engineering of pharmaceutical particles - diva
TRANSCRIPT





CONTENTS
PAPERS DISCUSSED 8
INTRODUCTION 9
PREPARATION OF GRANULES BY WET GRANULATION 10 Drying of granules 11
The influence of drying conditions on structural and functionalproperties of granules 11Contraction of granules during drying 11
Other factors influencing structural properties of granules 12
PREPARATION OF PARTICLES BY SPRAY DRYING 12Amorphous solids 13
Definitions 13Pharmaceutically advantageous properties of amorphous material 14The glass transition temperature 14Amorphous materials and water 14Crystallisation of amorphous materials 15
Stabilisation of amorphous materials 15
COMPRESSIBILITY AND COMPACTABILITY OF POWDERS 16Tabletting of granulated materials 17 Tabletting of amorphous materials 18
CONCLUDING REMARKS CONCERNINGTHE PERTINENT LITERATURE 19
AIMS OF THE THESIS 20
MATERIALS 20
EXPERIMENTAL SECTION 22 Particle preparation 22
Preparation of pellets 22Drying of pellets 22Assessment of the porosity, contraction, densification anddegree of liquid saturation of pellets during drying 22Drying of pellets under different conditions 23Preparation of spray-dried particles 23
Characterisation of the prepared particles 24 Morphology 24Circularity 24Apparent particle density 24Assessment of pellet porosity 25Determination of poured bulk density and voidage of poured pellet bed 25
5

Surface area of powders 25Moisture content 25Crystallinity 25Heat-induced transformations 25Moisture-induced isothermal transformations 26Long-term stability 26
Preparation of tablets 27 Determination of porosity, tensile strength and surface area of tablets 27Calculation of compression parameters 27
Heckel parameter 27Kawakita parameters 28Elastic recovery 28
INFLUENCE OF CONTRACTION AND DENSIFICATION DURINGCONVECTIVE DRYING ON THE POROSITY,COMPRESSION SHEAR STRENGTH AND TABLET-FORMINGABILITY OF MICROCRYSTALLINE CELLULOSE PELLETS 28
The importance of the composition of the agglomeration liquid oncontraction and densification during drying 28
The degree of liquid saturation during drying 30Modulation of densification, porosity, compression shear strength and tablet forming ability of mcc pellets with the drying rate 33
Drying under different conditions 33The effect of drying rate on densification, porosity and the shape of pellets 34The effect of porosity and drying rateon the compression shear strength of pellets 35The effect of pellet porosity and drying rate on tablet tensile strength 36
MODULATION OF SOLID-STATE STABILITYAND TABLETTING BEHAVIOUR OF AMORPHOUS LACTOSEBY CO-SPRAY DRYING WITH POLYMERS 37
Properties of the spray-dried amorphous composite particlescontaining lactose and different polymers 37
Morphology 37Crystallinity 38Apparent particle density 38
The effect of the content and molecular weight of PVP onthe physical stability of amorphous lactose 38
Resistance to transformations induced by heat and moisture 38The effect on the relationship between critical RH for crystallisationand storage time 41
The effect of incorporation of stabilising polymers and a surfactanton the tabletting behaviour of amorphous lactose 42
Compression behaviour 42Tablet forming ability 45
6

SUMMARY AND CONCLUSIONS 46
ACKNOWLEDGEMENTS 49
REFERENCES 51
7

PAPERS DISCUSSED
This thesis is based on the following papers, which will be referred to by their Romannumerals in the text.
I Berggren, J. and Alderborn, G., 2001. Drying behaviour of two sets of microcrystalline cellulose pellets. Int. J. Pharm. 219, 113-126. Reproduced with permission. © 2001 Elsevier Science.
II Berggren, J. and Alderborn, G., 2001. Effect of drying rate on porosity and tabletting behaviour of cellulose pellets. Int. J. Pharm. 227, 81-96. Reproduced with permission. © 2001 Elsevier Science.
III Berggren, J. and Alderborn, G., 2003. Effect of polymer content and molecularweight on the morphology and heat- and moisture-induced transformations of spray-dried composite particles of amorphous lactose and poly(vinylpyrrolidone). Pharm. Res. Accepted for publication. Reproduced with permission. © 2003 Plenum Publishing Corp. Kluwer Academic.
IV Berggren, J. and Alderborn, G., 2003. Long-term stabilisation potential of poly(vinylpyrrolidone) for amorphous lactose in spray-dried composites.Submitted.
V Berggren, J. and Alderborn, G., 2003. Compression behaviour and tablet forming ability of spray-dried amorphous composite particles. Submitted.
8

INTRODUCTION
The tablet is today the dominating pharmaceutical dosage form, for many reasons, and not least because it offers a convenient and safe way for the patient to administer a drug. The manufacturing procedure facilitates accurate dosing of the drug and tablets can be mass-produced with robust production procedures at relatively low manufacturing costs. Drug release from tablets can be modified to meet biopharmaceutical and pharmacological requirements. Furthermore, tablets are generally more stable than other dosage forms such as liquid preparations.
The production of tablets involves different processing steps including operations where particles are engineered with the intention of optimising functional properties such as the technical performance during tabletting and drug release properties. The manufacturing of tablets requires certain qualities of the powder, low segregation tendency, good flowability and compactability being examples. It is also important that the materials constituting a tablet, i.e. drug and excipients, are chemically and physically stable during processing and storage. The concept of engineering materials to improve their functionality is applied within many disciplines and knowledge of relationships between structure and functional properties will clearly lead to a morerational approach to engineer materials. The term structure, used here, covers a wide range of features, from those on a macroscale, visible to the eye, down to those on a nanoscale, corresponding to interatomic distances.
In the production of pharmaceutical tablets it is common to prepare particles with the aid of a liquid that is subsequently removed by drying. In this context, the most common particle preparation method is wet granulation, in which fine particles (often drug and excipient) are aggregated under convective mixing and by addition of an agglomeration liquid. Liquid bridges will be created that hold the fine particles togetherand, when the liquid is removed by drying, cohered aggregated particles remain.Another frequently used particle preparation technique is precipitation from a liquid, e.g. crystallisation and spray drying, where the material is dissolved or suspended in a liquid, which then is removed by drying.
Preparation of pharmaceutical particles using procedures of the kind described above will subject the materials to different processing stresses (Fig. 1), such as exposure to liquids, mechanical stress and heat. These stresses may significantly affect and change particle structural properties (i.e., physical and solid-state properties), with consequences on properties like tabletting behaviour, drug release and stability. Stressesoccurring during the removal of a liquid by drying may, for instance, produce amorphous materials and influence on the porosity of granules, with subsequent effects on the dosage form. Thus, obtaining mechanistic knowledge regarding relationships between processing stresses, structural and functional properties of particles is of obvious interest to be able to predict and optimise functionality of solid dosage forms.In this context, there is a need for increased understanding of relationships between stresses during the removal of a liquid by drying and structural properties of particles critical for functional behaviour, and an awareness of how these stresses can be used to engineer particles with desired pharmaceutical properties.
9

Processing stresses e.g. temperature, pressure, mechanical stress, radiation, exposure
to liquids, gases, vapours
Particlepreparatione.g. crystallisation, precipitation,milling, spray drying, size enlargement, drying, coating of particles
Tablet manufacturinge.g. mixing, filling, compression
PREPARATION OF GRANULES BY WET GRANULATION
Figure 1. Processing stresses involved during particle preparation and tablet manufacturing (modified from York (1992)).
In the production of pharmaceutical tablets it is common to agglomerate fine particles with the intention to improve technical and biopharmaceutical properties. Aggregates are, in general, more porous than the primary particles of which they consist. The mostfrequent procedure for the preparation of aggregates or granules in this context is wet granulation, where an agglomeration liquid is added during agitation that will contributeto the formation of liquid bridges and, consequently, to hold the fine particles together. Water, alcohols or mixtures of water and an alcohol are frequently used as agglomeration liquids, often in combination with a polymeric binder. The size-enlargement process involves nucleation of particles followed by coalescence of agglomerated particles (Kristensen, 1988). It is also generally believed that the liquid saturation of the granules, i.e. the ratio of the volume of agglomeration liquid to the volume of the intragranular pores, increases during the granulation procedure in parallel with granule growth (Kristensen et al., 1984). The wet-massing procedure can be combined with an extrusion-spheronisation process to obtain spherical granules, often denoted pellets (Vervaet et al., 1995). Pellets have excellent flowability and the shape and smooth surface texture facilitate coating with a polymer film for controlled drug delivery (Vervaet et al., 1995). Capsules may be filled with pellets or the pellets may be tabletted to form so-called multiple unit pellet systems. The preparation of pellets by extrusion-spheronisation requires a critical balance between plasticity and brittleness of the wet mass. Excipents that give the wet mass the required properties are, therefore, added to the formulation. Microcrystalline cellulose (mcc) has been found to be a particularly useful pelletisation aid in this respect because it is able to “hold” water in a way crucial for the process (Fielden et al., 1988). In addition, it has been suggested that the extrusion process results in complete filling of the pore space between the primaryparticles with liquid, irrespective of the liquid content (Jerwanska et al., 1995).
10

Drying of granules
After the size-enlargement process the agglomeration liquid is subsequently removed by drying, and cohered granules remain. Common drying methods in pharmaceuticalindustries are convective methods like tray drying and fluid-bed drying. The use of microwave and vaccum drying has also been reported for pharmaceutical granulations(van Scoik et al., 1991). During the first stage of convective drying the rate of evaporation per unit area of the drying surface is independent of time, with the evaporation rate being close to that from an open dish containing liquid. This first stage of drying is called the constant rate period (Brinker and Scherer, 1990). During this period the rate of heat transfer and the rate of mass transfer are balanced (van Scoik et al., 1991) and the rate of drying depends on factors such as the air temperature, the rate of air flow, the latent heat of vaporization and the relative humidity of the drying gas (van Scoik et al., 1991). The constant rate period continues until a critical point is reached, after which the drying rate reduces progressively, i.e. drying proceeds at a falling rate. When this period begins the rates of heat and mass transfer are no longer balanced, because the surfaces of the granules are no longer kept completely wetted and the liquid front descends into the pores of the granules (van Scoik et al., 1991). Hence, the drying rate hereon also depends on the movement of liquid inside the granules. Thedrying progresses until a critical liquid content is reached after which no more liquid can be removed under the given drying conditions.
The influence of drying conditions on structural and functional properties of granules It has been demonstrated that changes in structural and functional properties of granulated particles may be related to the drying conditions and the choice of drying method. It has been suggested, for example, that drying in a microwave oven gives rise to more porous granules with a lower fracture strength than drying in a convective dryer (Bataille et al., 1993). However, in contradiction, it has also been observed thatmicrowave drying may give granules of similar structure as conventional tray drying (Mandal, 1995). Furthermore, several articles have communicated differences in granule properties depending on the type of convective dryer used. Static bed drying has been reported to give granules of a higher strength, faster drug dissolution (Dyer et al., 1994), higher friability and bulk density (Remon and Schwartz, 1987) compared to granulesdried in a fluid-bed dryer. The effect of the drying method as such on the tabletting properties of granules and pellets has also been communicated in the literature (Chatrath and Staniforth, 1990). In addition, it has been shown that freeze-drying may give extremely porous granules, (Kleinebudde, 1994; Habib and Shangraw, 1997; Habib et al., 2002) with a significantly increased deformability and tablet-forming ability, compared with other drying methods (Habib and Shangraw, 1997). It should also be pointed out in this context that the drying of solids, e.g. the removal of crystal water (Hüttenrauch and Keiner, 1979) and spray drying (Corrigan et al., 1984), may result in solid-state transformations and in the production of amorphous materials, which will be discussed further below.
Contraction of granules during dryingIt has been recognised that pharmaceutical granules containing cellulose may contract and densify considerably during the drying process (Kleinebudde, 1994). However, the phenomenon of granule contraction has received limited attention in the pharmaceutical
11

literature. There are, however, a large number of reports in the ceramics literature(Scherer, 1990) since contraction and cracking during drying of ceramics have been identified as critical factors for their structure and functionality. The contraction of a wet plastic body is thought to be driven by a compressive force developed due to gradients in, e.g., capillary and osmotic pressure in the liquid in the pores, which drags particles in the material together during drying (Newitt and Coleman, 1952; Scherer,1990; Hasatani et al., 1993). Hence, the material will densify, i.e. the porosity decreasesduring the drying process. Most of the contraction is believed to occur during the constant rate period of drying and the volume of contraction of ceramics generally equates the volume of water removed by drying (Scherer, 1990). In addition, it has also been suggested that an increased drying rate increases the degree of contraction (Hasatani et al., 1993). Thus, contraction of granules during drying may be an importantfeature for the structure, e.g. porosity, and functional behaviour of the dry granules.
Other factors influencing structural properties of granules
The processing conditions during the wet massing procedure can markedly affect the properties of granules. It is widely considered that granules are densified during the wet granulation procedure and variables such as the intensity of the mechanical treatment(Holm et al., 1983; Jaegerskou et al., 1984) can affect the degree of densification during the process and thus the resultant granule porosity. Furthermore, the conditions and the equipment settings during extrusion-spheronisation, such as the diameter of the perforations, the thickness of extrusion screen and the extrusion and spheronisation speed may affect, for instance, pellet shape, size and bulk density (Vervaet et al., 1995). Bataille and co-workers (1993) reported a decreased intragranular porosity of pellets produced with an increased spheronisation speed.
The size of the primary particles constituting the granules (Hunter and Ganderton, 1972;Chalmers and Elworthy 1976) and the amount of agglomeration liquid (Jaegerskou et al., 1984; Tapper and Lindberg, 1986) have been reported to affect granule porosity as well. Furthermore, it has been suggested that densification of granules during wet granulation is influenced by the surface tension of the agglomeration liquid (Wells and Walker, 1983; Ritala et al., 1988) and it has been shown that the porosity and pore structure of granules can be significantly changed by varying the ratio of ethanol and water in the agglomeration liquid (Millili and Schwartz, 1990; Johansson et al., 1995).
PREPARATION OF PARTICLES BY SPRAY DRYING
Spray drying is a particle preparation technique that is widely used within the food, chemical, biochemical and pharmaceutical industries (Broadhead et al., 1992) for different applications. During spray drying, a solution or suspension is atomized and converted to solid particles by rapid evaporation in a one step process. Spray drying encompasses four consecutive stages: (i) atomization of the feed solution into a spray;(ii) spray-air contact; (iii) convective drying of the spray; and (iv) separation of the dried product from the drying gas (Broadhead et al., 1992). There are many different types of spray driers, however they generally consist of a feed solution delivery system,an atomizer, a heated air supply, a drying chamber, a solid-gas separator and a product
12

collection system (Corrigan, 1995). The atomizing process provides a large drying surface area for heat and mass transfer, which promotes rapid drying. The actual dryingtime of a droplet is in fact only a fraction of a second, with the consequence that amorphous or partially amorphous materials may be produced (Corrigan, 1995). However, the temperature of the solid particles is kept low by the evaporative cooling,making spray drying suitable for heat-sensitive materials, e.g. proteins (Forbes et al., 1998). Spray-dried particles also tend to be spherical and have a narrow particle size distribution (Broadhead et al.; 1992, Aulton, 2002). The spherical shape facilitatesfilling of such particles into capsules or dies in tablet presses. They may also be hollowand sometimes with open pores which arise from the drying process (Broadhead et al., 1992; Meenan et al., 1997; Aulton, 2002; Eissens et al., 2002). By changing the spray drying conditions it is possible to alter and control properties such as particle shape, particle size distribution, bulk and particle density, flowability, solid-state propertiesand moisture content (Broadhead et al., 1992; Corrigan, 1995), making it a suitable method for preparation of pharmaceutical particles. The use of spray drying for a wide range of pharmaceutical applications, such as tablet excipient production (Gunsel and Lachman, 1963; Fell and Newton, 1971; Eissens et al., 2002.), microencapsulation(Voellmy et al., 1977), enhancement of dissolution rate (Corrigan et al., 1984) and dry powder aerosol formulation (Dunbar et al., 1998; Forbes et al., 1998) have been reported. Spray-dried lactose was the first direct compression excipient introduced and,in this context, is probably the most well known example (Gunsel and Lachman, 1963).
Amorphous solids
DefinitionsThe terms “disordered”, “non-crystalline”, “activated” or “glass” are sometimes in the literature also used to denote amorphous material. Furthermore, “glass” is often used to describe an amorphous material that is below the glass transition temperature, whilst “super-cooled liquid” or “rubber” is used when it is above the glass transition temperature (Hancock and Zografi, 1997). Solid materials may be crystalline, amorphous or partially amorphous. In amorphous solid materials the position of the molecules relative to one another is random as in liquids, and there is a lack of the three-dimensional long-range order typical for crystalline materials (Hancock and Zografi, 1997). Nevertheless, these materials exhibit short-range order over a few molecular dimensions. An amorphous material has quite different properties in relation to the crystalline counterpart, e.g., it has a higher internal energy, increased molecularmobility and larger free volume. Free volume is the empty space in a material that is not occupied by molecules, and since it is greater in the amorphous state than in the crystalline state, because of the differences in molecular packing, the amorphous formof a solid has a lower density (Hüttenrauch, 1978).
Amorphous solids may be formed by rapid precipitation from a solution, e.g. lyophilisation (Shamblin et al., 1996; Craig et al., 1999), spray drying (Corrigan et al., 1984; Elamin et al., 1995); mechanical treatment of crystals, e.g. milling (Hüttenrauch, 1978; Elamin et al., 1994; Willart et al., 2001), compaction (Hüttenrauch, 1978; Ek et al., 1995); supercooling from a melt (Mosharaff and Nyström, 1999) and the removal of crystal solvents (Hüttenrauch and Keiner, 1979; Willart et al., 2002).
13

