effects of early clozapine treatment on remission rates in
TRANSCRIPT

EARLY- Protocol final Version 5.0 AM 3.0 - Date: 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 1 of 71
BEZIRKSKLINIKEN SCHWABEN
STUDY PROTOCOL
Effects of early clozapine treatment on remission
rates in acute schizophrenia (EARLY)
Sponsor:
Bezirkskliniken Schwaben
Dr.-Mack-Straße 4 - D – 86156 Augsburg
Sponsor Delegated Person (SDP):
Prof. Dr. med. Alkomiet Hasan
Bezirkskrankenhaus Augsburg, Klinik für Psychiatrie, Psychotherapie und Psychosomatik der Universität Augsburg
Dr.-Mack-Straße 1 - D – 86156 Augsburg
EudraCT Number: 2018-001514-15 Study Code: EARLY_KUM_PSY
Version: Final 5.0 - AM 3.0
Date 30.11.2020
Confidential
This document is confidential and should serve as a source of information for Investigators and
other personnel involved in this clinical study, consultants and ethics committees and
regulatory authorities. The contents of this document shall only be disclosed to others in
agreement with the coordinating investigator and/or sponsor.


EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 3 of 71
Table of Contents
Inhalt
Signatures ...................................................................................................................... 2
Responsibility ................................................................................................................. 5
Study-Synopsis .............................................................................................................. 6
Summary ..................................................................................................................... 10
1. Introduction and aims ............................................................................................... 11
1.1 Background, initial situation and medical problem ................................................................. 11
1.2 Rationale for the EARLY study .................................................................................................. 12
2. Objectives ................................................................................................................ 13
3. Study design ............................................................................................................ 14
4. Study endpoints ....................................................................................................... 15
4.1 Primary endpoint ......................................................................................................................... 15
4.2 Secondary endpoints .................................................................................................................. 16
5. Study population ...................................................................................................... 18
5.1 Inclusion criteria .......................................................................................................................... 18
5.2 Exclusion criteria ......................................................................................................................... 20
5.3 Sample size calculation .............................................................................................................. 20
6. Treatment of subjects .............................................................................................. 22
6.1 Investigational products ............................................................................................................. 22
6.2 Titration schemes ........................................................................................................................ 22
6.3 Use of concomitant medication ................................................................................................. 26
6.4 Treatment after the end of the double-blind treatment phase ................................................ 27
7. Investigational product ............................................................................................. 28
7.1 Contraindications ........................................................................................................................ 28
7.2 Interactions .................................................................................................................................. 29
7.3 Manufacturing of Trial Medication ............................................................................................ 31
7.4 Storage, Formulation, packaging and labelling ....................................................................... 31
7.5 Emergency unblinding ................................................................................................................ 32
8. Methods ................................................................................................................... 33
8.1 Randomisation ............................................................................................................................. 33
8.2 Description of study visits.......................................................................................................... 33
8.3 Special safety aspects during the intervention period ............................................................ 36
8.4 Naturalistic extension study....................................................................................................... 37
8.5 Description of rating scales and assessments ........................................................................ 37
8.6 Discontinuation of the study ...................................................................................................... 41
8.7 Study flow chart ........................................................................................................................... 43
9. Statistical Analysis ................................................................................................... 45

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 4 of 71
11. Feasibility of patient recruitment ............................................................................ 49
12. Safety reporting ..................................................................................................... 50
12.1 AE, SAE and Susar definitions and reporting procedures ................................................... 50
12.2 Follow-up of adverse events .................................................................................................... 53
12.3 Unblinding .................................................................................................................................. 55
12.4 Pregnancy .................................................................................................................................. 55
12.5 Overdose .................................................................................................................................... 56
12.6 Annual safety report (Safety Update Report (DSUR)) ............................................................ 56
12.7 Safety Monitoring Board (SMB) ............................................................................................... 56
13. Ethical and regulatory aspects ............................................................................... 57
13.1 Sponsor‘s and investigator’s responsibilities........................................................................ 57
13.2 Ethics committees and health authorities .............................................................................. 57
13.3 Ethical performance of the study ............................................................................................ 57
13.4 Informed consent of the study participants ........................................................................... 57
14. Study administration .............................................................................................. 59
14.1 Documentation and Data collection ........................................................................................ 59
14.2 Database management ............................................................................................................. 59
14.3 Audits and inspections ............................................................................................................. 59
14.4 Monitoring .................................................................................................................................. 60
14.5 Archiving .................................................................................................................................... 60
14.6 Reporting .................................................................................................................................... 60
14.7 Insurance for subjects participating in clinical studies ........................................................ 61
14.8 Data Privacy Protection and Confidentiality protection ........................................................ 62
14.9 Protocol amendments or changes in trial conduct ............................................................... 62
15. Abbreviation ........................................................................................................... 64
16. Annex ..................................................................................................................... 65
16.1 Declaration of Helsinki .............................................................................................................. 65
16. Reference List ........................................................................................................ 66
EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 5 of 71
Responsibility
S
po
nso
r
Sponsor
Bezirkskliniken Schwaben
Dr.-Mack-Straße 4 - D – 86156 Augsburg
Tel.: (0821) 4803 - 2727
Fax.: (0821) 4803 - 2702
E-Mail: [email protected]
Pro
toco
l C
om
mit
tee
Sponsor Delegated Person
Project Leader
Prof. Dr. med. Alkomiet Hasan
Director
Department of Psychiatry, Psychotherapy and
Psychosomatics, Medical Faculty, University of
Augsburg, BKH Augsburg
Bezirkskliniken Schwaben Dr.-Mack-Straße 1 - D – 86156 Augsburg Tel.: (0821) 4803-1001 Fax: (0821) 4803-1002 E-Mail: [email protected]
Co-Project Leader
Prof. Dr. med. Stefan Leucht
Deputy Director
Department of Psychiatry and Psychotherapy,
TU Munich,
Ismaningerstr. 22, 81675 München,
Tel.: +49-89/4140-4249
E-Mail: [email protected]
Clinical Project Management
Dr. med. Wolfgang Strube
Department of Psychiatry, Psychotherapy and
Psychosomatics, Medical Faculty, University of
Augsburg, BKH Augsburg
Bezirkskliniken Schwaben Dr.-Mack-Straße 1 - D – 86156 Augsburg Tel.: (0821) 4803-3225 Fax: (0821) 4803-1002 E-Mail: [email protected]
IME
DIS
Statistican
PD Dr. rer. nat. Alexander Hapfelmeier
Institute of Medical Informatics, Statistics and
Epidemiology
Technische Universität München
Ismaningerstr. 22, 81675 München
Tel.: +49-89/4140-4340
Fax.: +49-89/4140-4350
E-Mail: [email protected]
MS
Z
Project Management
Monitoring
Data Management
Safety Management
Technische Universität München
School of Medicine
Münchner Studienzentrum
Ismaninger Straße 22; 81675 München
E-Mail: [email protected]
Tel.+49-89/4140-6321
Fax:+49-89/4140-6322
EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 6 of 71
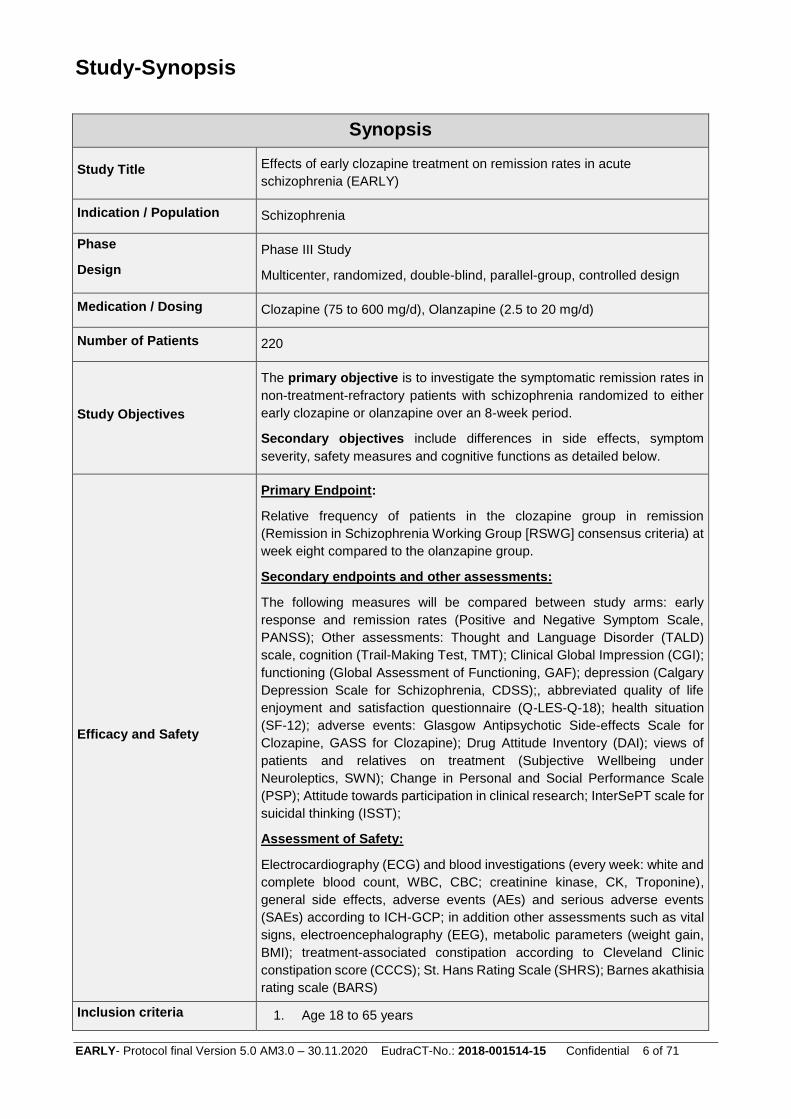
Study-Synopsis
Synopsis
Study Title Effects of early clozapine treatment on remission rates in acute
schizophrenia (EARLY)
Indication / Population Schizophrenia
Phase
Design
Phase III Study
Multicenter, randomized, double-blind, parallel-group, controlled design
Medication / Dosing Clozapine (75 to 600 mg/d), Olanzapine (2.5 to 20 mg/d)
Number of Patients 220
Study Objectives
The primary objective is to investigate the symptomatic remission rates in
non-treatment-refractory patients with schizophrenia randomized to either
early clozapine or olanzapine over an 8-week period.
Secondary objectives include differences in side effects, symptom
severity, safety measures and cognitive functions as detailed below.
Efficacy and Safety
Primary Endpoint:
Relative frequency of patients in the clozapine group in remission
(Remission in Schizophrenia Working Group [RSWG] consensus criteria) at
week eight compared to the olanzapine group.
Secondary endpoints and other assessments:
The following measures will be compared between study arms: early
response and remission rates (Positive and Negative Symptom Scale,
PANSS); Other assessments: Thought and Language Disorder (TALD)
scale, cognition (Trail-Making Test, TMT); Clinical Global Impression (CGI);
functioning (Global Assessment of Functioning, GAF); depression (Calgary
Depression Scale for Schizophrenia, CDSS);, abbreviated quality of life
enjoyment and satisfaction questionnaire (Q-LES-Q-18); health situation
(SF-12); adverse events: Glasgow Antipsychotic Side-effects Scale for
Clozapine, GASS for Clozapine); Drug Attitude Inventory (DAI); views of
patients and relatives on treatment (Subjective Wellbeing under
Neuroleptics, SWN); Change in Personal and Social Performance Scale
(PSP); Attitude towards participation in clinical research; InterSePT scale for
suicidal thinking (ISST);
Assessment of Safety:
Electrocardiography (ECG) and blood investigations (every week: white and
complete blood count, WBC, CBC; creatinine kinase, CK, Troponine),
general side effects, adverse events (AEs) and serious adverse events
(SAEs) according to ICH-GCP; in addition other assessments such as vital
signs, electroencephalography (EEG), metabolic parameters (weight gain,
BMI); treatment-associated constipation according to Cleveland Clinic
constipation score (CCCS); St. Hans Rating Scale (SHRS); Barnes akathisia
rating scale (BARS)
Inclusion criteria 1. Age 18 to 65 years

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 7 of 71
2. Signed informed consent
3. DSM-V diagnosis of schizophrenia confirmed by the Mini international
Neuropsychiatric Interview
4. At least one documented prior hospitalization due to the illness in the
medical history (the current hospitalization can be considered as
“prior” hospitalization if its ≥ 4 weeks) at screening
5. For treatment-naïve patients (defined as no previous antipsychotic treatment or a maximum of 30 days of treatment), an antipsychotic treatment attempt of at least 30 days with an antipsychotic in a therapeutic dose according to local guidelines other than clozapine and olanzapine before the screening phase is needed. For non-treatment-naïve patients (defined as having been treated for more than 30 days with an antipsychotic), a discontinuation of a foregoing antipsychotic treatment prior to the screening phase within a maximum of six months (=180 days) is possible (corresponding to the estimated average time for an antipsychotic washout phase and the expected time to develop a relapse of the disease). For patients being treated with a long-acting antipsychotic (other than PP3M), an inclusion is possible if inclusion date corresponds to the planned date of the next injection plus five to seven days. For patients being treated with oral olanzapine, an inclusion is possible if this treatment has lasted for no longer than 2 weeks prior to inclusion and if exclusion criteria 8 is not fulfilled.
6. Clinical need for a medication switch because of clinical inefficacy or side-effects or clinical need for a reintroduction of an antipsychotic treatment after treatment discontinuation prior to the screening phase (see 5.)
7. Moderate symptomatology on the PANSS, defined as a score ≥ 4 for
two or more symptoms from P1-P7 or a score of ≥ 6 for one symptom
from P1-P7 (minimum threshold definition) at screening
8. Male participants and female participants who are not capable of
bearing children or who use a method of contraception that is
medically approved by the health authority of the respective country
at screening
This includes:
A woman who is not capable of bearing a child is defined as follows:
post-menopausal (12 months natural (spontaneous) amenorrhea or
6 months spontaneous amenorrhea with serum-FSH-values (follicle-
stimulating hormone) of >40 mIU/mL); 6 weeks after a bilateral
ovariectomy with or without hysterectomy or sterilization by means of
tubal ligation
A woman capable of bearing child is defined as follows: a woman who
is physiologically capable of becoming pregnant, including women
whose occupation, lifestyle or sexual orientation exclude sexual
intercourse with a male partner and women whose partners have
been sterilized by vasectomy or other measures.
Medically-approved methods of contraception can include the
following: hormonal contraceptives, intrauterine device and double
barrier method. Acceptable preventive measures can include total
abstinence at the discretion of the investigator, in cases where
compliance is ensured because of the study participant’s age,
occupation, lifestyle or sexual orientation. Periodical abstinence (e.g.
calendar, ovulation, symptothermal methods or abstinence until the
4th day after the ovulation) as well as coitus interruptus are not
acceptable methods of contraception.
A reliable method of contraception (CTFG guideline) must be used
for the entire duration of the study.

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 8 of 71
Exclusion criteria
1. Patients who are not suitable for the study in the opinion of the
investigator
2. Patients who are unable to give informed consent
3. Coercive treatment at the time of study inclusion
4. White blood cell count (WBC) at inclusion not meeting the
requirements for clozapine use in Germany. Patients must have
normal leukocyte findings (white blood cell count ≥ 3500/mm3 (≥
3.5x109/l), and Absolute Neutrophil Count (ANC) ≥ 2000/mm3 (≥
2.0x109/l) at the screening visit
5. The presence of one or more of the contraindications against any of the study drugs as mentioned in the SmPC
6. Treatment-naïve or treatment-resistant schizophrenia. Treatment-
naïve will be defined as no previous antipsychotic treatment or a
maximum of 30 days of treatment. Treatment resistance is defined as
2 antipsychotic trials (with antipsychotics from two different chemical
classes) for a period of ≥ 6 weeks with CPZ equivalent doses ≥ 600
mg/day, both of which took place immediately before the screening
phase
7. Diagnosis of primary substance dependency other than nicotine
8. Documented previous non-response to an 8-week drug trial with
olanzapine or any documented previous treatment with clozapine
9. Intolerance to one of the study drugs
10. Pregnancy (incl. positive blood pregnancy test) / lactation (female
patients)
Study Procedures
Experimental intervention/index test: Application of double-blind, early
clozapine in acute schizophrenia patients not meeting the criteria for
treatment resistance
Control intervention/reference test: Application of double-blind olanzapine
in acute schizophrenia patients not meeting the criteria for treatment
resistance
Schedule / Follow up
Inclusion of the first patient (FPFV): approx. quarter II 2019
Inclusion of the last patient (LPFV): approx. quarter I 2022
End of the study (LPLV): approx. quarter IV 2022
End of naturalistic follow-up: approx. quarter IV 2024
Integrated final report of primary outcome: approx. quarter IV 2023
Statistics Sample size
To be assessed for eligibility (n = 880)
To be allocated to trial (n = 220)
To be analysed (n = 220)
Statistics
Efficacy/test accuracy: Relative frequency of patients in remission at week
eight in both study groups
Description of the primary efficacy/test accuracy analysis and population:
The primary efficacy endpoint will be compared between groups with a
Mantel-Haenszel test with centres as strata on a two-sided 5% significance
level. This analysis will be performed in the intention-to-treat (ITT)
population.
Safety: Absolute and relative frequencies of categorical safety outcomes will
be assessed along the course of the trial. Differences between groups will
also be tested for statistical significance by Fisher’s exact test. Continuous

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 9 of 71
safety outcomes will be presented by descriptive statistics and compared
with t-tests or Mann-Whitney U tests.
Secondary endpoints: Depending on the data distribution, continuous and
categorical secondary endpoints will be compared between groups by linear
or binary logistic regression analysis including a factor variable for centres.
Baseline values will be included as another covariate if existing. In case of
non-normally distributed residuals in the linear model, a van Elteren test with
centres as strata will be applied in an additional analysis. Corresponding
descriptive statistics and confidence intervals will also be given. All tests will
be computed for the ITT and per protocol (PP) populations in an explorative
manner at a two-sided 5% significance level.
Rationale
Achieving symptomatic remission quickly after the onset of psychotic
symptoms is the critical objective in schizophrenia treatment and determines
the subsequent disease course. In this context, only every second patient
with acute schizophrenia achieves symptomatic remission within three
months of initiating antipsychotic treatment, meaning that half of patients do
not achieve this key objective. Increasing the likelihood to achieve
symptomatic remission in acute schizophrenia will improve the overall
outcome, reduce disease-associated burden and potentially prevent mid-
and long-term disease chronicity. With this randomized, double-blind,
parallel-group multicentre trial we aim to provide evidence for the superior
efficacy of the ‘last resort’ antipsychotic clozapine compared to one of the
most effective second-generation antipsychotics (SGAs), olanzapine, in
acute schizophrenia patients who do not meet the criteria for treatment-naive
or treatment-resistant schizophrenia. Our target population represents the
largest group of schizophrenia patients; these patients are frequently
hospitalized and receive ~20% of all psychiatric inpatient treatments in
Germany. A total of 220 patients from several departments of psychiatry with
acute schizophrenia will be randomized to a double-blind, eight-week
treatment with either clozapine or olanzapine. The primary endpoint is the
number of patients in symptomatic remission at the end of week eight
according to the international consensus criteria (‘Andreasen criteria’).
Secondary endpoints and other assessments include e.g. symptom severity,
disease severity, global functioning, cognition, side-effect dimensions and
patients’ and relatives’ view on treatment.

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 10 of 71
Summary
Achieving symptomatic remission quickly after the onset of psychotic symptoms is the critical
objective in schizophrenia treatment and determines the subsequent disease course. In this context,
only every second patient with acute schizophrenia achieves symptomatic remission within three
months of initiating antipsychotic treatment, meaning that half of patients do not achieve this key
objective. Increasing the likelihood to achieve symptomatic remission in acute schizophrenia will
improve the overall outcome, reduce disease-associated burden and potentially prevent mid- and
long-term disease chronicity. With this randomized, double-blind, parallel-group multicentre trial we
aim to provide evidence for the superior efficacy of the ‘last resort’ antipsychotic clozapine compared
to one of the most effective second-generation antipsychotics (SGAs), olanzapine, in acute
schizophrenia patients who do not meet the criteria for treatment-naive or treatment-resistant
schizophrenia. Our target population represents the largest group of schizophrenia patients; these
patients are frequently hospitalized and receive ~20% of all psychiatric inpatient treatments in
Germany. A total of 220 patients from several departments of psychiatry and psychotherapy with
acute schizophrenia will be randomized to a double-blind, eight-week treatment with either clozapine
or olanzapine. The primary endpoint is the number of patients in symptomatic remission at the end
of week eight according to the international consensus criteria (‘RSWG criteria’, ‘Andreasen criteria’).
Secondary endpoints and other assessments include e.g. symptom severity, disease severity, global
functioning, cognition, side-effect dimensions and patients’ and relatives’ view on treatment.

