물리학자를 위한 mpi -...
TRANSCRIPT

물리학자를 위한 MPI
물리학자를 위한 MPI
한국표준과학연구원
2003년 3월 3일 출발합니다.
MPI (Message Passing Interface) 란 무엇인가?
MPI 자체는 병렬 라이브러리들에 대한 표준규약이다. (125개의 서브 프로그램들로 구성되어 있
다.) MPI로 만들어진 병렬 라이브러리를 사용한다면 작성된 응용프로그램이 source level의 호환성
을 보장받을 수 있다. MPI는 약 40개 기관이 참여하는 MPI forum에서 관리되고 있으며, 1992년 MPI 1.0 을 시작으로 현재 MPI 2.0까지 버전 업된 상태이며 이들 MPI를 따르는 병렬 라이브러리로는 Ohio supercomputer center 에서 개발한 Lam-MPI 와 Argonne National Laboratory 에서 개발한 MPICH
가 널리 사용되고 있다. MPI 2.0에서는 동원되는 프로세스의 수를 시간에 따라서 바꿀 수도 있다. 또한, 병렬 I/O를 지원한다. 메시지 = 데이터+송신지와 수신지 주소
병렬 프로그래밍의 모델들로는 아래와 같은 것들이 있다.PVM MPI OpenMP : 공유메모리 병렬 컴퓨팅의 사실상의 표준이다.UPC HPF
왜 MPI인가?
전산물리학을 하는 입장에서 간단하게 말하면, 뭐, 업계의 standard라고 하니 따르겠습니다. 컴퓨터
의 기종에 관계없이 일반적인 병렬처리를 위해서는 MPI를 사용하는 것이 일반적으로 유리하다. 물론, OpenMP와 같이 거의 자동적으로 소스 코드를 병렬처리에 맞도록 재설계해 주는 경우 (kapf90 -conc -psyntax=openmp prog.f)도 있지만 SMP (여러 개의 프로세서가 버스를 통
하여 하나의 거대한 메모리에 연결된 것. 공유메모리. 이와 같은 장비는 상대적으로 고가일 수밖에 없다. 프로세서 숫자의 확장에 상대적으로 심각한 제한이 있다. Compaq ES40, Sun E10000, HP N-class)와 같은 환경에서만 작동하며 일반적인 적용이 불가능하다. 위에서 언급한 방식으로는 유저
가 원하는대로 자동으로 병렬화가 안된다. SMP기종과 달리 일반적인 클러스터 장비들에서는 모든
메모리가 모든 프로세서들에게 연결되어 있지 않다. C, (C++)언어나 포트란언어에서 같은 방식으
로 사용되는 MPI를 이용하는 것이 병렬계산의 기본이다. 유저에 따라서는 PVM과 MPI를 동시에 사
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (1 of 73)2005-11-22 오후 4:09:44

물리학자를 위한 MPI
용하는 아주 전문적인 유저들 (병렬 계산 스페셜 리스트라고 할 만하다.)도 있다. 지역적으로 분산
된 장비들을 사용하는 분산컴퓨팅 (distributed computing, cluster computing 과는 구별되는 것이다.)
방식에서도 MPI를 이용할 수 있다. http://www.myri.com : MPICH-GM, http://www.niu.edu/mpi : MPICH-G2
Top 10 Reasons to Prefer MPI Over PVM 1. MPI has more than one freely available, quality implementation. (LAM,MPICH,CHIMP) 2. MPI defines a 3rd party profiling mechanism. 3. MPI has full asynchronous communication. 4. MPI groups are solid and efficient. 5. MPI efficiently manages message buffers. 6. MPI synchronization protects 3rd party software. 7. MPI can efficiently program MPP and clusters. 8. MPI is totally portable. 컴퓨터의 기종에 상관없이 사용될 수 있어야 한다.9. MPI is formally specified. 10. MPI is a standard.
물리학에서 왜 필요한가?
많은 전산 물리학 문제들이 병렬 알고리즘을 이용한 병렬처리를 하면 상대적으로 쉽게 풀리어진다. 그렇지 못한 경우도 물론 있다. 컴퓨터의 기원은 순차 알고리즘을 근간으로 한다. 이점을 생각하면, 격세지감을 느낀다. 바야흐로, 순차 알고리즘뿐만 아니라 병렬 알고리즘을 동시에 생각할 때가 된 것이다. 물론, 상당한 대가를 치르지만, 그 비용보다 얻는 것이 더 많다면 우리는 병렬계산을 한다. 잘 아시다시피 물리문제가 일반적으로 병렬처리가 용이하도록 정해져있지는 않다. 원천적으로 불가능한 것들도 많다. 많은 경우 전체 계산의 일부분은 병렬처리가 가능하다. 예를 들어 x %가 병렬 처리 가능하다면 병렬 컴퓨터 P대를 사용하면 한 대를 사용할 때보다도 100/{x/P+(100-x)} 배 정도 전체 계산이 빨라진다. 실제 계산에서는 병렬계산을 위하여 정보의 교환이 이루어진다. 이러한 절차 때문에 위의 식에서 표현되는 것보다는 효율이 나오지 못한다. 즉, 프로세서 간 정보의 교환이 병렬화의 효율을 떨어뜨린다. 실제의 계산에서는 하나의 프로그램에 여러 가지 데이터를 각각의 CPU가 자기에게 할당된 자료들을 구별하여 독자적으로 처리하는 형태로 프로그램이 완성된다. 이를 SPMD (single program multiple data)라고 한다. 모든 프로세스가 동일한 하나의 프로그램을 실행한다. 데이터를 분해하여 수행할 수도 있고, 서로 다른 함수들을 나누어서 실행할 수도 있다. 결국 실제 응용 프로그래밍하기가 훨씬 더 어려워 져 버리고 말았다. 결국, 가장 시간이 많이 소모되는 hot spot을 병렬화 할 수 있는가가 관건이다. 병렬 컴퓨터의 발달: 1990년대 후반부터는 단일 프로세서를 이용한 컴퓨터는 www.top500.org (컴퓨터 성능 세계랭킹 500)에서 발견하기가 힘들어짐. 이들은 모두가 500위 이하로 밀려남. 초고성능의 단일 CPU 제작은 상당히 비경제적이다. 다시 말해서, 지금 수준의 단일 CPU들을 동시에 사용하는 것이 좋은 아이디어로 보임.병렬컴퓨터 (베어울프, Beowulf 1994 년 여름) {Thomas Sterling and Donald Becker, CESDIS, NASA}*16개의 486 DX4, 100MHz 프로세서 사용 *16 MB of RAM each, 256 MB (total) * channel bonded ethernet (2x10Mbps) Beowulf형으로 최초로 슈퍼 컴퓨터 성능 랭킹 www.top500.org에 등재된 컴퓨터 = Avalon: 140 alpha
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (2 of 73)2005-11-22 오후 4:09:44

물리학자를 위한 MPI
21164A, 256MB/node, fast ethernet, 12 port Gigabit ethernet, 1998년 Bell Price/Performance prize (저비용 고효율) 블루진 (Bleu Gene) 컴퓨터는 100만개의 CPU를 이용하려고 한다. 이러한 목표의 시험용 버전인 Blue Gene/L은 32,768(=215)개의 CPU를 이용한다. 2004년 현존하는 최고의 컴퓨터이다. 와, 215개 이쯤되면 거의 막가자는 것이죠! 2004년 6월 Intel Itanium2, Tiger4 1.4GHz, Quadrics 4096 로 만든 클러스터 Thunder (LLNL)가 www.top500.org에서 2위에 랭크되었다. 뿐만 아니라 클러스터 형태의 컴퓨터가 슈퍼 컴퓨터 성능 상위 500위 내에서의 계속해서 점유도를 확장해가고 있다. 일단 병렬컴퓨터의 디자인과 실질적인 사용이 널리 보급된 지금 사실상 대부분의 컴퓨터 센터들을 병렬 컴퓨터를 구비하고 있다. 많은 유저들이 순차프로그램을 사용할 경우 그들에게 적당한 CPU를 할당해주면 되기 때문에 센터입장에서는 효율적으로 유저들을 지원하는 것이다. 유저들 입장에서도 변화가 일어나고 있다. 많은 슈퍼 컴퓨터 사용자들은 계속해서 제공되는 거대한 컴퓨터의 사용을 통해서 자신의 고유한 문제를 해결할 것으로 믿어 왔으나, 최근 동향은 그렇게 전개되고 있지 않다. 한 때 사장되었던 병렬 알고리즘이 리바이블되고 각 그룹마다 새로운 형식의 병렬 컴퓨터 계산이 득세하고 있다. 일반 PC사용자 수준에서는 변화가 없었다고 하더라도 컴퓨터를 이용한 연구 개발 프로젝트에서는 중대한 변화가 일고 있다. http://www.nersc.gov/ 분산컴퓨팅 (distributed computing, cluster computing 과는 구별되는 것이다.) 방식으로 유명한 프로젝트는 seti@home, folding@home을 들 수 있다. 이러한 계산 방식은 미리 셋업을 해둔 컴퓨터들(클러스터)을 사용하는 것이 아니라 연구 그룹 이외의 자원하여 컴퓨터를 제공한 (물론, 인터넷을 통한 연결) 사용자의 컴퓨터를 이용하는 것이다. 위에서 언급한 프로젝트들에서는 윈도우, 맥, 리눅스, 유닉스를 가리지 않고 자원하여 제공한 일반 컴퓨터들을 사용하여 과학적 계산 결과들을 얻어낸다. 컴퓨터의 수가 엄청나게 많기 때문에 (1000 대 이상 사용) 많은 CPU시간을 확보할 수 있다. free cluster (management) softwares:http://oscar.openclustergroup.org/tiki-index.phphttp://rocks.npaci.edu/Rocks/http://www.mosix.org/http://www.openpbs.org/http://www.fysik.dtu.dk/CAMP/pbs.htmlhttp://nfs.sourceforge.net/http://www-unix.mcs.anl.gov/petsc/petsc-2/
병렬화 항상 필요한가?/병렬화 언제 필요한가?/병렬화의 이득과 비용은?
실제 병렬계산은 특정한 시스템에서 최적화되도록 만들 수밖에 없다. 보통의 이더넷을 통신장비로 사용할 경우 그 통신에 들어가는 시간이 과도하게 많은 경우가 많다. 즉, 계산을 해 버리는 경우가 더 낫을 수 있다. 왜냐하면, 최근에 사용되고 있는 CPU들의 성능이 컴퓨터 간 통신속도에 비해서 충분히 좋기 때문이다. 따라서 통신을 빨리할 수 있는 값싼 장비가 나와야 병렬 계산은 더욱 활성화될 것이다. processor들 사이의 통신 기술이 병렬 컴퓨팅 기법의 핵심 기술 사항이라는 것이다.
통신 장비는 데이터를 주고 받기 전 단계에 소모되는 latency (microsecond)시간 (당연히 짧을수록 좋
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (3 of 73)2005-11-22 오후 4:09:44

물리학자를 위한 MPI
다.)과 정보가 전해질 때 한꺼번에 얼마나 많이/빨리 전해질 수 있는가 (bandwidth; communication capacity; bits/sec) 라는 두 가지 특성이 있다. (전화를 건다고 생각할 때, 전화를 걸면 상대가 전화를 걸자마자 받지 않는다. 약간은 기다려야 한다:latency. 상대가 전화를 받아도 주어진 시간에 얼마나 많은 정보를 전달할 수 있는 가는 상황에 따라 다르다:bandwidth.) 데이터의 분할, 기능적 분할등을 따져 볼 수 있다.
적은 크기의 데이터를 여러 번 주고 받는 것 보다는 한번에 모아서 주고 받는 것이 유리하다. 이는 데이터 통신시 필요한 latency시간을 줄인다는 의미이다. 최악의 경우 (자주 일어나는 경우이다.) 두 개의 CPU를 사용하여 한 대의 CPU를 사용하는 경우보다 더 느린 계산을 할 수 있다. 고속 통신 장비는 고가이며 CPU 장비와 거의 동일한 가격을 요구한다. 즉, {CPU 16 대 +이더넷(저속 통신 장비) }이 {CPU 8대 + 미리넷(고속통신 장비 이름)}과 얼추 비슷한 견적을 낸다. 보통 유선 인터넷의 bandwidth가 100 Mbps이다. 기가비트 이더넷이 bandwidth에서 패스트 이더넷보다 우의를 보여서 병렬계산에 유리할 것이라고 생각되지만, 많은 경우 latency 때문에 별 효과를 못보는 경우가 많다. 대량의 정보교환이 자주 일어나지 않는 경우에는 오케이다. 일반적인 컴퓨터 코드에서 확실한 병렬 효율성을 확보하려면 결국 고가 장비 (Myrinet)를 사용해야 한다.
Fast Ethernet Gigabit Ethernet Myrinet Quadrics 2
latency 120 microsecond 120 microsecond 7 microsecond 0.5 microsecond
bandwidth 100 Mbps 1 Gbps 1.98 Gbps 1 Gbyte/second
정말로 병렬화해야 되는가? 그렇다면, 다음의 두 가지 항목으로 견적을 내보자! 로드밸런싱과 스피
드업
원하는 시기, 정확한 시기(각 CPU마다 원하는 시기)에 원하는 데이터의 송수신 이것이 MPI구현의 핵심이다. 한 가지 더 추가하면 알고리즘을 바꿀 필요가 있을 수도 있다는 것이다. 결과적으로 같은 일을 하여도 병렬화가 가능한 알고리즘과 그렇지 못한 알고리즘이 상존할 수 있다. 또 상황에 따라
서는 알고리즘의 효율성이 다소 나쁘더라도 확실한 병렬화의 장점 때문에 병렬계산에서 대우받고 사용되는 알고리즘들도 많이 있다. 이러한 상황의 경우 그렇게 해야 한다면 반드시 따져봐야 할 항목이 있다. 결국, 가능한 한 CPU간 통신들을 줄이고 CPU중심의 계산들이 주축이 되도록 알고리즘
을 만들 수 있는 가이다.
마지막으로, 다중처리장비와 소프트웨어 (MPI를 이용한 프로그램)를 통해서 소위 speedup을 원하
는 수준까지 향상시킬 수 있는 가이다. 여기서 speedup은, x축을 사용한 CPU 수, y축을 하나의 계산
을 수행할 때 CPU시간이 아닌 wall clock시간으로 소요된 전체계산 시간의 역수로 그래프를 그렸을 때, 많은 CPU를 사용하여 실질적인 전체 계산소요 시간의 단축된 정도를 의미한다. 예를 들어, 8대
의 CPU를 사용하여 단일 CPU로 계산할 때 보다 7배의 정도 빨리 계산했다면 87.5 %의 speedup을 확보한 경우이다. 이 정도면 일주일에 걸쳐서 할 일을 하루 안(기본 가정: 하나의 CPU를 사용하여 24시간 계산해야 할 경우, 적당히 큰 계산, 큰 작업량 (? 여전히 좋은 표현은 아닌데)으로 봐 줄 수 있겠
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (4 of 73)2005-11-22 오후 4:09:44

물리학자를 위한 MPI
다.)에 할 수 있다는 것이다. (많은 사람들은 이정도면 만족하는데, 그렇지 못한 상황도 있을 수 있다. 즉, 일의 크기, 한 번 일을 할 때의 소요되는 계산 시간이 결국은 문제이다. 또 다른 형태의 문제
로는 한 프로그램 수행에 있어서 메모리가 많이 잡히는 상황이다. 한 CPU에서 처리가 아예 불가능
한 경우도 있을 수 있겠다.) 이 정도면 아주 성공적인 병렬계산을 수행한 경우라고 말 할 수 있겠다. 물론, 사용하는 알고리즘이 허용하는 경우에 한해서이다.
또한 기술적으로 (하드웨어 적으로) 고속 통신망을 사용하느냐 안하느냐에 따라서 결정적으로 위의 퍼포먼스는 달라 질 수 있다. 황당한 경우이지만 2개의 CPU를 사용했는데 전체 계산 시간이 1개
의 CPU를 사용하는 경우보다 느려지는 경우가 있다. 과도한 CPU간 통신들이 고성능 CPU의 발목
을 잡고 있는 경우이다. 이 보다 더 나쁠 순 없다. 병렬계산 최악의 상황이라고 할 수 있다. 고속통신 장비가 필수불가결한 경우이다. 물론, 이 때 다시한번 따져봐야 할 것이 load balancing (CPU 들에게
얼마나 골고루 일들이 균일하게 분배되었는가? 거의 동시에 CPU중심의 계산이 마무리되는가?)일 것이다.
통신에 연관된 각 프로세서들은 어떠한 시기에, 어떠한 데이터 형태를, 얼마만한 사이즈로 받아야 혹은 주어야 하는지를 완전히 알고 있다. 그리고 각각의 CPU는 묵묵히 계산들을 수행한다, 그야말
로 독립적으로.......; 그런데, 어떤 CPU는 할당된 일이 많지 않아서 일을 다 끝내고 놀고 있고 어떤 CPU는 아직도 일을 다 못끝내고 아직도 계산을 수행하는 경우가 있을 수 있는데.....엄청난 CPU계
산 속도를 고려할 때 상당한 자원의 낭비가 있다. 왜냐하면 결국 계산은 가장 느린(혹은 능력에 비해
서 가장 일을 많이 하는 단 하나의 CPU에 의해서 결정되기 때문이다.) 다시 말해서 load balancing이
잘 될수록 병렬계산은 효율적으로 진행될 수 있다.
가장 확실한 방법 중 하나는 각 계산노드에 들어가서 현재 계산을 실행하고 있는 노드들의 계산시
간을 분석해 보는 것이다. 각 노드로 ssh를 통해서 들어간다. 그 다음 top명령어를 이용하여 현재 사용한 CPU시간을 노드별로 분석해 본다. 실제 흘러가는 시간 (wall clock)과 더불어서 노드들의 CPU시간 증가를 확인해야 한다. load balancing (모든 노드들에서 CPU시간을 골고루 잘 증가함 )이 잘 되
어 보인다고 하더라도 실제 시간의 흐름과 마찬가지로 계산에 사용된 CPU시간의 증가도 확인해야 한다. 주의해야 할 것은 특정노드로부터의 계산결과를 기다리는 시간이 있으면 좋지 않다는 것이
다. 그 시간에 그 노드에서도 계산을 할 수 있다면 해야 한다. 어떠한 이유에서든지 각 노드에서 계산이 쉬는 것은 좋지 않다. 물론 고려해야 할 것이 있다. 계산보다 통신에 시간이 더 걸리는 것은 그 자리에서 해결하는 것이 좋다.
top명령어에서 관찰할 때 CPU의 사용 현황이 % 단위로 표시된다. 계산하지 않고 있으면 0.0 %가 된다. 계산을 집중적으로 할 때 당연히 99.0 % 처럼 나온다. 그 중간의 통신 상황에서는 다양한 퍼쎈트 대의 값이 표시된다. 한 번 0.에서 99.0 까지 올라간 다음 높은 CPU 점유율이 유지되는 시간이 길수
록 CPU중심의 계산이 잘 되고 있다는 것을 의미한다. 가능한 한 이렇게 만들어야 좋은 병렬 효율성
을 얻을 수 있다. 한 노드에 두 개의 CPU를 사용하여 병렬 계산을 할 때의 예. (물론, 다른 노드에서
도 유사한 자료를 확인할 수 있다.) 일을 여러 노드에 나누는 것이 목적이 아니다, wall clock시간 기준으로 일을 빨리 처리하기 위해서 병렬계산을 할 뿐이다. 물론 빠른 처리를 위해서는 일을 여러 개의 노드들에 잘 분담해서 처리해야 한다. 나누는 것이 중요하다. 하지만, 더 중요한 것은 빨리 계산
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (5 of 73)2005-11-22 오후 4:09:44

물리학자를 위한 MPI
하는 것이다.
