final progress report synthesis of novel phenoxazine derivatives...
TRANSCRIPT

Final Progress Report
Synthesis of novel phenoxazine derivatives as potent
anti-hyperglycaemic and hypo-lipidemic agents

SYNOPSIS
1
Synthesis of novel phenoxazine derivatives as potent anti-hyperglyceimic
and hypolipidemic agents
Recent advances in high throughput screening have thrown a challenge to synthetic organic/
medicinal chemists for rapid synthesis of structurally diverse and complex small molecules.
The growing demand for pharmacological probes and New Chemical Entities (NCE) for drug
discovery campaign has fuelled this interest while new approaches such as Diversity
Oriented Synthesis have improved access to small molecule libraries.
The present investigation targets two diseases i.e. diabetes and inflammation. A recent survey
reveals that India will have maximum number of diabetics in any one country (approximately
57.2 millions) and Type-2 diabetes constitutes more than 95% of diabetic patients in our
country. Since their introduction some 40 years ago, sulphonamides and sulfonylureas have
been main stays of Type-2 diabetes management and phenoxazine heterocyclic system is
unexplored till today in the search of new anti-diabetic agents. It is therefore planned to
discover new anti-diabetic agents that act as new insulin secretagogues and insulin
sensitizers.
During last decade several investigations have demonstrated that modulation of inflammatory
cells activation by selective PDE-4 inhibitors and established that an elevation of cAMP will
inhibit inflammatory process. It is therefore felt to design a molecular frame work containing
sulfonamido moiety with phenoxazine heterocyclic systems for the use as PDE-4 inhibitors to
treat inflammatory diseases like Chronic Obstructive Pulmonary Diseases (COPD) and
asthma.
The present investigation carried out is divided into five chapters.
Chapter I
Introduction of Phenoxazine.
Chapter II
Section A: Synthesis and characterisation of N-(alkyl/aryl/heteryl)-1-nitro-10H-phenoxazin-
3-sulfonamides.
Section B: Synthesis and characterisation of N-(1-nitro-10H-phenoxazin-3-sulfonyl)-N’-
alkyl/aryl and heteryl ureas.

SYNOPSIS
2
Chapter III
Synthesis and characterisation of N-alkyl/aryl-1-[4-(2,4-thiazolidindione-5-ylmethyl)-
benzene sulfonyl amino]-10H-phenoxazine-3-sulfonamides.
Chapter IV
Synthesis and characterisation of 1-(aryl)- imidazo [4,5,1-k,l]phenoxazine-4-sulfonamides.
Chapter V
Evaluation of the synthesized derivatives for pharmacological activities.
CHAPTER I
A brief introduction about the phenoxazine moiety is presented in this chapter. The structure,
methods of preparation, chemical and spectral properties and various biological activities of
phenoxazine derivatives have been discussed. Finally the objectives of the present
investigation are presented in detail.
CHAPTER II
Sulphonamide substituted heterocyclic compounds exhibits a wide spectrum of
pharmacological activities and hence it may not be surprising to find more than 30 drugs
containing this functionality. Phenoxazine skeleton as such shows a wide range of biological
activities and there is no single report in literature with the pharmacophore containing
sulfonamido moiety para to the nitro of phenoxazine.
Most of the phenoxazines reported till now are water insoluble and this factor is responsible
for hindering the development of phenoxazine derivatives as pharmacological agents. It is
therefore planned to introduce a polar substituent like nitro to enhance solubility for the
molecule and test for hypoglycaemic, anti hyperglycaemic and anti-diabetic character of the
new phenoxazines.

SYNOPSIS
3
This chapter is divided into two sections, section A and section B
Section A
This section describes the synthesis and characterisation of N-(alkyl/aryl/heteryl)-1-nitro-
10H-phenoxazin-3-sulfonamides and the steps involved are
1. Synthesis of potassium salt of 4-chloro-3,5-dinitrobenzenesulfonate (2)
2. Synthesis of potassium salt of 1-nitro-10H-phenoxazine-3-sulfonate (3)
3. Synthesis of 1-nitro-10H-phenoxazine-3-sulfonylchloride (4)
4. Synthesis of N-(alkyl/aryl/heteryl)-1-nitro-10H-phenoxazine-3-sulfonamides (5a-5u)
Potassium salt of 1-nitro-10H-phenoxazine-3-sulfonate (3) has been synthesised from the
condensation of Potassium salt of 4-chloro-3,5-dinitrobenzenesulfonate (2) and o-
aminophenol in ethanol in presence of NaOH. Then the conversion of sulfonic acid group to
sulfonyl chloride was taken up adopting different conditions using several reagents and
solvents. Finally it was accomplished by treating the sulfonic acid with POCl3 (1:7) ratio at
110°C to get the sulfonyl chloride (4) as cherry red solid. The sulfonylchloride was further
condensed with various alkyl/aryl/heteryl amines in chloroform in presence of TEA to form
the N-(alkyl/aryl/heteryl)-1-nitro-10H-phenoxazine-3-sulfonamides (5a-5u). Potassium salt of
4-chloro-3,5-dinitrobenzenesulphonate (1) was prepared from chlorobenzene. The
intermediates and the final compounds have been characterised by IR, 1H-NMR,
13C-NMR
and Mass spectral data.

SYNOPSIS
4
Section B
This section represents the synthesis of N-(1-nitro-10H-phenoxazine-3-sulfonyl)-N’-
alkyl/aryl/heteryl ureas (6a-6p) by two methods. In method A, 1-nitro-10H-phenoxazine-3-
sulfonamide (5a) was treated with alkyl/aryl isocyanates in presence of KOH in
dimethylformamide. In method B, 1-nitro-10H-phenoxazine-3-sulfonylcarbamate (7) was
prepared by the reaction of 1-nitro-10H-phenoxazin-3-sulfonamide (5a) and
ethylchloroformate in presence of potassium carbonate in acetone. Then the carbamate 7 was
treated with various alkyl/aryl/heteryl amines in toluene to get corresponding N-(1-nitro-10H-
phenoxazin-3-sulphonyl)-N’-alkyl/aryl/heterylureas. The intermediate and the final
compounds have been characterised by IR, 1H-NMR,
13C-NMR and Mass spectral data.

SYNOPSIS
5

SYNOPSIS
6
N-(Alkyl/aryl/heteryl)-1-nitro-10H-phenoxazine-3-sulfonamides (5a-u)

SYNOPSIS
7
N-(1-Nitro-10H-phenoxazine-3-sulfonyl)-N’-(aryl/alkyl/heteryl)ureas (6a-p)

SYNOPSIS
8
CHAPTER III
The objective of the project is to address the prevalence of Type-2 diabetes as this is an
asymptomatic disease and the apathy for treatment caused macro vascular complications and
lead to cardiovascular problems. The discovery of peroxisome proliferator activated receptor
(PPAR) and its subtypes lead to discovery of new generation of drugs with thiazolidinedione
moiety. This class of oral insulin sensitising agents will be expected to possess improved
compliance and reduced side effects.
There is only one single report on thiazolidinedione moiety with phenoxazine
skeleton. Therefore it is thought worthwhile to synthesise phenoxazine thiazolidinediones
bearing sulfonamido moiety for the first time and subject them to in vitro screening for anti-
diabetic activity.
This chapter deals with the synthesis of N-alkyl/aryl-1-[4-(2,4-thiazolidindione-5-ylmethyl)-
benzenesulphonylamino]-10H-phenoxazin-3-sulfonamides.
This was achieved in the following steps.
1. Synthesis of N-(alkyl/aryl)-1-amino-10H-phenoxazine-3-sulfonamides (9a-9p)
2. Synthesis of N-alkyl/aryl-1-[4-(2,4-thiazolidindione-5-ylidinemethyl)-benzene
sulfonyl-amino]-10H-phenoxazine-3-sulfonamides (10a-10p)
3. Synthesis of N-alkyl/aryl-1-[4-(2,4-thiazolidindione-5-ylmethyl)-benzenesulfonyl-
amino]-10H-phenoxazine-3-sulfonamides (11a-11p)
The required 5-(4-chlorosulfonylbenzylidine)-2,4-thiazolidinedione (xiii) was
prepared from the reaction of benzylidine-2,4-thiazolidinedione (xii) with
chlorosulfonic acid, which intern prepared from 2,4-thiazolidinedione (xi). N-
(alkyl/aryl)-1-amino-10H-phenoxazine-3-sulfonamides (9a-p) were prepared by the
reduction of N-(alkyl/aryl)-1-nitro-10H-phenoxazine-3-sulfonamides (5a-i, s-u, 8v-y).
5-(4-chlorosulphonylbenzylidine)-2,4-thiazolidinedione and N-(alkyl/aryl)-1-amino-
10H-phenoxazine-3-sulfonamides were condensed in pyridine to yield the
corresponding N-alkyl/aryl-1-[4-(2,4-thiazolidindione-5-ylidinemethyl)-benzene
sulfonylamino]-10H-phenoxazine-3-sulfonamides (10a-i, 10s-y). Finally the
exocyclic double bond of the thiazolidinedione was reduced to get the corresponding
N-alkyl/aryl-1-[4-(2,4-thiazolidindione-5-ylmethyl)-benzenesulfonyl-amino]-10H-
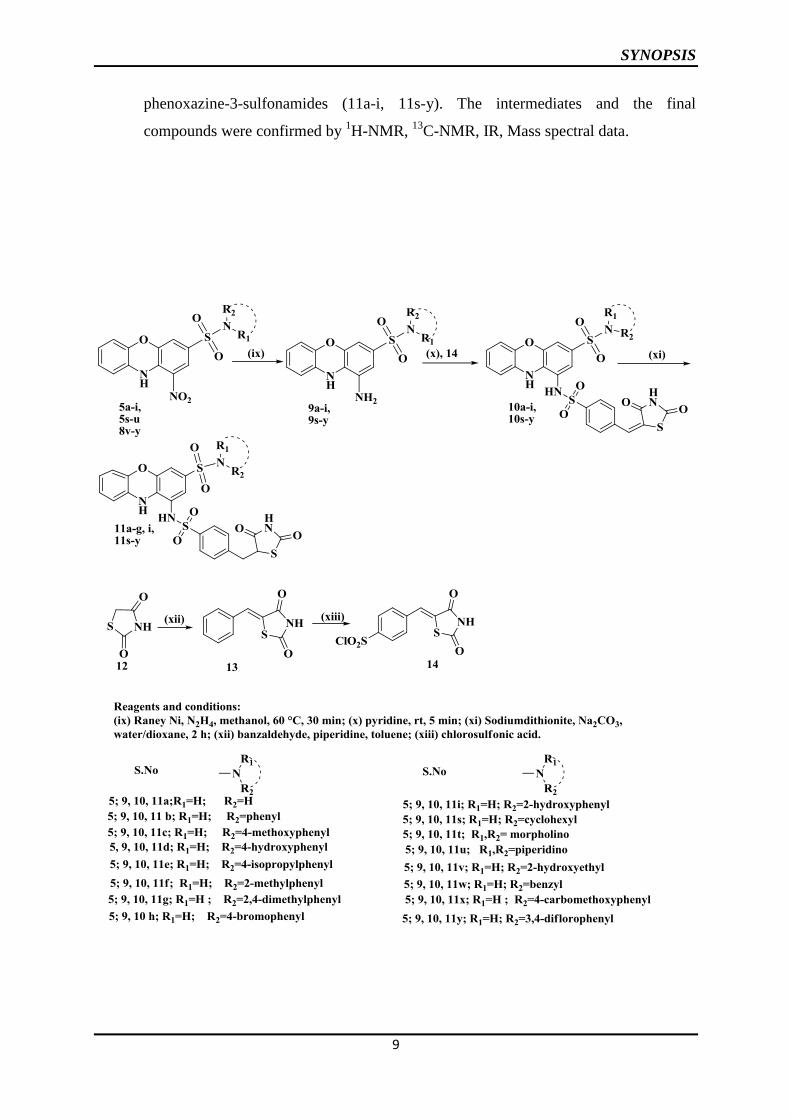
SYNOPSIS
9
phenoxazine-3-sulfonamides (11a-i, 11s-y). The intermediates and the final
compounds were confirmed by 1H-NMR,
13C-NMR, IR, Mass spectral data.

SYNOPSIS
10
N-(Alkyl/aryl)-1-[4-(2,4-thiazolidinedione-5-ylmethyl)-benzenesulfonylamino]-10H-
phenoxazine-3-sulfonamides (11a-g; 11i-y).

SYNOPSIS
11
CHAPTER IV
A recent report that phenoxazine nucleus is associated with potent anti-proliferative activity
and interest in identifying novel PDE-4 inhibiting property prompted us to synthesise a new
heterocyclic system.
Benzo[d]imidazole based carbon skeleton is known to exhibit PDE-4 inhibitory activity. We
therefore designed a heterocyclic skeleton in which a phenoxazine ring is incorporated into
benzo[d]imidazo structure with an anticipation that this new heterocyclic system might
induce anti-inflammatory as well as drug like property within the molecule.
To the best of our knowledge it is for the first time that PDE-4 inhibitor based on this
pharmacophore are attempted in the literature. A sulfonamido group was introduced at C-4 of
the resulting imidazo[4,5,1-k,l]phenoxazine moiety which enhance solubility and
pharmacokinetics of this new chemical entity.
The objective is achieved in the following steps
1. Synthesis of potassium salt of 1-amino-10H-phenoxazine-3-sulfonate (15)
2. Synthesis of 1-(aryl)-imidazo[4,5,1-k,l]phenoxazine-4-sulfonates (16a-e)
3. Synthesis of 1-phenylimidazo[4,5,1-k,l]phenoxazine-4-sulfonylchloride (17)
4. Synthesis of 1-phenylimidazo[4,5,1-k,l]phenoxazine-4-sulfonamide (18a-p)
5. Synthesis of 1-amino-10H-phenoxazine-3-sulfonamide (9a)
The synthesis was carried in two methods. In method 1 potassium salt of 1-nitro-10H-
phenoxazine-3-sulfonate (3) was reduced to get potassium-1-amino-10H-phenoxazine-3-
sulfonate (15) using Raney Ni and hydrazine hydrate in methanol. Compound 15 was treated
with various aryl aldehydes in dimethylformamide to get corresponding potassium salt of 1-
(aryl)-imdazo[4,5,1-k,l]phenoxazine-4-sulfonates (16a-e). Potassium salt of 1-phenyl-
imidazo[4,5,1-k,l]phenoxazine-4-sulfonate (16a) is treated with phosphorous oxychloride to
get 1-phenyl-imidazo[4,5,1-k,l]phenoxazine-4-sulfonylchloride (17). Compound 17 was
treated with aqueous ammonia in tetrahydrofuran to get 1-phenyl-imidazo [4,5,1-
k,l]phenoxazine-4-sulfonamide (18a).

SYNOPSIS
12
In method 2 compound 5a was reduced to get 1-amino-10H-phenoxazine-3-sulfonamide (9a)
using Raney Ni and hydrazine hydrate in methanol. Compound 9a was further treated with
different aromatic aldehydes to get the final compounds 18a-p. The intermediates and final
compounds were purified and characterised by analytical and spectral (Mass, IR, PMR, 13
C-
NMR) data.
Scheme 4
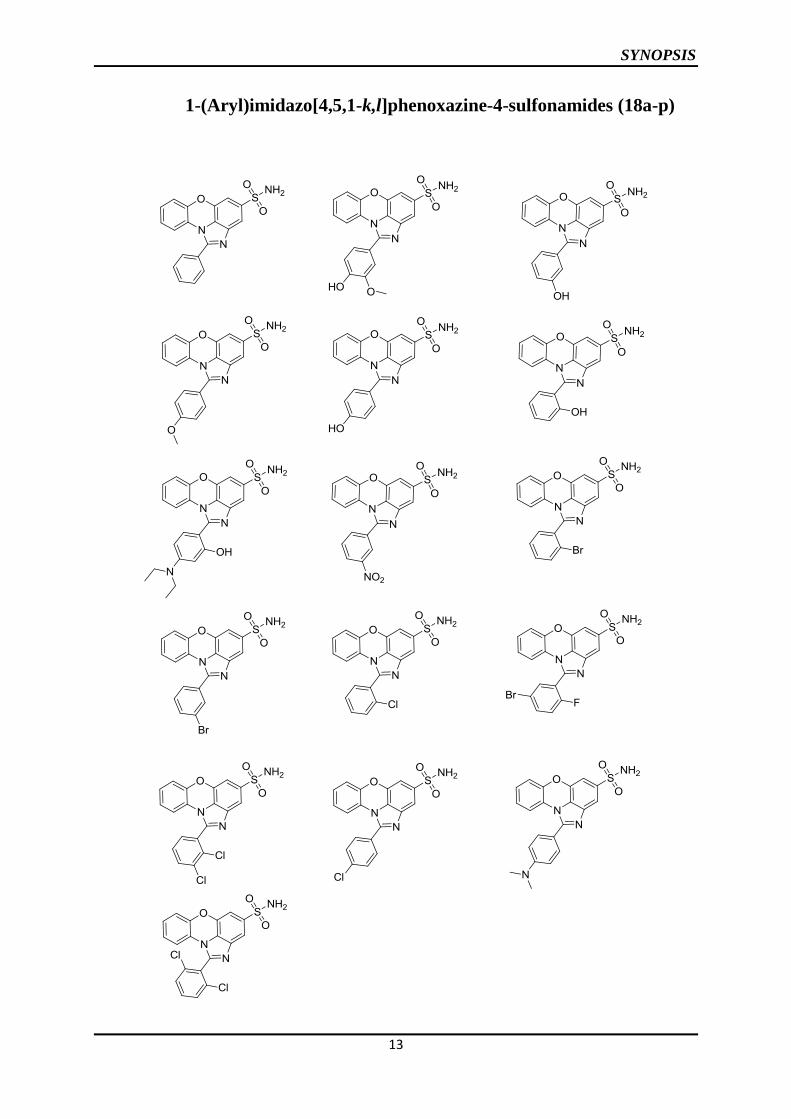
SYNOPSIS
13
1-(Aryl)imidazo[4,5,1-k,l]phenoxazine-4-sulfonamides (18a-p)

SYNOPSIS
14
CHAPTER V
Chapters II, III and IV contain molecules with phenoxazine skeleton and pharmacophores
with specific purpose. These well designed small molecules were screened for different
pharmacological activities based on the pharmacophore. These results are presented in 4
sections.
The procurement of animals, materials and methods adopted in vivo screening are presented
in this chapter. The results obtained are presented in tabular form depicting various
parameters and an attempt is made to correlate the structure with activity. The compounds
used in the screening of high purity and were characterised without leaving a doubt about the
structure of the compound.
This Chapter is divided into 4 sections, section A, section B, section C and Section D.
Section A and B
This section describes the pharmacological activities of N-(alkyl/aryl/heteryl)-1-nitro-10H-
phenoxazine-3-sulfonamides (5a-u) and N-(1-nitro-10H-phenoxazine-3-sulfonyl)-N’-
alkyl/aryl/ heterylureas (6a-p). All the synthesised compounds were evaluated for
hypoglycaemic, intraperitoneal glucose tolerance test and anti-diabetic activities. In addition
serum glutamic oxaloacetic transaminase and serum glutamate pyruvate transaminase
enzyme levels, serum triglycerides, cholesterol levels, the changes in body weights, and
insulin levels were tested. Finally histology of pancreas in normal and diabetic rats were
examined with the test compounds using 10 mg per kg body weight of the rats and the results
were compared with standard glibenclamide.
Section C
N-alkyl/aryl-1-[4-(2,4-thiazolidindione-5-ylmethyl)-benzenesulphonylamino]10H-
phenoxazine-3-sulphonamides (11a-i, s-y) were screened for in silico human PPARγ activity.
Section D
This section deals with the anti inflammatory activity of 1-(aryl)-imidazo[4,5,1-
k,l]phenoxazine-4-sulfonamides (18a-p). All the synthesised compounds were evaluated for
PDE-4B enzyme inhibitory activity by in vitro and in silico. IC50 values were also
determined and the results were compared with standard drug Rolipram.

Construction of phenoxazine rings containing nitroand sulfonic acid groups leading to phenoxazine-3-sulfonamide derivatives: their evaluation as noveland potential insulin secretagogues†
Seelam Venkata Reddy,a Gangula Mohan Rao,a Baru Vijaya Kumar,*a
Koppela Naresh Reddy,b Konda Sravya,b Puchchakayala Goverdhan,b
Vandana Rathore,c Girdhar Singh Deorad and Manojit Pal*c
A series of N-(alkyl/aryl/heteroaryl)-1-nitro-10H-phenoxazine-3-sulfonamides was designed, synthesized
and evaluated for its hypoglycemic, hyperglycemic and oral anti-diabetic activities. These compounds
were prepared via the construction of a phenoxazine ring containing nitro and sulfonic acid groups in a
single step followed by further transformations. One of these compounds exhibited promising anti-
diabetic activities comparable to glibenclamide and increased serum insulin levels indicating its potential
as a novel insulin secretagogue.
Type 2 Diabetes Mellitus (T2-DM) is effectively controlled by anapproach which is polypharmaceutical1 in nature, targeting theeffects of insulin sensitivity and related dyslipidemia andtherefore cardiovascular diseases. However, this approach isnot encouraged due to its potential additional risks.2 It istherefore necessary to search for newer agents to circumvent therisks involved in combination therapy. Accordingly, an attemptwas made to design a unique pan agonist3a of PPARs (peroxi-some proliferator-activated receptors) that has a bulkier hetero-cyclic scaffold with a sulfonamide side chain possessing thenecessary geometry and electrostatics which are complemen-tary to PPARs. Notably, PPAR a, g and d have been the targets ofintense preclinical research to treat dyslipidemia.3b The inves-tigation revealed that these three receptors are closely relatedand the design of a molecule which competently activates themis an intellectual challenge.4 Indeglitazar (A, Fig 1), a sulfon-amide based PPAR pan-active anti-diabetic agent, has beendiscovered using a process that couples low-affinity biochemicalscreening with high-throughput co-crystallography.5 The
phenoxazine moiety on the other hand has been explored in thediscovery and development of a dual PPAR agonist, DRF 2725,that has shown potent antihyperglycemic and lipid modulatingproperties.6 This, and our continued interest in novel anti-dia-betic agents,7–10 prompted us to design a series of new sulfon-amides, represented by B (Fig. 1), bearing phenoxazinemoieties. The key structural features of A were partly main-tained in B with the hope that the resulting analogues wouldshow in vivo pharmacological properties similar to A. Moreover,some of the structural features of glibenclamide (C, Fig. 1), awell known anti-diabetic drug, were also incorporated in B.Introduction of the R group into the sulfonamide moiety of Ballowed the generation of a diverse library of small moleculesfor pharmacological studies. Thus a series of sulfonamides,bearing bulky phenoxazine moieties, related to B were preparedand evaluated for hypoglycemic, hyperglycemic and anti-dia-betic activities, body weight changes, serum lipid proles, SGOT(serum glutamic oxaloacetic transaminase) and SGPT (serumglutamic pyruvic transaminase) levels and the histology of thepancreas. To the best of our knowledge evaluation of this classof molecules as potential anti-diabetic agents has not beenpreviously reported. Herein we report our preliminary results ofthis study.
Several syntheses11–15 of substituted phenoxazines have beenattempted but the incorporation of two substituents, such asnitro and sulfonic acid groups, in a single step followed bymodication of these substituents remains unexplored. Arecent report16 shows the synthesis of phenoxazine derivativesstarting from 2-aminophenol and substituted diuorobenzenefollowed by the introduction of a sulfonic acid group at the C-3position of the phenoxazine nucleus D (Fig. 2).
aMedicinal Chemistry Laboratory, Research Centre, C.K.M. Arts and Science College,
Warangal 506 006, Andhra Pradesh, India. E-mail: [email protected];
Tel: +91 8008098787bDiabetes and Aging Research Division, Department of pharmacology, Vaagdevi
College of Pharmacy, Kakatiya University, Warangal 506 006, Andhra Pradesh, IndiacDr. Reddy's Institute of Life Sciences, University of Hyderabad Campus, Gachibowli,
Hyderabad 500 046, India. E-mail: [email protected]; Tel: +91 40 6657
1500dThe University of Queensland, School of Pharmacy, Brisbane, Qld 4072, Australia
† Electronic supplementary information (ESI) available: Experimental procedures,spectral data for all new compounds, results of pharmacological studies. See DOI:10.1039/c3md00377a
Cite this:Med. Chem. Commun., 2014,5, 587
Received 9th December 2013Accepted 23rd January 2014
DOI: 10.1039/c3md00377a
www.rsc.org/medchemcomm
This journal is © The Royal Society of Chemistry 2014 Med. Chem. Commun., 2014, 5, 587–592 | 587
MedChemComm
CONCISE ARTICLE
Publ
ishe
d on
24
Janu
ary
2014
. Dow
nloa
ded
by N
atio
nal D
ong
Hw
a U
nive
rsity
Lib
rary
on
14/1
1/20
14 0
5:18
:10.
View Article OnlineView Journal | View Issue

