dspace cover page - research collection28006/eth-28006-02.pdf · doctoral thesisethno. 15998...
TRANSCRIPT

Research Collection
Doctoral Thesis
Plasma-induced graft polymerization of organophosphorusmonomers: a novel approach to flame retard polyacrylonitrileand cotton textiles
Author(s): Tsafack, Marie Jérôme
Publication Date: 2005
Permanent Link: https://doi.org/10.3929/ethz-a-005012411
Rights / License: In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For moreinformation please consult the Terms of use.
ETH Library

Doctoral Thesis ETHNo. 15998
Plasma-Induced Graft Polymerization of Organophosphorus Monomers:
A Novel Approach to Flame Retard Polyacrylonitrile and Cotton Textiles
A dissertation submitted to the
SWISS FEDERAL INSTITUTE OF TECHNOLOGY ZURICH
for the degree of
Doctor of Sciences
presented by
Marie Jérôme Tsafack
DEA, University of Yaounde, Cameroon
born 09.07.1973
citizen of Cameroon
accepted on the recommendation of
Prof. Dr. H. Grützmacher, examiner
Prof. Dr. J. Levalois-Grützmacher, co-examiner
Prof. Dr. R. De Jaeger, co-examiner
Zurich 2005

Parts of this work were published:
1) M.J. Tsafack, F. Hochart, J. Levalois-Grützmacher, Polymerization and surface
modification by low pressure plasma technique, Eur. Phys. J. A.ppl. Phys. 26 (2004) 215.
2) M.J. Tsafack, J. Levalois-Grützmacher, Plasma-induced graft-polymerization offlame
retardant monomers onto PANfabrics, Surf. Coat. Technol. in press.
3) M.J. Tsafack, J. Levalois-Grützmacher, Flame retardancy of polyacrylonitrile fabrics
treated with (meth)acrylate phosphates and phosphonates by application of a low pressure
plasma process, presented at FRPM 03 (Fire Retardancy and Protection of Materials) Lille,
September 17-19th 2003.
4) M.J. Tsafack, J. Levalois-Grützmacher, Flame retardancy of cotton fabrics by plasma-
induced graft-polymerization of phosphorus containing monomers, presented at the 16l
th
International Conference on Phosphorus Chemistry, Birmingham, July 4-9 2004.

Remerciements
Les travaux de recherche présentés dans ce mémoire de thèse ont été effectués à
l'Institut de Chimie Inorganique à l'Ecole Polytechnique de Zurich sous la direction du
Professeur Docteur Hansjörg Grützmacher. Je voudrais tout d'abord lui adresser mes plus
sincères remerciements ainsi que toute ma reconnaissance pour m'avoir accueillir au sein de
son équipe et pour m'avoir offert la possibilité de faire une thèse de doctorat.
Je tiens à exprimer ma profonde gratitude à Madame Joëlle Levalois-Grützmacher,
Professeur à 1' Université des Antilles et de la Guyane, dont la contribution à l'élaboration de
cette thèse est indéniable. Je la remercie très sincèrement de m'avoir fait profiter de son
expérience et de ses compétences scientifiques. Et aussi pour ses conseils et sa constante
disponibilité.
J'ai été très honorée de la présence de Monsieur Roger DeJaeger, Professeur à
l'Université des Sciences et Techniques de Lille, que je remercie de s'être intéressé à ce
travail et d'avoir accepté d'être membre du jury.
J'adresse également mes remerciements au Dr. Christian Mensing pour les analyses
thermogravimétriques des polymères et des tissus.
Un grand merci au Dr. Frank Krumeich pour son aide précieuse en MEB.
Je tiens à remercier le Dr. Hartmut Schönberg pour les photos.
Je tiens à remercier Joe et Dominique pour avoir participer aux corrections de ce
mémoire.
Je voudrais également remercier tous les membres du groupe et de l'institut qui m'ont
accompagné pendant ce doctorat. En particulier mes compagnons du Labo 136 Urs, Catherine
et Fatou, et Betta, Francesca et Karin avec qui j'ai partagé mes repas de midi.

Table of content
Résumé
Summary
Chapter I: Introduction
1 Mechanism of flame retardancy
1.1 Combustion process
1.2 Mode ofaction offlame retardants
1.2.1 Physical action
1.2.2 Chemical action
2 Important flame retardants
2.1 Halogen-containingflame retardants
2.2 Phosphorusflame retardants
2.3 Nitrogen-containingflame retardants
2.4 Inorganicflame retardants
2.4.1 Metal hydroxides
2.4.2 boron-containing compounds
3 Different approaches to reduce polymer flammabiiity
4 Cold plasma technique
4.1 Definition ofplasma
4.2 Principle ofcoldplasma technique
4.3 Interaction ofcoldplasma with a polymer surface
4.3.1 Ablation or etching
4.3.2 Crosslinking
4.3.3 Activation
4.3.4 Polymerization
4.3.5 Plasma grafting
4.4 Applications ofcoldplasma technique
4.5 Advantages and disadvantages ofcoldplasma techniq

Table ofcontent
5 Flame retardant testing methods 15
5.1 Underwriters Laboratories (UL) 94 15
5.2 Limiting oxygen index (LOI) 16
5.3 Heat release tests (cone calorimeter) 16
6 Flame retardancy of polymeric materials by cold plasma technique 16
6.1 Plasma polymerization of volatile compounds 17
6.2 Grafting ofnon-volatile monomers andpolymers 17
7 Characterization of the grafted copolymers 18
7.1 Infrared attenuated reflection spectroscopy (IR (ATR)) 18
7.2 X-ray photoelectron spectroscopy (XPS) 18
7.3 Scanning electron microscopy (SEM) 18
7.4 Thermal analysis 19
8 Objectives of the thesis 19
9 References 21
Chapter II: Synthesis and argon plasma-induced polymerization 25
of acrylic monomers containing phosphorus
1 Introduction 25
2 Synthesis of acrylic monomers containing phosphorus 25
2.1 Synthesis ofaerylate phosphates 25
2.2 Synthesis ofacrylate phosphonates 26
2.3 Synthesis ofnew acrylate phosphoramidates 27
2.3.1 Synthesis Diethyl (acryloyloxy) ethylphosphoramidate 27
2.3.2 Synthesis Acryloyloxy-1,3-Bis(diethylphosphoramidate)-propan 28
3 Polymerization of acrylic monomers containing phosphorus 29
3.1 Principle ofplasma-state polymerization 29
3.2 Principle ofplasma-inducedpolymerization 30
3.3 Argon plasma-inducedpolymerization ofacrylic monomers containing 32
phosphorus
3.3.1 Procedure 32
3.3.2 Effect ofthe presence and the nature ofphotoinitiators 32

Table ofcontent
3.3.3 Influence ofthe nature ofthe monomer 36
4 Characterization of acrylic polymers containing phosphorus 37
4.1 IR (ATR) spectroscopy 37
4.2 1H NMR spectroscopy 38
4.3 Thermogravimetric analysis (TGA) 39
4.4 Comparison ofTG curves ofthe polymers with untreated cotton and PAN 41
fabrics
5 Conclusions 42
6 References 44
Chapter III: Flame retardancy of polyacrylonitrile and cotton 47
fabrics
1 Introduction 47
2 Flame retardancy of polyacrylonitrile (PAN) textiles 50
2.1 graft polymerization ofacrylate phosphate (DEMEP, DEAEP) and 51
phosphonate (DEAMP, DMAMP) monomers onto PANfabrics
2.1.1 Graft-polymerization procedure 51
2.1.2. Effect ofthe concentration ofDEAEP on the grafting yield 52
2.1.3 Effect ofthe crosslinking agent (EGDMA) 54
2.1.4 Effect ofthe nature ofthe crosslinking agent 55
2.1.5 Effect ofthe nature ofthe monomer 57
2.2 Surface characterization of the grafted PANfabrics 59
2.2.1 IR (ATR) 59
2.2.2 SEM 60
2.3 Evaluation oftheflame retardancy of the grafted PANfabrics 60
2.3.1 LOI measurements 60
2.3.2. Burning rates 62
2.3.3 Thermogravimetric analyses 63
2.3.4 Burning behavior 64
2.4 Durability oftheflame retardant treatment 64
2.5 Attempts to improve wash-resistance and LOI value with multilayer 65

Table ofcontent
treatments
2.6 Conclusions 68
3 Flame retardancy of cotton fabrics 69
3.1 Plasma-induced grafting andpolymerization ofDEMEP, DEAEP, 70
DEAMP, DMAMP, DEAEPN and BisDEAEPN onto cottonfabrics
3.1.1 Graft-polymerization procedure 70
3.1.2. Effect ofthe concentration ofthe crosslinking agent on the grafting 71
yield
3.1.3 Effect ofthe concentration ofthe monomer on the grafting yield 73
3.2 Surface characterization ofthe grafted PANfabrics 74
3.2.1 IR (ATR) 74
3.2.2 SEM 75
3.3 Evaluation oftheflame retardancy ofthe grafted cotton fabrics 76
3.3.1 LOI measurements 76
3.3.2 Thermogravimetric analyses 79
3.3.3 Effect oftheflame retardants on LOI values and char residues 80
3.3.4 Burning behavior 81
3.4 Durability oftheflame retardant treatment 81
3.5 Effect ofthe surface area ofthe cotton fabrics on the grafting yield and 83
LOI values
3.6 Conclusions 86
4 Comparison of the flame retardant effect on PAN and cotton fabrics 87
5 References 89
Chapter IV: Compatibility of flame retardants with water- 91
repellent treatment
1 Introduction 91
2 CF4 plasma treatment of flame retarded cotton fabrics with DEAEP, 93
DEAEPN, and BisDEAEPN
3 Plasma-induced graft polymerization of AC8 on flame retarded fabrics 96
with DEAEP, DEAEPN, and BisDEAEPN.

Table ofcontent
4 Plasma-induced graft copolymerization of ACS and DEAEPN monomers. 98
5 Ageing tests 101
6 Conclusions 101
7 References 103
Chapter V: General Conclusions 105
Chapter VI: Experimental part 109
1 General comments 109
1.1 Workingprocedures 109
1.2 Analytical techniques 109
2 Preparation and characterization of the monomers 110
3 Plasma-induced polymerization of the monomers under argon microwave 118
plasma
4 References 121
Appendix 123
1 List of Abbreviations 123
2. Kinetic of homopolymerization of the acrylic monomers containing 125
phosphorus
3 TGA and DTG data of the polymers and DTG curves of untreated and 126
treated PAN and cotton fabrics with the acrylic monomers containing
phosphorus
4 Schmerber tests: Determination of resistance to water penetration 128
5 Curriculum Vitae 129

Résumé
Résumé
La modification chimique et superficielle des polymères constitue un enjeu important
dans le domaine de la chimie des polymères. Elle permet non seulement de synthétiser de
nouveaux matériaux mais aussi d'améliorer les propriétés de ceux déjà existants. Ce travail de
thèse décrit la modification superficielle des textiles synthétiques (polyacrylonitrile) et
naturels (coton) dans le but de leur conférer des propriétés ignifuges durables. Pour y
parvenir, nous avons développé une nouvelle approche basée sur l'utilisation de la technique
plasma froid. Le protocole employé vise, sous l'effet d'un gaz plasmagène à greffer et à
polymériser simultanément des monomères lourds tels que des acrylates phosphores ou
fluorés à la surface de divers substrats imprégnés de ceux-ci.
Tout d'abord trois différentes classes de monomères organophosphorés ont été
synthétisées: des acrylates phosphates et phosphonates, tous deux bien connus pour leurs
propriétés de retardateur de flamme. Nous avons également mis au point la synthèse de deux
nouveaux composés, les acrylates phosphoramidates (DEAEPN, BisDEAEPN) qui
contiennent à la fois des atomes d'azote et de phosphore.
% „OEt / ^\yoEt
o o \ ß
DEAEPN BisDEAEPN
Dans un second temps, la polymérisation induite par un plasma d'argon des différents
monomères a été étudiée. Cette étude a montré que les acrylates phosphores synthétisés
peuvent être facilement polymérisés en présence d'un photoinitiateur (Irgacure 819) dans un
plasma d'argon. Le suivi de la réaction de polymérisation par RMN "H a permis de démontrer
que la polymérisation induite par plasma argon des monomères obéit à un schéma classique
de réaction radicalaire. Les analyses thermogravimetriques (ATG) sous atmosphère d'argon
ont montré que les polymères organophosphorés se décomposent avant les tissus de
polyacrylonitrile (PAN) et de coton, et qu'ils sont thermiquement stables à plus de 600°C.
Les conditions expérimentales utilisées pour les réactions d'homopolymérisatlon ont
ensuite été appliquées pour le greffage et la polymérisation des acrylates phosphores sur les
tissus de PAN et de coton. L'influence de la concentration du monomère et d'un agent
réticulant (ethylene glycol diacrylate) sur le taux de greffage a été étudiée. La confirmation du

Résumé
greffage et de la polymérisation des monomères à la surface des tissus de PAN et de coton a
été démontrée par IR (ATR) et par MEB. Les mesures d'indice d'oxygène limite (IOL) et les
analyses thermogravimetriques (ATG) ont montré que des propriétés ignifuges ont été
conférées aux tissus de PAN et de coton traités, et que la stabilité thermique de ces derniers
est améliorée comparé aux tissus non traités. La durabilité du caractère ignifuge a été étudiée
en soumettant les tissus à une solution de lavage à 95°C. Les résultats obtenus ont montré une
persistance du caractère ignifuge ce qui est une confirmation d'un greffage de forte énergie
des polymères à la surface des tissus.
Pour finir, nous nous sommes intéressés à la compatibilité des propriétés ignifuges et
hydrophobes. Pour ce faire, trois types de traitement utilisant la technique plasma ont été
initiés: (i) le traitement par un plasma CF4, (ii) le greffage d'un acrylate fluoré à la surface des
tissus préalablement ignifugés, et (iii) la copolymérisation des acrylates phosphores et fluorés.
Les mesures d'indice d'oxygène limite (IOL) et de pression Schmerber ont permis de
démontrer qu'il était possible de conférer des propriétés hydrophobes aux tissus traités sans
altérer leur caractère ignifuge.

Summary
Summary
Chemical modification of polymeric materials to synthesize new materials or to
improve the chemical and surface properties of the existing polymers is still an important
challenge in polymer chemistry. This thesis describes the surface modification of synthetic
(polyacrylonitrile) and natural (cotton) textiles to impart durable flame retardant properties.
For this purpose a novel procedure which exploits the low-pressure technique has been
developed. This new approach based on the use of a gas plasma, simultaneously grafts and
polymerizes heavy monomers containing phosphorus or fluorine onto the surface of various
materials previously impregnated with the monomers.
Firstly, three different classes of organophosphorus monomers were synthesized:
acrylate phosphates and phosphonates which are known for their fire retardant properties. The
synthesis oftwo new acrylate phosphoramidate monomers (DEAEPN, BisDEAEPN) was also
developed.
°\\ X)Et
o o
DEAEPN BisDEAEPN
Secondly, the argon plasma-induced polymerization of the different monomers was
investigated. This study has shown that the monomers can be easily polymerized under argon
plasma in presence of Irgacure 819 as a photoinitiator. Monitoring the polymerization reaction
using H NMR spectroscopy allowed us to confirm that the plasma-induced polymerization of
the acrylic monomers containing phosphorus proceeds via a radical mechanism. The
thermogravimetric analysis (TGA) under an argon atmosphere of the polymers showed that
they decompose at lower temperatures than cotton and polyacrylonitrile (PAN) fabrics, and
that they are thermally stable above 600°C. The experimental conditions used for
homopolymerization were applied for the grafting and polymerization of the monomers onto
PAN and cotton fabrics. The effect of the concentration of the monomer and a crosslinking
agent (ethylene glycol diacrylate) on the grafting yield was studied. The grafting and
polymerization of the monomers onto the surface of PAN and cotton fabrics were confirmed
by IR (ATR) and SEM analyses. The limiting oxygen index (LOI) measurements showed that
fire retardant properties were conferred on PAN and cotton textiles, and TGA data an

Summary
enhancement of the thermal stability of the treated fabrics over the untreated. The durability
of the flame retardant treatment under washing at 95°C was investigated. The results obtained
showed a persistence of the flame retardant character. These results confirm that the acrylic
polymers containing phosphorus are covalently grafted onto the surface of the fabrics.
Finally the compatibility of the fire retardant and water-repellent properties was
studied. Three different ways using the cold plasma technique were investigated: the plasma-
induced graft polymerization of an organophosphorus monomer followed by (i) a CF4 plasma
treatment, by (ii) the plasma-induced graft polymerization of a fluorinated acrylate monomer,
and (iii) the plasma-induced graft copolymerization of organophosphorus and fluorinated
monomers. The LOI and Schmerber pressure measurements allowed us to demonstrate that it
was possible to confer water repellent properties to treated fabrics without altering the flame
retardant character.

Chapter I Introduction
Chapter I: Introduction
Natural and synthetic polymeric materials are used in a wide variety of applications
such as fabrics, appliances, home furnishing, buildings, transportation, electronics and
electrical engineering. However, a major problem arises because most polymers used for
these applications are flammable. Therefore, fire hazards associated with the use of these
polymeric materials have to be reduced or eliminated. This concern has led to efforts for
finding ways to reduce combustibility of the polymers, flame spread, heat production and
smoke or toxic fume emission by the use of flame retardants. The use of flame retardants in
the manufacture of electronic equipment, upholstered furniture, construction materials and
textiles has prevented the loss of many human lives. Traditionally, flame retardation of
polymers is achieved through the use of additives: inorganic, organic, halogen-containing or
phosphorus containing compounds [1,2]. Many of them, in particular halogenated additives,
produced toxic gases or dense and suffocating smokes when burned. Moreover, additives are
required in very high concentrations in order to be fully effective, leading to undesirable
changes in physical and mechanical properties of the polymer. Consequently, there is a
continual searching for flame retardant systems that do not suffer from these disadvantages.
The purpose of this work is to investigate new routes to flame retard textile fabrics
with permanent effect (laundry resistance) without the disadvantages mentioned above. The
method applied in this study is the grafting of halogen-free flame retardants compounds onto
the surface of synthetic and natural textiles using the cold plasma technique. In the first part of
chapter one the mechanism of flame retardation, the main families of fire retardants and their
mode of action, and the different approaches to reduce flammabiiity in polymeric materials
are presented. In the second part the cold plasma technique and its applications are described.
1 Mechanism of flame retardancy
There are many different fire retardant systems and they act in different ways. In order to
understand how flame retardants reduce the flammabiiity of polymeric materials, it is
necessary to explain the mechanism of polymer combustion.
1

Chapter I Introduction
1.1 Combustion process
Fire is a gas phase reaction requiring three components: heat, oxygen and fuel
(generated by the pyrolysis of the polymer). In order for a substance to burn, it must first
become a gas. When polymeric materials are exposed to intense heat or flame, they must be
decomposed in the condensed phase (pyrolysis) to release flammable gases. Then, an
adequate ratio between these gases and oxygen in the air leads to ignition of the polymer. The
heat produced by the combustion is spread out (flame) and then transferred back to the
polymer. This heat feedback again pyrolyses the polymer and maintains the combustion
process as shown in Figure 1.
Smoke and
Gaseous species
Heat ^ FlameA
* Oxygen (air)
Flammable
volatiles
Polymer
pyrolysis
Figure 1: Schematic representation of the polymer combustion cycle from Ref. [3].
To inhibit the burning cycle one or more of the three components needed to sustain
combustion has to be removed. Thus, flame retardants have to interact with any of the three
components in order to inhibit or even suppress the combustion process.
1.2 Mode ofaction offlame retardants
Depending on their nature, flame retardants for synthetic and natural polymers can act
chemically and/or physically in the solid (condensed) or gas (vapor) phase by interfering with
2

Chapter I Introduction
one or more stages of the combustion process: heating, decomposition, ignition, flame spread,
or smoke process [1-6].
1.2.1 Physical action
There are several ways in which the combustion process can be retarded by physical
action:
(a) By cooling.
The degradation reactions of the fire retardant can influence the energy balance of the
combustion. The flame retardant can degrade endothermally which cools the substrate to a
temperature below the one required for sustaining the combustion process (e.g. metal
hydroxides).
(b) By formation of a protective layer.
The condensed combustible layer can be shielded from the gaseous phase with a solid
or gaseous protective layer. Thus the solid phase is cooled, smaller quantities of pyrolysis
gases are evolved, the oxygen necessary for the combustion is excluded, and heat transfer is
impeded (e.g. phosphorus and boron compounds).
(c) By dilution.
The incorporation of inert substances (e.g. fillers) and flame retardant additives (which
evolve as inert gases on decomposition) dilutes the gases feeding the flame so that the lower
ignition limit of the gas mixture is not reached (e.g. metal hydroxides).
1.2.2 Chemical action
The chemical reactions interfering with the combustion process take place in the solid and
gas phases.
(a) Reactions in the gas phase.
The free radical mechanism of the combustion process can be interrupted by a flame
retardant. The exothermic processes, which occur in the flame, are inhibited, the system cools
and the supply of flammable gases is reduced and eventually completely eliminated (e.g.
halogenated flame retardants).
3

Chapter I Introduction
(b) Reactions in the solid phase.
(i) The breakdown of a polymer (thermoplastic) can be accelerated by flame
retardants, causing pronounced flow or drip of the molten polymer and, hence, its withdrawal
from the environment of the flame.
(ii) Flame retardants can form a layer of carbon (charring) on the surface of the
polymer upon combustion. This process can occur, for example through the dehydrating
action of the flame retardant generating double bonds in the polymer (usually in polymers
containing hydroxyl groups). These processes form a carbonaceous layer via cyclizing and
cross-linking (e.g. phosphorus compounds).
(iii) Another mechanism of flame retardation in the condensed phase is intumescence.
When exposed to heat in which materials swell to form foam, usually carbonaceous, which in
turn acts as a barrier to heat, air and pyrolysis products. Intumescent systems are based on
three basic ingredients: a catalyst, a charring agent and a foaming (spumific) agent.
2 Important flame retardants
The main flame retardant systems for polymers currently in use are based on
halogenated, phosphorus, nitrogen, and inorganic compounds [1,2,4-6]. Typically, these flame
retardants systems inhibit or even suppress the combustion process by chemical or physical
action in the gas or condensed phase.
2.1 Halogen-containingflame retardants [1-4]
Halogen-containing flame retardants are one of the largest groups of additives in the
plastic industry. They are used primarily in polymers for the electronic and building industries
and are known for their performance in styrenic copolymers, engineering thermoplastics, and
epoxy resins. There are three types of halogen-containing compounds that are used as flame
retardants: derivatives of compounds with aliphatic, cycloaliphatic, and aromatic structures.
The type of halogen atom is varied in each class.
Halogenated flame retardants act by inhibiting the radical mechanism which takes
place during the combustion (eqs. 1.1 and 1.2). In the gas phase, high-energy OH» and H«
radicals are formed by chain branching:
Ff. + 02 > OH* + O* (1.1)
O* + H2 OH* + H» (1.2)
4

Chapter I Introduction
The main exothermic reaction involves OH» radicals:
OH» + CO C02 + H» (1.3)
To slow down or stop combustion, it is imperative to hinder the chain-branching reactions
(1.1) and (1.2).
When exposed to high temperatures, halogenated flame retardants decompose to release
halogen, as free radicals X» (1.4). These radicals react with hydrocarbon molecules to give the
hydrogen halide HX (1.5). Then the high-energy radicals OH» and H* are removed by
reaction with HX and replaced by low-energy X» radicals (1.6 and 1.7). The actual flame
retardant effect is thus produced by HX. The hydrogen halide consumed is regenerated by
reaction with hydrocarbon.
RX R» + X» (1.4)
X» + RH R» + HX (1.5)
HX + H» H2 + X» (1.6)
HX + OH» H20 + X» (1.7)
The effectiveness of halogenated flame retardants depends on the quantity of the
halogen atoms they contain and also, very strongly on the control of halogen release. Flame-
inhibition studies on halogens have shown that the effectiveness increases in the order F < Cl
< Br < I. Bromine and chlorine compounds are generally used because iodine compounds are
thermally unstable at polymer processing temperatures, while fluorine compounds are too
stable.
To be more effective, some halogenated flame retardants require the presence of
antimony oxide (Sb203) as a synergistic catalyst. It acts by facilitating the breakdown of
halogenated flame retardants to active molecules. Sb203 also reacts with the halogens to
produce volatile antimony species (antimony halides or antimony oxyhalides), which are
capable of interrupting the combustion process by removing OH» and H» radicals.
Although halogenated compounds (chlorine and bromine) form some of the most
widely employed flame retardant materials, they have clear disadvantages; the potential to
corrode metal components, and the toxicity of hydrogen halides formed during combustion.
Thus, there is a growing demand to replace halogen-containing flame retardants.
5

Chapter I Introduction
2.2 Phosphorusflame retardants [1-4]
Phosphorus-containing compounds are used as flame retardants for thermoplastics,
thermosets, textiles, paper, coatings and mastics. They include elemental red phosphorus,
organic and inorganic phosphorus compounds. Some products contain both phosphorus and
halogen (chlorine or bromine) or nitrogen.
The flame-retardant mechanism for phosphorus depends on the type of phosphorus
compound used and on the chemical structure of the polymer. Phosphorus-containing flame
retardants mainly act in the condensed phase. The flame retardant is converted by thermal
decomposition to phosphoric or polyphosphoric acid. These acids act as dehydrating agents
(extracting water from the pyrolysing substrate), altering the thermal degradation of the
polymer, and promoting the formation of char. The char insulates the polymer substrate from
heat, flame, and oxygen.
A key feature of phosphorus flame retardants is intumescence [2,3,7]. Intumescent
coatings are made from a combination of products, which are applied to a surface like paint.
The products involved contain: a carbonific (char former) such as a polyol, an acid source or a
catalyst (phosphorus compounds), a spumific compound (amines or amides which liberate
non-flammable gases such as NH3 or C02 when heated), and a resin binder. The mechanism
of intumescence involves the decomposition of the phosphorus compound to phosphoric acid,
esteriflcation of the polyol to form polyol phosphate and char formation through a series of
elimination steps.
Like halogenated compounds some phosphorus compounds can act in the gas phase,
through the formation of PO» radicals. Flame-inhibition reactions similar to the halogen
radical trap mechanism have also been proposed [1,7]:
H3PO4 HP02, HPO, PO» (1.8)
PO» + H» HPO (1.9)
HPO + H» H2 + PO» (1.10)
PO» + OH» + H2 HPO + H2O (1.11)
Although phosphorus compounds are highly effective flame retardants and an
alternative to halogenated compounds, they are not effective in all types of polymers. They
work well in oxygen- or nitrogen-containing polymers but unsatisfactory in polymers which
do not char (polyolefin, styrenic resins).
6

Chapter I Introduction
2.3 Nitrogen-containingflame retardants [1,2,4,8]
Nitrogen-containing flame retardants are a small but rapidly growing group of flame
retardants. Nitrogen-based compounds can be employed in flame retardant systems or form a
part of intumescent flame retardant formulations. They are mainly found in polymers such as
Polyurethane and polyamides. Melamine-based products such as melamine, melamine
phosphate, and melamine cyanurate are currently the most widely used nitrogen flame
retardants. The chemical structure of melamine (2,4,6-triamino-l,3,5 triazine) is shown in
Scheme 1.
NH21
N"^N
A AH2N N NH2
Scheme 1 Chemical structure of melamine
The mechanisms of nitrogen-containing flame retardants are not fully understood, but
it is thought that they have several effects:
a) Formation of cross-linked molecular structures in the treated material. These are relatively
stable at high temperatures, thus physically inhibiting the decomposition of materials to
flammable gases (needed to feed flames).
b) Release of nitrogen gas which dilutes the flammable gases and thus reduces flames.
c) Synergy with phosphorus-containing flame retardants by reinforcing their function.
The main advantages of nitrogen-containing flame retardants are their low toxicity,
their solid state under standard conditions, and in case of fire, the absence of dioxin and
halogen acids as combustion products and their low evolution of smoke. Thus, they are
environmentally friendly compared to halogenated compounds.
2.4 Inorganicflame retardants [1-4]
A number of inorganic compounds are used as flame retardants, interfering by various
physical actions with the combustion process: release of water or non-flammable gases which
dilute the gases feeding the flame, absorption of heat energy thus cooling the substrate, or
production of a non-flammable and resistant layer on the surface material. Inorganic flame
retardants include metal hydroxides and boron compounds.
7

Chapter I Introduction
2.4.1 Metal hydroxides
Metal hydroxides are an important class of flame retardants. They are used in almost
every class of polymers such as polyolefin, thermosets, and in electronic, wire and cable
applications. The most widely employed metal hydroxides are aluminium trihydroxide (ATH)
and magnesium hydroxide
Metal hydroxides used as flame retardants interfere with the combustion process at
many levels. They first decompose endothermally to metal oxide (which forms a protective
non-flammable layer on the substrate surface) and to water:
2A1(0H)3 > A1203 + 3H20 (1.12)
Mg(OH)2 MgO + H20 (1.13)
The water (as steam) forms a layer of non-flammable gas near the substrate surface inhibiting
flames. The endothermic decomposition absorbs heat energy to cool the substrate and slow
down the burning. All hydroxides are relatively non toxic, but for meeting fire performance
requirements, extremely high loadings are necessary which can affect the properties of the
polymers.
2.4.2 boron-containing compounds
Common uses of borates are in mixture of boric acids and borax as flame retardants
for cellulose and of zinc borate for PVC and some engineering plastics. Boron-containing
compounds also act by releasing water in a heat absorbing reaction, and by forming a
protective glassy layer on the substrate surface. They can release boric acid, which also
induces charring of the substrate, and thus reduces flammable gases similar to phosphorus fire
retardants. They also reduce smoke emission or act synergistically to increase the
effectiveness of halogenated flame retardants.
3 Different approaches to reduce polymer flammabiiity
There are several ways to achieve flame retardancy in polymeric materials, for
example: Incorporation of flame retardant additives by physical means (additive approach)
[1,2] is the most classical way. However, this approach has several disadvantages such as
poor compatibility of the systems and leaching from the polymer through normal service and
ageing. The latter reduces the flame retardant's effect and can pose environmental threats.
8

