fÁjdalomcsillapÍtÓ hatÁsÚ nr2b altÍpusszelektÍv …
TRANSCRIPT

Budapesti Mőszaki és Gazdaságtudományi Egyetem
FÁJDALOMCSILLAPÍTÓ HATÁSÚ NR2B
ALTÍPUSSZELEKTÍV NMDA ANTAGONISTÁK KUTATÁSA
PhD ÉRTEKEZÉS
Borza István
Témavezetı:
Dr. Domány György
Richter Gedeon Nyrt. Gyógyszerkémiai Kutatólaboratórium I
Konzulens:
Dr. Keglevich György
BME Vegyészmérnöki Kar, Szerves Kémia és Technológia Tanszék
Budapest, 2010

1
Nyilatkozat
Alulírott Borza István kijelentem, hogy ezt a doktori értekezést saját magam készítettem meg
nem engedett segítség nélkül, és csak a megadott forrásokat használtam fel. Minden olyan
részt, amelyet szó szerint vagy azonos értelemben, de átfogalmazva más forrásból átvettem,
egyértelmően a forrás megadásával megjelöltem.
Budapest, 2010
……………………
Borza István

2
TARTALOMJEGYZÉK
RÖVIDÍTÉSEK
1. BEVEZETÉS, CÉLKITŐZÉS .........................................................................................6
2. IRODALMI ÁTTEKINTÉS.............................................................................................8
2.1. A fájdalom érzékelése ....................................................................................................8 2.1.1. Fájdalomtípusok és mechanizmusok ........................................................................................... 8 2.1.2. A fájdalom vázlatos élettana ...................................................................................................... 10
2.1.2.1. A fájdalominger útvonala ................................................................................................... 10 2.1.2.2. Kapuzárásos fájdalom kontroll (wind up).......................................................................... 12 2.1.2.3. Centrális fájdalom kontroll (analgetikus leszálló pályarendszer)...................................... 13
2.1.3. Receptorok, ioncsatornák és mediátorok a gerincvelı hátsó szarvában ................................... 14
2.2. Glutaminsav receptorok .............................................................................................. 15 2.2.1. Nem-NMDA glutaminsav receptorok és élettani jelentıségük ................................................. 15 2.2.2. NMDA receptorok élettani jelentısége ..................................................................................... 17 2.2.3. NMDA receptorok felépítése és mőködése................................................................................ 18 2.2.4. Az NMDA receptorok expressziója........................................................................................... 20
2.3. Az NMDA receptorok mint terápiás célpontok.......................................................... 20 2.3.1. Glicin antagonisták .................................................................................................................... 21 2.3.2. Glutaminsav antagonisták (kompetitív NMDA receptor antagonisták) ................................... 22 2.3.3. NMDA csatorna blokkolók (nem kompetitív NMDA receptor antagonisták).......................... 22 2.3.4. NR2B szelektív NMDA antagonisták........................................................................................ 23
2.3.4.1. Ifenprodil típusú NR2B szelektív NMDA antagonisták ................................................... 23 2.3.4.2. Nem ifenprodil típusú NR2B szelektív NMDA antagonisták ........................................... 35
3. KÍSÉRLETI MODELLEK, VIZSGÁLATI MÓDSZEREK ........................................ 40
3.1. Farmakológiai szőrırendszer...................................................................................... 40
3.2. In vitro vizsgálatok....................................................................................................... 42 3.2.1. Kötıdési tesztek......................................................................................................................... 42 3.2.2. Funkcionális tesztek .................................................................................................................. 42
3.2.2.1. Funkcionális teszt primer neokortikális tenyészeten.......................................................... 42 3.2.2.2. Funkcionális teszt NR1a/NR2B és NR1/NR2A alegység összetételő rekombináns NMDA receptorokon ...................................................................................................................... 43
3.3. In vivo tesztek............................................................................................................... 44 3.3.1. Egér formalin teszt ..................................................................................................................... 44 3.3.2. Neuropátiás fájdalom Seltzer modellje (antihiperalgézia) ........................................................ 44 3.3.3. Neuropátiás fájdalom CCI (chronic constriction injury) modellje (antiallodínia).................... 44 3.3.4. Diabéteszes neuropátia modell ................................................................................................... 45
3.4. Nemkívánatos mellékhatásokra utaló tesztek............................................................. 45 3.4.1. Irwin teszt................................................................................................................................... 45 3.4.2. Spontán lokomotoros aktivitás mérése rágcsálókon.................................................................. 45 3.4.3. Rotarod teszt .............................................................................................................................. 45 3.4.4. QT-megnyúlás ............................................................................................................................ 46
4. SAJÁT EREDMÉNYEK................................................................................................ 47
4.1. Fahéjsavamidok ........................................................................................................... 50 4.1.1. Tiraminszármazékok ................................................................................................................. 51 4.1.2. Oktopaminszármazékok ............................................................................................................ 52 4.1.3. Fahéjsavamidok elıállítása ........................................................................................................ 55

3
4.2. Indol-2-karbonsav-származékok................................................................................. 57 4.2.1. Hidroxilcsoport helyzete az indolgyőrőn ................................................................................... 58 4.2.2. Az X-csoport természete ............................................................................................................ 60 4.2.3. A piperidingyőrő szubsztituensének helyzete........................................................................... 60 4.2.4. Az Y összekötı lánc helyzete ..................................................................................................... 61 4.2.5. A Z-csoport természete és helyzete ........................................................................................... 61 4.2.6. Indol-2-karbonsav-származékok elıállítása ............................................................................. 62
4.3. Benzimidazol-2-karbonsav-származékok ................................................................... 65 4.3.1. Az indol pirrolrészének helyettesítése más karbo-, illetve heterociklusokkal .......................... 65 4.3.2. Benzimidazol-2-karboxamidok optimalizációja ....................................................................... 66 4.3.3. Benzimidazol-2-karbonsav-származékok elıállítása ................................................................ 67 4.3.4. Indol- és benzimidazol-származékok in vivo aktivitása ............................................................ 70
4.4. Oxálsavdiamidok ......................................................................................................... 71 4.4.1. Eredmények, szerkezet-hatás összefüggések............................................................................. 71 4.4.2. Oxálsavszármazékok elıállítása................................................................................................ 76
4.5. Kinurénsav amidszármazékok .................................................................................... 78 4.5.1. Eredmények, szerkezet-hatás összefüggések............................................................................. 78 4.5.2. Kinurénsav amidszármazékainak elıállítása ........................................................................... 82
4.6. Benzoil-karbamid-származékok.................................................................................. 83 4.6.1. Eredmények, szerkezet-hatás összefüggések............................................................................. 83 4.6.2. Benzoil-karbamid-származékok elıállítása .............................................................................. 88
4.7. Indol-2-karboxamidin-származék............................................................................... 89 4.7.1. Eredmények, szerkezet-hatás összefüggések............................................................................. 89 4.7. 2. Indol-2-karboxamidin-származékok elıállítása........................................................................ 93
4.8. Benzilidén-piperidin-származékok.............................................................................. 94 4.8.1. Eredmények, szerkezet-hatás összefüggések az indol- és benzimidazol- ...................................... származékok körében .......................................................................................................................... 95 4.8.2. Indol- és benzimidazol-származékok elıállítása ....................................................................... 98 4.8.3. Eredmények, szerkezet-hatás összefüggések az oxálsavdiamidok körében.............................. 98 4.8.4. Oxálsavdiamidok elıállítása..................................................................................................... 100
5. A PROJEKT BEFEJEZÉSE.......................................................................................101
6. ÖSSZEFOGLALÁS.....................................................................................................102
7. TÉZISEK .....................................................................................................................109
8. MOLEKULALISTA....................................................................................................111
9. IRODALOMJEGYZÉK..............................................................................................118
10. KÖSZÖNETNYILVÁNÍTÁS......................................................................................126

4
RÖVIDÍTÉSEK
Ac acetil
BA biohasznosulás (Bioavailability)
Bn benzil
cAMP ciklikus adenozin-monofoszfát
CC test patkány hipokampális neuronok primer tenyészetében meghatározott
glutaminsav-indukált sejthalál gátlás
CFA komplett Freund adjuváns (Complete Freund's Adjuvant)
CGRP kalcitonin gén-rokon peptid (Calcitonin gene related peptide)
COX ciklooxigenáz
CYP citokróm P450
DAU dopamin felvétel
DEAD dietil-aza-dikarboxilát
DIC diizopropil-karbodiimid
DIPEA diizopropil-etil-amin
DMAP 4-dimetil-amino-piridin
DRG hátsó gyöki ganglionsejtek (Dorsal root ganglion)
ED effektív dózis
GABA γ-amino-vajsav
HBTU 2-(1H-benzotriazol-1-il)-1,1,3,3-tetrametilurónium-hexafluorofoszfát
hERG Kv11.1 káliumcsatorna (Human Ether-à-go-go Related Gene)
HPCD hidroxipropil-β-ciklodextrin
HTS nagy áteresztı-képességő szőrés (High-throughput Screening)
i.p. intraperitoneális
IP3 inozitol-trifoszfát
i.v. intravénás
IVM FM% in vitro metabolizmus, becsült maximális felszívódás
LMA lokomotoros aktivitás
LTP hosszú távú aktiváció kialakulása (Long-term potentiation)
mCPBA meta-klór-perbenzoesav
MCP-1 monocita kemoattraktáns protein-1 (Monocyte chemotactic protein-1)
MED minimum effektív dózis
Mez mezil

5
mGluR metabotróp glutaminsav receptor
NCS N-klór-szukcinimid
N.D. nincs meghatározva (not determined)
NK-1 neurokinin 1 receptor
NMDA N-metil-D-aszparaginsav
NOS nitrogén-monoxid szintáz
PCP 1-(1-fenil-ciklohexil)piperidin
PK farmakokinetika
p.o. per os, szájon át
QT a szív elektromos ciklusának a Q hullám elejétıl a T hullám végéig tartó
idıintervalluma
SAR szerkezet-hatás összefüggés (Structure-activity relationship)
s.c. szubkután, bır alá
t. termelés
TFA trifluorecetsav
Toz tozil

6
1. BEVEZETÉS, CÉLKITŐZÉS
Bár a fájdalomcsillapítás a legısibb orvosi feladatok közé tartozik, a krónikus és neurogén
fájdalmak a mai napig megoldatlan terápiás problémát jelentenek. Neuropátiás fájdalom az
idegrendszer tartós sérülése vagy mőködéscsökkenése, például baleseti sérülés, mőtét,
kemoterápia és különbözı betegségek (AIDS, szklerózis multiplex, artritisz, cukorbetegség)
következtében alakulhat ki.1 Tünetei a körülményektıl függıen különbözıek lehetnek, de
leggyakrabban allodíniában (fájdalmatlan inger által kiváltott fájdalom) és hiperalgéziában
(felfokozódott fájdalomérzékenység) nyilvánulnak meg. A fájdalom krónikussá válásában
fıleg a perifériás idegek és a gerincvelı, ritkábban agyi területek is érintettek. Kezelésében a
közismert fájdalomcsillapító gyógyszerek hatása elégtelen, ennek ellenére a neuropátiás
fájdalomban szenvedık többségénél még mindig ezeket a szereket alkalmazzák. A
gyógyszeres és egyéb terápiák a krónikus neurogén fájdalomban szenvedı betegek felének
csillapítják csak a fájdalmát, ezért égetı szükség van új, hatékony gyógyszerekre.2 Az a tény,
hogy hagyományos analgetikumokra ez a fájdalomforma alig reagál, más terápiás területek
(antidepresszánsok, antiepileptikumok) meglévı gyógyszereinek „off label” alkalmazásához
vezetett. Késıbb ezek közül a leghatásosabbakat törzskönyvezték az új indikációra, ennek
ellenére a triciklusos antidepresszánsok és az arany standardnak tekintett gabapentin és
pregabalin a betegek nagy részénél gyenge hatékonyságot mutatnak.3 Az elmúlt néhány
évtizedben bebizonyosodott, hogy a glutaminsav receptorok fontos szerepet töltenek be a
perifériás szövetek és idegek sérüléseivel kapcsolatos fájdalomérzékelésben.3-5 A glutaminsav
a központi idegrendszer fı serkentı neurotranszmittere, számos metabotróp és három
ionotróp receptort aktivál (NMDA, AMPA és kainsav). Az ionotróp glutaminsav receptorok
serkentı szinaptikus jelátvitelt közvetítenek a központi idegrendszerben. Normális, nociceptív
fájdalom esetén a gerincvelı hátsó szarvában az afferens idegekbıl érkezı serkentı jelet
elsısorban a gyors aktivációra és deszenzibilizációra képes AMPA és kainsav receptorok
közvetítik. A hosszabb ideig tartó, intenzívebb, fájdalmas ingerek akkumulálódó, elnyújtott,
lassan depolarizáló szinaptikus potenciált alakítanak ki. Ezeket a krónikus és neuropátiás
fájdalomra jellemzı ingereket a lassabban reagáló NMDA receptor-ioncsatornák közvetítik.
Az idegi sérülés helyén neuroma, egy hiperérzékeny struktúra keletkezik, mely intenzív
fájdalom bemenetet ad a gerincvelı hátsó szarvába. Az ottlévı idegek a megnövekedett
bemenet miatt túlérzékeny állapotba kerülnek, csökken az ingerküszöbük, a nem fájdalmas
ingereket is fájdalomként továbbítják (allodínia), illetve a fájdalmas ingereket abnormálisan
felerısítik (hiperalgézia). Ezt a gerincvelıben jelentkezı hiperérzékeny állapotot ( „wind-up”)

7
már 1966-ban leírták.6 Az NMDA antagonisták lehetséges szerepének felvetését a neuropátiás
fájdalom enyhítésében 1987-re tehetjük amikor kimutatták, hogy NMDA antagonisták
gátolják a „wind-upot”.7, 8 Ezután számos in vivo modellben sikerült igazolni, hogy az NMDA
receptor antagonisták hatékonyan csillapítják mind a neurogén9, 10, mind a gyulladásos
fájdalmat11. Az NMDA antagonista hatás mellett egyéb receptoriális aktivitással is
rendelkezı, forgalomban lévı gyógyszerekkel szerzett klinikai tapasztalatok is megerısítik
használhatóságukat fájdalomcsillapításban, Parkinson- és Alzheimer-kórban (pl. ketamin12-14,
dextrometorfán15, memantin16). Ezek a vegyületek nem kompetitív antagonisták, az
ioncsatorna belsejében lévı csatornát blokkoló kötıhelyhez, az ún. fenciklidin kötıhelyhez
kapcsolódva fejtik ki hatásukat. Hatékony terápiás használatukat elsısorban az NMDA
receptor által mediált szinapszisokat túlzottan blokkoló hatásuk gátolta. Ennek ellenére a
kutatások elsı szakaszában népszerő célpont volt a fenciklidin kötıhely de az újonnan
elıállított antagonisták fejlesztése is abbamaradt tipikus mellékhatásaik (pszichotomimetikus,
szedatív, memóriarontó) miatt.17
Jelentıs elırelépést jelentettek az NR2B alegység altípusra szelektív antagonisták
megjelenése, melyek mentesek ezektıl a mellékhatásoktól. Elsıként egy forgalomban lévı
vérnyomáscsökkentıt, az ifenprodilt azonosítottak NR2B szelektív NMDA antagonista hatású
vegyületként. Az ifenprodil és analogonjai (pl. az eliprodil) vegyes profilú anyagok,
alkalmazásukat olyan mellékhatások korlátozták melyek nem függnek össze az NMDA
hatásmechanizmussal. Idıközben számos NR2B szelektív NMDA antagonista vegyületet
írtak le és védtek szabadalmi bejelentésekben, melyeket fıleg neuroprotektív területen
vizsgáltak. Késıbb merült fel fájdalomcsillapításra való felhasználásuknak a lehetısége, és
mivel mellékhatásaik egyéb receptoriális aktivitásuknak köszönhetı, okkal remélhettük, hogy
egy tiszta profilú, szelektív vegyület mellékhatásmentesen csillapíthatja a neuropátiás
fájdalmat. A kielégítetlen gyógyászati igényt és a hatásmechanizmus újdonságát mérlegelve,
Társaságunk a Richter Gedeon Gyógyszervegyészeti Gyár 1999-ben NR2B szelektív NMDA
antagonista hatásmechanizmuson alapuló új fájdalomcsillapító kutatási projekt elindítása
mellett döntött. Terápiás célunk egy orálisan adható fájdalomcsillapító volt, krónikus és
neurogén fájdalmak enyhítésére. Ennek érdekében ifenprodil típusu, NR2B szelektív
antagonista hatású molekulák elıállítását és tesztelését határoztuk el. Mivel abban az idıben a
receptor szerkezete nem volt teljesen felderítve és egy nagy áteresztıképességő biológiai
szőrésnek sem volt meg a feltétele, kémiai munkánkat a versenytársak ismert anyagainak a
szerkezetére alapoztuk.

8
2. IRODALMI ÁTTEKINTÉS
2.1. A fájdalom érzékelése
2.1.1. Fájdalomtípusok és mechanizmusok
A Nemzetközi Fájdalom Társaság (IASP) definíciója szerint a fájdalom: „ Kellemetlen érzéki
és emócionális élmény, amely valódi vagy lehetséges szövetkárosodással jár, vagy ehhez
hasonló panaszokat okoz. A fájdalom mindig szubjektív és emiatt mindig érzelmi töltése is
van”. A fájdalom funkcióját mind a mai napig Hippokratész fogalmazta meg a legtalálóbban:
„A fájdalom a szervezet házırzı kutyája”. Vagyis a fájdalom egy olyan életfontosságú
biológiai jelzırendszer, mely egyrészt segít az ártalmas ingerek elkerülésében , másrészt jelzi
a szervek, szövetek károsodását vagy megbetegedését. A fájdalom komplex fogalom, testi,
lelki, értelmi, szociális és spirituális összetevıi vannak.18 A fájdalomnak kórtani alapon
három típusa különíthetı el: nociceptív, gyulladásos és neuropátiás fájdalom (1. ábra).1 A
nociceptív fájdalom sérülést követıen többnyire akutan alakul ki. Biológiailag hasznos védı
szerepet ellátó fiziológiai jelenség. Figyelmeztet a kialakult vagy fenyegetı vázizomrendszeri
és zsigeri szövetkárosodásra, a károsodás elhárítását célzó magatartást, illetve reflexeket vált
ki. Az okozott fájdalom általában jól kezelhetı hagyományos fájdalomcsillapító szerekkel. Az
1.a. ábra egy normál, fiziológiás állapotú fájdalomérzékelést illusztrál. Alacsony intenzitású
inger, pl. érintés, alacsony küszöbértékő afferenseket (Aβ) aktivál fájdalom nélküli,
ártalmatlan érzetet keltve. Egy nagy intenzitású, fájdalmat elıidézı ártalmas inger pl. hı (az
ábrán tő), magasküszöbértékő (C és Aδ) afferenseket aktivál. A gyulladásos fájdalmat
perifériás szöveti sérülés, trauma, fertızés, mőtéti beavatkozás, égés okozta gyulladás vagy
gyulladásos betegségek , pl. artritisz váltanak ki. A gyulladt, károsodott szövetben számos
fájdalomkeltı mediátor válhat szabaddá: szerotonin, ATP, monoaminok, citokinek, kininek,
kemokinek, prosztaglandinok és növekedési faktorok. Ezek a mediátor anyagok különbözı
intracelluláris jelátalakító folyamatokon keresztül aktiválják, érzékenyítik a helyi nociceptív
mechanizmusokat. Például a prosztaglandin E2 (PGE2) aktiválja az EP prosztanoid receptort
amely aktiválja a protein kináz A-t (PKA), kiváltva ezzel a tetrodotoxin (TTX) rezisztens Na-
csatorna foszforilálódását, mely kizárólag a primer fájdalomérzı idegekben (afferens
nociceptorok) expresszálódik.

9
1. ábra. Normál, gyulladásos és neuropátiás fájdalom érzékelésének folyamata.1
Az 1.b. ábra olyan perifériás szövetkárosodást vagy gyulladást követı fájdalomérzékelési
folyamatokat mutat be, ahol ártalmas és nem ártalmas ingerekre is abnormális válasz
indukálódik. A periféria érzékennyé válik, csökken a magas ingerküszöbő (C és Aδ) rostok
ingerküszöbe, fájdalmas ingerek abnormálisan megnövekedett fájdalmérzetet okoznak a
sérülés, gyulladás helyén (primer hiperalgézia). Az érzékenyített területekrıl induló
nociceptív rostok képesek érzékenyíteni a közeli intakt területek nociceptorait is, így a
hiperalgézia ezeket a területeket is érinti (szekunder hiperalgézia). Az érzékennyé vált
nociceptorokból induló, felerısödött C-rost impulzus hiperérzékennyé teszi a gerincvelı és az
agy fájdalmat közvetítı és érzékelı területeit (centrális szenzitizáció). Ennek eredményeként
az alacsony küszöbértékő Aβ rostok is fájdalomérzést váltanak ki, nem fájdalmas inger (az
ábrán tollpihe) is fájdalmat okoz (allodínia).
A neuropátiás fájdalom az idegrendszer primer károsodása, vagy mőködészavara által
kiváltott vagy fenntartott fájdalom. A neuropátiás fájdalomnak nincs védı szerepe. Gyakran
válik krónkussá, a betegek értelmetlen szenvedését okozó fájdalommá. A neuropátiás
fájdalom kiváltásában toxikus, vaszkuláris, traumás, gyulladásos, tumoros, metabolikus és
iatrogén tényezık egyaránt szerepet játszhatnak. Az idegsérülés helyén keletkezı neurómákon
és a vele kapcsolatban lévı hátsó gyöki ganglionsejteken (DRG) rendellenes kisülések
keletkeznek, a sérülés helyén égı fájdalom jelentkezik (1.c. ábra). A megnövekedett

10
impulzusáram a gerincvelı hiperexcitibilitását és centrális szenzitizálását eredményezi
aminek következtében hiperalgézia és allodínia alakul ki.1
A különbözı humán fájdalomformák az akut, gyulladásos és neurológiai fájdalomtípusok
valamelyikébe besorolhatók, bár sok esetben a fájdalomnak gyulladásos és neuropátiás
komponense is van egyszerre (2. ábra).
akut fájdalom perzisztens/gyulladásos fájdalom
krónikus/neuropátiás fájdalom
neuropátiás fájdalom
-vénás szúrás/ tő okozta fájdalom -égés -posztoperatív azonnali fájdalom -poszttraumás azonnali fájdalom -akut betegségek (hasnyálmirigy gyulladás, infarktus, stb.) -alhasi fájdalom
-artritisz -posztoperatív fájdalom -irritábilis bél szindróma -viszcerális fájdalom -égési fájdalom -traumás szövetkárosodás -migrén -derékfájdalom -endometriózis -diszmenorrea -urológiai fájdalom
-oszteoartritisz -derékfájás (isiász, lumbágó) -fibromialgia -rákos csontfájdalom
perifériás neuropátia -posztherpeszes neuralgia -toxikus neuropátia -fokális traumás neuropátia -fantom és csonk fájdalom centrális neuropátiák -iszkémiás cerebrovaszkuláris sérülés (stroke) -szklerózis multiplex okozta fájdalom -gerincvelı sérülés -Parkinzon kór -amiotrópiás laterális szklerózis vegyes neuropátiák -diabéteszes neuropátiák -szimpatikus rendszer által fenntartott fájdalom
2. ábra. Humán fájdalomtípusok.
2.1.2. A fájdalom vázlatos élettana
2.1.2.1. A fájdalominger útvonala
A fájdalmat érzékelı receptoroktól az inger döntıen a gerincvelıben futó afferens rostokon
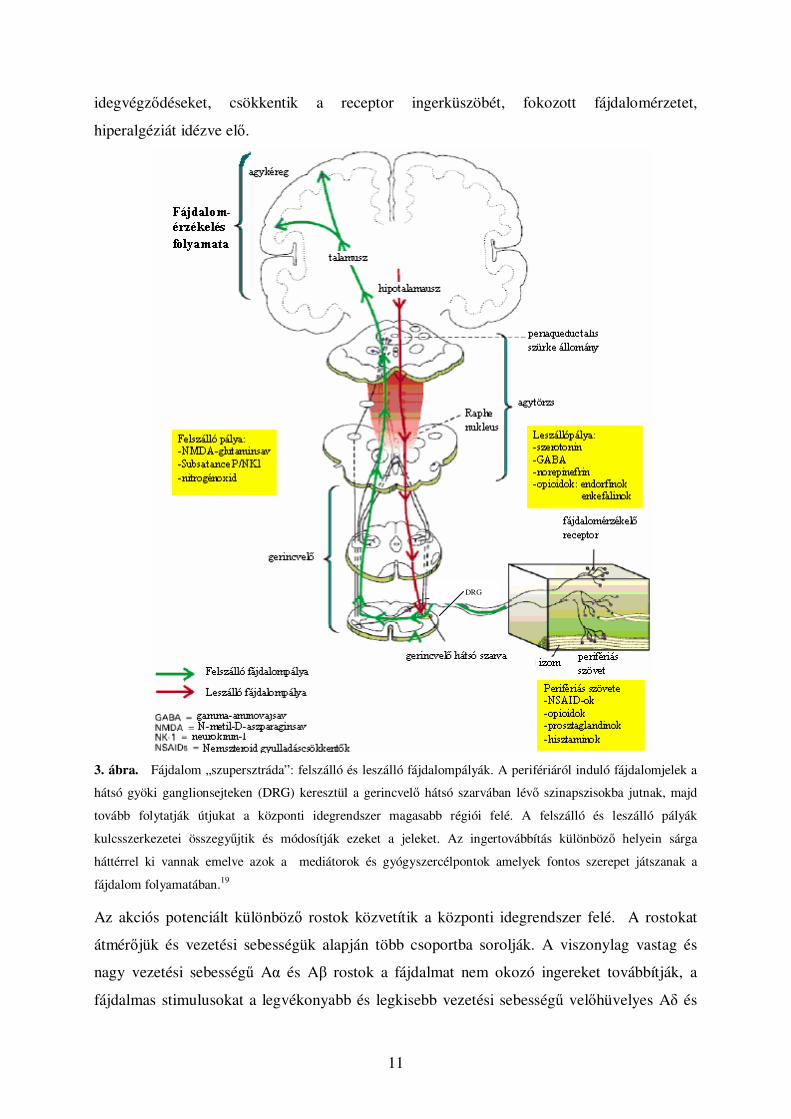
keresztül jut a központi idegrendszerbe, ahol kialakul a fájdalomérzet (3. ábra).19 A központi
idegrendszer válaszreakcióját az efferens rostok közvetítik. A fájdalomérzı receptorok,
másnéven nociceptorok az agyszövet kivételével mindenütt jelen vannak. A nociceptorokat
elsıdlegesen hı, mechanikai és kémiai ingerek aktiválják. Az inger hatására megváltozik a
receptor extracelluláris részének konformációja, ennek következtében az érzıidegsejt
kationcsatornája megnyílik. Generátorpotenciál képzıdik, amelynek amplitúdója a behatás
erısségétıl függ. Ha a generátorpotenciál eléri a közeli Na+ csatorna küszöbét, akkor
tovaterjedı akciós potenciál keletkezik. Külsı hı, mechanikai és kémiai ingerek mellett
vannak olyan szövetkárosodáskor felszabaduló molekulák („gyulladásos leves”: hisztamin,
szerotonin, bradikinin, stb.) amelyek önmagukban is kiváltják a nociceptor ingerületét, sıt
jelentıs ingerküszöb fölötti stimulust idézhetnek elı. Míg más felszabaduló faktorok
(leukotriének, prosztaglandinok, substance-P, CGRP, NO, stb) csak érzékenyítik a nociceptív
11
idegvégzıdéseket, csökkentik a receptor ingerküszöbét, fokozott fájdalomérzetet,
hiperalgéziát idézve elı.
3. ábra. Fájdalom „szupersztráda”: felszálló és leszálló fájdalompályák. A perifériáról induló fájdalomjelek a
hátsó gyöki ganglionsejteken (DRG) keresztül a gerincvelı hátsó szarvában lévı szinapszisokba jutnak, majd
tovább folytatják útjukat a központi idegrendszer magasabb régiói felé. A felszálló és leszálló pályák
kulcsszerkezetei összegyőjtik és módosítják ezeket a jeleket. Az ingertovábbítás különbözı helyein sárga
háttérrel ki vannak emelve azok a mediátorok és gyógyszercélpontok amelyek fontos szerepet játszanak a
fájdalom folyamatában.19
Az akciós potenciált különbözı rostok közvetítik a központi idegrendszer felé. A rostokat
átmérıjük és vezetési sebességük alapján több csoportba sorolják. A viszonylag vastag és
nagy vezetési sebességő Aα és Aβ rostok a fájdalmat nem okozó ingereket továbbítják, a
fájdalmas stimulusokat a legvékonyabb és legkisebb vezetési sebességő velıhüvelyes Aδ és
DRG

12
velıtlen C rostok szállítják a gerincvelıbe, illetve a trigeminus érzı magvába. Az Aδ rostok
az azonnal fellépı, éles, szúró jellegő fájdalmat közvetítik, míg a C rostok a fájdalom
második fázisából eredı, lassú, de tartós, égetı jellegő ingert szállítják.20 A nociceptív
afferensek többsége a gerincvelı hátsó szarvába fut be, ahol három fı idegcsoporttal, serkentı
és gátló interneuronokkal és projekciós neuronokkal szinaptizálnak. A gerincvelı hátsó
szarvának mind a hat rétege tartalmazza ezeket a fájdalomingert továbbító, nociceptív
struktúrákat. A projekciós neuronok a bejövı szenzoros információkat agyi központokba
szállítják. A serkentı interneuronok a projekciós neuronok felé közvetítik a szenzoros
információkat. Az inhibitorikus interneuronok a bejövı szenzoros információkat
szabályozzák. A projekciós neuronok lehetnek nociceptív specifikusak, amelyek csak az Aδ
és C rostoktól kapnak ingereket és lehetnek széles-dinamikus sávú idegek, melyek mind a
fájdalmat közvetítı Aδ és C rostoktól, mind pedig a fájdalmatlan ingereket továbbító Aα és
Aβ rostoktól kaphatnak információt. A fájdalomérzetet perifériás (spinális) és centrális
(supraspinális) mechanizmusok szabályozzák. Mindkét gátló mechanizmusban a hátsó szarvi
gátló interneuronok játszanak kulcsszerepet.20
2.1.2.2. Kapuzárásos fájdalom kontroll (wind up)
Miközben a C típusú rostok a fájdalmas ingereket, az Aα és Aβ rostok a fájdalmatlan
ingereket a projekciós neuronok felé továbbítják, ugyanazzal a projekciós neuronnal
kapcsolatban lévı gátló interneuronhoz is csatlakoznak (4. ábra) Az így létrejövı kör
szabályozza perifériásan a fájdalomérzetet. A C rostok gátló neurotranszmittereket, az Aα és
Aβ rostok serkentı neurotranszmittereket bocsájtanak ki az inhibíciós interneuronok felé.
A B 4. ábra. A fájdalominger spinális gátlása: A kapu nyitása, B kapu zárása.21
A perifériás idegsérülések miatt felszabaduló mediátorok (bradikinin, szerotonin, hisztamin,
prosztaglandin, acetilkolin, substance-P stb.) a környezı niciceptorokat direkt aktivációval
vagy az ingerlési küszöb csökkentésével hiperérzékennyé teszik. A nociceptorok állandó
ingerlésének következtében a C rostok folyamatosan tüzelnek. Ennek hatására a C rostokból

13
felszabaduló gátló transzmitterek blokkolják a gátló interneuronokat, a projekciós neuron
felszabadul a gátlás alól és a fájdalomérzés akadálytalanul továbbítódik (kapu nyitása) (4.A
ábra). Ha ugyanakkor más, fıleg mechanoreceptorokból származó ingerület érkezik a hátsó
szarvba, az Aα és Aβ rostok hatása érvényesül: a felszabaduló serkentı neurotranszmitterek
(fıként glutaminsav) serkentik a gátló interneuronokat, amik így gátolják a projekciós
neuronokat (gátlás gátlása), csökkentve vagy megszüntetve a továbbítandó fájdalom
ingerületét (kapu zárása) (4.A ábra).20
A kapu nyitásakor a C rostok végzıdésein a projekciós neuronok irányába ugyanakkor
serkentı transzmitter, glutaminsav szabadul fel, aminek hatására gyors excitatorikus
posztszinaptikus potenciál jön létre a projekciós neuronokon. Posztszinaptikusan a
glutaminsavat a projekciós neuronokon elhelyezkedı AMPA, kainsav és NMDA receptorok
fogadják. Az AMPA és kainsav receptorok gyors és éles fájdalmat, az NMDA receptorok
elhúzódó fájdalmat közvetítenek. (Lassú excitatorikus posztszinaptikus potenciált létrehozó
neurotranszmitterek is felszabadulnak, legismertebb közülük a substance-P és a CGRP.) A
perifériás idegsérülés miatt folyamatosan tüzelı C rostok végzıdésein felszabaduló
nagymennyiségő glutaminsav hatására a projekciós neuronokon lévı AMPA és kainsav
receptorok háttérbe szorulnak és az NMDA receptorok tartós aktivitása alakul ki. Az NMDA
receptorok által folyamatosan aktivált projekciós neuronok ráadásul szelektíven toxikusak
lehetnek az inhibíciós interneuronokra, így a leírt szabályozó körben az inhibíciós
interneuronok kiiktatásával megszünhet a fájdalominger negatív szabályozása. Az íly módon
progresszíven fokozódó fájdalominger következménye az agyi struktúrák szenzitizációja, az
allodínia és a hiperalgézia megjelenése.22 A folyamatban számos egyéb receptor (prosztanoid,
kemokin, COX, NOS, stb.) és mechanizmus is részt vesz, az NMDA receptornak azonban
központi szerepe van a centrális szenzitizáció kialakításában.
2.1.2.3. Centrális fájdalom kontroll (analgetikus leszálló pályarendszer)20
A dorzális szarv öt Rexed laminájából öt különféle idegpálya továbbítja a fájdalomingerületet
a felsıbb agyi struktúrákhoz. Ezek a pályák az agy különféle területein végzıdnek, amelyek a
fájdalom ingerre eltérı válaszokat adnak. A talamuszban történik a fájdalom lokalizálása, a
limbikus kéregben történik a fájdalom affektív feldolgozása (emlékezés a fájdalomra, a
fájdalom tolerálása). A nyúltvelı noredrenerg sejtjeiben végzıdı idegpályák a központi
noradrenerg rendszert aktiválják, a hipotalamuszban végzıdık endokrin választ váltanak ki.

14
Az ötödik pályának a fájdalom központi gátlásában van szerepe. A felszálló pálya a hátsó
szarvból közvetlenül a középagy substancia grisea centralisába fut, ahol enkefalin, dinorfin, β-
endorfin és szerotonin tartalmú idegsejtek foglalnak helyet. Az innen visszainduló, a
nyúltvelın keresztülmenı és a hátsó szarvi gátló interneuronokig leszálló pályák vesznek
részt a fájdalom supraspinális gátlásában. A fájdalom centrális gátlása végeredményben
ugyanazokon a hátsó szarvi gátló interneuronokon valósul meg, mint a perifériás
fájdalomcsillapítás. A leszálló rendszer pályája két irányba fut (5. ábra). Az enkefalintartalmú
rostok leszállnak a nyúltvelıbe, ahol
GABAerg interneuronokon végzıdnek,
gátolják azok (gátló)mőködését. A
GABA sejtek a ventrális nyúltvelı
szerotonin, subtance-P és thyreotrop-
releasing hormon tartalmú sejteire
fejtenek ki tónikus gátlást. Fájdalom
esetén a középagyban felszabaduló
enkefalin gátolja a GABA sejteket,
melyeknek gátló hatása alól felszabaduló
5HT-SP-TRH neuronok serkentı hatást
továbbítanak a hátsó szarv enkefalin tartalmú gátló interneuronjaihoz, melyek gátolják a
fájdalomingert felfelé továbbító hátsó szarvi projekciós neuronokat. A leszálló rendszer másik
pályáján dinorfin tartalmú rostok szállnak le a nyúltvelıbe, ahol noradrenerg sejtekbe
végzıdnek. A noradrenerg sejtek axonjai kapcsolatba kerülnek a hátsó szarv gátló
interneuronjaival, aktiválják azokat, gátolva a projekciós neuronok fájdalomtovábbító
képességét.
2.1.3. Receptorok, ioncsatornák és mediátorok a gerincvelı hátsó szarvában
A 6. ábrán a gerincvelı hátsó szarvában megvalósuló perifériás és centrális
fájdalomszabályozás fıbb receptoriális és molekuláris történései láthatók. A nociceptív
afferensek végzıdésein fájdalom inger hatására nagymennyiségő glutaminsav és substance-P
szabadul fel. Ezeket a mediátorokat a posztszinaptikus projekciós neuronok megfelelı
glutaminsav illetve NK-1 receptorai fogadják. Az ábrán látható módon a felszálló pálya
GABAerg és glicinerg inhibíciós interneuronjai, valamint a leszálló pálya noradrenerg
neuronjai képesek a fájdalomingert gátolni. A fent leírt folyamatokat kiegészíti, hogy az
idegrendszer immunrendszerének legfontosabb eleme, a mikroglia különbözı gyulladásos
5. ábra: A fájdalominger supraspinális gátlása Dy=dinorfin-; és ENK=enkefalin-tartalmú idegsejtek a substantia grisea centralisban (SGC); NE=noradrenalin; 5HT-SP-TRH=szerotonin-substanceP-thyreotrop releasing hormon-tartalmú idegsejtek a nyúltvelõ rostroventrális részében
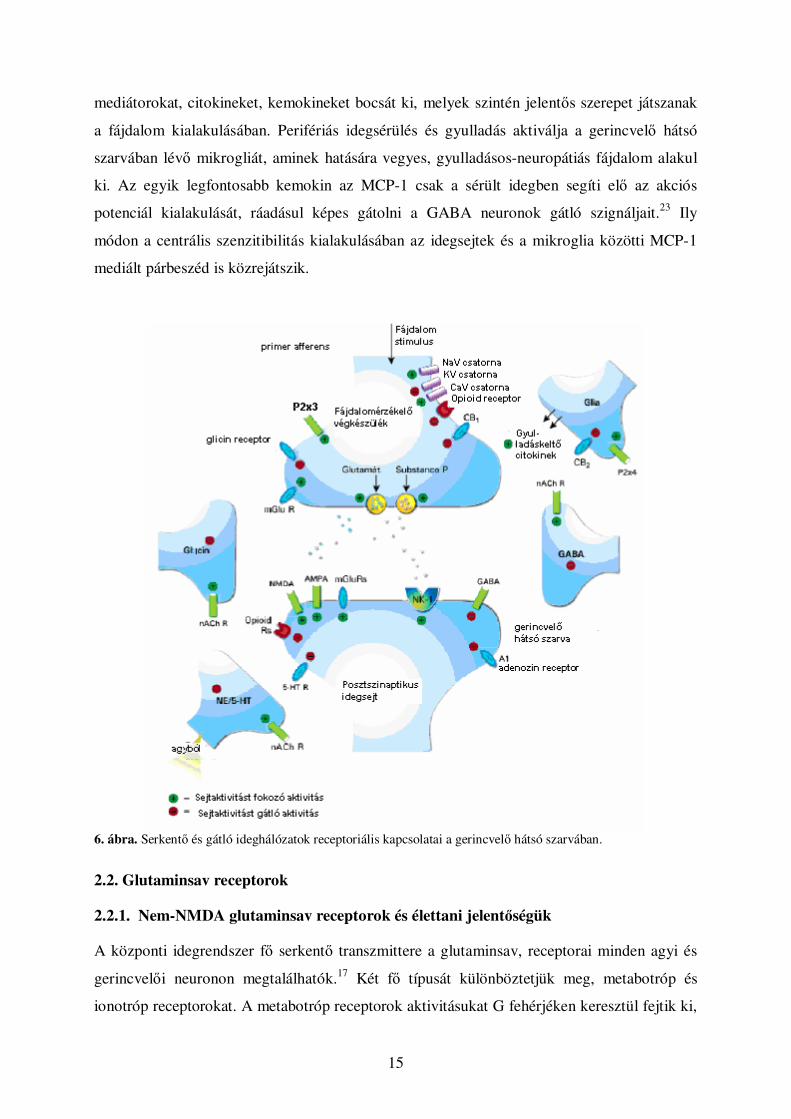
15
mediátorokat, citokineket, kemokineket bocsát ki, melyek szintén jelentıs szerepet játszanak
a fájdalom kialakulásában. Perifériás idegsérülés és gyulladás aktiválja a gerincvelı hátsó
szarvában lévı mikrogliát, aminek hatására vegyes, gyulladásos-neuropátiás fájdalom alakul
ki. Az egyik legfontosabb kemokin az MCP-1 csak a sérült idegben segíti elı az akciós
potenciál kialakulását, ráadásul képes gátolni a GABA neuronok gátló szignáljait.23 Ily
módon a centrális szenzitibilitás kialakulásában az idegsejtek és a mikroglia közötti MCP-1
mediált párbeszéd is közrejátszik.
6. ábra. Serkentı és gátló ideghálózatok receptoriális kapcsolatai a gerincvelı hátsó szarvában.
2.2. Glutaminsav receptorok
2.2.1. Nem-NMDA glutaminsav receptorok és élettani jelentıségük
A központi idegrendszer fı serkentı transzmittere a glutaminsav, receptorai minden agyi és
gerincvelıi neuronon megtalálhatók.17 Két fı típusát különböztetjük meg, metabotróp és
ionotróp receptorokat. A metabotróp receptorok aktivitásukat G fehérjéken keresztül fejtik ki,

16
családjuk három csoportból áll. Az I. csoport (mGluR1, mGluR5) ingerlése a foszfolipáz C-
IP3 jelátviteli utat aktiválja. Az endoplazmatikus retikulum Ca2+ csatornáinak nyitásával
növelik a citoszol Ca2+ koncentrációját. A II. (mGluR2, 3) és III. csoport (mGluR4, 6, 7, 8) tagjai
az adenil cikláz enzim gátlásával csökkentik a cAMP szintézisét. A három altípusba sorolható
ionotróp glutaminsav receptorokat erıs exogén agonistáik alapján nevezték el : NMDA (N-
metil-D-aszparaginsav), kainsav és AMPA (alfa-amino-3-hidroxi-5-metil-izoxazolpropion-
sav). Minhárom receptor glutaminsav receptor és ioncsatorna is egyben (7. ábra).
7. ábra. Glutaminsav receptorok osztályozása filogenetikai fa alapján.25
Szerkezetük közös jellemzıje, hogy négy alegységbıl felépőlı hetero-tetramerek. Az egyes
alegységek három transzmembrán régiót és egy a membránon teljesen át nem menı, a citoszol
felé vissza-kanyarodó régiót tartalmaznak. Nélkülözhetetlenek a szinaptikus plaszticitásban,
több posztszinap-tikus membrán szinaptikus transz-missziójában. Az AMPA receptoroknak
négy altípusát különböz-tetik meg (GluR1-4). Az aktivált AMPA receptorok K+, Na+, kisebb
mértékben Ca2+ ionokra permeábilisak. A GluR2 alegység tartalmú receptorok Ca2+ ionokra
nem permeábilisak, így védve a sejteket a túlzott Ca2+ ion beáramlásból adódó
citotoxicitástól.24 Az AMPA receptorok gyors szinaptikus ingerületátvitelt tesznek lehetıvé.

