excited state intramolecular proton transfer in 1 ... · pdf fileexcited state intramolecular...
TRANSCRIPT

Molecular Physics, Vol. 104, Nos. 5–7, 10 March–10 April 2006, 943–955
Excited state intramolecular proton transfer
in 1-(trifluoroacetylamino)naphthaquinone:a CASPT2//CASSCF computational studyy
ALESSANDRO CEMBRAN and JIALI GAO*
University of Minnesota, USA
(Received 24 April 2005; in final form 9 August 2005)
The excited state intramolecular proton transfer (ESIPT) in 1-(trifluoroacetylamino)-
naphthaquinone (TFNQ) has been investigated using the CASSCF and CASPT2 computa-
tional approaches with the 6-31G(d) basis set. The structures and relative energies of critical
points along the proton transfer reaction coordinate were optimized and the associated
spectroscopic and electrostatic properties obtained. Combined quantum mechanical and
molecular mechanical (QM/MM) Monte Carlo simulations were performed to elucidate
solvent effects on the vertical excitation S0!S1. It was found that the ESIPT reaction is a
barrierless process that takes place on a very flat potential energy surface (PES) and the
tautomeric structure of the reaction product is the only minimum on the excited state surface.
The PES for both the ground and excited state from accurate electronic structure calcualtions
will be used to parameterize empirical force fields in subsequent molecular dynamics
simulations of the reaction in solution.
1. Introduction
Excited state intramolecular proton transfer (ESIPT)
reactions were observed by Weller [1] in methyl salicy-
late and later by Kasha [2] in 3-hydroxyflavone. These
reactions have been a subject of great interest both
experimentally [3–9] and theoretically [10–13] thanks to
the potential applications in lasers [14], energy and
data storage devices, optical switches [14–20], Raman
filters [21], polymers photostabilizers [22], triplet
quenchers [23, 24] and LED materials [24]. In many
cases, the donor atom (to which the hydrogen is initially
bonded) is an oxygen atom and the reaction is driven
by the redistribution of the charge density to the
proton acceptor. In 1-aminoanthraquinones, however,
the donor group consists of a nitrogen atom, forming
an intramolecular hydrogen bond with the acceptor
quinonoid oxygen [3]. The compound is forced to be
planar by the rigidity of the anthraquinone framework.Smith et al. [3] studied the excited state properties
of the four 1-(acylamino)anthraquinones derivatives
shown in figure 1 by static fluorescence, time-resolved
emission and transient absorption experiments. After
photoexcitation to the spectroscopic �!�* excited
state, two fluorescence bands are observed, providingevidence for the existence of two different emitting
tautomeric structures. The short wavelength emission
(SWE), centred around 510 nm (235 kJmol�1), hasbeen attributed to the ‘normal’ structure N, in which
the hydrogen is bonded to the nitrogen. The long
wavelength emission (LWE), centered around 630 nm
(190 kJmol�1), is from the tautomeric form T, wherethe proton is transferred to the oxygen. The relative
intensities of these two bands depend on the sub-
stituents, in particular HPAQ displays mostly SWE,while only LWE is observed for TFAQ. This means that
electron-withdrawing groups favour the ESIPT by
shifting the equilibrium towards the T structure.CAAQ and DCAQ show both bands and their relative
intensity ratio is influenced by the solvent. In particular,
polar solvents yield strong SWE bands with respect tothat of the LWE, by greater stabilization of the N
structure than the T configuration. The roles played
by substituents on the two structures are reflectedin the ESIPT reaction rate. While for TFAQ, the
ESIPT is nearly instantaneous, a rate constant of
110 fs was observed for compounds with a lesselectron-withdrawing substituent like CAAQ [25].
*Corresponding author. Email: [email protected]
yThis paper is dedicated to Professor Michael Robb on the occasion
of his 60th birthday.
Molecular PhysicsISSN 0026–8976 print/ISSN 1362–3028 online � 2006 Taylor & Francis
http://www.tandf.co.uk/journalsDOI: 10.1080/00268970500417556

In the other extreme, HPAQ does not undergo fastproton transfer (PT). The lifetime of these moleculesranges from 80 to 700 ps. In connection with the rateconstant of intersystem crossing kisc for 1-acetylaminoanthraquinone [26], which is 1.43�109 s�1,of similar time scale to the slow ESIPT reactions, itsuggests that the decay to the ground state takes placeby intersystem crossing.Using Raman spectrscopic techniques, Blank
et al. [27] investigated the ESIPT reaction by probingthe solvent (acetonitrile) response directly. Their resultsconfirmed that HPAQ does not undergo ESIPT, whileTFAQ proceeds with a ‘ballistic’ and complete ESIPT.Interestingly, they found that initially CAAQ undergoesfast ESIPT, but the reaction returns to the N structurein about 1 ps, suggesting that the potential energysurface of the excited state evolves as the solventreorganizes in response to the charge distributionof the excited state species.In association with this work, we seek to understand
the mechanism of the ESIPT process in 1-(acylamino)anthraquinones, TFAQ is the most suitable derivativeto be examined thanks to its fast and complete PTmechanism. In the following, we first summarize thecomputational details employed in the present study.Due to the computational cost of the CASPT2//CASSCF approach [28, 29], we will show in the thirdsection that the smaller model compound 1, lacking oneof the aromatic rings (see figure 2) is a reasonablesimplification of the anthracene system. Then, wepresent the computational results and discussionin the third section. We conclude this paper witha summary of the major findings of this work.
2. Computational details
2.1. Electronic structure calculations
The ground state S0 and first excited state S1 potential
energy surfaces (PESs) for the ESIPT process have beendetermined by full optimizations at the CASSCF/6-31G(d) level, which is followed by single-pointCASPT2 computations on the optimized structures.
All the optimizations have been performed with singlestate calculations, with the exception of MT –S1
(see figures 2 and 3) for which a state averagewavefunction with equal weights for S0 and S1 has
been used. The 6-31G(d) basis set is used throughout.All computations have been carried out using theGaussian 98 [30] and the MOLCAS-5.2 [31] quantum
chemistry programs.In order to apply the CASPT2//CASSCF approach
using an adequate number of orbitals, it is necessary toreduce the dimension of the system. The HOMO andLUMO orbitals involved in the �!�* excitation are
located, respectively, in the amide-bearing ring andin the quinone ring and no important orbitals are foundin the third aromatic ring for the reaction studied here.Therefore, it is a reasonable approximation to remove
this fragment and use the reduced model compound 1,1-(trifluoroacetylamino)-naphthaquinone (TFNQ).
The selection of the active space is a crucial stepin CASSCF calculations. In this system, there are 16�electrons in 15� orbitals that can be potentially included
into the active space. In addition, it might be necessaryto include the � orbitals for the N–H bond and the lonepair on the oxygen to which the proton is transferred.In all, the target active space could be as large as
20 electrons in 17 orbitals, which is too large to befeasible computationally. By examining the occupanciesof different orbitals in various tests, we found
that a balanced, smaller active space that includes
RHPAQ n-C6H13CAAQ CH2ClDCAQ CHCl2TFAQ CF3
N T
N
O
OH
O
R
N
O
OH
O
R
Figure 1. Normal (N) and tautormer (T) structures forthe four different 1-(acylamino)anthraquinones of reference [3]1-(heptanoylamino)anthraquinone (HPAQ), 1-(chloro-acetylamino)anthraquinone (CAAQ), 1-(dichloroacety-lamino)anthraquinone (DCAQ) and 1-(trifluoroacetylamino)anthraquinone (TFAQ).