While the synthesis of 1-nitro-10H-phenoxazine-3-sulfonicacid E (Fig. 2) or 2 (Scheme 1) was reported17 by Ullmann et al. in1909, the conversion of the sulfonic acid group to sulfonamideremained a challenge.18,19 However, the use of phosphorusoxychloride in the absence of any solvent afforded the expectedsulfonyl chloride 5 (Scheme 1).20 Thus, the key starting material,i.e. 4-chloro-3,5-dinitrobenzenesulfonic acid (4), was synthe-sized from chlorobenzene (3) according to a reported procedure(Scheme 1).21 Compound 4 was then condensed with 2-amino-phenol (1) to give 1-nitro-10H-phenoxazine-3-sulfonic acid (2)which was converted to the corresponding sulfonyl chloride (5).
Compound 5was then condensed with ammonia20 or variousalkyl/aryl and heteroaryl amines to afford 6a–u.22
All compounds were characterized by IR, NMR and MSspectral data. Compounds 6a (ref. 20) and 6b–u generallyshowed a singlet near d 9.7 (NH group) and two doublets near d7.9 and 7.0 ppm (with J¼ 2.0 Hz) due to the C-2 and C-4 protonsof phenoxazine, respectively, in their 1H-NMR spectra. Theremaining protons of phenoxazine (H8, H7, H6 and H9)appeared as multiplets in the d 7.2–7.1 and 6.8–6.7 ppm regions.
The hypoglycemic activities of all synthesized compoundswere initially evaluated at a dose of 20 mg kg�1. Glibenclamideis oen used as a standard anti-diabetic drug in streptozotocin-induced moderate diabetes to compare the efficacy of a varietyof hypoglycemic agents.23 The effect of the test compounds i.e.6a–u on fasting blood sugar levels was assessed in normal ratsat various time intervals as shown in Table 1. Some of thecompounds caused signicant maximum reductions in bloodglucose levels in normal rats when tested at 10 mg kg�1 alongwith 10 mg kg�1 of glibenclamide aer 2 h of treatment e.g. 6a(�35%), 6m (�54%), 6o (�47%), 6p (�24%), 6r (�27%), 6s(�25%) 6t (�28%) and 6u (�32%). The compounds thatproduced encouraging reductions in blood glucose levels aer 6h are 6a (�50%), 6b (�40%), 6d (�25%), 6m (�43%), 6n(�44%), 6o (�12%), 6r (�21%) and 6u (�30%). Some of these
promising and representative compounds e.g. 6a, 6b, 6d, 6g, 6l,6m, 6o and 6s were taken for further screening e.g. intraperi-toneal glucose tolerance test (IPGTT).
When the selected compounds were administered to glucoseloaded normal rats which had fasted for 18 h, hypoglycemiceffects were observed aer 30 min as shown in Table 2. In thecases of compounds 6g, 6l and 6m, the decline in blood sugarlevels reached a maximum at 90 min. Nevertheless, compounds6m and 6g were evaluated further for their anti diabetic effects.A sub acute study was also performed on 6m. The differenceobserved between the initial and nal fasting plasma glucoselevels of different groups under investigation revealed a signif-icant elevation in blood glucose in the diabetic control groupcompared with normal animals at 0 days as shown in Table 3and at the end of the 14-day experimental period as shown inTable 4. These results indicated the promising effects ofcompound 6m in maintaining the blood glucose levels instreptozotocin–nicotinamide induced diabetic rats. Adminis-tration of 6m to diabetic rats showed a signicant decrease inthe blood glucose levels. A marked increase in the totalcholesterol and triglyceride levels has been observed inuntreated diabetic rats. Under normal circumstances insulinactivates the enzyme lipoprotein lipase and hydrolyses triglyc-erides. An insulin deciency results in failure to activate the
Fig. 1 The design of new molecules, B, based on the known PPARpan-active anti-diabetic agent indeglitazar A and another anti-diabeticdrug glibenclamide C.
Fig. 2 Phenoxazine derivatives D and E.
Scheme 1 Reagents and conditions: (a) oleum, conc. H2SO4, KNO3,130 �C, 2 h; (b) 2-aminophenol (1), NaOH, ethanol, reflux, 4 h; (c)POCl3, reflux, 3 h; (d) aqueous NH3, THF, 0 �C, 30 min; (e) aryl/alkyl/heteroaryl amines, chloroform, Et3N, 60 �C, 30 min.
588 | Med. Chem. Commun., 2014, 5, 587–592 This journal is © The Royal Society of Chemistry 2014
MedChemComm Concise Article
Publ
ishe
d on
24
Janu
ary
2014
. Dow
nloa
ded
by N
atio
nal D
ong
Hw
a U
nive
rsity
Lib
rary
on
14/1
1/20
14 0
5:18
:10.
View Article Online

enzymes, thereby causing hypertriglyceridemia. The signicantlowering of total cholesterol (Table 5) and triglyceride (Table 6)levels observed with compound 6m is a desirable biochemicalstate for the prevention of atherosclerosis and ischemic condi-tions.24 The observed hypolipidemic effect may be caused bydecreased cholesterogenesis and fatty acid synthesis.
It is known that diabetes raises the serum activity levels ofthe liver enzymes AST (aspartate aminotransferase) and ALT
(alanine transaminase). Elevated activities of serum amino-transferases are a common sign of liver and cardiovasculardiseases and are observed more frequently among people withdiabetes than in the general population. Such alterations totransaminase activity in the tissues are explicable causes ofenergy metabolism, as these enzymes play a role in gluconeo-genesis.25 Compound 6m has been found to reverse theincreased SGPT and SGOT activities towards near normality,
Table 1 Hypoglycemic effects of the test compounds 6a
Test groupDose (mg kg�1 ofbody weight (b. wt))
Blood glucose (mg dl�1)
Pre treatmentPost treatment
0 h 2 h 4 h 6 h
Control 0.5% gum acacia 102.23 � 4.03 101.03 � 1.93 97.13 � 2.89 101.01 � 1.60Glibenclamide 10 98.13 � 7.23 90.2 � 4.50* 63.73 � 11.50** 69.01 � 5.93***6a 10 91.14 � 42.84 65.62 � 40.45** 69.79 � 33.20* 51.04 � 19.90***6b 10 89.58 � 24.50 106.25 � 16.88 91.66 � 27.36 60.93 � 16.62***6c 10 80.20 � 20.50 86.45 � 25.89 114.06 � 18.19 84.37 � 20.056d 10 115.62 � 4.41 116.66 � 7.03 120.31 � 16.50 74.47 � 17.27**6e 10 110.93 � 7.05 102.60 � 9.14 100 � 7.90 118.22 � 3.076f 10 113.54 � 8.30 102.60 � 5.38 102.08 � 4.70 85.93 � 9.006g 10 106.25 � 17.56 95.31 � 9.42* 85.42 � 14.87** 89.58 � 9.19**6h 10 129.17 � 13.35 122.92 � 20.02 113.54 � 17.64 118.75 � 9.686i 10 96.43 � 10.55 93.55 � 7.92 89.88 � 12.94 85.42 � 4.106j 10 99.58 � 10.84 90.22 � 6.78 85.63 � 6.94 90.20 � 7.566k 10 103.79 � 6.04 95.45 � 4.97 87.88 � 4.69 93.94 � 4.696l 10 105.30 � 3.42 96.21 � 3.42* 86.36 � 2.87* 100.76 � 4.466m 10 71.76 � 1.44 46.34 � 4.87*** 44.65 � 3.61*** 66.14 � 4.50**6n 10 72.23 � 6.03 59.35 � 4.50 61.56 � 9.29 65.27 � 3.406o 10 82.11 � 7.07 52.43 � 2.58** 73.33 � 12.81 87.93 � 6.606p 10 102.22 � 5.06 75.48 � 6.68 88.70 � 7.90 98.70 � 4.476q 10 96.38 � 7.95 78.26 � 5.42 77.74 � 11.08 94.28 � 9.756r 10 80.27 � 16.08 72.67 � 20.95 69.35 � 6.74 78.41 � 6.306s 10 95 � 8.48 74.28 � 10.49 86.45 � 9.58 91.74 � 11.466t 10 89.72 � 12.63 71.38 � 8.81 85.0 � 8.18 88.88 � 13.996u 10 73.61 � 6.21 67.5 � 7.12 63.05 � 8.59 69.44 � 4.91
a Values are mean � SD (standard deviation), n ¼ 6 in each group, *p < 0.05, **p < 0.01 and ***p < 0.001 when compared with the vehicle treatedgroup (Dunnett's test).
Table 2 Intraperitoneal glucose tolerance test on selected compoundsa
Test compound Dose (mg kg�1 b. wt)
Blood glucose (mg dl�1)
Before glucoseadministration
Aer glucose administration
0 min 30 min 60 min 90 min 120 min
Control 0.5% gum acacia 82.21 � 5.58 109.28 � 20.53 106.07 � 13.98 94.44 � 6.99 101.07 � 8.98Glibenclamide 10 76.75 � 12.02 107.85 � 12.91** 89.0 � 8.07*** 77.5 � 12.66*** 91.78 � 23.33**6a 10 98.50 � 16.87 124.20 � 7.74 112.31 � 16.31 105.72 � 29.27 122.85 � 8.316b 10 75.70 � 18.79 100.36 � 23.30 106.23 � 20.93 93.77 � 17.86 76.90 � 3.356d 10 102.33 � 12.69 130.35 � 29.63 96.07 � 7.82 91.42 � 9.31 107.14 � 27.286g 10 83.57 � 18.44 148.35 � 5.02 111.4 � 17.38** 137.5 � 13.71 119.64 � 20.706l 10 75.94 � 5.52 115.43 � 13.42 81.89 � 17.08 80.0 � 22.74 83.89 � 21.116m 10 91.89 � 14.17 102.43 � 11.52** 95.40 � 14.41*** 78.10 � 25.91*** 98.91 � 25.37**6o 10 74.55 � 15.03 123.21 � 16.10 100.44 � 9.79 97.32 � 20.28 100.0 � 19.616s 10 72.33 � 14.22 124.33 � 31.17 96.66 � 10.20 98.0 � 23.13 106.0 � 23.82
a Values are mean � SD, n ¼ 5 in each group, *p < 0.05, **p < 0.01 and ***p < 0.001 when compared with the vehicle treated group (Dunnett's test).
This journal is © The Royal Society of Chemistry 2014 Med. Chem. Commun., 2014, 5, 587–592 | 589
Concise Article MedChemComm
Publ
ishe
d on
24
Janu
ary
2014
. Dow
nloa
ded
by N
atio
nal D
ong
Hw
a U
nive
rsity
Lib
rary
on
14/1
1/20
14 0
5:18
:10.
View Article Online
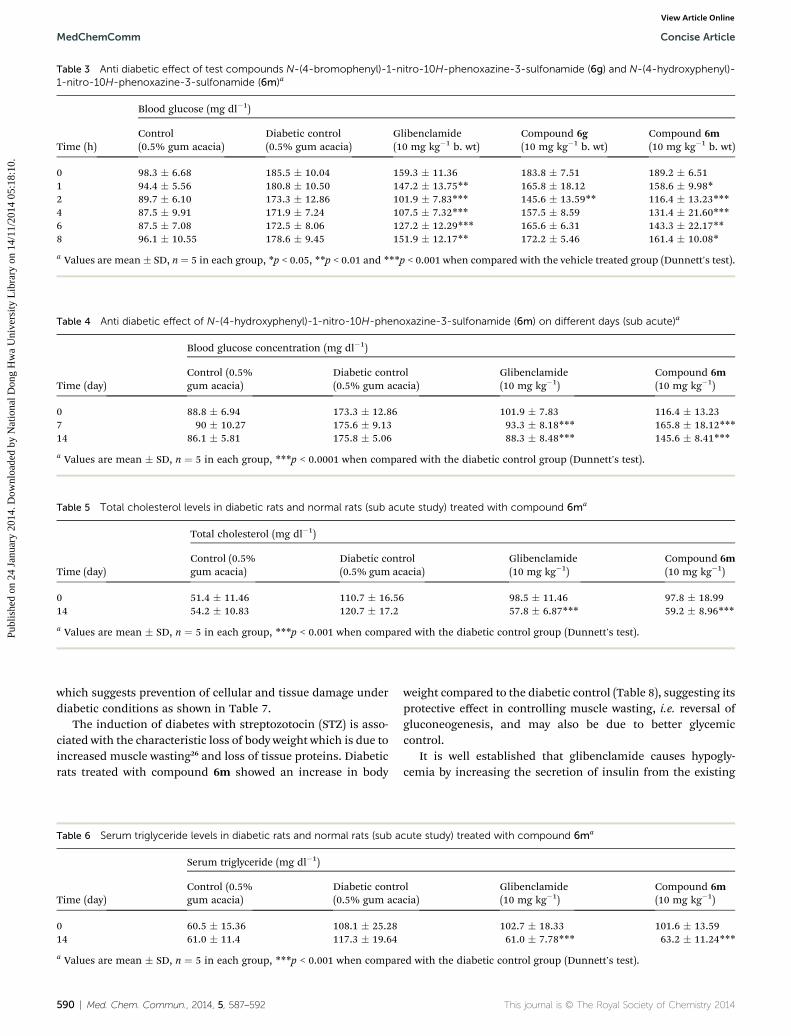
which suggests prevention of cellular and tissue damage underdiabetic conditions as shown in Table 7.
The induction of diabetes with streptozotocin (STZ) is asso-ciated with the characteristic loss of body weight which is due toincreased muscle wasting26 and loss of tissue proteins. Diabeticrats treated with compound 6m showed an increase in body
weight compared to the diabetic control (Table 8), suggesting itsprotective effect in controlling muscle wasting, i.e. reversal ofgluconeogenesis, and may also be due to better glycemiccontrol.
It is well established that glibenclamide causes hypogly-cemia by increasing the secretion of insulin from the existing
Table 3 Anti diabetic effect of test compounds N-(4-bromophenyl)-1-nitro-10H-phenoxazine-3-sulfonamide (6g) and N-(4-hydroxyphenyl)-1-nitro-10H-phenoxazine-3-sulfonamide (6m)a
Time (h)
Blood glucose (mg dl�1)
Control(0.5% gum acacia)
Diabetic control(0.5% gum acacia)
Glibenclamide(10 mg kg�1 b. wt)
Compound 6g(10 mg kg�1 b. wt)
Compound 6m(10 mg kg�1 b. wt)
0 98.3 � 6.68 185.5 � 10.04 159.3 � 11.36 183.8 � 7.51 189.2 � 6.511 94.4 � 5.56 180.8 � 10.50 147.2 � 13.75** 165.8 � 18.12 158.6 � 9.98*2 89.7 � 6.10 173.3 � 12.86 101.9 � 7.83*** 145.6 � 13.59** 116.4 � 13.23***4 87.5 � 9.91 171.9 � 7.24 107.5 � 7.32*** 157.5 � 8.59 131.4 � 21.60***6 87.5 � 7.08 172.5 � 8.06 127.2 � 12.29*** 165.6 � 6.31 143.3 � 22.17**8 96.1 � 10.55 178.6 � 9.45 151.9 � 12.17** 172.2 � 5.46 161.4 � 10.08*
a Values are mean � SD, n ¼ 5 in each group, *p < 0.05, **p < 0.01 and ***p < 0.001 when compared with the vehicle treated group (Dunnett's test).
Table 4 Anti diabetic effect of N-(4-hydroxyphenyl)-1-nitro-10H-phenoxazine-3-sulfonamide (6m) on different days (sub acute)a
Time (day)
Blood glucose concentration (mg dl�1)
Control (0.5%gum acacia)
Diabetic control(0.5% gum acacia)
Glibenclamide(10 mg kg�1)
Compound 6m(10 mg kg�1)
0 88.8 � 6.94 173.3 � 12.86 101.9 � 7.83 116.4 � 13.237 90 � 10.27 175.6 � 9.13 93.3 � 8.18*** 165.8 � 18.12***14 86.1 � 5.81 175.8 � 5.06 88.3 � 8.48*** 145.6 � 8.41***
a Values are mean � SD, n ¼ 5 in each group, ***p < 0.0001 when compared with the diabetic control group (Dunnett's test).
Table 5 Total cholesterol levels in diabetic rats and normal rats (sub acute study) treated with compound 6ma
Time (day)
Total cholesterol (mg dl�1)
Control (0.5%gum acacia)
Diabetic control(0.5% gum acacia)
Glibenclamide(10 mg kg�1)
Compound 6m(10 mg kg�1)
0 51.4 � 11.46 110.7 � 16.56 98.5 � 11.46 97.8 � 18.9914 54.2 � 10.83 120.7 � 17.2 57.8 � 6.87*** 59.2 � 8.96***
a Values are mean � SD, n ¼ 5 in each group, ***p < 0.001 when compared with the diabetic control group (Dunnett's test).
Table 6 Serum triglyceride levels in diabetic rats and normal rats (sub acute study) treated with compound 6ma
Time (day)
Serum triglyceride (mg dl�1)
Control (0.5%gum acacia)
Diabetic control(0.5% gum acacia)
Glibenclamide(10 mg kg�1)
Compound 6m(10 mg kg�1)
0 60.5 � 15.36 108.1 � 25.28 102.7 � 18.33 101.6 � 13.5914 61.0 � 11.4 117.3 � 19.64 61.0 � 7.78*** 63.2 � 11.24***
a Values are mean � SD, n ¼ 5 in each group, ***p < 0.001 when compared with the diabetic control group (Dunnett's test).
590 | Med. Chem. Commun., 2014, 5, 587–592 This journal is © The Royal Society of Chemistry 2014
MedChemComm Concise Article
Publ
ishe
d on
24
Janu
ary
2014
. Dow
nloa
ded
by N
atio
nal D
ong
Hw
a U
nive
rsity
Lib
rary
on
14/1
1/20
14 0
5:18
:10.
View Article Online

pancreatic b-cells.27 The hypoglycemic effect of 6m is generallydependent upon the degree of b-cell destruction. Treatment ofmoderately diabetic rats with 6m resulted in the stimulationof b-cells of the islets of Langerhans. Histopathological studiesof the pancreas revealed that test compound 6m signicantlyimproved the histological architectures of the islets of Langer-hans (Fig. 3). Groups treated with 6m showed greater isletpersistence and a smaller degree of necrotic changes comparedto the untreated STZ diabetic rats.
To rationalize the anti-diabetic activity of this class ofmolecules we performed in vitro transactivation28 of PPARg with6m and compared it with the known PPARg specic activatorrosiglitazone. PPARs (a group of nuclear receptor proteins thatfunction as transcription factors regulating the expression ofgenes) play essential roles in the regulation of cellular differ-entiation and in the development and metabolism (carbohy-drate, lipid and protein) of higher organisms. PPARg is themolecular target of antidiabetic drugs such as TZDs (thiazoli-dinediones). Compound 6m and rosiglitazone showed 15.48 �1.32 and 18.5� 1.17 [values are expressed as mean� SD (n¼ 4)]fold activation of PPARg respectively when tested at 1.0 mM,indicating that 6m exerts its anti-diabetic effects through ago-nising the nuclear receptor. Since PPARg agonists are known tobe cytotoxic to rat primary hepatocytes in a time and dosedependent manner,29 compound 6m along with rosiglitazonewere tested for cytotoxicity to rat primary hepatocytes. Prelimi-nary data, i.e. IC50 values of 343 mM for 6 h and 274 mM for 16 htreatment in the case of compound 6m and 225 mM for 6 h and165 mM for 16 h treatment in the case of rosiglitazone, indicatedthat 6m may have advantages over rosiglitazone.
In conclusion, we report for the rst time thedesign, synthesis and evaluation of a novel series of N-(alkyl/aryl/heteroaryl)-1-nitro-10H-phenoxazine-3-sulfonamides asinsulin secretagogues. These compounds were prepared from
1-nitro-10H-phenoxazine-3-sulfonic acid via 1-nitro-1H-phe-noxazine-3-sulfonyl chloride and the synthetic method adopteddoes not require the use of any expensive reagents or catalystsand can be used to prepare a library of compounds. All thesynthesized compounds were evaluated for their hypoglycemic,hyperglycemic and oral anti-diabetic activities. Normoglycemicand STZ-nicotinamide induced diabetic rats were treated withthe sulfonamides under investigation at concentrations of10 mg kg�1 of body weight and signicant (p < 0.001) reductions
Table 7 SGOT and SGPT levels after 14 days in diabetic rats and normal rats (sub acute study) treated with compound 6ma
Liver transaminase
Serum SGOT, SGPT levels (U dl�1)
Control (0.5%gum acacia)
Diabetic control(0.5% gum acacia)
Glibenclamide(10 mg kg�1)
Compound 6m(10 mg kg�1)
SGOT 41.0 � 4.47 86.0 � 5.47 45.8 � 5.89*** 47.2 � 5.26***SGPT 38.4 � 7.92 87.4 � 10.67 42.0 � 7.51*** 44.8 � 5.84***
a Values are mean � SD, n ¼ 5 in each group, ***p < 0.001 when compared with the diabetic control group (Dunnett's test).
Table 8 Body weights of diabetic rats and normal rats (sub acute study) treated with compound 6ma
Time (day)
Body weight (g)
Control (0.5%gum acacia)
Diabetic control(0.5% gum acacia)
Glibenclamide(10 mg kg�1)
Compound 6m(10 mg kg�1)
0 200 � 25.00 220 � 44.72 212 � 22.95 217 � 28.6214 248 � 17.53** 189 � 36.81 244 � 16.85* 242 � 28.07*
a Values are mean � SD, n ¼ 5 in each group, *p < 0.05, **p < 0.01 when compared with the diabetic control group (Dunnett's test).
Fig. 3 Histology of the pancreas in experimental rats after 14 days oftreatment with 6m (10 mg kg�1). (A) Normal control – presence ofnormal pancreatic islet cells. (B) Diabetic control – degranulated anddilated islet cells. (C) Diabetic + glibenclamide (10 mg kg�1) – granu-lated, absence of dilation and prominent hyperplasticity. (D) Diabetic +6m (10 mg kg�1 b. wt) granulated pancreatic islets, showing prominenthyperplasticity.
This journal is © The Royal Society of Chemistry 2014 Med. Chem. Commun., 2014, 5, 587–592 | 591
Concise Article MedChemComm
Publ
ishe
d on
24
Janu
ary
2014
. Dow
nloa
ded
by N
atio
nal D
ong
Hw
a U
nive
rsity
Lib
rary
on
14/1
1/20
14 0
5:18
:10.
View Article Online

in fasting blood glucose levels were observed. In addition,changes in body weight, serum lipid proles, and SGOT andSGPT levels were assessed for 14 days. Signicant results wereobserved in the estimated parameters when comparing thediabetic control and normal animals. Of the 21 compoundsN-(4-hydroxyphenyl)-1-nitro-10H-phenoxazine-3-sulfonamide(6m) exhibited pronounced anti-diabetic activities comparableto glibenclamide. The histology of the pancreas of testcompound 6m substantiated the cytoprotective action of thedrug. Additionally, the increase in serum insulin levels aertreatment with compound 6m proved that the compound actsas an insulin secretagogue. Overall, our research has identied6m as a promising novel non-TZD agent for the potentialtreatment of diabetes.
BVK thanks UGC, New Delhi, India for the Major ResearchProject (F. no. 35-151/2008). The authors thank the Manage-ment of C.K.M arts and Science College and Vaagdevi College ofPharmacy for providing the necessary facilities.
Notes and references
1 R. P. Austin, Diabetes Spectrum, 2006, 19, 13.2 (a) J. L. Evans, J. L. Lin and I. D. Goldne, Curr. Diabetes Rev.,2005, 1, 299; (b) Some of the adverse effects involved in theuse of common agents e.g. insulin and sulfonylureasinclude hypoglycemia and weight gain. Gastointestinaldisturbances are the common side effects associated withthe use of metformin, acarbose and GLP-1 analogues (inaddition to lactic acidosis for acarbose and nausea,abdominal pain and weight loss for GLP-1 analogues). Theuse of TZDs e.g. pio- and rosiglitazone showed weight gain,edema and anemia. However, several TZDs have shown anincreased risk of cardiovascular events (e.g. rosiglitazone)or bladder cancer (e.g. pioglitazone) or drug-inducedhepatitis (e.g. troglitazone) and are either kept underselling restrictions or have been withdrawn from the market.
3 (a) For a review, see: P. L. Feldman, M. H. Lambert andB. R. Henke, Curr. Top. Med. Chem., 2008, 8, 728; (b)T. M. Wilson, P. J. Brown, D. D. Sternbach andB. R. Henke, J. Med. Chem., 2000, 43, 527.
4 R. W. Grant, N. G. Devita, D. E. Singer and J. B. Meigs,Diabetes Care, 2003, 26, 1408.
5 D. R. Artis, J. J. Lin, C. Zhang, W. Wang, U. Mehra,M. Perreault, D. Erbe, H. I. Krupka, B. P. England,J. Arnold, A. N. Plotnikov, A. Marimuthu, H. Nguyen,S. Will, M. Signaevsky, J. Kral, J. Cantwell, C. Settachatgull,D. S. Yan, D. Fong, A. Oh, S. Shi, P. Womack, B. Powell,G. Habets, B. L. West, K. Y. J. Zhang, M. V. Milburn,G. P. Vlasuk, K. P. Hirth, K. Nolop, G. Bollag, P. N. Ibrahimand J. F. Tobin, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 262.
6 B. B. Lohray, V. B. Lohray, A. C. Bajji, S. Kalchar, R.R. Poondra,S. Padakanti, R. Chakrabarti, R. K. Vikramadithyan, P. Misra,S. Juluri, N. V. Mamidi and R. Rajagopalan, J. Med. Chem.,2001, 44, 2675.
7 (a) S. H. Havale and M. Pal, Bioorg. Med. Chem., 2009, 17,1783; (b) N. Mulakayala, U. Reddy CH, J. Iqbal and M. Pal,Tetrahedron, 2010, 66, 4919.
8 R. Gupta, S. S. Walunj, R. K. Tokala, K. V. L. Parsa, S. K. Singhand M. Pal, Curr. Drug Targets, 2009, 10, 71.
9 M. Pal, Drug Discovery Today, 2009, 14, 784.10 M. Pal, Curr. Med. Chem., 2009, 16, 3858.11 A. Bernthsen, Ber., 1887, 20, 942.12 F. Kehrmann, Liebigs Ann. Chem., 1902, 322, 1.13 F. Kehrmann and A. A. Neil, Ber., 1914, 47, 3102.14 N. M. Cullinane, H. G. Davey and H. J. H. Padeld, J. Chem.
Soc., 1934, 716.15 H. Gilman and L. O. Moore, J. Am. Chem. Soc., 1957, 79,
3485.16 H. Priaz, B. Chamasmani, K. Vogel, K. J. Beohm, B. Aicher,
M. Gerlach, E. G. Geunther, P. Amon, I. Ivanov andK. Meuller, J. Med. Chem., 2011, 54, 4247.
17 F. Ullmann, G. Engi, N. Wosnessensky, E. Kuhn andE. Herre, Justus Liebigs Ann. Chem., 1909, 366, 78.
18 G. Blotny, Tetrahedron Lett., 2003, 44, 1499.19 (a) L. De Luca and G. Giacomelli, J. Org. Chem., 2008, 73,
3967; (b) S. S. Pandit, V. U. Pandit and B. P. Bandgar,J. Sulfur Chem., 2008, 29, 619.
20 S. V. Reddy, G. M. Rao, B. V. Kumar, C. L. T. Meda,G. S. Deora, K. S. Kumar, K. V. L. Parsa and M. Pal, Bioorg.Med. Chem., 2013, 21, 1952.
21 F. D. Gunstone and S. H. Tucker, J. Appl. Chem., 1952, 2, 204.22 M. P. Olmsted, P. N. Craig, J. J. Lafferty, A. M. Pavloff and
C. L. Zirkel, J. Org. Chem., 1961, 26, 1901.23 G. P. Kumar, P. Arulselvan, D. S. Kumar and
S. P. Subramanian, J. Health Sci., 2006, 52, 283.24 A. Shirwaikar,K.RajendranandB.Rakesh, J. Ethnopharmacol.,
2006, 107, 285.25 A. A. H. Fernandes, E. L. B. Novelli, K. Okoshi, M. P. Okoshi,
B. P. D. Muzio, F. Julliano, C. Guimaraes and A. F. Junior,Biomed. Pharmacother., 2010, 64, 214.
26 S. K. Swanston-Flat, C. Day, C. J. Bailey and P. R. Flatt,Diabetologia, 1990, 33, 462.
27 P. Proks, F. Reimann, N. Green, F. Gribble and F. Ashcro,Diabetes, 2002, 51, S368.
28 J. M. Lehman, L. B. Moore, T. A. Oliver-Smith,T. M. Wilkinson and S. A. Kilewer, J. Biol. Chem., 1995,270, 12953.
29 L. Guo, L. Zhang, Y. Sun, L. Muskhelishvili, E. Blann, S. Dial,L. Shi, G. Schroth and Y. P. Dragan, Mol. Diversity, 2006, 10,349.
592 | Med. Chem. Commun., 2014, 5, 587–592 This journal is © The Royal Society of Chemistry 2014
MedChemComm Concise Article
Publ
ishe
d on
24
Janu
ary
2014
. Dow
nloa
ded
by N
atio
nal D
ong
Hw
a U
nive
rsity
Lib
rary
on
14/1
1/20
14 0
5:18
:10.
View Article Online

1
SYNTHESIS, CHARACTERIZATION AND ANTI-DIABETIC ACTIVITY OF NEW N-(1-NITRO-10H-
PHENOXAZINE-3-SULFONYL)-N’-ALKYL/ARL/HETERYLUREAS
Seelam Venkata Reddya, Gangula Mohan Rao
a, Baru Vijaya Kumar
a *, Dawood Shake
b , Koppela
Naresh Reddyb, Amrez
b, Goverdhan Puchchakayala
b.
aMedicinal Chemistry Laboratory, Research Centre, C.K.M. Arts and Science College, Warangal,
506006, Andhra Pradesh, India.
bDiabetes and Aging Research Division, Department of pharmacology, Vaagdevi College of Pharmacy,
Kakatiya University, Warangal 506001,Andhra pradesh, India.
*Corresponding Author:
Tel.: +91 8008098787, E-mail address: [email protected].
Abstract: A series of 16 new N-(1-nitro-10H-phenoxazine-3-sulfonyl)-N’alkyl/aryl/heterylureas were
synthesized from 1-nitro-10H-phenoxazine-3-sulfonamide and all the compounds were characterized by
spectral data and were evaluated for their hypoglycemic, hyperglycemic and anti-diabetic activity.
Normoglycemic and stz-nicotinamide induced diabetic rats were treated with sulfonyl ureas under
investigation at a concentration of 10 mg/kg body weight and significant (p< 0.001) reduction in fasting
blood glucose levels were observed. In addition, changes in body weights, serum lipid profiles, SGOT,
SGPT levels were assessed for 14 days. Significant results were observed in the estimated parameters in
comparison with diabetic control and normal animals. Out of the 16 compounds compound N-
Piperidinocarbonyl-1-nitro-10H-3-phenoxazinesulfonamide (2l) exhibited pronounced anti-diabetic
activity.
Key words: Phenoxazine, Sulfonyl urea, Streptozotocin, Nicotinamide, Diabetes.
INTRODUCTION:
Diabetes mellitus, a pervasive and multifactorial metabolic syndrome, is characterized by imperfection in
insulin secretion and insulin receptor or post receptor events with derangement in carbohydrate, protein
and lipid metabolism and results in chonic hyperglycaemia, a clinical hallmark of diabetes.1 A chonic
hyperglycaemia itself manifests adverse effects on b-cell insulin secretion and on insulin resistance. This
process leads to long-term damage, dysfunction, and failure of various organs, especially the eyes,
kidneys, nerves, heart, and blood vessels, and creates a huge economic burden related to the management
of diabetic complications.2
There are two major forms of diabetes. Type 1 or Insulin dependent diabetes mellitus is an autoimmune
genetic disease resulting from an absolute deficiency of insulin due to destruction of pancreatic β- cells.
Type 2 or Non insulin dependent diabetes mellitus is a multifactorial disease which is characterized by
insulin resistance associated not only with hyperinsulinaemia and hyperglycaemia but also with
atherosclerosis, hypertension and abnormal lipid profile, collectively called syndrome-x.3

