expression of cyclin e renders cyclin d-cdk4 dispensable ... · prb by the g1 cdk-cyclin complexes...
TRANSCRIPT

Expression of Cyclin E Renders Cyclin D-CDK4 Dispensable for Inactivation of the Retinoblastoma Tumor Suppressor Protein (pRB),
Activation of E2F, and G1-S Phase Progression
Susan M. Keenan§¶, Nathan H. Lents§, and Joseph J. Baldassare* The Department of Pharmacological and Physiological Sciences, Saint Louis University School of Medicine, Saint Louis, Missouri 63104 §SMK and NHL contributed equally to this work. *To whom correspondence should be addressed: Department of Pharmacological and Physiological Sciences, Saint Louis University School of Medicine, Saint Louis, Missouri 63104, Tel: 314-577-8543, E-mail: [email protected] ¶ Present Address: Department of Pharmacology, University of Medicine and Dentistry of New Jersey, Robert Wood Johnson Medical School, 675 Hoes Ln., Piscataway, NJ 08854 Keywords – cell cycle, retinoblastoma protein, E2F, CDK, cyclin-dependent kinase, LY294002, cyclin D, cyclin E Running Title: CDK2-cyclin E alone inactivates pRb Abbreviations used are CDK, cyclin-dependent kinase; PI3-kinase, phosphatidylinositol 3-OH kinase; ORF, open reading frame; pRb, retinoblastoma protein
Copyright 2003 by The American Society for Biochemistry and Molecular Biology, Inc.
JBC Papers in Press. Published on November 25, 2003 as Manuscript M310383200 by guest on July 17, 2019
http://ww
w.jbc.org/
Dow
nloaded from

Summary
The activation of CDK2-cyclin E in late G1 phase has been shown to play a critical role
in pRb inactivation and G1-S phase progression of the cell cycle. The PI3-Kinase inhibitor
LY294002 has been shown to block cyclin D1 accumulation, CDK4 activity, and thus G1
progression in α-thrombin stimulated IIC9 cells (Chinese Hamster Embryonic Fibroblasts). Our
previous results show that expression of cyclin E rescues S phase progression in α-thrombin-
stimulated IIC9 cells treated with LY294002, arguing that cyclin E renders CDK4 activity
dispensable for G1 progression. In this work, we investigate the ability of α-thrombin-induced
CDK2-cyclin E activity to inactivate pRb in the absence of prior CDK4-cyclin D1 activity. We
report that in the absence of CDK4-cyclin D1 activity, CDK2-cyclin E phosphorylates pRb in
vivo on at least one residue, and abolishes pRb binding to E2F response elements. We also find
that expression of cyclin E rescues E2F activation and cyclin A expression in cyclin D-kinase-
inhibited, α-thrombin-stimulated cells. Furthermore, the rescue of E2F activity, cyclin A
expression, and DNA synthesis by expression of E can be blocked by the expression of either
CDK2(D145N) or Rb∆CDK, a constitutively active mutant of pRb. However, restoring four
known cyclin E-CDK2 phosphorylation sites to Rb∆CDK renders it susceptible to inactivation in
late G1, as assayed by E2F activation, cyclin A expression, and S phase progression. These data
indicate that CDK2-cyclin E, without prior CDK4-cyclin D activity, can phosphorylate and
inactivate pRb, activate E2F, and induce DNA synthesis.
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

Introduction
The mammalian cell cycle is controlled by two important families of proteins, the cyclins
and the cyclin-dependent kinases (CDKs) (Reviewed in(1,2)). Progression through the cell cycle
is governed by the kinase activities of specific CDKs, which are regulated by association with
the regulatory cyclin subunits. The sequential activation of early G1 CDK activity – CDK4 or
CDK6, together with cyclin D1, D2, or D3 – and the late G1 CDK activity – CDK2, together
with cyclin E1 or E2 – is believed to be required for progression through G1 and into S phase.
Because IIC9 cells contain CDK4 but not CDK6, and cyclin D1 but not D2 or D3, CDK4-cyclin
D1 activation in early G1 is required for the expression of cyclin E, CDK2 activity, and G1-S
phase progression (3-5).
In most cell types, inactivation of the retinoblastoma (pRb) protein is essential for
passage through G1 and transition of cells into S phase (2,6-9). pRb regulates this progression
by its association with the E2F family of transcription factors (10-13). In quiescent cells (G0
phase), pRb is unphosphorylated; in early- to mid-G1, pRb is hypophosphorylated by the D-type
CDKs (14,15). This hypophosphorylated form of pRb, which binds to and inhibits E2F
transcription factors, has been shown in vivo to be phosphorylated on 13 of 16 potential CDK
phosphorylation sites suggesting that hypophosphorylated pRb may consist of multiple
phosphoisoforms (16-18). The hypophosphorylation of pRb in early G1 stimulates the release of
HDAC1 and the recruitment of SWI/SNF family members to the pRb-containing chromatin
remodeling complexes, thus allowing the expression of cyclin E (19-21). In late G1 and S phase,
pRb is hyperphosphorylated by CDK2-cyclin E, and later, CDK2-cyclin A (22-26). The
hyperphosphorylated form of pRb is inactivated because it loses affinity for, and therefore fails
to inhibit, the E2F transcription factors (7,23,27). Numerous proteins which are essential for
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

growth, such as the cyclins E and A, and proteins essential for DNA replication, such as DNA
polymerase α, thymidine kinase, dihydrofolate reductase, and histone H2A are controlled at
least, in part, by E2F-responsive promoters (28-35). pRb inhibits these promoters by either
directly blocking the activation domain of E2F, or by acting as a member of a repression
complex (21,36-41). Therefore, the major role of the G1 CDKs, CDK4/6-cyclin D and CDK2-
cyclin E in controlling G1-S phase progression is the inactivation of pRb.
While the role of CDK4-cyclin D in the inactivation of pRb is well established, the role
of CDK2-cyclin E in the inactivation of pRb is less clear. CDK2-cyclin E, in the absence of
prior phosphorylation by CDK4-cyclin D, is able to phosphorylate pRb in vitro (16) and
overexpression of cyclin E or A can overcome pRb-mediated suppression of proliferation (42).
Furthermore, Ezhevsky et al. show that CDK2-cyclin E activity phosphorylates pRb in vivo (18).
In agreement with these data, Lundberg and Weinberg also found that CDK2-cyclin E activity
was necessary for phosphorylation-induced inactivation of pRb (43). However, these authors
and others suggest that phosphorylation of pRb by CDK2-cyclin E requires pRb to be
hypophosphorylated, and thus the in activation of pRb involves sequential phosphorylation by
cyclin D-CDK4/6 and cyclin E-CDK2 (24,44). Conversely, the report of a cyclin E→D1
"knockin" mouse offers argument against the strict requirement for sequential phosphorylation of
pRb by the G1 CDK-cyclin complexes (45). These mice, in which the coding sequence of the
cyclin D1 gene is replaced with the coding sequence for cyclin E, reveal that cyclin E expression,
which results in a 20% change in the phosphorylation state of pRb, rescues the phenotypic
deficiencies found in the cyclin D1-ablated mouse (46). Although many tissues are unaffected
by the loss of cyclin D1 because of compensatory functions of cyclin D2 and D3, several tissues,
including retinal and breast tissue are severely deficient in growth, presumably because D2 and
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

D3 are not expressed in these tissues and could not compensate. Implicit in the discovery that
cyclin E expression under the cyclin D1 promoter reverses the cyclin D1-/- phenotypes is the
understanding that, in the rescued tissues, pRb is inactivated by cyclin E-kinase activity without
prior hypophosphorylation by cyclin D-kinase, with the caveat that the genes for cyclin D2 and
D3 are still present in these animals.
Previously, we reported that inhibition of PI3-kinase by LY294002 inhibits cyclin D1
accumulation, CDK4-cyclin D1 activity, and passage through G1 into S phase in IIC9 cells (3).
Surprisingly, expression of cyclin E in the presence of LY294002 rescues cyclin E-CDK2
activity and G1 progression (47). Under these conditions, cyclin D-kinase activation is inhibited,
while cyclin E-kinase activity is rescued. Therefore, this offers a unique opportunity to question
the necessity of prior phosphorylation of Rb by cyclin D-dependent kinases, by examining
whether CDK2-cyclin E activity alone can phosphorylate and inactivate pRb. Here we show that
expression of cyclin E recovers CDK2, but not CDK4 activation in LY294002-treated cells.
Under these conditions in which CDK2 can be activated without prior CDK4 activation, we
report that the phosphorylation of pRb on Ser795, the release of pRb from E2F response
elements, the activation of E2F, and G1-S phase progression all occur normally. Furthermore,
we report that the cyclin E-mediated rescue of E2F activity can be prevented by co-expression of
CDK2(D145N) or Rb∆CDK. Finally, we restore four CDK2 phosphorylation sites on Rb∆CDK and
show that expression of this construct, Rb∆+K2, does not block E2F activation or DNA synthesis.
Taken together, these data indicate that, in IIC9 cells, prior phosphorylation of pRb by cyclin D-
CDK4/6 is not necessary for the phosphorylation of pRb by cyclin E-CDK2 and the subsequent
activation of E2F and entry into S phase of the cell cycle.
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

