effect of temperature and adsorption potential on the
TRANSCRIPT

36 Electrochemistry
1 IntroductionThe electro-oxidation of carbon monoxide is one of the
most important catalytic reactions relevant to fuel cellsoperating on reformed gas or methanol. CO is known asa catalyst poison, strongly adsorbing on the surface of Pt.The binary PtRu system is one of the most widely stud-ied CO-tolerant anode catalyst.1-17)In the PtRu alloy, Ruacts as a promoter for the oxidation of adsorbed CO(COad).18)The promoting effect has often been discussedbased on the so-called “bifunctional effect” 19,20)or “ligandeffect” 21)or a mixture of both. The electrocatalytic oxi-dation of COad in acid takes place on the PtRu surfacevia a two-electron reaction which can be simplified asbelow:
Pt-COad+Ru-H2Oad → Pt-COad+Ru-OHad+H++e− →Pt+Ru+2H++2e−+CO2
CO-stripping voltammetry is a useful technique toprobe the COad electro-oxidation behavior of the catalyst.The COad electro-oxidation capability provides, at least inpart, information on the CO-tolerance and methanol oxi-dation ability of the catalyst. The electrochemicallyactive metal surface area can be estimated by measuringthe charge associated with the electro-oxidation of COad
(CO-stripping) upon potential sweep assuming full mono-layer coverage of linear CO. However, full monolayercoverage is often not accomplished, thereby complicatingthe accurate assessment of the electrochemically activesurface area.
The COad oxidation behavior on pure Pt is known tobe reflected by many factors. The CO coverage is oneparameter that is known to influence the onset potential,peak potential, and shape of the COad oxidation peak.22-37)
Factors such as the CO adsorption potential (the poten-tial that the electrode is kept during the adsorption ofCO dissolved in the solution) and cell temperature willinfluence the CO coverage. In contrast to the well-stud-
ied Pt, most studies on the CO oxidation behavior on Pt-Ru systems are limited to the effect of the alloy composi-tion and structure, and studies on the effect of tempera-ture38-42)or sub-monolayer CO coverage is limited.42-46)Ina recent communication,42)we reported the effect of tem-perature on the COad electro-oxidation behavior for car-bon supported Pt, Ru, and PtRu. The temperature depen-dence of the peak potential for COad oxidation exhibiteda non-linear behavior for Ru/C and PtRu/C when theadsorption potential was 300 mV vs. RHE, which wasdue to sub-monolayer COad coverage. Thus, understand-ing the effect of the COad adsorption potential is essentialfor investigating the effect of temperature on the COad
oxidation behavior of PtRu/C, analyzing the onset andpeak potential of COad oxidation, and to estimate theeffective surface area. Here we report a thorough studyon the COad electro-oxidation behavior of PtRu/C. TheCOad electro-oxidation behavior as a function of theadsorption potential and cell temperature was studied.The current transient during the CO adsorption processat adsorption potentials more positive than the onset ofCO oxidation was evaluated in order to probe the COad
oxidation behavior during the CO adsorption process.The thermodynamics of the COad oxidation reaction wasstudied by changing the reaction temperature. In addi-tion, it will be shown that saturated coverage can be cal-culated by combining the current transient measuredduring the CO adsorption process and the CO strippingcurrent.
2 ExperimentalCarbon supported Pt, Ru, and PtRu (30 mass% metal)
electrocatalysts were prepared by an impregnationmethod reported previously.10)Briefly, the electrocata-lysts were prepared by introducing appropriate amountsof Vulcan XC-72R into ethanolic solutions of Pt(NH3)2
― Article―
Effect of Temperature and Adsorption Potential on the Electro-oxidationof Adsorbed Carbon Monoxide on Carbon Supported PtRu
Tomoyuki KAWAGUCHI, Wataru SUGIMOTO,* and Yoshio TAKASU
Department of Fine Materials Engineering, Faculty of Textile Science and Technology, Shinshu University (3-15-1Tokida, Ueda 386-8567, Japan)
Received September 28, 2009 ; Accepted November 6, 2009
The effect of temperature and adsorption potential towards the electro-oxidation of pre-adsorbed carbon monoxide(COad) on carbon supported platinum-ruthenium alloy catalyst was studied by COad stripping voltammetry andchronoamperometry. The current transient during the CO adsorption process was analyzed at various adsorptionpotential to understand the catalytic reaction during the CO adsorption process. A clear correlation with thecharge associated with CO oxidation during the CO adsorption process and potential was obtained. A lineardecrease in the COad oxidation potential with a slope of −2.7 mV K−1 and an apparent activation energy of 126 kJmol−1 was observed with increasing temperature when full CO coverage is achieved.
