our findings
suggest that both the neutral and anionic nitro aromatic probe compounds applied
here also react exclusively with the external reactive sites in GR-SO4 Supposedly
the neutral and monovalent charge states of the NACs hinder their access to the
GR-SO4 interlayer A divalent anionic nitro aromatic probe compound might
exchange with the interlayer sulphate more readily and gain access to the inner
Fe(II) reactive sites in GR-SO4 only divalent anionic NACs are not commercially
available
44 Conclusions
This work demonstrates that NACs are completely reduced to their corresponding
anilines by GR-SO4 The surface area-normalised pseudo 1 order rate constants
obtained for the reduction of the neutral and anionic NACs by GR-SO4 under
90 Chapter 4
various experimental conditions did not differ significantly from each other despite
their different charges Neither mass transfer control nor surface saturation kinetics
could account for the similarity of the pseudo 1 order rate constants obtained for
the NACs These observations suggest that the anionic NACs do not have an
enhanced access to inner or outer Fe(II)-GR reactive sites as compared to the
neutral NACs Based on our estimations of the molecular sizes of the NACs we
propose that the charge and not the size of the NACs controls their access to the
internal reactive sites in GRs Hence the reaction between NAC and GR-SO4 takes
place primarily at the external reactive Fe(II) sites This work further demonstrated
that the reduction of the NACs by GR-SO4 only followed pseudo 1 order kinetics
throughout the whole reaction at high initial Fe(II)GR concentrations At low initial
Fe(II)GR concentrations the NACs were not reduced completely within the reaction
time observed though according to reaction stoichiometry the total Fe(II)-GR
present should be sufficient to reduce the whole amount of NAC This means that
at some point during the reaction the external reactive Fe(II) sites were depleted
and the regeneration of new external reactive sites was much slower than the
reduction of the NACs by GR-SO4 The reduction of 4-CNB by GR-SO4 reported
here was 10-100 times slower than its reduction by other Fe(II)-Fe(III) systems
such as goethite lepidocrocite and magnetite suspensions amended with Fe(II)
(Elsner et al 2004)
The results obtained in this work infer that under natural geochemical conditions
where GR-SO4 presumably forms in low concentrations the rate of regeneration of
external Fe(II) reactive sites may control the overall reductive transformation of
fast reacting pollutants by GR-SO4 Thus not only abiotic processes such as
interchanging redox conditions created by water level alterations but also the direct
microbial formation through Fe(III)-reducing bacteria may govern the formation of
GRs and the renewal of external Fe(II) reactive sites in GRs This holds both for
natural systems like iron-rich suboxic soils and sediments as well as engineered
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 91
systems like permeable reactive barriers of zero-valent iron implemented for on-
site remediation of organic and inorganic contaminants
Acknowledgments
We would like to thank Henrik T Andersen for performing the NB kinetic experiments and
Hanne Nancke-Krogh for technical assistance in the laboratory
References
Al-Agha MR Burley SD Curtis CD Esson J (1995) Complex cementation textures and authigenic mineral assemblages in recent concretions from the Lincolnshire Wash (east coast UK) driven by Fe(0) to Fe(II) oxidation Journal of the Geological Society 152 157-171 Bernal JD Dasgupta DR Mackay AL (1959) The oxides and hydroxides of iron and their structural inter-relationships Clay Minerals Bulletin 4 15-30 Bond DL Fendorf S (2003) Kinetics and structural constraints of chromate reduction of green rusts Environmental Science and Technology 37 2750-2757 Brunauer S Emmett PH Teller E (1938) Adsorption of gases in multimolecular layers Journal of American Chemical Society 60 309-319 Charlet L Silvester E Liger E (1998) N-compound reduction and actinide immobilisation in surficial fluids by Fe(II) The surface FeIIIFeIIOH0 species as major reductant Chemical Geology 151 85-93 Chacirctelet L Bottero JY Yvon J Bouchelaghem A (1996) Competition between monovalent and divalent anions for calcined and uncalcined hydrotalcite anion exchange and adsorption sites Colloids and Surfaces A Physicochemical and Engineering Aspects 111 167-175 Chaudhuri SK Lack JG Coates JD (2001) Biogenic magnetite formation through anaerobic biooxidation of Fe(II) Applied and Environmental Microbiology 67 2844-2848 Cui D Spahiu K (2002) The reduction of U(VI) on corroded iron under anoxic conditions Radiochemica Acta 90 623-628 Elsner M Haderlein SB Schwarzenbach RP (2004) Reactivity of Fe(II)-bearing minerals towards reductive transformation of organic contaminants Environmental Science and Technology 38 799-807 Erbs M Hansen HCB Olsen CE (1999) Reductive dechlorination of carbon tetrachloride using iron(II)iron(III)-hydroxide-sulphate (green rust) Environmental Science and Technology 33 307-311 Fadrus H Maly J (1975) Suppression of iron(III) interference in the determination of iron(II) in water by the 110-phenanthroline method The Analyst 100 549-554 Fredrickson JK Zachara JM Kennedy DW Dong H Onstott TC Hinman NW Li S
92 Chapter 4 (1998) Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium Geochimica et Cosmochimica Acta 62 3239-3257 Geacutenin JMR Bourrieacute G Trolard F Abdelmoula M Jaffrezic A Refait Ph Maitre V Humbert B Herbillon A (1998) Thermodynamic equilibria in aqueous suspensions of synthetic and natural Fe(II)-Fe(III) green rusts Occurrences of the mineral in hydromorphic soils Environmental Science and Technology 32 1058-1068
Geacutenin JMR Olowe AA Benbouzid-Rollet ND Prieur D Confente M Resiak B (1991) The simultaneous presence of green rust 2 and sulfate reducing bacteria in the corrosion of steel sheet piles in a harbour area Hyperfine Interactions 69 875-878 Glasauer S Weidler PG Langley S Beveridge TJ (2003) Controls on Fe reduction and mineral formation by a subsurface bacterium Geochimica et Cosmochimica Acta 67 1277- 1288 Hansen HCB (1989) Composition stabilization and light absorption of Fe(II)Fe(III) hydroxycarbonate (green rust) Clay Minerals 24 663-669 Hansen HCB Borggaard OK Soslashrensen J (1994) Evaluation of the free energy of formation of iron(II)iron(III)-hydroxidesulphate (Green Rust) and its reduction of nitrite Geochimica et Cosmochimica Acta 58 2599-2608 Hansen HCB Guldberg S Erbs M Koch CB (2001) Kinetics of nitrate reduction by green rusts ndash effects of interlayer anion and Fe(II)Fe(III) ratio Applied Clay Science 18 81-91 Hansen HCB Koch CB (1998) Reduction of nitrate to ammonium by sulphate green rust Activation energy and interlayer reaction mechanism Clay Minerals 33 87-101 Hansen HCB Koch CB Nancke-Krogh H Borggaard OK Soerensen J (1996) Abiotic nitrate reduction to ammonium Key role of green rust Environmental Science and Technology 30 2053-2056 Hansen HCB Poulsen IF (1999) Interaction of synthetic sulphate green rust with phosphate and the crystallization of vivianite Clays and Clay Minerals 47 312-318 Hansen HCB Taylor RM (1991) The use of glycerol intercalates in the exchange of CO3
2- with SO4
2- NO3- or Cl- in pyroaurite-type compounds Clay Minerals 26 311-327
Heasman DM Sherman DM Ragnarsdottir KV (2003) The reduction of aqueous Au3+ by sulfide minerals and green rust phases American Mineralogist 88 725-738 Hermosin MC Pavlovic I Ulibarri MA Cornejo J (1993) Trichlorophenol adsorption on layered double hydroxide a potential sorbent Journal of Environmental Science and Health A28 1875-1888 Hofstetter TB Heijmann CG Haderlein SB Holliger C Schwarzenbach RP (1999) Complete reduction of TNT and other (poly)nitroaromatic compounds under iron-reducing subsurface conditions Environmental Science and Technology 33 1479-1487 Hofstetter TB Schwarzenbach RP Haderlein SB (2003) Reactivity of Fe(II) species associated with clay minerals Environmental Science and Technology 37 519-528
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 93
Klausen J Troumlber SP Haderlein SB Schwarzenbach RP (1995) Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions Environmental Science and Technology 29 2396-2404 Koch CB Hansen HCB (1997) Reduction of nitrate to ammonium by sulphate green rust Advances in GeoEcology 30 373-393 Kukkadapu RK Zachara JM Smith SC Fredrickson JK Liu C (2001) Dissimilatory bacterial reduction of Al-substituted goethite in subsurface sediments Geochimica et Cosmochimica Acta 65 2913-2924 Lee W Batchelor B (2002a) Abiotic reductive dechlorination of chlorinated ethylenes by iron- bearing soil minerals 1 Pyrite and magnetite Environmental Science and Technology 36 5147- 5154 Lee W Batchelor B (2002b) Abiotic reductive dechlorination of chlorinated ethylenes by iron-bearing soil minerals 2 Green rust Environmental Science and Technology 36 5348- 5354 Liger E Charlet L Van Cappellen P (1999) Surface catalysis of uranium (VI) reduction by iron(II) Geochimica et Cosmochimica Acta 63 2939-2955 Loyaux-Lawniczak S Refait Ph Ehrhardt J Lecomte P Geacutenin JMR (2000) Trapping of Cr by formation of ferrihydrite during the reduction of chromate ions by Fe(II)-Fe(III) hydroxysalt green rusts Environmental Science and Technology 34 438-443 Loyaux-Lawniczak S Refait Ph Lecomte P Ehrhardt J Geacutenin JMR (1999) The reduction of chromate ions by Fe(II) layered hydroxides Hydrology and Earth System Sciences 3 593-599 Mendiboure A Schoumlllhorn A (1986) Formation and anion exchange reactions of layered transition metal hydroxides [Ni1-xMx](OH)2(CO3)x2(H2O)z (M = Fe Co) Revue de Chimie Mineacuterale 23 819-827 Miyata S (1983) Anion-exchange properties of hydrotalcite-like compounds Clays and Clay Minerals 31 305-311 Myneni SCB Tokunaga TK Brown Jr GE (1997) Abiotic selenium redox transformations in the presence of Fe(IIIII) oxides Science 278 1106-1109 Nielsen A (1976) Hvid groslashn og sort rust Beskrivelse af en korrosionsskade paring et svoslashmmebassin Nordisk Betong 2 21-24 OLoughlin EJ Burris DR (2004) Reduction of halogenated ethanes by green rust Environmental Toxicology and Chemistry 23 41-48 OLoughlin EJ Kelly SD Cook RE Csencsits R Kemner KM (2003a) Reduction of uranium(VI) by mixed iron(II)iron(III) hydroxide (green rust) Formation of UO2 nanoparticles Environmental Science and Technology 37 721-727
94 Chapter 4 OLoughlin EJ Kelly SD Kemner KM Csencsits R Cook RE (2003b) Reduction of AgI AuIII CuII and HgII by FeIIFeIII hydroxysulfate green rust Chemosphere 53 437-446 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002) Iron (IIIII) hydroxycarbonate green rust formation and stabilization from lepidocrocite bioreduction Environmental Science and Technology 36 16-20 Parmar N Gorby YA Beveridge TJ Ferris FG (2001) Formation of green rust and immobilization of nickel in response to bacterial reduction of hydrous ferric oxide Geomicrobiology Journal 18 375-385 Pepper SE Bunker DJ Bryan ND Livens FR Charnock JM Pattrick RAD Collison D (2003) Treatment of radioactive wastes An X-ray absorption spectroscopy study of the reaction of technetium with green rust Journal of Colloid and Interface Science 268 408- 412 Randall SR Sherman DM Ragnarsdottir KV (2001) Sorption of As(V) on green rust (Fe4(II)Fe2(III)(OH)12SO4
3H2O) and lepidocrocite (γ-FeOOH) Surface complexes from EXAFS spectroscopy Geochimica et Cosmochimica Acta 65 1015-1023 Roh Y Lee SY Elless MP (2000) Characterization of corrosion products in the permeable reactive barriers Environmental Geology 40 184-194 Schultz CA Grundl TJ (2000) pH dependence on reduction rate of 4-Cl-nitrobenzene by Fe(II)montmorillonite systems Environmental Science and Technology 34 3641-3648 Simon L Francois M Refait Ph Renaudin G Lelaurain M Geacutenin JMR (2003) Structure of the Fe(II-III)-layered double hydroxysulphate green rust two from Rietveld analysis Solid State Sciences 5 327-334 Tamaura Y (1986) Ni(II)-bearing green rust II and its spontaneous transformation into Ni(II)- bearing ferrites Bulletin of the Chemical Society of Japan 59 1829-1832 Tamaura Y (1985) ZnII-bearing green rust II and its spontaneous transformation into ZnII- bearing ferrite in aqueous solution Bulletin of the Chemical Society of Japan 58 2951-2954 Trolard F Abdelmoula M Bourrieacute G Humbert B Geacutenin JMR (1996) Mise en eacutevidence dun constituant de type rouilles vertes dans les sols hydromorphes Proposition de lexistence dun nouveau mineacuteral la fougeacuterite Geacuteosciences de surface Comptes Rendus de LrsquoAcademie des Sciences 323 1015-1022 Tuovinen OH Button KS Vuorinen A Carlson L Mair DM Yut LA (1980) Bacterial chemical and mineralogical characteristics of tubercles in distribution pipelines Journal of the American Water Works Association 72 626-635 Ulibarri MA Pavlovic I Barriga C Hermosin MC Cornejo J (2001) Adsorption of anionic species on hydrotalcite-like compounds effect of interlayer anion and crystallinity Applied Clay Science 18 17-27 Ulibarri MA Pavlovic I Hermosin MC Cornejo J (1995) Hydrotalcite-like compounds as potential sorbents of phenols from water Applied Clay Science 10 131-145
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 95
Williams AGB Scherer MM (2001) Kinetics of chromate reduction by carbonate green rust Environmental Science and Technology 35 3488-3494
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 97
5 Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems
Abstract
Trichloroacetate (TCA) is a widespread environmental contaminant with proven
phytotoxicity and suspected human carcinogenicity In order to assess the global
cycling of TCA and to predict its fate in subsurface environments information
regarding the reactivity and product distribution of TCA degradation is needed
Due to the high oxidation state of TCA conditions for oxidative transformation
pathways in soils and groundwater are unfavorable However in suboxic soils and
sediments Fe(II)-bearing minerals are potential reactants for reductive
dehalogenation reactions of TCA as has been demonstrated for other halogenated
contaminants We examined the reactivity of various Fe(II)-Fe(III) mineral
systems towards TCA and dichloroacetate (DCA) its expected transformation
product in laboratory batch experiments imitating natural conditions ie low
initial Fe(II) Fe(III) and TCADCA concentrations and no artificial buffer The
Fe(II)-Fe(III)-systems investigated included sulfate green rust (GR-SO4) carbonate
green rust (GR-CO3) magnetite Fe(II)goethite and Fe(II)lepidocrocite
Trichloroacetate was readily reduced to DCA by all Fe(II)-bearing minerals The
reactions generally followed pseudo 1 order kinetics with respect to TCA The
surface area-normalised pseudo 1 order rate constants obtained (035ndash76middot10-5 min-
1middotm-2middotL at [Fe(II)]0 = 020ndash122 mM [TCA]0 = 15ndash1000 microM and pH 70ndash87)
showed no striking differences regarding product distribution and surface area-
normalised reaction rate constants between the Fe(II)-Fe(III)-systems The
stoichiometrically formed DCA was not further reduced to monochloroacetate
(MCA) or acetate in any of the systems within the time frame in our experiments
To our knowledge this is the first published report on abiotic transformation of
TCA by Fe(II)-bearing minerals Our results imply that processes involving
reactive Fe(II)-bearing minerals may play a significant role in controlling the fate
98 Chapter 5
of TCA in natural subsurface environments and that DCA found in the subsurface
may be formed by such processes
51 Introduction
Trichloroacetic acid (TCA) has been applied as a herbicide for many years until its
use was banned in the late 1980acutes (Berg et al 2000) Today TCA is mainly used
as an etching agent in the metal industry as a swelling solvent in the plastic
production and as a bleaching agent in the paper and pulp manufacture (Muumlller et
al 1996) Other anthropogenic sources include formation of TCA as a result of the
chlorine based disinfecting process used in drinking water treatment and the
atmospheric photooxidation of chlorinated solvents including tetrachloroethene
and 111-trichloroethane (McCulloch 2002) Only very little information is
available on the TCA production volumes and even less is known about the
amount of TCA released into the environment as a result of its industrial
applications Due to its low volatility and high aqueous solubility TCA is easily
washed out of the atmosphere into the aquatic and terrestrial biospheres As TCA
is found in almost every ecosystem around the globe including non-urban and
non-industrial sites the relative contributions from anthropogenic and natural
sources are currently being debated (McCulloch 2002 Ahlers et al 2003)
Trichloroacetic acid is omnipresent in soils and the concentrations reported are
very variable ranging from lt005 microgkg to 380 microgkg (Euro Chlor 2001
McCulloch 2002 Ahlers et al 2003) Both abiotic and enzymatically catalyzed
formation of TCA from humic acids have been demonstrated in laboratory studies
(Haiber et al 1996 Hoekstra et al 1999b Fahimi et al 2003) Furthermore the
in situ natural formation of TCA from anthropogenic or natural tetrachloroethene
or 111-trichloroethane in biota has been suggested (Hoekstra et al 1999a
McCulloch 2002) Such natural sources may explain part of the TCA
concentrations found in soils but their environmental significance is still unknown
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 99
On account of its phytotoxicity suspected human carcinogenicity and widespread
occurrence TCA is of considerable environmental concern especially in the
terrestrial compartment The TCA concentrations found in soil air and water in
pre-industrial times were far below the present ones (Jordan amp Frank 1999 Ahlers
et al 2003) Based on the current TCA concentrations detected in soils the
European Commission proposed risk reduction measures concerning
tetrachloroethene - a precursor of TCA - to be taken immediately (Ahlers et al
2003 and references therein) Occurrences of monochloroacetic acid (MCA) and
dichloroacetic acid (DCA) reported include surface waters marine waters
precipitation ice (glaciers) and air (Reimann et al 1996 Berg et al 2000 Scott et
al 2000 Scott et al 2002) Based on the concentrations reported for the aquatic
environments it is reasonable to assume that MCA and DCA are omnipresent in
soils as well Sources of MCA and DCA include production in the chemical
industry photooxidation of chlorinated aliphatics in the atmosphere and reductive
transformation of TCA (Reimann et al 1996 Ahlers et al 2003 and references
therein) MCA and DCA are also toxins and suspected human carcinogens (Kuumlhn
amp Pattard 1990) hence not only TCA but also its daughter compounds are
pollutants of environmental concern
In subsurface environments TCA may be removed by sorption seepage chemical
transformation microbial degradation and plant uptake followed by metabolic
degradation andor physical removal at harvest (Foy 1975) There is little or no
evidence of abiotic transformations of TCA in the literature Only one recent study
demonstrated the reductive dechlorination of TCA to MCA by Fe(0) (Hozalski et
al 2001) It has been reported that the degradation of TCA in soil is slow and
mainly mediated by microorganisms but only little is known about the bacteria and
processes involved (Lignell et al 1984) Biodegradation of TCA has been found at
both oxic and anoxic conditions An aerobic microorganism capable of growing on
TCA as the sole carbon and energy source has been characterised (Yu amp Welander
1995) Moreover anaerobic bacteria coupling co-metabolic growth to reductive
100 Chapter 5
dechlorination of TCA have been isolated (Weightman et al 1992 De Wever et
al 2000) However more information regarding the abiotic and biotic
transformation of TCA is needed in order to assess the fate and transport of TCA in
natural subsurface environments
It is well-known that Fe(II) present in minerals or associated with mineral surfaces
is a much stronger reductant than Fe(II) in solution The enhanced reactivity of a
structural or surface-bound Fe(II) center can be rationalized by the increased
electron density donated by hydroxyl ligands and a stabilization of the Fe(III)
oxidation state by the hydroxyl ligands (Luther 1990) Fe(II)-bearing minerals
including layered Fe(II)-Fe(III) hydroxides (green rusts) magnetite (Fe3O4)
siderite (FeCO3) Fe(II) sulfides as well as Fe(II)-carrying Fe(III) oxides and clay
minerals have also been shown to reduce a range of organic and inorganic
contaminants such as nitro aromatic compounds chlorinated aliphatics chromate
uranyl pertechnetate nitrate monochloramine and carbamate pesticides (Chapter
4 this work Klausen et al 1995 Cui amp Eriksen 1996 Erbs et al 1999 Liger et
al 1999 Loyaux-Lawniczak et al 1999 Amonette et al 2000 Hansen et al
2001 Pecher et al 2002 Vikesland amp Valentine 2002 Hofstetter et al 2003
OrsquoLoughlin and Burris 2003 OrsquoLoughlin et al 2003a amp 2003b Strathmann amp
Stone 2003 Elsner et al 2004a) Laboratory and field studies showed that even in
geochemically highly heterogeneous anoxic aquifer sediments Fe(II) adsorbed to
Fe(III) (hydr)oxide surfaces was the dominant reductant of nitroaromatic and
halogenated contaminants (Ruumlgge et al 1998 Hofstetter et al 1999 Kenneke amp
Weber 2003) Only little is known about the nature of the Fe(II) species associated
with Fe(III) oxide surfaces but reactive hydroxylated Fe(II)-Fe(III)-hydroxo
surface complexes associated with hematite and magnetite above pH 65 have been
proposed (Charlet et al 1998aampb Liger et al 1999) Due to the presence of
structural Fe(II) within the mineral lattice the reactivity of Fe(II) associated with
mixed valent Fe(II)-Fe(III) minerals such as green rusts magnetite and reduced
ferruginous clay minerals may hold another reactivity than Fe(II) associated with
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 101
pure Fe(III) oxides However Fe(II) adsorbed on Fe(III) oxides such as goethite
hematite and lepidocrocite may also hold different reactivities as the Fe(III) oxides
contain different crystal and surface structures
Since chlorinated ethanes and ethenes such as hexachloroethane 111-
trichloroethane tetrachloroethene and trichloroethene are susceptible to chemical
reduction by a range of Fe(II)-bearing minerals including magnetite GR-SO4
Fe(II) sulfides and Fe(II)-carrying Fe(III) oxides (Butler amp Hayes 1998 amp 1999
Hwang amp Batchelor 2000 Gander et al 2002 Lee amp Batchelor 2002aampb Elsner
et al 2004a) we hypothesized that TCA may be transformed by Fe(II)-bearing
minerals as well The main goals of this work were to study such reactions and
establish product distribution and surface area-normalised reaction rates for the
reductive dechlorination of TCA by Fe(II)-Fe(III) mineral systems common in
nature
5 2 Materials and methods
No synthetic buffers were applied and iron concentrations were kept low The
calcareous systems were pH-controlled at 76 through a natural buffer system
(CaCO3(s) + 995 N205 CO2(g)) All handling and sampling of solutions and
suspensions were carried out under strict anoxic conditions Goethite (acicular
particles with size 01 times 06 microm specific surface area 16 m2g) and lepidocrocite
(acicular particles with size 005 times 03 microm specific surface area 18 m2g) were
purchased as fine powders from Bayer (Bayferrox 910 and 943) Calcite (grain size
170-350 microm Pluumlss-Staufer AG) was used as a buffer or as a Fe(III)-oxide-bearing
mineral In order to simulate natural conditions the iron minerals were applied as
coatings on calcite particles (model system for calcareous soils) in some
experiments Trichloroacetic acid dichloroacetic acid and monochloroacetic acid
were pa quality (Fluka)
102 Chapter 5
521 Synthesis of GRs and magnetite
GR-CO3 was synthesized by controlled air oxidation of an FeCl2 solution at a
constant pH of 700 (titrated with 1 M Na2CO3) according to the procedure given
by Hansen amp Koch (1997) 05 M aqueous stock solutions of FeCl2 were prepared
in 100 mL glass flasks by reacting 65 mmol of iron powder (particle size 10 microm
Merck) with 100 mL deoxygenated 10 M HCl The solutions were magnetically
stirred and heated (~80degC) during reaction until the H2(g) production had ceased (ge
2 hours) The FeCl2 solutions were stored in the dark under a small Ar
overpressure at 5degC The GR-CO3 suspensions were washed with deoxygenated
deionised water (DIW) separated on a folding filter (medium filtration rate cotton
linterhigh alpha pulp Schleicher amp Schuell) and redispersed in deoxygenated
DIW Washing separation and redispersion of the GR-CO3 suspension were
conducted in an anoxic glove box (92 N28 H2 Coy Laboratory Products Inc)
All suspensions and solutions were deoxygenated by Ar-purging (999998 Ar
Carbagas) Magnetite was synthesized by further aerial oxidation of GR-CO3 at pH
700 until consumption of 1 M Na2CO3 ceased GR-SO4 was synthesized by
controlled air oxidation of an FeSO4 solution at a constant pH of 70 according to
the procedure given by Koch amp Hansen (1997) The GR-SO4 suspension was
washed with deoxygenated DIW separated on a glass filter funnel (pore size 4
Duran) and redispersed in deoxygenated DIW Washing separation and
redispersion of the GR-SO4 suspension were conducted in an anoxic glove bag
(999995 Ar Aldrich)
522 Preparation of iron oxide coatings
Two grams of goethite (goe) or lepidocrocite (lep) and 100 g calcite were
combined with 200 mL DIW in a 500 mL polyethylene flask The suspension was
gently agitated on a reciprocating shaker for 24 h and left to stand for another 24 h
Excess Fe(III) oxides and salts were removed from the coated material by repeated
decantation and washing with DIW in polyethylene flasks until clear runoff
Finally the coatings were collected on folding filters and air dried The amount of
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 103
goethite and lepidocrocite coated onto calcite after washing and drying was
quantified to 10-11 mg Fe(III)g calcite
523 Mineral characterisation
The identity and purity of the GR-CO3 GR-SO4 and magnetite suspensions were
examined by means of X-ray diffraction (XRD) The XRD analyses were
performed on a Scintag XDS 2000 using Co Kα radiation (45 kV 40 mA) or a
Siemens D5000 XRD applying Co Kα radiation (40 kV 40 mA) Glycerol smears
made according to Hansen (1989) were scanned between 6 and 80 deg2θ with a scan
speed of 1 deg2θmin The specific surface area (SSA) of calcite was determined by
the BET multi-point method using N2 adsorption (Brunauer et al 1938) Powder
samples were filled into sample burettes in the glove box and the generously
lubricated stopcocks closed Samples and burettes were evacuated prior to
connecting them to the BET-instrument (Sorptomatic 1990 Fisons)
524 Kinetic experiments
All reactions were carried out in 25-100 mL serum vials sealed with stoppers
(Viton or Teflon coated rubber) and aluminum crimp caps Kinetic experiments
were conducted with GR-SO4 GR-CO3 magnetite Fe(II)goethite and
Fe(II)lepidocrocite at room temperature In most cases pH was controlled through
the carbonate-bicarbonate buffer system by adding calcite to suspensions
containing the iron minerals solely or by adding the iron minerals as coatings on
calcite Furthermore the calcite containing suspensions were deoxygenated with
05 CO2995 N2(g) thereby attaining an initial pH of 76-77 The GR-CO3 and
magnetite suspensions were deoxygenated with 100 N2(g) and no additional pH
buffer was added The goethite and lepidocrocite suspensions were amended with
300-1000 microM FeCl2(aq) and equilibrated gt 20 h prior to TCADCA addition See
Table 51 for more details on the experimental conditions To start the reaction 50
microM - 1 mM TCA or DCA was added to the mineral suspensions from aqueous
anoxic stock solutions The reaction vials were agitated gently on a roller apparatus
104 Chapter 5
or a shaking table (35 rpm) in order to minimize abrasion of the iron oxide mineral
coatings At appropriate time intervals suspension samples were withdrawn using
Ar(g)- 100 N2(g)- or 995 N205 CO2(g)-flushed sterile disposable syringes
and hypodermic needles The suspension samples were filtered (02 microm Teflon)
and collected for quantification of chloride and the chlorinated acetic acids The
samples were stored at -20degC and analysed without further treatment
525 Analytical methods
Total and aqueous Fe(II) were determined using a modified phenanthroline method
(Fadrus and Maly 1975) For determining [Fe(II)aq] and [Fe(II)total] 1 mL filtered
(02 microm Teflon) and 1 mL unfiltered mineral suspension were added to 18 mL 01
M HCl respectively and allowed to dissolve for 30 min From these acid digests
01 mL was added to 05 mL Fe(II)-phenanthroline-buffer-reagent and 19 mL
DIW added up Estimates of the structural or adsorbed Fe(II) in the Fe(II)-Fe(III)
mineral systems were estimated as the difference [Fe(II)solid] = [Fe(II)total] -
[Fe(II)aq] The total amount of Fe(III) coated on calcite was determined by atomic
absorption spectroscopy following dissolution in 6 M HCl(aq) for 24 h At low
initial TCA concentrations (le 50 microM) the chlorinated acetic acids were quantified
by means of a modified ion interaction (or paired-ion) chromatographic method
(Sarzanini et al 1999) Separation was performed on a LiChrospher 100 RP-18 (5
microm 125 times 4 mm ID) reversed-phase column coupled with a LiChroCART 100 RP-
18 (4 times 4 mm ID) precolumn Analytical conditions were isocratic and the eluent
consisted of 50 aqueous solution of 35 mM cetyltrimethylammonium chloride
(pH 50) and 50 CH3CN The injection volume was 20 microL and the flow-rate 10
mLmin HPLC analyses of the chloroacetates were performed using a Gynkotek
Pump M480 Gynkotek Gina 50 auto sampler and a diode array UV detector (340s
Gynkotek) UV-VIS detection was carried out at 200 nm At higher initial TCA
concentrations the chlorinated acetic acids were quantified by a modified HPLC
method (Husain et al 1992) Separation was performed on a ChromSphere C-18
(10 microm 250 times 46 mm ID) reversed-phase column Analytical conditions were
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 105
isocratic and the eluent consisted of 015 M (NH4)2SO4(aq) pH 55 The injection
volume was 20 microL and the flow-rate 10 mLmin HPLC analyses were performed
using a Series 10 Liquid Chromatographic Pump (Perkin-Elmer) and a SPD-10 A
VP UV-VIS detector (Shimadzu) UV-VIS detection was carried out at 210 nm
Chloride was determined in the GR-SO4 kinetic experiments using a flow injection
system with spectrophotometric detection (Cheregi amp Danet 1997)
53 Results and discussion
531 Product formation and reaction kinetics
Trichloroacetate was readily reduced to DCA by all the Fe(II)-bearing minerals
examined Only DCA was detected within the reaction time in all the Fe(II)-Fe(III)
mineral systems Experiments conducted with the various Fe(II)-Fe(III) mineral
systems and DCA confirmed that no significant reduction of DCA took place (data
not shown) Hence it is reasonable to assume that the further hydrogenolysis of
DCA to MCA is too slow to be detected within the experimental time frame here
The mass balance of TCA and DCA was almost complete in all suspensions ruling
out any alternative reaction pathways to reductive dechlorination Decarboxylation
of TCA producing chloroform and carbon dioxide requires high temperatures and
is therefore assumed not to take place at the experimental conditions applied here
(Atkins et al 1984) Based on these results we propose that the reductive
dechlorination of TCA by Fe(II)-bearing minerals proceeds via hydrogenolysis
(replacement of halogen by hydrogen) as reported for the transformation of TCA
by zero-valent iron (Hozalski et al 2001) Thus in order to reduce TCA to DCA
2 electrons corresponding to 2 Fe(II) are needed (Figure 51)
106 Chapter 5
Cl3CC
O
O- Cl2HCC
O
O-
2e- H+ Cl-
TCA DCA Figure 51 Proposed reductive transformation pathway of TCA
In the Fe(II)goe and Fe(II)lep systems we detected no TCA transformation in the
absence of either aqueous Fe(II) or pure or calcite-associated goethite and
lepidocrocite These results strongly indicate that reactive Fe(II) species associated
with the goethite and lepidocrocite surfaces are the reductants for TCA in these
systems The Fe(III) phases forming in the mineral suspensions were not
characterised and therefore the reaction stoichiometry cannot be assessed
At initial Fe(II) concentrations in large excess of initial TCA concentration we
found a pseudo 1 order rate law for the degradation of TCA by Fe(II)
[ ] [ ] [ b a TCAFe(II) TCA
sdotsdot=minus kdt
d ]
where a = 1 b = 1 and the observed pseudo 1 order rate constant kobs = k middot [Fe(II)]
At all [Fe(II)]0[TCA]0 ratios studied (6-738) TCA was transformed almost
quantitatively into DCA and the reaction kinetics followed pseudo 1 order kinetics
with respect to TCA (Figure 52) The observed pseudo 1 order rate constants for
the transformation of TCA by the various Fe(II)-Fe(III) mineral systems were
calculated as initial rates (ie max first two half-lives) from linear fits of (time ln
[TCA]t[TCA]0)-plots (Table 51) The amount of chloride produced during
reaction with GR-SO4 was always equivalent to the amount of TCA transformed
into DCA (Figure 52c) This also indicates that no significant further reduction of
DCA took place in GR-SO4 suspensions
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 107
Figure 52 Time course of TCA consumption and DCA and chloride production for a)
Fe(II)Goe ([Fe(II)tot]0 = 095 mM) b) Fe(II)Lep ([Fe(II)tot]0 = 091 mM) c) GR-SO4
([Fe(II)GR]0 = 962 mM) d) GR-CO3 ([Fe(II)tot]0 = 633 mM) and e) Magnetite ([Fe(II)tot]0 = 350
mM) Solid lines represent 1 order kinetic fits whereas symbols and dotted lines represent actual
data = TCA = DCA = Clndash
T
able
51
Exp
erim
enta
l con
ditio
ns a
nd p
seud
o 1
ord
er ra
te c
onst
ants
for t
he re
duct
ive
trans
form
atio
n of
TC
A b
y va
rious
Fe(
II)-
Fe(I
II) c
onta
inin
g m
iner
al sy
stem
s
Syst
em
Susp
ensi
on a
ge
(d)
[Fe(
II)] s
olid
a (m
M)
[Fe(
II)] a
q b
(mM
) [T
CA
] 0 (micro
M)
pHin
itcpH
endd
k obs
e (min
-1)
Surf
ace
area
(m
2 L)
k obs
f
(min
-1m
-2middotL
)
Fe(I
I)aq
1
0
030
434
nd
76
gn
dn
dn
d
Fe(I
I)G
oe
1
002
024
429
nd
78
g1
021
0-47
1 i
143
10-5
Fe(I
I)G
oe
1
013
094
543
77
70
225
10-4
71
i3
161
0-5
Fe(I
I)G
oe
coat
ing
1
023
40
066
484
765
80
g6
401
0-454
0 j
119
10-5
Fe(I
I)G
oe
coat
ing
1
0
150
8048
6n
d7
6 g12
43
10-4
540
j2
301
0-5
Fe(I
I)L
ep
1
0
020
2315
7n
d7
8 g0
751
0-48
0 i
094
10-5
Fe(I
I)L
ep
coat
ing
1
016
30
137
470
765
80
g2
821
0-454
0 j
052
10-5
Fe(I
I)L
ep
coat
ing
1
0
100
8141
7n
d7
7 g8
311
0-454
0 j
154
10-5
Fe3O
41
3
380
1251
38
107
8g
830
10-4
16
k5
311
0-4
Fe3O
477
112
56
556
70
70
153
10-4
52
k2
951
0-5
GR
-CO
31
5
940
3950
37
658
4g
761
10-4
419
l1
821
0-5
GR
-CO
32
7
60
147
88
568
2940
81
0-453
6 l
761
10-5
GR
-CO
332
73
003
563
85
80
490
10-4
515
l0
951
0-5
GR
-CO
314
2
3
530
005
629
nd
87 g
513
10-4
249
l2
061
0-5
GR
-SO
41
5
17-1
217
086
-13
910
5n
dn
d3
601
0-492
6 m
039
10-5
GR
-SO
41
6
22-1
027
093
-14
527
0n
dn
d3
761
0-488
1 m
043
10-5
GR
-SO
41
7
05-1
014
077
-17
950
0n
dn
d3
741
0-4 9
18
m0
411
0-5
GR
-SO
41
5
17-1
051
060
-16
510
00n
dn
d2
891
0-483
7 m
035
10-5
n
d =
not
det
ecte
d a
Ini
tial s
truct
ural
or a
dsor
bed
Fe(I
I) e
stim
ated
as [
Fe(I
I)to
tal]
ndash [F
e(II
) aq]
b In
itial
dis
solv
ed F
e(II
) mea
sure
d c
Sus
pens
ion
pH p
rior t
o TC
A a
dditi
on d
Sus
pens
ion
pH a
t rea
ctio
n
te
rmin
atio
n e
Pse
udo
1 o
rder
rate
con
stan
ts fo
r the
con
sum
ptio
n of
TC
A c
alcu
late
d fr
om in
itial
rate
s (m
ax f
irst t
wo
half-
lives
) f
Surf
ace
area
-nor
mal
ised
pse
udo
1 o
rder
rate
con
stan
ts g
pH
con
trol
th
roug
h pu
re C
aCO
3 and
05
C
O2(g
) h
pH
con
trol t
hrou
gh F
e(II
I) o
xide
-coa
ted
calc
ite a
nd 0
5
CO
2(g)
i Es
timat
ed u
sing
the
SSA
of t
he F
e(II
I) o
xide
app
lied
j E
stim
ated
usi
ng th
e SS
A o
f cal
cite
~1
m2 g
k E
stim
ated
ass
umin
g SS
A =
4 m
2 g (S
chw
ertm
ann
amp C
orne
ll 1
991)
frac12middot[
Fe(I
I) sol
id] 0middot
232
gmiddotm
ol-1
middot4 m
2 middotg-1
l E
stim
ated
ass
umin
g SS
A =
47
m2 g
(Will
iam
s amp S
cher
er 2
001)
frac14middot[F
e(II
) GR] 0middot
600
gmiddotm
ol-1
middot47
m2 middotg
-1 m
Est
imat
ed a
s in l
but u
sing
SSA
= 7
12
m2 middotg
-1 (C
hapt
er 4
thi
s wor
k)
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 109
532 Comparing rate constants obtained for the various Fe(II)-Fe(III) mineral
systems
Data for the systems containing iron oxide coated calcite were very similar to the
data obtained for the pure iron oxides (not shown in Figure 53) Since no SSA was
determined for magnetite in this study a SSA of 4 m2g was assumed
(Schwertmann amp Cornell 1991) However it should be noted that the magnetite
synthesized by Schwertmann and Cornell (1991) was prepared differently (ie
oxidation of Fe(II) by nitrate in a heated alkaline solution) from the magnetite
applied in this study The surface area-normalised pseudo 1 order kobs values
obtained for GR-CO3 GR-SO4 Fe(II)goethite and Fe(II)lepidocrocite were all
within the same order of magnitude (Figure 53a)
Figure 53 Average surface area-normalised pseudo 1 order rate constants for the degradation of
a) TCA (this work) b) hexachloroethane (Elsner et al 2004a) and c) carbon tetrachloride
(Amonette et al 2000 Pecher et al 2002 OrsquoLoughlin et al 2003c Elsner et al 2004b) by
GR-SO4 GR-CO3 (suspension age 1 d) Fe3O4 Fe(II)α-FeOOH and Fe(II)γ-FeOOH
Experimental conditions applied in this work [Fe(II)tot]0 = 025-107 mM in the goethite and
lepidocrocite suspensions [Fe(II)tot]0 = 025-116 mM in the GR-SO4 and GR-CO3 suspensions
pH 70-86 71-926 m2 mineral surface areaL Experimental conditions applied by Elsner et al
1 mM aqueous Fe(II) 25 m2 mineral surface areaL Experimental conditions applied in
references employed in c) [Fe(II)tot]0 = 1-83 mM 25-275 m2 mineral surface areaL GR-SO4 =
110 Chapter 5
sulfate green rust GR-CO3 = carbonate green rust Fe3O4 = magnetite α-FeOOH = goethite γ-
FeOOH = lepidocrocite
When comparing the rate constants for the Fe(II)-Fe(III) mineral systems found for
reduction of TCA in this study (Figure 2a) mixed valent Fe(II)-Fe(III) minerals
such as green rusts and magnetite containing structural Fe(II) within the mineral
lattice do not seem to be significantly more reactive than Fe(II)-Fe(III) mineral
systems containing Fe(II) associated with pure Fe(III) oxides Unlike most other
iron oxides GRs contain not only external Fe(II) reactive sites at the surface but
also internal sites in the space between consecutive Fe(II)-Fe(III) hydroxide layers
The GR interlayer thickness is a function of both the size and the charge of the
interlayer anion For solutes the Fe(II) within the GR hydroxide layer is accessible
at the outside basal planes and at the edges as well as through the interlayer under
certain conditions (see Figure 42 Chapter 4 this work) Due to electrostatic
forces oxidants holding different charge properties (anions cations neutral
molecules) may exhibit different affinities for the various reactive Fe(II) sites
present in GR As the reactive sites are located inat the Fe(II)-Fe(III) hydroxide
layers the rate of reaction depends on the hydroxide layer area which can be
accessed by the oxidant If the oxidant is able to exchange with the interlayer
anion reaction can take place both at outer and inner surfaces of the GR particles
and in total more reactive sites are available for the reaction Thus oxidant size
and charge primarily control its access to the internal sites in GRs XRD
characterization of the GR-SO4 crystals during reaction with TCA demonstrated
that the GR-SO4 interlayer spacing did not vary during reaction (Table 52) This
may indicate that TCA did not enter the GR-SO4 interlayers
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 111 Table 52 Diffraction angle d-spacing and width at half peak height (Wfrac12) for the 001 GR-SO4
diffraction peak as a function of time during reaction with TCA ([Fe(II)GR]0 = 4 mM [TCA]0 = 1
mM)
Time (min) Angle (deg2θ)
d001-spacing(nm)
Wfrac12(degθ)
0 9483 10821 0273
10 9494 10809 0287
215 9522 10777 0263
330 9550 10745 0273
510 9524 10775 0277
855 9509 10791 0253
1160 9467 10839 0268
We roughly estimated the molecular size of TCA by summing the covalent radii of
the individual atoms (see Supporting Information 74) When comparing the
molecular size of TCA with the GR-SO4 interlayer spacing of 061 nm it can be
concluded that only when the C-C bond is oriented perpendicular to the interlayer
plane does the size of TCA exceed the GR-SO4 interlayer spacing In contrast the
size of TCA exceeds the GR-CO3 interlayer spacing (026 nm) regardless of its
orientation Hence if TCA was intercalated in the GR-CO3 interlayer we would
expect the interlayer spacing to expand The same holds for intercalation of a
vertically oriented TCA in the GR-SO4 interlayer Supposedly both the low charge
and the size of TCA impeded its access to the GR-SO4 and GR-CO3 interlayers ie
the divalent sulphate and carbonate in the GR interlayers did not readily exchange
with the monovalent TCA since GR interlayers generally have a higher affinity for
divalent anions than for monovalent anions (Miyata 1983) Thus TCA did neither
access nor react with internal Fe(II) reactive sites in GR-SO4 which means that the
reaction between TCA and GR-SO4 took place at the external reactive Fe(II) sites
solely It is reasonable to assume that the same holds for the reaction between TCA
and GR-CO3 No significant aging effects eg rate constants varying as a function
of GR age were observed within 142 days (see Table 51) However the SSAs of
112 Chapter 5
the GR suspensions holding ages up to 142 days were not measured but estimated
assuming that the GR SSA did not decrease within the time frame
According to the Scherrer formula the width at half peak height (Wfrac12) of a
diffraction peak is inversely proportional to the average crystal dimension
perpendicular to the given crystal plane (Klug amp Alexander 1974) The average
GR-SO4 particle thickness perpendicular to the basal plane (Wfrac12 Table 52) was
constant during reaction implying that TCA reacts only at the edges and not at the
basal planes Assuming that the platy hexagonal GR-SO4 and GR-CO3 crystals
hold an average width of 1 microm and an average particle thickness of 35 nm (Hansen
and Koch 1998) the ratio of edge surface area to outer surface area is AedgeAouter ~
130 for GR-SO4 and 121 for GR-CO3 (see Supporting Information 73) This
means that only 3 of the outer surface area in GR-SO4 and 5 of the outer
surface area in GR-CO3 are available at the edges Assuming that TCA reacts at the
edges only and if employing the AedgeAouter in the estimation of the rate constants
the surface area-normalised pseudo 1 order rate constants for GR-SO4 and GR-
CO3 would be 20-30 times higher than the rate constants depicted in Figure 53a
Thus the reactivity of GRs normalised to their reactive surface area is much higher
than the reactivity normalised to their total surface area
533 Comparing with rate constants obtained for other chlorinated aliphatic
compounds
Though care must be taken when comparing kinetic parameters obtained at
different experimental conditions (eg pH [Fe(II)]0[TCA]0 ratios surface area to
volume ratios etc) it is interesting to compare our results to those reported for
hexachloroethane (Figure 53b data from Elsner et al 2004a) The reductive
transformation of hexachloroethane was investigated for various Fe(II)-bearing
minerals including Fe(II)goethite Fe(II)lepidocrocite and GR-SO4 in the presence
of 1 mM dissolved Fe(II) and 25 m2 mineral surface areaL at pH 72 except for the
GR-SO4 suspensions in which the dissolved Fe(II) concentrations were slightly
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 113
higher and pH = 8 The pseudo 1 order rate constants reported for
hexachloroethane are in the range 18middot10-4 ndash 75middot10-3 h-1middotm-2middotL (Elsner et al 2004a)
When comparing Figure 53a with Figure 53b it can be seen that the differences
in intrinsic reactivity of the Fe(II)-bearing mineral systems are more pronounced
for hexachloroethane than for TCA
Caution should also be advised to the different reaction mechanisms by which
hexachloroethane and TCA react The transfer of a single electron and the
formation of an alkyl radical upon removal of a chlorine atom constitute the first
and in most cases the rate-limiting step in the reduction of chlorinated aliphatic
compounds (Vogel et al 1987) Depending on the chemical structure of the
chlorinated aliphatic compound the resulting free alkyl radical may undergo
hydrogenolysis chloroelimination or dimerizationcoupling In the case of TCA
the free dichloroacetate radical most likely undergoes hydrogenolysis The almost
quantitative transformation of TCA to DCA confirms that hydrogenolysis is the
prevalent reaction mechanism in our mineral systems The pentachloroethyl radical
formed from hexachloroethane may undergo hydrogenolysis (producing
pentachloroethane) or dichloroelimination (producing tetrachloroethene) Elsner et
al (2004a) found that hexachloroethane was transformed quantitatively into
tetrachloroethylene for all minerals which strongly indicates that
dichloroelimination was the dominating reaction mechanism Another
polychlorinated aliphatic compound transformed mainly by hydrogenolysis under
reducing conditions is carbon tetrachloride Several studies have investigated the
reductive dechlorination of carbon tetrachloride by various Fe(II)-bearing minerals
including Fe(II)goethite and GR-SO4 and reported pseudo 1 order rate constants
in the order 152middot10-4 ndash 640middot10-4 h-1middotm-2middotL for Fe(II)goethite and 864middot10-4 h-1middotm-2middotL
for GR-SO4 (Amonette et al 2000 Pecher et al 2002 OLoughlin et al 2003c
Elsner et al 2004b) When comparing Figure 53a with Figure 53c it can be seen
that the range of magnitude of the rate constants and the differences in intrinsic
114 Chapter 5
reactivity of the Fe(II)-bearing mineral systems are similar for carbon tetrachloride
and TCA
534 Factors controlling the reactivity of surface-bound Fe(II)
The reactivity of an oxidant towards Fe(II) surface species cannot be predicted
from the reduction potentials of the redox couple alone In heterogeneous systems
processes such as mass transfer and adsorptiondesorption may have a rate-limiting
effect on the overall reaction rate If the adsorption follows a saturation-type
sorption isotherm (eg Langmuir) the sorbate (oxidant) concentration at the
surface will vary non-linearly with the total amount of oxidant added This
dependence will have to be taken into account when establishing rate laws for the
heterogeneous reactions and when testing the hypothesis that the reaction rates
depend on the sorbed concentration of the oxidants pH has a strong impact on the
sorption and thereby on the availability of ionizable oxidants At the pH values
applied here the chloroacetates are fully dissociated (pKa (TCA) = 066 pKa
(DCA) = 135 pKa (MCA) = 287) However we found the sorption of TCA to be
negligible in suspensions of pure calcite goethitecalcite and lepidocrocitecalcite
at pH 76-77 Moreover the mass balance of TCA and DCA was almost complete
in all suspensions and therefore loss of TCA or DCA due to adsorption at mineral
surfaces or incorporation in the GR anion interlayers can be ruled out Calcite has a
much lower adsorption capacity than most iron oxides hence we anticipate that
goethite and lepidocrocite control the adsorption of TCA and DCA in both the pure
FeOOH and the FeOOHcalcite suspensions This was supported by our
experimental results demonstrating that the presence of a calcite surface - either
pure or as a support for goethite and lepidocrocite coatings - did not exert any
noticeable effect on the reaction rates (see Table 51) In addition the surface area-
normalised rate constants for mineral systems containing goethite or lepidocrocite
in pure form and mineral systems containing goethite or lepidocrocite as coatings
on calcite were very similar In heterogeneous reactions mass transfer in bulk
solution becomes the rate-limiting step when the surface reaction is much faster
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 115
than the diffusion of the reacting species to the reactive surface However at the
low rate constants obtained here the reaction of TCA with the Fe(II)-bearing
minerals is not likely to be mass transfer limited (see Supporting Information 72)
One very important factor affecting heterogeneous redox reactions is pH which
influences the speciation of the complexes in solution and at mineral surfaces as
well as the stability of the more soluble Fe(II)-containing minerals such as GRs In
contrast to aqueous Fe(II) complexes it is not possible to predict the reactivity of
Fe(II) surface species as their reduction potentials are unknown In the absence of
specifically adsorbing solutes other than H+ the surface charge of the Fe(III)
oxides goethite and lepidocrocite is determined by the surface densities of the
charged surface species equivFeOH2+ and equivFeOndash whereas the surface charge of calcite
is determined by the density of the surface species equivCO3ndash equivCaOH2
+ and equivCaOndash
(Stumm 1992 Van Cappellen et al 1993) The point of zero charge (pHpzc) of
pure calcite is in the pH range 7-11 and depends on the partial pressure of carbon
dioxide pCO2 The higher the pCO2 the lower the pHpzc At the experimental
conditions applied here (05 = 0005 atm CO2(g)) the pHpzc = 82 for calcite
(Table 53) As only 10-11 mg Fe(III) of goethite and lepidocrocite was coated
onto calcite we assumed a pHpzc of 82 for the goethite and lepidocrocite coated
calcite particles as well The pHpzc values for green rusts are unknown
116 Chapter 5
Table 53 Specific surface areas and point of zero charge of the various iron minerals in pure form as well as goethite and lepidocrocite coated onto calcite
Mineral Structural formula SSA (m2g) pHpzc
GR-SO4 FeII4FeIII
2(OH)12SO4middot3H2O 71 a -
GR-CO3 FeII4FeIII
2(OH)12CO3middot3H2O 47 b -
Magnetite Fe3O4 - 69 e
Goethite α-FeOOH 16 c 85 f
Lepidocrocite γ-FeOOH 18 c 73 e
Calcite CaCO3 le 1 d 82 g
Goe coating - le 1 d 82 h
Lep coating - le 1 d 82 h
a Chapter 4 this work b Williams amp Scherer 2001 c Product information by Bayer d The SSA of calcite was
quantified to le 1 m2g The detection limit of our BET method was 1 m2g e Charlet et al 1998a f Liger et al
1999 g Van Cappellen et al 1993 h Same as for calcite
The surface hydroxyl groups on iron oxides may be both singly (equivFe-OH) doubly
(equivFe2-OH) triply (equivFe3-OH) and geminally (equivFe-(OH)2) coordinated (Cornell amp
Schwertmann 1996 Stumm 1992) The differently coordinated surface hydroxyl
groups are not equally reactive Adsorption reactions involve only singly
coordinated surface groups and therefore only this kind of hydroxyl groups on iron
oxides will be considered here (Cornell amp Schwertmann 1996) Hence the
predominant surface sites available for adsorption in pure suspensions of Fe(III)
oxides are equivFeOH0 equivFeOH2+ and equivFeOndash In the presence of dissolved Fe(II)
equivFeIIIOFeIIOH0 equivFeIIIOFeIIOndash and equivFeIIIOFeII+ constitute the main reactive sites at
the Fe(III) oxide surfaces (Liger et a 1999) Assuming that Fe2+ and other cationic
Fe(II) species are the dominating adsorbates on the mineral surfaces in our
experiments we expect the actual pHpzc to be higher than the pHpzc of the pure
oxides listed in Table 53 Hence at pHlt82 where most of our experiments were
conducted all the mineral surfaces presumably carry net positive charges
At pH 70 where Fe2+ is still the predominant Fe(II) species in solution (~50) we
expect that equivFeIIIOFeIIOH equivFeIIIOFeIIOndash and equivFeIIIOFeII+ constitute the main
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 117
reactive sites at the Fe(III) oxide surfaces as suggested by Liger et a 1999 As pH
increases from 70 to 87 the Fe(II) carbonate complexes become increasingly
important in solution at the expense of the Fe2+ FeCl+ FeSO40 and FeOH+ species
(King 1998) Fe(II) carbonate complexes do not bind at the oxide surface as
readily as the aquo or hydroxo complexes of Fe(II) but carbonate itself sorbs
readily to Fe(III) oxide surfaces through which the Fe(III) oxide surface is coated
by inner-sphere monodentate equivFeIIIOCOOH0 surface complexes (Villalobos amp
Leckie 2000 amp 2001) The presence of carbonate shifted the sorption edge for the
Fe(II) adsorption on goethite from pH 58 to 78 and the authors hypothesized this
to be a result of the formation of aqueous and surface Fe(II)-carbonate complexes
and to competition between carbonate and Fe(II) for Fe(III) oxide surface sites
(Vikesland amp Valentine 2002) Similarly monodentate surface complexes like
equivFeIIICl0 and equivFeIIIOSO3ndash as well as ternary monodentate surface complexes like
equivFeIIIOFeIICl0 and equivFeIIIOFeIIOSO3ndash and ternary bidentate surface complexes such
as (equivFeIIIO)2FeIIOSO3 may form at Fe(III) oxide surfaces when Fe(II) chloride and
sulfate are present in solution (Ostergren et al 2000 Kim et al 2004) However
the effects of anionic ligands such as chloride and sulfate on Fe(II) adsorption at
Fe(III) oxide surfaces and the reactivity of Fe(II) carbonate chloride and sulfate
surface sites are still unknown and need to be evaluated (see Supporting
Information 75) Thus we do not know whether chloride and sulfate decrease or
increase the Fe(II) sorption in our mineral systems We can only report that we did
not detect any significant differences in the rate of TCA transformation between
the mineral suspensions containing carbonate chloride and sulfate respectively
Hence we anticipate that equivFeIIIOFeIIOH equivFeIIIOFeIIOndash and equivFeIIIOFeII+ constitute
the main reactive sites at the Fe(III) oxide surfaces within the whole pH range 70-
87 This might also explain why we did not detect any obvious systematic pH
effect in the Fe(II)-Fe(III)-systems (see Table 51) In the case of
hexachloroethane the reactivity order GR-SO4gtgoethitegtmagnetitegtlepidocrocite
may be rationalized by the variations in surface site densities and total amount of
118 Chapter 5
Fe(II) sorbed on the iron minerals (see Supporting Information 75) as well as the
different speciations and reactivities of the Fe(II) surface sites on the iron minerals
535 Comparison with biotic and other abiotic systems
Only one report on abiotic transformation of TCA is found in the literature and the
study demonstrates the reductive dechlorination of TCA to MCA by Fe(0)
(Hozalski et al 2001) The authors reported a pseudo 1 order rate constant of
60middot10-4 min-1middotm-2middotL for the transformation of TCA to DCA and a pseudo 1 order
rate constant of 225middot10-4 min-1middotm-2middotL for the transformation of DCA to MCA at
[Fe(0)]0 = 025 M [TCA]0 = 100-200 microM and pH 36-62 The rate constant for
TCA reduction by Fe(0) is 10-300 times faster than the rate constants reported for
the Fe(II)-bearing mineral systems here
There is abundant evidence that soil microorganisms and fungi can dechlorinate
TCA but only little is known about the bacteria and processes involved in the
biodegradation of TCA Biotransformation of TCA has been found at both oxic
and anoxic conditions Most of the microorganisms isolated grow feebly on TCA
as a sole source of carbon (Foy 1975 Weightman et al 1992 De Wever et al
2000) Only one bacterium capable of growing on TCA as the sole carbon and
energy source has been characterized (Yu amp Welander 1995) In addition
anaerobic bacteria coupling co-metabolic growth to reductive dechlorination of
TCA have been isolated (Weightman et al 1992 De Wever et al 2000) The
inability to grow on the less chlorinated acids DCA and MCA is a notable feature
of both the aerobic and anaerobic bacteria Complete transformation of TCA to
methane and carbon dioxide has only been found when abiotic and biotic processes
were combined (Egli et al 1989) The abiotic transformation of TCA to DCA
occurred spontaneously in the presence of sterile activated charcoal whereas the
DCA formed was further degraded to methane and carbon dioxide by a mixed
culture of methanogenic bacteria However the abiotic reductant(s) responsible for
the transformation of TCA to DCA was not reported (Egli et al 1989)
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 119
The rate constants obtained in this work suggest that the Fe(II)-bearing mineral
systems may be important reductants of TCA in natural suboxic environments In
natural iron-rich soils holding specific surface areas of 22 m2g (Kenneke amp
Weber 2003) average bulk densities of 265 gcm3 and porosities of 25 and
containing 2 iron oxides a rough estimation of the half-life of TCA amounts to
47 minutes when applying the average surface area-normalised rate constant
obtained for all the Fe(II)-Fe(III) mineral systems in this work (1middot10-3 h-1middotm-2middotL)
This estimation is based on the assumption that enough reactive Fe(II) is available
in these soils The natural iron-reducing sediment investigated by Kenneke and
Weber (2003) contained 80 microM Fe(II) in the soil solution and 315 micromole Fe(II) per
g sediment At such low Fe(II) concentrations the overall rate of abiotic
transformation of TCA in natural soils and sediments is most likely limited by the
regeneration of reactive Fe(II) Hence the continuous regeneration of reactive
Fe(II) surface sites by adsorption of abiotically or microbially produced Fe(II) may
further the long-term abiotic transformation of TCA in such environments
54 Conclusions
This work demonstrates that various Fe(II)-Fe(III) minerals systems including GR-
SO4 GR-CO3 magnetite Fe(II)goethite and Fe(II)lepidocrocite readily transform
TCA to DCA Dichloroacetate was not further reduced to MCA or acetate by any
of the Fe(II)-bearing minerals The surface area-normalised pseudo 1 order rate
constants obtained for the reductive transformation of TCA by the various Fe(II)-
bearing minerals did not differ significantly from each other The results obtained
in this work infer that under natural geochemical conditions Fe(II)-bearing mineral
systems may play an important role in the overall transformation of TCA Thus
not only microbial degradation but also abiotic reductive transformation of TCA by
Fe(II)-bearing minerals may govern the fate of TCA in natural subsurface
environments This holds both for natural systems like iron-rich suboxic soils and
sediments as well as engineered systems like permeable reactive barriers of zero-
120 Chapter 5
valent iron implemented for on-site remediation where both Fe(0) and solid or
surface-bound Fe(II) corrosion intermediates may transform TCA
Acknowledgments
We would like to thank Susanne Guldberg for performing the experimental work comprising
GR-SO4
References
Ahlers J Regelmann J Riedhammer C (2003) Environmental risk assessment of airborne trichloroacetic acid - a contribution to the discussion of the significance of anthropogenic and natural sources Chemosphere 52 531-537 Amonette JE Workman DJ Kennedy DW Fruchter JS Gorby YA (2000) Dechlorination of carbon tetrachloride by Fe(II) associated with goethite Environmental Science and Technology 34 4606-4613 Atkins PJ Gold V Marsh R (1984) The decarboxylation of trichloroacetic acid and the reactions of the trichloromethyl anion with 135-trinitrobenzene and with hydrogen ions kinetic measurements in dimethyl sulphoxide solution Journal of the Chemical Society Perkin Transactions 2 7 1239-1245 Berg M Muumlller SR Muumlhlemann J Wiedmer A Schwarzenbach RP (2000) Concentrations and mass fluxes of chloroacetic acids and trifluoroacetic acid in rain and natural waters in Switzerland Environmental Science and Technology 34 2675-2683 Brunauer S Emmett PH Teller E (1938) Adsorption of gases in multimolecular layers Journal of American Chemical Society 60 309-319 Butler EC Hayes KF (1998) Effects of solution composition and pH on the reductive dechlorination of hexachloroethane by iron sulfide Environmental Science and Technology 32 1276-1284 Butler EC Hayes KF (1999) Kinetics of the transformation of trichloroethylene and tetrachloroethylene by iron sulfide Environmental Science and Technology 33 2021-2027 Charlet L Liger E Gerasimo P (1998a) Decontamination of TCE- and U-rich water by granular iron Role of sorbed Fe(II) Journal of Environmental Engineering 124 25-30 Charlet L Silvester E Liger E (1998b) N-compound reduction and actinide immobilisation in surficial fluids by Fe(II) the surface FeIIIFeIIOH0 species as major reductant Chemical Geology 151 85-93 Cheregi M Danet AF (1997) Flow injection determination of chloride ions with spectrophotometric detection Analytical Letters 30 2847-2858 Cornell RM Schwertmann U (1996) The iron oxides Structure properties reactions occurrence and uses VCH Verlagsgesellschaft mbH Weinheim
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 121 Cui D Eriksen TE (1996) Reduction of pertechnetate by ferrous iron in solution influence of sorbed and precipitated Fe(II) Environmental Science and Technology 30 2259-2262
Egli C Thuumler M Suter D Cook AM Leisinger T (1989) Monochloro- and dichloroacetic acids as carbon and energy sources for a stable methanogenic mixed culture Archives of Microbiology 152 218-223 Elsner M Haderlein SB Schwarzenbach RP (2004a) Reactivity of Fe(II)-bearing minerals towards reductive transformation of organic contaminants Environmental Science and Technology 38 799-807 Elsner M Haderlein SB Kellerhals T Luzi S Zwank L Angst W Schwarzenbach RP (2004b) Mechanisms and products of surface-mediated reductive dehalogenation of carbon tetrachloride by Fe(II) on goethite Environmental Science and Technology 38 2058-2066 Erbs M Hansen HCB Olsen CE (1999) Reductive dechlorination of carbon tetrachloride using iron(II)iron(III)-hydroxide-sulphate (green rust) Environmental Science and Technology 33 307-311 Euro Chlor (2001) Trichloroacetic acid in the environment a dossier Euro Chlor Brussels and the European Chlorinated Solvent Association Fadrus H Maly J (1975) Suppression of iron(III) interference in the determination of iron(II) in water by the 110-phenanthroline method The Analyst 100 549-554 Fahimi IJ Keppler F Schoumller HF (2003) Formation of chloroacetic acids from soil humic acid and phenolic moieties Chemosphere 52 513-520 Foy CL (1975) The chlorinated aliphatic acids In Herbicides Chemistry degradation and mode of action Kearney PC Kaufman DD (eds) Marcel Dekker Inc 399-452 Gander JW Parkin GF Scherer MM (2002) Kinetics of 111-trichloroethane transformation by iron sulfide and a methanogenic consortium Environmental Science and Technology 36 4540-4546 Haiber G Jacob G Niedan V Nkusi G Schoumller HF (1996) The occurrence of trichloroacetic acid (TCAA) ndash indications of a natural production Chemosphere 33 839-849
Hansen HCB (1989) Composition stabilization and light absorption of Fe(II)Fe(III) hydroxycarbonate (green rust) Clay Minerals 24 663-669 Hansen HCB Guldberg S Erbs M Koch CB (2001) Kinetics of nitrate reduction by green rusts ndash effects of interlayer anion and Fe(II)Fe(III) ratio Applied Clay Science 18 81-91 Hansen HCB Koch CB (1997) A comparison of nitrate reduction by carbonate and sulphate forms of green rust Kodama H Mermut A R Torrance J K (eds) Proceedings of the 11th International Clay Conference Ottawa Canada Clays for our future 11 295-302 Hoekstra EJ de Leer EWB Brinkman UATh (1999a) Mass balance of trichloroacetic acid in the soil top layer Chemosphere 38 551-563 Hoekstra EJ de Leer EWB Brinkman UATh (1999b) Findings supporting the natural
122 Chapter 5
formation of trichloroacetic acid in soil Chemosphere 38 2875-2883 Hofstetter TB Heijman CG Haderlein SB Holliger HC Schwarzenbach RP (1999) Complete reduction of TNT and other (poly)nitroaromatic compounds under iron-reducing subsurface conditions Environmental Science and Technology 33 1479-1487 Hofstetter TB Schwarzenbach RP Haderlein SB (2003) Reactivity of Fe(II) species associated with clay minerals Environmental Science and Technology 37 519-528 Hozalski RM Zhang L Arnold WA (2001) Reduction of haloacetic acids by Fe0 Implications for treatment and fate Environmental Science and Technology 35 2258-2263 Husain S Narsimha R Alvi SN Rao RN (1992) Monitoring the effluents of the trichloroacetic acid process by high-performance liquid chromatography Journal of Chromatography 600 316-319 Hwang I Batchelor B (2000) Reductive dechlorination of tetrachloroethylene by Fe(II) in cement slurries Environmental Science and Technology 34 5017-5022 Jordan A Frank H (1999) Trifluoroacetate in the environment Evidence for sources other than HFCHCFCs Environmental Science and Technology 33 522-527 Kenneke JF Weber EJ (2003) Reductive dehalogenation of halomethanes in iron- and sulfate-reducing sediments 1 reactivity pattern analysis Environmental Science and Technology 37 713-720 Kim CS Rytuba JJ Brown Jr GE (2004) EXAFS study of mercury(II) sorption to Fe- and Al-(hydr)oxides II Effects of chloride and sulphate Journal of Colloid and Interface Science 270 9-20 King DW (1998) Role of carbonate speciation on the oxidation rate of Fe(II) in aquatic systems Environmental Science and Technology 32 2997-3003 Klausen J Troumlber SP Haderlein SB Schwarzenbach RP (1995) Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions Environmental Science and Technology 29 2396-2404 Klug PH Alexander LE (1974) X-ray diffraction procedures John Wiley amp Sons Inc Koch CB Hansen HCB (1997) Reduction of nitrate to ammonium by sulphate green rust Advances in GeoEcology 30 373-393 Kuumlhn R Pattard M (1990) Results of the harmful effects of water pollutants to green algae (Scenedesmus subspicatus) in the cell multiplication inhibition test Water Research 24 31-38 Lee W Batchelor B (2002a) Abiotic reductive dechlorination of chlorinated ethylenes by iron- bearing soil minerals 1 Pyrite and magnetite Environmental Science and Technology 36 5147- 5154 Lee W Batchelor B (2002b) Abiotic reductive dechlorination of chlorinated ethylenes by iron-bearing soil minerals 2 Green rust Environmental Science and Technology 36 5348- 5354
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 123 Liger E Charlet L Van Cappellen P (1999) Surface catalysis of uranium (VI) reduction by iron(II) Geochimica et Cosmochimica Acta 63 2939-2955 Lignell R Heinonen-Tanski H Uusi-Rauva A (1984) Degradation of trichloroacetic acid (TCA) in soil Acta Agriculturae Scandinavia 34 3-8 Loyaux-Lawniczak S Refait Ph Lecomte P Ehrhardt J Geacutenin JMR (1999) The reduction of chromate ions by Fe(II) layered hydroxides Hydrology and Earth System Sciences 3 593-599 Luther III GW (1990) The Frontier-Molecular-Orbital theory approach in geochemical processes in W Stumm Ed Aquatic Chemical kinetics John Wiley and Sons New York pp 173-198 McCulloch A (2002) Trichloroacetic acid in the environment Chemosphere 47 667-686 Miyata S (1983) Anion-exchange properties of hydrotalcite-like compounds Clays and Clay Minerals 31 305-311 Muumlller SR Zweifel H-R Kinnison DJ Jacobsen JA Meier MA Ulrich MM Schwarzenbach RP (1996) Occurrence sources and fate of trichloroacetic acid in Swiss lakes Environmental Toxicology and Chemistry 15 1470-1478 OLoughlin EJ Burris DR (2003) Reduction of halogenated ethanes by green rust Environmental Toxicology and Chemistry 23 41-48 OLoughlin EJ Kelly SD Cook RE Csencsits R Kemner KM (2003a) Reduction of uranium(VI) by mixed iron(II)iron(III) hydroxide (green rust) Formation of UO2 nanoparticles Environmental Science and Technology 37 721-727 OLoughlin EJ Kelly SD Kemner KM Csencsits R Cook RE (2003b) Reduction of AgI AuIII CuII and HgII by FeIIFeIII hydroxysulfate green rust Chemosphere 53 437-446 OLoughlin EJ Kemner KM Burris DR (2003c) Effects of AgI AuIII and CuII on the reductive dechlorination of carbon tetrachloride by green rust Environmental Science and Technology 37 2905-2912 Ostergren JD Brown Jr GE Parks GA Persson P (2000) Inorganic ligand effects on Pb(II) sorption to goethite (α-FeOOH) II Sulfate Journal of Colloid and Interface Science 225 483-493 Pecher K Haderlein SB Schwarzenbach RP (2002) Reduction of polyhalogenated methanes by surface-bound Fe(II) in aqueous suspensions of iron oxides Environmental Science and Technology 36 1734-1741 Reimann S Grob K Frank H (1996) Chloroacetic acids in rainwater Environmental Science and Technology 30 2340-2344 Ruumlgge K Hofstetter TB Haderlein SB Bjerg PL Knudsen S Zraurig C Mosbaeligk H Christensen TH (1998) Characterization of predominant reductants in an anaerobic leachate- affected aquifer by nitroaromatic probe compounds Environmental Science and Technology 32
124 Chapter 5
23-31 Sarzanini C Bruzzoniti MC Mentasti E (1999) Preconcentration and separation of haloacetic acids by ion chromatography Journal of Chromatography A850 197-211 Schwertmann U Cornell RM (1991) Iron oxides in the laboratory Preparation and characterization VCH Verlagsgesellschaft mbH Weinheim Scott BF Mactavish DC Spencer C Strachan WMJ Muir DCG (2000) Haloacetic acids in Canadian lake waters and precipitation Environmental Science and Technology 34 4266-4272 Scott BF Spencer C Marvin CH Mactavish DC Muir DCG (2002) Distribution of haloacetic acids in the water columns of the Laurentian Great Lakes and Lake Malawi Environmental Science and Technology 36 1893-1898 Strathmann TJ Stone AT (2003) Mineral surface catalysis of reactions between FeII and oxime carbamate pesticides Geochimica et Cosmochimica Acta 67 2775-2791 Stumm W (1992) Chemistry of the solid-water interface John Wiley amp Sons Inc Van Cappellen P Charlet L Stumm W Wersin P (1993) A surface complexation model of the carbonate mineral-aqueous solution interface Geochimica et Cosmochimica Acta 57 3505- 3518 Vikesland PJ Valentine RL (2002) Iron oxide surface-catalyzed oxidation of ferrous iron by monochloramine implications of oxide type and carbonate on reactivity Environmental Science and Technology 36 512-519 Villalobos M Leckie JO (2000) Carbonate adsorption on goethite under closed and open CO2 conditions Geochimica et Cosmochimica Acta 64 3787-3802 Villalobos M Leckie JO (2001) Surface complexation modeling and FTIR study of carbonate adsorption to goethite Journal of Colloid and Interface Science 235 15-32 Vogel TM Criddle CS McCarty PL (1987) Transformations of halogenated aliphatic compounds Environmental Science and Technology 21 722-736 Weightman AL Weightman AJ Slater JH (1992) Microbial dehalogenation of trichloroacetic acid World Journal of Microbiology and Biotechnology 8 512-518 De Wever H Cole JR Fettig MR Hogan DA Tiedje JM (2000) Reductive dehalogenation of trichloroacetic acid by Trichlorobacter thiogenes gen nov spnov Applied and Environmental Microbiology 66 2297-2301 Williams AGB Scherer MM (2001) Kinetics of chromate reduction by carbonate green rust Environmental Science and Technology 35 3488-3494 Yu P Welander T (1995) Growth of an aerobic bacterium with trichloroacetic acid as the sole source of energy and carbon Applied Microbiology and Biotechnology 42 769-774
Conclusions and Outlook 125
6 Conclusions and Outlook The work presented in this dissertation adds to the understanding of how Fe(II)-
bearing minerals like green rusts (GRs) vivianite (Fe2(PO4)2sdot8H2O) magnetite
(Fe3O4) and Fe(II) associated with goethite and lepidocrocite may form and react in
nature In order to elucidate the role of bacteria in the formation of GRs in natural
soils and sediments we studied the iron mineral phases forming as a result of the
activity of iron-respiring bacteria In chapter 2 the Fe-containing products formed
by anaerobic autotrophic denitrifying Fe(II)-oxidizing bacteria (FeOB) were
examined The culture medium applied contained high levels of bicarbonate and
phosphate and is typically used in this kind of studies as it provides excellent
conditions for the nitrate-reducing FeOB Fe(II) was present initially as a whitish
solid Fe(II) hydroxy phosphate (vivianite) and as soluble Fe(II) The results
obtained demonstrate that the denitrifying FeOB produce poorly crystalline
goethite via a greenish Fe(III)-enriched vivianite intermediate Moumlssbauer
spectroscopic analyses provided no evidence of green rust formation At low
phosphate concentrations where vivianite does not control the Fe(II) activity it is
reasonable to assume that siderite (FeCO3) precipitates initially and that carbonate
GR phases may form during biooxidation At low bicarbonate concentrations we
would expect Fe(II) sulfate or chloride species to dominate initially (depending on
the Fe(II) source applied) and sulfate GR or chloride GR to form during
biooxidation In chapter 3 we investigated the Fe-containing products formed
during reduction of common Fe(III) oxides by the anaerobic dissimilatory Fe(III)-
reducing microorganism Shewanella algae BrY S algae BrY reduced substantial
amounts of the initial Fe(III) and green and blackish mineral phases were produced
within 1-2 weeks after inoculation Moumlssbauer spectroscopic analyses showed that
the green and black precipitates consisted of green rust and vivianite
We studied the reactivity of synthetic GRs towards reducible organic pollutants in
order to asses the potential significance of GR phases for the fate of such
126 Chapter 6
compounds To this end we used nitroaromatic compounds (NACs) and
chlorinated acetates as suitable model compounds for studying redox reactions
potentially relevant in the environment In chapter 4 we investigated the relative
reactivity of outer and inner Fe(II) reactive sites in synthetic sulfate green rust
(GR-SO4) by using a series of structurally closely related compounds with different
charge properties as ldquoreactive probesrdquo The probe compounds included
nitrobenzene 2-nitrophenol 4-nitrotoluene 4-chloronitrobenzene and 4-
nitrophenylacetic acid Our results demonstrated that NACs are completely
reduced to their corresponding anilines by GR-SO4 The reactions followed pseudo
1 order kinetics with respect to NAC and the surface area-normalised pseudo 1
order rate constants obtained were 016ndash465middot10-4 s-1middotm-2middotL at [Fe(II)GR]0 = 103-
1260 mM [NAC]0 = 20-102 microM and pH 84-86 Neither mass transfer control nor
surface saturation kinetics could account for the similarity of the surface-
normalised pseudo 1 order rate constants obtained for the reduction of the neutral
and anionic NACs by GR-SO4 These observations suggest that the reaction
between NAC and GR-SO4 takes place at the external reactive Fe(II) sites At low
initial Fe(II)GR concentrations the external reactive Fe(II) sites were depleted and
the regeneration of new external reactive sites eventually controlled the reduction
of the NACs by GR-SO4 In chapter 5 we examined the reactivity of various
Fe(II)-Fe(III) mineral systems towards trichloroacetic acid (TCA) and
dichloroacetate (DCA) in laboratory batch experiments imitating natural
conditions The Fe(II)-Fe(III)-systems investigated included GR-SO4 carbonate
green rust magnetite Fe(II)goethite and Fe(II)lepidocrocite TCA was readily
reduced to DCA by all Fe(II)-containing minerals The reactions followed pseudo
1 order kinetics with respect to TCA and the surface area-normalised pseudo 1
order rate constants obtained were 033ndash76middot10-5 min-1middotm-2middotL at [Fe(II)]0 = 025ndash
116 mM [TCA]0 = 15ndash1000 microM and pH 70ndash87 Our results showed no
significant differences regarding product distribution and surface area-normalised
reaction rate constants between the Fe(II)-Fe(III)-systems DCA was not further
Conclusions and Outlook 127
reduced to monochloroacetate (MCA) or acetate in any of the systems within the
time frame in our experiments
As suggested in chapters 2 and 3 sufficient evidence must be provided and caution
should be exercised when proclaiming new biogenic minerals The study of
microbially produced GRs is still in its infancy and more research is needed in
order to elucidate the role of bacteria in the formation of GRs in natural soils and
sediments The results presented in chapter 2 indicate that microbiological
processes may be responsible for the oxidation of vivianite and metavivianite
((FeII3-xFeIII
x)(PO4)2(OH)xmiddot(8-x)H2O x gt 12) in natural subsurface environments
In chapter 3 we demonstrated that GRs may be produced microbially at conditions
including low carbon and Fe(III) concentrations as well as the exclusion of
synthetic electron shuttles and pH buffers The role of microbial processes in the
redox cycling of iron in the subsurface and the ways in which these processes can
be coupled to contaminant remediation are currently active areas of research Zero-
valent iron has been the most extensively studied reductant for the treatment of
many inorganic and organic contaminants and is currently the most commonly
used material for the construction of permeable reactive barriers (PRB) but a
detailed understanding of the processes involved in the reduction of these
pollutants by Fe(0) is lacking (Scherer et al 2000) Potentially reactive Fe(II)-
bearing corrosion products identified in iron metal columns and barriers include
magnetite siderite Fe(II) sulfides green rusts as well as Fe(II) sorbed to mineral
surfaces (Gu et al 1999 Roh et al 2000) The formation of reactive Fe(II)-
bearing minerals like GRs may explain the effective long-term operation of zero-
valent iron PRBs despite the formation of thick oxide films Thus natural in situ
PRBs might be created by stimulating the activity of anaerobic dissimilatory
Fe(III)-reducing bacteria and the subsequent formation of Fe(II) species such as
GRs Furthermore suspensions of synthetic GRs which are easily prepared from
relatively inexpensive commodity chemicals may also be injected and dispersed
into the subsurface
128 Chapter 6
The reductive transformation of NACs and TCA by GRs is relevant to
understanding the processes responsible for their degradation in the subsurface and
the development of innovative technologies for their remediation The results
obtained in chapters 4 and 5 indicate that GRs may play a significant role in the
reductive transformation of NACs and TCA in natural subsurface environments
Furthermore our results suggest that mainly the outer Fe(II) sites in GRs are
utilized in the reaction with neutral and monovalent anionic compounds and that
these sites may be replenished eg by reduction of the oxidized surface sites or
adsorption of Fe(II) from solution The continuous restoration of Fe(II) surface
sites in GRs may promote their long-term reactivity towards reducible
contaminants
References
Gu B Phelps TJ Liang L Dickey MJ Roh Y Kinsall BL Palumbo AV Jacobs GK (1999) Biochemical dynamics in zero-valent iron columns Implications for permeable reactive barriers Environmental Science and Technology 33 2170-2177 Roh Y Lee SY Elless MP (2000) Characterization of corrosion products in the permeable reactive barriers Environmental Geology 40 184-194 Scherer MM Richter S Valentine RL Alvarez PJJ (2000) Chemistry and microbiology of permeable reactive barriers for In Situ groundwater clean up Critical Reviews in Environmental Science and Technology 30 363-411
Supporting Information I
7 Supporting Information
71 Estimation of the one-electron reduction potential for 4-NPA
The one-electron reduction potential of the half-reaction for a given NAC 1hE
ArNO2 + e- ArNO2
can be used for comparing reduction rates of different NACs in a given system
The formation of the nitroaryl radical is the rate-determining step in the overall rate
of the reduction of a NAC to the corresponding aniline The difference between the
of a NAC and a given reductant is proportional to the change in standard free
energy for the transfer of the first electron ∆G
1hE
1degrsquo If a linear relationship between
the free energy of activation and ∆G1degrsquo is assumed the values of various NACs
can be a measure of their relative reactivity with a given reductant
1hE
As neither the one-electron reduction potential for 4-nitrophenylacetic acid (4-
NPA) nor the Hammett constant for the acetic acid substituent could be found in
the literature the one-electron reduction potential for 4-NPA was estimated by
application of a linear free energy relationship (LFER) to experimental data
Kinetic experiments were conducted in order to obtain the pseudo 1 order rate
constant for the reduction of 4-NPA by a model hydroquinone (reduced
juglone (8-hydroxy-14-naphthoquinone) in the presence of HS
minusHJUGk
ndash) The reduction of
a NAC by juglone follows the rate law
[ ] [ ] [ ] [ ] [ ] [ ]2222 ArNOJUGfkArNOHJUGkArNOk
dtArNOd
totHJUGHJUGHJUGobs sdotsdotsdot=sdotsdot=sdot=minus minusminusminusminus
and the was deducted from a LFER 1hE
II Chapter 7
bEak hHJUG +sdot=minus 059160
log1
for which a and b values have been established for a range of NACs with known
values (Hofstetter et al 1999) An excellent correlation of and log
has been found to exist over a range of 250 mV corresponding to more than 5 order
of magnitude for This is due to the fact that the actual transfer of the first
electron is the rate-determining step under the experimental conditions chosen
1hE 1
hE minusHJUGk
minusHJUGk
For comparison experiments with 4-nitrotoluene (4-NT) were also conducted The
pseudo 1 order rate constants for the reduction of 4-NPA with juglone were
corrected for the reduction of 4-NPA with only HSndash (control experiments
containing no juglone)
[ ]minuslowast
minusminus
minus
minus=
HJUGkk
k HSHJUGHJUG
where (MminusHJUGk -1middots-1) is the rate constant for a compound in the presence of only
juglone (slowastminusHJUGk -1) is the pseudo 1 order rate constant for a compound in the
presence of both juglone and HSndash (sminusHSk -1) is the pseudo 1 order rate constant for
the control reaction in the presence of only HSndash and [HJUGndash] (M) is the
concentration of the reactive dissociated HJUG- form (nondissociated
hydroquinone species are very nonreactive as compared to the monophenolate
species)
Supporting Information III
OH 0 OH OH
+ e- + H+ = + e- + H+ =
0 0
pl(( ox) = 8 00 PK1 (red) = 6 60
JUG HJUG
OH OH
OH
pKa2(red) = 10 60
Figure 7 1 Oxidized and reduced juglone fo1m s
Kinetic experiments in homogeneous anoxic aqueous solutions contained 5 mM
HS- 20 microM total juglone 50 mM KH2P04 buffer and were conducted at pH =
660 corresponding to a concentration of the reactive dissociated juglone form
[HJUG] = 10 microM
By using the LFER
E1 logkHJUG- = 125 middot 005~16 + 923
the following values were obtained
(Hofstetter et al 1999)
NAC k (M-1 -1) HJUG- middots log kHJUG- E~ (mV)
4-NT
4-NPA
311 middot10middot7
164middot10middot7
847middot 10-8
116middot 10middot7
226middot10middot2
489middot10middot3
-165
-231
-515
-546
IV Chapter 7
lowast
minusHJUGk -values are averages of triplicates whereas -values are averages of
duplicates The determined for 4-NT in this work (-515 mV) differs 3 from
the -value of -500 mV reported in the literature (Meisel amp Neta 1975
Wardman 1989) Hence it is assumed that the -value determined for 4-NPA
also differs by 3
minusHSk
1hE
1hE
1hE
Note that even for NACs holding very different values the difference in their
reactivities are much less pronounced in Fe(II)-Fe(III) systems such as the
Fe(II)goethite system (LFER slope a = 06 Hofstetter et al 1999) and the
Fe(II)magnetite system (LFER slope a = 034 Klausen et al 1995) as compared
to the jugloneH
1hE
2S system (a = 125) Furthermore it should be noted that all
LFERs mentioned here were established for neutral NACs and in this work we
have simply assumed that the LFERs are also valid for anionic NACs
72 The rate-limiting step
The overall rate of a reaction is equal to the rate of the slowest step in the
