familial b-100: enhanced mb47to - pnas · alsobeenidentified in asecond,unrelatedsubject(w.s.)and...
TRANSCRIPT

Proc. Nati. Acad. Sci. USAVol. 85, pp. 9758-9762, December 1988Medical Sciences
Familial defective apolipoprotein B-100: Enhanced binding ofmonoclonal antibody MB47 to abnormal low density lipoproteins
(hypercholesterolemia/genetic disease/atherosclerosis/apolipoprotein B-100 polymorphism)
KARL H. WEISGRABER*t, THOMAS L. INNERARITY*, YVONNE M. NEWHOUSE*, STEPHEN G. YOUNG*,KAY S. ARNOLD*, RONALD M. KRAuSSt, GLORIA L. VEGA§, SCOTT M. GRUNDY§,AND ROBERT W. MAHLEY**Gladstone Foundation Laboratories for Cardiovascular Disease, Cardiovascular Research Institute, Departments of Pathology and Medicine, University ofCalifornia, San Francisco, CA 94140-060§; tDonner Laboratory, Lawrence Berkeley Laboratory, University of California, Berkeley, CA 94720; and §Centerfor Human Nutrition, Departments of Internal Medicine, Biochemistry, and Clinical Nutrition, University of Texas Health Science Center, Dallas, TX 75235
Communicated by Joseph L. Goldstein, September 2, 1988 (received for review July 20, 1988)
ABSTRACT Familial defective apolipoprotein (apo) B-100is a recently described genetic disorder that appears to resultfrom a mutation in the apoB-100 gene. This disorder ischaracterized by hypercholesteroledia resulting from elevatedplasma conce ntrations of low density lipoprotein LDL. Thedisorder was fist detected in three members ofone family. TheLDL from affected subjects binds defectively (:30% of nor-mal) to LDL receptors, retarding the clearance of LDL fromplasma. In the present study, two otflbr members of the affectedfamily were found to possess abnormal LDL. In addition,abnormal LDL with a similar binding defect were found in asecond, unrelated family. In both families, the defect istransmitted over three generations as an autosomal codomi-nant trait and all affected members are heterozygotes. Sincethere is only one apoB-100 molecule per LDL particle, theabnormal LDL in heterozygous subjects is made up of twoplpulations of particles: one that has normal binding activityto receptors and one that binds defectively. To localize themutation in apoB-100, the binding of five apoB-100-specificmonoclonal antibodies to abnormal LDL was assesd in asolid-phase RIA. Only antibody MB47, whose epitipe isbetween residues 3350 and 3506, distinguished abnormal LDLfrom normal LDL isolated from control subjects with normallipid levels; MB47 bound with a higher affinity (by =60%) toabnormal LDL. In every individual with abnormal LDL, theMB47 antibody bound with a higher affinity. The convenienceof this assay will facilitate screening of large populations todetermine the frequency of this disorder.
In humans, most plasma cholesterol -is contained in the lowdensity lipoprotein (LDL) density fraction. Plasma concentra-tions of LDL are regulated by the LDL receptor pathway (1),and the interaction of LDL with the receptor is mediated byapolipoprotein (apo) B-100 (2), the predominant apolipoproteincomponent of LDL. The structure of the Mr 550,000 apoB-100has recently been determined (3-6). Epidemiological studieshave established an association between increased plasmaconcentrations ofLDL and an increased incidence of coronaryheart disease (7, 8). An extreme example ofthis association, andone that underscores the importance of the LDL receptor inregulating plasma LDL concentrations, is the genetic disorderfamilial hypercholesterolemia (for review, see ref. 9). Subjectsheterozygous for familial hypercholesterolemia have elevatedplasma LDL and develop premature atherosclerosis. The dis-order results from several different mutations at the gene locusof the LDL receptor that lead to expression ofnonfunctional ordysfunctional receptors (1).
