extrachromosomal recombination in mammalian cells as studied
TRANSCRIPT

MOLECULAR AND CELLULAR BIOLOGY, Jan. 1987, p. 129-140 Vol. 7, No. 10270-7306/87/010129-12$02.00/0
Extrachromosomal Recombination in Mammalian Cells as Studiedwith Single- and Double-Stranded DNA Substrates
FWU-LAI M. LIN,* KAREN M. SPERLE, AND NAT L. STERNBERGCentral Research and Development Department, Experimental Station, Molecular Biology, E. I. Du Pont de Nemours
and Company, Inc., Wilmington, Delaware 19898
Received 30 May 1986/Accepted 26 September 1986
We have previously proposed a model to account for the high levels of homologous recombination that canoccur during the introduction ofDNA into mammalian cells (F.-L. Lin, K. Sperle, and N. Sternberg, Mol. Cell.Biol. 4:1020-1034, 1984). An essential feature of that model is that linear molecules with ends appropriatelylocated between homologous DNA segments are efficient substrates for an exonuclease that acts in a 5'-)3'direction. That process generates complementary single strands that pair in homologous regions to produce anintermediate that is processed efficiently to a recombinant molecule. An alternative model, in which stranddegradation occurs in the 3'-+5' direction, is also possible. In this report, we describe experiments that testedseveral of the essential features of the model. We first confirmed and extended our previous results withdouble-stranded DNA substrates containing truncated herpesvirus thymidine kinase (tk) genes (tkA5' andtkA3'). The results illustrate the importance of the location of double-strand breaks in the successfulreconstruction of the tk gene by recombination. We next transformed cells with pairs of single-stranded DNAscontaining truncated tk genes which should anneal in cells to generate the recombination intermediatespredicted by the two alternative models. One of the intermediates would be the favored substrate in our original5'- 3' degradative model and the other would be the favored substrate in the alternative 3'--5' degradativemodel. Our results indicate that the intermediate favored by the 3'--5' model is 10 to 20 times more efficientin generating recombinant tk genes than is the other intermediate.
DNA introduced into cultured mammalian cells, either asa calcium phosphate precipitate or by direct microinjectioninto nuclei, is capable of undergoing efficient homologousrecombination before it becomes integrated into chromo-somes (11, 13, 18, 22, 28, 32, 34, 35, 41, 42). Frequencies ofhomologous recombination in the range of 10 to 20% arecommon in these sorts of experiments (22). Recent in vitrorecombination studies with nuclear extracts indicate that theenzymes necessary to execute homologous recombinationare indeed present in mammalian cells (9, 15, 17, 19).Two general mechanistic models have been proposed to
account for the process of recombination during the intro-duction of DNA into mammalian cells. One, the double-strand (ds)-break repair model, is analogous to that proposedto explain homologous recombination in yeasts (37). Thesecond, the single-strand (ss) annealing model (22), is anal-ogous to a model originally proposed by Broker and Lehman(4) to explain recombination by bacteriophage T4 and onelater proposed by Lai and Nathans (20) to explain thegeneration of deletions after the introduction of simian virus40 DNA into mammalian cells. In the ds-break repair model,recombination is initiated by a ds break or gap in one DNAmolecule which is repaired faithfully with a second unbrokencopy of the homologous DNA as a template. A requirementof the model is that ends of the repaired DNA molecule belocated in a region of homology shared by both DNAmolecules. While this model can account for the high level ofrecombination between certain of the DNA substrates intro-duced into mammalian cells (3, 18, 36), it does not effectivelyexplain other efficient recombination events generated byintroducing into cells DNAs with ends located in nonhomol-
* Corresponding author.
129
ogous regions (1, 22, 40) nor does it explain why recombi-nation between DNAs introduced into mammalian cells islargely nonconservative (1, 6, 22, 40). To account for theseresults, we proposed the ss annealing model (22). An essen-tial feature of this model is that ends generated by a ds cut inDNA are substrates for a strand-specific exonuclease thatproduces complementary ss regions. Those regions arepaired to generate intermediate structures that are subse-quently processed to recombinant molecules (scored asintact genes). The success of this reaction depends on theoriginal location of the ds cut and the nature of the pairedrecombination intermediate produced after strand degrada-tion. In contrast to the ds-break repair model, the ss anneal-ing model does not require that the ds break be in a region ofshared homology, only that it be proximal to the recombin-ing homologous regions. In a recent report (40) a modifiedversion of the model was proposed to account for recombi-nation in simian virus 40 DNA molecules. In that model thess involved in the pairing process are generated by DNAunwinding from free ends rather than by strand-specificexonuclease degradation.To gain more insight into the mechanism of extrachromo-
somal recombination in mammalian cells, we transformedcells with pairs of ss DNAs that will generate the interme-diates proposed by the ss annealing model and measured theability of those intermediates to produce recombinant mol-ecules. The results allow us to confirm certain features of thess annealing model and refine others.
MATERIALS AND METHODSCell strains and media. The growth of LMtk- aprt- mouse
cells has been previously described (24). Hypoxanthine-aminopterin-thymidine (HAT) medium (38) was used forselection of thymidine kinase-positive (Tk+) transformants.

130 LIN ET AL.
A
Ix _I
tkmopLlr LOtCNCM
+ r tk st ructural
0o
+
+ Nq
FIG. 1. DNA substrates used to assay intramolecular homologous recombination. In panel A some of the landmarks of the tk gene ofherpes simplex virus type 1 are depicted. The NT positions of these landmarks are shown in parentheses and are from sequencing data ofWagner et al. (39) and acrylamide gel analysis of restriction enzyme digests (unpublished data). The exact position of deletion endpoints wasdetermined by dideoxy sequencing (33) in our laboratory. NT 1 in this scheme has been assigned to the starting NT of the mRNA with NTsupstream of that point designated with a minus prefix and those downstream with a plus prefix. Two in-frame ATG codons (NTs +108 and+243) are shown. Removal of both is necessary to completely inactivate the tk gene. In panel B the plasmids used to assay recombinationare described. Construction of plasmid ptk2DR is described by Lin et al. (22), and construction of the other plasmids is described in theMaterials and Methods. Each of the plasmids contains two defective tk genes. The two regions designated PH (proximal to homology) andDH (distal to homology) are also shown. The PH region is so designated because it is closer to the tk deletion junctions and to the shared tkhomology than it is to the tk sequences not shared by the two defective tk genes. The DH region has just the opposite properties. All plasmidscontain the ampicillin resistance gene and the pBR322 origin ofDNA replication. Symbols: -, pBR and non-herpesvirus DNA; ED , non-tkherpesvirus DNA; _, shared tk homology; ci>=, tkA&3' DNA from the mRNA start site to the region of shared homology; c> I , tkA&5' DNAfrom the region of shared tk homology to the tk termination codon. The arrowheads indicate the direction of tk transcription. Abbreviationsused: Bm, BamHI; C, ClaI; H, HindIII; K, KpnI; N, NcoI; Sc, SacI; Sl, Sail; Ori, origin.
Escherichia coli HB101 (2) and JM1O1 (27) were used toprepare plasmid and M13 DNAs, respectively.
Plasmid-tk DNAs. ptk contains the herpesvirus 3.4-kilobase (kb) BamHI tk fragment cloned in pBR322 at theplasmid BamHI site (12). ptk2.0 contains the 2.0-kb PvuII tkfragment with SalI linker ends cloned in pBR322 at theplasmid Sall site. The direction of the transcription isopposite that of the tetracycline gene. Plasmid pStk is the
2.0-kb PvuII tk fragment cloned between the unique AatIIand PvuII sites of pBR322 with XhoI linkers. The construc-tion of plasmid ptk2DR has been previously described (22). Itis shown in Fig. 1. It contains pBR322 with inserts of twotruncated tk genes, tkA3' and tkA5'. The two mutant genesshare 320 base pairs (bp) of homology and are oriented in thesame counterclockwise direction. The two genes are sepa-rated on one side by about 1.5 kb and on the other side by 5.8
gene - l
+ E :+
IZ I K
-0I I-
=01
I I4.