mechanism In heterogeneous reactions eg a compound reacting at the surface of
suspended particles in bulk solution the overall process by which the
heterogeneous reactions proceed may be broken down into a sequence of
individual diffusion steps and reaction steps 1) Mass transfer (diffusion) of the
reactant from the bulk fluid to the external surface of the solid phase 2)
Adsorption of reactant onto the solid surface 3) Reaction on the solid surface 4)
Desorption of the products from the solid surface 5) Mass transfer of the products
from the external solid surface to the bulk fluid Hence the rate of reaction of a
compound reacting at the surface of suspended particles in bulk solution may be
either mass transfer adsorptiondesorption or surface reaction limited When the
diffusion steps are much faster than the reaction steps the mass transfer or
diffusion steps do not affect the overall reaction rate However if the reaction steps
Supporting Information V
are very fast compared with the diffusion steps mass transport affects the reaction
rate Here only the external mass transfer is considered ie the diffusion of
reactants or products between the bulk fluid and the external surface of the solid
phase The additional internal mass transfer resistance for particles containing
substantial internal surface area is not addressed
721 Mass transfer (diffusion) limited kinetics
The overall rate constant can be represented by a system of resistances in series
(Fogler 1999 Arnold et al 1999)
⎟⎟⎠
⎞⎜⎜⎝
⎛+=
minusgeomSALobs kkak1111
where kobs is the observed rate constant kL is the mass transfer coefficient (mmiddots-1) a
is the ratio of the external (geometric) specific surface area to volume of solution
(m-1) and kSA-geom is the intrinsic rate constant of the reaction normalized to the
external specific surface area rather than the BET specific surface area By
comparing kLmiddota with kobs one can estimate the role of mass transfer on the rate of
reaction Thus if kLmiddota gtgt kobs mass transfer is so fast that it has no impact on the
reaction rate whereas if kLmiddota le kobs mass transfer is the rate limiting step
In fluid dynamics the Reynolds number Re is used for determining whether a
flow is laminar or turbulent
νtp ud sdot
=Re
where dp is the particle diameter (m) ut is the terminal particle settling velocity
(mmiddots-1) and ν is the kinematic fluid viscosity (m2middots-1) ν = η ρ where η is the
(absolute) dynamic fluid viscosity in centipoise (1 centipoise = 1 mPamiddots = 10-3
kgmiddotm-1middots-1) and ρ is the fluid density (kgmiddotm-3)
VI Chapter 7
At Re lt 1 we can apply Stokersquos particle settling velocity Stokersquos law is an
equation relating the terminal settling velocity of a smooth rigid sphere in a
viscous fluid of known density and viscosity to the diameter of the sphere when
subjected to a known force field
( )η
ρρsdot
minussdotsdot=
18
2pp
t
dgu (mmiddots-1)
where g = 981 mmiddots-2 is the gravitational constant ρp is the particle density (kgmiddotm-3)
The Sherwood number is the main parameter for prediction of the mass transfer
process
in fluid dynamics
3121 ScRe602Sh sdotsdot+=sdot
=lowast
lowast
iw
pL
Ddk
where Diw is the diffusion coefficient of the compound i in water (m2middots-1) is the
minimum (uncorrected) value of the mass transfer coefficient and Sc is the
Schmidt number This relation is often referred to as the Froumlssling correlation The
particle diameter is a key parameter in the Froumlssling correlation and the external
mass transfer coefficient varies with square of the particle size for smaller
particles
lowastLk
The Schmidt number is the ratio of the kinematic fluid viscosity and the diffusion
coefficient of the compound i in water
iwDν
=Sc
Supporting Information VII
According to Harriott (1962) the actual mass transfer coefficient kL is 15 times
greater than the minimum value of the mass transfer coefficient The
uncertainty in k
lowastLk
Lmiddota associated with particle sphericity and roughness issues are
believed not to exceed a factor of 2
The diffusion coefficient of a compound i in water can be estimated as (Hayduk amp
Laudie 1974)
5890141
9102613
iiw
VD
sdot
sdot=
minus
η (m2middots-1)
where iV is the molar volume of the compound i (cm3middotmol-1) estimated according
to Fuller et al 1966
Assuming spherical particles the external (geometric) specific surface area and the
particle diameter are calculated from the measured BET specific surface area Atot
assuming that our GR-SO4 has a AtotAouter ~ 30 similar to the one reported by
Hansen amp Koch (1998)
( ) ( ) ( )pppp
p
pp
p
dd
dV
SAAρρπ
πρ 1000
6
100061100030
SA3
2tot
geom sdot=
sdotsdot
sdot=
sdot== (m2middotg-1)
In our aqueous GR-SO4 system the density ρ = 1000 kgmiddotm-3 the absolute dynamic
viscosity η = 10-3 Pamiddots and the kinematic viscosity ν = 10-6 m2middots-1 for water The
GR-SO4 particle specific parameters used is found below
VIII Chapter 7
GR-SO4
Atot (m2g) 712
SAgeom (m2g) 237
Particle density ρp (kgm3) 1500
Particle diameter dp (m) 169middot10-6
Settling velocity ut (ms) 778middot10-7
Reynolds number 132middot10-6
The molar volumes the diffusion coefficients in water and the Schmidt numbers
for the NACs including 4-chloronitrobenzene (4-CNB) and nitrobenzene (NB)
were
Compound iV (cm3middotmol-1) Diw (m2middots-1) Sc
4-NT 1260 768middot10-10 1302
4-CNB 1230 779middot10-10 1284
4-NPA 1535 684middot10-10 1462
NB 1055 853middot10-10 1173
The ratio of the external (geometric) specific surface area to volume of solution
were calculated for GR-SO4 at the various concentrations applied
[Fe(II)GR]0 (mM) a (m-1)
103 366middot102
63 224middot103
126 448middot103
60 213middot103
The uncorrected mass transfer coefficients were estimated for the NACs using the
Froumlssling correlation
Supporting Information IX
[Fe(II)GR]0 (mM) lowastLk (mmiddots-1)
4-NT 912middot10-4
4-CNB 925middot10-4
4-NPA 812middot10-4
NB 101middot10-3
Finally kLmiddota was calculated and compared with the experimental 1 order rate
constants kobs obtained for the NACs
Compound [Fe(II)GR]0 (mM) kLmiddota (s-1) kobs (s-1) a
4-NT 103 050 420middot10-4
63 307 140middot10-3
126 613 590middot10-3
4-CNB 103 051 740middot10-4
63 311 170middot10-3
126 622 460middot10-3
4-NPA 103 045 640middot10-4
63 273 109middot10-3
126 546 473middot10-3
NB 60 324 137middot10-3 b
a Experimental pseudo 1 order rate constant at 50 microM [Ar-NO2]0 b Experimental pseudo 1 order rate constant at 10 microM [Ar-NO2]0
When comparing kLmiddota with kobs it can be seen that the rates of mass transfer for all
3 NACs exceed the observed rate constants by at least 3 or 4 orders of magnitude
at every initial Fe(II)GR concentration Thus the reaction of the given NACs with
GR-SO4 is not subject to mass transfer limitations under the experimental
conditions applied here
722 Surface saturation limited kinetics
More than 75 of all heterogeneous reactions that are not diffusion-limited are
surface-reaction-limited rather than adsorption- or desorption-limited We now
X Chapter 7
look at the reaction A = B = C where an intermediate B is formed In our system
A = Ar-NO2 B = Ar-NHOH and C = Ar-NH2 In this case the surface reaction is
assumed to be a single-site mechanism where only the site S on which A or B is
adsorbed is involved in the reaction forming B or C
KA
Adsorption 1 A + S = AmiddotS
kS1
Surface reaction 1 AmiddotS = BmiddotS
KB-1
Desorption 1 BmiddotS = B + S
KB
Adsorption 2 B + S = BmiddotS
kS2
Surface reaction 2 BmiddotS = CmiddotS
KC-1
Desorption 2 CmiddotS = C + S
The rate law for this surface-reaction limited single-site mechanism involving an
intermediate follows Langmuir-Hinshelwood kinetics (adopted from Fogler 1999)
CCBBAA
AAsitesSA
CKCKCKCKCk
dtdC
sdot+sdot+sdot+sdotsdotsdot
=minus1
1
Supporting Information XI
where kS1 is the intrinsic rate constant of the surface reaction transforming A into
the intermediate B Csites is the concentration of reactive sites S on the solid
surface KA KB and KC are the adsorption constants for A B and C at the reactive
surface sites and CA CB and CC are the concentrations of A B and C in the bulk
fluid Two major assumptions of the Langmuir isotherm imply that there is a fixed
number of localised surface sites present on the surface and that the activity of the
surface towards adsorption desorption or surface reaction is independent of
surface coverage
Hence fitting -∆CA∆t to CA CB and CC using a nonlinear curve fitting software
such as SigmaPlot may provide one with the intrinsic rate constant and the
adsorptions constants If KB and KC gtgt KA the intermediate and the product are
strongly competing with the reactant for vacant reactive surface sites
Our data was not fitted successfully by the Langmuir-Hinshelwood rate law
(regression results not shown) Simplifying the rate law by excluding either the
term KCmiddotCC or KBmiddotCB or both (assuming that the aniline product or the
hydroxylaniline intermediate or both did not compete for the reactive sites) did not
improve the regression The Langmuir-Hinshelwood rate law for a dual-site
mechanism did not fit our data either Thus Langmuir-Hinshelwood kinetics
cannot explain the reaction mechanism of the given NACs in our GR-SO4 system
73 External surface area of GR-SO4 and GR-CO3
The GR-SO4 unit cell consists of one double layer (d001 = 11 nm) ie one
hydroxide layer (049 nm ) and one interlayer (061 nm) Hexagonal GR-SO4
particles holding an average width of 1 microm (Figure 72) an average particle
thickness of 35 nm (Hansen amp Koch 1998) and a hydroxide layer thickness of
049 nm have a surface area of the basal plane
Abasal = 1 microm middot 1 microm ndash 2 middot 05 microm middot 025 microm = 075 microm2
XII Chapter 7
and a surface area of the edges
Aedge = (2 middot 05 microm + 4 middot 056 microm) middot 000049 microm = 00016 microm2
Figure 72 The hexagonal platy morphology of GR particles holding an average width of 1 microm
The particle thickness is the mean crystal thickness perpendicular to the 003 plane
as determined from the 003 reflections in an X-ray diffractogram A GR-SO4
particle holding a thickness of 35 nm contains 35 nm11 nm = 318 double layers
The GR-CO3 unit cell consists of one double layer (d001 = 075 nm) ie one
hydroxide layer (049 nm ) and one interlayer (026 nm) Hence a GR-CO3 particle
holding a thickness of 35 nm contains 35 nm075 nm = 467 double layers
The outer surface area of a GR-SO4 particle including outer basal planes and
edges is
Aouter = 222 microm 155microm 00016318microm 0752 =sdot+sdot
and the total surface area of a GR-SO4 particle including both inner and outer
basal planes as well as edges is
Supporting Information XIII
Atot = 222 microm 478)microm 00016microm 0752(318 =+sdotsdot
Hence the ratio of outer surface area to total surface area is
131microm 478microm 155
AA
2
2
tot
outer asymp=
Furthermore the ratio of edge surface area to outer surface area is
130microm 155
microm 00016318AA
2
2
outer
edge asympsdot
=
For GR-CO3 the outer surface area including outer basal planes and edges is
Aouter = 222 microm 157microm 00016746microm 0752 =sdot+sdot
and the total surface area of a GR-CO3 particle including both inner and outer
basal planes as well as edges is
Atot = 222 microm 701)microm 00016microm 0752(467 =+sdotsdot
Hence the ratio of outer surface area to total surface area is
145microm 701microm 157
AA
2
2
tot
outer asymp=
Furthermore the ratio of edge surface area to outer surface area is
121microm 157
microm 00016467AA
2
2
outer
edge asympsdot
=
XIV Chapter 7
74 Van der Waals radii
The size of polyatomic molecules can be estimated by summing the van der Waals
radii of the
individual atoms Van der Waals radii or nonbonded radii can be pictured as the
radii of hard spherical atoms (Figure 73)
Figure 73 Schematic of neighboring nonbonded atoms with van der Waals radii rA and rB
Assuming that the spheres of neighboring nonbonded atoms just touch (Figure
73) the highest possible ion or molecule size Ms can be estimated as the sum of
the van der Waals radii
Ms = 2middotrA + 2middotrB + (1)
Taking Paulingrsquos rule for nonmetals into account we can estimate the real size of
polyatomic ions bound by covalent bonds (Pauling 1960) The van der Waals
radius is larger than the covalent radius because it involves the interposition of two
electron pairs between the atoms rather than one The rule states that the van der
Waals radius of an atom exceeds its covalent radius by ~008 nm (overlap in
Figure 74)
Figure 74 Schematic of atoms undergoing covalent bonding
Supporting Information XV
Thus the size of polyatomic ions bound by covalent bonds Ms can now be
estimated as the sum of the van der Waals radii subtracted by 008 nm
Ms = 2middot(rA - 008 nm) + 2middot(rB - 008 nm) + (2)
We estimated the molecular size of the NACs (Table 72) by means of equation (2)
and the van der Waals radii of the atoms in Table 71
Table 71 Van der Waals radii of various atoms Values from Pauling 1960
Atom vdW radii (nm)
H 0120
O 0140
N 0150
C 0170
Cl 0181
S 0185
In order to make the calculations it was assumed that all atoms were spherical and
that all bond angles were 90deg or 180deg (linear structures) In addition no distinctions
were made between single and double bonds The molecular sizes of the NACs
were estimated with the benzene ring representing the xy plane
Table 72 Molecular sizes of the NACs a Thickness z of the xy plane
Compound Ms (x) (nm) Ms (y) (nm) Ms (z) (nm)a
NB 054 080 036
4-NT 054 106 036
4-CNB 054 100 036
4-NPA 054 136 036
XVI Chapter 7
Note that the molecular sizes in Table 72 are only rough estimations
For comparison with the GR-SO4 interlayer spacing (061 nm) we consider three
possible orientations of the NACs in the GR-SO4 interlayer 1) The NAC xyz
coordination is equivalent to the crystal abc coordination (z = c = 036 nm) 2) the
NAC xy plane is parallel to the crystal bc plane (z = a = 054 nm) and 3) the NAC
xy plane is parallel to the crystal ac plane (z = b = 080-136 nm) Hence the sizes
of the NACs do not hinder their access to the GR-SO4 interlayer Only when
oriented vertically do the sizes of the NACs (z = b = 080-136 nm) exceed the GR-
SO4 interlayer spacing
The molecular size of trichloroacetate (TCA) was also estimated by means of
equation (2) and the atomic van der Waals radii in Table 71 When the TCA
aliphatic chain is assumed to represent the x direction (Ms (x) = 066 nm) the
molecular size in the y and z directions ranges from 045-053 nm depending on the
free rotation of the C-C bond Thus only if the C-C bond is oriented perpendicular
to the crystal ab plane does the size of TCA exceed the GR-SO4 interlayer spacing
(061 nm) In contrast the size of TCA exceeds the GR-CO3 interlayer spacing
(026 nm) regardless of its orientation
75 Adsorption of Fe(II) onto Fe(III) oxides
As seen from the Fe(II) sorption isotherms Fe(II) sorption varies widely between
the Fe(III) oxides as a function of solution pH (Figure 75) Average surface
densities of approximately 2 singly coordinated sitesnm2 iron oxide have been
suggested for goethite and lepidocrocite (Cornell amp Schwertmann 1996) The
similar surface site densities of goethite and lepidocrocite might explain their
similar Fe(II) adsorption isotherms (Figure 75)
Supporting Information XVII
Figure 75 Fe(II) adsorption edges for ferrihydrite goethite hematite lepidocrocite and
magnetite in the absence of other specifically adsorbing cations and anions (from Vikesland amp
Valentine 2002 and references therein) The total number of surface sites was in excess of the
total Fe(II) concentrations in all experiments
Dissolved cations or anions may specifically adsorb at the calcite and Fe(III) oxide
surfaces by exchanging for H+ or OHndash at the equivCO3H0 equivCaOH0 equivFeOH0 and
equivFeIIIOFeIIOH0 surface sites At the experimental conditions applied here within a
pH range 70-87 the dominant species of interest in solution are Fe2+ HCO3ndash
CO32ndash Clndash SO4
2ndash (only in the GR-SO4 systems) and the anionic TCA and DCA In
addition Fe2+ readily forms aqueous complexes with hydroxide carbonate
chloride and sulfate whereby the species FeOH+ FeHCO3+ Fe(OH)(CO3)ndash
FeCO30 Fe(CO3)2
2ndash FeCl+ and FeSO40 may occur (Millero amp Hawke 1992) At
pH 70-87 we expect the Fe(II) species Fe2+ FeCO30 Fe(OH)(CO3)ndash FeOH+ and
Fe(CO3)22ndash to dominate in the GR-CO3 and CaCO3(s)CO2(g) buffered magnetite
suspensions In the goecalcite and lepcalcite suspensions we expect the FeCl+
species to dominate as well whereas the Fe2+ FeSO40 and FeOH+ species most
XVIII Chapter 7
likely dominate in the GR-SO4 suspensions Anionic inorganic ligands like
carbonate chloride and sulfate can lower or enhance the adsorption of Fe(II) due to
a) formation of stable nonadsorbing Fe(II) ligand aqueous complexes b) formation
of Fe(II) ligand Fe(III) oxide surface complexes which can lead to surface
precipitation at high Fe(II) and ligand concentrations c) competitive ligand
sorption to the Fe(III) oxide surface blocking reactive sorption sites at the surface
and d) diminution of the positive charge at the Fe(III) oxide surface (at pH levels
below the point of zero charge (pHpzc) of the Fe(III) oxide) thereby decreasing the
electrostatic repulsion of cations by the Fe(III) oxide surface Specifically adsorbed
cations increase the pHpzc whereas specifically adsorbed anions decrease the pHpzc
References Arnold WA Ball WP Roberts AL (1999) Polychlorinated ethane reaction with zero-valent zinc Pathways and rate control Journal of Contaminant Hydrology 40 183-200 Cornell RM Schwertmann U (1996) The iron oxides Structure properties reactions occurrence and uses VCH Verlagsgesellschaft mbH Weinheim Fogler HS (1999) Elements of chemical reaction engineering 3rd ed Prentice Hall Fuller EN Schettler PD Giddings JC (1966) A new method for prediction of binary gas-phase diffusion coefficients Industrial and Engineering Chemistry 58 19-27 Hansen HCB Koch CB (1998) Reduction of nitrate to ammonium by sulphate green rust Activation energy and interlayer reaction mechanism Clay Minerals 33 87-101 Harriott P (1962) Mass transfer to particles Part I Suspended in agitated tanks AIChE Journal 8 93-102 Hayduk W Laudie H (1974) Prediction of diffusion coefficients for nonelectrolytes in dilute aqueous solutions AIChE Journal 20 611-615 Hofstetter TB Heijmann CG Haderlein SB Holliger C Schwarzenbach RP (1999) Complete reduction of TNT and other (poly)nitroaromatic compounds under iron-reducing subsurface conditions Environmental Science and Technology 33 1479-1487 Klausen J Troumlber SP Haderlein SB Schwarzenbach RP (1995) Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions Environmental Science and Technology 29 2396-2404 Meisel D Neta P (1975) One-electron redox potentials of nitro compounds and radiosensitizers Correlation with spin densities of their radical anions Journal of the American Chemical Society 97 5198-5203
Supporting Information XIX
Millero FJ Hawke DJ (1992) Ionic interactions of divalent metals in natural waters Marine Chemistry 40 19-48 Pauling L (1960) The nature of the chemical bond 3rd ed Cornell University Press Ithaca Vikesland PJ Valentine RL (2002) Iron oxide surface-catalyzed oxidation of ferrous iron by monochloramine implications of oxide type and carbonate on reactivity Environmental Science and Technology 36 512-519 Wardman P (1989) Reduction potentials of one-eletron couples involving free radicals in aqueous solution The Journal of Physical Chemistry Reference Data 18 1637-1755
Curriculum Vitae 13011973 Born in Haderslev Denmark 1988-1991 Mathematical high school Haderslev Katedralskole Denmark 1991-1992 Sabbatical year 1992-1995 B Sc in environmental chemistry University of Copenhagen
(KU) Denmark B Sc thesis 1995 ldquoMethane oxidizing bacteria in soilrdquo
1995-1998 M Sc in environmental chemistry University of Copenhagen
Denmark 1997-1998 diploma thesis ldquoReductive dechlorination of carbon tetrachloride and chloroform in presence of iron(II)iron(III)-hydroxides (green rust)rdquo
1998-1999 Research and teaching assistant at the Chemistry Department
The Royal Veterinary amp Agricultural University (KVL) Denmark
1999-2004 PhD in environmental sciences Swiss Federal Institute of
Technology Zuumlrich (ETHZ) and Swiss Federal Institute for Environmental Science and Technology (EAWAG) Switzerland Docoral thesis ldquoFormation and redox reactions of green rusts under geochemical conditions found in natural soils and sedimentsrdquo
2000-2002 Teaching assistent at the Swiss Federal Institute of Technology
Zuumlrich and supervision of diploma students 2002 Microbial Diversity summer course (7 weeks) at the Marine
Biological Laboratory Woods Hole Massachusetts USA

Table of Contents
525 Analytical methods 104 53 Results and discussion 105
531 Product formation and reaction kinetics 105 532 Comparing rate constants obtained for the various Fe(II)-Fe(III)
mineral systems 109 533 Comparing with rate constants obtained for other chlorinated
aliphatic compound 112 534 Factors controlling the reactivity of surface-bound Fe(II) 114 535 Comparison with biotic and other abiotic systems 118
54 Conclusions 119 References 120 6 Conclusions and Outlook 125 References 128 7 Supporting Information I 71 Estimation of the one-electron reduction potential for 4-NPA I 72 The rate-limiting step IV 721 Mass transfer (diffusion) limited kinetics V
722 Surface saturation limited kinetics IX 73 External surface area of GR-SO4 and GR-CO3 XI 74 Van der Waals radii XIV 75 Adsorption of Fe(II) onto Fe(III) oxides XVI References XVIII Curriculum Vitae
Zusammenfassung I
Zusammenfassung Geschichtete Fe(II)-Fe(III)-Hydroxide (Gruumlner Rost) gehoumlren zur Gruppe der
Fe(II)-haltigen Mineralsysteme (zB Magnetit (Fe3O4) Siderit (FeCO3) Vivianit
(Fe2(PO4)2sdot8H2O) Fe(II)-Sulfide sowie an die Oberflaumlche von Fe(III)-Oxiden und
Tonmineralien gebundenes zweiwertiges Eisen) die die Aktivitaumlt von Fe(II) in
suboxischen und anoxischen Boumlden und Sedimenten kontrollieren Gruumlner Rost
Phasen (GRs) bestehen aus planaren positiv geladenen trioktaedrischen Fe(II)-
Fe(III)-Hydroxidschichten die durch hydratisierte Anionen in den
Zwischenschichten ausgeglichen werden Ihre generelle Zusammensetzung ist
[FeII(6-x)FeIII
x(OH)12]x+[(A)xnmiddotyH2O]x- wobei x = 09 - 42 ist A entspricht einem n-
valenten Anion (zB CO32- Clndash oder SO4
2-) und y repraumlsentiert die Anzahl
Wassermolekuumlle in der Zwischenschicht GRs sind wichtige intermediaumlre Phasen
die durch unvollstaumlndige Oxidation von Fe(II) oder teilweise Reduktion von Fe(III)
gebildet werden koumlnnen Sie koumlnnen in suboxischen nicht-sauren eisenhaltigen
natuumlrlichen wie auch technischen Systemen auftreten so wie in Wasser gesaumlttigten
Boumlden und interstitiellen Sedimenten Rohrleitungen in der
Trinkwasserversorgung Stahlpfosten in marinen Sedimenten Stahlbeton und in
reaktiven durchlaumlssigen Waumlnden aus nullwertigem Eisen zur in-situ Sanierung von
Altlasten und Aquiferen Aufgrund ihrer Schichtstruktur den anionischen
Zwischenschichten und der hohen spezifischen Oberflaumlchen sind GRs reaktive
Ionentauscher und Sorbentien von Anionen Des Weiteren wurde gezeigt dass
GRs eine Reihe anorganischer und organischer Schadstoffe reduzieren koumlnnen
Durch Immobilisierung und Transformation koumlnnen GRs somit eine wichtige Rolle
fuumlr das Abbauverhalten und den Transport solcher Schadstoffe in suboxischen
Boumlden und Sedimenten spielen Die Resultate dieser Dissertation tragen zum
Verstaumlndnis uumlber die Bildung und Reaktivitaumlt von Fe(II)-haltigen Mineralsystemen
wie GRs Vivianit Magnetit und an Goethit (α-FeOOH)- und Lepidokrozit (γ-
FeOOH)-Oberflaumlchen gebundenes Fe(II) in der Natur bei
II Zusammenfassung
Um die Rolle von Bakterien bei der Bildung von GRs in natuumlrlichen Boumlden und
Sedimenten aufzuklaumlren wurden Eisenminerale untersucht die als Folge der
Aktivitaumlt von eisenrespirierenden Bakterien gebildet wurden Kapitel 2 beschreibt
die Untersuchungen von eisenhaltigen Produkten die von anaeroben autotrophen
denitrifizierenden Fe(II)-oxidierenden Bakterien (FeOB) gebildet wurden Ein
Bikarbonat- und Phosphat-reiches Kulturmedium bot den nitratreduzierenden
FeOB optimale Bedingungen Fe(II) lag zu Anfang der Reaktion als weisses
Fe(II)-Hydroxyphosphat (Vivianit) und als geloumlstes Fe(II) vor Die Ergebnisse
zeigten dass die denitrifizierenden FeOB amorphen Goethit via ein gruumlnes Fe(III)-
angereichertes Vivianit-Zwischenprodukt bildeten Die Analyse mit Moumlssbauer
Spektroskopie deutet nicht auf eine Bildung von GR hin
In Kapitel 3 werden jene Eisenmineralien beschrieben die waumlhrend der Reduktion
verbreiteter Fe(III)-Oxide durch anaerobe dissimilative Fe(III)-reduzierende
Mikroorganismen Shewanella algae BrY gebildet wurden Um natuumlrliche
Zustaumlnde zu simulieren wurden Fe(III)-Oxide als Beschichtungen auf
Silikatpartikel (Modellsystem fuumlr Sandboumlden) oder Calcitpartikel (CaCO3
Modellsystem fuumlr kalkhaltige Boumlden) aufgetragen sowie synthetische
Elektronencarrier und hochkonzentrierte kuumlnstliche pH-Puffer ausgeschlossen
Die erforschten Mineralsysteme umfassten GoethitCalcit- LepidokrozitCalcit-
und FerrihydritSand-Suspensionen S algae BrY reduzierte beachtliche Mengen
des eingesetzten Fe(III) und es bildeten sich gruumlne und schwarze Festphasen
innerhalb von 1-2 Wochen nach der Animpfung Moumlssbauer Spektroskopie der
gruumlnen und schwarzen Praumlzipitate zeigte dass sich diese aus GR und Vivianit
zusammensetzen
Die Reaktivitaumlt synthetischer GRs gegenuumlber reduzierbaren organischen
Schadstoffen wurde erkundet um die potentielle Bedeutung von GR-Phasen fuumlr
das Schicksal solcher Verbindungen abzuschaumltzen Zu diesem Zweck wurden
Nitroaromaten (NACs) und Chloracetate als Modellverbindungen benutzt um
Zusammenfassung III
umweltrelevante Redoxreaktionen zu studieren In Kapitel 4 wurde die relative
Reaktivitaumlt von aumlusseren und inneren reaktiven Stellen in synthetischem Sulfat-
Gruumlnem Rost (GR-SO4) anhand von strukturaumlhnlichen ldquoreaktiven
Sondenmolekuumllenrdquo mit unterschiedlichen Ladungen untersucht Als reaktive
Sondenmolekuumlle wurden Nitrobenzen 2-Nitrophenol 4-Nitrotoluen 4-
Chlornitrobenzen und 4-Nitrophenylessigsaumlure verwendet Die Ergebnisse zeigen
dass GR-SO4 die NACs vollstaumlndig zu den entsprechenden Anilinen reduzierte
Die Reaktionen folgten einer pseudo 1 Ordnungs Kinetik bezuumlglich NAC und die
auf Oberflaumlche normalisierten pseudo 1 Ordnungs Geschwindigkeitskonstanten
(Anfangsraten) waren 016ndash465middot10-4 s-1middotm-2middotL fuumlr [Fe(II)GR]0 = 103-1260 mM
[NAC]0 = 20-102 microM und pH 84-86 Weder durch Einbezug von
Massentransferlimitierung noch von Oberflaumlchensaumlttigungskinetik war es moumlglich
die aumlhnlichen Oberflaumlchennormalisierten pseudo 1 Ordnungs
Geschwindigkeitskonstanten fuumlr die Reduktion der neutralen und anionischen
NACs durch GR-SO4 zu erklaumlren Dieser Umstand laumlsst vermuten dass die
Reaktion zwischen NAC und GR-SO4 an den externen reaktiven Fe(II)-Stellen
stattfindet Bei niedrigen Fe(II)GR-Anfangskonzentrationen wurden die externen
reaktiven Fe(II)-Stellen aufgebraucht und die Regenerierung von neuen externen
reaktiven Stellen haben schliesslich die Geschwindigkeit der Reduktion von NACs
durch GR-SO4 kontrolliert
In Kapitel 5 wurde die Reaktivitaumlt von verschiedenen umweltrelevanten Fe(II)-
Fe(III)-Mineralsystemen gegenuumlber Trichloressigsaumlure (TCA) und
Dichloressigsaumlure (DCA) in Batchexperimenten die natuumlrliche Bedingungen
imitierten untersucht Die Fe(II)-Fe(III)-Systeme umfassten Sulfat-Gruumlner Rost
Carbonat-Gruumlner Rost Magnetit Fe(II)Goethit und Fe(II)Lepidokrozit TCA
wurde von allen Fe(II)-haltigen Mineralien zu DCA reduziert Die Reaktionen
folgten einer pseudo 1 Ordnungs Kinetik bezuumlglich TCA und die auf Oberflaumlche
normalisierten pseudo 1 Ordnungs Geschwindigkeitskonstanten betrugen 033ndash
76middot10-5 min-1middotm-2middotL bei [Fe(II)]0 = 025ndash116 mM [TCA]0 = 15ndash1000 microM und pH
IV Zusammenfassung
70ndash87 Die Ergebnisse zeigen keine signifikanten Unterschiede zwischen den
verschiedenen Fe(II)-Fe(III)-Systemen bezuumlglich Produkteverteilung und
oberflaumlchen-normalisierten pseudo 1 Ordnungs Geschwindigkeits-konstanten In
keinem der Systeme wurde DCA innerhalb des experimentellen Zeitraums zu
Monochloressigsaumlure oder Essigsaumlure weiter reduziert
Die Ergebnisse die in dieser Dissertation praumlsentiert werden zeigen dass
mikrobiologische Prozesse fuumlr die Oxidation von Vivianit-Phasen im Untergrund
verantwortlich sein koumlnnen Zudem wurde nachgewiesen dass GRs bei tiefen
Kohlenstoff- und Fe(III)-Konzentrationen sowie durch Ausschluss von
kuumlnstlichen Elektronencarriern und pH-Pufferung mikrobiell gebildet werden
koumlnnen Ferner zeigten Befunde dass GRs eine bedeutende Rolle fuumlr die reduktive
Transformation von NACs und TCA in natuumlrlichen Boumlden und Sedimenten spielen
koumlnnen
Summary V
Summary
Layered iron(II)-iron(III)-hydroxides (green rusts) belong to the group of Fe(II)-
bearing mineral systems eg magnetite (Fe3O4) siderite (FeCO3) vivianite
(Fe2(PO4)2sdot8H2O) Fe(II) sulfides as well as Fe(II) associated with Fe(III) oxide
and clay mineral surfaces that control the Fe(II) activity in suboxic and anoxic
soils and sediments Green rusts (GRs) consist of plane positively charged
trioctahedral Fe(II)-Fe(III) hydroxide sheets balanced by hydrated anions in the
interlayers and hold the general formula [FeII(6-x)FeIII
x(OH)12]x+[(A)xnmiddotyH2O]x-
where x = 09 - 42 A is an n-valent anion eg CO32- Clndash or SO4
2- and y is the
number of water molecules in the interlayer GRs are important intermediate
phases formed by partial oxidation of Fe(II) or partial reduction of Fe(III) and they
have been found in suboxic non-acid iron-rich natural environments such as
hydromorphic soils and intertidal sediments and in engineering systems including
pipeline distribution systems for drinking water steel sheet piles in marine
sediments reinforced concrete and permeable reactive barriers of zero-valent iron
implemented for on-site remediation of contaminants Due to their layered
structures anionic interlayers and high specific surface areas GRs represent
reactive ion exchangers and sorbents of anions In addition GRs have been shown
to reduce a range of inorganic and organic pollutants Thus through sequestration
and reductive transformation GRs may play an important role in the fate and
transport of contaminants in suboxic soils and sediments The work presented in
this dissertation adds to the understanding of how Fe(II)-bearing minerals like
GRs vivianite magnetite and Fe(II) associated with goethite (α-FeOOH) and
lepidocrocite (γ-FeOOH) may form and react in nature
In order to elucidate the role of bacteria in the formation of GRs in natural soils
and sediments we studied the iron mineral phases forming as a result of the
activity of iron-respiring bacteria In the study described in chapter 2 the Fe-
containing products formed by anaerobic autotrophic denitrifying Fe(II)-oxidizing
VI Summary
bacteria (FeOB) were examined The culture medium applied contained high levels
of bicarbonate and phosphate and is typically used in this kind of studies as it
provides excellent conditions for the nitrate-reducing FeOB Fe(II) was present
initially as a whitish solid Fe(II) hydroxy phosphate (vivianite) and as soluble
Fe(II) The results obtained demonstrate that the denitrifying FeOB produce poorly
crystalline goethite via a greenish Fe(III)-enriched vivianite intermediate
Moumlssbauer spectroscopic analyses provided no significant evidence of green rust
formation
In chapter 3 the Fe-containing products formed during reduction of common
Fe(III) oxides by the anaerobic dissimilatory Fe(III)-reducing microorganism
Shewanella algae BrY are discussed In order to simulate natural conditions
Fe(III) oxides were applied as coatings on silica (model system for sandy soils) or
calcite (CaCO3) particles (model system for calcareous soils) and synthetic
electron shuttles as well as highly concentrated artificial pH buffers were excluded
The mineral systems studied include goethitecalcite lepidocrocitecalcite and
hydrous ferric oxidesand suspensions S algae BrY reduced substantial amounts
of the initial Fe(III) and green and blackish mineral phases were produced within
1-2 weeks after inoculation Moumlssbauer spectroscopic analyses showed that the
green and black precipitates consisted of GR and vivianite
The reactivity of synthetic GRs towards reducible organic pollutants was
investigated in order to asses the potential significance of GR phases for the fate of
such compounds To this end we used nitroaromatic compounds (NACs) and
chlorinated acetates as suitable model compounds for studying environmentally
relevant redox reactions In the work described in chapter 4 the relative reactivity
of outer and inner Fe(II) reactive sites in synthetic sulfate green rust (GR-SO4) was
studied using a series of structurally closely related compounds with different
charge properties as ldquoreactive probesrdquo The probe compounds included
nitrobenzene 2-nitrophenol 4-nitrotoluene 4-chloronitrobenzene and 4-
Summary VII
nitrophenylacetic acid The results show that NACs are completely reduced to their
corresponding anilines by GR-SO4 The reactions followed pseudo 1 order
kinetics with respect to NAC and the surface area-normalised pseudo 1 order rate
constants (initial rates) obtained were 016ndash465middot10-4 s-1middotm-2middotL at [Fe(II)GR]0 = 103-
1260 mM [NAC]0 = 20-102 microM and pH 84-86 Neither mass transfer control nor
surface saturation kinetics could explain the similarity of the surface-normalised
pseudo 1 order rate constants obtained for the reduction of the neutral and anionic
NACs by GR-SO4 These observations suggest that the reaction between NAC and
GR-SO4 takes place at the external reactive Fe(II) sites At low initial Fe(II)GR
concentrations the external reactive Fe(II) sites were depleted and the regeneration
of new external reactive sites eventually controlled the reduction of the NACs by
GR-SO4
Finally the reactivity of various Fe(II)-Fe(III) mineral systems towards
trichloroacetic acid (TCA) and dichloroacetate (DCA) has been investigated in
laboratory batch experiments imitating natural conditions (Chapter 5) The Fe(II)-
Fe(III)-systems investigated included GR-SO4 carbonate green rust magnetite
Fe(II)goethite and Fe(II)lepidocrocite TCA was readily reduced to DCA by all
Fe(II)-containing minerals The reactions followed pseudo 1 order kinetics with
respect to TCA and the surface area-normalised pseudo 1 order rate constants
obtained were 033ndash76middot10-5 min-1middotm-2middotL at [Fe(II)]0 = 025ndash116 mM [TCA]0 =
15ndash1000 microM and pH 70ndash87 Our results showed no significant differences
regarding product distribution and surface area-normalised reaction rate constants
between the Fe(II)-Fe(III)-systems DCA was not further reduced to
monochloroacetate (MCA) or acetate in any of the systems within the time frame
in our experiments
The results presented in chapter 2 indicate that microbiological processes may be
responsible for the oxidation of vivianite phases in natural subsurface
environments In chapter 3 we demonstrated that GRs may be produced
VIII Summary
microbially at conditions including low carbon and Fe(III) concentrations as well
as the exclusion of synthetic electron shuttles and pH buffers The results obtained
in chapter 4 and 5 show that GRs transform NACs and TCA readily The reductive
transformation of NACs and TCA by GRs is relevant to understanding the
processes responsible for their degradation in the subsurface and the development
of innovative technologies for their remediation
General Introduction 1
1 General Introduction
11 Iron cycling in the subsurface
Iron is the fourth most abundant element (4-5 mass) and the most abundant redox
sensitive element in the Earthrsquos crust It is found as Fe(II) and Fe(III) in a number
of minerals in rocks soils and sediments Under anoxic conditions solid Fe(III)-
containing minerals can be reduced to soluble Fe(II) once the more energetically
favoured electron donors - nitrate and manganese(IV) oxides - have been
consumed Dissolved Fe(II) can be reoxidized to insoluble Fe(III) microbially or
abiotically upon exposure to oxygen Due to this ready alternation between the
Fe(II) and Fe(III) redox states iron plays a major role in controlling the redox
potential and the carbon cycling in subsurface environments (Nealson amp Saffarini
1994)
Nonenzymatic processes were previously considered to account for most of the
Fe(III) reduction in subsurface environments The significance of bacteria in the
biogeochemical cycling of iron has been broadly recognized over the past two
decades Dissimilatory Fe(III)-reducing bacteria (DIRB) that gain energy by
coupling the oxidation of hydrogen or organic compounds to the reduction of
Fe(III) oxides have been known for many years but their biogeochemical
importance was only widely acknowledged about a decade ago (reviewed by
Lovley 1997) Fe(III) bioreduction accounts for a major fraction of the carbon
oxidation in many different anoxic environments and in the presence of sufficient
amounts of reactive Fe(III) microbial Fe(III) reduction may even inhibit sulphate
reduction and methanogenesis (King 1990 Lovley amp Phillips 1986) In fact most
of the Fe(III) reduction in the Fe(III) reduction zone of aquatic sediments and
aquifers is enzymatically catalyzed by microorganisms (Lovley et al 1991) A
wide diversity of DIRB distributed among several different phylogenetic groups
2 Chapter 1 is known today The two most studied DIRB are the obligate anaerobic Geobacter
spp and the facultatively anaerobic Shewanella spp (Figure 11)
Aerobic oxidation of Fe(II)-containing minerals by lithotrophic acidophilic and
neutrophilic bacteria has been known for many years but their broad significance
in the biogeochemical cycling of iron has only been recognized over the past two
decades Both acidophilic (Thiobacillus ferrooxidans) and neutrophilic
(Gallionella ferruginea Leptothrix ochracea Sphaerotilus natans) aerobic Fe(II)-
oxidizing bacteria (FeOB) have been isolated (Hanert 1992 Kuenen et al 1992
Mulder amp Deinema 1992)
Figure 11 The microbial iron cycle
Anaerobic Fe(II) oxidation by phototrophic purple non-sulfur bacteria utilizing
Fe(II) as an electron donor in the light was recognized only a decade ago (Widdel
et al 1993) Subsequently it was demonstrated that the biological oxidation of
Fe(II) in the absence of oxygen is possible by light-independent chemotrophic
microorganisms using nitrate as the electron acceptor (Straub et al 1996) Thus
the microbial iron cycle includes anaerobic Fe(III)-reducing microorganisms and
aerobic as well as anaerobic Fe(II)-oxidizing bacteria (Figure 11)
General Introduction 3
12 Green rusts
Iron oxides iron hydroxides and iron oxyhydroxides (collectively termed iron
oxides or Fe(III) oxides) are ubiquitous in the pedosphere where they originate
from aerobic weathering of surface magmatic rocks such as ferromagnesium
silicates and pyrite (Cornell amp Schwertmann 1996) Goethite (α-FeOOH)
lepidocrocite (γ-FeOOH) ferrihydrite (Fe5HO8sdot4H2O) hematite (α-Fe2O3)
magnetite (Fe3O4) maghemite (γ-Fe2O3) and akageneite (β-FeOOH) constitute the
most important iron oxides in soils and sediments (Schwertmann amp Cornell 1991)
The formation and transformation of iron oxides depend on pH solution
composition redox potential temperature rate of oxidationreduction and degree
and rate of hydrationdehydration Iron oxides are important to many soil
properties such as colour pH and redox buffer capacity aggregation with other
soil particles as well as retention of anions and cations (Cornell amp Schwertmann
1996) A number of Fe(II)-bearing minerals including Fe(II)-containing clays (eg
smectites vermiculites and micas) magnetite siderite (FeCO3) vivianite
(Fe2(PO4)2sdot8H2O) Fe(II) sulphides and green rusts (layered Fe(II)-Fe(III)
hydroxides) may be present in soils and sediments under suboxic and anoxic
conditions Green rusts are believed to play a central role as metastable
intermediates in the redox cycling of iron at circumneutral pH in aquatic and
terrestrial environments
Green rusts (GRs) are layered iron(II)-iron(III)-hydroxides consisting of plane
positively charged trioctahedral Fe(II)-Fe(III) hydroxide sheets balanced by
hydrated anions in the interlayers (cf Figure 41 this work) GRs belong
structually to the pyroaurite-sjoumlgrenite group of layered hydroxides and they hold
the general formula [FeII(6-x)FeIII
x(OH)12]x+[(A)xnmiddotyH2O]x- where x = 09 - 42 A is
an n-valent anion eg CO32- Clndash or SO4
2- and y is the number of water molecules
in the interlayer The three most common and investigated green rust forms include
chloride GR (GR-Cl) sulphate GR (GR-SO4) and carbonate GR (GR-CO3)
Generally GRs are crystallographically classified into the GRI (rhombohedral
4 Chapter 1 GR-Cl and GR-CO3) and GRII (hexagonal GR-SO4) crystal systems The GR
interlayer thickness is a function of both the size and the charge of the interlayer
anion Tetrahedrally coordinated anions like sulphate lead to larger interlayer
distances than smaller monoatomic anions like chloride or planar ions like
carbonate (Mendiboure amp Schoumlllhorn 1986) Not only size but also charge density
plays a role for the interlayer spacing That is for anions having the same number
of valence electrons anions with smaller ionic radii (higher electron density) are
bound more strongly and therefore result in smaller interlayer spacings The
interlayer in GR-SO4 is composed of two consecutive planes of anions and water
whereas GR-Cl and GR-CO3 interlayers consist of only one single plane (Simon et
al 2003)
GRs are important intermediate phases formed by partial oxidation of Fe(II) or
partial reduction of Fe(III) In neutral and weakly alkaline solutions the oxidation
of dissolved Fe(II) always passes through solid GR phases (Bernal et al 1959)
GRs may also form during oxidation of zero-valent iron and as a result of the
combination of Fe(II) and Fe(III) at circumneutral pH (Figure 12)
Figure 12 Formation and transformation of GRs Fe3O4 = magnetite γ-Fe2O3 = maghemite α-
FeOOH = goethite γ-FeOOH = lepidocrocite akageneite = β-FeOOH
General Introduction 5
Oxidation of GR-CO3 usually produces goethite and magnetite-maghemite
whereas GR-Cl and GR-SO4 transform into lepidocrocite and magnetite-
maghemite depending on pH and oxidation rate (Bernal et al 1959 Taylor 1980
Carlson amp Schwertmann 1990) The brown δ-FeOOH is formed by vigorous
oxidation of GR using air or a 30 aqueous solution of hydrogen peroxide (Bernal
et al 1959 Misawa et al 1974) Black ferromagnetic magnetite forms by slow
oxidation of GR whereas lepidocrocite forms at high oxidation rates (Misawa et
al 1974) The presence of chloride is a prerequisite for the formation of
akageneite (Bernal et al 1959 Refait amp Genin 1997)
A substantial amount of work has been conducted in order to estimate the free
energies of formation of green rusts The free energies of formation reported for
the carbonate and sulphate GRs fall in the range 4234ndash4384 kJsdotmol-1 as determined
from solution data monitored during anoxic alkalimetric titrations and from
reduction potential (Eh) and pH recordings monitored during oxidation of GRs in
aqueous solution (Hansen et al 1994 Drissi et al 1995 Genin et al 1996) The
free energies of formation provided allow for estimation of the stability domains of
GRs in Eh-pH phase diagrams (Drissi et al 1995 Genin et al 1996) As
evidenced from such diagrams (Figure 13) the stability domain of GR-SO4 lies
within pH 6-8 and Eh -700 ndash -400 mV depending on the activities of Fe(II) and
sulphate (compare Figures 13aampb) This agrees with the natural GR occurrences
found in suboxic non-acid iron-rich environments such as hydromorphic soils and
intertidal sediments (Al-Agha et al 1995 Trolard et al 1996 Genin et al 1998)
In addition GRs have been found as corrosion products in numerous engineering
systems including a pipeline distribution system for drinking water steel sheet
piles in marine sediments reinforced concrete (ferro-concrete) and permeable
reactive barriers of zero-valent iron implemented for on-site remediation of organic
and inorganic contaminants (Tuovinen et al 1980 Nielsen 1976 Genin et al
1991 Roh et al 2000)
6 Chapter 1
Figure 13 Eh-pH phase diagrams of GR-SO4 a) a = 10+2Fe-3 = 10minus2
4SOa -3 and b) a = 10+2Fe-2
= 10minus24SOa -1
The stability domains of GR-Cl and GR-CO3 are similar to the stability domain of
GR-SO4 At Fe(II) and sulphate activities lower than depicted in Figure 13b the
stability domain of GR-SO4 will be situated at higher pH and lower Eh Other
dissolved species present at anoxic conditions such as phosphate sulphide
carbonate and organic ligands may exert considerable effects on the availability of
Fe(II) and Fe(III) At anoxic and circumneutral conditions vivianite
(Fe2(PO4)2sdot8H2O) controls the Fe(II) activity even at very low phosphate
concentrations (Nriagu amp Dell 1974) The formation of solid Fe(II) sulphides and
siderite (FeCO3) as well as the complexation of Fe(II) and Fe(III) by organic
ligands may also control the activity of Fe(II) in the subsurface and thereby
interfere with the formation of GRs
Due to their layered structures anionic interlayers and high specific surface areas
GRs represent reactive ion exchangers and sorbents of environmentally concerning
anions eg arsenate and selenate (Myneni et al 1997 Randall et al 2001) In
addition GRs may incorporate divalent transition metal cations like Ni2+ Zn2+
Cd2+ Co2+ and Mg2+ by isomorphic substitution for Fe2+ in the hydroxide layers
General Introduction 7
(Tamaura 1985 Tamaura 1986 Refait et al 1994 Parmar et al 2001 Refait et
al 2001) Furthermore GRs have been shown to reduce a range of inorganic
contaminants such as nitrite nitrate selenate chromate uranyl pertechnetate and
the transition metals AgI AuIII CuII and HgII as well as organic pollutants
including halogenated ethanes ethenes and methanes (Hansen et al 1994 Hansen
et al 1996 Myneni et al 1997 Erbs et al 1999 Loyaux-Lawniczak et al 1999
Cui amp Spahiu 2002 Lee amp Batchelor 2002b Heasman et al 2003 OrsquoLoughlin et
al 2003aampb Pepper et al 2003 Elsner et al 2004 OrsquoLoughlin amp Burris 2004)
Thus through sequestration and reductive transformation GRs may play an
important role in the fate and transport of contaminants in suboxic soils and
sediments It should be noted that the rate constants reported for the reduction of
these inorganic and organic pollutants by GRs cannot be directly compared as the
various studies were conducted at very different experimental conditions
13 Microbial formation of green rusts
Generally one would expect that biogenic minerals have chemical compositions
and crystal habits similar to those produced by nonenzymatic processes as they are
governed by the same equilibrium principles In fact since the latter stages of
mineralization are abiotically driven and since the secondary Fe(II)-containing
minerals are formed indirectly by electron transfer outside the bacterial cell and not
directly inside the bacterial cell the type of iron mineral formed is a function of the
environmental conditions in which the bacteria live ie the same microorganism
form different minerals in different environments
The microbial formation of GRs resulting from bioreduction of various Fe(III)
oxides including ferrihydrite goethite and lepidocrocite by strains of the
anaerobic dissimilatory DIRB Shewanella putrefaciens has been reported
repeatedly over the last years (Fredrickson et al 1998 Kukkadapu et al 2001
Parmar et al 2001 Ona-Nguema et al 2002aampb Glasauer et al 2003)
However no evidence of biogenic formation of GRs at natural geochemical
8 Chapter 1 conditions have been offered and it is still unknown whether this process may take
place at natural conditions comprising low nutrient levels low iron concentrations
and the absence of synthetic electron shuttles and highly concentrated artificial pH
buffers Moreover the biotic formation of GRs by anaerobic denitrifying Fe(II)-
oxidizing bacteria has been suggested but the phases still need to be properly
identified (Chaudhuri et al 2001) In order to elucidate the role of bacteria in the
formation of GRs in natural soils and sediments we studied the iron mineral
phases forming as a result of the activity of iron-respiring bacteria (Chapters 2 and
3)
14 Redox reactions of green rusts
Fe(II) is one of the most abundant reductants present in aquatic and terrestrial
environments under suboxic and anoxic conditions (Lyngkilde amp Christensen
1992 Ruumlgge et al 1998) In these environments Fe(II) may be present as soluble
organic and inorganic complexes as surface complexes and as a host of Fe(II)-
bearing minerals Although aqueous Fe(II) complexes may reduce a number of
contaminants Fe(II) associated with mineral surfaces and structural Fe(II) present
in the mineral lattice in Fe(II)-containing minerals are often more powerful
reductants Fe(II)-bearing minerals including GRs magnetite siderite Fe(II)
sulphides as well as Fe(II)-carrying Fe(III) oxide and clay mineral surfaces have
been shown to reduce a number of organic and inorganic contaminants such as
nitroaromatic compounds chlorinated aliphatics chromate uranyl pertechnetate
nitrate monochloramine and carbamate pesticides (Klausen et al 1995 Cui amp
Eriksen 1996 Butler amp Hayes 1998amp1999 Erbs et al 1999 Liger et al 1999
Loyaux-Lawniczak et al 1999 Amonette et al 2000 Hwang amp Batchelor 2000
Hansen et al 2001 Gander et al 2002 Lee amp Batchelor 2002aampb Pecher et al
2002 Vikesland amp Valentine 2002 Hofstetter et al 2003 OrsquoLoughlin et al
2003aampb Strathmann amp Stone 2003 Elsner et al 2004 OrsquoLoughlin amp Burris
2004) However only few comparative studies on the reactivity of Fe(II)-bearing
minerals exist (Lee amp Batchelor 2002b Elsner et al 2004) When examining the
General Introduction 9
reaction rates of the reductive transformation of NACs and chlorinated aliphatics
by GRs and other Fe(II)-bearing minerals reported in these studies the rate
constants for GRs are mostly among the highest rates reported and in some cases
even higher than the rate constants for Fe(II) sulphides Thus GRs may play an
important role in the transformation of reducible contaminants in the subsurface
Nitroaromatic compounds (NACs) are widely applied as explosives herbicides
insecticides solvents and intermediates in the synthesis of dyes and pesticides
(Hartter 1985 Rosenblatt et al 1991) NACs are ubiquitous in the subsurface
environment and pose a health risk due to their toxicity (Rickert 1985) In anoxic
environments reduction of the nitro group is generally the first step during abiotic
or microbial transformation of the NACs (Macalady et al 1986) The
transformation reaction generally produces the corresponding aromatic amines and
minor amounts of intermediates (hydroxylamines and nitroso compounds) as well
as coupling products (azo and azoxy compounds) These products may be of
similar or even greater environmental concern
Trichloroacetic acid (TCA) is ubiquitous in soils and the concentrations reported
range from lt005 to 380 microgkg (Euro Chlor 2001 McCulloch 2002 Ahlers et al
2003) On account of its phytotoxicity suspected human carcinogenicity and
widespread occurrence TCA is of considerable environmental concern especially
in the terrestrial compartment (Ahlers et al 2003) Moreover the daughter
compounds of TCA - dichloroacetic acid (DCA) and monochloroacetic acid
(MCA) - are also toxins and suspected human carcinogens as well as widespread in
the environment (Reimann et al 1996 Berg et al 2000 Ahlers et al 2003 and
references therein) In this work the reactivity of synthetic green rusts towards
nitroaromatic compounds (NACs) and the reactivity of various Fe(II)-Fe(III)
mineral systems including synthetic GRs towards chlorinated acetates have been
studied (Chapters 4 and 5)
10 Chapter 1 15 Outline of the thesis
An examination of the Fe-containing products produced during solid state
oxidation of vivianite by anaerobic autotrophic denitrifying Fe(II)-oxidizing
bacteria is presented in chapter 2 The Fe(II)-oxidizing bacteria were cultured in a
mineral medium containing high levels of bicarbonate and phosphate which is
typically used in this kind of studies as it provides excellent conditions for the
nitrate-reducing FeOB The solid iron phases forming were investigated by
transmission Moumlssbauer spectroscopy infrared spectroscopy and scanning electron
microscopy
Chapter 3 includes a study on the Fe-containing products formed during reduction
of common Fe(III) oxides by the anaerobic dissimilatory Fe(III)-reducing
microorganism Shewanella algae BrY In order to simulate natural conditions
Fe(III) oxides were applied as coatings on silica (model system for sandy soils) or
calcite particles (model system for calcareous soils) and synthetic electron shuttles
as well as highly concentrated artificial pH buffers were excluded The mineral
systems studied include goethitecalcite lepidocrocitecalcite and hydrous ferric
oxidesand suspensions The solid iron phases produced were examined by
transmission Moumlssbauer spectroscopy
A study on the relative reactivity of outer and inner Fe(II) sites in synthetic GR-
SO4 by using a series of structurally closely related compounds with different
charge properties as ldquoreactive probesrdquo is presented in chapter 4 The probe
compounds included nitrobenzene 2-nitrophenol 4-nitrotoluene 4-
chloronitrobenzene and 4-nitrophenylacetic acid
In chapter 5 an investigation of the reactivity of various Fe(II)-Fe(III) mineral
systems towards TCA and DCA is presented The study included laboratory batch
experiments imitating natural conditions The Fe(II)-Fe(III)-systems investigated
included GR-SO4 carbonate green rust magnetite Fe(II)goethite and
General Introduction 11
Fe(II)lepidocrocite The reactivities of the Fe(II)-Fe(III) mineral systems were
examined by comparing their surface-normalized rate constants
The results and environmental implications of this work are summarized in chapter
6 References Ahlers J Regelmann J Riedhammer C (2003) Environmental risk assessment of airborne trichloroacetic acid - a contribution to the discussion of the significance of anthropogenic and natural sources Chemosphere 52 531-537 Al-Agha MR Burley SD Curtis CD Esson J (1995) Complex cementation textures and authigenic mineral assemblages in recent concretions from the Lincolnshire Wash (east coast UK) driven by Fe(0) to Fe(II) oxidation Journal of the Geological Society 152 157-171 Amonette JE Workman DJ Kennedy DW Fruchter JS Gorby YA (2000) Dechlorination of carbon tetrachloride by Fe(II) associated with goethite Environmental Science and Technology 34 4606-4613 Berg M Muumlller SR Muumlhlemann J Wiedmer A Schwarzenbach RP (2000) Concentrations and mass fluxes of chloroacetic acids and trifluoroacetic acid in rain and natural waters in Switzerland Environmental Science and Technology 34 2675-2683 Bernal JD Dasgupta DR Mackay AL (1959) The oxides and hydroxides of iron and their structural inter-relationships Clay Minerals Bulletin 4 15-30 Butler EC Hayes KF (1998) Effects of solution composition and pH on the reductive dechlorination of hexachloroethane by iron sulfide Environmental Science and Technology 32 1276-1284 Butler EC Hayes KF (1999) Kinetics of the transformation of trichloroethylene and tetrachloroethylene by iron sulfide Environmental Science and Technology 33 2021-2027 Carlson L Schwertmann U (1990) The effect of CO2 and oxidation rate on the formation of goethite versus lepidocrocite from an Fe(II) system at pH 6 and 7 Clay Minerals 25 65-71 Chaudhuri SK Lack JG Coates JD (2001) Biogenic magnetite formation through anaerobic biooxidation of Fe(II) Applied and Environmental Microbiology 67 2844-2848 Cornell RM Schwertmann U (1996) The iron oxides Structure properties reactions occurrence and uses VCH Verlagsgesellschaft mbH Weinheim Cui D Eriksen TE (1996) Reduction of pertechnetate by ferrous iron in solution influence of sorbed and precipitated Fe(II) Environmental Science and Technology 30 2259-2262
Cui D Spahiu K (2002) The reduction of U(VI) on corroded iron under anoxic conditions Radiochemica Acta 90 623-628
12 Chapter 1 Drissi SH Refait Ph Abdelmoula M Geacutenin JMR (1995) The preparation and thermodynamic properties of Fe(II)-Fe(III) hydroxide-carbonate (green rust I) Pourbaix diagram of iron in carbonate-containing aqueous media Corrosion Science 37 2025-2041 Elsner M Haderlein SB Schwarzenbach RP (2004) Reactivity of Fe(II)-bearing minerals towards reductive transformation of organic contaminants Environmental Science and Technology 38 799-807 Erbs M Hansen HCB Olsen CE (1999) Reductive dechlorination of carbon tetrachloride using iron(II)iron(III)-hydroxide-sulphate (green rust) Environmental Science and Technology 33 307-311 Euro Chlor (2001) Trichloroacetic acid in the environment a dossier Euro Chlor Brussels and the European Chlorinated Solvent Association Fredrickson JK Zachara JM Kennedy DW Dong H Onstott TC Hinman NW Li S (1998) Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium Geochimica et Cosmochimica Acta 62 3239-3257 Gander JW Parkin GF Scherer MM (2002) Kinetics of 111-trichloroethane transformation by iron sulfide and a methanogenic consortium Environmental Science and Technology 36 4540-4546 Geacutenin JMR Bourrieacute G Trolard F Abdelmoula M Jaffrezic A Refait Ph Maitre V Humbert B Herbillon A (1998) Thermodynamic equilibria in aqueous suspensions of synthetic and natural Fe(II)-Fe(III) green rusts Occurrences of the mineral in hydromorphic soils Environmental Science and Technology 32 1058-1068 Geacutenin JMR Olowe AA Benbouzid-Rollet ND Prieur D Confente M Resiak B (1991) The simultaneous presence of green rust 2 and sulfate reducing bacteria in the corrosion of steel sheet piles in a harbour area Hyperfine Interactions 69 875-878 Geacutenin JMR Olowe AA Refait Ph Simon L (1996) On the stoichiometry and Pourbaix diagram of Fe(II)-Fe(III) hydroxy-sulphate of sulphate-containing green rust 2 An electrochemical and Moumlssbauer spectroscopy study Corrosion Science 38 1751-1762 Glasauer S Weidler PG Langley S Beveridge TJ (2003) Controls on Fe reduction and mineral formation by a subsurface bacterium Geochimica et Cosmochimica Acta 67 1277- 1288 Hanert HH (1992) The genus Gallionella In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 4082-4088 Hansen HCB Borggaard OK Soslashrensen J (1994) Evaluation of the free energy of formation of iron(II)iron(III)-hydroxidesulphate (Green Rust) and its reduction of nitrite Geochimica et Cosmochimica Acta 58 2599-2608 Hansen HCB Guldberg S Erbs M Koch CB (2001) Kinetics of nitrate reduction by green rusts ndash effects of interlayer anion and Fe(II)Fe(III) ratio Applied Clay Science 18 81-91
General Introduction 13
Hansen HCB Koch CB Nancke-Krogh H Borggaard OK Soslashrensen J (1996) Abiotic nitrate reduction to ammonium Key role of green rust Environmental Science and Technology 30 2053-2056 Hartter DR (1985) The use and importance of nitroaromatic chemicals in the chemical industry In Toxicity of nitroaromatic compounds Rickert DE (ed) Hemisphere Publishing Corporation 1-13 Heasman DM Sherman DM Ragnarsdottir KV (2003) The reduction of aqueous Au3+ by sulfide minerals and green rust phases American Mineralogist 88 725-738 Hofstetter TB Schwarzenbach RP Haderlein SB (2003) Reactivity of Fe(II) species associated with clay minerals Environmental Science and Technology 37 519-528 Hwang I Batchelor B (2000) Reductive dechlorination of tetrachloroethylene by Fe(II) in cement slurries Environmental Science and Technology 34 5017-5022 King GM (1990) Effects of added manganic and ferric oxides on sulfate reduction and sulfide oxidation in intertidal sediments FEMS Microbiology Ecology 73 131-138 Klausen J Troumlber SP Haderlein SB Schwarzenbach RP (1995) Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions Environmental Science and Technology 29 2396-2404 Kuenen JG Robertson LA Tuovinen OH (1992) The genera Thiobacillus Thiomicrospira and Thiosphaera In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 2618-2624
Kukkadapu RK Zachara JM Smith SC Fredrickson JK Liu C (2001) Dissimilatory bacterial reduction of Al-substituted goethite in subsurface sediments Geochimica et Cosmochimica Acta 65 2913-2924 Lee W Batchelor B (2002a) Abiotic reductive dechlorination of chlorinated ethylenes by iron- bearing soil minerals 1 Pyrite and magnetite Environmental Science and Technology 36 5147- 5154 Lee W Batchelor B (2002b) Abiotic reductive dechlorination of chlorinated ethylenes by iron-bearing soil minerals 2 Green rust Environmental Science and Technology 36 5348- 5354 Liger E Charlet L Van Cappellen P (1999) Surface catalysis of uranium (VI) reduction by iron(II) Geochimica et Cosmochimica Acta 63 2939-2955 Lovley DR (1997) Microbial Fe(III) reduction in subsurface environments FEMS Microbiology Reviews 20 305-313 Lovley DR Phillips EJP (1986) Organic matter mineralization with reduction of ferric iron in anaerobic sediments Applied and Environmental Microbiology 51 683-689 Lovley DR Phillips EJP Lonergan DJ (1991) Enzymatic versus nonenzymatic mechanisms for Fe(III) reduction in aquatic sediments Environmental Science and Technology 25 1062-1067
14 Chapter 1 Loyaux-Lawniczak S Refait Ph Lecomte P Ehrhardt J Geacutenin JMR (1999) The reduction of chromate ions by Fe(II) layered hydroxides Hydrology and Earth System Sciences 3 593-599 Lyngkilde J Christensen TH (1992) Redox zones of a landfill leachate pollution plume (Vejen Denmark) Journal of Contaminant Hydrology 10 273-289 Macalady DL Tratnyek PG Grundl TJ (1986) Abiotic reduction reactions of anthropogenic organic chemicals in anaerobic systems A critical review Journal of Contaminant Hydrology 1 1-28 McCulloch A (2002) Trichloroacetic acid in the environment Chemosphere 47 667-686 Mendiboure A Schoumlllhorn A (1986) Formation and anion exchange reactions of layered transition metal hydroxides [Ni1-xMx](OH)2(CO3)x2(H2O)z (M = Fe Co) Revue de Chimie Mineacuterale 23 819-827 Misawa T Hashimoto K Shimodaira S (1974) The mechanism of formation of iron oxide and oxyhydroxides in aqueous solutions at room temperature Corrosion Science 14 131-149 Mulder EG Deinema MH (1992) The sheathed bacteria In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 2618-2624 Myneni SCB Tokunaga TK Brown Jr GE (1997) Abiotic selenium redox transformations in the presence of Fe(IIIII) oxides Science 278 1106-1109 Nealson KH Saffarini D (1994) Iron and manganese in anaerobic respiration Environmental significance physiology and regulation Annual Review of Microbiology 48 311-343 Nielsen A (1976) Hvid groslashn og sort rust Beskrivelse af en korrosionsskade paring et svoslashmmebassin Nordisk Betong 2 21-24 Nriagu JO Dell CI (1974) Diagenetic formation of iron phosphates in recent lake sediments American Mineralogist 59 934-946 OLoughlin EJ Burris DR (2004) Reduction of halogenated ethanes by green rust Environmental Toxicology and Chemistry 23 41-48 OLoughlin EJ Kelly SD Cook RE Csencsits R Kemner KM (2003a) Reduction of uranium(VI) by mixed iron(II)iron(III) hydroxide (green rust) Formation of UO2 nanoparticles Environmental Science and Technology 37 721-727 OLoughlin EJ Kelly SD Kemner KM Csencsits R Cook RE (2003b) Reduction of AgI AuIII CuII and HgII by FeIIFeIII hydroxysulfate green rust Chemosphere 53 437-446 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002a) Iron (IIIII) hydroxycarbonate green rust formation and stabilization from lepidocrocite bioreduction Environmental Science and Technology 36 16-20 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002b) Microbial reduction of lepidocrocite γ-FeOOH by Shewanella putrefaciens The
General Introduction 15
formation of green rust Hyperfine Interactions 139140 231-237 Parmar N Gorby YA Beveridge TJ Ferris FG (2001) Formation of green rust and immobilization of nickel in response to bacterial reduction of hydrous ferric oxide Geomicrobiology Journal 18 375-385 Pecher K Haderlein SB Schwarzenbach RP (2002) Reduction of polyhalogenated methanes by surface-bound Fe(II) in aqueous suspensions of iron oxides Environmental Science and Technology 36 1734-1741 Pepper SE Bunker DJ Bryan ND Livens FR Charnock JM Pattrick RAD Collison D (2003) Treatment of radioactive wastes An X-ray absorption spectroscopy study of the reaction of technetium with green rust Journal of Colloid and Interface Science 268 408- 412 Randall SR Sherman DM Ragnarsdottir KV (2001) Sorption of As(V) on green rust (Fe4(II)Fe2(III)(OH)12SO4
3H2O) and lepidocrocite (γ-FeOOH) Surface complexes from EXAFS spectroscopy Geochimica et Cosmochimica Acta 65 1015-1023 Refait Ph Abdelmoula M Trolard F Geacutenin JMR Ehrhardt JJ Bourrieacute G (2001) Moumlssbauer and XAS study of a green rust mineral the partial substitution of Fe2+ by Mg2+ American Mineralogist 86 731-739 Refait Ph Drissi SH Marie Y Geacutenin JMR (1994) The substitution of Fe2+ ions by Ni2+ ions in green rust one compounds Hyperfine Interactions 90 389-394 Refait Ph Geacutenin JMR (1997) The mechanisms of oxidation of ferrous hydroxychloride β- Fe2(OH)3Cl in aqueous solution The formation of akaganeite vs goethite Corrosion Science 39 539-553 Reimann S Grob K Frank H (1996) Chloroacetic acids in rainwater Environmental Science and Technology 30 2340-2344 Rickert DE (1985) Toxicity of nitroaromatic compounds Hemisphere Publishing Corporation 1-13 Roh Y Lee SY Elless MP (2000) Characterization of corrosion products in the permeable reactive barriers Environmental Geology 40 184-194 Rosenblatt DH Burrows EP Mitchell WR Parmer DL (1991) Organic explosives and related compounds In The Handbook of Environmental Chemistry Anthropogenic compounds Hutzinger O (Ed) Springer-Verlag 195-234 Ruumlgge K Hofstetter TB Haderlein SB Bjerg PL Knudsen S Zraurig C Mosbaeligk H Christensen TH (1998) Characterization of predominant reductants in an anaerobic leachate- affected aquifer by nitroaromatic probe compounds Environmental Science and Technology 32 23-31 Schwertmann U Cornell RM (1991) Iron oxides in the laboratory Preparation and characterization VCH Verlagsgesellschaft mbH Weinheim Simon L Francois M Refait Ph Renaudin G Lelaurain M Geacutenin JMR (2003)
16 Chapter 1 Structure of the Fe(II-III)-layered double hydroxysulphate green rust two from Rietveld analysis Solid State Sciences 5 327-334 Strathmann TJ Stone AT (2003) Mineral surface catalysis of reactions between FeII and oxime carbamate pesticides Geochimica et Cosmochimica Acta 67 2775-2791 Straub KL Benz M Schink B Widdel F (1996) Anaerobic nitrate-dependent microbial oxidation of ferrous iron Applied and Environmental Microbiology 62 1458-1460 Tamaura Y (1985) ZnII-bearing green rust II and its spontaneous transformation into ZnII- bearing ferrite in aqueous solution Bulletin of the Chemical Society of Japan 58 2951-2954 Tamaura Y (1986) Ni(II)-bearing green rust II and its spontaneous transformation into Ni(II)- bearing ferrites Bulletin of the Chemical Society of Japan 59 1829-1832 Taylor RM (1980) Formation and properties of Fe(II)Fe(III)-hydroxycarbonate and its possible significance in soil formation Clay Minerals 15 369-382 Trolard F Abdelmoula M Bourrieacute G Humbert B Geacutenin JMR (1996) Mise en eacutevidence dun constituant de type rouilles vertes dans les sols hydromorphes Proposition de lexistence dun nouveau mineacuteral la fougeacuterite Geacuteosciences de surface Comptes Rendus de LrsquoAcademie des Sciences 323 1015-1022 Tuovinen OH Button KS Vuorinen A Carlson L Mair DM Yut LA (1980) Bacterial chemical and mineralogical characteristics of tubercles in distribution pipelines Journal of the American Water Works Association 72 626-635 Vikesland PJ Valentine RL (2002) Iron oxide surface-catalyzed oxidation of ferrous iron by monochloramine implications of oxide type and carbonate on reactivity Environmental Science and Technology 36 512-519 Widdel F Schnell S Heising S Ehrenreich A Assmus B Schink B (1993) Ferrous iron oxidation by anoxygenic phototrophic bacteria Nature 362 834-836
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 17
2 Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria
Abstract
This work investigated the Fe-containing products formed by anaerobic
autotrophic denitrifying Fe(II)-oxidizing bacteria in a specific bicarbonate buffered
(30 mM HCO3- pH 70) culture media containing 10 mM Fe(II) 4 mM nitrate and
4 mM phosphate Fe(II) was present initially as a whitish vivianite-like
(Fe3(PO4)2middot8H2O) precipitate and as soluble Fe(II) The initial phase of the
oxidation produced a greenish metavivianite-like ((FeII3-xFeIII
x)(PO4)2(OH)xmiddot(8-
x)H2O x gt 12) phase In the late oxidation phase a reddish precipitate of poorly
crystalline goethite (α-FeOOH) dominated the colour of the media in coexistence
with Fe(II)-containing siderite (FeCO3) The increasing amounts of Fe(III) present
in the ldquovivianiterdquo and ldquometavivianiterdquo structures were accompanied by an
increasing intensity in the green colour as the Fe(II) biooxidation progressed This
colour development has produced the idea of biogenic green rusts (layered Fe(II)-
Fe(III) hydroxides) in several studies on nitrate-dependent Fe(II) biooxidation
However in this work no evidence of green rust formation mediated by anaerobic
denitrifying Fe(II)-oxidizing bacteria was obtained
21 Introduction
Aerobic oxidation of Fe(II)-containing minerals by lithotrophic acidophilic and
neutrophilic bacteria has been known for many years but their broad significance
in the biogeochemical cycling of iron has only been recognized over the past two
decades Aerobic Fe(II)-oxidizing bacteria (FeOB) have been isolated from acidic
ecosystems (Thiobacillus ferrooxidans) neutral eutrophic systems (Sphaerotilus
natans Leptothrix ochracea) and neutral oligotrophic systems (Gallionella
ferruginea) (Hanert 1992 Kuenen et al 1992 Mulder amp Deinema 1992) At
neutral pH Fe(II) is unstable in the presence of oxygen and is rapidly oxidized to
the insoluble Fe(III) Hence the only pH neutral environments where soluble
18 Chapter 2
Fe(II) is available for aerobic FeOB are at interfaces between oxic and anoxic
conditions The aerobic neutrophilic FeOB (Leptothrix ochracea Gallionella
ferruginea and Sphaerotilus natans) live at such interfaces and are usually
associated with the yellowishreddish ferric deposits formed there
Over the past several years there has been a growing recognition that other less
readily detectable types of bacteria are involved in Fe(II) oxidation in ecosystems
at circumneutral pH For example it has been reported that neutrophilic FeOB are
abundant at the Loihi seamount hydrothermal vents and play a major role in the
Fe(III) oxide deposition (Emerson amp Moyer 2002) Similarly unidentified
neutrophilic obligate lithotrophic FeOB have been isolated from the rhizosphere of
wetlands plants where they are closely associated with deposits of amorphous
Fe(III) oxides (Emerson et al 1999) It was previously believed that Fe(III) oxide
deposits associated with sheaths were produced biologically whereas Fe(III) oxide
deposits not associated with cells were produced abiotically Recently the
formation of amorphous Fe(III) oxide in gradient tubes has been attributed to the
action of FeOB (Sobolev amp Roden 2001) The authors attribute 90 of the
oxidation to biological processes and indicated that the organisms seem to produce
a mobile form of Fe(III) that diffuses away from the cells before being
precipitated thereby avoiding encrustation of the cells They suggest that such
soluble Fe(III) complexes might be substrates for closely associated Fe(III)-
reducing bacteria Such an arrangement might allow close coupling between
microbial Fe(II) oxidation and Fe(III) reduction within millimeters of the oxic-
anoxic interface
Anaerobic Fe(II) oxidation by phototrophic purple non-sulfur bacteria utilizing
Fe(II) as an electron donor in the light was recognized only a decade ago (Widdel
et al 1993) Subsequently it was demonstrated that the biological oxidation of
Fe(II) in the absence of oxygen is possible by light-independent chemotrophic
microbial activity using nitrate as the electron acceptor (Straub et al 1996) In
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 19
addition studies conducted in gradient cultures revealed that nitrate-reducing
strains could also oxidize Fe(II) with molecular oxygen (Benz et al 1998) Hence
these Fe(II)-oxidizing strains may use nitrate as well as oxygen as electron
acceptors The microbial oxidation of Fe(II) was coupled to stoichiometric
reduction of nitrate to N2 and only one strain produced traces of N2O as a by-
product (Straub et al 1996 Benz et al 1998) The authors proposed the formation
of 2-line ferrihydrite as the end product of Fe(II) biooxidation The chemical
reduction of nitrate by Fe(II) requires a catalyst eg at least 10 microM Cu2+ in order
to take place at significant rates and may thus be considered insignificant under the
conditions applied in our study (Moraghan amp Buresh 1976) The chemical
oxidation of Fe(II) with nitrous oxide has not been observed However nitrite can
oxidize Fe(II) chemically (Moraghan amp Buresh 1977 Straub et al 1996) but this
process is considered insignificant at the conditions applied here No denitrifying
Fe(II)-oxidizing enrichment culture has been found to produce ammonium from
nitrate
Both lithoheterotrophic (depending on organic cosubstrates such as acetate) and
strictly lithoautotrophic nitrate-reducing FeOB have been found in various marine
and freshwater sediments However most isolates depend on organic cosubstrates
for cell biosynthesis (Benz et al 1998) Most probable number estimations
showed that denitrifying FeOB accounted for 00006-08 of the acetate-oxidizing
denitrifying microbial population Lithotrophic FeOB accounted for less than
00001 of the total bacterial community Attempts to isolate CO2-fixing nitrate-
dependent FeOB from lithotrophic cultures have failed (Straub amp Buchholz-
Cleven 1998) Mixotrophic FeOB accounted for 0004-004 of the total bacterial
community In addition microbial nitrate-dependent Fe(II) oxidation was
demonstrated in a flooded paddy soil as well as in activated sludge from a
wastewater treatment plant (Nielsen amp Nielsen 1998 Ratering amp Schnell 2001)
Since the activity is not restricted to sunlight exposed habitats microbial nitrate-
dependent Fe(II) oxidation is supposedly more important on a global scale than
20 Chapter 2
anaerobic Fe(II) oxidation by phototrophic bacteria Furthermore it has been
reported that anaerobic denitrifying FeOB aptly oxidize biogenic Fe(II) minerals
formed by bioreduction of synthetic goethite and ferrihydrite and that anaerobic
Fe(III)-reducing bacteria readily reduce Fe(III) minerals formed by biooxidation of
Fe(II) (Weber et al 2001 Straub et al 1998) Hence autotrophic denitrifying
FeOB may play a significant role in the nitrogen and iron cycles in subsurface
environments where the nitrate and the Fe(II) zones overlap and organic carbon
supply is limited (Figure 21)
Figure 21 The microbial iron cycle linking the carbon and nitrogen cycles
Phosphate is released into the environment through natural processes such as rock
weathering and decomposition of dead organic material and anthropogenic
activities eg wastewater effluents and application of manure and fertilizers in
horti- and agriculture In anoxic soils and sediments phosphate may be sequestered
by sorption onto Fe(III) oxides (Williams et al 1971 Patrick amp Khalid 1974)
Phosphate strongly influences the type morphology and properties of Fe(III)
oxides formed by oxidation and hydrolysis of Fe(II) salts as well as the degree of
their transformation (Kandori et al 1992 Cumplido et al 2000 Benali et al
2001) Phosphate may also be retained by precipitation of Fe(II) phosphates such
as the monoclinic vivianite (Fe3(PO4)2middot8H2O) which is the most important stable
Fe(II) orthophosphate solid encountered in the subsurface under most conditions
(Nriagu 1972) At anoxic and circumneutral conditions the whitish vivianite
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 21
controls the Fe(II) activity even at very low phosphate concentrations (Nriagu amp
Dell 1974) Vivianite occurs as a secondary mineral in the gossans of metallic ore
deposits and as a weathering product of primary iron-manganese phosphates in
pegmatites (Gaines et al 1997) Moreover natural vivianite occurrences have
been identified in a number of lake and river sediments (Zwaan amp Kortenbout van
der Sluys 1971 Nriagu amp Dell 1974 Postma 1981 Nembrini et al 1983
Henderson et al 1984 Dodd et al 2003 House 2003 and references therein)
Vivianite is also found in sewage sludge as a result of the wastewater treatment
where iron salts are added in order to remove phosphate (Seitz et al 1973) It is
however still indefinite how ubiquitous vivianite is in nature Furthermore only
little is known about the mechanism of vivianite formation and the role played by
sedimentary Fe(III) oxides Anaerobic Fe(III)-reducing microorganisms may
reduce Fe(III) oxides thereby releasing the iron as soluble Fe(II) and mobilizing
the phosphate adsorbed to the Fe(III) oxides (Lovley 1997) It has been suggested
that vivianite is formed by precipitation following reductive dissolution of Fe(III)
oxides (Manning et al 1981 Manning amp Jones 1982) However it has also been
proposed that the transformation of Fe(III) oxides to vivianite occurs topotactically
and not via reductive dissolution (Nembrini et al 1983) Vivianite was shown to
form microbially as a result of the activity of the anaerobic Fe(III)-reducing
bacteria Shewanella putrefaciens in the presence of high Fe(III)-citrate and
phosphate concentrations (Jorand et al 2000) Moreover vivianite formation by
bioreduction of Fe(III) in hydrous ferric oxide and in smectite has been reported
(Fredrickson et al 1998 Dong et al 2003)
Only little is known about the oxidation products of vivianite Metavivianite a
greenish triclinic iron hydroxy phosphate mineral was first described by Ritz et al
(1974) and it was later found to coexist with vivianite in several natural sediment
samples (Henderson et al 1984) Once the Fe(III) content became evident the true
composition of metavivianite ((FeII3-xFeIII
x)(PO4)2(OH)x
22 Chapter 2
middot(8-x)H2O x gt 12)) was established (Rodgers amp Johnston 1985 Rodgers 1986
and references therein) The formation of intermediate greenish precipitates during
oxidation of fluffy colourless Fe(II) precipitates by anoxic phototrophic
microorganisms and nitrate-dependent FeOB have been reported (Ehrenreich amp
Widdel 1994 Chaudhuri et al 2001) Since both studies were conducted in
bicarbonate buffered mineral media (22-30 mM HCO3ndash pH 70-72) containing
37-5 mM phosphate we assume that the initial fluffy whitish precipitates
consisted mainly of vivianite Chaudhuri et al (2001) proposed that the
intermediate green phases produces by the denitrifying FeOB consist of carbonate
green rust (GR-CO3) but no convincing evidence of this biogenic GR-CO3 has been
provided yet The major objective of this work was to examine the Fe-containing
products forming during the course of biooxidation of vivianite by non-
phototrophic anaerobic denitrifying Fe(II)-oxidizing bacteria
2 2 Materials and methods
All handling and sampling of solutions and suspensions were carried out under
sterile and strict anoxic conditions All chemicals were pa quality
221 Microorganisms and media
Enrichment cultures of nitrate-reducing FeOB taken from town ditches (Bremen
Germany) were grown in anoxic bicarbonate-buffered (30 mM HCO3ndash 90
N210 CO2 pH 70) mineral media containing 4 mM phosphate as well as
essential trace elements and vitamins (Table 21 Straub amp Buchholz-Cleven
1998) Ammonium was omitted from the media in order to facilitate detection of
ammonium possibly produced by reduction of nitrate The techniques used for
preparation of media and cultivation of bacteria under anoxic conditions have been
described by Widdel amp Bak (1992) 05 M aqueous stock solutions of FeCl2 or
FeSO4 were prepared in 100 mL glass flasks by reacting 65 mmol of iron powder
(particle size 10 microm Merck) with 100 mL deoxygenated 10 M HCl or 05 M
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 23
H2SO4 respectively The solutions were magnetically stirred and heated (~80degC)
during reaction until the H2(g) production had ceased (ge 1 hour) The FeCl2 and
FeSO4 stock solutions were stored under a small Ar overpressure at 5degC
Table 21 Composition of the mineral medium (adopted from Straub amp Buchholz-Cleven
(1998))
Components Concentration (M) KH2PO4 15middot10-3
K2HPO4 25middot10-3
MgSO4middot7H2O 10middot10-3
CaCl2middot2H2O 50middot10-4
H3BO3 56middot10-5
ZnSO4middot7H2O 10middot10-6
Na2MoO4middot2H2O 40middot10-6
CuSO4middot5H2O 20middot10-7
MnSO4middotH2O 10middot10-6
Na2SeO4 12middot10-5
CoCl2middot6H2O 50middot10-6
NiCl2middot6H2O 80middot10-6
NaCl 10middot10-5
NaHCO3ndash 30middot10-2
Cyanocobalamine (vitamin B12) 37middot10-8
p-aminobenzoic acid (vitamin Hrsquo) 36middot10-7
D(+)-biotin (vitamin H) 41middot10-8
Nicotinic acid (Niacin) 81middot10-7
Ca-D(+)-pantothenate (vitamin B5) 52middot10-8
Pyridoxamine dihydrochloride 96middot10-7
Thiaminechloridehydrochloride (vitamin B1) 15middot10-7
NaNO3 40middot10-3
FeSO4 or FeCl2 0010
222 Characterisation of precipitates
In order to optimize the characterization and distinction between the spectral
components transmission Moumlssbauer spectra were obtained at temperatures
between 5 K and 250 K and in external magnetic fields of 4 T (parallel to the γ-ray
direction) using a conventional constant acceleration spectrometer and a source of 57Co in Rh The spectrometer was calibrated using a 125 microm foil of α-Fe at room
temperature and isomer shifts are given relative to the centroid of the spectrum of
this absorber The spectra were fitted using simple Lorentzian line shape Infrared
(IR) spectra were obtained using a Perkin Elmer FT-IR 2000 spectrometer and the
24 Chapter 2
KBr pellet technique Scanning electron microscopy (SEM) was carried out in
order to study the morphology and composition of the precipitates Specimens for
SEM were prepared by depositing suspended particles onto an aluminum stub
coated with a carbon sticker The stub was quickly transferred into a sputtering
chamber and coated with a thin Pt film (~20 nm) In order to avoid interfering Pt
signals in the energy dispersive spectra the stubs were in some cases not coated
with Pt but quickly transferred to the SEM chamber for evacuation Measurements
were performed using a Philips XL30 equipped with a LaB6 source and an
accelerating voltage of 20 kV and an EDAX eDXi X-ray dispersive spectrometer
223 Biooxidation experiments
The biooxidation experiments were conducted in 50-400 mL butyl rubber
stoppered bottles with a 90 N210 CO2 headspace constituting 10 of the total
volume Prior to inoculation 4 mM NaNO3 was added as the electron acceptor and
10 mM Fe2+ (as chloride or sulphate) as the electron donor to the mineral media
Control experiments were performed in the same media only they were not
inoculated Addition of ferrous iron to the media induced an immediate
precipitation of a solid whitish material The whitish precipitate was collected on
022 microm polyvinylidendifluorid (Durapore Millipore) filters and stored in an
anoxic atmosphere until further measurements Old outgrown media suspensions
that had turned reddish in color due to a precipitate produced by the denitrifying
FeOB were used as inocula Inoculum volume was kept lt1 of the culture volume
