drugs for the treatment of respiratory diseasesasthma and copd and to their prevention. whilst the...
TRANSCRIPT

Drugs for theTreatment ofRespiratoryDiseases
Domenico SpinaThe Sackler Institute of Pulmonary Pharmacology,King’s College London, UK
Clive P. PageThe Sackler Institute of Pulmonary Pharmacology,King’s College London, UK
William J. MetzgerNational Jewish Medical and Research Center, Denver, CO, USA
Brian J. O’ConnorThe Sackler Institute of Pulmonary Pharmacology,King’s College London, UK

The Pitt Building, Trumpington Street, Cambridge, United Kingdom
The Edinburgh Building, Cambridge CB2 2RU, UK
40 West 20th Street, New York, NY 10011–4211, USA
477 Williamstown Road, Port Melbourne, VIC 3207, Australia
Ruiz de Alarcón 13, 28014 Madrid, Spain
Dock House, The Waterfront, Cape Town 8001, South Africa
http://www.cambridge.org
© Cambridge University Press 2003
This book is in copyright. Subject to statutory exception
and to the provisions of relevant collective licensing agreements,
no reproduction of any part may take place without
the written permission of Cambridge University Press.
First published 2003
Printed in the United Kingdom at the University Press, Cambridge
Typeface Utopia 8½/12 pt. System QuarkXPress® [ ]
A catalogue record for this book is available from the British Library
Library of Congress Cataloguing in Publication data
Drugs for the treatment of respiratory diseases / edited by Domenico Spina . . . [et. al.].
p. ; cm.
Includes bibliographical references and index.
ISBN 0 521 77321 0 (hardback)
1. Lungs – Diseases – Chemotherapy. 2. Respiratory agents. I. Spina, Domenico.
[DNLM: 1. Respiratory Tract Diseases – drug therapy. 2. Anti-Allergic Agents – therapeutic
use. 3. Respiratory System Agents – therapeutic use. WF 145 D7947 2002]
RC756 .D784 2002
616.2�4061–dc21 2002023390
0 521 77321 0 hardback
Every effort has been made in preparing this book to provide accurate and up-to-
date information which is in accord with accepted standards and practice at the
time of publication. Nevertheless, the authors, editors and publisher can make no
warranties that the information contained herein is totally free from error, not least
because clinical standards are constantly changing through research and
regulation. The authors, editors and publisher therefore disclaim all liability for
direct or consequential damages resulting from the use of material contained in this
book. Readers are strongly advised to pay careful attention to information provided
by the manufacturer of any drugs or equipment that they plan to use.

Contents
List of contributors viiPreface xi
Part I Asthma and COPD
1 Pathology of asthma and COPD: 3inflammation and structurePeter K. Jeffery
2 Glucocorticosteroids 32Peter J. Barnes
3 ß2-adrenoceptor agonists 56Domenico Spina, Clive P. Page and
Brian J. O’Connor
4 Anticholinergic bronchodilators 105Jeremy M. Segal and Nicholas J. Gross
5 Antiallergic drugs 119Masakazu Ichinose
6 Drugs affecting the synthesis and action 124of leukotrienesPaul M. O’Byrne
7 Theophylline and selective 136phosphodiesterase inhibitors in thetreatment of respiratory diseaseNeil A. Jones, Domenico Spina and Clive P. Page
8 Potential therapeutic effects of 172potassium channel openers in respiratorydiseasesAhmed Z. El-Hashim
9 Tachykinin and kinin antagonists 184Pierangelo Geppetti
v