II
DH07kb
MOL. CELL. BIOL.

EXTRACHROMOSOMAL RECOMBINATION IN MAMMALIAN CELLS 131
kb of plasmid and non-tk-herpesvirus DNA. The shorter ofthese two segments was reduced to 180 bp in ptk2DRABC(Fig. 1) by first deleting the Sall-KpnI fragment and then theBamHI-ClaI fragment from pt&2DR DNA. A BamHI linkerwas ligated to the end generated by the BamHI-ClaI junc-tion. Figure 1 also shows the structure of plasmid ptk2DRA.It was constructed as follows. The starting plasmid is ptkA3'.It has the tkA3' gene (22) cloned into plasmid pBR327 at theBamHI site with the tk gene oriented in the same direction asthe tetracycline gene. Into that plasmid we inserted betweenthe unique ClaI and HindlIl sites of the vector a portion ofthe tk gene (tkA5') extending from the SphI site at nucleotide+493 (NT +493) to the PvuII site at NT +1839 (Fig. 1A).The insertion destroys the ClaI site. A unique SacI site wascreated at the edge of the tkA5' deletion. The two truncatedtk genes in ptk2DRA are oriented in the clockwise directionand share 410 bp of homology. They are separated on oneside by 1.7 kb and on the other by 4.1 kb of plasmid andnon-tk-herpesvirus DNA. To reduce the shorter non-tk DNAsegment, we deleted the NcoI fragments located in thisregion. This generates a plasmid, ptk2DRAAN (Fig. 1), withonly 685 bp between the tk genes and with unique BamHIand SalI sites downstream of tkA3' DNA.M13-tk DNAs. Two sets of mutant tk genes were cloned
into M13mplO and M13mpll vectors. During the earlystages of this work, we used a set of M13mplO clones thatcontains tkA3' and tkA5' genes sharing 410 bp of homology.The clones mplOtkA3'NC and mplOtkA3'C were constructedwith the 1.76-kb BamHI tkA3' fragment (22) cloned at theBamHI site of M13mplO in either of the two possibleorientations. In mplOtkA3'NC the direction of tk transcrip-tion is the same as that of the lacZ gene, while inmplOtkA3'C it is opposite that of lacZ. Here NC and Cdenote that the noncoding (nontranscribed) strand or coding(transcribed) strand of tk, respectively, is present in the M13DNA. The other two clones in this set, mplOtkA5'NC andmplOtkA5'C, contain the A5' SphI (NT +493)-PvuII (NT+ 1839) tk fragment cloned in both orientations at the SmaIsite of M13mplO. A second set of M13 clones was con-structed with deletions generated by Bal 31 digestion of tkDNA. The tkA5'm deletion removes sequences upstream oftk NT +257, and the tkA3'm deletion removes sequencesdownstream of tk NT + 1142. The two mutant tk genes share885 bp of homology. Oligonucleotide linkers with SacI orBamHI site were inserted at the deletion junctions of tkA5'mand tkA3'm, respectively. The tkA3'm fragment, bounded bya SailI site (generated from the PvuII site at -202; Fig. 1) anda BamHI site, was cloned between the Sail and BamHI sitesof M13mplO or M13mpll to create mplOtkA3'mC andmplltkA3'mNC, respectively. Similarly, the tkA5'm frag-ment, bounded by a Sall site (generated from the PvuII siteat + 1839; Fig. 1) and a Sacl site, was cloned between theSailI and SacI sites of M13mplO and M13mpll to createmplOtkA5'mNC and mplltkA5'mC, respectively.
Preparation of plasmid and bacteriophage DNAs. PlasmidDNA was prepared as previously described (7). ss DNAswere isolated from phage particles grown in JM101 inNZCYM medium (25). Phage were concentrated and washedas described by Hines and Ray (14) and then banded in CsClgradients. Purified phage particles were then digested withproteinase K (100 .g/ml for 1 h at 37°C), and the DNA wasextracted several times with phenol and ethanol precipi-tated. ds DNA concentrations were determined by fluores-cence in the presence of Hoechst 33258 (5). ss DNA con-centrations were determined by the A260 assuming that a37-,ug/ml solution has an A260 = 1. All DNAs were stored at
-20 or -70°C. Under these conditions over 90% of the dsDNA retains its superhelicity and over 90% of the ss DNAremains circular for at least a year.DNA transformation. LMtk- aprt- cells were prepared for
transformation as described by Lin and Stemnberg (24) andwere transformed with DNA precipitates prepared by amodification of a method used to transform cells withbacteriophage lambda (16). Briefly, various DNAs werepipetted into 0.5 ml of a solution containing 10 mM Trishydrochloride (pH 7.6), 1 mM EDTA, 250 mM CaC12, andcarrier DNA (10 ,ug of sheared LMtk-). To that mixture weadded 0.5 ml of a solution containing 50 mM PIPES[piperazine-N,N'-bis(2-ethanesulfonic acid)]-NaOH (pH6.9), 280 mM NaCl, and 1.5 mM sodium phosphate. A fineDNA precipitate formed almost immediately and was addedto the cells within 10 min. Unless specified, when two ssDNAs were used as a pair, they were simply pipetted intothe CaCl2 solution individually without prior treatment.After transformation, TK+ transformants were selected inHAT medium as described by Lin and Sternberg (24). dsDNAs were digested with restriction enzymes (from Be-thesda Research Laboratories, Inc., Gaithersberg, Md., NewEngland BioLabs, Inc., Beverly, Mass., or BoehringerMannheim Biochemicals, Indianapolis, Ind.) according tovendor specifications. Each digestion was checked byagarose gel electrophoresis.DNA hybridization. The presence of tk DNA in trans-
formed cells was analyzed by Southern blotting as describedby Lin and Stemnberg (24). The only change was the use ofBiodyne nylon filter paper (from Pall Ultrafine FiltrationCorp.) instead of nitrocellulose filter paper.
RESULTS
Effect of ds DNA breaks on recombination. In a previousstudy we used the plasmid ptk2DR (Fig. 1) to study theeffects of ds DNA breaks on recombination (22). One of theconclusions from that work was that breaks play a centralrole in the initiation of recombination and that their positionwithin the DNA substrate is important. To extend thosestudies, we constructed three additional tki2 plasmids:ptk2DRABC, ptk2DRA, and ptk2DRAAN (Fig. 1). Bothptk2DR and ptk2DRA contains 3'- and 5'-deleted tk genes indirectly repeated orientation. They differ in that the genesare oriented counterclockwise for ptk2DR and clockwise forptk2DRA. In addition, the amount of DNA between the tkgenes differs in the two plasmids. The distance between thetk deletion junctions, the segment of DNA proximal to theregion of shared tk homology (PH; Fig. 1), is 1.5 kb forptk2DR and 4.1 kb for ptk2DRA. The distance between the 5'end of tkA3' and the 3' end of tkAS', the segment of DNAdistal to the shared region of tk homology (DH; Fig. 1), is 5.8kb for ptk2DR and 1.7 kb for ptk2DRA. In ptk2DRABC thePH segment has been shortened to 180 bp, and inptk2DRAAN the DH distance has been shortened to 685 bp.The results of transformation with these plasmid DNAs areshown in Table 1. When supercoiled plasmid DNAs wereused, the transformation frequency was low in all cases.Compared with transformation with an intact tk gene (ptk),the efficiency ranges from <0.01% for ptk2DR to 0.1% forptk2DRAAN. In contrast, if ds breaks are introduced in vitrointo the PH region of the ptk2 DNAs at restriction sitessymmetrically located between the truncated tk genes, thenthere is a pronounced stimulation of transformation. Forptk2DR, this was achieved by cutting the DNA at the uniqueHindIII site. This created a ds break 0.48 and 1.0 kb from the
VOL. 7, 1987

132 LIN ET AL.
TABLE 1. Effects of ds breaks on intramolecular recombinationof ptk2 DNAs in LMtk- cells
DNA site(s) cleaved DNA colonies/plate
ptk2DR None 50 0.7 ± 0.6HindIII 50 409 ± 59Sall 50 3.7 ± 0.6Ncol 50 0
ptk2DRABC None 50 5 ± 1.7BamHI 50 2,160 ± 62
ptk2DRAAN None 50 16 ± 4.4Ncol 50 0BamHI 50 293 ± 42BamHI + Sacl 50 1,720 ± 35
ptk None 3 1,380 ± 1710 2,890 ± 156
BamHI 3 457 ± 4010 1,717 ± %
Ncol 3 61 ± 510 179 ± 24
PvuII 10 171 ± 31a Each datum point represents the average of three transformation plates.