in order to prevent the reddish inoculum from dominating over the initial whitish
precipitate Cultures were incubated in the dark at room temperature and gently
agitated once every day Typical color successions for the media were initial
whitish precipitates turning first more and more greenish over time and then finally
turning reddish (see Fig 22) The color developed uniformly without any
indications of multiple phases in the precipitate At different time intervals
suspension samples were withdrawn using 90 N210 CO2-flushed polyethylene
syringes The precipitates were collected on 022 microm polyvinylidendifluorid
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 25
(Durapore Millipore) filters and analyzed by Moumlssbauer spectroscopy and SEM
Nonfiltered suspension samples were digested in 01 M HCl and chemically
analyzed for Fe(II) NO3- and NH4
+
224 Analytical methods
Fe2+ was determined using a modified phenanthroline method (Fadrus amp Maly
1975) Nitrate was quantified by ion chromatography (Morales et al 2000) and
ammonium was measured photometrically using the indophenol reaction (Rossum
amp Villarruz 1963)
23 Results and discussion
231 Identification of solid iron-containing phases
In most cases the mineral media for cultivating denitrifying FeOB contained 10
mM FeCl2 or FeSO4 4 mM NO3ndash 4 mM total phosphorus and 30 mM HCO3
- at pH
70 ([HCO3-] = 2138middot[CO3
2-] at pH 70) Whitish flocs precipitated immediately at
these initial conditions when Fe(II) was added to the media (Figure 22a) Such
colourless flocs have been reported to precipitate in similar mineral media (10 mM
Fe(II) 37-5 mM phosphate 22-30 mM HCO3ndash pH 70-72) (Ehrenreich amp
Widdel 1994 Chaudhuri et al 2001) Our Fe(II) measurements showed that 20-
50 of the total Fe(II) added was present in this initial white precipitate
26 Chapter 2
a
210- Figure 22 Colour of suspended material in the growth media during Fe(II) biooxidation a)
Initial whitish precipitate prior to inoculation b) Inte1mediate greenish phase fo1med within 2-3
days after inoculation c) reddish precipitate at late stage of biooxidation (gt5-6 days)
The precipitates were filtered and investigated by Mossbauer and IR spectroscopy
The transmission Mossbauer spectra obtained for the initial whitish precipitate at
temperatures between 20 and 250 K are shown in Figure 23 The spectrum
measured at 250 K consists of two fairly well-resolved Fe(II) doublets (see
parameters in Table 22) The change in line-overlap with decreasing temperature
is primarily ascribed to differences in the temperature dependence of the
quadrupole splitting of the two components From the spectra at 10 and 6 K
(Figure 24) it can be concluded that magnetic ordering takes place between these
two temperatures and that only one transition occurs (indicating the presence of
only one phase) The parameters of one of the Fe(II) doublets at 250 K (designated
B in Table 22) are in very good agreement with previously published values for
the vivianite Fe(Il)8 site at room temperature (eg McCammon amp Burns 1980)
and the ordering temperature also agrees well with an assignment as vivianite
(Forsyth et al 1970) However the second Fe(II) doublet in the initial colourless
precipitate (Table 22) has parameters that deviate from previously reported values
by having a smaller quadrupole splitting (indicating a less distorted coordination)
and a significantly higher relative intensity and line width These effects might be
due to the presence of numerous defects in the vivianite crystal lattice particularly
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 27
affecting the Fe(II)A sites It should be noted that further components may be added
to the fit in order to improve its statistics Nevertheless we decided to include no
further components as suggested by the finding of one magnetic ordering only
Accordingly our interpretation of the Moumlssbauer results for the initial white
precipitate suggests a highly defective vivianite having a distribution of local
coordination environments particular in the A site This assignment is further
supported by a major absorption band due to phosphate anions in the infrared
spectrum at approximately 1000 cm-1 and the absence of other complex anions
(data not shown) Thus the whitish precipitate is referred to as a vivianite-like
(ldquovivianiterdquo) precipitate
0
3 5
3 0
2 5
2 0
1 5
1 0
0 5
0 0
20 K
Velocity (mms)
Rel
ativ
e ab
sorp
tion
()
3
2
1
0
80 K
3 5
3 0
2 5
2 0
1 5
1 0
0 5
0 0
150 K
-5 -4 -3 -2 -1 0 1 2 3 4 5
2 5
2 0
1 5
1 0
0 5
0 0
250 K
Figure 23 Transmission Moumlssbauer spectra measured between 250 and 20 K of the initial
whitish precipitate prior to inoculation (see Fig 22a) Fitting components (and their sum) are
shown as full lines
28 Chapter 2
Table 22 Selected Mossbauer parameters of the doublet components in the spectra obtained for
different precipitates
Precipitates Temperature Isomer shift Quadrupole Line width Area (K) (mms-1) splitting (mms-1) (mms-1) ()
Whitish Fe(II)B 250 127 309 035 38
Fe(II)A 250 128 181 051 62
Dark greenish Fe(II)B 250 126 305 023 17
Fe(II)A 250 132 238 051 44
Fe(III) 250 036 085 040 38
Reddish-orange 40 138 244 094 67 Fe(II) Hyperfine parameters are generally given with uncertainties of 003 mms- the spectral area with an uncertainty of 3
1005
1000
0995
0990
- 0985 ~ e c 0980 0
-~ 0975 E c nl b 1000 g ~ Qi 0995 0
0990
0985
0980 -12 -8
~ -~
bullbull bull Ibull bull bullbull bull bull ~ bull bull ~
It
10 K
bull bull bull bull (J ~ i~ 6K bullmiddotf bull bull
~ bull bull bull
-4 0 4 8 12
Velocity (mmls)
Figure 24 Transmission Mossbauer spectra measured at 10 and 6 K of the initial whitish
precipitate prior to inoculation (see Fig 22a)
In general the color of the media suspension changed from whitish into light green
color within 2-3 days after inoculation (Figure 22b ) This transformation occurred
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 29
without dissolving the initial whitish precipitate or preserving the whitish
precipitate as a separate phase implying some kind of solid state transformation
Two identical media suspensions were inoculated simultaneously but at the
sampling time they had distinctly different intensities of the green color
designated light and dark green respectively Moumlssbauer spectra of the dark green
sample are shown in Figure 25 The spectra of this sample are all fitted using three
doublet components (two Fe(II) and one Fe(III)) and the parameters of the
spectrum measured at 250 K are given in Table 22
30
25
20
15
10
05
00
20 K
Velocity (mms)
Rel
ativ
e ab
sorp
tion
()
35
30
25
20
15
10
05
00
80 K
30
25
20
15
10
05
00
150 K
-5 -4 -3 -2 -1 0 1 2 3 4 5
25
20
15
10
05
00
250 K
Figure 25 Transmission Moumlssbauer spectra measured between 250 and 20 K of the dark
greenish precipitate formed during biooxidation Fitting components (and their sum) are shown
as full lines
No magnetic ordering of the dark green precipitate was observed at temperatures
above 20 K but ordering occurred around 10 K (not shown - due to very thin
30 Chapter 2
samples this was not investigated in details) The two greenish samples had very
similar parameters only differing in the relative intensity of Fe(III) 26 and 38
in the light greenish and dark greenish samples respectively Assuming the
spectral area of a component to be proportional to the abundance of the species in
the solid these results indicate a correlation between the intensity of the green
color and the content of Fe(III) in the precipitate The parameters of the Fe(II)
doublets in the dark green precipitate (Table 22) were in very good agreement
with previously published values for vivianite with a non-negligible Fe(III) content
(McCammon amp Burns 1980) whereas the Fe(III) component in particular had a
higher quadrupole splitting The observation that magnetic ordering of both Fe(II)
and Fe(III) occurred at similar temperature for the light green phase (Figure 26) is
a strong indication that they both belong to the same phase The absence of the
component with the low quadrupole splitting in the spectra of both green samples
might indicate that the initial vivianite-like phase crystallized into a more well-
defined vivianite over time However freshly prepared and long-term aged (gt1
year) suspensions of the initial vivianite-like precipitate did not differ significantly
Hence we suggest that the recrystallization of the vivianite-like precipitate can be
explained by Fe(II) biooxidation
Solid State Oxidation ofFe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria
0
2
3
- 4
~ 5 c ~ 6 e-0 7 -2 nl Q) 00 gt ~ 05 Qi 0 10
15
20
25
30
35
bullbullbull hi 6~
bullbullbullbull bull bull bullbull bull 10 K bull bull
bull bull bull bull bull bull bull
~~ ~~ lf 6K bull bull bullbull middot bull bull r bull
bull bull bull bull bull bullbull bull bull bull - 12 -a -4 0 4 8 12
Velocity (mms)
31
Figure 26 Transmission Mossbauer spectrn measured at 6 and 10 K of the light greenish
precipitate fonned during biooxidation
The solid state oxidation of monoclinic vivianite to triclinic metavivianite is well-
known (McCammon amp Bums 1980 Pratesi et al 2003 and references therein)
The vivianite crystal structure contains Fe(II) ions in both isolated Fe(II)A and
paired Fe(Il)8 octahedra Mossbauer spectroscopic analyses have shown that the
Fe(Il)8 Fe(II)A ratio increases with increasing Fe(III) concentration suggesting that
the remaining Fe(II)A ions are more readily oxidized than the Fe(II)a ion of an
Fe(Il)8 -Fe(III)8 pair (McCammon amp Bums 1980) The mechanism of oxidation of
Fe(II) in vivianite involves conversion of H20 ligands to OH- ions producing a
progressive collapse of the vivianite structure due to the elimination of hydrogen
bonds (Moore 1971) The exact oxidation limits between which the triclinic lattice
is stable are somewhat disputed as the results obtained for synthetic and natural
vivianites oxidized chemically in the laboratory and naturally oxidized natural
vivianite specimens do not agree completely (Rodgers 1986 and references
therein) Taking all reports into account the monoclinic structure of vivianite is
supposedly maintained until 40-50 of total iron is oxidized Further oxidation
32 Chapter 2
leads to the formation of the triclinic metavivianite in which the FeA site is fully
oxidized whereas the oxidation of the FeB ranges from 20 to almost 100 Thus
the triclinic metavivianite structure persists close to complete oxidation of total
iron The Moumlssbauer results obtained in this study are consistent with the vivianite
solid state oxidation mechanism reported by McCammon amp Burns (1980) Thus
we propose that the intermediate greenish precipitate is a metavivianite-like
(ldquometavivianiterdquo) phase It should be noted that a minor oxidation of dissolved
Fe(II) may have occurred even though the solid state oxidation of Fe(II) was
predominant
Within 5-6 days after inoculation the greenish intermediate was transformed into a
reddish product (Figure 22c) The magnetically ordered sextet in the spectrum of
the red phase (Figure 27) measured at 40 K was due to goethite (α-FeOOH)
(magnetic hyperfine field of 470 T and a quadrupole shift of -01 mms-1 cp
Moslashrup et al 1983) The sextet deviated from ideal goethite by its asymmetric line
shape and its low ordering temperature (around 100 K ndash data not shown) and thus
the goethite was poorly crystalline It is very likely that the presence of phosphate
in the media retarded the crystal growth of goethite The unusual reddish colour of
the goethite might also be explained by the presence of phosphate The spectrum at
40 K was however dominated by a Fe(II) doublet (Table 22) that ordered
magnetically between 40 and 20 K (Figure 27) The hyperfine parameters and the
magnetic ordering temperature indicated that this component was due to siderite
(FeCO3) having a magnetic ordering temperature of 38 K (Jacobs 1963) The
siderite component may have formed as a result of the microbial activity changing
the chemistry of the solution and precipitating a major part of the remaining
dissolved Fe(II) at this stage The characteristic vivianite Fe(II) doublet with large
quadrupole splitting was not detected in this sample The reddish precipitate
contained considerably less Fe(III) than the greenish precipitate (only 33 as
estimated from the spectral area) None of the components in the reddish sample
Solid State Oxidation ofFe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 33
could be detected in freshly inoculated samples indicating that carryover of mineral
precipitates by inoculation of the culture media was negligible
0
2
- 3
~4 c
Q 5
e 6 0
~ 7 g
0 3l Q)
a 1
2
3
4
5
-12 a
40K
bull bull (
4 0 4 8 12 Velocity (mms)
Figure 27 Transmission Mossbauer spectra measured at 20 and 40 K of the reddish precipitate
fo1med during the late biooxidation stage Fitting components (and their sum) are shown as full
lines
Strengite (FeP04middot2H20) was not detected at any time during oxidation
Santabarbaraite a new amorphous F e(III) hydroxy phosphate mineral
(Fe3(P04)i(OH)3middot5H20) was reported in a recent study (Pratesi et al 2003) The
brownish mineral was a result of the solid state oxidation of vivianite through
metavivianite However no Mossbauer data have been provided for this new
mineral yet and therefore we cannot give an account of whether santabarbaraite
forms in our system or not Thus goethite was the dominating end product and we
propose the reaction path depicted in Figure 28 for the nitrate-dependent
biooxidation of Fe(II) in our systems
34 Chapter 2
Fe3(PO4)2middot8H2O (FeII3-xFeIII
x)(PO4)2(OH)xmiddot(8-x)H2O α-FeOOH
NO3- N2 NO3
- N2
ldquoVivianiterdquo ldquoMetavivianiterdquo Goethite
Figure 28 Proposed reaction path and iron-containing minerals forming during solid state
oxidation of vivianite by denitrifying FeOB at the experimental conditions applied in this study
The biotic formation of layered Fe(II)-Fe(III) hydroxides (green rusts) by
anaerobic denitrifying Fe(II)-oxidizing bacteria has been suggested but proper
identification of these phases still lacks (Chaudhuri et al 2001) We cannot rule
out that small amounts of green rusts (GRs) perhaps a phosphate intercalated GR
(Hansen amp Poulsen 1999) might have been present here during the greenish
intermediate ldquometavivianiterdquo oxidation stage When present in low concentrations
especially in mixtures including other iron minerals it is very difficult to identify
GRs even with Moumlssbauer spectroscopy At least two complementary methods
such as X-ray diffraction (XRD) and Moumlssbauer spectroscopy are required for
proper identification and characterization of GRs However the precipitates
collected in this work were poorly crystalline and did not allow for XRD analysis
Electron micrographs including energy dispersive X-ray spectroscopy suspension
colour and mineral stability calculations do not suffice as evidence Hence no
convincing evidence of GR formation facilitated by denitrifying FeOB has been
provided so far The blue-green colours of metavivianite and green rust minerals
originate from Fe(II)-Fe(III) charge transfer between adjacent Fe(II) and Fe(III)
ions in edge-shared octahedra (Faye et al 1968) The greenish suspension colour
occurring during the intermediate phase has incited the idea of biogenic GRs in
studies on nitrate-dependent Fe(II) biooxidation (Chaudhuri et al 2001 Lack et
al 2002aampb) However our results indicate that this reasoning is misleading
232 Factors controlling the rate and extent of Fe(II) biooxidation
Generally it was found that maximally 20-64 of the initial Fe(II) amount was
oxidized to Fe(III) (Figure 29) This indicates some limitations in the accessibility
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 35
of Fe(II) in the system Based solely on stoichiometry considerations the
microorganisms are expected to oxidize 5 mol Fe(II) for every 1 mol nitrate
reduced to dinitrogen However as exemplified in Figure 28 this ratio was mostly
lt4 which can be explained by the consumption of nitrogen as a result of microbial
growth At initial [Fe(II)][NO3ndash] ratios lt5 nitrate is in excess and should not limit
the extent of the biooxidation Thus the lack of complete biooxidation could not be
due to exhaustion of nitrate Furthermore all growth essential nutrients were more
than sufficiently applied hence the incomplete Fe(II) biooxidation was not caused
by lack of nutrients The most reasonable explanation therefore seems to be that an
increasingly limited access to the electron donor over time inhibits complete long-
term Fe(II) biooxidation At least four mechanisms may cause this inhibition 1)
the Fe(II) becomes isolated within the structure of the mixed Fe(II)-Fe(III)
minerals forming during biooxidation or underneath a passive Fe(III)-bearing
surface film on the initial Fe(II) precipitates 2) the FeOB cell surface becomes
covered with a passive Fe(III)-bearing surface film 3) the Fe(II) biooxidation is
controlled by the rate of dissolution of the initial Fe(II) minerals or 4) the reaction
proceeds primarily by biooxidation of dissolved Fe(II) whose concentration
gradually decreases due to changes in solid phase composition The actual
mechanisms whereby the surface-associated Fe(III) can inhibit Fe(II) biooxidation
are unknown but they may involve both kinetic and thermodynamic constraints on
the electron transfer The Moumlssbauer results obtained in this work strongly suggest
that the Fe(II) biooxidation occurred mainly in the solid state of the initial
ldquovivianiterdquo phase However we cannot rule out that some dissolved Fe(II) was
oxidized as well
36 Chapter 2
Figure 29 Concentration profiles of total Fe(II) and nitrate as a function of time during Fe(II)
biooxidation
No Fe(II) oxidation took place in cultures where nitrate had been omitted
confirming that the microbial Fe(II) oxidation is nitrate-dependent (data not
shown) No Fe(II) oxidation was detected in the non-inoculated control
experiments within the duration of the experiments and thus the chemical
oxidation of dissolved Fe(II) by nitrate catalyzed by vitamins or trace elements
(eg Cu(II)) can be neglected Ammonium did not form in detectable amounts
during Fe(II) biooxidation (data not shown) and therefore dinitrogen was assumed
to be the end product as reported previously (Straub et al 1996 Benz et al 1998)
The absence of ammonium formation does indirectly support the absence of
biologically induced green rust formation as synthetic green rust is known to
convert nitrate into ammonium in purely chemical reactions (Hansen et al 1996)
It was visually observed that the phosphate concentration in the media exerted a
control on the microbial Fe(II)-oxidation At phosphorus concentrations le 2 mM
no Fe(II)-oxidation took place However the solubility product for vivianite (Ksp =
171middot10-36 at 25degC (Al-Borno et al 1994)) was still by far exceeded under these
conditions It is not known whether this phosphate limiting effect is due to growth
constraints in the mixed bacterial community or whether specific Fe(II) phosphate
Solid State Oxidation ofFe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 37
precipitates are prerequisites of the Fe(II) biooxidation to take place Experiments
are currently underway in our laboratory in order to elucidate the role of specific
initial Fe(II) precipitates It should be noted that the growth of the denitrifying
FeOB could not be estimated as they were present in highly heterogeneous
suspensions containing both solid iron phases as well as other bacteria (enrichment
culture)
233 Morphology of solid iron phases
The morphology of the various precipitates was studied by SEM The initial
whitish precipitate consisted of a web-like structure (Figure 21 Oa and background
in Figure 21 Ob) whereas more distinct hexagonally shaped rosettes with particle
size ~20 microm (Figure 2lObampc) formed during Fe(II) biooxidation The energy
dispersive spectroscopic element analyses showed that other than iron the initial
whitish precipitate and the rosettes contained mainly phophorus
Figure 210 Scanning electron micro graphs of precipitates fo1med at various stages of the
experiment a Initial whitish precipitate bampc Rosettes observed in the intennediate greenish
precipitate d Reddish precipitate sampled during the late biooxidation stage
38 Chapter 2
These observations are interpreted as vivianite forming a web-like morphology in
the initial whitish precipitate and partly transforming into hexagonal particles in
the greenish colored stage The interpretations are supported by similar vivianite
morphologies reported including pseudo-hexagonal vivianite crystals of low
symmetry resulting from microbial Fe(III) reduction of HFO and platy rosettes of
vivianite crystals formed during bioreduction of Fe(III) in smectite (Fredrickson et
al 1998 Dong et al 2003) It was not possible to associate the morphology
observed in the reddish precipitate with the minerals identified in this phase
(Figure 210d)
24 Conclusions
This work demonstrated that anaerobic autotrophic denitrifying Fe(II)-oxidizing
bacteria produce poorly crystalline goethite by solid state oxidation of ldquovivianiterdquo
via a ldquometavivianiterdquo intermediate The increasing amount of Fe(III) forming in the
vivianite structure was accompanied by an increasing intensity in the green colour
as the Fe(II) biooxidation progressed Moumlssbauer spectroscopic analyses provided
no significant evidence of green rust formation The finding of microbially
oxidized vivianite in this study raises the question of the oxidation state of
vivianite specimens from natural sediments Vivianite is generally believed to be
an ideal Fe(II) hydroxy phosphate mineral and the presence of Fe(III) is explained
by aerial oxidation upon sampling The results presented here indicate that
microbiological processes may be responsible for the oxidation of vivianite and
metavivianite in natural subsurface environments Acknowledgments
We would like to thank Dr K Straub for providing and advising us how to culture the nitrate-
reducing FeOB Furthermore we thank Dr C B Koch for performing the Moumlssbauer analyses
and Dr D Mavrocordatos for performing the SEM analyses
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 39
References Al-Borno A Tomson MB (1994) The temperature dependence of the solubility product constant of vivianite Geochimica et Cosmochimica Acta 58 5373-5378 Benali O Abdelmoula M Refait Ph Geacutenin JMR (2001) Effect of orthophosphate on the oxidation products of Fe(II)-Fe(III) hydroxycarbonate The transformation of green rust to ferrihydrite Geochimica et Cosmochimica Acta 65 1715-1726 Benz M Brune A Schink B (1998) Anaerobic and aerobic oxidation of ferrous iron at neutral pH by chemohetorotrophic nitrate-reducing bacteria Archives of Microbiology 169 159-165 Chaudhuri SK Lack JG Coates JD (2001) Biogenic magnetite formation through anaerobic biooxidation of Fe(II) Applied and Environmental Microbiology 67 2844-2848 Cumplido J Barron V Torrent J (2000) Effect of phosphate on the formation of nanophase lepidocrocite from Fe(II) sulfate Clays and Clay Minerals 48 503-510 Dodd J Large DJ Fortey NJ Kemp S Styles M Wetton P Milodowski A (2003) Geochemistry and petrography of phosphorus in urban canal bed sediment Applied Geochemistry 18 259-267 Dong H Kostka JE Kim J (2003) Microscopic evidence for microbial dissolution of smectite Clays and Clay Minerals 51 502-512 Ehrenreich A Widdel F (1994) Anaerobic oxidation of ferrous iron by purple bacteria a new type of phototrophic metabolism Applied and Environmental Microbiology 60 4517-4526 Emerson D Moyer CL (2002) Neutrophilic Fe-oxidizing bacteria are abundant at the Loihi seamount hydrothermal vents and play a major role in Fe oxide deposition Applied and Environmental Microbiology 68 3085-3093 Emerson D Weiss JV Megonigal JP (1999) Iron-oxidizing bacteria are associated with ferric hydroxide precipitates (Fe-plaque) on the roots of wetland plants Applied and Environmental Microbiology 65 2758-2761 Fadrus H Maly J (1975) Suppression of iron(III) interference in the determination of iron(II) in water by the 110-phenanthroline method The Analyst 100 549-554 Faye GH Manning PG Nickel EH (1968) The polarized optical absorption spectra of tourmaline cordierite chloritoid and vivianite ferrous-ferric electronic interaction as a source of pleochroism American Mineralogist 53 1174-1201 Forsyth JB Johnson CE Wilkonson C (1970) The magnetic structure of vivianite Fe3(PO4)2middot8H2O Journal of Physics Part C Solid State Physics 3 1127-1139 Fredrickson JK Zachara JM Kennedy DW Dong H Onstott TC Hinman NW Li S (1998) Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium Geochimica et Cosmochimica Acta 62 3239-3257 Gaines RV Skinner HCW Foord EE Mason B Rosenzweig A (1997) Danas new
40 Chapter 2 mineralogy 8th ed John Wiley amp Sons Inc Hanert HH (1992) The genus Gallionella In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 4082-4088 Hansen HCB Koch CB Nancke-Krogh H Borggaard OK Soerensen J (1996) Abiotic nitrate reduction to ammonium Key role of green rust Environmental Science and Technology 30 2053-2056 Hansen HCB Poulsen IF (1999) Interaction of synthetic sulphate green rust with phosphate and the crystallization of vivianite Clays and Clay Minerals 47 312-318 Henderson GS Black PM Ridgers KA Rankin PC (1984) New data on New Zealand vivianite and metavivianite New Zealand Journal of Geology and Geophysics 27 367-378 House WA (2003) Geochemical cycling of phosphorus in rivers Applied Geochemistry 18 739-748 Jacobs IS (1963) Metamagnetism of siderite (FeCO3) Journal of Applied Physics 34 1106-1107 Jorand F Appenzeller BMR Abdelmoula M Refait Ph Block J-C Geacutenin JMR (2000) Assessment of vivianite formation in Shewanella putrefaciens culture Environmental Technology 21 1001-1005 Kandori K Uchida S Kataoka S Ishikawa T (1992) Effects of silicate and phosphate ions on the formation of ferric oxide hydroxide particles Journal of Materials Science 27 719-728 Kuenen JG Robertson LA Tuovinen OH (1992) The genera Thiobacillus Thiomicrospira and Thiosphaera In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 2618-2624 Lack JG Chaudhuri SK Chakraborty R Achenbach LA Coates JD (2002a) Anaerobic biooxidation of Fe(II) by Dechlorosoma suillum Microbial Ecology 43 424-431 Lack JG Chaudhuri SK Kelly SD Kemner KM OConnor SM Coates JD (2002b) Immobilization of radionuclides and heavy metals through anaerobic bio-oxidation of Fe(II) Applied and Environmental Microbiology 68 2704-2710 Lovley DR (1997) Microbial Fe(III) reduction in subsurface environments FEMS Microbiology Reviews 20 305-313 Manning PG Birchall T Jones W (1981) Ferric hydroxides in surficial sediments of the great lakes and their role in phosphorus availability a Moumlssbauer spectral study Canadian Mineralogist 19 525-530 Manning PG Jones W (1982) The binding capacity of ferric hydroxides for non-apatite inorganic phosphorus in sediments of the depositional basins of Lakes Erie and Ontario Canadian Mineralogist 20 169-176 McCammon CA Burns RG (1980) The oxidation mechanism of vivianite as studied by Moumlssbauer spectroscopy American Mineralogist 65 361-366 Moore PB (1971) The Fe2+
3(H2O)n(PO4)2 homologous series crystal-chemical relationships
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 41
and oxidized equivalents American Mineralogist 56 1-17 Moraghan JT Buresh RJ (1976) Chemical reduction of nitrate by ferrous iron Journal of Environmental Quality 5 320-325 Moraghan JT Buresh RJ (1977) Chemical reduction of nitrite and nitrous oxide by ferrous iron Journal of American Soil Science Society 40 47-50 Morales JA de Graterol LS Mesa J (2000) Determination of chloride sulfate and nitrate in groundwater samples by ion chromatography Journal of Chromatography A 884 185-190 Mulder EG Deinema MH (1992) The sheathed bacteria In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 2618-2624 Moslashrup S Madsen MB Franck J Villadsen J Koch CJW (1983) A new interpretation of Moumlssbauer spectra of microcrystalline goethiterdquosuper-ferromagnetismrdquo of ldquosuper-spin-glassrdquo behaviour Journal of Magnetism and Magnetic Materials 40 163-174 Nembrini GP Capobianco JA Viel M Williams AF (1983) A Moumlssbauer and chemical study of the formation of vivianite in sediments of Lago Maggiore (Italy) Geochimica et Cosmochimica Acta 47 1459-1464 Nielsen JL Nielsen PH (1998) Microbial nitrate-dependent oxidation of ferrous iron in activated sludge Environmental Science and Technology 32 3556-3561 Nriagu JO (1972) Stability of vivianite and ion-pair formation in the system Fe3(PO4)2-H3PO4- H2O Geochimica et Cosmochimica Acta 36 459-470 Nriagu JO Dell CI (1974) Diagenetic formation of iron phosphates in recent lake sediments American Mineralogist 59 934-946 Patrick Jr WH Khalid RA (1974) Phosphate release and sorption by soils and sediments Effect of aerobic and anaerobic conditions Science 186 53-55 Postma D (1981) Formation of siderite and vivianite and the pore-water composition of a recent bog sediment in Denmark Chemical Geology 31 225-244 Pratesi G Cipriani C Giuli G Birch WD (2003) Santabarbaraite a new amorphous phosphate mineral European Journal of Mineralogy 15 185-192 Ratering S Schnell S (2001) Nitrate-dependent iron(II) oxidation in paddy soil Environmental Microbiology 3 100-109 Ritz C Essene EJ Peacor DR (1974) Metavivianite Fe3(PO4)2middot8H2O a new mineral American Mineralogist 59 896-899 Rodgers KA (1986) Metavivianite and kerchenite a review Mineralogical Magazine 50 687- 691 Rodgers KA Johnston JH (1985) Type metavivianite Moumlssbauer evidence for a revised composition Neues Jahrbuch fuumlr Mineralogie-Monatshefte 12 539-542
42 Chapter 2 Rossum JR Villarruz PA (1963) Determination of ammonia by the indophenol method Journal of American Water Works Association 55 657-658 Seitz MA Riedner RJ Malhotra SK Kipp RJ (1973) Iron-phosphate compound identification in sewage sludge residue Environmental Science and Technology 7 354-357 Sobolev D Roden EE (2001) Suboxic deposition of ferric iron by bacteria in opposing gradients of Fe(II) and oxygen at circumneutral pH Applied and Environmental Microbiology 67 1328-1334 Straub KL Benz M Schink B Widdel F (1996) Anaerobic nitrate-dependent microbial oxidation of ferrous iron Applied and Environmental Microbiology 62 1458-1460 Straub KL Buchholz-Cleven BEE (1998) Enumeration and detection of anaerobic ferrous iron-oxidizing nitrate-reducing bacteria from diverse European sediments Applied and Environmental Microbiology 64 4846-4856 Straub KL Hanzlik M Buchholz-Cleven BEE (1998) The use of biologically produced ferrihydrite for the isolation of novel iron-reducing bacteria Systematic and Applied Microbiology 21 442-449 Weber KA Picardal FW Roden EE (2001) Microbially catalyzed nitrate-dependent oxidation of biogenic solid-phase Fe(II) compounds Environmental Science and Technology 35 1644-1650 Widdel F Bak F (1992) Gram-negative mesophilic sulfate-reducing bacteria In The Prokaryotes (Balows A Truumlper HG Dworkin M Harder W Schleifer K-H (eds)) Springer 2nd ed 3352-3378
Widdel F Schnell S Heising S Ehrenreich A Assmus B Schink B (1993) Ferrous iron oxidation by anoxygenic phototrophic bacteria Nature 362 834-836 Williams JDH Syers JK Shukla SS Harris RF Armstrong DE (1971) Levels of inorganic and total phosphorus in lake sediments as related to other sediment parameters Environmental Science and Technology 5 1113-1120
Zwaan PC Kortenbout van der Sluys G (1971) Vivianite crystals from Hare Noord Brabant Province The Netherlands Scripta Geology 6 1-7
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 43
3 Formation of Layered Iron Hydroxides by Microbial Fe(III)
Reduction Abstract
Many inorganic and organic pollutants may be degraded by microorganisms in the
subsurface However a wide range of contaminants including chromate nitrate
radionuclides nitroaromatic compounds chlorinated aliphatics and carbamate
pesticides may also be chemically transformed by reduction reactions involving
layered iron(II)-iron(III)-hydroxides (green rusts) Hence green rusts (GRs) may
play a potentially important role in the fate and transport of pollutants in iron-rich
suboxic soils and sediments Yet only little is known about the formation of GRs
in these environments The biotic formation of GRs mediated by the anaerobic
dissimilatory Fe(III)-reducing bacteria Shewanella spp has been reported and
proposed in several studies However the experimental conditions applied were
mostly not natural and the evidence of GR formation provided may be questioned
This work investigated the Fe-containing products formed by the facultatively
anaerobic Fe(III)-reducing microorganism Shewanella algae BrY in culture
media containing 4-10 mM formate or lactate and 8-27 mM Fe(III) In order to
simulate natural conditions Fe(III) oxides were applied as coatings on silica
(model system for sandy soils) or calcite particles (model system for calcareous
soils) and synthetic electron shuttles as well as highly concentrated artificial pH
buffers were excluded S algae BrY reduced 19-72 of the initial Fe(III) when
grown in goethitecalcite lepidocrocitecalcite or hydrous ferric oxidesand mineral
systems and green or blackish mineral phases were produced within 1-2 weeks
after inoculation Moumlssbauer spectroscopic analyses indicated that the green and
blackish precipitates were dominated by vivianite (Fe3(PO4)2sdot8H2O) and green rust
44 Chapter 3
31 Introduction
The significance of bacteria in the biogeochemical cycling of iron has been broadly
recognized over the past two decades Chemical processes were previously
considered to account for most of the Fe(III) reduction in subsurface environments
Dissimilatory Fe(III)-reducing bacteria (DIRB) that gain energy by coupling the
oxidation of hydrogen or organic compounds to the reduction of Fe(III) oxides
have been known for many years but their biogeochemical importance was
acknowledged only a decade ago (reviewed by Lovley 1997) DIRB transfer
electrons to extracellular Fe(III) without assimilating the iron Fe(III) bioreduction
accounts for a major fraction of the carbon oxidation in many different
environments and in the presence of high amounts of reactive Fe(III) microbial
Fe(III) reduction may even inhibit sulfate reduction and methanogenesis (King
1990 Lovley amp Phillips 1986) In fact most of the Fe(III) reduction in the Fe(III)
reduction zone of aquatic sediments and aquifers is thought to be enzymatically
catalyzed by microorganisms (Lovley et al 1991) However the relative
importance of microbial and chemical processes involved in the Fe(III) reduction
are still somewhat disputed among microbiologists and geochemists
A wide diversity of Fe(III)-reducing bacteria which fall in a number of different
phylogenetic groups is known today Both organisms growing by respiration and
by fermentation have been isolated and identified (Lovley 1991 Nealson amp
Saffarini 1994) Hydrogen short- and long-chained fatty acids amino acids
sugars and aromatic compounds may serve as electron donors for Fe(III)
bioreduction The enzymes responsible for dissimilatory Fe(III) reduction are outer
membrane associated ferric reductases (Lower et al 2001 and references therein)
Iron reducing bacteria may utilize alternative electron acceptors such as O2 nitrate
S0 sulfate humic substances contaminant metals and metalloids as well as
chlorinated solvents The first organism shown to couple respiratory growth to
dissimilatory iron reduction was Pseudomonas ferrireductans now known as
Shewanella oneidensis but previously classified as Alteromonas putrefaciens and
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 45
Shewanella putrefaciens (Venkateswaran et al 1999) Various DIRB including
the obligate anaerobic Geobacter sp and the facultatively anaerobic Shewanella
sp have been isolated from both marine and freshwater sediments soil and
aquifers (Thamdrup 2000 and references therein)
The redox potentials of oxidized and reduced iron couples and thus the energy
yield available from Fe(III) reduction depend strongly on the specific iron phases
involved In soil and aquatic environments Fe(III) oxides mainly occur in
association with other sediment particles as aggregates or coatings Amorphous
and poorly crystalline Fe(III) oxides usually make up 20 or less of the iron
content in a sediment (Thamdrup 2000) They are the main products of abiotic and
biotic Fe(II) oxidation in sediments and they constitute the most important phases
for microbial Fe(III) reduction Until recently it was generally believed that DIRB
reduced insoluble Fe(III) oxides only by direct contact with the Fe(III) oxide
thereby allowing electron transfer from the cell to the Fe(III) oxide surface
However over the past several years there has been a growing recognition that
DIRB may use different strategies in order to access the solid Fe(III) oxides These
strategies include solubilization of Fe(III) by synthetic or natural Fe(III) chelators
and Fe(III) reduction via electron shuttling with soluble humic substances or
microbially produced electron shuttles (Nevin amp Lovley 2002 and references
therein Turick et al 2003) The Fe(III) complexing agents may also stimulate
Fe(III) oxide reduction indirectly by chelation and thus removal of Fe(II) from
the cell and the Fe(III) oxide surfaces Both chelated Fe(III) and soluble electron
shuttles are more accessible to Fe(III) reductases than solid Fe(III) oxides In
contrast to Geobacter metallireducens S algae produces and releases extracellular
electron shuttling compounds (Nevin amp Lovley 2000) However in the absence of
soluble electron shuttles reversible adhesion is required for reduction of solid
Fe(III) oxides by S algae BrY (Das amp Caccavo 2000) Shewanella algae BrY
adheres readily and preferentially to a range of solid Fe(III) oxides such as
ferrihydrite goethite and hematite (Das amp Caccavo 2001) The adhesion
46 Chapter 3
mechanisms are not completely understood but recent results suggest that the
adhesion is mediated by cell surface proteins and independent of cell motility
(Caccavo amp Das 2002)
The microbial formation of GRs resulting from bioreduction of various Fe(III)
oxides including ferrihydrite goethite and lepidocrocite by strains of the
anaerobic DIRB Shewanella putrefaciens has been reported repeatedly over the
last years (Fredrickson et al 1998 Kukkadapu et al 2001 Liu et al 2001
Parmar et al 2001 Ona-Nguema et al 2002aampb Glasauer et al 2003)
However no evidence of biogenic formation of GRs at natural geochemical
conditions have been offered and it is still unknown whether this process may take
place at natural conditions comprising low carbon and iron concentrations as well
as the absence of synthetic electron shuttles and highly concentrated artificial pH
buffers GRs are layered iron(II)-iron(III)-hydroxides with anionic interlayers and
they hold the general formula [FeII(6-x)FeIII
x(OH)12]x+[(A)xnmiddotyH2O]x- where x = 09 -
42 A is an n-valent anion eg CO32- Clndash or SO4
2- and y is the number of water
molecules in the interlayer In circumneutral solutions the oxidation of dissolved
Fe(II) always passes through solid GR phases (Bernal et al 1959) This agrees
with the natural GR occurrences found in suboxic nonacid iron-rich environments
such as hydromorphic soils and intertidal sediments (Al-Agha et al 1995 Trolard
et al 1996 Genin et al 1998) In addition GRs have been found as corrosion
products in numerous engineering systems eg in a pipeline distribution system
for drinking water steel sheet piles in marine sediments reinforced concrete
(ferro-concrete) and permeable reactive barriers of zero-valent iron implemented
for on-site remediation of organic and inorganic contaminants (Tuovinen et al
1980 Nielsen 1976 Genin et al 1991 Roh et al 2000) Through sequestration
and reductive transformation GRs may play an important role in the fate and
transport of organic and inorganic pollutants in suboxic iron-rich soils and
sediments (see Chapters 4 amp 5 in this work and references therein)
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 47
The major goal of this work was to examine the iron minerals forming during the
course of Fe(III) bioreduction of hydrous ferric oxide goethite and lepidocrocite
Two model systems simulating sandy and calcareous soils in subsurface
environments were designed in order to investigate the formation of iron minerals
at conditions including low carbon levels low Fe(III) concentrations applied as
Fe(III) oxide coatings on sand or calcite no electron shuttle and no synthetic pH
buffers
32 Materials and methods
All handling and sampling of solutions and suspensions were carried out at strict
anoxic conditions Standard sterile techniques were used throughout (Hungate
1969 Miller amp Wolin 1974) Only the iron oxide coatings were not autoclaved in
order to avoid the iron oxides from transforming Goethite (acicular particles with
size 01 times 06 microm specific surface area 16 m2g) and lepidocrocite (acicular
particles with size 005 times 03 microm specific surface area 18 m2g) were purchased as
fine powders from Bayer (Bayferrox 910 and 943) Calcite (grain size 170-350 microm
Pluumlss-Staufer AG) and sea sand (dominantly quartz grain size 01-03 mm Riedel-
de Haeumln) were used as Fe(III) oxide coating bearing minerals
321 Preparation of iron oxide coatings
Two grams of goethite (goe) or lepidocrocite (lep) and 100 g calcite were
combined with 200 mL deionized water (DIW) in a 500 mL polyethylene flask
Hydrous ferric oxide (HFO) was synthesized by dissolving 4 g Fe(NO3)3middot9H2O in
70 mL DIW followed by slow neutralization under magnetic stirring till pH 7 with
approximately 30 mL 1 M NaOH (method modified after Schwertmann amp Cornell
1991) The HFO coating was made by combining 100 mL freshly precipitated
HFO with 900 mL deionized water and 50 g sea sand in a polyethylene bottle The
suspensions containing the iron oxide coatings were gently agitated on a
reciprocating shaker for 24 h and left to stand for another 24 h Excess Fe(III)
oxides and salts were removed from the coated material by repeated decantation
48 Chapter 3
and washing with 003 M NaNO3 followed by washing with DIW until clear
runoff Finally the coatings were collected on folding filters and air dried The
amount of HFO goethite and lepidocrocite coated onto sand and calcite after
washing and drying was quantified to 7-11 mg Fe(III)g sand or calcite
322 Mineral characterisation
The identity and purity of the HFO synthesized were examined by means of X-ray
diffraction (XRD) measurements The XRD analyses were performed on a Scintag
XDS 2000 using Co Kα radiation (45 kV 40 mA) using divergence scatter and
receiving slits of 1deg 05deg and 02 mm respectively Samples were scanned
between 6 and 80 deg2θ with a scan speed of 1 deg2θmin Mineral suspension samples
for transmission Moumlssbauer spectroscopic analysis were collected on 02 microm filters
in an anoxic glove box (Coy Laboratory Products Inc) transferred to Perspex
capsules and stored in liquid nitrogen until measurement Moumlssbauer spectra were
obtained between 250 and 5 K using a conventional constant acceleration
spectrometer and a source of 57Co in Rh The spectrometer was calibrated using a
125 microm foil of α-Fe at room temperature and isomer shifts are given relative to
the centroid of this absorber The spectra were fitted using simple Lorentzian line
shape and it was assumed that all positions have identical f-factors
323 Culture conditions and cell preparation
Shewanella algae BrY is a motile gram-negative rod which was isolated first from
anoxic estuary sediments (Caccavo et al 1992) S algae BrY was grown
aerobically in tryptic soy broth (30 gL CASO-bouillon Merck) at 28degC on a rotary
shaker at 150 rpm for 16-18 h Cells were harvested by centrifugation (6000 rpm times
g 4ordmC 15 min) during the late exponential ndash early stationary growth phase at
OD660 ~ 06 Optimal Fe(III) reductase activity is expressed at this stage of growth
(Roden amp Zachara 1996) The cells were washed twice in oxic 50 mM PIPES
[piperazine-NNacute-bis(2-ethanesulfonic acid)] buffer (pH 70) and resuspended in
culture medium containing no Fe(III) and no carbon source Washed cell
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 49
suspensions were used as inocula for Fe(III) reduction experiments Oxygen was
expelled from the inoculum by extensive purging with 100 N2(g) (9999999
purity) Working stock cultures of S algae BrY were maintained aerobically on
tryptic soy agar plates at ambient temperature
324 Bioreduction experiments
All anaerobic incubations were carried out in anoxic serum vials (25 mL) or test
tubes (13 mL) sealed with thick (10-13 mm) butyl rubber stoppers and aluminum
crimp caps or plastic screw caps The basal culture medium (Table 31) was
prepared according to Kostka amp Nealson (1998) but with a phosphate
concentration of 2 mM and the exclusion of Fe(II) and EDTA
(ethylenediaminetetraacetic acid) The medium was amended with 4-10 mM
lactate or formate and 8-27 mM Fe(III) The Fe(III) was applied as Fe(III) oxide
coatings on sand or calcite The suspensions were purged extensively with 100
N2(g) (HFOsand suspensions) or 995 N205 CO2(g) (goecalcite and
lepcalcite suspensions) prior to inoculation The calcareous systems were buffered
at pH ~ 76 through a natural buffer system (CaCO3(s) + 995 N205 CO2(g))
whereas the sandy systems contained no pH buffer (100 N2(g) pH 55-60)
Inoculum size made up 5 of the total volume Cultures were incubated dark at
room temperature and gently agitated once every day At different time intervals
suspension samples for Fe(II) and Moumlssbauer analysis were withdrawn from the
reaction mixture using 100 N2(g) or 995 N205 CO2(g)-flushed sterile
disposable syringes and hypodermic needles Suspension samples for Fe(II)
analysis were digested in 01 M HCl for 30 min
50 Chapter 3
Table 31 Composition of the mineral medium (modified from Kostka amp Nealson (1998))
Components Concentration (M)
(NH4)2SO4 00143 KH2PO4 73middot10-4
K2HPO4 13middot10-3
MgSO4middot7H2O 10middot10-3
CaCl2middot2H2O 50middot10-4
H3BO3 56middot10-5
ZnSO4middot7H2O 10middot10-6
Na2MoO4middot2H2O 40middot10-6
CuSO4middot5H2O 20middot10-7
MnSO4middotH2O 10middot10-6
Na2SeO4 12middot10-5
CoCl2middot6H2O 50middot10-6
NiCl2middot6H2O 80middot10-6
NaCl 10middot10-5
L-arginine 11middot10-4
L-serine 19middot10-4
L-glutamic acid 14middot10-4
Lactate or formate 4-10middot10-3
Fe(III) 8-27middot10-3
325 Analytical methods
Fe(II) was determined using a modified phenanthroline method (Fadrus amp Maly
1975) The total amount of Fe(III) coated on calcite and sand was determined by
atomic absorption spectroscopy following dissolution in 6 M HCl(aq) for 24 h
33 Results and discussion
331 Fe(II) production and suspension colour changes
Strongly chelating agents such as EDTA were omitted from the culture medium in
order to prevent complexation of Fe(II) and Fe(III) which interferes with
precipitation of Fe(II) and Fe(II)-Fe(III) mineral phases Within 1-2 weeks after
inoculation Shewanella algae BrY produced green mineral phases in media
suspensions containing lepidocrocite and goethite as coatings on calcite and 4-10
mM formate or lactate (Figure 31) The formation of the green precipitates was
generally slower for the lepidocrocite coating than for the goethite coating The
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 51
blue-green colours of the phases produced most likely originate from Fe(II)-Fe(III)
charge transfer between adjacent Fe(II) and Fe(III) ions in edge-shared octahedra
(Faye et al 1968) Dark brown and blackish products were formed when the
bacteria were inoculated on HFO coated sand (Figure 32)
Figure 31 Culture tubes containing a) goethite and b) lepidocrocite coated calcite in culture
medium The left tubes of the pair were not inoculated whereas the right tubes were
photographed 5 months after inoculation with S algae BrY Experimental conditions [formate]0
= 4 mM [Fe(III)]0 = 8 mM 995 N205 CO2(g) pH 76
52 Chapter 3
Figure 32 Culture tubes containing HFO coated sand in culture medium Tubes 1 and 2 to the
left were not inoculated whereas tubes 3-5 to the right were photographed a) 13 days and b) 21
days after inoculation with S algae BrY Experimental conditions [lactate]0 = 10 mM [Fe(III)]0
= 25 mM 100 N2(g) pH 55-60
The green and black colours did not change to other colours (observed for gt1
year) indicating that the microbial Fe(III) reduction ceased at these mineral stages
The concentrations of dissolved ferrous iron (Fe(II)sol) estimated during Fe(III)
bioreduction were generally low (Figure 33) When comparing the final Fe(II)sol
amounts produced and the slopes of the Fe(II)sol formation curves for HFO
goethite and lepidocrocite in Figure 33 it can be seen that the final Fe(II)sol
amount and the Fe(II)sol production rate both follow the order HFO gt goethite gt
lepidocrocite at similar cell densities regardless of the carbon source applied This
suggests that bioreduction by S algae BrY is more facile for HFO than for goethite
and lepidocrocite at the experimental conditions employed here It should be noted
that the final Fe(II)sol amounts and the Fe(II)sol production rates reported in this
work have not been normalised with respect to the specific surface areas of the iron
oxides and coating-bearing solids applied The reactivity trend is consistent with
previous findings demonstrating higher reducibility of natural and poorly
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 53
crystalline Fe(III) oxides as compared to synthetic crystalline Fe(III) oxides
(Zachara et al 1998) The authors ascribed these differences in reducibility to
differences in particle size surface area and crystal defects of the Fe(III) oxides In
some cases the dissolved Fe(II) concentration decreased again with time (Figure
33 b-d) This indicates that the Fe(II) formed was incorporated into solid phases
forming andor adsorbed onto the calcite sand or Fe(III) oxide surfaces The solid
Fe(II) concentrations were not estimated spectrophotometrically The solid
material was generally low in total iron and therefore saving it for Moumlssbauer
spectroscopic analysis was given highest priority
54 Chapter 3
Figure 33 Time course of dissolved Fe(II) production during bioreduction of HFOsand
goethitecalcite and lepidocrocitecalcite by S algae BrY Experimental conditions [Fe(III)]0 =
25 mM [formate]0 = 10 mM (a-c) or [lactate]0 = 10 mM (d-f)
No color change and no Fe(II) production were observed in mineral suspensions
lacking either a carbon source or S algae BrY cells (data not shown)
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 55
332 Identification of solid iron phases
The purity of the Fe(III) oxides used in the experiments were investigated by
transmission Mossbauer spectroscopy (Figure 34) Single (or strongly dominating)
sextets in the spectra with magnetic hyperfine fields of 48 1 505 and 45 5 Tat 5 K
demonstrated the purity of the HFO goethite and lepidocrocite samples
respectively A minor impurity of goethite in the lepidocrocite sample was
resolved in the spectrum measured at 80 K (not shown for pure sample but can be
seen as a magnetically ordered sextet in Figure 35c) No Fe(II)-containing
components were detected
a)
c)
middot 12 -8 -4 4 8 12
Velocity (mmls
bull middot12 -8 -4 0 4 12
Velocity (mmls)
b)
middot12 -8 -4 4 8 12
Velocity (mmls
Figure 34 Transmission Mossbauer spectra measured at 5 K of a) HFO (magnetic hyperfine
field of 481 T isomer shift of 048 1nrns-1 negligible quadrupole shift and line width of outer
lines 110 rmns-1) b) goethite (magnetic hyperfine field of 505 T isomer shift of 049 rmns-1
quadmpole shift of -013 1nrns-1 and line width of outer lines 042 rnrns-1
) and c) lepidocrocite
56 Chapter 3
(magnetic hyperfine field of 455 T isomer shift of 050 mms-1 quadrupole shift of ndash001 mms-1
and line width of outer lines 060 mms-1) prior to inoculation Simple Lorenztian fits are shown
The oxidation state and coordination of Fe in the microbially reduced HFO
goethite and lepidocrocite samples were also examined by transmission Moumlssbauer
spectroscopy (Figure 35) The bioreduced HFO goethite and lepidocrocite samples
cultured on formate contained Fe(II) holding similar coordination as inferred from
the similarity of the hyperfine parameters (see legend in Figure 35) but different
relative intensities (72 19 and 71 respectively) The major part of the Fe(III)
remaining in the bioreduced samples were coordinated similarly to the Fe(III)
present in the initial Fe(III) oxide The coordination of Fe(II) in the bioreduced
lepidocrocite samples cultured on lactate was slightly different (a smaller
quadrupole splitting of 288 mms-1 for the ferrous component dominates ndash data not
shown) The exact mineralogy of the Fe(II) present in the green phases was not
fully resolved but its coordination is very akin to one of the Fe(II) sites in vivianite
(see Chapter 2 this work) and synthetic green rusts (Koch 1998) These findings
agree with other reports on the bioformation of vivianite and green rusts by
Shewanella putrefaciens CN32 although the evidence provided may be discussed
(Fredrickson et al 1998 Glasauer et al 2003 Parmar et al 2001) Our
Moumlssbauer data on the green phases did not allow for a detailed account of the type
of green rust produced However when considering solution composition (see
Table 31) and the high affinity of GR interlayers for carbonate it is reasonable to
assume that carbonate GR was formed (Hansen amp Taylor 1991) Due to the high
amounts of Fe(III) in the oxides present in the experiments it was difficult to probe
a possible content of Fe(III) in the vivianite with certainty The differences in the
number of Fe(II) positions in the Moumlssbauer spectra and particular the different
temperatures at which magnetic ordering takes place can be employed in order to
distinguish between green rust and vivianite Preliminary Moumlssbauer data obtained
for the blackish precipitates formed in the HFOsand suspensions indicate that they
hold no resemblance to magnetite eventhough the colour suggests so On the
contrary the black precipitates seemed to be more similar to synthetic green rusts
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 57
Mossbauer spectroscopic measurements are currently underway in order to resolve
the Fe(II) coordinations in the greenish and blackish phases
a) b)
c c g Q e- e 0 0 1l 1l
-~ bull ~
iii ~ Qi bull a bull
-12 -8 4 0 4 12
4 -3 -2 -1 0 1 2 4 Velocity (mmts) Velocity ( rmis)
c)
-12 -8 4 8 12
v elocity (m mis)
Figure 35 Transmission Mossbauer spectra of the black and green phases fo1med within 1-2
weeks after inoculation of a) HFO (measured at 130 K) b) goethitecalcite (measured at 80 K)
and c) lepidocrocitecalcite (measured at 80 K) with S algae BrY Experimental conditions
[fo1m ate]0 = 4 mM [Fe(III)]o = 8 mM 995 Ni05 C02(g) pH 76 The quadrnpole
splittings and isomer shifts for the Fe(II) components in the three systems are a) 293 nnns-1 and
126 mmsmiddot1 b) 308 rmnsmiddot1 and131 rmnsmiddot1 and c) 322 mmsmiddot1 and 132 mmsmiddot1 Simple Lorenztian
fits are shown
The evidence provided in many of the studies proposing biogenic GRs is not all
too convincing but it strongly suggests the probability of microbially produced GR
being present The challenge encountered is that when present in low
58 Chapter 3
concentrations especially in mixtures including other iron minerals it is very
difficult to identify GRs using conventional solid phase analysis methods even
with Moumlssbauer spectroscopy At least two complementary methods such as XRD
and Moumlssbauer spectroscopy are required for proper identification and
characterization of GRs However in this work the solid materials were generally
too low in total iron to allow for XRD analysis Moreover the highly
heterogeneous suspensions were dominated by the coating-bearing sand and calcite
solids Electron micrographs including energy dispersive X-ray spectroscopy
suspension colour and mineral stability calculations do not suffice as evidence The
most convincing evidence provided so far involves an atypical GR-CO3 with an
Fe(II)Fe(III) ratio of 1 (Ona-Nguema et al 2002aampb) This GR-CO3 was formed
as a result of lepidocrocite reduction by Shewanella putrefaciens CIP 8040 at
conditions comprising high nutrient levels (50-75 mM formate) high Fe(III)
concentrations (80-300 mM) and a synthetic electron shuttle (100 microM
anthraquinone-26-disulfonate (AQDS)) at initial pH 75 Hence the results
reported during recent years suggest that microbial formation of GR may be
possible The results presented here indicate that GRs may be produced
microbially at conditions including low carbon and Fe(III) concentrations as well
as the exclusion of synthetic electron shuttles and pH buffers
333 Factors controlling the identity of the secondary iron minerals
In general one would expect that biogenic minerals have chemical compositions
and crystal habits similar to those produced by nonenzymatic processes as they are
governed by the same equilibrium principles In fact since the latter stages of
mineralization are inorganically driven and the secondary Fe(II)-containing
minerals are formed indirectly by electron transfer outside the bacterial cell and not
directly inside the bacterial cell the type of iron mineral formed is a function of the
environmental conditions in which the bacteria live ie the same microorganism
form different minerals in different environments The key factors controlling the
identity of the secondary iron minerals include medium composition electron
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 59
donor and electron acceptor concentrations mineral aging as well as adsorbed ions
(Zachara et al 2002) The main factor controlling the nature of the secondary
mineral products are the respiration-driven biogenic Fe(II) supply rate and
magnitude and its surface reaction with the residual oxide and other sorbed ions
(Zachara et al 2002) Especially solution and medium composition have a strong
impact on the nature of the Fe(II)-containing biomineralization products forming
Accordingly siderite (FeCO3) and magnetite (Fe3O4) were the secondary solid
phases resulting from the bioreduction of ferrihydrite by Shewanella putrefaciens
CN32 in bicarbonate buffered medium (pH 71) containing no phosphate whereas
siderite and vivianite were the secondary iron minerals dominating in bicarbonate
buffered medium (pH 74) containing 4 mM phosphate (Zachara et al 2002) This
is explained by the inhibiting effect of phosphate on crystallization of magnetite
(Couling amp Mann 1985 Fredrickson et al 1998)
334 Factors controlling the rate and extent of Fe(III) bioreduction
In this study the extent of Fe(III) bioreduction was estimated to 19-72 by
transmission Moumlssbauer measurements In fact complete microbial reduction of
crystalline Fe(III) minerals has never been observed in laboratory batch culture
studies (Roden amp Urrutia 2002) It has been found that Fe(II) does not inhibit
Fe(III) reductase activity through an enzyme inhibition mechanism (Roden amp
Urrutia 2002) Hence other chemical andor physiologic factors control the
bioavailability of solid Fe(III) phases and thus the extent of their microbial Fe(III)
reduction The initial rate and long-term extent of microbial reduction of
amorphous and crystalline Fe(III) oxides including HFO goethite and hematite
were linearly correlated with oxide surface area (Roden amp Zachara 1996)
Association of biogenic Fe(II) with Fe(III) oxide and DIRB cell surfaces reduced
the long-term extent of crystalline Fe(III) oxide bioreduction (Roden amp Urrutia
2002) These results were explained by Fe(II) surface complexes andor
precipitates creating a passive Fe(II)-bearing surface film providing direct physical
interference with the electron transfer from the DIRB cells to Fe(III) However the
60 Chapter 3
real mechanisms whereby the surface-associated Fe(II) inhibits Fe(III) oxide
bioreduction are unclear but they most likely involve both kinetic and
thermodynamic constraints on the electron transfer Culture medium composition
in particular the presence and the concentration of phosphate as well as Fe(II)
chelating ligands also exert an influence on the extent of the microbial reduction
of Fe(III) oxides The extent of Fe(III) bioreduction was inhibited by high
phosphate concentrations which favoured surfacebulk precipitation processes
(Urrutia et al 1998) The carbon sources most frequently applied in Fe(III)
bioreduction studies include malate citrate and other di- and tricarboxylic acids
which are not only easily metabolizable carbon sources but also eminent Fe(II) and
Fe(III) chelators In this study we employed formate and lactate as carbon sources
since they are the weakest complexing agents of Fe(II) and Fe(III) among the C1-
C3 monocarboxylic acids (Martell 1964) Thus we expect less dissolution of
prevailing precipitates by complexation as compared to other studies
34 Conclusions
This work demonstrated that Shewanella algae BrY reduced 19-72 of initial
Fe(III) when grown in culture media containing 4-10 mM formate or lactate and 8-
27 mM Fe(III) applied as goethite or lepidocrocite coatings on calcite (pH 76) or
HFO coatings on sand (pH 55-60) Within 1-2 weeks after inoculation green
mineral phases were produced in the goethitecalcite and lepidocrocitecalcite
mineral systems whereas black precipitates formed in the HFOsand suspensions
Moumlssbauer spectroscopic analyses indicated that the greenish and blackish phases
most likely were mineral mixtures dominated by vivianite and green rust Thus the
results indicate that GRs may be produced microbially at conditions including low
carbon and Fe(III) concentrations as well as the exclusion of synthetic electron
shuttles and pH buffers
Acknowledgments
We would like to thank Dr R Gerlach for providing us the Shewanella algae BrY culture and
Dr C B Koch for performing the Moumlssbauer analyses
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 61
References Al-Agha MR Burley SD Curtis CD Esson J (1995) Complex cementation textures and authigenic mineral assemblages in recent concretions from the Lincolnshire Wash (east coast UK) driven by Fe(0) to Fe(II) oxidation Journal of the Geological Society 152 157-171 Bernal JD Dasgupta DR Mackay AL (1959) The oxides and hydroxides of iron and their structural inter-relationships Clay Minerals Bulletin 4 15-30 Caccavo Jr F Blakemore RP Lovley DR (1992) A hydrogen-oxidizing Fe(III)-reducing microorganism from the Great Bay Estuary New Hampshire Applied and Environmental Microbiology 58 3211-3216 Caccavo Jr F Das A (2002) Adhesion of dissimilatory Fe(III)-reducing bacteria to Fe(III) minerals Geomicrobiology Journal 19 161-177 Couling SB Mann S (1985) The influence of inorganic phosphate on the crystallization of magnetite (Fe3O4) from aqueous solution Journal of the Chemical Society Chemical Communications 1713-1715 Das A Caccavo Jr F (2000) Dissimilatory Fe(III) oxide reduction by Shewanella alga BrY requires adhesion Current Microbiology 40 344-347
Das A Caccavo Jr F (2001) Adhesion of the dissimilatory Fe(III)-reducing bacterium Shewanella alga BrY to crystalline Fe(III) oxides Current Microbiology 42 151-154 Fadrus H Maly J (1975) Suppression of iron(III) interference in the determination of iron(II) in water by the 110-phenanthroline method The Analyst 100 549-554 Faye GH Manning PG Nickel EH (1968) The polarized optical absorption spectra of tourmaline cordierite chloritoid and vivianite ferrous-ferric electronic interaction as a source of pleochroism American Mineralogist 53 1174-1201 Fredrickson JK Zachara JM Kennedy DW Dong H Onstott TC Hinman NW Li S (1998) Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium Geochimica et Cosmochimica Acta 62 3239-3257 Geacutenin JMR Olowe AA Benbouzid-Rollet ND Prieur D Confente M Resiak B (1991) The simultaneous presence of green rust 2 and sulfate reducing bacteria in the corrosion of steel sheet piles in a harbour area Hyperfine Interactions 69 875-878 Geacutenin JMR Bourrieacute G Trolard F Abdelmoula M Jaffrezic A Refait Ph Maitre V Humbert B Herbillon A (1998) Thermodynamic equilibria in aqueous suspensions of synthetic and natural Fe(II)-Fe(III) green rusts Occurrences of the mineral in hydromorphic soils Environmental Science and Technology 32 1058-1068 Glasauer S Weidler PG Langley S Beveridge TJ (2003) Controls on Fe reduction and mineral formation by a subsurface bacterium Geochimica et Cosmochimica Acta 67 1277- 1288 Hansen HCB Taylor RM (1991) The use of glycerol intercalates in the exchange of CO3
2- with SO4
2- NO3- or Cl- in pyroaurite-type compounds Clay Minerals 26 311-327
62 Chapter 3
Hungate RE (1969) A roll tube method for cultivation of strict anaerobes Methods in Microbiology 3B 117-132 King GM (1990) Effects of added manganic and ferric oxides on sulfate reduction and sulfide oxidation in intertidal sediments FEMS Microbiology Ecology 73 131-138 Koch CB (1998) Structures and properties of anionic clay minerals Hyperfine Interactions 117 131 -157 Kostka J Nealson KH (1998) Isolation cultivation and characterization of iron- and manganese reducing bacteria In Techniques in Microbial Ecology Burlage RS Atlas R Stahl D Geesey G Sayler G (eds) Oxford University Press Inc 58-78 Kukkadapu RK Zachara JM Smith SC Fredrickson JK Liu C (2001) Dissimilatory bacterial reduction of Al-substituted goethite in subsurface sediments Geochimica et Cosmochimica Acta 65 2913-2924
Liu C Zachara JM Gorby YA Szecsody JE Brown CF (2001) Microbial reduction of Fe(III) and sorptionprecipitation of Fe(II) on Shewanella putrefaciens strain CN32 Environmental Science and Technology 35 1385-1393 Lovley DR (1991) Dissimilatory Fe(III) and Mn(IV) reduction Microbiological Reviews 55 259-287 Lovley DR (1997) Microbial Fe(III) reduction in subsurface environments FEMS Microbiology Reviews 20 305-313 Lovley DR Phillips EJP (1986) Organic matter mineralization with reduction of ferric iron in anaerobic sediments Applied and Environmental Microbiology 51 683-689 Lovley DR Phillips EJP Lonergan DJ (1991) Enzymatic versus nonenzymatic mechanisms for Fe(III) reduction in aquatic sediments Environmental Science and Technology 25 1062-1067 Lower SK Hochella Jr MF Beveridge TJ (2001) Bacterial recognition of mineral surfaces Nanoscale interactions between Shewanella and α-FeOOH Science 292 1360-1363 Martell AE (1964) Stability constants of metal-ion complexes Part 2 Organic including macromolecule ligands The Chemical Society London 2 ed Miller TL Wolin MJ (1974) A serum bottle modification of the Hungate technique for cultivating obligate anaerobes Applied Microbiology 27 985-987 Nealson KH Saffarini D (1994) Iron and manganese in anaerobic respiration Environmental significance physiology and regulation Annual Review of Microbiology 48 311-343 Nevin KP Lovley DR (2000) Lack of production of electron-shuttling compounds or solubilization of Fe(III) during reduction of insoluble Fe(III) oxide by G metallireducens Applied and Environmental Microbiology 66 2248-2251 Nevin KP Lovley DR (2002) Mechanisms for Fe(III) oxide reduction in sedimentary
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 63
environments Geomicrobiology Journal 19 141-159 Nielsen A (1976) Hvid groslashn og sort rust Beskrivelse af en korrosionsskade paring et svoslashmmebassin Nordisk Betong 2 21-24 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002a) Iron (IIIII) hydroxycarbonate green rust formation and stabilization from lepidocrocite bioreduction Environmental Science and Technology 36 16-20 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002b) Microbial reduction of lepidocrocite γ-FeOOH by Shewanella putrefaciens The formation of green rust Hyperfine Interactions 139140 231-237 Parmar N Gorby YA Beveridge TJ Ferris FG (2001) Formation of green rust and immobilization of nickel in response to bacterial reduction of hydrous ferric oxide Geomicrobiology Journal 18 375-385 Roden EE Urrutia MM (2002) Influence of biogenic Fe(II) on bacterial crystalline Fe(III) oxide reduction Geomicrobiology Journal 19 209-251 Roden EE Zachara JM (1996) Microbial reduction of crystalline iron(III) oxides Influence of oxide surface area and potential for cell growth Environmental Science and Technology 30 1618-1628 Roh Y Lee SY Elless MP (2000) Characterization of corrosion products in the permeable reactive barriers Environmental Geology 40 184-194 Schwertmann U Cornell RM (1991) Iron oxides in the laboratory Preparation and characterization VCH Verlagsgesellschaft mbH Weinheim Thamdrup B (2000) Bacterial manganese and iron reduction in aquatic sediments In Advances in Microbial Ecology (Schink B ed) Kluwer AcademicPlenum Publishers New York 41-84 Trolard F Abdelmoula M Bourrieacute G Humbert B Geacutenin JMR (1996) Mise en eacutevidence dun constituant de type rouilles vertes dans les sols hydromorphes Proposition de lexistence dun nouveau mineacuteral la fougeacuterite Geacuteosciences de surface Comptes Rendus de LrsquoAcademie des Sciences 323 1015-1022 Tuovinen OH Button KS Vuorinen A Carlson L Mair DM Yut LA (1980) Bacterial chemical and mineralogical characteristics of tubercles in distribution pipelines Journal of the American Water Works Association 72 626-635 Turick CE Caccavo Jr F Tisa LS (2003) Electron transfer from Shewanella algae BrY to hydrous ferric oxide is mediated by cell-associated melanin FEMS Microbiology Letters 220 99-104 Urrutia MM Roden EE Fredrickson JK Zachara JM (1998) Microbial and surface chemistry controls on reduction of synthetic Fe(III) oxide minerals by the dissimilatory iron- reducing bacterium Shewanella alga Geomicrobiology 15 269-291 Venkateswaran K Moser DP Dollhopf ME Lies DP Saffarini DA MacGregor BJ Ringelberg DB White DC Nishijima M Sano H Burghardt J Stackebrandt E
64 Chapter 3
Nealson KH (1999) Polyphasic taxonomy of the genus Shewanella and description of Shewanella oneidensis sp nov International Journal of Systematic Bacteriology 49 705-724 Zachara JM Fredrickson JK Li S Kennedy DW Smith SC Gassman PL (1998) Bacterial reduction of crystalline Fe3+ oxides in single phase suspensions and subsurface materials American Mineralogist 83 1426-1443
Zachara JM Kukkadapu RK Fredrickson JK Gorby YA Smith SC (2002) Biomineralization of poorly crystalline Fe(III) oxides by dissimilatory metal reducing bacteria (DMRB) Geomicrobiology Journal 19 179-207
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 65
4 Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust The Effect of Probe Compound Charge
Abstract
Layered iron(II)-iron(III)-hydroxides (green rusts) may play an important role in
controlling the fate and transport of many organic and inorganic contaminants in
iron-rich suboxic soils and sediments Unlike most other iron oxides green rusts
(GRs) contain not only external Fe(II) reactive sites at the basal planes and at the
edges but also internal sites in the space between consecutive Fe(II)-Fe(III)
hydroxide layers The GR interlayer thickness is a function of both the size and the
charge of the interlayer anion Whether a given oxidant has access to the internal
sites in GRs is dependent on its charge We investigated the reductive
transformation of nitroaromatic compounds (NACs) by GR-SO4 and studied the
effect of NAC charge on the reactivity towards GR-SO4 A series of structurally
closely related compounds with different charge properties including nitrobenzene
4-nitrotoluene 4-chloronitrobenzene and 4-nitrophenylacetic acid were used as
probe compounds The NACs were completely reduced to their corresponding
anilines by GR-SO4 The reactions followed pseudo 1 order kinetics with respect
to NAC and the surface area-normalised pseudo 1 order rate constants obtained
were 016ndash465middot10-4 s-1middotm-2middotL at [Fe(II)GR]0 = 103-1260 mM [NAC]0 = 20-102
microM and pH 84-86 Neither mass transfer control nor surface saturation kinetics
could account for the rather unexpected similarity of the surface area-normalised
pseudo 1 order rate constants obtained for the reduction of the neutral and anionic
NACs by GR-SO4 These observations suggest that the anionic NACs did not have
an enhanced access to the inner or outer Fe(II)-GR reactive sites as compared to
the neutral NACs Hence the reaction between NAC and GR-SO4 primarily took
place at the edges of GR-SO4
66 Chapter 4
41 Introduction
Layered iron(II)-iron(III)-hydroxides (green rusts) are intermediate phases formed
by partial oxidation of Fe(II) or partial reduction of Fe(III) In neutral and weakly
alkaline solutions the oxidation of dissolved Fe(II) always passes through solid
green rust (GR) phases (Bernal et al 1959) This agrees with the natural GR
occurrences found in suboxic non-acid iron-rich environments such as
hydromorphic soils and intertidal sediments (Al-Agha et al 1995 Trolard et al
1996 Genin et al 1998) In addition GRs have been found as corrosion products
in numerous engineered systems ie a pipeline distribution system for drinking
water steel sheet piles in marine sediments reinforced concrete (ferro-concrete)
and permeable reactive barriers of zero-valent iron implemented for on-site
remediation of organic and inorganic contaminants (Tuovinen et al 1980 Nielsen
1976 Genin et al 1991 Roh et al 2000) Furthermore the microbial formation
of GRs resulting from bioreduction of Fe(III) oxides by strains of the anaerobic
dissimilatory Fe(III) reducing bacteria Shewanella putrefaciens has been reported
increasingly over the last 5 years (Fredrickson et al 1998 Kukkadapu et al 2001
Parmar et al 2001 Ona-Nguema et al 2002 Glasauer et al 2003) Moreover
the biotic formation of GRs by anaerobic denitrifying Fe(II) oxidizing bacteria has
been proposed but proper identification of the GR phases still lacks (Chaudhuri et
al 2001) All these indications of microbial GR formation infer the importance of
GRs as a link between geochemical and biological processes in natural systems
GRs form platy crystals with the general formula [FeII(6-
x)FeIIIx(OH)12]x+[(A)xnmiddotyH2O]x- where x = 09 - 42 A is an n-valent anion eg
CO32- Clndash or SO4
2- and y is the number of water molecules in the interlayer The
crystal structure consists of positively charged hydroxide sheets with Fe(II) and
Fe(III) cations having octahedral hydroxyl coordination The Fe(III) in the
hydroxide layers creates a net positive charge which is balanced by hydrated
anions in the interlayers (Figure 41) The interlayers have a higher affinity for
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 67
divalent anions than for monovalent anions (Miyata 1983) Among the 3 most
common GR forms the affinity follows the order CO32- gt SO4
2- gt Clndash The extreme
preference shown for carbonate hinders further access and exchange except under
certain conditions (Hansen amp Taylor 1991) Non-carbonate forms are readily
exchanged with other anions when dispersed in a solution containing the
exchanging anion (Mendiboure amp Schoumlllhorn 1986)
c
ba
Figure 41 Green rust layer structure The hydroxide layers and the interlayers are connected by
hydrogen bonds (not shown) The GR-SO4 crystal structure is characterised by the hexagonal
unit cell having a = b = 055 nm and c = 110 nm (Simon et al 2003) The unit cell consists of
one double layer (a double layer is a hydroxide layer and an interlayer) ie the hydroxide layer
constitutes 049 nm and the interlayer 061 nm in GR-SO4
The GR interlayer thickness (extending in the c axis direction Figure 41) is a
function of both the size and the charge of the interlayer anion Tetrahedrally
coordinated anions like sulphate lead to larger interlayer distances than smaller
monoatomic anions like chloride or planar ions like carbonate (Mendiboure amp
Schoumlllhorn 1986) Not only size but also charge density plays a role for the
interlayer spacing That is for anions having the same number of valence
electrons anions with smaller ionic radii (higher electron density) are bound more
strongly and therefore result in smaller interlayer spacings
Due to their layered structures anionic interlayers and high specific surface areas
GRs represent reactive ion exchangers and sorbents of anions eg arsenate
selenate and phosphate (Myneni et al 1997 Hansen amp Poulsen 1999 Randall et
al 2001) In addition GR may incorporate heavy metal cations by isomorphic
68 Chapter 4
substitution into the GR hydroxide layers (Tamaura 1985 Tamaura 1986)
Furthermore GRs have been shown to reduce a range of inorganic contaminants
such as nitrite nitrate selenate chromate uranyl pertechnetate and the transition
metals AgI AuIII CuII and HgII as well as organic pollutants including halogenated
ethanes ethenes and methanes (Hansen et al 1994 Hansen et al 1996 Myneni et
al 1997 Erbs et al 1999 Loyaux-Lawniczak et al 1999 Cui amp Spahiu 2002
Lee amp Batchelor 2002b Heasman et al 2003 OrsquoLoughlin et al 2003a amp 2003b
Pepper et al 2003 Elsner et al 2004 OrsquoLoughlin amp Burris 2004) Thus through
sequestration and reductive transformation GRs may play an important role in
controlling the fate and transport of contaminants in suboxic soils and sediments
In a previous study the effects of interlayer anion and Fe(II)Fe(III) ratio in GRs
on the reduction rate of nitrate were investigated (Hansen et al 2001) It was
found that the rate of nitrate reduction to ammonium increased with increasing
Fe(II)Fe(III) ratio and decreased when exchanging a monovalent interlayer anion
(chloride) with a divalent anion (sulphate) The results suggest that for anionic
oxidants like nitrate Fe(II) within the hydroxide layer is available from the outside
basal planes and from the edges as well as through the interlayer under certain
conditions (Figure 42) However oxidants with different charge properties
(cations neutral molecules) may exhibit different affinities for the various reactive
Fe(II) sites present in GR
As the reactive sites are located inat the Fe(II)-Fe(III) hydroxide layers the rate of
reaction depends on the hydroxide layer area which can be accessed by the
oxidant If the oxidant can exchange with the interlayer anion reaction can take
place both at outer and inner surfaces of the GR particles and in total more
reactive sites are available for the reaction However it was found that nitrate
cannot penetrate the interlayer when carbonate or sulphate constitutes the
interlayer anions (Hansen amp Koch 1998) This agrees with the fact that the
interlayers have a lower affinity for monovalent anions than for divalent anions
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 69
(Miyata 1983) However when nitrate was forced into the interlayer by extracting
the interlayer sulphate through precipitation of barium sulphate outside the GR
particles the observed 40 fold increase in rate of nitrate reduction almost equalled
the increase in exposed surface area of the Fe(II)-Fe(III) hydroxide layers (Hansen
amp Koch 1998) From these observations it is expected that the rate of reaction
depends on the particular GR form the crystallite size and the ease with which an
oxidant can exchange with An- in the GR interlayer (Figure 42) Due to
electrostatic interactions we expect anions to be attracted to the positively charged
outer and inner surfaces to a higher degree than cations and neutral compounds If
this theory holds we may expect oxidants with similar intrinsic reactivity (similar
one-electron reduction potentials) to react in the following order anionic gt non-
charged gt cationic (Figure 43) granting that we do not normalise the rate
constants with respect to the amount of oxidant sorbed
Figure 42 Reaction of a probe compound at basal planes at edges and in the interlayer of GR
The hypothesis only holds in cases where the oxidants possess the same intrinsic
reactivities If the relative reactivities of the probe compounds differ greatly from
what would be expected when considering only their reduction potentials
70 Chapter 4
compound specific effects such as charge properties might explain this and the
relative reactivities may follow a pattern like the one depicted in Figure 43
Figure 43 Hypothetical plot of observed reaction rate constants for the reactions between
cationic neutral and anionic probe compounds and GR-SO4 assuming that the oxidant charge
controls its reactivity towards GR
In this work we investigated the reductive transformation of NACs by GR-SO4
Furthermore the effect of NAC charge on the rate of reaction and the possible
access to the internal reactive sites in GR-SO4 were assessed When quantifying
Fe(II) in GRs by means of acid digestion it is not possible to distinguish between
the reactive sites accessible from the outside (at the basal planes or at the edges) or
through the interlayer However we designed an indirect method to gain insight
into the relative importance of the various reactive sites by using a series of
structurally closely related compounds with different charge properties as ldquoreactive
probesrdquo Neutral and anionic probes were needed in order to access all Fe(II)
reactive sites According to our hypothesis cationic and non-charged oxidants
should provide information about the reactivity of the outer Fe(II) reactive sites in
GR whereas the anionic oxidants should provide information about the reactivity
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 71
of both outer and inner Fe(II) reactive sites We chose five nitro aromatic
compounds (NACs) - representing an important group of reducible organic
pollutants - as probe compounds (Figure 43) This class of compounds is not only
of great environmental concern but also comprises suitable model compounds for
studying redox reactions potentially relevant in the environment Moreover they
react readily with Fe(II) surface species associated with iron oxides or clay
minerals transforming them into well-defined easily detected products allowing
mass and electron balances to be established (Hofstetter et al 2003 Klausen et al
1995 Schultz amp Grundl 2000) Our main goals were to establish the rate law and
estimate the surface area-normalised reaction rates for the reaction of the probe
compounds with GR-SO4 in order to assess the importance of the Fe(II) reactive
sites accessible through the interlayer relative to the Fe(II) reactive sites accessible
at the outer surface in GR-SO4
4 2 Materials and methods
All handling and sampling of solutions and suspensions were carried out under
strict anoxic conditions All chemicals were pa quality or better Methanolic stock
solutions (5 mM) of nitrobenzene (NB) 4-nitrotoluene (4-NT) 4-
chloronitrobenzene (4-CNB) and 4-nitrophenylacetic acid (4-NPA) were prepared
in deoxygenated methanol Several attempts to synthesize the cationic probe
compound 4-(NNN-trimethylammonium)-nitrobenzene failed and therefore the
study had to be carried out with only neutral and anionic oxidants The sulphate
GR form was chosen as it is the most stable form and thus the easiest to work
with in the lab
421 Synthesis of GR-SO4
GR-SO4 was synthesized by controlled air oxidation of an FeSO4 solution at a
constant pH of 700 according to the procedure given by Koch amp Hansen (1997)
The GR-SO4 suspension was washed with deoxygenated deionised water and
72 Chapter 4
separated on a folding filter redispersed in deoxygenated 25 mM Na2SO4(aq) in
order to stabilize the GR-SO4 and prevent it from transforming into magnetite
spontaneously Washing separation and redispersion of the GR-SO4 suspension
were conducted in an anoxic glove box (Coy Laboratory Products Inc) All
suspensions and solutions were deoxygenated by Ar-purging (999998 Ar
Carbagas)
422 Mineral characterisation
The identity and purity of the GR-SO4 suspensions were examined by means of X-