N
O
OH
O
CF3
1
12
3445
6
78 8a
1
2
Figure 2. 1-(Trifluoroacetylamino)naphthaquinone (TFNA),1, used in all the calculations as a reduced model of TFAQ.
944 A. Cembran and J. Gao

10 electrons and 10 orbitals (10e/10o) is adequate forthe proton transfer process in CASSCF optimizations.The optimized geometries are further refined by singlepoint (12e/12o) state averaged (equal weight for S0and S1) CASSCF calculations, followed by perturbationcorrections at the CASPT2 level. Interestingly, the two �orbitals are not essential in these calculations for thereaction studied here since even in the transitionstate region their occupation numbers are very closeto two.For each optimized structure, the dipole moment
and partial atomic charges are determined using theelectrostatic potential (ESP) fitting scheme at theCASSCF(12e/12o) and the CASSCF(10e/10o) level.To assess the efficiency and probability of radiativetransition between S0 and S1, the RASSI [32, 33]approach is used to compute the oscillator strength f
of the vertical transitions for the optimized structures.
2.2. Combined QM/MM Monte Carlo simulations
In order to evaluate solvent effects on the S0!S1transition energy, statistical Monte Carlo simulations
are carried out using a combined quantum mechanical
and molecular mechanical (QM/MM) potential for 1
in tetragonal boxes of different solvents, including
CH2Cl2 [34], CH3CN [35] and TIP4P [36] H2O. The
boxes dimensions are approximately 30� 30� 46 A3
for CH2Cl2, 29� 29� 43 A3 for CH3CN and 20� 20�
30 A3 for H2O, containing a total of 380 solvent
molecules in each system. The solute molecule electronic
structure in the ground and first excited states is
described by a configuration-interaction with single
excitations (CIS) within an active space of six doubly
occupied and six unoccupied molecular orbital obtained
from the semiempirical Austin Model 1 (AM1) wave-
function [37, 38]. All Monte Carlo simulations are
executed in an isothermal-isobaric (NPT) ensemble at
25�C and 1 atm. A spherical cutoff distance of 12 A is
used for both solvent–solvent and solute–solvent inter-
actions based on solute heavy atom and water oxygen
distances. The van der Waals parameters used for the
solute molecule are taken from reference [39]. In all
simulations, the molecular geometry of 1 is kept fixed
at the configuration optimized at the HF/6-31G* level
in the gas phase.
S0
Reaction coordinate (RNH–ROH) (Å)
−1 −0.5 0 0.5 1
Ene
rgy
(kJ
mol
−1)
MN-S0
TS–S1
MT–S0
MT–S1
MN–S1
TS–S0
0
100
200
300
400
323.50.080
268.40.115
135.20.141
187.20.176
S1
O N ONH
N T
MN–S0
0
100
200
300
400
323.50.080
268.40.115
135.20.141
187.20.176
H
Figure 3. S0 and S1 PES for the proton transfer reaction in 1 along the reaction coordinate defined as the difference between theN–H and O–H bond distances. All the structures are optimized at the CASSCF[10e,10o]/6-31G(d). The CASSCF[12e,12o]/6-31G(d)energy profiles are reported with dashed lines while the CASPT2 corrected profiles are shown with solid lines. In dashedarrows the absorption from the S0 minimum and the fluorescence energies from S1 are reported together with the oscillatorstrength value in italic.
Excited state intramolecular proton transfer in 1-(trifluoroacetylamino)anthraquinone 945

In each case, 106 configurations of Monte Carlosimulations were performed to equilibrate the system,which was followed by additional 106 configurationsfor data averaging. New configurations were generatedby randomly translating a randomly selected moleculein the three Cartesian directions and rotating it arounda randomly chosen axis. To facilitate convergenceof solvent near the solute molecular, Owicki–Scheraga[40, 41] preferential sampling technique was usedsuch that solvent moves are made proportional to1/(R2
þW), where W¼ 250 A2 for CH2Cl2 andW¼ 200 A2 for CH3CN and H2O. Volume moveswere attempted on every 2375 configurations with amaximum allowed variation of �585 A3, and the solutemolecule was moved in every 90 configurations.Different ranges for the rotation and translations wereused for the different solvents and for the solute: forthe solvent molecules the ranges are from �0.15to �0.25 A and from � 15� to �23�, while for thesolute they span from �0.10 to �0.13 A and from �7�
to �10�. These options yielded an acceptance rateof about 45% on all attempted moves. All simulationswere performed using the MCQUM [42] programinterfaced with the MOPAC [43] package.
3. Results and discussion
3.1. Validation of the model compound
In order to check if compound 1 is a good model forthe larger system, both molecules were optimized at theHF/6-31G(d) level in the ground state and verticalexcitations energies were computed at the semiempiricalZINDO and CIS/6-31G(d) levels.The transition energies for 1 at the ZINDO
and CIS level are, respectively, 356.9 kJmol�1 and478.6 kJmol�1, in reasonable agreement with valuesof 369.4 kJmol�1 and 484.9 kJmol�1 obtained at thesame levels for TFAQ. These results confirm thatthe third aromatic ring has little participation in the�!�* transition and that the reduced model 1, TFNQ,can be used to approximate the electronic structuresof TFAQ used in experiments.