10 Drugs affecting IgE (synthesis inhibitors 195and monoclonal antibodies)Lawrence G. Garland and Alan G. Lamont
11 Drugs targeting cell signalling 218Brydon L. Bennett, Yoshitaka Satoh and
Alan J. Lewis
Part II Diffuse parenchymal lung disease
12 Current approaches to the treatment of 247parenchymal lung diseasesJoseph P. Lynch III and Michael Keane
13 Drug treatments of the future in fibrotic 336lung diseaseAthol U. Wells
Part III Infection
14 Current and future management of 363pneumoniaMario Cazzola and Maria G. Matera
15 Current treatment of chronic bronchial 403suppurationRobert Wilson
16 Current and future treatment of 428cystic fibrosisR.G. Gary Ruiz, Hilary H. Wyatt and John F. Price
Part IV Pulmonary vascular diseases
17 Pathophysiology of pulmonary vascular 453diseaseSanjay Mehta and David G. McCormack
18 Current treatment of pulmonary vascular 475diseasesTarek Saba and Andrew Peacock
19 Future treatment of pulmonary vascular 504diseasesNorbert F. Voelkel, Mark W. Geraci and
Steven Abman
Part V Lung cancer
20 Molecular pathology of lung cancer 519Ignacio I. Wistuba and Adi F. Gazdar
21 Small cell lung cancer 535Desmond N. Carney
Part VI Cough
22 Mechanisms of cough 553John J. Adcock
23 Current treatment of cough 565Peter V. Dicpinigaitis
Index 579
Colour plate section between pp. 12 and 13.
vi Contents

Introduction
It is widely recognized that neither asthma norCOPD are disease entities but rather each is acomplex of inflammatory conditions that have incommon airflow limitation (syn. obstruction) whosereversibility varies (Fig. 1.1). The characteristics anddistinctions between mild stable asthma and COPDhave been reviewed1,2. However, these differencesbecome less clear when the conditions becomesevere or there are exacerbations due to infection orother cause. An understanding of whether or notthere are fundamental differences of inflammationand airway/lung structure between these two condi-tions is relevant to clinical decisions regarding bothinitiation and long-term treatment and to patientmanagement during exacerbations. In the longerterm it is of value to the design of specific therapy forasthma and COPD and to their prevention. Whilstthe definitions of asthma and COPD highlight thediffering degrees of airflow variability and reversibil-ity3,4, there is a prevailing clinical impression that,with age, there is often overlap and a progressionfrom the reversible airflow obstruction of the youngasthmatic to the more irreversible or ‘fixed’ obstruc-tion of the older patient with COPD. The Dutchhypothesis encompasses the idea that both condi-tions are extreme ends of a single condition5. In theauthor’s opinion it may, in the future, be less relevantto be concerned with the clinical labels of ‘asthma’ or‘COPD’ and more important to ascertain and targettreatment to the predominant pattern of inflamma-tion and structural change that prevails in eachpatient.
Asthma may be divided into extrinsic (also calledallergic or atopic), intrinsic (late onset or non-atopic) and occupational forms. At this time thepathologist cannot distinguish between these dis-tinct clinical forms of asthma: there are alterationsthat appear to be common to all forms. COPD isassociated, usually, with the smoking habit as therelationship between cigarette smoking and COPDis a strong one statistically. Three conditions cancontribute, the degree varying in each patient, to theclinical expression of COPD: chronic bronchitis(syn. mucus hypersecretion), chronic bronchiolitis(syn. small airways disease) and emphysema,inflammatory conditions broadly affecting bronchi(airways with cartilage in their wall), bronchioli(membranous or non-cartilaginous airways) andlung parenchyma respectively. In both asthma andCOPD, the persistence of distinct inflammatory cellsinitiated by allergen or cigarette smoke, respectively,is probably responsible for most of the structuralchange and usually referred to as ‘remodelling’:interactions with the effects of acute and chronicinfection and genetic predisposition are clearlyimportant also.
The chapter focuses on the patterns of infiltratinginflammatory cells in asthma and COPD and theassociated remodelling of the airway wall. First,airway wall thickening is considered, particularly inasthma, remodelling is defined and the relationshipbetween inflammation and remodelling discussedbriefly. Lumenal secretions obtained as sputum orlavage and asphyxic plugging of the airways withmixtures of mucus and inflammatory exudate arediscussed briefly. The chapter then divides into two
3
1
Pathology of asthma and COPD :inflammation and structure
Peter K JefferyImperial College at the Royal Brompton Hospital, London, UK

