effects of ca2+ channel blockers on ca2+ translocation ... · inhibit ca2' translocation...
TRANSCRIPT

Journal of Neurochemistry Raven Press. New York 0 I986 International Society for Neurochcmistry
Effects of Ca2+ Channel Blockers on Ca2+ Translocation Across Synaptosomal Membranes
C. A. M. Carvalho, 0. P. Coutinho, and A. P. Carvalho
Center for Cell Biology, Department of Zoology, University of Coimbra, Coimbra, Portugal
Abstract: The binding of ['Hlnimodipine to purified synap tic plasma membranes (SPM) isolated from sheep brain cor- tex was characterized, and the effects of nimodipine, nifedi- pine, and (+)+erapamil on the ['Hlnimodipine binding were compared to the effects on 45Ca2+ translocation under conditions that separate 45Ca2' fluxes through Ca" chan- nels fiom 4sCa2' uptake via Na'/Ca2' exchange. [3H]Ni- modipine labels a single class of sites in SPM, with a KD of 0.64 f 0.1 nM, a Em of 16 1 2 27 fmol . mg-' protein, and a Hill slope of 1.07, at 25'C. Competition of ['Hlnimodipine binding to purified SPM with unlabelled Ca2' channel blockers shows that: ( I ) dedipine and nimodipine are po- tent competitors, with IC, values of 4.7 nM and 5.9 nM, respectively; (2) verapamil and (-)-D 600 are partial com- petitors, with biphasic competition behavior. Thus, (+)-ver- apamil shows an IC, of 708 &for the higher af5nity com- ponent and the maximal inhibition is 50% of the specific binding, whereas for (-)+erapamil the IC, is 120 nM, and the maximal inhibition is 30%; (-)-D 600 is even less potent than verapamil in inhibiting [3H]nimdpine binding (ICs0 = 430 nM). However, (+)-verapamil, nifedipine, and ni- modipine are less potent in inhibiting depolarization-in- d u d 45Ca2' influx into synaptosomes in the absence of Na+/Ca2' exchange than in competing for ['Hlnimodipine binding. Thus, (+)-verapamil inhibits Ca" influx by 50%
at about 500 p M , whereas it inhibits 50% of the binding at concentrations 200-fold lower, and the discrepancy is even larger for the dihydropyridines. The Na'/Ca2' exchange and the ATP-dependent Ca2+ uptake by SPM vesicles are also inhibited by the Ca2+ channel blockers verapamil, nifedipine, and d-cis-diltiazem, with similar ICso values and in the same concentration range (10-5-10-3 M) at which they inhibit Caz+ influx through Ca2' channels. We con- clude that high-affinity binding of the Ca2' blockers by SPM is not correlated with inhibition of the Ca2' fluxes through channels in synaptosomes under conditions of minimal Na'/Ca2+ exchange. Furthermore, the relatively high con- centrations of blockers required to block the channels also inhibit Ca2' translocation through the Ca2+-ATPase and the Na'/Ca2' exchanger. In this study, clear differentiation is made of the effects of the Ca2+ channel blockers on these three mechanisms of moving Ca2+ across the synaptosomal membrane, and particular care is taken to separate the con- tribution of the Na+/Ca2+ exchange from that of the Ca2+ channels under conditions of K+ depolarization. Key Words: Synaptosomes-Synaptic membranes-Ca" chan- nel bIockers-['H]Nimodipine-Na+/Ca2+ exchange- Ca2' transport. Carvalho C. A. M. et al. Effects of Ca" channel blockers on Ca2+ translocation across synapto- soma1 membranes. J. Neurochem. 47,1774- I784 ( 1986).
The entry of Ca2' into nerve terminals and axons is controlled mainly by voltage-sensitive Ca'' channels (Baker, 1972; Llink et al., 1972; Baker et al., 1973; Kostyuk, 198 1). Membrane depolarization opens the Ca2+ channels and causes a transient increase in the [ca2+~ that, in turn, leads to neurotransmitter release (Katz and M i l d , 1970; Llink et al., 1972; Hagiwara and Byerly; 198 1).
The Ca channel blockers potently block Ca2' in- flux into smooth and cardiac muscle cells (Fleck-
enstein, 1977; Triggle, 1981), which possess high- affinity binding sites, as determined from studies in muscle membrane fractions using radiolabelled Ca2+ channel blockers (Bellemann et al., 198 1,1982; Ehlert et al., 1982; Glossmann et al., 1982; Bolger et al., 1983). High-affinity binding sites for Ca2+ channel blockers have also been found in homogenates pre- sumably rich in brain membranes (Ferry and Gloss- mann, 1982; Glossmann et al., 1982; Gould et al., 1982; Murphy and Snyder, 1982; Marangos et al.,
Received January21,1986; revised June 14,1986; accepted June 14,1986.
Address correspondence and reprint requests to Lk. C. A, M. Car- valho at Center for cell Biology, Department of Zoology, Univer- sity of Cairnbra, 3049 Coimbra Codex, Portugal.
Abbreviations used: ChCI, choline chloride; HEPES, N-2-hy- droxyethylpiperazine-N'-2-ethanesulfonic acid; SPM, synaptic plasma membrane.
1774

EFFECTS OF Ca2' BLOCKERS ON SYNAPTIC MEMBRANES 1775
1982; Lee et al., 1984). However, in neuronal tissue the presence of voltage-operated Ca2+ channels, sensi- tive to 1 ,Cdihydropyridines, has been clearly demon- strated only in certain neuronal cell lines (Toll, 1982; Takahashi and Ogura, 1983; Albus et al., 1984; Freed- man et al., 1984). In isolated synaptosomes, it was ini- tially shown that depolarization-induced "Ca2' influx is insensitive to dihydropyridines (Nachshen and Blaustein, 1979; Daniel1 et al., 1983). However, the results of a recent study (Turner and Goldin, 1985) indicate that under certain conditions organic cal- cium channel blockers also inhibit 45Ca2+ influx into synaptosomes. Moreover, Middlemiss and Spedding ( 1985) in a recent report show that the agonist Bay K 8644 increases the Ca2+-dependent neurotransmitter release in brain slices, and the effect is reversed by Ca2+ channel blockers. These various results indicate that the dihydropyridine binding site in brain may have a functional correlate, but that there is some difficulty in observing it consistently, especially in iso- lated brain functions.
It must be pointed out that Ca2+ may also enter the nerve cell by Na+/Ca2+ exchange and that increasing the external K+ in substitution for Na' during K+ de- polarization of synaptosomes increases the Ca2+ in- flux through Na'/Ca2+ exchange (Coutinho et al., 1984). The CaZ+ entries through channels and through Na+/Ca2+ exchange often are not separated, and this has added confusion to the field. There is at least one recent report showing that verapamil inhib- its the Na+/Ca2+ exchange in synaptic plasma mem- brane (SPM) vesicles (Erdreich et al., 1983), so that this adds further to the uncertainty regarding the ac- tion of Ca2+ channel blockers on Ca" fluxes studied in brain fractions.
In an attempt to clarify some of these problems in synaptosomes, we first studied the effects of various classes of Ca2' channel blockers on the binding of [ 3H]nimodipine to purified SPM since most previous studies have been performed with gross homogenates, and we then investigated the effects of the blockers on the CaZ+ influx in synaptosomes through the Ca2+ channels, under conditions of minimal Nac/Ca2+ ex- change. Furthermore, we investigated the effect of the Ca" blockers on the Na+/Ca2+ exchange and on the ATP-dependent Ca" transport which regulate the Caz+ concentration in the nerve cell (Baker, 1972; Blaustein, 1977; DiPolo and Beaugk, 1980; Gill et al., 1981; Coutinho et al., 1983, 1984). We conclude that under carefully controlled experimental conditions, which separate depolanzation-induced 45Ca2+ influx from Na+/Ca2' exchange in synaptosomes, voltage- sensitive CaZ+ influx is inhibited only by relatively high concentrations of Ca2+ blockers, and that these concentrations also inhibit the Na+/Ca2+ exchange and the ATP-dependent Ca2+ transport, Therefore, the inherent difficulty in showing in isolated brain fractions an action of Ca" channel blockers on the Ca" channels at nanomolar concentrations of these
drugs remains unresolved and recent results showing the contrary must be viewed with caution (Turner and Goldin, 1985).
