electronic, optical and lattice dynamic properties of the novel diamond-like semiconductors...
TRANSCRIPT

Electronic, optical and lattice dynamic properties of the novel diamond-like semiconductors
Li2CdGeS4 and Li2CdSnS4
This article has been downloaded from IOPscience. Please scroll down to see the full text article.
2011 J. Phys.: Condens. Matter 23 225401
(http://iopscience.iop.org/0953-8984/23/22/225401)
Download details:
IP Address: 128.248.155.225
The article was downloaded on 02/06/2012 at 20:59
Please note that terms and conditions apply.
View the table of contents for this issue, or go to the journal homepage for more
Home Search Collections Journals About Contact us My IOPscience

IOP PUBLISHING JOURNAL OF PHYSICS: CONDENSED MATTER
J. Phys.: Condens. Matter 23 (2011) 225401 (11pp) doi:10.1088/0953-8984/23/22/225401
Electronic, optical and lattice dynamicproperties of the novel diamond-likesemiconductors Li2CdGeS4 andLi2CdSnS4
Yanlu Li1, Weiliu Fan2,3, Honggang Sun1, Xiufeng Cheng1, Pan Li1
and Xian Zhao1,3
1 State Key Laboratory of Crystal Materials, Shandong University, Jinan 250100,People’s Republic of China2 Department of Chemistry and Chemical Engineering, Shandong University, Jinan 250100,People’s Republic of China
E-mail: [email protected] and [email protected]
Received 19 February 2011, in final form 21 April 2011Published 16 May 2011Online at stacks.iop.org/JPhysCM/23/225401
AbstractLi2CdGeS4 and Li2CdSnS4 are novel quaternary diamond-like semiconductors (DLSs) whichhave been synthesized recently. We present first-principles calculations of their electronic,optical and lattice dynamic properties with the plane-wave pseudopotential method. We havefound an indirect band gap of 2.78 eV for Li2CdGeS4 and a direct band gap of 2.50 eV forLi2CdSnS4. The serious stretching vibrations of the Ge/Sn–S and Li–S bonds may enhance theirphonon energies, and cause them to exhibit high heat capacities and Debye temperatures, whichare promising for nonlinear optical applications. Compared with Cu-based DLSs, Li plays a keyrole in enlarging the band gaps and increasing the lattice phonon energies, which wouldincrease the thermal conductivity accompanied by an increase of the optical damage threshold.
S Online supplementary data available from stacks.iop.org/JPhysCM/23/225401/mmedia
(Some figures in this article are in colour only in the electronic version)
1. Introduction
Diamond-like semiconductors (DLSs) are technologicallyuseful and have been investigated for a variety of applicationsin photovoltaics [1], nonlinear optics [2, 3], thermoelectrics [4]and spintronics [5]. Many efforts have been made on binaryand ternary DLSs, which already have a wide range of practicalapplications. However, quaternary DLSs possess increasedflexibility and functionality in compositions and physicalproperties, which may bring out more applications in the areasof nonlinear optics, solar cells and light-emitting diodes [6].
Much research on the quaternary I2–II–IV–VI4 (I = Cu,Ag; II = Cd, Zn; IV = Ge, Sn; VI = S, Se) compoundshas been reported [4, 7–16], while there have been fewreports concerning Li2–II–IV–VI4 compounds [17–19]. A
3 Authors to whom any correspondence should be addressed.
novel diamond-like Li2CdGeS4 was first synthesized in bothsingle crystal and polycrystalline forms by Lekse et al [18],who also reported the synthesis of novel Li2CdSnS4 crystalwhich has been previously prepared by Suseela Devi andVidyasagar [19]. It is pointed out that the replacement ofCu or Ag by Li may enlarge the band gap of these materials,thereby increasing the laser damage thresholds. This is of greatimportance in the potential for second harmonic generation(SHG) application. Additionally, the lighter Li atoms canenhance the lattice vibration frequency, which ensures goodpossibilities for high thermal conductivity and their applicationin nonlinear conversion devices pumped by high-power lasers.However, relevant theoretical investigations are very immatureand scarce. The theoretical investigation of band structure iscrucial for understanding the physical properties and predictingthe optical susceptibilities. The prediction of fundamental
0953-8984/11/225401+11$33.00 © 2011 IOP Publishing Ltd Printed in the UK & the USA1

J. Phys.: Condens. Matter 23 (2011) 225401 Y Li et al
optical parameters can provide a guide for the further studyof the nonlinear optical susceptibilities and bring us importantinsights in understanding the origin of the electronic behaviors.In addition, the knowledge of the lattice dynamics plays akey role in understanding the stability of the structure, phasetransition, Raman and infrared vibrations, thermodynamicproperties, as well as phenomena related to the electron–phonon interaction. In particular, quaternary DLSs containingLi may exhibit some interesting electronic, optical, vibrationaland thermodynamic properties.
In this paper, we present first-principles investigations onthe electronic, optical and lattice dynamic properties, includingthe electronic band structure, the density of states, thecomplex dielectric function, the optical absorption coefficient,the refractive index, the extinction coefficient, the energy-loss spectrum, the phonon dispersion curves, the densityof phonon states, the �-point vibrational frequency, the IRspectrum and some thermodynamic properties, of Li2CdGeS4
and Li2CdSnS4 DLSs. We also take Li2CdGeS4 as an exampleto confirm and further illustrate the role of Li cations bycomparing its physical properties with those of Cu2CdGeS4
from the point of view of theoretical calculations.