Chapter I Introduction
Furthermore, to be effective the additive is often required in high loadings, which may result
in undesirable changes of the polymer's physical and mechanical properties.
To overcome these problems flame retardant groups can be incorporated into the
material (reactive approach) [2,6], via copolymerization with monomers or by chemical
modification of existing polymers. By introducing fire retardant units into the polymer back
bone or as pendant groups, leaching and volatilization of the flame retardant is prevented.
Since the incorporation of even a few weight percent of the flame retardant units into the
polymer chain can lead to an acceptable level of flame retardancy, the original physical and
mechanical properties of the polymers are maintained. This strategy has been employed with
polyesters [6,9-11], polyurethanes, [12-14], poly(meth)acrylates [15,16], epoxy resins [5,19-
21] and demonstrating effectiveness of the modification. However, it is difficult to apply the
chemical incorporation method on an industrial scale [22]. Furthermore, this approach is
limited to the synthetic polymers.
An alternative approach is grafting of flame retardant compounds onto the surface of
the polymer (surface approach). Since flammabiiity is mainly controlled by surface properties,
modification of substrate surfaces and proper grafting of flame-retardant groups could alter
flammabiiity. The surface approach is often the only way to flame retard polymeric materials
such as wood and natural fibers. Various surface grafting methods such as UV [23-25],
gamma irradiation [26-29], and cold plasma technique [22,30-39] have been used to confer
fire retardancy to polymeric materials. Among these methods, the cold plasma technique is a
new technology which permits covalent grafting of small functional groups and
macromolecular compounds onto the surface of polymeric materials. Furthermore, cold
plasma has the advantage compared to other radiation methods to modify only the surface
properties of the polymer without altering its bulk properties [40,41].
The present study focuses on the use of cold plasma technique to graft and polymerize
flame retardant monomers onto the surface of polymeric materials (textiles). The cold plasma
technique and its applications are described in more details in the next section.
4 Cold plasma technique
4.1 Definition ofplasma
The word "plasma" was first used by Langmuir in 1929 to describe an ionized gas
[42,43]. A plasma is a partially or fully ionized gas with roughly an equal number of
9

Chapter I Introduction
positively and negatively charged particles. Plasmas occur over a wide range of temperatures
and pressures. There are two types of plasmas: hot plasmas also known as equilibrium or
thermal plasmas, and cold plasmas or non-equilibrium or non-thermal plasmas.
In a hot plasma, full ionization takes place and the pressure of the gases is relatively
high, raising the number of collisions between particles (neutral, charges, excited, non-
excited) and thereby promoting the transfer of energy among particles. The result is a plasma,
thermally equilibrated where all the particles have the same high energy. This type of plasma
can be artificially generated with a high voltage and high temperature arc (e.g electric arcs,
rockets jets).
In comparison, the degree of ionization in a cold plasma is small and the pressure of
the gases is reduced (ca 1-104 Pa). The neutral and positively charged species have low
energies, while the electrons have relatively high energies. These electrical discharges are
non-equilibrium plasmas and, owing to the low energy levels of the particles composing the
plasma, reactions may proceed at temperatures close to 25°C. Consequently, cold plasma is
suitable for the surface modification of organic materials.
4.2 Principle ofcoldplasma technique [44]
The cold plasma process is initiated by placing a polymer in a closed evacuated
chamber. The selected gas to be ionized is then released into the chamber under a partial
vacuum and subjected to an electromagnetic field radio frequency (RF) or microwave (MW).
Within the RF or MW field, the gas molecules are excited to free electrons, ions, radicals,
metastables and neutral atoms with a broad distribution of energy levels. Upon relaxing to
their ground state the electrons emitted UV and visible radiations in the plasma. All the active
species (radicals, ions, various neutral particles, and photons) have sufficient energy to break
carbon-carbon and carbon-hydrogen bonds in the polymer Table 1.
10

Chapter I Introduction
Table 1 Energies available in a plasma and some typical bond energies from Ref. [41, 45]
Glow discharge Energy (eV)
Electrons 0-20
Ions 0-2
Metastables 0-20
UV/Visible 3-40
Bond Energy (eV)
H —H 4.5
H —F 5.8
F —F 1.6
C —H 4.3
C —N 2.9
C —F 4.4
C —Cl 3.4
C —Br 2.9
C—I 2.3
C —C 3.6
C = 0 8.0
C = C 6.1
C = C 8.4
4.3 Interaction ofcoldplasma with a polymer surface
When a substrate is in contact with gas plasmas, its surface is bombarded by the excited
species (ions, radicals, metastables, and photons). The energy of these excited species is
transferred from the plasma to the substrate (Figure 2) and then dissipated within the material
by a variety of chemical and physical processes resulting in surface modification.
11

( 'hupler 1 Introduction
Microwave generator
2.45.GIIZ
&\
Gas
inletuElectrons Photons
o° Radicals o
Vacuum
chamber
o
Metastable species© o O ^
ZIV- I 1 4*
p<lTorrSubstrate
Vacuum
pump
Figure 2 Schematic representation ol" plasma-polymer interaction.
Depending on Ihe chemical nature of the gas plasma and of the polymer, various
processes can occur:
4.3 1 Ablation or etching [46-49]
Ablation is the removal of molecular layers and organic contaminants from the surface of
a polymer. 1 his effect is due to the continuous bombardment of a surface by the energetic
plasma particles. Ablation of a polymer surface can occur by physical and/or chemical etching
principles. Physical ablation occurs when the energy transfer from the plasma particles to a
surface exceed the binding or cohesive energy o[ the material. These particles break the
covalcnt bonds of the polymer backbone, resulting in fragmented polymer chains of lower
molecular weight. The chemical ablation of a polymer surface occurs through reactions of the
organic contaminants with the reactive species created in the plasma. Oxygen plasma and
oxygen- and fluorine-containing plasmas arc frequently used for the etching of polymers.
4 3 2 Cross!inking 147-49J
Crosslinking occurs when two or more radicals combine to form a covalent link. The
bombardment and the radiation produced by plasma particles cause the cleavage of polymer
12

Chapter I Introduction
macromolecules and the concurrent creation of free radicals. In this way a cross-linked
polymeric layer is rapidly formed, which is characterized by a higher molecular weight and
higher melting temperature compared the non-treated layer. Inert gases such as argon or
helium are used for crosslinking of a polymer surface.
4.3.3 Activation [48,49]
Activation is the addition of different atoms or chemical groups from the plasma to the
surface of a polymeric substrate. As with ablation, surface bombardment by high-energy
species breaks the polymer chain or extracts pendant groups or atoms, to form free radicals.
With activation, the surface energy of a polymer can be increased by employing an oxygen-
rich process gas, or can be decreased by employing a gas with high fluorine content.
4.3.4 Polymerization [44,46,50]
Polymerization occurs when organic vapors are introduced into plasma or when a plasma
of the organic vapor is created (without addition of plasma gas), resulting in the deposition of
a polymer film. If the polymer deposition is allowed to occur onto an appropriate polymer
substrate, the method provides means of surface coating or surface grafting.
Two types of polymerization reactions have been described [41,46]: the plasma-state
polymerization and the plasma-induced polymerization. In the former case, polymerization
occurs in a plasma in which electrons and other reactive species have enough energy to break
any bond (see table 1). Any organic compound, including those without a conventional
polymerizable structure, can be used in the plasma-state polymerization. In the latter case, the
plasma of an organic vapor or an inert gas initiates polymerization at the surface of liquid or
solid monomers. For this to occur, monomers must contain polymerizable functional groups
such as double bonds, triple bonds or cyclic structures.
4.3.5 Plasma grafting [49,51 ]
Surface activation of a polymer followed by a treatment with the monomer, usually in
vapor state is called plasma grafting. For example, when a surface is treated with an inert gas
plasma the surface is left rich with free radicals. If this surface is then exposed to an
unsaturated compound without additional RF or MW activation. The substrate react (couple)
with the free radicals on the polymer and grafting occurs.
13

Chapter I Introduction
4.4 Applications ofcoldplasma technique
Cold plasma processes have been successfully used to enhance or to replace
conventional wet finishing processes in several industrial applications such as packaging,
aerospace, biomedicine, microelectronics, automotives, and textiles. Some applications of
plasma for modification of polymer surfaces are listed in Table 2.
Table 2 Some applications of cold plasma technique
Area of
applicationPurposes Substrates
Gases or
monomersRef
Biomedical Heparin bonding for
improved blood
compatibility
PP, PVC, PTFE, PC,
PU, PMMA
NH3 (or N2+H2) [50]
Reduce leaching of
small molecules from
polymer into body
PP, PET, PVC, PMA C2H4, Ar [50]
Chemical
processing
Gas separation of
02/N2
Natural rubber,
polydimethylsiloxane4-vinyl pyridine,
2-vinyl pyridine,
[45]
Water vapour barrier Silicon rubber CH4 [41]
Diffusion barrier PVC CH4, C2H2 [41]
Surface
modification
Improve adhesion of
epoxy resins
PE, PTFE, PVC, PVF C2H2 [45]
Surface hardness PP sheet TMS [41]
Abrasion resistance PC VinylTMOS+02 [50]
Electrical
uses
Improvement of
electrical conductivity
PPS Air/I2 [52]
Optical uses Anti reflection
coating
PMMA Perfluoerbutene-
2
[45]
Textile Water repellency Silk SF6 [53]
Water repellency Cotton C3F6, CF4 [54]
Water repellency PAN Perfluroacrylate
compounds
[51,55]
Flame retardancy Rayon, PAN Phosphorus
compounds
[30-32]
Abbreviation of polymers and monomers: PP: polypropylene, PVC: polyvinyl chloride),PTFE: polytetrafluoroethylene, PC: polycarbonate, PU: polyurethane, PMMA: poly(methylmethacrylate), PET: poly(ethylene terephthalate), PMA: poly(methyl acrylate), PE:
polyethylene, PVF: poly(vinylidene fluoride), PPS: polyphenylene sulphide, PAN:
polyacrylonitrile, TMS: tetramethylsilane, TMOS: tetramethoxysilane.
14

Chapter I Introduction
4.5 Advantages and disadvantages ofcoldplasma technique
The advantages of the cold plasma technique are numerous [41]. Modification can be
confined to the surface layer without altering the bulk properties of the polymer. The cold
plasma processes can modify the surfaces of all kind of polymers, regardless of their structure
and chemical reactivity. By selecting different gases, it is possible to choose the type of
chemical modification for the polymer surface. Furthermore, the modification is fairly
uniform over the entire surface. The use of a gas plasma can avoid the problems encountered
in wet chemical techniques such as residual solvent on the surface and swelling of the
polymer. Surface modification via cold plasma is an environmentally clean process and
allows the treatment of large surface areas.
However, one disadvantage of many plasma systems is that they only operate at low-
pressures. Several examples of atmospheric non-equilibrium plasma processes have been
reported as alternative to low pressure plasma processes [56]. Due to its extreme complexity,
the mechanism of how cold plasma induces surface modification is not fully understood.
However, investigations are currently underway to accurately describe all plasma processes.
5 Flame retardant testing methods
There are five standards of polymer combustibility [2,6]: ignitability tests (or UL 94),
flame spread tests, limiting oxygen index (LOI), heat release tests (cone calorimeter), and
smoke tests. A brief description of the most common tests is given below.
5.1 Underwriters Laboratories (UL) 94
This standard incorporates a number of different test methods; the most common used
is the UL 94 vertical burning test [1,6,57]. The specimen 13 mm wide by 125mm long is
suspended vertically above the burner tube for two 10-s ignitions. A 50-W methane flame is
used. The flammabiiity is rated from V-0 to V-2. The best flame retardancy rating of V-0 is
achieved when the after-ignition burn time is less than 10 s on each ignition and no drips are
allowed. The UL 94 is a full-scale test fire when small items such as electric switch insulators
are concerned. When the fire performance of larger objects must be considered, the results of
UL 94 cannot access solely the fire performance of the material.
15

Chapter I Introduction
5.2 Limiting oxygen index (LOI)
The LOI measures the minimum oxygen concentration (in a flowing mixture of
oxygen and nitrogen gas) required to support candle-like downward flame combustion [1,2,6].
It measures the ease of extinction of the materials. The specimen size depend on the
application: cellular plastics (such as foams) use specimens 125 mm long, 12.5 mm wide and
12.5 mm thick, while films and fabrics require samples 140 by 52 mm, and use thickness. The
specimen is placed vertically inside a glass column and ignites at the top with a small gas
flame. This method is suitable as a semi-qualitative indicator of the effectiveness of flame
retardants during the research and development stage. Polymeric materials having LOI value
of 21% or below ignite easily and burn rapidly in the air (containing 20.8% of oxygen). Those
with LOI values above 21 ignite and burn more slowly and generally, when LOI values rise
above approximately 26-28, the polymers may be considered to be flame retarded [3].
However, this test method is not appropriate as a predictor of real scale fire performance
mainly because of the low heat input and the simulated high oxygen concentration.
5.3 Heat release tests (cone calorimeter)
The cone calorimeter is a small scale instrument that measures rate of heat release
(RHR) of materials using the oxygen consumption principle [1,57]. This empirical principle is
based on the observation that, generally, the net heat of combustion of any organic material is
directly related to the amount of oxygen required for combustion. Approximately 13.1 MJ of
heat are released per kilogram of oxygen consumed. A square sample of 100 mm x 100 mm,
with the thickness varying from 6 to 50 mm is heated in the shape of a truncated cone. The
irradiance of the specimen can be set by to any desired value from 0 to 100kW/m2. The mass
of the specimen is recorded continuously through the used of a load cell. The heat release
magnitudes determined are the heat release rates per unit area and the total heat release per
unit area. The rate of heat release (RHR) is a measure of flammabiiity which is relevant to
real fires [1].
6 Flame retardancy of polymeric materials by cold plasma technique
The application of cold plasma technique to flame retard textiles and polymers has
been investigated as an alternative to traditional methods. Different procedures have been
developed.
16

Chapter I Introduction
6.1 Plasmapolymerization of volatile compounds
Akovali et al. [32,33] studied the flammabiiity of polyacrylonitrile (PAN) fibers and
polyester fabrics treated with different volatile monomers. Hexamethyldisiloxane (HMDS),
was used at various plasma powers and exposure time. The results showed a slight decrease of
the oxygen index (OI) values of PAN fibers and polyester fabrics compared to the untreated
one. When PAN fibers were treated with ethyldichlorophosphate (EDCP) and tris
(butoxyethyl) phosphate (TBEP) an increase of the oxygen index values compared to the
untreated one was observed.
Jama et al. [37] investigated the flame retardancy of polyamide-6 (PA-6) and
polystyrene (PS) substrates coated with thin film of 1,1,3,3-tetramethyldisiloxane (TMDS).
These films were obtained from the polymerization of TMDS monomer doped with oxygen
using CNRP (cold remote nitrogen plasma) process. The rate of heat release (RHR) of coated
PA-6 and PS were decreased by about 40% and the limiting oxygen index (LOI) values were
also improved compared to virgin polymers.
Laishun [34] has developed a CF4/CH4 plasma induced surface modification approach
to impart flame retardant properties to polypropylene (PP) and poly(ethylene terephthalate)
(PET) polymers. The flame retardancy of PP and PET film pretreated with CH4 to deposit a
barrier layer and then with CF4/CH4 plasma was improved.
6.2 Grafting ofnon-volatile monomers andpolymers
Simionescu et al.[30,31] investigated the flame retardation of rayon fibers and fabrics
treated with phosphorus and halogen-containing monomers and polymers under RF nitrogen
plasma. The fabrics and fibers were previously impregnated with pure organophosphorus
compounds or their concentrated solutions. Although the phosphorus content of the fiber was
low (0.13-0.66 %) the flame retardancy was improve over untreated rayon. The best flame
retardant properties were obtained with triphenyl phosphite, 2-phenoxy-1,3,2-
dioxaphospholane and triethyl phosphite. Importantly, the macroscopic aspect of the fabrics
was not changed. However, the physico-mechanical properties of the fibers were deteriorated.
The grafting took place through aromatic ring opening mechanism and/or through
dehydrogenation and dehydrochlorination reactions.
Laishun [35,36] improve the flame retardancy of ethylene-vinyl acetate (EVA)
copolymer by incorporating acrylic monomers (acrylic acid, methacrylic acid, acrylamide) to
the substrate. The EVA copolymer samples were treated by argon plasma (to form radicals)
17

Chapter I Introduction
and then immersed in an aqueous solution containing the acrylic monomers, at boiling
temperature. An increase of LOI value from 19 to 24 was achieved with acrylic acid.
Errifai et al.[39] decreased the flammabiiity of polyamide-6 (PA-6) by grafting and
polymerization of a fluorinated acrylate monomer (AC8) onto the PA-6 surface. PA-6 plates
pretreated with an oxygen plasma and immersed at room temperature in a petroleum ether
solution of AC8 were treated with an argon plasma. A decrease of 50%> of the peak value of
the rate of heat release (RHR) of the coated PA-6 in comparison to uncoated samples was
obtained.
7 Characterization of the grafted copolymers
Several techniques are used characterize surface and chemical modification of the
polymers. A brief description is given below.
7.1 Infrared attenuated reflection spectroscopy (IR (ATR))
IR spectroscopy is used for functional group identification. The principle of the
technique involves detecting the IR absorption bands of the grafted polymer layer by
comparing IR (ATR) spectra of graft copolymer with the original substrate. The sampling
depth can be up to a micrometer or more. IR allows also following the polymerization. This
method is the most convenient one because the characterization can be quickly carried out.
7.2 X-ray photoelectron spectroscopy (XPS)
The principle of the technique is the determination of the binding energy (BE) of a
core electron in an atom. With this technique it is possible to obtain the chemical composition
of the grafted copolymers surface up to 10 nm. The information available from the technique
includes semi quantitative elemental analysis of the surface (% of elements, except H),
chemical environment around the probed atom (for example information on nearest and next-
nearest neighbors) and some structural analysis.
7.3 Scanning electron microscopy (SEM)
The grafting of a polymer onto a substrate often leads to the alteration of the polymer
morphology which can be observed by (SEM). The morphology of the treated and untreated
polymers is compared and this may bring additional proof for the polymer deposition.
18

Chapter 1 Introduction
7,4 Thermal analysis
The coating of a polymer substrate may change its thermal characteristics, which can
be studied by thermogravimetric analysis (TGA). The thermal decomposition studies can be
used to predict mechanisms of action and potential effectiveness of fire retardants.
8 Objectives of the thesis
This work is a part of an ongoing project to study the effectiveness of the plasma-
induced graft polymerization (PIGP) procedure as a novel method to confer new and durable
properties to synthetic and natural textiles, without altering their bulk properties. With the
PIGP procedure, an argon microwave (MW) plasma is used to induce, in one step, the
grafting and the polymerization of acrylic monomers containing the functional groups needed
to impart the desired properties. One advantage of PIGP procedure is that the polymerization
occurs with structure retention of the monomer. Furthermore, the desired properties are
obtained by creating covalent bonds between the substrate and the growing polymer on the
surface (Figure 3) leading to a durable effect.
Polymerizablefunction Functionality
Polymerization occurs with
structure retention of the
©r-O
monomer
K>Argon plasma
Substrate
^m ^m ^m
Grafted polymer
Figure 3 PIGP procedure
In this study, the PIGP procedure is used to impart durable flame retardant properties
to polyacrylonitrile (PAN) and cotton textiles using acrylic monomers containing phosphorus.
Firstly, we describe the synthesis and the plasma-induced polymerization of acrylate
phosphate, phosphonate and new phosphoramidate monomers used. The monomers differ in
their phosphorus content, in their polymerizable functional groups, and in the structure of the
phosphorus groups. It is thus possible to compare the flame retardant effect of different
phosphorus containing monomers on PAN and cotton textiles. Acrylate phosphate and
19

Chapter I Introduction
phosphonate monomers have already been used as additives (in the polymeric form) and as
comonomers to produced flame-resistant acrylic fibres [58]. Electron-beam and gamma
radiations have also been employed to graft a methacrylate phosphate monomer onto cotton
fabrics to decrease flammabiiity [29]. So far, no studies have been reported on the flame
retardation of PAN and cotton textiles by grafting and polymerization of acrylic monomers
containing phosphorus using the cold plasma technique.
Secondly, the grafting and polymerization of these monomers onto PAN and cotton
fabrics are carried out. Surface and thermal analyses of untreated and treated fabrics will be
presented and discussed. The flame retardant properties of untreated and treated fabrics are
evaluated by the LOI method. The durability of the flame retardant treatment to washing is
tested according to the accelerated laundering method proposed McSherry et al.[59].
Finally the compatibility of the fire retardant treatment with other finishes is
investigated. Flame retardant and water-repellent properties are combined via three different
ways: the plasma-induced graft polymerization of an organophosphorus monomer followed
by (i) a CF4 plasma treatment, by (ii) the plasma-induced graft polymerization of a fluorinated
acrylate monomer (AC8), and (iii) the plasma-induced graft copolymerization of
organophosphorus and AC8 monomers
20

Chapter I Introduction
9 References
[1] A.F. Grand, CA. Wilkie, editors, Fire retardancy of polymeric materials, Marcel
Dekker, Inc., New York (2000).
[2] G.E. Zaikov, S.M. Lomakin, Modern polymer flame retardancy, VSP,Utrecht (2003).
[3] A.R. Horrocks, D.Price, editors, Fire retardant materials, Woodhead publishing Ltd
and CRC press LLC, Cambridge (2001).
[4] J.H. Troitzsch, Chem Oggi-Chem Today 16 (1998).
[5] G.E. Zaikov, S.M. Lomakin, J. Appl. Polym. Sei. 86 (2002) 2449.
[6] S.Y. Lu, I. Hamerton, Prog. Polym. Sei. 27 (2002) 1661.
[7] M. Le Bras, G. Camino, S. Bourbigot, R. Delobel, editors, Fire retardancy of
polymers. The use of intumescence, Royal Chemistry Society, Cambridge (1998).
[8] H. Horacek, R. Grabner, Polym. Degrad. Stab. 54 (1996) 205.
[9] C. S. Wang, J. Y. Shieh, Y. M. Sun, J. Appl. Polym. Sei. 70 (1998) 1959.
[10] C. S. Wang, C. H. Lin, C. Y. Chen, J. Appl. Polym. Sei., Part A, Polym. Chem. 36
(1998)3051.
[111 L. S. Wang, X. L. Wang, G. L. Yan, Polym. Degrad. Stab. 69 (2000) 127.
[12] C. Sivriev, L. Zabski, Eur. Polym. J. 30 (1994) 509.
[13] D. J. Liaw, S. P. Lin, Eur. Polym. J. 32 (1996) 1377.
[14] Y. L. Liu, G. H. Hsiue, C. W. Lan, Y.S. Chiu, J. Appl. Polym. Sei., Part A, Polym.
Chem. 35(1997)1769.
[15] J.R. Ebdon, D. Price, B.J. Hunt, P. Joseph, F. Gao, G.J Milnes, L.K. Cunliffe, Polym.
Degrad. Stab., 69 (2000) 267.
[16] D. Price, K. Pyrah, G.J Milnes, J.R. Ebdon, B.J. Hunt, P. Joseph, Polym. Degrad. Stab.
77 (2002) 227.
[17] M. R. Buckinghama, A. J. Lindsaya, D. E. Stevenson, G. Müller, E. Morel, B. Coates,
Y. Henry, Polym. Degrad. Stab. 54 (1996) 311.
[18] D. Derouet, F. Morvan, J. C. Brosse, J. Appl. Polym. Sei. 62 (1996) 1855.
[19] Y. L. Liu, G. H. Hsiue, Y. S. Chiu, J. Polym. Sei., Part A, Polym. Chem. 35 (1997)
565.
[20] C. S. Wang, J. Y. Shieh, Polymer 39 (1998), 5819.
[21] Y. L. Liu, C. S. Wu, K. Y. Hsu, T. C. Chang, J. Polym. Sei., Part A, Polym. Chem. 40
(2002)2329.
21

Chapter I Introduction
[22] S. Bourbigot, C. Jama, M. Le Bras, R. Delobel, O. Dessaux, P. Goudmand, Polym.
Degrad. Stab. 66(1999)153.
J.A. Harris, C. J. Keating, W.R. Goynes, J. Appl. Polym. Sei. 25 (1980) 2295.
Y.W. Chen-Yang, J. R. Chuang, Y. C. Yang, C. Y. Li, Y.S. Chui, J. Appl. Polym. Sei.
69(1998)115.
T. Randoux, J. C. Vanovervelt, H. Van den Bergen, G. Camino, Progress in Organic
Coatings 45 (2002) 281.
R. Liepins, J. R. Surles, N. Morosoff, V. T. Stannett, J. Appl. Polym. Sei. 21 (1977)
2529.
R. Liepins, J. R. Surles, N. Morosoff, V. T. Stannett, J. J. Duffy, F. H. Day, J. Appl.
Polym. Sei. 22 (1978) 2403.
A. Mey-Marom, D. Behar, J. Appl. Polym. Sei. 25 (1980) 691.
N. Shiraishi, J. L. Williams, V. Stannett, Radiât. Phys. Chem. 19 (1982) 79.
C. I. Simionescu, F. Denes, M. M. Macoveanu, G. Cazacu, M. Totolin, S. Percec,
D. Balaur, Cell. Chem. Technol. 13 (1979) 475.
C. I. Simionescu, F. Denes, M. M. Macoveanu, G. Cazacu, M. Totolin, S. Percec, D.
Balaur, Cell. Chem. Technol. 14 (1980) 869.
G. Akovali, F. Takrouri, J. Appl. Polym. Sei. 41 (1990) 2011.
G. Akovali, F. Takrouri, J. Appl. Polym. Sei. 42 (1991) 2717.
L. Shi, J. Polym. Eng. 19 (1999) 445.
L. Shi, Eur. Polym. J. 36 (2000) 2611.
L. Shi, React. Funct.Polymers. 45 (2000) 85.
C. Jama, A. Quede, H. Sadiki, O. Dessaux, P. Goudmand, R. Delobel, M. Le Bras,
Recent Advances in Flame Retardancy of Polymeric Materials, 12 (2001) 127.
A. Quede, J.Cardoso, M. Le Bras, R. Delobel, P. Goudmand, O. Dessaux, C. Jama, J.
Mater. Sei. 37 (2002) 1395.
I. Errifai, C. Jama, M. Le Bras, R. Delobel, L. Gengembre, A. Mazzah, R. De Jaeger,
Surf. Coat. Technol. 180-181 (2004) 297.
H. Yasuda, Plasma Polymerization, Academic Press, INC, New York (1985).
C. M. Chan, Polymer surface modification and characterization, Hanser/Gardner
publications, Inc.(1994).
F. Denes, TRIP, 5 (1997) 23.
A.R. Denes, M.A. Tshabalala, R. Rowell, F. Denes, R.A. Young, Holzforshung, 53
(1999)318.
22

Chapter I Introduction
[44] H. Yasuda, Radiât. Phys. Chem. 9 (1977) 805.
[45] N. Morosoff, in R. d'Agostino (editor), Plasma deposition, treatment, and etching of
polymers, Academic press, INC, Boston a.o (1990).
[46] H. Yasuda in J. R. Ebdon, G. C. Eastmond (editors) New methods of polymer
synthesis, Vol 2, Blackie Academic and professional, London a.o (1995).
[47] M. R. Wertheimer, A. C. Fozza, A. Holländer, Nucl. Instr. and Meth. in Phys. Res. B
151 (1999).
[48] L. Carrino, G. Moroni, W. Polini,, J. Mater. Process. Technol. 58 (1996) 96.
[49] S. Kaplan, Surf. Coat. Technol. 155 (2002) 11.
[50] H. Biederman, Y. Osada in Plasma chemistry of polymers, G and S.Olive, editors.
(1990) 59.
[51] U. Vohrer, M. Müller, C. Oehr, Surf. Coat. Technol. 98 (1998) 1128.
[52] D. M. Tu, G. P. Zhuang, K. C. Kao, J. Appl. Polym. Sei. 43 (1991) 1625.
[53] E. Selli, C. Riccardi, M. R. Massafra, B. Marcandally, Macromol. Chem. Phys. 202
(2001)1672.
[54] M. G. McCord, Y. J. Hwang, Y. Qiu, L. K. Hughes, M. A. Bourham, J. Appl. Polym.
Sei. 88 (2003) 2038.
[55] F. Hochart, R. De Jaeger, J. Levalois-Grützmacher, Surf. Coat. Technol. 165 (2001)
201.
[56] M. J. Shenton, G. C. Stevens, J. Phys.D : Appl. Phys. 34 (2001) 2761.
[57] S. V. Levchik, E. D. Weil, Polym. Int 49 (2000), 1033.
[58] H. Herlinger, G. Hardtmann, F. Hermanutz, R. Schneider, U. Einsele, Melliand
Textilber. 72 (1991) E141.
[59] W. F. McSherry, G. L. Drake, A.B. Cooper, A. R. Markezich, Am. Dyest. Rep. 63
(1974) 52.
23