2
The dramatic increase in the prevalence of diabetes can be attributed to several factors. Globally, diabetes
has shadowed the spread of modern lifestyle and can be linked to an increasingly overweight and
sedentary population.4
The International Diabetes Federation (IDF), 2011 has just released some startling new figures on the
escalating diabetes epidemic. Global studies reveal that a 366 million people across the world are dealing
with diabetes. The disease is responsible for 4.6 million deaths a year and related health care costs have
reached $465 billion in US dollars.5
Type-2 DM is more prevalent and account for about 90% to 95% of all diagnosed cases of diabetes. With
an increasing incidence worldwide, DM will be a leading cause of morbidity and mortality in the near
future.6 So there is growing need of effective therapies to achieve optimal glycaemic control in
management of diabetes. Number of orally administrated anti-hyperglycaemic agents has increased
significantly in last decade. However current therapies to reduce plasma glucose level have inherent
problems including compliance, ineffectiveness and occurrences of hypoglycaemic episode. Therefore
there is a agents that will both normalize glucose and insulin level.7
Sulfonylureas are the most widely used oral hypoglycemic agents. These agents act on pancreatic β-cells
stimulating insulin secretion.8 The sulfonylureas are blood glucose lowering agents, although all of these
are not necessarily influenceive in every patient. The side-influences in most persons have proven
negligible and usually disappear when the dose is lowered or discontinued. No real toxicity has been
shown after several decades of therapeutic use in millions of people.9 Based on these observations we for
the first time reporting sulfonylurea functionality on phenoxazine.
CHEMISTRY
The steps involved in the synthesis are shown in scheme 1. The compounds are synthesized using two
methods, 1-Nitro-10H-phenoxazine-3-sulfonamide10
(1) was treated with alkyl/aryl isocyantes in the
presence of potassium hydroxide to give the corresponding sulfonylureas in the method A. In method B,
compound-1 was converted to the 1-nitro-10H-phenoxazine-3-sulfonyl carbamate (3) using ethyl
chloroformate and potassium carbonate, later it was reacted with various amines such as alkyl/aryl/heteryl
amines in toluene to give the corresponding sulfonylureas (2a-2p).
According to the method A for example N-(1-Nitro-10H-phenoxazine-3-sulfonyl)-N’-4-chlorophenylurea
(2b) was prepared by reacting 1-Nitro-10H-phenoxazine-3-sulfonamide (1) with 4-chlorophenyl isocyante
in dimethylformamide in the presence of potassium hydroxide at 60 °C for 3 h. The formation of the
compound was confirmed from its IR, NMR and Mass spectral data. The IR spectrum of the compound
(2b) showed the characteristic absorptions at 3317 (NH), 3238 (CONH), 1697 (C=O), 1533 & 1288
(NO2), 1324 & 1146 (SO2) cm-1
respectively. The 1H-NMR spectrum showed the following signals 11.0
(brs, 1H, SO2NH), 9.72 (s, 1H, NH), 9.07 (s, 1H, CONH), 8.02 (d, 1H, J= 2.0 Hz, H-2), 7.41 (d, 2H, J=
8.8 Hz, Ar H-2 & H-6), 7.28 (d, 2H, J= 8.8 Hz, Ar H-3 & H-5), 7.23 (d, 1H, J=2.0 Hz, H-4), 7.21-7.20
(m, 1H. H-6), 6.86-6.84 (m, 2H, H-8 & H-7), 6.76-6.75 (m, 1H, H-9). And finally the LC mass spectrum
showed the molecular ion peak at m/z 459 (M-H, 100%) further confirming the compound.
In method B for example N-(1-Nitro-10H-phenoxazine-3-sulfonyl)-N’-4-isopropylphenylurea (2f) was
prepared by the reaction of compound 1 with ethylchloroformate in refluxing acetone in the presence of

3
potassium carbonate to give 1-nitro-10H-phenoxazine-3-sulfonyl cabamate (3). When 3 was treated with
4-isopropylaniline and refluxing in toluene for 18 h gave 2f.
NH
O
NO2
S
NH
O
NO2
S
2a-p
O
O
NH2
O
O
HN
O
OEt
NH
O
NO2
S
O
O
HN
O
NR2
(i)
(ii)
iii
Method A
Method B
1
3
R1
2a R1=H; R2=phenyl
2b R1=H; R2=4-chlorophehyl
2c R1=H; R2=3-fluorophenyl
2d R1=H; R2=4-hydroxyphehyl
2e R1=H; R2=benzyl
2f R1=H; R2=4-isopropylphenyl
2g R1=H; R2=3,4-difluorophenyl
2h R1=H; R2=thiophene-2yl-methyl
Reagents and conditions:
(i) R-NCO, KOH, DMF, 70 °C, 3 h;
(ii) ethylchloroformate, K2CO3, acetone, reflux, 12 h;
iii) alkyl/aryl/heteryl amines, toluene, reflux 12 h.
2i R1=H; R2=2-methylphenyl
2j R1=H; R2=4-methoxyphenyl
2k R1=H; R2=N-ethylpyrrolidin-2yl-methyl
2l R1, R2=piperidino
2m NR1,NR2=piperzino
2n NR1,NR2=isopropyl
2o NR1,NR2=morpholino
2p R1,H; R2=4-carbomethoxyphenyl
Scheme 1
The formation of the compounds was established from their IR, NMR and mass spectral data. The IR
spectrum of the compound-3 showed the characteristic absorption bands at 3296 (NH), 3224 (SO2NH),
1748 (C=O), 1538 & 1285 (NO2), 1357 & 1148 (SO2) cm-1
respectively. The 1H-NMR spectrum of the
compound showed the following proton chemicals shifts at δ 12.0 (SO2NH), 9.81 (s, 1H, NH), 7.97 &
7.08 (d, 2H, J=2.0 Hz, H-2 & H-4), 7.25-7.22 (m, 1H, H-8), 6.88-6.86 (m, 2H, H-6 & H-7), 6.77-6.75 (m,
1H, H-9), 4.05 (q, 2H, J=7.2 Hz, N-CH2), 1.15 (t, 3H, J=7.2 Hz, CH3) ppm respectively. The mass
spectrum of the compound showed the molecular ion peak at m/z 378 (M-H, 100%) further confirming
the compound as 1-nitro-10H-phenoxazine-3-sulfonyl cabamate (3). The IR spectrum of the compound
N-(1-Nitro-10H-phenoxazine-3-sulfonyl)-N’-4-isopropylphenylurea (2f) showed the following
characteristic absorptions at 3325 (NH), 3225 (SO2NH), 1694 (C=O), 1533 & 1285 (NO2), 1323 & 1151
(SO2) cm-1
respectively. The 1H-NMR spectrum showed the chemical shifts at 10.8 (brs,1H, SO2NH),
9.78 (s, 1H, NH), 8.90 (s, 1H, CONH), 8.04 (d, 1H, J=2.0 Hz, H-2), 7.27-7.21 (m, 4H, Ar H-2 & H-6;
phenoxazine H-8 & H-4), 7.13 (d, 2H, J=8.8 Hz, Ar H-3 & H-5), 6.87-6.85 (m, 2H, H-6 & H-7), 6.77-

4
6.75 (m, 1H, H-9), 2.81 (septet, 1H, CH), 1.15 (d, 6H, J=6.8 Hz, CH3) ppm resectively. The 13
C-NMR
spectrum showed the chemical shifts at 149.4 (C=O), 144.9 (C4a), 143.4 (Ar C4), 142.5 (C5a), 135.6 (Ar
C1), 135.1 (C1), 130.0 (C9a), 129.1 (C10a), 126.8 (C3), 126.4 (Ar C3 & C5), 124.7 (C8), 124.5 (C4), 120.6
(C6), 119.3 (Ar C2 & C6), 117 (C7), 116.0 (C9), 115.1 (C2), 32.7 (CH), 23.8 ( CH3). Finally the mass
spectrum of the compound showed the molecular ion peak at m/z 467 (M-H, 100%) further confirming
the compound.
EXPERIMENTAL SECTION
All the chemicals and solvents used for the synthesis were of commercial grade and procured from SD
Fine/Merck chemical company. All the reactions were performed under nitrogen atmosphere. Reactions
were monitored by thin layer chromatography (TLC) on silica gel plates (60 F254), visualizing with
ultraviolet light. Column chromatography was performed on silica gel (60-120 mesh) using methanol and
chloroform. 1H NMR and
13C-NMR spectra were determined in DMSO-d6 solution by using Bruker
Biospin, Advance-III 400 MHz Fourier Transform Digital NMR Spectrometer. Proton chemical shifts (δ)
are relative to tetramethylsilane (TMS, δ = 0.00) as internal standard and expressed in ppm and spin
multiplicities are given as s (singlet), brs (broad singlet), d (doublet), t (triplet), q (quartet) and m
(multiplet). Coupling constants (J) were given in hertz. Infrared spectra were recorded on a FT-IR, Bruker
Vertex-70 spectrometer using KBr pellets. Melting points were determined using Polmon digital melting
point apparatus-m96 and are uncorrected. Mass spectra were recorded on Agilent 6300 Ion Trap LC/MS
System.
General procedure for the synthesis of N-(1-Nitro-10H-phenoxazine-3-sulfonyl)-N’-alky/ arylureas
(2a, b; Method A)
To a solution of potassium hydroxide (4.8 mmol ) in dimethylformamide (20 ml) was added 1-nitro-10H-
phenoxazine-3-sulfonamide (3.2 mmol) followed by arylisocyanate (4.8 mmol) at room temperature and
the reaction mixture was stirred at 60 °C for 3 h. The progress of the reaction was monitored by TLC (5%
methanol in chloroform) and after disappearance of the starting material, the reaction was discontinued
and poured in cold dilute 1N HCl solution. The reddish brown solid separated was filtered and washed
with cold water, dried in vacuo and purified by column chromatography using 2% methanol in
chloroform.
N-(1-Nitro-10H-phenoxazine-3-sulfonyl)-N’-phenylurea (2a):
2a was synthesized according to the general procedure (Method A) using 1-nitro-10H-phenoxazine-3-
sulfonamide (1.0 g, 3.2 mmol), phenylisocyanate (0.58 g, 4.8 mmol) and potassium hydroxide (0.27 g,
4.8 mmol) in dimethylformamide (20 ml) to yield 1.0 g (72%), reddish brown solid, mp 259 °C
decomposition; 1H-NMR, 400 MHz (DMSO-d6):
10.89 (brs, 1H), 9.79 (s, 1H), 8.91 (s, 1H), 8.01 (d, 1H,
J=1.6 Hz), 7.32 (d, 2H, J=7.6 Hz), 7.29 (t, 3H, J=7.6 Hz), 7.27 (m, 1H), 7.16 (d, 1H, J=1.6 Hz), 6.88 -
6.85 (m, 2H), 6.73-6.71 (m, 1H).
N-(1-Nitro-10H-phenoxazine-3-sulfonyl)-N’-4-cholorophenylurea (2b):
2b was synthesized according to the general procedure given in method A using 1-nitro-10H-
phenoxazine-3-sulfonamide (1.0 g, 3.2 mmol), 4-chlorophenylisocyanate (0.75 g, 4.8 mmol) and
potassium hydroxide (0.27 g, 4.8 mmol) in dimethylformamide (20 ml) to yield 1.2 g (80%), reddish

5
brown solid, mp 285 °C decomposition; Mass: calculated for C19H13Cl N4O6S 460, found 459 (M-H); IR
(KBr, cm-1
): v 3317, 3238, 1697, 1533, 1354, 1288, 1146; 1H-NMR, 400 MHz (DMSO-d6): 11.0 (brs,
1H), 9.72 (s, 1H), 9.07 (s, 1H), 8.02 (d, 1H, J=2.0 Hz), 7.41 (d, 2H, J=8.8 Hz), 7.28 (d, 2H, J=8.8 Hz),
7.23 (d, 1H, J=2.0 Hz), 7.21-7.20 (m, 1H), 6.86-6.84 (m, 2H), 6.76-6.75 (m, 1H).
Method B
1-Nitro-10H-phenoxazine-3-sulfonylcarbamate (3):
To a solution of 1-nitro-10H-phenoxazine-3-sulfonamide (4.1 g, 13 mmol) in acetone (40 ml) was added
potassium carbonate (2.76 g, 20 mmol) and cooled to 0-5 °C. Then ethyl chloroformate (2.17 g, 20 mmol)
was added drop wise and the resulting mixture was refluxed for 12 h. The progress of the reaction was
monitored by TLC (50% ethyl acetate in hexane) and after completion of the reaction the reaction mass
was cooled to room temperature and the solid was filtered and dissolved in water, acidified using 6N HCl.
The solid separated was filtered and washed with water and dried in vacuo to yield 4 g (80%), reddish
brown solid, mp 225 °C; Mass: calculated for C15H13N3O7S 379, found 378 (M-H); IR (KBr, cm-1
): v
3296, 3224, 1748, 1538, 1357, 1285, 1148; 1H-NMR, 400 MHz (DMSO-d6): 12.0 (brs, 1H), 9.81
(s,1H), 7.97 (d, 1H, J=2.0 Hz), 7.25-7.22 (m, 1H), 7.08 (d, 1H, J=2.0 Hz), 6.88-6.86 (m, 2H), 6.78-6.75
(m, 1H), 4.05 (q, 2H. J=7.2 Hz), 1.15 (t, 3H, J=7.2 Hz).
General procedure for the preparation of N-(1-Nitro-10H-phenoxazine-3-sulfonyl)-N’-
alkyl/arylureas (2c-p; Method B):
To a suspension of 1-nitro-10H-phenoxazine-3-sulfonyl carbamate (2.0 mmol) in toluene (30 ml) was
added alkyl/aryl/heteryl amines (3.0 mmol) and the resulting mixture was refluxed for 12 h. The progress
of the reaction was monitored by TLC (5% methanol in chloroform) and after completion of the reaction,
the reaction mixture was cooled to room temperature; the solid separated out was filtered and purified
from silica gel column chromatography by eluting with 2% methanol in chloroform.
N-(1-Nitro-10H-phenoxazine-3-sulfonyl)-N’-3-fluorophenylurea (2c):
2c was synthesized according to the general procedure, method B using 1-nitro-10H-phenoxazine-3-
sulfonyl carbamate (1.0 g, 2.0 mmol) and 3-fluoroaniline (0.35 g, 3.0 mmol) in toluene to yield 0.8 g
(68%), reddish brown solid, mp 252 °C decomposition; 1H-NMR, 400 MHz (DMSO-d6): 10.78 (brs,
1H), 9.75 (s, 1H), 9.1 (s, 1H), 7.96 (d, 1H, J=2.0 Hz), 7.35-7.17 (m, 4H), 7.16-6.99 (m, 2H), 6.82-6.76
(m, 2H), 6.69-6.65 (m, 1H).
N-(1-Nitro-10H-phenoxazine-3-sulfonyl)-N’-4-hydroxyphenylurea (2d):
2d was synthesized according to method B using 1-nitro-10H-phenoxazine-3-sulfonyl carbamate (1.0 g,
2.60 mmol) and 4-aminophenol (0.34 g, 3.0 mmol) in toluene (40 ml) to yield 0.8 g (68%), brown solid,
mp 298 °C decomposition; 1H-NMR, 400 MHz (DMSO-d6): 11.02 (brs, 1H), 9.80 (s, 1H), 9.78 (s, 1H),
9.02 (s, 1H), 7.91 (d, 1H, J=2.0 Hz), 7.35 (d, 2H, J=8.4 Hz), 7.27-7.20 (m, 1H), 6.89-6.86 (m, 2H), 6.76-
6.72 (m, 1H), 6.65 (d, 2H, J=8.4 Hz).

6
N-(1-Nitro-10H-phenoxazine-3-sulfonyl)-N’-benzylurea (2e):
2e was synthesized according to method B using 1-nitro-10H-phenoxazine-3-sulfonyl carbamate (0.9 g,
2.60 mmol) and benzylamine (0.27 g, 2.60 mmol) in toluene (40 ml) to yield 1.0 g (86%), reddish brown
solid, mp 262 °C decomposition; Mass calculated for C20H16N4O6S 440, found 439 (M-H); IR (KBr, cm-
1): v
3346, 3327, 3109, 1656, 1538, 1354, 1288, 1150;
1H-NMR, 400 MHz (DMSO-d6): 10.80 (brs,
1H), 9.78 (s, 1H), 7.99 (s, 1H), 7.27-7.17 (m, 8H), 6.87-6.74 (m, 3H), 4.18 (s, 2H); 13
C-NMR, 100 MHz
(DMSO-d6): 151.5, 145.0, 142.0, 139.0, 135.0, 130.0, 129.2, 128.1, 127.0, 126.8, 124.7, 124.5, 120.3,
117.0, 115.8, 115.1, 42.7.
N-(1-Nitro-10H-phenoxazine-3-sulfonyl)-N’-4-isopropylphenylurea (2f):
2f was synthesized according to method B using 1-nitro-10H-phenoxazine-3-sulfonyl carbamate (1.0 g,
2.60 mmol) and 4-isopropylaniline (0.47 g, 3.10 mmol) in toluene (40 ml) to yield 0.88 g (71%), reddish
brown solid, mp 269 °C decomposition; Mass: calculated for C22H20N4O6S 468, found 467 (M-H); IR
(KBr, cm-1
): v 3325, 3225, 1649, 1533, 1323, 1285, 1323, 1151;
1H-NMR, 400 MHz (DMSO-d6): 10.8
(brs, 1H), 9.78 (s, 1H), 8.90 (s, 1H), 8.04 (d, 1H, J=2.0 Hz), 7.27-7.21 (m, 4H), 7.13 (d, 2H, J=8.8 Hz),
6.87-6.85 (m, 2H), 6.77-6.75 (m, 1H), 2.81 (m, 1H), 1.15 (d, 6H, J=6.8 Hz); 13
C-NMR, 100 MHz
(DMSO-d6): 149.4, 144.9, 143.4, 142.5, 135.6, 135.1, 130.0, 129.1, 126.8, 126.4, 124.7, 124.5, 120.6,
119.3, 117, 116.0, 115.1, 32.7, 23.8.
N-(1-Nitro-10H-phenoxazine-3-sulfonyl)-N’-3,4-difluorophenylurea (2g):
2g was synthesized according to the general procedure in method B using 1-nitro-10H-phenoxazine-3-
sulfonyl carbamate (1.0 g, 2.60 mmol) and 3,4-difluoroaniline (0.4 g, 3.10 mmol) in toluene (40 ml) to
yield 0.9 g (75%), reddish brown solid, mp 294 °C decomposition; 1H-NMR, 400 MHz (DMSO-d6)
10.8 (brs, 1H), 9.77 (s, 1H), 9.02 (s, 1H), 7.98 (d, 1H, J=2.0 Hz), 7.25-7.22 (m, 3H), 7.19-7.08 (m, 2H),
6.89-6.73 (m, 2H), 6.79-6.73 (m, 1H).
1-Nitro-3-[([(2-thienylmethyl)amino]carbonylamino)sulfonyl]-10H-phenoxazine (2h):
2h was synthesized according to the general procedure, method B using 1-nitro-10H-phenoxazine-3-
sulfonyl carbamate (1.0 g, 2.60 mmol) and thiophene-2yl-methylamine (0.325 g, 3.10 mmol) in toluene
(40 ml) to yield 1.0 g (85%), reddish brown solid, mp 229 °C decomposition; 1H-NMR, 400 MHz
(DMSO-d6): 11.0 (brs, 1H), 9.73 (s, 1H), 8.01 (t, 1H, NH, J=4.2 Hz), 7.95 (d, 1H, J=2.0 Hz), 7.32-
7.226 (m, 1H), 7.23-7.21 (m, 1H), 7.19 (d, 1H, J=2.0 Hz), 6.93-6.76 (m, 4H), 6.77-6.75 (m, 1H), 4.21 (d,
2H, J=4.2 Hz).
N-(1-Nitro-10H-phenoxazine-3-sulfonyl)-N’-2-methylphenylurea (2i):
2i was synthesized according method B using 1-nitro-10H-phenoxazine-3-sulfonyl carbamate (1.0 g, 2.6
mmol) and o-toluedine (0.36 g, 3.0 mmol) in toluene (40 ml) to yield 1.0 g (86%), reddish brown solid,
mp 241 °C decomposition; Mass: calculated for C20H16N4O6S 440, found 439 (M-H); IR (KBr, cm-1
): v
3336, 3327, 3290, 1685, 1532, 1356, 1277, 1144; 1H-NMR, 400 MHz (DMSO-d6): 10.8 (brs, 1H), 9.79
(s, 1H), 8.18 (s, 1H), 8.04 (d, 1H, J=2.0 Hz), 7.56 (d, 1H, J=7.6 Hz), 7.24-7.22 (m, 2H), 7.17 (d, 1H,
J=7.6 Hz), 7.13 (t, 1H, J=7.6 Hz), 7.01 (t, 1H, J=7.6 Hz), 6.87-6.85 (m, 2H), 6.77-6.74 (m, 1H), 2.15 (s,
3H).

7
N-(1-Nitro-10H-phenoxazine-3-sulfonyl)-N’-4-methoxyphenylurea (2j):
2j was synthesized according to the general procedure, method B using 1-nitro-10H-phenoxazine-3-
sulfonyl carbamate (1.0 g, 2.60 mmol) and p-anisidine (0.38 g, 3.0 mmol) in toluene (40 ml) to yield 0.84
g (70%), reddish brown solid, mp 297 °C decomposition; 1H-NMR, 400 MHz (DMSO-d6): 10.89 (brs,
1H), 9.75 (s, 1H), 9.01 (s, 1H), 8.02 (d, 1H, J=2.0 Hz), 7.21-7.20 (m, 1H, 7.15 (d, 2H, J=8.8 Hz), 6.97 (d,
1H, J=2.0 Hz), 6.88-6.86 (m, 4H), 6.76-6.72 (m, 1H), 3.65 (s, 3H).
3-[([(1-Ethyltetrahydro-1H-3-pyrrolyl)methyl]aminocarbonyl)amino]sulfonyl-1-nitro-10H-
phenoxazine (2k):
2k was synthesized according to method B, using 1-nitro-10H-phenoxazine-3-sulfonyl carbamate (1.0 g,
2.60 mmol) and N-ethylpyrrolidine-2yl-methylamine (0.51 g, 3.9 mmol) in toluene (40 ml) to yield 0.8 g
(66%), brown solid, mp 212 °C decomposition; 1H-NMR, 400 MHz (DMSO-d6): 10.78 (brs, 1H), 9.78
(s, 1H), 8.01 (s, 1H), 7.79 (d, 1H, J=2.0 Hz), 7.21-7.19 (m, 1H), 7.09 (d, 1H, J=2.0 Hz), 6.89-6.85 (m,
2H), 6.77-6.74 (m, 1H), 3.22 (d, 2H), 2.99 (m, 1H), 2.67 (q, 2H, J=6.4 Hz), 2.42-2.39 (m, 2H), 1.98-1.75
(m, 4H), 1.25 (t, 3H, J=6.4 Hz).
N-Piperidinocarbonyl-1-nitro-10H-3-phenoxazinesulfonamide (2l):
2l was synthesized according to the given procedure in method B using 1-nitro-10H-phenoxazine-3-
sulfonyl carbamate (1.0 g, 2.60 mmol) and piperidine (5.0 ml) in toluene to yield 0.8 g (67%), reddish
brown solid, mp 253 °C decomposition; 1H-NMR, 400 MHz (DMSO-d6): 11.0 (brs, 1H), 9.75 (s, 1H),
8.03 (d, 1H, J=2.0 Hz), 7.25 (m, 1H), 6.88-6.85 (m, 2H), 6.78-6.76 (m, 1H), 3.02 (m, 4H), 1.96-1.43 (m,
6H).
N-Piperazinocarbonyl-1-nitro-10H-3-phenoxazinesulfonamide (2m):
2m was synthesized according to the general procedure, method B using 1-nitro-10H-phenoxazine-3-
sulfonyl carbamate (1.0 g, 2.60 mmol) and piperizine (0.27 g, 3.1 mmol) in toluene (40 ml) to yield 0.8 g
(72%), brown solid, mp 303 °C decomposition; 1H-NMR, 400 MHz (DMSO-d6): δ 10.98 (brs, 1H), 9.72
(s, 1H), 7.98 (d, 1H, J=2.0 Hz), 7.23-7.19 (m, 1H), 7.07 (d, 1H, J=2.0 Hz), 6.87-6.84 (m, 2H), 6.76-6.74
(m, 1H), 3.42 (t, 4H, J=5.2 Hz), 2.98 (t, 4H, J=5.2 Hz).
3-([(Diisopropylamino)carbonyl]aminosulfonyl)-1-nitro-10H-phenoxazine (2n):
2n was synthesized according to the general procedure, method B using 1-nitro-10H-phenoxazine-3-
sulfonyl carbamate (1.0 g, 2.60 mmol) and diisopropylamine (0.37 g, 3.9 mmol) in toluene (400 ml) to
yield 0.8 g (70%), brown solid, mp 295 °C decomposition; 1H- NMR, 400 MHz (DMSO-d6): 10.8 (brs,
1H), 9.75 (s, 1H), 8.00 (d, 1H, J=2.0 Hz), 7.27-7.23 (m, 1H), 7.08 (d, 1H, J=2.0 Hz), 6.89-6.84 (m, 2H),
6.78-6.74 (m, 1H), 3.64 (s, 2H, J=6.6 Hz), 1.24 (d, 12H, J=6.6 Hz).
N-Morpholinocarbonyl-1-nitro-10H-3-phenoxazinesulfonamide (2o):
2o was synthesized according to the general procedure, method B using 1-nitro-10H-phenoxazine-3-
sulfonyl carbamate (1.0 g, 2.60 mmol) and morpholine (0.46 g, 5.2 mmol) in toluene (40 ml) to yield 0.7
g (63%), reddish brown solid, mp 218 °C decomposition; Mass: calculated for C17H16N4O7S 420, found

8
419 (M-H); IR (KBr, cm-1
): v 3331, 1690, 1533, 1336, 1284, 1155;
1H-NMR, 400 MHz (DMSO-d6): δ
10.8 (brs, 1H), 9.76 (s, 1H), 8.01 (d, 1H, J=2.0 Hz), 7.24-7.21 (m, 1H), 7.17 (d, 1H, J=2.0 Hz), 6.88-6.85
(m, 2H), 6.77-6.75 (m, 1H), 3.52 (t, 4H, J=5.4 Hz), 3.33 (t, 4H, J=5.4 Hz).
N-(1-Nitro-10H-phenoxazine-3-sulfonyl)-N’-4-carbomethoxyphenylurea (2p):
2p was synthesized according to the general procedure, method B using 1-nitro-10H-phenoxazine -3-
sulfonyl carbamate (1.40 g, 3.6 mmol) and 4-aminobenzoic acid methyl ester (0.67 g, 4.40 mmol) in
toluene (40 ml) to yield 1.2g (67%), reddish brown solid, mp 246 °C decomposition; 1H-NMR, 400 MHz
(DMSO-d6): δ 11.0 (brs, 1H), 9.79 (s, 1H), 9.02 (s, 1H), 7.98 (d, 1H, J=2.0Hz), 7.69 (d, 2H, J=8.7 Hz),
7.27-7.08 (m, 4H), 6.87-6.85 (m, 2H), 6.77-6.74 (m, 1H), 3.76 (s, 3H).
PHARMACOLOGY:
MATERIALS AND METHODS
Drugs and Chemicals:
Streptozotocin, Glibenclamide, Nicotinamide were procured from Sigma Aldrich labs, Glucose oxidase-
Peroxidase (GOD-POD) kits, Serum Glutamic Oxaloacetic Transaminase (SGOT), serum Glutamic-
Pyruvic Transaminase (SGPT) kits, lipid profile kits were procured from Excel Diagnostics Ltd,
Hyderabad and all were of analytical grade
Experimental Animals:
Male Wister albino rats of weighing 180-200g, used for study were procured from Teena Biolabs Pvt.
Ltd. (Reg, no. 177/99 CPCSEA), Hyderabad. All animals were maintained under standard laboratory
conditions [temperature (22± 2 °C) and humidity 50 ± 15%] with 12 h day and 12 h night cycle. The
animals were fed with normal laboratory diet and allowed to drink water ad libitum. The experimental
protocol has been approved by the Institutional Animal Ethical Committee of Vaagdevi College of
Pharmacy, (2012/10/1/2) and by the regulatory body of Government of India.
Acute toxicity studies:
Acute oral toxicity study was performed as per organisation for economic cooperation and development
(OECD) guidelines 423.11
After the oral administration of compounds, animals were observed
individually at least once during the first 30 min and periodically during the first 24 h, with special
attention given during the first 4 h and daily thereafter for total of 14 days.
Hypoglycemic activity:
All the animals were fasted for 18 h, before experimentation, but allowed free access to water and the
animals were divided into 18 groups (n=6), group I rats served as normal-control
and received 5% gum acacia, group II rats served as standard and received standard drug Glibenclamide
(10 mg/kg body weight), Group III-XVIII rats were administered with compounds 2a-p (10 mg/kg body
weight) orally. Blood samples were collected for the measurement of blood glucose by puncture at retro-
orbital plexus during 0, 1, 2, 4 and 6 h after feeding the compounds. The blood glucose levels were
measured by GOD-POD method.