Key Words : Electrocatalytic Oxidation, Carbon Monoxide, Anode Catalyst, CO Stripping Voltammetry, PtRu

78,No. 1(2010) 37
(NO2)2, Ru(NO3)3, or a 1 : 1 molar ratio of Pt(NH3)2(NO2)2and Ru(NO3)3. After thorough mixing, the precursor solu-tion was allowed to dry at 60℃ to a powder. The driedpowder was then reduced in a tube furnace under flow-ing H2(10%)-N2(90%) gas for 2 h at 200℃. Detailedanalysis of the structure of the catalysts, i.e., particlesize and alloying state were confirmed by X-ray diffrac-tion (XRD, Rigaku RINT-2550 with monochromatedCuKα radiation at 40 kV and 50 mA) in the 2θ range of15-95°at a scanning speed of 2°/min and detailed pro-files in the 2θ range of 62-75°were examined with astep scan of 0.02°and counting time of 5 s. Particle sizeand distribution was analyzed utilizing high-resolutionscanning electron microscopy (HR-SEM, Hitachi S-5000).The synthetic conditions applied in this study produceswell-alloyed catalysts with average particle size of 3 nmthat are highly dispersed on the carbon support, result-ing in high activity towards CO and methanol oxida-tion.10)
A beaker-type electrochemical cell equipped with theworking electrode, a platinum mesh counter electrode,and an Ag/AgCl/KCl(sat.) reference electrode was used.The reference electrode was kept at the same tempera-ture as the working electrode. A Luggin capillary facedthe working electrode at a distance of 2 mm.Electrochemical measurements were conducted at 0, 25,30, 40, 50, and 60℃. Electrode potentials are referred tothe RHE scale corrected for the temperature effect(RHE(t)) according to the following equation:
E vs. RHE(t)=E vs Ag/AgCl+(224 − T )×
− ×pH×2.303
where R is the gas constant (8.314 J K−1 mol−1), T is thetemperature, n is the number of electrons involved in thereaction (in this case, n=1), F is the Faraday constant(96,485 C mol−1) and ∂T/∂E is 0.0010 K V−1.47)The acti-vation energies of the COad oxidation were obtained bythe intercept in the plot of the peak potential of CO elec-tro-oxidation versus the absolute temperature. The fol-lowing equation was used to convert measured poten-tials to the standard hydrogen electrode at 298 K:
E vs. SHE(298)=E vs RHE(t) − 0.021 V+(T − 298)×
where ∂T/∂E is 0.00084 K V−1.48,49)
The working electrode was a thin film electrode50,51)
composed of a mirror polished Glassy Carbon rod (0.196cm2 exposed surface) modified with 40 µg of the activematerial (12 µg metal). 20 µL of a 1 wt% Nafion®
ionomer was dropped onto the electrode surface to stabi-lize the electrocatalysts to the Glassy Carbon rod.
CO-stripping voltammetry was conducted in the fol-lowing manner. After initial surface cleaning in de-aerat-ed 0.5 M H2SO4 (100 cycles at 50 mV s−1 between 50 to1200 mV for Pt/C and 800 mV for PtRu/C and Ru/C),the potential was stopped during the anodic scan. 100%gaseous CO was then purged into the cell for 40 minuteswhile maintaining a constant voltage (adsorption poten-
∂T∂E
RTnF
∂T∂E
tial, Ead=50-300 mV) to allow CO to adsorb onto theelectrocatalyst surface. Dissolved CO in the electrolytewas then purged out with N2 for 40 minutes. The electro-chemically active surface area was estimated by integra-tion of the COad stripping peak, corrected for the electricdouble-layer capacitance. For simplicity, a monolayer oflinearly adsorbed CO and the Coulombic charge neces-sary for oxidation as 420 µC cm−2 was assumed. As it isknown that bridge adsorbed CO is also present to someextent on the PtRu surface,52)the absolute q values (inunits of Coulombs per gram of PtRu) may be slightlyunderestimated, possibly by 10%. However, the overalltrend in the CO oxidation charge should not be affectedby the value used for normalization.
