editando espectros de rmn com o software mestrenova: u m...
TRANSCRIPT

Rev. Virtual Quim. |Vol 9| |No. 6| |2650-2672| 2650
Artigo
Editando Espectros de RMN com o Software MestReNova: Um
Guia Prático
Forezi, L. S. M.;a,
* Castelo-Branco, F. S.b
Rev. Virtual Quim., 2017, 9 (6), 2650-2672. Data de publicação na Web: 13 de novembro de 2017
http://rvq.sbq.org.br
Editing NMR Spectra with MestReNova Software: A Practical Guide
Abstract: The Nuclear Magnetic Resonance (NMR) is a technique that has many applications in
chemistry, biochemistry and medical sciences. Within the chemistry, the NMR spectra allow elucidation
and characterization from small molecules of synthetic or natural origin, to macromolecules and their
interactions with ligands. However, in many cases, the edition of the spectra is restricted to proprietary
software of workstations that are part of the NMR system, being performed by the operator of the
equipment, which undoubtedly can delay the performance of new analyzes, as well as generate spectra
that lacks some of the information that the user needs. Thus, independent editing by the users through
spectral editing software, which can be installed on their personal computers, is highly useful, which can
streamline the process, as well as allowing the user to extract all the relevant information of the
spectrum. Thus, this manuscript describes a practical guide to editing 1H NMR, COSY,
13C / APT, HSQC
and HMBC NMR spectra from FID files through MestReNova 6.0 software, which is one of the most used
in the world for this purpose. In addition to being intuitive and easy to operate, it offers a large variety
of tools that can help students, professors and researchers in their NMR analysis.
Keywords: Spectroscopic techniques; 1H-RMN,
13C-APT-RMN; HSQC; HMBC; FID.
Resumo
A Ressonância Magnética Nuclear (RMN) é uma técnica que apresenta inúmeras aplicações, seja na
química, bioquímica e ciências médicas. Dentro da química, os espectros de RMN possibilitam a
elucidação e caracterização desde pequenas moléculas de origem sintética ou natural, até
macromoléculas e suas interações com ligantes. No entanto, muitas vezes a edição dos espectros fica
restrita à softwares proprietários exclusivos das estações de trabalho integrantes do sistema de RMN,
sendo realizadas pelo próprio operador do equipamento, o que, sem dúvidas, pode atrasar a realização
de novas análises, bem como não gerar espectros com todas as informações que o usuário necessita.
Assim, a edição independente por parte do próprio usuário através de software de edição de espectros,
que podem ser instalados em seu próprio computador, se mostra altamente útil, podendo agilizar o
processo, além de permitir que o usuário extraia todas as informações que julgar pertinentes do
espectro. Desta forma, este manuscrito descreve um guia prático de edições de espectros de RMN de 1H, COSY,
13C/APT, HSQC e HMBC, a partir de arquivos FID, através do software MestReNova 6.0, que é
um dos mais usados no mundo para este propósito e que, além de ser intuitivo e de fácil operação,
apresenta inúmeras ferramentas que podem ajudar a alunos, professores e pesquisadores em suas
análises por RMN.
Palavras-chave: Técnicas espectroscópcas; 1H-RMN,
13C-APT-RMN; HSQC; HMBC; FID.
* Universidade Federal Fluminense, Departamento de Química Orgânica, Campus do Valonguinho, CEP 24020-150,
Niterói-RJ, Brasil.
DOI: 10.21577/1984-6835.20170155

Volume 9, Número 6
Revista Virtual de Química
ISSN 1984-6835
Novembro-Dezembro 2017
2651 Rev. Virtual Quim. |Vol 9| |No. 6| |2650-2672|
Editando Espectros de RMN com o Software MestReNova: um
Guia Prático
Luana S. M. Forezi,a,
* Frederico S. Castelo-Brancob,
* a Universidade Federal Fluminense, Departamento de Química Orgânica, Campus do
Valonguinho, CEP 24020-150, Niterói-RJ, Brasil.
b
Fundação Oswaldo Cruz – Fiocruz, Instituto de Tecnologia em Fármacos – Farmanguinhos,
Departamento de Síntese de Fármacos, Laboratório de Síntese 1, Manguinhos, 21041-250, Rio
de Janeiro-RJ, Brasil.
Recebido em 6 de novembro de 2017. Aceito para publicação em 12 de novembro de 2017
1. Introdução
2. Guia do Software MestreNova®
2.1. RMN de Hidrogênio (1H)
2.2. Espectroscopia de Correlação (COSY)
2.3. RMN de Carbono 13 (13
C)/APT
2.4. Espectroscopia Heteronuclear de Coerência de Quantum Simples (HSQC)
2.5. Espectroscopia hereronuclear de correlação entre múltiplas ligações (HMBC)
1. Introdução
A espectroscopia de Ressonância
Magnética Nuclear (RMN) é uma técnica
poderosa e versátil, largamente empregada
na solução de problemas químicos, físicos e
biológicos. Suas aplicações incluem: a
caracterização de substâncias orgânicas de
origem sintética ou natural, bem como a
determinação de sua estrutura
tridimensional, a localização dos sítios ativos
de biomoléculas ou de moléculas adsorvidas
na superfície de catalisadores e a
interpretação de seus respectivos
mecanismos de ação, entre muitas outras.1,2
A RMN passou por uma verdadeira revolução
nos últimos anos.
Com o advento da informática, a partir
dos anos 2000, a edição dos espectros de
RMN foi popularizada e tornou-se uma
ferramenta fundamental e indispensável para
a elucidação e apresentação dos espectros de
RMN nas teses, dissertações e publicações
científicas a partir de softwares executáveis
em computadores domésticos, não sendo
mais restritos as estações de trabalhos
anexas aos equipamentos de RMN.
Os aparelhos atuais de RMN utilizam a
técnica pulsada, na qual um campo
magnético é aplicado aos núcleos analisados,
através de pulsos de radiação de
radiofrequência, o que os excita. Ao voltar ao
estado fundamental, estes núcleos irradiam
energia, a qual é detectada. A soma da
energia irradiada por todos o núcleos é então

Forezi, L. S. M.; Castelo-Branco, F. S.
Rev. Virtual Quim. |Vol 9| |No. 6| |2650-2672| 2652
somada, gerando o decaimento de indução
livre (FID, do inglês Free Induction Decay).3
Desta forma, os softwares de edição de
espectros de RMN fazem o processamento
dos arquivos contendo o FID e, após a
transformada de Fourier (FT, do inglês
Fourier Transform) é gerado o espectro tal
qual conhecemos, e a edição poderá ser
realizada.
Assim, este artigo consiste em um guia
prático para utilização do software
MestReNova® versão 6.0 (2009) de edição de
espectros de RMN, visando a correta
utilização de suas ferramentas por
estudantes, professores e pesquisadores.
Este Software é amplamente usado em todo
mundo, sendo o mais conhecido e um dos
mais completos para este propósito, além de
ser intuitivo e de fácil operação.4 Assim, o
usuário poderá fazer a edição de seus
espectros de forma autônoma e
independente, visualizando com mais
detalhes as regiões de interesse do espectro,
bem como apresentar as informações da
forma mais pertinente, além de contar com
todas as facilidades que o programa oferece.