Most pharmaceutical materials in solid dosage forms are crystalline and often is a lot of effort made to ensure that the active ingredient is highly crystalline for reasons of stability. However, amorphous or partially amorphous drugs and excipients may be found in formulations, for instance because many tablet excipients, such as binders, fillers and film forming agents are amorphous or partially amorphous.
Pharmaceutically advantageous properties of amorphous material Amorphous materials have certain properties that can be of advantage in drug formulation. It might for example be beneficial to render a sparingly soluble drug amorphous, since it will have a lower energetic barrier to overcome when entering a solution than the crystalline counterpart. This can result in increased solubility(Hancock and Parks, 2000), dissolution rate (Corrigan et al., 1984) and bioavailability (Fukuoka et al., 1987). It has also been shown that amorphous sugars provide stability to proteins in spray-dried or freeze-dried formulations (Suzuki et al., 1998) and that thetabletting properties of a material can be significantly improved if it is made amorphous(Hüttenrauch and Keiner, 1976; Vromans et al., 1986; Sebhatu and Alderborn, 1999).
The glass transition temperatureThe glass transition temperature (Tg) is a characteristic property of an amorphousmaterial. At Tg a significant change is observed in the molecular mobility and the freevolume. The amorphous material undergoes a change from a rigid glassy state with high viscosity to a more flexible rubbery state with a much lower viscosity (Hancock and Zografi, 1997). The molecular mobility of an amorphous material is significantly higherabove Tg resulting in a higher reactivity and propensity to crystallise. However, it has been reported that amorphous solids exhibit marked molecular mobility even well below the glass transition temperature (Hancock et al., 1995). The Tg of an amorphousmaterial can be determined with differential scanning calorimetry by heating the material and observing any change in the heat capacity. The Tg is associated with a change in heat capacity and sometimes also with a relaxation endotherm, (Hancock et al., 1995) which may arise from a mismatch between heating and cooling rates or as a consequence of the amorphous material not being in a state of equilibrium, enabling it to relax over time (Craig et al., 1999). The Tg is a dynamic property of a material and can depend on, for instance, the heating rate and the moisture content.
Amorphous materials and water It is generally considered that water can interact with crystalline solids by adsorption of water vapour to the solid-air interface, crystal hydrate formation, deliquescence and capillary condensation (Ahlneck and Zografi, 1990). However, amorphous solids absorb water into the structure with the consequence that a significant quantity of water can be taken up by the material. The amount of water that can be absorbed is related to the total mass of solid rather than the surface area available for sorption, such as for crystallinematerials (Ahlneck and Zografi, 1990). The water that is absorbed into the amorphousmaterial increases the free volume and reduces the degree of bonding between molecules in the solid, resulting in an increased molecular mobility (Oksanen and Zografi, 1993) and depressed Tg (Hancock and Zografi, 1994).
14

Crystallisation of amorphous materials A clear disadvantage of low molecular weight amorphous materials is that they are thermodynamically and physically unstable. Moisture and heat may increase the molecular mobility to such an extent that a conversion to the crystalline state spontaneously occurs under certain conditions. Thus, an amorphous material is less stable than the crystalline form of the same chemical entity.
The crystallisation of amorphous materials involves the formation of nuclei and crystal growth from the nuclei produced (Schmitt et al., 1999). Crystallisation kinetics may be evaluated for amorphous substances with the Johnson-Mehl-Avrami (JMA) theory, which describes the exponential growth of the crystalline fraction of a sample over time.Its use has been reported for the determination of isothermal crystallisation kinetics of amorphous lactose in the presence and absence of seed crystals (Schmitt et al., 1999).
It has been suggested that the rate of crystallisation of an amorphous material is highest somewhere between the Tg and the melting temperature (Jolley, 1970). However, it is also believed that amorphous materials can exhibit sufficient molecular mobility (Hancock et al., 1995) to crystallise at temperatures even below the Tg (Yoshioka et al., 1994).
Stabilisation of amorphous materials
Amorphous solids have unique properties that, clearly, can be of advantage in drug formulation. However, the underlying tendency of such materials to crystallise needs to be circumvented if they are intended to be used in pharmaceutical preparations becauseany phase-transformation during processing and storage may cause severe changes in functional properties such as the dissolution and bioavailability. Ensuring that the materials do not crystallise can be achieved by storage at a temperature significantly below Tg and by avoiding exposure to substances that depress Tg, e.g. water. However, for practical reasons, this strategy may not always be possible. Interest to modulate the solid-state reactivity of amorphous substances by other means has therefore arisen. A challenging alternative in this context is to co-process the amorphous material with a small amount of a water-soluble polymer with a high glass transition temperature to produce a more stable composite material (Fig. 2). In this context, a compositeamorphous material is defined as one in which the compounds form a solid solution and which consists mainly of a low molecular weight substance with a smaller fraction of a polymer. The most frequently used preparation methods for composite materials are freeze drying (Shamblin et al., 1996; Shamblin et al., 1998; Taylor and Zografi, 1998; Shamblin and Zografi, 1999; Zeng et al., 2001) and precipitation from organic solvents (Yoshioka et al., 1995; Matsumoto and Zografi, 1999). It has also been demonstratedthat spray drying can be a feasible method for the preparation of such materials(Corrigan and Holohan, 1984; Corrigan et al., 1985; Takeuchi et al., 2000a; Takeuchi et al., 2000b), while other methods may fail to solidify the materials in the amorphousform (Corrigan and Crean, 2002).
Polymers that have been recognised as stabilising agents for amorphous materialsinclude poly(vinylpyrrolidone) (PVP) (Corrigan and Holohan, 1984; Corrigan et al., 1985; Yoshioka et al., 1995; Shamblin and Zografi, 1999; Takeuchi et al., 2000a),
15

hydroxypropylcellulose (Takeuchi et al., 2000a), methacrylate polymers (Kotiyan and Vavia, 2001) and sodium alginate (Takeuchi et al., 2000a; Takeuchi et al., 2000b).
Figure 2. Schematic illustration showing inhibition of crystallisation of an amorphous material by incorporation of a stabilising polymer.
The stability of composites has been evaluated in terms of their resistance to transformations induced by heat- and moisture, such as the glass transition temperature,crystallisation temperature, crystallisation under high relative humidities and also by enthalpy relaxation measurements (Shamblin and Zografi, 1998). The mechanism of stabilisation is believed to be related to a general increase in Tg of the system, which can be predicted by assumptions of perfect volume additivity at Tg (Gordon and Taylor, 1952) or based on thermodynamic considerations (Couchman and Karasz, 1978). Hence, incorporation of a polymer with a low Tg may promote crystallisation, which has been shown to be the case for spray-dried lactose/polyethylene systems (Chidavaenzi et al.; 2001, Corrigan et al., 2002). However, it has been reported that stability may be significantly improved despite almost unchanged Tg of the system, which has led to the assumption that other factors than just a general increase in Tg, such as specificinteractions (Yoshioka et al., 1995; Shamblin et al., 1996; Matsumoto and Zografi, 1999; Shamblin and Zografi, 1999), are also of importance for the enhanced stability.
COMPRESSIBILITY AND COMPACTABILITY OF POWDERS
Critical characteristics of pharmaceutical tablets are an adequate mechanical strength to withstand stresses during processing and transportation, and release of the active in a preordained manner. Tablets are usually prepared by uniaxial compression using eccentric presses or rotary tabletting machines. When a particle bed in a die is subjectedto an external load its volume is reduced and the interparticulate porosity is decreased.The term compressibility is often used to describe the volume reduction ability of a material, whilst the compactability is the ability of a material to form a tablet of a specified mechanical strength (Leuenberger, 1982). The mechanism of volume
16

reduction involves an initial rearrangement of the particles, followed by fragmentationand deformation (Duberg and Nyström, 1986). The dominating volume reduction mechanism during the compression event depends on the properties of the material. In addition, the sequence of volume reduction mechanisms has been suggested to be different for granules than for non-porous particles (van der Zwan and Siskens, 1982; Johansson, 1999), which will be discussed below. During compression, the particles will be brought together into such close proximity that interparticulate bonds can be created, resulting in a tablet of a certain strength. It has been suggested that the mechanical strength is governed by the interparticulate bonding mechanism and the area over which these bonds interact (Nyström et al., 1993).
The strength of tablets is commonly determined by diametral compression testing and subsequent calculation of the radial tensile strength (Fell and Newton, 1970). The prerequisite for the calculation of the radial tensile strength is that the tablet fails in tension during testing. Factors affecting the strength of tablets have been discussed considerably in the literature and it has been suggested that properties of the materials,such as the particle size and shape (Alderborn and Nyström, 1982ab; Alderborn et al., 1988), plastic deformation (Roberts and Rowe, 1987; Hiestand, 1991), the fragmentation propensity, elastic deformation (Nyström et al., 1993), the moisturecontent (Sebhatu et al., 1997) and processing factors like the applied load and the time of loading (Roberts and Rowe, 1987) have an effect upon the mechanical strength of tablets. Several models that describe the strength of tablets have been proposed in the literature. These models are generally based on two different theoretical approaches, i.e., bond summation (Rumpf, 1962) or kinematic crack propagation (Mullier et al., 1987; Kendall, 1988).
Tabletting of granulated materials
Granules are often prepared with the intention of improving tabletting properties such as the compactability of a material. Granules are generally porous, therefore tablets of granulated particles contain both inter and intragranular pores. It has been proposed that the volume reduction process of granules (van der Zwan and Siskens, 1982) includes four successive stages: (i) particle rearrangement and the filling of intergranular pores, (ii) fragmentation and plastic deformation of the granules, (iii) filling of the intragranular pores with primary particles, i.e., the densification of the granule, and (iv) fragmentation and plastic deformation of the primary particles. It is believed that microcrystalline cellulose granules fragment to a limited extent during compression (Johansson et al., 1995) and experiments have revealed that the incorporation of a hard material into such granules did not change the fragmentation propensity (Nicklasson et al., 1999a).
The intragranular porosity has been found to have a significant influence on tablet strength, irrespective of the granule composition (Millili and Schwartz, 1990; Wikbergand Alderborn, 1991; Wikberg and Alderborn, 1993; Johansson et al., 1995; Nicklasson et al., 1999b). Furthermore, it has been suggested that increased intragranular porosity increases the propensity of the granules to fragment facilitating the formation of stronger tablets (Wikberg and Alderborn, 1991). For granules less prone to fragment it has been proposed that the intragranular porosity is critical for the compressibility of the
17

granules and for the structure and tensile strength of the tablets formed. An increased intragranular porosity increased the degree of deformation, resulting in formation of a closer intergranular pore structure during compression and, therefore, stronger tablets (Johansson et al., 1995). The compression shear strength of granules during confined compression has also been found to be related to the intragranular porosity (Nicklasson and Alderborn, 2000).
The size and shape of the granules may also influence the tabletting behaviour, e.g. decreasing the granule size has been claimed to facilitate the formation of stronger tablets (Li and Peck, 1990; Riepma et al., 1993). However, for mcc granules it has also been shown that the compactability was independent of granule size at moderate applied pressures and slightly higher for larger granules at a higher applied pressure (Johansson et al., 1998). In addition, it has been found that irregularly shaped granules formstronger tablets than spherical ones of similar intragranular porosity (Johansson and Alderborn, 2001).
The properties of the primary particles comprising a granule may also affect thetabletting behaviour, examples being granule deformability, mode of deformability andcompactability (Nicklasson et al., 1999a; Nicklasson and Alderborn, 1999). In this context, polymeric binders are frequently incorporated into granules to improve the granule and tablet strength, and factors such as type, amount and distribution of the binder (Wikberg and Alderborn, 1993) may influence the strength. The addition of materials, like lubricants and surface-active agents (Femi-Oyewo and Spring, 1982), have been reported to decrease tablet strength. However, the reduction in strength caused by a lubricant is related to the degree of fragmentation that takes place (Nyströmet al., 1993).
Tabletting of amorphous materials
It is generally accepted that the tabletting properties of a material may be changed andthe compactability improved if it is made amorphous. However, the effect may depend on the type of material, e.g. relatively small differences in mechanical properties between the crystalline and the amorphous form of a drug have been reported (Hancock et al., 2002).
The concept of mechanical activation to increase the compactability of materials hasbeen presented by Hüttenrauch and Keiner (1976), who found that the tablet strength increased with milling time and the degree of disorder. Furthermore, it has been suggested that induced disorder increased the surface reactivity and plastic deformability, resulting in a sinter-like reaction between particles during compaction,facilitating interparticulate bonding, and tablet strength (Hüttenrauch, 1983).
Commercially available spray-dried lactose is partially amorphous and the particles consist of -lactose monohydrate crystals glued together by amorphous lactose (Bolhuis and Chowhan, 1996). The spray-dried quality has been reported to have a markedlyhigher compactability than -lactose monohydrate, which has been attributed to the amorphous part of the spray-dried lactose (Vromans et al., 1986). Rendering a materialamorphous can change the volume reduction behaviour in such way that it may be more
18

readily deformed and less prone to fragment than the crystalline counterpart (Vromanset al., 1986; Sebhatu and Alderborn, 1999) and increased moisture content may increase the particle deformability and tablet tensile strength (Vromans et al., 1986; Sebhatu etal., 1997) owing to the increased particle contact area during compression (Sebhatu et al., 1997). Furthermore, it has been proposed that the difference in compactabilitybetween the amorphous and a crystalline form of lactose is mainly related to a difference in particle-particle adhesivity (Sebhatu and Alderborn, 1999).
Incorporation of substances such as stabilising polymers into amorphous materials has been reported to significantly change their deformability and tablet-forming ability (Takeuchi et al., 1999), in addition to which, there are also indications in the literature that the observed effect on the compactability is related to the type of polymer(Takeuchi et al., 2000a).
CONCLUDING REMARKS CONCERNING THE PERTINENT LITERATURE
The literature survey presented here indicates that the drying conditions and choice of drying method may significantly affect structural and functional properties, such as tabletting behaviour, of pharmaceutical particles. Thus, it seems plausible to control and optimise functional properties by the drying process. However, this requires a fundamental knowledge of relationships between stresses during the drying process and particle structural properties critical for their behaviour. In this light, for the two particle types discussed here, i.e. wet-granulated particles and spray-dried particles, someimportant structural properties can be identified. These are discussed below.
Firstly, it seems that the intragranular porosity is an important characteristic for the compression behaviour and compactability of granules. Hence, factors controlling the intragranular porosity are of considerable interest for the engineering of granules with optimal tabletting properties. It has been found that varying the ratio of ethanol and water in the agglomeration liquid may result in granules with a markedly different intragranular porosity. However no explanation has been given for these observations and the relative importance of the drying and the wet-agglomeration steps has not been addressed. One possible explanation is that differences in granule contraction during drying can explain the differences in porosity. This would imply that the drying rate, in turn, could be a factor controlling the porosity and thereby the tabletting properties ofgranules because it has been reported that the drying rate may influence the contraction.
Secondly, it has also been recognised that amorphous materials may have advantageous functional properties, e.g., increased compactability in comparison to their crystallinecounterparts, and that the dramatic evaporation that occurs during spray drying can enable the formation of amorphous material. However, the use of amorphous materialsin pharmaceutical preparations is limited because of their metastable character. Thus, the incorporation of a water-soluble polymer in an amorphous substance to form an amorphous composite material represents an attractive way to increase the stability of amorphous materials. In this context, spray drying is a pharmaceutically relevant method by which to prepare such particles that seems to facilitate the formation of an intimate mix of the low molecular weight substance with the polymer. Amongst stabilising polymers, PVP is the most frequently used in the literature, however despite
19