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 11 of 71
1. Introduction and aims
1.1 Background, initial situation and medical problem
Despite the significant social and healthcare burden caused by the difficult therapy of schizophrenia,
no novel antipsychotic concepts have been established in psychiatric care since the mid-1990s.
Even worse, the industry is increasingly withdrawing from the field of schizophrenia research (Insel
and Sahakian 2012). Therefore, urgently needed new antipsychotics with the capacity to improve
outcomes and remission rates in schizophrenia are unlikely to be introduced in the coming years.
Remission rates in schizophrenia trials have been reported to be in the range of ~30% to 50%
(Leucht, Beitinger et al. 2007, Boter, Peuskens et al. 2009, Lambert, Karow et al. 2010, AlAqeel and
Margolese 2012) and one in two patients is discussed to be at risk of a chronic disease course (An
der Heiden and Hafner 2010). One recently published meta-analysis investigated 167 double-blind
randomized-controlled trials comparing antipsychotics to placebo with 28102 schizophrenia patients
and was able to show a minimal response in 51% of the patients treated with antipsychotics (30%
with placebo) and a good response, which is principally related to remission, in only 23% of all
patients (14% with placebo) (Leucht, Leucht et al. 2017). However, the most effective antipsychotic
clozapine was excluded from this meta-analysis. A related meta-analysis investigated the response
rates in first-episode schizophrenia patients and showed a minimal response in 81.3% and a good
response in 51.9% of all patients (Zhu, Li et al. 2017). These sources of evidence show that
schizophrenia patients have a high likelihood of response during their first-episode, but that response
rates drop after the first relapse. As many patients do not remit after the first relapse and as remission
is related to good functional outcome and improved quality of life (Lambert, Karow et al. 2010), new
evidence-based strategies are urgently needed to increase remission rates. An increase in remission
will not only improve the aforementioned outcome domains, but is likely to reduce the risks for
hospitalization and treatment discontinuation.
In this context, the early application of clozapine, the most effective antipsychotic, has been
discussed now for years as one possibility to improve outcome in relapsing schizophrenia
(Remington, Agid et al. 2013). This issue of a potential indication extension of clozapine from being
the ‘last-resort antipsychotic’ to being applied at an earlier stage is supported by several lines of
evidence (Remington, Agid et al. 2013), but respective clinical trials to test these hypotheses are
lacking. This application of clozapine as a second-line treatment (Remington, Agid et al. 2013) is
discussed on the basis of the observations that (1) contrary to treatment with the first antipsychotic,
the response rates for consecutive treatments with non-clozapine antipsychotics are low, (2)
clozapine is the most effective antipsychotic for treatment-resistant cases and (3) clozapine
treatment is associated with earlier and longer remission intervals.
As indicated above, sufficiently powered clinical trials investigating the proposed superior efficacy of
early clozapine application compared to standard treatment are lacking. A recently published
retrospective cohort study confirmed the hypothesized superior effectiveness of clozapine compared
to standard antipsychotics with regard to time to hospitalization and risk for treatment discontinuation
(Stroup, Gerhard et al. 2015), and clozapine is still the gold standard in treatment-refractory cases
(DGPPN 2006, Buchanan, Kreyenbuhl et al. 2010, Hasan, Falkai et al. 2012). Moreover, one
nationwide cohort-study investigated the real-world effectiveness of 29823 patients with
schizophrenia and showed that clozapine treatment was associated with the lowest rates of
treatment failure compared to all other oral antipsychotics (Tiihonen, Mittendorfer-Rutz et al. 2017).
In contrast to guideline recommendations (DGPPN 2006, Buchanan, Kreyenbuhl et al. 2010, Hasan,
Falkai et al. 2012), in clinical practice clozapine use is delayed by many years, making clear that
more convincing evidence is needed to overcome this discrepancy (Howes, Vergunst et al. 2012).

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 12 of 71
The EARLY trial will
(1) allow for novel evidence-based recommendations,
(2) foster the future risk-benefit evaluation of clozapine use in acute schizophrenia and
(3) help to decrease the time lag before clozapine is introduced.
(4) will test whether low-dosages of clozapine are effective in achieving symptomatic
remission
(5) will provide a comprehensive risk-benefit-assessment of the early application of
clozapine
We hypothesize that the early introduction of clozapine in patients with acute schizophrenia will result
in higher remission frequencies and thus reduce the risk to develop unfavourable disease outcomes.
In summary, there is broad evidence that clozapine is an effective antipsychotic that can be safely
used in non-treatment-refractory patients and that has superior effectiveness in real-world settings.
However, high-quality clinical trials investigating the role of clozapine in acute relapsing
schizophrenia are lacking (second-line treatment). Such an approach also has to specifically address
the potentially high levels of side effects and AEs to allow for a comprehensive risk-benefit
evaluation.
1.2 Rationale for the EARLY study
There is a pressing need for new treatments that increase remission rates in patients with relapsing
schizophrenia. Symptomatic remission is associated with better social functioning, a better quality of
life, a more stable disease course and better cognitive functioning (Lambert, Karow et al. 2010). The
possible earlier and broader application of clozapine to reach this objective is extensively discussed
in the field (Remington, Agid et al. 2013, Schooler, Marder et al. 2016), but as detailed in the
paragraph above until now adequately powered and designed trials with a comprehensive
assessment of outcome and burden are lacking. All foregoing trials in non-refractory patients need
to be considered as proof-of-concept trials with no clear rationale to investigate whether clozapine
should be introduced before treatment resistance and no extensive assessment of side effects and
risks. Therefore, our trial is needed to answer the clinically meaningful question whether clozapine
really is superior to other SGAs in non-treatment-refractory schizophrenia patients. Because we will
compare clozapine with olanzapine, one of the most effective SGAs, superiority of clozapine would
impact guideline recommendations and clinical practice. The individual patient and the patient
population will have several benefits from our trial:
(1) in case of superior remission rates in the clozapine arm, a novel treatment option will emerge
that will significantly change guideline recommendations (earlier clozapine application),
(2) increased remission rates will reduce the disease-associated burden of patients and families
and
(3) the broad assessment of side effects and treatment-associated burden will allow an improved
risk-benefit assessment of clozapine.
An increase in the likelihood of remission has the potential to prevent treatment resistance and long-
standing disability in schizophrenia patients with a relapsing disease course and thus to reduce the
overall socioeconomic burden.
Poor remission rates and frequent relapses in schizophrenia lead to the highest direct costs of all
brain disorders in Europe, totaling 29 billion Euros per year (Gustavsson, Svensson et al. 2011).

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 13 of 71
Furthermore, early disease chronicity entails physical and mental comorbidity, enduring social and
vocational exclusion and excess mortality, which alone in Europe amount to annual indirect costs of
65 billion Euros (Gustavsson, Svensson et al. 2011). An estimation of the societal benefit of
increased remission rates highlights the importance of our approach. First, Zeidler et al. (Zeidler,
Slawik et al. 2012) investigated the direct costs generated by patients with unstable (U; N=1449) or
stable (S; N=8497, U/S ratio=15%) courses of schizophrenia and found that direct costs amounted
to €24,377,976 and €34,234,413, respectively. In this context, a 10% reduction of unstable disease
courses would hypothetically translate into a €1,906,455 reduction of direct treatment costs. Second,
clozapine treatment has been linked to faster remission rates (Lieberman, Phillips et al. 2003) and
less treatment discontinuation and hospitalization (Stroup, Gerhard et al. 2015, Schooler, Marder et
al. 2016), making clear that a clozapine-related increase in remission rates will have a direct
socioeconomic benefit. In accordance with the DFG guidelines, we will involve a patient-relatives
organisation (German Federal Association of the Relatives of Mentally-ill Patients (BApK) in our
project. The organization participated in the application process and will assist in investigating the
patients’ view on treatment and also serve as peer-to-peer consultants for study patients. The aspect
of the patients’ view on treatment is especially important, because some studies have found that
psychiatrists and patients have different valuations of clozapine side effects (Hodge and Jespersen
2008), and one Cochrane review states that former trials have neglected patients’ attitude towards
clozapine (Essali, Al-Haj Haasan et al. 2009). Our trial offers the unique possibility for an in-depth
investigation of this important treatment moderator and will help to increase the acceptance of
clozapine by patients, relatives and professionals. Because neither study drug is under patent
protection, no commercial interest for any pharmaceutical company exists. Therefore, this trial has
the potential to change guideline recommendation in an area where no new development from the
perspective of the pharmaceutical industry can be expected.
Moreover, this trial will also allow investigating whether low-dosages of those highly effective
antipsychotics are sufficient to reach symptomatic remission following the discussions of minimal
effective dose treatment for an optimal risk-benefit evaluation. The latter will be possible, because
the EARLY trial includes the so far most extensive side-effect evaluation of clozapine and olanzapine
in a controlled design going far beyond the safety assessment in clinical practice.
In medication-naïve and first-episode patients (first line treatment), an outstanding superiority of
clozapine could not be established (Remington, Agid et al. 2013) and thus the application is restricted
following the risk/benefit evaluation. In treatment-resistant schizophrenia patients (Kane, Honigfeld
et al. 1988) (third line treatment), clozapine is more effective than other antipsychotics, but due to
the progressed stage of the illness, remission rates are not satisfying. Thus, an early clozapine
application as proposed in the EARLY trial in disease stages after the first episode and before
treatment resistance (second line treatment) has the potential to increase remission rates and to
prevent poor disease outcomes and chronicity.
2. Objectives
With this randomized, double-blind, parallel-group, controlled multicentre trial we aim to provide
evidence for the superior efficacy of the ‘last resort’ antipsychotic clozapine compared to one of the
most effective second-generation antipsychotics (SGAs), olanzapine, in acute schizophrenia
patients who do not meet the criteria for treatment-naive or treatment-resistant schizophrenia. The
primary endpoint (see paragraph 4.1) is the number of patients in symptomatic remission at the end
of week eight according to the international consensus criteria (‘RSWG-criteria’). Secondary
endpoints and other assessments (see paragraph 4.2) include e.g. symptom severity, disease
severity, global functioning, cognition, side-effect dimensions and patients’ and relatives’ view on
treatment. Moreover, we will provide a comprehensive safety assessment and we will provide

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 14 of 71
measures to disentangle the relationship between early clozapine response and biological factors
(e.g. brain morphology)
3. Study design
The Effects of Early Clozapine Treatment on Remission Rates in Acute Schizophrenia (EARLY) trial
is a double-blind, randomized, controlled parallel group multicentre trial investigating the
symptomatic remission rates in non-treatment-refractory patients with schizophrenia randomized to
either early clozapine or olanzapine over an 8-week period.
Number of patients:
N = 880 schizophrenia patients will be assessed for eligibility
N = 220 schizophrenia patients will be allocated to trial
N = 220 will be analysed (N = 110 in each study group)
Timelines:
Inclusion of the first patient (FPFV): approx. quarter II 2019
Inclusion of the last patient (LPFV): approx. quarter I 2022
End of the study (LPLV): approx. quarter IV 2022
End of naturalistic follow-up: approx. quarter IV 2024
Integrated final report of primary outcome: approx. quarter IV 2023
Operating requirements for the site:
The following requirements must be available:
Enough place for medication and study records
Access to an emergency room/or and emergency unit
Possibility to refer patients to a department of internal medicine
Condition precedent is an adequate number of patients and sufficient infrastructure for treatment at the study site
EARLY is a randomized, double-blind controlled parallel-group multicenter trial to compare the
relative frequency of patients in the clozapine group in remission (Remission in Schizophrenia
Working Group (RSWG) consensus criteria,) (Andreasen, Carpenter et al. 2005) at week eight
compared to the olanzapine group. 220 acute schizophrenia patients not meeting the criteria for
treatment-naïve or treatment-resistant schizophrenia will be recruited from inpatient and outpatients
units of the participating centers. After providing informed consent, patients will be screened for
eligibility to the study. Potential contraindications concerning the use of any of the study drugs will to
be ruled out before starting treatment in the study. Diagnosis and psychiatric comorbidities will be
investigated with the MINI interview. Physical health will be checked in a physical examination.
Several demographic and clinical variables will be assessed, including year of birth, sex, educational
level, previous psychiatric disorders and duration of untreated psychosis. Thus, this phase will serve
the dual purpose of conducting or completing such examinations and, if necessary, gradually
reducing medication. The screening phase will be completed before the baseline visit and will
preferably not be longer than two weeks. At baseline, the standard rating scales will be used
including PANSS, CGI, CDSS and other scales as displayed in the study visit flow-chart.

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 15 of 71
Figure 1: Schematic Study Flow-Chart. Please see paragraph 8.7 and the Study Flow-Chart there for a detailed visit
plan and paragraph 8.2 for the detailed visit descriptions. Legend: BL= baseline.
4. Study endpoints
For all aims of the study (described in paragraph 1), the primary and secondary endpoints are defined below. The assessment of primary and secondary endpoints will be documented in the eCRF as detailed below.
4.1 Primary endpoint
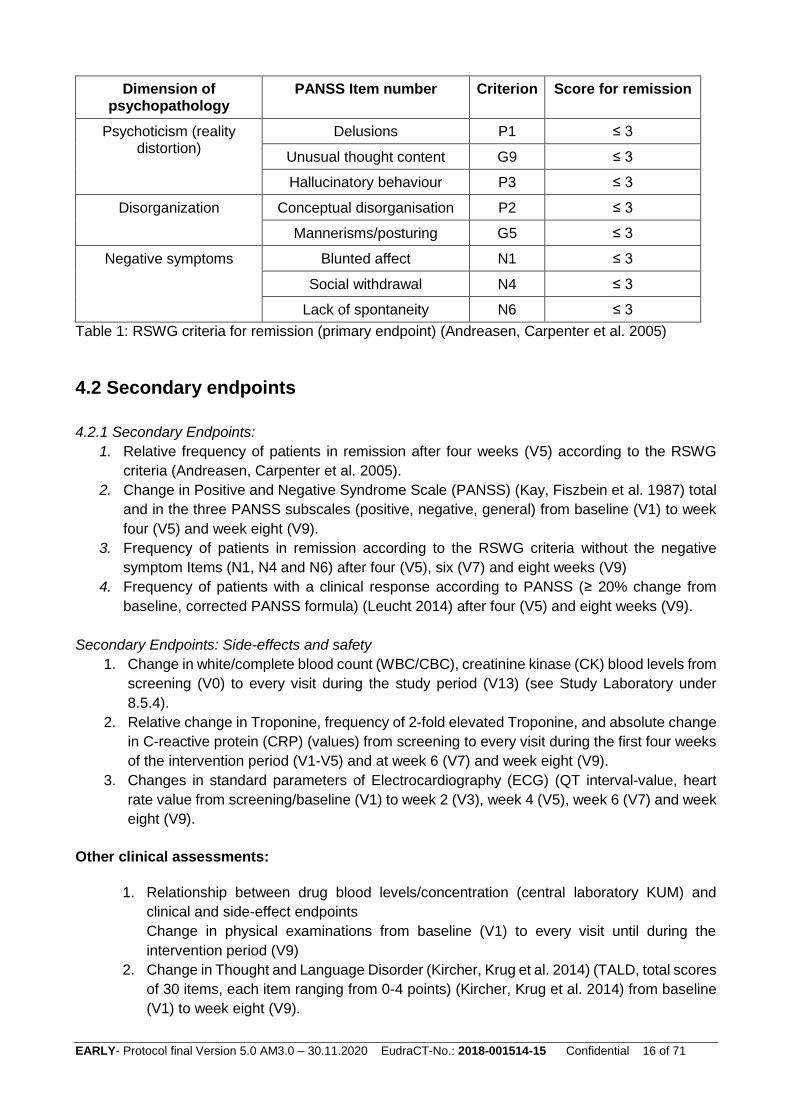
The primary endpoint is the relative frequency of patients in remission after eight weeks according
to the RSWG criteria (Andreasen, Carpenter et al. 2005), without the time criterion (Gorwood,
Peuskens et al. 2012, Leucht 2014). The RSWG criteria (Andreasen, Carpenter et al. 2005) link
DSM-IV symptoms of schizophrenia with items of the Positive and Negative Syndrome Scale
(PANSS) (Kay, Fiszbein et al. 1987). As detailed elsewhere (Andreasen, Carpenter et al. 2005,
Leucht, Beitinger et al. 2007, Beitinger, Lin et al. 2008), three symptom clusters need to be
considered: (1) psychoticism/reality distortion (PANSS items: delusions, unusual thought content
and hallucinatory behaviour), (2) disorganisation (PANSS items: conceptual disorganisation and
mannerisms/posturing), and (3) negative symptoms (PANSS items: blunted affect, social
withdrawal, lack of spontaneity). According to the RSWG criteria, the symptomatic criterion states
that to achieve symptomatic remission all items (see table 1) must be rated as absent or present
only to a mild degree (PANSS value ≤ 3). All eight items of the primary endpoint will be recorded in
the eCRF (scores).
EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 16 of 71
Dimension of psychopathology
PANSS Item number Criterion Score for remission
Psychoticism (reality distortion)
Delusions P1 ≤ 3
Unusual thought content G9 ≤ 3
Hallucinatory behaviour P3 ≤ 3
Disorganization Conceptual disorganisation P2 ≤ 3
Mannerisms/posturing G5 ≤ 3
Negative symptoms Blunted affect N1 ≤ 3
Social withdrawal N4 ≤ 3
Lack of spontaneity N6 ≤ 3
Table 1: RSWG criteria for remission (primary endpoint) (Andreasen, Carpenter et al. 2005)
4.2 Secondary endpoints
4.2.1 Secondary Endpoints:
1. Relative frequency of patients in remission after four weeks (V5) according to the RSWG
criteria (Andreasen, Carpenter et al. 2005).
2. Change in Positive and Negative Syndrome Scale (PANSS) (Kay, Fiszbein et al. 1987) total
and in the three PANSS subscales (positive, negative, general) from baseline (V1) to week
four (V5) and week eight (V9).
3. Frequency of patients in remission according to the RSWG criteria without the negative
symptom Items (N1, N4 and N6) after four (V5), six (V7) and eight weeks (V9)
4. Frequency of patients with a clinical response according to PANSS (≥ 20% change from
baseline, corrected PANSS formula) (Leucht 2014) after four (V5) and eight weeks (V9).
Secondary Endpoints: Side-effects and safety
1. Change in white/complete blood count (WBC/CBC), creatinine kinase (CK) blood levels from
screening (V0) to every visit during the study period (V13) (see Study Laboratory under
8.5.4).
2. Relative change in Troponine, frequency of 2-fold elevated Troponine, and absolute change
in C-reactive protein (CRP) (values) from screening to every visit during the first four weeks
of the intervention period (V1-V5) and at week 6 (V7) and week eight (V9).
3. Changes in standard parameters of Electrocardiography (ECG) (QT interval-value, heart
rate value from screening/baseline (V1) to week 2 (V3), week 4 (V5), week 6 (V7) and week
eight (V9).
Other clinical assessments:
1. Relationship between drug blood levels/concentration (central laboratory KUM) and
clinical and side-effect endpoints
Change in physical examinations from baseline (V1) to every visit until during the
intervention period (V9)
2. Change in Thought and Language Disorder (Kircher, Krug et al. 2014) (TALD, total scores
of 30 items, each item ranging from 0-4 points) (Kircher, Krug et al. 2014) from baseline
(V1) to week eight (V9).

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 17 of 71
3. Change in Clinical Global Impression scale (CGI) (Endicott, Spitzer et al. 1976) score
(scores from 1-7) from baseline (V1) to week four (V5) and week eight (V9) and to the
follow-up visits.
4. Change in Personal and Social Performance scale (PSP) (total score from 1-100)
(Nasrallah, Morosini et al. 2008) from baseline (V1) to week eight (V9) and to the follow-
up visits.
5. Change in Global Assessment of Functioning scale (GAF) scores (total score from 0-100)
from baseline (V1) to week four (V5) and week eight (V9).
6. Change in Calgary Depression Scale for Schizophrenia (CDSS) (Addington, Addington
et al. 1993) total score (total score from 0-27) from baseline (V1) to week four (V5) and
week eight (V9).
7. Trail-Making Test A and B (total scores for speed and errors). Change in total TMT scores
from baseline (V1) to week eight (V9).
8. Change in suicidal ideations based on the InterSePT scale (ISST) (total score)
(Lindenmayer, Czobor et al. 2003) from baseline (V1) to week eight (V9).
9. Change in quality of life measures according to the abbreviated quality of life enjoyment
and satisfaction questionnaire (Q-LES-Q-18) (total score) (Ritsner, Kurs et al. 2005) from
baseline (V1) to week eight (V9) and to the follow-up visits.
10. Change in SF-12 score items (12 separate items) (Ware, Kosinski et al. 1996) from
baseline (V1) to week eight (V9).
11. Change in Drug Attitude Inventory (DAI10) (total score) (Stjernsward, Persson et al. 2013)
scores from baseline (V1) to week eight (V9).
12. Change in Subjective Wellbeing under Neuroleptics short form (SWN-K) (Naber 1995)
(Naber 1995) total score (from baseline (V1) to week eight (V9)
13. Attitude of patients towards participation in clinical research and towards the treatment
with olanzapine and clozapine at baseline (self-developed questionnaire) in collaboration
with BApK (a relatives-driven pan-organisation that represents the interests of relatives
and persons affected by mental illness) at screening. Here, a likert-scale will be used to
present descriptive statistics.
Other assessments- Side effects and Safety:
1. Changes in blood pressure and heart rate (values) from baseline (V1) to every visit during
the intervention period (V9).
2. Changes in Glasgow Antipsychotic Side-effect Scale for Clozapine (GASS for Clozapine)
(Hynes, Keating et al. 2015) scores from baseline (V1) to week 2 (V3), week 4 (V5) and
week eight (V9).
3. Changes in the St Hans Rating Scale (SHRS) (Gerlach, Korsgaard et al. 1993) (Gerlach,
Korsgaard et al. 1993) scores from baseline (V1) to week eight (V9).
4. Week eight Changes in Cleveland Clinic Constipation Score (CCCS) (Agachan, Chen et
al. 1996) scores from baseline (V1) to week 2 (V3), week 4 (V5) and week eight (V9).
5. Changes in the Barnes Akathisia Rating Scale (BARS) (Barnes 1989) scores from
baseline (V1) and week eight (V9)
6. Change in fasting glucose, cholesterol, HDL (values) from baseline (V1) to week 4 (V5)
and week eight (V9).
7. Change in weight and BMI from baseline (V1) to week four (V5) and week eight (V9)
8. Change in number of cigarettes from baseline (V1) to week two (V3), week four (V5),
week five (V6) and week eight (V9).