17
Aktivációjuk és deaktivációjuk gyors és rövid idejő (< 1msec). Tartós agonista jelenlét a
receptor deszenzitizációját okozhatja.26 A kainsav receptorok fıként K+ és Na+ ionokra
permeábilisak, Ca2+ ionokra kevésbé. Ioncsatornájának nyitása és zárása az AMPA
receptoréhoz hasonlóan gyors és rövid idejő. A kainsav receptorok kis affinitású GluR5-7 és
nagy affinitású KA1-2 alegységekbıl épülhetnek fel. Elıbbiek elsısorban a dominsav exogén
agonistára specifikusabbak, míg az utóbbiak a kainsavra. Az AMPA receptorral ellentétben
kevésbé a szinaptikus jelátvitelért, mint inkább a szinaptikus plaszticitásért felelısek. Fıleg
preszinaptikusan fordulnak elı, neurotranszmitterek kibocsátását szabályozzák.27
2.2.2. NMDA receptorok élettani jelentısége28
Az NMDA receptor a központi idegrendszer leggyakoribb glutaminsavfüggı ioncsatornája.
Számos neurológiai betegségben szerepet játszik, ezért intenzíven kutatott, jól jellemzett
terápiás célpont.
Az NMDA receptor, két olyan fontos, a többi ionotróp glutaminsav receptortól eltérı
sajátsággal rendelkezik melyek alapvetıen meghatározzák farmakológiai és funkcionális
tulajdonságait. Az NMDA receptor-ioncsatorna aktivációja az agonista glutaminsavra
lassabban következik be, mivel az ioncsatorna a nyugalmi membránpotenciálon Mg2+ ionok
által blokkolt. Depolarizáció hatására ez a feszültségfüggı blokád megszőnik az ioncsatorna
maximális aktivitását eredményezve.29 Fontos tulajdonsága, hogy különösen nagy
áteresztıképességő Ca2+ ionokra, így a legjobb Ca2+ vezetıképességgel rendelkezik. A
csatorna nyitvatartási idıtartama is nagy, akár több száz msec is lehet. E tulajdonságai miatt
az NMDA receptorok fiziológiás körülmények között fontos szerepet játszanak a szinaptikus
plaszticitás jelenségeiben (idegsejtek migrációja, túlélése, szinaptikus kapcsolatok kialakítása,
fenntartása) és a szinaptikus jelátvitel tartós serkentésében (LTP), melyek fontos elemei a
tanulás és a hosszútávú memória mechanizmusának.30 Az NMDA receptoroknak patológiás
állapotokban is kulcsszerepet tulajdonítanak. Különbözı neurodegeneratív betegségek
mechanizmusának végsı közös eleme a glutamáterg rendszer kóros túlmőködése. Akut
rendellenességek pl. stroke, agyi, gerincvelıi iszkémia vagy epilepsziás roham során az
idegsejtek környezetében a glutaminsav koncentrációja megnı, ami elsısorban az NMDA
receptorok hiperaktivitását váltja ki. Az idegsejtekbe a receptor-ioncsatornán keresztül túlzott
mértékben beáramló Ca2+ ionok az idegsejtek károsodásához és pusztulásához vezetı
biokémiai folyamatokat indítanak be.31 Krónikus betegségek, pl. Parkinson kór, Alzheimer
kór, skizofrénia, krónikus fájdalom esetén is hasonló excitotoxikus állapotok alakulhatnak ki,

18
ezért az NMDA receptorok a neuroprotekció mellett ezen betegségek kiemelkedı molekuláris
célpontjaivá is váltak.
2.2.3. NMDA receptorok felépítése és mőködése
Az N-metil-D-aszparaginsav receptor heteromer alegységekbıl álló tetramer felépítéső
receptor. Kutatásunk kezdetekor még két fajta alegységet azonosítottak, az NR1 és NR2
alegységeket. Késıbb számoltak be az NR3 alegység létezésérıl.31 Az NR1 alegységnek egy
gén által kódolt egy izoformája létezik. Az alegységnek 3 exon eltérı expressziója révén 8
splice variánsa (1-4a, 1-4b) lehetséges, leggyakoribb ezek közül az 1a. Az NR2 alegységnek
négy különbözı gén által kódolt altípusa NR2A, NR2B, NR2C, NR2D van (8. ábra).25 Az
emlısök központi idegrendszerében lévı NMDA receptorban az NR1 alegység mellett
legalább az egyik NR2 alegység altípusnak jelen kell lennie, hogy a receptor mőködıképes
legyen.32
8. ábra. Az NMDA receptor NR1/NR2B heterotetramer formája a módosító kötıhelyekkel.28
Mindegyik alegység három hidrofób transzmembrán régióból (I, III, IV) és a membránban a
citoszol felé hajtőkanyarszerően visszakanyarodó régióból (II) áll. Az ioncsatorna pórusát ez
utóbbi (II) szegmens alkotja. A fehérje extracelluláris N-terminálisán többféle specifikus
affinitással rendelkezı kötıhely, míg az intracelluláris C-terminálisán a receptor
mőködésének sejten belüli szabályozásában résztvevı foszforilációs helyek találhatók. Az
ioncsatorna pórusa Mg2+ ion által blokkolt, depolarizálódás hatására ez a tónikus blokk
megszőnik. A receptor aktiválódásához még két ko-agonista, a glicin és a glutaminsav

19
egyidejő kötése is szükséges, ekkor az ioncsatorna Na+, K+, és Ca2+ ionokra permeábilis lesz.
Az NR1NR2 összetételő receptorokban glicin kötıhelye az NR1 alegység N terminálisán, míg
a ko-agonista glutaminsav kötıhelye az NR2 alegységek N-terminális részén helyezkednek el.
Az agonista kötıhelyeken kívül az NR2 alegységek több allosztérikus modulátor és
antagonista kötıhelyet is tartalmaznak. A különbözı NR2 alegységösszetételő receptorok
eltérı érzékenységgel és farmakológiai profillal viszonyulnak a különféle modulátorokhoz. A
Zn2+ ionok az N terminálison lévı kötıhelyükhöz kapcsolódva feszültségfüggetlen módon az
NR2A tartalmú receptorokat antagonizálják. Nagyobb koncentrációban feszültségfüggı
mechanizmus szerint az ioncsatorna Mg2+ kötıhelyéhez kapcsolódva már az NR2B tartalmú
receptorokat is gátolja. Feszültségfüggetlen módon befolyásolja a receptor mőködését a
környezetében lévı pH és redoxpotenciál is. A pH bázikus irányba történı kismértékő
eltolása vagy redukáló hatású környezet a receptor aktivitását fokozza míg, a pH kismértékő
savas irányban történı eltolása vagy oxidáló hatású környezet a receptor aktivitását gátolja.33
Endogén poliaminok, pl. a spermin és spermidin az NR2B és NR2A alegységeken lévı
poliamin kötıhelyhez kapcsolódva alacsony koncentrációban, feszültségfüggetlen
mechanizmussal fokozzák a receptorok aktivitását. NR2B alegységtartalmú receptorok
esetében az endogén és exogén poliaminok a csatorna nyitvatartási frekvenciájának
növelésével serkentik a receptor mőködését. NR2A alegységtartalmú receptoroknál a
poliaminok közvetve, a glicin affinitásának növelésével fokozzák a receptor aktivitását. Míg
magasabb koncentrációban vélhetıen az ioncsatorna Mg2+ kötıhelyéhez kapcsolódva
feszültségfüggı módon mindkét alegységtartalmú receptor mőködését antagonizálják.25 Az
1980-as évek végén fedezték fel, hogy az amúgy neuroprotektív és értágító hatású alfa-
adrenerg antagonista ifenprodil sperminszenzitiv módon blokkolja az NMDA receptort, így
vélhetıen kapcsolódik a poliamin kötıhelyhez. Késıbb igazolták, hogy az ifenprodil nem a
poliamin kötıhelyhez, hanem tıle független helyhez, az NR2B alegységen megtaláható, róla
ifenprodil kötıhelynek nevezett aktív helyhez kapcsolódik. Az ifenprodil kötıhely
allosztérikus kölcsönhatásban áll a poliamin kötıhellyel és a glicin kötıhellyel.34 A nyitott
ioncsatorna belsejében lévı csatornablokkoló helyhez használatfüggı módon („use-
dependence”, azaz gátló hatásuk az agonista koncentrációjának növekedésével fokozódik)
nemkompetitív antagonisták kötıdhetnek. Ezt az aktív helyet a hozzá nagy affinitással kötıdı
1-(1-fenil-ciklohexil)piperidinrıl PCP-kötıhelynek nevezzük. A PCP kötıhelyhez kis
affinitású vegyületek (pl. a memantin) is kapcsolódnak. Az újonnan felfedezett NR3
alegységnek két altípusa ismert az NR3A, és NR3B. Az NR1/NR3A és NR1/NR3B
receptorokra jellemzı, hogy glutaminsavra érzéketlenek, glicin hatására aktiválódnak.

20
Csökkentik az ioncsatorna vezetıképességét, a Ca2+ ionokat nem engedik át és a Mg2+ ionok
sem blokkolják a csatorna pórusát. A csatorna nem kompetitív antagonistái (MK-801,
memantin) nem gátolják a receptor mőködését.35 A glicin mindkét alegységhez kötıdik, az
NR3A alegységhez azonban nagyságrendekkel nagyobb affinitással vonzódik, mint az NR1
alegységhez. Ezért NR3 nagy affinitású glicin kötıhelyet és NR1 kis-affinitású glicin
kötıhelyet különböztünk meg. Az NR3A alegységtartalmú receptorok farmakológiai
jelentısége még kevéssé ismert, de tekintve a glicin hozzá való kötıdésének szelektivitását
egy NR3A szelektív kompetitív antagonista vagy allosztérikus inhibitor terápiásan értékes
lehet.28
2.2.4. Az NMDA receptorok expressziója
Az NMDA receptor alegységek térben és idıben eltérı génexpressziós mintázatot mutatnak.
In situ hibridizációs technikával bizonyították, hogy rágcsálók agyában leggyakrabban az
NR1 alegység fordul elı.36 NR2 alegységek közül az NR2A altípus expresszálódik a
legnagyobb mértékben, fıként az agykéregben, hippokampuszban, a kisagy kérgében és a
szaglószervi gumókban fordul elı. NR2B alegység leginkább az agykéregben, kisebb
mértékben a köztiagyban és az agytörzsben figyelhetı meg. NR2C alegység elsısorban a
kisagykéreg szemcsesejtjeiben expresszálódik, az NR2D alegység a köztiagy és az agytörzs
régiójában figyelhetı meg.30
Az egyedfejlıdés során az egyes alegységek idıbeli megjelenése jelentısen eltér. Míg az NR1
alegység folyamatosan jelen van, az NR2 alegységek expressziója folyamatosan változik.
Patkány embrionális agyban fıként az NR2B kisebb részben NR2D alegység van jelen.
NR2B alegység az egész agyban kifejezıdik, NR2D alegység csak a köztiagyban és az
agykéregben található meg. Embrionális stádiumban NR2A és NR2C alegység nem
expresszálódik, születés után jelennek meg különbözı agyterületeken. Az alegységek
expressziójának idıbeli változása és agyi régiók szerinti megoszlása az alegységek különbözı
funkciójára utalnak.
2.3. Az NMDA receptorok mint terápiás célpontok
A 2.1.1. fejezetben leírt betegségek neuropátiás fájdalomkomponensének patomechanizmusa
az NMDA receptorok kóros túlmőködésével hozhatók összefüggésbe, ezért a receptor
mőködését gátló anyagok alkalmazásával valószínőleg csökkenthetık, megállíthatók a fenti
kórfolyamatok. Az NMDA receptorok blokkolásának lehetséges célpontjai a 2.2.3. fejezetben
számbavett valamennyi kötıhely:

21
- agonista kötıhelyek: glicin, glutaminsav
- ioncsatorna pórusa
- allosztérikus modulátor régiók (poliamin kötıhely, ifenprodil kötıhely, Zn2+ kötıhely, pH
és redox érzékeny régió)
A felsorolt kötıhelyek terápiás felhasználhatósága eltérı, ezért csak a gyógyszerkutatás által
ígéretesnek tartott célpontokat és azok gátló szereit ismertetem.28
2.3.1. Glicin antagonisták
Az NR1NR2 összetételő receptorokban a glicin kötıhely az NR1 alegységen helyezkedik el,
de a glicin és a glicin agonisták receptorhoz való affinitását az NR2 alegységek is
befolyásolják. A glicin és glicin agonisták legkevésbé az NR2A, leginkább az NR2B
alegységet tartalmazó receptorokhoz kötıdnek. Így a glicin antagonisták is az NR2B
alegységet tartalmazó receptorokon fejtik ki leginkább gátló hatásukat. A glicin kötıhely
egyetlen ismert endogén antagonistája a triptofán egyik metabolikus terméke, a kinurénsav.
Neuroprotektív hatása és a fájdalomcsillapításban betöltött szerepe közismert, hatástartama
gyors kiürülése miatt azonban rövid. A triptofán két különbözı metabolikus úton, két
ellentétes hatású anyaggá, kinurénsavvá és az NMDA agonista kvinolinsavvá alakul.37 A
kutatások elsı idıszakában a kompetitív lebomlási útvonal enziminhibitorokkal való
gátlásával próbálták növelni az endogén kinurénsav koncentrációját. Az elképzelés nem
váltotta be a hozzá főzött reményeket, exogén antagonisták hatékonyabbnak bizonyultak. Elsı
képviselıjük az 5,7-diklór-kinurénsav volt (DCKA). A Glaxo egy 4,6-diklórindol-2-
karbonsav-származékot (GV-150526A), a CoCensys a Novartissal együttmőködve egy 6,7-
diklór-kinoxalin-származékot (ACEA-1021) fejlesztett neuroprotektív indikációval, de a
klinikai vizsgálatok III. fázisában megbuktak. A sikeres állatkísérletes eredmények ellenére
emberben a placebóval összehasonlítva nem voltak kellıképpen hatásosak.
N
O
O
OH
HN
O
O
OH
Cl
ClH H
N
O
OHCl
Cl
O
NH
N
N O
O
NO2
Cl
Cl
H
HN
O
O
OH
OH
kinurénsav kvinolinsav DCKA GV-150526A ACEA-1021

22
Több nagy gyógyszergyár is bekapcsolódott a glicin antagonisták kutatásába de egy idı után
valamennyien abbahagyták a hasonló vegyületek fejlesztését.
2.3.2. Glutaminsav antagonisták (kompetitív NMDA receptor antagonisták)
Az NR2 alegység glutaminsav kötıhelyén ható kompetitív antagonisták szerkezete a
glutatminsavból vezethetı le. A glutaminsav távolabbi karboxil csoportját foszfonsav csoport
helyettesíti. A molekulák a glutaminsavtól általában néhány metiléncsoporttal hosszabbak (D-
AP5). Merevebb szerkezető származékokkal nagyobb aktivitást és szelektivitást értek el. A
legelırehaladottabb munka a Novartis selfotel és az SDZ-EAA-494 jelő vegyületével folyt,
stroke és traumás agysérülés indikációval klinikai vizsgálatokat is folytattak.
O
OH
P
O
OH
OH
H2N
O
OH
P
O
OH
OH
NH
N
O
OHN
P
O
OH
OH
HO
OH
O
H2N
HO
glutaminsav D-AP5 selfotel SDZ-EAA-494
2.3.3. NMDA csatorna blokkolók (nem kompetitív NMDA receptor antagonisták)
Az NMDA csatorna blokkolók nem kompetitív antagonisták, nem a glutaminsav
kötıhelyéhez kapcsolódnak. A nyitott csatornában használatfüggı módon (use dependence)
alakít ki blokádot. Kis és nagy affinitású képviselıiket különböztetjük meg. A kis-közepes
affinitású vegyületek (pl. ketamin, dextrometorfan, memantin) gyors blokkoló kinetiájuknak
és erıs feszültségfüggésüknek köszönhetıen általában jobb terápiás mutatókkal rendelkeznek
mint a nagy affinitású, ám lassú kinetikájú csatorna blokkolók (pl. PCP, MK-801, aptiganel).
A kis affinitású, gyors kinetikával rendelkezı vegyületek egy fiziológiai aktiváció átmenete
alatt gyorsan elhagyják az ioncsatornát millimoláris szinaptikus glutaminsav koncentráció
mellett is, míg patológiás körülmények között, már a mikromoláris glutaminsav koncentráció
hatására kialakuló folyamatos aktivációt is blokkolják.
NH2
H3C CH3
N
CH3O
CH3
H
Cl
HN
O
CH3
ketamin dextrometorfan memantin

23
N
CH3
NHN N
NH
CH3
CH3H
PCP (+)-MK-801 aptiganel
2.3.4. NR2B szelektív NMDA antagonisták
2.3.4.1. Ifenprodil típusú NR2B szelektív NMDA antagonisták38
Az elsıként azonosított NR2B altípus szelektív NMDA anatagonista hatású molekula az
ifenprodil volt. Az ifenprodil (4-[2-(4-benzil-piperidin-1-il)-1-hidroxi-propil]-fenol) tartarát
sója piacon lévı vérnyomáscsökkentık hatóanyaga (Cerocral, Dilvax, Vadilex).39 Az 1973-
ban forgalomba kerülı ifenprodilt α-1 adrenoceptor antagonista hatása alapján fejlesztették a
Synthelabo kutatói.40 Késıbb kiderült, hogy vegyes profilú, számos receptor és ioncsatorna,
többek között az NMDA receptor mőködését is gátolja.41 Nem sokkal ezután kimutatták, hogy
az ifenprodil nem kompetitív, NR2B altípus szelektív antagonistája az NMDA receptornak.42
Az ezen a területen megindult kutatásokban leggyakrabban az ifenprodilt választották
kiindulópontként, aminek eredményeként számos hatásos, hozzá hasonló farmakofórral
rendelkezı molekulát és vegyületcsaládot fedeztek fel.43-45 A Pfizer kutatói megvizsgálták a
két aszimmetriacentrumot tartalmazó ifenprodil sztereoizomer formáinak eltérı aktivitását.46
Az α-1 adrenerg affinitás meghatározásához a standard [3H]-prazozin kötıdési tesztet
használták. Az NMDA antagonizmust egy funkcionális teszt eredményével jellemezték,
melynek során glutaminsav-indukált sejthalál gátlásának mértékét határozták meg patkány
hipokampális neuronok primer tenyészetében (CC). Az eredmények azt mutatták, hogy a (+)-
treo-ifenprodil hatásosabb a CC tesztben, mint a (+)-eritro-ifenprodil, ugyanakkor kisebb az
α-1 adrenoceptorhoz való affinitása.
HO
OH
CH3
N
HO
OH
CH3
N
(+)-eritro-ifenprodil (+)-treo-ifenprodil CC-IC50: 263 nM CC-IC50: 55 nM α-1-IC50: 100 nM α-1-IC50: 843 nM
A (+)-eritro-ifenprodilt kivéve izolálták és megvizsgálták az egyes diasztereomereket is. Azt
találták, hogy a legnagyobb NMDA antagonista hatása a (-)-treo-ifenprodilnak van, amely α-

24
1 receptorokhoz képest kb. ötvenszeres szelektivitással rendelkezik (CC-IC50: 13,3 nM; α-1-
IC50: 629 nM). Az ifenprodilból kiinduló kutatások alapvetı törekvése az NMDA antagonista
hatás erısítése mellett az α-1 receptorokhoz való affinitás csökkentése volt. Érdekes módon
ezt az elvet, miszerint a több támadáspontú szereket szelektívvé lehet tenni, többek között
úgy, hogy a fıhatás kitervezésével egy mellékhatást erısítünk fel új fıhatásnak, csak a
közelmúltban fogalmazták meg mint lehetséges stratégiát (selective optimization of side
activities = SOSA).47 Szerkezet-hatás összefüggések (SAR) felderítése céljából a Pfizer
kutatói ifenprodilszármazékokat állítottak elı. Az ifenprodilon véghezvitt módosításaik
gyengébb NMDA antagonistákat eredményeztek, ráadásul az NMDA és az α-1 receptorokon
való aktivitáskülönbség sem javult a (-)-treo-ifenprodilhoz képest. A baloldali benzolgyőrő
hidroxilcsoportjának hiánya (1) jelentısen csökkentette mindkét teszten az aktivitást, míg a
piperidingyőrő 4-es helyzetében lévı benzilcsoport elhagyása mindkét receptoriális hatás
teljes eltőnésével járt (2). A 3-as vegyület eredményei jelzik, hogy bár az alkil összekötı
láncnak jelentıs szerepe van, a rajta lévı benzil-helyzető hidroxilcsoport jelenléte nem
elengedhetetlen egyik hatás szempontjából sem.
OH
CH3
N
HO
OH
CH3
N
HOCH3
N
1 2 3 CC-IC50: 3700 nM CC-IC50: >10000 nM CC-IC50: 153 nM α-1-IC50: 3400 nM α-1-IC50: >10000 nM α-1-IC50: 130 nM
A Synthélabo Recherche kutatói az ifenprodil sztereoizomerek NMDA receptor
alegységszelektivitását vizsgálták. Az egyes antipódok inhibíciós képességét NR1a/NR2A és
NR1a/NR2B alegységeket tartalmazó rekombináns NMDA receptorokat expresszáló Xenopus
oocytában határozták meg patch-clamp technikával.48 Méréseik szerint mind a négy
sztereoizomer jóval hatásosabb antagonistája az NR1a/NR2B alegységet tartalmazó
receptornak (IC50: < 0,8 µM), mint az NR1a/NR2A receptornak (IC50: > 100 µM). Az
NR1a/NR2B receptoron a (+) eritro- és (-)-treo-ifenprodil (IC50: 0,21 µM és 0,22 µM)
négyszer potensebb volt mint a (-)-eritro- és (+)-treo-ifenprodil (IC50: 0,81 µM és 0,76 µM).
Ezek az eredmények azt mutatják, hogy az ifenprodil sztereoizomerjei bár kismértékő, de
szignifikáns sztereoszelektivitási különbséget mutatnak az NR1a/NR2B receptorokon.
25
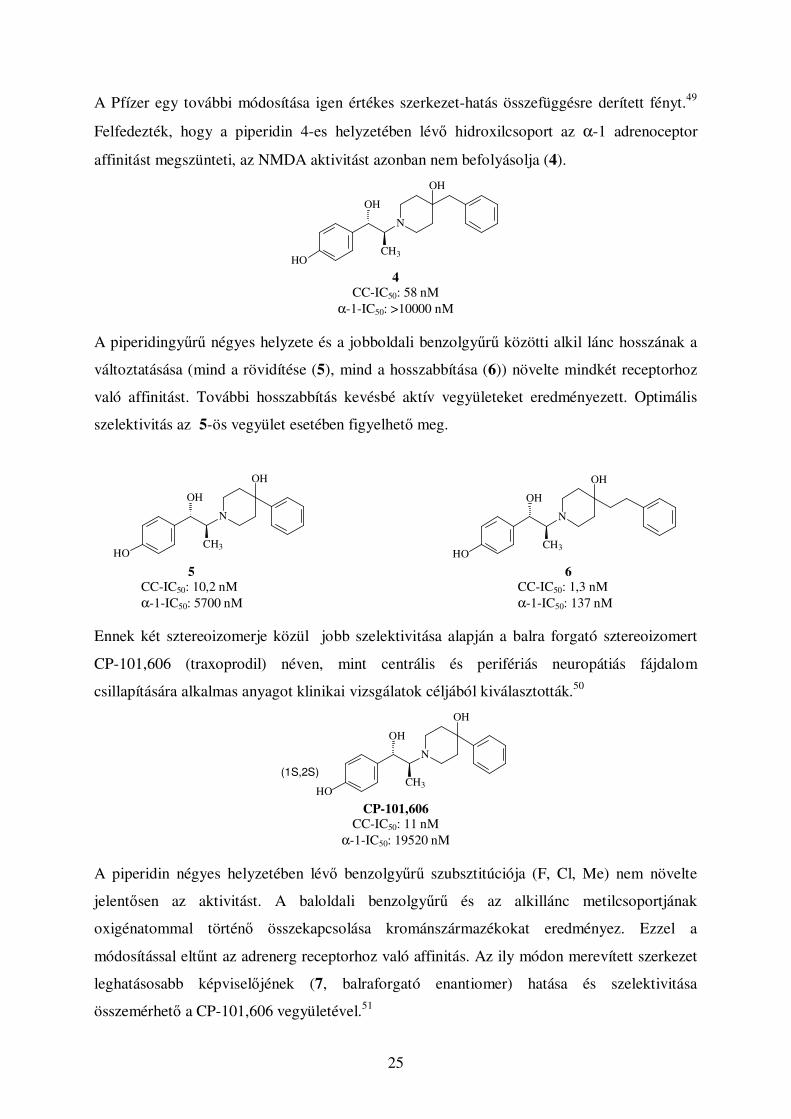
A Pfízer egy további módosítása igen értékes szerkezet-hatás összefüggésre derített fényt.49
Felfedezték, hogy a piperidin 4-es helyzetében lévı hidroxilcsoport az α-1 adrenoceptor
affinitást megszünteti, az NMDA aktivitást azonban nem befolyásolja (4).
OH
CH3
N
OH
HO 4
CC-IC50: 58 nM α-1-IC50: >10000 nM
A piperidingyőrő négyes helyzete és a jobboldali benzolgyőrő közötti alkil lánc hosszának a
változtatásása (mind a rövidítése (5), mind a hosszabbítása (6)) növelte mindkét receptorhoz
való affinitást. További hosszabbítás kevésbé aktív vegyületeket eredményezett. Optimális
szelektivitás az 5-ös vegyület esetében figyelhetı meg.
OH
CH3
N
OH
HO
OH
CH3
N
OH
HO 5 6
CC-IC50: 10,2 nM CC-IC50: 1,3 nM α-1-IC50: 5700 nM α-1-IC50: 137 nM
Ennek két sztereoizomerje közül jobb szelektivitása alapján a balra forgató sztereoizomert
CP-101,606 (traxoprodil) néven, mint centrális és perifériás neuropátiás fájdalom
csillapítására alkalmas anyagot klinikai vizsgálatok céljából kiválasztották.50
OH
CH3
N
OH
HO
(1S,2S)
CP-101,606
CC-IC50: 11 nM α-1-IC50: 19520 nM
A piperidin négyes helyzetében lévı benzolgyőrő szubsztitúciója (F, Cl, Me) nem növelte
jelentısen az aktivitást. A baloldali benzolgyőrő és az alkillánc metilcsoportjának
oxigénatommal történı összekapcsolása krománszármazékokat eredményez. Ezzel a
módosítással eltőnt az adrenerg receptorhoz való affinitás. Az ily módon merevített szerkezet
leghatásosabb képviselıjének (7, balraforgató enantiomer) hatása és szelektivitása
összemérhetı a CP-101,606 vegyületével.51

26
OH
N
OH
HO O
F
7
CC-IC50: 58 nM
Az eddigi adatok alapján a következıképpen összegezhetık az ifenprodil típusú NMDA
antagonisták farmakofór jellemzıi: két benzolgyőrőt egy meghatározott hosszúságú lánc köt
össze. Az egyik benzolgyőrőn egy hidroxilcsoport jelenléte szükséges. Bár az összekötı lánc
szubsztituenseit és azok sztereokémiáját részletesen vizsgálták, egyértelmő következtetés nem
vonható le az eredményekbıl.
A Merck KGaA kutatói kiemelt vegyületük (8, EMD 95885) és az ifenprodil összehasonlító
receptor kötıdési adatait tették közzé egyik tanulmányukban.52 Patkány cortexben
radioligandként [3H]-ifenprodilt használva 8 vegyület nagyobb affinitással kötıdött az
ifenprodil kötıhelyhez (IC50: 3,9 nM), mint maga az ifenprodil (IC50: 23,3 nM).
O
NO
O
NF
H
8
Mint késıbb kiderült ez a vegyület fontos mérföldkı volt az ifenprodil farmakofór
fejlıdésében. Megmutatta, hogy nem szükséges királis szénatom jelenléte az összekötı
láncban. A vegyületnek gyenge az affinitása az α-1 adrenoceptorhoz (IC50: 332 nM).38
Ráadásul az eddig alkalmazott fenolos hidroxilcsoportot sikerült helyettesíteni a
metabolikusan stabilabb, hasonlóan hidrogénkötés donor tulajdonságú NH-csoportot
tartalmazó benzoxazolinon győrővel. Ez a vegyület olyan 1-szubsztituált benzil-piperidin
NR2B altípusszelektív NMDA antagonisták prototípusává vált, ahol egy hidrogénkötés
donorcsoportot tartalmazó benzolgyőrő három atom hosszúságú láncon keresztül kapcsolódik
a piperidin nitrogénjéhez .
Az F. Hoffmann-La Roche Ltd. kutatói szintén sok energiát fektettek az ifenprodil típusú
farmakofór fejlesztésébe. Egyik elsı vezérmolekulájuk az ifenprodil homológ 9 (Ro 25-6981,
((1R,2S)-3-(4-benzil-piperidin-1-il)-1-(4-hidroxi-fenil)-2-metil-1-propanol), számos in vitro
és in vivo tesztben jobbnak bizonyult az ifenprodilnál.53 Racém formájának triciált analogonja
az NR2B szelektív NMDA antagonisták azonosítására szolgáló kötıdési teszt leggyakrabban
alkalmazott radioligandja lett.54

27
HO
N
OH
CH3
9
Hasonló aktivitást mutat 10-es számú kiemelt vegyületük (Ro 8-4304). A molekula
hosszúsága megegyezik 9 hosszúságával, de a baloldali benzolgyőrő és a piperidin nitrogénje
között lévı lánc egy oxigén atommal hosszabb, kompenzálva a piperidin 4-es helyzete és a
jobboldali benzolgyőrő közötti metiléncsoport elhagyását. Hasonló affinitással rendelkezik az
NR2B tartalmú NMDA receptorokhoz, mint az ifenprodil, de gyorsabb on/off kinetika
jellemzi.55
O
NH2
O N
F
OH
10
A 9-es vegyület in vitro aktivitása az ifenprodiléhoz hasonló, azonban in vivo
kardiovaszkuláris mellékhatás jelentkezik, amely szignifikáns α-1 adrenoceptor antagonista
hatásával (Ki: 0,18 µM [3H]-prazozin kötıdési tesztben56) állhat összefüggésben. Az NMDA
és az α-1 aktivitás szétválasztását célul kitőzı optimalizálási programjuk kiindulási pontja 9
vezérmolekula volt.57 Kitartó munkájuk eredménye a 11-es vegyület, melynek NR2B
receptorhoz való affinitása nem javult ugyan számottevıen a 9-es vegyülethez képest (Ki: 4,9
nM szemben a 5,6 nM értékkel a [3H]-Ro 25-6981 kötıdési tesztben54), az α-1
adrenoceptorhoz való affinitása azonban jelentısen lecsökkent (Ki: 7,30 µM). A két aktivitás
közötti különbség íly módon történı növelése ebben az esetben is a piperidin 4-es helyzetében
lévı hidroxilcsoport jelenlétével magyarázható.
HO
NCH3
OH
OH
11
További módosítások vezettek a 12 (Ro 63-1908) vegyülethez, melyet kisebb NR2B és α-1
aktivitásbeli különbséget jellemez (IC50: 10 nM a [3H]-Ro 25-6981, és IC50: 3,5 µM a [3H]-
prazozin kötıdési tesztben), de egyéb tulajdonságai miatt részletes in vitro és in vivo

28
vizsgálatoknak vetették alá.58, 59 Sajnos ez a vegyület hatásosan blokkolja a hERG kálium-
csatornát (EC50: 0,6 µM), mely fontos szerepet játszik a szív akciós potenciál repolarizációs
fázisában. Blokkolása QT megnyúlást és szívritmuszavart okoz.60
Az éteres O helyettesítése SO2-csoporttal, továbbá a OH-csoport áthelyezése a piperidin 4-es
helyzetébıl a hármas helyzetbe egy aktívabb és szelektívebb NMDA antagonistát, a 13
vegyületet eredményezte, mely még inkább elkülönítette az NMDA (14 nM) és a hERG (24
µM) aktivitást.61
HO
ON
CH3
OH
HO
SN
CH3O O
OH
12 13
Az ifenprodil típusú farmakofór modell fejlıdéséhez különösen értékes módon járultak hozzá
az University of Oregon, a CoCensys Inc. és a Parke-Davis Pharm. Res. együttmőködı
kutatói. Bebizonyították, hogy a piperidingyőrő és az alkil lánc szubsztitúciója nem feltétele
sem a receptorhoz való kötıdés mértékének, sem a szelektivitásnak. Emellett ık írták le az
elsı farmakofór modellt. Az ifenprodilból és tercier helyett szekunder amint tartalmazó
analogonjából, a nylidrinbıl kiindulva állapították meg a hatás szempontjából kritikus
részeket (9. ábra). Klónozott patkány NMDA receptor alegységek bináris kombinációit
(NR1a-NR2A, NR1a-NR2B, NR1a-NR2C) expresszáló Xenopus oocytákban vizsgálták az
aktivitást és a szelektivitást.62 Az alkoholos hidroxilcsoportok és a metilcsoportok elhagyása
(14, 15) az IC50 értékek nagyságrendi megváltozását nem eredményezték, jelezve, hogy a lánc
szubsztituálása nem alapvetı követelmény. A 15 származék fenolos hidroxilcsoportjának
elhagyása az ifenprodiltól tízszer gyengébben ható molekulát eredményezett. További
egyszerősítéseket hajtottak végre a molekulákon annak érdekében, hogy meghatározzák mely
elemek szükségesek a hatás szempontjából. Elıállították a piperidingyőrő 3. és 4. szénatomja
közötti kötés felbontásával levezethetı molekulát (17). A győrő felnyitása csak kis mértékben
csökkentette az aktivitást, vagyis a konformációs kényszer megszüntetése nem zárja ki a
szelektív NR2B antagonista hatást. Legaktívabb származékok a szekunder aminok voltak. A
nitrogén atom és a receptor közötti elektrosztatikus kölcsönhatás nagyon fontos, oxigén atom
beépítése a nitrogén helyére (14 → 16) mintegy negyvenszeres aktivitáscsökkenést
eredményezett. Különbözı hosszúságú szénlánc beépítésével változtatták az aromás győrők
és a nitrogén közötti távolságot.
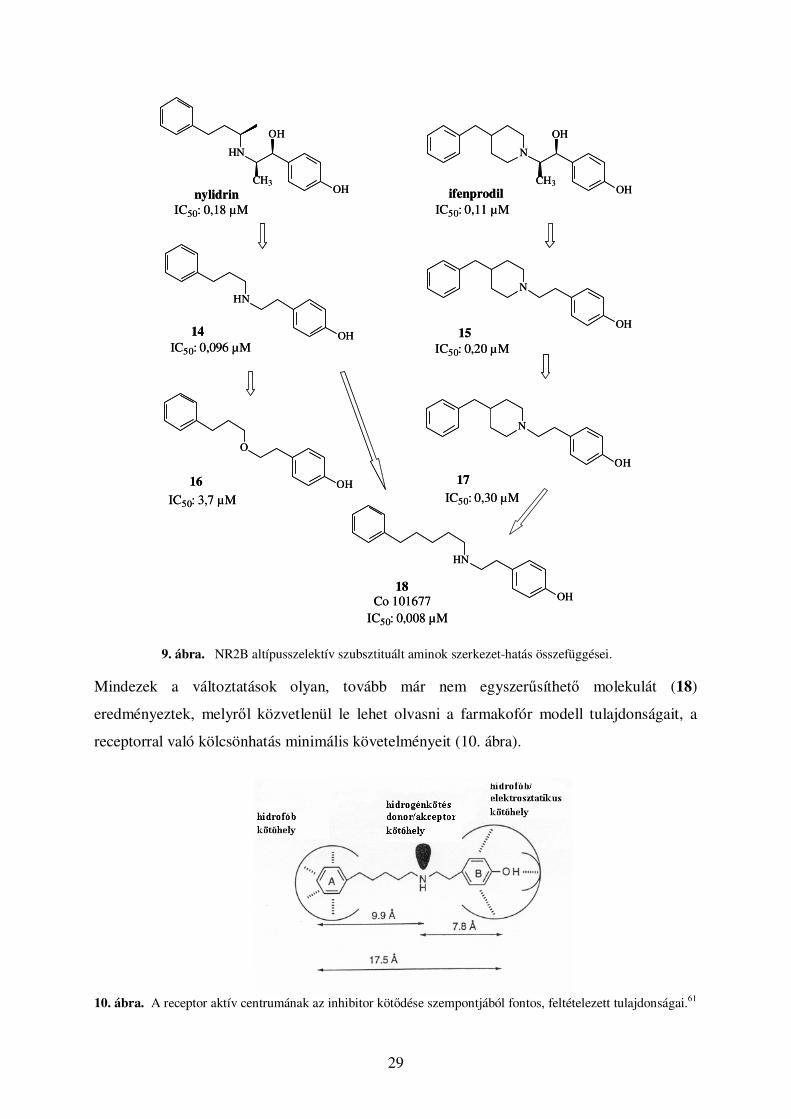
29
OH
OH
CH3
N
OH
OH
CH3
HN
nylidrinIC50: 0,18 µM IC50: 0,11 µM
ifenprodil
OH
HN
OH
N
IC50: 0,20 µMIC50: 0,096 µM
OH
O
IC50: 3,7 µM
OH
N
IC50: 0,008 µM
OH
HN
14 15
16 17
18
IC50: 0,30 µM
Co 101677
OH
OH
CH3
N
OH
OH
CH3
HN
nylidrinIC50: 0,18 µM IC50: 0,11 µM
ifenprodil
OH
HN
OH
N
IC50: 0,20 µMIC50: 0,096 µM
OH
O
IC50: 3,7 µM
OH
N
IC50: 0,008 µM
OH
HN
14 15
16 17
18
IC50: 0,30 µM
Co 101677
9. ábra. NR2B altípusszelektív szubsztituált aminok szerkezet-hatás összefüggései. Mindezek a változtatások olyan, tovább már nem egyszerősíthetı molekulát (18)
eredményeztek, melyrıl közvetlenül le lehet olvasni a farmakofór modell tulajdonságait, a
receptorral való kölcsönhatás minimális követelményeit (10. ábra).
10. ábra. A receptor aktív centrumának az inhibitor kötıdése szempontjából fontos, feltételezett tulajdonságai.61

30
Ezek szerint kellı nagyságú NR2B aktivitást és szelektivitást el lehet érni fenilalkil
csoportokkal helyettesített szekunder aminokkal. Az aromás győrők feltételezhetıen a
receptor egy-egy hidrofób zsebébe illeszkednek, míg a nitrogén elektrosztatikus
kölcsönhatásba lép a receptor megfelelı részével. A fenolos hidroxilcsoport jelenléte az egyik
aromás győrőn esszenciális a hatás szempontjából, vélhetıen hidrogénhidas kötéssel
kapcsolódik a receptor megfelelı részéhez. A fenolos hidroxilcsoport oxigénje és az A győrő
para-helyzető szénatomja közötti optimális távolság 17,5 Å, ami 3 Å-mel rövidebb az
ifenprodil esetében számított értéknél. Ezen belül az oxigén és nitrogén távolsága 7,8 Å.
Az egyszerősítések irányába haladó, egyfajta optimalizáció volt ez a tanulmány, melynek
szerkezet-hatás összefüggései a maximális hatás minimális követelményeit leíró farmakofór
modellt határozták meg. Az optimalizáció mindemellett az abban az idıben a leghatásosabb
NR2B szelektív NMDA antagonista hatású anyagot is szolgáltatta (18). A vegyületcsaládot
szabadalmi bejelentésben nem védték, aminek vélhetıen az volt az oka, hogy bár az NMDA
receptoron szelektíven fejti ki hatását, több más receptoron és ioncsatornán is aktív lehet.
Méréseink szerint a 18 bisz-arilalkil-származék az α-1 és az 5HT2A receptorokon is hat. Ez is
megerısíti azt az általános felfogást, miszerint egy merevebb, kevesebb konformerrel
rendelkezı struktúra specifikusabban viselkedik biológiai rendszerekben.
Ennek a minimalista struktúrának a merevített változatai (19, 20) is hasonlóan aktívak.63
N
HO HO
N
19 20
NR2B-IC50: 17 nM NR2B-IC50: 22 nM
A szerkezet további merevítése (21 és 22) az aktivitás kismértékő csökkenését
eredményezte.64
H
HO
NN HO N
N
H
21 22
NR2B-IC50: 87 nM NR2B-IC50: 50 nM
Bár az University of Oregon, a CoCensys Inc. és a Parke-Davis Pharm. Res. kutatóinak közös
tanulmánya jelentıs elırelépés volt a farmakofór modell fejlıdésében, megfelelıen szelektív,

31
fejleszthetı molekulákat nem eredményezett. Annak ellenére, hogy a modell a 4-benzil-
piperidinnek nem tulajdonít különös jelentıséget, csak mint a benzolgyőrő és a nitrogén
közötti megfelelı távolságot biztosító szerkezetként értékeli, új NR2B szelektív NMDA
antagonisták elıállításakor a 4-benzil-piperidinek a továbbiakban is kedvenc építıelemek
maradtak. Érdekes megjegyezni, hogy a Roche által felfedezett és Ro 63-1908 számon
fejlesztett 12-es számú vegyületet a CoCensys/Parke-Davis kutatói szintén azonosították,
ráadásul védelmére hét hónappal korábban tettek szabadalmi bejelentést (Co 101244/PD
174494).65 Ebben leírják, hogy a kívánt elınyös tulajdonságok az összekötı láncban lévı
sztereocentrumok nélkül is elérhetık.
A 23-as molekulából (Co 101071) kiindulva alternatív hidrogénkötés donor tulajdonságú
csoportokkal, benzimidazolonokkal és hidantoinokkal próbálkoztak.66 Mindkét származék
esetében megvizsgálták a különbözı hosszúságú összekötı láncot tartalmazó homológok
aktivitását. Optimális lánchosszúságú képviselıik alapján elmondható, hogy a
benzimidazolon (24) képes helyettesíteni a fenolt, míg hidantoin (25) nem. Ez újabb
bizonyíték arra, hogy az NR2B receptor aktivitáshoz az aromás győrőre és a hidrogénkötés
donorra is szükség van.
HO
ON
23
NR2B-IC50: 25 nM
N N
NOH
N
NOH
O
N
24 25
NR2B-IC50: 95 nM NR2B-IC50: 1000 nM
Másik közleményükben tioéterekbıl kiinduló hasonló kísérleteikrıl adnak számot.67
Néhány heterociklus és kondenzált heterobiciklus mellett a 27 aminotriazol-származék is
aktívabbnak bizonyult a kiinduló 26 fenolszármazéknál.
HHO
SN
SN
NN
NH2N
26 27
NR2B-IC50: 100 nM NR2B-IC50: 35 nM
A CoCensys Inc. és a Warner-Lambert együttmőködésében elıállított 28-as számú
vegyületben (CI-1041, besonprodil, (+)-6-[2-[4-(4-fluorbenzil)piperidin-1-il]etilszulfinil]-

32
benzoxazol-2(3H)-on) a 8-as számú rokonvegyülethez hasonlóan a benzoxazolon játsza a
fenol szerepét.68
H
SN
O
FO
NO
28
NR2B-IC50: 6,6 nM Kísérleteket végeztek a piperidin és a baloldali benzolgyőrő közötti lánc acetiléncsoporttal
történı merevítésével. A 29-es számú, viszonylag gyenge aktivitású NR2B szelektív
antagonistát egy vegyülettár in vitro szkrínelésekor találták.69
N
29
NR2B-IC50: 4,7 µM
Ahogy várható volt, a baloldali benzolgyőrő kiegészítése hidrogénkötés donor tulajdonságú
csoporttal aktívabb vegyületeket eredményezett. A legaktívabb származékok a 4-hidroxi 30 és
31 analogonok voltak. Sajnos ez a vegyületcsalád az α-1 adrenoceptorok és a dopamin D2
receptorokhoz is affinitást mutatott. A rövidebb, három atom hosszúságú összekötı láncot
tartalmazó 31 származékban válik szét leginkább a fıhatás a mellékhatástól.
N
HOHO
N
30 31
NR2B-IC50: 0,17 µM NR2B-IC50: 0,10 µM α-1-IC50: 0,58 µM; D2-IC50: 0,29 µM α-1-IC50: 1,5 µM; D2-IC50: 1,7 µM
Fenolos hidroxilcsoport helyett heterociklusos fenol bioizosztérek alkalmazásával próbálták
növelni a metabolikus stabilitást.70 A heterobiciklusos származékok közül a benzimidazolinon
(32) volt a legaktívabb, ugyanakkor szelektív és orálisan felszívódó vegyület.

