evaluation of the carbonization of thermo-stabilized ... · pdf filedepartment of physics,...
TRANSCRIPT

Department of Physics, Chemistry and Biology
Master’s Thesis
Evaluation of the Carbonization of Thermo-Stabilized
Lignin Fibers into Carbon Fibers
Henrik Kleinhans
5th July 2015
LITH-IFM-A-EX--15/3112--SE
Linköping University Department of Physics, Chemistry and Biology
581 83 Linköping

Department of Physics, Chemistry and Biology
Evaluation of the Carbonization of Thermo-Stabilized
Lignin Fibers into Carbon Fibers
Henrik Kleinhans
Thesis work performed at INNVENTIA AB
5th July 2015
Supervisors
Professor Lennart Salmén, INNVENTIA AB
Daniel Aili, Linköping University
Examiner
Dr. Thomas Ederth, Linköping University
Linköping University Department of Physics, Chemistry and Biology
581 83 Linköping

Abstract
Thermo-stabilized lignin fibers from pH-fractionated softwood kraft lignin were carbonized to
various temperatures during thermomechanical analysis (TMA) under static and increasing
load and different rates of heating. The aim was to optimize the carbonization process to
obtain suitable carbon fiber material with good mechanical strength potential (high tensile
strength and high E-modulus). The carbon fibers were therefore mainly evaluated of
mechanical strength in Dia-Stron uniaxial tensile testing.
In addition, chemical composition, in terms of functional groups, and elemental (atomic)
composition was studied in Fourier transform infrared spectroscopy (FTIR) and in energy-
dispersive X-ray spectroscopy (EDS), respectively. The structure of carbon fibers was imaged
in scanning electron microscope (SEM) and light microscopy. Thermogravimetrical analysis
was performed on thermo-stabilized lignin fibers to evaluate the loss of mass and to calculate
the stress-changes and diameter-changes that occur during carbonization.
The TMA-analysis of the deformation showed, for thermo-stabilized lignin fibers, a
characteristic behavior of contraction during carbonization. Carbonization temperatures above
1000°C seemed most efficient in terms of E-modulus and tensile strength whereas rate of
heating did not matter considerably. The E-modulus for the fibers was improved significantly
by slowly increasing the load during the carbonization. The tensile strength remained however
unchanged.
The FTIR-analysis indicated that many functional groups, mainly oxygen containing,
dissociate from the lignin polymers during carbonization. The EDS supported this by showing
that the oxygen content decreased. Accordingly, the relative carbon content increased
passively to around 90% at 1000°C. Aromatic structures in the carbon fibers are thought to
contribute to the mechanical strength and are likely formed during the carbonization.
However, the FTIR result showed no evident signs that aromatic structures had been formed,
possible due to some difficulties with the KBr-method.
In the SEM and light microscopy imaging one could observe that porous formations on the
surface of the fibers increased as the temperature increased in the carbonization. These
formations may have affected the mechanical strength of the carbon fibers, mainly tensile
strength.
The carbonization process was optimized in the sense that any heating rate can be used. No
restriction in production speed exists. The carbonization should be run to at least 1000°C to
achieve maximum mechanical strength, both in E-modulus and tensile strength. To improve
the E-modulus further, a slowly increasing load can be applied to the lignin fibers during
carbonization. The earlier the force is applied, to counteract the lignin fiber contraction that
occurs (namely around 300°C), the better. However, in terms of mechanical performance, the
lignin carbon fibers are still far from practical use in the industry.

Abbreviations
DLaTGS Deuterated L-alanine doped Triglycene Sulphate
DSC Differential Scanning Calorimetry
EDS Energy Dispersive X-ray Spectrometer
E-modulus Elastic modulus, Young’s modulus
FTIR Fourier Transform Infrared Spectroscopy
G Guaiacyl
GC Gas chromatograph
H P-hydroxyphenyl (P=para)
IR Infrared
Mn Number Average Molecular Weight
MPP Mesophase pitch
Mw Average Molecular Weight
PAN Polyacrylonitrile
PDI Polydispersity Index
S Syringyl
SEM Scanning Electron Microscope
TE Thermo Electric
Tg Glass Transition Temperature
TGA Thermogravimetric Analysis
TMA Thermomechanical Analysis
Ts Softening Point
v Version
WD Working Distance

i
Contents
1 Introduction ......................................................................................................................... 1
1.1 Aim ............................................................................................................................. 1
1.2 Background ................................................................................................................ 1
1.2.1 Polymer properties ............................................................................................... 3
1.2.2 Carbon fibers ........................................................................................................ 4
1.2.2.1 PAN-based carbon fibers .............................................................................. 4
1.2.2.2 Pitch-based carbon fibers .............................................................................. 5
1.2.2.3 Structure of carbon fibers .............................................................................. 6
1.2.3 Lignin polymer ..................................................................................................... 7
1.2.3.1 Refining of lignin ........................................................................................ 10
1.2.3.2 Lignin-based carbon fibers.......................................................................... 10
1.2.3.3 Production of lignin carbon fiber ................................................................ 11
2 Experimental ..................................................................................................................... 15
2.1 Stabilization .............................................................................................................. 15
2.2 Carbonization and Thermomechanical Analysis (TMA) ......................................... 16
2.2.1 Principles ............................................................................................................ 16
2.2.2 Method ............................................................................................................... 17
2.3 Uniaxial Tensile Testing (with Dia-Stron) ............................................................... 20
2.3.1 Principles ............................................................................................................ 20
2.3.2 Method ............................................................................................................... 20
2.4 Thermogravimetric Analysis (TGA) ........................................................................ 22
2.4.1 Principles ............................................................................................................ 22
2.4.2 Method ............................................................................................................... 22
2.5 Fourier Transform Infrared Spectroscopy (FTIR) ................................................... 23
2.5.1 Principles ............................................................................................................ 24
2.5.2 Method ............................................................................................................... 25
2.6 Scanning Electron Microscope (SEM) ..................................................................... 27
2.6.1 Principles ............................................................................................................ 27
2.6.2 Method ............................................................................................................... 28
2.7 Light Microscopy ..................................................................................................... 29
3 Results ............................................................................................................................... 30
3.1 TMA ......................................................................................................................... 30

ii
3.1.1 Static measurement ............................................................................................ 30
3.1.2 Measurements with increasing force .................................................................. 35
3.2 Dia-stron ................................................................................................................... 39
3.2.1 Evaluation of temperature and heating rate ........................................................ 39
3.2.2 Evaluation of applied force during carbonization .............................................. 45
3.3 TGA .......................................................................................................................... 47
3.4 FTIR ......................................................................................................................... 52
3.5 SEM .......................................................................................................................... 60
3.5.1 Imaging ............................................................................................................... 60
3.5.2 Energy-dispersive X-ray spectroscopy ............................................................... 64
3.6 Light Microscopy ..................................................................................................... 69
4 Discussion ......................................................................................................................... 74
4.1 Analysis of the main results ..................................................................................... 74
4.1.1 TMA ................................................................................................................... 74
4.1.2 Dia-Stron ............................................................................................................ 74
4.1.3 TGA .................................................................................................................... 76
4.1.4 FTIR ................................................................................................................... 76
4.1.5 SEM .................................................................................................................... 78
4.1.6 Microscopy ......................................................................................................... 78
4.2 Impact in a broad sense ............................................................................................ 78
4.3 Ethical implications in a broad sense ....................................................................... 79
4.4 Future perspectives ................................................................................................... 79
5 Conclusion ........................................................................................................................ 81
6 Acknowledgements ........................................................................................................... 82
7 References ......................................................................................................................... 83
8 Appendix ........................................................................................................................... 87
8.1 Process ...................................................................................................................... 87
8.1.1 Timetable ............................................................................................................ 88
8.1.2 Planning .............................................................................................................. 90
8.1.3 Process results .................................................................................................... 91
8.1.4 Comprehensive analysis of the process .............................................................. 95
8.2 Data .......................................................................................................................... 95
8.2.1 TMA ................................................................................................................... 95

iii
8.2.2 TGA .................................................................................................................... 96
8.2.2.1 Calibration ................................................................................................... 96
8.2.2.2 Calculations of diameter and stress change ................................................ 98
8.2.3 Dia-stron ............................................................................................................. 98
8.2.3.1 Compliance ................................................................................................. 98
8.2.3.2 Data of carbonized fiber samples under load ............................................ 105
8.2.4 Light microscopy .............................................................................................. 106
8.2.4.1 Carbonization series .................................................................................. 106
8.2.4.2 Comparison with SEM series .................................................................... 110

1
1 Introduction Aim and background of the project are discussed in this section.
All information regarding the process is found in appendix 8.1.
1.1 Aim The aim of this project was to optimize the carbonization process of lignin to obtain suitable
carbon fiber material with good mechanical properties (high tensile strength and
high E-modulus).
In order to optimize the process the following parameters were being looked at:
Temperature
Rate of temperature elevation
Applied force
Several tests were conducted to provide information about mechanical properties as well as
structural formation (visual morphologies and chemical structures), for each setting of
parameters, to determine the optimal carbonization.
The elemental composition of carbon fibers was identified by energy dispersive X-ray
spectrometer (EDS). To be considered ‘carbon fibers’, the content of carbon must be
at least 90 % of the weight.
The structure and morphology of the carbon fibers was studied in light microscopy
and scanning electron microscopy (SEM). Defects and irregularities affect the tensile
strength. Structures like skin-core may also affect strength properties.
The change in molecular structure was studied, mainly by looking at disappearing
functional groups and molecules in the carbonization, by Fourier transform infrared
spectroscopy (FTIR). The loss of functional groups, during carbonization, should
result in a graphite skeleton, with little to none non-carbon compounds.
Mechanical properties, e.g. stress, strain, elastic modulus and tensile strength, of the
carbon fiber were tested using uniaxial tensile testing.
1.2 Background Carbon fibers are defined by the relatively high content of carbon which must be at least 90%
wt. (Liu, Kumar 2012)
Carbon fibers have remarkable properties and are often used in composite materials. The
mechanical properties are characterized by high tensile strength (2-7 GPa), good compressive
strength (up to 3 GPa) and high tensile modulus (200-900 GPa). The density (1.75-2.18
g/cm3) is 4 times lower than steel, whereas the strength of the carbon fiber is much higher.
(Liu, Kumar 2012)

2
The high modulus of carbon fibers can be explained by high crystallinity and high alignment
of crystals in the fiber direction. The tensile strength of carbon fibers is however affected by
defects and crystalline morphologies in the carbon fibers. (Huang 2009)
Additional attributes are good temperature resistance, low thermal expansion, excellent
electrical and thermal conductivity and good chemical resistance.(Liu, Kumar 2012) The
thermal as well as electric conductivity is due to the high content of delocalized π electrons
and the parallel alignment of graphene layers along the fiber axis. The thermal conductivity
coefficient is close to that of metals. (Huang 2009)
The market for carbon fibers has been growing steadily over the past 20 years, an average
increase of 12% per year. It is estimated to be a 1.6 billion US dollar market by 2017. Some
notable manufacturers of carbon fiber are AKSA (AKSACA), Cytec (Thornel®), Formosa
Plastics (Tairyfil), Hexcel (HexTowR®,HexForceR®), Mitsubishi Rayon (PYROFIL™),
SGL (SIGRAFILR®©), Teijin (TENAX®), Toho Tenax (TenaxR®), Toray
(TORAYCAR®), and Zoltek (PANEXR® ). (Liu, Kumar 2012)
Carbon fibers are today exclusively used in certain types of industry such as aerospace (e.g.
aircraft and space systems), military, automobile and sporting goods, to name a few. (Huang
2009) The benefits of using carbon fibers other than using it as a strong composite is to
decrease weight in for example vehicles. Lower weight also means lower fuel consumption.
Composites reinforced with carbon fibers could offer as much as 60 % part weight reduction.
The cost would, however, be ten times higher, due to the expensive raw material of carbon
fibers. (Baker, Rials 2013)
In today’s commercial carbon fiber production, polyacrylonitrile (PAN) is the most
domination precursor used. More than 90 % of commercial carbon fibers are manufactured
from PAN precursors. The rest are made from pitch, foremost mesophase pitch (MPP). (Liu,
Kumar 2012)
Both PAN and pitch precursors are derived from raw fossil materials, such as petroleum or
oil. Therefore, the manufacturing cost of carbon fiber is highly dependent on the oil price. In
other words, carbon fibers are not cheap. As for the total cost of PAN-based carbon fibers,
51% stands for the cost of the precursor, 18% for utilities, 12% depreciation, 10% labor and
the rest is represented by other fixed costs. (Baker, Rials 2013)
Lignin is on the other hand an abundant, cheaper and renewable material found throughout the
nature. Around 30 % of the content in wood is lignin and it can be extracted in bio refineries
during the pulping process, kraft pulp is often used. (Sjöström 1993) Lignin is a complex
polymer that can be used as a cheap precursor alternative for carbon fiber manufacturing.
The interest of manufacture carbon fibers out of lignin has been growing over the recent
years, as depicted by Figure 1. It illustrates the rising frequency of published articles since the
year 1964. (Baker, Rials 2013) But the idea of producing carbon fibers from lignin is nothing
new. In the early 1970’s carbon fiber production from lignin had a brief period of small-scale
commercialization under the name Kayacarbon by the Nippon Kayaku. Unfortunately, the

3
Kayacarbon could not compete with other precursor materials and had to leave the market due
to the high competition. (Baker, Rials 2013)
Figure 1: Frequency of journal articles (dark) and patents (light) about lignin-based carbon fibers by year to
20130131. (Baker, Rials 2013)
Even today, the tensile strength and modulus do not meet the requirement for proper use in
the industry. PAN and MPP are still superior regarding strength and mechanical properties.
(Baker, Rials 2013)
The standards, set in the automotive industry, require carbon fibers to have tensile strength of
1.72 GPa, and E-modulus of 172 GPa. (Baker, Rials 2013) Current modified lignin carbon
fibers, with average strength of 1.07 GPa, E-modulus of 82.7 GPa and extensibilities of
2.03%, do not meet these criteria. (Baker, Rials 2013)
Because of this, much of the lignin carbon fiber research has been focused on improving the
mechanical properties. (Baker, Rials 2013)
1.2.1 Polymer properties
Polymers are large macromolecules composed of repeated subunits called monomers. They
can be divided into two groups: natural and synthetic polymers. PAN is an example of
synthetic polymer whereas lignin is a natural polymer. (McCrum, Bucknall & Buckley 1997)
The polymer size and length is often expressed as molecular weight. However,
polymerization yields a product with a wide range of molecular weights. Therefore it is
preferred to express the weight statistically in order to describe the distribution of polymer
weights in the product. Average molecular weight and number average molecular weight is
denoted Mw and Mn, respectively. The ratio, Mw/Mn, between those is termed polydispersity
index (PDI). (McCrum, Bucknall & Buckley 1997)
Often considered as amorphous material, due to the lack of crystallinity order, polymers
exhibit a glass transition temperature. The glass transition temperature (Tg) is commonly
defined as the inflection point (onset) of the slope in a differential scanning calorimetry
(DSC) plot or in a dilatometry plot where the polymer material transform from a glassy (solid)
to a rubbery state when the temperature is increased. Unlike idealistic solid materials a
distinct melting temperature cannot be determined for polymers. Instead of a direct phase
transition from solid to liquid at a critical temperature, the viscosity of amorphous polymers
continues to decrease upon heating as the molecules gain movement. This defines the rubbery
state. (McCrum, Bucknall & Buckley 1997)

4
The Tg is impacted by the length of the polymer chain. Along with viscosity the Tg tend to
increase with increasing length. (McCrum, Bucknall & Buckley 1997)
Polymers possess viscoelastic properties and exhibit hysteresis during tensile testing.
1.2.2 Carbon fibers
Carbon fibers derived of PAN and MPP precursors dominates the current market. Although
they differ in composition the production of carbon fibers is fairly similar.
Extrusion and spinning of polymers into fibers are made after the preparations of the
precursors. The fibers are then stabilized by thermo-oxidation, in oxidative environment
(oxygen rich gas/atmosphere), transforming it from a thermoplastic into a thermoset material.
The final step is carbonization where the stabilized fiber is heated in high temperatures.
(Huang 2009)
1.2.2.1 PAN-based carbon fibers
PAN, containing 68% carbon, are made from acrylonitrile (AN) through free radical
polymerization (Bajaj, Sreekumar & Sen 2001), with the use of initiators such as peroxides
and azo-compounds (Huang 2009).
In the stabilization process, the PAN polymers undergo cyclization, dehydrogenation and
finally oxidation. The cyclization is preferred to happen before the oxidative treatment since
the oxidation reaction prefers to take place with cyclized PAN chains. (Liu, Kumar 2012)
Although the mechanism involved in the cyclization is not fully understood, different models
of the final cyclized structure have been proposed (Figure 2). (Huang 2009) Furthermore, the
PAN fibers must be stabilized under tension/stretching of the fiber. Otherwise the fiber length
will shrink in the process. (Liu, Kumar 2012)
Figure 2: Linear PAN chains are proposed to cyclize into the right hand structure.
The stretching is also often used during the carbonization as well. However, the importance is
still under debate. (Huang 2009) Since the stabilized fiber is considered a thermoset, the
dimensions should not be affected by temperature changes at this point.
The strength of a carbon fiber is dependent on the carbonization temperature and the
maximum strength is observed at around 1,500°C. (Huang 2009)
In the early carbonization, crosslinking reactions take place in the oxidized PAN and the
cyclized structure starts to link up sideways during dehydration and denitrogenation. A planar
structure is formed along the fiber axis (Figure 3). (Huang 2009)

5
Figure 3: A proposed model of crosslinking and formation of PAN-polymers into graphite structure through
dehydrogenation and denitrogenation.
The oxidative stabilization is controlled by oxygen diffusion through the fiber. Due to the
insufficient diffusion through the entire fiber a skin-core structure is often formed on the
surface after stabilization. The oxygen content in this structure is higher and the form is more
compact and ordered than the core. The skin-core in stabilized fibers is also present in the
final carbon fiber. (Liu, Kumar 2012)
The excellent mechanical properties of the PAN-based carbon fibers can be explained by
intermolecular interactions between the polar nitrile groups (placement shown in Figure 2) in
PAN chains. (Chatterjee et al. 2014) The interaction also contributes to a high melting point
and the polymer tends to degrade before melting, Therefore melt-spinning is not suitable for
PAN and wet-spinning is thus preferred. (Huang 2009)
Today, conventional PAN precursors provide carbon fiber with tensile strengths of 1.03 GPa.
(Baker, Rials 2013)
1.2.2.2 Pitch-based carbon fibers
Pitch precursors contains 80% carbon, are isotropic and can be produced in two ways: either
by destructive distillation of petroleum and coal or pyrolysis of synthetic polymers. The
former is attractive in commercial perspective where petroleum pitch is preferred over coal.
Coal derived pitch filaments break more easily during extrusion and thermal treatments due to
the higher content of particles. (Huang 2009)
Another common way to produce carbon fibers is by the use of anisotropic pitch, such as
mesophase pitch. Mesophase means that the material contains a liquid crystalline phase. In
addition, mesophase pitch also contains appreciable amount of anisotropic and isotropic
phase. (Liu, Kumar 2012) The mesophase pitch is obtained from heating coal or petroleum at
specific temperatures, or by transforming isotropic pitch. (Huang 2009)
The isotropic pitch contains thousands of aromatic hydrocarbons that form spheres and larger
aromatic rings during heat treatment. Further heat treatment fuses the spheres together,
resulting in a formation with large anisotropic domains combined with isotropic parts, i.e.
mesophase. (Matsumoto 1985)

6
The aromatic ring network in the mesophase pitch structure is stacked through π-electron
interaction. Due to this structure, a highly oriented basal plane structure can be achieved
through carbonization. (Matsumoto 1985)
The graphitic structure in mesophase pitch-based (MPP) carbon fibers gives a higher modulus
than PAN-based carbon fibers. Due to the amount of anisotropic phase, and in contrast to
isotropic pitch, mesophase pitch exhibit much higher tensile properties. (Liu, Kumar 2012)
Although mesophase pitch-based carbon fibers have a higher modulus and elastic modulus, its
compressive and tensile strength is lower than PAN. (Liu, Kumar 2012, Huang 2009)
The processing of isotropic and mesophase pitch precursor fibers does not differ much from
the conventional processing of PAN precursor. Melt-spinning, thermo-stabilization and
carbonization are the same. (Huang 2009) The reactions in pitch during the stabilization
process include oxidation, dehydrogenation, cyclization, elimination, condensation and cross-
linking. (Matsumoto, Mochida 1992)
The three-dimensional crosslinking in pitch based carbon fibers are accounted by the
introduction of oxygen containing groups and the formation of hydrogen bonding between
molecules. (Huang 2009)
Stabilization of MPP, as well as PAN, has the challenge of achieving homogenous
stabilization throughout the entire fiber. Skin-core structures have been observed in MPP, just
like in PAN. The structure prevents oxygen from penetrating deeper into the structure. This is
thought to be caused by the competition of oxidative reactions on the surface of the fiber and
the diffusion of oxygen. (Lü et al. 1998, Brodin et al. 2012)
In the pitch based carbon fibers sheet-like morphology can be observed, where the sheets are
tens of nanometers thick while the length is considerably longer, 10-100 microns. (Liu,
Kumar 2012)
1.2.2.3 Structure of carbon fibers
The layer planes in carbon fibers may be either in turbostratic, graphitic, or a hybrid form
depending on precursor and manufacturing process. (Huang 2009)
In graphitic structure, the planes of layers are stacked parallel in a regular and crystalline
fashion (Figure 4). The covalent double bonds between carbon atoms are sp2 hybridized
while the sheets are held together by relatively weak Van der Waals forces and π-stacking.
(Steed, Turner & Wallace 2007) The distance between two graphene layers is about 0.335 nm.
(Huang 2009)
In contrast, the graphene sheets are stacked irregular or haphazardly folded, tilted or split in
turbostratic structure (Figure 4). The bonds are instead sp3 hybridized which increase the
spacing to 0.344 nm. (Huang 2009)

7
Due to the crystallites in MPP carbon fibers tend to be more graphitic and the spacing is in the
range of 0.337–0.340 nm. PAN carbon fibers contain mainly turbostratic crystals. (Huang
2009)
Figure 4: Turbostratic and graphitic ordering.
1.2.3 Lignin polymer
Lignin is present in all vascular plants with few exceptions. In fact, lignin is the second, to
cellulose, most abundant bio-macromolecule in the world. It is mainly located in the
secondary cell wall of plant cells where it provides stability and support to the plant.(Becker
2008) It also plays a vital role in transportation of water and protects the cell wall from
harmful enzymatic degradation. (Gellerstedt, Ek & Henriksson 2009)
The lignin macromolecule is biosynthesized in the plant from three different monolignol
monomers: p-coumaryl alcohol, coniferyl alcohol, and sinapyl alcohol. They are incorporated
and linked together into the lignin polymer as p-hydroxyphenyl (H), guaiacyl (G) and syringyl
(S), respectively (Figure 5). (Freudenberg 1959, Baker, Rials 2013)
Figure 5: The 3 building components of lignin.
R1=OMe, R2=H: Coniferyl alcohol
R1=R2=OMe: Sinapyl alcohol
R1=R2=H: p-Coumaryl alcohol
R1=OMe, R2=H: Guaiacyl unit
R1=R2=OMe: Syringyl unit
R1=R2=H: p-hydroxyphenyl unit

