energy conversion: solid oxide fuel cells -...
TRANSCRIPT

6Energy Conversion: Solid Oxide
Fuel CellsFirst-Principles Modeling of Elementary Processes
E.A. Kotomin1,2, R. Merkle1, Y. A. Mastrikov2,3, M.M. Kuklja3, and J. Maier1
1Max Planck Institute for Solid State Research, Stuttgart, Germany2Institute for Solid State Physics, University of Latvia, Riga, Latvia
3Materials Science and Engineering Department, University of Maryland, College Park, USA
6.1 Introduction
Fuel cells are electrochemical devices that directly transform the chemical freeenergy of combustion (e.g., H2 + O2 and CHx + O2) into electrical energy.The avoidance of a thermal detour guarantees high theoretical efficiency. As faras the temperature regimes are concerned, we distinguish between high tempera-ture ceramic fuel cells, intermediate-temperature fuel cells, and low temperature(i.e., only slightly above room temperature) fuel cells. The high temperature fuelcells are usually based on oxide components (ternary transition metal oxides ascathodes, Ni or Cu cermets as anodes, and acceptor-doped zirconia or ceria aselectrolytes). The high temperature necessary for ion conduction (though givingrise to problems concerning materials durability and compatibility) is favorablefor the electrode reaction kinetics and additionally allows direct electrochemicalconversion of hydrocarbons. Low temperature fuel cells basically rely on the reac-tion between H2 and O2 and typically use polymeric proton conductors (ionomers)([1] and references cited there). A major materials challenge is the selection of the
Computational Approaches to Energy Materials, First Edition. Edited by Aron Walsh, Alexey A. Sokol and C. Richard A. Catlow.
© 2013 John Wiley & Sons, Ltd. Published 2013 by John Wiley & Sons, Ltd.

150 Computational Approaches to Energy Materials
electrocatalysts, in particular on the cathode side. Even though intense research isunderway on alternative materials, noble metals are still the candidates of choice.A particularly challenging task is the development of direct methanol (or ethanol)fuel cells at these temperatures. A good compromise, as far as the dilemma betweenmaterials problems and electrochemical kinetics is concerned, is to be expectedfrom intermediate temperature fuel cells. Here, mostly highly protonically conduct-ing oxides, phosphates, and silicates are envisaged as electrolytes [2–4]. However,the development of properly adapted, catalytically active electrodes for this deviceclass is still in its infancy.A great deal of modeling work has been performed in the context of low and
intermediate temperature fuel cells, in terms of understanding the conductionmech-anisms of the electrolytes (e.g., see [5–9] and references therein), as well as in termsof elucidating the electrochemical reactions (e.g., see [10–15] and references citedtherein). As it is impossible to take account of all of this progress in a single chap-ter, we concentrate on high temperature fuel cells here and lay emphasis on thoseissues which atomistic modeling is most helpful for.Solid oxide fuel cells (SOFCs) have been under intensive investigation since the
1980s (for an overview see [16–20]). The detailed studies of SOFCs raise a numberof questions that are interesting also from a fundamental point of view, such asthe kinetics of the electrode reactions, the oxygen transport rate in solids, andthe thermodynamic stability of the involved phases and interfaces. With the helpof modeling one can vary parameters and handle adjusting screws that are ofteninaccessible by experiments. So, a major part of this chapter consists of showinghow one can extract a detailed reaction mechanism for the oxygen reaction at thecathode, in particular, when the conclusions are complemented by experimentalresults. As the point defects are the key species, we begin with modeling them, andat the end of the chapter we will also briefly tackle transport in the electrolyte andfuel reduction at the anode.Not least owing to the kinetic stability of the oxygen molecule, one of the
main limiting contributions to the overall cell performance comes from the oxygenreduction and incorporation reaction at the cathode, which can be addressed byoptimization of electrode morphology and choice of electrode material. ABO3-type perovskites and perovskite-related oxides with adequate electronic as wellas ionic (oxygen vacancy) conductivities are widely applied as cathode materi-als. The rate constants for oxygen incorporation into these materials vary widely,as shown in Figure 6.1 for (La,Sr)MnO3 (LSM), (La,Sr)(Co,Fe)O3 (LSCF), and(Ba,Sr)(Co,Fe)O3 (BSCF) perovskites. To really reflect the intrinsicmaterials prop-erties and avoid influences (e.g., from varying morphologies in porous films or gasdiffusion limitations), these data were measured on dense films (prepared by pulsedlaser deposition (PLD)). Without going into details, the sheer fact that the materialwith the highest concentration of oxygen vacancies (V ..
O, Kroger–Vink notation[21]) exhibits the highest rate constant indicates that point defects play a decisiverole in the electrode reactions.
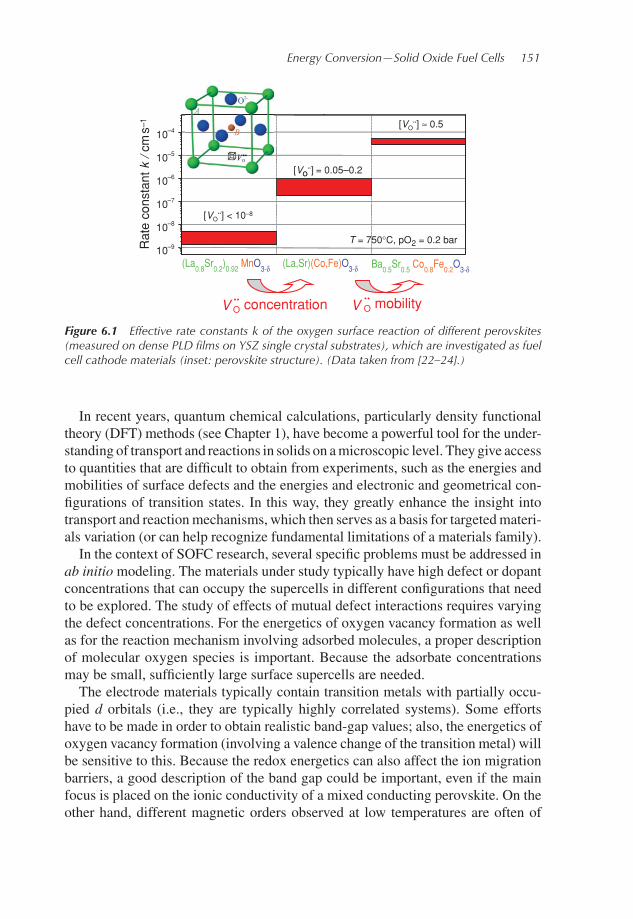
Energy Conversion—Solid Oxide Fuel Cells 151
V O mobilityV O concentration.. ..
Rat
e co
nsta
nt k
/ cm
s–1
10–9
10–8
10–7
10–6
10–5
10–4
(La0.8
Sr0.2
)0.92 MnO
3-δ
[VO..] ≈ 0.5
[VO..]
[V ] < 10
O = 0.05–0.2
O.. –8
T = 750°C, pO2 = 0.2 bar
Ba0.5
Sr0.5 Co
0.8Fe
0.2O
3-δ(La,Sr)(Co,Fe)O
3-δ
Figure 6.1 Effective rate constants k of the oxygen surface reaction of different perovskites(measured on dense PLD films on YSZ single crystal substrates), which are investigated as fuelcell cathode materials (inset: perovskite structure). (Data taken from [22–24].)
In recent years, quantum chemical calculations, particularly density functionaltheory (DFT) methods (see Chapter 1), have become a powerful tool for the under-standing of transport and reactions in solids on amicroscopic level. They give accessto quantities that are difficult to obtain from experiments, such as the energies andmobilities of surface defects and the energies and electronic and geometrical con-figurations of transition states. In this way, they greatly enhance the insight intotransport and reactionmechanisms, which then serves as a basis for targetedmateri-als variation (or can help recognize fundamental limitations of a materials family).In the context of SOFC research, several specific problems must be addressed in
ab initio modeling. The materials under study typically have high defect or dopantconcentrations that can occupy the supercells in different configurations that needto be explored. The study of effects of mutual defect interactions requires varyingthe defect concentrations. For the energetics of oxygen vacancy formation as wellas for the reaction mechanism involving adsorbed molecules, a proper descriptionof molecular oxygen species is important. Because the adsorbate concentrationsmay be small, sufficiently large surface supercells are needed.The electrode materials typically contain transition metals with partially occu-
pied d orbitals (i.e., they are typically highly correlated systems). Some effortshave to be made in order to obtain realistic band-gap values; also, the energetics ofoxygen vacancy formation (involving a valence change of the transition metal) willbe sensitive to this. Because the redox energetics can also affect the ion migrationbarriers, a good description of the band gap could be important, even if the mainfocus is placed on the ionic conductivity of a mixed conducting perovskite. On theother hand, different magnetic orders observed at low temperatures are often of

152 Computational Approaches to Energy Materials
minor importance because their energy difference is much smaller than the reactionenthalpies and reaction barriers considered, and the ion transfer processes are notdirectly influenced by the magnetic state. All of these issues are discussed in thischapter.Because the SOFCs are operated at 600–1000 ◦C, the effect of elevated
temperatures must be taken seriously (e.g., for the identification of the stablesurface terminations). This is briefly discussed in Section 6.3.1. The stability ofdifferent terminations does not only depend on temperature and gas pressure,but may also be affected by the presence (or even segregation) of dopants at therespective surface. For many perovskites studied as cathode materials, the simplesurface terminations are also polar.An increase in perovskite complexity when going from LaMnO3 (LMO) to
solid solutions with mixed occupancy on both cation sites ((Ba,Sr)(Co,Fe)O3−δ
and (La,Sr)(Co,Fe)O3−δ) is accompanied by an increase in material instabilitywith respect to disorder and phase transformations. The bulk (and surface) oxygenvacancy formation energy strongly depends on the actual oxidation state of the tran-sition metals and thus on the exact materials stoichiometry (dopant content as wellas oxygen deficiency), which is often not clearly addressed, not only theoreticallybut also in experimental studies. These issues are discussed in Section 6.3.2.Finally, in addition to point defects, higher dimensional defects can influence
the electrical properties, in particular of the electrolyte materials. Most elec-trolytes exhibit a more or less pronounced blocking character of the grain bound-aries for ion (oxygen vacancy) transfer. Ab initio calculations of model grainboundaries are thus highly desirable, but owing to the many degrees of freedom(mutual crystal and grain boundary plane orientation, but also local deviations fromaverage stoichiometry and electroneutrality), they are also extremely challenging(see Section 6.4).In this chapter, we focus on key SOFC topics, such as stable surface terminations,
bulk and surface point defect formation and migration energetics, and in particularthe mechanism of the oxygen incorporation surface reaction mainly based onthe results of studies obtained in our research groups. Other topics, such as theeffect of strain on defect mobility and ab initio treatments of grain boundaries, arediscussed on the basis of recent results from literature. Experimental studies willbe cited when necessary for the comparison with the theoretical findings (a fullcoverage of experimental research would go far beyond the scope of this chapter).This chapter exemplarily illustrates the range and scope of contemporary methodsbased on DFT in materials research.
6.2 Computational Details
Basic computational approaches to calculations of the atomic and electronic struc-ture of perfect and defective solids are discussed in Chapter 1 of this book (see

Energy Conversion—Solid Oxide Fuel Cells 153
also [25]). In most of our SOFC calculations of LSM and BSCF cathodes, weemployed the ab initio DFT computer code VASP, which combines a plane-wavebasis set and an exchange–correlation functional based on the generalized gra-dient approximation (GGA). Spin polarized calculations of the LMO bulk andthe (001) and (110) surfaces are in good agreement with experiments (whereavailable) [26]. In particular, for the low-temperature orthorhombic structure, theA-type antiferromagnetic configuration is the energetically most favorable, whichis in agreement with experiment. The lattice constant of both the cubic (stableabove 750K) and orthorhombic phases exceeds the experimental value by only0.5%. The calculated cohesive energy of 30.7 eV is close to the experimental value(31 eV).The calculated surface relaxations and surface energies show aweak dependence
on the magnetic configuration [26, 27]. For slabs, the ferromagnetic configurationhas the lowest energy; thus, we performed all further calculations with collinearspins. We must also keep in mind that the relevant magnetic effects (≈0.1 eV)are much smaller than the adsorption and migration energies under study (one toseveral electron volts). However, a complete neglect of spin polarization (“closedshell” calculation) results in considerable errors in material properties. We studiedboth orthorhombic and cubic phases for the oxygen adsorption and thermodynamicstability of surfaces with adsorbates, but focused on the orthorhombic phase in theanalysis of intermediates and transition states for the kinetics treatment. Calcu-lations for the defects/adsorbates in the high-temperature cubic phase [28] givequalitatively similar results; in particular, the atomic O adsorption energy atop Mnincreases by approximately 3% only. Nevertheless, the use of symmetry restrictionsmay lead to artifacts from rearrangements in the bulk structure if these restrictionshave to be released, for example, for low-symmetrical transition states (all resultspresented here were obtained without symmetry restrictions).For surface models, convergence with respect to the number of atomic layers
should always be checked. We found that seven- and eight-plane slabs are thickenough to show convergence of the main properties. The periodically repeatedslabs were separated by a vacuum gap of 15.8 A. All atomic coordinates in a slabwere allowed to relax. The eight-layer slab (LaOMnO2)4 has the proper cation ratioand oxygen stoichiometry (all Mn atoms are in the formal + 3 oxidation state), butexhibits a nonzero slab dipole moment and is terminated by different planes (MnO2and LaO). This is avoided in the symmetric seven-layer slab MnO2(LaOMnO2)3,but this slab has a Mn excess relative to La and a higher oxygen content. Thisslightly affects the energy for dissociative oxygen adsorption on the MnO2(001)surface,
O2 + 2MnxMn � 2O−ad + 2Mn.
Mn, (6.1)
which is −2.7 eV for the (LaOMnO2)4 slab and −2.2 eV for the symmetricalMnO2(LaOMnO2)3 slab [28]. On the other hand, the average Mn oxidation stateof more than 3 implicitly mimics the situation in Sr-doped LaMnO3, which is