major sections considering first inflammation andthen remodelling in asthma and COPD. The resultsof examination of the conducting airways by flexiblefibre-optic bronchoscopy are included as this tech-nique has provided the means by which the earlyinflammatory and structural alterations of asthmaand COPD have been compared, free from the com-plications of end stage disease6.
Airway wall thickening
The airway walls in asthma are thickened by theremodelling process by between 50 and 300% ofnormal and there is lumenal narrowing, which isfurther compromised by excessive mucus admixedwith an inflammatory exudate (Fig. 1.2, see colourplate section). In cases of fatal asthma, the longer theduration of asthma, the thicker becomes the airwaywall7. However, it has been suggested that airwaywall thickness per se is not a requirement forasphyxic fatality as a group of relatively young asth-matics (i.e. with a relatively short history of asthma)had an airway wall thickness not significantly differ-ent from that of non-asthma controls. Lumenalsecretions and plugging are likely the greater contri-bution to asthmatic death in these young cases of
fatal asthma7. All tissue structural components, aswell as inflammatory cell infiltration and edema,can contribute to the observed thickening; however,in the last mentioned study it is thickening of the(outer) adventitial layers that was most pronouncedin the older group with the longest duration ofdisease. The airway walls are also thickened inCOPD. One systematic study has described changesin large airway dimensions in relation to the lungfunction of patients with COPD and found wall areainternal to the muscle to be significantly thickenedover the entire range of cartilaginous airways meas-ured8. The relative contributions of the airway wallcomponents contributing to the thickening,however, vary with airway generation.
Inflammation and remodelling
Acute inflammation is the response of vascularizedtissue to injury: the inflammatory reaction isdesigned to protect the host and to restore tissue andits function to normal. One generally accepted pro-posal is that the accelerated decline in forced expir-atory flow over time in COPD, and that which occursalso in an important subset of asthmatics, is thedirect result of a switch from acute, episodic, tochronic inflammation and to consequent airwayand parenchymal remodelling1. The proposal is
4 P.K. Jeffery
Asthma?COPD
Irreversible
Emphysema
Chronicbronchiolitis
Chronicbronchitis
Reversible
Fig. 1.1 Venn diagram illustrating the overlap between asthma
and COPD.

attractive but, as yet, there is no convincing evidencethat the remodelling process is dependent upon theprior development of chronic inflammation. It isequally plausible that the processes responsible forthe development of chronic inflammation are dis-tinct to those responsible for remodelling. The lastconsideration has important implications for thedesign of disease modifying therapy: thus thoseagents that are effective antiinflamatory com-pounds will not necessarily prevent or attenuate theprocess of remodelling for which new classes ofdrugs will be required.
Definition
The concept of ‘remodelling’ implies that a processof ‘modelling’ must have preceded it. The lung, inutero, undergoes extensive modelling and remodel-ling yet these processes are entirely appropriate tothe normal process of lung development. Many ofthe cytokines and growth factors thought to be pro-inflammatory in asthma and in COPD are alsoexpressed normally without detriment to the devel-oping lung; these include: members of the fibroblastgrowth factor family, the transforming growth factorfamily, epithelial-derived growth factor, granulo-cyte–macrophage colony stimulating factor,platelet-derived growth factor, vascular endothelialgrowth factor and hepatocyte growth factor1,9.Accordingly the working definition of remodellingproposed herein recognizes that the process ofremodelling per se is not of necessity abnormal. It is:an alteration in size, mass or number of tissue struc-tural components that occurs during growth or inresponse to injury and/or inflammation. It may beappropriate, as in normal lung development or thatwhich occurs during acute reaction to injury, or‘inappropriate’ when it is chronic and associatedwith abnormally altered tissue structure and func-tion as, for example, in asthma or COPD.
In wound healing (in the skin) the components ofan appropriate response include: clot formation,swelling/edema, rapid restitution of the denudedareas by epithelial dedifferentiation, proliferationand migration from the margins of the wound. This
is normally associated with an inflammatory reac-tion, i.e. early infiltration of the injured tissue byneutrophils and later by lymphocytes and macro-phages. Reticulin is deposited within days and thismay mature to form interstitial collagen, a scar,within 2–3 weeks. In addition, healing may involvecontraction of the surrounding tissue (in the case ofan open wound), by myofibroblasts that may pro-liferate transiently in relatively large numbers10.Vasodilatation, congestion and mucosal oedema arealso cardinal signs of acute inflammation and theangiogenesis of the granulation tissue is an integralpart of the reparative response. Thus, normal tissuearchitecture and function is restored consequent toan entirely appropriate inflammatory reaction withwhich there has been an associated remodellingprocess. Each of these stages in normal woundhealing and many of the inflammatory cell types andcytokines involved appear also in asthma and inCOPD, but in these last two conditions both theinflammation and remodelling persist and result inexaggerated remodelling inappropriate to the main-tenance of normal (airway) function. The reasons forthe persistence of the inflammation are unknownbut may be the result of repeated inhalation of aller-gen or exposure to high concentrations of allergen,irritation (e.g. by tobacco smoke) or persistent infec-tion or a genetically influenced abnormal hostinflammatory response or a defective repair process.
Lumenal secretions
Sputum and bronchoalveolar lavage
The examination of spontaneously produced orsaline-induced sputum has become a much usedand relatively non-invasive method for determiningthe extent of inflammation in the asthmaticairway11,12 (Fig. 1.3(a), 1.2(b), see colour platesection). Corkscrew-shaped twists of condensedmucus (Curshmann’s spirals), clusters of surfaceairway epithelial cells (referred to as Creola bodiesand named after the first patient in whom they weredescribed)13, and the presence of Charcot–Leyden
Pathology of asthma and COPD 5