However, familial hypercholesterolemia heterozygosityaccounts for only a fraction of the high plasma cholesterollevels in the population (10). Most individuals with elevatedLDL appear to have receptors with a normal structure or withno detectable dysfunction (11-13). Therefore, other factorsmust contribute to increased plasma levels of LDL in mosthypercholesterolemic individuals (13, 14). To determinewhether other genetic abnormalities of LDL catabolismresult in hypercholesterolemia, attention has recently beendirected to'moderately hypercholesterolemic subjects whodo not have classic familial hypercholesterolemia heterozy-gosity. Vega and Grundy (15) have shown that in somehypercholesterolemic subjects the catabolic rate for autolo-gous LDL is significantly slower than the rate for normalheterologous LDL. These results suggested that LDL interactabnormally with the LDL receptor, a possibility confimed inone of these subjects, G.R., by using an in vitro receptorbinding assay (16). The LDL from'G.R. and two of his threebrothers bound to receptors with -30% the activity of normalLDL. Since the interaction ofLDL with the LDL receptor ismediated through apoB-100 (2) and the defect appears to beassociated with the apoB-100 molecule, this disorder has beendesignated familial defective apoB-100 (16). Because there isonly one molecule of apoB-100 per LDL particle (17); theabnormal LDL in heterozygous subjects would be a mixture,containing a population of particles with normal bindingactivity to receptors and a population with defective binding.The current study was undertaken to localize the region of
apoB-100 responsible for the defect. Our approach was to testthe binding of selected apoB-specific monoclonal antibodies toabnormal and normal LDL to determine whether any boundwith altered affinity to abnormal LDL from familial defectiveapoB-100 subjects. Four of the five antibodies selected wereknown to inhibit the binding of LDL to LDL receptors. Onlythe MB47 antibody described by Young et al. (18) bound toabnormal LDL with a higher apparent affinity than it bound tonormal LDL. The high-affinity binding of the MB47 antibodycosegregates with defective-binding LDL in all R. familymembers examined thus far. Familial defective apoB-100 hasalso been identified in a second, unrelated subject (W.S.) andfour family members. The high-affinity binding of the MB47antibody is also linked to abnormal LDL in this second family.
MATERIALS AND METHODSSubject Material. The probands (G.R. and W.S.) were
recruited from a population of subjects being treated for
Abbreviations: apo, apolipoprotein; LDL, low density lipopro-tein(s).tTo whom reprint requests should be addressed at: GladstoneFoundation Laboratories for Cardiovascular Disease, P.O. Box40608, San Francisco, CA 94140-0608.
9758
The publication costs of this article were defrayed in part by page chargepayment. This article must therefore be hereby marked "advertisement"in accordance with 18 U.S.C. §1734 solely to indicate this fact.

Proc. Natl. Acad. Sci. USA 85 (1988) 9759
primary hypercholesterolemia at the University of TexasCenter for Human Nutrition. Subject G.R. was previouslydescribed (16), and subject W.S. was identified in a receptorbinding assay. Normal, control subjects were volunteers withnormal plasma lipid levels. Test subjects for verification ofthe antibody RIA were recruited from a church in WalnutCreek, CA.Plasma (from blood treated with EDTA, 1 mg/ml) obtained
from subjects who had fasted overnight was used for lipo-protein analysis or LDL preparation. Total plasma choles-terol and LDL cholesterol concentrations were determinedwith a spectrophotometric assay kit (Boehringer Mannheim);LDL cholesterol concentrations were measured using theGilford 400E analyzer after precipitation of plasma withheparin and manganese chloride (19, 20).
General Methods. LDL (p = 1.02-1.05 g/ml) were isolatedfrom plasma by sequential ultracentrifugation (40C) at 59,000rpm in a Beckman 60 Ti rotor and were recentrifuged at p =1.05 g/ml. The protein content of LDL was determinedaccording to the method of Lowry et al. (21) using bovineserum albumin as a standard. The binding of unlabeled LDLsamples to LDL receptors on cultured human fibroblasts wasdetermined in competitive receptor binding assays as de-scribed (22, 23). The concentration of unlabeled test LDLrequired to displace 50% of control 125I-labeled LDL wascalculated by linear regression analysis of the logarithm ofconcentration (,ug/ml) vs. probits. Probits were obtainedfrom a probit transformation table (24).
Antibodies. Four of the five apoB-100-specific monoclonalantibodies used block the binding of LDL to LDL receptors:4G3, at residues 3029-3132 (5, 25); 3F5, at 3029-3132 (5);5E11, at 3249-3636 (5, 25); and MB47, at 3350-3506 (5).Antibody MB3, which does not block LDL binding andwhose epitope is located in the amino-terminal thrombolyticfragment (T4, residues 1-1297), wqs used as a control antibody(26, 27).