2. Computational details
First-principles density functional theory (DFT) calcula-tions [20] were employed in the Cambridge Sequential TotalEnergy Package (CASTEP) code [21] with a plane-wave basisset for the electronic wavefunctions and periodic boundaryconditions. The interaction between the valence electronsand the core electrons was described by a norm-conservingpseudopotential [22], which represented 2s1, 3d104s1, 4d105s2,4s24p2, 5s25p2 and 3s23p4 electron configurations for Li,Cu, Cd, Ge, Sn and S atoms, respectively. The exchangeand correlation potential was described both in the local-density approximation (LDA) of Ceperley–Alder [23] and thegeneralized gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE) [24]. Typically, the LDA underestimateslattice constants relative to the experimental values while theopposite is true for the GGA. However, from the supplemen-tary table S1 (available at stacks.iop.org/JPhysCM/23/225401/mmedia) we can see that the GGA overestimates the latticea value and underestimates the lattice b and c values of bothLi2CdGeS4 and Li2CdSnS4, and by comparing the calculatedratios (a:b:c) with the experimental values, we find that thestructures optimized by the LDA could keep the proportionof lattice axis and crystal symmetry better. Considering thatthe results of phonon spectra are strongly dependent on thestability and deformation of optimized structures, we chosethe LDA to relax the structures and calculate all the physicalproperties of these two compounds. The Monkhorst–Packscheme k-point sampling was used for integration over the firstBrillouin zone [25], and the Kohn–Sham energy function wasdirectly minimized via the conjugate-gradient method. Underthe restriction of the symmetry, the total energy was minimizedvia varying the cell parameters and the atomic positions toobtain the structure optimization. The lattice constants andinternal positions in primitive cells at various volumes were
Figure 1. Crystal structures of Li2CdGeS4 and Li2CdSnS4.
fully optimized. The kinetic energy cutoff values were set to660 eV and the k-point meshes were 3 × 4 × 4 for Li2CdGeS4,Li2CdSnS4 and Cu2CdGeS4. The convergence criteria forstructure optimization and energy calculation were set to ultra-fine quality with tolerance for self-consistent field, energy,maximum force, maximum displacement, and maximumstress set to be 5.0 × 10−7 eV/atom, 5.0 × 10−6 eV/atom,0.01 eV A
−1, 5.0 × 10−4 A, and 0.02 GPa, respectively. The
lattice dynamic and thermodynamic properties were calculatedwithin the framework of density functional perturbation theory.Technical details of the computation of the response to smallatomic displacements can be found in [26].
3. Results and discussion
3.1. Crystalline structures
Li2CdGeS4 crystallizes in the orthorhombic space groupPmn21 and exhibits a three-dimensional framework, whichis corner-connected by three types of tetrahedra, LiS4, CdS4
and GeS4, as shown in figure 1. Li2CdSnS4 is isostructuralwith Li2CdGeS4. By minimizing the crystal total energy,the equilibrium lattice constants and some other structuralparameters have been computed. The results are givenin table 1 with experimental values. The LDA structuralrelaxation results are somewhat smaller than the measuredones, but still in good agreement with experiment withdeviations of around 2%. After relaxation, the GeS4 and SnS4
tetrahedra are fairly regular with nearly the same bond lengths(2.14 A for the Ge–S bond and 2.34 A for the Sn–S bond)while the LiS4 and CdS4 polyhedra exhibit greater distortionof 1.68% (1.26%) for the Li–S bond and 1.14% (0.63%) forthe Cd–S bond in Li2CdGeS4 (Li2CdSnS4). The bond lengthvariations of the Cd–S and Li–S bonds are more apparent inLi2CdGeS4 than in Li2CdSnS4.
3.2. Band structures, densities of states and electron chargedensity
The calculated electronic band structures of Li2CdGeS4 andLi2CdSnS4 are shown in figure 2. The band structures ofthe compounds are similar. Li2CdGeS4 possesses an indirectband gap of 2.78 eV with the valence band maximum atthe X-point and the conduction band minimum at the �-point of the Brillouin zone. The smallest direct band gap
2

J. Phys.: Condens. Matter 23 (2011) 225401 Y Li et al
Figure 2. Electronic band structures of Li2CdGeS4 and Li2CdSnS4.
Figure 3. Total and projected densities of states for Li2CdGeS4 (a) and Li2CdSnS4 (b).
Table 1. Lattice constants, charge transfer, bond types, bond populations and bond lengths of Li2CdGeS4 and Li2CdSnS4.
Lattice constant (A)
Compound Present Exp. [18] Charge (e) Bond Population Length (A)
Li2CdGeS4 a = 7.601(−1.75%) a = 7.737 Li: 0.90 (Li–S)1–4 0.03–0.07 2.334–2.374b = 6.654(−2.86%) b = 6.850 Cd: 0.26 (Cd–S)1–3 0.43–0.55 2.514–2.543c = 6.253(−1.82%) c = 6.369 Ge: 0.78 (Ge–S)1–3 0.65–0.80 2.140–2.143
S: −0.71Li2CdSnS4 a = 7.856(−1.25%) a = 7.956 Li: 0.89 (Li–S)1–4 0.04–0.07 2.352–2.382
b = 6.799(−2.48%) b = 6.968 Cd: 0.45 (Cd–S)1–3 0.45–0.49 2.521–2.537c = 6.394(−1.48%) c = 6.489 Sn: 0.86 (Ge–S)1–3 0.64–0.71 2.342–2.346
S: −0.77
of Li2CdGeS4 is 2.83 eV and agrees well with the measureddata of 3.10 eV [18]. In contrast, Li2CdSnS4 shows a �-pointdirect band gap of 2.50 eV, which is 0.76 eV smaller than theexperimental value [18]. The usual underestimation of DFT isobserved.
The calculated total and partial densities of states ofLi2CdGeS4 and Li2CdSnS4 are shown in figure 3. The resultsdemonstrate, overall, a rather similar energy distribution of theeigenstates for these two compounds. For both compounds,Li s electronic states mainly lie in the energy range above
3

J. Phys.: Condens. Matter 23 (2011) 225401 Y Li et al
Figure 4. Projected electronic density distribution corresponding to the energy ranges of −3.27 to −2.23 eV (a1) and 3.94–5.95 eV (a2) forLi2CdGeS4 and −3.57 to −2.47 eV (b1) and 3.61–5.72 eV (b2) for Li2CdSnS4.
2.5 eV with a small contribution in the upper valence band.This indicates that the Li occur as ions that provide chargebalance to the [CdGeS4]2− polyanion, and, consequently, theelectrostatic interactions between the Li atoms and polyanionscontribute to the stability of the structures. There are threeintervals in the valence band: the lowest-lying states (below−10 eV) mainly stem from the Ge sp/Sn sp and S 3s states withsmall contributions of Cd spd states; the bands located at −10–−5 eV are isolated ones with strong Cd 4d and Ge 4s/Sn 5scharacters; the electronic structures of the upper valence band(−5–0 eV) originate predominantly from Cd sp, Ge 4p/Sn 5pand S 3p states. The conduction bands mainly consist of Li 2s,Cd sp and Ge sp/Sn sp states.