crystals, composed of eosinophil cell and granulemembrane lysophospholipase (Fig. 1.3(a), (b), seecolour plate section14), together with eosinophilsand metachromatic cells, are characteristic featuresof sputa obtained from asthmatic, but not bron-chitic patients15. Sputum eosinophilia has, however,also been reported in non-asthmatics in the absenceof the airways hyper-responsiveness (AHR) charac-teristic of asthma16. In contrast, sputa from bron-chitic patients may be mucoid or, during infectiveexacerbations, purulent when neutrophils may bepresent in large numbers. BAL in mild (allergic)asthma demonstrates the presence of sloughed epi-thelial cells, the numbers of which show an associa-tion with AHR,17 and of eosinophils and their highlycharged secreted products (such as eosinophil cat-ionic protein (ECP) and major basic protein(MBP))18. In contrast, in smoker’s bronchitis, macro-phages are the most usually reported cell type andneutrophils are numerous as are their secreted prod-ucts.
Airway plugging
Examination, postmortem, of cases of fatal asthmahas shown that the lungs are hyperinflated andremain so on opening the pleural cavities due to thewidespread presence of markedly tenacious airway‘plugs’ in both large (segmental) and small bronchi(Fig. 1.4(a)), see colour plate section). Even intra-bronchial inflation with fixative to a 1.5-metre headof pressure hardly moves these sticky lumenalplugs19. Histologically the airway plugs in asthma arecomposed predominantly of inflammatory exudatetogether with mucus in which lie: eosinophils, lym-phocytes and desquamated surface epithelial cells.The arrangement of the eosinophilic elements of theplug is often as concentric, multiple lamellae sug-gesting that several episodes of inflammation haveled to their formation rather than a single terminalevent (Fig. 1.4(b), see colour section). The non-mucinous, proteinaceous contribution is the resultof increased vascular permeability and includes afibrin. Electrostatic interaction of positively charged(cationic) eosinophil products and serum constitu-
ents and negatively charged (due to carboxyl andsulfate groups) mucin likely contributes to the par-ticular stickiness of the airway plug. There are,however, reports of sudden death in asthmatics inwhich intraluminal plugs are absent20 but these arerare. In the absence of a history of smoking, emphy-sema in fatal asthma and right ventricular hypertro-phy is uncommon. However, areas of atelectasis andpetechial hemorrhages may be present in asthmadue to bronchial obstruction, reabsorption collapseand repeated forced inspiratory efforts. The asth-matic who has smoked will likely have featureswhich overlap between asthma and COPD and, inthese cases, there may be focal evidence of centriac-inar (i.e.bronchocentric) alveolar destruction (seeFig. 1.4(a), colour plate section).
Inflammation
To the physiologist, inflammation is characterizedby cardinal signs: redness, heat, swelling, pain andloss of normal function. To the pathologist, inflam-mation is recognized in tissue sections as congestionof vessels together with the recruitment (i.e. margi-nation within and emigration from vessels) of avariety of morphologically and immuno-phenotypi-cally distinct inflammatory cells. It is now recog-nized that both asthma21,22 and COPD areinflammatory conditions albeit the relative magni-tude and site of the inflammatory infiltrate and thepredominant inflammatory cell phenotype differs.
Asthma
Studies of biopsies obtained by fibreoptic bronchos-copy or at open lung biopsy in asthma demonstratethe presence of an inflammatory cell infiltrate evenin patients with newly diagnosed asthma23. The infil-trate comprises CD3 immuno-positive (T) lympho-cytes of the CD4 (i.e. T-helper) subset andeosinophils17,24–26. An increase in leukocytes, includ-ing lymphocytes and eosinophils, occurs in rela-tively mild atopic, occupational and intrinsicasthma and it is associated with an increase of ‘acti-vation’ markers for both lymphocytes (CD25�cells)
6 P.K. Jeffery