RIAs. The ability of individual LDL preparations to bind tothe various apoB-100 monoclonal antibodies and the concen-trations of apoB-100 in plasma were determined in solid-phase competition RIAs according to methods prev puslydescribed (26). Removawell plates (Dynatech, Cq tilly,VA) were coated at roorq temperature for 2 hr with tffd ofa phosphate-buffered saline buffer (0.15 M Naq:l cont ining0.022 M Na2HPO4 and 0.015 M Na2HPO4,9fI 7.4) containing10 ,ug of control LDL per ml. The plates were wvshed fivetimes with washing buffer (phosphpte-buffered saline con-taining 0.3 mM EDTA, 0.02% NaN3, 0.05% Tween 20,0.04%aprotinin, and 0.1% bovine serum albumin). Wells wereblocked by incubation with 200,ul ofblocking buffer (washingbuffer containing 4% bovine serum albumin) for 1 hr at roomtemperature and then were washed five times with washingbuffer. A standard curve for the LDL binding assays wasconstructed by diluting control LDL in dilution buffer(washing buffer containing 31% bovine serum albumin) toconcentrations ranging from 0.4 to 100 jig ofLDL protein perml. A range of 0.16-50 ,ug/ml was used for determining theapoB concentrations in plasma. Twenty-five microliters ofstandard LDL, unknown LDL samples, or plasma dilutions(1:100, 1:200, and 1:300) was loaded into each LDL-coatedwell, followed by 25 pA of a fixed concentration of antibody(either ascites fluid or IgG that was prepared by protein Apurification) in dilution buffer. Optimal antibody concentra-tion was determined in a separate assay. The plates were thenincubated overnight at 4°C and washed five times withwashing buffer, and the amount of antibody bound wasdetermined after a 4-hr incubation at 40C with I251-labeledsheep anti-mouse IgG (Amersham). In the plasma assay, useof the Pro/Pette System (Perkin-Elmer) improved pipettingprecision compared with manual pipetting.
RESULTS
We previously described familial defective apoB-100 (16) inthe R. family. By using in vitro receptor binding assays, wedetermined that LDL from two of the proband's (G.R.)brothers bound defectively to LDL receptors, whereas LDLfrom a third brother and his son bound normally (16). Byusing a competitive fibroblast binding assay, we have ex-tended the study to include four additional first-degreerelatives of one of G.R.'s affected brothers (Fig. 1). Two ofthese subjects were found to have an LDL binding deficiencysimilar to that of the other affected family members: =30% ofnormal [an average of 7.4 pug of LDL protein per ml wasrequired for 50% inhibition of 1251-labeled LDL binding tonormal fibroblasts compared with 2.6 ,ug/ml for normal LDL(Table 1)]. In agreement with previous results (16), theadditional subjects with abnormal LDL had a higher plasmacholesterol concentration than age- and sex-matched con-trols, and this hypercholesterolemia is associated with in-creased LDL cholesterol (Table 1).
