exercises (sequence databases, sequence alignment
TRANSCRIPT
Exercises (Sequence databases, sequence alignment, sequence database similarity search)
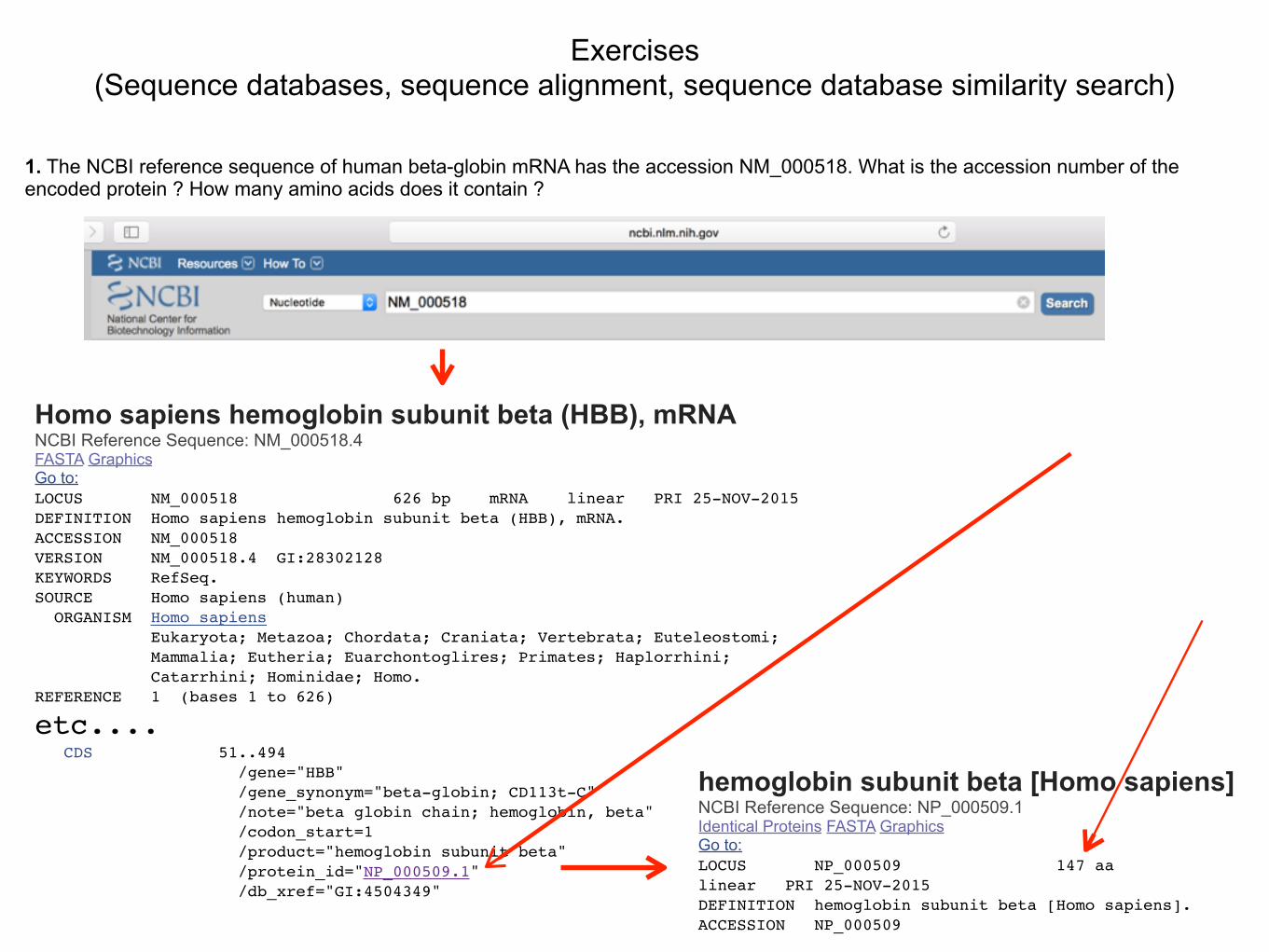
1. The NCBI reference sequence of human beta-globin mRNA has the accession NM_000518. What is the accession number of the encoded protein ? How many amino acids does it contain ?