this fact, the role of the molecular weight of PVP for the stability of the amorphousmaterial is not clear (Matsumoto and Zografi, 1999; Zeng et al., 2001). Furthermore,there are some very relevant questions that need to be addressed, in terms of the effect of the incorporation of polymers in amorphous material on the long-term stability and tabletting behaviour.
The examples given above clearly show the need for greater knowledge concerning drying-related effects on particle structural characteristics decisive for thecompressibility, tablet-forming ability and the solid-state stability. This would lead to better prediction of the functional behaviour of particles and enable the drying process to be used to control pharmaceutical functional properties.
AIMS OF THE THESIS
The general objective of this thesis was to deepen the understanding of relationships between stresses arising during the drying process and structural properties of particles important for their functionality, and of the use of the drying process to engineer particles with respect to tabletting properties and solid-state stability.Two common pharmaceutical preparation methods (i.e., wet granulation and spray drying) that involve particle formation with the aid of a liquid, subsequently removedby convective drying, were selected for the current investigation. The overall aim was consequently subdivided into the following specific aims:
To investigate the importance of the composition of the agglomeration liquid on contraction, densification and the degree of liquid saturation of microcrystallinecellulose pellets during static convective drying (Paper I).
To study the relative importance of the drying and wet granulation phases, and the effect of the rate of drying on the porosity, compression shear strength and tablet-forming ability of microcrystalline cellulose pellets (Papers I-II).
To investigate the effect of polymer content and molecular weight on the morphology, resistance to transformations induced by heat and moisture, and the relationship between the critical relative humidity for crystallisation and thestorage time of amorphous lactose in spray-dried composite particles withpoly(vinylpyrrolidone) (Papers III-IV).
To illustrate how the compression behaviour and tablet-forming ability of spray-dried lactose can be modulated by addition of stabilising polymers and a surfactant to the feed solution during spray drying (Paper V).
MATERIALS
All the test materials used in this thesis are pharmaceutical excipents commonly used in solid oral dosage forms.
20

Microcrystalline cellulose (Avicel PH101, FMC, Ireland, with an apparent particle density of 1.58 g/cm3), ethanol (Finsprit 95% w/w, Solveco Chemicals AB, Sweden) and deionised water were used for the preparation of the pellets in Papers I and II. Cellulose is a linear polymer consisting of -D-glucosopyranoside units (Fig. 3), and mcc particles are composed of fibrils, which are believed to have both paracrystallineand crystalline regions (Mathur, 1994; Bolhuis and Chowhan, 1996). Mcc has excellent binding properties and is widely used as a binder and filler in oral formulations (Bolhuisand Chowhan, 1996). It has also been found to be a valuable extrusion/spheronisationaid that is crucial for the preparation of pellets (Fielden et al., 1988).
-lactose monohydrate (Pharmatose 200M, DMV, The Netherlands, with an apparent particle density of 1.54 g/cm3) was used for preparation of spray-dried particles in Papers III-V. Lactose is a naturally occurring disaccharide consisting of one galactoseand one glucose unit (Fig. 3) that exists in two isomeric forms, -and -lactose. Lactoseis widely used as a filler in tablets, capsules and lyophilised products and as a drug carrier in inhalation products (Goodhart, 1994; Bolhuis and Chowhan, 1996). Amorphous lactose may be readily produced by, e.g., spray drying, and has often been used in the literature as a model substance for amorphous materials.
Poly(vinylpyrrolidone) (PVP) with a viscosity-average molecular weight of 8200 (PVPK17) and a glass transition temperature of 140 C (Kollidon 17 PF, BASF, Germany, with an apparent particle density of 1.18 g/cm3) and PVP with a viscosity-average molecular weight of 1 100 000 (PVPK90) and a glass transition temperature of 176 C (Kollidon 90 F, BASF, Germany, with an apparent particle density of 1.23 g/cm3) were used for the preparation of spray-dried composite particles in Papers III-V. PVP is a linear polymer consisting of 1-vinyl-2-pyrrolidinone units (Fig. 3) which is frequently used in both solid and liquid formulations, e.g. as a binder in tablets and a viscosity-increasing agent in suspensions (Walkling, 1994).
Polysorbate 80 (Polysorbatum 80, Apoteket AB, Sweden) was used for the preparation of spray-dried composite particles in Paper V. Polysorbate 80 or polyoxyethylene sorbitan fatty acid ester is a non-ionic surfactant that is used as an emulsifying,solubilising and wetting agent in topical, oral and parenteral formulations (Leyland, 1994).
OCH2OH
HH
H
OH
OH
HH
O
OO
H
OHHCH2OH
H
H
OH
H
n
OCH2OH
HOH
HH
OH
OH
HH
OCH2OH
HH
H
OH
OH
HOH
H
O N OCH CH2
n
Figure 3. Structural formula of -D-glucosopyranoside unit (left), -lactose(middle) and 1-vinyl-2-pyrrolidinone (right).
21

EXPERIMENTAL SECTION
Particle preparation
Preparation of pellets The pellets of microcrystalline cellulose in Papers I and II were prepared by an extrusion/spheronisation technique. Mixtures containing different proportions of water and ethanol were used as the agglomeration liquid. The powder was dry mixed for 1 min at 500 rpm in a high shear mixer (model QII, Donsmark Process Technology, Denmark), thereafter the agglomeration liquid was added by atomisation at a flow rate of 100 g/min, whereupon wet mixing took place for 1 min at 500 rpm. The wet powder mass was then extruded in a radial screen extruder (model E140, Nica System, Sweden) equipped with a 1.2 mm thick screen with circular openings of diameter 1.0 mm. The extrudate was spheronised for 3 min at 875 rpm on a radial plate spheroniser (modelS320, Nica System, Sweden). The pellets were then immediately placed in closed containers and stored at room temperature.
Drying of pelletsA comparison between parallel changes in the volume and liquid content of the pellets during drying (Paper I), was made possible by placing pellets on microscope slides to dry by leaving the slides in the open atmosphere in the laboratory (about 20 Ctemperature and 40 % RH) without any forced convection. Pellets agglomerated with water were placed in wells in a specially constructed microscope slide. At set intervals,the pellets were photographed in an optical light microscope (model Vanox, Olympus,Japan) at 1.3 times magnification and weighed on an analytical balance immediatelyafter each photograph. The pellets agglomerated with a mixture of water and ethanolwere randomly placed on an ordinary microscope slide and some of them were photographed in the optical light microscope at set intervals. Drying profiles were obtained by drying an approximately equal number of pellets on a microscope slide on an analytical balance. The volume of the pellets and their liquid content were calculated from the microscopy and weight data. The pellet volume (Vp) was calculated from the projected area diameter of the pellets, i.e. the diameter of a circle of equal projected area to that of the pellet (Dp), by assuming a perfect spherical shape, i.e.:
Vp = Dp3 / 6 (1)
The liquid content of the pellets was calculated as the mass of liquid divided by the mass of solid and as the volume of liquid divided by the mass of solid (Ht).
Assessment of the porosity, contraction, densification and degree of liquid saturation of pellets during dryingThe initial porosity and the porosity at different times during the drying process of each individual pellet (Et) was in calculated from the following equation (Paper I):
Et (%) = (1-(Vdry edry / Vt a
dry)) x 100 (2)
where Vdry and Vt are the volumes of the dry pellet and the wet pellet (at time t), respectively. e
dry and adry are the effective particle density of the dry pellet (obtained
22

by mercury pycnometry as described below) and the apparent particle density respectively. The contraction and densification of each pellet during the drying processwere calculated as follows (Papers I and II):
Contraction (%) = (1-(Vt / V0)) x 100 (3)
Densification (%) = (1-(Et / E0)) x 100 (4)
where V0 is the initial pellet volume and Vt is the pellet volume at time t. E0 and Et are the initial pellet porosity and the pellet porosity at time t, respectively. The ratio of the pore volume occupied by the agglomeration liquid to the total volume of the pores, i.e. the degree of liquid saturation (Kristensen et al., 1984) St, was calculated for each individual pellet at set times during the drying process using the following equation (Paper I):
St (%) = (Ht a (1- Et / Et)) x 100 (5)
where Ht is the liquid content at time t (volume liquid/mass of solid), Et is the porosity at time t (from Eq. 1) and a is the apparent particle density.
Drying of pellets under different conditionsIn Paper II a convective static dryer was constructed with the intention of investigatingthe effect of the drying rate on the densification of the pellets during drying. Four different sets of pellets (agglomerated with different agglomeration liquids) were dried under six different drying conditions (21-60 C, 15-58 l/min) in the dryer and under ambient conditions as well. The weight loss on drying was determined by removing the pellets from the dryer and placing them on an analytical balance at given time intervals. Drying was continued until the weight of the pellets was constant with time. From the drying rate profiles, a drying rate constant (kD) was calculated as the gradient of the relationship between ln mt / m0 and the time, i.e.:
ln mt / m0 = kD t (6)
where mt and m0 are the amount of removable liquid at time t and the total amount of removable liquid, respectively. It has been suggested that during tray drying of a bed of granular material, the reduction in liquid content with time commonly obeys a log-lin relationship, i.e. ln mt / m0 relates linearly to the drying time (Carstensen and Zoglio, 1982). The weight of the pellets before and after drying was determined and their liquid content calculated as the percentage mass of liquid/mass of solid. After drying, these pellets were stored in a desiccator (40% RH) for at least 10 days prior to characterisation and tabletting.
Preparation of spray-dried particlesParticles of only lactose and composite particles of lactose and different contents of PVPK17 or PVPK90 (two-component particles) were prepared by spray drying (Papers III-V). Three-component composite particles with 0.1% w/w polysorbate 80 (as a proportion of the dry mass) were also prepared (Paper V). Water solutions of the materials were spray-dried in a spray dryer (Niro Atomizer, Niro A/S, Denmark),
23

equipped with a rotary atomizer. The liquid feed rate was 17 ml/min (peristaltic pump,Watson Marlow 505S, Watson Marlow Ltd., England), inlet and outlet temperatures 170 5 C and 95 5 C, respectively. The particles were stored in a desiccator over P2O5
(0% RH) for at least 7 days, after which a size fraction 5-15 m was prepared by air classification (Alpine 100 MZR, Alpine AG, Germany). Finally, the particles were stored at room temperature and 0% RH for 7 days and then for a further 7 days at 40 Cand 0% RH. Thereafter the particles were stored at room temperature and 0% RH. Particles used for the preparation of tablets were pre-stored in 33% RH until a constant weight was reached.
Characterisation of the prepared particles
MorphologySuspensions of spray-dried particles in liquid paraffin were prepared and sonicated until complete deaggregation was achieved. The particles were then inspected in an opticallight microscope (model Vanox, Olympus, Japan) at 40 times magnification (Papers III and V). Images of spray-dried particles, pellets and also of the upper surfaces of tablets formed from spray-dried particles were taken with the aid of a scanning electron microscope (Leo 1530 Gemini, Leo, UK (Papers III and V) and Philips SEM 525, The Netherlands (Paper II)).
CircularityIn Paper II, pellets were manually dispersed on microscope slides and photographed in the light microscope at two times magnification. The photographs were digitalised and the perimeter (P) and projected area (A) were determined by image analysis (NIH Image 1.61, USA, available on the Internet at www.nih.gov) The pixel resolution was 5.3-5.2 µm/pixel. A steel sphere (SKF, Sweden, with a diameter of 1 mm), with an assumed circularity of 1.00 (Eriksson et al., 1997), was used as a reference in each picture. The circularity (C) of the pellets was calculated from the following equation (Cox, 1927):
C= 4 A / P2 (7)
Apparent particle density The apparent particle density of the raw materials (Papers I-V) and the spray-driedparticles (Papers III and V) was determined using a helium pycnometer (AccuPyc 1330, Micromeritics, USA). The density of mixtures of materials (Papers III and V) was estimated according to the following equation:
= (w1 + w2) / ((w1 / 1) + (w2 / 2)) (8)
where w1 and w2 are the weight fractions and 1 and 2 the densities of the components.
24

Assessment of pellet porosityThe porosity of the dried pellets (Papers I and II) was calculated from the apparent particle density of the microcrystalline cellulose powder and the effective particle (or pellet) density, according to:
E (%) =(1-( e / a)) x 100 (9)
where e and a are the effective particle density of the dry pellet and the apparent particle density, respectively. The effective particle density was determined by mercurypycnometry at 101 kPa (Paper I) or 90.3 kPa (Paper II) using a mercury porosimeter(Autopore III, Micromeritics, USA). As a plateau was observed around this pressure in the graph showing the cumulative volume of mercury intruded plotted as a function of pressure, it was concluded that the intergranular pores were filled at this point.
Determination of poured bulk density and voidage of poured pellet bed Pellets (Paper II) or spray-dried particles (Paper V) were poured into a 10 ml cylinder (with an inner diameter of 10 mm) to a volume of 9 ml and the poured bulk density was calculated from the volume and weight of the pellets. The voidage of the poured pellet bed, i.e., the relative volume of the intergranular pores, was calculated from the poured bulk density and the effective particle density.
Surface area of powders The surface area of the powders was determined by air permeametry (Paper V). The samples were poured into the sample container and compressed manually to a porosity of approximately 40% whereupon the permeability was determined using a Blaine permeameter. The surface area was calculated by the Kozeny-Carman equation, including a slip flow correction term (Alderborn et al., 1985).
Moisture contentSamples (500 5 mg) of the different materials were weighed in 5 ml glass beakers and then stored in a desiccator over P2O5 until the weight change was less than 1 mg/24 h. Thereafter, the materials were transferred to a desiccator with 33% RH and sampleswere periodically removed to be weighed on an analytical balance until the weight change was less than 1 mg in 24 h. The moisture content was calculated as the percentage mass of liquid/mass of dry solid (Paper V).
CrystallinityThe different spray-dried particles (Papers III and IV) were analysed with a Diffractor D5000 (Siemens, Germany), equipped with a scintillation detector, using Cu-Kradiation, 45 kV and 40 mA. Bragg-Brentano focusing geometry was used and the samples were scanned in steps of 0.02 from 5 to 35 (2 ).
Heat-induced transformationsHeat-induced transformations, i.e. the glass transition, crystallisation, melting and heat of crystallisation were determined (Papers III-V) using a differential scanningcalorimeter (DSC 220, SSC/5200H, Seiko, Japan). The materials were placed in open aluminium DSC pans and analysed under dry nitrogen purge. The samples were heated to 20 C above their glass transition temperature, then cooled with liquid nitrogen to
25