Materials and Methods
Cell Culture and Transient Transfection – IIC9 cells, a subclone of Chinese Hamster
Embryo Fibroblasts (48,49), were maintained as previously described (50). Quiescent cells were
established by washing subconfluent (80%) cells once with phosphate buffered saline (PBS)
followed by a 48 hour incubation with alpha-MEM media containing 2mM L-glutamine
(BioWhittaker) supplemented with 100U/ml penicillin and 100mg/ml of streptomycin (basal
media). For transient transfections, IIC9 cells were grown to subconfluency (80%). The cells
were transfected as previously described (51). After 12 hours the cells were serum-arrested for
48 hours prior to stimulation. >80% transfection efficiency was determined by co-transfection of
GFP. LY294002 (Calbiochem, San Diego, CA) was added 30 minutes prior to stimulation to a
final concentration of 10µM. Growth arrested IIC9 cells were stimulated with 1 unit(U)/ml of
human α-thrombin and incubated for the indicated times.
Constructs – Human cyclin E, HU4 fragment, was generously provided by James
Roberts; pGL3-TATA-6xE2F-Luc was generously provided by Kristian Helin; human
CDK2(D145N) was generously provided by Jim Koh; murine p16INK4a cDNA was generously
provided by Martine Roussel and Charles Sherr; human wild-type Rb and Rb∆CDK was
generously provided by J. Wade Harper and all mutations were confirmed by sequencing
analysis. Rb∆+K2 was generated from Rb∆CDK by site-directed mutagenesis restoring Thr-373,
Thr-612, Ser-795, and Thr-821 individually, following manufacturer’s protocol (QuikChange™
kit, Stratagene). Restoration of the threonine or serine codons was confirmed by automated
capillary sequencing following manufacturer’s protocol (Beckman-Coulter).
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

Cyclin-dependent Kinase Activity Assays – CDK4-cyclin D1 and CDK2-cyclin E assays
were performed as previously described (3,47,50,52). Briefly, following transfection and serum-
starvation, cells were stimulated with 1U/mL α-thrombin for eight hours (for CDK4) or
seventeen hours (for CDK2). Lysates (100µg) were immunoprecipitated with monoclonal
antibodies to cyclin D1, or polyclonal antibodies to CDK4 or CDK2, as indicated.
Immunoprecipitates were analyzed for ability to phosphorylate GST-Rb (for CDK4 or cyclin D1)
or histone H1 (for CDK2) in vitro and 32P-phosphate incorporation was quantified using a
phosphorimager (Molecular Dynamics). Data is presented as fold-activation over basal level.
Western Blot Analysis – Asynchronously growing IIC9 cells or human HL60 cells were
washed twice with cold PBS and lysed in cold lysis buffer (52). Lysates were sonicated briefly
and insoluble material pelleted by microfugation at 14,000rpm at 4°C for 2 minutes. 40µg of
protein lysate was resolved by SDS-polyacrylamide gel electrophoresis and transferred to a
polyvinylidene difluoride membrane (Millipore Corp, Boston, MA) as recommended by the
manufacturer. Membranes were probed individually with polyclonal antibodies to CDK6, cyclin
D1, cyclin D2, or cyclin D3 (all from Santa Cruz Biotechnology, Santa Cruz, CA).
Immunoreactive bands were visualized by enhancer chemiluminescence (ECL) detection
(Amersham, Arlington Heights, IL) as recommended by the manufacturer.
Growth-arrested IIC9 cells were incubated in the presence or absence of 1U/ml α-
thrombin for 19 hours after pretreatment in the presence or absence of 10µM LY294002 for 30
minutes. Cells were then washed twice with cold PBS and lysed in cold lysis buffer (52).
Lysates were prepared for Western analysis as described and probed with polyclonal antibodies
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

to phosphorylated Ser780- or Ser 795-pRb (Cell Signaling Technology, Beverly, MA), or cyclin
A (Santa Cruz Biotechnology, Santa Cruz, CA).
Luciferase Reporter Assay – IIC9 cells were transiently transfected with 50ng/ml pGL3-
TATA-6xE2F-Luc, 200ng/ml β-Galactosidase, and 1.5-2.0µg/ml of cyclin E, CDK2(D145N),
Rb∆CDK, or Rb∆+K2 as indicated in figure legends. Basal cells were stimulated with 1U/ml α-
thrombin where indicated either in the presence or absence of 30 min. pre-incubation with 10µM
LY294002. Lysate protein was prepared for luciferase activity assay as recommended by
manufacturer (Promega, Madison, WI) and 10µl of room temperature lysate was mixed with
90µl of room temperature Luciferase Assay Buffer-Reagent (Promega) and placed in an
OpticompII luminometer (MCM Instruments, Baltimore, MD). Light produced was measured
and normalized to transfection efficiency by dividing relative light units by optical density units
obtained from β-galactosidase activity. β-galactosidase activity was measure as previously
described(5).
Northern Blot Analysis – Quiescent IIC9 cells were incubated in the presence or absence
of 1U/ml α-thrombin for indicated lengths of time after pre-incubation in the presence or
absence of 10µM LY294002. At the indicated times, total RNA was isolated using TRIZOL
reagent (Life Technologies, Inc.) according to the manufacturer's protocol. RNA (20 µg) was
resolved in a 2%(w/v) agarose-formaldehyde gel. After electrophoresis, formaldehyde was
removed from the gel by washing in 0.5% ammonium acetate. RNA was then transferred to a
Hybond N+ nylon membrane. (Amersham Pharmacia Biotech) using a TurboblotterTM system
(Schleicher & Schuell, Keene, NH) and cross-linked onto the membrane using an ultraviolet
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

cross-linker (Amersham Pharmacia Biotech) as recommended by the manufacturer. Randomly
labeled [α-32P]dCTP cDNA probes were made using the Random Primed DNA Labeling Kit
(Roche Molecular Biochemicals, Indianapolis, IN). Membranes were pre-incubated with rapid
hybridization buffer (Amersham Pharmacia Biotech) for 1 hour at 65°C and then probed
simultaneously with cyclin A and glyceraldehyde-3-phosphate dehydrogenase probes for 2 hours
at 65 °C. After hybridization, the membrane was washed twice with 5XSSPE (20mM EDTA,
1M NaCl, 50mM NaH2PO4-H2O) with 0.1%(w/v) SDS at room temperature and once with
1XSSPE, 0.1% SDS at 65 °C. The membrane was developed using a PhosphorImagerTM
(Molecular Dynamics)
EMSA – Slightly adapted from established method (12,39). Nuclear Fractionation: IIC9 cells
were quickly scraped in 3ml/100-mm2 plate of ice cold 10mM Tris/HCl, pH 7.5 containing
10mM NaCl, 1mM EDTA (Buffer A). The cells were homogenized 20 times in a Potter-
Elvehjem homogenizer and spun at 500g for 7 minutes. The nuclear pellet was suspended in
Buffer A and homogenized 15 times in a Dounce homogenizer with a pestle, layered over 45%
sucrose in Buffer A and centrifuged at 1660g for 15 minutes. The nuclei were washed once and
resuspended in 10mM Tris/HCl, pH 7.5 containing 10mM NaCl, 1mM MgCl2 and 10% sucrose
and sonicated to lyse the nuclei. Nuclear proteins (4µg) were immunoprecipitated by incubating
with 1µg of anti-RB monoclonal antibody (Calbiochem). Immunoprecipitated proteins were
recovered on protein G-Sepharose beads and dissociated by treatment with deoxycholate (12,39).
Electrophoretic mobility shift assays (EMSAs) were performed as described previously using
pRb immunoprecipitates and an end-radiolabeled double-stranded DNA fragment (1.5–2 x 105
cpm/assay) containing a single E2F consensus binding site derived from the dihydrofolate
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

reductase (DHFR) promoter (sc-2507; Santa Cruz Biotechnology), termed E2FRE (12,39). For
competition studies, the DNA binding assays also included 10ng of unlabeled E2FRE, mutant
E2FRE double-stranded oligonucleotide with a CG to AT substitution at the E2F binding motif
(E2FREmut, sc-2508) as a nonspecific competitor. To identify proteins in complex with the E2F
consensus site, extracts were pre-incubated with rabbit polyclonal antibodies (0.5 µg each) to
E2F1 (sc-193 X) and E2F4 (sc-1083 X) from Santa Cruz Biotechnology prior to EMSA.
Thymidine Incorporation Assay – Performed as previously described((53)).
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

Results
Expression of Cyclin E Rescues CDK2, but not CDK4 Activity in LY294002-treated
cells. PI3-kinase activity is essential for the accumulation of cyclin D protein and CDK4-cyclin
D activity (54). In most cell types, including IIC9 cells (3), activation of CDK4-cyclin D in early
G1 results in expression of cyclin E, and the activation of CDK2-cyclin E in late G1. To
determine whether ectopic expression of cyclin E rescues CDK2 activity in the absence of CDK4
activity, we perform in vitro kinase assays with histone H or recombinant pRb, respectively, as
the substrate (fig.1). As expected, the activity of CDK4 and CDK2 is negligible in quiescent
cells and α-thrombin stimulates a marked activation of both CDK4 (fig. 1A) and CDK2 (fig.
1B). However, pre-treatment with LY294002 prevents the activation of both CDK4 and CDK2.
These data are consistent with the prevailing notion that CDK4-cyclin D activity is essential for
pRb phosphorylation and the subsequent expression of cyclin E and CDK2-cyclin E activation.
Interestingly, ectopic expression of cyclin E rescues the α-thrombin-induced activation of CDK2
(fig. 1B), but not CDK4 (fig. 1A). Because IIC9 cells do not express CDK6 or cyclins D2 or D3
(fig. 1C), these conditions represent a G1 cell in which CDK2 is activated without the prior
activation of D-type CDK activity.
In addition to providing an ideal system for studying the ability of CDK2-cyclin E alone
to phosphorylate and inactivate pRb, these data argue that cyclin D-CDK4/6 activity is
dispensable for CDK2 activity and G1 progression in the event of unscheduled cyclin E
expression ((47) and fig. 7). According to the current model, prior activation of cyclin D-CDK
complexes has two roles in the activation of cyclin E-CDK2 activation. First, the
hypophosphorylation of pRb is required for the expression of cyclin E. This role is easily
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