PID USER PRI NI SIZE RSS SHARE STAT %CPU %MEM TIME COMMAND
14683 ihlee 15 0 11376 11M 1380 S 63.2 1.1 2:27 action_lbfgs.x14671 ihlee 13 0 11372 11M 1380 S 62.8 1.1 2:27 action_lbfgs.x14715 ihlee 9 0 1052 1052 852 R 0.2 0.1 0:00 top당연히 하나의 CPU를 하나의 실행화일이 CPU 중심의 계산을 수행할 때 CPU 점유율이 99.0 %이상 나온다. #!/bin/csh -fset nodes = (hpc1 hpc2 hpc3 hpc4 hpc5 hpc6 hpc7 hpc8 \ hpc9 hpc10 hpc11 hpc12 hpc13 hpc14 hpc15 hpc16 \ hpc17 hpc18 hpc19 hpc20 hpc21 hpc22 hpc23 hpc24 \ hpc25 hpc26 hpc27 hpc28 hpc29 hpc30 hpc31 hpc32 \ hpc33 hpc34 hpc35 hpc36 hpc37 hpc38 hpc39 hpc40 \ hpc41 hpc42 hpc43 hpc44 hpc45 hpc46 hpc47 hpc48 \ hpc49 hpc50 hpc51 hpc52 hpc53 hpc54 hpc55 hpc56 \ hpc57 hpc58 hpc59 hpc60 hpc61 hpc62 hpc63 hpc64 \ hpc65 hpc66 hpc67 hpc68 hpc69 hpc70 hpc71 hpc72 \ hpc73 hpc74 hpc75 hpc76 hpc77 hpc78 hpc79 hpc80 \ hpc81 hpc82 hpc83 hpc84 hpc85 hpc86 hpc87 hpc88 \ hpc89 hpc90 hpc91 hpc92 hpc93 hpc94 hpc95 hpc96 \ hpc97 hpc98 hpc99 hpc100 hpc101 hpc102 hpc103 hpc104 \ hpc105 hpc106 hpc107 hpc108 hpc109 hpc110 hpc111 hpc112 \ hpc113 hpc114 hpc115 hpc116 hpc117 hpc118 hpc119 hpc120 \ hpc121 hpc122 hpc123 hpc124 hpc125 hpc126 hpc127 hpc128 ) foreach n ($nodes) echo '----------' $n '-----------' rsh $n $*end 만약 61개의 노드에 계산이 분포된 경우 앞에서 이야기한 확인이 결코 쉽지 않다. 이럴 경우 위에서 제시한 스크립트 (이름을 pexec라고 하면. 또한 노드들의 이름을 위에서와 같이 hpc*처럼 정의한 경우.)를 이용하여 아래와 같은 명령을 주면된다. 병렬 계산 중에 아래의 명령어를 실행한다. 실제 각 노드들에서 소모된 CPU 시간들을 체크할 수 있다. 아래와 같이 그 결과가 나왔다면, 로드 밸런싱이 잘 되어 있다고 말할수 있다. rsh를 통하여 노드에 들어가고 프린트하는 시간이 포함되기 때문에 이와 관련된 시간에 의한 오차는 무시한 경우 이다. 완전히 동일한 시간을 소모한다면 오름차순으로 나올 것 이다. 프로그램 개발 단계와 응용 단계에서 로드 밸런싱 결함의 심각한 문제를 빨리 체크해 낼 수 있다.
$ pexec ps |grep admd>summary_file
10487 ? 00:08:04 admd.x 5696 ? 00:08:04 admd.x 5174 ? 00:08:05 admd.x 5166 ? 00:08:05 admd.x 5159 ? 00:08:05 admd.x
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (6 of 73)2005-11-22 오후 4:09:44

물리학자를 위한 MPI
5150 ? 00:08:05 admd.x 5150 ? 00:08:05 admd.x 5150 ? 00:08:05 admd.x 5150 ? 00:08:05 admd.x 5223 ? 00:08:06 admd.x 9241 ? 00:08:06 admd.x 5502 ? 00:08:06 admd.x 5339 ? 00:08:06 admd.x 5392 ? 00:08:06 admd.x 5355 ? 00:08:06 admd.x 5639 ? 00:08:07 admd.x 5336 ? 00:08:07 admd.x12646 ? 00:08:07 admd.x 5296 ? 00:08:07 admd.x 5543 ? 00:08:08 admd.x 8310 ? 00:08:08 admd.x 5790 ? 00:08:08 admd.x 5360 ? 00:08:08 admd.x 5360 ? 00:08:08 admd.x 5360 ? 00:08:08 admd.x 3118 ? 00:08:08 admd.x 5336 ? 00:08:09 admd.x 5334 ? 00:08:09 admd.x 5334 ? 00:08:09 admd.x 5334 ? 00:08:09 admd.x 9306 ? 00:08:09 admd.x 5736 ? 00:08:09 admd.x 위의 경우와 같이 각 노드들에서 8분씩만 계산을 하였다고 해도, 8분/노드 *61노드 => 총 CPU 사용시간 488 분이 되는 것이다. 즉, 8분만에 488 분 (=8.1333 시간)의 CPU시간을 소모한 계산이다. 다시 말해서, 병렬효율이 높은 경우, 8분만에 8시간 짜리 작업을 완성할 수 있다는 것이다. 이 정도되면 가히 고효율이라고 할 수 있겠다. 컴퓨터 사용료는 통상 사용한 총 CPU시간 기준이다. 단위 시간당 많은 CPU 시간을 사용했기 때문에 돈도 많이 내어야 하는 것은 당연하다.
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (7 of 73)2005-11-22 오후 4:09:44

물리학자를 위한 MPI
스피드업 계산 예를 표시했다. wall clock 시간 기준으로 얼마나 빨리 계산을 할 수 있는가를 표시한다. 단일 노드를 활용할 경우 약 30분 정도 소요 (wall clock기준되는 계산의 예를 표시했다. 상당히 병렬화가 잘 된 경우의 예로 받아 들일 수 있는 경우이다. Embarrassingly parallel 알고리즘의 경우 이상적인 스피드업 값 (대각선으로 표시된 값에 접근하는 경우이다. 소위 observed speedup이란 (wall-clock time of serial execution)와 (wall-clock time of parallel execution)의 비율을 말한다. 즉, CPU시간 기준이 아니라 wall-clock 시간 기준으로 빨라지는 것으로 정의되는 것이다.
통상의 구조는 어떠한가?
MPI 함수 125개 중 아래의 6개만 사용하는 프로그램도 상당히 많다. 아주 행복한 계산들을 수행하
는 경우이다. 실제로 이러한 함수만을 이용하는 응용프로그램들이 많이 존재한다.
MPI_INIT: MPI 환경 초기화하기 : 유저 수준에서 바꿀 것이 사실상 없음. 모든 CPU에서 공통으로 불리어진다.
MPI_COMM_SIZE: 사용 중인 processor 숫자 반환 : 유저 수준에서 바꿀 것이 사실상 없음.MPI_COMM_RANK: 현 CPU의 번호 (rank라도 함. processor 갯수가 nproc일 때, 가능한 rank 값은
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (8 of 73)2005-11-22 오후 4:09:44

물리학자를 위한 MPI
0,1,2,3....nproc-1이다.) : 유저 수준에서 바꿀 것이 사실상 없음.
두 개의 processor 간 통신: rank값들을 사용하여서 현재 processor 번호를 확인하고 준비된 데이터
를 원하는 processor로 전송한다. 마찬가지로 현재의 processor번호를 확인하고 전송되어 올 데이터
를 받는다. 물론, 병렬계산은 "짜고 치는 고스톱이기 때문에 우리는 어떤 processor로부터 데이터가 오는지 그리고 어떤 processor가 이 데이터를 받아야 하는지 다 알고 있다. 정보가 특정한 노드로 보내어지는데, 그 노드가 받지 않으면 일이 안됩니다. 반드시 받아야 다음의 일들이 진행됩니다. 즉, 프로그래밍 작업중에, deadlock (교착)에 걸리는지 안 걸리는 지를 점검해야 합니다. 최소 두 대의
CPU들간에 다른 CPU로 부터의 데이터 송신이 지속적으로 발생한 경우.통신, 계산 순서 의존성, 동기화, 그리고 교착 상황의 체크가 병렬 프로그래밍의 주요 항목이라고 할 수 있다. 통상, 순차 프로
그램의 완성, 최적화 이후에 병렬 프로그래밍에 착수 한다.
MPI_SEND: 원하는 processor에게 데이터 전송시 사용 : 유저의 구체적인 목적이 적용됨 (원하는 데이터 형, 사이즈,...)
MPI_RECV: 원하는 processor로부터 데이터 전송받을 때 사용 : 유저의 구체적인 목적이 적용됨 (원하는 데이터 형, 사이즈,...)
MPI_FINALIZE: MPI 환경 종료하기: 유저 수준에서 바꿀 것이 사실상 없음. 모든 CPU에서 공통으
로 불리어진다.
MPI 함수들은 포트란 버전과 C 버전으로 나누어져 있다. 구체적인 함수 모양은 언어의 특성을 고려
하다보니 다르게 생겼지만, 수행하는 일은 사실상 같다. 실제 포트란에서 사용될 때의 모습. 잘 알려
진 것처럼 포트란에서는 소문자/대문자 구별이 없다.즉, mpi_init나 MPI_init나 같은 함수를 지칭한
다.
program testUSE important_module, ONLY : variables, sub_program_namesimplicit noneinclude "mpif.h"integer istatus(MPI_STATUS_SIZE) ! MPI_STATUS_SIZE는 위에서 선언한 include문으로 불러들인 내용에서 이미 정의된 것들이다. ...................... ............................integer nproc,myid,ierr,idestination,isource,iroot,kount INTEGER itemp,itemq,irateCHARACTER*8 fnnd ; CHARACTER*10 fnnt .........................................
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (9 of 73)2005-11-22 오후 4:09:44

물리학자를 위한 MPI
...........메시지 = 데이터+송신지와 수신지 주소커뮤티케이터 = 서로 통신할 수 있는 프로세스들의 집합. (MPI 핸들이다.) MPI_COMM_WORLD는 기본 커뮤티케이터이다. 헤더파일에서 정의된다. 사용자가 특별한 프로세스들만으로 구성되는 커뮤티케이터를 만들 수 있다................... call MPI_Init(ierr) call MPI_Comm_size(MPI_COMM_WORLD,nproc,ierr) call MPI_Comm_rank(MPI_COMM_WORLD,myid,ierr) if(myid == 0)then ! -----[ PROCESS ID = 0 CALL DATE_AND_TIME(date=fnnd,time=fnnt) write(6,'(1x,a10,2x,a8,2x,a10)') 'date,time ', fnnd,fnnt CALL SYSTEM_CLOCK(itemp,irate) endif ! -----] PROCESS ID = 0 if(myid == 0)then ! -----[ PROCESS ID = 0 각종 입력들...... endif ! -----] PROCESS ID = 0읽어 들인 정보 중에서 모든 노드에게 "방송할 필요가 있는 경우 kount=1 ; iroot=0 call MPI_BCAST(natom,kount,MPI_INTEGER,iroot,MPI_COMM_WORLD,ierr) myid는 processor번호를 나타낸다. (myid=0,1,2,3,....nproc-1 중의 하나 값을 가진다. 각 노드마다 다른 값을 가진다.)nproc는 현재 몇 개의 processor가 살아있는지를 나타낸다. (모든 노드에서 같은 값을 가진다.) 즉, SPMD에 따라서, 모든 컴퓨터에서 같은 프로그램을 수행하기 때문에 모든 컴퓨터에서 현재 살아있는 컴퓨터의 숫자 (nproc)는 같다. 각 컴퓨터마다 자신의 번호 (myid 값)는 컴퓨터마다 다르다. 따라서, 병렬 계산은 nproc와 myid값을 가지고 주어진 문제에 대한 분업/병렬 작업들을 설계할 수 있다. 당연히, 메모리 할당도 노드별로 다르게, 또는 동시에 같은 크기로 잡을 수 있다. 미리 선언해 두는 부분은 모든 노드에서 같이 해 두어야 한다. 예를 들면, real*8, allocatable :: abcd(:,:,:)처럼. 당연히 한 노드에서만 메모리 할당이 될 경우도 있을 수 있다. 병렬 프로그래밍은 기본적으로 노드별 작업을 설계해야 하기 때문에 순차 프로그래밍 보다 더 난이도가 높다. 보다 많은 테스트 작업들이 필요하다. 일반으로 순차 프로그램을 끝내고 그 다음 병렬 프로그램을 작성한다. itag=19idestination=1call MPI_Send(real_array_user,n_array_length,MPI_REAL8,idestination,itag,MPI_COMM_WORLD,ierr)real*8 형태의 데이터가 가야할 곳 지정해주어야 한다. 물론, 이 데이터의 크기도 보내는 곳에서 지정해줘야 한다. 특정 노드에서 정보를 보내기 때문에 위 함수는 특정 노드에서 불려져야 한다. itag=19isource=0call MPI_Recv(real_array_user,n_array_length,MPI_REAL8,isource,itag,MPI_COMM_WORLD,istatus,ierr)
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (10 of 73)2005-11-22 오후 4:09:44

물리학자를 위한 MPI
데이터를 받는 쪽에서는 그 형태와 크기를 알고 있어야 하며, 어디에서부터 출발했는지를 알아야 한다. MPI_REAL (실수, 싱글 프리시전)과 MPI_REAL8 (실수, 더블 프리시전)는 엄연히 다른 값들 임을 유의해야 한다. MPI_REAL8보다는 MPI_DOUBLE_PRECISION을 사용하는 것이 좋다. 왜냐하면, LAM에서도 사용가능하기때문이다. MPI_REAL8는 MPICH에서만 사용가능한 것이다. ! ........................................................................... master/slave 형식으로 일할 경우, 일들을 나누어서 수행하는 경우: myid=0에서 일들을 분배하고, 취합 한다. myid=0에서도 일부/균등 일들을 수행한다. 해야 할 총 일들의 단위가 (np+1)인 경우이고, 동원된 노드의 수는 nproc (> 1) 이다. myid=1,2,....nproc-2 :이들 노드들은 균등한 일의 양을 수행한다. myid=0 노드는 상황에 따라서 위의 노드들 보다 적은 양을 일들을 수행한다. 물론, 균등한 일의 양을 취급하는 경우도 있다. nblk=(np+1)/nproc+1 ; if( np+1 == (nblk-1)*nproc) nblk=nblk-1 allocate(wk_input(natom,3,nblk),wk_output(natom,3,nblk),wk_output2(nblk)) wk_input=0.0d0 ; wk_output=0.0d0 ; wk_output2=0.0d0! if(myid == 0)then ! -----[ PROCESS ID = 0! 정보를 나누어서 전달하기 do loop=0,nproc-2 jj=0 do j=(loop)*nblk,(loop+1)*nblk-1 jj=jj+1 ; wk_input(:,:,jj)=qq(:,:,j) enddo kount=3*natom*nblk ; idest=loop+1 ; itag=1 call MPI_SEND(wk_input,kount,MPI_DOUBLE_PRECISION,idest,itag,MPI_COMM_WORLD,ierr) enddo! 적당히 할당된 일하기, myid=0에서 do j=(nproc-1)*nblk,np call sma_energy_force(qq(1,1,j),force(1,1,j),vofqj(j)) enddo! myid /=0에서 보내온 정보를 나누어서 받아들이기 do loop=0,nproc-2 kount=3*natom*nblk ; isour=loop+1 ; itag=3 call MPI_RECV(wk_output,kount,MPI_DOUBLE_PRECISION,isour,itag,MPI_COMM_WORLD,istatus,ierr) jj=0 do j=(loop)*nblk,(loop+1)*nblk-1 jj=jj+1 ; force(:,:,j)=wk_output(:,:,jj) enddo kount=nblk ; isour=loop+1 ; itag=4 call MPI_RECV(wk_output2,kount,MPI_DOUBLE_PRECISION,isour,itag,MPI_COMM_WORLD,istatus,ierr) jj=0 do j=(loop)*nblk,(loop+1)*nblk-1
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (11 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
jj=jj+1 ; vofqj(j)=wk_output2(jj) enddo enddo! else ! -----| PROCESS ID = 0! myid=0에서 보내온 정보 받아들이기 kount=3*natom*nblk ; isour=0 ; itag=1 call MPI_RECV(wk_input,kount,MPI_DOUBLE_PRECISION,isour,itag,MPI_COMM_WORLD,istatus,ierr)! 할당된 일하기, myid /=0에서 do jj=1,nblk call sma_energy_force(wk_input(1,1,jj),wk_output(1,1,jj),wk_output2(jj)) enddo! 정보를 myid=0로 보내기, myid/=0에서 보내기임 kount=3*natom*nblk ; idest=0 ; itag=3 call MPI_SEND(wk_output,kount,MPI_DOUBLE_PRECISION,idest,itag,MPI_COMM_WORLD,ierr) kount=nblk ; idest=0 ; itag=4 call MPI_SEND(wk_output2,kount,MPI_DOUBLE_PRECISION,idest,itag,MPI_COMM_WORLD,ierr)! endif ! -----] PROCESS ID = 0 .................................................................... elapsed (or wall) clock : DOUBLE PRECISION MPI_WTIME()t1=mpi_wtime().....null input 형식으로 입력이 없다.노드에 따라 달리 시작한다.------------code to be timed ---------- t2=mpi_wtime() if(myid == 0) write(6,*) t2-t1,' sec' if(myid == 0)then ! -----[ PROCESS ID = 0 각종 출력들...... endif ! -----] PROCESS ID = 0 .......................... if(myid == 0)then ! -----[ PROCESS ID = 0 CALL SYSTEM_CLOCK(itemq) write(6,'(2e15.4,2x,a9)') float(itemq-itemp)/float(irate)/60.,float(itemq-itemp)/float(irate)/3600.,' min or h' endif ! -----] PROCESS ID = 0 call MPI_Finalize(ierr)stopend program testMPI_Wtime()함수를 사용할 수 있다. double precision변수이다. wall-clock-time 측정에 사용된다. microsecond 수준의 분해능을 가지고 C/C++/Fortran에서 사용가능하다. 물론, 컴퓨터 기종에 상관없
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (12 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
이 사용가능하다.s1=MPI_WTIME()......s2=MPI_WTIME()s2-s1 sec단위로 출력됨 !234567890 subroutine equal_load(n1,n2,nproc,myid,istart,ifinish) implicit none integer nproc,myid,istart,ifinish,n1,n2 integer iw1,iw2 iw1=(n2-n1+1)/nproc ; iw2=mod(n2-n1+1,nproc) istart=myid*iw1+n1+min(myid,iw2) ifinish=istart+iw1-1 ; if(iw2 > myid) ifinish=ifinish+1 ! print*, n1,n2,myid,nproc,istart,ifinish return end !234567890 program equal_load_sum implicit none include 'mpif.h' integer nn real*8, allocatable :: aa(:) integer nproc,myid,ierr,istart,ifinish integer i real*8 xsum,xxsum nn=10000 call MPI_INIT(ierr) call MPI_COMM_SIZE(MPI_COMM_WORLD, nproc, ierr) call MPI_COMM_RANK(MPI_COMM_WORLD, myid, ierr) call equal_load(1,nn,nproc,myid,istart,ifinish) allocate(aa(istart:ifinish)) ! 단순한 인덱스의 분할 뿐만아니라 메모리의 분할이 이루어지고 있다. 노드별로 do i=istart,ifinish aa(i)=float(i) enddo xsum=0.0d0 do i=istart,ifinish xsum=xsum+aa(i) enddo call MPI_REDUCE(xsum,xxsum,1,MPI_DOUBLE_PRECISION,MPI_SUM,0,
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (13 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
MPI_COMM_WORLD,ierr) xsum=xxsum if(myid == 0)then write(6,*) xsum,' xsum' endif deallocate(aa) call MPI_FINALIZE(ierr) end program equal_load_sum위와 같은 경우: 블록 (block) 분할이라고 한다.순환 (cyclic) 분할은 위와 대비되는 것이다.do i=n1,n2-------enddo위의 루프가 아래처럼 바뀐다.do i=n1+myid,n2,nproc-------enddo block-cyclic 분할: nblk의 사이즈만큼씩 처리한다. do j=n1+myid*nblk,n2,nproc*nblk do i=j,min(j+nblk-1,n2) .......... enddoenddo 병렬계산의 이면: 실제 컴퓨터계산에서는 유한한 정밀도를 사용한다. 따라서, aa(1)+aa(2)+aa(3)+,,,,aa(100)과 같이 순차 프로그램에의해서, 순차적으로 계산된 양은 병렬 프로그램에의해서, 즉, [aa(1)+aa(2)+aa(3)]+[aa(4)+aa(5)+aa(6)]+[aa(7)+aa(8)+aa(9)]+....처럼,부분적으로 합해진 합들의 합이며, 순서 또한 순차프로램의 것과 차이가 있다. 이 결과들로 인해서 정밀도 근처의 오차가 있을 수 있다. 결국 MPI_REDUCE를 사용할 경우 rounding error가 있다는 것이다. 통상 실전에서 이것이 문제가 되는 경우는 그리 많지 않다. 블록킹과 논-블로킹 통신: MPI_Send, MPI_Recve들은 통신이 완료될 때까지 호출한 프로세스들을 블로킹해둔다. 블로킹 통신의 경우 교착이 발생할 수 있다. 교착은 일종의 프로그래밍 에러이다. 송, 수신 연산의 초기화와 종료를 분리한 형식의 호출을 통한 통신이 논-블로킹 통신이다. 두 호출사이에 프로그램이 다른 일들을 할 수 있다는 장점이 있다. 논-블로킹 통신을 호출해서 초기화하는 것을 포스팅(posting)이라고 한다. 실제 MPI프로그램에서는 각각의 CPU를 이용한 일 처리 전후에 일어나는 데이터의 교환이 핵심이다. 따라서 프로그래머가 원하는 대로 데이터가 적절한 시기에 적절한 CPU로 전파되는지를 프린트를 통해서 확인할 필요가 있다. 원하는 시기에 원하는 노드로의 정확한 데이터 송신 및 수신 이것을 반드시 테스트해야 한다. 이것이야 말로 모든 MPI 구현의 핵심이기 때문이다. 하나의 프로그램에서 각기 다른 CPU상에서 일어나는 일들을 다같이 점검해야 한다. 물론, 허용한다면, 너무 자주 컴퓨터 간 통신을 하지 않을수록 병렬 효율성은 좋다.