154 Computational Approaches to Energy Materials
actually used in SOFCs. Due to the reduced computation time for the symmetricalconfiguration and the fact that the symmetrical slab allows decoupling effects ofdifferent surface terminations, all further calculations refer to the seven-layer slabs.The choice of the 2
√2× 2√2 (001) surface supercell for the calculation of
adsorbates/surface defects corresponds to a surface coverage of 12.5%. This con-centration is quite large compared to the actual adsorbate coverage at operatingconditions (cf. Section 6.3.1.2), but is still acceptable for semiquantitative conclu-sions. Several test calculations were performed for a smaller coverage of 5%. Theuse of even larger defect concentrations and/or highly polar and oxygen-deficient(110) surfaces could lead to reaction enthalpies that are much too exothermic andoxygen incorporation reaction barriers that are too low (or even missing) [29–32].The reaction and ion migration barriers (also for the other perovskites studied inSections 6.3.2 and 6.3.3) were calculated using the nudged elastic band method[33,34]. The effective charges on the ions were determined according to the Bader(topological) analysis [35]. The effective charges both in the bulk and on the sur-faces of LaMnO3 are quite different from the formal charges, namely, + 2.13 e0 forLa, + 1.85 e0 for Mn, and−1.29 e0 for O [26,27]. The considerable contribution ofthe covalence effects inMn−O chemical bondingmakes classical force-field calcu-lations of these systems, and especially of diffusion-controlled processes therein, anontrivial task.In calculations of oxygen-exchange related processes, the proper description of
molecular oxygen is important. In plane-wave calculations, the O2 dissociationenergy Ediss and bond length d depend considerably on both the O pseudopotentialand the exchange–correlation functional used. The VASP calculations with theso-called standard pseudopotential considerably overestimate Ediss = 5.9 eV butgive a reasonable d = 1.23 A (experimental: Ediss = 5.12 eV and d = 1.21 A [36]),whereas “soft” O pseudopotentials yield a better Ediss = 5.24 eV but overestimated = 1.29 A. Several procedures were suggested to correct for such DFT errors inmetal oxide and O2 calculations [28, 37, 38].For comparison with VASP plane-wave modeling, a series of calculations were
performed using the CRYSTAL computer code [39] with a linear combinationof atomic orbital (LCAO) basis set. Its advantages are the efficient incorporationof the so-called hybrid exchange–correlation functionals, which permit a moreaccurate calculation of the band gap (see Chapter 1), and the absence of theartificial translation of slabs along the z-axis in surface calculations, which isunavoidable in plane-wave calculations. The O2 dissociation energy and bondlength are reproduced quite well: Ediss = 5.30 eV and d = 1.20 A.Lastly, we note that in oxygen vacancy calculations, the supercells used are
always neutral (no compensating “background charge”). The charge distributionbetween the O vacancy and its environment adjusts itself while minimizing thetotal energy of the system. For reducible transition metals, the electrons are typi-cally transferred to the nearest cations (see Section 6.3.2 and 6.3.3 on LSCF andBSCF).

Energy Conversion—Solid Oxide Fuel Cells 155
6.3 Cathode Materials and Reactions
6.3.1 Surfaces: LaMnO3 and (La,Sr)MnO3 Perovskites
The prerequisite for investigating the oxygen incorporation reaction mechanismis to identify the stable surface terminations of the mixed conducting perovskites.Effects of doping (and potential dopant segregation to the surface) have to be con-sidered as well as the elevated operating temperature (thermodynamic treatment,Section 6.3.1.1). Owing to the high operating temperature (allowing for rearrange-ments also in the cation lattice), the perovskite is expected to expose mainly themost stable surface terminations. Nevertheless, the overall reaction rate may bedominated by a contribution from a slightly less stable surface with intrinsicallyhigher rate (see Section 6.3.1.3).
6.3.1.1 Surface Termination, Surface Point Defects
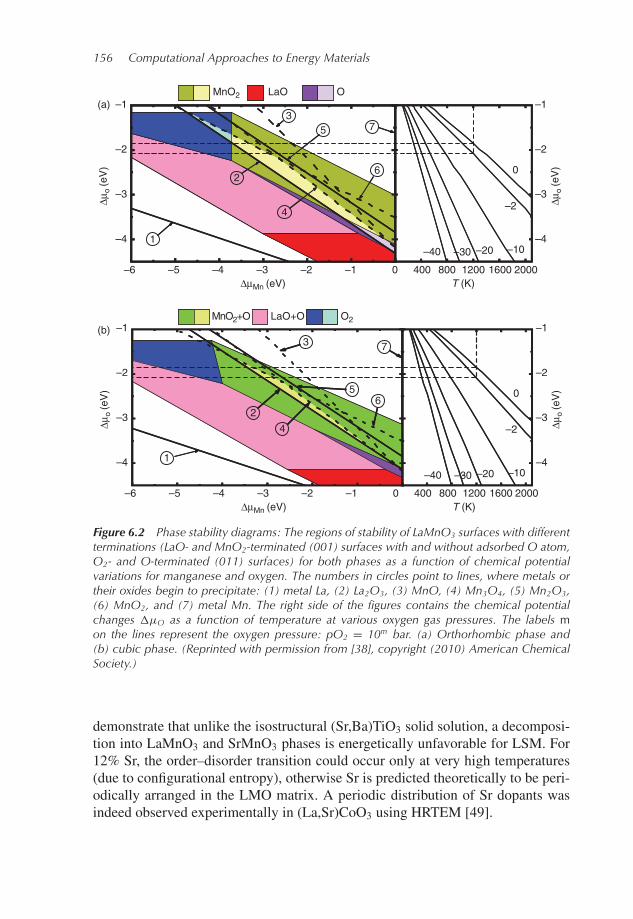
We performed a series of calculations for the structure of LaMnO3 (001) and (110)surfaces using different methods: pair potentials [40], Hartree–Fock LCAO [41],hybrid B3PW LCAO [27], and GGA [42] density functional. The results for theatomic relaxations obtained using these different methods are in reasonable agree-ment. The next step is the thermodynamic analysis of surface stabilities based onthe ab initio calculations [26, 43, 44]. The diagram of the LMO (001) and (110)surface stabilities in the cubic (high temperature) and orthorhombic (low tempera-ture) phases is presented in Figure 6.2. The most stable termination depends on theoxygen chemical potential shown for different temperatures and gas pressures onthe right side of the diagram. In particular, under usual SOFC operating conditions(T = 1200K, oxygen pressure in the range 10−2–1 atm, shown by dashed lines), themost stable cubic phase termination is MnO2 (001) with adsorbed oxygen atoms,whereas the hypothetical orthorhombic phase (unstable at such high temperatures)would be adsorbate free. Note also that the LaO-terminated (001) surface couldbe stable only at very low oxygen partial pressure whereas the O-terminated (110)surface stability range lies in between these two terminations. Additionally, thestability of LMO surfaces is strongly restricted by the surface decomposition intometals and simple oxides (numbered lines in Figure 6.2). In a study of the LaFeO3(010) surface stability FeO2 terminations were similarly predicted to be stable athigh temperatures [45].The cathode material actually used is LaMnO3 typically doped with 20% Sr
on the La site (LSM). This acceptor doping has a twofold effect: it increases thep-type electronic conductivity, and it brings the nominal oxygen excess (which isaccommodated by cation vacancies, and can reach up to 0.2 O per formula unit)down to almost zero, thereby also increasing the oxygen vacancy concentration[46,47].The thermodynamic stability of LSM has been analyzed using CRYSTAL and
WIEN-2k (all-electron DFT) computer codes [48]. Both types of calculations
156 Computational Approaches to Energy Materials
–4
–6 –5 –4 –3 –2 –1 0 400 800 1200 1600 2000
–3
–2
–1
–4–40
1–30 –20 –10
–2
0
–3
–2
–1
2
4
MnO2Δμ
o (e
V)
Δμo
(eV
)
ΔμMn (eV) T (K)
LaO(a)
(b)
O
MnO2+O LaO+O O2
6
753
–4
–6 –5 –4 –3 –2 –1 0 400 800 1200 1600 2000
–3
–2
–1
–4–40
1
–30 –20 –10
–2
0
–3
–2
–1
2
Δμo
(eV
)
Δμo
(eV
)
ΔμMn (eV) T (K)
4
6
7
5
3
Figure 6.2 Phase stability diagrams: The regions of stability of LaMnO3 surfaces with differentterminations (LaO- and MnO2-terminated (001) surfaces with and without adsorbed O atom,O2- and O-terminated (011) surfaces) for both phases as a function of chemical potentialvariations for manganese and oxygen. The numbers in circles point to lines, where metals ortheir oxides begin to precipitate: (1) metal La, (2) La2O3, (3) MnO, (4) Mn3O4, (5) Mn2O3,(6) MnO2, and (7) metal Mn. The right side of the figures contains the chemical potentialchanges �μO as a function of temperature at various oxygen gas pressures. The labels mon the lines represent the oxygen pressure: pO2 = 10m bar. (a) Orthorhombic phase and(b) cubic phase. (Reprinted with permission from [38], copyright (2010) American ChemicalSociety.)
demonstrate that unlike the isostructural (Sr,Ba)TiO3 solid solution, a decomposi-tion into LaMnO3 and SrMnO3 phases is energetically unfavorable for LSM. For12% Sr, the order–disorder transition could occur only at very high temperatures(due to configurational entropy), otherwise Sr is predicted theoretically to be peri-odically arranged in the LMO matrix. A periodic distribution of Sr dopants wasindeed observed experimentally in (La,Sr)CoO3 using HRTEM [49].

Energy Conversion—Solid Oxide Fuel Cells 157
The electronic structure and thermodynamic stability of LSM (001) surfaces wasalso calculated using the LCAO approach and hybrid functionals [50, 51]. Twelvepercent Sr doping transforms the semiconducting LMO into a half-metal (in agree-ment with the experimentally observed decreased activation energy of electronicconductivity [46]). Sr exhibits a tendency to segregate to the surface, as exper-imentally observed for epitaxial thin films prepared by PLD [52, 53]. A relatedeffect of Sr doping is to increase the stability of the (La,Sr)O (100) termination.At room temperature, MnO2 and (La,Sr)O terminations are comparably stable, butat higher temperatures, the (La,Sr)O termination gradually occupies a larger por-tion of the stability region, and at typical SOFC operating temperatures (1100K),this termination becomes predominant. In other words, Sr stabilizes the (La,Sr)Otermination with respect to MnO2 (100). The decrease of the surface dipole (cf.the nominal excess positive charge in an LaO layer) upon Sr doping certainly con-tributes to this. These developments are shown in Figure 6.3 [43] along with thepredicted precipitation of La2O3 and MnO at high temperatures. This is consistentwith experimental observations [54]. Thus, Sr doping introduced to increase theoxygen vacancy concentration (and thus the ionic conductivity through the cath-ode) has the side effect of changing the thermodynamic stability of various surfaceterminations, which will also affect cathode performance.The calculation results predict a considerable segregation of oxygen vacancies
toward theMnO2 (001) surface because the O vacancy formation energy is reduced
–5–10ΔμMn(eV)
0
400 2
5
6
4
3
800
1200
1600
2000
0
T (
K)
(La,Sr)O MnO2
Figure 6.3 LCAO-calculated cross-sections of the surface stability diagram for (La,Sr)MnO3
(001) surface structures along the LaMnO3 precipitation lines for an O2 pressure of 0.2 atm.The shaded area corresponds to the perovskite stability area. Light red denotes the LaSrOtermination, and green MnO2. The numbers in the circles indicate precipitation lines for (2)La2O3, (3) MnO, (4) Mn3O4, (5) Mn2O3, and (6) MnO2 (cf. Figure 6.2).

158 Computational Approaches to Energy Materials
by 0.91 eV in the surface layer compared to the bulk value for the followingreaction:
OxO + 2MnxMn � 0.5O2 + V ..O + 2Mn′
Mn. (6.2)
On the other hand, there is no O vacancy accumulation on the LaO-terminatedsurface [28, 38] because there its formation energy is larger by 0.73 eV than in thebulk. This stems from the higher ionicity of the LaO-terminated surface as com-pared to the MnO2-terminated surface. As shown in more detail in Section 6.3.1.3(and in agreement with the experimental findings, cf. Figure 6.1), a high concentra-tion of surface oxygen vacancies is beneficial for the fast incorporation of oxygeninto the cathode and its further path to the electrolyte and finally to the anode.Another important factor determining cathode performance is the surface oxygen
vacancy migration energy (cf. Section 6.3.1.3). The GGA-DFT calculations [28]on the MnO2 (001) surface estimate it as low as 0.67 eV, significantly smaller thanin the bulk (0.95 eV, close to the typical values measured for ABO3 perovskitessuch as SrTiO3 [55], (La,Sr)FeO3 and (La,Sr)CoO3 [56] or (La,Ca)AlO3) [57]).(The orthorhombic LMO phase has a set of barriers for bulk migration, which varyby ≈0.2 eV, as shown in pair potential calculations [58].) Lastly, it is not a prioriobvious, whether or not the incorporation of the adsorbed O into a neighboring sur-face vacancy has a reaction barrier. Explicit calculations (Figure 6.4) indicate sucha barrier to be absent. Oxygen atom incorporation into the vacancy is associatedwith an energy gain of 1.4 eV.
6.3.1.2 Oxygen Adsorption and Diffusion
For the investigation of oxygen adsorption (and O incorporation in the next sec-tion), we focused on the MnO2 (001) termination of LaMnO3, which was identifiedabove as the most stable surface for the undoped material (and still one of the ter-minations with comparably high stability in case of Sr doping). Oxygen moleculesadsorb on LaMnO3 preferentially atop surface Mn ions. All adsorption config-urations involve some electron transfer from the perovskite to the adsorbate (aphysisorbed oxygen molecule has a negligible adsorption enthalpy, for example,
(1)
(2)
(3)
1.64Å
1.88Å
2.05Å2.15Å 2.14Å
1.96Å
1.92Å
1.91Å
1.98Å
Figure 6.4 View of the O atom (1) adsorbed atop surface Mn atom (2) on the orthorhombicMnO2-terminated (001) surface just before dropping into oxygen vacancy (3). Some inter-atomic distances are indicated.