The nature of the chemical bonding can be clearlyelucidated from the total and partial densities of states. Wefind that strong hybridizations between Ge 4p/Sn 5p and S 3pstates as well as Cd 5s and S 3p states locate at −4.5 to −2 eV,corresponding to the σ bonding states. The correspondingσ ∗ antibonding states locate at the upper conduction bands,and are also from the hybridizations between Ge 4p/Sn 5pand S 3p states as well as Cd 5s and S 3p states. Toinvestigate the electronic states in greater detail, we plottedthe projected electronic density distribution of the (110) planeat energies of −3.27 to −2.23 and 3.94 to 5.95 eV for
Li2CdGeS4 as well as −3.57 to −2.47 and 3.61 to 5.72 eVfor Li2CdSnS4 in figure 4. Figures 4(a1) and (b1) correspondto the bonding states of Li2CdGeS4 and Li2CdSnS4 whilefigures 4(a2) and (b2) correspond to the antibonding statesof Li2CdGeS4 and Li2CdSnS4. For Li2CdGeS4, there is noobvious electron density accumulation at the Li–S bondingregions, and thus we obtain an ionic-like bonding picture ofthe Li–S bonds. The Ge–S bonds, which show a distinctdegree of covalence, are the main originators of the strongσ bonding states of Li2CdGeS4, while the Cd–S bonds showweaker covalence and also have contributions to the bondingstates (figure 4(a1)). The contributions to the antibondingstates are almost the same, while the Cd–S hybridization inthe antibonding states is a little larger than in the bondingstates (figure 4(a2)). The bonding and antibonding electronicstates of Li2CdSnS4 are similar to those of Li2CdGeS4, but theelectron distribution is more dispersed in Li2CdSnS4, and moreelectrons around Sn atoms distribute along the Sn–S bondsinstead of accumulating around Sn atoms.
This picture can be strengthened by Mulliken populationanalysis, as shown in table 1. For Li2CdGeS4, the Mullikencharges are determined to be 0.90e for Li, 0.26e for Cd, 0.78efor Ge and −0.71e for S, indicating that electrons transfer fromLi, Cd and Ge to S atoms. The Li atoms contribute almost
4

J. Phys.: Condens. Matter 23 (2011) 225401 Y Li et al
Figure 5. The imaginary part ε2(ω) and the real part ε1(ω) of the dielectric functions for Li2CdGeS4 ((a), (b)) and Li2CdSnS4 ((a′), (b′)).
all the outer electrons acting as fairly idealized monovalentions, and the Li–S bond exhibits a highly ionic nature witha Mulliken overlap population (MOP) of 0.03–0.07. TheMOP values of the Cd–S bonds are in the range 0.43–0.55,which is smaller than the range 0.65–0.80 for the Ge–S bonds,indicating that the Cd–S bonds are less covalent than the Ge–S bonds. These results agree with the analysis of electronicstructures above. The same phenomenon can be found forLi2CdSnS4, while the MOP values of the Sn–S bonds are alittle smaller than those of the Ge–S bonds. The replacementof the anion Ge by Sn has little influence on the electronicproperties of the crystal.
3.3. First-order (linear) optical susceptibilities
The linear optical properties are determined by the complexdielectric function ε(ω) = ε1(ω) + iε2(ω), describingthe polarization response of the system to an externalelectromagnetic field with a small wavevector. The electricfield of the photon leads to the transition between occupiedand unoccupied states, including plasmons and single particleexcitations. The excitation spectra can be described as ajoint density of states between the valence and conductionbands. The intraband transition cannot be considered becauseit is crucial only for metals. We also neglect the indirectinterband transitions involving scattering of phonons assumingthat they give a small contribution to the frequency-dependentdielectric functions. The imaginary part ε2(ω) of the dielectricfunction can be obtained from the momentum matrix elementsbetween the occupied and unoccupied wavefunctions and is
given by [27]
ε2(ω) = 2π2e2
�ε0
∑
i∈c. f ∈v
∑
k
|〈�ck |μ∧ · r |�v
k 〉|2δ
× [Eck − Ev
k − hω].The real part ε1(ω) can be evaluated from ε2(ω) using theKramers–Kronig relations and is given by [28]
ε1(ω) = 1 +(
2
π
) ∫ ∞
0dω′ ω′2ε2(ω
′)ω′2 − ω2
.
All the other optical properties, including the absorptioncoefficient α(ω), the refractive index n(ω), the extinctioncoefficient k(ω), and the energy-loss spectrum L(ω) can bedirectly calculated from ε1(ω) and ε2(ω) [27, 29].
By comparing the calculated band gaps with experimentalvalues, scissor operators of 0.27 and 0.76 eV are applied forthe calculation of the optical properties of Li2CdGeS4 andLi2CdSnS4, respectively. Figure 5 depicts the variation of theimaginary part ε2(ω) and the real part ε1(ω) of the frequency-dependent dielectric function for Li2CdGeS4 and Li2CdSnS4.The orthorhombic symmetry leads to three components of thedielectric functions of Li2CdGeS4 and Li2CdSnS4. The onsetsto the response of ε2(ω) reflect the band gap energy, andfollowing the ε2(ω) spectra one can find that Li2CdGeS4 showslarger anisotropy (ε
yy2 ≈ εzz
2 > εxx2 ) than Li2CdSnS4 in the
vicinity of the onset. For Li2CdGeS4, the principal peaks aresituated at energies around 4.2 and 6.8–7.5 eV for εxx
2 (ω), 6.5–8.0 eV for ε
yy2 (ω) and 5.0–7.0 eV for εzz
2 (ω), and mainly arisefrom the electron transition from S 3p states to Ge sp and Cd spstates with a small contribution from the transition betweenCd sp and Ge sp states. The highest peak of ε2(ω) is along
5

J. Phys.: Condens. Matter 23 (2011) 225401 Y Li et al
Figure 6. The absorption coefficient α(ω), refractive index n(ω), extinction coefficient k(ω) and energy-loss function L(ω) for Li2CdGeS4
((a)–(c)) and Li2CdSnS4 ((a′)–(c′)).
the y direction, and the two peaks at about 6.5 and 7.5 eV arealso from the electron transition from Se p states in the uppervalence bands to the Ge sp and Cd sp states in the conductionbands. The ε2(ω) spectrum of Li2CdSnS4 is similar to thatof Li2CdGeS4. However, owing to the different dispersionof the topmost valence bands, Li2CdSnS4 displays only onemain peak in the ε2(ω) spectrum along the y direction, whichoriginates from the electron transition from S 3p states to Sn spand Cd sp states.