20 C, and subsequently reheated to 300 C. Heating and cooling rates of 10 C/min were used. To determine the effect of absorbed moisture on Tg, the materials were placed in coated aluminium DSC pans that were hermetically sealed and scanned according to the procedure described above. To verify that the moisture content was kept constant, the weight of the samples was monitored before and after each cycle. The calorimeter was temperature and heat calibrated with indium, tin, gallium and mercury.
The theoretical glass transition temperatures of the composite particles were calculated (Paper III) with the Gordon-Taylor equation (Gordon and Taylor, 1952). If the glass transition temperature (Tg), true density ( ) and weight fraction (w) of the componentsare known, the Tg of the mixture can be calculated as follows:
Tgmix = ((w1 Tg1)+(K w2 Tg2)) / (w1+(K w2)) (10)
The constant K was estimated from the true densities and the glass transition temperatures of the two components:
K= ( 1 Tg1) / ( 2 Tg2) (11)
Moisture-induced isothermal transformations To study moisture-induced isothermal (25 C) crystallisation of the materials, a microcalorimeter (2277 Thermal Activity Monitor, Thermometic AB, Sweden) equipped with a humidity control device (perfusion cell) was used (Papers III and IV). Dry nitrogen gas was pumped by means of a peristaltic pump into the humidity control device and mixed with different proportions of nitrogen gas saturated water to obtaindifferent controlled relative humidities. The perfusion cell was calibrated with saturatedsalt solutions (NaCl, CuCl2, Mg(NO3)2 and NaI) according to a method described by Buckton (2000). First, the material was dried with dry nitrogen gas, until no endothermal signal was observed. Thereafter the humidity control device was set to the desired relative humidity (RH), and the heat flow was monitored. This method was used to determine the critical RH for crystallisation within 10 days, time to moisture-inducedcrystallisation and heat of crystallisation at different relative humidities. Moisture-induced isothermal crystallisation was investigated gravimetrically as well (Paper III). Samples (500 5 mg) of the different materials were weighed in 5 ml glass beakers and then stored in a desiccator over P2O5 (0% RH) until the weight change was less than 1 mg in 24 h. Thereafter the materials were moved to desiccators (25 C) with a certain RH and the samples were periodically taken out of the desiccators and weighed on an analytical balance.
Long-term stability In Paper IV samples were placed in desiccators (n=2) with different relative humiditiesand stored at 25 C for up to 6 months. After 3 and 6 months samples were taken out, placed in open aluminium DSC pans and analysed with DSC (as described above). TheDSC thermograms were compared with the thermograms obtained before storage to check for the existence of a glass transition event and a crystallisation exotherm to determine whether the materials were crystalline or amorphous. For those samples that still were amorphous at the end of the six months period, Tg was also determined as a function of relative humidity (as described above).
26

Preparation of tablets
Tablets of pellets (Paper II) were prepared with an instrumented single punch press (Korsch EK 0, Germany) at an applied pressure of 200 MPa. Tablets of spray-dried particles (Paper V) were prepared using the same press at applied pressures of 25, 50, 75, 100, 150, 200 and 275 MPa. The press was equipped with flat-faced punches with a diameter of 11.3 mm. The particles were poured manually into the die, which was lubricated before each compaction with a 1% w/w magnesium stearate in ethanol suspension (Paper II) or magnesium stearate powder (Paper V). The weight of the material was kept constant and the upper punch pressure was controlled by adjusting the position of the lower punch (Paper II). In Paper V the upper punch pressure was controlled by adjusting the amount of material in the die and the distance between the punches was kept constant (3.00 mm).
Determination of porosity, tensile strength and surface area of tablets
After compaction, the weight and dimensions of the tablets were determined.Thereafter, the tablet porosity was calculated from the weight, dimensions and apparent particle density. Within 60 s of compaction, the tablets were also compresseddiametrically at 4 mm/min (Paper II) or 1 mm/min (Paper V) in a tablet-testinginstrument (Holland C50, UK) and the tensile strength was calculated from the diameter, thickness of the compact and the force required to fracture the tablet (Fell and Newton, 1970). Judging from the appearance of the fractured tablets, it was concluded that they failed in tension during testing. In addition (Paper V), tablets were formed in special dies with the instrumented single punch press at an applied pressure of 75 MPa. The permeability of the tablets was determined after compaction using a Blaine permeameter and the slip flow-corrected surface area was calculated from the modifiedKozeny–Carman equation as described by Alderborn et al. (1985).
Calculation of compression parameters
Heckel parameterUsing the Heckel equation (Heckel, 1961a), a compression parameter can be derived that is generally considered to reflect the deformability of the particles in terms of a yield strength or yield pressure (Heckel, 1961b). The Heckel equation describes the relationship between the Heckel number and the applied pressure during compression,as follows:
ln (1 / e) = Pa / Py + intercept (12)
where e is the porosity of the tablet, Pa the applied pressure and Py the yield pressure. Heckel numbers, ln (1 / e), were determined for a series of compaction pressuresbetween 25 and 200 MPa in Paper V. They were determined on ejected tablets, to avoid any influence of particle elastic deformation on the calculated yield pressures (Sun and Grant, 2001).
27

Kawakita parameters The Kawakita equation (Lüdde and Kawakita, 1966) can be used to derive two compression parameters. This equation describes the relationship between the degree of compression (C) of a bed of particles in a die and the applied pressure during compression (P), as follows:
P / C = 1 / ab + P / a (13)
The compression parameter, denoted a in the equation, reflects the total degree of volume reduction of the powder at infinite applied pressure. It has been suggested that the reciprocal of the compression parameter b (i.e. 1/b) represents an indication of the stress at which particles deform or fail during confined compression (Kawakita et al., 1977; Adams and McKeown, 1996; Nicklasson and Alderborn, 2000). The degree ofcompression (C) was calculated from the tablet height and the height before compression, which was itself calculated from the poured bulk density. The height of the bed of pellets in-die at pressure was estimated from the in-die displacement data after correction for punch deformation (Paper II). In Paper V the tablet height was determined “out of die”.
Elastic recoveryThe elastic recovery (ER) of the particles during decompression was also determined in Paper V, using the equation:
ER (%) = ((he – hpmax ) / hpmax) x 100 (14)
where he is the height of the ejected tablet and hpmax the in-die height of the tablet at maximum pressure (calculated from the minimum distance between the punches at zero pressure (3.00 mm) and the punch deformation at the given pressure).
INFLUENCE OF CONTRACTION AND DENSIFICATION DURING CONVECTIVE DRYING ON THE POROSITY, COMPRESSION SHEAR STRENGTH AND TABLET-FORMING ABILITY OF MICROCRYSTALLINECELLULOSE PELLETS
The importance of the composition of the agglomeration liquid on contraction anddensification during drying
Previously it has been reported (Millili and Schwartz, 1990; Johansson et al., 1995) thatsignificant differences in the porosity of mcc granules can by obtained by varying the ratio of water and ethanol in the agglomeration liquid. Thus, to investigate (Paper I) the importance of the drying phase in relation to the wet granulation phase of such granules, mcc pellets were prepared with two different agglomeration liquids, i.e. only water (W)and a mixture of water and ethanol (W/E) (25/75% w/w), and contraction and densification was determined during static convective drying.
Drying of a single layer of pellets on microscope slides facilitated rapid drying for both types of pellets. The initial and final liquid content was similar and the drying profiles
28

0
10
20
30
40
50
60
Con
trac
tion
(%)
1 10 100 1000 10000
Time (min)
100/0% w/w water/ethanol
25/75% w/w water/ethanol
0
25
50
75
100
Den
sific
atio
n (%
)
1 10 100 1000 10000
Time (min)
a.
b.
(i.e., the liquid content as a function of time) were non-linear for both types of pellet with the rate of drying decreasing throughout the drying process. The absence of a constant drying rate period (i.e. one in which the rate of drying per unit area of surface is independent of time) may be explained in terms of a reduction in the area from which liquid was evaporated during drying, as the pellets contracted or due to differences in the drying rate of individual pellets.
Figure 4. Contraction (a) and densification (b) during drying of two mcc pellet types prepared with different agglomeration liquids. Symbols are defined in the graph. Error bars show 95% confidence limits of arithmetic mean values.
The absolute reduction in pellet volume and the relative change in pellet volume (i.e. the contraction) (Fig. 4a) were considerably higher for the pellets agglomerated with water only. However, both pellet types contracted by a significant amount: 54% and 40% for the W and W/E pellets, respectively. The final porosity determined by mercury pycnometry on dry pellets was 7.85 and 33.8%, respectively, for the W and W/E, while the initial porosity (calculated from Eq. 2) of the wet pellets was similar for both types
29

0
10
20
30
40
50
60
Con
trac
tion
(%)
020406080100120
Degree of liquid saturation (%)
25/75% w/w water/ethanol
100/0% w/w water/ethanol
0
10
20
30
40
50
60
70
80
90
Den
sific
atio
n (%
)
020406080100120
Degree of liquid saturation (%)
a.
b.
(57.3 and 60.0%, respectively, for the W and W/E pellets). Thus, the densification during drying was strongly influenced by the type of agglomeration liquid used (Fig. 4b).
Figure 5. Contraction (a) and densification (b) of two mcc pellet types prepared with different agglomeration liquids as a function of degree of liquid saturation during drying. Symbols are defined in the graph. Error bars show 95% confidence limits of arithmetic mean values.
The degree of liquid saturation during drying The mechanical treatment to which the pellets were subjected during preparation was similar and seemed to densify both sets of pellets to a maximum, i.e. to a degree of liquid saturation of approximately 100% (Fig. 5), which is consistent with previous findings on extruded cylinders (Jerwanska et al., 1995). Thus, the pores inside the pellets were completely filled with liquid at the beginning of the drying process. Consequently, the difference in the final porosity between the two types seemed to be
30

0
0,1
0,2
0,3
0,4
0,5
Cum
ulat
ive
decr
ease
in p
ore
volu
me/
pelle
t (m
m3 )
0 0,1 0,2 0,3 0,4
Cumulative volume liquid evaporated/pellet (mm 3 )
25/75% w/w water/ethanol
100/0% w/w water/ethanol
the result of a difference in densification, due to contraction, during drying rather than a difference in the degree of densification during the pelletisation procedure. For the pellets prepared with water, the volume of contraction was approximately equal to the volume of liquid removed (Fig. 6), thus the degree of liquid saturation of the pellets was kept high for a considerable fraction of the drying process (Fig. 5). It seems that the drying front was located at the exterior of the pellets for a significant time during drying and the system strives to keep the solid pore surfaces wetted. In contrast, for the pellets prepared with water and ethanol, the volume of contraction was less than the volume of removed liquid (Fig. 6), so the degree of liquid saturation decreased whilst drying took place (Fig. 5). It seems therefore, that the drying front moved gradually towards the interior of these pellets throughout the drying process.
Figure 6. Mean cumulative decrease in pore volume as a function of mean cumulative volume of liquid evaporated during drying for two mcc pellet types prepared with different agglomeration liquids. Symbols are defined in the graph. Error bars show 95% confidence limits of arithmetic mean values.
Each solid particle in a liquid filled granule can be described to be surrounded by a liquid film, separating the particles from each other. Removal of this film during dryingleads to the particles approaching one another, i.e., as it is progressively reduced in thickness, the interparticulate separation distance and granule volume are reduced. It has been suggested that the driving force drawing the particles together, originates frompressure gradients in the liquid in the pores caused by capillary pressure and osmoticpressure (Newitt and Coleman, 1952; Scherer, 1990; Hasatani et al., 1993). Since the pellets contained no water-soluble components, it is reasonable to assume that capillary pressure was the driving force behind the contraction of the pellets studied here. It has been suggested (Scherer, 1990) that the maximum capillary pressure in a pore is related to the contact angle between the liquid and the solid and the liquid/vapour interfacial energy (or surface tension). The change in granulation liquid from water to the water/ethanol mixture will alter the interfacial energies of the system (Wells and
31

0
10
20
30
40
50
60
70
80
90
100
Liq
uid
cont
ent (
% w
/w d
ry b
asis
)
0 10 20 30 40 50 60 70 80 90
Time (min)
Pellet denom. III 58 l/min, 21oC
Pellet denom. III 15 l/min, 21oC
-6
-5
-4
-3
-2
-1
0
ln m
t/m0
(-)
0 10 20 30 40 50 60 70 80
Time (min)
b.
a.
Walker, 1983) and capillary forces of different magnitude might explain the differences in contraction between the pellets prepared with different liquids.
Figure 7. Examples of drying profiles of a bed of mcc pellets prepared with 50/50% w/wwater/ethanol, dried under two different rates of air flow (a) and plots of ln mt / m0 vs. time, where mt and m0 are the amount of removable liquid at time t and the total amount of removable liquid, respectively (b). Symbols are defined in the graph.
It is also possible that bonding between the particles will counteract their movement and the progressive reduction in the interparticulate separation distance. It has previously been shown (Karehill and Nyström, 1990; Olsson et al., 1996) that the tensile strength of microcrystalline cellulose compacts varied markedly depending on the liquid in the pores of the compacts, i.e., the bonds between the primary particles were affected by the medium surrounding the particles. The drying behaviour of the pellets, in terms of localisation of the drying front and the contraction of the granules, will thus depend on the balance of the driving and the counteracting forces. Both of these factors will probably differ for the two sets of pellets and can explain the different contraction behaviour during drying. Thus, the differences observed previously in the porosity of
32

mcc granules prepared with different water/ethanol agglomeration liquids could be related to differences in densification during drying, rather than differences in densification during the pelletisation procedure. Hence the drying behaviour of these pellets may be decisive for their structure and functional tabletting behaviour.
Modulation of densification, porosity, compression shear strength and tablet forming ability of mcc pellets with the drying rate
Since there are indications in the literature that the drying rate may affect contraction and densification (Hasatani et al., 1993), the influence of the drying rate on the densification during drying and on the tabletting behaviour of mcc pellets was investigated in Paper II. Pellets prepared with four agglomeration liquids containing different proportions of water and ethanol, were subjected to a number of drying conditions and thereafter characterised in terms of their porosity, shape and tabletting behaviour. For the pellets dried with the lowest and highest drying rate, the compressionshear strength and tablet forming ability were determined.
10
15
20
25
30
35
40
Pelle
t por
osity
(%)
0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9
Drying rate constant (min -1 )
Pellet denom. IV
Pellet denom. III
Pellet denom. II
Pellet denom. I
Drying under different conditions
Figure 8. Pellet porosity as a function of drying rate constant for mcc pellets preparedwith four different agglomeration liquids (0, 25, 50, 75% w/w ethanol, denoted I, II, III and IV respectively) dried by convective drying. Results from linear regression are included in the graph. Error bars shows 95% confidence limits of arithmetic mean values.
The transfer of heat and mass during convective drying is controlled by variables such as the air temperature and the rate of air flow (Nonhebel and Moss, 1971), so pellets were dried with different air temperatures and with different rates of air flow to vary the drying rate. Examples of drying profiles are shown in Fig. 7. As can be seen in this
33

15
20
25
30
35
40
45
1/b
(MPa
)
0 10 20 30 40 5
Pellet p orosity (%)
Pellet denom. I L/H drying rate constant
Pellet denom. II L/H drying rate constant
Pellet denom. III L/H drying rate constant
Pellet denom. IV L/H drying rate constant
0
figure, the relationships between ln mt / m0 and the drying time were generally linear (r2= 0.93-1). Thus, the slope (i.e., the drying rate constant) determined from these relationships could be used as measures of the drying rate of the pellets. An increasedrate of air flow, air temperature and proportion of ethanol in the agglomeration liquid generally decreased the drying time and increased the drying rate constant of the pellets. In addition, the different drying conditions used gave a considerable variation in thedrying rate.
Figure 9. Kawakita 1/b values as a function of pellet porosity for mcc pellets preparedwith four different agglomeration liquids (0, 25, 50, 75% w/w ethanol, denoted I, II, III and IV respectively) dried under convective drying by a low (L) and high (H) drying rate. The lowest pellet porosity value in each set represents pellets dried with the lowest dryingrate constant. Error bars shows 95% confidence limits of arithmetic mean values.
The effect of drying rate on densification, porosity and the shape of the pellets A greater proportion of ethanol in the agglomeration liquid and a higher drying rate generally increased the porosity of the dried pellets (Fig. 8). However, no simplecorrelation was obtained between the drying rate constant and the pellet porosity, although, the porosity of the pellets prepared with ethanol in the agglomeration liquid was significantly (unpaired t-test, p<0.05) higher after drying with the highest drying rate constant than after drying with the lowest drying rate constant. In addition, the rate of air flow had a more marked effect on the pellet porosity than the air temperature.Since the porosity of the pellets before drying can be assumed to be similar as discussed above, the results can be interpreted in terms of an effect of the drying rate on pellet densification during drying. It has been proposed that an increased drying rate increases the driving force for contraction (Hasatani et al., 1993), which may lead to the formationof cracks within the agglomerates (Scherer, 1990), reflected as an apparently higher granule porosity. However, no major cracks could be observed on the surfaces of the pellets by inspecting SEM images.
34