3 Results and DiscussionFigure 1 shows the CO-stripping voltammograms for
PtRu/C with various CO adsorption potential Ead at 25,40, and 60℃. At 25℃, the change in Ead does not affectthe CO-stripping voltammograms; the COad oxidationcharge, peak potential, and peak onset are insensitive toEad. A decrease in the COad oxidation charge is observedwhen Ead>280 at 40℃ and Ead>200 mV at 60℃. Theseobservations are an indication of a decrease in CO cover-age at high Ead. The lower CO coverage on the PtRu sur-face at high Ead is due to partial oxidative removal of COduring the CO adsorption procedure.
A negative shift in the onset potential of COad oxida-tion on PtRu/C was observed with increased Ead, i.e.lower CO coverage. This trend follows that observed forPt/C [for example, refs. 23, 24, 29, 37, 53]. In the case ofRu/C, the onset potential does not change when Ead isincreased (low CO coverage conditions).42)If two neigh-boring Ru sites were responsible for the initial COad oxi-dation on PtRu, then the trend in the onset potential forCO oxidation as a function of the CO coverage forPtRu/C should follow that of Ru/C. On the other hand, iftwo neighboring Pt sites were accountable, then theonset of COad oxidation would be observed at a morepositive potential (near that of Pt/C). Thus, the most like-ly explanation for the negative shift in the COad onsetpotential observed for PtRu/C would be the reactionbetween Pt-COad and Ru-OHad, which is the well-knownbi-functional mechanism.19,20)As the onset potential ofCOad oxidation on PtRu/C at low CO coverage is lowerthan that of Ru/C, an electronic effect should also beaccounted for in addition to the bi-functional mechanism.
In terms of the change in the peak potential of COad
oxidation on PtRu/C as a function of CO coverage, a pos-itive shift is observed at lower coverage, which is similarto the behavior of Ru/C,42)and opposite to the trendobserved for Pt/C.29,37)A tentative explanation for thisbehavior may be given on the basis of the difference inthe mobility of the reactive species on Pt, Ru, and PtRu.It is postulated that the mobility of the species on Pt atthe COad oxidation potential (>0.6 V vs. RHE) is ratherhigh, allowing such species to move to more reactivesites. On the other hand for Ru and PtRu, the mobility ismore restrained, possibly as a consequence of the lowerpotential applied, resulting in a tailing of the COad oxida-

38 Electrochemistry
tion peak to positive potentials. Overall, the trend in thechange in the onset potentials of CO oxidation withdecreasing CO coverage follows that of Pt/C, while theshift in peak potentials for PtRu/C follows that of Ru/C,thus showing a mixed behavior between Pt/C and Ru/C.
Figure 2 shows the current transients observed dur-ing the CO adsorption procedure as a function of time atvarious Ead at 60℃ . An initial cathodic current isobserved during the first few minutes of the CO adsorp-
tion process regardless of the potential applied. Similarresults have been observed for Pt/C and have beeninterpreted as the non-faradaic current discharge due tothe change in the value of the metal double layer capaci-tance upon adsorption of CO.29)Following this initialcathodic current, the current rises and settles to asteady-state current. The current transients from t=0to 40 min represents bulk CO oxidation. The steady-stateanodic current observed during the CO adsorptionprocess (t≤40 min) is attributed to continuous CO oxida-tion and CO adsorption onto the free catalysts surface.The kinetics for the continuous CO oxidation naturallyincreases when the applied potential is increased, result-ing in an increase in the steady-state current at higherEad. At t=40 min, CO gas is changed to N2 gas to driveout dissolved CO in the electrolyte. As the concentrationof dissolved CO in the electrolyte decreases, the proba-bility of CO adsorption decreases, resulting in a drop incurrent. After a critical point where COad oxidation andCO adsorption from the electrolyte reaches equilibrium,an anodic current is observed since there is no more dis-solved CO to adsorb onto the free metal sites. After thecritical point, CO oxidation should represent currenttransients observed for monolayer COad oxidation at therespective potentials. The anodic current reaches a max-imum current jmax at tmax and settles to a steady-statecurrent once all the weakly bound CO is oxidized. Theanodic peak is asymmetric; that is the decay at t> tmax isslower than the rise at t< tmax. The observation of maxi-
Fig. 1 The COad stripping voltammograms for PtRu/C at(a) 25, (b) 40, (c) 60℃. The adsorption potentials are shownin the figure in units of mV vs. RHE(t).