From figure 5(b), we can see that Li2CdGeS4 still showslarger anisotropy in the real part ε1(ω) of the dielectric functionthan Li2CdSnS4. In addition, Li2CdGeS4 shows a larger high-frequency dielectric constant than that of Li2CdSnS4. This isin accordance with the smaller band gap of Li2CdGeS4 and canbe explained by the Penn model ε1(0) ≈ 1 + (hωp/Eg)
2 [30],but, overall, the compounds show rather comparable spectra.For Li2CdGeS4, the calculated static dielectric constants ε1(0)
are 5.43 for εxx1 (0), 5.56 for ε
yy1 (0) and 5.70 for εzz
1 (0),while the three components of ε1(0) are all equal to 5.00for Li2CdSnS4. The maximum of uniaxial anisotropy (δε =(εzz
1 (0) − εxx1 (0))/εtot
1 (0); εtot1 (0) = (εxx
1 (0) + εyy1 (0) +
εzz1 (0))/3) is 0.05 for Li2CdGeS4. Note that we do not include
phonon contributions to the dielectric screening. The ε1(0)
corresponds to the static optical dielectric constant, and thephonon contributions will be discussed in section 3.4.
For more details about the spectral features of the opticalsusceptibilities of Li2CdGeS4 and Li2CdSnS4, we investigatedthe spectral dependences for the absorption coefficient α(ω),the refractive index n(ω), the extinction coefficient k(ω), andthe energy-loss spectrum L(ω), as shown in figure 6. Theabsorption coefficient represents the linear optical responsefrom the valence bands to the lowest conduction bands.Since the absorption is obtained directly from the dielectricfunction, similarities in the polarization response for the twocompounds are reflected also in the absorption coefficient.Thus, the compounds have comparable absorption, althoughwith different photon energies for the onset of absorption.Experimentally, it has been reported [18] that Li2CdGeS4
and Li2CdSnS4 have large band-edge absorptions of 3.10 and3.26 eV, which have been confirmed by the present calculatedvalues of 2.75–3.00 eV and 3.10–3.23 eV, respectively.Such a large optical band gap is expected because the
6

J. Phys.: Condens. Matter 23 (2011) 225401 Y Li et al
Table 2. �-point phonon frequencies of Li2CdGeS4 and Li2CdSnS4.
Frequency (cm−1)
Mode (active) A1 (IR/Raman) A2 (Raman) B1 (IR/Raman) B2 (IR/Raman)
Li2CdGeS4 59.40 46.87 77.02 69.1584.34 80.13 153.4 91.83 (TO)
108.2 134.1 171.5 (TO) 119.0 (LO)169.8 147.7 190.7 (LO) 198.6251.1 191.4 234.7 250.8269.7 238.3 366.2 270.2283.4 376.4 386.0 285.7368.9 (TO) 383.9 400.7 348.9380.5 (LO) 403.7 410.4 381.1385.1 410.3 395.1395.4 413.3411.2 420.8418.6 435.8
Li2CdSnS4 57.11 42.32 80.28 61.5169.06 79.28 126.3 83.1894.09 109.9 146.6 97.76
139.0 129.9 187.7 180.0244.5 182.2 236.6 236.4257.4 240.9 345.0 255.7267.6 355.1 371.2 (TO) 269.6351.7 (TO) 368.7 375.0 (LO) 336.4355.8 (LO) 373.9 403.7 363.2363.0 403.7 375.1 (TO)369.4 376.3 (LO)375.7 382.6401.3 417.4
laser damage threshold of a material is dependent upon thecompound’s band gap, with larger gaps leading to increasedthresholds [31], which makes them promising for nonlinearoptical applications.
From the spectra of the refractive index and the extinctioncoefficient for Li2CdGeS4 and Li2CdSnS4 (figures 6(b)and (b′)), we find that in the infrared region, the refractive indexand the extinction coefficient increase with the increase ofphoton energy: the increase in refractive index with the photonenergy shows normal dispersion behavior of the materials; theincrease in extinction coefficient with the photon energy showsthat the fraction of light lost due to scattering and absorbancedecreases. Although the spectra of the refractive index andthe extinction coefficient still exhibit anisotropy, the staticrefractive indices are hardly distinguished, and are equal to2.36 for Li2CdGeS4 and 2.25 for Li2CdSnS4.
The energy-loss spectrum L(ω) describes the energy lostby an electron passing through a homogeneous dielectricmaterial. The main peak, which shows the collective excitationof the loosely bound valence electrons into the unoccupiedenergy levels in the conduction bands, is generally definedas the bulk plasma oscillation frequency ωp, whose positioncorresponds to the zero crossing of ε1(ω). From figures 6(c)and (c′) we cannot see distinct maxima in the low energy range,and the main peaks in the L(ω) are located at 19.2 and 19.9 eVfor Li2CdGeS4 and Li2CdSnS4, respectively. These high peaksare associated with the transitions from the hybridization statesof Cd 5s, Ge 4p/Sn 5p and S 3p to the empty Cd sp states. Forthe slightly stronger hybridization effect of Cd 5s, Sn 5p andS 3p states, the plasma peak of Li2CdSnS4 is a little sharper andhigher than that of Li2CdGeS4. As there are few experimental
or theoretical optical spectra results available for Li2CdGeS4
and Li2CdSnS4, we hope that our work will stimulate furtherworks. However, the calculated optical properties are limitedby considering direct transitions only, and at high temperaturesphonon-assisted transitions will give an additional contributionthat will somewhat broaden the spectra.