Homo sapiens hemoglobin subunit beta (HBB), mRNA NCBI Reference Sequence: NM_000518.4 FASTA Graphics Go to: LOCUS NM_000518 626 bp mRNA linear PRI 25-NOV-2015DEFINITION Homo sapiens hemoglobin subunit beta (HBB), mRNA.ACCESSION NM_000518VERSION NM_000518.4 GI:28302128KEYWORDS RefSeq.SOURCE Homo sapiens (human) ORGANISM Homo sapiens Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi; Mammalia; Eutheria; Euarchontoglires; Primates; Haplorrhini; Catarrhini; Hominidae; Homo.REFERENCE 1 (bases 1 to 626)
etc.... CDS 51..494 /gene="HBB" /gene_synonym="beta-globin; CD113t-C" /note="beta globin chain; hemoglobin, beta" /codon_start=1 /product="hemoglobin subunit beta" /protein_id="NP_000509.1" /db_xref="GI:4504349"
hemoglobin subunit beta [Homo sapiens] NCBI Reference Sequence: NP_000509.1 Identical Proteins FASTA Graphics Go to: LOCUS NP_000509 147 aa linear PRI 25-NOV-2015DEFINITION hemoglobin subunit beta [Homo sapiens].ACCESSION NP_000509
Exercises (Sequence databases, sequence alignment, sequence database similarity search)
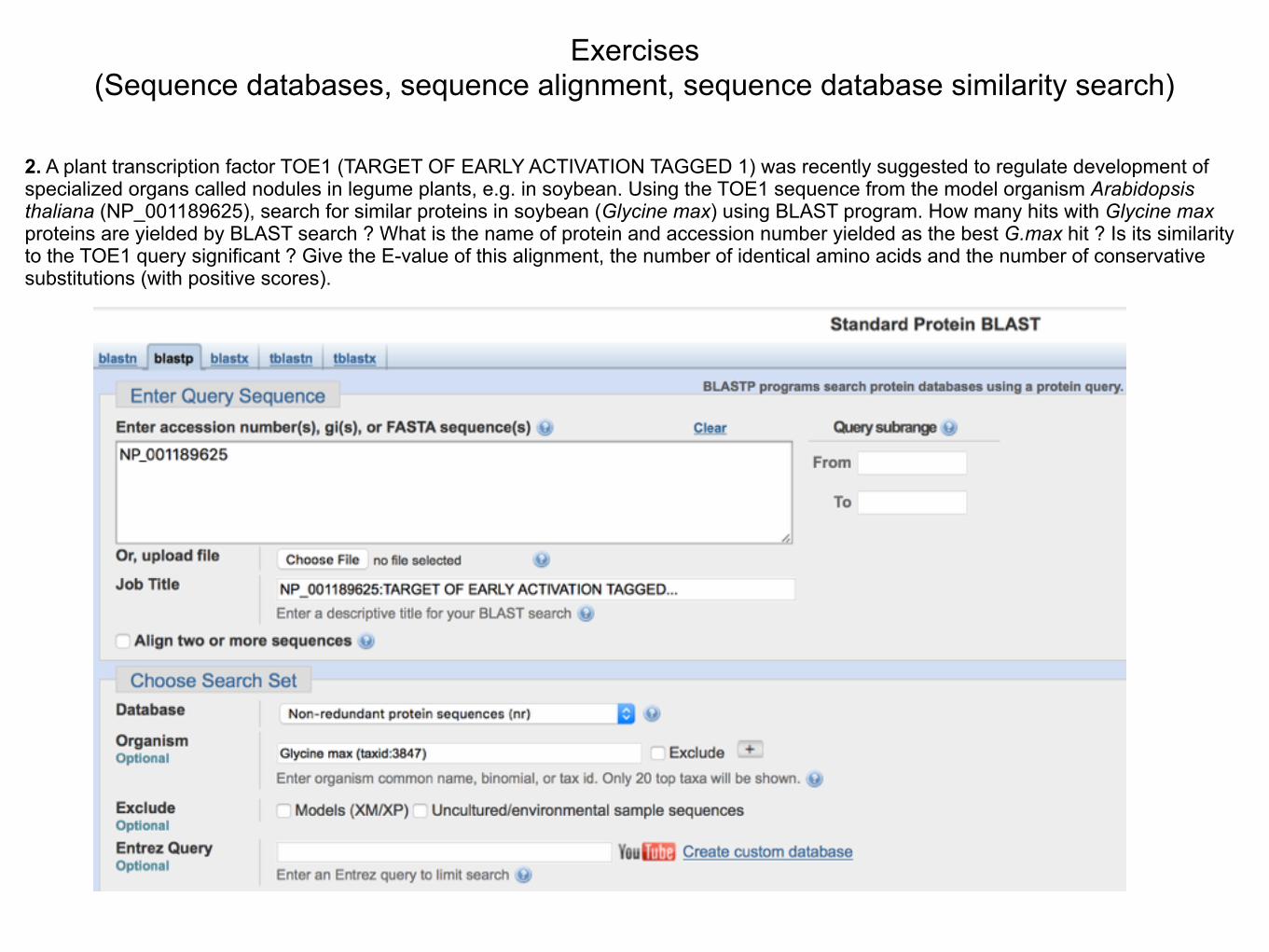
2. A plant transcription factor TOE1 (TARGET OF EARLY ACTIVATION TAGGED 1) was recently suggested to regulate development of specialized organs called nodules in legume plants, e.g. in soybean. Using the TOE1 sequence from the model organism Arabidopsis thaliana (NP_001189625), search for similar proteins in soybean (Glycine max) using BLAST program. How many hits with Glycine max proteins are yielded by BLAST search ? What is the name of protein and accession number yielded as the best G.max hit ? Is its similarity to the TOE1 query significant ? Give the E-value of this alignment, the number of identical amino acids and the number of conservative substitutions (with positive scores).

