final navigating multiple clinical trial requirements for the us
TRANSCRIPT

Navigating Multiple Clinical Trial Requirements for the US Market to
Ensure Compliance & Drive Exports
Dr. Bhaswat S. ChakrabortySenior Vice President & Chair
R&D Core Committee, Cadila Pharmaceuticals Ltd., Ahmedabad
Presented at CPhI India Conference, Mumbai, December 2, 2011

Content Guidelines• Devising effective strategies to manage the US
approval process and meet stringent global protocols
• Understanding the documentation requirements
• Overcoming the complexities around stability studies and batch designs to ensure a robust clinical trials procedure
• Examining best practice on conducting effective clinical trials in the US to establish a roadmap for success and swift future approval

Approval Process• Complex – Main goal is to evaluate Safety, Efficacy
& CMC decisively
• Data driven– Large volume, scientific, experimental
• Multi-disciplinary– Medical, chemistry, pharmaceutics, toxicology,
pharmacology, biopharmaceutics, statistics, microbiology….
• Team work & sponsor-regulator communication
• Expertise and technicality required

Drug Development• Drug development is ideally a logical, stepwise procedure in
which information from small early studies is used to support and plan later larger, more definitive studies.
• To develop new drugs efficiently, it is essential to identify characteristics of the investigational medicine in the early stages.
• Initial trials provide an early evaluation of short-term safety and tolerability and can provide PK & PD information to choose a suitable dosage range and administration schedule for exploratory therapeutic trials.
• Later confirmatory studies are generally larger and longer and include a more diverse patient population.

• Dose-response information should be obtained at all stages of development, from early tolerance studies, to studies of PD effects, to large efficacy studies (see ICH E4).
• Throughout development, new data may suggest the need for additional studies (part of an earlier phase).
– For example, blood level data in a late trial may suggest a need for a drug-drug interaction study,
– ADRs may suggest the need for further dose finding and/or additional nonclinical studies.
• In addition, to support a new marketing application approval for the same drug, e.g., for a new indication, pharmacokinetic or therapeutic exploratory studies are considered to be in Phase I or Phase II of development.
Drug Development..

Source: Dr. R. Kane, USFDA
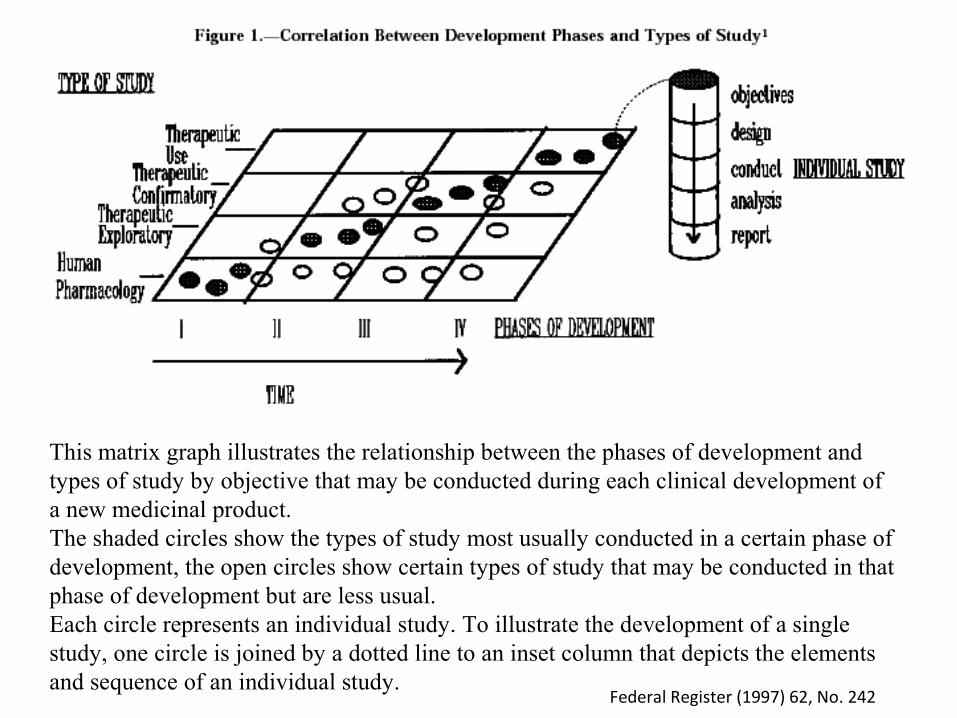
This matrix graph illustrates the relationship between the phases of development and types of study by objective that may be conducted during each clinical development of a new medicinal product. The shaded circles show the types of study most usually conducted in a certain phase of development, the open circles show certain types of study that may be conducted in that phase of development but are less usual. Each circle represents an individual study. To illustrate the development of a single study, one circle is joined by a dotted line to an inset column that depicts the elements and sequence of an individual study.
Federal Register (1997) 62, No. 242