0
1
2
3
4
5
6
7
8
9
10
Tab
let t
ensi
le st
reng
th (M
N/m
2 )
5 10 15 20 25 30 35 40 45 5
Pellet porosity (%)
Data from Johansson et al., 1995
Pellet denom. IV L/H drying rate constant
Pellet denom. III L/H drying rate constant
Pellet denom. II L/H drying rate constant
Pellet denom. I L/H drying rate constant
0
It can be assumed that the liquid descendings into, at least for a fraction of, the pores during the drying phase. The formation of dry pore surfaces may counteract contractionand it can be hypothesised that the formation of dry pore surfaces is dependent on the drying rate in such a way that the evaporation of liquid is not balanced by a contraction of the pore system. This might be due to that all pores of the agglomerate are not emptied in parallel, i.e. the larger pores will be emptied first and liquid will flow fromthe larger pores to the smaller ones because of their higher capillary pressure. It is possible that with an increased drying rate, evaporation will be faster than the internal liquid flow.
Figure 10. Tensile strength of tablets compacted at 200 MPa as a function of pellet porosity for mcc pellets prepared with four different agglomeration liquids (0, 25, 50, 75% w/w ethanol, denoted I, II, III and IV respectively) dried under convective drying by a low (L) and high (H) drying rate. The lowest pellet porosity value in each set represents pellets dried with the lowest drying rate constant. Error bars shows 95% confidence limits of arithmetic mean values.
The shape (from the circularity determination) of the pellets was not affected by the drying rate (Table 1). However, the poured bulk density of beds of pellets varied between the sets of pellets and also within each set. This variation in bulk density could not be explained by differences in voidage of the beds, which was generally similar. The voidage reflects the packing of the pellets, and is related to their shape and surface geometry. The variation in bulk density might, instead, be related to the differences inthe porosity of the pellets.
The effect of porosity and drying rate on the compression shear strength of pelletsA measure of the yield strength of particles during compression is most frequentlycalculated from the tablet porosity and compaction pressure data, described with the
35

Heckel function. However, this procedure has been questioned when applied to granules rather than to individual, dense particles (Wikberg and Alderborn, 1990; Adams and McKeown, 1996; Johansson and Alderborn, 1996). It has been proposed that the Kawakita equation can be used, by which a compression parameter (1/b) can be derived that is considered (Adams and McKeown, 1996; Nicklasson and Alderborn, 2000), to reflect the stress at which the granules fail during confined compression. For cellulose pellets, the failure mechanism is thought to be deformation rather than fracturing (Nicklasson and Alderborn, 2000). Thus the parameter derived reflects the pellet shear strength during compression. Generally, the 1/b values decreased with an increased pellet porosity and they appeared to obey a single relationship (Fig. 9). An increased drying rate resulted in a different compression behaviour of the pellets and, generally, in a lower compression shear strength, i.e. increased deformability.
Table 1Porosity, poured bulk density, voidage and circularity of mcc pellets prepared with differentagglomeration liquids and subjected to a high and low drying rate
Denominationa Porosityb
(%)(n=3)
Poured bulk density of
pelletsb
(g/cm3)(n=3)
Voidage ofpoured pellet
bed(%)
(n=3)
Circularityb
(-)(n=50)
I/L 8.51 (3.2) 0.80 (1.1) 45 0.97 (1.7)I/H 10.1 (2.2) 0.82 (0.070) 42 0.97 (1.8)II/L 12.9 (2.2) 0.78 (1.0) 44 0.96 (2.4)II/H 17.4 (2.2) 0.73 (0.32) 44 0.95 (2.7)III/L 17.1 (1.2) 0.71 (1.7) 46 0.96 (1.9)III/H 27.6 (2.2) 0.63 (1.2) 45 0.96 (2.1)IV/L 35.2 (2.2) 0.54 (1.7) 47 0.94 (3.0)IV/H 40.9 (0.51) 0.50 (0.23) 47 0.91 (4.9)
a The denominations I, II, III and IV used here correspond to 0, 25, 50, 75% w/w ethanol in theagglomeration liquid, respectively. L and H denote a low and high drying rate constant, respectively.b Mean values. The numbers in parentheses refer to the relative standard deviations in per cent.
The effect of pellet porosity and drying rate on tablet tensile strength An increased degree of deformation of the pellets owing to increased porosity gave rise to an increased tablet-forming ability and tablet tensile strength (Fig. 10). For pellets prepared with ethanol in the agglomeration liquid, the drying rate related change in porosity significantly increased the tensile strength of the tablets (unpaired t-test, p<0.05). Thus, differences in the contraction and densification of granules during drying and the influence of the drying rate on the densification may considerably affect the tensile strength of tablets formed from granules.
36

MODULATION OF SOLID-STATE STABILITY AND TABLETTINGBEHAVIOUR OF AMORPHOUS LACTOSE BY CO-SPRAY DRYING WITH POLYMERS
Figure 11. SEM images of amorphous lactose particles (a); composite particles with 25% PVPK17 (b);composite particles with 25% PVPK17 and polysorbate 80 (c); composite particles with 25% PVPK90 (d); andcomposite particles with 25% PVPK90 and polysorbate 80 (e).
Properties of the spray-dried amorphous composite particles containing lactoseand different polymers
MorphologyIn general, all the types of spray-dried particles were spherical and the surface of the pure amorphous lactose particles (Fig. 11) was smooth. However, incorporation of PVP changed the surface geometry markedly in such a way that the presence of PVPK17 resulted in a folded surface, while PVPK90 produced large cavities. The number and depth of the folds and cavities generally increased with increasing concentration of PVP (Paper III). Furthermore, addition of polysorbate 80 did not seem to affect the particle
37

morphology (Paper V). The differences in surface geometry might be explained in termsof an influence of the concentration and the molecular weight of PVP on surface viscosity during the drying process.
CrystallinityX-ray powder diffraction analysis was used to confirm that the prepared particles were amorphous (Papers III and IV). All X-ray powder diffraction patterns obtained showed broad and diffuse maxima with no sharp peaks, suggesting that the particles were completely amorphous.
Apparent particle density The apparent particle density of the pure amorphous lactose was slightly lower than of
-lactose monohydrate and the density of the composite particles decreased with increasing PVP content (Table 2) (Paper III). The presence of polysorbate 80 did not affect the particle density (Paper V). The measured particle densities of the compositeparticles compared favourably with the calculated densities, suggesting that there was no significant volume of pores impermeable to He, in comparison with the pure amorphous lactose and PVP particles.
Table 2. Apparent particle density and glass transition temperature (Tg) of amorphous lactoseand composite particles with PVPK90 and PVPK17
Material Apparent particledensity(g/cm3)
Calculateddensity(g/cm3)
Tg(ºC)
TheoreticalTg
(ºC)
Amorphous lactose 1.53 (0.059) - 116.8 (0.386) -5% PVPK17 1.50 (0.048) 1.51 117.2 (0.0492) 118.25% PVPK90 1.50 (0.057) 1.51 117.5 (0.123) 120.010% PVPK17 1.48 (0.013) 1.48 117.2 (0.215) 119.710% PVPK90 1.49 (0.051) 1.49 117.8 (0.255) 123.125% PVPK17 1.43 (0.038) 1.42 117.7 (0.298) 123.425% PVPK90 1.43 (0.057) 1.44 119.7 (0.174) 132.5
Mean values (n=3), relative standard deviation in per cent in parentheses.
The effect of the content and molecular weight of PVP on the physical stability of amorphous lactose
The importance of the molecular weight of PVP for the physical stability of amorphousmaterials needs to be clarified (Matsumoto and Zografi, 1999; Zeng et al., 2001). Furthermore, the possibility of preparing amorphous composite particles of lactose and PVP with different molecular weight by spray drying has not been addressed previously. Hence, amorphous spray-dried composite particles of lactose and PVP were prepared and the effect of the polymer content and molecular weight on the physical stability investigated in Papers III and IV.
Resistance to transformations induced by heat and moisture Polymer-related effects on the resistance to heat-induced transformations of the different particles were investigated by DSC (Papers III-V), and the glass transitiontemperatures (Tg) and crystallisation temperatures (Tc) were used as indicators of the
38

physical stability of the particles (Paper III). The Tg of the pure amorphous lactose particles was approximately 117 C and the shape of the glass transition became slightly broader with increased content and molecular weight of PVP. The observed glass transition temperatures were generally lower than predicted from the Gordon–Taylorequation (Table 2), which may reflect non-ideal mixing between lactose and PVP (Hancock and Zografi, 1997). The effect of the additives on Tg was limited, however a tendency for Tg to increase with the content and molecular weight of PVP was found. Only in one case, the 25% PVPK90 system, was a significant increase in Tg obtained compared with amorphous lactose (unpaired t-test, p<0.05). Although polymer content and molecular weight had a minor effect on the Tg, the Tc clearly depended on the content and the molecular weight (Figs. 12 and 13). Thus, incorporation of PVP into amorphous lactose particles changed the thermal behaviour of the materials in such a way that Tg was almost unchanged, while Tc increased significantly.
80 100 120 140 160 180 200 220 240 260
Temperature (oC)
Hea
t flo
w
Tc, particles with 25% PVPK17
Tc, particles with25% PVPK90
Tc, pure amorphouslactose particles
Tc, particles with 10% PVPK17Tc, particles with 10% PVPK90
5 m
W
Exo
Endo
Another means to get an indication of the potential of the polymers to stabiliseamorphous lactose was to investigate the resistance to moisture provoked crystallisation. The microcalorimetry heat flow curves (Fig. 14) for the differentmaterials showed an immediate exothermic reaction just after introduction of humid gas to the sample, which lasted for several hours. This first exotherm is related to the absorption of water and was followed by a second exotherm caused by crystallisation of the amorphous lactose (Sebhatu et al., 1994). It was found that the time to the crystallisation peak was greater for the composite particles than for pure amorphous lactose and it was generally increased with increased molecular weight of PVP and decreased RH (Fig. 14). The specific heat of crystallisation of amorphous lactose was similar to reported values (Sebhatu et al., 1994), suggesting that the amorphous lactose in this study was completely amorphous and that complete crystallisation occurred at the different RHs. For the composite particles crystallised at 75% RH the specific heat of crystallisation corresponded reasonably well with the value for pure amorphous lactose, suggesting that complete crystallisation occurred at this RH. However, the specific heat of crystallisation decreased with decreased RH for the composites, which might indicate that only partial crystallisation occurred under these conditions.
Figure 12. DSC thermograms of pure amorphous lactose particles and composite particles with 10% and 25% PVPK17 and PVPK90.
39
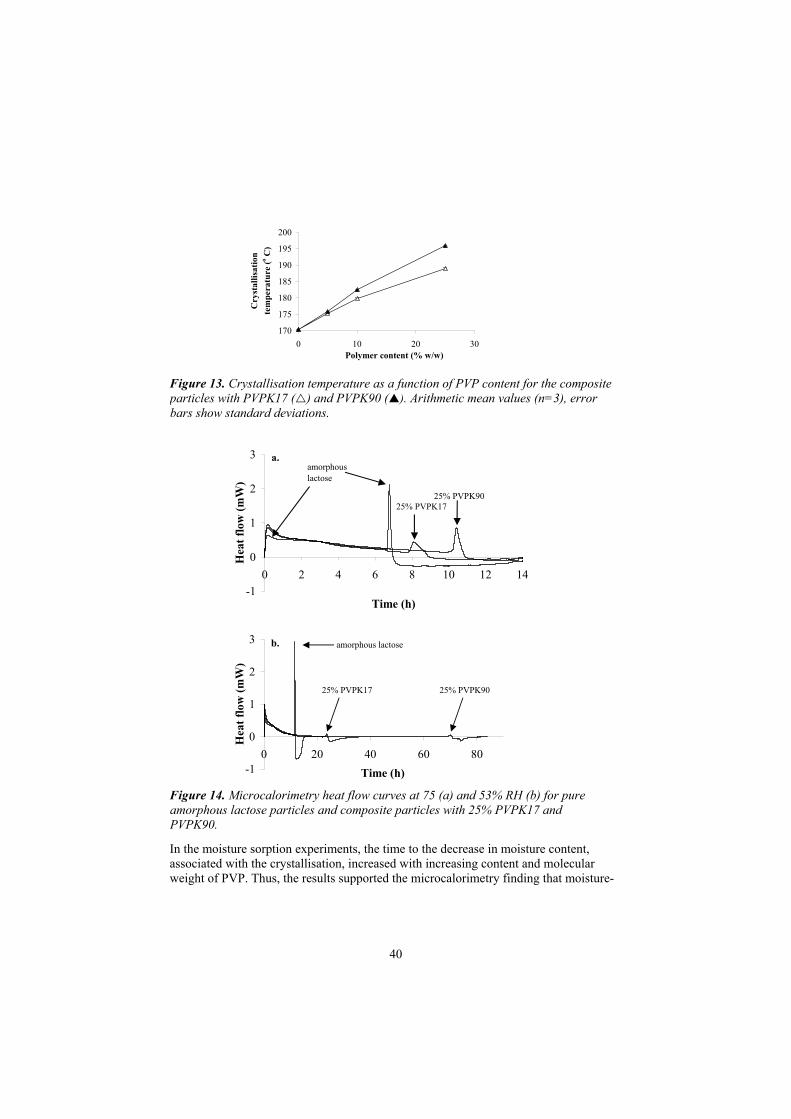
170
175
180
185
190
195
200
0 10 20Polymer content (% w/w)
Cry
stal
lisat
ion
tem
pera
ture
(o C)
30
Figure 13. Crystallisation temperature as a function of PVP content for the composite particles with PVPK17 ( ) and PVPK90 ( ). Arithmetic mean values (n=3), error bars show standard deviations.
-1
0
1
2
3
0 2 4 6 8 10 12 14
Time (h)
Hea
t flo
w (m
W)
amorphouslactose
25% PVPK1725% PVPK90
a.
-1
0
1
2
3
0 20 40 60 80Time (h)
Hea
t flo
w (m
W)
amorphous lactose
25% PVPK17 25% PVPK90
b.
In the moisture sorption experiments, the time to the decrease in moisture content, associated with the crystallisation, increased with increasing content and molecular weight of PVP. Thus, the results supported the microcalorimetry finding that moisture-
Figure 14. Microcalorimetry heat flow curves at 75 (a) and 53% RH (b) for pure amorphous lactose particles and composite particles with 25% PVPK17 and PVPK90.
40

induced crystallisation of the composites was affected by the molecular weight of the PVP.
Despite the fact that Tg was only slightly changed or was unchanged by the addition of PVP in the composites, increased resistance to heat- and moisture-inducedcrystallisation could be obtained by increasing the content and molecular weight of the polymer. The small changes observed in Tg with content and molecular weight of PVP, and thus a general increase in viscosity, might contribute to the increased stability of the composites. However, it is more likely that this is related to specific interactions formedbetween monomers and lactose molecules, such as hydrogen bonds. This explanation is supported by earlier suggestions that hydrogen bonds may be formed between lactose and PVP (Taylor and Zografi, 1998) and that inhibition of moisture-inducedcrystallisation of co-lyophilised sucrose-polymer systems was related to coupling of sucrose mobility to the motions of the polymer through hydrogen bonding (Shamblinand Zografi, 1999). This suggestion may also be substantiated by the fact that the Tg of the composite with 25% PVPK90 was reduced to only a few degrees above the storagetemperature, owing to the absorbed water, although, crystallisation was inhibited up to 6 months (Paper IV). On the basis of enthalpy relaxation measurements it has been suggested that a pure pharmaceutical amorphous solid must be stored at least fifty temperature degrees below the glass transition temperature (Tg) to reduce molecularmobility of the material to such an extent that it is physically stable for a significant period of time (Hancock et al., 1995). However, it seems that crystallisation in a composite material can be inhibited for a significant time period at a storagetemperature only a few degrees below the Tg. It has also been shown that the enthalpy relaxation of co-lyophilised PVP-sucrose was significantly less than for sucrose alone, although Tg was similar to sucrose (Shamblin and Zografi, 1998).
Table 3. Crystallisation of amorphous lactose and composite particlescontaining 5 and 25% PVP as a function of RH after 3 and 6 months ofstorage
Material 51% RH 43% RH 33% RH 22% RH
Amorphous lactose C C C A5% PVPK17 C C A A5% PVPK90 C C A A25% PVPK17 C C A A25% PVPK90 C A A A
C=Crystalline, A=Amorphous.
The effect on the relationship between critical RH for crystallisation and storage time By using microcalorimetry it was found that the critical RH for crystallisation within 10days for amorphous lactose was between 45 and 43 % RH and between 53 and 51% RH for the composite particles with 25% PVPK17 or PVPK90 (Paper IV). The critical RH generally decreased with time and the materials were stable at a significantly lower RH after 3 and 6 months (Table 3). However, independent of the storage time, the additionof PVP stabilised the amorphous lactose by increasing the critical RH. The relationshipbetween the critical RH for crystallisation and the storage time differed between the materials and there was a greater difference in the critical RH with increased storage
41