Exercises (Sequence databases, sequence alignment, sequence database similarity search)
2. A plant transcription factor TOE1 (TARGET OF EARLY ACTIVATION TAGGED 1) was recently suggested to regulate development of specialized organs called nodules in legume plants, e.g. in soybean. Using the TOE1 sequence from the model organism Arabidopsis thaliana (NP_001189625), search for similar proteins in soybean (Glycine max) using BLAST program. How many hits with Glycine max proteins are yielded by BLAST search ? What is the name of protein and accession number yielded as the best G.max hit ? Is its similarity to the TOE1 query significant ? Give the E-value of this alignment, the number of identical amino acids and the number of conservative substitutions (with positive scores).

Exercises (Sequence databases, sequence alignment, sequence database similarity search)
2. A plant transcription factor TOE1 (TARGET OF EARLY ACTIVATION TAGGED 1) was recently suggested to regulate development of specialized organs called nodules in legume plants, e.g. in soybean. Using the TOE1 sequence from the model organism Arabidopsis thaliana (NP_001189625), search for similar proteins in soybean (Glycine max) using BLAST program. How many hits with Glycine max proteins are yielded by BLAST search ? What is the name of protein and accession number yielded as the best G.max hit ? Is its similarity to the TOE1 query significant ? Give the E-value of this alignment, the number of identical amino acids and the number of conservative substitutions (with positive scores).
Exercises (Sequence databases, sequence alignment, sequence database similarity search)
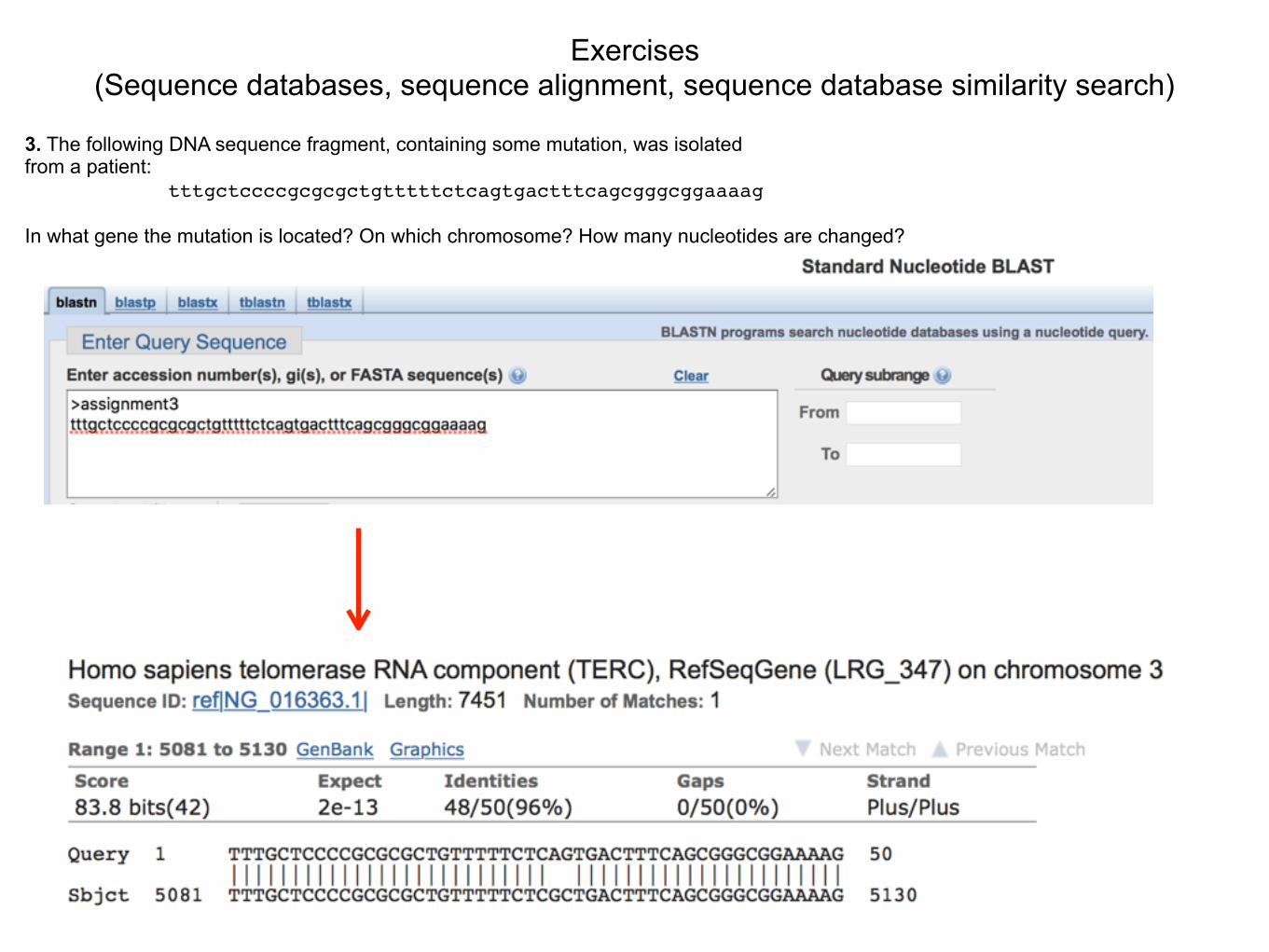
3. The following DNA sequence fragment, containing some mutation, was isolated from a patient: tttgctccccgcgcgctgtttttctcagtgactttcagcgggcggaaaag In what gene the mutation is located? On which chromosome? How many nucleotides are changed?