Source: Dr. R. Kane, USFDA

10
Review Process• Once a new drug application is filed
– an FDA review team evaluates whether the studies the sponsor submitted show that the drug is safe and effective for its proposed use
• Team consists of medical doctors, chemists, statisticians, microbiologists, pharmacologists, and other experts
• No drug is absolutely safe; all drugs have side effects– "Safe" in this sense above means that the benefits of the drug appear to
outweigh the risks.• The review team
– analyzes study results– looks for possible issues with the application
• e.g., weaknesses of the study design or analyses• may agree with the sponsor's results and conclusions, or may need
additional information to make a decision• Each reviewer prepares a written evaluation containing conclusions and
recommendations about the application• These evaluations are then considered by team leaders, division directors,
and office directors, depending on the type of application

The Quantity of Evidence to Support Effectiveness
• Mainly three scenarios:
• Effectiveness of a new use may be extrapolated entirely from existing efficacy studies.
• A single adequate and well-controlled study of a specific new use can be supported by information from other related adequate and well-controlled studies
– e.g., studies in other phases of a disease, in closely related diseases, of other conditions of use (different dose, duration of use, regimen), of different dosage forms, or of different endpoints.
– a single multicenter study, without supporting information from other adequate and well-controlled studies
– when a single study is used, there should be hardly any room for study imperfections or contradictory (non-supportive) information.
• Two or more adequate and well-controlled studies.

Documentation of the Quality of Evidence
• Two main themes:1. Completeness of the documentation and
2. The ability to access the primary study data and the original study-related records (e.g., subjects’ medical records, drug accountability records) for the purposes of verifying the data submitted as evidence.
• These interrelated elements bear on a determination of whether a study is adequate and well-controlled.
• In practice, to achieve a high level of documentation, studies supporting claims are ordinarily conducted in accordance with good clinical practices (GCPs).
• Sponsors routinely monitor all clinical sites, and FDA routinely has access to the original clinical protocols, primary data, clinical site source documents for on-site audits, and complete study reports.

Quality by Design• Quality by Design is understanding the manufacturing process and
identifying the key steps for obtaining and assuring a pre-defined final product quality.
• FDA is constantly working to identify ways to improve the manufacturing process to ensure consistent product quality throughout the shelf life as well as to identify when contamination or other production failures may occur.
• Improved quality by design will also lower product development and manufacturing costs by reducing the likelihood of production failures during a long run and by providing opportunities for continuous improvement.
• As part of its Quality by Design effort, FDA is now working on three new areas to support increased manufacturing quality.
– The first is a continuous processing where materials constantly flow in and out of equipment.
– The second is the use of process analytical technology to monitor and control processes, as opposed to the current method of just testing products.
– The third is the development of new statistical approaches to detect changes in process or product quality.

New Drug approved by USFDA in 2011
Incomplete list

Case Study Icatibant (FirazyrTM)

Icatibant
Icatibant (FirazyrTM) is a peptidomimetic drug consisting of ten amino acids, which is a selective and specific antagonist of bradykinin B2 receptors. It has been approved by USFDA & EMA for the symptomatic treatment of acute attacks of hereditary angioedema (HAE) in adults (with C1-esterase-inhibitor deficiency).

http://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/022150_firazyr_toc.cfm

• 34 USFDA officers, half of whom were reviewers took part in the approval process of this submission.

19
What does FDA Look for in Clinical Portion?
• FDA approves a drug application based on– Substantial evidence of efficacy & safety from
“adequate and well-controlled investigations”
– A valid comparison to a control
– Quantitative assessment of the drug’s effect • (21 CFR 314.126.)
• The design and data quality of trials intended to support drug approval is very important

Proof of Efficacy & Safety Using a Valid Control
• The original NDA was submitted in October 2007, and a Not Approval action was taken in April 2008, because substantial evidence of efficacy was not demonstrated in two pivotal studies.
• The NDA included one placebo-controlled study that did not show efficacy.
• Another tranexamic acid (TA) active-controlled study that showed efficacy.
• Demonstration of efficacy in the TA active-controlled study was not considered adequate for approval because TA is not approved for the treatment of acute attacks of HAE.
• The sponsor has adequately addressed the efficacy deficiency with data submitted from a new placebo controlled study that shows efficacy.