2. Guia do Software MestReNova®
2.1. RMN de Hidrogênio (1H)
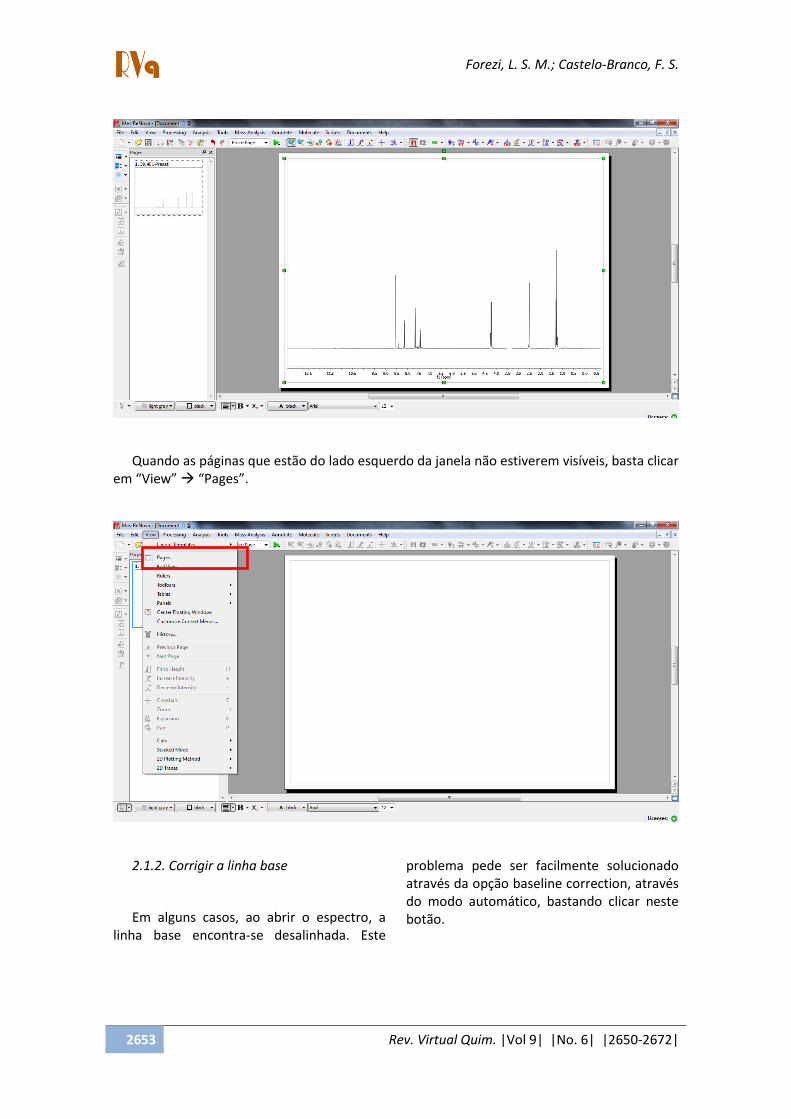
2.1.1. Abrir um FID
Para abrir um determinado FID, com o
MestreNova já aberto, segue-se os seguintes
passos:
- Clique em File Open Busque a
pasta que contém o FID da amostra a ser
analisar, dê um duplo clique no arquivo FID.
Se o arquivo estiver zipado, basta dar um
duplo click sobre ele.
(*) Mesmo que o FID esteja compactado
em arquivo zip, este também pode ser
arrastado até a janela já aberta e soltado.
Assim, aparecerá imediatamente o espectro.
- Aparecerá o espectro:
arrastar até essa janela
Forezi, L. S. M.; Castelo-Branco, F. S.
2653 Rev. Virtual Quim. |Vol 9| |No. 6| |2650-2672|
Quando as páginas que estão do lado esquerdo da janela não estiverem visíveis, basta clicar
em View Pages .
2.1.2. Corrigir a linha base
Em alguns casos, ao abrir o espectro, a
linha base encontra-se desalinhada. Este
problema pede ser facilmente solucionado
através da opção baseline correction, através
do modo automático, bastando clicar neste
botão.

Forezi, L. S. M.; Castelo-Branco, F. S.
Rev. Virtual Quim. |Vol 9| |No. 6| |2650-2672| 2654
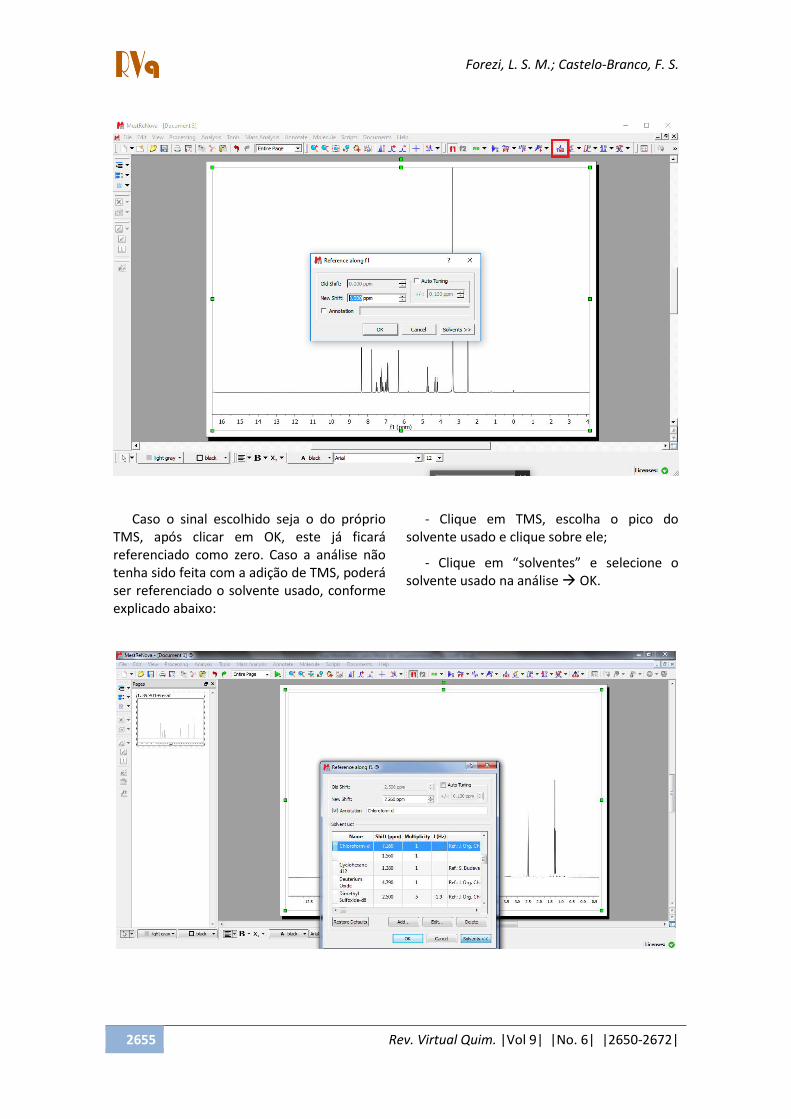
2.1.3. Referenciar o espectro
- Antes de iniciar a edição do espectro
deve-se fazer a referência dos sinais. Esta
poderá ser feita pelo tetrametilsilano (TMS),
comumente usado em RMN como a
referência do zero ppm, ou, na falta deste,
poderá ser referenciado o solvente utilizado.
- Clique no botão TMS e direcione o
cursor para o pico a ser referenciado e clique
sobre ele (o cursor estará com uma linha
vermelha vertical).
- Ao clicar aparecerá a seguinte imagem:
Forezi, L. S. M.; Castelo-Branco, F. S.