MATERIALS AND METHODS Isolation of subcellular fractions from sheep brain
Synaptosomes were isolated from sheep brain cortex ho- mogenates according to the method described by Haj& (1975), with some modifications (Carvalho and Carvalho, 1979). The final synaptosomal pellets were washed by resus- pension in 0.32 M sucrose buffered with 10 mM N-2-hy- droxyethylpiperazine-N'-2-ethanesulfonic acid (HEPES) Tris, pH 7.4, and centrifugation at 20,000 g for 30 min, and, finally, the synaptosomes were resuspended in the same me- dium. This synaptosomal suspension was immediately uti- lized for measuring 45Ca2+ influx due to K+ depolarization, or was submitted to a short preincubation in ionic medium, as described in a subsequent section.
Some synaptosomal pellets were submitted to hypotonic lysis, and SPM were partially purified from the lysate, as described previously by Coutinho et al. (1983). The SPM fractions used for enzymatic characterization were resus- pended in 0.32 M sucrose buffered with 10 mM HEPES Tris, pH 7.4, whereas those used for Ca2+ uptake (Na'/Ca2' exchange or ATPdependent Ca2+ uptake) were washed and resuspended in ionic medium, containing 150 mMNaCl or KCl, 1 mM MgClz, 10 mM glucose, and 10 m M HEPES Tris, pH 7.4, at a protein concentration of about 20 mg/ml, as determined by the biuret method (Layne, 1957).
The microsomal fraction was prepared by centrifugation of the supernatant of the crude mitochondrial fraction at 40,000 g for 30 min, and the pellet was resuspended in 0.25 M sucrose buffered with 50 mM Tris-C1, pH 7.4. This frac- tion, the crude microsomal pellet, was submitted to subfrac- tionation in a sucrose density gradient, as follows: 5-ml por- tions were overlayed on a discontinuous sucrose gradient with the following composition: 8 ml each of 4070, 351, 30%, and 25% (wt/wt) sucrose in buffer (50 MTris-Cl, pH 7.4). The gradients were centrifuged at 60,000 g, for 2 h, at 0-TC, in the swinging bucket rotor of the International B 60 ultracentrifuge. Fractions F, through F4 (see Fig. 1) were collected from the interfaces, diluted five times with icecold 50 mM Tris-C1, pH 7.4, and centrifuged for 30 min at 40,000 g. The pellets used for enzymatic characterization were resuspended in 0.32 M sucrose plus 10 mM HEPES Tris, pH 7.4, whereas the pellets used for ['H]nimodipine binding were resuspended in 50 mM Tris-CI, pH 7.4, di- vided into aliquots, frozen in liquid nitrogen, and kept at -60°C for further analysis. All the experiments were per- formed within 4 weeks after isolation, and the [3H]nimodi- pine binding characteristics were preserved during this pe- riod of storage. [3H]Nimodipine binding
Prior to an experiment on ['Hlnimodipine binding, the purified membrane fractions were thawed at room tempera- ture, and were homogenized at OXC, by using a Polytron appartus (Kinematica GmbH), at setting 5 , with two 10-s bursts at intervals of I5 s. The suspension was then diluted with buffer (50 mM Tris-C1, pH 7.4), to obtain a protein concentration of I mgjml, and was immediately utilized in the binding assay.
The [3H]nimodipine binding assay was performed as fol- lows: 0.6 mg of membrane protein were equilibrated in 1.8 ml of standard buffer (50 mM Tris-C1, pH 7.4) containing
J . Neurochem , Vol 47. N o 6 . 1986

1776 C. A . M. CARVALHO ET AL.
the indicated concentrations of [-'H]nimodipine in the pres- ence (nonspecific binding) and in the absence (total binding) of 5 pM unlabelled nimodipine. The specific [-'H]nimodi- pine binding is defined as the difference in binding under the two conditions. For saturation kinetics ['HHlnirnodipine was varied between 0.05 and 1.5 nM. The competition ex- periments were normally performed with 0.35 nM [-'H]ni- modipine, in the presence of unlabelled drugs (8-10 differ- ent concentrations). In all cases 50-pl aliquots of the incuba- tion mixture were taken for measurement of the concentration of tritiated drug present. Assays were nor- mally performed in triplicate and under yellow dim light because of the light sensitivity of the dihydropyridines.
After the incubation p" nod ' , (45 , min, , normally), mem- brane-bound and free [ Hlnimodipine were separated by rapid vacuum filtration of 0.5-ml samples through What- man GF/B glass fiber filters prewashed with 5 ml of ice-cold (0-4'C) buffer (50 mM Tris-C1, pH 7.4). Filtration of the sample was followed by a 10-ml washing with the same me- dium. Filters, as well as samples of radioactive solutions, were placed in glass vials containing 8 ml of scintillation fluid {composition per liter of toluene: 7.3 g 2,5di- phenyloxazole (PPO), I76 mg pbis-[2-(5-phenyloxazolyl)] benzene (POPOP), and 250 ml Triton X- IOO), and the ra- dioactivity was counted in a Packard TriCarb liquid scintil- lation spectrometer, model 460-CD. The quenching of the radioactivity in the samples was corrected automatically by using an efficiency correlation curve obtained for 3H- quenched standards, by the external standardization method.
Calcium uptake studies Calcium uptake due to K + depolarization. Sheep brain
cortex synaptosomes isolated in 0.32 M sucrose buffered with 10 mM HEPES-Tris, pH 7.4, were diluted 20-fold (fi- nal protein concentration, 1 mg/ml) into Ca" uptake media containing 150 mM KCI (or 60 mM KCI, 73 mM NaCl), 1 mM MgCl2, 10 mM glucose, 1 mM CaC12 supplemented with 45CaC12 (2.5 pCi/pmol), 10 mMHEPES-Tns, pH 7.4, and increasing concentrations of the calcium blockers. The reaction was conducted at 3VC, and was terminated by fil- tration of 0.5-mI samples through Watman GF/B filters pre- washed with 10 ml of 0.32 M ice-cold (0-4'C) sucrose bufferedwith 10 mMTris-Cl, pH 7.4. The samples retained on the filters were washed with 10 ml of buffered sucrose containing 1 mM LaClx, to remove the surface-bound 45Ca2+. The filters were treated and radioactivity was counted as described above. The amount of Ca2+ retained by the synaptosomes in a control medium with the same composition as described above, except that 150 mMNaCl (or 128 mM NaCl, 5 mM KCI) was present instead of KCI, was determined for all the drug concentrations tested and was subtracted from total Ca" taken up to obtain the Ca" uptake due to K+ depolarization (Fig. 4).
In the experiments shown in Figs. 3 and 5, the synapto- somal suspension was preincubated in a Na-rich medium (128 mMNacI, 5 mMKCI; 1.2 mMMgCI2; 10 mMglu- COX; 10 mM HEPES-Tris, pH 7.4; 1 mM Na2HP04) for 15 min, at 3VC, prior to the Ca" uptake assays. After the incubation, the suspension was centrifuged for 15 min at 20,000 g, and the pellet was resuspended in ionic medium containing 128 W c h o l i n e chloride (ChCl); 5 mM KCI; 1 mMM&h; 10 mMglucose; 10 mMHEPES-Tris, pH 7.4, at 20 mg of protein/ml and was utilized for studying 45Ca2+ uptake in the next 1-2 h.
Na+/Ca'+ exchange and ATP-dependent Ca" uptake. The effects of calcium blockers (verapamil, nifedipine, and d-cisdiltiazem) on Ca" uptake by SPM under conditions that permit Na+/Ca2+ exchange were studied as described previously by Coutinho et al. (1983). by diluting membrane vesicles preloaded with Na+ overnight (1 50 mM NaCI, 1
into C j + uptake media [ 150 mM KCI or 150 mM NaCI; 10 mMglucose; 10 mMHEPES-Tris, pH 7.4; and20pMCaClz + 45Ca (2.5 pCi/pmol)] at a final protein concentration of 0.5 mg/ml. The reaction media also contained increasing concentrations of either nifedipine, verapamil, or d-cis-dilti- azem. The reaction proceeded at 30°C and, after 3 min, was terminated by filtering 0.5-ml samples, as described above.
