finding supertrees using distance methods by stephen j. willson mathematics department iowa state...
Post on 21-Dec-2015
215 views
TRANSCRIPT

Finding Supertrees Using Distance Methods
by Stephen J. Willson
Mathematics Department
Iowa State University

Trees are such that each vertex has degree 1 (leaf) or 3.
Example. A tree T with leaf set S = {1,2,3,4,5,6}.
1
2
3 4
5
6

1 pygmy chimp
2 common chimp
3 human
4 gorilla
5 orangutan
8 grey seal
9 harbor seal
11 horse
10 cow
12 whale
13 rabbit
14 guinea pig
6 mouse
7 rat
16 hedgehog
15 opossum
100
100
100
100
100
10082
81
77
58
100
100
61

Sequences of length 3857 letters for 16 taxa. Complete protein-coding region for mitochondrial DNA.
pygmychimp TPMTNLLLLI VPVLIAMAFL MLTERKILGYcommonchim TPMTNLLLLI VPILIAMAFL MLTERKILGYhuman MPMANLLLLI VPILIAMAFL MLTERKILGYgorilla MSMANLLLLI VPILIAMAFL MLTERKILGYorangutan MPVINLLLLT MSILIAMAFL MLTERKILGYmouse ---INILTLL VPILIAMAFL TLVERKILGYrat VYFINILTLL IPILIAMGLL TLVERKILGYgreyseal MFMINIISLI IPILLAVAFL TLVERKVLGYharborseal MFMINIISLI IPILLAVAFL TLVERKVLGYcow MFMINILMLI IPILLAVAFL TLVERKVLGYhorse MFMINVLLLI VPILLAVAFL TLVERKVLGYwhale MFMINILTLI LPILLAVAFL TLVERKILGY rabbit MFLINTLLLI LPVLLAMAFL TLVERKILGYguineapig MFMINLLLLI IPILLAMAFL TLTERKILGY opossum MFLINLLMYI IPILLAVAFL TLVERKVLGYhedgehog MLMINILCLI IPILLAVAFL TLIERKVLGY

If T is a tree with leaf set S and S' is a subset of S, then T | S' consists of T from which all leaves not in S' have been successively removed (together with their edges) and vertices of degree 2 have been deleted. T | S' is the subtree with leaf set S'.
1
2
3 4
5
6
1
2
4
5
T | {1,2,4,5}T

Suppose that tree Ti has leaves Si for i = 1,...,k. A supertree for T1, T2, …, Tk is a tree T with leaf set S = S1 S2 ... Sk such that T | Si = Ti for all i.

1
2
4
5
1 4
36
3
5
6
2
Example: (Steel, Dress, and Böcker 2000)
Initial trees

1
2
3 4
5
6
2
3
4 5
1
6
There are exactly two supertrees:

Supertree methods
• Researcher 1 studies taxa S1 and uses evidence to find a tree T1 for S1.
• Researcher 2 studies taxa S2 and finds a tree T2 for S2.
• ...
• Researcher k studies taxa Sk and finds a tree Tk for Sk.
• Now a supertree T is found for T1, ..., Tk.

Advantages
• Independent studies can be combined into a single tree.
• Initial trees can be based on different kinds of data: DNA of different genes, morphology, behavior, DNA-DNA hybridization, .…
• Initial trees can be obtained by different methodologies: maximum parsimony, maximum likelihood, neighbor-joining;

More advantages
• Initial trees often have been selected from competing trees by professional judgment.
• There are most likely no common data for all species in S.
• Methods such as maximum likelihood would not be computationally tractable on such a large dataset as all S.

MRP
• Currently the most commonly used method is Matrix representation with parsimony (MRP)
• Baum 1992, Ragan 1992
• Sanderson et al 1998
• software RADCON (Thorley and Page 2000)

MRP
• Form a large matrix M as follows: The rows correspond to the taxa in S. For each nontrivial (nonleaf) edge e of each tree Ti
there is a column with each entry 0, 1, or ? Remove e from Ti; the remaining taxa in Si split into two sets A and B. The column has 0 in row for a taxon t if t is in A, 1 if t is in B, ? if t is not in Si.