8
Lignin polymerization is initiated by oxidation of the phenyl hydroxyl group of the
monolignols by enzymatic dehydrogenation, forming resonance-stabilized free radicals. The
radicals then undergo radical coupling reactions, producing dimers called dilignols connected
through a variety of linkages (Table 1). The dilignols continue to link to each other in an end-
wise polymerization. (Freudenberg 1959)
Table 1: Linkages found in softwood lignin.
Linkage
Type
Dimer Structure Percent of Total
Linkages (%)
β-O-4 Phenylpropane β−aryl
ether
45-50
5-5 Biphenyl and
Dibenzodioxocin
18-25
β -5 Phenylcoumaran 9-12
β-1 1,2-Diaryl propane 7-10
α-O-4 Phenylpropane α−aryl
ether
6-8
4-O-5 Diaryl ether 4-8
β - β β-β-linked structures 3
Lignin polymers are extracted and separated during the pulping process in biorefineries,
mainly from wood. The main component in wood, along with cellulose and hemicellulose, is
namely lignin. (Sjöström 1993)
The composition of lignin may vary depending on the plant species. Wood can be divided into
two main categories: softwood (gymnosperm, i.e. evergreen trees) and hardwood
(angiosperm, i.e. deciduous trees). Gymnosperms produce their seeds without covering and
just release the seeds as they are, whereas the seeds of angiosperms are protected with a
covering. Ex. Apple seeds are protected inside the apple, thus apple trees belong to
angiosperm. (HowStuffWorks 2001, Mongeau, Brooks 2001)
Softwood lignin is composed almost entirely of G with small amounts of H. An example of
chemical structure can be seen in Figure 6. In contrast, hardwood lignin have a blend of G, S
and very little of H. (Sjöström 1993)
The syringyl in addition to guaiacyl units contributes to a more linear structure in hardwood.
Whereas softwood lignin is more branched and/or cross-linked. (Kubo et al. 1997)
The level of crosslinking has an effect on properties such as glass-transition temperature (Tg).
Comparing lignin of softwood and hardwood with the same molecular weight and
distribution, the branched and/or cross-linked nature of softwood lignin results in a slightly
higher Tg compared to that of hardwood. (Baker, Rials 2013)

9
Figure 6: A possible structure of softwood lignin polymer. Some of the three most occurring linkages and structures
are indicated by blue circles. (Gellerstedt, Ek & Henriksson 2009)
β-O-4
β-5
Dibenzodioxocin
structure
5-5

10
1.2.3.1 Refining of lignin
Lignin is produced and recovered as a byproduct in the bio refinery industry, i.e. in the paper
pulping process or the cellulosic ethanol fuel production. As for today, Kraft pulping is the
most commonly used process to extract lignin. (Huang 2009) Along with hemicellulose lignin
is dissolved and separated from cellulose fibers into a so called black liquor solution using
strong alkali reagents and sodium sulfite catalysts. (Sjöström 1993) Therefore lignin
molecules often contain traces of sulfur.
Considered waste, lignin is used to supply with low-value energy and is simply burned in the
refinery. Burning the black liquor also regenerates inorganic substances. (Baker, Rials 2013)
The lignin can be extracted from black liquor through pH fractioning. The lignin molecules
precipitate by lowering the pH, in the alkali solution, below 10 pH. (Helander et al. 2013)
Organosolv, which is a cleaner alternative to kraft pupling, can also be used to fractionate
biomass feedstock into the different lignocellulosic components. (Baker, Rials 2013)
Developed by Innventia AB, Lignoboost is another process of lignin extraction. It is designed
to work alongside traditional kraft pulping and allows for high recovery of high purity lignin.
The process involves the treatment of industrial black liquor with carbon dioxide which
causes precipitation of lignin, which is slurried, washed with dilute acid and recovered.
(Baker, Rials 2013)
Through cleaner and purer fractioning the biofuel market has become competitive with oil
refining and several products have even been identified as direct replacement for petroleum
derived chemicals. That includes: polymer blends, epoxy resins, conduction polymers and
antioxidants. The possibility to manufacture carbon fibers from lignin is of particular interest.
(Baker, Rials 2013)
1.2.3.2 Lignin-based carbon fibers
Lignin carbon fibers do have advantages over other precursors (MPP and PAN) in the sense
of cost and that lignin is a renewable material. (Baker, Rials 2013) Another feature of lignin is
that it is already substantially oxidized, which might be beneficial in the carbon fiber
manufacturing. (Baker, Rials 2013)
The best lignin-based carbon fiber samples produced to date have an average strength of 1.07
GPa, modulus of 82.7 GPa, and extensibilities of 2.03%. (Baker, Rials 2013) As mentioned
above, these values are, however, inferior to those of PAN and MPP.
The mechanical strength of lignin carbon fibers need to be increased. It is however restricted
by the unavailability of suitable and pure lignin. Most industrial lignin contains significant
amounts of impurities, which may induce inconsistent rheological and ill-defined properties
that hinder processing. (Chatterjee et al. 2014) (Lignoboost is a workaround/solution that
produces pure lignin) The lignin needs to meet certain requirement to allow multifilament
melt spinning and conversion trials. For instance, the melt-spinning requires a relatively low
Tg whereas the stabilization requires high Tg and this causes complications. In the economic

11
perspective of production, the fibers need to be converted into carbon fibers rapidly and at
low expenses. (Baker, Rials 2013)
Processing conditions such as stabilization and carbonization have a great influence on the
mechanical properties of carbon fibers and should thus be considered to improve the fiber
quality. (Baker, Rials 2013)
Furthermore, narrow molecular weight distribution would ensure uniform increases in
molecular weight throughout the material during oxidative thermostabilization, and
consequently provide for a more beneficial isotropic/uniform structure during carbonization.
(Baker, Rials 2013)
1.2.3.3 Production of lignin carbon fiber
The process of manufacturing carbon fibers from lignin polymers is similar to that of PAN
and MPP.
After the extraction and pre-processing, lignin is usually melt spun due to its thermoplastic
properties. Pure softwood can be dry or wet spun. (Baker, Rials 2013) Then the produced
fiber undergoes thermostabilization to convert it to a thermoset. In the last step non-carbon
elements are removed as the fiber is carbonized. (Norberg et al. 2013)
During spinning extrusion, the temperature is often maintained at approximatively 200°C, and
in the following stabilization step, the temperature should be between 200-300°C and lastly
the carbonization is performed at temperatures higher than 1000°C. (Brodin, Sjöholm &
Gellerstedt 2010)
The process flow is illustrated in Figure 7.
Figure 7: Process flow of lignin into carbon fibers. The energy use is different in each of the steps. Carbonization and
graphitization tend to take a lot of energy.
It has been stated that increased alkoxy contents and/or carbon contents enhance lignin-based
carbon fibers. (Baker, Rials 2013) Accounting for this, softwood lignin has a better potential

12
in carbon fiber manufacturing than hardwood, since its carbon yield is higher. However, the
more cross-linked nature of softwood does not allow melt spinning without prior
modification, e.g. blending lignin polymers with plasticizers or softening agents. (Huang
2009)
Thermostabilization
In the thermostabilization of lignin, the sample is often treated in oxidative environment and
the temperature is ramped from around 200°C to above 300°C. It is important to maintain the
temperature below the Tg so that the integrity of the polymer is kept unchanged and does not
start to degrade or fuse. Oxidative stabilization prior to carbonization causes the lignin to have
an increased resistance to further thermal treatment, mainly due to the formation of crosslinks.
(Baker, Rials 2013)
In the oxidative thermostabilization, oxygen contents increase as the temperature rise to 250-
260°C. This implies that oxidation reactions take place to create oxygenic function groups.
After further heating, beyond 260°C, the oxygen content decrease. (Baker, Rials 2013)
Meanwhile, the hydrogen content decrease as water, methanol and methane are released upon
heating. Passively, due to the decrease of other elements, the carbon content percentage
becomes higher and formation of C-C bonds increases. (Li et al. 2013)
In the condensation of carboxylic acid groups, ester and anhydride moieties are formed. (Li et
al. 2013) The esters and anhydrides are formed as the thermostabilization temperature rises.
This indicates a cross-linking reaction taking place within the lignin macromolecules. (Li et
al. 2013)
Also involved in the stabilization is the β-O-4 linkage. It accounts for nearly 48-60% of the
total inter-unit linkages of lignin. (Braun, Holtman & Kadla 2005) Homolytic cleavage of the
β-O-4 bond, during the oxidative stabilization, is likely the first step in which radicals are
formed followed by a series of rearrangement reactions (Figure 8). (Braun, Holtman & Kadla
2005) This is thought to be sufficient to stabilize lignin and transform it into a thermoset.
(Norberg 2012) A more aromatic structure is believed to be formed in the lignin molecule. (Li
et al. 2013)
Figure 8: Homolysis of the β-O-4 bond in the stabilization. The formed radicals react further with the lignin.

13
Studies have shown that Tg is dependent on both cross-linking and incorporation of oxidized
groups. Low temperature and low heating rates favor oxidative weight gain which result in
formation of crosslinks. (Braun, Holtman & Kadla 2005)
Softwood lignin is more susceptible to the oxidizing treatment than hardwood. (Norberg et al.
2013), and possesses more cross-linked structure than hardwood lignin. The molecular
structure of softwood lignin is considered to be more branched than hardwood lignin. (Heber
S. et al. 2009) The level of crosslinking contributes to a faster stabilization (Lin et al. 2014).
On a sidenote, softwood lignin structure contains high amount of oxygen as compared with
petroleum pitch and PAN. It might be sufficient for autocatalytic stabilization to take place
using only heat. (Norberg et al. 2013)
In order to determine whether the material is thermoset or not, differential scanning
calorimetry (DSC) can be used to look for a glass-transition. If no transition or Tg can be
found, it is an indication that the fibers were fully stabilized (i.e., the fusible thermoplastic
lignin fiber has become an infusible thermoset structure). (Norberg et al. 2013)
As for lignin, a skin-core structure has also been identified in the fiber. The structure is
formed by oxidative reactions near the surface. Formation prevents further diffusion of
oxygen into the fiber. As shown for pitch, fibers with thicker diameters have a greater
difficulty in achieving full stabilization during the oxidative stabilization. The same may
apply to lignin fibers. (Norberg et al. 2013, Matsumoto, Mochida 1992) It is not known how
the skin-core affects mechanical properties of the final carbon fiber. It may have negative
complications. (Norberg 2012)
Carbonization
The stabilization is followed by the carbonization process. The sample is treated under
pyrolytic conditions in inert atmosphere (N2) at temperatures between 1000-2000°C. The rate
of heating may vary; 5-10°C/min is preferred. The high temperatures cause removal of
undesired heteroatoms (non-carbon elements) from the fiber and formation of ordered
graphitical structure, and thus improving mechanical properties as well as electrical and
thermal properties. Involved in the carbonization is dehydration, decarboxylation,
aromatization and condensation reactions (Chatterjee et al. 2014).
The outcome of finalized carbon fibers is highly dependent on the thermostabilization.
Generally speaking, the yield increases as the stabilization temperature reaches 260°C and it
has been shown that the best yield of carbon fiber can be attained from fibers stabilized in
between 260-290°C, due to the high amount of aromatic structure formed in the stabilization.
Above 290°C the yield tend to decrease as the stabilization temperature goes higher. (Li et al.
2013)
Raman spectroscopy can be used to identify the degree of structure ordering in carbon fiber
material. (Li et al. 2013) showed that carbonized fibers, in a temperature of 1400°C, had low
ordering when the sample had been thermostabilized below 260°C. Above 290°C the amount
of C-C structures were higher but the aromatic ordering was disrupted due to the pyrolysis or

14
thermostabilization. A competition may occur between ordering and disruption in the range
260-290°C. Compared to PAN, lignin does not have the same good graphitic structure when
treated under the same conditions. (Li et al. 2013)
Furthermore, in their studies the Raman spectra also indicate that the disordered carbon
structure exist mainly as sp3 hybridization rather than sp2 hybridization. (Li et al. 2013)
There is a possibility that pores and defects could be formed due to the elimination of
heteroatoms in heat treatment. (Huang 2009) Regarding the mechanical strength of carbon
fiber, it is a well-known fact that voids and defects weaken the carbon fibers. (Lin et al. 2014)
conclude that improved mechanical properties are more likely due to the reduction of voids
and defects rather than modification of the microstructure. They also state that anisotropic or
oriented form is preferred over isotropic form.
To determine if carbon fibers has been formed, SEM or X-ray analysis can be utilized.

15
2 Experimental The entire project and its studies were conducted at Innventia AB in Stockholm.
Short overview: Lignin fibers were stabilized prior to carbonization. After measuring the
deformation during carbonization in the TMA, the carbonized fibers were evaluated in Dia-
Stron, TGA, FTIR, SEM and light microscopy analyses (Figure 9).
Figure 9: Schematic overview of the process.
2.1 Stabilization Prior the start of the project, pH-fractionated softwood kraft lignin (Mw= 51 kDa, Mn = 11
kDa and PDI= 4.6) from Bäckhammar had been extruded and formed to fibers through
multifilament melt spinning. The lignin was pre-treated in vacuum, 24 hours at 80°C followed
by 2 hours at 160°C and lastly in nitrogen gas at 180°C for 1.5 hours prior extrusion. A
multifilament extruder (Alex James & associates Inc., USA) was used.
In order to work with the fibers during the project they were stabilized, increasing the glass
transition temperature due to cross-linking and thus retaining a thermoset material.
The stabilization was then carried out in a gas chromatograph (GC) oven (Hewlett Packard
5890 Series II, Agilent Technologies Inc.).
In course of the project three batches of stabilized fibers were made. The first two were
stabilized in the same way and the third batch was processed slightly different.
In the preparation of the two first batches, lignin fibers were hinged, with clamps serving as
counterweights in each end, over a glass rod put inside a glass bowl. The counterweights
purpose was to suppress the dimensional changes of the fibers, i.e. to retain the original
length. The entire setup was then covered in aluminum foil to protect it from the turbulent fan
inside the GC oven.
Stabilization
TMA Carbonization
Dia-Stron Tensile-testing
TGA Mass loss analysis
FTIR Composition analysis
SEM Imaging and composition
analysis
Light microscopy Imaging

16
In the making of the third batch the glass rod was left out and the fibers was simply placed on
the bowls aluminum foil covered bottom (Figure 10). Although with glass rod missing, the
clamps were still attached to serve as counterweights. However, in this case and position they
were probably less effective.
Figure 10: Not yet stabilized lignin fibers placed on the foil covered bottom.
The entire setup was then inserted into the oven. Starting at 25°C, the temperature was
increased to 140°C at 3°C/min, then up to 250°C at 0.2°C/min where it remained for 1 hour.
The oven completed the program in 10.8 hours after which the temperature passively
decreased.
2.2 Carbonization and Thermomechanical Analysis (TMA) The principles of TMA and carbonization methods are described in the following sections.
2.2.1 Principles
In static force TMA, the specimen is strapped into a holder in the instrument, subjected to a
small/negligible load. A dilatometer then measure the deformation of the specimen as the
temperature continuously increases in a linear fashion (Figure 11). This gives information
about the thermal and force dependent expansion or contraction (elongation) of the material.
(Brown 2001)
Figure 11: Classic thermo-dilatometry of expansion. Note that the arrow can be in the opposite direction, in which
case the material contract. The specimen is fixated in the top as indicated by the dot. A small force is applied via the
pushrod of the dilatometer to the specimen.
For length measurements, the linear expansion or dimensional change (∆L/L0, where ∆L is the
length difference L-L0 and L0 is initial length) is related to the temperature changes (∆T) by a
linear expansion coefficient (α) and can be expressed as:
L0
∆L
Positive
direction
F

17
𝛥𝐿
𝐿0= 𝛼𝛥𝑇
The linear expansion coefficient is thermomechanically characteristic for a material and is
determined by composition and structure of the solid. (Brown 2001)
The TMA can also be used to determine glass-transition temperatures Tg and softening points
(Ts) for certain materials, polymers in particular. (Brown 2001)
2.2.2 Method
A multiple of fibers were glued together into a bundle by applying carbon paint suspension
(SPI Supplies®) in both ends of the fibers (Figure 12 a). After a brief moment of curing, in
normal atmosphere (1 at) and room temperature (20°C), clamps of alumina (Al2O3) (Model
TMA40200A06.022-00, NETZSCH GmbH, Germany) were attached to the glued part of the
fibers with the help of a mounting station (Model TMA40200A06.023-00, NETZSCH GmbH,
Germany) (Figure 12 b).
Figure 12: In a), a bundle of stabilized lignin fibers are placed on two stacked microscope slides. Both ends are glued
together. The mounting station in b) was used for alignment fixture of the sample in the clamps (white pieces).
The sample of fibers was then mounted in the TMA 402 F1 Hyperion® (NETZSCH GmbH,
Germany) header connected to an alumina pushrod (Model TMA40200A07.023-00,
NETZSCH GmbH, Germany) (Figure 13), to be carbonized at high temperatures. The
furnace was lowered upon the header, centering it in the chamber, sealing off the outside
environment in total isolation. A thermometer thread was positioned close to the sample,
measuring the temperature. The freely moving pushrod was connected to a linear variable
displacement transducer (LVDT) in the interior of the instrument. As the sample deformed
during the carbonization, the sensor recorded the absolute displacement from the point of
origin.
a) b)

18
Figure 13: The TMA-system. The furnace is in the elevated state above the header. Magnification of the header
depicts an inserted sample of fibers. Sprints are holding the setup in place. The lower clamp is connected to the
pushrod.
Cooling and regulating temperatures below room temperature was accomplished by a F25-
MA Refrigerated/Heating Circulator modulus (Julabo GmbH, Germany) connected to the
TMA.
For control and acquisition of the TMA-measurements the NETZSCH Measurement software
(v.6.1.0, NETZSCH GmbH, Germany) was employed.
Prior to carbonization the chamber was evacuated of atmosphere, to a pressure of approximate
4 mbar (indicated as 100 % vacuum by the TMA monitor), by a diaphragm pump MPC 105 7
(NETZSCH GmbH, Germany). The chamber was shortly after filled again with pure nitrogen
gas from a Nitrogen 5.0 LAB LINE® N2 (N2 ≥ 99.999%) tube (Strandmöllen AB, Sweden) by
opening the TMA by-pass valve. The by-pass valve was left open until the atmosphere gained
a slight overpressure and was then closed.
When the system were set up and the run program had been added via the NETZSCH
Measurement software (v.6.1.0, NETZSCH GmbH, Germany), the carbonization could take
place. The main inlets were opened; purge 2 (connected to the TMA chamber) and the
protective inlet (protecting the LVDT sensor) along with the main outlet. Opening the main
outlet normalized the pressure once again. The N2 gas flow was set to 75 ml/min for each inlet
and remained constant throughout the run.
The carbonization was carried out differently for each phase in the project:

19
In the first phase, the bundle of fibers (samples) underwent carbonization from 20°C
to set temperatures between 300-1200°C in steps of 100°C for each bundle, increasing
the temperature 1K/min (or 1°C/min) at a load of 5 mN.
In phase two, the increasing of temperature was varied, testing 10, 20 and 40 K/min, to
set temperatures (900-1300°C, in steps of 100°C). Same load as before.
During the third and last phase, heating of 10 K/min and a final carbonization
temperature of 1000°C were selected as fixed parameters in order to test different
forces. The initial load was increased to 10 mN.
However, since the dimension of the fibers changed, both in length and diameter, due to the
mass loss and evaporation of substances, as the temperature rose during the carbonization, the
above stated preparations of the fibers (i.e. the use of carbon paint) and attachment could not
be applied in the force measurements (last phase). As the fibers shrunk they were simply
drawn out from the clamps when the force increased.
The fibers had to be fixated in a better way so that they would not move at all. Plates of
graphite were used, instead of the clamps, onto which the fibers were placed, one plate for
each end, and adhered with graphite adhesive (Figure 14). A thin layer of graphite bonding
agent (Graphi-Bond 551-RN, Aremco Products Inc.) was applied over the fibers and plate.
The adhesive was then cured in an oven (Gas Chromatograph, Hewlett Packard 5890 Series
II, Agilent Technologies Inc, USA) in three steps; first dried at 25°C for 4 hours, secondly the
temperature was increased to 130°C at 25°C/min and was held there for 4 hours and thirdly
the temperature was increased further up to 260°C at 3°C/min where it remained for 2 hours.
The curing was performed in normal atmosphere (1 at). After the curing the plates could be
inserted/mounted in the TMA sample holder.
Figure 14: Plates of graphite onto which stabilized lignin fibers were adhered.
The TMA measured the deformation (in this case linear expansion/contraction) through a
dilatometer, at an acquisition rate of 25 points per minute, which could only operate in the
range of 5000 µm (5 mm). Reaching the upper limit resulted in an underflow error and lower
limit in an overflow error. When the fibers sometimes snapped/ruptured the monitor would
always show an underflow error. After completing a run the computer software transferred the
data directly to another program (NETZSCH Proteus – Thermal Analysis v.6.1.0) for
processing.

20
Each sample of carbonized lignin fibers were put in small glass jars sealed with a plastic cap
and marked on the side with date, sample number etc. The samples prepared in the TMA were
then examined in FTIR-spectroscopy, tensile tested in the Dia-stron and imaged by SEM.
2.3 Uniaxial Tensile Testing (with Dia-Stron)
The principles of uniaxial tensile testing and used methods are described in the following
sections.
2.3.1 Principles
A commonly used method to evaluate a material is tensile testing. The principle behind the
test is gripping opposite ends of a specimen/sample in a test machine. Force is exerted on the
object by the machine, resulting in gradual elongation and ultimately a fracture of the sample.
(Davis 2004)
During the process, force-extension data is recorded, which is a measure how the sample
deforms under applied force. This data gives information about important mechanical
properties of the sample. Elastic deformation (Young’s modulus or E-modulus), yield strength
and ultimate tensile strength are such properties. (Davis 2004)
Stress (s) is defined as the force that acts over a given cross-sectional area and the unit is
N/m2 or Pa. Stress allows for strength comparison between materials. (Davis 2004)
𝑠 =𝐹
𝐴
Strain (e) expressed as the ratio of total dimensional change to the initial dimension which
describes the deformation. (Davis 2004)
𝑒 =𝛥𝐿
𝐿0=
𝐿 − 𝐿0
𝐿0
The Young’s modulus or E-modulus (E) is the slope of the linear part of the stress-strain
curve. It is a measure of rigidity and is derived from the stress to strain ratio, which is also
expressed in Pa units. (Davis 2004)
𝐸 =𝑠
𝑒=
𝐹 ∕ 𝐴0
𝛥𝐿 ∕ 𝐿0=
𝐹𝐿0
𝐴0𝛥𝐿
In this project the stress and strain are referred to as engineering stress and strain. (The true
stress and strain is defined respectively by the instantaneous change of area and length.)
(Davis 2004)
2.3.2 Method
The tensile testing was carried out on an automated system, called Dia-stron (Dia-stron
Limted, Andover, UK). The system is composed of several modules; an automated sample
loader (ALS1500), a linear extensometer (LEX820) and a laser diffraction system (LDS0200)
(Figure 15). Pneumatically driven, the moving arm of the automated sample loader was
provided with air from a PG28L-Dual purifier (PEAK Sientific Instruments Ltd., Renfrew,

21
UK) connected to the system, controlled and regulated by a PU1100 Pneumatic unit (Dia-
stron Limted, Andover, UK). The system was controlled through a UV1000 control unit (Dia-
stron Limted, Andover, UK).
Figure 15: Modules constituting the Dia-Stron instrument.
The tests were performed on stabilized and carbonized lignin fibers made in the TMA.
Prior the strength measurement, plastic plates were placed in designated slots on a cassette, 20
slots in total. Each slot consisted of two plastic plates, in between the distance or gauge length
was 12, 20 or 30 mm depending on the cassette. For each slot single fibers were placed, one
end in the well of one plate (Figure 16). The wells were then filled with UV-adhesive
(DyMAX 3193, Dymax Corporation, USA) with a Performus™ III dispenser (Nordson EFD,
Westlake, USA) set to 3.36 Bar and 0.335 seconds flow. The adhesive was then curated for
roughly 4 seconds with a pE-100 ultraviolet pen (CoolLED Limited, Andover, UK) set to an
intensity of 50% and a wavelength of 400 nm.
Figure 16: Cassette of gauge length 12 mm with plastic plates. The lines in the magnification indicate the alignment
and placement of the fibers.
Automated
sample loader
Cassette
platform
Linear extensometer
Laser diffraction system