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 18 of 71
9. Changes in standard parameters of Electroencephalography (EEG) (e.g. frequency
bands) from baseline (V1) to week eight (V9).
Optional activities
Additional facultative assessments: (these data are not part of the study database, see optional activities 8.5.6)
Changes in parameters of transthoracic echocardiography (TTE) from baseline (V1) to week
eight (V9) (optional) (such as, e.g. change in left ventricular ejection fraction (LVEF) in %,
inter-ventricular septum and posterior wall thickness in mm, chamber dimensions
(sizes/diameters of left ventricle, left atrium, right ventricle in cm), RV function (tricuspid
annular plane systolic excursion: TAPSE)
Changes in 30 min ECG parameters from baseline (V1) to week eight (V9) (optional) (change
in 30min-heart rate variability scores)
MRI/rsMRI (facultative)
Changes in brain morphology and functions (MRI, rs-fMRI) from baseline (V1) to week eight
(V9) (optional). Longitudinal MRI/rs-fMRI analyses will be performed using the most recent
version of SPM to detect volumetric (Redlich, Opel et al. 2016, Hasan, Wobrock et al. 2017)
and functional network changes (Hahn, Kircher et al. 2015) over time. Moreover, baseline
MRI/fMRI will be used to develop single-subject predictors based on MVPA approaches
(Hahn, Kircher et al. 2015, Koutsouleris, Wobrock et al. 2017, Redlich, Burger et al. 2017).
For MRI/rsMRI, TTE and 30 min ECG monitor, we will document in the eCRF whether these
measures have been performed or not. All optional activities can be performed with a range of –max.
7 days before baseline, as long as informed consent was given a priori.
Missing data of variables defined as other assessments and optional activities will not result in a
protocol violation.
Investigators will be encouraged to keep patients in the trial for the full of 8 weeks study period and
to perform V9 regardless of them deciding to stay on the study medication or to discontinue the
study. Patients do not automatically drop out from the study if they discontinue medication, they will
be invited for the same visits as patients who did not discontinue medication (with the exclusion of
patients who have withdrawn consent).
5. Study population
5.1 Inclusion criteria
1. Age 18 to 65 years
2. Signed informed consent
3. DSM-V diagnosis of schizophrenia confirmed by the Mini International Neuropsychiatric
Interview

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 19 of 71
4. At least one documented prior hospitalization due to the illness in the medical history (the
current hospitalization can be considered as ’prior’ hospitalization if its ≥ 4 weeks) at
screening
5. For treatment-naïve patients (defined as no previous antipsychotic treatment or a maximum
of 30 days of treatment), an antipsychotic treatment attempt of at least 30 days with an
antipsychotic in a therapeutic dose according to local guidelines other than clozapine and
olanzapine before the screening phase is needed. For non-treatment-naïve patients (defined
as having been treated for more than 30 days with an antipsychotic), a discontinuation of a
foregoing antipsychotic treatment prior to the screening phase within a maximum of six
months (=180 days) is possible (corresponding to the estimated average time for an
antipsychotic washout phase and the expected time to develop a relapse of the disease). For
patients being treated with a long-acting antipsychotic (other than PP3M), an inclusion is
possible if inclusion date corresponds to the planned date of the next injection plus five to
seven days. For patients being treated with oral olanzapine, an inclusion is possible if this
treatment has lasted for no longer than 2 weeks prior to inclusion and if exclusion criteria 8
is not fulfilled.
6. Clinical need for a medication switch because of clinical inefficacy or side-effects or clinical
need for a reintroduction of an antipsychotic treatment after treatment discontinuation prior
to the screening phase (see 5.)
7. Moderate symptomatology on the PANSS, defined as a score ≥ 4 for two or more symptoms
from P1-P7 or a score of ≥ 6 for one symptom from P1-P7 (minimum threshold definition) at
screening
8. Male participants and female participants who are not capable of bearing children or who use
a method of contraception that is medically approved by the health authority of the respective
country at screening
This includes:
a. A woman who is not capable of bearing a child is defined as follows: post-menopausal
(12 months natural (spontaneous) amenorrhea or 6 months spontaneous
amenorrhea with serum-FSH-values (follicle-stimulating hormone) of >40 mIU/mL); 6
weeks after a bilateral ovariectomy with or without hysterectomy or sterilization by
means of tubal ligation
b. A woman capable of bearing child is defined as follows: a woman who is
physiologically capable of becoming pregnant, including women whose occupation,
lifestyle or sexual orientation exclude sexual intercourse with a male partner and
women whose partners have been sterilized by vasectomy or other measures
c. Medically-approved methods of contraception can include the following: hormonal
contraceptives, intrauterine device and double barrier method. Acceptable preventive
measures can include total abstinence at the discretion of the investigator, in cases
where compliance is ensured because of the study participant’s age, occupation,
lifestyle or sexual orientation. Periodical abstinence (e.g. calendar, ovulation,
symptothermal methods or abstinence until the 4th day after the ovulation) as well as
coitus interruptus are not acceptable methods of contraception
d. A reliable method of contraception (CTFG guideline) must be used for the entire
duration of the study

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 20 of 71
5.2 Exclusion criteria
1. Patients who are not suitable for the study in the opinion of the investigator (including acutely
suicidal patients)
2. Patients who are unable to give informed consent
3. Coercive treatment at the time of study inclusion
4. White blood cell count (WBC) at inclusion not meeting the requirements for clozapine use in
Germany according to the SmPC. Patients must have normal leukocyte findings (white blood
cell count ≥ 3500/mm3 (≥ 3.5x109/l), and Absolute Neutrophil Count (ANC) ≥ 2000/mm3 (≥
2.0x109/l) at the screening visit
5. The presence of one or more of the contraindications against any of the study drugs as
mentioned in the SmPC
6. Treatment-naïve or treatment-resistant schizophrenia. Treatment-naïve will be defined as no
previous antipsychotic treatment or a maximum of 30 days of treatment. Treatment
resistance is defined as two antipsychotic trials (with antipsychotics from two different
chemical classes) for a period of ≥ 6 weeks with CPZ equivalent doses ≥ 600 mg/day (Howes,
McCutcheon et al. 2017), both of which took place immediately before the screening phase
7. Diagnosis of primary substance dependency other than nicotine: exclusion alcohol
dependency via AUDIT-screening (Bohn, Babor et al. 1995, Babor 2001) and DSM V criteria
(MINI-interview); exclusion of other drug dependencies other than alcohol and nicotine: drug
screening of urine and DSM V criteria (MINI-interview: patient fulfilling early (> 3 months) or
sustained (>12 months) remission criteria and/or with low severity of substance use disorder
according to MINI (DSM-V) are eligible for the study)
8. Documented previous non-response to an 8-week drug trial with olanzapine or any
documented previous treatment with clozapine
9. Documented intolerance to one of the study drugs
10. Pregnancy (incl. positive blood pregnancy test) / lactation (female patients)
Existing legal representatives for health care issues have to be informed and will also sign the IC or
will give their agreement via speaking to the responsive study doctor. An agreement (signed IC) from
the respective legal representative for health care issues can also obtained via fax or as a scan via
e-mail. This contact will be documented as a “Note to file”.
5.3 Sample size calculation
The sample size calculation was performed on the basis of previous studies, meta-analyses and
systematic reviews (Leucht, Beitinger et al. 2007, Galderisi, Davidson et al. 2009, Lambert, Karow
et al. 2010, AlAqeel and Margolese 2012), which indicate remission rates of approximately 30% after
four weeks (Gorwood, Peuskens et al. 2012) for antipsychotics in acute schizophrenia and mean
remission rates of ~ 50% after 8 to 12 weeks. Because olanzapine is one of the most effective SGAs
and its efficacy is similar to or even higher than that of amisulpride (Lieberman, Stroup et al. 2005,
Kahn, Fleischhacker et al. 2008, Leucht, Cipriani et al. 2013), we assume a remission rate of 55%
in the non-clozapine group after eight weeks.
Assuming that 55% (= p2) of acute schizophrenia patients will remit after treatment with olanzapine
and assuming a superiority of clozapine in the range of 10% to 30% in achieving remission (Kane,

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 21 of 71
Honigfeld et al. 1988) in severely affected patients, we expect that clozapine will be associated with
a ≥20% (= p1 – p2) increased likelihood of achieving remission compared to non-clozapine
treatment. A two-group Chi² test (α = 0.05, two-sided, power [1-β]: 80%) will detect a difference
between groups after adjusting p1 (clozapine) to 0.74 and p2 (olanzapine) to 0.56 (odds ratio of
2.236) when the sample size in each group is 110 (computed by nQuery Advisor 7.0). Thus, the total
required sample size is 220. The adjustment of relative frequencies is due to the conservative
assessment of the primary outcome (as described in the next paragraph) assuming less than 2%
losses to follow-up with no primary outcome data. As we are aiming at evaluating the primary
outcome in every participant, irrespective whether he or she discontinues treatment or not, the
estimated rate of 2% losses to follow-up/drop-outs is realistic. As in other double-blind schizophrenia
trials with an intervention period of 8 weeks, we expect that 20% to 40% patients discontinue
treatment due to any reasons.
The analysis will be performed in the ITT population, which will include all patients as randomized.
To ensure a conservative assessment of the primary outcome, losses to follow-up in the
experimental group will be rated as failures, while those in the control group will be rated as
successes. This approach is designed to avoid a beneficial bias for the experimental group which
might be induced by non-compliance and losses to follow-up. As a consequence, there is no need
to include further participants or increase sample size. Study participants will be allowed to
discontinue the study at any time and without any reason, in accordance with national and
international guidelines for the handling of missing data (EMA 2010, BfArM 2011). We will assess
the primary outcome criterion for study participants that did not drop out but discontinued study
treatment as long as the visit corresponding to V9 (with an obligatory assessment of the RSWG
criteria) is performed as defined in paragraph 8.2. In our clozapine sample we considered the
probability of potential life-threatening events, i.e. agranulocytosis and myocarditis. Agranulocytosis
has an estimated event rate in the literature of 1% (0.01) and myocarditis of 0.1% (0.001) (De Fazio,
Gaetano et al. 2015). Consequently, the probability to not observe any of these events in our sample
of 110 patients is 0.99110 = 33% for agranulocytosis and 0.999110 = 90% for myocarditis. We used
the sample size of the clozapine group (n=110) as the exponent and the literature-based likelihood
for the complement of the incidence as basis for this calculation.

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 22 of 71
6. Treatment of subjects
6.1 Investigational products
Subjects participating in the current study will be randomized to one of two arms:
- Clozapine or
- Olanzapine
Treatment following randomization is double-blind to reduce the risk for expectation effects and to
main the highest quality of head-to-head studies. Study medication will be provided centrally by the
pharmacy of the University of Heidelberg. The dose ranges of the study medication in acutely ill
patients with schizophrenia are well documented. The dose range of clozapine will be 75 – 600
mg/day and for olanzapine the dose range will be 2.5 – 20 mg/day. The distribution of the doses
over the day will be at the discretion of the investigators, but it is recommended to give study
medication at least twice a day according to the description below.
Olanzapine as comparator
As the objective of the EARLY trial is to compare early clozapine-treatment to non-clozapine
treatment in a clinically relevant setting, we will use one highly effective SGA with a comparable side-
effect profile to maintain the blind. Clozapine and olanzapine are very comparable in terms of weight-
gain/appetite increase, motor-side effects, sedation and obstipation – these side-effects may allow
unblinding and by comparing those drugs, this risk is reduced. In the population of acute and chronic
schizophrenia, olanzapine can be considered as one of the most effective antipsychotic agents. In
the CATIE-trial, olanzapine had compared to three other SGAs and one FGA the longest time to
treatment discontinuation (Lieberman, Stroup et al. 2005, Swartz, Stroup et al. 2008). In the largest
available network meta-analysis, olanzapine received a top ranking in terms of overall efficacy and
all-cause treatment discontinuation (Leucht, Cipriani et al. 2013). Evidence-based treatment
guidelines also confirm the high efficacy of olanzapine in acute schizophrenia (DGPPN 2006,
Buchanan, Kreyenbuhl et al. 2010, Hasan, Falkai et al. 2012). As detailed below, clozapine and
olanzapine have a comparable side-effect profile and one previous trial using both agents showed
that a double-blind procedure for this head-to-head comparison is feasible (Meltzer, Bobo et al.
2008).
6.2 Titration schemes
The titration phase starts at day 1 (V1) and may last until day 14 (V3). During this phase, prestudy
antipsychotics have to be tapered down and study medication will be started at day 1. It is
recommended to be at least at stage 2 at the end of the titration phase (exceptions are described in
the text below). Dosages higher than stage 8 are not allowed. Please see Table 2, Table 3 and Table
4 for the schedule of capsules. According to the 2018 published (Konsultationsfassung) national
schizophrenia guideline (DGPPN 2018), a cross-taper strategy with reduction of the foregoing
antipsychotic medication is recommended. According to the guideline, this is particular important for
the start and slow up-titration of clozapine. This guideline-based procedure is recommended to
reduce the risk of antipsychotic-withdrawal-symptoms or a worsening of psychosis. Moreover, it
allows a slow up-titration of the new antipsychotics, which is from particular importance for clozapine.
Furthermore, Galling et al. (Galling, Roldan et al. 2017) analyzed data of 20 clinical trials that
investigated clozapine combination strategies with a first- or second-generation antipsychotic. In
these trials, clozapine was combined with a second antipsychotic. As a consequence, parallel

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 23 of 71
antipsychotic medication alongside clozapine has been extensively investigated in international
clinical trials. In EARLY, no combination of two antipsychotics is allowed beyond V3 – From V1 to
V3 the combination is needed for the guideline-recommended cross-taper strategy. The add-on
treatment with haloperidol in the first two weeks of the study (see 6.3) as rescue medication is
established in clinical practice and was used in previous clinical trials using clozapine (Meltzer, Bobo
et al. 2008).
Clozapine will be started with 12.5 mg/day and dosages should be increased a maximum of 1
capsule per day of the titration blister (25 mg/capsule) from the day after start of titration during the
first week. The dose has to be increased by using capsules of the titration blister until stage 1 (75
mg/day) has been reached, which should be preferably 3 to 7 days after day 1. Next, dosage should
be up-titrated until stage 2 (150 mg/day) has been reached. Stage 2 is the minimal target dosage in
this trial and dosage below stage 2 (namely stage 1) is only permitted due to tolerability reasons and
this down titration must be documented. Stage 2 should be reached 3 to 7 days after stage 1. From
stage 2, a dose increase to the next stage is possible by using titration capsules (up to 2/day) and
increasing the dosage by maximal 1 capsule/day). Between each stage a minimum of 3 days and a
maximum of 7 days for up-titration should be fulfilled. It is not allowed to use the capsules of the next
stages without the aforementioned up-titration steps. The hierarchy of the following rules is:
1. From stage 2 onwards, the use of at least two capsules per day is obligatory.
2. In general, the lowest possible amount of capsules should be used if tolerated
3. The titration capsules are not meant for maintenance treatment and should only be used
for up-titration
4. Titration can be slowed or stopped below the target dose if subjects cannot tolerate the
standard titration schedule because of adverse effects
5. If a dose reduction is needed, a dose reduction to the stage below is recommended
without down-titration.
A daily dosage of < 75mg (stage 1) for > 7 consecutive days constitutes a protocol violation. A
protocol deviation must be recorded. The visit corresponding to V9 must be scheduled for week eight
where RSWG criteria must be assessed. There is no overall target dose, but as indicated above
stage 2 (150 mg/day) should be the minimal dose and it is not allowed to give a dosage above stage
8 (600 mg/day). In our study, a daily dosage over 600mg/d for > 3 consecutive days constitutes a
protocol violation. A protocol deviation must be recorded. The visit corresponding to V9 must be
scheduled for week eight where RSWG criteria must be assessed. 600 mg/day is below that of trials
conducted in treatment-refractory patients, which allowed up to 900 mg/day. However, the mean
dose actually reached in the double-blind treatment-refractory patients trials were e.g. 400 mg
(Meltzer, Bobo et al. 2008), 304 mg (Tollefson, Birkett et al. 2001) or 291 mg (Bondolfi, Dufour et al.
1998) and one recent meta-analysis analyzed trials of treatment-resistant schizophrenia patients
and showed that the mean clozapine dose was only 392 mg/d (Samara, Dold et al. 2016).
Furthermore, the patients in our study are not treatment resistant, so we do not expect them to
require such high doses as the treatment-resistant patients in the earlier studies.
Dosage can be adjusted to higher or lower dosages if a patient fails to improve or if patients develop
relevant side-effects.
Olanzapine will be started with 0 mg/day (placebo) to maintain the blind between both treatment
arms. Dosages should be increased a maximum of 1 capsule/day of the titration blister from the day
after start of titration during the first week. The dose has to be increased by using capsules of the
titration blister (0mg/capsule, placebo) until stage 1 (2.5 mg/day) has been reached, which should
be preferably 3 to 7 days after day 1. Next, dosage should be up-titrated until stage 2 (5 mg/day)
has been reached. Stage 2 is the minimal target dosage in this trial and dosage below stage 2

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 24 of 71
(namely stage 1) is only permitted due to tolerability reasons and this down titration must be
documented. Stage 2 should be reached 3 to 7 days after stage 1. From stage 2, a dose increase
to the next stage is possible by using titration capsules (up to 2/day) and increasing the dosage by
maximal 1 capsule/day). Between each stage a minimum of 3 days and a maximum of 7 days for
up-titration should be fulfilled. It is not allowed to use the capsules of the next stages without the
aforementioned up-titration steps. There is no overall target dose, but as indicated above stage 2 (5
mg/day) should be the minimal dose and it is not allowed to give a dosage above stage 8 (20
mg/day). Dosage can be adjusted to higher or lower dosages if a patient fails to improve or if patients
develop relevant side-effects. Titration can be slowed or stopped below the target dose if subjects
cannot tolerate the standard titration schedule because of adverse effects. For uptitrating the
capsules of the titration blister have to be used according to the described procedures, but there is
no need for down-titrating. If a dose reduction is needed, the capsules of the lower stage can be
used instead of those of the current stage. A daily dosage of <2.5mg (stage 1) for > 7 consecutive
days constitutes a protocol violation. A protocol deviation must be recorded. The visit corresponding
to V9 must be scheduled for week eight where RSWG criteria must be assessed. A daily dosage
over 20 mg/d for > 3 consecutive days constitutes a protocol violation. A protocol deviation must be
recorded. The visit corresponding to V9 must be scheduled for week eight where RSWG criteria
must be assessed.
The hierarchy of the following rules is:
1. From stage 2 onwards, the use of at least two capsules per day is obligatory
2. In general, the lowest possible amount of capsules should be used if tolerated
3. The titration capsules are not meant for maintenance treatment and should only be used
for up-titration
4. Titration can be slowed or stopped below the target dose if subjects cannot tolerate the
standard titration schedule because of adverse effects
5. If a dose reduction is needed, a dose reduction to the stage below is recommended
without down-titration.
Thus, the titrations schemes of both drugs are the same to maintain the blind. Blisters will be color-
coded to improve the usability. The recommended dose range in this trial is stage 4 (300 mg
clozapine or 10 mg olanzapine). Patient-specific used and unused blisters should be collected at
V9/Early Termination Visit. For this reason, it is recommended that the study staff collects remaining
(both used and unused) blisters.
Table 2: Stages and dose ranges of the trial. The rules for using the lowest dosages (clozapine 75 mg/ olanzapine 2.5
mg) in cases of lacking tolerability are indicated in the text above.
Stage 1 75 mg Clozapine or 2.5 mg Olanzapine, earliest timepoint: day 4
Stage 2 150 mg Clozapine or 5 mg Olanzapine, earliest timepoint: day 7
Stage 3 225 mg Clozapine or 7.5 mg Olanzapine, earliest timepoint: day 10
Stage 4 300 mg Clozapine or 10 mg Olanzapine, earliest timepoint: day 13
Stage 5 375 mg Clozapine or 12.5 mg Olanzapine, earliest timepoint: day 16
Stage 6 450 mg Clozapine or 15 mg Olanzapine, earliest timepoint: day 19
Stage 7 525 mg Clozapine or 17,5 mg Olanzapine, earliest timepoint: day 22

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 25 of 71
Stage 8 600 mg Clozapine or 20 mg Olanzapine, earliest timepoint: day 25
Table 3: Minimal and maximal dose ranges for the study visits. The rules for using the lowest dosages (clozapine 75 mg/ olanzapine 2.5 mg) in cases of lacking tolerability are indicated in the text above. Maximal doses can be higher than the given dose ranges (but must be always according to defined study medication titration scheme), if visits are performed within the range of max. + 2 working days.
V1 (day 0-1) 12,5 mg Clozapine or 0 mg Olanzapine
V2 (day 7) 75-150 mg Clozapine or 2.5-5 mg Olanzapine
V3 (day 14) 150-325 mg Clozapine or 5-10 mg Olanzapine
V4 (day 21) 150-500 mg Clozapine or 5-15 mg Olanzapine
V5 (day 28) 150-600 mg Clozapine or 5-20 mg Olanzapine
V6 (day 35) 150-600 mg Clozapine or 5-20 mg Olanzapine
V7 (day 42) 150-600 mg Clozapine or 5-20 mg Olanzapine
V8 (day 49) 150-600 mg Clozapine or 5-20 mg Olanzapine
V9 (day 56) 150-600 mg Clozapine or 5-20 mg Olanzapine
Table 4: Description of capsule and blisters
Blister Contains To be used for
Starting blister Red-Label
12.5 mg clozapine or placebo
First day of the treatment phase (first day with study medication only!)
Titration blister Blue-Label
25 mg clozapine or placebo
Titration at the start of the study and uptitration between stages
Maintenance blister I Yellow-Label
75 mg clozapine or 2.5 olanzapine
Stage 1, Stage 2, Stage 3, Stage 5 and Stage 7
Maintenance blister II White-Label
150 mg clozapine or 5 mg olanzapine
Stage 3, Stage 4, Stage 5, Stage 6, Stage 7, Stage 8
The blood level of clozapine (and desmethylclozapine) and olanzapine (desmethylolanzapine) will
be measured 2 and 4 weeks after randomization at visits V3 and V5 at the central laboratory of the
Klinikum der Universität München. If the assessment of the blood levels is not possible at these
visits, the respective blood draw for blood level analyses should be performed at the next planned
visit. The reason for the delayed blood draw must be documented in the eCRF. All sites will send
blood to this central laboratory via the Department of Psychiatry and Psychotherapy at the Klinikum
der Universität München. If the reference ranges of the trial drugs have not been reached and
patients have not obtained remission criteria (assessed at visit V5 and visit V7), the dose should be
increased in order to reach blood levels within the recommended range. To avoid unblinding
investigators, patients and raters will only receive blinded qualitative information about the drug
levels. Thus, the rater will be informed if the patient’s blood level is within the therapeutic range or
not and the actual blood level of the investigational drug will be graded according to the following
ranges:

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 26 of 71
1) below (<) recommended therapeutic range
2) lower third within recommended therapeutic range
3) middle third within recommended therapeutic range
4) upper third within recommended therapeutic range
5) above (>) recommended therapeutic range, but below warning threshold
6) warning threshold)
The quantitative information of the drug levels will be stored blinded at the laboratory and included
post hoc in the study data-base after the end of the study for further statistical analyses. In the eCRF
we will document whether blood has been taken for this procedures and the qualitative rating as
indicated above. Investigators will only receive the descriptive information detailed above.
6.3 Use of concomitant medication
Non-permissible medication: The use of any antipsychotic in addition to the study medication is not
permitted from one day after V3 until V9. During the two-week titration phase, the on-demand use of
haloperidol (max. 10 mg/day) is permitted as rescue medication in accordance with previous trials
(Meltzer, Bobo et al. 2008). Pre-study antipsychotics will be tapered down during the titration phase
and from day 15 onwards only study antipsychotics are permitted. The use of haloperidol as a rescue
medication has to be considered as standard care in Germany (especially during clozapine titration
to avoid high doses of benzodiazepines) and the permitted use during the titration phase will reduce
the disease-associated stress and increase the willingness of patients with acute schizophrenia to
participate.
Concomitant medication: Benzodiazepines are allowed (in accordance with the German
recommendation for their use in combination with clozapine (Benkert and Hippius 2013), The
following concomitant medication is allowed:
Lorazepam of max. 4 mg/d, diazepam of max 40 mg/d and oxazepam of max 80 mg/d are
permitted; Lorazepam expidet is not permitted due to possible clozapine interactions.
Biperiden and pirenzepine: The permitted maximum daily dose of biperiden is 4mg/day and
of pirenzepine is 50 mg/day to reduce the risk of anticholinergic side-effects.
Zopiclone up to 7.5 mg/d and zolpidem up to 10mg/d and doxepine (or trimipramine) up to
50 mg/d or mirtazapine (up to 15 mg/day) for sleep disturbances. Investigators should be
aware of a possible increase in anticholinergic side-effects when using doxepine /
trimipramine and of a possible increase in weight gain when using mirtazapine.
An antidepressant treatment is only permitted in cases of clinically relevant depression
(CDSS > 6, depressive symptoms for a minimum of 2 weeks) and only if antidepressants are
used that do not have relevant interactions with any of the study drugs.
The introduction of mood-stabilizers during the trial is not permitted. Pretrial mood-stabilizers
can be continued if the used substances do not have relevant interactions with any of the
study drugs.
All drugs to treat somatic conditions (e.g. weight gain, high blood pressure, tachycardia,
constipation) are permitted if the used substances do not have relevant interactions with any
of the study drugs.