Time
Cri
tical
RH
for
crys
talli
satio
n
amorphous lactose
composites with a low Mw of PVP
composites with a high Mw of PVP
time, i.e. the stabilising potential of the polymer, including the importance of molecularweight, was more evident after a longer time, which is schematically shown in Fig. 15. In addition, the stability indicators discussed above, i.e. crystallisation temperature and time to crystallisation after exposure to moist gas, seem to reflect differences in the potential of the polymers to inhibit crystallisation during long-term storage.
Figure 15. Schematic illustration of the critical relative humidity for provokedcrystallisation of amorphous lactose and composites with two different molecular weights of PVP as a function of storage time.
The effect of the incorporation of stabilising polymers and a surfactant on the tabletting behaviour of amorphous lactose
Dispersion of a stabilising polymer into an amorphous solid might significantly alter its mechanical properties and tablet-forming ability. However, only a few reports exist in the literature that have addressed this topic. It has, for example, been reported that the tablet-forming ability could be significantly changed depending on the polymerincorporated (Takeuchi et al., 1999; Takeuchi et al., 2000a). In Paper V, the implications of incorporation of the stabilising polymers discussed above on the compression behaviour and tablet-forming ability of amorphous lactose were addressed.Furthermore, it was also demonstrated how the tabletting properties and particle-particle adhesivity can be modulated by the addition of a small amount of a surfactant to suchparticles.
Compression behaviourSEM images (Fig. 16) of the upper surfaces of tablets of amorphous lactose, two- and three-component composite particles revealed cohering individual particles that were slightly deformed at the contact points. Cracks originating at the surfaces of the particles and propagating towards their centers could also be observed. The surface areaof the tablets relative to the surface area of the powders was only slightly increased(Table 4). It is generally thought that amorphous materials deform during compressionand that fragmentation of such materials is limited. The results for the materials hereseem to support this view and the dominating compression mechanism was permanentdeformation. The presence of the polymers did not induce a change in response to the applied pressure in terms of a change in the propensity to fragment.
42

Table 4. Powder and tablet surface area, moisture content and compression parameters (Py, 1/b, a, elasticrecovery) of amorphous lactose and composite particles with PVP and polysorbate 80
Material Powdersurface area
(cm-1)b
Tabletsurface area
(cm-1)b
Moisturecontent
(% w/w)b
Py(MPa)
Kawakita1/b
(MPa)
Kawakitaa
(-)
Elasticrecovery
at 275MPa(%)
Amorphous lactose 5340 (2.3) 4640 (2.6) 6.4 (2) 88 12 0.69 1025%PVPK90 6760 (4.0) 8520 (0.95) 6.8 (2) 110 12 0.72 11
25%PVPK90+Pa 6100 (2.5) 6770 (1.6) 6.6 (2) 130 12 0.71 1125%PVPK17 5900 (1.2) 6140 (4.7) 6.7 (0.4) 94 13 0.68 10
25%PVPK17+Pa 5330 (0.91) 6680 (4.3) 6.7 (0.8) 97 11 0.70 10
a Polysorbate 80. b Mean values (n=3), relative standard deviation in per cent in parentheses.
The compressibility at infinite applied pressure (Kawakita parameter, a) was similar for the different types of particles (Table 4). However, the particles with the highermolecular weight PVP gave slightly more porous tablets, within the range of applied pressures used (Fig. 17a). Since the elastic recovery of the tablets was generally comparable (Table 4), this can be interpreted as an effect of the polymer on the plastic or viscous deformability of the particles. The Py values obtained from the Heckel profiles (Fig. 17b) supported this idea and indicated that incorporation of PVP with a high molecular weight slightly decreased the particle deformability, while the lower molecular weight PVP resulted in particles with similar deformability as the pure lactose particles (Table 4). In addition, the surfactant had a limited effect (PVPK17) or slightly decreased the particle deformability (PVPK90). It is known that the amount of moisture absorbed into an amorphous material may affect the deformability (Sebhatu et al., 1997), however the moisture content of the spray-dried particles was similar (Table 4).
It is generally believed that spray drying may produce particles with pores (Meenan et al., 1997; Eissens et al., 2002). Thus, it is possible that the particles here have a certain intra-particulate porosity, which may vary between the different types of particles, and which could, therefore, affect the derived Py values and contribute to an apparent difference in Py between the particles (Wikberg and Alderborn, 1990; Adams and McKeown, 1996).
For cellulose granules the 1/b compression parameter is thought to reflect compressionshear strength (Nicklasson and Alderborn, 2000), which is supported by a relationship between the granule porosity and 1/b, as discussed above. The 1/b values for the spray-dried particles were nearly the same also markedly lower than the Py values (Fig. 17c, Table 4). The discrepancy between the Py and 1/b values may be explained by the fact that these parameters indicate different physical characteristics of particles. It has been suggested that 1/b reflects the stress at which particles fail during powder compression(Adams and McKeown, 1996). Accordingly, it cannot be ruled out that the 1/b parameter indicates the stress at which particles crack since cracks were observed on the particles (Fig. 16). This stress was thus significantly lower than that required to initiatedeformation of the particles, i.e., the Py values, and consequently it was independent of their composition.
43

Figure 16. SEM images of upper surfaces of tablets of different amorphous particles compacted at 100 MPa: amorphous lactose particles (a); composite particles with 25% PVPK17 (b); composite particles with 25% PVPK17 and polysorbate 80 (c);composite particles with 25% PVPK90 (d); and composite particles with 25% PVPK90 and polysorbate 80 (e).
It is also plausible that, since the spray-dried particles were of such fineness, considerable powder compression probably occurred by particle rearrangement at low applied pressures. Meaning that, since the Kawakita values indicate degree of compression in relation to the initial height of the bed of powder, it cannot be excluded that the Kawakita approach is unsatisfactory in terms of sensitivity to detect differences in deformation of particles for which considerable volume reduction occurs by particle rearrangement.
44
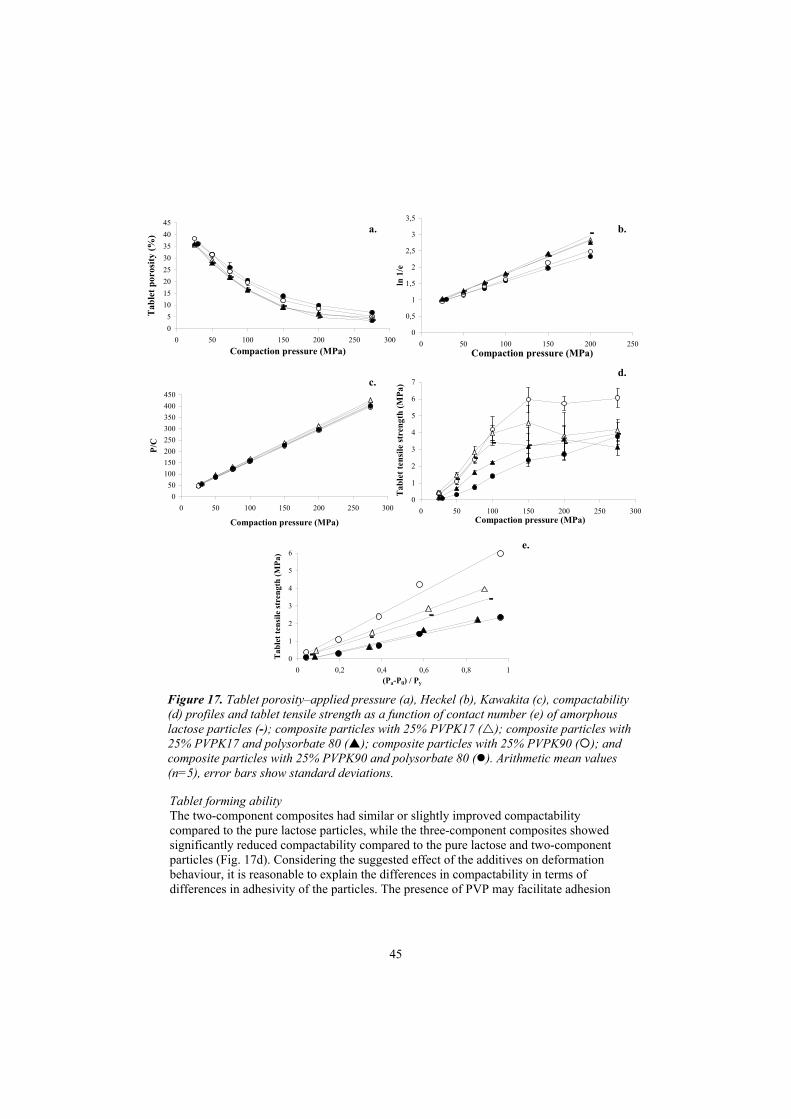
05
1015202530354045
0 50 100 150 200 250 300Compaction pressure (MPa)
Tab
let p
oros
ity (%
) a.
0
0,5
1
1,5
2
2,5
3
3,5
0 50 100 150 200 250Compaction pressure (MPa)
ln 1
/e
b.
0
1
2
3
4
5
6
7
0 50 100 150 200 250 300Compaction pressure (MPa)
Tab
let t
ensi
le st
reng
th (M
Pa)
d.c.
050
100150200250300350400450
0 50 100 150 200 250 300
Compaction pressure (MPa)
P/C
0
1
2
3
4
5
6
0 0,2 0,4 0,6 0,8 1(Pa-P0) / Py
Tab
let t
ensi
le st
reng
th (M
Pa)
e.
Tablet forming ability
Figure 17. Tablet porosity–applied pressure (a), Heckel (b), Kawakita (c), compactability (d) profiles and tablet tensile strength as a function of contact number (e) of amorphous lactose particles (-); composite particles with 25% PVPK17 ( ); composite particles with 25% PVPK17 and polysorbate 80 ( ); composite particles with 25% PVPK90 ( ); and composite particles with 25% PVPK90 and polysorbate 80 ( ). Arithmetic mean values(n=5), error bars show standard deviations.
The two-component composites had similar or slightly improved compactabilitycompared to the pure lactose particles, while the three-component composites showed significantly reduced compactability compared to the pure lactose and two-componentparticles (Fig. 17d). Considering the suggested effect of the additives on deformationbehaviour, it is reasonable to explain the differences in compactability in terms of differences in adhesivity of the particles. The presence of PVP may facilitate adhesion
45

between the particles in the tablets, while the surfactant on the surfaces of the particlesmay reduce particle-particle adhesion, i.e. the tablets will be less resistant to fracturingwhen stressed. In an effort to substantiate this explanation, an adhesion parameter was derived which has been suggested to indicate the stress needed to break an inter-particulate joint of unit dimension (Sebhatu and Alderborn, 1999). This parameterrepresents the relationship between the tablet tensile strength ( t) and a contact number((Pa – P0) / Py) as follows:
t = ad ((Pa – P0) / Py) (15)
where ad is the adhesion parameter, Pa is the applied pressure and P0 is the criticalformation pressure (i.e., the pressure needed to form a coherent compact). The criticalformation pressure was obtained by extrapolation of the linear part of the compactabilityprofile to zero tablet strength. In Fig. 17e, relationships between t and ((Pa – P0) / Py)are given and the slopes (i.e., the adhesion parameters) were found to differ in a way that is consistent with the suggestions given above about regarding the adhesivity.
SUMMARY AND CONCLUSIONS
In this thesis the relationships between stresses during the drying process, structural and functional properties of particles have been addressed. In addition, the possibility of utilising the drying process to modulate tabletting properties and solid-state stability has also been demonstrated. Two different particle types, prepared by common pharmaceutical techniques, i.e. wet-granulation and spray drying, were used for the present investigation. Although these preparation methods are quite different, they are similar in the sense that particles are formed with the aid of a liquid that is subsequently removed by convective drying. The drying process involved in these preparation procedures significantly affected the structure of the particles formed and, consequently, their functional properties. The structural properties investigated were on two different levels as shown in Table 5.
The importance of the composition of the agglomeration liquid on the contraction and densification of microcrystalline cellulose pellets during drying and the relative importance of the drying and wet granulation phases for their porosity were investigated first. Water alone or a mixture of water and ethanol were used as the agglomerationliquid. The pelletisation procedure utilised facilitated a complete filling of the pore space with liquid so that the degree of liquid saturation was around 100% at the beginning of the drying process and the initial porosity of the two types of pellets was similar. For the pellets agglomerated with water, the pores inside the pellets remainedfilled with liquid for a significant part of the drying process, i.e., the volume of water that evaporated was equivalent to the volume by which the pellets contracted. For the pellets agglomerated with an ethanol/water mixture, on the other hand, the degree of liquid saturation decreased throughout the drying process, i.e. the volume of liquid that evaporated was not balanced by an equal amount of contraction. Thus, the drying behaviour of the two sets of pellets differed. A difference in the degree of densification during the drying process caused by differences in the interaction between the solid and the liquid could explain the significant difference in the porosity of the dry pellets.
46