1
2
4
53
T1
e1 e21
3
4
6
e3
T2
M: e1 e2 e3
1 1 1 12 1 1 ?3 0 1 14 0 0 05 0 0 ?6 ? ? 0

Give the matrix M to a standard software package (such as PAUP* by Swofford) and find the maximum parsimony trees. Select some consensus tree.

Advantages of MRP
• Easy to do for all trees
• No alignment difficulties
• Software exists (RADCON)

Disadvantages of MRP
• Maximum parsimony is NP-hard with lots of heuristics.
• Maximum parsimony often has many different optimal trees.
• There is no conceptual basis for why maximum parsimony is the right criterion.

Kennedy and Page, 2002, started with 7 published trees of Procellariiform seabirds:
• 1. Austin 1996: 307 bases of cytochrome b, • 2. Bretagnolle et al. 1998: 496 bases of cytochrome b• 3. Heidrich et al. 1998: 1043 bases of cytochrome b• 4. Imber 1985: morphology and life history information• 5. Nunn and Stanley 1998: 1143 bases of cytochrome b
for 90 taxa• 6. Paterson et al. 1995: 12SrRNA, isozyme and
behavioral life history data• 7. Sibley and Ahlquist 1990: DNA-DNA hybridization
data


Kennedy and Page used MRP on these seven trees with a total of 122 taxa. The trees were combined into a single matrix M with a total of 188 characters. Exploratory searches suggested there were 100,000 equally parsimonious trees. These were broken into samples of 10,000 MP trees of 214 steps. They found the strict consensus trees and the Adams consensus trees for each block of 10,000 MP trees. The strict consensus trees were identical, but there was some deviation in the Adams consensus trees. These were reported and compared with an independent tree from mitochondrial DNA for all the taxa.

Additive distance functions
• A distance function d on the set S of leaves is a nonnegative function d: S S R such that d(x,y) = d(y,x) and d(x,x) = 0.
• It is additive for the tree T if d(i,j) is the sum of the edge lengths on the path from i to j.

1
2
3
4
t
u
v
x
w
Ex: t+v+w = d(1,3)

Theorem. (rough statement) An additive distance function on S for T determines the tree T uniquely.
Theorem. (exact statement) Suppose d is an additive distance function on S for T whose branchlength function w is positive for each edge.
(1) The branchlength function w is determined by d.(2) If the tree W, W≠T, has the same set S of leaves,
then d is not additive for W.

Idea of (1): In the example, t = (d(1,2) + d(1,3) - d(2,3))/2u = (d(2,1)+d(2,3)-d(1,3))/2Remove 1 and 2 and their edges so 5 becomes a
leaf.d(5,3) = d(1,3)-t = d(2,3)-ud(5,4) = d(1,4)-t = d(2,4)-uUse induction. This proves (1).
1
2
3
4
t
u
v
x
w
5 6
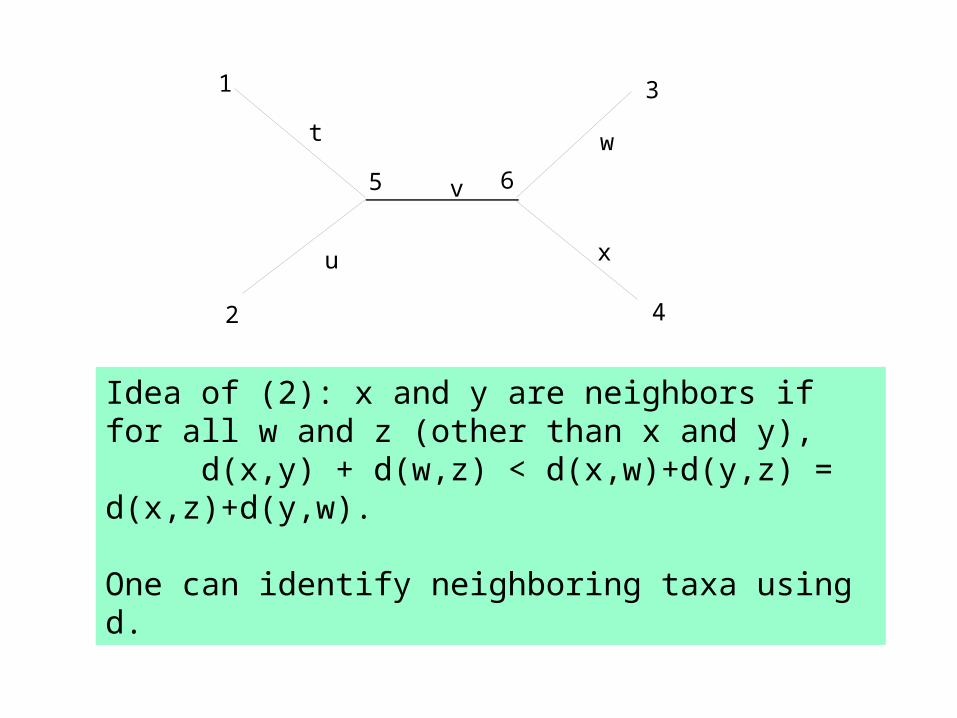
Idea of (2): x and y are neighbors if for all w and z (other than x and y),
d(x,y) + d(w,z) < d(x,w)+d(y,z) = d(x,z)+d(y,w).
One can identify neighboring taxa using d.
1
2
3
4
t
u
v
x
w
5 6