To study the effects of calcium blockers on the ATP-de- pendent Ca" uptake by SPM, the membrane vesicles iso- lated in KCI medium, as described above, were diluted 20- fold into a reaction medium of the following composition:
HEPES-Tris, pH 7.4; 20 pM CaCI2 + 45Ca (2.5 wCi/pmol) at a final protein concentration of 0.5 mg/ml, and supple- mented with various concentrations of the various blockers. Ca2+ uptake in the absence and in the presence of 1 mM ATP-Mg was determined, and the reaction was followed at 3VC, for 3 min, after which duplicate samples were filtered, as described above.
Materials All chemicals used were of highest purity available com-
mercially. The radiotabelled 1 ,4-dihydropyridine utilized in this study, [-'H]nimodipine (150.2 Ci/mmol), and unla- belled nimodipine were provided by Prof. Hoffmeister, from Bayer AG, Wuppertal, F.R.G. Nifedipine and d-cis- diltiazem were provided by Prof. T. Macedo, Faculty of Medicine, Coimbra, Portugal. Verapamil (racemic mixture) was obtained from Knoll Lusitana, Portugal. Both (-)- and (+)-verapamil and (-)-D 600 were provided by Prof. H. Glossmann, Rudolf-Bucheim-Institut fur Pharmakologie, Giessen, F.R.G. The 1,4-dihydropyridines were first dis- solved in absolute ethanol at 1 mM and then were diluted to the appropriate concentrations with buffer (50 mM Tris- HCl, pH 7.4). Due to the extreme lability ofthe 1 ,4-dihydro- pyridines to light, all solutions were stored light-protected at -20°C.
mM M C12, 10 ~ M ~ ~ u c o s ~ , 10 mM HEPES-Tris, PH 7.4)
150 mM KCl; 1 mM MgCI2; 10 mM glucose; 10 mM
RESULTS Distribution of [3H]nimodipine binding sites in sheep brain membrane fractions
High-affinity binding sites for [3H]nimodipine have been reported previously in brain (Ferry and Gloss- mann, 1982, 1983; Bellemann et al., 1982; Gloss- mann et al., 1983). However, most binding studies were performed with total membrane preparations (homogenates), and there is some uncertainty about subcellular membrane localization of the 1 ,Cdihy- dropyridine binding sites in brain tissue (Bellemann et al., 1982). To clarify this issue, we studied the bind- ing of [3H]nimodipine to purified sheep brain mem- brane subfractions obtained from synaptosomal and microsomal preparations, and correlated the binding with the Na+,K+-ATPase activity in the various frac- tions (Fig. 1).
J. Neurochem., Vol. 47, No. 6. 1986

EFFECTS OF Cd' BLOCKERT ON SYNAPTIC MEMBRANES 1777
FIG. 2. Equilibrium binding of [%]ni- modipine by SPM fraction as a function of increasing concentrations [3~]nimod- ipine. A: The membranes were incu- bated, as described in Materials and Methods, with concentrations of [%]ni- modipine varying between 0.05 and 1.4 nM. Nonspecific binding was measured in the presence of 5 pJ.4 unlabellad ni- modipine. B Scatchard plot of the spe- cifically bound [3H]nimodipine. The data are means of three experiments, each performed in triplicate. and K. and S, values obtained by linear regression analysis are 0.64 f 0.1 nM and 161 ? 27 frnd . mg-' protein. respectively. C Hill transformation of the specific binding data (slope = 1.07).
1
FIG. 1. Distribution of ATPase activities and [3H]nimodipine specific binding in sheep brain membrane fractions. The Mg2+-ATPase is the ATPase activity not inhibited by 1 .O mM ouabain and the Na'.K+-ATPase was taken as the activ- ity inhibited by 1 .O mM ouabain. [3H]Nimodipine binding was determined at a free [3H]nimodipine concentration of 0.25 nM, in 50 mM Tris-CI. pH 7.4, in the absence (total binding) or in the pres- ence (nonspecific) of 5 pM unlabelled nimodi- pine. The specific binding is taken as the differ- ence between total and nonspecific binding. Val- ues for the ATPase activities are means of the values obtained in two different experiments, and agreed within 10%. Data for [3H]nimodipine binding are means * SD from three different ex- periments, each performed in triplicate.
Mg? ATPore (No:K')-ATPore 'H -NIMODIPINE BINDING
I& PM M F, F2 F, F,
As shown in Fig. 1, the SPM obtained after osmotic lysis of synaptosomes followed by partial purification (Coutinho et al., 1983) are enriched in [3H]nimodi- pine binding sites (about 70 fmol [3H]nimodipine bound/mg protein) and in Na+,K+-ATPase activity (about 300 nmol ATP hydrolyzed/min/mg protein). Therefore, it is assumed that the 1 ,Cdihydropyridine binding sites are localized predominantly at the plasma membrane of nerve terminals. Figure 1 also shows that the brain microsomal fraction has some binding sites for [3H]nimodipine, but there is no fur- ther significant purification of these binding sites when the crude microsomal fraction (M) is subfrac- tionated into four fractions (F, through F4) by sucrose gradient centrifugation. Therefore, the SPM fraction was preferentially utilized in subsequent studies to characterize the [3H]nimodipine binding sites and to study the effects of various Ca2+ channel blockers on Ca2+ translocation in brain.
300
200
100
0 SPM
Affinities of several Ca2+ blockers for ['HJnimodipine binding sites in purified SPM
The purified SPM fraction isolated from sheep brain contains high-affinity binding sites for t3H]ni- modipine, as shown in Fig. 2, which shows the results of saturation experiments. [3H]Nimodipine labels a single class of sites in SPM, with an apparent KD of 0.64 f 0.1 nM and a B,, of 161 f 27 fmol.mg-' protein, as determined from Scatchard analysis of spe- cific binding (Fig. 2B), measured at 25°C. The Hill slope is 1.07 (Fig. 2C), which implies that one homo- geneous group of binding sites is present.
The specific binding of [3H]nimodipine was defined as that displaced by an excess of unlabelled nimodi- pine (Fig. 2A), as described previously by other inves- tigators for gross membrane homogenates (Belle- mann et al., 1982; Ferry and Glossmann, 1982; Gloss- mann et al., 1983). In the present experiments, specific binding accounted for 50-80% of the total
1 . 0 5 - 'P - P200-
- P I-k NIMODIPINE.IMI
100 -
50 v] 2 AA\ ,
O 0 2 a4 0.6 0.8 10 1.2 1.4 100 2 0 0
'H-NIMODIPINE , ("MI 'H-NIMODIPINE BOUND ( h o i mg-'protalnj
J. Neurochem.. Vol. 47, No. 6, 1986
I778 C. A . M. CARVALHO ET AL.
binding, and it decreased with increasing concentra- tion of radioligand. At 0.6 nM [3H]nimodipine, which is about the KD value, the specific binding is 73% of the total binding.
We also determined the relative affinities of various Ca2+ channel blockers for the [3H]nimodipine bind- ing sites in SPM by performing competition studies in which the membranes were labelled with ['Hlnimodi- pine and then increasing concentrations of various unlabelled Ca" blockers were added to displace the bound [3H]nimodipine. The results, summarized in Table I, show that: (1) nimodipine and nifedipine are potent competitors for the [3H]nimodipine binding sites, with ICso values of4.7 and 5.8 nM, respectively, as determined by Hill transformation of the inhibi- tion-binding data. The Hill slopes for these com- pounds are 0.93 and 0.83, respectively, indicating competition for an homogeneous class of [3H]nimodi- pine binding sites. (2) On the other hand, (+)- and (-)- verapamil and (-)-I2 600 are weak competitors, and display a biphasic competition behavior as described previously by Ferry and Glossmann ( 1982) and Gloss- mann et al. (1983); thus, for (+)-verapamil, the ICsO for the higher affinity component is 708 nM, the Hill slope is 0.73, and the maximal inhibition is 50% of the specific binding, whereas for (-)-verapamil the ICsO is lower (120 nM), the Hill slope is 0.57, but the maxi- mal inhibition is only 30% of the specific binding, at concentrations Of 3 X lo-' M of unlabelled drug (Ta- ble I).
These results indicate that there is some stereoselec- tivity in the interaction of phenylalkylamines with the [3H]nimodipine binding sites in SPM, as shown pre- viously in other systems (Ferry and Glossmann, 1982; Glossmann et al., 1983). The stereoselectivity for the IP-dihydropyridines in inhibiting binding has also been shown previously by Bellemann et al. (1 98 1) and by Glossmann et al. (1 982).