Energy Conversion—Solid Oxide Fuel Cells 159
O2 on the SrTiO3 (001) surface shows a negligible charge transfer and only aslightly exothermic adsorption energy of−0.25 eV [59]). The O−O bond length ishelpful for assigning the molecular oxygen adsorbates (alternatively also the O–Ostretch vibration frequencies can be used, e.g., see [29]) because the Bader atomiccharges are usually smaller than the formal charges. We can identify an adsorbedsuperoxide O2− (Eads = −1.1 eV, Bader charge = −0.42 e0, d(O−O) = 1.36 A,cf. HO2 radical 1.33 A [60]) as well as peroxide O22− (Eads = −0.9 eV, charge =−0.65 e0, d(O–O) = 1.43 A cf. H2O2 1.48 A [60]), which are both attached toMn cations. Atomically adsorbed oxygen (also bound to Mn) can be regarded as“O−” (charge = −0.62 e0 per O−, Eads = −2.2 eV for 2 O−). This very negativechemisorption energy for atomic oxygen, typical for transition metal oxides withvariable oxidation state, allows for an exothermic dissociation of O2 moleculeson the defect-free LMO surface, in contrast to large band-gap perovskites suchas SrTiO3. The electron redistribution, mainly from the neighboring Mn atom, isillustrated in Figure 6.5.The adsorption enthalpies given above refer to a surface coverage of 12.5%
(2√2× 2√2 surface supercell). For a lower coverage of 5%, Eads(2 O−) increases
to−2.5 eV, indicating some defect interaction already at these moderate degrees ofcoverage. To calculate adsorbate coverage under SOFC operating conditions, weused as a reasonable approximation adsorption entropies of about −200 J/mol Kmeasured for gas adsorption on inert oxides, such as ZnO, SiO2, and Al2O3. Thisstrongly negative �adsS0 is one reason for the resulting adsorbate coverage beingcomparably low. The other is that all adsorbed species are charged, leading torepulsive interactions, as it becomes obvious in the more negative Eads at lowercoverage (see above), and as it is described in a phenomenological model [61].
(a) (b)
Mn OlatticeOad
Figure 6.5 The total (a) and difference (b) electron density maps for O atoms adsorbed atopMn on the symmetrical seven-plane slab with MnO2 (100) termination. Red and blue indicatethe electron density deficiency and excess. The density increment is 0.05 e A−3, and the blackdash-dot line is the zero level. Notice the rather local character of lattice perturbation by thedefect. (Reproduced from [28] by permission of the PCCP Owner Societies.)

160 Computational Approaches to Energy Materials
The resulting coverage values are below 1% for the molecular adsorbates O2− andO22−, and about 10% for O− (the more exothermic Eads favors higher coveragethan for O2− and O22−, but the mutual adsorbate repulsion leads to a leveling-offat ≈10% [38]).The VASP modeling of atomic oxygen migration along the MnO2 (001) surface
[28, 38] shows that it occurs along the Mn−O−Mn direction. First, the Oad atommoves from its position atop Mn toward the neighboring surface O, forming atilted O–O species, which requires 1.6 eV and which can be regarded as a peroxidesticking upright in an oxygen vacancy. This dumbbell turns vertical (requiringanother 0.4 eV), and then the migrating O moves toward the next surface Mnion. Thus, the migration energy of adsorbed O atoms turns out to be quite high,2 eV, as compared with 1 eV in SrTiO3 [59]. The energy difference stems fromdifferent equilibrium positions: a strong O bonding atop surface Mn in LMO andattachment atop O in SrTiO3 in the form of a less stable peroxide species. In otherwords, adsorbed O ions are quite immobile at the LMO surface, which will becomeimportant for the mechanism of O incorporation in the next section.While the experimental techniques are making strong progress toward observa-
tion of surface defects, an investigation under fuel cell operating conditions remainschallenging. Recently, adsorbed atomic oxygen species could be studied by scan-ning tunneling microscopy on the (Pr,Sr)O termination of PrSr2Mn2O7 and evensome O migration be observed [62], nevertheless the experiments are restricted totemperatures of maximum 200K, vacuum conditions and those surfaces accessibleby single crystal cleaving.Dissociative oxygen adsorption on the LaO-terminated (001) polar surface was
found to be highly exothermic (−4.57 eV) using the GGA approach [63]. However,as discussed above, a very low oxygen vacancy concentration is expected on thissurface; thus, it cannot be active for oxygen incorporation. Detailed GGA+ U cal-culations of oxygen adsorption on a series ofMn-based perovskites were performedrecently [37]. The findings are similar to our calculations as discussed above, inparticular, the prediction of quite a low oxygen surface coverage of the MnO2surface and the reduced oxygen vacancy formation energy at the MnO2 surfaceas compared to the bulk. On the other hand, the choice of the optimal Hubbard Uparameter is not unique. It is usually fitted to one of several experimental properties(e.g., lattice constant, band gap, and material formation enthalpy). In particular,Lee et al. [37] used aUeff fitted to the formation energies of a series of 3d transitionmetal binary oxides [64], which does not necessarily ensure an optimal choice forternary oxides such as perovskites, keeping in mind, for example, the exceptionallypronounced covalency in the transition metal−oxygen bonds.
6.3.1.3 Rate-Determining Step of the Surface Reaction
The oxygen incorporation reaction, converting oxygen molecules into regular lat-tice oxide ions, is a multistep chemical reaction. While adsorption on the perfect

Energy Conversion—Solid Oxide Fuel Cells 161
O–O
O Mn O
O–
Mn O Mn
Mn O Mn
O2
Mn Mn
O2
O–
Mn Mn
O O––––
Mn O Mn
VO..
O22–
VO
O–
.. Mn O2–Mn
O–
Mn O–Mn
VO
O
..
–
Figure 6.6 Reaction network for oxygen incorporation on the MnO2 (001) termination ofthe LaMnO3 perovskite. Green arrows indicate reaction steps, blue arrows migration steps ofadsorbed oxygen species or oxygen vacancies. The reaction begins with adsorption of O2 eitheron the defect-free surface (top rail) or into an oxygen vacancy (bottom rail). The dissociationcan occur as well with or without V ..
O assistance, and a crossover between the rails can takeplace if beneficial for the overall reaction rate. For the approach of V ..
O and adsorbed oxygenspecies (blue arrows) either the V ..
O or the adsorbates migrate.
surface (including the transfer of one or two electrons) can be assumed to be fast,the slowest of the following steps (the rate-determining step) will limit the overallreaction rate. Naturally, the reaction can proceed in various ways (e.g., the dissoci-ation can occur with or without assistance of a V ..
O), and the mechanistic landscapeis complex. Ab initio calculations offer an excellent tool to explore the reactionnetwork (Figure 6.6) and in particular to find the slowest step in the fastest parallelbranch [38].The black lines in Figure 6.7 show the energetic profile for one of the possible
reaction pathways. After exothermic adsorption the dissociation of the molecularoxygen species without assistance of V ..
O has a barrier of 0.6 eV. For completionof the reaction, the atomic oxygen species (O−) and oxygen vacancies have tomeet. The calculations yield a barrier for migration parallel to the surface of2.0 eV for O−, but only 0.7 eV for the oxygen vacancies in the surface layer (seeSection 6.3.1.1). Thus, it is the (very few) oxygen vacancies that approach theadsorbed O− rather than vice versa. The subsequent incorporation of O− into adirectly neighboring V ..
O occurs fast (without barrier). This purely energetic pictureis not yet sufficient for identification of the rate-determining step and estimationof reaction rates. One has to keep in mind that the oxygen incorporation has astrongly negative overall reaction entropy (loss of translational/rotational degreesof freedom of the O2 molecule), amounting to T�rS0 ≈ 2 eV at 1000K. The bluelines in Figure 6.7 correspond to a (semiquantitative) estimation of the Gibbs freeenthalpy profile, most of the negative�rS0 occurring already in the adsorption stepand resulting in comparably low adsorbate concentrations (cf. Section 6.3.1.2). Inthis free enthalpy profile, now the two maxima of similar height (dissociation andV ..O approach) represent the rate-determining steps, and it depends on the specificconditions (e.g., pO2) which one becomes limiting.

162 Computational Approaches to Energy Materials
ΔG
ΔH
–1.1 eV
Mn O Mn
O–
O
Mn O2–Mn
Mn O2–Mn
O O
Mn O Mn
+0.6 eV–1.6 eV
O2
Mn O Mn
O–
Mn Mn
–O O–
Mn O Mn O Mn
+0.7eV+0.7 eV
Mn O Mn
O–
O
O O
Mn O Mn
–O O–
Mn O Mn O Mn
O–
Mn Mn
+0.7eV+0.7 eVO2
Mn O Mn
ΔHo
x0
-T. Δ
So
x0≈
2eV
(fre
e) e
ntha
lpy
-2.1.5eV–2.1.5 eV
ΔGo
x0
DissociationChemisorption Approach of VO..
Incorporation
ΔGo
x0
Transition state
Figure 6.7 One of the possible reaction pathways for oxygen incorporation on the MnO2
(001) termination of the LaMnO3 perovskite. The black lines are the energy profile (�E ≈ �Hsince the pV term for 1/2 O2 amounts to less than 0.04 eV even at 1000 K), and the red linesare a (semiquantitative) free energy profile (�G) including the effect of the overall negativereaction entropy.
Similarly, two other reaction pathways were studied: one starting with O2adsorption directly into a V ..
O and the other where the dissociation occurs withassistance of a V ..
O. Based on the energies of the intermediates, correspondingadsorbate coverages, and reaction barriers (combined with standard attempt fre-quencies), reaction rates were estimated as a function of the concentration ofmolecular adsorbates and surface oxygen vacancies. The O− adsorbate concen-tration is assumed constant (≈0.1, cf. Section 6.3.1.2). Considering parameterstypical for the La0.8Sr0.2MnO3−δ composition actually used in SOFCs (bulk V ..
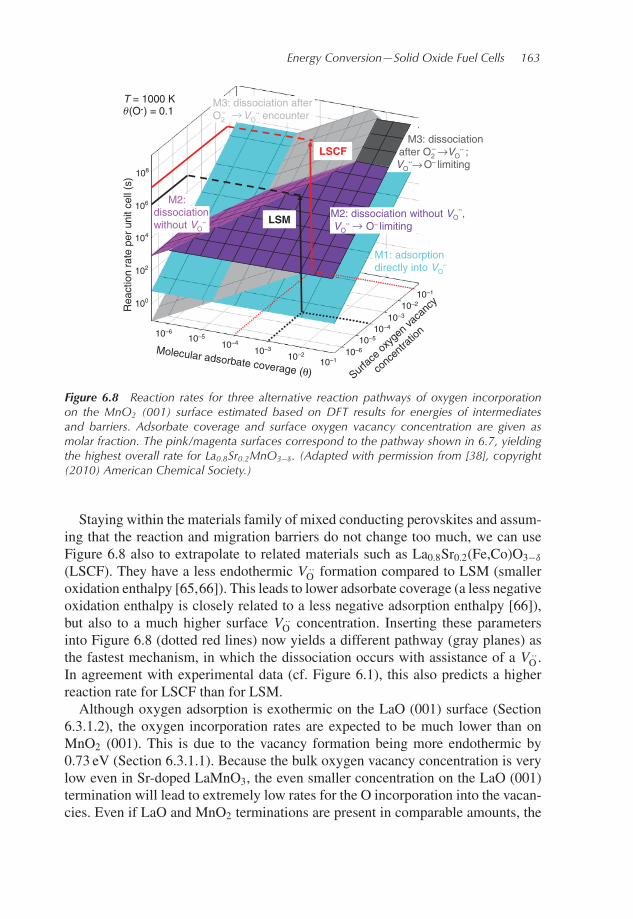
Oconcentration from experimental data and enhanced at surface as discussed inSection 6.3.1.1, adsorbate concentration estimated in Section 6.3.1.2, cf. [38]), asindicated by the black dotted lines in Figure 6.8, shows that the pathway depicted inFigure 6.7 does indeed yield the highest overall reaction rate. Within this pathway,at higher molecular adsorbate coverage (i.e., at high pO2) theV ..
O approach to O−
is rate determining, while at low coverage (low pO2) the dissociation step is thebottleneck. The mechanism with O2 adsorption directly into a V ..
O is unfavorablefor La0.8Sr0.2MnO3−δ due to its very low bulk and surface oxygen vacancy concen-tration. In all of the estimates of reaction rates, we assume that electron transferis faster than the other reaction steps, which involve either ion motion or bondbreaking. This can be justified by the broad electron energy bands that are typicalfor perovskites.
Energy Conversion—Solid Oxide Fuel Cells 163
Surfac
eox
ygen
vaca
ncy
conc
entra
tion10–6
Rea
ctio
n ra
te p
er u
nit c
ell (
s)
T = 1000 Kθ(O ) = 0.1-
10–6
10–510–4
10–3
10–210–1
10–5
10–4Molecular adsorbate coverage (θ)
10–310–2
10–1
100
102
104
106
108
M1: adsorption directly into VO
..
M3: dissociation after O2
- ? VO.. encounter
M3: dissociation after O2
– VO.. encounter
M3: dissociationafter O2
–
–VO
.. ;VO
.. O limiting
M2:dissociation without VO
..
M2:dissociation without VO
..
LSCF
LSM M2: diss. without VVO
.. ? O- limitingM2: dissociation without VO
..,
VO .. O– limiting→
→→
→
Figure 6.8 Reaction rates for three alternative reaction pathways of oxygen incorporationon the MnO2 (001) surface estimated based on DFT results for energies of intermediatesand barriers. Adsorbate coverage and surface oxygen vacancy concentration are given asmolar fraction. The pink/magenta surfaces correspond to the pathway shown in 6.7, yieldingthe highest overall rate for La0.8Sr0.2MnO3−δ. (Adapted with permission from [38], copyright(2010) American Chemical Society.)
Staying within the materials family of mixed conducting perovskites and assum-ing that the reaction and migration barriers do not change too much, we can useFigure 6.8 also to extrapolate to related materials such as La0.8Sr0.2(Fe,Co)O3−δ
(LSCF). They have a less endothermic V ..O formation compared to LSM (smaller
oxidation enthalpy [65,66]). This leads to lower adsorbate coverage (a less negativeoxidation enthalpy is closely related to a less negative adsorption enthalpy [66]),but also to a much higher surface V ..
O concentration. Inserting these parametersinto Figure 6.8 (dotted red lines) now yields a different pathway (gray planes) asthe fastest mechanism, in which the dissociation occurs with assistance of a V ..
O.In agreement with experimental data (cf. Figure 6.1), this also predicts a higherreaction rate for LSCF than for LSM.Although oxygen adsorption is exothermic on the LaO (001) surface (Section
6.3.1.2), the oxygen incorporation rates are expected to be much lower than onMnO2 (001). This is due to the vacancy formation being more endothermic by0.73 eV (Section 6.3.1.1). Because the bulk oxygen vacancy concentration is verylow even in Sr-doped LaMnO3, the even smaller concentration on the LaO (001)termination will lead to extremely low rates for the O incorporation into the vacan-cies. Even if LaO and MnO2 terminations are present in comparable amounts, the