9
Intraperitoneal glucose tolerance test:
The animals which were fasted over night for this study were loaded with glucose (2 g/kg body weight
though intraperitoneal route), 30 min after the administration of test compounds.12
The animals were divided into nine groups (n=6), group I rats served as normal-control and received 5 %
gum acacia, group II rats served as standard and received standard drug Glibenclamide (10 mg/kg body
weight) and group III-IX rats were administered (10 mg/kg body weight) orally with compounds 2a, b, e,
l, n, o, p respectively. The blood samples were collected during 0, 30, 60, 90, 120 min time intervals. The
blood glucose levels were determined by using GOD-POD method.13
Induction of Type-2 diabetes:
Type-2 diabetes was induced 14 to overnight fasted adult wistar strain albino male rats weighing 180–
200g by a single intraperitoneal injection of streptozotocin (60 mg/kg body weight), 15 min after the
intraperitoneal administration of nicotinamide (120 mg/kg body weight). Streptozotocin (STZ) was
dissolved in citrate buffer (pH 4.5) and nicotinamide was dissolved in normal saline. The threshold value
of fasting plasma glucose to diagnose diabetes was taken as >126 mg/dl.15
The rats with type-2-diabetes
were used to perform the anti-diabetic activity.
Anti-diabetic activity:
The rats were divided into six groups (n=6), group I rats served as normal-control and receive 5% gum
acacia, group II diabetic rats received 5% gum acacia served as diabetic control, group III diabetic rats
served as standard and received standard drug Glibenclamide (10 mg/kg body weight) and group IV-VI
rats were administered orally with compounds 2l, n, o (10 mg/kg body weight).
After an overnight fast, compounds were suspended in 5 % gum acacia was fed by oral gavage with the
syringe. Blood samples were collected by puncture at retro-orbital plexus during 0, 1, 2, 4, 6 and 8 h and
the blood glucose levels were determined by using GOD-POD method.16
Sub acute study:
The compound 2l and standard compounds were administered for 14 days. Blood samples were collected
by Retro-orbital puncture on 1st , 7th and 14
th day during 0, 1, 2, 4, 6 and 8 h and the glucose levels were
estimated by GOD-POD kit. On 14 th day, total cholesterol, triglycerides, SGOT and SGPT enzyme
levels including insulin levels were estimated by using biochemical kits.17
Statistical analysis:
The results were expressed as mean ± SEM comparison between the groups was made by analysis of
variance (ONE WAY ANOVA), followed by Dunnet’s test.
RESULTS AND DISCUSSION
Acute toxicity study:
From the acute toxicity studies lethal dose was found to be 200 mg/kg and the dose selected is the 1/20th
of lethal dose.

10
Hypoglycaemic activity of compounds 2a-p in normal rats:
The influence of compounds 2a-p on fasting blood sugar levels were calculated in normal rats at various
time intervals (Table 1). Significant (p<0.01) reduction observed in case of compounds 2 a, b, e, l, n, o
and p, the glucose levels were reduced to 28.90±3.94, 11.56±6.54, 16.49±5.09, 20.15±6.91, 16.76±9.96,
25.77±13.93 and 31.55±2.48 respectively.
Intraperitoneal glucose tolerance test:
Glucose levels were increased with in 30 min after the glucose load. Significant (p<0.001) decrease in
glucose levels were observed for the compounds 2l, 2n, 2o, 2p when compared with normal group and
after 120 min of compound treatment the per centage of reduction of blood glucose levels were observed
as 10.15±10.96, 7.55±4.98, 5.24± 3.25 and 6.69±3.77 respectively (Table 2).
Anti-diabetic activity:
Significant (P<0.001) decrease in the glucose levels were observed for the compounds 2l, 2n, 2o after 2
hours of the treatment in diabetic rats and the per centage reduction was found to be 27.57±4.88,
17.76±9.96, 22.56± 3.98 respectively (Table 3).
Sub acute study:
On the 7th and 14
th day of the treatment compound showed the significant (p<0.001) decrease in the
glucose levels when compared to the diabetic control and the per centage reduction was found to be
39.52± 10.21 and 16.64± 11.65 respectively (Table 4).
Effect of compound 2l on SGOT, SGPT, Lipid profiles and body weights:
Compound 2l effectively control the SGOT, SGPT, cholesterol, triglycerides and when compared diabetic
control rats (Table 5, 6 and 7). In addition it also showed significant (p<0.05) increase in body weight as
compared to the diabetic control animals (Table 8).
Influence on insulin secretion:
Test compound 2l showed significant (p<0.001) increase in insulin as compared to the diabetic control
animals (Table 9).
DISCUSSION
From the acute toxicity studies lethal dose was found to be 200mg/kg and the dose selected is the 1/20th of
lethal dose.
The Influence of compounds 2a-p on fasting blood sugar level was calculated in normal rats at various
time intervals. Significant (p<0.01) reduction of blood glucose levels were observed in case of
compounds 2 a, b, e, l, n, o and p, the glucose levels were reduced to 28.90±3.94, 11.56±6.54,
16.49±5.09, 20.15±6.91, 16.76±9.96, 25.77±13.93 and 31.55±2.48 respectively when compared with
normal rats after 2h of compounds treatment. The compounds, which are shown good hypoglycaemic
activity were taken for further intraperitoneal glucose tolerance screening. Glucose levels in rats were
increased with in 30 min after the glucose load and after 120 min of compound treatment significant

11
(p<0.001) decrease in glucose levels were observed for the compounds 2l, n, o, p when compared with
normal group and the per centage of reduction of blood glucose levels were 10.15±10.96, 7.55±4.98,
5.24± 3.25 and 6.69±3.77 respectively (Table 2). Compounds 2l, n, o were selected for the anti diabetic
activity. The diabetic induced rats were treated with compounds II.66 l, n, o and after 2 h of the treatment
significant (P<0.001) decrease in blood glucose levels were observed and the per centage of reduction
was found to be 27.57±4.88, 17.76±9.96, 22.56± 3.98 respectively (Table 3).
Compound 2l was selected as best compound from the above studies and it was taken for further sub
acute studies. The compound showed good anti-diabetic activity and significant (p<0.001) decrease in the
glucose levels were observed when compared to the diabetic control on the 7th and 14
th day of the
treatment and the percentage of reduction was found to be 39.52± 10.21 and 16.64± 11.65% respectively
in diabetic rats (Table 4). It also shows the significant reduction (p<0.001) in the levels of SGOT, SGPT
(Table 5), total cholesterol (Table 6), triglycerides (Table 7) and increase body weight (Table 8) when
compared diabetic control animals. Compound 2l was tested for its effect in inducing the insulin release
and it was found to be increase insulin levels as compared with diabetic control animals.
The proposed mechanism of action may be by promoting unrestricted endogenous insulin action and
further effect β-cells to release insulin and activate the insulin receptors to absorb the blood sugar.
Liver was necrotized in diabetic rats.18
Therefore an increase in the activities of SGOT and SGPT in
plasma might be mainly due to the leakage of these enzymes from the liver cytosol into the blood stream 19
which gives an indication of the hepato toxic effect of STZ. Treatment of the diabetic rats with
Glibenclamide and test compound 2l caused reduction in the activity of these enzymes in plasma
compared to the diabetic control group
Lipid profile, which is altered in the serum of the diabetic rats, appears to be a vital factor in the
development of atherosclerosis characterized by elevated levels of serum triglycerides and total
cholestero.20, 21
In this study, the compound 2l treated group significantly recovered the levels of serum
lipid profile in treated diabetic rats when compared to diabetic control rats.
Test compound 2l potentiated insulin secretion from surviving β- cells. The increase in insulin secretion
and consequent decrease in blood glucose level may lead to inhibition of lipid peroxidation and control of
lipolytic hormones (Table 9). Histopathological studies of pancreas showed that compound 2l
significantly improved the histology of the islets of Langerhans (Figure 1).The groups treated with test
compounds showed greater persistence of the islets and lesser degree of necrotic changes as compared to
the untreated diabetic rats.
CONCLUSION:
In conclusion, we report for the first time the design, synthesis and evaluation of novel series of N-(1-
nitro-10H-phenoxazine-3-sulfonyl)-N’-alkyl/aryl/heterylureas as anti-diabetic agents. All the synthesized
compounds were evaluated for their hypoglycemic, hyperglycemic and oral anti-diabetic activities.
Fianlly our research has identified N-Piperidinocarbonyl-1-nitro-10H-3-phenoxazine sulfonamide (2l) as
a promising non-TZD and novel agent for the potential treatment of diabetes.

12
Ackonoledgements:
BVK thanks UGC, New Delhi, India for Major Research Project (F. No. 35-151/2008). Authors thank the
Management of C.K.M arts and Science College and Vaagdevi College of Pharmacy for providing the
necessary facilities.
Table 1: Hypoglycemic activity:
Groups Treatment Dose Blood glucose levels (mg/dl)
0 1 h 2 h 4 h 6 h
I Normal control
5% gum
acacia 85.5±2.90 87±9.99 103.7±8.23 91.4±8.18 82.2±2.02
II
Standard
10 mg/kg 98.1±7.23 90.2±4.50 63.73±11.5 74.1±8.53 69.2±5..92 (Glibenclamide)
IV 2a 10 mg/kg 81.88.85 87.4±5.28* 64±7.95 69.2±3.20* 81.8±5.28*
VI 2b 10 mg/kg 85.9±4.13 67.4±9.96* 73.3±7.07** 77.7±12.4** 67±3.13
III 2c 10 mg/kg 98.5±3.51 91.1±9.69 85.9±10.1 74.1±8.53 69.2±3.20
XIII 2d 10 mg/kg 81.8±2.17 96.8±9.13 102.3±8.31 75.4±8.31 89.4±3.20
VII 2e 10 mg/kg 91.1±7.76 75.5±4.45* 68.8±8.23* 76.2±9.28** 65.5±2.47
VIII 2f 10 mg/kg 81.8±14.91 77.7±3.34 81.8±10.33 94±8.68 102.9±8.31
IX 2g 10 mg/kg 103.3±9.96 81.9±4.98 64.3±4.13 86.5±7.57 92.1±10.19
X 2h 10 mg/kg 82.5±2.57 65.9±2.81* 65.9±2.37 80.7±10.13 95.1±7.42
XI 2i 10 mg/kg 107.4±5.71 111.1±8.26 104.8±7.29 91.4±8.36 82.2±2.02
XII 2j 10 mg/kg 89.2±4.02 102.5±9.59 80.3±8.82* 81.8±4.02 71.4±16.21
XIV 2k 10 mg/kg 92.2±10.37 113.3±12.90* 94.4±7.18* 84.8±5.79* 72.9±3.12
V 2l 10 mg/kg 90.74±4.55 67.5±1.97*** 75.8±4.98** 75.4±4.98** 86.6±2.86*
XV 2m 10 mg/kg 74.6±6.28 47.9±4.63 47.6±3.04* 47.9±3.77* 55.9±3.12
XVI 2n 10 mg/kg 81.2±7.48 34.8±4.95* 36.8±3.43* 46.9±4.82* 55.4±5.74
XVII 2o 10 mg/kg 73.5±5.62 50.2±6.50* 41.8±4.78* 51.2±1.94* 51.6±5.37
XVIII 2p 10 mg/kg 80±6.88 63.3±4.57 54.4±4.11* 61.5±3.26* 61.3±3.86
Comparison: Group I vs Group II, III, IV, V, VI, VII,VIII,IX, X, XI, XII, XIII, XIV, XV, XVI, XVII & XVIII. Statistical
significance test for comparisons were done by one way ANOVA, followed by Dunnet’s multiple test. n=6; values are expressed
in mean ± SEM compared to normal rats P< 0.05, P<0.01, P<0.001.

13
Table 2: Intraperitoneal glucose tolerance test.
Groups Treatment Dose Blood glucose levels (mg/dl)
0 min 30 min 60 min 90 min 120 min
I Normal control 5% gum acasia 85.8±2.23 115.4±9.7 112±6.60 98.7±3.66 106.7±4.24
II Standard (Glibenclamide)
10 mg/kg 80.1±5.44 113.9±6.10 92.93.64 81.85.98 96.911.02
III 2a 10 mg/kg 76.25.18 104.1±3.39 88.34.75 81.5±6.86 81.14.99*
IV 2l 10 mg/kg 80.3±5.77 87.5±15.24 73.9±9.15** 73.9±8.46 * 72.16.16***
V 2b 10 mg/kg 89.8±5.64 93.2±7.29 76.6±8.46** 82.2±3.89 62.2±2.60***
VI 2e 10 mg/kg 86±6.49 91.3±5.01 89±8.21 72±7.01* 64.1±6.95***
VII 2n 10 mg/kg 85.3±3.11 103.0±2.76 89.9±3.71** 82.0±4.50* 82.8±4.09*
VIII 2o 10 mg/kg 84.5±2.76 92.0±3.28* 88.5±2.19** 82.2±3.35* 83.0±2.90*
IX 2p 10 mg/kg 89.5±3.28 86.7±3.22** 83.2±2.36*** 83.5±2.06 86.7±2.08*
Comparison: Group I vs Group II, III, IV, V, VI, VII, VIII & IX. Statistical significance test for comparisons were done by one
way ANOVA, followed by Dunnet’s multiple test. n=6; values are expressed in mean ± SEM compared to normal rats P< 0.05,
P<0.01, P<0.001
Table 3: Anti diabetic activity
Comparison: Group II vs Group I, III, IV, V & VI. Statistical significance test for comparisons were done by one way ANOVA,
followed by Dunnet’s multiple test. n=6; values are expressed in mean ± SEM compare to diabetic rats P< 0.05, P<0.01,
P<0.001.
Groups Treatment Dose
Blood glucose levels (mg/dl)
0 h 1 h 2 h 4 h 6 h 8 h
I Normal control 5% gum acacia 98.3±2.55 94.4±2.05 89.7±2.55 87.5±4.71 87.5±3.45 96.1±4.62
II Diabetic Control 5% gum acacia 185.5±5.09 180.8±5.37 173.3±6.60 171.9±2.92 172.5±3.80 178.6±4.66
III
Standard
(Glibenclamide)
10 mg/kg 159.3±5.85 147.2±6.91***
101.9±2.11*** 107.5±3.73*** 127.2±5.76*** 151.9±4.63**
IV 2l 10 mg/kg 161.4±4.94 125±6.77*** 102.6±8.88*** 109.8±7.42*** 125.8±4.94*** 145.1±3.60***
V 2n 10 mg/kg 143.5±6.45 132.9±10.3
0** 99.3±9.88*** 111.2±8.26*** 126.1±3.44*** 129.6±5.82***
VI 2o 10 mg/kg 145.1±7.14 123.2±6.33**
96.1±4.84*** 113.2±2.77** 122.5±10.5** 130.6±4.32**
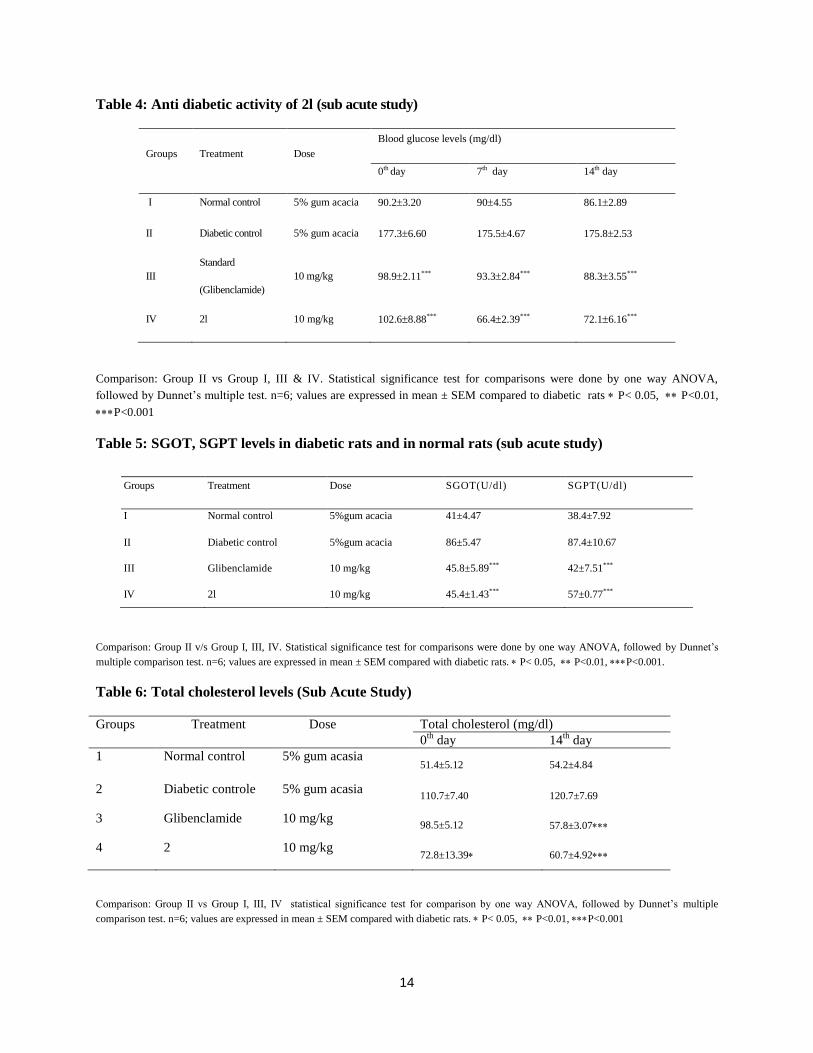
14
Table 4: Anti diabetic activity of 2l (sub acute study)
Groups Treatment Dose
Blood glucose levels (mg/dl)
0th day 7th day 14th day
I Normal control 5% gum acacia 90.23.20 904.55 86.12.89
II Diabetic control 5% gum acacia 177.36.60 175.54.67 175.82.53
III
Standard
(Glibenclamide)
10 mg/kg 98.92.11*** 93.32.84*** 88.33.55***
IV 2l 10 mg/kg 102.68.88*** 66.42.39*** 72.16.16***
Comparison: Group II vs Group I, III & IV. Statistical significance test for comparisons were done by one way ANOVA,
followed by Dunnet’s multiple test. n=6; values are expressed in mean ± SEM compared to diabetic rats P< 0.05, P<0.01,
P<0.001
Table 5: SGOT, SGPT levels in diabetic rats and in normal rats (sub acute study)
Comparison: Group II v/s Group I, III, IV. Statistical significance test for comparisons were done by one way ANOVA, followed by Dunnet’s
multiple comparison test. n=6; values are expressed in mean ± SEM compared with diabetic rats. P< 0.05, P<0.01, P<0.001.
Table 6: Total cholesterol levels (Sub Acute Study)
Groups Treatment Dose Total cholesterol (mg/dl)
0th
day 14th
day
1 Normal control 5% gum acasia 51.4±5.12 54.2±4.84
2 Diabetic controle 5% gum acasia 110.7±7.40 120.7±7.69
3 Glibenclamide 10 mg/kg 98.5±5.12 57.8±3.07
4 2 10 mg/kg 72.8±13.39 60.7±4.92
Comparison: Group II vs Group I, III, IV statistical significance test for comparison by one way ANOVA, followed by Dunnet’s multiple
comparison test. n=6; values are expressed in mean ± SEM compared with diabetic rats. P< 0.05, P<0.01, P<0.001
Groups Treatment Dose SGOT(U/dl) SGPT(U/dl)
I Normal control 5%gum acacia 41±4.47 38.4±7.92
II Diabetic control 5%gum acacia 86±5.47 87.4±10.67
III Glibenclamide 10 mg/kg 45.8±5.89*** 42±7.51***
IV 2l 10 mg/kg 45.4±1.43*** 57±0.77***

15
Table 7: Triglycerides levels (Sub Acute Study)
Groups Treatment Dose
Serum triglycerids (mg/dl)
0th day 14th day
I Normal control 5%gum acacia 60.5±6.86 61.08±5.09
II Diabetic control 5%gum acacia 108.1±11.30 117.2±8.78
III Glibenclamide 10 mg/kg 102.7±8.19 61±3.48
IV 2l 10 mg/kg 90.8±4.32 60±5.75
Comparison: Group II vs Group I, III & IV Statistical significance test for comparisons were done by one way ANOVA, followed by Dunnet’s
multiple comparison test. n=6; values are expressed in mean ± SEM compared with diabetic rats. P< 0.05, P<0.01, P<0.001
Table 8: Effect of compound 2l on body weights (Sub Acute Study):
Groups Treatment Dose
Body Weights (g)
0th day 14th day
I Normal control 5% gum acacia 200±11.18 248±7.84
II Diabetic control 5% gum acacia 220±20 189±16.46
III Glibenclamide 10 mg/kg 212±10.67 244±8.86
IV 2l 10 mg/kg 200±15.08 225±15.75
Comparison: Group II v/s Group I, III & IV Statistical significance test for comparisons were done by one way ANOVA, followed by Dunnet’s
multiple comparison test.n=6; values are expressed in mean ± SEM compared with diabetic rats. P< 0.05, P<0.01, P<0.001
Table 9: Insulin Levels
Groups Treatment Dose Insulin(IU/l)
I Normal control 5%gum acacia 17.66±0.21
II Diabetic control 5%gum acacia 7.72±0.32
III Glibenclamide 10 mg/kg 15.38±0.19***
IV 2l 10 mg/kg 14.10±0.96***
Comparison: Group II v/s Group I, III, IV. Statistical significance test for comparisons were done by one way ANOVA, followed by Dunnet’s
multiple comparison test. n=6; values are expressed in mean ± SEM compared with diabetic rats. P< 0.05, P<0.01, P<0.001

16
Figure 1. Histology of pancreas in experimental rats after 14 days of treatment of 2l
(A) (C)
(B) (D)
(A) Normal control – presence of normal pancreatic islet cells. (B) Diabetic control –degranulated and
dilated islet cells. (C) ) Diabetic + glibenclamide (10 mg/kg) – granulated, absence of dilation and
prominent hyperplasticity (D) Diabetic + 2l (10 mg/kg)–granulated pancreatic islets, showing prominent
hyperplasticity

17
REFERENCES
1. Palsamy P., Sivakumar S., Subramanian S., Chem. Biol. Interact., 2010, 186, 200-210.
2. Palsamy P., Subramanian S., Biomed. Pharmcother., 2008, 62, 598-605.
3. Kumar R. V, Uma Ramachandran, Curr. Sci., 2005, 88, 241-249.
4. Chander A. P., Reddy A. R., Goverdhan P., Eur. J. Biol. Sci., 2011, 3, 126-130.
5. International Diabetes Federation. Diabetes Atlas, 5, 2011.
6. Husain G.M., Singh P.N., Kumar V., Drug. Discov. Ther., 2009, 3, 88-92.
7. Ishan P., Bibhuranjan P., Kamal M., Patel C.N., J. Chem. Pharm. Res., 2010, 2, 609-617.
8. Malamas M. S., Sredy J., Mc Caleb M., Gunawan I., Mihan B., Sullivan D., Eur. J. Med. Chem.,
2001, 36, 31-42.
9. Krall, L. P. World Book of Diabetes in Practice, 1988, 3, 134-141.
10. S. V. Reddy, G. M. Rao, B. V. Kumar, C. L. T. Meda, G. S. Deora, K. S. Kumar, K. V. L. Parsa and M. Pal,
Bioorg. Med. Chem. 2013, 21, 1952.
11. OECD guideline for testing of chemicals 423, Acute oral toxicity (acute toxic class method)
December 2001.
12. Nicholls H. Standard Operating Procedure #39, AMREP AEC, 2008.
13. Trinder P., J. Clin Path., 1969, 22, 246.
14. Shirwaikar A., Rajendran K., Punitha I. S. R., J. Ethnopharmcol. 2005, 97, 369–374.
15. Shirwaikar A., Rajendran K., Rakesh B., J. Ethnopharmacol., 2006, 107, 285–290.
16. Gandhi G. R., Ignacimuthu S., Paulraj M. G., Food. Chem. Toxicol, 2011, 49, 2725-2733.
17. Kavalali G., Tuncel H., Goksel S., Hatemi M. H., J. Ethnopharmacol., 2002, 84, 241–245.
18. Ohaeri O. C., Biosci. Report., 2001, 21, 19–24.
19. Navarro M. C., Montilla M. P., Martín A., Jimenez J., Utrilla M. P., Planta. Med. 1993, 59, 312–314.
20. Chattopadhyay, R.R., Bandyopadhyay M., A fr. J. Biomed. Research. 2005, 8, 101-104.
21. Yadav J. P., Saini S., Kalia A.N., Dangi A.S., Ind. J. Pharm. 2008, 40, 23-27.