In fact, if d is an additive distance function on S for T, then there exists a fast algorithm to construct T.

Supertrees with distances
• Given: trees Ti with leaf sets Si, i = 1, ..., k. Assume given an additive distance function di on Si for Ti.
• Problem: Find a supertree T with additive distance function d such that for all i, d | Si Si = di.

Theorem. Suppose that d is additive on S for T. Let Si, i = 1,..., k be subsets of S and let Ti = T | Si, di = d | Si Si. Suppose that for all pairs (x,y) from S there exists i so both x and y are in Si. Then T is uniquely determined and can be reconstructed easily.

Quick estimate
• Suppose S has 200 taxa.
• Suppose each given tree Ti with leaf set Si has 50 taxa.
• What is the minimum possible number k of given trees such that one might know d(x,y) for all pairs x and y?

Ballpark solution
• Let C(a,b) be the binomial coefficient C(a,b) = a!/(b! (a-b)!).
• For S there are C(200,2) distances given.
• Each Ti provides C(50,2) distances.
• If they were all distinct we would need C(200,2)/C(50,2) trees.
• Hence k ≥ C(200,2)/C(50,2) = (200) (199)/ [(50) (49)] = 16.245. So k≥17.

The quartet trees
1
3 4
2 3
2 4
1 4
2 3
((1 2)(3 4)) ((1 3)(2 4)) ((1 4)(2 3))
1
For 4 taxa there are only 3 different topologies.

What if we use only topology?
• For purely topological methods (like MRP) what it the minimum number k of subtrees that must be given to tell all the quartet trees?

Quick estimate• Suppose S has 200 taxa.
• Suppose each given tree Ti with leaf set Si has 50 taxa.
• Then T has C(200,4) quartet trees.
• Each Ti provides C(50,4) quartet trees.
• If they were all distinct, we would need C(200,4)/C(50,4) trees.
• Hence k ≥ C(200,4)/C(50,4) = 280.87
• Hence k ≥ 281.

Main idea
• There is so much redundancy that one can reconstruct the branchlengths knowing d(x,y) for only some pairs (x,y). There is so much redundancy that one can reconstruct the topology knowing the quartet trees for only some quartets.
• We expect to need many fewer subtrees if we use distances rather than topology.

Supertrees using distances
• Part A. (Projection) For each given tree Ti on leaf set Si, find a good additive distance function di on Si
• Part B. (Supertree construction) Use the distance functions di to build a supertree.

Background
Typically, for a tree Ti on leaf set Si there is DNA data or protein data which provides some evidence. Then the tree Ti is selected using various criteria with some judgment and with some consensus techniques.