2655 Rev. Virtual Quim. |Vol 9| |No. 6| |2650-2672|
Caso o sinal escolhido seja o do próprio
TMS, após clicar em OK, este já ficará
referenciado como zero. Caso a análise não
tenha sido feita com a adição de TMS, poderá
ser referenciado o solvente usado, conforme
explicado abaixo:
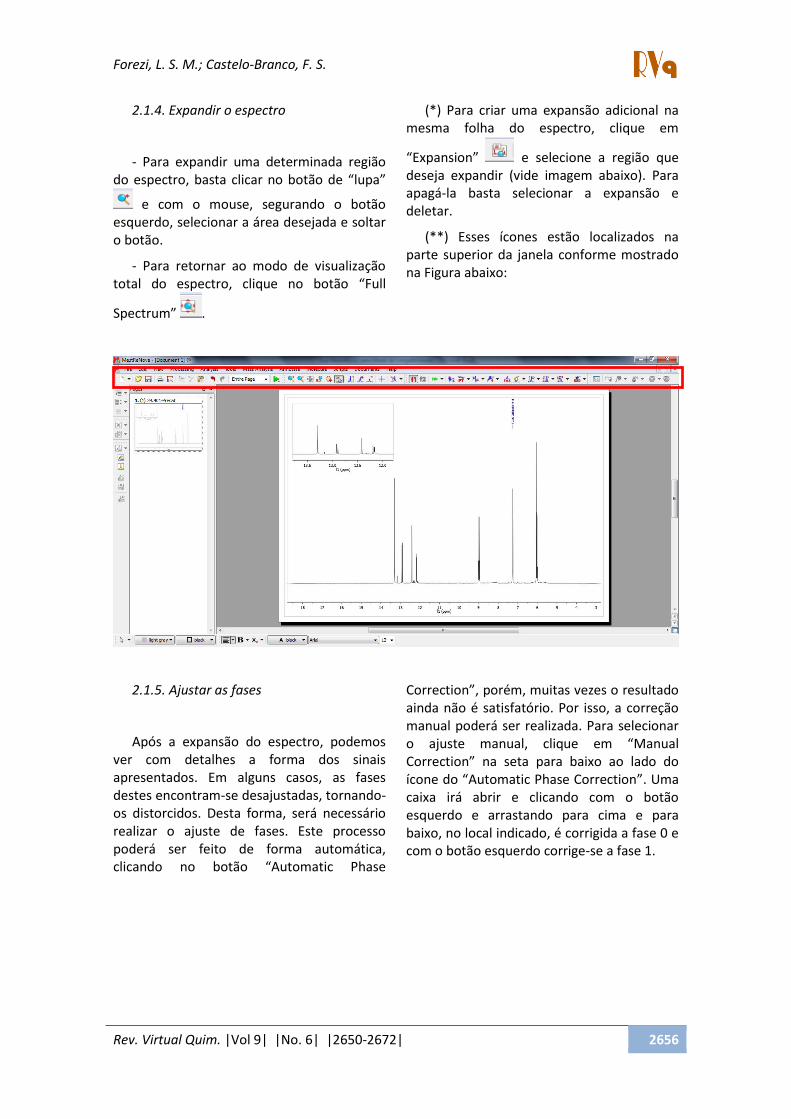
- Clique em TMS, escolha o pico do
solvente usado e clique sobre ele;
- Clique em solventes e selecione o
solvente usado na análise OK.
Forezi, L. S. M.; Castelo-Branco, F. S.
Rev. Virtual Quim. |Vol 9| |No. 6| |2650-2672| 2656
2.1.4. Expandir o espectro
- Para expandir uma determinada região
do espectro, basta clicar no botão de lupa
e com o mouse, segurando o botão
esquerdo, selecionar a área desejada e soltar
o botão.
- Para retornar ao modo de visualização
total do espectro, clique no botão Full
Spectrum .
(*) Para criar uma expansão adicional na
mesma folha do espectro, clique em
Expansion e selecione a região que
deseja expandir (vide imagem abaixo). Para
apagá-la basta selecionar a expansão e
deletar.
(**) Esses ícones estão localizados na
parte superior da janela conforme mostrado
na Figura abaixo:
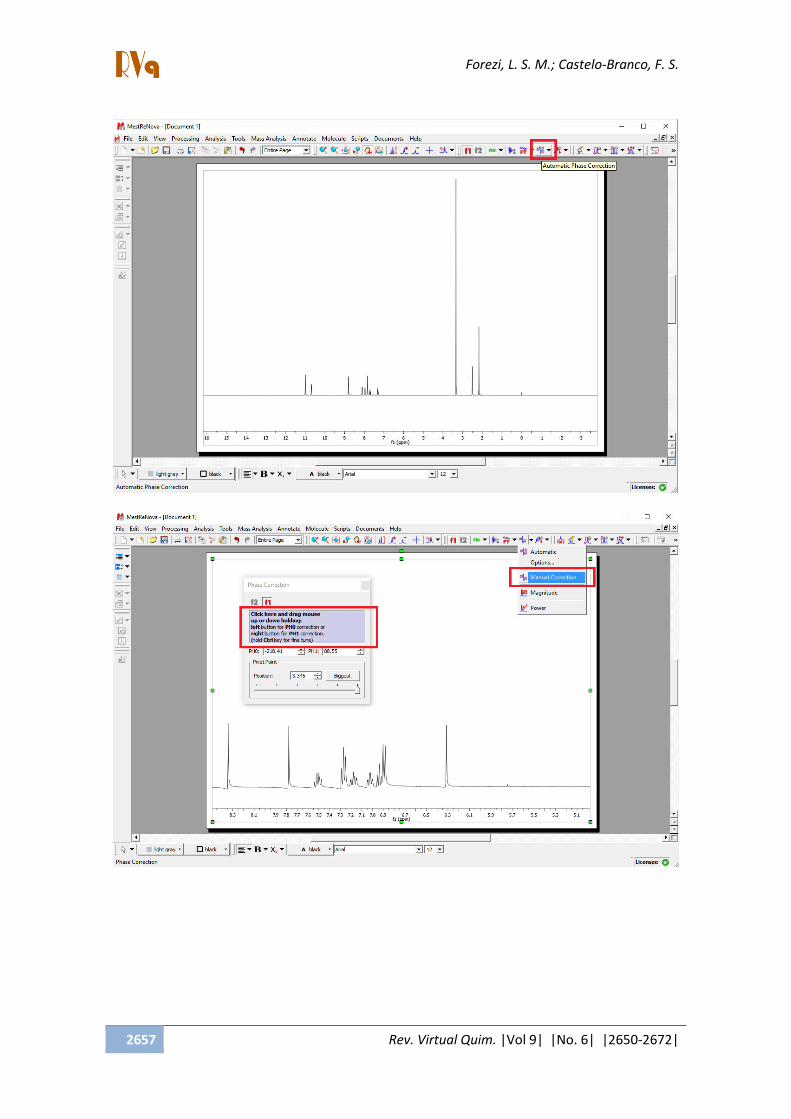
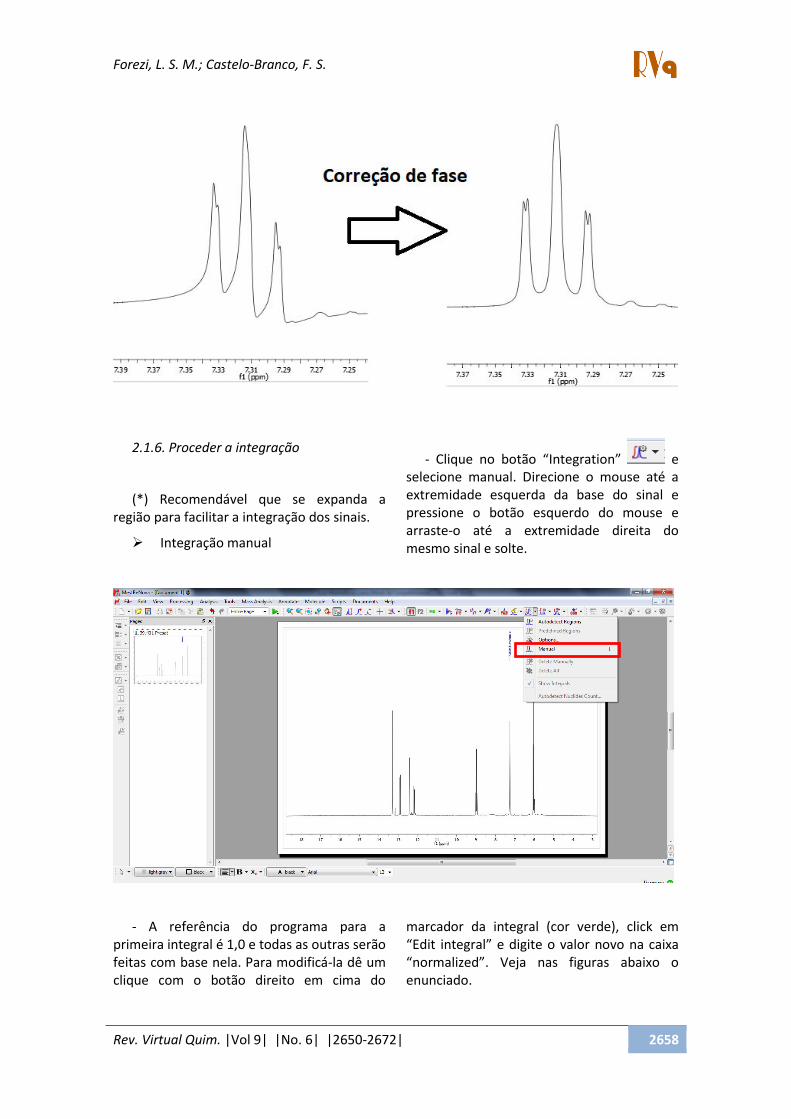
2.1.5. Ajustar as fases
Após a expansão do espectro, podemos
ver com detalhes a forma dos sinais
apresentados. Em alguns casos, as fases
destes encontram-se desajustadas, tornando-
os distorcidos. Desta forma, será necessário
realizar o ajuste de fases. Este processo
poderá ser feito de forma automática,
clicando no botão Automatic Phase
Correction , porém, muitas vezes o resultado
ainda não é satisfatório. Por isso, a correção
manual poderá ser realizada. Para selecionar
o ajuste manual, clique em Manual
Correction na seta para baixo ao lado do
ícone do Automatic Phase Correction . Uma
caixa irá abrir e clicando com o botão
esquerdo e arrastando para cima e para
baixo, no local indicado, é corrigida a fase 0 e