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 27 of 71
Prior to the introduction of concomitant medication, the possibility of clinically relevant drug-
interactions with the medicinal products has to be checked. Other drugs that have relevant
interactions with the study drugs are not permitted. Daily doses of concomitant medication including
start and stop dates need to be documented throughout the study.
6.4 Treatment after the end of the double-blind treatment phase
Unblinding patients at the end of the double-blind treatment phase (until V9) is sensitive, as it is not
acceptable that the advantages of a double blind study are diminished by the possibility to change
previously collected data after the treatment is unblinded. Therefore, a SOP will be implemented
how to continue treatment after the end of the double-blind treatment phase. After visit 9 (day 56),
the physicians being responsible for the further treatment have the possibility to receive an envelope
with the information in which arm the patient has been allocated (unblinding according to protocol).
People involved in the trial will not receive this information. For patients who drop out prior to visit 9,
unblinding according to protocol will also only be possible following the aforementioned principles.
For patients who meet the remission criteria at week eight, it is recommended to stay on the same
drug. For patients who do not meet remission criteria at week eight and being treated with clozapine,
it is recommended to carry on treatment and perhaps increase the dose as clinical knowledge is
available that the effect of clozapine may occur with a delay. For patients being treated with
olanzapine not meeting remission criteria, it is recommended to increase the dose and if not
successful, to switch to clozapine.
However, the decision of any further treatment after visit 9 will be made between the treating
physicians and the patients following national guideline recommendations. The study team will not
be involved in this decision. Clozapine should not be stopped abruptly but tapered down according
to clinical standard procedures. Therefore, our SOP is written to assure that patients at the end of
the double-blind treatment phase will have enough study medication until the next meeting with their
treating physician. However, we are aiming at coordinating the meeting with the treating physician
at the same day of V9. This means that all centers have to have study doctors and must either have
treating physicians or be in contact with those persons.

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 28 of 71
7. Investigational product
Name of the investigational products:
Clozapine
Olanzapine
Placebo (Mannitol/Siliciumdioxid 99.8/0.2 %)
Clozapine general information
Clozapine is an antipsychotic that is used for the treatment and maintenance treatment of acute and
chronic schizophrenia, but usually the use is restricted treatment-resistant schizophrenia and
psychosis during the course of Parkinson’s disease. Please see the SmPC for further information.
Olanzapine general information
Olanzapine is an antipsychotic that is used for the treatment of acute and chronic schizophrenia, for
the treatment of mania and for maintenance treatment of schizophrenia and mania. Please see the
SmPC for further information.
Placebo
Placebo are capsules containing lactose-monohydrate, magnesium stearate and cellulose powder.
Placebo is considered to be without a direct clinical relevant physiological mechanism except the
“placebo effect”.
7.1 Contraindications
Clozapine: According to the SmPC, the following contraindications need to be considered:
Hypersensitivity to the active substance or to any of the excipients listed in section 6.1 of the SmPC
Patients unable to undergo regular blood tests.
History of toxic or idiosyncratic granulocytopenia/agranulocytosis (with the exception of granulocytopenia/agranulocytosis from previous chemotherapy).
History of Clozapine-induced agranulocytosis.
Impaired bone marrow function.
Uncontrolled epilepsy
Alcoholic and other toxic psychoses, drug intoxication, comatose conditions
Severe renal or cardiac disorders (e.g. myocarditis).
Active liver disease associated with nausea, anorexia or jaundice; progressive liver disease, hepatic failure
Paralytic ileus
Treatment with clozapine must not be started concurrently with substances known to have a substantial potential for causing agranulocytosis; concomitant use of depot antipsychotics is to be discouraged.

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 29 of 71
Olanzapine: According to the SmPC, the following contraindications need to be considered:
Hypersensitivity to the active substance or to any of the excipients listed in section 6.1. of the SmPC
Patients with known risk for narrow-angle glaucoma.
Placebo: Contraindications may be known allergic reactions or intolerance of ingredients (mostly cellulose and/or magnesium stearat and/or lactulose).
7.2 Interactions
Please see SmPC’ s for a full description of possible drug interactions.
For clozapine the SmPC describes the following interactions with other medicinal products and
other forms of interactions:
Substances known to have a substantial potential to depress bone marrow function must not
be used concurrently with clozapine
Long-acting depot antipsychotics (which have myelosuppressive potential) must not be used
concurrently with clozapine because these cannot be rapidly removed from the body in
situations where this may be required
Alcohol should not be used concomitantly with Clozapine due to possible potentiation of
sedation.
Clozapine may enhance the central effects of CNS depressants such as narcotics,
antihistamines and benzodiazepines. Particular caution is advised when clozapine therapy
is initiated in patients who are receiving a benzodiazepine or any other psychotropic agent.
These patients may have an increased risk of circulatory collapse, which, on rare occasions,
can be profound and may lead to cardiac and/or respiratory arrest. It is not clear whether
cardiac or respiratory collapse can be prevented by dose adjustment.
Because of the possibility of additive effects, caution is essential in the concomitant
administration of substances possessing anticholinergic, hypotensive, or respiratory
depressant effects.
Owing to its anti-alpha-adrenergic properties, clozapine may reduce the blood-pressure-
increasing effect of norepinephrine or other predominantly alpha-adrenergic agents and
reverse the pressor effect of epinephrine.
Concomitant administration of substances known to inhibit the activity of some cytochrome
P450 isozymes may increase the levels of clozapine, and the dose of clozapine may need to
be reduced to prevent undesirable effects. This is more important for CYP 1A2 inhibitors such
as caffeine (see below), perazine and the selective serotonin reuptake inhibitor fluvoxamine.
Some of the other serotonin reuptake inhibitors such as fluoxetine, paroxetine, and, to a
lesser degree, sertraline, are CYP 2D6 inhibitors and, as a consequence, major
pharmacokinetic interactions with clozapine are less likely. Similarly, pharmacokinetic
interactions with CYP 3A4 inhibitors such as azole antimycotics, cimetidine, erythromycin
and protease inhibitors are unlikely, although some have been reported. Hormonal
contraceptives (including combinations of estrogen and progesterone or progesterone only)
are CYP 1A2, CYP 3A4 and CYP 2C19 inhibitors. Therefore, initiation or discontinuation of
hormonal contraceptives, may require dose adjustment of clozapine according to the

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 30 of 71
individual medical need. Because the plasma concentration of clozapine is increased by
caffeine intake and decreased by nearly 50% following a 5-day caffeine-free period, dosage
changes of clozapine may be necessary when there is a change in caffeine-drinking habit.
In cases of sudden cessation of smoking, the plasma clozapine concentration may be
increased, thus leading to an increase in adverse effects.
Cases have been reported of an interaction between citalopram and clozapine, which may
increase the risk of adverse events associated with clozapine. The nature of this interaction
has not been fully elucidated.
Concomitant administration of substances known to induce cytochrome P450 enzymes may
decrease the plasma levels of clozapine, leading to reduced efficacy. Substances known to
induce the activity of cytochrome P450 enzymes and with reported interactions with clozapine
include, for instance, carbamazepine (not to be used concomitantly with clozapine, due to its
myelosuppressive potential), phenytoin and rifampicin. Known inducers of CYP1A2, such as
omeprazole, may lead to decreased clozapine levels. The potential for reduced efficacy of
clozapine should be considered when it is used in combination with these substances
Concomitant use of lithium or other CNS-active agents may increase the risk of development
of neuroleptic malignant syndrome (NMS).
Rare but serious reports of seizures, including onset of seizures in non-epileptic patients, and
isolated cases of delirium where clozapine was co-administered with valproic acid have been
reported. These effects are possibly due to a pharmacodynamic interaction, the mechanism
of which has not been determined.
Caution is called for in patients receiving concomitant treatment with other substances which
are either inhibitors or inducers of the cytochrome P450 isozymes. With tricyclic
antidepressants, phenothiazines and type 1C anti-arrhythmics, which are known to bind to
cytochrome P450 2D6, no clinically relevant interactions have been observed thus far.
As with other antipsychotics, caution should be exercised when clozapine is prescribed with
medicines known to increase QTc interval, or causing electrolyte imbalance.
For olanzapine the SmPC describes the following interactions with other medicinal products and
other forms of interactions:
Caution should be exercised in patients who consume alcohol or receive medicinal products
that can induce hypotension, bradycardia, respiratory or central nervous system depression
Since olanzapine is metabolised by CYP1A2, substances that can specifically induce or
inhibit this isoenzyme may affect the pharmacokinetics of olanzapine.
o Induction of CYP1A2: The metabolism of olanzapine may be induced by smoking and
carbamazepine, which may lead to reduced olanzapine concentrations. Only slight to
moderate increase in olanzapine clearance has been observed. The clinical
consequences are likely to be limited, but medical monitoring is recommended and
an increase of olanzapine dose may be considered if necessary
o Inhibition of CYP1A2: Fluvoxamine, a specific CYP1A2 inhibitor, has been shown to
significantly inhibit the metabolism of olanzapine. The mean increase in olanzapine
Cmax following fluvoxamine was 54 % in female nonsmokers and 77% in male
smokers. The mean increase in olanzapine AUC was 52 % and 108% respectively.
A lower starting dose of olanzapine should be considered in patients who are using
fluvoxamine or any other CYP1A2 inhibitors, such as ciprofloxacin. A decrease in the

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 31 of 71
dose of olanzapine should be considered if treatment with an inhibitor of CYP1A2 is
initiated.
Activated charcoal reduces the bioavailability of oral olanzapine by 50 to 60 % and should
be taken at least 2 hours before or after olanzapine. Fluoxetine (a CYP2D6 inhibitor), single
doses of antacid (aluminium, magnesium) or cimetidine have not been found to significantly
affect the pharmacokinetics of olanzapine.
Olanzapine may antagonise the effects of direct and indirect dopamine agonists
Olanzapine does not inhibit the main CYP450 isoenzymes in vitro (e.g. 1A2, 2D6, 2C9, 2C19,
3A4). Thus no particular interaction is expected as verified through in vivo studies where no
inhibition of metabolism of the following active substances was found: tricyclic antidepressant
(representing mostly CYP2D6 pathway), warfarin (CYP2C9), theophylline (CYP1A2) or
diazepam (CYP3A4 and 2C19).
Olanzapine showed no interaction when co-administered with lithium or biperiden.
Therapeutic monitoring of valproate plasma levels did not indicate that valproate dosage
adjustment is required after the introduction of concomitant olanzapine.
The concomitant use of olanzapine with anti-Parkinsonian medicinal products in patients with
Parkinson’s disease and dementia is not recommended
Placebo
Side effects are expected to be limited to allergic reactions on ingredients of the applied placebos.
7.3 Manufacturing of Trial Medication
For the production of the verum capsules, the approved medicinal product Clozapine or Olanzapine
tablets will be modified by encapsulation and packing in blister packaging. The tablets used remain
undamaged in this process, apart from the manufacturing of the 12.5 mg Clozapine starting dose
(here clozapine 25 mg tablets will be bisected); the quality of the tablets is not altered by the
encapsulation. The manufacturing process is described in the sIMPDs.
7.4 Storage, Formulation, packaging and labelling
Clozapine and Olanzapine hard capsules as well as placebo hard capsules are preserved when
stored in well-closed, light-resistant containers in a cool place at room temperature not more than
30°.
Study medication will be provided by the central pharmacy (University of Heidelberg).
Labeling of Study Medication
According to GCP-V § 5 (see trial application documents).
Shipping Department
The pharmacist of the study medication is:
Apotheke Universitätsklinikum Heidelberg / Pharmacy University
Hospital Heidelberg Im Neuenheimer Feld 670
69120 Heidelberg

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 32 of 71
Germany
Tel: +49-(0)6221- 56-32819
Fax: +49-(0)6221- 56-5413
Handling Placebo
The placebo capsules will contain mannitol / silica 99.8 / 0.2% (equivalent to the capsule filler
according to DAC / NRF). This will also be included in the Verum capsules in addition to the tablets
for refilling.
7.5 Emergency unblinding
Unblinding of study medication before database hardlock may only occur on an individual basis if
the information can help treat an (S)AE and for safety reasons. The decision to unblinding is at the
discretion of the investigator. For this purpose, investigators will have sealed envelopes comprising
the information on the type of medication stored used for a given randomization number. All sites
will have one sealed envelope for each randomization number. In the case of a medical emergency
as described below, the envelope must be opened and the treatment assignment of a patient will be
unblinded.
Emergency unblinding should be only performed in the following situations:
In case of an emergency treatment of the subject at the investigators discretion
In cases where unblinding is required by local laws or regulations (in case of SUSAR)
In cases where the Safety Monitoring Board decides that unblinding is necessary
When a study site is closed all closed emergency envelopes will be collected and returned to the
sponsor. Opened emergency envelopes will be kept in the Investigator Site File at the site.

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 33 of 71
8. Methods
Patients with a schizophrenia diagnoses will be prescreened for possible participation in this trials.
No study related procedures will take place prior to the informed consent process. Please see below
for a description of all scales and investigations and the Study Flow-chart for the detailed visit plan.
8.1 Randomisation
The study will be performed in a controlled, randomized, double-blind design. Patients will be
randomized at the end of the screening visit to one of the two treatment arms (clozapine or
olanzapine). The randomization lists will be created by MSZ using RANCODE 2015 professional
2015. Randomization will be performed block wise and for each centre. The randomization list will
then be forwarded to the pharmacy for distributing and labelling of concealed medication.
8.2 Description of study visits
Screening (Day -14 to 0): After providing informed consent, patients will be screened for eligibility
to the study. Potential contraindications concerning the use of any of the study drugs (clozapine,
olanzapine, placebo) will be ruled out before starting study treatment. Diagnosis and psychiatric
comorbidities will be investigated with the MINI interview. Physical health will be checked in a
physical examination. An AUDIT interview will be performed. Psychotic symptoms will be rated with
PANSS assessments (PANNS, PANSS RSWG items). Study laboratory will be taken for the first
time to check for contraindication in terms of the use of clozapine and olanzapine. Pre-study
medication will be assessed and stopped before visit 3 (V3). An ECG will be performed. Several
demographic and clinical variables will be assessed, including year of birth, sex, educational level,
marital status, occupation, living circumstances, previous psychiatric disorders and admissions,
duration of untreated psychosis and other variables, (medical history, cardiovascular risk factors). A
pregnancy test will be performed among female patients. Furthermore, a drug screening will be
performed. The screening phase will be completed before the baseline visit and should preferably
not be longer than one week. Randomization will be performed during screening and baseline visit
(V1). Screening visit and baseline visit (V1) can be performed together on the same day. If screening
visit and baseline visit are performed together within a maximum of 7 days, only one ECG is sufficient
instead of two. From baseline visit onwards, a study discontinuation check will be performed to raise
the safety standard of the clinical trial.
Baseline visit (Visit 1- Day 0-1): At baseline, the standard rating scales will be used for the first
time, including CGI, GAF, TALD, PSP, CDSS and other scales as displayed in the Study Flow-Chart,
EEG and ECG will be performed and cognitive functioning assessed. EEG can be performed with a
range of +/-5 working days around day 1. Fagerström-test will be performed (smokers). Number of
cigarettes will be assessed. A physical examination will be performed. Concomitant medication will
be assessed. An interaction check (e.g. via textbooks, tables or commercial software) of current
medication with our study medication will be performed (documented as performed: yes/no in the
eCRF). Optional investigations (MRI, echocardiography, 30 min ECG) will be performed (see the
Study Flow-Chart). Study medication will be dispensed for the first time at day 1. One study
laboratory at Screening is sufficient if the number of days between Screening visit and Baseline visit
is ≤ 3 days.
Visit 2 (Day 7): This visit has the main aim of assessing several safety parameters 1 week after start
of medication (especially WBC). Blood pressure and heart rate will be assessed. A physical

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 34 of 71
examination will be performed. Concomitant medication will be assessed. Enough study medication
will be (re-)dispensed until next visit.
Visit 3 (Day 14): This visit has the main aim of assessing several safety parameters 2 weeks after
start of medication (especially WBC). GASS and CCCS are performed. Moreover, an ECG is
performed within a possible range of +/-2 working days. in the same week of the visit. Number of
cigarettes will be assessed. Moreover, additional blood is taken for blinded drug-level analyses.
Blood pressure and heart rate will be assessed. A physical examination will be performed.
Remaining pre-study medication will be stopped. Concomitant medication will be assessed. Enough
study medication will be (re-)dispensed until next visit.
Visit 4 (Day 21): This visit has the main aim of assessing several safety parameters 3 weeks after
start of medication (especially WBC). Blood pressure and heart rate will be assessed. A physical
examination will be performed. Concomitant medication will be assessed. Enough study medication
will be (re-)dispensed until next visit.
Visit 5 (Day 28): This visit is the first outcome parameter visit and many scales/questionnaires
(CCCS, GASS, CDSS, GAF, CGI, PANSS, PANSS RSWG items) have to be used to determine the
outcome four weeks after the start of the study medication. Moreover, safety parameters are
assessed including WBC, ECG and other parameters. ECG can be performed within a possible
range of +/-2 working days. Moreover, additional blood is taken for blinded drug-level analyses.
Number of cigarettes, weight and BMI, blood pressure and heart rate will be assessed. A physical
examination will be performed. Concomitant medication will be assessed. Enough study medication
will be (re-)dispensed until next visit.
Visit 6 (V6 - Day 35): This visit has the main aim of assessing several safety parameters 5 weeks
after start of medication (especially WBC). Number of cigarettes will be assessed. Blood pressure
and heart rate will be assessed. A physical examination will be performed. Concomitant medication
will be assessed. Enough study medication will be (re-)dispensed until next visit.
Visit 7 (V7 - Day 42): This visit has the main aim of assessing several safety parameters 6 weeks
after start of medication (especially WBC). Moreover, an ECG is performed within a possible range
of +/-2 working days. PANSS RSWG items will be assessed. Blood pressure and heart rate will be
assessed. A physical examination will be performed. Concomitant medication will be assessed.
Enough study medication will be (re-)dispensed until next visit.
Visit 8 (V8 - Day 49): This visit has the main aim of assessing several safety parameters 7 weeks
after start of medication (especially WBC). Blood pressure and heart rate will be assessed. A
physical examination will be performed. Concomitant medication will be assessed. Enough study
medication will be (re-)dispensed until next visit.
Visit 9 (V9 - Day 56): This is the primary endpoint visit and this visit marks the end of the double-
blind treatment phase. The standard rating scales will be used, including PANSS, CGI, TALD, CDSS,
St. Hans Rating Scale (SHRS) and other scales as displayed in the Study Flow-Chart. EEG and
ECG will be performed and cognitive functioning assessed. EEG can be performed with a range of
+/-5 working days around Visit 9. Number of cigarettes, blood pressure and heart rate will be
assessed. A physical examination will be performed. Concomitant medication will be assessed.
Optional investigations (MRI, echocardiography, 30 min ECG) will be performed (see Study Flow-
Chart). Participants and responsible study doctors will be asked to estimate whether the given
participant was in the clozapine or olanzapine treatment arm. Unused and used patient-specific study
medication will be returned.
Safety follow-up I (V10 - Day 63): This visit has the main aim of safety parameters for all patients,
because even when clozapine has been stopped at Visit 9 (which should not be done, see above),
WBC has to be assessed for four 4 weeks after drug withdrawal on a weekly basis. At this visit, only