Thus, replacing the cellulose with another material might result in a change in the interaction between the solid and the liquid and also the contraction and densification during drying.
Table 5. Summary of the particle formation processes of the two particle types, structural andfunctional properties studied in this thesis
Granulated particles Spray-dried particles
Particleformationprocess
Wet agglomeration and convective drying
Rapid precipitation from solutionby convective drying
Macro- and mesostructuralproperties
Particle size, shape, morphology and porosity
Particle size, shape and morphology
Micro- and nanostructuralproperties
Crystallinity, glass solution of low Mw substance and polymer, and surface composition
Functionalproperties
Compression behaviour and tablet-forming ability
Solid-state stability, compressionbehaviour and tablet-formingability
The possibility of controlling the intragranular porosity with the drying rate was also investigated and it was found that, in general, an increased drying rate decreased the degree of densification during drying and hence resulted in more porous pellets. The importance of the drying behaviour and drying rate on the compression shear strength and tablet-forming ability of the pellets was shown. The increased porosity due to decreased densification during drying resulted in stronger tablets and lower pellet compression shear strength. The compression shear strength or the deformability during confined compression was evaluated by deriving a compression parameter (1/b) fromthe Kawakita equation and a relationship was found between this parameter and the intragranular porosity.
The next part of this thesis dealt with the modulation of solid-state stability and the tabletting behaviour of an amorphous substance by incorporation of different polymers.Spray drying was found to be a feasible method with which to prepare composite particles of amorphous lactose and PVP, facilitating the formation of a single-phaseamorphous solid system with different contents and molecular weights of PVP. Incorporation of the polymers significantly affected the morphology of the particles in terms of surface geometry. Furthermore, increased content and molecular weight of PVP resulted in an increased resistance to heat and moisture provoked crystallisation. Content and molecular weight of PVP also influenced the critical RH for crystallisationand the relationship between the critical RH and the storage time. On the other hand, Tgof the composite particles was almost unaffected by the incorporation of the polymer.Consequently, the use of Tg as a stability indicator for these types of materials can be questioned. The stabilisation mechanism seems, therefore, mainly to be related to a
47

specific interaction between the lactose molecules and polymer monomer units. However, the slightly increased Tg may also play a part in the increased stability.
The implications of the incorporation of a stabilising polymer and a surfactant, by spray drying, on the compression behaviour and tablet-forming ability of an amorphousmaterial were also addressed. The presence of the polymers resulted in particles with somewhat higher Py values, i.e. decreased deformability. On the other hand, the Kawakita 1/b values were unaffected by incorporation of the different polymers and the values were generally lower than the Py values, which may imply that these parametersindicate different physical characteristics of the particles. It is plausible that, for the spray-dried particles, the 1/b parameter is related to the stress at which the particlescrack, in contrast to the granulated particles where it seems to reflect the stress at which the primary particles of the granules start to flow. It is possible that the Kawakitaapproach has insufficient sensitivity to detect differences in deformation of particles for which a considerable volume reduction occurs by particle rearrangement.
The compactability of amorphous lactose could be changed by incorporation of the various polymers in such way that PVP slightly increased the compactability, whilst the surfactant markedly decreased it. The different spray-dried particles represented interesting model systems because they were comparable in terms of their size, shape, moisture content, dominating volume reduction mechanism and particle deformability,however the particle-particle adhesivity seemed to differ. This could clearly be shown by using a relationship, between the tablet tensile strength, a contact number and an adhesion parameter, proposed previously, and also of the applicability of using this relationship to discriminate between particles with different particle-particle adhesivity.
In conclusion, this thesis has contributed to an increased mechanistic understanding ofrelationships between stresses during the drying process and particle structuralproperties critical for their functionality. It has also been shown how the drying process can be taken advantage of to engineer particles with respect to tabletting properties and solid-state stability. The knowledge generated may lead to better prediction and optimisation of functional properties of pharmaceutical particles, which is of the utmostimportance in the development and production of solid dosage forms.
48

ACKNOWLEDGEMENTS
The studies in this thesis were carried out at the Department of Pharmacy, Faculty of Pharmacy, Uppsala University.
I wish to express my sincere gratitude to:
Professor Göran Alderborn, Head of the Department and my supervisor, for providing excellent working facilities, enthusiastic guidance and for sharing your scientific knowledge with me. I am also grateful for the stimulating collaboration, your encouragement and support, and for your friendship.
Professor Christer Nyström and Professor Sven Engström for inspiring and interesting scientific discussions.
Denny Mahlin, for sharing my interest in solid-state transformations and for valuable discussions regarding crystallisation of amorphous lactose, and for your friendship.
Nils-Olof Ersson at the Department of Materials Chemistry, the Ångström Laboratory for your most valuable help with the X-ray powder diffraction analyses.
Dr. Anna Hillgren and Anna Imberg for your kind help with the DSC apparatus.
Johan Gråsjö for always being positive and helpfully solving mathematical problems.
My two office-mates during the years at the department, Albert Miharanyan and Dr. Helena Nicklasson for scientific discussions, nice chats, laughter, great company and for your friendship.
Ilgar Baghainia, Måns Ahlén, Johan Unga and Francesca Caprani for experimentallycontributing to this thesis as under-graduate students.
Former and present “powder”colleagues: Dr. Åsa Adolfsson, Gunilla Andersson,Susanne Bredenberg, Elisabet Börjesson, Dr. Johan Carlfors, Leif Dahlberg, Dr. Ragnar Ek, Frauke Fichtner, Dr. Göran Frenning, Dr. Christina Gustafsson, Mina Heidarian, Josefina Hellström, Dr. Barbro Johansson, Dr. Sofia Matsson, Albert Mihranyan, Dr. Mitra Mosharaff, Dr. Helena Nicklasson, Dr. Fredrik Nicklasson, Martin Nilsson, Dr. Maria Strömme, Åsa Tunón and Sitaram Velaga for fruitful scientific discussions, valuable comments on my papers, support and friendship.
Gunilla Andersson for excellent experimental assistance.
Eva Nises Ahlgren, Ulla Wästberg-Galik, Eva Lide, Christin Magnusson and Harriet Pettersson for always being kind and helpful in practical matters.
Dr. Tesfai Sebhatu and Dr. Seth Björk at AstraZeneca R&D Södertälje for performingX-ray powder diffraction analyses.
49

Suzanne Lidström, Martin Naylor and Antona Wagstaff for skilful and rapid linguistic revision of the papers and this thesis, especially Suzanne Lidström who revised this thesis with very short notice in less than two days in between fetching children at school, planting trees and revising manuscripts. I am impressed!
The staff at the BMC forskningsverkstad and fotoavdelning for always being friendly and helpful.
Christel Bergström, Dr. Erik Björk, Susanne Bredenberg, Gunilla Bäckström, Dr. Christina Gustafsson, Dr. Sofia Matsson, Denny Mahlin, Albert Mihranyan, Dr. Mattias Paulsson, Niclas Petri, Eva Ragnarsson, Niclas Rydell, Christer Tannergren, Staffan Tavelin, Dr. Karin Östh for very pleasant travel company at international meetings and for sharing fun and strange experiences. I will never forget The Capital Room in Indianapolis.
All former and present PhD-students at the department, especially Dr. Eva “Evur” Krondahl, Per Nydert, Helena Engman, Elisabet Gullberg, Björn Jansson, Helene Hägerström, Denny Mahlin, Christel Bergström, Magnus Köping-Höggård, Lena Strindelius, Carola Wassvik, for your support, being great friends and much joy, also outside the department. Tobias Bramer, who persistently tries to drag me out to the golf course. I promise that I will play more golf now.
All my other colleagues at the department for contributing to a friendly and open atmosphere.
All my friends outside the department for filling my life with much more besides powders.
My parents Jan and Monica, and all my other relatives for your belief in me and for your warm and generous support.
Mari-Anne, Bengt and Peggy for your pleasant hospitality in Södertälje.
Eric and Yvonne for your kind interest in my work.
Sofia, my sweet wife, for your loving support, words of encouragement, all your help, and most of all for your love.
AstraZeneca AB, NUTEK (Swedish National Board for Industrial and Technical Development), Pharmacia AB, CR Leffman Scholarship Fund, IF Foundation for Pharmaceutical Research, CD Carlsson Scholarship Fund for financial support.
50

REFERENCES
Adams, M. J. and McKeown, R., 1996. Micromechanical analyses of the pressure-volume relationships for powders under confined uniaxial compression. Powder Technol., 88, 155-163.
Ahlneck, C. and Zografi, G., 1990. The molecular basis of moisture effects on the physical and chemical stability of drugs in the solid state. Int. J. Pharm., 62, 87-95.
Alderborn, G., Börjesson, E., Glazer, M. and Nyström, C., 1988. Studies on direct compression of tablets XIX. The effect of particle size and shape on the mechanical strength of sodium bicarbonate tablets. Acta Pharm. Suec., 25, 31-40.
Alderborn, G., Duberg, M. and Nyström, C., 1985. Studies on direct compression of tablets X. Measurements of tablet surface area by permeametry. Powder Technol., 41, 49-56.
Alderborn, G. and Nyström, C., 1982a. Studies on direct compression of tablets III. The effect on tablet strength of changes in particle shape and texture obtained by milling. Acta Pharm. Suec., 19, 147-156.
Alderborn, G. and Nyström, C., 1982b. Studies on direct compression of tablets IV. The effect of particle size on the mechanical strength of tablets. Acta Pharm. Suec., 19, 381-390.
Aulton, M. E., 2002. Drying. In Aulton, M. E. (Ed.), Pharmaceutics: The science of dosage form design. Hartcourt Publishers Ltd., London, pp. 389-390.
Bataille, B., Ligarski, K., Jacob, M., Thomas, C. and Duru, C., 1993. Study of the influence of spheronization and drying conditions on the physico-mechanicalproperties of neutral spheroids containing Avicel PH 101 and lactose. Drug Dev. Ind. Pharm., 19, 653-671.
Bolhuis, G. K. and Chowhan, Z. T., 1996. Materials for direct compression. In Alderborn, G. and Nyström, C. (Eds.), Pharmaceutical powder compactiontechnology. Marcel Dekker Inc, New York.
Brinker, C. J. and Scherer, G. W., 1990. Drying. In Sol-gel science. Academic Press Ltd., London.
Broadhead, J., Rouan, E. S. K. and Rhodes, C. T., 1992. The spray drying of pharmaceuticals. Drug Dev. Ind. Pharm., 18, 1169-1206.
Buckton, G., 2000. Isothermal microcalorimetry water sorption experiments: calibration issues. Thermochim. Acta, 347, 63-71.
Carstensen, J. T. and Zoglio, M. A., 1982. Tray drying of pharmaceutical wet granulations. J. Pharm. Sci., 71, 35-39.
Chalmers, A. A. and Elworthy, P. H., 1976. Oxytetracycline tablet formulations: the effect of wet mixing time, particle size and batch variation on granule and tablet properties. J. Pharm. Pharmacol., 28, 239-243.
Chatrath, M. and Staniforth, J.M., 1990. The relative influence of dielectric and other drying techniques on the physico-mechanical properties of a pharmaceutical tablet excipient. Part I: Compaction characteristics. Drying Technol., 8, 1089-1109.
Chidavaenzi, O. C., Buckton, G. and Koosha, F., 2001. The effect of co-spray drying with polyethylene glycol 4000 on the crystallinity and physical form of lactose.Int. J. Pharm., 216, 43-49.
51

Corrigan, O. I., 1995. Thermal analysis of spray dried products. Thermochim. Acta, 248, 245-258.
Corrigan, O. I. and Crean, A. M., 2002. Comparative physicochemical properties of hydrocortisone-PVP composites prepared using supercritical carbon dioxide by the GAS anti-solvent recrystallisation process, by coprecipitation and by spray drying. Int. J. Pharm., 245, 75-82.
Corrigan, D. O., Healy, A. M. and Corrigan, O. I., 2002. The effect of spray drying solutions of polyethylene glycol (PEG) and lactose/PEG on their physicochemicalproperties. Int. J. Pharm., 235, 193-205.
Corrigan, O. I. and Holohan, E. M., 1984. Amorphous spray-dried hydroflumethiazide-polyvinylpyrrolidone systems: physicochemical properties. J. Pharm. Pharmacol.,36, 217-221.
Corrigan, O. I., Holohan, E. M. and Reilly, M. R., 1985. Physicochemical properties ofindomethacin and related compounds co-spray dried with polyvinylpyrrolidone. Drug Dev. Ind. Pharm., 11, 677-695.
Corrigan, O. I., Holohan, E. M. and Sabra, K., 1984. Amorphous forms of thiazide diuretics prepared by spray-drying. Int. J. Pharm., 18, 195-200.
Couchman, P. R. and Karasz, F. E., 1978. A classical thermodynamic discussion of the effect of composition on glass-transition temperatures. Macromolecules, 11, 117-119.
Cox, E. P., 1927. A method of assessing numerical and percentage values to the degree of roundness of sand grains. J. Paleontol., 1, 179-183.
Craig, D. Q. M., Royall, P. G., Kett, V. L. and Hopton, M. L., 1999. The relevance of the amorphous state to pharmaceutical dosage forms: glassy drugs and freeze dried systems. Int. J. Pharm., 179, 179-207.
Duberg, M. and Nyström, C., 1986. Studies on direct compression of tablets. XVII. Porosity-pressure curves for the characterization of volume reduction mechanismsin powder compression. Powder Technol., 46, 67-75.
Dunbar, C. A., Concessio, N. M. and Hickey, A. J, 1998. Evaluation of atomizerperformance in production of respirable spray-dried particles. Pharm. Dev.Technol., 3, 433-441.
Dyer, A. M., Khan, K. A. and Aulton, M. E., 1994. Effect of the drying method on the mechanical and drug release properties of pellets prepared by extrusion-spheronization. Drug Dev. Ind. Pharm., 20, 3045-3068.
Eissens, A. C., Bolhuis, G. K., Hinrichs, W. L. J. and Frijlink, H. W., 2002. Inulin as filler-binder for tablets prepared by direct compaction. Eur. J. Pharm. Sci., 15, 31-38.
Ek, R., Wormald, P., Östelius, J., Iversen, T. and Nyström, C., 1995. Crystallinity index of microcrystalline cellulose particles during compression of tablets. Int. J. Pharm., 125, 257-264.
Elamin, A. A., Ahlneck, C., Alderborn, G. and Nyström, C., 1994. Increased metastablesolubility of milled griseofulvin, depending on the formation of a disorderedsurface structure. Int. J. Pharm., 111, 159-170.
Elamin, A. A., Sebhatu, T. and Ahlneck, C., 1995. The use of amorphous modelsubstances to study mechanically activated materials in the solid state. Int. J. Pharm., 119, 25-36.
52

Eriksson, M., Alderborn, G., Nyström, C., Podczeck, F. and Newton, J. M., 1997. Comparison between and evaluation of some methods for the assessment of the sphericity of pellets. Int. J. Pharm., 148, 149-154.
Fell, J. T. and Newton, J. M., 1970. Determination of tablet strength by the diametral-compression test. J. Pharm. Sci., 59, 688-691.
Fell, J. T. and Newton, J. M., 1971. The production and properties of spray dried lactose. Part 3. The compaction properties of samples of spray dried lactose produced on an experimental drier. Pharm. Acta Helv., 46, 441-447.
Femi-Oyewo, M. N. and Spring, M. S., 1982. The effect of surface active agents on granules and compacts containing 5% sulphanilamide. Int. J. Pharm. Technol.Prod. Manuf., 3, 73-75.
Fielden, K.E., Newton, J. M., O`Brien, P. and Rowe, R. C., 1988. Thermal studies on the interaction of water and microcrystalline cellulose. J. Pharm. Pharmacol., 40, 674-678.
Forbes, R. T., Davis, K. G., Hindle, M., Clarke, J. G and Maas, J., 1998. Water vapour sorption studies on the physical stability of a series of spray-dried protein/sugarpowders for inhalation. J. Pharm. Sci., 87, 1316-1321.
Fukuoka, E., Makita, M., Yamamura, S., 1987. Glassy state of pharmaceuticals. II. Bioinequivalence of glassy and crystalline indomethacin. Chem. Pharm. Bull., 35, 2943-2948.
Goodhart, F. W., 1994. Lactose. In Wade, A. and Weller, P. J. (Eds.), Handbook of pharmaceutical excipients, The Pharmaceutical Press, London.
Gordon, M. and Taylor, J. S., 1952. Ideal copolymers and the second-order transitions of synthetic rubbers. I. Non-crystalline copolymers. J. Appl. Chem., 2, 493-500.
Gunsel, W. C. and Lachman, L., 1963. Comparative evaluation of tablet formulationsprepared from conventionally-processed and spray-dried lactose. J. Pharm. Sci., 52, 178-182.
Habib, Y. S., Augsburger, L. L. and Shangraw, R. F., 2002. Production of inert cushioning beads: effect of excipients on the physicomechanical properties of freeze-dried beads containing microcrystalline cellulose produced by extrusion-spheronization. Int. J. Pharm., 233, 67-83.
Habib, Y. S. and Shangraw, R. F., 1997. Effect of different drying techniques on the physico-mechanical properties of beads containing microcrystalline cellulose (mcc) produced by extrusion spheronization. Pharm. Res., 14, S14.
Hancock, B. C., Carlson, G. T., Ladipo, D. D., Langdon, B. A. and Mullarney, M. P., 2002. Comparison of the mechanical properties of the crystalline and amorphousform of a drug substance. Int. J. Pharm., 241, 73-85.
Hancock, B. C. and Parks, M., 2000. What is the true solubility advantage for amorphous pharmaceuticals? Pharm. Res., 17, 397-404.
Hancock, B. C., Shamblin, S. L., Zografi, G., 1995. Molecular mobility of amorphouspharmaceutical solids below their glass transition temperatures. Pharm. Res., 12, 799-806.
Hancock, B. C. and Zografi, G., 1994. The relationship between the glass transition temperature and the water content of amorphous pharmaceutical solids. Pharm.Res., 11, 471-477.
Hancock, B. C. and Zografi, G., 1997. Characteristics and significance of the amorphous state in pharmaceutical systems. J. Pharm. Sci., 86, 1-12.
53