Anti-diabetic agents with dual pharmacophores: Synthesis and insilico
screening of some new phenoxazine derivatives
Seelam Venkata Reddya,
Gangula Mohan Rao
a, Baru Vijaya Kumar
a,*, Girdhar Singh Deora.
b
aMedicinal Chemistry Laboratory, Research Centre, C. K. M Arts and Science College,
Warangal 506006, Andhra Pradesh, India.
bInstitute of Life Sciences, University of Hyderabad Campus, Gachibowli, Hyderabad 500 046,
India.
Abstract: In continuation of our work we have developed a series of new phenoxaxazine derivatives
bearing two pharmacophores, sulfonamide and thiazolidinedione moiety based on insilico screening
against human PPARγ and characterized them from their spectral data.
Key words: Phenoxazine, sulfonamide, thiazolidinedione, in silico screening, PPARγ.
*Corresponding author. Tel.+91 8008098787 E-mail address: [email protected]

Type-2 Diabetes Mellitus (T2DM) prevalence is increasing constantly and reaching epidemic proportions.
This asymptomatic disease leads to dreaded micro and macro vascular complications.1 the main classes of
anti-diabetic drugs that are heterogeneous in their mechanism of action include the agents that stimulate
insulin secretion (sulfonamides and sufonylureas),2 reduce hepatic glucose production (biguanides),
3 delay
digestion and absorption of intestinal carbohydrates (α-glycosidase inhibitors),4 or improve insulin action
(thiazolidinedione).5 Phenoxazine a part of antibiotic actinomycin-D shows diverse biological activities. It
is found to exhibit antitumor,6-9
immunosuppressant,10
anti inflammatory,11
and transthyretin fibril
inhibitory actions.12
[quite a few derivatives possessing phenoxazine skeleton have shown PPARα,
PPARδ and PPARγ agonistic activity13
]. Several N10
-substituted phenoxazines are potent inhibitors of
AKT signaling.14
Our recent investigation on phenoxazine derivatives containing sulfonamido moiety have shown15
that
they act as insulin secretagogues such as sulfonyl ureas and non-sulfonyl urea derivatives. Sulfonyl ureas
bind to β-Cell sulfonyl urea receptor (SUR-1), part of trans membrane complex with adenosine -5’-
triphosphate sensitive Kir 6.2 potassium channels (KATP channels).16,17
This binding results in the release
of pre formed insulin granules (first phase) followed by extended release of second phase of insulin.18
If
β-cells are fully functional this increase in release of insulin continues as long as drug stimulation occurs.
Thiazolidinediones decrease hepatic glucose output and increase peripheral glucose utilization by
improving insulin sensitivity at hepatic and muscular sites.19
They stimulate PPARγ receptors expressed
at highest levels in adipose tissue. They enhance the effect of insulin in skeletal muscle, adipose and
hepatic tissues and decrease blood glucose levels in diabetic subjects.20
Of late therapeutic approach to
T2DM has been polypharmacetical21
in nature with several patients on four or more concomitant drugs.
This poly pharmacy approach has been a subject of controversy as it has potential additional risk factors.22
It is observed23
that 53% of Type-2 diabetic patients had a fasting plasma glucose concentration more
than 140 mg and irrespective of mode of therapy a steady increase in HbA1C was seen over nine years
follow up. Precincts of available anti-diabetic agents for controlling HbA1C has enthused search on new
anti-diabetic line of attack. Of these a strategy would lie in designing a single agent containing more than
one pharmacophore that has the affinity for stimulation or inhibition of related targets. As a step in this
direction it is proposed to design a phenoxazine nucleus that contains two pharmacophores i.e. a
sulfonamide moiety at 3rd
position and thiazolidinedione group with a spacer at 1st position. There is only
one report in which unsubstituted phenoxazine is joined to thiazolidinedione moiety for the purpose of
anti-diabetic screening.24
The synthetic protocol involved five steps
1. Synthesis of 1-nitro-10H-phenoxazine-3-sulfonylchloride (2)
2. Synthesis of N-(alkyl/aryl)-1-nitro-10H-phenoxazine-3-sulfonamides (3a-o)
3. Synthesis of N-(alkyl/aryl)-1-amino-10H-phenoxazine-3-sulfonamides (4a-o)
4. Synthesis of 4-(2,4-thiazolidinedidione-5-ylidine methyl)-benznesulfonylchloride (5)
5. Condensation of 4-a-p and 5 to form N-(alkyl/aryl)-1-[4-(2,4-thiazolidinedione-5-ylidine methyl)
benzene sulfonylamino]-10H-phenoxazine-3-sulfonamides (6a-o)
6. Reduction of 6a-p to N-(alkyl/aryl)-1-[4-(2,4-thiazolidinedione-5-ylmethyl) benzenesulfonyl
amino]-10H-phenoxazine-3-sulfonamides (7a-o)
Scheme 1

S
NH
O
O
ClO2S NH
O S
HNS
O
O
S
HN
OO
NH
O S
HNS
O
O
S
HN
OO
NH
O
NO2
S
NO
O
R1
R2
NH
O
NH2
S
NO
O
R1
R2
O
O
NR2
R1O
O
N
R1
R2
NH
O SO3-K
+
NO2
NH
O SO2Cl
NO2
a b
d e
1 2 3a-o 4a-o
5 6a-o 7a-o
4a-p
c
N
R1
R2
S.No
3/4/6/7a R1=H; R2=H
S.No N
R1
R2
3/4/6/7b R1=H; R2=benzyl
3/4/6/7c R1=H; R2=phenyl
3/4/6/7d R1=H R2=4-carbomethoxyphenyl
3/4/6/7e R1=H; R2=cyclohexyl
3/4/6/7f R1=H; R2=4-methoxyphenyl
3/4/6/7g R1=H; R2=4-isopropylphenyl
3/4/6/7h R1=H R=2,4-dimethylphenyl
3/4/6/7i R1=H; R2=4-bromophenyl
3/4/6/7/j R1,R2= morpholino
3/4/6/7k R1,R2=piperidino
3/4/6/7l R1=H; R2=2-hydroxyethyl
3/4/6/7m R1=H; R2=4-hydroxyphenyl
3/4/6/7n R1=H; R2=2-methylphenyl
3/4/6/7o R1=H; R2=3,4-diflorophenyl
Reagents and conditions
(a) phosphorous oxychloride, reflux, 3 hours, 84%; (b) aqueous ammonia, tetrahydrofuran, 0 °C (3a),
80%; alkyl/aryl amines, triethylamine, chloroform, reflux, 3 hours (3b-p), 65-90%; (c) Raney Ni,
hydrazinehydrate, reflux, 30 minutes, 65-96%; (d) pyridine, room temperature, 5 minutes, 40-60%; (e)
Sodium carbonate, sodium dithionite, water, dioxane, 80°C, 1hour, 40-70%.
The steps involved in the synthesis are depicted in scheme-1 and the synthesis of the target molecules
starts from the preparation of 1-nitro-10H-phenoxazine-3-sulfonyl chloride (2) from potassium salt of 1-
nitro-10H-phenoxazine-3-sulfonamide (1) using phosphorous oxychloride. 1 was prepared according to
the reported procedure25
. 2 was condensed with aqueous ammonia in tetrahydrofuran or various alkyl/
aryl amines in chloroform using triethylamine to afford sulfonamides (3a-o). The nitro group of the
compounds 3a-o was reduced to the corresponding N-(alkyl/aryl)-1-amino-10H-phenoxazine-3-
sulfonamides (4a-o) using Raney Ni and hydrazine hydrate in methanol. The structure determination of
the synthesized compounds was established from IR, 1H-NMR and Mass spectral data. The stretching
vibrations of NH and NH2 groups in IR spectra were occurred between 3385-3252 cm-1
. The NH bending
vibrations were appeared between 1640-1610 cm-1
and the asymmetric and symmetric stretching
vibrations of sulfonyl group were observed at 1340-1330 and 1150-1140 cm-1
respectively. The 1H-NMR

spectra of the compounds (4a-o) showed the chemical shift between δ 7.7-7.5 ppm as singlet represents
the absorptions of NH proton of the phenoxazine. The two absorptions appeared between δ 6.75-6.60 and
6.40-6.20 ppm with a meta coupling constant 2.0 Hz due to the 4th and 2
nd protons of phenoxazine. The
multiplet between δ 7.77-6.73 ppm was due to the 8th proton of the phenoxazine nucleus. The remaining
protons observed as multiplet and doublet between δ 6.62-6.50 ppm. The compounds 4a-p were
condensed with 4-chlorosulfonylbenzylidene-2,4-thiazolidinedione (5) in pyridine to afford N-
(alkyl/aryl)-1-[4-(2,4-thiazolidindione-5-ylidinemethyl)-benzenesulfonylamino]-10H-phenoxazine-3-
sulfonamides (6a-o). Compound 5 was prepared according to the reported procedure.26
The IR spectra of
the compounds showed the stretching vibrations of the secondary amines between 3400-3150 cm-1
. The
benzylidine CH and C=C stretching vibration at 5th position of the thiazolidinedione was occurred
between 3075-3050 cm-1
and 1610-1600 cm-1
respectively. The two carbonyl stretching vibrations of the
thiazolidinedione were observed between 1760-1690 cm-1
. The asymmetric and symmetric vibrations of
the sulfonyl group were occurred between 1340-1315 and 1170-1140 cm-1
respectively. The 1H-NMR
spectra of the compounds showed the NH proton of the thiazolidinediones high downfield from TMS and
appeared as broad singlet between δ 12.9-12.6 ppm. The NH proton of the sulfonamide at 3rd
position of
the phenoxazine was observed between δ 10.2-9.70 ppm as singlets. The absorptions of the NH protons of
the sulfonamide at 1st
position of the phenoxazine resonated between δ 9.70-8.0 ppm and the NH proton
of the phenoxazine was occurred between δ 8.20-7.90 ppm. The benzylidine proton was observed
between δ7.780-7.60 ppm and the four protons of the phenyl ring between thiazolidinedione and
phenoxazine were resonated as singlet or doublet between δ 7.77-7.60 ppm.
NH
O S
HNS
O
O
S
NH
O
O
7
7
7
7
MgMethanol
Reflux
24 h
Fe,
AcOH/HCl
Reflux
24 h
Zn,
AcOH/HCl
24 h
Pd/C 5%
Ammonium
formate
THF/MeOH
reflux 24 h
7
Zn, NH4Cl
Water/Methanol
reflux
7Na2S2O4
water
Dioxane
80°C
Na2CO3
6
O
O NR2
R1
Fig 1
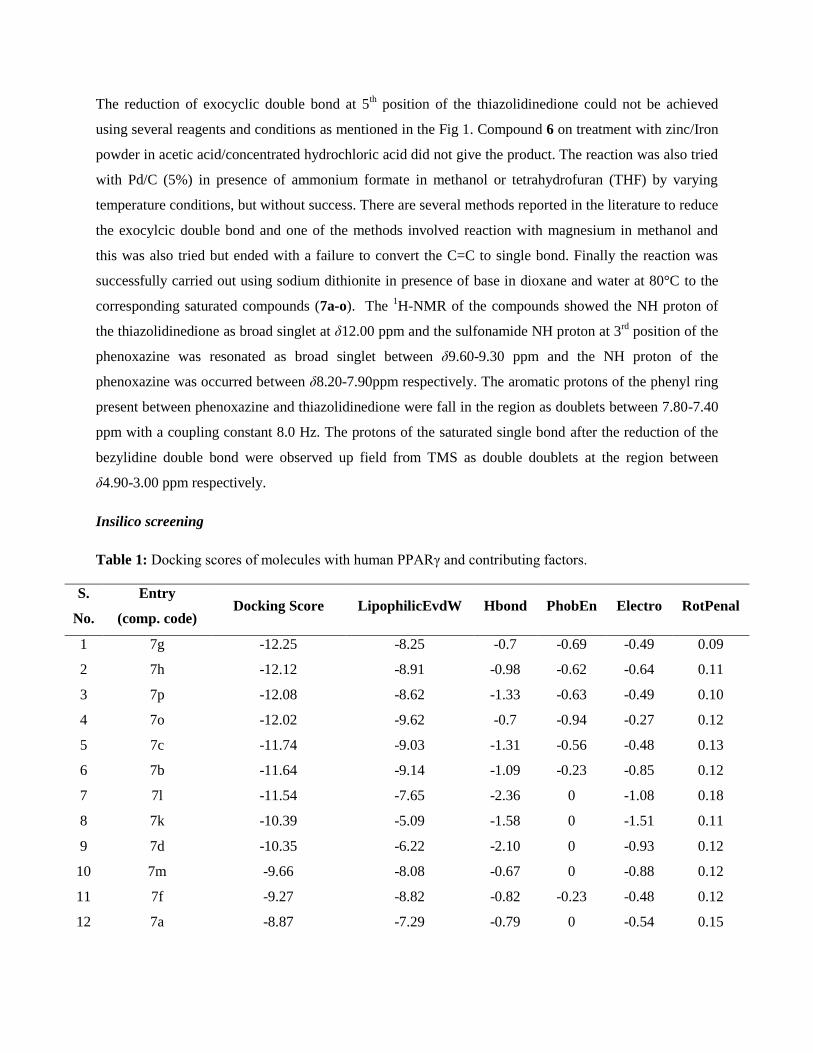
The reduction of exocyclic double bond at 5th position of the thiazolidinedione could not be achieved
using several reagents and conditions as mentioned in the Fig 1. Compound 6 on treatment with zinc/Iron
powder in acetic acid/concentrated hydrochloric acid did not give the product. The reaction was also tried
with Pd/C (5%) in presence of ammonium formate in methanol or tetrahydrofuran (THF) by varying
temperature conditions, but without success. There are several methods reported in the literature to reduce
the exocylcic double bond and one of the methods involved reaction with magnesium in methanol and
this was also tried but ended with a failure to convert the C=C to single bond. Finally the reaction was
successfully carried out using sodium dithionite in presence of base in dioxane and water at 80°C to the
corresponding saturated compounds (7a-o). The 1H-NMR of the compounds showed the NH proton of
the thiazolidinedione as broad singlet at δ12.00 ppm and the sulfonamide NH proton at 3rd
position of the
phenoxazine was resonated as broad singlet between δ9.60-9.30 ppm and the NH proton of the
phenoxazine was occurred between δ8.20-7.90ppm respectively. The aromatic protons of the phenyl ring
present between phenoxazine and thiazolidinedione were fall in the region as doublets between 7.80-7.40
ppm with a coupling constant 8.0 Hz. The protons of the saturated single bond after the reduction of the
bezylidine double bond were observed up field from TMS as double doublets at the region between
δ4.90-3.00 ppm respectively.
Insilico screening
Table 1: Docking scores of molecules with human PPARγ and contributing factors.
S.
No.
Entry
(comp. code) Docking Score LipophilicEvdW Hbond PhobEn Electro RotPenal
1 7g -12.25 -8.25 -0.7 -0.69 -0.49 0.09
2 7h -12.12 -8.91 -0.98 -0.62 -0.64 0.11
3 7p -12.08 -8.62 -1.33 -0.63 -0.49 0.10
4 7o -12.02 -9.62 -0.7 -0.94 -0.27 0.12
5 7c -11.74 -9.03 -1.31 -0.56 -0.48 0.13
6 7b -11.64 -9.14 -1.09 -0.23 -0.85 0.12
7 7l -11.54 -7.65 -2.36 0 -1.08 0.18
8 7k -10.39 -5.09 -1.58 0 -1.51 0.11
9 7d -10.35 -6.22 -2.10 0 -0.93 0.12
10 7m -9.66 -8.08 -0.67 0 -0.88 0.12
11 7f -9.27 -8.82 -0.82 -0.23 -0.48 0.12
12 7a -8.87 -7.29 -0.79 0 -0.54 0.15
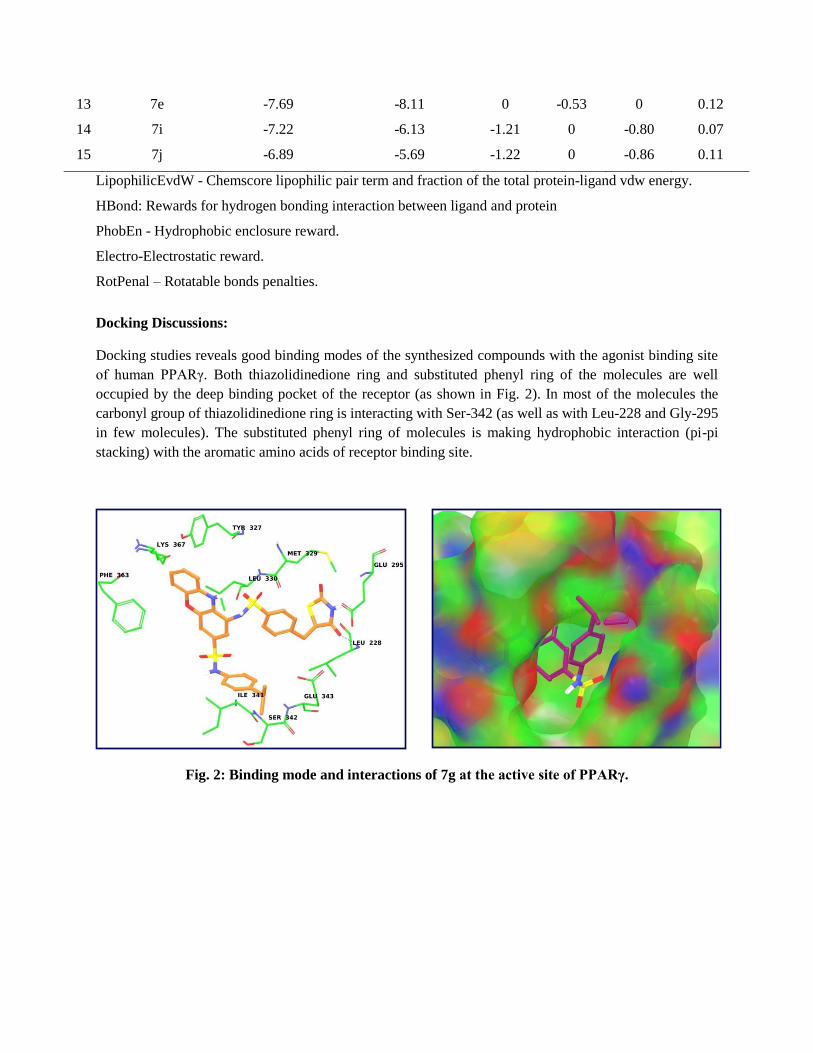
13 7e -7.69 -8.11 0 -0.53 0 0.12
14 7i -7.22 -6.13 -1.21 0 -0.80 0.07
15 7j -6.89 -5.69 -1.22 0 -0.86 0.11
LipophilicEvdW - Chemscore lipophilic pair term and fraction of the total protein-ligand vdw energy.
HBond: Rewards for hydrogen bonding interaction between ligand and protein
PhobEn - Hydrophobic enclosure reward.
Electro-Electrostatic reward.
RotPenal – Rotatable bonds penalties.
Docking Discussions:
Docking studies reveals good binding modes of the synthesized compounds with the agonist binding site
of human PPARγ. Both thiazolidinedione ring and substituted phenyl ring of the molecules are well
occupied by the deep binding pocket of the receptor (as shown in Fig. 2). In most of the molecules the
carbonyl group of thiazolidinedione ring is interacting with Ser-342 (as well as with Leu-228 and Gly-295
in few molecules). The substituted phenyl ring of molecules is making hydrophobic interaction (pi-pi
stacking) with the aromatic amino acids of receptor binding site.
Fig. 2: Binding mode and interactions of 7g at the active site of PPARγ.

Fig. 3: Binding mode and interactions of 7h at the active site of PPARγ.
Fig. 4: Binding mode and interactions of 7p at the active site of PPARγ.
Fig. 5: Binding mode and interactions of 7n at the active site of PPARγ.
Fig. 6: Binding mode and interactions of 7c at the active site of PPARγ.
Fig. 7: Binding mode and interactions of 7b at the active site of PPARγ.

Fig. 8: Binding mode and interactions of 7l at the active site of PPARγ.
Fig. 9: Binding orientation and interactions of 7g at the active site of PPARγ.

Experimental section
All the chemicals and solvents used for the synthesis were of commercial grade and procured from SD
Fine/Merck India chemical company. All the reactions were monitored by thin layer chromatography
(TLC) on silica gel plates (60 F254), visualizing with ultraviolet light or iodine spray. Column
chromatography was performed on silica gel (60-120 mesh) using hexane, ethyl acetate, dichloromethane
and methanol. 1H NMR and
13C NMR spectra were determined in DMSO-d6 solution by using Bruker
Biospin, Advance-III 400 MHz Fourier Transform Digital NMR spectrometer. Proton chemical shifts (δ)
were relative to tetramethylsilane (TMS, δ = 0.00) as internal standard and expressed in ppm. Spin
multiplicities are given as s (singlet), br s (broad singlet), d (doublet), t (triplet), q (quartet) and m
(multiplet). Coupling constants (J) were given in hertz. Infrared spectra were recorded on a FT-IR, Bruker
Vertex-70 spectrometer using KBr pellets. Melting points were determined using Polmon digital melting
point apparatus-m96 and are uncorrected. Mass spectra were recorded on Agilent 6300 Ion Trap LC/MS
System.
1-Nitro-10H-phenoxazine-3-sulfonylchloride (2): Potassium;1-nitro-10H-phenoxazine-3-sulfonate (10
g) was added portion wise to in phosphorous oxychloride (70 mL) and the mixture was refluxed for 3
hours. After completion of the reaction excess POCl3 was removed by distillation and the crude reddish
brown colour solid was poured in crushed ice and filtered under reduced pressure. The solid was washed
with cold water (100 mL), cold methanol (50 mL) and dried to yield 8 g (84%), reddish brown solid, mp.
204°C decomposition. IR (KBr cm
-1) v: 3340, 1499 & 1379 , 1281 & 1162 .
1H-NMR, 400 MHz (DMSO-
d6): 9.41 (s, 1H), 7.72 (d, 1H, J=2.0 Hz), 7.15-7.13 (m, 1H), 6.95 (d, 1H, J=2.0 Hz), 6.86-6.78 (m, 2H),
6.75-6.73 (m, 1H); MS m/z (%): 327 (M+H, 100).
1-Nitro-10H-phenoxazine-3-sulfonamide (3a): 1-Nitro-10H-phenoxazine-3-sulfonylchloride (8 g) was
added portion wise to a solution of aqueous ammonia (10 mL) in tetrahydrofuran (50 mL) at 0°C. The
reaction mixture was stirred for 30 minutes at same temperature. Completion of the reaction was
monitored by TLC (30% ethyl acetate in hexane) and excess solvent was removed by distillation under
reduced pressure. The product was extracted in ethyl acetate (300 mL), and washed with water (300 mL),
dilute hydrochloric acid (100 mL) followed by water (100 mL), saturated sodium chloride solution (100
mL). The organic phase was dried over anhydrous sodium sulphate, filtered and distilled in vacuo to yield
6 g (80%), reddish brown solid. The compound was purified by column chromatography using 30% ethyl
acetate in hexane, mp 282-283°C. IR (KBr cm-1
) v: 3388, 3335 & 3255, 1536 & 1337, 1291 & 1146; 1H-
NMR 400 MHz (DMSO-d6): 9.75 (s, 1H), 7.91 (d, 1 H, J= 2.0 Hz), 7.43 (s, 2H), 7.24-7.21 (m, 1 H),
7.12 (d, 1H, J= 2.0 Hz), 6.89-6.72 (m, 3H); 13
C-NMR, 100 MHz (DMSO-d6): δ 145.2, 142.5), 134.0,
133.7, 129.9, 127.1, 124.5, 124.4, 117.7, 116.9, 115.2, 115.1; MS m/z (%): 306 (M-H+, 100).

General Procedure for preparation of N-(alkyl/aryl/heteryl)-1-nitro-10H-phenoxazine-3-
sulfonamides (3b-o): 1-Nitro-10H-phenoxzine-3-sulfonylchloride (6.0 mmol) was added to a solution of
alkyl/aryl/heteryl amine (6.0 mmol) and triethylamine (12.0 mmol) in chloroform (30 mL). The resulting
reaction mixture was refluxed for 30 minutes and completion of the reaction was monitored by TLC (50%
chloroform in hexane). The excess solvent was removed under reduced pressure and the crude solid was
dissolved in ethyl acetate (200 mL). The organic layer was washed with water (100 mL), dilute
hydrochloric acid (100 mL), water (50 mL) and saturated sodium chloride solution (50 mL). Then the
organic layer was dried over sodium sulphate, filtered and concentrated under reduced pressure to yield
brown/ reddish brown colour solid. The compound was purified by column chromatography using 60–
120 silica gel in 30 % chloroform in hexane.
N-(Benzyl)-1-nitro-10H-phenoxazine-3-sulfonamide (3b): yield 77%, brown solid, mp 222-224°C.
4-(1-Nitro-10H-phenoxazine-3-sulfonylamino)-benzoic acid (3d): yield 65%, reddish brown solid, mp
>300°C.
N-(2-Hydroxyethyl)-1-nitro-10H-phenoxazine-3-sulfonamide (3l): yield 70%, reddish brown solid mp
218-219°C.
N-(3,4-Difluorophenyl)-1-nitro-10H-phenoxazine-3-sulfonamide (3o): yield 75%, brown solid, mp
258-260°C.
General procedure for the synthesis of N-(aryl/alkyl)-1-amino-10H-phenoxazine-3-sulfonamides
(4a-o)
To a suspension of N-(aryl/alkyl)-1-amino-10H-phenoxazine-3-sulfonamide (3 g), and Raney Ni (1.8 g)
in methanol (200 ml) was added hydrazine hydrate (3 ml) drop wise over a period of 20 min at 65°C and
refluxed for 15 min. Completion of the reaction was monitored by TLC (40% ethyl acetate in hexane).
The reaction mass was cooled and filtered by passing through cilite pad and the filtrate was concentrated
under reduced pressure to afford 2.0g of off white solid.
1-amino-10H-phenoxazine-3-sulfonamide (4a): yield 85%; off white solid; mp 176 °C
(decomposition); IR (KBr cm-1
) v: 3316 & 3268, 1636, 1337&1145.; 1H NMR (400 MHz, DMSO-d6) δ
7.56 (s, 1H), 7.01 (s, 2H), 6.77-6.75 (m, 1H), 6.74 (s, 1H), 6.70 (d, J = 4 Hz, 2H), 6.55-6.53 (m, 1H),
6.37 (s, 1H), 5.12 (s, 2H); m/z (CI) 276 (M-1, 100)

N-(benzyl)-1-amino-10H-phenoxazine-3-sulfonamide (4b): yield 80%, ash colour solid, mp 165°C
decomposition
N-(phenyl)-1-amino-10H-phenoxazine-3-sulnamide (4c): yield 93%, ash colour solid, mp 200°C
decomposition
4-(1-amino-10H-phenoxazine-3-sulfonylamino)-benzoicacid (4d): yield 75%, off white solid, mp
>300°C.
N-(cyclohexyl)-1-amino-10H-phenoxazine-3-sulfonamide (4e): yield 75%, light brown colour solid,
mp 188-190°C.
N-4-(methoxyphenyl)-1-amino-10H-phenoxazine-3-sulfonamide (4f): yield 87%, mp 194-196°C. IR
(KBr) cm-1:
3457, 3373, 3252, 1641, 1338, 1147. 1H-NMR, 400 MHz (DMSO-d6): 9.59 (s, 1H), 7.60 (s,
1H), 6.98 (d, 2H, J=8.8 Hz), 6.82 (d, 2H, J=8.8 Hz), 6.77-6.72 (m, 1H), 6.61-6.60 (m, 2H), 6.57 (1H,
J=2.0Hz), 6.52 (d, 1H, J=8.0 Hz), 6.23 (1H), 5.11 (s, 2H), 3.67 (s, 3H). Mass: calculated for C19H17N3O4S
383, found 382 (M-H+, 100).
N-4-isopropylphenyl-1-amino-10H-phenoxazine-3-sulfoanmide (4g): yield 89%, off white solid, mp
180-183°C.
N-(2,4-dimethylphenyl)-1-amino-10H-phenoxazine-3-sulfonamide (4h): yield 88%, oof white solid,
mp 170°C decomposition.
N-(4-bromophenyl)-1-amino-10H-phenoxazine-3-sulfonamide (4i): yield 95%, off white solid, mp
225°C decomposition. IR (KBr) cm
-1: 3371, 3269, 1610, 1330, 1143.
1H-NMR, 400 MHz (DMSO-d6)
10.15 (s, 1H), 7.65 (s, 1H), 7.43 (d, 2H, J=8.8 Hz), 7.03 (d, 2H, J=8.8 Hz), 6.76-6.73 (m, 1H), 6.64 (1H,
J=2.0Hz), 6.61-6.60 (m, 2H), 6.52 (d, 1H, J=7.2 Hz), 6.23 (1H), 5.16 ( s , 2H). Mass: calculated for
C18H14BrN3O3S 432, found 430(M-2H, 100).
3-(Morpholinosulfonyl)-1-amino-10H--phenoxazine (4j): yield 68%, light brown colour solid, mp
178°C decomposition.
3-piperidinosulfonyl-1-amino-10H-phenoxazine (4k): yield 85%, off white solid, mp 249°C
decomposition.
N-(2-hydroxyethyl)-1-amino-10H-phenoxazine-3-sulfonamide (4l): yield 82%, off whiter solid,
162°C decomposition.