Fig. 2 Current transients recorder during CO adsorptionon PtRu/C at 60℃. The CO adsorption potentials are (a)100, (b) 200, (c) 250, (d) 260, (e) 270, (f) 280, (g) 290 and (h)300 mV vs. RHE(t).
78,No. 1(2010) 39
mum current suggests COad oxidation proceeds via aLangmuir-Hinshelwood reaction. The tailing currentdecay indicates that COad diffusion to OHad nucleationsites is rate determining.
A quantitative assessment of the CO oxidation processduring the course of CO adsorption was conducted bycalculating the charge associated with the oxidation ofCOad from the critical point to t=80 min, denoted as q1(shown as the shaded portion in Fig. 3). The currenttransient between t=40 min to the critical point falls toa steady-state current, which is the same as the steady-state current at t=80 min. This allows calculation of q1,which represents the current transients observed formonolayer COad oxidation at the respective potentials
immediately following the point where CO adsorptionfrom the electrolyte and COad oxidation reaches equilibri-um. It should be noted that for Ead=290 and 300 mV, thecurrent transient of dissolved CO slightly overlaps thatof q1. However, the margin of error for q1 should be verysmall compared to the absolute q1 value, thus peak sepa-ration was not attempted for sake of simplicity.
The charge obtained from CO-stripping (q2) and thecharge accumulated during the COad process (q1) and the
Fig. 3 Current transients recorded during CO adsorptionon PtRu/C at 60℃. The shaded portion shows the chargeassociated with the oxidation of COad during the adsorptionprocedure, q1. The CO adsorption potentials are (a) 300 and(b) 100 mV vs. RHE(t).
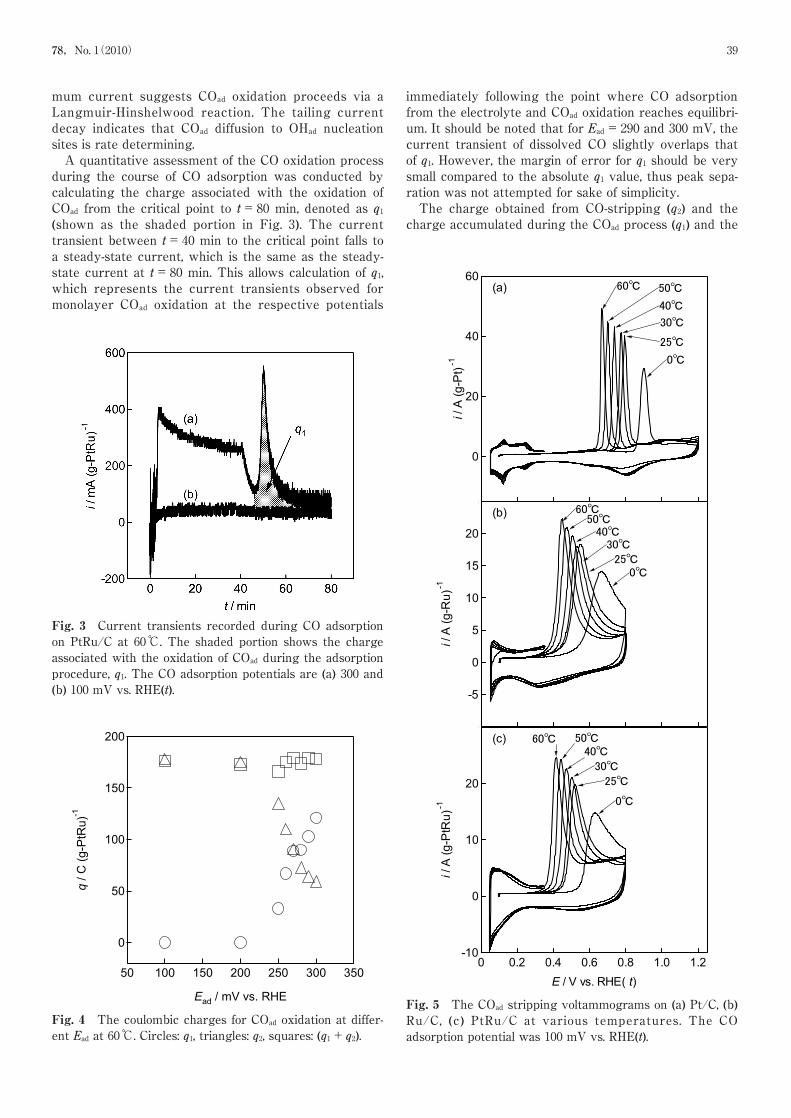
Fig. 5 The COad stripping voltammograms on (a) Pt/C, (b)Ru/C, (c) PtRu/C at various temperatures. The COadsorption potential was 100 mV vs. RHE(t).