3.2. CASPT2//CASSCF study
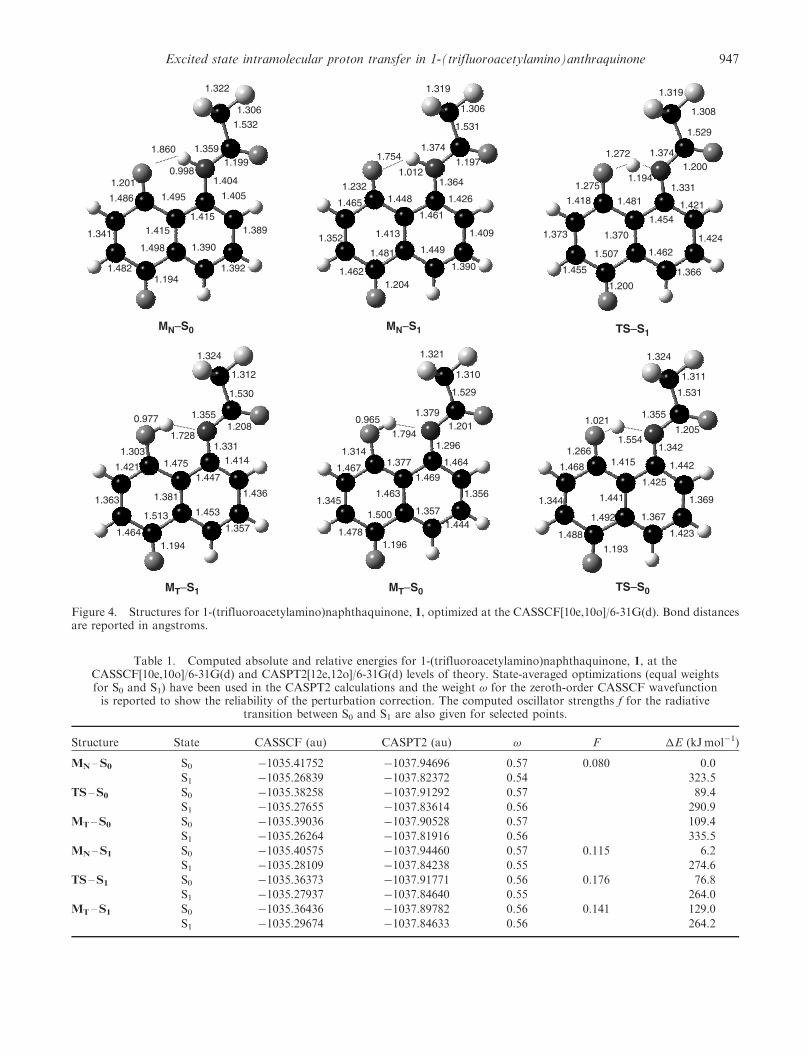
We have determined the S0 and S1 energy profiles alongthe proton transfer reaction coordinate at the CASSCFand CASPT2 levels of theory using the 6-31G(d) basisset, which are shown in figure 3. The optimizedstructures are depicted in figure 4 along with keygeometrical parameters, and table 1 summarizes theabsolute and relative energies computed using thesemethods.
At the CASSCF level, following excitation fromthe ground state minimum (MN –S0) to the ��*state S1, the system relaxes to a minimum (MN –S1) inwhich the proton is still bonded to the nitrogen atom.A transition state (TS –S1) connecting this minimumto the tautomeric structure (MT –S1) was located, whichhas a barrier height of 4.5 kJmol�1. The tautomerminimum lies about 41 kJmol�1 below the Franck–Condon state. Accompanying the proton transferreaction, geometrical changes are found in a numberof bond distances, particularly those involving atomsdirectly associated with the proton transfer reaction.While in the ground state, the proton is bonded to theamide nitrogen with a bond distance of 0.998 A, it formsa covalent bond with the oxygen atom at a distanceof 0.977 A in the excited state tautomer structureMT –S1. In the transition state structure, by slightlyreducing the bending angles, the nitrogen and oxygenatoms are closer than that in the equilibrium statesto minimize the bond distances with the proton. Thiscooperative process assists the PT reaction by reducingits barrier with donor–acceptor geometrical distortions.
In going from the ground state MN –S0 geometry tothe excited state product form MT –S1, the N–C bondshortens from 1.40 to 1.33 A and the O–C bondlengthens from 1.20 to 1.30 A, indicating alterationin single and double bond characters. Although the�-electrons are fully delocalized, there are no detectablechanges in single and double bonding characters overthe aromatic rings.
We also constructed the ground state adiabaticpotential energy profile for the proton transfer from 1
(MN –S0) to the tautomer structure MT –S0, which is alocal minimum at the CASSCF level. The barrier at thetransition state, TS –S0, separating these two structuresis 89.4 kJmol�1 from MN –S0 to MT –S0.
When dynamic correlation corrections are includedat the CASPT2 level, both the ground and excitedstate potential energy surfaces change dramatically.First, figure 3 shows that the entire S1 PES is shifteddownward by 80 kJmol�1, relative to the globalminimum of the ground state, MN –S0. Furthermore,electron correlation effects affect the Frank–Condonstate MN –S1 and the transition state TS –S1 more thanthe charge (proton) transfer state MT –S1. The S1 PEShas flattened out, resulting in a potential surface thatis barrierless for the proton transfer from theFrank–Condon region to the final product. Thus, boththe MN –S1 and TS –S1 structures are no longerstationary points at the CASPT2 level of theory, andthe single minimum structure is the proton transferproduct state, MT –S1, just 2.5 kJmol�1 below theenergy at the fixed MN –S1 configuration. The groundstate PES has also undergone major changes. The main
946 A. Cembran and J. Gao
TS–S1
TS–S0
0.998
1.860
1.4041.2011.495
1.415
1.3591.199
1.532
1.306
1.322
1.405
1.389
1.392
1.390
1.415
1.498
1.1941.482
1.341
1.486
1.754
1.0121.364
1.374
1.197
1.531
1.319
1.306
1.2321.448
1.461
1.426
1.409
1.390
1.449
1.413
1.481
1.2041.462
1.352
1.465
1.272
1.1941.331
1.454
1.481
1.275
1.3741.200
1.529
1.308
1.319
1.421
1.424
1.366
1.462
1.370
1.507
1.200
1.455
1.373
1.418
0.977
1.7281.331
1.3551.208
1.530
1.312
1.324
1.447
1.414
1.436
1.357
1.453
1.381
1.4751.303
1.421
1.363
1.4641.194
1.513
0.965
1.7941.296
1.3791.201
1.529
1.310
1.321
1.469
1.3771.314
1.464
1.356
1.4441.357
1.463
1.467
1.345
1.478
1.500
1.196
1.021
1.554
1.355
1.205
1.342
1.531
1.311
1.324
1.425
1.4151.266
1.442
1.369
1.423
1.367
1.441
1.492
1.193
1.488
1.344
1.468
MN–S1MN–S0
MT–S1 MT–S0
Figure 4. Structures for 1-(trifluoroacetylamino)naphthaquinone, 1, optimized at the CASSCF[10e,10o]/6-31G(d). Bond distancesare reported in angstroms.
Table 1. Computed absolute and relative energies for 1-(trifluoroacetylamino)naphthaquinone, 1, at theCASSCF[10e,10o]/6-31G(d) and CASPT2[12e,12o]/6-31G(d) levels of theory. State-averaged optimizations (equal weightsfor S0 and S1) have been used in the CASPT2 calculations and the weight ! for the zeroth-order CASSCF wavefunctionis reported to show the reliability of the perturbation correction. The computed oscillator strengths f for the radiative
transition between S0 and S1 are also given for selected points.