22
The whole cassette was then placed on the cassette holder of the automated tensile testing
system Dia-stron (Dia-stron Limted, Andover, UK). The automated sample loader picked up
the couple of plates and moved the sample to the linear extensometer modulus coupled with
the laser diffraction system. Through light diffraction the diameter was measured, in which
the modulus “rocked” back and forth to find the position where the signal/intensity of the
diffraction was strongest. The laser diffraction system was very sensitive and the software
could not detect/calculate any diameter from the diffraction pattern if the samples were
irregular in form, insufficiently round or transparent. Regardless of whether the diameter
measurements were successful or not the LEX modulus performed, afterwards, an extension
of the fiber till it ultimately broke. From the test the UvWin software (v. 3.35.0000 build 2,
Dia-stron Limted) plotted the stress versus strain in a graph. By using the analytical tool
provided by the same software, the corrected E-modulus could be determined. (The
mathematical models are given above, in section 2.3.1).
The compliance for the system was calculated according the appendix (section 8.2.3.1).
However, the compliance was neglected in the calculations of E-modulus.
The LDS was set to a tension force of 3 gram force (gmf) prior to measurement and to an
extension rate of 0.1 mm/sec. The linear extensometer had a gauge force of 2 gmf prior to
measurement and extended the sample to 7% of the gauge length at 0.1 mm/sec. The
threshold of breakage was set to 5 gmf and the max force allowed was 500 gmf. When the
sample snapped/broke the instrument informed the user by sending out a loud beep noise
where after the automated sample loader arm moved and discarded the sample in the
originating slot and continued to the next slot.
2.4 Thermogravimetric Analysis (TGA) The principles of TGA and used methods are described in the following sections.
2.4.1 Principles
TGA is a thermal analysis in which the specimen is subjected to elevated temperatures. In this
fashion the chemical and physical properties can be determined by measuring the change of
weight as a function of increasing temperature. This provides information about thermal
stability, degradation, decomposition and vaporization of substances. (Coats, Redfern 1963)
In particular, for this project, decomposition in the form of pyrolysis (in inert atmosphere) is
the most interesting aspect.
2.4.2 Method
During carbonization of the stabilized lignin fibers mainly the oxygen and hydrogen content
of the fibers will be reduced, a pyrolysis, meaning that different oxygen containing substances
will be released from the fibers. In order to monitor the change in mass a thermogravimetric
analysis was carried out in a TGA 7 thermogravimetric analyser (PerkinElmer Inc, Waltham,
USA).
A few stabilized fibers were hacked into small pieces and put into a little platinum dish. In
turn the dish was placed on the internal balance of the TGA. The glass cylinder tube was then
elevated so it would contain the sample. In the elevated state gas of pure nitrogen from a

23
Nitrogen 5.0 LAB LINE® N2 (N2 ≥ 99.999%) tube (Strandmöllen AB, Sweden) flowed
through the system. Afterwards the balance was tared and the instrument was then left
untouched alone in the room for a brief moment so that the balance would adjust and stabilize
itself. (The sensitive balance fluctuated and would react to even the slightest movement in the
room.)
The instrument was connected to a TAC 7/DX thermal analysis control unit (PerkinElmer Inc,
Waltham, USA). Via a computer, measurement data of the weight loss was collected by the
Pyris software (v. 8.0.0.0172, PerkinElmer Inc., Waltham, USA) over a set temperature range.
The program was set to run the TGA from 20°C to 1000°C at 10°C/min as the weight was
measured at each point. However, the TGA managed only to reach 885°C before cooling. The
initial mass was 0.6290 mg.
However, TGA-instrument was very unreliable since it provided illogical readings (e.g. some
measurements showed negative mass). This was probably caused by air leaking into the
system, resulting in combustion of the stabilized lignin fibers. In other word the instrument
was not sealed completely and the environment inside the system was not kept inert or oxygen
free.
As a measure, a reference carbonization was done in the TMA-instrument for comparison. 30
fibers, glued together with carbon paint, were weighted on an AE200 balance (Mettler
Toledo, USA) before and after carbonization up to temperatures of 300, 600 and 900°C
(10K/min). Initial masses were 2.7, 2.2 and 2.7 mg respectively for each carbonization
temperature. The results were thought to serve as calibration point for the mass-loss curve in
the TGA. The weight loss graph was corrected accordingly.
As a second measure of obtaining a decent measurement of weight loss, stabilized lignin
fibers were sent to the NETZSCH Applications Laboratory, section of thermal analysis, in
Selb, Germany for investigation, in mission of Innventia AB (Stockholm, Sweden).
Two measurements were performed with the thermogravimetric analyzer, thermo-
microbalance TG 209 F1 Libra® (NETZSCH GmbH, Selb, Germany). Precise internal mass
flow controllers of the vacuum tight instrument and a resolution of 0.1 µg enabled high
precision measurements. For control and data acquisition the PROTEUS® software
(NETZSCH GmbH, Selb, Germany) was employed.
The samples were placed into an Al2O3 crucible, and the system was evacuated and refilled
with nitrogen (20 ml/min) before measurement. The program was set to run from 20°C to
1100°C at 10°C/min as the weight was measured at each point. Initial masses of the both
samples were 1.972 and 2.038 mg.
2.5 Fourier Transform Infrared Spectroscopy (FTIR) The principles of FTIR and used methods are described in the following sections.

24
2.5.1 Principles
Infrared spectroscopy works on the principle that molecules and atoms interact with
electromagnetic radiation. Infrared light causes atoms and groups of atoms to vibrate with
increased amplitude about the connecting covalent bond. (Solomons, Fryhle 2011a)
The vibration or oscillation occurs only at certain frequencies, meaning that the vibrational
states are quantized energy levels. A transition between states can only occur when the
frequency of the radiation match the transition energy (i.e. the frequencies resonate), in which
case the radiation is absorbed by the compound. (Solomons, Fryhle 2011a)
Since different functional groups and molecules are arranged in specific ways, light will be
absorbed at frequencies that correspond to the molecules and functional groups. The
frequency is related to the masses of the bonded atoms and the stiffness of the bond.
(Solomons, Fryhle 2011a)
In order for a molecule to be ‘IR-active’, the dipole moment must change as the vibration
occurs. Thus symmetric molecules, such as homodiatomic molecules (e.g. N2 and O2), does
not absorb IR-light. (Solomons, Fryhle 2011a)
The vibration can occur in a variety of ways: symmetric stretching, asymmetric stretching,
scissoring and twisting etc. (Solomons, Fryhle 2011a)
An IR-spectrometer operates by irradiating a sample with IR-light. The transmittance of the
radiation is compared to the transmittance in absence of sample, a so called reference. The
comparison indicates the frequencies where the sample absorbed light. (Solomons, Fryhle
2011a)
FTIR spectrometers employ a Michelson interferometer. The radiation is split into two beams,
by a beam splitter. The beams are reflected back on two mirrors, of which one is mobile, and
recombine which causes interference. The radiation passes through the sample to the detector
which records the signal as an ‘interferogram’. Through a Fourier transform the interferogram
is then converted into a spectrum. (Solomons, Fryhle 2011a)
The absorption can be observed as a peak in the spectrum where the position is specified in
units of wavenumbers, which is the reciprocal of the wavelength, often expressed as cm-1
.
(Solomons, Fryhle 2011a)
�̅� =1
𝜆
This is due to the energy (∆E) of absorption being directly proportional to the frequency (f)
according the equation:
𝛥𝐸 = ℎ𝑓 =∕ 𝑓 =𝑐
𝜆∕=
ℎ𝑐
𝜆= �̅�ℎ𝑐

25
2.5.2 Method
Fiber samples were carbonized in the TMA prior analysis in FTIR-spectroscopy. The
carbonization temperature and rate was 1000°C and 10 K/min, respectively.
By using an agat mortar the carbonized fibers were grinded thoroughly. Small fragments of
the fibers spurt away when grinding, thus the mortar had to be contained within a hand folded
dome of aluminum foil. This prevented fiber fragments from escaping.
Depending on the degree of carbonization different amount of sample was used when
grinding. The more carbonized the fibers were the less amount had to be used in order to get
distinctive spectrum. The weight of the fiber sample was determined by using a highly
sensitive balance (Ultramicro Type 4504 MP8-1, Sartorius AG, Goettingen, Germany),
measuring weights less than 1 mg, 4 digits precision.
Previously dried for 30 min in a Memmert Type U30 oven (Gemini BV, Apeldoorn,
Netherlands), set to 105°C, potassium bromide (KBr) powder (Uvasol®, Merck Millipore,
Billerica, USA) was added and mixed/grinded together with the sample. The amount was
roughly the same for each sample, i.e. approximately 300 mg.
The mixture was then transferred to a hydraulic press to form a slightly transparent thin black
disc-like pellet, 13 mm in diameter and approximatively 2 mm thick, under pressure of 250
kg/cm2 (recalculated to nearly 250 bar) and slight vacuum (Figure 17). Pellets of lignin
powder, stabilized or carbonized lignin fibers up to 300°C were, instead, somewhat brown in
color. The reference pellet was made in a similar manner with solely KBr powder, 300 mg,
resulting in a clear transparent pellet.
Figure 17: Hydraulic press (top) with pieces (bottom) used to form disc-shaped pellets.

26
Due to the hygroscopic nature of KBr (van der Maas, J. H., Tolk 1962, Abo 2010) the full
procedure had to be done quickly. The pellet was then rapidly inserted with the holder into the
FTIR-spectrometer, since the pellet had the tendency to become “cloudy” rather fast due to it
soaking up moisture directly from the atmosphere.
A Varian 680-IR FTIR Spectrometer (Varian Incorporate, Palo Alto, USA) was used for this
study (Figure 18).
Figure 18: The Varian spectroscopy instrument.
During the FTIR-spectrometry the atmosphere in the sample compartment was continuously
purged with dry nitrogen gas, eliminating substances like carbon dioxide that may interfere
with the spectra. The level of moisture in the gas was regulated by an external filter connected
to the device keeping the atmosphere as dry as possible.
Since the compartment was opened when the sample was inserted into the pellet holder, air
leaked in. By looking at the live spectra, the scan was manually started when the carbon
dioxide peak had disappeared. The time between insertion and spectral scan was estimated to
60 sec.
The spectral resolution was set to 2 cm-1
and the sample was scanned in range 4000-600 cm-1
.
A thermo electric (TE) cooled DLaTGS (deuterated L-alanine doped triglycene sulphate)
detector was used to collect the absorbance data at 5 kHz frequency. The samples were
illuminated by a mid-infrared source (MIR) and the beam splitter was of KBr.
Both the background and samples were scanned 32 times each. One background spectra was
made (150115) and used for all the samples throughout the study. However, it should be noted
that the correct procedure is to collect a background spectrum for each measurement,
preferable as close to the measurement as possible.
The collected spectrums were visualized in the Varian Resolutions Pro software (v.5.1.0.829,
Varian Incorporate, Palo Alto, USA).

27
2.6 Scanning Electron Microscope (SEM) The principles of SEM and used methods are described in the following sections.
2.6.1 Principles
The principle behind electron microscope is bombarding a surface of a specimen with a
stream of electrons. The scattering of the electrons can be used to generate an image of the
scanned area.
The electrons are produced by an electron gun which consists of an electron source (usually
tungsten), called cathode, and an electron accelerator. There are different methods to generate
the electrons from the cathode. In a field emission gun, the principle of field emission is
utilized. I.e. the electrostatic field at the tip of the cathode is increased to the point that the
potential barrier becomes small enough to allow electrons to escape through this barrier by
quantum-mechanical tunneling. After emission the electrons are accelerated in an electric
field, parallel to the optic axis, to a final kinetic energy. (Egerton 2005)
An objective in the SEM condenses the incident beam into a so called electron probe,
typically 10 nm in diameter. The beam is scanned over a rectangular area of the specimen,
known as raster scanning. (Egerton 2005)
Figure 19: Elastic and inelastic scattering. The electrons are incident from above, indicated by arrows.
Primary electrons from the incident beam penetrate the specimen and scatter both elastically
and inelastically (Figure 19). The respective scattering is caused by interaction with the
nucleus or with electrons surrounding the nucleus. Primary electrons that backscatter (i.e.
scattering through an angle greater than 90° to the incident beam) can be collected by a
backscattering detector placed above the specimen. The signal increases with atomic number
and thus contrast between chemical compositions can be observed in the imaging of materials.
(Egerton 2005)

28
Through inelastic interaction and transfer of kinetic energy, primary electrons cause ejection
of electrons orbiting around the nucleus. As moving particles, the secondary electrons
themselves will interact with other atomic electrons and scatter inelastically, gradually losing
kinetic energy until most of them are brought to rest within the material. Secondary electrons
that instead escape close to the surface are collected for imaging by an Everhart-Thornley
detector. Hence the image is a property of the surface structure (topography) and displays
topographical contrast. (Egerton 2005)
The same inelastic effect that ‘knock out’ electrons (in other words causes the electrons to
undergo transition to a higher energy state) in the inner shell (e.g. K-shell), results in a
vacancy in the shell. An electron in a higher shell de-excite, in a downward energy transition,
to fill the hole. The transition causes release/emission of energy in form of photons with
wavelengths in the X-ray spectrum. Since the X-rays are characteristic to atomic species, it
allows for measurement of the elemental composition of the specimen. A silicon drift detector
is often used to detect the X-rays. (Egerton 2005)
Transition to the innermost shell is called K-emission and is denoted α if the electron de-
excite from the L shell, β from the M shell etc. Similarly, transition between the M and L
shell would result in Lα-emission. (Egerton 2005)
2.6.2 Method
A few stabilized and carbonized lignin fibers from the TMA carbonization process were
selected for imaging and X-ray analyses. The analysis was carried out using a scanning
electron microscope (Quanta FEG 650, FEI™, USA) combined with an energy dispersive X-
ray spectrometer (EDS) (Figure 20). The instrument was located in the Department of
Geological Sciences at the Stockholm University.
Figure 20: The scanning electron microscope, Quanta FEG 650.
Main components of the instrument include a field emission gun (FEG), a concentric
backscattered electron detector (CBS) and an EDS with a silicon drift detector (SDD) (X-Max

29
silicon X-ray, Oxford Instruments). The instrument was operated through the AZtec software
(Oxford Instruments).
Prior the study, the fiber samples were attached to the sample holders with double coated
PELCO carbon tape (Ted Pella Inc., Redding, USA). Parts of the axial fiber surface and fiber
cross-section was imaged and analyzed of content. In the former, the fibers were positioned
horizontally on the holder and in the latter the fibers were positioned vertically in a special
holder.
The following day the fiber samples were placed inside the SEM chamber which was
evacuated of atmosphere to a pressure of approximatively 1 mbar. The system operated at an
accelerating voltage of 15 kV and a working distance (WD) of 10 mm and the fibers could be
observed in 1000-5000 times magnification.
2.7 Light Microscopy Prior and after carbonization treatment in the TMA a light microscope (Axioplan 2 Imaging,
Carl Zeiss) was used to look at the structure, integrity, to measure diameter changes and to
spot defects. Placed on top of the microscope was a CT5 camera (ProgRes®, Jenoptik AG,
Jena, Germany) which was used to capture images of the fibers in the computer software
Capture Pro 2.8.8 (ProgRes®, Jenoptik AG, Jena, Germany).
The fibers were simply put on a microscope slide which was placed under the microscope and
illuminated from above. The magnification was 10, 20 or 50x and the camera captured the
images in resolution of 2592x1944 and 1296x972 2x Bin. In the software the diameters was
be measured by drawing a straight perpendicular line between two opposite points of the fiber
contour.

30
3 Results In the following section the outcome of each experiment is described.
3.1 TMA The TMA provided information about the behavior of stabilized lignin fibers in temperature
changes and under static or varied load during carbonization. Each sample is represented by
one carbonization in the TMA instrument.
The data from the carbonization is presented as linear dimensional change over temperature or
time.
3.1.1 Static measurement
By studying the expansion/contraction (red curve) in Figure 21, where the slope/derivative
(blue curve) is represented as the linear expansion coefficient α, we can observe a slight
expansion up to 250°C, followed by a fast shrinkage. Thereafter the rate of contraction
diminishes slightly around 420°C and continues in a linear fashion until it begins to disappear
around 700°C. After 1100°C the slope cease completely, as illustrated in Figure 22 and Figure
23, where some of the samples have been carbonized further.
Figure 21: Deformation of approximatively 50 fibers, 20 mm each, during carbonization to 1000°C at 1 K/min and 5
mN load. The red curve represents the length changes, while the blue curve represents the slope/alpha value of the
blue curve.
Hereafter, the carbonization in Figure 21 will be referred to as the standard carbonization,
since it represents the general deformation pattern of all fibers. It is color marked as red in the
following charts, allowing for easy comparisons.
-0.0010
-0.0005
0.0000
0.0005
-25%
-20%
-15%
-10%
-5%
0%
5%
0 100 200 300 400 500 600 700 800 900 1000 1100 1200
α (
1/K
)
∆L/
L 0
Temperature (°C)

31
Carbonizations of stabilized fibers with different lengths are compared in Figure 22. The
curves follow approximatively the same path. There is no clear difference between 15 and 20
mm fibers. However, the curve of 10 mm deviates from the path and possess a nick around
600°C (possibly due to a fiber or multiple fiber breaking). The final difference, after
completion, is around 2 percentage units compared to the 15 and 20 mm fiber samples.
Figure 22: Comparison of samples with varying lengths. The number of fiber in the samples, corresponding to the
blue, green and red curve, was respectively 10, 20 and 50. The blue curve possesses a nick around 600°C. The
carbonization was performed at 1 K/min, under load of 5 mN.
The following Figure 23 is compiled of sample data with varied amount of fibers. We can see
that the majority of the curves fall into the same path.
-25%
-20%
-15%
-10%
-5%
0%
5%
0 100 200 300 400 500 600 700 800 900 1000 1100 1200
∆L/
L 0
Temperature (°C)
10 mm
15 mm
20 mm

32
Figure 23: Carbonizations of samples with varying amount of fibers. The triangles indicate where the fibers broke,
slipped out from the clamps or where the length exceeded the TMAs limit of measurement. The missing segments
between 200-300°C are also due to the latter. The red curve (50 fibers) is from Figure 21 and is here represented as a
reference. The carbonization was performed at 1 K/min, under load of 5 mN.
In particular, samples with 50 fibers lie close together, whereas the 20- and 10-fiber-samples
are more likely to deviate from the path. The standard carbonization curve (Figure 21) is
drawn into the middle.
Figure 24 depicts the elongation of samples at different rates of increasing temperature, 1, 10,
20 and 40 K/min. The respective deformation at 1 K/min and 40 K/min differ little to each
other. However, the curve of 40 K/min may be a little smoother than that of 1 K/min. It is
apparent that the curves follow the same pattern, although the pairs (1 and 40 K/min
respective 10 K/min and 20 K/min) have an offset to each other.
-30%
-25%
-20%
-15%
-10%
-5%
0%
5%
0 200 400 600 800 1000 1200
∆L/
L 0
Temperature (°C)
10 fibers
20 fibers
50 fibers

33
Figure 24: Comparison between carbonizations at 1, 10, 20 and 40 K/min under load of 5 mN. The sample amount
was 50 fibers, 20 mm in length. The blue curve of 40 K/min may be slightly smoother than the red curve of 1K/min.
The green curve of 10 K/min and orange curve of 20 K/min have no noticeable differences, except that the latter has
shrunk slightly more. The curves of 10 and 20 K/min respective 1 and 40 K/min are offset to each other. The missing
segments is due to the length signal exceeding the TMAs limit of measurement, hence the data is missing.
Put into a different perspective, the curves of 10, 20 and 40 K/min are illustrated as
deformation over time in Figure 25, accompanied with the change of temperature over time.
The heating appears to be faster in the beginning despite the specified rate in the
carbonization program. Not until after 200°C had the rate been corrected to match the
program. However, the different rates of heating seemingly do not affect the deformation of
the fiber samples.
-25%
-20%
-15%
-10%
-5%
0%
5%
0 100 200 300 400 500 600 700 800 900 1000 1100 1200 1300
∆L/
L 0
Temperature (°C)
1 K/min
10 K/min
20 K/min
40 K/min

34
Figure 25: The elongation as well as temperature in the TMA sample holder is visualized over time. The rate of
heating is faster prior 200°C.
0
100
200
300
400
500
600
700
800
900
1000
1100
1200
1300
-25%
-20%
-15%
-10%
-5%
0%
5%
0 20 40 60 80 100 120 140
Tem
pe
ratu
re (
°C)
∆L/
L 0
Time (min)
10 K/min
20 K/min
40 K/min

35
3.1.2 Measurements with increasing force
The result of increasing the force applied to the fiber samples are presented in this section.
Each sample of 5 fibers was carbonized at 10 K/min up to 1000°C. They were attached with
graphitic adhesive to graphitic plates, serving as clamps. The entire setup, of plates, adhesive
and fibers, were put in an oven for curing of the adhesive.
The samples (Table 2) are marked with letters since we will evaluate the strength performance
of these in the section below (3.2.2). The TMA-deformation curve of sample Z can be found
in the appendix (8.2.1).
Table 2: Displaying the range of temperature and force of sample A-L and Z along with amount of fiber.
Sample Force range (N)
Temperature (°C)
Rate (mN/K) Fiber amount
A 0.01-0.3 375-975 0.48 5
B 0.01-0.4 350-950 0.65 5
C 0.01-0.5 350-950 0.82 5
D 0.01-0.1 450-550 0.90 4
E 0.01-0.2 450-550 1.9 5
F 0.01-0.2 400-600 0.95 5
G 0.01-0.35 300-900 0.57 5
H 0.01-0.07-0.35 300-390-850 0.67, 0.61 5
I 0.01-0.12-0.6 300-420-820 0.92, 1.2 10
J 0.01-0.083-0.6 300-390-770 0.81, 1.4 10
K 0.01-0.083-0.6 300-390-770 0.81, 1.4 10
L 0.01-0.07-0.35 300-420-820 0.5, 0.7 5
Z 0.005-0.4 500-700 2.0 10
In the following Figure 26, the carbonization and elongation of three samples (A-C), under
same parameters (10 K/min and 5 fibers in each sample), is shown. The force is continuously
increased from 10 mN to 30, 40 and 50 mN, starting at 375°C, 350°C and 350°C and ending
at 975°C, 950°C and 950°C, respectively for sample A-C. As illustrated by the figure, the
contraction of the fiber is inhibited by applying an increasing amount of force. If compared to
the standard carbonization curve (Figure 21), which had a final contraction of nearly 21 % of
the initial length the contraction here is significantly smaller. The stabilized fibers of sample
A-C shrunk to 9, 7 and 3 %, respectively.