com o botão esquerdo corrige-se a fase 1.
Forezi, L. S. M.; Castelo-Branco, F. S.
2657 Rev. Virtual Quim. |Vol 9| |No. 6| |2650-2672|
Forezi, L. S. M.; Castelo-Branco, F. S.
Rev. Virtual Quim. |Vol 9| |No. 6| |2650-2672| 2658
2.1.6. Proceder a integração
(*) Recomendável que se expanda a
região para facilitar a integração dos sinais.
Integração manual
- Clique no botão Integration e
selecione manual. Direcione o mouse até a
extremidade esquerda da base do sinal e
pressione o botão esquerdo do mouse e
arraste-o até a extremidade direita do
mesmo sinal e solte.
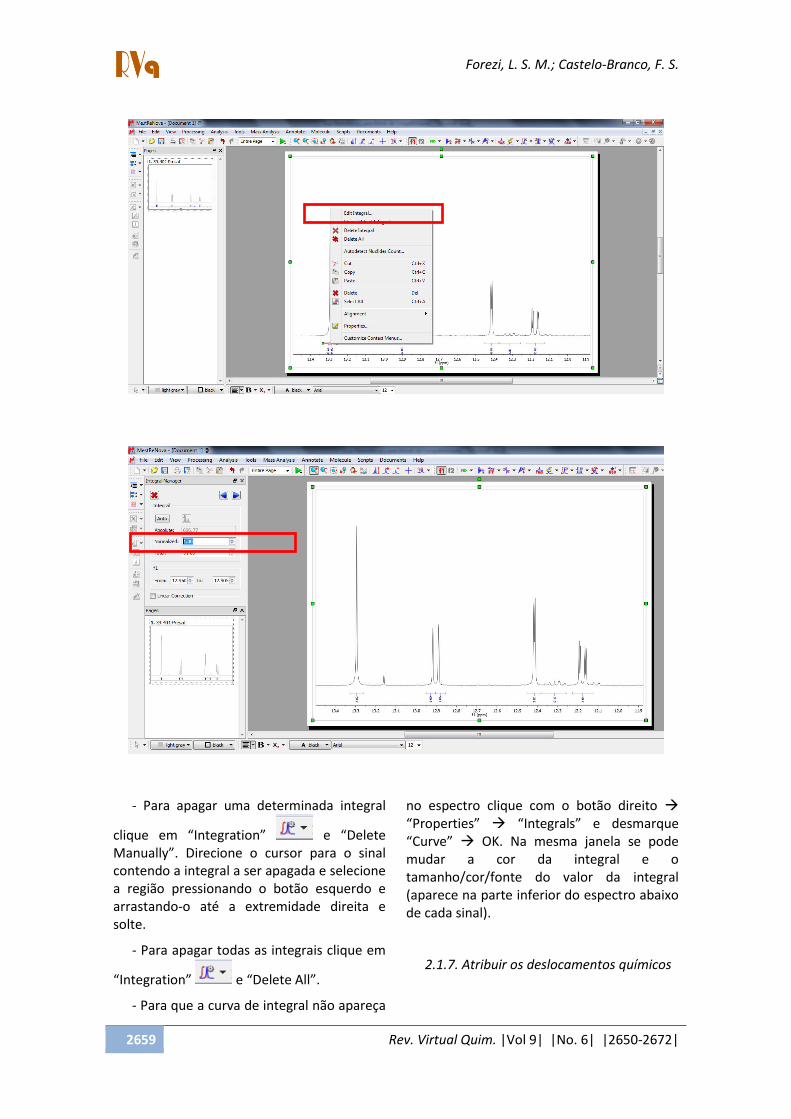
- A referência do programa para a
primeira integral é 1,0 e todas as outras serão
feitas com base nela. Para modificá-la dê um
clique com o botão direito em cima do
marcador da integral (cor verde), click em
Edit integral e digite o valor novo na caixa
normalized . Veja nas figuras abaixo o
enunciado.
Forezi, L. S. M.; Castelo-Branco, F. S.
2659 Rev. Virtual Quim. |Vol 9| |No. 6| |2650-2672|
- Para apagar uma determinada integral
clique em Integration e Delete
Manually . Direcione o cursor para o sinal
contendo a integral a ser apagada e selecione
a região pressionando o botão esquerdo e
arrastando-o até a extremidade direita e
solte.
- Para apagar todas as integrais clique em
Integration e Delete All .
- Para que a curva de integral não apareça
no espectro clique com o botão direito
Properties Integrals e desmarque
Curve OK. Na mesma janela se pode
mudar a cor da integral e o
tamanho/cor/fonte do valor da integral
(aparece na parte inferior do espectro abaixo
de cada sinal).
2.1.7. Atribuir os deslocamentos químicos

Forezi, L. S. M.; Castelo-Branco, F. S.
Rev. Virtual Quim. |Vol 9| |No. 6| |2650-2672| 2660
- Se desejar atribuir os valores em um pico
de cada vez clique em Peak Picking
Peak by peak . Clicar sobre cada pico e o
deslocamento aparecerá na parte superior.