ray diffraction measurements The XRD analyses were performed on a Scintag
XDS 2000 using Cu Kα radiation (45 kV 40 mA) Glycerol smears made
according to Hansen (1989) were scanned between 6 and 80 deg2θ with a scan speed
of 1 deg2θmin
423 Lyophilization and determination of specific surface area
Simple air-drying of the GR mineral in the glove box resulted in big flakes with
very low surface areas hence a more suitable lyophilization method was adopted
from Elsner et al (2004) The GR-SO4 suspensions were lyophilised using
Schlenk-type glassware The set-up consisted of a 1 L round bottom flask and a
200 mL glass finger connected by a crescent-shaped bridge equipped with an
evacuation outlet and a stopcock All ground joints and fittings were attached using
high-vacuum grease The washed and resuspended GR-SO4 suspensions were
filled into the glass finger and the freeze-drying apparatus was assembled and
closed before taking it out of the glove box The suspension was frozen by
carefully submerging the lower part of the glass finger into liquid nitrogen for a
few hours Subsequently the evacuation outlet was connected to a vacuum pump
by a metal hose Following a short evacuation of the metal hose the lyophilization
apparatus was evacuated for several minutes by gently opening the stopcock The
evacuation was terminated by closing the stopcock and disconnecting the vacuum
pump The apparatus position was now reversed by removing the glass finger from
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 73
and immersing the round bottom flask into liquid nitrogen As any other
lyophilization method this method depends on sublimation of the ice from the
frozen sample and its recondensation on a cool surface in this case the round
bottom flask Generally it took 1-2 d for the mineral to dry The apparatus was
disassemled in the glove box and the fine powder stored under anoxic conditions
The specific surface area (SSA) of GR-SO4 was determined by the BET multi-
point method using N2 adsorption (Brunauer et al 1938) Powder samples were
filled into sample burettes in the glove box and the generously greased stopcocks
closed Samples and burettes were evacuated prior to connecting them to the BET-
instrument (Sorptomatic 1990 Fisons)
424 Estimation of the one-electron reduction potential for 4-NPA
Kinetic experiments in 100 mL Viton stoppered and alu-crimp capped serum vials
were carried out under the exclusion of oxygen as described by Hofstetter et al
(1999) The homogeneous aqueous solutions contained 50 mM KH2PO4 buffer
(pH = 660) 5 mM Na2S redox buffer and 20 microM juglone (8-hydroxy-14-
naphthoquinone) added as deoxygenated 20 mM methanolic stock solution The
solutions were equilibrated at least one day prior to 4-NPA addition To start the
reaction 50 microM 4-NPA was added as deoxygenated 20 mM methanolic stock
solution The vials were agitated on a roller apparatus in the dark at 21ordmC Control
experiments were prepared similarly except for the addition of juglone At
different time intervals aqueous samples were withdrawn with a syringe and
collected in 18 mL HPLC vials containing 100 microL 1 M HCl The sample vials
were sealed with Teflon-coated silicone septa and plastic screw caps and vortexed
for 10 s The samples were stored at -20degC and analysed without further treatment
For comparison experiments with 4-NT were also conducted See Supporting
Information 71 for more information on the one-electron reduction potentials
74 Chapter 4
425 Kinetic experiments
All reactions took place at pH 84-86 where GR-SO4 tends to stabilize and buffer
itself Samples for Fe(II) and XRD analysis were withdrawn prior to reaction Due
to the fast reactions the experiments were conducted in 10 mL single-use
polyethylene syringes (BD Plastipak) in the glove box To start reaction 40-200
microL 5 mM methanolic stock solutions of NAC were quickly added to 10 mL GR-
SO4 suspension (1-12 mM Fe(II)GR) washed and resuspended in 25 mM
Na2SO4(aq) A Teflon filter (25 mm x 02 microm BGB Analytik) was quickly
mounted on the tip of the syringe and the syringe was vigorously shaken between
sampling At different time intervals filtered suspension samples were collected in
18 mL HPLC vials The HPLC vials were sealed with Teflon-coated silicone septa
and plastic screw caps The samples were stored at -20degC and analysed without
further treatment Absorption of NAC in the syringe and in the Teflon filter
evaluated in blank experiments with NAC added to 25 mM Na2SO4(aq) was found
to be negligible
426 Analytical methods
Initial total and aqueous Fe(II) were determined using a modified phenanthroline
method (Fadrus and Maly 1975) In order to determine [Fe(II)aq] and [Fe(II)total] 1
mL filtered (022 microm) and 1 mL unfiltered GR-SO4 suspension samples were
withdrawn and each treated with 18 mL 01 M HCl for at least 30 min From these
acid digests 01 mL was added to 05 mL Fe(II)-reagent and 19 mL deionised
water (DIW) added up The Fe(II) content in GR-SO4 was estimated as the
difference [Fe(II)GR] = [Fe(II)total] - [Fe(II)aq] The NACs and their corresponding
intermediates and products formed during reduction by GR-SO4 were quantified by
reversed-phase HPLC Separation was performed on a LiChrospher 100 RP-18 (5
microm 125 times 4 mm ID) reversed-phase column coupled with a LiChroCART 100 RP-
18 (4 times 4 mm ID) precolumn Analytical conditions were isocratic and the eluent
consisted of 10 mM hydroxylammonium chloride in various DIWCH3OH
mixtures (vv 3565 and pH 70 for 4-NT and 4-CNB 955 and pH 60 for
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 75
4-NPA) The injection volume was 20 microLand the flow-rate 10 mLmin HPLC
analyses were performed using a Gynkotek High Precision Pump M480 Gynkotek
Gina 50 autosampler and a diode array UV detector (340s Gynkotek) UV-VIS
detection was carried out at the wavelengths of maximum absorption for the
various nitro aromatic and aniline analytes
43 Results and discussion
431 Productformation and reaction kinetics
The reduction of the aromatic nitro group occurs via nitroso- and hydroxylamino-
intermediates where 2 electrons are transferred in each reaction step (Figure 44)
0 --0 H OH H H --0 N N N N
2e- 2H+ H20 + 2e-~ 2e- 2H+ H20
~ ~ R R R
Nitro benzene Nitrosobenzene Hydroxylamine Aniline
Figure 44 Reductive transfonnation pathway of NA Cs
Thus in order to reduce 1 Ar-N02 completely to Ar-NH2 6 electrons
corresponding to 6 mol Fe(II) are needed As magnetite was the major iron phase
formed during reaction (XRD results not shown) we assume the following
reaction stoichiometry
The aniline product was not formed at the same rate as the nitro compound
degraded which is consistent with the detection of early eluting hydroxylamine
intermediates during the course of the reaction (Figure 45a amp 45c) No traces of
76 Chapter 4
nitrosobenzene intermediates or side products such as azoxy- azo- or
hydrazobenzene were found In Figure 45 pseudo 1 order kinetic plots and ln
[Ar-NO2]t[Ar-NO2]0) versus time plots for the neutral probe compounds 4-CNB
and 4-NT are shown as examples The plots for NB and 4-NPA look similar
Figure 45 a Concentration versus time plots for reaction of GR-SO4 with 4-CNB ([Fe(II)GR]0 =
126 mM [4-CNB]0 = 30 microM) b ln [Ar-NO2]t[Ar-NO2]0) versus time plots for reaction of GR-
SO4 with 4-CNB ([Fe(II)GR]0 = 126 mM + [4-CNB]0 = 30 microM [Fe(II)GR]0 = 63 mM + [4-
CNB]0 = 50 microM) c Concentration versus time plots for reaction of GR-SO4 with 4-NT
([Fe(II)GR]0 = 131 mM [4-NT]0 = 20 microM) d ln [Ar-NO2]t[Ar-NO2]0) versus time plots for
reaction of GR-SO4 with 4-NT ([Fe(II)GR]0 = 131 mM [4-NT]0 = 20 microM [Fe(II)GR]0 = 131
mM [4-NT]0 = 50 microM) The hydroxylamino intermediate shown in microM equals the deficit in the
mass balance and in abs equals the detector response (peak area) Solid lines represent 1 order
kinetic fits (a amp c) and ln [Ar-NO2]t[Ar-NO2]0) versus time fits (d) whereas symbols and dotted
lines represent actual data
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 77
At intial Fe(II)GR concentrations in large excess of initial Ar-NO2 concentration
we found a pseudo 1 order rate law for the degradation of Ar-NO2 by GR-SO4
[ ] [ ] [ b 2
a GR
2 ArNOFe(II) ArNOsdotsdot=minus k
dtd ]
where a = 1 b = 1 and the observed pseudo 1 order rate constant kobs = k middot
[Fe(II)GR] At high [Fe(II)GR]0[Ar-NO2]0 ratios the nitro compound was
transformed completely into the aniline product within reaction duration and the
degradation curves of the nitro compound were shaped according to pseudo 1
order kinetics (data points follow solid line in Figure 45a) In some instances ie
at low [Fe(II)GR]0[Ar-NO2]0 ratios the reactions did not follow pseudo 1 order
kinetics for the whole duration of reaction (data points deviate from solid line in
Figure 45c) Hence in order to allow comparison all the pseudo 1 order rate
constants were calculated as initial rates (ie max first two half-lives) from linear
fits of (time ln [Ar-NO2]t[Ar-NO2]0)-plots (Figure 45b amp 45d) Surface area-
normalised pseudo 1 order rate constants are shown in Table 41
Tabl
e 4
1 S
urfa
ce a
rea-
norm
alis
ed p
seud
o 1
ord
er ra
te c
onst
ants
for t
he re
duct
ive
trans
form
atio
n of
4-n
itrot
olue
ne (4
-NT)
4-
chlo
roni
trobe
nzen
e (4
-CN
B) a
nd 4
-nitr
ophe
nyla
cetic
aci
d (4
-NPA
) by
GR
-SO
4
Exp
erim
ent
Age
GR
(d
) [F
e(II
) GR] 0
(mM
) [N
AC
] 0 (micro
M)
[Fe(
II) G
R] 0
[N
AC
] 0∆[
ArN
O2]
(microM
) af b
k obs
(s-1
) ck o
bs (s
-1middotm
-2middotL
) d
GR
-SO
4 + 4
-NT
3 1
103
20
51
5
109
54
5
7
65middot1
0-46
95middot1
0-5
GR
-SO
4 + 4
-NT
3 1
103
50
20
6
174
34
8
7
41middot1
0-46
74middot1
0-5
GR
-SO
4 + 4
-NT
3 1
103
10
0 10
3
214
21
4
2
63middot1
0-42
39middot1
0-5
GR
-SO
4 + 4
-CN
B 1
1
103
55
18
7
177
32
2
4
21middot1
0-43
83middot1
0-5
GR
-SO
4 + 4
-CN
B 1
1
103
10
2 10
1
165
16
2
2
37middot1
0-42
15middot1
0-5
GR
-SO
4 + 4
-NPA
1
1 1
03
25
412
9
7 38
8
4
82middot1
0-44
38middot1
0-5
GR
-SO
4 + 4
-NPA
1
1 1
03
46
224
9
9 21
5
6
37middot1
0-45
79middot1
0-5
GR
-SO
4 + 4
-NPA
1
1 1
03
100
103
13
7
137
196
middot10-4
178
middot10-5
GR
-SO
4 + 4
-NT
2 15
1
31
20
655
5
4 27
0
6
74middot1
0-44
82middot1
0-5
GR
-SO
4 + 4
-NT
2 15
1
31
50
262
9
7 19
4
5
89middot1
0-44
21middot1
0-5
GR
-SO
4 + 4
-NT
4 2
126
0 50
25
2 49
1
982
110
middot10-2
817
middot10-5
GR
-SO
4 + 4
-NT
4 2
630
50
12
6 42
6
852
186
middot10-3
276
middot10-5
GR
-SO
4 + 4
-CN
B 2
2
126
0 30
42
0 29
0
967
925
middot10-3
687
middot10-5
GR
-SO
4 + 4
-CN
B 2
2
630
50
12
6 38
0
760
136
middot10-3
202
middot10-5
GR
-SO
4 + 4
-NPA
2
2 12
60
40
315
371
92
8
5
96middot1
0-34
43middot1
0-5
GR
-SO
4 + 4
-NPA
2
2 6
30
45
140
273
60
7
1
09middot1
0-31
62middot1
0-5
a A
mou
nt o
f NA
C re
duce
d by
GR
-SO
4 at r
eact
ion
term
inat
ion
b F
ract
ion
of in
itial
ly a
dded
NA
C tr
ansf
orm
ed b
y G
R-S
O4 a
t rea
ctio
n te
rmin
atio
n c
Pse
udo
1
orde
r rat
e co
nsta
nts c
alcu
late
d as
initi
al ra
tes
ie m
ax f
irst t
wo
half-
lives
d S
urfa
ce a
rea-
norm
alis
ed p
seud
o 1
ord
er ra
te c
onst
ants
The
are
a of
GR
-SO
4 per
L
su
spen
sion
was
cal
cula
ted
as frac14
middot[Fe
(II)
GR] 0middot
600
gmiddotm
ol-1
middot71
2 m
2 middotg-1
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 79
432 Comparison of rate constants for the different NACs
Even for NACs holding very different one-electron transfer reduction potentials
( ) their reactivities differed only little in Fe(II)-Fe(III) systems such as the
Fe(II)goethite system (slope a = 06 for linear free energy relationship (LFER)
between k
1hE
obs and Hofstetter et al 1999) and the Fe(II)magnetite system
(LFER slope a = 034 Klausen et al 1995) When considering only the for the
reductive transformation reactions of the NACs applied in this study (Table 42)
we expect the surface area-normalised pseudo 1 order rate constants for the
reduction of the NACs to follow the order 4-CNB gt NB gt 4-NT gt 4-NPA Based
on log k
1hE
1hE
obs versus correlations obtained in Fe(II)goethite systems we expect 4-
CNB to react 6 times faster than 4-NPA (Hofstetter et al 1999)
1hE
Table 42 One-electron reduction potentials and relative reactivities in Fe(II)-magnetite and GR-
SO4 systems for the nitro aromatic probe compounds
Compound pKa Eh1
acute (mV) krel (Fe3O4) cd krel (GR-SO4) ce
4-Chlornitrobenzene - -450 a 122 148
Nitrobenzene - -486 a 1 1
4-Nitrotoluene - -500 a 057 176
4-Nitrophenylacetic acid 385 -543 b - 123 a Values from references cited in Hofstetter et al 1999 b Estimated at pH 660 using a LFER (Hofstetter et al 1999 see Supporting Information 71) c Reactivity relative to NB d Values from Klausen et al 1995 e Values from this work
A comparison of the relative rate constants of the NACs obtained for their
transformation by GR-SO4 (this work) and by magnetite (Klausen et al 1995)
shows that they do not differ significantly from each other in any of the mineral
systems (Table 42) When considering charge effects we expect the anionic probe
compounds to react faster with GR-SO4 than the neutral probe compounds
provided that they sorb preferentially within the GR-SO4 interlayers and that Fe(II)
in the interlayers are equally or more reactive than external Fe(II) sites Still the
surface area-normalised kobs values obtained for NB 4-NT 4-CNB and 4-NPA
under various experimental conditions did not differ significantly from each other
80 Chapter 4
(Figure 46 Table 41) The anionic probe compound 4-NPA did not react
significantly faster with GR-SO4 than the neutral probe compounds NB 4-NT and
4-CNB This may indicate that 4-NPA does not significantly interact with reactive
Fe(II) sites in the interlayer Alternatively the negative charge carried by 4-NPA
may be compensating for the lower intrinsic reactivity as compared to the neutral
probe compounds thus explaining the similarity in rate constants for 4-NPA and
the neutral probe compounds Finally other factors than intrinsic reactivity or
charge of the probe compounds such as regeneration of reactive sites or formation
of the magnetite phases may control the overall reactivity of the system
Figure 46 Actual plot of surface area-normalised pseudo 1 order rate constants for the reactions
between neutral and anionic probe compounds and GR-SO4
In heterogeneous reactions mass transfer in bulk solution becomes the rate-
limiting step when the surface reaction is much faster than the diffusion of the
reacting species to the reactive surface In cases where mass transfer controls the
overall rate of reaction the observed pseudo 1 order rate constant kobs ge kLmiddota
where kL is the calculated mass transfer coefficient (mmiddots-1) and a is the ratio of the
external (geometric) specific surface area to volume of solution (m-1) (see
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 81
Supporting Information 72) Mass transfer controlled reactions between GR-SO4
particles and the NACs in bulk solution would explain the similar pseudo 1 order
rate constants obtained for the NACs in this work However when comparing our
estimates of kLmiddota with kobs (see Supporting Information 72) we found that the rates
of mass transfer for all 4 NACs exceed the observed rate constants by at least 3
orders of magnitude at every initial Fe(II)GR concentration Thus the reactions of
the given NACs with GR-SO4 are not likely to be mass transfer limited under the
experimental conditions applied here
Since mass transfer in bulk solution does not control the reaction between GR-SO4
and NACs the overall reaction rate may be surface saturation controlled During
the reductive transformation of NACs not only the parent compound but also
various intermediates forming may compete for the restricted number of reactive
sites present in GR-SO4 This competition may constitute the rate limiting step in
the overall reactivity and may even be enhanced if the number of reactive sites is
depleted during reaction However surface saturation kinetics would not explain
the unexpected similarity of the pseudo 1 order rate constants obtained for the
NACs but it could explain the bent curves observed at low initial Fe(II)GR
concentrations (Figure 45d) The kinetically deviating cases at low [Fe(II)GR]0
were evaluated according to Langmuir-Hinshelwood kinetics (see Supporting
Information 72) Our experimental data did not agree with the Langmuir-
Hinshelwood rate law for any of the NACs (regression results not shown)
Simplifying the rate law by assuming that the aniline product or the
hydroxylamino intermediate or both did not compete for the reactive sites did not
improve the regression Thus the Langmuir-Hinshelwood model cannot explain
the deviations from pseudo 1 order kinetics observed at [Fe(II)GR]0 in our GR-SO4
system and it does not suffice as the correct reaction mechanism nor as the rate-
limiting step
82 Chapter 4
If the adsorption follows a saturation-type sorption isoterm (eg Langmuir) the
sorbate (oxidant) concentration at the surface will vary non-linearily with the total
amount of oxidant added This dependence will have to be taken into account when
establishing rate laws for the heterogeneous reactions and when testing the
hypothesis that the reaction rates depend on the sorbed concentration of the
oxidants However at the high reaction rates observed here we could not quantify
sorption Since the measured initial NAC concentrations corresponded to the
nominal amount of NAC added we assume that transformation and not sorption
was responsible for the consumption of NAC
433 Factors influencing the reaction rate
In general numerous compound- and system-specific factors influence redox
reactions One very important factor is pH which influences the speciation of
dissociable compounds as well as the stability of GR and the formation of other
iron minerals in the system pH has a strong impact on the sorption and therefore
the availability of ionisable oxidants such as carboxylic acids At pH ~ 84 where
our experiments were conducted 4-NPA (pKa = 385) is completely dissociated
Our experiments conducted with GR-SO4 and NB showed that pH was constant
during reaction In addition solution pH has an effect on the surface speciation
From other Fe(II)-Fe(III) systems such as Fe(II) surface species associated with
iron oxides or clay minerals it is well-known that other reactive hydroxylated
Fe(II)-Fe(III)-hydroxo surface complexes can form at higher pH (Charlet et al
1998 Liger et al 1999) Williams amp Scherer (2001) reported a small decrease (5
fold) in the reduction rate of chromate with GR-CO3 when increasing pH from 50
to 90 This decrease may be due to the alternating speciation of the Fe(II) surface
sites on GR-CO3 and of chromate in solution (pKa (H2CrO4) = 08 pKa (HCrO4ndash) =
65) when raising pH (Williams amp Scherer 2001) In contrast other studies have
reported small increases (4 fold) in the reduction rates of nitrate and
trichloroethene with GR-SO4 when increasing pH from 71 to 84 and from 68 to
101 respectively (Koch amp Hansen 1997 Lee amp Batchelor 2002b)
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 83
In this work all experiments were carried out in the presence of 25 mM
Na2SO4(aq) in order to minimize GR-SO4 dissolution and spontaneous
transformation into magnetite Preliminary results from experiments conducted
with NB show that the bulk concentration of Na2SO4 has only a very small impact
on the rate ie increasing the concentration of Na2SO4(aq) in the GR-SO4
suspension from 5 to 25 mM reduced the observed rate constant by a factor of 2
At Na2SO4 concentrations above 25 mM the effect leveled off and therefore
[Na2SO4] = 25 mM was chosen for this work Portions of the same GR-SO4
suspension were used for kinetic experiments over a period of two weeks No
significant aging effects eg rate constants decreasing as a function of GR age
were observed within this time frame
434 Comparison with rate constants obtained for other Fe(II) containing
mineral systems
A recent study compared the reactivity of various Fe(II) containing iron mineral
systems towards organic probe compounds representing different classes of
pollutants (Elsner et al 2004) The reductive transformation of 4-CNB was
investigated for the Fe(III) minerals goethite (α-FeOOH) lepidocrocite (γ-
FeOOH) and hematite (α-Fe2O3) as well as for the Fe(II)-Fe(III) oxide magnetite
(Fe3O4) All experiments were conducted in the presence of 1 mM dissolved Fe(II)
at pH 72 As seen from the surface area-normalised pseudo 1 order rate constants
in Figure 47 the reduction of 4-CNB by the Fe(II)-amended goethite
lepidocrocite and magnetite systems was up to 40 times faster than its reduction by
GR-SO4 The reduction rate obtained for the Fe(II)hematite system was only
slightly higher than the rate for the blank containing no iron mineral but aqueous
Fe(II) solely
84 Chapter 4
Figure 47 Surface area-normalised pseudo 1 order rate constants for the degradation of 4-CNB
by GR-SO4 (open square this work) and various Fe(II) containing mineral systems (solid circles
Elsner et al 2004) Experimental conditions applied by Elsner et al 1 mM aqueous Fe(II) pH
72 25 m2 mineral surface areaL GR-SO4 = green rust sulphate α-FeOOH = goethite Fe3O4 =
magnetite γ-FeOOH = lepidocrocite α-Fe2O3 = hematite
The experiments with GR-SO4 in our study were carried out at pH 84 whereas the
experiments comprising the other systems in Figure 47 were conducted at pH 72
As the reactivity of GR is expected to increase with pH (Lee amp Batchelor 2002b
Koch amp Hansen 1997) the lower of GR-SO4 compared to other Fe(II) systems
cannot be explained by differences in pH values GR-SO4 might just contain fewer
or less reactive surface sites than Fe(II)-amended goethite lepidocrocite and
magnetite suspensions These findings contrast those of other studies which found
higher surface area-normalised pseudo 1 order rate constants for dechlorination
reactions for GR-SO4 than for magnetite (Lee amp Batchelor 2002a amp 2002b Elsner
et al 2004) The different reactivity orders of the Fe(II)-bearing minerals found
for chlorinated aliphatics and nitro aromatics suggest that effects other than pH and
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 85
the intrinsic reduction potentials of the reacting species play a role for the
reactivity of these Fe(II)-bearing minerals
435 Depletion of reactive sites
Assuming that the platy hexagonal GR-SO4 crystals hold an average width of 1 microm
and an average particle thickness of 35 nm (Hansen amp Koch 1998) the ratio of
outer surface area to total surface area AouterAtotal ~ 131 (see Supporting
Information 73) This means that only 3 of the total surface area in GR-SO4 is
available at the external surface Thus as the interlayer sulphate in GR-SO4 is not
readily exchanged with the anionic NAC applied we conclude that 4-NPA and
other NACs only react with the Fe(II) sites at the external GR-SO4 surface In
Table 43 the actual amounts of NAC reduced by GR-SO4 during reaction is
compared with the amount of NAC which theoretically can be reduced by the
initial amount of external reactive Fe(II) sites in GR-SO4 at AouterAtot ~ 131
Table 43 The actual amounts of NAC reduced by GR-SO4 during reaction ∆[NAC]act
compared with the amount of NAC which stoichiometrically should be reduced by the initial
amount of external reactive Fe(II) sites in GR-SO4 ∆[Ar-NO2]theory (calculated as
[Fe(II)GR]0(31middot6) assuming an even distribution of Fe(II) throughout the GR-SO4 structure)
Calculated for [NAC]0 ~ 50 microM
[Fe(II)GR]0 (mM)
∆[Ar-NO2]theory(microM)
∆[4-NT]act (microM)
∆[4-CNB]act (microM)
∆[4-NPA]act (microM)
103 55 174 177 99
63 339 426 380 273
As seen in Table 43 the actual amounts of NAC reduced by GR-SO4 during
reaction are in most cases higher than the amount of NAC which should be
reduced at the given [Fe(II)GR]0 according to reaction stoichiometry This indicates
that new external reactive sites were regenerated eg the Fe(III) phases produced
peel off the GR surface exposing new Fe(II) sites or that outermost internal
86 Chapter 4
reactive sites in close vicinity to the edges are available for reaction as well Lee
and Batchelor (2000b) also found the experimentally observed reduction capacity
of GR-SO4 for chlorinated ethylenes to be 2-3 orders of magnitudes lower than the
estimated reduction capacity including all Fe(II) in GR-SO4
At low initial Fe(II)GR concentrations only a fraction of NAC was reduced within
the reaction time observed (Figure 45c) even though there was stoichiometric
excess of Fe(II)-GR present The fraction of initial Ar-NO2 reduced by GR-SO4 at
reaction termination decreased as [Fe(II)GR]0 decreased (Table 41) and was
accompanied by a change in apparent rate laws with time (compare Figures 45b amp
d) In order to explain these observations we propose that the NACs react only at
external reactive Fe(II) sites and that the regeneration of new external reactive sites
is much slower than the reduction of NAC by GR-SO4 Thus the fast reduction of
NAC taking place at the external reactive sites represents the pseudo 1 order
behaviour whereas depletion of external reactive sites and their slow regeneration
are represented by the second bent part of the (time ln [Ar-NO2]t[Ar-NO2]0)-
curves deviating from pseudo 1 order kinetics Hence at low [Fe(II)GR]0 the
regeneration of reactive sites will eventually control the overall reaction rate
Depletion of available Fe(II) was also observed during the fast reduction of
chromate by GR-CO3 when the initial chromate concentration was increased or
when the GR-CO3 suspension was respiked with chromate repeatedly (Williams amp
Scherer 2001)
436 The role of external and internal reactive sites
It is reasonable to assume that GRs hold adsorption properties similar to other
layered double hydroxides such as hydrotalcites The sorption of 246-
trinitrophenol (TNP) and 245-trichlorophenol (TCP) on chloride and carbonate
intercalated hydrotalcites (HT-Cl = Mg3Al(OH)8ClmiddotyH2O HT-CO3 =
Mg6Al2(OH)16CO3middotyH2O) has been investigated (Hermosin et al 1993 Ulibarri et
al 1995 Ulibarri et al 2001) The authors found that the adsorption of TCP on
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 87
HT-CO3 was very low and that TCP adsorbs only on the external surface sites of
HT-CO3 (Hermosin et al 1993) Furthermore is was reported that the adsorption
of TNP on HT was dramatically affected by the nature of the interlayer anion ie
the adsorption of TNP was considerably higher on HT-Cl than on HT-CO3
(Ulibarri et al 2001) For HT-Cl interlayer anion exchange of chloride with TNP
was detected by XRD analysis and an expansion of the characteristic basal d003
spacing from 79 Ǻ to 132 Ǻ confirmed the presence of TNP in the HT interlayer
(Ulibarri et al 1995) Collating the results reported for HTs with GRs it is not
likely that the divalent SO42- in GR-SO4 is exchanged with the monovalent 4-NPA
Chacirctelet et al (1996) investigated the adsorption of mono- and divalent anions
onin the outer and inner adsorption sites in HT by varying the zetapotential with
pH in the presence of various electrolytes The authors found that SO42- adsorbs on
the external HT surfaces by formation of outer-sphere complexes whereas chloride
hardly adsorbed on HT Moreover it was reported that the adsorption of sulphate
onto HT was not strongly affected by the presence of chloride while sulphate on
the contrary inhibited the adsorption of chloride on HT Studies applying
spectroscopic analyses have investigated the sorption of oxyanions at external and
internal GR-SO4 surfaces (Myneni et al 1997 Randall et al 2001) Selenate was
adsorbed only on the outer GR-SO4 surface when added after GR formation
whereas it was primarily coprecipitated into the interlayer when present during GR
formation Thus for selenate its presence during GR formation is a prerequisite of
its incorporation in the GR interlayer Selenate is readily reduced by GR-SO4 and
the rates of reduction of coprecipitated selenate were very similar to the reduction
rates of selenate adsorbed at the outer GR surface (Myneni et al 1997) This
finding suggests that the outer and inner reactive Fe(II) sites in GR-SO4 hold
similar reactivities
Results based on electron microscopy reported that the reduction of uranyl took
place primarily at the edges of hexagonal GR-SO4 particles (OrsquoLoughlin et al
2003a) In another recent study XRD characterization of the GR-SO4 crystals
88 Chapter 4
during reaction with trichloroacetate (TCA) indicated that TCA did not enter the
GR-SO4 interlayer during reaction (Chapter 5 this work) The average GR-SO4
particle thickness perpendicular to the basal plane was constant during reaction
implying that TCA reacts only at the edges and not at the basal planes Assuming
that the platy hexagonal GR-SO4 crystals hold an average width of 1 microm and an
average particle thickness of 35 nm (Hansen amp Koch 1998) the ratio of edge
surface area to outer surface area is AedgeAouter ~ 130 (see Supporting Information
73) Hence only 3 of the outer surface area in GR-SO4 is available at the edges
Once more the regeneration of new external reactive sites is strongly inferred as
the actual amounts of NAC reduced by GR-SO4 during reaction are much higher
than the amount of NAC which may be reduced by the reactive edge sites present
initially Assuming that the NACs react at the edges only and if employing the
AedgeAouter in the estimation of the rate constants the surface area-normalised
pseudo 1 order rate constants for GR-SO4 would be 30 times higher than the rate
constants depicted in Figure 47 Thus the reactivity of GR-SO4 normalised to its
reactive surface area is higher than the reactivity normalised to its outer surface
area determined by the BET method (N2 adsorption)
The reduction of chromate has been examined in the presence of all the common
GR forms (Bond amp Fendorf 2003 Loyaux-Lawniczak et al 1999 Loyaux-
Lawniczak et al 2000 Williams amp Scherer 2001) The results reported by Bond
amp Fendorf (2003) confirm that not only the surface area of GR but also the
interlayer spacing (interlayer anion size) and interlayer anion charge play an
important role for the reaction rate Hence it follows that coordination (size) and
charge of the oxidant determine its access to the internal sites in GRs
The results obtained for all 4 NACs support what has been reported for nitrate and
TCA At [Fe(II)GR]0 = 2-10 mM and [NO3-]0 = 143 mM pseudo 1 order rate
constants for the reduction of nitrate by GR-SO4 were 158middot10-7 s-1middotm-2middotL (Hansen
et al 2001) This reaction rate increased 40 times by adding barium nitrate instead
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 89
of sodium nitrate thereby precipitating the interlayer sulphate as barium sulphate
and enhancing access to the interlayer Though barium addition changes the GR-
SO4 system dramatically it indicates the importance of interlayer anion exchange
(Hansen amp Koch 1998) The rate constant reported for nitrate (no barium added) is
100-1000 smaller than the rate constants obtained for the NACs in this work
Moreover the reaction kinetics for nitrate did not deviate from pseudo 1 order
kinetics At [Fe(II)GR]0 = 025-104 mM and [TCA]0 = 50 microM-1 mM pseudo 1
order rate constants for the reduction of TCA by GR-CO3 or GR-SO4 were 65middot10-7
s-1middotm-2middotL (Chapter 5 this work) The rate constant for TCA is 10-1000 smaller than
the rate constants for the NACs and the reaction kinetics for TCA did not deviate
from pseudo 1 order kinetics This suggests that the overall reductive
transformation of slowly reacting oxidants such as nitrate and TCA is not
controlled by the rate of regeneration of external Fe(II) reactive sites Altogether
the results reported for selenate chromate and nitrate clearly demonstrate that
these anionic oxidants react primarily with external reactive sites in GR-SO4 Only
under certain conditions ie adding the oxidant prior to GR-SO4 formation or
extracting the interlayer sulphate through precipitation with barium outside the
GR-SO4 particles do the oxidants have access to the interlayer Our findings
suggest that both the neutral and anionic nitro aromatic probe compounds applied
here also react exclusively with the external reactive sites in GR-SO4 Supposedly
the neutral and monovalent charge states of the NACs hinder their access to the
GR-SO4 interlayer A divalent anionic nitro aromatic probe compound might
exchange with the interlayer sulphate more readily and gain access to the inner
Fe(II) reactive sites in GR-SO4 only divalent anionic NACs are not commercially
available
44 Conclusions
This work demonstrates that NACs are completely reduced to their corresponding
anilines by GR-SO4 The surface area-normalised pseudo 1 order rate constants
obtained for the reduction of the neutral and anionic NACs by GR-SO4 under
90 Chapter 4
various experimental conditions did not differ significantly from each other despite
their different charges Neither mass transfer control nor surface saturation kinetics
could account for the similarity of the pseudo 1 order rate constants obtained for
the NACs These observations suggest that the anionic NACs do not have an
enhanced access to inner or outer Fe(II)-GR reactive sites as compared to the
neutral NACs Based on our estimations of the molecular sizes of the NACs we
propose that the charge and not the size of the NACs controls their access to the
internal reactive sites in GRs Hence the reaction between NAC and GR-SO4 takes
place primarily at the external reactive Fe(II) sites This work further demonstrated
that the reduction of the NACs by GR-SO4 only followed pseudo 1 order kinetics
throughout the whole reaction at high initial Fe(II)GR concentrations At low initial
Fe(II)GR concentrations the NACs were not reduced completely within the reaction
time observed though according to reaction stoichiometry the total Fe(II)-GR
present should be sufficient to reduce the whole amount of NAC This means that
at some point during the reaction the external reactive Fe(II) sites were depleted
and the regeneration of new external reactive sites was much slower than the
reduction of the NACs by GR-SO4 The reduction of 4-CNB by GR-SO4 reported
here was 10-100 times slower than its reduction by other Fe(II)-Fe(III) systems
such as goethite lepidocrocite and magnetite suspensions amended with Fe(II)
(Elsner et al 2004)
The results obtained in this work infer that under natural geochemical conditions
where GR-SO4 presumably forms in low concentrations the rate of regeneration of
external Fe(II) reactive sites may control the overall reductive transformation of
fast reacting pollutants by GR-SO4 Thus not only abiotic processes such as
interchanging redox conditions created by water level alterations but also the direct
microbial formation through Fe(III)-reducing bacteria may govern the formation of
GRs and the renewal of external Fe(II) reactive sites in GRs This holds both for
natural systems like iron-rich suboxic soils and sediments as well as engineered
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 91
systems like permeable reactive barriers of zero-valent iron implemented for on-
site remediation of organic and inorganic contaminants
Acknowledgments
We would like to thank Henrik T Andersen for performing the NB kinetic experiments and
Hanne Nancke-Krogh for technical assistance in the laboratory
References
Al-Agha MR Burley SD Curtis CD Esson J (1995) Complex cementation textures and authigenic mineral assemblages in recent concretions from the Lincolnshire Wash (east coast UK) driven by Fe(0) to Fe(II) oxidation Journal of the Geological Society 152 157-171 Bernal JD Dasgupta DR Mackay AL (1959) The oxides and hydroxides of iron and their structural inter-relationships Clay Minerals Bulletin 4 15-30 Bond DL Fendorf S (2003) Kinetics and structural constraints of chromate reduction of green rusts Environmental Science and Technology 37 2750-2757 Brunauer S Emmett PH Teller E (1938) Adsorption of gases in multimolecular layers Journal of American Chemical Society 60 309-319 Charlet L Silvester E Liger E (1998) N-compound reduction and actinide immobilisation in surficial fluids by Fe(II) The surface FeIIIFeIIOH0 species as major reductant Chemical Geology 151 85-93 Chacirctelet L Bottero JY Yvon J Bouchelaghem A (1996) Competition between monovalent and divalent anions for calcined and uncalcined hydrotalcite anion exchange and adsorption sites Colloids and Surfaces A Physicochemical and Engineering Aspects 111 167-175 Chaudhuri SK Lack JG Coates JD (2001) Biogenic magnetite formation through anaerobic biooxidation of Fe(II) Applied and Environmental Microbiology 67 2844-2848 Cui D Spahiu K (2002) The reduction of U(VI) on corroded iron under anoxic conditions Radiochemica Acta 90 623-628 Elsner M Haderlein SB Schwarzenbach RP (2004) Reactivity of Fe(II)-bearing minerals towards reductive transformation of organic contaminants Environmental Science and Technology 38 799-807 Erbs M Hansen HCB Olsen CE (1999) Reductive dechlorination of carbon tetrachloride using iron(II)iron(III)-hydroxide-sulphate (green rust) Environmental Science and Technology 33 307-311 Fadrus H Maly J (1975) Suppression of iron(III) interference in the determination of iron(II) in water by the 110-phenanthroline method The Analyst 100 549-554 Fredrickson JK Zachara JM Kennedy DW Dong H Onstott TC Hinman NW Li S
92 Chapter 4 (1998) Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium Geochimica et Cosmochimica Acta 62 3239-3257 Geacutenin JMR Bourrieacute G Trolard F Abdelmoula M Jaffrezic A Refait Ph Maitre V Humbert B Herbillon A (1998) Thermodynamic equilibria in aqueous suspensions of synthetic and natural Fe(II)-Fe(III) green rusts Occurrences of the mineral in hydromorphic soils Environmental Science and Technology 32 1058-1068
Geacutenin JMR Olowe AA Benbouzid-Rollet ND Prieur D Confente M Resiak B (1991) The simultaneous presence of green rust 2 and sulfate reducing bacteria in the corrosion of steel sheet piles in a harbour area Hyperfine Interactions 69 875-878 Glasauer S Weidler PG Langley S Beveridge TJ (2003) Controls on Fe reduction and mineral formation by a subsurface bacterium Geochimica et Cosmochimica Acta 67 1277- 1288 Hansen HCB (1989) Composition stabilization and light absorption of Fe(II)Fe(III) hydroxycarbonate (green rust) Clay Minerals 24 663-669 Hansen HCB Borggaard OK Soslashrensen J (1994) Evaluation of the free energy of formation of iron(II)iron(III)-hydroxidesulphate (Green Rust) and its reduction of nitrite Geochimica et Cosmochimica Acta 58 2599-2608 Hansen HCB Guldberg S Erbs M Koch CB (2001) Kinetics of nitrate reduction by green rusts ndash effects of interlayer anion and Fe(II)Fe(III) ratio Applied Clay Science 18 81-91 Hansen HCB Koch CB (1998) Reduction of nitrate to ammonium by sulphate green rust Activation energy and interlayer reaction mechanism Clay Minerals 33 87-101 Hansen HCB Koch CB Nancke-Krogh H Borggaard OK Soerensen J (1996) Abiotic nitrate reduction to ammonium Key role of green rust Environmental Science and Technology 30 2053-2056 Hansen HCB Poulsen IF (1999) Interaction of synthetic sulphate green rust with phosphate and the crystallization of vivianite Clays and Clay Minerals 47 312-318 Hansen HCB Taylor RM (1991) The use of glycerol intercalates in the exchange of CO3
2- with SO4
2- NO3- or Cl- in pyroaurite-type compounds Clay Minerals 26 311-327
Heasman DM Sherman DM Ragnarsdottir KV (2003) The reduction of aqueous Au3+ by sulfide minerals and green rust phases American Mineralogist 88 725-738 Hermosin MC Pavlovic I Ulibarri MA Cornejo J (1993) Trichlorophenol adsorption on layered double hydroxide a potential sorbent Journal of Environmental Science and Health A28 1875-1888 Hofstetter TB Heijmann CG Haderlein SB Holliger C Schwarzenbach RP (1999) Complete reduction of TNT and other (poly)nitroaromatic compounds under iron-reducing subsurface conditions Environmental Science and Technology 33 1479-1487 Hofstetter TB Schwarzenbach RP Haderlein SB (2003) Reactivity of Fe(II) species associated with clay minerals Environmental Science and Technology 37 519-528
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 93
Klausen J Troumlber SP Haderlein SB Schwarzenbach RP (1995) Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions Environmental Science and Technology 29 2396-2404 Koch CB Hansen HCB (1997) Reduction of nitrate to ammonium by sulphate green rust Advances in GeoEcology 30 373-393 Kukkadapu RK Zachara JM Smith SC Fredrickson JK Liu C (2001) Dissimilatory bacterial reduction of Al-substituted goethite in subsurface sediments Geochimica et Cosmochimica Acta 65 2913-2924 Lee W Batchelor B (2002a) Abiotic reductive dechlorination of chlorinated ethylenes by iron- bearing soil minerals 1 Pyrite and magnetite Environmental Science and Technology 36 5147- 5154 Lee W Batchelor B (2002b) Abiotic reductive dechlorination of chlorinated ethylenes by iron-bearing soil minerals 2 Green rust Environmental Science and Technology 36 5348- 5354 Liger E Charlet L Van Cappellen P (1999) Surface catalysis of uranium (VI) reduction by iron(II) Geochimica et Cosmochimica Acta 63 2939-2955 Loyaux-Lawniczak S Refait Ph Ehrhardt J Lecomte P Geacutenin JMR (2000) Trapping of Cr by formation of ferrihydrite during the reduction of chromate ions by Fe(II)-Fe(III) hydroxysalt green rusts Environmental Science and Technology 34 438-443 Loyaux-Lawniczak S Refait Ph Lecomte P Ehrhardt J Geacutenin JMR (1999) The reduction of chromate ions by Fe(II) layered hydroxides Hydrology and Earth System Sciences 3 593-599 Mendiboure A Schoumlllhorn A (1986) Formation and anion exchange reactions of layered transition metal hydroxides [Ni1-xMx](OH)2(CO3)x2(H2O)z (M = Fe Co) Revue de Chimie Mineacuterale 23 819-827 Miyata S (1983) Anion-exchange properties of hydrotalcite-like compounds Clays and Clay Minerals 31 305-311 Myneni SCB Tokunaga TK Brown Jr GE (1997) Abiotic selenium redox transformations in the presence of Fe(IIIII) oxides Science 278 1106-1109 Nielsen A (1976) Hvid groslashn og sort rust Beskrivelse af en korrosionsskade paring et svoslashmmebassin Nordisk Betong 2 21-24 OLoughlin EJ Burris DR (2004) Reduction of halogenated ethanes by green rust Environmental Toxicology and Chemistry 23 41-48 OLoughlin EJ Kelly SD Cook RE Csencsits R Kemner KM (2003a) Reduction of uranium(VI) by mixed iron(II)iron(III) hydroxide (green rust) Formation of UO2 nanoparticles Environmental Science and Technology 37 721-727
94 Chapter 4 OLoughlin EJ Kelly SD Kemner KM Csencsits R Cook RE (2003b) Reduction of AgI AuIII CuII and HgII by FeIIFeIII hydroxysulfate green rust Chemosphere 53 437-446 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002) Iron (IIIII) hydroxycarbonate green rust formation and stabilization from lepidocrocite bioreduction Environmental Science and Technology 36 16-20 Parmar N Gorby YA Beveridge TJ Ferris FG (2001) Formation of green rust and immobilization of nickel in response to bacterial reduction of hydrous ferric oxide Geomicrobiology Journal 18 375-385 Pepper SE Bunker DJ Bryan ND Livens FR Charnock JM Pattrick RAD Collison D (2003) Treatment of radioactive wastes An X-ray absorption spectroscopy study of the reaction of technetium with green rust Journal of Colloid and Interface Science 268 408- 412 Randall SR Sherman DM Ragnarsdottir KV (2001) Sorption of As(V) on green rust (Fe4(II)Fe2(III)(OH)12SO4
3H2O) and lepidocrocite (γ-FeOOH) Surface complexes from EXAFS spectroscopy Geochimica et Cosmochimica Acta 65 1015-1023 Roh Y Lee SY Elless MP (2000) Characterization of corrosion products in the permeable reactive barriers Environmental Geology 40 184-194 Schultz CA Grundl TJ (2000) pH dependence on reduction rate of 4-Cl-nitrobenzene by Fe(II)montmorillonite systems Environmental Science and Technology 34 3641-3648 Simon L Francois M Refait Ph Renaudin G Lelaurain M Geacutenin JMR (2003) Structure of the Fe(II-III)-layered double hydroxysulphate green rust two from Rietveld analysis Solid State Sciences 5 327-334 Tamaura Y (1986) Ni(II)-bearing green rust II and its spontaneous transformation into Ni(II)- bearing ferrites Bulletin of the Chemical Society of Japan 59 1829-1832 Tamaura Y (1985) ZnII-bearing green rust II and its spontaneous transformation into ZnII- bearing ferrite in aqueous solution Bulletin of the Chemical Society of Japan 58 2951-2954 Trolard F Abdelmoula M Bourrieacute G Humbert B Geacutenin JMR (1996) Mise en eacutevidence dun constituant de type rouilles vertes dans les sols hydromorphes Proposition de lexistence dun nouveau mineacuteral la fougeacuterite Geacuteosciences de surface Comptes Rendus de LrsquoAcademie des Sciences 323 1015-1022 Tuovinen OH Button KS Vuorinen A Carlson L Mair DM Yut LA (1980) Bacterial chemical and mineralogical characteristics of tubercles in distribution pipelines Journal of the American Water Works Association 72 626-635 Ulibarri MA Pavlovic I Barriga C Hermosin MC Cornejo J (2001) Adsorption of anionic species on hydrotalcite-like compounds effect of interlayer anion and crystallinity Applied Clay Science 18 17-27 Ulibarri MA Pavlovic I Hermosin MC Cornejo J (1995) Hydrotalcite-like compounds as potential sorbents of phenols from water Applied Clay Science 10 131-145
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 95
Williams AGB Scherer MM (2001) Kinetics of chromate reduction by carbonate green rust Environmental Science and Technology 35 3488-3494
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 97
5 Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems
Abstract
Trichloroacetate (TCA) is a widespread environmental contaminant with proven
phytotoxicity and suspected human carcinogenicity In order to assess the global
cycling of TCA and to predict its fate in subsurface environments information
regarding the reactivity and product distribution of TCA degradation is needed
Due to the high oxidation state of TCA conditions for oxidative transformation
pathways in soils and groundwater are unfavorable However in suboxic soils and
sediments Fe(II)-bearing minerals are potential reactants for reductive
dehalogenation reactions of TCA as has been demonstrated for other halogenated
contaminants We examined the reactivity of various Fe(II)-Fe(III) mineral
systems towards TCA and dichloroacetate (DCA) its expected transformation
product in laboratory batch experiments imitating natural conditions ie low
initial Fe(II) Fe(III) and TCADCA concentrations and no artificial buffer The
Fe(II)-Fe(III)-systems investigated included sulfate green rust (GR-SO4) carbonate
green rust (GR-CO3) magnetite Fe(II)goethite and Fe(II)lepidocrocite
Trichloroacetate was readily reduced to DCA by all Fe(II)-bearing minerals The
reactions generally followed pseudo 1 order kinetics with respect to TCA The
surface area-normalised pseudo 1 order rate constants obtained (035ndash76middot10-5 min-
1middotm-2middotL at [Fe(II)]0 = 020ndash122 mM [TCA]0 = 15ndash1000 microM and pH 70ndash87)
showed no striking differences regarding product distribution and surface area-
normalised reaction rate constants between the Fe(II)-Fe(III)-systems The
stoichiometrically formed DCA was not further reduced to monochloroacetate
(MCA) or acetate in any of the systems within the time frame in our experiments
To our knowledge this is the first published report on abiotic transformation of
TCA by Fe(II)-bearing minerals Our results imply that processes involving
reactive Fe(II)-bearing minerals may play a significant role in controlling the fate
98 Chapter 5
of TCA in natural subsurface environments and that DCA found in the subsurface
may be formed by such processes
51 Introduction
Trichloroacetic acid (TCA) has been applied as a herbicide for many years until its
use was banned in the late 1980acutes (Berg et al 2000) Today TCA is mainly used
as an etching agent in the metal industry as a swelling solvent in the plastic
production and as a bleaching agent in the paper and pulp manufacture (Muumlller et
al 1996) Other anthropogenic sources include formation of TCA as a result of the
chlorine based disinfecting process used in drinking water treatment and the
atmospheric photooxidation of chlorinated solvents including tetrachloroethene
and 111-trichloroethane (McCulloch 2002) Only very little information is
available on the TCA production volumes and even less is known about the
amount of TCA released into the environment as a result of its industrial
applications Due to its low volatility and high aqueous solubility TCA is easily
washed out of the atmosphere into the aquatic and terrestrial biospheres As TCA
is found in almost every ecosystem around the globe including non-urban and
non-industrial sites the relative contributions from anthropogenic and natural
sources are currently being debated (McCulloch 2002 Ahlers et al 2003)
Trichloroacetic acid is omnipresent in soils and the concentrations reported are
very variable ranging from lt005 microgkg to 380 microgkg (Euro Chlor 2001
McCulloch 2002 Ahlers et al 2003) Both abiotic and enzymatically catalyzed
formation of TCA from humic acids have been demonstrated in laboratory studies
(Haiber et al 1996 Hoekstra et al 1999b Fahimi et al 2003) Furthermore the
in situ natural formation of TCA from anthropogenic or natural tetrachloroethene
or 111-trichloroethane in biota has been suggested (Hoekstra et al 1999a
McCulloch 2002) Such natural sources may explain part of the TCA
concentrations found in soils but their environmental significance is still unknown
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 99
On account of its phytotoxicity suspected human carcinogenicity and widespread
occurrence TCA is of considerable environmental concern especially in the
terrestrial compartment The TCA concentrations found in soil air and water in
pre-industrial times were far below the present ones (Jordan amp Frank 1999 Ahlers
et al 2003) Based on the current TCA concentrations detected in soils the
European Commission proposed risk reduction measures concerning
tetrachloroethene - a precursor of TCA - to be taken immediately (Ahlers et al
2003 and references therein) Occurrences of monochloroacetic acid (MCA) and
dichloroacetic acid (DCA) reported include surface waters marine waters
precipitation ice (glaciers) and air (Reimann et al 1996 Berg et al 2000 Scott et
al 2000 Scott et al 2002) Based on the concentrations reported for the aquatic
environments it is reasonable to assume that MCA and DCA are omnipresent in
soils as well Sources of MCA and DCA include production in the chemical
industry photooxidation of chlorinated aliphatics in the atmosphere and reductive
transformation of TCA (Reimann et al 1996 Ahlers et al 2003 and references
therein) MCA and DCA are also toxins and suspected human carcinogens (Kuumlhn
amp Pattard 1990) hence not only TCA but also its daughter compounds are
pollutants of environmental concern
In subsurface environments TCA may be removed by sorption seepage chemical
transformation microbial degradation and plant uptake followed by metabolic
degradation andor physical removal at harvest (Foy 1975) There is little or no
evidence of abiotic transformations of TCA in the literature Only one recent study
demonstrated the reductive dechlorination of TCA to MCA by Fe(0) (Hozalski et
al 2001) It has been reported that the degradation of TCA in soil is slow and
mainly mediated by microorganisms but only little is known about the bacteria and
processes involved (Lignell et al 1984) Biodegradation of TCA has been found at
both oxic and anoxic conditions An aerobic microorganism capable of growing on
TCA as the sole carbon and energy source has been characterised (Yu amp Welander
1995) Moreover anaerobic bacteria coupling co-metabolic growth to reductive
100 Chapter 5
dechlorination of TCA have been isolated (Weightman et al 1992 De Wever et
al 2000) However more information regarding the abiotic and biotic
transformation of TCA is needed in order to assess the fate and transport of TCA in
natural subsurface environments
It is well-known that Fe(II) present in minerals or associated with mineral surfaces
is a much stronger reductant than Fe(II) in solution The enhanced reactivity of a
structural or surface-bound Fe(II) center can be rationalized by the increased
electron density donated by hydroxyl ligands and a stabilization of the Fe(III)
oxidation state by the hydroxyl ligands (Luther 1990) Fe(II)-bearing minerals
including layered Fe(II)-Fe(III) hydroxides (green rusts) magnetite (Fe3O4)
siderite (FeCO3) Fe(II) sulfides as well as Fe(II)-carrying Fe(III) oxides and clay
minerals have also been shown to reduce a range of organic and inorganic
contaminants such as nitro aromatic compounds chlorinated aliphatics chromate
uranyl pertechnetate nitrate monochloramine and carbamate pesticides (Chapter
4 this work Klausen et al 1995 Cui amp Eriksen 1996 Erbs et al 1999 Liger et
al 1999 Loyaux-Lawniczak et al 1999 Amonette et al 2000 Hansen et al
2001 Pecher et al 2002 Vikesland amp Valentine 2002 Hofstetter et al 2003
OrsquoLoughlin and Burris 2003 OrsquoLoughlin et al 2003a amp 2003b Strathmann amp
Stone 2003 Elsner et al 2004a) Laboratory and field studies showed that even in
geochemically highly heterogeneous anoxic aquifer sediments Fe(II) adsorbed to
Fe(III) (hydr)oxide surfaces was the dominant reductant of nitroaromatic and
halogenated contaminants (Ruumlgge et al 1998 Hofstetter et al 1999 Kenneke amp
Weber 2003) Only little is known about the nature of the Fe(II) species associated
with Fe(III) oxide surfaces but reactive hydroxylated Fe(II)-Fe(III)-hydroxo
surface complexes associated with hematite and magnetite above pH 65 have been
proposed (Charlet et al 1998aampb Liger et al 1999) Due to the presence of
structural Fe(II) within the mineral lattice the reactivity of Fe(II) associated with
mixed valent Fe(II)-Fe(III) minerals such as green rusts magnetite and reduced
ferruginous clay minerals may hold another reactivity than Fe(II) associated with
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 101
pure Fe(III) oxides However Fe(II) adsorbed on Fe(III) oxides such as goethite
hematite and lepidocrocite may also hold different reactivities as the Fe(III) oxides
contain different crystal and surface structures
Since chlorinated ethanes and ethenes such as hexachloroethane 111-
trichloroethane tetrachloroethene and trichloroethene are susceptible to chemical
reduction by a range of Fe(II)-bearing minerals including magnetite GR-SO4
Fe(II) sulfides and Fe(II)-carrying Fe(III) oxides (Butler amp Hayes 1998 amp 1999
Hwang amp Batchelor 2000 Gander et al 2002 Lee amp Batchelor 2002aampb Elsner
et al 2004a) we hypothesized that TCA may be transformed by Fe(II)-bearing
minerals as well The main goals of this work were to study such reactions and
establish product distribution and surface area-normalised reaction rates for the
reductive dechlorination of TCA by Fe(II)-Fe(III) mineral systems common in
nature
5 2 Materials and methods
No synthetic buffers were applied and iron concentrations were kept low The
calcareous systems were pH-controlled at 76 through a natural buffer system
(CaCO3(s) + 995 N205 CO2(g)) All handling and sampling of solutions and
suspensions were carried out under strict anoxic conditions Goethite (acicular
particles with size 01 times 06 microm specific surface area 16 m2g) and lepidocrocite
(acicular particles with size 005 times 03 microm specific surface area 18 m2g) were
purchased as fine powders from Bayer (Bayferrox 910 and 943) Calcite (grain size
170-350 microm Pluumlss-Staufer AG) was used as a buffer or as a Fe(III)-oxide-bearing
mineral In order to simulate natural conditions the iron minerals were applied as
coatings on calcite particles (model system for calcareous soils) in some
experiments Trichloroacetic acid dichloroacetic acid and monochloroacetic acid
were pa quality (Fluka)
102 Chapter 5
521 Synthesis of GRs and magnetite
GR-CO3 was synthesized by controlled air oxidation of an FeCl2 solution at a
constant pH of 700 (titrated with 1 M Na2CO3) according to the procedure given
by Hansen amp Koch (1997) 05 M aqueous stock solutions of FeCl2 were prepared
in 100 mL glass flasks by reacting 65 mmol of iron powder (particle size 10 microm
Merck) with 100 mL deoxygenated 10 M HCl The solutions were magnetically
stirred and heated (~80degC) during reaction until the H2(g) production had ceased (ge
2 hours) The FeCl2 solutions were stored in the dark under a small Ar
overpressure at 5degC The GR-CO3 suspensions were washed with deoxygenated
deionised water (DIW) separated on a folding filter (medium filtration rate cotton
linterhigh alpha pulp Schleicher amp Schuell) and redispersed in deoxygenated
DIW Washing separation and redispersion of the GR-CO3 suspension were
conducted in an anoxic glove box (92 N28 H2 Coy Laboratory Products Inc)
All suspensions and solutions were deoxygenated by Ar-purging (999998 Ar
Carbagas) Magnetite was synthesized by further aerial oxidation of GR-CO3 at pH
700 until consumption of 1 M Na2CO3 ceased GR-SO4 was synthesized by
controlled air oxidation of an FeSO4 solution at a constant pH of 70 according to
the procedure given by Koch amp Hansen (1997) The GR-SO4 suspension was
washed with deoxygenated DIW separated on a glass filter funnel (pore size 4
Duran) and redispersed in deoxygenated DIW Washing separation and
redispersion of the GR-SO4 suspension were conducted in an anoxic glove bag
(999995 Ar Aldrich)
522 Preparation of iron oxide coatings
Two grams of goethite (goe) or lepidocrocite (lep) and 100 g calcite were
combined with 200 mL DIW in a 500 mL polyethylene flask The suspension was
gently agitated on a reciprocating shaker for 24 h and left to stand for another 24 h
Excess Fe(III) oxides and salts were removed from the coated material by repeated
decantation and washing with DIW in polyethylene flasks until clear runoff
Finally the coatings were collected on folding filters and air dried The amount of
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 103
goethite and lepidocrocite coated onto calcite after washing and drying was
quantified to 10-11 mg Fe(III)g calcite
523 Mineral characterisation
The identity and purity of the GR-CO3 GR-SO4 and magnetite suspensions were
examined by means of X-ray diffraction (XRD) The XRD analyses were
performed on a Scintag XDS 2000 using Co Kα radiation (45 kV 40 mA) or a
Siemens D5000 XRD applying Co Kα radiation (40 kV 40 mA) Glycerol smears
made according to Hansen (1989) were scanned between 6 and 80 deg2θ with a scan
speed of 1 deg2θmin The specific surface area (SSA) of calcite was determined by
the BET multi-point method using N2 adsorption (Brunauer et al 1938) Powder
samples were filled into sample burettes in the glove box and the generously
lubricated stopcocks closed Samples and burettes were evacuated prior to
connecting them to the BET-instrument (Sorptomatic 1990 Fisons)
524 Kinetic experiments
All reactions were carried out in 25-100 mL serum vials sealed with stoppers
(Viton or Teflon coated rubber) and aluminum crimp caps Kinetic experiments
were conducted with GR-SO4 GR-CO3 magnetite Fe(II)goethite and
Fe(II)lepidocrocite at room temperature In most cases pH was controlled through
the carbonate-bicarbonate buffer system by adding calcite to suspensions
containing the iron minerals solely or by adding the iron minerals as coatings on
calcite Furthermore the calcite containing suspensions were deoxygenated with
05 CO2995 N2(g) thereby attaining an initial pH of 76-77 The GR-CO3 and
magnetite suspensions were deoxygenated with 100 N2(g) and no additional pH
buffer was added The goethite and lepidocrocite suspensions were amended with
300-1000 microM FeCl2(aq) and equilibrated gt 20 h prior to TCADCA addition See
Table 51 for more details on the experimental conditions To start the reaction 50
microM - 1 mM TCA or DCA was added to the mineral suspensions from aqueous
anoxic stock solutions The reaction vials were agitated gently on a roller apparatus
104 Chapter 5
or a shaking table (35 rpm) in order to minimize abrasion of the iron oxide mineral
coatings At appropriate time intervals suspension samples were withdrawn using
Ar(g)- 100 N2(g)- or 995 N205 CO2(g)-flushed sterile disposable syringes
and hypodermic needles The suspension samples were filtered (02 microm Teflon)
and collected for quantification of chloride and the chlorinated acetic acids The
samples were stored at -20degC and analysed without further treatment
525 Analytical methods
Total and aqueous Fe(II) were determined using a modified phenanthroline method
(Fadrus and Maly 1975) For determining [Fe(II)aq] and [Fe(II)total] 1 mL filtered
(02 microm Teflon) and 1 mL unfiltered mineral suspension were added to 18 mL 01
M HCl respectively and allowed to dissolve for 30 min From these acid digests
01 mL was added to 05 mL Fe(II)-phenanthroline-buffer-reagent and 19 mL
DIW added up Estimates of the structural or adsorbed Fe(II) in the Fe(II)-Fe(III)
mineral systems were estimated as the difference [Fe(II)solid] = [Fe(II)total] -
[Fe(II)aq] The total amount of Fe(III) coated on calcite was determined by atomic
absorption spectroscopy following dissolution in 6 M HCl(aq) for 24 h At low
initial TCA concentrations (le 50 microM) the chlorinated acetic acids were quantified
by means of a modified ion interaction (or paired-ion) chromatographic method
(Sarzanini et al 1999) Separation was performed on a LiChrospher 100 RP-18 (5
microm 125 times 4 mm ID) reversed-phase column coupled with a LiChroCART 100 RP-
18 (4 times 4 mm ID) precolumn Analytical conditions were isocratic and the eluent
consisted of 50 aqueous solution of 35 mM cetyltrimethylammonium chloride
(pH 50) and 50 CH3CN The injection volume was 20 microL and the flow-rate 10
mLmin HPLC analyses of the chloroacetates were performed using a Gynkotek
Pump M480 Gynkotek Gina 50 auto sampler and a diode array UV detector (340s
Gynkotek) UV-VIS detection was carried out at 200 nm At higher initial TCA
concentrations the chlorinated acetic acids were quantified by a modified HPLC
method (Husain et al 1992) Separation was performed on a ChromSphere C-18
(10 microm 250 times 46 mm ID) reversed-phase column Analytical conditions were
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 105
isocratic and the eluent consisted of 015 M (NH4)2SO4(aq) pH 55 The injection
volume was 20 microL and the flow-rate 10 mLmin HPLC analyses were performed
using a Series 10 Liquid Chromatographic Pump (Perkin-Elmer) and a SPD-10 A
VP UV-VIS detector (Shimadzu) UV-VIS detection was carried out at 210 nm
Chloride was determined in the GR-SO4 kinetic experiments using a flow injection
system with spectrophotometric detection (Cheregi amp Danet 1997)
53 Results and discussion
531 Product formation and reaction kinetics
Trichloroacetate was readily reduced to DCA by all the Fe(II)-bearing minerals
examined Only DCA was detected within the reaction time in all the Fe(II)-Fe(III)
mineral systems Experiments conducted with the various Fe(II)-Fe(III) mineral
systems and DCA confirmed that no significant reduction of DCA took place (data
not shown) Hence it is reasonable to assume that the further hydrogenolysis of
DCA to MCA is too slow to be detected within the experimental time frame here
The mass balance of TCA and DCA was almost complete in all suspensions ruling
out any alternative reaction pathways to reductive dechlorination Decarboxylation
of TCA producing chloroform and carbon dioxide requires high temperatures and
is therefore assumed not to take place at the experimental conditions applied here
(Atkins et al 1984) Based on these results we propose that the reductive
dechlorination of TCA by Fe(II)-bearing minerals proceeds via hydrogenolysis
(replacement of halogen by hydrogen) as reported for the transformation of TCA
by zero-valent iron (Hozalski et al 2001) Thus in order to reduce TCA to DCA
2 electrons corresponding to 2 Fe(II) are needed (Figure 51)
106 Chapter 5
Cl3CC
O
O- Cl2HCC
O
O-
2e- H+ Cl-
TCA DCA Figure 51 Proposed reductive transformation pathway of TCA
In the Fe(II)goe and Fe(II)lep systems we detected no TCA transformation in the
absence of either aqueous Fe(II) or pure or calcite-associated goethite and
lepidocrocite These results strongly indicate that reactive Fe(II) species associated
with the goethite and lepidocrocite surfaces are the reductants for TCA in these
systems The Fe(III) phases forming in the mineral suspensions were not
characterised and therefore the reaction stoichiometry cannot be assessed
At initial Fe(II) concentrations in large excess of initial TCA concentration we
found a pseudo 1 order rate law for the degradation of TCA by Fe(II)
[ ] [ ] [ b a TCAFe(II) TCA
sdotsdot=minus kdt
d ]
where a = 1 b = 1 and the observed pseudo 1 order rate constant kobs = k middot [Fe(II)]
At all [Fe(II)]0[TCA]0 ratios studied (6-738) TCA was transformed almost
quantitatively into DCA and the reaction kinetics followed pseudo 1 order kinetics
with respect to TCA (Figure 52) The observed pseudo 1 order rate constants for
the transformation of TCA by the various Fe(II)-Fe(III) mineral systems were
calculated as initial rates (ie max first two half-lives) from linear fits of (time ln
[TCA]t[TCA]0)-plots (Table 51) The amount of chloride produced during
reaction with GR-SO4 was always equivalent to the amount of TCA transformed
into DCA (Figure 52c) This also indicates that no significant further reduction of
DCA took place in GR-SO4 suspensions
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 107
Figure 52 Time course of TCA consumption and DCA and chloride production for a)
Fe(II)Goe ([Fe(II)tot]0 = 095 mM) b) Fe(II)Lep ([Fe(II)tot]0 = 091 mM) c) GR-SO4
([Fe(II)GR]0 = 962 mM) d) GR-CO3 ([Fe(II)tot]0 = 633 mM) and e) Magnetite ([Fe(II)tot]0 = 350
mM) Solid lines represent 1 order kinetic fits whereas symbols and dotted lines represent actual
data = TCA = DCA = Clndash
T
able
51
Exp
erim
enta
l con
ditio
ns a
nd p
seud
o 1
ord
er ra
te c
onst
ants
for t
he re
duct
ive
trans
form
atio
n of
TC
A b
y va
rious
Fe(
II)-
Fe(I
II) c
onta
inin
g m
iner
al sy
stem
s
Syst
em
Susp
ensi
on a
ge
(d)
[Fe(
II)] s
olid
a (m
M)
[Fe(
II)] a
q b
(mM
) [T
CA
] 0 (micro
M)
pHin
itcpH
endd
k obs
e (min
-1)
Surf
ace
area
(m
2 L)
k obs
f
(min
-1m
-2middotL
)
Fe(I
I)aq
1
0
030
434
nd
76
gn
dn
dn
d
Fe(I
I)G
oe
1
002
024
429
nd
78
g1
021
0-47
1 i
143
10-5
Fe(I
I)G
oe
1
013
094
543
77
70
225
10-4
71
i3
161
0-5
Fe(I
I)G
oe
coat
ing
1
023
40
066
484
765
80
g6
401
0-454
0 j
119
10-5
Fe(I
I)G
oe
coat
ing
1
0
150
8048
6n
d7
6 g12
43
10-4
540
j2
301
0-5
Fe(I
I)L
ep
1
0
020
2315
7n
d7
8 g0
751
0-48
0 i
094
10-5
Fe(I
I)L
ep
coat
ing
1
016
30
137
470
765
80
g2
821
0-454
0 j
052
10-5
Fe(I
I)L
ep
coat
ing
1
0
100
8141
7n
d7
7 g8
311
0-454
0 j
154
10-5
Fe3O
41
3
380
1251
38
107
8g
830
10-4
16
k5
311
0-4
Fe3O
477
112
56
556
70
70
153
10-4
52
k2
951
0-5
GR
-CO
31
5
940
3950
37
658
4g
761
10-4
419
l1
821
0-5
GR
-CO
32
7
60
147
88
568
2940
81
0-453
6 l
761
10-5
GR
-CO
332
73
003
563
85
80
490
10-4
515
l0
951
0-5
GR
-CO
314
2
3
530
005
629
nd
87 g
513
10-4
249
l2
061
0-5
GR
-SO
41
5
17-1
217
086
-13
910
5n
dn
d3
601
0-492
6 m
039
10-5
GR
-SO
41
6
22-1
027
093
-14
527
0n
dn
d3
761
0-488
1 m
043
10-5
GR
-SO
41
7
05-1
014
077
-17
950
0n
dn
d3
741
0-4 9
18
m0
411
0-5
GR
-SO
41
5
17-1
051
060
-16
510
00n
dn
d2
891
0-483
7 m
035
10-5
n
d =
not
det
ecte
d a
Ini
tial s
truct
ural
or a
dsor
bed
Fe(I
I) e
stim
ated
as [
Fe(I
I)to
tal]
ndash [F
e(II
) aq]
b In
itial
dis
solv
ed F
e(II
) mea
sure
d c
Sus
pens
ion
pH p
rior t
o TC
A a
dditi
on d
Sus
pens
ion
pH a
t rea
ctio
n
te
rmin
atio
n e
Pse
udo
1 o
rder
rate
con
stan
ts fo
r the
con
sum
ptio
n of
TC
A c
alcu
late
d fr
om in
itial
rate
s (m
ax f
irst t
wo
half-
lives
) f
Surf
ace
area
-nor
mal
ised
pse
udo
1 o
rder
rate
con
stan
ts g
pH
con
trol
th
roug
h pu
re C
aCO
3 and
05
C
O2(g
) h
pH
con
trol t
hrou
gh F
e(II
I) o
xide
-coa
ted
calc
ite a
nd 0
5
CO
2(g)
i Es
timat
ed u
sing
the
SSA
of t
he F
e(II
I) o
xide
app
lied
j E
stim
ated
usi
ng th
e SS
A o
f cal
cite
~1
m2 g
k E
stim
ated
ass
umin
g SS
A =
4 m
2 g (S
chw
ertm
ann
amp C
orne
ll 1
991)
frac12middot[
Fe(I
I) sol
id] 0middot
232
gmiddotm
ol-1
middot4 m
2 middotg-1
l E
stim
ated
ass
umin
g SS
A =
47
m2 g
(Will
iam
s amp S
cher
er 2
001)
frac14middot[F
e(II
) GR] 0middot
600
gmiddotm
ol-1
middot47
m2 middotg
-1 m
Est
imat
ed a
s in l
but u
sing
SSA
= 7
12
m2 middotg
-1 (C
hapt
er 4
thi
s wor
k)
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 109
532 Comparing rate constants obtained for the various Fe(II)-Fe(III) mineral
systems
Data for the systems containing iron oxide coated calcite were very similar to the
data obtained for the pure iron oxides (not shown in Figure 53) Since no SSA was
determined for magnetite in this study a SSA of 4 m2g was assumed
(Schwertmann amp Cornell 1991) However it should be noted that the magnetite
synthesized by Schwertmann and Cornell (1991) was prepared differently (ie
oxidation of Fe(II) by nitrate in a heated alkaline solution) from the magnetite
applied in this study The surface area-normalised pseudo 1 order kobs values
obtained for GR-CO3 GR-SO4 Fe(II)goethite and Fe(II)lepidocrocite were all
within the same order of magnitude (Figure 53a)
Figure 53 Average surface area-normalised pseudo 1 order rate constants for the degradation of
a) TCA (this work) b) hexachloroethane (Elsner et al 2004a) and c) carbon tetrachloride
(Amonette et al 2000 Pecher et al 2002 OrsquoLoughlin et al 2003c Elsner et al 2004b) by
GR-SO4 GR-CO3 (suspension age 1 d) Fe3O4 Fe(II)α-FeOOH and Fe(II)γ-FeOOH
Experimental conditions applied in this work [Fe(II)tot]0 = 025-107 mM in the goethite and
lepidocrocite suspensions [Fe(II)tot]0 = 025-116 mM in the GR-SO4 and GR-CO3 suspensions
pH 70-86 71-926 m2 mineral surface areaL Experimental conditions applied by Elsner et al
1 mM aqueous Fe(II) 25 m2 mineral surface areaL Experimental conditions applied in
references employed in c) [Fe(II)tot]0 = 1-83 mM 25-275 m2 mineral surface areaL GR-SO4 =
110 Chapter 5
sulfate green rust GR-CO3 = carbonate green rust Fe3O4 = magnetite α-FeOOH = goethite γ-
FeOOH = lepidocrocite
When comparing the rate constants for the Fe(II)-Fe(III) mineral systems found for
reduction of TCA in this study (Figure 2a) mixed valent Fe(II)-Fe(III) minerals
such as green rusts and magnetite containing structural Fe(II) within the mineral
lattice do not seem to be significantly more reactive than Fe(II)-Fe(III) mineral
systems containing Fe(II) associated with pure Fe(III) oxides Unlike most other
iron oxides GRs contain not only external Fe(II) reactive sites at the surface but
also internal sites in the space between consecutive Fe(II)-Fe(III) hydroxide layers
The GR interlayer thickness is a function of both the size and the charge of the
interlayer anion For solutes the Fe(II) within the GR hydroxide layer is accessible
at the outside basal planes and at the edges as well as through the interlayer under
certain conditions (see Figure 42 Chapter 4 this work) Due to electrostatic
forces oxidants holding different charge properties (anions cations neutral
molecules) may exhibit different affinities for the various reactive Fe(II) sites
present in GR As the reactive sites are located inat the Fe(II)-Fe(III) hydroxide
layers the rate of reaction depends on the hydroxide layer area which can be
accessed by the oxidant If the oxidant is able to exchange with the interlayer
anion reaction can take place both at outer and inner surfaces of the GR particles
and in total more reactive sites are available for the reaction Thus oxidant size
and charge primarily control its access to the internal sites in GRs XRD
characterization of the GR-SO4 crystals during reaction with TCA demonstrated
that the GR-SO4 interlayer spacing did not vary during reaction (Table 52) This
may indicate that TCA did not enter the GR-SO4 interlayers
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 111 Table 52 Diffraction angle d-spacing and width at half peak height (Wfrac12) for the 001 GR-SO4
diffraction peak as a function of time during reaction with TCA ([Fe(II)GR]0 = 4 mM [TCA]0 = 1
mM)
Time (min) Angle (deg2θ)
d001-spacing(nm)
Wfrac12(degθ)
0 9483 10821 0273
10 9494 10809 0287
215 9522 10777 0263
330 9550 10745 0273
510 9524 10775 0277
855 9509 10791 0253
1160 9467 10839 0268
We roughly estimated the molecular size of TCA by summing the covalent radii of
the individual atoms (see Supporting Information 74) When comparing the
molecular size of TCA with the GR-SO4 interlayer spacing of 061 nm it can be
concluded that only when the C-C bond is oriented perpendicular to the interlayer
plane does the size of TCA exceed the GR-SO4 interlayer spacing In contrast the
size of TCA exceeds the GR-CO3 interlayer spacing (026 nm) regardless of its
orientation Hence if TCA was intercalated in the GR-CO3 interlayer we would
expect the interlayer spacing to expand The same holds for intercalation of a
vertically oriented TCA in the GR-SO4 interlayer Supposedly both the low charge
and the size of TCA impeded its access to the GR-SO4 and GR-CO3 interlayers ie
the divalent sulphate and carbonate in the GR interlayers did not readily exchange
with the monovalent TCA since GR interlayers generally have a higher affinity for
divalent anions than for monovalent anions (Miyata 1983) Thus TCA did neither
access nor react with internal Fe(II) reactive sites in GR-SO4 which means that the
reaction between TCA and GR-SO4 took place at the external reactive Fe(II) sites
solely It is reasonable to assume that the same holds for the reaction between TCA
and GR-CO3 No significant aging effects eg rate constants varying as a function
of GR age were observed within 142 days (see Table 51) However the SSAs of
112 Chapter 5
the GR suspensions holding ages up to 142 days were not measured but estimated
assuming that the GR SSA did not decrease within the time frame
According to the Scherrer formula the width at half peak height (Wfrac12) of a
diffraction peak is inversely proportional to the average crystal dimension
perpendicular to the given crystal plane (Klug amp Alexander 1974) The average
GR-SO4 particle thickness perpendicular to the basal plane (Wfrac12 Table 52) was
constant during reaction implying that TCA reacts only at the edges and not at the
basal planes Assuming that the platy hexagonal GR-SO4 and GR-CO3 crystals
hold an average width of 1 microm and an average particle thickness of 35 nm (Hansen
and Koch 1998) the ratio of edge surface area to outer surface area is AedgeAouter ~
130 for GR-SO4 and 121 for GR-CO3 (see Supporting Information 73) This
means that only 3 of the outer surface area in GR-SO4 and 5 of the outer
surface area in GR-CO3 are available at the edges Assuming that TCA reacts at the
edges only and if employing the AedgeAouter in the estimation of the rate constants
the surface area-normalised pseudo 1 order rate constants for GR-SO4 and GR-
CO3 would be 20-30 times higher than the rate constants depicted in Figure 53a
Thus the reactivity of GRs normalised to their reactive surface area is much higher
than the reactivity normalised to their total surface area
533 Comparing with rate constants obtained for other chlorinated aliphatic
compounds
Though care must be taken when comparing kinetic parameters obtained at
different experimental conditions (eg pH [Fe(II)]0[TCA]0 ratios surface area to
volume ratios etc) it is interesting to compare our results to those reported for
hexachloroethane (Figure 53b data from Elsner et al 2004a) The reductive
transformation of hexachloroethane was investigated for various Fe(II)-bearing
minerals including Fe(II)goethite Fe(II)lepidocrocite and GR-SO4 in the presence
of 1 mM dissolved Fe(II) and 25 m2 mineral surface areaL at pH 72 except for the
GR-SO4 suspensions in which the dissolved Fe(II) concentrations were slightly
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 113
higher and pH = 8 The pseudo 1 order rate constants reported for
hexachloroethane are in the range 18middot10-4 ndash 75middot10-3 h-1middotm-2middotL (Elsner et al 2004a)
When comparing Figure 53a with Figure 53b it can be seen that the differences
in intrinsic reactivity of the Fe(II)-bearing mineral systems are more pronounced
for hexachloroethane than for TCA
Caution should also be advised to the different reaction mechanisms by which
hexachloroethane and TCA react The transfer of a single electron and the
formation of an alkyl radical upon removal of a chlorine atom constitute the first
and in most cases the rate-limiting step in the reduction of chlorinated aliphatic
compounds (Vogel et al 1987) Depending on the chemical structure of the
chlorinated aliphatic compound the resulting free alkyl radical may undergo
hydrogenolysis chloroelimination or dimerizationcoupling In the case of TCA
the free dichloroacetate radical most likely undergoes hydrogenolysis The almost
quantitative transformation of TCA to DCA confirms that hydrogenolysis is the
prevalent reaction mechanism in our mineral systems The pentachloroethyl radical
formed from hexachloroethane may undergo hydrogenolysis (producing
pentachloroethane) or dichloroelimination (producing tetrachloroethene) Elsner et
al (2004a) found that hexachloroethane was transformed quantitatively into
tetrachloroethylene for all minerals which strongly indicates that
dichloroelimination was the dominating reaction mechanism Another
polychlorinated aliphatic compound transformed mainly by hydrogenolysis under
reducing conditions is carbon tetrachloride Several studies have investigated the
reductive dechlorination of carbon tetrachloride by various Fe(II)-bearing minerals
including Fe(II)goethite and GR-SO4 and reported pseudo 1 order rate constants
in the order 152middot10-4 ndash 640middot10-4 h-1middotm-2middotL for Fe(II)goethite and 864middot10-4 h-1middotm-2middotL
for GR-SO4 (Amonette et al 2000 Pecher et al 2002 OLoughlin et al 2003c
Elsner et al 2004b) When comparing Figure 53a with Figure 53c it can be seen
that the range of magnitude of the rate constants and the differences in intrinsic
114 Chapter 5
reactivity of the Fe(II)-bearing mineral systems are similar for carbon tetrachloride
and TCA
534 Factors controlling the reactivity of surface-bound Fe(II)
The reactivity of an oxidant towards Fe(II) surface species cannot be predicted
from the reduction potentials of the redox couple alone In heterogeneous systems
processes such as mass transfer and adsorptiondesorption may have a rate-limiting
effect on the overall reaction rate If the adsorption follows a saturation-type
sorption isotherm (eg Langmuir) the sorbate (oxidant) concentration at the
surface will vary non-linearly with the total amount of oxidant added This
dependence will have to be taken into account when establishing rate laws for the
heterogeneous reactions and when testing the hypothesis that the reaction rates
depend on the sorbed concentration of the oxidants pH has a strong impact on the
sorption and thereby on the availability of ionizable oxidants At the pH values
applied here the chloroacetates are fully dissociated (pKa (TCA) = 066 pKa
(DCA) = 135 pKa (MCA) = 287) However we found the sorption of TCA to be
negligible in suspensions of pure calcite goethitecalcite and lepidocrocitecalcite
at pH 76-77 Moreover the mass balance of TCA and DCA was almost complete
in all suspensions and therefore loss of TCA or DCA due to adsorption at mineral
surfaces or incorporation in the GR anion interlayers can be ruled out Calcite has a
much lower adsorption capacity than most iron oxides hence we anticipate that
goethite and lepidocrocite control the adsorption of TCA and DCA in both the pure
FeOOH and the FeOOHcalcite suspensions This was supported by our
experimental results demonstrating that the presence of a calcite surface - either
pure or as a support for goethite and lepidocrocite coatings - did not exert any
noticeable effect on the reaction rates (see Table 51) In addition the surface area-
normalised rate constants for mineral systems containing goethite or lepidocrocite
in pure form and mineral systems containing goethite or lepidocrocite as coatings
on calcite were very similar In heterogeneous reactions mass transfer in bulk
solution becomes the rate-limiting step when the surface reaction is much faster
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 115
than the diffusion of the reacting species to the reactive surface However at the
low rate constants obtained here the reaction of TCA with the Fe(II)-bearing
minerals is not likely to be mass transfer limited (see Supporting Information 72)
One very important factor affecting heterogeneous redox reactions is pH which
influences the speciation of the complexes in solution and at mineral surfaces as
well as the stability of the more soluble Fe(II)-containing minerals such as GRs In
contrast to aqueous Fe(II) complexes it is not possible to predict the reactivity of
Fe(II) surface species as their reduction potentials are unknown In the absence of
specifically adsorbing solutes other than H+ the surface charge of the Fe(III)
oxides goethite and lepidocrocite is determined by the surface densities of the
charged surface species equivFeOH2+ and equivFeOndash whereas the surface charge of calcite
is determined by the density of the surface species equivCO3ndash equivCaOH2
+ and equivCaOndash
(Stumm 1992 Van Cappellen et al 1993) The point of zero charge (pHpzc) of
pure calcite is in the pH range 7-11 and depends on the partial pressure of carbon
dioxide pCO2 The higher the pCO2 the lower the pHpzc At the experimental
conditions applied here (05 = 0005 atm CO2(g)) the pHpzc = 82 for calcite
(Table 53) As only 10-11 mg Fe(III) of goethite and lepidocrocite was coated
onto calcite we assumed a pHpzc of 82 for the goethite and lepidocrocite coated
calcite particles as well The pHpzc values for green rusts are unknown
116 Chapter 5
Table 53 Specific surface areas and point of zero charge of the various iron minerals in pure form as well as goethite and lepidocrocite coated onto calcite
Mineral Structural formula SSA (m2g) pHpzc
GR-SO4 FeII4FeIII
2(OH)12SO4middot3H2O 71 a -
GR-CO3 FeII4FeIII
2(OH)12CO3middot3H2O 47 b -
Magnetite Fe3O4 - 69 e
Goethite α-FeOOH 16 c 85 f
Lepidocrocite γ-FeOOH 18 c 73 e
Calcite CaCO3 le 1 d 82 g
Goe coating - le 1 d 82 h
Lep coating - le 1 d 82 h
a Chapter 4 this work b Williams amp Scherer 2001 c Product information by Bayer d The SSA of calcite was
quantified to le 1 m2g The detection limit of our BET method was 1 m2g e Charlet et al 1998a f Liger et al
1999 g Van Cappellen et al 1993 h Same as for calcite
The surface hydroxyl groups on iron oxides may be both singly (equivFe-OH) doubly
(equivFe2-OH) triply (equivFe3-OH) and geminally (equivFe-(OH)2) coordinated (Cornell amp
Schwertmann 1996 Stumm 1992) The differently coordinated surface hydroxyl
groups are not equally reactive Adsorption reactions involve only singly
coordinated surface groups and therefore only this kind of hydroxyl groups on iron
oxides will be considered here (Cornell amp Schwertmann 1996) Hence the
predominant surface sites available for adsorption in pure suspensions of Fe(III)
oxides are equivFeOH0 equivFeOH2+ and equivFeOndash In the presence of dissolved Fe(II)
equivFeIIIOFeIIOH0 equivFeIIIOFeIIOndash and equivFeIIIOFeII+ constitute the main reactive sites at
the Fe(III) oxide surfaces (Liger et a 1999) Assuming that Fe2+ and other cationic
Fe(II) species are the dominating adsorbates on the mineral surfaces in our
experiments we expect the actual pHpzc to be higher than the pHpzc of the pure
oxides listed in Table 53 Hence at pHlt82 where most of our experiments were
conducted all the mineral surfaces presumably carry net positive charges
At pH 70 where Fe2+ is still the predominant Fe(II) species in solution (~50) we
expect that equivFeIIIOFeIIOH equivFeIIIOFeIIOndash and equivFeIIIOFeII+ constitute the main