bypassed in our system by transfection of cyclin E. An additional function of cyclin D in the
activation of cyclin E is that the formation of cyclin D-CDK complexes is thought to titrate and
sequester CDK inhibitors, such as p21CIP1/WAF1 and p27KIP1, away from CDK2 complexes.
Transfection of cyclin E also bypasses this second function of cyclin D complexes because
cyclin E protein levels are ectopically maintained at levels above the threshold required for
formation of active cyclin E-CDK2 complexes and subsequent destruction of p27KIP1 by
ubiquitin-mediated proteolysis, as indicated by the observed activation of CDK2 in figure 1B. In
any event, we generate conditions in which CDK2 complexes can be activated without prior
activation of CDK4 complexes and it is these conditions that we utilize to address the
inactivation of pRb by cyclin E-CDK2 alone.
Expression of Cyclin E Rescues LY294002- and p16INK4a-inhibited E2F Activation.
Ectopic cyclin E expression restores DNA synthesis in LY294002-treated cells (47), presumably
because E2F activation occurs normally in these cells and ushers cells into S phase. To examine
E2F activity under these conditions, we make use of an E2F reporter plasmid termed pGL3-
TATA-6xE2F-Luc, which is a construct containing six E2F response elements in tandem driving
the expression of the firefly luciferase ORF (38,55). An assay of relative luciferase activity from
pGL3-TATA-6xE2F-transfected cells reveals that in seventeen hours, α-thrombin stimulates a
~6-fold increase in E2F activity over that of quiescent cells (fig. 2A). Not surprisingly,
LY294002 abolishes this stimulation presumably by inhibiting CDK activation, and thus, pRb
phosphorylation and subsequent E2F activation. Expression of cyclin E, however, completely
reverses this inhibition, as the level of E2F activity is similar to that of uninhibited cells (fig.
2A). This argues that E2F activation is normal in these cells despite CDK4-cyclin D inhibition.
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

In addition to the luciferase reporter of E2F activity, we also examine the steady-state
mRNA levels of cyclin A, a gene that is known to contain an E2F response element and is
transcriptionally regulated by E2F1 (56). Cyclin A mRNA levels reach a maximum after cells
have entered S phase, and, by northern blotting, we observe a 9-fold induction of cyclin mRNA
(normalized for GAPDH) over basal levels after 17 hours of α-thrombin stimulation (fig. 2B).
Interestingly, both cyclin A mRNA accumulation (fig. 2B) and E2F reporter activation (fig. 2A)
display G1 time-course kinetics suggestively similar to CDK2-cyclin E activation (47,50,57,58).
Also in concurrence with the data from the E2F reporter, we find that the α-thrombin-stimulated
induction of cyclin A message levels can be largely blocked by pre-incubation with LY294002
(fig. 2C). However, as also seen with the E2F reporter, this attenuation can be reversed by
ectopic expression of cyclin E.
To ensure that these observations regarding mRNA levels translate to steady-state protein
expression, we also assess cyclin A protein levels by western blotting. In accordance with our
observations of mRNA levels, we find that cyclin E expression completely restores the
LY294002-attenuated, α-thrombin-stimulated induction of cyclin A protein expression (fig. 2D).
Although treatment of IIC9 cells with LY294002 attenuates the activity of CDK4-cyclin D1 to
the basal level of detection (figure 1A), we utilize a potent inhibitor of D-type-CDKs, p16INK4a
(59), to ensure complete inhibition of cyclin D1-CDK4. As expected, p16 expression prevents
α-thrombin-stimulated cyclin A protein induction (fig. 2D). However, cyclin E expression
restores cyclin A accumulation in p16-expressing, LY294002-treated cells. These data, taken
together, demonstrate that cyclin E expression renders cyclin D-CDK4 activity dispensable in the
initiation of E2F activity.
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

GW8510 or Co-expression of CDK2(D145N) Eliminates the Ability of Cyclin E to
Rescue E2F Activity. Because cyclin E is not known to bind to or activate other CDKs, the
ability of ectopic cyclin E expression to rescue E2F activity in LY294002-treated cells is
presumably through the activation of CDK2. To test this, we make use of a dominant-negative
(dnCDK2) construct, CDK2(D145N) (60). We find that co-expression of dnCDK2 prevents the
rescue of E2F activity by cyclin E in LY294002-treated, α-thrombin-stimulated cells, as
measured by the luciferase reporter (fig. 3A) and cyclin A steady-state mRNA levels (fig. 3B).
Similarly, pre-incubation with 7.5µM GW8510 reduces E2F activity in stimulated cells to levels
below that of basal cells, regardless of ectopic cyclin E expression (fig. 2A, lane8-9). GW8510
(61) is a non-selective CDK inhibitor which, when pre-incubated at 7.5µM, inhibits CDK2
activation by 95-100% and CDK4 activation by 40-60% in IIC9 cells (data not shown). These
data, together with a time-course of E2F activation which closely mirrors CDK2 activation (fig.
2A, lane1-4; fig. 3A) (50,57,58,62), strongly support the notion of a function for CDK2 in the
activation of E2F during G1 progression. We cannot rule out the possibility that, as may be
suggested by recent reports, an additional pRb-kinase activity exists (see Discussion). However,
the kinase activity in our system requires PI3-kinase for activation, is activated by cyclin E
expression, and is sensitive to inhibition by GW8510 and dnCDK2, compelling evidence that
CDK2 is the pRb-kinase responsible for E2F activation in this study.
CDK2-cyclin E Alone Can Phosphorylate and Inactivate pRb in vivo. The implication
of our observation that cells in which α-thrombin stimulates E2F activation and G1 progression
despite CDK4 inhibition is that CDK2-cyclin E is able to phosphorylate and inactivate pRb
without prior phosphorylation by CDK4-cyclin D1. However, the possibility remains that the
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

activation of E2F observed under these conditions is somehow independent of pRb
phosphorylation. In order to investigate the phosphorylation of pRb in cells lacking CDK4
activation, we made use of two phospho-specific pRb antibodies. One recognizes only pRb that
is phosphorylated on position Ser-795, which can be phosphorylated by either CDK4 or CDK2;
and the other recognizes pRb that is phosphorylated on position Ser-780, which is only
phosphorylated by CDK4, and not CDK2 (7,16,18,43). Not surprisingly, very little
phosphorylation of these residues is detected in lysates from serum-starved cells (fig. 4A) as pRb
is thought to be unphosphorylated in quiescence (17,18,24,43,44). The minimal phosphorylation
seen in our blots is most likely due to a small population of cells that escape serum starvation. In
contrast, robust phosphorylation of pRb is observed with both phospho-specific antibodies after
seventeen hours of α-thrombin stimulation. As expected, pre-incubation with LY294002
diminishes phosphorylation at both sites. While having no effect on the LY294002-inhibited
phosphorylation of Ser-780, dramatically, ectopic cyclin E expression completely restores the
level of phosphorylation of Ser-795 to uninhibited levels. Thus, a CDK4-specific
phosphorylation site remains unphosphorylated, while a site that can be phosphorylated by
CDK2-cyclin E is indeed phosphorylated. Whilst far from a complete analysis of pRb
phosphorylation, this observation demonstrates that at least a subset of potential CDK2
phosphorylation sites on pRb is phosphorylated without prior hypophosphorylation of pRb by
CDK4.
Our evidence of E2F activation under conditions of CDK4 inhibition substantiates the
assertion that phosphorylation of pRb by CDK2-cyclin E alone can trigger pRb inactivation
(42,45). To more directly test this supposition, we next conducted an electromobility shift assay,
EMSA, according to an established protocol utilizing the E2F response element as the probe
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

(12,39). Figure 4B, lane 2 shows electromobility shift of the probe when incubated with pRb
immunoprecipitates from quiescent nuclei. Specificity was ensured by failure of pRb to shift a
mutated probe (not shown) and by competition with unlabelled probe (lane3). Interestingly,
when pRb immunoprecipitates from α-thrombin stimulated nuclei are incubated with the probe,
electromobility shift is strikingly reduced (lane6), an effect that is both blocked by LY294002
(lane7) and rescued by ectopic expression of cyclin E (lane8). Because pRb is not thought to
directly bind to DNA, we interpret these data as an indirect observation of the ability of pRb to
bind to E2F-family transcription complexes.
Our interpretation is supported by the observation that antibodies to E2F1 (lane4) and
E2F4 (lane5) super-shift the pRb-retarded band. Failure of saturating amounts of these
antibodies to induce complete super-shift is consistent with the notion that, in quiescent cells,
pRb exists in multiple complexes (7,63). Antibodies to E2F1 and E2F4 were chosen for study
because they are thought to be binding partners of pRb in quiescent cells (7,64-66). When
coalesced with our observations regarding E2F activation, these data regarding pRb
phosphorylation and E2F-RE binding make a strong case that hypophosphorylation by cyclin D-
dependent kinase, once thought to be prerequisite, is actually dispensable for the phosphorylation
and inactivation of pRb by cyclin E-dependent kinase under the condition of constitutive cyclin
E expression.
Expression of Rb∆CDKBlocks the Activation of E2F by CDK2-Cyclin E. Given our data
regarding activation of E2F and release of pRb from E2F transcription complexes in the absence
of cyclin D1-CDK4 activity, we reasoned that expression of Rb∆CDK, a hyper-repressive mutant
of pRb, would block cyclin E-dependent rescue of E2F activation in our system. Rb∆CDK has
fourteen of the sixteen known CDK phosphorylation sites mutated to alanine and therefore
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