3.4. Lattice dynamics
Since the primitive unit cells of Li2CdGeS4 and Li2CdSnS4
contain 16 atoms, there are 48 phonon branches in the vibrationspectrum. The irreducible representation of the C2v groupat the �-point is decomposed by A1 + B1 + B2 for threeacoustic modes, and 13A1 + 10A2 + 13B1 + 9B2 for 45 opticalmodes. The calculated �-point phonon frequencies of all theoptical modes for Li2CdGeS4 and Li2CdSnS4 are listed intable 2. All modes are Raman-active, and the modes withsymmetries A1, B1 and B2 are also active in the IR spectra.All the A1, B1 and B2 modes belong to the vector transformingrepresentation, and the inclusion of the long-range polarizationinteraction results in splitting of these modes into transverseoptical (TO) and longitudinal optical (LO) components giving35 polar vibrations.
The difference between the LO and TO phononfrequencies, which characterizes the change in electronicpolarization with the ionic displacements, is defined by theBorn effective charge tensors Z∗ and the electronic permittivitytensor ε∞ through nonanalytical contribution to the forceconstants. We list the dynamical effective charge tensors ofLi2CdGeS4 and Li2CdSnS4 in table 3 as well as their static andhigh-frequency dielectric tensor components in table 4. Due
7

J. Phys.: Condens. Matter 23 (2011) 225401 Y Li et al
Table 3. Born effective charges Z∗ of Li2CdGeS4 and Li2CdSnS4.X denotes Ge or Sn, respectively.
Li2CdGeS4 Li2CdSnS4
Z∗Li
(0.91 0.05 0.09
−0.04 1.17 −0.14−0.07 0.07 1.05
) (0.93 0.06 0.08
−0.03 1.14 −0.14−0.07 0.08 1.04
)
Z∗Cd
(1.95 0.00 0.000.00 1.31 −0.300.00 0.08 1.79
) (1.64 0.00 0.000.00 1.12 −0.290.00 0.10 1.50
)
Z∗X
(3.02 0.00 0.000.00 2.26 −0.510.00 0.17 2.86
) (3.16 0.00 0.000.00 2.48 −0.410.00 0.15 3.06
)
Z∗S1
(−2.18 0.15 0.180.27 −1.36 −0.03
−0.25 −0.15 −1.44
) (−2.02 0.06 0.100.17 −1.38 −0.01
−0.16 −0.09 −1.48
)
Z∗S2
(−1.21 0.00 0.000.00 −1.87 −0.370.00 −0.24 −1.66
) (−1.30 0.00 0.000.00 −1.70 −0.260.00 −0.18 −1.70
)
to the orthorhombic symmetry of Li2CdGeS4 and Li2CdSnS4,the Born effective charge tensors of the Li, Cd, X and Satoms have three independent components. The Z∗ diagonalcomponents are close to the expected values of ionic chargesLi (+1), Cd (+2), X (+4) and S (−2) from the chemicalpoint of view. We note that from Li2CdGeS4 to Li2CdSnS4
there occurs an increase of the Z∗X diagonal components and
a decrease of the Z∗Cd diagonal components. Meanwhile, the
Z∗Li diagonal component along the x axis increases with the
decrease of those along the y and z directions, and the Z∗S
diagonal components along the x and z directions increase withthe decrease of that along the y direction. This results in asmaller deviation from the ideal ionic charges in Li2CdSnS4
than Li2CdGeS4, and can be understood by the dynamicaltransfer of the charge due to the modification of the bondhybridization during the atomic displacement. As seen fromthe data presented, for Li2CdGeS4, the largest anisotropyoccurs along the x and y directions with −22.2% for Li,49.0% for Cd, 33.6% for Ge, 60.5% for S1, and −35.2 forS2. Li2CdSnS4 shows the same trend but smaller anisotropyalong the x and y directions. It is noted that the shift of theS anion atoms creates eight different configurations for theseatoms and the resulting effective charge tensor elements can bedivided into two classes according to the shift direction alongx or y.
Contrary to the effective charges, the form of the dielectrictensor is determined by the symmetry of the crystal and isexpected to be diagonal for Li2CdGeS4 to Li2CdSnS4, asshown in table 4: the optical-frequency and low-frequencydielectric tensors of Li2CdGeS4 to Li2CdSnS4 all have threeindependent components ε
↔ xx , ε↔ yy and ε
↔ zz due to theorthorhombic symmetry. From the dielectric theory, the static
dielectric tensor ε↔
(0) is usually composed of the electroniccontribution ε
↔ω→∞ and the lattice vibration contribution
ε↔
ω=0, namely ε↔
(0) = ε↔
ω→∞ + ε↔
ω=0. Qualitatively,the lattice component depends quadratically on the ratio ofeffective dynamical charges to the frequency of IR activemodes [32, 33]. From table 4 we can obtain that forLi2CdGeS4, the three independent components of the optical-frequency dielectric constant are 4.90, 4.37 and 4.69 for εxx
ω→∞,ε
yyω→∞ and εzz
ω→∞, respectively, with an average value εω→∞ of4.65, while they are 4.58, 4.26 and 4.48 for εxx
ω→∞, εyyω→∞ and
εzzω→∞ with an average value εω→∞ of 4.44 for Li2CdSnS4.
The calculated low-frequency dielectric constants along the x ,y, and z axes are a little larger than the corresponding opticalcomponents. The εxx
ω=0, εyyω=0 and εzz
ω=0 yield 7.27, 6.15 and7.00 with an average value εω=0 of 6.81 for Li2CdGeS4 and6.77, 6.01 and 6.72 with an average value εω=0 of 6.50 forLi2CdSnS4, respectively. It should be noted that the DFTmethod always overestimates the lattice component of thestatic dielectric tensor [34], so the actual dielectric constantmay be a little smaller than our calculated results. Besides,the anisotropy of the optical and dc dielectric constants issmall for both Li2CdGeS4 and Li2CdSnS4, with the dielectriccomponent along the x direction a little larger.