TABLE I. Binding-inhibition constants ~f/~H]nimodipine binding to SPM by various Cd+ channel blockers
Maximal inhibition (nw nH (% of total binding)
Nimodipine 4.7 L 0.50 0.93 & 0.60 70 Nifedipine 5.8 + 0.45 0.83 c 0.07 70 (+)-Verapamil 708" 0.73 50' (-)-Verapamil 120" 0.57 30' (-kD 600 430" - 40'
ICm is the concentration of drug causing 50% inhibition of spe- cific ['Hlnimodipine binding, as calculated from linear regression analysis of data transformed into the Hill equation. nH is the Hill slope. Binding-inhibition curves were performed with a fixed ['H]nimodipine concentration (0.37 nM), and increasing concen- trations of the competing unlabelled drug. The data are means of three experiments, each performed in triplicate. Examples of bind- ing-inhibition experiments are shown in Fig. 4.
a Higher affinity component. 'At concentrations = 3 X M.
0 30 60 PO 120 30 60 Po 120
INCUBATION TIME (rec)
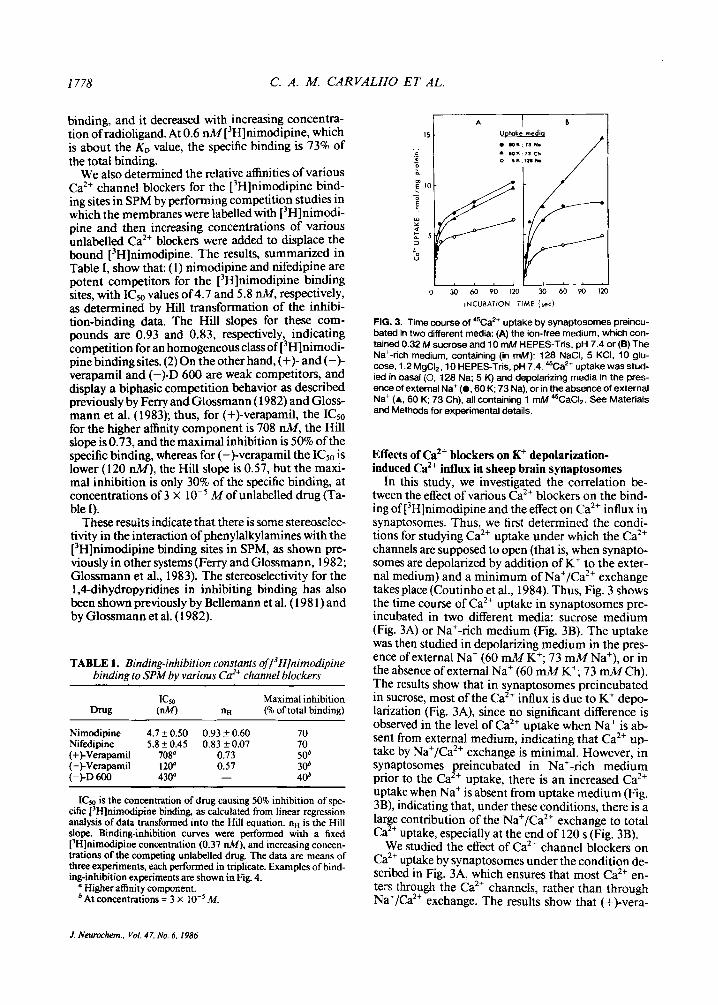
FIG. 3. Time course of '%a2+ uptake by synaptosomes preincu- bated in two different media: (A) the ion-free medium, which con- tained 0.32 M sucrose and 10 mM HEPES-Tris. pH 7.4 or (B) The Naf-rich medium, containing (in mM): 128 NaCI, 5 KCI, 10 glu- cose, 1.2 MgCI,, 10 HEPES-Tris, pH 7.4. 45Ca2+ uptake was stud- ied in basal (0, 128 Na; 5 K) and depolarizing media in the pres- ence of external Na+ (0.60 K; 73 Na), or in the absence of external Na+ (A, 60 K; 73 Ch). all containing 1 mM 45CaCI,. See Materials and Methods for experimental details.
Effects of Ca*+ blockers on K+ depolarization- induced Ca2+ influx in sheep brain synaptosomes
In this study, we investigated the correlation be- tween the effect of various Ca2+ blockers on the bind- ing of [3H]nimodipine and the effect on Ca2+ influx in synaptosomes. Thus, we first determined the condi- tions for studying Ca2+ uptake under which the Ca2+ channels are supposed to open (that is, when synapto- somes are depolarized by addition of K+ to the exter- nal medium) and a minimum of Na+/Ca2+ exchange takes place (Coutinho et al., 1984). Thus, Fig. 3 shows the time course of Ca2+ uptake in synaptosomes pre- incubated in two different media: sucrose medium (Fig. 3A) or Na+-rich medium (Fig. 3B). The uptake was then studied in depolarizing medium in the pres- ence of external Na+ (60 mM K+; 73 mM Na+), or in the absence of external Na+ (60 mM K+; 73 mM Ch). The results show that in synaptosomes preincubated in sucrose, most of the Ca2+ influx is due to K+ depo- larization (Fig. 3A), since no significant difference is observed in the level of Ca2+ uptake when Na+ is ab- sent from external medium, indicating that Ca2+ u p take by Na+/Ca2+ exchange is minimal. However, in synaptosomes preincubated in Na+-rich medium prior to the Ca2+ uptake, there is an increased Ca2+ uptake when Na+ is absent from uptake medium (Fig. 3B), indicating that, under these conditions, there is a lar e contribution of the Naf/Ca2+ exchange to total
We studied the effect of Ca2+ channel blockers on Ca2+ uptake by synaptosomes under the condition de- scribed in Fig. 3A, which ensures that most Ca2+ en- ters through the Ca2+ channels, rather than through Na+/Ca2+ exchange. The results show that (+)-Vera-
Ca !+ uptake, especially at the end of 120 s (Fig. 3B).
1. Neurochem., Vol. 47. No. 6. I986

EFFECTS OF Cd' BLOCKERS ON SYNAPTIC MEMBRANES
-
I - 0 z
pamil, nifedipine, and nimodipine are much less po- tent in inhibiting Ca2+ influx than in inhibiting the binding of [3H]nimodipine to the purified membranes (Fig. 4). Thus, (+)-verapamil inhibits Ca2+ influx in- duced by K+ depolarization by 50% at about 500 pM, whereas it inhibits 50% ofthe [3H]nimodipine binding at concentrations 200-fold lower. The discrepancy is even larger for nimodipine and nifedipine (Fig. 4). Therefore, the results show that, under our experi- mental conditions, there is no close correlation be- tween the affinities of the Ca2+ blockers for the SPM binding sites and their potency for blocking Ca2+ in- flux due to K+ depolarization in synaptosomes under conditions of minimal Na+/Ca2+ exchan e. These re-
are at variance with those recently reported by Turner and Goldin ( 1 985). See later for discussion.
We further examined this problem by studying the effect of verapamil on the total Ca" that enters the synaptosomes both by Ca2+ influx due to K+ depolar- ization and by Na+/Ca2+ exchange. As indicated above (Fig. 3), Ca2+ influx by both mechanisms occurs in synaptosomes that have been preincubated in a Na+-rich medium prior to utilization in studies of Ca2+ influx, as described also in the legend to Fig. 5. This preincubation in Na+-rich medium has been normal practice by other investigators who, neverthe- less, assumed they were studying only Ca2+ influx due
sults, obtained in the absence of Na+/Ca R exchange,
- c
d l NlFEDlPlNE 2 0 m 8 . VERAPAMIL Z
- 0 . NIMODIPINE 2
I D R U G # , M
FIG. 4. Relationship of the relative inhibition of the specific bind- ing of [3H]nimodipine by nifedipine (0). nimodipine (A), and (+)- verapamil (m) with their inhibition of %a2+ influx into sheep brain synaptosomes. [3H]Nimodipine binding was performed at a fixed [3H]nimodipine concentration (0.37 nM), in the presence of in- creasing concentrations of each of the competing unlabelled drug, and maximal specific binding is defined as that displaced by 5 pM unlabelled nimodipine. The %a2' uptake experiments were performed by transferring synaptosomes isolated in 0.32 M su- crose, 10 mM HEPES-Tris, pH 7.4, to depolarizing media contain- ing 150 mM KCI, 1 mM MgCI,, 10 mM glucose, 10 mM HEPES- Tris. pH 7.4, 100 pM CaCI, supplemented with '5CaC12 (2.5 pCi/ pmol). and increasing concentrations of nimodipine. nifedipine. or (+bverapamil. The amount of Caz+ retained in control media, con- taining 150 mM NaCl instead of KCI. was determined and was subtracted for all drug concentrations tested. The binding data are means of three independent experiments performed in tripli- cate. The Ca" influx data are means of two experiments per- formed in duplicate.