Familial defective apoB-100 has also been identified inthree generations in a second, unrelated family by using thecompetitive receptor binding assay. Testing showed that 4 of15 relatives of the proband W.S. have defective-binding LDL(Fig. 2, Table 2). As in the R. family, the defective LDL ofS. family members has =30% of normal binding activity.Hypercholesterolemia and elevated LDL are also associatedwith the presence of abnormal LDL in this family, except forLe.S. (Table 2). Although Le.S. had hypercholesterolemia(280 mg/dl in 1985), his present total cholesterol and LDLcholesterol levels are in the normal range. However, he hasrecently developed diabetes and is presently hypertriglycer-idemic. Several unaffected S. family members also havehypercholesterolemia, but its basis is not yet clear.Because the molecular defect associated with familial
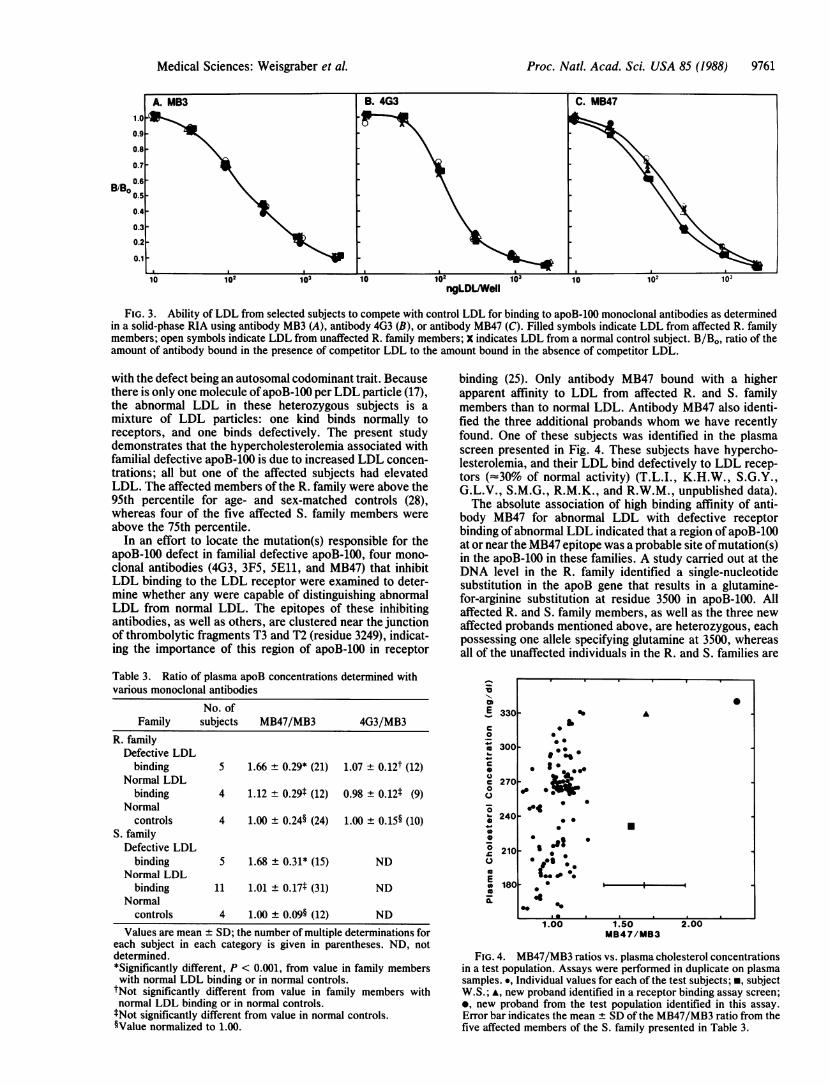
defective apoB-100 appears to reside in apoB-100 (16), wesought to localize the region of apoB-100 that contained themutation(s). Because the mutation(s) affects receptor bind-ing, we examined the binding to LDL of four apoB-100-specific monoclonal antibodies (4G3, 3F5, SEll, andMB47), all of which block LDL binding to the LDL receptor.A fifth antibody, MB3, which does not block receptor bindingof LDL and whose epitope is in the amino-terminal region ofapoB, was used as a control. As shown in Fig. 3 A and B,antibofjes MB3 and 4G3 bound equally well to all LDLsamples. Similar results were obtained with antibodies 5E11and :F5 (data not shown). However, with antibody MB47(Fig. 3C), the binding curves of abnormal LDL were dis-placed to the left compared with those from a normal subjector from unaffected family members. This displacement tolowgr LDL concentrations was reproducible in three sepa-rate assays and probably reflects a higher binding affinity ofabnopmal LDL for antibody MB47 than normal LDL display.The fPL from affected subjects P.B. and E.S. were also
Lo ¼LL
G.R. Sta.R. W.R.
Ste.R.
C.R. M.R.
J.P. M.P. PB.
E.S.
FIG. 1. Pedigree of the R. family. Subjects whose LDL havebeen examined for receptor binding activity are indicated by initialsbelow the symbols; the arrow indicates the proband. Half-filledsymbols indicate subjects whose LDL bound defectively to apoB,E(LDL) receptors, and unfilled symbols indicate subjects whose LDLbound normally. Crosses indicate that the subject is deceased.
Medical Sciences: Weisgraber et al.