functions as a constitutively active cell cycle repressor immune to the inactivating effects of the
G1 cyclin-CDKs (67). This construct, when transfected into IIC9 cells, prevents the α-thrombin-
induced activation of the E2F-luciferase reporter plasmid (not shown). Interestingly, expression
of Rb∆CDK further prevents the ectopic cyclin E-mediated rescue of E2F activation in LY294002-
treated cells (fig. 5A), while expression of wild-type pRb is ineffective in blocking E2F
activation, presumably because wild-type pRb is subject to inactivation by the CDKs as cells
progress through G1 (fig.6A, and data not shown). In accordance with this result, Rb∆CDK
significantly attenuates the cyclin E rescue of cyclin A induction in LY294002-treated cells (fig.
5B). Because this non-phosphorylatable mutant of pRb blocks the function of cyclin E in these
experiments, these data add further evidence that the role of ectopic cyclin E in the rescue of E2F
activity is to facilitate the phosphorylation of pRb. This result, when coupled with the
observations that dnCDK2 (fig.3), and GW8510 (fig.2) also reverse the cyclin E rescue, strongly
implicates CDK2-cyclin E in the observed inactivation of pRb.
Restoration of Four CDK2 phosphorylation Sites to Rb∆CDK renders it susceptible to
Inactivation by Cyclin E-CDK2. By all accounts, the phosphorylation of pRb by CDK2 is less
extensive than the phosphorylation of pRb by CDK4 (16). Therefore, when CDK4 is inhibited,
but E2F is activated nonetheless by expression of cyclin E, it can be argued that pRb is
phosphorylated on a relatively small number of residues. In order to demonstrate that this
minimal phosphorylation of pRb is sufficient to relieve pRb-mediated repression of E2F activity,
we created a construct termed Rb∆+K2. This construct was generated by restoring the following
four reported in vitro CDK2-cyclin E phosphorylation sites to Rb∆CDK by site-directed
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

mutagenesis: Thr-373, Ser-612, Ser-795, and Thr-821. It should be noted that, of these, Thr-373
and Ser-795 can also be phosphorylated by CDK4-cyclin D in vitro (16,68).
In order to ascertain whether Rb∆+K2 is inactivated by cyclin E-CDK2 activity, we first
examine the ability of cyclin E-CDK2 to activate E2F in the presence of Rb∆+K2. In contrast to
Rb∆CDK, expression of Rb∆+K2 does not significantly block the activation of E2F (fig.6A) in the
condition of constitutive cyclin E expression. To confirm that CDK2-cyclin E alone can
inactivate Rb∆+K2, we again generate conditions in which cyclin D-dependent kinases are
inhibited by pre-incubation with LY294002 but cyclin E expression is provided (fig. 6B). Under
these conditions, expression of Rb∆+K2 fails to repress α-thrombin-stimulated E2F activation.
Furthermore, expression of Rb∆+K2 does not prevent the accumulation of cyclin A steady-state
protein level, as does Rb∆CDK (fig.6C). These results argue that the phosphorylation of Rb∆+K2 by
CDK2-cyclin E on just four residues is sufficient to neutralize pRb-mediated repression of E2F
activity.
Ultimately, we examine the effect of Rb∆CDK and Rb∆+K2 on G1 progression. First, we
confirm our earlier observation that LY294002-inhibited S phase entry can be rescue by ectopic
expression of cyclin E in α-thrombin-stimulated cells (fig.7A). In addition to PI3-kinase
inhibition by LY294002, we express p16INK4a to ensure that cyclin D-CDK complexes are
inhibited. Confirming our previous report (47), we find that unscheduled cyclin E expression
rescues S phase progression in LY294002-treated, p16INK4a-expressing cells. Not surprisingly,
expression of Rb∆CDK blocks α-thrombin-stimulated DNA synthesis, despite cyclin E expression,
because it is a constitutive repressor of E2F activity, immune to inactivation by the CDKs
(fig.7B). However, Rb∆+K2 is not dominantly repressive, as levels of 3H-thymidine incorporation
in α-thrombin-stimulated cells expressing this construct are similar to those expressing wild-type
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

pRb. Again, our interpretation of this observation is that the phosphorylation of pRb on the four
CDK2-preferred sites is sufficient to mediate release of pRb from E2F transcription complexes,
and thus, functionally inactivate pRb, allowing for E2F-mediated transcription of S-phase genes
and the initiation of DNA synthesis.
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

Discussion
Unscheduled Cyclin E Expression Renders Cyclin D-CDK Activity Dispensable for G1
Progression. CDK2-cyclin E activity has been long thought to be essential for the progression
from G1 to S phase of the cell cycle (2,69). In cells expressing pRb, hyperphosphorylation of
pRb by the G1 CDKs, which occurs in a cell cycle-dependent manner and results in pRb
inactivation, is important for progression from G1 to S phase. While the direct phosphorylation
of pRb by CDK4-cyclin D in vivo is not disputed, the role CDK2-cyclin E plays in the
phosphorylation of pRb in vivo has remained unclear. In this work, we have examined the role
of cyclin E-CDK2 in the phosphorylation and inactivation of pRb. We have used antibodies to
specific forms of phosphorylated pRb and found that CDK2-cyclin E activity can phosphorylate
pRb in the absence of CDK4-cyclin D1 activity, suggesting that this phosphorylation of pRb by
CDK2-cyclin E is not dependent on prior phosphorylation by CDK4-cyclin D. These data are
consistent with in vitro experiments showing the ability of CDK2-cyclin E to phosphorylate
unphosphorylated pRb (16) and with studies in the E→D1 “knockin" mouse (45) that found a
significant increase in the phosphorylation state of pRb in the absence of cyclin D1 activity.
These researchers reported that CDK2-cyclin E activity restores all the deficiencies associated
with the ablation of cyclin D1 in the cyclin D1 knockout mouse (46). In addition to
demonstrating that cyclin E-CDK2 can directly phosphorylate pRb in vivo in the absence of prior
phosphorylation by cyclin D-CDK4, we also found that this phosphorylation results in the
activation of E2F and the expression of cyclin A, which are essential for S-phase progression.
Our results further the understanding of the recognized role that deregulation of cyclin E
plays in tumorigenesis. In addition to the observation that many cancer cell lines overexpress
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

full-length cyclin E or an amino-truncated variant (70-73), deregulated cyclin E has been
reported in primary tumors of many different cell types (74). The clinical impact of cyclin E
expression appears most dramatic in the pathology of breast cancer. A recent report from
Keyomarsi, et al. argues convincingly that cyclin E expression levels are the single most
powerful clinical parameter in predicting long-term prognosis in breast cancer patients, with high
levels of cyclin E having an even higher hazard ratio than the classic prognostic indicators
cyclins D1 and D3, HER-2/neu, ER/PR status, and lymph node metastases (75). Supplementing
that argument, Span et al. recently reported that the expression level of cyclin E is a strong
predictor of endocrine therapy failure in breast cancer patients (76), a result that builds upon
mounting that evidence that cyclin E is a critical target of endocrine signaling in promoting
steroid hormone-dependent tumor growth (77). Finally, ten percent of mice that express a
human cyclin E transgene develop carcinoma (78), and cyclin E knockout MEFs are strikingly
resistant to transformation with oncogenic Ras ((79), see next paragraph). The data reported here
is consistent with the notion that the oncogenic potential of atypical cyclin E expression is due to
inappropriate cell cycle re-entry through the inactivation of pRb.
Two recent controversial reports seemingly call into question the necessity of CDK2-
cyclin E activity for cell cycle progression. The two reports that CDK2 knockout mice develop
normally and, despite meiotic defects and sterility, are viable and healthy (80,81); and that cyclin
E1/E2 double knock-outs can be born alive by placental rescue of cyclin E expression (79)
suggest that CDK2-cyclin E activity is dispensable for cell-autonomous murine embryogenesis
and cell proliferation. These findings, while intriguing, and no doubt warrant re-examination of
current cell cycle dogma, do not challenge the observations reported here. This work is
particularly concerned with what unscheduled cyclin E expression can do, in terms of pRb
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

phosphorylation, E2F activation, and cell cycle progression. The observation that cyclin E-null
MEFs cannot be stimulated to proliferate following serum-starvation suggests a crucial role for
cyclin E in cell cycle re-entry from quiescence, an event which may be critical during in vivo
tumorigenesis (79). In any event, whether or not CDK2-cyclin E is the normal mediator of E2F
activation awaits a more comprehensive study and is beyond the scope addressed in the present
study. However, given our findings, a strong case can be made for the ability of aberrant cyclin
E expression to drive the inactivation of pRb, and thus G1 progression, despite CDK4-cyclin D
inhibition, a finding that augments the known role of cyclin E in tumor progression and
prognosis.
Interestingly, another recent report has challenged previous interpretations regarding the
role of pRb in G1 cell cycle control. By using “acute” conditional loss of pRb to cells in culture,
a system more accurately representing the sporadic inactivation of RB alleles seen in the genesis
of a tumor, Sage et al. convincingly reaffirm that pRb is the critical mediator of the senescence
program in cell culture-induced cell cycle exit in MEFs (82). By escaping the complications of
developmental compensation and plasticity, this report further highlights the tumor-promoting
potential of pRb inactivation in quiescent somatic cells, that is, cells that have not recalibrated
their senescence regulators to compensate for the loss of pRb. In light of these results, aberrant
cyclin E expression could provide just the window of opportunity needed to initiate tumor
formation in vivo.
Going further, it would be interesting to analyze the phosphorylation status of all of the
CDK2-preferred sites on pRb in CDK2 or cyclin E knock out cells. Provocatively, in cyclin
E1/E2-null MEFs, pRb does appear to become phosphorylated sometime in late G1, suggesting
the compensatory, or perhaps redundant, activity of a cyclin E-independent G1 kinase (79).
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