tkA3' and tkA&5' deletion junctions, respectively. Digestion ofptk2DRABC DNA with BamHI or digestion of ptk2DRAANwith BamHI and Sacl also created ds breaks in the PHregion close to the tk deletion junctions. In the latter case,recombination was stimulated about 100-fold, while in theformer two cases the stimulation was about 400-fold. If theDNA was cut at a site not symmetrically located in the PHregion, e.g., ptk2DRAAN cut with BamHI, the stimulatoryeffect of the cut was reduced relative to that observed withDNA cut in a more symmetric position, but it was still quitesignificant (18-fold for BamHI-cut ptk2DRAAN DNA com-pared with 100-fold for BamHI-SacI-cut ptk2DRAAN DNA).On the other hand, if ds breaks were introduced into theDNA in an area distal to the tk homology (the DH segment),there was either very little stimulation of recombination(Sail-cut ptk2DR DNA) or an inhibition of recombination(NcoI-cut ptk2DR DNA or NcoI-cut ptk2DRAAN DNA),relative to uncut DNA. DNA digested with NcoI is aninteresting case. There is one NcoI site 286 bp upstream ofthe second distal transcriptional signal of tk (26) (396 bp fromthe beginning of the tk mRNA) and two others 289 and 377bp downstream of the tk termination codon. Thus, when anyone of the ptk2 substrates is digested with Ncol the DNA iscleaved fairly symmetrically between the 3' and 5' ends ofthe tk genes in the DH segment (Fig. 1). If this DNA is usedto transform cells, recombinants are not detected (Table 1).We suspect that part of this reduction in transformationefficiency is due to an interference with tk expression byNcoI digestion. To test this hypothesis, we digested plasmidptk, which contains an intact tk gene, with various restrictionenzymes and then measured the transforming efficiency ofthe products. Digestion of ptk with BamHI, NcoI, and PvuIIgenerated DNA fragments with intact tk genes of 3.4, 1.9,and 2.0 kb, respectively (Fig. 1) (39). BamHI digestionreduced transformation efficiency about threefold relative touncut ptk. Ncol or PvuII digestion reduced the number oftransformants by another 10-fold. On the basis of theseresults we presume that there are two factors acting torepress TK+ transformation with NcoI-digested ptk2 DNA.
One is the lack of stimulation of tk recombination by DNAcleavage in the DH region of the plasmids, and the other isthe negative effect of Ncol cleavage on tk expression. Inrecent experiments we have digested ptk2DRA in the DHregion with HindlIl and have observed as much as a 100-foldreduction of transformation relative to uncut DNA (data notshown). The magnitude of this effect cannot be explainedsimply by an effect on tk expression but rather suggests thatdigestion within the DH region of this plasmid can actuallysuppress recombination. We conclude from the results inTable 1 that the cleavage of ptk2 plasmids in the regionproximal to the shared tk homology stimulates recombina-tion, while cutting in the region distal to that homology doesnot, and can even suppress recombination. It should benoted that the ds breaks need not be made in the homologoustk sequences, or at the edge of those sequences, to have astimulatory effect on recombination.
In addition to the intramolecular recombination studiesdescribed above, we also carried out intermolecular recom-bination experiments by transforming cells with ds comple-mentary defective tk DNAs (tkA5' and tkA3') cloned intoM13 vectors (see Materials and Methods). The results ofthose experiments indicate that cleavage of both DNAs inthe PH region generates many more transformants than doescleavage of both DNAs in the DH region. The difference canexceed 5,000-fold (data not shown).
All the above results are completely consistent with ourpreviously published observations (22). On the basis of thoseobservations we proposed a model (Fig. 2A) in which a 5'exonuclease acts on ds DNA ends to generate complemen-tary ss. After those strands have paired, a gap-repair processgenerates a recombinant tk gene. The model illustrates whyds breaks made in the PH region of ptk2DR DNA withHindIlI or in the PH region of ptk2DRAAN DNA withBamHI and Sacl stimulate recombination. Moreover, it alsoillustrates (Fig. 2B) why recombination levels are low whencuts are made in the DH region of ptk2DRA DNA withHindIll or NcoI or in the DH region of ptk2DR DNA withSall or Nco. Important recombination intermediates in themodel are shown in the dashed boxes in schemes A and B ofFig. 2. For scheme A which efficiently generates TK+recombinants, it is the tkA3'NC strand paired with thetkA5'C strand; and for scheme B, which does not efficientlygenerate those recombinants, it is the tkA3'C strand pairedwith the tkA5'NC strand. Based on the data presentedabove, the ability to generate TK+ recombinants from eachof the two intermediates can differ by 3 orders of magnitude.
Transformation with ss, truncated tk genes. To assess themodel shown in Fig. 2, we transformed cells with ss DNAscapable of generating either of the two recombination inter-mediates shown in Fig. 2 and measured the number of TK+transformants produced. Truncated tk genes (tkA3' andtkA5') were cloned in M13mplO and M13mpll vectors togenerate ss DNAs containing either the coding or noncodingtk strands (see Materials and Methods). Those strands wereisolated from purified virions and used in pairwise combina-tions to transform LMtk- cells. The number of colonies thatsurvive HAT selection is a measure of the efficiency ofrecombination. When individual ss were used we could notdetect any HAT-resistant colonies even with as much as 1 ,ugof DNA (data not shown). In contrast, transformations thatemployed pairwise combinations of ss DNAs readily pro-duced TK+ tranformants (Table 2). Two sets of experimentsare shown. The experiments in set 1 employ substrates thatshare 410 bp of tk homology, and the experiments in set 2employ substrates that share 885 bp of tk homology. In two
MOL. CELL. BIOL.

EXTRACHROMOSOMAL RECOMBINATION IN MAMMALIAN CELLS 133
AA3'NC Hind A5N
A3,c A~t5'C 5
( 1 ) tIDouble Strand Break
5; 3'51
(5' Exonuclease
(2) |iDegradation
3~~~3N35' 5 >g3'
(3) lSingle Strand Annealing
F3NC _3
(4) | Nuclease
BtA3'C Nco I AWC
53=A3NC t 5INC(1) |Double Strand Break
5' A3'C 3'5' A5'C '
A3'NC 5'3' A5NC(2) 15' Exonuclease
Degradation
5 3' 3'3'= 5' 65'NC(3) |Single Strand Annealing
r-
i &3'C,,*'O~~' 5'_ 5
_ _ _ _ _ _ ___N_uJ
(4 ) | Nuclease
3,-.~~~3
(5) Gap Repair
3't<5""3,- ¢' 7/$ 5'
I DNA Ligation
Reconstructed tk
3
tk not Reconstructed
FIG. 2. Our previously proposed model for homologous recombination during the introduction of DNA into mammalian cells. The modelis illustrated for HindIll-cut (A) or NcoI-cut (B) ptk2DR plasmid DNA. The specific steps in the model are described in the text and in ourprevious report (22). Symbols: >>>>, DNA polymerase action to restore duplex DNA. Other symbols are as described in the legend to Fig.1. Presumptive recombination intermediates are enclosed in the dashed boxes.