9760 Medical Sciences: Weisgraber et al.
Table 1. Characterization of R. family membersCholesterol,
Family Age, mg/dl LDL IC50,*member Sex yr Plasma LDL Ag/ml
Defective LDL bindingG.R. d 68 311 202 9.3W.R. d 72 279 206 6.9C.R. d 70 336 256 7.1P.B. 9 41 324 265 5.6E.S. 9 22 250 195 8.0
7.4 ± 1.4tNormal LDL binding
Sta.R. d 64 181 ND 2.9Ste.R. 34 236 158 3.0J.P. 9 42 138 73 2.3M.P. 9 40 149 68 2.4
2.6 ± 0.4t
ND, not determined.*Concentration of LDL required to inhibit 50% of I251-labeled LDLbinding to normal cultured human fibroblasts.tx± SD.
consistently -50% more effective in binding to antibodyMB47 than were LDL from unaffected subjects J.P. and M.P.(data not shown) Qr from normal, control subjects.The foregoing data demonstrate that immunoassays using
antibody MB47 can distinguish abnormal LDL from normalLDL. Because this assay requires prior isolation of LDL byultracentrifugation, a more direct assay was developed,based on a method used to study the polymorphism detectedby antibody MB19 (26). The "apparent" plasma concentra-tion of apoB was determined by using antibody MB47 in acompetitive RIA. The value is referred to as apparentbecause the higher affinity of abnormal LDL for the antibodywould be expected to result in an artificially high plasmavalue in subjects with abnormal LDL. The "true" concen-tration of apoB was determined in a parallel assay usingantibody MB3, and the results from both assays wereexpressed as a ratio. The MB47/MB3 ratio from normalcontrol subjects was normalized to 1.00 because the absolutevalue of this ratio varies slightly from assay to assay. Theratios for unaffected and affected family members were thenexpressed relative to this normalized value. As shown inTable 3 for the R. family, the ratios for normal controls andunaffected family members were essentially identical, where-as the ratio for family members with abnormal LDL wassignificantly higher (-60%). This higher ratio reflects thehigher binding affinity of antibody MB47 for abnormal LDL.In a similar assay, the 4G3/MB3 ratios for plasma fromcontrol, unaffected, and affected subjects were not siglifi-cantly different (Table 3). In the S. family also, the MB47/MB3ratio was significantly higher in the 5 affected subjects than inthe 11 unaffected family members (1.68 vs. 1.01, Table 3). Theratio is quite reproducible for a given individual. For example,the normalized values determined on 5 separate days for the
Table 2. Characterization of S. family members
Cholesterol,
Family Age, mg/dl LDL IC5o,*member Sex yr Plasma LDL Ag/ml
Defective LDL bindingW.S. d 66 231 183 9.9J.L. 9 42 247 171 9.4K.L. 9 19 253 190 7.4M.S. d 52 257 168 5.4Le.S. d 68 197 122 6.6
7.7 ± 1.9tNormal LDL binding
S.St. 9 38 182 105 2.2T.N. 9 21 141 78 2.7B.S. d 37 243 178 2.1R.W. 23 219 147 2.4K.J. 9 21 174 115 3.2S.J. 9 18 154 86 2.8J.J. d 19 171 98 2.4L.S. d 64 172 102 1.8R.S. d 42 213 146 2.3C.L. 9 39 210 136 2.8J.M. 9 40 188 112 1.9
2.4 0.4t*As defined in Table 1.tx± SD.
proband W.S. were 1.56, 1.95, 1.82, 1.60, and 1.91 (x ± SD -1.77 ± 0.18).To test the usefulness of this assay as a screening tool and
to rule out the possibility that the high MB47 value wassimply an artifact associated with hypercholesterolemia, wetested 110 subjects. The assay was done in duplicate (in ourexperience, duplicate analyses are sufficient to distinguishsubjects with familial defective apoB-100 from unaffectedsubjects), and subject W.S. and another affected proband,identified in a receptor assay screen, were included aspositive controls. The MB47/MB3 ratio was determined foreach subject, was normalized to 1.00 for subjects with aplasma cholesterol of <240 mg/dl, and was compared withthe value in subjects with levels >240 mg/dl. There was nosignificant difference between the ratios of the two groups(<240 mg/dl, x ± SD = 1.00 ± 0.11, n = 42; >240 mg/dl,x + SD = 1.07 ± 0.10, n = 67). The MB47/MB3 ratio vs.plasma cholesterol concentration is presented in Fig. 4.Fig. 4 also demonstrates that the MB47/MB3 values forthe two positive controls (m, A) clearly lie outside the rangeof values for the test subjects. In addition, one test subjectwith a cholesterol level of 355 mg/dl was found to have aMB47/MB3 ratio of 2.37, indicating that this subject wasaffected with familial defective apoB-100. This was con-firmed in a receptor binding assay that showed that thissubject's LDL bound defectively to LDL receptors (34% ofnormal).