I779
1 7
External m d i w
4 K ' , k* 7.5 - Ch*, NC -
0
- E 5 - 0 m
0
I
5 2.5
- YI X 4 t
0 U
0 lo' lo' I d I d 10'
(VERAPAMIL), M
FIG. 5. Effect of verapamil on Ca" uptake induced by K+ (curve c - curve b), or by Na+/Caz+ exchange (curve b - curve a), in sheep brain synaptosomes. Synaptosomes were preincubated for 15 min. at 30°C, in Na+-rich medium. as described in the Mate- rials and Methods, and the Ca2+ uptake was initiated by transfer- ring preincubated synaptosomes (0.5 mg/ml, final protein concen- tration) to three different media: 128 mM NaCl and 5 mM KCI (0); 60 mM ChCl and 73 mM NaCl(0); 60 mM KCI and 73 mM NaCl (A), all containing. additionally, I mM MgC12, 10 mM glucose. 1 mM Na2HP04. 10 mM HEPES-Tris. pH 7.4, 1 mM CaCI, supple- mented with %aCIZ (2.5 pCi/pmol). and increasing Concentra- tions of verapamil. Ca" uptake was terminated by filtration after 2 min of reaction at 3OOC.
to K+ depolarization (Blaustein, 1977; Nachshen and Blaustein, 1979, 1980, 1982). The preincubation is usually for a period of 15 min, at 30"C, followed by addition of K+ to the external medium, to obtain membrane depolarization and opening of the Ca2+ channels. The addition of K+ is accompanied with an equimolar reduction in the Na+ concentration in the outside medium, to maintain the osmolarity constant. In our experience, this procedure leads to Ca" influx not only due to K+ depolarization but also by Na+/ Ca2+ exchange (Coutinho et al., 1984), since there is a favorable Na+ gradient from inside to the out- side, which drives Ca2+ uptake by Na+/Ca2+ exchange (Fig. 3B).
As shown in Fig. 5, when synaptosomes preincu- bated for 15 min, at 30"C, in a Na+-rich medium (128 mM NaCl; 5 mM KCl), were transferred to a Ca2+ uptake medium of the same ionic composition, there is a basal level of Ca2+ binding (about 3.5 nmol Ca2+/ mg protein; Fig. 5, curve a). However, when the exter- nal medium contains part of the NaCl substituted for ChCl(60 mMChC1; 73 mMNaC1; 5 mMKCl), aNa+ gradient from inside to outside is created, Ca2+ uptake in exchange for Na+ is induced, and it is observed that 5 nmol of Ca2+ are taken up per milligram of protein (Fig. 5, curve b). Furthermore, if part of the NaCl in the external medium is substituted for KCI (60 mM KCl; 73 mMNaCl), we simultaneously depolarize the membrane (by increasing K+ outside) and create a Na+ gradient, which results in Ca2+ uptake simulta- neously through Ca2+ channels and by Na+/Ca2+ ex-
J. Neurmhem.. Vol. 47. No. 6. 1986

1780 C. A. M. CARVALHO ET AL.
change. Consequently, total Ca2+ uptake increases in this situation, to about 6.5 nmol Ca2+/mg protein (Fig. 5, curve c). These results indicate that the condi- tions reported in the literature for K+ depolarization also induce some Na+/Ca2+ exchange, and most pub- lished data must be reviewed, if care was not taken to distinguish Ca2+ entry through the channels from Ca2' entry through Nat/Ca2+ exchange (Nachshen and Blaustein, 1980, 1982; Daniel1 et al., 1983; Rampe et al., 1984).
When the effect of verapamil on Caz+ uptake by synaptosomes by these mechanisms (Fig. 5) was deter- mined, it was observed that this Ca2+ blocker inhibits the Ca2+ uptake due both to K+ depolarization and to Na+/Ca2+ exchange, for concentrations higher than
Mofthe drug, and that the potency for inhibiting either Ca2+ uptake system is similar. Therefore, in the concentration range studied there is no selective effect of verapamil on Caz+ channels. We observed similar results for the inhibition of Ca2+ uptake in synapto- somes by other Ca2+ blockers, such as d-cis-diltiazem and nifedipine (results not shown), i.e., the Ca2+ blockers inhibit not only the K+ depolarization-in- duced Ca" uptake, but also the Ca2+ uptake that oc- curs through Na+/Ca2+ exchange. This was further studied in SPM, and the results are presented in the next section.
Comparative effect of GI*+ blockers on the Ca2+ transport systems in SPM
We further studied the effects of various Ca2+ chan- nel blockers on the Caz+transport in membrane vesi- cles derived from SPM, in which the ionic gradients can be controlled. Thus, SPM vesicles were preloaded with NaCl or KCI medium, as described in Materials and Methods, and Ca2+ uptake by Na+/Ca2+ exchan e (in the NaCl-loaded vesicles) or ATP-dependent Ca'+ uptake (in the KCl-loaded vesicles) was studied in the presence of increasing concentrations of the Ca2+ blockers (Figs. 6 and 7).
FIG. 6. Effects of verapamil, nifedi- pine, and d-cis-diltiazem on Na'/Ca2' exchange in SPM. The SPM were loaded overnight at 0-4OC in medium containing 150 mM NaU, 1 mM MgCh. 10 mM glucose. and 10 mM HEPESTris, pH 7.4, to load the vesi- des with Na+. The Na'/Ca2+ ex- change was assayed by transferring samples of the SPM suspension into media mtaining 150 mM NaCl (Na,,) w 150 mM KCI (&) and 20 jd.4 CaCI2. supplemented with %aCI2 (2.5 pCi/ rmol) and the various drug Concentra- tions tested. The reaction was cok ducted for 3 min. at 3OoC, and was terminated by filtration of duplicate samples (0.5 ml, containing 0.25 mg protein), as described in Materials and Methods. Data are means of three in- dependent experiments.
The results in Fig. 6 show that the three types of Ca2+ blockers studied (verapamil, nifedipine, and d- cis-diltiazem) inhibit the Na+/Ca2+ exchanger in SPM. The concentrations required to inhibit 50% of the Ca2+ uptake by this mechanism are 4.6 X M, 3 x M, and M for verapamil, nifedipine, and d-cisdiltiazem, respectively. The values for Ca2+ uptake shown in Fig. 6 are taken after 3 min of reac- tion, and therefore represent total Ca2+ uptake. How- ever, we also determined the effects of the Ca2+ block- ers on the initial rate of Na+/Ca2+ exchange, and the same percent effect is observed in both the rate and total Ca2+ uptake (results not shown).
The ATP-dependent Ca" uptake in SPM isolated from sheep brain is also inhibited by high concentra- tions of the Ca2+ blockers, as shown in Fig. 7. The concentrations of the drugs required to inhibit 50% of the Ca2+ uptake are 8.7 X M, and 1 0-3 M verapamil, nifedipine, and d-cis-diltiazem, re- spectively. The significance of the effects of the Ca2+ blockers on the Ca" transport systems in the brain is referred to in the Discussion.