33
H N
N
NO
H 32
NR2B-IC50: 0,0053 µM α-1-IC50: 0,5 µM; D2-IC50: 2,6 µM
A vegyületcsalád további optimalizációja során az NR2B receptorhoz való affinitást [3H]-ifenprodil radioligand kötıdési tesztben határozták meg natív patkány agyat felhasználva.71
H N
N
NO
F
N
N
N
NO
F
H H
H
33 34
IC50: 3 nM IC50: 2 nM
A nagy affinitással és NR2B szelektivitással rendelkezı 33-as vegyület hatékonynak
mutatkozott Parkinson-kór és neuropátiás fájdalom modellekben. A molekula további
módosítása hasonlóan potens vegyületeket eredményezett (pl. 34).
Az University of Oregon, CoCensys, Parke-Davis közös kutatócsoportja azt a további értékes
megfigyelést tette, hogy a bázikus nitrogén jelenléte nem feltétele az NMDA receptor
antagonizmusnak. N-(2-feniletil)-fahéjsavamidokat állítottak elı melyeket NMDA
antagonistaként teszteltek Xenopus oocytában minhárom altípusra (NR1a/2A-C). A 35-ös
vegyület potens és NR2B szelektív antagonistának bizonyult.72
H
HO
N
O
Cl
35
NR2B-IC50: 170 nM
A vegyületcsalád optimalizációja során még aktívabb analogonokat is találtak, például a 36 és
37 vegyületeket.73
HN
O
HO
N
O
HO 36 37
NR2B-IC50: 77 nM NR2B-IC50: 120 nM
Funkcionális tesztben a 36-os vegyület az ifenprodilnál (IC50: 110 nM) potensebbnek
mutatkozott, a (+)-5 (CP-101,606) vegyülettel (IC50: 73 nM) azonos aktivitással bírt. A 37-es

34
vegyület az ifenprodil fejlıdésének új szintjét képviseli. Benzil-piperidin három atom
hosszúságú láncon keresztül kapcsolódik egy fenol négyes helyzetéhez olymódon, hogy a
molekulában lévı nitrogén nem bázikus. A jelentısége ennek a fejleménynek abból a
megfigyelésbıl ered, hogy a bázikus nitrogént tartalmazó természetes ligandumok (pl.
adrenalin, dopamin, szerotonin stb.) receptorai a bázikus nitrogént tartalmazó vegyületeket
részesítik elınyben. A bázikus nitrogén jelenléte sok esetben elıfeltétele a jó affinitásnak
ezekhez a mellékhatásokért felelıs receptorokhoz, vagyis a farmakofór modelljük tartalmazza
a bázikus nitrogént. Ebbıl az következik, hogy ha egy NR2B aktív vegyületcsalád nem
tartalmaz bázikus nitrogént, nagy valószínőséggel kevesebb mellékhatással fog rendelkezni,
mint a bázikus nitrogént tartalmazók.
A Merck Research Laboratories kutatói szintén állítottak elı ifenprodil farmakofórral
rendelkezı molekulákat, eredményeiknek azonban csak egy részét közölték. Vegyülettárukat
tesztelve egy benzimidazol származékot (38) azonosítottak.74 Másik vezérmolekulájukat (39)
egy száztagú benzimidazol könyvtárból azonosították. Egyik molekula sem tartalmaz
hidrogénkötés donorcsoportot a terminális benzolgyőrőkön. Az optimalizáció során a
baloldali benzolgyőrőt kiegészítették hidrogénkötés donorcsoporttal. A vegyületeket NR2B
szelektív kötıdési tesztben és sejt alapú Ca2+ influxot meghatározó funkcionális tesztben75
mérve mindkét vegyületcsaládban nagyon potens képviselıkhöz jutottak.
H
N
NN
N
N
O
H 38 39 NR2B kötıdés Ki: 420 nM; Ca2+-IC50: 710 nM NR2B kötıdés Ki: 260 nM; Ca2+-IC50: 200 nM hERG-IP: 1400 nM; α-1-IC50: 2800 nM hERG-IP: 2000 nM; α-1-IC50: 4200 nM
A legaktívabbak a 40 és 41-es vegyületek voltak, melyek ráadásul jó szelektivitással
rendelkeztek az NMDA receptor NR2A, NR2C és NR2D altípusával, a hERG csatorna
aktivitással és az α-1 adrenerg kötıdéssel szemben. A 41-es vegyület kiválóan aktív volt a
karragén-indukált mechanikai hiperalgézia tesztben patkányban és jó farmakokinetikai
tulajdonságokkal bírt kutyában.
N
NN
HO
F
N
N
ONS
CH3
O O
H
H
H 40 41 NR2B kötıdés Ki: 0,85 nM; Ca2+-IC50: 9,7 nM NR2B kötıdés Ki: 0,68 nM; Ca2+-IC50: 0,72 nM hERG-IP: 2900 nM; α-1-IC50: 730 nM hERG-IP: 120 nM; α-1-IC50: 4000 nM

35
Azt a jelenséget, miszerint a bázikus nitrogén nem feltétele az altípusszelektív NMDA
receptor antagonisták hatásosságának a Merck & Co. Inc. is vizsgálta. A egyik szabadalmi
bejelentésükben 42 és 43 jellemzı példákkal N-benziloxikarbonil-piperidin-származékokat
védenek.76 Biológiai adatokat nem közölnek, saját méréseink szerint77 ezek a vegyületek
hatásos NR2B receptor antagonisták.
H
O
N
N
O
OHO
O
N
N
O
ONO
H
H
42 43 NR2B-IC50: 4,8 nM NR2B-IC50: 6,0 nM
Az irodalom áttekintésével az a következtetés vonható le, hogy az elmúlt tíz évben több
kutatóegység is óriási erıfeszítéseket tett annak érdekében, hogy az ifenprodilt olyan NR2B
altípusszelektív NMDA receptor antagonistává alakítsák, mely más receptorokhoz nem
kötıdik számottevıen. Tanulmányaik alapján a következı megállapítások tehetık az
ifenprodil farmakofór modellt illetıen:
A leghatásosabb szerkezetek két terminális benzolgyőrőt tartalmaznak, melyek közül az egyik
egy metabolikusan stabil hidrogénkötés donorcsoportot visel. A két benzolgyőrőt elınyösen
bázikus csoportot nem tartalmazó, különbözı hosszúságú és heteroatom-tartalmú lánc köti
össze. Ez az összekötı lánc nem egyszerően távtartó, hanem fontos szerepet tölt be a
receptorral való kölcsönhatásban.
Az elmúlt idıszakban számos, az ifenprodil farmakofór modellbe illeszkedı in vitro és in vivo
aktív, orálisan is hatásos, biztonságos vegyület állítottak elı, melyek közül a közeljövıben új
gyógyszer forgalombahozatalát is remélhetjük.
2.3.4.2. Nem ifenprodil típusú NR2B szelektív NMDA antagonisták
Az elmúlt években több, az ifenprodil farmakofór modellbe nem illeszkedı molekulatípust is
azonosítottak. A legtöbb adatot velük kapcsolatban szőkszavú szabadalmi bejelentésekben
tették közzé. Részletes szerkezet-hatás összefüggések ismerete nélkül nehéz egy közös
farmakofór modellt felállítani, ennek ellenére néhány egyedi kivételtıl (pl. fehérjék)
eltekintve több hasonló szerkezeti elem is felfedezhetı ezekben a molekulákban. Nem
tartalmaznak hidrogénkötés donorcsoportot és a legtöbb esetben hiányzik belılük az
ifenprodil típusú NR2B szelektív NMDA antagonistákra korábban jellemzı bázikus nitrogén.
Meglehetısen kompakt molekulák, általában két terminális arilcsoportot tartalmaznak,

36
melyeket egy sokszor győrőbe zárt, merev, rövid lánc köt össze. A legszembetőnıbb közös
szerkezeti elem az amidincsoport, az összes molekula leírható 44 általános képlettel.
N R4
R3N
R2
R1
44
Az elsı NR2B szelektív NMDA antagonista hatású amidin-típusú vegyületcsaládot a Merck
Sharp & Dohme kutatói azonosították. Új vegyületek tesztelése során az E-N1-
(benzil)fahéjsav-amidin (45) jelentıs affinitást mutatott az NR2B-tartalmú receptorokhoz.78
Mivel ez a molekula az ifenprodil farmakofór modelltıl eltérı szerkezető, ezért az NR2B
szelektív NMDA antagonista hatású vegyületek új típusú, reménykeltı jelöltje lehet. Ez a
felismerés egy új gyógyszerkémiai programot indított el a Mercknél. A vegyületcsalád
optimalizációja során legaktívabbnak a 2-metoxi-benzil-származék (46) adódott.
N
NH
HN
NH O
H
45 46 NR2B kötıdés Ki: 9 nM NR2B kötıdés Ki: 0,7 nM
Mivel a molekula sztiril szerkezeti része potenciális Michael akceptor, rögtön megindult az
ezt a lehetıséget kizáró rokon szerkezetek keresése. Így találtak rá a benzil-benzamidin
vegyületcsaládra, amelynek szubsztituálatlan képviselıje (47) hatástalan volt, megfelelıen
helyettesített benzolgyőrőkkel viszont aktív származékokat sikerült elıállítaniuk.79 A
vegyületek aktivitását [3H]-ifenprodil-kötıdési tesztben és teljes sejtes patch-clamp technikán
alapuló funkcionális esszében határozták meg. Funkcionális tesztben leghatásosabb a
benzilcsoport 2-es helyzetében szintén metoxicsoporttal szubsztituált 48 származék volt.
H HN
NH
N
NH O
CF3O
47 48 NR2B kötıdés Ki > 15000 nM NR2B kötıdés Ki: 5,7 nM NR2B funkcionális teszt IC50 : 4,1 nM
A vegyületcsalád aktív tagjai általában jó orális farmakokinetikával rendelkeztek, és a
karragén-indukált hiperalgézia tesztben rágcsálókban hatásosak, ugyanakkor a nemszelektív
NMDA antagonistákra jellemzı lokomotoros mellékhatástól mentesek voltak. A

37
farmakokinetikai paraméterek különösen szubsztituensfüggı jellemzıknek bizonyultak. A
kötıdési teszten legaktívabb 49 származék klóratomjait metilcsoportra cserélve (50) a felezési
idı kevesebb mint negyedére, a biohasznosulás a töredékére csökkent. Ugyanakkor az 51
vegyület trifluormetil-csoportja szokatlan módon 100 %-os biohasznosulást kölcsönzött a
nagyon poláris amidincsoportot tartalmazó molekulának is. A közölt adatok ismét
rávilágítanak arra, hogy a biohasznosulásnak (BA) milyen fontos szerepe van a jó in vivo
hatás elérésében. A funkcionális tesztben közepes hatékonyságú (IC50 : 24 nM), 68 %
biohasznosulást elérı 49 származék a karragén-indukált hiperalgézia tesztben is közepes
hatású (ED50: 16 mg/kg). Az in vitro aktívabb (IC50 : 4,2 nM) de alig felszívódó (BA : 4%) 50
in vivo hatástalan. Míg az in vitro fele olyan aktív 51 a teljes felszívódásának köszönhetıen
kiváló in vivo aktivitással rendelkezik.
H HN
NH
CF3O
Cl
Cl
N
NH CF3
CF3O
HN
NH
CF3O
CH3
CH3 49 50 51 NR2B Ki: 0,6 nM; IC50 : 24 nM NR2B Ki: 1,2 nM; IC50 : 4,2 nM NR2B Ki: 72 nM; IC50 : 47 nM BA: 68 %; T1/2 : 420 perc, BA: 4 %; T1/2 : 92 perc, BA: 100 %; T1/2 : 147 perc ED50: 16 mg/kg ED50: >30 mg/kg ED50: 5,5 mg/kg
Hamarosan a Hoffmann La Roche is jelentkezett egy az amidin szerkezeti elemet rejtetten
tartalmazó molekulacsaláddal. 52 triazolszármazékuk a [3H]-Ro 25-6981 kötıdési tesztben
kiváló affinitást mutatott.80 A vegyületcsaládot jobban megvizsgálva derült ki, hogy a triazol
összekötı elem különbözı heterociklusokkal, pl. piridazinnal (53) vagy imidazollal (54) való
helyettesítése is aktív molekulákat eredményez.81, 82
N
N
N
N
Cl
CH3
H2N
O H3C
NN
N
NH2 NN
N N
Cl
CH3
CH3
H
52 53 54 NR2B kötıdés IC50 : 8,3 nM NR2B kötıdés IC50 : 7 nM NR2B kötıdés IC50 : 1 nM
ON
NHN
ON
NHN
55 56
NR2B kötıdés Ki: 60 nM NR2B kötıdés Ki: 7,5 nM

38
Kiemelkedı in vitro aktivitásaik ellenére a vegyületeket nem fejlesztették tovább.
A Roche kutatói egy [3H]-Ro 25-6981 kötıdési teszten alapuló HTS vizsgálat során egy
NR2B aktív imidazolin származékot azonosítottak (55).83 A vegyületcsalád optimalizálásakor
az egyik kritikus paraméternek a molekulák pKa értékét feltételezve a bázicitást jelentısen
befolyásoló szerkezeti módosításokat is végrehajtottak. Megfigyeléseik alapján fontos, hogy a
vegyület fiziológiai pH-n protonálódjon, ugyanakkor a túlzott bázicitás sem elınyös. A
legaktívabb 56 származék pKa értéke (9,9) az optimálishoz közeli lehet, mind a bázicitás
csökkentése, mind a növelése kevésbé aktív vegyületeket eredményezett. 56 egy
antikonvulzív tulajdonságot vizsgáló egér audiogén roham modellben, i.p. adagolást követıen
elfogadható agyi penetrációt mutatott. Valószínő, hogy egy kedvezı pKa érték nemcsak a
receptorral való kölcsönhatás szempontjából fontos, de befolyásolhatja a sejtmembránokon,
így az vér-agy gáton való átjutást is.
A Roche vegyülettárának [3H]-Ro 25-6981 kötıdési teszt alapú HTS szőrésekor egy 4-
aminokinolin származékot (57) is NR2B szelektív NMDA antagonistaként azonosítottak.84
H H
N
N OH
OH
N
N
N
OH
57 58
NR2B kötıdés Ki: 260 nM NR2B kötıdés Ki: 3,5 nM
A vegyületcsalád optimalizálása során megállapították, hogy a 4-es helyzető aminocsoporthoz
képest vicinális helyzetben legalább egy hidrogénkötés donorcsoport jelenléte szükséges.
Elınyös, ha a C2 helyzetben lévı lipofil aromás győrő nem közvetlenül kapcsolódik a
kinolingyőrőhöz. Valamennyi aktív származék protonálódik fiziológiás pH-n (pKa > 8,4),
valószínősítve, hogy ez a bioaktív forma. A leghatásosabb származékban a kinolingyőrő 2-es
helyzető szénatomja egy 3,4-dihidro-1H-izokinolin nitrogénatomjához kapcsolódik (58). A
vegyület egér audiogén roham modellben i.p. adagolás mellett in vivo is hatékonynak
bizonyult (ED50<12 mg/kg).84,85 A kinolinszármazékok α1 és muszkarin M1 receptorral
szemben mutatott jelentıs affinitása miatt az optimalizációt tovább folytatták. Kinolingyőrő
helyett piridint alkalmazva az NR2B aktivitás megtartása mellett sikerült jelentıs mértékben
növelni az ezekkel a receptorokkal szembeni szelektivitást. Az NR2B receptorokhoz közel
azonos affinitással rendelkezik az 59 4-aminopiridin, a 60 2-aminopiridin, és a mindkét
aminocsoporton szubsztituált 61 származék is.86-88

39
N N
NH2
N
N
NH2
N N
NOH
H
59 60 61 NR2B kötıdés IC50 : 8 nM NR2B kötıdés IC50 : 4 nM NR2B kötıdés IC50 : 4 nM
Többek között a mi, késıbb ismertetett, hasonló vegyületek körében elért eredményeinkre
hivatkozva a Merck kutatói folytatták az amidin típusú molekulákkal korábban megkezdett
munkájukat. Öt, hat és héttagú ciklikus benzamidineket állítottak elı, melyek közül NR2B
aktivitással csak az öttagúak rendelkeztek.89 Korábbi 48 benzamidin-származékuk szerkezetét
kétféleképpen merevítették. Az amidincsoport szubsztituálatlan nitrogénje és a benzilcsoport
alifás szénatomja közzé beépített metiléncsoport 4,5-dihidro-1H-imidazol származékokat
eredményezett. A vegyületcsalád leghatásosabb, egy klóratommal is szubsztituált
származékának (62) az aktivitása elmarad a 48 anyavegyület aktivitásától. A benzamidin
szubsztituált nitrogénje és orto-helyzető szénatomja közé egy metiléncsoport beépítésével
levezethetı izoindolin-1-imin (63) az anyavegyületnél hatásosabbnak bizonyult.
HN
NH OCH3
CF3O CF3O
N
NH OCH3
CF3O
N
N
Cl
CH3O
H
62 48 63 NR2B kötıdés Ki: 75 nM NR2B kötıdés Ki: 5,7 nM NR2B kötıdés Ki: 3,6 nM
A vegyület viszonylag gyenge farmakokinetikai sajátságokkal bír: 5 % biohasznosulás, 2 óra
plazma féléletidı, 215 nM cmax. Orálisan aktív, a karragén-indukált hiperalgézia tesztben
meghatározott ED50 értéke 20 mg/kg. Ennél a dózisnál 50 %-os hiperalgézia válasz
csökkenést eredményezett a kontrollhoz képest.
A Merck ezeken kívül több 64 reprezentatív molekulával jellemezhetı iminopirimidin
származékot védett szabadalmi bejelentésben.90
N N
NH
64

40
3. KÍSÉRLETI MODELLEK, VIZSGÁLATI MÓDSZEREK
3.1 Farmakológiai szőrırendszer
Az elıállított vegyületek biológiai hatását gondosan összeválogatott, egymásra épülı
tesztsorozatban vizsgáltuk. Ez a kaszkádszerően felépített farmakológiai szőrırendszer négy
jól elkülönülı részbıl állt. Egyik lépcsıbıl a következıbe csak az elıre felállított
kritériumoknak megfelelı vegyületek juthattak át. A projekt négy éves idıtartama alatt a
kaszkádrendszer számos pontját megváltoztattuk. Menet közben új módszereket fejlesztettünk
ki, az egyes teszteket átrangsoroltuk, legtöbbször a szelekciós kritériumokat módosítottuk.
Eredményeinket, lehetıségeinket és a szők keresztmetszeteket figyelembe véve szigorítottuk,
vagy lazítottuk a továbbjutás feltételeit. Az 11. ábrán tesztrendszerünk egy jellemzı állapotát
mutatom be.
NMDA antagonizmus
1. lépcsı IC50 < 25 nM
In vitro metabolizmus
Szelektivitás >100
4. lépcsı
Farmakokinetika (PK) orális biohasznosulás (BA) (patkány)
Orális hatás neuropátiás fájdalom modellben ED50
QT megnyúlás nyúl szíven
BA > 20% 3. lépcsı
2. lépcsı >100 x szelektivitás, stabilitás (FM > 70 %)
Orális ED50 < 5 mg/kg
Egér formalin teszt Egyszeri 10 mg/kg dózis, Inh > 60 % →ED50
Irwin teszt egérben – minimális effektív dózis (MED) p.o.
Mellékhatást, szelektivitást feltérképezı tesztek
α, 5ΗΤ2Α, NR2A, DAfelvétel Met.stab. patkány és humán mikroszómán (Fm % > 70)
Fluorometriás [Ca2+] meghatározás
Egér lokomotoros aktivitás (LMA) MED p.o.
MED/ ED50 > 10
Patány lokomotoros aktivitás (LMA)
Nincs effektus az ED50 - nél
Nincs effektus 1 µM-nál
Fı hatások: Seltzer modell (antihiperalgézia) Gyulladásos fájdalom modell Termális hiperalgézia Ismételt adás Elektrofiziológiás wind up
Biztonság/fejleszthetıség Irwin teszt patkányban CYP2D6 metabolizmus Kognitív teszt Toxicitás Kardiovaszkuláris mellékhatások
Rotarod teszt P450 enzim indukció NR2B szelektivitás AMES teszt
11. ábra. Az NR2B szelektív NMDA antagonisták projekt farmakológiai tesztrendszere

41
Az 1. lépcsıben a hatékony NMDA antagonistákat válogattuk ki, egy, a kortikális neuron
tenyészeteken az NMDA intracelluláris Ca2+-szintet növelı hatásának antagonizmusa alapján
mőködı funkcionális teszt segítségével. E teszt eredménye elsı közelítésben a kémiai munka
megfelelı és gyors visszacsatolásának bizonyult. Hatásos vegyületek esetében a
legvalószínőbb mellékhatások feltérképezése céljából α1 és 5HT2A receptor kötıdési
teszteket, a szelektivitás alátámasztása céljából további funkcionális teszteket végeztünk
NR1a/NR2B és NR1/NR2A alegység összetételő rekombináns NMDA receptorokon, illetve
egy további kötıdési tesztet melyben egy NR2B szelektív referenciavegyület tríciált
változatát (3H-Ro 256981 ) használtuk fel radioligandként.
Az 1. lépcsı része volt még az in vitro metabolizmus, melynek során a hatásos vegyületek
metabolikus stabilitását patkány és humán mikroszómán mértük. Az in vitro kellıen
hatékony, szelektív és metabolikusan stabil vegyületekkel a kaszkád 2. lépcsıjében
megkezdıdtek az in vivo vizsgálatok. A fájdalomcsillapító fıhatást egér formalin tesztben
határoztuk meg, míg a lehetséges CNS mellékhatásokat Irwin teszt és egér lokomotoros
aktivitást (LMA) mérı teszt alapján próbáltuk meg feltérképezni. Ha a mellékhatásokra és a
fıhatásra jellemzı dózisok közötti különbség, vagyis a terápiás ablak, illetve a kettı
hányadosa a terápiás index kellıen nagy (MED/ED50>10) és a vegyület kielégítı
fájdalomcsillapító hatással rendelkezett, akkor a 3. lépcsıbe került. Ebben a fázisban a
továbbjutott vegyületekrıl részletes farmakokinetikai és farmakodinámiás jellemzés készült.
Az ADME tulajdonságokat leíró farmakokinetikai jellemzık, a kiürülés, féléletidı,
megoszlási térfogat, cmax, biohasznosulás, agyi penetráció közül a biohasznosulás (BA)
szelekciós tényezı volt. A hatékonyságot CCI neuropátiás fájdalommodellben, míg a
mellékhatást patkány lokomotoros aktivitást (LMA) mérı modellben vizsgáltuk tovább. A
CNS mellékhatásokat jelzı LMA méréseket a szív arritmiájával kapcsolatba hozható QT
megnyúlás mérésével egészítettük ki. Ebben a fázisban kerül sor az adásmódok
összehasonlítására. Rosszul felszívódó vagy a first-pass metabolizmuson lebomló vegyületek
sok esetben nem mutatnak per os hatékonyságot, de más adásmóddal (i.v., s.c., i.p.) még
hatékonyak lehetnek. A rosszul oldódó és felszívódó vegyületek biohasznosulását, ezáltal in
vivo hatékonyságát különbözı komplexképzık és oldószerek segítségével, vagy a hatóanyag
mikronizálásával több esetben jelentısen sikerült javítani. Az utolsó lépcsıben még több a fı
hatást és a lehetséges mellékhatásokat feltáró vizsgálatnak vetettük alá az ide kerülı
vegyületeket. Bár új molekulákat elsısorban az in vitro eredmények alapján terveztünk, a
kaszkád elsı három lépcsıjének teszteredményei is hatással voltak a kémiai munkára. A
tervezett molekulák szerkezeti, fiziko-kémiai paraméterein (oldékonyság, PAMPA, clogP, pK

42
predikciók) keresztül eredményesen tudtuk befolyásolni a felszívódást, biohasznosulást, agyi
penetrációt, ezeken keresztül pedig az orális hatékonyságot. Gyógyszerkémiai szabályokat
alkalmazva több esetben pozitívan alakítottuk a metabolikus stabilitást. A 4. lépcsı sem
teljesen fekete doboz, a benne lévı tesztekre vonatkozóan is alkalmazhatunk racionális
megfontolásokat, különbözı predikciós módszereket. Ebben a fázisban fontoltuk meg a
prodrug startégia lehetıségét. Túl sok paraméterre azonban nem lehet optimalizálni így a 4.
lépcsı teszteredményei már csak rangsorolják az idáig jutott vegyületeket. A fıhatás,
biztonság és fejleszthetıség szempontjait alaposan mérlegelve közülük választottuk ki a
továbbfejlesztésre alkalmas jelölteket. Mivel a kémiai tervezımunkát az 1. lépcsı
meghatározó in vitro vizsgálatai mellett a következı két lépcsı in vivo teszteredményei is
befolyásolták, és dolgozatomban a szerkezet-hatás összefüggések elemzésekor felhasználom
ezeket az erdményeket, röviden ismertetem az elsı három lépcsı tesztjeit. A kaszkád
felépítése és a kritériumrendszer az 11. ábrán kísérhetı figyelemmel.
3.2. In vitro vizsgálatok
3.2.1. Kötıdési tesztek
Az NMDA antagonisták mellékhatásaiért elsısorban az α1 és az 5-HT2A receptorok felelısek,
ezért már korai szakaszban fontos, hogy ismerjük a vegyületek affinitását ezekhez a
receptorokhoz. Az α1 kötıdési tesztben56 [3H]-prazozint, az 5-HT2A kötıdési tesztben91 [3H]-
ketanszerint használtunk radioligandként.
A funkcionális tesztadatok mellett elsısorban QSAR módszerek kiindulási adataként egy
NR2B kötıdési tesztre54 is szükségünk volt. Méréseink során radioligandként egy nagy NR2B
szelektivitással rendelkezı triciummal jelzett referenciavegyületet, a [3H]-Ro 256981-et
alkalmaztuk.
3.2.2. Funkcionális tesztek
3.2.2.1. Funkcionális teszt primer neokortikális tenyészeten
Az elıállított új vegyületek NMDA antagonista hatását a következı, intracelluláris Ca2+
koncentráció mérésen alapuló funkcionális tesztben vizsgáltuk.76 Az NMDA receptorok az
agonista NMDA alkalmazását követıen ingerült állapotba kerülve Ca2+-ra permeábilisak.
Funkcionális tesztünkben a vegyületeknek az NMDA által kiváltott intracelluláris Ca2+-
koncentráció emelkedésre gyakorolt gátló hatását határoztuk meg fluoreszcenciás Ca2+
méréssel.

43
A méréshez 17 napos Charles River-patkány embriókból származó, standard 96-lyukú
szövettenyésztı lemezekre szélesztett primer neokortikális tenyészeteket használtunk. Mivel a
patkány és az ember NMDA receptorainak nagyon nagy a szekvencia hasonlósága (99, 95, 97
% az NR1, NR2A illetıleg az NR2B alegységek esetén), farmakológiai érzékenységükben
nagyon kicsi a különbség, ha van egyáltalán. Ezért azok az eredmények, amelyeket patkány
NMDA receptorokon kaptunk, reményeink szerint jól extrapolálhatók az emberre is.
Mérés elıtt a sejteket feltöltöttük Fluo-4 fluoreszcens Ca2+ indikátorral, majd a tesztelendı
vegyületeket végül NMDA-t adagoltunk a sejtekhez. A Ca2+ koncentráció mérését egy plate
reader fluoriméter készülékkel végeztük.
A vegyület gátló hatását különbözı koncentrációknál a kalcium felszabadulás csökkenésének
mérésével határoztuk meg. Egy konkrét koncentrációnál a vegyület gátló hatását a kontrol
NMDA válasz gátlás százalékában fejezzük ki. Ezt az értéket elosztva az ifenprodil esetében
mért százalékos gátlással egy relatív értékhez jutunk. A vegyület 0,1 µM koncentrációnál
meghatározott gátló hatását osztottuk a 10 µM ifenprodil gátló hatásával. Tanulmányomban a
receptor gátlását a közvetlenül mérhetı abszolút százalékos adat helyett ezzel a relatív
értékkel jellemzem (Inh %). A tesztrendszerben adott százalékos érték felett IC50 értéket is
meghatároztunk, így a hatásosabb vegyületeket IC50 értékkel jellemzem. Az idegsejtek
NMDA receptor alegység összetétele a születést követı fejlıdés során változik. Hasonló
változás figyelhetı meg neuronális sejttenyészetekben.92 Az irodalmi adatok, és saját
immunocitokémiai vizsgálataink szerint a 4 - 7 napig in vitro tenyészetben tartott neuronok
túlnyomórészt NR2B alegységet expresszálnak az NR1 mellett. Így az NMDA antagonizmus
ilyen sejteken végzett funkcionális tesztje elsısorban az NR2B alegység tartalmú
receptorokon való hatást mutatja.
3.2.2.2. Funkcionális teszt NR1a/NR2B és NR1/NR2A alegység összetételő rekombináns
NMDA receptorokon
Bár az elızı megfontolások alapján a natív idegsejteken meghatározott funkcionális teszt
elsısorban az NR2B alegység tartalmú receptorok gátlását fejezi ki, a vegyületek egyértelmő
jellemzése miatt a mérést NR1a/NR2B alegység összetételő NMDA receptorokat stabilan
expresszáló sejtvonalakon is végrehajtottuk. A Ca2+ koncentráció fent leírt fluoreszcenciás
meghatározását patkány NR1a és NR2B NMDA receptor alegység kombinációt stabilan
expresszáló HEK293 sejteken hajtottuk végre. Vegyületeink NR2B szelektív hatásának
alátámasztására a mérést NR1/NR2A alegység összetételő rekombináns NMDA receptorokat
stabilan expresszáló sejtvonalakon is végrehajtottuk.

44
3.3. In vivo tesztek
3.3.1. Egér formalin teszt
Egerek hátsó lábába injektált higított formalin kétfázisú, fájdalommal kapcsolatos viselkedést
vált ki, amely a sérült láb nyalakodási/harapdálási idejével mérhetı. Az elsı fázis az azonnal
jelentkezı nociceptív válasz. A második fázis az azon fájdalommal kapcsolatos események,
amelyek a formalin injektálását követıen 15-60 percen belül jelentkeznek. Az NMDA
receptorok a formalin injektálásra adott válasz második fázisában játszanak szerepet, és ez a
viselkedési válasz az NMDA receptorok gátlására érzékeny.93 Ezért mi a formalin teszt
második - “ hiperalgéziás” - fázisát használtuk, hogy a vegyületek in vivo hatékonyságát
jellemezzük. A válasz második fázisának gátlása a kémiailag indukált állandó fájdalommal
szembeni analgetikus hatás mértékének tekinthetı.94 A vizsgálandó vegyületeket orálisan
adagoltuk 15 perccel a formalin injektálás elıtt. Az injektált láb nyalásával és harapdálásával
töltött idıt mértük a formalin injektálástól számított 20-25. percben. Az eredményeket az
azonos napon megfigyelt kontrollcsoportban mért nyalakodási idıkhöz viszonyítva %-os
gátlásban fejeztük ki, hatásos vegyületek esetén ED50 értéket határoztunk meg.
3.3.2. Neuropátiás fájdalom Seltzer modellje (antihiperalgézia)
Egerek egyik oldali nervus ischiadicusát az állat combján részlegesen kipreparáltuk és
szorosan elkötöttük szilikonos varróanyaggal, majd a sebet lezártuk. Ez az ideglekötés a láb
spontán fájdalmát és mechanonociceptív küszöbének csökkenését idézi elı.95 A kialakuló
mechanikai hiperalgézia mértékét egy analgeziméterrel határoztuk meg és grammban fejeztük
ki. Az állatok lábát egyenletesen fokozódó erıhatásnak tettük ki. A fájdalomküszöb
elérésekor az állat kirántja a lábát a mőszerbıl, így a fájdalomküszöb grammban kifejezhetı.
ED50 érték helyett egy minimális effektív dózist (MED) állapítottunk meg ahol a vegyület
statisztikailag szignifikánsan visszafordítja a hiperalgéziát.
3.3.3. Neuropátiás fájdalom CCI (chronic constriction injury) modellje (antiallodínia)
Az idegsérülés hatását ebben a modellben a jobboldali ülıidegnek egy kettéhasított polietilén
csıbe való zárásával modelleztük. Az operációt követı 30. napon stabil allodínia alakul ki,
amely még két hónapig fennáll. A fájdalom küszöbértékét a hátsó láb Von Frey
filamentumokkal való érintésével határoztuk meg. A fájdalom küszöbértékének annak a Von
Frey szálnak a grammokban kifejezett értékét tekintettük, ahol az állat a lábát visszahúzta a

45
fájdalom miatt (láb-visszahúzási küszöbérték). A mért értéket a hatóanyag helyett csak
vivıanyaggal kezelt kontrollcsoport adataihoz viszonyítottuk.
3.3.4. Diabéteszes neuropátia modell
Patkányokban sztreptozotocin egyszeri adásával két hét alatt diabéteszt idéztünk elı. További
hat hét alatt diabéteszes neuropátia alakult ki. A láb-visszahúzási küszöbértéket Von Frey
filamentumokkal való mechanikus ingerléssel határoztuk meg. A mért értéket a hatóanyag
helyett csak vivıanyaggal kezelt kontrollcsoport adataihoz viszonyítottuk.
3.4 Nemkívánatos mellékhatásokra utaló tesztek
3.4.1. Irwin teszt
A központi idegrendszerre ható szerek gyors szőrésre alkalmas aktivitásvizsgálat, amely az
állatok spontán magatartásváltozásáról (pl. szedáció, excitáció, sztereotípia, agresszivitás,
motoros inkoordináció, izomtónus, stb) ad kvalitatív jellegő felvilágosítást. Az egyes állatokat
minden egyes tesztben vizuálisan megfigyeltük, és a tesztben mutatott reakcióját ( pl.
önápolás, hangadás, remegés, görcsök, passzivitás, fájdalomreakció, ingerlékenység stb.)
pontokkal osztályoztuk, a kapott pontokat összehasonlítottuk a kontroll azonos tesztben
mutatott pontjaival. A teszt eredményeként minimális effektív dózis (MED) került
meghatározásra.
3.4.2. Spontán lokomotoros aktivitás mérése rágcsálókon
A spontán lokomotoros aktivitást egy, a mozgási aktivitást fotocella sorral automatikusan
detektáló készülékben vizsgáltuk. Harminc perccel a tesztvegyület vagy a vivıanyag orális
adagolása után az állatok horizontális és vertikális mozgását határoztuk meg a fénysugár
megszakítások számaként egy órán keresztül 15 perces intervallumokban.
A mérést a horizontális aktivitás adatainak kontrollcsoporthoz (vivı anyaggal kezelt)
viszonyított százalékos változása jellemzi. Egy vegyület akkor lokomotoros stimuláló hatású,
ha 50 %-nál nagyobb emelkedést váltott ki a fénysugár megszakításokban. Vagyis,
definíciószerően a lokomotoros aktivitás mentes (LMAfree) dózisok 50 %-nál kisebb
emelkedést mutattak.
3.4.3. Rotarod teszt
A rotarod teszt széleskörben használt egyszerő módszer a vegyületek motoros
mellékhatásának detektálására. A teszt során a rágcsálót forgó rúdra állítottuk, ezzel motoros

46
aktivitásra kényszerítettük. A forgó rúdon való tartózkodási idı arányos az állat
egyensúlyozási, koordinációs képességével. A meghatározott ED50 érték a vizsgált
vegyületnek ezekre a képességekre, motoros funkciókra gyakorolt hatását jellemzi.
3.4.4. QT-megnyúlás
A kísérlet során azt vizsgáltuk, hogy izolált nyúlszíven vagy érzéstelenített kutyában
i.v.adagolt tesztvegyület meghosszabbítja-e az EKG-görbe QT tartamát. QT megnyúlás esetén
fatális kamrai tachycardia, ún. torsades de pointes típusú ritmuszavar jelentkezhet. A Q
hullám a kamrai depolarizációt (gyors Na+ beáramlás), a T hullám a repolarizációt tükrözi. A
repolarizáció több ionáram egymás utáni vagy egyidejő mőködésének az eredménye,
legnagyobb részét a human Ether-à-go-go Related Gene (hERG) gén által kódolt K+
ioncsatornán keresztüláramló kifelé irányúló késıi egyenirányító K+ áram határozza meg. Így
sok esetben a QT megnyúlás arányos a vegyület hERG K+ csatornára gyakorolt gátló
hatásával. A QT megnyúlását msec-ban vagy a kontrollhoz viszonyítva, százalékosan adják
meg.

47
4. SAJÁT EREDMÉNYEK
Az NR2B projekt indulásakor ifenprodil típusú molekulák elıállítását határoztuk el. Mivel
abban az idıben cégünknél még nem voltak meg a feltételei egy nagy áteresztıképességő
biológiai szőrésnek, kémiai munkánkat a versenytársak ismert molekuláira alapoztuk, és a
mai szakirodalomban lead-hoppingnak nevezett módszer segítségével reméltünk új, hatásos
szerkezeteket találni. A lead-hopping vagy scaffold-hopping során egy hatásos molekulából
kiindulva hasonló aktivitású, de eltérı szerkezető molekulát azonosítunk. Ma már különféle
számítógépes technikák generálnak ezen az elven új szerkezeteket.
A dolgozatomban feldolgozott, általunk azonosított NR2B aktivitással rendelkezı
vegyületcsaládok fejlıdését, logikai kapcsolatát, a vegyületcsaládokat jellemzı molekulák
alapján a 12. ábrán foglalom össze. Kémiai munkánk kiindulópontja az egyik versenytárs a
CoCensys abban az idıben legújabb eredménye, a 35-ös számú fahéjsavamid volt. A
molekula az akkor már jól ismert farmakofor modellt azzal az új elemmel egészítette ki, hogy
nem szükséges bázikus nitrogén jelenléte az NR2B aktivitás eléréséhez. Feltételezésünk
szerint a mellékhatások döntı részéért éppen az ifenprodil analogonokban lévı bázikus
nitrogén a felelıs. A 35 fahéjsavamid amin komponensének elınyös cseréjével egy aktívabb
származékhoz jutottam (69). Közben az University of Oregon, a CoCensys és a Parke Davis
közös kutatócsoportja újabb közleményben számolt be a fahéjsavamidok körében végzett
további munkájáról. Megfigyelték, hogy a klóratom és a hidroxilcsoport helyzetének
felcserélése szintén hatásos molekulát (65) eredményezett. Ennek alapján a szerzık azt a
következtetést vonták le, hogy a molekulák a receptorzsebbel minkét orientációból képesek
kapcsolatot létesíteni. A 4-hidroxi-fahéjsavamid (65) amidkomponensét 4-benzil-piperidinre
cserélve egy közel háromszor aktívabb molekulához jutottak (37). Korábban az ifenprodil
analogonok szekunder amin része kizárólag 4-es helyzetben szubsztituált piperidinek voltak.
Elhatároztam, hogy felderítem, milyen szekunder aminok eredményeznek még hatásos
fahéjsavamidokat. Másik célom a fenolos hidroxilcsoport cseréje vélhetıen metabolikusan
stabilabb csoportra. A két célkitőzést kombinatorikus kémia eszközökkel, a molekula két
részének egyidejő szerkezeti változtatásával próbáltam egyszerre elérni. A megtervezett
vegyülettár létrehozásához elıször a fenolos hidroxilcsoporttal izosztér, heterociklusokkal
kondenzált fahéjsavakat állítottam elı, melyekben a hidrogénkötés donor szerepet egy NH-
csoport tölti be. A fahéjsavakkal, szilárd fázishoz kötött kapcsolószer segítségével, válogatott
szekunder aminokat acileztem. A fahéjsavamidok körében végzett kombinatorikus kémiai
tanulmány eredményeként a 37 anyavegyülethez képest négyszer aktívabb molekulához

48
jutottam (81). Munkánkban alapvetı fordulatot a fahéjsavszerkezet merevítésére tett
erıfeszítéseink sikere hozott. A fahéjsav α-szénatomjának összekötése az orto-helyzető
aromás szénatommal egy hétszer aktívabb molekulához, a 82 indol-2-karboxamid-
származékhoz vezetett. A 82 vezérmolekula optimalizációja számos hasonlóan aktív
molekulát eredményezett. A molekula összes pontján megvizsgáltam a lehetséges szerkezeti
módosítások hatását. Legeredményesebb változtatás az indol 3-as helyzetében lévı CH-
csoportnak nitrogénatomra történı cseréje volt (142). Az ilymódon levezethetı benzimidazol-
2-karboxamidok általában tízszer aktívabbak voltak a megfelelı indol-2-karboxamidoknál.
Mind az indol, mind a benzimidazol-származékok körében számos in vivo is hatásos vegyület
található. Az indolgyőrő bıvítésével egy szintén aktív vegyületcsaládot, a
kinurénsavamidokat azonosítottuk (230). Az indol és benzimidazol-2-karboxamidok körében
megismert szerkezet-hatás összefüggések a kinurénsavamidok gyors optimalizációját tették
lehetıvé. A projekt igazi sikerét az indolgyőrő felnyitásával levezethetı oxálsavdiamidok
(183) hozták meg. Az in vivo leghatásosabb molekulák közülük kerültek ki. A vegyületcsalád
tagjait hatás-mellékhatás szempontjából egymással összehasonlítva a 185-ös vegyületet
választottuk ki klinikai vizsgálatok céljából. Az oxalilcsoport eredményességét követıen
elhatároztam, hogy más háromtagú összekötı láncot is megvizsgálok. Számos,
gyógyszerszerő molekulát eredményezı háromtagú lánccal kötöttem össze a baloldali aromás
győrőt a piperidin nitrogénjével. Ezek közül legaktívabb vegyületcsaládnak a benzoil-
karbamidok bizonyultak (274). Dolgozatomban az egyes vegyületcsaládokat, a molekulák
egymásra épülését, logikai kapcsolatát érzékeltetve, felfedezésük idırendjében mutatom be.
Így a benzilidén-piperidin-származékokat az utolsó fejezetben ismertetem, annak ellenére,
hogy szerkezetük alapján az ifenprodil típusú molekulákat leíró korábbi fejezetekhez
kapcsolódnak szorosan. Elınyös tulajdonságaikat a projekt végén ismertük fel. A korábban
elıállított leghatásosabb molekulák szekunder amin része valamennyi esetben szubsztituált 4-
benzil-piperidin volt. Ezek benzilidén-piperidin megfelelıi (312, 321) hasonlóan aktívnak
bizonyultak, ráadásul jóval nagyobb terápiás ablakkal rendelkeztek. Az indol- és
benzimidazol-2-karbonsavak benzilidén-piperidinekkel képezett amidjait hagyományos
módon, míg az oxálsavdiamidokat oldatfázisú parallel szintézissel állítottam elı. A nem
ifenprodil típusú molekulák kutatását egy korábbi Merck közleményt követıen indítottuk el,
melyben NR2B receptoron ható amidinekrıl számoltak be (46, 48) 119 (13. ábra). A Merck
kutatóinak eredményeit felhasználva, korábbi tapasztalatom alapján új típusú,
gyógyszerszerőbb szerkezetet, indol-2-karboxamidineket terveztem (299). A vegyületeket
kombinatorikus kémia segítségével állítottam elı.

49
O
N
Cl
OH
H
O
N
HO
Cl
H
O
N
HO 35 (Co 101526) 65 37 NR2B-IC50 : 319 nM (irodalmi: 170 nM) NR2B-IC50 : 385 nM (irodalmi: 330 nM) NR2B-IC50: 131 nM (irodalmi: 120 nM)
O
N
Cl
OH
OHH
N
O
N HO
H
O
NO
NO
H 69 82 81 NR2B-IC50: 130 nM NR2B-IC50: 18 nM NR2B-IC50: 28 nM
N
N
O
N HO
H
N
O
N
O
HO
H HN
N
HO
O
O
N N
HO
OO
H
142 230 183 274 NR2B-IC50: 2,2 nM NR2B-IC50: 9,1 nM NR2B-IC50: 11 nM NR2B-IC50: 24 nM
H
N
N
O
N HO
HN
N
O
O
O
NO
F
H
H
HN
N
O
O
O
NO
312 185 (radiprodil) 321 NR2B-IC50: 3,3 nM NR2B-IC50: 4 nM NR2B-IC50: 21 nM
12. ábra. Ifenprodil típusú NR2B szelektív NMDA antagonista hatású vezérmolekulák fejlıdése.
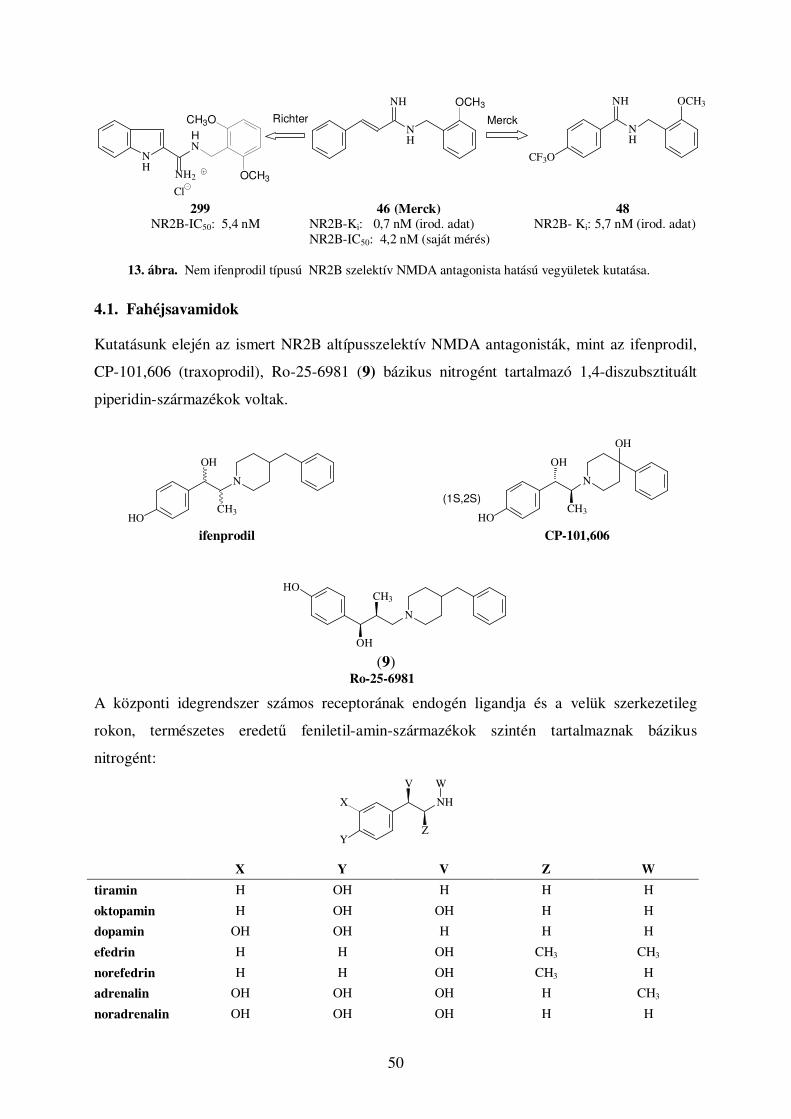
50
H
Cl
N
NH2
N
CH3O
OCH3
HN
NH OCH3
H HN
NH OCH3
CF3O
Merck
Richter
299 46 (Merck) 48 NR2B-IC50: 5,4 nM NR2B-Ki: 0,7 nM (irod. adat) NR2B- Ki: 5,7 nM (irod. adat) NR2B-IC50: 4,2 nM (saját mérés)
4.1. Fahéjsavamidok
Kutatásunk elején az ismert NR2B altípusszelektív NMDA antagonisták, mint az ifenprodil,
CP-101,606 (traxoprodil), Ro-25-6981 (9) bázikus nitrogént tartalmazó 1,4-diszubsztituált
piperidin-származékok voltak.
HO
N
OH
CH3
OH
CH3
N
OH
HO
(1S,2S)
ifenprodil CP-101,606
HO
N
OH
CH3
(9)
Ro-25-6981
A központi idegrendszer számos receptorának endogén ligandja és a velük szerkezetileg
rokon, természetes eredető feniletil-amin-származékok szintén tartalmaznak bázikus
nitrogént:
Y
NH
WV
Z
X
X Y V Z W
tiramin H OH H H H
oktopamin H OH OH H H
dopamin OH OH H H H
efedrin H H OH CH3 CH3
norefedrin H H OH CH3 H
adrenalin OH OH OH H CH3
noradrenalin OH OH OH H H
13. ábra. Nem ifenprodil típusú NR2B szelektív NMDA antagonista hatású vegyületek kutatása.