Se desejar atribuir os deslocamentos de uma
única vez clique em Peak Picking
Manual Threshold . Com o botão esquerdo
pressionado, arraste o cursor até passar com
a linha horizontal vermelha por todos os
sinais desejados e solte. Esta é a opção mais
prática e que apresenta os melhores
resultados.
- Para aumentar ou diminuir as casas
decimais dos valores dos deslocamentos
químicos clique no espectro com o botão
direito Properties Peaks
Decimals .
2.1.8. Editar a intensidade dos picos
-Para aumentar ou diminuir a altura dos
picos, clique no ícone ou .
Também se pode utilizar a roda (scroll) do
mouse para frente (para aumentar a
intensidade) e para trás (para diminuir a
intensidade).
2.1.9. Mudar a aparência do espectro
- Clique no espectro com o botão direito
Properties . Lá se pode modificar as
cores do espectro, tamanho e fonte das
scales , integrals e peaks .
- Para retirar o quadriculado do espectro
clique em Properties Scales Grid
desmarque as caixas show horizontal e
show vertical OK.
(*) Show frame pode deixar marcado,
pois é a borda do espectro.
2.1.10. Melhorar a resolução e a
qualidade do espectro
Muitas vezes, dada a resolução do
espectrômetro de RMN, os sinais têm
multiplicidade difícil de se determinar. Em
alguns casos, as técnicas de processamento
do MestreNova podem melhorar a resolução
dos sinais, auxiliando a sua atribuição.
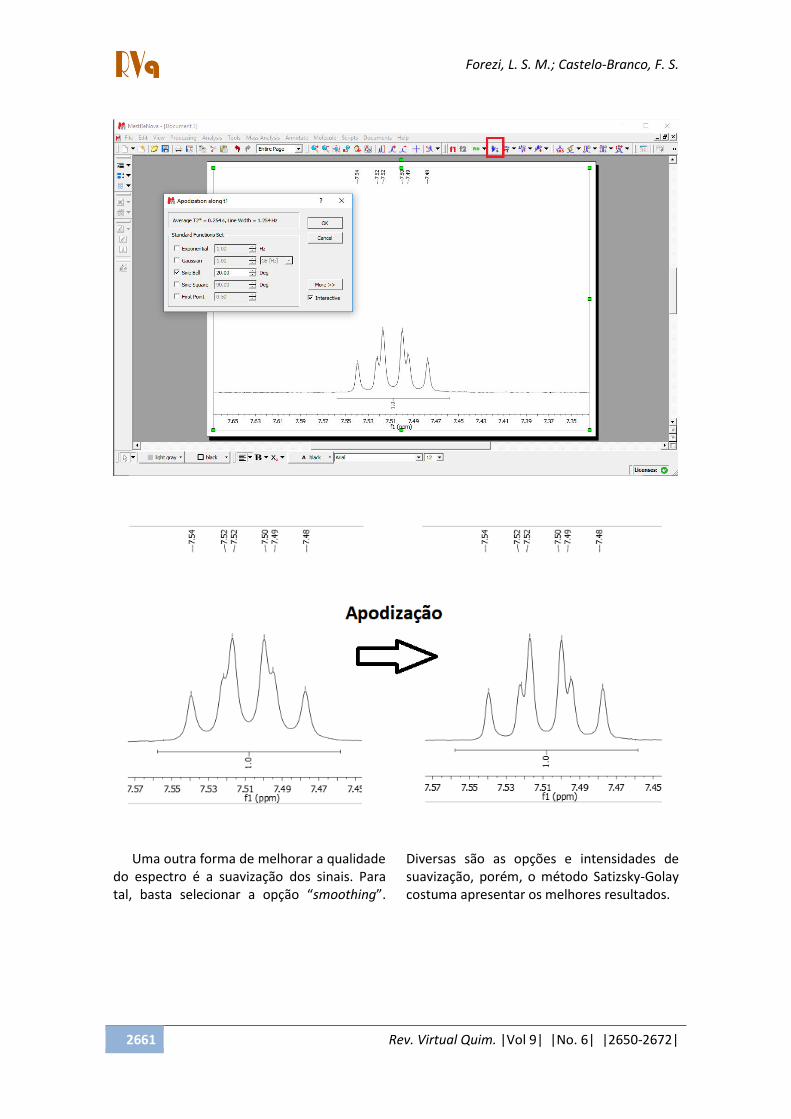
Uma das técnicas é a apodização, que
pode ser aplicada por diferentes métodos.
Para selecioná-la, basta clicar em
Apodization e, em seguida, selecionar um
dos diferentes métodos. De forma geral, o
método sine bell costuma apresentar bons
resultados e, após aplicação, necessita de
reajuste na intensidade do sinal.
No exemplo a seguir, após a apodização,
pode-se notar a multiplicidade de forma mais
fácil do que antes da técnica, quando o sinal,
que é um tripleto de dupletos, poderia ser
confundido com um quarteto.
Forezi, L. S. M.; Castelo-Branco, F. S.
2661 Rev. Virtual Quim. |Vol 9| |No. 6| |2650-2672|
Uma outra forma de melhorar a qualidade
do espectro é a suavização dos sinais. Para
tal, basta selecionar a opção smoothing .
Diversas são as opções e intensidades de
suavização, porém, o método Satizsky-Golay
costuma apresentar os melhores resultados.

Forezi, L. S. M.; Castelo-Branco, F. S.
Rev. Virtual Quim. |Vol 9| |No. 6| |2650-2672| 2662
2.1.11. Determinar a constante de
acoplamento (J) e análise de multipletos
- Para se medir a constante de
acoplamento clique em Multiplets Analysis
Manual . Direcione o cursor para o sinal
desejado e selecione a região pressionando o
botão esquerdo e arrastando-o até a
extremidade direita e solte. Faça o mesmo
processo para todos os sinais que deseja
determinar a constante de acoplamento J .
Na parte superior de cada sinal aparecerá
uma pequena caixa com o valor do
deslocamento químico do sinal e sua
multiplicidade.

Forezi, L. S. M.; Castelo-Branco, F. S.
2663 Rev. Virtual Quim. |Vol 9| |No. 6| |2650-2672|
- Após esse passo clique em Multiplets
Analysis report . Os valores dos J
aparecerão para cada sinal, em texto colado
no próprio espectro. Os valores de J já são
apresentados em Hz. No report da análise de
multipletos também é reportada a frequência
do aparelho utilizado na análise, o número de
hidrogênios, os valores dos deslocamentos
químicos, bem como a multiplicidade de cada
sinal, além de discriminar o solvente
empregado na análise.
Uma opção ainda mais útil é a de poder
copiar o texto da análise de multipletos e
colá-lo diretamente em um documento, tal
como um relatório, artigo ou tese. Para tal,
basta fazer a análise de multipletos para
todos os sinais e em seguida clicar em

Forezi, L. S. M.; Castelo-Branco, F. S.
Rev. Virtual Quim. |Vol 9| |No. 6| |2650-2672| 2664
Multiplets Analysis copy . O texto com
todas as informações do espectro poderá ser
colado onde o usuário desejar.
2.1.12. Retirar o texto do espectro
- Para retirar a caixa de texto com as
informações da análise, clique com o botão
direito Properties Spectrum
clique em Common e desmarque a caixa
title OK. A caixa fica localizada no canto
superior esquerdo.
2.1.13. Simular um espectro teórico
- Desenhe a estrutura desejada no
ChemDraw copie e cole-a em uma nova
página do MestreNova selecione a
estrutura e clique em Molecule Predict 1H Spectrum .