164 Computational Approaches to Energy Materials
overall oxygen incorporation rate is predicted to be dominated by the kinetics onthe MnO2 termination.In the investigation of the oxygen incorporation in the surface reaction mech-
anism on mixed conducting perovskites, the DFT calculations made consider-able contributions, ranging from obtaining basic quantities, such as adsorptionenthalpies, to elucidating the concentrations and mobilities of surface defects, andfinally reaction barriers. The emerging mechanistic picture is in good agreementwith the trends observed for the experimentally measured reaction rate constants.It is a fortunate point in the consideration of a realistic mechanistic pathway thatthe relative differences in the reaction rates of possible alternatives are greater thanthe uncertainties of the modeling.Of course, one has to be aware that the calculations usually refer to an ideal-
ized surface model that neglects, for example, higher dimensional defects (e.g.,steps and kinks), and may also not correctly represent dopant segregation (unlessexplored in very extensive calculations). Recent experiments and calculations onstrained (La,Sr)MnO3−δ and LaCoO3−δ films have shown that moderate tensilestrain favors Sr surface segregation as well as oxygen vacancy formation andoxygen chemisorption [67,68], which may in turn change surface rate constants.The estimate of rate constants from the ab initio input (energies of interme-
diates and transition states) can be also improved beyond the model of a singlerate-determining step used here as the first approximation. The respective tools aremicrokinetic modeling (typically a mean field approach) and kinetic Monte Carlosimulations (as discussed in Chapters 1 and 3 of this book) [69–71]. For a fuel cellunder electrical bias (current flow), one further has to consider that the relationbetween overpotential and electrical potential drops at the decisive solid–gas inter-face may be more complex than assumed for Butler–Volmer kinetics at solid/liquidelectrodes [72].
6.3.2 Bulk Properties of Multicomponent Perovskites
6.3.2.1 Oxygen Vacancy Formation in (Ba,Sr)(Co,Fe)O3−δ
(Ba,Sr)(Co,Fe)O3−δ (BSCF) perovskites were first optimized to have a high oxygendiffusivity for oxygen permeation membranes [73], but were soon discovered toalso exhibit a fast oxygen surface reaction [74, 75]. The most studied compositionis Ba0.5Sr0.5Co0.8Fe0.2O3−δ, which combines a high mobility of oxygen vacancieswith a high vacancy concentration that can exceed δ = 0.5 without formation of V ..
O-ordered superstructures such as the brownmillerite structure. Before investigatingthe oxygen vacancy mobility in the next section, we first deal with the energeticsof oxygen vacancy formation. As oxygen removal implies a gradual reduction ofthe material, this also requires a detailed inspection of the electronic structure.Recently, several quantummechanical studies on oxygen vacancymigration bar-
riers were reported [76–79], mostly for the composition Ba0.5Sr0.5Co0.8Fe0.2O3−δ,which experimentally exhibits the highest diffusivity and surface exchange rate.The basis for the following discussion of the special properties of BSCF perovskites

Energy Conversion—Solid Oxide Fuel Cells 165
SrCo1-yFeyO2.875
Fe fraction (y)1.000.750.500.250.00
Vac
ancy
form
atio
n en
ergy
(eV
)
1.0
1.5
2.0
2.5
Ba0.5Sr0.5Co1-yFeyO2.875
Co-VO..-Co
Co-VO..-Fe
Co-VO..-Co
Co-VO..-Fe
Fe-VO..-Fe
Figure 6.9 Oxygen vacancy formation energy EV (at 0 K) as a function of iron content y inBa0.5Sr0.5Co1−yFeyO2.875 (open symbols) and SrCo1−yFeyO2.875 (solid symbols). (Reproducedwith permission from [80], copyright 2012, The Electrochemical Society.)
and their dependence on cation composition is DFT VASP calculations [80–83]with 40-atom supercells (Ba/Sr arrangement, cf. Figure 6.11a) containing oneoxygen vacancy. We also modeled Ba0.5Sr0.5CoO3−δ (BSC) and Ba0.5Sr0.5FeO3−δ
(BSF) with a cubic perovskite structure (but allowing for distortions within thesupercell), although they adopt a hexagonal structure [84]. This can be justifiedby the fact that both can be prepared as cubic perovskites by a small additionaldoping (5% Ce on Fe site) or as metastable cubic perovskites by oxidation from ahighly oxygen-deficient brownmillerite phase. Because the connectivity within thestructure is completely different (corner-sharing vs. edge-sharing octahedra), nophase transformation will occur when relaxing the structure within the supercell.The oxygen vacancy formation energy EV (at 0 K) in Ba0.5Sr0.5Co1−yFeyO2.875
is shown in Figure 6.9. For a given Fe content, the EV values vary by approx-imately 0.1 eV depending on the vacancy location, either between two Co ions,between Fe and Co, or between two Fe ions. Over the whole composition range,EV increases linearly with iron content from 1.2 eV for BSC up to 2.2 eV for BSF.This smooth increase (not changing stepwise from predominant Co reduction atlow y to Fe reduction at high y) reflects the pronounced electron density delocal-ization between Co, Fe, and O orbitals. This is in qualitative agreement with X-rayabsorption spectroscopy (XAS) results [85], revealing a charge redistribution toCo and Fe as well as O ions. The vacancy formation is more endothermic (by0.3 eV) for SrCo0.75Fe0.25O3−δ (SCF) than for BSCF with the same iron content,in agreement with the smaller oxygen deficiency measured for SCF (in the cubicperovskite structure [86]). The effect of the barium content on the redox energeticscan be related to the lattice expansion caused by the large Ba ions (SCF: a = 3.84 A,BSCF: 3.90 A) leading to an increased size mismatch for cobalt. Because a changeto a lower formal oxidation state increases the ionic radius (4+ to 3+ by 0.08 A,

166 Computational Approaches to Energy Materials
(a)
O
Energy (eV)43210–1–2–3–4–5–6–7
–20
–10
0
10
20
Arb
. uni
tsA
rb. u
nits
Arb
. uni
ts
–10
0
10
Fe
–5
0
5
Co Spin-up
Spin-down
(b)
E O Co
Fe
E F (δ > 0)
E F (δ = 0)
Figure 6.10 (a) Spin-projected DOS for Ba0.5Sr0.5Co0.75Fe0.25O2.875 derived from the GGAcalculations. The oxygen vacancy is located between two Co ions. (b) Schematic DOS foroxygen-stoichiometric BSCF. The Fermi energy is indicated by the dashed line, and by the dottedline for oxygen-deficient BSCF. Spin-down DOS to the left, spin-up to the right. (Reproducedwith permission from [80], copyright 2012, The Electrochemical Society.)
3+ to 2+ by≈0.1 A [87]), the lattice expansion then favors the formation of lowerCo oxidation states. Indeed, the highest oxygen deficiency is observed experimen-tally [88, 89] for Co-rich materials in the presence of a significant Ba content.The experimental oxidation enthalpies for Ba0.5Sr0.5Co1−yFeyO3−δ perovskites
(−58 kJ/mol for BSF at δ = 0.34 and −47 kJ/ mol for Ba0.5Sr0.5Co0.8Fe0.2O3−δ atδ = 0.52 [90]) vary perceptibly with oxygen deficiency. This is more pronouncedfor Co-rich compositions, similarly to the behavior in La0.6Sr0.4Co1−yFeyO3−δ
perovskites [91]. Although an extrapolation to δ = 1/8 (used in our calculations)has quite a high numerical uncertainty, the qualitative trend of a less endothermicvacancy formation for the Co-rich materials is supported by the experimental data(larger oxygen deficiency for BSCF than for BSF).The variation of the vacancy formation energywith Fe content can be rationalized
by an analysis of the density of states (DOS) shown in Figure 6.10a, which exhibitsa behavior similar to half-metals (insulator in spin-up and conductor in spin-down;note that the small but nonzero DOS at the Fermi level for spin-up electrons is dueto the Fe and Co eg orbitals [80], which correspond to metal-oxygen σ -type bondsonly weakly contributing to conductivity). This is consistent with the experimentalfinding of a comparably modest electronic conductivity [92]. Owing to the strongcovalency, Co, Fe, and O states cover a broad energy range of approximately 10 eV.This is in contrast with the DOS for typical perovskites with a large band gap (e.g.,SrTiO3 [93,94]), which is an insulator with an O2p bandwidth of 6 eV below the topedge of the valence band, occupied Ti states at−3 eV to−6 eV, and empty Ti and Srstates in the lower and upper parts of the conduction band. The exact nature of thetwoB-type ions nearest to the vacancy (Co-V ..
O-Co or Co-V..O-Fe configuration) does
not noticeably affect the DOS pattern as the remaining electrons of the removed

Energy Conversion—Solid Oxide Fuel Cells 167
O atom are delocalized in the whole supercell. Co exhibits a considerable DOSaround the Fermi level, and thus Co4+ can easily transform into Co3+ . On theother hand, Fe shows a peak of about 1 eV higher than the Fermi level (2 eV higherthan the occupied Fe states), which makes a change of its oxidation state much lessenergetically favorable. This is a direct consequence of the Fe high spin state andCo intermediate spin state. The resulting schematic DOS for BSCF (Figure 6.10b)is more precise than that suggested byMueller et al. [85] based on semiquantitativearguments.
6.3.2.2 Oxygen Vacancy Migration in (Ba,Sr)(Co,Fe)O3−δ
The oxygenmobility in acceptor-doped perovskites is determined by the bottleneckin the O atom passage through the “critical triangle” formed by the B cation andthe two A cations surrounding the migrating O∗ in the transition state (see Figure6.11c). Allowing the O∗ to pass requires an outward relaxation of the A and Bcations in the critical triangle, which is on the other hand limited by the other ionsin the lattice, but also leads to slight rearrangements in the whole supercell (cf.Figure 6.11d). A failure to obtain these relaxations throughout the whole supercellin a consistent way for initial as well as transition states will lead to grossly wrongbarriers. The calculation of the O migration barriers using the nudged elastic band
(c)
BaSr
CoO (d)
(a) (b)
Sr*
Ba*
O*Co*
Ba**
C
Co**
CoCo
Sr**
Co
Figure 6.11 (a) Initial state for Ba0.5Sr0.5CoO2.875 with alternating arrangement of Ba and Sr;the square indicates the oxygen vacancy. (b) Initial state with modified Ba/Sr configuration. (c)Transition state for BSC; red color indicates the significantly shortened Co∗−O∗ and Co∗−Obonds. (d) Electron density map of BSC in the (110) plane for the transition state of O∗
migration. The black lines emphasize the structure deformations caused by O∗ migration; thewhite lines indicate the “critical triangle” of one B and two A cations around the migrating O∗
in the transition state. (Adapted from [83] by permission of ECS, The Electrochemical Society.)
168 Computational Approaches to Energy Materials
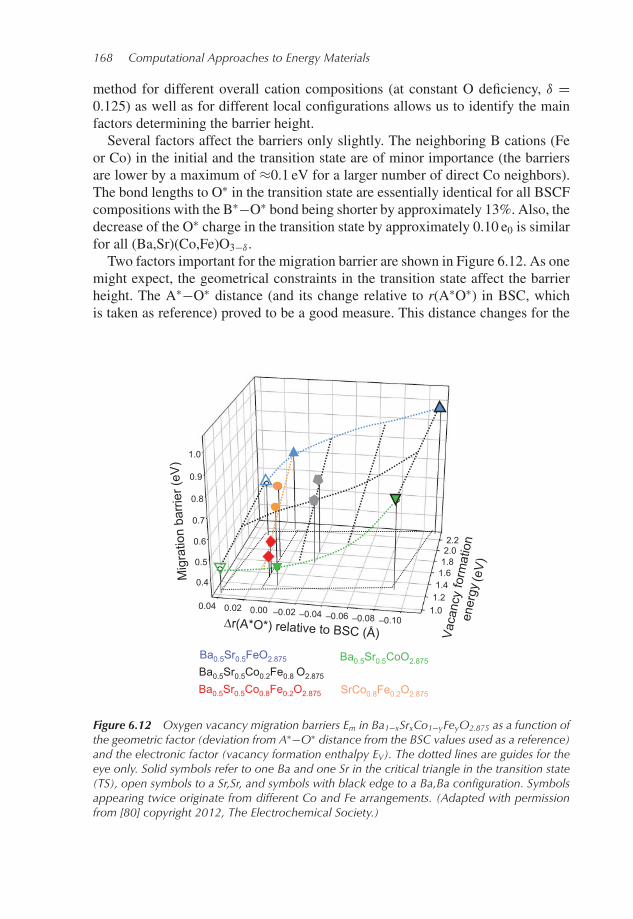
method for different overall cation compositions (at constant O deficiency, δ =0.125) as well as for different local configurations allows us to identify the mainfactors determining the barrier height.Several factors affect the barriers only slightly. The neighboring B cations (Fe
or Co) in the initial and the transition state are of minor importance (the barriersare lower by a maximum of ≈0.1 eV for a larger number of direct Co neighbors).The bond lengths to O∗ in the transition state are essentially identical for all BSCFcompositions with the B∗−O∗ bond being shorter by approximately 13%. Also, thedecrease of the O∗ charge in the transition state by approximately 0.10 e0 is similarfor all (Ba,Sr)(Co,Fe)O3−δ .Two factors important for the migration barrier are shown in Figure 6.12. As one
might expect, the geometrical constraints in the transition state affect the barrierheight. The A∗−O∗ distance (and its change relative to r(A∗O∗) in BSC, whichis taken as reference) proved to be a good measure. This distance changes for the
0.4
0.5
0.6
0.7
0.8
0.9
1.0
1.0
1.2
1.4
1.61.82.02.2
–0.10–0.08–0.06–0.04–0.020.000.020.04
Mig
ratio
nbarr
ier
(eV
)
Δr(A*O*) relative to BSC (Å) Vaca
ncy
form
atio
nenerg
y(e
V)
Ba0.5Sr0.5FeO2.875
Ba0.5Sr0.5Co0.8Fe0.2O2.875
Ba0.5Sr0.5CoO2.875
SrCo0.8Fe0.2O2.875
Ba0.5Sr0.5Co0.2Fe0.8 O2.875
Figure 6.12 Oxygen vacancy migration barriers Em in Ba1−xSrxCo1−yFeyO2.875 as a function ofthe geometric factor (deviation from A∗−O∗ distance from the BSC values used as a reference)and the electronic factor (vacancy formation enthalpy EV). The dotted lines are guides for theeye only. Solid symbols refer to one Ba and one Sr in the critical triangle in the transition state(TS), open symbols to a Sr,Sr, and symbols with black edge to a Ba,Ba configuration. Symbolsappearing twice originate from different Co and Fe arrangements. (Adapted with permissionfrom [80] copyright 2012, The Electrochemical Society.)