Chapter I Introduction
Seite Leer /
Blank leaf
24

Chapter II Synthesis andpolymerization ofacrylic monomers
Chapter II: Synthesis and argon plasma-induced polymerization of acrylic
monomers containing phosphorus
1 Introduction
Organophosphorus polymers have been the subject of several studies because of their
large domain of applications [1]. For example, they can be used as adhesion promoters for
paints, and lacquers. They are also used for artificial glasses, for fibers and films with a high
mechanical resistance, as ion exchangers and as lubricants. Phosphorus groups have also
plasticizing properties. They can lower the glass transition temperature and play a role in
some medical applications. In the present work, we are interested in organophosphorus
polymers because of their flame retardant properties.
As already mentioned in the previous chapter phosphorus-containing fire retardants
are widely used in plastics and textiles [2-4]. They may be incorporated into the polymer
chains through blending, homopolymerization, copolymerization, or surface modification.
These compounds mostly perform their flame retardant function in the solid phase of burning
materials by increasing the amount of carbonaceous residue or char and reducing flammable
by-products.
In this chapter, the synthesis and plasma-induced polymerization of acrylate
phosphate, phosphonate and phosphoramidate monomers are described. The kinetics of
homopolymerization of the different monomers under microwave (MW) argon plasma is
investigated. Acrylic monomers were chosen because it is known that they are easier to
polymerize than allyl and vinyl compounds [5], The polymers obtained are characterized by1 ^ 1
H and ~ P NMR, and IR(ATR) spectroscopy. Finally, the thermal decomposition of the
different polymers is investigated by thermogravimetric analysis (TGA).
2 Synthesis of acrylic monomers containing phosphorus
2.1 Synthesis of acrylate phosphates: Diethyl-2-(methacryloyloxy) ethyl phosphate
(DEMEP) and Diethyl (acryloyoxy) ethylphosphate (DEAEP)
DEMEP (1) and DEAEP (2) were synthesized following, with minor adaptation, the
method of Clouet et al. [6,7]. DEMEP and DEAEP were prepared by condensation of
25
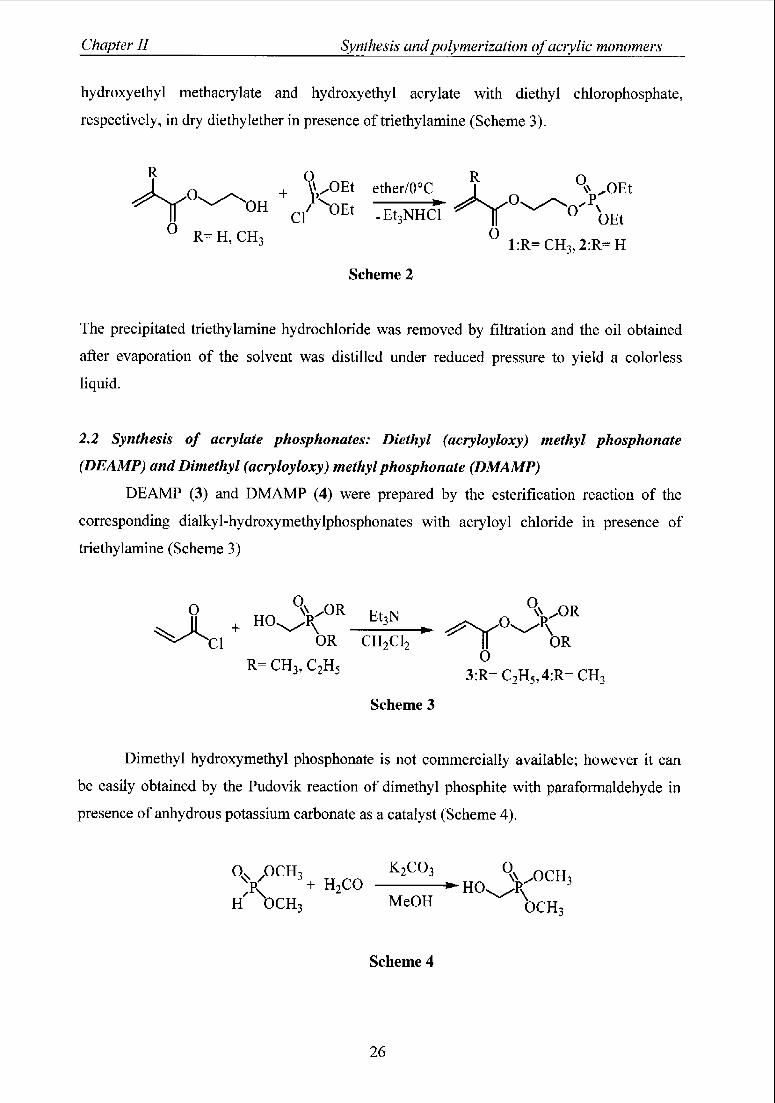
Chapter II Synthesis andpolymerization ofacrylic monomers
hydroxyethyl methacrylate and hydroxyethyl acrylate with diethyl chlorophosphate,
respectively, in dry diethylether in presence of Methylamine (Scheme 3).
*rQ „„
R
oU* OEt
o- - "
0Et
R=H'CH3 °1:R=CH3,2:R=H
Scheme 2
R OR
J^ O ^ + V0Et ether/o°c I_
The precipitated triethylamine hydrochloride was removed by filtration and the oil obtained
after evaporation of the solvent was distilled under reduced pressure to yield a colorless
liquid.
2.2 Synthesis of acrylate phosphonates: Diethyl (acryloyloxy) methyl phosphonate
(DEAMP) and Dimethyl (acryloyloxy) methylphosphonate (DMAMP)
DEAMP (3) and DMAMP (4) were prepared by the esteriflcation reaction of the
corresponding dialkyl-hydroxymethylphosphonates with acryloyl chloride in presence of
triethylamine (Scheme 3)
OR
R=CH3,C2H53:R=C2H5,4:R=CH3
M HOVU* Et3N
^ 0N + HU^FX 3
^ ^yü^^^"Cl OR CH2C12 [I
Scheme 3
Dimethyl hydroxymethyl phosphonate is not commercially available; however it can
be easily obtained by the Pudovik reaction of dimethyl phosphite with paraformaldehyde in
presence of anhydrous potassium carbonate as a catalyst (Scheme 4).
Ck DCH3 K2C03 00CH
V 3+ H2CO -HOOVH OCH3 MeOH N0CH3
Scheme 4
26

Chapter II Synthesis andpolymerization ofacrylic monomers
This procedure was recently developed by Jeanmaire et al. [8] and allows the
formation of a pure product in excellent yield after 1 hour at room temperature.
2.3 Synthesis of new acrylate phosphoramidates: Diethyl (acryloyloxy) ethyl
phosphoramidate (DEAEPN) and (acryloyloxy)-l,3-Bis(diethylphosphoramidate)-propan
(BisDEAEPN),
2.3.1 Synthesis Diethyl (acryloyloxy) ethyl phosphoramidate DEAEPN (5)
The synthesis of DEAEPN (5) which was developed in our laboratory was performed
in a one-pot procedure as shown in (Scheme 5).
HO ^^OEt Et3N TT/T-v/NH./?
*NH2 + ?C_ ^HO ^ P^
d 0Et CH2C12 5a 0'EtOtt
CH2Cl2/Et3N
O
CI
^y°W ^OEt
,NH\)Et
0 5
Scheme 5
First, diethyl-N-(hydrohyethyl) phosphoramidate (5a) is prepared by the reaction of 2-
aminoethanol with diethyl chlorophosphate in the presence of triethylamine as acid scavenger
in dichloromethane. 5a is selectively produced due to the stronger nucleophilic character of
nitrogen towards oxygen. The reaction is conveniently monitored by 31P NMR. After 2 hours
of stirring, the signal corresponding to diethyl chlorophosphate (5.4 ppm) is shifted at 9.6
ppm. This new signal is attributed to the phosphoramidate functional group in compound 5a.
The filtrate containing the crude product obtained after filtration of triethylamine
hydrochloride salt is used directly for the following esteriflcation reaction with acryloyl
chloride to give DEAEPN (5).
The monomer 5 was analyzed by !H, 13C, 3,P NMR in CDC13, IR spectroscopy and by
mass spectrometry. The 31P NMR analysis of the compound 5 shows a singlet at ö = 9.0 ppm.
Furthermore, the presence of polymerizable acrylic unit is supported by the observation of
27

Chapter II Synthesis andpolymerization ofacrylic monomers
three doublets assignable to the vinyl protons at Ô = 5.60, 5.87, and 6.18 in the ]H NMR
spectrum, or by signal arising from the double bond of carbon atom at about 5 = 127 and 130
ppm, and from the carbonyl group at 5 = 160 ppm in the 13C NMR spectrum. The IR spectrum
shows a single broad signal at about 3217 cm"1 corresponding to the N-H stretching vibration,
and peaks at 1238 cm" and 1030 cm" corresponding to the P=0 and P-O-C bonds
respectively. In mass spectrometry analysis a peak at m/z = 252 corresponding to [M+l]+ was
found. All of this confirmed the identity of the product, which was isolated in good yield (62-
65%) as pale yellow viscous oil after distillation.
2.3.2 Synthesis Acryloyloxy-l,3-Bis(diethylphosphoramidale)-propan: BisDEAEPN (6)
BisDEAEPN (6) was prepared in two steps (Scheme 6) following the same procedure
described for the synthesis ofDEAEPN in the previous section.
HO-(\—i
NH2 O
+ V0EtEtiN
O /0Et
/^OEt
NHuiit
NH2 c',\)Et CH2C12Ho/
6a oAu
OEt
CH2Cl2/Et3N
M
o
CI
VNH
OEt
sOEt
NHoc*
\_^OEt
0'AOEt
Scheme 6
l,3-Bis(diethylphosphoramidate)-propan-2-ol (6a), was first prepared by the reaction
of 1,3-diaminopropan-2-ol with diethyl chlorophosphate in presence of triethylamine in
dichloromethane. On the contrary to the previous reaction leading to compound 5a, this
reaction was slow. Instead of 2 hours of stirring at room temperature, the reaction mixture was
28

Chapter II Synthesis andpolymerization ofacrylic monomers
first kept for 5 hours at 0°C, and then stirred overnight at room temperature to allow the
formation of the triethylamine hydrochloride salt which displaces the equilibrium toward the
compound 6a. The crude product obtained was not pure according to 31P NMR analysis, but
could be purified by precipitation in hexane and washing with a saturated solution of
NaHC03. After purification the 31P NMR spectrum shows a single signal at 5 = 9.9 ppm and
in mass spectrometry analysis a peak at m/z = 363 corresponding to [M+l]+ was found. The
product 6a was isolated as highly viscous colorless oil in a good yield (60-70 %).
Compound 6a was then reacted with acrylolyl chloride in presence of triethylamine, to
give the monomer 6. The P analysis of the compound 6 in CDC13 shows a peak at 5 = 9.0
ppm, and in mass spectrometry analysis a molecular peak at m/z = 417 corresponding to
[M+l]+ is observed. Monomer 6 was isolated in fairly good yield (40-50%) as highly viscous
yellow oil.
3 Polymerization of acrylic monomers containing phosphorus
Acrylic polymers containing phosphorus can be obtained by free radical
polymerization in bulk or in solution in presence of AIBN or benzoyl peroxide as initiator. A
great deal of work concerns the copolymerization of acrylic monomers containing phosphorus
with other acrylic monomers such as methyl methacrylate (MMA) [9-13], styrene [9-11],
acrylonitrile [5,10], and acrylamide [10], in order to improve the flame retardancy of the
corresponding polymers. So far, no studies on the polymerization of acrylic monomers
containing phosphorus using cold plasma technique have been reported. Cold plasma can be
utilized in the polymerization of organic monomers in vapor, liquid, and solid phase. Two
types of plasma polymerization can be distinguished depending on the vapor tension of the
monomer [14]: the plasma (state) polymerization and the plasma-induced polymerization.
3.1 Principle ofplasma-state polymerization [14,15]
The monomer is an organic compound in the vapor state and constitutes partially or
totally the plasma. The polymerization occurs in a plasma in which electrons and other
reactive species have enough energy to break any bond. With this method, any organic
compound and even those without a polymerizable structure, needed for conventional type of
polymerization can be used. The polymers formed by plasma-state polymerization are
significantly different from conventional polymers, due to the fragmentation of monomer
molecules, followed by recombination of the fragments and deposition. Furthermore the
29

Chapter II Synthesis andpolymerization ofacrylic monomers
processes are highly system dependent and no unique correlation exists between the starting
material (monomer) and the product (polymer). Consequently, the mechanism of
polymerization by which an organic vapor polymerizes under plasma is quite complex and
can not be specifically described for the general case.
3.2 Principle ofplasma-inducedpolymerization
Plasma-induced polymerization process concerns monomers having a low vapour
tension. The plasma of an organic vapor or an inert gas initiates polymerization at the surface
of liquid or solid monomers, which must contain polymerizable functions, such as double
bonds, triple bonds or cyclic structures. The propagation of polymer chain takes place in bulk
via the conventional addition polymerization mechanism initiated by active species of plasma.
This type of reaction is comparable to the polymerization induced by other ionizing radiations
such as UV, gamma or electron-beam. However, in plasma-induced polymerization the
plasma contacts the monomer in either liquid or solid phase directly, and consequently a
transfer of some excited species from the plasma phase to the monomer phase takes place.
This is a significant difference with ordinary radiation polymerization, in which only energy
is transferred to a monomer phase to create reactive species, such as ions or free radicals of
the monomer. On contrary of the plasma-state polymerization, which occurs only under
plasma conditions, the plasma-induced polymerization can proceed after the plasma is
extinguished as long as reactive sites (e.g. free radicals) are available in the monomer [15,16].
In the present study the plasma-induced polymerization process is used for the
polymerization of the acrylic monomers containing phosphorus. Two types of plasma-induced
polymerization have been described.
In the first procedure developed by Osada et al.[17,18] the vapor phase of a liquid
monomer in a sealed ampoule is used to create a plasma. The monomer is introduced into an
evacuated ampoule (13-13xl0"3 Pa), and then the ampoule is sealed and inserted between a
pair of parallel-plate electrodes. Due to the vacuum conditions, the monomer is partially
evaporated and a glow discharge is applied in the vapor phase for a short period (generally a
few seconds). After plasma exposure, the ampoule is shaken in order to mix plasma-induced
reactive species (they act as initiators of polymerization) with the monomer and is kept at a
constant temperature (polymerization temperature) for a prolonged period of time. Numerous
studies have been carried out using this approach to initiate chain polymerization of vinyl and
acrylic monomers in bulk or in solution [15-19], Polymerization characteristics and properties
of polymers formed by plasma-induced polymerization strongly resemble thermal
30

Chapter II Synthesis andpolymerization ofacrylic monomers
polymerization of the corresponding monomer. The molecular weight of polymer increases
with the polymerization time, which is distinctively different from the free radical
polymerization. Consequently after a long reaction time, polymers with exceptionally high
molecular weight can be synthesized by plasma-induced polymerization. However, monomers
that can be polymerized by plasma-induced polymerization are limited and not all monomers
(for free radical addition polymerization) can be polymerized by this method. Attempts to
polymerize for example higher alkyl methacrylates other than MMA in the bulk by plasma-
initiated polymerization have been shown to yield very small amounts of the polymer [16].
In the second type of plasma-initiated polymerization, instead of the monomer vapor,
another gas is used to create the plasma. In the studies carried out by Kuzuya et al.[20,21], the
polymerization of several methacrylate monomers was investigated by exposing the effluent
gas resulting from the plasma state of the initiator such as methyl isobutyrate (MIB), 4-
methyl-1-pentene (MP), or 1,5-hexadiene (HD) to the monomer. The plasma state was
generated at the upper part of the sealed ampoule (containing the monomer and an initiator) at
so low a temperature that the gaseous phase was composed mostly of the vapor of an initiator.
With this method, higher alkyl acrylates were polymerized efficiently.
Another procedure was developed by Hirotsu [22] for the plasma-induced
polymerization of high molecular weight acrylates (tridecylmethacrylate and
octadecylmethacrylate) by direct irradiation of argon or helium plasma onto the monomer
spread on a slide glass. With these heavy monomers the evaporation in the vacuum is
minimized. The polymer yields were of 30 to 50% depending on the plasma conditions.
This approach was further developed by Epaillard et al.[23-27]. They investigated the
plasma-induced polymerization of various high molecular weight hydrogenated acrylate
monomers (multifunctional acrylate, such as trimethylpropane triacrylate, and
polyethyleneglycole diacrylate) in the liquid phase. The monomers were spread on an
aluminum foil which had been treated before under an oxygen plasma, and irradiated with
plasma of various gases such as argon, helium, nitrogen, carbon dioxide, oxygen and CF4.
They showed that the rate of polymerization depends mainly on monomer functionality,
plasma parameters (nature of the gas used to generate the plasma, power of irradiation), and
the thickness of the monomer layer. Furthermore, the addition of radical initiators increased
the polymerization rate as well as the maximum of conversion. The efficiency of the initiator
also depended on the plasma parameters. They concluded that oxygen and C02 plasmas
inhibit the polymerization. In nitrogen plasma, the polymerization is mostly induced by UV-
visible radiations emitted by the excited species present in plasma. But in He, CF4, and Ar
31

Chapter II Synthesis andpolymerization ofacrylic monomers
plasmas, competitive reactions to the polymerization are observed. Additions, terminations,
and degradations lead to a decrease of the polymerization rate and they are predominant at
critical values of power depending on the gas nature. The plasma-induced polymerization is
therefore described in terms of direct or indirect energy transfer from the plasma to the
monomer. The direct transfer corresponds to the active species bombardment on the polymer
surface which leads to competitive reactions. The indirect one is related to the absorption of
UV-visible radiations by the monomer which leads to polymerization reactions.
Hochart et al. [28] studied the polymerization of perfluroalkyl acrylate monomers
(AC8 and MAC8) induced by an Ar MW plasma. The monomers mixed with a photoinitiator
(Darocurll73) were coated onto glass plates to obtain liquid films of variable thickness. The
results show that the polymerization proceeds via a radical mechanism and that the reactivity
follows the order AC8 >MAC8.
This last procedure is used in the present work for the plasma-induced polymerization
of the acrylic monomers containing phosphorus (DEAEP, DEMEP, DEAMP, DMAMP,
DEAEPN and BisDEAEPN).
3.3 Argon plasma-inducedpolymerization ofacrylic monomers containingphosphorus
3.3.1 Procedure
The monomers (0.5 g) without or with 5% (w/w) of a photoinitiator are coated onto
glass plates to give a liquid film. The coated plates are then introduced in the plasma chamber.
Before treatment, the reactor is evacuated (pressure: 40 Pa) and the gas flow rate adjusted (FAr
= 125 seem). The discharge is then initiated by adjusting the power of the generator (100 W).
After the treatments for specific periods of time (5 to 20 min), the reactor is opened and the
films obtained are removed from the glass plates.
3.3.2 Effect ofthe presence and the nature ofphotoinitiators
In order to investigate and to improve the polymerization of acrylate monomers
containing phosphorus induced by an Ar MW plasma, it was worth studying, the influence of
the presence and of the nature of photoinitiators on the kinetic of homopolymerization. It has
already been demonstrated that the UV-visible radiations emitted from excited species of the
plasma play an important role in the plasma-induced polymerization of acrylic monomers
[27]. The effect of two photoinitiators: Darocur 1173 (2-hydroxy-2-methyl-l-phenyl-propan-
1-one) and Irgacure 819 (bis(2,4,6-trimethylbenzoyl)-phenylphosphine oxide) on the kinetics
32
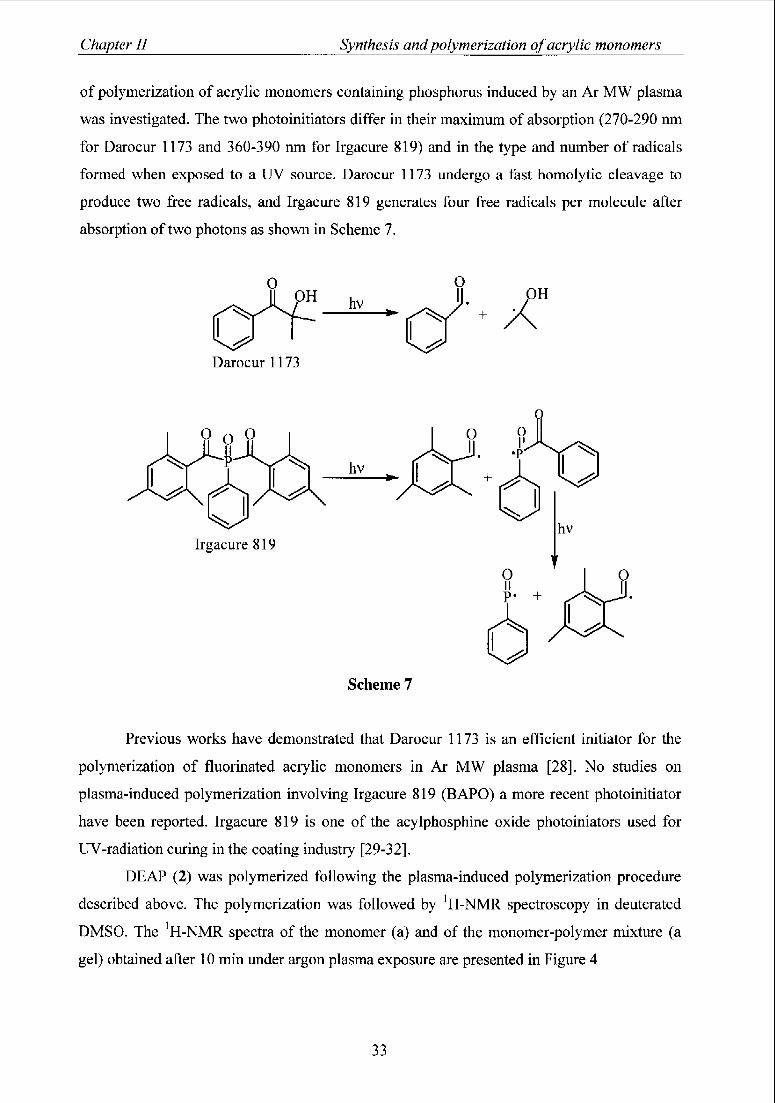
Chapter II Synthesis andpolymerization ofacrylic monomers
of polymerization of acrylic monomers containing phosphorus induced by an Ar MW plasma
was investigated. The two photoinitiators differ in their maximum of absorption (270-290 nm
for Darocur 1173 and 360-390 nm for Irgacure 819) and in the type and number of radicals
formed when exposed to a UV source. Darocur 1173 undergo a fast homolytic cleavage to
produce two free radicals, and Irgacure 819 generates four free radicals per molecule after
absorption oftwo photons as shown in Scheme 7.
Darocur 1173
Irgacure 819
Scheme 7
Previous works have demonstrated that Darocur 1173 is an efficient initiator for the
polymerization of fluorinated acrylic monomers in Ar MW plasma [28]. No studies on
plasma-induced polymerization involving Irgacure 819 (BAPO) a more recent photoinitiator
have been reported. Irgacure 819 is one of the acylphosphine oxide photoiniators used for
UV-radiation curing in the coating industry [29-32],
DEAP (2) was polymerized following the plasma-induced polymerization procedure
described above. The polymerization was followed by H-NMR spectroscopy in deuterated
DMSO. The H-NMR spectra of the monomer (a) and of the monomer-polymer mixture (a
gel) obtained after 10 min under argon plasma exposure are presented in Figure 4
33

Chapter II Synthesis andpolymerization ofacrylic monomers
(a) -CH3
-OCH2
H2C=CH
iJjL lil J
(b)
„jJUdL JL'" 1"''
6.5 2.5 2 0 15 10 ppm55 50 45 40 35 30
Figure 4 'H-NMR spectra of (a) DEAEP and (b) DEAEP after 10 min under a MW argon
plasma (FAr = 125 seem, P = 100 W)
A diminution of the intensity of the protons of the vinyl group (H?C=CH) of the
monomer can be observed in the'H-NMR spectrum of monomer-polymer (b), while the
intensity of the protons of methylene (-OCH2) and methyl groups (-CH3) remain constant
during the polymerization. It can be seen that broad signals appear between 2.4 and 1.5 ppm,
which is an indication of the presence of the polymer. For the various treatment times, the
ratio R between the relative intensities of the vinyl protons H2C=CH and the methyl groups in
-P(0)(OCH2CHj)2 was calculated. The latter was used as internal reference because it remains
unaffected during the reaction. This procedure allows us to evaluate the conversion rate p of
the monomer to the polymer and the amount \-p of remaining acrylates: \-p = 2R (Appendix
2).
34

Chapter II Synthesis andpolymerization ofacrylic monomers
Figure 5 shows the evolution of the remaining acrylate function versus the time of
treatment of DEAP (a) without initiator, (b) with 5% (w/w) of Darocurl 173 and (c) with 5%
(w/w) of Irgacure 819.
0 -,—
0 5 10 15 20 25
Time (min)
Figure 5 Kinetics of homopolymerization induced by an argon MW plasma of (a) DEAEP,
(b) DEAEP+ Darocur 1173 (5% (w/w)), (c) DEAEP+ Irgacure 819 (5% (w/w))
The results clearly show that the addition of a photoinitiator increases the conversion
rates of polymerization of DEAEP. After 10 min under argon plasma exposure, the percentage
of conversion is 43% without initiator, and 70% and 83%) with Darocur 1173 and Irgacure
819, respectively. This result proves the role of the vacuum UV radiations in the plasma.
Irgacure 819 (BAPO) appears to be the most effective for the polymerization of DEAEP in Ar
MW plasma. This difference in efficiency can be attributed either to the high reactivity of the
phosphinoyl radicals generated by Irgacure 819 compared to the benzoyl radicals, or to a
faster decomposition of the photoinitiator under argon plasma. The decomposition of a
photoinitaitor is mainly due to the wave emission from the plasma ambient gas, in this case
argon. Previous studies [25] have shown that a RF argon plasma (P 100 W, pressure 1.6 torr)
emit UV radiations in the range of 386-414 nm. This region corresponds to the strongest
absorption of Irgacure 819. The difference of efficiency between Irgacure 819 and Darocur
1173 has already been observed when they were used to initiate photopolymerization of
acrylate monomers [29-31]. The great reactivity of the of phosphinoyl radicals toward the
35
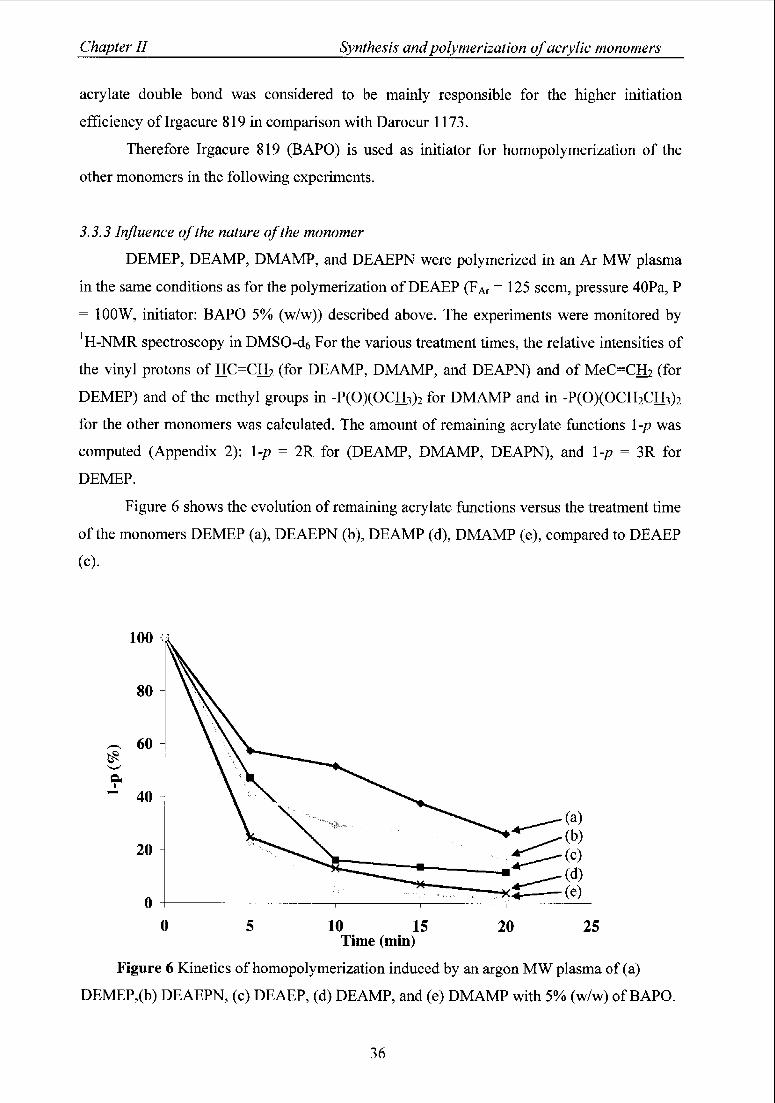
Chapter II Synthesis andpolymerization ofacrylic monomers
acrylate double bond was considered to be mainly responsible for the higher initiation
efficiency of Irgacure 819 in comparison with Darocur 1173.
Therefore Irgacure 819 (BAPO) is used as initiator for homopolymerization of the
other monomers in the following experiments.
3.3.3 Influence ofthe nature ofthe monomer
DEMEP, DEAMP, DMAMP, and DEAEPN were polymerized in an Ar MW plasma
in the same conditions as for the polymerization of DEAEP (FAr = 125 seem, pressure 40Pa, P
= 100W, initiator: BAPO 5% (w/w)) described above. The experiments were monitored by1H-NMR spectroscopy in DMSO-d6 For the various treatment times, the relative intensities of
the vinyl protons of HC=CH2 (for DEAMP, DMAMP, and DEAPN) and of MeC=CH2 (for
DEMEP) and of the methyl groups in -P(0)(OCHj)2 for DMAMP and in -P(0)(OCH2CH3)2
for the other monomers was calculated. The amount of remaining acrylate functions \-p was
computed (Appendix 2): \-p = 2R for (DEAMP, DMAMP, DEAPN), and 1-/7 = 3R for
DEMEP.
Figure 6 shows the evolution of remaining acrylate functions versus the treatment time
of the monomers DEMEP (a), DEAEPN (b), DEAMP (d), DMAMP (e), compared to DEAEP
(c).
0 5 10 15 20 25Time (min)
Figure 6 Kinetics of homopolymerization induced by an argon MW plasma of (a)
DEMEP,(b) DEAEPN, (c) DEAEP, (d) DEAMP, and (e) DMAMP with 5% (w/w) of BAPO.
36

Chapter II Synthesis andpolymerization ofacrylic monomers
The results clearly show that all the monomers polymerized under an Ar MW plasma
in presence of BAPO. The conversion rates is higher for the acrylate monomers (DEAEPN,
DEAEP, DEAMP, DMAMP) than for the methacrylate monomer (DEMEP (a)). After 10 min
of treatment, more than 70% of the acrylate monomers are converted into polymers, while for
DEMEP only 50% are converted. The observation that a methacrylate monomer is more
difficult to homopolymerize than an acrylate monomer has already been made [28,32,33], In
comparison with DEAEP (c), the degrees of conversion of the acrylate phosphonate
monomers (DEAMP and DMAMP) are higher. After 15 min, more than 90% of the
phosphonate monomers are converted into polymers. The percentage of conversion of the
acrylate phosphoramidate (DEAEPN) is lower than DEAEP. This difference could be
attributed to the viscosity of DEAEPN monomer which is higher than DEAEP. With
increasing time of polymerization the viscosity of the polymer-monomer mixture raises
leading to mobility restrictions of the polymer radicals, and consequently to a prematurely end
of the polymerization.
The plasma-induced polymerization of BisDEAEPN could not be carried out in bulk
in presence of BAPO. This monomer is highly viscous oil and it could not be mixed with
BAPO. In solution in methanol, the degree of conversion was about 40% after 20 min. The
low conversion can be attributed to the fact that under vacuum the solvent is evaporated and
the polymerization stops early because of the high viscosity of the monomer and related
mobility restrictions.
4 Characterization of acrylic polymers containing phosphorus
The acrylic polymers containing phosphorus obtained after 20 min of treatment in Ar
MW plasma under the conditions described in the previous section were characterized by
spectroscopic methods (IR and 'H-NMR) and by thermogravimetric analysis (TGA). Rubbery
polymer films were obtained with DEAEP, DEAMP, DMAMP, DEAEPN, and BisDEAEPN,
and a glassy polymer film with DEMEP. PolyDEAMP and polyBisDEAEPN were soluble in
ethanol, methanol and in DMSO. The other polymers formed gels in DMSO in the exception
ofpolyDEMEP, which was not soluble.
4.1 IR (ATR) spectroscopy
The infrared spectroscopy was used to observe the changes of the chemical structure
during polymerization of the different monomers. The IR spectra of the monomers and of the
37