1. Select some good distance estimations di' from the DNA data for Si:
Jukes-CantorKimuraHKY 1985log determinant
2. "Project" di' onto Ti to find an additive distance function di on Si for Ti.
A. Projection

e
W
X
Y
Z
Example: Estimate the branchlength e.

e
W
X
Y
Z
If w is in W, x in X, y in Y, z in Z obtain an estimate for the length e by
estwxyz = ((d(w,y) + d(x,z) - d(w,x) - d(y,z) )+ (d(w,z)+d(x,y) - d(w,x) - d(y,z)) )/4

Select the average of these values for all choices of w, x, y, z.

Difficulties: Projection
• The procedure may lead to negative branchlengths.
• One can crudely zero out negative branchlengths, but is there a better tractable method?
• Least squares have the same difficulties.
• Are distances from different datasets comparable?

Jukes Cantor distances
0
0.5
1
1.5
2
2.5
0 0.5 1 1.5 2 2.5
Length 3000
Length 1000
Comparison of length estimates on simulated data.

Jukes Cantor distances
00.020.040.060.080.10.120.140.160.180.2
0 0.020.040.060.080.10.120.140.160.18Cytochrome b
12S rRNA
Comparison of distances: real data, 12SrRNA vs. Cytochrome b.

B. Supertree construction
• Given various di(x,y), accept d'(x,y) as the average of the values di(x,y) which are known. For some x,y, d'(x,y) is not defined.

Suppose we have constructed T' on leaf set S' = {1,2,3,..., m-1}. We try to add taxon m.
Maybe there are taxa a and b from S' such that(i) d'(a,m) is known(ii) d'(b,m) is known(iii) On the path P from a to b , the location x at distance A from a
A := (d'(a,m)+d'(a,b)-d'(m,b))/2is not a vertex of T'.
Usual situation

We expect
a
b
m
A
B
M
The lengths A, B, and M are estimated.

a
bx
A
a
bx
A
mM

Theorem. Suppose that the distance function d is additive for the tree T such that each edge has positive branchlength. Suppose d(x,y) is input for each pair of taxa x and y. Then the procedure reconstructs T with the same distance function.
(This is true even if T is not completely resolved, for an extension of the procedure.)

Difficulties if there are errors
• Different choices of a and b may lead to slightly different attachments.
• The additive distances d’ in the new tree involving m may need to be modified.
• What is the tolerance for accepting that "x is not at a vertex"?
• There may be no such pair a and b.
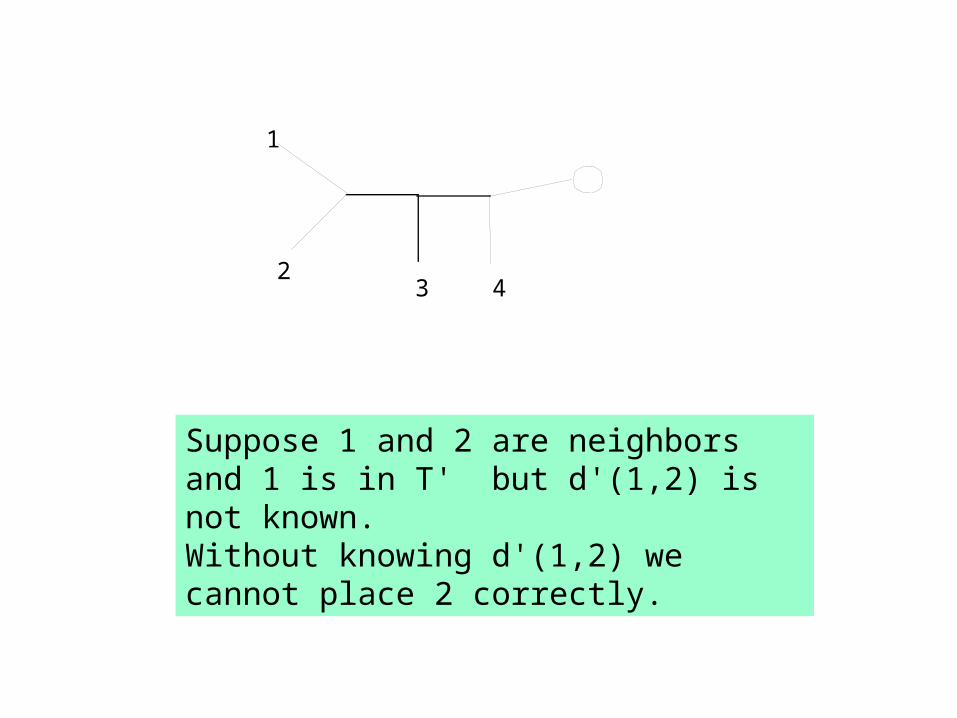
1
23 4
Suppose 1 and 2 are neighbors and 1 is in T' but d'(1,2) is not known. Without knowing d'(1,2) we cannot place 2 correctly.