Exercises (Sequence databases, sequence alignment, sequence database similarity search)
4. A special option of BLAST for pairwise alignment of two sequences (bl2seq) is sometimes a quick way to determine similarity between two closely related sequences. (a) For instance, determine identity percentage in the alignment of two genomes of Zika virus: the one of an isolate from Suriname, October 2015 (accession KU312312) and the reference genome (NC_012532). - Also, using bl2seq for proteins, determine identities and positives in the alignment of polyproteins encoded by these two genomes. Locate the amino positions of gaps in the alignment: what are the residues inserted (or deleted) in the Suriname isolate as compared to the reference genome? - Are there insertions or deletions in the Suriname isolate polyprotein as compared to the polyprotein of Zika virus from a French Polynesia outbreak in 2013 (KJ776791) ?
(b) Is this BLAST option also optimal for the proteins of assignment 2: A.thaliana TOE1 (NP_001189625) and its homolog from G.max ? What is the alternative algorithm/program in this case ? Determine the percentage of similarity between these two proteins.
Suriname isolate polyprotein vs. polyprotein encoded by the reference genome: Identities: 3300/3423(96%); Positives 3369/3423(98%); Gaps 4/3423
Query 421 QPENLEYRIMLSVHGSQHSGMIVNDTGHETDENRAKVEITPNSPRAEATLGGFGSLGLDC 480 QPENLEYRIMLSVHGSQHSGMI G+ETDE+RAKVE+TPNSPRAEATLGGFGSLGLDCSbjct 421 QPENLEYRIMLSVHGSQHSGMI----GYETDEDRAKVEVTPNSPRAEATLGGFGSLGLDC 476
No indels as compared to the polyprotein of the Polynesian 2013 Zika oubreak

Exercises (Sequence databases, sequence alignment, sequence database similarity search)
(b) Is this BLAST option also optimal for the proteins of assignment 2: A.thaliana TOE1 (NP_001189625) and its homolog from G.max ? What is the alternative algorithm/program in this case ? Determine the percentage of similarity between these two proteins.
- BLASTP alignment does not cover full lengths of these proteins (local alignment) ! - It is necessary to use a global alignment program, e.g. Needleman-Wunsch Global
Sequence Alignment Tool provided by BLAST page.
Exercises (Sequence databases, sequence alignment, sequence database similarity search)
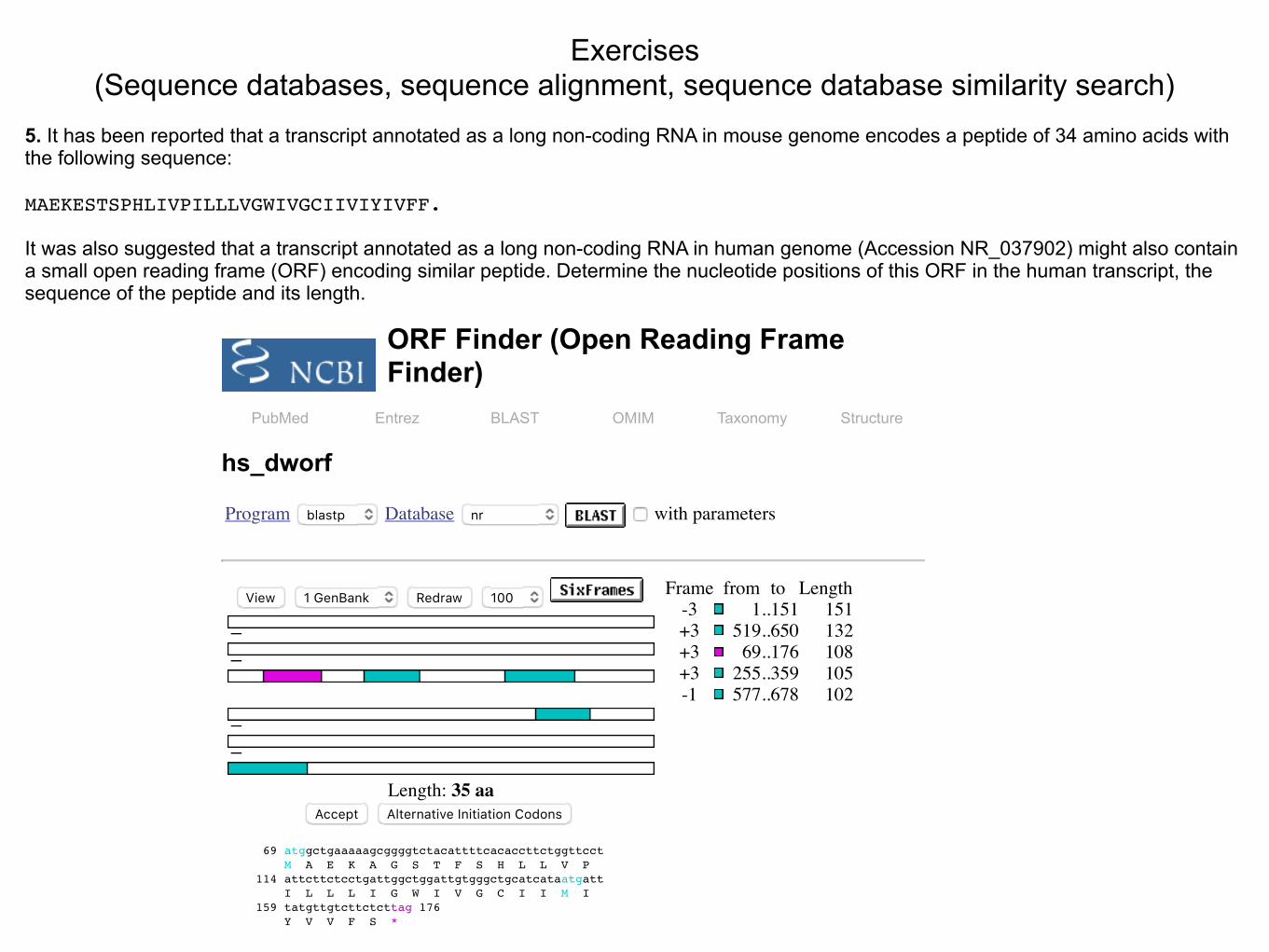
5. It has been reported that a transcript annotated as a long non-coding RNA in mouse genome encodes a peptide of 34 amino acids with the following sequence:
MAEKESTSPHLIVPILLLVGWIVGCIIVIYIVFF.
It was also suggested that a transcript annotated as a long non-coding RNA in human genome (Accession NR_037902) might also contain a small open reading frame (ORF) encoding similar peptide. Determine the nucleotide positions of this ORF in the human transcript, the sequence of the peptide and its length.
29/01/16 12:04ORF Finder
Page 1 of 1http://www.ncbi.nlm.nih.gov/gorf/orfig.cgi
ORF Finder (Open Reading FrameFinder)
PubMed Entrez BLAST OMIM Taxonomy Structure
hs_dworf
Program blastp Database nr with parameters
View 1 GenBank Redraw 100
Length: 35 aaAccept Alternative Initiation Codons
69 atggctgaaaaagcggggtctacattttcacaccttctggttcct M A E K A G S T F S H L L V P 114 attcttctcctgattggctggattgtgggctgcatcataatgatt I L L L I G W I V G C I I M I 159 tatgttgtcttctcttag 176 Y V V F S *
Frame from to Length-3 1..151 151+3 519..650 132+3 69..176 108+3 255..359 105-1 577..678 102

Exercises (Sequence databases, sequence alignment, sequence database similarity search)
5. It has been reported that a transcript annotated as a long non-coding RNA in mouse genome encodes a peptide of 34 amino acids with the following sequence:
MAEKESTSPHLIVPILLLVGWIVGCIIVIYIVFF.
It was also suggested that a transcript annotated as a long non-coding RNA in human genome (Accession NR_037902) might also contain a small open reading frame (ORF) encoding similar peptide. Determine the nucleotide positions of this ORF in the human transcript, the sequence of the peptide and its length.
Alternative solution:
The alignment of mouse peptide to the translated NR_037902 sequence by tblastn identifies the homologous ORF location, but yields a partial sequence (1 amino acid shorter):
Homo sapiens uncharacterized LOC100507537 (LOC100507537), long non-coding RNA Sequence ID: ref|NR_037902.1|Length: 795Number of Matches: 1
Query 1 MAEKE-STSPHLIVPILLLVGWIVGCIIVIYIVF 33 MAEK ST HL+VPILLL+GWIVGCII+IY+VFSbjct 69 MAEKAGSTFSHLLVPILLLIGWIVGCIIMIYVVF 170

Exercises (Sequence databases, sequence alignment, sequence database similarity search)
6. Using ENTREZ Gene database, determine the differences between alternative splicing isoforms of the human microtubule-associated protein tau (MAPT, GeneID 4137). How many exons are contained in the tau gene according to the RefSeqGene data? How many exons do alternative transcripts lack?