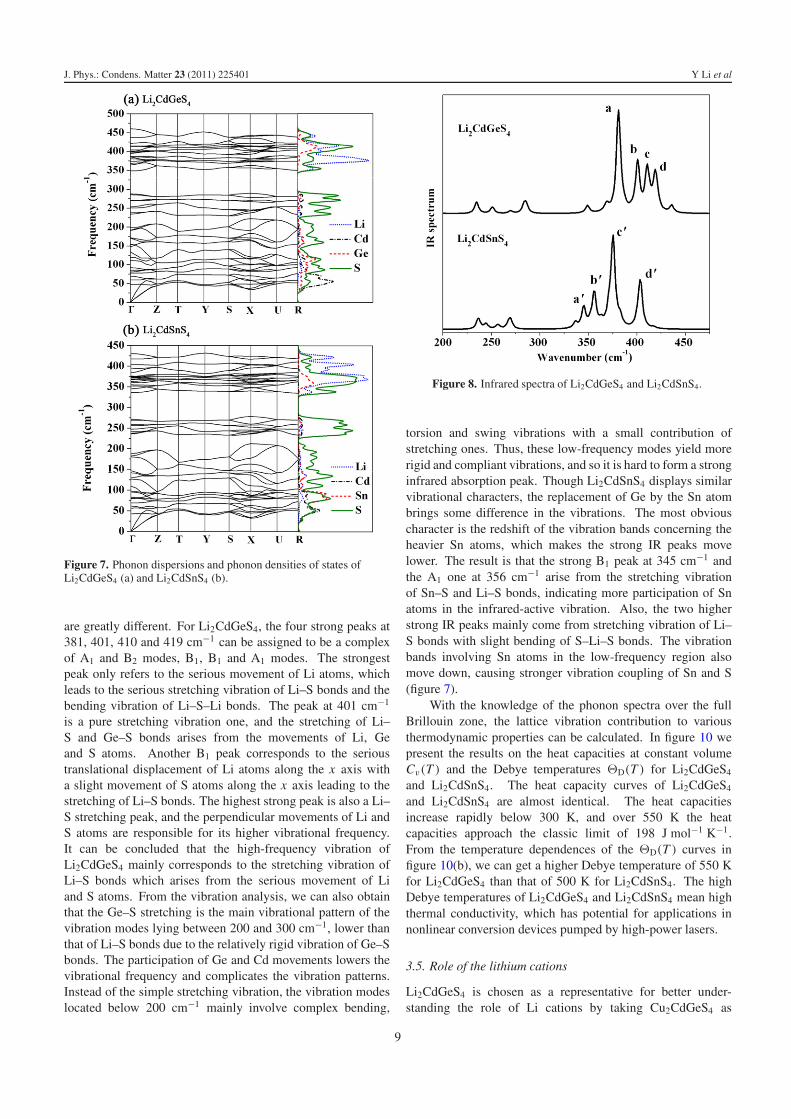
The phonon dispersion curves calculated along the high-symmetry lines of the Brillouin zone and the correspondingdensities of phonon states of Li2CdGeS4 and Li2CdSnS4 areshown in figure 7. It can be seen that the calculated phonondispersion relations have no soft modes at any wavevectors,indicating the stability of Li2CdGeS4 and Li2CdSnS4. Thereis a more considerable energy overlap between some opticmodes and the acoustic modes for Li2CdSnS4 than Li2CdGeS4,because the masses of the Cd and Sn atoms are much moresimilar than those of the Cd and Ge atoms in the unit cell.The overlap means that energy transfer between these opticand acoustic modes is easy. With anharmonic effects, theselow-frequency optic modes will strongly scatter the acousticmodes, which carry the heat flow and may lead to low latticethermal conductivity. The density of phonon states results tracewell the main features of the phonon spectrum. As expected,the contribution of Cd and Ge/Sn movements dominates atlower frequency, and the movement of the lighter Li lies athighest frequency, while the vibration of S atoms spans throughthe whole vibration region. It can be seen from figure 7that, compared with the lighter Ge atoms, the two vibrationalbands of the heavier Sn atoms (below 150 cm−1 and above350 cm−1) all downshift, and further lead to the change of thecorresponding vibrational patterns. This can also be reflectedfrom the calculated IR spectrum in figures 8 and 9. Fromfigure 8 we can find four weak IR peaks below 300 cm−1
in both Li2CdGeS4 and Li2CdSnS4, whereas the main peaks
Table 4. Optical-frequency (ω → ∞) and low-frequency (ω = 0) dielectric tensor components of Li2CdGeS4 and Li2CdSnS4.
ε (ω → ∞) ε (ω = 0)
εxxω→∞ εyy
ω→∞ εzzω→∞ εω→∞ εxx
ω=0 εyyω=0 εzz
ω=0 εω=0
Li2CdGeS4 4.90 4.37 4.69 4.65 7.27 6.15 7.00 6.81Li2CdSnS4 4.58 4.26 4.48 4.44 6.77 6.01 6.72 6.50
8
J. Phys.: Condens. Matter 23 (2011) 225401 Y Li et al
Figure 7. Phonon dispersions and phonon densities of states ofLi2CdGeS4 (a) and Li2CdSnS4 (b).
are greatly different. For Li2CdGeS4, the four strong peaks at381, 401, 410 and 419 cm−1 can be assigned to be a complexof A1 and B2 modes, B1, B1 and A1 modes. The strongestpeak only refers to the serious movement of Li atoms, whichleads to the serious stretching vibration of Li–S bonds and thebending vibration of Li–S–Li bonds. The peak at 401 cm−1
is a pure stretching vibration one, and the stretching of Li–S and Ge–S bonds arises from the movements of Li, Geand S atoms. Another B1 peak corresponds to the serioustranslational displacement of Li atoms along the x axis witha slight movement of S atoms along the x axis leading to thestretching of Li–S bonds. The highest strong peak is also a Li–S stretching peak, and the perpendicular movements of Li andS atoms are responsible for its higher vibrational frequency.It can be concluded that the high-frequency vibration ofLi2CdGeS4 mainly corresponds to the stretching vibration ofLi–S bonds which arises from the serious movement of Liand S atoms. From the vibration analysis, we can also obtainthat the Ge–S stretching is the main vibrational pattern of thevibration modes lying between 200 and 300 cm−1, lower thanthat of Li–S bonds due to the relatively rigid vibration of Ge–Sbonds. The participation of Ge and Cd movements lowers thevibrational frequency and complicates the vibration patterns.Instead of the simple stretching vibration, the vibration modeslocated below 200 cm−1 mainly involve complex bending,
Figure 8. Infrared spectra of Li2CdGeS4 and Li2CdSnS4.
torsion and swing vibrations with a small contribution ofstretching ones. Thus, these low-frequency modes yield morerigid and compliant vibrations, and so it is hard to form a stronginfrared absorption peak. Though Li2CdSnS4 displays similarvibrational characters, the replacement of Ge by the Sn atombrings some difference in the vibrations. The most obviouscharacter is the redshift of the vibration bands concerning theheavier Sn atoms, which makes the strong IR peaks movelower. The result is that the strong B1 peak at 345 cm−1 andthe A1 one at 356 cm−1 arise from the stretching vibrationof Sn–S and Li–S bonds, indicating more participation of Snatoms in the infrared-active vibration. Also, the two higherstrong IR peaks mainly come from stretching vibration of Li–S bonds with slight bending of S–Li–S bonds. The vibrationbands involving Sn atoms in the low-frequency region alsomove down, causing stronger vibration coupling of Sn and S(figure 7).