36
Figure 26: Carbonization with ramped force. The solid line is the elongation of the sample (A-C) while the dashed line
is the force applied to each sample. The blue, red and green curves belong respectively to sample A, B and C. The rate
of heating was 10 K/min and the amount of fibers was 5.
The effects of starting temperatures and rate of force ramp are studied in Figure 27. The
samples (D-G), of 5 fibers each, underwent carbonization at 10 K/min. For each carbonization
the force ramp was successively put at an earlier temperature, 450, 400, 400 and 300°C for
sample D-G, respectively.
Although, the final force for sample D-F is lower than that for sample A-C in Figure 26, the
deformation is seemingly the same at 12, 6 and 3 % for sample D, E and F, respectively. This
is because the temperature interval wherein the force can increase is kept small, and the slope
of the ramp is bigger, hence the force is already reasonably high at early temperatures
compared to sample A-C where the same forces are reached at higher temperatures.
However, one difference is that this setting of force results in a broad peak in the curves
around 550°C, in particular the curves E and F. In a usual fashion, the fibers expand up to
250°C and then contract. But progressively, the fibers internal force of contraction cannot
compete with the applied force of the TMA anymore and thus expand. After 550°C the
internal force becomes the winning competitor of the forces again and the fibers start to
contract once again. The contraction is not only dependent on the tension (stress) but also on
the stiffness (E-modulus) of the fibers. Initially the stiffness is relatively low, but as it starts to
increase around 400°C, as the temperature in the carbonization is continuously increased, the
contraction will be more and more affected by the increasing stiffness.
This behavior is also visible in the curve of sample D, although the force of the TMA is not
high enough to withstand the contraction we can see that the applied force suppress the
contraction by a moderate amount.
0.00
0.10
0.20
0.30
0.40
0.50
0.60
-15%
-10%
-5%
0%
5%
0 100 200 300 400 500 600 700 800 900 1000 1100
Forc
e (
N)
∆L/
L 0
Temperature (°C)
C B A
0.01-0.05 N 0.01-0.04 N 0.01-0.03 N

37
In the curve of sample G the force ramp is similar to those in Figure 26 in terms of rate and
final value. In contrast to the previous carbonizations of samples A-F the ramp starts earlier at
300°C which causes a longer expansion of the fibers. The nick in the curve, due to a fiber
breaking, may tell us that the applied force is too strong in this moment. However, due to this
fiber breakage, the remaining fibers does not contract more than the equivalent amount of the
expansion, the fibers stays close to 0 % deformation. When accounting for the force per fiber
the stress increases for the remaining fibers when one fiber breaks.
Figure 27: The ramping of force at different starting temperatures. The samples D-G, of 5 fibers each, were
carbonized at 10 K/min Earlier starting temperatures seems to suppress the shrinkage. The nick in curve of G around
500°C is possibly due to a fiber breaking.
The samples A, G and H are compared in Figure 28. The force ramps are almost the same for
each sample. However, the ramping of the force starts earlier in the carbonization of G and H.
This results in contractions that are less than that of sample A.
The force is stronger in the carbonization of sample G than H. Even so, the curve of sample G
is kept above curve H. In reality, curve G should follow the same path since the force differs
only slightly between the carbonizations. The breakage of one fiber, observed as a nick in the
G curve, results in a higher stress on each remaining fiber which might explain why curve G
is above H.
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
-15%
-10%
-5%
0%
5%
0 100 200 300 400 500 600 700 800 900 1000 1100
Forc
e (
N)
∆L/
L 0
Temperature (°C)
G F E D
0.01-0.35 N 0.01-0.2 N 0.01-0.2 N 0.01-0.1 N

38
Figure 28: Comparison between samples A, G and H. The force ramp is similar for each carbonization. However, the
ramp starts earlier in the carbonization of sample G and H. The force ramp in carbonization of sample H was divided
into two parts, first up to 0.07 N between 300 and 390°C then to 0.35 between 390°C and 850°C.
The next figure depicts the deformation of 10 fibers (Figure 29). The force was doubled to
match the carbonization of 5 fibers. We can observe a pattern similar to that of sample H in
both curves in Figure 28. The final deformation stays around the same at 6 % contraction of
the initial length, whereas sample H ends at 7 %. The force ramp was divided into two parts,
for sample I the force was increased to 70 mN at 420°C whereas the force in sample J was
increased to 83 mN at 390°C. In the second stage the force was increased to 600 mN at 820°C
for sample I and 770°C for sample J.
Figure 29: Carbonizations of samples with 10 stabilized fibers. Both curves differ little to each other. However, curve
J deviates unexpected from the path around 700°C, possible due to a fiber breaking. A nick around 300°C can also be
observed.
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
-10%
-5%
0%
5%
0 100 200 300 400 500 600 700 800 900 1000 1100
Forc
e (
N)
∆L/
L 0
Temperature (°C)
G
A
H
0.01-0.07-0.35 N 0.01-0.35 N
0.00
0.10
0.20
0.30
0.40
0.50
0.60
0.70
-10%
-5%
0%
5%
0 100 200 300 400 500 600 700 800 900 1000 1100
Forc
e (
N)
∆L/
L 0
Temperature (°C)
J I
0.01-0.07-0.6 N 0.01-0.083-0.6 N

39
The sample K and L contains 10 respectively 5 fibers. Figure 30 illustrates the difficulty of
applying force on the samples. The force ramp was divided into two parts, for sample K the
force was increased to 83 mN, 390°C, whereas the force in sample L was increased to 70 mN,
420°C. In the second stage the force was increased to 600 mN, 770°C, and 350 mN, 820°C,
respectively for sample K and L.
Stabilized fibers are in the region of 300-500°C very susceptible to force, this is the reason
they easily break when ramping the force. The curve of sample K is almost straight up till the
breakage.
Figure 30: Successful carbonizations compared to failed carbonizations when applying force. The triangles indicate
where the fiber bundle broke.
3.2 Dia-stron Uniaxial tensile testing was performed on the lignin fibers, carbonized in the TMA-instrument
through different programs, by the Dia-Stron instrument.
3.2.1 Evaluation of temperature and heating rate
In terms of mechanical performance, the significance of final temperature (i.e. 300°C, 400°C,
and so on up to 1300°C) and heating rates (1, 10, 20 and 40 K/min) of the lignin fiber
carbonization is evaluated in this section.
Starting with the carbonization temperatures, we can observe in Figure 31 that the fibers
become stronger with increasing carbonization temperature. The rate of heating was kept at
10 K/min during the carbonization and the gauge length in the Dia-Stron measurement was 12
mm.
0.00
0.10
0.20
0.30
0.40
0.50
0.60
0.70
-10%
-5%
0%
5%
0 100 200 300 400 500 600 700 800 900 1000 1100
Forc
e (
N)
∆L/
L 0
Temperature (°C)
K L
I J
0.01-0.083-0.6 N 0.01-0.07-0.6 N 0.01-0.07-0.6 N 0.01-0.07-0.35 N

40
Stabilized lignin fibers (non-carbonized) and fibers carbonized to 500°C tend have a low
tensile strength below 200 MPa. The fibers are however still more susceptible to
deformation/extension before breaking. Plastic behavior is also hinted by the curvature of
these fibers in the diagram.
As the temperature increases we observe that the fibers become stiffer and the curves become
more and more linear in shape. The slope, which account for the E-modulus, increases as well
and reaches its maximum above 900°C. At which point the tensile strength is above 600 MPa.
Figure 31: Strain versus stress. The dip indicates the breakpoint of the fiber samples, at which point the stress account
for the ultimate tensile strength. Each sample was carbonized at 10 K/min and the gauge length was 12 mm.
Figure 32 present the E-modulus taken from measurements of lignin fibers carbonized at 1
K/min to different final temperatures (300°C, 400°C… and 1200°C). At least 4 points of data
are plotted in each temperature. The data at 25°C mark stabilized lignin fibers (non-
carbonized).
The red curve in the diagram provides a picture about how the average E-modulus changes.
As the carbonization temperature increases, the E-modulus increases. The maximum is
observed at 1000°C and thereafter the E-modulus inclines slightly. Consider, however, that
the data in each temperature may be insufficient for an accurate picture since the points are
widely spread, especially at 1000°C and forward.
0
100
200
300
400
500
600
700
800
0 0.5 1 1.5 2 2.5 3 3.5 4 4.5
Stre
ss (
MP
a)
Extension (%)
400°C 300°C
1300°C 1200°C 1100°C 1000°C 900°C 800°C 700°C 600°C 500°C
Stabilized Lignin

41
Figure 32: Representation of how the E-modulus increases with temperature. A (red) line is drawn between the
average points, calculated from the data points (blue) in each temperature. The error bars in each temperature
represent the sample standard deviation. The tested fibers were carbonized at 1 K/min.
Figure 33 is informative regarding how the tensile strength improves as the carbonization
temperature increases. The data of tensile strength was acquired from the same measurement
as above. In contrast to the E-modulus, the increasing of tensile strength may not be as clear.
The average line in red fail to show a smooth curve, compared to the E-modulus of the points.
The tensile strength is kept at relatively low values up to 600°C. Then it makes a drastic jump
at 700°C. For the following temperatures it is difficult to evaluate whether the tensile strength
increases or not, since the points are widely spread, even more than in the previous figure.
0
10
20
30
40
50
60
0 100 200 300 400 500 600 700 800 900 1000 1100 1200
E-m
od
ulu
s (G
Pa)
Temperature (°C)

42
Figure 33: Tensile strength plotted against temperature. A (red) line is drawn between the average points, calculated
from the data points (blue) in each temperature. The error bars in each temperature represent the sample standard
deviation. The tested fibers were carbonized at 1 K/min.
Similarly, a series was made to evaluate the influence of 10 K/min on E-modulus and tensile
strength (Figure 34, Figure 35).
Although a fewer data points are used in the diagrams, the same behavior can be observed; the
E-modulus increase with increasing temperature and the tensile strength also becomes higher
(However, with an increasing margin of error as previously).
0
100
200
300
400
500
600
700
800
900
0 100 200 300 400 500 600 700 800 900 1000 1100 1200
Ten
sio
n S
tren
gth
(M
Pa)
Temperature (°C)

43
Figure 34: E-modulus plotted against temperature. A (red) line is drawn between the average points, calculated from
the data points (blue) in each temperature. The error bars in each temperature represent the sample standard
deviation. The tested fibers were carbonized at 10 K/min.
Figure 35: Tensile strength plotted against temperature. A (red) line is drawn between the average points (red
squares), calculated from the data points (blue) in each temperature. The error bars in each temperature represent
the sample standard deviation. The tested fibers were carbonized at 10 K/min.
0
10
20
30
40
300 400 500 600 700 800 900 1000 1100 1200 1300
E-m
od
ulu
s (G
Pa)
Temperature (°C)
0
100
200
300
400
500
600
700
800
900
1000
300 400 500 600 700 800 900 1000 1100 1200 1300
Ten
sile
Str
en
gth
(M
Pa)
Temperature (°C)

44
The E-modulus and tensile strength values with averages from every carbonization (at 1, 10,
20 and 40 K/min) are displayed in the following charts (Figure 36, Figure 37).
Figure 36: E-modulus plotted against temperature. Contains all data points from all samples carbonized in every rate.
The plotted lines illustrate how the average E-modulus changes over temperature. The error bars in each temperature
and for each heating rate represent the sample standard deviation.
Figure 37: Tensile strength plotted against temperature. Contains all data points from all samples carbonized in every
rate. The drawn lines illustrate how the average tensile strength progresses over carbonization temperature. The
error bars in each temperature and for each heating rate represent the sample standard deviation.
0
10
20
30
40
50
0 100 200 300 400 500 600 700 800 900 1000 1100 1200 1300
E-m
od
ulu
s (
GP
a)
Temperature (°C)
1K/min
10K/min
20K/min
40K/min
0
100
200
300
400
500
600
700
800
900
300 400 500 600 700 800 900 1000 1100 1200 1300
Te
nsi
le S
tre
ngt
h (
MP
a)
Temperature (°C)
1K/min
10K/min
20K/min
40K/min

45
The progression of tensile strength over temperature is still hard to evaluate, since the points
in each temperature have a large spread. The spread of points cause a large margin of error
and evaluation must be considered with caution.
The averages of each rate are summarized in the tables below (Table 3, Table 4).
Table 3: An overview of the average E-modulus for the most interesting carbonization temperatures. The values are
displayed in units of GPa. The sample standard deviation is displayed in parenthesis.
1 K/min 10 K/min 20 K/min 40 K/min
900°C 34 (2.2) 34 (1.9) 30 (N/A) 31 (0.86)
1000°C 37 (3.7) 35 (3.2) 36 (0.83) 35 (1.4)
1100°C 37 (9.4) 34 (2.5) 35 (2.4) 36 (2.1)
1200°C 35 (4.1) 32 (0.37) 32 (2.5) 34 (0.88)
1300°C N/A 33 (2.9) 36 (3.3) 38 (1.2)
Table 4: An overview of the average tensile strength for the most interesting carbonization temperatures. The values
are displayed in units of MPa. The sample standard deviation is displayed in parenthesis.
1 K/min 10 K/min 20 K/min 40 K/min
900°C 420 (76) 550 (59) 430 (N/A) 470 (74)
1000°C 600 (240) 640 (80) 590 (88) 570 (51)
1100°C 500 (76) 700 (230) 650 (130) 300 (69)
1200°C 460 (190) 590 (74) 550 (56) 620 (6.9)
1300°C N/A 660 (64) 550 (130) 650 (220)
3.2.2 Evaluation of applied force during carbonization
In terms of mechanical performance, the significance of applied force on the stabilized lignin
fiber during carbonization is evaluated in this section. The samples, mainly A-H, that
underwent carbonization, as shown in section 3.1.2, were tested in the Dia-Stron system and
the result is presented here. (The deformation of sample Z during carbonization can be found
in appendix 8.2.1). Fibers that were initially broken prior carbonization served as reference in
the Dia-Stron testing. They showed strength properties characteristic with samples carbonized
under static load.
In short, strong connections between the strength and deformation can be drawn.

46
Table 5: An overview of the interesting samples with average E-modulus and tensile strengths. The sample standard
deviation is displayed in parenthesis. See Table 2 in section 3.1.2 for additional information. The TMA data of the
carbonization of Z can be found in the appendix.
Sample Force range (N)
Temperature (°C)
Average E-mod (GPa)
Average Breakpoint (MPa)
A 0.01-0.3 375-975 49 (2.4) 160 (57)
B 0.01-0.4 350-950 47 (4.0) 760 (150)
C 0.01-0.5 350-950 51 (0.96) 660 (120)
D 0.01-0.1 450-550 39 (2.2) 570 (120)
E 0.01-0.2 450-550 49 (0.40) 550 (100)
F 0.01-0.2 400-600 49 (1.5) 660 (140)
G 0.01-0.35 300-900 51 (6.3) 820 (36)
H 0.01-0.07-0.35 300-390-850 46 (1.1) 600 (73)
Z 0.005-0.4 500-700 38 (7.9) 490 (84)
The first diagram, Figure 38, provides a quick graphical overview of the E-modulus data
shown in Table 5. All samples except for D and Z show a higher value if compared to the E-
modulus without increasing load (recall Table 3 and Table 4).
Figure 38: E-modulus for sample A-H and Z.
If the E-modulus is plotted against tensile strength of every sample, including the samples
carbonized at static load, the samples can be categorized into groups. The following figure
shows all data obtained from the tensile testing (Figure 39). (Note however that tensile
strength and E-modulus in general do not correlate, in other words they do not necessarily
relate to each other. The figure is merely to compare the values of each sample and categorize
them.)
0
10
20
30
40
50
60
A B C D E F G H Z
E-m
od
ulu
s (G
Pa)

47
Figure 39: All obtained data from samples carbonized under static respectively non-static carbonization. The static
carbonized samples are indicated by rate 1, 10, 20 and 40 K/min. All samples were carbonized to 1000°C and the non-
static samples in a rate of 10 K/min.
Although the data from non-static carbonization (letters) is more spread it clearly belongs to a
different group, farther away from the static carbonization cluster (diamonds, ♦). An
exception to that is the D and Z sample, which show no mechanical improvements in contrast
to the other samples and can thus be grouped with the static carbonization. The fibers of
sample A show surprisingly low tensile strength whereas the E-modulus is better than that of
the static carbonized samples. Samples of G and C are relatively close to each other and are
placed farthest away from the static carbonized group.
3.3 TGA The thermogravimetric analysis provided information about the weight loss of stabilized
lignin fibers during carbonization.
Figure 40 displays the measurement made at Innventia AB, where the pale blue line
represents the original measurement and the darker blue line the corrected version. The data
was corrected according calibration points provided by three carbonizations in the TMA-
instrument (300, 600 and 900°C at 10K/min each). Complete calculations and calibrations can
be found in appendix (8.2.2).
Nicks, dents and sudden steps in the curves are evident and are probably caused by vibrations
and motions near the instrument, since the balance was extremely sensitive. The result must
be considered with caution because combustion occurred of the stabilized lignin fibers, due to
air leaking in.
A
A
B
B
B
C
C
C
C
C
D
D
D
D
D
E
E F
F
F
G
G G
H
H H
H
H
Z
Z
I
Y
0
100
200
300
400
500
600
700
800
900
1000
30 35 40 45 50 55 60
Ten
sile
Str
en
gth
(M
Pa)
E-modulus (GPa)
1K/min
10K/min
20K/min
40K/min

48
Aside from that, the figure provides an approximate view of the weight loss over temperature.
Although a residual mass below 30% is not reasonable. The corrected curve is more reliable,
showing a residual mass of 53 %.
Figure 40: TGA measurement of stabilized lignin fibers. (Pale blue) original curve and (strong blue) corrected curve.
The initial mass was 0.6290 mg.
To get a more reliable TGA measurement, stabilized lignin fibers were sent to NETZSCH
GmbH in Selb, Germany, for investigation. The result of the two measurements is shown in
Figure 41.
The data suggest that the weight loss occur in three steps. The first step shows a mass loss of
around 25 % of both samples. The maximum loss rate for this step can be observed in the
derivative curve (DTG, derivative thermogravimetry) around 425°C. The following step is
around 15% mass loss and the derivate peaks at around 560°C. The last step is about 3 % and
the DTG peak is found at 785°C. The residue mass for both samples is about 55%,
corresponding nicely with the corrected curve of the TGA-measurement at Innventia AB.
0
10
20
30
40
50
60
70
80
90
100
0 100 200 300 400 500 600 700 800 900 1000
We
igh
t (%
)
Temperature (°C)
Corrected Weight loss
Weight loss Residue mass:
53% 25%

49
Figure 41: TGA measurements of two samples (blue and green). The TG (thermogravimetry) curve shows the mass
loss over temperature while the derivative of this curve is provided by the DTG (derivative thermogravimetry) curve.
The initial mass of the blue and green sample was 1.972 and2.038 mg, respectively.
In comparison, our TGA-measurement shows a convex shape during the mass loss (between
450 and 750°C) whereas the NETSCH measurement is completely sigmoidal.
Combining the data from the TGA and TMA, the change of diameter of single fibers can be
calculated, assuming that the volume changes in the same manner as the mass. The
assumption is however not entirely true, since the volume change is also caused by sintering
of the stabilized lignin fibers. But it provides with an approximate understanding how the
diameter progresses over carbonization temperature (Figure 42).
The diameter change is compared with static and non-static TMA carbonization. Recall the
deformation of fiber sample G (Figure 28) and the deformation in the standard carbonization
curve (Figure 21). The fibers of sample G are subjected to an increasing force and the
diameter changes more than that of the fibers carbonized under static load. The pattern are
derived from the TGA measurements, at Innventia AB respectively NETZSCH (Figure 40 and
Figure 41).

50
Figure 42: The change of diameter during carbonization of sample G (blue) and the sample of the standard
carbonization curve (red). (Diamonds and triangles) indicate data calculated from the TGA measurement at
NETZSCH while (X) are calculated from the TGA measurement at Innventia AB. The lines represent the force
applied on each sample. The standard carbonization and carbonization of sample G was performed at 1 respectively
10 K/min to 1000°C. Initial diameter was 47 µm.
In turn, the diameter can be recalculated into cross-sectional area of single fibers. By using the
area and force, the change of individual stress of the fibers, during the carbonization in the
TMA-instrument, can be calculated for sample G and the standard carbonization, respectively
(Figure 43 and Figure 44). As mentioned, the full calculations are found in appendix 8.2.2.2.
The breakage of one fiber in the carbonization of sample G (occurs around 500°C, recall
Figure 27) has been taken into consideration when calculating the stress in Figure 43.
Note that the stress scale differs in Figure 43 and Figure 44.
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
60%
70%
80%
90%
100%
0 100 200 300 400 500 600 700 800 900 1000 1100
Forc
e (
N)
Dia
me
ter
Temperature (°C)
Blue: carbonization G Red: standard carbonization

51
Figure 43: Change of stress during the carbonization of sample G under increasing force. (Diamonds) indicate data
calculated from the TGA measurement at NETZSCH while (X) are calculated from the TGA measurement at
Innventia AB. The breakage of one fiber at 500°C has been taken into account. The carbonization was performed at
10 K/min to 1000°C.
Figure 44 Change of stress during the standard carbonization. (Diamonds) indicate data calculated from the TGA
measurement at NETZSCH while (X) are calculated from the TGA measurement at Innventia AB. The carbonization
was performed at 1 K/min to 1000°C.
0.00E+00
2.00E+07
4.00E+07
6.00E+07
8.00E+07
1.00E+08
0 100 200 300 400 500 600 700 800 900 1000 1100
Stre
ss (
Pa)
Temperature (°C)
4.00E+04
6.00E+04
8.00E+04
1.00E+05
0 100 200 300 400 500 600 700 800 900 1000 1100
Stre
ss (
Pa)
Temperature (°C)

52
3.4 FTIR Infrared spectroscopy was used to study the molecular changes that take place within lignin
fibers during carbonization. Samples that was carbonized to temperatures of 300, 400…
1200°C at 10 K/min were analyzed.
Since many volatile substances evaporate during pyrolysis, the study will mainly focus on the
loss of functional groups and molecules. This will hopefully give an idea of how the
structure/conformation is affected.
Figure 45 shows FTIR-spectra of pulverized and stabilized lignin, respectively. (Lignin
powder is the starting material when spinning fibers. It is melted and extruded into lignin
fibers, followed by stabilization.) Since no valuable information exists in the region of 1800-
2800 cm-1
, all spectra have been cropped. The section above 3000 cm-1
has also been removed
where a broad band 3100-3700 cm-1
with strong intensity was found. The band at that region
is attributed to O-H vibrations which probably indicate that water is present in the KBr-pellet.
It is also coupled with the peak at 1640 cm-1
. Furthermore, the absorption intensities in each
spectrum have been divided by the amount of sample used and all spectra have been baseline
corrected.
The spectroscopic study was hindered or limited by the fact that carbon fibers have a high
tendency of absorbing light (Boccara et al. 1997). The higher degree of carbonization, the
more the lignin fibers absorbed. In fact, amounts higher than 0.3 mg resulted in absorption
levels above 50 % for all frequencies at 900°C, which covered interesting peaks and features
completely. An implication of this was to use extremely low sample amounts as the
carbonization temperature rose. Unfortunately, the spectra become noisy at higher
temperatures as a result from this.
Figure 45: FTIR-spectra of pulverized and stabilized lignin, 3 mg sample per 300 mg KBr.
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
7009001100130015001700190021002300250027002900
Ab
sorp
tio
n
cm-1
Stabilized Lignin
Pulverized Lignin

53
Table 6: Assignment of bands of softwood lignin in the middle infrared region.
Peak (no.) Frequency of
absorption bands (cm-1)
Assignment/Interpretation
1 1695-1720 C=O stretch in unconjugated ketones, carbonyl, and in ester groups (Derkacheva, Sukhov 2008); conjugated aldehydes and carboxylic acids around and below 1700
cm-1 (Faix 1991) 2 1600 Aromatic skeletal vibration + C=O stretch (Faix 1991, Hergert 1960, Pandey 1999)
3 1515 Aromatic skeletal vibrations (Faix 1991, Hergert 1960)
4 1450-1465 C-H deformation, asymmetric deformation in -CH3 and -CH2- (Faix 1991)
5 1430 Aromatic ring stretching combined with C-H in plane deformation (Faix 1991)
6 1365-1370 Bending vibrations of C–H bond in C–CH3, in-plane bending vibrations of phenolic O–H groups (Faix 1991, Derkacheva, Sukhov 2008)
7 1270 Stretching of guaiacyl rings with C=O groups (Faix 1991, Derkacheva, Sukhov 2008, Agarwal, McSweeny & Ralph 2011)
8 1210-1220 C-C, C-O and C=O stretch; aromatic phenol (Faix 1991, Pandey 1999)
9 1205 Vibrations of aryl–O in aryl–OH and aryl–O–CH3 (Karmanov, Derkacheva 2013); vibrations of ring of guaiacyl ring with C=O mode (Derkacheva, Sukhov 2008,
Agarwal, McSweeny & Ralph 2011) 10 1150 Aromatic C-H in-plane deformation in the guaiacyl ring (Derkacheva, Sukhov 2008,
Kubo, Kadla 2005) 11 1136 A mode of coniferaldehyde (Agarwal, McSweeny & Ralph 2011)
12 1080 C-O deformation in secondary alcohols and aliphatic ethers (Pandey 1999, Faix 1991)
13 1033 C-O vibrations of aryl-O-CH3 and aryl-OH (Agarwal, McSweeny & Ralph 2011)
13 1030-1035 Aromatic C-H in-plane deformation of guaiacyl rings, C-O stretch in primary alcohols, unconjugated C=O stretch (Derkacheva, Sukhov 2008)
14 855 C-H out-of-plane stretch in position 2 of G-units (Norberg et al. 2013, Faix 1991)
15 822 C-H out-of-plane stretch in position 5 and 6 of G-units (Norberg et al. 2013, Faix 1991)
3000-2800 C-H Stretch in methyl and methylene groups
16 2940 C-H stretch in O-CH3, asymmetric (Karmanov, Derkacheva 2013)
17 2845 C-H stretch in O-CH3, symmetric (Karmanov, Derkacheva 2013)
Guaiacyl group (G-unit)