Search for Gene 4137"Go to reference sequence details": -> Genomic NG_007398 RefSeqGene "sequence viewer (graphics)" : 14 exons or "Genbank" exons are numbered in Features: 14
"mRNA and Protein(s)" transcript variants (isoforms) listed; missing exons are indicated as compared to isoform 6.-lacks four internal exons- three- five- one- six

Exercises (Sequence databases, sequence alignment, sequence database similarity search)
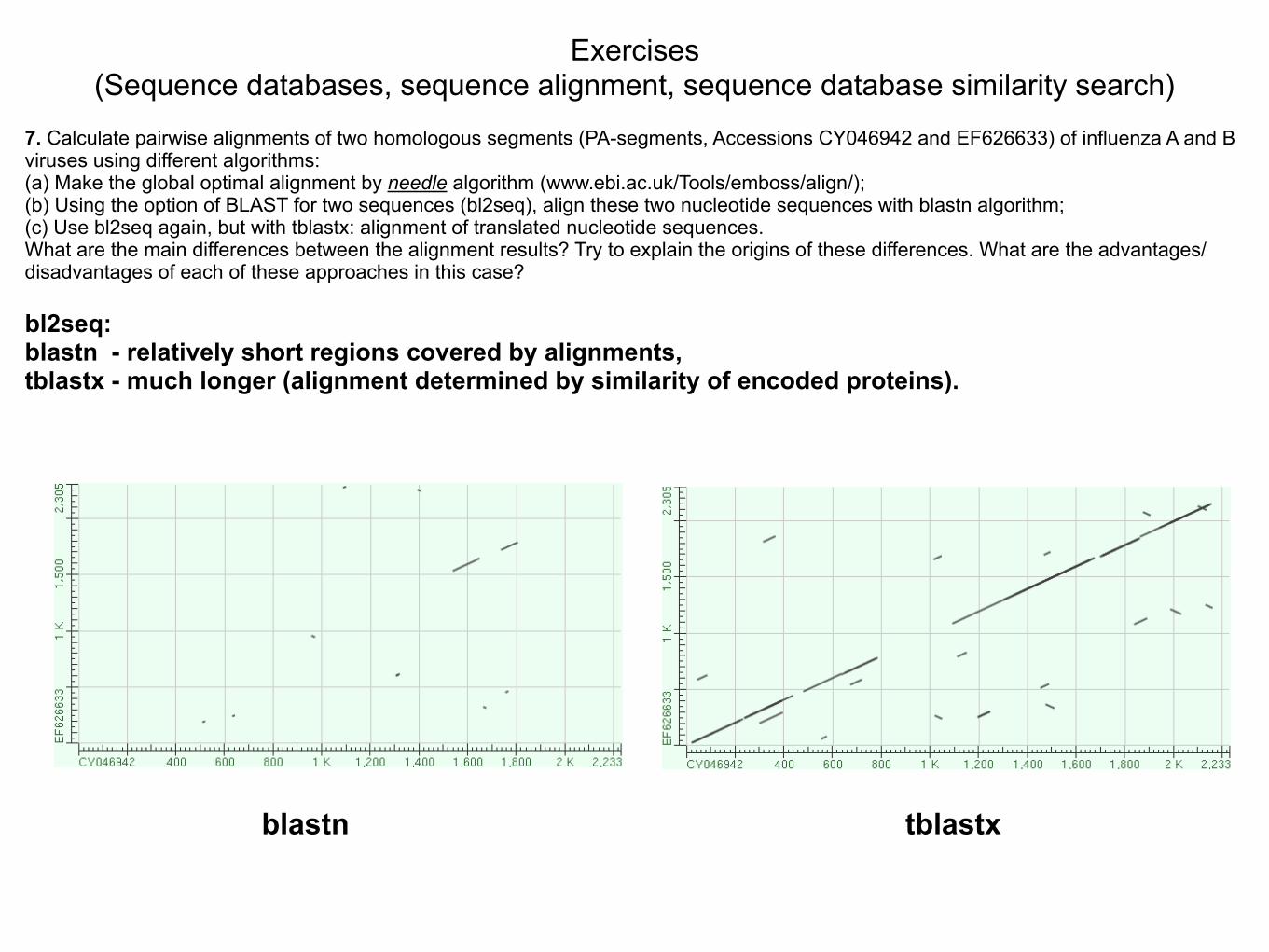
7. Calculate pairwise alignments of two homologous segments (PA-segments, Accessions CY046942 and EF626633) of influenza A and B viruses using different algorithms: (a) Make the global optimal alignment by needle algorithm (www.ebi.ac.uk/Tools/emboss/align/); (b) Using the option of BLAST for two sequences (bl2seq), align these two nucleotide sequences with blastn algorithm; (c) Use bl2seq again, but with tblastx: alignment of translated nucleotide sequences. What are the main differences between the alignment results? Try to explain the origins of these differences. What are the advantages/disadvantages of each of these approaches in this case?