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (14 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
많이 쓰는 것 한 가지 더 추가하면 mpi_bcast를 이야기할 수 있겠다. 이것은 broadcast를 의미한다. 특별히, 모든 노드에게 알릴 때 사용한다. 알리고자 하는 정보의 근원지를 지정한다.물론, 정보의 크기와 형태를 지정해줘야 한다.
iroot=0 ; kount=1 call MPI_BCAST(l_pbc,kount,MPI_LOGICAL,iroot,MPI_COMM_WORLD,ierr)
정보가 공유되도록 방송하는 관계로 모든 노드에서 동시에 위의 함수가 불려져야 한다. 특정한 노드에서만 변수값이 새로 읽어지거나 계산되었을 때, 그리고 이것이 모든 노드들에게 알려질 필요가 있을 때 사용한
다.
point-to-point 통신이 아닌 집단적인 통신이다. collective communication master/slave 형식: master 노드에서 중요한 일들을 수행하고 그 이외의 노드들은 master의 지휘하에 계산들을 수행하는 형식. 알고리즘 구현에서 유리한 경우가 있다. 물론, 그렇지 못한 경우도 많다. master노드가 거의 하는 일이 없어지면 병렬 효율성이 떨어지기 마련이다. 물론, master도 일정한 slave노드처럼 일정한 계산들을 수행함으로써 전체 병렬 효율성을 높일 수 있다. master/slave형식의 경우 대개 메모리의 한계는 문제가 되질 않은 경우이다. 하지만, 거대 계산의 경우 메모리 할당 문제 때문에 변수들을 여러 노드들에 걸쳐서 표현할 수밖에 없는 경우도 매우 많이 있다. 이러한 경우는 master/slave노드 개념의 프로그래밍은 좋은 아이디어가 아니다. Introduction to MPI http://www.llnl.gov/computing/tutorials/workshops/workshop/mpi/MAIN.html http://www.netlib.org/utk/papers/intro-mpi/intro-mpi.html http://www.nas.nasa.gov/Groups/SciCon/Tutorials/MPIintro/toc.html http://www-unix.mcs.anl.gov/mpi/tutorial/mpiintro/ http://www-unix.mcs.anl.gov/mpi/tutorial/mpibasics/ http://www.ats.ucla.edu/at/hpc/parallel_computing/mpi-intro.htm http://www.gre.ac.uk/~selhpc/announcements/mpi-24-06-96.html Introduction to MPI 2 http://acrl.cs.unb.ca/php/training/mpi/cmacphee-intro_to_mpi2/index.html Cornell theory center의 Code Examples http://www.tc.cornell.edu/Services/Docs/Examples/
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (15 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
간단한 예제들--===============
프로그램 분석을 위해서 프로그램을 프린터할 때 a2ps -o output.ps <input.f90 처럼 a2ps프로그램을 이용하면 보기 좋은(포트란 키워드는 진하게 나타난다.) PS파일이 생긴다. http://www.gnu.org/software/a2ps/
예제 (1) http://www.dartmouth.edu/~rc/classes/ PROGRAM hello IMPLICIT NONE INCLUDE "mpif.h" CHARACTER(LEN=12) :: inmsg,message INTEGER i,ierr,me,nproc,itag INTEGER istatus(MPI_STATUS_SIZE) call MPI_Init(ierr) call MPI_Comm_size(MPI_COMM_WORLD,nproc,ierr) call MPI_Comm_rank(MPI_COMM_WORLD,me,ierr) if(me == 0 .and. nproc == 1) write(6,*) nproc, 'is alive' if(me == 0 .and. nproc >1 ) write(6,*) nproc,'are alive' itag = 100 if (me == 0) then message = "Hello, world" do i = 1,nproc-1 call MPI_Send(message,12,MPI_CHARACTER,i,itag,MPI_COMM_WORLD,ierr) end do write(6,*) "process", me, ":", message else call MPI_Recv(inmsg,12,MPI_CHARACTER,0,itag,MPI_COMM_WORLD,istatus,ierr) write(6,*) "process", me, ":", inmsg end if call MPI_Finalize(ierr)END PROGRAM hello이 프로그램을 사용하여 MPICH 또는 LAM이 시스템에 제대로 인스톨되어 있는지를 확인할 수 있다. 물론, 이러한 작업들을 통해서, 병렬 컴파일러의 설치와 이용 (MPICH와 LAM 사이에 차이점이 존재한다. 두 가지 모두 설치하는 경우가 많다.)이 자동으로 테스트될 것이다. PBS를 사용하여 job를 submit하는 것을 테스트 해볼 수 있다. 거의 장난하는 수준의 프로그램이지만, 이것이 제대로 동작하면 그 시스템은 병렬계산을 위한 준비가 되었다는 큰 의미를 가지게 해주는 프로그램이다. 예를 들면 아래와 같이 설치된 파일들을 이용할 수 있다./usr/local/lam/bin/mpif90/usr/local/lam/lib /usr/local/mpich/bin/mpichf90/usr/local/mpich/lib DQS
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (16 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
#!/bin/bash#$ -cwd#$ -l qty.eq.16#$ -N Test#$ -A ihlee/usr/local/mpich/bin/mpirun -np $NUM_HOSTS -machinefile $HOSTS_FILE ../image_parallel_sma.x <input_fileecho "End of Job" #!/bin/bash# This is an example DQS script for running a parallel MPI job# on MAIDROC cluster using gigabit ethernet interface## start in the directory where the job was# submitted#$ -cwd## specify number of processors and which set of nodes to use# we can request up to 48 processors because we use gigabit ethernet# Specify which set of nodes to use: fastnet_1 - for nodes 1 - 24# fastnet_2 - for nodes 25 - 48# here we request 48 processors on nodes 1-24#$ -l qty.eq.12,fastnet_2## name of the job#$ -N Tlam## User specified environment variables are set with -v#$ -v NCPUS=12# commands to be executed# type your commands below #use mdo_mpi_fast command to run mpi job on gigabit ethernet interfacemdo_mpi_fast ../admd_lam_sma.x < admd.i#NOTE: mdo_mpi_fast starts and runs your mpi program automagically.#Don't try to call mpirun yourself!#mpirun -np 2 ./a.out PBS #!/bin/sh ### Job name #PBS -N AM_A_1### Declare job non-rerunable #PBS -r n ### Output files #PBS -j oe ### Mail to user #PBS -m ae ### Queue name (n2, n4, n8, n16, n32)
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (17 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
#PBS -q n4 # This job's working directory echo Working directory is $PBS_O_WORKDIR cd $PBS_O_WORKDIR echo Running on host `hostname` echo Time is `date` echo Directory is `pwd` echo This jobs runs on the following processors: echo `cat $PBS_NODEFILE` # Define number of processors NPROCS=`wc -l < $PBS_NODEFILE` echo This job has allocated $NPROCS nodes # your job # Run the parallel MPI executable "a.out"# mpirun -v -machinefile $PBS_NODEFILE -np $NPROCS a.outmpirun -machinefile $PBS_NODEFILE -np $NPROCS -nolocal action_lbfgs.x > out1 #!/bin/bash#$ -cwd#$ -l qty.eq.12#$ -N Tmpich#$ -A jun/usr/local/mpich/bin/mpirun -np $NUM_HOSTS -machinefile $HOSTS_FILE ../admd_mpich_sma.x < admd.iecho "End of Job" qstatgonza w-1400-2v maido04 173177 0:3 r RUNNING 02/10/05 14:24: 1 gonza w-0808-1v maido11 173174 0:1 r RUNNING 02/10/05 14:05:56 sha mcmp.sh maido13 11397 0:1 r RUNNING 12/05/04 03:18:48 gonza w-0808-2v maido13 173178 0:4 r RUNNING 02/10/05 14:26:56 colax bench-zab maido17 173170 0:1 r RUNNING 02/10/05 05:48:55 gonza w-1400-1v maido24 173176 0:2 r RUNNING 02/10/05 14:08: 3 ihlee Tlam maido25 173179 0:1 r RUNNING 02/10/05 15:17:37 ihlee Tlam maido26 173179 0:1 r RUNNING 02/10/05 15:17:37 ihlee Tlam maido30 173179 0:1 r RUNNING 02/10/05 15:17:37 ihlee Tlam maido32 173179 0:1 r RUNNING 02/10/05 15:17:37 ihlee Tlam maido33 173179 0:1 r RUNNING 02/10/05 15:17:37 ihlee Tlam maido34 173179 0:1 r RUNNING 02/10/05 15:17:37 ihlee Tlam maido36 173179 0:1 r RUNNING 02/10/05 15:17:37 ihlee Tlam maido38 173179 0:1 r RUNNING 02/10/05 15:17:37 ihlee Tlam maido40 173179 0:1 r RUNNING 02/10/05 15:17:37 ihlee Tlam maido42 173179 0:1 r RUNNING 02/10/05 15:17:37 ihlee Tlam maido43 173179 0:1 r RUNNING 02/10/05 15:17:37 ihlee Tlam maido45 173179 0:1 r RUNNING 02/10/05 15:17:37 예제 (2) http://www.dartmouth.edu/~rc/classes/ ! Program hello.ex1.f
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (18 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
! Parallel version using MPI calls! Modified from basic version so that workers send back! a message to the master, who prints out a message for each workerprogram hello implicit none integer, parameter:: DOUBLE=kind(1.0d0), SINGLE=kind(1.0) include "mpif.h" character(LEN=12) :: inmsg,message integer :: i,ierr,me,nproc,itag,iwrank integer, dimension(MPI_STATUS_SIZE) :: istatus ! call MPI_Init(ierr) call MPI_Comm_size(MPI_COMM_WORLD,nproc,ierr) call MPI_Comm_rank(MPI_COMM_WORLD,me,ierr) tag = 100 ! if (me == 0) then message = "Hello, world" do i = 1,nproc-1 call MPI_Send(message,12,MPI_CHARACTER,i,itag,MPI_COMM_WORLD,ierr) end do write(*,*) "process", me, ":", message do i = 1,nproc-1 call MPI_Recv(iwrank,1,MPI_INTEGER,MPI_ANY_SOURCE,itag,MPI_COMM_WORLD, istatus, ierr) write(*,*) "process", iwrank, ":Hello, back" end do else call MPI_Recv(inmsg,12,MPI_CHARACTER,0,itag,MPI_COMM_WORLD,istatus,ierr) call MPI_Send(me,1,MPI_INTEGER,0,tag,MPI_COMM_WORLD,ierr) end if call MPI_Finalize(ierr)end program hello
http://www.dartmouth.edu/~rc/classes/
! Program hello.ex2.f!! Parallel version using MPI calls.! Modified from basic version so that workers send back a message to the ! master, who prints out a message for each worker. In addition, the ! master now sends out two messages to each worker, with two different ! tags, and the worker receives the messages in reverse order.!! Note that this solution works only because the messages are small,! and can fit into buffers. A later talk will provide details on! how buffers are used in MPI_SEND and MPI_RECEIVE,!program hello implicit none integer, parameter:: DOUBLE=kind(1.0d0), SINGLE=kind(1.0) include "mpif.h"
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (19 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
character(LEN=12) :: inmsg,message integer :: i,mpierr,me,nproc,itag,itag2,iwrank integer isatus(MPI_STATUS_SIZE) ! call MPI_Init(ierr) call MPI_Comm_size(MPI_COMM_WORLD,nproc,ierr) call MPI_Comm_rank(MPI_COMM_WORLD,me,ierr) itag = 100 itag2 = 200 ! if (me == 0) then message = "Hello, world" do i = 1,nproc-1 call MPI_Send(message,12,MPI_CHARACTER,i,itag,MPI_COMM_WORLD,ierr) call MPI_Send(message,12,MPI_CHARACTER,i,itag2,MPI_COMM_WORLD,ierr) end do write(*,*) "process",me, ":", message do i = 1,nproc-1 call MPI_Recv(iwrank,1,MPI_INTEGER,MPI_ANY_SOURCE,itag,MPI_COMM_WORLD, istatus, ierr) write(*,*) "process", iwrank, ":Hello, back" end do else call MPI_Recv(inmsg,12,MPI_CHARACTER,0,itag2,MPI_COMM_WORLD,istatus,ierr) call MPI_Recv(inmsg,12,MPI_CHARACTER,0,itag,MPI_COMM_WORLD,istatus,ierr) call MPI_Send(me,1,MPI_INTEGER,0,itag,MPI_COMM_WORLD,ierr) end if call MPI_Finalize(ierr)end program hello! 예제 (3) http://www.dartmouth.edu/~rc/classes/ program karp ! ! This simple program approximates pi by computing pi = integral ! from 0 to 1 of 4/(1+x*x)dx which is approximated by sum from ! k=1 to N of 4 / (1+((k-.5)/N)**2). The only input data required is N. ! ! NOTE: Comments that begin with "cspmd" are hints for part b of the ! lab exercise, where you convert this into an MPI program. ! !spmd Each process could be given a chunk of the interval to do. ! ! RLF 3/21/97 Change floats to real*8 ! SHM 8/29/97 Change input to read from a file to accommodate ! VW Companion ! SHM 8/29/97 Replaced goto with do while ! Nils Smeds Aug 14, 2000 Converted to F90 ! implicit none integer, parameter:: DOUBLE=kind(1.0d0), SINGLE=kind(1.0) integer n,i real(DOUBLE) :: err,pi,sum,w,x intrinsic atan pi = 4.0 * atan(1.0) open (unit = 20,file = "values") !