Chapter II Synthesis andpolymerization ofacrylic monomers
polymers are compared. As examples the IR spectra of DEAEP (a) compared to polyDEAEP
(b), and DEAEPN (c) compared to polyDEAEPN (d) are shown in Figure 7.
3600 3100 2600 2100 1600 1100 600
Wavenumber (cm" )
Figure 7 IR spectra of (a) DEAEP, (b) polyDEAEP, (c) DEAEPN, (d) polyDEAEPN
In the IR spectra of polyDEAEP (b) and polyDEAEPN (d), it can be observed that the
characteristic absorption bands of acrylic group v (C=C) at 1636 cm"1 and 5 (=CH2) at 1410
cm"1 present in the spectra of DEAEP (a) and DEAEPN (b) have disappeared. The similar
observation (disappearance of the acrylic group) in the spectra was made for all of other
polymers. Furthermore the other functional groups of the monomer are present in the
polymer. So, the plasma-induced polymerization of acrylic monomers containing phosphorus
occurs with the retention of the properties of the monomer and no fragmentation of the
monomer molecules take place. These facts suggest that vinyl-type polymerization has mainly
proceeded.
4.2 HNMR spectroscopy
The H NMR was used to determine the monomer conversion; it can be also used to
compare the tacticity of the polymer obtained by plasma and the one obtained by UV. Figure
8 shows the 'H NMR of DEAMP (a), and polyDEAMP (b) obtained by plasma and
polyDEAMP (c) obtained by UV.
38
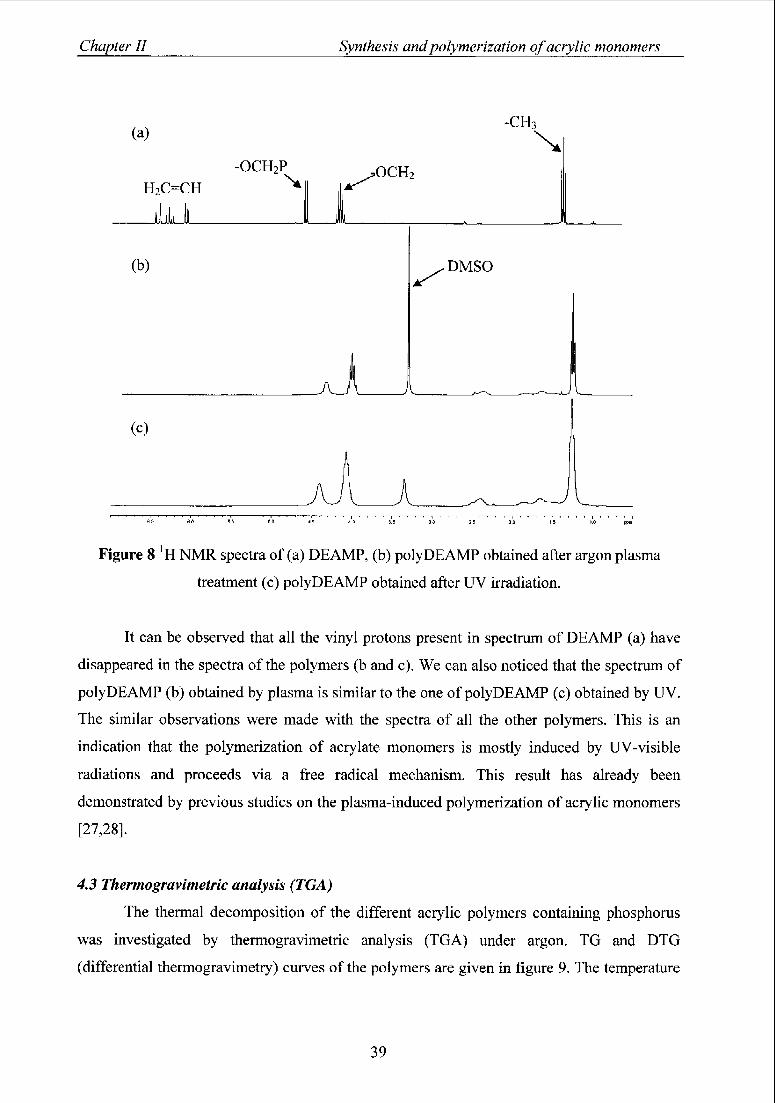
Chapter II Synthesis andpolymerization ofacrylic monomers
(a)
H2C=CH
_jliJl_
(b)
-CH3
X-OCH2P
^iOCH,
L-
J^J
SDMSO
A
(c)
Figure 8 H NMR spectra of (a) DEAMP, (b) polyDEAMP obtained after argon plasma
treatment (c) polyDEAMP obtained after UV irradiation.
It can be observed that all the vinyl protons present in spectrum of DEAMP (a) have
disappeared in the spectra of the polymers (b and c). We can also noticed that the spectrum of
polyDEAMP (b) obtained by plasma is similar to the one of polyDEAMP (c) obtained by UV.
The similar observations were made with the spectra of all the other polymers. This is an
indication that the polymerization of acrylate monomers is mostly induced by UV-visible
radiations and proceeds via a free radical mechanism. This result has already been
demonstrated by previous studies on the plasma-induced polymerization of acrylic monomers
[27,28].
4.3 Thermogravimetric analysis (TGA)
The thermal decomposition of the different acrylic polymers containing phosphorus
was investigated by thermogravimetric analysis (TGA) under argon. TG and DTG
(differential thermogravimetry) curves of the polymers are given in figure 9. The temperature
39

Chapter II Synthesis andpolymerization ofacrylic monomers
at which the rate of volatilization reached a maximum is designated by Tmax- The TGA and
DTG data of the polymers are given in Appendix 3.
(a) PolyDMAMP
PolyDEAEPN
PolyDEAMP
PolyDEAEP
PolyBisDEAEPN
PolyDEMEP
15 115 215 315 415
Temperature (°C)515
(b)
0
-5
's -10
B
£-\5oH
Q -20
-25
-30
-35
piM<^»»»i^»wim»yymmp iMM^BW^W
15 115 215 315 415
Temperature (°C)
515
Figure 9 (a) TG and (b) DTG curves of polyDMAMP, polyDEAEPN, polyDEAMP,
polyDEAEP, polyBisDEAEPN, and polyDEMEP.
40

Chapter II Synthesis andpolymerization ofacrylic monomers
PolyDEMEP and polyDEAEP start to decompose around 230°C and 235°C,
respectively. The TG and DTG curves show two significant areas of weight loss. For
polyDEMEP at Tmax=260°C (weight loss 42.4%) and 310°C (18.9%). For polyDEAEP, the
first stage of weight loss occurs at Tmax=266°C (29.3%) and the second at 310°C (29.3%).
Previous studies [34] have demonstrated that for polyDEMEP, the first region of weight loss
correspond to the decomposition of the phosphate linkage and the second region to the main-
chain random scission and char formation. This mechanism can also be attributed to
polyDEAEP, since both polymers exhibit almost the same DTG peaks. Char yields of about
19.5% and 31.2% for polyDEMEP and polyDEAEP, respectively are recorded at 570°C.
The decomposition of polyDEAMP and polyDMAMP begin around 240°C and
244°C, respectively. In the case of polyDEAMP, two stages of weight loss were identified: at
Tmax=290°C (33.6%) and 310°C (8.8%). On the TG and DTG curves of polyDMAMP only
one peak (Tmax=324°C) is observed and the weigh loss is 37.2%. The percentage of char at
570°C is about 32.1% and 40.0% for polyDEAMP and polyDMAMP, respectively.
For the phosphoramidate polymers, the decomposition starts around 215°C for
polyDEAEPN and 204°C for polyBisDEAEPN. Three stages of weight loss were found for
polyDEAEPN. The first one at Tmax= 240°C (25.7%) is the most important and the two
following at Tmax= 304°C (13.97%) and 368°C (5.5%). For polyBisDEAEPN, the
decomposition occurs in two steps. The first step at Tmax= 231°C (25.9%) and the second one
at Tmax= 266°C (14.0%). The percentage of remaining residue at 570°C is about 34.2%
(polyDEAEPN) and 29.6% (polyBisDEAEPN).
From the results of the TG analysis, we can conclude that the acrylate
phosphoramidate polymers have a lower decomposition temperature than the acrylate
phosphate and phosphoanate polymers. The methacrylate polymer (PolyDEMEP) gave the
lowest weight of residue.
4.4 Comparison ofTG curves ofthe polymers with untreated cotton and PANfabrics
The TG thermograms of PAN, cotton, and the different acrylic polymers containing
phosphorus of this study are shown in figure 10.
41
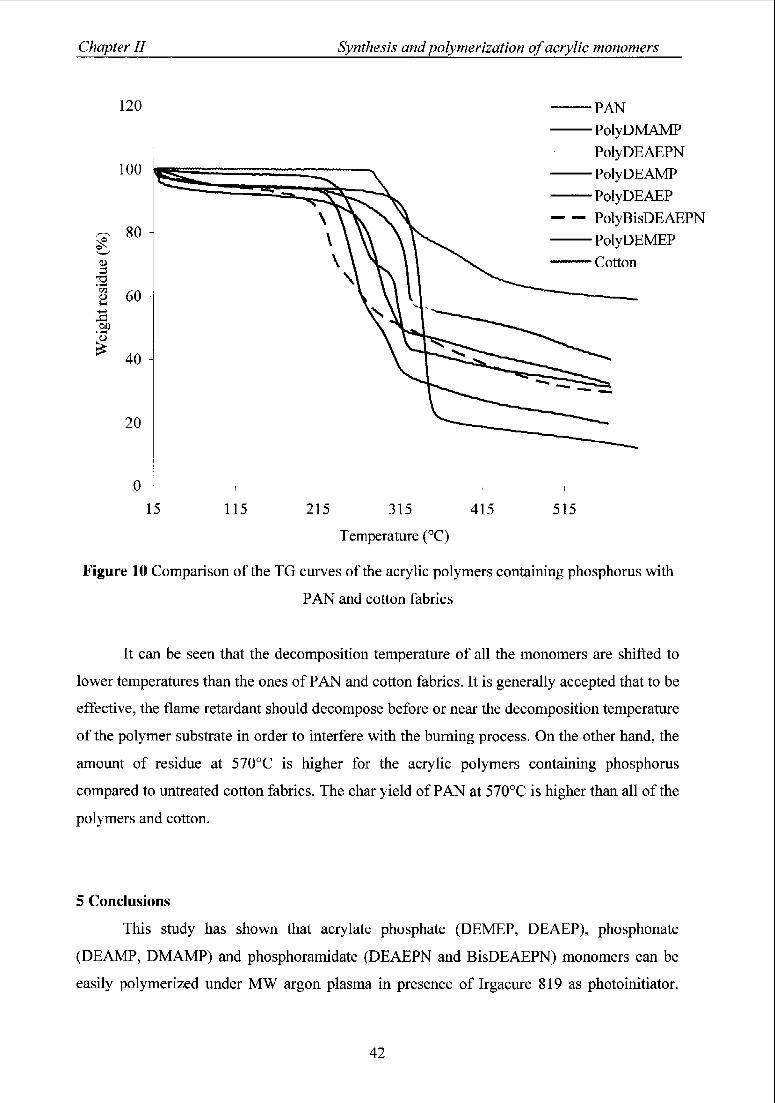
Chapter II Synthesis andpolymerization ofacrylic monomers
120
100
80
T3
I 60
I40
20
0
15 115
PAN
PolyDMAMP
PolyDEAEPN
PolyDEAMP
PolyDEAEP
PolyBisDEAEPN
PolyDEMEP
Cotton
515215 315 415
Temperature (°C)
Figure 10 Comparison of the TG curves of the acrylic polymers containing phosphorus with
PAN and cotton fabrics
It can be seen that the decomposition temperature of all the monomers are shifted to
lower temperatures than the ones of PAN and cotton fabrics. It is generally accepted that to be
effective, the flame retardant should decompose before or near the decomposition temperature
of the polymer substrate in order to interfere with the burning process. On the other hand, the
amount of residue at 570°C is higher for the acrylic polymers containing phosphorus
compared to untreated cotton fabrics. The char yield of PAN at 570°C is higher than all of the
polymers and cotton.
5 Conclusions
This study has shown that acrylate phosphate (DEMEP, DEAEP), phosphonate
(DEAMP, DMAMP) and phosphoramidate (DEAEPN and BisDEAEPN) monomers can be
easily polymerized under MW argon plasma in presence of Irgacure 819 as photoinitiator.
42

Chapter II Synthesis andpolymerization ofacrylic monomers
The kinetic of homopolymerization of the monomers were monitored by H NMR
spectroscopy in DMSO-dö. The results showed that the order of the reactivity is DEMEP <
DEAEPN < DEAEP < DEAMP < DMAMP. BisDEAEPN could not be polymerized in bulk
due to its high viscosity. H NMR and IR (ATR) analyses allowed us to confirm that the
polymerization of the acrylic monomers containing phosphorus proceeds by a radical
mechanism and that the polymerization under MW argon plasma is mostly induced by UV-
Visible radiations. The thermal behavior of the polymers was studied by thermogravimetric
analysis (TGA). The TG thermograms of the polymers showed that all the polymers
decompose at lower temperatures than cotton and PAN fabrics. For all the polymers, the
percentage of remaining residue at 570°C was higher compared to cotton, and lower than for
PAN. These results indicated that the polymers are thermally stable and can be potential flame
retardant for PAN and cotton fabrics.
43

Chapter II Synthesis andpolymerization ofacrylic monomers
6 References
[I] U. Quittmann, L. Lecamp, W. El Khatib, B. Youssef, C. Brunei, Macromol. Chem.
Phys. 202(2001)628.
[2] A.F. Grand, CA. Wilkie, editors, Fire retardancy of polymeric materials, Marcel
Dekker, Inc., New York (2000).
[3] G.E. Zaikov, S.M. Lomakin, Modern polymer flame retardancy, VSP,Utrecht (2003).
[4] A.R. Horrocks, D.Price, editors, Fire retardant materials, Woodhead publishing Ltd
and CRC press LLC, Cambridge (2001).
[5] H. Herlinger, G. Hardtmann, F. Hermanutz, R. Schneider, U. Einsele, Melliand
Textilber. 72 (1991) E141.
[6] N. C. Reghunadhan, G. Clouet, J. Brossas, J. Polym. Sei. Part A: Polym. Chem. 26 (7)
(1988)791.
[7] N. C. Reghunadhan, G. Clouet, Eur. Polym. J. 25 (3) ( 1989) 251.
[8] T. Jeanmaire, Y. Hervaud, B. Hervaud, Phosphorus Sulfur Silicon Relat. Elem. 177 (5)
(2002)1137.
[9] N. C. Reghunadhan, G. Clouet, Y. Guilbert, Polym. Degrad. Stab. 26 (1989) 305.
[10] M. Banks, J. R. Ebdon, M. Johnson, polymer 35 (16) (1994) 3470.
[II] J. R. Ebdon, D. Price, B.J. Hunt, P. Joseph, F. Gao, G. J. Milnes, L. K. Cunliffe,
Polym. Degrad. Stab. 69 (2000) 267.
[12] D. Price, K. Pyrah, R. Hull, G. J. Milne, J. R. Ebdon, ,B.J. Hunt, P. Joseph, Polym.
Degrad. Stab. 77 (2002) 227.
[13] A. Gentilhomme, M. Cochez, M. Ferriol, N. Oget, J. L. Mieloszynski, , Polym.
Degrad. Stab. 82 (2003) 347.
[14] C. M. Chan, Polymer surface modification and characterization, Hanser/Gardner
publications, Inc.(1994).
[15] H. Yasuda in New methods of polymer synthesis, J. R. Ebdon, G. C. Eastmond
(editors) Vol 2, Blackie Academic and professional, London a.o (1995).
[16] H. Yasuda, Plasma Polymerization, Academic Press, INC, New York (1985).
[17] Y. Osada, A. T. Bell, M. Shen, J. polym. Sei., polym. Lett. Ed. 16 (1978) 309.
[18] Y. Osada, M. Takase, J. polym. Sei., polym. Lett. Ed. 21 (1983) 643
[19] M. Kuzuya, K. Kamiya, T. Kawaguchi, T. Okuda, J. polym. Sei., polym. Lett. Ed. 21
(1983)509.
44

Chapter II Synthesis andpolymerization ofacrylic monomers
[20] M. Kuzuya, T. Kawaguchi, T. Daikyo, T. Okuda, J. polym. Sci., polym. Lett. Ed. 21
(1983)515.
[21] M. Kuzuya, T. Kawaguchi, Y. Yanagihara, S. Nakai, T. Okuda, J. Polym. Sci., Part A,
Polym. Chem. 24 (1986) 707.
[22] T. Hirotsu, J. polym. Sci., polym. Lett. Ed. 21 (1983) 688.
[23] J.C. Brosse, F. Epaillard, G. Legeay, Eur. Polym. J. 19 (5) (1983) 381.
[24] J.C. Brosse, F. Epaillard, G. Legeay, Eur. Polym. J. 19 (9) (1983) 743.
[25] J.C. Brosse, F. Epaillard, G. Legeay, Eur. Polym. J. 19 (9) (1983) 749.
[26] F. Epaillard, J.C. Brosse, G. Legeay, Eur. Polym. J. 23 (3) (1987) 233.
[27] F. Epaillard, J.C. Brosse, J. Appl. Polym. Sei. 38 (1989) 887.
[28] F. Hochart, J. Levalois-Mitjavilie, R. De Jaeger, Polymer 41 (2000) 3159.
[29] C. Decker, K. Zahouily, D. Decker, T. Nguyen, T. Viet, Polymer 42 (2001) 7551.
[30] C. Decker, Macromol. Rapid. Commun. 23 (2002) 1067.
[31] X. Wu, H. A. Reed, L. R. Rhodes, E. Elce, R. Ravikiran, R. A. Shick, C. L.
Henderson, S. A. Bidstrup Allen, P. A. Kohi, J. Appl. Polym. Sci. 88 (2003) 1186.
[32] K. Studer, C. Decker, E. Beck, R. Schwalm, Progress in Organic Coatings 48 (2003)
101.
[33] O. Soppera, C. Croutxé-Barghorn, J. Polym. Sci., Part A, Polym. Chem. 41 (2003)
831.
[34] L. H. Perng, C. J. Tsai, Y. C. Ling, S. D. Wang, C. Y. Hsu, J. Appl. Polym. Sci. 85
(2002) 821.
45

Chapter II Synthesis andpolymerization ofacrylic monomers
Seite Leer /
Blank leaf

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
Chapter III: Flame retardancy of polyacrylonitrile and cotton fabrics
1 Introduction
Looking into the history, most fire accidents have their origin in the domestic
environment [1,2].Therefore, natural and man-made fibers, which are widely used for clothing
and home furnishings (e.g., furniture, coverings, curtain material, wall hangings, carpets),
should be flame retarded. It is generally accepted that textiles with LOI value greater than 26
are flame retardants [1]. The limiting oxygen index (LOI) values of several textiles are given
in Table 3. It can be seen that, most of the textiles listed in this table are flammable in the air.
Table 3 Limiting oxygen index (LOI) of the more commonly used fibers from Ref [1]
Fiber LOI (%)
Acrylic 18.2
Cotton 18.4
Polypropylene 18.6
Viscose 18.9
Nylon 6 and 6.6 20-21.5
Polyester 20-21
Wool 25
Modacrylic 29-30
Meta-aramid (Nomex) 29-30
Para-aramid (Kevlar) 29
Flame retardancy may be conferred on synthetic and natural textile fabrics by various
routes as shown in Figure 11. Synthetic fibers may be rendered flame retardant by one of the
following three ways: (i) copolymerization of flame retardant monomers into the basic fiber-
forming polymer chains, (ii) incorporation of a flame retardant additive in the polymer melt or
solution prior to extrusion, or (iii) chemical after-treatments which include surface treatments,
coatings and functional finishes which become a part of the final fiber structure, whereas
natural fibers may be flame retarded mainly by chemical after-treatments.
47

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
Monomer < Use of FR comonomers (i)
Polymerization *
Polymer* Use of additives (ii)
Spinning
Fibers Modification by grafting (iii)
Fabric < FR Finishing (iii)
1Flame retardant articles
Figure 11 Flame retardant (FR) finishes, (from Ref [2])
Synthetic fibers produce by methods (i) and/or (ii) are often said to be inherently flame
retardant fibers. They are the most durable to laundering and service although additive
leaching may be a problem. However, these fibers are difficult to produce because of
problems of compatibility during polymerization or extrusion, which limit considerably the
range of flame retardant monomers and compounds which can be used.
For chemically after-treated textiles, the flame retardants are generally applied by
conventional padding methods such as pad-dry, pad-dry-(heat or chemical cure) and by back-
coating methods [1,3,4]. The durability depends on the strength with which the flame
retardant adheres or bonds to the fiber/fabric surface. Topical applications and coatings,
which require the presence of binding agents or resins, are often not durable. A high level of
durability can be achieved with functional finishes if there is a reaction with or
polymerization within the fiber structure.
Most of the emphasis is currently on flame retardant treatments which are durable to
multiple launderings. Attempts have been made to modify conventional and to develop new
processing technological procedures in order to improve efficiency, reduce energy
requirements, and reduce effluents problems [3].The surface grafting of flame retardant
compounds on the surface of textile fibers or fabrics as an alternative approach to the
conventional processes has been investigated. Various grafting techniques such as UV [5,6],
gamma irradiation [7-10], and redox systems [11] have been mostly applied on natural fibers.
48

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
As already described in the introduction chapter, the cold plasma technique is a new
technology which allows the covalent grafting of small functional groups and macromolecular
compounds onto the surface of polymeric materials. The grafting and polymerization of a
monomer onto polymeric substrates such as textile fabrics using cold plasma technique can be
carried out by two different ways as summarized in Figure 12.
JsLPlasma M'
Reactive gas
SgP
^b)
Inert gas plasma Plasma M'
Deposited or graftedpolymer
S: Substrate
M: non volatile
monomer
M': volatile
compound
j.*A _.*ï*x* ,' > *
UV
Activation Dipping in a
solution ofM
S*+M
Impregnation
Figure 12 Surface grafting using cold plasma technique
The first approach (a) is the direct modification of the substrate in plasma of a reactive
gas or a volatile monomer. The polymer deposits or grafts onto the surface of the substrate.
This approach is solvent-free and environmentally friendly. The grafted polymer obtained
with this procedure is highly crosslinked. However, the disadvantage is that there is no
retention of the monomer structure due to the fragmentation of the monomer molecules during
the excitation and deposition processes. Thus if the monomer bears a property which depends
on its structure, this property will be deactivated. Another disadvantage of this method is that
it is limited to monomer that can be volatilized and can not be applied to heavy monomers.
The second way involves a two-step procedure (b). Plasma of inert gases such as
argon, nitrogen, helium or oxygen is used for activation of the substrate to form free radicals
on the substrate surface. The activated polymer substrate is then treated with the monomer
which is either in vapor state or as a solution in a suitable solvent. In the latter case the
grafting is subsequently performed either thermally or by UV irradiation. Most of the work
carried out using this latter approach concern vinyl or acrylic non volatile monomers [12-14].
Since plasma of inert gas like argon can induce polymerization of monomers containing
49

Chapter III Flame retardancy ofpolyacrylonitrile and cotton fabrics
polymerizable functional groups, the question is why not performing the activation and the
polymerization steps in the same plasma reactor. This novel approach is the plasma-induced
graft polymerization procedure (PIGP). The simultaneous grafting and polymerization of
acrylic monomer is performed using the cold plasma technique.
A few studies on the use of cold plasma technique to flame retard textiles have been
reported (see Chapter I. Section 6). Simionescu et al.[15,16] investigated the flame retardancy
of rayon fibers and fabrics grafted under RF nitrogen plasma with non-volatile phosphorus
and halogen-containing compounds. This procedure is similar to the way (a), even the
compounds are non volatile. The "monomers" were aromatic or simple organic (without
polymerizable structure) compounds and the polymers were formed by plasma-state
polymerization leading to fragmentation of "monomer" molecules. Akovali et al. [17,18]
studied the flame retardancy of polyacrylonitrile (PAN) fibers and polyester fabrics treated
with several volatile monomers (way a) such as hexamethyldisiloxane (HMDS) at various
plasma powers and exposure time.
Thus far, the flame retardancy of PAN and cotton fabrics by grafting and
polymerization of acrylic monomers containing phosphorus using the cold plasma technique
has not been investigated.
2 Flame retardancy of polyacrylonitrile (PAN) textiles
Acrylic fibers, which are widely used in the clothing and home textile sectors, have a
limiting oxygen index (LOI) of 18.2% which is the lowest among the commonly used textile
fibers. The high flammabiiity of acrylic textiles under burning conditions derives from the
intensive exothermic pyrolysis reaction which occurs at about 300°C for most commercial
variants [5]. This reaction gives rise to formation of flammable nitriles and carbon monoxide
and oligomerisation of the adjacent pendant nitrile groups to a carbonaceous char. Any
effective flame retardant should therefore reduce the volatilization tendency and enhance char
formation. Phosphorus compounds, which act mainly by promoting char formation, have been
demonstrated to be effective as flame retardants for acrylic fibers [1,4,19], During burning,
they decompose to polyphosphoric acids, which act as nucleophilic agents to promote
oligomerisation of the pendant nitrile groups, leading to cyclization and char formation.
Various phosphorus compounds such as ammonium phosphates, polyphosphates, and
acrylic monomers containing phosphorus have been used to flame retard acrylic textiles
[2,19]. They can be introduced either (i) as comonomers during polymerization, (ii) as
50

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
additives in the spinning dope prior to extrusion or (iii) use for chemical treatments of fibers
or fabrics.
Flame retardant acrylic textiles are often difficult to produce by methods (i) and/or (ii)
because of problems of compatibility during polymerization or spinning. Most of the
available literature is on modacrylics (copolymers of acrylonitrile and vinylchloride
/vinylidene chloride in the ratio of 60:40). They provide sufficient flame retardancy, however,
because of the presence of halogen, they cannot be claimed to be safe anymore for the use in
confined places due to their toxicity when burning. The surface treatments are often not
resistant to washing [1,20]. In spite of many efforts there is still no satisfactory solution to
flame retard acrylic textiles with permanent effect (laundry resistance).
In this section, the surface modification of acrylic fabrics by plasma-induced graft
polymerization of acrylate phosphate (DEMEP, DEAEP) and phosphonate (DEAMP,
DMAMP) monomers is investigated. DEMEP and DMAMP have already been used as
additives (in the polymeric form) and as comonomers to produced flame-resistant acrylic
fibers by Herlinger et al. [20]. Therefore this study allows for a comparison of the surface
approach with the additive one.
2.1 graft polymerization of acrylate phosphate (DEMEP, DEAEP) and phosphonate
(DEAMP, DMAMP) monomers onto PANfabrics
2.1.1 Graft-polymerization procedure
The optimum conditions (¥\t = 125 seem, pressure 40 Pa, P = 100 W, initiator: BAPO
5% (w/w)) found for the plasma-induced polymerization of acrylic monomers containing
phosphorus in the previous chapter are applied for the graft polymerization of DEMEP,
DEAEP, DEAMP and DMAMP on PAN fabrics. The experiments are performed using the
one step (PIGP) procedure depicted in Figure 13.
rs.!
• Dipping( ' in solution of
M
Figure 13 Procedure for the Ar plasma-induced graft-polymerization of monomers
2. Ar
plasma
3. Washing
4. Drying
51

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
In a first step, pieces of woven PAN fabrics (52 x 140 mm) are weighed and then
immersed at room temperature for one minute in 20 ml of an ethanol solution containing 100
g/L to 300 g/L of DEMEP, DEAEP, DEAMP, or DMAMP in the presence of 5% (w/w) of the
photoinitiator (BAPO) and various amounts (0, 10 and 20% (w/w)) of the cross linking agent.
These impregnated fabrics are then pressed to evacuate the excess of the solution, placed onto
glass plates and submitted to a MW argon plasma (FAr= 125 seem, base pressure= 40 Pa, P =
100 W, t = 15 min). After treatment the samples are washed for 24 hours in a soxhlet
apparatus with ethanol to remove the monomer and non-grafted homopolymer, then washed
in water for 24h (in a soxhlet) and then air-dried. The grafting yield is evaluated as the weight
increase of the sample after washing and air-drying; it is expressed as the percentage increase
of weight and calculated as follows:
where, Wo and Wg are the weights of the PAN fabric samples before and after grafting (after
washing and air-drying), respectively.
In order to find the optimum conditions for plasma-induced graft polymerization of
acrylate phosphate and phosphonate monomers onto PAN fabrics, the effects of the monomer
concentration, of the amount and nature of a crosslinking agent on the grafting yield is
investigated.
Note that a phosphorus content of about 3 % (w/w) on the fabric is generally used to
impart adequate flame retardancy to PAN textiles [2,20], To attain this value, a minimum
amount in the range of 20 to 25 % (w/w) of grafted polymer is necessary, depending on the
monomer phosphorus content. Preliminary studies are carried out using DEAEP.
2.1.2. Effect ofthe concentration ofDEAEP on the grafting yield
Samples of PAN fabrics impregnated with ethanolic solutions containing 100g/L,
200g/L or 300 g/L of DEAEP, 5% (w/w) of BAPO and 10% (w/w) of crosslinking agent
(EGDMA) (Scheme 8) are submitted under an argon microwave plasma for 15 minutes.
EGDMA (ethylene glycol dimethacrylate) is a difunctional methacrylate monomer.
52

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
O CH3
Ethylene glycol dimethacrylate
Scheme 8 Chemical structure ofEGDMA
The effect of the monomer concentration on the amount of deposited and grafted
polymer is shown in Figure 14.
D Before washing
Washed with EtOH
Washed with H20
ü
35
30
^ 25
20
15
£ 10
5
0
100 200 300
[DEAEP] g/L
Figure 14 Effect of the concentration ofDEAEP on the percentage of grafting on PAN
fabrics treated in a MW argon plasma.
It can be observed that, the amount of deposited polymer before and after washing and
air-drying, increases linearly with the monomer concentration. After washing with ethanol
(soxhlet 24h), which is the solvent of the monomer and air-drying, the weight loss is about
43.5% for monomer concentrations of 100 and 200g/L and 30.4% for a concentration of
300g/L. In order to identify the products present in the solution after washing, H NMR
analysis was performed. The H NMR spectrum of the washing ethanol solution reveals only
the presence of the signals corresponding to the polymer. This result demonstrates that the
53

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
weight loss after washing in ethanol is mainly due to the non-grafted homopolymer. So, the
amount of polymer which remains on the PAN fabric after washing with ethanol is the grafted
polymer. The grafting yields increase from 7.5% to 22.9% with monomer concentrations of
100 and 300g/L, respectively.
After washing the grafted PAN fabric samples with water (soxhlet 24h), no noticeable
weight loss is observed. This result indicates the stability of the grafted polymer to hydrolysis.
Finally, it is interesting to note that the grafting yields are reproducible for each monomer
concentration since similar values were obtained in a series of tests.
2.1.3 Effect ofthe crosslinking agent (EGDMA)
The effect of the crosslinking agent and of its concentration on the graft
polymerization of DEAEP onto PAN fabrics is also investigated. The different experiments
are performed with the crosslinking agent EGDMA (10 and 20% (w/w)) and without at
monomer concentration of 200g/L, using the graft polymerization procedure described above.
The grafting yields obtained after washing (soxhlet ethanol and water) and air-drying are
shown in Figure 15.
^oN
40
35
30
25
| 2°
3 15
10
5
0
Before washing
Washed with EtOH
0 10 20
%wt ofEGDMA
30
Figure 15 Effect of the amount ofEGDMA on the percentage of grafting ofDEAEP on PAN
fabrics treated in a MW argon plasma
54