Even more provocatively, pRb appears phosphorylated on Thr-821 in CDK2-/- MEFs, a site
thought to be phosphorylated by CDK2, suggesting that the crucial inactivation of pRb is
somehow endogenously recovered in these cells (80). Further complicating this issue is the fact
that a mutant form of cyclin E that cannot bind to CDK2 transforms rat embryo fibroblasts when
expressed with oncogenic H-Ras (83), a result not easily explained by current understanding of
cell cycle dynamics. Clearly, further studies are required to elucidate the role of CDK2, cyclin
E, and possibly an undiscovered G1 kinase in normal cell proliferation and in tumorigenesis.
However, the reality remains that cyclin E expression and pRb inactivation are intimately
connected and most likely have a causal relationship to the development and morbidity of human
cancer.
Acknowledgements – The authors would like to thank James Roberts, Kristian Helin, Jim Koh,
Martine Roussel, Charles Sherr, and J. Wade Harper for their generous supply of cDNAs. NHL
would like to thank Jaqueline Lees and J. Alan Diehl for encouragement and helpful discussions,
and Leroy Wheeler for critical reading of the manuscript.
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

Figure Legends
Figure 1. Expression of cyclin E recovers CDK2, but not CDK4 activity in LY294002-
treated cells. A-B.) IIC9 cells transiently transfected with cyclin E, followed by serum
deprivation for 48-60 hours. Cells were incubated in the presence or absence of 10µM
LY294002 30 minutes prior to stimulation. A.) Cells were stimulated with α-thrombin at 1U/mL
for eight hours. Lysates were assayed and quantified for CDK4-cyclin D activity as described in
materials and methods. B.) Cells were stimulated with α-thrombin at 1U/mL for seventeen
hours. Lysates were assayed and quantified for CDK2-cyclin E activity as described in materials
and methods. Error bars represent standard deviation (n=3) and data are representative of three
independent experiments. C.) Lysates from asynchronously growing IIC9 or HL60 cells were
analyzed for CDK6, cyclin D1, cyclin D2, or cyclin D3 in separate western blots.
Figure 2. CDK2-cyclin E activity drives E2F activation in the absence of CDK4-cyclin D
activity. A.) IIC9 cells were transiently transfected with pGL3-TATA-6xE2F, β-Galactosidase,
and, where indicated, cyclin E followed by serum deprivation for 48-60 hours. Quiescent cells
were incubated in the absence or presence of 10µM LY294002 or 7.5µM GW8510, followed by
stimulation with α-thrombin for 17 hours. Lysates were assayed for luciferase and β-
galactosidase activity, and relative light units were divided by units of optical density,
respectively, for quantification and normalization. B.) Quiescent IIC9 cells were stimulated with
α-thrombin for indicated lengths of time. RNA was isolated and analyzed for cyclin A and
GAPDH mRNA and quantified as described in materials and methods; cyclin A arbitrary units
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

were divided by GAPDH arbitrary units for normalization. Error bars represent standard
deviation (n=3). C-D.) IIC9 cells were transiently transfected with cyclin E and/or p16INK4a,
where indicated, followed by 48-60 hours of serum deprivation. Cells were incubated in the
presence or absence of 10µM LY29004 or 7.5µM GW8510 for 30 minutes, then stimulated with
1U/mL α-thrombin for C.) seventeen hours or D.) nineteen hours. C.) RNA was isolated and
analyzed for cyclin A mRNA and quantified, normalizing for GAPDH mRNA for normalization.
D.) Protein lysates were analyzed for cyclin A protein levels by western blotting, as described in
methods. All data are representative of at least three independent experiments.
Figure 3. Dominant-negative CDK2 blocks the cyclin E rescue of E2F activity in LY29004-
treated cells. A.) IIC9 cells were transiently transfected with pGL3-TATA-6xE2F, β-
Galactosidase, and, where indicated, cyclin E and/or CDK2(D145N) followed by serum
deprivation for 48-60 hours. Quiescent cells were incubated in the absence or presence of 10µM
LY294002, followed by stimulation with α-thrombin for 17 hours. Lysates were assayed for
luciferase and β-galactosidase activity, and relative light units were divided by units of optical
density, respectively, for quantification and normalization. B.) IIC9 cells were transiently
transfected with cyclin E and/or CDK2(D145N), where indicated, followed by 48-60 hours of
serum deprivation. Cells were incubated in the presence or absence of 10µM LY29004 for 30
minutes, then stimulated with 1U/mL α-thrombin for seventeen hours. Lysates were analyzed
for cyclin A and GAPDH mRNA and quantified; cyclin A arbitrary units were divided by
GAPDH arbitrary units for normalization. All data are representative of three independent
experiments.
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

Figure 4. CDK2-cyclin E phosphorylates and inactivates pRb in vivo in the absence of
prior CDK4-cyclin D activity. IIC9 cells transiently transfected with cyclin E, where indicated,
followed by serum deprivation for 48-60 hours. Cells were incubated in the presence or absence
of 10µM LY29004 30 minutes prior to stimulation with 1U/mL α-thrombin for seventeen hours.
A.) Lysates were assayed for phospho-pRb(Ser-780) and phospho-pRb(Ser795) by western
blotting. B.) pRb-immunoprecipitates were prepared from isolated nuclei as described in
methods and assayed for their ability to retard electro-migration of a 32P-labelled E2F-response
element probe by EMSA. Lane 1 = free probe, Lane 2-5 = probe incubate with pRb-
immunoprecipitates from basal nuclei, Lane 3 = competition with unlabelled probe, Lane 4-5 =
super-shift with antibodies to E2F1 (Lane 4) and E2F4 (Lane 5), Lane 6-8 = probe incubated
with pRb-immunoprecipitates from α-thrombin stimulated nuclei. Data are representative of at
least two independent experiments.
Figure 5. Rb∆CDK blocks the cyclin E-mediated rescue of E2F activation in LY29004-
treated cells. A.) IIC9 cells were transiently transfected with pGL3-TATA-6xE2F, β-
Galactosidase, and where indicated, cyclin E and/or Rb∆CDK followed by serum deprivation for
48-60 hours. Quiescent cells were incubated in the absence or presence of 10µM LY294002,
followed by stimulation with α-thrombin for 17 hours. Lysates were assayed for luciferase and
β-galactosidase activity, and relative light units were divided by units of optical density,
respectively, for quantification and normalization. B.) IIC9 cells were transiently transfected
with cyclin E and/or Rb∆CDK, where indicated, followed by 48-60 hours of serum deprivation.
Cells were incubated in the presence or absence of 10µM LY29004 for 30 minutes, then
stimulated with 1U/mL α-thrombin for seventeen hours. Lysates were analyzed for cyclin A and
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

GAPDH mRNA and quantified; cyclin A arbitrary units were divided by GAPDH arbitrary units
for normalization. Error bars represent standard deviation (n=3) and data are representative of
two independent experiments.
Figure 6. Expression of Rb∆+K2 does not block E2F activation in α-thrombin stimulated,
cyclin E-expressing cells. A-B.) IIC9 cells were transiently transfected with pGL3-TATA-
6xE2F, β-Galactosidase, and, where indicated, pcDNA3.1 (vector), Rb, Rb∆CDK, Rb∆+K2 , and/or
cyclin E followed by serum deprivation for 48-60 hours. Quiescent cells were incubated in the
absence or presence of 10µM LY294002 or 7.5µM GW8510, followed by stimulation with α-
thrombin for 17 hours. Lysates were assayed for luciferase and β-galactosidase activity, and
relative light units were divided by units of optical density, respectively, for quantification and
normalization. C.) IIC9 cells were transiently transfected with cyclin E, Rb∆CDK, and/or Rb∆+K2,
where indicated, followed by 48-60 hours of serum deprivation. Cells were incubated in the
presence or absence of 10µM LY29004 for 30 minutes, then stimulated with 1U/mL α-thrombin
for nineteen hours. Lysates were harvested and analyzed for cyclin A protein levels by western
blotting, as described in methods. All data are representative of at least three independent
experiments.
Figure 7. Cyclin E-CDK2, without prior cyclin D-CDK activity, is sufficient for entry into
S phase. IIC9 cells were transiently transfected with p16INK4a, cyclin E, pcDNA3.1, pRb,
Rb∆CDK, and/or Rb∆+K2 where indicated, followed by serum deprivation for 48-60 hours. Cells
were incubated in the presence or absence of 10µM LY29004 or 7.5µM GW8510 30 minutes
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

prior to stimulation with 1U/mL α-thrombin for seventeen hours. Cells were then pulsed with
0.5µCi/mL 3H-thymidine for four hours and assayed for TCA-precipitable counts as described.
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