With the knowledge of the phonon spectra over the fullBrillouin zone, the lattice vibration contribution to variousthermodynamic properties can be calculated. In figure 10 wepresent the results on the heat capacities at constant volumeCv(T ) and the Debye temperatures �D(T ) for Li2CdGeS4
and Li2CdSnS4. The heat capacity curves of Li2CdGeS4
and Li2CdSnS4 are almost identical. The heat capacitiesincrease rapidly below 300 K, and over 550 K the heatcapacities approach the classic limit of 198 J mol−1 K−1.From the temperature dependences of the �D(T ) curves infigure 10(b), we can get a higher Debye temperature of 550 Kfor Li2CdGeS4 than that of 500 K for Li2CdSnS4. The highDebye temperatures of Li2CdGeS4 and Li2CdSnS4 mean highthermal conductivity, which has potential for applications innonlinear conversion devices pumped by high-power lasers.
3.5. Role of the lithium cations
Li2CdGeS4 is chosen as a representative for better under-standing the role of Li cations by taking Cu2CdGeS4 as
9

J. Phys.: Condens. Matter 23 (2011) 225401 Y Li et al
Figure 9. Atomic displacement patterns which correspond to the main peaks of the IR spectra of Li2CdGeS4 ((a)–(d)) and Li2CdSnS4
((a′)–(d′)).
Figure 10. Temperature dependence of the constant volume heat capacities Cv(T ) (a) and the Debye temperatures �D(T ) (b) for Li2CdGeS4
and Li2CdSnS4.
a comparison. Cu2CdGeS4 is also a novel diamond-likecrystal, which has the same orthorhombic structure Pmn21
as Li2CdGeS4. Using the same calculation method, we getlattice constants of a = 7.624, b = 6.484, c = 6.260 Afor Cu2CdGeS4, which are within 1% deviations from theexperimental values of a = 7.692, b = 6.555, c =6.299 A [13].
The calculated band structures and partial densities ofstates of both the compounds are given in the supplementaryfigures S2 and S3 (available at stacks.iop.org/JPhysCM/23/225401/mmedia). Unlike Li2CdGeS4, Cu2CdGeS4 is a directband gap semiconductor with an Eg of 1.23 eV, smaller than theexperimental value of 2.05 eV [13] due to the underestimationof DFT. We notice that Li2CdGeS4 has a much larger bandgap (Eg(indirect) = 2.78 eV; Eg(direct) = 2.83 eV) thanCu2CdGeS4. As discussed above, in Li2CdGeS4, Li has almostno contribution in the valence bands, and the hybridizationof Ge 4p and Cd 5s with S 3p states contributes to thebonding interaction. Such p–p hybridization interaction canreduce the energy of the highest occupied valence bandsand increase the energy of the lowest unoccupied conductionbands, and thus enlarges the energy gap of Li2CdGeS4.From the supplementary figure S3 (available at stacks.iop.org/JPhysCM/23/225401/mmedia) we can see that the mainbonding interaction of Cu2CdGeS4 is the hybridization ofCu 3d and S 3p states as well as a mixing of Ge 4p withS 3p states in the upper valence bands. The repulsion of
S 3p and Cu-3d levels in the upper valence bands increases theenergy of the highest occupied valence bands and contributesto the smaller direct energy gap. The larger band gap allowspumping at relatively short wavelengths with less tendency totwo-photon absorption and leads to an increased laser damagethreshold, meaning that it is more promising in nonlinearoptical applications.
Besides, Li2CdGeS4 with a larger band gap yieldsdifferent optical properties. The most obvious is that all theoptical property curves have redshifts with the increase of bandgap (see supplementary figure S4 available at stacks.iop.org/JPhysCM/23/225401/mmedia). Following the Penn model,ε1(0) ≈ 1 + (hωp/Eg)
2, the static dielectric constants alongthe three polarization directions all decrease with the increaseof the band gap. And, consequently, other electronic-relatedoptical properties also change, such as a reduction of therefractive index and the extinction coefficient as well as a sharpplasma peak with the increase of the band gap.
In addition, our calculations show a considerableinfluence of Li cations on the lattice dynamic properties (seesupplementary figure S5 and S6 available at stacks.iop.org/JPhysCM/23/225401/mmedia). The higher ionicity of the Li–S bond contributes to the larger LO/TO splitting of the polarphonon modes in Li2CdGeS4. And the serious vibration oflighter Li atoms enhances the high-frequency optical bandsin the phonon spectrum, leading to the lower lattice heatcapacity and higher Debye temperature. Therefore, the thermal
10

J. Phys.: Condens. Matter 23 (2011) 225401 Y Li et al
conductivity is expected to be much larger than that ofCu2CdGeS4, and in turn is accompanied by an increase of theoptical damage threshold. At equivalent residual absorptionloss, thermal lensing effects should hence be minimized.Thus, Li2CdGeS4 may have broader application prospects innonlinear optics than Cu2CdGeS4.