FDA and Sponsor-FDA Meetings
• The Agency and the applicant had various interactions dating back to 2003.– teleconferences
• The FDA had Advisory Committee meetings.
• The FDA had many milestone review meetings

Validity of The Primary Efficacy Endpoint (often the Main Issue)
• Subsequent to the Not Approval action to the original NDA, the Sponsor met with the Division on December 15, 2008, to clarify the clinical deficiencies outlined.
• The Sponsor agreed to conduct a third, controlled study in patients with HAE to assess efficacy.
• Subsequently, the Sponsor submitted a request on February 12, 2009, for a Special Protocol Assessment for the third study.
• Although no agreement was reached, the Division informed the Sponsor that a trial that was generally similar in design to the two previous studies would be acceptable for addressing the clinical deficiencies.

Novelty, Understanding the Mechanism of Action and Differences from the Existing Drugs
• At present Berinert and Kalbitor are approved in the US and elsewhere in the world for treatment of acute attacks of HAE.
– Both of these products require administration by a healthcare professional and carry a risk of anaphylaxis.
• Icatibant is a new molecular entity proposed for the treatment of acute attacks of HAE (hereditary angioedema) .
– The putative mechanism of action of icatibant is inhibition of the bradykinin pathway by blocking the bradykinin type 2 receptor.
– The bradykinin pathway is not directly responsible etiologically for HAE, but is thought to play an important role in causing the symptoms of HAE when the complement pathway is activated due to deficiency of C1 inhibitor (C1-INH) in these patients.

Chemistry, Manufacturing, and Controls• The proposed expiry period of 18 months for drug product
when stored at 2-25°C is supported by the submitted stability data.
• All manufacturing and testing sites related to this product have acceptable inspection status.
• Regarding syringe-needle compatibility, there were concerns raised by the Agency. These concerns, however, were not of a magnitude to preclude approval.
• Additional testing of the syringe needle compatibility that is statistically relevant may be pursued post-approval with the applicant since there are no safety or device performance issues identified so far.

Stability Data Required
• In general, 12-month real time stability data is recommended (ICH Guidance Q1A).
• Assignment of expiry dating period for the drug product may depend upon the real time stability data.
• And also on the totality of the NDA submission (e.g., analytical, validation, impurity, etc.).
• These were the data asked for Icatibant.

Global Protocols and Reviews are Influenced by Experiences Elsewhere
• ….Worldwide post-marketing experience with Firazyr (Icatibant), which was approved in Europe three years ago and is now marketed in 37 countries overseas.
• As of June 30, 2011, a total of 2,044 injections have been administered during clinical trials and syringe/needle units have been sold, including for patient self-administration.
• To date, there have been no reports of device failure in the clinical trials, including a designated self-administration trial in 95 patients, and no post-marketing adverse events associated with device failure.

Nonclinical Pharmacology and Toxicology
• Nonclinical toxicology studies lasting 6 months in rats and 9 months in dogs. – The primary toxicities were injection site irritation, testicular and
uterine atrophy, and delay in sexual maturation.
– Injection site irritation was not of concern because it can be monitored in humans.
• The reproductive toxicities observed in animals would not preclude approval given the severity of HAE disease and the fact that animals were dosed daily, whereas humans will receive icatibant intermittently.
• To address this finding further, a human clinical study to evaluate icatibant effects on reproductive hormones is currently ongoing.

Nonclinical Pharmacology and Toxicology..
• A complete genetic toxicology program was conducted, which was negative.
• A complete battery of reproductive toxicology studies was conducted and the results support a Pregnancy Category C designation. Although there were no observed teratogenic effects, there were signs of embryotoxicity, and dose-related decreases in post-implantations and total number of live fetuses.
• Additionally, icatibant prolonged gestation, resulting in spontaneous abortions and litter deaths.
• The Sponsor has initiated carcinogenicity studies in rats and mice, which will be completed as post-marketing required (PMR) studies.
• The Sponsor will also complete a post-marketing commitment study (PMC) to qualify impurities occurring at concentrations higher than defined thresholds.

Clinical Pharmacology and Biopharmaceutics
• The application is recommended for Approval from a Clinical Pharmacology perspective, and there are no outstanding clinical pharmacology issues.
• The application included results from a comprehensive clinical pharmacology program, which included studies to assess protein binding and metabolism in vitro, single- and multiple-dose pharmacokinetics, effect of hepatic impairment, the effect of renal impairment in hepatorenal syndrome, QTc effect, and effect on CYP540 isoenzymes.

Concluding Remarks• Options & factors for Sponsor’s strategies well considered• Therapeutic context
– NCE/NBE– Orphan designation– NDDS– Generics
• Approval tracks– Unmet needs– Fast track– Priority review – 6 months– One study (high quality)– Standard review – 10 months
• Protocol Assessments• Meetings with FDA• Thorough knowledge of data requirements, review process, milestones of
the review process, how to respond to FDA queries, carry out additional (required studies), gain final approval and negotiate the post-marketing commitments

Thank you Very Much