Structure State CASSCF (au) CASPT2 (au) ! F �E (kJmol�1)
MN –S0 S0 �1035.41752 �1037.94696 0.57 0.080 0.0S1 �1035.26839 �1037.82372 0.54 323.5
TS –S0 S0 �1035.38258 �1037.91292 0.57 89.4S1 �1035.27655 �1037.83614 0.56 290.9
MT –S0 S0 �1035.39036 �1037.90528 0.57 109.4
S1 �1035.26264 �1037.81916 0.56 335.5MN –S1 S0 �1035.40575 �1037.94460 0.57 0.115 6.2
S1 �1035.28109 �1037.84238 0.55 274.6
TS –S1 S0 �1035.36373 �1037.91771 0.56 0.176 76.8S1 �1035.27937 �1037.84640 0.55 264.0
MT –S1 S0 �1035.36436 �1037.89782 0.56 0.141 129.0
S1 �1035.29674 �1037.84633 0.56 264.2
Excited state intramolecular proton transfer in 1-(trifluoroacetylamino)anthraquinone 947

effect is that the minimum on the tautomer side
disappears, resulting in an energy profile that directly
leads to the ground state minimum at MN –S0 without
transition state.Our best estimate of the S0!S1 transition energy
is 323.5 kJmol�1 (3.35 eV) at the CASPT2(12,12)/
6-31G(d)//CASCF(10,10)/6-31G(d) level. This is in
good agreement with the experimental value of
310.0 kJmol�1 (3.21 eV) for TFAQ in cyclohexane,
since studies [44–46] have shown that there is essentially
no spectral shift in going from the gas phase to non-
polar organic solvents [3]. The computed oscillator
strength f is 0.080, which is consistent with a molar
extinction coefficient of 6070Lmol�1 cm�1 determined
experimentally in hexaney.The CASPT2 potential energy surfaces are
consistent with the experimental finding of fast
proton transfer following photoexcitation to the S1Franck–Condon state, which is about 60 kJmol�1
higher than the tautomeric structure. Further, there
is no barrier separating the Franck–Condon region
and the proton transfer product. For comparison, the
excited state intramolecular proton transfer reaction
of TFAQ is an ultrafast process, with an upper
(instrumentation) limit of 80 fs.The experimental fluorescence maximum is centered
at 188.4 kJmol�1 in cyclohexane [3]. Since the PES
in the TS –S1/MT –S1 region is flat, it is not possible
to attribute the decay to a specific stable structure
on the excited state surface. It is interesting to notice
that the vertical emission energy from the TS –S1
structure to the ground state is in perfect agreement
with the experimental value; however, the fluorescence
transition can originate from any location along the
proton transfer coordinate between TS –S1 andMT –S1.
The smallest energy gap between the S1 and S0 states
is 135 kJmol�1, which basically rules out the possibility
of finding a low-energy conical intersection between
these two states that could provide a mechanism
for fast decay. This finding is in agreement with the
experimental data in that the observed lifetime of TFAQ
is over 100 ps and the decay takes place by intersystemcrossing [26].
To gain insights into the charge transfer process,we monitored the variation of atomic partial chargesdetermined by fitting the quantum mechanical electro-static potentials (ESP) (see supporting information),along the reaction coordinate. Although the quantitativeresults depend on the specific method used in derivingthese charges, the qualitative trends are reasonable usingthe ESP charges. It can be seen that the S0!S1transition involves a gradual charge migration fromthe aromatic ring to which the amide group is attachedto the quinonoid moiety. The net charge transferis �0.35 e from the ESP procedure. Since the dominantfactor in the �!�* transition is charge transfer fromthe HOMO into the LUMO of the compound, thisobservation is consistent with the fact that the formerorbital is primarily localized on the aromatic ring,whereas the LUMO is largely on the quinone ring.The negative charge continues flowing to the quinonering along the reaction coordinate, so that at the TS –S1
structure the quinone moiety gains about �0.60 e.Concomitant to the electronic charge transfer, theproton is also transferred from the amide nitrogenon to the quinone oxygen. These two compensatingresult in a net charge transfer of �0.27 e to the quinonesystem in the MT –S1 structure.
The charge shift upon photoexcitation is reflected bya change in the dipole moment both in magnitude andin its directionality as shown in table 2. The S0!S1
Table 2. The CASSCF/6-31G* (12e/12o) dipole momentsvectors and their modules (Debye units) are reported forboth S0 and S1. The structures to which they refer can befound in the supporting information. The XY plane is the
mirror plane in the CS symmetry group to which thismolecule belongs, therefore the dipole moment componentalong Z is always zero. Since the orientation of the moleculedoes not change along the energy profile, it is meaningfulto report the angle of twisting � of the dipole moment with
respect the ground state of the MN –S0 structure.
Structure State X Y Total �
MN –S0 S0 2.61 �0.41 2.64 0.0
S1 0.14 1.62 1.63 94.1TS –S0 S0 2.82 �0.05 2.82 8.1
S1 2.91 0.04 2.91 9.8MT –S0 S0 1.34 0.48 1.42 28.8
S1 3.48 �0.03 3.48 8.6MN –S1 S0 2.51 �0.47 2.56 1.6
S1 �0.71 2.48 2.58 115.0
TS –S1 S0 3.87 �1.27 4.08 �9.1S1 0.01 2.81 2.81 98.9
MT –S1 S0 2.59 �0.06 2.59 7.6
S1 4.50 �0.58 4.54 1.7
yFrom reference [47], for a broadband excitation, the oscillator
strength f can be related to the molar extinction coefficient " by the
formula:
f ¼ 4:33� 10�9
Z"d ~v
in which the integral runs over the band shape and ~v is the frequency in
cm�1. From reference [3] we can estimate an absorption band width of
8000 cm�1. By describing the band as a Gaussian function
" ~�ð Þ ¼ 6070 expð�� ~�2Þ with an � value of 3� 10�7 (which makes the
function decay to zero around �4000 cm�1) and integrating, it turns
out that f should be around 0.085.
948 A. Cembran and J. Gao

excitation leads to a 90� rotation in the direction ofthe molecular dipole moment, which lies in the planeof the conjugated ring system. The magnitude of thedipole moment is slightly reduced in the Franck–Condon region. It is interesting to note that afterthe proton transfer is complete in the MT –S1 structure,the excited state (S1) dipole is again aligned in thesame direction as on the ground state minimumMN –S0. However, the magnitude of the dipole isgreatly increased, a reflection of the charge transfereffect noted above.It is interesting to examine solvation effects on
the excited state proton transfer reaction. Since thetimescale required for solvent reorganization is muchgreater than the ESIPT process (a few picoseconds vs.less than 80 fs), the Franck–Condon state will not besignificantly stabilized by the slow solvent response,which is further exacerbated by a tilt in dipole orien-tation. Remarkably, the equilibrium solvent configura-tion that stabilizes the ground state species is perfectlysuited for stabilizing the proton transfer productin the excited state both because of an enhanced dipolemoment of the chromophore molecule and the restora-tion of the dipole orientation to the initial groundstate direction. This suggests that in solution theMT –S1 structure is immediately (in less than 80 fs)stabilized by solvent configuration that is in equilibriumwith the donor state in the ground state electronicsurface. Furthermore, even over a longer timescale,the excited state MT –S1 structure will be the moststabilized configuration by solvation, rather than othergeometries, along the S1 proton transfer coordinatebecause the magnitude of the dipole moment is thelargest. Therefore, it can be inferred that in the solvatedsystem, the S1 PES would show a more pronouncedwell in the MT –S1 region than the system in gas phase.