DISCUSSION
R.W. K.J. S.J. J.J. T.N.
FIG. 2. Pedigree of the S. family (legend as in Fig. 1,
Familial defective apoB-100 represents a recently describedgenetic disorder that is characterized by hypercholesterol-emia and by abnormal LDL that bind poorly to LDLreceptors (16). The functional defect in the LDL appears tobe caused by a mutation(s) in apoB-100 (16). This results inincreased concentrations of LDL cholesterol in these sub-jects because of a retarded clearance rate for plasma LDL(15). We have examined two kindreds (R. and S.) with this
K.L. disorder and have demonstrated transmittance through threegenerations (Figs. 1 and 2). The pattern of inheritance, in
).which the affected subjects are heterozygotes, is consistent
Proc. Natl. Acad. Sci. USA 85 (1988)
Proc. Natl. Acad. Sci. USA 85 (1988) 9761
B/B.0.4 -
0.3 -
0.2 -
0.1 -I~| a I _ I \S.a I
10 102 103 10 102 103 10 102 lo,ngLDL/Well
FIG. 3. Ability of LDL from selected subjects to compete with control LDL for binding to apoB-100 monoclonal antibodies as determinedin a solid-phase RIA using antibody MB3 (A), antibody 4G3 (B), or antibody MB47 (C). Filled symbols indicate LDL from affected R. familymembers; open symbols indicate LDL from unaffected R. family members; X indicates LDL from a normal control subject. B/Bo, ratio of theamount of antibody bound in the presence of competitor LDL to the amount bound in the absence of competitor LDL.
with the defect being an autosomal codominant trait. Becausethere is only one molecule ofapoB-100 per LDL particle (17),the abnormal LDL in these heterozygous subjects is amixture of LDL particles: one kind binds normally toreceptors, and one binds defectively. The present studydemonstrates that the hypercholesterolemia associated withfamilial defective apoB-100 is due to increased LDL concen-trations; all but one of the affected subjects had elevatedLDL. The affected members of the R. family were above the95th percentile for age- and sex-matched controls (28),whereas four of the five affected S. family members wereabove the 75th percentile.
In an effort to locate the mutation(s) responsible for theapoB-100 defect in familial defective apoB-100, four mono-clonal antibodies (4G3, 3F5, 5Ell, and MB47) that inhibitLDL binding to the LDL receptor were examined to deter-mine whether any were capable of distinguishing abnormalLDL from normal LDL. The epitopes of these inhibitingantibodies, as well as others, are clustered near the junctionof thrombolytic fragments T3 and T2 (residue 3249), indicat-ing the importance of this region of apoB-100 in receptor
Table 3. Ratio of plasma apoB concentrations determined withvarious monoclonal antibodies
No. ofFamily subjects MB47/MB3 4G3/MB3
R. familyDefective LDL
binding 5 1.66 ± 0.29* (21) 1.07 ± 0.12t (12)Normal LDL
binding 4 1.12 ± 0.29t (12) 0.98 ± 0.12t (9)Normal
controls 4 1.00 ± 0.24§ (24) 1.00 ± 0.15§ (10)S. family
Defective LDLbinding 5 1.68 ± 0.31* (15) ND
Normal LDLbinding 11 1.01 ± 0.17t (31) ND
Normalcontrols 4 1.00 ± 0.09§ (12) ND
Values are mean ± SD; the number of multiple determinations foreach subject in each category is given in parentheses. ND, notdetermined.*Significantly different, P < 0.001, from value in family memberswith normal LDL binding or in normal controls.tNot significantly different from value in family members withnormal LDL binding or in normal controls.tNot significantly different from value in normal controls.§Value normalized to 1.00.
binding (25). Only antibody MB47 bound with a higherapparent affinity to LDL from affected R. and S. familymembers than to normal LDL. Antibody MB47 also identi-fied the three additional probands whom we have recentlyfound. One of these subjects was identified in the plasmascreen presented in Fig. 4. These subjects have hypercho-lesterolemia, and their LDL bind defectively to LDL recep-tors (==30% of normal activity) (T.L.I., K.H.W., S.G.Y.,G.L.V., S.M.G., R.M.K., and R.W.M., unpublished data).The absolute association of high binding affinity of anti-
body MB47 for abnormal LDL with defective receptorbinding of abnormal LDL indicated that a region ofapoB-100at or near the MB47 epitope was a probable site ofmutation(s)in the apoB-100 in these families. A study carried out at theDNA level in the R. family identified a single-nucleotidesubstitution in the apoB gene that results in a glutamine-for-arginine substitution at residue 3500 in apoB-100. Allaffected R. and S. family members, as well as the three newaffected probands mentioned above, are heterozygous, eachpossessing one allele specifying glutamine at 3500, whereasall of the unaffected individuals in the R. and S. families are
0Er-
._.