54
The peaks and band are pointed out as numbers in Figure 46 and are assigned vibrational
mode in Table 6. When comparing spectra of carbonized samples, the figure will serve as a
guide in this essay.
Although the softwood characteristics of the powder form are retained in the finger print
region (700-1800 cm-1
) after extrusion and stabilization, the spectra show some differences in
absorption. For instance, the first peak at 1715 cm-1
, attributed to vibrational stretching of
C=O in unconjugated carbonyl groups (Derkacheva, Sukhov 2008), has increased.
Skeletal breathing of aromatic rings is indicated by peak 2 and 3, at 1600 respectively 1515
cm-1
, typically for unconjugated guaiacyl units (Faix 1991, Hergert 1960, Pandey 1999). A
significant drop is observed for the latter peak in the stabilized lignin. The fifth peak can also
be assigned to skeletal vibration combined with stretching of the C-H bond in the same plane.
In between and around 1460 cm-1
, stretch in C-H bonds, asymmetrical for -CH3 and -CH2-
(Faix 1991), represented by peak 4.
In-plane deformation of phenolic O-H groups and vibrations of C-H in methyl groups is
associated with peak 6 and the band at 1365-1370 cm-1
(Faix 1991, Derkacheva, Sukhov
2008). The peak is found as a small but distinguishable shoulder in the stabilized lignin
spectrum. It is followed by another peak, no. 7 at 1270 cm-1
, that can be attributed to breathing
of the guaiacyl ring with a C=O group (Faix 1991, Derkacheva, Sukhov 2008, Agarwal,
McSweeny & Ralph 2011). In stabilized lignin this band has decreased.
The peak at no. 8, around 1210-1215 cm-1
, interpreted as deformations in C-C, C-O and C=O
bonds (Faix 1991, Pandey 1999), has disappeared completely in the stabilized lignin
spectrum. The same peak may also be attributed to phenolic units in the guaiacyl ring
(Hergert 1960). The next peak, no. 9 around 1200 cm-1
, is also likely associated with the
guaiacyl ring and phenolic modes (Derkacheva, Sukhov 2008, Agarwal, McSweeny & Ralph
2011).
The peak, no. 10, around 1150 cm-1
(Derkacheva, Sukhov 2008, Kubo, Kadla 2005) is
attributed to in-plane deformations of C-H bonds in guaiacyl rings and has increased for the
stabilized lignin. Similarly, the absorption intensity at peak no. 11, 1136 cm-1
, has increased.
However, little information is given in the literature about this peak. It may represent a mode
of coniferaldehyde (Agarwal, McSweeny & Ralph 2011).
The peak/shoulder no. 12 at 1080 cm-1
is probably caused by stretching in C-O bonds of
secondary alcohols and aliphatic ethers (Pandey 1999, Faix 1991) and is in higher absorption
intensity for stabilized lignin. No. 13, 1030-1035 cm-1
, is an indication of the aromaticity in
guaicyl rings as well as primary alcohols and C=O groups, and has also increased
(Derkacheva, Sukhov 2008).
Peak no. 14, at 855 cm-1
, and no. 15, at 822 cm-1
, are attributed to the out-of-plane C-H stretch
at position 2, respectively 5 and 6 of the guaiacyl ring (Norberg et al. 2013, Faix 1991).
A region of peaks is located between 2800 and 3000 cm-1
outside the fingerprint region. These
are associated with C-H stretching methyl and methylene groups, -CH3 and -CH2-. More

55
specifically, peak 16 and 17 belong to asymmetric respectively symmetric stretching of the C-
H bond in O-CH3 residues.
Figure 46: Spectrum of lignin in powder and stabilized fiber form. The peaks are interpreted in Table 6.
As the carbonization temperature increases, features and characteristics of the lignin spectrum
disappear (Figure 47, Figure 48 and Figure 49). Some decrease in intensity faster than others.
For instance, peak no. 1 diminishes steadily but still exists in the range between 300-500°C
and disappears completely after 500°C. In contrast, peak no. 3 is completely gone already at
500°C.
Peak no. 2 shifts slightly to a higher wavenumber. The redshift is possible due to
conformational changes in the aromatic rings.
The intensity of peak no. 4 falls in the same rate as no. 1. However, a faint signal is still
present at 600°C and becomes indistinguishable from noise at higher temperatures, indicated
by the bracket at 1300-1500 cm-1
in Figure 49 and Figure 50. Similar behavior is seen from
peak no. 5, which stays at the same level between 300 and 400°C.
Peak no. 7 diminishes into a shoulder at 500°C. The intensity of the band remains unchanged
thereafter but disappears completely at 900°C. The phenolic and methyl bending at peak 6
remains principally unchanged during the whole carbonization. It disappears however
temporary at 500°C.
The intensity of peak no. 9 decreases slow at first and becomes covered in absorption from
other residues at 500°C. Afterwards it continues to decrease along with adjacent bands. Peak
no. 10, 11 and 12 disappears in the same fashion, and then the intensity in the region starts to
increase after 600°C. Features of peak 10 and 11 are visible till 900°C. The spectrum of
280029003000
16
17
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
700800900100011001200130014001500160017001800
Ab
sorp
tio
n
cm-1
Stabilized Lignin
Pulverized Lignin1
2
3
4
5
6
7
8
9
10
11 12
13
14 15

56
900°C shows temporary a shoulder feature that represents the peak of 10. Somewhere around
900°C and forward the intensity of peak 12 is above that of stabilized lignin. Consider
however that the increasing intensity may arise from an uneven or faulty baseline correction,
resulting in a magnification of the peak.
Peak no. 13, as with no. 10, 11 and 12, finds a stationary point between 300 and 400°C. At
500°C a new peak (A) arises, but it may belong to peak 13, just shifted slightly.
The features of peak no. 14 and 15 remain fairly constant throughout the whole carbonization.
The intensity drops between the stabilized state and 300°C then stays principally unchanged.
No. 15 shifts however slightly to 800 cm-1
at 700°C. Note, that the region of 700-900 cm-1
,
indicated by the bracket in Figure 49 and Figure 50, is very noisy at high carbonization
temperatures and thus becomes hard to evaluate.
Figure 47: Significant changes in intensity of peaks indicated by lines. For the spectra of 300, 400 and 500°C, 0.33,
0.28 and 0.35 mg fiber sample was used, respectively.
280029003000
0
0.1
0.2
700800900100011001200130014001500160017001800
Ab
sorp
tio
n
cm-1
Stabilized Lignin 300°C 400°C 500°C
1 3 4
5
7
9
10 11
12 13
14 15
A
2
6
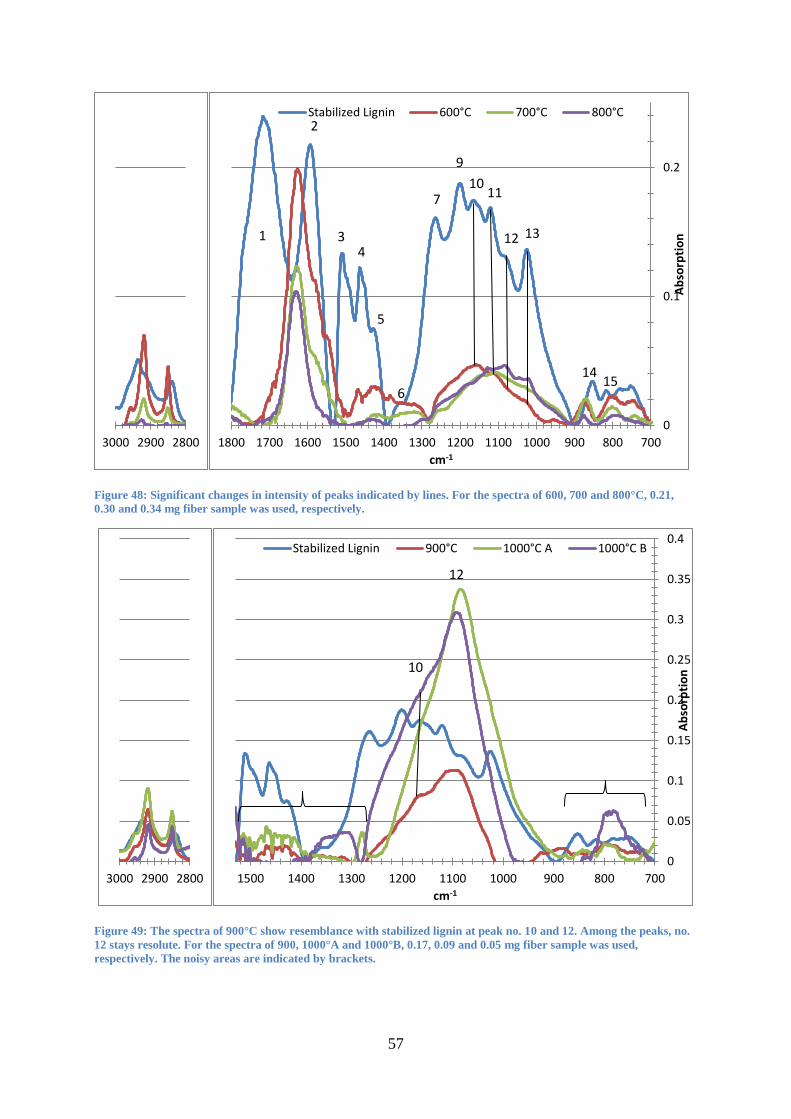
57
Figure 48: Significant changes in intensity of peaks indicated by lines. For the spectra of 600, 700 and 800°C, 0.21,
0.30 and 0.34 mg fiber sample was used, respectively.
Figure 49: The spectra of 900°C show resemblance with stabilized lignin at peak no. 10 and 12. Among the peaks, no.
12 stays resolute. For the spectra of 900, 1000°A and 1000°B, 0.17, 0.09 and 0.05 mg fiber sample was used,
respectively. The noisy areas are indicated by brackets.
280029003000
0
0.1
0.2
700800900100011001200130014001500160017001800
Ab
sorp
tio
n
cm-1
Stabilized Lignin 600°C 700°C 800°C
1 3 4
5
7
9
10 11
12 13
14 15
2
6
280029003000
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
0.4
700800900100011001200130014001500
Ab
sorp
tio
n
cm-1
Stabilized Lignin 900°C 1000°C A 1000°C B
12
10
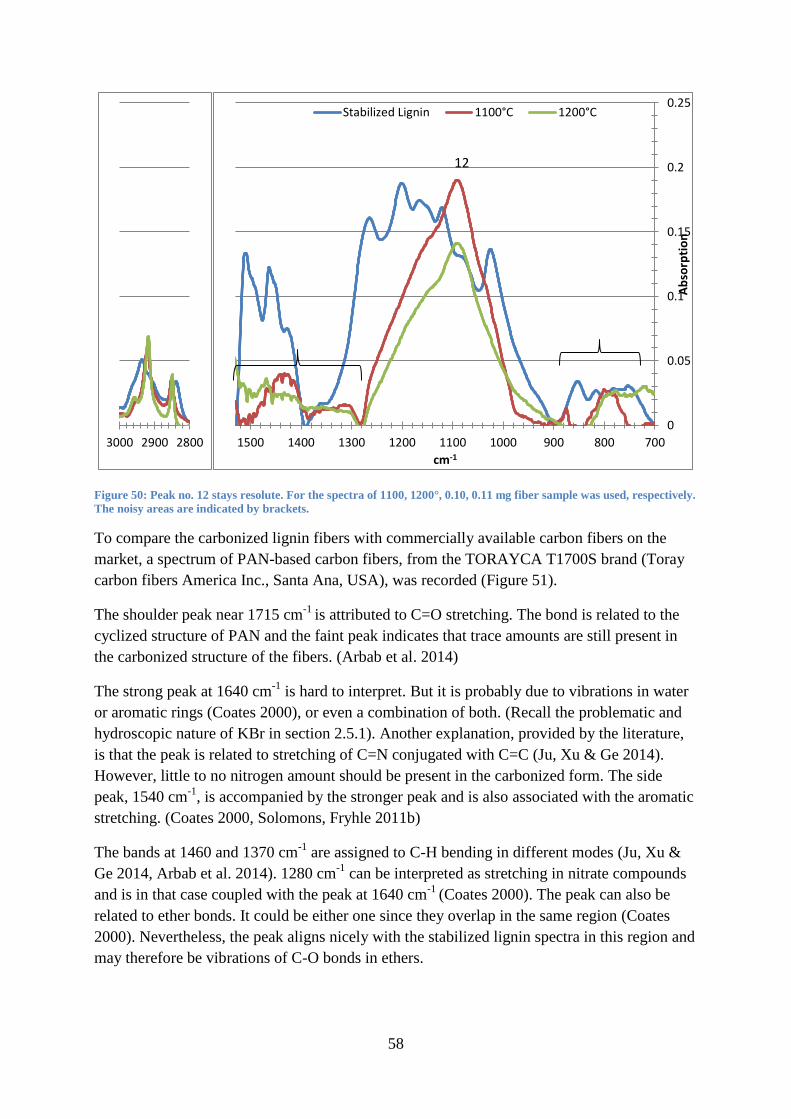
58
Figure 50: Peak no. 12 stays resolute. For the spectra of 1100, 1200°, 0.10, 0.11 mg fiber sample was used, respectively.
The noisy areas are indicated by brackets.
To compare the carbonized lignin fibers with commercially available carbon fibers on the
market, a spectrum of PAN-based carbon fibers, from the TORAYCA T1700S brand (Toray
carbon fibers America Inc., Santa Ana, USA), was recorded (Figure 51).
The shoulder peak near 1715 cm-1
is attributed to C=O stretching. The bond is related to the
cyclized structure of PAN and the faint peak indicates that trace amounts are still present in
the carbonized structure of the fibers. (Arbab et al. 2014)
The strong peak at 1640 cm-1
is hard to interpret. But it is probably due to vibrations in water
or aromatic rings (Coates 2000), or even a combination of both. (Recall the problematic and
hydroscopic nature of KBr in section 2.5.1). Another explanation, provided by the literature,
is that the peak is related to stretching of C=N conjugated with C=C (Ju, Xu & Ge 2014).
However, little to no nitrogen amount should be present in the carbonized form. The side
peak, 1540 cm-1
, is accompanied by the stronger peak and is also associated with the aromatic
stretching. (Coates 2000, Solomons, Fryhle 2011b)
The bands at 1460 and 1370 cm-1
are assigned to C-H bending in different modes (Ju, Xu &
Ge 2014, Arbab et al. 2014). 1280 cm-1
can be interpreted as stretching in nitrate compounds
and is in that case coupled with the peak at 1640 cm-1
(Coates 2000). The peak can also be
related to ether bonds. It could be either one since they overlap in the same region (Coates
2000). Nevertheless, the peak aligns nicely with the stabilized lignin spectra in this region and
may therefore be vibrations of C-O bonds in ethers.
280029003000
0
0.05
0.1
0.15
0.2
0.25
700800900100011001200130014001500
Ab
sorp
tio
n
cm-1
Stabilized Lignin 1100°C 1200°C
12

59
The region between 1000 and 1200 cm-1
is a result of stretching in alcohols and aliphatic
ethers (Solomons, Fryhle 2011b). Regardless, the peak at 1068 cm-1
is attributed to C-H
bending (Arbab et al. 2014).
Similarly as lignin, peaks are located around 835 cm-1
in the spectrum. They represent out-of-
plane deformation in C-H bonds in mono or disubstituted aromatic rings (Coates 2000,
Solomons, Fryhle 2011b).
The area between 2800 and 3000 cm-1
are related to stretching vibrations of C-H. .
Figure 51: Spectrum of PAN-based carbon fibers. Interesting peaks are indicated by wavenumber (cm-1). 0.09 mg
sample was used.
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
7009001100130015001700190021002300250027002900
Ab
sorp
tio
n
cm-1
TORAYCA T1700S
2850
2918
1716
1643
1540
1460 1370
1280
1068
835

60
3.5 SEM SEM enabled high resolution imaging of a few selected fibers. All fibers pictured below were
also analyzed in ESD. For comparison, the light microscopy images from the same batch as
the following fibers are found in appendix 8.2.4.2.
The interaction volume is a couple of micrometers deep into the material from the surface.
3.5.1 Imaging
The aim of the analysis was to inspect the fiber structure prior and after carbonization to
certain temperatures as well as the effect of the heating rate and applied load. The images was
acquisitioned by a backscatter detector, therefore (as mentioned in 2.6) contrasts indicate
different chemical composition.
To compare the carbonized lignin fibers with commercially available carbon fibers, an image
of TORAYCE T1700S (Toray carbon fibers America Inc., Santa Ana, USA), was acquired.
These PAN-derived carbon fibers are presented in Figure 52.
Contamination (bright spots) is clearly visible on the surface. Analysis of the particles is
presented below, in section 3.5.2. Nevertheless, the fibers seem very uniform in shape and
have no visible defects whatsoever.
Figure 52: SEM image of TORAYCE T1700S carbon fibers. The diameters are (upper) 7.07 µm and (lower) 6.99 µm.
In Figure 53 below, the fiber axis and the cross-section of stabilized lignin is pictured. The
intensity from the axial surface is brighter in comparison with T1700S. The higher intensity
might be caused by higher proportions of oxygen in the surface compared to carbon, since O
has a higher atomic number than C. The slightly noisy appearance may be caused by uneven
distribution of different compounds across the surface.
A few contaminants (white dots) are also found, although smaller in size. When handling the
lignin fibers, carbonized as stabilized, they became easily charged with static electricity. As a
consequence they probably attracted small dust particles, hence the contamination seen in the
following images.

61
Figure 53: SEM images of stabilized lignin fibers. Image a) and b) shows the surface of fibers in the axial direction
while c) shows the cross-section. The diameter of the fiber is in a) 50.7 µm b) 55.6 µm and c) 39.9 µm.
The intensity is still very bright after carbonization to 300°C, indicating that the material
changed little in composition (Figure 54).
Protuberance is often seen in the images, for example in Figure 54 and Figure 56 a. It is an
artifact produced in the extrusion and results unfortunately in fibers that are not entirely
uniform in shape.
Figure 54: SEM image of a fiber carbonized at 1 K/min to 300°C. The image shows the surface of fibers in the axial
direction. The smaller diameter (to the left) of the fiber is 41.4 µm while the protuberance (to the right) is 50.3 µm.
The intensity has decreased in the 500°C carbonization sample and the fiber has become
darker (Figure 55). Furthermore, the static seems to have disappeared and the signal from the
surface is more uniform in intensity (excluding contaminating particles).
The cross-section, Figure 55 b, displays a fiber that appears to be very grainy.
a) b)
c)

62
Figure 55: SEM images of fibers carbonized at 1 K/min to 500°C. Image a) shows the surface of fibers in the axial
direction while b) shows the cross-section. The diameter of the fiber is in a) 50.2 µm and b) 50.6 µm.
As the temperature increases to 1000°C we start to see porosities in the fibers (Figure 56). An
extreme case is displayed in Figure 56 a), where the fiber is covered with deep pinholes.
Image b) and c), on the other hand, shows minor distribution of pores across the surfaces.
Figure 56: SEM images of fibers carbonized at 1 K/min to 1000°C. Image a), b) and c) shows the surface of fibers in
the axial direction while d) shows the cross-section. The diameter of the fiber (and bulge in parenthesis) is in a) 39.3
µm (70.0 µm), b) 35.9 µm, c) 37.1 µm (43.4 µm) and d) 37.2 µm.
The pores become more distinguishable when the carbonization temperature reaches 1200°C
(Figure 57). When comparing the heating of 10 and 40 K/min in b) and c), respectively, no
differences are apparent. In contrast, the slowest heating, 1 K/min in a), appears to have
affected the fiber structure the most.
a) b)
a) b)
c) d)

63
Figure 57: SEM images of fibers carbonized at a) 1 K/min, b) 10 K/min and c) 40 K/min to 1200°C. Image a), b) and c)
shows the surface of fibers in the axial direction while d) shows the cross-section. The diameter of the fiber (and bulge
in parenthesis) is in a) 33.0 µm (43.7 µm), b) 30.6 µm (38.5 µm) and c) 42.4 µm.
Next, we evaluate fibers from sample A and B (recall section 3.1.2) in Figure 58. Almost no
pores are found in the fibers which retain a uniform shape and composition. The cross-section
c) hints of a slightly bright circle in the periphery. This might indicate a skin-core formation
that contains more oxygen than the interior.
a) b)
c)

64
Figure 58: SEM images of fibers carbonized at 10 K/min to 1000°C under increasing load from 10 mN to 300 mN
between 375-975°C in a) and to 400 mN between 350-950°C in b) and c). Image a) and b) shows the surface of fibers
from sample A respectively B in the axial direction while c) shows the cross-section of a fiber from sample B. The
diameter of the fiber is in a) 26.6 µm, b) 36.4 µm (left) 37.1 µm (right) and c) 26.8 µm.
3.5.2 Energy-dispersive X-ray spectroscopy
The EDS-scan was performed over three areas:
The fiber axis, rectangle Figure 59 a).
The whole cross-section, circle in Figure 59 b).
The inner section of the cross-section, square in Figure 59 b).
Figure 59: a) The scanned area along the fiber axis is indicated by the rectangle. b) The circle indicates the scan over
the whole cross-section and the square the inner section. The fiber in a) and b) is the same as in b) resp. c) in Figure
56.
a) b)
c)
a) b)

65
In this fashion, spectra were gathered from all fibers pictured in the previous section 3.5.1 and
some that were purely for the EDS-study. A few examples of spectra are found in Figure 60,
which were collected from the fibers in Figure 56.
Figure 60: EDS-spectra generated from the scanned areas above, Figure 59, showing Kα-energies from emission in
various elements in the fiber. Spectrum 8 belongs to the axial scan, spectrum 36 to the cross-sectional area and
spectrum 37 the inner section.
The main elements found in the fibers were carbon, oxygen and sulfur. The O and C relate to
the lignin molecule itself, whereas S residues are remains from the kraft pulping process
(section 1.2.3.1).
In some instances, trace amount of inorganic species, among silicon, alumina, chlorine and
potassium (rare), was also identified, which rarely exceeded 0.5 wt%. The Al originates
probably from alumina foil that was used to protect and store lignin fibers, i.e. foil was
wrapped around the fibers. The K and Cl are probably constituents in salts and are not part of
the lignin molecule. The salts could originate from the skin when handling the fibers, but this
is highly speculative.

66
X-ray analysis of the particles that cover the surface of the PAN-derived carbon fibers in
Figure 52 shows that main constituents are potassium and chlorine. Without any doubt the
particles are composed of potassium chloride (KCl) crystals.
Elemental analysis of the contaminant on the cross-section in Figure 58 shows a large variety
of elements, among Si, Al, Ca and Na.
From the spectral analysis along the fiber axis, the average elemental composition of the
fibers, sorted by carbonization temperature, was put into the chart seen in Figure 61. The
proportion of oxygen is observed to decrease with carbonization temperature. Meanwhile, the
relative carbon content increases passively. Note that the amounts of carbon do not increase
during the carbonization but rather increase proportionally to the entire content. This means
that the majority of the carbon structure stays resolute to temperature changes whereas
unstable, weakly bonded or volatile compounds are eliminated from the polymer
macromolecules and evaporate. Oxygen containing compounds are likely to disappear from
the fiber.
The relative sulfur content changed little during the carbonization and may indicate that the S
atoms are strongly attached to the polymer macromolecule with covalent bonds.
In order to be classified as carbon fibers, the amount of carbon must at least be 90 wt%
(section 1.2). The lignin carbon fibers do not quite reach the bar. On the other hand, the PAN-
derived carbon fibers barely meet the criterion themselves. The chemical composition is
specified to 93 % in the datasheet of T1700S. According to the analysis of the atomic weight
this appears to be true.