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 35 of 71
blood is taken. An objective value of WBC will be assessed The concomitant medication is only
documented at a new laboratory-related AE after V9/Early Termination or any SAE.
Safety follow-up II (V11 - Day 70): This visit has the main aim of safety parameters for all patients, because even when clozapine has been stopped at Visit 9 (which should not be done, see above), WBC has to be assessed for four 4 weeks after drug withdrawal on a weekly basis.
At this visit, only blood is taken. An objective value of WBC will be assessed The concomitant medication is only documented at a new laboratory-related AE after V9/Early Termination or any SAE.
Safety follow-up III (V12 - Day 77): This visit has the main aim of safety parameters for all patients, because even when clozapine has been stopped at Visit 9 (which should not be done, see above), WBC has to be assessed for four 4 weeks after drug withdrawal on a weekly basis. At this visit, only blood is taken. An objective value of WBC will be assessed. The concomitant medication is only documented at a new laboratory-related AE after V9/Early Termination or any SAE.
Safety follow-up IV (V13 - EOS - Day 84): This visit has the main aim of assessing safety parameters for all patients, because even when clozapine has been stopped at Visit 9 (which should not be done, see above), WBC has to be assessed weekly for 4 continuous weeks after drug withdrawal. At this visit, only blood is taken. An objective value of WBC will be assessed.
The concomitant medication is only documented at a new laboratory-related AE after V9/ Early Termination or any SAE.
The double-blind study phase ends with Visit 9 and patients will receive regular clinical treatment.
There is some flexibility in the visit window intervals and visits can be performed with a range of +/-
2 working days as long as it is ensured that WBC is controlled on a weekly basis (once a week),
dose ranges of study medication are not violated (see Table 2) and enough study medication is
available. If one of the questionnaires or interviews that are not required for inclusion/exclusion
criteria or the primary endpoint are not performed, this does not constitute a protocol violation. If a
patient has to discontinue the study for any reason, the primary outcome visit (V9) should be
scheduled for week eight (+/- 1 week) corresponding to V9. In case the patient has relevant side-
effects (e.g. agranulozytosis) or decides to leave the study earlier, a protocol deviation must be
recorded. (). The visit corresponding to V9 can be shortened if necessary, but the primary outcome
(8 PANSS items for calculating remission score) should be assessed and recorded in the eCRF. If
the patient has no recorded V9 visit (No RSWG criteria at V9), the patient is declared as a drop-out.
All patients leaving the trial prior to V9, but accepting the assessment of RSWG criteria at the
timepoint of V9 (+ 1 week), are no drop-outs. For patients who receive study medication below or
above the defined dose ranges for a certain time period (see paragraph 6.2), a protocol deviation
must be recorded. If patients cannot be discharged from inpatient treatment at week eight for medical
reasons, it must be assured that the treating physician is not the responsible study doctor. Patient
who have a valid V9 visit, are invited to participate in the naturalistic follow-up.
Please note exception for SAE:
A SAE will not be reported if patients are re-hospitalized for psychiatric reasons between V9 and
V13
Please note exceptions for safety FU V10-V13:
We strongly recommend that the safety FU-visits including laboratory tests (V10-V13) take place,
within the given timeframe (+/- 2 working days). V10 to V13 can be performed via telephone interview
as long as the necessary laboratory results are available for the investigators. It is allowed that
patients bring their own laboratory measures (e.g. from the attending physician or from an outpatient
unit) for V10 to V13. A serious protocol violation has to be documented if the WBC test of visits 10
to 13 is missing (exception: if patients do not appear to the scheduled visits or are not available for
a telephone visit, this will be documented as minor protocol violation)

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 36 of 71
8.3 Special safety aspects during the intervention period
Blood count / agranulocytosis
Patients will be informed at the screening visit that they must contact the study doctors or any other
physician immediately if any kind of infection begins to develop. According to the SmPC of clozapine,
particular attention must be paid to flu-like complaints such as fever or sore throat and to other
evidence of infection, which may be indicative of neutropenia. The SmPC further states that patients
and their caregivers (if patients agree) must be informed that, in the event of any of these symptoms,
they must have a blood cell count performed immediately. Due the double-blind nature of the study,
all patients and will receive an emergency card with the study information and the potential risks
associated with clozapine treatment.
Study medication has to be immediately discontinued if either the WBC count is less than 3000/mm3
(3.0x109/l) or the ANC is less than 1500/mm3 (1.5x109/l). In these cases, the clinical work-up
according to the SmPC of clozapine has be performed and the study ends.
In the event of eosinophilia (eosinophil count ≥ 3000/mm³ (3.0 x 109/l) or thrombocytopenia (platelet
count ≤ 5000/mm³ (5 x 109/l) study medication has to be discontinued. In these cases, the clinical
work-up according to the SmPC of clozapine has be performed and the study ends
If WBC drops by 3000/mm³ (3.0x109/l) between two visits or within three weeks or if the WBC is
between 3000/mm3 to 3500 mm³ (3.0 to 3.5 x109/l), WBC and ANC have to be controlled twice a
week.
In case of relevant changes in white blood count or relevant thrombocytopenia or eosinophilia
leading to fulfilment of discontinuation criteria (please see paragraph 8.6 for details), study
medication will be discontinued and the patient will be excluded from the study. According to the
SmPC of Clozapine, weekly blood tests will be performed for at least for four continuous weeks and
until normalization of the respective blood parameters after the treatment was discontinued (see
Safety Follow-up visits).
Myocarditis
According to the clozapine SmPC, myocarditis or cardiomyopathy should be suspected in patients
who experience persistent tachycardia at rest, especially in the first 2 months of treatment, and/or
palpitations, arrhythmias, chest pain and other signs and symptoms of heart failure (e.g. unexplained
fatigue, dyspnoea, tachypnoea) or symptoms that mimic myocardial infarction. At screening, CRP,
troponine T in the study laboratory, a physical examination and an ECG will be assessed and
performed in order to exclude a myocarditis and/or a cardiomyopathy. In the case that a myocarditis
and/or cardiomyopathy is assumed by these diagnostic procedures, the patient should be referred
to a cardiologist in order to perform echocardiography diagnostics. During the double-blind study
phase CRP and troponine T are assessed on a weekly basis during the first four weeks, and on a
two-weekly basis until the end of the double-blind treatment phase. If troponine T increases > 2 fold
of the reference level or CRP increases > 100 mg/l (>10 mg/dl), study medication should be
discontinued and patients should be referred to a cardiologist. If the heart-frequency is > 120/min or
has increased by >30/min between visits or CRP is between 50 to 100 mg/l (5 to 10 mg/dl) or
troponine T has been increased, but is ≤2-fold of the reference level, study medication should not
be elevated until normalisation. Also in these cases, the patient will be referred to a cardiologist. This
is in accordance with national and international myocarditis screening and monitoring
recommendations for clozapine (Benkert and Hippius 2013) (Ronaldson, Fitzgerald et al. 2011). If
myocarditis or cardiomyopathy are clinically suspected already by a study doctor, treatment will be
promptly discontinued and the patient immediately referred to a cardiologist. If troponine T is not
available as parameter at the specific study center, troponine I can be assessed. The same
thresholds as mentioned above for troponine T are applicable.

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 37 of 71
Other side-effects
Both study drugs can be associated with relevant side-effects as indicated in the SmPC. This trial
has a comprehensive assessment of all relevant side-effects that goes far beyond the surveillance
procedures of the clinical routine. Please see below (paragraph 8.6) regarding criteria for withdrawal
of individuals due to side-effects.
8.4 Naturalistic extension study
Visits of the naturalistic extension study are scheduled after the double-blind pharmacotherapy
phase is completed as defined above (valid V9 visit, see paragraph 8.2), to assess the current status
of the patient after the double-blind part of the study. The following visits (N1 to N3) are not part of
the primary study and will not be monitored as they are part of a naturalistic extension study. Patients
have to sign an additional consent form at the screening visit for this naturalistic extension study.
Naturalistic visit 1 (N1): This is the first naturalistic visit and will be performed 26 weeks (1/2 year)
after the primary outcome assessment. This visit can only be performed if the patients have already
been monitored and the data will be entered to an additional CRF. During this visit year of birth, sex,
living circumstances, working status, the PANSS, the PANSS-RSWG items and a limited amount of
clinical scales will be used (PANSS, PANSS RSWG items, CGI, GAF, PSP, Q-LES-Q-18, SF12).
Dates of hospitalizations and discharges from V13 until N1 will be recorded. Data of what psychiatric
medications patients were taking from V13 until N1 will be recorded. Loss of follow-up in the
naturalistic extension study and causes will be documented.
Naturalistic visit 2 (N2): This is the second naturalistic visit and will be performed 52 weeks (1 year)
after the primary outcome assessment and the data will be entered to an additional CRF. During this
visit living circumstances, working status, the PANSS, the PANSS-RSWG items and a limited
amount of clinical scales will be used (PANSS, PANSS RSWG items, CGI, GAF, PSP, Q-LES-Q-18,
SF12). Dates of hospitalizations and discharges from N1 until N2 will be recorded. Data of what
psychiatric medications patients were taking from N1 until N2 will be recorded. Loss of follow-up in
the naturalistic extension study and causes will be documented.
Naturalistic visit 3 (N3): This is the third naturalistic visit and will be performed 104 weeks (2 years)
after the primary outcome assessment and the data will be entered to an additional CRF. During this
visit living circumstances, working status, the PANSS, the PANSS-RSWG items and a limited
amount of clinical scales will be used (PANSS, PANSS RSWG items, CGI, GAF, PSP, Q-LES-Q-18,
SF12). Dates of hospitalizations and discharges from N2 until N3 will be recorded. Data of what
psychiatric medications patients were taking from N2 until N3 will be recorded. Loss of follow-up in
the naturalistic extension study and causes will be documented.
There is some flexibility in the visit window intervals of the naturalistic extension study and visits can
be performed with a range of +/- 1 month.
8.5 Description of rating scales and assessments
8.5.1 Clinical rating scales
Alcohol Use Disorders Screening Test (AUDIT) (Babor 2001): 10-item core questionnaire for
screening of alcohol use disorders. The range is from 0-40 points.
Fagerström Test for cigarette dependence (Fagerstrom and Schneider 1989): 6-item questionnaire
for screening of nicotine dependence. The range is from 0-10 points.

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 38 of 71
Mini International Neuropsychiatric Interview (MINI) for DSM-V is a structured interview for
classification of axis-I disorders according to DSM-V and ICD-10.
Positive and Negative Syndrome Scale (PANSS) (Kay, Fiszbein et al. 1987): PANSS is a 30-item
rating scale that has been developed to investigate the severity of psychopathology in schizophrenia
patients (total score). The scale is composed of three subscales (positive, negative, general), but
from a principle point of view five symptom components, positive, negative, depression, agitation-
excitement, and disorganisation, can be investigated.
PANSS RSWG items: According to the RSWG criteria, the symptomatic criterion states that to
achieve symptomatic remission all eight PANSS items (P1, P2, P3, N1, N4, N6, G5, G9) must be
must be rated as absent or present only to a mild degree (PANSS value ≤ 3, see table 1).
Thought and Language Disorder Scale (TALD) (Kircher, Krug et al. 2014): TALD is a 30-item rating
scale for formal thought disorders representing a core syndrome of schizophrenia. Principally, each
of the items can be investigated separately, total minimum score is 0 and total maximum score is
120 points.
Clinical global impression scale (CGI) (Guy W 1976): CGI is a scale to rate disease severity on two
7 point scales (ranging from 0-7). There is one for the present state (severity scale: CGI-S) and one
for the change in severity (improvement scale: CGI-I), both ranging from 0-7 respectively. Both
scales are used in this trial.
Global assessment of functioning (GAF) (Jones, Thornicroft et al. 1995): GAF is numeric scale with
a range from 1 (severe impairments) to 100 (high functioning, no symptoms). It is used to globally
rate social, occupational, and psychological functioning of adults with mental disorders.
Personal and Social Performance scale (PSP) (Nasrallah, Morosini et al. 2008): PSP is a clinician-
reported measure of severity of personal and social dysfunction being composed of a numeric scale
with a range from 1 to 100.
Calgary Depression Scale for Schizophrenia (CDSS) (Addington, Addington et al. 1993): This 9-item
scale (0-3 points for each item) is designed to assess depression in patients with schizophrenia
without overlap with negative symptoms and extrapyramidal symptoms. The range of CDSS is from
0 to 27.
The InterSePT scale for suicidal thinking (ISST) (Lindenmayer, Czobor et al. 2003) is a 12-item rating
scale (0-2 points for each item) that has been specifically developed to measure suicidal ideations
in patients with schizophrenia or schizoaffective disorder. The range of ISST is from 0-24 points.
Quality of life enjoyment and satisfaction questionnaire (Q-LES-Q-18) (Ritsner, Kurs et al. 2005): Q-
LES-Q-18 is an 18-item short-form self-rating scale to assess quality of life in persons with severe
mental illness. The domains are physical health (items 1-4), subjective feelings (items 5-9), leisure
time activity (items 10-12), social relationships (items 13-17), satisfaction with medication (item 18).
Scoring is from 1 to 5. Principally, each domain can be analyzed separately, but total score is
primarily investigated.
SF-12 Questionnaire (SF-12) (Ware, Kosinski et al. 1996): SF-12 is a 12-item short-form health
survey: each item can be ranked from 1-2, 1-3, 1-5 or 1-6. Each item is analyzed separately.
8.5.2 Side-effect and treatment-related rating scales
Drug Attitude Inventory (DAI) (Hogan, Awad et al. 1983, Stjernsward, Persson et al. 2013): The DAI
is self-rating questionnaire that has been originally developed based on 30 questions, but now being
used as a short version with 10 questions (+ I agree/- I disagree) that have been validated. The scale
is used to measured adherence (ranging from -10 to +10).

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 39 of 71
Subjective Wellbeing under Neuroleptics short form (SWN-K) (Naber 1995): The SWN-K is a self-
rating scale developed to measure mentally ill patients’ well-being under the antipsychotic drug
treatment. The SWN-K is compromised of 20 questions, each of which is rated using a 6-point scale
ranging from 1 to 6. Possible scores range from 20 to 120, with higher scores implying higher
subjective well-being.
Glasgow Antipsychotic Side-effect Scale for Clozapine (GASS) (Hynes, Keating et al. 2015): The
GASS is a 16-item self-rating scale that has been developed to captures specifically clozapine-
related side-effects. Each item is rated using a 4-point scale ranging from 0-3 points (thus total scores
are ranging from 0-48 points). Higher scores indicate higher clozapine side-effects.. Moreover, the
frequency of severe or distressing side effects will be documented.
St. Hans Rating Scale (SHRS) (Gerlach, Korsgaard et al. 1993) The SHRS is a multidimensional
rating scale. The SHRS consists of 4 main components (neuroleptic-induced hyperkinesia,
parkinsonism, akathisia and dystonia). The hyperkinesia subscale consists of 8 items, the
parkinsonian subscale also consists of 8 items. Dystonia is only represented by a global score. The
akathisia component consists of a global score for psychic and motor akathisia. Each item is rated
from 0 (not present) to 6 (present to an extreme degree). This gives a total score from 0 to 48 for
hyperkinesia and for parkinsonism.
Barnes Akathisia Rating Scale (BARS) (Barnes 1989): BARS is scored as follows: objective
akathisia, subjective awareness of restlessness and subjective distress related to restlessness are
rated on a 4-point scale from 0-3 and are summed yielding a total score ranging from 0 to 9. The
Global Clinical Assessment of Akathisia uses a 5-point scale from 0-4.
Cleveland Clinic Constipation (Agachan, Chen et al. 1996) (CCCS) CCCS is a 8-item self-report
questionnaire. Items are rated on a 1-item Likert scale from 0-2 (symptom absent) to 4 (very
severe).Total score ranges from 0-30 points (item 6 of the CCCS questionnaire with a Likert-scale
from 0-2)-
8.5.3 Cognition
Trail-Making Test A and B (Tombaugh 2004) is a cognitive test to investigate complex visual
scanning, motor speed, and the ability to shift strategies. The time needed to draw lines to
connect either consecutively numbered circles (TMT-A) or to connect consecutively
alternating numbered and letters circles (TMT-B) in seconds, as well as the amount of errors
while doing this, are the outcome parameters.
8.5.4 Study laboratory
Regular study laboratory:
White blood count (WBC) including neutrophils, lymphocytes, monocytes, eosinophils and
basophils, absolute and relative neutrophile counts, red blood cell count (RBC), platelet count
(at all visits from Screening to V13)
Electrolytes (sodium, potassium), Creatinine, Creatinkinase (CK total), ASAT and ALAT (at
all visits from Screening to V10)
C-reactive protein (CRP) (at Screening, V1, V2, V3, V4, V5, V7 and V9)
Troponine T (at Screening, V1, V2, V3, V4, V5, V7 and V9). Only if local laboratories are not
able to measure troponine T, troponine I can be assessed.

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 40 of 71
Drug screening (urine) at Screening
Beta-HCG (serum) for female patients at Screening
Additional study laboratory (other assessment) (fasting blood collection at Screening, V1, V5, V9,
V13) includes:
Glucose, Triglycerides, LDL, HDL, cholesterol (only at Screening, V1, V5, V9 and V13)
8.5.5 Therapeutic drug monitoring
Blinded blood levels of clozapine or olanzapine and of their metabolites will be performed at week
two and week four (V3 and V5).
8.5.6 Additional assessments
Assessment of weight gain and BMI. The weight and the BMI are documented in the eCRF.
Participants and responsible study doctors will be asked to estimate whether the given participant
was in the clozapine or olanzapine treatment arm to allow for a post-hoc testing of blinding integrity
and this assessment will be documented in the eCRF.
Electrocardiography (ECG): Convention 12-channel ECG to evaluate rate and rhythm, axis,
amplitudes and intervals. The QTc time and whether the ECG is abnormal or not will be documented.
Electroencephalography (EEG): Convention, preferentially 64-channel, EEG to evaluate activity,
rhythms and bands (wave patterns). We will document whether the EEG was normal or not as well
as the information about activity, rhythms and wave patterns.
30 min ECG (optional): 30 minute-ECG to evaluate rate, rhythm and heart rate variability. We
document in the eCRF whether the 30 min ECG assessment was performed or not (these data is
not part of the study database)
Transthoracic echocardiography (TTE) to evaluate e.g. change in left ventricular ejection fraction
(LVEF) in %, inter-ventricular septum and posterior wall thickness in mm, chamber dimensions
(sizes/diameters of left ventricle, left atrium, right ventricle in cm), RV function (tricuspid annular
plane systolic excursion: TAPSE). We document in the eCRF whether the TTE monitor assessment
was performed or not (these data is not part of the study database)
8.5.7 Magnetic resonance imaging (MRI/ Optional)
Structural (sMRI) and resting-state (rsMRI) will consist of T1-weighted, T2-weighted and EPI1
sequences. MRI will only be offered to patients at sites with an access to an MRI scanner, preferably,
but not necessarily 3T. 1.5T scanners may also be used. Parameters will be certified in a MRI-
scanning protocol. Data will be analyzed with regard to volumetric changes over time, to the
predictive accuracy of baseline images and to the neurofunctional changes over time. It is planned
to perform a calibration study based on the PRONIA calibration protocol (https://www.pronia.eu/) or
other established protocols. We will document in the eCRF whether the MRI has been performed or
not (these data is not part of the study database)

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 41 of 71
8.6 Discontinuation of the study
The patient has the right to withdraw the informed consent at any time with no reason given, without
risk of being penalized and is declared a drop-out. The investigator, on the other hand, has the right
to exclude a patient from the study in the event of concomitant disease, adverse events, therapy
failure or any other reason or condition that warrants withdrawal in the interest of the patient. Such
patients are not primarily defined as drop-outs. If the patient has no recorded V9 visit (No RSWG
criteria at V9), the patient is declared as a drop-out (see also paragraph 8.2): If a patient has to
discontinue the study for any reason (relevant AEs/SAEs, intolerance to study drug, non-response,
incompliance regarding study drug intake or violation of study drug dose ranges as described in
paragraph 6.2, relevant concomitant disease), the primary outcome visit (V9) should be scheduled
for week eight (+/- 1 week) corresponding to V9. A protocol deviation must be recorded. The visit
corresponding to V9 can be shortened if necessary, but the primary outcome (8 PANSS items for
calculating remission score) should be assessed and recorded in the eCRF. All patients leaving the
trial prior to V9, but accepting the assessment of RSWG criteria at the timepoint of V9 (+ 1 week),
are no drop-outs.
Patient who have a valid V9 visit are invited to participate in the naturalistic follow-up.
We strongly recommend after discontinuation:
When for any reason study treatment is discontinued before Visit 9 or patients leave the study before visit 13, patients will be informed again about the necessity to have WBC controls for further four weeks. This clarifying consultation will be documented as being performed in the source data. Patients will have the possibility to receive WBC testing in the outpatient units of the participating study sites, but are free to consult other doctors such as their general practitioner or their psychiatrist. If a patient becomes a lost to FU after V9, the same procedure is used as described above.Every case of premature study drug treatment termination must be evaluated by the investigator and reasons for the termination assessed. The entire documentation must be as complete as possible. The final safety assessments should follow the protocol specifications. Any cases of premature termination must be documented in the eCRF, additional information is to be added, if applicable.
The patient’s study drug treatment will be terminated when
The patient withdraws her/his consent
The patient can no longer give informed consent according to the assessment of the study
doctor or due to any SAE
Intolerance to the study drug or non-tolerable side-effects
The nature of the patients treatment is changed to coercive treatment (based on judicial
ruling)
In particular, treatment needs to be discontinued due to changes in WBC/ANC (WBC count
less than 3000/mm3 or 3.0 x 109/l or the ANC is less than 1500/mm3 (1.5 x 109/l) see
paragraph 8.3 for details) or when a myocarditis and/or cardiomyopathy is assumed
according to protocol
Furthermore, treatment needs to be discontinued in case of eosinophilia (eosinophil count ≥
3000/mm³ (3.0 x 109/l) and should be restarted only after the eosinophil count has fallen
below 1000/mm³ (1.0 x 109/l)
Treatment needs to discontinued in case of thrombocytopenia (platelet count ≤ 5000/mm³
(5 x 109/l))
Treatment needs to be discontinued in case of paralytic ileus

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 42 of 71
Treatment needs to be discontinued in case of epileptic seizure(s)
“Treatment needs to be discontinued if the patient suffers from relevant QT prolongation
(QTcF > 500msec and/or QTcF prolongation > 60msec compared to QTcF at baseline
Treatment needs to be discontinued in cases where hepatitis (including hepatocellular,
cholestatic or mixed liver injury) has been diagnosed
Treatment needs to be discontinued if a patient develops signs and symptoms indicative of
Neuroleptic Malignant Syndrome (NMS), or presents with unexplained high fever without
additional clinical manifestations of NMS
Patient becomes pregnant or initiates lactation
The investigator considers a patient’s continued participation in the study to be unjustifiable
on medical grounds (i.e., because of side-effects or unusual risks)
If an individual patient is discontinued due to one of the above mentioned reasons, this
patient will be treated as usual in normal clinical practice.
The sponsor has the right to terminate this study at any time. Reasons for terminating the study
may include but are not limited to the following:
The incidence or severity of AEs in this or other studies indicates a potential health hazard
to patients;
Unsatisfactory patient enrollment;
The continuation of study is unethical or it has been proven that the therapy has a clearly
negative influence;
Unforeseen complications arise that no longer justify a continuation of the study.
The sponsor will notify the investigator of a decision to discontinue the study. The sponsor has the
right to close a site at any time.
Reasons for closing a site may include, but are not limited to, the following:
Excessively slow recruitment (<5 inclusions/year)
Poor protocol adherence;
Inaccurate or incomplete data recording;
Non-compliance with the ICH-GCP guideline;
No study activity (i.e. all patients have completed and all obligations have been fulfilled);
If a study site discontinues the study on behalf of one of the above mentioned reasons, the patients
that have already been included will be followed up, but no new inclusions will be made.
The investigator can discontinue the clinical study at his site at any time if he no longer considers
the continuation of the study, for example if there are ethical and/or medical concerns.