3.3. Solvatochromic shifts
The computed solvatochromic spectral shifts for 1
in various solvents are listed in table 1. The S0!S1
(�!�*) energy difference is computed to be343.0 kJmol�1 from a configuration interaction calcu-lation that includes single excitations only using thesemiempirical Austin Model 1 (AM1) model. Thesecalculations utilized the HF/6-31G(d) optimized struc-ture and six electrons and six active orbitals are includedin the CIS calculation. Despite the rough approxima-tion, the AM1 value is in good accord with that(356.9 kJmol�1) obtained using the ZINDO method,which is also a CIS procedure. For comparison, theCASPT2 vertical excitation energy is 323.5 kJmol�1
using the structure optimized at the CASSCF level.Thus, it is suggested that it is reasonable to use thesemiempical CIS method, coupled with statisticalQM/MM Monte Carlo simulations, to access solventeffects on the excitation energies. The computedtransition energy is in good agreement also with theexperimental data of 310.0 kJmol�1 in cyclohexanefor TFAQ [3].
Experimentally, Barbara et al. [3] observed a verysmall blue shift of the absorption band for TFAQin going from non-polar to polar aprotic solvents(6.6 kJmol�1 from cyclohexane to acetonitrile). Thecomputed spectral shift in going from the gas phaseto water from QM-CIS/MM simulations is less than2 kJmol�1, in reasonable accord with experiment.The results presented here confirm that solvent effectson the absorption maximum for TFAQ is almostnegligible. We attribute the small spectral shift to thesmall change in dipole moment (less than 1 Debye).Although the change in dipole orientation in the excitedstate is about 90�, it is not sufficient to cause significantdestabilization to the excited state solvation relativeto that in the ground state.
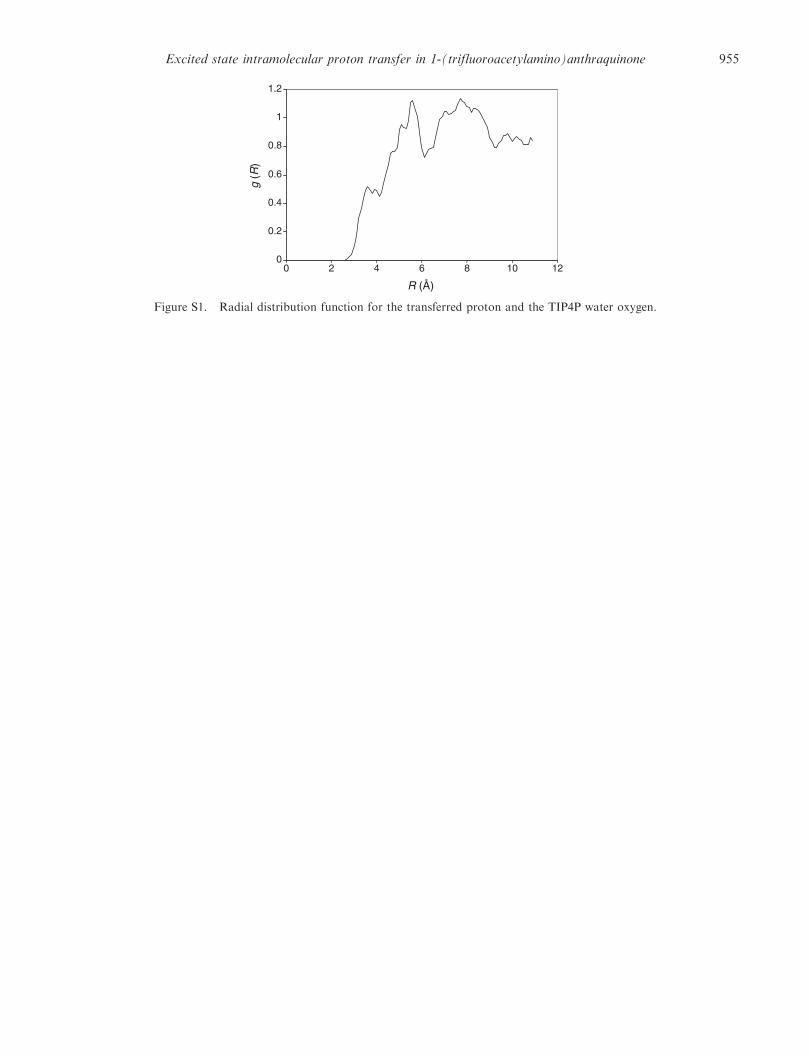
We also recorded the radial distribution functionbetween the transferred proton with the oxygen atomof water (see supporting information). There are nopeaks that can be identified as hydrogen-bond interac-tions, indicating that hydrogen atom is fully utilizedin forming an intramolecular hydrogen bond. Thisobservation supports the picture that the protontransfer is primarily an intramolecular process, inwhich the solvent does not actively participate andconfirms that the reaction path obtained in the gasphase is a reasonable model for the proton transferprocess in solution.
4. Conclusions
The excited state intramolecular proton transfer reactionof a model compound for TFAQ on the spectroscopic��* state S1 has been studied in gas phase at theCASPT2//CASSCF level. The results show that the
Table 3. Solvatochromic shifts for TFNQ, 1. The S0–S1(�!�*) transition energies are given in kJmol�1 together
with statistical errors. Experimental values are listedwhere available.
Solvent QM/MM Exp.
Gas Phase 343.0 310.0a
CH2Cl2 344.5� 0.7 312.4CH3CN 343.8� 0.5 316.6
H2O 344.6� 0.4 n.a.
a Solvent cyclohexane.
Excited state intramolecular proton transfer in 1-(trifluoroacetylamino)anthraquinone 949

reaction is a barrierless process in the excited state,
consistent with the experimental results that the proton
transfer takes place in less than 80 fs. While an extended
energy plateau in the S1 state has been found inthe gas phase along the proton transfer coordinate,
supporting the view that the proton is weakly bound
to neither the acceptor nor the donor atoms and can
undergo large amplitude oscillations, solvent effects can
stabilize the proton-transfer tautomer due to its larger
dipole moment along the excited state potentialenergy surface.The computed excitation energy is in good agreement
with the experimental data. Furthermore, the large
energy gap between the S1 and S0 state along the
entire reaction coordinate excludes the possibility of
a fast decay through low-energy conical intersections.A comparison of the gas phase computed S1/S0 energy
gaps with the experimental fluorescence maximum
suggests that the radiative decay could take place from
a structure in which the O–H bond is stretched and
the hydrogen is about halfway between the nitrogen
and oxygen atoms.Statistical Monte Carlo QM/MM (AM1-CIS) simu-
lations have been performed to investigate the effect
of various solvents on the absorption maximum. It is
found that even in a polar protic solvent like water the
spectral shift is still very small (less than 2 kJmol�1).
This is consistent with the experimental observationof little solvatochromic spectral shifts of TFAQ from
non-polar to polar solvents. In all, the present quantum
chemical analysis of the ESIPT reaction provided
a framework to construct a molecular mechanics
force field that can be used in dynamics simulations to
investigate the mutual effects of solute proton transferand solvent reorganization dynamics in the excited state.
Acknowledgements
The authors thank the National Institutes of Health
for partial support of this research.