Energy Conversion—Solid Oxide Fuel Cells 169
different A-cation configurations in the critical triangle (SrSr, BaSr, or BaBa; cf.cation configurations in Figures 6.11a and 6.11b). As shown in Figure 6.12, thepresence of more Ba cations enforces a decrease of this distance, in parallel with apronounced increase of the migration barrier, from 0.4 eV to 0.75 eV in BSC andfrom 0.72 eV to 0.97 eV in BSF.On the other hand, the distance r(A∗O∗) is approximately constant for all O∗
jumpswith oneBa and one Sr in the critical triangle, while the barriers increase from0.40 eV (BSC) to 0.72 eV (BSF). In parallel, the V ..
O formation energy increasesfromBSC to BSF (cf. Section 6.3.2.1); thus, obviously themigration barrier and thevacancy formation energy are coupled in the BSCF perovskites. This correlationcan be understood from the fact that a certain electron transfer fromO∗ to B∗ occursin the migration transition state, and the oxygen vacancy formation (involving thetransfer of two electrons) gives a measure for the energetic cost of this transfer.This correlation between vacancy formation and vacancy migration energies maynot naively be generalized to all mixed conducting perovskites; for example, for(La,Sr)(Co,Fe)O3−δ the charge transfer in the transition state is less pronouncedand thus a correlation to EV not obvious [98]. The lowest EV is found in the BSCFfamily for the perovskites with simultaneously high Ba and Co contents (cf. Section6.3.2.1), which are also the materials with the largest size mismatch between theA and B cations. This suggests that a further improvement of the V ..
O mobilitywill be difficult because a too strong size mismatch, a too strong deviation of theGoldschmidt factor
t = rA + rO√2(rB + rO)
(6.3)
from unity, will ultimately lead to the formation of a hexagonal instead of cubicperovskite structure. The perovskites from the (La,Sr)(Fe,Co,Mn)O3−δ family [56,57] have a higher barrier, which seems to be related to their smaller lattice parameterand probably also to a less pronounced electron redistribution in the migrationtransition state.The calculated migration barriers for the Ba,Sr transition state configuration are
in fair agreementwith the activation energy of the vacancy diffusion coefficient DV..O
experimentally obtained for Ba0.5Sr0.5Co0.8Fe0.2O3−δ (0.47 eV [95]), BSF (1.0 eV[24]), and SCF (ranging from 0.65 eV [96] to 1.0 eV [97]). Our results for barriersin Ba0.5Sr0.5Co0.75Fe0.25O2.875 also agree with other DFT calculations [77, 79]. Acomparison with calculated vacancy migration barriers in La1–xSrxCo1–yFeyO2.875perovskites is given byMastrikov et al. [98]. For a complete study of ion conductiv-ity in a multicomponent perovskite such as (Ba,Sr)(Co,Fe)O3−δ , the different localenvironments and the corresponding distribution of migration barriers (as obtainedfrom DFT) should be considered in detail. Approaches such as the cluster expan-sion method (e.g., see [99,100] for treating the spatial distribution of dopants) andkinetic Monte Carlo simulations (e.g., see [101] to extract the overall diffusivity ina landscape of varying barriers) may be applied.

170 Computational Approaches to Energy Materials
6.3.2.3 Disorder and Cation Rearrangement in (Ba,Sr)(Co,Fe)O3−δ
The composition Ba0.5Sr0.5Co0.8Fe0.2O3−δ is known to lie at the borderline of thecubic perovskite stability region, which shows up in a slow decomposition leadingto the formation of an additional hexagonal perovskite observed in the temperaturerange of 800–1000 ◦C [102–104]. As the size mismatch between Co and Ba influ-ences the oxygen vacancy formation and indirectly also the vacancy migration, asshown in the previous section, the favorable oxygen transport properties of BSCFare not independent of its limited stability in the cubic perovskite structure.Wewill briefly explore this structural instability based on recent GGAVASP cal-
culations [105, 106]. The energetics of Frenkel defect pairs (vacancy–interstitial),Schottky defects (vacancies), and cation sublattice disorder were studied alongwith a set of possible solid-state decomposition reactions into several phases forBa0.5Sr0.5Co0.75Fe0.25O3, which is simulated most frequently due to its high Odiffusivity and exchange rate constant.Among Frenkel defects, the oxygen vacancy–interstitial pairs have the lowest
formation energy of 1.2–1.5 eV per defect, with some preference for having theoxygen vacancy between Co and Fe rather than between two Co atoms. Thisformation energy turns out to be much smaller than in other perovskites with smalllattice constants, for example, in SrTiO3 (≈10 eV [107]) with a more denselypacked lattice. The dumbbell configuration of these split-oxygen interstitials isshown in Figure 6.13. TheO−Odistance of 1.41 A is comparable to the bond lengthin adsorbed peroxide on the surface of LSM (cf. Section 6.3.1.2). While the oxygenFrenkel (vacancy–interstitial) and oxygen partial Schottky disorder (formation ofvacancy + 1/2 O2, endothermic by 1.3 eV, cf. Figure 6.9) have comparable defectformation energies in BSCF, at elevated temperatures the positive reaction entropyfor the latter will favor the oxygen vacancy formation over Frenkel pairs.In most calculations on BSCF and other perovskite solid solutions, a periodic
arrangement of cations is assumed. In exploring how important the order in sucha composition is, we simulated possible rearrangements in the A- and B-cation
1.41 Å
Fe
Co O
1.85 Å1.85 Å
Figure 6.13 Schematic view of the split-oxygen interstitial (“peroxide”) predicted inBa0.5Sr0.5Co0.75Fe0.25O3.

Energy Conversion—Solid Oxide Fuel Cells 171
SrBa
(a) (b)
Figure 6.14 The clustering effect caused by Sr ↔ Ba exchange in Ba0.5Sr0.5Co0.75Fe0.25O3. Inthe original structure (a), the central A-cation site is occupied by Ba, while in the “defective”supercell (b), the site is occupied by Sr (For simplicity, only the A-cation sublattice of the ABO3
perovskite structure is shown in a fragment of the larger 4 × 4 × 4 supercell; other parts willhave a Ba excess to maintain the overall stoichiometry).
sublattices. For example, exchanging one Ba with one Sr in the supercell causes aclustering of Sr (and Ba), as illustrated in Figure 6.14. This can be regarded as amanifestation of self-segregation in the cation sublattice.Energies describing the cation sublattice disorder are summarized in Table 6.1.
A perturbation caused by the redistribution in either the A or B-cation sublatticedoes not require a significant excess energy (reactions I and II). From the ener-getic point of view, this implies that both A metals (Ba and Sr) and B metals
Table 6.1 Energies of cation structural disorder (within the cubic BSCF phase)and decomposition of cubic BSCF into several perovskite phases
No. Disorder reaction Energy (eV)
Cation exchangeI Ba ↔ Sr −0.20II Co ↔ Fe 0.08III Ba ↔ Fe 7.87IV Sr ↔ Co 3.79V Sr ↔ Fe 5.70VI Ba ↔ Co 6.16
Phase DecompositionVII 4 BSCF ↔ 2 BaCo0.75Fe0.25O3 (cub)
+ 2 SrCo0.75Fe0.25O3 (cub)−0.01
VIII 4 BSCF ↔ 3 Ba0.5Sr0.5CoO3 (hex)+ Ba0.5Sr0.5FeO3 (cub)
−0.56
IX 4 BSCF ↔ 2BaCoO3 (hex)+ SrCoO3 (cub) + SrFeO3 (cub)
−0.18
Calculations are performed for a single exchange pair of cations placed in a large 4 × 4 × 4supercell, which consists of 64 BSCF formula units. Reactions (I) and (II) represent the clusteringeffect in A and B sublattices, reactions (III) and (VI) describe the formation of antisite defects,and reactions (VII) to (IX) refer to decomposition into two or three phases.

172 Computational Approaches to Energy Materials
(Co and Fe) can be randomly distributed over the respective sites. This conclu-sion is additionally confirmed by the negative energies obtained for decompo-sition reactions into two phases (VII) and (VIII) described in Table 6.1. Equa-tion (VII) may be interpreted as a complete separation of Ba0.5Sr0.5Co0.75Fe0.25O3into BaCo0.75Fe0.25O3 and SrCo0.75Fe0.25O3, both in the cubic phase. The decom-position of Ba0.5Sr0.5Co0.75Fe0.25O3 into hexagonal Ba0.5Sr0.5CoO3 and cubicBa0.5Sr0.5FeO3 perovskites according to equation (VIII) is even more exothermicdue to a contribution from the cubic to hexagonal phases. Equation (IX) describesthe exothermic decomposition of BSCF into perovskites with either the larger(BaCoO3) or smaller (SrFeO3) cations. In summary, the instability of the BSCFlattice can largely be attributed to the A- and B-site cation size mismatch. A moredetailed thermodynamic analysis of phase separation is in progress.Further, we predict that antisite substitutions (a pair of defects, in which an A
metal occupies a B position of the ABO3 lattice and the corresponding B metal fillsthe A position, e.g., Sr↔Co) are in principle possible. However, as expected due tothe different ion size, charge, and preferred coordination, they require significantlyhigher energies than the exchange of cations within the same sublattice. Table 6.1shows that a single A ↔ B cation exchange in a large supercell requires 3.79–7.87 eV. This energy corresponds to an extremely small concentration of suchantisite defects and turns out to be comparable to the cation vacancy formationenergies.Strictly speaking, a direct comparison of the calculated energies to available
experiments is inappropriate, although tempting, for at least three reasons. First,thermodynamic analysis at realistic temperatures is necessary, which requires quitetime-consuming phonon calculations. Second, the calculated crystals are materialswithout O deficiency, which cannot be expected from real samples. Third, withoutmaking speculative a priori assumptions on compositions, an additional uncertaintycomplicates the analysis evenmore, as the relative stabilities of possible phases willstrongly depend not only on defect content but also on their chemical compositions.Nevertheless, the findings are consistent with X-ray diffraction (XRD) results andtransmission electron microscopy (TEM) analysis, which demonstrates that below1073K in air, BSCF decomposes into hexagonal and cubic perovskite phases withdifferent cation composition [102, 103, 108]. In summary, both DFT calculationsand experiments indicate a phase instability for BSCF, which will be a seriousbottleneck for its application in permeation membranes and SOFC cathodes. Asimilar analysis of LSCF, which appears to be more stable, is in progress.Of course, the thermodynamic treatment of phase (in)stability has to be comple-
mented with an investigation of cation mobilities, in order to obtain the completepicture. Experimentally, cation diffusivities in perovskites are known to be muchlower than oxygen diffusivity, and thus measurements are quite challenging andscarce. Even more important is the modeling of cation migration in perovskitespreferably at the ab initio level of theory. A challenge is that the exact mechanismof cation migration for the two sublattices has to be resolved (e.g., the B cation canmigrate along [101] or [111] or evenwith assistance ofA-site vacancies) [109–112].

Energy Conversion—Solid Oxide Fuel Cells 173
The experimental observation of comparable diffusivities for A and B cations inBaTiO3 [113] and LaMnO3+ δ [114, 115] supports B-cation migration with theassistance of A vacancies. A combination of classical methods (exploratory stud-ies) and quantum mechanical techniques (refinement of barriers) was employedfor the modeling of Sr migration in SrTiO3 [116]. Sr migration is enhanced by thepresence of an oxygen vacancy, which decreases the barrier for migration of Sr to aneighboring Sr vacancy from 3.7 eV to 2.9 eV. Nevertheless, this migration energyis much higher than the typical value for oxygen migration in ABO3 perovskitesof 0.9 eV.
6.3.3 Defects in (La,Sr)(Co,Fe)O3−δ
Let us consider another mixed conducting SOFC cathode material (La,Sr)(Co,Fe)O3−δ (LSCF). First calculations on La1−xSrxCo0.75Fe0.25O3−δ were per-formed recently using the GGA+ U approach implemented into the VASP code[117]. Another method, the hybrid functional PBE0 successfully reproduces thenonmagnetic behavior of LaCoO3 in contrast to the standard GGA functionals[118]. The PBE0 results suggest that LaCoO3 is a semiconducting material with aband gap of approximately 2 eV. The DFT+ Umethod confirmed the nonmagneticnature of LaCoO3 with a very narrow band gap of approximately 0.2 eV, in agree-ment with experiment [119] (the value of the Hubbard U parameter was optimizedhere as 1.5 eV [120] using the Dudarev-type of exchange–correlation functional[121]). Although the PBE0 functional overestimates the band gap, this functionalreproduces the experimental phonon frequencies much better than the DFT+ U[122], which is important for the calculations of thermodynamic properties.In the next step [120], standard DFT functionals were used to calculate
LSCF with and without oxygen deficiency. The distortion from the cubicstructure depends significantly on the stoichiometry [123]. Thus, a particularLa0.875Sr0.125Co0.25Fe0.75O3 composition was studied in a 40-atom orthorhombicsupercell. Different configurations for the Co/Fe and La/Sr sublattices were calcu-lated as suggested by Fuks et al. [48]. The minimum perovskite formation energywas found for the superstructure with two Co atoms being direct neighbors, andSr at largest distance from Co. This superstructure was then used for calculationswith O vacancies. All degrees of freedom were relaxed, including spin magneticmoments of Co and Fe (all calculations were performed for the ferromagnetic struc-ture). The defect formation energies were obtained using the formation enthalpyof MgO as a reference for the oxygen chemical potential [124,125].The free enthalpy of vacancy formation is shown in Figure 6.15 as a function
of temperature for two Sr concentrations, x = 0.125 and 0.5, and fixed oxygendeficiency, δ = 0.125, in La1−xSrxCo0.25Fe0.75O3−δ (one vacancy per 40-atomsupercell). The temperature dependence of the gas-phase O2 chemical potential istaken from experimental data [60]. These two dopant concentrations correspondto different transition metal oxidation states: for x = 0.5, Co and Fe have a formaloxidation state between 4+ and 3+ in both the oxygen-stoichiometric perovskite

174 Computational Approaches to Energy Materials
0 300 600 900 1200 1500
0 300 600 900 1200 1500
1.0
1.5
2.0
2.5
3.0
3.5
4.0
1.0
1.5
2.0 La0.5Sr0.5Co0.25Fe0.75O3
La0.875Sr0.125Co0.25Fe0.75O3
T (K)
ΔGF
(eV
)2.5
3.0
3.5
4.0
Figure 6.15 The standard free enthalpy �G0 of oxygen vacancy formation inLa1−xSrxCo0.75Fe0.25O2.875 for two Sr concentrations as a function of temperature (data below100 K are extrapolated), calculated within GGA-DFT.
and after introducing one oxygen vacancy into the supercell. For the lower Sr dop-ing x = 0.125 without oxygen vacancies (La0.875Sr0.125Co0.25Fe0.75O3), the Fe/Cooxidation state remains between 4+ and 3+ ; however, due to introduction ofvacancies (La0.875Sr0.125Co0.25Fe0.75O2.875), it decreases to values between 3+ and2+ . Oxygen vacancy formation in the Sr-poor perovskites involves a reduction ofthe transitionmetals below the 3+ oxidation state. As one can see fromFigure 6.15,the reduction in the 3+ /2+ regime (Sr-poor sample) is energetically more costlythan in the 4+ /3+ regime. The difference in vacancy formation enthalpy is quitelarge (≈0.7 eV) and practically temperature independent. This is a direct result ofthe difference in the transition metal oxidation states, in agreement with exper-iments (oxidation enthalpy for LaCoO3 is more negative by ≈0.8 eV comparedto La0.5Sr0.5CoO3−δ [126]; an even greater difference occurs for La1−xSrxFeO3−δ
[127]). This demonstrates how carefully experimental conditions should be ana-lyzed when comparing to theory (see also [128]).An increase in the temperature from 300K to 1200K leads to a drop (≈1 eV)
in the free enthalpy of vacancy formation �G0 due to the considerable T�S0 termof the gas-phase O2. The calculated oxygen vacancy formation energy �G0 at0 K in La0.875Sr0.125Co0.25Fe0.75O3 is close to the experimental reaction enthalpyof 3.3 eV for La0.9Sr0.1FeO3−δ , averaging over the values for 4+ /3+ and 3+ /2+valence change [127]. Also for the DFT+ U approach [37], the vacancy formationenergies for 3+ /2+ valence change in LaFeO3−δ and LaCoO3−δ are fairly closeto experiment. For La0.5Sr0.5Co0.25Fe0.75O3 the agreement is less good (the exper-imental enthalpies are only 1.0–1.1 eV for La0.5Sr0.5CoO3−δ and La0.5Sr0.5FeO3−δ
[126, 127]); some improvement might be achieved by including phonons (entropycontributions in the solid phase).