N-(4-hydroxyphenyl)-1-amino-10H-phenoxazine-3-sulfonamide (4m) yield 81%, off white solid mp
290-291°C decomposition
N-(2-methyl)-1-amino-10H-phenoxazine-3-sulfonamide (4n): yield 96%, off white solid, mp 178°C
decomposition.
N-(3,4-difluorophenyl)-1-amino-10H-phenoxazine-3-sulfonamide (4o): yield 95%, off white solid,
mp 253°C decomposition.
Synthesis of 4'-chlorosulfonyl-benzylidene 2, 4-thiazolidinedione(5):
Chlorosulfonic acid (18 gm, 0.155 mol) was added drop wise to benzylidine-2, 4-thiazolidinedione (8 gm,
0.0388 mol) at room temperature and the mixture was stirred at 100°C for 1 hour. The reaction was
monitored by TLC and after completion of the reaction the mixture was cooled and poured in crushed ice.
The solid separated was filtered and dried. The product was purified by recrystallization from acetone to
yield 68%, m.p. 180-181°C. 1H NMR (400 MHz, DMSO-d6) δ 12.62 (brs, 1H), 7.78 (s, 1H), 7.72 (d, 2 H,
J=8.0 Hz), 7.57 (d, 2H, J=8.0 Hz).
III. General procedure for the condensation of N-(alkyl/aryl)-1-amino-10H-phenoxazine-3-
sulfonamides (4a-p) and 4-(2, 4-thiazolidindione-5-ylidenemethyl)-benzenesulfonylchloride (5)
To a solution of N-(alkyl/aryl)-1-amino-10H-phenoxazine-3-sulfonamide (0.0039 mol) in pyridine (15
ml), 4-(2, 4-thiazolidindione-5-ylidenemethyl)-benzenesulfonylchloride (0.0046 mol ) was added and the
resulting mixture was stirred for 5 minutes. Completion of the reaction was monitored by TLC (50% ethyl
acetate in hexane) and the reaction mass was poured in cold dilute hydrochloric acid solution. The solid
separated out was filtered and dried and purified by crystallisation from acetic acid.
1-[4-(2,4-thiazolidindione-5-ylidinemethyl)-benzenesulfonylamino]-10H-phenoxazin-3-sulfonamide
(6a): yield 43%, off white solid, mp > 305°C.. IR (KBr) cm-1
: 3397, 3372 & 3340, 3261 & 3221, 1761 &
1707, 3061, 1607, 1538 & 1503, 1326 & 1159. 1H NMR (400 MHz, DMSO-d6) δ 12.73 (brs, 1H), 9.56
(brs, 1H), 7.85 (d, 2H, J= 8.0 Hz), 7.78 (s, 1H), 7.76 (d, 2H, J= 8.0 Hz), 7.15 (s, 2H), 6.85 (s, 1H), 6.76-
6.74 (m, 2H), 6.68-6.61 (m, 3H). Mass calculated for C22H16N4O7S3 544, found 543 (M-1, 100).
N-(benzyl)-1-[4-(2,4-thiazolidindione-5-ylidinemethyl)-benzenesulfonylamino]-10H-phenoxazin-3-
sulfonamides (6b): yield 58%, off white solid, 293°C decomposition.
N-(phenyl)-1-[4-(2,4-thiazolidindione-5-ylidinemethyl)-benzenesulfonylamino]-10H-phenoxazin-3-
sulfonamides (6c): yield 56%, mp 270°C decomposition. IR (KBr) cm
-1 3410, 3289, 3191, 1747, 1696,
1603, 1315, 1148.1H NMR (400 MHz, DMSO-d6) δ 12.70 (brs, 1H), 10.10 (s, 1H), 9.52 (brs, 1H), 8.10 (s,

1H), 7.78 (s, 1H), 7.68 (s, 4H), 7.25 (t, 2H, J=8.0 Hz), 7.05 (t, 2H, J= 8.0 Hz), 7.0 (d, 1H, J=7.6 Hz),
6.77-6.71 (m, 3H), 6.64-6.57 (m, 3H). Mass calculated for C28H20N4O7S3 620, found 619 (M-1, 100).
4-[(1-[(4-[(2,4-thiazolidinedione-5-ylidine)methyl]phenylsulfonyl)amino]-10H-3-phenoxazinyl-
sulfonyl)amino]benzoic acid (6d); yield 57%, off white solid, mp >300°C.
N-(cyclohexyl)-1-[4-(2,4-thiazolidindione-5-ylidinemethyl)-benzenesulfonylamino]-10H-phenoxazin-
3-sulfonamides (6e): yield 58%, off white solid, mp 283°C decomposition.
N-(4-methoxyphenyl)-1-[4-(2,4-thiazolidindione-5-ylidinemethyl)-benzenesulfonylamino]-10H-
phenoxazin-3-sulfonamides (6f): yield 59%, off white solid, mp 288°C decomposition. IR (KBr) cm
-1:
3427, 3221, 3183, 3058, 1748, 1698, 1607, 1337, 1162.1H NMR (400 MHz, DMSO-d6) δ 12.78 (brs, 1H,
NH), 9.74 (s, 1H, SO2NH), 9.51(brs, 1H, SO2NH), 8.07 (s, 1H, Phe NH), 7.77 (s, 1H, =CH), 7.70(s, 4H),
6.90(d, 2H, J=8.8 Hz), 6.82(d, 2H, J= 8.8 Hz), 6.74-6.64 (m, 6H), 3.67 ( s, 3H). 13C-NMR (100 MHz,
DMSO-d6): δ 167.5, 166.9, 156.3, 143.1, 141.9, 139.5, 137.4, 133.0, 130.2, 130.0, 129.5, 129.3, 127.6,
126.9, 124.3, 122.9, 122.1, 119.5, 115.0, 114.5, 114.2, 111.4, 55. Mass calculated for C29H22N4O8S3:650,
found 649 (M-1, 100).
N-(4-isopropylphenyl)-1-[4-(2,4-thiazolidindione-5-ylidinemethyl)-benzenesulfonylamino]-10H-
phenoxazin-3-sulfonamides (6g): yield 58%, off white solid, mp 270°C decomposition.
N-(2,4-dimethylphenyl)-1-[4-(2,4-thiazolidindione-5-ylidinemethyl)-benzenesulfonylamino]-10H-
phenoxazin-3-sulfonamides (6h): yield 55%, off white solid, mp 298°C decomposition.
N-(4-bromophenyl)-1-[4-(2,4-thiazolidindione-5-ylidinemethyl)-benzenesulfonylamino]-10H-
phenoxazin-3-sulfonamides (6i): yield 61%, off white solid, mp 293°C decomposition. IR (KBr) cm-1
:
3429, 3221, 3167, 1757, 1690, 1608, 1338, 1163. 1
H NMR (400 MHz, DMSO-d6) δ 12.75 (brs, 1H),
10.28 ( s, 1H), 9.55 (brs, 1H), 8.11 (s, 1H), 7.78 (s, 1H), 7.69 (s,4H), 7.43 (d, 24, J=8.8Hz), 6.94 (d,2H,
J=8.8 Hz), 6.76-7.72 (m, 3H), 6.64-6.59 (m, 3H). 13c-NMR (100 MHz, DMSO-d6) δ 137.4, 16639, 143.2,
141.9, 139.5, 137.4, 137.0, 133.2, 131.9, 130.2, 129.5, 129.4, 128.37, 127.6, 127.0, 124.4, 122.2, 121.5,
121.4, 119.6, 115.9, 115.0, 114.0, 111, Mass calculated for C28H19BrN4O7S3:698, found 699 (M-1, 75%)
& 701(25%)..
N-1-[3-(morpholinosulfonyl)-10H-1-phenoxazinyl]-4-[(2,4-dioxo-1,3-thiazolan-5-yliden)methyl]-1-
benzenesulfonamide (6j): yield 56%, off white solid, mp 288°C decomposition.
N-1-[3-(piperidinosulfonyl)-10H-1-phenoxazinyl]-4-[(2,4-thiazolidinedione-5-ylidine)methyl]-1-
benzenesulfonamide (6k): yield 55%, off white solid, mp >300°C

N-(2-hydroxyethyl)-1-[4-(2,4-thiazolidindione-5-ylidinemethyl)-benzenesulfonylamino]-10H-
phenoxazin-3-sulfonamides (6l) yield 85%, off white solid, mp >270°C.
N-(4-hydroxyphenyl)-1-[4-(2,4-thiazolidindione-5-ylidinemethyl)-benzenesulfonylamino]-10H-
phenoxazin-3-sulfonamides (6m): yield 57%, off white solid, mp 293°C decomposition.
N-(2-methylphenyl)-1-[4-(2,4-thiazolidindione-5-ylidinemethyl)-benzenesulfonylamino]-10H-
phenoxazin-3-sulfonamides (6n): yield 57%, off white solid, mp 290°C decomposition.
N-(3,4-difluorophenyl)-1-[4-(2,4-thiazolidindione-5-ylidinemethyl)-benzenesulfonylamino]-10H-
phenoxazin-3-sulfonamides (6o): yield 59%, off white solid, mp 295°C.
General procedure for the reduction of N-(alkyl/aryl)-1-[4-(2,4-thiazolidindione-5-ylidinemethyl)-
benzenesulfonylamino]-10H-phenoxazin-3-sulfonamides to N-(alkyl/aryl)-1-[4-(2,4-thiazolidine-
dione-5-ylmethyl)-benzenesulfonylamino]-10H-phenoxazin-3-sulfonamides (7a-p)
To a solution of N-(alkyl/aryl)-1-[4-(2,4-thiazolidindione-5-ylidinemethyl)-benzenesulfonylamino]-10H-
phenoxazin-3-sulfonamide (0.0045mol) in 1,4-dioxane (30 ml) and water(15 ml) was added sodium
carbonate (0.055 mol) and heated to 80°C. Then a solution of sodium dithionite (0.05 mol) in 15 ml of
water was added drop wise and the resulting reaction mixture was stirred at same temperature for 1 h.
Completion of the reaction was monitored by TLC using 10% methanol in dichloromethane. Then the
reaction mass was cooled to 10°C and the pH of the reaction mass was adjusted to 6 using acetic acid and
stirred for 30 min and the solid separated out was filtered and dried. The product was purified using
column chromatography by eluting with 5% methanol in dichloromethane.
1-[4-(2,4-thiazolidindione-5-ylmethyl)-benzenesulfonylamino]-10H-phenoxazin-3-sulfonamide (7a):
yield 56%, off white solid, mp 230°C decomposition 1H NMR (400 MHz, DMSO-d6) δ 12.09 (br s,1H,
NH), 9.49 ( br s, 1H, NH), 7.91(s, 1H, NH ), 7.70 (d, 2H, J=8.0 Hz), 7.45 (d, 2H, J=8.0 Hz), 7.14 (s, 2H),
6.83 - 6.61 (m, 6H), 4.86 (dd, 1H, J=4.0, 9.6 Hz), 3.43 (dd,1H, J= 4.4, 14 Hz), 3.15 (dd, 1H, J=9.6, 14.0
Hz). 13
C- NMR (100 MHz, DMSO-d6) δ 175.4, 171.4, 143, 142.7, 142.0, 137.9, 134.5, 131.8, 129.9,
129.8, 127.1, 124.3, 121.9, 119.9, 119.7, 115.0, 114.5, 110.5, 51.8, 37 .0.
N-(benzyl)-1-[4-(2,4-thiazolidindione-5-ylmethyl)-benzenesulfonylamino]-10H-phenoxazin-3-
sulfonamides (7b): yield 60%, off white solid, mp 216°C decomposition 1H-NMR (400 MHz, DMSO-
d6) δ 12.07 (br s, 1H, CONH), 9.53 (br s, 1H, NH Phen), 8.14 (s, 1H, NHSO2), 7.87 (t, 1H, SO2NH, J=
4.4 & 6.4), 7.73 (d, 2H, J=8.0 Hz, Ar H2 & H6), 7.41 (d, 2H, J=8.0 Hz, Ar H3 & H5), 7.33-7.21 (m, 5H,
benzyl Hs), 6.81-6.75 (m, 3H, phen H8,H4,H6), 6.68-6.61 (m, 3H, phen H5,H9,H2), 4.77 (dd, 1H, J=9.6 &

4.8 Hz), 4.03 (dd, 1H, J= 14.4 & 7.2 Hz), 3.68 (d, 2H, J=6.0 Hz), 3.01 (dd, 1H, J=14.0 9.6 Hz). 13
C-
NMR (100 MHz, DMSO-d6) δ 175.2, 171.4, 143.4, 142.7, 142.1, 137.7, 137.4, 133.1, 129.9, 129.8,
128.2, 127.6, 127.2, 127.1, 124.3, 119.7, 115.0, 114.6, 111.2, 59.7, 51.7, 46.0.
N-(phenyl)-1-[4-(2,4-thiazolidindione-5-ylmethyl)-benzenesulfonylamino]-10H-phenoxazin-3-
sulfonamides (7c): yield 66%, off white solid, 225°C decomposition
4-[(1-[(4-[(2,4-thiazolidinedione-5-ylmethyl]phenylsulfonyl)amino]-10H-3-phenoxazinylsulfonyl)-
amino] benzoic acid (7d): yield 50%, off white solid, 250°C decomposition.
N-(cyclohexyl)-1-[4-(2,4-thiazolidindione-5-ylmethyl)-benzenesulfonylamino]-10H-phenoxazin-3-
sulfonamides (7e): yield 65%, off white solid, 237°C decomposition .
N-(4-methoxyphenyl)-1-[4-(2,4-thiazolidindione-5-ylmethyl)-benzenesulfonylamino]-10H-
phenoxazin-3-sulfonamides (7f): yield 66%, off white solid, 234°C decomposition . 1H-NMR (400
MHz, DMSO-d6) δ 12.09 (br s, 1H, NH), 9.75 (1H, NH), 9.32 (br s, 1H, NH), 8.00 (s, 1H, NH), 7.55 (d,
2H, J=8.0 Hz), 7.38 (d, 2H, J=8.0 Hz), 6.95(d, 2H, J=8.6Hz, ArH), 6.85 (d, 2H, J= 8.6 Hz, ArH), 6.75-
6.58 (m, H), 4.85 (dd, 1H, J= 9.6 & 4.4 Hz H5 Thiazolidine), 3.69 (s, 3H, OCH3), 3.41 (dd, 1H, J= 14.4,
& 4.4 Hz), 3.14 (dd, 1H, J=14.0 & 9.6 Hz). 13
C-NMR (400 MHz, DMSO-d6) δ 175.3, 171.3, 156.4, 143.0,
142.7, 141.9, 137.6, 132.6, 130.0, 129.7, 129.6, 129.2, 127.0, 124.3, 123.0, 122.1, 121.4, 119.8, 115.0,
114.3, 111.2, 55.1, 51.8, 37.0.
N-(4-isopropylphenyl)-1-[4-(2,4-thiazolidindione-5-ylmethyl)-benzenesulfonylamino]-10H-
phenoxazin-3-sulfonamides (7g): yield 69%, off white solid, 219°C decomposition.
N-(2,4-dimethylphenyl)-1-[4-(2,4-thiazolidindione-5-ylmethyl)-benzenesulfonylamino]-10H-
phenoxazin-3-sulfonamides (7h): yield 70%, off white solid, 256°C decomposition.
N-(4-bromophenyl)-1-[4-(2,4-thiazolidindione-5-ylmethyl)-benzenesulfonylamino]-10H-phenoxazin-
3-sulfonamides (7i): yield 44%, off white solid, mp 232°C decomposition.
N-[3-(morpholinosulfonyl)-10H-1-phenoxazinyl]-4-[(2,4-thiazolidine-5-ylmethyl]-1-benzene-
sulfonamide (7j): yield 66%, off white solid, 255°C decomposition.
N-[3-(piperidinosulfonyl)-10H-1-phenoxazinyl]-4-[(2,4-thiazolidinedione-5-ylmethyl]-1-
benzenesulfonamide (7k): yield 70%, off white solid, 230°C decomposition . 1H-NMR (400 MHz,
DMSO-d6) δ 12.09 (br s, 1H, NH), 9.57 (br s, 1H, NH), 8.26 (s,1H,NH), 7.75 (d, 2H, J=8.0 Hz), 7.48 (d,

2H, J= 8.0 Hz), 6.81-6.78 (m, 2H), 6.69-6.61(m, 3H), 6.43 (d,1H, J=2. Hz), 4.91(dd, 1H, J=9.6 & 4.2
Hz), 3.44 (dd, 1H, J= 14.0 & 4.4 Hz), 3.19 (dd, 1H, J=14.0, 9.6 Hz), 2.50 (m, 2H), 1.49-1.35 (m, 8H).
N-(2-hydroxyethyl)-1-[4-(2,4-thiazolidindione-5-ylmethyl)-benzenesulfonylamino]-10H-phenoxazin-
3-sulfonamides (7l): yield 57%, off white solid, 232°C decomposition . 1H-NMR (400 MHz, DMSO-d6)
δ 12.09(br s,1H, NH), 9.53( br s, 1H,NH), 8.09(s, 1H,NH), 7.70(d, 2H, d, J=8.0 Hz), 7.30 (d, 2H, J=8.0
Hz), 7.29 (t,1H, NH, J= 5.6 Hz), 6.79-6.62(m, 6H), 4.87 (dd,1H, J=9.6 & 4.4 Hz), 4.69 (br s, 1H, OH),
3.45 (dd,1H, 14.0 & 4.4), 3.32(t, 2H, J=5.6 Hz), 3.17 ( dd, 1H, J= 14.0 & 9.6 Hz), 2.55 (q, 2H, J=5.6 Hz).
13C-NMR (100 MHz, DMSO-d6) δ 175.3, 171.3, 143.3, 142.7, 142.1, 137.7, 133.0, 129.9, 127.2, 124.3,
122.0, 121.3, 119.7, 115.0, 114.6, 111.2, 59.7, 51.8, 44.9, 37.0.
N-(4-hydroxyphenyl)-1-[4-(2,4-thiazolidindione-5-ylmethyl)-benzenesulfonylamino]-10H-
phenoxazin-3-sulfonamides (7m): yield 66%, off white solid, 239°C decomposition.
N-(2-methylphenyl)-1-[4-(2,4-thiazolidindione-5-ylmethyl)-benzenesulfonylamino]-10H-
phenoxazin-3-sulfonamides (7n): yield 61%, off white solid, 249°Cdecomposition.
N-(3,4-difluorophenyl)-1-[4-(2,4-thiazolidindione-5-ylmethyl)-benzenesulfonylamino]-10H-
phenoxazin-3-sulfonamid (7o): yield 69%, off white solid , mp 228°C decomposition.
V. Molecular Modeling Studies (TZD-PPAR γ):
The docking analysis of molecules was performed using Maestro, version 9.227
implemented from
Schrödinger molecular modeling suite.
Ligands preparation:
All molecules were sketched in 3D format using build module of maestro and LigPrep module was used
to produce low-energy conformers of the molecules. LigPrep produced a single, low-energy, 3D structure
with corrected chiralities for each successfully processed input structure.
Protein Preparation:
The crystal structure coordinates of Human PPARγ (PDB ID: 2PRG)28
were obtained from the protein
data bank. The protein was prepared by giving preliminary treatment like adding hydrogen, adding
missing residues, refining the loop with prime and finally minimized by using OPLS-2005 force field.
The grids for molecular docking were generated with bound co-crystallized ligand and extended up to 20
Å.
Docking study:

Molecules were docked using Glide in extra-precision mode29
with up to three poses saved per molecule.
Throughout the docking studies, ligands were kept flexible by producing the ring conformations and by
penalizing non-polar amide bond conformations, whereas the receptor was kept rigid. Finally, the
minimized poses are re-scored using Schrödinger’s proprietary GlideScore scoring function. The lowest
energy conformation was selected and, the ligand interactions (hydrogen bonding and hydrophobic
interaction) with the agonistic site of PPARγ were determined.
Conclusion:
Acknowledgments
The authors are thankful to Principal and Management of CKM Arts and Science College for
encouragement. The authors are also thankful to UGC, New Delhi for financial assistance.
References
1. UK Prospective Diabetic Study (UKPDS) Groups VIII. Diabetologia 1991, 34, 877.
2. Evans, A.J., Krentz, A.J, Prescriber 1999, 10, 51.
3. Spillar, H.A., Drug saf. 1998, 19, 411; Bailey C.J. Turner R.C. N.Engl.J.Med. 1996, 334, 574.
4. Hanefeld, M., J. diabetes complicat. 1998, 12, 228.; Yee H.S., Fong, N.T., Pharmacother. 1996,
16, 792.
5. (a) C.R. Kahn, L. Chen, S.E. Cohen., J. Clin. Invest. 2000, 106, 1305; (b) Imran, M., Ilyas, B.,
Depanjali, Khan, S.A., J. Sci. Ind. Res. 2007, 66, 99.; (c) Pastoromas, S., Koulouris, S., Hellinic J
Cardiol. 2006, 47, 352.
6. Cincotta, L., Foley, J.W., Cincotta, A. H., Cancer Res., 1993, 53, 2571.
7. Ruan, J. W., Huang. Z. S., Huang, J. F., Du C.J., Huang, S.L., Shi, Z., Fu L.W., Gu L. Q.,
Chinese Chem. Lett. 2006, 17,, 1141.
8. Bolognese, A., Correale, G., Manfra, M., Lavecchia, A., Novellino, E., Pepe, S., J. Med. Chem.,
2006, 49, 5110.
9. Fumika, S., Ken, H., Mariko, I., Gunnar, W., Kristin, S., Masami, K., Noboru, M., Hiroshi, S.,
Anticancer Res. 2007, 7, 4233..
10. Sanyang, G., Tomoko, T., Kiyonao, S., Jinsong, H., Chiseko, N., Naoko, H., Akio, T., Hirohei Y.,
B. J. Pharmacol. 2002, 137, 749.
11. Benjamin, B., Lawrence, L.B., J. Med. Chem. 1968, 11, 807.
12. Klabunde, T., Petrassi, H.M., Oza, V.B., Raman, P., Kelly, J.W., Sacchettini, J.C., Nat. Struct.
Biol. 2000, 7, 312.
13. Soren, E., Ingrid, P., Hanne, B. R., Heinz, J. D., Anette, F. J., Steen, B. M., Jan, F., Lone, P.,
Lars, N., Per ,S., J. Med. Chem., 2003, 46, 1306.

14. Thimmaiah, K.N, Easton, J.B., Germain, G.S, Morton, C.L., Kamath, S., Buolamwini, J.K.,
Houghton, P.J. J. Biol.Chem. 2005, 280, 31924.
15.
16. Ashcroft, F.M., Gribble, F.M., Diabetologia 1991, 42, 903.
17. Gribble, F.M., Reimann, F., Bio. Chem. Soc. Trans. 2002, 30, 333.
18. Groop, L.C., Diabetes Care 1992, 15, 1737.
19. Saltiel, A.R., Olefsky, J.M., Diabetes 1996, 45, 1661.
20. Henry, R.R., Endocrinol. Metab. Clin. North. Am. 1997, 26, 553.
21. Grant, R.N., Devita, N.G., Singer, D.E., Meigs, G.B., Diabetes care 2003, 26, 1408.
22. Autin, R.P., Diabetes Spectrum 2006, 19, 13.
23. UK Prospective Diabetes Study 17. Ann. Intem. Med., 1996, 124 , 136.
24. Sattigeri, Jitendra , A., PCT, WO 2005/058813 A2, 2005.
25. Reddy, S.V., Rao, G.M., Kumar, B.V., Meda, C.L., Deora, G.S., Kumar, K.S., Parsa, K.V., Pal,
M. Bioorg.Med.Chem. 2013, 21, 1952.
26. Pattan, S.R., Kekare, P., Dighe, N.S., Bhawar, S.B., Nikalje, A., Pati, A., Hole, M.B., Asian J.
Res. Chem. 2009, 2, 123.
27. Maestro, version 9.2; Schrodinger, LLC: New York, NY, 2012.
28. Robert, T. N., Bruce, W.G., Stefan, W., Jeffery, E. C., Millard, H. L., Riki, K., Michael, G. R.,
Timothy, M. W., Christopher, K. G., Michael, V. M., Nature 1998, 395, 137.
29. Glide, version 5.7; Schrodinger, LLC: New York, NY, 2012.

This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/copyright

Author's personal copy
Novel imidazophenoxazine-4-sulfonamides: Their synthesisand evaluation as potential inhibitors of PDE4
Seelam Venkata Reddy a, Gangula Mohan Rao a, Baru Vijaya Kumar a,⇑, Chandana L. T. Meda b,Girdhar Singh Deora b, K. Shiva Kumar b, Kishore V. L. Parsa b, Manojit Pal b,⇑a Medicinal Chemistry Laboratory, Research Centre, C.K.M. Arts and Science College, Warangal 506 006, Andhra Pradesh, Indiab Institute of Life Sciences, University of Hyderabad Campus, Gachibowli, Hyderabad 500 046, India
a r t i c l e i n f o
Article history:Received 4 December 2012Revised 11 January 2013Accepted 12 January 2013Available online 26 January 2013
Keywords:ImidazophenoxazineSulfonamidePDE4Docking
a b s t r a c t
A number of novel imidazophenoxazine-4-sulfonamides have been designed as potential inhibitors ofPDE4. All these compounds were readily prepared via an elegant multi-step method involving the initialconstruction of 1-nitro-10H-phenoxazine ring and then fused imidazole ring as key steps. Some of thesecompounds showed promising PDE4B and D inhibition when tested in vitro and good interactions withthese proteins in silico. Three of these compounds showed dose dependent inhibition of PDE4B with IC50
value of 3.31 ± 0.62, 1.23 ± 0.18 and 0.53 ± 0.18 lM.� 2013 Elsevier Ltd. All rights reserved.
1. Introduction
Due to their interesting pharmacological properties, compoundscontaining the phenoxazine framework (A, Fig. 1) have attractedconsiderable attention in medicinal chemistry. A number of com-pounds based on A have been reported to be potent anti-prolifera-tive agents.1 For example 5H-pyrido[2,3-a]phenoxazin-5-one (B,Fig. 1) that belongs to this class has shown promising anti-prolifer-ative activities when tested against human neoplastic cell lines.2 Aphenoxazine based dual PPAR agonist for example, DRF 2725 (C,Fig. 1) has shown potent antihyperglycemic and lipid modulatingproperties.3 In view of the pharmacological importance of phenox-azine derivatives and our long standing interest in identification ofnovel phosphodiesterase 4 (PDE4) inhibitors4 we became inter-ested in assessing PDE4 inhibiting properties of small moleculesbased on phenoxazine fused with an imidazole ring for example,imidazo[4,5,1-kl]phenoxazine. In inflammatory and immune cells,the inhibition of cellular responses, including the productionand/or release of proinflammatory mediators, cytokines, and activeoxygen species, is associated with elevated levels of cAMP. PDE4exists in four different isoforms for example, PDE4A, B, C and Dand plays a key role in the hydrolysis of cAMP to AMP.5 Thus, inhi-
bition of PDE4 results in elevated levels of cAMP in the airway tis-sues and cells thereby suppressing inflammatory cell functions.This is supported by the fact that PDE4 inhibitors have been foundto be beneficial for the treatment of inflammatory and immunolog-ical diseases including asthma and chronic obstructive pulmonarydisease (COPD). Notably, rolipram the first-generation PDE4 inhib-itor showed adverse effects such as nausea and vomiting.5 More re-cently, cardiovascular effects of PDE4 inhibitors have beenreported.6 While these dose-limiting side effects were reduced bysecond-generation inhibitors like cilomilast7a (Ariflo) and roflumi-last, their therapeutic index has delayed market launch so far.While roflumilast (Daxas�, Nycomed) has been launched in Europefor the treatment of COPD recently it is however, necessary to de-vote a continuing effort in exploring new class of compounds fortheir PDE4 inhibitory potential. Additionally, the improvement offasting blood glucose and hemoglobin A1C levels shown by roflu-milast during its clinical studies in patients with type 2 diabetes7b
and the recent report7c of resveratrol-PDE link have generated re-newed interest in the discovery and development of new PDE4inhibitor. The design of our target compounds (E, Fig. 2) was per-formed by incorporating a phenoxazine ring (A) into thebenzo[d]imidazol based known inhibitors8a of PDE4 (D, Fig. 2). Asulfonamide group was introduced at C-4 of the resulting imi-dazo[4,5,1-kl]phenoxazine moiety with the anticipation that thisgroup might induce anti-inflammatory8b as well as favorable druglike properties8c within the molecule. Herein we report imi-dazo[4,5,1-kl]phenoxazine-4-sulfonamide as a new template for
0968-0896/$ - see front matter � 2013 Elsevier Ltd. All rights reserved.http://dx.doi.org/10.1016/j.bmc.2013.01.023
⇑ Corresponding authors. Tel.: +91 8008098787; fax: +91 40 6657 1581 (B.V.K.);tel.: +91 40 6657 1500; fax: +91 40 6657 1581 (M.P.).
E-mail addresses: [email protected] (B.V. Kumar), [email protected] (M. Pal).
Bioorganic & Medicinal Chemistry 21 (2013) 1952–1963
Contents lists available at SciVerse ScienceDirect
Bioorganic & Medicinal Chemistry
journal homepage: www.elsevier .com/locate /bmc
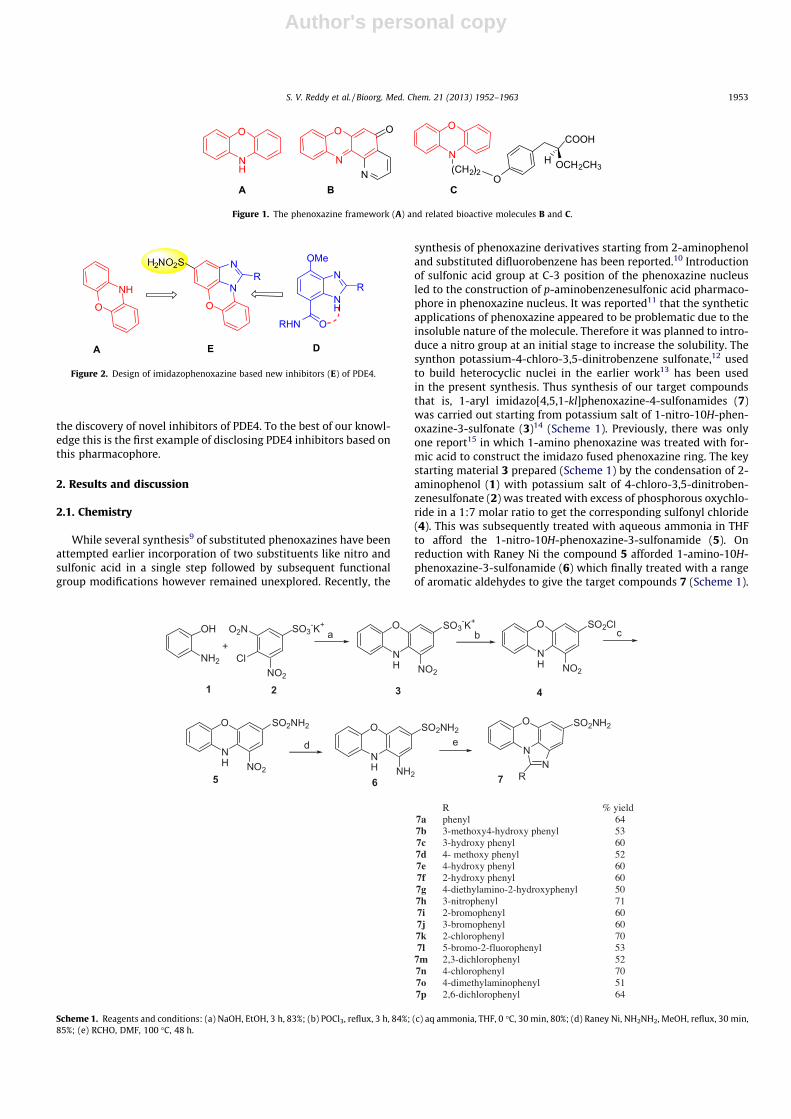
Author's personal copy
the discovery of novel inhibitors of PDE4. To the best of our knowl-edge this is the first example of disclosing PDE4 inhibitors based onthis pharmacophore.
2. Results and discussion
2.1. Chemistry
While several synthesis9 of substituted phenoxazines have beenattempted earlier incorporation of two substituents like nitro andsulfonic acid in a single step followed by subsequent functionalgroup modifications however remained unexplored. Recently, the
synthesis of phenoxazine derivatives starting from 2-aminophenoland substituted difluorobenzene has been reported.10 Introductionof sulfonic acid group at C-3 position of the phenoxazine nucleusled to the construction of p-aminobenzenesulfonic acid pharmaco-phore in phenoxazine nucleus. It was reported11 that the syntheticapplications of phenoxazine appeared to be problematic due to theinsoluble nature of the molecule. Therefore it was planned to intro-duce a nitro group at an initial stage to increase the solubility. Thesynthon potassium-4-chloro-3,5-dinitrobenzene sulfonate,12 usedto build heterocyclic nuclei in the earlier work13 has been usedin the present synthesis. Thus synthesis of our target compoundsthat is, 1-aryl imidazo[4,5,1-kl]phenoxazine-4-sulfonamides (7)was carried out starting from potassium salt of 1-nitro-10H-phen-oxazine-3-sulfonate (3)14 (Scheme 1). Previously, there was onlyone report15 in which 1-amino phenoxazine was treated with for-mic acid to construct the imidazo fused phenoxazine ring. The keystarting material 3 prepared (Scheme 1) by the condensation of 2-aminophenol (1) with potassium salt of 4-chloro-3,5-dinitroben-zenesulfonate (2) was treated with excess of phosphorous oxychlo-ride in a 1:7 molar ratio to get the corresponding sulfonyl chloride(4). This was subsequently treated with aqueous ammonia in THFto afford the 1-nitro-10H-phenoxazine-3-sulfonamide (5). Onreduction with Raney Ni the compound 5 afforded 1-amino-10H-phenoxazine-3-sulfonamide (6) which finally treated with a rangeof aromatic aldehydes to give the target compounds 7 (Scheme 1).
NH
NR
OMe
ORHN
D
N
O
NR
E
NH
O
A
Figure 2. Design of imidazophenoxazine based new inhibitors (E) of PDE4.
N
O
NH
O O
NN
O
(CH2)2O
H OCH2CH3
COOH
A CB
Figure 1. The phenoxazine framework (A) and related bioactive molecules B and C.
ClNO2
O2N SO3-K+
NH
O SO3-K+
NO2
+NH
O SO2Cl
NO2
N
O SO2NH2
NR
1 2 3 4
6
a b c
dNH
O SO2NH2
NO25
NH
O SO2NH2
NH2
e
7
OH
NH2
yield%R7a 46phenyl7b 3-methoxy4-hydroxy phenyl 53 7c 3-hydroxy phenyl 60 7d 4- methoxy phenyl 52 7e 4-hydroxy phenyl 60 7f 2-hydroxy phenyl 60 7g 4-diethylamino-2-hydroxyphenyl 50 7h 17lynehportin-37i 06lynehpomorb-27j 06lynehpomorb-37k 07lynehporolhc-27l 5-bromo-2-fluorophenyl 53
7m 2,3-dichlorophenyl 52 7n 70lynehporolhc-47o 4-dimethylaminophenyl 51 7p 2,6-dichlorophenyl 64
Scheme 1. Reagents and conditions: (a) NaOH, EtOH, 3 h, 83%; (b) POCl3, reflux, 3 h, 84%; (c) aq ammonia, THF, 0 �C, 30 min, 80%; (d) Raney Ni, NH2NH2, MeOH, reflux, 30 min,85%; (e) RCHO, DMF, 100 �C, 48 h.
S. V. Reddy et al. / Bioorg. Med. Chem. 21 (2013) 1952–1963 1953