EARLY- Protocol final Version 5.0 AM 3.0 - Date: 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 43 of 71
8.7 Study flow chart
Study Visit Screening Baseline 1
2
3
4
5
6
7
8
9
10
11
12
13
Day -14 to 0 0-1 7 14 21 28 35 42 49 56
Endpoints* 63 70 77 84 (EOS)
Intervention of medication Administration from day 1 to day 56
Informed Consent X ****
Inclusion/exclusion criteria X
Drug Screening X
AUDIT interview and MINI X
Fagerström Test (Smoker) X Demographic data/ Medical / Psychiatric History X
Pre-study medication X X X X
Physical examination X X X X X X X X X X
Study laboratory X X X X X X X X X X X X X X
Pregnancy Test X
Number of cigarettes X X X X X
Interaction check****** X X X X X X X X X
PANSS *** X X X X
PANSS RSWG items X X X X X
CGI and GAF X X X
PSP, ISST, Trail-Making-Test (TMT) X X
CDSS X X X
Q-LES-Q-18, SF-12, DAI10, SWN-K, TALD X X
Attitude towards clinical trials X
BARS X X
GASS X X X X
St. Hans Rating Scale (SHRS) X X
CCCS X X X X
Assessment blood pressure and heart rate X X X X X X X X X
Assessment of weight and BMI X X X
EEG X X
ECG X X X X X X
Study medication (re-)dispensing X X X X X X X X *
Study medication return X
Blood draw Drug levels ** X X
Assessment of blinding integrity X
Discontinuation check X X X X X X X X
SAE/AE X X*****
Concomitant medication X X*****
* one additional blister of study medication can be distributed to the patient if the patient is discharged from inpatient treatment (lasting for around 4 days) **documentation of amount and type of capsules related to the timepoint of blood draw for drug level assessment; If a blood draw is not possible on the planned visits, the assessment of drug levels can be performed at the next planned visit. ***if psychopathological worsening is suspected, additional PANSS ratings should be performed in those visits where PANSS rating is not planned. **** also in case of relevant psychopathological worsening (increase in PANSS total score of at least 20%) or an emerging SAE.
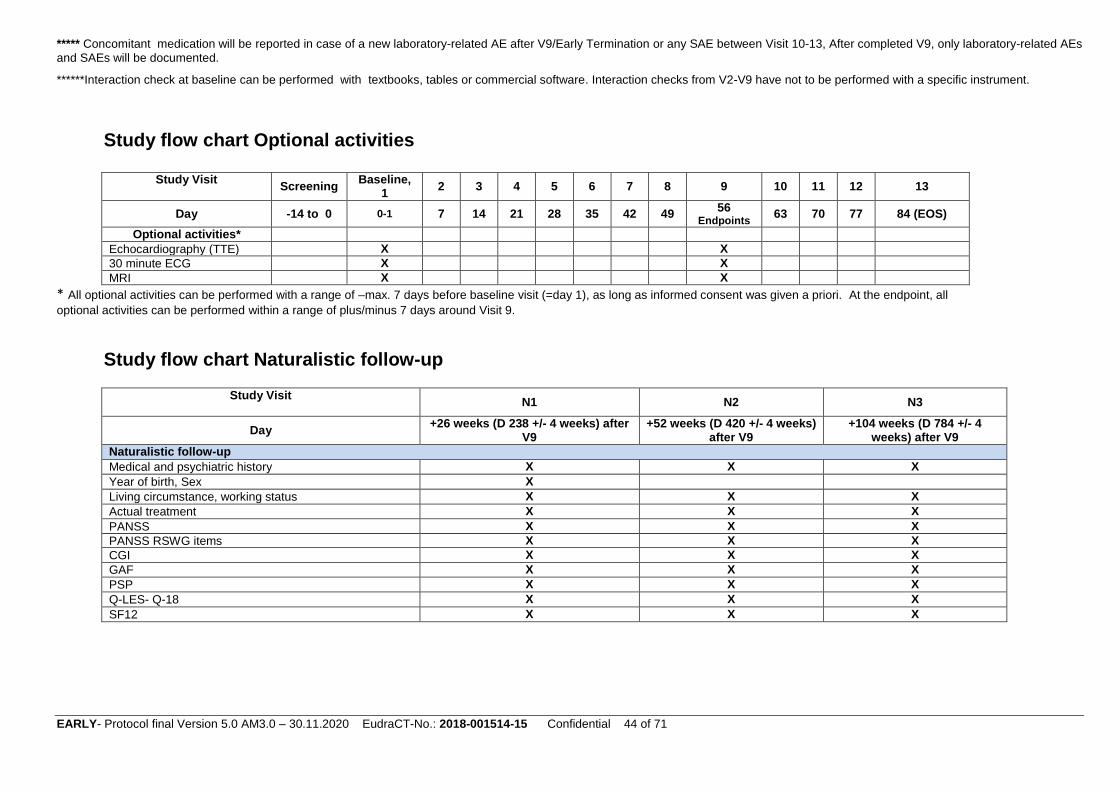
EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 44 of 71
***** Concomitant medication will be reported in case of a new laboratory-related AE after V9/Early Termination or any SAE between Visit 10-13, After completed V9, only laboratory-related AEs
and SAEs will be documented.
******Interaction check at baseline can be performed with textbooks, tables or commercial software. Interaction checks from V2-V9 have not to be performed with a specific instrument.
Study flow chart Optional activities
Study Visit
Screening Baseline,
1 2 3 4 5 6 7 8 9 10 11 12 13
Day -14 to 0 0-1 7 14 21 28 35 42 49 56
Endpoints 63 70 77 84 (EOS)
Optional activities*
Echocardiography (TTE) X X
30 minute ECG X X
MRI X X
* All optional activities can be performed with a range of –max. 7 days before baseline visit (=day 1), as long as informed consent was given a priori. At the endpoint, all
optional activities can be performed within a range of plus/minus 7 days around Visit 9.
Study flow chart Naturalistic follow-up
Study Visit
N1 N2 N3
Day +26 weeks (D 238 +/- 4 weeks) after
V9 +52 weeks (D 420 +/- 4 weeks)
after V9 +104 weeks (D 784 +/- 4
weeks) after V9
Naturalistic follow-up
Medical and psychiatric history X X X
Year of birth, Sex X
Living circumstance, working status X X X
Actual treatment X X X
PANSS X X X
PANSS RSWG items X X X
CGI X X X
GAF X X X
PSP X X X
Q-LES- Q-18 X X X
SF12 X X X

EARLY- Protocol final Version 5.0 AM 3.0 - Date: 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 45 of 71
9. Statistical Analysis
The statistical analyses will be performed after data-base hardlock (end of study), but before the
end of the naturalistic follow-up. The latter is a secondary analysis that will be performed at the
end of the naturalistic follow-up period.
General statistical considerations
Descriptive statistics will be provided for all data broken down by treatment group, by visit and
by country. Mean, median, standard deviation, range, interquartile range and number of
observations will describe continuous variables. Absolute and relative frequencies will describe
categorical variables. All statistical tests will be carried out two-tailed; the alpha (level of
significance) is 5%. -
Baseline comparisons
The randomization procedure will be evaluated by a comparison of the two treatment groups for
all relevant variables recorded at baseline.
Primary analyses
The primary efficacy endpoint in the ITT population will be compared between groups by a
Mantel-Haenszel test, with centres as strata on a confirmatory two-sided significance level of
5%.
An additional subgroup analysis will be performed by logistic regression using the primary
efficacy endpoint as outcome and the factor variables group, centre and the categorized number
of previous lifetime treatments (cNLT categories: 0, 1, >1) as covariates. An interaction effect
between group and the cNLT subgroups will be included in the model. Parameter estimates of
the model will be used to obtain point and interval estimates (95% confidence intervals) of the
treatment effect, represented by Odds Ratios, within cNLT subgroups. The hypothesis test on
the model’s interaction effect equals the test for differences of the treatment effects between
subgroups. This subgroup analysis is exploratory and none of the hypothesis tests of effects
within and between subgroups is powered by the study’s sample size calculation. The major
objective is effect estimation instead.
Secondary analyses
Depending on the data distribution, group comparisons of continuous and categorical secondary
endpoints will be performed by linear or binary logistic regression analysis that includes a factor
variable for centres. Baseline values will be included as another covariate if existing. In case of
non-normally distributed residuals of the linear model, a van Elteren test (= stratified Wilcoxon
test) with centres as strata will be applied in an additional analysis. Corresponding descriptive
statistics and confidence intervals will also be given. Absolute and relative frequencies of
categorical safety outcomes will be assessed along the course of the trial (safety population).
Differences between groups will also be tested for statistical significance with Fisher’s exact test.
Likewise, continuously distributed safety outcomes (safety population) will be presented by
descriptive statistics and compared with t-tests or Mann-Whitney U tests, as appropriate. All
tests of secondary endpoints will be computed in the ITT and per protocol (PP) populations in
an explorative manner on a two-sided significance level of 5%.
Other assessments
Exploratory analyses will be performed as appropriate for other assessments.

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 46 of 71
Study populations
The intention-to-treat (ITT) population includes all patients as randomized. The per protocol (PP)
population will include all participants without major protocol violations. The safety population
will include all participants who received at least one study drug and will be analysed as treated.
The allocation of patients to the study populations will finally be determined in a blinded data
review meeting (BDRM). All analyses will be performed in full accordance with the ICH
Guidelines E9.

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 47 of 71
10. Analysis of risks and benefits
Participation in the EARLY trial implies that subjects contribute to the development of medical
knowledge of which they and other patients could benefit. An advantage of participation could
be that subjects will be more thoroughly followed and examined, and that therefore effects and
side effects are better measured. Moreover, patients may have an increased likelihood for
remission that will reduce the symptom burden. Patients will be treated with antipsychotics that
are used in clinical practice at this stage (especially olanzapine) with a far more sophisticated
side-effect evaluation increasing the individual safety. Clozapine is usually introduced at the
stage of treatment-resistance (Hasan, Falkai et al. 2012), but as indicated above in most cases
far too late. The participation in the EARLY trial increases the likelihood for an introduction of
clozapine before treatment resistance to reduce the risk of clozapine non-response.
As the inclusion criteria include a moderate symptomatology on the PANSS, defined as a score
≥ 4 for two or more symptoms from P1-P7 or a score of ≥ 6 for one symptom from P1-P7, it must
be concluded that these patients need antipsychotic treatment and both study drugs are
amongst the most effective antipsychotics (Leucht, Cipriani et al. 2013).
Use clozapine or olanzapine, the study drugs, will imply that there is a risk of drug-specific side
effects. From a principle point of view, the use of antipsychotics in this population is indicated
anyway in the treatment of subjects who are eligible for this study. However, clozapine is usually
applied a bit later in the disease stage so that the clozapine-associated risks of using clozapine
have to be considered as increased risk for 50% of the study participants. The potential benefits
are the expected higher likelihood for short-term and long-term response, the reduced risks of
motor-side effects and the reduced risk of a potential re-hospitalization. Moreover, the strict
safety protocol addressed all clozapine-associated risks and goes far beyond the recommended
safety procedures described in the SmPC increasing the patient safety. Another limitation all
subjects face within this study is that they cannot choose a side effect profile associated with an
individual antipsychotic, as they are randomized. In addition, participation of this trial will take
more of the subject’s time than regular treatment.
Clozapine is safe in both early-episode and chronically ill patients if the necessary monitoring
(especially complete and white blood cell count) is applied (Agid, Arenovich et al. 2011, Meltzer
2012, Remington, Agid et al. 2013, Sanz-Fuentenebro, Taboada et al. 2013). Thus, application
as second-line treatment can be warranted, especially in schizophrenia patients who experience
a relapse or who have not responded to an antipsychotic trial. Such patients are at high risk for
an unfavorable disease outcome and the experimental intervention with early clozapine is likely
to improve remission rates in these patients. By excluding first-episode patients who have a very
high likelihood to response to any kind of antipsychotic treatment (Zhu, Krause et al. 2017, Zhu,
Li et al. 2017), we reduce the risk of treating patients with drugs that are highly effective on the
one hand, but where the side-effects do not warrant the use in first-episode patients.
All available antipsychotics carry the risk of specific side effects, which need to be weighed
against the benefits. Clozapine is associated with a small risk of agranulocytosis and
myocarditis. On the basis of the literature, in our study we may have ~1 case of agranulocytosis
and ~0.1 case of myocarditis. However, as recently outlined in a literature review the absolute
risk for both complications is low and can be mitigated by regular monitoring and timely
interventions (Citrome, McEvoy et al. 2016). In addition to the regular monitoring of blood counts
and CK levels as well of troponine T, we will implement several levels of safety measures and
surveillance that go far beyond the usual assessments in other schizophrenia trials or clinical
practice. Moreover, we will perform 5 ECGs during the intervention phase and we will perform

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 48 of 71
two EEGs to monitor for potential epileptic activity and thus reduce the risk for clozapine-
associated seizures.
According to German guidelines, treatment will be discontinued immediately if leucocytes are
<3000/µl or neutrophil granulocytes are <1500/µl or both. Before study medication is dispensed,
a WBC must confirm leucocyte levels >3500/µl. If leucocytes drop to levels between 3000/µl and
3500/µl, the WBC will be controlled twice a week, in accordance with German guidelines. Study
medication can only be increased if WBC > 3500/µl and neutrophil granulocytes are > 1500/µl.
If a patient reports clinical signs of myocarditis (e.g. chest pain, palpitations, dyspnoea, and
discomfort) or shows a relevant CK elevation (assessed on a weekly basis) or respective ECG
abnormalities or changes in troponine T (Müller and Benkert 2017), treatment will be
discontinued and cardiac echography will be performed by a cardiologist. Please see paragraphs
8.3 (Special safety aspects during the intervention period) and 8.5 (Description of rating scales
and assessments) for a complete description of our safety procedures that will be implemented
in addition to those procedures necessary for every trial (see paragraph 12, Safety reporting).

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 49 of 71
11. Feasibility of patient recruitment
Although there are no pilot studies for the current trial, the study design resembles that of
previous randomised studies in the field of schizophrenia research (Lieberman, Phillips et al.
2003, Lieberman, Stroup et al. 2005, Meltzer, Bobo et al. 2008). In addition, the registration trials
for the antipsychotics in the current design (especially olanzapine) have recruited large patient
samples. Given that these studies have already been successfully completed, it seems
reasonable to assume that with respect to sample size, the chosen statistical methods, and the
estimated number of patients who will drop out, the present study can be realised. The present
study will not use any medication which is yet to be approved and does not ask many extra
activities of the patients, making recruitment all the easier.
Because participant compliance is the most critical point for study success, when designing
the study, we made great efforts to provide a setting for good compliance and took into account
our study we made great efforts to provide a setting for good compliance and took into account
our experience with previous large-scale multicentre trials in schizophrenia patients. We aim to
conduct the trial for eight weeks and focus on the main research question whether clozapine is
superior to olanzapine in achieving remission. This short trial period will have much higher
compliance rates than 26- or 52-week trials. The following design aspects will improve the
compliance rate:
Minimal exclusion and broad inclusion criteria
Different dose steps rather than a fixed-dose strategy, minimal number of needed
capsules
Possibility to add haloperidol as rescue medication during the titration phase
Possibility to add a broad array of rescue medication during the complete study phase
(e.g. lorazepam, zopiclone)
Slow titration of study drugs to reduce risk for side effects
No placebo control – two active comparators
No use of non-approved drugs and use of two highly effective SGAs
The expected loss to follow-up is low because patients who drop out of the study will still be
treated at the study centres. We will follow a strict intention-to-treat principle and still evaluate
patients at the primary outcome visit (unless they withdraw consent), even if patients are put on
another medication during the trial. Therefore, the primary outcome will be assessed at standard
routine visits during daily clinical work. We believe that we will have less than 1% total missing
values for the primary endpoint. To protect against missing values that might still occur we will
perform statistical analyses conservatively and include all randomized patients. The drop-out
rates of short-term clinical trials comparing two antipsychotics have been reported in the range
of 5% to 50%. Waiving a placebo arm and using SGAs are the main factors to decrease drop-
out rates (Kemmler, Hummer et al. 2005, Martin, Perez et al. 2006) and we addressed both
factors in our trial design. We are aware that drop-outs cannot be completely avoided. As
outlined above we will assess the primary outcome in nearly all patients, irrespective of whether
they are a completer or drop-out. Therefore, drop-outs are not lost patients. However, clinical
trial experience has shown us that despite all efforts we might lose some patients, and we have
therefore addressed this issue by adjusting our power analysis regarding the expected rates of
total missing values.

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 50 of 71
12. Safety reporting
12.1 AE, SAE and Susar definitions and reporting procedures
Adverse Event
An Adverse Event (AE) is defined as any new untoward medical occurrence or worsening of a
pre-existing medical condition in a patient or clinical investigation subject administered an
investigational (medicinal) product and that does not necessarily have a causal relationship with
this treatment. An AE can therefore be any unfavourable and unintended sign (including an
abnormal laboratory finding, for example), symptom, or disease temporally associated with the
use of investigational product, whether or not considered related to the investigational product.
Adverse events can be spontaneously reported or elicited during open-ended questioning,
examination, or evaluation of a subject (In order to prevent reporting bias, subjects should not
be questioned regarding the specific occurrence of one or more adverse events). Adverse
events will be recorded in the eCRF in relation to stage of the study drug. Furthermore, it will be
assessed if the patient was in a titration phase or not when the adverse event occurred.
Serious Adverse Event
A Serious Adverse Event (SAE) is any untoward medical occurrence at any dose that:
results in death
is life-threatening (defined as an event in which the subject was at risk of death at the time of the event; it does not refer to an event which hypothetically might have caused death if it were more severe)
requires inpatient hospitalization or causes prolongation of existing hospitalization (see NOTE*: below for exceptions)
results in persistent or significant disability/incapacity is a congenital anomaly/birth defect
is an important medical event, defined as a medical event that may not be immediately life-threatening or result in death or hospitalization but, based on appropriate medical and scientific judgment, may jeopardize the subject or may require intervention (e.g., medical, surgical) to prevent one of the other serious outcomes listed above. Examples of such events include but are not limited to intensive treatment in an emergency department or at home for allergic bronchospasm; blood dyscrasias or convulsions that do not result in hospitalization.
Suspected transmission of an infectious agent (e.g., pathogenic or non-pathogenic) via the study drug is an SAE.
Although pregnancy, overdose, and cancer are not always serious by regulatory definition, these events must be handled as SAEs.
The following hospitalizations are not considered SAEs:
Pre-planned hospitalizations, i.e. before enrolment into the study and which are not related to the underlying condition.
Hospitalization due to social reasons (e.g. homelessness) in absence of an AE

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 51 of 71
A planned hospitalization for a pre-existing condition, or a procedure required by the Clinical Protocol, without a serious deterioration in health.
Surgery or procedure planned before entry into the study.
re-hospitalized patient between V9 and V13 for psychiatric reasons.
Serious Adverse Reaction
Serious Adverse Reaction (SAR) is defined as an adverse drug reaction that is serious and at
least possibly related to the investigational medicinal product. In case of a SAR the investigator
has to provide a case narrative.
Suspected Unexpected Serious Adverse Reaction (SUSAR)
SUSARs are defined as Suspected Unexpected Serious Adverse Reactions, i.e. all suspected
adverse reactions related to the tested investigational medicinal product that is both: unexpected
and serious. SUSARs require expedited reporting
An event qualifies as a SUSAR when:
the criteria for an SAE are met and
it is plausible that the event is caused by the study medication and
the event is unexpected, meaning that the nature, or severity, is not consistent with the
applicable product information (i.e. Summary of Product Characteristics).
Second Assessment of Serious Adverse Events
All SAE will be subject to a second assessment by a medical expert, who will be independent
from the reporting investigator and the coordinating investigator.
The second assessor will monitor each SAE following the conditions below:
I. known SAR with unexpected outcome
II. assessment of relationship between SAE and IMP
III. assessment of expectedness of SAE (derived from IB)
IV. statement if the benefit/ risk assessment for the trial did change as a result of SAE (or
of an unexpected outcome of an expected SAR).
V. Statement, if there are any safety issues, which are sufficient to consider changes in
the conduct of the trial.
An “unexpected” adverse event is one the nature or severity of which is not consistent with the
applicable product information, e.g. Investigator Brochure (IB), device manual or scientific
literature.

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 52 of 71
Assessment of Adverse Events
Assessment of intensity
The intensity of adverse events will be graded using the CTCAE, Version 4.0 of the US NCI
(e.g. Website:https://www.eortc.be/services/doc/ctc/CTCAE_4.03_2010-06-
14_QuickReference_5x7.pdf). For events not reported in the CTCAE Version 4.0, the
investigator will use the Grade or adjectives:
Grade I Mild; asymptomatic or mild symptoms; clinical or diagnostic observations only; intervention not indicated.
Grade II Moderate; minimal, local or noninvasive intervention indicated; limiting age-appropriate instrumental Activities of Daily Living (ADL)
Grade III Severe or medically significant but not immediately life-threatening; hospitalization or prolongation of hospitalization indicated; disabling; limiting self care ADL
Grade IV Life-threatening consequences; urgent intervention indicated
Grade V Death related to the adverse event
Table 5; Grading of adverse events
Note the distinction between the gravity and the intensity of an adverse event. Severe is a
measure of intensity. Thus, a severe reaction is not necessarily a serious reaction. For example,
a headache may be severe in intensity but would not be classified as serious unless it meets
one of the criteria for serious events listed above.
Assessment of causality
The investigator must make a causality assessment for all AEs. The relationship of an adverse
event to the study treatment regimen and the device has to be recorded on the eCRF and defined
as not related, unlikely, possible related, probable related or highly probable/definite:
Relatedness Description
Highly
probable/definite
An event that follows an established temporal sequence from the
study treatment regimen, the re-challenge is positive, or there is a
reoccurring pattern of characteristics during onset and cessation of
event.
Probably related An event that follows a reasonable temporal sequence from the
study treatment regimen and that is not easily explained by
another cause such as known characteristics of the subject´s
clinical state or the treatment
Possibly related An event that follows a reasonable temporal sequence from the
study treatment regimen but that may be due to another cause
Unlikely An event that follows such a temporal sequence from the study
treatment regimen that a relationship is not likely, and is likely to

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 53 of 71
be due to cause such as the subject`s clinical state or other
treatment
Not related The event is definitely not associated with the study treatment
regimen in the study but is judged clearly and incontrovertibly due
to causes other than the study treatment
The categories “Highly probable/definite”, “Probably related” and “Possibly related” will be
assumed as related, the categories “Unlikely” and “Not related” are assumed as not related.
12.2 Follow-up of adverse events
Period of Adverse Event reporting
All Adverse Events must be collected that occur during visit 1 (start of study treatment) and within
30 days of discontinuing dosing or until all drug-related toxicities are resolved, whichever is later,
or until the investigators assess AEs as “chronic” or “stable”. Open AE's and SAE's are only
observed until end of study (EOS). The investigator should report any Adverse Event occurring
after these time periods that is believed to be related to study drug or protocol-specified
procedure.
If the investigator believes that an SAE is not related to study drug, but is potentially related to
the conditions of the study (such as withdrawal of previous therapy, or a complication of a study
procedure), the relationship should be specified in the narrative section of the SAE Report Form.
SAEs, whether related or unrelated to the study drug, and pregnancies must be reported to the
MSZ immediately (latest within 24hours). SAEs must be recorded on the SAE Report Form;
Pregnancies on a Pregnancy Report Forms.
Münchner Studienzentrum (MSZ)
Klinikum rechts der Isar
Technische Universität München
Ismaningerstr. 22
81675 München
MSZ E-Mail: [email protected]
MSZ Fax Number: +49/89/4140-6480
If only limited information is initially available, follow-up reports are required.
If an ongoing SAE changes in its intensity or relationship to study drug or device or if new
information becomes available, a follow-up SAE report should be sent immediately (latest within
24 hours) to the MSZ using the same procedure used for transmitting the initial SAE report. All
SAEs should be followed to resolution or stabilization.