Energy Conversion—Solid Oxide Fuel Cells 175
6.4 Ion Transport in Electrolytes: Recent Studies
Oxygen vacancy migration in SOFC electrolyte materials (mostly Y-stabilized zir-conia (YSZ) but also Gd-doped Ceria (CGO) and to some degree the perovskite(La,Sr)(Ga,Mg)O3−z (LSGM) and apatite-relatedmaterials) has been studied exten-sively mainly with interatomic potential methods but increasingly also at the abinitio level (e.g., see [129–133] and related work on battery electrodes in Chapter4 of this book). Here, we want to mention some recent studies based on DFTapproaches and/or focusing on interface effects. DFT calculations for acceptor-doped ceria [134] and zirconia [135] showed that depending on the actual arrange-ment of dopants around the migrating oxygen the barrier might change by up to0.5 eV. For both of these fluorites, the highest barrier is found when the migratingO has to squeeze through the space between two dopants (and the barrier heightincreases with the dopant radius). The interaction and eventual ordering of oxy-gen vacancies was the focus of a molecular dynamics study using pair potentialsderived from ab initio calculations [136]. Combining ab initio results with a clusterexpansion approach, the dopant distribution in YSZ was investigated [137]. Forzirconia doped with 8% Y2O3, a strong clustering of Y into nanodomains wasfound, which was less pronounced for higher Y content.The effect of biaxial strain on vacancy mobility in YSZ was studied by DFT and
kinetic Monte Carlo simulations in [138]. Tensile strain up to 4% was found toincrease oxygen diffusivity, while larger strain leads to a severe structural distortion(Figure 6.16) and lower mobility. The increase of diffusivity at 4% strain is strongerat lower temperature (by almost 4 orders ofmagnitude at 400K).While experimentsalso show increased diffusivity under tensile strain, the effects are much smaller(also the achievable strain was much lower [139]).In oxide ions conducting electrolytes with fluorite as well as perovskite struc-
tures, grain boundaries typically exhibit amore or less pronounced blocking charac-ter for oxygen transport. Theoretical investigations of grain boundaries (a bicrystalbeing the simplest model) are challenging due to the large number of degrees offreedom to be considered (relative orientation of the crystal slabs as well as of theboundary plane, but also rigid body shifts and potential compositional variation).Particularly interesting are changes in defect formation energies in the core of thegrain boundary because they can give rise to defect segregation and space chargezones [140]. For YSZ, studies were mostly performed with pair potentials (e.g.,see [141]), but recently also on the ab initio level [142]. The obtained segregationenergies depend on the dopant (more negative for Al than Y [142]), and segrega-tion of dopant–oxygen vacancy pairs is found to be beneficial [141, 142]. Dopantsegregation is also found for the surface of YSZ [143]. For YSZ films on a single-crystalline MgO substrate, a simulated amorphization and crystallization approachsuggests an increased ionic conductivity in the interfacial layer [144].For the perovskite electrolyte LSGM, ab initio calculations were performed
for the defect formation [145] but the oxygen migration has only been studied

176 Computational Approaches to Energy Materials
Oxygen plane
ε = 0.02
ε = 0.05
ε = 0.07
0.0 0.3Å–3
Bonding plane
Figure 6.16 Changes in electron density in YSZ under tensile biaxial strain ε. Gray spheres,Zr; red, O; blue, oxygen vacancy. The white ellipse indicates open space for migration. (Repro-duced from [138] by permission of The Royal Society of Chemistry.)
based on pair potentials so far [146]. The comparison of different cation config-urations around the migrating O shows that geometrical constraints are important(as expressed by the “critical radius” [147] describing the available space for themigrating O). For Ca doping a “charge-weighted ionic radius” [146] was found toimprove the correlation. While the lowest migration barriers in LSGM (0.64 eV[146]) are lower than the value of 0.8–0.9 eV found for many perovskites [56,57],the reason for the low barriers is different than for BSCF.While for BSCF the redoxproperties of the B cation (related to a partial electron transfer in the transition stateto Fe and Co) play a decisive role (see Section 6.3.2.2), this is expected to be lesspronounced in LSGM and thus the steric constraints dominate (the “critical radius”is significantly larger in LSGM than in BSCF, facilitating fast oxygen migration).
6.5 Reactions at SOFC Anodes
Having demonstrated the case of atomistic modeling in detail for the example ofcathode materials, we want to touch upon anode reactions only briefly. A completediscussion would by far exceed the length of this chapter by far. SOFC anodescan roughly be divided into two types (e.g., see [19, 148, 149]): cermets fromelectrolytes (doped zirconia or ceria) andmetallic particles (mostlyNi, occasionallyalso Cu) where each phase has to form a connected network, and anodes made ofelectronically conducting (sometimes also mixed conducting) oxides for which adecreased sulfur poisoning and carbon deposition is expected. We will not further

Energy Conversion—Solid Oxide Fuel Cells 177
discuss the second case as theoretical studies for the reactions on ceramic anodesare scarce. Even for hydrogen as fuel, the experimental indications on the reactionmechanism for cermet anodes are controversial. For Ni thin films on YSZ, thereaction seems to proceed by spillover of H from Ni to the YSZ surface wherewater formation occurs [150, 151]. On the other hand, for porous cermets basedon different metals, a volcano-shaped correlation between anode reaction rate andformation energy of the bulk metal oxide suggests that the reaction proceeds viaoxygen spillover to the metal surface [152].To confirm the O spillover mechanism, the measured reaction rates were plotted
versus ab initio calculated adsorption energies for atomic O on the metal [153], alsoyielding a volcano curve, although only energies of intermediates (not transitionstates) were considered. Recent ab initio studies deal explicitly with the YSZ/Nitwo-phase system and also determine some reaction and migration barriers (e.g.,see [154]). Nevertheless, to get a full and reliable picture, a combination of ab initiodetermined barriers and modeling of diffusive and reaction steps is required, whichalso properly includes the effect of the electrical potential (the oxygen requiredfor fuel oxidation is supplied in the form of oxide ions migrating through theelectrolyte and electrons being withdrawn through the metal; and field effects oncharge transfer barriers also have to be considered).
6.6 Conclusions
As demonstrated in this chapter, in recent years, first-principles materials modelingbecame a powerful tool for understanding atomistic details of complex processes inSOFC and permeation membranes. However, despite the great progress achievedin computer simulations, a larger number of precise large-scale calculations arerequired to model processes under realistic operating conditions for the relevantmaterials. This implies taking seriously the temperature effects and the thermo-chemical conditions.The modeling results can give insight into the atomistic details of the relevant
transport and reaction processes, which are difficult or impossible to address byexperiments. Key examples for mixed conducting perovskites used as SOFC cath-ode materials are surface reaction and surface migration barriers, which revealthe important role of oxygen vacancy mobility for the actual surface reaction. Forthe oxygen vacancy migration in perovskites, the barriers for the different indi-vidual cation configurations can be inspected, helping to identify the parametersresponsible for the low migration barriers.Far from being able to compete with experiments for specific experimentally
testable questions, modeling, though, offers an excellent complementation asit can exploit “degrees of freedom” that are not (or not readily) accessible byexperimental procedures. While for some phenomena, such as conductivity in pureion conductors, semi-empirical approaches such as pair potentials may largelycover the physics of the system, for reactions involving electron transfer—and

178 Computational Approaches to Energy Materials
sometimes even for ion migration in redox-active materials—ab initio methodsare indispensable.
Acknowledgments
Authors are greatly indebted to D. Fuks, R.A. Evarestov, E. Heifets, D. Gryaznov,S. Piskunov, W. Meulenberg, J.M. Serra, R. Dronskowski, and M. Lumey for manystimulating discussions. The research leading to these results received partial fund-ing from the German-Israeli Foundation (GIF) (grant number 1025-5.10/2009),the European Union’s COST CM1104 project, and the US National Science Foun-dation (NSF, grant CMMI-1132451). Authors thank NSF for its support throughTeraGrid resources provided by the Texas Advanced Computing Center (TACC)and the National Center for Supercomputing Applications (NCSA) under grantnumber TG-DMR100021. This study was also supported by a grant of computertime at the EMS Laboratory at PNNL (Project No 42498). MMK is grateful to theOffice of the Director of NSF for support under the IRD Program. Any appearanceof findings, conclusions, or recommendations expressed in this material are thoseof the authors and do not necessarily reflect the views of NSF.
References
1. Vielstich, W., Lamm, A. and Gasteiger, H.A. (eds) (2003) Handbook of Fuel Cells,Vol. 3 and vol. 5 (2009), John Wiley & Sons, Ltd, Chichester.
2. Kreuer, K.D. (2003) Proton-conducting oxides, Annu. Rev. Mater. Res. 33, 333–359.3. Coors, W.G. (2003) Protonic ceramic fuel cells for high-efficiency operation withmethane, J. Power Sources 118, 150–156.
4. Orera, A. and Slater, P.R. (2010) New chemical systems for solid oxide fuel cells,Chem. Mater. 22, 675–690.
5. Munch, W., Kreuer, K.D., Seifert, G. and Maier, J. (2000) Proton diffusion in per-ovskites: comparison between BaCeO3, BaZrO3, SrTiO3, and CaTiO3 using quantummolecular dynamics, Solid State Ion. 136, 183–189.
6. Islam, M.S., Davies, R.A. and Gale, J.D. (2001) Hop, skip or jump? Proton transportin the CaZrO3 perovskite oxide, Chem. Mater. 13, 2049–2055.
7. Kreuer, K.D., Paddison, S.J., Spohr, E. and Schuster, M. (2004) Transport in protonconductors for fuel-cell applications: Simulations, elementary reactions, and phe-nomenology, Chem. Rev. 104, 4637–4678.
8. Berkelbach, T.C., Lee, H.S. and Tuckerman, M.E. (2009) Concerted hydrogen-bonddynamics in the transport mechanism of the hydrated proton: a first-principles molec-ular dynamics study, Phys. Rev. Lett. 103, 238302.
9. Malavasi, L., Fisher, C.A.J. and Islam, M.S. (2010) Oxide-ion and proton conductingelectrolytematerials for clean energy applications: structural andmechanistic features,Chem. Soc. Rev. 39, 4370–4387.
10. Liu, P., Logadottir, A. and Norskov, J.K. (2003) Modeling the electro-oxidation ofCO and H2/CO on Pt, Ru, PtRu and Pt3Sn, Electrochim. Acta 48, 3731–3742.

Energy Conversion—Solid Oxide Fuel Cells 179
11. Norskov, J.K., Rossmeisl, J., Logadottir, A. et al. (2004) Origin of the overpotentialfor oxygen reduction at a fuel-cell cathode, J. Phys. Chem. B 108, 17886–17892.
12. Goddard, W., Merinov, B., van Duin, A. et al. (2006) Multi-paradigm multi-scalesimulations for fuel cell catalysts and membranes, Molec. Sim. 32, 251–268.
13. Gavartin, J.L., Sarwar, M., Papageorgopoulos, D.C. et al. (2009) Exploring fuel cellcathode materials: a High throughput calculation approach theory, anode, cathode,and oxygen evolution catalysis, ECS Transact. 25(1), 1335–1344.
14. Keith, J.A., Jerkiewicz, G. and Jacob, T. (2010) Theoretical investigations of theoxygen reduction reaction on Pt(111), Chem. Phys. Chem. 11, 2779–2794.
15. Mashio, T., Malek, K., Eikerling, M. et al. (2010) Molecular dynamics study ofionomer and water adsorption at carbon support materials, J. Phys. Chem. C 114,13739–13745.
16. Minh, N.Q. (1993) Ceramic fuel-cells, J. Am. Ceram. Soc. 76, 563–588.17. Fleig, J. (2003) Solid oxide fuel cell cathodes: polarization mechanisms and modeling
of the electrochemical performance, Annu. Rev. Mat. Res. 33, 361–382.18. Adler, S.B. (2004) Factors governing oxygen reduction in solid oxide fuel cell cath-
odes, Chem. Rev. 104, 4791–4843.19. Jiang, S.P. and Chan, S.H. (2004) A review of anode materials development in solid
oxide fuel cells, J. Mater. Sci. 39, 4405–4439.20. Jacobson, A.J. (2010) Materials for solid oxide fuel cells, Chem. Mater. 22, 660–674.21. Kroger, F.A. (1974)The Chemistry of Imperfect Crystals, North-Holland,Amsterdam.22. Baumann, F.S., Fleig, J., Cristiani, G. et al. (2007) Quantitative comparison of mixed
conducting SOFC cathode materials by means of thin film model electrodes, J. Elec-trochem. Soc. 154, B931–B941.
23. Fleig, J., Kim, H.R., Jamnik, J. and Maier, J. (2008) Oxygen reduction kinetics oflanthanum manganite (LSM) model cathodes: partial pressure dependence and rate-limiting steps, Fuel Cells 8, 330–337.
24. Wang, L., Merkle, R. and Maier, J. (2010) Surface kinetics and mechanism of oxygenincorporation into Ba1-xSrxCoyFe1-yO3-δ SOFCmicroelectrodes, J. Electrochem. Soc.157, B1802–B1808.
25. Catlow, C.R.A. and Kotomin, E.A. (eds) (2001) Computational Materials Science,IOS Publishers, Oxford, Amsterdam.
26. Mastrikov, Y.A., Heifets, E., Kotomin, E.A. and Maier, J. (2009) Atomic, electronicand thermodynamic properties of cubic and orthorhombic LaMnO3 surfaces, Surf.Sci. 603, 326–335.
27. Evarestov, R.A., Kotomin, E.A., Mastrikov, Y.A. et al. (2005) Comparative density-functional LCAO and plane-wave calculations of LaMnO3 surfaces, Phys. Rev. B 72,214411.
28. Kotomin, E.A.,Mastrikov, Y.A., Heifets, E. andMaier, J. (2008) Adsorption of atomicand molecular oxygen on the LaMnO3(001) surface: ab initio supercell calculationsand thermodynamics, Phys. Chem. Chem. Phys. 10, 4644–4649.
29. Choi, Y.M., Lin, M.C. and Liu, M. (2007) Computational study on the catalyticmechanism of oxygen reduction on La0.5Sr0.5MnO3 in solid oxide fuel cells, Angew.Chem. Int. Ed. 46, 7214–7219.
30. Choi, Y.M., Mebane, D.S., Lin, M.C. and Liu, M.L. (2007) Oxygen reduction onLaMnO3-based cathode materials in solid oxide fuel cells, Chem. Mater. 19, 1690–1699.