M, 8.7 X
DISCUSSION
In the present study, we investigated the affinities of the Ca2+ channel blockers for their binding sites in purified brain membranes and also the effects of the blockers on the Ca" channels and the Ca2+ transport systems of the plasma membrane, namely the Na+/ Ca2+ exchange and ATP-dependent Ca2+ transport. To study the a5nity of the Ca2+ channel blockers for the binding sites, we utilized mostly the SPM fraction, enriched in Na+,K+-ATPase activity (Coutinho et al., 1983) and in f3H]nimodipine binding sites (Fig. 1). The value of the dissociation constant for [3H]nimo- dipine binding to SMP, of 0.6 &(Fig. 2), indicates a high-affinity binding, and the competition of the vari- ous classes of Ca2+ channel blockers for the [3H]ni- modipine binding (Table I, Fig. 4) is similar to that
J. &wcxhem.., VoI. 47, No. 6,1986

EFFECTS OF Cd' BLOCKERS ON SYNAPTIC MEMBRANES 1781
A
' ATP ATP I AT P
T O b & r ( ; - 6 10 10 10 10 10 lb-5 i0-4 1:
( V E R A P A M I L ) . M (NIFEDIPINE). M (d.c,s.Dl LTl A 2 E M ), M
reported in previous studies carried out with whole brain homogenates (Bellemann et al., 1982; Ferry and Glossmann, 1982; Glossmann et al., 1983), indicating drug specificity for the binding sites.
However, a clear selective effect of the Ca2' channel blockers on the Ca2' fluxes through Ca" channels at the concentrations that saturate the binding sites of SPM has not yet been clearly demonstrated by previ- ous studies in synaptosomes. In fact, studies on the effect of Ca2+ channel blockers on the depolarization- induced Ca2+ fluxes in brain synaptosomes (Nach- shen and Blaustein, 1979; Akerman and Nicholls, 198 1) show that verapamil and D 600 only partially inhibit Ca" fluxes, even at concentrations of the or- der of 10-5-10-4 M. Furthermore, nifedipine ( M) was also shown to be ineffective (Nachshen and Blaustein, 1979), and other dihydropyridines, such as nitrendnpine, nimodipine, and nisoldipine, at con- centrations up to M, did not alter Ca2+ influx into brain synaptosomes (Daniel1 et al., 1983). More recently, it was also shown that BAY K 8644, a 1,4- dihydropyridine Ca2+ channel activator in smooth and cardiac muscles, is also without effect on Ca2+ up- take in brain synaptosomes (Rampe et al., 1984), but Middlemiss and Spedding (1985) reported that BAY K 8644 can augment the K+-stimulated release of 5- hydroxytryptamine (serotonin) from rat cortex slices and that this effect can be antagonized by Ca2' antag- onists.
There is only a recent study (Turner and Goldin, 1985) showing partial inhibition of depolarization- stimulated Ca2' uptake in synaptosomes by nitren- dipine, nifedipine, verapamil, D 600, and diltiazem in the nanomolar concentration range of these drugs. These workers tried to eliminate the Na'/Ca2' ex- change component in their experiments, working in choline medium in the absence of Na+, but we have shown, previously, under similar conditions, that Kt
FIG. 7. Effects of verapamil. nifedipine. and d-cisdiltiazem of the ATP-depen- dent Ca" uptake by SPM. The mfjm- brane vesicles were incubated overnight at 0-4OC in medium containing 150 mM
mM HEPESTris, pH 7.4. ATP-deependent Ca" uptake was assayed by transferring samples of SPM suspension to a similar KCI medium containing. additionally, 20 pl.4 CaCI2 supplemented with *CaCI2 (2.5 pCi/pmol), 0.1 mM dinitrophenol. 0.1 mM azide, 1 .O pg/ml olygomycin, and the vari- ous drug Concentrations tested. The r e action was initiated by addition of 1 mM ATP-Mg, was conducted for 3 min at 3OoC, and was terminated by filtration. The effects of the various drug concen- trations on the retention of Ca" by SPM in a control medium without ATP (operr symbols) were also tested. The data are means of three independent experi- ments.
KCI, 1 mM MgC12.10 mM glUCOSe, and 10
stimulates the Na+/Ca2+ exchange in SPM (Coutinho et al., 1983). Thus, some of the effect of the Ca" blockers on Ca2' fluxes in synaptosomes reported by Turner and Goldin (1985) may reflect the effect of the blockers in the K+-dependent Na+/Ca2' exchange (Coutinho et aI., 1983). The fact that Turner and Gol- din (1985) did not find a corresponding "Na+ efflux may reflect the inherent difficulty of carrying out this type of experiment with 22Na (K. Phillipson, personal communication).
In most studies previously designed to determine the effects of Ca2+ channel blockers on Ca" uptake, synaptosomes were submitted to a preincubation in a Na+-rich medium, which allows some accumulation of Na' (Coutinho et al., 1984) and subsequently, when the synaptosomes were transferred to the K+- rich medium containing 45Ca, there was 45Ca entry through Na'/Ca2' exchange that is superimposed on the Ca2+ entry through the Ca" channels. Thus, it is important to differentiate between the effects of Ca2+ channel blockers on the two Ca" entry mechanisms.
In this study we utilized synaptosomes isolated in buffered sucrose, not exposed to Na+, to study the effect of nimodipine, nifedipine, and (+)+erapamil on the Ca2+ influx when membrane depolarization by high K+ was induced (Fig. 4). It is observed that, under these experimental conditions, in which there is mini- mal Na'/Caz' exchange and the Ca2+ influx takes place through CaZ+ channels, there is inhibition of Ca2+ entry only for very high concentrations of the drugs ( 10-4-10-3 M). This does not correlate with the effect of the Ca2' channel blockers on their inhibition of the binding of [3H]nimodipine as shown in Fig. 4. Under conditions in which Ca" influx occurs both through the Ca2' channels and by Na+/Ca2+, there is a similar sensitivity of both mechanisms to inhibition by verapamil (Fig. 5) . Previous studies have also dem- onstrated inhibition by Ca" channel blockers of Na+/
J. Neurochem.. Vol. 47. No. 6. 1986

I782 C. A. M. CARVALHO ET AL.
Ca2+ exchange in heart and brain mitochondria (Vighy et al., 1982; Matlib and Schwartz, 1983), as well as in brain microsomes (Liron et al., 1985), and in SPM (Erdreich et al., 1983), and in all cases rela- tively high concentrations of the blockers are neces- sary.
Our studies with isolated SPM also show that the uptake of Ca" by Na+/Ca2+ exchange and by the Ca2+-ATPase systems is inhibited by verapamil, nifedipine, and d-cisdiltiazem at concentrations above los4 M(Figs. 6 and 7).
In our attempt to dissociate the depolarizationde- pendent Ca2+ entry into synaptosomes from the Na+/ Ca" exchange we resorted to using synaptosomes not exposed previously to Na+ and to carry out the K+ depolarization in the absence of choline because in both instances there occurs Na+/Ca2' exchange. However, our results are at variance with those re- ported by Turner and Goldin (1985), since under con- ditions that would ensure 45Ca2+ influx through only the Ca2' channels the Ca" blockers tested are not effective at concentrations at which they saturate the membrane binding sites (Figs. 2 and 4).
The Na'/Ca2' exchange and ATPdependent Ca2+ uptake are important mechanisms in regulating intra- cellular Ca2+ concentration in nerve (Baker, 1972; Blaustein and Nelson, 1982; Gill et al., 1981; Cou- tinho et al., 1983) and muscle cells (Hurwitz et al., 1983; van Breemen et al., 1979; ozalu and Urakawa, 1979; Morel et al., 198 1 ; Verbist et al., 1984), but the finding that Ca" channel blockers can inhibit these transport systems at relatively high concentrations (> M) (Figs. 6 and 7) probably is of little signifi- cance under normal conditions. However, when high doses of the Ca2+ channel blockers are administered to patients with cardiovascular diseases some of these drugs may reach concentrations in membranes that are much higher than their extracellular level, because of their high lipid solubility (Pang and Sperelakis, 1984). We are currently determining the partition co- efficients of various Ca2' channel blockers in mem- branes isolated from brain, heart, and smooth muscle to estimate the maximal concentration of these drugs in the lipid phase of the membranes.
It is of interest that the binding affinities and the inhibition of Ca2' fluxes by Ca2+ channel blockers are correlated in certain neuronal cell lines. In fact, in PC12 cells the Ca2+ currents and the Ca2+ uptake through the channels are inhibited by low concentra- tions of Ca" channel blockers (Toll, 1982); also, neu- rotransmitter release and K+-induced Ca" uptake in some neuronal cell lines are sensitive to low (nanomo- lar) concentrations of Ca2' channel blockers (Taka- hashi and Ogura, 1983; Albus et al., 1984; Freedman et al., 1984). Thus, since neuronal cells in culture are sensitive to Ca2+ channel blockers, it was suggested (Spedding and Middlemiss, 1985) that, under culture conditions, it is possible that the cells lose a factor pro- tecting from blockade by Ca2+ channel blockers in
brain tissue. The search for such a factor is of great interest for the elucidation of the discrepancies ob- served between the behavior ofcells in culture and ho- mogenated fractions of brain tissue.