Author's personal copy
One of the target compounds 7a was also prepared via an alter-native route (Scheme 2). Thus, the compound 3 was reduced to thepotassium salt of 1-amino-10H-phenoxazine-3-sulfonate (8) usingRaney Ni, which was treated with benzaldehyde to give the potas-sium salt of 1-phenylimidazo[4,5,1-kl]phenoxazine-4-sulfonates(9). On treating with POCl3 the compound 9 provided the corre-sponding chloro derivative 10 which on treatment with aqueousammonia in THF afforded the compound 7a.16
2.2. Pharmacology
Having synthesized a range of 1-substituted imidazo[4,5,1-kl]phenoxazine-4-sulfonamides many of these compounds weretested initially for their PDE4B inhibitory properties in vitro at30 lM using PDE4B enzyme assay17 (Table 1). Rolipram18 was usedas a reference compound in this assay. Except 7g (along with 7m and7o) all other compounds showed significant inhibition of PDE4B andcompounds 7f, 7j and 7l showed superior inhibitions in compared toother molecules when tested at 30 lM (Table 1). The initial com-pound 7a showed good inhibition of PDE4B which though was notaffected by the addition of OH and OMe substituents to the C-1 ben-zene ring for example, compound 7b (Table 1, entry 2) but improvedupon addition of m-OH group to the C-1 benzene ring for example,compound 7c (Table 1). However, a p-MeOC6H4 moiety at C-1 de-creased the activity for example, compound 7d (Table 1, entry 4)whereas a p-HOC6H4 or o-HOC6H4 moiety at C-1 restored the activityfor example, compound 7e and 7f (Table 1, entries 5 and 6). A bulkiergroup that is, p-(Et2N)-o-(HO)C6H3 at C-1 was not tolerated forexample, compound 7g (Table 1, entry 7) and a m-NO2C6H4 moietyat C-1 decreased the activity (Table 1, entry 8). Interestingly, both
the o-BrC6H4 and m-BrC6H4 group at C-1 was well tolerated forexample, compounds 7i and 7j (Table 1, entries 9 and 10). Whileo-ClC6H4 group at C-1 decreased the activity marginally for example,compound 7k (Table 1, entry 11) a 2-F-5-BrC6H3 or p-ClC6H4 or 2,6-dichloro phenyl at the same position was well tolerated (Table 1,entries 12–14). Based on their promising inhibitory properties doseresponse studies were carried out using most active compounds 7f,7j and 7l. All of them showed dose dependent inhibition of PDE4with IC50 value of 3.31 ± 0.62, 1.23 ± 0.18 and 0.53 ± 0.18 lM,respectively (Figs. 3–5) in compared to rolipram’s IC50 value of0.941 ± 0.24 lM (Fig. 6). Thus the compound 7l was identified asthe most potent compound in this series. To assess the other subtypeinhibitory potential of 7 few selected compounds were testedagainst PDE4D when compounds 7f, 7g, 7h, and 7l showed 88.4%,16.9%, 68.6%, and 85.3% inhibition, respectively.
2.3. Docking studies
In order to understand the nature of interactions of these mol-ecules with PDE4B docking studies were carried out using com-pounds 7f, 7j and 7l. The XP (extra precision) docking wasperformed for all the molecules using glide module of Schrödinger2011. The glide scores and other parameters obtained after dockingof these molecules into the PDE4B protein are summarized in Ta-ble 2. The data shown in Table 2 suggests that these molecules bindwell with PDE4B. The interaction of compound 7f with the PDE4Bprotein (Fig. 7) was mainly contributed by (i) a H-bonding betweenthe amino group of 7f and His-278, (ii) a H-bonding between thehydroxyl group of 7f and Asp-392 and (iii) two P–P stacking inter-actions between the benzoimidazole moiety and His234 of the pro-tein. Similarly, the interaction of compound 7j with the PDE4Bprotein (Fig. 8) was contributed by (i) H-bonding between theNH2-group of 7j and Thr-345 as well as Asp-392, (ii) P–P stackinginteraction between the central 1,4-oxazine ring and tyrosine(Tyr233) and (iii) P–P stacking interaction between the phenylgroup of 7j and phenylalanine (Phe446). The interaction of com-pound 7l with the PDE4B protein (Fig. 9) was mainly contributedby (i) a H-bonding between the amino group of 7l and Asp-275and (ii) P–P stacking interaction between aromatic ringsystems of 7l and Tyr233, His234 and Phe446. Overall, the presentimidazophenoxazine-4-sulfonamides showed good interactions
NH
O SO3-K+
NH2
3
8
a b
N
O SO3-K+
NPh
9
N
O SO2Cl
NPh
c d
10
7a
Scheme 2. Reagents and conditions: (a) Raney Ni, NH2NH2, MeOH, reflux, 30 min, 77%; (b) PhCHO, DMF, 100 �C, 24 h, 52%; (c) POCl3, reflux, 3 h; (d) aq ammonia, THF, 0 �C,30 min, 50%.
Table 1Inhibition of PDE4B by compound 7 at 30 lM
N
O SO2NH2
NR
7
Entry Compounds R = Average %inhibition
SD
1 7a Phenyl 79.65 1.162 7b 3-Methoxy4-hydroxy
phenyl82.43 1.48
3 7c 3-Hydroxy phenyl 87.58 0.544 7d 4-Methoxy phenyl 66.53 4.165 7e 4-Hydroxy phenyl 88.80 0.926 7f 2-Hydroxy phenyl 89.28 3.317 7g 4-Diethylamino-2-
hydroxyphenyl19.66 1.43
8 7h 3-Nitrophenyl 66.81 2.569 7i 2-Bromophenyl 81.02 1.1710 7j 3-Bromophenyl 89.69 1.2311 7k 2-Chlorophenyl 76.32 0.8612 7l 5-Bromo-2-fluorophenyl 92.86 0.5313 7n 4-Chlorophenyl 80.35 1.3214 7p 2,6-Dichlorophenyl 80.23 2.15
SD = standard deviation. Figure 3. Dose dependent inhibition of PDE4B by compound 7f.
1954 S. V. Reddy et al. / Bioorg. Med. Chem. 21 (2013) 1952–1963

Author's personal copy
with PDE4B protein where the central phenoxazine ring and sul-fonamide moiety played a key role in these interactions. Notably,the observed dissimilar orientation of binding of 7f compared to7j and 7l was possibly aided by the strong H-bonding betweenthe hydroxyl group of 7f and Asp-392 of the PDE4B protein. Dock-ing of all these molecules into the PDE4D suggested that they bindwell with this protein (see Supplementary data) which correlatedthe results of their PDE4D inhibitions in vitro. Thus, the aminogroup of sulfonamide moiety of 7f interacted with Asp-201 and318 and the hydroxyl group of 7f formed an H-bond with Asn321. Aromatic rings of molecules 7j participated in P–P stackingwith Tyr-159, His-160 and Phe-372. The amino group of same mol-ecule formed a hydrogen bond with Thr-271. In case of molecule 7l
P–P stacking with Tyr-159 and hydrogen bonding with Asp-318and Thr-271 was observed (see Figs. in Supplementary data).
3. Conclusions
In conclusion, a number of novel imidazophenoxazine-4-sulfon-amides have been designed and synthesized as potential inhibitorsof PDE4. All these compounds were readily prepared via a multi-step method starting from potassium-4-chloro-3,5-dinitro ben-zene sulfonate involving the construction of 1-nitro-10H-phenoxa-zine ring and then fused imidazole ring as key steps. Some of thesecompounds showed promising PDE4B and D inhibition whentested in vitro and good interactions with these proteins in silico.The docking studies indicated that the central phenoxazine ringand sulfonamide moiety played a key role in these interactions.In a dose response study three compounds showed dose dependentinhibition of PDE4B with IC50 value of 3.31 ± 0.62, 1.23 ± 0.18 and0.53 ± 0.18 lM. Overall, the imidazophenoxazine-4-sulfonamideframework presented here could be a new template for the identi-fication of small molecule based novel inhibitors of PDE4.
4. Experimental
4.1. Chemistry
4.1.1. General methodsUnless stated otherwise, reactions were performed under nitrogen
atmosphere. Reactions were monitored by thin layer chromatography(TLC) on silica gel plates (60 F254), visualizing with ultraviolet light oriodine spray. Flash chromatography was performed on silica gel (60–120 mesh) using hexane, ethyl acetate, dichloromethane, methanol.1H NMR and 13C NMR spectra were determined in DMSO-d6 solutionby using 400 and 100 MHz spectrometers, respectively. Proton chem-ical shifts (d) are relative to tetramethylsilane (TMS, d = 0.00) as inter-nal standard and expressed in ppm. Spin multiplicities are given as s(singlet), d (doublet), t (triplet) and m (multiplet) as well as b (broad).Coupling constants (J) are given in hertz. Infrared spectra were re-corded on a FT-IR spectrometer. Melting points were determined usingmelting point apparatus and are uncorrected. MS spectra were ob-tained on a mass spectrometer.
4.1.2. Potassium-4-chloro-3,5-dinitrobenzenesulfonate (2)
ClNO2
O2N SO3-K+
Chlorobenzene (50 ml) was added to a mixture of fuming sulfuricacid (260 ml) and sulfuric acid (60 ml) at 70 �C. The mixture wasstirred at same temperature for 1 h. Potassium nitrate (50 g) wasadded portion wise to the reaction mixture for about 15 min, andthen fuming sulfuric acid (130 ml) was added followed by 2 more
Figure 4. Dose dependent inhibition of PDE4B by compound 7j.
Figure 5. Dose dependent inhibition of PDE4B by compound 7l.
Figure 6. Dose dependent inhibition of PDE4B by rolipram.
Table 2Glide scores and other parameters of compounds after docking with PDE4B
Entry Compound Dock score E-1a E-2b E-3c E-4d
1 7f �6.18 �2.95 �1.4 �0.59 �1.052 7j �7.84 �3.82 0 �2.88 �0.973 7l �8.19 �3.99 �0.22 �2.82 �0.88
a Chemscore lipophilic pair term and fraction of the total protein–ligand vdwenergy reward.
b Hydrophobic enclosure reward.c Electrostatic reward.d Rewards for hydrogen bonding interaction between ligand and protein.
S. V. Reddy et al. / Bioorg. Med. Chem. 21 (2013) 1952–1963 1955

Author's personal copy
portions of potassium nitrate (50 + 50 g) portion wise. The reactionmass was stirred at 130 �C for 1 h, cooled to room temperature,poured in excess crushed ice and allowed to stand for 12 h. The solidwas filtered and washed with cold water (3 � 200 ml), dried andwashed three times with hot toluene (3 � 200 ml) and dried, yield56% (80 g) of pure product; mp 298–300 �C (lit.12 290 �C).
4.1.3. Potassium-1-nitro-10H-phenoxazine-3-sulfonate (3)
NH
O SO3-K+
NO2
To a solution of sodium hydroxide (7.5 g, 0.187 mol) inethanol (500 ml) was added 2-aminophenol (17 g, 0.156mol) and potassium-4-chloro-3,5-dinitrobenzenesulfonate (50 g,0.156 mol). The reaction mixture was refluxed for 1 h. Then asolution of sodium hydroxide (3.75 g) in water (5 ml) was addedto the reaction mass and refluxed for two more hours. Completionof the reaction was monitored by TLC. The reaction mass was cooledto room temperature and the solid was filtered and washed twicewith ethanol (2 � 200 ml) and dried, yield 83% (45 g); mp >300 �C;m/z (CI) 307 (M-K, 100); 1H NMR (400 MHz, DMSO-d6) d 9.45 (s,1H, NH), 7.73 (s, 1H), 7.20 (dd, J = 2 and 6.8 Hz, 1H,), 7.01–6.9 (m,1H), 6.94–6.36 (m, 3H); IR (KBr, cm�1) vmax: 3341, 1531, 1203,1052, 745.
Figure 7. Docking of 7f at the active site of PDE4B.
1956 S. V. Reddy et al. / Bioorg. Med. Chem. 21 (2013) 1952–1963

Author's personal copy
4.1.4. 1-Nitro-10H-phenoxazine-3-sulfonylchloride (4)
NH
O SO2Cl
NO2
Potassium-1-nitro-10H-phenoxazine-3-sulfonate (10 g) inPOCl3 (70 ml) was refluxed for 3 h. Completion of the reactionwas monitored by TLC. The excess of POCl3 was removed by distil-lation, the crude cherry red coloured solid was poured in crushedice and filtered, washed with water, cold methanol and dried,
and yield 8 g (84%); mp 204 �C (decomposition) and the productwas immediately used for the next step.
4.1.5. 1-Nitro-10H-phenoxazine-3-sulfonamide (5)
NH
O SO2NH2
NO2
To a solution of aqueous ammonia (10 ml) in tetrahydrofuran(50 ml) was added 1-nitro-10H-phenoxazine-3-sulfonylchloride
Figure 8. Docking of 7j at the active site of PDE4B.
S. V. Reddy et al. / Bioorg. Med. Chem. 21 (2013) 1952–1963 1957
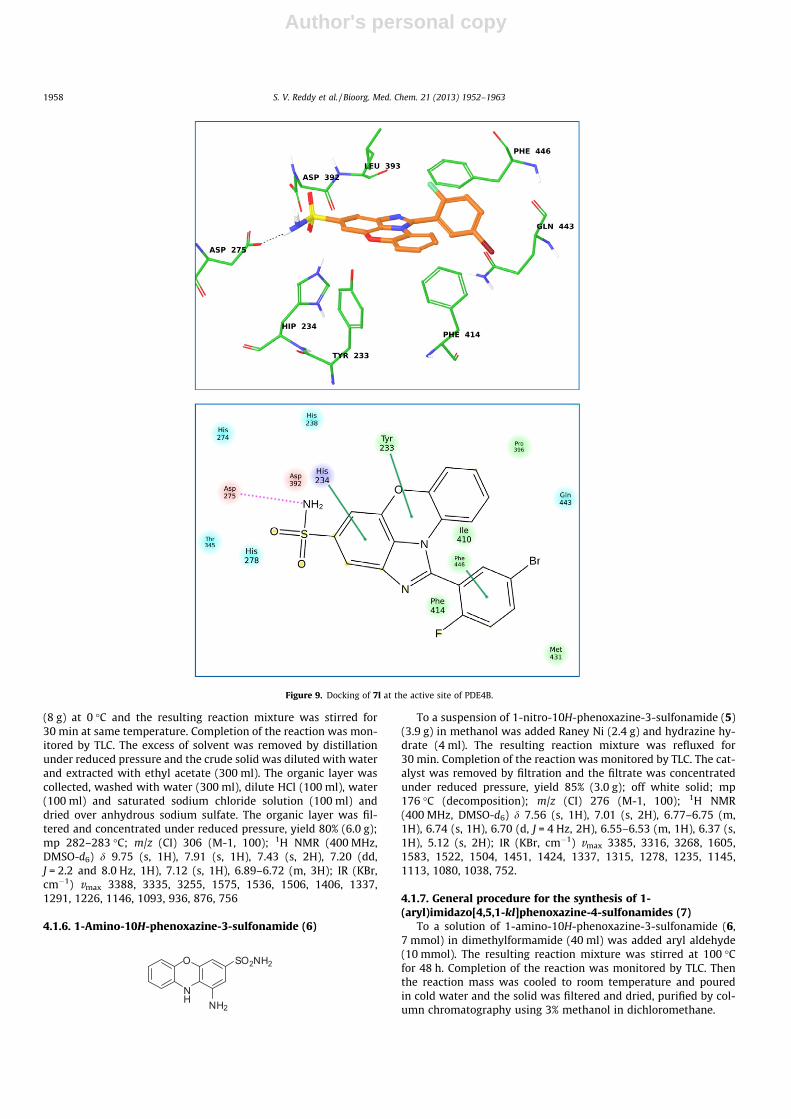
Author's personal copy
(8 g) at 0 �C and the resulting reaction mixture was stirred for30 min at same temperature. Completion of the reaction was mon-itored by TLC. The excess of solvent was removed by distillationunder reduced pressure and the crude solid was diluted with waterand extracted with ethyl acetate (300 ml). The organic layer wascollected, washed with water (300 ml), dilute HCl (100 ml), water(100 ml) and saturated sodium chloride solution (100 ml) anddried over anhydrous sodium sulfate. The organic layer was fil-tered and concentrated under reduced pressure, yield 80% (6.0 g);mp 282–283 �C; m/z (CI) 306 (M-1, 100); 1H NMR (400 MHz,DMSO-d6) d 9.75 (s, 1H), 7.91 (s, 1H), 7.43 (s, 2H), 7.20 (dd,J = 2.2 and 8.0 Hz, 1H), 7.12 (s, 1H), 6.89–6.72 (m, 3H); IR (KBr,cm�1) vmax 3388, 3335, 3255, 1575, 1536, 1506, 1406, 1337,1291, 1226, 1146, 1093, 936, 876, 756
4.1.6. 1-Amino-10H-phenoxazine-3-sulfonamide (6)
NH
O SO2NH2
NH2
To a suspension of 1-nitro-10H-phenoxazine-3-sulfonamide (5)(3.9 g) in methanol was added Raney Ni (2.4 g) and hydrazine hy-drate (4 ml). The resulting reaction mixture was refluxed for30 min. Completion of the reaction was monitored by TLC. The cat-alyst was removed by filtration and the filtrate was concentratedunder reduced pressure, yield 85% (3.0 g); off white solid; mp176 �C (decomposition); m/z (CI) 276 (M-1, 100); 1H NMR(400 MHz, DMSO-d6) d 7.56 (s, 1H), 7.01 (s, 2H), 6.77–6.75 (m,1H), 6.74 (s, 1H), 6.70 (d, J = 4 Hz, 2H), 6.55–6.53 (m, 1H), 6.37 (s,1H), 5.12 (s, 2H); IR (KBr, cm�1) vmax 3385, 3316, 3268, 1605,1583, 1522, 1504, 1451, 1424, 1337, 1315, 1278, 1235, 1145,1113, 1080, 1038, 752.
4.1.7. General procedure for the synthesis of 1-(aryl)imidazo[4,5,1-kl]phenoxazine-4-sulfonamides (7)
To a solution of 1-amino-10H-phenoxazine-3-sulfonamide (6,7 mmol) in dimethylformamide (40 ml) was added aryl aldehyde(10 mmol). The resulting reaction mixture was stirred at 100 �Cfor 48 h. Completion of the reaction was monitored by TLC. Thenthe reaction mass was cooled to room temperature and pouredin cold water and the solid was filtered and dried, purified by col-umn chromatography using 3% methanol in dichloromethane.
Figure 9. Docking of 7l at the active site of PDE4B.
1958 S. V. Reddy et al. / Bioorg. Med. Chem. 21 (2013) 1952–1963