Em Prediction Options pode-se mudar
diversos parêmetros de predição, tal como a
frequência do aparelho simulado (300 MHz,
500 Mhz etc.) e o solvente.
Vale ressaltar que o espectro simulado
poderá ser editado da mesma forma que um
espectro experimental (real).
Forezi, L. S. M.; Castelo-Branco, F. S.
2665 Rev. Virtual Quim. |Vol 9| |No. 6| |2650-2672|
(*) Para criar uma nova página em branco
clique em Edit Create new page .
Também pode-se usar o atalho ctrl+M .
2.1.14. Colar o espectro
Para colar um espectro no Word,
PowerPoint etc, clique uma vez no espectro
com o botão direito, copie e em seguida cole
no lugar de destino.
2.1.15. Adicionar figuras e estruturas ao
espectro
- Para adicionar figuras ou estruturas ao
espectro basta copiar e colar no espectro.
(*) Recomenda-se copiar o espectro do
MestreNova e colar o mesmo no arquivo
Word desejado e em seguida anexar a
estrutura sobre a imagem em uma caixa de
texto.
2.2. Espectroscopia de Correlação (COSY)
2.2.1. Abrir um FID de COSY
- Clique em File Open Busque a
pasta que contém o FID que deseja analisar,
dê um duplo clique no arquivo FID. Se o
arquivo estiver zipado, basta dar um duplo
click sobre ele.
(*) Repare que a pasta é a cosy.fid e não 1H.fid.
(**) Se o FID estiver zipada também pode-
se arrastar até a janela já aberta e soltar.
Assim aparecerá imediatamente o espectro
COSY.
- O espectro COSY aparecerá da seguinte
forma:
Forezi, L. S. M.; Castelo-Branco, F. S.
Rev. Virtual Quim. |Vol 9| |No. 6| |2650-2672| 2666
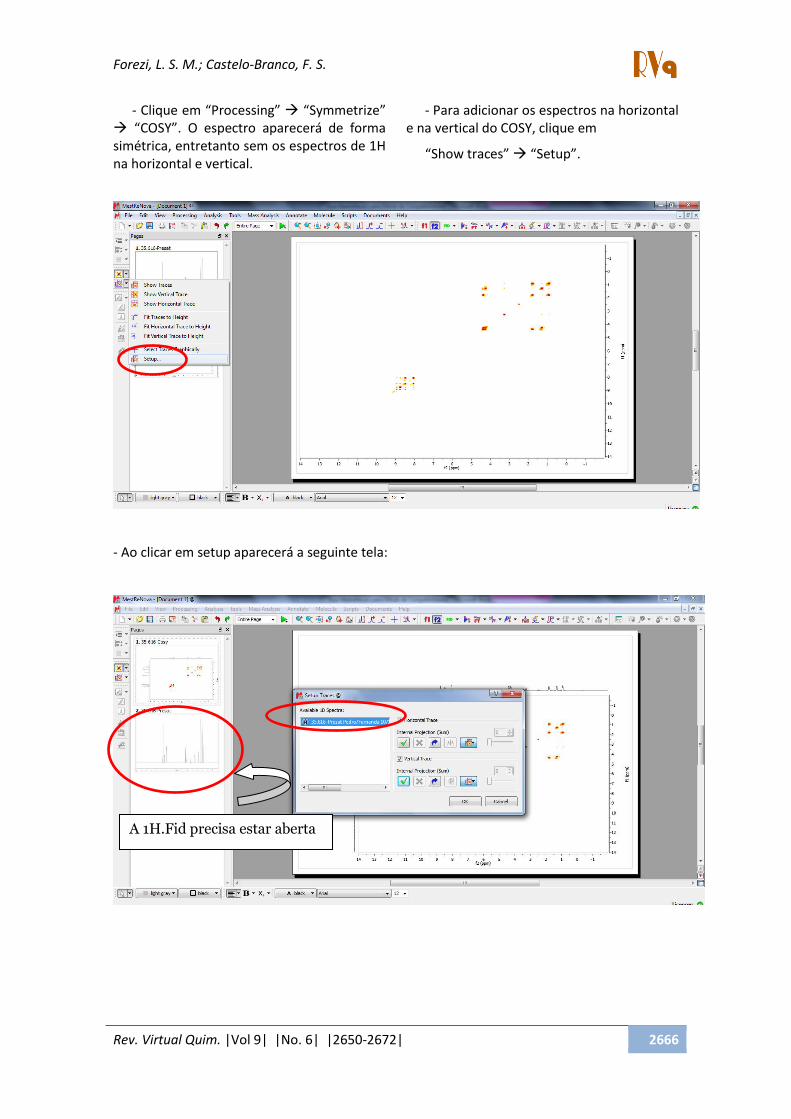
- Clique em Processing Symmetrize
COSY . O espectro aparecerá de forma
simétrica, entretanto sem os espectros de 1H
na horizontal e vertical.
- Para adicionar os espectros na horizontal
e na vertical do COSY, clique em
Show traces Setup .
- Ao clicar em setup aparecerá a seguinte tela:
A 1H.Fid precisa estar aberta
Forezi, L. S. M.; Castelo-Branco, F. S.
2667 Rev. Virtual Quim. |Vol 9| |No. 6| |2650-2672|
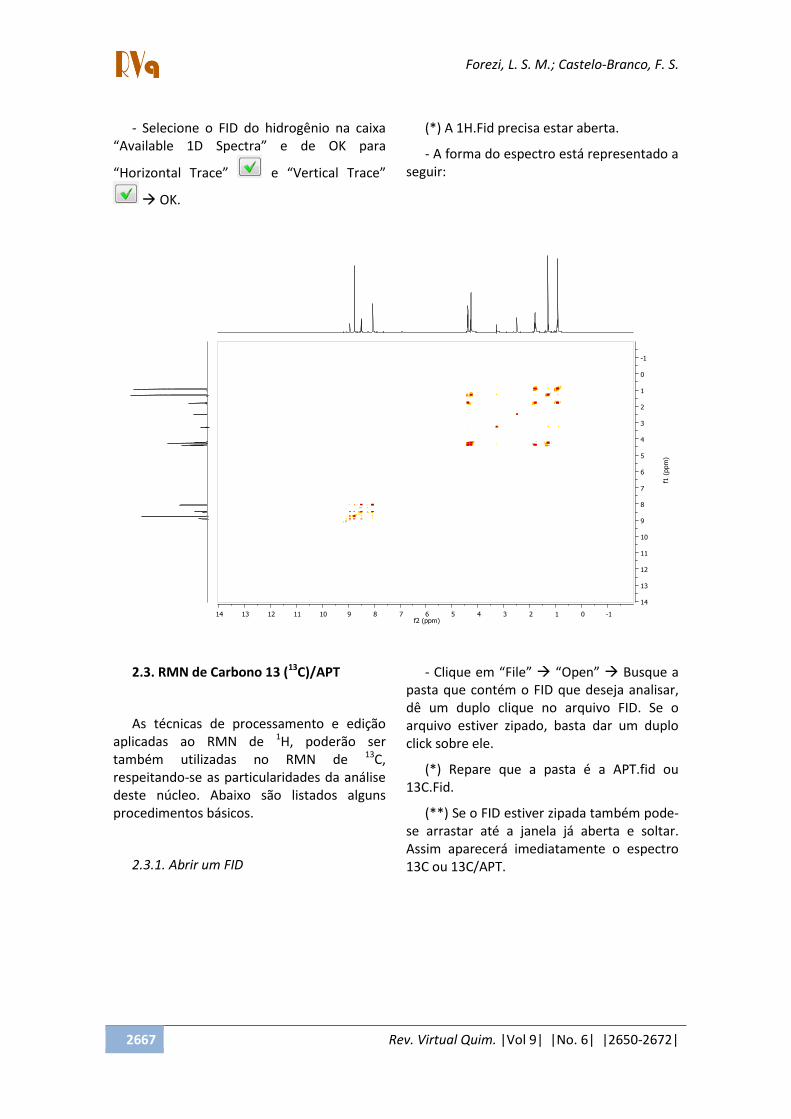
- Selecione o FID do hidrogênio na caixa
Available 1D Spectra e de OK para
Horizontal Trace e Vertical Trace
OK.