Furthermore, a recent study (Middlemiss and Spedding, 1985) shows that, under certain specific conditions, Ca2+ channel blockers can directly affect neurotransmitter release in brain slices. In these stud- ies, the agonist Bay K 8644 increased neurotransmit- ter release, in a &'+-dependent manner, and the effect was, in turn, inhibited by Ca2+ antagonists at low (1 pM) concentrations. Therefore, the conclusion from these observations is that Ca2+ channel blockers can have effects on neuronal Ca2+ channels, but only subsequent to certain forms of activation of the chan- nels. Thus although there are indications of a func- tional correlate of the 1,4dihydropyridine binding sites in brain tissue, it is not easy to demonstrate this correlation, especially in isolated brain fractions. For instance, in isolated brain synaptosomes, Ca2+ uptake due to K" depolarization is not sensitive to Bay K 8644 (Rampe et al., 1984), which is not the case in brain slices (Middlemiss and Spedding, 1985). As re- ferred to earlier we cannot explain the recent report by Turner and Goldin (1985) that nitrendipine inhibits depolarization-stimulated Ca2+ uptake and [3H]nor- epinephrine release from synaptosomes at low nitren- dipine concentrations (Kam of 56 nM in the presence of Na+ and Kam of 1.7 nM in the absence of Na').
In another development Bean (1984) and Sangui- netti and Kass (1 984) reported that the binding of the 1,4dihydropyridines is voltagedependent. Thus, Bean ( 1984) showed that nitrendipine blocks cardiac Ca2+ currents very potently (KD, 0.36 nM) at holding potentials at which most CaZ+ channels are inacti- vated and, at more negative holding potentials, the block is less potent by a factor of > 1 ,OOO. Thus, it is suggested that nitrendipine binds tightly to the inacti- vated state of the Ca2+ channel and much more weakly to the resting state. These electrophysiological studies were complemented by radioligand binding (Reuter et al., 1985), and it was shown that the bind- ingof ['Hjnimodipine to living cardiac cells is voltage- dependent. Schwartz et al. (1985) also showed that, in skeletal muscle fibers, depolarization increases the number of binding sites in the high-affinity state. The authors (Schwartz et al., 1985) also compare the bind- ing results with the voltage-clamp measurements of Ca2+ channel blockade and conclude that less than a few percent of the binding sites in skeletal muscle rep- resent functional Ca2+ channels. Similar voltage de- pendence binding studies of Ca2+ channel blockers in brain cells are not available to our knowledge. This type of experiment should also be feasible with synap tosomes and we are currently exploring this possibil- ity. Our preliminary results show a slight difference in the amount of nitrendipine bound to synaptosomal membranes in the presence of K' or Na+, indicating that the membrane potential may in fact be an impor-
1. Neurmhem.. Vol. 47, No. 6. 1986

EFFECTS OF Cd’ BLOCKERS ON SYNAPTIC MEMBRANES 1783
tant factor in determining the availability of the bind- ing sites.
In summary, the results reported in this study show that, although purified SPM are enriched in high- affinity binding sites for [’Hlnimodipine (& in the na- nomolar range), the various types of Ca2+ channel blockers (verapamil, nifedipine, and d-cis-diltiazen) inhibit Ca” influx through Caz+ channels, the Na+/ Caz+ exchange, and the ATP-dependent Ca2+ uptake, in a range of concentrations ( 10-5-10-4 M ) that are much higher than the concentrations that saturate the 1 ,Cdihydropyridine binding sites in the brain mem- branes. In this stud clear differentiation is made of the effects of the Ca channel blockers on these three mechanisms of moving Ca” across the synaptosomal membrane, and particular care is taken to separate the contribution of the Na+/Ca2+ exchange from that of the influx through Ca” channels under conditions of K+ depolarization. Under carefully defined condi- tions, we confirm the inherent difficulty in showing in isolated brain fractions an action of Ca” channel blockers on the Ca” channels at the nanomolar con- centrations required for the drugs to saturate the membrane binding sites, and recent results claiming the contrary must be viewedx+ith caution (Turner and Goldin, 1985).
K
Acknowledgment: The present work was supported by INIC, The Portuguese National Institute for Scientific Re- search.
REFERENCES Akerman K. E. 0. and Nicholls D. G. (1981) Ca” transport by
intact synaptosomes: voltagedependent Ca” channel and a re- evaluation of the role of scdium/calcium exchange. Eur. J. Bi- ochem. 117,491-497.
Albus U., Habermann E., Ferry D. R., and Glossmann H. (1984) Novel 1,4dihydropyridine (Bay K 8644) facilitates calcium- dependent [’Hlnoradrenaline release from PC 12 cells. J. Neu- rochem. 42,1186- 1 189.
Baker P. F. (1972) Transport and metabolism of calcium ions in nerve. Prog. Biophys. Mol. Biol. 24, 177-223.
Baker P. F., Meves H., and Ridgway E. B. (1973) Calcium entry in response to maintained depolarization of squid axons. J. Phy- siol. (Lond.) 231,527-548.
Bean B. P. (1984) Nitrendipine block of cardiac calcium channels: high-affinity binding to the inactivated state. Proc. Natl. Acad. Sci. USA 81,6388-6392.
Bellemann P., Ferry D. R., Lubbecke F., and Glossmann H. (198 I ) [’HI-Nitrendipine, a potent calcium antagonist, binds with high affinity to cardiac membranes. Arzneim. Forsch./Drug Res. 31,2964-2067.
Bellemann P., Ferry D., Liibbecke F., and Glossmann H. (1982) [3H]-Nimodipine and [’HI-nitrendipine as tools to directly identify the sites of action of 1,4dihydropyridine calcium an- tagonist in guinea pig tissues. Arzneim. Forsch./Drug Res. 32,
Blaustein M. P. (1977) Effects of internal and external cations and of ATP on sodium-calcium and calcium-calcium exchange in squid axons. Biophys. J. 20,79- I 1 I.
Blaustein M. P. and Nelson M. T. (1982) Sodium-calcium ex- change: its role in the regulation of cell calcium, in Membrane Transport ofCalcium (Carafoli E., ed), pp. 2 17-236. Academic Press. New York.
361-363.
Bolger G. T., Gengo P., Klockowski R., Luchowski E., Siege1 H., Janis R. A., Triggle A. M., and Triggle D. G. ( 1983) Character- ization of binding of the Ca” channel antagonist, [3H]-nitren- dipine, to guinea-pig ileal smooth muscle. J. Pharmacol. Exp. Ther. 225,291-309.
Carvalho C. A. M. and Carvalho A. P. (1979) Effect of temperature and ionophores on the permeability of synaptosomes. J. Neu- rochem. 33,309-317.
Coutinho 0. P., Carvalho A. P., and Carvalho C. A. M. (1983) Effect of monovalent cations on Na+/Ca*+ exchange and ATP- dependent Ca2+ transport in synaptic plasma membranes. J. Neurochem. 41,610-676.
Coutinho 0. P., Carvalho C. A. M., and Carvalho A. P. (1984) Cal- cium uptake related to K+-depolarhtion and Na+/Ca2+ ex- change in sheep brain synaptosomes. Brain Res. 290,26 1-27 1.
Daniel1 L. C., Barr E. M., and Leslie S. W. (1983) ‘%Za2+ uptake into rat whole brain synaptosomes unaltered by dihydropyri- dine calcium antagonists. J. Neurochern. 41,1455-1459.
DiPolo R. and Beaugb L. (1980) Mechanisms of calcium transport in the giant axon of the squid and their physiological role. Cell Calcium 1, 147-169.
Ehlert F. J., Itago E., Roeske W. R., and Yamamura H. I. (1982) The interaction on [’HI-nitrendipine with receptors for @- cium antagonists in the cerebral cortex and heart of rats. B i e chem. Biophys. Res. Commun. 104,937-943.
Erdreich A., Spanier R., and Rahamimoff H. (1983) The inhibition of Na-dependent of Ca-uptake by verapamil in synaptic plasma membrane vesicles. Eur. J. Phunnucol. 90,193-202.