of the combinations of ss, tkA3'NC plus tkA5'NC and tkA3'Cplus tkA5'C, the ss tk DNAs are not complementary, and thetransformation efficiency is low and similar in the two cases.With increasing DNA concentration the number of trans-formants increases more than proportionately. For the othertwo DNA combinations, tkA3'NC plus tkA5'C (combinationA) and tkA3'C plus tkA5'NC (combination B), the tk strandsare complementary and can anneal once they enter the cellto generate the intermediates shown in Fig. 3. Note that theintermediate shown in Fig. 3A is generated by DNA combi-nation A and is analogous to the intermediate shown in Fig.2A. The intermediate shown in Fig. 3B is generated by DNAcombination B and is analogous to the intermediate shown inFig. 2B. Both DNA combinations transform cells to TK+much better than do the noncomplementary DNA combina-tions. For example, in the experiment with set 2 DNAs at 10ng per plate, we obtained an average of 35 and 470 coloniesper plate with combination A and B DNAs, respectively. Asnoted above, the noncomplementary pairs generated notransformants under these conditions. The model in Fig. 2predicts that combination A DNAs should generate TK+transformants more efficiently than combination B DNAs. Itwas surprising, therefore, that the data shown in Table 2indicate that just the opposite is true. Combination B DNAstransform cells to TK+ 10 to 20 times better than combina-tion A DNAs when the DNA is not saturating for transfor-mation. This result is true for both sets of experimentsshown in Table 2. In Fig. 4 the data in Table 2 have beenplotted to more clearly illustrate the dependence of transfor-mation efficiency on DNA concentration. With both combi-
nations of DNAs the transformation reaction appears toreach saturation at about 1,000 to 2,000 transformants perplate. In control experiments in which cells were trans-formed with an intact tk gene cloned in pBR322 (ptk2.0),approximately 400 to 1,300 colonies were obtained per plateper ng of DNA (Table 2). Thus, the efficiency of transfor-mation with the B combination of DNAs is approximately 4to 8% that of a circular DNA molecule containing an intactds tk gene. All the experiments shown in Table 2 involvetransformations of LMtk- cells with a large excess (10 ,ug) ofhomologous carrier DNA. When similar experiments wereperformed in the absence of carrier DNA, the number oftransformants was generally 10- to 50-fold less for eachcombination ofDNAs at a given DNA concentration, but therelative efficiencies of transformation with the differentDNAs were the same as in the experiment with carrier (datanot shown).
It should be noted that in our first experiments performedwith ss DNAs (preliminary results reported in reference 23),we observed that the A combination of strands transformedcells better than the B combination of strands. This result isthe opposite of that reported here. Subsequent analysis ofthe ss DNAs used in those early experiments indicated thatthe tkA&5'NC strand that was used was largely degraded. Thiswould account for the reduced level of transformation withthe B combination of DNAs. In the experiments reportedhere the ss DNAs routinely were stored at -20°C rather than4°C, as was the case in the early experiments, and the DNAswere checked before each transformation. In all cases theywere judged to be intact.
3'5'
3;5 .
3
VOL. 7, 1987
'i - - j51 31-t=:;.'Z' r Iu
I3, 1 31z -5
513-1,4- 51
5131
5J. 3I
/.f 51
134 LIN ET AL.
TABLE 2. Transformation of LMtk- cells with two defective,ss tk genesa
TK+ colonies/plateDNA 1 (ng) DNA 2 (ng)
Set lb Set 2b
ss tkA3'NC ss tkA5'NC10 10 0 0 0 030 30 1 3 4 6100 100 14 21 25 31300 300 135 194 168 200
1,000 1,000 293 303 509
ss tkA3'NC ss tkA5'C10 10 12 15 30 3930 30 90 119 144 166100 100 349 384 598 690300 300 1,000 1,000 1,230 1,430
1,000 1,000 960 980 920 1,530
ss tkA3'C ss tkA5'NC10 10 133 127 400 54630 30 846 1,140 2,080 2,110100 100 3,520 3,560 3,300 3,320300 300 4,790 5,300 3,580 5,560
1,000 1,000 5,000 5,000 4,600 4,630
ss tkA3'C ss tkAS'C10 10 0 0 0 030 30 1 3 10 15100 100 35 94 102300 300 84 158 275
1,000 1,000 175 212 682 736
ds ptk2.02 820 850 2,77010 3,000 3,000 6,480
a The two defective ss DNAs were not preannealed before being added tothe cells.
b The DNAs in the set 1 and set 2 experiments share 410 and 885 bp of tkhomology, respectively.
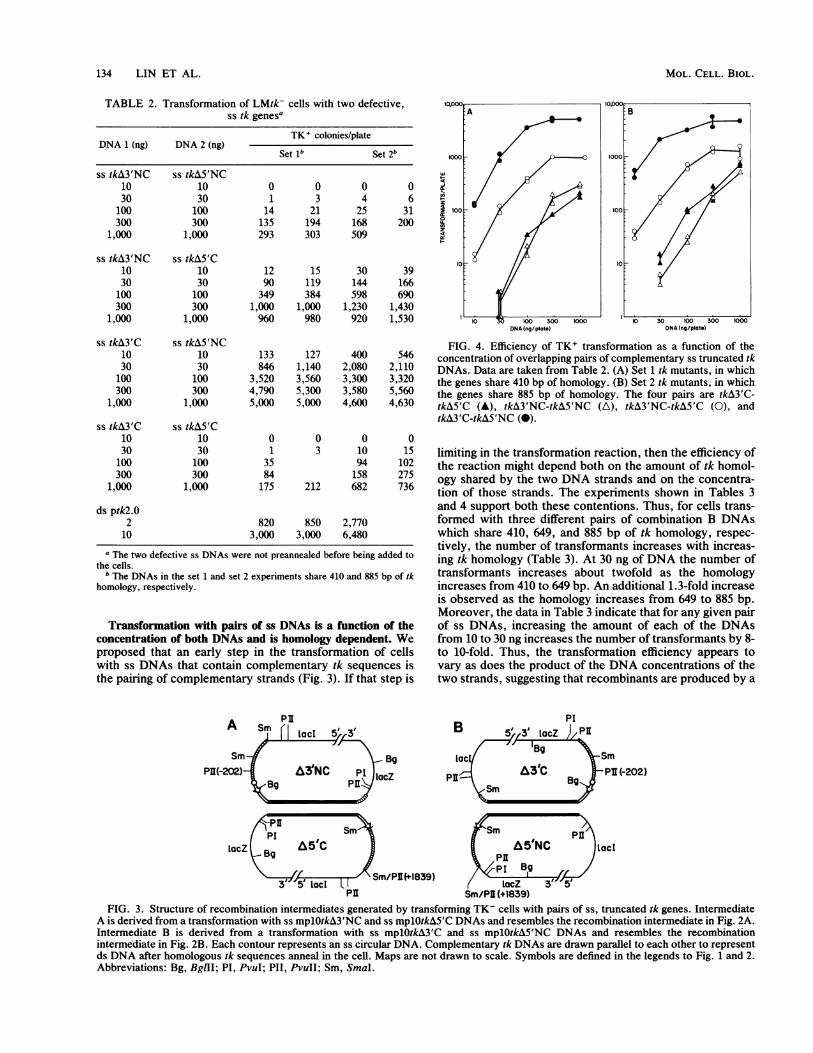
Transformation with pairs of ss DNAs is a function of theconcentration of both DNAs and is homology dependent. Weproposed that an early step in the transformation of cellswith ss DNAs that contain complementary tk sequences isthe pairing of complementary strands (Fig. 3). If that step is
PHASm icl 5' 3'
Sm BgPH(-2 13NC P lacZ
Bg P11
100. 100
10 10
10 100 300 1000 10 30 100 300 1000DNA(ng/plote) DNA (ng/plate)
FIG. 4. Efficiency of TK+ transformation as a function of theconcentration of overlapping pairs of complementary ss truncated tkDNAs. Data are taken from Table 2. (A) Set 1 tk mutants, in whichthe genes share 410 bp of homology. (B) Set 2 tk mutants, in whichthe genes share 885 bp of homology. The four pairs are tkA3'C-tkA5'C (A), tkA3'NC-tkA&5'NC (A), tkA3'NC-tkA&5'C (0), andtkA3'C-tkA5'NC (0).