Reference List
1. Obaya, A. J. and Sedivy, J. M. (2002) Cell Mol.Life Sci. 59, 126-142
2. Sherr, C. J. (1996) Science 274, 1672-1677
3. Phillips-Mason, P. J., Raben, D. M., and Baldassare, J. J. (2000) J.Biol.Chem. 275, 18046-18053
4. Weber, J. D., Raben, D. M., Phillips, P. J., and Baldassare, J. J. (1997) Biochem.J. 326 ( Pt 1), 61-68
5. Weber, J. D., Cheng, J., Raben, D. M., Gardner, A., and Baldassare, J. J. (1997) J.Biol.Chem. 272, 17320-17326
6. Bartek, J., Bartkova, J., and Lukas, J. (1996) Curr.Opin.Cell Biol. 8, 805-814
7. Weinberg, R. A. (1995) Cell 81, 323-330
8. Chen, P. L., Scully, P., Shew, J. Y., Wang, J. Y., and Lee, W. H. (1989) Cell 58, 1193-1198
9. DeCaprio, J. A., Ludlow, J. W., Lynch, D., Furukawa, Y., Griffin, J., Piwnica-Worms, H., Huang, C. M., and Livingston, D. M. (1989) Cell 58, 1085-1095
10. Bandara, L. R., Adamczewski, J. P., Hunt, T., and La Thangue, N. B. (1991) Nature 352, 249-251
11. Cao, L., Faha, B., Dembski, M., Tsai, L. H., Harlow, E., and Dyson, N. (1992) Nature 355, 176-179
12. Chellappan, S. P., Hiebert, S., Mudryj, M., Horowitz, J. M., and Nevins, J. R. (1991) Cell 65, 1053-1061
13. Dyson, N. (1998) Genes Dev. 12, 2245-2262
14. Ewen, M. E., Sluss, H. K., Sherr, C. J., Matsushime, H., Kato, J., and Livingston, D. M. (1993) Cell 73, 487-497
15. Kato, J., Matsushime, H., Hiebert, S. W., Ewen, M. E., and Sherr, C. J. (1993) Genes Dev. 7, 331-342
16. Mittnacht, S., Lees, J. A., Desai, D., Harlow, E., Morgan, D. O., and Weinberg, R. A. (1994) EMBO J. 13, 118-127
17. Ezhevsky, S. A., Nagahara, H., Vocero-Akbani, A. M., Gius, D. R., Wei, M. C., and Dowdy, S. F. (1997) Proc.Natl.Acad.Sci.U.S A 94, 10699-10704
18. Ezhevsky, S. A., Ho, A., Becker-Hapak, M., Davis, P. K., and Dowdy, S. F. (2001) Mol.Cell Biol. 21, 4773-4784
19. Brehm, A., Miska, E. A., McCance, D. J., Reid, J. L., Bannister, A. J., and Kouzarides, T. (1998) Nature 391, 597-601
20. Magnaghi-Jaulin, L., Groisman, R., Naguibneva, I., Robin, P., Lorain, S., Le Villain, J. P., Troalen, F., Trouche, D., and Harel-Bellan, A. (1998) Nature 391, 601-605
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

21. Zhang, H. S., Gavin, M., Dahiya, A., Postigo, A. A., Ma, D., Luo, R. X., Harbour, J. W., and Dean, D. C. (2000) Cell 101, 79-89
22. Akiyama, T., Ohuchi, T., Sumida, S., Matsumoto, K., and Toyoshima, K. (1992) Proc.Natl.Acad.Sci.U.S.A 89, 7900-7904
23. Dynlacht, B. D., Flores, O., Lees, J. A., and Harlow, E. (1994) Genes Dev. 8, 1772-1786
24. Harbour, J. W., Luo, R. X., Dei, S. A., Postigo, A. A., and Dean, D. C. (1999) Cell 98, 859-869
25. Zarkowska, T. and Mittnacht, S. (1997) J.Biol.Chem. 272, 12738-12746
26. Lin, B. T., Gruenwald, S., Morla, A. O., Lee, W. H., and Wang, J. Y. (1991) EMBO J. 10, 857-864
27. Adams, P. D., Li, X., Sellers, W. R., Baker, K. B., Leng, X., Harper, J. W., Taya, Y., and Kaelin, W. G., Jr. (1999) Mol.Cell Biol. 19, 1068-1080
28. Botz, J., Zerfass-Thome, K., Spitkovsky, D., Delius, H., Vogt, B., Eilers, M., Hatzigeorgiou, A., and Jansen-Durr, P. (1996) Mol.Cell Biol. 16, 3401-3409
29. Henglein, B., Chenivesse, X., Wang, J., Eick, D., and Brechot, C. (1994) Proc.Natl.Acad.Sci.U.S.A 91, 5490-5494
30. Oswald, F., Dobner, T., and Lipp, M. (1996) Mol.Cell Biol. 16, 1889-1895
31. Pearson, B. E., Nasheuer, H. P., and Wang, T. S. (1991) Mol.Cell Biol. 11, 2081-2095
32. Dou, Q. P., Markell, P. J., and Pardee, A. B. (1992) Proc.Natl.Acad.Sci.U.S A 89, 3256-3260
33. Hiebert, S. W., Blake, M., Azizkhan, J., and Nevins, J. R. (1991) J.Virol. 65, 3547-3552
34. Blake, M. C. and Azizkhan, J. C. (1989) Mol.Cell Biol. 9, 4994-5002
35. Ohtani, K., DeGregori, J., and Nevins, J. R. (1995) Proc.Natl.Acad.Sci.U.S A 92, 12146-12150
36. Chow, K. N., Starostik, P., and Dean, D. C. (1996) Mol.Cell Biol. 16, 7173-7181
37. Flemington, E. K., Speck, S. H., and Kaelin, W. G., Jr. (1993) Proc.Natl.Acad.Sci.U.S.A 90, 6914-6918
38. Helin, K., Harlow, E., and Fattaey, A. (1993) Mol.Cell Biol. 13, 6501-6508
39. Hiebert, S. W., Chellappan, S. P., Horowitz, J. M., and Nevins, J. R. (1992) Genes Dev. 6, 177-185
40. Zhang, H. S. and Dean, D. C. (2001) Oncogene 20, 3134-3138
41. Zhang, H. S., Postigo, A. A., and Dean, D. C. (1999) Cell 97, 53-61
42. Hinds, P. W., Mittnacht, S., Dulic, V., Arnold, A., Reed, S. I., and Weinberg, R. A. (1992) Cell 70, 993-1006
43. Lundberg, A. S. and Weinberg, R. A. (1998) Mol.Cell Biol. 18, 753-761
44. Hatakeyama, M., Brill, J. A., Fink, G. R., and Weinberg, R. A. (1994) Genes Dev. 8, 1759-1771
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

45. Geng, Y., Whoriskey, W., Park, M. Y., Bronson, R. T., Medema, R. H., Li, T., Weinberg, R. A., and Sicinski, P. (1999) Cell 97, 767-777
46. Sicinski, P., Donaher, J. L., Parker, S. B., Li, T., Fazeli, A., Gardner, H., Haslam, S. Z., Bronson, R. T., Elledge, S. J., and Weinberg, R. A. (1995) Cell 82, 621-630
47. Keenan, S. M., Bellone, C., and Baldassare, J. J. (2001) J.Biol.Chem. 276, 22404-22409
48. Low, D. A., Scott, R. W., Baker, J. B., and Cunningham, D. D. (1982) Nature 298, 476-478
49. Wright, T. M., Rangan, L. A., Shin, H. S., and Raben, D. M. (1988) J.Biol.Chem. 263, 9374-9380
50. Lents, N. H., Keenan, S. M., Bellone, C., and Baldassare, J. J. (2002) J.Biol.Chem. 277, 47469-47475
51. Hu, W., Bellone, C. J., and Baldassare, J. J. (1999) J.Biol.Chem. 274, 3396-3401
52. Weber, J. D., Hu, W., Jefcoat, S. C., Jr., Raben, D. M., and Baldassare, J. J. (1997) J.Biol.Chem. 272, 32966-32971
53. Goel, R., Phillips-Mason, P. J., Raben, D. M., and Baldassare, J. J. (2002) J.Biol.Chem. 277, 18640-18648
54. Diehl, J. A., Cheng, M., Roussel, M. F., and Sherr, C. J. (1998) Genes Dev. 12, 3499-3511
55. Lukas, J., Herzinger, T., Hansen, K., Moroni, M. C., Resnitzky, D., Helin, K., Reed, S. I., and Bartek, J. (1997) Genes Dev. 11, 1479-1492
56. DeGregori, J., Kowalik, T., and Nevins, J. R. (1995) Mol.Cell Biol. 15, 4215-4224
57. Dulic, V., Lees, E., and Reed, S. I. (1992) Science 257, 1958-1961
58. Koff, A., Giordano, A., Desai, D., Yamashita, K., Harper, J. W., Elledge, S., Nishimoto, T., Morgan, D. O., Franza, B. R., and Roberts, J. M. (1992) Science 257, 1689-1694
59. Serrano, M., Hannon, G. J., and Beach, D. (1993) Nature 366, 704-707
60. van den, H. S. and Harlow, E. (1993) Science 262, 2050-2054
61. Davis, S. T., Benson, B. G., Bramson, H. N., Chapman, D. E., Dickerson, S. H., Dold, K. M., Eberwein, D. J., Edelstein, M., Frye, S. V., Gampe Jr, R. T., Griffin, R. J., Harris, P. A., Hassell, A. M., Holmes, W. D., Hunter, R. N., Knick, V. B., Lackey, K., Lovejoy, B., Luzzio, M. J., Murray, D., Parker, P., Rocque, W. J., Shewchuk, L., Veal, J. M., Walker, D. H., and Kuyper, L. F. (2001) Science 291, 134-137
62. Rosenblatt, J., Gu, Y., and Morgan, D. O. (1992) Proc.Natl.Acad.Sci.U.S.A 89, 2824-2828
63. Nevins, J. R. (1992) Science 258, 424-429
64. Ferreira, R., Magnaghi-Jaulin, L., Robin, P., Harel-Bellan, A., and Trouche, D. (1998) Proc.Natl.Acad.Sci.U.S A 95, 10493-10498
65. Li, J. M., Hu, P. P., Shen, X., Yu, Y., and Wang, X. F. (1997) Proc.Natl.Acad.Sci.U.S A 94, 4948-4953
66. Wang, D., Russell, J. L., and Johnson, D. G. (2000) Mol.Cell Biol. 20, 3417-3424
67. Leng, X., Connell-Crowley, L., Goodrich, D., and Harper, J. W. (1997) Curr.Biol. 7, 709-712
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