67
Figure 61: Relative content of carbon (blue), oxygen (red), and sulfur (green) after carbonization is indicated as a) the
weight percentage (Wt %) and b) atomic percentage (At %).
Data from compositional analysis of the cross-sections is gathered in Figure 62. For all
carbonization states we observe that the carbon content is greater in the interior of the fibers,
followed by slightly less in the whole cross-section. The conclusion is that the oxygen is
generally higher on the edges or in the periphery of the fiber cross-section. In all instances the
0
10
20
30
40
50
60
70
80
90
100
StabilizedLignin
300°C 500°C 1000°C 1200°C TORAYCAT1700S
Wt
%
C O S
a)
0
10
20
30
40
50
60
70
80
90
100
StabilizedLignin
300°C 500°C 1000°C 1200°C TORAYCAT1700S
At
%
C O S
b)

68
highest amount of oxygen is found on the fiber axis surface along with the lowest amount of
carbon.
The amount of carbon in the interior of the 1000°C carbonized fibers actually meets the
requirement of 90 wt% carbon.
Figure 62: Relative content of carbon (blue), oxygen (red), and sulfur (green). The full colored pile represents the
composition of the axial surface of the fiber, horizontally striped piles the cross-section and diagonally dashed piles
the inner square.
Compositional analysis was also carried out for the fibers carbonized under increasing force
(Figure 63). Although the fiber of sample B contains slightly more carbon than the fiber of
sample A, the content is essentially the same for both. The level at the axis surface is however
higher, in both cases, than that of fibers carbonized under static load. The interior carbon
content is noticeable, a few percent, higher as well.
The fiber of sample B fulfills the 90 wt% carbon requirement in all aspects.
0
10
20
30
40
50
60
70
80
90
100
Stabilized Lignin 500°C 1000°C
Wt
%
C O S Axial Cross-section Inner Square

69
Figure 63: Relative content of carbon (blue), oxygen (red), and sulfur (green) in lignin fibers carbonized under
increasing load.
3.6 Light Microscopy The lignin fibers were studied in microscope in order to observe the degradation and
structural change that took place during carbonization. The structural integrity of the surface
correlates to strength. So any information regarding defects would for instance explain
premature rupture in tensile tests (i.e. low tensile strength) or even in the TMA-carbonization
itself. A newer series can be found appendix 8.2.4.1.
If not stated otherwise all images were taken with a magnification of x50 with focus adjusted
to the top of the fiber. However, since the microscope was configured with an improper
calibration when measuring the fibers, the scales indicated in the images are somewhat
misleading. In reality the length, represented by the scale, should be about 20-25 µm less than
indicated (estimated from the newer series).
Nevertheless, the same incorrect calibration and resolution was used for all measurements and
images, hence the diameter differences between carbonized and stabilized lignin fiber could
be calculated (Table 7)
When evaluating the images, the surface of the stabilized fibers is moderately glossy and
reflects plenty of light that is incident from above. The fibers also appear grainy but the
surface is still smooth (Figure 64).
0
10
20
30
40
50
60
70
80
90
100
A Axial B Axial B Outer Cross-section
B Inner Square Axial 1000°C
Wt
%
C O S

70
Figure 64: Individual stabilized lignin fibers. The magnification in the image to the right is x20.
The appearance is maintained during carbonization up to 500-600°C where small cavities or
craters start to form (Figure 65 and Figure 66). The craters gradually increase in amount and
at 800-900°C they overwhelm the surface which seems porous and rough at this point (Figure
67 and Figure 68). Although plenty in number, the craters do not appear to be very deep. The
fibers also lose the glossy appearance and reflect less light, in other words they become
darker.
Figure 65: Individual lignin carbon fibers carbonized to 500°C.
Figure 66: Individual lignin carbon fibers carbonized to 600°C.
100 µm 300 µm
100 µm
100 µm 100 µm
100 µm 100 µm

71
Figure 67: Individual lignin carbon fibers carbonized to 800°C.
Figure 68: Individual lignin carbon fibers carbonized to 900°C.
At 1000°C and onward the contrast of the fibers is extremely dark (Figure 69, Figure 70 and
Figure 71). The craters are still present, although larger in diameter.
Figure 69: Individual lignin carbon fibers carbonized to 1000°C.
100 µm 100 µm
100 µm 100 µm
100 µm 100 µm

72
Figure 70: Individual lignin carbon fibers carbonized to 1100°C.
Figure 71: Individual lignin carbon fibers carbonized to 1200°C.
An explanation for the crater formations may be that the surface reacts with something during
the carbonization which causes gouging. For example, presence of oxygen in the TMA-
system or contamination of particles on the fiber surface.
Never the less, the size of the fiber deteriorates with increasing temperature (Table 7). At first
the diameter remains principally the same. After 500°C the change is starting to get
noticeable.
If compared with the values that were calculated from the TGA-measurements, the change in
percent roughly match each other, give or take a few units (Figure 42; Red triangles).
100 µm 100 µm
100 µm 100 µm

73
Table 7: Displaying average diameters of stabilized respectively carbonized lignin fibers, sample standard deviation
and number of samples used in the measurements. The percentage relate to the remaining diameter after
carbonization. Since the microscope was not correctly calibrated during the diameter measurement of the fibers
carbonized to 400, 500 and 1000°C, the data for those are not displayed. However, the same, yet wrong, calibration
was used thus the residual diameter could be calculated.
Temperature (°C)
Heating (K/min)
Average Diameter (µm)
Sample Standard Deviation (µm)
Residual Diameter
(%)
Data Points
Stabilized Lignin
0 47.1 5.91 100 30
300 10 46.6 5.35 99.0 30
400 1 N/A N/A 95.1 6
500 1 N/A N/A 93.4 3
600 10 39.8 3.47 84.5 29
900 10 38.1 4.91 80.9 29
1000 1 N/A N/A 75.8 14

74
4 Discussion Previous results, interesting aspects of the future, importance in broad sense and ethics are
discussed in this section.
4.1 Analysis of the main results The results are discussed in detail in each topic, ordered after method and analysis.
4.1.1 TMA
The TMA measurement of the carbonization shows that the fiber deformation is only
temperature dependent. Different heating rates, at least for values between 1 and 40 K/min, do
not seem to affect the deformation pattern (Figure 24). The deformation per temperature
neither increases nor decreases when varying the rate of heating.
The deformation occurs in three steps as shown by the curve in Figure 21. The extension
coefficient α curve indicates maximum deformation at 375, 530 and 650°C. Analogous to the
TGA measurement the loss of mass also occur in three steps, where the derivative maximums
are found at 425, 560 and 785°C (Figure 41).
Variations of the deformation were observed (Figure 23). Samples with 50 fibers tended to
follow the same pattern whereas samples with fewer fibers did not. Logically, the samples
with fewer fibers would contract less compared to the samples with higher amount, due to the
static force exerted on the fibers. Small variations are possible since lignin fibers are
amorphous, but these strong variations have other explanations. The behavior could for
instance be attributed to mounting and/or sample preparation.
Furthermore, the deformation comes to a standstill above 1000°C. Probably a sign that the
fiber is fully carbonized, meaning that remaining molecular compounds in the fiber are stable
enough to withstand the temperature (i.e. further evaporation of volatile substances and
compounds will not occur) and that the structure of the lignin polymers is sufficiently cross-
linked and has retained a strong conformation that takes up as little space as possible (i.e. the
fiber will not contract more), preferable aromatic in nature.
4.1.2 Dia-Stron
The overall stability of the carbonized lignin fibers is by this assumption stronger than the
non-carbonized lignin fiber. The Dia-Stron results support this by showing the improved
mechanical properties of the fibers. Compare the E-modulus and tensile strength (around 36
GPA, and 600 MPa, respectively) of fibers that underwent carbonization to 1000°C to the E-
modulus and tensile strength (around 5 GPa and 150 MPa, respectively) of the stabilized
lignin fibers.
Along with deformation, the rate of heating did not affect the mechanical strength properties
either. The E-modulus and tensile strength remained principally the same for all rates,
although 1 K/min were slightly better. Since rate did not matter much, the carbonization rate
was chosen to 10 K/min for practical production of samples to, for instance, the Dia-Stron and
FTIR-spectroscopy. A carbonization to 1000°C at 1 K/min would take approximatively 16
hours whereas a 10 K/min carbonization would take only 2 hours (including cooling).

75
When the load was increased to compensate for the contraction, the carbonized lignin fibers
showed result of improved E-modulus, they became stiffer in other words. Compared to the
average value for carbonization under static load, 36 GPa, the E-modulus reached values
around 50 GPa. As for the tensile strength, the change was too insignificant to be considered
an improvement.
Depending on the force and interval, where the load was applied, the stiffness of the carbon
fibers could be adjusted (Table 5). Not necessarily did higher loads mean higher E-modulus.
On the contrary, fibers that had been applied an slowly increasing force over a long interval
(300-900°C) to a high final load had approximatively the same E-modulus as fibers that had
been applied a rapidly increasing force over a small interval (400-600°C) to a low final load
(Compare sample G & H against E & F, Table 5).
By studying the deformation, fibers that had contracted least showed better E-modulus than
those who had contracted more. The best result was achieved for fibers that stayed at positive
length during the entire carbonization (above 0% in Figure 27). It seems that the restriction of
the fiber contraction during the carbonization has a larger impact rather than the force itself.
In this regard, the ramping of force was preferable started early in the carbonization (around
300°C) where the fibers still underwent expansion and were susceptible to load. This
compensated for the tendency to contract early on and resulted in a lower final contraction.
Another way to regulate the contraction was to successively increase the force elevation in
steps as soon as the fibers tended to contract (Figure 30).
It may even be of interest to test applied load as early as the stabilization, since some sintering
takes place there as well.
Regarding the load, the fibers were very sensitive to increasing stress during the
carbonization. Many TMA carbonizations failed due to this. Figure 30 shows two examples.
The E-modulus of those fibers that ruptured mid carbonization was essentially the same as
that of fibers that had been carbonized under low static force. Low rates of increasing force
are therefore necessary.
The following discussion regarding improved mechanical properties is only speculative from
my behalf. The improved stiffness may be attributed to increased alignment of the lignin
polymer aromatic chains. Initially the lignin polymers are somewhat randomly distributed in
the fiber, i.e. the order is random and the fiber is considered amorphous. However, it is not
excluded that some molecular orientation may be present. During the carbonization,
aromatization or cyclization of the polymer chains is thought to occur and due to the random
organization the resulting carbon fiber will achieve turbostratic ordering.(Beste 2014) By
instead applying an increasing load on the fibers in the process, the polymers are forced into a
more aligned structure as they react and undergo conformational changes, facilitating cross-
linking and aromatization along the direction of the fiber axis. Compare with a tangled metal
chain that unfolds and becomes straightened as you pull it in both ends. The fibers are
probably still turbostratic although to a less extent.

76
In comparison, graphitic and ordered MPP has generally higher E-modulus compared to the
turbostratic PAN (Liu, Kumar 2012, Huang 2009). This suggests that the lignin carbon fibers
have obtained a conformation that is similar to graphitic.
Under almost negligible force, the fibers contract unhindered during carbonization, mainly
due to the conformational rearrangement that occurs intra- and intermolecularly in the
polymer molecule structure and polymer network, respectively. Participating in the
rearrangement are free-radical, cross-linking, condensation, cyclization and aromatization
reactions. The molecular structure becomes more tightly bonded and more stable in every
direction, i.e. in and between polymers constituting the fibers (Figure 72 a).
By increasing the load successively the fiber contraction is hindered. The applied load in the
direction of the fiber axis straightens out the polymer chains, facilitating the forming of
favorable conformations along the chains and the fiber axis whereas adjacent conformational
reactions are unfavorable and thus inhibited by the force (Figure 72 b).
Figure 72: Simple depiction of the intramolecular conformational changes. Thick lines represent polymer chains while
thin lines represent molecular structures (e.g. cross-links, conjugated aromatic and cyclic structures, aliphatic ethers
etc.) that form during carbonization and bind the chain together into a new conformation. In a) conformational
reactions occur freely in every direction whereas in b), the reactions are only favored in the direction of the force. In
reality one has to account for the random ordering and branching of the polymers as well as the entire polymer
network.
Another theory is that the force breaks apart weak structures during the carbonization. The
strongest remain while the broken structures, which cannot undergo further cross-linking,
simply decompose by the heat and evaporate.
4.1.3 TGA
Without any doubt partial combustion occurred during the TGA measurement of the
carbonization at Innventia AB. Since combustion is caused by oxidation reactions that take
place within the fiber the mass temporarily increases slightly (oxygen react and form
temporary intermediates). This can be observed as a convex shape or bulge in the curve
between 450 and 720°C in Figure 40, where the mass still decrease although slightly less.
(Vaimakis 2013)
4.1.4 FTIR
The FTIR-study proved to be quite difficult to implement, since carbon fibers are in general a
material with high absorption. Even a little amount of carbon fiber resulted in low
transmittance.
a) b) Force

77
In order to work around this problem, extremely small amounts of sample were used in this
study. In contrast to the normal ratio of 3 mg sample per 300 mg KBr in the pellet,
approximatively a tenth of the recommended sample amount was used. Implications resulting
from this were that the signal became indistinguishable from noise at some frequencies and
the intensity of water became dominant in the spectra, especially at higher degree of
carbonization (i.e. higher carbonization temperature) where even lower amounts were used.
The peak at 1600 cm-1
, attributed to aromatic skeleton, became masked by the dominant
intensity of water around 1640 cm-1
in this respect. (Most likely water, since a broad band
with large intensity was found at 3100-3700 cm-1
in all spectra.)
According the literature, main compounds released during the carbonization are CH4, water,
CO2 and CH3OH. (Li et al. 2013) Mainly oxygen containing compounds are released during
the carbonization. Water is probably released in the early stage of carbonization, accounting
for the largest loss of mass.
The carbonization involves dehydrogenation and decarboxylation reactions in combination
with condensation and aromatization, releasing nearly all non-carbon elements (Chatterjee et
al. 2014). Other weakly bonded side groups of the lignin polymer is probably eliminated
through homolysis and evaporated during the carbonization.
Homolytic cleavage of linkages constituting the polymer chain also occurs, resulting in
radical formations that allow for rearrangement of the lignin molecule conformation. For
instance, the most commonly and also one of the weakest linkages, the β-O-4 linkage is likely
involved in the radical reactions. (Beste 2014)
Furthermore, oxygen containing functional groups and linkages in lignin may supposedly
facilitate cyclization steps that transform weak ether and rotatable aliphatic linkages into
stable and stiff aromatic units. (Beste 2014)
In the FTIR series from 300 to 1200°C, nearly all features characteristic to lignin vanished as
the carbonization temperature increased. Although many compounds dissociated from the
originally stabilized lignin, some compounds endured the temperature changes and was
removed slower than other.
The decarboxylation and release of CO2 is indicated by the fast decreasing intensity of 1710
cm-1
between 300°C and 500°C (Peak 1 Figure 47) (Huang, Ma & Zhao 2015). As indicated
by the reduced bands at 1365-1370, 1210-1220, 1080 and 1030-1035 cm-1
, phenolic hydroxyl,
hydroxymethyl and ordinary hydroxyl groups were consumed in condensation reactions
during the early carbonization (Peak 1 Figure 47), contributing to release of water and ether
formation. The increasing absorbance intensity of 1080 cm-1
, attributed to aliphatic ethers
(Peak 12 Figure 49-Figure 50), demonstrates that ether structures increase after 900°C,
connecting aromatic rings together. (Huang, Ma & Zhao 2015) Some ether bond probably
originated from linkages (β-O-4 and 4-O-5 for instance), which may be the reason that the
intensity never faded out in the region. (Beste 2014)

78
Another spectral feature that remained after full carbonization to 1000°C, was in-plane
deformation of C-H in guaiacyl rings at 1150 cm-1
and 1030-1035 cm-1
(Peak 10 and 13 in
Figure 47-Figure 50). The aromatic structure is thought to remain and even increase during
the carbonization and the result points in this direction. Peak 10 and 13 actually increases in
absorbance after 900°C.
However, other results points in the opposite direction. The intensity of the band regions at
1515 cm-1
and 1430 cm-1
, attributed to breathing of aromatic rings with contribution of in-
plane deformation of C-H in the latter, decreased (Peak 3 and 5, respectively, in Figure 47-
Figure 50). The first band decreased faster than the second until it had disappeared completely
at 600°C whereas the second band still remained, but may be attributed to noise, after 600°C.
As for the peak at 1600 cm-1
, also attributed to breathing of aromatic rings, it is concealed
under the dominant water intensity in the region and thus cannot be fully evaluated.
4.1.5 SEM
The EDS analysis along with FTIR confirms the fact many that oxygen containing
compounds dissociate and disappear though evaporation during carbonization, as indicated by
the decreasing amount of oxygen.
SEM imaging shows that the fibers darken during the carbonization. Pinholes formations
appear at high carbonization temperature which may be the same formations shown in the
light microscope imaging.
The surface of the fibers seems oxygen rich in content, as indicated by comparisons between
inner cross-sections and edges/surfaces (Figure 62). Skin-core structures are therefore most
likely present in the fibers which was stated in the literature (section 1.2.3.3).
4.1.6 Microscopy
Defects and the seemingly porous surface probably weakened the carbonized lignin fibers.
The wide spread in the tensile strength graphs are most likely a testament to this (Figure 37).
According the literature pores and defects could arise from elimination of heteroatoms
(section 1.2.3.3). In our case the SEM-study imply that the fiber surface is rich in oxygen
content which would explain the porous appearance after carbonization.
4.2 Impact in a broad sense The performance of lignin carbon fibers is not nearly close to that of commercially available
carbon fibers. In the sense of progress towards obtaining carbon fibers of lignin suitable for
the industrial use, a long way still remains.
However, the improved mechanical performance that was achieved and presented, in this
work, for stabilized lignin fibers that underwent carbonization under increasing load is a step
in the right direction. The information is beneficial regarding future work in discovering
better parameters in order to improve the mechanical strengths. In that aspect, this work
would serve as a starting point.
In the sole purpose of the project, the carbonization was focused on small scale production
where a few fibers were carbonized at a time. In mass-production, however, where large

79
quantities of fibers are carbonized, one has to consider that the heat and energy needs to be
high enough for carbonization to take place. The speed of the carbonization is also of great
importance.
The information regarding heating rate is therefore of value in industrial applications. As
indicated, the rate of heating does not matter considerably for the mechanical strength
properties. If one wants to produce large quantities of carbon fibers from lignin, the speed of
the process is only limited by heating rate and carbonization temperature, meaning that the
process can be made as fast as the heating allows for.
When and if the problems of the production of lignin carbon fibers are solved, the prices of
carbon fiber will plummet, since there is an abundance of lignin in the pulping process. In this
case strong reinforced carbon fiber composites could be manufactured cheaply and would, for
instance, greatly benefit production of low weight vehicles which in turn would lower the fuel
consumption, where economical resources can be saved. Lower fuel consumption would also
lead to a healthier ecological environment. Needless to say, the transport industry would
greatly benefit from this.
4.3 Ethical implications in a broad sense As mentioned above, carbon fiber production from lignin would have a lot of positive
consequences. However, and this is highly speculative from my part, the demand of low cost
carbon fibers would most likely increase as a result. In that case one has to consider the risk of
increased deforestation in order to supply with lignin for carbon fiber production. Even
though lignin is considered a by-product today, it may be the main product from wood
tomorrow.
In the worst case scenario, the pulping process would be so focused on extracting lignin that
the other components, e.g. for paper production, would simply be disregarded as waste. The
tables would have been turned. The full potential of wood would not be utilized and a lot of
resources would go to waste.
Deforestation affects the nature and ecological systems. The topic is always under debate,
especially when talking about the rainforests.
The ever increasing use of tablets, smartphones etc. in today’s society, will render
newspapers, books, papers etc. obsolete in the near future. The trend will probably cause the
production of paper materials to decrease by the looks of it. Therefore it would be imperative
to find another uses for the pulping process. If lignin carbon fibers can be improved, it would
solve the problem.
4.4 Future perspectives
More research must be put into improving the mechanical performance of lignin carbon
fibers. In order to do so, the carbonization process must be fully understood. Today, little
information is known about the chemical reactions and conformational rearrangements that
take place which would be an interesting aspect to study in future research. The FTIR study in
this work did not fully answer the questions.

80
Further testing of load is another interesting aspect. The force program must be optimized so
that unnecessary rupture of the fibers is inhibited. In addition, large expansion of the fibers is
something to consider in respect to how the mechanical performance would be affected.
Moreover, it would be interesting to experiment with different loads during the stabilization
as well.

81
5 Conclusion The main goal of the project was to optimize the carbonization process (compare section 8.1.2
and 1.1) and to obtain carbon fibers with good mechanical properties. Recommendations for
the parameters of interest are as follows:
Since the rate of temperature elevation did not affect the strength properties
considerably much, i.e. changing the rate neither improved nor impaired the
mechanical strength, any rate can be selected solely in the purpose and suitability of
the production. In this work 10 K/min proved useful but an increased rate would
benefit faster carbon fiber production from stabilized lignin. However, 1 K/min may
be to slow and may induce defects in the carbon fibers as seen in the light microscopy
and SEM imaging. For small scale production 10 K/min is recommended.
Regarding carbonization temperature, the E-modulus and tensile strength reached its
maximum around 1000°C. The fiber contraction also stopped at that temperature.
Carbonization to higher temperatures is possible, but there is a risk that the fibers may
become severely damaged, especially at the surface, affecting the tensile strength. A
temperature around 1000°C is therefore recommended.
The E-modulus was affected considerably by applying an increasing load during the
carbonization, which hindered the contraction of the fibers. When the dimensional
change was close to 0 % the carbonized lignin fibers had and E-modulus of about 50
GPa, an improvement of 14 units from 36 GPa. The recommendation here is to apply a
force early in the carbonization process that inhibits the contraction as much as
possible. For instance, from 0.01 to 0.35 N between 300 and 900°C, for approximately
5 fibers.
Although the mechanical strength was improved, the lignin carbon fibers are still not suitable
for industrial use. The improvement is however a step in the right direction and would serve
as a guideline for further load testing during carbonization. Nevertheless, these
recommendations should optimize the production of lignin carbon fibers regardless of
mechanical strength. In sense of optimization, the goal was achieved!
The EDS analysis revealed that much oxygen was removed from the lignin during the
carbonization process. The FTIR analysis supported this by showing that several oxygen
containing compounds were driven out by the heat. The formation of aromatic structure was
however difficult to observe because peaks, representing aromatic structures, were masked by
the dominant water intensity in the FTIR spectra.
The SEM in combination with the EDS showed the presence of skin-core structures in the
fibers. Furthermore, defects like “porous surface” were observed in the SEM and light
microscopy imaging. These irregularities affected the tensile strength of the fibers.

82
6 Acknowledgements This work was carried out in the Lignin Carbon Fiber group at Innventia AB under
supervision of Professor Lennart Salmén.
I wish to express my sincere gratitude to Professor Lennart Salmén for introducing me to the
area of research regarding lignin and carbon fibers and also allowing me to work with this
project. His door was always open throughout the project and in dire need he would always
provide with advices and suggestions.
I would also like to thank Anne-Mari Olsson for introducing me to the practical work at the
start of the project, helping me solve the “mysteries” of the TMA-instrument and showing me
how to operate the TGA. Whenever there was a practical or technical issue regarding
equipment or preparations, she always had a solution at hand.
I was able to operate the FTIR-equipment at Innventia AB thanks to Jasna Stevanic-Srndovic
and her instructions.
My gratitude also goes to Joanna Hornatowska who always was kind and considerate. She
helped me with the SEM-study. And thanks to her instructions I could fully utilize the light
microscope.
Many thanks to the members of the Lignin Carbon Fiber group for your support. I hope this
work will contribute in your further work with lignin carbon fibers.
I would like to thank Mårten Åkerström for “serving me”, as he himself would put it, in
solving the adhesive problems, ordering graphitic components (adhesive and plates) and
providing me with stabilized lignin fibers. Thank you for meaningful discussions.
Thanks to Anders Uhlin for showing me how to perform the stabilization on my own when all
stabilized lignin fibers were consumed.
To Dr. Thomas Edert and Daniel Aili, at Linköping University, I wish to express my
appreciation that you accepted the role of being examiner and internal supervisor for this
thesis. Your suggestions and advices are greatly acknowledged.
At last, but not least, I would like to thank my family for providing support and temporary
accommodation during the difficult time of train service in Sweden. I always think about you
and appreciate you even though it may not seem like that sometimes.