1
23 4
Suppose 1 and 2 are neighbors and 1 is in T' but d'(1,2) is not known. If d'(2,3) and d'(2,4) are known, we infer the correct attachment edge for 2 but not how far along it to break the edge.

1
23 4
Worse, suppose neither d'(2,1) nor d'(2,3) is known but d'(2,4) and d'(2,5) are known, where 5 is in the circle. Then there are three edges where 2 might be attached.

In such situations, there is an option to pick an edge at random from among the plausible edges and to attach the new vertex at the midpoint of that edge.

Simulations
• Build a model tree T with n taxa and random branchlengths.
• Select k different subtrees with s taxa selected randomly.
• Try to reconstruct T. Let T^ be the reconstruction.

Measuring topological accuracy
• v = the number of quartets {w,x,y,z} in T^ whose quartet tree in T^ differs from the quartet tree in T
• i = the number of quartets {w,x,y,z} in T^ whose quartet tree in T^ is the same as the quartet tree in T
• Q = v/(v+i) = quartet fit error

Q = 0 means every computed quartet topology was right
Q = 1 means every computed quartet topology was wrong

Measuring metric accuracy
• C = # (x,y) : x and y are distinct taxa in T^
• E = √ (∑ (d(x,y)-d^(x,y))2 / C)

k Q StDevQ E StDevE
4 0.158 0.109 13.24 8.138 0.00266 0.0053 1.01 0.70012 0.0000567 0.00025 0.1175 0.19116 0.0000000 0.00000 0.0000 0.000
Simulations with 32 taxa, 20 subtrees, each with k taxa. Results of 50 replicates.

f Q StDevQ E StDevE
.125 0.158 0.109 13.24 8.13
.25 0.00266 0.0053 1.01 0.700
.375 0.0000567 0.00025 0.1175 0.191
.5 0.0000000 0.00000 0.0000 0.000
Simulations with 32 taxa, 20 subtrees, each with 32f taxa. Results of 50 replicates.

32 taxa, 50 replications with 20 subtrees

Simulation with 96 taxa, 50 replications, 20 subtrees each with k = 32f taxa.
f k Q StDevQ E EStDev
.0625 6 0.325 0.174 31.0 14.4
.125 12 0.0612 0.083 11.34 8.85
.25 24 .00020 0.00022 0.688 0.313
.375 36 0.0000061 0.000017 0.0902 0.113
.5 48 .000000 00000 .00118 .00838

Compare some results using MRP only (Piaggio-Talice, Burleigh, Eulenstein to appear).
96 taxa, 20 subtrees with 96f taxa
f Q(MRP) Q(dis).25 0.1109 0.00020041.5 0.0708 0.00000000

Advantages over MRP
• MRP adds all taxa. Distance method adds only taxa for which there is evidence of a particular placement.
• Fast implementation of distance algorithm.
• The heuristics are plausible.
• Should require many fewer subtrees.

Disadvantages over MRP
• Requires distance estimates for all trees. And distances should be comparable.
• MRP adds all taxa. Distance method adds only taxa for which there is evidence of a particular placement.

Thanks for your attention.