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 54 of 71
Reporting requirement
Each adverse event is to be classified by the investigator as SERIOUS or NON-SERIOUS. This
classification of the gravity of the event determines the reporting procedures to be followed. If a
serious adverse event occurs, reporting will follow local and international regulations, as
appropriate. If a serious adverse event occurs, the Sponsor’s Drug Safety Officer are to be
notified, using the designated SAE Report form, immediately (latest within 24 hours of
awareness of the event) by the investigator. The respective Fax-number and E-Mail address will
be provided to the investigator as part of the ISF.
The initial report is to be followed by submission of a more detailed adverse event information
(only if new case information is available) as soon as possible. Reporting requirements for
adverse events are summarized in the following table.
Gravity Reporting time Type of Report
Serious Immediately (latest
within 24 hours)
Initial/Follow up report on
designated serious adverse event
form
Non-Serious Per case report form
submission procedure Appropriate case report forms
Table 6: Reporting of adverse events
Any AE that meets any criterion for an SAE requires the completion of an SAE Report Form in addition to being recorded on the AE page of the eCRF. AEs and SAEs will be recorded after first drug intake onwards and will be followed-up until end of study (EOS). After completed V9, only laboratory-related AEs and SAEs will be documented.
It should be noted that the form for collection of serious adverse event information is not the
same as the adverse event case report form. Where the same data are collected, the forms must
be completed in a consistent manner. For example, the same adverse event term should be
used on both forms.
In the rare case that the investigator does not immediately become aware of the occurrence of
a serious adverse event, the investigator must report the event immediately (latest within 24
hours after having been informed about it) and document his/her first awareness of that adverse
event.
Non-Serious-Adverse-Events are to be reported on the adverse event case report forms (eCRF)
which are to be submitted as specified in the adverse event report submission procedure for this
protocol.
The investigator will be requested to supply as much detailed information as possible regarding
the event that is available at the time of the initial contact. The investigator is also required to
complete missing or requested information and to submit follow-up reports to the person
mentioned above until the SAE or SUSAR has resolved or, in the case of permanent impairment,
until the SAE has stabilized.
The response time for queries to SAE reports is expected to be no more than five (5) business
days. Urgent queries (e.g. missing causality assessment) may be handled by phone.

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 55 of 71
MSZ as representative of the sponsor will ensure the reporting of all SUSARs and other safety
issues which require expedited reporting to the responsible authorities according to the legal
requirements. MSZ will report all SUSARs to the competent authority and Ethics Committee (EC)
within 15 calendar days after the Drug Safety Officer was first aware of the minimum criteria for
expedited reporting.
MSZ will report all fatal or life-threatening SUSARs to the competent authority and EC within 7
calendar days after the Drug Safety Officer has first knowledge of the minimum criteria for
expedited reporting.
In addition to reporting to competent authority and EC the MSZ will inform all investigators
concerned on findings that could adversely affect the safety of study patients.
Responsibilities of the investigator
A SAE-report form (initial report) must be sent immediately (latest within 24 hours of onset or
acknowledgement of the SAE) to the Sponsor’s Safety Officer.
Follow-up reports must be sent when further information becomes available using the route of
transmission described above.
Other documents must be submitted upon request (e.g., histology findings, case report forms).
All documents must be blinded with respect to the patient’s name. The confidentiality of patient
information is ensured by revealing only a patient number. All these documents will be forwarded
by fax as described above.
Although such information is not routinely sought or collected by the sponsor, SAE that occurred
after the patient had completed the clinical trial (including any protocol-required post-treatment
follow-up) will possibly be reported by an investigator to the sponsor. Such cases will be
regarded for expedited reporting purposes as though they were study reports. Therefore a
causality assessment and determination of expectedness will be obtained for a decision on
whether or not expedited reporting is required.
12.3 Unblinding
Unblinding of a patient in the case of emergency or in case of SUSAR reporting is ensured by a
set of special emergency envelopes which are available on site. For the blinded data review
meeting specific SOPs will be implemented for these cases.
12.4 Pregnancy
If, following initiation of the investigational product, it is subsequently discovered that a study
subject is pregnant or may have been pregnant at the time of investigational product exposure,
including during at least 5 half-lives after product administration, the investigational product will
be permanently discontinued in an appropriate manner (e.g., dose tapering if necessary for
subject safety).
The investigator must immediately notify MSZ of this event via the ”Report on the drug exposure
during pregnancy” immediately (latest within 24 hours) and in accordance with SAE reporting
procedures.

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 56 of 71
Follow-up information regarding the course of the pregnancy, including perinatal and neonatal
outcome and, where applicable, offspring information must be reported on a “Report on the
pregnancy outcome during drug exposure”.
Any pregnancy occurring in a female partner of a male study participant the investigator
becomes aware of should be reported to the MSZ. Information on this pregnancy may also be
collected on the pregnancy reporting forms.
12.5 Overdose
An overdose is defined as the accidental or intentional administration of any dose of a product
that is considered both excessive and medically important. All occurrences of overdose must be
reported as SAEs
12.6 Annual safety report (Safety Update Report (DSUR))
In addition to the expedited reporting, sponsors shall submit, once a year throughout the clinical
study or on request a safety report to the competent authority and the EC of the concerned
Member States, taking into account all new available safety information received during the
reporting period. The aim of the safety report is to concisely describe all new safety information
relevant for one or several clinical study(s) and to assess the safety conditions of subjects
included in the concerned study. The annual safety report should be the same for the competent
authorities concerned and the EC concerned.
12.7 Safety Monitoring Board (SMB)
The safety of the study will be judged by an independent committee of experts on regular basis,
at a frequency of at least once a year via personal meeting or telephone conference. The
members of this board will have access to the unblinded data, to all SAEs and SUSARs and to
the inclusion and drop-out rates. This Safety Monitoring Board (SMB) will suggest changes to
the protocol or provide an altered judgement of feasibility if information from the annual safety
report or new information about the applied study medication has become available

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 57 of 71
13. Ethical and regulatory aspects
13.1 Sponsor‘s and investigator’s responsibilities This study is conducted in compliance with all applicable laws and regulations and also the
Declaration of Helsinki. The sponsor has the overall responsibility for the ethical and scientific
conduct of the study. All participating investigators agree to adhere to the instructions and
procedures described in the study protocol and thereby to adhere to the principles of GCP that
it conforms too.
The ethics committee and BfArM will review the final study documents. The ethics committee's
and BfArM’s decision concerning the conduct of the study will be communicated in written form
to the sponsor. The sponsor will assure submission of required progress reports, annual safety
reports and substantial amendments for approval to the ethics committee and BfArM. Before
initiating the study, the sponsor must submit any required amendments to BfArM for review and
acceptance to begin the trial according to § 42 AMG. Furthermore, the sponsor has to inform the
ethics committee and BfArM within 90 days about completion of the trial and provide a brief
report of its outcome 1 year after completion of the trial.
13.2 Ethics committees and health authorities
Prior to the start of this study, the protocol and other required documents have to be reviewed
and approved by the ethics committee and BfArM before study initiation. Any amendments to
the protocol, other than administrative ones (of which the ethics committee and BfArM will merely
be informed), must be reviewed and approved by both.
Before inclusion of the first patient the federal state authorities (zuständige Regierungsbehörden
der Länder) will be informed about the study. A copy of this report needs to be forwarded to
BfArM and needs to be filed in the ISF and TMF.
13.3 Ethical performance of the study
The study is conducted according to the ethical principles as defined in the Declaration of
Helsinki. The present clinical study is conducted in accordance with principles published in the
Good Clinical Practice Guideline (ICH-GCP) and the applicable legal regulations (AMG, GCP-
V). These principles concern ethics committee procedures, patient information and informed
consent procedures, adherence to the protocol, administrative documents, documentation of the
study medication, data collection, patient records (source documents), recording and reporting
of AEs/SAEs, preparation of inspections and audits as well as storage and safekeeping of the
documents. All the investigators and personnel involved in the study have been informed that
the competent authorities and the sponsor are authorised to review the study documents and
patient files.
13.4 Informed consent of the study participants
A patient can only be included in the study, if he provides written consent after being informed
by a GCP-trained investigator (orally and in writing) about the nature, significance and scope of
the clinical study in an appropriate and understandable way. Additionally, a doctor that is not
associated to the study (board certified psychiatrist/Facharzt für Psychiatrie und Psychotherapie)

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 58 of 71
has to confirm and sign in the document that the patient can give informed consent in the
informed consent document. The assessment of the clinician (board certified
psychiatrist/Facharzt für Psychiatrie und Psychotherapie) is considered to be the gold standard
in the checking the capability of consent among schizophrenia patients (Cordes, Wölwer et al.
2013). The investigator must fully explain the purpose of the study to the patient or his/her
guardian prior to entering the patient into the study. The investigator is responsible for obtaining
written informed consent from each patient. The person signing the consent form will receive an
original of the signed form. By providing such consent the patient is declaring that he
understands and accepts the recording of data that is part of the study and its verification by
authorised monitors or federal authorities. The patient will be educated about the potential
benefits and complications of the IMP used in the study. It must be clear to him/her that he/she
can withdraw his/her consent at any point of time without any disadvantages to his/her further
treatment. One original of the written ICF will be kept in the study folder (ISF) of the study site.
Another original of the written patient information and ICF will be given to the patient.
Additionally, copies of both documents will be filed in the patient’s medical file. The patient
information and ICF will be submitted to the responsible ethics committee for assessment before
the study will be initiated.
During the study visits of the double-blind treatment phase, potential psychopathological
worsening of the patient is documented. Psychopathological worsening is defined as an increase
of at least 20 % in the PANSS total score. If psychopathological worsening is suspected, an
additional PANSS rating should be performed. The 20% threshold was derived from the
response criteria published by Leucht 2014. In the case of a psychopathological worsening or in
case of any SAE, a clinician that is not associated to the study (board certified
psychiatrist/Facharzt für Psychiatrie und Psychotherapie) has to reconfirm and document that
the patient continues to be capable to consent. If not, the patient has to be excluded from the
study.

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 59 of 71
14. Study administration
14.1 Documentation and Data collection
The documentation of the study data in adherence to the GCP-guidelines and the clinical trial
protocol is the responsibility of the investigator. Original data (source documents) remain in
hospital medical record and information on the eCRF must be traceable and consistent with the
original data. Source documents are e.g. laboratory results. Original written informed consent
signed by the patient is kept by the investigator and a signed copy will be given to the patient.
No information in source documents about the identity of the patients will be disclosed. All data
collected in this study must be entered in an eCRF which has to be completed by the investigator
or authorized trial personnel and signed by the investigator. This also applies for those patients
who do not complete the study. If a patient withdraws from the study, the reason must be
recorded on the eCRF. The investigator is responsible for ensuring the accuracy, completeness,
and timeliness of all data reported to the sponsor in the eCRFs and in all required reports.
After database lock, the principal investigator will receive data of the investigational site for
archiving in the Investigator Site File (ISF).
14.2 Database management
Data are administered and processed by data management of the MSZ with the support of a
study database (eCRF) according to the SOPs of the MSZ. Data of the optional assessments
will not be added to the eCRF – we will only document whether these procedures were
performed or not.
A description of the study specific processes is given in the Data Management Plan that details
the key planning and control elements for the data management component of the study.
The evaluation of the data takes place by programmed validity- and consistency checks. In
addition a manual/visual evaluation of plausibility is performed in accordance to the requirements
of GCP. Queries may occur, which will be visualized on the study database. The investigator
has to resolve all data discrepancies in the study database.
After entry of all collected data and clarification of all queries, the database will be closed at the
completion of the study. The database closure has to be documented.
Data and results electronically recorded will be archived according to legal guidelines. Separate
databases for the main study (V1-V13) and for the naturalistic follow-up study will be created.
All pseudonymized data can be shared between the main study and the naturalistic follow-up
study via a secure procedure fulfilling all necessities of data protection.
14.3 Audits and inspections
As part of quality assurance according to GCP, the sponsor and the competent health authorities
have the right to audit/inspect the study sites and any other institutions involved in the trial. The
aim of an audit/inspection is to verify the validity, accuracy and completeness of data, to establish
the credibility of the clinical trial, and to check whether the trial subject’s rights and trial subject
safety are being maintained. The sponsor may assign these activities to persons otherwise not
involved in the trial (auditors). These persons as well as inspectors are allowed to access all trial

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 60 of 71
documentation (especially the trial protocol, eCRFs, trial subjects’ medical records, drug
accountability documentation, and trial-related correspondence).
The sponsor and all investigators of the participating study sites undertake all efforts to support
auditors and inspections by the competent authorities at all times and to allow the persons
charged with these duties access to the necessary original documentation.
All persons conducting audits undertake to keep all trial subject data and other trial data
confidential.
After each external audit the investigator receives an audit confirmation from the responsible
auditor. This confirmation has to be stored in the ISF in order to provide access to it in case of
an inspection by the competent authorities. The audit report is provided to the sponsor for
control. At the end of the study an audit certificate is added to the final report. A respective
inspection report is provided to the trial centers for notification.
14.4 Monitoring
Monitoring activities are performed to ensure that the trial is conducted in accordance with the
trial protocol, the principles of GCP and local legislation. A monitoring plan describing the scope
of the monitoring activities in detail will be prepared.
The responsible monitor will contact the investigator and will be allowed, on request, to inspect
the various records of the trial (eCRF and other pertinent data) provided that patient
confidentiality is maintained in accord with local requirements. The monitor should have access
to patient records, any information needed to verify the entries in the eCRF and all necessary
information and essential study documents. The investigator agrees to cooperate with the
monitor to ensure that any problems detected in the course of these monitoring visits are
resolved. A monitoring visit report is prepared for each visit describing the progress of the clinical
trial and all identified problems. Monitoring will take place exclusively for the main study (until
V13).
14.5 Archiving
At the end of the clinical study all study-relevant data must be archived as required by law and
when indicated in addition according to the Clinical Trial Agreement. All documentation forms,
ICFs and other essential study documents must be retained as required by law. Patient ID lists
and patient files are retained in the respective study sites separately. The ICFs are kept in with
the study documents.
14.6 Reporting
Final report
Within one year of the completion of the trial, BfArM and the ethics committee will be supplied
with a summary of the final report on the clinical trial containing the principle results. The sponsor
delegated person is responsible for the generation of the final report.

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 61 of 71
Publications
The trial protocol will be published after initiation of the first study center in a peer-reviewed
scientific journal following the CONSORT guideline (www.consort-statement.org)..
We will build up a trial website with a section in layman’s language for patients and relatives,
involving national patient and relative societies from the beginning, and actively maintain social
media accounts (e.g. science-to-public [Facebook], science-to-science [LinkedIn,
ResearchGate], and science-to-press [Twitter]). During the trial, we will continuously inform
professionals and the public about the current progress via our website and social media
accounts.
Upon availability of the final results, we will publish all findings, positive or negative, in
international peer-reviewed journals and thereby use open-access options if possible to the
largest possible extent (e.g. according to the CONSORT statement). No secondary endpoint
result will be published prior to the publication of the primary endpoint.
Financing of the study
The study is funded by the Deutsche Forschungsgemeinschaft (DFG), grant number: HA
6091/4-1, project number: 316096538 (http://gepris.dfg.de/gepris/projekt/316096538).
Public Register
Before the study begins, the clinical study will be filed at an international public register (e.g.
clinicaltrials.gov or WHO clinical trials).
Data Sharing
Is not planned.
14.7 Insurance for subjects participating in clinical studies
In the clinical study of a drug all the participants are insured in accordance with the German
Medicines Act. The scope of the insurance coverage is derived from the insurance documents
that are included in the investigator’s folder. Before inclusion, the insurance conditions shall be
submitted to the patient for review without request to do so.
The insurance conditions should be furnished to the patient to take with him before being
included in the study on request and after inclusion in any case.
Insurance cover age is being provided by the following insurance provider:
HDI Global SE
Niederlassung Düsseldorf
Am Schönenkamp 45 – D-40599 Düsseldorf
Tel.: 0211/7482-199
E-Mail: [email protected]
Insurance Number: 76294201 03011
represented by
ECCLESIA Versicherungsdienst GmbH
Ecclesiastraße 1-4 - 32758 Detmold
Tel.: 05231/603-307

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 62 of 71
Fax: 05231/603-60307
www.em-hospital.de
If an insured event is suspected to have occurred, the principal investigator of the clinical study
is to be notified of such event immediately. He shall notify the insurance provider about the
damages immediately.
The patient receives a copy of the notification to the insurance provider.
The patient may also inform the insurance provider by bypassing the personnel involved in the
study and reporting any claims. He must be told about both options. Furthermore, he should also
be informed that he shall inform the principal investigator of the clinical study in the event of any
notification sent by him on his own.
Moreover, a home-to-clinic accident insurance has been concluded:
SV SparkassenVersicherung AG
Bahnhofstr. 69, D-65021 Wiesbaden
Tel.: 0611/178-100
Fax: 0611/178-109
Insurance Number: 50074652021
Client Number: 0 049 107 784-4
represented by
ECCLESIA Versicherungsdienst GmbH
Ecclesiastraße 1-4 – D-32758 Detmold
Tel.: 05231/603-307
Fax: 05231/603-60307
www.em-hospital.de
14.8 Data Privacy Protection and Confidentiality protection
The applicable local regulations of data privacy protection will be followed. The patients will be
informed that any patient-related data and materials will be appropriately made pseudonymous
(pursuant to § 12 and § 13 of the GCP Regulations) and that these data may be used for analysis
and publication purposes. Furthermore, the patients will be informed that their data may be
inspected by representatives of BfArM or of the sponsor for the purpose of validation of a proper
study conduct. Patients who do not provide consent for transmission of their data, according to
the data protection agreement included in the ICF, will not be included in the clinical study.
14.9 Protocol amendments or changes in trial conduct
In order to insure comparable conditions in all study sites and in the interest of standardized
evaluations of the trial, changes in this protocol are not foreseen. However, changes in trial
conduct are possible. Any change (besides administrative changes) of this protocol requires a
written protocol amendment that must be reviewed by the sponsor before implementation.
Furthermore, consent needs to be obtained by the investigator of each participating site.
Amendments that significantly affect the safety of subjects, the scope of the investigation or the
scientific quality of the study, additionally require approval of the ethics committee and the
investigator at each study site. Examples of amendments requiring such approval are:

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 63 of 71
Significant changes in the study design;
Increases in the number of invasive procedures.
However, these requirements for approval should in no way prevent the investigator or sponsor
to take any immediate action in the interests of preserving patient safety. If the investigator feels
an immediate change to the protocol is necessary and is implemented for safety reasons, the
sponsor, ethics committee and BfArM must be informed immediately. Amendments affecting
only administrative aspects of the study do not require formal protocol amendments or ethics
committee and BfArM approval. However, the ethics committee and BfArM must still be notified
about the changes.

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 64 of 71
15. Abbreviation
AE Adverse Event
BfArM Bundesinstitut für Arzneimittel und Medizinprodukte
BW Body Weight
CRF Case Report Form
eCRF electronic Case Report Form
FPFV First Patient First Visit
GCP Good Clinical Practice
ITT Intention to Treat
kg Kilogramm
LPFV Last Patient First Visit
LPLV Fast Patient Last Visit
MSZ Münchner Studienzentrum
PP Per Protocol
PLC Placebo
SAE Serious Adverse Event

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 65 of 71
16. Annex
16.1 Declaration of Helsinki
The declaration of Helsinki it its recent version is attached to the protocol.