References
[1] A. Weller, Naturwissenschaften 42, 175 (1955).[2] P. K. Sengupta and M. Kasha, Chem. Phys. Lett.
68, 382 (1979).[3] T. P. Smith, K. A. Zaklika, K. Thakur, G. C. Walker,
K. Tominaga, and P. F. Barbara, J. Phys. Chem. 95, 10465(1991).
[4] T. Elsaesser, W. Kaiser, and W. Luttke, J. Phys. Chem.90, 2901 (1986).
[5] D. Legourrierec, S. M. Ormson, and R. G. Brown,Prog. Reaction Kinetics 19, 211 (1994).
[6] S. M. Ormson and R. G. Brown, Prog. Reaction Kinetics19, 45 (1994).
[7] P. Mitchell, Nature (London) 191, 144 (1961).[8] L. G. Arnaut and S. J. Formosinho, J. Photochem.
Photobiol. A 75, 1 (1993).[9] F. V. R. Neuwahl, P. Foggi, and R. G. Brown, Chem.
Phys. Lett. 319, 157 (2000).[10] S. Mitra, R. Das, S. P. Bhattacharyya, and S. Mukherjee,
J. Phys. Chem. A 101, 293 (1997).[11] S. Moller, K. B. Andersen, J. Spanget-Larsen, and
J. Waluk, Chem. Phys. Lett. 291, 51 (1998).[12] F. R. Prieto, M. C. R. Rodriguez, M. M. Gonzalez, and
M. A. R. Fernandez, J. Phys. Chem. 98, 8666 (1994).[13] S. Maheshwari, A. Chowdhury, N. Sathyamurthy,
H. Mishra, H. B. Tripathi, M. Panda, andJ. Chandrasekhar, J. Phys. Chem. A 103, 6257 (1999).
[14] P. T. Chou, D. McMorrow, T. J. Aartsma, andM. Kasha, J. Phys. Chem. 88, 4596 (1984).
[15] N. P. Ernsting and B. Nikolaus, Appl. Phys. B-LasersOpt. 39, 155 (1986).
[16] M. L. Ferrer, A. U. Acuna, F. Amatguerri, A. Costela,J. M. Figuera, F. Florido, and R. Sastre, Appl Opt.33, 2266 (1994).
[17] G. Jones and M. A. Rahman, J. Phys. Chem.98, 13028 (1994).
[18] M. Liphardt, A. Goonesekera, B. E. Jones, S. Ducharme,J. M. Takacs, and L. Zhang, Science 263, 367 (1994).
[19] A. Douhal and R. Sastre, Chem. Phys. Lett. 219, 91(1994).
[20] K. Kuldova, A. Corval, H. P. Trommsdorff, andJ. M. Lehn, J. Phys. Chem. A 101, 6850 (1997).
[21] M. L. Martinez, W. C. Cooper, and P. T. Chou, Chem.Phys. Lett. 193, 151 (1992).
[22] H. J. Heller and H. R. Blattmann, Pure Appl. Chem.36, 141 (1973).
[23] T. Werner, J. Phys. Chem. 83, 320 (1979).[24] R. M. Tarkka, X. L. Chen, and S. A. Jenekhe, in Photonic
And Optoelectronic Polymers, edited by S. A. Jenekheand K. J. Wynne (American Chemical Society,Washington, D.C., 1997), p. 475.
[25] F. V. R. Neuwahl, L. Bussotti, R. Righini, andG. Buntinx, Phys. Chem. Chem. Phys. 3, 1277 (2001).
[26] V. J. P. Srivatsavoy and B. Venkataraman, Chem. Phys.Lett. 174, 406 (1990).
[27] S. J. Schmidtke, D. F. Underwood, and D. A. Blank,J. Am. Chem. Soc. 126, 8620 (2004).
[28] K. Andersson, P.-A Malmqvist, and B. O. Roos, J. Chem.Phys. 96, 1218 (1992).
[29] B. O. Roos, in Ab Initio Methods in Quantum Chemistry II(John Wiley & Sons, Chichester, 1987), p. 399.
[30] M. J. Frisch, G. W. Trucks, H. B. Schlegel,G. E. Scuseria, M. A. Robb, J. R. Cheeseman,V. G. Zakrzewski, J. A. Montgomery, R. E. Stratmann,J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels,K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi,V. Barone, M. Cossi, R. Cammi, B. Mennucci,C. Pomelli, C. Adamo, S. Clifford, J. Ochterski,G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma,D. K. Malick, A. D. Rabuck, K. Raghavachari,J. B. Foresman, J. Cioslowski, J. V. Ortiz,B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz,I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox,T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara,C. Gonzalez, M. Challacombe, P. M. W. Gill,
950 A. Cembran and J. Gao

B. G. Johnson, W. Chen, M. W. Wong, J. L. Andres,M. Head-Gordon, E. S. Replogle, and J. A. Pople,in Gaussian 98, A1 (Gaussian Inc., Pittsburgh, PA, 1998).
[31] K. Andersson, M. Barysz, A. Bernhardsson,M. R. A. Blomberg, Y. Carissan, D. L. Cooper,M. Cossi, T. Fleig, M. P. Fulscher, L. Gagliardi,C. deGraaf, B. A. Hess, G. Karlstrom, R. Lindh,P.-A Malmqvist, P. Neogrady, J. Olsen, B. O. Roos,B. Schimmelpfennig, M. Schutz, L. Seijo, L. Serrano-Andres, P. E. M. Siegbahn, J. Stalring, T. Thorsteinsson,V. Veryazov, M. Wierzbowska, and P.-O. Widmark,in MOLCAS, 5.2 (Department of Theoretical Chemistry,Chemistry Center, University of Lund, Lund,Sweden, 2001).
[32] P.-A. Malmqvist, Int. J. Quantum Chem. 30, 479 (1986).[33] P.-A. Malmqvist and B. O. Roos, Chem. Phys. Lett.
155, 189 (1989).[34] W. L. Jorgensen, J. M. Briggs, and M. L. Contreras,
J. Phys. Chem. 94, 1683 (1990).[35] W. L. Jorgensen and J. M. Briggs, Mol. Phys. 63, 547
(1988).[36] W. L. Jorgensen, J. Chandrasekhar, J. D. Madura,
R. W. Impey, and M. Klein, J. Chem. Phys. 79, 926(1983).
[37] M. J. S. Dewar, Z. E. G. Zoebisch, E. F. Healy, andJ. J. P. Stewart, J. Am. Chem. Soc. 107, 3902 (1985).
[38] J. J. P. Stewart, J. Comput.-aided Mol. Design 4, 1 (1990).[39] J. Gao, in Modeling the Hydrogen Bond, edited by
D. A. Smith (American Chemical Society, Washington,D.C., 1994), p. 8.
[40] J. C. Owicki and H. A. Scheraga, Chem. Phys. Lett.47, 600 (1977).
[41] J. C. Owicki, in Computer Modeling of Matter edited byP. Lykos (American Chemical Society, Washington, D.C.,1978), p. 159.
[42] J. Gao, in MCQUM (University of Minnesota,Minneapolis, 2001).