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
It can be seen that without crosslinking agent the grafting yield is very low (about
2.5%o) and weight loss is around 89 %. This result indicates that homopolymerization and
grafting are concurrent processes, and that in the absence of EGDMA the
homopolymerization prevails significantly over the grafting reaction. In the presence of the
crosslinking agent, the percentage of grafting is substantially enhanced to 13.7% with 10%
(w/w) of EGDMA. It is clear that the presence of a crosslinking agent is necessary for the
grafting, because it is a coupling agent between the growing polymer and the fiber. The
amount of grafted polymer also increases from 13.5% to 23% with the concentration of
EGDMA of 10% and 30%, respectively. However in presence of high amounts of the
crosslinking agent, the stiffness of the treated PAN fabrics can be affected.
2.1.4 Effect ofthe nature ofthe crosslinking agent
The previous results show that it is necessary to use a crosslinking agent for an
efficient grafting of DEAEP onto PAN fabrics. Therefore, the effect of two other crosslinking
agents EGDA (ethylene glycol diacrylate) and TTEGDA (tetra (ethylene glycol) diacrylate)
on graft polymerization of DEAEP on PAN fabrics is investigated and compared with the
results obtained with EGDMA. EGDA and TTEGDA (Scheme 9) are difunctional acrylate
monomers which differ by the length of the space between the two acrylate functions.
HO HO
OH OH
Ethylene glycol diacrylate Tetra(ethylene glycol)diacrylate
Scheme 9 Chemical structure ofEGDA and TTEGDA
Different experiments are performed with 20% (w/w) of the crosslinking agents at
monomer concentration of 200g/L, 5% (w/w) of BAPO. The grafting yields obtained are
shown in Figure 16.
55

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
45
40
35
30
25
5 20
C5^
%
D Before washing
D Washed with EtOH
15
10
5
0
EGDMA EGDA TTEGDA
crosslinking agent
Figure 16 Effect of the nature of crosslinking agent on the percentage of grafting of DEAEP
on PAN fabrics treated in a MW argon plasma
It can be observed that the amount of deposited polymer (before washing) is almost
the same for all the three crosslinking agents. However, after washing with ethanol, the
amounts of grafted polymer vary: 24.7% with EGDA, 21.7% with TTEGDA, and 16.9% with
EGDMA and the percentage of weight loss obtained with EGDA (20%) is less than the 33%
and 48 % obtained with TTEGDA and EGDMA, respectively. The variation of grafting yields
among the three crosslinking agents can be explained by the difference of reactivity in
presence of DEAEP. The smallest amount of grafted polymer obtained with EGDMA can be
attributed to the lower reactivity of the methacrylate compared to the acrylate (EGDA and
TTEGDA. The difference of the amount of grafted polymer between EGDA and TTEGDA
can be explained by the difference of solubility of the monomer and the crosslinking agents.
From the results obtained in this preliminary study we can conclude that the presence
of a crosslinking agent is necessary for the grafting of DEAEP onto PAN fabrics. With the
plasma-induced graft polymerization (PIGP) procedure it is possible to attain about 24% of
grafted polymer onto PAN fabrics with DEAEP. This level of grafting can be reached by
increasing either the monomer concentration or the amount of crosslinking agent. EGDA is
the most efficient crosslinking agent in this study and therefore it is used as crosslinking agent
for the plasma-induced graft polymerization of the other monomers.
56

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
2.1.5 Effect ofthe nature ofthe monomer
In this section the influence of the nature of the monomer on graft polymerization onto
PAN fabrics is investigated. Different tests are performed at various monomer concentrations
(200g/L and 300g/L) in presence of the crosslinking agent EGDA (10% (w/w)) following the
PIGP procedure described above. The grafting yields obtained after washing (soxhlet ethanol
and water) and air-drying are shown in Figure 17.
30 -.
D 200 g/L
25 -
H 300 g/L
ùû 20 -
Ö
a
2oo 15
o
DEMEP DEAEP DEAMP DMAMP
Monomers
Figure 17 Effect of the concentration of DEMEP, DEAEP, DEAMP and DMAMP on the
percentage of grafting on PAN fabrics treated in MW argon plasma after washing and air-
drying.
It can be observed that the amount of grafted polymer increases slightly with the
monomer concentration. The maximum amount of grafted polymer is reached at about 24%
for all the monomers with exception of methacrylate (DEMEP), which gives only about 10%
of grafting yield independently of the monomer concentration. This result can be explained by
the homopolymerization rate which is slower for DEMEP than for the acrylate monomers.
Note that even when 24% of polymer is grafted, the texture of the PAN fabrics is not visibly
affected. In order to increase the percentage of grafting, the effect of the amount of EGDA is
also investigated.
57

Chapter III Flame retardancy ofpolyacrylonitrile and cotton fabrics
Figure 18 represents the percentage of grafting on PAN fabrics treated with EGDA (0, 10 and
20% (w/w)) at a monomer concentration of 200 g/L, after washing (soxhlet ethanol and then
water) and air-drying following the PIGP procedure.
0% EGDA
10% EGDA
20% EGDA
30
25
20
15
o
^10
5
0
DEMEP DEAEP DEAMP DMAMP
Monomers
Figure 18 Comparison of the percentage of grafting of the monomers (DEMEP, DEAEP,
DEAMP and DMAMP) on PAN fabrics treated with or without EGDA in MW argon plasma
after washing and air-drying
As we can see the grafting yields increase with the amount of EGDA for all the four
monomers and depend on the nature of the monomer. Without crosslinking agent, the amount
of grafted polymer is undetectable on PAN fabrics treated with DEMEP and DEAMP, and
very low with DEAEP (2.5 %) and DMAMP (3.9 %). In presence of 10% to 20% (w/w) of
EGDA the grafting yields augment from 10.6% to 19.2% for DEMEP, from 17.5% to 24.7%
for DEAEP, from 17.7% to 22.5% for DEAMP, and from 19.6% to 28.0% for DMAMP,
respectively. So, for all the monomers, the presence of a crosslinking agent is necessary for an
efficient grafting. The highest percentage of grafting found for DMAMP and the lowest for
DEMEP correlate with the homopolymerization rate which is faster for DMAMP than for the
other monomers (see Chapter II, Section 3).
Like for DEAEP these results indicate that the application of the novel plasma-induced
graft-polymerization procedure allows a minimum amount of about 20% of acrylate
58

Chapter III Flame retardancy ofpolyacrylonitrile and cotton fabrics
phosphate and phosphonate monomers to be grafted and polymerized onto PAN fabrics in
presence ofEGDA as a crosslinking agent.
Further confirmations supporting the presence of the graft polymer onto PAN fabrics
will be obtained by studying the changes of surface characteristics after plasma-induced graft-
polymerization of the different monomers.
2.2 Surface characterization ofthe grafted PANfabrics
2.2.1 IR (ATR)
Both untreated and treated PAN samples were submitted to IR (ATR) analysis. Figure
19 shows the IR (ATR) spectra of untreated (a) and treated (b, c, d, e) PAN samples, after
washing and air-drying.
3
at
(a)
(b)'—^^S
«-v(ON)
(c)
(d)
(e)
900 6002700 2400 2100 1S00 1500 1200
Waverumber(cm )
Figure 19 IR spectra ofPAN fabrics (a) not treated; (b) treated with ethanol solution
containing DEMEP, (c) DEAEP, (d) DMAMP and (e). DEAMP [Monomer] = 200 g/L, 5 %(
w/w) of BAPO, 10 %( w/w) of EGDA, after washing and air-drying.
For these treated samples, one can easily identify the characteristic IR absorptions at
v = 1242, 1026 and 979 cm"1 for DEMEP (b), at v = 1256, 1025 and 978 cm"1 for DEAEP (c),
at v = 1238, 1025 cm"1 for DMAMP (d) and at v = 1242, 1017 and 979 cm"1 for DEAMP (e)
59

C liapter III Flume retardancy of polyat rylonitrite and cotton fabrics
corresponding to the stretching vibrations P_0 and P-O-C respectively These bands indicate
the presence ol a phosphorus containing polymer on the surlace of the PAN fabric. Note that
the stretching vibration of the C-N group of the PAN is still visible on the treated sample
This indicates that the thickness of the deposited polymer layer is less than 1 urn.
222 ShM
1 he grafting of a thin polymer film layer could be easily demonstrated by comparing
ShM photomicrograph of a virgin PAN sample (Hgure20.a) with one treated with DEAEP loi
example (figure 20.b).
Figure 20 SEM photomicrographs of PAN fabrics (a) not tieated; (b) treated with a Dl AFP
(200g/L), 10 % (w/w) of KiDA and 5% (w/w) of BAPO after washing and air-drying.
As shown in Figure 20 a, the surface morphology along the fiber is giooved which
contrasts with the one of the treated sample (1 igure 20.b). It can be observed that the tieated
fiber is covered with polyDLALP. Note that space around the fiber stays free of polymer,
which guarantees the breathability of the fabric.
2.3 Evaluation oftheflame retardancy of the grafted PAN fabrics
2 3 1 LOI measurements
The flame retardancy oi the untreated and treated PAN fabrics is assessed by limiting
oxygen index (LOI) measurements The results obtained are presented in Table 4. I he I OI
values augment when the monomer concentration and the percentage of crosslinking agent are
60

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
increased whereby the amount of grafted phosphorus increases. The results clearly indicate
that the LOI value of the virgin PAN 18.5 increases up to 4 units when treated with the
phosphate monomers (DEMEP and DEAEP), and up to 5 and 8 units with DEAMP and
DMAMP, respectively. Only the fabric treated with the phosphonate (DMAMP) with a LOI
value of 26.5 and phosphorus content of 3.9% can be considered to be self-extinguishing. The
LOI values under 26 obtained with the other monomers can be attributed to the fact that, the
minimum amount of 3% (w/w) of grafted phosphorus necessary to impart adequate flame
retardancy to PAN fabrics could not be reached.
Table 4 LOI values of treated PAN fabrics as a function of monomer type, of the treatment
conditions and percentage of grafting after washing and air-drying
Monomer Treatment conditions %G
%P (w/w)measured on
PAN
LOI (%)
Untreated PAN - - - 18.5
DEMEP 200 g/L, 10% EGDA 10.6 - 20.8
300 g/L, 10% EGDA 11.3 - 21.5
200 g/L, 20% EGDA 19.2 - 21.5
300 g/L, 20% EGDA 20.6 1.6 22
DEAEP 200 g/L, 10% EGDA 17.5 - 21.5
300 g/L, 10% EGDA 23.5 - 22.3
200 g/L, 20% EGDA 24.2 2.0 22
300 g/L, 20% EGDA 26 - 22.3
DEAMP 200 g/L, 10% EGDA 17.7 - 21.8
300 g/L, 10% EGDA 21.9 - 22.5
200 g/L, 20% EGDA 22.5 - 22.5
300 g/L, 20% EGDA 28.5 2.7 23.5
DMAMP 200 g/L, 10% EGDA 20.1 2.3 23.3
300 g/L, 10% EGDA 24.2 - 24
200 g/L, 20% EGDA 28 3.0 24.5
300 g/L, 20% EGDA 39.7 3.9 26.5
61

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
2.3.2. Burning rates
For a better evaluation of the efficiency of the fire retardancy, we have measured the
burning rates of a virgin PAN sample and the polymer grafted samples. The virgin PAN starts
to burn very slowly at 18% percent of oxygen and an increase of the oxygen concentration
increases dramatically the burning rate as shown in Figure 21.
0 4 1 1 !
19.5 20.5 21.5 22.5 23.5 24.5 25.5
% oxygen
Figure 21 Burning rates of untreated PAN and PAN treated fabrics with DEMEP, DEAEP,
DEAMP and DMAMP as a function of the oxygen content in a O2/N2 gas mixture.
[Monomer] = 200 g/L with 5 %( w/w) ofBAPO and 20 %( w/w) of EGDA, after washing and
air-drying.
This behavior changes significantly with the polymer coated samples. At can be
observed, they start to burn at 21%, 22%, 22.5% and 24.5% percent of oxygen for
polyDEMEP, polyDEAEP, polyDEAMP and polyDMAMP grafted fabrics, respectively.
Under these LOI values they are self extinguishing. The fire retardant coating does not only
affect the LOI but also decrease the burning rates. In fact, when the amount of oxygen is
increased, the slope of the curves in Figure 21 is flatter when compared to untreated PAN.
62

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
2.3.3 Thermogravimetric analyses
The thermal behavior of the untreated and treated samples was investigated by
thermogravimetric analysis (TGA) under argon. Typical TGA curves of untreated and PAN
treated with DEAEP, DEMEP, DEAMP and DMAMP are shown in Figure 22.
110
50 4 ,—
-t—
20 220 420 620
Temperature (°C)
Figure 22 TGA thermograms of (a) untreated and treated PAN fabrics with (b) DEMEP, (c)
DEAEP, (d) DEAMP and (e) DMAMP. [Monomer] = 200 g/L with 5% (w/w) of BAPO and
20% (w/w) of EGDA, after washing and air-drying
It can be seen that the untreated PAN fabric starts to decompose around 280°C and the
TG curve show two significant areas of weight loss below and above 350°C. These steps
correspond to oligomerisation or cyclization of nitrile groups and dehydrogenation of PAN
[20]. For the treated PAN the onset of the thermal decomposition is shifted to a lower
temperature (228°C for PAN and DEMEP, 235°C for PAN and DEAEP, 240°C for PAN and
DEAMP, 245°C for PAN and DMAMP) than the untreated one. This corresponds to the
degradation of the polymer coating (polyphosphate or polyphosphonate) which occurs before
the decomposition of the PAN fabric and then reduces the PAN decomposition rate. This
effect can be easily visualized in the 280 to 400°C region where the weight loss curve for the
untreated PAN fabric drops dramatically, while the decomposition of the treated fabrics is
slower. The percentage of remaining residues of treated PAN is about 61%) (DEMEP,
63

( 'hapter III Flame retardancy of polyacrylonitrile and cotton fabrics
DEAEP) and 65% (DEAMP, DMAMP) compared to 58% for virgin PAN at 650°C. These
two factors (reduction of degradation temperature and enhanced thermal stability) indicate
that flame retardant character of the treated labrics was improved
2 3 4 Burning behavior
I he photograph in figure 23 shows the behavior of a treated sample compared to the
untreated one during the LOI test. I he virgin PAN (a) burns completely while most of the
burned material is retained in case of the treated sample
Figure 23 Burning behavior of (a) an untreated and (b) a treated PAN fabric
I his char layer is characteristic for a homogeneously grafted phosphorus containing
polymer on the surface of the substrate
2.4 Durability of theflame retardant treatment
In order to evaluate the durability of the coating to washing, the treated samples were
submitted to the accelerated laundering method proposed by McSherry et al. [21]. They
describe a 4h boiling procedure of llame retardant fabrics with tribasic sodium phosphate
solution as being equivalent to the 50 wash cycle, which is an important criterion for the
durability of the finish. 1 he results obtained for the fabrics treated with DEAEP, DEMLP.
DFAMP and DMAMP are given in I able 5.
I he results show that the I 01 values of the fabrics drop slightly for the samples
treated with the phosphate monomers DEMEP and DEAFP and more for those treated with
the phosphonate monomers DEAMP and DVIAMP, I he unexpected significant drop of the
64

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
LOI values of the fabrics treated with the phosphonate monomers can be attributed to the fact
that under the laundering conditions the grafted polymer nevertheless has certain water-
solubility. The similar observation have been reported when DEMEP and DMAMP were used
as additives and co-monomers to flame retard PAN fibers, the treated fibers were not wash
resistant at 60°C and 95 °C [21]. On the other hand, the burning rates decrease considerably,
in comparison with the virgin PAN which has at LOI = 21% a value of 1.2 mm/s; these results
demonstrate that the polymers were tightly grafted on the PAN fabrics.
Table 5 LOI values and burning rates of treated fabrics before LOI] and after LOI2 the
accelerated laundering method: [Monomer] = 200 g/L (DEMEP, DEAEP) and 300 g/L
(DEAEMP, DMAMP), 5 %( w/w) of BAPO, 20 %( w/w) of EGDA.
Monomers LOIi (burning rates) LOI2 (burning rates)
DEMEP 21.5(0.47 mm/s) 21 (0.44 mm/s)
DEAEP 22.0 (0.44 mm/s) 20 (0.29 mm/s)
DEAMP 23.5 (0.57) mm/s) 20 (0.40 mm/s)
DMAMP 26.5 (0.72 mm/s) 21 (0.44 mm/s)
2.5 Attempts to improve wash-resistance and LOI value with multilayer treatments
The best LOI value (26.5) was obtained with DMAMP at monomer concentration of
300g/L and in presence of 20% of EGDA. However the permanence of the flame retardant
effect needs to be improved. An attempt was therefore made to enhance the wash-resistance
of PAN fabrics treated with DMAMP and the LOI value by performing a multilayer treatment
The experiments are carried out several times on the same sample following the PIGP
procedure at a monomer concentration of 100 g/L with 10% (w/w) ofEGDA and 5% (w/w) of
BAPO. Table 6 shows the grafting yields after washing (soxhlet ethanol) and air-drying for
each treatment.
It can be observed that the total grafting yield increases quasi-linearly with the number
of layers. However, as the number of layer increases, the amount of polymer grafted after
each new treatment decreases. This can be attributed to the fact that, if the layers are above
each other and if the crosslinking agent has no affinity with the grafted polyDMAMP, it can
not act efficiently as coupling agent. This fact can lead to a decrease of the percentage of
grafting. Another possible explanation can be the reduction of the available grafting sites on
the surface of the PAN fabrics after each treatment. Hence, the grafting rate is slower and the
65

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
homopolymerization predominates. The results show that multiple treatments with the PIGP
procedure are possible and allow efficient grafting onto PAN fabrics. It was possible to attain
the same amount of grafted polymer obtained at DMAMP concentration of 300g/L and in
presence of 20% of EGDA, with four different treatments at 100 g/L and 10% of EGDA.
Table 6 Percentage of grafting after multilayer treatment [DMAMP] = 100 g/L, 5 %( w/w) of
BAPO, 10 %( w/w) ofEGDA
layersAdditional Total
grafting percentage grafting percentage
1st layer 10.9 10.9
2nd layer 9.4 21.4
3rd layer 8.9 32.3
4th layer 5.4 39.4
The IR(ATR) spectra and SEM photomicrographs of monolayer and multilayer treated
PAN samples are compared to see if there is major differences.
Figure 24 shows the IR (ATR) spectra of untreated (a), and PAN samples treated with
DMAMP monolayer (b) and multilayer (c), after washing and air-drying.
2600 2100 1600
Wavenumber (cm-1)
1100 600
Figure 24 IR spectra of PAN fabrics (a) untreated, and (b) treated monolyer and (c)
multilayer.
66

Chapter III Flame retardancy of polyacrylonitrile and cotton fabrics
Compared to the monolayer treated sample (b), the multilayer treated sample (c)
shows the same characteristic IR absorption bands at v -~ 1238 and 1025 cm"1 corresponding
to the stretching vibrations of P=() and P-O-C, respectively, fhesc bands indicate the
presence of a phosphorus containing polymer on the surface of the PAN fabric. Furthermore
the stretching vibration of the C N group of the PAN is still visible on the multilayer treated
sample.
SEM photomicrographs of untreated and treated PAN fabrics are shown in Figure 25.
The surface morphology along the untreated PAN (Figure 25.a) is rough. Compared to the
PAN fabrics treated with DMAMP in a single treatment (Figure 25.b), the multilayer treated
sample (Figure25.c) is also completely surrounded by the grafted polyDMAMP and the space
between the fibers stays free of polymer, liiere is no noticeable difference between the
surface morphology of a monolayer and multilayer coated PAN fabrics.
Figure 25 SEM photomicrographs of PAN labrics (a) not treated; (b) monolayer coated, and
(c) multilayer coated with DMAMP after washing and air-drying.
The aim of multilayer was to improve the grafting rate, the wash-resistance and LOI
value of PAN fabrics treated with DMAMP, LOI values of multilayer treated labrics before
(LOI i) and after the accelerated laundry method (LOI?) are reported in I able 7.
67

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
Table 7 LOI (%) values of multilayer treated PAN fabrics before
accelerated laundering method (LOI2)
(LOI,) and after the
Layers Total
grafting
percentage
%P (w/w)measured on
PAN
LOI, LOI2
1st layer 10.9
2nd layer 21.4 2.4 22.5
3rd layer 32.3 - 25
4th layer 39.4 4.0 26 21
The LOI values augment when the number of layers and the percentage of grafting are
increased whereby the amount of grafted phosphorus increases. The results show that the LOT
value of the untreated PAN 18.5 increases by 4 units after two treatments, by 6 and 8 units
after the third and the fourth treatment, respectively. In comparison with the monolayer
treatment (%G 39.7, %P 3.9 and LOI 26.5), the LOI value of 26 could be also achieved with
approximately the same amount of grafted polymer and phosphorus content (%G 39.4, %P
4.0).
The LOI value of 21 is obtained after the accelerated laundering procedure, this result
is similar with the one obtained with the monolayer treatment. Thus, with the multilayer
treatment there is no improvement of the wash-resistance of the treated PAN fabrics.
2.6 Conclusions
In this section, the plasma-induced graft-polymerization of acrylate phosphate
(DEMEP, DEAEP) and phosphonate (DEAMP, DMAMP) monomers onto PAN fabrics have
been studied. The grafting yields and LOI values were found to vary with type of monomers
and the presence of crosslinking agents. It was possible to improve the flame retardancy of
PAN textiles by using this novel graft polymerization procedure. The LOI value of 26.5 was
obtained with DMAMP with a phosphorus content of 3.9 % (w/w) on the fabric. Despite the
fact the LOI values of the treated fabrics drop after the McSherry procedure, the burning rates
were considerably reduced in comparison with untreated fabrics. We could demonstrate that
this surface treatment can seriously compete with the other types of treatment using
organophosphorus compounds. Indeed, with the other classical approaches (reactive and
additive) the best LOI values obtained so far reaches 28, for about 23% of polymer loading as
68

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
additives with 3.6 % of phosphorus content in the fiber, and the fire retardant character was
not resistant at 95°C as in our case. The major achievement of PIGP procedure is the fact that
it allows to graft and to polymerize various monomers onto the surface of fabrics in one-step.
By this way, the polymer is covalently linked to the surface of the fabric, which guarantees
the resistance of the coating towards the washing, even at higher temperatures. Therefore, it is
worth considering the PIGP procedure as a new approach to flame retard permanently textile
fabrics on which only a surface treatment can be applied like natural fibers.
In the next section the PIGP procedure is applied to cotton fabrics.
3 Flame retardancy of cotton fabrics
Overall textile fibers cotton is the most commonly used and being also one of the most
flammable materials (LOI 18.4%), cotton fibers have to be flame retard.
Phosphorus-based compounds are the main flame retardant used on cotton textiles.
Numerous studies [1,3,22] have shown that, when present in cotton they reduce volatile
formation and catalyze char formation. They act in this double capacity because, on heating
they release polyphosphoric acid, which phosphorylates the C(6) hydroxyl group of the
cellulose in the anhydroglucopyranose moiety, and simultaneously acts as an acidic catalyst
for dehydration of these repeat units. The first reaction prevents formation of laevogluscosan,
the precursor of flammable volatile compounds and this ensures that the competing char-
forming reaction is now the favored pyrolysis route. The acidic catalytic effect of the released
polyacid further increases the rate of this favored route.
Among the compounds which are effective in producing these effects, many of them
are found to contain both phosphorus and nitrogen. This observation has led to proposal of
synergistic interaction involving the two elements. Several studies [23-26] have demonstrated
that compounds containing nitrogen caused synergistic enhancement in the efficiency of
phosphorus based-flame retardant by further increasing char-forming tendencies. However,
not all nitrogen-containing compounds are effective; compounds containing nucleophilic
nitrogen atoms such as amides and amines are the only ones that are effective.
Flame retardant cottons are usually produced by after-treating fabrics chemically as a
textile finishing process which, depending on chemical character, yields flame-retardant
properties having varying degrees of durability to various laundering processes. The fire
retardants may be (i) simple soluble salts (e.g. ammonium phosphates, polyphosphate and
bromide; borate-boric acid mixtures) to give non-durable finishes; (ii) chemically reactive,
69

Chapter III Flame retardancy ofpolyacrylonitrile and cotton fabrics
usually functional finishes (e.g. organophosphorus and nitrogen containing monomers such as
alkylphosphonamide derivatives, or polycondensates such as tetrakis (hydroxyl methyl)
phosphonium salt) to give durable flame retardancy, or (iii) back-coating, which usually
comprise a resin-bonded antimony-bromine flame retardant system.
Durable flame retardant properties are more complex and more difficult to achieve
than the non-durable. Many techniques for imparting durable fire retardant properties to
cotton fabrics such as pad-dry (-cure), exhaust, spray-dry (-cure), of coat-dry (-cure) have
been described in the literature [1,3,27]. However; relatively few of them are practiced today,
either because of the safety concerns, or process control issues. To overcome such difficulties,
we thought that the grafting of acrylic monomers containing phosphorus by our novel plasma-
induced graft-polymerization (PIGP) procedure described in the previous section, might lead
to durable flame retardant effect.
For this purpose the acrylate phosphate (DEMEP and DEAEP), phosphonate (DEAMP
and DMAP) and phosphoramidate (DEAEPN and BisDEAEPN) monomers are investigated.
The acrylate phosphate and phosphonate monomers have already been used in the previous
section to flame retard PAN fabrics. DEAEPN and BisDEAEPN are acrylic monomers
containing phosphorus and nitrogen. It has already been demonstrated that the presence of
nitrogen contributes to improve the efficiency of phosphorus-based flame retardant
compounds of cotton fibers. Flame retardancy of cotton fabrics by surface grafting ofDEMEP
using gamma and electron-beam irradiation technique has already been reported [10].
In this section, the plasma-induced graft polymerization of the acrylate phosphate,
phosphonate and phosphoramidate monomers on cotton fabrics is carried out, the flame
retardant effect of acrylic monomers containing phosphorus and nitrogen (DEAEPN and
BisDEAEPN) are compared with the acrylate phosphate and phosphonate monomers, finally
the durability of the treatment is investigated.
3.1 Plasma-induced grafting and polymerization ofDEMEP, DEAEP, DEAMP, DMAMP,
DEAEPNand BisDEAEPN onto cottonfabrics
3.1.1 Graft-polymerization procedure
The optimum conditions (FAr =125 seem, pressure 40 Pa, P - 100 W, initiator:
BAPO 5% (w/w)) found for the plasma-induced polymerization of acrylic monomers
70

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
containing phosphorus are applied for the graft polymerization of DEMEP, DEAEP, DEAMP
DMAMP, DEAEPN and BisDEAEPN onto cotton fabrics. The experiments are performed
using the one step (PIGP) procedure depicted in Figure 13.
In a first step, pieces (52 x 140 mm) of bleached cotton fabrics (120g/m2) are weighed
and then immersed at room temperature for one minute in 20 ml of a methanol solution
containing 100g/L, 200g/L or 300 g/L of the monomers (DEMEP, DEAEP, DEAMP,
DMAMP, DEAEPN or BisDEAEPN) in the presence of 5% (w/w) of BAPO and various
amounts (0, 5, 10 and 20% (w/w)) of the cross linking agent (EGDA). These impregnated
fabrics are then pressed to evacuate the excess of the solution, placed onto glass plates and
submitted to a MW argon plasma (FAr= 125 seem, base pressure= 40 Pa, P = 100 W, t = 15
min). After treatment the samples are washed in a soxhlet apparatus with methanol (24h) to
remove the non-grafted homopolymer and monomer, then in water (24h) and air-dried.
Levels of fire retardant to be applied depend upon the degree of flame retardancy
required and the area density and structure of the fabric. Generally, a loading of 1.5 to 4%
(w/w) of phosphorus on the fabric is required [1,3]. For this a minimum of 10 to 15% (w/w)
of grafted phosphorus polymer, depending on the monomer is necessary. In the previous
section, it has been demonstrated that the presence of a crosslinking agent is necessary for an
efficient grafting of acrylate phosphate and phosphonate monomers on PAN fabrics.
Therefore the influence of the crosslinking agent and the monomer concentration is
investigated in order to find the optimum conditions for plasma-induced graft polymerization
of the acrylic monomers containing phosphorus onto cotton fabrics.
3.1.2. Effect ofthe concentration ofthe crosslinking agent on the grafting yield
The different experiments are performed with the crosslinking agent EGDA (10 and
20%) (w/w)) and without at monomer concentration of 200g/L, using the PIGP procedure. The
grafting yields obtained after washing (soxhlet methanol and water) and air-drying are shown
in Figure 26.
71

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
60
50
40
30
20
10
0
0% EGDA
D 10% EGDA
20% EGDA
Figure26 Effect of the mount of crosslinking agent (EGDA) on the percentage of grafting of
the monomers (DEMEP, DEAEP, DEAMP and DMAMP) on cotton fabrics treated in MW
argon plasma after washing and air-drying.
It can be observed that without EGDA the percentage of grafting is undetectable with
DEMEP, is very low with DEAMP (1.7%), DMAMP (3.3%), and BisDEAEPN (3.9). With
DEAEP and DEAEPN an amount of grafted polymer of 6.9% and 20.9%, is attained,
respectively. Cotton has C-H backbone chain with -OH functional group attached to the
backbone which can formed hydrogen bonds with oxygen or nitrogen atoms. The higher
percentage of grafting (20.9%) obtained with DEAEPN without crosslinking agent can be
attributed to the formation of hydrogen bonds between the -OH functional group of the cotton
with the nitrogen of the phosphoramidate unit. However, compare to DEAEPN the amount of
grafted polymer without EGDA on fabrics treated with BisDEAEPN is low. This fact can be
explained by the difference of structure between DEAEPN and BisDEAEPN. When 10%
(w/w) of EGDA is added in the monomer solution, the amount of grafted polymer is less than
10% for DEMEP, between 22 and 28% for DEAEP, DEAMP and DMAMP, and more than
30% for DEAEPN and BisDEAEPN. The highest grafting yield of 34.7% is attained with
BisDEAEPN. Because of the high grafting rate of acrylate phosphoramidate monomers with
72

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
10% of EGDA not tests were performed with 20% of EGDA. When the concentration of the
crosslinking agent is augmented from 10% to 20%), the amount of grafted polymer increases
considerably for all the monomers tested (from 6.3% to 17.8% for DEMEP, from 23.8% to
46.2% with DEAMP, from 29 to 47.6%) excepted for DEAEP. With this monomer, the
grafting yields increases slightly (from 24% to 27%). It is clear that the presence of
crosslinking agent is necessary to achieve a good grafting of all the monomers on cotton
fabrics excepted for DEAEPN. On the other hand with the high amount of grafting achieved
with DEAMP (46.2%) and DMAMP (47.6%) the stiffness of the fabrics is visibly affected.
Therefore for the cotton fabrics it is not necessary to use a high amount of crosslinking agent
since with 10% of EGDA and a concentration of 200g/L the percentage of grafting attained is
more than 20% for all the monomers with the exception of DEMEP. In order to increase the
grafting yield without affecting the stiffness of the fabrics, the effect of increasing monomer
concentration is investigated.
3.1.3 Effect ofthe concentration ofthe monomer on the grafting yield
The different experiments are carried out with the crosslinking agent EGDA (10%
(w/w)) at monomer concentration of 200g/L and 300 g/L, using the PIGP procedure. The
grafting yields obtained after washing (soxhlet methanol and water) and air-drying are shown
in Figure 27.
The results clearly indicate that the amount of grafted polymer increases with the
monomer concentration. The lowest grafting yield is obtained with DEMEP. This observation
has already been made when DEMEP was grafted and polymerized onto PAN fabrics using
the PIGP procedure. The highest grafting is attained at about 42% with DEAP, DEAMP,
DMAMP and DMAMP. These results indicate that it is not needed to use 10% (w/w) of
EGDA at monomer concentration of 300 g/L for these monomers. With fewer amounts of
crosslinking agent it can be possible to reach the minimum amount of grafted polymer
required to impart adequate flame retardancy to cotton fabrics. Therefore, treatments were
performed with 5% (w/w) of EGDA for DEAEP, DEAMP, DMAMP and DEAEPN, at
monomer concentration of 200g/L or 300 g/L.
73