Fig. 4 The coulombic charges for COad oxidation at differ-ent Ead at 60℃. Circles: q1, triangles: q2, squares: (q1+q2).

40 Electrochemistry
total charge (qtotal=q1+q2) are plotted in Fig. 4 as afunction of Ead. From Figs. 3 and 4, Ead=100 mV at 60℃gives maximum CO coverage under the present experi-mental conditions. Figure 4 also shows that one can easi-ly obtain the maximum CO charge for PtRu even at anon-optimized Ead value by monitoring the current tran-sient during the CO adsorption process.
The temperature dependence of COad on PtRu/C iscompared with Pt/C and Ru/C in Fig. 5 at Ead=100 mV.With increasing temperature, the CO oxidation peak cur-rent increases and the peak width narrows. The narrow-ing of the CO oxidation peak with increasing tempera-
ture is due to the increase in surface diffusion of theadsorbed species. The exposed metal surface area esti-mated from the CO oxidation charge is 39, 55, and 50 m2
(g-metal)−1, for Pt, Ru, and PtRu/C, respectively at 25℃.The surface area tends to decrease at elevated tempera-tures for all three catalysts (Fig. 6). The decrease in COoxidation charge for Pt/C is in agreement with earlierstudies on Pt/C37 ,52)as well as single crystals54)andPtRu/C.52)As a reference, the specific surface area calcu-lated from the hydrogen adsorption region for Pt/C isshown in Fig. 6. It can be seen that the specific surfacearea obtained from CO-stripping and hydrogen adsorp-tion closely matches. The decrease in apparent specificsurface area may be due to the decrease in the stabilityof the adsorbed species, i.e. thermal desorption of Had
and COad.A linear relationship between peak potential and tem-
perature is obtained with a slope of approximately −3.1, −2.7, −2.7 mV K−1 and activation energy of 163, 130, and126 kJ mol−1 for Pt, Ru, and PtRu, respectively (Fig. 7).Note that the CO oxidation peak potential for PtRu/C is~30 mV negative compared to Ru, which can be inter-preted as the effect of the change in the electronic stateof Pt (and also Ru) due to the alloying (ligand effect) inaddition to the bi-functional mechanism as discussedabove.
4 ConclusionsThe COad oxidation behavior on PtRu/C as a function
of the operating temperature and adsorption potentialwas studied by COad stripping voltammetry. When theadsorption potential is more positive than the CO electro-oxidation onset potential, full CO coverage is not accom-plished. The onset of CO electro-oxidation shifts to nega-tive potentials for sub-monolayer CO coverage. By moni-toring the current transients during the CO adsorptionprocedure, the number of free metal sites formed duringthe CO adsorption procedure could be quantitativelydetermined. The optimized adsorption potential at 60℃was 100 mV vs. RHE. Under optimized conditions, thepeak potential of the COad oxidation on PtRu/C wasobserved at a more negative potential than on Ru/C forthe temperature range studied (0-60℃). A linear rela-tionship between peak potential and temperature isobtained with a slope of approximately −3.1, −2.7, −2.7mV K−1 and activation energy of 163, 130, and 126 kJmol−1 for Pt, Ru, and PtRu, respectively.
AcknowledgementsThe authors are grateful to Ishifuku Metal Industry
Co. for kindly supplying Pt(NH3)2(NO2)2. This work wassupported in part by the Polymer Electrolyte Fuel CellProgram from the New Energy and IndustrialTechnology Development Organization (NEDO) of Japanand a Grant-in-Aid for Scientific Research (18685026)from Ministry of Education, Science, Sports, and Culture(MEXT).