68. Connell-Crowley, L., Harper, J. W., and Goodrich, D. W. (1997) Mol.Biol.Cell 8, 287-301
69. Tsai, L. H., Lees, E., Faha, B., Harlow, E., and Riabowol, K. (1993) Oncogene 8, 1593-1602
70. Harwell, R. M., Porter, D. C., Danes, C., and Keyomarsi, K. (2000) Cancer Res. 60, 481-489
71. Kitahara, K., Yasui, W., Kuniyasu, H., Yokozaki, H., Akama, Y., Yunotani, S., Hisatsugu, T., and Tahara, E. (1995) Int.J.Cancer 62, 25-28
72. Courjal, F., Louason, G., Speiser, P., Katsaros, D., Zeillinger, R., and Theillet, C. (1996) Int.J.Cancer 69, 247-253
73. Keyomarsi, K. and Pardee, A. B. (1993) Proc.Natl.Acad.Sci.U.S A 90, 1112-1116
74. Schraml, P., Bucher, C., Bissig, H., Nocito, A., Haas, P., Wilber, K., Seelig, S., Kononen, J., Mihatsch, M. J., Dirnhofer, S., and Sauter, G. (2003) J.Pathol. 200, 375-382
75. Keyomarsi, K., Tucker, S. L., Buchholz, T. A., Callister, M., Ding, Y., Hortobagyi, G. N., Bedrosian, I., Knickerbocker, C., Toyofuku, W., Lowe, M., Herliczek, T. W., and Bacus, S. S. (2002) N.Engl.J.Med. 347, 1566-1575
76. Span, P. N., Tjan-Heijnen, V. C., Manders, P., Beex, L. V., and Sweep, C. G. (2003) Oncogene 22, 4898-4904
77. Dhillon, N. K. and Mudryj, M. (2002) Oncogene 21, 4626-4634
78. Bortner, D. M. and Rosenberg, M. P. (1997) Mol.Cell Biol. 17, 453-459
79. Geng, Y., Yu, Q., Sicinska, E., Das, M., Schneider, J. E., Bhattacharya, S., Rideout, W. M., Bronson, R. T., Gardner, H., and Sicinski, P. (2003) Cell 114, 431-443
80. Ortega, S., Prieto, I., Odajima, J., Martin, A., Dubus, P., Sotillo, R., Barbero, J. L., Malumbres, M., and Barbacid, M. (2003) Nat.Genet. 35, 25-31
81. Berthet, C., Aleem, E., Coppola, V., Tessarollo, L., and Kaldis, P. (2003) Curr.Biol. 13, 1775-1785
82. Sage, J., Miller, A. L., Perez-Mancera, P. A., Wysocki, J. M., and Jacks, T. (2003) Nature 424, 223-228
83. Geisen, C. and Moroy, T. (2002) J.Biol.Chem. 277, 39909-39918
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

Figure 1AFo
ld-a
ctiv
atio
n
LY294002 - - + +Cyclin E - - - +
α-Thrombin
0
2
4
6
8
10
12
AEndogenous CDK4 Activity
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

Figure 1BFo
ld-a
ctiv
atio
n
0
2
46
8
10
1214
16
LY294002 - - + +Cyclin E - - - +
α-Thrombin
BEndogenous CDK2 Activity
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

Figure 1C
C IIC9 HL60
CDK6
Cyclin D1
Cyclin D2
Cyclin D3
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

Figure 2A
1.001.26
1.61
4.80
6.28
1.33
6.05
0.79 0.86
0
1
2
3
4
5
6
7
Fold
-incr
ease
in R
LU/B
-gal
α-thrombin - 5h 8h 11h 17h 17h 17h 17h 17hCyclin E - - - - - - + - +
LY294002
GW8510
E2F Reporter ActivityA
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

Figure 2BFo
ld In
crea
se
0
2
4
6
8
10
12
13 hrs
17 hrs
Cyclin A
GAPDH
α-Thrombin
Endogenous Cyclin A mRNAB
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

Figure 2C
Fold
Incr
ease
0
2
4
6
8
10
12
Cyclin A
Endogenous Cyclin A mRNAC
GAPDHLY294002 - - + + Cyclin E - - - +
α-Thrombin
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

Figure 2D
D Endogenous Cyclin A Protein
α-Thrombin
LY294002 - - + + - - + +Cyclin E - - - + - - - +p16INK4a - - - - + + + + by guest on July 17, 2019
http://ww
w.jbc.org/
Dow
nloaded from

Figure 3A
1.00
5.80
1.61
4.97
1.30 1.400.92
0
1
2
3
4
5
6
7
Fold
-incr
ease
in R
LU/B
-gal
Cyclin E - - - + - - +
LY294002
LY294002
dnCDK2
E2F Reporter Activity
α-Thrombin
A
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

Figure 3B
Fold
Incr
ease
0
2
4
6
8
10Endogenous Cyclin A mRNA
B
Cyclin A
GAPDH
LY294002 - - + + + Cyclin E - - - + + dnCDK2 - - - - +
α-Thrombin
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from
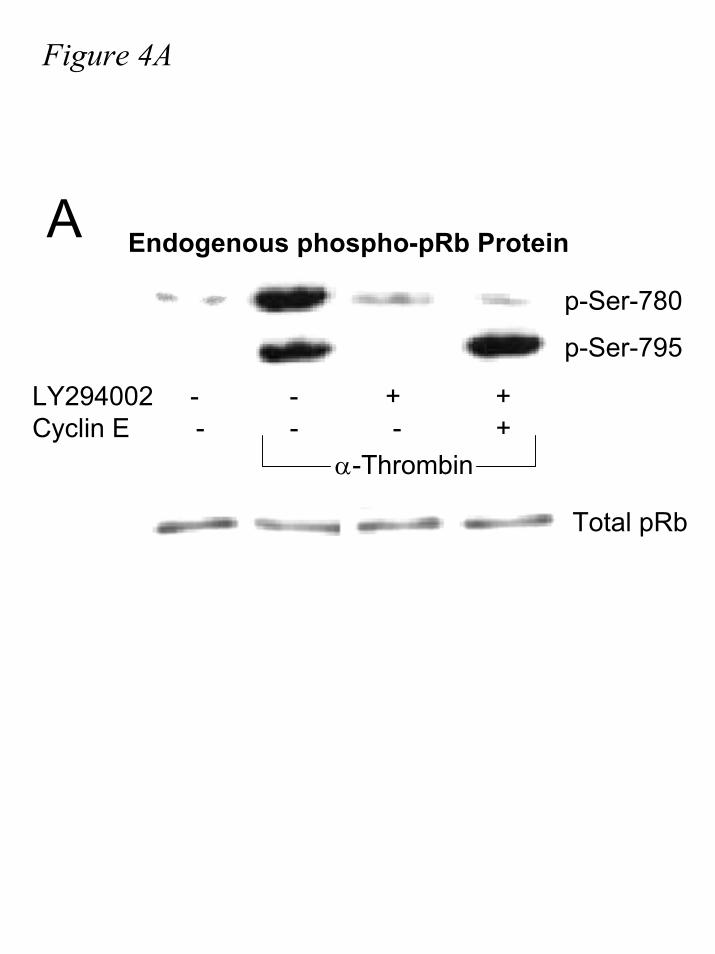
Figure 4A
A Endogenous phospho-pRb Protein
LY294002 - - + + Cyclin E - - - +
α-Thrombin
p-Ser-780
p-Ser-795
Total pRb
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

Figure 4B
B1 2 3 4 5 6 7 8
LY294002 - + +Cyclin E - - +
α-Thrombin
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