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (20 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
!spmd call startup routine that returns the number of tasks and the !spmd taskid of the current instance. ! ! Now read in a new value for N. When it is 0, then you should depart. ! read(20,*) n print *, "Number of approximation intervals = ", n ! do while (n > 0) w = 1.0 / n sum = 0.0 do i = 1,n sum = sum + f((i-0.5)*w) end do sum = sum * w err = sum - pi print *, "sum = ", sum, " err =", err ! read (20,*) n print *, "Number of approximation intervals = ", n ! end do ! close (unit = 20) contains real(DOUBLE) function f(x) implicit none real(DOUBLE), intent(in) :: x f = 4.0 / (1.0+x*x) end function fend program karp 예제 (4) http://www.dartmouth.edu/~rc/classes/ program karp ! karp.soln.f ! This simple program approximates pi by computing pi = integral ! from 0 to 1 of 4/(1+x*x)dx which is approximated by sum from ! k=1 to N of 4 / (1+((k-.5)/N)**2). The only input data required is N. ! ! 10/11/95 RLF MPI Parallel version 1 ! 3/7/97 RLF Replace nprocs and mynum with size and rank ! 3/21/97 RLF Change floats to real*8 ! SHM 8/29/97 Change input to read from a file to accommodate ! VW Companion ! SHM 8/29/97 Replaced goto with do while ! Nils Smeds Aug 14, 2000 Converted to F90 ! ! Uses only the 6 basic MPI calls ! implicit none integer, parameter:: DOUBLE=kind(1.0d0), SINGLE=kind(1.0) include "mpif.h" integer :: n,i,mpierr,rank,size,tag real(DOUBLE) :: err,pi,sum,w,x
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (21 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
integer, dimension(MPI_STATUS_SIZE) :: status intrinsic atan pi = 4.0 * atan(1.0) tag = 111 open (unit = 20,file = "values") ! ! All processes call the startup routine to get their rank call MPI_Init(mpierr) call MPI_Comm_size(MPI_COMM_WORLD,size,mpierr) call MPI_Comm_rank(MPI_COMM_WORLD,rank,mpierr) ! ! ------- Each new approximation to pi begins here. ------------------- ! (Step 1) Get first value of N call solicit(n,size,rank) ! ! (Step 2): do the computation in N steps ! Parallel Version: there are "size" processes participating. Each ! process should do 1/size of the calculation. Since we want ! i = 1..n but rank = 0, 1, 2..., we start off with rank+1. do while (n > 0) w = 1.0 / n sum = 0.0 do i = rank+1,n,size sum = sum + f((i-0.5)*w) end do sum = sum * w ! ! (Step 3): print the results ! (Parallel version: collect partial results and let master process print it) if (rank == 0) then print *, "host calculated x=", sum do i = 1,size-1 call MPI_Recv(x,1,MPI_DOUBLE_PRECISION,i,tag,MPI_COMM_WORLD,status, mpierr) print *, "host got x=", x sum = sum + x end do err = sum - pi print *, "sum, err =", sum, err else call MPI_Send(sum,1,MPI_DOUBLE_PRECISION,0,tag,MPI_COMM_WORLD,mpierr) end if ! Get a new value of N call solicit(n,size,rank) end do ! call MPI_Finalize(mpierr) close (unit = 20) !contains ! real(DOUBLE) function f(x) implicit none real(DOUBLE), intent(in) :: x f = 4.0 / (1.0+x*x) end function f subroutine solicit(n,nprocs,mynum) ! Get a value for N, the number of intervals in the approximation
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (22 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
! (Parallel versions: master process reads in N and then ! sends N to all the other processes) ! Note: A single broadcast operation could be used instead, but ! is not one of the 6 basics calls. implicit none ! Get a value for N, the number of intervals in the approximation ! (Parallel versions: master process reads in N and then ! sends N to all the other processes) ! Note: A single broadcast operation could be used instead, but ! is not one of the 6 basics calls. include "mpif.h" integer, intent(inout) :: n integer, intent(in) :: mynum,nprocs integer :: i,mpierr,tag integer, dimension(MPI_STATUS_SIZE) :: status tag = 112 if (mynum == 0) then read (20,*) n print *, "Number of approximation intervals = ", n do i = 1,nprocs-1 call MPI_Send(n,1,MPI_INTEGER,i,tag,MPI_COMM_WORLD,mpierr) end do else call MPI_Recv(n,1,MPI_INTEGER,0,tag,MPI_COMM_WORLD,status,mpierr) end if end subroutine solicitend program karp 예제 (5) http://www.dartmouth.edu/~rc/classes/ !#########################################!#!# This is an MPI example that solves Laplace's equation by using Jacobi!# iteration on a 1-D decomposition. Non-blocking communications routines!# are used.!# It demonstrates the use of :!#!# * MPI_Init!# * MPI_Comm_rank!# * MPI_Comm_size!# * MPI_Cart_create!# * MPI_Cart_shift!# * MPI_Cart_shift!# * MPI_Bcast!# * MPI_Allreduce!# * MPI_Isend!# * MPI_Irecv!# * MPI_Finalize!#!#################################################### program onedovlp include "mpif.h" integer maxn parameter (maxn = 128) double precision a(maxn,maxn), b(maxn,maxn), f(maxn,maxn) double precision diff, diffnorm, diffw
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (23 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
integer nx, ny, myid, numprocs, comm1d integer nbrbottom, nbrtop, s, e, it, ierr call MPI_INIT( ierr ) call MPI_COMM_RANK( MPI_COMM_WORLD, myid, ierr ) call MPI_COMM_SIZE( MPI_COMM_WORLD, numprocs, ierr ) print *, "Process ", myid, " of ", numprocs, " is alive" if (myid .eq. 0) then!! Get the size of the problem! print *, 'Enter nx' read *, nx! nx = 110 endif call MPI_BCAST(nx,1,MPI_INTEGER,0,MPI_COMM_WORLD,ierr) ny = nx!!! Get a new communicator for a decomposition of the domain! call MPI_CART_CREATE( MPI_COMM_WORLD, 1, numprocs, .false., .true., comm1d, ierr )!! Get my position in this communicator, and my neighbors! call MPI_COMM_RANK( comm1d, myid, ierr ) call MPI_Cart_shift( comm1d, 0, 1, nbrbottom, nbrtop, ierr )!! Compute the decomposition! call MPE_DECOMP1D( ny, numprocs, myid, s, e )!! Initialize the right-hand-side (f) and the initial solution guess (a)! call onedinit( a, b, f, nx, s, e )!! Actually do the computation. Note the use of a collective operation to! check for convergence, and a do-loop to bound the number of iterations.! do 10 it=1, 200! call nbexchng1( a, nx, s, e, comm1d, nbrbottom, nbrtop, 0 ) call nbsweep( a, f, nx, s, e, b ) call nbexchng1( a, nx, s, e, comm1d, nbrbottom, nbrtop, 1 ) call nbsweepend( a, f, nx, s, e, b )! call nbexchng1( b, nx, s, e, comm1d, nbrbottom, nbrtop, 0 ) call nbsweep( b, f, nx, s, e, a ) call nbexchng1( b, nx, s, e, comm1d, nbrbottom, nbrtop, 1 ) call nbsweepend( b, f, nx, s, e, a )! diffw = diff( a, b, nx, s, e ) call MPI_Allreduce( diffw, diffnorm, 1, MPI_DOUBLE_PRECISION, MPI_SUM, comm1d, ierr ) if (diffnorm .lt. 1.0e-5) goto 2010 continue if (myid .eq. 0) print *, 'Failed to converge'20 continue
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (24 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
if (myid .eq. 0) then print *, 'Converged after ', it, ' Iterations' do i = 1,nx do j = 1,nx print *,"i,j,b=",i,j,b(i,j) end do end do endif !! call MPI_FINALIZE(ierr) end
예제 (6) http://www.dartmouth.edu/~rc/classes/ !**********************************************************************! matmul.f - matrix - vector multiply, simple self-scheduling version!************************************************************************ Program Matmult!########################################!#!# This is an MPI example of multiplying a vector times a matrix!# It demonstrates the use of :!#!# * MPI_Init!# * MPI_Comm_rank!# * MPI_Comm_size!# * MPI_Bcast!# * MPI_Recv!# * MPI_Send!# * MPI_Finalize!# * MPI_Abort!#!##################################### program main include 'mpif.h' integer MAX_ROWS, MAX_COLS, rows, cols parameter (MAX_ROWS = 1000, MAX_COLS = 1000, MAX_PROCS =32) double precision a(MAX_ROWS,MAX_COLS), b(MAX_COLS), c(MAX_COLS) double precision buffer(MAX_COLS), ans integer procs(MAX_COLS), proc_totals(MAX_PROCS) integer myid, master, numprocs, ierr, status(MPI_STATUS_SIZE) integer i, j, numsent, numrcvd, sender, job(MAX_ROWS) integer rowtype, anstype, donetype call MPI_INIT( ierr ) call MPI_COMM_RANK( MPI_COMM_WORLD, myid, ierr ) call MPI_COMM_SIZE( MPI_COMM_WORLD, numprocs, ierr ) if (numprocs .lt. 2) then print *, "Must have at least 2 processes!"
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (25 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
call MPI_ABORT( MPI_COMM_WORLD, 1 ) stop else if (numprocs .gt. MAX_PROCS) then print *, "Must have 32 processes or less." call MPI_ABORT( MPI_COMM_WORLD, 1 ) stop endif print *, "Process ", myid, " of ", numprocs, " is alive" rowtype = 1 anstype = 2 donetype = 3 master = 0 rows = 100 cols = 100 if ( myid .eq. master ) then! master initializes and then dispatches! initialize a and b do 20 i = 1,cols b(i) = 1 do 10 j = 1,rows a(i,j) = I 10 continue 20 continue numsent = 0 numrcvd = 0 ! send b to each other process call MPI_BCAST(b, cols, MPI_DOUBLE_PRECISION, master, MPI_COMM_WORLD, ierr)! send a row to each other process do 40 i = 1,numprocs-1 do 30 j = 1,cols buffer(j) = a(i,j) 30 continue call MPI_SEND(buffer, cols, MPI_DOUBLE_PRECISION, i, rowtype, MPI_COMM_WORLD, ierr) job(i) = I numsent = numsent+1 40 continue do 70 i = 1,rows call MPI_RECV(ans, 1, MPI_DOUBLE_PRECISION, MPI_ANY_SOURCE, anstype, MPI_COMM_WORLD, status, ierr) sender = status(MPI_SOURCE) c(job(sender)) = ans procs(job(sender))= sender proc_totals(sender+1) = proc_totals(sender+1) +1 if (numsent .lt. rows) then do 50 j = 1,cols buffer(j) = a(numsent+1,j) 50 continue call MPI_SEND(buffer, cols, MPI_DOUBLE_PRECISION, sender, rowtype, MPI_COMM_WORLD, ierr) job(sender) = numsent+1 numsent = numsent+1 else call MPI_SEND(1, 1, MPI_INTEGER, sender, donetype, MPI_COMM_WORLD, ierr) endif 70 continue
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (26 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
! print out the answer do 80 i = 1,cols print *,"c(", i,") = ", c(i)," computed by proc #",procs(i) 80 continue do 81 i=1,numprocs write(6,810) i-1,proc_totals(i) 810 format('Total answers computed by processor #',i2,' were ',i3) 81 continue else! slaves receive b, then compute dot products until done message call MPI_BCAST(b, cols, MPI_DOUBLE_PRECISION, master, MPI_COMM_WORLD, ierr) 90 call MPI_RECV(buffer, cols, MPI_DOUBLE_PRECISION, master, MPI_ANY_TAG, MPI_COMM_WORLD, status, ierr) if (status(MPI_TAG) .eq. donetype) then go to 200 else ans = 0.0 do 100 i = 1,cols ans = ans+buffer(i)*b(i) 100 continue call MPI_SEND(ans, 1, MPI_DOUBLE_PRECISION, master, anstype, MPI_COMM_WORLD, ierr) go to 90 endif endif 200 call MPI_FINALIZE(ierr) stop end 예제 (7)http://www.dartmouth.edu/~rc/classes/ Program Example1 implicit none integer n, p, i, j,num real h, result, a, b, integral, pi real my_a,my_range pi = acos(-1.0) !! = 3.14159... a = 0.0 !! lower limit of integration b = pi*1./2. !! upper limit of integration p = 4 !! number of processes (partitions) n = 100000 !! total number of increments h = (b-a)/n !! length of increment num= n/p !! number of calculations done by each process result = 0.0 !! stores answer to the integral do i=0,p-1 !! sum of integrals over all processes my_range = (b-a)/p my_a = a + i*my_range result = result + integral(my_a,num,h) enddo print *,'The result =',result
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (27 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
stop end real function integral(a,n,h) implicit none integer n, i, j real h, h2, aij, a real fct, x fct(x) = cos(x) !! kernel of the integral integral = 0.0 !! initialize integral h2 = h/2. do j=0,n-1 !! sum over all "j" integrals aij = a+j*h !! lower limit of "j" integral integral = integral + fct(aij+h2)*h enddo return end 예제 (7)http://www.dartmouth.edu/~rc/classes/ Program Example2!################################################!#!# This is an MPI example on parallel integration!# It demonstrates the use of :!#!# * MPI_Init!# * MPI_Comm_rank!# * MPI_Comm_size!# * MPI_Recv!# * MPI_Send!# * MPI_Finalize!# * MPI_WTime!#!############################################## implicit none integer n, p, i, j, ierr, master,num real h, result, a, b, integral, pi double precision MPI_WTime,start_time,end_time include "mpif.h" !! This brings in pre-defined MPI constants, ... integer Iam, source, dest, tag, status(MPI_STATUS_SIZE) real my_result real my_a, my_range data master/0/ !! 0 is defined as the master processor !! which will be responsible for collecting !! integral sums ...!! Placement of executable statements before MPI_Init is not!! advisable as the side effect is implementation-dependent pi = acos(-1.0) !! = 3.14159... a = 0.0 !! lower limit of integration b = pi*1./2. !! upper limit of integration n = 10000000 !! total number of increments across all processors
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (28 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
dest = master !! define the process that computes the final result tag = 123 !! set the tag to identify this particular job!**Starts MPI processes ... call MPI_Init(ierr) !! starts MPI call MPI_Comm_rank(MPI_COMM_WORLD, Iam, ierr) !! get current process id call MPI_Comm_size(MPI_COMM_WORLD, p, ierr) !! get # procs from env print*,'Process #',Iam, ' out of ',p,' total process' start_time = MPI_Wtime() !! variable or command line h = (b-a)/n !! length of increment num=n/p !! number of increments for each processor my_range = (b-a)/p my_a = a + Iam*my_range my_result = integral(my_a,num,h) !! compute local sum write(*,"('Process ',i2,' has the partial result of',f10.6)") Iam,my_result if(Iam .eq. master) then result = my_result !! initialize final result to master's do source=1,p-1 !! loop on sources (serialized) to collect local sum call MPI_Recv(my_result, 1, MPI_REAL, source, tag, MPI_COMM_WORLD, status, ierr) result = result + my_result enddo print *,'The result =',result end_time = MPI_Wtime() print *, 'elapsed time is ',end_time-start_time,' seconds' else call MPI_Send(my_result, 1, MPI_REAL, dest, tag, MPI_COMM_WORLD, ierr) !! send my_result to intended dest. endif call MPI_Finalize(ierr) !! let MPI finish up ... stop end real function integral(a,n,h) implicit none integer n, i, j real h, h2, aij, a real fct, x fct(x) = cos(x) !! kernel of the integral integral = 0.0 !! initialize integral h2 = h/2. do j=0,n-1 !! sum over all "j" integrals aij = a+j*h !! lower limit of "j" integral integral = integral + fct(aij+h2)*h enddo return end 예제 (8)http://www.dartmouth.edu/~rc/classes/ Program Example2!################################################!#!# This is an MPI example on parallel integration!# It demonstrates the use of :
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (29 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
!#!# * MPI_Init!# * MPI_Comm_rank!# * MPI_Comm_size!# * MPI_Recv!# * MPI_Send!# * MPI_Finalize!# * MPI_WTime!################################################ implicit none integer n, p, i, j, ierr, master,num real h, result, a, b, integral, pi double precision MPI_WTime,start_time,end_time include "mpif.h" !! This brings in pre-defined MPI constants, ... integer Iam, source, dest, tag, status(MPI_STATUS_SIZE) real my_result real my_a, my_range data master/0/ !! 0 is defined as the master processor !! which will be responsible for collecting !! integral sums ...!! Placement of executable statements before MPI_Init is not!! advisable as the side effect is implementation-dependent pi = acos(-1.0) !! = 3.14159... a = 0.0 !! lower limit of integration b = pi*1./2. !! upper limit of integration n = 10000000 !! total number of increments across all processors dest = master !! define the process that computes the final result tag = 123 !! set the tag to identify this particular job!**Starts MPI processes ... call MPI_Init(ierr) !! starts MPI call MPI_Comm_rank(MPI_COMM_WORLD, Iam, ierr) !! get current process id call MPI_Comm_size(MPI_COMM_WORLD, p, ierr) !! get # procs from env print*,'Process #',Iam, ' out of ',p,' total process' start_time = MPI_Wtime() !! variable or command line h = (b-a)/n !! length of increment num=n/p !! number of increments for each processor my_range = (b-a)/p my_a = a + Iam*my_range my_result = integral(my_a,num,h) !! compute local sum write(*,"('Process ',i2,' has the partial result of',f10.6)") Iam,my_result if(Iam .eq. master) then result = my_result !! initialize final result to master's do source=1,p-1 !! loop on sources (serialized) to collect local sum call MPI_Recv(my_result, 1, MPI_REAL, source, tag, MPI_COMM_WORLD, status, ierr) result = result + my_result enddo print *,'The result =',result end_time = MPI_Wtime() print *, 'elapsed time is ',end_time-start_time,' seconds' else call MPI_Send(my_result, 1, MPI_REAL, dest, tag, MPI_COMM_WORLD, ierr) !! send my_result to intended dest. endif call MPI_Finalize(ierr) !! let MPI finish up ... stop
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (30 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
end real function integral(a,n,h) implicit none integer n, i, j real h, h2, aij, a real fct, x fct(x) = cos(x) !! kernel of the integral integral = 0.0 !! initialize integral h2 = h/2. do j=0,n-1 !! sum over all "j" integrals aij = a+j*h !! lower limit of "j" integral integral = integral + fct(aij+h2)*h enddo return end 예제 (9) http://www.dartmouth.edu/~rc/classes/ Program Example5!################################!#!# This is an MPI example on parallel integration!# It demonstrates the use of :!#!# * MPI_Init!# * MPI_Comm_rank!# * MPI_Comm_size!# * MPI_Bcast!# * MPI_Reduce!# * MPI_SUM!# * MPI_Finalize!# * MPI_WTime!#!################################################ implicit none integer n, p, i, j, ierr, master,num real h, result, a, b, integral, pi real my_a, my_range double precision MPI_WTime,start_time,end_time include "mpif.h" !! This brings in pre-defined MPI constants, ... integer Iam, source, dest, tag, status(MPI_STATUS_SIZE) real my_result data master/0/ !**Starts MPI processes ... call MPI_Init(ierr) !! starts MPI call MPI_Comm_rank(MPI_COMM_WORLD, Iam, ierr) !! get current process id call MPI_Comm_size(MPI_COMM_WORLD, p, ierr) !! get number of processes pi = acos(-1.0) !! = 3.14159... a = 0.0 !! lower limit of integration b = pi*1./2. !! upper limit of integration