(*) A 1H.Fid precisa estar aberta.
- A forma do espectro está representado a
seguir:
2.3. RMN de Carbono 13 (13
C)/APT
As técnicas de processamento e edição
aplicadas ao RMN de 1H, poderão ser
também utilizadas no RMN de 13
C,
respeitando-se as particularidades da análise
deste núcleo. Abaixo são listados alguns
procedimentos básicos.
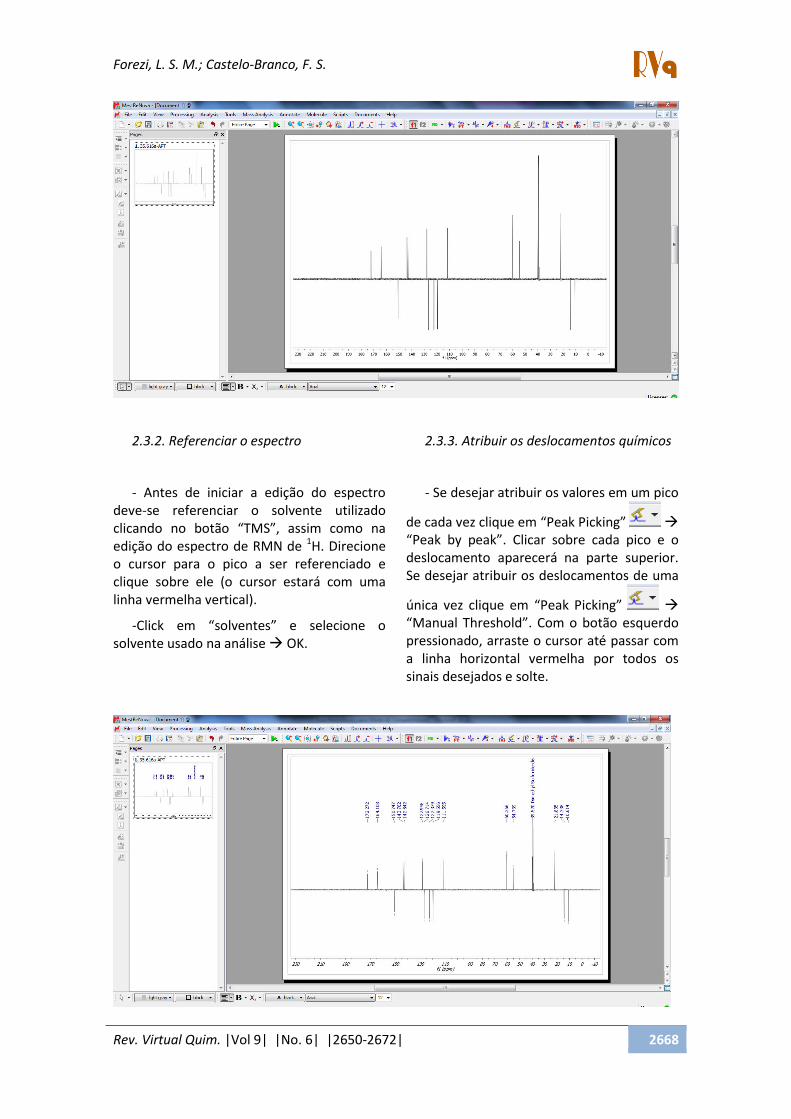
2.3.1. Abrir um FID
- Clique em File Open Busque a
pasta que contém o FID que deseja analisar,
dê um duplo clique no arquivo FID. Se o
arquivo estiver zipado, basta dar um duplo
click sobre ele.
(*) Repare que a pasta é a APT.fid ou
13C.Fid.
(**) Se o FID estiver zipada também pode-
se arrastar até a janela já aberta e soltar.
Assim aparecerá imediatamente o espectro
13C ou 13C/APT.
Forezi, L. S. M.; Castelo-Branco, F. S.
Rev. Virtual Quim. |Vol 9| |No. 6| |2650-2672| 2668
2.3.2. Referenciar o espectro
- Antes de iniciar a edição do espectro
deve-se referenciar o solvente utilizado
clicando no botão TMS , assim como na
edição do espectro de RMN de 1H. Direcione
o cursor para o pico a ser referenciado e
clique sobre ele (o cursor estará com uma
linha vermelha vertical).
-Click em solventes e selecione o
solvente usado na análise OK.
2.3.3. Atribuir os deslocamentos químicos
- Se desejar atribuir os valores em um pico
de cada vez clique em Peak Picking
Peak by peak . Clicar sobre cada pico e o
deslocamento aparecerá na parte superior.
Se desejar atribuir os deslocamentos de uma
única vez clique em Peak Picking
Manual Threshold . Com o botão esquerdo
pressionado, arraste o cursor até passar com
a linha horizontal vermelha por todos os
sinais desejados e solte.

Forezi, L. S. M.; Castelo-Branco, F. S.
2669 Rev. Virtual Quim. |Vol 9| |No. 6| |2650-2672|
2.3.4. Simular um espectro teórico
- Desenhe no ChemDraw copie a
estrutura e cole em uma nova pagina do
MestreNova selecione a estrutura e clique
em Molecule Predict 13
C Spectrum .
2.4. Espectroscopia Heteronuclear de
Coerência de Quantum Simples (HSQC)
Esta técnica correlaciona dois espectros
com núcleos distintos, tal como 1H e
13C,
conforme mostrado no exemplo a seguir.
2.4.1. Abrir um FID de HSQC
- Clique em File Open Busque a
pasta que contém o FID que deseja analisar,
dê um duplo clique no arquivo fid. Se o
arquivo estiver zipado, basta dar um duplo
click sobre ele.
(*)Repare que a pasta é a HSQC.fid.
(**) Se o FID estiver zipada também pode-
se arrastar até a janela já aberta e soltar.
Assim aparecerá imediatamente o espectro
do HSQC.
- Para adicionar os espectros na horizontal
e na vertical do HSQC, clique em
Show traces Setup Selecione o
FID do hidrogênio na caixa Available 1D
Spectra e de OK para Horizontal Trace
e selecione o FID do carbono na caixa
Available 1D Spectra e de OK Vertical
Trace OK.
(*) As 1H.Fid e 13C.Fid (ou APT.Fid)
precisam estar abertas.
- A forma do espectro de HSQC está
representada a seguir:
Forezi, L. S. M.; Castelo-Branco, F. S.