Figure 5A
LY294002 - - + + +Cyclin E - - - + + Rb∆CDK - - - - +
α-Thrombin
0
1
2
3
4
5
Fold
-incr
ease
in R
LU/B
-gal
E2F Reporter ActivityA
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from
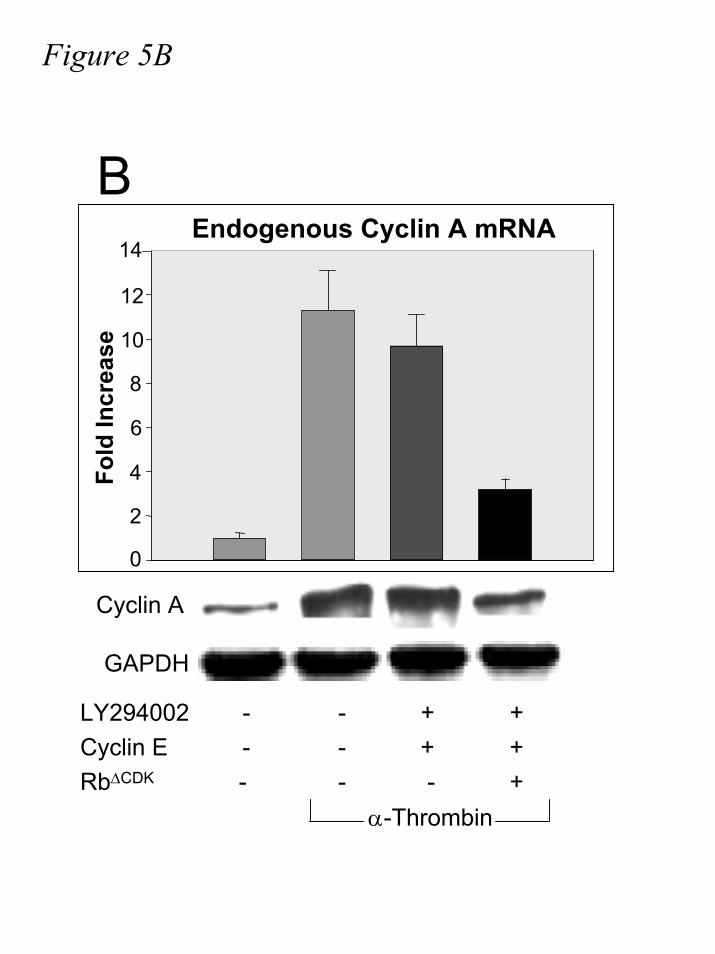
Figure 5BFo
ld In
crea
se
0
2
4
6
8
10
12
14Endogenous Cyclin A mRNA
B
Cyclin A
GAPDH
LY294002 - - + + Cyclin E - - + + Rb∆CDK - - - +
α-Thrombin
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from
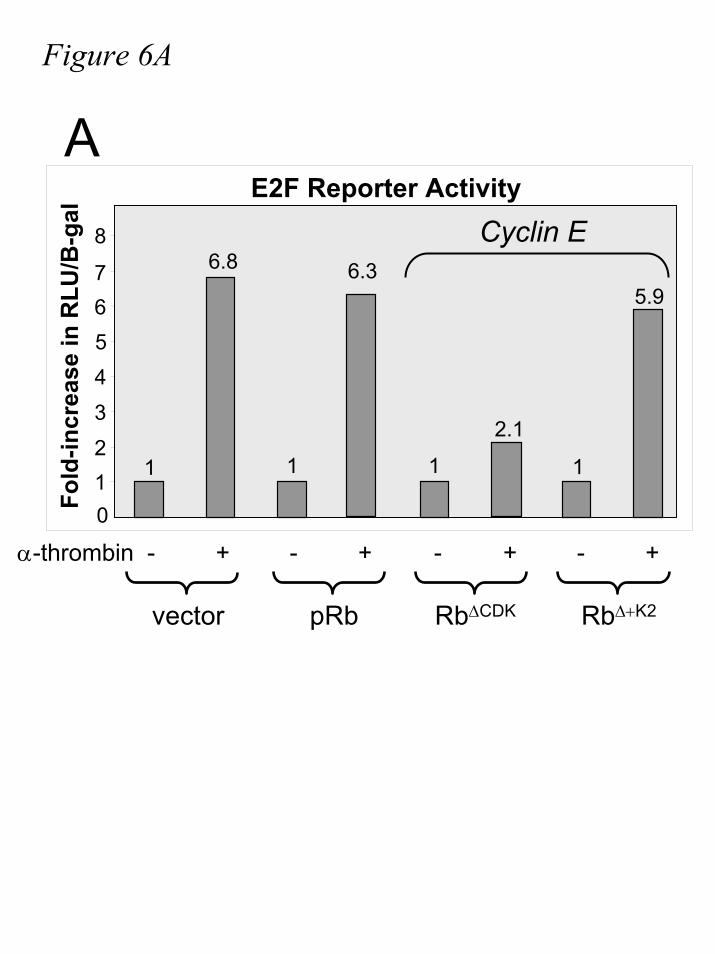
Figure 6A
1
6.8
1
2.1
1
6.3
1
5.9
1234567
8
0
pRb Rb∆CDKvector Rb∆+K2
Fold
-incr
ease
in R
LU/B
-gal
E2F Reporter ActivityCyclin E
α-thrombin - + - + - + - +
A
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

Figure 6B
1.00
4.92
1.26
4.60
0.93 0.87
0
1
2
3
4
5
6E2F Reporter Activity
Fold
-incr
ease
in R
LU/B
-gal
LY294002
GW8510
Cyclin E - - - + - +α-Thrombin
Rb∆+K2
B
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from
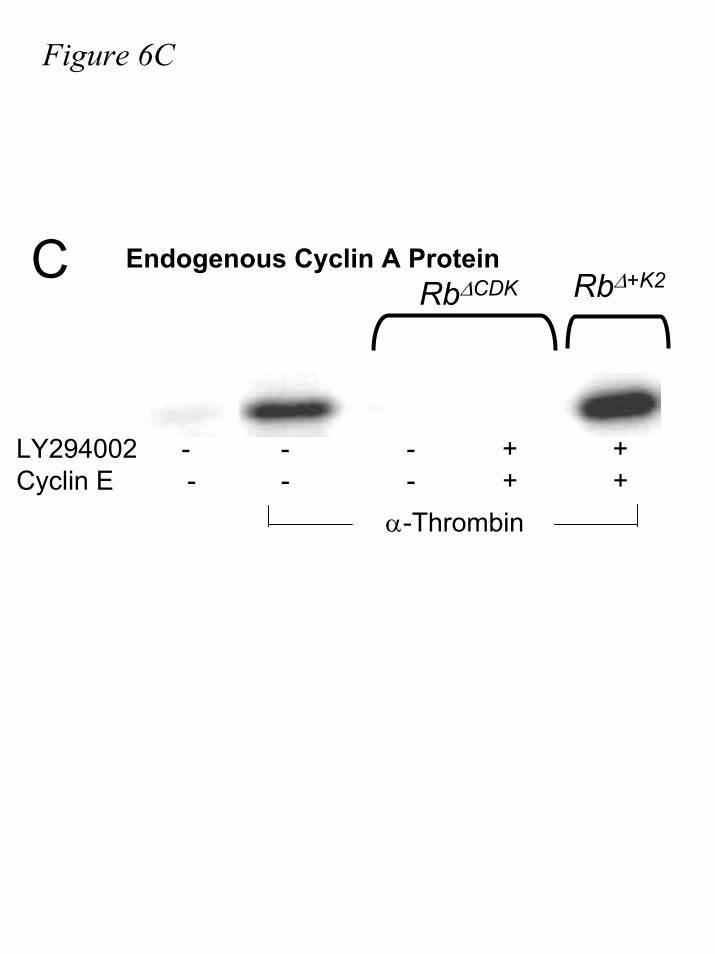
Figure 6C
CRb∆CDK Rb∆+K2
Endogenous Cyclin A Protein
LY294002 - - - + +Cyclin E - - - + +
α-Thrombin
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from
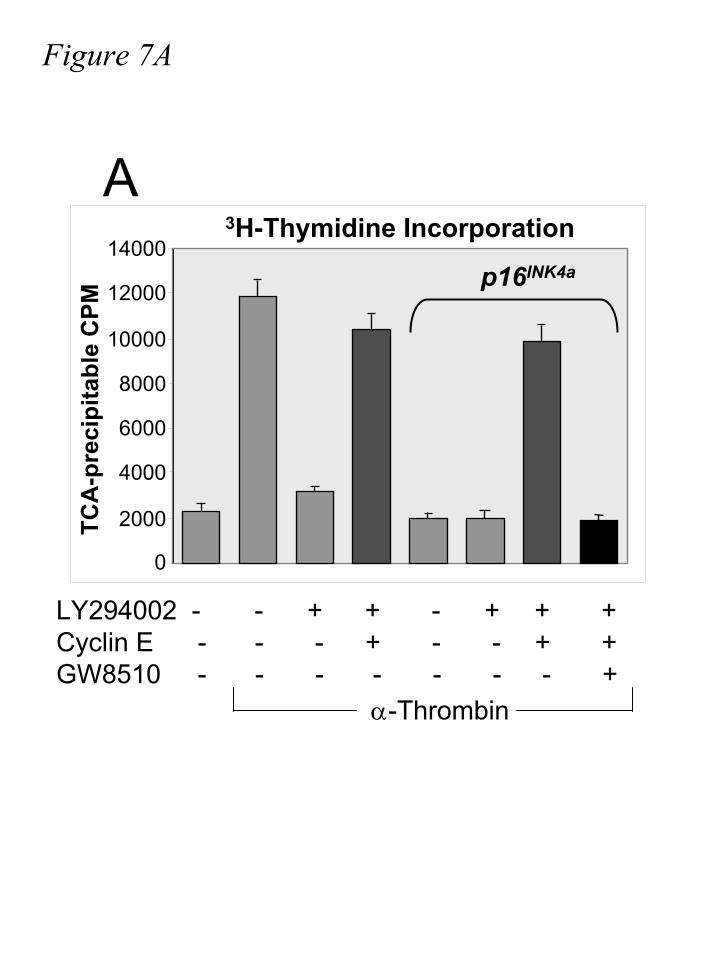
Figure 7A
LY294002 - - + + - + + + Cyclin E - - - + - - + +GW8510 - - - - - - - +
α-Thrombin
0
2000
4000
6000
8000
10000
12000
140003H-Thymidine Incorporation
TCA
-pre
cipi
tabl
e C
PM
p16INK4a
A
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

Figure 7B
pRb Rb∆CDKvector Rb∆+K2
0
2000
4000
6000
8000
10000
12000
14000
TCA
-pre
cipi
tabl
e C
PM
3H-Thymidine IncorporationB
α-Thrombin / Cyclin E
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from

Susan M. Keenan, Nathan H. Lents and Joseph J. Baldassareprogression
retinoblastoma tumor suppressor protein (pRB), activation of E2F, and G1-S phase Expression of cyclin E renders cyclin D-CDK4 dispensable for inactivation of the
published online November 25, 2003J. Biol. Chem.
10.1074/jbc.M310383200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on July 17, 2019http://w
ww
.jbc.org/D
ownloaded from