180 Computational Approaches to Energy Materials
31. Choi, Y.M., Lynch, D.E., Lin,M.C. and Liu,M.L. (2009) Prediction of O2 dissociationkinetics on LaMnO3-based cathode materials for solid oxide fuel cells, J. Phys. Chem.C 113, 7290–7297.
32. Chen, H.-T., Raghunath, P. and Lin, M.C. (2011) Computational investigation of O2reduction and diffusion on 25% Sr-doped LaMnO3 cathodes in solid oxide fuel cells,Langmuir 27, 6787–6793.
33. Henkelman, G., Uberuaga, B.P. and Jonsson, H. (2000) A climbing image nudgedelastic band method for finding saddle points and minimum energy paths, J. Chem.Phys. 113, 9901–9904.
34. Henkelman, G., Johannesson, G. and Jonsson, H. (2000) Progress on TheoreticalChemistry and Physics (ed. S.D. Schwartz), Kluwer, New York.
35. Henkelman, G., Arnaldsson, A. and Jonsson, H. (2006) A fast and robust algorithmfor Bader decomposition of charge density, Comp. Mater. Sci. 36, 354–360.
36. Lyde, D.L. (1993) CRC Handbook of Chemistry and Physics, 74th edn, CRC Press,Boca Raton, FL.
37. Lee, Y.-L., Kleis, J., Rossmeisl, J. and Morgan, D. (2009) Ab initio energetics ofLaBO3 (001) (B=Mn, Fe, Co, and Ni) for solid oxide fuel cell cathodes, Phys. Rev. B80, 224101.
38. Mastrikov, Y.A., Merkle, R., Heifets, E. et al. (2010) Pathways for oxygen incor-poration in mixed conducting perovskites: a DFT-based mechanistic analysis for(La, Sr)MnO3-δ , J. Phys. Chem. C, 114, 3017–3027.
39. Dovesi, R., Saunders, V.R., Roetti, C. et al. (2009) CRYSTAL09 User’s Manual,University of Torino, Italy.
40. Kotomin, E.A., Heifets, E., Maier, J. and Goddard, W.A. III (2003) Atomistic sim-ulations of the LaMnO3 (110) polar surface, Phys. Chem. Chem. Phys. 5, 4180–4184.
41. Evarestov, R.A., Kotomin, E.A., Fuks, D. et al. (2004) Ab initio calculations of theLaMnO3 surface properties, Appl. Surf. Sci. 238, 457–463.
42. Kotomin, E.A., Evarestov, R.A., Mastrikov, Y.A. and Maier, J. (2005) DFT planewave calculations of the atomic and electronic structure of LaMnO3(001) surface,Phys. Chem. Chem. Phys. 7, 2346–2350.
43. Heifets, E., Kotomin, E.A.,Mastrikov,Y.A. et al. (2011)Thermodynamics-InteractionStudies, InTech Open Access Publishers, pp. 491–518.
44. Ahmad, E.A., Liborio, L., Kramer, D. et al. (2011) Thermodynamic stability ofLaMnO3 and its competing oxides: a hybrid density functional study of an alkalinefuel cell catalyst, Phys. Rev. B 84, 085137.
45. Lee, C.-W., Behera, R.K., Wachsman, E.D. et al. (2011) Stabilization mechanisms ofLaFeO3 (010) surfaces determined with first principles calculations, Phys. Rev. B 83,115418.
46. Mizusaki, J., Yonemura, Y., Kamata, H. et al. (2000) Electronic conductivity, Seebeckcoefficient, defect and electronic structure of nonstoichiometric La1-xSrxMnO3, SolidState Ion. 132, 167–180.
47. Mizusaki, J., Mori, N., Takai, H. et al. (2000) Oxygen nonstoichiometry and defectequilibrium in the perovskite-type oxides La1-xSrxMnO3+ δ , Solid State Ion. 129,163–177.
48. Fuks, D., Bakaleinikov, L., Kotomin, E.A. et al. (2006) Thermodynamic stability anddisordering in LacSr1-cMnO3 solid solutions, Solid State Ion. 177, 217–222.

Energy Conversion—Solid Oxide Fuel Cells 181
49. Wang, Z.L. and Zhang, J. (1996) Tetragonal domain structure and magnetoresistanceof La1-xSrxCoO3, Phys. Rev. B 54, 1153–1158.
50. Piskunov, S., Spohr, E., Jacob, T. et al. (2007) Electronic and magnetic structure ofLa0.875Sr0.125MnO3 calculated by means of hybrid density-functional theory, Phys.Rev. B 76, 012410.
51. Piskunov, S., Heifets, E., Jacob, T. et al. (2008) Electronic structure and thermody-namic stability of LaMnO3 and La1-xSrxMnO3 (001) surfaces: ab initio calculations,Phys. Rev. B 78, 121406.
52. Fister, T.T., Fong, D.D., Eastman, J.A. et al. (2008) In situ characterization of stron-tium surface segregation in epitaxial La0.7Sr0.3MnO3 thin films as a function of oxygenpartial pressure, Appl. Phys. Lett. 93, 151804.
53. Herger, R., Willmott, P.R., Schleputz, C.M. et al. (2008) Structure determination ofmonolayer-by-monolayer grown La1-xSrxMnO3 thin films and the onset of magne-toresistance, Phys. Rev. B 77, 085401.
54. Kuo, J.H., Anderson, H.U. and Sparlin, D.M. (1989) Oxygen reduction behavior ofundoped and Sr-doped LaMnO3 nonstoichiometry and defect structure, J. Solid StateChem. 83, 52–60.
55. Denk, I., Munch, W. and Maier, J. (1995) Partial conductivities in SrTiO3: bulk polar-ization experiments, oxygen concentration cell measurements, and defect-chemicalmodeling, J. Am. Ceram. Soc. 78, 3265–3272.
56. Ishigaki, T., Yamauchi, S., Kishio, K. et al. (1988) Diffusion of oxide ion vacanciesin perovskite-type oxides, J. Solid State Chem. 73, 179–187.
57. Mizusaki, J., Yasuda, I., Shimoya, J. et al. (1993) Electrical-conductivity, defect equi-librium and oxygen vacancy diffusion-coefficient of La1-xCaxAlO3-δ single-crystals,J. Electrochem. Soc. 140, 467–471.
58. Woodley, S.M., Gale, J.D., Battle, P.D. and Catlow, C.R.A. (2003) Oxygen ionmigration in orthorhombic LaMnO3-δ , J. Chem. Phys. 119, 9737–9744.
59. Alexandrov, V., Piskunov, S., Zhukovskii, Y. et al. (2011) First-principles modeling ofoxygen interaction with SrTiO3(001) surface: comparative density-functional LCAOand plane-wave study, Integr. Ferroel. 123, 10–17.
60. American Chemical Society (2006) NIST Computational Chemistry Comparison andBenchmark Database, 14th edn, American Chemical Society, Washington, DC.
61. Fleig, J., Merkle, R. and Maier, J. (2007) The p(O2) dependence of oxygen surfacecoverage and exchange current density of mixed conducting oxide electrodes: modelconsiderations, Phys. Chem. Chem. Phys. 9, 2713–2723.
62. Bryant, B., Renner, C., Tokunaga, Y. et al. (2011) Imaging oxygen defects and theirmotion at a manganite surface, Nature Comm. 2, 212.
63. Pilania, G. and Ramprasad, R. (2010) Adsorption of atomic oxygen on cubic PbTiO3and LaMnO3 (001) surfaces: A density functional theory study, Surf. Sci. 604, 1889–1893.
64. Wang, L., Maxisch, T. and Ceder, G. (2006) Oxidation energies of transition metaloxides within the GGA+U framework, Phys. Rev. B 73, 195107.
65. Bakken, E., Norby, T. and Stolen, S. (2002) Redox energetics of perovskite-relatedoxides, J. Mater. Chem. 12, 317–323.
66. Choi, Y.M., Lin,M.C. andLiu,M.L. (2010)Rational design of novel cathodematerialsin solid oxide fuel cells using first-principles simulations, J. Power Sources 195, 1441–1445.

182 Computational Approaches to Energy Materials
67. Jalili, H., Han, J.W., Kuru, Y. et al. (2011) New insights into the strain couplingto surface chemistry, electronic structure, and reactivity of La0.7Sr0.3MnO3, J. Phys.Chem. Lett. 2, 801–907.
68. Kushima, A., Yip, S. and Yildiz, B. (2010) Competing strain effects in reactivity ofLaCoO3 with oxygen, Phys. Rev. B 82, 115435.
69. Stoltze, P. (2000) Microkinetic simulation of catalytic reactions, Prog. Surf. Sci. 65,65–150.
70. Hellman, A. and Honkala, K. (2007) Including lateral interactions into microkineticmodels of catalytic reactions, J. Chem. Phys. 127, 194704.
71. Kang, H.C. and Weinberg, W.H. (1995) Weinberg, Modeling the kinetics of hetero-geneous kinetics, Chem. Rev. 95, 667–676.
72. Fleig, J. (2005) On the current-voltage characteristics of charge transfer reactionsat mixed conducting electrodes on solid electrolytes, Phys. Chem. Chem. Phys. 7,2027–2037.
73. Shao, Z.P., Yang,W.S., Cong, Y. et al. (2000) Investigation of the permeation behaviorand stability of a Ba0.5Sr0.5Co0.8Fe0.2O3-δ oxygen membrane, J. Membr. Sci. 172,177–188.
74. Shao, Z. and Haile, S.M. (2004) A high-performance cathode for the next generationof solid-oxide fuel cells, Nature 431, 170–173.
75. Baumann, F.S., Fleig, J., Habermeier, H.-U. and Maier, J. (2006)Ba0.5Sr0.5Co0.8Fe0.2O3-δ thin film microelectrodes investigated by impedancespectroscopy, Solid State Ion. 177, 3187–3191.
76. Ganopadhyay, S., Inerbaev, T.,Masunov,A.E. et al. (2009) Structural characterizationcombined with the first principles simulations of barium/strontium cobaltite/ferrite aspromising material for solid oxide fuel cells cathodes and high-temperature oxygenpermeation membranes, Appl. Mater. Interfaces 1, 1512–1519.
77. Ganopadhyay, S., Masunov, A.E., Inerbaev, T. et al. (2010) Understanding oxygenvacancy migration and clustering in barium strontium cobalt iron oxide, Solid StateIon. 181, 1067–1073.
78. Stoffel, R.P., Wessel, C., Lumey, M.-W. and Dronskowski, R. (2010) Ab initio ther-mochemistry of solid-state materials, Angew. Chem. Int. Ed. 49, 5242–5266.
79. Wessel, C., Lumey, M.-W. and Dronskowski, R. (2011) First-principleselectronic-structure calculations on the stability and oxygen conductivity inBa0.5Sr0.5Co0.8Fe0.2O3-δ , J. Membr. Sci. 366, 92–96.
80. Merkle, R., Mastrikov, Y.A., Kotomin, E.A. et al. (2012) First principles calculationsof oxygen vacancy formation and migration in Ba1-xSrxCo1-yFeyO3-δ perovskites,J. Electrochem. Soc. 159, B219–226.
81. Kotomin, E.A., Merkle, R., Mastrikov, Y.A. et al. (2011) First principles cal-culations of oxygen vacancy formation and migration in mixed conductingBa0.5Sr0.5Co1-yFeyO3-δ perovskites, Solid State Ion. 188, 1–5.
82. Mastrikov, Y.A., Kuklja, M.M., Kotomin, E.A. and Maier, J. (2010) First-principlesmodelling of complex perovskite (Ba1-xSrx)(Co1-yFey)O3-δ for solid oxide fuel celland gas separation membrane applications, Energy Environm. Sci. 10, 1544–1550.
83. Kotomin, E.A., Merkle, R., Mastrikov, Y.A. et al. (2010) First principles modelingof oxygen mobility in perovskite SOFC cathode and oxygen permeation membranematerials, ECS Transact. 35(1), 823–830.