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (31 of 73)2005-11-22 오후 4:09:45
물리학자를 위한 MPI
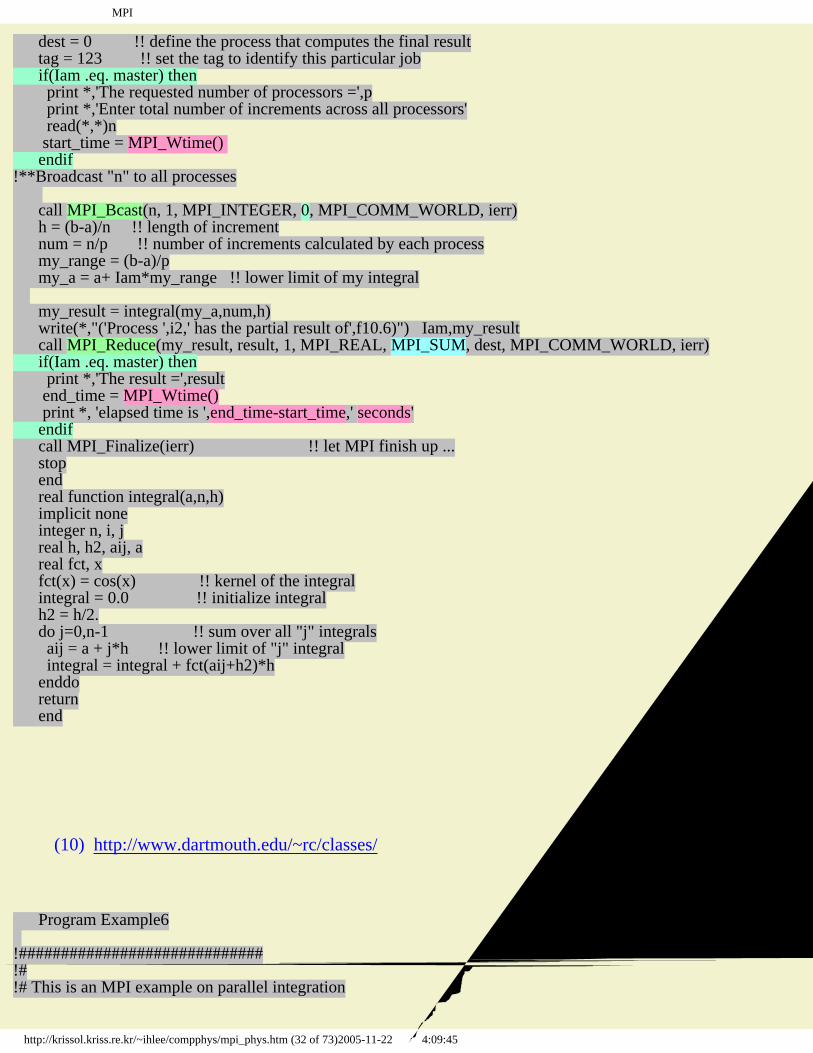
dest = 0 !! define the process that computes the final result tag = 123 !! set the tag to identify this particular job if(Iam .eq. master) then print *,'The requested number of processors =',p print *,'Enter total number of increments across all processors' read(*,*)n start_time = MPI_Wtime() endif!**Broadcast "n" to all processes call MPI_Bcast(n, 1, MPI_INTEGER, 0, MPI_COMM_WORLD, ierr) h = (b-a)/n !! length of increment num = n/p !! number of increments calculated by each process my_range = (b-a)/p my_a = a+ Iam*my_range !! lower limit of my integral my_result = integral(my_a,num,h) write(*,"('Process ',i2,' has the partial result of',f10.6)") Iam,my_result call MPI_Reduce(my_result, result, 1, MPI_REAL, MPI_SUM, dest, MPI_COMM_WORLD, ierr) if(Iam .eq. master) then print *,'The result =',result end_time = MPI_Wtime() print *, 'elapsed time is ',end_time-start_time,' seconds' endif call MPI_Finalize(ierr) !! let MPI finish up ... stop end real function integral(a,n,h) implicit none integer n, i, j real h, h2, aij, a real fct, x fct(x) = cos(x) !! kernel of the integral integral = 0.0 !! initialize integral h2 = h/2. do j=0,n-1 !! sum over all "j" integrals aij = a + j*h !! lower limit of "j" integral integral = integral + fct(aij+h2)*h enddo return end 예제 (10) http://www.dartmouth.edu/~rc/classes/ Program Example6 !#############################!#!# This is an MPI example on parallel integration
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (32 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
!# It demonstrates the use of :!#!# * MPI_Init!# * MPI_Comm_rank!# * MPI_Comm_size!# * MPI_Pack!# * MPI_Unpack!# * MPI_Reduce!# * MPI_SUM, MPI_MAXLOC, and MPI_MINLOC!# * MPI_Finalize!#!############################## implicit none integer n, p, i, j, ierr, m, master real h, result, a, b, integral, pi include "mpif.h" !! This brings in pre-defined MPI constants, ... integer Iam, source, dest, tag, status(MPI_STATUS_SIZE),num real my_result(2), min_result(2), max_result(2) real my_a, my_range double precision MPI_WTime,start_time,end_time integer Nbytes parameter (Nbytes=1000, master=0) character scratch(Nbytes) !! needed for MPI_pack/MPI_unpack; counted in bytes integer index, minid, maxid!**Starts MPI processes ... call MPI_Init(ierr) !! starts MPI call MPI_Comm_rank(MPI_COMM_WORLD, Iam, ierr) !! get current process id call MPI_Comm_size(MPI_COMM_WORLD, p, ierr) !! get number of processes pi = acos(-1.0) !! = 3.14159... dest = 0 !! define the process that computes the final result tag = 123 !! set the tag to identify this particular job if(Iam .eq. 0) then print *,'The requested number of processors =',p print *,'Enter the total # of intervals over all processes' read(*,*)n print *,'enter a & m' print *,' a = lower limit of integration' print *,' b = upper limit of integration' print *,' = m * pi/2' read(*,*)a,m start_time = MPI_Wtime() b = m * pi / 2.!**to be efficient, pack all things into a buffer for broadcast index = 1 call MPI_Pack(n, 1, MPI_INTEGER, scratch, Nbytes, index, MPI_COMM_WORLD, ierr) call MPI_Pack(a, 1, MPI_REAL, scratch, Nbytes, index, MPI_COMM_WORLD, ierr) call MPI_Pack(b, 1, MPI_REAL, scratch, Nbytes, index, MPI_COMM_WORLD, ierr) call MPI_Bcast(scratch, Nbytes, MPI_PACKED, 0, MPI_COMM_WORLD, ierr) else call MPI_Bcast(scratch, Nbytes, MPI_PACKED, 0, MPI_COMM_WORLD, ierr)!**things received have been packed, unpack into expected locations index = 1 call MPI_Unpack(scratch, Nbytes, index, n, 1, MPI_INTEGER, MPI_COMM_WORLD, ierr) call MPI_Unpack(scratch, Nbytes, index, a, 1, MPI_REAL, MPI_COMM_WORLD, ierr) call MPI_Unpack(scratch, Nbytes, index, b, 1, MPI_REAL, MPI_COMM_WORLD, ierr) endif
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (33 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
h = (b-a)/n !! length of increment num= n/p !! number of iterations on each processor my_range = (b-a)/p my_a = a + Iam*my_range my_result(1) = integral(my_a,num,h) my_result(2) = Iam write(*,"('Process ',i2,' has the partial result of',f10.6)") Iam,my_result(1) call MPI_Reduce(my_result, result, 1, MPI_REAL, MPI_SUM, dest, MPI_COMM_WORLD, ierr) !! data reduction by way of MPI_SUM call MPI_Reduce(my_result, min_result, 1, MPI_2REAL, MPI_MINLOC, dest, MPI_COMM_WORLD, ierr) !! data reduction by way of MPI_MINLOC call MPI_Reduce(my_result, max_result, 1, MPI_2REAL, MPI_MAXLOC, dest, MPI_COMM_WORLD, ierr) !! data reduction by way of MPI_MAXLOC if(Iam .eq. master) then print *,'The result =',result end_time = MPI_Wtime() print *, 'elapsed time is ',end_time-start_time,' seconds' maxid = max_result(2) print *,'Proc',maxid,' has largest integrated value of', max_result(1) minid = min_result(2) print *,'Proc',minid,' has smallest integrated value of', min_result(1) endif call MPI_Finalize(ierr) !! let MPI finish up ... stop end real function integral(a,n,h) implicit none integer n, i, j real h, h2, aij, a real fct, x fct(x) = cos(x) !! kernel of the integral integral = 0.0 !! initialize integral h2 = h/2. do j=0,n-1 !! sum over all "j" integrals aij = a + j*h !! lower limit of "j" integral integral = integral + fct(aij+h2)*h enddo return end 예제 (10) ! ------------------------------------------------------------------------! pi_send.f! FILES: pi_send.f, dboard.f, make.pi.f! DESCRIPTION: MPI pi calculation example program. Fortran version.! This program calculates pi using a "dartboard" algorithm. See
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (34 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
! Fox et al.(1988) Solving Problems on Concurrent Processors, vol.1! page 207. All processes contribute to the calculation, with the! master averaging the values for pi. ! ! SPMD Version: Conditional statements check if the process is the! master or a worker. ! This version uses low level sends and receives to collect results ! AUTHOR: Roslyn Leibensperger (C program for PVM).! REVISED: 05/11/93 Blaise Barney Ported to Fortran.! 05/24/93 R. Leibensperger Ported to API.! 01/10/94 S. Pendell Changed API to MPL.! 05/18/94 R. Leibensperger Non-blocking send.! CONVERTED TO MPI: 11/12/94 by Xianneng Shen.! ------------------------------------------------------------------------!Explanation of constants and variables used in this program:! DARTS = number of throws at dartboard ! ROUNDS = number of times "DARTS" is iterated ! MASTER = task ID of master task! mytid = task ID of current task! nproc = number of tasks! homepi = value of pi calculated by current task! pi = average of pi for this iteration! avepi = average pi value for all iterations ! pirecv = pi received from worker! pisum = sum of workers' pi values ! seednum = seed number - based on mytid! source = source of incoming message! mtype = message type ! sbytes = size of message being sent! nbytes = size of message successfully sent! rbytes = size of message received! ------------------------------------------------------------------------ program pi_send include 'mpif.h' integer DARTS, ROUNDS, MASTER parameter(DARTS = 5000) parameter(ROUNDS = 10) parameter(MASTER = 0) integer ierr, status(MPI_STATUS_SIZE), request integer mytid, nproc, source, mtype, msgid, sbytes, rbytes, i, n real*4 seednum real*8 homepi, pi, avepi, pirecv, pisum, dboard! Obtain number of tasks and task ID call mpi_init(ierr) call mpi_comm_rank(MPI_COMM_WORLD, mytid, ierr) call mpi_comm_size(MPI_COMM_WORLD, nproc, ierr) write(*,*)'MPI task id = ', mytid ! Use the task id to set the seed number for the random number generator. seednum = real(mytid) call srand(seednum) avepi = 0 do 40 i = 1, ROUNDS! Calculate pi using dartboard algorithm homepi = dboard(DARTS)
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (35 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
! ******************** start of worker section ***************************! All workers send result to master. Steps include: ! -set message type equal to this round number! -set message size to 8 bytes (size of real8)! -send local value of pi (homepi) to master task! -a non-blocking send followed by mpi_wait is used! this is safe programming practice if (mytid .ne. MASTER) then mtype = I sbytes = 8 call mpi_isend(homepi, 1, MPI_DOUBLE_PRECISION, MASTER, i, MPI_COMM_WORLD, request, ierr) call mpi_wait(request, status, ierr)! ******************** end of worker section ***************************** else! ******************** start of master section **************************! ! Master receives messages from all workers. Steps include:! -set message type equal to this round ! -set message size to 8 bytes (size of real8)! -receive any message of type mytpe! -keep running total of pi in pisum! Master then calculates the average value of pi for this iteration ! Master also calculates and prints the average value of pi over all ! iterations mtype = I sbytes = 8 pisum = 0 do 30 n = 1, nproc-1 call mpi_recv(pirecv, 1, MPI_DOUBLE_PRECISION, MPI_ANY_SOURCE, mtype, MPI_COMM_WORLD, status, ierr) pisum = pisum + pirecv 30 continue pi = (pisum + homepi)/nproc avepi = ((avepi*(i-1)) + pi) / I write(*,32) DARTS*i, avepi 32 format(' After',i6,' throws, average value of pi = ',f10.8) ! ********************* end of master section **************************** endif 40 continue call mpi_finalize(ierr) end 예제 (11) C ------------------------------------------------------------------------! pi_reduce.f! FILES: pi_reduce.f, dboard.f, make.pi.fC DESCRIPTION: MPI pi calculation example program. Fortran version.! This program calculates pi using a "dartboard" algorithm. See
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (36 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
! Fox et al.(1988) Solving Problems on Concurrent Processors, vol.1! page 207. All processes contribute to the calculation, with the! master averaging the values for pi. ! ! SPMD Version: Conditional statements check if the process is the! master or a worker.!! This version uses mpi_reduce to collect results! ! AUTHOR: Roslyn Leibensperger (C program for PVM).! REVISED: 05/11/93 Blaise Barney Ported to Fortran.! 06/01/93 R. Leibensperger Ported to API.! 01/10/94 S. Pendell !hanged API to MPL.! 05/18/94 R. Leibensperger Correction to comments.! CONVERTED TO MPI: 11/12/94 by Xianneng Shen.! ------------------------------------------------------------------------! Explanation of constants and variables used in this program:! DARTS = number of throws at dartboard ! ROUNDS = number of times "DARTS" is iterated ! MASTER = task ID of master task! mytid = task ID of current task ! nproc = number of tasks! homepi = value of pi calculated by current task! pisum = sum of tasks' pi values ! pi = average of pi for this iteration! avepi = average pi value for all iterations ! seednum = seed number - based on mytid! sbytes = size of message being sent! ------------------------------------------------------------------------ program pi_reduce include 'mpif.h' integer DARTS, ROUNDS, MASTER parameter(DARTS = 5000) parameter(ROUNDS = 10) parameter(MASTER = 0) integer ierr integer mytid, nproc, sbytes, I real*4 seednum real*8 homepi, pi, avepi, pisum, dboard! Obtain number of tasks and task ID call mpi_init(ierr) call mpi_comm_rank(MPI_COMM_WORLD, mytid, ierr) call mpi_comm_size(MPI_COMM_WORLD, nproc, ierr) write(*,*)'MPI task id = ', mytid ! Use the task id to set the seed number for the random number generator. seednum = real(mytid) call srand(seednum) avepi = 0 do 40 i = 1, ROUNDS! Calculate pi using dartboard algorithm homepi = dboard(DARTS)! Use mpi_reduce to sum values of homepi across all task! Master will store the accumulated value in pisum! - homepi is the send buffer! - pisum is the receive buffer (used by the receiving task only)! - sbytes is the size of the message
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (37 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
! - MASTER is the task that will receive the result of the reduction! operation sbytes = 8 call mpi_reduce(homepi, pisum, 1, MPI_DOUBLE_PRECISION, . MPI_SUM, MASTER, MPI_COMM_WORLD, ierr)! Master computes average for this iteration and all iterations if (mytid .eq. MASTER) then pi = pisum/nproc avepi = ((avepi*(i-1)) + pi) / I write(*,32) DARTS*i, avepi 32 format(' After',i6,' throws, average value of pi = ',f10.8) endif 40 continue call mpi_finalize(ierr) end
! ------------------------------------------------------------------------! dboard.f! see pi_send.f and pi_reduce.f! ------------------------------------------------------------------------! Explanation of constants and variables used in this function:! darts = number of throws at dartboard! score = number of darts that hit circle! n = index variable! r = random number between 0 and 1 ! x_coord = x coordinate, between -1 and 1 ! x_sqr = square of x coordinate! y_coord = y coordinate, between -1 and 1 ! y_sqr = square of y coordinate! pi = computed value of pi! ------------------------------------------------------------------------ real*8 function dboard(darts) integer darts, score, n real*4 r real*8 x_coord, x_sqr, y_coord, y_sqr, pi score = 0! Throw darts at board. Done by generating random numbers! between 0 and 1 and converting them to values for x and y ! coordinates and then testing to see if they "land" in ! the circle." If so, score is incremented. After throwing the! specified number of darts, pi is calculated. The computed value! of pi is returned as the value of this function, dboard. ! Note: the seed value for rand() is set in pi_send.f or pi_reduce.f. do 10 n = 1, darts r = rand() x_coord = (2.0 * r) - 1.0 x_sqr = x_coord * x_coord r = rand() y_coord = (2.0 * r) - 1.0 y_sqr = y_coord * y_coord if ((x_sqr + y_sqr) .le. 1.0) then score = score + 1 endif 10 continue
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (38 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
pi = 4.0 * score / darts dboard = pi end 예제 (12) ----------------------------------------------------------- program int_pi1cc For APIcc This simple program approximates pi by computing pi = integralc from 0 to 1 of 4/(1+x*x)dx which is approximated by sum fromc k=1 to N of 4 / ((1 + (k-1/2)**2 ). The only input data required is N.cc Parallel version #1: All instances are started at load time, but no c messages or division of workc RLF 4/4/93 16:02c revised: 6/4/93 riordanc Converted to MPI: 11/12/94 Xianneng Shenc include "mpif.h" integer ierr, status(MPI_STATUS_SIZE) parameter (maxproc=100) real err, f, pi, sum, w integer i, N, nprocs, mynum f(x) = 4.0/(1.0+x*x) pi = 4.0*atan(1.0)!c All instances call the startup routine to get their instance number (mynum) call MPI_INIT(ierr) call MPI_COMM_RANK(MPI_COMM_WORLD, mynum, ierr) call MPI_COMM_SIZE(MPI_COMM_WORLD, nprocs, ierr)c ------- Each new approximation to pi begins here. -------------------5 continuec Step (1): get a value for N, the number of intervals in the approximationc (Parallel versions: the initial instance, or master, reads this in.)c This would be a good place to add message-passing, so that the masterc can send N to the other nodes, and the other nodes can get N from thec master. if (mynum .eq. 0) then print *,'Enter number of approximation intervals:(0 to exit)' read *, N else N=0 endifc Step (2): check for exit condition. if (N .le. 0) then print *,'node (',mynum, ') left' call exit
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (39 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
endifc Step (3): do the computation in N stepsc (Ultimately, this work should be divided up among the processes) w = 1.0/N sum = 0.0 do i = 1,N sum = sum + f((i-0.5)*w) enddo sum = sum * wc Step (4): print the results c (Ultimately, partial results will have to be sent to the master,c who will then print the answer) err = sum - pi print *, 'sum, err =', sum, err go to 5 call MPI_FINALIZE(ierr) end------------------------------------------------------ program int_pi2!! For API!! This simple program approximates pi by computing pi = integral! from 0 to 1 of 4/(1+x*x)dx which is approximated by sum from! k=1 to N of 4 / ((1 + (k-1/2)**2 ). The only input data required is N.!! Parallel version number 2: (int_pi2.f)! RLF 4/6/93 17:00! revised: 6/4/93 riordan! Completely parallelized version! Converted to MPI: 11/12/94 by Xianneng Shen! include "mpif.h" integer ierr, status(MPI_STATUS_SIZE) real err, f, pi, sum, w integer i, N, info, mynum, nprocs, source, dest, type integer nbytes, len, dontcare, nbuf(4) f(x) = 4.0/(1.0+x*x) pi = 4.0*atan(1.0) dest = 0 type = 2 len = 4 nbytes = 0! All instances call the startup routine to get their instance number (mynum) call MPI_INIT(ierr) call MPI_COMM_RANK(MPI_COMM_WORLD, mynum, ierr) call MPI_COMM_SIZE(MPI_COMM_WORLD, nprocs, ierr)! ------- Each new approximation to pi begins here. -------------------! (Step 1) Get value N for a new run 5 call solicit (N,nprocs,mynum)! Step (2): check for exit condition.