51
Ezekkel az aminokkal további szerkezeti hasonlóság is megfigyelhetı. Közös elem a 4-
hidroxi-feniletil-amin rész is, bár az NMDA antagonisták körében az etil lánc hosszabb is
lehet egy metiléncsoporttal (9), vagy egy heteroatommal (12). A szerkezeti hasonlóság
eredményeként ezek a vegyületek nemcsak az NMDA receptorhoz kötıdnek, hanem pl. az α-
1 adrenerg és a szerotonin receptorokhoz. A feniletil szerkezeti elem mellett a biogén aminok
bázikus nitrogénjének alapvetı szerepe van a receptorukhoz való kötıdésben. Munkánk
kezdetekor tették közzé az University of Oregon, a CoCensys és a Parke-Davis
kutatócsoportjai közös munkájukban azt a megfigyelést, miszerint a bázikus nitrogén jelenléte
nem feltétele az NMDA receptor antagonizmusnak. Az általuk elıállított 35-ös N-(2-
feniletil)fahéjsavamid-származék hatásos és NR2B szelektív NMDA antagonistának
bizonyult.72 Más receptorokhoz mutatott affinitásáról az irodalom nem közölt adatokat,
feltételeztük azonban, hogy ez a bázikus nitrogént nem tartalmazó molekula nem, vagy jóval
kisebb mértékben kötıdik a biogén aminok receptoraihoz. Hipotézisünk igazolása céljából
megvizsgáltuk a 35-ös fahéjsavszármazékot. Natív NMDA receptorokon végzett funkcionális
teszt 319 nM-os IC50 értéke hasonló moláris hatékonyságot mutatott az irodalmi
rekombináns NR2B receptorokon mért 170 nM-os értékkel. Sejtésünknek megfelelıen a
vegyület egyáltalán nem kötıdött az α-1 adrenerg és a szerotonin 5HT2a receptorokhoz. Az
eredményeken felbuzdulva újabb fahéjsavamid-származékokat és analogonokat terveztünk és
állítottunk elı további szerkezet-hatás összefüggések felderítése érdekében. Az elıállított
vegyületek biológiai aktivitását elsısorban NR2B receptorokat expresszáló natív idegsejteket
felhasználó funkcionális esszében határoztuk meg, melynek során az NMDA által kiváltott
intracelluláris Ca2+ szint növekedésének gátlását mértük.
4.1.1. Tiraminszármazékok
Kémiai munkám kiindulópontja a CoCensys 35-ös számú molekulája. Ahogy az ifenprodil
esetében, úgy a 35 fahéjsavszármazéknál is egy fenolos hidroxilcsoport szubsztituálja 4-es
helyzetben az egyik benzolgyőrőt. Az University of Oregon, a CoCensys, Inc. és a Parke-
Davis Pharm. Res. kutatói közös tanulmányukban72 szerkezet-hatás összefüggésben elemezték
a molekulát. A fenolos hidroxilcsoport para helyzetbıl meta helyzetbe történı áthelyezése egy
nagyságrenddel csökkentette az aktivitást, míg elhagyása a hatás teljes elvesztésével járt. A
klóratom és a hidroxilcsoport helyzetének felcserélésével közel azonos hatsú molekulákhoz
jutottak (35, 65). A fahéjsavamid mindkét végének hidroxilcsoporttal történı helyettesítése a
hatás jelentıs csökkenésével járt (NR2B-IC50 : 21 µM).

52
N
HOO
Cl
H
N
OCl
OH
H
35 (Co 101526) 65 NR2B-IC50 : 319 nM (irodalmi: 170 nM) NR2B-IC50 : 385 nM (irodalmi: 330 nM)
Saját méréseink nem mutatták az irodalomban közölt meggyızı különbséget, a továbbiakban
új molekulák tervezésekor azért az aktívabb, 35 molekula szerkezetét részesítettem elınyben.
A molekula baloldali részét változatlanul hagyva, 4-hidroxi-feniletil-amin, triviális nevén
tiraminszármazékokat állítottam elı. Fahéjsavamid helyett vele azonos hosszúságú,
heteroatomot is tartalmazó amid- (66) illetve karbamidszármazék (67) teljesen hatástalannak
bizonyult.
N
HO
O
O
Cl
H
N
HO
N
O
Cl
H H
66 67
A tiramin etilén láncának egy oxigén atommal való meghosszabbítása (68) szintén a hatás
elvesztésével járt.
HO
ON
O
Cl
H
68 NR2B-IC50 : 3520 nM
4.1.2. Oktopaminszármazékok
A 35 vegyület tiramin részének 1-(4-hidroxi-fenil)-1-hidroxi-etil-aminnal (triviális nevén
oktopaminnal) való kicserélése az anyavegyületnél kétszer hatékonyabb vegyületet
eredményezett (69). Az α1 és 5-HT2A receptorokhoz nem mutatott affinitást. Ez volt az elsı
hatást javító változtatás az anyavegyületen, ezért további alapvetı szerkezetmódosításokkal
járó, új molekulák elıállítása mellett a 69 molekula optimalizációját határoztuk el (1.
táblázat).

53
1. táblázat. Oktopaminszármazékok flourimetriás módszerrel meghatározott NMDA antagonista hatása patkány kortikális sejteken.
N
HOO
XOH
H
vegyület
X
NMDA-kiváltott ∆∆∆∆[Ca2+]i
a
inhibíció (%)b IC50 (nM)
69 4-Cl 130 nM
70 4-H 1220 nM
71 4-F 978 nM
72 4-CH3O 513 nM
73 4-OH 10,1 %
a NMDA-kiváltott intracelluláris Ca2+ koncentrációváltozás. b Inhibíció (%) a vegyület 0,1 µM koncentrációjánál meghatározva.
A fahéjsav klór szubsztituense valószínőleg optimális, más csoportokra való kicserélése (70,
71, 72) ugyanis a hatás nagymértékő csökkenésével járt. Hidroxilcsoportok egyidejő jelenléte
a két benzolgyőrőn (73) az oktopaminszármazékok körében is az aktivitás teljes elvesztését
eredményezte. Egy fenolos hidroxilcsoport jelenléte mindenképpen szükséges, hiánya (74) a
hatás elvesztését vonta maga után.
N
O
ClOH
H
74 NR2B-inhibíció: 31,3 %
A 69-es molekula optimalizálása során merész lépésnek tőnt más hidroxilcsoportot is
tartalmazó természetes feniletil-amin felhasználása oktopamin analogonok elıállítása
céljából, de bízva munkahipotézisünkben, miszerint a biogén aminok acilezésüket követıen
elvesztik természetes receptoraik iránti affinitásukat, a hozzáférhetı 4-hidroxi-feniletil-
aminokat rendre acileztem 4-klór-fahéjsavval. A 4-hidroxi-norefedrinbıl (75), szinefrinbıl
(76), adrenalinból (77) és a tirozin metilészterébıl (78) képzett 4-klór-fahéjsavamidok
azonban hatástalannak bizonyultak.

54
N
HOO
ClOH
CH3
H
N
O
ClOH CH3
HO 75 76
NR2B-IC50 : 516 nM NR2B-inhibíció.: 68,5 %
N
O
ClOH CH3
HO
HO
N
HOO
Cl
CH3O O
H
77 78
NR2B-IC50 : 4880 nM NR2B-IC50 : 8860 nM
A 69 anyavegyület benzil-helyzető hidroxilcsoportjának oxidációja ketocsoporttá (79) az
aktivitás kismértékő javulását, míg a nagyobb térkitöltéső triazolcsoporttal való kicserélése
(80) az aktivitás jelentıs csökkenését eredményezte.
N
HOO
ClO
H
N
O
ClN
HO
NN
H
79 80
NR2B-IC50 : 105 nM NR2B-IC50 : 443 nM
A projekt elsı évében (1999) közel 250 vegyületet szintetizáltunk és teszteltünk. A
bevezetıben leírt stratégiánk alapján elsısorban bázikus nitrogént nem tartalmazó
molekulákat terveztünk. Kisebb mértékben azért a hagyományos ifenprodil struktúrából
kiinduló kutatást is folytattunk. Kutatásunk elejét jellemzı merész próbálkozásaink, bár nem
eredményeztek az irodalomban leírt fahéjsavszármazéknál hatásosabb molekulát, a tanulási
folyamatunk fontos részévé váltak. Egy év elteltével véglegesen elvetettük a bázikus nitrogént
tartalmazó ifenprodil-analogonok szintézisét. A CoCensys cég újtípusú, hatásos
molekulájából (35) kiindulva, szerkezetét módosítva, egy aktívabb vegyületcsaládhoz,
oktopaminszármazékokhoz jutottunk. Ekkor számolt be újra az University of Oregon, a
CoCensys és a Parke-Davis közös kutatócsoportja a fahéjsavamidok körében végzett
munkájuk legfrissebb eredményeirıl.73 Közleményükben részletesen bemutatták 35
vezérmolekulájuk optimalizációját. Az egyik leghatékonyabb molekulának bizonyúló 37
piperidinszármazékuk termékenyítıleg hatott munkánkra.

55
O
N
HO 37 NR2B-IC50: 131 nM (irodalmi: 120 nM)
Elhatároztuk, hogy feltérképezzük, milyen, az NR2B szelektív NMDA antagonisták körében
közkedvelt piperidinektıl eltérı szekunder aminnal képezett fahéjsavamid mutat még kellı
hatékonyságot.
4.1.3. Fahéjsavamidok elıállítása
Néhány fahéjsavszármazék hagyományos módon történı elıállítása után a kombinatorikus
megközelítés mellett döntöttünk. Idıközben ugyanis felmerült a fenolos hidroxilcsoport
metabolikusan stabilabb csoportra történı cseréjének szükségessége is. Így a kombinatorikus
kémia alkalmazhatóságának feltétele, a minimum két diverzitási pont, adott volt a molekulán
(14. ábra). Egyik változó a szekunder amin komponens, másik variálási lehetıség a fenolos
hidroxilcsoport helyett, hozzá hasonlóan hidrogénkötés donor sajátságú, NH-elemet
tartalmazó különféle csoportok, heterociklusok alkalmazása. Ennek érdekében elıször tíz
ilyen, vélhetıen metabolikusan stabilabb szerkezeti részt, többnyire benzolgyőrővel
kondenzált heterociklust tartalmazó fahéjsavat szintetizáltam (15. ábra). A fahéjsavakat a
megfelelı aldehidbıl kiindulva Knoevenagel-szintézissel állítottam elı. A fahéjsavakkal 24
piperidin és piperazinszármazékot acileztem a peptidkémiában használatos pentafluorfenolos
kapcsolási módszer szilárd fázisra adaptált megfelelıjével. A fahéjsavból elıször Argonaut
PS-TFP gyanta fenol funkciójával aktív észtert képeztem diizopropil karbodiimid
segítségével, majd szőrés, mosás után az aktív észterrel 0,9 ekvivalens szekunder amint
acileztem dimetilformamidban. A gyanta kiszőrése után a reakcióelegyben csak a termék
maradt. Ha az amin sósavsóként volt hozzáférhetı, a reakcióelegyhez adott, gyantához kötött
trimetil-amin hidrokarbonáttal szabadítottam fel a bázist. 24 győrős szekunder amint
acileztem ílymódon (16. ábra). A célmolekulák harmadát sikerült izolálni, a kiindulási
fahéjsav és a termékek rendkívül rossz oldékonysága miatt. A rosszul oldódó fahéjsavakból
nem képzıdött aktív észter, míg a rosszul oldódó termékek a gyantával együtt kiszőrésre
kerültek.

56
AB
C
O AB
C
O
OHCOOH
COOH
piperidin+
CH2 N C
O
F F
F F
O C
O
CH CH C
BA
H
HNR1
R2HN
R1
R2 HCl
,N(CH3)3x
HCO3 O
N
R2
R1A
BC
0,1 mmol
0,09 mmol
piridin
DMF, 5 óra szobahõfokon
3 óra reflux
CH2 N C
O
F F
F F
OHH
Argonaut PS-TFP 1,1 mmol/g
DIC, DMAP
DMF, DCM , 5 óra szobahõfokon
14. ábra. Fahéjsavamidok szilárd fázisú, kombinatorikus kémiai elıállítása .
O
N O
O
OH
H
O
OHO
NOH
O
N O
O
OHH
O
NO
O
OH
H
O
N S
O
OH
H
O
N
O
OH S
H
N
N O
O
OH
H
H
HN Ac
O
OH
HNMez
O
OH
N O
O
OH
H
15. ábra. Fahéjsavamidok szilárd fázisú, kombinatorikus kémiai elıállításához felhasznált fahéjsavak.

57
CH2 N NH
CH2 NH
CH2 NHF
CH2 NHCH3
CH2 NHCl
CH2 NH
CH2 NHCH3O
CH2
NH
CH3O
CH2
HN
CH3O
NH
HO
Cl
NH
N NH
OCH3
NBr NH
NCH3 NH
CH3
CH2 CH2 NHF
O NHCl
N NH
F3C
H
CH2 N NHH
NCl NH
CH3
CH2 O NH
CH CH CH2 NH
N
NNH
H
NNH
H
N NH
16. ábra. Fahéjsavamidok szilárd fázisú, kombinatorikus kémiai elıállításához felhasznált szekunder aminok.
A biológiailag tesztelt 74 vegyület közül a benzoxazolonnal kondenzált fahéjsavval acilezett
4-benzil-piperidin-származék (81) bizonyult a leghatásosabbnak.
HO
O
N
O
NO
NO
H 37 81 NR2B-IC50: 131 nM NR2B-IC50: 28 nM
4.2. Indol-2-karbonsav-származékok
Kutatásunk elsı idıszakára az útkeresés volt jellemzı. A CoCensys elsı publikációi után a fı
csapásirányt a bázikus nitrogént nem tartalmazó, savamid jellegő vegyületek irányában
határoztuk meg. A fahéjsavamidok körében végzett munkám utólag egy tanulási folyamatként
fogható fel. Kisebb-nagyobb lépésekkel próbáltam tágítani az addig ismert farmakofór modell
szők kereteit. A fenolos hidroxilcsoport helyett metabolikusan stabilabb hidrogénkötés donor

58
tulajdonságú csoportokkal szubsztituáltam az aromás győrőt. Az NR2B szelektív NMDA
antagonistákra addig kizárólag jellemzı 4-fenil-piperidin és 4-benzil-piperidin szekunder
aminkomponens helyett más aminokat is felhasználtam. Törekedtem a fahéjsav sztiril
részének eliminálására, mivel biológiai rendszerekben potenciálisan elektrofil támadás
célpontja lehet. Módosításaimmal nem sikerült kellıképpen eltávolodni a CoCensys 35 és 37
számú fahéjsavamidjaitól. Az elsı hatásos molekulát eredményezı jelentıs változtatás aztán
alapvetıen új irányt szabott munkánknak. Feltételeztük, hogy egy újabb hidrogénkötés donor
tulajdonságú csoporttal történı merevítéssel növelhetjük az aktivitást. A fahéjsav α
szénatomja és a benzolgyőrő közé beépített nitrogén 6-hidroxi-2-indolkarboxamid-
származékot eredményezett (82) mely hétszer hatásosabb volt 37 anyavegyületnél.
N
O
N HO
H 1
2
34
5
67
W
V XN
QZ
Y
82 NR2B-IC50: 18 nM A
Vezérmolekulánknak a 82 indol-2-karboxamidot választottuk és elhatároztuk, hogy a
molekulát optimalizáljuk, származékok képzésével feltárjuk, milyen kapcsolat van a biológiai
aktivitás és az A általános képleten jelölt, következı szerkezeti változtatások között:
Hidroxilcsoport helyzete a benzolgyőrőn (4.2.1.).
Az X-csoport természete (4.2.2.).
A piperidingyőrő szubsztituensének a helyzete (4.2.3.).
Az Y összekötı lánc helyzete (4.2.4.).
A Z-csoport természete és helyzete (4.2.5.).
Az indol pirrolrészének lehetséges helyettesítése más karbo- ill. heterociklusokkal (4.2.6.).
Vezérmolekulánkat a farmakológiai szőrırendszerünk 1. lépcsıjében lévı funkcionális
NMDA teszt alapján optimalizáltuk. Már a molekulák tervezésekor figyelemmel voltunk a
szabadalmaztathatóságra. Az optimalizálás során az in vitro aktivitás növelése mellett a
gyógyszerszerőség (Lipinsky szabályok, stb.) és az ADME tulajdonságok (logP, oldhatóság,
stb.) javítására is törekedtünk.
4.2.1. Hidroxilcsoport helyzete az indolgyőrőn
Elsıként az indolgyőrő hidroxiszubsztituensének szerepét tanulmányoztam. Azt vizsgáltam,
hogy a hidroxilcsoport helyének és számának változtatása hogyan befolyásolja a biológiai
aktivitást. Az eredmények a 2. táblázatban azt mutatják, hogy a 82 vegyület 5- és 4-hidroxi-

59
analogonjai (83 és 84) hasonló aktivitással rendelkeznek, míg a 7-hidroxi-analogonnak (85)
kisebb az aktivitása. Ez azt jelzi, hogy valószínőleg egynél több hidrogénkötés akceptor
csoport van a receptor megfelelı helyein. Feltételezésünket megerısíti az a megfigyelés,
miszerint a 4,6-dihidroxi-származéknak (86) nagyobb az aktivitása a monohidroxi-
származékoktól, a vegyület azonos hatású volt a legaktívabb referenciavegyülettel a
besonprodillal (28) (3.táblázat). A 85 vegyület hatásának a csökkenése a 82-84 vegyületekhez
képest, valamint a 87 vegyület kisebb aktivitása a 86-os vegyülethez képest a 7-es helyzető
hidroxilcsoport gyenge kölcsönhatását tükrözi a receptorral. Ennek oka valószínőleg az, hogy
ez a hidroxilcsoport nem a receptorral, hanem az indol NH-csoportjával létesít
intramolekuláris hidrogénkötést.
2. táblázat. Hidroxilcsoportok helyének és számának hatása a biológiai aktivitásra, funkcionális és kötıdési tesztadatok alapján.
NN
O
Q
H
vegyület
Q
NMDA-kiváltott ∆ [Ca2+]i
a
IC50 ± S.E.M. (nM)
n
[3H]Ro-25,6981 kötıdés
IC50 ± S.E.M. (nM)
n
82 6-OH 18 ± 4 13 12±2 4
83 5-OH 25 ± 8 3 31±10 3
84 4-OH 16 ± 2 8 18±5 4
85 7-OH 165 ± 30 4 380±141 3
86 4,6-di-OH 6,5 ± 0,7 3 6±1 3
87 5,7-di-OH 31 ± 3 3 106±19 3 aNMDA-kiváltott intracelluláris Ca2+ koncentrációváltozás patkány kortikális sejteken meghatározva.
3. táblázat. Referenciavegyületek in vitro biológiai adatai.
aNMDA-kiváltott intracelluláris Ca2+ koncentrációváltozás patkány kortikális sejteken meghatározva.
referenciavegyületek NMDA-kiváltott
∆∆∆∆[Ca2+]ia
IC50 ± S.E.M. (nM)
n
[3H]Ro-25,6981 kötıdés
IC50 ± S.E.M. (nM)
n
CP-101,606 41 ± 5 7 7 ± 1 6
28 (besonprodil) 6,6 ± 1,2 3 4 ± 1 3
9 (Ro-256981) 159 ± 29 3 6 ± 1 3
37 131 ± 10 2 196 ± 43 3

60
4.2.2. Az X-csoport természete
Megvizsgáltam a 82 molekula karbonsavamid funkciójának szerepét. Cseréje
karbonsavamidinre aktívabb analogont (88) eredményezett, míg a CO-csoport cseréje CH2-
vagy SO2-csoportra (89 és 90) a hatás elvesztésével járt (4. táblázat).
4. táblázat. A 82 molekula különbözı X-csoportot tartalmazó származékainak funkcionális és kötıdési tesztadatai.
N XN
HOH
vegyület
X
NMDA-kiváltott ∆ [Ca2+]i
a
IC50 ± S.E.M. (nM)
n
[3H]Ro-25,6981 kötıdés
IC50 (nM)
n
88 C=NH 8,5 ± 1,7 5 10 2
89 CH2 1158 ± 231 3 N.D. -
90 SO2 > 8000 1 N.D. - aNMDA-kiváltott intracelluláris Ca2+ koncentrációváltozás patkány kortikális sejteken meghatározva.
4.2.3. A piperidingyőrő szubsztituensének helyzete
A benzilcsoport piperidingyőrőn való helyzetének szerepét a 4-metoxi-benzil-származékok
esetében tanulmányoztam (91-93). Ebben az esetben mind a 3-as (92) mind a 2-es helyzet
(93) szubsztitúciója a hatás alapvetı csökkenésével járt (5. táblázat).
5. táblázat. A 82 molekula piperidingyőrőjén lévı benzilcsoport helyzetének in vitro biológiai hatása funkcionális és kötıdési tesztadatok alapján.
.
N
O
N HO
OMe
23
4
H
vegyület
szubsztitúció
NMDA-kiváltott ∆ [Ca2+]i
a
IC50 ± S.E.M. (nM)
n
[3H]Ro-25,6981 kötıdés
IC50 (nM)
n
91 4-(4-MeO)Bn 80 ± 14 3 17 2
92 3-(4-MeO)Bn 2206 ± 406 7 N.D. -
93 2-(4-MeO)Bn > 8000 ± 2 2 N.D. - aNMDA-kiváltott intracelluláris Ca2+ koncentrációváltozás patkány kortikális sejteken meghatározva.

61
4.2.4. Az Y összekötı lánc helyzete
A 82 molekula másik érzékeny pontja a terminális benzolgyőrőt és a piperidingyőrő 4-es
helyzető szénatomját összekötı lánc. A metiléncsoport elhagyása (94) és egy
metiléncsoporttal való megnyújtása (95) kevésbé aktív vegyületeket eredményezett. A hatás
hasonló csökkenése figyelhetı meg, amikor a CH2-csoportot CO-, CHOH- vagy NH-
csoportra cseréljük (99-101). Meglepı módon a benziloxi-piperidin- és a fenoximetil-
piperidin-származékok aktivitása (97 és 98) közelebb áll a 82 vegyület aktivitásához, mint a
geometriailag hozzá hasonlóbb 95 feniletil-piperidin-származékéhoz (6.táblázat). Ez egy
nyilvánvalóan új példája a Friedman paradoxonnak.96
6. táblázat. A 82 molekula különbözı Y-csoportot tartalmazó származékainak in vitro biológiai hatása funkcionális és kötıdési tesztadatok alapján.
N
O
N
Y
HOH
vegyület
Y
NMDA-kiváltott ∆[Ca2+]i
a
IC50 ± S.E.M. (nM)
n
[3H]Ro-25,6981 kötıdés
IC50 (nM)
n
94 - > 8000 ± 2 2 N.D. -
95 CH2CH2 875 ± 187 5 359 ± 93 3
96 O 107 ± 27 3 89 2
97 OCH2 16 ± 3 9 47 ± 4 3
98 CH2O 36 ± 6 7 19 ± 2 3
99 CO 744 ± 131 4 N.D. -
100 CHOH 111 ± 23 4 60 2
101 NH 348 ± 75 6 1084 2 aNMDA-kiváltott intracelluláris Ca2+ koncentrációváltozás patkány kortikális sejteken meghatározva.
4.2.5. A Z-csoport természete és helyzete
A 82 molekula benzil-piperidin részének benzolgyőréjét helyettesítı csoportok minıségének
és helyzetének aktivitásra gyakorolt hatását a 7. táblázatban foglalom össze. Az elıállított
néhány származék (102-105) kisebb aktivitással rendelkezik a kiindulási 82 anyavegyülethez
képest.

62
7. táblázat. A 82 molekula különbözı Z-csoportot tartalmazó származékainak in vitro biológiai hatása funkcionális és kötıdési tesztadatok alapján.
HN
O
N HO
Z
23
4
vegyület
Z
NMDA-kiváltott ∆ [Ca2+]i
a
IC50 ± S.E.M. (nM)
n
[3H]Ro-25,6981 kötıdés
IC50 (nM)
n
102 4-F 90 ± 15 6 12 ± 3 3
103 4-Me 208 ± 34 3 28 2
104 2-MeO 249 ± 55 3 N.D. -
105 3-F 38 ± 8 6 31 1 aNMDA-kiváltott intracelluláris Ca2+ koncentrációváltozás patkány kortikális sejteken meghatározva.
4.2.6. Indol-2-karbonsav-származékok elıállítása
A 82-85, 91-105 monohidroxiszármazékokat a megfelelı indol-2-karbonsavakból (106-109)
és piperidinszármazékokból állítottam elı egy ma már gyakori amid kapcsolási reakciót
felhasználva.97 A kapcsolási reakcióban karbonsavból 2-(1H-benztriazol-1-il)-1,1,3,3-
tetrametilurónium-hexafluorofoszfáttal (HBTU) és trietil-aminnal képzett aktív észter szerepel
acilezı ágensként (17. ábra).
+
HN
O
OHQ
YZ
i
106 Q = 6-OH107 Q = 5-OH108 Q = 4-OH109 Q = 7-OH
82, 91-105 Q = 6-OH (t.82: 71 %)83 Q = 5-OH (t.: 31 %)84 Q = 4-OH (t.: 90 %)85 Q = 7-OH (t.: 25 %)
HN
YZ
O
N
HN
Q
17. ábra. Monohidroxiszármazékok elıállítása. Reagensek és reakciókörülmények: (i) HBTU, Et3N, DMF, 0°C - szobahımérséklet, 16 óra.
A kereskedelmi forgalomban nem lévı és az irodalomban sem ismertetett indol-2-
karbonsavak egy részének metilésztereit (114, 115) Hemetsberger-Knittel szerint állítottam
elı.98 A 86 és 87 dihidroxiszármazékokhoz a megfelelı dibenziloxiszármazékok (116 és 117)
katalitikus hidrogenolízisével jutottam (18. ábra).

63
(t.: 38 %)(t.: 70 %)
(t.: 63 %)(t.: 27 %)
CHO
N3
O
OCH3
N
O
OCH3Q Q Q
N
O
NQ
110 Q = 2,4-di-BnO
111 Q = 3,5-di-BnO
i ii iii, iv
H
H
116 Q = 4,6-di-BnO
86 Q = 4,6-di-OH
117 Q = 5,7-di-BnO
87 Q = 5,7-di-OH
v
114 Q = 4,6-di-BnO
115 Q = 5,7-di-BnO
112 Q = 2,4-di-BnO 113 Q = 3,5-di-BnO
v
(t.: 43 %)
(t.: 60 %)
(t.: 21 %)
18. ábra. Dihidroxiszármazékok elıállítása. Reagensek és reakciókörülmények: (i) metil-azidoacetát, NaOMe, MeOH, 0°C, 4 óra (ii) forralás xilolban; (iii) KOSi(CH3)3, THF, reflux, 1 óra; (iv) 4-benzil-piperidin, HBTU, Et3N, DMF, 0°C - szobahımérséklet, 16 óra; (v) H2, 10 % Pd/C, MeOH, szobahımérséklet.
A 88 amidint 6-benziloxi-1H-indol-2-karbonsavból (118) kiindulva nitrilen (119) keresztül
Pinner reakcióban99 alakítottam ki (19. ábra).
(t.: 57 %)
HN
O
OHBnO
HN CNBnO N
NHO
NH
118 119 88
i, ii iii, iv
H
(t.: 4,5 %) 19. ábra. 88 karbonsavamidin elıállítása. Reagensek és reakciókörülmények: (i) 1. SOCl2, CHCl3, reflux, 1 óra; 2. NH3; (ii) POCl3 CHCl3, reflux, 2 óra; (iii) HCl, MeOH, 5 óra; (iv) 4-benzil-piperidin.
A 89 vegyületet 82 karbonsavamid lítium-alumíniumhidrides redukciójával állítottam elı (20.
ábra).
HNHO
N
OHNHO
N
82 89
i
(t.: 15 %)
HNHO
N
OHNHO
N
82 89
i
(t.: 15 %) 20. ábra. 89 származék elıállítása. Reagensek és reakciókörülmények: (i) LiAlH4, THF, szobahımérséklet, 1óra.
90 szulfonamid elıállításakor 6-benziloxi-1H-indolból (120) indultam ki. Többlépéses
reakciót egy edényben végrehajtva alakítottam ki 121 szulfonsav-kloridot. Az amidképzés
után a benzil védıcsoport hidrogenolízissel történı eltávolítása eredményezte 90
célvegyületet (21. ábra).

64
(ti.: 99 %) (tii+iii.:17 %)
HNBnO
H HN SHO
N
O O
i, ii iii, iv
120 121 90
N SBnOCl
O O
(tiv.:12 %)
21. ábra. 90 származék elıállítása. Reagensek és reakciókörülmények: (i) t-Boc2O, DMAP, CH2Cl2, szobahımérséklet, 2óra; (ii) 1. n-BuLi, SO2, THF, -70°C - szobahımérséklet; 2. NCS, CH2Cl2, 5°C; (iii) 4-benzil-piperidin, szobahımérséklet, 24 óra; (iv) H2, 10 % Pd/C, MeOH, szobahımérséklet.
A szintézisek során felhasznált piperidinszármazékok kereskedelmi forgalomban
beszerezhetıek vagy az irodalomból ismert eljárás szerint elıállíthatók. 4-fenoxi-
piperidineket 4-klór-piridinbıl és fenolokból képzett 4-fenoxi-piridin-származékok két
lépésben történı telítésével állítottam elı (22. ábra).100
HN
O
Y
(i) (ii)OH
Y N
Cl
+N
O
YN
O
YBr
N
O
Y
(iv)
124 tY = H.:79 % 125 tY = H.:99 %
126 tY = H.:69 % 127 tY = H.:99 %
122 123
(iii)
22. ábra . 4-fenoxi-piperidinek elıállítása. Reagensek és reakciókörülmények: (i) kat. 1-(4-piridil)piridinium- klorid hidroklorid, 220 oC, 6 óra; (ii) benzil-bromid, dietil éter, 20 oC, 48 óra; (iii) NaBH4, EtOH; (iv) H2, 10% Pd/C, MeOH, szobahımérséklet.
Fenoximetil-piperidineket etil-izonipekotátból kiindulva ismert módszer alapján állítottam elı
(23. ábra).101 A nitrogén védése után az észtercsoportot litium-alumíniumhidriddel a
megfelelı alkohollá redukáltam. Az alkoholból metánszulfonil-kloriddal mezilátot képeztem,
mellyel ekvivalens nátrium-hidroxiddal deprotonált fenolokat O-alkileztem. A Boc
védıcsoportot sósavas etilacetáttal távolítottam el.
129 ti.: 94 %, tii.: 78 %, t iii.: 93 %
N
O
Y
Boc
(i) (ii) (iv)(iii)
HN
O
Y
N
COOEt
(v)
N
OMez
Boc
131 Y = F : tiv, v.: 78 %H
128 130
23. ábra. 4-fenoximetil-piperidinek elıllítása. Reagensek és reakciókörülmények: (i) Boc2O, EtOAc, 22 óra, 0 oC - szobahımérséklet; (ii) LiAlH4, THF, 0 oC - szobahımérséklet; (iii) MezCl, Et3N, CH2Cl2, 0 oC – szobahımérséklet; (iv) szubsztituált fenolok, NaOH, DMF, 2 óra reflux; (v) 1. 10% HCl/EtOAc, 18 óra, 2. NaHCO3.

65
4-benziloxi-piperidineket N-Boc-4-piperidinol102 O-benzilezésével állítottam elı (24. ábra.).
(i)
N
OH
BocN
O
Y
BocHN
O
Y
(ii)
133 (Y = H : t.: 58 %) 134 (Y = H : t.: 36 %)132
24. ábra. 4-benziloxi-piperidinek elıállítása. Reagensek és reakciókörülmények: (i) NaH, DMF, benzilkloridok, 2 óra, szobahımérséklet; (ii) 1. 30% TFA - CH2Cl2, 1 óra szobahımérséklet, 2. NaHCO3.
4-benzoil-piperidint egy új, eddig nem publikált eljárással állítottam elı. Nitrogénen nem
védett etil-izonipekotátot Grignard reakcióban ekvivalens fenil-magnézium-bromiddal
reagáltatva jó termeléssel 4-benzoil-piperidint eredményezett. A piperidin nitrogénjének Boc-
csoporttal történı védelme után a karbonilcsoport nátrium-borohidrides redukciójával, majd a
védıcsoport eltávolításával a megfelelı alkoholhoz jutottam (25. ábra).
135 ( t.: 70 %) 136 ( tii, iii, iv.: 54 %)
(i)
N
O
OEt (ii) (iii) (iv)
H H N
O
H N
OH
128 25. ábra. 4-benzoil-piperidin elıállítása és redukciója. Reagensek és reakciókörülmények: (i) PhMgBr, THF, 1óra reflux; (ii) Boc2O, Et3N, dioxán-víz, 18 óra, szobahımérséklet; (iii) NaBH4, MeOH, 18 óra, szobahımérséklet; (iv) ) 1. 10% HCl/EtOAc, 18 óra, 2. NaHCO3.
2-, 3- és 4-(szubsztituált-benzil)piperidineket egy új és hatékony eljárás szerint állították elı
mőegyetemi partnereink.103
4.3. Benzimidazol-2-karbonsav-származékok
4.3.1. Az indol pirrolrészének helyettesítése más karbo-, illetve heterociklusokkal
A továbbiak során a 82 molekula NH-csoportjának fontosságát mértük fel. Az N-Me analogon
(137) inaktív volt. Kutatótársaimmal benztiazol, benzofurán és benztiofén analogonokat
állítottunk elı. A megfelelı benzofurán (138) és benztiazol (139) származékoknak szintén
gyenge volt az aktivitásuk. Az 5-hidroxi-benzofurán izomer (141) és az 5-hidroxi-benztiofén-
származék (140) hasonlóan inaktívnak bizonyultak. Az eredmények azt jelzik, hogy a 82
vegyület NH-csoportjának fontos szerepe van a receptorral való kölcsönhatásban. Ezt a
megfigyelést támasztja alá az a tény, hogy a 142 benzimidazol-2-karboxamid szintén hatásos
66
vegyület. Egy nitrogénatom beépítése a 82 molekulába kilencszeres aktivitásnövekedéssel az
egyik leghatásosabb NR2B szelektív NMDA antagonistát eredményezte (142) (8. táblázat).
8. táblázat. Az indol pirrolrészének helyettesítése különbözı V- és W-csoportokkal.
W
V
O
N HO
45
67
vegyület
szubsztituens
V
W
NMDA-kiváltott ∆∆∆∆[Ca2+]i
a
IC50 ± S.E.M. (nM)
n
[3H]Ro-25,6981 kötıdés
IC50 (nM)
n
137 6-OH NMe CH 5773 ± 958 4 N.D. -
138 6-OH O CH 6920 ± 1280 4 >10000 1
139 6-OH S N 2597 ± 369 3 N.D. -
140 5-OH S CH 5613 ± 1066 5 >10000 1
141 5-OH O CH 1901 ± 295 5 >10000 1
142 5(6)-OH NH N 2,2 ± 0,4 7 4 2 aNMDA-kiváltott intracelluláris Ca2+ koncentrációváltozás patkány kortikális sejteken meghatározva. 142 benzimidazol-származékot, mivel funkcionális teszteredménye csaknem egy
nagyságrenddel jobb volt mint a 82 anyavegyületé, új vezérmolekulává választottuk és
megkezdtük a molekula optimalizálását.
4.3.2. Benzimidazol-2-karboxamidok optimalizációja
A benzimidazol és indolgyőrők szerkezeti hasonlósága miatt remélhettük, hogy a molekula
többi részére érvényesek lesznek az indolszármazékoknál megfigyelt szerkezet-hatás
összefüggések. Néhány benzimidazol analogon elıállítása és tesztelése után feltételezésünk
igazolódott, a két heterociklus az NR2B receptoron bioizosztér párként viselkedik, ráadásul a
legtöbb esetben érvényesült a 142 és 82 vegyületpárnál megfigyelhetı egy nagyságrendbeli in
vitro aktivitásbeli különbség a benzimidazol-származékok javára. Ezek után elsısorban a
hatásos indolszármazékok benzimidazol megfelelıjét állítottam elı, jelentısen lecsökkentve
az optimalizáció idejét. Az elıállított és tesztelt vegyületek közül néhányat a 9. táblázatban
mutatok be. A 4(7)-hidroxi-izomer (143) azonos hatású a 142 vezérmolekulával. Az OH-
csoport cseréje NH2-csoporttal kevésbé aktív analogont eredményezett (144). A megfelelı
mezilamino-származék (145) egy kicsit hatásosabb a 142 anyavegyületnél. A 142
molekulában az OH helyettesítése egy a benzimidazollal kondenzált oxazolinongyőrő NH-
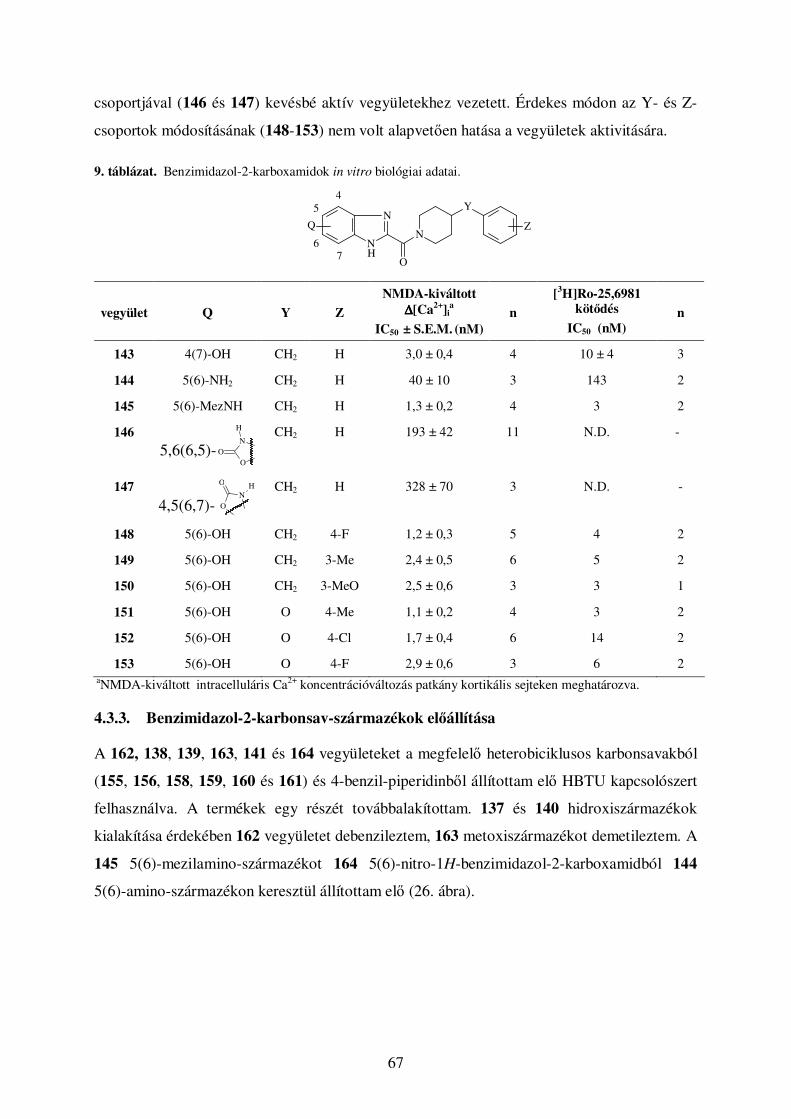
67
csoportjával (146 és 147) kevésbé aktív vegyületekhez vezetett. Érdekes módon az Y- és Z-
csoportok módosításának (148-153) nem volt alapvetıen hatása a vegyületek aktivitására.
9. táblázat. Benzimidazol-2-karboxamidok in vitro biológiai adatai.
H
N
N
O
N
Y
Q Z
45
67
vegyület
Q
Y
Z
NMDA-kiváltott ∆∆∆∆[Ca2+]i
a
IC50 ± S.E.M. (nM)
n
[3H]Ro-25,6981 kötıdés
IC50 (nM)
n
143 4(7)-OH CH2 H 3,0 ± 0,4 4 10 ± 4 3
144 5(6)-NH2 CH2 H 40 ± 10 3 143 2
145 5(6)-MezNH CH2 H 1,3 ± 0,2 4 3 2
146
O
N O
H
5,6(6,5)-
CH2 H 193 ± 42 11 N.D. -
147 O
NH O
4,5(6,7)-
CH2 H 328 ± 70 3 N.D. -
148 5(6)-OH CH2 4-F 1,2 ± 0,3 5 4 2
149 5(6)-OH CH2 3-Me 2,4 ± 0,5 6 5 2
150 5(6)-OH CH2 3-MeO 2,5 ± 0,6 3 3 1
151 5(6)-OH O 4-Me 1,1 ± 0,2 4 3 2
152 5(6)-OH O 4-Cl 1,7 ± 0,4 6 14 2
153 5(6)-OH O 4-F 2,9 ± 0,6 3 6 2 aNMDA-kiváltott intracelluláris Ca2+ koncentrációváltozás patkány kortikális sejteken meghatározva.
4.3.3. Benzimidazol-2-karbonsav-származékok elıállítása
A 162, 138, 139, 163, 141 és 164 vegyületeket a megfelelı heterobiciklusos karbonsavakból
(155, 156, 158, 159, 160 és 161) és 4-benzil-piperidinbıl állítottam elı HBTU kapcsolószert
felhasználva. A termékek egy részét továbbalakítottam. 137 és 140 hidroxiszármazékok
kialakítása érdekében 162 vegyületet debenzileztem, 163 metoxiszármazékot demetileztem. A
145 5(6)-mezilamino-származékot 164 5(6)-nitro-1H-benzimidazol-2-karboxamidból 144
5(6)-amino-származékon keresztül állítottam elı (26. ábra).
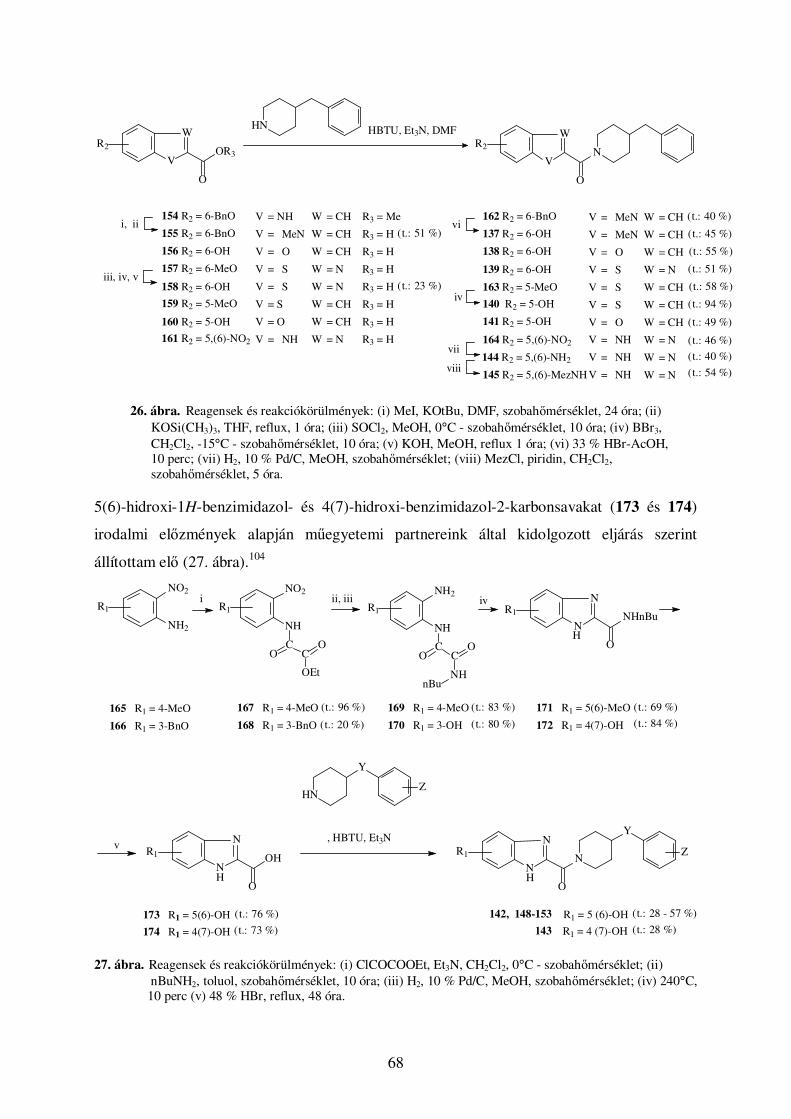
68
(t.: 40 %)
V
W
O
NR2
V
W
O
OR3R2
HN HBTU, Et3N, DMF
V1= NH
V1= MeN
V1= O
V1= S
V1= S
V1= S
V1= O
V1= NH
i, ii 154 R2 = 6-BnO
155 R2 = 6-BnO
156 R2 = 6-OH
157 R2 = 6-MeO
158 R2 = 6-OH
159 R2 = 5-MeO
160 R2 = 5-OH
161 R2 = 5,(6)-NO2
W1= CH
W1= CH
W1= CH
W1= N
W1= N
W1= CH
W1= CH
W1= N
R3 = Me
R3 = H
R3 = H
R3 = H
R3 = H
R3 = H
R3 = H
R3 = H
iii, iv, v
vi
viii
162 R2 = 6-BnO
137 R2 = 6-OH
138 R2 = 6-OH
139 R2 = 6-OH
163 R2 = 5-MeO
140 R2 = 5-OH
141 R2 = 5-OH
164 R2 = 5,(6)-NO2
144 R2 = 5,(6)-NH2
145 R2 = 5,(6)-MezNH
V1= MeN
V1= MeN
V1= O
V1= S
V1= S
V1= S
V1= O
V1= NH
V1= NH
V1= NH
W1= CH
W1= CH
W1= CH
W1= N
W1= CH
W1= CH
W1= CH
W1= N
W1= N
W1= N
iv
vii
(t.: 45 %)
(t.: 55 %)
(t.: 51 %)
(t.: 58 %)
(t.: 94 %)
(t.: 49 %)
(t.: 46 %)
(t.: 40 %)
(t.: 54 %)
(t.: 51 %)
(t.: 23 %)
26. ábra. Reagensek és reakciókörülmények: (i) MeI, KOtBu, DMF, szobahımérséklet, 24 óra; (ii) KOSi(CH3)3, THF, reflux, 1 óra; (iii) SOCl2, MeOH, 0°C - szobahımérséklet, 10 óra; (iv) BBr3, CH2Cl2, -15°C - szobahımérséklet, 10 óra; (v) KOH, MeOH, reflux 1 óra; (vi) 33 % HBr-AcOH, 10 perc; (vii) H2, 10 % Pd/C, MeOH, szobahımérséklet; (viii) MezCl, piridin, CH2Cl2, szobahımérséklet, 5 óra.
5(6)-hidroxi-1H-benzimidazol- és 4(7)-hidroxi-benzimidazol-2-karbonsavakat (173 és 174)
irodalmi elızmények alapján mőegyetemi partnereink által kidolgozott eljárás szerint
állítottam elı (27. ábra).104
142, 148-153
NO2
NH2
NO2
NH
CO C
O
OEt
NH2
NH
CO C
O
NHnBu
HN
N
O
NHnBuR1 R1 R1R1
v
HN
N
O
OH
HN
Y
Z
, HBTU, Et3N
HN
N
O
N
Y
ZR1 R1
165 R1 = 4-MeO
166 R1 = 3-BnO
171 R1 = 5(6)-MeO
172 R1 = 4(7)-OH
173 R1 = 5(6)-OH
174 R1 = 4(7)-OH
i ii, iii iv
167 R1 = 4-MeO
168 R1 = 3-BnO
169 R1 = 4-MeO
170 R1 = 3-OH
R1 = 5 (6)-OH
R1 = 4 (7)-OH
(t.: 96 %)
(t.: 20 %)
(t.: 83 %)
(t.: 80 %)
(t.: 69 %)
(t.: 84 %)
(t.: 76 %)
(t.: 73 %)
(t.: 28 - 57 %)
(t.: 28 %)143 27. ábra. Reagensek és reakciókörülmények: (i) ClCOCOOEt, Et3N, CH2Cl2, 0°C - szobahımérséklet; (ii) nBuNH2, toluol, szobahımérséklet, 10 óra; (iii) H2, 10 % Pd/C, MeOH, szobahımérséklet; (iv) 240°C, 10 perc (v) 48 % HBr, reflux, 48 óra.