Rev. Virtual Quim. |Vol 9| |No. 6| |2650-2672| 2670
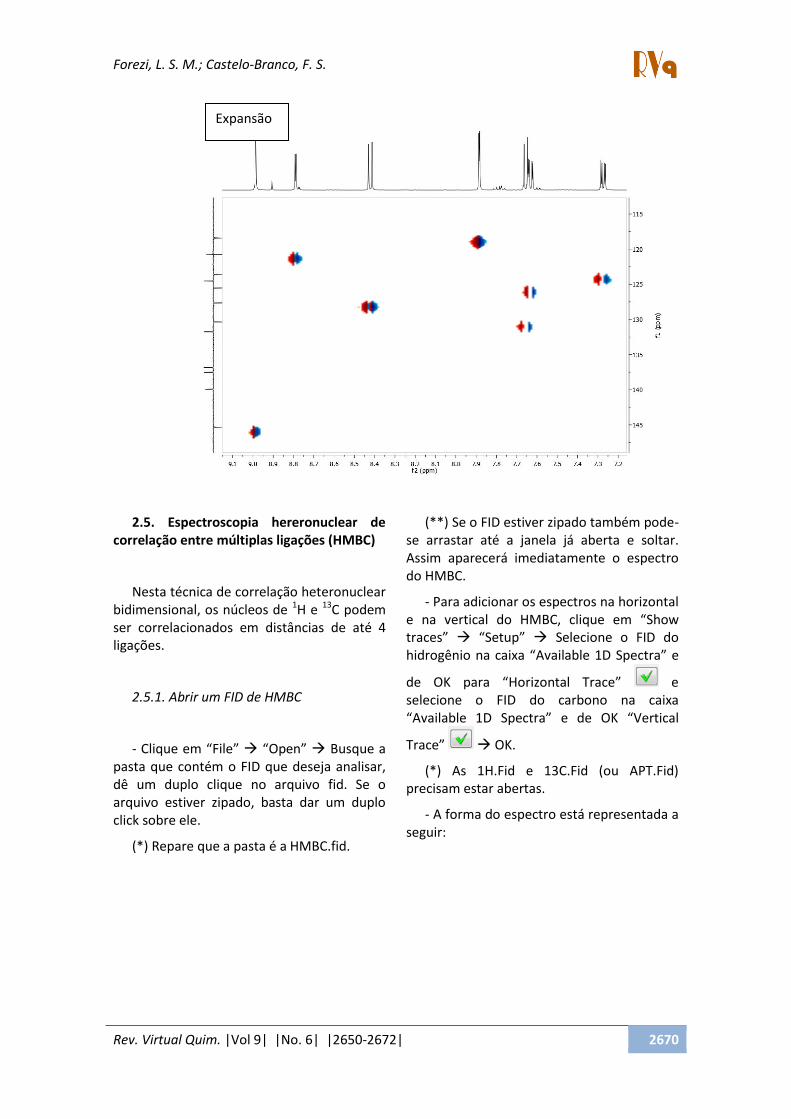
2.5. Espectroscopia hereronuclear de
correlação entre múltiplas ligações (HMBC)
Nesta técnica de correlação heteronuclear
bidimensional, os núcleos de 1H e
13C podem
ser correlacionados em distâncias de até 4
ligações.
2.5.1. Abrir um FID de HMBC
- Clique em File Open Busque a
pasta que contém o FID que deseja analisar,
dê um duplo clique no arquivo fid. Se o
arquivo estiver zipado, basta dar um duplo
click sobre ele.
(*) Repare que a pasta é a HMBC.fid.
(**) Se o FID estiver zipado também pode-
se arrastar até a janela já aberta e soltar.
Assim aparecerá imediatamente o espectro
do HMBC.
- Para adicionar os espectros na horizontal
e na vertical do HMBC, clique em Show
traces Setup Selecione o FID do
hidrogênio na caixa Available 1D Spectra e
de OK para Horizontal Trace e
selecione o FID do carbono na caixa
Available 1D Spectra e de OK Vertical
Trace OK.
(*) As 1H.Fid e 13C.Fid (ou APT.Fid)
precisam estar abertas.
- A forma do espectro está representada a
seguir:
Expansão
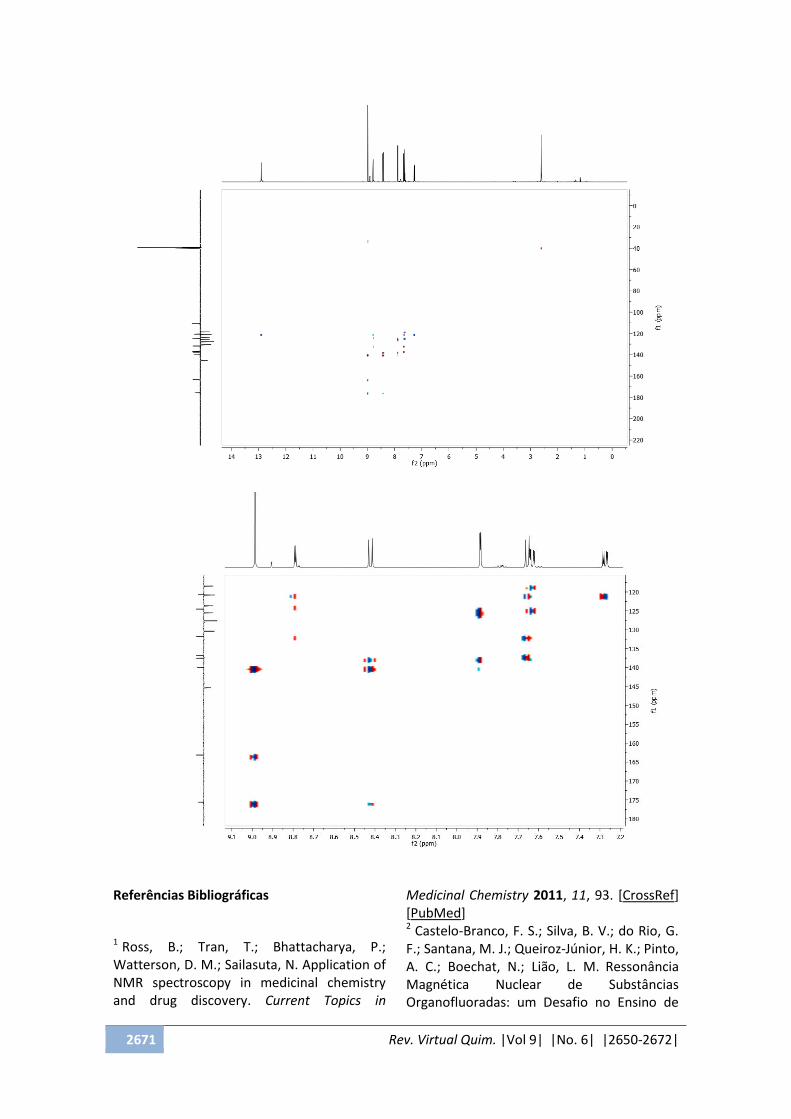
2671 Rev. Virtual Quim. |Vol 9| |No. 6| |2650-2672|
Referências Bibliográficas
1 Ross, B.; Tran, T.; Bhattacharya, P.;
Watterson, D. M.; Sailasuta, N. Application of
NMR spectroscopy in medicinal chemistry
and drug discovery. Current Topics in
Medicinal Chemistry 2011, 11, 93. [CrossRef]
[PubMed] 2 Castelo-Branco, F. S.; Silva, B. V.; do Rio, G.
F.; Santana, M. J.; Queiroz-Júnior, H. K.; Pinto,
A. C.; Boechat, N.; Lião, L. M. Ressonância
Magnética Nuclear de Substâncias
Organofluoradas: um Desafio no Ensino de

Rev. Virtual Quim. |Vol 9| |No. 6| |2650-2672| 2672
Espectroscopia. Química Nova 2015, 38,
1237. [CrossRef] 3 Silverstein, R. M.; Webster, F. X.; Kiemle, D.
J.; Identificação espectrofotométrica de
compostos orgânicos, 7a. ed., LTC: Rio de
Janeiro, 2006 4 Willcott, M. R. MestRe Nova. Journal of The
American Chemical Society 2009, 131, 13180.
[CrossRef]