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (40 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
if (N .le. 0) then call exit endif! Step (3): do the computation in N steps! Parallel Version: there are "nprocs" instances participating. Each! instance should do 1/nprocs of the calculation. Since we want! i = 1..n but mynum = 0, 1, 2..., we start off with mynum+1. w = 1.0/N sum = 0.0 do i = mynum+1,N,nprocs sum = sum + f((i-0.5)*w) enddo sum = sum * w! Step (4): print the results ! (Parallel version: collect partial results and let master instance print it) if (mynum.eq.0) then print *,'host calculated x=',sum do i = 1,nprocs-1 nbytes = len call MPI_RECV(x, 1, MPI_REAL, MPI_ANY_SOURCE, type, MPI_COMM_WORLD, status, ierr) print *,'host got x=',x sum=sum+x enddo err = sum - pi print *, 'sum, err =', sum, err! Other instances just send their sum and go back to 5 and wait for more input else call MPI_SEND(sum, 1, MPI_REAL, dest, type, MPI_COMM_WORLD, ierr) print *,'instance',mynum,' sent partial sum',sum, ' to instance 0' endif go to 5 call MPI_FINALIZE(ierr) end-------------------------------------------------- subroutine solicit (N,nprocs,mynum)c Get a value for N, the number of intervals in the approximationc (Parallel versions: master instance reads in N and thenc broadcasts N to all the other instances of the program) include "mpif.h" integer ierr integer source, msglen,nbuf(4) source = 0 msglen = 4 if (mynum .eq. 0) then print *,'Enter number of approximation intervals:(0 to exit)' read *, N endif call MPI_BCAST(N, 1, MPI_INTEGER, source, MPI_COMM_WORLD, ierr) return end
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (41 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
예제 (13)
C *****************************************************************************C FILE: mpl.ex1.fC DESCRIPTION:C In this simple example, the master task initiates numtasks-1 number ofC worker tasks. It then distributes an equal portion of an array to eachC worker task. Each worker task receives its portion of the array, andC performs a simple value assignment to each of its elements. The valueC assigned to each element is simply that element's index in the array+1.C Each worker task then sends its portion of the array back to the masterC task. As the master receives back each portion of the array, selectedC elements are displayed.C AUTHOR: Blaise BarneyC LAST REVISED: 6/10/93C LAST REVISED: 1/10/94 Changed API to MPL Stacy PendellC CONVERTED TO MPI: 11/12/94 by Xianneng ShenC ************************************************************************** program example1_master include 'mpif.h' integer status(MPI_STATUS_SIZE) integer ARRAYSIZE parameter (ARRAYSIZE = 60000) parameter (MASTER = 0) integer numtask, numworkers, taskid, dest, index, i, & arraymsg, indexmsg, source, chunksize, & int4, real4 real*4 data(ARRAYSIZE), result(ARRAYSIZE)C ************************ initializations ***********************************C Find out how many tasks are in this partition and what my task id is. ThenC define the number of worker tasks and the array partition size as chunksize.C Note: For this example, the MP_PROCS environment variable should be setC to an odd number...to insure even distribution of the array to numtasks-1C worker tasks.C ***************************************************************************** call mpi_init(ierr) call mpi_comm_rank(MPI_COMM_WORLD, taskid, ierr) call mpi_comm_size(MPI_COMM_WORLD, numtasks, ierr) write(*,*)'taskid =',taskid numworkers = numtasks-1 chunksize = (ARRAYSIZE / numworkers) arraymsg = 1 indexmsg = 2 int4 = 4 real4 = 4 C *************************** master task ************************************* if (taskid .eq. MASTER) then print *, '*********** Starting MPI Example 1 ************'C Initialize the array do 20 i=1, ARRAYSIZE
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (42 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
data(i) = 0.0 20 continueC Send each worker task its portion of the array index = 1 do 30 dest=1, numworkers write(*,*) 'Sending to worker task', dest call mpi_send(index, 1, MPI_INTEGER, dest, 0, . MPI_COMM_WORLD, ierr) call mpi_send(data(index), chunksize, MPI_REAL, dest, 0, . MPI_COMM_WORLD, ierr) index = index + chunksize 30 continueC Now wait to receive back the results from each worker task and print C a few sample values do 40 i=1, numworkers source = i call mpi_recv(index, 1, MPI_INTEGER, source, 1, . MPI_COMM_WORLD, status, ierr) call mpi_recv(result(index), chunksize, MPI_REAL, source, 1, . MPI_COMM_WORLD, status, ierr) print *, '---------------------------------------------------' print *, 'MASTER: Sample results from worker task ', source print *, ' result[', index, ']=', result(index) print *, ' result[', index+100, ']=', result(index+100) print *, ' result[', index+1000, ']=', result(index+1000) print *, ' ' 40 continue print *, 'MASTER: All Done!' endifC *************************** worker task ************************************ if (taskid .gt. MASTER) thenC Receive my portion of array from the master task */ call mpi_recv(index, 1, MPI_INTEGER, MASTER, 0, . MPI_COMM_WORLD, status, ierr) call mpi_recv(result(index), chunksize, MPI_REAL, MASTER, 0, . MPI_COMM_WORLD, status, ierr) C Do a simple value assignment to each of my array elements do 50 i=index, index + chunksize result(i) = i + 1 50 continueC Send my results back to the master call mpi_send(index, 1, MPI_INTEGER, MASTER, 1, . MPI_COMM_WORLD, ierr) call mpi_send(result(index), chunksize, MPI_REAL, MASTER, 1, . MPI_COMM_WORLD, ierr) endif call mpi_finalize(ierr) end-------------------------------------------- 예제 (14)
C******************************************************************************
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (43 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
C FILE: mm.fC DESCRIPTION:C In this template code, the master task distributes a matrix multiplyC operation to numtasks-1 worker tasks.C NOTE1: C and Fortran versions of this code differ because of the wayC arrays are stored/passed. C arrays are row-major order but FortranC arrays are column-major order.C AUTHORS of MPL version: Ros Leibensperger / Blaise Barney C LAST MPL version REVISED: 6/10/93 bbarneyC COVERTED TO MPI: 11/12/94 by Xianneng Shen C****************************************************************************** program mm1_master parameter (NRA = 62) parameter (NCA = 15) parameter (NCB = 7) parameter (MASTER = 0) parameter (FROM_MASTER = 1) parameter (FROM_WORKER = 2) include 'mpif.h' integer status(MPI_STATUS_SIZE), ierr integer numtasks,taskid,numworkers,source,dest,nbytes,mtype, & i4size,r8size,cols,avecol,extra, offset,i,j,k real*8 a(NRA,NCA), b(NCA,NCB), c(NRA,NCB) i4size = 4 r8size = 8 call MPI_INIT(ierr) call MPI_COMM_RANK(MPI_COMM_WORLD, taskid, ierr) call MPI_COMM_SIZE(MPI_COMM_WORLD, numtasks, ierr) numworkers = numtasks-1 C *************************** master task ************************************* if (taskid .eq. MASTER) thenC Initialize A and B do 30 i=1, NRA do 30 j=1, NCA a(i,j) = (i-1)+(j-1) 30 continue do 40 i=1, NCA do 40 j=1, NCB b(i,j) = (i-1)*(j-1) 40 continueC Send matrix data to the worker tasks avecol = NCB/numworkers extra = mod(NCB, numworkers) offset = 1 mtype = FROM_MASTER do 50 dest=1, numworkers if (dest .le. extra) then cols = avecol + 1 else cols = avecol endif write(*,*)' sending',cols,' cols to task',dest call MPI_SEND(offset,1, MPI_INTEGER, dest, mtype, . MPI_COMM_WORLD, ierr) call MPI_SEND(cols, 1, MPI_INTEGER, dest, mtype,
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (44 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
. MPI_COMM_WORLD, ierr) call MPI_SEND(a, NRA*NCA, MPI_DOUBLE_PRECISION, dest, . mtype, MPI_COMM_WORLD, ierr) call MPI_SEND(b(1,offset), cols*NCA, MPI_DOUBLE_PRECISION, . dest, mtype, MPI_COMM_WORLD, ierr) offset = offset + cols 50 continue C Receive results from worker tasks mtype = FROM_WORKER do 60 i=1, numworkers source = I call MPI_RECV(offset, 1, MPI_INTEGER, source, mtype, . MPI_COMM_WORLD, status, ierr) call MPI_RECV(cols, 1, MPI_INTEGER, source, mtype, . MPI_COMM_WORLD, status, ierr) call MPI_RECV(c(1,offset), cols*NRA, MPI_DOUBLE_PRECISION, . source, mtype, MPI_COMM_WORLD, status, ierr) 60 continueC Print results do 90 i=1, NRA do 80 j = 1, NCB write(*,70)c(i,j) 70 format(2x,f8.2,$) 80 continue print *, ' ' 90 continue end C *************************** worker task ************************************* if (taskid > MASTER) thenC Receive matrix data from master task mtype = FROM_MASTER call MPI_RECV(offset, 1, MPI_INTEGER, MASTER, . mtype, MPI_COMM_WORLD, status, ierr) call MPI_RECV(cols, 1, MPI_INTEGER, MASTER, . mtype, MPI_COMM_WORLD, status, ierr) call MPI_RECV(a, NRA*NCA, MPI_DOUBLE_PRECISION, . MASTER, mtype, MPI_COMM_WORLD, status, ierr) call MPI_RECV(b, cols*NCA, MPI_DOUBLE_PRECISION, . MASTER, mtype, MPI_COMM_WORLD, status, ierr)C Do matrix multiply do 100 k=1, cols do 100 i=1, NRA c(i,k) = 0.0 do 100 j=1, NCA c(i,k) = c(i,k) + a(i,j) * b(j,k) 100 continueC Send results back to master task mtype = FROM_WORKER call MPI_SEND(offset, 1, MPI_INTEGER, MASTER, . mtype, MPI_COMM_WORLD, ierr) call MPI_SEND(cols, 1, MPI_INTEGER, MASTER, . mtype, MPI_COMM_WORLD, ierr) call MPI_SEND(c, cols*NRA, MPI_DOUBLE_PRECISION, . MASTER, mtype, MPI_COMM_WORLD, ierr) endif
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (45 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
call MPI_FINALIZE(ierr) end
예제 (15)
C -----------------------------------------------------------------------C This program implements the concurrent wave equation described C in Chapter 5 of Fox et al., 1988, Solving Problems on ConcurrentC Processors, vol 1. CC A vibrating string is decomposed into points. Each processor is C responsible for updating the amplitude of a number of points overC time.C C At each iteration, each processor exchanges boundary points withC nearest neighbors. This version uses low level sends and receivesC to exchange boundary points.CC AUTHOR: Roslyn Leibensperger (C program for MPL)C REVISED: 06/07/93 R. Leibensperger Ported to FortranC CONVERTED to MPI: 11/12/94 by Xianneng ShenC ------------------------------------------------------------------------ C ------------------------------------------------------------------------ C Explanation of constants and variables used in common blocks andC include filesC MASTER = task ID of masterC INTSIZE = size of integer in bytesC REAL8SIZE = size of real*8 in bytesC E_OUT1, E_OUT2 = message typesC taskid = task IDC nproc = number of tasksC tpoints = total points along waveC nsteps = number of time stepsC npoints = number of points handled by this taskC first = index of first point handled by this taskC values(0:1001) = values at time tC oldval(0:1001) = values at time (t-dt)C newval(0:1001) = values at time (t+dt)C ------------------------------------------------------------------------ program wave implicit none include 'mpif.h' include 'parameters.h' integer ierr integer taskid, nproc common/config/taskid, nproc integer left, right, nbuf(4)C Learn number of tasks and taskid
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (46 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
call mpi_init(ierr) call mpi_comm_rank(MPI_COMM_WORLD, taskid, ierr) call mpi_comm_size(MPI_COMM_WORLD, nproc, ierr) write (*,5) taskid 5 format (I5, ': Wave Program running')C Learn value of allgrp and dontcareC Determine left and right neighbors if (taskid .eq. nproc-1) then right = 0 else right = taskid + 1 end if if (taskid .eq. 0) then left = nproc - 1 else left = taskid - 1 end ifC Get program parameters and initialize wave values if (taskid .eq. MASTER) then call init_master else call init_workers end if call init_lineC Update values along the wave for nstep time steps call update(left, right)C Master collects results from workers and prints if (taskid .eq. MASTER) then call out_master else call out_workers end if call mpi_finalize(ierr) end ---------------------------------------------------------------C ------------------------------------------------------------------------C Master obtains input values from userC ------------------------------------------------------------------------ subroutine init_master implicit none include 'parameters.h' include 'mpif.h' integer ierr integer taskid, nproc common/config/taskid, nproc integer tpoints, nsteps common/inputs/tpoints, nsteps integer MAXPOINTS, MAXSTEPS parameter (MAXPOINTS = 1000) parameter (MAXSTEPS = 10000) integer buffer(2), msglen tpoints = 0 nsteps = 0 do while ((tpoints .lt. nproc) .or. (tpoints .gt. MAXPOINTS)) write (*,*)'Enter number of points along vibrating string'
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (47 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
read (*,*) tpoints if ((tpoints .lt. nproc) .or. (tpoints .gt. MAXPOINTS)) & write (*,*) 'enter value between ',nproc,' and ',MAXPOINTS end do do while ((nsteps .lt. 1) .or. (nsteps .gt. MAXSTEPS)) write (*,*) 'Enter number of time steps' read (*,*) nsteps if ((nsteps .lt. 1) .or. (nsteps .gt. MAXSTEPS)) & write (*,*) 'enter value between 1 and ', MAXSTEPS end do write (*,10) taskid, tpoints, nsteps 10 format(I5, ': points = ', I5, ' steps = ', I5)C Broadcast total points, time steps buffer(1) = tpoints buffer(2) = nsteps call mpi_bcast(buffer, 2, MPI_INTEGER, 0, MPI_COMM_WORLD,ierr) endC -------------------------------------------------------------------------C Workers receive input values from masterC ------------------------------------------------------------------------- subroutine init_workers implicit none include 'parameters.h' include 'mpif.h' integer ierr integer taskid, nproc common/config/taskid, nproc integer tpoints, nsteps common/inputs/tpoints, nsteps integer buffer(2), msglenC Receive time advance parameter, total points, time steps call mpi_bcast(buffer, 2, MPI_INTEGER, 0, MPI_COMM_WORLD, ierr) tpoints = buffer(1) nsteps = buffer(2) endC ------------------------------------------------------------------------C Initialize points on lineC ----------------------------------------------------------------------- subroutine init_line implicit none integer taskid, nproc common/config/taskid, nproc integer tpoints, nsteps common/inputs/tpoints, nsteps integer npoints, first common/decomp/npoints, first real*8 values(0:1001), oldval(0:1001), newval(0:1001) common/data/values, oldval, newval real*8 PI parameter (PI = 3.14159265) integer nmin, nleft, npts, i, j, k real*8 x, facC Calculate initial values based on sine curve nmin = tpoints/nproc nleft = mod(tpoints, nproc) fac = 2.0 * PI
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (48 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
k = 0 do i = 0, nproc-1 if (i .lt. nleft) then npts = nmin + 1 else npts = nmin endif if (taskid .eq. i) then first = k + 1 npoints = npts write (*,15) taskid, first, npts 15 format (I5, ': first = ', I5, ' npoints = ', I5) do j = 1, npts x = float(k)/float(tpoints - 1) values(j) = sin (fac * x) k = k + 1 end do else k = k + npts end if end do do i = 1, npoints oldval(i) = values(i) end do endC -------------------------------------------------------------------------C Calculate new values using wave equationC ------------------------------------------------------------------------- subroutine do_math(i) implicit none integer i integer tpoints, nsteps common/inputs/tpoints, nsteps real*8 values(0:1001), oldval(0:1001), newval(0:1001) common/data/values, oldval, newval real*8 dtime, c, dx, tau, sqtau dtime = 0.3 c = 1.0 dx = 1.0 tau = (c * dtime / dx) sqtau = tau * tau newval(i) = (2.0 * values(i)) - oldval(i) & + (sqtau * (values(i-1) - (2.0 * values(i)) + values(i+1))) endC -------------------------------------------------------------------------C Update all values along line a specified number of times C ------------------------------------------------------------------------- subroutine update(left, right) implicit none include 'mpif.h' integer left, right integer ierr, status(MPI_STATUS_SIZE), request include 'parameters.h' integer npoints, first common/decomp/npoints, first integer tpoints, nstep