[43] J. J. P. Stewart, J. Compt.-aided Desg., 4, 1 (1990).[44] M. M. Carrabba, J. E. Kenny, W. R. Moomaw,
J. Cordes, and M. Denton, J. Phys. Chem. 89, 674(1985).
[45] K. K. Innes, H. D. J. McSwiney, J. D. Simmons, andS. G. Tilford, J. Mol. Spectrosc. 31, 76 (1969).
[46] M. N. Pisanias, L. G. Christophorou, C. J. G. Carter, andM. D. L. McCorkle, J. Chem. Phys. 58, 2110 (1972).
[47] G. C. Shatz and M. A. Ratner, in Quantum Mechanicsin Chemistry (Dover Publications Inc., Mineola,NY, 2002), p. 259.
Excited state intramolecular proton transfer in 1-(trifluoroacetylamino)anthraquinone 951
Supporting Information:
Excited State Intramolecular Proton Transfer in
1-(trifluoroacetylamino)anthraquinone: a CASPT2//CASSCF
Computational Study
ALESSANDRO CEMBRAN and JIALI GAO
Department of Chemistry, University of Minnesota, Minneapolis, MN 55455, USA
The geometries for the optimized structures are reportedtogether with the ESP charges for S0 and S1 for each structure.
Geometries (Cartesian coordinates in angstroms):
MN –S0
1 H1 6.203831 5.590033 0.0000002 H2 1.946793 7.594354 0.0000003 H3 �1.853402 4.972858 0.000000
4 H4 �1.694711 �1.936280 0.0000005 H5 5.622437 �5.558546 0.0000006 H6 9.446509 �2.791984 0.000000
7 C1 0.133661 1.444091 0.0000008 C2 2.547508 0.293964 0.0000009 C3 4.723073 1.848855 0.00000010 C4 4.518034 4.467536 0.000000
11 C5 2.130499 5.572617 0.00000012 C6 �0.037911 4.093917 0.00000013 C7 2.891388 �2.509929 0.000000
14 C8 7.330234 0.743600 0.00000015 C9 �6.302200 �1.620836 0.00000016 C10 5.505952 �3.533064 0.000000
17 C11 7.559170 �2.047532 0.00000018 C12 �4.500113 0.645487 0.00000019 N1 �2.037780 �0.081413 0.00000020 O1 1.163463 �3.981928 0.000000
21 O2 9.172993 2.045015 0.00000022 O3 �5.344617 2.747284 0.00000023 F1 �5.912238 �3.046311 2.014261
24 F1 �5.912238 �3.046311 �2.01426125 F2 �8.657550 �0.883663 0.000000
TS –S0
1 H1 6.469052 5.087260 0.000000
2 H2 2.375711 7.419652 0.0000003 H3 �1.637933 5.178167 0.0000004 H4 �1.063921 �2.746972 0.000000
5 H5 4.697889 �5.952609 0.0000006 H6 8.831859 �3.675031 0.0000007 C1 0.000000 1.424560 0.0000008 C2 2.359761 0.126534 0.000000
9 C3 4.715087 1.494004 0.00000010 C4 4.712980 4.076619 0.00000011 C5 2.365334 5.388779 0.000000
12 C6 0.096028 4.147327 0.00000013 C7 2.429887 �2.546253 0.000000
14 C8 7.174229 0.112854 0.00000015 C9 �6.421657 �1.227111 0.000000
16 C10 4.833770 �3.929239 0.00000017 C11 7.055137 �2.696858 0.00000018 C12 �4.488029 0.924804 0.00000019 N1 �2.099291 0.001604 0.000000
20 O1 0.469860 �3.918307 0.00000021 O2 9.168265 1.166569 0.00000022 O3 �5.248827 3.071759 0.000000
23 F1 �6.131129 �2.683417 2.01427224 F1 �6.131129 �2.683417 �2.01427225 F2 �8.747426 �0.372941 0.000000
MT –S0
1 H1 6.679868 4.962066 0.0000002 H2 2.619699 7.375935 0.0000003 H3 �1.428516 5.235363 0.000000
4 H4 �0.891572 �3.117352 0.0000005 H5 4.877336 �5.980888 0.0000006 H6 9.021301 �3.751672 0.0000007 C1 0.114922 1.406975 0.000000
8 C2 2.512793 0.009437 0.0000009 C3 4.901055 1.402966 0.00000010 C4 4.915025 3.967398 0.000000
11 C5 2.559105 5.346189 0.00000012 C6 0.282788 4.169386 0.00000013 C7 2.600375 �2.590952 0.000000
14 C8 7.375482 0.033130 0.00000015 C9 �6.334051 �1.018029 0.00000016 C10 5.012888 �3.956992 0.00000017 C11 7.253785 �2.757531 0.000000
18 C12 �4.365153 1.096987 0.00000019 N1 �1.956098 0.101175 0.00000020 O1 0.636898 �4.112517 0.000000
21 O2 9.352829 1.126785 0.00000022 O3 �5.063453 3.256352 0.00000023 F1 �6.060714 �2.466052 2.016086
24 F1 �6.060714 �2.466052 �2.01608625 F2 �8.637875 �0.111125 0.000000
MN –S1
1 H1 6.215668 5.575026 0.000000
2 H2 1.986762 7.690806 0.000000
(continued)
952 A. Cembran and J. Gao

Geometries (Cartesian coordinates in angstroms):
3 H3 �1.884014 5.041029 0.000000
4 H4 �1.555507 �1.893154 0.0000005 H5 5.540042 �5.536904 0.0000006 H6 9.370571 �2.822610 0.000000
7 C1 0.107148 1.487178 0.0000008 C2 2.556866 0.211961 0.0000009 C3 4.719046 1.778595 0.00000010 C4 4.496577 4.507769 0.000000
11 C5 2.140974 5.670158 0.00000012 C6 �0.062986 4.176639 0.00000013 C7 2.828607 �2.511504 0.000000
14 C8 7.309307 0.716504 0.00000015 C9 �6.236522 �1.661545 0.00000016 C10 5.409817 �3.511896 0.000000
17 C11 7.498875 �2.040282 0.00000018 C12 �4.487177 0.641822 0.00000019 N1 �1.979029 �0.027601 0.00000020 O1 1.020246 �3.978010 0.000000
21 O2 9.128078 2.085077 0.00000022 O3 �5.347667 2.733479 0.00000023 F1 �5.811925 �3.066008 2.015285
24 F1 �5.811925 �3.066008 �2.01528525 F2 �8.602453 �0.961209 0.000000
TS –S1
1 H1 6.333672 5.297023 0.000000
2 H2 2.222280 7.602362 0.0000003 H3 �1.784599 5.176727 0.0000004 H4 �1.279488 �2.117597 0.000000
5 H5 4.869754 �5.837779 0.0000006 H6 8.910786 �3.400681 0.0000007 C1 �0.009212 1.515972 0.000000
8 C2 2.395313 0.184968 0.0000009 C3 4.582837 1.568155 0.00000010 C4 4.555908 4.331732 0.000000