e-0
0C0
-
S
0Si
E
330[
300[
270
240
2101
180
0
S
* 3*s
S :0
0.
1.00 1.50MB47/MB3
FIG. 4. MB47/MB3 ratios vs. plasma cholesterol concentrationsin a test population. Assays were performed in duplicate on plasmasamples. ., Individual values for each of the test subjects; *, subjectW.S.; A, new proband identified in a receptor binding assay screen;*, new proband from the test population identified in this assay.Error bar indicates the mean + SD of the MB47/MB3 ratio from thefive affected members of the S. family presented in Table 3.
2.00
A
U
Medical Sciences: Weisgraber et al.

9762 Medical Sciences: Weisgraber et al.
homozygous, with both alleles specifying arginine at 3500(29). Thus, to date there is a perfect correlation amongdefective receptor binding activity, increased MB47 affinity,and a glutamine-for-arginine substitution at residue 3500 in allsubjects with familial defective apoB-100. Therefore, itseems likely that the glutamine substitution at residue 3500 isresponsible for the enhanced binding of antibody MB47 byabnormal LDL. Residue 3500 is within the MB47 epitope (5)and, compared with published sequences of apoB-100 (3-6),this substitution is the only difference in this region of theapoB gene from affected subject G.R. (29).Determining the frequency of familial defective apoB-100
will require extensive screening studies. Our recent identifi-cation of three additional probands with familial defectiveapo-B100 indicates that this mutation may be common andtherefore may contribute significantly to primary hypercho-lesterolemia in the general population. The contribution ofthis disorder to increasing the risk of atherosclerosis inaffected subjects is another important question that can onlybe answered by identifying a large group of subjects forfurther study. The convenience of the parallel immunoassayson plasma using antibodies MB47 and MB3 should allow theefficient screening of large populations. The identification ofan additional proband with familial defective apoB-100 in ourtest population demonstrates the effectiveness of this assayas a screening tool.
We thank Maureen Balestra and Martha Kuehneman for excellenttechnical assistance. We also thank Kerry Humphrey and SylviaRichmond for manuscript preparation, James X. Warger for graphicart, and Al Averbach and Sally Gullatt Seehafer for editorialassistance. We are grateful to Drs. Joseph Witztum and LindaCurtiss for generously supplying antibody MB47 and to Drs. RossMilne and Yves Marcel for providing antibodies 4G3, 3F5, and SE11.Special thanks are extended to members of the R. and S. families,who graciously participated in these studies. This work was sup-ported by Grant HL 36701 from the National Institutes of Health.
1. Brown, M. S. & Goldstein, J. L. (1986) Science 232, 34-47.2. Mahley, R. W. & Innerarity, T. L. (1983) Biochim. Biophys.
Acta 737, 197-222.3. Chen, S.-H., Yang, C.-Y., Chen, P.-F., Setzer, D., Tanimura,
M., Li, W.-H., Gotto, A. M., Jr., & Chan, L. (1986) J. Biol.Chem. 261, 12918-12921.
4. Cladaras, C., Hadzopoulou-Cladaras, M., Nolte, R. T., Atkin-son, D. & Zannis, V. I. (1986) EMBO J. 5, 3495-3507.
5. Knott, T. J., Pease, R. J., Powell, L. M., Wallis, S. C., Rall,S. C., Jr., Innerarity, T. L., Blackhart, B., Taylor, W. H.,Marcel, Y., Milne, R., Johnson, D., Fuller, M., Lusis, A. J.,McCarthy, B. J., Mahley, R. W., Levy-Wilson, B. & Scott, J.(1986) Nature (London) 323, 734-738.
6. Law, S. W., Grant, S. M., Higuchi, K., Hospattankar, A.,Lackner, K., Lee, N. & Brewer, H. B., Jr. (1986) Proc. Nati.Acad. Sci. USA 83, 8142-8146.