References1)B. D. McNicol and R. T. Short, J. Electroanal. Chem. 81,
Fig. 6 The temperature dependence of the exposed metalsurface area obtained by COad-stripping for Pt/C (circles),Ru/C (squares), and PtRu/C (triangles) and the metalsurface area obtained from the hydrogen adsorption chargefor Pt/C (X).
Fig. 7 The COad oxidation potential for Pt/C (circles),Ru/C (squares), and PtRu/C (triangles) as a function oftemperature.

78,No. 1(2010) 41
249 (1977). 2)M. Watanabe, M. Uchida, and S. Motoo, J. Electroanal.
Chem., 229, 395 (1987).3)J. B. Goodenough, A. Hamnett, B. J. Kennedy, R.
Manohara, and S. A. Weeks, J. Electroanal. Chem., 240,133 (1988).
4)A. Hamnett, S. A. Weeks, B. J. Kennedy, G. Troughton,and P. A. Christensen, Ber. Bunsen-Ges. Phys. Chem., 94,1014 (1990).
5)A. S. Aricò, P. Creti, H. Kim, R. Mantegna, H. Giordano,and V. Antonucci, J. Electrochem. Soc., 143, 3950 (1996).
6)C. He, H. R. Kunz, and J. M. Fenton, J. Electrochem. Soc.,144, 970 (1997).
7)A. S. Aricò, A. K. Shukla, K. M. El-Khatib, P. Creti, andV. Antonucci, J. Appl. Electrochem., 29, 671 (1999).
8)T. J. Schmidt, H. A. Gasteiger, and R. J. Behm,Electrochem. Commun., 1, 1 (1999).
9)A. S. Aricò, P. Creti, E. Modica, G. Monforte, V. Baglio,and V. Antonucci, Electrochim. Acta, 45, 4319 (2000).
10)Y. Takasu, T. Fujiwara, Y. Murakami, K. Sasaki, M.Oguri, T. Asaki, and W. Sugimoto, J. Electrochem. Soc.,147, 4421 (2000).
11)H. N. Dinh, X. Ren, F. H. Garzon, P. Zelenay, and S.Gottesfeld, J. Electroanal. Chem., 491, 222 (2000).
12)Y. Takasu, H. Itaya, T. Iwazaki, R. Miyoshi, T.Ohnuma, W. Sugimoto, and Y. Murakami, Chem.Commun., 2001, 341 (2001).
13)A. V. Tripkovíc, K. D. Popovíc, B. N. Grgur, B.Blizanac, P. N. Ross, and N. M. Markovíc, Electrochim.Acta, 47, 3707 (2002).
14)C. Roth, N. Marty, F. Hahn, J.-M. Léger, C. Lamy, andH. Fuess, J. Electrochem. Soc., 149, E433 (2002).
15)Y. Takasu, H. Itaya, T. Kawaguchi, W. Sugimoto, andY. Murakami, Stud. Surf. Sci. Catal., 145, 279 (2003).
16)Y. Takasu, T. Kawaguchi, W. Sugimoto, and Y.Murakami, Electrochim. Acta, 48, 3861 (2003).
17)Y. Takasu, W. Sugimoto, and Y. Murakami, Catal.Surveys Asia, 7, 21 (2003).
18)J. O’M. Bockris and H. Wroblowa, J. Electroanal. Chem.,7, 428 (1964).
19)M. Watanabe and S. Motoo, Denki Kagaku (presentlyElectrochemistry) 41, 190 (1973).
20)M. Watanabe and M. Motoo, J. Electroanal. Chem., 60,267 (1975).
21)T. Frelink, W. Visscher, and J. A. R. van Veen, Surf.Sci., 335, 353 (1995).
22)T. Iwasita and U. Vogel, Electrochim. Acta, 33, 557(1988).
23)L. Palaikis, D. Zurawski, M. Hourani, and A.Wieckowski, Surf. Sci., 199, 183 (1988).
24)D. Zurawski, M. Wasberg, and A. Wieckowski, J. Phys.Chem., 94, 2076 (1990).
25)J. A. Caram and C. Gutiérrez, J. Electroanal. Chem., 305,259 (1991).