Hasatani, M., Itaya, Y., Muroie, K. and Taniguchi, S., 1993. Contraction characteristics of molded ceramics during drying. Drying Technol., 11, 815-830.
Heckel, R. W., 1961a. Density-pressure relationships in powder compaction. Trans. Metall. Soc. AIME, 221, 671-675.
Heckel, R. W., 1961b. An analysis of powder compaction phenomena. Trans. Metall. Soc. AIME, 221, 1001-1008.
Hiestand, E. N., 1991. Tablet bond. I. A theoretical model. Int. J. Pharm., 67, 217-229. Holm, P., Jungersen, O., Schaefer, T. and Kristensen, H.G., 1983. Granulation in high
speed mixers. Part I: Effects of process variables during kneading. Pharm. Ind., 45, 806-811.
Hunter, B. M. and Ganderton, D., 1972. The effect of particle size on the granulation of lactose by massing and screening. J. Pharm. Pharmacol., 24, 17P-24P.
Hüttenrauch, R., 1978. Molekulargalenik als Grundlage moderner Arzneiformung. Acta Pharm. Technol., APV Informationsdienst, Suppl. 6, 55-127.
Hüttenrauch, R., 1983. Modification of starting materials to improve tabletting properties. Pharm. Ind., 45, 435-440.
Hüttenrauch, R. and Keiner, I., 1976. Molekulargalenik. 6. Über Komprimatbildungenaus aktivierten Grundstoffen am Beispiel der Lactose. Pharmazie, 31, 331.
Hüttenrauch, R. and Keiner, I., 1979. Producing lattice defects by drying process. Int. J. Pharm., 2, 59-60.
Jaegerskou, A., Holm, P., Schaefer, T. and Kristensen, H. G., 1984. Granulation in high speed mixers. Part 3: Effects of process variables on the intragranular porosity. Pharm. Ind., 46, 310-314.
Jerwanska, E., Alderborn, G., Newton, J. M. and Nyström, C., 1995. The effect of water content on the porosity and liquid saturation of extruded cylinders. Int. J. Pharm.,121, 65-71.
Johansson, B., 1999. Tabletting behaviour of aggregates. Mechanistical conception and modulation by variations in aggregate physical properties. Ph.D. Thesis, Uppsala University, Sweden.
Johansson, B. and Alderborn, G., 2001. The effect of shape and porosity on the compression behaviour and tablet forming ability of granular materials formedfrom microcrystalline cellulose. Eur. J. Pharm. Biopharm., 52, 347-357.
Johansson, B., Nicklasson, F. and Alderborn, G., 1998. Effect of pellet size on degree of deformation and densification during compression and on compactability of microcrystalline cellulose pellets. Int. J. Pharm., 163, 35-48.
Johansson, B., Wikberg, M., Ek, R. and Alderborn, G., 1995. Compression behaviour and compactability of microcrystalline cellulose pellets in relationship to their pore structure and mechanical properties. Int. J. Pharm., 117, 57-73.
Jolley, J. E., 1970. The microstructure of photographic gelatin binders. Photogr. Sci. Eng., 14, 169-177.
Karehill, P. G. and Nyström, C., 1990. Studies on direct compression of tablets XXI. Investigation of bonding mechanisms of some directly compressed materials by strength characterization in media with different dielectric constants (relativepermittivity). Int. J. Pharm., 61, 251-260.
Kawakita, K., Hattori, I. and Kishigami, M., 1977. Characteristic constants in Kawakita’s powder compression equation. J. Powder Bulk Solids Technol., 1, 3-8.
Kendall, K., 1988. Agglomerate strength. Powder Metallurgy, 31, 28-31.
54

Kleinebudde, P., 1994. Shrinking and swelling properties of pellets containingmicrocrystalline cellulose and low substituted hydroxpropylcellulose: I. Shrinkingproperties. Int. J. Pharm., 109, 209-219.
Kotiyan, P. N. and Vavia, P. R., 2001. Eudragits: Role as crystallisation inhibitors in drug-in-adhesive transdermal systems of estradiol. Eur. J. Pharm. Biopharm., 52, 173-180.
Kristensen, H. G.,1988. Agglomeration of powders. Acta Pharm. Suec., 25, 187-204. Kristensen, H. G., Holm, P., Jaegerskou, A. and Schaefer, T., 1984. Granulation in high
speed mixers. Part 4: Effect of liquid saturation on the agglomeration. Pharm.Ind., 46, 763-767.
Leuenberger, H., 1982. The compressibility and compactability of powder systems. Int. J. Pharm., 12, 41-55.
Leyland, R. L., 1994. Polyoxyethylene sorbitan fatty acid esters. In Wade, A. and Weller, P. J. (Eds.), Handbook of pharmaceutical excipients, The PharmaceuticalPress, London.
Li, L. C. and Peck, G. E., 1990. The effect of agglomeration methods on the micromeritic properties of a maltodextrin. Drug Dev. Ind. Pharm., 16, 1491-1503.
Lüdde, K.-H. and Kawakita, K., 1966. Die Pulverkompression. Pharmazie, 21, 393-403. Mandal, T. K., 1995. Evaluation of microwave drying for pharmaceutical granulations.
Drug Dev. Ind. Pharm., 21, 1683-1688. Mathur, L., 1994. Microcrystalline cellulose. In Wade, A. and Weller, P. J. (Eds.),
Handbook of pharmaceutical excipients, The Pharmaceutical Press, London.Matsumoto, T. and Zografi, G., 1999. Physical properties of solid molecular dispersions
of indomethacin with poly(vinylpyrrolidone) and poly(vinylpyrrolidone-co-vinyl-acetate) in relation to indomethacin crystallization. Pharm. Res., 11, 1722-1728.
Meenan, P., Roberts, K. J., Knight, P. C. and Yuregir, K., 1997. The influence of spray drying conditions on the particle properties of recrystallized burkeite(Na2CO3*(Na2SO4)2). Powder Technol., 90, 125-130.
Millili, G. P. and Schwartz, J. B., 1990. The strength of microcrystalline cellulose pellets: the effect of granulating with water/ethanol mixtures. Drug. Dev. Ind. Pharm., 16, 1411-1426.
Mosharaff, M. and Nyström, C., 1999. Effect of dry mixing on the apparent solubility of hydrophobic, sparingly soluble drugs. Eur. J. Pharm. Sci., 9, 145-156.
Mullier, M. A., Seville, J. P. K. and Adams, M. J., 1987, A fracture mechanics approachto the breakage of particle agglomerates. Chem. Eng. Sci., 42, 667-677.
Newitt, D. M. and Coleman, M., 1952. The mechanism of drying of solids. Part III. Thedrying characteristics of china clay. Trans. Instn. Chem. Eng., 30, 8-45.
Nicklasson, F. and Alderborn, G., 1999. Modulation of the tabletting behaviour of microcrystalline cellulose pellets by the incorporation of polyethylene glycol. Eur. J. Pharm. Sci., 9, 57-65.
Nicklasson, F. and Alderborn, G., 2000. Analysis of the compression mechanics of pharmaceutical agglomerates of different porosity and composition using the Adams and Kawakita equations. Pharm. Res., 17, 947-952.
Nicklasson, F., Johansson, B. and Alderborn, G., 1999a. Occurrence of fragmentationduring compression of pellets prepared from a 4 to 1 mixture of dicalciumphosphate dihydrate and microcrystalline cellulose. Eur. J. Pharm. Sci., 7, 221-229.
55

Nicklasson, F., Johansson, B. and Alderborn, G., 1999b. Tabletting behaviour of pellets of two series of porosities-a comparison between pellets of two different compositions. Eur. J. Pharm. Sci., 8, 11-17.
Nonhebel, G. and Moss, A. A. H., 1971. Drying of solids in the chemical industry. Butterworth & Co Ltd., London.
Nyström, C., Alderborn, G., Duberg, M. and Karehill, P. G., 1993. Bonding surface area and bonding mechanism-two important factors for the understanding of powder compactability. Drug Dev. Ind. Pharm., 19, 2143-2196.
Oksanen, C. A. and Zografi, G., 1993. Molecular mobility of absorbed water and poly(vinylpyrrolidone). Pharm. Res., 11, 791-799.
Olsson, H., Adolfsson, Å. and Nyström, C., 1996. Compaction and measurement of tablets in liquids with different dielectric constants for determination of bonding mechanisms-evaluation of the concept. Int. J. Pharm., 143, 233-245.
Remon, J. P. and Schwartz, J. B., 1987. Effect of raw materials and processing on the quality of granules prepared from microcrystalline cellulose-lactose mixtures.Drug Dev. Ind. Pharm., 13, 1-14.
Riepma, K. A., Vromans, H., Zuurman, K. and Lerk, C. F., 1993. The effect of dry granulation on the consolidation and compaction of crystalline lactose. Int. J. Pharm., 97, 29-38.
Ritala, M., Holm, P., Schaefer, T. and Kristensen, H. G., 1988. Influence of liquid bonding strength on power consumption during granulation in a high shear mixer.Drug Dev. Ind. Pharm., 14, 1041-1060.
Roberts, R. J. and Rowe, R. C., 1987. The compaction of pharmaceutical and other model materials-a pragmatic approach. Chem. Eng. Sci., 42, 903-911.
Rumpf, H., 1962. The strength of granules and agglomerates. In Knepper, W. A (Ed.),Agglomeration, Interscience, New York, pp. 379-418.
Scherer, G. W., 1990. Theory of drying. J. Am. Ceram. Soc., 73, 3-14. Schmitt, E. A., Law, D. and Zhang, G. G. Z., 1999. Nucleation and crystallization
kinetics of hydrated amorphous lactose above the glass transition temperature. J. Pharm. Sci., 88, 291-296.
van Scoik, K. G., Zoglio, M. A. and Carstensen, J. T., 1991. Drying and driers. In Boylan, J. C. and Swarbrick, J. (Eds.), Encyclopedia of pharmaceutical technology, Marcel Dekker Inc., New York.
Sebhatu, T., Ahlneck, C. and Alderborn, G., 1997. The effect of moisture content on the compression and bond-formation properties of amorphous lactose particles. Int. J. Pharm., 146, 101-114.
Sebhatu, T. and Alderborn, G., 1999. Relationships between the effective interparticulate contact area and the tensile strength of tablets of amorphous and crystalline lactose of varying particle size. Eur. J. Pharm. Sci., 8, 235-242.
Sebhatu, T., Angberg, M. and Ahlneck, C., 1994. Assessment of the degree of disorder in crystalline solids by isothermal microcalorimetry. Int. J. Pharm., 104, 135-144.
Shamblin, S. L., Huang, S. Y. and Zografi, G., 1996. The effects of co-lyophilized polymeric additives on the glass transition temperature and crystallization ofamorphous sucrose. J. Thermal Anal., 47, 1567-1579.
Shamblin, S. L., Taylor, L. S. and Zografi, G., 1998. Mixing behavior of colyophilized binary systems. J. Pharm. Sci., 87, 694-701.
Shamblin, S. L. and Zografi, G., 1998. Enthalpy relaxation in binary amorphousmixtures containing sucrose. Pharm. Res., 15, 1828-1834.
56

Shamblin, S. L. and Zografi, G., 1999. The effects of absorbed water on the properties of amorphous mixtures containing sucrose. Pharm. Res., 16, 1119-1124.
Sun, C. and Grant, D. J. W., 2001. Influence of elastic deformation of particles on Heckel analysis. Pharm. Dev. Technol., 6, 193-200.
Suzuki, T., Imamura, K., Fujimoto, H. and Okazaki, M., 1998. Relation between thermal stabilizing effect of sucrose on LDH and sucrose-LDH hydrogen bond. J. Chem. Eng. Japan, 31, 565-570.
Takeuchi, H., Yasuji, T., Hino, T., Yamamoto, H. and Kawashima, Y., 1999. Compaction properties of composite particles consisting of lactose with sodiumalginate prepared by spray-drying. Pharm. Res., 16, 1193-1198.
Takeuchi, H., Yasuji, T., Hino, T., Yamamoto, H. and Kawashima, Y., 2000a. Temperature-induced crystallization and compactability of spray dried compositeparticles composed of amorphous lactose and various types of water-soluble polymer. Chem. Pharm. Bull., 48, 585-588.
Takeuchi, H., Yasuji, T., Hino, T., Yamamoto, H. and Kawashima, Y., 2000b. Temperature-and moisture-induced crystallization of amorphous lactose in composite particles with sodium alginate prepared by spray-drying. Pharm. Dev. Technol., 5, 355-363.
Tapper, G. I. and Lindberg, N.O., 1986. Granulation of some lactose qualities with different particle size distributions in a domestic type mixer. Acta Pharm. Suec., 23, 47-56.
Taylor, L. S. and Zografi, G., 1998. Sugar-polymer hydrogen bond interactions in lyophilized amorphous mixtures. J. Pharm. Sci., 87, 1615-1621.
Walkling, W. D., 1994. Povidone. In Wade, A. and Weller, P. J. (Eds.), Handbook of pharmaceutical excipients, The Pharmaceutical Press, London.
Wells, J. I. and Walker, C. V., 1983. The influence of granulating fluids upon granule and tablet properties: the role of secondary binding. Int. J. Pharm., 15, 97-111.
Vervaet, C., Baert, L. and Remon, J. P., 1995. Extrusion-spheronisation. A literaturereview. Int. J. Pharm., 116, 131-146.
Wikberg, M. and Alderborn, G., 1990. Compression characteristics of granulated materials II. Evaluation of granule fragmentation during compression by tablet permeability and porosity measurements. Int. J. Pharm., 62, 229-241.
Wikberg, M. and Alderborn, G., 1991. Compression characteristics of granulated materials. IV. The effect of granule porosity on the fragmentation propensity and compactability of some granulations. Int. J. Pharm., 69, 239-253.
Wikberg, M. and Alderborn, G., 1993. Compression characteristics of granulated materials. VII. The effect of intragranular binder distribution on the compactability of some lactose granulations. Pharm. Res., 10, 88-94.
Willart, J. F., De Gusseme, A., Hemon, S., Descamps, M., Leveiller, F. and Rameau,A., 2002. Vitrification and polymorphism of trehalose induced by dehydration of trehalose dihydrate. J. Phys. Chem. B, 106, 3365-3370.
Willart, J. F., De Gusseme, A., Hemon, S., Odou, G., Danede, F. and Descamps, M., 2001. Direct crystal to glass transformation of trehalose induced by ball milling.Solid State Commun., 119, 501-505.
Voellmy, C., Speiser, P. and Solvia, M., 1977. Microencapsulation of Phenobarbital by spray polycondensation. J. Pharm. Sci., 66, 631-634.
57

Vromans, H., Bolhuis, G. K. and Lerk, C. F., 1986. Studies on tableting properties of lactose. VI. Consolidation and compaction of spray dried amorphous lactose. Acta Pharm. Suec., 23, 231-240.
York, P., 1992. Crystal engineering and particle design for the powder compaction process. Drug Dev. Ind. Pharm., 18, 677-721.
Yoshioka, M., Hancock, B. C. and Zografi, G., 1994. Crystallization of indomethacinfrom the amorphous state below and above its glass transition temperature. J. Pharm. Sci., 83, 1700-1705.
Yoshioka, M., Hancock, B. C. and Zografi, G., 1995. Inhibition of indomethacincrystallization in poly(vinylpyrrolidone) coprecipitates. J. Pharm. Sci., 84, 983-986.
Zeng, X. M., Martin, G. P. and Marriott, C., 2001. Effects of molecular weight of polyvinylpyrrolidone on the glass transition and crystallization of co-lyophilized sucrose. Int. J. Pharm., 218, 63-73.
van der Zwan, J. and Siskens, C. A. M., 1982. The compaction and mechanicalproperties of agglomerated materials. Powder Technol., 33, 43-54.
58