Ferry D. R. and Glossmann H. ( 1982) Evidence for multiple r e p - tor sites within the putative calcium channel. Naunyn Schrnie- debergs Arch. Pharmacol. 321,80-83.
Ferry D. R. and Glossmann H. (1983) Tissue-specific regulation of [’HI-nimodipine binding to putative calcium channels by the biologically active isomer of diltiazem. Br. J. Pharmacol. 78, 81.
Ferry D. R., Rombusch M., Go11 A,, and Glossmann H. ( 1984) Pho- toaffinity labelling of Ca2+ channels with [3H]-azidopine. FEES Lett. 169, I 12- I 18.
Fleckenstein A. (1977) Specific pharmacology of calcium in my* cardial cardiac pacemakers and vascular smooth muscle. Annu. Rev. Pharmacol. Toxicol. 17,149-166.
Freedman S . B., Dawson G., Villereal M. L., and Miller R. J. (1984) Identification and characterization of voltage-sensitive cal- cium channels in neuronal clonal cell lines. J. Neurosci. 4, 1453-1467.
Gill D. L., Grollman E. F., and Khon L. D. (198 I ) Calcium trans port mechanisms in membrane vesicles from guinea pig brain synaptosomes. J. Biol. Chern. 256,184-192.
Glossmann H., Ferry D. R., Lubbecke F., Mewes R., and Hoffmann F. (1982) Calcium channels: direct identification with radioli- gand binding studies. Trends Pharmucol. Sci. 3,43 1-437.
Glossmann H., Feny D. R., Lubbecke F., Mewes R., and Hoffmann F. (1983) Identification of voltage operated calcium channels by binding studies: differentiation of subclasses of calcium an- tagonist drugs with ’H-nimodipine radioligand binding. J. Re- cept. Res. 3, 1 77- 190.
Gould R. J., Murphy K. M. M., and Snyder S. (1982) [3H]-Nitren- dipine-labeled calcium channels discriminate inorganic cal- cium agonists and antagonists. Proc. Natl. Acad. Sci. USA 79, 3656-3660.
Hagiwara S. and Byerly L. (1981) Calcium channel. Annu. Rev. Neurosci. 4,69-125.
Haj6s F. (1975) An improved method for the preparation of synap tosomal fraction in high purity. Bruin Res. 93,485-489.
Hunvitz L., Fitzpatrick D., Debbas G., and Landon E. (1973) Lu- calization of Ca pump activity in smooth muscle. Science 179, 384-385.
Katz B. and Miledi R. (1970) Further study of the role of calcium in synaptic transmission. J. Physiol. (Lond.) 207,789-80 I.
Kostyuk P. G. (1981) Calcium channels in neuronal membranes. Biochim. Biophys. Acfa 650, 128-150.
Layne E. ( 1957) Spectrophotometric and turbidimetric method for
I Neurochem.. Vol. 47. No. 6. 1986

1784 C. A . M. CARVALHO ET AL.
measuring proteins, in Methods in Enzymolom, Vol. 3 (Col- owick S. P. and Kaplan N. O., eds), pp. 447-451. Academic Press, New York.
Lee H. R., R m k e W. R., and Yamamura H. 1. (1984) High affinity specific [3H] (+) PN 200-1 10 binding to dihydropyridine recep tors associated with calcium channels in rat cerebral cortex and heart. LifeSci. 35,721-732.
Liron 2.. Roberts E., and Wong E. (1985) Verapamil is a competi- tive inhibitor of yaminobutyric acid and calcium uptake by mouse brain subcellular particles. LifeSci. 36,32 1-327.
Llinik R., Blinks J. R., and Nicholson C. (1972) Calcium transient in presynaptic terminal of squid giant synapse: detection with aequorin. Science176, 1127-1129.
Marangos P. J., Patel J., Miler C., and Martino A. M. ( 1 982) Specific calcium antagonist binding sites in brain. Life Sci. 31, 1575- 1585.
Matlib M. A. and Schwartz A. ( 1983) Selective effects of diltiazem, a benzothiazepine calcium channel blocker, and diazepam, and other benzodiapines on Na+/Ca2+ exchange carrier system of heart and brain mitochondria. Life Sci. 32,2837-2842.
Middlemiss D. N. and Spedding M. (1985) A functional correlate for the dihydropyridine binding site in rat brain. Nature 314, 94-96.
Morel N., Wibo M., and Godfraind T. A. ( 198 1) Calmodulin-stim- ulated Ca2+ pump in rat aorta plasma membranes. Eiochim. Biophys. Acta 644,82-88.
Murphy K. M. M. and Snyder S. H. (1982) Calcium antagonist re- ceptor binding sites labeled with [3H]-nitrendipine. Eur. J. Pharmacol. 77,201-202.
Nachshen D. A. and Blaustein M. P. (1979) The effects of some organic “calcium antagonists” on calcium influx in presynap tic nerve terminals. Mol. Pharmacol. 16,579-586.
Nachshen D. A. and Blaustein M. P. (1980) Some properties of po- tassium-stimulated calcium influx in presynaptic nerve end- ings. J. Gen. Physiol. 76,709-728.
Nachshen D. A. and Blaustein M. P. (1982) Influx ofcalcium, stron- tium and barium in pmynaptic nerve endings. J. Gen. Physiol.
Ozaki H. and Urakawa N. (1979) NaCa exchange and tension de- velopment in guinea pig aorta. Naunyn Schmiedebergs Arch. Pharmacol. 309,171-178.
Pang D. C. and Sperelakis N. ( 1984) Uptake of calcium antagonistic
79,1065-1087.
drugs into muscles as related to their lipid solubilities. Bio- chem. Pharmacol. 33,82 1-826.
Rampe D., Janis R. A,, and Triggle D. J. (1984) BAY K 8644, a I ,4dihydropyridine Ca2+ channel activator: dissociation of binding and functional effects in brain synaptosomes. J. Neuro- chem. 43,1688- 1692.
Reuter H., Po& H., Kokubun S., and Prod‘hom B. (1985) 1,4- Dihydropyridines as tools in the study of Ca2+ channels. Trends Neurosci. 8,396-400.
Sanguinetti M. C. and Kass R. S. (1984) Dihydropyridine deriva- tives: voltagedependent modulation of calcium channel cur- rent. Biophys. J. 45,394a.
Schwartz L. M., McCleskey E. W., and Almers W. (1985) Dihydro- pyridine receptors in muscle are voltagedependent but most are not functional calcium channels. Nature 314,747-75 I.
Spedding M. and Middlemiss D. N. (1985) Central effects of Ca2+ antagonists. Trends Pharmacol. Sci. 6,309-3 10.
Takahashi M. and Ogura A. (1983) Dihydropyridines as potent cal- cium channel blockers in neuronal cells. FEES Lett. 152,19 l - 194.
Toll L. (1982) Calcium antagonist. High-affinity binding and inhi- bition of calcium transport in a clonal cell line. J. Biol. Chem.
Triggle D. J. (198 1) Calcium antagonists: some basic chemical and pharmacologic aspects, in New Perspectives on Calcium Antag- onists (Weiss G. B., ed), pp. 1-18. American Physiological So- ciety, Bethesda, Maryland.
Turner T. J. and Goldin S. M. (1985) Calcium channels in rat brain synaptosomes: identification and pharmacological character- ization. High affinity blockade by organic Ca2+ channel block- ers. J. Neurosci. 5,841-849.
Vighy P. L., Johnson J. D., Matlib M. A., Wang T., and Shwartz A. ( 1982) Selective inhibition of Na+-induced Ca2+ release from heart mitochondria by diltiazem and certain other Ca2+ antag- onist drugs. J. Biol. Chem. 257,6000-6002.
van Breemen C., Aarouson P., and Loutzenhiser R. (1979) Na-Ca interactions in mammalian smooth muscle. Pharmacol. Rev. 30, 167-208.
Verbist J., Wuytack F., De Schutter G., Raeymaekers L., and Cas- teels R. (1984) Reconstitution of the purified calmodulinde- pendent (Ca2+ + Mg2+)-ATPase from smooth muscle. Cell Calcium 5,253-263.
257, I3 189-13 192.
J. Neurochem.. Vol. 47. No. 6, 1986