69
Az piperidinszármazékokat 173 és 174 savakkal acilezve 142, 143 és 148-153 hidroxi-
benzimidazol-2-karboxamidokhoz jutottam. A kapcsolási reakcióban HBTU-t alkalmaztam
(27. ábra). 146 heterociklusos karbonsavamidot a 25. ábrán bemutatott benzimidazol-
szintézishez hasonló módon állítottam elı 178 anilinszármazékból és 176 savkloridból. Ez
utóbbiakat 6-aminobenzoxazolinonból (177) és 4-benzil-piperidinbıl (175) alakítottam ki (28.
ábra).
(t.: 92 %)
(t.: 78 %)
(t.: 46 %) (t.: 5,7 %)
HN NCl
O
O
v
i, ii, iii
iv
vi, viiN
OO
NO2
N N
O
O
H
HN
N
O
N
N
OO
H
H
175 176
177 178
179 146
N
OO
NH2
HN
OO
NO2
NH2
H
28. ábra. Reagensek és reakciókörülmények: (i) ClCOCOOEt, DIPEA, CH2Cl2, 0°C; (ii) 1.KOH, MeOH, szobahımérséklet; 2. HCl; (iii) SOCl2, reflux, 2 óra; (iv) NaNO3, TFA, szobahımérséklet; (v) CHCl3, Et3N, szobahımérséklet; (vi) H2, 10 % Pd/C, MeOH, szobahımérséklet; (vii) 240°C, 10 perc.
147 heterociklusos karbonsavamid elıállításakor 5(6)-hidroxi-1H-benzimidazol-2-
karbonsavból (173) indultam ki. A 4(7) helyzetben történı nitrálás és 4-benzil-piperidinnel
való kapcsolás 182 karbonsavamidot eredményezte. A nitrocsoport redukcióját követı
karbonildiimidazolos győrőzárás vezetett a kívánt végtermékhez (29. ábra).
(t.: 68 %) (t.: 65 %)
(t.: 54 %) (t.: 93 %)
i ii iii
173 180 181
iv
182 147
HN
N
O
OH
HO
HN
N
OH
NO2
HO
OHN
N
O
N
NO2
HO
HN
N
O
N
NH2
HO
HN
N
O
N
NHO
O
29. ábra. Reagensek és reakciókörülmények: (i) NaNO3, TFA, szobahımérséklet; (ii) 4-benzil-piperidin, HBTU, Et3N, DMF; (iii) H2, 10 % Pd/C, THF, szobahımérséklet; (iv) 1,1’-karbonildiimidazol, THF, szobahımérséklet.
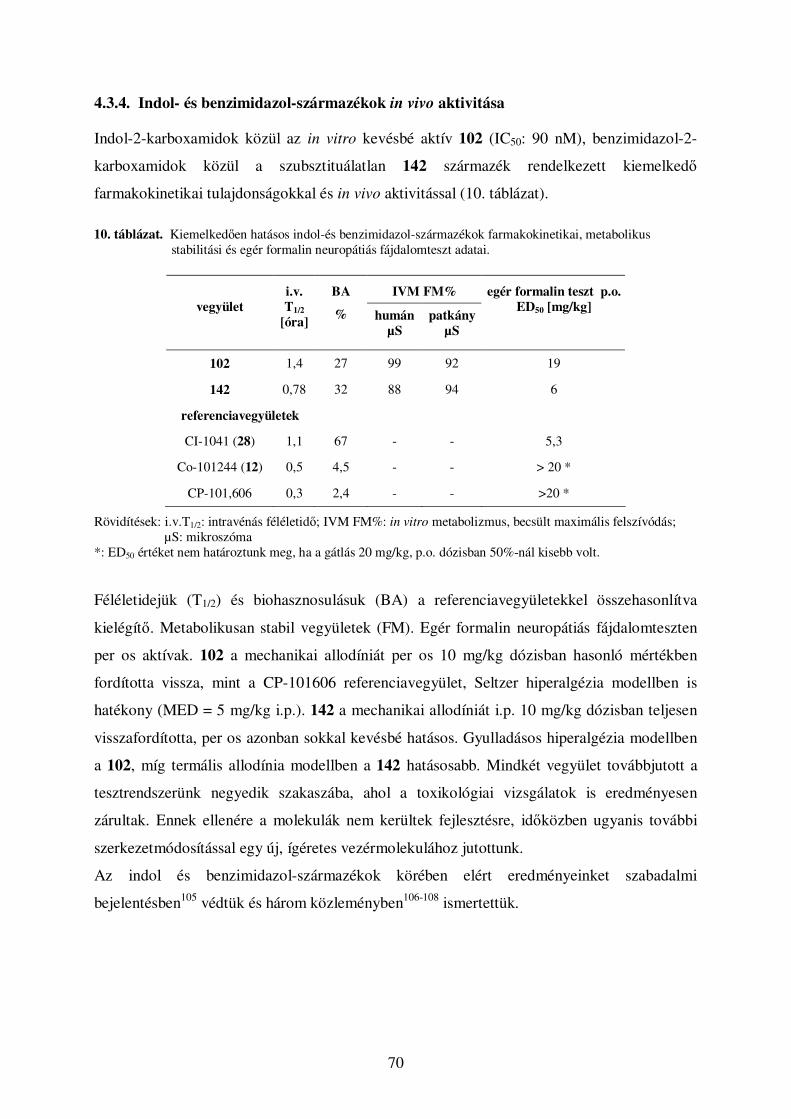
70
4.3.4. Indol- és benzimidazol-származékok in vivo aktivitása
Indol-2-karboxamidok közül az in vitro kevésbé aktív 102 (IC50: 90 nM), benzimidazol-2-
karboxamidok közül a szubsztituálatlan 142 származék rendelkezett kiemelkedı
farmakokinetikai tulajdonságokkal és in vivo aktivitással (10. táblázat).
10. táblázat. Kiemelkedıen hatásos indol-és benzimidazol-származékok farmakokinetikai, metabolikus
stabilitási és egér formalin neuropátiás fájdalomteszt adatai.
Rövidítések: i.v.T1/2: intravénás féléletidı; IVM FM%: in vitro metabolizmus, becsült maximális felszívódás; µS: mikroszóma *: ED50 értéket nem határoztunk meg, ha a gátlás 20 mg/kg, p.o. dózisban 50%-nál kisebb volt.
Féléletidejük (T1/2) és biohasznosulásuk (BA) a referenciavegyületekkel összehasonlítva
kielégítı. Metabolikusan stabil vegyületek (FM). Egér formalin neuropátiás fájdalomteszten
per os aktívak. 102 a mechanikai allodíniát per os 10 mg/kg dózisban hasonló mértékben
fordította vissza, mint a CP-101606 referenciavegyület, Seltzer hiperalgézia modellben is
hatékony (MED = 5 mg/kg i.p.). 142 a mechanikai allodíniát i.p. 10 mg/kg dózisban teljesen
visszafordította, per os azonban sokkal kevésbé hatásos. Gyulladásos hiperalgézia modellben
a 102, míg termális allodínia modellben a 142 hatásosabb. Mindkét vegyület továbbjutott a
tesztrendszerünk negyedik szakaszába, ahol a toxikológiai vizsgálatok is eredményesen
zárultak. Ennek ellenére a molekulák nem kerültek fejlesztésre, idıközben ugyanis további
szerkezetmódosítással egy új, ígéretes vezérmolekulához jutottunk.
Az indol és benzimidazol-származékok körében elért eredményeinket szabadalmi
bejelentésben105 védtük és három közleményben106-108 ismertettük.
IVM FM%
vegyület i.v. T1/2
[óra]
BA
% humán µS
patkány µS
egér formalin teszt p.o. ED50 [mg/kg]
102 1,4 27 99 92 19
142 0,78 32 88 94 6
referenciavegyületek
CI-1041 (28) 1,1 67 - - 5,3
Co-101244 (12) 0,5 4,5 - - > 20 *
CP-101,606 0,3 2,4 - - >20 *

71
4.4. Oxálsavdiamidok
4.4.1. Eredmények, szerkezet-hatás összefüggések
Eddigi kutatásunk során N-acilezett 4-benzil-piperidin-származékokkal, indol és
benzimidazol-2-karboxamidokkal sikerült bizonyítanunk hipotézisünk helyességét, miszerint
bázikus nitrogén jelenléte nem alapfeltétele az aktivitásnak. Megállapítottuk továbbá, hogy az
indol és benzimidazolgyőrő szubsztituálatlan NH-csoportja fontos a hatás szempontjából. Új
szerkezetek tervezésekor olyan módosításokat hajtottunk végre, amelyek nem érintik ezeket a
farmakofór jellemzıket. Egyik ilyen szerkezeti módosítás eredménye az 183 oxálsavdiamid-
származék lett, mely kétszer olyan hatásos volt, mint a 83 indolszármazék, mind a natív
patkány idegsejteken mért funkcionális, mind a kötıdési teszten.
HN
N
HO
O
O
HN
N
HO
O 83 183
NR2B-IC50: 25 nM NR2B-IC50: 11 nM [3H]Ro-25,6981-IC50: 31 nM [3H]Ro-25,6981-IC50: 16 nM
Elhatároztuk, hogy a fenolos hidroxilcsoportot, kedvezıtlen metabolikus tulajdonságai miatt
legalább egy NH-csoportot tartalmazó kondenzált heterociklussal helyettesítjük, ezzel pótolva
a hidroxilcsoport hidrogénkötés donor sajátságát. Célunk volt továbbá a piperidin 4-es
helyzetét a jobboldali benzolgyőrővel összekötı lánc és a benzolgyőrő szubsztitúciójának
optimalizálása. Reprezentatív oxamidszármazékok (183-188), valamint a
referenciavegyületek biológiai adatait az 11. táblázat tartalmazza. Az elıállított vegyületek
biológiai aktivitását [3H]Ro-25,6981 kötıdési tesztben és a rekombináns NR1/NR2B
receptorokat expresszáló sejteket felhasználó funkcionális esszében határoztuk meg, melynek
során az NMDA által kiváltott intracelluláris Ca2+ szint növekedésének gátlását mértük.
Szelektivitásukat az NR1/NR2A alegységeket tartalmazó NMDA rceptorokhoz képest
ugyanebben a funkcionális esszében határoztuk meg, rekombináns NR1/NR2A receptorokat
expresszáló sejteket használva. In vivo analgetikus hatást a hosszantartó fájdalmat modellezı
egér formalin tesztben mértük.

72
11. táblázat. Referenciavegyületek és néhány reprezentatív oxálsavdiamid-származék (183-188) kötıdési és funkcionális tesztjeinek eredményei valamint in vivo aktivitási adatai.
H
N
X
YN
O
O
Q
a Átlagértékek ± szórás. A kísérletek száma [3H]Ro-25,6981 kötıdési teszt esetében 3-4, NR1/NR2B funkcionális teszt esetében 4-5, NR1/NR2A esetében 1-2. b NMDA-kiváltott intracelluláris Ca2+ koncentrációváltozás rekombináns NR1/NR2B receptorokat expresszáló sejteken meghatározva. c Inhibíció (%) a vegyület 15 µM koncentrációjánál meghatározva. d Klinikai fejlesztésre kiemelt vegyület. A fenolos hidroxilcsoport helyére a versenytársak által is alkalmazott heterociklusokat
beépítve rendkívől aktív és szelektív benzoxazolon (184, 185), benzimidazolon (186),
benztiazolon- (187) és indanon- (188) származékokat sikerült elıállítani. Ezek a vegyületek in
vitro és in vivo is felülmúlják az anyavegyületek (83, 183) és a referenciavegyületek
aktivitását. A sikereken felbuzdulva folytattuk az oxálsavdiamidok körében végzett kémiai
munkát, hiszen egyrészt a tesztrenszerben elırehaladva számos vizsgálatnál elbukhattak még
ezek az igen aktív vegyületek is, másrészt közepes farmakokinetikai paramétereik javításától
vegyület
Q
X
Y oldé-
konyság [µg/ml]
[3H]Ro-25,6981 kötıdésa
IC50 (nM)
NMDA-kiváltott
∆∆∆∆[Ca2+]ia,b
IC50 (nM)
NR1/NR2Aa inhibícióc
(%)
egér formalin teszt, p.o.
ED50 (mg/kg)
83
156 31±10 42±13 18 1,4
183 HO
CH2 H - 16±2 31±6 20 9,4
184 O
N O
H
CH2 H 13 8±2 3±1 9 0,9
185d O
N O
H
CH2 F 15,2 6±1 4±1 5 7,7
186 N
N O
H
H CH2 H 12 4±1 6±1 -1 14
187 S
N O
H
CH2 H 26,5 17±4 9±2 17 0,4
188 N O
H
CH2 CH3 23,3 54±6 15±2 7 0,5
Ro-25,6981 (9) 6±1 57±5 1 p.o. inaktív 5,1 (i.p.)
Co-101,244 (12) 4±1 5±1 -8,7 p.o. inaktív 5,9 (i.p.)
EMD 95885 (8) 8±1 48 0,1 3,7
CP-101,606
7±1 30±4 2,5 >20 (s.c és p.o.)
CI-1041(28)
4±1 8±1 21,0 2,4

73
in vivo még hatásosabb származékokat reméltünk. Iparjogvédelmi szempontból is kívánatos
volt minél többféle származék elıállítása.
A vegyületcsalád kutatásába bekapcsolódva két irányba indultam el. Egyik kitőzött célom a
versenytársak által még nem alkalmazott, egyéb területen is ritkán elıforduló, de azért
gyógyszerszerő heterociklusokkal kondenzált anilinek, majd ezekbıl oxálsavdiamidok
elıállítása volt. Másik célom az indolszármazékoknál szerzett pozitív tapasztalatok alapján a
4-benzil-piperidin helyett olyan analogonok elıállítása amelyben a piperidint és a
benzolgyőrőt összekötı lánc oxigént vagy kenet tartalmaz. Elsıként ez utóbbi területen elért
eredményeimet foglalom össze. Mivel a leghatásosabb molekulák általában benzoxazolon
győrőrendszert tartalmaznak, ezért a 12. táblázatban csak a benzoxazolon-származékok natív
idegsejteken mért funkcionális teszteredményeit mutatom be.
12. táblázat : A piperidin 4-es helyzetében oxigén- és kéntartalmú összekötı láncot tartalmazó benzoxazolon- származékok (189-199) NMDA antagonista hatása patkány kortikális sejteken.
H
HR1
NR2N
O
O
O
NO
vegyület 189 190 191 192 193 194 195 196 197 198 199
R1
NR2
N
O
Cl
N
S
N
SO
O
N
O
N
O
F
N
S
N
SO
O
N
O
N
O
N
SO
N
S O
NMDA-kiváltott ∆∆∆∆[Ca2+]i
a inhib. %b IC50 (nM)
7 3,9 85 11 9 9,9 11,7 % 26 12 220 25,7 %
a NMDA-kiváltott intracelluláris Ca2+ koncentrációváltozás. b Inhibíció (%) a vegyület 0,1 µM koncentrációjánál meghatározva.
Érdekes módon az oxigéntartalmú összekötı lánc hosszára az aktivitás nem igazán érzékeny.
A lánc egy-egy metiléncsoporttal történı hosszabbítása az aktivitás kismértékő csökkenését
idézi csak elı (189, 192, 197). A háromtagú összekötı lánc esetében már az oxigén láncon
belül elfoglalt helyzete is meghatározó. A 197, melyben az oxigénatom a piperidinhez
közelebb van, kétszer aktívabb mint a 196. A kénanalogonok hasonlóan aktívak, metabolikus
stabilitásuk azonban nem kielégítı, (190 humán mikroszóma 50 %, patkány mikroszóma 58
%; 194 humán mikroszóma 87 %, patkány mikroszóma 64 %). A kén oxidációjával az
aktivitás nagymértékben lecsökken (198, 191, 199, 195).

74
Számos oxigén és kéntartalmú összekötılánccal rendelkezı molekula továbbjutott az elsı in
vitro szőrıteszteken. A kaszkád második tesztrendszerében a legfontosabb szelekciós
kritérium a biohasznosulás és az egér formalin teszt elfogadható eredménye. Innen a harmadik
tesztrendszerbe csak a 193 jutott tovább.
Egér formalin tesztben ED50 értéke 1,4 mg/kg. Gyenge, 7 %-os biohasznosulását
hidroxipropil-ciklodextrinnel (HPCD) képzett komplexe révén sikerült 20 %-ra megemelni.
Ezt a formulációt alkalmazva egér neuropátiás modellben 0,25 mg/kg dózisban 60,2 %-os
választ váltott ki. Bennett-Xie patkány mechanikai allodíniás modellben hasonló nagyságú
választ (54%) csak 16 mg/kg dózisban sikerült elérni. Diabéteszes neuropátiás
fájdalomtesztben is hatásos, 8 mg/kg dózisban 46 % a válasz. 193 a gyulladásos
fájdalommodellekben a leghatékonyabb. Patkány gyulladásos artritisz modellben 0,2 mg/kg
dózisban hatásos. A komplett Freund adjuvánssal (CFA) indukált gyulladásos egér
fájdalomtesztben kialakúló allodíniát pedig már 0,1 mg/kg dózisban teljes mértékben gátolta.
Neuropátiás fájdalomtesztben tolerancia nem alakult ki. A vegyület jelentısebb, nagy
kockázatú mellékhatásoktól mentes. Nem gátolja a hERG csatornákat, nincs
kardiovaszkuláris mellékhatása. A CYP enzimekkel végzett vizsgálatok is eredményesen
zárultak. Ames teszt alapján nem mutagén. A Morris vízlabirintus tesztben nincs hatása a
tanulási folyamatokra. Terápiás dózisban a lokomotoros aktivitást nem növelte. Patkány
toxikológiai vizsgálatban 150 mg/kg/nap dózist is jól toleráltak az állatok. A reménykeltı
képet azonban néhány kedvezıtlen tulajdonság árnyalta. Ismételt adáskor kismértékben
csökkent az expozíció és az analgetikus dózishoz közel szedatív típusú mellékhatások
jelentkeztek. A vegyület inkább a gyulladásos fájdalmat csillapítja mint a célúl kitőzött
neuropátiás fájdalmat. Ellene szólt a kis biohasznosulási értéke is, emiatt HPCD komplexre
vagy valamilyen hatásos formulációra minden bizonnyal a humán alkalmazás során is
szükség lenne. Végül a vegyület nem került továbbfejlesztésre.
Az oxálsavdiamidok körében végzett munkám másik célkitőzése új kondenzált heterociklusok
alkalmazása a molekula baloldali részén. Ennek érdekében elıször a farmakofór modellbe
illeszkedı anilineket kellett elıállítani. Mivel a vegyületcsaládra jellemzı viszonylag gyenge
biohasznosulás oka a rossz oldékonyságból eredı elégtelen felszívódás, ezért új vegyületek
tervezésekor elsısorban az oldékonyság növelésére törekedtem, bár iparjogvédelmi
szempontból, a vegyületkör minél teljesebb lefedése érdekében bármilyen, a megfelelı
pozícióban NH-csoportot tartalmazó heterociklussal kondenzált anilinszármazék elıállítása
kívánatos volt. Az oldékonyság és ezen keresztül a többi ADME paraméter érdemi javulását

75
leginkább a molekula ezen részének megfelelı változtatásától remélhettük. Eredményeimet a
13. táblázatban foglalom össze.
Legrosszabb oldékonysággal éppen az igen hatásos benzimidazolon-származékok
rendelkeztek (186, 11. táblázat). A két hidrogénkötés donor NH-csoport egyikének
metilezésével (200) sikerült a kristályrács energia csökkentésén keresztül az oldékonyságot
növelni, az aktivitás azonban a 186 anyavegyülethez képest egy nagyságrendet romlott. Az
oxigén bázikus jelleget kölcsönzı aminocsoporttal (207) való helyettesítésével szintén javult
az oldékonyság, a receptorhoz való affinitás azonban csökkent. Az indanon- (188) és
benzoxazolon- (184) származékok karbonilcsoportjának szulfocsoportra történı cseréje az
oldékonyság várt mértékő növelése mellett az aktivitás drasztikus mértékő csökkenéséhez
(201), illetve teljes hatástalansághoz (208) vezetett. A még jó oldékonysággal rendelkezı 206
is hatástalan. Anyavegyületeiknél jóval gyengébb a 184 benzoxazolon-származék
benzoxazoltion analogonja (203) és a 186 benzimidazolon-származékból levezethetı
trifluormetil-csoportot tartalmazó molekula (204) is.
13. táblázat. Különbözı, NH-tartalmú heterociklusokkal kondenzált anilinszármazékok (200-208)
NMDA antagonista hatása patkány kortikális sejteken.
H
NN
O
O
Q
a NMDA-kiváltott intracelluláris Ca2+ koncentrációváltozás. b Inhibíció (%) a vegyület 0,1 µM koncentrációjánál meghatározva. c Kinetikai oldékonyság pH=7,4-en és szobahımérsékleten meghatározva.
Annak ellenére, hogy az oxazol és az imidazolgyőrők egy metiléncsoporttal történı
bıvítésével levezethetı heterociklusokat tartalmazó származékok (205, 206) in vitro
eredményei nem érik el az anyavegyületek (184, 186) megfelelı értékeit, 205 in vivo igen
hatásosnak bizonyult. Egér formalinos fájdalomtesztben elért ED50 < 0,1 mg/kg a
legalacsonyabb a projektben meghatározott értékek között. A gyógyszerkémiai gyakorlatban
vegyület 200 201 202 203 204 205 206 207 208
Q NN
O
CH3H
HS
N
O O H
N
N O
ON
S
H
NN
CF3
H
O
N
O
H
N
N
O
H
H NN
NH2
H
H O
SN
O O
Oldékonyságc [µg/ml] 135 >200 71.3 12,7 3,5 5,4 - >200 >200
NMDA-kiváltott ∆∆∆∆[Ca2+]ia
inhibíció %b; IC50 (nM) 42 99 15,2 % 88 48,1 % 29 62 % 99 4 %

76
nem példanélküli eredmény, miszerint egy vegyület in vivo aktivitása jóval nagyobb, mint az
a mért in vitro adatok alapján várható lenne, arra figyelmeztet, hogy az elsıdleges szőrésre
használt in vitro adatokat csak kellı körültekintéssel, több adat összevetésével szabad a
vegyületek kiválasztására használni. Sajnos 205 analgetikus hatása nem volt dózisarányos,
ezért nem került továbbfejlesztésre.
4.4.2. Oxálsavszármazékok elıállítása
A 189 – 197 és 200 – 206 vegyületeket a 30. ábrán látható módon állítottam elı. A
piperidineket trietil-amin jelenlétében etil-oxalil-kloriddal acileztem. Az észtercsoportot
lúgosan hidrolizáltam, majd a sósavval felszabadított karbonsavval (210) HBTU kapcsolószer
segítségével az anilineket acileztem (30. ábra).
H
H
209 210 189 - 197, 200 - 206
N
X
YN
X
YHO
O
O
(i) (ii)
N
X
YN
O
O
Q
190 iii 198194 199iii
30. ábra. Oxálsavdiamidok elıállítása. Reagensek és reakciókörülmények: (i) (1) ClCOCOOEt, Et3N, CHCl3, 10oC, 2 óra; (2) KOH, EtOH/H2O, szobahımérséklet, 2óra; (3) 6N HCl, 10oC, 2 óra; (ii) anilinek, HBTU, Et3N, DMF, szobahımérséklet, 18 óra; (iii) H2O2, ecetsav, szobahımérséklet, 18 óra.
A 198 és 199 szulfoxidszármazékokat a megfelelı tiovegyületek (190 és 194)
hidrogénperoxidos oxidációjával állítottam elı ecetsavban, szobahımérsékleten.
A 207 2-amino-benzimidazol-származékhoz a 30. ábra szerint elıállított 1-es helyzetben Boc-
csoporttal védett származék védıcsoportjának savas eltávolításával jutottam. A 208
származékot 2-amino-5-nitrofenolból kiindulva öt lépésen keresztül a 31. ábrának
megfelelıen alakítottam ki.
NO2
H2N
HO NN
O
ONS
O
O
O
H
H
iii, iv,v
208 (ti.: 34 %) (tii.: 87 %) (tiii.: 66 %) (tiv.: 46 %) (tv.: 40 %)
213211
NO2
HN
HO
Toz
NH2
HN
HO
Toz
i ii
212
31. ábra. 208 elıállítása. Reagensek és reakciókörülmények: (i) TozCl, CaCO3, CH3CN, reflux, 8 óra; (ii)
H2, 10 % Pd/C, CH3OH - THF, szobahımérséklet, 6 óra; (iii) (4-benzil-piperidin-1-il)-oxo-ecetsav, HBTU, Et3N, DMF, szobahımérséklet, 6 óra; (iv) SO2Cl2 CH2Cl2, Et3N, -78°C – szobahımérséklet; (v) NaN3, víz – CH3CN szobahımérséklet, 12 óra.
4-feniltiometil-piperidint (216) tiofenol N-Boc-4-meziloximetil-piperidinnel (129) történı S-
alkilezésével állítottam elı. A kén meta-klór-perbenzoesavval történı oxidációjával a
megfelelı szulfonszármazékhoz (217) jutottam (32. ábra).

77
(ti.: 95 %)
N
X
Boc
(i)
HN
X
N
OMez
Boc
(iii)
SO
O
216 X = S
217 X =
(ii)S
O
O
214 X = S
215 X =
(tiii.: 99 %)
(tii, iii.: 87 %)
129
32. ábra. 4-feniltiometil-piperidin és oxidált származékának elıllítása. Reagensek és reakciókörülmények: (i) NaH, szubsztituált tiofenolok, DMF, 3 óra szobahımérséklet; (ii) mCPBA, 1 óra szobahımérséklet; (iii) 1. 10% HCl/EtOAc, 18 óra, 2. NaHCO3.
4-feniltio-piperidint (220) N-Boc-4-piperidinolból (132) kiindulva ismert eljárás szerint
állítottam elı.109 A kén meta-klórperbenzoesavval történı oxidációjával a megfelelı
szulfonszármazékhoz jutottam (221) (33. ábra).
(ti, iii.: 69 %)
(ti - iii.: 95 %)
N
X
Boc
(i)
N
OH
Boc
(iii)
HN
X
(ii)S
O
O
218 X = S
219 X =
132
SO
O
220 X = S
221 X =
33. ábra. 4-feniltio-piperidin és oxidált származékának elıllítása. Reagensek és reakciókörülmények: (i) (nBu)3P, PhSSPh, THF, 6 óra reflux; (ii) mCPBA, 1 óra szobahımérséklet; (iii) 1. 10% HCl/EtOAc, 18 óra, 2. NaHCO3. 4-Benziloximetil-piperidint (224) N-Boc-4-hidroxi-metil-piperidin (222) O-benzilezésével
(34. ábra), míg 4-fenoxietil-piperidint (227) fenol N-Boc-4-meziloxietil-piperidinnel történı
alkilezésével állítottam elı (35. ábra).
(i)
NBoc
OH (ii)
NBoc
O
HN
O
(t.: 38 %) (t.: 67 %)222 223 224
34. ábra. 4-benziloximetil-piperidin elıllítása. Reagensek és reakciókörülmények: (i) NaH, DMF, benzil- bromid, 6 óra szobahımérséklet (ii) 1. 10% HCl/EtOAc, 18 óra, 2. NaHCO3.
(t.: 83 %) (t.: 77 %)225 226 227
(i)
NBoc
OH(iii)
NBoc
O
HN
O(ii)
35. ábra. 4-fenoxietil-piperidin elıllítása. Reagensek és reakciókörülmények: (i) CH3SO2Cl, Et3N, CH2Cl2, 18 óra szobahımérséklet; (ii) fenol, K2CO3, DMF 5 óra reflux; (iii) 1. 10% HCl/EtOAc, 18 óra, 2. NaHCO3. Eredményeinket szabadalmi bejelentésben110 védtük és egy közleményben111 ismertettük.

78
4.5. Kinurénsav amidszármazékok
4.5.1. Eredmények, szerkezet-hatás összefüggések
Miután a fahéjsavszerkezetet sikeresen transzformáltuk merevebb indol- és benzimidazol
győrőrendszerré, elhatároztuk, hogy megvizsgáljuk az ezekkel rokon, hasonlóan merev
szerkezetek NR2B aktivitását. Az indol és benzimidazol heterociklusos részének egy
karbonilcsoport beépítésével történı hipotetikus győrőbıvítésével 1-H-kinolin-4-on és 1H-
kinazolin-4-on analogonokhoz jutunk:
X
N
O
N
W
Z
Y
H
X
N
O
N
W
O
ZH
Y
82 X = CH, Y = 6-OH 83 X = CH, Y = 5-OH 142 X = N, Y = 5,(6)-OH
Ténylegesen elıállítva ezeket a szerkezeteket elsısorban a 1-H-kinolin-4-on
savszármazékának, a kinurénsavnak piperidinekkel alkotott amidjai voltak a hatásosak. A
vegyületcsaládot az indol és benzimidazol-származékok körében szerzett tapasztalatok
alapján rövid idı alatt, mindössze két tucat vegyület elıállításával sikerült optimalizálni.
Az elıállított vegyületek biológiai aktivitását natív patkány idegsejteket és rekombináns
NR1/NR2B receptorokat expresszáló sejteket felhasználó funkcionális esszében is
meghatároztuk. Ha a vegyület az NMDA receptorcsaládon belül csak az NR2B altípust
gátolja, és az NMDA-n kívül más receptoron keresztül nem befolyásolja az intracelluláris
Ca2+ koncentrációt, úgy a két teszt eredményeinek jó egyezést kell mutatniuk.
Szelektivitásukat az NR2A alegységeket tartalmazó NMDA rceptorokhoz képest ugyanebben
a funkcionális esszében határoztuk meg, rekombináns NR1/NR2A receptorokat expresszáló
sejteket felhasználva. In vivo analgetikus hatást a hosszantartó fájdalmat modellezı egér
formalin tesztben mértük. Szerkezet-hatás összefüggéseket reprezentáló molekulák
funkcionális esszé eredményeit az 14. táblázatban válogattam össze. A natív patkány
kortikális sejteken és a rekombináns NR1a/NR2B receptorokat expresszáló sejteken
végrehajtott funkcionális esszé eredményei, ha számszerően nem is, de nagyságrendileg jó
egyezést mutatnak. Bár hasonló szerkezet-hatás összefüggés olvasható ki mindkét teszt
eredményeibıl, következtetéseimet a leggyorsabban rendelkezésünkre álló natív patkány
kortikális sejteken mért adatok alapján vonom le. Rekombináns NR1a/NR2B és NR1/NR2A
receptorokat expresszáló sejteken csak az elsı szőrın aktív vegyületeket mértük. A csoport
leghatásosabb képviselıje egy 6-hidroxi-kinurénsav-származék, a 236.
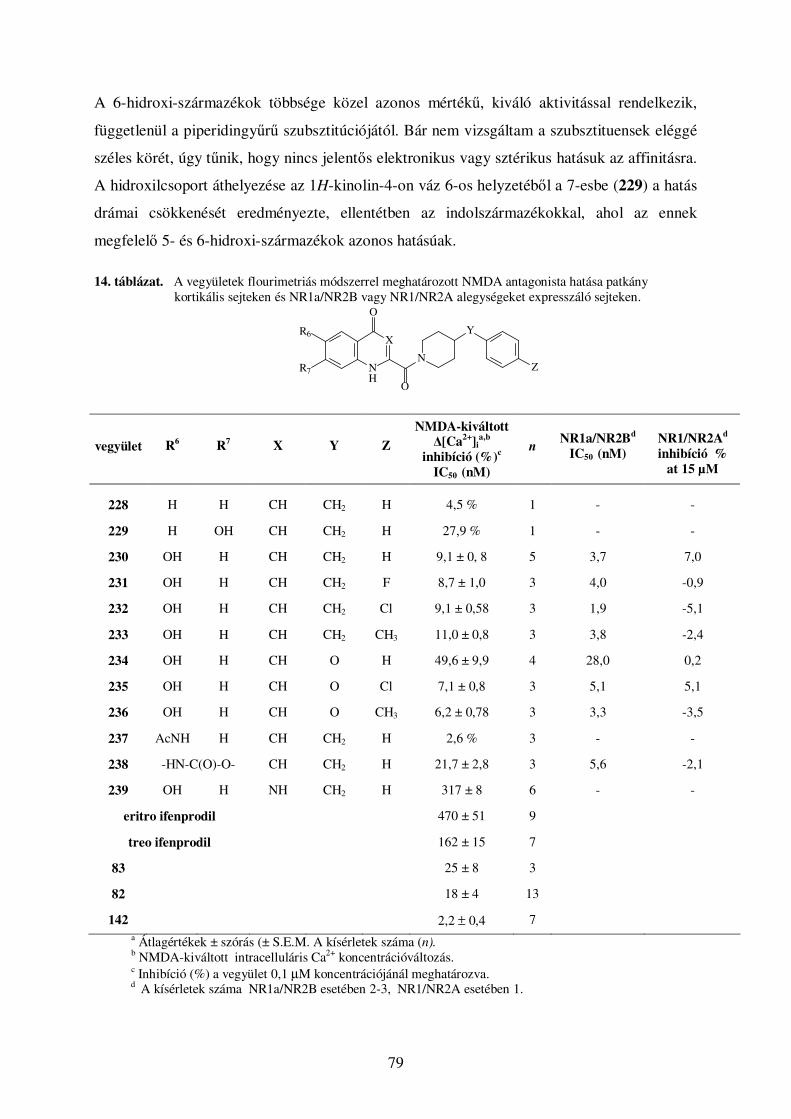
79
A 6-hidroxi-származékok többsége közel azonos mértékő, kiváló aktivitással rendelkezik,
függetlenül a piperidingyőrő szubsztitúciójától. Bár nem vizsgáltam a szubsztituensek eléggé
széles körét, úgy tőnik, hogy nincs jelentıs elektronikus vagy sztérikus hatásuk az affinitásra.
A hidroxilcsoport áthelyezése az 1H-kinolin-4-on váz 6-os helyzetébıl a 7-esbe (229) a hatás
drámai csökkenését eredményezte, ellentétben az indolszármazékokkal, ahol az ennek
megfelelı 5- és 6-hidroxi-származékok azonos hatásúak.
14. táblázat. A vegyületek flourimetriás módszerrel meghatározott NMDA antagonista hatása patkány kortikális sejteken és NR1a/NR2B vagy NR1/NR2A alegységeket expresszáló sejteken.
X
N
O
N
Y
R7
R6
O
ZH
vegyület
R6
R7
X
Y
Z
NMDA-kiváltott ∆[Ca2+]i
a,b
inhibíció (%)c IC50 (nM)
n NR1a/NR2Bd
IC50 (nM) NR1/NR2Ad
inhibíció % at 15 µM
228 H H CH CH2 H 4,5 % 1 - -
229 H OH CH CH2 H 27,9 % 1 - -
230 OH H CH CH2 H 9,1 ± 0, 8 5 3,7 7,0
231 OH H CH CH2 F 8,7 ± 1,0 3 4,0 -0,9
232 OH H CH CH2 Cl 9,1 ± 0,58 3 1,9 -5,1
233 OH H CH CH2 CH3 11,0 ± 0,8 3 3,8 -2,4
234 OH H CH O H 49,6 ± 9,9 4 28,0 0,2
235 OH H CH O Cl 7,1 ± 0,8 3 5,1 5,1
236 OH H CH O CH3 6,2 ± 0,78 3 3,3 -3,5
237 AcNH H CH CH2 H 2,6 % 3 - -
238 -HN-C(O)-O- CH CH2 H 21,7 ± 2,8 3 5,6 -2,1
239 OH H NH CH2 H 317 ± 8 6 - -
eritro ifenprodil 470 ± 51 9
treo ifenprodil 162 ± 15 7
83 25 ± 8 3
82 18 ± 4 13
142 2,2 ± 0,4 7 a Átlagértékek ± szórás (± S.E.M. A kísérletek száma (n). b NMDA-kiváltott intracelluláris Ca2+ koncentrációváltozás. c Inhibíció (%) a vegyület 0,1 µM koncentrációjánál meghatározva. d A kísérletek száma NR1a/NR2B esetében 2-3, NR1/NR2A esetében 1.
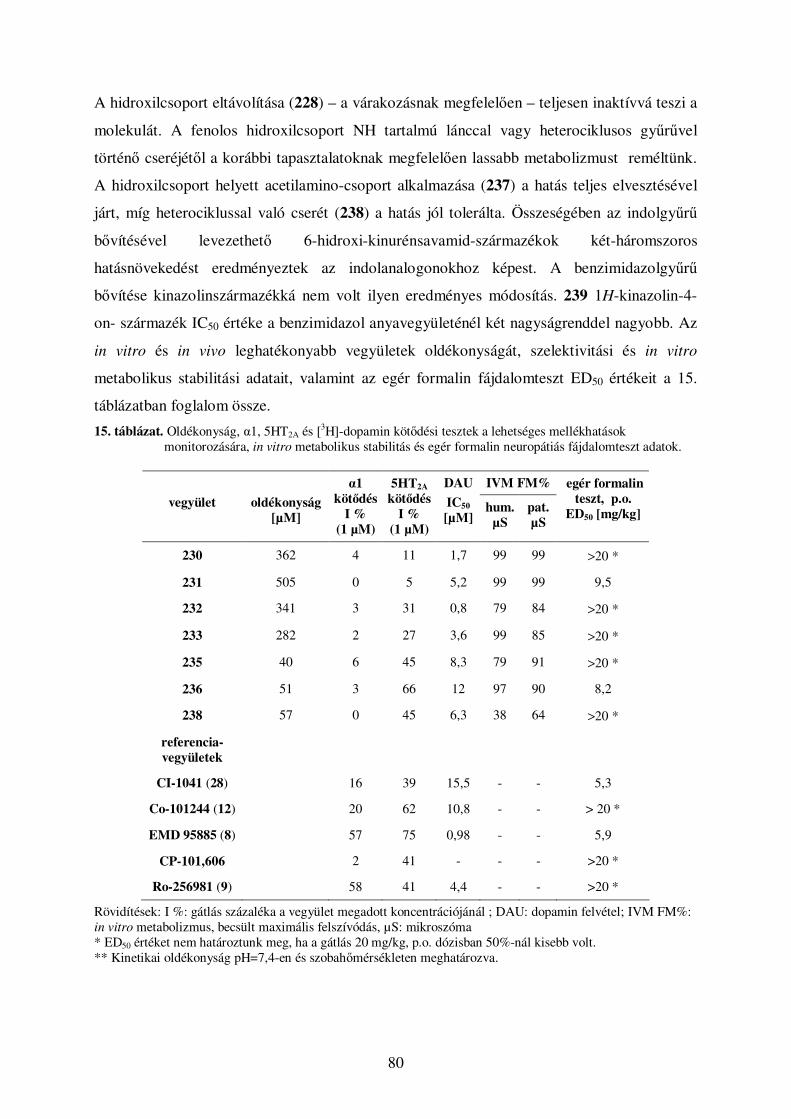
80
A hidroxilcsoport eltávolítása (228) – a várakozásnak megfelelıen – teljesen inaktívvá teszi a
molekulát. A fenolos hidroxilcsoport NH tartalmú lánccal vagy heterociklusos győrővel
történı cseréjétıl a korábbi tapasztalatoknak megfelelıen lassabb metabolizmust reméltünk.
A hidroxilcsoport helyett acetilamino-csoport alkalmazása (237) a hatás teljes elvesztésével
járt, míg heterociklussal való cserét (238) a hatás jól tolerálta. Összeségében az indolgyőrő
bıvítésével levezethetı 6-hidroxi-kinurénsavamid-származékok két-háromszoros
hatásnövekedést eredményeztek az indolanalogonokhoz képest. A benzimidazolgyőrő
bıvítése kinazolinszármazékká nem volt ilyen eredményes módosítás. 239 1H-kinazolin-4-
on- származék IC50 értéke a benzimidazol anyavegyületénél két nagyságrenddel nagyobb. Az
in vitro és in vivo leghatékonyabb vegyületek oldékonyságát, szelektivitási és in vitro
metabolikus stabilitási adatait, valamint az egér formalin fájdalomteszt ED50 értékeit a 15.
táblázatban foglalom össze.
15. táblázat. Oldékonyság, α1, 5HT2A és [3H]-dopamin kötıdési tesztek a lehetséges mellékhatások monitorozására, in vitro metabolikus stabilitás és egér formalin neuropátiás fájdalomteszt adatok.
Rövidítések: I %: gátlás százaléka a vegyület megadott koncentrációjánál ; DAU: dopamin felvétel; IVM FM%: in vitro metabolizmus, becsült maximális felszívódás, µS: mikroszóma * ED50 értéket nem határoztunk meg, ha a gátlás 20 mg/kg, p.o. dózisban 50%-nál kisebb volt. ** Kinetikai oldékonyság pH=7,4-en és szobahımérsékleten meghatározva.
IVM FM%
vegyület
oldékonyság [µM]
α1 kötıdés
I % (1 µM)
5HT2A kötıdés
I % (1 µM)
DAU
IC50 [µM]
hum. µS
pat. µS
egér formalin teszt, p.o.
ED50 [mg/kg]
230 362 4 11 1,7 99 99 >20 *
231 505 0 5 5,2 99 99 9,5
232 341 3 31 0,8 79 84 >20 *
233 282 2 27 3,6 99 85 >20 *
235 40 6 45 8,3 79 91 >20 *
236 51 3 66 12 97 90 8,2
238 57 0 45 6,3 38 64 >20 *
referencia-vegyületek
CI-1041 (28) 16 39 15,5 - - 5,3
Co-101244 (12) 20 62 10,8 - - > 20 *
EMD 95885 (8) 57 75 0,98 - - 5,9
CP-101,606 2 41 - - - >20 *
Ro-256981 (9) 58 41 4,4 - - >20 *

81
A 6-hidroxi-kinurénsav benzilpiperidinekkel alkotott származékai (230-233) rendkívül
oldékonyak, míg a fenoxipiperidinekkel alkotott származékok (235, 236) és a heterociklust
tartalmazó 238 oldékonysága az elıbbiekétıl egy nagyságrenddel roszabbak. A nem szelektív
NMDA antagonisták mellékhatásaiért leginkább felelıssé tehetı α1 receptorokhoz egyáltalán
nem kötıdnek és az 5-HT2A receptorokhoz sem mutatnak nagy affinitást. A dopamin felvételt
(DAU) azonban viszonylag nagymértékben gátolják. In vitro metabolizmusuk mértékét
humán és patkány mikroszómán meghatározva a 238 heterociklust tartalmazó származék
kivételével metabolikusan stabil vegyületek. In vivo, egér formalin tesztben leghatásosabb
származék az in vitro is legaktívabb 236. A vegyület szelektív, nem kötıdik az NR1/NR2A,
az α1 és 5HT2A receptorokhoz. A dopamin felvételt (DAU) is ez gátolja a legkevésbé.
Összefoglalva, hatásos indol és benzimidazol-származékokból kiindulva az NR2B
altípusszelektív NMDA receptor antagonisták új osztályát, kinurénsav-származékokat
azonosítottam. A vegyületcsalád in vitro legaktívabb tagja a 236, egyben in vivo is a
leghatásosabb. A vegyület további fejlesztését gyenge farmakokinetikai jellemzıi, 158ng/ml
cmax és 0,8 óra plazma féléletidı, de legfıképpen 8 %-os biohasznosulási adata hiúsította meg.
Emellett az idegrendszeri mellékhatásokat jelzı Irwin tesztben már 30 mg/kg dózisnál
detektálható tünetek jelentkeztek. Ezt a számot elosztva a formalin tesztben mért 8,2 mg/kg
ED50 értékkel, négynél kisebb, kockázatosnak ítélhetı terápiás index adódik.