26)M. Bergelin, J. M. Feliu, and M. Wasberg, ElectrochimActa, 44, 1069 (1998).
27)N. M. Markovic, B. N. Grgur, C. A. Lucas, and P. N.Ross, J. Phys. Chem. B, 103, 487 (1999).
28)E. Herrero, J. M. Feliu, S. Blais, Z. Radovic-Hrapovic,and G. Jerkiewicz, Langmuir, 16, 4779 (2000).
29)B. Rush, J. A. Reimer, and E. J. Cairns, J. Electrochem.Soc., 148, A137 (2001).
30)Z. Jusys and R. J. Behm, J. Phys. Chem. B, 105, 10874(2001).
31)K. Jambunathan and A. C. Hillier, J. Electroanal. Chem.524-525, 144 (2002).
32)A. López-Cudero, A. Cuesta, and C. Gutiérrez,Electrochem. Commun., 6, 395 (2004).
33)A. López-Cudero, A. Cuesta, and C. Gutiérrez, J.Electroanal. Chem., 579, 1 (2005).
34)T. Smolinka, M. Heinen, Y.-X. Chen, Z. Jusys, W.Lehnert, and R. J. Behm, Electrochim. Acta, 50, 5189(2005).
35)A. López-Cudero, A. Cuesta, and C. Gutiérrez, J.Electroanal. Chem., 586, 204 (2006).
36)A. Cuesta, A. Couto, A. Rincón, M. C. Pérez, A. López-Cudero, and C. Gutiérrez, J. Electroanal. Chem., 586, 284(2006).
37)R. J. Behm and Z. Jusys, J. Power Sources, 154, 327(2006).
38)H. N. Dinh, X. Ren, F. H. Garzon, P. Zelenay, and S.Gottesfeld, J. Electroanal. Chem., 491, 222 (2000).
39)H. Nonaka and Y. Matsumura, J. Electroanal. Chem.,520, 101 (2002).
40)A. S. Aricò, V. Baglio, A. Di Blasi, E. Modica, P. L.Antonucci, and V. Antonucci, J. Electroanal. Chem., 557,167 (2003).
41)M. Watanabe, T. Sato, K. Kunimatsu, and H. Uchida,Electrochim. Acta, 53, 6928 (2008).
42)T. Kawaguchi, W. Sugimoto, Y. Murakami, and Y.Takasu, Electrochem. Commun., 6, 480 (2004).
43)P. Waszczuk, G.-Q. Lu, A. Wieckowski , C. Lu, C. Rice,and R. I. Masel, Electrochim. Acta, 47, 3637 (2002).
44)N. Yee, G. S. Chottiner, and D. A. Scherson, J. Phys.Chem. B, 109, 5707 (2005).
45)T. Vidakovic, M. Christov, and K. Sundmacher,Electrochim. Acta, 52, 5606 (2007).
46)F. Colmati Jr., W. H. Lizcano-Valbuena, G. A. Camara,E. A. Ticianelli, and E. R. Gonzalez, J. Braz. Chem. Soc.,13, 474 (2002).
47)D. J. G. Ives and G. J. Janz, Reference Electrodes,Academic Press, New York, NY, p. 189 (1961).
48)B. E. Conway, H. Angerstein-Kozlowska, and W. B. A.Sharp, J. Chem. Soc., Faraday Trans. I, 74, 1373 (1978).
49)S. Blais, G. Jerkiewicz, E. Herrero, and J. M. Feliu,Langmuir, 17, 3030 (2001).
50)T. J. Schmidt, M. Noeske, H. A. Gasteiger, R. J. Behm,P. Britz, and H. Bønnemann, J. Electrochem. Soc., 145,925 (1998).
51)T. J. Schmidt, H. A. Gasteiger, G. D. Stäb, P. M. Urban,D. M. Kolb, and R. J. Behm, J. Electrochem. Soc., 145,2354 (1998).
52)M. Watanabe, T. Sato, K. Kunimatsu, and H. Uchida,Electrochim. Acta, 53, 6928 (2008).
53)K. Jambunathan and A. C. Hillier, J. Electroanal. Chem.,524-525, 144 (2002).
54)E. Herrero, B. Álvarez, J. M. Feliu, S. Blais, Z. Radovic-Hrapovic, and G. Jerkiewicz, J. Electrocanal. Chem., 567,139 (2004).