limiting in the transformation reaction, then the efficiency ofthe reaction might depend both on the amount of tk homol-ogy shared by the two DNA strands and on the concentra-tion of those strands. The experiments shown in Tables 3and 4 support both these contentions. Thus, for cells trans-formed with three different pairs of combination B DNAswhich share 410, 649, and 885 bp of tk homology, respec-tively, the number of transformants increases with increas-ing tk homology (Table 3). At 30 ng of DNA the number oftransformants increases about twofold as the homologyincreases from 410 to 649 bp. An additional 1.3-fold increaseis observed as the homology increases from 649 to 885 bp.Moreover, the data in Table 3 indicate that for any given pairof ss DNAs, increasing the amount of each of the DNAsfrom 10 to 30 ng increases the number of transformants by 8-to 10-fold. Thus, the transformation efficiency appears tovary as does the product of the DNA concentrations of thetwo strands, suggesting that recombinants are produced by a
PIB 5z3' lacZ Pi
loclt 7 Bg Sm
Pi A3'C PR (-202)Sm
Bg
PRPI Sm Sm l
LacZ Bg A5'C A5'NC LadlPR1PI BIg
3 If' Laci 1m/PH1839) CZ
PH Sm/PH (+1839)FIG. 3. Structure of recombination intermediates generated by transforming TK- cells with pairs of ss, truncated tk genes. Intermediate
A is derived from a transformation with ss mplOtkA&3'NC and ss mplOtkA5'C DNAs and resembles the recombination intermediate in Fig. 2A.Intermediate B is derived from a transformation with ss mplOtkA3'C and ss mplOtkA5'NC DNAs and resembles the recombinationintermediate in Fig. 2B. Each contour represents an ss circular DNA. Complementary tk DNAs are drawn parallel to each other to representds DNA after homologous tk sequences anneal in the cell. Maps are not drawn to scale. Symbols are defined in the legends to Fig. 1 and 2.Abbreviations: Bg, BglII; PI, PvuI; PII, PvuII; Sm, SmaI.
1000
MOL. CELL. BIOL.

EXTRACHROMOSOMAL RECOMBINATION IN MAMMALIAN CELLS 135
strand-annealing process that is a limiting step in the reac-tion. It should be noted that a similar conclusion can bedrawn from the experiments with set 1, combination BDNAs shown in Table 2. Less of an effect, however, is seen
in that same table with the set 2, combination B DNAs.While the reason for the difference is not clear, it may berelated to the higher transformation frequencies seen in theexperiments of Table 2 compared with those in Table 3. Atthe high levels of transformation seen with set 2, combina-tion B DNAs in Table 2 as little as 30 ng of DNA is nearlysaturating for transformation. Accordingly, the effect ontransformation frequency of increasing the amount of DNAused from 10 to 30 ng may be diminished relative thatobserved under nonsaturating conditions. It should be notedthat if the transforming DNA contains an intact tk gene,either ss (mplOtkC) or ds (ptk2.0), the efficiency of transfor-mation is directly proportional to the amount DNA used(Table 3).
Additional support for the arguments made above comefrom experiments in which the strands are preannealedbefore being mixed in the transformation solution (Table 4).If the amount of preannealed DNAs is increased from 10 to30 ng per plate, transformation efficiency increases 2.5-fold.In the same experiment a 3-fold increase in the amount ofunannealed ss produces an 8.4-fold increase in transforma-tion efficiency. The major effect of preannealing the strandsis seen at the lower DNA concentration (10 ng). At thatconcentration preannealed DNAs transformed cells eighttimes better than unannealed DNAs. In contrast, at thehigher DNA concentration (30 ng), preannealing the DNAsproduced only a twofold effect. Thus, at least at the lowerDNA concentration used in this experiment, strand anneal-ing is a limiting step in transformation.
Southern analysis of transformants. Seven TK+ transform-ants obtained with either combination A or combination BDNAs from the first set of experiments in Table 2 werecloned. Clones Al to A7 were all selected from plates with
TABLE 3. Dependence of TK+ transformation on DNAhomology and DNA concentration with ss mutant tk genes
tk TK colonies/DNA 1 (ng)a DNA 2 (ng)a homology plate (± SD)b
(bp)
ss mplOtkA3'C ss mplOtkA5'NC10 10 410 7 2(4)30 30 410 67 14 (4)
ss mplOtkA3'mC ss mplOtkA5'NC10 10 649 15 3 (4)30 30 649 155 11 (4)
ss mplOtkA3'mC ss mplOtkAS'mNC10 10 885 27 7(4)30 30 885 215 11 (4)
ss mplOtkCc10 124 3 (2)
100 1,625 50 (2)
ds ptk2.03 810 141 (2)10 2,450 71 (2)
a DNAs were added to cells without preannealing.b The numbers in parentheses indicate the number of plates scored in each
experiment.c mplOtkC is the tk PvuII fragment (NT -202 to + 1839) cloned at the SalI
site ofM13mplO with a Sall linker. Transcription of tk is opposite that of lacZ.
TABLE 4. Effect of preannealing ss, truncated tk genes onTK+ transformation
TK+ Rela-
DNA 1 (ng) DNA 2 (ng) Prean- colonies/ tiveDNA1(ng) DNA 2 (ng) nealinga plate effi-(± SD)b ciency
ss mplOtkA3'mC ss mplOtkAS'mNC10 10 No 54 ± 10 130 30 No 452 ± 20 8.410 10 Yes 430 ± 16 8.030 30 Yes 1,101 ± 53 20.4a For the in vitro annealing reaction the two ss DNAs were mixed at 20
p.g/ml in annealing buffer (100 mM NaCl, 10 mM Tris hydrochloride [pH 7.6],1 mM EDTA), heated to 80°C for 2 min, and then cooled to 68°C and allowedto anneal for 1 h. The sample was then slowly cooled to room temperature.
b Five plates were scored in each experiment.
combination A DNAs at a concentration of 30 ng ofDNA perplate, and clones Bi to B7 were all selected from plates withcombination B DNA at a concentration of 10 ng ofDNA perplate. High-molecular-weight genomic DNA was isolatedfrom each of the clones and then digested with the restrictionenzymes BglII, SmaI, and PvuII either singly or in combi-nation. The digested DNAs were analyzed by agarose gelelectrophoresis and filter-blot hybridization. We illustratethe results of those analyses for DNAs digested with BglIIand SmaI (Fig. 5).
Since the DNA that encodes the tk transcriptional unitcontains a BgIII site and a SmaI site separated by 1.16 kb ofDNA (Fig. 1), we expected to find that all the TK+ trans-formants would have that fragment. Indeed, they all did (Fig.5). Moreover, six of the seven combination B clones (Fig.5A) and four of the seven combination A clones (Fig. SB)contained only that fragment. B6 had a fragment that mi-grated in the gel at a size of about 10 kb, and A3, A6, and A7had several fragments that migrated slower than the ex-pected fragment. The copy number of the 1.16-kb BglII-SmaI fragment in each of the transformants was about one totwo copies per diploid genome. We also analyzed SmaI andPvuII digests of DNA from the transformants. Those dataare summarized in Table 5 and indicate that there is DNArearrangement in some of the clones in the region immedi-ately flanking either the 5' or the 3' portion of the tkstructural gene. DNA upstream of the tk gene seemed to bebetter preserved than the DNA downstream of the gene.There was only one clone (B5), of the total of 14, that did notretain at least 300 bp upstream of the tk structural gene (allexcept B5 have the upstream SmaI site; Fig. 1), and all butfour clones (A2, A5, Bi, and B4) had a 600-bp regiondownstream of tk (they have the downstream PvuII site).Finally, some of the transformants (A3, A6, and A7) alsocontained DNA derived from the unrecombined defective tkgenes (tkA5' or tkA3').