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 117
reactive sites at the Fe(III) oxide surfaces as suggested by Liger et a 1999 As pH
increases from 70 to 87 the Fe(II) carbonate complexes become increasingly
important in solution at the expense of the Fe2+ FeCl+ FeSO40 and FeOH+ species
(King 1998) Fe(II) carbonate complexes do not bind at the oxide surface as
readily as the aquo or hydroxo complexes of Fe(II) but carbonate itself sorbs
readily to Fe(III) oxide surfaces through which the Fe(III) oxide surface is coated
by inner-sphere monodentate equivFeIIIOCOOH0 surface complexes (Villalobos amp
Leckie 2000 amp 2001) The presence of carbonate shifted the sorption edge for the
Fe(II) adsorption on goethite from pH 58 to 78 and the authors hypothesized this
to be a result of the formation of aqueous and surface Fe(II)-carbonate complexes
and to competition between carbonate and Fe(II) for Fe(III) oxide surface sites
(Vikesland amp Valentine 2002) Similarly monodentate surface complexes like
equivFeIIICl0 and equivFeIIIOSO3ndash as well as ternary monodentate surface complexes like
equivFeIIIOFeIICl0 and equivFeIIIOFeIIOSO3ndash and ternary bidentate surface complexes such
as (equivFeIIIO)2FeIIOSO3 may form at Fe(III) oxide surfaces when Fe(II) chloride and
sulfate are present in solution (Ostergren et al 2000 Kim et al 2004) However
the effects of anionic ligands such as chloride and sulfate on Fe(II) adsorption at
Fe(III) oxide surfaces and the reactivity of Fe(II) carbonate chloride and sulfate
surface sites are still unknown and need to be evaluated (see Supporting
Information 75) Thus we do not know whether chloride and sulfate decrease or
increase the Fe(II) sorption in our mineral systems We can only report that we did
not detect any significant differences in the rate of TCA transformation between
the mineral suspensions containing carbonate chloride and sulfate respectively
Hence we anticipate that equivFeIIIOFeIIOH equivFeIIIOFeIIOndash and equivFeIIIOFeII+ constitute
the main reactive sites at the Fe(III) oxide surfaces within the whole pH range 70-
87 This might also explain why we did not detect any obvious systematic pH
effect in the Fe(II)-Fe(III)-systems (see Table 51) In the case of
hexachloroethane the reactivity order GR-SO4gtgoethitegtmagnetitegtlepidocrocite
may be rationalized by the variations in surface site densities and total amount of
118 Chapter 5
Fe(II) sorbed on the iron minerals (see Supporting Information 75) as well as the
different speciations and reactivities of the Fe(II) surface sites on the iron minerals
535 Comparison with biotic and other abiotic systems
Only one report on abiotic transformation of TCA is found in the literature and the
study demonstrates the reductive dechlorination of TCA to MCA by Fe(0)
(Hozalski et al 2001) The authors reported a pseudo 1 order rate constant of
60middot10-4 min-1middotm-2middotL for the transformation of TCA to DCA and a pseudo 1 order
rate constant of 225middot10-4 min-1middotm-2middotL for the transformation of DCA to MCA at
[Fe(0)]0 = 025 M [TCA]0 = 100-200 microM and pH 36-62 The rate constant for
TCA reduction by Fe(0) is 10-300 times faster than the rate constants reported for
the Fe(II)-bearing mineral systems here
There is abundant evidence that soil microorganisms and fungi can dechlorinate
TCA but only little is known about the bacteria and processes involved in the
biodegradation of TCA Biotransformation of TCA has been found at both oxic
and anoxic conditions Most of the microorganisms isolated grow feebly on TCA
as a sole source of carbon (Foy 1975 Weightman et al 1992 De Wever et al
2000) Only one bacterium capable of growing on TCA as the sole carbon and
energy source has been characterized (Yu amp Welander 1995) In addition
anaerobic bacteria coupling co-metabolic growth to reductive dechlorination of
TCA have been isolated (Weightman et al 1992 De Wever et al 2000) The
inability to grow on the less chlorinated acids DCA and MCA is a notable feature
of both the aerobic and anaerobic bacteria Complete transformation of TCA to
methane and carbon dioxide has only been found when abiotic and biotic processes
were combined (Egli et al 1989) The abiotic transformation of TCA to DCA
occurred spontaneously in the presence of sterile activated charcoal whereas the
DCA formed was further degraded to methane and carbon dioxide by a mixed
culture of methanogenic bacteria However the abiotic reductant(s) responsible for
the transformation of TCA to DCA was not reported (Egli et al 1989)
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 119
The rate constants obtained in this work suggest that the Fe(II)-bearing mineral
systems may be important reductants of TCA in natural suboxic environments In
natural iron-rich soils holding specific surface areas of 22 m2g (Kenneke amp
Weber 2003) average bulk densities of 265 gcm3 and porosities of 25 and
containing 2 iron oxides a rough estimation of the half-life of TCA amounts to
47 minutes when applying the average surface area-normalised rate constant
obtained for all the Fe(II)-Fe(III) mineral systems in this work (1middot10-3 h-1middotm-2middotL)
This estimation is based on the assumption that enough reactive Fe(II) is available
in these soils The natural iron-reducing sediment investigated by Kenneke and
Weber (2003) contained 80 microM Fe(II) in the soil solution and 315 micromole Fe(II) per
g sediment At such low Fe(II) concentrations the overall rate of abiotic
transformation of TCA in natural soils and sediments is most likely limited by the
regeneration of reactive Fe(II) Hence the continuous regeneration of reactive
Fe(II) surface sites by adsorption of abiotically or microbially produced Fe(II) may
further the long-term abiotic transformation of TCA in such environments
54 Conclusions
This work demonstrates that various Fe(II)-Fe(III) minerals systems including GR-
SO4 GR-CO3 magnetite Fe(II)goethite and Fe(II)lepidocrocite readily transform
TCA to DCA Dichloroacetate was not further reduced to MCA or acetate by any
of the Fe(II)-bearing minerals The surface area-normalised pseudo 1 order rate
constants obtained for the reductive transformation of TCA by the various Fe(II)-
bearing minerals did not differ significantly from each other The results obtained
in this work infer that under natural geochemical conditions Fe(II)-bearing mineral
systems may play an important role in the overall transformation of TCA Thus
not only microbial degradation but also abiotic reductive transformation of TCA by
Fe(II)-bearing minerals may govern the fate of TCA in natural subsurface
environments This holds both for natural systems like iron-rich suboxic soils and
sediments as well as engineered systems like permeable reactive barriers of zero-
120 Chapter 5
valent iron implemented for on-site remediation where both Fe(0) and solid or
surface-bound Fe(II) corrosion intermediates may transform TCA
Acknowledgments
We would like to thank Susanne Guldberg for performing the experimental work comprising
GR-SO4
References
Ahlers J Regelmann J Riedhammer C (2003) Environmental risk assessment of airborne trichloroacetic acid - a contribution to the discussion of the significance of anthropogenic and natural sources Chemosphere 52 531-537 Amonette JE Workman DJ Kennedy DW Fruchter JS Gorby YA (2000) Dechlorination of carbon tetrachloride by Fe(II) associated with goethite Environmental Science and Technology 34 4606-4613 Atkins PJ Gold V Marsh R (1984) The decarboxylation of trichloroacetic acid and the reactions of the trichloromethyl anion with 135-trinitrobenzene and with hydrogen ions kinetic measurements in dimethyl sulphoxide solution Journal of the Chemical Society Perkin Transactions 2 7 1239-1245 Berg M Muumlller SR Muumlhlemann J Wiedmer A Schwarzenbach RP (2000) Concentrations and mass fluxes of chloroacetic acids and trifluoroacetic acid in rain and natural waters in Switzerland Environmental Science and Technology 34 2675-2683 Brunauer S Emmett PH Teller E (1938) Adsorption of gases in multimolecular layers Journal of American Chemical Society 60 309-319 Butler EC Hayes KF (1998) Effects of solution composition and pH on the reductive dechlorination of hexachloroethane by iron sulfide Environmental Science and Technology 32 1276-1284 Butler EC Hayes KF (1999) Kinetics of the transformation of trichloroethylene and tetrachloroethylene by iron sulfide Environmental Science and Technology 33 2021-2027 Charlet L Liger E Gerasimo P (1998a) Decontamination of TCE- and U-rich water by granular iron Role of sorbed Fe(II) Journal of Environmental Engineering 124 25-30 Charlet L Silvester E Liger E (1998b) N-compound reduction and actinide immobilisation in surficial fluids by Fe(II) the surface FeIIIFeIIOH0 species as major reductant Chemical Geology 151 85-93 Cheregi M Danet AF (1997) Flow injection determination of chloride ions with spectrophotometric detection Analytical Letters 30 2847-2858 Cornell RM Schwertmann U (1996) The iron oxides Structure properties reactions occurrence and uses VCH Verlagsgesellschaft mbH Weinheim
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 121 Cui D Eriksen TE (1996) Reduction of pertechnetate by ferrous iron in solution influence of sorbed and precipitated Fe(II) Environmental Science and Technology 30 2259-2262
Egli C Thuumler M Suter D Cook AM Leisinger T (1989) Monochloro- and dichloroacetic acids as carbon and energy sources for a stable methanogenic mixed culture Archives of Microbiology 152 218-223 Elsner M Haderlein SB Schwarzenbach RP (2004a) Reactivity of Fe(II)-bearing minerals towards reductive transformation of organic contaminants Environmental Science and Technology 38 799-807 Elsner M Haderlein SB Kellerhals T Luzi S Zwank L Angst W Schwarzenbach RP (2004b) Mechanisms and products of surface-mediated reductive dehalogenation of carbon tetrachloride by Fe(II) on goethite Environmental Science and Technology 38 2058-2066 Erbs M Hansen HCB Olsen CE (1999) Reductive dechlorination of carbon tetrachloride using iron(II)iron(III)-hydroxide-sulphate (green rust) Environmental Science and Technology 33 307-311 Euro Chlor (2001) Trichloroacetic acid in the environment a dossier Euro Chlor Brussels and the European Chlorinated Solvent Association Fadrus H Maly J (1975) Suppression of iron(III) interference in the determination of iron(II) in water by the 110-phenanthroline method The Analyst 100 549-554 Fahimi IJ Keppler F Schoumller HF (2003) Formation of chloroacetic acids from soil humic acid and phenolic moieties Chemosphere 52 513-520 Foy CL (1975) The chlorinated aliphatic acids In Herbicides Chemistry degradation and mode of action Kearney PC Kaufman DD (eds) Marcel Dekker Inc 399-452 Gander JW Parkin GF Scherer MM (2002) Kinetics of 111-trichloroethane transformation by iron sulfide and a methanogenic consortium Environmental Science and Technology 36 4540-4546 Haiber G Jacob G Niedan V Nkusi G Schoumller HF (1996) The occurrence of trichloroacetic acid (TCAA) ndash indications of a natural production Chemosphere 33 839-849
Hansen HCB (1989) Composition stabilization and light absorption of Fe(II)Fe(III) hydroxycarbonate (green rust) Clay Minerals 24 663-669 Hansen HCB Guldberg S Erbs M Koch CB (2001) Kinetics of nitrate reduction by green rusts ndash effects of interlayer anion and Fe(II)Fe(III) ratio Applied Clay Science 18 81-91 Hansen HCB Koch CB (1997) A comparison of nitrate reduction by carbonate and sulphate forms of green rust Kodama H Mermut A R Torrance J K (eds) Proceedings of the 11th International Clay Conference Ottawa Canada Clays for our future 11 295-302 Hoekstra EJ de Leer EWB Brinkman UATh (1999a) Mass balance of trichloroacetic acid in the soil top layer Chemosphere 38 551-563 Hoekstra EJ de Leer EWB Brinkman UATh (1999b) Findings supporting the natural
122 Chapter 5
formation of trichloroacetic acid in soil Chemosphere 38 2875-2883 Hofstetter TB Heijman CG Haderlein SB Holliger HC Schwarzenbach RP (1999) Complete reduction of TNT and other (poly)nitroaromatic compounds under iron-reducing subsurface conditions Environmental Science and Technology 33 1479-1487 Hofstetter TB Schwarzenbach RP Haderlein SB (2003) Reactivity of Fe(II) species associated with clay minerals Environmental Science and Technology 37 519-528 Hozalski RM Zhang L Arnold WA (2001) Reduction of haloacetic acids by Fe0 Implications for treatment and fate Environmental Science and Technology 35 2258-2263 Husain S Narsimha R Alvi SN Rao RN (1992) Monitoring the effluents of the trichloroacetic acid process by high-performance liquid chromatography Journal of Chromatography 600 316-319 Hwang I Batchelor B (2000) Reductive dechlorination of tetrachloroethylene by Fe(II) in cement slurries Environmental Science and Technology 34 5017-5022 Jordan A Frank H (1999) Trifluoroacetate in the environment Evidence for sources other than HFCHCFCs Environmental Science and Technology 33 522-527 Kenneke JF Weber EJ (2003) Reductive dehalogenation of halomethanes in iron- and sulfate-reducing sediments 1 reactivity pattern analysis Environmental Science and Technology 37 713-720 Kim CS Rytuba JJ Brown Jr GE (2004) EXAFS study of mercury(II) sorption to Fe- and Al-(hydr)oxides II Effects of chloride and sulphate Journal of Colloid and Interface Science 270 9-20 King DW (1998) Role of carbonate speciation on the oxidation rate of Fe(II) in aquatic systems Environmental Science and Technology 32 2997-3003 Klausen J Troumlber SP Haderlein SB Schwarzenbach RP (1995) Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions Environmental Science and Technology 29 2396-2404 Klug PH Alexander LE (1974) X-ray diffraction procedures John Wiley amp Sons Inc Koch CB Hansen HCB (1997) Reduction of nitrate to ammonium by sulphate green rust Advances in GeoEcology 30 373-393 Kuumlhn R Pattard M (1990) Results of the harmful effects of water pollutants to green algae (Scenedesmus subspicatus) in the cell multiplication inhibition test Water Research 24 31-38 Lee W Batchelor B (2002a) Abiotic reductive dechlorination of chlorinated ethylenes by iron- bearing soil minerals 1 Pyrite and magnetite Environmental Science and Technology 36 5147- 5154 Lee W Batchelor B (2002b) Abiotic reductive dechlorination of chlorinated ethylenes by iron-bearing soil minerals 2 Green rust Environmental Science and Technology 36 5348- 5354
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 123 Liger E Charlet L Van Cappellen P (1999) Surface catalysis of uranium (VI) reduction by iron(II) Geochimica et Cosmochimica Acta 63 2939-2955 Lignell R Heinonen-Tanski H Uusi-Rauva A (1984) Degradation of trichloroacetic acid (TCA) in soil Acta Agriculturae Scandinavia 34 3-8 Loyaux-Lawniczak S Refait Ph Lecomte P Ehrhardt J Geacutenin JMR (1999) The reduction of chromate ions by Fe(II) layered hydroxides Hydrology and Earth System Sciences 3 593-599 Luther III GW (1990) The Frontier-Molecular-Orbital theory approach in geochemical processes in W Stumm Ed Aquatic Chemical kinetics John Wiley and Sons New York pp 173-198 McCulloch A (2002) Trichloroacetic acid in the environment Chemosphere 47 667-686 Miyata S (1983) Anion-exchange properties of hydrotalcite-like compounds Clays and Clay Minerals 31 305-311 Muumlller SR Zweifel H-R Kinnison DJ Jacobsen JA Meier MA Ulrich MM Schwarzenbach RP (1996) Occurrence sources and fate of trichloroacetic acid in Swiss lakes Environmental Toxicology and Chemistry 15 1470-1478 OLoughlin EJ Burris DR (2003) Reduction of halogenated ethanes by green rust Environmental Toxicology and Chemistry 23 41-48 OLoughlin EJ Kelly SD Cook RE Csencsits R Kemner KM (2003a) Reduction of uranium(VI) by mixed iron(II)iron(III) hydroxide (green rust) Formation of UO2 nanoparticles Environmental Science and Technology 37 721-727 OLoughlin EJ Kelly SD Kemner KM Csencsits R Cook RE (2003b) Reduction of AgI AuIII CuII and HgII by FeIIFeIII hydroxysulfate green rust Chemosphere 53 437-446 OLoughlin EJ Kemner KM Burris DR (2003c) Effects of AgI AuIII and CuII on the reductive dechlorination of carbon tetrachloride by green rust Environmental Science and Technology 37 2905-2912 Ostergren JD Brown Jr GE Parks GA Persson P (2000) Inorganic ligand effects on Pb(II) sorption to goethite (α-FeOOH) II Sulfate Journal of Colloid and Interface Science 225 483-493 Pecher K Haderlein SB Schwarzenbach RP (2002) Reduction of polyhalogenated methanes by surface-bound Fe(II) in aqueous suspensions of iron oxides Environmental Science and Technology 36 1734-1741 Reimann S Grob K Frank H (1996) Chloroacetic acids in rainwater Environmental Science and Technology 30 2340-2344 Ruumlgge K Hofstetter TB Haderlein SB Bjerg PL Knudsen S Zraurig C Mosbaeligk H Christensen TH (1998) Characterization of predominant reductants in an anaerobic leachate- affected aquifer by nitroaromatic probe compounds Environmental Science and Technology 32
124 Chapter 5
23-31 Sarzanini C Bruzzoniti MC Mentasti E (1999) Preconcentration and separation of haloacetic acids by ion chromatography Journal of Chromatography A850 197-211 Schwertmann U Cornell RM (1991) Iron oxides in the laboratory Preparation and characterization VCH Verlagsgesellschaft mbH Weinheim Scott BF Mactavish DC Spencer C Strachan WMJ Muir DCG (2000) Haloacetic acids in Canadian lake waters and precipitation Environmental Science and Technology 34 4266-4272 Scott BF Spencer C Marvin CH Mactavish DC Muir DCG (2002) Distribution of haloacetic acids in the water columns of the Laurentian Great Lakes and Lake Malawi Environmental Science and Technology 36 1893-1898 Strathmann TJ Stone AT (2003) Mineral surface catalysis of reactions between FeII and oxime carbamate pesticides Geochimica et Cosmochimica Acta 67 2775-2791 Stumm W (1992) Chemistry of the solid-water interface John Wiley amp Sons Inc Van Cappellen P Charlet L Stumm W Wersin P (1993) A surface complexation model of the carbonate mineral-aqueous solution interface Geochimica et Cosmochimica Acta 57 3505- 3518 Vikesland PJ Valentine RL (2002) Iron oxide surface-catalyzed oxidation of ferrous iron by monochloramine implications of oxide type and carbonate on reactivity Environmental Science and Technology 36 512-519 Villalobos M Leckie JO (2000) Carbonate adsorption on goethite under closed and open CO2 conditions Geochimica et Cosmochimica Acta 64 3787-3802 Villalobos M Leckie JO (2001) Surface complexation modeling and FTIR study of carbonate adsorption to goethite Journal of Colloid and Interface Science 235 15-32 Vogel TM Criddle CS McCarty PL (1987) Transformations of halogenated aliphatic compounds Environmental Science and Technology 21 722-736 Weightman AL Weightman AJ Slater JH (1992) Microbial dehalogenation of trichloroacetic acid World Journal of Microbiology and Biotechnology 8 512-518 De Wever H Cole JR Fettig MR Hogan DA Tiedje JM (2000) Reductive dehalogenation of trichloroacetic acid by Trichlorobacter thiogenes gen nov spnov Applied and Environmental Microbiology 66 2297-2301 Williams AGB Scherer MM (2001) Kinetics of chromate reduction by carbonate green rust Environmental Science and Technology 35 3488-3494 Yu P Welander T (1995) Growth of an aerobic bacterium with trichloroacetic acid as the sole source of energy and carbon Applied Microbiology and Biotechnology 42 769-774
Conclusions and Outlook 125
6 Conclusions and Outlook The work presented in this dissertation adds to the understanding of how Fe(II)-
bearing minerals like green rusts (GRs) vivianite (Fe2(PO4)2sdot8H2O) magnetite
(Fe3O4) and Fe(II) associated with goethite and lepidocrocite may form and react in
nature In order to elucidate the role of bacteria in the formation of GRs in natural
soils and sediments we studied the iron mineral phases forming as a result of the
activity of iron-respiring bacteria In chapter 2 the Fe-containing products formed
by anaerobic autotrophic denitrifying Fe(II)-oxidizing bacteria (FeOB) were
examined The culture medium applied contained high levels of bicarbonate and
phosphate and is typically used in this kind of studies as it provides excellent
conditions for the nitrate-reducing FeOB Fe(II) was present initially as a whitish
solid Fe(II) hydroxy phosphate (vivianite) and as soluble Fe(II) The results
obtained demonstrate that the denitrifying FeOB produce poorly crystalline
goethite via a greenish Fe(III)-enriched vivianite intermediate Moumlssbauer
spectroscopic analyses provided no evidence of green rust formation At low
phosphate concentrations where vivianite does not control the Fe(II) activity it is
reasonable to assume that siderite (FeCO3) precipitates initially and that carbonate
GR phases may form during biooxidation At low bicarbonate concentrations we
would expect Fe(II) sulfate or chloride species to dominate initially (depending on
the Fe(II) source applied) and sulfate GR or chloride GR to form during
biooxidation In chapter 3 we investigated the Fe-containing products formed
during reduction of common Fe(III) oxides by the anaerobic dissimilatory Fe(III)-
reducing microorganism Shewanella algae BrY S algae BrY reduced substantial
amounts of the initial Fe(III) and green and blackish mineral phases were produced
within 1-2 weeks after inoculation Moumlssbauer spectroscopic analyses showed that
the green and black precipitates consisted of green rust and vivianite
We studied the reactivity of synthetic GRs towards reducible organic pollutants in
order to asses the potential significance of GR phases for the fate of such
126 Chapter 6
compounds To this end we used nitroaromatic compounds (NACs) and
chlorinated acetates as suitable model compounds for studying redox reactions
potentially relevant in the environment In chapter 4 we investigated the relative
reactivity of outer and inner Fe(II) reactive sites in synthetic sulfate green rust
(GR-SO4) by using a series of structurally closely related compounds with different
charge properties as ldquoreactive probesrdquo The probe compounds included
nitrobenzene 2-nitrophenol 4-nitrotoluene 4-chloronitrobenzene and 4-
nitrophenylacetic acid Our results demonstrated that NACs are completely
reduced to their corresponding anilines by GR-SO4 The reactions followed pseudo
1 order kinetics with respect to NAC and the surface area-normalised pseudo 1
order rate constants obtained were 016ndash465middot10-4 s-1middotm-2middotL at [Fe(II)GR]0 = 103-
1260 mM [NAC]0 = 20-102 microM and pH 84-86 Neither mass transfer control nor
surface saturation kinetics could account for the similarity of the surface-
normalised pseudo 1 order rate constants obtained for the reduction of the neutral
and anionic NACs by GR-SO4 These observations suggest that the reaction
between NAC and GR-SO4 takes place at the external reactive Fe(II) sites At low
initial Fe(II)GR concentrations the external reactive Fe(II) sites were depleted and
the regeneration of new external reactive sites eventually controlled the reduction
of the NACs by GR-SO4 In chapter 5 we examined the reactivity of various
Fe(II)-Fe(III) mineral systems towards trichloroacetic acid (TCA) and
dichloroacetate (DCA) in laboratory batch experiments imitating natural
conditions The Fe(II)-Fe(III)-systems investigated included GR-SO4 carbonate
green rust magnetite Fe(II)goethite and Fe(II)lepidocrocite TCA was readily
reduced to DCA by all Fe(II)-containing minerals The reactions followed pseudo
1 order kinetics with respect to TCA and the surface area-normalised pseudo 1
order rate constants obtained were 033ndash76middot10-5 min-1middotm-2middotL at [Fe(II)]0 = 025ndash
116 mM [TCA]0 = 15ndash1000 microM and pH 70ndash87 Our results showed no
significant differences regarding product distribution and surface area-normalised
reaction rate constants between the Fe(II)-Fe(III)-systems DCA was not further
Conclusions and Outlook 127
reduced to monochloroacetate (MCA) or acetate in any of the systems within the
time frame in our experiments
As suggested in chapters 2 and 3 sufficient evidence must be provided and caution
should be exercised when proclaiming new biogenic minerals The study of
microbially produced GRs is still in its infancy and more research is needed in
order to elucidate the role of bacteria in the formation of GRs in natural soils and
sediments The results presented in chapter 2 indicate that microbiological
processes may be responsible for the oxidation of vivianite and metavivianite
((FeII3-xFeIII
x)(PO4)2(OH)xmiddot(8-x)H2O x gt 12) in natural subsurface environments
In chapter 3 we demonstrated that GRs may be produced microbially at conditions
including low carbon and Fe(III) concentrations as well as the exclusion of
synthetic electron shuttles and pH buffers The role of microbial processes in the
redox cycling of iron in the subsurface and the ways in which these processes can
be coupled to contaminant remediation are currently active areas of research Zero-
valent iron has been the most extensively studied reductant for the treatment of
many inorganic and organic contaminants and is currently the most commonly
used material for the construction of permeable reactive barriers (PRB) but a
detailed understanding of the processes involved in the reduction of these
pollutants by Fe(0) is lacking (Scherer et al 2000) Potentially reactive Fe(II)-
bearing corrosion products identified in iron metal columns and barriers include
magnetite siderite Fe(II) sulfides green rusts as well as Fe(II) sorbed to mineral
surfaces (Gu et al 1999 Roh et al 2000) The formation of reactive Fe(II)-
bearing minerals like GRs may explain the effective long-term operation of zero-
valent iron PRBs despite the formation of thick oxide films Thus natural in situ
PRBs might be created by stimulating the activity of anaerobic dissimilatory
Fe(III)-reducing bacteria and the subsequent formation of Fe(II) species such as
GRs Furthermore suspensions of synthetic GRs which are easily prepared from
relatively inexpensive commodity chemicals may also be injected and dispersed
into the subsurface
128 Chapter 6
The reductive transformation of NACs and TCA by GRs is relevant to
understanding the processes responsible for their degradation in the subsurface and
the development of innovative technologies for their remediation The results
obtained in chapters 4 and 5 indicate that GRs may play a significant role in the
reductive transformation of NACs and TCA in natural subsurface environments
Furthermore our results suggest that mainly the outer Fe(II) sites in GRs are
utilized in the reaction with neutral and monovalent anionic compounds and that
these sites may be replenished eg by reduction of the oxidized surface sites or
adsorption of Fe(II) from solution The continuous restoration of Fe(II) surface
sites in GRs may promote their long-term reactivity towards reducible
contaminants
References
Gu B Phelps TJ Liang L Dickey MJ Roh Y Kinsall BL Palumbo AV Jacobs GK (1999) Biochemical dynamics in zero-valent iron columns Implications for permeable reactive barriers Environmental Science and Technology 33 2170-2177 Roh Y Lee SY Elless MP (2000) Characterization of corrosion products in the permeable reactive barriers Environmental Geology 40 184-194 Scherer MM Richter S Valentine RL Alvarez PJJ (2000) Chemistry and microbiology of permeable reactive barriers for In Situ groundwater clean up Critical Reviews in Environmental Science and Technology 30 363-411
Supporting Information I
7 Supporting Information
71 Estimation of the one-electron reduction potential for 4-NPA
The one-electron reduction potential of the half-reaction for a given NAC 1hE
ArNO2 + e- ArNO2
can be used for comparing reduction rates of different NACs in a given system
The formation of the nitroaryl radical is the rate-determining step in the overall rate
of the reduction of a NAC to the corresponding aniline The difference between the
of a NAC and a given reductant is proportional to the change in standard free
energy for the transfer of the first electron ∆G
1hE
1degrsquo If a linear relationship between
the free energy of activation and ∆G1degrsquo is assumed the values of various NACs
can be a measure of their relative reactivity with a given reductant
1hE
As neither the one-electron reduction potential for 4-nitrophenylacetic acid (4-
NPA) nor the Hammett constant for the acetic acid substituent could be found in
the literature the one-electron reduction potential for 4-NPA was estimated by
application of a linear free energy relationship (LFER) to experimental data
Kinetic experiments were conducted in order to obtain the pseudo 1 order rate
constant for the reduction of 4-NPA by a model hydroquinone (reduced
juglone (8-hydroxy-14-naphthoquinone) in the presence of HS
minusHJUGk
ndash) The reduction of
a NAC by juglone follows the rate law
[ ] [ ] [ ] [ ] [ ] [ ]2222 ArNOJUGfkArNOHJUGkArNOk
dtArNOd
totHJUGHJUGHJUGobs sdotsdotsdot=sdotsdot=sdot=minus minusminusminusminus
and the was deducted from a LFER 1hE
II Chapter 7
bEak hHJUG +sdot=minus 059160
log1
for which a and b values have been established for a range of NACs with known
values (Hofstetter et al 1999) An excellent correlation of and log
has been found to exist over a range of 250 mV corresponding to more than 5 order
of magnitude for This is due to the fact that the actual transfer of the first
electron is the rate-determining step under the experimental conditions chosen
1hE 1
hE minusHJUGk
minusHJUGk
For comparison experiments with 4-nitrotoluene (4-NT) were also conducted The
pseudo 1 order rate constants for the reduction of 4-NPA with juglone were
corrected for the reduction of 4-NPA with only HSndash (control experiments
containing no juglone)
[ ]minuslowast
minusminus
minus
minus=
HJUGkk
k HSHJUGHJUG
where (MminusHJUGk -1middots-1) is the rate constant for a compound in the presence of only
juglone (slowastminusHJUGk -1) is the pseudo 1 order rate constant for a compound in the
presence of both juglone and HSndash (sminusHSk -1) is the pseudo 1 order rate constant for
the control reaction in the presence of only HSndash and [HJUGndash] (M) is the
concentration of the reactive dissociated HJUG- form (nondissociated
hydroquinone species are very nonreactive as compared to the monophenolate
species)
Supporting Information III
OH 0 OH OH
+ e- + H+ = + e- + H+ =
0 0
pl(( ox) = 8 00 PK1 (red) = 6 60
JUG HJUG
OH OH
OH
pKa2(red) = 10 60
Figure 7 1 Oxidized and reduced juglone fo1m s
Kinetic experiments in homogeneous anoxic aqueous solutions contained 5 mM
HS- 20 microM total juglone 50 mM KH2P04 buffer and were conducted at pH =
660 corresponding to a concentration of the reactive dissociated juglone form
[HJUG] = 10 microM
By using the LFER
E1 logkHJUG- = 125 middot 005~16 + 923
the following values were obtained
(Hofstetter et al 1999)
NAC k (M-1 -1) HJUG- middots log kHJUG- E~ (mV)
4-NT
4-NPA
311 middot10middot7
164middot10middot7
847middot 10-8
116middot 10middot7
226middot10middot2
489middot10middot3
-165
-231
-515
-546
IV Chapter 7
lowast
minusHJUGk -values are averages of triplicates whereas -values are averages of
duplicates The determined for 4-NT in this work (-515 mV) differs 3 from
the -value of -500 mV reported in the literature (Meisel amp Neta 1975
Wardman 1989) Hence it is assumed that the -value determined for 4-NPA
also differs by 3
minusHSk
1hE
1hE
1hE
Note that even for NACs holding very different values the difference in their
reactivities are much less pronounced in Fe(II)-Fe(III) systems such as the
Fe(II)goethite system (LFER slope a = 06 Hofstetter et al 1999) and the
Fe(II)magnetite system (LFER slope a = 034 Klausen et al 1995) as compared
to the jugloneH
1hE
2S system (a = 125) Furthermore it should be noted that all
LFERs mentioned here were established for neutral NACs and in this work we
have simply assumed that the LFERs are also valid for anionic NACs
72 The rate-limiting step
The overall rate of a reaction is equal to the rate of the slowest step in the
mechanism In heterogeneous reactions eg a compound reacting at the surface of
suspended particles in bulk solution the overall process by which the
heterogeneous reactions proceed may be broken down into a sequence of
individual diffusion steps and reaction steps 1) Mass transfer (diffusion) of the
reactant from the bulk fluid to the external surface of the solid phase 2)
Adsorption of reactant onto the solid surface 3) Reaction on the solid surface 4)
Desorption of the products from the solid surface 5) Mass transfer of the products
from the external solid surface to the bulk fluid Hence the rate of reaction of a
compound reacting at the surface of suspended particles in bulk solution may be
either mass transfer adsorptiondesorption or surface reaction limited When the
diffusion steps are much faster than the reaction steps the mass transfer or
diffusion steps do not affect the overall reaction rate However if the reaction steps
Supporting Information V
are very fast compared with the diffusion steps mass transport affects the reaction
rate Here only the external mass transfer is considered ie the diffusion of
reactants or products between the bulk fluid and the external surface of the solid
phase The additional internal mass transfer resistance for particles containing
substantial internal surface area is not addressed
721 Mass transfer (diffusion) limited kinetics
The overall rate constant can be represented by a system of resistances in series
(Fogler 1999 Arnold et al 1999)
⎟⎟⎠
⎞⎜⎜⎝
⎛+=
minusgeomSALobs kkak1111
where kobs is the observed rate constant kL is the mass transfer coefficient (mmiddots-1) a
is the ratio of the external (geometric) specific surface area to volume of solution
(m-1) and kSA-geom is the intrinsic rate constant of the reaction normalized to the
external specific surface area rather than the BET specific surface area By
comparing kLmiddota with kobs one can estimate the role of mass transfer on the rate of
reaction Thus if kLmiddota gtgt kobs mass transfer is so fast that it has no impact on the
reaction rate whereas if kLmiddota le kobs mass transfer is the rate limiting step
In fluid dynamics the Reynolds number Re is used for determining whether a
flow is laminar or turbulent
νtp ud sdot
=Re
where dp is the particle diameter (m) ut is the terminal particle settling velocity
(mmiddots-1) and ν is the kinematic fluid viscosity (m2middots-1) ν = η ρ where η is the
(absolute) dynamic fluid viscosity in centipoise (1 centipoise = 1 mPamiddots = 10-3
kgmiddotm-1middots-1) and ρ is the fluid density (kgmiddotm-3)
VI Chapter 7
At Re lt 1 we can apply Stokersquos particle settling velocity Stokersquos law is an
equation relating the terminal settling velocity of a smooth rigid sphere in a
viscous fluid of known density and viscosity to the diameter of the sphere when
subjected to a known force field
( )η
ρρsdot
minussdotsdot=
18
2pp
t
dgu (mmiddots-1)
where g = 981 mmiddots-2 is the gravitational constant ρp is the particle density (kgmiddotm-3)
The Sherwood number is the main parameter for prediction of the mass transfer
process
in fluid dynamics
3121 ScRe602Sh sdotsdot+=sdot
=lowast
lowast
iw
pL
Ddk
where Diw is the diffusion coefficient of the compound i in water (m2middots-1) is the
minimum (uncorrected) value of the mass transfer coefficient and Sc is the
Schmidt number This relation is often referred to as the Froumlssling correlation The
particle diameter is a key parameter in the Froumlssling correlation and the external
mass transfer coefficient varies with square of the particle size for smaller
particles
lowastLk
The Schmidt number is the ratio of the kinematic fluid viscosity and the diffusion
coefficient of the compound i in water
iwDν
=Sc
Supporting Information VII
According to Harriott (1962) the actual mass transfer coefficient kL is 15 times
greater than the minimum value of the mass transfer coefficient The
uncertainty in k
lowastLk
Lmiddota associated with particle sphericity and roughness issues are
believed not to exceed a factor of 2
The diffusion coefficient of a compound i in water can be estimated as (Hayduk amp
Laudie 1974)
5890141
9102613
iiw
VD
sdot
sdot=
minus
η (m2middots-1)
where iV is the molar volume of the compound i (cm3middotmol-1) estimated according
to Fuller et al 1966
Assuming spherical particles the external (geometric) specific surface area and the
particle diameter are calculated from the measured BET specific surface area Atot
assuming that our GR-SO4 has a AtotAouter ~ 30 similar to the one reported by
Hansen amp Koch (1998)
( ) ( ) ( )pppp
p
pp
p
dd
dV
SAAρρπ
πρ 1000
6
100061100030
SA3
2tot
geom sdot=
sdotsdot
sdot=
sdot== (m2middotg-1)
In our aqueous GR-SO4 system the density ρ = 1000 kgmiddotm-3 the absolute dynamic
viscosity η = 10-3 Pamiddots and the kinematic viscosity ν = 10-6 m2middots-1 for water The
GR-SO4 particle specific parameters used is found below
VIII Chapter 7
GR-SO4
Atot (m2g) 712
SAgeom (m2g) 237
Particle density ρp (kgm3) 1500
Particle diameter dp (m) 169middot10-6
Settling velocity ut (ms) 778middot10-7
Reynolds number 132middot10-6
The molar volumes the diffusion coefficients in water and the Schmidt numbers
for the NACs including 4-chloronitrobenzene (4-CNB) and nitrobenzene (NB)
were
Compound iV (cm3middotmol-1) Diw (m2middots-1) Sc
4-NT 1260 768middot10-10 1302
4-CNB 1230 779middot10-10 1284
4-NPA 1535 684middot10-10 1462
NB 1055 853middot10-10 1173
The ratio of the external (geometric) specific surface area to volume of solution
were calculated for GR-SO4 at the various concentrations applied
[Fe(II)GR]0 (mM) a (m-1)
103 366middot102
63 224middot103
126 448middot103
60 213middot103
The uncorrected mass transfer coefficients were estimated for the NACs using the
Froumlssling correlation
Supporting Information IX
[Fe(II)GR]0 (mM) lowastLk (mmiddots-1)
4-NT 912middot10-4
4-CNB 925middot10-4
4-NPA 812middot10-4
NB 101middot10-3
Finally kLmiddota was calculated and compared with the experimental 1 order rate
constants kobs obtained for the NACs
Compound [Fe(II)GR]0 (mM) kLmiddota (s-1) kobs (s-1) a
4-NT 103 050 420middot10-4
63 307 140middot10-3
126 613 590middot10-3
4-CNB 103 051 740middot10-4
63 311 170middot10-3
126 622 460middot10-3
4-NPA 103 045 640middot10-4
63 273 109middot10-3
126 546 473middot10-3
NB 60 324 137middot10-3 b
a Experimental pseudo 1 order rate constant at 50 microM [Ar-NO2]0 b Experimental pseudo 1 order rate constant at 10 microM [Ar-NO2]0
When comparing kLmiddota with kobs it can be seen that the rates of mass transfer for all
3 NACs exceed the observed rate constants by at least 3 or 4 orders of magnitude
at every initial Fe(II)GR concentration Thus the reaction of the given NACs with
GR-SO4 is not subject to mass transfer limitations under the experimental
conditions applied here
722 Surface saturation limited kinetics
More than 75 of all heterogeneous reactions that are not diffusion-limited are
surface-reaction-limited rather than adsorption- or desorption-limited We now
X Chapter 7
look at the reaction A = B = C where an intermediate B is formed In our system
A = Ar-NO2 B = Ar-NHOH and C = Ar-NH2 In this case the surface reaction is
assumed to be a single-site mechanism where only the site S on which A or B is
adsorbed is involved in the reaction forming B or C
KA
Adsorption 1 A + S = AmiddotS
kS1
Surface reaction 1 AmiddotS = BmiddotS
KB-1
Desorption 1 BmiddotS = B + S
KB
Adsorption 2 B + S = BmiddotS
kS2
Surface reaction 2 BmiddotS = CmiddotS
KC-1
Desorption 2 CmiddotS = C + S
The rate law for this surface-reaction limited single-site mechanism involving an
intermediate follows Langmuir-Hinshelwood kinetics (adopted from Fogler 1999)
CCBBAA
AAsitesSA
CKCKCKCKCk
dtdC
sdot+sdot+sdot+sdotsdotsdot
=minus1
1
Supporting Information XI
where kS1 is the intrinsic rate constant of the surface reaction transforming A into
the intermediate B Csites is the concentration of reactive sites S on the solid
surface KA KB and KC are the adsorption constants for A B and C at the reactive
surface sites and CA CB and CC are the concentrations of A B and C in the bulk
fluid Two major assumptions of the Langmuir isotherm imply that there is a fixed
number of localised surface sites present on the surface and that the activity of the
surface towards adsorption desorption or surface reaction is independent of
surface coverage
Hence fitting -∆CA∆t to CA CB and CC using a nonlinear curve fitting software
such as SigmaPlot may provide one with the intrinsic rate constant and the
adsorptions constants If KB and KC gtgt KA the intermediate and the product are
strongly competing with the reactant for vacant reactive surface sites
Our data was not fitted successfully by the Langmuir-Hinshelwood rate law
(regression results not shown) Simplifying the rate law by excluding either the
term KCmiddotCC or KBmiddotCB or both (assuming that the aniline product or the
hydroxylaniline intermediate or both did not compete for the reactive sites) did not
improve the regression The Langmuir-Hinshelwood rate law for a dual-site
mechanism did not fit our data either Thus Langmuir-Hinshelwood kinetics
cannot explain the reaction mechanism of the given NACs in our GR-SO4 system
73 External surface area of GR-SO4 and GR-CO3
The GR-SO4 unit cell consists of one double layer (d001 = 11 nm) ie one
hydroxide layer (049 nm ) and one interlayer (061 nm) Hexagonal GR-SO4
particles holding an average width of 1 microm (Figure 72) an average particle
thickness of 35 nm (Hansen amp Koch 1998) and a hydroxide layer thickness of
049 nm have a surface area of the basal plane
Abasal = 1 microm middot 1 microm ndash 2 middot 05 microm middot 025 microm = 075 microm2
XII Chapter 7
and a surface area of the edges
Aedge = (2 middot 05 microm + 4 middot 056 microm) middot 000049 microm = 00016 microm2
Figure 72 The hexagonal platy morphology of GR particles holding an average width of 1 microm
The particle thickness is the mean crystal thickness perpendicular to the 003 plane
as determined from the 003 reflections in an X-ray diffractogram A GR-SO4
particle holding a thickness of 35 nm contains 35 nm11 nm = 318 double layers
The GR-CO3 unit cell consists of one double layer (d001 = 075 nm) ie one
hydroxide layer (049 nm ) and one interlayer (026 nm) Hence a GR-CO3 particle
holding a thickness of 35 nm contains 35 nm075 nm = 467 double layers
The outer surface area of a GR-SO4 particle including outer basal planes and
edges is
Aouter = 222 microm 155microm 00016318microm 0752 =sdot+sdot
and the total surface area of a GR-SO4 particle including both inner and outer
basal planes as well as edges is
Supporting Information XIII
Atot = 222 microm 478)microm 00016microm 0752(318 =+sdotsdot
Hence the ratio of outer surface area to total surface area is
131microm 478microm 155
AA
2
2
tot
outer asymp=
Furthermore the ratio of edge surface area to outer surface area is
130microm 155
microm 00016318AA
2
2
outer
edge asympsdot
=
For GR-CO3 the outer surface area including outer basal planes and edges is
Aouter = 222 microm 157microm 00016746microm 0752 =sdot+sdot
and the total surface area of a GR-CO3 particle including both inner and outer
basal planes as well as edges is
Atot = 222 microm 701)microm 00016microm 0752(467 =+sdotsdot
Hence the ratio of outer surface area to total surface area is
145microm 701microm 157
AA
2
2
tot
outer asymp=
Furthermore the ratio of edge surface area to outer surface area is
121microm 157
microm 00016467AA
2
2
outer
edge asympsdot
=
XIV Chapter 7
74 Van der Waals radii
The size of polyatomic molecules can be estimated by summing the van der Waals
radii of the
individual atoms Van der Waals radii or nonbonded radii can be pictured as the
radii of hard spherical atoms (Figure 73)
Figure 73 Schematic of neighboring nonbonded atoms with van der Waals radii rA and rB
Assuming that the spheres of neighboring nonbonded atoms just touch (Figure
73) the highest possible ion or molecule size Ms can be estimated as the sum of
the van der Waals radii
Ms = 2middotrA + 2middotrB + (1)
Taking Paulingrsquos rule for nonmetals into account we can estimate the real size of
polyatomic ions bound by covalent bonds (Pauling 1960) The van der Waals
radius is larger than the covalent radius because it involves the interposition of two
electron pairs between the atoms rather than one The rule states that the van der
Waals radius of an atom exceeds its covalent radius by ~008 nm (overlap in
Figure 74)
Figure 74 Schematic of atoms undergoing covalent bonding
Supporting Information XV
Thus the size of polyatomic ions bound by covalent bonds Ms can now be
estimated as the sum of the van der Waals radii subtracted by 008 nm
Ms = 2middot(rA - 008 nm) + 2middot(rB - 008 nm) + (2)
We estimated the molecular size of the NACs (Table 72) by means of equation (2)
and the van der Waals radii of the atoms in Table 71
Table 71 Van der Waals radii of various atoms Values from Pauling 1960
Atom vdW radii (nm)
H 0120
O 0140
N 0150
C 0170
Cl 0181
S 0185
In order to make the calculations it was assumed that all atoms were spherical and
that all bond angles were 90deg or 180deg (linear structures) In addition no distinctions
were made between single and double bonds The molecular sizes of the NACs
were estimated with the benzene ring representing the xy plane
Table 72 Molecular sizes of the NACs a Thickness z of the xy plane
Compound Ms (x) (nm) Ms (y) (nm) Ms (z) (nm)a
NB 054 080 036
4-NT 054 106 036
4-CNB 054 100 036
4-NPA 054 136 036
XVI Chapter 7
Note that the molecular sizes in Table 72 are only rough estimations
For comparison with the GR-SO4 interlayer spacing (061 nm) we consider three
possible orientations of the NACs in the GR-SO4 interlayer 1) The NAC xyz
coordination is equivalent to the crystal abc coordination (z = c = 036 nm) 2) the
NAC xy plane is parallel to the crystal bc plane (z = a = 054 nm) and 3) the NAC
xy plane is parallel to the crystal ac plane (z = b = 080-136 nm) Hence the sizes
of the NACs do not hinder their access to the GR-SO4 interlayer Only when
oriented vertically do the sizes of the NACs (z = b = 080-136 nm) exceed the GR-
SO4 interlayer spacing
The molecular size of trichloroacetate (TCA) was also estimated by means of
equation (2) and the atomic van der Waals radii in Table 71 When the TCA
aliphatic chain is assumed to represent the x direction (Ms (x) = 066 nm) the
molecular size in the y and z directions ranges from 045-053 nm depending on the
free rotation of the C-C bond Thus only if the C-C bond is oriented perpendicular
to the crystal ab plane does the size of TCA exceed the GR-SO4 interlayer spacing
(061 nm) In contrast the size of TCA exceeds the GR-CO3 interlayer spacing
(026 nm) regardless of its orientation
75 Adsorption of Fe(II) onto Fe(III) oxides
As seen from the Fe(II) sorption isotherms Fe(II) sorption varies widely between
the Fe(III) oxides as a function of solution pH (Figure 75) Average surface
densities of approximately 2 singly coordinated sitesnm2 iron oxide have been
suggested for goethite and lepidocrocite (Cornell amp Schwertmann 1996) The
similar surface site densities of goethite and lepidocrocite might explain their
similar Fe(II) adsorption isotherms (Figure 75)
Supporting Information XVII
Figure 75 Fe(II) adsorption edges for ferrihydrite goethite hematite lepidocrocite and
magnetite in the absence of other specifically adsorbing cations and anions (from Vikesland amp
Valentine 2002 and references therein) The total number of surface sites was in excess of the
total Fe(II) concentrations in all experiments
Dissolved cations or anions may specifically adsorb at the calcite and Fe(III) oxide
surfaces by exchanging for H+ or OHndash at the equivCO3H0 equivCaOH0 equivFeOH0 and
equivFeIIIOFeIIOH0 surface sites At the experimental conditions applied here within a
pH range 70-87 the dominant species of interest in solution are Fe2+ HCO3ndash
CO32ndash Clndash SO4
2ndash (only in the GR-SO4 systems) and the anionic TCA and DCA In
addition Fe2+ readily forms aqueous complexes with hydroxide carbonate
chloride and sulfate whereby the species FeOH+ FeHCO3+ Fe(OH)(CO3)ndash
FeCO30 Fe(CO3)2
2ndash FeCl+ and FeSO40 may occur (Millero amp Hawke 1992) At
pH 70-87 we expect the Fe(II) species Fe2+ FeCO30 Fe(OH)(CO3)ndash FeOH+ and
Fe(CO3)22ndash to dominate in the GR-CO3 and CaCO3(s)CO2(g) buffered magnetite
suspensions In the goecalcite and lepcalcite suspensions we expect the FeCl+
species to dominate as well whereas the Fe2+ FeSO40 and FeOH+ species most
XVIII Chapter 7
likely dominate in the GR-SO4 suspensions Anionic inorganic ligands like
carbonate chloride and sulfate can lower or enhance the adsorption of Fe(II) due to
a) formation of stable nonadsorbing Fe(II) ligand aqueous complexes b) formation
of Fe(II) ligand Fe(III) oxide surface complexes which can lead to surface
precipitation at high Fe(II) and ligand concentrations c) competitive ligand
sorption to the Fe(III) oxide surface blocking reactive sorption sites at the surface
and d) diminution of the positive charge at the Fe(III) oxide surface (at pH levels
below the point of zero charge (pHpzc) of the Fe(III) oxide) thereby decreasing the
electrostatic repulsion of cations by the Fe(III) oxide surface Specifically adsorbed
cations increase the pHpzc whereas specifically adsorbed anions decrease the pHpzc
References Arnold WA Ball WP Roberts AL (1999) Polychlorinated ethane reaction with zero-valent zinc Pathways and rate control Journal of Contaminant Hydrology 40 183-200 Cornell RM Schwertmann U (1996) The iron oxides Structure properties reactions occurrence and uses VCH Verlagsgesellschaft mbH Weinheim Fogler HS (1999) Elements of chemical reaction engineering 3rd ed Prentice Hall Fuller EN Schettler PD Giddings JC (1966) A new method for prediction of binary gas-phase diffusion coefficients Industrial and Engineering Chemistry 58 19-27 Hansen HCB Koch CB (1998) Reduction of nitrate to ammonium by sulphate green rust Activation energy and interlayer reaction mechanism Clay Minerals 33 87-101 Harriott P (1962) Mass transfer to particles Part I Suspended in agitated tanks AIChE Journal 8 93-102 Hayduk W Laudie H (1974) Prediction of diffusion coefficients for nonelectrolytes in dilute aqueous solutions AIChE Journal 20 611-615 Hofstetter TB Heijmann CG Haderlein SB Holliger C Schwarzenbach RP (1999) Complete reduction of TNT and other (poly)nitroaromatic compounds under iron-reducing subsurface conditions Environmental Science and Technology 33 1479-1487 Klausen J Troumlber SP Haderlein SB Schwarzenbach RP (1995) Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions Environmental Science and Technology 29 2396-2404 Meisel D Neta P (1975) One-electron redox potentials of nitro compounds and radiosensitizers Correlation with spin densities of their radical anions Journal of the American Chemical Society 97 5198-5203
Supporting Information XIX
Millero FJ Hawke DJ (1992) Ionic interactions of divalent metals in natural waters Marine Chemistry 40 19-48 Pauling L (1960) The nature of the chemical bond 3rd ed Cornell University Press Ithaca Vikesland PJ Valentine RL (2002) Iron oxide surface-catalyzed oxidation of ferrous iron by monochloramine implications of oxide type and carbonate on reactivity Environmental Science and Technology 36 512-519 Wardman P (1989) Reduction potentials of one-eletron couples involving free radicals in aqueous solution The Journal of Physical Chemistry Reference Data 18 1637-1755
Curriculum Vitae 13011973 Born in Haderslev Denmark 1988-1991 Mathematical high school Haderslev Katedralskole Denmark 1991-1992 Sabbatical year 1992-1995 B Sc in environmental chemistry University of Copenhagen
(KU) Denmark B Sc thesis 1995 ldquoMethane oxidizing bacteria in soilrdquo
1995-1998 M Sc in environmental chemistry University of Copenhagen
Denmark 1997-1998 diploma thesis ldquoReductive dechlorination of carbon tetrachloride and chloroform in presence of iron(II)iron(III)-hydroxides (green rust)rdquo
1998-1999 Research and teaching assistant at the Chemistry Department
The Royal Veterinary amp Agricultural University (KVL) Denmark
1999-2004 PhD in environmental sciences Swiss Federal Institute of
Technology Zuumlrich (ETHZ) and Swiss Federal Institute for Environmental Science and Technology (EAWAG) Switzerland Docoral thesis ldquoFormation and redox reactions of green rusts under geochemical conditions found in natural soils and sedimentsrdquo
2000-2002 Teaching assistent at the Swiss Federal Institute of Technology
Zuumlrich and supervision of diploma students 2002 Microbial Diversity summer course (7 weeks) at the Marine
Biological Laboratory Woods Hole Massachusetts USA

Zusammenfassung I
Zusammenfassung Geschichtete Fe(II)-Fe(III)-Hydroxide (Gruumlner Rost) gehoumlren zur Gruppe der
Fe(II)-haltigen Mineralsysteme (zB Magnetit (Fe3O4) Siderit (FeCO3) Vivianit
(Fe2(PO4)2sdot8H2O) Fe(II)-Sulfide sowie an die Oberflaumlche von Fe(III)-Oxiden und
Tonmineralien gebundenes zweiwertiges Eisen) die die Aktivitaumlt von Fe(II) in
suboxischen und anoxischen Boumlden und Sedimenten kontrollieren Gruumlner Rost
Phasen (GRs) bestehen aus planaren positiv geladenen trioktaedrischen Fe(II)-
Fe(III)-Hydroxidschichten die durch hydratisierte Anionen in den
Zwischenschichten ausgeglichen werden Ihre generelle Zusammensetzung ist
[FeII(6-x)FeIII
x(OH)12]x+[(A)xnmiddotyH2O]x- wobei x = 09 - 42 ist A entspricht einem n-
valenten Anion (zB CO32- Clndash oder SO4
2-) und y repraumlsentiert die Anzahl
Wassermolekuumlle in der Zwischenschicht GRs sind wichtige intermediaumlre Phasen
die durch unvollstaumlndige Oxidation von Fe(II) oder teilweise Reduktion von Fe(III)
gebildet werden koumlnnen Sie koumlnnen in suboxischen nicht-sauren eisenhaltigen
natuumlrlichen wie auch technischen Systemen auftreten so wie in Wasser gesaumlttigten
Boumlden und interstitiellen Sedimenten Rohrleitungen in der
Trinkwasserversorgung Stahlpfosten in marinen Sedimenten Stahlbeton und in
reaktiven durchlaumlssigen Waumlnden aus nullwertigem Eisen zur in-situ Sanierung von
Altlasten und Aquiferen Aufgrund ihrer Schichtstruktur den anionischen
Zwischenschichten und der hohen spezifischen Oberflaumlchen sind GRs reaktive
Ionentauscher und Sorbentien von Anionen Des Weiteren wurde gezeigt dass
GRs eine Reihe anorganischer und organischer Schadstoffe reduzieren koumlnnen
Durch Immobilisierung und Transformation koumlnnen GRs somit eine wichtige Rolle
fuumlr das Abbauverhalten und den Transport solcher Schadstoffe in suboxischen
Boumlden und Sedimenten spielen Die Resultate dieser Dissertation tragen zum
Verstaumlndnis uumlber die Bildung und Reaktivitaumlt von Fe(II)-haltigen Mineralsystemen
wie GRs Vivianit Magnetit und an Goethit (α-FeOOH)- und Lepidokrozit (γ-
FeOOH)-Oberflaumlchen gebundenes Fe(II) in der Natur bei
II Zusammenfassung
Um die Rolle von Bakterien bei der Bildung von GRs in natuumlrlichen Boumlden und
Sedimenten aufzuklaumlren wurden Eisenminerale untersucht die als Folge der
Aktivitaumlt von eisenrespirierenden Bakterien gebildet wurden Kapitel 2 beschreibt
die Untersuchungen von eisenhaltigen Produkten die von anaeroben autotrophen
denitrifizierenden Fe(II)-oxidierenden Bakterien (FeOB) gebildet wurden Ein
Bikarbonat- und Phosphat-reiches Kulturmedium bot den nitratreduzierenden
FeOB optimale Bedingungen Fe(II) lag zu Anfang der Reaktion als weisses
Fe(II)-Hydroxyphosphat (Vivianit) und als geloumlstes Fe(II) vor Die Ergebnisse
zeigten dass die denitrifizierenden FeOB amorphen Goethit via ein gruumlnes Fe(III)-
angereichertes Vivianit-Zwischenprodukt bildeten Die Analyse mit Moumlssbauer
Spektroskopie deutet nicht auf eine Bildung von GR hin
In Kapitel 3 werden jene Eisenmineralien beschrieben die waumlhrend der Reduktion
verbreiteter Fe(III)-Oxide durch anaerobe dissimilative Fe(III)-reduzierende
Mikroorganismen Shewanella algae BrY gebildet wurden Um natuumlrliche
Zustaumlnde zu simulieren wurden Fe(III)-Oxide als Beschichtungen auf
Silikatpartikel (Modellsystem fuumlr Sandboumlden) oder Calcitpartikel (CaCO3
Modellsystem fuumlr kalkhaltige Boumlden) aufgetragen sowie synthetische
Elektronencarrier und hochkonzentrierte kuumlnstliche pH-Puffer ausgeschlossen
Die erforschten Mineralsysteme umfassten GoethitCalcit- LepidokrozitCalcit-
und FerrihydritSand-Suspensionen S algae BrY reduzierte beachtliche Mengen
des eingesetzten Fe(III) und es bildeten sich gruumlne und schwarze Festphasen
innerhalb von 1-2 Wochen nach der Animpfung Moumlssbauer Spektroskopie der
gruumlnen und schwarzen Praumlzipitate zeigte dass sich diese aus GR und Vivianit
zusammensetzen
Die Reaktivitaumlt synthetischer GRs gegenuumlber reduzierbaren organischen
Schadstoffen wurde erkundet um die potentielle Bedeutung von GR-Phasen fuumlr
das Schicksal solcher Verbindungen abzuschaumltzen Zu diesem Zweck wurden
Nitroaromaten (NACs) und Chloracetate als Modellverbindungen benutzt um
Zusammenfassung III
umweltrelevante Redoxreaktionen zu studieren In Kapitel 4 wurde die relative
Reaktivitaumlt von aumlusseren und inneren reaktiven Stellen in synthetischem Sulfat-
Gruumlnem Rost (GR-SO4) anhand von strukturaumlhnlichen ldquoreaktiven
Sondenmolekuumllenrdquo mit unterschiedlichen Ladungen untersucht Als reaktive
Sondenmolekuumlle wurden Nitrobenzen 2-Nitrophenol 4-Nitrotoluen 4-
Chlornitrobenzen und 4-Nitrophenylessigsaumlure verwendet Die Ergebnisse zeigen
dass GR-SO4 die NACs vollstaumlndig zu den entsprechenden Anilinen reduzierte
Die Reaktionen folgten einer pseudo 1 Ordnungs Kinetik bezuumlglich NAC und die
auf Oberflaumlche normalisierten pseudo 1 Ordnungs Geschwindigkeitskonstanten
(Anfangsraten) waren 016ndash465middot10-4 s-1middotm-2middotL fuumlr [Fe(II)GR]0 = 103-1260 mM
[NAC]0 = 20-102 microM und pH 84-86 Weder durch Einbezug von
Massentransferlimitierung noch von Oberflaumlchensaumlttigungskinetik war es moumlglich
die aumlhnlichen Oberflaumlchennormalisierten pseudo 1 Ordnungs
Geschwindigkeitskonstanten fuumlr die Reduktion der neutralen und anionischen
NACs durch GR-SO4 zu erklaumlren Dieser Umstand laumlsst vermuten dass die
Reaktion zwischen NAC und GR-SO4 an den externen reaktiven Fe(II)-Stellen
stattfindet Bei niedrigen Fe(II)GR-Anfangskonzentrationen wurden die externen
reaktiven Fe(II)-Stellen aufgebraucht und die Regenerierung von neuen externen
reaktiven Stellen haben schliesslich die Geschwindigkeit der Reduktion von NACs
durch GR-SO4 kontrolliert
In Kapitel 5 wurde die Reaktivitaumlt von verschiedenen umweltrelevanten Fe(II)-
Fe(III)-Mineralsystemen gegenuumlber Trichloressigsaumlure (TCA) und
Dichloressigsaumlure (DCA) in Batchexperimenten die natuumlrliche Bedingungen
imitierten untersucht Die Fe(II)-Fe(III)-Systeme umfassten Sulfat-Gruumlner Rost
Carbonat-Gruumlner Rost Magnetit Fe(II)Goethit und Fe(II)Lepidokrozit TCA
wurde von allen Fe(II)-haltigen Mineralien zu DCA reduziert Die Reaktionen
folgten einer pseudo 1 Ordnungs Kinetik bezuumlglich TCA und die auf Oberflaumlche
normalisierten pseudo 1 Ordnungs Geschwindigkeitskonstanten betrugen 033ndash
76middot10-5 min-1middotm-2middotL bei [Fe(II)]0 = 025ndash116 mM [TCA]0 = 15ndash1000 microM und pH
IV Zusammenfassung
70ndash87 Die Ergebnisse zeigen keine signifikanten Unterschiede zwischen den
verschiedenen Fe(II)-Fe(III)-Systemen bezuumlglich Produkteverteilung und
oberflaumlchen-normalisierten pseudo 1 Ordnungs Geschwindigkeits-konstanten In
keinem der Systeme wurde DCA innerhalb des experimentellen Zeitraums zu
Monochloressigsaumlure oder Essigsaumlure weiter reduziert
Die Ergebnisse die in dieser Dissertation praumlsentiert werden zeigen dass
mikrobiologische Prozesse fuumlr die Oxidation von Vivianit-Phasen im Untergrund
verantwortlich sein koumlnnen Zudem wurde nachgewiesen dass GRs bei tiefen
Kohlenstoff- und Fe(III)-Konzentrationen sowie durch Ausschluss von
kuumlnstlichen Elektronencarriern und pH-Pufferung mikrobiell gebildet werden
koumlnnen Ferner zeigten Befunde dass GRs eine bedeutende Rolle fuumlr die reduktive
Transformation von NACs und TCA in natuumlrlichen Boumlden und Sedimenten spielen
koumlnnen
Summary V
Summary
Layered iron(II)-iron(III)-hydroxides (green rusts) belong to the group of Fe(II)-
bearing mineral systems eg magnetite (Fe3O4) siderite (FeCO3) vivianite
(Fe2(PO4)2sdot8H2O) Fe(II) sulfides as well as Fe(II) associated with Fe(III) oxide
and clay mineral surfaces that control the Fe(II) activity in suboxic and anoxic
soils and sediments Green rusts (GRs) consist of plane positively charged
trioctahedral Fe(II)-Fe(III) hydroxide sheets balanced by hydrated anions in the
interlayers and hold the general formula [FeII(6-x)FeIII
x(OH)12]x+[(A)xnmiddotyH2O]x-
where x = 09 - 42 A is an n-valent anion eg CO32- Clndash or SO4
2- and y is the
number of water molecules in the interlayer GRs are important intermediate
phases formed by partial oxidation of Fe(II) or partial reduction of Fe(III) and they
have been found in suboxic non-acid iron-rich natural environments such as
hydromorphic soils and intertidal sediments and in engineering systems including
pipeline distribution systems for drinking water steel sheet piles in marine
sediments reinforced concrete and permeable reactive barriers of zero-valent iron
implemented for on-site remediation of contaminants Due to their layered
structures anionic interlayers and high specific surface areas GRs represent
reactive ion exchangers and sorbents of anions In addition GRs have been shown
to reduce a range of inorganic and organic pollutants Thus through sequestration
and reductive transformation GRs may play an important role in the fate and
transport of contaminants in suboxic soils and sediments The work presented in
this dissertation adds to the understanding of how Fe(II)-bearing minerals like
GRs vivianite magnetite and Fe(II) associated with goethite (α-FeOOH) and
lepidocrocite (γ-FeOOH) may form and react in nature
In order to elucidate the role of bacteria in the formation of GRs in natural soils
and sediments we studied the iron mineral phases forming as a result of the
activity of iron-respiring bacteria In the study described in chapter 2 the Fe-
containing products formed by anaerobic autotrophic denitrifying Fe(II)-oxidizing
VI Summary
bacteria (FeOB) were examined The culture medium applied contained high levels
of bicarbonate and phosphate and is typically used in this kind of studies as it
provides excellent conditions for the nitrate-reducing FeOB Fe(II) was present
initially as a whitish solid Fe(II) hydroxy phosphate (vivianite) and as soluble
Fe(II) The results obtained demonstrate that the denitrifying FeOB produce poorly
crystalline goethite via a greenish Fe(III)-enriched vivianite intermediate
Moumlssbauer spectroscopic analyses provided no significant evidence of green rust
formation
In chapter 3 the Fe-containing products formed during reduction of common
Fe(III) oxides by the anaerobic dissimilatory Fe(III)-reducing microorganism
Shewanella algae BrY are discussed In order to simulate natural conditions
Fe(III) oxides were applied as coatings on silica (model system for sandy soils) or
calcite (CaCO3) particles (model system for calcareous soils) and synthetic
electron shuttles as well as highly concentrated artificial pH buffers were excluded
The mineral systems studied include goethitecalcite lepidocrocitecalcite and
hydrous ferric oxidesand suspensions S algae BrY reduced substantial amounts
of the initial Fe(III) and green and blackish mineral phases were produced within
1-2 weeks after inoculation Moumlssbauer spectroscopic analyses showed that the
green and black precipitates consisted of GR and vivianite
The reactivity of synthetic GRs towards reducible organic pollutants was
investigated in order to asses the potential significance of GR phases for the fate of
such compounds To this end we used nitroaromatic compounds (NACs) and
chlorinated acetates as suitable model compounds for studying environmentally
relevant redox reactions In the work described in chapter 4 the relative reactivity
of outer and inner Fe(II) reactive sites in synthetic sulfate green rust (GR-SO4) was
studied using a series of structurally closely related compounds with different
charge properties as ldquoreactive probesrdquo The probe compounds included
nitrobenzene 2-nitrophenol 4-nitrotoluene 4-chloronitrobenzene and 4-
Summary VII
nitrophenylacetic acid The results show that NACs are completely reduced to their
corresponding anilines by GR-SO4 The reactions followed pseudo 1 order
kinetics with respect to NAC and the surface area-normalised pseudo 1 order rate
constants (initial rates) obtained were 016ndash465middot10-4 s-1middotm-2middotL at [Fe(II)GR]0 = 103-
1260 mM [NAC]0 = 20-102 microM and pH 84-86 Neither mass transfer control nor
surface saturation kinetics could explain the similarity of the surface-normalised
pseudo 1 order rate constants obtained for the reduction of the neutral and anionic
NACs by GR-SO4 These observations suggest that the reaction between NAC and
GR-SO4 takes place at the external reactive Fe(II) sites At low initial Fe(II)GR
concentrations the external reactive Fe(II) sites were depleted and the regeneration
of new external reactive sites eventually controlled the reduction of the NACs by
GR-SO4
Finally the reactivity of various Fe(II)-Fe(III) mineral systems towards
trichloroacetic acid (TCA) and dichloroacetate (DCA) has been investigated in
laboratory batch experiments imitating natural conditions (Chapter 5) The Fe(II)-
Fe(III)-systems investigated included GR-SO4 carbonate green rust magnetite
Fe(II)goethite and Fe(II)lepidocrocite TCA was readily reduced to DCA by all
Fe(II)-containing minerals The reactions followed pseudo 1 order kinetics with
respect to TCA and the surface area-normalised pseudo 1 order rate constants
obtained were 033ndash76middot10-5 min-1middotm-2middotL at [Fe(II)]0 = 025ndash116 mM [TCA]0 =
15ndash1000 microM and pH 70ndash87 Our results showed no significant differences
regarding product distribution and surface area-normalised reaction rate constants
between the Fe(II)-Fe(III)-systems DCA was not further reduced to
monochloroacetate (MCA) or acetate in any of the systems within the time frame
in our experiments
The results presented in chapter 2 indicate that microbiological processes may be
responsible for the oxidation of vivianite phases in natural subsurface
environments In chapter 3 we demonstrated that GRs may be produced
VIII Summary
microbially at conditions including low carbon and Fe(III) concentrations as well
as the exclusion of synthetic electron shuttles and pH buffers The results obtained
in chapter 4 and 5 show that GRs transform NACs and TCA readily The reductive
transformation of NACs and TCA by GRs is relevant to understanding the
processes responsible for their degradation in the subsurface and the development
of innovative technologies for their remediation
General Introduction 1
1 General Introduction
11 Iron cycling in the subsurface
Iron is the fourth most abundant element (4-5 mass) and the most abundant redox
sensitive element in the Earthrsquos crust It is found as Fe(II) and Fe(III) in a number
of minerals in rocks soils and sediments Under anoxic conditions solid Fe(III)-
containing minerals can be reduced to soluble Fe(II) once the more energetically
favoured electron donors - nitrate and manganese(IV) oxides - have been
consumed Dissolved Fe(II) can be reoxidized to insoluble Fe(III) microbially or
abiotically upon exposure to oxygen Due to this ready alternation between the
Fe(II) and Fe(III) redox states iron plays a major role in controlling the redox
potential and the carbon cycling in subsurface environments (Nealson amp Saffarini
1994)
Nonenzymatic processes were previously considered to account for most of the
Fe(III) reduction in subsurface environments The significance of bacteria in the
biogeochemical cycling of iron has been broadly recognized over the past two
decades Dissimilatory Fe(III)-reducing bacteria (DIRB) that gain energy by
coupling the oxidation of hydrogen or organic compounds to the reduction of
Fe(III) oxides have been known for many years but their biogeochemical
importance was only widely acknowledged about a decade ago (reviewed by
Lovley 1997) Fe(III) bioreduction accounts for a major fraction of the carbon
oxidation in many different anoxic environments and in the presence of sufficient
amounts of reactive Fe(III) microbial Fe(III) reduction may even inhibit sulphate
reduction and methanogenesis (King 1990 Lovley amp Phillips 1986) In fact most
of the Fe(III) reduction in the Fe(III) reduction zone of aquatic sediments and
aquifers is enzymatically catalyzed by microorganisms (Lovley et al 1991) A
wide diversity of DIRB distributed among several different phylogenetic groups
2 Chapter 1 is known today The two most studied DIRB are the obligate anaerobic Geobacter
spp and the facultatively anaerobic Shewanella spp (Figure 11)
Aerobic oxidation of Fe(II)-containing minerals by lithotrophic acidophilic and
neutrophilic bacteria has been known for many years but their broad significance
in the biogeochemical cycling of iron has only been recognized over the past two
decades Both acidophilic (Thiobacillus ferrooxidans) and neutrophilic
(Gallionella ferruginea Leptothrix ochracea Sphaerotilus natans) aerobic Fe(II)-
oxidizing bacteria (FeOB) have been isolated (Hanert 1992 Kuenen et al 1992
Mulder amp Deinema 1992)
Figure 11 The microbial iron cycle
Anaerobic Fe(II) oxidation by phototrophic purple non-sulfur bacteria utilizing
Fe(II) as an electron donor in the light was recognized only a decade ago (Widdel
et al 1993) Subsequently it was demonstrated that the biological oxidation of
Fe(II) in the absence of oxygen is possible by light-independent chemotrophic
microorganisms using nitrate as the electron acceptor (Straub et al 1996) Thus
the microbial iron cycle includes anaerobic Fe(III)-reducing microorganisms and
aerobic as well as anaerobic Fe(II)-oxidizing bacteria (Figure 11)
General Introduction 3
12 Green rusts
Iron oxides iron hydroxides and iron oxyhydroxides (collectively termed iron
oxides or Fe(III) oxides) are ubiquitous in the pedosphere where they originate
from aerobic weathering of surface magmatic rocks such as ferromagnesium
silicates and pyrite (Cornell amp Schwertmann 1996) Goethite (α-FeOOH)
lepidocrocite (γ-FeOOH) ferrihydrite (Fe5HO8sdot4H2O) hematite (α-Fe2O3)
magnetite (Fe3O4) maghemite (γ-Fe2O3) and akageneite (β-FeOOH) constitute the
most important iron oxides in soils and sediments (Schwertmann amp Cornell 1991)
The formation and transformation of iron oxides depend on pH solution
composition redox potential temperature rate of oxidationreduction and degree
and rate of hydrationdehydration Iron oxides are important to many soil
properties such as colour pH and redox buffer capacity aggregation with other
soil particles as well as retention of anions and cations (Cornell amp Schwertmann
1996) A number of Fe(II)-bearing minerals including Fe(II)-containing clays (eg
smectites vermiculites and micas) magnetite siderite (FeCO3) vivianite
(Fe2(PO4)2sdot8H2O) Fe(II) sulphides and green rusts (layered Fe(II)-Fe(III)
hydroxides) may be present in soils and sediments under suboxic and anoxic
conditions Green rusts are believed to play a central role as metastable
intermediates in the redox cycling of iron at circumneutral pH in aquatic and
terrestrial environments
Green rusts (GRs) are layered iron(II)-iron(III)-hydroxides consisting of plane
positively charged trioctahedral Fe(II)-Fe(III) hydroxide sheets balanced by
hydrated anions in the interlayers (cf Figure 41 this work) GRs belong
structually to the pyroaurite-sjoumlgrenite group of layered hydroxides and they hold
the general formula [FeII(6-x)FeIII
x(OH)12]x+[(A)xnmiddotyH2O]x- where x = 09 - 42 A is
an n-valent anion eg CO32- Clndash or SO4
2- and y is the number of water molecules
in the interlayer The three most common and investigated green rust forms include
chloride GR (GR-Cl) sulphate GR (GR-SO4) and carbonate GR (GR-CO3)
Generally GRs are crystallographically classified into the GRI (rhombohedral
4 Chapter 1 GR-Cl and GR-CO3) and GRII (hexagonal GR-SO4) crystal systems The GR
interlayer thickness is a function of both the size and the charge of the interlayer
anion Tetrahedrally coordinated anions like sulphate lead to larger interlayer
distances than smaller monoatomic anions like chloride or planar ions like
carbonate (Mendiboure amp Schoumlllhorn 1986) Not only size but also charge density
plays a role for the interlayer spacing That is for anions having the same number
of valence electrons anions with smaller ionic radii (higher electron density) are
bound more strongly and therefore result in smaller interlayer spacings The
interlayer in GR-SO4 is composed of two consecutive planes of anions and water
whereas GR-Cl and GR-CO3 interlayers consist of only one single plane (Simon et
al 2003)
GRs are important intermediate phases formed by partial oxidation of Fe(II) or
partial reduction of Fe(III) In neutral and weakly alkaline solutions the oxidation
of dissolved Fe(II) always passes through solid GR phases (Bernal et al 1959)
GRs may also form during oxidation of zero-valent iron and as a result of the
combination of Fe(II) and Fe(III) at circumneutral pH (Figure 12)
Figure 12 Formation and transformation of GRs Fe3O4 = magnetite γ-Fe2O3 = maghemite α-
FeOOH = goethite γ-FeOOH = lepidocrocite akageneite = β-FeOOH
General Introduction 5
Oxidation of GR-CO3 usually produces goethite and magnetite-maghemite
whereas GR-Cl and GR-SO4 transform into lepidocrocite and magnetite-
maghemite depending on pH and oxidation rate (Bernal et al 1959 Taylor 1980
Carlson amp Schwertmann 1990) The brown δ-FeOOH is formed by vigorous
oxidation of GR using air or a 30 aqueous solution of hydrogen peroxide (Bernal
et al 1959 Misawa et al 1974) Black ferromagnetic magnetite forms by slow
oxidation of GR whereas lepidocrocite forms at high oxidation rates (Misawa et
al 1974) The presence of chloride is a prerequisite for the formation of
akageneite (Bernal et al 1959 Refait amp Genin 1997)
A substantial amount of work has been conducted in order to estimate the free
energies of formation of green rusts The free energies of formation reported for
the carbonate and sulphate GRs fall in the range 4234ndash4384 kJsdotmol-1 as determined
from solution data monitored during anoxic alkalimetric titrations and from
reduction potential (Eh) and pH recordings monitored during oxidation of GRs in
aqueous solution (Hansen et al 1994 Drissi et al 1995 Genin et al 1996) The
free energies of formation provided allow for estimation of the stability domains of
GRs in Eh-pH phase diagrams (Drissi et al 1995 Genin et al 1996) As
evidenced from such diagrams (Figure 13) the stability domain of GR-SO4 lies
within pH 6-8 and Eh -700 ndash -400 mV depending on the activities of Fe(II) and
sulphate (compare Figures 13aampb) This agrees with the natural GR occurrences
found in suboxic non-acid iron-rich environments such as hydromorphic soils and
intertidal sediments (Al-Agha et al 1995 Trolard et al 1996 Genin et al 1998)
In addition GRs have been found as corrosion products in numerous engineering
systems including a pipeline distribution system for drinking water steel sheet
piles in marine sediments reinforced concrete (ferro-concrete) and permeable
reactive barriers of zero-valent iron implemented for on-site remediation of organic
and inorganic contaminants (Tuovinen et al 1980 Nielsen 1976 Genin et al
1991 Roh et al 2000)
6 Chapter 1
Figure 13 Eh-pH phase diagrams of GR-SO4 a) a = 10+2Fe-3 = 10minus2
4SOa -3 and b) a = 10+2Fe-2
= 10minus24SOa -1
The stability domains of GR-Cl and GR-CO3 are similar to the stability domain of
GR-SO4 At Fe(II) and sulphate activities lower than depicted in Figure 13b the
stability domain of GR-SO4 will be situated at higher pH and lower Eh Other
dissolved species present at anoxic conditions such as phosphate sulphide
carbonate and organic ligands may exert considerable effects on the availability of
Fe(II) and Fe(III) At anoxic and circumneutral conditions vivianite
(Fe2(PO4)2sdot8H2O) controls the Fe(II) activity even at very low phosphate
concentrations (Nriagu amp Dell 1974) The formation of solid Fe(II) sulphides and
siderite (FeCO3) as well as the complexation of Fe(II) and Fe(III) by organic
ligands may also control the activity of Fe(II) in the subsurface and thereby
interfere with the formation of GRs
Due to their layered structures anionic interlayers and high specific surface areas
GRs represent reactive ion exchangers and sorbents of environmentally concerning
anions eg arsenate and selenate (Myneni et al 1997 Randall et al 2001) In
addition GRs may incorporate divalent transition metal cations like Ni2+ Zn2+
Cd2+ Co2+ and Mg2+ by isomorphic substitution for Fe2+ in the hydroxide layers
General Introduction 7
(Tamaura 1985 Tamaura 1986 Refait et al 1994 Parmar et al 2001 Refait et
al 2001) Furthermore GRs have been shown to reduce a range of inorganic
contaminants such as nitrite nitrate selenate chromate uranyl pertechnetate and
the transition metals AgI AuIII CuII and HgII as well as organic pollutants
including halogenated ethanes ethenes and methanes (Hansen et al 1994 Hansen
et al 1996 Myneni et al 1997 Erbs et al 1999 Loyaux-Lawniczak et al 1999
Cui amp Spahiu 2002 Lee amp Batchelor 2002b Heasman et al 2003 OrsquoLoughlin et
al 2003aampb Pepper et al 2003 Elsner et al 2004 OrsquoLoughlin amp Burris 2004)
Thus through sequestration and reductive transformation GRs may play an
important role in the fate and transport of contaminants in suboxic soils and
sediments It should be noted that the rate constants reported for the reduction of
these inorganic and organic pollutants by GRs cannot be directly compared as the
various studies were conducted at very different experimental conditions
13 Microbial formation of green rusts
Generally one would expect that biogenic minerals have chemical compositions
and crystal habits similar to those produced by nonenzymatic processes as they are
governed by the same equilibrium principles In fact since the latter stages of
mineralization are abiotically driven and since the secondary Fe(II)-containing
minerals are formed indirectly by electron transfer outside the bacterial cell and not
directly inside the bacterial cell the type of iron mineral formed is a function of the
environmental conditions in which the bacteria live ie the same microorganism
form different minerals in different environments
The microbial formation of GRs resulting from bioreduction of various Fe(III)
oxides including ferrihydrite goethite and lepidocrocite by strains of the
anaerobic dissimilatory DIRB Shewanella putrefaciens has been reported
repeatedly over the last years (Fredrickson et al 1998 Kukkadapu et al 2001
Parmar et al 2001 Ona-Nguema et al 2002aampb Glasauer et al 2003)
However no evidence of biogenic formation of GRs at natural geochemical
8 Chapter 1 conditions have been offered and it is still unknown whether this process may take
place at natural conditions comprising low nutrient levels low iron concentrations
and the absence of synthetic electron shuttles and highly concentrated artificial pH
buffers Moreover the biotic formation of GRs by anaerobic denitrifying Fe(II)-
oxidizing bacteria has been suggested but the phases still need to be properly
identified (Chaudhuri et al 2001) In order to elucidate the role of bacteria in the
formation of GRs in natural soils and sediments we studied the iron mineral
phases forming as a result of the activity of iron-respiring bacteria (Chapters 2 and
3)
14 Redox reactions of green rusts
Fe(II) is one of the most abundant reductants present in aquatic and terrestrial
environments under suboxic and anoxic conditions (Lyngkilde amp Christensen
1992 Ruumlgge et al 1998) In these environments Fe(II) may be present as soluble
organic and inorganic complexes as surface complexes and as a host of Fe(II)-
bearing minerals Although aqueous Fe(II) complexes may reduce a number of
contaminants Fe(II) associated with mineral surfaces and structural Fe(II) present
in the mineral lattice in Fe(II)-containing minerals are often more powerful
reductants Fe(II)-bearing minerals including GRs magnetite siderite Fe(II)
sulphides as well as Fe(II)-carrying Fe(III) oxide and clay mineral surfaces have
been shown to reduce a number of organic and inorganic contaminants such as
nitroaromatic compounds chlorinated aliphatics chromate uranyl pertechnetate
nitrate monochloramine and carbamate pesticides (Klausen et al 1995 Cui amp
Eriksen 1996 Butler amp Hayes 1998amp1999 Erbs et al 1999 Liger et al 1999
Loyaux-Lawniczak et al 1999 Amonette et al 2000 Hwang amp Batchelor 2000
Hansen et al 2001 Gander et al 2002 Lee amp Batchelor 2002aampb Pecher et al
2002 Vikesland amp Valentine 2002 Hofstetter et al 2003 OrsquoLoughlin et al
2003aampb Strathmann amp Stone 2003 Elsner et al 2004 OrsquoLoughlin amp Burris
2004) However only few comparative studies on the reactivity of Fe(II)-bearing
minerals exist (Lee amp Batchelor 2002b Elsner et al 2004) When examining the
General Introduction 9
reaction rates of the reductive transformation of NACs and chlorinated aliphatics
by GRs and other Fe(II)-bearing minerals reported in these studies the rate
constants for GRs are mostly among the highest rates reported and in some cases
even higher than the rate constants for Fe(II) sulphides Thus GRs may play an
important role in the transformation of reducible contaminants in the subsurface
Nitroaromatic compounds (NACs) are widely applied as explosives herbicides
insecticides solvents and intermediates in the synthesis of dyes and pesticides
(Hartter 1985 Rosenblatt et al 1991) NACs are ubiquitous in the subsurface
environment and pose a health risk due to their toxicity (Rickert 1985) In anoxic
environments reduction of the nitro group is generally the first step during abiotic
or microbial transformation of the NACs (Macalady et al 1986) The
transformation reaction generally produces the corresponding aromatic amines and
minor amounts of intermediates (hydroxylamines and nitroso compounds) as well
as coupling products (azo and azoxy compounds) These products may be of
similar or even greater environmental concern
Trichloroacetic acid (TCA) is ubiquitous in soils and the concentrations reported
range from lt005 to 380 microgkg (Euro Chlor 2001 McCulloch 2002 Ahlers et al
2003) On account of its phytotoxicity suspected human carcinogenicity and
widespread occurrence TCA is of considerable environmental concern especially
in the terrestrial compartment (Ahlers et al 2003) Moreover the daughter
compounds of TCA - dichloroacetic acid (DCA) and monochloroacetic acid
(MCA) - are also toxins and suspected human carcinogens as well as widespread in
the environment (Reimann et al 1996 Berg et al 2000 Ahlers et al 2003 and
references therein) In this work the reactivity of synthetic green rusts towards
nitroaromatic compounds (NACs) and the reactivity of various Fe(II)-Fe(III)
mineral systems including synthetic GRs towards chlorinated acetates have been
studied (Chapters 4 and 5)
10 Chapter 1 15 Outline of the thesis
An examination of the Fe-containing products produced during solid state
oxidation of vivianite by anaerobic autotrophic denitrifying Fe(II)-oxidizing
bacteria is presented in chapter 2 The Fe(II)-oxidizing bacteria were cultured in a
mineral medium containing high levels of bicarbonate and phosphate which is
typically used in this kind of studies as it provides excellent conditions for the
nitrate-reducing FeOB The solid iron phases forming were investigated by
transmission Moumlssbauer spectroscopy infrared spectroscopy and scanning electron
microscopy
Chapter 3 includes a study on the Fe-containing products formed during reduction