11 C5 2.294528 5.575508 0.00000012 C6 �0.018187 4.201033 0.00000013 C7 2.477854 �2.611752 0.000000
14 C8 7.116799 0.268010 0.00000015 C9 �6.320999 �1.504424 0.00000016 C10 4.874658 �3.808731 0.000000
17 C11 7.104480 �2.481140 0.00000018 C12 �4.515003 0.749964 0.00000019 N1 �2.025979 0.012363 0.000000
20 O1 0.402676 �3.835902 0.00000021 O2 8.999710 1.532689 0.00000022 O3 �5.326104 2.867684 0.00000023 F1 �5.937007 �2.918096 2.016120
24 F1 �5.937007 �2.918096 �2.01612025 F2 �8.674281 �0.748042 0.000000
MT –S1
1 H1 6.721370 5.003915 0.0000002 H2 2.698763 7.462094 0.0000003 H3 �1.390638 5.209048 0.000000
4 H4 �0.882624 �3.007279 0.0000005 H5 4.874778 �6.033710 0.0000006 H6 9.022620 �3.725843 0.000000
7 C1 0.154532 1.470068 0.0000008 C2 2.511512 0.082485 0.0000009 C3 4.778051 1.377707 0.000000
10 C4 4.902661 4.120671 0.00000011 C5 2.702425 5.432901 0.00000012 C6 0.317478 4.137198 0.00000013 C7 2.630342 �2.701876 0.000000
14 C8 7.282810 �0.000668 0.00000015 C9 �6.299785 �1.038208 0.00000016 C10 4.976582 �4.008199 0.000000
17 C11 7.238269 �2.766488 0.00000018 C12 �4.310413 1.058798 0.00000019 N1 �1.943399 0.081212 0.000000
20 O1 0.605229 �4.101527 0.00000021 O2 9.200285 1.190134 0.00000022 O3 �5.011375 3.231801 0.000000
23 F1 �6.050148 �2.499167 2.01488224 F1 �6.050148 �2.499167 �2.01488225 F2 �8.600960 �0.116908 0.000000
Excited state intramolecular proton transfer in 1-(trifluoroacetylamino)anthraquinone 953

MN –S0 TS –S0 MT –S0 MN –S1 TS –S1 MT –S1
ESP charges:S01 H 0.167511 0.158320 0.156043 0.178809 0.187828 0.1759582 H 0.158224 0.142611 0.145159 0.164945 0.154536 0.1494413 H 0.230088 0.239035 0.245932 0.231392 0.235399 0.241965
4 H 0.371946 0.457685 0.445797 0.372356 0.452239 0.4522815 H 0.185219 0.200038 0.196429 0.179196 0.183931 0.2044826 H 0.169832 0.179706 0.181640 0.168287 0.173586 0.1870947 C 0.567965 0.962558 1.021366 0.537045 0.718590 0.946600
8 C �0.461321 �0.657902 �0.626332 �0.461959 �0.585516 �0.6186979 C �0.002650 0.018135 0.003465 0.021670 0.080588 0.06110010 C �0.170269 �0.167234 �0.151753 �0.211536 �0.261468 �0.188315
11 C �0.108656 �0.009027 �0.017167 �0.096513 �0.038686 �0.03388412 C �0.306797 �0.465232 �0.499360 �0.312588 �0.397392 �0.45921913 C 0.837721 0.721835 0.609620 0.833557 0.835639 0.649128
14 C 0.656594 0.667137 0.670355 0.651804 0.643964 0.64293115 C 0.599355 0.581605 0.619205 0.610489 0.607978 0.63633616 C �0.315490 �0.260205 �0.220337 �0.294708 �0.270486 �0.258965
17 C �0.206607 �0.247482 �0.279523 �0.205656 �0.194096 �0.26039218 C 0.643909 0.734982 0.729535 0.621312 0.654985 0.70048119 N �0.766447 �0.949812 �0.910032 �0.733162 �0.865846 �0.88455420 O �0.583653 �0.615687 �0.625730 �0.600788 �0.644360 �0.618614
21 O �0.536797 �0.542235 �0.547413 �0.538824 �0.536149 �0.54434122 O �0.501652 �0.503393 �0.480576 �0.488203 �0.499539 �0.49482723 F �0.207796 �0.210439 �0.215189 �0.205883 �0.206307 �0.223639
24 F �0.207796 �0.210439 �0.215189 �0.205883 �0.206307 �0.22363925 F �0.212434 �0.224559 �0.235945 �0.215160 �0.223110 �0.238708
S11 H 0.145515 0.169474 0.178637 0.154531 0.172584 0.196231
2 H 0.156967 0.152643 0.148335 0.167170 0.162501 0.1516253 H 0.223582 0.226766 0.232390 0.221825 0.213311 0.2308284 H 0.362883 0.454911 0.471408 0.357886 0.420022 0.471639
5 H 0.194813 0.219493 0.221377 0.192885 0.208052 0.2224956 H 0.183073 0.187905 0.183499 0.185750 0.196511 0.1875237 C 0.587663 0.584153 0.690769 0.550908 0.543646 0.6688508 C �0.370958 �0.434303 �0.552553 �0.353873 �0.367836 �0.539274
9 C 0.090937 0.159335 0.250903 0.108164 0.200473 0.30131410 C �0.074512 �0.240129 �0.324413 �0.091004 �0.182269 �0.36223011 C �0.174332 �0.086518 �0.024804 �0.180737 �0.146196 �0.033183
12 C �0.288163 �0.325636 �0.346992 �0.261594 �0.245458 �0.34338013 C 0.644850 0.687138 0.748611 0.595541 0.525201 0.72844214 C 0.576931 0.629615 0.619897 0.580477 0.589934 0.601198
15 C 0.617633 0.620413 0.659685 0.629674 0.642547 0.66697416 C �0.301436 �0.341254 �0.361951 �0.282855 �0.302734 �0.35675217 C �0.264440 �0.273058 �0.255342 �0.295657 �0.336795 �0.24764218 C 0.628924 0.650946 0.645912 0.603217 0.598388 0.637807
19 N �0.654004 �0.725338 �0.821388 �0.575524 �0.553833 �0.81711420 O �0.621932 �0.627356 �0.653088 �0.656258 �0.686588 �0.64213621 O �0.549038 �0.519432 �0.511100 �0.554411 �0.536701 �0.503994
22 O �0.475580 �0.489566 �0.498025 �0.456757 �0.452979 �0.50716223 F �0.211800 �0.226241 �0.231718 �0.210419 �0.217117 �0.23577324 F �0.211800 �0.226241 �0.231718 �0.210419 �0.217117 �0.235773
25 F �0.215775 �0.227718 �0.238334 �0.218520 �0.227544 �0.240514
954 A. Cembran and J. Gao
0
0.2
0.4
0.6
0.8
1
1.2
0 2 4 6 8 10 12
R (Å)
g (R
)
Figure S1. Radial distribution function for the transferred proton and the TIP4P water oxygen.
Excited state intramolecular proton transfer in 1-(trifluoroacetylamino)anthraquinone 955