DISCUSSION
Two aspects of our previously proposed strand-annealingmodel (Fig. 2) for recombination were examined in thisreport. First, we assessed the effect of the position of the dsbreak on recombination with a variety of DNA substrates,and then we determined which of the two proposed recoim-bination intermediates shown in Fig. 2 is more efficientlyconverted by the cell to an intact gene.
Location of the ds break. Our studies with the three DNAsubstrates shown in Fig. 1 (ptk2DR, ptk2DRABC, andptk2DRAAN) support the basic contention of the model that
VOL. 7, 1987

136 LIN ET AL.
A 9cNY+e g2 ,</?brA/;
kb I16.5--71,
3.6-
B .kk
9bIf Q-'6.5-7.1-
3.6-.9-
1.67-1.41 _-1.16- _
FIG. 5. Southern analyses of DNAs from TK+ transformants. The DNAs in panel A were isolated from seven TK+ transformants (Bi toB7) obtained with the complementary pairs of ss, truncated tk genes shown in Fig. 3B, and the DNAs in panel B were isolated from sevenTK+ transformants (Al to A7) obtained with the complementary pairs of ss, truncated tk genes shown in Fig. 3A. The first and last lanescontain pStk DNA. All the DNAs were digested with BglII and SmaI. The pStk DNA contains 2 pg of the tk fragment or about two copiesof the tk gene per 5 ,ug of genomic DNA (21). Size markers are from bacteriophage P1 DNA digested with EcoRI (data not shown). The probeused in these analyses is the 1.16-kb BglII-SmaI tk fragment (Fig. 1).
ds breaks within the PH region of the plasmids stimulaterecombination during the transfer of those DNAs into mam-malian cells. Thus, recombination between complementarytruncated tk genes on a ptk2 plasmid was elevated at least100-fold, relative to uncut DNA, when ptk2DR was digestedwith HindlIl; or ptkDRA&BC was digested with BamHI, orptk2DRAAN was digested with both BamHI and Sacl (Table1). The DNAs that were cleaved symmetrically between thedefective tk genes in the PH region (BamHI-SacI-digestedptk2DRAAN) recombined more efficiently than DNAscleaved assymmetrically within that region (BamHI-digested
TABLE 5. Restriction fragments present in TK+ cells generatedby transforming LMtk- cells with complementary pairs of
ss truncated tk genesaRestriction fragments detected by Southern
hybridization analysis
TK+ trans- 216r Nonrecombinantfortnant 1.16-kb 1.64-kb 2.23-kb fragments
BglII-SmaI SmaI PvuII From Fromfragment fragment fragments tkA5' tkA3'
A1 + + + _ _2 + + _ _ _3 + + + + _4 + + + _ _5 + + _ _ _6 + + + + +7 + + + + +
B1 + + _ _ _2 + + + _ _3 + + + _ _4 + + _ _ _5 + - + _ _6 + + + _ _7 + + + _ _
a This table is a summary of the tk fragments detected in the seven clonesderived from transformation with the combination A DNAs and seven clonesderived from transformation with combination B DNAs (Fig. 3). The data arebased on the Southern analyses shown in Fig. 5 and on Southern analyses notshown here. The positions of the restriction fragments are shown in Fig. 1 and3.
ptk2DRAAN) (Table 1). This result has been interpreted (22)to mean that degradation from each of the two DNA endsmust reach the homologous tk sequences at about the sametime to permit efficient complementary strand pairing (Fig.2A, step 3). This interpretation is supported by the experi-ments of Anderson and Eliason (1). Unlike results withcleavage in the PH region, when the ds break is made in theDH region of the ptk2 plasmids (ptk2DR digested with Sall orNcoI; ptk2-DRAAN digested with NcoI) there is either littleor no stimulation of recombination or there is a significantreduction in recombination relative to that observed withuncut plasmids. The effects of ds breaks on intermolecularrecombination between tkAY3 and tkA5' genes separatelycloned in Ml3mplO and Ml3mpll vectors was also as-sessed. The results are completely consistent with thoseobtained with the intramolecular ptk2 substrates. ds breaksin both molecules proximal to the homologous tk sequencesstimulated recombination 50- to 100-fold relative to uncutDNA, while breaks distal to the homologous regions inhib-ited recombination relative to uncut DNA (data not shown).Differences of over 5,000-fold in recombination frequencycould be produced by simply varying the position of the endsin the substrate DNAs. It should be noted that these resUltsare not unique to the calcium phosphate method of DNAtransfer as similar results have been obtained by theelectroporation of DNA (30) into mammalian cells (data notshown).One of the questions posed by these results is whether the
low level of recombination detected with uncut DNA sub-strates is due to the chance introduction of a ds break at theappropriate location in the DNA after its introduction intocells. If those breaks are produced randomly in uncutcircular DNA, the efficiency of recombination should simplyvary with the relative size of the PH segment in the sub-strate. For example, one would expect a higher level ofrecombination with uncut p MDR plasmid (PH segment is1.5 kb) than with uncut ptk2DRABC (PH segment is 180 bp).Both uncut DNAs recombine with about the same efficiency.Indeed, if any difference exists the pt&DRABC plasmid ismore efficient. This suggests that the recombinants producedwith circular plasmid DNA substrates are generated by apathway different from that proposed for the linear DNAsubstrates. This interpretation assumes that the rate-limiting
MOL. CELL. BIOL.

EXTRACHROMOSOMAL RECOMBINATION IN MAMMALIAN CELLS 137
step in recombination with ds circular plasmid DNAs is thecleavage of that DNA within the cell.
Effi,ciency of processing recombination intermediates. Todetermine which recombination intermediates are the pre-ferred substrates for the resolution steps in mammalian cellrecombination, we transformed cells with pairs of ss DNAscontaining complementary defective tk genes. We proposethat once the DNAs enter the cells complementary ss tkregions pair to generate the two DNA structures shown inFig. 3. Our previous model predicts that structure A shouldbe more efficiently processed to produce an intact tk genethan should structure B. The data from the ds break exper-iments indicate that this difference should be about 1,000- to10,000-fold. The results reported here are the opposite ofwhat we expected; structure B transforms LMtk- cells toTK+ 10 to 20 times more efficiently than does structure A(Table 2).