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (49 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
common/inputs/tpoints, nsteps real*8 values(0:1001), oldval(0:1001), newval(0:1001) common/data/values, oldval, newval integer E_RtoL, E_LtoR parameter (E_RtoL = 10) parameter (E_LtoR = 20) integer i, j, id_rtol, id_ltor, nbytes, msglenC Update values for each point along string do i = 1, nstepsC Exchange data with "left-hand" neighbor if (first .ne. 1) then call mpi_isend(values(1), 1, MPI_DOUBLE_PRECISION, left, . E_RtoL, MPI_COMM_WORLD, request, ierr) call mpi_wait(request, status, ierr) call mpi_recv(values(0), 1, MPI_DOUBLE_PRECISION, left, . E_LtoR, MPI_COMM_WORLD, status, ierr) end ifC Exchange data with "right-hand" neighbor if (first+npoints-1 .ne. tpoints) then call mpi_isend(values(npoints), 1, MPI_DOUBLE_PRECISION, . right, E_LtoR, MPI_COMM_WORLD, request, ierr) call mpi_wait(request, status, ierr) call mpi_recv(values(npoints+1), 1, MPI_DOUBLE_PRECISION, . right, E_RtoL, MPI_COMM_WORLD, status, ierr) end ifC Update points along line do j = 1, npointsC Global endpoints if ((first+j-1 .eq. 1).or.(first+j-1 .eq. tpoints))then newval(j) = 0.0 else call do_math(j) end if end do do j = 1, npoints oldval(j) = values(j) values(j) = newval(j) end do end do endC ------------------------------------------------------------------------C Receive results from workers and printC ------------------------------------------------------------------------ subroutine out_master implicit none include 'parameters.h' include 'mpif.h' integer ierr, status(MPI_STATUS_SIZE), request integer taskid, nproc common/config/taskid, nproc integer tpoints, nsteps common/inputs/tpoints, nsteps integer npoints, first common/decomp/npoints, first real*8 values(0:1001), oldval(0:1001), newval(0:1001) common/data/values, oldval, newval integer i, start, npts, buffer(2), tpts
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (50 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
real*8 results(1000) C Store worker's results in results array do i = 1, nproc - 1C Receive number of points and first point call mpi_recv(buffer, 2, MPI_INTEGER, MPI_ANY_SOURCE, . E_OUT1, MPI_COMM_WORLD, status, ierr) start = buffer(1) npts = buffer(2)C Receive results call mpi_recv(results(start), npts, MPI_DOUBLE_PRECISION, . MPI_ANY_SOURCE, E_OUT2, MPI_COMM_WORLD, status, ierr) end doC Store master's results in results array do i = first, first+npoints-1 results(i) = values(i) end do if (tpoints .lt. 10) then tpts = tpoints else tpts = 10 end if write (*,200) tpts, (results(i), i = 1, tpts) 200 format('first ', I5, ' points (for validation):'/ & 10(f4.2, ' ')) endC -------------------------------------------------------------------------C Send the updated values to the masterC ------------------------------------------------------------------------- subroutine out_workers implicit none include 'mpif.h' integer ierr, status(MPI_STATUS_SIZE), request include 'parameters.h' integer npoints, first common/decomp/npoints, first real*8 values(0:1001), oldval(0:1001), newval(0:1001) common/data/values, oldval, newval integer buffer(2) C Send first point and number of points handled to master buffer(1) = first buffer(2) = npoints call mpi_isend(buffer, 2, MPI_INTEGER, 0, E_OUT1, . MPI_COMM_WORLD, request, ierr) call mpi_wait(request, status, ierr) C Send results to master call mpi_isend(values(1), npoints, MPI_DOUBLE_PRECISION, . 0, E_OUT2, MPI_COMM_WORLD, request, ierr) call mpi_wait(request, status, ierr) end http://www.tc.cornell.edu/Services/Docs/Examples/code.htm#example5
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (51 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
MPI/Fortran Examples : Cornell Theory Center
Example 5 This program calculates the value of pi, using numerical integration with parallel processing. The user selects the number of points of integration. By selecting more points you get more accurate results at the expense of additional computation. C 1996 Saleh Elmohamed and the old npac staff.C 2001 [email protected]**********************************************************************c This program calculates the value of pi, using numerical integrationc with parallel processing. The user selects the number of points ofc integration. By selecting more points you get more accurate resultsc at the expense of additional computationc c This version is written using p4 calls to handle message passingc It should run without changes on most workstation clusters and MPPs.c c Each node: c 1) receives the number of rectangles used in the approximation.c 2) calculates the areas of it's rectangles.c 3) Synchronizes for a global summation.c Node 0 prints the result.c c Constants:c c SIZETYPE initial message to the cubec ALLNODES used to load all nodes in cube with a node processc INTSIZ four bytes for an integerc DBLSIZ eight bytes for double precisionc c Variables:c c pi the calculated resultc n number of points of integration. c x midpoint of each rectangle's intervalc f function to integratec sum,pi area of rectanglesc tmp temporary scratch space for global summationc i do loop indexc********************************************************************** program main include 'mpif.h' double precision PI25DT parameter (PI25DT = 3.141592653589793238462643d0) integer INTSIZ , DBLSIZ, ALLNODES, ANYNODE
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (52 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
parameter(INTSIZ=4,DBLSIZ=8,ALLNODES=-1,ANYNODE=-1) double precision pi, h, sum, x, f, a, temp integer n, myid, numnodes, i, rc integer sumtype, sizetype, masternode integer status(3) c function to integrate f(a) = 4.d0 / (1.d0 + a*a) call MPI_INIT( ierr ) call MPI_COMM_RANK( MPI_COMM_WORLD, myid, ierr ) call MPI_COMM_SIZE( MPI_COMM_WORLD, numnodes, ierr )c print *, "Process ", myid, " of ", numnodes, " is alive" sizetype = 10 sumtype = 17 masternode = 0 10 if ( myid .eq. 0 ) then write(6,98) 98 format('Enter the number of intervals: (0 quits)') read(5,99)n 99 format(i10) do i=1,numnodes-1 call MPI_SEND(n,1,MPI_INTEGER,i,sizetype,MPI_COMM_WORLD,rc) enddo else call MPI_RECV(n,1,MPI_INTEGER,masternode,sizetype, + MPI_COMM_WORLD,status,rc) endif c check for quit signal if ( n .le. 0 ) goto 30 c calculate the interval size h = 1.0d0/n sum = 0.0d0 do 20 i = myid+1, n, numnodes x = h * (dble(i) - 0.5d0) sum = sum + f(x) 20 continue pi = h * sum if (myid .ne. 0) then call MPI_SEND(pi,1,MPI_DOUBLE_PRECISION,masternode,sumtype,
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (53 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
+ MPI_COMM_WORLD,rc) else do i=1,numnodes-1 call MPI_RECV(temp,1,MPI_DOUBLE_PRECISION,i,sumtype, + MPI_COMM_WORLD,status,rc) pi = pi + temp enddo endif c node 0 prints the answer. if (myid .eq. 0) then write(6, 97) pi, abs(pi - PI25DT) 97 format(' pi is approximately: ', F18.16, + ' Error is: ', F18.16) endif goto 10 30 call MPI_FINALIZE(rc) end
Example 6 Implementation of Fast Fourier Transforms (FFT). C ----------------------------------C 1996 Saleh Elmohamed and the old npac staff.C 2001 [email protected] ---------------------------------- program fft_with_MPI parameter(indx = 15) parameter(nx = 2**indx,maxiters=1000) integer comm,myid,nnodes,ierr double complex,allocatable,dimension(:) :: f,g,sct,a,b,t integer,allocatable,dimension(:) :: mixup double precision time_begin,time_end include 'mpif.h' comm = MPI_COMM_WORLD call MPI_init(ierr) call MPI_COMM_RANK(comm,myid,ierr) call MPI_COMM_SIZE(comm,nnodes,ierr) kxp = nx/nnodes allocate(f(kxp)) allocate(g(kxp)) allocate(sct(kxp)) allocate(mixup(kxp)) do i=1,kxp
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (54 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
f(i) = cmplx(real(i),real(myid+1)) enddo isign = 1 kblok = nnodes time_begin = MPI_Wtime() call bitrv1(mixup,isign,indx,kxp,kblok,sct,myid) !do iter =1,maxiters call permute(f,g,mixup,indx,kxp,kblok,comm) call fft(f,g,isign,mixup,sct,indx,kxp,kblok,comm,myid) !enddo time_end = MPI_Wtime() print *,'fft of 1-d with size= ',nx,' took ' print *,time_end-time_begin,' seconds for ',maxiters,' iterations' call MPI_FINALIZE(ierr) end !kblok is the number of processors!g is the input vector whose fft will be taken!kxp is the length of local data subroutine fft(f,g,isign,mixup,sct,indx,kxp,kblok,comm,myprocid) double complex f,g,sct dimension f(kxp),g(kxp) dimension sct(kxp),mixup(kxp) integer myprocid,comm include 'mpif.h' integer dest,source,nproc,dest_id,lind,lb,ub,sign,info double complex t1 integer status(MPI_STATUS_SIZE) nproc = kblok nx = 2**indx nxh = nx/2 indx1 = indx !-1 kbs = nxh/kxp dnx = 6.28318530717959/float(nx) sign = isign temp= log10(float(kxp))/log10(2.0) lind = temp C ---------------------------
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (55 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
C inverse fourier transformC bit-reverse array elements to temporaryC --------------------------- do l = 1, indx1 nxs = 2**(l - 1) kxs = nxs/kxpC ---------------------------C local calculationC --------------------------- if (kxs.eq.0) then do j = 1, kxp lb = (j - 1)/nxs if (lb.eq.(2*(lb/2))) then f(j) = g(j+nxs) else f(j) = g(j-nxs) endif enddo C ---------------------------C perform reductionC --------------------------- km = kxp/nxs do j = 1, kxp kb = j - 1 lb = kb/nxs if (sign .eq. -1) then t1 = sct(1+km*(kb-nxs*lb)) else if (sign .eq.1) then t1 = conjg(sct(1+km*(kb-nxs*lb)) ) endif if (lb.eq.(2*(lb/2))) then g(j) = g(j) + t1*f(j) else g(j) = f(j) - t1*g(j) endif enddo elseC ---------------------------C copy dataC --------------------------- if (btest(myprocid,l-lind-1) .eqv. .false.) then dest = ibset(myprocid,l-lind-1) call MPI_SEND(g,kxp,MPI_DOUBLE_COMPLEX,dest,0,comm,ierr) else source = ibclr(myprocid,l-lind-1) call MPI_RECV(f,kxp,MPI_DOUBLE_COMPLEX,source,0,comm,status,ierr) endif
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (56 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
if (btest(myprocid,l-lind-1) .eqv. .true.) then dest = ibclr(myprocid,l-lind-1) call MPI_SEND(g,kxp,MPI_DOUBLE_COMPLEX,dest,0,comm,ierr) else source = ibset(myprocid,l-lind-1) call MPI_RECV(f,kxp,MPI_DOUBLE_COMPLEX,source,0,comm,status,ierr) endif C ------------------C perform reductionC ------------------ km = nxh/nxs dns = dnx*float(km) kb = myprocid llb = kb/kxs kb = kxp*(kb - kxs*llb) - 1 llb = llb - 2*(llb/2) do j = 1, kxp arg = dns*float(j + kb) t1 = cmplx(cos(arg),sign*sin(arg)) if (llb.eq.0) then g(j ) = g(j) + t1*f(j) else g(j ) = f(j) - t1*g(j) endif enddo endif enddoC do l=1,indx1 return end subroutine bitrv1(mixup,isign,indx,kxp,kblok,sct,myprocnum) double complex sct(kxp) integer mixup(kxp) integer isign,indx,kxp,kblok,myprocnum integer llb,ub real arg,dnx nx = 2**indx nxh = nx/2 dnx = 6.28318530717959/float(nx) C ---------------------------------
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (57 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
C prepare bit-reverse index tableC --------------------------------- koff = kxp*myprocnum - 1 do j = 1, kxp lb = j + koff ll = 0 do l = 1, indx jb = lb/2 it = lb - 2*jb lb = jb ll = 2*ll + it enddo mixup(j) = ll + 1 enddo do j = 1, kxp arg = dnx*float((nxh*(j - 1))/kxp) sct(j) = cmplx(cos(arg),-sin(arg)) enddo return end subroutine permute(f,g,mixup,indx,kxp,kblok,comm) double complex f,g dimension f(kxp),g(kxp) dimension mixup(kxp) integer comm double complex global_f(kxp,kblok) include 'mpif.h' C ---------------------------C inverse fourier transformC bit-reverse array elements to temporaryC --------------------------- call MPI_ALLGATHER(f,kxp,MPI_DOUBLE_COMPLEX,global_f,kxp,& MPI_DOUBLE_COMPLEX,comm,ierr) do j = 1, kxp ll = mixup(j) kk = (ll - 1)/kxp + 1 jj = ll - kxp*(kk - 1) g(j) = global_f(jj,kk) enddo return end
Example 7 Implementation of the two concepts of gather and scatter with MPI.
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (58 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
!---------------------------------! 1996 Saleh Elmohamed! 2001 [email protected]! --------------------------------- program gather_vector include 'mpif.h' integer ndims,xmax,ymax,nnodes,myid,totelem parameter(ndims=2) parameter(xmax=100,ymax=100) parameter(niters=10) parameter (totelem=xmax*ymax) integer comm,ierr integer status(MPI_STATUS_SIZE) double precision,allocatable,dimension(:,:) :: A,gA double precision,allocatable,dimension(:) :: V,dindex1,dindex2,gV integer,allocatable,dimension(:) :: index1,index2 integer,allocatable,dimension(:) :: lindex1,lindex2 !distribute each array into nnodes processors except gV and index1 and index2!these last 3 arrays are global arrays. gv will hold the all local V's!index1 and index2 will hold all the local lindex1's and lindex2's !respectively integer xb,yb,i,j,nitems,xbv comm=MPI_COMM_WORLD call MPI_init(ierr) call MPI_COMM_RANK(comm,myid,ierr) call MPI_COMM_SIZE(comm,nnodes,ierr) !initialize the input matrixes A and allocate necessary spaces for A,B!partition the matrix a in (block ,*) way xb = xmax/nnodes xbv = totelem/nnodes!allocate necessary arrays allocate(A(xb,ymax)) allocate(gA(xmax,ymax)) allocate(V(xbv)) allocate(gV(totelem)) allocate(index1(totelem)) allocate(index2(totelem)) allocate(dindex1(totelem)) allocate(dindex2(totelem)) allocate(lindex1(xbv)) allocate(lindex2(xbv))
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (59 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
!last processor is generating random index vectors index1 and index2!and partition them onto processors and send to correspoding processor!also last processor is generating random data vector V for each processor if (myid == nnodes -1) then call random_number(dindex1) call random_number(dindex2) index1 = xmax*dindex1 + 1 index2 = ymax*dindex2 + 1 do np = 0,nnodes-1 call random_number(A) A = 1000000.0*A if (np < nnodes-1) then call MPI_SEND(A,xb*ymax,MPI_DOUBLE_PRECISION,np,0,comm,ierr) call MPI_SEND(index1(xbv*np+1),xbv,MPI_INTEGER,np,0,comm,ierr) call MPI_SEND(index1(xbv*np+1),xbv,MPI_INTEGER,np,0,comm,ierr) endif enddo lindex1 = index1((nnodes-1)*xbv+1:totelem) lindex2 = index2((nnodes-1)*xbv+1:totelem) else call MPI_RECV(A,xb*ymax,MPI_DOUBLE_PRECISION,nnodes-1,0,comm,& status,ierr) call MPI_RECV(lindex1,xbv,MPI_INTEGER,nnodes-1,0,comm,status,ierr) call MPI_RECV(lindex1,xbv,MPI_INTEGER,nnodes-1,0,comm,status,ierr) endif !start timer ..... time_begin = MPI_Wtime() do iter = 1,niters!collect all the local A's in gA call all_to_all_float(myid,nnodes,comm,A,gA,xb,xmax,ymax)!collect all the local arrays index1's and index2's in index1 and index2 call all_to_all_int(myid,nnodes,comm,lindex1,index1,xbv,totelem) call all_to_all_int(myid,nnodes,comm,lindex2,index2,xbv,totelem) ilb = myid*xbv+1 iub = (myid+1)*xbv do i=1,totelem !If I am holding the vector index 'i' in my local array!I will get the gA(index1(i),index2(i)) if ((i.ge.ilb).and.(i.le.iub)) then
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (60 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
V(i-ilb+1) = gA(index1(i),index2(i)) endif enddo enddo!Stop timer time_end = MPI_Wtime() if (myid == 0) then print *,'Elapsed time ',niters,'iterations for scatter' print *,'For matrix with dimensions',xmax,ymax ,'is' print *,time_end-time_begin ,'seconds' endif deallocate(a) deallocate(v) deallocate(gv) deallocate(index1) deallocate(index2) deallocate(lindex1) deallocate(lindex2) deallocate(dindex1) deallocate(dindex2) call MPI_FINALIZE(ierr) end SUBROUTINE all_to_all_float(myid,nnodes,comm,fx,global_fx,xb,xmax,ymax) integer xb,xmax,ymax integer fx(xb,ymax),global_fx(xmax,ymax) integer myid,nnodes,comm include 'mpif.h' integer dest,source,nproc,dest_id,ierr integer status(MPI_STATUS_SIZE) global_fx(xb*myid+1:xb*(myid+1),:) = fx nproc = nnodes kcnt = myid dest = mod(myid+1,nproc) source = mod(myid-1+nproc,nproc) do i=1,nproc-1 if (mod (myid,2) .eq. 0) then call MPI_SEND(global_fx(kcnt*xb+1:(kcnt+1)*xb,:),xb,& MPI_INTEGER,dest,0,comm,ierr) else ikcnt = mod(kcnt-1+nproc,nproc) call MPI_RECV(global_fx(ikcnt*xb+1:(ikcnt+1)*xb,:),xb, & MPI_INTEGER,source,0,comm,status,ierr)