Author's personal copy
4.1.8. 1-Phenylimidazo[4,5,1-kl]phenoxazine-4-sulfonamide (7a)
N
O S
N
OO
NH2
Compound 7a was prepared by using 1-amino-10H-phenoxa-zine-3-sulfonamide (6) (1.4 g, 5 mmol) and benzaldehyde (0.64 g,6 mmol) in DMF according to the general procedure as an ash col-oured solid; yield 64% (1.2 g); mp 316–317 �C; m/z (CI) 364 (M+1,100); 1H NMR (400 MHz, DMSO-d6) d 7.81 (d, J = 6.8 Hz, 2H), 7.68–7.62 (m, 4H), 7.39 (s, 2H), 7.27 (d, J = 8 Hz, 1H), 7.18 (t, J = 8 Hz,1H,), 7.13 (s, 1H), 6.94 (t, J = 8 Hz, 1H), 6.89 (d, J = 8 Hz, 1H); 13CNMR (100 MHz, DMSO-d6) d 150.2, 144.8, 141.7, 140.8, 130.8,130.2, 129.5, 129.0, 128.8, 127.6, 126.1, 124.9, 124.4, 118.5,116.1, 111.2, 102.4; IR (KBr, cm�1) vmax 3344, 3024, 1662, 1494,1454, 1380, 1336, 1299, 1267, 1148, 1096, 751.
4.1.9. 1-(4-Hydroxy-3-methoxyphenyl)imidazo[4,5,1-kl]phenoxazine-4-sulfonamide (7b)
N
O SOO
NH2
HO OCH3
N
Compound 7b was prepared by using 1-amino-10H-phenoxazine-3-sulfonamide (6) (1.4 g, 5 mmol) and 4-hydroxy-3-methoxybenzal-dehyde (1.12 g, 7.5 mmol) in DMF according to the general procedureas an off white solid, yield 53% (1.23 g); mp 294–296 �C; m/z (CI) 408(M-1, 100); 1H NMR (400 MHz, DMSO-d6) d 9.77 (s, 1H), 7.64 (s, 1H),7.38 (s, 2H), 7.34 (d, J = 2 Hz, 1H), 7.26–7.16 (m, 3H), 7.114 (s, 1H),7.075–6.981 (m, 3H), 3.80 (s, 3H); 13C NMR (100 MHz, DMSO-d6) d150.8, 148.9, 147.6, 144.9, 141.8, 141.6, 140.8, 127.5, 126.1, 125.2,124.5, 123.5, 122.7, 120.6, 118.4, 116.4, 115.6, 113.4, 111.8, 102.3,55.8; IR (KBr, cm�1) vmax 3343, 3064, 1662, 1499, 1459, 1424,1328, 1303, 1235, 1151, 1032, 936, 875, 787, 760.
4.1.10. 1-(3-Hydroxyphenyl)imidazo[4,5,1-kl]phenoxazine-4-sulfonamide (7c)
N
O S
N
O O
NH2
OH
Compound 7c was prepared by using 1-amino-10H-phenoxa-zine-3-sulfonamide (6) (1.4 g, 5 mmol) and 3-hydroxy benzalde-hyde (0.98 g, 8.1 mmol) in DMF according to the generalprocedure as an off white solid, yield 60% (1.2 g); mp 273–276 �C;m/z (CI) 378 (M-1, 100); 1H NMR (400 MHz, DMSO-d6) d 9.94 (s,1H, OH), 7.66 (s, 1H), 7.43 (t, J = 8.0 Hz, 1H,), 7.38 (s, 2H, SO2NH2),7.26 (d, J = 7.6 Hz, 1H), 7.19–7.12 (m, 4H), 7.05 (dd, J = 1.6 and8.0 Hz, 1H,), 6.98 (m, 2H); IR (KBr, cm�1) vmax 3324, 3246, 3115,1662, 1582, 1493, 1453, 1382, 1308, 1266, 1163,1151, 1102, 755.
4.1.11. 1-(4-Methoxyphenyl)imidazo[4,5,1-kl]phenoxazine-4-sulfonamide (7d)
N
O S
N
O O
NH2
H3CO
Compound 7d was prepared by using 1-amino-10H-phenoxa-zine-3-sulfonamide (6) (2.0 g, 7.2 mmol) and 4-methoxybenzalde-hyde (1.5 g, 10 mmol) in DMF according to the general procedureas an off white solid; yield 52% (1.5 g); mp 302–303 �C; m/z (CI)392 (M-1, 100); 1H NMR (400 MHz, DMSO-d6) d 7.73 (d,J = 8.8 Hz, 2H), 7.66 (s, 1H), 7.39 (s, 2H, SO2NH2), 7.23 (d,J = 8.0 Hz, 1H), 7.18–7.13 (m, 3H), 7.12 (s, 1H), 6.99–6.93 (m, 2H),3.89 (3H); 13C NMR (100 MHz, DMSO-d6) d 161.0, 150.4, 145.0,141.9, 141.6, 140.9, 131.1, 127.6, 126.2, 125.1, 124.5, 122.2,118.5, 116.2, 114.3, 111.0, 102.3, 55.4; IR (KBr, cm�1) vmax 3385,3116, 1664, 1613, 1584, 1495, 1378, 1301, 1229, 1208, 1128,1079, 750.
4.1.12. 1-(4-Hydroxyphenyl)imidazo[4,5,1-kl]phenoxazine-4-sulfonamide (7e)
N
O S
N
O O
NH2
HO
Compound 7e was prepared by using 1-amino-10H-phenoxa-zine-3-sulfonamide (6) (1.5 g, 5.4 mmol) and 4-hydroxybenzalde-hyde (0.99 g, 8.1 mmol) in DMF according to the generalprocedure as an off white solid, yield 60% (1.2 g); mp 323–325 �C; m/z (CI) 378 (M-1, 100); 1H NMR (400 MHz, DMSO-d6)d 10.16 (s, 1H), 7.63–7.60 (m, 3H), 7.37 (s, 2H), 7.26–7.18 (m,2H), 7.10 (s, 1H), 7.02–6.97 (m, 4H); IR (KBr, cm�1) vmax 3323,3243, 3117, 1663, 1588, 1497, 1452, 1382, 1308, 1266, 1151,1102, 795.
4.1.13. 1-(2-Hydroxyphenyl)imidazo[4,5,1-kl]phenoxazine-4-sulfonamide (7f)
N
O S
N
O O
NH2
OH
Compound 7f was prepared by using 1-amino-10H-phenoxazine-3-sulfonamide (6) (1.4 g, 5.4 mmol) and 2-hydroxy-benzaldehyde (0.98 g, 8.1 mmol) in DMF according to thegeneral procedure as an off white solid, yield 60% (1.2 g); mp308–310 �C; m/z (CI) 378 (M-1, 100); 1H NMR (400 MHz,DMSO-d6) d 10.25 (s, 1H), 7.66 (s, 1H), 7.53–7.50 (m, 2H), 7.38(s, 2H), 7.25–7.23 (m, 1H), 7.18–6.95 (m, 5H), 6.82 (d,J = 7.6 Hz, 1H); IR (KBr, cm�1) vmax 3329, 3236, 3119, 1661,1572, 1463, 1463, 1392, 1318, 1266, 1151, 1102, 1023, 962,853,789, 744, 667.
S. V. Reddy et al. / Bioorg. Med. Chem. 21 (2013) 1952–1963 1959
Author's personal copy
4.1.14. 1-(4-(Diethylamino)-2-hydroxyphenyl)imidazo[4,5,1-kl]phenoxazine-4-sulfonamide (7g)
N
O S
N
O O
NH2
OH
N
Compound 7g was prepared by using 1-amino-10H-phenoxa-zine-3-sulfonamide (6) (1.5 g, 5.4 mmol) and 4-diethylamino-2-hydroxybenzaldehyde (1.2 g, 6.4 mmol) in DMF according to thegeneral procedure as an off white solid; yield 50% (1.2 g); mp280–282 �C; m/z (CI) 449 (M-1, 100); 1H NMR (400 MHz, DMSO-d6) d 9.69 (1H), 7.65 (s, 1H), 7.35 (s, 2H), 7.31 (d, J = 7.6 Hz, 1H),7.19–7.12 (m, 4H), 6.51–6.4 (m, 3H), 3.4 (q, J = 7.2 Hz, 4H), 1.0 (t,J = 7.2 Hz, 6H); IR (KBr, cm�1) vmax 3332, 3233, 3117, 1661, 1562,1473, 1389, 1321, 1257, 1151, 1102, 1015, 755.
4.1.15. 1-(3-Nitrophenyl)imidazo[4,5,1-kl]phenoxazine-4-sulfonamide (7h)
N
O S
N
O O
NH2
NO2
Compound 7h was prepared by using 1-amino-10H-phenoxa-zine-3-sulfonamide (6) (1.45 g, 5.2 mmol) and 3-nitrobenzalde-hyde (1.18 g, 7.8 mmol) in DMF according to the generalprocedure as an off white solid, yield 71% (1.5 g); mp 332–335 �C; m/z (CI) 407 (M-1, 100); 1H NMR (400 MHz, DMSO-d6) d8.67 (s, 1H), 8.52 (d, J = 9.6 Hz, 1H), 8.29 (d, J = 8.0 Hz, 1H), 7.94(t, J = 8.0 Hz, 1H), 7.70 (s, 1H), 7.41 (s, 2H), 7.30–7.17 (m, 3H),6.97–6.89 (m, 2H); IR (KBr, cm�1) vmax 3344, 3024, 1654, 1540,1491, 1350, 1304, 1195, 1077, 1036, 734.
4.1.16. 1-(2-Bromophenyl)imidazo[4,5,1-kl]phenoxazine-4-sulfonamide (7i)
N
O S
N
OO
NH2
Br
Compound 7i was prepared by using 1-amino-10H-phenoxa-zine-3-sulfonamide (6) (1.5 g, 5.4 mmol) and 2-bromobenzalde-hyde (1.2 g, 6.4 mmol) in DMF according to the generalprocedure as an off white solid, yield 60% (1.4 g); mp 310–312 �C; m/z (CI) 440 (M-2, 100); 1H NMR (400 MHz, DMSO-d6) d7.94–7.92 (m, 1H), 7.80–7.77 (m, 1H), 7.71–7.66 (m, 3H), 7.40 (s,2H), 7.29–7.27 (m, 1H), 7.21–7.16 (m, 2H), 6.95 (t, J = 8.0 Hz, 1H),6.41 (d, J = 8.0 Hz, 1H); IR (KBr, cm�1) vmax 3343, 3069, 1662,1483, 1476, 1339, 1234, 1179, 1137, 1085, 847, 766, 647.
4.1.17. 1-(3-Bromophenyl)imidazo[4,5,1-kl]phenoxazine-4-sulfonamide (7j)
N
O S
N
OO
NH2
Br
Compound 7j was prepared by using 1-amino-10H-phenoxa-zine-3-sulfonamide (6) (1.5 g, 5.4 mmol) and 3-bromobenzalde-hyde (1.5 g, 8.1 mmol) in DMF according to the generalprocedure as an off white solid; yield 60% (1.4 g); mp 310–312 �C; m/z (CI) 440 (M-2, 100); 1H NMR (400 MHz, DMSO-d6) d7.69 (s, 1H), 7.49 (t, J = 8.8 Hz, 1H,), 7.34 (s, 2H), 7.24 (d,J = 8.8 Hz, 1H,), 7.12–7.08 (m, 4H), 7.05 (d, J = 8.0 Hz, 1H), 6.98(m, 2H); IR (KBr, cm�1) vmax 3342, 3029, 1663, 1483, 1455, 1307,1262, 1192, 1136, 1083, 837, 756, 657.
4.1.18. 1-(2-Chlorophenyl)imidazo[4,5,1-kl]phenoxazine-4-sulfonamide (7k)
N
O S
N
OO
NH2
Cl
Compound 7k was prepared by using 1-amino-10H-phenoxa-zine-3-sulfonamide (6) (1.5 g, 5.4 mmol) and 2-chlorobenzalde-hyde (1.14 g, 8.1 mmol) in DMF according to the generalprocedure as an off white solid; yield 70% (1.4 g); mp 327–330 �C; m/z (CI) 396 (M-1, 100); 1H NMR (400 MHz, DMSO-d6)d 7.82–7.73 (m, 3H), 7.71 (s, 1H), 7.66–7.64 (m, 1H), 7.40 (s,2H), 7.29–7.21 (m, 1H), 7.21–7.19 (m, 2H), 6.96 (t, J = 8.0 Hz,1H), 6.45 (d, J = 7.8 Hz, 1H); IR (KBr, cm�1) vmax 3342,3029, 1663, 1483, 1455, 1307, 1262, 1192, 1136, 1083, 837,756, 657.
4.1.19. 1-(5-Bromo-2-fluorophenyl)imidazo[4,5,1-kl]phenoxazine-4-sulfonamide (7l)
N
O S
N
OO
NH2
FBr
Compound 7l was prepared by using 1-amino-10H-phenoxa-zine-3-sulfonamide (6) (1.4 g, 5 mmol) and 5-bromo-2-florobenz-aldehyde(1.12 g, 7.5 mmol) in DMF according to the generalprocedure as a light brown solid; yield 53% (1.23 g); mp 315–317 �C; m/z (CI) 458 (M-1, 100); 1H NMR (400 MHz, DMSO-d6) d8.07–7.99 (m, 1H), 7.99–7.96 (m, 1H), 7.71 (s, 1H), 7.55 (t,J = 9.6 Hz, 1H), 7.42–7.31 (m, 2H), 7.29–7.21 (m, 2H), 7.17 (s, 1H),7.04 (t, J = 7.2 Hz, 1H), 6.78 (d, J = 8.4 Hz, 1H); IR (KBr, cm�1) vmax
3342, 3032, 1665, 1497, 1459, 1329, 1301, 1225, 1188, 1080,1038, 737, 690, 679.
1960 S. V. Reddy et al. / Bioorg. Med. Chem. 21 (2013) 1952–1963

Author's personal copy
4.1.20. 1-(2,3-Dichlorophenyl)imidazo[4,5,1-kl]phenoxazine-4-sulfonamide (7m)
N
O S
N
O O
NH2
Cl
Cl
Compound 7m was prepared by using 1-amino-10H-phenoxa-zine-3-sulfonamide (6) (1.5 g, 5.4 mmol) and 2,3-dichlorobenzal-dehyde (1.42 g, 8.1 mmol) in DMF according to the generalprocedure as an ash colour solid, yield 52% (1.2 g); mp 316–320 �C; m/z (CI) 430 (M-1, 100); 1H NMR (400 MHz, DMSO-d6) d7.65 (s, 1H), 7.38 (s, 2H), 7.32 (s, 1H), 7.30–7.25 (m, 3H), 7.15 (s,1H), 7.01–6.92 (m, 3H); IR (KBr, cm�1) vmax 3344, 3024, 1662,1499, 1484, 1380, 1336, 1299, 1267, 1158, 1096, 955, 821, 785,725.
4.1.21. 1-(4-Chlorophenyl)imidazo[4,5,1-kl]phenoxazine-4-sulfonamide (7n)
N
O S
N
OO
NH2
Cl
Compound 7n was prepared by using 1-amino-10H-phenoxa-zine-3-sulfonamide (6) (1.5 g, 5.4 mmol) and 4-chlorobenzalde-hyde (1.140 g, 8.1 mmol) in DMF according to the generalprocedure as an off white solid; yield 70% (1.4 g); mp 335–337 �C; m/z (CI) 396 (M+1, 100); 1H NMR (400 MHz, DMSO-d6) d7.86–7.84 (m, 2H), 7.73–7.67 (m, 3H), 7.39 (s, 2H), 7.29–7.27 (m,1H), 7.20 (t, J = 7.2 Hz, 1H), 7.14 (s, 1H), 7.00 (t, J = 8.2 Hz, 1H),6.90 (d, J = 7.6 Hz, 1H); IR (KBr, cm�1) vmax 3344,, 3037, 1663,1483, 1455, 1307, 1262, 1192, 1136, 756.
4.1.22. 1-(4-Dimethylaminophenyl)imidazo [4,5,1-kl]phenoxazine-4-sulfonamide (7o)
N
O S
N
OO
NH2
N
Compound 7o was prepared by using 1-amino-10H-phenoxa-zine-3-sulfonamide (6) (1.4 g, 5 mmol) and 4-diethylaminobenzal-dehyde (1.56 g, 10 mmol) in DMF according to the generalprocedure as an off white solid; yield 51% (1.5 g); mp 260–262 �C; m/z (CI) 405 (M-1, 100); 1H NMR (400 MHz, DMSO-d6) d7.62–7.60 (m, 3H), 7.36 (s, 2H), 7.26–7.24 (m, 1H), 7.19–7.16 (m,2H), 7.09 (s, 1H), 7.02–6.98 (m, 1H), 6.88 (d, J = 8.2 Hz, 2H), 3.04
(s, 6H); IR (KBr, cm�1) vmax 3332, 3233, 3117, 1662, 1562, 1473,1389, 1321, 1257, 1151, 1102, 1090, 1015, 928, 866, 765, 684.
4.1.23. 1-(2,6-Dichlorophenyl)imidazo[4,5,1-kl]phenoxazine-4-sulfonamide (7p)
N
O S
N
OO
NH2
Cl
Cl
Compound 7p was prepared by using 1-amino-10H-phenoxa-zine-3-sulfonamide (6) (1.5 g, 5.4 mmol) and 2,6-dichlorobenzal-dehyde (1.13 g, 6.4 mmol) in DMF according to the generalprocedure as an off white solid; yield 64% (1.5 g); mp 288–290 �C; m/z (CI) 430 (M-1, 100); 1H NMR (400 MHz, DMSO-d6) d7.62–7.60 (m, 2H), 7.36 (s, 2H), 7.26–7.24 (m, 1H), 7.19–7.16 (m,2H), 7.09 (s, 1H), 7.00 (t, J = 7.6 Hz, 1H), 6.88 (d, J = 8.0 Hz, 2H);IR (KBr, cm�1) vmax 3344, 3024, 1662, 1494, 1454, 1380, 1336,1299, 1267, 1148, 1096, 823, 765, 741,720.
4.1.24. Potassium-1-amino-10H-phenoxazine-3-sulfonate (8)
NH
O SO3-K+
NH2
To a solution of potassium 1-nitro-10H-phenoxazine-3-sulfo-nate (3) (5 g) in methanol (200 ml) was added Raney Ni (5 g) andhydrazine hydrate (2 ml). The resulting mixture was stirred for30 min at 65 �C. Completion of the reaction was monitored byTLC. The catalyst was removed by filtration under reduced pressureand concentrated under reduce pressure obtained as a light ashcolour solid; yield 77% (3.50 g); mp >300 �C; m/z (CI) 315 (M-1,100); 1H NMR (400 MHz, DMSO-d6) d 7.32 (s, 1H), 6.82–6.65 (m,1H), 6.64–6.45 (m, 4H), 6.21(s, 1H), 4.78 (s, 2H); IR (KBr, cm�1) vmax
3338, 1611, 1500, 1442, 1417, 1329, 1188, 1108, 1085, 1044, 848.
4.1.25. Potassium-1-phenylimidazo[4,5,1-kl]phenoxazine-4-sulfonate (9)
N
O SO3-K+
NPh
To a solution of potassium-1-amino-10H-phenoxazine-3-sulfo-nate (8) (3.0 g, 9.43 mmol) in DMF (80 ml) was added benzalde-hyde (1.0 g, 9.43 mmol) and the resulting reaction mixture wasstirred for 24 h at 100 �C. Completion of the reaction was moni-tored by TLC. The solvent was removed from the reaction under re-duced pressure, then the crude solid was triturated with ethylacetate, filtered and purified by column chromatography; yield52% (2 g), mp >300 �C; m/z (CI) 365 (M-K, 100); 1H NMR(400 MHz, DMSO-d6) d 7.79 (d, 2H, J = 7.9 Hz), 7.64–7.59 (m, 3H),7.42 (s, 1H), 7.22 (d, 1H, J = 8.0 Hz), 7.12 (t, 1H, J = 8.0 Hz), 6.91–6.83 (m, 3H); IR (KBr, cm�1) vmax 3492, 1661, 1494, 1455, 1302,1195, 1078, 1039, 700.
S. V. Reddy et al. / Bioorg. Med. Chem. 21 (2013) 1952–1963 1961

Author's personal copy
4.1.26. 1-Phenylimidazo[4,5,1-kl]phenoxazine-4-sulfonylchloride (10)
N
O SO2Cl
NPh
Potassium-1-phenylimidazo[4,5,1-kl]phenoxazine-4-sulfonate(9) (2 g) was added to POCl3 (14 ml) and refluxed for 3 h. Comple-tion of the reaction was monitored by TLC. Excess of POCl3 was re-moved by distillation and the crude solid was poured in excesscrushed ice and the solid was filtered and used immediately forthe next step.
4.1.27. Alternative preparation of 1-Phenylimidazo[4,5,1-kl]phenoxazine-4-sulfonamide (7a)
To a solution of aqueous ammonia (5 ml) in tetrahydrofuran(20 ml) was added 1-phenylimidazo [4,5,1-kl]phenoxazine-4-sul-fonylchloride (10) at 0 �C. The resulting reaction mixture was stir-red for 30 min at same temperature. Completion of the reactionwas monitored by TLC and the excess solvent was removed underreduced pressure, the crude solid was acidified with dilute hydro-chloric acid and the solid was filtered and dried. Compound ismatching with the prepared by another method, yield 50% (0.8 g).
5. Pharmacology
5.1. Materials and methods
5.1.1. Cells and reagentsSf9 cells were obtained from ATCC (Washington DC, USA) and
were routinely maintained in Grace’s supplemented medium(Invitrogen) with 10% FBS. cAMP was purchased from SISCO Re-search Laboratories (Mumbai, India). PDElight HTS cAMP phospho-diesterase assay kit was procured from Lonza (Basel, Switzerland).PDE4B1 clone was procured from OriGene Technologies (Rockville,MD, USA). PDE4D2 enzyme was purchased from BPS Bioscience(San Diego, CA, USA).
5.1.2. PDE4B protein production and purificationPDE4B1 cDNA was sub-cloned into pFAST Bac HTB vector (Invit-
rogen) and transformed into DH10Bac (Invitrogen) competentcells. Recombinant bacmids were tested for integration by PCRanalysis. Sf9 cells were transfected with bacmid using Lipofect-amine 2000 (Invitrogen) according to manufacturer’s instructions.Subsequently, P3 viral titer was amplified, cells were infected and48 h post infection cells were lysed in lysis buffer (50 mM Tris–HClpH 8.5, 10 mM 2-mercaptoethanol, 1% protease inhibitor cocktail(Roche), 1% NP40). Recombinant His-tagged PDE4B protein waspurified as previously described elsewhere.17 Briefly, lysate wascentrifuged at 10,000 rpm for 10 min at 4 �C and supernatantwas collected. Supernatant was mixed with Ni-NTA resin (GE LifeSciences) in a ratio of 4:1 (v/v) and equilibrated with binding buffer(20 mM Tris–HCl pH 8.0, 500 mM-KCl, 5 mM imidazole, 10 mM 2-mercaptoethanol and 10% glycerol) in a ratio of 2:1 (v/v) and mixedgently on rotary shaker for 1 h at 4 �C. After incubation, lysate–Ni–NTA mixture was centrifuged at 4500 rpm for 5 min at 4 �C and thesupernatant was collected as the flow-through fraction. Resin waswashed twice with wash buffer (20 mM Tris–HCl pH 8.5, 1 M KCl,10 mM 2-mercaptoethanol and 10% glycerol). Protein was elutedsequentially twice using elution buffers (Buffer I: 20 mM Tris–HCl pH 8.5, 100 mM KCl, 250 mM imidazole, 10 mM 2-mercap-toethanol, 10% glycerol, Buffer II: 20 mM Tris–HCl pH 8.5,
100 mM KCl, 500 mM imidazole, 10 mM 2-mercaptoethanol, 10%glycerol). Eluates were collected in four fractions and analyzedby SDS–PAGE. Eluates containing PDE4B protein were pooled andstored at �80 �C in 50% glycerol until further use.
5.1.3. PDE4 enzymatic assayThe inhibition of PDE4 enzyme was measured using PDElight
HTS cAMP phosphodiesterase assay kit (Lonza) according to man-ufacturer’s recommendations. Briefly, 10 ng of in house purifiedPDE4B1 or 0.5 ng commercially procured PDE4D2 enzyme waspre-incubated either with DMSO (vehicle control) or compoundfor 15 min before incubation with the substrate cAMP (5 lM) for1 h. The reaction was halted with stop solution and reaction mixwas incubated with detection reagent for 10 min in dark. Dose re-sponse studies were performed at 9 different concentrations rang-ing from 100 lM to 0.01 lM. Luminescence values (RLUs) weremeasured by a Multilabel plate reader (Perklin Elmer 1420 Multi-label counter).The percentage of inhibition was calculated usingthe following formula and the IC50 values were determined by anonlinear regression analysis from dose response curve usingGraphpad Prism software (San Diego, USA). IC50 values are ex-pressed as mean ± SD.
%Inhibition ¼ ½ðRLU of vehicle control
� RLU of inhibitorÞ=ðRLU of vehicle controlÞ�� 100
6. Docking study
6.1. With PDE4B
Docking simulations of molecules were performed using Schro-dinger software suite (Maestro, version 9.2).19 The Protein (PDE4B)for docking studies was retrieved from protein data bank with PDBID: 3O0J.20 The protein was prepared by giving preliminary treat-ment like adding hydrogen, adding missing residues, refining theloop with prime and finally minimized by using OPLS 2005 forcefield. The search grid was generated by picking the co-crystal li-gands and extended up to 20 Å. The hydroxyl groups of search areawere kept flexible during grid generation process.
6.2. With PDE4D
Docking simulations of molecules were performed using Schro-dinger software suite (Maestro, version 9.2).19 The Protein coordi-nates (PDE4D) for docking studies was retrieved from protein databank with PDB ID: 1XOR.20 The protein was prepared by givingpreliminary treatment like adding hydrogen, adding missing resi-dues, refining the loop with prime and finally minimized by usingOPLS 2005 force field. The search grid was generated by picking theco-crystal ligands and extended up to 20 Å. The hydroxyl groups ofsearch area were kept flexible during grid generation process.
All molecules were minimized by using MacroModel21 applica-tion. Molecules were docked by using glide XP (extra precision)docking mode.22 We performed flexible docking by allowing sam-ple ring conformations and sample nitrogens to move to possibleextent in docking. The docking results are shown in tables andfigures.
Acknowledgments
One of the authors (B.V.K.) is thankful to UGC, New Delhi, Indiafor Major Research Project (F. No. 35-151/2008). The authors arethankful to Principal and Management of C.K.M. Arts and Science
1962 S. V. Reddy et al. / Bioorg. Med. Chem. 21 (2013) 1952–1963

Author's personal copy
College for encouragement and obliged to Dr. D. Mohan Rao,SYMED Laboratories Ltd, Hyderabad for analytical support.
Supplementary data
Supplementary data associated with this article can be found, inthe online version, at http://dx.doi.org/10.1016/j.bmc.2013.01.023.
References and notes
1. Suzuki, F.; Hashimoto, K.; Ishihara, M.; Westman, G.; Samuelsson, K.; Kawase,M.; Motohashi, N.; Sakagami, H. Anticancer Res. 2007, 27, 4233.
2. Bolognese, A.; Correale, G.; Manfra, M.; Lavecchia, A.; Mazzoni, O.; Novellino,E.; Barone, V.; La Colla, P.; Loddo, R. J. Med. Chem. 2002, 45, 5217.
3. Lohray, B. B.; Lohray, V. B.; Bajji, A. C.; Kalchar, S.; Poondra, R. R.; Padakanti, S.;Chakrabarti, R.; Vikramadithyan, R. K.; Misra, P.; Juluri, S.; Mamidi, N. V.;Rajagopalan, R. J. Med. Chem. 2001, 44, 2675.
4. (a) Reddy, G. R.; Reddy, T. R.; Joseph, S. C.; Reddy, K. S.; Reddy, L. S.; Kumar, P.M.; Krishna, G. R.; Reddy, C. M.; Rambabu, D.; Kapavarapu, R.; Lakshmi, C.;Meda, T.; Priya, K. K.; Parsa, K. V. L.; Pal, M. Chem. Commun. 2011, 47, 7779; (b)Kodimuthali, A.; Gupta, R.; Parsa, K. V. L.; Prasunamba, P. L.; Pal, M. Lett. DrugDes. Disc. 2010, 7, 402; (c) Kumar, K. S.; Kumar, S. K.; Sreenivas, B. Y.; Gorja, D.R.; Kapavarapu, R.; Rambabu, D.; Krishna, G. R.; Reddy, C. M.; Rao, M. V. B.;Parsa, K. V. L.; Pal, M. Bioorg. Med. Chem. 2012, 20, 2199; (d) Pal, S.; Durgadas, S.;Nallapati, S. B.; Mukkanti, K.; Kapavarapu, R.; Lakshmi, C.; Parsa, K. V. L.; Pal, M.Bioorg. Med. Chem. Lett. 2011, 21, 6573; (e) Kumar, P. M.; Kumar, K. S.;Mohakhud, P. K.; Mukkanti, K.; Kapavarapu, R.; Parsa, K. V. L.; Pal, M. Chem.Commun. 2012, 48, 431; (f) Kumar, P. M.; Kumar, K. S.; Meda, C. L. T.; Reddy, G.R.; Mohakhud, P. K.; Mukkanti, K.; Krishna, G. R.; Reddy, C. M.; Rambabu, D.;Kumar, K. S.; Priya, K. K.; Chennubhotla, K. S.; Banote, R. K.; Kulkarni, P.; Parsa,K. V. L.; Pal, M. Med. Chem. Commun. 2012, 3, 667; (g) Adepu, R.; Rambabu, D.;Prasad, B.; Meda, C. L. T.; Kandale, A.; Krishna, G. R.; Reddy, C. M.; Chennuru, L.N.; Parsa, K. V. L.; Pal, M. Org. Biomol. Chem. 2012, 10, 5554; (h) Kumar, T. B.;Sumanth, Ch.; Vaishaly, S.; Rao, M. S.; Sekhar, K. B. C.; Meda, C. L. T.; Kandale,A.; Rambabu, D.; Krishna, G. R.; Reddy, C. M.; Kumar, K. S.; Parsa, K. V. L.; Pal, M.Bioorg. Med. Chem. Lett. 2012, 22, 5639.
5. Kodimuthali, A.; Jabaris, S. S. L.; Pal, M. J. Med. Chem. 2008, 51, 5471.6. Rao, Y. J.; Xi, L. Acta Pharmacol. Sin. 2009, 30, 1.7. (a) Dastidar, S. G.; Rajagopal, D.; Ray, A. Curr. Opin. Invest. Drugs 2007, 85, 364;
(b) Field, S. K. Clin Med. Insights Circ. Respir. Pulm. Med. 2011, 5, 57; (c) Chung, J.H. Aging 2012, 4, 144.
8. (a) Regan, J.; Bruno, J.; Mc Garry, D.; Poli, G.; Hanney, B.; Bower, S.; Travis, J.;Sweeney, D.; Miller, B.; Souness, J.; Djuric, S. Bioorg. Med. Chem. Lett. 1998, 8,2737; (b) Roberts, D. E.; Curd, J. G. Arthritis Rheum. 1990, 33, 1590; (c) Lajiness,M. S.; Vieth, M.; Erickson, J. Curr. Opin. Drug. Disc. Dev. 2004, 7, 470.
9. (a) Bernthsen, A. Ber. Dtsh. Chem. Ges. 1887, 20, 942; (b) Kehrmann, F. Ann.Chem. 1902, 322, 1; (c) Kehrmann, F.; Neil, A. A. Ber. Dtsch. Chem. Ges. 1914, 47,3102; (d) Cullinane, N. M.; Davey, H. G.; Padfield, H. J. H. J. Chem. Soc. 1934, 716;(e) Gilman, H.; Moore, L. O. J. Am. Chem. Soc. 1957, 79, 3485.
10. Prinz, H.; Chamasmani, B.; Vogel, K.; Beohm, K. J.; Aicher, B.; Gerlach, M.;Geunther, E. G.; Amon, P.; Ivanov, I.; Meuller, K. J. Med. Chem. 2011, 54, 4247.
11. (a) Motohashi, N. Yakugaku Zasshi 1983, 103, 364; (b) Shimamoto, T.; Tomoda,A.; Ishida, R. Clin. Cancer Res. 2001, 7, 704.
12. Gunstone, F. D.; Tucker, S. H. J. Appl. Chem. 1952, 2, 204.13. (a) Kumar, B. V.; Rathore, H. G. S.; Reddy, V. M. J. Indian Chem. 1982, 21B, 1126;
(b) Kumar, B. V.; Reddy, V. M. Indian Drugs 1985, 23, 94; (c) Kumar, B. V.; Reddy,V. M. J. Indian Chem. 1985, 24B, 1098; (d) Rao, V. M.; Kumar, B. V.; Reddy, M. M.;Reddy, V. M. Indian Drugs 1988, 25, 304.
14. Ullmann, F.; Engl, G.; Wosnessensky, N.; Kuhn, E.; Herre, E. Justus Liebig Ann.Chem. 1909, 366, 79.
15. Shirai, H.; Hayazaki, T. Yakugaku Zasshi 1970, 90, 588.16. This route is amenable for the preparation of sulfonamide derivatives
possessing various substituents on the sulfonamide nitrogen depending onthe amines used in place of aqueous ammonia in the last step. We thank thereviewer for pointing out this.
17. Wang, P.; Myers, J. G.; Wu, P.; Cheewatrakoolpong, B.; Egan, R. W.; Billah, M. M.Biochem. Biophys. Res. Commun. 1997, 234, 320.
18. Demnitz, J.; LaVecchia, L.; Bacher, E.; Keller, T. H.; Muller, T.; Schurch, F.;Weber, H. P.; Pombo-Villar, E. Molecules 1998, 3, 107.
19. Maestro, version 9.2, Schrodinger, LLC: New York, NY, 2012.20. Card, G. L.; England, B. P.; Suzuki, Y.; Fong, D.; Powell, B.; Lee, B.; Luu, C.;
Tabrizizad, M.; Gillette, S.; Ibrahim, P. N.; Artis, D. R.; Bollag, G.; Milburn, M. V.;Kim, S.-H.; Schlessinger, J.; Zhang, K. Y. J. Structure 2004, 12, 2233.
21. MacroModel, version 9.9, Schrödinger, LLC: New York, NY, 2011.22. Glide, version 5.7, Schrodinger, LLC: New York, NY, 2012.
S. V. Reddy et al. / Bioorg. Med. Chem. 21 (2013) 1952–1963 1963