Modified recombination model. The DNA intermediateshown in Fig. 3B cannot generate a tk gene by the modelshown in Fig. 2. If the endonuclease proposed in that modelcut the structure at positions b and c (Fig. 6A), importantinformation necessary for tk reconstruction would be sev-ered from the tk gene. If the cuts were made at positions aand d, the DNA ends that would have to be used to primesynthesis to reconstruct the truncated parts of the tk genewould be 5' ends and, therefore, could not be used for thatpurpose (Fig. 2B). Because of these results we modified ouroriginal model in a way that accounts for all the data. Toexplain why the intermediate shown in Fig. 3B is moreefficient than that shown in Fig. 3A, we propose that anessential step in the processing of these intermediates is aDNA polymerization step with ss DNA as a template. It isknown that eucaryotic polymerases can initiate and propa-gate DNA synthesis on unprimed ss DNA templates effi-ciently (31). Alternatively, DNA fragments generated bydegradation during the transformation process might be ableto serve as primers for a polymerase. In any case, DNAsynthesis on the ss M13 DNA of the intermediate shown inFig. 3B would generate a molecule like that shown in Fig.6B. To complete the processing reaction one need simplyargue that when the polymerization reaches the junction ofpaired tk sequences, the lone ss at that junction is cleaved(Fig. 6C), and the newly synthesized DNA strand is thenligated to the 5' end generated by the cleavage (Fig. 6D). Asimilar mechanism applied to the DNA intermediate in Fig.3A fails to reconstruct the tk gene because the polymeriza-tion step doesn't copy the tk sequences and the cleavage stepcuts those sequences away from the rest of the gene.To extend this proposal to the ds DNA cutting experi-
ments, the model shown in Fig. 2 needs to be modified in twoways (Fig. 7). First, instead of a 5' exonuclease,we proposethat a 3' exonuclease acts at the ends of the ds breaks (Fig.7, step 2). Like the 5' exonuclease it specifically degradesone strand of the DNA and exposes complementary tksequences for the subsequent strand-annealing step (Fig. 7,step 3). At some point after the strands have annealed wepropose that the 3' strand ceases to be degraded andbecomes a primer for a resynthesis reaction (Fig. 7, step 4).This leads to a structure similar to that shown in Fig. 6Bwhich is processed to generate an intact tk gene as wediscussed in Fig. 6C and D. Unlike the original model, themodified model predicts that ds breaks within the regionproximal to the homologous tk sequences (e.g., ptk2DRDNA digested with HindlIl; Fig. 7A) generate a recombina-tion intermediate similar to that shown in Fig. 3B. Since thatintermediate is efficiently processed by the cell, recombina-
(A)
(B)
(C)
5/ 3
A3' 5
IDNA Synthesis
EndonucleaseCleavage
5/ 31 >>> »>>>>'>--
¾-~~~~~~~~45, 31 51,
I DNA Ligation
5'/3 »>>>> >>
/~~~~~~~~~~~~~~~(D)_____________~~~~~~~~~~~~~~~~~
AA,t<,z««« 31 51~~~~~~~~
FIG. 6. Generation of an intact tk gene from the recombinationintermediate shown- in Fig. 3B. (A) Four ss, ds DNA joints, a, b, c,and d. Cutting the DNA at b and c or a and d will not generaterecombinant molecules (see Discussion). However, polymeraseaction along the ss DNA will lead to the structure shown in panel B.Cleavage of the DNA at the lone ss, indicated by the position of thearrows, generates the structure in panel C. Ligation of the resultingnick in the ds DNA produces an intact tk gene (D).
tion levels are high. In contrast, breaks introduced in theregion distal to the homologous tk sequences (ptk2DRADNA digested with NcoI; Fig. 7B) produce a recombinationintermediate (dashed box, Fig. 7B) similar to that shown inFig. 3A. This intermediate is less efficiently processed by the
VOL. 7, 1987

138 LIN ET AL.
(A)W3'NC /ind m t&5'NC
A3'C tM5'C(1) Double Strand Break
5'r3't= X 5 '
3' Exonuclease(2) a Degradation
5 3 5' &~~~~5'NC3'%t3 >¢ ~~A3'C 5 35'
(3) Single Strand Annealing
(4) | Resynthesis from 3' End
3158f3,,,,,,,,,,,,,,,,,5,>~ ~ 3, 5s3(5) I Nuclease
3
DNA Ligation
Reconstructed tk
(B)N'co I-~' IC I~ 5'
A3'NC &5'N'C(1) |Double Strand Break
5' 3"~~~5'33'$ == - 5'
(2) 3' ExonucleaseDegradation
5I~~3 51 A5'C
A3'NC 5 5'
(3) Single Strand Annealing'553$3~~~~~~~~~ 3't3
5'_I_(4) t Resynthesis from 3' End
5'3X+> ______________________ 3,3"''~~~~~~~~~~~~3
5'(5) jNuclease
5
________ _ _ _ _ ~ ,i~Lf5
5'
tk Not Reconstructed
FIG. 7. Modified model for homologous recombination after the introduction of ds DNA substrates into mammalian cells. The model isillustrated for HindIII-cut (A) or NcoI-cut (B) ptk2DR plasmid DNA. For symbols and discussion see the legends to Fig. 1 and 2 and theDiscussion section of the text.
cell, and recombination levels are low. In support of the newmodel it should be noted that precedent exists for a poly-merase capable of switching from a 3'->5' hydrolysis modeto a 5'-*3' polymerization mode without leaving the primertemplate (10, 29).
Qualitatively, the scheme shown in Fig. 7A can accountfor the transformation data with both linearized ds tk2substrates and the ss recombination intermediates. Quanti-tatively, however, a discrepancy exists. The ds experimentsindicate that differences of 1,000-fold or more should beexpected depending on where the break is made and, there-fore, which intermediate is produced. Yet, the difference intransformation for the two intermediates shown in Fig. 3 isonly 10- to 20-fold. Perhaps alternative recombination path-ways exist that can distinguish between the intermediatesshown in Fig. 3A and 7B and can process the former moreefficiently then they can the latter. This would account forthe quantitative discrepancy between ds and paired ss sub-strates.Two additional results obtained with ss DNAs deserve
further consideration. First, the efficiency of transformationwith unannealed complementary ss DNAs appears to vary asthe product of the concentration of those DNAs (Tables 3and 4). In contrast, if the ss are preannealed before beingadded to the transformation solution, then the efficiency oftransformation varies as a linear function of DNA concen-tration (Table 4). At the lower DNA concentration,used,transformation with the preannealed DNAs is eight times
greater than with unannealed DNAs. The simplest interpre-tation of these results is that the generation of TK+ trans-formants with overlapping pairs of complementary ss tkDNAs involves the interaction of those DNAs (presumablythe annealing of complementary regions on each strand) in astep that is limiting for the transformation process. That stepmight be limiting if, as we have proposed previously (22),there is a competition in cells between the annealing of ssand their degradation. Further experiments are needed toevaluate this possibility.Another result of interest is the observation that transfor:
mation with pairs of overlapping ss DNAs containing thesame, rather than complementary, tk strands generates TK+transformants (Table 2). The efficiency of that transforma-tion is 0.5 to 10% that observed with complementary ikstrands and presumably requires the replication of at leastone of the ss DNAs in the cell to generate the complemen-tary tk sequences needed for recombination. Consistent withthis possibility is the observation that ss DNA containing anintact tk gene can tranform cells with an efficiency that isabout 5 to 10% that observed with a comparable ds DNA(Table 3). Note that these and other re-sults presented herepreclude the possibility that any significant fraction of therecombinants generated with complementary ss DNAs oc-curs by a pathway that first involves the conversion of the ssto ds.The model proposed here to account for recombination
between DNAs used to transform mammalian cells makes
MOL. CELL. BIOL.

EXTRACHROMOSOMAL RECOMBINATION IN MAMMALIAN CELLS 139
several important predictions about enzymes that might beinvolved in the process. Included among those enzymes area 3' exonuclease, a possible DNA-pairing protein analogousto the E. coli RecA or ss DNA-binding protein, a DNApolymerase, an ss endonuclease, and a DNA ligase. With theavailability of an in vitro system from mammalian cells thatappears to be able to catalyze at least some of the steps inhomologous recombination (9, 15, 17, 19; N. Stemnberg andS. L. Brenner, unpublished data), it should soon be possibleto test the involvement of these proteins in the recombina-tion process. Recent experiments of Hsieh et al. (15) andCox and Lehman (8) have identified proteins from mamma-lian cells that could be RecA and ssb analogs.
ACKNOWLEDGMENTSWe thank S. Brenner, J. Eliason, and R. Zagursky for critical
reading of this manuscript.
LITERATURE CITED1. Anderson, R., and S. L. Eliason. 1986. Recombination of homol-
ogous DNA fragments transfected into mammalian cells occurspredominantly by terminal pairing. Mol. Cell. Biol.6:3246-3252.
2. Boyer, H. W., and D. Roulland-Dussoix. 1969. A complementa-tion analysis of the restriction and modification of DNA inEscherichia coli. J. Mol. Biol. 41:459-472.
3. Brenner, D. A., A. C. Smigocki, and R. D. Camerini-Otero.1985. Effect of insertions, deletions, and double-strand breakson homologous recombination in mouse cells. Mol. Cell. Biol.5:684-691.
4. Broker, T. R., and I. R. Lehman. 1971. Branched DNA mole-cules: intermediates in T4 recombination. J. Mol. Biol.60:131-149.