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 66 of 71
16. Reference List
Addington, D., J. Addington and E. Maticka-Tyndale (1993). "Assessing depression in schizophrenia: the Calgary Depression Scale." Br J Psychiatry Suppl(22): 39-44.
Agachan, F., T. Chen, J. Pfeifer, P. Reissman and S. D. Wexner (1996). "A constipation scoring system to simplify evaluation and management of constipated patients." Dis Colon Rectum 39(6): 681-685.
Agid, O., T. Arenovich, G. Sajeev, R. B. Zipursky, S. Kapur, G. Foussias and G. Remington (2011). "An algorithm-based approach to first-episode schizophrenia: response rates over 3 prospective antipsychotic trials with a retrospective data analysis." J Clin Psychiatry 72(11): 1439-1444.
AlAqeel, B. and H. C. Margolese (2012). "Remission in schizophrenia: critical and systematic review." Harv Rev Psychiatry 20(6): 281-297.
An der Heiden, W. and H. Hafner (2010). Course and Outcome. Chichester, Wiley-Blackwell.
Andreasen, N. C., W. T. Carpenter, Jr., J. M. Kane, R. A. Lasser, S. R. Marder and D. R. Weinberger (2005). "Remission in schizophrenia: proposed criteria and rationale for consensus." Am J Psychiatry 162(3): 441-449.
Babor, T. F. (2001). "AUDIT-The Alcohol Use Disorders Identification Test-Guidelines for Use in Primary Care." Second Edition.
Barnes, T. R. (1989). "A rating scale for drug-induced akathisia." Br J Psychiatry 154: 672-676.
Beitinger, R., J. Lin, W. Kissling and S. Leucht (2008). "Comparative remission rates of schizophrenic patients using various remission criteria." Prog Neuropsychopharmacol Biol Psychiatry 32(7): 1643-1651.
Benkert, O. and H. Hippius (2013). Kompendium der Psychiatrischen Pharmakotherapie, Springer.
BfArM, B. (2011). "Anforderungen zur Statistik bei Anträgen auf Genehmigung einer klinischen Prüfung mit Medizinprodukten." from http://www.bfarm.de/DE/Medizinprodukte/klinPrMP/ao-kp-gen_anlage.html.
Bohn, M. J., T. F. Babor and H. R. Kranzler (1995). "The Alcohol Use Disorders Identification Test (AUDIT): validation of a screening instrument for use in medical settings." J Stud Alcohol 56(4): 423-432.
Bondolfi, G., H. Dufour, M. Patris, J. P. May, U. Billeter, C. B. Eap and P. Baumann (1998). "Risperidone versus clozapine in treatment-resistant chronic schizophrenia: a randomized double-blind study. The Risperidone Study Group." Am J Psychiatry 155(4): 499-504.
Boter, H., J. Peuskens, J. Libiger, W. W. Fleischhacker, M. Davidson, S. Galderisi, R. S. Kahn and E. s. group (2009). "Effectiveness of antipsychotics in first-episode schizophrenia and schizophreniform disorder on response and remission: an open randomized clinical trial (EUFEST)." Schizophr Res 115(2-3): 97-103.
Buchanan, R. W., J. Kreyenbuhl, D. L. Kelly, J. M. Noel, D. L. Boggs, B. A. Fischer, S. Himelhoch, B. Fang, E. Peterson, P. R. Aquino, W. Keller and T. Schizophrenia Patient Outcomes Research (2010). "The 2009 schizophrenia PORT psychopharmacological treatment recommendations and summary statements." Schizophr Bull 36(1): 71-93.
Citrome, L., J. P. McEvoy and S. R. Saklad (2016). "A Guide to the Management of Clozapine-Related Tolerability and Safety Concerns." Clin Schizophr Relat Psychoses.
Cordes, J., W. Wölwer, A. Lowe and W. Gaebel (2013). Ethische Probleme in der Schizophrenieforschung Ethik psychiatrischer Forschung. H. Helmchen. Berlin Heidelberg, Springer. 1: 170.

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 67 of 71
De Fazio, P., R. Gaetano, M. Caroleo, G. Cerminara, F. Maida, A. Bruno, M. R. Muscatello, M. J. Moreno, E. Russo and C. Segura-Garcia (2015). "Rare and very rare adverse effects of clozapine." Neuropsychiatr Dis Treat 11: 1995-2003.
DGPPN (2006). S3 Praxisleitlinien in Psychiatrie und Psychotherapie. Band 1 - Behandlungsleitlinie Schizophrenie. Darmstadt, Steinkopff-Verlag.
DGPPN (2018). "S3-Leitlinie Schizophrenie Stand 29.08.2018."
EMA. (2010). "Guideline on Missing Data in Confirmatory Clinical Trials ", from www.ema.europa.eu.
Endicott, J., R. L. Spitzer, J. L. Fleiss and J. Cohen (1976). "The global assessment scale. A procedure for measuring overall severity of psychiatric disturbance." Arch Gen Psychiatry 33(6): 766-771.
Essali, A., N. Al-Haj Haasan, C. Li and J. Rathbone (2009). "Clozapine versus typical neuroleptic medication for schizophrenia." Cochrane Database Syst Rev(1): CD000059.
Fagerstrom, K. O. and N. G. Schneider (1989). "Measuring nicotine dependence: a review of the Fagerstrom Tolerance Questionnaire." J Behav Med 12(2): 159-182.
Galderisi, S., M. Davidson, R. S. Kahn, A. Mucci, H. Boter, M. D. Gheorghe, J. K. Rybakowski, J. Libiger, S. Dollfus, J. J. Lopez-Ibor, J. Peuskens, L. G. Hranov, W. W. Fleischhacker and E. group (2009). "Correlates of cognitive impairment in first episode schizophrenia: the EUFEST study." Schizophr Res 115(2-3): 104-114.
Galling, B., A. Roldan, K. Hagi, L. Rietschel, F. Walyzada, W. Zheng, X. L. Cao, Y. T. Xiang, M. Zink, J. M. Kane, J. Nielsen, S. Leucht and C. U. Correll (2017). "Antipsychotic augmentation vs. monotherapy in schizophrenia: systematic review, meta-analysis and meta-regression analysis." World Psychiatry 16(1): 77-89.
Gerlach, J., S. Korsgaard, P. Clemmesen, A. M. Lauersen, G. Magelund, U. Noring, U. J. Povlsen, P. Bech and D. E. Casey (1993). "The St. Hans Rating Scale for extrapyramidal syndromes: reliability and validity." Acta Psychiatr Scand 87(4): 244-252.
Gorwood, P., J. Peuskens and R. i. S. European Group On Functional Outcomes (2012). "Setting new standards in schizophrenia outcomes: symptomatic remission 3 years before versus after the Andreasen criteria." Eur Psychiatry 27(3): 170-175.
Gustavsson, A., M. Svensson, F. Jacobi, C. Allgulander, J. Alonso, E. Beghi, R. Dodel, M. Ekman, C. Faravelli, L. Fratiglioni, B. Gannon, D. H. Jones, P. Jennum, A. Jordanova, L. Jonsson, K. Karampampa, M. Knapp, G. Kobelt, T. Kurth, R. Lieb, M. Linde, C. Ljungcrantz, A. Maercker, B. Melin, M. Moscarelli, A. Musayev, F. Norwood, M. Preisig, M. Pugliatti, J. Rehm, L. Salvador-Carulla, B. Schlehofer, R. Simon, H. C. Steinhausen, L. J. Stovner, J. M. Vallat, P. Van den Bergh, J. van Os, P. Vos, W. Xu, H. U. Wittchen, B. Jonsson, J. Olesen and C. D. Group (2011). "Cost of disorders of the brain in Europe 2010." Eur Neuropsychopharmacol 21(10): 718-779.
Guy W, B. R. (1976). CGI: Clinical Global Impressions., National Institute of Mental Health.
Hahn, T., T. Kircher, B. Straube, H. U. Wittchen, C. Konrad, A. Strohle, A. Wittmann, B. Pfleiderer, A. Reif, V. Arolt and U. Lueken (2015). "Predicting treatment response to cognitive behavioral therapy in panic disorder with agoraphobia by integrating local neural information." JAMA Psychiatry 72(1): 68-74.
Hasan, A., P. Falkai, T. Wobrock, J. Lieberman, B. Glenthoj, W. F. Gattaz, F. Thibaut, H. J. Moller and S. World Federation of Societies of Biological Psychiatry Task Force on Treatment Guidelines for (2012). "World Federation of Societies of Biological Psychiatry (WFSBP) Guidelines for Biological Treatment of Schizophrenia, part 1: update 2012 on the acute treatment of schizophrenia and the management of treatment resistance." World J Biol Psychiatry 13(5): 318-378.

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 68 of 71
Hasan, A., T. Wobrock, B. Guse, B. Langguth, M. Landgrebe, P. Eichhammer, E. Frank, J. Cordes, W. Wolwer, F. Musso, G. Winterer, W. Gaebel, G. Hajak, C. Ohmann, P. E. Verde, M. Rietschel, R. Ahmed, W. G. Honer, P. Dechent, B. Malchow, M. F. U. Castro, D. Dwyer, C. Cabral, P. M. Kreuzer, T. B. Poeppl, T. Schneider-Axmann, P. Falkai and N. Koutsouleris (2017). "Structural brain changes are associated with response of negative symptoms to prefrontal repetitive transcranial magnetic stimulation in patients with schizophrenia." Mol Psychiatry 22(6): 857-864.
Hodge, K. and S. Jespersen (2008). "Side-effects and treatment with clozapine: a comparison between the views of consumers and their clinicians." Int J Ment Health Nurs 17(1): 2-8.
Hogan, T. P., A. G. Awad and R. Eastwood (1983). "A self-report scale predictive of drug compliance in schizophrenics: reliability and discriminative validity." Psychol Med 13(1): 177-183.
Howes, O. D., R. McCutcheon, O. Agid, A. de Bartolomeis, N. J. van Beveren, M. L. Birnbaum, M. A. Bloomfield, R. A. Bressan, R. W. Buchanan, W. T. Carpenter, D. J. Castle, L. Citrome, Z. J. Daskalakis, M. Davidson, R. J. Drake, S. Dursun, B. H. Ebdrup, H. Elkis, P. Falkai, W. W. Fleischacker, A. Gadelha, F. Gaughran, B. Y. Glenthoj, A. Graff-Guerrero, J. E. Hallak, W. G. Honer, J. Kennedy, B. J. Kinon, S. M. Lawrie, J. Lee, F. M. Leweke, J. H. MacCabe, C. B. McNabb, H. Meltzer, H. J. Moller, S. Nakajima, C. Pantelis, T. Reis Marques, G. Remington, S. L. Rossell, B. R. Russell, C. O. Siu, T. Suzuki, I. E. Sommer, D. Taylor, N. Thomas, A. Ucok, D. Umbricht, J. T. Walters, J. Kane and C. U. Correll (2017). "Treatment-Resistant Schizophrenia: Treatment Response and Resistance in Psychosis (TRRIP) Working Group Consensus Guidelines on Diagnosis and Terminology." Am J Psychiatry 174(3): 216-229.
Howes, O. D., F. Vergunst, S. Gee, P. McGuire, S. Kapur and D. Taylor (2012). "Adherence to treatment guidelines in clinical practice: study of antipsychotic treatment prior to clozapine initiation." Br J Psychiatry 201(6): 481-485.
Hynes, C., D. Keating, S. McWilliams, K. Madigan, A. Kinsella, I. Maidment, C. Feetam, R. J. Drake, P. M. Haddad, F. Gaughran, M. Taylor and M. Clarke (2015). "Glasgow Antipsychotic Side-effects Scale for Clozapine - Development and validation of a clozapine-specific side-effects scale." Schizophr Res 168(1-2): 505-513.
Insel, T. R. and B. J. Sahakian (2012). "Drug research: a plan for mental illness." Nature 483(7389): 269.
Jones, S. H., G. Thornicroft, M. Coffey and G. Dunn (1995). "A brief mental health outcome scale-reliability and validity of the Global Assessment of Functioning (GAF)." Br J Psychiatry 166(5): 654-659.
Kahn, R. S., W. W. Fleischhacker, H. Boter, M. Davidson, Y. Vergouwe, I. P. Keet, M. D. Gheorghe, J. K. Rybakowski, S. Galderisi, J. Libiger, M. Hummer, S. Dollfus, J. J. Lopez-Ibor, L. G. Hranov, W. Gaebel, J. Peuskens, N. Lindefors, A. Riecher-Rossler, D. E. Grobbee and E. s. group (2008). "Effectiveness of antipsychotic drugs in first-episode schizophrenia and schizophreniform disorder: an open randomised clinical trial." Lancet 371(9618): 1085-1097.
Kane, J., G. Honigfeld, J. Singer and H. Meltzer (1988). "Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine." Arch Gen Psychiatry 45(9): 789-796.
Kay, S. R., A. Fiszbein and L. A. Opler (1987). "The positive and negative syndrome scale (PANSS) for schizophrenia." Schizophr Bull 13(2): 261-276.
Kemmler, G., M. Hummer, C. Widschwendter and W. W. Fleischhacker (2005). "Dropout rates in placebo-controlled and active-control clinical trials of antipsychotic drugs: a meta-analysis." Arch Gen Psychiatry 62(12): 1305-1312.
Kircher, T., A. Krug, M. Stratmann, S. Ghazi, C. Schales, M. Frauenheim, L. Turner, P. Fahrmann, T. Hornig, M. Katzev, M. Grosvald, R. Muller-Isberner and A. Nagels (2014). "A rating scale for the assessment of objective and subjective formal Thought and Language Disorder (TALD)." Schizophr Res 160(1-3): 216-221.

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 69 of 71
Koutsouleris, N., T. Wobrock, B. Guse, B. Langguth, M. Landgrebe, P. Eichhammer, E. Frank, J. Cordes, W. Wolwer, F. Musso, G. Winterer, W. Gaebel, G. Hajak, C. Ohmann, P. E. Verde, M. Rietschel, R. Ahmed, W. G. Honer, D. Dwyer, F. Ghaseminejad, P. Dechent, B. Malchow, P. M. Kreuzer, T. B. Poeppl, T. Schneider-Axmann, P. Falkai and A. Hasan (2017). "Predicting Response to Repetitive Transcranial Magnetic Stimulation in Patients With Schizophrenia Using Structural Magnetic Resonance Imaging: A Multisite Machine Learning Analysis." Schizophr Bull.
Lambert, M., A. Karow, S. Leucht, B. G. Schimmelmann and D. Naber (2010). "Remission in schizophrenia: validity, frequency, predictors, and patients' perspective 5 years later." Dialogues Clin Neurosci 12(3): 393-407.
Leucht, S. (2014). "Measurements of response, remission, and recovery in schizophrenia and examples for their clinical application." J Clin Psychiatry 75 Suppl 1: 8-14.
Leucht, S., R. Beitinger and W. Kissling (2007). "On the concept of remission in schizophrenia." Psychopharmacology (Berl) 194(4): 453-461.
Leucht, S., A. Cipriani, L. Spineli, D. Mavridis, D. Orey, F. Richter, M. Samara, C. Barbui, R. R. Engel, J. R. Geddes, W. Kissling, M. P. Stapf, B. Lassig, G. Salanti and J. M. Davis (2013). "Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis." Lancet 382(9896): 951-962.
Leucht, S., C. Leucht, M. Huhn, A. Chaimani, D. Mavridis, B. Helfer, M. Samara, M. Rabaioli, S. Bacher, A. Cipriani, J. R. Geddes, G. Salanti and J. M. Davis (2017). "Sixty Years of Placebo-Controlled Antipsychotic Drug Trials in Acute Schizophrenia: Systematic Review, Bayesian Meta-Analysis, and Meta-Regression of Efficacy Predictors." Am J Psychiatry: appiajp201716121358.
Lieberman, J. A., M. Phillips, H. Gu, S. Stroup, P. Zhang, L. Kong, Z. Ji, G. Koch and R. M. Hamer (2003). "Atypical and conventional antipsychotic drugs in treatment-naive first-episode schizophrenia: a 52-week randomized trial of clozapine vs chlorpromazine." Neuropsychopharmacology 28(5): 995-1003.
Lieberman, J. A., T. S. Stroup, J. P. McEvoy, M. S. Swartz, R. A. Rosenheck, D. O. Perkins, R. S. Keefe, S. M. Davis, C. E. Davis, B. D. Lebowitz, J. Severe, J. K. Hsiao and I. Clinical Antipsychotic Trials of Intervention Effectiveness (2005). "Effectiveness of antipsychotic drugs in patients with chronic schizophrenia." N Engl J Med 353(12): 1209-1223.
Lindenmayer, J. P., P. Czobor, L. Alphs, A. M. Nathan, R. Anand, Z. Islam, J. C. Chou and P. T. S. G. InterSe (2003). "The InterSePT scale for suicidal thinking reliability and validity." Schizophr Res 63(1-2): 161-170.
Martin, J. L., V. Perez, M. Sacristan, F. Rodriguez-Artalejo, C. Martinez and E. Alvarez (2006). "Meta-analysis of drop-out rates in randomised clinical trials, comparing typical and atypical antipsychotics in the treatment of schizophrenia." Eur Psychiatry 21(1): 11-20.
Meltzer, H. Y. (2012). "Clozapine: balancing safety with superior antipsychotic efficacy." Clin Schizophr Relat Psychoses 6(3): 134-144.
Meltzer, H. Y., W. V. Bobo, A. Roy, K. Jayathilake, Y. Chen, A. Ertugrul, A. E. Anil Yagcioglu and J. G. Small (2008). "A randomized, double-blind comparison of clozapine and high-dose olanzapine in treatment-resistant patients with schizophrenia." J Clin Psychiatry 69(2): 274-285.
Müller, M. J. and O. Benkert (2017). Antipsychotika. Kompendium der Psychiatrischen Pharmakotherapie,. O. Benkert and H. Hippius. Berlin, Heidelberg, Springer Verlag. 11: 269 - 488.
Naber, D. (1995). "A self-rating to measure subjective effects of neuroleptic drugs, relationships to objective psychopathology, quality of life, compliance and other clinical variables." Int Clin Psychopharmacol 10 Suppl 3: 133-138.

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 70 of 71
Nasrallah, H., P. Morosini and D. D. Gagnon (2008). "Reliability, validity and ability to detect change of the Personal and Social Performance scale in patients with stable schizophrenia." Psychiatry Res 161(2): 213-224.
Redlich, R., C. Burger, K. Dohm, D. Grotegerd, N. Opel, D. Zaremba, S. Meinert, K. Forster, J. Repple, R. Schnelle, C. Wagenknecht, M. Zavorotnyy, W. Heindel, H. Kugel, M. Gerbaulet, J. Alferink, V. Arolt, P. Zwanzger and U. Dannlowski (2017). "Effects of electroconvulsive therapy on amygdala function in major depression - a longitudinal functional magnetic resonance imaging study." Psychol Med 47(12): 2166-2176.
Redlich, R., N. Opel, D. Grotegerd, K. Dohm, D. Zaremba, C. Burger, S. Munker, L. Muhlmann, P. Wahl, W. Heindel, V. Arolt, J. Alferink, P. Zwanzger, M. Zavorotnyy, H. Kugel and U. Dannlowski (2016). "Prediction of Individual Response to Electroconvulsive Therapy via Machine Learning on Structural Magnetic Resonance Imaging Data." JAMA Psychiatry 73(6): 557-564.
Remington, G., O. Agid, G. Foussias, M. Hahn, N. Rao and M. Sinyor (2013). "Clozapine's role in the treatment of first-episode schizophrenia." Am J Psychiatry 170(2): 146-151.
Ritsner, M., R. Kurs, A. Gibel, Y. Ratner and J. Endicott (2005). "Validity of an abbreviated quality of life enjoyment and satisfaction questionnaire (Q-LES-Q-18) for schizophrenia, schizoaffective, and mood disorder patients." Qual Life Res 14(7): 1693-1703.
Ronaldson, K. J., P. B. Fitzgerald, A. J. Taylor, D. J. Topliss and J. J. McNeil (2011). "A new monitoring protocol for clozapine-induced myocarditis based on an analysis of 75 cases and 94 controls." Aust N Z J Psychiatry 45(6): 458-465.
Samara, M. T., M. Dold, M. Gianatsi, A. Nikolakopoulou, B. Helfer, G. Salanti and S. Leucht (2016). "Efficacy, Acceptability, and Tolerability of Antipsychotics in Treatment-Resistant Schizophrenia: A Network Meta-analysis." JAMA Psychiatry 73(3): 199-210.
Sanz-Fuentenebro, J., D. Taboada, T. Palomo, M. Aragues, S. Ovejero, C. Del Alamo and V. Molina (2013). "Randomized trial of clozapine vs. risperidone in treatment-naive first-episode schizophrenia: results after one year." Schizophr Res 149(1-3): 156-161.
Schooler, N. R., S. R. Marder, K. N. Chengappa, G. Petrides, D. Ames, W. C. Wirshing, M. McMeniman, R. W. Baker, H. Parepally, D. Umbricht and J. M. Kane (2016). "Clozapine and risperidone in moderately refractory schizophrenia: a 6-month randomized double-blind comparison." J Clin Psychiatry 77(5): 628-634.
Stjernsward, S., K. Persson, R. Nielsen, E. Tuninger and S. Levander (2013). "A modified Drug Attitude Inventory used in long-term patients in sheltered housing." Eur Neuropsychopharmacol 23(10): 1296-1299.
Stroup, T. S., T. Gerhard, S. Crystal, C. Huang and M. Olfson (2015). "Comparative Effectiveness of Clozapine and Standard Antipsychotic Treatment in Adults With Schizophrenia." Am J Psychiatry: appiajp201515030332.
Swartz, M. S., T. S. Stroup, J. P. McEvoy, S. M. Davis, R. A. Rosenheck, R. S. Keefe, J. K. Hsiao and J. A. Lieberman (2008). "What CATIE found: results from the schizophrenia trial." Psychiatr Serv 59(5): 500-506.
Tiihonen, J., E. Mittendorfer-Rutz, M. Majak, J. Mehtala, F. Hoti, E. Jedenius, D. Enkusson, A. Leval, J. Sermon, A. Tanskanen and H. Taipale (2017). "Real-World Effectiveness of Antipsychotic Treatments in a Nationwide Cohort of 29823 Patients With Schizophrenia." JAMA Psychiatry 74(7): 686-693.
Tollefson, G. D., M. A. Birkett, G. M. Kiesler, A. J. Wood and G. Lilly Resistant Schizophrenia Study (2001). "Double-blind comparison of olanzapine versus clozapine in schizophrenic patients clinically eligible for treatment with clozapine." Biol Psychiatry 49(1): 52-63.
Tombaugh, T. N. (2004). "Trail Making Test A and B: normative data stratified by age and education." Arch Clin Neuropsychol 19(2): 203-214.

EARLY- Protocol final Version 5.0 AM3.0 – 30.11.2020 EudraCT-No.: 2018-001514-15 Confidential 71 of 71
Ware, J., Jr., M. Kosinski and S. D. Keller (1996). "A 12-Item Short-Form Health Survey: construction of scales and preliminary tests of reliability and validity." Med Care 34(3): 220-233.
Zeidler, J., L. Slawik, J. Fleischmann and W. Greiner (2012). "The costs of schizophrenia and predictors of hospitalisation from the statutory health insurance perspective." Health Econ Rev 2(1): 9.
Zhu, Y., M. Krause, M. Huhn, P. Rothe, J. Schneider-Thoma, A. Chaimani, C. Li, J. M. Davis and S. Leucht (2017). "Antipsychotic drugs for the acute treatment of patients with a first episode of schizophrenia: a systematic review with pairwise and network meta-analyses." Lancet Psychiatry.
Zhu, Y., C. Li, M. Huhn, P. Rothe, M. Krause, I. Bighelli, J. Schneider-Thoma and S. Leucht (2017). "How well do patients with a first episode of schizophrenia respond to antipsychotics: A systematic review and meta-analysis." Eur Neuropsychopharmacol.