Needle: #=======================================## Aligned_sequences: 2# 1: PA_fluA# 2: PA_fluB# Matrix: EDNAFULL# Gap_penalty: 10.0# Extend_penalty: 0.5## Length: 2717# Identity: 1390/2717 (51.2%)# Similarity: 1390/2717 (51.2%)# Gaps: 896/2717 (33.0%)# Score: 2792.5# ##=======================================
PA_fluA 1 AGCAAAAGCAGGTAC---TGAT---CCAAAATGGAAGACTTTGTGCGACA 44 ||||.|||| |||.| |||| .||.||||| |.||||| |...|||PA_fluB 1 AGCAGAAGC-GGTGCGTTTGATTTGTCATAATGG-ATACTTT-TATTACA 47
PA_fluA 45 ATG---CTTC---AATCCAATGAT-CGTCGAGCTTGCGGAAAAGG----- 82 | | |||| |.|.||||.|| | |||||| PA_fluB 48 A-GAAACTTCCAGACTACAATAATAC-------------AAAAGGCCAAA 83
...... global alignment till the 3’ends:
PA_fluA 2222 -----CTTGTTTCTACT 2233 |.||||||||||PA_fluB 2289 ATGCACGTGTTTCTACT 2305
#---------------------------------------
Exercises (Sequence databases, sequence alignment, sequence database similarity search)
7. Calculate pairwise alignments of two homologous segments (PA-segments, Accessions CY046942 and EF626633) of influenza A and B viruses using different algorithms: (a) Make the global optimal alignment by needle algorithm (www.ebi.ac.uk/Tools/emboss/align/); (b) Using the option of BLAST for two sequences (bl2seq), align these two nucleotide sequences with blastn algorithm; (c) Use bl2seq again, but with tblastx: alignment of translated nucleotide sequences. What are the main differences between the alignment results? Try to explain the origins of these differences. What are the advantages/disadvantages of each of these approaches in this case?
bl2seq: blastn - relatively short regions covered by alignments, tblastx - much longer (alignment determined by similarity of encoded proteins).
05/02/14 15:25NCBI Blast:gb|CY046942| (2233 letters)
Page 2 of 4http://blast.ncbi.nlm.nih.gov/Blast.cgi
Plot of gi|258589187|gb|CY046942.1| vs gi|148509292|gb|EF626633.1|
Sequences producing significant alignments:
Influenza B virus (B/Vienna/1/99) segment 3 polymerase PA (PA) gene, complete cdsSequence ID: gb|EF626633.1| Length: 2305 Number of Matches: 11Range 1: 1719 to 1787
Score Expect Identities Gaps Strand Frame
37.4 bits(40) 3e-05() 51/70(73%) 1/70(1%) Plus/Plus
Features:
Query 1738 AAGATCAAGATGAAATGGGGCATGGAACTGAGGCGCTGCCTTCTTCAGTCTCTTCAGCAG 1797 |||||| | ||||||||||| |||||| || | || || ||||| || | || || Sbjct 1719 AAGATCCAAATGAAATGGGGAATGGAAGCTAGAAGATGTCTGCTTCAATCAATGCAACAA 1778
Query 1798 ATTGAGAGCA 1807 || || ||||Sbjct 1779 ATGGA-AGCA 1787
Range 2: 1533 to 1645
Score Expect Identities Gaps Strand Frame
35.6 bits(38) 1e-04() 76/113(67%) 2/113(1%) Plus/Plus
Features:
Query 1540 AAAGGAAGGTCTCATTTGAGAAATGATACTGATGTGGTGAACTTTGTAAGTATGGAGTTC 1599
Dot Matrix View
[?]
Descriptions
Description Maxscore
Totalscore
Querycover
Evalue
Ident Accession
Influenza B virus (B/Vienna/1/99) segment 3polymerase PA (PA) gene, complete cds
37.4 277 12% 3e-05 73% EF626633.1
Alignments
05/02/14 15:26NCBI Blast:gb|CY046942| (2233 letters)
Page 2 of 9http://blast.ncbi.nlm.nih.gov/Blast.cgi
Plot of gi|258589187|gb|CY046942.1| vs gi|148509292|gb|EF626633.1|
Sequences producing significant alignments:
Influenza B virus (B/Vienna/1/99) segment 3 polymerase PA (PA) gene, complete cdsSequence ID: gb|EF626633.1| Length: 2305 Number of Matches: 42Range 1: 1086 to 1667
Score Expect Method Identities Positives Gaps Frame
186 bits(400) 2e-140(7) 77/194(40%) 119/194(61%) 0/194(0%) +1/+3
Features:
Query 1093 TKNMKRTSQLKWALGENMAPEKVDFDDCKDVGDLKQYDSDEPEPRSLASWVQNEFNKACE 1272 + +++T+ KWA G+ + +K+ + D + Q + P +A+WVQ E N Sbjct 1086 SNELQKTNYAKWATGDGLTYQKIMKEVAIDDETMCQEEPKIPNKCRVAAWVQTEMNLLST 1265
Query 1273 LTDSSWIELDEIGEDVAPIEHIASMRRNYFTAEVSHCRATEYIMKGVYINTALLNASCAA 1452 LT ++L EIG DVAP+EH+ S RR YF E+++C+A+ +MK V +T+LLN S A+Sbjct 1266 LTSKKALDLPEIGPDVAPVEHVGSERRKYFVNEINYCKASTVMMKYVLFHTSLLNESNAS 1445
Query 1453 MDDFQLIPMISKCRTKEGRRKTNLYGFIIKGRSHLRNDTDVVNFVSMEFSLTDPRLEPHK 1632 M +++IP+ ++ ++G LYG +KG+SHLR DTDVV V+ EFS TDPR++ KSbjct 1446 MGKYKVIPITNRVVNEKGESFDMLYGLAVKGQSHLRGDTDVVTVVTFEFSSTDPRVDSGK 1625
Query 1633 WEKYCVLEIGDMLL 1674 W KY V IG + +Sbjct 1626 WPKYTVFRIGSLFV 1667
Range 2: 1857 to 2150
Dot Matrix View
[?]