of common Fe(III) oxides by the anaerobic dissimilatory Fe(III)-reducing
microorganism Shewanella algae BrY In order to simulate natural conditions
Fe(III) oxides were applied as coatings on silica (model system for sandy soils) or
calcite particles (model system for calcareous soils) and synthetic electron shuttles
as well as highly concentrated artificial pH buffers were excluded The mineral
systems studied include goethitecalcite lepidocrocitecalcite and hydrous ferric
oxidesand suspensions The solid iron phases produced were examined by
transmission Moumlssbauer spectroscopy
A study on the relative reactivity of outer and inner Fe(II) sites in synthetic GR-
SO4 by using a series of structurally closely related compounds with different
charge properties as ldquoreactive probesrdquo is presented in chapter 4 The probe
compounds included nitrobenzene 2-nitrophenol 4-nitrotoluene 4-
chloronitrobenzene and 4-nitrophenylacetic acid
In chapter 5 an investigation of the reactivity of various Fe(II)-Fe(III) mineral
systems towards TCA and DCA is presented The study included laboratory batch
experiments imitating natural conditions The Fe(II)-Fe(III)-systems investigated
included GR-SO4 carbonate green rust magnetite Fe(II)goethite and
General Introduction 11
Fe(II)lepidocrocite The reactivities of the Fe(II)-Fe(III) mineral systems were
examined by comparing their surface-normalized rate constants
The results and environmental implications of this work are summarized in chapter
6 References Ahlers J Regelmann J Riedhammer C (2003) Environmental risk assessment of airborne trichloroacetic acid - a contribution to the discussion of the significance of anthropogenic and natural sources Chemosphere 52 531-537 Al-Agha MR Burley SD Curtis CD Esson J (1995) Complex cementation textures and authigenic mineral assemblages in recent concretions from the Lincolnshire Wash (east coast UK) driven by Fe(0) to Fe(II) oxidation Journal of the Geological Society 152 157-171 Amonette JE Workman DJ Kennedy DW Fruchter JS Gorby YA (2000) Dechlorination of carbon tetrachloride by Fe(II) associated with goethite Environmental Science and Technology 34 4606-4613 Berg M Muumlller SR Muumlhlemann J Wiedmer A Schwarzenbach RP (2000) Concentrations and mass fluxes of chloroacetic acids and trifluoroacetic acid in rain and natural waters in Switzerland Environmental Science and Technology 34 2675-2683 Bernal JD Dasgupta DR Mackay AL (1959) The oxides and hydroxides of iron and their structural inter-relationships Clay Minerals Bulletin 4 15-30 Butler EC Hayes KF (1998) Effects of solution composition and pH on the reductive dechlorination of hexachloroethane by iron sulfide Environmental Science and Technology 32 1276-1284 Butler EC Hayes KF (1999) Kinetics of the transformation of trichloroethylene and tetrachloroethylene by iron sulfide Environmental Science and Technology 33 2021-2027 Carlson L Schwertmann U (1990) The effect of CO2 and oxidation rate on the formation of goethite versus lepidocrocite from an Fe(II) system at pH 6 and 7 Clay Minerals 25 65-71 Chaudhuri SK Lack JG Coates JD (2001) Biogenic magnetite formation through anaerobic biooxidation of Fe(II) Applied and Environmental Microbiology 67 2844-2848 Cornell RM Schwertmann U (1996) The iron oxides Structure properties reactions occurrence and uses VCH Verlagsgesellschaft mbH Weinheim Cui D Eriksen TE (1996) Reduction of pertechnetate by ferrous iron in solution influence of sorbed and precipitated Fe(II) Environmental Science and Technology 30 2259-2262
Cui D Spahiu K (2002) The reduction of U(VI) on corroded iron under anoxic conditions Radiochemica Acta 90 623-628
12 Chapter 1 Drissi SH Refait Ph Abdelmoula M Geacutenin JMR (1995) The preparation and thermodynamic properties of Fe(II)-Fe(III) hydroxide-carbonate (green rust I) Pourbaix diagram of iron in carbonate-containing aqueous media Corrosion Science 37 2025-2041 Elsner M Haderlein SB Schwarzenbach RP (2004) Reactivity of Fe(II)-bearing minerals towards reductive transformation of organic contaminants Environmental Science and Technology 38 799-807 Erbs M Hansen HCB Olsen CE (1999) Reductive dechlorination of carbon tetrachloride using iron(II)iron(III)-hydroxide-sulphate (green rust) Environmental Science and Technology 33 307-311 Euro Chlor (2001) Trichloroacetic acid in the environment a dossier Euro Chlor Brussels and the European Chlorinated Solvent Association Fredrickson JK Zachara JM Kennedy DW Dong H Onstott TC Hinman NW Li S (1998) Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium Geochimica et Cosmochimica Acta 62 3239-3257 Gander JW Parkin GF Scherer MM (2002) Kinetics of 111-trichloroethane transformation by iron sulfide and a methanogenic consortium Environmental Science and Technology 36 4540-4546 Geacutenin JMR Bourrieacute G Trolard F Abdelmoula M Jaffrezic A Refait Ph Maitre V Humbert B Herbillon A (1998) Thermodynamic equilibria in aqueous suspensions of synthetic and natural Fe(II)-Fe(III) green rusts Occurrences of the mineral in hydromorphic soils Environmental Science and Technology 32 1058-1068 Geacutenin JMR Olowe AA Benbouzid-Rollet ND Prieur D Confente M Resiak B (1991) The simultaneous presence of green rust 2 and sulfate reducing bacteria in the corrosion of steel sheet piles in a harbour area Hyperfine Interactions 69 875-878 Geacutenin JMR Olowe AA Refait Ph Simon L (1996) On the stoichiometry and Pourbaix diagram of Fe(II)-Fe(III) hydroxy-sulphate of sulphate-containing green rust 2 An electrochemical and Moumlssbauer spectroscopy study Corrosion Science 38 1751-1762 Glasauer S Weidler PG Langley S Beveridge TJ (2003) Controls on Fe reduction and mineral formation by a subsurface bacterium Geochimica et Cosmochimica Acta 67 1277- 1288 Hanert HH (1992) The genus Gallionella In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 4082-4088 Hansen HCB Borggaard OK Soslashrensen J (1994) Evaluation of the free energy of formation of iron(II)iron(III)-hydroxidesulphate (Green Rust) and its reduction of nitrite Geochimica et Cosmochimica Acta 58 2599-2608 Hansen HCB Guldberg S Erbs M Koch CB (2001) Kinetics of nitrate reduction by green rusts ndash effects of interlayer anion and Fe(II)Fe(III) ratio Applied Clay Science 18 81-91
General Introduction 13
Hansen HCB Koch CB Nancke-Krogh H Borggaard OK Soslashrensen J (1996) Abiotic nitrate reduction to ammonium Key role of green rust Environmental Science and Technology 30 2053-2056 Hartter DR (1985) The use and importance of nitroaromatic chemicals in the chemical industry In Toxicity of nitroaromatic compounds Rickert DE (ed) Hemisphere Publishing Corporation 1-13 Heasman DM Sherman DM Ragnarsdottir KV (2003) The reduction of aqueous Au3+ by sulfide minerals and green rust phases American Mineralogist 88 725-738 Hofstetter TB Schwarzenbach RP Haderlein SB (2003) Reactivity of Fe(II) species associated with clay minerals Environmental Science and Technology 37 519-528 Hwang I Batchelor B (2000) Reductive dechlorination of tetrachloroethylene by Fe(II) in cement slurries Environmental Science and Technology 34 5017-5022 King GM (1990) Effects of added manganic and ferric oxides on sulfate reduction and sulfide oxidation in intertidal sediments FEMS Microbiology Ecology 73 131-138 Klausen J Troumlber SP Haderlein SB Schwarzenbach RP (1995) Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions Environmental Science and Technology 29 2396-2404 Kuenen JG Robertson LA Tuovinen OH (1992) The genera Thiobacillus Thiomicrospira and Thiosphaera In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 2618-2624
Kukkadapu RK Zachara JM Smith SC Fredrickson JK Liu C (2001) Dissimilatory bacterial reduction of Al-substituted goethite in subsurface sediments Geochimica et Cosmochimica Acta 65 2913-2924 Lee W Batchelor B (2002a) Abiotic reductive dechlorination of chlorinated ethylenes by iron- bearing soil minerals 1 Pyrite and magnetite Environmental Science and Technology 36 5147- 5154 Lee W Batchelor B (2002b) Abiotic reductive dechlorination of chlorinated ethylenes by iron-bearing soil minerals 2 Green rust Environmental Science and Technology 36 5348- 5354 Liger E Charlet L Van Cappellen P (1999) Surface catalysis of uranium (VI) reduction by iron(II) Geochimica et Cosmochimica Acta 63 2939-2955 Lovley DR (1997) Microbial Fe(III) reduction in subsurface environments FEMS Microbiology Reviews 20 305-313 Lovley DR Phillips EJP (1986) Organic matter mineralization with reduction of ferric iron in anaerobic sediments Applied and Environmental Microbiology 51 683-689 Lovley DR Phillips EJP Lonergan DJ (1991) Enzymatic versus nonenzymatic mechanisms for Fe(III) reduction in aquatic sediments Environmental Science and Technology 25 1062-1067
14 Chapter 1 Loyaux-Lawniczak S Refait Ph Lecomte P Ehrhardt J Geacutenin JMR (1999) The reduction of chromate ions by Fe(II) layered hydroxides Hydrology and Earth System Sciences 3 593-599 Lyngkilde J Christensen TH (1992) Redox zones of a landfill leachate pollution plume (Vejen Denmark) Journal of Contaminant Hydrology 10 273-289 Macalady DL Tratnyek PG Grundl TJ (1986) Abiotic reduction reactions of anthropogenic organic chemicals in anaerobic systems A critical review Journal of Contaminant Hydrology 1 1-28 McCulloch A (2002) Trichloroacetic acid in the environment Chemosphere 47 667-686 Mendiboure A Schoumlllhorn A (1986) Formation and anion exchange reactions of layered transition metal hydroxides [Ni1-xMx](OH)2(CO3)x2(H2O)z (M = Fe Co) Revue de Chimie Mineacuterale 23 819-827 Misawa T Hashimoto K Shimodaira S (1974) The mechanism of formation of iron oxide and oxyhydroxides in aqueous solutions at room temperature Corrosion Science 14 131-149 Mulder EG Deinema MH (1992) The sheathed bacteria In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 2618-2624 Myneni SCB Tokunaga TK Brown Jr GE (1997) Abiotic selenium redox transformations in the presence of Fe(IIIII) oxides Science 278 1106-1109 Nealson KH Saffarini D (1994) Iron and manganese in anaerobic respiration Environmental significance physiology and regulation Annual Review of Microbiology 48 311-343 Nielsen A (1976) Hvid groslashn og sort rust Beskrivelse af en korrosionsskade paring et svoslashmmebassin Nordisk Betong 2 21-24 Nriagu JO Dell CI (1974) Diagenetic formation of iron phosphates in recent lake sediments American Mineralogist 59 934-946 OLoughlin EJ Burris DR (2004) Reduction of halogenated ethanes by green rust Environmental Toxicology and Chemistry 23 41-48 OLoughlin EJ Kelly SD Cook RE Csencsits R Kemner KM (2003a) Reduction of uranium(VI) by mixed iron(II)iron(III) hydroxide (green rust) Formation of UO2 nanoparticles Environmental Science and Technology 37 721-727 OLoughlin EJ Kelly SD Kemner KM Csencsits R Cook RE (2003b) Reduction of AgI AuIII CuII and HgII by FeIIFeIII hydroxysulfate green rust Chemosphere 53 437-446 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002a) Iron (IIIII) hydroxycarbonate green rust formation and stabilization from lepidocrocite bioreduction Environmental Science and Technology 36 16-20 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002b) Microbial reduction of lepidocrocite γ-FeOOH by Shewanella putrefaciens The
General Introduction 15
formation of green rust Hyperfine Interactions 139140 231-237 Parmar N Gorby YA Beveridge TJ Ferris FG (2001) Formation of green rust and immobilization of nickel in response to bacterial reduction of hydrous ferric oxide Geomicrobiology Journal 18 375-385 Pecher K Haderlein SB Schwarzenbach RP (2002) Reduction of polyhalogenated methanes by surface-bound Fe(II) in aqueous suspensions of iron oxides Environmental Science and Technology 36 1734-1741 Pepper SE Bunker DJ Bryan ND Livens FR Charnock JM Pattrick RAD Collison D (2003) Treatment of radioactive wastes An X-ray absorption spectroscopy study of the reaction of technetium with green rust Journal of Colloid and Interface Science 268 408- 412 Randall SR Sherman DM Ragnarsdottir KV (2001) Sorption of As(V) on green rust (Fe4(II)Fe2(III)(OH)12SO4
3H2O) and lepidocrocite (γ-FeOOH) Surface complexes from EXAFS spectroscopy Geochimica et Cosmochimica Acta 65 1015-1023 Refait Ph Abdelmoula M Trolard F Geacutenin JMR Ehrhardt JJ Bourrieacute G (2001) Moumlssbauer and XAS study of a green rust mineral the partial substitution of Fe2+ by Mg2+ American Mineralogist 86 731-739 Refait Ph Drissi SH Marie Y Geacutenin JMR (1994) The substitution of Fe2+ ions by Ni2+ ions in green rust one compounds Hyperfine Interactions 90 389-394 Refait Ph Geacutenin JMR (1997) The mechanisms of oxidation of ferrous hydroxychloride β- Fe2(OH)3Cl in aqueous solution The formation of akaganeite vs goethite Corrosion Science 39 539-553 Reimann S Grob K Frank H (1996) Chloroacetic acids in rainwater Environmental Science and Technology 30 2340-2344 Rickert DE (1985) Toxicity of nitroaromatic compounds Hemisphere Publishing Corporation 1-13 Roh Y Lee SY Elless MP (2000) Characterization of corrosion products in the permeable reactive barriers Environmental Geology 40 184-194 Rosenblatt DH Burrows EP Mitchell WR Parmer DL (1991) Organic explosives and related compounds In The Handbook of Environmental Chemistry Anthropogenic compounds Hutzinger O (Ed) Springer-Verlag 195-234 Ruumlgge K Hofstetter TB Haderlein SB Bjerg PL Knudsen S Zraurig C Mosbaeligk H Christensen TH (1998) Characterization of predominant reductants in an anaerobic leachate- affected aquifer by nitroaromatic probe compounds Environmental Science and Technology 32 23-31 Schwertmann U Cornell RM (1991) Iron oxides in the laboratory Preparation and characterization VCH Verlagsgesellschaft mbH Weinheim Simon L Francois M Refait Ph Renaudin G Lelaurain M Geacutenin JMR (2003)
16 Chapter 1 Structure of the Fe(II-III)-layered double hydroxysulphate green rust two from Rietveld analysis Solid State Sciences 5 327-334 Strathmann TJ Stone AT (2003) Mineral surface catalysis of reactions between FeII and oxime carbamate pesticides Geochimica et Cosmochimica Acta 67 2775-2791 Straub KL Benz M Schink B Widdel F (1996) Anaerobic nitrate-dependent microbial oxidation of ferrous iron Applied and Environmental Microbiology 62 1458-1460 Tamaura Y (1985) ZnII-bearing green rust II and its spontaneous transformation into ZnII- bearing ferrite in aqueous solution Bulletin of the Chemical Society of Japan 58 2951-2954 Tamaura Y (1986) Ni(II)-bearing green rust II and its spontaneous transformation into Ni(II)- bearing ferrites Bulletin of the Chemical Society of Japan 59 1829-1832 Taylor RM (1980) Formation and properties of Fe(II)Fe(III)-hydroxycarbonate and its possible significance in soil formation Clay Minerals 15 369-382 Trolard F Abdelmoula M Bourrieacute G Humbert B Geacutenin JMR (1996) Mise en eacutevidence dun constituant de type rouilles vertes dans les sols hydromorphes Proposition de lexistence dun nouveau mineacuteral la fougeacuterite Geacuteosciences de surface Comptes Rendus de LrsquoAcademie des Sciences 323 1015-1022 Tuovinen OH Button KS Vuorinen A Carlson L Mair DM Yut LA (1980) Bacterial chemical and mineralogical characteristics of tubercles in distribution pipelines Journal of the American Water Works Association 72 626-635 Vikesland PJ Valentine RL (2002) Iron oxide surface-catalyzed oxidation of ferrous iron by monochloramine implications of oxide type and carbonate on reactivity Environmental Science and Technology 36 512-519 Widdel F Schnell S Heising S Ehrenreich A Assmus B Schink B (1993) Ferrous iron oxidation by anoxygenic phototrophic bacteria Nature 362 834-836
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 17
2 Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria
Abstract
This work investigated the Fe-containing products formed by anaerobic
autotrophic denitrifying Fe(II)-oxidizing bacteria in a specific bicarbonate buffered
(30 mM HCO3- pH 70) culture media containing 10 mM Fe(II) 4 mM nitrate and
4 mM phosphate Fe(II) was present initially as a whitish vivianite-like
(Fe3(PO4)2middot8H2O) precipitate and as soluble Fe(II) The initial phase of the
oxidation produced a greenish metavivianite-like ((FeII3-xFeIII
x)(PO4)2(OH)xmiddot(8-
x)H2O x gt 12) phase In the late oxidation phase a reddish precipitate of poorly
crystalline goethite (α-FeOOH) dominated the colour of the media in coexistence
with Fe(II)-containing siderite (FeCO3) The increasing amounts of Fe(III) present
in the ldquovivianiterdquo and ldquometavivianiterdquo structures were accompanied by an
increasing intensity in the green colour as the Fe(II) biooxidation progressed This
colour development has produced the idea of biogenic green rusts (layered Fe(II)-
Fe(III) hydroxides) in several studies on nitrate-dependent Fe(II) biooxidation
However in this work no evidence of green rust formation mediated by anaerobic
denitrifying Fe(II)-oxidizing bacteria was obtained
21 Introduction
Aerobic oxidation of Fe(II)-containing minerals by lithotrophic acidophilic and
neutrophilic bacteria has been known for many years but their broad significance
in the biogeochemical cycling of iron has only been recognized over the past two
decades Aerobic Fe(II)-oxidizing bacteria (FeOB) have been isolated from acidic
ecosystems (Thiobacillus ferrooxidans) neutral eutrophic systems (Sphaerotilus
natans Leptothrix ochracea) and neutral oligotrophic systems (Gallionella
ferruginea) (Hanert 1992 Kuenen et al 1992 Mulder amp Deinema 1992) At
neutral pH Fe(II) is unstable in the presence of oxygen and is rapidly oxidized to
the insoluble Fe(III) Hence the only pH neutral environments where soluble
18 Chapter 2
Fe(II) is available for aerobic FeOB are at interfaces between oxic and anoxic
conditions The aerobic neutrophilic FeOB (Leptothrix ochracea Gallionella
ferruginea and Sphaerotilus natans) live at such interfaces and are usually
associated with the yellowishreddish ferric deposits formed there
Over the past several years there has been a growing recognition that other less
readily detectable types of bacteria are involved in Fe(II) oxidation in ecosystems
at circumneutral pH For example it has been reported that neutrophilic FeOB are
abundant at the Loihi seamount hydrothermal vents and play a major role in the
Fe(III) oxide deposition (Emerson amp Moyer 2002) Similarly unidentified
neutrophilic obligate lithotrophic FeOB have been isolated from the rhizosphere of
wetlands plants where they are closely associated with deposits of amorphous
Fe(III) oxides (Emerson et al 1999) It was previously believed that Fe(III) oxide
deposits associated with sheaths were produced biologically whereas Fe(III) oxide
deposits not associated with cells were produced abiotically Recently the
formation of amorphous Fe(III) oxide in gradient tubes has been attributed to the
action of FeOB (Sobolev amp Roden 2001) The authors attribute 90 of the
oxidation to biological processes and indicated that the organisms seem to produce
a mobile form of Fe(III) that diffuses away from the cells before being
precipitated thereby avoiding encrustation of the cells They suggest that such
soluble Fe(III) complexes might be substrates for closely associated Fe(III)-
reducing bacteria Such an arrangement might allow close coupling between
microbial Fe(II) oxidation and Fe(III) reduction within millimeters of the oxic-
anoxic interface
Anaerobic Fe(II) oxidation by phototrophic purple non-sulfur bacteria utilizing
Fe(II) as an electron donor in the light was recognized only a decade ago (Widdel
et al 1993) Subsequently it was demonstrated that the biological oxidation of
Fe(II) in the absence of oxygen is possible by light-independent chemotrophic
microbial activity using nitrate as the electron acceptor (Straub et al 1996) In
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 19
addition studies conducted in gradient cultures revealed that nitrate-reducing
strains could also oxidize Fe(II) with molecular oxygen (Benz et al 1998) Hence
these Fe(II)-oxidizing strains may use nitrate as well as oxygen as electron
acceptors The microbial oxidation of Fe(II) was coupled to stoichiometric
reduction of nitrate to N2 and only one strain produced traces of N2O as a by-
product (Straub et al 1996 Benz et al 1998) The authors proposed the formation
of 2-line ferrihydrite as the end product of Fe(II) biooxidation The chemical
reduction of nitrate by Fe(II) requires a catalyst eg at least 10 microM Cu2+ in order
to take place at significant rates and may thus be considered insignificant under the
conditions applied in our study (Moraghan amp Buresh 1976) The chemical
oxidation of Fe(II) with nitrous oxide has not been observed However nitrite can
oxidize Fe(II) chemically (Moraghan amp Buresh 1977 Straub et al 1996) but this
process is considered insignificant at the conditions applied here No denitrifying
Fe(II)-oxidizing enrichment culture has been found to produce ammonium from
nitrate
Both lithoheterotrophic (depending on organic cosubstrates such as acetate) and
strictly lithoautotrophic nitrate-reducing FeOB have been found in various marine
and freshwater sediments However most isolates depend on organic cosubstrates
for cell biosynthesis (Benz et al 1998) Most probable number estimations
showed that denitrifying FeOB accounted for 00006-08 of the acetate-oxidizing
denitrifying microbial population Lithotrophic FeOB accounted for less than
00001 of the total bacterial community Attempts to isolate CO2-fixing nitrate-
dependent FeOB from lithotrophic cultures have failed (Straub amp Buchholz-
Cleven 1998) Mixotrophic FeOB accounted for 0004-004 of the total bacterial
community In addition microbial nitrate-dependent Fe(II) oxidation was
demonstrated in a flooded paddy soil as well as in activated sludge from a
wastewater treatment plant (Nielsen amp Nielsen 1998 Ratering amp Schnell 2001)
Since the activity is not restricted to sunlight exposed habitats microbial nitrate-
dependent Fe(II) oxidation is supposedly more important on a global scale than
20 Chapter 2
anaerobic Fe(II) oxidation by phototrophic bacteria Furthermore it has been
reported that anaerobic denitrifying FeOB aptly oxidize biogenic Fe(II) minerals
formed by bioreduction of synthetic goethite and ferrihydrite and that anaerobic
Fe(III)-reducing bacteria readily reduce Fe(III) minerals formed by biooxidation of
Fe(II) (Weber et al 2001 Straub et al 1998) Hence autotrophic denitrifying
FeOB may play a significant role in the nitrogen and iron cycles in subsurface
environments where the nitrate and the Fe(II) zones overlap and organic carbon
supply is limited (Figure 21)
Figure 21 The microbial iron cycle linking the carbon and nitrogen cycles
Phosphate is released into the environment through natural processes such as rock
weathering and decomposition of dead organic material and anthropogenic
activities eg wastewater effluents and application of manure and fertilizers in
horti- and agriculture In anoxic soils and sediments phosphate may be sequestered
by sorption onto Fe(III) oxides (Williams et al 1971 Patrick amp Khalid 1974)
Phosphate strongly influences the type morphology and properties of Fe(III)
oxides formed by oxidation and hydrolysis of Fe(II) salts as well as the degree of
their transformation (Kandori et al 1992 Cumplido et al 2000 Benali et al
2001) Phosphate may also be retained by precipitation of Fe(II) phosphates such
as the monoclinic vivianite (Fe3(PO4)2middot8H2O) which is the most important stable
Fe(II) orthophosphate solid encountered in the subsurface under most conditions
(Nriagu 1972) At anoxic and circumneutral conditions the whitish vivianite
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 21
controls the Fe(II) activity even at very low phosphate concentrations (Nriagu amp
Dell 1974) Vivianite occurs as a secondary mineral in the gossans of metallic ore
deposits and as a weathering product of primary iron-manganese phosphates in
pegmatites (Gaines et al 1997) Moreover natural vivianite occurrences have
been identified in a number of lake and river sediments (Zwaan amp Kortenbout van
der Sluys 1971 Nriagu amp Dell 1974 Postma 1981 Nembrini et al 1983
Henderson et al 1984 Dodd et al 2003 House 2003 and references therein)
Vivianite is also found in sewage sludge as a result of the wastewater treatment
where iron salts are added in order to remove phosphate (Seitz et al 1973) It is
however still indefinite how ubiquitous vivianite is in nature Furthermore only
little is known about the mechanism of vivianite formation and the role played by
sedimentary Fe(III) oxides Anaerobic Fe(III)-reducing microorganisms may
reduce Fe(III) oxides thereby releasing the iron as soluble Fe(II) and mobilizing
the phosphate adsorbed to the Fe(III) oxides (Lovley 1997) It has been suggested
that vivianite is formed by precipitation following reductive dissolution of Fe(III)
oxides (Manning et al 1981 Manning amp Jones 1982) However it has also been
proposed that the transformation of Fe(III) oxides to vivianite occurs topotactically
and not via reductive dissolution (Nembrini et al 1983) Vivianite was shown to
form microbially as a result of the activity of the anaerobic Fe(III)-reducing
bacteria Shewanella putrefaciens in the presence of high Fe(III)-citrate and
phosphate concentrations (Jorand et al 2000) Moreover vivianite formation by
bioreduction of Fe(III) in hydrous ferric oxide and in smectite has been reported
(Fredrickson et al 1998 Dong et al 2003)
Only little is known about the oxidation products of vivianite Metavivianite a
greenish triclinic iron hydroxy phosphate mineral was first described by Ritz et al
(1974) and it was later found to coexist with vivianite in several natural sediment
samples (Henderson et al 1984) Once the Fe(III) content became evident the true
composition of metavivianite ((FeII3-xFeIII
x)(PO4)2(OH)x
22 Chapter 2
middot(8-x)H2O x gt 12)) was established (Rodgers amp Johnston 1985 Rodgers 1986
and references therein) The formation of intermediate greenish precipitates during
oxidation of fluffy colourless Fe(II) precipitates by anoxic phototrophic
microorganisms and nitrate-dependent FeOB have been reported (Ehrenreich amp
Widdel 1994 Chaudhuri et al 2001) Since both studies were conducted in
bicarbonate buffered mineral media (22-30 mM HCO3ndash pH 70-72) containing
37-5 mM phosphate we assume that the initial fluffy whitish precipitates
consisted mainly of vivianite Chaudhuri et al (2001) proposed that the
intermediate green phases produces by the denitrifying FeOB consist of carbonate
green rust (GR-CO3) but no convincing evidence of this biogenic GR-CO3 has been
provided yet The major objective of this work was to examine the Fe-containing
products forming during the course of biooxidation of vivianite by non-
phototrophic anaerobic denitrifying Fe(II)-oxidizing bacteria
2 2 Materials and methods
All handling and sampling of solutions and suspensions were carried out under
sterile and strict anoxic conditions All chemicals were pa quality
221 Microorganisms and media
Enrichment cultures of nitrate-reducing FeOB taken from town ditches (Bremen
Germany) were grown in anoxic bicarbonate-buffered (30 mM HCO3ndash 90
N210 CO2 pH 70) mineral media containing 4 mM phosphate as well as
essential trace elements and vitamins (Table 21 Straub amp Buchholz-Cleven
1998) Ammonium was omitted from the media in order to facilitate detection of
ammonium possibly produced by reduction of nitrate The techniques used for
preparation of media and cultivation of bacteria under anoxic conditions have been
described by Widdel amp Bak (1992) 05 M aqueous stock solutions of FeCl2 or
FeSO4 were prepared in 100 mL glass flasks by reacting 65 mmol of iron powder
(particle size 10 microm Merck) with 100 mL deoxygenated 10 M HCl or 05 M
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 23
H2SO4 respectively The solutions were magnetically stirred and heated (~80degC)
during reaction until the H2(g) production had ceased (ge 1 hour) The FeCl2 and
FeSO4 stock solutions were stored under a small Ar overpressure at 5degC
Table 21 Composition of the mineral medium (adopted from Straub amp Buchholz-Cleven
(1998))
Components Concentration (M) KH2PO4 15middot10-3
K2HPO4 25middot10-3
MgSO4middot7H2O 10middot10-3
CaCl2middot2H2O 50middot10-4
H3BO3 56middot10-5
ZnSO4middot7H2O 10middot10-6
Na2MoO4middot2H2O 40middot10-6
CuSO4middot5H2O 20middot10-7
MnSO4middotH2O 10middot10-6
Na2SeO4 12middot10-5
CoCl2middot6H2O 50middot10-6
NiCl2middot6H2O 80middot10-6
NaCl 10middot10-5
NaHCO3ndash 30middot10-2
Cyanocobalamine (vitamin B12) 37middot10-8
p-aminobenzoic acid (vitamin Hrsquo) 36middot10-7
D(+)-biotin (vitamin H) 41middot10-8
Nicotinic acid (Niacin) 81middot10-7
Ca-D(+)-pantothenate (vitamin B5) 52middot10-8
Pyridoxamine dihydrochloride 96middot10-7
Thiaminechloridehydrochloride (vitamin B1) 15middot10-7
NaNO3 40middot10-3
FeSO4 or FeCl2 0010
222 Characterisation of precipitates
In order to optimize the characterization and distinction between the spectral
components transmission Moumlssbauer spectra were obtained at temperatures
between 5 K and 250 K and in external magnetic fields of 4 T (parallel to the γ-ray
direction) using a conventional constant acceleration spectrometer and a source of 57Co in Rh The spectrometer was calibrated using a 125 microm foil of α-Fe at room
temperature and isomer shifts are given relative to the centroid of the spectrum of
this absorber The spectra were fitted using simple Lorentzian line shape Infrared
(IR) spectra were obtained using a Perkin Elmer FT-IR 2000 spectrometer and the
24 Chapter 2
KBr pellet technique Scanning electron microscopy (SEM) was carried out in
order to study the morphology and composition of the precipitates Specimens for
SEM were prepared by depositing suspended particles onto an aluminum stub
coated with a carbon sticker The stub was quickly transferred into a sputtering
chamber and coated with a thin Pt film (~20 nm) In order to avoid interfering Pt
signals in the energy dispersive spectra the stubs were in some cases not coated
with Pt but quickly transferred to the SEM chamber for evacuation Measurements
were performed using a Philips XL30 equipped with a LaB6 source and an
accelerating voltage of 20 kV and an EDAX eDXi X-ray dispersive spectrometer
223 Biooxidation experiments
The biooxidation experiments were conducted in 50-400 mL butyl rubber
stoppered bottles with a 90 N210 CO2 headspace constituting 10 of the total
volume Prior to inoculation 4 mM NaNO3 was added as the electron acceptor and
10 mM Fe2+ (as chloride or sulphate) as the electron donor to the mineral media
Control experiments were performed in the same media only they were not
inoculated Addition of ferrous iron to the media induced an immediate
precipitation of a solid whitish material The whitish precipitate was collected on
022 microm polyvinylidendifluorid (Durapore Millipore) filters and stored in an
anoxic atmosphere until further measurements Old outgrown media suspensions
that had turned reddish in color due to a precipitate produced by the denitrifying
FeOB were used as inocula Inoculum volume was kept lt1 of the culture volume
in order to prevent the reddish inoculum from dominating over the initial whitish
precipitate Cultures were incubated in the dark at room temperature and gently
agitated once every day Typical color successions for the media were initial
whitish precipitates turning first more and more greenish over time and then finally
turning reddish (see Fig 22) The color developed uniformly without any
indications of multiple phases in the precipitate At different time intervals
suspension samples were withdrawn using 90 N210 CO2-flushed polyethylene
syringes The precipitates were collected on 022 microm polyvinylidendifluorid
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 25
(Durapore Millipore) filters and analyzed by Moumlssbauer spectroscopy and SEM
Nonfiltered suspension samples were digested in 01 M HCl and chemically
analyzed for Fe(II) NO3- and NH4
+
224 Analytical methods
Fe2+ was determined using a modified phenanthroline method (Fadrus amp Maly
1975) Nitrate was quantified by ion chromatography (Morales et al 2000) and
ammonium was measured photometrically using the indophenol reaction (Rossum
amp Villarruz 1963)
23 Results and discussion
231 Identification of solid iron-containing phases
In most cases the mineral media for cultivating denitrifying FeOB contained 10
mM FeCl2 or FeSO4 4 mM NO3ndash 4 mM total phosphorus and 30 mM HCO3
- at pH
70 ([HCO3-] = 2138middot[CO3
2-] at pH 70) Whitish flocs precipitated immediately at
these initial conditions when Fe(II) was added to the media (Figure 22a) Such
colourless flocs have been reported to precipitate in similar mineral media (10 mM
Fe(II) 37-5 mM phosphate 22-30 mM HCO3ndash pH 70-72) (Ehrenreich amp
Widdel 1994 Chaudhuri et al 2001) Our Fe(II) measurements showed that 20-
50 of the total Fe(II) added was present in this initial white precipitate
26 Chapter 2
a
210- Figure 22 Colour of suspended material in the growth media during Fe(II) biooxidation a)
Initial whitish precipitate prior to inoculation b) Inte1mediate greenish phase fo1med within 2-3
days after inoculation c) reddish precipitate at late stage of biooxidation (gt5-6 days)
The precipitates were filtered and investigated by Mossbauer and IR spectroscopy
The transmission Mossbauer spectra obtained for the initial whitish precipitate at
temperatures between 20 and 250 K are shown in Figure 23 The spectrum
measured at 250 K consists of two fairly well-resolved Fe(II) doublets (see
parameters in Table 22) The change in line-overlap with decreasing temperature
is primarily ascribed to differences in the temperature dependence of the
quadrupole splitting of the two components From the spectra at 10 and 6 K
(Figure 24) it can be concluded that magnetic ordering takes place between these
two temperatures and that only one transition occurs (indicating the presence of
only one phase) The parameters of one of the Fe(II) doublets at 250 K (designated
B in Table 22) are in very good agreement with previously published values for
the vivianite Fe(Il)8 site at room temperature (eg McCammon amp Burns 1980)
and the ordering temperature also agrees well with an assignment as vivianite
(Forsyth et al 1970) However the second Fe(II) doublet in the initial colourless
precipitate (Table 22) has parameters that deviate from previously reported values
by having a smaller quadrupole splitting (indicating a less distorted coordination)
and a significantly higher relative intensity and line width These effects might be
due to the presence of numerous defects in the vivianite crystal lattice particularly
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 27
affecting the Fe(II)A sites It should be noted that further components may be added
to the fit in order to improve its statistics Nevertheless we decided to include no
further components as suggested by the finding of one magnetic ordering only
Accordingly our interpretation of the Moumlssbauer results for the initial white
precipitate suggests a highly defective vivianite having a distribution of local
coordination environments particular in the A site This assignment is further
supported by a major absorption band due to phosphate anions in the infrared
spectrum at approximately 1000 cm-1 and the absence of other complex anions
(data not shown) Thus the whitish precipitate is referred to as a vivianite-like
(ldquovivianiterdquo) precipitate
0
3 5
3 0
2 5
2 0
1 5
1 0
0 5
0 0
20 K
Velocity (mms)
Rel
ativ
e ab
sorp
tion
()
3
2
1
0
80 K
3 5
3 0
2 5
2 0
1 5
1 0
0 5
0 0
150 K
-5 -4 -3 -2 -1 0 1 2 3 4 5
2 5
2 0
1 5
1 0
0 5
0 0
250 K
Figure 23 Transmission Moumlssbauer spectra measured between 250 and 20 K of the initial
whitish precipitate prior to inoculation (see Fig 22a) Fitting components (and their sum) are
shown as full lines
28 Chapter 2
Table 22 Selected Mossbauer parameters of the doublet components in the spectra obtained for
different precipitates
Precipitates Temperature Isomer shift Quadrupole Line width Area (K) (mms-1) splitting (mms-1) (mms-1) ()
Whitish Fe(II)B 250 127 309 035 38
Fe(II)A 250 128 181 051 62
Dark greenish Fe(II)B 250 126 305 023 17
Fe(II)A 250 132 238 051 44
Fe(III) 250 036 085 040 38
Reddish-orange 40 138 244 094 67 Fe(II) Hyperfine parameters are generally given with uncertainties of 003 mms- the spectral area with an uncertainty of 3
1005
1000
0995
0990
- 0985 ~ e c 0980 0
-~ 0975 E c nl b 1000 g ~ Qi 0995 0
0990
0985
0980 -12 -8
~ -~
bullbull bull Ibull bull bullbull bull bull ~ bull bull ~
It
10 K
bull bull bull bull (J ~ i~ 6K bullmiddotf bull bull
~ bull bull bull
-4 0 4 8 12
Velocity (mmls)
Figure 24 Transmission Mossbauer spectra measured at 10 and 6 K of the initial whitish
precipitate prior to inoculation (see Fig 22a)
In general the color of the media suspension changed from whitish into light green
color within 2-3 days after inoculation (Figure 22b ) This transformation occurred
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 29
without dissolving the initial whitish precipitate or preserving the whitish
precipitate as a separate phase implying some kind of solid state transformation
Two identical media suspensions were inoculated simultaneously but at the
sampling time they had distinctly different intensities of the green color
designated light and dark green respectively Moumlssbauer spectra of the dark green
sample are shown in Figure 25 The spectra of this sample are all fitted using three
doublet components (two Fe(II) and one Fe(III)) and the parameters of the
spectrum measured at 250 K are given in Table 22
30
25
20
15
10
05
00
20 K
Velocity (mms)
Rel
ativ
e ab
sorp
tion
()
35
30
25
20
15
10
05
00
80 K
30
25
20
15
10
05
00
150 K
-5 -4 -3 -2 -1 0 1 2 3 4 5
25
20
15
10
05
00
250 K
Figure 25 Transmission Moumlssbauer spectra measured between 250 and 20 K of the dark
greenish precipitate formed during biooxidation Fitting components (and their sum) are shown
as full lines
No magnetic ordering of the dark green precipitate was observed at temperatures
above 20 K but ordering occurred around 10 K (not shown - due to very thin
30 Chapter 2
samples this was not investigated in details) The two greenish samples had very
similar parameters only differing in the relative intensity of Fe(III) 26 and 38
in the light greenish and dark greenish samples respectively Assuming the
spectral area of a component to be proportional to the abundance of the species in
the solid these results indicate a correlation between the intensity of the green
color and the content of Fe(III) in the precipitate The parameters of the Fe(II)
doublets in the dark green precipitate (Table 22) were in very good agreement
with previously published values for vivianite with a non-negligible Fe(III) content
(McCammon amp Burns 1980) whereas the Fe(III) component in particular had a
higher quadrupole splitting The observation that magnetic ordering of both Fe(II)
and Fe(III) occurred at similar temperature for the light green phase (Figure 26) is
a strong indication that they both belong to the same phase The absence of the
component with the low quadrupole splitting in the spectra of both green samples
might indicate that the initial vivianite-like phase crystallized into a more well-
defined vivianite over time However freshly prepared and long-term aged (gt1
year) suspensions of the initial vivianite-like precipitate did not differ significantly
Hence we suggest that the recrystallization of the vivianite-like precipitate can be
explained by Fe(II) biooxidation
Solid State Oxidation ofFe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria
0
2
3
- 4
~ 5 c ~ 6 e-0 7 -2 nl Q) 00 gt ~ 05 Qi 0 10
15
20
25
30
35
bullbullbull hi 6~
bullbullbullbull bull bull bullbull bull 10 K bull bull
bull bull bull bull bull bull bull
~~ ~~ lf 6K bull bull bullbull middot bull bull r bull
bull bull bull bull bull bullbull bull bull bull - 12 -a -4 0 4 8 12
Velocity (mms)
31
Figure 26 Transmission Mossbauer spectrn measured at 6 and 10 K of the light greenish
precipitate fonned during biooxidation
The solid state oxidation of monoclinic vivianite to triclinic metavivianite is well-
known (McCammon amp Bums 1980 Pratesi et al 2003 and references therein)
The vivianite crystal structure contains Fe(II) ions in both isolated Fe(II)A and
paired Fe(Il)8 octahedra Mossbauer spectroscopic analyses have shown that the
Fe(Il)8 Fe(II)A ratio increases with increasing Fe(III) concentration suggesting that
the remaining Fe(II)A ions are more readily oxidized than the Fe(II)a ion of an
Fe(Il)8 -Fe(III)8 pair (McCammon amp Bums 1980) The mechanism of oxidation of
Fe(II) in vivianite involves conversion of H20 ligands to OH- ions producing a
progressive collapse of the vivianite structure due to the elimination of hydrogen
bonds (Moore 1971) The exact oxidation limits between which the triclinic lattice
is stable are somewhat disputed as the results obtained for synthetic and natural
vivianites oxidized chemically in the laboratory and naturally oxidized natural
vivianite specimens do not agree completely (Rodgers 1986 and references
therein) Taking all reports into account the monoclinic structure of vivianite is
supposedly maintained until 40-50 of total iron is oxidized Further oxidation
32 Chapter 2
leads to the formation of the triclinic metavivianite in which the FeA site is fully
oxidized whereas the oxidation of the FeB ranges from 20 to almost 100 Thus
the triclinic metavivianite structure persists close to complete oxidation of total
iron The Moumlssbauer results obtained in this study are consistent with the vivianite
solid state oxidation mechanism reported by McCammon amp Burns (1980) Thus
we propose that the intermediate greenish precipitate is a metavivianite-like
(ldquometavivianiterdquo) phase It should be noted that a minor oxidation of dissolved
Fe(II) may have occurred even though the solid state oxidation of Fe(II) was
predominant
Within 5-6 days after inoculation the greenish intermediate was transformed into a
reddish product (Figure 22c) The magnetically ordered sextet in the spectrum of
the red phase (Figure 27) measured at 40 K was due to goethite (α-FeOOH)
(magnetic hyperfine field of 470 T and a quadrupole shift of -01 mms-1 cp
Moslashrup et al 1983) The sextet deviated from ideal goethite by its asymmetric line
shape and its low ordering temperature (around 100 K ndash data not shown) and thus
the goethite was poorly crystalline It is very likely that the presence of phosphate
in the media retarded the crystal growth of goethite The unusual reddish colour of
the goethite might also be explained by the presence of phosphate The spectrum at
40 K was however dominated by a Fe(II) doublet (Table 22) that ordered
magnetically between 40 and 20 K (Figure 27) The hyperfine parameters and the
magnetic ordering temperature indicated that this component was due to siderite
(FeCO3) having a magnetic ordering temperature of 38 K (Jacobs 1963) The
siderite component may have formed as a result of the microbial activity changing
the chemistry of the solution and precipitating a major part of the remaining
dissolved Fe(II) at this stage The characteristic vivianite Fe(II) doublet with large
quadrupole splitting was not detected in this sample The reddish precipitate
contained considerably less Fe(III) than the greenish precipitate (only 33 as
estimated from the spectral area) None of the components in the reddish sample
Solid State Oxidation ofFe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 33
could be detected in freshly inoculated samples indicating that carryover of mineral
precipitates by inoculation of the culture media was negligible
0
2
- 3
~4 c
Q 5
e 6 0
~ 7 g
0 3l Q)
a 1
2
3
4
5
-12 a
40K
bull bull (
4 0 4 8 12 Velocity (mms)
Figure 27 Transmission Mossbauer spectra measured at 20 and 40 K of the reddish precipitate
fo1med during the late biooxidation stage Fitting components (and their sum) are shown as full
lines
Strengite (FeP04middot2H20) was not detected at any time during oxidation
Santabarbaraite a new amorphous F e(III) hydroxy phosphate mineral
(Fe3(P04)i(OH)3middot5H20) was reported in a recent study (Pratesi et al 2003) The
brownish mineral was a result of the solid state oxidation of vivianite through
metavivianite However no Mossbauer data have been provided for this new
mineral yet and therefore we cannot give an account of whether santabarbaraite
forms in our system or not Thus goethite was the dominating end product and we
propose the reaction path depicted in Figure 28 for the nitrate-dependent
biooxidation of Fe(II) in our systems
34 Chapter 2
Fe3(PO4)2middot8H2O (FeII3-xFeIII
x)(PO4)2(OH)xmiddot(8-x)H2O α-FeOOH
NO3- N2 NO3
- N2
ldquoVivianiterdquo ldquoMetavivianiterdquo Goethite
Figure 28 Proposed reaction path and iron-containing minerals forming during solid state
oxidation of vivianite by denitrifying FeOB at the experimental conditions applied in this study
The biotic formation of layered Fe(II)-Fe(III) hydroxides (green rusts) by
anaerobic denitrifying Fe(II)-oxidizing bacteria has been suggested but proper
identification of these phases still lacks (Chaudhuri et al 2001) We cannot rule
out that small amounts of green rusts (GRs) perhaps a phosphate intercalated GR
(Hansen amp Poulsen 1999) might have been present here during the greenish
intermediate ldquometavivianiterdquo oxidation stage When present in low concentrations
especially in mixtures including other iron minerals it is very difficult to identify
GRs even with Moumlssbauer spectroscopy At least two complementary methods
such as X-ray diffraction (XRD) and Moumlssbauer spectroscopy are required for
proper identification and characterization of GRs However the precipitates
collected in this work were poorly crystalline and did not allow for XRD analysis
Electron micrographs including energy dispersive X-ray spectroscopy suspension
colour and mineral stability calculations do not suffice as evidence Hence no
convincing evidence of GR formation facilitated by denitrifying FeOB has been
provided so far The blue-green colours of metavivianite and green rust minerals
originate from Fe(II)-Fe(III) charge transfer between adjacent Fe(II) and Fe(III)
ions in edge-shared octahedra (Faye et al 1968) The greenish suspension colour
occurring during the intermediate phase has incited the idea of biogenic GRs in
studies on nitrate-dependent Fe(II) biooxidation (Chaudhuri et al 2001 Lack et
al 2002aampb) However our results indicate that this reasoning is misleading
232 Factors controlling the rate and extent of Fe(II) biooxidation
Generally it was found that maximally 20-64 of the initial Fe(II) amount was
oxidized to Fe(III) (Figure 29) This indicates some limitations in the accessibility
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 35
of Fe(II) in the system Based solely on stoichiometry considerations the
microorganisms are expected to oxidize 5 mol Fe(II) for every 1 mol nitrate
reduced to dinitrogen However as exemplified in Figure 28 this ratio was mostly
lt4 which can be explained by the consumption of nitrogen as a result of microbial
growth At initial [Fe(II)][NO3ndash] ratios lt5 nitrate is in excess and should not limit
the extent of the biooxidation Thus the lack of complete biooxidation could not be
due to exhaustion of nitrate Furthermore all growth essential nutrients were more
than sufficiently applied hence the incomplete Fe(II) biooxidation was not caused
by lack of nutrients The most reasonable explanation therefore seems to be that an
increasingly limited access to the electron donor over time inhibits complete long-
term Fe(II) biooxidation At least four mechanisms may cause this inhibition 1)
the Fe(II) becomes isolated within the structure of the mixed Fe(II)-Fe(III)
minerals forming during biooxidation or underneath a passive Fe(III)-bearing
surface film on the initial Fe(II) precipitates 2) the FeOB cell surface becomes
covered with a passive Fe(III)-bearing surface film 3) the Fe(II) biooxidation is
controlled by the rate of dissolution of the initial Fe(II) minerals or 4) the reaction
proceeds primarily by biooxidation of dissolved Fe(II) whose concentration
gradually decreases due to changes in solid phase composition The actual
mechanisms whereby the surface-associated Fe(III) can inhibit Fe(II) biooxidation
are unknown but they may involve both kinetic and thermodynamic constraints on
the electron transfer The Moumlssbauer results obtained in this work strongly suggest
that the Fe(II) biooxidation occurred mainly in the solid state of the initial
ldquovivianiterdquo phase However we cannot rule out that some dissolved Fe(II) was
oxidized as well
36 Chapter 2
Figure 29 Concentration profiles of total Fe(II) and nitrate as a function of time during Fe(II)
biooxidation
No Fe(II) oxidation took place in cultures where nitrate had been omitted
confirming that the microbial Fe(II) oxidation is nitrate-dependent (data not
shown) No Fe(II) oxidation was detected in the non-inoculated control
experiments within the duration of the experiments and thus the chemical
oxidation of dissolved Fe(II) by nitrate catalyzed by vitamins or trace elements
(eg Cu(II)) can be neglected Ammonium did not form in detectable amounts
during Fe(II) biooxidation (data not shown) and therefore dinitrogen was assumed
to be the end product as reported previously (Straub et al 1996 Benz et al 1998)
The absence of ammonium formation does indirectly support the absence of
biologically induced green rust formation as synthetic green rust is known to
convert nitrate into ammonium in purely chemical reactions (Hansen et al 1996)
It was visually observed that the phosphate concentration in the media exerted a
control on the microbial Fe(II)-oxidation At phosphorus concentrations le 2 mM
no Fe(II)-oxidation took place However the solubility product for vivianite (Ksp =
171middot10-36 at 25degC (Al-Borno et al 1994)) was still by far exceeded under these
conditions It is not known whether this phosphate limiting effect is due to growth
constraints in the mixed bacterial community or whether specific Fe(II) phosphate
Solid State Oxidation ofFe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 37
precipitates are prerequisites of the Fe(II) biooxidation to take place Experiments
are currently underway in our laboratory in order to elucidate the role of specific
initial Fe(II) precipitates It should be noted that the growth of the denitrifying
FeOB could not be estimated as they were present in highly heterogeneous
suspensions containing both solid iron phases as well as other bacteria (enrichment
culture)
233 Morphology of solid iron phases
The morphology of the various precipitates was studied by SEM The initial
whitish precipitate consisted of a web-like structure (Figure 21 Oa and background
in Figure 21 Ob) whereas more distinct hexagonally shaped rosettes with particle
size ~20 microm (Figure 2lObampc) formed during Fe(II) biooxidation The energy
dispersive spectroscopic element analyses showed that other than iron the initial
whitish precipitate and the rosettes contained mainly phophorus
Figure 210 Scanning electron micro graphs of precipitates fo1med at various stages of the
experiment a Initial whitish precipitate bampc Rosettes observed in the intennediate greenish
precipitate d Reddish precipitate sampled during the late biooxidation stage
38 Chapter 2
These observations are interpreted as vivianite forming a web-like morphology in
the initial whitish precipitate and partly transforming into hexagonal particles in
the greenish colored stage The interpretations are supported by similar vivianite
morphologies reported including pseudo-hexagonal vivianite crystals of low
symmetry resulting from microbial Fe(III) reduction of HFO and platy rosettes of
vivianite crystals formed during bioreduction of Fe(III) in smectite (Fredrickson et
al 1998 Dong et al 2003) It was not possible to associate the morphology
observed in the reddish precipitate with the minerals identified in this phase
(Figure 210d)
24 Conclusions
This work demonstrated that anaerobic autotrophic denitrifying Fe(II)-oxidizing
bacteria produce poorly crystalline goethite by solid state oxidation of ldquovivianiterdquo
via a ldquometavivianiterdquo intermediate The increasing amount of Fe(III) forming in the
vivianite structure was accompanied by an increasing intensity in the green colour
as the Fe(II) biooxidation progressed Moumlssbauer spectroscopic analyses provided
no significant evidence of green rust formation The finding of microbially
oxidized vivianite in this study raises the question of the oxidation state of
vivianite specimens from natural sediments Vivianite is generally believed to be
an ideal Fe(II) hydroxy phosphate mineral and the presence of Fe(III) is explained
by aerial oxidation upon sampling The results presented here indicate that
microbiological processes may be responsible for the oxidation of vivianite and
metavivianite in natural subsurface environments Acknowledgments
We would like to thank Dr K Straub for providing and advising us how to culture the nitrate-
reducing FeOB Furthermore we thank Dr C B Koch for performing the Moumlssbauer analyses
and Dr D Mavrocordatos for performing the SEM analyses
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 39
References Al-Borno A Tomson MB (1994) The temperature dependence of the solubility product constant of vivianite Geochimica et Cosmochimica Acta 58 5373-5378 Benali O Abdelmoula M Refait Ph Geacutenin JMR (2001) Effect of orthophosphate on the oxidation products of Fe(II)-Fe(III) hydroxycarbonate The transformation of green rust to ferrihydrite Geochimica et Cosmochimica Acta 65 1715-1726 Benz M Brune A Schink B (1998) Anaerobic and aerobic oxidation of ferrous iron at neutral pH by chemohetorotrophic nitrate-reducing bacteria Archives of Microbiology 169 159-165 Chaudhuri SK Lack JG Coates JD (2001) Biogenic magnetite formation through anaerobic biooxidation of Fe(II) Applied and Environmental Microbiology 67 2844-2848 Cumplido J Barron V Torrent J (2000) Effect of phosphate on the formation of nanophase lepidocrocite from Fe(II) sulfate Clays and Clay Minerals 48 503-510 Dodd J Large DJ Fortey NJ Kemp S Styles M Wetton P Milodowski A (2003) Geochemistry and petrography of phosphorus in urban canal bed sediment Applied Geochemistry 18 259-267 Dong H Kostka JE Kim J (2003) Microscopic evidence for microbial dissolution of smectite Clays and Clay Minerals 51 502-512 Ehrenreich A Widdel F (1994) Anaerobic oxidation of ferrous iron by purple bacteria a new type of phototrophic metabolism Applied and Environmental Microbiology 60 4517-4526 Emerson D Moyer CL (2002) Neutrophilic Fe-oxidizing bacteria are abundant at the Loihi seamount hydrothermal vents and play a major role in Fe oxide deposition Applied and Environmental Microbiology 68 3085-3093 Emerson D Weiss JV Megonigal JP (1999) Iron-oxidizing bacteria are associated with ferric hydroxide precipitates (Fe-plaque) on the roots of wetland plants Applied and Environmental Microbiology 65 2758-2761 Fadrus H Maly J (1975) Suppression of iron(III) interference in the determination of iron(II) in water by the 110-phenanthroline method The Analyst 100 549-554 Faye GH Manning PG Nickel EH (1968) The polarized optical absorption spectra of tourmaline cordierite chloritoid and vivianite ferrous-ferric electronic interaction as a source of pleochroism American Mineralogist 53 1174-1201 Forsyth JB Johnson CE Wilkonson C (1970) The magnetic structure of vivianite Fe3(PO4)2middot8H2O Journal of Physics Part C Solid State Physics 3 1127-1139 Fredrickson JK Zachara JM Kennedy DW Dong H Onstott TC Hinman NW Li S (1998) Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium Geochimica et Cosmochimica Acta 62 3239-3257 Gaines RV Skinner HCW Foord EE Mason B Rosenzweig A (1997) Danas new
40 Chapter 2 mineralogy 8th ed John Wiley amp Sons Inc Hanert HH (1992) The genus Gallionella In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 4082-4088 Hansen HCB Koch CB Nancke-Krogh H Borggaard OK Soerensen J (1996) Abiotic nitrate reduction to ammonium Key role of green rust Environmental Science and Technology 30 2053-2056 Hansen HCB Poulsen IF (1999) Interaction of synthetic sulphate green rust with phosphate and the crystallization of vivianite Clays and Clay Minerals 47 312-318 Henderson GS Black PM Ridgers KA Rankin PC (1984) New data on New Zealand vivianite and metavivianite New Zealand Journal of Geology and Geophysics 27 367-378 House WA (2003) Geochemical cycling of phosphorus in rivers Applied Geochemistry 18 739-748 Jacobs IS (1963) Metamagnetism of siderite (FeCO3) Journal of Applied Physics 34 1106-1107 Jorand F Appenzeller BMR Abdelmoula M Refait Ph Block J-C Geacutenin JMR (2000) Assessment of vivianite formation in Shewanella putrefaciens culture Environmental Technology 21 1001-1005 Kandori K Uchida S Kataoka S Ishikawa T (1992) Effects of silicate and phosphate ions on the formation of ferric oxide hydroxide particles Journal of Materials Science 27 719-728 Kuenen JG Robertson LA Tuovinen OH (1992) The genera Thiobacillus Thiomicrospira and Thiosphaera In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 2618-2624 Lack JG Chaudhuri SK Chakraborty R Achenbach LA Coates JD (2002a) Anaerobic biooxidation of Fe(II) by Dechlorosoma suillum Microbial Ecology 43 424-431 Lack JG Chaudhuri SK Kelly SD Kemner KM OConnor SM Coates JD (2002b) Immobilization of radionuclides and heavy metals through anaerobic bio-oxidation of Fe(II) Applied and Environmental Microbiology 68 2704-2710 Lovley DR (1997) Microbial Fe(III) reduction in subsurface environments FEMS Microbiology Reviews 20 305-313 Manning PG Birchall T Jones W (1981) Ferric hydroxides in surficial sediments of the great lakes and their role in phosphorus availability a Moumlssbauer spectral study Canadian Mineralogist 19 525-530 Manning PG Jones W (1982) The binding capacity of ferric hydroxides for non-apatite inorganic phosphorus in sediments of the depositional basins of Lakes Erie and Ontario Canadian Mineralogist 20 169-176 McCammon CA Burns RG (1980) The oxidation mechanism of vivianite as studied by Moumlssbauer spectroscopy American Mineralogist 65 361-366 Moore PB (1971) The Fe2+
3(H2O)n(PO4)2 homologous series crystal-chemical relationships
Solid State Oxidation of Fe(II) in Vivianite by Anaerobic Denitrifying Fe(II)-Oxidizing Bacteria 41
and oxidized equivalents American Mineralogist 56 1-17 Moraghan JT Buresh RJ (1976) Chemical reduction of nitrate by ferrous iron Journal of Environmental Quality 5 320-325 Moraghan JT Buresh RJ (1977) Chemical reduction of nitrite and nitrous oxide by ferrous iron Journal of American Soil Science Society 40 47-50 Morales JA de Graterol LS Mesa J (2000) Determination of chloride sulfate and nitrate in groundwater samples by ion chromatography Journal of Chromatography A 884 185-190 Mulder EG Deinema MH (1992) The sheathed bacteria In The Prokaryotes Balows A Truper HG Dworkin M Harder W Schleifer KH (eds) Springer Verlag 2618-2624 Moslashrup S Madsen MB Franck J Villadsen J Koch CJW (1983) A new interpretation of Moumlssbauer spectra of microcrystalline goethiterdquosuper-ferromagnetismrdquo of ldquosuper-spin-glassrdquo behaviour Journal of Magnetism and Magnetic Materials 40 163-174 Nembrini GP Capobianco JA Viel M Williams AF (1983) A Moumlssbauer and chemical study of the formation of vivianite in sediments of Lago Maggiore (Italy) Geochimica et Cosmochimica Acta 47 1459-1464 Nielsen JL Nielsen PH (1998) Microbial nitrate-dependent oxidation of ferrous iron in activated sludge Environmental Science and Technology 32 3556-3561 Nriagu JO (1972) Stability of vivianite and ion-pair formation in the system Fe3(PO4)2-H3PO4- H2O Geochimica et Cosmochimica Acta 36 459-470 Nriagu JO Dell CI (1974) Diagenetic formation of iron phosphates in recent lake sediments American Mineralogist 59 934-946 Patrick Jr WH Khalid RA (1974) Phosphate release and sorption by soils and sediments Effect of aerobic and anaerobic conditions Science 186 53-55 Postma D (1981) Formation of siderite and vivianite and the pore-water composition of a recent bog sediment in Denmark Chemical Geology 31 225-244 Pratesi G Cipriani C Giuli G Birch WD (2003) Santabarbaraite a new amorphous phosphate mineral European Journal of Mineralogy 15 185-192 Ratering S Schnell S (2001) Nitrate-dependent iron(II) oxidation in paddy soil Environmental Microbiology 3 100-109 Ritz C Essene EJ Peacor DR (1974) Metavivianite Fe3(PO4)2middot8H2O a new mineral American Mineralogist 59 896-899 Rodgers KA (1986) Metavivianite and kerchenite a review Mineralogical Magazine 50 687- 691 Rodgers KA Johnston JH (1985) Type metavivianite Moumlssbauer evidence for a revised composition Neues Jahrbuch fuumlr Mineralogie-Monatshefte 12 539-542
42 Chapter 2 Rossum JR Villarruz PA (1963) Determination of ammonia by the indophenol method Journal of American Water Works Association 55 657-658 Seitz MA Riedner RJ Malhotra SK Kipp RJ (1973) Iron-phosphate compound identification in sewage sludge residue Environmental Science and Technology 7 354-357 Sobolev D Roden EE (2001) Suboxic deposition of ferric iron by bacteria in opposing gradients of Fe(II) and oxygen at circumneutral pH Applied and Environmental Microbiology 67 1328-1334 Straub KL Benz M Schink B Widdel F (1996) Anaerobic nitrate-dependent microbial oxidation of ferrous iron Applied and Environmental Microbiology 62 1458-1460 Straub KL Buchholz-Cleven BEE (1998) Enumeration and detection of anaerobic ferrous iron-oxidizing nitrate-reducing bacteria from diverse European sediments Applied and Environmental Microbiology 64 4846-4856 Straub KL Hanzlik M Buchholz-Cleven BEE (1998) The use of biologically produced ferrihydrite for the isolation of novel iron-reducing bacteria Systematic and Applied Microbiology 21 442-449 Weber KA Picardal FW Roden EE (2001) Microbially catalyzed nitrate-dependent oxidation of biogenic solid-phase Fe(II) compounds Environmental Science and Technology 35 1644-1650 Widdel F Bak F (1992) Gram-negative mesophilic sulfate-reducing bacteria In The Prokaryotes (Balows A Truumlper HG Dworkin M Harder W Schleifer K-H (eds)) Springer 2nd ed 3352-3378
Widdel F Schnell S Heising S Ehrenreich A Assmus B Schink B (1993) Ferrous iron oxidation by anoxygenic phototrophic bacteria Nature 362 834-836 Williams JDH Syers JK Shukla SS Harris RF Armstrong DE (1971) Levels of inorganic and total phosphorus in lake sediments as related to other sediment parameters Environmental Science and Technology 5 1113-1120
Zwaan PC Kortenbout van der Sluys G (1971) Vivianite crystals from Hare Noord Brabant Province The Netherlands Scripta Geology 6 1-7
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 43
3 Formation of Layered Iron Hydroxides by Microbial Fe(III)
Reduction Abstract
Many inorganic and organic pollutants may be degraded by microorganisms in the
subsurface However a wide range of contaminants including chromate nitrate
radionuclides nitroaromatic compounds chlorinated aliphatics and carbamate
pesticides may also be chemically transformed by reduction reactions involving
layered iron(II)-iron(III)-hydroxides (green rusts) Hence green rusts (GRs) may
play a potentially important role in the fate and transport of pollutants in iron-rich
suboxic soils and sediments Yet only little is known about the formation of GRs
in these environments The biotic formation of GRs mediated by the anaerobic
dissimilatory Fe(III)-reducing bacteria Shewanella spp has been reported and
proposed in several studies However the experimental conditions applied were
mostly not natural and the evidence of GR formation provided may be questioned
This work investigated the Fe-containing products formed by the facultatively
anaerobic Fe(III)-reducing microorganism Shewanella algae BrY in culture
media containing 4-10 mM formate or lactate and 8-27 mM Fe(III) In order to
simulate natural conditions Fe(III) oxides were applied as coatings on silica
(model system for sandy soils) or calcite particles (model system for calcareous
soils) and synthetic electron shuttles as well as highly concentrated artificial pH
buffers were excluded S algae BrY reduced 19-72 of the initial Fe(III) when
grown in goethitecalcite lepidocrocitecalcite or hydrous ferric oxidesand mineral
systems and green or blackish mineral phases were produced within 1-2 weeks
after inoculation Moumlssbauer spectroscopic analyses indicated that the green and
blackish precipitates were dominated by vivianite (Fe3(PO4)2sdot8H2O) and green rust
44 Chapter 3
31 Introduction
The significance of bacteria in the biogeochemical cycling of iron has been broadly
recognized over the past two decades Chemical processes were previously
considered to account for most of the Fe(III) reduction in subsurface environments
Dissimilatory Fe(III)-reducing bacteria (DIRB) that gain energy by coupling the
oxidation of hydrogen or organic compounds to the reduction of Fe(III) oxides
have been known for many years but their biogeochemical importance was
acknowledged only a decade ago (reviewed by Lovley 1997) DIRB transfer
electrons to extracellular Fe(III) without assimilating the iron Fe(III) bioreduction
accounts for a major fraction of the carbon oxidation in many different
environments and in the presence of high amounts of reactive Fe(III) microbial
Fe(III) reduction may even inhibit sulfate reduction and methanogenesis (King
1990 Lovley amp Phillips 1986) In fact most of the Fe(III) reduction in the Fe(III)
reduction zone of aquatic sediments and aquifers is thought to be enzymatically
catalyzed by microorganisms (Lovley et al 1991) However the relative
importance of microbial and chemical processes involved in the Fe(III) reduction
are still somewhat disputed among microbiologists and geochemists
A wide diversity of Fe(III)-reducing bacteria which fall in a number of different
phylogenetic groups is known today Both organisms growing by respiration and
by fermentation have been isolated and identified (Lovley 1991 Nealson amp
Saffarini 1994) Hydrogen short- and long-chained fatty acids amino acids
sugars and aromatic compounds may serve as electron donors for Fe(III)
bioreduction The enzymes responsible for dissimilatory Fe(III) reduction are outer
membrane associated ferric reductases (Lower et al 2001 and references therein)
Iron reducing bacteria may utilize alternative electron acceptors such as O2 nitrate
S0 sulfate humic substances contaminant metals and metalloids as well as
chlorinated solvents The first organism shown to couple respiratory growth to
dissimilatory iron reduction was Pseudomonas ferrireductans now known as
Shewanella oneidensis but previously classified as Alteromonas putrefaciens and
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 45
Shewanella putrefaciens (Venkateswaran et al 1999) Various DIRB including
the obligate anaerobic Geobacter sp and the facultatively anaerobic Shewanella
sp have been isolated from both marine and freshwater sediments soil and
aquifers (Thamdrup 2000 and references therein)
The redox potentials of oxidized and reduced iron couples and thus the energy
yield available from Fe(III) reduction depend strongly on the specific iron phases
involved In soil and aquatic environments Fe(III) oxides mainly occur in
association with other sediment particles as aggregates or coatings Amorphous
and poorly crystalline Fe(III) oxides usually make up 20 or less of the iron
content in a sediment (Thamdrup 2000) They are the main products of abiotic and
biotic Fe(II) oxidation in sediments and they constitute the most important phases
for microbial Fe(III) reduction Until recently it was generally believed that DIRB
reduced insoluble Fe(III) oxides only by direct contact with the Fe(III) oxide
thereby allowing electron transfer from the cell to the Fe(III) oxide surface
However over the past several years there has been a growing recognition that
DIRB may use different strategies in order to access the solid Fe(III) oxides These
strategies include solubilization of Fe(III) by synthetic or natural Fe(III) chelators
and Fe(III) reduction via electron shuttling with soluble humic substances or
microbially produced electron shuttles (Nevin amp Lovley 2002 and references
therein Turick et al 2003) The Fe(III) complexing agents may also stimulate
Fe(III) oxide reduction indirectly by chelation and thus removal of Fe(II) from
the cell and the Fe(III) oxide surfaces Both chelated Fe(III) and soluble electron
shuttles are more accessible to Fe(III) reductases than solid Fe(III) oxides In
contrast to Geobacter metallireducens S algae produces and releases extracellular
electron shuttling compounds (Nevin amp Lovley 2000) However in the absence of
soluble electron shuttles reversible adhesion is required for reduction of solid
Fe(III) oxides by S algae BrY (Das amp Caccavo 2000) Shewanella algae BrY
adheres readily and preferentially to a range of solid Fe(III) oxides such as
ferrihydrite goethite and hematite (Das amp Caccavo 2001) The adhesion
46 Chapter 3
mechanisms are not completely understood but recent results suggest that the
adhesion is mediated by cell surface proteins and independent of cell motility
(Caccavo amp Das 2002)
The microbial formation of GRs resulting from bioreduction of various Fe(III)
oxides including ferrihydrite goethite and lepidocrocite by strains of the
anaerobic DIRB Shewanella putrefaciens has been reported repeatedly over the
last years (Fredrickson et al 1998 Kukkadapu et al 2001 Liu et al 2001
Parmar et al 2001 Ona-Nguema et al 2002aampb Glasauer et al 2003)
However no evidence of biogenic formation of GRs at natural geochemical
conditions have been offered and it is still unknown whether this process may take
place at natural conditions comprising low carbon and iron concentrations as well
as the absence of synthetic electron shuttles and highly concentrated artificial pH
buffers GRs are layered iron(II)-iron(III)-hydroxides with anionic interlayers and
they hold the general formula [FeII(6-x)FeIII
x(OH)12]x+[(A)xnmiddotyH2O]x- where x = 09 -
42 A is an n-valent anion eg CO32- Clndash or SO4
2- and y is the number of water
molecules in the interlayer In circumneutral solutions the oxidation of dissolved
Fe(II) always passes through solid GR phases (Bernal et al 1959) This agrees
with the natural GR occurrences found in suboxic nonacid iron-rich environments
such as hydromorphic soils and intertidal sediments (Al-Agha et al 1995 Trolard
et al 1996 Genin et al 1998) In addition GRs have been found as corrosion
products in numerous engineering systems eg in a pipeline distribution system
for drinking water steel sheet piles in marine sediments reinforced concrete
(ferro-concrete) and permeable reactive barriers of zero-valent iron implemented
for on-site remediation of organic and inorganic contaminants (Tuovinen et al
1980 Nielsen 1976 Genin et al 1991 Roh et al 2000) Through sequestration
and reductive transformation GRs may play an important role in the fate and
transport of organic and inorganic pollutants in suboxic iron-rich soils and
sediments (see Chapters 4 amp 5 in this work and references therein)
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 47
The major goal of this work was to examine the iron minerals forming during the
course of Fe(III) bioreduction of hydrous ferric oxide goethite and lepidocrocite
Two model systems simulating sandy and calcareous soils in subsurface
environments were designed in order to investigate the formation of iron minerals
at conditions including low carbon levels low Fe(III) concentrations applied as
Fe(III) oxide coatings on sand or calcite no electron shuttle and no synthetic pH
buffers
32 Materials and methods
All handling and sampling of solutions and suspensions were carried out at strict
anoxic conditions Standard sterile techniques were used throughout (Hungate
1969 Miller amp Wolin 1974) Only the iron oxide coatings were not autoclaved in
order to avoid the iron oxides from transforming Goethite (acicular particles with
size 01 times 06 microm specific surface area 16 m2g) and lepidocrocite (acicular
particles with size 005 times 03 microm specific surface area 18 m2g) were purchased as
fine powders from Bayer (Bayferrox 910 and 943) Calcite (grain size 170-350 microm
Pluumlss-Staufer AG) and sea sand (dominantly quartz grain size 01-03 mm Riedel-
de Haeumln) were used as Fe(III) oxide coating bearing minerals
321 Preparation of iron oxide coatings
Two grams of goethite (goe) or lepidocrocite (lep) and 100 g calcite were
combined with 200 mL deionized water (DIW) in a 500 mL polyethylene flask
Hydrous ferric oxide (HFO) was synthesized by dissolving 4 g Fe(NO3)3middot9H2O in
70 mL DIW followed by slow neutralization under magnetic stirring till pH 7 with
approximately 30 mL 1 M NaOH (method modified after Schwertmann amp Cornell
1991) The HFO coating was made by combining 100 mL freshly precipitated
HFO with 900 mL deionized water and 50 g sea sand in a polyethylene bottle The
suspensions containing the iron oxide coatings were gently agitated on a
reciprocating shaker for 24 h and left to stand for another 24 h Excess Fe(III)
oxides and salts were removed from the coated material by repeated decantation
48 Chapter 3
and washing with 003 M NaNO3 followed by washing with DIW until clear
runoff Finally the coatings were collected on folding filters and air dried The
amount of HFO goethite and lepidocrocite coated onto sand and calcite after
washing and drying was quantified to 7-11 mg Fe(III)g sand or calcite
322 Mineral characterisation
The identity and purity of the HFO synthesized were examined by means of X-ray
diffraction (XRD) measurements The XRD analyses were performed on a Scintag
XDS 2000 using Co Kα radiation (45 kV 40 mA) using divergence scatter and
receiving slits of 1deg 05deg and 02 mm respectively Samples were scanned
between 6 and 80 deg2θ with a scan speed of 1 deg2θmin Mineral suspension samples
for transmission Moumlssbauer spectroscopic analysis were collected on 02 microm filters
in an anoxic glove box (Coy Laboratory Products Inc) transferred to Perspex
capsules and stored in liquid nitrogen until measurement Moumlssbauer spectra were
obtained between 250 and 5 K using a conventional constant acceleration
spectrometer and a source of 57Co in Rh The spectrometer was calibrated using a
125 microm foil of α-Fe at room temperature and isomer shifts are given relative to
the centroid of this absorber The spectra were fitted using simple Lorentzian line
shape and it was assumed that all positions have identical f-factors
323 Culture conditions and cell preparation
Shewanella algae BrY is a motile gram-negative rod which was isolated first from
anoxic estuary sediments (Caccavo et al 1992) S algae BrY was grown
aerobically in tryptic soy broth (30 gL CASO-bouillon Merck) at 28degC on a rotary
shaker at 150 rpm for 16-18 h Cells were harvested by centrifugation (6000 rpm times
g 4ordmC 15 min) during the late exponential ndash early stationary growth phase at
OD660 ~ 06 Optimal Fe(III) reductase activity is expressed at this stage of growth
(Roden amp Zachara 1996) The cells were washed twice in oxic 50 mM PIPES
[piperazine-NNacute-bis(2-ethanesulfonic acid)] buffer (pH 70) and resuspended in
culture medium containing no Fe(III) and no carbon source Washed cell
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 49
suspensions were used as inocula for Fe(III) reduction experiments Oxygen was
expelled from the inoculum by extensive purging with 100 N2(g) (9999999
purity) Working stock cultures of S algae BrY were maintained aerobically on
tryptic soy agar plates at ambient temperature
324 Bioreduction experiments
All anaerobic incubations were carried out in anoxic serum vials (25 mL) or test
tubes (13 mL) sealed with thick (10-13 mm) butyl rubber stoppers and aluminum
crimp caps or plastic screw caps The basal culture medium (Table 31) was
prepared according to Kostka amp Nealson (1998) but with a phosphate
concentration of 2 mM and the exclusion of Fe(II) and EDTA
(ethylenediaminetetraacetic acid) The medium was amended with 4-10 mM
lactate or formate and 8-27 mM Fe(III) The Fe(III) was applied as Fe(III) oxide
coatings on sand or calcite The suspensions were purged extensively with 100
N2(g) (HFOsand suspensions) or 995 N205 CO2(g) (goecalcite and
lepcalcite suspensions) prior to inoculation The calcareous systems were buffered
at pH ~ 76 through a natural buffer system (CaCO3(s) + 995 N205 CO2(g))
whereas the sandy systems contained no pH buffer (100 N2(g) pH 55-60)
Inoculum size made up 5 of the total volume Cultures were incubated dark at
room temperature and gently agitated once every day At different time intervals
suspension samples for Fe(II) and Moumlssbauer analysis were withdrawn from the
reaction mixture using 100 N2(g) or 995 N205 CO2(g)-flushed sterile
disposable syringes and hypodermic needles Suspension samples for Fe(II)
analysis were digested in 01 M HCl for 30 min
50 Chapter 3
Table 31 Composition of the mineral medium (modified from Kostka amp Nealson (1998))
Components Concentration (M)
(NH4)2SO4 00143 KH2PO4 73middot10-4
K2HPO4 13middot10-3
MgSO4middot7H2O 10middot10-3
CaCl2middot2H2O 50middot10-4
H3BO3 56middot10-5
ZnSO4middot7H2O 10middot10-6
Na2MoO4middot2H2O 40middot10-6
CuSO4middot5H2O 20middot10-7
MnSO4middotH2O 10middot10-6
Na2SeO4 12middot10-5
CoCl2middot6H2O 50middot10-6
NiCl2middot6H2O 80middot10-6
NaCl 10middot10-5
L-arginine 11middot10-4
L-serine 19middot10-4
L-glutamic acid 14middot10-4
Lactate or formate 4-10middot10-3
Fe(III) 8-27middot10-3
325 Analytical methods
Fe(II) was determined using a modified phenanthroline method (Fadrus amp Maly
1975) The total amount of Fe(III) coated on calcite and sand was determined by
atomic absorption spectroscopy following dissolution in 6 M HCl(aq) for 24 h
33 Results and discussion
331 Fe(II) production and suspension colour changes
Strongly chelating agents such as EDTA were omitted from the culture medium in
order to prevent complexation of Fe(II) and Fe(III) which interferes with
precipitation of Fe(II) and Fe(II)-Fe(III) mineral phases Within 1-2 weeks after
inoculation Shewanella algae BrY produced green mineral phases in media
suspensions containing lepidocrocite and goethite as coatings on calcite and 4-10
mM formate or lactate (Figure 31) The formation of the green precipitates was
generally slower for the lepidocrocite coating than for the goethite coating The
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 51
blue-green colours of the phases produced most likely originate from Fe(II)-Fe(III)
charge transfer between adjacent Fe(II) and Fe(III) ions in edge-shared octahedra
(Faye et al 1968) Dark brown and blackish products were formed when the
bacteria were inoculated on HFO coated sand (Figure 32)
Figure 31 Culture tubes containing a) goethite and b) lepidocrocite coated calcite in culture
medium The left tubes of the pair were not inoculated whereas the right tubes were
photographed 5 months after inoculation with S algae BrY Experimental conditions [formate]0
= 4 mM [Fe(III)]0 = 8 mM 995 N205 CO2(g) pH 76
52 Chapter 3
Figure 32 Culture tubes containing HFO coated sand in culture medium Tubes 1 and 2 to the
left were not inoculated whereas tubes 3-5 to the right were photographed a) 13 days and b) 21
days after inoculation with S algae BrY Experimental conditions [lactate]0 = 10 mM [Fe(III)]0
= 25 mM 100 N2(g) pH 55-60
The green and black colours did not change to other colours (observed for gt1
year) indicating that the microbial Fe(III) reduction ceased at these mineral stages
The concentrations of dissolved ferrous iron (Fe(II)sol) estimated during Fe(III)
bioreduction were generally low (Figure 33) When comparing the final Fe(II)sol
amounts produced and the slopes of the Fe(II)sol formation curves for HFO
goethite and lepidocrocite in Figure 33 it can be seen that the final Fe(II)sol
amount and the Fe(II)sol production rate both follow the order HFO gt goethite gt
lepidocrocite at similar cell densities regardless of the carbon source applied This
suggests that bioreduction by S algae BrY is more facile for HFO than for goethite
and lepidocrocite at the experimental conditions employed here It should be noted
that the final Fe(II)sol amounts and the Fe(II)sol production rates reported in this
work have not been normalised with respect to the specific surface areas of the iron
oxides and coating-bearing solids applied The reactivity trend is consistent with
previous findings demonstrating higher reducibility of natural and poorly
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 53
crystalline Fe(III) oxides as compared to synthetic crystalline Fe(III) oxides
(Zachara et al 1998) The authors ascribed these differences in reducibility to
differences in particle size surface area and crystal defects of the Fe(III) oxides In
some cases the dissolved Fe(II) concentration decreased again with time (Figure
33 b-d) This indicates that the Fe(II) formed was incorporated into solid phases
forming andor adsorbed onto the calcite sand or Fe(III) oxide surfaces The solid
Fe(II) concentrations were not estimated spectrophotometrically The solid
material was generally low in total iron and therefore saving it for Moumlssbauer
spectroscopic analysis was given highest priority
54 Chapter 3
Figure 33 Time course of dissolved Fe(II) production during bioreduction of HFOsand
goethitecalcite and lepidocrocitecalcite by S algae BrY Experimental conditions [Fe(III)]0 =
25 mM [formate]0 = 10 mM (a-c) or [lactate]0 = 10 mM (d-f)
No color change and no Fe(II) production were observed in mineral suspensions
lacking either a carbon source or S algae BrY cells (data not shown)
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 55
332 Identification of solid iron phases
The purity of the Fe(III) oxides used in the experiments were investigated by
transmission Mossbauer spectroscopy (Figure 34) Single (or strongly dominating)
sextets in the spectra with magnetic hyperfine fields of 48 1 505 and 45 5 Tat 5 K
demonstrated the purity of the HFO goethite and lepidocrocite samples
respectively A minor impurity of goethite in the lepidocrocite sample was
resolved in the spectrum measured at 80 K (not shown for pure sample but can be
seen as a magnetically ordered sextet in Figure 35c) No Fe(II)-containing
components were detected
a)
c)
middot 12 -8 -4 4 8 12
Velocity (mmls
bull middot12 -8 -4 0 4 12
Velocity (mmls)
b)
middot12 -8 -4 4 8 12
Velocity (mmls
Figure 34 Transmission Mossbauer spectra measured at 5 K of a) HFO (magnetic hyperfine
field of 481 T isomer shift of 048 1nrns-1 negligible quadrupole shift and line width of outer
lines 110 rmns-1) b) goethite (magnetic hyperfine field of 505 T isomer shift of 049 rmns-1
quadmpole shift of -013 1nrns-1 and line width of outer lines 042 rnrns-1
) and c) lepidocrocite
56 Chapter 3
(magnetic hyperfine field of 455 T isomer shift of 050 mms-1 quadrupole shift of ndash001 mms-1
and line width of outer lines 060 mms-1) prior to inoculation Simple Lorenztian fits are shown
The oxidation state and coordination of Fe in the microbially reduced HFO
goethite and lepidocrocite samples were also examined by transmission Moumlssbauer
spectroscopy (Figure 35) The bioreduced HFO goethite and lepidocrocite samples
cultured on formate contained Fe(II) holding similar coordination as inferred from
the similarity of the hyperfine parameters (see legend in Figure 35) but different
relative intensities (72 19 and 71 respectively) The major part of the Fe(III)
remaining in the bioreduced samples were coordinated similarly to the Fe(III)
present in the initial Fe(III) oxide The coordination of Fe(II) in the bioreduced