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
50
45
40
3501)
30
f°1—
00 25<+-<
o
0s-
20
15
10
D 200 g/L
H 300 g/L
flIIÉ1DEMEP DEAEP DEAMP DMAMP DEAEPN
Monomers
Figure 27 Effect of the concentration of DEMEP, DEAEP, DEAMP and DMAMP on the
percentage of grafting of cotton fabrics (120g/m2) treated in MW argon plasma after washing
and air-drying.
From the results obtained, it can be concluded that: by using the PIGP procedure it is
possible to graft quantitatively acrylic monomers containing phosphorus onto the surface of
cotton fabrics (120g/m2) in presence of EGDA as a crosslinking agent. Furthermore in
comparison with PAN fabrics less amount ofEGDA is required.
Further evidence supporting the occurrence of the graft-polymerization onto cotton
fabrics will be obtained by studying the changes of surface characteristics after plasma-
induced graft-polymerization of the different monomers.
3.2 Surface characterization ofthe grafted PANfabrics
3.2.1 IR(ATR)
IR(ATR) spectra of untreated (a) and treated (b, c, d, e, f, g) cotton fabrics are shown
in Figure 28. Comparing the IR (ATR) spectra of the untreated and treated cotton fabrics,we
can observed on the spectra of the treated fabrics the presence of a new absorption band at
around 1730 cm" attributed to the carbonyl stretching vibration of the acrylic polymer
containing phosphorus grafted onto the cotton fabrics. It was not easy to identify the P=0 and
74

Chapter III Flame retardancy ofpolyacrylonitrile and cotton fabrics
P-O-C vibrations of the grafted polymers on the treated fabrics, since the absorption bands of
the primary and secondary OH deformation of the cotton appear in the same region at about
1057, 1373, and at 1162.cm"\ Note that the -OH stretching vibration present on untreated
cotton is still visible on the treated fabrics; this is an indication of a superficial grafting.
3600 3100 2600 2100 1600 1100 600
Wavenumber (cm"1)
Figure 28 IR spectra of cotton fabrics (a) not treated; (b) treated with methanol solution
containing DEMEP, (c) DEAEP, (d) DEAMP, (e) DMAMP, (f) DEAEPN and (g) BisDEAPN
[Monomer] = 200 g/L, 5 %( w/w) of BAPO, 10 %( w/w) of EGDA, after washing and air-
drying.
3.2.2 SEM
SEM photomicrographs of untreated (a) and treated with DEAEP (b) are shown in
Figure 29. The surface morphology of the untreated cotton along the fiber is rough while the
one of the fabric treated with DEAEP is smooth and entirely covered with the polymer. This
75

( 'hapter III Flame retardancy of polyacrylonitrile and cotton fabrics
is also evidence that grafting is localized on the surface. Note that the space around the fiber
stays free of polymer, which guarantees the breathability of the fabric.
Figure 29 SEM photomicrographs of cotton fabrics (a) not treated; (b) treated with a DEAFP
(200g/L), 10 % (w/w) of EGDA and 5% (w/w) of BAPO after washing and air-drying.
3.3 Evaluation oftheflame retardancy ofthe grafted cotton fabrics
3.3.1 LOI measurements
fhe flame retardancy of the untreated and treated cotton fabrics is assessed by limiting
oxygen index (LOI) measurements. The results obtained are given in Table 8. They clearly
indicate that the initial LOI value of the untreated cotton ( 19) increases up to 4 units when
treated with DEAMP. up to 7 units when treated with DEMEP, DEAEP, and DMAMP, up to
9 and 10 units when treated with Dl^AEPN and BisDEAEPN.. respectively.
It can be also noticed that when the monomer concentration augment from 200g/L to
3()()g/L with 10% of EGDA. despite the fact the amount of grafted polymer (for fabrics
treated with DEAFP, DEAMP DMAMP, DFAEPN) is considerably increased the LOI values
augment slightly. This fact can be attributed to the law of diminishing effectiveness. Indeed,
most flame retardants when applied at higher add-on levels exhibit this law. there is no linear
improvement in llame retardancy and a ceiling value of LOI is thus approached as the add-on
increased.
The results also show that the fabrics treated with the acrylate phosphoramidate
monomers give better flame retardant effect than the fabrics treated with acrylate phosphate
and phosphonate monomers. While a LOI value of 26 is achieved with DEAEP at phosphorus
content of 2.75%, a LOI value of 27.5 is obtained with DEAEPN at approximately the same P
content of 2.77%. Comparing the LOI values obtained with DEAEPN and the one obtained
76

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
with the acrylate phosphonate monomers, it can be observed that whereas a LOI value of 28.5
is obtained with DEAEPN at phosphorus content of 3.16%, LOI values of 25.5 and 23 is
obtained for DMAMP and DEAMP, respectively, at approximately the same P content of
3.27%i. The LOI values indicate that the effectiveness of the acrylic monomers containing
phosphorus studied follow the order BisDEAEPN > DEAEPN > DEAEP > DEMEP >
DMAMP > DEAMP. This difference in efficiency between the acryalate phosphoramidate
monomers and the acrylate phosphate and phosphonate monomers can be attributed to the fact
that phosphorus-nitrogen containing compounds are better phosphorylating agents than are
the related compounds without nitrogen [23-26], And, it has been reported that an increasing
rate of phosphorylation of cellulose hydroxyl functional groups produce a corresponding
increase in the flame retardant efficiency [25],
From these results, it can be concluded that the flame retardant effect depend greatly
on the chemical structure of the compound, because the same phosphorus content on the
fabrics can lead to different LOI values. With all the monomers studied, it was possible to
attain LOI values greater than 25 with exception of DEAMP. The highest LOI values are
recorded at 28.5 and 29.5 for fabrics treated with DEAEPN and BisDEAEPN, respectively
77

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
Table 8 LOI values of treated cotton fabrics as a function of monomer type, of the treatment
conditions and percentage of grafting after washing and air-drying
%P (w/w)Monomer Treatment conditions %>G measured on LOI (%)
cotton
Untreated cotton - - - 19
DEMEP 200 g/L, 0% EGDA - - 19
200 g/L, 10% EGDA 6.3 - 21
300 g/L, 10% EGDA 19.3 1.96 24.5
300 g/L, 20% EGDA 29.2 - 25.5-26
DEAEP 200 g/L, 0% EGDA 6.9 - 21.5
200 g/L, 10% EGDA 24.4 - 25.5
300 g/L, 5% EGDA 28.6 2.75 26.0
300 g/L, 10% EGDA 42.7 - 26.5
DEAMP 200 g/L, 0% EGDA 1.7 - 19
200 g/L, 10% EGDA 23.8 - 22.5-23
300 g/L, 5% EGDA 34.7 3.28 23.0
300 g/L, 10% EGDA 44.3 - 23-23.5
DMAMP 200 g/L, .0% EGDA 3.3 - 20.0
200 g/L, 10% EGDA 29.0 3.27 25.5-26
300 g/L, 5% EGDA 37.8 4.10 26.0
300 g/L, 10% EGDA 41.2 - 26.0
DEAEPN 200 g/L, 0% EGDA 20.9 - 26.5
200 g/L, 5% EGDA 24.0 - 26.5
200 g/L, 10% EGDA 32.4 2.77 27.5
300 g/L, 0% EGDA 36.2 3.16 28.5
300 g/L, 5% EGDA 38.6 3.36 28.5
300 g/L, 10% EGDA 42.5 - 28.0
BisDEAEPN 100 g/L, 10% EGDA 13.0 1.48 25.0
200 g/L, 5% EGDA 29.7 3.29 29.5
200 g/L, 10% EGDA 34.7 - 29.5
78
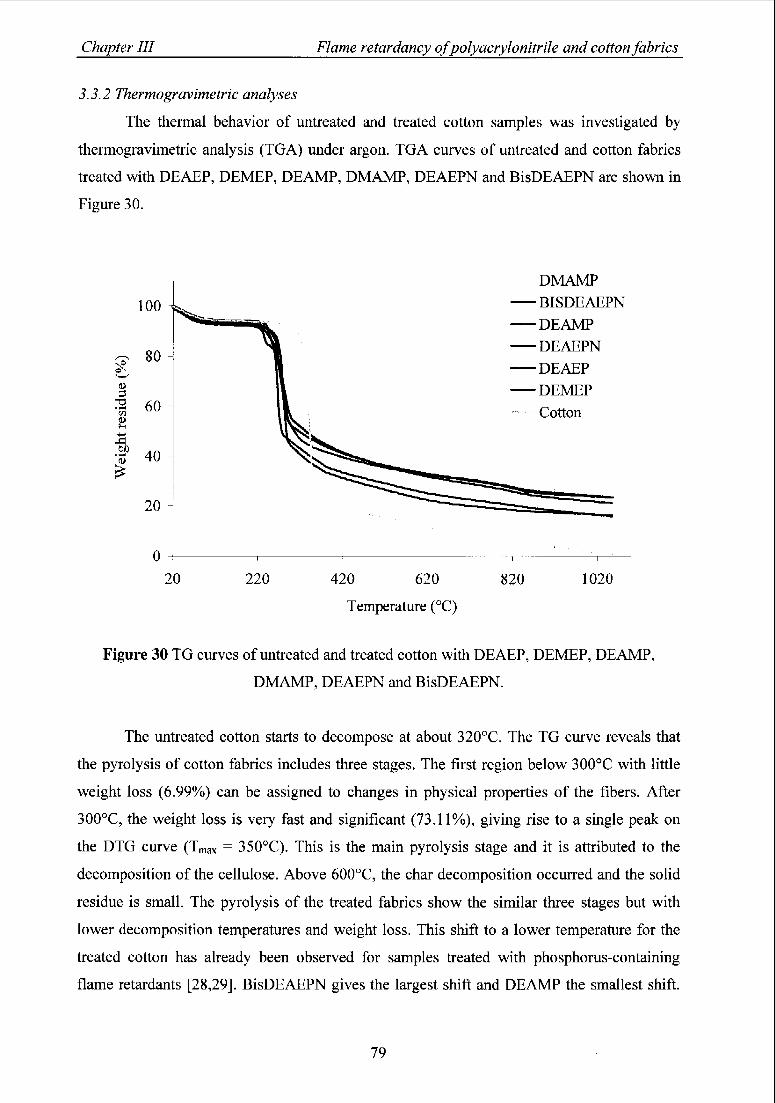
Chapter III Flame retardancy ojpolyacrylonitrile and cottonfabrics
3.3.2 Thermogravimetric analyses
The thermal behavior of untreated and treated cotton samples was investigated by
thermogravimetric analysis (TGA) under argon. TGA curves of untreated and cotton fabrics
treated with DEAEP, DEMEP, DEAMP, DMAMP, DEAEPN and BisDEAEPN are shown in
Figure 30.
u
3
at)
I
20 220 420 620
Temperature (°C)
DMAMP
BISDEAEPN
DEAMP
DEAEPN
DEAEP
DEMEP
Cotton
820 1020
Figure 30 TG curves of untreated and treated cotton with DEAEP, DEMEP, DEAMP,
DMAMP, DEAEPN and BisDEAEPN.
The untreated cotton starts to decompose at about 320°C. The TG curve reveals that
the pyrolysis of cotton fabrics includes three stages. The first region below 300°C with little
weight loss (6.99%) can be assigned to changes in physical properties of the fibers. After
300°C, the weight loss is very fast and significant (73.11%>), giving rise to a single peak on
the DTG curve (Tmax = 350°C). This is the main pyrolysis stage and it is attributed to the
decomposition of the cellulose. Above 600°C, the char decomposition occurred and the solid
residue is small. The pyrolysis of the treated fabrics show the similar three stages but with
lower decomposition temperatures and weight loss. This shift to a lower temperature for the
treated cotton has already been observed for samples treated with phosphorus-containing
flame retardants [28,29]. BisDEAEPN gives the largest shift and DEAMP the smallest shift.
79

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
The percentage of remaining residue at 1060°C of treated cotton fabrics is higher than
that of the untreated cotton (0.72). DMAMP gives the highest char yield (25.8%), followed by
BisDEAEPN and DEAMP (23.4%), DEAEPN (21.1%), and DEMEP and DEAEP (16.3%).
3.3.3 Effect oftheflame retardants on LOI values and char residues
The LOI values and the TGA data (onset of degradation temperature and char yield) of
cotton fabrics treated with the acrylic monomers containing phosphorus are given in Table 9.
Table 9 Comparison of the LOI values and TGA data
%G%P (w/w)measured
LOI (%)
Onset of
degradation
temperature
(°C)
% Weightresidues at
1060°C
Untreated - - 19 320 0.72
DEAEP 28.6 2.75 26 240 16.3
DEAMP 34.7 3.28 23 250 23.4
DMAMP 37.8 4.10 26 245 25.8
DEAEPN 32.4 2.77 27.5 232 21.1
BisDEAEPN 29.7 3.29 29.5 223 23.4
The LOI values and char residues of all the treated fabrics are increased compared to
the untreated. The results clearly show that among all the monomers studied, BisDEAEPN
has the lowest decomposition temperature (223°C) and the highest LOI value (29.5), where as
DEAMP has the highest temperature of degradation (250°C) and the lowest LOI value (23).
Note that, the phosphorus content (about 3.29%) and the percentage of remaining residue
(about 23.4%) is the same for both. From these observations we can assume that for all the
monomers studied there is no strong correlation between the amount of char produced and the
LOI values. Indeed, DMAMP, which promotes the greatest amount of char residue, does not
give the highest LOI value. The results also show that the effectiveness of the flame retardants
follow the order BisDEAEPN > DEAEPN > DEAEP > DMAMP > DEAMP, which correlates
with the increasing onset decomposition temperature. We can therefore suppose that the
effectiveness of a flame retardant for cotton is related to the ease of producing polyphosphoric
acids to phosphorylate cellulose.
80

( "hapter III Flame retardancy of polyacrylonitrile and cotton fabric s
3 3 4 Burning behavior
The photograph in Figure 31 shows the behavior of a treated sample compared to the
untreated one during the LOI test 1 he virgin cotton bums by producing a small amount of
char whereas the treated sample burned by torming a layer of char. Ihe amount of char is thus
increases in treated sample compared to the untied ted.
Figure 31 Burning behavior of (a) untreated and (b) treated cotton fabric
This char layer is characteristic for a homogeneously grafted phosphorus containing
polymer on the surface of the substrate
3.4 Durability of the flame retardant treatment
In order to evaluate the durability of the coating to washing, the treated samples were
submitted to the accelerated laundering method proposed by McSherry et al. |21 | Ihe results
obtained for the fabrics treated with DFAFP, DMAMP, DFAFPN, and BisDEAEPN arc
given in Table 10
Ihe results also indicate that the LOI values alter the washing procedure depend on
the amount of crosslinking agent added Without EGDA the LOI value of Dl AFPN drops
from 26 5 to 22.5, where as with 10% of EGDA the LOI value drops only from 27.5 to 24.5
In the case of DF ALP the I 01 value decreases from 26 to 23.5 with 5% of EGDA and from
26.5 to 25 5 with 10% of I GDA, at monomer concentration of 300g/L The LOI values of
about 25 obtained with DFAhP, DhAhPN, and BisDEAF.PN, demonstrate that the polymers
were covalcntly grafted on the cotton fabrics
81

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
Table 10 LOI values of treated fabrics before LOI i
method
and after LOI2 the accelerated laundering
Monomer Treatment conditions %Gi LOI, %G2 LOI2
DEAEP 300 g/L, 5% EGDA
300 g/L, 10% EGDA
30.6
43.2
26
26.5
17
35.5
23.5
25.5
DMAMP 200 g/L, 10% EGDA 31.1 26.0 24.2 22.5
DEAEPN 200 g/L, 0% EGDA 20.9 26.5 8.7 22.5
200 g/L, 10% EGDA 34.0 27.5 23.6 24.5
300 g/L, 0% EGDA 38.1 28.5 17.8 25.0
300 g/L, 5% EGDA 38.6 28.5 25.4 25.5
300 g/L, 10% EGDA 43.5 28.0 34.2 26.0
BisDEAEPN 200 g/L, 5% EGDA 29.8 29.5 26.7 25.0
Further evidence supporting the presence of the grafted polymer on treated fabrics
after the McSherry procedure can be obtained by the IR (ATR) analysis. The different spectra
are shown in Figure 32.
Comparing the IR spectra of untreated and treated cotton fabrics after the washing
procedure, we can observed that on the spectra of treated washed fabrics the characteristic
absorption band of the carbonyl stretching at around 1730 cm"1 attributed to the grafted
acrylic polymer containing phosphorus is still visible. This result demonstrates that the
coating is resistant to washing at elevated temperatures and it is a confirmation of a covalent
grafting on the surface of the fabrics.
82

Chapter III Flame retardancy ofpolyacrylonitrile and cotton fabrics
3600 3100 2600 2100 1600 1100 600
Wavenumber (cm-1)
Figure 32 IR spectra of cotton fabrics (a) not treated; (b) treated with DEAEP, (c) DEAEPN
and (d) BisDEAEPN after the accelerated laundering method and air-drying.
3.5 Effect ofthe surface area ofthe cotton fabrics on the grafting yield and LOI values
The influence of the area density of the fabrics on the grafting of acrylic monomers
containing phosphorus using the PIGP procedure is investigated. The grafting yields obtained
with bleached cotton fabrics (210g/m2) and (120g/m2) treated with DEAEP, DMAMP and
DEAPN, in presence of EGDA (10 %> (w/w)) at monomer concentration of 200 g/L, after
washing (soxhlet methanol and then water) and air-drying are compared. The Figure 33 show
the results obtained.
83

Chapter III Flame retardancy ofpolyacrylonitrile and cotton fabrics
35
30
25
a
S 202ao
o 15
10
5
0
D Cotton 120
ffl Cotton 210
DEAEP DMAMP
Monomers
DEAEPN
Figure 33 Comparison between the percentage of grafting on cotton 120 (120g/m ) and
cotton 210 (210g/m2) treated in MW argon plasma after washing and air-drying.
It can be seen that the amount of grafted polymer is lower for cotton 210 fabrics with
surface area of 21 Og/m2 than that of cotton 120. The percentage of grafting is less than 20%
for all the monomers tested. The decrease of grafting yield observed with cotton 210 can be
attributed to the fact that the heavier fabrics are more compact than the lighter one. Because of
the compactness of the fabric, the surface contact between the polymer (cotton 210) and the
solution of the monomer is weaker than that of lighter fabrics.
The LOI values of the treated cotton 210 fabrics with DEAEP, DMAMP and DEAPN
are given in Table 11.
84

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
Table 11 LOI values of treated cotton 210 (LOIi) fabrics as a function of monomer type, of
the treatment conditions and percentage of grafting after washing and air-drying in
comparison with LOI values of cotton 120 (LOI2).
t + *%pi
T%p2 t
iv4Treatment
0/r, , , \ LOIi a/^ , ,\ LOI2Monomer
MnAiiinne %Gi(w/w)
(0J%G2
(w/w)
Untreated
cotton
DEAEP J00g/L,10% 17.2 - 25 24.4 - 25.5
conditions
v v»"v
(0/o)
-^ K
'A (%)measured measured
19 - 19
EGDA
300 g/L, 10%2?8 26g _ 265
EGDA
DMAMP I^L'10% 17.4 - 25.5 29.0 3.27 25.5EGDA
300ig/L, 10%231 237
EGDA41.2 - 26.0
DEAEPN ï°^h10% 18.5 1.92 27.5 32.4 2.77 27.5EGDA
300 g/L, 10%_ 2g
EGDA
The results show that the untreated cotton 210 has a LOI value of 19 like untreated
cotton 120. The burning rate of heavier fabrics is 0.78 mm/s, while cotton 120 has a burning
rate of 1.5mm/s. The difference of the burning rates between cotton 120 and 210 can be
explained by the fact that they have different specific surface areas. Indeed the rate at which
pyrolytic formation of volatile compounds and subsequent combustion occur, is related to the
specific surface area of the material [4].
The LOI values (LOIi of cotton 210) augment with the monomer concentration
whereby the amount of phosphorus content on the fabrics increased for DEAEP and
DEAEPN, while for DMAMP, there is no change. Flame retardant fabrics are obtained with
DEAPN at LOI, values of 27.5 and 28.5 at phosphorus content of 1.92% and 2.63%,
respectively. The same LOI values were also attained with lighter fabrics but with more
phosphorus content (LOI2 values of 27.5 and 28.5 were achieved at a phosphorus content of
2.77% and 3.2%, respectively). Lighter fabrics have a high specific surface area compare to
heavier, and for this reason they require relatively more fire retardant.
85

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
The durability of the coating to washing was investigated using the McSherry
procedure, the results obtained with DEAEP and DEAEPN are shown in Table 12. The LOI
values after the accelerated laundering methods drop slightly the decrease is less with DEAEP
(from 26.5 to 25) than with DEAPN (from 27.5 to 24, and from 28.5 to 24.5). This difference
can be attributed to the higher solubility of polyDEAEPN compared to polyDEAEP under the
washing conditions.
Table 12 LOI values of treated fabrics before LOIi and after LOI2 the accelerated laundering
method
Monomer Treatment conditions %Gi LOIi %G2 LOI2
DEAEP 300 g/L, 10% EGDA 29.7 26.5 23.6 25.0
DEAEPN 200 g/L, 10% EGDA
300 g/L, 10% EGDA
18.8
29.0
27.5
28.5
14.8
22.9
24.0
24.5
3.6 Conclusions
In this section the flame retardation of cotton fabrics by grafting and polymerization of
acrylic monomers containing phosphorus using the novel PIGP procedure has been
investigated. The grafting yields were found to vary with the type of monomers, the amount
of the crosslinking agent and area density of the cotton fabrics. The flame retardancy the
fabrics treated with acrylate phosphate, phosphonate and phosphoramidate monomers was
improved (DEMEP, DEAMP, DMAMP, DEAEPN, and BisDEAEPN). The results show that
the flame retardant effect depends greatly on the chemical structure of the monomer and not
on the amount of grafted phosphorus. The highest LOI values of 28.5 and 29.5 were obtained
with DEAEPN and BisDEAEPN, repectively. The good flame retardant properties of the
phosphoramidate monomers could be attributed to the presence of nitrogen which cause
synergistic enhancement in the efficiency of phosphorus-based flame retardant. After the
accelerated laundering procedure the LOI values of the treated fabrics decreased. On the other
hand a LOI value of about 25 is still retain with DEAEP and the acrylate phosphoramidate
monomers. This result demonstrated that the resistance of the coating towards washing at
elevated temperature and the presence of covalent bond between the fabrics and the acrylic
monomer containing phosphorus.
86

Chapter HI Flame retardancy ofpolyacrylonitrile and cotton fabrics
4 Comparison of the flame retardant effect on PAN and cotton fabrics
In the previous sections, the flame retardancy of PAN and cotton fabrics by grafting
and polymerization of acrylic monomers containing phosphorus using the PIGP procedure
have been investigated. In this section the results obtained with both fabrics are compared.
The grafting yields, the phosphorus content on the fabrics, the LOI values and char yield are
summarized in Table 13.
Table 13 Comparison of the flame retardant effect on PAN (300 g/m ) and cotton (210g/m )
fabrics
Monomer Fabrics %G %P (w/w)measured on
cotton
LOI (%) Char yieldat 650°C
PAN
cotton
- - 18.5
19.0
58.3
DEAEPPAN
cotton
24.2
27.8
2.00
2.68
22.0
26.5
61.0
DMAMPPAN
cotton
28.0
23.1
3.0
2.37
24.5
25.5
65.0
DEAEPNPAN
cotton
34.4
31.0
2.78
2.63
23.5
27.5
-
It can be observed that the flame retardants (DEAEP, DMAMP, and DEAEPN) are
more effective when treated with cotton (210g/m ) compare to PAN (300g/m ). The
difference of efficiency can be attributed to the fact that the most effective flame retardants
for acrylic should promote highest residual char, while the effectiveness of fire retardants for
cotton is related to the rate of phosphorylation of cellulose. The results clearly demonstrated
that there is no significant increase of the char yield for the treated PAN compared to
untreated. The fact that the polymers do not produce a high amount of char compared to the
untreated PAN (see Chapter II, Section 3.4) can explained why the monomers used in this
study are not highly efficient when grafted and polymerized onto PAN fabrics. In fact the
highest char yield among the polymers studied (about 40%) is obtained with polyDMAMP at
570°C, while with the untreated PAN the amount of char residue is about 58% at 650°C.
We can therefore conclude that the performance of a particular phosphorus compound
depends not only on its chemical structure but also strongly on the nature of the polymeric
87

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
material treated. For this reason, it is not possible to make generalized statements concerning
the relative effectiveness of two phosphorus compounds without indicating the polymer with
which they are used.
88

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
5 References
[I] A.R. Horrocks in Fire retardant materials, A.R. Horrocks and D.Price, editors,
Woodhead publishing Ltd and CRC press LLC, Cambridge (2001) 128.
[2] P. Bajaj, AK. Agrawal, A. Dhand, N. Kasturia, Hansraj, J. Macromol. Sci, Rev.
Macromol. Chem. Phys. C40 (4) (2000) 309
[3] A.R. Horrocks, Rev. Prog. Coloration, 16 (1986)
[4] A.R. Horrocks, Polym. Degrad. Stab. 54 (1996) 143
[5] J.A. Harris, C. J. Keating, W.R. Goynes, J. Appl. Polym. Sei. 25 (1980) 2295.
[6] T. Randoux, J. C. Vanovervelt, H. Van den Bergen, G. Camino, Progress in Organic
Coatings 45 (2002) 281.
[7] R. Liepins, J. R. Surles, N. Morosoff, V. T. Stannett, J. Appl. Polym. Sci. 21 (1977)
2529.
[8] R. Liepins, J. R. Surles, N. Morosoff, V. T. Stannett, J. J. Duffy, F. H. Day, J. Appl.
Polym. Sci. 22 (1978) 2403.
[9] A. Mey-Marom, D. Behar, J. Appl. Polym. Sci. 25 (1980) 691.
[10] N. Shiraishi, J. L. Williams, V. Stannett, Radiât. Phys. Chem. 19 (1982) 79.
[II] F. A. Abdel-Mohdy, J. Appl. Polym. Sci. 89 (2003) 2573.
[12] M. Suzuki, A. Kishida, H. Iwata, Y. Ikada, Macromolecules, 19 (1986) 1804.
[13] Zubaidi, T. Hirotsu, J. Appl. Polym. Sci. 61 (1996) 1579.
[14] N. Abidi, E. Hequet, J. Appl. Polym. Sci. 93 (2004) 145.
[15] C. I. Simionescu, F. Denes, M. M. Macoveanu, G. Cazacu, M. Totolin, S. Percec,
D. Balaur, Cell. Chem. Technol. 13 (1979) 475.
[16] C. I. Simionescu, F. Denes, M. M. Macoveanu, G. Cazacu, M. Totolin, S. Percec, D.
Balaur, Cell. Chem. Technol. 14 (1980) 869.
[17] G. Akovali, F. Takrouri, J. Appl. Polym. Sci. 41 (1990) 2011.
[18] G. Akovali, F. Takrouri, J. Appl. Polym. Sci. 42 (1991) 2717.
[19] M. E. Hall, J. Zhang, A. R. Horrocks, Fire and materials, 18 ( 1994) 231.
[20] H. Herlinger, G. Hardtmann, F. Hermanutz, R. Schneider, U. Einsele, Melliand
Textilber.72(1991)E141.
[21] W. F. McSherry, G. L. Drake, A.B. Cooper, A. R. Markezich, Am. Dyest. Rep. 63
(1974)52.
[22] W. E. Franklin, S. P. Rowland, J. Appl. Polym. Sci. 24 (1979) 1281.
[23] J. E. Hendrix, G. L. Drake, J. Appl. Polym. Sci. 16 (1972) 257.
89

Chapter III Flame retardancy ofpolyacrylonitrile and cottonfabrics
[24] J. T. Langley, M. J. Drews, R. H. Barker, J. Appl. Polym. Sci. 25 (1980) 243.
[25] T. E. Lawler, M. J. Drews, R. H. Barker, J. Appl. Polym. Sci. 30 (1985) 2263.
[26] M. Lewin, Journal of Fire Sciences, 17 (1999).
[27] P. J. Wakelyn, W. Rearick, J. Turner, Am. Dyest. Rep. (1998) 13.
[28] S. Nakanishi, F. Masuko, T. Hashimoto, J. Appl. Polym. Sci. 71 (1999) 975.
[29] P. Zhu, S. Sui, B. Wang, K. Sun, G. Sun, J. Anal. Appl. Pyrolysis 71 (2004) 645.
90