82
4.5.2. Kinurénsav amidszármazékainak elıállítása
228-238 vegyületeket szubsztituált kinurénsav és piperidinszármazékok reakciójával
állítottam elı a szokásos HBTU-t felhasználó kapcsolási eljárás szerint (36. ábra).7
(t.: 60 %)
HN
O
O
OCH3
HNH2
COOCH3
COOCH3N
O
OCH3
O
OCH3+
HN
O
O
OH
R R R
R
i
HN
O
O
N
Y
Z
R
ii
iii, iv
v, vi viii
vii
R = 4-NO2
N
OO
H
6,7-
R =
R =
H249
252
254
R = 253 6-AcNH
N
OO
H
6,7-
R =
R =
H255
261
R = 260 6-AcNHN
OO
H
6,7-
R =
R =
H228
238
R = 237 6-AcNH
R = 3-BnO,R = 4-NO2 N
OO
H
4,5-
R =
R =
H240241a242
243 R = 4-NO2N
OO
H
4,5-
R =
R =
H244
247
248
R = 4-BnO241b R = 3-BnO245 R = 4-BnO246
R = 7-BnO250
R = 6-BnO 251
R = 7-BnO256
R = 6-BnO 258 R = 7-OH257
R = 6-OH 259
R = 7-OH 229
R = 6-OH230-236
vii
(t.: 62 %)
(t.: 88 %)
(t.: 73 %)
(t.: 76 %)
(t.: 91 %)(t.: 32 %)
(t.: 77 %)
(t.: 87 %)
(t.: 99 %)
(t.: 70 %)
(t.: 99 %)(t.: 83 %)
(t.: 99 %)
(t.: 99 %)
(t.: 57 %)
(t.: 32 - 57 %)
(t.: 52 %)
(t.: 55 %)
(t.: 42 %)
36. ábra. Kinurénsav amidszármazékainak elıállítása. Reagensek és reakciókörülmények: (i) MeOH, reflux, 2 óra; (ii) Dowtherm, 240 °C,10 perc; (iii) H2, 10 % Pd/C, MeOH, szobahımérséklet; (iv) Ac2O, reflux; (v) NaOH, MeOH, H2O, szobahımérséklet; (vi) HCl; vii) H2, 10 % Pd/C, MeOH, szobahımérséklet; (viii) piperidinek, HBTU, Et3N, DMF, szobahımérséklet. A kinurénsav intermediereket anilinszármazékokból kiindulva módosított Conrad-Limpach
módszer alapján szintetizáltam.112 253 acetilamino-szubsztituált kinurénsav észtert a
nitroszármazék (252) katalitikus redukciójával képzıdött amino intermedier acetilezésével
állítottam elı. Az észterek alkalikus hidrolízisével szubsztituált kinurénsavakhoz jutottam.
Hidroxiszubsztituált kinurénsavakat O-benzilszármazékuk katalitikus hidrogenolízisével
állítottam elı.
A kinazolinszármazék (239) szintézisekor a kereskedelmi forgalomban beszerezhetı 4-
hidroxi-antranilsavból (262) indultam ki és a 37. ábrának megfelelıen folytattam. 265
antranilamidot hat lépésben a szokásos reakciókkal állítottam elı. A kinazolingyőrőt 266
oxálsavdiamid termális kondenzációjával alakítottam ki. A végtermékekhez az O-benzil
védıcsoport katalitikus hidrogenolízisével jutottam.

83
HO
NH2
C
O
OH HO
N
C
O
OCH3
AcH
BnO
NH2
C
O
OCH3
BnO
NH2
C
O
NH2 BnO
N
C
O
NH2
C
O
C
O
NH
N
NHO
O
O
N
H
i, ii
v, vi vii
viii
iii,iv
262 263 264
265 266
239
(t.: 70 %) (t.: 88 %)
(t.: 12 %)
(t.: 21 %) 37. ábra: Kinazolinszármazék (239) elıállítása. Reagensek és reakciókörülmények: (i) SOCl2, MeOH, -20°C - szobahımérséklet; (ii) 1. Ac2O, 50°C; 2. Na2CO3 aq, szobahımérséklet; (iii) benzil-bromid, K2CO3, aceton, reflux, 4 óra; (iv) HCl, MeOH, reflux, 1 óra; (v) 1. 10 % NaOH, MeOH, reflux, 1 óra; 2. HCl; (vi) SOCl2, CHCl3, 1csepp DMF, reflux, 2 óra; 2. NH3, víz, 0°C; (vii) (4-benzil-piperidin-1-il)- oxo-ecetsav, HBTU, Et3N, DMF; (viii) 1. 250°C, 1,5 óra; 2. H2, 10 % Pd/C, THF, szobahımérséklet. Eredményeinket szabadalmi bejelentésben113 védtük és egy közleményben114 ismertettük.
4.6. Benzoil-karbamid-származékok
4.6.1. Eredmények, szerkezet-hatás összefüggések
A fahéjsavamidok, indol- és benzimidazol-2-karboxamidok és az oxálsavdiamidok körében
végzett kutatásunk alapján a következı megállapítások tehetık az ifenprodil farmakofór
modellt illetıen. A leghatásosabb szerkezetek két terminális benzolgyőrőt tartalmaznak,
melyek közül az egyik egy metabolikusan stabil hidrogénkötés donorcsoportot visel. A két
benzolgyőrőt elınyösen bázikus csoportot nem tartalmazó, különbözı hosszúságú és
heteroatomtaralmú lánc köti össze. Kémiai munkánk kiinduló pontja az University of Oregon,
a CoCensys és a Parke-Davis 37-es fahéjsavamid-származéka volt. A fahéjsav α szénatomja
és a benzolgyőrő közé beépített nitrogén indol-2-karboxamid-származékot eredményezett
(82), mely hétszer hatásosabb volt 37 anyavegyületnél. Az indol-2-karboxamid
pirrolgyőrőjének újbóli képzeletbeli felnyitásával és az így képzıdött metiléncsoport oxigénre
való cseréjével levezethetı oxálsavszármazék (183) kétszer aktívabb volt in vitro a 82 indol-
2-karboxamidnál. Bár ezek az intuitív szerkezeti módosítások eredményesnek bizonyultak, a
rendelkezésünkre álló kémiai tér teljesebb körő feltérképezéséhez, újabb hatásos

84
vegyületcsaládok azonosításához a következı analitikus megközelítésre volt szükség. A
tervezett új vegyületekben a farmakofór modell legbiztosabb elemeit, a hidrogénkötés
donorcsoportot tartalmazó benzolgyőrőt és a leggyakrabban elıforduló 4-benzil-piperidint
állandónak hagytam, csak az ezeket összekötı, a leghatásosabb molekulákra jellemzı
háromtagú láncot változtattam. A hidrogénkötés donor praktikus okokból vagy
hidroxilcsoport vagy benzolgyőrővel kondenzált oxazolon volt, attól függıen, hogy melyik
származékot lehetett egyszerőbben, gyorsabban elıállítani. A hidrogénkötés donorcsoporthoz
képest para-helyzető aromás szénatomot a 4-benzil-piperidin nitrogénjével számos stabilnak
és gyógyszerszerőnek tőnı háromtagú lánccal kötöttem össze. Az elıállított vegyületek
patkány kortikális sejteken meghatározott funkcionális tesztjének eredményeit a képletek alatt
tüntetem fel (38. ábra).
AB
CN
HO
N
O
HN
S
O O O
HO
O
O
O
O
OOH
N
O O
H
26832 %
269100 nM
267511 nM
N
O
O
18311 nM
O
37 131 nM
H
273-2,8 %
27424 nM
272461 nM
271354 nM
270 366 nM
38. ábra. 4-hidroxi-benzol és 4-benzil-piperidin nitrogénjét összekötı háromtagú A-B-C összekötı láncok.
Az összekötı lánc nem egyszerően távtartó, hanem fontos szerepet tölt be a receptorral való
kölcsönhatásban. Ezt támasztja alá az a megfigyelés, miszerint a lánc elemei, ahogy azt a
példák is mutatják, nem kombinálhatók szabadon. Vizsgálatunk etalonja a CoCensys
fahéjsavamidja (37) és a kutatócsoportunk oxálsavdiamid vezérmolekulája (183). A fahéjsav-
amid (37) kettıs kötésének telítése (268) a hatás elvesztésével járt. A savamid jelleg
megszüntetésével (269) a hatás kismértékben javult de nem közelítette meg az oxálsavdiamid
vezérmolekuláét. A 267, 270, 271 és 272 három atom hosszúságú acilszármazékok gyenge in
vitro aktivitást mutattak, míg a 273 szulfamidszármazék teljesen inaktív volt. A 274 benzoil-
karbamid-származékot viszont optimalizálásra alkalmasnak találtuk. Fenol helyett 3,4-
benzoxazolont tartalmazó modellvegyületek esetében is hasonló eredményre jutottam (39.
ábra). Az oxálsavdiamid anyavegyület (184) aktivitását egyik származék sem közelítette meg,

85
a korábban optimalizált fahéjsavamid-származékunk (81) teszteredményét azonban három
molekula is (275, 279, 281). A 147 oxálsavdiamid kén tartalmú megfelelıje (275) tizenötször
kevésbé hatásos az anyavegyületnél (184). Szerkezete alapvetıen nem különbözik tıle,
drasztikus hatásnövekedést az optimalizációjától nem várhatunk, ezért több kéntartalmú
származékot nem állítottam elı. A 279 szerkezete már jelentısen eltér az oxálsavdiamid
anyavegyületekétıl. Tartalmaz azonban egy bázikus nitrogént, amit el szerettünk volna
kerülni. Ily módon a 3,4-benzoxazolont tartalmazó modellvegyületek funkcionális
teszteredményei alapján is a benzoil-karbamid-származék (281) bizonyult optimalizációra
alkalmas molekulának. Újabb származékok képzése elıtt a két benzoil karbamidot (274 és
281) további biológiai vizsgálatoknak vetettük alá. Meglepı módon a heterociklussal
kondenzált benzoil-karbamid-származék (281) hatása az anyavegyületekéhez képest nem volt
olyan meggyızı, mint a hidroxianalogon (274) esetében.
HN
O AB
CN
O
H
184 2,4 nM
275 38 nM
81 28 nM
N
O
CH3H
N
NH
OH
N
NOH
OH
N
O
CH3
N
NH
H
N
O
O
HN
S
O
HO
N
OO
H
277 1800 nM
278 345 nM
276 395 nM
280 162 nM
281 59 nM
279 39 nM
39. ábra. 3,4-benzoxazolont és 4-benzil-piperidin nitrogénjét összekötı különbözı háromtagú láncok hatása a vegyületek funkcionális tesztjeinek eredményére.
A kielégítı in vitro aktivitás és a szerkezeti újdonság miatt további származékok képzését
határoztam el. Az eddig kutatott vegyületcsaládoktól eltérıen a hidroxiszármazék hatásosabb
volt a 3,4-benzoxazolon analogonnál, ezért elsısorban hidroxiszármazékokat állítottam elı. A
benzoil-karbamidok biológiai aktivitását mindhárom funkcionális esszében meghatároztuk. E
három funkcionális esszé eredményeit, szerkezet-hatás összefüggéseket is reprezentáló
molekulák esetében, valamint a referenciavegyületek NR1a/NR2B és NR1/NR2A
alegységeket expresszáló sejteken meghatározott aktivitását a 16. táblázatban foglaltam
össze.

86
16. táblázat. Benzoil-karbamid-származékok NMDA antagonista aktivitása natív patkány kortikális sejteken, valamint NR1a/NR2B és NR1/NR2A alegységeket expresszáló sejteken fluorometriás módszerrel meghatározva.
H
C
O
NC
N
Y
O
X Z
vegyület
X
Y
Z
NMDA-kiváltott ∆ [Ca2+]ia,b
inhibíció (%)c IC50 (nM)
NR1a/NR2Bd IC50 (nM)
NR1/NR2Ad inhibíció %
at 15 µM
282 4-OH CH2 Cl 14 5,3 1,8
283 4-OH CH2 4-CH3 13 6,2 8,7
284 4-OH CH2 4-OCH3 16 7,6 -5,7
285 4-OH CH2 4-CF3 37 8,3 19,7
286 4-OH CH2 F 23 19,3 4,9
274 4-OH CH2 H 24 28,0 2,6
287 4-OH CH2 3-OCH3 46 58,9 -0,9
288 4-OH CH2CH2 4-CH3 42 14,3 11,4
289 4-OH CH2S H 35 59,1 -2,9
referenciavegyületek
CI-1041 (28) 8,4 21,0
Co-101244 (12) 4,8 -8,7
EMD 95885 (8) 48 0,1
CP 101,606 30 2,5
Ro 25.6981 (9) 57 1,0
eritro ifenprodil 459 -2,7
MK-801 43 IC50=386 nM a A kísérletek száma natív patkány kortikális sejteken 4-6 b NMDA-kiváltott intracelluláris Ca2+ koncentrációváltozás. c Inhibíció (%) a vegyület 0,1 µM koncentrációjánál meghatározva. d A kísérletek száma NR1a/NR2B esetében 2, NR1/NR2A esetében 1.
Bár következtetéseimet az NR1a/NR2B receptorokat expresszáló sejteken meghatározott
adatok alapján vonom le, a nagyfokú NR2B szelektivitásnak köszönhetıen a leírt
összefüggések a natív patkány kortikális sejteken mért adatok alapján is helytállóak. A
fenilgyőrő 4-es helyzetének különféle szubsztitúciója nem befolyásolja alapvetıen az
aktivitást, elektronküldı (283, 284) és elektronszívó (282, 285) csoportok is kiváló hatású,
szelektív vegyületeket eredményeznek. A 4-fluorfenil (286) és fenil (274) származékok
ezekhez képest 3 - 4-szeres NR1a/NR2B aktivitáscsökkenést mutatnak. Az aktivitási sorrend

87
arra utal, hogy a nagyobb affinitás elérésében a hidrofób kölcsönhatásnak nagyobb szerepe
lehet, mint az elektronikus faktoroknak. A metoxicsoport 3-as helyzetbe történı áthelyezése
(287) a hatás egyértelmő csökkenését vonta maga után. Az Y összekötı lánc 283 molekula
esetében egy metiléncsoporttal (288), 174 molekula esetében egy kén atommal (289) történı
meghosszabbításával az aktivitás a felére csökkent. Az összes vegyület magas NR1a/NR2B
szelektivitást mutat, az NR1/NR2A receptorokon inaktívak.
A leghatásosabb benzoil-karbamid-származékok elérik a legaktívabb referenciavegyületek
megfelelı in vitro értékeit, a klinikai fázisba került CP-101,606 hatását pedig többszörösen
felülmúlják. In vivo analgetikus hatást a hosszantartó fájdalmat modellezı egér formalin
tesztben mértük (17. táblázat). Leghatásosabb, orálisan is felszívódó származék a szubsztituá-
latlan 274 analogon, jelezve, hogy a benzoil-karbamidok nem csak in vitro aktivitásban és
szelektivitásban de in vivo aktivitásban is összemérhetıek a referenciavegyületekkel.
17. táblázat. Egér formalin neuropátiás fájdalomteszt adatok.
vegyület ED50 (mg/kg p.o. vagy jelezve)
274 1,6
286 27
referenciavegyületek
CI-1041 (28) 2,4
Co-101244 (12) >20* (5,9 mg/kg i.p.)
EMD 95885 (8) 3,7
CP-101,606 >20*
Ro-256981 (9) >20* (5,1 mg/kg i.p.)
*: ED50 értéket nem lettek meghatározva, ha a gátlás kevesebb mint 50 % 20 mg/kg, p.o..
A 274 vegyület egér formalin tesztben kiváló hatással rendelkezett, ráadásul a projekt
vegyületei közül egyedülálló módon nem növelte a lokomotoros aktivitást (15 mg/kg dózisig
nincs effektus). Jó oldékonysága (44 µg/ml) és metabolikus stabilitása (95 % humán
mikroszóma) ellenére a molekula nem szívódott fel kellı mértékben, biohasznosulása csak 11
%. A többi farmakokinetikai paramétere sem kielégítı: alacsony cmax (652 ng/ml) és féléletidı
(0,6 óra), ugyanakkor magas kiürülés (31,7 ml/perc/kg) jellemzi. Egér neuropátiás fájdalom
modellben nem mutat antiallodíniás hatást. Ennek oka valószínőleg a kismértékő felszívódás
mellett a gyenge agyi penetráció. Ez lehet az oka a lokomotoros aktivitást nem befolyásoló
mellékhatás-mentességének is. A vegyület nem került továbbfejlesztésre.

88
4.6.2. Benzoil-karbamid-származékok elıállítása
A tervezett vegyülettár minél gyorsabb létrehozása érdekében szilárd fázisú parallel reakciók
kivitelezése mellett döntöttem. A gyorsaság mellett célszerő is volt ez a megközelítés, mivel a
kiindulási 4-hidroxi-benzoesavamidot a fenolos hidroxilcsoporton keresztül könnyen szilárd
fázisú gyantához lehetett kötni. Megközelítıleg 50 vegyületet állítottam elı ily módon (40.
ábra). A 4-hidroxi-benzamidot (290) Mitsunobu körülmények között Wang gyantához
kötöttem, majd a benzamidot (291) oxalil-kloriddal benzoil-izocianáttá (292) alakítottam.115 A
benzoil-karbamid-származékokat a benzoil-izocianát (292) és különbözı piperidinek addícós
reakciójában állítottam elı. Végül a 4-hidroxi-benzoil-karbamid célvegyületeket TFA 10 %
diklórmetános oldatával hasítottam le a gyantáról.
H
290
OH
CO NH2
iii, ivC
O
NC
N
X
O
HO
i iiY
O
CO NH2
O
CO NCO
291 292 274, 282-289 (t.: 5-10 %) 40. ábra. 4-hidroxi-benzoil-karbamid-származékok elıállítása szilárd fázison. Reagensek és reakciókörülmé- nyek: (i) Wang gyanta, Ph3P, DEAD, THF, 0°C - szobahımérséklet, 24 óra; (ii) ClCOCOCl, 1,2-diklóretán, 75°C, 0,5 óra; (iii) piperidinszármazékok, 1,2-diklóretán, DIEA, szobahımérséklet, 1 óra; (iv) 10 % TFA - CH2Cl2, 2 óra, szobahımérséklet.
A bepárlási maradék oszlopkromatográfiás tisztítását követıen a termék azonosságát és
tisztaságát HPLC-MS módszerrel határoztuk meg. A hatásos vegyületeket újraszintetizáltuk,
szerkezetüket igazoltuk (IR,1-H NMR, HRMS). Az újraszintézist 4-benziloxi-benzamidból
kiindulva a szilárd fázisú eljárással analogon módon végeztem el.
Iparjogvédelmi szempontból a szintézisutat az irodalomban elıforduló másik gyakori benzoil-
izocianát elıállítási módszerrel is kidolgoztam (41. ábra).116 4-benziloxi-benzoesavból
tionilkloriddal savkloridot állítottam elı, majd a savkloridból nátrium-cianáttal ón-tetraklorid
jelenlétében izocianátot képeztem. A végtermékeket a megfelelı benziloxiszármazékok
katalitikus hidrogenolízisével, hidrogénezésre érzékeny csoportok jelenléte esetén
hidrogénbromidos hasításával állítottam elı.

89
293 294 274, 282-289
H
BnO
C
O
OH i, ii
BnO
C
O
NCO iii, iv
HO
C
O
NC
N
R2
R1
O
(t.: 56 %)(t.: 38 %)
41. ábra. 4-hidroxi-benzoil-karbamid-származékok elıállítása. Reagensek és reakciókörülmények: (i) SOCl2, reflux, 3 óra; (ii) NaOCN, SnCl4, acetonitril, benzol, reflux, 3 óra; (iii) szekunder amin, szobahımérséklet, 1óra; (iv) H2, 10 % Pd/C, MeOH, szobahımérséklet vagy 33 % HBr/ecetsav, 1 óra, szobahımérséklet.
Eredményeinket szabadalmi bejelentésben117 védtük és egy közleményben118 ismertettük.
4.7. Indol-2-karboxamidin-származék
4.7.1. Eredmények, szerkezet-hatás összefüggések
A Merck Sharp & Dohme kutatói új vegyületek tesztelése során az E-N1-(benzil)fahéjsav-
amidint (45) az NR2B-tartalmú receptorokhoz erısen kötıdı molekulaként azonosították.119
A molekula teljesen új szerkezet, eltér az ifenprodil farmakofór modelltıl, hiányzik belıle a
fenolos hidroxilcsoport és a benzil-piperidin, ezért reménykeltı jelöltként egy új
gyógyszerkémiai programot iniciált a Mercknél. Az optimalizáció során az egyik legaktívabb
származéknak a 2-metoxi-benzil-származék (46) adódott.
N
NH
HN
NH O
H
45 46
NR2B-Ki: 9 nM (irod. adat) NR2B-Ki: 0,7 nM (irod. adat) NR2B-IC50: 4,2 nM (saját mérés)
Gyógyszerszerőbb származékokat keresve találtak rá a benzil-benzamidin vegyületcsaládra,
amelynek szubsztituálatlan képviselıje (47) hatástalan volt, megfelelıen helyettesített
benzolgyőrőkkel viszont aktív származékokat sikerült elıállítaniuk.79 Az egyik leghatásosabb
képviselı a benzilcsoport 2-es helyzetében szintén metoxicsoporttal szubsztituált (48). A
vegyületcsalád aktív tagjai jó orális farmakokinetikával rendelkeztek és a karragén-indukált
hiperalgézia tesztben rágcsálókban hatásosak, ugyanakkor a nemszelektív NMDA
antagonistákra jellemzı lokomotoros mellékhatástól mentesek voltak.

90
H HN
NH
N
NH O
CF3O
47 48 Ki > 15000 nM (irod. adat) Ki = 5,7 nM (irod. adat)
Kutatásunk elején a Cocensys fahéjsavamid (37) sztirol részének merevítésével jutottunk
az elsı vezérmolekulánkhoz, a (82) indol-2-karboxamid-származékhoz.
HN
O
N HOHO
O
N
37 82 NR2B-IC50: 131 nM NR2B-IC50: 28 nM
Feltételezésem, miszerint ez a transzformáció az amidinek esetében is sikeres lesz, helyesnek
bizonyult. Indol-2-karboxamidineket (295-309) állítottam elı, melyek hatásosan gátolták az
NR2B receptorokat. A vegyületcsalád optimalizációját megkönnyítették a Merck
közleményei119, 79. E eredményeiket felhasználva határoztam meg az elıállítandó molekulák
szubsztituenseit.
Az elıállított vegyületek biológiai aktivitását funkcionális esszében határoztuk meg, melynek
során az NMDA által kiváltott intracelluláris Ca2+ szint növekedésének gátlását mértük
patkány kortikális sejteken. Szelektivitásukat az NR2A alegységeket tartalmazó NMDA
rceptorokhoz képest ugyanebben a funkcionális esszében határoztuk meg, rekombináns
NR1/NR2A receptorokat expresszáló sejteket felhasználva. Az in vivo analgetikus hatást a
hosszantartó fájdalmat modellezı egér formalin tesztben mértük. A szerkezet-hatás
összefüggéseket reprezentáló molekulák funkcionális esszé eredményeit a 18. táblázatban
válogattam össze.
A benzilcsoport szubsztitúciója azt mutatja, hogy a hatás nem viseli el a 3-as és a 4-es
helyzetek helyettesítését. A 3-metoxi (307) és 4-metoxi (308) analogonok teljesen
hatástalanok a funkcionális teszten. A 2-metoxi-származék (295) azonban igen aktív, míg más
csoportok ebben a pozícióban (305, 306) kevésbé hatásos molekulákat eredményeznek. A 2-
es és 6-os helyzetben metoxicsoportokkal diszubsztituált származékok aktivitása hasonló
(300, 302), vagy nagyobb (299, 301) a monoszubsztituált analogonokhoz (296, 298 és 295,
297) képest. Más diszubsztitúció (303 és 304) gyengébb vegyületeket eredményezett.

91
18. táblázat. Indol-2-karboxamidin-származékok flourimetriás módszerrel meghatározott NMDA antagonista hatása patkány kortikális sejteken.
H
HCl
N
NH2
NR
1R
2
vegyület R1 R2 k’d
NMDA-kiváltott ∆∆∆∆[Ca2+]i
a,b
inhibíció (%)c
IC50 (nM)
n
295 H 2-OCH3
5,23 35 ± 5 5
296 5-MeO 2-OCH3
5,56 19 ± 3 3
297 5-F 2-OCH3
5,63 33 ± 6 3
298 5-Cl 2-OCH3
6,55 64 ± 12 3
299 H 2,6-(OCH3)2 3,67 5,4 ± 0,8 3
300 5-MeO 2,6-(OCH3)2 3,61 26 ± 5 5
301 5-F 2,6-(OCH3)2 3,80 17 ± 3 3
302 5-Cl 2,6-(OCH3)2 3,90 69 ± 13 4
303 H 2,6-di-F 3,52 143 ± 27 4
304 H 3,5-di-Cl 3,91 159 ± 17 3
305 H 2-CH3 3,73 170 ± 34 4
306 H 2-F 3,54 232 ± 36 3
307 H 3-OCH3 2,61 57,0 % 2
308 H 4-OCH3 2,58 10,5 % 1
309 H H 2,46 32,8 % 1
Referenciavegyületek
eritro ifenprodil
470 ± 51 9
37 131 ± 10 2
82 18 ± 4 13
46 6,6 ± 1,1 3 a Az értékek az átlagot és a szórást (± S.E.M) képviselik. A kísérletek száma (n). b NMDA-kiváltott intracelluláris Ca2+ koncentrációváltozás. c Inhibíció (%) a vegyület 0,1 µM koncentrációjánál meghatározva. d HPLC-MS kapacitás faktora.
Valószínőleg azért vezet hatásos vegyületekhez az orto-szubsztitúció és méginkább a nagyobb
kiterjedéső 2,6-diszubsztitució, mivel az orto-szubsztitúció képes az aromás B győrőt
kifordítani a benzamidincsoport síkjából. A 2-metoxi R2 csoportot konstansként tartva az

92
indolgyőrő szubsztitúciójának jelentıs hatása figyelhetı meg az affinitásra. Az aktivitás az R1
szubsztituens elektron küldı sajátságától függ, az 5-klórtól kiindulva az 5-fluoron át az 5-
metoxicsoportig nı. Leghatásosabb vegyület az indolrészen nem szubsztituált, 2,6-
dimetoxibenzil-származék (299). A nem szubsztituált analogon (309) inaktív, ami azt jelzi,
hogy önmagában a váz nem elég az aktivitás eléréséhez. Az in vitro és in vivo leghatékonyabb
vegyületek oldékonyságát, szelektivitási és in vitro metabolikus stabilitási adatait a 19.
táblázatban, az egér formalin fájdalomteszt ED50 értékeit a 20. táblázatban foglalom össze.
19. táblázat. Oldékonyság, α1, 5HT2A és [3H]-dopamin kötıdési tesztek a lehetséges mellékhatások monitorozására, NR1/NR2A szelektivitási adatok, in vitro metabolikus stabilitás és egér formalin neuropátiás fájdalomteszt adatok.
Rövidítések: I %: gátlás százaléka a vegyület megadott koncentrációjánál; DAU: dopamin felvétel; IVM FM%:
in vitro metabolizmus, becsült maximális felszívódás; µS: mikroszóma; Old.: kinetikai oldékonyság pH=7,4-en
és szobahımérsékleten meghatározva.
Az indol-2-karboxamidinek a projekt legoldékonyabb vegyületei. A nem tiszta profilú NR2B
szelektív NMDA antagonisták mellékhatásaiért leginkább felelıssé tehetı α1 és 5-HT2A
receptorokhoz nem mutatnak affinitást, ellenben a dopamin felvételt (DAU) viszonylag
nagymértékben gátolják. A humán és patkány mikroszómán mért in vitro metabolikus
stabilitási értékek jelentıs eltérése ellenére, a relevánsabb humán adatok alapján
metabolikusan stabil vegyületek. Bár a csoport metabolikusan legkevésbé stabil képviselıje
éppen az in vivo, egér formalin fájdalomteszten leghatékonyabb 295 származék. Ebben a
tesztben az 295 vegyület jó közepes hatást mutatott, ED50 értéke a legjobb
referenciavegyületek értékének alig háromszorosa, míg a többi NR2B szelektív
referenciavegyület nem hatékony.
IVM FM%
vegyület
R1
R2
Old. [µM]
α1 kötıdés
I % (1 µM)
5HT2A kötıdés
I % (1 µM)
DAU IC50 [µM]
NR1/NR2A I %
(15 µM) humán µS
patkány µS
295 H 2-OCH3
200 40 10 2.5 - 68 93
297 5-F 2-OCH3
599 16 18 31 - 99 67
299 H 2,6-(OCH3)2 578 39 48 1,2 32,1 99 61
301 5-F 2,6-(OCH3)2
550 42 47 0,8 - 99 68

93
20. táblázat. Egér formalin neuropátiás fájdalomteszt adatok.
*: ED50 értéket nem határoztunk meg, ha a gátlás 20 mg/kg, p.o. dózisban 50%-nál kisebb volt. Összefoglalva, új indol-2-karboxamidin-származékokat állítottam elı, amelyek hatásos és
NR2B szelektív antagonistái az NMDA receptornak. A vegyületcsalád körében végzett
vizsgálatainkból az a következtetés vonható le, hogy az ilyen típusú vegyületek aktivitása az
aromás győrők szubsztituenseinek karakterétıl és helyzetétıl szokatlanul nagymértékben
függ. A legjobb orális hatása egér formalin tesztben az 295 vegyületnek volt. Eredményeinket
szabadalmi bejelentésben120 védtük és egy közleményben121 ismertettük. Az általam, a Roche
és a Merck kutatói által elıállított aciklusos és ciklusos amidinek in vitro igen aktív és NR2B
szelektív, in vivo a referenciavegyületekhez képest elfogadható hatású vegyületek, az újabb
generációs leghatásosabb ifenprodilszerő anyagokkal szemben azonban nem állják ki az
összehasonlítás próbáját, sem farmakodinámiai, sem farmakokinetikai szempontból.
4.7. 2. Indol-2-karboxamidin-származékok elıállítása
Az indol 2-es helyzetében korábban még senki sem alakított ki amidincsoportot A tervezett
vegyületeket oldatfázisú parallel szintézissel állítottam elı (42. ábra).
310
HN
O
OHR1
N CN
R1i, ii, iii iv, v, vi
Cl
N
NH2
NR1
R2
H H
H
ti, ii.: 90 - 99 %; tiii.: 41- 84 % tiv.: 82 - 99 %; tv, vi.:3 - 59 %
311 295-309
42. ábra. Indol-2-karboxamidin-származékok elıállítása. Reagensek és reakciókörülmények: (i) SOCl2, CHCl3, reflux 2 óra; (ii) NH4OH; (iii) POCl3, CHCl3, reflux 2 óra; (iv) HCl, EtOH, szobahımérséklet, 2 óra; (v) benzilaminok, szobahımérséklet, 6 óra; (vi) HCl.
vegyület R1 R2
egér formalin teszt, p.o. ED50 [mg/kg]
295 H 2-OCH3
15
297 5-F 2-OCH3
>20*
299 H 2,6-(OCH3)2 >20*
301 5-F 2,6-(OCH3)2
-
referenciavegyületek
CI-1041 (28) 5,3
Co-101244 (12) >20* (5,9 mg/kg i.p.)
EMD 95885 (8) 5,9
CP-101,606 >20*
Ro-256981(9) >20* (5,1 mg/kg i.p.)

94
Közel 200 tagú vegyülettárat hoztam létre nemszubsztituált és 5-szubsztituált-indol-2-
karbonsavnitrilekbıl kiindulva benzilaminokkal, Pinner szintézisen keresztül. Az indol-2-
karbonsavnitrileket a kereskedelmi forgalomból beszerezhetı indol-2-karbonsavakból
állítottam elı a szokásos eljárásokkal. A nitrileket Pinner reakcióban alakítottam a megfelelı
imidátokká8. Ezeket benzilaminokkal reagáltatva jutottam az amidin végtermékekhez. A
nyerstermék oszlopkromatográfiás tisztítását követıen a termék azonosságát valamint
tisztaságát HPLC-MS módszerrel határoztuk meg, és k' értékkel (tisztaság-kapacitás faktor)
jellemeztük. A k' faktorokat a következı képlet szerint határoztuk meg: k' = (tR - t0) / t0
ahol k’= kapacitás faktor, tR = retenciós idı és t0 = az eluens retenciós ideje.
A hatásos vegyületeket újraszintetizáltuk, szerkezetüket igazoltuk (IR,1H-NMR, HRMS).
4.8. Benzilidén-piperidin-származékok
Az NR2B szelektív NMDA antagonisták várhatóan csak kis mértékben, vagy egyáltalán nem
rendelkeznek azokkal a mellékhatásokkal, amelyek a nem szelektív NMDA antagonistákra
jellemzıek. A nem szelektív NMDA antagonista fenciklidin és ketamin pszichotomimetikus
hatásokat, hallucinációt, rosszullétet, katatóniát, amnéziát idéznek elı. Ezek a komoly
mellékhatások gátolják gyógyszerként való használatukat. Az ebbe a csoportba tartozó
vegyületek állatoknál is viselkedési rendellenességeket okoznak, például erısen stimulálják a
motoros aktivitást, amnéziát és csökkent motoros koordinációt váltanak ki. NR2B altípus
szelektív antagonistáknál kevés humán vizsgálat áll rendelkezésre, így ennek hiányában az
állatokban tapasztalt mellékhatásokat tekintjük meghatározónak. Az NR2B szelektív
vegyületek a nem szelektív NMDA antagonistákra jellemzı komoly mellékhatásokat nem
mutatják, állat viselkedés tanulmányokban azonban a legtöbbnél lokomotoros aktivitást
fokozó hatást figyeltek meg. A referenciavegyületek közül az Ro 25-6981-nél tapasztaltak
ilyen hatást122, a CP-101,606 vegyületnél még nagyobb dózisokben sem123. Az irodalom
alapján a CP-101,606 jelő vegyület az egyetlen NR2B szelektív antagonista, mely nem
rendelkezik mozgást stimuláló hatással. Mivel a CP-101,606 jelő vegyület gyenge orális
hatékonysággal rendelkezik és a közölt adatok alapján embereknek csak intravénásan
adagolták, valamint polimorf CYP2D6 mediált metabolizmusa van124, nagy szükség van egy
új NR2B szelektív antagonistára, mely kevés mellékhatással (magas terápiás index) és jó
orális hatékonysággal (biológiai hasznosíthatóság) rendelkezik és terápiás célokra, fıleg orális
kezelésekre, jól fejleszthetı. Munkánk során azt tapasztaltuk, hogy ha az eddig általunk
azonosított hatásos szerkezetekben a benzil-piperidin részt benzilidén-piperidinre cseréljük
szintén hatásos NR2B szelektív NMDA antagonistákhoz jutunk, amelyek in vivo

95
fájdalomcsillapító hatással rendelkeznek, hasonlóan a telített benzil-piperidin analogonokhoz.
Ráadásul míg az utóbbi molekulák analgetikus dózisukban kisebb-nagyobb mértékben
fokozták a lokomotoros aktivitást, addig a benzilidén-piperidin-származékok nem
rendelkeznek mozgást stimuláló hatással a hatékony dózis legalább 6-szoros mennyiségénél
sem. Ezen tulajdonságuk miatt terápiásan kedvezıbbek, mint az alacsonyabb terápiás index-
szel rendelkezı NR2B szelektív NMDA antagonisták. Benzilidén-piperidin analogonokat a
leghatásosabb vegyületcsaládokban, az indol, benzimidazol és oxálsavdiamidok körében
állítottam elı.
4.8.1. Eredmények, szerkezet-hatás összefüggések az indol- és benzimidazol-
származékok körében
Az NR2B és rekombináns NR1/NR2A receptorokat expresszáló sejteken meghatározott
funkcionális esszék eredményeit a 21. táblázatban foglaltam össze. Az 5(6)-hidroxi-
benzimidazol-2-karbonsav 4-benzilidén-piperidinnel képezett amidja (312) az in vitro
funkcionális tesztben ugyanolyan hatásos, mint a korábban elıállított telített analogonja. Bár
a 4-fluor-származék (314) IC50 értéke egy nagyságrenddel nagyobb a telített változatétól, még
így is igen aktív vegyület.
21. táblázat. A benzimidazol- és indolszármazékok flourimetriás módszerrel meghatározott NMDA antagonista hatása patkány kortikális sejteken és NR1/NR2A transzfektált sejteken.
H
Y
N
O
N HO X
a Zárójelben a telített analogon száma. b NMDA-kiváltott intracelluláris Ca2+ koncentrációváltozás. c Zárójelben a telített analogon IC50 értéke.
vegyületa
Y
X
NMDA-kiváltott ∆∆∆∆ [Ca2+]i
b
IC50 (nM) c
NR1/NR2A I % (15 µM)
312 (142) N H
3,3 (2,2) 33%
313 N CH3
28 34%
314 (148) N F
11 (1,2) <30%
315 N Cl
21 <30%.
316 N CH3O 10 52%
317 (82) CH H 77 (18) <30%

96
A metoxi-, klór- és metilcsoporttal helyettesített származékok (316, 315, 313) is kellı
hatékonyságot mutattak ezen a teszten. A 6-hidroxi-indol-2-karbonsav 4-benzilidén-
piperidinnel képezett amidja (317) négyszer kevésbé aktív, mint a telített (82) analogon.
Szelektivitásuk az NR2A alegységeket tartalmazó NMDA receptorokhoz képest kielégítı.
Affinitásuk az NR2A receptorokhoz szokatlanul nagy. Bár a 316 származék 52 %-os gátlása
NR2A transzfektált sejteken a projektben mért legmagasabb hasonló érték, a szelektivitás
még így is kb. 100. A benzilidén-piperidin-származékok a többi NR2B szelektív NMDA
anatagonista vegyületcsalád közül azzal tőnnek ki, hogy az in vivo analgetikus tesztekben
meghatározott hatékony dózisai a leginkább elkülönülnek azoktól a dózisoktól, ahol már
mellékhatások figyelhetık meg. A farmakon biztonságosságát a két dózis hányadosával az ún.
terápiás index-szel jellemzzük. Minél nagyobb ez az érték, a szer annál inkább biztonságos, a
dozírozási lehetıségek nem lesznek szők korlátok közé szorítva. A vegyületek in vivo
analgetikus hatását a tesztrendszerünknek megfelelıen elıször egér formalin tesztben mértük,
a többi analgetikus modellt csak a tesztrendszer következı szintjein alkalmaztuk. Az elsı
CNS mellékhatást jelzı teszt a lokomotoros aktivitás (LMA) mérése, így a terápiás ablakot
elıször e két teszt alapján határozhatjuk meg. Egy vegyületet akkor tekintettünk lokomotor
stimuláló hatásúnak, ha 50 %-nál nagyobb emelkedést váltott ki a mozgékonyságban. Vagyis,
definíciószerően a lokomotoros aktivitás mentes (LMAfree) dózisok 50 %-nál kisebb
emelkedést mutattak. A terápiás indexet a LMAfree és a formalin teszt ED50 értékének
hányadosaként értelmezhetjük.
A 22. táblázatban két jellemzı benzilidén-piperidin (312, 317) valamint azok telített
analogonjainak analgetikus és lokomotoros aktivitás tesztben meghatározott eredményeit
tüntettem fel. Referenciavegyületek hasonló értékeit a 23. táblázat tartalmazza. Látható, hogy
míg a telített analogonok esetében nincs különbség az analgetikus dózis és a lokomotoros
aktivitást stimuláló dózis között, addig a benzilidén-piperidin-származékok nem okoznak
hiperaktivitást az analgetikus dózisok 6-szorosában sem. Ennyire különbözı hatásprofil nem
volt várható ilyen látszólag minimális szerkezeti módosítás esetén. A referenciavegyületek
közül a nem szelektív NMDA receptor antagonista MK-801 erısen növeli a lokomotoros
aktivitást a farmakológiailag hatékony dózis tartományban (23. táblázat). A szelektív NR2B
antagonista vegyületek közül a CI-1041 jelő referencia vegyület esetében sincs nagy
különbség a hatékony dózis és a lokomotoros aktivitást fokozó dózis között. A CP-101,606 és
az Ro-256981 az egér formalin tesztben nem volt hatásos.