Energy Conversion—Solid Oxide Fuel Cells 183
84. Yang, Z., Harvey, A.S., Infortuna, A. and Gauckler, L.J. (2009) Phase relations in theBa-Sr-Co-Fe-O system at 1273 K in air, J. Appl. Cryst. 42, 153–160.
85. Mueller, D.N., De Souza, R.A., Brendt, J. et al. (2009) Oxidation states ofthe transition metal cations in the highly nonstoichiometric perovskite-type oxideBa0.1Sr0.9Co0.8Fe0.2O3-δ , J. Mater. Chem. 19, 1960–1963.
86. Liu, L.M., Lee, T.H., Qiu, L. et al. (1996) A thermogravimetric study of the phasediagram of strontium cobalt iron oxide, SrCo0.8Fe0.2O3-δ , Mat. Res. Bull. 31, 29–35.
87. Shannon, R.D. (1976) Revised effective ionic-radii and systematic studies of inter-atomic distances in halides and chalcogenides, Acta Crystallogr. A 32, 751–767.
88. Harvey, A.S., Litterst, F.J., Yang, Z. et al. (2009) Oxidation states of Co and Fe inBa1-xSrxCo1-yFeyO3-δ (x, y=0.2-0.8) and oxygen desorption in the temperature range300-1273 K, Phys. Chem. Chem. Phys. 11, 3090–3098.
89. Wang, L. (2009) PhD thesis, University of Stuttgart, Germany.90. Wang, L., Merkle, R., Cristiani, G. et al. (2008) PLD-deposited (Ba,Sr)(Co,Fe)O3-d
thin film microelectrodes: structure aspects and oxygen incorporation kinetics, ECSTransact. 13(26), 85–95.
91. Bucher, E., Sitte, W., Caramann, G.B. et al. (2006) Defect equilibria and partial molarproperties of (La,Sr)(Co,Fe)O3-δ , Solid State Ion. 177, 3109–3115.
92. Chen, Z.H., Ran, R., Zhou, W. et al. (2007) Assessment of Ba0.5Sr0.5Co1-yFeyO3-δ(y=0.0-1.0) for prospective application as cathode for IT-SOFCs or oxygen permeatingmembrane, Electrochim. Acta 52, 7343–7351.
93. Kowalczyk, S.P., McFeely, F.R., Ley, L. et al. (1977) Electronic structure of SrTiO3and some simple related oxides (MgO. Al2O3, SrO, TiO2), Solid State Commun. 23,161–169.
94. Piskunov, S., Heifets, E., Eglitis, R. and Borstel, G. (2004) Bulk properties andelectronic structure of SrTiO3, BaTiO3, PbTiO3 perovskites: an ab initio HF/DFTstudy, Comput. Mater. Sci. 29, 165–178.
95. Wang, L., Merkle, R., Maier, J. et al. (2009) Oxygen tracer diffusion in denseBa0.5Sr0.5Co0.8Fe0.2O3-δ films, Appl. Phys. Lett. 94, 071908.
96. Teraoka, Y., Zhang, H.M., Okamoto, K. and Yamazoe, N. (1988) Mixed ionic-electronic conductivity of La1-xSrxCo1-yFeyO3-δ perovskite-type oxides, Mater. Res.Bull. 23, 51–58.
97. Lee, T.H., Yang, Y.L., Jacobson, A.J. et al. (1997) The activation energy of 1.0eV measured for Dd is expected to be also close to the DVOactivation energy(i.e., negligible T-dependence of the thermodynamic factor). Solid State Ion. 100,77–85.
98. Mastrikov, Y.A, Merkle, R., Kotomin, E.A. et al. (2013) Formation and migrationof oxygen vacancies in La1-xSrxCo1-yFeyO3-δ perovskites: insight from ab initio cal-culations and comparison with Ba1-xSrxCo1-yFeyO3-δ , Phys. Chem. Chem. Phys. 15,911–918.
99. Sanchez, J.M., Ducastelle, F. and Gratias, D. (1984) Generalized cluster descriptionof multicomponent systems, Physica 128A, 334–350.
100. Ducastelle, F. (1994) Order and Phase Stability in Alloys, Elsevier, New York.101. Murray, A.D., Murch, G.E. and Catlow, C.R.A. (1986) A new hybrid scheme of
computer-simulation based on HADES and Monte-Carlo – applications to ionic-conductivity in Y3+ doped CeO2, Solid State Ion. 18, 196–202.

184 Computational Approaches to Energy Materials
102. Svarcova, S., Wiik, K., Tolchard, J. et al. (2008) Structural instability of cubic per-ovskite BaxSr1-xCo1-yFeyO3-δ , Solid State Ion. 178, 1787–1791.
103. Mueller, D.N., De Souza, R.A., Weirich, T.E. et al. (2010) Mayer and M. Mar-tin, A kinetic study of the decomposition of the cubic perovskite-type oxideBaxSr1-xCo0.8Fe0.2O3-δ (BSCF) (x=0.1 and 0.5),Phys. Chem. Chem. Phys. 12, 10320–10328.
104. Efimov, K., Xu, Q. and Feldhoff, A. (2010) Transmission electron microscopy studyof Ba0.5Sr0.5Co0.8Fe0.2O3-δ perovskite decomposition at intermediate temperatures,Chem. Mater. 22, 5866–5875.
105. Kuklja, M.M., Mastrikov, Y.A., Rashkeev, S.N. and Kotomin, E.A. (2011) The Struc-tural disorder and lattice stability of (Ba,Sr)(Co,Fe)O3 complex perovskites cathodematerials, processing and performance, ECS Transact. 35(1), 2077–2084.
106. Kulkja, M.M., Mastrikov, Y.A., Jansang, B. and Kotomin, E.A. (2012) The intrinsicdefects, disordering, and structural stability of BaxSr1-xCoyFe1-yO3-δ perovskite solidsolutions, J. Phys. Chem. C 116, 18605–18611.
107. Thomas, B.S.,Marks,N.A. andBegg, B.D. (2007)Defects and threshold displacementenergies in SrTiO3 perovskite using atomistic computer simulations, Nucl. Instrum.Meth. B 254, 211–218.
108. Mueller, D.N., de Souza, R.A., Yoo, H.-I. and Martin, M. (2012) phase stability andoxygen nonstoichiometry of highly oxygen-deficient perovskite-type oxides: a casestudy of (Ba,Sr)(Co,Fe)O3-δ , Chem. Mater. 24, 269–274.
109. De Souza, R.A., Islam, M.S. and Ivers-Tiffee, E. (1999) Formation and migration ofcation defects in the perovskite oxide LaMnO3, J. Mater. Chem. 9, 1621–1627.
110. De Souza, R.A. and Maier, J. (2003) A computational study of cation defects inLaGaO3, Phys. Chem. Chem. Phys. 5, 740–748.
111. Jones, A. and Islam,M.S. (2008) Atomic-scale insight into LaFeO3 perovskite: defectnanoclusters and ion migration, J. Phys. Chem. C 112, 4455–4462.
112. Kilo, M., Taylor, M.A., Argirusis, C. et al. (2004) Modeling of cation diffu-sion in oxygen ion conductors using molecular dynamics, Solid State Ion. 175,823–827.
113. Koerfer, S., De Souza, R.A., Yoo, H.-I. and Martin, M. (2008) Diffusion of Sr and Zrin BaTiO3 single crystals, Solid State Sci. 10, 725–734.
114. Palcut, M., Christensen, J.S., Wiik, K. and Grande, T. (2008) Impurity diffusion ofPr-141 in LaMnO3, LaCoO3 and LaFeO3 materials, Phys. Chem. Chem. Phys. 10,6544–6552.
115. Miyoshi, S. andMartin,M. (2009)B-site cation diffusivity ofMn andCr in perovskite-type LaMnO3 with cation-deficit nonstoichiometry, Phys. Chem. Chem. Phys., 11,3063–3070.
116. Walsh, A., Catlow, C.R.A., Smith, A.G.H. et al. (2011) Strontium migration assistedby oxygen vacancies in SrTiO3 from classical and quantum mechanical simulations,Phys. Rev. B 83, 220301.
117. Lee, Y.L., Kleis, J., Rossmeisl, J. et al. (2011) Prediction of solid oxide fuel cellcathode activity with first-principles descriptors, Energy Environm. Sci. 4, 3966–3970.
118. Ernzerhof, M. and Scuseria, G.E. (1999) Assessment of the Perdew-Burke-Ernzerhofexchange-correlation functional, J. Chem. Phys. 110, 5029–5036.

Energy Conversion—Solid Oxide Fuel Cells 185
119. Arima, T., Tokura, Y. and Torrance, J.B. (1993) Variation of optical gaps in perovskite-type 3d transition-metal oxides, Phys. Rev. B, 48, 17006–17009.
120. Gryaznov,D. and Finnis,M.W. (2010)HPC-Europa2 Research Highlights, www.hpc-europa.eu/files/SSCinEurope/CD2010/index.html (accessed 12 December 2012).
121. Dudarev, S.L., Botton, G.A., Savrasov, S.Y. et al. (1998) Electronic structure andelastic properties of strongly correlated metal oxides from first principles: LSDA+U,SIC-LSDA and EELS study of UO2 and NiO, Phys. Status Solidi A 166, 429–443.
122. Gryaznov, D., Evarestov, R.A. and Maier, J. (2010) Hybrid density-functional calcu-lations of phonons in LaCoO3, Phys. Rev. B 82, 224501.
123. Swierczek, K., Dabrowksi, B., Suescun, L. and Kolesnik, S. (2009) Crystal structureand magnetic properties of high-oxygen pressure annealed Sr1-xLaxCo0.5Fe0.5O3-δ(0 <= x <= 0.5), J. Solid State Chem. 182, 280–288.
124. Finnis, M.W., Lozovoi, A.Y. and Alavi, A. (2005) The oxidation of NiAl: what canwe learn from ab initio calculations?, Annu. Rev. Mater. Res. 35, 167–207.
125. He, J., Behera, R.K., Finnis, M.W., Li, X., Dickey, E.C. and Phillpot, S.R. (2007)Prediction of high-temperature point defect formation in TiO2 from combined abinitio and thermodynamic calculations Acta Mater. 55, 4325–4337.
126. Mizusaki, J.,Mima,Y., Yamauchi, S. et al. (1989)Nonstoichiometry of the perovskite-type oxides La1-xSrxCoO3-δ , J. Solid State Chem. 80, 102–111.
127. Mizusaki, J., Yoshihiro, M., Yamauchi, S. and Fueki, K. (1987) Thermody-namic quantities and defect equilibrium in the perovskite-type oxide solid-solutionLa1-xSrxFeO3-δ , J. Solid State Chem. 67, 1–8.
128. Gryaznov, D., Finnis, M., Evarestov, R.A. and Maier, J. (2013) Oxygen vacancyformation energies in Sr-doped complex perovskites: ab initio thermodynamic study.Submitted to Solid State Ion.
129. Khan, M.S., Islam, M.S. and Bates, D.R. (1998) Cation doping and oxygen diffusionin zirconia: a combined atomistic simulation and molecular dynamics study, J. Mater.Chem. 8, 2299–2307.
130. Wilde, P.J. and Catlow, C.R.A. (1998) Defects and diffusion in pyrochlore structuredoxides, Solid State Ion. 112, 173–183.
131. Islam, M.S. (2000) Ionic transport in ABO3 perovskite oxides: a computer modellingtour, J. Mater. Chem. 10, 1027–1038.
132. Zacate, M.O., Minervini, L., Bradfield, D.J. et al. (2000) Defect cluster formation inM2O3-doped cubic ZrO2, Solid State Ion. 128, 243–254.
133. Chroneos, A., Yildiz, B., Tarancon, A. and Kilner, J.A. (2011) Oxygen diffusionin solid oxide fuel cell cathode and electrolyte materials: mechanistic insights fromatomistic simulations, Energy Environm. Sci. 4, 2774–2789.
134. Nakayama, M. and Martin, M. (2009) First-principles study on defect chemistry andmigration of oxide ions in ceria doped with rare-earth cations, Phys. Chem. Chem.Phys. 11, 3241–3249.
135. Pornprasertsuk, R., Ramanarayanan, P., Musgrave, C.B. and Prinz, F.B. (2005) Pre-dicting ionic conductivity of solid oxide fuel cell electrolyte from first principles,J. Appl. Phys. 98, 103513.
136. Marrocchelli, D., Madden, P.A., Norberg, S.T. and Hull, S. (2011) Structural disorderin doped zirconias, Part II: vacancy ordering effects and the conductivity maximum,Chem. Mater. 23, 1365–1373.

186 Computational Approaches to Energy Materials
137. Dalach, P., Ellis, D.E. and van der Walle, A. (2010) First-principles thermody-namic modeling of atomic ordering in yttria-stabilized zirconia, Phys. Rev. B 82,144117.
138. Kushima, A. and Yildiz, B. (2010) Oxygen ion diffusivity in strained yttria stabilizedzirconia: where is the fastest strain?, J. Mater. Chem. 20, 4809–4819.
139. Araki, W., Kuribara, M. and Arai, Y. (2011) Effect of uniaxial stress on ionic con-ductivity of 14 mol%-yttria-stabilized zirconia single crystal, Solid State Ion. 193,5–10.
140. Jamnik, J., Maier, J. and and Pejovnik, S. (1995) Interfaces in solid state ionicconductors – equilibrium and small-signal picture, Solid State Ion. 75, 51–58.
141. Yoshiya, M. and Oyama, T. (2011) Impurity and vacancy segregation at symmetrictilt grain boundaries in Y2O3-doped ZrO2, J. Mater. Sci. 46, 4176–4190.
142. Marinopoulos, A.G. (2011) First principles study of segregation to the�5(310) grainboundary of cubic zirconia, J. Phys.: Condens. Matter. 23, 085005.
143. Lee, H.B., Prinz, F.B. and Cai,W. (2010) Atomistic simulations of surface segregationof defects in solid oxide electrolytes, Acta Mater. 58, 2197–2206.
144. Sankaranarayanan, S.K.R. and Ramanathan, S. (2011) Interface proximity effects onionic conductivity in nanoscale oxide-ion conducting yttria stabilized zirconia: anatomistic simulation study, J. Chem. Phys. 134, 064703.
145. Kuwabara, A. and Tanaka, I. (2004) First principles calculation of defect formationenergies in Sr- and Mg-doped LaGaO3, J. Phys. Chem. B 108, 9168–9172.
146. De Souza, R.A. and Martin, M. (2009) An atomistic simulation study of oxygen-vacancy migration in perovskite electrolytes based on LaGaO3, Monatsh. Chemie140, 1011–1015.
147. Kilner, J.A. and Brook, R.J. (1982) A study of oxygen ion conductivity in dopednonstoichiometric oxides, Solid State Ion. 6, 237–252.
148. Gorte, R.J. and Vohs, J.M. (2009) Nanostructured anodes for solid oxide fuel cells,Curr. Opin. Colloid Interface Sci. 14, 236–244.
149. Cowin, P.I., Petit, C.T.G., Lan, R. et al. (2011) Recent progress in the developmentof anode materials for solid oxide fuel cells, Adv. Energy Mater. 1, 314–322.
150. Vogler,M., Bieberle-Hutter, A.,Gauckler, L.J. et al. (2009)Modelling study of surfacereactions, diffusion, and spillover at a Ni/YSZ patterned anode, J. Electrochem. Soc.156, B663–672.
151. Goodwin, D.G., Zhu, H., Colclasure, A.M. and Kee, R.J. (2009) Modeling electro-chemical oxidation of hydrogen on Ni-YSZ pattern anodes, J. Electrochem. Soc. 156,B1004–B1021.
152. Setoguchi, T., Okamoto, K., Eguchi, K. and Arai, H. (1992) Effects of anode materialand fuel on anodic reaction of solid oxide fuel-cells, J. Electrochem. Soc. 139, 2875–2880.
153. Rossmeisl, J. and Bessler, W.G. (2008) Trends in catalytic activity for SOFC anodematerials, Solid State Ion. 178, 1694–1700.
154. Shishkin, M. and Ziegler, T. (2009) Oxidation of H2, CH4, and CO molecules at theinterface between nickel and yttria-stabilized zirconia: a theoretical study based onDFT, J. Phys. Chem. C 113, 21667–21678.