Descriptions
Description Maxscore
Totalscore
Querycover
Evalue
N Accession
Influenza B virus (B/Vienna/1/99) segment 3polymerase PA (PA) gene, complete cds
186 1524 80% 2e-140 Sumn7 EF626633.1
Alignments
blastn tblastx

Exercises (Sequence databases, sequence alignment, sequence database similarity search)
1. The NCBI reference sequence of human beta-globin mRNA has the accession NM_000518. What is the accession number of the encoded protein ? How many amino acids does it contain ?
2. A plant transcription factor TOE1 (TARGET OF EARLY ACTIVATION TAGGED 1) was recently suggested to regulate development of specialized organs called nodules in legume plants, e.g. in soybean. Using the TOE1 sequence from the model organism Arabidopsis thaliana (NP_001189625), search for similar proteins in soybean (Glycine max) using BLAST program. How many hits with Glycine max proteins are yielded by BLAST search ? What is the name of protein and accession number yielded as the best G.max hit ? Is its similarity to the TOE1 query significant ? Give the E-value of this alignment, the number of identical amino acids and the number of conservative substitutions (with positive scores).
3. The following DNA sequence fragment, containing some mutation, was isolated from a patient: tttgctccccgcgcgctgtttttctcagtgactttcagcgggcggaaaag In what gene the mutation is located? On which chromosome? How many nucleotides are changed?
4. A special option of BLAST for pairwise alignment of two sequences (bl2seq) is sometimes a quick way to determine similarity between two closely related sequences. (a) For instance, determine identity percentage in the alignment of two genomes of Zika virus: the one of an isolate from Suriname, October 2015 (accession KU312312) and the reference genome (NC_012532). - Also, using bl2seq for proteins, determine identities and positives in the alignment of polyproteins encoded by these two genomes. Locate the amino positions of gaps in the alignment: what are the residues inserted (or deleted) in the Suriname isolate as compared to the reference genome? - Are there insertions or deletions in the Suriname isolate polyprotein as compared to the polyprotein of Zika virus from a French Polynesia outbreak in 2013 (KJ776791) ?
(b) Is this BLAST option also optimal for the proteins of assignment 2: A.thaliana TOE1 (NP_001189625) and its homolog from G.max ? What is the alternative algorithm/program in this case ? Determine the percentage of similarity between these two proteins.

Exercises (Sequence databases, sequence alignment, sequence database similarity search)
5. It has been reported that a transcript annotated as a long non-coding RNA in mouse genome encodes a peptide of 34 amino acids with the following sequence:
MAEKESTSPHLIVPILLLVGWIVGCIIVIYIVFF.
It was also suggested that a transcript annotated as a long non-coding RNA in human genome (Accession NR_037902) might also contain a small open reading frame (ORF) encoding similar peptide. Determine the nucleotide positions of this ORF in the human transcript, the sequence of the peptide and its length.
6. Using ENTREZ Gene database, determine the differences between alternative splicing isoforms of the human microtubule-associated protein tau (MAPT, GeneID 4137). How many exons are contained in the tau gene according to the RefSeqGene data? How many exons do alternative transcripts lack?
7. Calculate pairwise alignments of two homologous segments (PA-segments, Accessions CY046942 and EF626633) of influenza A and B viruses using different algorithms: (a) Make the global optimal alignment by needle algorithm (www.ebi.ac.uk/Tools/emboss/align/); (b) Using the option of BLAST for two sequences (bl2seq), align these two nucleotide sequences with blastn algorithm; (c) Use bl2seq again, but with tblastx: alignment of translated nucleotide sequences. What are the main differences between the alignment results? Try to explain the origins of these differences. What are the advantages/disadvantages of each of these approaches in this case?