83
7 References
Abo, H. 2010, The ABSs of Measurement Methods: KBr Pellet Method, vol. 14 edn, FTIR
Talk Letter, Shimadzu Corp.
Agarwal, U.P., McSweeny, J.D. & Ralph, S.A. 2011, "FT-Raman Investigation of Milled-
Wood Lignins: Softwood, Hardwood, and Chemically Modified Black Spruce Lignins",
Journal of Wood Chemistry & Technology, vol. 31, no. 4, pp. 324.
Arbab, S., Mirbaha, H., Zeinolebadi, A. & Nourpanah, P. 2014, "Indicators for evaluation of
progress in thermal stabilization reactions of polyacrylonitrile fibers", Journal of Applied
Polymer Science, vol. 131, no. 11, pp. n/a.
Bajaj, P., Sreekumar, T.V. & Sen, K. 2001, "Effect of reaction medium on radical
copolymerization of acrylonitrile with vinyl acids", Journal of Applied Polymer Science,
vol. 79, no. 9, pp. 1640-1652.
Baker, D.A. & Rials, T.G. 2013, "Recent advances in low-cost carbon fiber manufacture from
lignin", Journal of Applied Polymer Science, vol. 130, no. 2, pp. 713-728.
Becker, W.M. 2008, The world of the cell, 7th edn, Pearson, Benjamin Cummings, San
Francisco.
Beste, A. 2014, "ReaxFF Study of the Oxidation of Lignin Model Compounds for the Most
Common Linkages in Softwood in View of Carbon Fiber Production", Journal of
Physical Chemistry A, vol. 118, no. 5, pp. 803-814.
Boccara, A.C., Fournier, D., Kumar, A. & Pandey, G.C. 1997, "Nondestructive evaluation of
carbon fiber by mirage-FTIR spectroscopy", Journal of Applied Polymer Science, vol.
63, no. 13, pp. 1785-1791.
Braun, J.L., Holtman, K.M. & Kadla, J.F. 2005, "Lignin-based carbon fibers: Oxidative
thermostabilization of kraft lignin", Carbon, vol. 43, no. 2, pp. 385-394.
Brodin, I., Gellerstedt, G., Sjöholm, E. & Ernstsson, M. 2012, "Oxidative stabilisation of kraft
lignin for carbon fibre production", Holzforschung, vol. 66, no. 2, pp. 141-147.
Brodin, I., Sjöholm, E. & Gellerstedt, G. 2010, "The behavior of kraft lignin during thermal
treatment", Journal of Analytical and Applied Pyrolysis, vol. 87, pp. 70-77.
Brown, M.E. 2001, Introduction to thermal analysis. [Elektronisk resurs] techniques and
applications, 2nd edn, Kluwer Academic, Dordrecht ; London.
Chatterjee, S., Johs, A., Jones, E.B., Rios, O., Clingenpeel, A.C., McKenna, A.M., McNutt,
N.W. & Keffer, D.J. 2014, "Conversion of lignin precursors to carbon fibers with
nanoscale graphitic domains", ACS Sustainable Chemistry and Engineering, vol. 2, no. 8,
pp. 2002-2010.
Coates, J. 2000, "Interpretation of Infrared Spectra, A Practical Approach" in Encyclopedia of
Analytical Chemistry, ed. R.A. Meyers, John Wiley & Sons Ltd., Chichester, pp. 10815.

84
Coats, A.W. & Redfern, J.P. 1963, "Thermogravimetric analysis. A review", Analyst, vol. 88,
no. 1053, pp. 906.
Davis, J.R. 2004, Tensile testing. [Elektronisk resurs], Materials Park, Ohio : ASM
International, 2004; 2nd ed.
Derkacheva, O. & Sukhov, D. 2008, "Investigation of lignins by FTIR spectroscopy",
Macromolecular Symposia, vol. 265, no. 1, pp. 61-68.
Egerton, R.F. 2005, Physical principles of electron microscopy. [Elektronisk resurs] : an
introduction to TEM, SEM, and AEM, New York, N.Y. : Springer, c2005.
Faix, O. 1991, "Classification of Lignins from Different Botanical Origins by FT-IR
Spectroscopy", Holzforschung: International Journal of the Biology, Chemistry, Physics,
& Technology of Wood, vol. 45, pp. 21.
Freudenberg, K. 1959, "Biosynthesis and Constitution of Lignin", Nature, vol. 183, no. 4669,
pp. 1152.
Gellerstedt, G., Ek, M. & Henriksson, G. 2009, , Wood Chemistry and Wood Biotechnology.
Heber S., A., João V.F., L., Regina P.W., P., Maria, B.O., Fábio A., A. & Kelysson F., A.
2009, "A supramolecular proposal of lignin structure and its relation with the wood
properties", Anais da Academia Brasileira de Ciências, , no. 1, pp. 137.
Helander, M., Theliander, H., Lawoko, M., Henriksson, G., Zhang, L. & Lindström, M.E.
2013, "Fractionation of technical lignin: Molecular mass and pH effects", BioResources,
vol. 8, no. 2, pp. 2270-2282.
Hergert, H.L. 1960, "Infrared spectra of lignin and related compounds. II. Conifer lignin and
model compounds", Journal of Organic Chemistry, vol. 25, no. 3, pp. 405-413.
HowStuffWorks 2001, 2001-03-21-last update, What is the difference between a hardwood
and a softwood? [Homepage of HowStuffWorks], [Online]. Available:
http://science.howstuffworks.com/life/genetic/question598.htm [2014, 10/31].
Huang, X. 2009, "Fabrication and Properties of Carbon Fibers", Materials, vol. 2, no. 4, pp.
2369.
Huang, Y., Ma, E. & Zhao, G. 2015, "Thermal and structure analysis on reaction mechanisms
during the preparation of activated carbon fibers by KOH activation from liquefied
wood-based fibers", Industrial Crops & Products, vol. 69, pp. 447-455.
Ju, A., Xu, H. & Ge, M. 2014, "Preparation and thermal properties of poly[acrylonitrile- co-(
ß-methylhydrogen itaconate)] used as carbon fiber precursor", Journal of Thermal
Analysis & Calorimetry, vol. 115, no. 2, pp. 1037-1047.
Karmanov, A.P. & Derkacheva, O.Y. 2013, "Application of fourier transform infrared
spectroscopy for the study of lignins of herbaceous plants", Russian Journal of
Bioorganic Chemistry, vol. 39, no. 7, pp. 677-685.

85
Kubo, S., Ishikawa, N., Uraki, Y. & and Sano, Y. 1997, "Preparation of Lignin Fibers from
Softwood Acetic Acid Lignin. Relationship between Fusability and the Chemical
Structure of Lignin", Mokuzai Gakkaishi, vol. 43, no. 8, pp. 655-662.
Kubo, S. & Kadla, J.F. 2005, "Hydrogen bonding in lignin: A fourier transform infrared
model compound study", Biomacromolecules, vol. 6, no. 5, pp. 2815-2821.
Li, Y., Cui, D., Tong, Y. & Xu, L. 2013, "Study on structure and thermal stability properties
of lignin during thermostabilization and carbonization", International journal of
biological macromolecules, vol. 62, pp. 663-669.
Lin, J., Koda, K., Uraki, Y., Kubo, S., Yamada, T. & Enoki, M. 2014, "Improvement of
mechanical properties of softwood lignin-based carbon fibers", Journal of Wood
Chemistry and Technology, vol. 34, no. 2, pp. 111-121.
Liu, Y. & Kumar, S. 2012, "Recent Progress in Fabrication, Structure, and Properties of
Carbon Fibers", Polymer Reviews, vol. 52, no. 3, pp. 234-258.
Lü, Y., Wu, D., Zha, Q., Liü, L. & Yang, C. 1998, "Skin-core structure in mesophase pitch-
based carbon fibers: causes and prevention", Carbon, vol. 36, no. 12, pp. 1719.
Matsumoto, T. 1985, "Mesophase pitch and its carbon fibers", Pure & Applied Chemistry,
vol. 57, no. 11, pp. 1553.
Matsumoto, T. & Mochida, I. 1992, "A structural study on oxidative stabilization of
mesophase pitch fibers derived from coaltar", Carbon, vol. 30, pp. 1041-1046.
McCrum, N.G., Bucknall, C.B. & Buckley, C.P. 1997, Principles of polymer engineering,
Oxford : Oxford Univ. Press, 1997; 2. ed.
Mongeau, R. & Brooks, S.P.J. 2001, "Chemistry and Analysis of Lignin", Food Science and
Technology -New York-Marcel Dekker-, , no. 113, pp. 321-374.
Norberg, I. 2012, Carbon Fibres from Kraft Lignin, KTH Royal Institute of Technology,
Stockholm.
Norberg, I., Nordstrom, Y., Drougge, R., Gellerstedt, G. & Sjo¨holm, E. 2013, "A new
method for stabilizing softwood kraft lignin fibers for carbon fiber production", Journal
of Applied Polymer Science, vol. 128, no. 6, pp. 3824-3830.
Pandey, K.K. 1999, "A Study of Chemical Structure of Soft and Hardwood and Wood
Polymers by FTIR Spectroscopy", Journal of Applied Polymer Science, vol. 71, no. 12,
pp. 1969-1975.
Sjöström, E. 1993, Wood chemistry: fundamentals and applications, Academic Press, Inc, San
Diego.
Solomons, T.W.G. & Fryhle, C.B. 2011a, Organic chemistry, Hoboken, N.J. : Wiley ;
Chichester : John Wiley distributor, cop. 2011; 10. ed., International student version.

86
Steed, J.W., Turner, D.R. & Wallace, K.J. 2007, Core concepts in supramolecular chemistry
and nanochemistry [Elektronisk resurs] / Jonathan W. Steed, David R. Turner, Karl J.
Wallace, Chichester : John Wiley, c2007.
Vaimakis, T.C. 2013, Thermogravimetry (TG) or Thermogravimetric Analysis (TGA),
Introduction edn, Summer School for Thermal Analysis Techniques, Faculty of Science,
Aristotle University of Thessaloniki.
van der Maas, J. H. & Tolk, A. 1962, "The influence of moisture on potassium bromide disks
used in infrared spectrometry", Spectrochimica Acta, vol. 18, no. 2, pp. 235-240.

87
8 Appendix Process, calibrations, calculations and data that may or may not be interesting regarding the
outcome of the project are presented here.
8.1 Process A planning report was established in the beginning of the project. Its purpose was to describe
the project, formulate problems, suggest solution methods and served as a guide for the student during
the project.
Timetable and Planning are taken directly from the original planning report and is presented in the
following sections.

88
8.1.1 Timetable
Figure 73: Deadlines of the original time plan.
4041
4243
4445
4647
4849
5051
521
23
45
67
89
1011
1213
1415
1617
18
Activite
s (ho
urs)
De
adlin
es
D1
Han
d-in
of p
lann
ing re
po
rt24-o
kt
D2
Han
d-in
of h
alf-time
rep
ort
D3
Half-tim
e p
rese
ntatio
n19-d
ec
D4
Han
d-in
of th
esis re
po
rt27-m
ar
D4:1
Han
d-in
of th
esis re
po
rt to e
xamin
er
27-mar
D4:2
Han
d-in
of th
esis re
po
rt to o
pp
on
ant
27-mar
D5
Re
po
rt pre
sen
tation
17-apr
D6
Final h
and
-in o
f rep
ort
27-apr
D7
Han
d-in
of re
flectio
n d
ocu
me
nt
27-apr
Mile
ston
es
M1
Pilo
t stud
y31-o
kt
M2
Expe
rime
nts d
on
e07-m
ar
M2:1
Expe
rime
nt p
erio
d 1
12-de
c
M2:2
Expe
rime
nt p
erio
d 2
30-jan
M2:3
Expe
rime
nt p
erio
d 3
27-feb
M3
Evaluate
resu
lts07-m
ar
Mars
Ap
ril
We
ek
Nr.
Mo
nth
Octo
be
rN
ove
mb
er
De
cem
be
rJan
uary
Feb
ruary

89
Figure 74: Original time plan of the project over 2014-2015.
4041
4243
4445
4647
4849
5051
521
23
45
67
89
1011
1213
1415
1617
18
Nr.
Christmas holiday
Buffer weeks
Easter
Mars
Ap
rilSu
mG
oal
Diffe
ren
ceW
ee
k
Mo
nth
Octo
be
rN
ove
mb
er
De
cem
be
rJan
uary
Feb
ruary
Activite
s (ho
urs)
Co
urse
relate
d
C1
Plan
nin
g rep
ort
26
66
2020
0
C2
Time
plan
22
35
1212
0
C3
Writin
g half-tim
e re
po
rt0
0
C4
Cre
ating h
alf-time
pre
sen
tation
66
618
180
C5
Writin
g the
sis rep
ort
1616
1648
480
C5:1
Aim
, intro
du
ction
, backgro
un
d title
s.8
88
428
280
C5:2
Me
tho
d, m
aterial, re
sults
48
88
2828
0
C5:3
Discu
ssion
, con
clusio
n, ab
stract16
1632
320
C6
Cre
ating th
esis p
rese
ntatio
n16
1616
0
C7
Co
mp
letin
g the
final th
esis re
po
rt12
1224
240
C8
Re
flectio
n d
ocu
me
nt
1010
2020
0
Stud
y relate
d0
0
S1P
ilot stu
dy
1116
1410
1162
620
S2D
eve
lop
me
nt o
f me
tho
d10
26
48
3030
0
S3Exp
erim
en
ts
S3:1Exp
erim
en
ts 114
1515
2017
1899
990
S3:2Exp
erim
en
ts 25
1417
1715
6868
0
S3:3Exp
erim
en
ts 317
1717
1667
670
S4Evalu
ation
of re
sults
55
810
55
1250
500
S5G
athe
r info
rmatio
n2
55
55
55
54
55
5151
0
Me
etin
g and
ed
ucatio
n0
0
E1A
pp
aratus train
ing/in
structio
ns
75
810
88
95
6060
0
E2M
ee
ting w
ith e
xamin
er
11
11
44
0
E3M
ee
ting w
ith su
pe
rvisors
12
11
11
11
11
11
11
11
11
120
200
E4A
dm
inistrative
tasks (e-m
ail, inst. So
ftware
, saftey re
gulatio
ns, tim
ecard
etc.)
95
32
22
11
11
11
11
11
11
11
138
380
E5Se
min
ars1
11
11
55
0
Nr.
Christmas holiday
Buffer weeks
Easter
Sum
Christmas holiday
Buffer weeks
Easter
3030
3030
3037
4040
4040
4020
2040
4040
4040
4040
302
1722
22800
8000
3030
3030
3037
4040
4040
4020
2040
4040
4040
4040
302
1722
22800
8000
00
00
00
00
00
00
00
00
00
00
00
00
00
00
Sum
Christmas holiday
Buffer weeks
Easter
Go
al
Diffe
rance

90
8.1.2 Planning
Main goal:
Optimize temperature programming and forces in the carbonization process to obtain suitable
carbon fiber material with good mechanical strength potential (high tensile strength and high
modulus).
The parameters that should be considered are:
Temperature
Temperature ramping
Run time
Applied force during the carbonization process
Determine the best suitable setting (optimal program) of parameters by following the
objectives.
The time window is about 4 months in which the main goal should be answered. This is done
prior the presentation of the project.
Objectives:
O1. Determine the carbon content in carbon fibers after carbonization and look at the
chemical elementary composition in the carbon fiber.
O2. Find and identify structures, such as skin-core, that may be present in the morphology
of the final carbon fiber.
O3. Study the molecular chain orientation and molecular structure of the lignin carbon
fiber.
O4. Test the strength properties of the carbon fiber, i.e. tension, strain, Young’s modulus
and breakage.
Preliminary description of chosen solution method
Thermostabilized lignin fibers, in semi-optimized conditions, will be provided for the
experiments.
These fibers will be carbonized in a thermo-mechanical analysis (TMA) apparatus during
different settings of temperature, ramping of temperature, applied force and run time.
The next step is to determine the strength properties as well as structural formation for each
setting of parameters, in order to find the best suitable program to manufacture the carbon
fibers. Each sub goal is a test that will provide with the necessary information to make the
decision.
Not only will the tests provide with information which is needed for the determination of best
parameters but also provide with a deeper understanding of the carbon fiber itself. For
example, how the structure/morphology and/or composition looks like.

91
O1. The content of carbon and/or compositions of other elements can be evaluated in
scanning electron microscope analysis (SEM).
O2. Morphology and structures can be visualized in SEM-analysis.
O3. The molecular chain orientation of a single lignin carbon fiber can be evaluated in
Infrared (IR) transmittance analysis. However, pre-experiments are necessary to find a
fiber with a small enough diameter, so that the irradiation of polarized infrared light
can pass through the fiber. By using IR light and by comparing the spectra in different
polarization angles one can see in which overwhelming direction certain bonds are
lying in, due to the vibration mode of molecules. Evaluation of the molecular structure
can also be done in IR spectroscopy. The fiber needs to be dissolved in hydrophobic
solution first. Pre-experiments are needed to identify a suitable hydrophobic liquid to
dissolve the fibers in.
O4. The strength mechanical properties can be measured in Dynamical Mechanical
Analysing (DMA). Noteworthy, is that this type of measurement destroys the sample
and can only be done once for each fiber string. Therefore, the objective is put last in
the list and will consequently be executed last.
8.1.3 Process results
Regarding the main goal, the listed parameters were tested accordingly in the TMA
carbonizations. However, by enlightenment and better understanding of the process, the ‘run
time’ was scratched out as a test parameter. Run time is dependent on rate of heating and final
temperature, thus making it an obsolete parameter.
An early obstacle was faced when performing TMA carbonizations in the beginning of the
project. The fibers that underwent carbonization in the instrument ruptured easily. By
investigating the problem it was concluded that the two part adhesive component WH-
Refractory cement 1500 (Morgan Technical Ceramics Haldenwanger, Waldkraiburg,
Germany) (a ceramic adhesive that was initially used to adhere ceramic clamps, made from
the same solution) weakened the fibers where it was applied. The ceramic adhesive was then
switched for the more reliable and practical carbon paint suspension (SPI Supplies®).
However, for the TMA carbonizations with increasing load graphitic adhesive had to be used.
In addition, the piping that provided the instrument with N2 gas probably leaked and air was
introduced into the instrument. This problem was fixed by replacing the plastic hoses with a
more tight metallic piping.
The objective in O3 had to be changed as well. Because, as already mentioned, the FTIR-
study was not a simple matter. It took some time to realize that extremely small amounts of
sample had to be used in order to get IR-spectra with distinguishably features. Furthermore,
the “suspension in hydrophobic liquid” was replaced by the KBr-method.
The KBr pelletizing was also practiced wrongly until it was realized. Initially, it was thought
that the analysis could be performed on whole fibers in the KBr-pellet. This is not the case.
On the contrary, it is required that the fibers are grinded to a suitable particle size in order to
be visible in the instrument. As a result, the orientation study was averted.

92
FTIR Imaging (PerkinElmer Spotlight 400) was first utilized to study the whole fibers. But
since carbon fibers have a high level of absorbance and the fiber diameter was not small
enough to compensate. The technique was abandoned for the KBr-method in the Varian 680-
IR instrument.
The implications of the TMA and FTIR procedure resulted in a delay of the project, the
deadlines had to be postponed a month and the time plan was modified accordingly (Figure
75).
In objective O4, the DMA technique was replaced with Dia-Stron. The main difference
between the instruments is that the Dia-Stron can do the same performance tests only more
reliably and practically.
The TGA measurement was added later in the project in which the mass loss of stabilized
lignin fibers was studied.
The project was divided into three experiment phases (noted as M2 in Figure 73 and Figure
75) where the parameters for the TMA carbonization were changed.
M2.1 In the first phase, each bundle of fibers (each sample) underwent carbonization from
20°C to set temperatures between 300-1200°C in steps of 100°C at 1K/min (or
1°C/min) under a load of 5 mN. After evaluation of the deformation, the carbonization
temperature was selected to 900-1300°C (in steps of 100°C) for further testing in the
following phase.
M2.2 In phase two, the heating rate was varied, testing 10, 20 and 40 K/min, to set
temperatures (900-1300°C, in steps of 100°C). Same load as before. After evaluation
of the deformation, the heating rate and final temperature was selected to 10 K/min
and 1000°C, respectively, for further testing in following phase. The parameters were
practical in the sense that they allowed fast carbonization that took mere 2 hours
(cooling included) whereas 1 K/min carbonizations took around 16 hours to complete.
M2.3 During the third and last phase, heating of 10 K/min and a final carbonization
temperature of 1000°C were selected as fixed parameters in order to test different
forces. The initial load was increased to 10 mN.
After said carbonizations the fibers were analyzed in Dia-Stron and light microscope.
A different batch was produced for the purpose of FTIR. The parameters were 10 K/min to
1000°C under load of 5 mN.
Both the FTIR and Dia-Stron tests destroyed the sample during analysis hence large quantities
of fibers were carbonized in the TMA. FTIR, in particular, required a lot due to the grinding
process, at least before discovering that small amounts were obligatory. The Dia-Stron did not
make it easier by occasionally not measuring the diameter of the fibers; hence identical
batches of carbonized fibers had to be produced to make the same tensile test all over again.
Late in the project when a lot of samples had been accumulated, the SEM analysis was carried
out during one workday. A few interesting samples were selected for this study.

93
Figure 75: Modified deadlines of the time plan with date of completion.
4041
4243
4445
4647
4849
5051
521
23
45
67
89
1011
1213
1415
1617
1819
2021
22
Activite
s (ho
urs)
De
adlin
es
D1
Han
d-in
of p
lann
ing re
po
rt24-o
kt24-o
ct
D2
Han
d-in
of h
alf-time
rep
ort
N/A
D3
Half-tim
e p
rese
ntatio
n04-fe
b04-fe
b
D4
Han
d-in
of th
esis re
po
rt30-ap
r24-m
ay
D4:1
Han
d-in
of th
esis re
po
rt to e
xamin
er
30-apr
24-may
D4:2
Han
d-in
of th
esis re
po
rt to o
pp
on
ant
09-maj
03-jun
D5
Re
po
rt pre
sen
tation
19-maj
17-jun
D6
Final h
and
-in o
f rep
ort
29-maj
05-jul
D7
Han
d-in
of re
flectio
n d
ocu
me
nt
29-maj
10-jul
Mile
ston
es
M1
Pilo
t stud
y31-o
kt31-o
ct
M2
Expe
rime
nts d
on
e27-m
ar27-m
ar
M2:1
Expe
rime
nt p
erio
d 1
30-jan30-jan
M2:2
Expe
rime
nt p
erio
d 2
27-feb
27-feb
M2:3
Expe
rime
nt p
erio
d 3
27-mar
27-mar
M3
Evaluate
resu
lts17-ap
r17-ap
r
Mars
Ap
rilM
ayD
on
eW
ee
k
Nr.
Mo
nth
Octo
be
rN
ove
mb
er
De
cem
be
rJan
uary
Feb
ruary

94
Figure 76: Final time plan.
4041
4243
4445
4647
4849
5051
521
23
45
67
89
1011
1213
1415
1617
1819
2021
2223
2425
2627
We
ek
Nr.
Christmas holiday
Buffer weeks
Easter
Difference
Mo
nth
Octo
be
rN
ove
mb
er
De
cem
be
rJan
uary
Feb
ruary
Mars
Ap
rilM
aySu
mG
oal
Jun
e
Activite
s (ho
urs)
Co
urse
relate
d
C1
Plan
nin
g rep
ort
7.252
132
24.2520
4.25
C2
Time
plan
18
4.51
34
21.512
9.5
C3
Writin
g half-tim
e re
po
rt5
55
C4
Cre
ating h
alf-time
pre
sen
tation
212
1327
189
C5
Writin
g the
sis rep
ort
46
1048
-38
C5:1
Aim
, intro
du
ction
, backgro
un
d title
s.4
1715
3628
8
C5:2
Me
tho
d, m
aterial, re
sults
13
34
143
2025
3038
16828
140
C5:3
Discu
ssion
, con
clusio
n, ab
stract36
3632
4
C6
Cre
ating th
esis p
rese
ntatio
n28
119
4816
32
C7
Co
mp
letin
g the
final th
esis re
po
rt36
3624
12
C8
Re
flectio
n d
ocu
me
nt
1616
20-4
Stud
y relate
d
S1P
ilot stu
dy
16.7516.58
1215.5
25.2586.08
6224.08
S2P
re-e
xpe
rime
nts an
d p
rep
aration
2516
9.512
712
78
59
109
810
124
4167.5
30137.5
S3Exp
erim
en
ts6
44
519
19
S3:1Exp
erim
en
ts 15
4.522
2010
56
277
106.599
7.5
S3:2Exp
erim
en
ts 220
616
2264
68-4
S3:3Exp
erim
en
ts 316
1618
218
180
6713
S4Evalu
ation
of re
sults
22
46
81
312
22
69
5750
7
S5G
athe
r info
rmatio
n2
23
82
92
52
44
410
5751
6
Me
etin
g and
ed
ucatio
n
E1A
pp
aratus train
ing/in
structio
ns7
510.5
4.58.5
62
43.560
-16.5
E2M
ee
ting w
ith e
xamin
er
11
24
-2
E3M
ee
ting w
ith su
pe
rvisors an
d d
iscussio
n2
21
31
12
31
12
11
2120
1
E4A
dm
inistrative
tasks (e-m
ail, inst. So
ftware
, saftey re
gulatio
ns, tim
ecard
etc.)
81
2.252.5
12
31
11
11
11
52
12
21
11
32
22
42
24
163.75
3825.75
E5Se
min
ars1
15
-4
Sum
Nr.
Christmas holiday
Buffer weeks
Easter
32.7526.83
29.2546
35.7533
3220.5
4032
4024
1422
3336
3632
4038
3234
3837
1611
1344
4047
4240
402
638
119
531196.08
800396.08
3030
3030
3037
4040
4040
4020
020
4040
4040
4040
4030
217
2222
800800
0
2.75-3.17
-0.7516
5.75-4
-8-19.5
0-8
04
142
-7-4
-4-8
0-2
-84
3837
1411
1327
1825
4240
402
638
119
53396.08
0396.08
Sum
Go
al
Diffe
rance