lepidocrocite samples cultured on lactate was slightly different (a smaller
quadrupole splitting of 288 mms-1 for the ferrous component dominates ndash data not
shown) The exact mineralogy of the Fe(II) present in the green phases was not
fully resolved but its coordination is very akin to one of the Fe(II) sites in vivianite
(see Chapter 2 this work) and synthetic green rusts (Koch 1998) These findings
agree with other reports on the bioformation of vivianite and green rusts by
Shewanella putrefaciens CN32 although the evidence provided may be discussed
(Fredrickson et al 1998 Glasauer et al 2003 Parmar et al 2001) Our
Moumlssbauer data on the green phases did not allow for a detailed account of the type
of green rust produced However when considering solution composition (see
Table 31) and the high affinity of GR interlayers for carbonate it is reasonable to
assume that carbonate GR was formed (Hansen amp Taylor 1991) Due to the high
amounts of Fe(III) in the oxides present in the experiments it was difficult to probe
a possible content of Fe(III) in the vivianite with certainty The differences in the
number of Fe(II) positions in the Moumlssbauer spectra and particular the different
temperatures at which magnetic ordering takes place can be employed in order to
distinguish between green rust and vivianite Preliminary Moumlssbauer data obtained
for the blackish precipitates formed in the HFOsand suspensions indicate that they
hold no resemblance to magnetite eventhough the colour suggests so On the
contrary the black precipitates seemed to be more similar to synthetic green rusts
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 57
Mossbauer spectroscopic measurements are currently underway in order to resolve
the Fe(II) coordinations in the greenish and blackish phases
a) b)
c c g Q e- e 0 0 1l 1l
-~ bull ~
iii ~ Qi bull a bull
-12 -8 4 0 4 12
4 -3 -2 -1 0 1 2 4 Velocity (mmts) Velocity ( rmis)
c)
-12 -8 4 8 12
v elocity (m mis)
Figure 35 Transmission Mossbauer spectra of the black and green phases fo1med within 1-2
weeks after inoculation of a) HFO (measured at 130 K) b) goethitecalcite (measured at 80 K)
and c) lepidocrocitecalcite (measured at 80 K) with S algae BrY Experimental conditions
[fo1m ate]0 = 4 mM [Fe(III)]o = 8 mM 995 Ni05 C02(g) pH 76 The quadrnpole
splittings and isomer shifts for the Fe(II) components in the three systems are a) 293 nnns-1 and
126 mmsmiddot1 b) 308 rmnsmiddot1 and131 rmnsmiddot1 and c) 322 mmsmiddot1 and 132 mmsmiddot1 Simple Lorenztian
fits are shown
The evidence provided in many of the studies proposing biogenic GRs is not all
too convincing but it strongly suggests the probability of microbially produced GR
being present The challenge encountered is that when present in low
58 Chapter 3
concentrations especially in mixtures including other iron minerals it is very
difficult to identify GRs using conventional solid phase analysis methods even
with Moumlssbauer spectroscopy At least two complementary methods such as XRD
and Moumlssbauer spectroscopy are required for proper identification and
characterization of GRs However in this work the solid materials were generally
too low in total iron to allow for XRD analysis Moreover the highly
heterogeneous suspensions were dominated by the coating-bearing sand and calcite
solids Electron micrographs including energy dispersive X-ray spectroscopy
suspension colour and mineral stability calculations do not suffice as evidence The
most convincing evidence provided so far involves an atypical GR-CO3 with an
Fe(II)Fe(III) ratio of 1 (Ona-Nguema et al 2002aampb) This GR-CO3 was formed
as a result of lepidocrocite reduction by Shewanella putrefaciens CIP 8040 at
conditions comprising high nutrient levels (50-75 mM formate) high Fe(III)
concentrations (80-300 mM) and a synthetic electron shuttle (100 microM
anthraquinone-26-disulfonate (AQDS)) at initial pH 75 Hence the results
reported during recent years suggest that microbial formation of GR may be
possible The results presented here indicate that GRs may be produced
microbially at conditions including low carbon and Fe(III) concentrations as well
as the exclusion of synthetic electron shuttles and pH buffers
333 Factors controlling the identity of the secondary iron minerals
In general one would expect that biogenic minerals have chemical compositions
and crystal habits similar to those produced by nonenzymatic processes as they are
governed by the same equilibrium principles In fact since the latter stages of
mineralization are inorganically driven and the secondary Fe(II)-containing
minerals are formed indirectly by electron transfer outside the bacterial cell and not
directly inside the bacterial cell the type of iron mineral formed is a function of the
environmental conditions in which the bacteria live ie the same microorganism
form different minerals in different environments The key factors controlling the
identity of the secondary iron minerals include medium composition electron
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 59
donor and electron acceptor concentrations mineral aging as well as adsorbed ions
(Zachara et al 2002) The main factor controlling the nature of the secondary
mineral products are the respiration-driven biogenic Fe(II) supply rate and
magnitude and its surface reaction with the residual oxide and other sorbed ions
(Zachara et al 2002) Especially solution and medium composition have a strong
impact on the nature of the Fe(II)-containing biomineralization products forming
Accordingly siderite (FeCO3) and magnetite (Fe3O4) were the secondary solid
phases resulting from the bioreduction of ferrihydrite by Shewanella putrefaciens
CN32 in bicarbonate buffered medium (pH 71) containing no phosphate whereas
siderite and vivianite were the secondary iron minerals dominating in bicarbonate
buffered medium (pH 74) containing 4 mM phosphate (Zachara et al 2002) This
is explained by the inhibiting effect of phosphate on crystallization of magnetite
(Couling amp Mann 1985 Fredrickson et al 1998)
334 Factors controlling the rate and extent of Fe(III) bioreduction
In this study the extent of Fe(III) bioreduction was estimated to 19-72 by
transmission Moumlssbauer measurements In fact complete microbial reduction of
crystalline Fe(III) minerals has never been observed in laboratory batch culture
studies (Roden amp Urrutia 2002) It has been found that Fe(II) does not inhibit
Fe(III) reductase activity through an enzyme inhibition mechanism (Roden amp
Urrutia 2002) Hence other chemical andor physiologic factors control the
bioavailability of solid Fe(III) phases and thus the extent of their microbial Fe(III)
reduction The initial rate and long-term extent of microbial reduction of
amorphous and crystalline Fe(III) oxides including HFO goethite and hematite
were linearly correlated with oxide surface area (Roden amp Zachara 1996)
Association of biogenic Fe(II) with Fe(III) oxide and DIRB cell surfaces reduced
the long-term extent of crystalline Fe(III) oxide bioreduction (Roden amp Urrutia
2002) These results were explained by Fe(II) surface complexes andor
precipitates creating a passive Fe(II)-bearing surface film providing direct physical
interference with the electron transfer from the DIRB cells to Fe(III) However the
60 Chapter 3
real mechanisms whereby the surface-associated Fe(II) inhibits Fe(III) oxide
bioreduction are unclear but they most likely involve both kinetic and
thermodynamic constraints on the electron transfer Culture medium composition
in particular the presence and the concentration of phosphate as well as Fe(II)
chelating ligands also exert an influence on the extent of the microbial reduction
of Fe(III) oxides The extent of Fe(III) bioreduction was inhibited by high
phosphate concentrations which favoured surfacebulk precipitation processes
(Urrutia et al 1998) The carbon sources most frequently applied in Fe(III)
bioreduction studies include malate citrate and other di- and tricarboxylic acids
which are not only easily metabolizable carbon sources but also eminent Fe(II) and
Fe(III) chelators In this study we employed formate and lactate as carbon sources
since they are the weakest complexing agents of Fe(II) and Fe(III) among the C1-
C3 monocarboxylic acids (Martell 1964) Thus we expect less dissolution of
prevailing precipitates by complexation as compared to other studies
34 Conclusions
This work demonstrated that Shewanella algae BrY reduced 19-72 of initial
Fe(III) when grown in culture media containing 4-10 mM formate or lactate and 8-
27 mM Fe(III) applied as goethite or lepidocrocite coatings on calcite (pH 76) or
HFO coatings on sand (pH 55-60) Within 1-2 weeks after inoculation green
mineral phases were produced in the goethitecalcite and lepidocrocitecalcite
mineral systems whereas black precipitates formed in the HFOsand suspensions
Moumlssbauer spectroscopic analyses indicated that the greenish and blackish phases
most likely were mineral mixtures dominated by vivianite and green rust Thus the
results indicate that GRs may be produced microbially at conditions including low
carbon and Fe(III) concentrations as well as the exclusion of synthetic electron
shuttles and pH buffers
Acknowledgments
We would like to thank Dr R Gerlach for providing us the Shewanella algae BrY culture and
Dr C B Koch for performing the Moumlssbauer analyses
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 61
References Al-Agha MR Burley SD Curtis CD Esson J (1995) Complex cementation textures and authigenic mineral assemblages in recent concretions from the Lincolnshire Wash (east coast UK) driven by Fe(0) to Fe(II) oxidation Journal of the Geological Society 152 157-171 Bernal JD Dasgupta DR Mackay AL (1959) The oxides and hydroxides of iron and their structural inter-relationships Clay Minerals Bulletin 4 15-30 Caccavo Jr F Blakemore RP Lovley DR (1992) A hydrogen-oxidizing Fe(III)-reducing microorganism from the Great Bay Estuary New Hampshire Applied and Environmental Microbiology 58 3211-3216 Caccavo Jr F Das A (2002) Adhesion of dissimilatory Fe(III)-reducing bacteria to Fe(III) minerals Geomicrobiology Journal 19 161-177 Couling SB Mann S (1985) The influence of inorganic phosphate on the crystallization of magnetite (Fe3O4) from aqueous solution Journal of the Chemical Society Chemical Communications 1713-1715 Das A Caccavo Jr F (2000) Dissimilatory Fe(III) oxide reduction by Shewanella alga BrY requires adhesion Current Microbiology 40 344-347
Das A Caccavo Jr F (2001) Adhesion of the dissimilatory Fe(III)-reducing bacterium Shewanella alga BrY to crystalline Fe(III) oxides Current Microbiology 42 151-154 Fadrus H Maly J (1975) Suppression of iron(III) interference in the determination of iron(II) in water by the 110-phenanthroline method The Analyst 100 549-554 Faye GH Manning PG Nickel EH (1968) The polarized optical absorption spectra of tourmaline cordierite chloritoid and vivianite ferrous-ferric electronic interaction as a source of pleochroism American Mineralogist 53 1174-1201 Fredrickson JK Zachara JM Kennedy DW Dong H Onstott TC Hinman NW Li S (1998) Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium Geochimica et Cosmochimica Acta 62 3239-3257 Geacutenin JMR Olowe AA Benbouzid-Rollet ND Prieur D Confente M Resiak B (1991) The simultaneous presence of green rust 2 and sulfate reducing bacteria in the corrosion of steel sheet piles in a harbour area Hyperfine Interactions 69 875-878 Geacutenin JMR Bourrieacute G Trolard F Abdelmoula M Jaffrezic A Refait Ph Maitre V Humbert B Herbillon A (1998) Thermodynamic equilibria in aqueous suspensions of synthetic and natural Fe(II)-Fe(III) green rusts Occurrences of the mineral in hydromorphic soils Environmental Science and Technology 32 1058-1068 Glasauer S Weidler PG Langley S Beveridge TJ (2003) Controls on Fe reduction and mineral formation by a subsurface bacterium Geochimica et Cosmochimica Acta 67 1277- 1288 Hansen HCB Taylor RM (1991) The use of glycerol intercalates in the exchange of CO3
2- with SO4
2- NO3- or Cl- in pyroaurite-type compounds Clay Minerals 26 311-327
62 Chapter 3
Hungate RE (1969) A roll tube method for cultivation of strict anaerobes Methods in Microbiology 3B 117-132 King GM (1990) Effects of added manganic and ferric oxides on sulfate reduction and sulfide oxidation in intertidal sediments FEMS Microbiology Ecology 73 131-138 Koch CB (1998) Structures and properties of anionic clay minerals Hyperfine Interactions 117 131 -157 Kostka J Nealson KH (1998) Isolation cultivation and characterization of iron- and manganese reducing bacteria In Techniques in Microbial Ecology Burlage RS Atlas R Stahl D Geesey G Sayler G (eds) Oxford University Press Inc 58-78 Kukkadapu RK Zachara JM Smith SC Fredrickson JK Liu C (2001) Dissimilatory bacterial reduction of Al-substituted goethite in subsurface sediments Geochimica et Cosmochimica Acta 65 2913-2924
Liu C Zachara JM Gorby YA Szecsody JE Brown CF (2001) Microbial reduction of Fe(III) and sorptionprecipitation of Fe(II) on Shewanella putrefaciens strain CN32 Environmental Science and Technology 35 1385-1393 Lovley DR (1991) Dissimilatory Fe(III) and Mn(IV) reduction Microbiological Reviews 55 259-287 Lovley DR (1997) Microbial Fe(III) reduction in subsurface environments FEMS Microbiology Reviews 20 305-313 Lovley DR Phillips EJP (1986) Organic matter mineralization with reduction of ferric iron in anaerobic sediments Applied and Environmental Microbiology 51 683-689 Lovley DR Phillips EJP Lonergan DJ (1991) Enzymatic versus nonenzymatic mechanisms for Fe(III) reduction in aquatic sediments Environmental Science and Technology 25 1062-1067 Lower SK Hochella Jr MF Beveridge TJ (2001) Bacterial recognition of mineral surfaces Nanoscale interactions between Shewanella and α-FeOOH Science 292 1360-1363 Martell AE (1964) Stability constants of metal-ion complexes Part 2 Organic including macromolecule ligands The Chemical Society London 2 ed Miller TL Wolin MJ (1974) A serum bottle modification of the Hungate technique for cultivating obligate anaerobes Applied Microbiology 27 985-987 Nealson KH Saffarini D (1994) Iron and manganese in anaerobic respiration Environmental significance physiology and regulation Annual Review of Microbiology 48 311-343 Nevin KP Lovley DR (2000) Lack of production of electron-shuttling compounds or solubilization of Fe(III) during reduction of insoluble Fe(III) oxide by G metallireducens Applied and Environmental Microbiology 66 2248-2251 Nevin KP Lovley DR (2002) Mechanisms for Fe(III) oxide reduction in sedimentary
Formation of Layered Iron Hydroxides by Microbial Fe(III) Reduction 63
environments Geomicrobiology Journal 19 141-159 Nielsen A (1976) Hvid groslashn og sort rust Beskrivelse af en korrosionsskade paring et svoslashmmebassin Nordisk Betong 2 21-24 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002a) Iron (IIIII) hydroxycarbonate green rust formation and stabilization from lepidocrocite bioreduction Environmental Science and Technology 36 16-20 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002b) Microbial reduction of lepidocrocite γ-FeOOH by Shewanella putrefaciens The formation of green rust Hyperfine Interactions 139140 231-237 Parmar N Gorby YA Beveridge TJ Ferris FG (2001) Formation of green rust and immobilization of nickel in response to bacterial reduction of hydrous ferric oxide Geomicrobiology Journal 18 375-385 Roden EE Urrutia MM (2002) Influence of biogenic Fe(II) on bacterial crystalline Fe(III) oxide reduction Geomicrobiology Journal 19 209-251 Roden EE Zachara JM (1996) Microbial reduction of crystalline iron(III) oxides Influence of oxide surface area and potential for cell growth Environmental Science and Technology 30 1618-1628 Roh Y Lee SY Elless MP (2000) Characterization of corrosion products in the permeable reactive barriers Environmental Geology 40 184-194 Schwertmann U Cornell RM (1991) Iron oxides in the laboratory Preparation and characterization VCH Verlagsgesellschaft mbH Weinheim Thamdrup B (2000) Bacterial manganese and iron reduction in aquatic sediments In Advances in Microbial Ecology (Schink B ed) Kluwer AcademicPlenum Publishers New York 41-84 Trolard F Abdelmoula M Bourrieacute G Humbert B Geacutenin JMR (1996) Mise en eacutevidence dun constituant de type rouilles vertes dans les sols hydromorphes Proposition de lexistence dun nouveau mineacuteral la fougeacuterite Geacuteosciences de surface Comptes Rendus de LrsquoAcademie des Sciences 323 1015-1022 Tuovinen OH Button KS Vuorinen A Carlson L Mair DM Yut LA (1980) Bacterial chemical and mineralogical characteristics of tubercles in distribution pipelines Journal of the American Water Works Association 72 626-635 Turick CE Caccavo Jr F Tisa LS (2003) Electron transfer from Shewanella algae BrY to hydrous ferric oxide is mediated by cell-associated melanin FEMS Microbiology Letters 220 99-104 Urrutia MM Roden EE Fredrickson JK Zachara JM (1998) Microbial and surface chemistry controls on reduction of synthetic Fe(III) oxide minerals by the dissimilatory iron- reducing bacterium Shewanella alga Geomicrobiology 15 269-291 Venkateswaran K Moser DP Dollhopf ME Lies DP Saffarini DA MacGregor BJ Ringelberg DB White DC Nishijima M Sano H Burghardt J Stackebrandt E
64 Chapter 3
Nealson KH (1999) Polyphasic taxonomy of the genus Shewanella and description of Shewanella oneidensis sp nov International Journal of Systematic Bacteriology 49 705-724 Zachara JM Fredrickson JK Li S Kennedy DW Smith SC Gassman PL (1998) Bacterial reduction of crystalline Fe3+ oxides in single phase suspensions and subsurface materials American Mineralogist 83 1426-1443
Zachara JM Kukkadapu RK Fredrickson JK Gorby YA Smith SC (2002) Biomineralization of poorly crystalline Fe(III) oxides by dissimilatory metal reducing bacteria (DMRB) Geomicrobiology Journal 19 179-207
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 65
4 Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust The Effect of Probe Compound Charge
Abstract
Layered iron(II)-iron(III)-hydroxides (green rusts) may play an important role in
controlling the fate and transport of many organic and inorganic contaminants in
iron-rich suboxic soils and sediments Unlike most other iron oxides green rusts
(GRs) contain not only external Fe(II) reactive sites at the basal planes and at the
edges but also internal sites in the space between consecutive Fe(II)-Fe(III)
hydroxide layers The GR interlayer thickness is a function of both the size and the
charge of the interlayer anion Whether a given oxidant has access to the internal
sites in GRs is dependent on its charge We investigated the reductive
transformation of nitroaromatic compounds (NACs) by GR-SO4 and studied the
effect of NAC charge on the reactivity towards GR-SO4 A series of structurally
closely related compounds with different charge properties including nitrobenzene
4-nitrotoluene 4-chloronitrobenzene and 4-nitrophenylacetic acid were used as
probe compounds The NACs were completely reduced to their corresponding
anilines by GR-SO4 The reactions followed pseudo 1 order kinetics with respect
to NAC and the surface area-normalised pseudo 1 order rate constants obtained
were 016ndash465middot10-4 s-1middotm-2middotL at [Fe(II)GR]0 = 103-1260 mM [NAC]0 = 20-102
microM and pH 84-86 Neither mass transfer control nor surface saturation kinetics
could account for the rather unexpected similarity of the surface area-normalised
pseudo 1 order rate constants obtained for the reduction of the neutral and anionic
NACs by GR-SO4 These observations suggest that the anionic NACs did not have
an enhanced access to the inner or outer Fe(II)-GR reactive sites as compared to
the neutral NACs Hence the reaction between NAC and GR-SO4 primarily took
place at the edges of GR-SO4
66 Chapter 4
41 Introduction
Layered iron(II)-iron(III)-hydroxides (green rusts) are intermediate phases formed
by partial oxidation of Fe(II) or partial reduction of Fe(III) In neutral and weakly
alkaline solutions the oxidation of dissolved Fe(II) always passes through solid
green rust (GR) phases (Bernal et al 1959) This agrees with the natural GR
occurrences found in suboxic non-acid iron-rich environments such as
hydromorphic soils and intertidal sediments (Al-Agha et al 1995 Trolard et al
1996 Genin et al 1998) In addition GRs have been found as corrosion products
in numerous engineered systems ie a pipeline distribution system for drinking
water steel sheet piles in marine sediments reinforced concrete (ferro-concrete)
and permeable reactive barriers of zero-valent iron implemented for on-site
remediation of organic and inorganic contaminants (Tuovinen et al 1980 Nielsen
1976 Genin et al 1991 Roh et al 2000) Furthermore the microbial formation
of GRs resulting from bioreduction of Fe(III) oxides by strains of the anaerobic
dissimilatory Fe(III) reducing bacteria Shewanella putrefaciens has been reported
increasingly over the last 5 years (Fredrickson et al 1998 Kukkadapu et al 2001
Parmar et al 2001 Ona-Nguema et al 2002 Glasauer et al 2003) Moreover
the biotic formation of GRs by anaerobic denitrifying Fe(II) oxidizing bacteria has
been proposed but proper identification of the GR phases still lacks (Chaudhuri et
al 2001) All these indications of microbial GR formation infer the importance of
GRs as a link between geochemical and biological processes in natural systems
GRs form platy crystals with the general formula [FeII(6-
x)FeIIIx(OH)12]x+[(A)xnmiddotyH2O]x- where x = 09 - 42 A is an n-valent anion eg
CO32- Clndash or SO4
2- and y is the number of water molecules in the interlayer The
crystal structure consists of positively charged hydroxide sheets with Fe(II) and
Fe(III) cations having octahedral hydroxyl coordination The Fe(III) in the
hydroxide layers creates a net positive charge which is balanced by hydrated
anions in the interlayers (Figure 41) The interlayers have a higher affinity for
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 67
divalent anions than for monovalent anions (Miyata 1983) Among the 3 most
common GR forms the affinity follows the order CO32- gt SO4
2- gt Clndash The extreme
preference shown for carbonate hinders further access and exchange except under
certain conditions (Hansen amp Taylor 1991) Non-carbonate forms are readily
exchanged with other anions when dispersed in a solution containing the
exchanging anion (Mendiboure amp Schoumlllhorn 1986)
c
ba
Figure 41 Green rust layer structure The hydroxide layers and the interlayers are connected by
hydrogen bonds (not shown) The GR-SO4 crystal structure is characterised by the hexagonal
unit cell having a = b = 055 nm and c = 110 nm (Simon et al 2003) The unit cell consists of
one double layer (a double layer is a hydroxide layer and an interlayer) ie the hydroxide layer
constitutes 049 nm and the interlayer 061 nm in GR-SO4
The GR interlayer thickness (extending in the c axis direction Figure 41) is a
function of both the size and the charge of the interlayer anion Tetrahedrally
coordinated anions like sulphate lead to larger interlayer distances than smaller
monoatomic anions like chloride or planar ions like carbonate (Mendiboure amp
Schoumlllhorn 1986) Not only size but also charge density plays a role for the
interlayer spacing That is for anions having the same number of valence
electrons anions with smaller ionic radii (higher electron density) are bound more
strongly and therefore result in smaller interlayer spacings
Due to their layered structures anionic interlayers and high specific surface areas
GRs represent reactive ion exchangers and sorbents of anions eg arsenate
selenate and phosphate (Myneni et al 1997 Hansen amp Poulsen 1999 Randall et
al 2001) In addition GR may incorporate heavy metal cations by isomorphic
68 Chapter 4
substitution into the GR hydroxide layers (Tamaura 1985 Tamaura 1986)
Furthermore GRs have been shown to reduce a range of inorganic contaminants
such as nitrite nitrate selenate chromate uranyl pertechnetate and the transition
metals AgI AuIII CuII and HgII as well as organic pollutants including halogenated
ethanes ethenes and methanes (Hansen et al 1994 Hansen et al 1996 Myneni et
al 1997 Erbs et al 1999 Loyaux-Lawniczak et al 1999 Cui amp Spahiu 2002
Lee amp Batchelor 2002b Heasman et al 2003 OrsquoLoughlin et al 2003a amp 2003b
Pepper et al 2003 Elsner et al 2004 OrsquoLoughlin amp Burris 2004) Thus through
sequestration and reductive transformation GRs may play an important role in
controlling the fate and transport of contaminants in suboxic soils and sediments
In a previous study the effects of interlayer anion and Fe(II)Fe(III) ratio in GRs
on the reduction rate of nitrate were investigated (Hansen et al 2001) It was
found that the rate of nitrate reduction to ammonium increased with increasing
Fe(II)Fe(III) ratio and decreased when exchanging a monovalent interlayer anion
(chloride) with a divalent anion (sulphate) The results suggest that for anionic
oxidants like nitrate Fe(II) within the hydroxide layer is available from the outside
basal planes and from the edges as well as through the interlayer under certain
conditions (Figure 42) However oxidants with different charge properties
(cations neutral molecules) may exhibit different affinities for the various reactive
Fe(II) sites present in GR
As the reactive sites are located inat the Fe(II)-Fe(III) hydroxide layers the rate of
reaction depends on the hydroxide layer area which can be accessed by the
oxidant If the oxidant can exchange with the interlayer anion reaction can take
place both at outer and inner surfaces of the GR particles and in total more
reactive sites are available for the reaction However it was found that nitrate
cannot penetrate the interlayer when carbonate or sulphate constitutes the
interlayer anions (Hansen amp Koch 1998) This agrees with the fact that the
interlayers have a lower affinity for monovalent anions than for divalent anions
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 69
(Miyata 1983) However when nitrate was forced into the interlayer by extracting
the interlayer sulphate through precipitation of barium sulphate outside the GR
particles the observed 40 fold increase in rate of nitrate reduction almost equalled
the increase in exposed surface area of the Fe(II)-Fe(III) hydroxide layers (Hansen
amp Koch 1998) From these observations it is expected that the rate of reaction
depends on the particular GR form the crystallite size and the ease with which an
oxidant can exchange with An- in the GR interlayer (Figure 42) Due to
electrostatic interactions we expect anions to be attracted to the positively charged
outer and inner surfaces to a higher degree than cations and neutral compounds If
this theory holds we may expect oxidants with similar intrinsic reactivity (similar
one-electron reduction potentials) to react in the following order anionic gt non-
charged gt cationic (Figure 43) granting that we do not normalise the rate
constants with respect to the amount of oxidant sorbed
Figure 42 Reaction of a probe compound at basal planes at edges and in the interlayer of GR
The hypothesis only holds in cases where the oxidants possess the same intrinsic
reactivities If the relative reactivities of the probe compounds differ greatly from
what would be expected when considering only their reduction potentials
70 Chapter 4
compound specific effects such as charge properties might explain this and the
relative reactivities may follow a pattern like the one depicted in Figure 43
Figure 43 Hypothetical plot of observed reaction rate constants for the reactions between
cationic neutral and anionic probe compounds and GR-SO4 assuming that the oxidant charge
controls its reactivity towards GR
In this work we investigated the reductive transformation of NACs by GR-SO4
Furthermore the effect of NAC charge on the rate of reaction and the possible
access to the internal reactive sites in GR-SO4 were assessed When quantifying
Fe(II) in GRs by means of acid digestion it is not possible to distinguish between
the reactive sites accessible from the outside (at the basal planes or at the edges) or
through the interlayer However we designed an indirect method to gain insight
into the relative importance of the various reactive sites by using a series of
structurally closely related compounds with different charge properties as ldquoreactive
probesrdquo Neutral and anionic probes were needed in order to access all Fe(II)
reactive sites According to our hypothesis cationic and non-charged oxidants
should provide information about the reactivity of the outer Fe(II) reactive sites in
GR whereas the anionic oxidants should provide information about the reactivity
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 71
of both outer and inner Fe(II) reactive sites We chose five nitro aromatic
compounds (NACs) - representing an important group of reducible organic
pollutants - as probe compounds (Figure 43) This class of compounds is not only
of great environmental concern but also comprises suitable model compounds for
studying redox reactions potentially relevant in the environment Moreover they
react readily with Fe(II) surface species associated with iron oxides or clay
minerals transforming them into well-defined easily detected products allowing
mass and electron balances to be established (Hofstetter et al 2003 Klausen et al
1995 Schultz amp Grundl 2000) Our main goals were to establish the rate law and
estimate the surface area-normalised reaction rates for the reaction of the probe
compounds with GR-SO4 in order to assess the importance of the Fe(II) reactive
sites accessible through the interlayer relative to the Fe(II) reactive sites accessible
at the outer surface in GR-SO4
4 2 Materials and methods
All handling and sampling of solutions and suspensions were carried out under
strict anoxic conditions All chemicals were pa quality or better Methanolic stock
solutions (5 mM) of nitrobenzene (NB) 4-nitrotoluene (4-NT) 4-
chloronitrobenzene (4-CNB) and 4-nitrophenylacetic acid (4-NPA) were prepared
in deoxygenated methanol Several attempts to synthesize the cationic probe
compound 4-(NNN-trimethylammonium)-nitrobenzene failed and therefore the
study had to be carried out with only neutral and anionic oxidants The sulphate
GR form was chosen as it is the most stable form and thus the easiest to work
with in the lab
421 Synthesis of GR-SO4
GR-SO4 was synthesized by controlled air oxidation of an FeSO4 solution at a
constant pH of 700 according to the procedure given by Koch amp Hansen (1997)
The GR-SO4 suspension was washed with deoxygenated deionised water and
72 Chapter 4
separated on a folding filter redispersed in deoxygenated 25 mM Na2SO4(aq) in
order to stabilize the GR-SO4 and prevent it from transforming into magnetite
spontaneously Washing separation and redispersion of the GR-SO4 suspension
were conducted in an anoxic glove box (Coy Laboratory Products Inc) All
suspensions and solutions were deoxygenated by Ar-purging (999998 Ar
Carbagas)
422 Mineral characterisation
The identity and purity of the GR-SO4 suspensions were examined by means of X-
ray diffraction measurements The XRD analyses were performed on a Scintag
XDS 2000 using Cu Kα radiation (45 kV 40 mA) Glycerol smears made
according to Hansen (1989) were scanned between 6 and 80 deg2θ with a scan speed
of 1 deg2θmin
423 Lyophilization and determination of specific surface area
Simple air-drying of the GR mineral in the glove box resulted in big flakes with
very low surface areas hence a more suitable lyophilization method was adopted
from Elsner et al (2004) The GR-SO4 suspensions were lyophilised using
Schlenk-type glassware The set-up consisted of a 1 L round bottom flask and a
200 mL glass finger connected by a crescent-shaped bridge equipped with an
evacuation outlet and a stopcock All ground joints and fittings were attached using
high-vacuum grease The washed and resuspended GR-SO4 suspensions were
filled into the glass finger and the freeze-drying apparatus was assembled and
closed before taking it out of the glove box The suspension was frozen by
carefully submerging the lower part of the glass finger into liquid nitrogen for a
few hours Subsequently the evacuation outlet was connected to a vacuum pump
by a metal hose Following a short evacuation of the metal hose the lyophilization
apparatus was evacuated for several minutes by gently opening the stopcock The
evacuation was terminated by closing the stopcock and disconnecting the vacuum
pump The apparatus position was now reversed by removing the glass finger from
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 73
and immersing the round bottom flask into liquid nitrogen As any other
lyophilization method this method depends on sublimation of the ice from the
frozen sample and its recondensation on a cool surface in this case the round
bottom flask Generally it took 1-2 d for the mineral to dry The apparatus was
disassemled in the glove box and the fine powder stored under anoxic conditions
The specific surface area (SSA) of GR-SO4 was determined by the BET multi-
point method using N2 adsorption (Brunauer et al 1938) Powder samples were
filled into sample burettes in the glove box and the generously greased stopcocks
closed Samples and burettes were evacuated prior to connecting them to the BET-
instrument (Sorptomatic 1990 Fisons)
424 Estimation of the one-electron reduction potential for 4-NPA
Kinetic experiments in 100 mL Viton stoppered and alu-crimp capped serum vials
were carried out under the exclusion of oxygen as described by Hofstetter et al
(1999) The homogeneous aqueous solutions contained 50 mM KH2PO4 buffer
(pH = 660) 5 mM Na2S redox buffer and 20 microM juglone (8-hydroxy-14-
naphthoquinone) added as deoxygenated 20 mM methanolic stock solution The
solutions were equilibrated at least one day prior to 4-NPA addition To start the
reaction 50 microM 4-NPA was added as deoxygenated 20 mM methanolic stock
solution The vials were agitated on a roller apparatus in the dark at 21ordmC Control
experiments were prepared similarly except for the addition of juglone At
different time intervals aqueous samples were withdrawn with a syringe and
collected in 18 mL HPLC vials containing 100 microL 1 M HCl The sample vials
were sealed with Teflon-coated silicone septa and plastic screw caps and vortexed
for 10 s The samples were stored at -20degC and analysed without further treatment
For comparison experiments with 4-NT were also conducted See Supporting
Information 71 for more information on the one-electron reduction potentials
74 Chapter 4
425 Kinetic experiments
All reactions took place at pH 84-86 where GR-SO4 tends to stabilize and buffer
itself Samples for Fe(II) and XRD analysis were withdrawn prior to reaction Due
to the fast reactions the experiments were conducted in 10 mL single-use
polyethylene syringes (BD Plastipak) in the glove box To start reaction 40-200
microL 5 mM methanolic stock solutions of NAC were quickly added to 10 mL GR-
SO4 suspension (1-12 mM Fe(II)GR) washed and resuspended in 25 mM
Na2SO4(aq) A Teflon filter (25 mm x 02 microm BGB Analytik) was quickly
mounted on the tip of the syringe and the syringe was vigorously shaken between
sampling At different time intervals filtered suspension samples were collected in
18 mL HPLC vials The HPLC vials were sealed with Teflon-coated silicone septa
and plastic screw caps The samples were stored at -20degC and analysed without
further treatment Absorption of NAC in the syringe and in the Teflon filter
evaluated in blank experiments with NAC added to 25 mM Na2SO4(aq) was found
to be negligible
426 Analytical methods
Initial total and aqueous Fe(II) were determined using a modified phenanthroline
method (Fadrus and Maly 1975) In order to determine [Fe(II)aq] and [Fe(II)total] 1
mL filtered (022 microm) and 1 mL unfiltered GR-SO4 suspension samples were
withdrawn and each treated with 18 mL 01 M HCl for at least 30 min From these
acid digests 01 mL was added to 05 mL Fe(II)-reagent and 19 mL deionised
water (DIW) added up The Fe(II) content in GR-SO4 was estimated as the
difference [Fe(II)GR] = [Fe(II)total] - [Fe(II)aq] The NACs and their corresponding
intermediates and products formed during reduction by GR-SO4 were quantified by
reversed-phase HPLC Separation was performed on a LiChrospher 100 RP-18 (5
microm 125 times 4 mm ID) reversed-phase column coupled with a LiChroCART 100 RP-
18 (4 times 4 mm ID) precolumn Analytical conditions were isocratic and the eluent
consisted of 10 mM hydroxylammonium chloride in various DIWCH3OH
mixtures (vv 3565 and pH 70 for 4-NT and 4-CNB 955 and pH 60 for
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 75
4-NPA) The injection volume was 20 microLand the flow-rate 10 mLmin HPLC
analyses were performed using a Gynkotek High Precision Pump M480 Gynkotek
Gina 50 autosampler and a diode array UV detector (340s Gynkotek) UV-VIS
detection was carried out at the wavelengths of maximum absorption for the
various nitro aromatic and aniline analytes
43 Results and discussion
431 Productformation and reaction kinetics
The reduction of the aromatic nitro group occurs via nitroso- and hydroxylamino-
intermediates where 2 electrons are transferred in each reaction step (Figure 44)
0 --0 H OH H H --0 N N N N
2e- 2H+ H20 + 2e-~ 2e- 2H+ H20
~ ~ R R R
Nitro benzene Nitrosobenzene Hydroxylamine Aniline
Figure 44 Reductive transfonnation pathway of NA Cs
Thus in order to reduce 1 Ar-N02 completely to Ar-NH2 6 electrons
corresponding to 6 mol Fe(II) are needed As magnetite was the major iron phase
formed during reaction (XRD results not shown) we assume the following
reaction stoichiometry
The aniline product was not formed at the same rate as the nitro compound
degraded which is consistent with the detection of early eluting hydroxylamine
intermediates during the course of the reaction (Figure 45a amp 45c) No traces of
76 Chapter 4
nitrosobenzene intermediates or side products such as azoxy- azo- or
hydrazobenzene were found In Figure 45 pseudo 1 order kinetic plots and ln
[Ar-NO2]t[Ar-NO2]0) versus time plots for the neutral probe compounds 4-CNB
and 4-NT are shown as examples The plots for NB and 4-NPA look similar
Figure 45 a Concentration versus time plots for reaction of GR-SO4 with 4-CNB ([Fe(II)GR]0 =
126 mM [4-CNB]0 = 30 microM) b ln [Ar-NO2]t[Ar-NO2]0) versus time plots for reaction of GR-
SO4 with 4-CNB ([Fe(II)GR]0 = 126 mM + [4-CNB]0 = 30 microM [Fe(II)GR]0 = 63 mM + [4-
CNB]0 = 50 microM) c Concentration versus time plots for reaction of GR-SO4 with 4-NT
([Fe(II)GR]0 = 131 mM [4-NT]0 = 20 microM) d ln [Ar-NO2]t[Ar-NO2]0) versus time plots for
reaction of GR-SO4 with 4-NT ([Fe(II)GR]0 = 131 mM [4-NT]0 = 20 microM [Fe(II)GR]0 = 131
mM [4-NT]0 = 50 microM) The hydroxylamino intermediate shown in microM equals the deficit in the
mass balance and in abs equals the detector response (peak area) Solid lines represent 1 order
kinetic fits (a amp c) and ln [Ar-NO2]t[Ar-NO2]0) versus time fits (d) whereas symbols and dotted
lines represent actual data
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 77
At intial Fe(II)GR concentrations in large excess of initial Ar-NO2 concentration
we found a pseudo 1 order rate law for the degradation of Ar-NO2 by GR-SO4
[ ] [ ] [ b 2
a GR
2 ArNOFe(II) ArNOsdotsdot=minus k
dtd ]
where a = 1 b = 1 and the observed pseudo 1 order rate constant kobs = k middot
[Fe(II)GR] At high [Fe(II)GR]0[Ar-NO2]0 ratios the nitro compound was
transformed completely into the aniline product within reaction duration and the
degradation curves of the nitro compound were shaped according to pseudo 1
order kinetics (data points follow solid line in Figure 45a) In some instances ie
at low [Fe(II)GR]0[Ar-NO2]0 ratios the reactions did not follow pseudo 1 order
kinetics for the whole duration of reaction (data points deviate from solid line in
Figure 45c) Hence in order to allow comparison all the pseudo 1 order rate
constants were calculated as initial rates (ie max first two half-lives) from linear
fits of (time ln [Ar-NO2]t[Ar-NO2]0)-plots (Figure 45b amp 45d) Surface area-
normalised pseudo 1 order rate constants are shown in Table 41
Tabl
e 4
1 S
urfa
ce a
rea-
norm
alis
ed p
seud
o 1
ord
er ra
te c
onst
ants
for t
he re
duct
ive
trans
form
atio
n of
4-n
itrot
olue
ne (4
-NT)
4-
chlo
roni
trobe
nzen
e (4
-CN
B) a
nd 4
-nitr
ophe
nyla
cetic
aci
d (4
-NPA
) by
GR
-SO
4
Exp
erim
ent
Age
GR
(d
) [F
e(II
) GR] 0
(mM
) [N
AC
] 0 (micro
M)
[Fe(
II) G
R] 0
[N
AC
] 0∆[
ArN
O2]
(microM
) af b
k obs
(s-1
) ck o
bs (s
-1middotm
-2middotL
) d
GR
-SO
4 + 4
-NT
3 1
103
20
51
5
109
54
5
7
65middot1
0-46
95middot1
0-5
GR
-SO
4 + 4
-NT
3 1
103
50
20
6
174
34
8
7
41middot1
0-46
74middot1
0-5
GR
-SO
4 + 4
-NT
3 1
103
10
0 10
3
214
21
4
2
63middot1
0-42
39middot1
0-5
GR
-SO
4 + 4
-CN
B 1
1
103
55
18
7
177
32
2
4
21middot1
0-43
83middot1
0-5
GR
-SO
4 + 4
-CN
B 1
1
103
10
2 10
1
165
16
2
2
37middot1
0-42
15middot1
0-5
GR
-SO
4 + 4
-NPA
1
1 1
03
25
412
9
7 38
8
4
82middot1
0-44
38middot1
0-5
GR
-SO
4 + 4
-NPA
1
1 1
03
46
224
9
9 21
5
6
37middot1
0-45
79middot1
0-5
GR
-SO
4 + 4
-NPA
1
1 1
03
100
103
13
7
137
196
middot10-4
178
middot10-5
GR
-SO
4 + 4
-NT
2 15
1
31
20
655
5
4 27
0
6
74middot1
0-44
82middot1
0-5
GR
-SO
4 + 4
-NT
2 15
1
31
50
262
9
7 19
4
5
89middot1
0-44
21middot1
0-5
GR
-SO
4 + 4
-NT
4 2
126
0 50
25
2 49
1
982
110
middot10-2
817
middot10-5
GR
-SO
4 + 4
-NT
4 2
630
50
12
6 42
6
852
186
middot10-3
276
middot10-5
GR
-SO
4 + 4
-CN
B 2
2
126
0 30
42
0 29
0
967
925
middot10-3
687
middot10-5
GR
-SO
4 + 4
-CN
B 2
2
630
50
12
6 38
0
760
136
middot10-3
202
middot10-5
GR
-SO
4 + 4
-NPA
2
2 12
60
40
315
371
92
8
5
96middot1
0-34
43middot1
0-5
GR
-SO
4 + 4
-NPA
2
2 6
30
45
140
273
60
7
1
09middot1
0-31
62middot1
0-5
a A
mou
nt o
f NA
C re
duce
d by
GR
-SO
4 at r
eact
ion
term
inat
ion
b F
ract
ion
of in
itial
ly a
dded
NA
C tr
ansf
orm
ed b
y G
R-S
O4 a
t rea
ctio
n te
rmin
atio
n c
Pse
udo
1
orde
r rat
e co
nsta
nts c
alcu
late
d as
initi
al ra
tes
ie m
ax f
irst t
wo
half-
lives
d S
urfa
ce a
rea-
norm
alis
ed p
seud
o 1
ord
er ra
te c
onst
ants
The
are
a of
GR
-SO
4 per
L
su
spen
sion
was
cal
cula
ted
as frac14
middot[Fe
(II)
GR] 0middot
600
gmiddotm
ol-1
middot71
2 m
2 middotg-1
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 79
432 Comparison of rate constants for the different NACs
Even for NACs holding very different one-electron transfer reduction potentials
( ) their reactivities differed only little in Fe(II)-Fe(III) systems such as the
Fe(II)goethite system (slope a = 06 for linear free energy relationship (LFER)
between k
1hE
obs and Hofstetter et al 1999) and the Fe(II)magnetite system
(LFER slope a = 034 Klausen et al 1995) When considering only the for the
reductive transformation reactions of the NACs applied in this study (Table 42)
we expect the surface area-normalised pseudo 1 order rate constants for the
reduction of the NACs to follow the order 4-CNB gt NB gt 4-NT gt 4-NPA Based
on log k
1hE
1hE
obs versus correlations obtained in Fe(II)goethite systems we expect 4-
CNB to react 6 times faster than 4-NPA (Hofstetter et al 1999)
1hE
Table 42 One-electron reduction potentials and relative reactivities in Fe(II)-magnetite and GR-
SO4 systems for the nitro aromatic probe compounds
Compound pKa Eh1
acute (mV) krel (Fe3O4) cd krel (GR-SO4) ce
4-Chlornitrobenzene - -450 a 122 148
Nitrobenzene - -486 a 1 1
4-Nitrotoluene - -500 a 057 176
4-Nitrophenylacetic acid 385 -543 b - 123 a Values from references cited in Hofstetter et al 1999 b Estimated at pH 660 using a LFER (Hofstetter et al 1999 see Supporting Information 71) c Reactivity relative to NB d Values from Klausen et al 1995 e Values from this work
A comparison of the relative rate constants of the NACs obtained for their
transformation by GR-SO4 (this work) and by magnetite (Klausen et al 1995)
shows that they do not differ significantly from each other in any of the mineral
systems (Table 42) When considering charge effects we expect the anionic probe
compounds to react faster with GR-SO4 than the neutral probe compounds
provided that they sorb preferentially within the GR-SO4 interlayers and that Fe(II)
in the interlayers are equally or more reactive than external Fe(II) sites Still the
surface area-normalised kobs values obtained for NB 4-NT 4-CNB and 4-NPA
under various experimental conditions did not differ significantly from each other
80 Chapter 4
(Figure 46 Table 41) The anionic probe compound 4-NPA did not react
significantly faster with GR-SO4 than the neutral probe compounds NB 4-NT and
4-CNB This may indicate that 4-NPA does not significantly interact with reactive
Fe(II) sites in the interlayer Alternatively the negative charge carried by 4-NPA
may be compensating for the lower intrinsic reactivity as compared to the neutral
probe compounds thus explaining the similarity in rate constants for 4-NPA and
the neutral probe compounds Finally other factors than intrinsic reactivity or
charge of the probe compounds such as regeneration of reactive sites or formation
of the magnetite phases may control the overall reactivity of the system
Figure 46 Actual plot of surface area-normalised pseudo 1 order rate constants for the reactions
between neutral and anionic probe compounds and GR-SO4
In heterogeneous reactions mass transfer in bulk solution becomes the rate-
limiting step when the surface reaction is much faster than the diffusion of the
reacting species to the reactive surface In cases where mass transfer controls the
overall rate of reaction the observed pseudo 1 order rate constant kobs ge kLmiddota
where kL is the calculated mass transfer coefficient (mmiddots-1) and a is the ratio of the
external (geometric) specific surface area to volume of solution (m-1) (see
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 81
Supporting Information 72) Mass transfer controlled reactions between GR-SO4
particles and the NACs in bulk solution would explain the similar pseudo 1 order
rate constants obtained for the NACs in this work However when comparing our
estimates of kLmiddota with kobs (see Supporting Information 72) we found that the rates
of mass transfer for all 4 NACs exceed the observed rate constants by at least 3
orders of magnitude at every initial Fe(II)GR concentration Thus the reactions of
the given NACs with GR-SO4 are not likely to be mass transfer limited under the
experimental conditions applied here
Since mass transfer in bulk solution does not control the reaction between GR-SO4
and NACs the overall reaction rate may be surface saturation controlled During
the reductive transformation of NACs not only the parent compound but also
various intermediates forming may compete for the restricted number of reactive
sites present in GR-SO4 This competition may constitute the rate limiting step in
the overall reactivity and may even be enhanced if the number of reactive sites is
depleted during reaction However surface saturation kinetics would not explain
the unexpected similarity of the pseudo 1 order rate constants obtained for the
NACs but it could explain the bent curves observed at low initial Fe(II)GR
concentrations (Figure 45d) The kinetically deviating cases at low [Fe(II)GR]0
were evaluated according to Langmuir-Hinshelwood kinetics (see Supporting
Information 72) Our experimental data did not agree with the Langmuir-
Hinshelwood rate law for any of the NACs (regression results not shown)
Simplifying the rate law by assuming that the aniline product or the
hydroxylamino intermediate or both did not compete for the reactive sites did not
improve the regression Thus the Langmuir-Hinshelwood model cannot explain
the deviations from pseudo 1 order kinetics observed at [Fe(II)GR]0 in our GR-SO4
system and it does not suffice as the correct reaction mechanism nor as the rate-
limiting step
82 Chapter 4
If the adsorption follows a saturation-type sorption isoterm (eg Langmuir) the
sorbate (oxidant) concentration at the surface will vary non-linearily with the total
amount of oxidant added This dependence will have to be taken into account when
establishing rate laws for the heterogeneous reactions and when testing the
hypothesis that the reaction rates depend on the sorbed concentration of the
oxidants However at the high reaction rates observed here we could not quantify
sorption Since the measured initial NAC concentrations corresponded to the
nominal amount of NAC added we assume that transformation and not sorption
was responsible for the consumption of NAC
433 Factors influencing the reaction rate
In general numerous compound- and system-specific factors influence redox
reactions One very important factor is pH which influences the speciation of
dissociable compounds as well as the stability of GR and the formation of other
iron minerals in the system pH has a strong impact on the sorption and therefore
the availability of ionisable oxidants such as carboxylic acids At pH ~ 84 where
our experiments were conducted 4-NPA (pKa = 385) is completely dissociated
Our experiments conducted with GR-SO4 and NB showed that pH was constant
during reaction In addition solution pH has an effect on the surface speciation
From other Fe(II)-Fe(III) systems such as Fe(II) surface species associated with
iron oxides or clay minerals it is well-known that other reactive hydroxylated
Fe(II)-Fe(III)-hydroxo surface complexes can form at higher pH (Charlet et al
1998 Liger et al 1999) Williams amp Scherer (2001) reported a small decrease (5
fold) in the reduction rate of chromate with GR-CO3 when increasing pH from 50
to 90 This decrease may be due to the alternating speciation of the Fe(II) surface
sites on GR-CO3 and of chromate in solution (pKa (H2CrO4) = 08 pKa (HCrO4ndash) =
65) when raising pH (Williams amp Scherer 2001) In contrast other studies have
reported small increases (4 fold) in the reduction rates of nitrate and
trichloroethene with GR-SO4 when increasing pH from 71 to 84 and from 68 to
101 respectively (Koch amp Hansen 1997 Lee amp Batchelor 2002b)
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 83
In this work all experiments were carried out in the presence of 25 mM
Na2SO4(aq) in order to minimize GR-SO4 dissolution and spontaneous
transformation into magnetite Preliminary results from experiments conducted
with NB show that the bulk concentration of Na2SO4 has only a very small impact
on the rate ie increasing the concentration of Na2SO4(aq) in the GR-SO4
suspension from 5 to 25 mM reduced the observed rate constant by a factor of 2
At Na2SO4 concentrations above 25 mM the effect leveled off and therefore
[Na2SO4] = 25 mM was chosen for this work Portions of the same GR-SO4
suspension were used for kinetic experiments over a period of two weeks No
significant aging effects eg rate constants decreasing as a function of GR age
were observed within this time frame
434 Comparison with rate constants obtained for other Fe(II) containing
mineral systems
A recent study compared the reactivity of various Fe(II) containing iron mineral
systems towards organic probe compounds representing different classes of
pollutants (Elsner et al 2004) The reductive transformation of 4-CNB was
investigated for the Fe(III) minerals goethite (α-FeOOH) lepidocrocite (γ-
FeOOH) and hematite (α-Fe2O3) as well as for the Fe(II)-Fe(III) oxide magnetite
(Fe3O4) All experiments were conducted in the presence of 1 mM dissolved Fe(II)
at pH 72 As seen from the surface area-normalised pseudo 1 order rate constants
in Figure 47 the reduction of 4-CNB by the Fe(II)-amended goethite
lepidocrocite and magnetite systems was up to 40 times faster than its reduction by
GR-SO4 The reduction rate obtained for the Fe(II)hematite system was only
slightly higher than the rate for the blank containing no iron mineral but aqueous
Fe(II) solely
84 Chapter 4
Figure 47 Surface area-normalised pseudo 1 order rate constants for the degradation of 4-CNB
by GR-SO4 (open square this work) and various Fe(II) containing mineral systems (solid circles
Elsner et al 2004) Experimental conditions applied by Elsner et al 1 mM aqueous Fe(II) pH
72 25 m2 mineral surface areaL GR-SO4 = green rust sulphate α-FeOOH = goethite Fe3O4 =
magnetite γ-FeOOH = lepidocrocite α-Fe2O3 = hematite
The experiments with GR-SO4 in our study were carried out at pH 84 whereas the
experiments comprising the other systems in Figure 47 were conducted at pH 72
As the reactivity of GR is expected to increase with pH (Lee amp Batchelor 2002b
Koch amp Hansen 1997) the lower of GR-SO4 compared to other Fe(II) systems
cannot be explained by differences in pH values GR-SO4 might just contain fewer
or less reactive surface sites than Fe(II)-amended goethite lepidocrocite and
magnetite suspensions These findings contrast those of other studies which found
higher surface area-normalised pseudo 1 order rate constants for dechlorination
reactions for GR-SO4 than for magnetite (Lee amp Batchelor 2002a amp 2002b Elsner
et al 2004) The different reactivity orders of the Fe(II)-bearing minerals found
for chlorinated aliphatics and nitro aromatics suggest that effects other than pH and
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 85
the intrinsic reduction potentials of the reacting species play a role for the
reactivity of these Fe(II)-bearing minerals
435 Depletion of reactive sites
Assuming that the platy hexagonal GR-SO4 crystals hold an average width of 1 microm
and an average particle thickness of 35 nm (Hansen amp Koch 1998) the ratio of
outer surface area to total surface area AouterAtotal ~ 131 (see Supporting
Information 73) This means that only 3 of the total surface area in GR-SO4 is
available at the external surface Thus as the interlayer sulphate in GR-SO4 is not
readily exchanged with the anionic NAC applied we conclude that 4-NPA and
other NACs only react with the Fe(II) sites at the external GR-SO4 surface In
Table 43 the actual amounts of NAC reduced by GR-SO4 during reaction is
compared with the amount of NAC which theoretically can be reduced by the
initial amount of external reactive Fe(II) sites in GR-SO4 at AouterAtot ~ 131
Table 43 The actual amounts of NAC reduced by GR-SO4 during reaction ∆[NAC]act
compared with the amount of NAC which stoichiometrically should be reduced by the initial
amount of external reactive Fe(II) sites in GR-SO4 ∆[Ar-NO2]theory (calculated as
[Fe(II)GR]0(31middot6) assuming an even distribution of Fe(II) throughout the GR-SO4 structure)
Calculated for [NAC]0 ~ 50 microM
[Fe(II)GR]0 (mM)
∆[Ar-NO2]theory(microM)
∆[4-NT]act (microM)
∆[4-CNB]act (microM)
∆[4-NPA]act (microM)
103 55 174 177 99
63 339 426 380 273
As seen in Table 43 the actual amounts of NAC reduced by GR-SO4 during
reaction are in most cases higher than the amount of NAC which should be
reduced at the given [Fe(II)GR]0 according to reaction stoichiometry This indicates
that new external reactive sites were regenerated eg the Fe(III) phases produced
peel off the GR surface exposing new Fe(II) sites or that outermost internal
86 Chapter 4
reactive sites in close vicinity to the edges are available for reaction as well Lee
and Batchelor (2000b) also found the experimentally observed reduction capacity
of GR-SO4 for chlorinated ethylenes to be 2-3 orders of magnitudes lower than the
estimated reduction capacity including all Fe(II) in GR-SO4
At low initial Fe(II)GR concentrations only a fraction of NAC was reduced within
the reaction time observed (Figure 45c) even though there was stoichiometric
excess of Fe(II)-GR present The fraction of initial Ar-NO2 reduced by GR-SO4 at
reaction termination decreased as [Fe(II)GR]0 decreased (Table 41) and was
accompanied by a change in apparent rate laws with time (compare Figures 45b amp
d) In order to explain these observations we propose that the NACs react only at
external reactive Fe(II) sites and that the regeneration of new external reactive sites
is much slower than the reduction of NAC by GR-SO4 Thus the fast reduction of
NAC taking place at the external reactive sites represents the pseudo 1 order
behaviour whereas depletion of external reactive sites and their slow regeneration
are represented by the second bent part of the (time ln [Ar-NO2]t[Ar-NO2]0)-
curves deviating from pseudo 1 order kinetics Hence at low [Fe(II)GR]0 the
regeneration of reactive sites will eventually control the overall reaction rate
Depletion of available Fe(II) was also observed during the fast reduction of
chromate by GR-CO3 when the initial chromate concentration was increased or
when the GR-CO3 suspension was respiked with chromate repeatedly (Williams amp
Scherer 2001)
436 The role of external and internal reactive sites
It is reasonable to assume that GRs hold adsorption properties similar to other
layered double hydroxides such as hydrotalcites The sorption of 246-
trinitrophenol (TNP) and 245-trichlorophenol (TCP) on chloride and carbonate
intercalated hydrotalcites (HT-Cl = Mg3Al(OH)8ClmiddotyH2O HT-CO3 =
Mg6Al2(OH)16CO3middotyH2O) has been investigated (Hermosin et al 1993 Ulibarri et
al 1995 Ulibarri et al 2001) The authors found that the adsorption of TCP on
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 87
HT-CO3 was very low and that TCP adsorbs only on the external surface sites of
HT-CO3 (Hermosin et al 1993) Furthermore is was reported that the adsorption
of TNP on HT was dramatically affected by the nature of the interlayer anion ie
the adsorption of TNP was considerably higher on HT-Cl than on HT-CO3
(Ulibarri et al 2001) For HT-Cl interlayer anion exchange of chloride with TNP
was detected by XRD analysis and an expansion of the characteristic basal d003
spacing from 79 Ǻ to 132 Ǻ confirmed the presence of TNP in the HT interlayer
(Ulibarri et al 1995) Collating the results reported for HTs with GRs it is not
likely that the divalent SO42- in GR-SO4 is exchanged with the monovalent 4-NPA
Chacirctelet et al (1996) investigated the adsorption of mono- and divalent anions
onin the outer and inner adsorption sites in HT by varying the zetapotential with
pH in the presence of various electrolytes The authors found that SO42- adsorbs on
the external HT surfaces by formation of outer-sphere complexes whereas chloride
hardly adsorbed on HT Moreover it was reported that the adsorption of sulphate
onto HT was not strongly affected by the presence of chloride while sulphate on
the contrary inhibited the adsorption of chloride on HT Studies applying
spectroscopic analyses have investigated the sorption of oxyanions at external and
internal GR-SO4 surfaces (Myneni et al 1997 Randall et al 2001) Selenate was
adsorbed only on the outer GR-SO4 surface when added after GR formation
whereas it was primarily coprecipitated into the interlayer when present during GR
formation Thus for selenate its presence during GR formation is a prerequisite of
its incorporation in the GR interlayer Selenate is readily reduced by GR-SO4 and
the rates of reduction of coprecipitated selenate were very similar to the reduction
rates of selenate adsorbed at the outer GR surface (Myneni et al 1997) This
finding suggests that the outer and inner reactive Fe(II) sites in GR-SO4 hold
similar reactivities
Results based on electron microscopy reported that the reduction of uranyl took
place primarily at the edges of hexagonal GR-SO4 particles (OrsquoLoughlin et al
2003a) In another recent study XRD characterization of the GR-SO4 crystals
88 Chapter 4
during reaction with trichloroacetate (TCA) indicated that TCA did not enter the
GR-SO4 interlayer during reaction (Chapter 5 this work) The average GR-SO4
particle thickness perpendicular to the basal plane was constant during reaction
implying that TCA reacts only at the edges and not at the basal planes Assuming
that the platy hexagonal GR-SO4 crystals hold an average width of 1 microm and an
average particle thickness of 35 nm (Hansen amp Koch 1998) the ratio of edge
surface area to outer surface area is AedgeAouter ~ 130 (see Supporting Information
73) Hence only 3 of the outer surface area in GR-SO4 is available at the edges
Once more the regeneration of new external reactive sites is strongly inferred as
the actual amounts of NAC reduced by GR-SO4 during reaction are much higher
than the amount of NAC which may be reduced by the reactive edge sites present
initially Assuming that the NACs react at the edges only and if employing the
AedgeAouter in the estimation of the rate constants the surface area-normalised
pseudo 1 order rate constants for GR-SO4 would be 30 times higher than the rate
constants depicted in Figure 47 Thus the reactivity of GR-SO4 normalised to its
reactive surface area is higher than the reactivity normalised to its outer surface
area determined by the BET method (N2 adsorption)
The reduction of chromate has been examined in the presence of all the common
GR forms (Bond amp Fendorf 2003 Loyaux-Lawniczak et al 1999 Loyaux-
Lawniczak et al 2000 Williams amp Scherer 2001) The results reported by Bond
amp Fendorf (2003) confirm that not only the surface area of GR but also the
interlayer spacing (interlayer anion size) and interlayer anion charge play an
important role for the reaction rate Hence it follows that coordination (size) and
charge of the oxidant determine its access to the internal sites in GRs
The results obtained for all 4 NACs support what has been reported for nitrate and
TCA At [Fe(II)GR]0 = 2-10 mM and [NO3-]0 = 143 mM pseudo 1 order rate
constants for the reduction of nitrate by GR-SO4 were 158middot10-7 s-1middotm-2middotL (Hansen
et al 2001) This reaction rate increased 40 times by adding barium nitrate instead
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 89
of sodium nitrate thereby precipitating the interlayer sulphate as barium sulphate
and enhancing access to the interlayer Though barium addition changes the GR-
SO4 system dramatically it indicates the importance of interlayer anion exchange
(Hansen amp Koch 1998) The rate constant reported for nitrate (no barium added) is
100-1000 smaller than the rate constants obtained for the NACs in this work
Moreover the reaction kinetics for nitrate did not deviate from pseudo 1 order
kinetics At [Fe(II)GR]0 = 025-104 mM and [TCA]0 = 50 microM-1 mM pseudo 1
order rate constants for the reduction of TCA by GR-CO3 or GR-SO4 were 65middot10-7
s-1middotm-2middotL (Chapter 5 this work) The rate constant for TCA is 10-1000 smaller than
the rate constants for the NACs and the reaction kinetics for TCA did not deviate
from pseudo 1 order kinetics This suggests that the overall reductive
transformation of slowly reacting oxidants such as nitrate and TCA is not
controlled by the rate of regeneration of external Fe(II) reactive sites Altogether
the results reported for selenate chromate and nitrate clearly demonstrate that
these anionic oxidants react primarily with external reactive sites in GR-SO4 Only
under certain conditions ie adding the oxidant prior to GR-SO4 formation or
extracting the interlayer sulphate through precipitation with barium outside the
GR-SO4 particles do the oxidants have access to the interlayer Our findings
suggest that both the neutral and anionic nitro aromatic probe compounds applied
here also react exclusively with the external reactive sites in GR-SO4 Supposedly
the neutral and monovalent charge states of the NACs hinder their access to the
GR-SO4 interlayer A divalent anionic nitro aromatic probe compound might
exchange with the interlayer sulphate more readily and gain access to the inner
Fe(II) reactive sites in GR-SO4 only divalent anionic NACs are not commercially
available
44 Conclusions
This work demonstrates that NACs are completely reduced to their corresponding
anilines by GR-SO4 The surface area-normalised pseudo 1 order rate constants
obtained for the reduction of the neutral and anionic NACs by GR-SO4 under
90 Chapter 4
various experimental conditions did not differ significantly from each other despite
their different charges Neither mass transfer control nor surface saturation kinetics
could account for the similarity of the pseudo 1 order rate constants obtained for
the NACs These observations suggest that the anionic NACs do not have an
enhanced access to inner or outer Fe(II)-GR reactive sites as compared to the
neutral NACs Based on our estimations of the molecular sizes of the NACs we
propose that the charge and not the size of the NACs controls their access to the
internal reactive sites in GRs Hence the reaction between NAC and GR-SO4 takes
place primarily at the external reactive Fe(II) sites This work further demonstrated
that the reduction of the NACs by GR-SO4 only followed pseudo 1 order kinetics
throughout the whole reaction at high initial Fe(II)GR concentrations At low initial
Fe(II)GR concentrations the NACs were not reduced completely within the reaction
time observed though according to reaction stoichiometry the total Fe(II)-GR
present should be sufficient to reduce the whole amount of NAC This means that
at some point during the reaction the external reactive Fe(II) sites were depleted
and the regeneration of new external reactive sites was much slower than the
reduction of the NACs by GR-SO4 The reduction of 4-CNB by GR-SO4 reported
here was 10-100 times slower than its reduction by other Fe(II)-Fe(III) systems
such as goethite lepidocrocite and magnetite suspensions amended with Fe(II)
(Elsner et al 2004)
The results obtained in this work infer that under natural geochemical conditions
where GR-SO4 presumably forms in low concentrations the rate of regeneration of
external Fe(II) reactive sites may control the overall reductive transformation of
fast reacting pollutants by GR-SO4 Thus not only abiotic processes such as
interchanging redox conditions created by water level alterations but also the direct
microbial formation through Fe(III)-reducing bacteria may govern the formation of
GRs and the renewal of external Fe(II) reactive sites in GRs This holds both for
natural systems like iron-rich suboxic soils and sediments as well as engineered
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 91
systems like permeable reactive barriers of zero-valent iron implemented for on-
site remediation of organic and inorganic contaminants
Acknowledgments
We would like to thank Henrik T Andersen for performing the NB kinetic experiments and
Hanne Nancke-Krogh for technical assistance in the laboratory
References
Al-Agha MR Burley SD Curtis CD Esson J (1995) Complex cementation textures and authigenic mineral assemblages in recent concretions from the Lincolnshire Wash (east coast UK) driven by Fe(0) to Fe(II) oxidation Journal of the Geological Society 152 157-171 Bernal JD Dasgupta DR Mackay AL (1959) The oxides and hydroxides of iron and their structural inter-relationships Clay Minerals Bulletin 4 15-30 Bond DL Fendorf S (2003) Kinetics and structural constraints of chromate reduction of green rusts Environmental Science and Technology 37 2750-2757 Brunauer S Emmett PH Teller E (1938) Adsorption of gases in multimolecular layers Journal of American Chemical Society 60 309-319 Charlet L Silvester E Liger E (1998) N-compound reduction and actinide immobilisation in surficial fluids by Fe(II) The surface FeIIIFeIIOH0 species as major reductant Chemical Geology 151 85-93 Chacirctelet L Bottero JY Yvon J Bouchelaghem A (1996) Competition between monovalent and divalent anions for calcined and uncalcined hydrotalcite anion exchange and adsorption sites Colloids and Surfaces A Physicochemical and Engineering Aspects 111 167-175 Chaudhuri SK Lack JG Coates JD (2001) Biogenic magnetite formation through anaerobic biooxidation of Fe(II) Applied and Environmental Microbiology 67 2844-2848 Cui D Spahiu K (2002) The reduction of U(VI) on corroded iron under anoxic conditions Radiochemica Acta 90 623-628 Elsner M Haderlein SB Schwarzenbach RP (2004) Reactivity of Fe(II)-bearing minerals towards reductive transformation of organic contaminants Environmental Science and Technology 38 799-807 Erbs M Hansen HCB Olsen CE (1999) Reductive dechlorination of carbon tetrachloride using iron(II)iron(III)-hydroxide-sulphate (green rust) Environmental Science and Technology 33 307-311 Fadrus H Maly J (1975) Suppression of iron(III) interference in the determination of iron(II) in water by the 110-phenanthroline method The Analyst 100 549-554 Fredrickson JK Zachara JM Kennedy DW Dong H Onstott TC Hinman NW Li S
92 Chapter 4 (1998) Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium Geochimica et Cosmochimica Acta 62 3239-3257 Geacutenin JMR Bourrieacute G Trolard F Abdelmoula M Jaffrezic A Refait Ph Maitre V Humbert B Herbillon A (1998) Thermodynamic equilibria in aqueous suspensions of synthetic and natural Fe(II)-Fe(III) green rusts Occurrences of the mineral in hydromorphic soils Environmental Science and Technology 32 1058-1068
Geacutenin JMR Olowe AA Benbouzid-Rollet ND Prieur D Confente M Resiak B (1991) The simultaneous presence of green rust 2 and sulfate reducing bacteria in the corrosion of steel sheet piles in a harbour area Hyperfine Interactions 69 875-878 Glasauer S Weidler PG Langley S Beveridge TJ (2003) Controls on Fe reduction and mineral formation by a subsurface bacterium Geochimica et Cosmochimica Acta 67 1277- 1288 Hansen HCB (1989) Composition stabilization and light absorption of Fe(II)Fe(III) hydroxycarbonate (green rust) Clay Minerals 24 663-669 Hansen HCB Borggaard OK Soslashrensen J (1994) Evaluation of the free energy of formation of iron(II)iron(III)-hydroxidesulphate (Green Rust) and its reduction of nitrite Geochimica et Cosmochimica Acta 58 2599-2608 Hansen HCB Guldberg S Erbs M Koch CB (2001) Kinetics of nitrate reduction by green rusts ndash effects of interlayer anion and Fe(II)Fe(III) ratio Applied Clay Science 18 81-91 Hansen HCB Koch CB (1998) Reduction of nitrate to ammonium by sulphate green rust Activation energy and interlayer reaction mechanism Clay Minerals 33 87-101 Hansen HCB Koch CB Nancke-Krogh H Borggaard OK Soerensen J (1996) Abiotic nitrate reduction to ammonium Key role of green rust Environmental Science and Technology 30 2053-2056 Hansen HCB Poulsen IF (1999) Interaction of synthetic sulphate green rust with phosphate and the crystallization of vivianite Clays and Clay Minerals 47 312-318 Hansen HCB Taylor RM (1991) The use of glycerol intercalates in the exchange of CO3
2- with SO4
2- NO3- or Cl- in pyroaurite-type compounds Clay Minerals 26 311-327
Heasman DM Sherman DM Ragnarsdottir KV (2003) The reduction of aqueous Au3+ by sulfide minerals and green rust phases American Mineralogist 88 725-738 Hermosin MC Pavlovic I Ulibarri MA Cornejo J (1993) Trichlorophenol adsorption on layered double hydroxide a potential sorbent Journal of Environmental Science and Health A28 1875-1888 Hofstetter TB Heijmann CG Haderlein SB Holliger C Schwarzenbach RP (1999) Complete reduction of TNT and other (poly)nitroaromatic compounds under iron-reducing subsurface conditions Environmental Science and Technology 33 1479-1487 Hofstetter TB Schwarzenbach RP Haderlein SB (2003) Reactivity of Fe(II) species associated with clay minerals Environmental Science and Technology 37 519-528
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 93
Klausen J Troumlber SP Haderlein SB Schwarzenbach RP (1995) Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions Environmental Science and Technology 29 2396-2404 Koch CB Hansen HCB (1997) Reduction of nitrate to ammonium by sulphate green rust Advances in GeoEcology 30 373-393 Kukkadapu RK Zachara JM Smith SC Fredrickson JK Liu C (2001) Dissimilatory bacterial reduction of Al-substituted goethite in subsurface sediments Geochimica et Cosmochimica Acta 65 2913-2924 Lee W Batchelor B (2002a) Abiotic reductive dechlorination of chlorinated ethylenes by iron- bearing soil minerals 1 Pyrite and magnetite Environmental Science and Technology 36 5147- 5154 Lee W Batchelor B (2002b) Abiotic reductive dechlorination of chlorinated ethylenes by iron-bearing soil minerals 2 Green rust Environmental Science and Technology 36 5348- 5354 Liger E Charlet L Van Cappellen P (1999) Surface catalysis of uranium (VI) reduction by iron(II) Geochimica et Cosmochimica Acta 63 2939-2955 Loyaux-Lawniczak S Refait Ph Ehrhardt J Lecomte P Geacutenin JMR (2000) Trapping of Cr by formation of ferrihydrite during the reduction of chromate ions by Fe(II)-Fe(III) hydroxysalt green rusts Environmental Science and Technology 34 438-443 Loyaux-Lawniczak S Refait Ph Lecomte P Ehrhardt J Geacutenin JMR (1999) The reduction of chromate ions by Fe(II) layered hydroxides Hydrology and Earth System Sciences 3 593-599 Mendiboure A Schoumlllhorn A (1986) Formation and anion exchange reactions of layered transition metal hydroxides [Ni1-xMx](OH)2(CO3)x2(H2O)z (M = Fe Co) Revue de Chimie Mineacuterale 23 819-827 Miyata S (1983) Anion-exchange properties of hydrotalcite-like compounds Clays and Clay Minerals 31 305-311 Myneni SCB Tokunaga TK Brown Jr GE (1997) Abiotic selenium redox transformations in the presence of Fe(IIIII) oxides Science 278 1106-1109 Nielsen A (1976) Hvid groslashn og sort rust Beskrivelse af en korrosionsskade paring et svoslashmmebassin Nordisk Betong 2 21-24 OLoughlin EJ Burris DR (2004) Reduction of halogenated ethanes by green rust Environmental Toxicology and Chemistry 23 41-48 OLoughlin EJ Kelly SD Cook RE Csencsits R Kemner KM (2003a) Reduction of uranium(VI) by mixed iron(II)iron(III) hydroxide (green rust) Formation of UO2 nanoparticles Environmental Science and Technology 37 721-727
94 Chapter 4 OLoughlin EJ Kelly SD Kemner KM Csencsits R Cook RE (2003b) Reduction of AgI AuIII CuII and HgII by FeIIFeIII hydroxysulfate green rust Chemosphere 53 437-446 Ona-Nguema G Abdelmoula M Jorand F Benali O Gehin A Block J-C Geacutenin JMR (2002) Iron (IIIII) hydroxycarbonate green rust formation and stabilization from lepidocrocite bioreduction Environmental Science and Technology 36 16-20 Parmar N Gorby YA Beveridge TJ Ferris FG (2001) Formation of green rust and immobilization of nickel in response to bacterial reduction of hydrous ferric oxide Geomicrobiology Journal 18 375-385 Pepper SE Bunker DJ Bryan ND Livens FR Charnock JM Pattrick RAD Collison D (2003) Treatment of radioactive wastes An X-ray absorption spectroscopy study of the reaction of technetium with green rust Journal of Colloid and Interface Science 268 408- 412 Randall SR Sherman DM Ragnarsdottir KV (2001) Sorption of As(V) on green rust (Fe4(II)Fe2(III)(OH)12SO4
3H2O) and lepidocrocite (γ-FeOOH) Surface complexes from EXAFS spectroscopy Geochimica et Cosmochimica Acta 65 1015-1023 Roh Y Lee SY Elless MP (2000) Characterization of corrosion products in the permeable reactive barriers Environmental Geology 40 184-194 Schultz CA Grundl TJ (2000) pH dependence on reduction rate of 4-Cl-nitrobenzene by Fe(II)montmorillonite systems Environmental Science and Technology 34 3641-3648 Simon L Francois M Refait Ph Renaudin G Lelaurain M Geacutenin JMR (2003) Structure of the Fe(II-III)-layered double hydroxysulphate green rust two from Rietveld analysis Solid State Sciences 5 327-334 Tamaura Y (1986) Ni(II)-bearing green rust II and its spontaneous transformation into Ni(II)- bearing ferrites Bulletin of the Chemical Society of Japan 59 1829-1832 Tamaura Y (1985) ZnII-bearing green rust II and its spontaneous transformation into ZnII- bearing ferrite in aqueous solution Bulletin of the Chemical Society of Japan 58 2951-2954 Trolard F Abdelmoula M Bourrieacute G Humbert B Geacutenin JMR (1996) Mise en eacutevidence dun constituant de type rouilles vertes dans les sols hydromorphes Proposition de lexistence dun nouveau mineacuteral la fougeacuterite Geacuteosciences de surface Comptes Rendus de LrsquoAcademie des Sciences 323 1015-1022 Tuovinen OH Button KS Vuorinen A Carlson L Mair DM Yut LA (1980) Bacterial chemical and mineralogical characteristics of tubercles in distribution pipelines Journal of the American Water Works Association 72 626-635 Ulibarri MA Pavlovic I Barriga C Hermosin MC Cornejo J (2001) Adsorption of anionic species on hydrotalcite-like compounds effect of interlayer anion and crystallinity Applied Clay Science 18 17-27 Ulibarri MA Pavlovic I Hermosin MC Cornejo J (1995) Hydrotalcite-like compounds as potential sorbents of phenols from water Applied Clay Science 10 131-145
Reduction of Nitroaromatic Probe Compounds by Sulphate Green Rust 95
Williams AGB Scherer MM (2001) Kinetics of chromate reduction by carbonate green rust Environmental Science and Technology 35 3488-3494
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 97
5 Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems
Abstract
Trichloroacetate (TCA) is a widespread environmental contaminant with proven
phytotoxicity and suspected human carcinogenicity In order to assess the global
cycling of TCA and to predict its fate in subsurface environments information
regarding the reactivity and product distribution of TCA degradation is needed
Due to the high oxidation state of TCA conditions for oxidative transformation
pathways in soils and groundwater are unfavorable However in suboxic soils and
sediments Fe(II)-bearing minerals are potential reactants for reductive
dehalogenation reactions of TCA as has been demonstrated for other halogenated
contaminants We examined the reactivity of various Fe(II)-Fe(III) mineral
systems towards TCA and dichloroacetate (DCA) its expected transformation
product in laboratory batch experiments imitating natural conditions ie low
initial Fe(II) Fe(III) and TCADCA concentrations and no artificial buffer The
Fe(II)-Fe(III)-systems investigated included sulfate green rust (GR-SO4) carbonate
green rust (GR-CO3) magnetite Fe(II)goethite and Fe(II)lepidocrocite
Trichloroacetate was readily reduced to DCA by all Fe(II)-bearing minerals The
reactions generally followed pseudo 1 order kinetics with respect to TCA The
surface area-normalised pseudo 1 order rate constants obtained (035ndash76middot10-5 min-
1middotm-2middotL at [Fe(II)]0 = 020ndash122 mM [TCA]0 = 15ndash1000 microM and pH 70ndash87)
showed no striking differences regarding product distribution and surface area-
normalised reaction rate constants between the Fe(II)-Fe(III)-systems The
stoichiometrically formed DCA was not further reduced to monochloroacetate
(MCA) or acetate in any of the systems within the time frame in our experiments
To our knowledge this is the first published report on abiotic transformation of
TCA by Fe(II)-bearing minerals Our results imply that processes involving
reactive Fe(II)-bearing minerals may play a significant role in controlling the fate
98 Chapter 5
of TCA in natural subsurface environments and that DCA found in the subsurface
may be formed by such processes
51 Introduction
Trichloroacetic acid (TCA) has been applied as a herbicide for many years until its
use was banned in the late 1980acutes (Berg et al 2000) Today TCA is mainly used
as an etching agent in the metal industry as a swelling solvent in the plastic
production and as a bleaching agent in the paper and pulp manufacture (Muumlller et
al 1996) Other anthropogenic sources include formation of TCA as a result of the
chlorine based disinfecting process used in drinking water treatment and the
atmospheric photooxidation of chlorinated solvents including tetrachloroethene
and 111-trichloroethane (McCulloch 2002) Only very little information is
available on the TCA production volumes and even less is known about the
amount of TCA released into the environment as a result of its industrial
applications Due to its low volatility and high aqueous solubility TCA is easily
washed out of the atmosphere into the aquatic and terrestrial biospheres As TCA
is found in almost every ecosystem around the globe including non-urban and
non-industrial sites the relative contributions from anthropogenic and natural
sources are currently being debated (McCulloch 2002 Ahlers et al 2003)
Trichloroacetic acid is omnipresent in soils and the concentrations reported are
very variable ranging from lt005 microgkg to 380 microgkg (Euro Chlor 2001
McCulloch 2002 Ahlers et al 2003) Both abiotic and enzymatically catalyzed
formation of TCA from humic acids have been demonstrated in laboratory studies
(Haiber et al 1996 Hoekstra et al 1999b Fahimi et al 2003) Furthermore the
in situ natural formation of TCA from anthropogenic or natural tetrachloroethene
or 111-trichloroethane in biota has been suggested (Hoekstra et al 1999a
McCulloch 2002) Such natural sources may explain part of the TCA
concentrations found in soils but their environmental significance is still unknown
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 99
On account of its phytotoxicity suspected human carcinogenicity and widespread
occurrence TCA is of considerable environmental concern especially in the
terrestrial compartment The TCA concentrations found in soil air and water in
pre-industrial times were far below the present ones (Jordan amp Frank 1999 Ahlers
et al 2003) Based on the current TCA concentrations detected in soils the
European Commission proposed risk reduction measures concerning
tetrachloroethene - a precursor of TCA - to be taken immediately (Ahlers et al
2003 and references therein) Occurrences of monochloroacetic acid (MCA) and
dichloroacetic acid (DCA) reported include surface waters marine waters
precipitation ice (glaciers) and air (Reimann et al 1996 Berg et al 2000 Scott et
al 2000 Scott et al 2002) Based on the concentrations reported for the aquatic
environments it is reasonable to assume that MCA and DCA are omnipresent in
soils as well Sources of MCA and DCA include production in the chemical
industry photooxidation of chlorinated aliphatics in the atmosphere and reductive
transformation of TCA (Reimann et al 1996 Ahlers et al 2003 and references
therein) MCA and DCA are also toxins and suspected human carcinogens (Kuumlhn
amp Pattard 1990) hence not only TCA but also its daughter compounds are
pollutants of environmental concern
In subsurface environments TCA may be removed by sorption seepage chemical
transformation microbial degradation and plant uptake followed by metabolic
degradation andor physical removal at harvest (Foy 1975) There is little or no
evidence of abiotic transformations of TCA in the literature Only one recent study
demonstrated the reductive dechlorination of TCA to MCA by Fe(0) (Hozalski et
al 2001) It has been reported that the degradation of TCA in soil is slow and
mainly mediated by microorganisms but only little is known about the bacteria and
processes involved (Lignell et al 1984) Biodegradation of TCA has been found at
both oxic and anoxic conditions An aerobic microorganism capable of growing on
TCA as the sole carbon and energy source has been characterised (Yu amp Welander
1995) Moreover anaerobic bacteria coupling co-metabolic growth to reductive
100 Chapter 5
dechlorination of TCA have been isolated (Weightman et al 1992 De Wever et
al 2000) However more information regarding the abiotic and biotic
transformation of TCA is needed in order to assess the fate and transport of TCA in
natural subsurface environments
It is well-known that Fe(II) present in minerals or associated with mineral surfaces
is a much stronger reductant than Fe(II) in solution The enhanced reactivity of a
structural or surface-bound Fe(II) center can be rationalized by the increased
electron density donated by hydroxyl ligands and a stabilization of the Fe(III)
oxidation state by the hydroxyl ligands (Luther 1990) Fe(II)-bearing minerals
including layered Fe(II)-Fe(III) hydroxides (green rusts) magnetite (Fe3O4)
siderite (FeCO3) Fe(II) sulfides as well as Fe(II)-carrying Fe(III) oxides and clay
minerals have also been shown to reduce a range of organic and inorganic
contaminants such as nitro aromatic compounds chlorinated aliphatics chromate