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (61 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
endif if (mod (myid,2) .eq. 1) then call MPI_SEND(global_fx(kcnt*xb+1:(kcnt+1)*xb,:),xb,& MPI_INTEGER,dest,0,comm,ierr) else ikcnt = mod(kcnt-1+nproc,nproc) call MPI_RECV(global_fx(ikcnt*xb+1:(ikcnt+1)*xb,:),xb, & MPI_INTEGER,source,0,comm,status,ierr) endif kcnt = ikcnt enddo return end SUBROUTINE all_to_all_int(myprocid,nnodes,comm,fx,global_fx,xbv,totelem) integer xbv,totelem integer fx(xbv),global_fx(totelem) integer myprocid,nnodes,comm include 'mpif.h' integer dest,source,nproc,dest_id,ierr integer status(MPI_STATUS_SIZE) do j=1,xbv global_fx(xbv*myprocid+1+j) = fx(j) enddo nproc = nnodes kcnt = myprocid dest = mod(myprocid+1,nproc) source = mod(myprocid-1+nproc,nproc) do i=1,nproc-1 if (mod (myprocid,2) .eq. 0) then call MPI_SEND(global_fx(kcnt*xbv+1),xbv,& MPI_INTEGER,dest,0,comm,ierr) else ikcnt = mod(kcnt-1+nproc,nproc) call MPI_RECV(global_fx(ikcnt*xbv+1),xbv, & MPI_INTEGER,source,0,comm,status,ierr) endif if (mod (myprocid,2) .eq. 1) then call MPI_SEND(global_fx(kcnt*xbv+1),xbv,& MPI_INTEGER,dest,0,comm,ierr) else ikcnt = mod(kcnt-1+nproc,nproc) call MPI_RECV(global_fx(ikcnt*xbv+1),xbv, & MPI_INTEGER,source,0,comm,status,ierr) endif
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (62 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
kcnt = ikcnt enddo return end…………………………………………………………………………………………..program scatter_vector include 'mpif.h' integer ndims,xmax,ymax,nnodes,myid,totelem parameter(ndims=2) parameter(xmax=1000,ymax=1000) parameter(niters=1) parameter (totelem=xmax*ymax) integer comm,ierr integer status(MPI_STATUS_SIZE) double precision,allocatable,dimension(:,:) :: A double precision,allocatable,dimension(:) :: V,dindex1,dindex2,gV integer,allocatable,dimension(:) :: index1,index2 integer,allocatable,dimension(:) :: lindex1,lindex2 !distribute each array into nnodes processors except gV and index1 and index2!these last 3 arrays are global arrays. gv will hold the all local V's!index1 and index2 will hold all the local lindex1's and lindex2's !respectively integer xb,yb,i,j,nitems,xbv comm=MPI_COMM_WORLD call MPI_init(ierr) call MPI_COMM_RANK(comm,myid,ierr) call MPI_COMM_SIZE(comm,nnodes,ierr) !initialize the input matrixes A and allocate necessary spaces for A,B!partition the matrix a in (block ,*) way xb = xmax/nnodes xbv = totelem/nnodes!allocate necessary arrays allocate(A(xb,ymax)) allocate(V(xbv)) allocate(gV(totelem)) allocate(index1(totelem)) allocate(index2(totelem)) allocate(dindex1(totelem)) allocate(dindex2(totelem)) allocate(lindex1(xbv)) allocate(lindex2(xbv))!last processor is generating random index vectors index1 and index2
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (63 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
!and partition them onto processors and send to correspoding processor!also last processor is generating random data vector V for each processor if (myid == nnodes -1) then call random_number(dindex1) call random_number(dindex2) index1 = xmax*dindex1 + 1 index2 = ymax*dindex2 + 1 do np = 0,nnodes-1 call random_number(V) V = 1000000.0*V if (np < nnodes-1) then call MPI_SEND(V,xbv,MPI_DOUBLE_PRECISION,np,0,comm,ierr) call MPI_SEND(index1(xbv*np+1),xbv,MPI_INTEGER,np,0,comm,ierr) call MPI_SEND(index1(xbv*np+1),xbv,MPI_INTEGER,np,0,comm,ierr) endif enddo lindex1 = index1((nnodes-1)*xbv+1:totelem) lindex2 = index2((nnodes-1)*xbv+1:totelem) else call MPI_RECV(V,xbv,MPI_DOUBLE_PRECISION,nnodes-1,0,comm,status,ierr) call MPI_RECV(lindex1,xbv,MPI_INTEGER,nnodes-1,0,comm,status,ierr) call MPI_RECV(lindex1,xbv,MPI_INTEGER,nnodes-1,0,comm,status,ierr) endif !start timer ..... time_begin = MPI_Wtime() do iter = 1,niters!collect all the local V's in gV call MPI_ALLGATHER(V,xbv,MPI_DOUBLE_PRECISION, & gV,xbv,MPI_DOUBLE_PRECISION,comm,ierr) !collect all the local arrays index1's and index2's in index1 and index2 call MPI_ALLGATHER(lindex1,xbv,MPI_INTEGER,index1,& xbv,MPI_INTEGER,comm,ierr) call MPI_ALLGATHER(lindex2,xbv,MPI_INTEGER,index2,& xbv,MPI_INTEGER,comm,ierr) ilb = myid*xbv+1 iub = (myid+1)*xbv do i=1,totelem!If I am holding the vector index 'index1(i)' in my local array!I will get the gV(i) if ((index1(i) .ge. ilb).and.(index1(i).le.iub)) then A(index1(i)-ilb+1,index2(i)) = gV(i) endif enddo
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (64 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
enddo!Stop timer time_end = MPI_Wtime() if (myid == 0) then print *,'Elapsed time ',niters,'iterations for scatter' print *,'For matrix with dimensions',xmax,ymax ,'is' print *,time_end-time_begin ,'seconds' endif deallocate(a) deallocate(v) deallocate(gv) deallocate(index1) deallocate(index2) deallocate(lindex1) deallocate(lindex2) deallocate(dindex1) deallocate(dindex2) call MPI_FINALIZE(ierr) end SUBROUTINE all_to_all_int(myprocid,nnodes,comm,fx,global_fx,xbv,totelem) integer xbv,totelem integer fx(xbv),global_fx(totelem) integer myprocid,nnodes,comm include 'mpif.h' integer dest,source,nproc,dest_id,ierr integer status(MPI_STATUS_SIZE) do j=1,xbv global_fx(xbv*myprocid+1+j) = fx(j) enddo nproc = nnodes kcnt = myprocid dest = mod(myprocid+1,nproc) source = mod(myprocid-1+nproc,nproc) do i=1,nproc-1 if (mod (myprocid,2) .eq. 0) then call MPI_SEND(global_fx(kcnt*xbv+1),xbv,& MPI_INTEGER,dest,0,comm,ierr) else ikcnt = mod(kcnt-1+nproc,nproc)
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (65 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
call MPI_RECV(global_fx(ikcnt*xbv+1),xbv, & MPI_INTEGER,source,0,comm,status,ierr) endif if (mod (myprocid,2) .eq. 1) then call MPI_SEND(global_fx(kcnt*xbv+1),xbv,& MPI_INTEGER,dest,0,comm,ierr) else ikcnt = mod(kcnt-1+nproc,nproc) call MPI_RECV(global_fx(ikcnt*xbv+1),xbv, & MPI_INTEGER,source,0,comm,status,ierr) endif kcnt = ikcnt enddo return end SUBROUTINE all_to_all_float(myprocid,nnodes,comm,fx,global_fx,xbv,totelem) integer xbv,totelem double precision fx(xbv),global_fx(totelem) integer myprocid,nnodes,comm include 'mpif.h' integer dest,source,nproc,dest_id,ierr integer status(MPI_STATUS_SIZE) do j=1,xbv global_fx(xbv*myprocid+1+j) = fx(j) enddo nproc = nnodes kcnt = myprocid dest = mod(myprocid+1,nproc) source = mod(myprocid-1+nproc,nproc) do i=1,nproc-1 if (mod (myprocid,2) .eq. 0) then call MPI_SEND(global_fx(kcnt*xbv+1),xbv,& MPI_DOUBLE_PRECISION,dest,0,comm,ierr) else ikcnt = mod(kcnt-1+nproc,nproc) call MPI_RECV(global_fx(ikcnt*xbv+1),xbv, & MPI_DOUBLE_PRECISION,source,0,comm,status,ierr) endif if (mod (myprocid,2) .eq. 1) then call MPI_SEND(global_fx(kcnt*xbv+1),xbv,& MPI_DOUBLE_PRECISION,dest,0,comm,ierr)
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (66 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
else ikcnt = mod(kcnt-1+nproc,nproc) call MPI_RECV(global_fx(ikcnt*xbv+1),xbv, & MPI_DOUBLE_PRECISION,source,0,comm,status,ierr) endif kcnt = ikcnt enddo return end
Example 8This program finds the force on each of a set of particles interacting via a long-range 1/r**2 force law. The number of processes must be even, and the total number of points must be exactly divisible by the number of processes. C -----------------------------------------C Comments and questions are forwarded to:C Saleh Elmohamed 1996-2001C [email protected] Design and version 1 is due to C David Walker, ORNL, March 1995.C -----------------------------------------c This program finds the force on each of a set of particles interactingc via a long-range 1/r**2 force law.cc The number of processes must be even, and the total number of pointsc must be exactly divisible by the number of processes.c program nbody implicit none include 'mpif.h' integer myrank, ierr, nprocs, npts, nlocal integer pseudohost, NN, MM, PX, PY, PZ, FX, FY, FZ real G parameter (pseudohost = 0) parameter (NN=10000, G = 1.0) parameter (MM=0, PX=1, PY=2, PZ=3, FX=4, FY=5, FZ=6) real dx(0:NN-1), dy(0:NN-1), dz(0:NN-1) real dist(0:NN-1), sq(0:NN-1) real fac(0:NN-1), tx(0:NN-1), ty(0:NN-1), tz(0:NN-1) real p(0:6,0:NN-1), q(0:6,0:NN-1) integer i, j, k, dest, src double precision timebegin, timeend integer status(MPI_STATUS_SIZE) integer newtype double precision ran integer irancc Initialize MPI, find rank of each process, and the number of
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (67 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
processesc call mpi_init (ierr) call mpi_comm_rank (MPI_COMM_WORLD, myrank, ierr) call mpi_comm_size (MPI_COMM_WORLD, nprocs, ierr)cc One process acts as the host and reads in the number of particlescc if (myrank .eq. pseudohost) thenc open (4,file='nbody.input',status='old',err=998) if (mod(nprocs,2) .eq. 0) then read (4,*) npts if (npts .gt. nprocs*NN) then print *,'Warning!! Size out of bounds!!' npts = -1 else if (mod(npts,nprocs) .ne. 0) then print *,'Number of processes must divide npts' npts = -1 end if else print *,'Number of processes must be even' npts = -1 end if end ifcc The number of particles is broadcast to all processesc call mpi_bcast (npts, 1, MPI_INTEGER, pseudohost, # MPI_COMM_WORLD, ierr)cc Abort if number of processes and/or particles is incorrectc if (npts .eq. -1) goto 999cc Work out number of particles in each processc nlocal = npts/nprocscc The pseudocode hosts initializes the particle data and sends each c process its particles.c if (myrank .eq. pseudohost) then iran = myrank + 111 do i=0,nlocal-1 p(MM,i) = sngl(ran(iran)) p(PX,i) = sngl(ran(iran)) p(PY,i) = sngl(ran(iran)) p(PZ,i) = sngl(ran(iran)) p(FX,i) = 0.0 p(FY,i) = 0.0 p(FZ,i) = 0.0 end do do k=0,nprocs-1
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (68 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
if (k .ne. pseudohost) then do i=0,nlocal-1 q(MM,i) = sngl(ran(iran)) q(PX,i) = sngl(ran(iran)) q(PY,i) = sngl(ran(iran)) q(PZ,i) = sngl(ran(iran)) q(FX,i) = 0.0 q(FY,i) = 0.0 q(FZ,i) = 0.0 end do call mpi_send (q, 7*nlocal, MPI_REAL, # k, 100, MPI_COMM_WORLD, ierr) end if end do else call mpi_recv (p, 7*nlocal, MPI_REAL, # pseudohost, 100, MPI_COMM_WORLD, status, ierr) end ifcc Initialization is now complete. Start the clock and begin work.c First each process makes a copy of its particles.c timebegin = mpi_wtime () do i= 0,nlocal-1 q(MM,i) = p(MM,i) q(PX,i) = p(PX,i) q(PY,i) = p(PY,i) q(PZ,i) = p(PZ,i) q(FX,i) = 0.0 q(FY,i) = 0.0 q(FZ,i) = 0.0 end docc Now the interactions between the particles in a single process arec computed.c do i=0,nlocal-1 do j=i+1,nlocal-1 dx(i) = p(PX,i) - q(PX,j) dy(i) = p(PY,i) - q(PY,j) dz(i) = p(PZ,i) - q(PZ,j) sq(i) = dx(i)**2+dy(i)**2+dz(i)**2 dist(i) = sqrt(sq(i)) fac(i) = p(MM,i) * q(MM,j) / (dist(i) * sq(i)) tx(i) = fac(i) * dx(i) ty(i) = fac(i) * dy(i) tz(i) = fac(i) * dz(i) p(FX,i) = p(FX,i)-tx(i) q(FX,j) = q(FX,j)+tx(i) p(FY,i) = p(FY,i)-ty(i) q(FY,j) = q(FY,j)+ty(i) p(FZ,i) = p(FZ,i)-tz(i)
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (69 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
q(FZ,j) = q(FZ,j)+tz(i) end do end docc The processes are arranged in a ring. Data will be passed in anC anti-clockwise direction around the ring.c dest = mod (nprocs+myrank-1, nprocs) src = mod (myrank+1, nprocs)cc Each process interacts with the particles from its nprocs/2-1c anti-clockwise neighbors. At the end of this loop p(i) in eachc process has accumulated the force from interactions with particlesc i+1, ...,nlocal-1 in its own process, plus all the particles from itsc nprocs/2-1 anti-clockwise neighbors. The "home" of the q array isC regarded as the process from which it originated. At the end ofc this loop q(i) has accumulated the force from interactions with C particles 0,...,i-1 in its home process, plus all the particles from theC nprocs/2-1 processes it has rotated to.c do k=0,nprocs/2-2 call mpi_sendrecv_replace (q, 7*nlocal, MPI_REAL, dest, 200, # src, 200, MPI_COMM_WORLD, status, ierr) do i=0,nlocal-1 do j=0,nlocal-1 dx(i) = p(PX,i) - q(PX,j) dy(i) = p(PY,i) - q(PY,j) dz(i) = p(PZ,i) - q(PZ,j) sq(i) = dx(i)**2+dy(i)**2+dz(i)**2 dist(i) = sqrt(sq(i)) fac(i) = p(MM,i) * q(MM,j) / (dist(i) * sq(i)) tx(i) = fac(i) * dx(i) ty(i) = fac(i) * dy(i) tz(i) = fac(i) * dz(i) p(FX,i) = p(FX,i)-tx(i) q(FX,j) = q(FX,j)+tx(i) p(FY,i) = p(FY,i)-ty(i) q(FY,j) = q(FY,j)+ty(i) p(FZ,i) = p(FZ,i)-tz(i) q(FZ,j) = q(FZ,j)+tz(i) end do end do end docc Now q is rotated once more so it is diametrically opposite its homec process. p(i) accumulates forces from the interaction with particlesc 0,..,i-1 from its opposing process. q(i) accumulates force from thec interaction of its home particles with particles i+1,...,nlocal-1 inc its current location.c if (nprocs .gt. 1) then
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (70 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
call mpi_sendrecv_replace (q, 7*nlocal, MPI_REAL, dest, 300, # src, 300, MPI_COMM_WORLD, status, ierr) do i=nlocal-1,0,-1 do j=i-1,0,-1 dx(i) = p(PX,i) - q(PX,j) dy(i) = p(PY,i) - q(PY,j) dz(i) = p(PZ,i) - q(PZ,j) sq(i) = dx(i)**2+dy(i)**2+dz(i)**2 dist(i) = sqrt(sq(i)) fac(i) = p(MM,i) * q(MM,j) / (dist(i) * sq(i)) tx(i) = fac(i) * dx(i) ty(i) = fac(i) * dy(i) tz(i) = fac(i) * dz(i) p(FX,i) = p(FX,i)-tx(i) q(FX,j) = q(FX,j)+tx(i) p(FY,i) = p(FY,i)-ty(i) q(FY,j) = q(FY,j)+ty(i) p(FZ,i) = p(FZ,i)-tz(i) q(FZ,j) = q(FZ,j)+tz(i) end do end docc In half the processes we include the interaction of each particle withc the corresponding particle in the opposing process.c if (myrank .lt. nprocs/2) then do i=0,nlocal-1 dx(i) = p(PX,i) - q(PX,i) dy(i) = p(PY,i) - q(PY,i) dz(i) = p(PZ,i) - q(PZ,i) sq(i) = dx(i)**2+dy(i)**2+dz(i)**2 dist(i) = sqrt(sq(i)) fac(i) = p(MM,i) * q(MM,i) / (dist(i) * sq(i)) tx(i) = fac(i) * dx(i) ty(i) = fac(i) * dy(i) tz(i) = fac(i) * dz(i) p(FX,i) = p(FX,i)-tx(i) q(FX,i) = q(FX,i)+tx(i) p(FY,i) = p(FY,i)-ty(i) q(FY,i) = q(FY,i)+ty(i) p(FZ,i) = p(FZ,i)-tz(i) q(FZ,i) = q(FZ,i)+tz(i) end do endifcc Now the q array is returned to its home process.c dest = mod (nprocs+myrank-nprocs/2, nprocs) src = mod (myrank+nprocs/2, nprocs) call mpi_sendrecv_replace (q, 7*nlocal, MPI_REAL, dest, 400, # src, 400, MPI_COMM_WORLD, status, ierr) end if
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (71 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
cc The p and q arrays are summed to give the total force on each particle.c do i=0,nlocal-1 p(FX,i) = p(FX,i) + q(FX,i) p(FY,i) = p(FY,i) + q(FY,i) p(FZ,i) = p(FZ,i) + q(FZ,i) end docc Stop clock and write out timingsc timeend = mpi_wtime () print *,'Node', myrank,' Elapsed time: ', # timeend-timebegin,' seconds'cc Do a barrier to make sure the timings are written out firstc call mpi_barrier (MPI_COMM_WORLD, ierr)cc Each process returns its forces to the pseudohost which prints them out.c if (myrank .eq. pseudohost) then open (7,file='nbody.output') write (7,100) (p(FX,i),p(FY,i),p(FZ,i),i=0,nlocal-1) call mpi_type_vector (nlocal, 3, 7, MPI_REAL, newtype, ierr) call mpi_type_commit (newtype, ierr) do k=0,nprocs-1 if (k .ne. pseudohost) then call mpi_recv (q(FX,0), 1, newtype, # k, 100, MPI_COMM_WORLD, status, ierr) write (7,100) (q(FX,i),q(FY,i),q(FZ,i),i=0,nlocal-1) end if end do else call mpi_type_vector (nlocal, 3, 7, MPI_REAL, newtype, ierr) call mpi_type_commit (newtype, ierr) call mpi_send (p(FX,0), 1, newtype, # pseudohost, 100, MPI_COMM_WORLD, ierr) end if cc Close MPIc 999 call mpi_finalize (ierr) stop 100 format(3e15.6) c
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (72 of 73)2005-11-22 오후 4:09:45

물리학자를 위한 MPI
c Abort if no input filec 998 print *,'input file nbody.input missing or invalid' call mpi_abort (ierr) end
http://krissol.kriss.re.kr/~ihlee/compphys/mpi_phys.htm (73 of 73)2005-11-22 오후 4:09:45