97
22. táblázat. Benzimidazol- és indol-2-karboxamidok körében a benzilidén-piperidin-származékok és telített analogonjainak jellemzése formalin tesztben és lokomotoros aktivitás (LMA) tesztben. Terápiás indexek (TI) számolása.
benzilidén-piperidinek
LMA vegyület
egér formalin teszt, p.o.
ED50 mg/kg dózis mg/kg %-os
növekedés
TI
LMAmentes/ED50
15 27 312 2,6
30 63 5,8
3,75 0
7,5 55 317 0,43
15 69
8,7
benzil-piperidinek
LMA vegyület
egér formalin teszt, p.o.
ED50 mg/kg dózis mg/kg
%-os növekedés
TI
LMAmentes/ED50
142 1,6 1,875 80 < 1,2
82 1,7 1,875 90 < 1,1
23. táblázat. NMDA antagonista referencia vegyületek, a nem szelektív NMDA antagonista MK-801 és szelektív NR2B antagonista CI-1041 jellemzése formalin tesztben és lokomotoros aktivitás (LMA) tesztben. Terápiás indexek (TI) számolása.
LMA
vegyület
egér formalin teszt, p.o.
ED50 mg/kg dózis mg/kg %-os növekedés
TI
LMAmentes/ED50
0,1 114 MK-801 0,15
0,3 217 <1
CI-1041 2,4 10 137 <4
Ro 25-6981 >20*
CP-101,606 >20*
* CP-101,606 és Ro-256981 20 mg/kg dózisban csak 38 %-os illetve 12 %-os gátlást mutatott a formalin
tesztben.
Eredményeinket szabadalmi bejelentésben125 védtük.

98
4.8.2. Indol- és benzimidazol-származékok elıállítása A 312-316 és 317 vegyületeket a megfelelı indol- és benzimidazol-2-karbonsavakból (106 és
173) és 4-benzilidén-piperidinekbıl (318) állítottam elı HBTU kapcsolószer segítségével (43.
ábra).
Y
N
O
N HO X
Y
N
O
OH HO
i+
312-316 Y = N (t.:19 - 41 %) 317 Y = CH (t.: 65 %)
173 Y = N 106 Y = CH
H H
HNX
318
43. ábra. A 312-316 és 317 vegyületek elıállítása. Reagensek és reakciókörülmények: (i) HBTU, Et3N, DMF, szobahımérséklet, 16 óra.
A nitrogénen benzilcsoporttal védett 4-benzilidén-piperidineket (318) 1-benzil-piperidin-4-
onból kiindulva Horner-Wadsworth-Emmons reakcióval állítottam elı (44. ábra). A
kereskedelmi forgalomban nem hozzáférhetı szubsztituált benzilfoszfonát reagensekhez
Arbuzov-reakcióval a megfelelı benzil-halogenid és trietil-foszfit hevítésével jutottam. A
piperidin benzil-védıcsoportjának 1-klóretil-kloroformáttal történı eltávolításával a termék
sósavsóként, tisztán és jó termeléssel izolálható volt. A kettıskötés a piperidingyőrőhöz
képest 100 %-ban exo-helyzető, míg benzil helyett Boc-védıcsoport alkalmazásakor a
védıcsoport sósavas eltávolítása exociklusos és endociklusos termékek 5 : 1 arányú keverékét
eredményezte. A benzil védıcsoporttal kialakított 4-benzilidén-piperidinben és
származékaiban a kettıskötés exo helyzete már stabil, savak, lúgok hatására sem változik. 4-
benzilidén-piperidin-származékok az irodalomban nem voltak ismert vegyületek. Benzil-
piperidinek szintézisekor alkalmazták ugyan a Horner-Wadsworth-Emmons reakciót, de a
telítetlen terméket senki sem izolálta, azonnali hidrogénezéssel a telített származékot nyerték
ki.
N
O
NX H
N
HX
i iiCl
(tx = H.: 70 %)319 320 318 44. ábra. 4-benzilidén-piperidinek elıllítása. Reagensek és reakciókörülmények: (i) dietil-benzilfoszfonátok, NaH, DMF, 0-20oC, 2óra (ii) (1) 1-klóretil-kloroformát, diklóretán, 0-10oC 1 óra, reflux 1 óra; (2) metanol, reflux 1 óra.
4.8.3. Eredmények, szerkezet-hatás összefüggések az oxálsavdiamidok körében
Az oxálsavdiamid vegyületcsalád benzil-piperidin szerkezeti részének benzilidén-piperidinre
való cseréje a terápiás index még nagyobb mérvő emelkedését eredményezte. In vitro

99
aktivitásuk a legtöbb estben elmarad a telített analogonokhoz képest in vivo aktivitásuk
azonban lényegesen nem változott. A lokomotoros aktivitást jelentısen emelı dózis viszont
jóval nagyobb, így a számított terápiás index is ezzel arányosan nı (24. táblázat).
24. táblázat. Oxálsavdiamidok körében a benzilidén-piperidin-származékok és telített analogonjainak
jellemzése formalin tesztben és lokomotoros aktivitás (LMA) tesztben. Terápiás indexek (TI)
számolása.
NN
O
O
QX
H
a NMDA-kiváltott intracelluláris Ca2+ koncentrációváltozás.
Az 321 benzilidén-piperidin-származék a vegyületcsalád egyik leghatásosabb vegyülete. A
terápiás dózisban a lokomotoros aktivitást nem növelte. Egyéb CNS mellékhatásokat jelzı
tesztekben, PCP és CCI modellben, rotarod tesztben sem mutat aktivitást. A Morris
vízlabirintus tesztben nincs hatása a tanulási folyamatokra. Ames teszt alapján nem mutagén.
Nem gátolja a hERG csatornákat, nincs kardiovaszkuláris mellékhatása. Patkány toxikológiai
vizsgálatban 150 mg/kg/nap dózist is jól toleráltak az állatok, a kutya toxigológia is
biztonságos profilt mutatott.
benzilidén-piperidinek
LMA
vegyület Q X NMDA-kiváltott
∆∆∆∆ [Ca2+]ia
IC50 (nM)
egér formalin teszt, p.o
ED50 mg/kg dózis mg/kg
%-os növekedés
TI LMAmentes
/ED50
60 35 321
N
OO
N
H
H H
21 1,3
120 69 46
322 N
O
N
H
H CH3
90 0,94 60 13 >64
benzil-piperidinek
LMA
vegyület Q X NMDA-kiváltott
∆∆∆∆ [Ca2+]ia
IC50 (nM)
egér formalin teszt, p.o
ED50 mg/kg dózis mg/kg
%-os növekedés
TI LMAmentes
/ED50
1 34 184
N
OO
N
H
H H
2,4 0,85
3 62 < 1,2
3,75 72 188
NO
N
H
H CH3
11 0,48
7,5 141 <8

100
Seltzer féle egér neuropátiás modellben 2 mg/kg dózisban 50,2 %-os választ váltott ki. A
karragén-indukált hiperalgézia tesztben patkányokban hatásos, akut és krónikus modellben
egyaránt. A vegyület magas terápiás indexe lehetıvé teszi, hogy a viszonylag gyenge 20 %-os
biohasznosulását egy nagyobb dózis kompenzálja. Magasabb dózisban azonban enyhe
tolerancia alakult ki. Eredményeinket szabadalmi bejelentésben126 védtük.
4.8.4. Oxálsavdiamidok elıállítása
A telítetlen oxálsavdiamidok kialakításakor a telített analogonoknál bevált reakcióutat
alkalmaztam. Elıször a benzilidén-piperidineket acileztem etil-oxalil-kloriddal. Majd az
észtercsoport hidrolízését követıen az oxálsav másik karboxilcsoportjával anilineket
acileztem. (45. ábra). A tervezett vegyületeket oldatfázisú parallel szintézissel állítottam elı
10, a telített analogonoknál bevált anilin és 5 szubsztituált benzilidén-piperidin
felhasználásával (46. ábra). A nagyobb mennyiségben elıállított oxálsav-félamiddal
párhuzamosan acileztem a 10 anilint. A nyerstermékek oszlopkromatográfiás tisztítását
követıen a termék azonosságát és tisztaságát HPLC-MS módszerrel határoztuk meg. A
hatásos vegyületeket újraszintetizáltuk, szerkezetüket igazoltuk (IR,1-H NMR, HRMS).
318 323 321, 322
H
i
HON
O
O
X NN
O
O
QX
iiN
XH
(tX = H.: 88 %) (t321.: 65 %) 45. ábra. Oxálsavdiamidok elıállítása. Reagensek és reakciókörülmények: (i) (1) ClCOCOOEt, Et3N, CHCl3, 10 - 20oC, 1 óra; (2) NaOH, EtOH/H2O, szobahımérséklet, 1 óra; (3) 1 N HCl; (ii) QNH2, HBTU, Et3N, DMF, szobahımérséklet, 18 óra.
N
NO
NH2
CH3
HN
NO
NH2
H
HNH2O
NOH H
NH2
NAcN
OO
NH2
H
N
SO
NH2
HO
NO
NH2H
H
NH2
NMezN
OS
NH2
HN
O
NH2
H
HN
X
X : H, CH3, F, Cl, CH3O
46. ábra. Telítetlen oxálsavdiamidok parallel szintézise során felhasznált 10 anilin és 5 benzilidén-piperidin származék.

101
5. A PROJEKT BEFEJEZÉSE
Az NR2B receptor antagonista hatású vegyületek kutatása nyolc éven át tartott (1998-2006).
Ezen idı alatt 2805 molekulát állítottunk elı és vizsgáltak meg farmakológus kollégáink. A
vegyületek döntı részét az elsı 4-5 évben szintetizáltuk, 1527-et hagyományos módon, 1278-
at a kombinatorikus kémia eszközeivel. Eközben 8 alapszabadalmi bejelentést nyújtottunk
be. A 47. ábrán a versenytársak szabadalmi bejelentéseit is összefoglaltam. 1989-tıl 2008-ig
88 bejelentést tettek a felsorolt gyógyszergyárak. Ebbıl 40-et a legintenzívebb idıszaknak
tekinthetı 1999 és 2001 között nyújtottak be. A terület kutatásába 1998-ban, a versenytársak
maximális aktivitásakor kapcsolódtunk be. A viszonylag kései indulás miatti hátrányunkat az
addigra felhalmozott tudásnak köszönhetıen hamar behoztuk. Elsı szabadalmi
bejelentésünket már 2000-ben benyújtottuk az indol és benzimidazol-2-karboxamidok
védelmére. A következı évben az oxálsavdiamid-származékokat védtük. 2004-ben egyszerre
hat szabadalmi bejelentést nyújtottunk be, az ismertetett vegyületcsaládok oltalmát kérve. Ezt
követıen a versenytársak szabadalmi aktivitása is megszőnt. Cégünk kutatási vezetése 2001
december 19-én döntött a 185-ös számú (RGH-896, radiprodil) oxálsavdiamid-származék
klinikai fejlésztését illetıen. Ezt követıen backup és follow-up kutatást végeztünk. A számos
in vitro aktív származék közül, valamennyi ismertetett vegyületcsaládban sikerült azonosítani
orálisan is felszívódó, in vivo hatékony vegyületeket.
Cég/év ‘89 ‘90 ‘91 ‘92 ‘93 ‘94 ‘95 ‘96 ‘97 ‘98 ‘99 ‘00 ‘01 ‘02 ‘03 ‘04 ’05 ‘06 ‘07 ‘08 Searle 1 Pfizer 4 1 2 3 1 1 1 2 Warner-L. 1 5 5 CoCensys 6 1 Pharmacia 1 Roche 1 4 1 1 4 4 3 1 Merck KGaA 1 2 2 1 2 Synthelabo 1 1 Merck& Co. 2 9 2 3 1 3 Alcon 1 Richter 1 1 6 Vertex 1 Shionogi 1 1
47. ábra. NR2B szelektív NMDA antagonista hatású vegyületek védelmére benyújtott alapszabadalmi bejelentések.

102
6. ÖSSZEFOGLALÁS
Doktori munkámban a Richter Gedeon Vegyészeti Gyárban folytatott NR2B szelektív NMDA
antagonisták körében végzett kutatás gyógyszerkémiai vonatkozásait mutatom be. Az NMDA
receptor az ionotróp glutaminsav receptorok egyik típusa. NR1 és NR2 alegységekbıl épül
fel, melyek közül az utóbbinak négy (A-D) altípusa ismert. Az NMDA antagonisták
alkalmazhatósága számos területen felmerült és kísérletes bizonyítást nyert neuroprotekció,
fájdalomcsillapítás, görcsgátlás, Parkinson modellek, drog- és alkohol-függıség esetén. A
tisztán NMDA antagonista hatású vegyületek fejlesztése tipikus mellékhatásaik miatt
(pszichotomimetikus, szedatív, memóriarontó) abbamaradtak. Az NR2B alegység altípusra
szelektív antagonisták mentesnek bizonyultak ezektıl a mellékhatásoktól. A krónikus és
neurogén fájdalmak csillapítása megoldatlan terápiás probléma. Társaságunk a kielégítetlen
gyógyászati igényt és a hatásmechanizmus újdonságát mérlegelve, 1999-ben döntött az NR2B
szelektív NMDA antagonista hatásmechanizmuson alapuló új fájdalomcsillapító kutatási
projekt elindításáról. Terápiás célunk egy orálisan adható fájdalomcsillapító volt krónikus és
neurogén fájdalmak enyhítésére. Ennek érdekében ifenprodil típusu, NR2B szelektív
antagonista hatású molekulák elıállítását és tesztelését határoztuk el. Egy az irodalomból
ismert közepes hatású molekulából kiindulva több, számos szerkezeti módosításra lehetıséget
adó, hatásos vegyületcsaládot azonosítottunk. Az egyes vegyületcsaládok vezérmolekuláit a
klasszikus gyógyszerkémia szabályainak megfelelıen optimalizáltuk. Az in vitro és in vivo
biológiai vizsgálati módszerek egymásra épülı rendszert, ún. szkrínkaszkádot alkottak. Új
molekulák tervezésekor elsısorban egy NMDA funkcionális teszt adatait vettük figyelembe.
Az elıállított molekulákat a farmakofor modellünk fejlıdését leginkább tükrözı idırendi
sorrend alapján mutatom be. Saját kutatásunk és a versenytársak reprezentatív eredményeit
47. ábrán egy idıskálán állítom párhuzamba. Az elsıként azonosított NR2B altípus szelektív
NMDA anatagonista hatású molekula az ifenprodil volt. Az ifenprodil azonban vegyes profilú
anyag, számos receptor és ioncsatorna mőködését is gátolja. Az eredetileg α-1 adrenoceptor
antagonista hatása alapján vérnyomáscsökkentésre kifejlesztett ifenprodilról késıbb mutatták
ki, hogy nem kompetitív antagonistája az NR2B altípust tartalmazó NMDA receptornak. Az
ezen a területen megindult kutatásokban leggyakrabban az ifenprodilt választották
kiindulópontként, aminek eredményeként számos hatásos, hozzá hasonló farmakoforral
rendelkezı molekulát és vegyületcsaládot fedeztek fel.

103
HO
OH
CH3
N
ifenprodil
Hosszú idın keresztül az ifenprodilhoz hasonlóan bázikus nitrogént is tartalmazó farmakofor
modell alapján folyt a vegyületek elıállítása. A kilencvenes évek elején társaságunknál is
nagyszámú ifenprodil analogon készült in vivo CNS vizsgálatok céljából. 1998-ban jelent meg
az irodalomban az a megfigyelés, miszerint a bázikus nitrogén nem alapfeltétele a szelektív
NMDA receptor antagonizmusnak. Az University of Oregon, a CoCensys és a Parke Davis
közös kutatócsoportja egy fahéjsavamidot (35) azonosított hatásos és szelektív NR2B
antagonistaként. Kémiai munkánk kiindulópontjaként választottuk ezt a vegyületet mivel
feltételezésünk szerint a mellékhatások döntı részéért éppen az ifenprodil analogonokban
lévı bázikus nitrogén a felelıs. A 35 fahéjsavamid aminkomponensének elınyös cseréjével
egy aktívabb származékhoz jutottam (69).
O
N
Cl
OH
H
O
N
Cl
OH
OHH
35 69
Közben az University of Oregon, a CoCensys és a Parke Davis közös kutatócsoportja egy
újabb közleményben további megfigyelésekrıl számolt be. Többek között leírták, hogy a
klóratom és a hidroxilcsoport helyzetének felcserélése szintén hatásos molekulát (65)
eredményezett, vagyis mindkét orientációjú molekula képes kötıdni a receptorhoz. A 4-
hidroxi-fahéjsavamid (65) 4-klór-feniletil-amin amidkomponensét 4-benzil-piperidinre
cserélve egy közel háromszor aktívabb molekulához jutottak (37).
O
N
HO
Cl
H
N
O
HO
65 37 Felismerve a bázikus nitrogént nem tartalmazó molekulák vélhetı elınyeit, kémiai
munkánkat erre a megfigyelésre alapoztuk, és sokáig egyedül a versenytársak közül savamid
típusú vegyületeket állítottunk elı. Az ifenprodilszerő anyagokat leíró farmakofor modell
esszenciális és a változtatásokat jobban toleráló részeinek megismerése után igazoltuk

104
feltételezésünket, miszerint a fahéjsav szerkezeti rész merevítésével még hatásosabb
molekulákhoz juthatunk. A fahéjsav α-szénatomjának és benzolgyőrőjének összekötése a
protondonor sajátságú NH-csoporttal egy hatszor hatásosabb vegyülethez a 6-hidroxi-indol-2-
karboxamidhoz (82) vezetett.
N
O
N HO
H 1
2
34
5
67
W
V XN
QZ
Y
82 A Az indolszármazékok optimalizációja során az összes lehetséges ponton megvizsgáltuk a
gyógyszerszerő molekulákat eredményezı változtatások biológiai hatását (A).
Tanulmányoztuk miként befolyásolja az aktivitást a hidroxilcsoportok száma és pozíciója.
Vizsgáltuk az indolgyőrő 1-es és 3-as helyzetének helyettesíthetıségét, a karbonilcsoport
szerepét. A farmakofor modell legkonzervatívabb részének a piperidingyőrő bizonyult, más
aminra való lecserélése a hatás elvesztésével járt. Változtattuk a piperidingyőrőn a
benzilcsoport helyét, az aromás rész szubsztituenseit, a piperidin 4-es helyzető szénatomja és
a jobboldali aromásgyőrőt összekötı láncot. A fahéjsav merevítésével levezethetı
indolszármazékok kedvezı farmakológiai eredménye után más heterobiciklusokat is
megvizsgáltunk. Az indolváznak a vele potenciálisan izoszter benzimidazol győrőrendszerrel
való helyettesítése még aktívabb származékot (142) eredményezett. A benzimidazol-
származékokat az indolszármazékok körében nyert tapasztalatok alapján optimalizáltuk.
N
N
O
N HO
H
142 Az indol- és benzimidazol-2-karboxamidokat védı szabadalmi bejelentésünk benyújtása után
egy évvel, egy Merck szabadalmi bejelentés nyilvánossá válásakor derült ki, hogy a Merck
kutatói velünk egyidıben szintén benzimidazol-származékokat vizsgáltak. Bár
példavegyületeikben a győrő kettes helyzető szénatomját karbonil helyett alkillánc köti össze
a piperidin nitrogénjével, a fél évvel hamarabb benyújtott bejelentésükben megfogalmazott túl
általános képlet rontotta a mi vegyületeink újdonságát. További iparjogvédelmi lépésekre nem
volt szükség, ugyanis idıközben egy még aktívabb vegyületcsaládot azonosítottunk. Az indol
pirrolrészének képzeletbeli győrőfelnyitásával egy új hatásos szerkezethez, oxálsavdiamidhoz
jutottunk (183). Sikeres stratégiát dolgoztunk ki a metabolikusan instabil fenolos

105
hidroxilcsoport helyettesítésére. A fenol molekularész lecserélése protondonor sajátságú NH-
csoportot tartalmazó heterobiciklusokra az ADME tulajdonságok mellett a hatás javulását is
eredményezte. A molekula optimalizálását követıen a legelınyösebb tulajdonságokkal
rendelkezı 185-ös származékot (RGH-896, radiprodil) választottuk ki klinikai vizsgálatok
céljából.
HN
N
HO
O
O
HN
N
O
O
O
NO
F
H
183 185 (RGH-896, radiprodil) Az indolgyőrő pirrolrészének egy karbonilcsoporttal történı képzeletbeli győrőbıvítésével
levezethetı kinurénsavszármazék (230) szintén hatásos molekula. A 230 vezérmolekulát az
indol- és benzimidazol-2-karboxamidok körében nyert tapasztalatok alapján rövid idın belül,
egy tucat származék képzésével sikerült optimalizálni.
N
O
N
O
HO
H
230 Az oxálsavdiamidok eredményességét követıen tíz olyan háromtagú láncot vizsgáltam meg
mellyel összekötve a baloldali aromásgyőrőt és a piperidin nitrogénjét, gyógyszerszerő
molekulához jutunk (B). Ezek közül legaktívabbnak egy benzoil-karbamid-származék
bizonyult (274). A molekula optimalizációját oldatfázisú kombinatorikus kémiai szintézissel
oldottam meg.
N
O
N
HO
O
H
AB
CN
HO
B 274 A nem ifenprodil típusú molekulák kutatását egy korábbi Merck közleményt követıen
indítottuk el 2003-ban, melyben NR2B receptoron ható amidinekrıl számoltak be (46, 48)119,
79. Szabadalmi védelmükrıl már, 1999-ben és 2000-ben gondoskodtak. A Merck kutatóinak
eredményeit felhasználva, korábbi tapasztalatom alapján új típusú, gyógyszerszerőbb

106
szerkezetet, indol-2-karboxamidineket (299) terveztem. A vegyületeket kombinatorikus
kémia módszerével állítottam elı.
H
Cl
N
NH2
N
CH3O
OCH3
HN
NH OCH3
H HN
NH OCH3
CF3O
Merck
Richter
299 46 (Merck) 48 Az utolsó fejezetben 4-benzilidén-piperidin-származékokat ismertetek. A korábban elıállított
hatásos molekulák többsége 4-benzil-piperidin-származék volt. Ezek benzilidén-piperidin
megfelelıi azon túlmenıen, hogy hasonló affinitással kötıdnek az NR2B receptorokhoz, még
egy elınyös tulajdonsággal rendelkeznek. A vizsgált mellékhatásokkal kapcsolatban, különös
tekintettel a lokomotoros aktivitás fokozására, jóval nagyobb a terápiás ablakuk. Ez a hatás
kiváltképpen a benzimidazol (312) és az oxálsavdiamid-származékok (321) esetében
figyelhetı meg.
H
N
N
O
N HO
H
HN
N
O
O
O
NO
312 321 Az indol- és benzimidazol-2-karbonsavak benzilidén-piperidinekkel képezett amidjait
hagyományos módon, míg az oxálsavdiamidokat oldatfázisú parallel szintézissel állítottam
elı.
Kutatómunkánk több preklinikai vizsgálatokig jutó ígéretes anyagot eredményezett, melyek
közül egy (RGH-896, radiprodil) klinikai vizsgálatokra került.
Munkánk eredményeirıl eddig 8 szabadalmi bejelentésben és 9 gyógyszerkémiai tárgyú
cikkben számoltunk be.
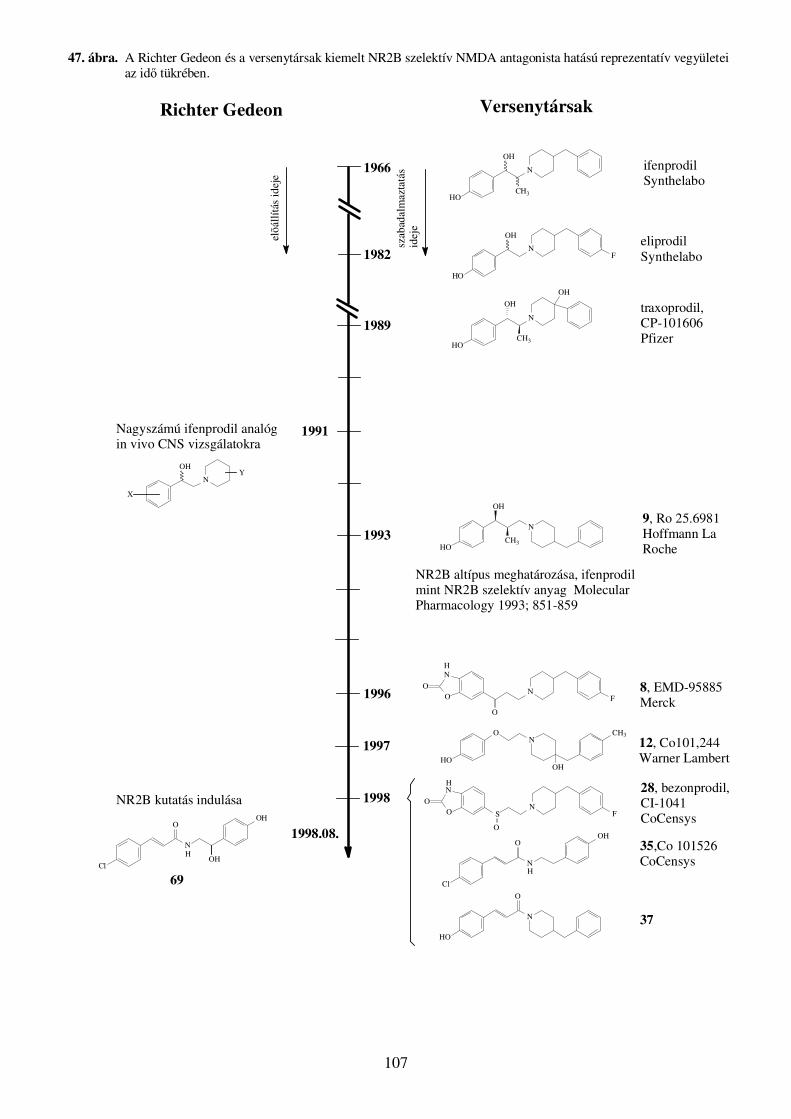
107
69
1966
1982
1989
1991
1993
1996
1997
1998
N
HO
OH
CH3
N
HO
F
OH
N
CH3HO
OH
OH
OH
CH3HO
N
O
HO
N
OH
CH3
O
N
HO
O
N
Cl
OH
OHH
NR2B kutatás indulása
Nagyszámú ifenprodil analóg in vivo CNS vizsgálatokra
NS
O
N
OO
F
H
O
N
Cl
OH
H
N
O
N
OO
F
H
Richter Gedeon Versenytársak
ifenprodilSynthelabo
eliprodilSynthelabo
traxoprodil, CP-101606Pfizer
9, Ro 25.6981Hoffmann La Roche
NR2B altípus meghatározása, ifenprodilmint NR2B szelektív anyag Molecular Pharmacology 1993; 851-859
8, EMD-95885Merck
12, Co101,244Warner Lambert
28, bezonprodil, CI-1041CoCensys
35,Co 101526CoCensys
N
OHY
X
37
1998.08.
elõá
llítá
s id
eje
szab
adal
maz
tatá
sid
eje
47. ábra. A Richter Gedeon és a versenytársak kiemelt NR2B szelektív NMDA antagonista hatású reprezentatív vegyületei az idı tükrében.

108
1999
2000
Richter Gedeon Versenytársak
12HoffmannLa Roche
1999.10.
2001
1999.11.
2003
2004
2003.07.
2002.11.
2002.04.
2001.01.
2000.07.
Pfizer
elõá
llítá
s id
eje
szab
adal
maz
tatá
sid
eje
N
O
NHO
H
2002
O
HO
N
OH
CH3OH
38 analógMerck82
N
N
O
NHO
H
142
HN
O
N
N
OO
OH
184
46Merck
HO
O
N
N O
O
H 42Merck
H
H
N
N
OO
O
NO
O
HO
O
N N
NH
H
NO
OH
N
OH
N
OEt
H
Merck
Pfizer
HN
O
N
N
OO
OH
321
N
O
N
HO
O
H274
HN
O
N
O
HO
230
HN
NH
N
CH3O
OCH3
H299
N
NH
CF3O
CF3
H
H
N N
NOH
61HoffmannLa Roche
51Merck
N
N
NN
H
N
NH
I
H

109
7. TÉZISEK
1. Felismerve a bázikus nitrogént nem tartalmazó molekulák vélhetı elınyeit egy, az
irodalomból ismert közepes hatású, ám nem bázikus nitrogént tartalmazó savamid típusú
molekulából kiindulva, in vitro aktív, NR2B szelektív NMDA antagonista hatású fahéjsav-
amidokat állítottam elı. Feltevésünk igazolódott, az NR2B szelektív NMDA antagonisták
mellékhatásaiért leginkább felelıssé tehetı α1 és 5-HT2A receptorokhoz a fahéjsavamidok
nem mutatnak affinitást.
2. Az University of Oregon, a CoCensys és a Parke Davis közös kutatócsoportjának a
fahéjsavamidok kutatásában elért eredményeit felhasználva feltérképeztem milyen, az
ifenprodil analogonok körében közkedvelt piperidinektıl eltérı szekunder aminnal képezett
fahéjsavamid mutat még kellı hatékonyságot. A kiválasztott szekunder aminokat,
kombinatorikus kémiai módszerrel több, a fenolos hidroxilcsoporttól metabolikusan stabilabb,
de hasonlóan protondonor csoporttal szubsztituált fahéjsavval acileztem. Ílymódon a
versenytársak és saját korábbi leghatásosabb vegyületeinknél is aktívabb, vélhetıen
metabolikusan stabilabb fahéjsavszármazékot sikerült elıállítanom.
3. Igazoltam feltételezésünket, miszerint a fahéjsav szerkezeti rész merevítése még aktívabb
molekulákat eredményez. A fahéjsav α-szénatomjának és benzolgyőrőjének protondonor
sajátságú NH-csoporttal való összekötésével egy, a fahéjsavamidoktól sokkal aktívabb
vegyületcsaládhoz, 6-hidroxi-indol-2-karboxamidokhoz jutottam. Több képviselıjük, in vivo
is hatásos, megfelnek egy gyógyszerrel szemben támasztott farmakokinetikai és
farmakodinámiai követelményeknek. A vezérmolekula optimalizálása során elıállított
nagyszámú származék szerkezet-hatás összefüggéseit felhasználva igyekeztem teljeskörően
felderíteni a farmakofor modellt, lehetıvé téve újabb hatásos vegyületcsaládok azonosítását,
azok minél hatékonyabb optimalizálását.105, 106, 108
4. A 6-hidroxi-indol-2-karboxamidok indolvázának benzimidazol győrőrendszerrel való
helyettesítésével általánosan egy nagyságrenddel aktívabb származékokat sikerült
elıállítanom. 105, 107, 108
5. A 6-hidroxi-indol-2-karboxamidokban az indol pirrolrészének képzeletbeli győrőfel-
nyitásával egy új hatásos szerkezethez, oxálsavdiamidhoz jutottunk. Sikeres, a fahéjsav-

110
amidok körében már bevált stratégiát dolgoztunk ki a metabolikusan instabil fenolos
hidroxilcsoport helyettesítésére. A fenol molekularész lecserélése protondonor sajátságú NH-
csoportot tartalmazó heterobiciklusokra az ADME tulajdonságok mellett a hatás javulását is
eredményezte.110, 111
6. A 6-hidroxi-indol-2-karboxamidokban az indol pirrolrészének egy karbonilcsoporttal
történı képzeletbeli győrőbıvítésével szintén aktív molekulacsaládhoz,
kinurénsavszármazékokhoz jutottam.113, 114
7. Az oxálsavdiamidok eredményességét követıen analitikus módon megvizsgálva és
változtatva a baloldali aromásgyőrőt és a piperidin nitrogénjét összekötı láncot, in vivo is
hatásos, NR2B szelektív NMDA antagonista hatású benzoil-karbamid-származékokat
azonosítottam.117, 118
8. Az NR2B szelektív NMDA antagonistákra jellemzı 4-benzil-piperidin aminkomponenst
az általunk korábban elıállított hatásos származékokban 4-benzilidén-piperidinre cserélve az
NR2B receptorokhoz hasonló affinitással kötıdı, de sokkal kedvezıbb mellékhatásprofillal
rendelkezı vegyületekhez jutottam. A 4-benzilidén-piperidinek gyógyszerkémiai
alkalmazását az általam kidolgozott elıállítás tette lehetıvé, melynek során a kettıskötés exo
helyzete stabil marad.125, 126
9. A nem ifenprodil típusú molekulák körében végzett munkám során a Merck kutatói által
azonosított E-N1-(benzil)fahéjsavamidint választva kiindulópontnak, felhasználva a korábban
tapasztalt fahéjsav-indol szerkezeti elemek bioizosztériáját, gyógyszerszerőbb szerkezetet,
indol-2-karboxamidineket terveztem. A vegyületeket kombinatorikus kémiai módszerrel
állítottam elı.120, 121

111
8. MOLEKULALISTA
Dolgozatom a Richterben végzett NR2B szelektív NMDA antagonista kutatás egészét átfogja.
A hagyományos módon elıállított vegyületek szerkezetét NMR és MS spektroszkópiával,
tisztaságát (min. 95 %) HPLC módszerrel igazoltuk. A kombinatorikus kémiai módon vagy
parallel reakciókkal elıállított vegyületeket HPLC-MS alapján azonosítottuk (tisztaság min.
85 %), a közülük hatásosnak bizonyúló molekulákat hagyományos módszerrel is elıállítottuk,
szerkezetüket igazoltuk. Munkánk eredményeirıl eddig a következı szabadalmi
bejelentésekben, cikkekben és elıadásokban számoltunk be.
Cikkek:
1./ 38. Borza, I.; Domány, Gy. NR2B Selective NMDA Antagonists: The Evolution of the Ifenprodil-Type Pharmacophore. Curr. Top. In Med. Chem. 2006, 6, 687-695. (IF: 4,40; I: 22)
2./ 106. Borza, I.; Kolok, S.; Gere, A.; Ágai-Csongor, É.; Ágai, B.; Tárkányi, G.; Horváth, Cs.; Barta-Szalai, G.; Bozó, É.; Kiss, Cs.; Bielik, A.; Nagy, J.; Farkas, S.; Domány, Gy. Indole-2-carboxamides as Novel NR2B Selective NMDA Receptor Antagonists. Bioorg. Med. Chem. Letters 2003, 13, 3859-3861. (IF: 2,182; I: 19) 3./ 107. Borza, I.; Kolok, S.;Gere, A.; Nagy, J.; Fodor, L.; Galgóczy, K.; Fetter, J.; Bertha, F.; Ágai, B.; Horváth, Cs.; Farkas, S.; Domány, Gy. Benzimidazole-2-carboxamides as Novel NR2B Selective NMDA Receptor Antagonists. Bioorg. Med. Chem. Lett.
2006, 16, 4638-4640. (IF: 2,538; I: 8) 4./ 108. Borza, I.; Bozó, É.; Barta-Szalai, G.; Kiss, Cs.; Tárkányi , Á.; Demeter Á.; Gáti T.; Háda, V.; Kolok, S.; Gere, A.; Fodor, L.; Nagy, J.; Galgóczy, K.; Magdó, I.; Ágai, B.; Fetter,J.; Bertha, F.; M. Keserő, Gy.; Horváth, Cs.; Farkas, S.; Greiner, I.; Domány, Gy. Selective NR1/2B N-Methyl-D-aspartate Receptor Antagonists among Indole-2-carboxamides and Benzimidazole-2-carboxamides. J. Med. Chem. 2007, 50, 901-914. (IF: 4,895; I: 5) 5./ 111. Barta-Szalai, G.; Borza, I.; Bozó, É.; Kiss, Cs.; Ágai, B.; Proszenyák, Á.; Keserő, Gy. M.; Gere, A.; Kolok, S.; Galgóczy, K.; Horváth, Cs.; Farkas, S.; Domány, Gy. Oxamides as novel NR2B selective NMDA receptor antagonists. Bioorg. Med.
Chem. Letters 2004, 14, 3953-3956. (IF: 2,333; I: 19)
6./ 114. Borza, I.; Kolok, S.; Horváth, Cs.; Farkas, S.; Domány, Gy. New kynurenic acid amides as novel NR2B selective NMDA receptor antagonists. Bioorg. Med. Chem.
Letters 2007, 17, 406-409. (IF: 2,604; I: 2) 7./ 118. Borza, I.; Greiner, I.; Kolok, S.; Galgóczy, K.; Ignácz-Szendrei, Gy.; Horváth, Cs.; Farkas, S.; Gáti, T.; Háda, V.; Domány, Gy. New benzoyl urea derivatives as

112
novel NR2B selective NMDA receptor antagonists. Die Pharmazie 2006 61(9), 799- 800. (IF: 0,606; I: 2) 8./ 121. Borza, I.; Kolok, S.; Ignácz-Szendrei, G.; Greiner, I.; Tárkányi, G.; Galgóczy K.; Horváth, Cs.; Farkas, S.; Domány, Gy. Indole-2-carboxamidines as novel NR2B selective NMDA receptor antagonists. Bioorg. Med. Chem. Letters 2005, 15, 5439- 5441. (IF: 2,478; I: 4) 9./ Wéber, Cs.; Bielik, A.; Demeter, Á.; Borza, I.; Ignácz-Szendrei, G.; Keserő, Gy.;
Greiner, I.; Solid-phase synthesis of 6-hydroxy-2,4-diaminoquinazolines. Tetrahedron 2005, 61, 9375-9380. (IF: 2,61; I: 3)
Szabadalmak: 1./ 105. Borza, I.; Bartáné Szalai, G.; Domány, Gy.; Horváth, Cs.; Farkas, S.; Nagy, J.; Kolok, S. Amide derivatives as NMDA receptor antagonists. WO 2002/34718A1. 2./ 110. Domány, Gy.; Horváth, Cs.; Farkas, S.; Barta-Szalai, G.; Nagy, J.; Kolok, S.; Bozó, É.; Borza, I.; Vágó, I.; Bielik,A.; Ignácz-Szendrei Gy.; Keserő, Gy. M. Piperidine derivatives as NMDA receptor antagonists. WO 2003010159A1. 3./ 113. Borza, I.; Horváth, Cs.; Farkas, S.; Nagy, J.; Kolok, S. Kynurenic acid amide derivatives as NR2B receptor antagonists. WO 2006010967A1. 4./ 117. Borza, I.; Bartáné Szalai, G.; Bozó, É.; Kis, Cs.; Horváth, Cs.; Farkas, S.; Nagy, J.; Kolok, S. New benzoyl urea derivatives. WO 2006010966A1. 5./ 120. Borza, I.; Horváth, Cs.; Farkas, S.; Nagy, J.; Kolok, S. Indole-2-carboxamidine derivatives as NMDA receptor antagonists. WO 2006010965A1. 6./ 125. Borza, I.; Horváth, Cs.; Farkas, S.; Gyertyán, I.; Nagy, J.; Kolok, S.; Galgóczy, K.; Sághy, K. New heterocyclic acid amide derivatives. WO 2006010969A1. 7./ 126. Borza, I.; Horváth, Cs.; Farkas, S.; Gyertyán, I.; Nagy, J.; Kolok, S.; Galgóczy, K.; Sághy, K. New 4-benzylidene-piperidine derivatives. WO 2006010964A1. 8./ Keserő, Gy. M.; Vágó, I.; Farkas, S.; Horváth, Cs.; Bielik, A.; Borza, I.; Wéber, Cs.; Kolok, S. New aryloxy acetic acid amide derivatives. WO 2006010968A1. Elıadások:
1./ Új, NR2B-szelektív NMDA antagonista hatású indol-2-karbonsav-amid és
benzimidazol-2-karbonsav-amid származékok Borza István, Tárkányi Gábor, Demeter Ádám, Kolok Sándor, Bartáné Szalai Gizella, Bozó Éva, Kis Csilla, Ágai Béla, Berta Ferenc, Fetter József, Domány György
2002. Balatonszemes, Heterociklusos Kémiai Munkabizottsági Őlés 2/ Új, NR2B-szelektív NMDA antagonista hatású indol-2-karbonsav-amid és
benzimidazol-2-karbonsav-amid származékok

113
Borza István, Tárkányi Gábor, Demeter Ádám, Kolok Sándor, Bartáné Szalai Gizella, Bozó Éva, Kis Csilla, Ágai Béla, Berta Ferenc, Fetter József, Domány György 2002. Visegrád, Gyógyszerkémiai és Gyógyszertechnológiai Szimpózium
3/ Fenol bioizosztér heterociklusok NR2B szelektív NMDA antagonisták körében
Borza István, Bozó Éva,Tárkányi Gábor, Demeter Ádám, Kolok Sándor, Kis Csilla, Domány György 2003. Eger, Gyógyszerkémiai és Gyógyszertechnológiai Szimpózium
4/ Újabb eredmények a heterobiciklusok körében Borza István 2003. Budapest, Flavonoidkémiai Munkabizottsági Őlés Poszterek: 1./ Indole-2-carboxamides as novel NR2B selective NMDA antagonists
Borza István, Tárkányi Gábor, Demeter Ádám, Kolok Sándor, Bartáné Szalai Gizella, Bozó Éva, Kis Csilla, Ágai Béla, Berta Ferenc, Fetter József, Domány György 2002. Barcelona, XVII th. International Symposium on Medicinal Chemistry
2./ Fenol bioizosztér heterociklusok NR2B szelektív NMDA antagonisták körében
Borza István, Bozó Éva,Tárkányi Gábor, Demeter Ádám, Kolok Sándor, Kis Csilla, Domány György 2003. Hajdúszoboszló, Vegyészkonferencia
3/ Benzimidazole-2-carboxamides as novel NR2B selective NMDA receptor antagonists
György Domány, István Borza, Sándor Kolok, Anikó Gere, Kornél Galgóczy, Ildikó Magdó, József Fetter, Ferenc Bertha, Béla Ágai, Csilla Horváth, Sándor Farkas, György M. Keserő 2006. San Francisco, American Chemical Society 232nd National Meeting and Exposition
4/ CoMSIA Study of Oxamide and Glycineamide-type NR2B Selective NMDA Receptor Antagonists Magdó Ildikó. - Borza István. - Bartáné Szalai Gizella. - Bozó Éva. - Gere Anikó. - Horváth Csilla. - Farkas Sándor. - Domány György. - Keserő György M. Austrian-German-Hungarian-Italian-Polish-Spanish Joint Meeting on Medicinal Chemistry (2005. június 20-23.) (Bécs, Ausztria)
5/ CoMSIA Study of Indole- and Benzimidazole-2-carboxamide-type NR2B Selective
NMDA Receptor Antagonists Magdó Ildikó. - Borza István. - Kolok Sándor. - Gere Anikó. - Horváth Csilla. - Bartáné Szalai Gizella. - Domány György. - Keserő György M. 15th European Symposium on QSAR (Euro-QSAR 2004) (2004. szeptember 5-1) (Isztanbul, Törökország)
6/ Antinociceptive Effect of NR2B Selective NMDA Receptor Antagonists in Chronic
Pain Models, in Comparison with Pregabalin. - 2003 Horváth Csilla. - Farkas Sándor. - Kedves Rita. - Galgóczy Kornél. - Egyed R. - Nagy

114
József, Kolok Sándor. - Borza István. - Domány György. - Szombathelyi Zsolt 23rd European Winter Conference on Brain Research (2003. március 8-15.) (Les Arcs, Franciaország)
7/ Kynurenic Acid Amides as Novel NR2B Selective NMDA Receptor Antagonists
István Borza, Sándor Kolok, Kornél Galgóczy, Csilla Horváth, Sándor Farkas, István Greiner, György Domány
11th Belgian Organic Synthesis Symposium (2008. július-13-18) (Ghent, Belgium)
Dolgozatomban a témában közölt kilenc kémiai tárgyú cikkbıl nyolcat, a benyújtott nyolc
alapszabadalmi bejelentésbıl hetet idézek. A Bioorganic & Medicinal Chemistry Lettersben
megjelentetett cikkekben közöltük a molekulák elıállításának módját, a Journal of Medicinal
Chemistry és a Pharmazie folyóiratokban, valamint a szabadalmi bejelentésekben részletes
receptekkel együtt mutattuk be a vegyületeket. Mivel a dolgozatomban szereplı vegyületek
jelentıs részét leírtuk, terjedelmi okokból a kémiai kísérleti részt a bemutatott molekulák
publikációs listájával helyettesítem.