uranyl pertechnetate nitrate monochloramine and carbamate pesticides (Chapter
4 this work Klausen et al 1995 Cui amp Eriksen 1996 Erbs et al 1999 Liger et
al 1999 Loyaux-Lawniczak et al 1999 Amonette et al 2000 Hansen et al
2001 Pecher et al 2002 Vikesland amp Valentine 2002 Hofstetter et al 2003
OrsquoLoughlin and Burris 2003 OrsquoLoughlin et al 2003a amp 2003b Strathmann amp
Stone 2003 Elsner et al 2004a) Laboratory and field studies showed that even in
geochemically highly heterogeneous anoxic aquifer sediments Fe(II) adsorbed to
Fe(III) (hydr)oxide surfaces was the dominant reductant of nitroaromatic and
halogenated contaminants (Ruumlgge et al 1998 Hofstetter et al 1999 Kenneke amp
Weber 2003) Only little is known about the nature of the Fe(II) species associated
with Fe(III) oxide surfaces but reactive hydroxylated Fe(II)-Fe(III)-hydroxo
surface complexes associated with hematite and magnetite above pH 65 have been
proposed (Charlet et al 1998aampb Liger et al 1999) Due to the presence of
structural Fe(II) within the mineral lattice the reactivity of Fe(II) associated with
mixed valent Fe(II)-Fe(III) minerals such as green rusts magnetite and reduced
ferruginous clay minerals may hold another reactivity than Fe(II) associated with
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 101
pure Fe(III) oxides However Fe(II) adsorbed on Fe(III) oxides such as goethite
hematite and lepidocrocite may also hold different reactivities as the Fe(III) oxides
contain different crystal and surface structures
Since chlorinated ethanes and ethenes such as hexachloroethane 111-
trichloroethane tetrachloroethene and trichloroethene are susceptible to chemical
reduction by a range of Fe(II)-bearing minerals including magnetite GR-SO4
Fe(II) sulfides and Fe(II)-carrying Fe(III) oxides (Butler amp Hayes 1998 amp 1999
Hwang amp Batchelor 2000 Gander et al 2002 Lee amp Batchelor 2002aampb Elsner
et al 2004a) we hypothesized that TCA may be transformed by Fe(II)-bearing
minerals as well The main goals of this work were to study such reactions and
establish product distribution and surface area-normalised reaction rates for the
reductive dechlorination of TCA by Fe(II)-Fe(III) mineral systems common in
nature
5 2 Materials and methods
No synthetic buffers were applied and iron concentrations were kept low The
calcareous systems were pH-controlled at 76 through a natural buffer system
(CaCO3(s) + 995 N205 CO2(g)) All handling and sampling of solutions and
suspensions were carried out under strict anoxic conditions Goethite (acicular
particles with size 01 times 06 microm specific surface area 16 m2g) and lepidocrocite
(acicular particles with size 005 times 03 microm specific surface area 18 m2g) were
purchased as fine powders from Bayer (Bayferrox 910 and 943) Calcite (grain size
170-350 microm Pluumlss-Staufer AG) was used as a buffer or as a Fe(III)-oxide-bearing
mineral In order to simulate natural conditions the iron minerals were applied as
coatings on calcite particles (model system for calcareous soils) in some
experiments Trichloroacetic acid dichloroacetic acid and monochloroacetic acid
were pa quality (Fluka)
102 Chapter 5
521 Synthesis of GRs and magnetite
GR-CO3 was synthesized by controlled air oxidation of an FeCl2 solution at a
constant pH of 700 (titrated with 1 M Na2CO3) according to the procedure given
by Hansen amp Koch (1997) 05 M aqueous stock solutions of FeCl2 were prepared
in 100 mL glass flasks by reacting 65 mmol of iron powder (particle size 10 microm
Merck) with 100 mL deoxygenated 10 M HCl The solutions were magnetically
stirred and heated (~80degC) during reaction until the H2(g) production had ceased (ge
2 hours) The FeCl2 solutions were stored in the dark under a small Ar
overpressure at 5degC The GR-CO3 suspensions were washed with deoxygenated
deionised water (DIW) separated on a folding filter (medium filtration rate cotton
linterhigh alpha pulp Schleicher amp Schuell) and redispersed in deoxygenated
DIW Washing separation and redispersion of the GR-CO3 suspension were
conducted in an anoxic glove box (92 N28 H2 Coy Laboratory Products Inc)
All suspensions and solutions were deoxygenated by Ar-purging (999998 Ar
Carbagas) Magnetite was synthesized by further aerial oxidation of GR-CO3 at pH
700 until consumption of 1 M Na2CO3 ceased GR-SO4 was synthesized by
controlled air oxidation of an FeSO4 solution at a constant pH of 70 according to
the procedure given by Koch amp Hansen (1997) The GR-SO4 suspension was
washed with deoxygenated DIW separated on a glass filter funnel (pore size 4
Duran) and redispersed in deoxygenated DIW Washing separation and
redispersion of the GR-SO4 suspension were conducted in an anoxic glove bag
(999995 Ar Aldrich)
522 Preparation of iron oxide coatings
Two grams of goethite (goe) or lepidocrocite (lep) and 100 g calcite were
combined with 200 mL DIW in a 500 mL polyethylene flask The suspension was
gently agitated on a reciprocating shaker for 24 h and left to stand for another 24 h
Excess Fe(III) oxides and salts were removed from the coated material by repeated
decantation and washing with DIW in polyethylene flasks until clear runoff
Finally the coatings were collected on folding filters and air dried The amount of
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 103
goethite and lepidocrocite coated onto calcite after washing and drying was
quantified to 10-11 mg Fe(III)g calcite
523 Mineral characterisation
The identity and purity of the GR-CO3 GR-SO4 and magnetite suspensions were
examined by means of X-ray diffraction (XRD) The XRD analyses were
performed on a Scintag XDS 2000 using Co Kα radiation (45 kV 40 mA) or a
Siemens D5000 XRD applying Co Kα radiation (40 kV 40 mA) Glycerol smears
made according to Hansen (1989) were scanned between 6 and 80 deg2θ with a scan
speed of 1 deg2θmin The specific surface area (SSA) of calcite was determined by
the BET multi-point method using N2 adsorption (Brunauer et al 1938) Powder
samples were filled into sample burettes in the glove box and the generously
lubricated stopcocks closed Samples and burettes were evacuated prior to
connecting them to the BET-instrument (Sorptomatic 1990 Fisons)
524 Kinetic experiments
All reactions were carried out in 25-100 mL serum vials sealed with stoppers
(Viton or Teflon coated rubber) and aluminum crimp caps Kinetic experiments
were conducted with GR-SO4 GR-CO3 magnetite Fe(II)goethite and
Fe(II)lepidocrocite at room temperature In most cases pH was controlled through
the carbonate-bicarbonate buffer system by adding calcite to suspensions
containing the iron minerals solely or by adding the iron minerals as coatings on
calcite Furthermore the calcite containing suspensions were deoxygenated with
05 CO2995 N2(g) thereby attaining an initial pH of 76-77 The GR-CO3 and
magnetite suspensions were deoxygenated with 100 N2(g) and no additional pH
buffer was added The goethite and lepidocrocite suspensions were amended with
300-1000 microM FeCl2(aq) and equilibrated gt 20 h prior to TCADCA addition See
Table 51 for more details on the experimental conditions To start the reaction 50
microM - 1 mM TCA or DCA was added to the mineral suspensions from aqueous
anoxic stock solutions The reaction vials were agitated gently on a roller apparatus
104 Chapter 5
or a shaking table (35 rpm) in order to minimize abrasion of the iron oxide mineral
coatings At appropriate time intervals suspension samples were withdrawn using
Ar(g)- 100 N2(g)- or 995 N205 CO2(g)-flushed sterile disposable syringes
and hypodermic needles The suspension samples were filtered (02 microm Teflon)
and collected for quantification of chloride and the chlorinated acetic acids The
samples were stored at -20degC and analysed without further treatment
525 Analytical methods
Total and aqueous Fe(II) were determined using a modified phenanthroline method
(Fadrus and Maly 1975) For determining [Fe(II)aq] and [Fe(II)total] 1 mL filtered
(02 microm Teflon) and 1 mL unfiltered mineral suspension were added to 18 mL 01
M HCl respectively and allowed to dissolve for 30 min From these acid digests
01 mL was added to 05 mL Fe(II)-phenanthroline-buffer-reagent and 19 mL
DIW added up Estimates of the structural or adsorbed Fe(II) in the Fe(II)-Fe(III)
mineral systems were estimated as the difference [Fe(II)solid] = [Fe(II)total] -
[Fe(II)aq] The total amount of Fe(III) coated on calcite was determined by atomic
absorption spectroscopy following dissolution in 6 M HCl(aq) for 24 h At low
initial TCA concentrations (le 50 microM) the chlorinated acetic acids were quantified
by means of a modified ion interaction (or paired-ion) chromatographic method
(Sarzanini et al 1999) Separation was performed on a LiChrospher 100 RP-18 (5
microm 125 times 4 mm ID) reversed-phase column coupled with a LiChroCART 100 RP-
18 (4 times 4 mm ID) precolumn Analytical conditions were isocratic and the eluent
consisted of 50 aqueous solution of 35 mM cetyltrimethylammonium chloride
(pH 50) and 50 CH3CN The injection volume was 20 microL and the flow-rate 10
mLmin HPLC analyses of the chloroacetates were performed using a Gynkotek
Pump M480 Gynkotek Gina 50 auto sampler and a diode array UV detector (340s
Gynkotek) UV-VIS detection was carried out at 200 nm At higher initial TCA
concentrations the chlorinated acetic acids were quantified by a modified HPLC
method (Husain et al 1992) Separation was performed on a ChromSphere C-18
(10 microm 250 times 46 mm ID) reversed-phase column Analytical conditions were
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 105
isocratic and the eluent consisted of 015 M (NH4)2SO4(aq) pH 55 The injection
volume was 20 microL and the flow-rate 10 mLmin HPLC analyses were performed
using a Series 10 Liquid Chromatographic Pump (Perkin-Elmer) and a SPD-10 A
VP UV-VIS detector (Shimadzu) UV-VIS detection was carried out at 210 nm
Chloride was determined in the GR-SO4 kinetic experiments using a flow injection
system with spectrophotometric detection (Cheregi amp Danet 1997)
53 Results and discussion
531 Product formation and reaction kinetics
Trichloroacetate was readily reduced to DCA by all the Fe(II)-bearing minerals
examined Only DCA was detected within the reaction time in all the Fe(II)-Fe(III)
mineral systems Experiments conducted with the various Fe(II)-Fe(III) mineral
systems and DCA confirmed that no significant reduction of DCA took place (data
not shown) Hence it is reasonable to assume that the further hydrogenolysis of
DCA to MCA is too slow to be detected within the experimental time frame here
The mass balance of TCA and DCA was almost complete in all suspensions ruling
out any alternative reaction pathways to reductive dechlorination Decarboxylation
of TCA producing chloroform and carbon dioxide requires high temperatures and
is therefore assumed not to take place at the experimental conditions applied here
(Atkins et al 1984) Based on these results we propose that the reductive
dechlorination of TCA by Fe(II)-bearing minerals proceeds via hydrogenolysis
(replacement of halogen by hydrogen) as reported for the transformation of TCA
by zero-valent iron (Hozalski et al 2001) Thus in order to reduce TCA to DCA
2 electrons corresponding to 2 Fe(II) are needed (Figure 51)
106 Chapter 5
Cl3CC
O
O- Cl2HCC
O
O-
2e- H+ Cl-
TCA DCA Figure 51 Proposed reductive transformation pathway of TCA
In the Fe(II)goe and Fe(II)lep systems we detected no TCA transformation in the
absence of either aqueous Fe(II) or pure or calcite-associated goethite and
lepidocrocite These results strongly indicate that reactive Fe(II) species associated
with the goethite and lepidocrocite surfaces are the reductants for TCA in these
systems The Fe(III) phases forming in the mineral suspensions were not
characterised and therefore the reaction stoichiometry cannot be assessed
At initial Fe(II) concentrations in large excess of initial TCA concentration we
found a pseudo 1 order rate law for the degradation of TCA by Fe(II)
[ ] [ ] [ b a TCAFe(II) TCA
sdotsdot=minus kdt
d ]
where a = 1 b = 1 and the observed pseudo 1 order rate constant kobs = k middot [Fe(II)]
At all [Fe(II)]0[TCA]0 ratios studied (6-738) TCA was transformed almost
quantitatively into DCA and the reaction kinetics followed pseudo 1 order kinetics
with respect to TCA (Figure 52) The observed pseudo 1 order rate constants for
the transformation of TCA by the various Fe(II)-Fe(III) mineral systems were
calculated as initial rates (ie max first two half-lives) from linear fits of (time ln
[TCA]t[TCA]0)-plots (Table 51) The amount of chloride produced during
reaction with GR-SO4 was always equivalent to the amount of TCA transformed
into DCA (Figure 52c) This also indicates that no significant further reduction of
DCA took place in GR-SO4 suspensions
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 107
Figure 52 Time course of TCA consumption and DCA and chloride production for a)
Fe(II)Goe ([Fe(II)tot]0 = 095 mM) b) Fe(II)Lep ([Fe(II)tot]0 = 091 mM) c) GR-SO4
([Fe(II)GR]0 = 962 mM) d) GR-CO3 ([Fe(II)tot]0 = 633 mM) and e) Magnetite ([Fe(II)tot]0 = 350
mM) Solid lines represent 1 order kinetic fits whereas symbols and dotted lines represent actual
data = TCA = DCA = Clndash
T
able
51
Exp
erim
enta
l con
ditio
ns a
nd p
seud
o 1
ord
er ra
te c
onst
ants
for t
he re
duct
ive
trans
form
atio
n of
TC
A b
y va
rious
Fe(
II)-
Fe(I
II) c
onta
inin
g m
iner
al sy
stem
s
Syst
em
Susp
ensi
on a
ge
(d)
[Fe(
II)] s
olid
a (m
M)
[Fe(
II)] a
q b
(mM
) [T
CA
] 0 (micro
M)
pHin
itcpH
endd
k obs
e (min
-1)
Surf
ace
area
(m
2 L)
k obs
f
(min
-1m
-2middotL
)
Fe(I
I)aq
1
0
030
434
nd
76
gn
dn
dn
d
Fe(I
I)G
oe
1
002
024
429
nd
78
g1
021
0-47
1 i
143
10-5
Fe(I
I)G
oe
1
013
094
543
77
70
225
10-4
71
i3
161
0-5
Fe(I
I)G
oe
coat
ing
1
023
40
066
484
765
80
g6
401
0-454
0 j
119
10-5
Fe(I
I)G
oe
coat
ing
1
0
150
8048
6n
d7
6 g12
43
10-4
540
j2
301
0-5
Fe(I
I)L
ep
1
0
020
2315
7n
d7
8 g0
751
0-48
0 i
094
10-5
Fe(I
I)L
ep
coat
ing
1
016
30
137
470
765
80
g2
821
0-454
0 j
052
10-5
Fe(I
I)L
ep
coat
ing
1
0
100
8141
7n
d7
7 g8
311
0-454
0 j
154
10-5
Fe3O
41
3
380
1251
38
107
8g
830
10-4
16
k5
311
0-4
Fe3O
477
112
56
556
70
70
153
10-4
52
k2
951
0-5
GR
-CO
31
5
940
3950
37
658
4g
761
10-4
419
l1
821
0-5
GR
-CO
32
7
60
147
88
568
2940
81
0-453
6 l
761
10-5
GR
-CO
332
73
003
563
85
80
490
10-4
515
l0
951
0-5
GR
-CO
314
2
3
530
005
629
nd
87 g
513
10-4
249
l2
061
0-5
GR
-SO
41
5
17-1
217
086
-13
910
5n
dn
d3
601
0-492
6 m
039
10-5
GR
-SO
41
6
22-1
027
093
-14
527
0n
dn
d3
761
0-488
1 m
043
10-5
GR
-SO
41
7
05-1
014
077
-17
950
0n
dn
d3
741
0-4 9
18
m0
411
0-5
GR
-SO
41
5
17-1
051
060
-16
510
00n
dn
d2
891
0-483
7 m
035
10-5
n
d =
not
det
ecte
d a
Ini
tial s
truct
ural
or a
dsor
bed
Fe(I
I) e
stim
ated
as [
Fe(I
I)to
tal]
ndash [F
e(II
) aq]
b In
itial
dis
solv
ed F
e(II
) mea
sure
d c
Sus
pens
ion
pH p
rior t
o TC
A a
dditi
on d
Sus
pens
ion
pH a
t rea
ctio
n
te
rmin
atio
n e
Pse
udo
1 o
rder
rate
con
stan
ts fo
r the
con
sum
ptio
n of
TC
A c
alcu
late
d fr
om in
itial
rate
s (m
ax f
irst t
wo
half-
lives
) f
Surf
ace
area
-nor
mal
ised
pse
udo
1 o
rder
rate
con
stan
ts g
pH
con
trol
th
roug
h pu
re C
aCO
3 and
05
C
O2(g
) h
pH
con
trol t
hrou
gh F
e(II
I) o
xide
-coa
ted
calc
ite a
nd 0
5
CO
2(g)
i Es
timat
ed u
sing
the
SSA
of t
he F
e(II
I) o
xide
app
lied
j E
stim
ated
usi
ng th
e SS
A o
f cal
cite
~1
m2 g
k E
stim
ated
ass
umin
g SS
A =
4 m
2 g (S
chw
ertm
ann
amp C
orne
ll 1
991)
frac12middot[
Fe(I
I) sol
id] 0middot
232
gmiddotm
ol-1
middot4 m
2 middotg-1
l E
stim
ated
ass
umin
g SS
A =
47
m2 g
(Will
iam
s amp S
cher
er 2
001)
frac14middot[F
e(II
) GR] 0middot
600
gmiddotm
ol-1
middot47
m2 middotg
-1 m
Est
imat
ed a
s in l
but u
sing
SSA
= 7
12
m2 middotg
-1 (C
hapt
er 4
thi
s wor
k)
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 109
532 Comparing rate constants obtained for the various Fe(II)-Fe(III) mineral
systems
Data for the systems containing iron oxide coated calcite were very similar to the
data obtained for the pure iron oxides (not shown in Figure 53) Since no SSA was
determined for magnetite in this study a SSA of 4 m2g was assumed
(Schwertmann amp Cornell 1991) However it should be noted that the magnetite
synthesized by Schwertmann and Cornell (1991) was prepared differently (ie
oxidation of Fe(II) by nitrate in a heated alkaline solution) from the magnetite
applied in this study The surface area-normalised pseudo 1 order kobs values
obtained for GR-CO3 GR-SO4 Fe(II)goethite and Fe(II)lepidocrocite were all
within the same order of magnitude (Figure 53a)
Figure 53 Average surface area-normalised pseudo 1 order rate constants for the degradation of
a) TCA (this work) b) hexachloroethane (Elsner et al 2004a) and c) carbon tetrachloride
(Amonette et al 2000 Pecher et al 2002 OrsquoLoughlin et al 2003c Elsner et al 2004b) by
GR-SO4 GR-CO3 (suspension age 1 d) Fe3O4 Fe(II)α-FeOOH and Fe(II)γ-FeOOH
Experimental conditions applied in this work [Fe(II)tot]0 = 025-107 mM in the goethite and
lepidocrocite suspensions [Fe(II)tot]0 = 025-116 mM in the GR-SO4 and GR-CO3 suspensions
pH 70-86 71-926 m2 mineral surface areaL Experimental conditions applied by Elsner et al
1 mM aqueous Fe(II) 25 m2 mineral surface areaL Experimental conditions applied in
references employed in c) [Fe(II)tot]0 = 1-83 mM 25-275 m2 mineral surface areaL GR-SO4 =
110 Chapter 5
sulfate green rust GR-CO3 = carbonate green rust Fe3O4 = magnetite α-FeOOH = goethite γ-
FeOOH = lepidocrocite
When comparing the rate constants for the Fe(II)-Fe(III) mineral systems found for
reduction of TCA in this study (Figure 2a) mixed valent Fe(II)-Fe(III) minerals
such as green rusts and magnetite containing structural Fe(II) within the mineral
lattice do not seem to be significantly more reactive than Fe(II)-Fe(III) mineral
systems containing Fe(II) associated with pure Fe(III) oxides Unlike most other
iron oxides GRs contain not only external Fe(II) reactive sites at the surface but
also internal sites in the space between consecutive Fe(II)-Fe(III) hydroxide layers
The GR interlayer thickness is a function of both the size and the charge of the
interlayer anion For solutes the Fe(II) within the GR hydroxide layer is accessible
at the outside basal planes and at the edges as well as through the interlayer under
certain conditions (see Figure 42 Chapter 4 this work) Due to electrostatic
forces oxidants holding different charge properties (anions cations neutral
molecules) may exhibit different affinities for the various reactive Fe(II) sites
present in GR As the reactive sites are located inat the Fe(II)-Fe(III) hydroxide
layers the rate of reaction depends on the hydroxide layer area which can be
accessed by the oxidant If the oxidant is able to exchange with the interlayer
anion reaction can take place both at outer and inner surfaces of the GR particles
and in total more reactive sites are available for the reaction Thus oxidant size
and charge primarily control its access to the internal sites in GRs XRD
characterization of the GR-SO4 crystals during reaction with TCA demonstrated
that the GR-SO4 interlayer spacing did not vary during reaction (Table 52) This
may indicate that TCA did not enter the GR-SO4 interlayers
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 111 Table 52 Diffraction angle d-spacing and width at half peak height (Wfrac12) for the 001 GR-SO4
diffraction peak as a function of time during reaction with TCA ([Fe(II)GR]0 = 4 mM [TCA]0 = 1
mM)
Time (min) Angle (deg2θ)
d001-spacing(nm)
Wfrac12(degθ)
0 9483 10821 0273
10 9494 10809 0287
215 9522 10777 0263
330 9550 10745 0273
510 9524 10775 0277
855 9509 10791 0253
1160 9467 10839 0268
We roughly estimated the molecular size of TCA by summing the covalent radii of
the individual atoms (see Supporting Information 74) When comparing the
molecular size of TCA with the GR-SO4 interlayer spacing of 061 nm it can be
concluded that only when the C-C bond is oriented perpendicular to the interlayer
plane does the size of TCA exceed the GR-SO4 interlayer spacing In contrast the
size of TCA exceeds the GR-CO3 interlayer spacing (026 nm) regardless of its
orientation Hence if TCA was intercalated in the GR-CO3 interlayer we would
expect the interlayer spacing to expand The same holds for intercalation of a
vertically oriented TCA in the GR-SO4 interlayer Supposedly both the low charge
and the size of TCA impeded its access to the GR-SO4 and GR-CO3 interlayers ie
the divalent sulphate and carbonate in the GR interlayers did not readily exchange
with the monovalent TCA since GR interlayers generally have a higher affinity for
divalent anions than for monovalent anions (Miyata 1983) Thus TCA did neither
access nor react with internal Fe(II) reactive sites in GR-SO4 which means that the
reaction between TCA and GR-SO4 took place at the external reactive Fe(II) sites
solely It is reasonable to assume that the same holds for the reaction between TCA
and GR-CO3 No significant aging effects eg rate constants varying as a function
of GR age were observed within 142 days (see Table 51) However the SSAs of
112 Chapter 5
the GR suspensions holding ages up to 142 days were not measured but estimated
assuming that the GR SSA did not decrease within the time frame
According to the Scherrer formula the width at half peak height (Wfrac12) of a
diffraction peak is inversely proportional to the average crystal dimension
perpendicular to the given crystal plane (Klug amp Alexander 1974) The average
GR-SO4 particle thickness perpendicular to the basal plane (Wfrac12 Table 52) was
constant during reaction implying that TCA reacts only at the edges and not at the
basal planes Assuming that the platy hexagonal GR-SO4 and GR-CO3 crystals
hold an average width of 1 microm and an average particle thickness of 35 nm (Hansen
and Koch 1998) the ratio of edge surface area to outer surface area is AedgeAouter ~
130 for GR-SO4 and 121 for GR-CO3 (see Supporting Information 73) This
means that only 3 of the outer surface area in GR-SO4 and 5 of the outer
surface area in GR-CO3 are available at the edges Assuming that TCA reacts at the
edges only and if employing the AedgeAouter in the estimation of the rate constants
the surface area-normalised pseudo 1 order rate constants for GR-SO4 and GR-
CO3 would be 20-30 times higher than the rate constants depicted in Figure 53a
Thus the reactivity of GRs normalised to their reactive surface area is much higher
than the reactivity normalised to their total surface area
533 Comparing with rate constants obtained for other chlorinated aliphatic
compounds
Though care must be taken when comparing kinetic parameters obtained at
different experimental conditions (eg pH [Fe(II)]0[TCA]0 ratios surface area to
volume ratios etc) it is interesting to compare our results to those reported for
hexachloroethane (Figure 53b data from Elsner et al 2004a) The reductive
transformation of hexachloroethane was investigated for various Fe(II)-bearing
minerals including Fe(II)goethite Fe(II)lepidocrocite and GR-SO4 in the presence
of 1 mM dissolved Fe(II) and 25 m2 mineral surface areaL at pH 72 except for the
GR-SO4 suspensions in which the dissolved Fe(II) concentrations were slightly
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 113
higher and pH = 8 The pseudo 1 order rate constants reported for
hexachloroethane are in the range 18middot10-4 ndash 75middot10-3 h-1middotm-2middotL (Elsner et al 2004a)
When comparing Figure 53a with Figure 53b it can be seen that the differences
in intrinsic reactivity of the Fe(II)-bearing mineral systems are more pronounced
for hexachloroethane than for TCA
Caution should also be advised to the different reaction mechanisms by which
hexachloroethane and TCA react The transfer of a single electron and the
formation of an alkyl radical upon removal of a chlorine atom constitute the first
and in most cases the rate-limiting step in the reduction of chlorinated aliphatic
compounds (Vogel et al 1987) Depending on the chemical structure of the
chlorinated aliphatic compound the resulting free alkyl radical may undergo
hydrogenolysis chloroelimination or dimerizationcoupling In the case of TCA
the free dichloroacetate radical most likely undergoes hydrogenolysis The almost
quantitative transformation of TCA to DCA confirms that hydrogenolysis is the
prevalent reaction mechanism in our mineral systems The pentachloroethyl radical
formed from hexachloroethane may undergo hydrogenolysis (producing
pentachloroethane) or dichloroelimination (producing tetrachloroethene) Elsner et
al (2004a) found that hexachloroethane was transformed quantitatively into
tetrachloroethylene for all minerals which strongly indicates that
dichloroelimination was the dominating reaction mechanism Another
polychlorinated aliphatic compound transformed mainly by hydrogenolysis under
reducing conditions is carbon tetrachloride Several studies have investigated the
reductive dechlorination of carbon tetrachloride by various Fe(II)-bearing minerals
including Fe(II)goethite and GR-SO4 and reported pseudo 1 order rate constants
in the order 152middot10-4 ndash 640middot10-4 h-1middotm-2middotL for Fe(II)goethite and 864middot10-4 h-1middotm-2middotL
for GR-SO4 (Amonette et al 2000 Pecher et al 2002 OLoughlin et al 2003c
Elsner et al 2004b) When comparing Figure 53a with Figure 53c it can be seen
that the range of magnitude of the rate constants and the differences in intrinsic
114 Chapter 5
reactivity of the Fe(II)-bearing mineral systems are similar for carbon tetrachloride
and TCA
534 Factors controlling the reactivity of surface-bound Fe(II)
The reactivity of an oxidant towards Fe(II) surface species cannot be predicted
from the reduction potentials of the redox couple alone In heterogeneous systems
processes such as mass transfer and adsorptiondesorption may have a rate-limiting
effect on the overall reaction rate If the adsorption follows a saturation-type
sorption isotherm (eg Langmuir) the sorbate (oxidant) concentration at the
surface will vary non-linearly with the total amount of oxidant added This
dependence will have to be taken into account when establishing rate laws for the
heterogeneous reactions and when testing the hypothesis that the reaction rates
depend on the sorbed concentration of the oxidants pH has a strong impact on the
sorption and thereby on the availability of ionizable oxidants At the pH values
applied here the chloroacetates are fully dissociated (pKa (TCA) = 066 pKa
(DCA) = 135 pKa (MCA) = 287) However we found the sorption of TCA to be
negligible in suspensions of pure calcite goethitecalcite and lepidocrocitecalcite
at pH 76-77 Moreover the mass balance of TCA and DCA was almost complete
in all suspensions and therefore loss of TCA or DCA due to adsorption at mineral
surfaces or incorporation in the GR anion interlayers can be ruled out Calcite has a
much lower adsorption capacity than most iron oxides hence we anticipate that
goethite and lepidocrocite control the adsorption of TCA and DCA in both the pure
FeOOH and the FeOOHcalcite suspensions This was supported by our
experimental results demonstrating that the presence of a calcite surface - either
pure or as a support for goethite and lepidocrocite coatings - did not exert any
noticeable effect on the reaction rates (see Table 51) In addition the surface area-
normalised rate constants for mineral systems containing goethite or lepidocrocite
in pure form and mineral systems containing goethite or lepidocrocite as coatings
on calcite were very similar In heterogeneous reactions mass transfer in bulk
solution becomes the rate-limiting step when the surface reaction is much faster
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 115
than the diffusion of the reacting species to the reactive surface However at the
low rate constants obtained here the reaction of TCA with the Fe(II)-bearing
minerals is not likely to be mass transfer limited (see Supporting Information 72)
One very important factor affecting heterogeneous redox reactions is pH which
influences the speciation of the complexes in solution and at mineral surfaces as
well as the stability of the more soluble Fe(II)-containing minerals such as GRs In
contrast to aqueous Fe(II) complexes it is not possible to predict the reactivity of
Fe(II) surface species as their reduction potentials are unknown In the absence of
specifically adsorbing solutes other than H+ the surface charge of the Fe(III)
oxides goethite and lepidocrocite is determined by the surface densities of the
charged surface species equivFeOH2+ and equivFeOndash whereas the surface charge of calcite
is determined by the density of the surface species equivCO3ndash equivCaOH2
+ and equivCaOndash
(Stumm 1992 Van Cappellen et al 1993) The point of zero charge (pHpzc) of
pure calcite is in the pH range 7-11 and depends on the partial pressure of carbon
dioxide pCO2 The higher the pCO2 the lower the pHpzc At the experimental
conditions applied here (05 = 0005 atm CO2(g)) the pHpzc = 82 for calcite
(Table 53) As only 10-11 mg Fe(III) of goethite and lepidocrocite was coated
onto calcite we assumed a pHpzc of 82 for the goethite and lepidocrocite coated
calcite particles as well The pHpzc values for green rusts are unknown
116 Chapter 5
Table 53 Specific surface areas and point of zero charge of the various iron minerals in pure form as well as goethite and lepidocrocite coated onto calcite
Mineral Structural formula SSA (m2g) pHpzc
GR-SO4 FeII4FeIII
2(OH)12SO4middot3H2O 71 a -
GR-CO3 FeII4FeIII
2(OH)12CO3middot3H2O 47 b -
Magnetite Fe3O4 - 69 e
Goethite α-FeOOH 16 c 85 f
Lepidocrocite γ-FeOOH 18 c 73 e
Calcite CaCO3 le 1 d 82 g
Goe coating - le 1 d 82 h
Lep coating - le 1 d 82 h
a Chapter 4 this work b Williams amp Scherer 2001 c Product information by Bayer d The SSA of calcite was
quantified to le 1 m2g The detection limit of our BET method was 1 m2g e Charlet et al 1998a f Liger et al
1999 g Van Cappellen et al 1993 h Same as for calcite
The surface hydroxyl groups on iron oxides may be both singly (equivFe-OH) doubly
(equivFe2-OH) triply (equivFe3-OH) and geminally (equivFe-(OH)2) coordinated (Cornell amp
Schwertmann 1996 Stumm 1992) The differently coordinated surface hydroxyl
groups are not equally reactive Adsorption reactions involve only singly
coordinated surface groups and therefore only this kind of hydroxyl groups on iron
oxides will be considered here (Cornell amp Schwertmann 1996) Hence the
predominant surface sites available for adsorption in pure suspensions of Fe(III)
oxides are equivFeOH0 equivFeOH2+ and equivFeOndash In the presence of dissolved Fe(II)
equivFeIIIOFeIIOH0 equivFeIIIOFeIIOndash and equivFeIIIOFeII+ constitute the main reactive sites at
the Fe(III) oxide surfaces (Liger et a 1999) Assuming that Fe2+ and other cationic
Fe(II) species are the dominating adsorbates on the mineral surfaces in our
experiments we expect the actual pHpzc to be higher than the pHpzc of the pure
oxides listed in Table 53 Hence at pHlt82 where most of our experiments were
conducted all the mineral surfaces presumably carry net positive charges
At pH 70 where Fe2+ is still the predominant Fe(II) species in solution (~50) we
expect that equivFeIIIOFeIIOH equivFeIIIOFeIIOndash and equivFeIIIOFeII+ constitute the main
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 117
reactive sites at the Fe(III) oxide surfaces as suggested by Liger et a 1999 As pH
increases from 70 to 87 the Fe(II) carbonate complexes become increasingly
important in solution at the expense of the Fe2+ FeCl+ FeSO40 and FeOH+ species
(King 1998) Fe(II) carbonate complexes do not bind at the oxide surface as
readily as the aquo or hydroxo complexes of Fe(II) but carbonate itself sorbs
readily to Fe(III) oxide surfaces through which the Fe(III) oxide surface is coated
by inner-sphere monodentate equivFeIIIOCOOH0 surface complexes (Villalobos amp
Leckie 2000 amp 2001) The presence of carbonate shifted the sorption edge for the
Fe(II) adsorption on goethite from pH 58 to 78 and the authors hypothesized this
to be a result of the formation of aqueous and surface Fe(II)-carbonate complexes
and to competition between carbonate and Fe(II) for Fe(III) oxide surface sites
(Vikesland amp Valentine 2002) Similarly monodentate surface complexes like
equivFeIIICl0 and equivFeIIIOSO3ndash as well as ternary monodentate surface complexes like
equivFeIIIOFeIICl0 and equivFeIIIOFeIIOSO3ndash and ternary bidentate surface complexes such
as (equivFeIIIO)2FeIIOSO3 may form at Fe(III) oxide surfaces when Fe(II) chloride and
sulfate are present in solution (Ostergren et al 2000 Kim et al 2004) However
the effects of anionic ligands such as chloride and sulfate on Fe(II) adsorption at
Fe(III) oxide surfaces and the reactivity of Fe(II) carbonate chloride and sulfate
surface sites are still unknown and need to be evaluated (see Supporting
Information 75) Thus we do not know whether chloride and sulfate decrease or
increase the Fe(II) sorption in our mineral systems We can only report that we did
not detect any significant differences in the rate of TCA transformation between
the mineral suspensions containing carbonate chloride and sulfate respectively
Hence we anticipate that equivFeIIIOFeIIOH equivFeIIIOFeIIOndash and equivFeIIIOFeII+ constitute
the main reactive sites at the Fe(III) oxide surfaces within the whole pH range 70-
87 This might also explain why we did not detect any obvious systematic pH
effect in the Fe(II)-Fe(III)-systems (see Table 51) In the case of
hexachloroethane the reactivity order GR-SO4gtgoethitegtmagnetitegtlepidocrocite
may be rationalized by the variations in surface site densities and total amount of
118 Chapter 5
Fe(II) sorbed on the iron minerals (see Supporting Information 75) as well as the
different speciations and reactivities of the Fe(II) surface sites on the iron minerals
535 Comparison with biotic and other abiotic systems
Only one report on abiotic transformation of TCA is found in the literature and the
study demonstrates the reductive dechlorination of TCA to MCA by Fe(0)
(Hozalski et al 2001) The authors reported a pseudo 1 order rate constant of
60middot10-4 min-1middotm-2middotL for the transformation of TCA to DCA and a pseudo 1 order
rate constant of 225middot10-4 min-1middotm-2middotL for the transformation of DCA to MCA at
[Fe(0)]0 = 025 M [TCA]0 = 100-200 microM and pH 36-62 The rate constant for
TCA reduction by Fe(0) is 10-300 times faster than the rate constants reported for
the Fe(II)-bearing mineral systems here
There is abundant evidence that soil microorganisms and fungi can dechlorinate
TCA but only little is known about the bacteria and processes involved in the
biodegradation of TCA Biotransformation of TCA has been found at both oxic
and anoxic conditions Most of the microorganisms isolated grow feebly on TCA
as a sole source of carbon (Foy 1975 Weightman et al 1992 De Wever et al
2000) Only one bacterium capable of growing on TCA as the sole carbon and
energy source has been characterized (Yu amp Welander 1995) In addition
anaerobic bacteria coupling co-metabolic growth to reductive dechlorination of
TCA have been isolated (Weightman et al 1992 De Wever et al 2000) The
inability to grow on the less chlorinated acids DCA and MCA is a notable feature
of both the aerobic and anaerobic bacteria Complete transformation of TCA to
methane and carbon dioxide has only been found when abiotic and biotic processes
were combined (Egli et al 1989) The abiotic transformation of TCA to DCA
occurred spontaneously in the presence of sterile activated charcoal whereas the
DCA formed was further degraded to methane and carbon dioxide by a mixed
culture of methanogenic bacteria However the abiotic reductant(s) responsible for
the transformation of TCA to DCA was not reported (Egli et al 1989)
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 119
The rate constants obtained in this work suggest that the Fe(II)-bearing mineral
systems may be important reductants of TCA in natural suboxic environments In
natural iron-rich soils holding specific surface areas of 22 m2g (Kenneke amp
Weber 2003) average bulk densities of 265 gcm3 and porosities of 25 and
containing 2 iron oxides a rough estimation of the half-life of TCA amounts to
47 minutes when applying the average surface area-normalised rate constant
obtained for all the Fe(II)-Fe(III) mineral systems in this work (1middot10-3 h-1middotm-2middotL)
This estimation is based on the assumption that enough reactive Fe(II) is available
in these soils The natural iron-reducing sediment investigated by Kenneke and
Weber (2003) contained 80 microM Fe(II) in the soil solution and 315 micromole Fe(II) per
g sediment At such low Fe(II) concentrations the overall rate of abiotic
transformation of TCA in natural soils and sediments is most likely limited by the
regeneration of reactive Fe(II) Hence the continuous regeneration of reactive
Fe(II) surface sites by adsorption of abiotically or microbially produced Fe(II) may
further the long-term abiotic transformation of TCA in such environments
54 Conclusions
This work demonstrates that various Fe(II)-Fe(III) minerals systems including GR-
SO4 GR-CO3 magnetite Fe(II)goethite and Fe(II)lepidocrocite readily transform
TCA to DCA Dichloroacetate was not further reduced to MCA or acetate by any
of the Fe(II)-bearing minerals The surface area-normalised pseudo 1 order rate
constants obtained for the reductive transformation of TCA by the various Fe(II)-
bearing minerals did not differ significantly from each other The results obtained
in this work infer that under natural geochemical conditions Fe(II)-bearing mineral
systems may play an important role in the overall transformation of TCA Thus
not only microbial degradation but also abiotic reductive transformation of TCA by
Fe(II)-bearing minerals may govern the fate of TCA in natural subsurface
environments This holds both for natural systems like iron-rich suboxic soils and
sediments as well as engineered systems like permeable reactive barriers of zero-
120 Chapter 5
valent iron implemented for on-site remediation where both Fe(0) and solid or
surface-bound Fe(II) corrosion intermediates may transform TCA
Acknowledgments
We would like to thank Susanne Guldberg for performing the experimental work comprising
GR-SO4
References
Ahlers J Regelmann J Riedhammer C (2003) Environmental risk assessment of airborne trichloroacetic acid - a contribution to the discussion of the significance of anthropogenic and natural sources Chemosphere 52 531-537 Amonette JE Workman DJ Kennedy DW Fruchter JS Gorby YA (2000) Dechlorination of carbon tetrachloride by Fe(II) associated with goethite Environmental Science and Technology 34 4606-4613 Atkins PJ Gold V Marsh R (1984) The decarboxylation of trichloroacetic acid and the reactions of the trichloromethyl anion with 135-trinitrobenzene and with hydrogen ions kinetic measurements in dimethyl sulphoxide solution Journal of the Chemical Society Perkin Transactions 2 7 1239-1245 Berg M Muumlller SR Muumlhlemann J Wiedmer A Schwarzenbach RP (2000) Concentrations and mass fluxes of chloroacetic acids and trifluoroacetic acid in rain and natural waters in Switzerland Environmental Science and Technology 34 2675-2683 Brunauer S Emmett PH Teller E (1938) Adsorption of gases in multimolecular layers Journal of American Chemical Society 60 309-319 Butler EC Hayes KF (1998) Effects of solution composition and pH on the reductive dechlorination of hexachloroethane by iron sulfide Environmental Science and Technology 32 1276-1284 Butler EC Hayes KF (1999) Kinetics of the transformation of trichloroethylene and tetrachloroethylene by iron sulfide Environmental Science and Technology 33 2021-2027 Charlet L Liger E Gerasimo P (1998a) Decontamination of TCE- and U-rich water by granular iron Role of sorbed Fe(II) Journal of Environmental Engineering 124 25-30 Charlet L Silvester E Liger E (1998b) N-compound reduction and actinide immobilisation in surficial fluids by Fe(II) the surface FeIIIFeIIOH0 species as major reductant Chemical Geology 151 85-93 Cheregi M Danet AF (1997) Flow injection determination of chloride ions with spectrophotometric detection Analytical Letters 30 2847-2858 Cornell RM Schwertmann U (1996) The iron oxides Structure properties reactions occurrence and uses VCH Verlagsgesellschaft mbH Weinheim
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 121 Cui D Eriksen TE (1996) Reduction of pertechnetate by ferrous iron in solution influence of sorbed and precipitated Fe(II) Environmental Science and Technology 30 2259-2262
Egli C Thuumler M Suter D Cook AM Leisinger T (1989) Monochloro- and dichloroacetic acids as carbon and energy sources for a stable methanogenic mixed culture Archives of Microbiology 152 218-223 Elsner M Haderlein SB Schwarzenbach RP (2004a) Reactivity of Fe(II)-bearing minerals towards reductive transformation of organic contaminants Environmental Science and Technology 38 799-807 Elsner M Haderlein SB Kellerhals T Luzi S Zwank L Angst W Schwarzenbach RP (2004b) Mechanisms and products of surface-mediated reductive dehalogenation of carbon tetrachloride by Fe(II) on goethite Environmental Science and Technology 38 2058-2066 Erbs M Hansen HCB Olsen CE (1999) Reductive dechlorination of carbon tetrachloride using iron(II)iron(III)-hydroxide-sulphate (green rust) Environmental Science and Technology 33 307-311 Euro Chlor (2001) Trichloroacetic acid in the environment a dossier Euro Chlor Brussels and the European Chlorinated Solvent Association Fadrus H Maly J (1975) Suppression of iron(III) interference in the determination of iron(II) in water by the 110-phenanthroline method The Analyst 100 549-554 Fahimi IJ Keppler F Schoumller HF (2003) Formation of chloroacetic acids from soil humic acid and phenolic moieties Chemosphere 52 513-520 Foy CL (1975) The chlorinated aliphatic acids In Herbicides Chemistry degradation and mode of action Kearney PC Kaufman DD (eds) Marcel Dekker Inc 399-452 Gander JW Parkin GF Scherer MM (2002) Kinetics of 111-trichloroethane transformation by iron sulfide and a methanogenic consortium Environmental Science and Technology 36 4540-4546 Haiber G Jacob G Niedan V Nkusi G Schoumller HF (1996) The occurrence of trichloroacetic acid (TCAA) ndash indications of a natural production Chemosphere 33 839-849
Hansen HCB (1989) Composition stabilization and light absorption of Fe(II)Fe(III) hydroxycarbonate (green rust) Clay Minerals 24 663-669 Hansen HCB Guldberg S Erbs M Koch CB (2001) Kinetics of nitrate reduction by green rusts ndash effects of interlayer anion and Fe(II)Fe(III) ratio Applied Clay Science 18 81-91 Hansen HCB Koch CB (1997) A comparison of nitrate reduction by carbonate and sulphate forms of green rust Kodama H Mermut A R Torrance J K (eds) Proceedings of the 11th International Clay Conference Ottawa Canada Clays for our future 11 295-302 Hoekstra EJ de Leer EWB Brinkman UATh (1999a) Mass balance of trichloroacetic acid in the soil top layer Chemosphere 38 551-563 Hoekstra EJ de Leer EWB Brinkman UATh (1999b) Findings supporting the natural
122 Chapter 5
formation of trichloroacetic acid in soil Chemosphere 38 2875-2883 Hofstetter TB Heijman CG Haderlein SB Holliger HC Schwarzenbach RP (1999) Complete reduction of TNT and other (poly)nitroaromatic compounds under iron-reducing subsurface conditions Environmental Science and Technology 33 1479-1487 Hofstetter TB Schwarzenbach RP Haderlein SB (2003) Reactivity of Fe(II) species associated with clay minerals Environmental Science and Technology 37 519-528 Hozalski RM Zhang L Arnold WA (2001) Reduction of haloacetic acids by Fe0 Implications for treatment and fate Environmental Science and Technology 35 2258-2263 Husain S Narsimha R Alvi SN Rao RN (1992) Monitoring the effluents of the trichloroacetic acid process by high-performance liquid chromatography Journal of Chromatography 600 316-319 Hwang I Batchelor B (2000) Reductive dechlorination of tetrachloroethylene by Fe(II) in cement slurries Environmental Science and Technology 34 5017-5022 Jordan A Frank H (1999) Trifluoroacetate in the environment Evidence for sources other than HFCHCFCs Environmental Science and Technology 33 522-527 Kenneke JF Weber EJ (2003) Reductive dehalogenation of halomethanes in iron- and sulfate-reducing sediments 1 reactivity pattern analysis Environmental Science and Technology 37 713-720 Kim CS Rytuba JJ Brown Jr GE (2004) EXAFS study of mercury(II) sorption to Fe- and Al-(hydr)oxides II Effects of chloride and sulphate Journal of Colloid and Interface Science 270 9-20 King DW (1998) Role of carbonate speciation on the oxidation rate of Fe(II) in aquatic systems Environmental Science and Technology 32 2997-3003 Klausen J Troumlber SP Haderlein SB Schwarzenbach RP (1995) Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions Environmental Science and Technology 29 2396-2404 Klug PH Alexander LE (1974) X-ray diffraction procedures John Wiley amp Sons Inc Koch CB Hansen HCB (1997) Reduction of nitrate to ammonium by sulphate green rust Advances in GeoEcology 30 373-393 Kuumlhn R Pattard M (1990) Results of the harmful effects of water pollutants to green algae (Scenedesmus subspicatus) in the cell multiplication inhibition test Water Research 24 31-38 Lee W Batchelor B (2002a) Abiotic reductive dechlorination of chlorinated ethylenes by iron- bearing soil minerals 1 Pyrite and magnetite Environmental Science and Technology 36 5147- 5154 Lee W Batchelor B (2002b) Abiotic reductive dechlorination of chlorinated ethylenes by iron-bearing soil minerals 2 Green rust Environmental Science and Technology 36 5348- 5354
Reductive Transformation of Trichloroacetate in Abiotic Fe(II)-Fe(III) Mineral Systems 123 Liger E Charlet L Van Cappellen P (1999) Surface catalysis of uranium (VI) reduction by iron(II) Geochimica et Cosmochimica Acta 63 2939-2955 Lignell R Heinonen-Tanski H Uusi-Rauva A (1984) Degradation of trichloroacetic acid (TCA) in soil Acta Agriculturae Scandinavia 34 3-8 Loyaux-Lawniczak S Refait Ph Lecomte P Ehrhardt J Geacutenin JMR (1999) The reduction of chromate ions by Fe(II) layered hydroxides Hydrology and Earth System Sciences 3 593-599 Luther III GW (1990) The Frontier-Molecular-Orbital theory approach in geochemical processes in W Stumm Ed Aquatic Chemical kinetics John Wiley and Sons New York pp 173-198 McCulloch A (2002) Trichloroacetic acid in the environment Chemosphere 47 667-686 Miyata S (1983) Anion-exchange properties of hydrotalcite-like compounds Clays and Clay Minerals 31 305-311 Muumlller SR Zweifel H-R Kinnison DJ Jacobsen JA Meier MA Ulrich MM Schwarzenbach RP (1996) Occurrence sources and fate of trichloroacetic acid in Swiss lakes Environmental Toxicology and Chemistry 15 1470-1478 OLoughlin EJ Burris DR (2003) Reduction of halogenated ethanes by green rust Environmental Toxicology and Chemistry 23 41-48 OLoughlin EJ Kelly SD Cook RE Csencsits R Kemner KM (2003a) Reduction of uranium(VI) by mixed iron(II)iron(III) hydroxide (green rust) Formation of UO2 nanoparticles Environmental Science and Technology 37 721-727 OLoughlin EJ Kelly SD Kemner KM Csencsits R Cook RE (2003b) Reduction of AgI AuIII CuII and HgII by FeIIFeIII hydroxysulfate green rust Chemosphere 53 437-446 OLoughlin EJ Kemner KM Burris DR (2003c) Effects of AgI AuIII and CuII on the reductive dechlorination of carbon tetrachloride by green rust Environmental Science and Technology 37 2905-2912 Ostergren JD Brown Jr GE Parks GA Persson P (2000) Inorganic ligand effects on Pb(II) sorption to goethite (α-FeOOH) II Sulfate Journal of Colloid and Interface Science 225 483-493 Pecher K Haderlein SB Schwarzenbach RP (2002) Reduction of polyhalogenated methanes by surface-bound Fe(II) in aqueous suspensions of iron oxides Environmental Science and Technology 36 1734-1741 Reimann S Grob K Frank H (1996) Chloroacetic acids in rainwater Environmental Science and Technology 30 2340-2344 Ruumlgge K Hofstetter TB Haderlein SB Bjerg PL Knudsen S Zraurig C Mosbaeligk H Christensen TH (1998) Characterization of predominant reductants in an anaerobic leachate- affected aquifer by nitroaromatic probe compounds Environmental Science and Technology 32
124 Chapter 5
23-31 Sarzanini C Bruzzoniti MC Mentasti E (1999) Preconcentration and separation of haloacetic acids by ion chromatography Journal of Chromatography A850 197-211 Schwertmann U Cornell RM (1991) Iron oxides in the laboratory Preparation and characterization VCH Verlagsgesellschaft mbH Weinheim Scott BF Mactavish DC Spencer C Strachan WMJ Muir DCG (2000) Haloacetic acids in Canadian lake waters and precipitation Environmental Science and Technology 34 4266-4272 Scott BF Spencer C Marvin CH Mactavish DC Muir DCG (2002) Distribution of haloacetic acids in the water columns of the Laurentian Great Lakes and Lake Malawi Environmental Science and Technology 36 1893-1898 Strathmann TJ Stone AT (2003) Mineral surface catalysis of reactions between FeII and oxime carbamate pesticides Geochimica et Cosmochimica Acta 67 2775-2791 Stumm W (1992) Chemistry of the solid-water interface John Wiley amp Sons Inc Van Cappellen P Charlet L Stumm W Wersin P (1993) A surface complexation model of the carbonate mineral-aqueous solution interface Geochimica et Cosmochimica Acta 57 3505- 3518 Vikesland PJ Valentine RL (2002) Iron oxide surface-catalyzed oxidation of ferrous iron by monochloramine implications of oxide type and carbonate on reactivity Environmental Science and Technology 36 512-519 Villalobos M Leckie JO (2000) Carbonate adsorption on goethite under closed and open CO2 conditions Geochimica et Cosmochimica Acta 64 3787-3802 Villalobos M Leckie JO (2001) Surface complexation modeling and FTIR study of carbonate adsorption to goethite Journal of Colloid and Interface Science 235 15-32 Vogel TM Criddle CS McCarty PL (1987) Transformations of halogenated aliphatic compounds Environmental Science and Technology 21 722-736 Weightman AL Weightman AJ Slater JH (1992) Microbial dehalogenation of trichloroacetic acid World Journal of Microbiology and Biotechnology 8 512-518 De Wever H Cole JR Fettig MR Hogan DA Tiedje JM (2000) Reductive dehalogenation of trichloroacetic acid by Trichlorobacter thiogenes gen nov spnov Applied and Environmental Microbiology 66 2297-2301 Williams AGB Scherer MM (2001) Kinetics of chromate reduction by carbonate green rust Environmental Science and Technology 35 3488-3494 Yu P Welander T (1995) Growth of an aerobic bacterium with trichloroacetic acid as the sole source of energy and carbon Applied Microbiology and Biotechnology 42 769-774
Conclusions and Outlook 125
6 Conclusions and Outlook The work presented in this dissertation adds to the understanding of how Fe(II)-
bearing minerals like green rusts (GRs) vivianite (Fe2(PO4)2sdot8H2O) magnetite
(Fe3O4) and Fe(II) associated with goethite and lepidocrocite may form and react in
nature In order to elucidate the role of bacteria in the formation of GRs in natural
soils and sediments we studied the iron mineral phases forming as a result of the
activity of iron-respiring bacteria In chapter 2 the Fe-containing products formed
by anaerobic autotrophic denitrifying Fe(II)-oxidizing bacteria (FeOB) were
examined The culture medium applied contained high levels of bicarbonate and
phosphate and is typically used in this kind of studies as it provides excellent
conditions for the nitrate-reducing FeOB Fe(II) was present initially as a whitish
solid Fe(II) hydroxy phosphate (vivianite) and as soluble Fe(II) The results
obtained demonstrate that the denitrifying FeOB produce poorly crystalline
goethite via a greenish Fe(III)-enriched vivianite intermediate Moumlssbauer
spectroscopic analyses provided no evidence of green rust formation At low
phosphate concentrations where vivianite does not control the Fe(II) activity it is
reasonable to assume that siderite (FeCO3) precipitates initially and that carbonate
GR phases may form during biooxidation At low bicarbonate concentrations we
would expect Fe(II) sulfate or chloride species to dominate initially (depending on
the Fe(II) source applied) and sulfate GR or chloride GR to form during
biooxidation In chapter 3 we investigated the Fe-containing products formed
during reduction of common Fe(III) oxides by the anaerobic dissimilatory Fe(III)-
reducing microorganism Shewanella algae BrY S algae BrY reduced substantial
amounts of the initial Fe(III) and green and blackish mineral phases were produced
within 1-2 weeks after inoculation Moumlssbauer spectroscopic analyses showed that
the green and black precipitates consisted of green rust and vivianite
We studied the reactivity of synthetic GRs towards reducible organic pollutants in
order to asses the potential significance of GR phases for the fate of such
126 Chapter 6
compounds To this end we used nitroaromatic compounds (NACs) and
chlorinated acetates as suitable model compounds for studying redox reactions
potentially relevant in the environment In chapter 4 we investigated the relative
reactivity of outer and inner Fe(II) reactive sites in synthetic sulfate green rust
(GR-SO4) by using a series of structurally closely related compounds with different
charge properties as ldquoreactive probesrdquo The probe compounds included
nitrobenzene 2-nitrophenol 4-nitrotoluene 4-chloronitrobenzene and 4-
nitrophenylacetic acid Our results demonstrated that NACs are completely
reduced to their corresponding anilines by GR-SO4 The reactions followed pseudo
1 order kinetics with respect to NAC and the surface area-normalised pseudo 1
order rate constants obtained were 016ndash465middot10-4 s-1middotm-2middotL at [Fe(II)GR]0 = 103-
1260 mM [NAC]0 = 20-102 microM and pH 84-86 Neither mass transfer control nor
surface saturation kinetics could account for the similarity of the surface-
normalised pseudo 1 order rate constants obtained for the reduction of the neutral
and anionic NACs by GR-SO4 These observations suggest that the reaction
between NAC and GR-SO4 takes place at the external reactive Fe(II) sites At low
initial Fe(II)GR concentrations the external reactive Fe(II) sites were depleted and
the regeneration of new external reactive sites eventually controlled the reduction
of the NACs by GR-SO4 In chapter 5 we examined the reactivity of various
Fe(II)-Fe(III) mineral systems towards trichloroacetic acid (TCA) and
dichloroacetate (DCA) in laboratory batch experiments imitating natural
conditions The Fe(II)-Fe(III)-systems investigated included GR-SO4 carbonate
green rust magnetite Fe(II)goethite and Fe(II)lepidocrocite TCA was readily
reduced to DCA by all Fe(II)-containing minerals The reactions followed pseudo
1 order kinetics with respect to TCA and the surface area-normalised pseudo 1
order rate constants obtained were 033ndash76middot10-5 min-1middotm-2middotL at [Fe(II)]0 = 025ndash
116 mM [TCA]0 = 15ndash1000 microM and pH 70ndash87 Our results showed no
significant differences regarding product distribution and surface area-normalised
reaction rate constants between the Fe(II)-Fe(III)-systems DCA was not further
Conclusions and Outlook 127
reduced to monochloroacetate (MCA) or acetate in any of the systems within the
time frame in our experiments
As suggested in chapters 2 and 3 sufficient evidence must be provided and caution
should be exercised when proclaiming new biogenic minerals The study of
microbially produced GRs is still in its infancy and more research is needed in
order to elucidate the role of bacteria in the formation of GRs in natural soils and
sediments The results presented in chapter 2 indicate that microbiological
processes may be responsible for the oxidation of vivianite and metavivianite
((FeII3-xFeIII
x)(PO4)2(OH)xmiddot(8-x)H2O x gt 12) in natural subsurface environments
In chapter 3 we demonstrated that GRs may be produced microbially at conditions
including low carbon and Fe(III) concentrations as well as the exclusion of
synthetic electron shuttles and pH buffers The role of microbial processes in the
redox cycling of iron in the subsurface and the ways in which these processes can
be coupled to contaminant remediation are currently active areas of research Zero-
valent iron has been the most extensively studied reductant for the treatment of
many inorganic and organic contaminants and is currently the most commonly
used material for the construction of permeable reactive barriers (PRB) but a
detailed understanding of the processes involved in the reduction of these
pollutants by Fe(0) is lacking (Scherer et al 2000) Potentially reactive Fe(II)-
bearing corrosion products identified in iron metal columns and barriers include
magnetite siderite Fe(II) sulfides green rusts as well as Fe(II) sorbed to mineral
surfaces (Gu et al 1999 Roh et al 2000) The formation of reactive Fe(II)-
bearing minerals like GRs may explain the effective long-term operation of zero-
valent iron PRBs despite the formation of thick oxide films Thus natural in situ
PRBs might be created by stimulating the activity of anaerobic dissimilatory
Fe(III)-reducing bacteria and the subsequent formation of Fe(II) species such as
GRs Furthermore suspensions of synthetic GRs which are easily prepared from
relatively inexpensive commodity chemicals may also be injected and dispersed
into the subsurface
128 Chapter 6
The reductive transformation of NACs and TCA by GRs is relevant to
understanding the processes responsible for their degradation in the subsurface and
the development of innovative technologies for their remediation The results
obtained in chapters 4 and 5 indicate that GRs may play a significant role in the
reductive transformation of NACs and TCA in natural subsurface environments
Furthermore our results suggest that mainly the outer Fe(II) sites in GRs are
utilized in the reaction with neutral and monovalent anionic compounds and that
these sites may be replenished eg by reduction of the oxidized surface sites or
adsorption of Fe(II) from solution The continuous restoration of Fe(II) surface
sites in GRs may promote their long-term reactivity towards reducible
contaminants
References
Gu B Phelps TJ Liang L Dickey MJ Roh Y Kinsall BL Palumbo AV Jacobs GK (1999) Biochemical dynamics in zero-valent iron columns Implications for permeable reactive barriers Environmental Science and Technology 33 2170-2177 Roh Y Lee SY Elless MP (2000) Characterization of corrosion products in the permeable reactive barriers Environmental Geology 40 184-194 Scherer MM Richter S Valentine RL Alvarez PJJ (2000) Chemistry and microbiology of permeable reactive barriers for In Situ groundwater clean up Critical Reviews in Environmental Science and Technology 30 363-411
Supporting Information I
7 Supporting Information
71 Estimation of the one-electron reduction potential for 4-NPA
The one-electron reduction potential of the half-reaction for a given NAC 1hE
ArNO2 + e- ArNO2
can be used for comparing reduction rates of different NACs in a given system
The formation of the nitroaryl radical is the rate-determining step in the overall rate
of the reduction of a NAC to the corresponding aniline The difference between the
of a NAC and a given reductant is proportional to the change in standard free
energy for the transfer of the first electron ∆G
1hE
1degrsquo If a linear relationship between
the free energy of activation and ∆G1degrsquo is assumed the values of various NACs
can be a measure of their relative reactivity with a given reductant
1hE
As neither the one-electron reduction potential for 4-nitrophenylacetic acid (4-
NPA) nor the Hammett constant for the acetic acid substituent could be found in
the literature the one-electron reduction potential for 4-NPA was estimated by
application of a linear free energy relationship (LFER) to experimental data
Kinetic experiments were conducted in order to obtain the pseudo 1 order rate
constant for the reduction of 4-NPA by a model hydroquinone (reduced
juglone (8-hydroxy-14-naphthoquinone) in the presence of HS
minusHJUGk
ndash) The reduction of
a NAC by juglone follows the rate law
[ ] [ ] [ ] [ ] [ ] [ ]2222 ArNOJUGfkArNOHJUGkArNOk
dtArNOd
totHJUGHJUGHJUGobs sdotsdotsdot=sdotsdot=sdot=minus minusminusminusminus
and the was deducted from a LFER 1hE
II Chapter 7
bEak hHJUG +sdot=minus 059160
log1
for which a and b values have been established for a range of NACs with known
values (Hofstetter et al 1999) An excellent correlation of and log
has been found to exist over a range of 250 mV corresponding to more than 5 order
of magnitude for This is due to the fact that the actual transfer of the first
electron is the rate-determining step under the experimental conditions chosen
1hE 1
hE minusHJUGk
minusHJUGk
For comparison experiments with 4-nitrotoluene (4-NT) were also conducted The
pseudo 1 order rate constants for the reduction of 4-NPA with juglone were
corrected for the reduction of 4-NPA with only HSndash (control experiments
containing no juglone)
[ ]minuslowast
minusminus
minus
minus=
HJUGkk
k HSHJUGHJUG
where (MminusHJUGk -1middots-1) is the rate constant for a compound in the presence of only
juglone (slowastminusHJUGk -1) is the pseudo 1 order rate constant for a compound in the
presence of both juglone and HSndash (sminusHSk -1) is the pseudo 1 order rate constant for
the control reaction in the presence of only HSndash and [HJUGndash] (M) is the
concentration of the reactive dissociated HJUG- form (nondissociated
hydroquinone species are very nonreactive as compared to the monophenolate
species)
Supporting Information III
OH 0 OH OH
+ e- + H+ = + e- + H+ =
0 0
pl(( ox) = 8 00 PK1 (red) = 6 60
JUG HJUG
OH OH
OH
pKa2(red) = 10 60
Figure 7 1 Oxidized and reduced juglone fo1m s
Kinetic experiments in homogeneous anoxic aqueous solutions contained 5 mM
HS- 20 microM total juglone 50 mM KH2P04 buffer and were conducted at pH =
660 corresponding to a concentration of the reactive dissociated juglone form
[HJUG] = 10 microM
By using the LFER
E1 logkHJUG- = 125 middot 005~16 + 923
the following values were obtained
(Hofstetter et al 1999)
NAC k (M-1 -1) HJUG- middots log kHJUG- E~ (mV)
4-NT
4-NPA
311 middot10middot7
164middot10middot7
847middot 10-8
116middot 10middot7
226middot10middot2
489middot10middot3
-165
-231
-515
-546
IV Chapter 7
lowast
minusHJUGk -values are averages of triplicates whereas -values are averages of
duplicates The determined for 4-NT in this work (-515 mV) differs 3 from
the -value of -500 mV reported in the literature (Meisel amp Neta 1975
Wardman 1989) Hence it is assumed that the -value determined for 4-NPA
also differs by 3
minusHSk
1hE
1hE
1hE
Note that even for NACs holding very different values the difference in their
reactivities are much less pronounced in Fe(II)-Fe(III) systems such as the
Fe(II)goethite system (LFER slope a = 06 Hofstetter et al 1999) and the
Fe(II)magnetite system (LFER slope a = 034 Klausen et al 1995) as compared
to the jugloneH
1hE
2S system (a = 125) Furthermore it should be noted that all
LFERs mentioned here were established for neutral NACs and in this work we
have simply assumed that the LFERs are also valid for anionic NACs
72 The rate-limiting step
The overall rate of a reaction is equal to the rate of the slowest step in the
mechanism In heterogeneous reactions eg a compound reacting at the surface of
suspended particles in bulk solution the overall process by which the
heterogeneous reactions proceed may be broken down into a sequence of
individual diffusion steps and reaction steps 1) Mass transfer (diffusion) of the
reactant from the bulk fluid to the external surface of the solid phase 2)
Adsorption of reactant onto the solid surface 3) Reaction on the solid surface 4)
Desorption of the products from the solid surface 5) Mass transfer of the products
from the external solid surface to the bulk fluid Hence the rate of reaction of a
compound reacting at the surface of suspended particles in bulk solution may be
either mass transfer adsorptiondesorption or surface reaction limited When the
diffusion steps are much faster than the reaction steps the mass transfer or
diffusion steps do not affect the overall reaction rate However if the reaction steps
Supporting Information V
are very fast compared with the diffusion steps mass transport affects the reaction
rate Here only the external mass transfer is considered ie the diffusion of
reactants or products between the bulk fluid and the external surface of the solid
phase The additional internal mass transfer resistance for particles containing
substantial internal surface area is not addressed
721 Mass transfer (diffusion) limited kinetics
The overall rate constant can be represented by a system of resistances in series
(Fogler 1999 Arnold et al 1999)
⎟⎟⎠
⎞⎜⎜⎝
⎛+=
minusgeomSALobs kkak1111
where kobs is the observed rate constant kL is the mass transfer coefficient (mmiddots-1) a
is the ratio of the external (geometric) specific surface area to volume of solution
(m-1) and kSA-geom is the intrinsic rate constant of the reaction normalized to the
external specific surface area rather than the BET specific surface area By
comparing kLmiddota with kobs one can estimate the role of mass transfer on the rate of
reaction Thus if kLmiddota gtgt kobs mass transfer is so fast that it has no impact on the
reaction rate whereas if kLmiddota le kobs mass transfer is the rate limiting step
In fluid dynamics the Reynolds number Re is used for determining whether a
flow is laminar or turbulent
νtp ud sdot
=Re
where dp is the particle diameter (m) ut is the terminal particle settling velocity
(mmiddots-1) and ν is the kinematic fluid viscosity (m2middots-1) ν = η ρ where η is the
(absolute) dynamic fluid viscosity in centipoise (1 centipoise = 1 mPamiddots = 10-3
kgmiddotm-1middots-1) and ρ is the fluid density (kgmiddotm-3)
VI Chapter 7
At Re lt 1 we can apply Stokersquos particle settling velocity Stokersquos law is an
equation relating the terminal settling velocity of a smooth rigid sphere in a
viscous fluid of known density and viscosity to the diameter of the sphere when
subjected to a known force field
( )η
ρρsdot
minussdotsdot=
18
2pp
t
dgu (mmiddots-1)
where g = 981 mmiddots-2 is the gravitational constant ρp is the particle density (kgmiddotm-3)
The Sherwood number is the main parameter for prediction of the mass transfer
process
in fluid dynamics
3121 ScRe602Sh sdotsdot+=sdot
=lowast
lowast
iw
pL
Ddk
where Diw is the diffusion coefficient of the compound i in water (m2middots-1) is the
minimum (uncorrected) value of the mass transfer coefficient and Sc is the
Schmidt number This relation is often referred to as the Froumlssling correlation The
particle diameter is a key parameter in the Froumlssling correlation and the external
mass transfer coefficient varies with square of the particle size for smaller
particles
lowastLk
The Schmidt number is the ratio of the kinematic fluid viscosity and the diffusion
coefficient of the compound i in water
iwDν
=Sc
Supporting Information VII
According to Harriott (1962) the actual mass transfer coefficient kL is 15 times
greater than the minimum value of the mass transfer coefficient The
uncertainty in k
lowastLk
Lmiddota associated with particle sphericity and roughness issues are
believed not to exceed a factor of 2
The diffusion coefficient of a compound i in water can be estimated as (Hayduk amp
Laudie 1974)
5890141
9102613
iiw
VD
sdot
sdot=
minus
η (m2middots-1)
where iV is the molar volume of the compound i (cm3middotmol-1) estimated according
to Fuller et al 1966
Assuming spherical particles the external (geometric) specific surface area and the
particle diameter are calculated from the measured BET specific surface area Atot
assuming that our GR-SO4 has a AtotAouter ~ 30 similar to the one reported by
Hansen amp Koch (1998)
( ) ( ) ( )pppp
p
pp
p
dd
dV
SAAρρπ
πρ 1000
6
100061100030
SA3
2tot
geom sdot=
sdotsdot
sdot=
sdot== (m2middotg-1)
In our aqueous GR-SO4 system the density ρ = 1000 kgmiddotm-3 the absolute dynamic
viscosity η = 10-3 Pamiddots and the kinematic viscosity ν = 10-6 m2middots-1 for water The
GR-SO4 particle specific parameters used is found below
VIII Chapter 7
GR-SO4
Atot (m2g) 712
SAgeom (m2g) 237
Particle density ρp (kgm3) 1500
Particle diameter dp (m) 169middot10-6
Settling velocity ut (ms) 778middot10-7
Reynolds number 132middot10-6
The molar volumes the diffusion coefficients in water and the Schmidt numbers
for the NACs including 4-chloronitrobenzene (4-CNB) and nitrobenzene (NB)
were
Compound iV (cm3middotmol-1) Diw (m2middots-1) Sc
4-NT 1260 768middot10-10 1302
4-CNB 1230 779middot10-10 1284
4-NPA 1535 684middot10-10 1462
NB 1055 853middot10-10 1173
The ratio of the external (geometric) specific surface area to volume of solution
were calculated for GR-SO4 at the various concentrations applied
[Fe(II)GR]0 (mM) a (m-1)
103 366middot102
63 224middot103
126 448middot103
60 213middot103
The uncorrected mass transfer coefficients were estimated for the NACs using the
Froumlssling correlation
Supporting Information IX
[Fe(II)GR]0 (mM) lowastLk (mmiddots-1)
4-NT 912middot10-4
4-CNB 925middot10-4
4-NPA 812middot10-4
NB 101middot10-3
Finally kLmiddota was calculated and compared with the experimental 1 order rate
constants kobs obtained for the NACs
Compound [Fe(II)GR]0 (mM) kLmiddota (s-1) kobs (s-1) a
4-NT 103 050 420middot10-4
63 307 140middot10-3
126 613 590middot10-3
4-CNB 103 051 740middot10-4
63 311 170middot10-3
126 622 460middot10-3
4-NPA 103 045 640middot10-4
63 273 109middot10-3
126 546 473middot10-3
NB 60 324 137middot10-3 b
a Experimental pseudo 1 order rate constant at 50 microM [Ar-NO2]0 b Experimental pseudo 1 order rate constant at 10 microM [Ar-NO2]0
When comparing kLmiddota with kobs it can be seen that the rates of mass transfer for all
3 NACs exceed the observed rate constants by at least 3 or 4 orders of magnitude
at every initial Fe(II)GR concentration Thus the reaction of the given NACs with
GR-SO4 is not subject to mass transfer limitations under the experimental
conditions applied here
722 Surface saturation limited kinetics
More than 75 of all heterogeneous reactions that are not diffusion-limited are
surface-reaction-limited rather than adsorption- or desorption-limited We now
X Chapter 7
look at the reaction A = B = C where an intermediate B is formed In our system
A = Ar-NO2 B = Ar-NHOH and C = Ar-NH2 In this case the surface reaction is
assumed to be a single-site mechanism where only the site S on which A or B is
adsorbed is involved in the reaction forming B or C
KA
Adsorption 1 A + S = AmiddotS
kS1
Surface reaction 1 AmiddotS = BmiddotS
KB-1
Desorption 1 BmiddotS = B + S
KB
Adsorption 2 B + S = BmiddotS
kS2
Surface reaction 2 BmiddotS = CmiddotS
KC-1
Desorption 2 CmiddotS = C + S
The rate law for this surface-reaction limited single-site mechanism involving an
intermediate follows Langmuir-Hinshelwood kinetics (adopted from Fogler 1999)
CCBBAA
AAsitesSA
CKCKCKCKCk
dtdC
sdot+sdot+sdot+sdotsdotsdot
=minus1
1
Supporting Information XI
where kS1 is the intrinsic rate constant of the surface reaction transforming A into
the intermediate B Csites is the concentration of reactive sites S on the solid
surface KA KB and KC are the adsorption constants for A B and C at the reactive
surface sites and CA CB and CC are the concentrations of A B and C in the bulk
fluid Two major assumptions of the Langmuir isotherm imply that there is a fixed
number of localised surface sites present on the surface and that the activity of the
surface towards adsorption desorption or surface reaction is independent of
surface coverage
Hence fitting -∆CA∆t to CA CB and CC using a nonlinear curve fitting software
such as SigmaPlot may provide one with the intrinsic rate constant and the
adsorptions constants If KB and KC gtgt KA the intermediate and the product are
strongly competing with the reactant for vacant reactive surface sites
Our data was not fitted successfully by the Langmuir-Hinshelwood rate law
(regression results not shown) Simplifying the rate law by excluding either the
term KCmiddotCC or KBmiddotCB or both (assuming that the aniline product or the
hydroxylaniline intermediate or both did not compete for the reactive sites) did not
improve the regression The Langmuir-Hinshelwood rate law for a dual-site
mechanism did not fit our data either Thus Langmuir-Hinshelwood kinetics
cannot explain the reaction mechanism of the given NACs in our GR-SO4 system
73 External surface area of GR-SO4 and GR-CO3
The GR-SO4 unit cell consists of one double layer (d001 = 11 nm) ie one
hydroxide layer (049 nm ) and one interlayer (061 nm) Hexagonal GR-SO4
particles holding an average width of 1 microm (Figure 72) an average particle
thickness of 35 nm (Hansen amp Koch 1998) and a hydroxide layer thickness of
049 nm have a surface area of the basal plane
Abasal = 1 microm middot 1 microm ndash 2 middot 05 microm middot 025 microm = 075 microm2
XII Chapter 7
and a surface area of the edges
Aedge = (2 middot 05 microm + 4 middot 056 microm) middot 000049 microm = 00016 microm2
Figure 72 The hexagonal platy morphology of GR particles holding an average width of 1 microm
The particle thickness is the mean crystal thickness perpendicular to the 003 plane
as determined from the 003 reflections in an X-ray diffractogram A GR-SO4
particle holding a thickness of 35 nm contains 35 nm11 nm = 318 double layers
The GR-CO3 unit cell consists of one double layer (d001 = 075 nm) ie one
hydroxide layer (049 nm ) and one interlayer (026 nm) Hence a GR-CO3 particle
holding a thickness of 35 nm contains 35 nm075 nm = 467 double layers
The outer surface area of a GR-SO4 particle including outer basal planes and
edges is
Aouter = 222 microm 155microm 00016318microm 0752 =sdot+sdot
and the total surface area of a GR-SO4 particle including both inner and outer
basal planes as well as edges is
Supporting Information XIII
Atot = 222 microm 478)microm 00016microm 0752(318 =+sdotsdot
Hence the ratio of outer surface area to total surface area is
131microm 478microm 155
AA
2
2
tot
outer asymp=
Furthermore the ratio of edge surface area to outer surface area is
130microm 155
microm 00016318AA
2
2
outer
edge asympsdot
=
For GR-CO3 the outer surface area including outer basal planes and edges is
Aouter = 222 microm 157microm 00016746microm 0752 =sdot+sdot
and the total surface area of a GR-CO3 particle including both inner and outer
basal planes as well as edges is
Atot = 222 microm 701)microm 00016microm 0752(467 =+sdotsdot
Hence the ratio of outer surface area to total surface area is
145microm 701microm 157
AA
2
2
tot
outer asymp=
Furthermore the ratio of edge surface area to outer surface area is
121microm 157
microm 00016467AA
2
2
outer
edge asympsdot
=
XIV Chapter 7
74 Van der Waals radii
The size of polyatomic molecules can be estimated by summing the van der Waals
radii of the
individual atoms Van der Waals radii or nonbonded radii can be pictured as the
radii of hard spherical atoms (Figure 73)
Figure 73 Schematic of neighboring nonbonded atoms with van der Waals radii rA and rB
Assuming that the spheres of neighboring nonbonded atoms just touch (Figure
73) the highest possible ion or molecule size Ms can be estimated as the sum of
the van der Waals radii
Ms = 2middotrA + 2middotrB + (1)
Taking Paulingrsquos rule for nonmetals into account we can estimate the real size of
polyatomic ions bound by covalent bonds (Pauling 1960) The van der Waals
radius is larger than the covalent radius because it involves the interposition of two
electron pairs between the atoms rather than one The rule states that the van der
Waals radius of an atom exceeds its covalent radius by ~008 nm (overlap in
Figure 74)
Figure 74 Schematic of atoms undergoing covalent bonding
Supporting Information XV
Thus the size of polyatomic ions bound by covalent bonds Ms can now be
estimated as the sum of the van der Waals radii subtracted by 008 nm
Ms = 2middot(rA - 008 nm) + 2middot(rB - 008 nm) + (2)
We estimated the molecular size of the NACs (Table 72) by means of equation (2)
and the van der Waals radii of the atoms in Table 71
Table 71 Van der Waals radii of various atoms Values from Pauling 1960
Atom vdW radii (nm)
H 0120
O 0140
N 0150
C 0170
Cl 0181
S 0185
In order to make the calculations it was assumed that all atoms were spherical and
that all bond angles were 90deg or 180deg (linear structures) In addition no distinctions
were made between single and double bonds The molecular sizes of the NACs
were estimated with the benzene ring representing the xy plane
Table 72 Molecular sizes of the NACs a Thickness z of the xy plane
Compound Ms (x) (nm) Ms (y) (nm) Ms (z) (nm)a
NB 054 080 036
4-NT 054 106 036
4-CNB 054 100 036
4-NPA 054 136 036
XVI Chapter 7
Note that the molecular sizes in Table 72 are only rough estimations
For comparison with the GR-SO4 interlayer spacing (061 nm) we consider three
possible orientations of the NACs in the GR-SO4 interlayer 1) The NAC xyz
coordination is equivalent to the crystal abc coordination (z = c = 036 nm) 2) the
NAC xy plane is parallel to the crystal bc plane (z = a = 054 nm) and 3) the NAC
xy plane is parallel to the crystal ac plane (z = b = 080-136 nm) Hence the sizes
of the NACs do not hinder their access to the GR-SO4 interlayer Only when
oriented vertically do the sizes of the NACs (z = b = 080-136 nm) exceed the GR-
SO4 interlayer spacing
The molecular size of trichloroacetate (TCA) was also estimated by means of
equation (2) and the atomic van der Waals radii in Table 71 When the TCA
aliphatic chain is assumed to represent the x direction (Ms (x) = 066 nm) the
molecular size in the y and z directions ranges from 045-053 nm depending on the
free rotation of the C-C bond Thus only if the C-C bond is oriented perpendicular
to the crystal ab plane does the size of TCA exceed the GR-SO4 interlayer spacing
(061 nm) In contrast the size of TCA exceeds the GR-CO3 interlayer spacing
(026 nm) regardless of its orientation
75 Adsorption of Fe(II) onto Fe(III) oxides
As seen from the Fe(II) sorption isotherms Fe(II) sorption varies widely between
the Fe(III) oxides as a function of solution pH (Figure 75) Average surface
densities of approximately 2 singly coordinated sitesnm2 iron oxide have been
suggested for goethite and lepidocrocite (Cornell amp Schwertmann 1996) The
similar surface site densities of goethite and lepidocrocite might explain their
similar Fe(II) adsorption isotherms (Figure 75)
Supporting Information XVII
Figure 75 Fe(II) adsorption edges for ferrihydrite goethite hematite lepidocrocite and
magnetite in the absence of other specifically adsorbing cations and anions (from Vikesland amp
Valentine 2002 and references therein) The total number of surface sites was in excess of the
total Fe(II) concentrations in all experiments
Dissolved cations or anions may specifically adsorb at the calcite and Fe(III) oxide
surfaces by exchanging for H+ or OHndash at the equivCO3H0 equivCaOH0 equivFeOH0 and
equivFeIIIOFeIIOH0 surface sites At the experimental conditions applied here within a
pH range 70-87 the dominant species of interest in solution are Fe2+ HCO3ndash
CO32ndash Clndash SO4
2ndash (only in the GR-SO4 systems) and the anionic TCA and DCA In
addition Fe2+ readily forms aqueous complexes with hydroxide carbonate
chloride and sulfate whereby the species FeOH+ FeHCO3+ Fe(OH)(CO3)ndash
FeCO30 Fe(CO3)2
2ndash FeCl+ and FeSO40 may occur (Millero amp Hawke 1992) At
pH 70-87 we expect the Fe(II) species Fe2+ FeCO30 Fe(OH)(CO3)ndash FeOH+ and
Fe(CO3)22ndash to dominate in the GR-CO3 and CaCO3(s)CO2(g) buffered magnetite
suspensions In the goecalcite and lepcalcite suspensions we expect the FeCl+
species to dominate as well whereas the Fe2+ FeSO40 and FeOH+ species most
XVIII Chapter 7
likely dominate in the GR-SO4 suspensions Anionic inorganic ligands like
carbonate chloride and sulfate can lower or enhance the adsorption of Fe(II) due to
a) formation of stable nonadsorbing Fe(II) ligand aqueous complexes b) formation
of Fe(II) ligand Fe(III) oxide surface complexes which can lead to surface
precipitation at high Fe(II) and ligand concentrations c) competitive ligand
sorption to the Fe(III) oxide surface blocking reactive sorption sites at the surface
and d) diminution of the positive charge at the Fe(III) oxide surface (at pH levels
below the point of zero charge (pHpzc) of the Fe(III) oxide) thereby decreasing the
electrostatic repulsion of cations by the Fe(III) oxide surface Specifically adsorbed
cations increase the pHpzc whereas specifically adsorbed anions decrease the pHpzc
References Arnold WA Ball WP Roberts AL (1999) Polychlorinated ethane reaction with zero-valent zinc Pathways and rate control Journal of Contaminant Hydrology 40 183-200 Cornell RM Schwertmann U (1996) The iron oxides Structure properties reactions occurrence and uses VCH Verlagsgesellschaft mbH Weinheim Fogler HS (1999) Elements of chemical reaction engineering 3rd ed Prentice Hall Fuller EN Schettler PD Giddings JC (1966) A new method for prediction of binary gas-phase diffusion coefficients Industrial and Engineering Chemistry 58 19-27 Hansen HCB Koch CB (1998) Reduction of nitrate to ammonium by sulphate green rust Activation energy and interlayer reaction mechanism Clay Minerals 33 87-101 Harriott P (1962) Mass transfer to particles Part I Suspended in agitated tanks AIChE Journal 8 93-102 Hayduk W Laudie H (1974) Prediction of diffusion coefficients for nonelectrolytes in dilute aqueous solutions AIChE Journal 20 611-615 Hofstetter TB Heijmann CG Haderlein SB Holliger C Schwarzenbach RP (1999) Complete reduction of TNT and other (poly)nitroaromatic compounds under iron-reducing subsurface conditions Environmental Science and Technology 33 1479-1487 Klausen J Troumlber SP Haderlein SB Schwarzenbach RP (1995) Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions Environmental Science and Technology 29 2396-2404 Meisel D Neta P (1975) One-electron redox potentials of nitro compounds and radiosensitizers Correlation with spin densities of their radical anions Journal of the American Chemical Society 97 5198-5203
Supporting Information XIX
Millero FJ Hawke DJ (1992) Ionic interactions of divalent metals in natural waters Marine Chemistry 40 19-48 Pauling L (1960) The nature of the chemical bond 3rd ed Cornell University Press Ithaca Vikesland PJ Valentine RL (2002) Iron oxide surface-catalyzed oxidation of ferrous iron by monochloramine implications of oxide type and carbonate on reactivity Environmental Science and Technology 36 512-519 Wardman P (1989) Reduction potentials of one-eletron couples involving free radicals in aqueous solution The Journal of Physical Chemistry Reference Data 18 1637-1755
Curriculum Vitae 13011973 Born in Haderslev Denmark 1988-1991 Mathematical high school Haderslev Katedralskole Denmark 1991-1992 Sabbatical year 1992-1995 B Sc in environmental chemistry University of Copenhagen
(KU) Denmark B Sc thesis 1995 ldquoMethane oxidizing bacteria in soilrdquo
1995-1998 M Sc in environmental chemistry University of Copenhagen
Denmark 1997-1998 diploma thesis ldquoReductive dechlorination of carbon tetrachloride and chloroform in presence of iron(II)iron(III)-hydroxides (green rust)rdquo
1998-1999 Research and teaching assistant at the Chemistry Department
The Royal Veterinary amp Agricultural University (KVL) Denmark
1999-2004 PhD in environmental sciences Swiss Federal Institute of
Technology Zuumlrich (ETHZ) and Swiss Federal Institute for Environmental Science and Technology (EAWAG) Switzerland Docoral thesis ldquoFormation and redox reactions of green rusts under geochemical conditions found in natural soils and sedimentsrdquo
2000-2002 Teaching assistent at the Swiss Federal Institute of Technology
Zuumlrich and supervision of diploma students 2002 Microbial Diversity summer course (7 weeks) at the Marine
Biological Laboratory Woods Hole Massachusetts USA