7. Kannel, W. B., Castelli, W. P. & Gordon, T. (1979) Ann.Intern. Med. 90, 85-91.
8. Heiss, G. & Tyroler, H. A. (1982) Proceedings ofthe Workshopon Apolipoprotein Quantification (U.S. Dept. of Health andHuman Services, National Institutes of Health, Bethesda,MD), NIH Publ. No. 83-1266, pp. 7-24.
9. Goldstein, J. L. & Brown, M. S. (1983) in The Metabolic BasisofInherited Disease, eds. Stanbury, J. B., Wyngaarden, J. B.,Fredrickson, D. S., Goldstein, J. L. & Brown, M. S. (Mc-Graw-Hill, New York), 5th Ed., pp. 672-712.
10. Expert Panel, National Cholesterol Education Program (1988)Arch. Intern. Med. 148, 36-69.
11. Wilson, P. W., Garrison, R. J., Castelli, W. P., Feinleib, M.,McNamara, P. M. & Kannel, W. B. (1980) Am. J. Cardiol. 46,649-654.
12. Lipid Research Clinics Program (1984) J. Am. Med. Assoc. 251,365-374.
13. Brown, M. S. & Goldstein, J. L. (1987) in Harrison's Principlesof Internal Medicine, eds. Braunwald, E., Isselbacher, K. J.,Petersdorf, R. G., Wilson, J. D., Martin, J. B. & Fauci, A. S.(McGraw-Hill, New York), 11th Ed., Vol. 2, pp. 1650-1661.
14. Grundy, S. M. & Vega, G. L. (1985) J. Lipid Res. 26, 1464-1475.
15. Vega, G. L. & Grundy, S. M. (1986) J. Clin. Invest. 78, 1410-1414.
16. Innerarity, T. L., Weisgraber, K. H., Arnold, K. S., Mahley,R. W., Krauss, R. M., Vega, G. L. & Grundy, S. M. (1987)Proc. Natl. Acad. Sci. USA 84, 6919-6923.
17. Elovson, J., Jacobs, J. C., Schumaker, V. N. & Puppione,D. L. (1985) Biochemistry 24, 1569-1578.
18. Young, S. G., Witztum, J. L., Casal, D. C., Curtiss, L. K. &Bernstein, S. (1986) Arteriosclerosis 6, 178-188.
19. Steele, B. W., Koehler, D. F., Azar, M. M., Blaszkowski,T. P., Kuba, K. & Dempsey, M. E. (1976) Clin. Chem. 22, 98-101.
20. Friedewald, W. T., Levy, R. I. & Fredrickson, D. S. (1972)Clin. Chem. 18, 499-502.
21. Lowry, 0. H., Rosebrough, N. J., Farr, A. L. & Randall, R. J.(1951) J. Biol. Chem. 193, 265-275.
22. Goldstein, J. L., Basu, S. K. & Brown, M. S. (1983) MethodsEnzymol. 98, 241-260.
23. Innerarity, T. L., Pitas, R. E. & Mahley, R. W. (1986) MethodsEnzymol. 129, 542-566.
24. Fisher, R. A. & Yates, F. (1953) Statistical Tables for Biolog-ical, Agricultural and Medical Research (Oliver & Boyd,Edinburgh), 4th Ed., p. 60.
25. Marcel, Y. L., Innerarity, T. L., Spilman, C., Mahley, R. W.,Protter, A. A. & Milne, R. W. (1987) Arteriosclerosis 7, 166-175.
26. Young, S. G., Bertics, S. J., Curtiss, L. K., Casal, D. C. &Witztum, J. L. (1986) Proc. Natl. Acad. Sci. USA 83, 1101-1105.
27. Curtiss, L. K. & Edgington, T. S. (1982) J. Biol. Chem. 257,15213-15221.
28. The Lipid Research Clinics (1980) Population Studies DataBook Vol. 1: The Prevalence Study (U.S. Dept. of Health andHuman Services, National Institutes of Health, Bethesda,MD), NIH Publ. No. 80-1527.
29. Soria, L. F., Ludwig, E. H., Clarke, H. R. G., Vega, G. L.,Grundy, S. M. & McCarthy, B. J. (1989) Proc. Natl. Acad. Sci.USA, in press.
Proc. Natl. Acad. Sci. USA 85 (1988)