4. Conclusions
In this paper, we present first-principles calculations of theelectronic, optical and lattice dynamic properties of novelLi2CdGeS4 and Li2CdSnS4 DLSs. The two compounds showsimilar physical and chemical properties due to having thesame crystal structures and chemical compositions. Thecalculations show an indirect band gap of 2.78 eV forLi2CdGeS4 and a direct band gap of 2.50 eV for Li2CdSnS4.The Li–S bond shows strong ionicity while the Cd–S andGe/Sn–S bonds are typical covalent ones. Hybridizationsbetween Ge 4p/Sn 5p and S 3p as well as Cd 5s and S 3pstates are responsible for the σ bonding states in the uppervalence bands and σ ∗ antibonding states in the conductionbands. The calculated average optical- and low-frequencydielectric constants are 4.65 and 6.81 for Li2CdGeS4 and 4.44and 6.50 for Li2CdSnS4. The lattice dynamics results illustratethat the high-frequency modes mainly arise from the stretchingvibration of the Li–S bonds for Li2CdGeS4 and the Sn–S andLi–S bonds for Li2CdSnS4. The Ge/Sn–S stretching and thecomplex of bending, torsion and swing vibrations contribute tothe bands at 200–300 cm−1 and below 200 cm−1. Li2CdGeS4
and Li2CdSnS4 show high heat capacities of 198 J mol−1 K−1
and Debye temperatures of 500 and 550 K. By comparing thephysical properties of Li2CdGeS4 with Cu2CdGeS4, we findthat the Li occur as ions that provide charge balance to the[CdGeS4]2− polyanion. Instead of the p–d hybridization inCu2CdGeS4, hybridizations between Ge p and S p levels arethe main bonding interactions and enlarge the energy gap ofLi2CdGeS4. The large band gap yields a redshift of the opticalspectrum and a reduction of the optical parameters. Besides,the replacement of Cu by the lighter Li also increases the latticephonon energy and Debye temperature, and is expected toincrease the thermal conductivity, accompanied by an increaseof the optical damage threshold, which makes it promising fornonlinear optical applications.
Acknowledgments
This work is supported by the National Natural ScienceFoundation of China (Grant Nos 50802056 and 91022034),the Excellent Youth Foundation of Shandong ScientificCommittee (Grant No. JQ201015), the Youth Scientist(Doctoral) Foundation of Shandong Province of China (GrantNo. BS2009CL038), the Independent Innovation Foundationof Shandong University (Grant No. 2009TS016), and theGraduate Independent Innovation Foundation of ShandongUniversity (Grant No. yzc10054).
References
[1] Goetzberger A, Hebling C and Schock H-W 2003 Mater. Sci.Eng. R 40 1
[2] Ohmer M C and Pandey R 1998 MRS Bull. 23 16[3] Catella G C and Burlage D 1998 MRS Bull. 23 28[4] Davidyuk G E, Parasyuk O V, Semenyuk S A and
Romanyuk Y E 2003 Inorg. Mater. 39 919[5] Pearton S J, Abernathy C R, Norton D P, Hebard A F,
Park Y D, Boatner L A and Budai J D 2003 Mater. Sci. Eng.R 40 137
[6] Shay J L, Bachmann K J, Buehler E and Wernick J H 1973Appl. Phys. Lett. 23 226
[7] Parthe E, Yvon K and Dietch R H 1969 Acta Crystallogr. B25 1164
[8] Schleich D M and Wold A 1977 Mater. Res. Bull. 12 111[9] Quintero M, Barreto A, Grima P, Tovar R, Quintero E,
Porras G S, Ruiz J, Woolley J C, Lamarche G andLamarche A M 1999 Mater. Res. Bull. 34 2263
[10] Mkrtchyan S A, Dovletov K, Zhukov E G, Melikdzhanyan A Gand Nuryev S 1988 Izv. Akad. Nauk SSSR Neorg. Mater.24 1094
[11] Konstantinova N N, Medvedkin G A, Polushina I K, Rud Yu V,Smirnova A D, Sokolova V I and Tairov M A 1989 Izv.Akad. Nauk SSSR Neorg. Mater. 5 1445
[12] Filonenko V V, Nechiporuk B D, Novoseletskii N E,Yukhimchuk V A and Lavorik Y F 1991 Izv. Akad. NaukSSSR Neorg. Mater. 27 1166
[13] Davydyuka G Y, Parasyukb O V, Romanyukb Y E,Semenyuka S A, Zarembac V I, Piskach L V, Kozioł J Jand Halka V O 2002 J. Alloys Compounds 339 40
[14] Parasyuk O V, Olekseyuk I D, Piskach L V, Volkov S V andPekhnyo V I 2005 J. Alloys Compounds 399 173
[15] Tsuji I, Shimodaira Y, Kato H, Kobayashi H and Kudo A 2010Chem. Mater. 22 1402
[16] Persson C 2010 J. Appl. Phys. 107 053710[17] Lekse J W, Leverett B M, Lake C H and Aitken J A 2008
J. Solid State Chem. 181 3217[18] Lekse J W, Moreau M A, McNerny K L, Yeon J,
Halasyamani P S and Aitken J A 2009 Inorg. Chem. 48 7516[19] Suseela Devi M and Vidyasagar K 2002 J. Chem. Soc., Dalton
Trans. 2002 2092[20] Payne M C, Teter M P, Allan D C, Arias T A and
Joannopoulos J D 1992 Rev. Mod. Phys. 64 1045[21] Clark S J, Segall M D, Pickard C J, Hasnip P J, Probert M J,
Refson K and Payne M C 2005 Z. Kristallogr. 220 567[22] Vanderbilt D 1990 Phys. Rev. B 41 7892[23] Ceperley D M and Alder B J 1980 Phys. Rev. Lett. 45 566[24] Perdew J P, Burke K and Ernzerhof M 1996 Phys. Rev. Lett.
77 3865[25] Monkhorst H J and Pack J D 1976 Phys. Rev. B 13 5188[26] Gonze X 1997 Phys. Rev. B 55 10337[27] Saha S, Sinha T P and Mookerjee A 2000 Phys. Rev. B 62 8828[28] Yu P Y and Cardona M 1996 Fundamentals of Semiconductors
(Berlin: Springer)[29] Cai M-Q, Yin Z and Zhang M-S 2003 Appl. Phys. Lett.
83 2805[30] Penn D R 1962 Phys. Rev. 128 2093[31] Jackson A G, Ohmer M C and LeClair S R 1997 Infrared Phys.
Technol. 38 233[32] Zhao X and Vanderbilt D 2002 Phys. Rev. B 65 075105
Zhao X and Vanderbilt D 2002 Phys. Rev. B 65 233106[33] Bruesch P 1986 Phonons: Theory and Experiments II (Berlin:
Springer)[34] Delugas P, Fiorentini V and Filippetti A 2005 Phys. Rev. B
71 134302
11