95
8.1.4 Comprehensive analysis of the process
In the beginning of the project, a lot of time was spent on solving the problems of the TMA
carbonizations. The right procedure behind the FTIR also took a lot of time to figure out. As a
result the time of pre-experiments and preparations (S2 Figure 76) greatly exceeded the goal.
On the contrary, the time spent on the experiment parts and the time goal approximatively
matched each other.
A lesson from this is to not underestimate unforeseen problems that will occur. The solution is
to set aside enough time in the planning to solve them. Rarely, a large project, like this,
proceeds like it was intended; Murphy’s Law has to be considered and unforeseen problems
have to be taken into account.
Another problem faced during the project was that the fibers were not sufficiently fixated by
carbon paint in the alumina clamps; they simply slid out when increasing the load. Leant from
the earlier mistakes the problem was solved prior the force measurement phase.
The pilot study was also a bit time consuming, mainly because the time of writing it was
underestimated (S1 Figure 76).
An extremely large amount of time was spent on the result in the writing of this thesis (C5:2
Figure 76). The main time consuming step was to create all charts, tables, images and
calculations and adjust their exhibition (font, size, axes etc.) in order to write the text to the
section. In hindsight, all data management should have been prepared during and
simultaneously with the evaluation of the results (S4). In which scenario the time goal of
evaluation should have been significantly increased. Then more time could have been moved
from the result to the evaluation. 48 hours were set aside as an additional time to write the
report (C5 Figure 76), but was not far enough to account for the results.
At the time of writing this, the hand in of the thesis report is several weeks overdue. To avoid
delays the report should not have been written at the very end, it would have been wiser to
start the writing earlier and distribute the time evenly over the course of the project. The
overall time of the project greatly exceeded the 800 hour goal, mainly because of the said
implications of FTIR and TMA carbonizations. And maybe the different analyses were a few
to many. The main goal was solved solely by the TMA and Dia-Stron measurements. The
others were only interesting in the aspect of knowing why the fibers became stronger. If, for
instance, the FTIR analysis had been cut from the project, more time could have been spent
on the other objectives.
8.2 Data Data, calculations and calibrations are presented in this section.
8.2.1 TMA
The carbonization of stabilized fibers attached to graphitic plates with non-curated graphitic
adhesive resulted in a strange deformation curve (Figure 77). As the force ramp begins the
fibers are expanded in a small step. When the force decreases (due to a faulty inputted
program) the deformation steps down. The fibers seem to have been affected despite the non-

96
curated adhesive. The behavior arises from the fact that the stabilized fibers were not properly
fixated in the thin layer of graphite adhesive; they simply slid out when the stronger force was
applied and moved back to the original positon as soon as the force was decreased.
Figure 77: Deformation curve of stabilized lignin fibers improperly fixated to the graphitic plates due to a non-
curated graphite adhesive. Initial sample length was 20 mm and the fibers were carbonized at 10 K/min to 1000°C.
For additional information see Table 2.
8.2.2 TGA
Data from the TGA are presented here.
8.2.2.1 Calibration
Figure 78: Curve of mass loss during “carbonization” in the TGA. The instrument was programed to heat stabilized
lignin fibers of mass 0.629 mg at 10 K/min to 1000°C. However, it did not manage to complete the full program. The
temperature of the sample reached a maximum of 885°C where the program was set to 925°C, followed by cooling.
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
-10%
-5%
0%
5%
0 100 200 300 400 500 600 700 800 900 1000 1100
Forc
e (
N)
∆L/
L0
Temperature (°C)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0 100 200 300 400 500 600 700 800 900 1000

97
Table 8: Weights before and after TMA carbonization. The initial fiber count was 30 for each sample.
Weight of carbon paint + 30 fibers (mg) Fibers
Initial Final Final
300 3.2 2.9 30
600 2.8 2 29
By using the values in Table 8 the mass loss was calculated. The result is shown as version 1
in Table 9. In version 2, the data at 300°C was corrected to the value of the TGA
measurement.
Table 9: The weight loss presented as percentage. Version 1 is solely from the TMA carbonization whereas in version
2 the value at 300°C is adjusted to the TGA measurement.
Temp. (°C) TMA Weight loss (%) version 1 TMA Weight loss (%) version 2
300 91 96
600 74 74
Version 2 served as a starting point for the following calculations and were plotted as ‘Trend
TMA Weight loss’ in Figure 79, and a tangent was drawn from the line: 𝑦1 = −0.0745𝑥 +
118.58. Another trend line, called ‘Trend TGA Weight loss’, was drawn between 300°C and
the final temperature of the Recorded TGA Weight loss curve: 𝑦2 = −0.1232𝑥 + 133.19.
The difference 𝑦1 − 𝑦2 = −0.0745𝑥 + 118.58 − (−0.1232 + 133.19) = 0.0487𝑥 −
14.61 was then added to the ‘Recorded TGA Weight loss’ curve after 300°C, resulting in the
‘Corrected Weight loss’ curve.
Figure 79: Illustration of trend lines, corrected and uncorrected weight loss of the TGA measurement. The trend lines
are drawn from 300°C to the final temperature.
y1 = -0.0745x + 118.58
y2 = -0.1232x + 133.19
0
20
40
60
80
100
120
140
0 200 400 600 800 1000
Recorded TGA Weight loss
Trend TMA Weight loss
Trend TGA Weight loss
Corrected Weight loss

98
8.2.2.2 Calculations of diameter and stress change
Following calculations are based on the assumption that the change of mass is the same as the
change of volume.
Initial values are listed in Table 10.
Table 10: Initial parameters of the fibers of sample G and in the standard carbonization.
Sample G Standard carbonization
Initial Length (m) 0.02 0.015
Initial Diameter (m) 4.70E-05 4.70E-05
Initial Volume (m3) 3.47E-11 2.6E-11
By simply multiply the change percentage of the TGA curve with the volume we get the
change of volume over temperature. From the TMA-measurements we have the change of
length.
The volume of the fibers, assumed that they are uniformly and cylindrical, is 𝑉 = 𝐴 ⋅ 𝑙, where
l is the length and A the cross-sectional area. Simple rearrangements allow us to obtain the
change of cross-sectional area through 𝐴 =𝑉
𝑙.
Familiarly the area is 𝐴 = 𝜋𝑟2 = 𝜋 (𝑑
2)
2
. Rearrangement into 𝑑 = 2 ⋅ √𝐴 ∕ 𝜋 provides the
diameter change.
The stress in each point is derived from 𝐹 ∕ 𝐴.
8.2.3 Dia-stron
8.2.3.1 Compliance
Data (Recordings = Rec) were collected from tensile testing with the Dia-stron using three
different gauge lengths (12, 20 and 30 mm). Linear regression was made for each recording.
The recordings were as follow:

99
y = 0.0019x - 23.376 R² = 0.9996
0
0.05
0.1
0.15
0.2
0.25
0.3
12000 12050 12100 12150 12200
Load
(N
)
Displacement (µm)
12mm
Rec 1
y = 0.0026x - 31.29 R² = 0.9998
0
0.1
0.2
0.3
0.4
0.5
0.6
11950 12000 12050 12100 12150 12200
Load
(N
)
Displacement (µm)
12mm
Rec 4
y = 0.0031x - 37.734 R² = 0.9999
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
12150 12200 12250 12300 12350 12400 12450
Load
(N
)
Displacement (µm)
12mm
Rec 6
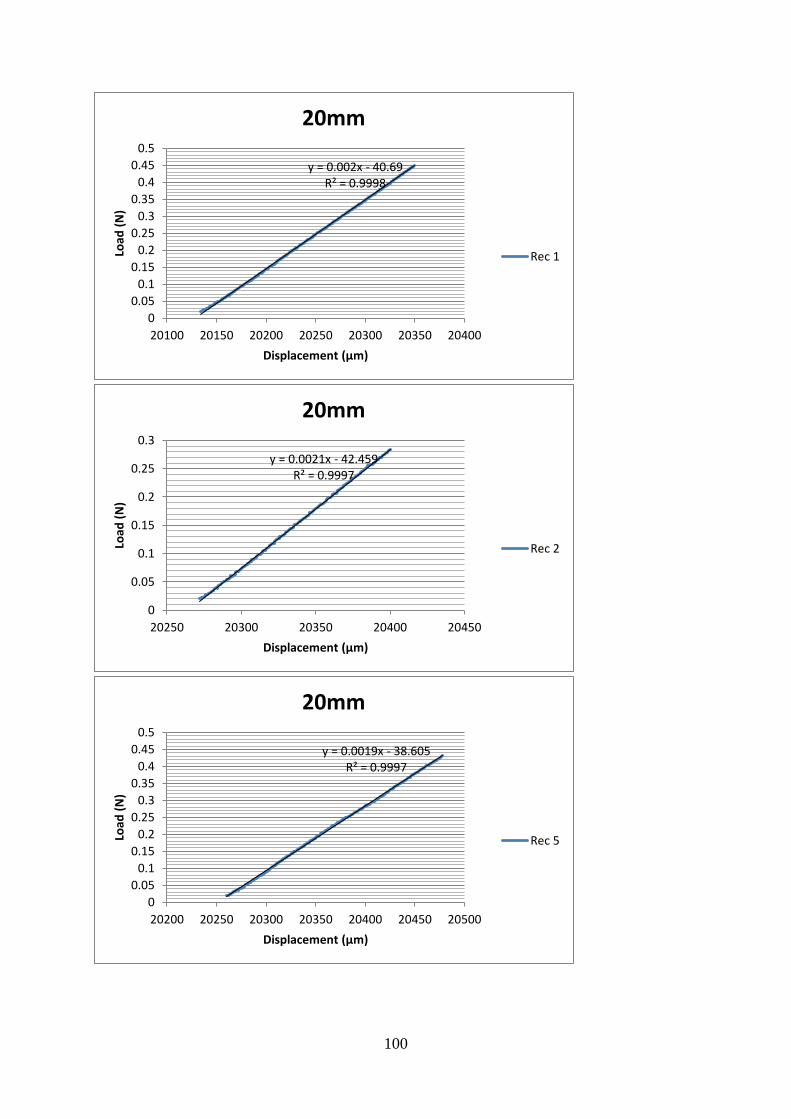
100
y = 0.002x - 40.69 R² = 0.9998
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
0.4
0.45
0.5
20100 20150 20200 20250 20300 20350 20400
Load
(N
)
Displacement (µm)
20mm
Rec 1
y = 0.0021x - 42.459 R² = 0.9997
0
0.05
0.1
0.15
0.2
0.25
0.3
20250 20300 20350 20400 20450
Load
(N
)
Displacement (µm)
20mm
Rec 2
y = 0.0019x - 38.605 R² = 0.9997
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
0.4
0.45
0.5
20200 20250 20300 20350 20400 20450 20500
Load
(N
)
Displacement (µm)
20mm
Rec 5

101
y = 0.0008x - 24.715 R² = 0.9999
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
30000 30100 30200 30300 30400 30500
Load
(N
)
Displacement (µm)
30mm
Rec 3
y = 0.0011x - 33.786 R² = 0.9999
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
0.4
30200 30300 30400 30500 30600
Load
(N
)
Displacement (µm)
30mm
Rec 4
y = 0.001x - 31.141 R² = 0.9999
0
0.05
0.1
0.15
0.2
0.25
0.3
30100 30150 30200 30250 30300 30350 30400
Load
(N
)
Discplacement (µm)
30mm
Rec 5

102
The slope and initial gauge length of each recording are summarized in Table 11.
Table 11: Values taken from the linear regression of each recording.
Record
12mm Slope F/displacement (N/µm)
Inverse (µm/N)
Initial length (µm)
Cross-Area (µm2)
L0/A (µm-1)
1 0.0031 322.5806452 12027 682.9 17.61166 4 0.0026 384.6153846 12007 897.9 13.37231 6 0.0031 322.5806452 12147 1037.3 11.71021
20mm 1 0.002 500 20134 1065.6 18.89452 2 0.0021 476.1904762 20272 1126.1 18.00195 5 0.0019 526.3157895 20248 1058 19.138
30mm
3 0.0008 1250 30044 643.9 46.65942 4 0.0011 909.0909091 30235 886 34.12528 5 0.001 1000 30126 774.4 38.90238
The inverse of the slopes from the recordings was plotted against the initial length over cross-
sectional area. Following graph was obtained:
Figure 80: Compliance graph.
y = 26.731x - 16.356 R² = 0.9753
0
200
400
600
800
1000
1200
1400
0 5 10 15 20 25 30 35 40 45 50
∆L/
F (µ
m/N
)
l0/A (µm-1)
Compliance

103
The Young’s modulus is given by eq. (1):
𝐸 =𝜎
𝜀 (1)
Where:
E = E-modulus, Pa and
σ = stress, Pa,
ε = strain.
𝜎 =𝐹
𝐴, 𝜀 =
𝛥𝑙
𝑙0 (2), (3)
Where:
F = force, N,
A = cross-sectional area (assumed to be constant throughout the testing), m2,
∆l = actual elongation of the gage length, m and
l0 = initial gage length, m.
Equation (1) and (3) provide:
𝜀 =𝛥𝑙
𝑙0=
𝜎
𝐸=
𝐹
𝐸𝐴 (4)
Rearrangement of (4) gives:
𝛥𝑙 =𝑙0𝐹
𝐸𝐴 (5)
The recorded displacement/elongation of the sample is shown by the following equation:
𝛥𝐿 = 𝛥𝑙 + 𝐶𝑠𝐹 (6)
Where:
∆L = recorded displacement/elongation, m and
Cs =system compliance, m/N.
Rearrangement of (6):
𝛥𝐿
𝐹=
𝛥𝑙
𝐹+ 𝐶𝑠 (7)
Insertion of eq. (5) into (7) provides:
𝛥𝐿
𝐹=
𝑙0
𝐸𝐴+ 𝐶𝑠 (8)
The 1/E and Cs, where l0/A is 0, is taken from the linear regression line in the compliance
graph and inserted into eq. (8):
1
𝐸= 26.731 µ
m
N𝑎𝑛𝑑 𝐶𝑠 = −16.356 µ
m
N

104
𝛥𝐿
𝐹= 26.731
𝑙0
𝐴− 16.356
Using eq. (6) the actual elongation can be calculated:
𝛥𝑙 = 𝛥𝐿 − 𝐶𝑠𝐹 (9)
Although the system compliance of -16.356 µm would lower the given values of E-modulus
in the result section, it is seemingly small and would not affect the calculations of E-modulus
relatively much. The compliance can thus be neglected and no correction has been made.

105
8.2.3.2 Data of carbonized fiber samples under load Table 12: Collected data from the tensile testing of fiber samples carbonized under load. The only data that was
obtained from sample Y was from a fiber that broke prior or during the carbonization hence the low values.
Sample Temp (°C) E-mod (GPa)
Break (MPa)
A 0.01N375-0.3N975 50.34 198
A 0.01N375-0.3N975 46.9 115
B 0.01N350-0.4N950 44.22 819.2
B 0.01N350-0.4N950 45.72 884.2
B 0.01N350-0.4N950 51.85 589.4
C 0.01N350°C-0.5N950°C 51.94 694
C 0.01N350°C-0.5N950°C 50.64 458.3
C 0.01N350°C-0.5N950°C 51.29 743.5
C 0.01N350°C-0.5N950°C 49.55 653.4
C 0.01N350°C-0.5N950°C 50 735.3
D 0.01N450-0.1N550 36.23 463.2
D 0.01N450-0.1N550 37.18 649.8
D 0.01N450-0.1N550 39.2 430.3
D 0.01N450-0.1N550 38.43 711
D 0.01N450-0.1N550 41.94 615.5
E 0.01N450-0.2N550 48.89 482
E 0.01N450-0.2N550 49.46 624.1
F 0.01N400-0.1N600 47.36 615.7
F 0.01N400-0.1N600 49.52 826.4
F 0.01N400-0.1N600 50.18 551.5
G 0.01N300°C-0.35N900°C 56.98 786.5
G 0.01N300°C-0.35N900°C 51.85 857.4
G 0.01N300°C-0.35N900°C 44.44 828.9
H 0.01N300°C-0.07N390°C-0.35N850°C
46.37 489.8
H 0.01N300°C-0.07N390°C-0.35N850°C
46.82 622.4
H 0.01N300°C-0.07N390°C-0.35N850°C
46.43 600.1
H 0.01N300°C-0.07N390°C-0.35N850°C
44.55 694.2
H 0.01N300°C-0.07N390°C-0.35N850°C
44.7 608.7
I 0.01N300°C-0.12N420°C-0.6N820°C
34.6 95.05
Y 0.01N300°C-0.07N420°C-0.35N820°C
32.19 308.9
Z 0.005N500-0.4N700 43.43 545.2
Z 0.005N500-0.4N700 32.21 426.4

106
Figure 81: Graphical overview of E-modulus in Table 12.
8.2.4 Light microscopy
Newly taken series of images regarding carbonization at 1 and 10 K/min as well as SEM
analysis in section 3.5.1 are presented here.
8.2.4.1 Carbonization series
Additional images from the microscopy evaluation, starting with stabilized lignin fibers
(Figure 82) followed by carbonized lignin fibers in the order of carbonization temperature
300, 400, 500… 1100, 1200°C. The left are carbonized at 1 K/min and the right 10 K/min.
The magnification is 50x and all images have the same scale.
Figure 82: Individual stabilized lignin fibers, second batch to the left and third to the right.
0
10
20
30
40
50
60
A A B B B C C C C C D D D D D E E F F F G G G H H H H H I Y Z Z
E-m
od
ulu
s (G
Pa)
Temperature (°C)

107
300°C
400°C
500°C
600°C

108
700°C
800°C
900°C
1000°C

109
Figure 83: Carbonized lignin fibers in the order of carbonization temperature; 300, 400, 500… 1000, 1100, 1200°C.
Carbonization rate in left column is 1 K/min and right 10 K/min (with exception for the right at 1100°C which was
carbonized at 20 K/min).
1100°C
1200°C
20 K/min

110
8.2.4.2 Comparison with SEM series
Figure 84: 300°C, 1 K/min.
Figure 85: 500°C, 1 K/min.

111
Figure 86: 1000°C, 1 K/min.
Figure 87: 1200°C 1 K/min.

112
Figure 88: 1200°C, 10 K/min.
Figure 89: Not from the same batch as the fibers analyzed in the SEM, just for comparison. 1300°C, 40 K/min.
Figure 90: : Carbonized at 10 K/min to 1000°C under increasing load from 10 mN to 300 mN between 375-975°C.

Linköping University Electronic Press
Upphovsrätt
Detta dokument hålls tillgängligt på Internet – eller dess framtida ersättare – från
publiceringsdatum under förutsättning att inga extraordinära omständigheter
uppstår.
Tillgång till dokumentet innebär tillstånd för var och en att läsa, ladda ner,
skriva ut enstaka kopior för enskilt bruk och att använda det oförändrat för icke-
kommersiell forskning och för undervisning. Överföring av upphovsrätten vid
en senare tidpunkt kan inte upphäva detta tillstånd. All annan användning av
dokumentet kräver upphovsmannens medgivande. För att garantera äktheten,
säkerheten och tillgängligheten finns lösningar av teknisk och administrativ art.
Upphovsmannens ideella rätt innefattar rätt att bli nämnd som upphovsman i
den omfattning som god sed kräver vid användning av dokumentet på ovan be-
skrivna sätt samt skydd mot att dokumentet ändras eller presenteras i sådan form
eller i sådant sammanhang som är kränkande för upphovsmannens litterära eller
konstnärliga anseende eller egenart.
För ytterligare information om Linköping University Electronic Press se för-
lagets hemsida http://www.ep.liu.se/
Copyright
The publishers will keep this document online on the Internet – or its possible
replacement –from the date of publication barring exceptional circumstances.
The online availability of the document implies permanent permission for
anyone to read, to download, or to print out single copies for his/hers own use
and to use it unchanged for non-commercial research and educational purpose.
Subsequent transfers of copyright cannot revoke this permission. All other uses
of the document are conditional upon the consent of the copyright owner. The
publisher has taken technical and administrative measures to assure authenticity,
security and accessibility.
According to intellectual property law the author has the right to be
mentioned when his/her work is accessed as described above and to be protected
against infringement.
For additional information about the Linköping University Electronic Press
and its procedures for publication and for assurance of document integrity,
please refer to its www home page: http://www.ep.liu.se/.
© Henrik Kleinhans

Date
2015-07-05
Division, Department
Chemistry
Department of Physics, Chemistry and Biology
Linköping University
URL for electronic version
http://urn.kb.se/resolve?urn=urn:nbn:se:liu:diva-120519
ISBN
ISRN: LITH-IFM-A-EX--15/3112--SE _________________________________________________________________
Title of series, numbering ISSN ______________________________
Language
Svenska/Swedish
Engelska/English
________________
Report category
Licentiatavhandling
Examensarbete
C-uppsats D-uppsats
Övrig rapport
_____________
Title
Evaluation of the Carbonization of Thermo-Stabilized Lignin Fibers into Carbon Fibers
Author
Henrik Kleinhans
Keyword
Lignin, Carbon Fibers, Carbonization, Stabilization, Uniaxial Tensile Testing, Thermomechanical Analysis (TMA), Thermogravimetric Analysis
(TGA), Fourier Transform Infrared Spectroscopy (FTIR), Scanning Electron Microscope (SEM), Energy Dispersive X-ray Spectrometer (EDS)
Abstract
Thermo-stabilized lignin fibers from pH-fractionated softwood kraft lignin were carbonized to various temperatures during thermomechanical
analysis (TMA) under static and increasing load and different rates of heating. The aim was to optimize the carbonization process to obtain suitable carbon fiber material with good mechanical strength potential (high tensile strength and high E-modulus). The carbon fibers were therefore mainly
evaluated of mechanical strength in Dia-Stron uniaxial tensile testing.
In addition, chemical composition, in terms of functional groups, and elemental (atomic) composition was studied in Fourier transform infrared
spectroscopy (FTIR) and in energy-dispersive X-ray spectroscopy (EDS), respectively. The structure of carbon fibers was imaged in scanning electron microscope (SEM) and light microscopy. Thermogravimetrical analysis was performed on thermo-stabilized lignin fibers to evaluate the
loss of mass and to calculate the stress-changes and diameter-changes that occur during carbonization.
The TMA-analysis of the deformation showed, for thermo-stabilized lignin fibers, a characteristic behavior of contraction during carbonization.
Carbonization temperatures above 1000°C seemed most efficient in terms of E-modulus and tensile strength whereas rate of heating did not matter considerably. The E-modulus for the fibers was improved significantly by slowly increasing the load during the carbonization. The tensile strength
remained however unchanged.
The FTIR-analysis indicated that many functional groups, mainly oxygen containing, dissociate from the lignin polymers during carbonization. The
EDS supported this by showing that the oxygen content decreased. Accordingly, the relative carbon content increased passively to around 90% at 1000°C. Aromatic structures in the carbon fibers are thought to contribute to the mechanical strength and are likely formed during the carbonization.
However, the FTIR result showed no evident signs that aromatic structures had been formed, possible due to some difficulties with the KBr-method.
In the SEM and light microscopy imaging one could observe that porous formations on the surface of the fibers increased as the temperature
increased in the carbonization. These formations may have affected the mechanical strength of the carbon fibers, mainly tensile strength.
The carbonization process was optimized in the sense that any heating rate can be used. No restriction in production speed exists. The carbonization should be run to at least 1000°C to achieve maximum mechanical strength, both in E-modulus and tensile strength. To improve the E-modulus
further, a slowly increasing load can be applied to the lignin fibers during carbonization. The earlier the force is applied, to counteract the lignin
fiber contraction that occurs (namely around 300°C), the better. However, in terms of mechanical performance, the lignin carbon fibers are still far
from practical use in the industry.