Chapter TV Compatibility offlame retardants with water-repellent treatment
Chapter IV: Compatibility of flame retardants with water-repellent
treatment
1 Introduction
In this chapter the compatibility of different flame retardants for cotton (DEAEP,
DEAEPN, and BisDEAEPN) with water repellent treatment is investigated. Water repellent
and flame retardant properties can be conferred on textiles either in a single or in a two-step
treatment. The purpose of this work is to combine both properties by using the cold plasma
technique.
Perfluorocarbon plasmas are well known to be effective for water proofing polymeric
substrates either by fluorination of the surface layer (e.g. CF4) or deposition of plasma
polymers (e.g. C3F6, and C(,¥\4) [1-7]. It is worth investigating a two-step treatment where the
cotton fabrics which have been flame retarded using the PIGP procedure are submitted in CF4
gas plasma treatment (Figure 33). The obtained fluorination of the surface layer of the grafted
phosphorus polymer may lead to an increase of the hydrophobic character of the flame
retarded fabrics.
(S)1. Dipping
in solution of
M
2£ys^ 3. Washing
2. Ar
plasma
SgP4. Drying c cVa
FR grafted,
4Grafted
f plasma
polymerr
polymer
Figure 33 Procedure of the two-step treatment with Ar plasma-induced graft polymerization
ofFR monomers followed by a CF4 gas plasma
Water repellent textiles can also be obtained by coating with fluorine-containing
compounds such as homo and copolymers of perfluoroalkyl acrylates [8,9]. In Chapter III of
this work, it has been demonstrated that the PIGP procedure can be applied for grafting and
polymerization of various acrylic monomers containing phosphorus onto PAN and cotton
fabrics in one or in multiple treatments. The PIGP procedure could therefore be used to
combine the flame retardant and water repellent properties in two different ways:
(a) Multiple treatments: grafting and polymerization of a fluorinated acrylate monomer
onto the flame retarded fabrics (bi-layer treatment). This approach is presented in figure 34.
91
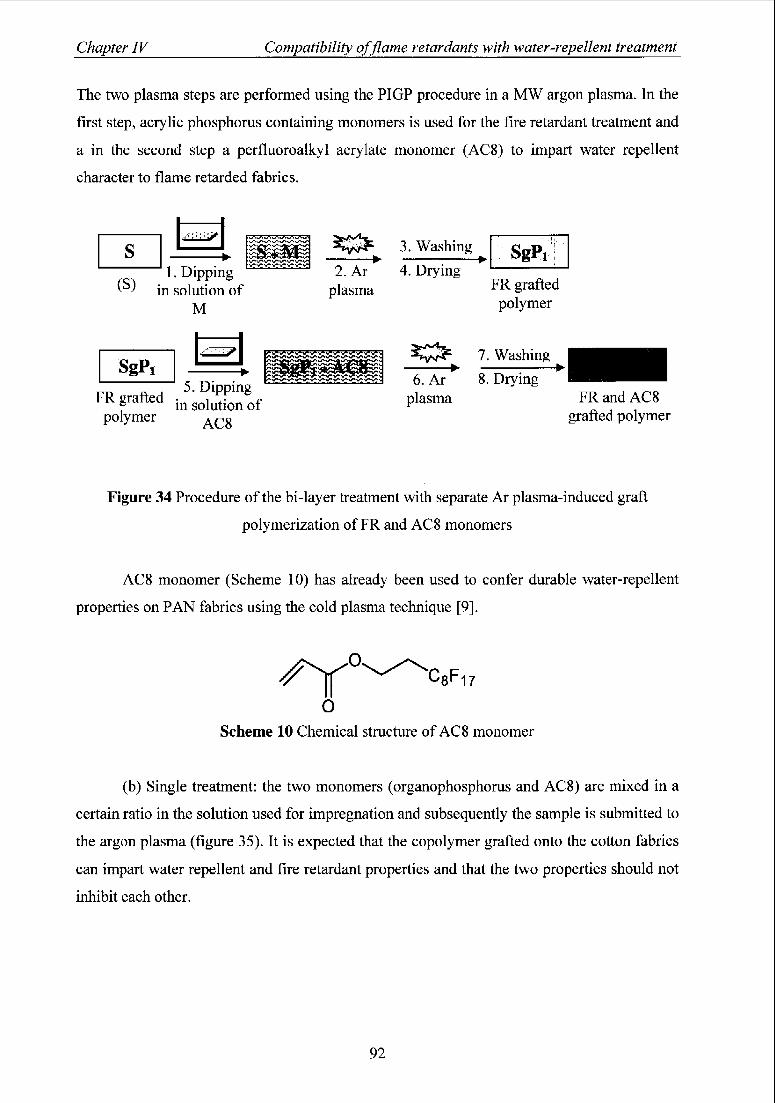
Chapter IV Compatibility offlame retardants with water-repellent treatment
The two plasma steps are performed using the PIGP procedure in a MW argon plasma. In the
first step, acrylic phosphorus containing monomers is used for the fire retardant treatment and
a in the second step a perfluoroalkyl acrylate monomer (AC8) to impart water repellent
character to flame retarded fabrics.
(S). Dipping
in solution of
M
% 'si<t4T- 3. Washing
SgPi
2. Ar
plasma
5. DippingFR grafted insolutionofpolymer ACg
4. Drying
6. Ar
plasma
SgPt
FR grafted
polymer
^vv^ 7. Washing
8. DryingFR and AC8
grafted polymer
Figure 34 Procedure of the bi-layer treatment with separate Ar plasma-induced graft
polymerization of FR and AC8 monomers
AC8 monomer (Scheme 10) has already been used to confer durable water-repellent
properties on PAN fabrics using the cold plasma technique [9].
C8F-|
Scheme 10 Chemical structure of AC8 monomer
(b) Single treatment: the two monomers (organophosphorus and AC8) are mixed in a
certain ratio in the solution used for impregnation and subsequently the sample is submitted to
the argon plasma (figure 35). It is expected that the copolymer grafted onto the cotton fabrics
can impart water repellent and fire retardant properties and that the two properties should not
inhibit each other.
92

Chapter TV Compatibility offlame retardants with water-repellent treatment
(S)1. Dipping
in solution of
M+AC8
Figure 35 Procedure for a single step treatment for the Ar plasma-induced graft
copolymerization of monomers (M+AC8)
In this study, the effectiveness of the fire retardant monomers (DEAEP, DEAEPN,
BisDEAEPN) combined with the different water-repellent treatments presented above is
investigated.
The LOI values and Schmerber pressures (PSCh) are measured to evaluate the flame
retardant and water-repellent character, respectively. Owing to the roughness and irregularity
of the textile surfaces, the commonly used contact angle measurements were not reliable for
the investigation of the wettability of the treated fabrics. Therefore they were replaced by
Schmerber tests. The Schmerber value recorded at the end of the test corresponds to the water
pressure (mbar) reached when water has penetrated through the fabric at three different
places. The principle of the Schmerber test is described in Appendix 4.
2 CF4 plasma treatment of flame retarded cotton fabrics with DEAEP, DEAEPN, and
BisDEAEPN
The untreated and treated cotton (120 g/m2) fabrics with (DEAEP, DEAEPN and
BisDEAEPN) were submitted to a CF4 plasma (Fcf4 = 36 seem, base pressure= 40 Pa, P =
300 W, t = 5 min). After the plasma treatment the Schmerber pressures (Psch2) were measured.
The results obtained are given in Table 15.
93

Chapter TV Compatibility offlame retardants with water-repellent treatment
Table 15 PSCh (mbar) and LOI values of untreated and treated cotton (120 g/m2) fabrics with
DEAEP, DEAEPN, and BisDEAEPN before (Pschi) and after CF4 plasma treatment (Psch2) and
heating 1 h at 100°C(Psch3)
MonomerTreatment
conditionsLOIi Pschi Psch2 Psch3 LOI2
Untreated
cotton- 19.0 0 2 - 19.0
DEAEP300 g/L, 5%
EGDA26.5 0 2 4 26.5
DEAEPN200 g/L, 10%
EGDA27.5 0 2 4 27.5
BisDEAEPN100 g/L, 10%EGDA
25.0 10 2 4 25.0
It can be observed that before the CF4 plasma treatment, the cotton fabrics treated with
BisDEAEPN after washing in methanol and water present a hydrophobic character (PSChi - 10
mbar), whereas the fabrics treated with DEAP and DEAEPN washed in the same conditions
are totally absorbent (Pschi = 0 mbar). Note that before washing (methanol and water) the
BisDEAEPN fabrics were also totally absorbent. The difference of wettability before and after
washing can be attributed to the fact that polyBisDEAEPN is soluble in methanol, which is
not the case for polyDEAEP and polyDEAEPN. The chain mobility of polyBisDEAEPN after
washing (methanol and water) thus increases provoking a migration of the non-polar groups
to the surface. This effect leads to a decrease in surface energy and an increase in the Psch
value.
After the CF4 plasma treatment, the PSCh values of the sample treated with
BisDEAEPN decrease dramatically from 10 to 2 mbar. This drop of the PSCh values can be
explained by the concurrent etching action of the fluorine species which occurs in plasma [5].
Indeed, plasma ablation competes with plasma polymerization in almost every case where
plasma is used to treat surface of solid materials. After the CF4 plasma treatment, the weight
of all the tested fabrics decreases by about 2%. For the fabrics treated with DEAEP and
DEAEPN, a slight increase of the PSCh values (from 0 to 2 mbar) is observed after the CF4
plasma treatment. Droplets of water remain but do not roll on the surface of the treated
fabrics, as can be seen in figure 36(b).
94

( 'hapter IV Compatibility of flame relardants with water-repellent treatment
It is known that the mobility of polymer chains increases with the temperature [2,9-
111. This effect was evaluated for the CT4 treated cotton fabrics After the CT4 plasma
treatment, the fabrics were heated loi lh at 100°C in an oven I he Psj, values were measured
alter cooling ol the samples to room temperature I he temperature of 10()°C is used because
at higher temperature the color of the fabrics is affected and they become yellow It can be
seen that after heating the Pslj, values double from 2 to 4 mbar I he droplets of water slightly
roll on the surface of the fabrics as can be seen in figure 36(c).
1 he water-repellent properties of the llame retarded cotton labrics with DI AI PN after
CT 1 plasma treatment arc shown in the following pictures (figure 36).
Figure 36 Water-repellent properties of cotton (120 g/mf ) fabrics treated with DEALPN, (a)
before, (b) after CT4 plasma treatment, and (c) alter heating
It can also be noticed that there are no changes of the LOI values before and after the
CT \ plasma and heating treatments
Wc can conclude that the treatment ol llame letarded labrics in CF4 gas plasma results
in surface fluorination, since a slight increase of the P^i, values can be observed The aspect ol
the droplets of water on the treated textiles also indicates a decrease in surface energy of the
95

Chapter IV Compatibility offlame retardants with water-repellent treatment
treated fabrics compare to the untreated. However, this treatment is not sufficient to impart a
good water repellent character.
3 Plasma-induced graft polymerization of AC8 on flame retarded fabrics with DEAEP,
DEAEPN, and BisDEAEPN.
In this approach the AC8 monomer is grafted and polymerized on the surface of cotton
(120 g/m2) fabrics flame retarded with DEAEP, DEAEPN and BisDEAEPN. The treated
fabrics were weighed and then immersed at room temperature for one minute in 20 ml of a
petroleum ether solution containing 50 g/L or 100 g/L of AC8 in the presence of 2.5% (w/w)
of the photoinitiator Darocur 1173 and 3.5% (w/w) of the cross linking agent EGDMA. These
impregnated fabrics were then pressed to evacuate the excess of the solution, placed onto
glass plates and submitted to a MW argon plasma (FAr = 125 seem, base pressure= 40 Pa, P =
100 W, t = 10 min). After treatment the samples were washed for 24 hours in a soxhlet
apparatus with chloroform to remove the monomer and non-grafted homopolymer and dried
for lhouratlOO°C.
The characteristic absorption bands of the polyAC8 at 1200 cnf'and 1150 cm"1
corresponding to the asymmetric and symmetric CF stretching could not be easily identified
on the IR(ATR) spectra of the bi-layer treated fabrics, since the absorption bands of the
primary and secondary OH deformation of cotton appear in the same region at about 1057,
1373, and 1162 cm". The grafting can also be confirmed by measurement of the PSCh values.
The percentage of grafting, the LOI and the Psch values obtained are given in Table 16.
Table 16 Psch (mbar) and LOI values of untreated and treated cotton (120 g/m ) fabrics with
DEAEP, DEAEPN, and BisDEAEPN before (%GFR, LOIi, Pschi) and after grafting and
polymerization of AC8) and drying 1 hour at 100°C (%Gacs, %>GFr+ac% LOI2, PSch2)
Monomer %GFR LOI, Pschi %GAC8 %Gfr+AC8 LOI2 PSch2
Untreated cotton - 19 0 3.03 3.03 19 11
DEAEPN24
23.46
26.5
26.5
0
0
3.50
10.48
29.36
36.41
27
27
15
17
DEAEP 30.86 26.0 0 3.74 35.76 26 10
BisDEAEPN 12.36 25.0 10 8.5 21.93 25 20
96

Chapter IV Compatibility of flame retardants with water-repellent treatment
Fhe results clearly indicate that the percentage of grafting and the Psj, values augment
after the plasma-induced graft polymerization of AC8 on llame retarded fabrics 1 he
significant increase of the P%,i values indicates the presence of a fluorinated polymer on the
surface of the fabrics, Ihe Pst,h values of cotton fabrics treated with DEAbP and DEAEPN
augment from 0 to 10 mbar, and to 15 mbar, respectively, with approximately the same
amount of grafted polyAC8. Note that this result is comparable to the Pst,i, value obtained with
similar amount of grafted polyAC8 on untreated cotton. For fabrics treated with DEAEPN, it
can be seen, that the PS(ji value augments from 15 to 17 mbar with an increase of grafted
fluorinated polymer from 3.5 to 10.5%. The Psch value of the sample treated with
BisDEAEPN doubles from 10 to 20 mbar.
It can be also observed that the Psül values of this bi-layer treatment depend on the
nature of the monomers used for the fiame retardation. The highest Pscii value is obtained with
the phosphoramidate monomers (DEAEPN, BisDEAEPN). Huorinatcd poly(meth)acrylates
arc comb-shaped polymers having fluorocarbon segments in the side chains oriented
perpendicularly to the longitudinal axis of the macromoleculc |9J. 1 his comb-shape
orientation is the key of a good water repellent character. The difference of PSL), \alues
between the samples treated with the phosphate and the phosphoramidate monomers could be
attributed to the assumption that the first layer of grafted polyphosphoramidatc polymer
(polyDEAEPN, polyBisDEAEPN) can permit a better parallel organization (comb-shaped) of
the fluorinated polyacrylate (polyAC8) chains on the surlace.
The water-repellent properties of the llame retarded cotton fabrics with Dl AhPN,
after grafting and polymerization of AC8 are presented in the following pictures (figure 37).
The droplets of water roll onto the surface of the bi-layer treated fabrics This observation
confirms the good water repellent character of the cotton textiles.
Figure 37 water-repellent properties of cotton ( 120 g/m") fabrics treated with DEAEPN (a)
before and (b) after grafting and polymerization of AC8, washing, and drying at 100°C
97

Chapter TV Compatibility offlame retardants with water-repellent treatment
The LOI values of the flame retarded fabrics remain almost the same after the grafting
of a layer of polyAC8. An increase of the amount of grafted polyAC8 from 3 to 10% does not
affect the LOI value of DEAEPN treated cotton. This can be explained by the fact that AC8
monomer is not a flame retardant for cotton.
From these results, it can be concluded that it is possible to confer good water
repellent properties to flame retarded cotton fabrics with DEAEP, DEAEPN, and
BisDEAEPN with 3%> of grafted polyAC8 using the PIGP procedure. Furthermore the
presence of the fluorinated polymer does not affect the flame retardant character of the
fabrics.
4 Plasma-induced graft copolymerization of AC8 and DEAEPN monomers.
The third approach investigated is the copolymerization of acrylic monomers
containing phosphorus (DEAEPN) and fluorine (AC8). In the previous Section 3, it has been
demonstrated that the best PSCh value is obtained with the phosphoramidate monomers.
Furthermore LOI values greater than 26 can be obtained with DEAEPN at monomer
concentration of 200g/L in presence of 10% of EGDA for cotton fabrics. Therefore cotton
"i -y
fabrics (120g/m and 210g/m ) were treated in 20 ml of methanol solution of concentration
300g/L containing DEAEPN and AC8 in the ratio 70:30 (w/w) which corresponds to a molar
ratio of 4:1, in the presence of 5% (w/w) of the photoinitiator (BAPO) and 10%) (w/w) EGDA
in a MW argon plasma (FAr= 125 seem, base pressure= 40 Pa, P = 100 W, t = 15 min). After
treatment the samples were washed for 24 hours in a soxhlet apparatus with chloroform to
remove the monomers and non-grafted copolymer, then in water and dried 1 hour at 100°C.
The surface grafting of the copolymer (polyDEAEPN and polyAC8) onto cotton
fabrics was evidenced by IR(ATR) analysis. The IR spectra of untreated and treated cotton
(120 g/m2) with DEAEPN and AC8 are shown in Figure 38.
98

Chapter TV Compatibility offlame retardants with water-repellent treatment
3600 3100 2600 2100 1600 1100 600
Wavenumber (cm" )
Figure 38 IR(ATR) spectra of cotton (120 g/m2) fabrics (a) untreated, (b) treated with
methanol solution containing DEAEPN and AC8 (70:30 (w/w)), after washing and drying 1 h
atl00°C.
One can easily identify the characteristic absorption band of the carbonyl stretching at
around 1730 cm" attributed to the grafted acrylate copolymer. However, it was not easy to
identify the asymmetric and symmetric CF and phosphate functional groups, as already
mentioned in Section 3. The results of the elemental analysis of the treated cotton shown in
Table 16 confirm the presence of polyDEAEPN and polyAC8 on the surface of the fabrics.
The amount of graft copolymer, the LOI and the PSCh values are given in Table 16.
Table 16 Percentage of grafting, LOI (%) and PSCh (mbar) values
Monomer %G %P (w/w)measured on
cotton
%F (w/w)measured on
cotton
LOI Psch
Untreated cotton
(120 and 210)- - - 19 0
DEAEPN (120) 39.57 2.8 2.29 27.5 16.0
DEAEPN (210) 33.01 2.3 2.83 28.0 22.0
The results show an increase of the LOI values from 19 to 28 and of the Psch values
from 0 to 16 for the lighter fabric, and from 0 to 22 for the heavier. The difference of the Psch
99

Chapter IV Compatibility of flame retardants with water-repellent treatment
values between the treated labrics can be attributed to the fact that the PSCh values depend on
the capillaries of the fabrics, which are related to the thickness
Ihe LOI values obtained arc similar to those obtained with the same phosphorus
content when the fabrics are treated only with DEAEPN (LOI value of 27.5 with a P content
of 2.77% for cotton 120, and LOI 28.5 with P content of 2.63 for cotton 210). I his is an
indication that the presence of the fluorinated polymer docs not affect the llame retardant
character of the treated fabrics. With the ratio 70:30 (w/w) of DEAEPN and AC8 it is possible
to confer simultaneously, water repellent and flame retardant properties on cotton fabrics.
Moreover, the low surface energy renders the treated fabrics also oil resistant as can be seen
from Figure 39.
Figure 39 (a) Water-and (b) oil-repellent properties of cotton (120 g/m2) fabrics treated with
DEAEPN and AC8, after washing and drying at 100°C
Ihe durability of the grafted copolymer (polyAC8 and polyDEAEPN) to washing was
investigated by the accelerated laundering method of McSherry et al.[12j. Ihe LOI value of
cotton 210 drops from 28 (%(i 34) to 24 (%Ci 26.4), and the Psth value decreases from 22 to 8
mbar. Ihe drop of the LOI value can be attributed to a slight ablation of the grafted
copolymer leading to a decrease of the phosphorus content. The significant decrease of the
Psch value after the McSherry procedure (95°C) can be attributed to the mutual variation of the
surface tension of polyAC8 and the washing solution. Indeed, when the temperature of the
water augments the surface tension decreases, where as the surlace tension of polyAC8 films
increases [9] The fluorinated chains tend to migrate in the direction of the surface increasing
the surface tension of the fabric. I his effect leads to the decrease of the Psch value. By drying
the fabrics 1 hour at 100°C after the McSherry procedure, the Psti» value increases from 8 to
20.
100

Chapter TV Compatibility offlame retardants with water-repellent treatment
5 Ageing tests
In order to test the stability of the water repellent treatment, the PSCh values of different
samples treated by the three procedures described above were left in the laboratory for 30
days in ambient air.
For the fabrics treated in CF4 plasma and not heated, a dramatic decrease of the
hydrophobic character is observed and the PSCh values can not be measured. For the fabrics
which were heated (100°C, 1 hour) a slight drop (from 4 to 2 mbar) is observed. However, the
Pseh values can be restored by heating again.
For the samples treated with AC8 monomer (bi-layer and copolymerization), the Psch
values remain almost the same. This demonstrates the durability of the coating due to the
formation of covalent bond between the cotton surface and the grafted polyAC8.
6 Conclusions
The aim of this study was to investigate if the flame retardants (DEAEP, DEAEPN,
BisDEAEPN) used in this work to flame retard cotton fabrics are compatible with a water
repellent treatment.
From the results obtained, we can conclude that the effectiveness of the flame
retardant is not affected by a CF4 plasma treatment, neither by grafting of a fluorinated
polymer. The increase of the hydrophobic character was much greater for the samples treated
with AC8 (bi-layer) than for those treated in CF4 plasma. The reason is that with AC8
compound, a fluorinated polymer is grafted onto the surface of the fabrics, while the treatment
in CF4 gas plasma resulted in surface fluorination [5-7], By copolymerization of DEAEPN
with AC8 in the ratio (70:30) we could demonstrate that by using the PIGP procedure it is
possible to confer in single treatment fire retardant and water repellent properties on cotton
fabrics. Furthermore there is no inhibition of the flame retardant effect by the water repellent
property.
Further investigations could be performed with the copolymerization and the bi-layer
treatment using the PIGP procedure, especially the possibility of creating polyfunctional
surfaces could be studied. The need to produce protective clothing, which combines various
properties such as flame retardant, soil-resistant, water repellent, antibacterial, weather
resistance, dyes resistance has increased. The PIGP procedure could be efficiently used for
these purposes. The choice between the single or the multilayer process will mainly depend
101

Chapter TV Compatibility offlame retardants with water-repellent treatment
on the kinetics of homo or copolymerization of each monomers used, on their compatibility,
and their mutual effects (they should not inhibit each other).
102

Chapter IV Compatibility offlame retardants with water-repellent treatment
7 References
[I] Y. Haque, B. D. Ratner, J. Appl. Polym. Sci. 42 (1986) 4369.
[2] Y. Iriyama, T. Yasuda, D. L. Cho, H. Yasuda, J. Appl. Polym. Sci. 39 (1990) 249.
[3] N. Inagaki, S. Tasaka, K. Mori, J. Appl. Polym. Sci. 43 (1991) 4369
[4] H. Z. Wang, M. W. Rembold, J. Q. Wang, J. Appl. Polym. Sci. 49 (1993) 701
[5] F. Hochart, J. Levalois-Mitjaville, R. De Jaeger, L. Gengembre, J. Grimblot, Appl.
Surf. Sci. 142(1999)574.
[6] J. Garbassi, E. Occhiello, J. Adhesion. Sei. Technol. 13 (1999), 65.
[7] M. G. McCord, Y. J. Hwang, Y. Qiu, L. K. Hughes, M. A. Bourham, J. Appl. Polym.
Sci. 88 (2003) 2038.
[8] U. Vorher, M. Müller, C. Oehr, Surf. Coat. Technol. 98 (1998) 1128.
[9] F. Hochart, R. De Jaeger, J. Levalois-Grützmacher, Surf. Coat. Technol. 165 (2001)
201.
[10] CM. Chan, Polymer surface modification and characterization, Hanser/Gardner
publications, Inc.(1994).
[II] E. Selli, G. Mazzone, C Oliva, F. Martini, C. Riccardi, R. Barni, B. Marcandalli, M.
R. Massafra, J. Mater. Chem. 11 (2001),
[12] W. F. McSherry, G. L. Drake, A.B. Cooper, A. R. Markezich, Am. Dyest. Rep. 63
(1974) 52.
103

Chapter IV Compatibility offlame retardants with water-repellent treatment
Seite Leer /
Blank leaf

Chapter V General conclusions
Chapter V : General conclusions
The aim of this thesis was to study the effectiveness of the plasma-induced graft
polymerization (PIGP) procedure as a novel method to impart durable flame retardant
properties to synthetic (polyacrylonitrile) and natural (cotton) textiles, without altering their
bulk properties. This new approach is based on the use of an argon microwave (MW) plasma
to induce in one step, the grafting and the polymerization of acrylic monomers containing
phosphorus onto the surface of various materials previously impregnated with the monomers.
Three different classes of organophosphorus monomers were synthesized: acrylate
phosphates (DEMEP, DEAEP) and phosphonates (DEAMP, DMAMP) which are known for
their fire retardant properties. The synthesis of two new acrylate phosphoramidate monomers
(DEAEPN, BisDEAEPN) was also developed.
The argon plasma-induced polymerization of the different monomers was investigated.
This study has shown that acrylate phosphate (DEMEP, DEAEP), phosphonate (DEAMP,
DMAMP) and phosphoramidate (DEAEPN, BisDEAEPN) monomers can be easily
polymerized under MW argon plasma in presence of Irgacure 819 as photoinitiator. The
kinetic of homopolymerization of the monomers were monitored by 'H NMR spectroscopy in
DMSO-d6. The results showed that the order of the reactivity is DEMEP < DEAEPN <
DEAEP < DEAMP < DMAMP. BisDEAEPN could not be polymerized in bulk due to its
high viscosity. *H NMR and IR (ATR) analyses allowed us to confirm that the polymerization
of the acrylic monomers containing phosphorus proceeds by a radical mechanism and that the
polymerization under MW argon plasma is mostly induced by UV-Visible radiations. The
thermal behavior of the polymers was studied by thermogravimetric analysis (TGA). The TG
thermograms of the polymers showed that all the polymers decompose at lower temperatures
than cotton and polyacrylonitrile (PAN) fabrics. For all the polymers, the percentage of
remaining residue at 570°C was higher compared to cotton, and lower than for PAN. These
results indicated that the polymers are thermally stable and can be potential flame retardant
for PAN and cotton fabrics.
105

Chapter V General conclusions
The experimental conditions used for homopolymerization were applied for the
grafting and polymerization of the monomers onto PAN and cotton fabrics. The grafting and
polymerization of the monomers onto the surface of PAN and cotton fabrics were confirmed
by IR (ATR) and SEM analyses. The grafting yields were found to vary with the type and
concentration of the monomers, and the amount of the crosslinking agent.
The limiting oxygen index (LOI) measurements showed that fire retardant properties
of treated PAN and cotton fabrics were improved compared to the untreated ones. For PAN
fabric the LOI value of 26.5 was obtained with DMAMP, and for cotton fabrics LOI values of
28.5 and 29.5 were obtained with DEAEPN and BisDEAEPN, respectively. The good flame
retardant properties of the phosphoramidate monomers could be attributed to the presence of
nitrogen which cause synergistic enhancement in the efficiency of phosphorus-based flame
retardant.
The comparison of the flame effect on PAN and cotton fabrics showed that the flame
retardants (DEAEP, DMAMP, and DEAEPN) are more effective when treated with cotton
compare to PAN. The difference of efficiency could be attributed to the fact that the most
effective flame retardants for acrylic should promote highest residual char possible, while the
effectiveness of fire retardants for cotton is related to the rate of phosphorylation of cellulose.
The durability of the flame retardant treatment under washing at 95°C was
investigated using the McSherry procedure. The results obtained showed a persistence of the
flame retardant character. These results confirm that .the acrylic polymers containing
phosphorus are covalently grafted onto the surface of the fabrics.
Using the PIGP procedure we could demonstrate that this surface treatment can
seriously compete with the other types of treatment using organophosphorus compounds.
Indeed, with the other classical approaches (reactive and additive) the best LOI value obtained
so far reaches 28 for PAN fabrics treated with DMAMP with about 23% of polymer loading
as additives and with 3.6 % of phosphorus content in the fiber. The major achievement of
PIGP procedure is the fact that it allows to graft and to polymerize various monomers onto the
surface of fabrics in one-step. By this way, the polymer is covalently linked to the surface of
the fabric, which guarantees the resistance of the coating towards the washing, even at higher
temperatures. Therefore, it is worth considering the PIGP procedure as a new approach to
permanently flame retard textile fabrics on which only a surface treatment can be applied like
on natural fibers.
106

Chapter V General conclusions
The compatibility of the fire retardant and water-repellent properties was also studied.
The aim was to investigate if the flame retardants DEAEP, DEAEPN, and BisDEAEPN are
compatible with a water repellent treatment. From the results obtained, we could conclude
that the effectiveness of the flame retardants is not affected by a CF4 plasma treatment, neither
by grafting of a fluorinated polymer. The increase of the hydrophobic character was much
greater for the samples treated with AC8 (bi-layer) than for those treated in CF4 plasma. The
reason is that with AC8, a fluorinated polymer is grafted onto the surface of the fabrics, while
the treatment in CF4 gas plasma resulted in surface fluorination. By copolymerization of
DEAEPN with AC8 in the ratio (70:30) we could demonstrate that by using the PIGP
procedure it is possible to confer fire retardant and water repellent properties on cotton fabrics
in a single treatment. Furthermore there is no inhibition of the flame retardant effect by the
water repellent property.
Further investigations could be performed with the copolymerization and the bi-layer
treatment using the PIGP procedure, especially the possibility of creating polyfunctional
surfaces could be studied. The need to produce protective clothing, which combines various
properties such as flame retardant, soil-resistant, water repellent, antibacterial, weather
resistance, dyes resistance has increased. The PIGP procedure could be efficiently used for
these purposes. The choice of the single or the multilayer process will mainly depend on the
kinetics of homo- or copolymerization of each monomers used, on their compatibility, and on
their mutual effects (they should not inhibit each other).
107

I'—
Seite Leer /
Blank leaf
108

Chapter VI Experimentalpart
Chapter VI: Experimental part
1 General comments
7.7 Working procedures
The synthesis of the monomers was performed under argon atmosphere with use of
standard schlenk technique. Polymerizations were carried out under MW argon plasma onto
glass plates coated with the liquid monomers. The argon was provided by PANGAS.
Solvents (from Fluka, Baker or Merck) were distilled under argon prior to use if
necessary, diethyl ether from sodium/benzophenone and dichloromethane from calcium
hydride.
Basic chemicals were purchased at ABCR, Acros, Aldrich, Fluka, or Lancaster and
used without further purification. The photoinitiator Irgacure 819 (BAPO) was obtained from
Ciba Specialty Chemicals.
Polyacrylonitrile (PAN) and cotton fabrics were kindly supplied by Dickson-Constant
Society and DJH International, respectively.
The following compounds were prepared by the literature methods: diethyl-2-
(methacryloyloxy) ethyl phosphate [1], diethyl (acryloyloxy) ethyl phosphate [2], diethyl
(acryloyloxy) methyl phosphonate [3], dimethyl (acryloyloxy) methyl phosphonate [3] and
dimethyl hydroxymethyl phosphoante [4].
1.2 Analytical techniques
NMR measurements were carried out on Bruker Avance 250 and 300 MHz
spectrometers. Chemicals shifts (Ô) are in ppm and are reported relative to external standards:
!H and13C NMR to TMS and 31P to H3PO4, multiplicities are abbreviated as singlet (s),
doublet (d), triplet (t), m (multiplet), br (broad). The absolute values of the coupling constants
are given in hertz (Hz).
IR spectra were recorded on a Perkin-Elmer FT-IR spectrometer in range 4000-600
cm",the ATR technique was applied. The absorptions bands are described as intensity (vw
(very weak), w (weak), m (medium), s (strong)), shape (broad (br) or sharp (sh)) and position
(cm"1).
109