5. Cesarone, C. F., C. Bolognesi, and L. Santi. 1979. Improvedmicrofluorometric DNA determination in biological materialusing 33258 Hoechst. Anal. Biochem. 100:188-197.
6. Chakrabarti, S., and M. Seidman. 1986. Intramolecular recom-bination between transfected repeated sequences in mammaliancells is nonconservative. Mol. Cell. Biol. 6:2520-2526.
7. Cleweli, D. B., and D. R. Helenski.. 1969. Supercoiled circularDNA-protein complex in E. coli: purification and inducedconversion to an open circular DNA form. Proc. Natl. Acad.Sci. USA 62:1159-1166.
8. Cox, M. M., and I. R. Lehman. 1981. Renaturation of DNA: anovel reaction of histones. Nucleic Acids Res. 9:389-400.
9. Darby, V., and F. Blattner. 1984. Homologous recombinationcatalyzed by mammalian cell extracts in vitro. Science226:1213-1215.
10. Das, S. K., and R. K. Fujimura. 1980. Mechanism of primer-template-dependent conversion of dNTP--dNMP by T5 DNApolymerase. J. Biol. Chem. 255:7149-7154.
11. DeSaint Vincent, B. R., and G. M. Wahl. 1983. Homologousrecombination in mammalian cells mediates formation of afunctional gene from two overlapping gene fragments. Proc.Natl. Acad. Sci. USA 80:2002-2006.
12. Enquist, L. W., G. F. Vande Woude, M. Wagner, J. R. Smiley,and W. C. Summers. 1979. Construction and characterization ofa recombinant plasmid encoding the gene for the thymidinekinase of herpes simplex type I virus. Gene 7:325-342.
13. Folger, K. R., E. A. Wong, G. Wahl, and M. R. Capecchi. 1982.Pattern of integration of DNA microinjected into culturedmammalian cells; evidence for homologous recombination be-tween injected plasmid DNA molecules. Mol. Cell. Biol.2:1372-1387.
14. Hines, J. C., and D. S. Ray. 1980. Construction and character-ization of new coliphage M13 cloning vectors. Gene 11:207-218.
15. Hsieh, P., M. S. Meyer, and R.D. Camerini-Otero. 1986. Partialpurification and characterization of a recombinase from humancells. Cell 44:885-894.
16. Ishiura, M., S. Hirose, T. Uchida, Y. Hamada, Y. Suzuki, and Y.Okada. 1982. Phage particle-mediated gene transfer to cultured
mammalian cell. Mol. Cell. Biol. 2:607-616.17. Kenne, K., and S. Ljungquist. 1984. A DNA-recombinogenic
activity in human cells. Nucleic Acids Res. 12:3057-3068.18. Kucherlapati, R. S., E. M. Eves, K.-Y. Song, B. S. Morse, and 0.
Smithies. 1984. Homologous recombination between plasmids inmammalian cells can be enhanced by treatment of input DNA.Proc. Natl. Acad. Sci. USA 81:3153-3157.
19. Kucherlapati, R. S., J. Spencer, and P. D. Moore. 1985. Homol-ogous recombination catalyzed by human cell extracts. Mol.Cell. Biol. 5:714-720.
20. Lai, C.-J., and D. Nathans. 1974. Deletion mutants of simianvirus 40 generated by enzymatic excision of DNA segmentsfrom the viral genome. J. Mol. Biol. 89:179-193.
21. Laird, C. D. 1971. Chromatid structure: relationship betweenDNA content and nucleotide sequence diversity. Chromosoma32:378-406.
22. Lin, F.-L., K. Sperle, and N. Sternberg. 1984. Model forhomologous recombination during transfer of DNA into mouseL cells: role for DNA ends in the recombination process. Mol.Cell. Biol. 4:1020-1034.
23. Lin, F.-L., K. Sperle, and N. Sternberg. 1984. Homologousrecombination in mouse L cells. Cold Spring Harbor Symp.Quant. Biol. 49:139-149.
24. Lin, F.-L., and N. Sternberg. 1984. Homologous recombinationbetween overlapping thymidine kinase gene fragments stablyinserted into a mouse cell genome. Mol. Cell. Biol. 4:852-861.
25. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecularcloning, a laboratory manual. Cold Spring Harbor Laboratory,Cold Spring Harbor, N.Y.
26. McKnight, S. L. 1982. Functional relationships between tran-scriptional control signals of the thymidine kinase gene ofherpes simplex virus. Cell 31:355-365.
27. Messing, J. 1983. New M13 vectors for cloning. MethodsEnzymol. 101:20-78.
28. Miller, C. K., and H. M. Temin. 1983. High-efficiency ligationand recombination of DNA fragments by vertebrate cells.Science 220:606-609.
29. Mizrahi, V., P. Benkovic, and S. J. Benkovic. 1986. Mechanismof DNA polymerase I: exonuclease/polymerase activity switchand DNA sequence dependence of pyrophosphorolysis andmisincorporation reactions. Proc. Natl. Acad. Sci. USA83:5769-5773.
30. Potter, H. L., L. Weir, and P. Leder. 1984. Enhancer-dependentexpression of human immunoglobulin genes introduced intomouse pre-B lymphocytes by electroporation. Proc. Natl. Acad.Sci. USA 81:7161-7165.
31. Riedel, H.-D., H. Konig, H. Stahl, and R. Knippers. 1982.Circular single-stranded phage M13 DNA as a template forDNA synthesis in protein extracts from Xenopus laevis eggs:evidence for a eukaryotic DNA priming activity. Nucleic AcidsRes. 10:5621-5635.
32. Rubnitz, J., and S. Subramani. 1985. Rapid assay forextrachromosomal homologous recombination in monkey cells.Mol. Cell. Biol. 5:529-537.
33. Sanger, F., S. Nicklen, and A. R. Coulson. 1977. DNA sequenc-ing with chain-terminating inhibitors. Proc. Natl. Acad. Sci.USA 74:5463-5467.
34. Shapira, G., J. L. Stachelek, A. Letsou, L. K. Soodak, and R. M.Liskay. 1983. Novel use of synthetic oligonucleotide insertionmutants for the study of homologous recombination in mamma-lian cells. Proc. Natl. Acad. Sci. USA 80:4827-4831.
35. Small, J., and G. Scangos. 1983. Recombination during genetransfer into mouse cells can restore the function of deletedgenes. Science 219:174-176.
36. Song, K.-Y., L. Chekuri, S. Rauth, S. Ehrlich, and R.Kucherlapati. 1985. Effect of double-stranded breaks on homol-ogous recombination in mammalian cells and extracts. Mol.Cell. Biol. 5:3331-3336.
37. Szostak, J. W., T. L. Orr-Weaver, R. J. Rothstein, and F. W.Stahl. 1983. The double-stranded-break repair model for recom-bination. Cell 33:25-35.
38. Szybalski, W., E. H. Szybalski, and G. Ragni. 1962. Geneticstudies with human cell lines. National Cancer Institute Mono-
VOL. 7, 1987

140 LIN ET AL. MOL. CELL. BIOL.
graph 7. National Cancer Institute, Bethesda, Md.39. Wagner, M. J., J. A. Sharp, and W. C. Summers. 1981.
Nucleotide sequences of the thymidine kinase gene of herpessimplex virus type I. Proc. Natl. Acad. Sci. USA 78:1441-1445.
40. Wake, C. T., F. Vernaleone, and J. H. Wilson. 1985. Topologicalrequirements for homologous recombination among DNA mol-ecules transfected into mammalian cells. Mol. Cell. Biol.
5:2080-2089.41. Wake, C. T., and J. H. Wilson. 1979. Simian virus 40 recombi-
nants are produced it high frequency during infection withgenetically mixed oligomeric DNA. Proc. Natl. Acad. Sci. USA76:2876-2880.
42. Wake, C. T., and J. H. Wilson. 1980. Defined oligomeric SV40DNA: a sensitive probe of general recombination in somaticcells. Cell 21:141-148.