115
Végtermékek listája, elıfordulásuk közleményekben, szabadalmi bejelentésekben.
vegyületszám vegyületszám a közleményben
Példaszám szabadalmi
bejelentésben
FAHÉJSAVSZÁRMAZÉKOK
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
INDOL-2-KARBOXAMIDOK
82 6a108 1105
83 7a108 3105
84 8a108 49105
85 9a108
86 10108 26105
87 11108 42105
88 23108
89 24108
90 25108
91 6b108
92 6c108 43105
93 6d108 33105
94 6e108 7105
95 6f108 6105
96 6g108 9105
97 6h108 4105
vegyületszám vegyületszám a közleményben
példaszám szabadalmi
bejelentésben
98 6i108 29105
99 6j108 19105
100 6k108
101 6l108 5105
102 6m108 12105
103 6n108 40105
104 6o108
105 6p108
137 55108
138 56108
139 57108
140 58108
141 59108
BENZIMIDAZOL-2-KARBOXAMIDOK
142 60a108 100105
143 61a108
144 62108
145 63108
146 64108
147 65108
148 60m108
149 60q108
150 60r108
151 60s108
152 60t108
153 60u108
OXÁLSAVDIAMIDOK
183 2111
184 3d111 8110
185 3e111 3110
186 3b111 7110
187 3g111 61110
188 3i111 101110
189 3f111 31110
190

116
vegyületszám vegyületszám a közleményben
Példaszám szabadalmi
bejelentésben
OXÁLSAVDIAMIDOK
191
192
193
194
195
196
197
198
199
200
201
202
203 32110
204 48110
205 3k111
206
207 39110
208
KINURÉNSAVAMIDOK
228 28
229 29114 12113
230 30a114 2113
231 30b114 1113
232 30c114 4113
233 30d114 3113
234 30e114 10113
235 30f114 7113
236 30g114 8113
237 31114
238 32114 9113
239 38114
BENZOIL-KARBAMIDOK
267
268
269
vegyületszám vegyületszám a közleményben
példaszám szabadalmi
bejelentésben
270
271
272
273
274 9118 1117
275
276
277
278
279
280
281 7117
282 4118 4117
283 5118 3117
284 6118 2117
285 7118 29117
286 8118 5117
287 10118 26117
288 11118 27117
289 12118 28117
INDOL-2-KARBOXAMIDINEK
295 5a121 42120
296 5b121 151120
297 5c121 76120
298 5d121 117120
299 5e121 36120
300 5f121 147120
301 5g121 72120
302 5h121 112120
303 5i121 31120
304 5j121 38120
305 5k121 32120
306 5l121 29120
307 5m121 17120
308 5n121 18120
309 5o121 1120
117
vegyületszám vegyületszám a közleményben
példaszám szabadalmi
bejelentésben
BENZILIDÉN-PIPERIDINEK
312 1125
313 2125
314 3125
315 4125
316 5125
317 6125
321 1126
322 24126

118
9. IRODALOMJEGYZÉK
1. Williams, M.; Kowaluk, E. A.; Arneric, S. P. Emerging Molecular Approaches to Pain
Therapy. J. Med. Chem. 1999, 42, 1481-1500. 2. Int. Assoc. For Study of Pain. 1994. Pain Vol. II. Issue3, Neuregenic Pain: Diagnosis and Treatment. 3. Sindrup, S. H.; Jensen, T. S. Efficacy of pharmacological treatments of neurophtic pain: an update and effect releated to mechanism of drug action. Pain 1999, 83, 389-400. 4. Chizh, B. A.; Headley, P. M. NMDA antagonists and neuropathic pain-multiple drug targets and uses. Cur. Pharm. Des. 2005, 11, 2977-2994. 5. Salter, M.W. Cellular signalling pathways of spinal pain neuroplasticity as targets for analgesic development. Cur. Top. Med. Chem. 2005, 5, 557-567. 6. Mendel, L. M., Physiological properties of unmyelinated fiber projections to the spinal cord. Exp. Neurol. 1966, 16, 316-332. 7. Davies, S. N.; Lodge, D. Evidence for involvement of N-methylaspartate receptors in”windup” of class 2 neurones in the dorsal horn of the rat. Brain Res. 1987, 424, 402- 406. 8. Dickenson, A. H.; Sullivan, A. F. Evidence for a role of the NMDA receptor in the frequency dependent potentiation of deep rat dorsal horn nociceptive neurones following C fibre stimulation. Neuropharmacology 1987, 26, 1235-1238. 9. Kim, Y. I.; Na, H. S., NMDA receptors are important for both mechanical and thermal allodynia from peripheral nerve injury in rats. Neuroreport 1997, 8, 2149-53. 10. Wong, C. S.; Cherng, C. H. Intrathecal administration of excitatory amino acid receptor antagonists or nitric oxide synthase inhibitor reduced automy behavior in rat. Anesth. Analg. 1998, 87, 605-608. 11. Ren, K.; Williams, G. N. The intrathecal administration of excitatory amino acid receptor antagonists selectively attenuated carrageenan-induced behavioral hyperalgesia in rats. Eur. J. Pharmacol. 1992, 219, 235-243. 12. Klepstad, P.; Maurset, A. Evidence of a role for NMDA receptors in pain perception. Eur. J. Pharmacol. 1990, 187, 513-518. 13. Nikolajsen, L.; Hansen, P. O. Oral ketamine treatment of postamputation stump pain. Acta
Anaesthesiol. Scand. 1997, 41, 427-429. 14. Eide, P. K.; Stubhag, A. Continuous subcutaneous administration of the N-methyl-D- aspartate receptor antagonist ketamine in the treatment of post-herpetic neuralgia. Pain 1995, 2, 221-228. 15. Price, D. D.; Mao, J. The N-methyl-D-aspartate receptor antagonist dextromethorphan selectively reduces temporal pain summation in man. Pain 1994, 59, 165-174. 16. Nikolajsen, L.; Gottrup, H.; Kristensen, A. G. D.; Jensen, T. S. Memantine (a N-Methyl- D-Aspartate Receptor Antagonist) in the Treatment of Neuropathic Pain After Amputation or Surgery: A Randomized, Double-Blinded, Cross-Over Study 17. Madsen, U.; Brauner-Osborne, H.; Greenwood, J. R.; Johansen, T. N.; Krogsgaard- Larsen, P.; Liljefors, T.; Nielsen, M.; Frolund, B. GABA and Glutamate Receptor Ligands and Their Therapeutic Potential in CNS Disorders. ed. Shayne Cox Gad, Drug Discovery Handbook, John Wiley & Sons Ltd, Chichester West Sussex, 2005: ch. 18, 797-907. 18. Borbényi, E.; Dank, M. Makó, E. Fájdalomcsillapítás a daganatos betegek kezelésében Magyar Onkológia 2001, 45, 81-88. 19. Kennedy, J. D. Neuropathic Pain: Molecular Complexity Underlies Continuing Unmet Medical Need J. Med. Chem. 2007, 50, 2547-2556. 20. Palkovits, M. Az agy és a fájdalom: az érzékelés és a válasz agypályái és transzmitterei. Orvosi Hetilap 2000, 141, 2231-2239.

119
21.http://cogsci.bme.hu/~ktkuser/KURZUSOK/BMEGT47MN10/DOC/05_fajdalom_latas_be vezeto.pdf 22. Schwartzman, R. J.; Grothusen, J.; Kiefer T. R.; Rohr, P. Arch Neuropathic Central Pain: Epidemiology, Etiology, and Treatment Options. Neurology 2001, 58, 1547-1550. 23. Zhang, J.; Hoffert, C.; Vu, H. K.; Groblewski, T.; Ahmad, S.; O’Donnell, D. Induction of CB2 receptor expression in the rat spinal cord of neuropathic but not inflammatory chronic pain models. Eur.J. Neurosci. 2003, 17, 2750-2754. 24. Kim, D. Y.; Kim, S. H.; Choi, H. B.; Min, C.; Gwag, B. J. High abundance of GluR1 mRNA and reduced Q/R editing of GluR2 mRNA in individual NADPH-diaphorase neurons. Mol. Cell. Neurosci. 2001, 17, 1025–33. 25. Parsons, C. G.; Danysz, W. Quack, G. Glutamate in CNS Disorders as a Target for Drug Development: An Update. Drug news and perspectives 1998, 11, 523-569. 26. Conti, F.; Weinberg, R.J.; Shaping excitation at glutamatergic synapses Trends Neurosci. 1999, 22, 451-458. 27. Huettner, J. E. Kainate receptors and synaptic transmission. Prog. Neurobiol. 2003, 70, 387–407. 28. Paoletti, P.; Neyton, J. NMDA receptor subunits: function and pharmacology. Current
Opinion in Pharmacology 2007, 7, 39-47. 29. Chen, P. E.; Wyllie, D. J. A. Pharmacological insights obtained from structure-function studies of ionotropic glutamate receptors. Br. J. Pharmacol. 2006, 147, 839-853. 30. Michaelis, E. K. Molecular biology of glutamate receptors in the central nervous system and their role in excitotoxicity, oxidative stress and aging. Prog. Neurobiol. 1998, 54, 369-415. 31. Chen, H. S. V.; Lipton, S. A. The chemical biology of clinically tolerated NMDA receptor antagonists. J. Neurochem. 2006, 97, 1611-1626. 32. a, Blahos, J.; Wenthold, R. J. Relationship betwen N-methyl-D-aspartate receptor NR1 splice variants and NR2 subunits. J. Biol. Chem. 1996, 271, 15669-15674. B, Chazot, P.; Coleman, S. K.; Cik, M..; Stephenson, F. A. Molecular characterization of N-methyl-D-aspartate receptors expressed in mammalian cells yields evidence for the coexistence of three subunit types within a discrete receptor molecule. J. Biol. Chem. 1994, 269, 24403-24409. 33. Dingledine, R.; Borges, K.; Bowie, D.; Traynelis, S. F. The glutamate receptor ion channels. Pharmacol. Rev. 1999, 51, 7-61. 34. Kew, J. N. C.; Kemp, J. A. An allosteric interaction between the NMDA receptor polyamine and ifenprodil sites in rat cultured cortical neurones. The Journal of
Physiology, 1998, 512, 17-28. 35. Chatterton, J. E.; Awobuluyi, M.; Premkumar, L. S.; Takahashi, H.; Talantova, M.; Shin, Y.; Cui, J.; Tu, S.; Sevarino, K. A.; Nakanishi, N.; Tong, G.; Lipton, S. A.; Zhang, D. Excitatory glycine receptors containing the NR3 family of NMDA receptor subunits. Nature, 2002, 415, 793-798. 36. a, Monyer, H.; Sprengel, R.; Schoepfer, R.; Herb, A.; Higuchi, M.; Lomeli, H.; Burnashev, N.; Sakmann, B.; Seeburg, P.H. Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science 1992, 256, 1217-1221. b, Ishii, T.; Moriyoshi, K.; Sugihara, H.; Sakurada, K.; Kadotani, H.; Yokoi, M.; Akazawa, C.; Shigemoto, R.; Mizuno, N.; Masu, M.; Nakanishi, S.; Molecular characterization of the family of the N-methyl-D-aspartate receptor subunits. J. Biol. Chem. 1993, 268, 2836-2843. 37. Kékesi, G.; Horváth, GY. A kinurénsav fájdalomcsillapító hatása. Ideggyógyászati Szemle 2002, 55, 313–322. 38. Borza, I.; Domány, Gy. NR2B Selective NMDA Antagonists: The Evolution of the

120
Ifenprodil-Type Pharmacophore. Curr. Top. In Med. Chem. 2006, 6, 687-695. 39. The Merck Index (Thirteenth Edition) Merck Research Laboratories Division of Merck
& Co., Inc., Whitehouse Station, NJ, 2001, page 878. 40. Carron, C.; Jullien, A.; Bucher, B. Synthesis and Pharmacological Properties of a Series of 2-Piperidino Alkanol Derivatives. Arzneim. Forsch.1971, 21, 1992-1998. 41. Scatton, B.; Carter, C.; Claustre, Y.; L’Heureux, R.; Arbilla, S.; Langer, S. Z.; Gotti, B.; Duverger, D.; MacKenzie, E. T. The Cerebral Anti-ischemic Agents, Ifenprodil and SL- 82.0715, Antagonise the Effects of NMDA. Proceedings of the Xth International Congress of Pharmacology, Sydney, Australia, 1987; Abstract No. 012.064. 42. Williams, K. Ifenprodil Discriminates Subtypes of the N-Methyl-D-aspartate Receptor: Selectivity and Mechanisms at Recombinant Heteromeric Receptors. Mol. Pharm. 1993, 44, 851-859. 43. Chenard, B. L.; Menniti, F. S. Antagoists Selective for NMDA Receptors Containing the NR2B Subunit. Curr. Pharm. Des. 1999, 5, 381-404. 44. Nikam, S. S.; Meltzer, L. NR2B Selective NMDA Receptor Antagonists. Curr. Pharm.
Des. 2002, 8, 845-855. 45. McCauley, J. A. NR2B Subtype-selective NMDA Receptor Antagonists: 2001-2004. Expert Opin. Ther. Patents 2005, 15, 389-407. 46. Chenard, B. L.; Shalaby, I. A.; Koe, B. K.; Ronau, R.T.; Butler, T. W.; Prochniak, M. A.; Schmidt, A. W.; Fox, C. B. Separation of α1 Αdrenergic and N-Methyl-D-aspartate Antagonist Activity in a Series of Ifenprodil Compounds. J. Med. Chem. 1991, 34, 3085- 3090. 47. Wermuth, C. G. Selective Optimization of Side Activities: Another Way for Drug Discovery. J. Med. Chem. 2004, 47, 1-12. 48. Avenet, P.; Léonardon, J.; Besnard, F.; Graham, D.; Frost, J.; Depoortere, H.; Langer, S.
Z.; Scatton, B. Antagonist Porperties of the Stereoisomers of ifenprodil at NR1a/NR2A and NR1a/NR2B subtypes of the NMDA receptor expressed in Xenopus oocytes. Eur. J.
Pharm. 1996, 296, 209-213. 49. Chenard, B. L.; Bordner, J.; Butler, T. W.; Chambers, L. K.; Collins, M. A.; De Costa, D.
L.; Ducat, M. F.; Dumont, M. L.; Fox, C. B.; Mena, E. E.; Menniti, F. S.; Nielsen, J.; Pagnozzi, M. J.; Richter, K. G. E.; Ronau, R. T.; Shalaby, I. A.; Stemple, J. Z.; White, W. F. (1S,2S)-1-(4-Hydroxyphenyl)-2-(4-hydroxy-4-phenylpiperidino)-1-propanol: A Potent New Neuroprotectant Which Blocks N-Methyl-D-aspartate Responses. J. Med. Chem. 1995, 38, 3138-3145.
50. Sang, C. N.; Weaver, J. J.; Jinga, L.; Wouden, J.; Salterelli, M. D. The NR2B subunit- selective NMDA receptor antagonist, CP-101,606, reduces spontaneous pain intensity in patients with central and peripheral neuropathic pain. Program No 814.9 2003 Abstract
Viewer/Itinerary Planner; Society for Neuroscience: Washington, DC, 2003. Online. 51. Butler, T. W.; Blake, J. F.; Bordner, J.; Butler, P.; Chenard, B. L.; Collins, M. A.;
DeCosta, D.; Ducat, M. J.; Eisenhard, M. E.; Menniti, F. S.; Pagnozzi, M. J.; Sands, S. B.; Segelstein, B. E.; Volberg, W.; White, W. F.; Zhao, D. (3R,4S)-3-[4-(4-Fluorophenyl)-4-hydroxypiperidin-1-yl]chroman-4,7-diol: A Conformationally Restricted Analogue of the NR2B Subtype-Selective NMDA Antagonist (1S,2S)-1-(4-Hydroxyphenyl)-2-(4-hydroxy-4-phenylpiperidino)-1-propanol. J. Med. Chem. 1998, 41, 1172-1184.
52. Leibrock, J.; Prücher, H.; Rautenberg, W. EMD 95885, a new eliprodil analogue with higher affinity for the N-methyl-D-aspartate (NMDA) receptor. Pharmazie 1997, 52, 479- 480. 53. Fischer, G.; Mutel, V.; Trube, G.; Malherbe, P.; Kew, J. N. C.; Mohacsi, E.; Heitz, M. P.;

121
Kemp, J. A. Ro 25-6981, a Highly Potent and Selective Blocker of N-Methyl-D-aspartate Receptors Containing the NR2B Subunit. Characterization in Vitro. J. Pharmacol. Exp.
Ther. 1997, 283, 1285-1292. 54. Mutel, V.; Buchy, D.; Klingelschmidt, A.; Messer, J.; Bleuel, Z.; Kemp, J. A.; Richards, J.
G. In Vitro Binding Properties in Rat Brain of [3H]-Ro 25-6981, a Potent and Selective Antagonist of NMDA Receptors Containing NR2B Subunits. J. Neurochem. 1998, 70, 2147-2155.
55. Kew, J. N. C.; Trube, G.; Kemp, J.A. State-dependent NMDA receptor antagonism by Ro 8-4304, a novel NR2B selective, non-competitive, voltage-independent antagonist. Brit. J.
Pharmacol. 1998, 123, 463-472. 56. Greengrass P.; Bremmer, R. Binding characteristics of 3H-Prazosin to rat brain α- adrenergic receptors. Eur. J. Pharmacol. 1979, 55, 323-326. 57. Pinard, E.; Alanine, A.; Bourson, A.; Büttelmann, B.; Gill, R.; Heitz, M.-P.; Jaeschke, G.;
Mutel, V.; Trube, G.; Wyler, R. Discovery of (R)-1-[2-Hydroxy-3-(4-hydroxy-phenyl)-propyl]-4-(4-methyl-benzyl)-piperidin-4-ol: a Novel NR1/2B Subtype Selective NMDA Receptor Antagonist. Bioorg. Med. Chem. Letters 2001, 11, 2173-2176.
58. Gill, R.; Alanine, A.; Bourson, B.; Buttelmann, B.; Fischer, G.; Heitz, M.-P.; Kew, J. N. C.; Levet-Trafit, B.; Lorez, H.-P.; Malherbe, P.; Miss, M.-T.; Pinard, E.; Roever, S.; Schmitt, M.; Trube, G.; Wybrecht, R.; Wyler, R.; Kemp, J. A. Pharmacological Characterization of Ro 63-1908 (1-[2-(4-Hydroy-phenoxy)-ethyl]-4-(4-methyl-benzyl)-pipridin-4-ol), a Novel Subtype-Selective N-Methyl-D-aspartate Antagonist. J.
Pharmacol. Exp. Ther. 2002, 302, 940-948. 59. Higgins, G. A.; Ballard, T. M.; Huwyler, J.; Kemp, J. A.; Gill, R. Evaluation of the
NR2B-selective NMDA receptor antagonist Ro 63-1908 on rodent behaviour: evidence for an involvement of NR2B NMDA receptors in response inhibition. Neuropharm. 2003, 44, 324-341.
60. Gill, R.; Lorez, H. P.; Wybrecht, R.; Miss, M.-T.; Bourson, A.; Fischer, G.; Kew, J.; Mutel, V.; Trube, G.; Roever, S.; Heitz, M.-P.; Kemp, J. A. Novel, Activity-dependent NMDA NR2B Subtype Selective Antagonists as Neuroprotective Agents. Abstracts book, XVIth International Symposium on Medicinal Chemistry, Bologna, 2000,: Abst ML-43.
61. Jaeschke, G.; Alanine, A.; Sleight, A. B.; Büttelman, B.; Fischer, H.; Gill, R.; Neidhart, M.-P. H.; Huwyler, J.; Kemp, J. A.; Kansy, M.; Mutel, V.; Pinard, E.; Trube, G.; Wyler, R. Synthesis and Biological Evaluation of β-Aminosulfones as Novel NMDA-NR1/2B Subtype Selective Antagonists. Drugs Fut. 2002, 27(Suppl. A): Abst C76.
62. Tamiz, A. P.; Whittemore, E. R.; Zhou, Z.-L.; Huang, J.-C.; Drewe, J. A.; Chen, J.-C.; Cai, S.-X.; Weber, E.; Woodward, R. M.; Keana, J. F. W. Structure-Activity Relationships for a Series of Bis(phenylalkyl)amines: Potent Subtype-Selective Inhibitors of N-Methyl-D-aspartate Receptors. J. Med. Chem. 1998, 41, 3499-3506.
63. Guzikowski, A. P.; Tamiz, A. P.; Acosta-Burruel, M.; Hong-Bae, S.; Cai, S. X.; Hawkinson, J. E.; Keana, J. F. W.; Kesten, S. R.; Shipp, C. T.; Tran, M.; Whittemore, E. R.; Woodward, R. M.; Wright, J. L.; Zhou, Z.-L. Synthesis of N-Substituted 4-(4-Hydroxy-phenyl)piperidines, 4-(4-Hydroxybenzyl)piperidines, and (+)-3-(4-Hydroxyphenyl)-pyrrolidines: Selective Antagonists at the 1A/2B NMDA Receptor Subtype. J. Med. Chem. 2000, 43, 984-994.
64. Tamiz, A. P.; Whittemore, E. R.; Woodward, R. M.; Upasani, R. B.; Keana, J. F. W. Structure-Activity Relationship for a Series of 2-Substituted 1,2,3,4-Tetrahydro-9H-pyrido[3,4-b]indoles: Potent Subtype-Selective Inhibitors of N-Methyl-D-aspartate (NMDA) Receptors. Bioorg. Med. Chem. Letters 1999, 9, 1619-1624.
65. Zhou, Z.-L.; Cai, S. X.; Whittemore, E. R.; Konkoy, C. S.; Espitia, S. A.; Tran, M.; Rock, D. M.; Coughenour, L. L.; Hawkinson, J. E.; Boxer, P. A.; Bigge, C. F.; Wise, L. D.;

122
Weber, E.; Woodward, R. M.; Keana, J. F. W. 4-Hydroxy-1-[2-(4- hydroxyphenoxy)ethyl]-4-(4-methylbenzyl)piperidine: A Novel, Potent, and Selective NR1/2B NMDA Receptor Antagonist. J. Med. Chem. 1999, 42, 2993-3000.
66. Shelkun, R. M.; Yuen, P., Serpa, K.; Meltzer, L. T.; Wise, L. D. Subtype-Selective N- Methyl-D-aspartate Receptor Antagonists: Benzimidazolone and Hydantoin as Phenol Replacements J. Med. Chem. 2000, 43, 1892-1897.
67. Gregory, T. F.; Wright, J. L.; Wise, L. D.; Meltzer, L. T.; Serpa, K. A.; Konkoy, C. S.; Whittemore, E. R.; Woodward, R. M. Parallel Synthesis of a Series of Subtype-Selective NMDA Receptor Antagonists Bioorg. Med. Chem. Letters 2000, 10, 527-529.
68. Wright, J. L.; Kesten, S. R.; Upasani, R. B.; Lan, N. C. 4-Benzylpiperidinylalkylsulfinyl- substituted heterocycles and their use as subtype-selective NMDA receptor antagonists. PCT Int. Appl. WO 2000/000197-A1, Jan 6, 2000; Chem. Abstr. 2000, 132, 64255.
69. Wright, J. L.; Gregory, T. F.; Bigge, C. F.; Boxer, P. A.; Serpa, K.; Meltzer, L. T.; Wise, L. D.; Cai, S. X.; Hawkinson, J. E.; Konkoy, C. S.; Whittemore, E. R.; Woodward, R. M.; Zhou, Z. L. Subtype-Selective N-Methyl-D-aspartate Receptor Antagonists: Synthesis and Biological Evaluation of 1-(Aralkynyl)-4-benzylpiperidines. J. Med. Chem. 1999, 42, 2469-2477.
70. Wright, J. L.; Gregory, T. F.; Kesten, S. R.; Boxer, P. A.; Serpa, K. A.; Meltzer, L. T.; Wise, L. D. Subtype-Selective N-Methyl-D-aspartate Receptor Antagonists: Synthesis and Biological Evaluation of 1-(Heteroarylalkynyl)-4-benzylpiperidines. J. Med. Chem. 2000, 43, 3408-3419.
71. Kornberg, B. E.; Nikam, S. S.; Wright, J. L.; Kesten, S. R.; Meltzer, L. T.; Coughenour, L.; Barr, B.; Serpa, K. A.; McCormick, J. Subtype selective NMDA receptor antagonists: evaluation of some novel alkynyl analogues. Bioorg. Med. Chem. Letters 2004, 14, 1213- 1216.
72. Tamiz, A. P.; Whittemore, E. R.; Schelkun, R. M.; Yuen, P.-W.; Woodward, R. M.; Cai, S.-X.; Weber, E.; Keana, J. F. W. N-(2-(4-Hydroxyphenyl)ethyl)-4-chlorocinnamide: a Novel Antagonist at the 1A/2B NMDA Receptor Subtype. Bioorg. Med. Chem. Letters
1998, 8, 199-200. 73. Tamiz, A. P.; Cai, S. X.; Zhou, Z.-L.; Yuen, P.-W.; Schelkun, R. M.; Whittemore, E. R.;
Weber, E.; Woodward, R. M.; Keana, J. F. W. Structure-Activity Relationship of N- (Phenyl-alkyl)cinnamides as Novel NR2B Subtype-Selective NMDA Receptor Antagonists. J. Med. Chem. 1999, 42, 3412-3420.
74. McCauley, J. A.; Theberge, C. R.; Romano, J. J.; Billings, S. B.; Anderson, K. D.; Claremon, D. A.; Freidinger, R. M.; Bednar, R. A.; Mosser, S. D.; Gaul, S. L.; Connolly, T. M.; Condra, C. L.; Xia, M. Cunningham, M. E.; Bednar, B.; Stump, G. L.; Lynch, J. J.; Macaulay, A.; Wafford, K. A.; Koblan, K. S.; Liverton, N. J. NR2B-Selective N-Methyl- D-aspartate Antagonists: Synthesis and Evaluation of 5-Substituted Benzimidazoles. J.
Med. Chem. 2004, 47, 2089-2096. 75. McCauley, J. A.; Theberge, C. R.; Liverton, N. J.; Claremon, D. A.; Claiborne, C. F. 2-
Benzyl and 2-Heteroaryl Benzimidazole NMDA/NR2B Antagonists. U.S. Patent 6,316,474 B1, November 13, 2001.
76. Liverton, N. J.; Butcher, J. W.; McIntyre C. J.; Claiborne, C. F.; Claremon, D. A.; McCauley, C. J.; Romano, J. J.; Thompson, W.; Munson, P.M. N-substituted nonaryl- heterocyclo amidyl NMDA/NR2B antagonists. PCT Int. Appl. WO 2002080928-A1, Apr 3, 2001. 77. Nagy, J.; Boros, A.; Dezsı, P.; Kolok, S.; Fodor, L. Inducible expression and
pharmacology of recombinant NMDA receptors, composed of rat NR1a/NR2B subunits. Neurochem. Int. 2003, 43, 19-29.
78. Curtis, N. R.; Diggle, H. J.; Kulagowski, J. J.; London, C.; Grimwood, S.; Hutson, P. H.;

123
Murray, F.; Richards, P.; Macaulay, A.; Wafford, K. A. Bioorg. Med. Chem. Letters
2003, 13, 693-696. 79. Curtis, N. R.; Diggle, H. J.; Kulagowski, J. J.; London, C.; Grimwood, S.; Hutson, P. H.;
Murray, F.; Richards, P.; MaCaulay, A.; Wafford, K. A. Bioorg. Med. Chem. Letters
2003, 13, 697-700. 80. US6265426 (2001) 81. WO03097637 (2003) 82. US6683097 (2004) 83. Alanine, A.; Bourson, A.; Buttelmann, B.; Gill, R.; Heitz, M.-P.; Mutel, V.; Pinard, E.;
Trube, G.; Wyler, R. 1-Benzyloxy-4,5-dihydro-1H-imidazol-2-yl-amines, a novel class of NR1/2B subtype selective NMDA receptor antagonists. Bioorg. Med. Chem. Letters. 2003, 13, 3155-3159.
84. Pinard, E.; Alanine, A.; Bourson, A.; Buttelmann, B.; Heitz, M.-P.; Mutel, R.G.; Trube G.; Wyler R. 4-Aminoquinolines as a novel class of NR1/2B subtype selective NMDA receptor antagonists. Bioorg. Med. Chem. Letters. 2002, 12, 2615-2619. 85. US6440995 (2002) 86. Büttelmann, B.; Alanine, A.; Bourson, A.; Gill, R.; Heitz, M. P.; Mutel, V.; Pinard, E.;
Trube, G.; Wyler, R. 4-(3,4-Dihydro-1H-isoquinolin-2yl)-pyridines and 4-(3,4-Dihydro- 1H-isoquinolin-2-yl)-quinolines as potent NR1/2B subtype selective NMDA receptor antagonists. Bioorg. Med. Chem. Letters. 2003, 13, 1759-1762.
87. Büttelmann, B.; Alanine, A.; Bourson, A.; Gill, R.; Heitz, M. P.; Mutel, V.; Pinard, E.; Trube, G.; Wyler, R. 2-(3,4-Dihydro-1H-isoquinolin-2yl)-pyridines as a novel class of NR1/2B subtype selective NMDA receptor antagonists. Bioorg. Med. Chem. Letters. 2003, 13, 829-832.
88. US6831087 (2004) 89. Nguyen, K. T.; Claiborne, C. F.; McCaulay, J. A.; Libby, B. E.; Claremon, D. A.; Bednar,
R. A.; Mosser, S. D.; Gaul, S. L.; Connolly, T. M.; Condra, C. L.; Bednar, B.; Stump, G. L.; Lynch, J. J.; Koblan, K. S.; Liverton, N. J. Cyclic benzamidines as orally efficacious NR2B-selective NMDA receptor antagonists. Bioorg. Med. Chem. Letters 2007, 17, 3997-4000.
90. US6440976 (2002) 91. Gozlan, H.; Emerit, M. B.; Hall, M. D.; Nielsen M.; Hamon, M. In situ molecular sizes of the various types of 5-HT binding sites in the rat brain Biochem. Pharmacol. 1986, 35, 1891–1897. 92. Li, J. H.; Wang, Y. H.; Wolfe, B. B.; Krueger, K. E.; Corsi, L.; Stocca, G.; Vicini, S. Developmental changes in localization of NMDA receptor subunits in primary cultures of cortical neurons. Eur. J. Neurosci. 1998, 10, 1704-1715. 93. Dickenson, A. and Besson J.-M. (Editors): Chapter 1, pp. 6-7: Animal models of Analgesia; and Chapter 8, pp. 180-183: Mechanism of Central Hypersensitivity: Excitatory Amino Acid Mechanisms and Their Control – In Pharmacology of Pain. Springer-Verlag (Berlin) 1997.]. 94. Hunskaar, S.; Fasmer, O. B.; Hole, K. Formalin Test in Mice, a Useful Technique for Evaluating Mild. Journal of Neuroscience Methods, 1985, 14, 69-76. 95. Seltzer, Z.; Dubner, R.; Shym, Y. A novel behavioral model of neurophaticpain
disorders produced in rats by partial sciatic nerve injury. Pain 1990, 43, 205-218. 96. a, Wermuth, C. G. In The Practice of Medicinal Chemistry, Wermuth, C. G., Ed.; Academic Press : London, San Diego, New York, Boston, Sydney, Tokyo, Toronto, 1996; p 228. b, Friedman, H. L. (1951) Influence of isosteric replacements upon biological activity. In Symposium on Chemical-Biological Correlation. National Research Council Publication,

124
Washington D.C. 97. Nicolaou, K. C.; Ramanjulu, Joshi M.; Natarajan, Swaminathan; Braese, Stefan; Li, Hui; et al. A Suzuki coupling-macrolactamization approach to the AB-COD bicyclic system of vancomycin. Chem. Commun. 1997, 19, 1899-1900. 98. (a) Kondo, K.; Morohoshi, S.; Mitsuhashi, M.; Murakami, Y. Chem. Pharm. Bull. 1999, 47, 1227. (b) Boger, D. L.; Cerbone, L. R.; Yohannes, D. Oxidative Coupling of Methyl 6-Hydroxyindole-2-carboxylate with Primary Amines: Preparation of 2-Substituted Methyl Pyrrolo[2,3-e]benzoxazole-5-carboxylates. J.Org. Chem. 1988, 53, 5163. 99. Doyle, F. P.; Ferrier, W.; Holland, D. O.; Mehta, M. D.; Nayler, J. H. C. Potential Antituberculosis Agents of the Indole Series. J. Chem. Soc. 1956, 78, 2853-2856. 100. Parke Davis; NL 6409825 1965; Chem. Abstr. 1965, 63, 11517d. 101. Lu, T.; Soll, R. M.; Illig, C. R.; Bone, R.; Murphy, L.; Spurlino, J.; Salemme, R. R.; Tomczuk, B. E. Structure-Activity and Crystallogrphic Analysis of a New Class of Non- amide-Based Thrombin Inhibitor. Bioorg. Med. Chem. Lett. 2000, 10, 79-82. 102. Xue, C. B.; Roderick, J.; Jackson, S.; Rafalski, M.; Rockwell, A.; Mousa, S.; Olson,
R.E.; DeGrado, W. F. Design, Synthesis, and In vitro Activities of Benzamide-Core Glycoprotein IIb/IIIa Antagonists: 2,3-Diaminopropionic Acid Derivatives as Surrogates of Aspartic Acid. Bioorg. Med. Chem. 1997, 5, 693-705. 103. (a) Ágai, B.; Nádor, A.; Proszenyák, Á.; Tárkányi, G.; Faigl, F. A facile synthesis of 3- (substituted benzyl)piperidines. Tetrahedron 2003, 59, 7897-7900. (b) Ágai, B.; Proszenyák, Á.; Tárkányi, G.; Vida, L.; Faigl, F. Convenient, benign and scalable synthesis of 2- and 4-substituted benzylpiperidines. Eur. J. Org. Chem. 2004, 3623-3632. (c) Proszenyák, Á.; Ágai, B.; Hegedős, L.; Faigl, F. Hydrogenation of a 4- benzylpiperidine derivative over supported precious metal catalysts. Applied Catalysis A 2004, 269, 249-253. 104. Crowther, A. F.; Curd, F. H. S.; Davey, D. G.; Stacey, G. J. Synthetic Antimalarials. Dialkylaminoalkylaminoquinoxalines. J. Chem. Soc. 1949, 1260. 105. Borza, I.; Bartáné Szalai, G.; Domány, Gy.; Horváth, Cs.; Farkas, S.; Nagy, J.; Kolok, S. Amide derivatives as NMDA receptor antagonists. WO 2002/34718A1. 106. Borza, I.; Kolok, S.; Gere, A.; Ágai-Csongor, É.; Ágai, B.; Tárkányi, G.; Horváth, Cs.; Barta-Szalai, G.; Bozó, É.; Kiss, Cs.; Bielik, A.; Nagy, J.; Farkas, S.; Domány, Gy. Indole-2-carboxamides as Novel NR2B Selective NMDA Receptor Antagonists Bioorg.
Med. Chem. Letters 2003, 13, 3859-3861. 107. Borza, I.; Kolok, S.; Gere, A.; Nagy, J.; Fodor, L.; Galgóczy, K.; Fetter, J.; Bertha, F.; Ágai, B.; Horváth, Cs.; Farkas, S.; Domány, Gy. Benzimidazole-2-carboxamides as Novel NR2B Selective NMDA Receptor Antagonists.Bioorg. Med. Chem. Lett. 2006, 16, 4638-4640. 108. Borza, I.; Bozó, É.; Barta-Szalai, G.; Kiss, Cs.; Tárkányi , Á.; Demeter Á.; Gáti T.; Háda, V.; Kolok, S.; Gere, A.; Fodor, L.; Nagy, J.; Galgóczy, K.; Magdó, I.; Ágai, B.; Fetter, J.; Bertha, F.; M. Keserő, Gy.; Horváth, Cs.; Farkas, S.; Greiner, I.; Domány, Gy. Selective NR1/2B N-Methyl-D-aspartate Receptor Antagonists among Indole-2- carboxamides and Benzimidazole-2-carboxamides. J. Med. Chem. 2007, 50, 901-914. 109. Gilligan, P. J.; Cain, G. A.; Christos, T. E.; Cook, L.; Drummond, S .; Johnson, A. L.; Kergay, A. A.; McElroy, J. F.; Rohrbach, K. W.; Schmidt, W. K.; Tam, S . W. Novel Piperidine s receptor ligands as potential antipsychotic drugs. J. Med. Chem., v.35, p.4344-4361, 19921. 110. Domány, Gy.; Horváth, Cs.; Farkas, S.; Barta-Szalai, G.; Nagy, J.; Kolok, S.; Bozó, É.; Borza, I.; Vágó, I.; Bielik,A.; Ignácz-Szendrei Gy.; Keserő, Gy. M. Piperidine derivatives as NMDA receptor antagonists. WO 2003010159A1. 111. Barta-Szalai, G.; Borza, I.; Bozó, É.; Kiss, Cs.; Ágai, B.; Proszenyák, Á.; Keserő, Gy.

125
M.; Gere, A.; Kolok, S.; Galgóczy, K.; Horváth, Cs.; Farkas, S.; Domány, Gy. Oxamides As novel NR2B selective NMDA receptor antagonists. Bioorg. Med. Chem. Letters
2004, 14, 3953-3956. 112. Jaen, J. C.; Laborde, E.; Bucsh, R. A.; Caprathe, B. W.; Sorenson, R. J.; Fergus, J.; Spiegel, K.; Marks, J.; Dickerson, M. R.; Davis, R. E. J. Med. Chem. 1995, 38, 4439- 4445. 113. Borza, I.; Horváth, Cs.; Farkas, S.; Nagy, J.; Kolok, S. Kynurenic acid amide derivatives as NR2B receptor antagonists. WO 2006010967A1. 114. Borza, I.; Kolok, S.; Horváth, Cs.; Farkas, S.; Domány, Gy. New kynurenic acid amides as novel NR2B selective NMDA receptor antagonists. Bioorg. Med. Chem. Letters 2007 17, 406-409. 115. Holland G.F. (1979) Heterocyclylcarbonyl derivatives of urea, agents for dissolution of gallstones. US 4,163 784 116. Deng, M. Z.; Caubere, P.; Senet, J. P.; Lecolier, S. Tetrahedron, 1988, 44, 6079 – 6086. 117. Borza, I.; Bartáné Szalai, G.; Bozó, É.; Kis, Cs.; Horváth, Cs.; Farkas, S.; Nagy, J.; Kolok, S. New benzoyl urea derivatives. WO 2006010966A1. 118. Borza, I.; Greiner, I.; Kolok, S.; Galgóczy, K.; Ignácz-Szendrei, Gy.; Horváth, Cs.; Farkas, S.; Gáti, T.; Háda, V.; Domány, Gy. New benzoyl urea derivatives as novel NR2B selective NMDA receptor antagonists. Die Pharmazie 2006 61(9), 799-800. 119. Curtis, N. R.; Diggle, H. J.; Kulagowski, J. J.; London, C.; Grimwood, S.; Hutson, P. H.; Murray, F.; Richards, P.; Macaulay, A.; Wafford, K. A. Bioorg. Med. Chem. Letters
2003, 13, 693-696. 120. Borza, I.; Horváth, Cs.; Farkas, S.; Nagy, J.; Kolok, S. Indole-2-carboxamidine derivatives as NMDA receptor antagonists. WO 2006010965A1. 121. Borza, I.; Kolok, S.; Ignácz-Szendrei, G.; Greiner, I.; Tárkányi, G.; Galgóczy K.; Horváth, Cs.; Farkas, S.; Domány, Gy. Indole-2-carboxamidines as novel NR2B selective NMDA receptor antagonists. Bioorg. Med. Chem. Letters 2005, 15, 5439-5441. 122. Chaperon, F.; Müller, W.; Auberson, Y. P.; Tricklebank, M. D.; Neijt, H. C. N-methyl- D-aspartate receptor antagonists: preferential involvement of the NR2B rather than NR2A subunit. Behavioural Pharmacology 2003, 14, 477-487. 123. Boyce, S.; Wyatt, A.; Webb, J. K.; O'Donnell, R.; Mason, G.; Rigby, M.; Sirinathsinghji, D.; Hill, R.G.; Rupniak, N. M. J. Selective NMDA NR2B antagonists induce antinociception without motor dysfunction: correlation with restricted localisation of NR2B subunit in dorsal horn. Neuropharmacology 1999, 38, 611-623. 124. Johnson, K.; Shah, A.; Jaw-Tsai, S.; Baxter, J.; Prakash, C. Metabolism, Pharmacokinetics, and Excretion of a Highly Selective N-Methyl-d-aspartate Receptor Antagonist, Traxoprodil, in Human Cytochrome P450 2D6 Extensive and Poor Metabolizers. Drug metabolism and disposition 2003, 31, 76-87. 125. Borza, I.; Horváth, Cs.; Farkas, S.; Gyertyán, I.; Nagy, J.; Kolok, S.; Galgóczy, K.; Sághy, K. New heterocyclic acid amide derivatives.WO 2006010969A1. 126. Borza, I.; Horváth, Cs.; Farkas, S.; Gyertyán, I.; Nagy, J.; Kolok, S.; Galgóczy, K.;
Sághy, K. New 4-benzylidene-piperidine derivatives.WO 2006010964A1

126
KÖSZÖNETNYILVÁNÍTÁS
Köszönettel tartozom valamennyi közremőködınek, akikkel együtt fáradhatatlanul
dolgoztunk, segítettük egymást közös célunk elérése érdekében. Külön köszönöm Domány
Györgynek a biztatást, iránymutatást és segítséget a kutatómunkában, a publikációk, valamint
a doktori dolgozat elkészítésében.
Hálás vagyok Bozó Évának, Bartáné Szalai Gizellának és Ignáczné Szendrei Györgyinek a
baráti biztatásért, szakmai tanácsaikért, pótolhatatlan segítségükért.
Köszönöm a családomnak a folyamatos, lelkes támogatást.
KÖZREMŐKÖDİK
KUTATÁSVEZETÉS Szombathelyi Zsolt Greiner István Tihanyi Károly Farkas Sándor PROJEKTVEZETİ Horváth Csilla GYÓGYSZERKÉMIA Domány György Ágainé Csongor Éva Bartáné Szalai Gizella Bielik Attila Borza István Bozó Éva Galambos János Greiner István Kiss Csilla Szentirmay Éva Vágó István Wágner Gábor Wéber Csaba BME Ágai Béla Bertha Ferenc Fetter József Proszenyák Ágnes MOLEKULATERVEZÉS Keserő György Miklós Kovári Zoltán Kósa János Magdó Ildikó SZERKEZETKUTATÁS Demeter Ádám Hegedős Béla Ignáczné Szendrei Györgyi Mák Mariann Szántay Csaba jr. Tárkányi Gábor KÖNYVTÁR Fazekas Andrea
VEGYÉSZTECHNIKUSOK András Judit Bozsokiné Téglás Ildikó Csomontányi Józsefné Csomós Erzsébet Csontosné Brunner Éva Demeter Lászlóné Huszár Lászlóné Jéney Erzsébet Kállayné Sohonyai Anna Mikó Istvánné Molnár Magdolna Stark Istvánné ANALITIKA Demeter Mária Meszlényi Gábor Meszlényiné Sipos Márta Rill Attila Tischler Ferenc NEUROFARMAKOLÓGIA Horváth Csilla Felmérai Laura Házi Zsolt Galgóczy Kornél Kis-Varga Ágnes Kordás Krisztina TOXIKOLÓGIA Hegyi Lászlóné Horváth Irén Karsai Edit Krajcsovicz Zoltán Selényi György Varga Márta ÁLTALÁNOS FARMAKOLÓGIA Dézsi László Hornok Katalin Komlódi Zsolt Makó Éva Orosz Szabolcs
SEJTBIOLÓGIA Dezsı Péter Kolok Sándor Krizsán Ágnes Nagy József Tomic Marianna VISELKEDÉS FARMAKOLÓGIA Gyertyán István Egyed Róbert Kedves Rita Laszy Judit Orosz Szabolcs Sághy Katalin Gál Krisztina IN VITRO METABOLIZMUS Dalmadi Balázs Leibinger János Tihanyi Károly Vastag Mónika Szeberényi Szabolcs MOLEKULÁRIS FARMAKOLÓGIA Gere Anikó Kiss Béla Schmidt Éva Szikra Judit Fazekas Károly FARMAKOKINETIKA Dallos Szvetlana Gémesi Larisza Kapás Margit Nagy Zoltán Trafikánt Gábor Terjéki Etelka ELEKTROFIZIOLÓGIA Fodor László Kocsis Pál Tarnawa István Boros András Kovács Gyula FORMULÁCIÓ Bódis Attila