Chapter VI Experimental part
Mass spectra of the monomers were recorded on a Finnigan MAT SSQ 7000 mass
spectrometer.
Boiling points were determined by distillation and are uncorrected.
Thermogravimertic analyses (TGA) were performed on a Netzsch STA 409C
apparatus. The sample weight was about 25 mg and the temperature range from 20 to 700°C
or to 1100°C, at heating rate of 1 OK/min under argon atmosphere.
Scanning electron microscopy (SEM) was performed on a LEO 1530 microscope with
a field gun emission operated at 1KV.
Elemental analyses were performed in the laboratory of organic chemistry of the ETH
Zürich, the Phosphorus content of the treated fabrics were determined by the
vanadomolybdophosphoric acid colorimetric method using an Uvikon 810, after perchloric
acid-sulfuric acid digestion. Carbon, hydrogen and nitrogen contents were determined using a
LECO CHN-900.
The flame retardancy of the fabrics was evaluated by limiting oxygen index (LOI) test
according to ISO 4589-2, using an oxygen index test apparatus from Fire Instrumentation
Research Equipment LTD with a digital readout of oxygen concentration to ±0.1%. The LOI
value corresponds to the minimum concentration of oxygen in the mixture of oxygen/nitrogen
just necessary to burn the sample (52x140mm) during 3 min or over a length of 80 mm.
The wettability of the fabrics was evaluated by Schmerber tests according to DIN
53886 using a Textest FX 3000 water impermeability II apparatus from Textest Instruments.
The Schmerber value recorded at the end of the test corresponds to the water pressure (mbar)
reached when water has penetrated through the fabric at three different places.
The durability of the treatment to washing was tested according to the accelerated
laundering method proposed by McSherry et al. [5], The samples were boiled for 4h in a
solution of 0.5% Na3P04.12H20 and 0.1 % triton X-100 at an approximate liquor ratio of
40:1.
2 Preparation and characterization of the monomers
Diethyl-2-(methacryloyloxy) ethylphosphate: DEMEP (I)
CH3 o
MF:CioHi906P 1 n^ V°Et
MW: 266.23 g/mol II OEt>Y°WC
110

Chapter VI Experimentalpart
To a cooled mixture of 13 g (0.10 mol) of 2-hydroxy ethyl methacrylate, 12.1 g (0.12
mol) of dry triethylamine and 0.1 g of CuCl in 100 ml of dry diethyl ether, 17.2 g (0.10 mol)
of diethylchlorophosphate was added dropwise while magnetically stirring under argon. The
system was then allowed to attain room temperature and stirred overnight. The precipitated
triethylamine chloride salt was filtered and washed with ether. The filtrate was washed with
ice cold aqueous solution ofNaOH (2%>), followed with distilled water then dried over
anhydrous MgS04 The ether was evaporated off and the residue was distilled under vacuum
after adding a small amount of hydroquinone. A colorless liquid was obtained; yield: 17 g,
64%. Bp: 90°C/ 0.1 mm Hg.
'H-NMR (300.1 MHz, CDCI3) 5 = 1.38 (t, 3Jmi = 7.8 Hz, 6H,-CH3), 1.98 (s, 3H, =C-CH3),
4.14-4.39 (m, 8H, -OCH2), 5.64 (s, 1H, =CH), 6.20 (s, 1H, =CH).
13C-NMR (75.5 MHz, CDC13) 5 = 16.3-18.4 (CH3), 64.0-65.3 (OCH2), 126.3(=CH2), 136.1
(H2C=C-), 167.1 (-C=0).
31P-NMR (121.5 MHz, CDC13) 5 = -1.14 (s)
MS m/z (fragment, intensity in %): 267 (M+1, 30), 221 (M-45, 40), 180 (M-C4H602, 30), 152
(M-C6Hio02, 40), 112 (M-C4H,,04P, 100), 99 (M-C5H12O4P, 50), 82 (M-184, 60), 69 (M-
C6H140sP, 85), 67 (M-199, 30).
IR (neat): 2984 (w) and 2904 (vw) (CH stretch), 1719 (s, C=0 stretch), 1636 (w, C=C
stretch), 1450 (m, CH bending), 1396 (w), 1269 (s, P=0 stretch), 1165 (s), 1025 (s) and 974
(s) (P-O-C stretch), 813 (m), 743(w), 657 (m).
Ill

Chapter VI Experimental part
Diethyl (acryloyoxy) ethylphosphate:DEAEP (2)
MF: C9H1706P^N^°
MW: 252.20 g/mol T
To a cooled (0-5°C) mixture of 11.6 g (0.10 mol) of 2-hydroxy ethyl acrylate, 12.1 g
(0.12 mol) of dry triethylamine and 0.1 g of CuCl in 100 ml of dry diethyl ether, 17.2 g (0.10
mol) of diethylchlorophosphate was added dropwise while magnetically stirring under argon.
The system was then allowed to attain room temperature and stirred overnight. The
precipitated was filtered and washed with ether. The filtrate was washed with ice cold
aqueous solution of NaOH (2%), followed with distilled water then dried over anhydrous
MgS04 The ether was evaporated off and the residue was distilled under vacuum after adding
a small amount of hydroquinone. A colorless liquid was obtained; yield: 15 g, 59.2%. Bp:
105°C/0.1 mmHg.
!H-NMR (300.1 MHz, CDCh) 8 = 1.34 (t, 3JHH = 7.0 Hz, 6H,-CH3), 3.83-4.40 (m, 8H, -
OCH2), 5.88 (dd,2JHH =1-7 Hz, 3JHH= 10.2 Hz, 1H, =CH eis), 6.11 (dd,3JHn= 17.3Hz,3Jh
10.2 Hz, 1H, HC=C), 6.48 (dd,2Jn,i =1.7 Hz, 3Jim = 17.3 Hz, 1H, =CH trans).
13C-NMR (75.5 MHz, CDC13) 5 = 16.2 (-CH3), 63.2-66.3 (-OCH2), 128.1 (=CH-), 131.5
(=CH2), 165.7 (-C=0).
3,P-NMR (121.5 MHz, CDC13) 5 = 0.08 (s)
MS m/z (fragment, intensity in %): 253 (M+1, 45), 209 (M-43, 100), 180 (M-C3H402, 75),
153 (M-C5H702, 75), 124 (M-128, 95), 99 (M-C4Hi0O4P, 85), 81 (M-171, 85), 81 (M-171,
85),69(M-184,60).
112

Chapter VI Experimentalpart
IR (neat): 2984 (m) and 2909 (vw) (CH stretch), 1727 (s, C=0 stretch), 1636 (w, C=C
stretch), 1446 (w, CH bending), 1410 (m, =CH2 bending), 1370 (vw), 1268 (s, P=0 stretch),
1192 (s), 1028 (s) and 976 (s) (P-O-C stretch), 807 (m), 744 (w), 669 (m)
Diethyl (acryloyloxy) methylphosphonate: DEAMP (3)
MFiCgHisOjP >. n VOC2H5
MW: 222.17 g/mol II OC2H5
9.05 g (0.10 mol) of acryloyl chloride dissolved in 200 ml of dry dichloromethane was
added dropwise to a stirred and cooled (0-5°C) mixture of 16.8g (0.10 mol) of diethyl
hydroxmethyl phosphonate and 12.1 g (0.12 mol) of dry triethylamine under argon while
magnetically stirring. The system was then allowed to attain room temperature and stirred
overnight. The precipitated was filtered and washed with dichloromethane. The filtrate was
washed with aqueous solution of Na2C03, the solvent was evaporated and the residue was
distilled under vacuum after adding a small amount of hydroquinone. A colorless liquid was
obtained; yield: 17 g, 76.5%. Bp: 90°C/ 0.1 mm Hg.
^-NMR (300.1 MHz, CDC13) 5 = 1.29 (t, 3JHH = 7 Hz, 6H,-CH3), 4.00-4.14 (m, 4H, -
POCH2), 4.37 (d,2JHP = 8.7 Hz, 2H, -OCH2P), 5.86 (dd, 2JHH =1.3 Hz, 3JHH = 10.4 Hz, 1H,
=CH eis), 6.10 (dd,3.IHH = 17.3Hz,3JHH = 10.4 Hz, 1H, HC=C), 6.40 (dd, 2JHH =1.3 Hz, 3JHH =
17.3 Hz, 1H,=CH trans).
°C-NMR (75.5 MHz, CDC13) 5 = 16.0 (-CH3), 55.5 and 57.8 (d, -OCH2P), 62.5 (-POCH2),
127.0 (-HC=), 131.9 (=CH2), 165.0 (-C=0).
31P-NMR (121.5 MHz, CDC13) 0= 18.8 (s)
113

Chapter VI Experimentalpart
IR (neat): 2984 (w) and 2935 (vw) (CH stretch), 1735 (s, C=0 stretch), 1634 (w, C=C
stretch), 1444 (vw, CH bending), 1405 (m, =CH2 bending), 1325 (vw), 1246 (s, P=0 stretch),
1177 (s), 1023 (s) and 971 (s) (P-O-C stretch), 826 (vw), 806 (w), 632 (w).
Dimethyl (acryloyloxy) methylphosphonate: DMAMP (4)
MF: CsHnOsP ^K^^J8^0^MW: 194.12 g/mol jj 0CH3
9.05 g (0.10 mol) of acryloyl chloride dissolved in 200 ml of dry dichloromethane was
added dropwise to a stirred and cooled (0-5°C) mixture of 19.4 g (0.10 mol) of dimethyl
hydroxmethyl phosphonate [4] and 12.1 g (0.12 mol) of dry triethylamine under argon while
magnetically stirring. The system was then allowed to attain room temperature and stirred
overnight. The precipitated was filtered and washed with dichloromethane. The filtrate was
washed with aqueous solution of Na2C03, the solvent was evaporated and the residue was
distilled under vacuum after adding a small amount of hydroquinone. A colorless liquid was
obtained; yield: 15 g, 77.3 %. Bp: 80°C/ 0.1 mm Hg.
'H-NMR (300.1 MHz, CDC13) 5 3.70 (d, 3JHP = 10.8 Hz, 6H,-OCH3), 4.40 (d, 2JHp = 8.7 Hz,
2H, -OCH2P), 5.83 (dd, 2JHH =1.3 Hz, 3Jim = 10.4 Hz, 1H, -CH eis), 6.08 (dd,3JHH = 17.3Hz,
3Jhh = 10.4 Hz, 1H, HC=C), 6.36 (dd, 2JHH =1.3 Hz, 3JHH = 17.3 Hz, 1H, =CH trans).
"C-NMR (75.5 MHz, CDC13) 5 53.1 (-OCH3), 54.3and 57.1 (d, -OCH2P), 126.5(-HC=),
132.2(=CH2), 165.8 (-C=0).
31P-NMR (121.5 MHz, CDC13) 5 21.5 (s)
IR (neat): 2959 (w) and 2856 (vw) (CH stretch), 1735 (s, C=0 stretch), 1633 (w, C=C
stretch), 1457 (w, CH bending), 1407 (m, =CH2 bending), 1328 (m), 1250 (s, P=0 stretch),
1174 (s), 1030 (s, P-O-C stretch), 837 (w), 809 (vw), 632 (w).
114

Chapter VI Experimentalpart
Diethyl (acryloyloxy) ethylphosphoramidate: DEAEPN (5)
°W ^OEtMF: C9H18N05P ^Y^^AiVtMW:251.21g/mol o
17.2 g (0.10 mol) of diethyl chlorophosphate was added dropwise to a mixture of 6.1
g (0.10 mol) of ethanolamine and 10.1 g (0.10 mol) of triethyamine in 100 ml
dichloromethane at 0°C and under argon. After the addition was complete, the mixture was
then warmed at room temperature and stirred for 2 hours. The precipitated triethylamine
hydrochloride was removed by filtration. The filtrate was then cooled to 0°C, and to this
stirred solution was added 12.1 g (0.12 mol) of dry triethylamine under an atmosphere of
argon. A solution of acryloyl chloride (9.05 g, 0.10 mol) in 100 ml of dry dichloromethane
was then introduced dropwise. The system was then allowed to attain room temperature and
stirred overnight. The precipitated was filtered and washed with dichloromethane. The filtrate
was washed with aqueous solution of Na2C03, the solvent was evaporated and the residue
was distilled under vacuum after adding a small amount of hydroquinone. Pale yellow viscous
oil was obtained; yield: 15.6g, 62%. Bp: 135°C/ 0.1 mm Hg.
'H-NMR (250.1 MHz, CDC13) 5 = 1.10 (t, 3JHH = 7.0 Hz, 6H,-CH3), 2.88-3.00 (m, 2H, -
CH2N), 3.73-3.97 (m, 7H, -OCH2andNH), 5.60 (dd,2JHH =1.7 Hz,3JHH = 10.5 Hz, 1H, =CH
eis), 5.87 (dd,3JHH = 17.3Hz,3JHH = 10.3 Hz, 1H, HC=C), 6.18 (dd,2JHn =1.7 Hz, 3JHH = 17.3
Hz, 1H,=CH trans).
13C-NMR (75.5 MHz, CDC13) Ô = 16.0 (-CH3), 40.0 (CH2N), 61.9 (POCH2), 64.4 (-OCH2),
127.9 (=CH-), 130.9 (=CH2), 165.7 (-C=0).
31P-NMR (121.5 MHz, CDC13) Ö = 9.0 (s)
MS m/z (fragment, intensity in %): 252 (M+1, 30), 179 (M-43, 100), 179 (M-C3H402,
45),166 (M-C4H502, 78), 152 (M-CsH702, 7), 138 (M- C5H702N, 35), 122 (M-129,16), 110
(M-141, 81), 81 (M-170, 19), 65 (M-186, 10), 55 (M-C3H30, 100).
115

Chapter VI Experimentalpart
IR (neat): 3217 (m, br, NH stretch), 2981 (m) and 2899 (vw) (CH stretch), 1724 (s, C=0
stretch), 1635 (w, C=C stretch), 1445 (w, CH bending), 1409 (m, =CH2 bending), 1238 (s,
P=0 stretch), 1193 (s), 1135 (w), 1030 (s) and 965 (s) (P-O-C stretch),, 806 (m).
l,3-Bis(diethylphosphoramidate)-propan-2-ol: BisDEAEPNOH (6a)
VOEt
MF: CiiH28N207P2 ^~NHHO
MW: 362.29 g/mol ^
' ,OEt>—1\
OEt
uOEt
17.2 g (0.10 mol) of diethyl chlorophosphate was added dropwise to a mixture of 4.5
g (0.05 mol) of l,3-diaminopropan-2-ol and 12.1 g (0.10 mol) of triethyamine in 100 ml of
dry dichloromethane at 0°C and under argon. After the addition was complete, the mixture
was kept at 0°C for 5 to 6 hours, and then stirred at room temperature overnight. The
precipitated triethylamine hydrochloride was removed by filtration and washed with ethyl
acetate. The combined organic phases were precipitate in hexane. The highly viscous oil
obtained was dissolved in dichloromethane, washed with a saturated solution of NaHC03 and
dried over MgS04, and then the solvent was evaporated. Colorless highly viscous oil was
obtained; yield 12.67 g, 70%.
XH-NMR (300.1 MHz, CDCh) 5 = 1.30 (t, 3Jim = 7.0 Hz, 6H,-CH3), 2.92-3.03 (m, 4H, -
CH2N), 3.65-3.78 (m, 3H, -CH and -NH), 3.99-4.08 (m, 8H, -OCH2), 4.38 (br, OH).
13C-NMR (75.5 MHz, CDC13) Ô = 16.1 (-CH3), 44.2 (CH2N), 62.5 (POCH2), 71.2 (-CH).
31P-NMR (121.5 MHz, CDC13) 5 = 9.9 (s).
116

Chapter VI Experimentalpart
MS m/z (fragment, intensity in %): 363 (M+1, 15), 344 (M-H20, 100), 317 (M-C2H50, 10),
207( M-155, 25), 196 (M-C5H13N03P, 100), 166 (M-C6H15N04P, 47), 154 (M-208, 22), 138
(M-224, 35), 122 (M-240,28), 110 (M-252, 77), 98 (M-264,15), 81 (M-281,16).
IR (neat) 3244 (m, br, NH and OH stretch), 298 l(m) and 2907 (w) (CH stretch), 1444 (w, CH
bending), 1392 (w), 1227 (s, P=0 stretch), 1027 (s) and 962 (s) (P-O-C stretch), 796 (m).
(Acryloyloxy)-l,3-Bis(diethylphosphoramidate)-propan: bisDEAEPN (6)
St\ /?
OEt
MF: Ci4H30N2O8P2O
' OFt
NHU£/l
MW: 416.34g/mol ^-NH__
s ^OEt
Ae,
2.94 g (0.0325 mol) of acryloyl chloride dissolved in 10 ml of dry dichloromethane
was added dropwise to a stirred and cooled (0-5°C) mixture of 9 g (0.0250 mol) of 1,3-
bis(diethylphosphoramidate)-propan-2-ol (6a) and 4.1 g (0.040 mol) of dry triethylamine
under argon while magnetically stirring. After the addition was complete, the mixture was
kept at 0°C for 2 to 3 hours, and then stirred at room temperature overnight. The precipitated
triethylamine hydrochloride was removed by filtration and washed with dichloromethane. The
filtrate was washed with aqueous solution of Na2C03, the solvent was evaporated and the
residue was purified by dissolution in ether. After filtration, the solvent was evaporated off
and yellow highly viscous oil was obtained; yield 5.1 g, 48.9%.
XH-NMR (300.1 MHz, CDC13) 5 = 1.32 (t, 3JHH = 7.0 Hz, 6H,-CH3), 3.16-3.30 (m, 6H, -
CH2N and NH), 4.0-4.16 (m, 8H, -OCH2), 4.91-4.96 (m, 1H, -CH), 5.87 (dd,2JHH =1.4 Hz,
3Jhh = 10.4 Hz, 1H, =CH eis), 6.10 (dd,3JHH = 17.3Hz,3JHH = 10.4 Hz, 1H, HC=C), 6.44 (dd,
2Jhh =L4 Hz, 3JHH = 17.3 Hz, 1H, =CH trans).
117

Chapter VI Experimentalpart
,3C-NMR (75.5 MHz, CDC13) 8 = 16.2 (-CH3), 41.1 (CH2N), 62.6 (POCH2), 73.4 (-CH), 128
(=CH-), 131.6 (=CH2), 165.4 (-C=0).
31P-NMR (121.5 MHz, CDC13) S = 9.0 (s).
MS m/z (fragment, intensity in %): 417 (M+1, 70), 371 (M-C2H50, 10), 344 (M-C3H402,
100), 315 (M-101, 40), 207( M-155, 25), 291 (M-191, 30), 263 (M-C4H12N03P, 70), 234 (M-
C6Hi5N04P, 37), 207 (M-209, 90), 196 (M-C8H16N04P, 77), 166 (M-C9H17N05P), 154 (M-
262, 35), 138 (M-278, 28), 110 (M-2(C4Hi2N03P), 49), 98 (M-321, 32), 81 (M-335, 27),
55(M-C„H27N2P2).
IR (neat) 3220 (m, br, NH stretch), 298 l(m) and 2906 (vw) (CH stretch), 1723 (s, C=0
stretch), 1633 (w, C=C stretch), 1444 (w, CH bending), 1407 (m, =CH2 bending), 1237 (s,
P=0 stretch), 1195 (m), 1098 (w), 1028 (s) and 964 (s) (P-O-C stretch), 799 (m).
3 Plasma-induced polymerization of the monomers under argon microwave plasma
The microwave plasma was generated by a Europlasma DC300PC system composed
of three parts: (i) a microwave generator (2.46 GHz) with a tunable power ranging from 0 to
600 W generates the argon glow discharge, (ii) the vacuum chamber (27 1) (aluminium based
container) in which the process takes place and (iii) a pumping system composed of a primary
pump (E2M28 PFPE, Edwards). The gas flow was regulated by unit mass flow controllers.
The monomers (0.5g) were mixed with 5% (w/w) of the photoinitiator (BAPO), put
onto glass plates to give liquid film of non-optimized thickness, and then they were
introduced in the plasma chamber. After plasma treatment (FAr = 125seem, P = 100 W, base
pressure= 40 Pa,) for 20 min, the films obtained were removed from the glass plates. The
polymers are washed in methanol (in ether for polyDEAMP and polyBisDEAEPN) to remove
the monomer and dried under vacuum at 100°C. PolyDEMEP was insoluble in DMSO and
could not be therefore characterized by NMR spectroscopy in solution.
118

Chapter VI Experimentalpart
Poly[diethyl-2-(methacryloyloxy) ethylphosphate]: PolyDEMEP
IR (neat) 2984 (w) and 2909 (vw) (CH stretch), 1724 (s, C=0 stretch), 1446 (w, CH
bending), 1392 (w), 1260 (s, P=0 stretch), 1153 (s), 1020 (s) and 969 (s) (P-O-C stretch), 854
(vw), 799 (m), 746 (w).
Poly[diethyl (acryloyoxy) ethylphosphate]: PolyDEAEP
'H-NMR (300.1 MHz, DMSO-d6) 6 = 1.25 (br, 6H,-CH3), 2.3-1.6 (br, 3H, CH2-CH), 4.03
(br, 6H -OCH2).
31P-NMR (121.5 MHz, DMSO-d6) Ô = -0.94 (s)
IR (neat) 2984 (m) and 2909 (vw) (CH stretch), 1732 (s, C=0 stretch), 1446 (w, CH
bending), 1391 (vw), 1260 (s, P=0 stretch), 1163(s), 1018 (s) and 966 (s) (P-O-C stretch),
799 (m), 744 (w).
Poly[diethyl (acryloyloxy) methyl phosphonate]: PolyDEAMP
'H-NMR (300.1 MHz, DMSO-d6) 5 = 1.28 (br, 6H,-CH3), 2.4-1.4 (br, 3H, CH2-CH), 4.1-4.0
(m, 4H, -OCH2), 4.4 (br, 2H, -OCH2P).
31P-NMR (121.5 MHz, DMSO-d6) 5 = 18.9 (s)
IR (neat) 2983 (w) and 2936 (vw) (CH stretch), 1741 (s, C=0 stretch), 1444 (w, CH
bending), 1391 (vw), 1246 (s, P=0 stretch), 1156 (s), 1019 (s) and 968 (s) (P-O-C stretch),
809 (m).
119

Chapter VI Experimental part
Poly[dimethyl (acryloyloxy) methylphosphonate]: PolyDMAMP
XH-NMR (300.1 MHz, DMS0-d6) 8 = 2.4-1.6 (br, 3H, CH2-CH), 3.70 (br, 6H,-CH3), 4.47
(br, 2H, -OCH2).
31P-NMR (121.5 MHz, DMSO-d6) 5 = 21.6 (s)
IR (neat): 2958 (w) and 2856 (vw) (CH stretch), 1738 (s, C=0 stretch), 1448 (w, CH
bending), 1324 (m), 1243 (s, P=0 stretch), 1150 (s), 1014 (s, P-O-C stretch), 914 (w), 805 (s).
Poly[diethyl (acryloyloxy) ethylphosphoramidate]: PolyDEAEPN
'H-NMR (300.1 MHz, DMSO-d6) 5 = 1.21 (br, 6H,-CH3), 2.2-1.9 (br, 3H, CH2-CH), 3.9 (br,
4H, -OCH2), 4.96 (br, 1H, NH).
31P-NMR (121.5 MHz, DMSO-d6) 8 = 9.6 (s).
IR (neat) 3214 (m, br, NH stretch), 2980 (m) and 2904 (vw) (CH stretch), 1730 (s, C=0
stretch), 1443 (m, CH bending), 1392 (w), 1226 (s, P=0 stretch), 1162 (m), 1135 (vw), 1022
(s) and 957 (s) (P-O-C stretch), 793 (m).
Poly [(acryloyloxy)-l,3-Bis(diethylphosphoramidate)-propan]: PolyBisDEAEPN
'H-NMR (300.1 MHz, DMSO-d6) S = 1.21 (br, 12H,-CH3), 3.1-2.7 (br, 3H, CH2-CH), 3.91
(br, 8H, -OCH2), 4.4-5.0 (br, -CH, NH, -CH2N).
31P-NMR (121.5 MHz, DMSO-d6) 5 = 9.96 (s)
IR (neat) 3209 (m, br, NH stretch), 2981 (m) and 2906 (vw) (CH stretch), 1726 (s, C=0
stretch), 1442 (m, CH bending), 1391 (m), 1228 (s, P=0 stretch), 1161 (m), 1098 (w), 1019(s)
and 954 (s) (P-O-C stretch), 794 (m), 749 (w).
120

Chapter VI Experimentalpart
4 References
[1] N. C. Reghunadhan, G. Clouet, J. Brossas, J. Polym. Sci. Part A: Polym. Chem. 26 (7)
(1988)791.
[2] N. C. Reghunadhan, G. Clouet, Eur. Polym. J. 25 (3) (1989) 251.
[3] R. Liepins, J. R. Surles, N. Morosoff, V. Stannett, JJ. Duffy, FH. Day, J. Appl. Polym.
Sci. 22(9) (1978) 2403.
[4] T. Jeanmaire, Y. Hervaud, B. Hervaud, Phosphorus Sulfur Silicon Relat. Elem. 177 (5)
(2002)1137.
[5] W. F. McSherry, G. L. Drake, A.B. Cooper, A. R. Markezich, Am. Dyest. Rep. 63(7)
(1974)52
121

Chapter V Experimental part
Seite Leer /
Blank leaf

Appendix
Appendix
1 List of Abbreviations
a.u
AC8
BAPO (Irgacure 819)
BisDEAEPN
Darocur 1173
DEAEP
DEAEPN
DEAMP
DEMEP
DMAMP
DMSO
DTG
EGDA
EGDMA
Et
Et3N
EtOH
FR
IR(ATR)
LOI
MAC8
Me
MeOH
MW
NMR
PAN
PIGP
Psch
RF
arbitrary unit
1,1,2,2-tetrahydroperfluorodecyl acrylate
bis(2,4,6-trimethylbenzoyl)-phenylphosphine oxide.
(Acryloyloxy)-1,3-Bis(diethylphosphoramidate)-propan
2-hydroxy-2-methyl-1 -phenyl-propan-1 -one
Diethyl (acryloyoxy) ethyl phosphate
Diethyl (acryloyloxy) ethyl phosphoramidate
Diethyl (acryloyloxy) methyl phosphonate
Diethyl-2-(methacryloyloxy) ethyl phosphate
Dimethyl (acryloyloxy) methyl phosphonate
Dimethylsulfoxide
Differential thermogravimetry
Ethylene glycol diacrylate
Ethylene glycol dimethacrylate
Ethyl
Triethylamine
Ethanol
Flame retardant
Infrared attenuated reflection spectroscopy
Limiting oxygen index
1,1,2,2-tetrahydroperfluorodecyl methacrylate
Methyl
Methanol
Microwave
Nuclear magnetic resonance
Polyacrylonitrile
Plasma-induced graft polymeriaztion
Schmerber pressure
Radio frequency
123

Appendix
SEM Scanning electron microscopy
TG Thermogravimetry
TGA Thermogravimetric analysis
TTEGDA Tetra (ethylene glycol) diacrylate
UV Ultraviolet
124

Appendix
2. Kinetic of homopolymerization of the acrylic monomers containing phosphorus
Kinetic ofhomopolymerization ofDEAEP, DEAMP, DMAMP, andDEAEPN
The experiments were monitored by *H NMR spectroscopy in DMSO-dö. For each
treatment time, the ratio R between the relative intensities of the vinyl protons HC=CH? and
the methyl groups in -P(0)(OCH2CH3)2 or in -P(0)(OCH3)2 for DMAMP was calculated. The
latter was used as internal reference because it remains unaffected during the reaction. This
procedure allows us to evaluate the amount remaining acrylates: \-p and the conversion rate/»
of the monomer to the polymer.
Ih(HC=CH2)/Iii(CH3)
I-/? = 2R
/»=1-2R
Kinetic ofhomopolymerization ofDEMEP
The ratio R between the relative intensities of the proton H2C=C(Me) and the methyl
groups in -P(0)(OCH2CHj)2 was calculated.
lH(H2C=C(Me))/IH(CH3)
1-/7 = 3R
/*=1-3R
125

Appendix
3 TGA and DTG data of the polymers and DTG curves of untreated and treated PAN
and cotton fabrics with the acrylic monomers containing phosphorus
TGA and DTG data ofthe polymers
Tablel7 Temperatures and percentage of weight residue for the thermal decomposition of
acrylic polymers containing phosphorus under argon atmosphere
polymer Sample
weight(mg)
Onset of
degradation
temperature
(°C)
1max
Stepl
from DTG
Step 2
(°C)
Step 3
Weightresidue at
570°C
polyDEMEP 13.9 228 260 310 - 19.5
polyDEAEP 12.8 237 266 310 - 31.2
polyDEAMP 13.5 240 290 310 - 32.1
polyDMAMP 13.1 244 324 - - 40.0
polyDEAEPN 17.4 215 240 304 368 34.2
polyBisDEAEPN 14.7 204 231 266 - 29.6
DTG curves of untreated and treated PANfabrics
0 \wy^fl^'li"<>"it
ö
| -4
&-
O -6
a
-8
10
-12
>i. i I m mW"1
15 115 215 315 415 515
Temperature (°C)
PAN
DMAMP
DEAMP
DEMEP
DEAEP
615 715
126

DTG curves of untreated and treated cotton fabrics
Appendix
15
• Cotton
DEAEP
•DEAEPN
DMAMP
•DEMEP
• DEAMP
BisDEAEPN
215 415
Temperature (°C)
615
127

Appendix
4 Schmerber tests: Determination of resistance to water penetration (DIN 53886)
Owing to the roughness and irregularity of the textile surfaces, the commonly used
contact angle measurements were not reliable for the investigation of the wettability of the
treated fabrics and were replaced by Schmerber tests.
The principle is the following: A column of water is connected from below to a piece
of textile stretched on a frame. Water is added progressively to the column, whereby the
textile is exposed to an increasing pressure of water. The Schmerber value noted corresponds
to the height of water (cm) reached when three drops of water went through the fabric. The
height of the water column is directly transformed to the water pressure, in our case, 1 cm of
water = 0.981 mbar. This pressure Pschmerber is proportional to the cosine of the contact angle
between a drop of water and the surface of the textile.
P = (2 yL/R) cos Q (4.1).
In which yL and R are determined by the nature of the textile and the liquid used, 9 is the
contact angle. When the values of the surface tension are used, for water at 25°C, (yL = 72.8,
d p 1
y i= 21.8 and y i= 51.0 mN.m ), equation 1 becomes:
P = (145.6 IR) cos 0 (4.2)
With P (mN.m"2) and R (m).
128

Appendix
5 Curriculum Vitae
Name: Marie Jérôme Tsafack
Born on: July 9th, 1973
In: Douala, Cameroon
Citizen of: Cameroon
08/1993:
09/1993-09/1996
09/1997-09/1999
09/1999-09/2000
02/2001-07/2001
Baccalauréat C
Licence de Chimie at the University of Douala
Maîtrise de Chimie at the University of Yaounde
DEA de Chimie at the University of Yaounde
Research internship at ETHZ in the group of Prof. Dr. H. Grützmacher
11/2001-2005: PhD thesis at the ETH Zurich, in the group of Prof. Dr. H. Grützmacher
129