and eosinophils (EG2�cells)21,24,26–28. In sympto-matic atopic asthmatics, in electron microscopicstudies, irregularly shaped lymphomononuclearcells appear and these may represent ultrastructuralforms of the CD25�(activated) lymphocyte. EG2 is amarker for the cleaved (‘secreted’) form of eosino-phil cationic protein that can be found both withineosinophils and diffusely in the wall, often in associ-ation with the epithelial reticular basement mem-brane. Eosinophil-derived products such as majorbasic protein29 together with toxic oxygen radicalsand proteases probably all contribute to the epithe-lial fragility described in asthma (see below).Eosinophil cytolysis or disintegration and release ofgranules30,31 and of cytokines may also stimulatenearby fibroblasts to produce additional reticulinand so induce thickening of the reticular basementmembrane.
In fatal asthma there is a marked infiltratethroughout the airway wall, in sputum and also inthe occluding plug. Compare Fig. 1.5(a) and (b), seecolour plate section, see Figs. 1.3(a) and 1.4(b), seecolour plate section: lymphocytes are abun-dant22,32,33 and (EG2�)eosinophils are characteristic(Fig. 1.6, see colour plate section)22,34,35. Neutrophilsare sparse in mild asthma21 albeit they are present inrelatively large numbers in sputa during infectiveexacerbations36, in biopsies of severe asthmaticsrefractory to high dose treatment with corticoster-oids37 and in status asthmaticus when death issudden (i.e. within 24 hours of the attack)38. It hasbeen suggested based on examination of biopsytissue that two forms of asthma may be usefully dis-tinguished: those with a relatively high eosinophilcount and those with predominant neutrophilia39.The inflammation of the airway wall may involvethe adjacent pulmonary artery33 and, in small(distal) airways, may spread to surrounding alveolarseptae40. Alveolar walls may thus show evidence ofeosinophilic infiltration40 and alveolar spaces maycontain a fibrillar-rich component, most likelyfibrin (author’s unpublished observations).However, destruction of the parenchyma (i.e.emphysema) is not a feature of asthma. Thus, bothsmall and large airways may be inflamed in asthma:
transbronchial biopsy studies of relatively severeasthma and studies of resection tissue in asthmaticshave demonstrated infiltration of bronchioli byeosinophils and lymphocytes40,41. There are alsorecent data in severe asthma that demonstrate theinner wall to be infiltrated by neutrophils innumbers considerably greater than in largerairways42. Thus the pattern of inflammation insevere asthma appears to be different from that inmild and, in order to be effective, treatment needs tobe tailored accordingly. The association of tissueeosinophilia and asthma is a strong one. However,the extent of tissue eosinophilia varies greatly witheach case and with the duration of the terminalepisode22,38,43. The variation may be due, in part, toeosinophil degranulation, which makes cell iden-tification difficult. In comparison with mild asthma,fatal asthma is reported to be associated with ahigher concentration of eosinophils in the largeairways and a reduction of lymphocytes in theperipheral (smaller) airways35.
The role of the activated T-helper (Th) lymphocytein controlling and perpetuating the chronic inflam-matory reaction in asthma has received much atten-tion24,44. The T-lymphocyte is thought to controlallergic inflammation via the selective release of theproinflammatory cytokines (interleukins) IL-4 andIL-5, which characterize the T-helper (type 2) phe-notype45. IL-5 gene expression has been shown to beincreased in bronchial biopsies from symptomaticatopic asthmatic subjects46 (Fig. 1.10), and this issupported by studies of cells obtained at broncho-alveolar lavage47,48 and peripheral blood49. IL5appears to be a key cytokine required to induce ter-minal differentiation of eosinophils and, togetherwith IL4, enhances their vascular retention and lon-gevity in tissues. It is also a key cytokine in the latephase reaction to allergen challenge48. IL4 is alsoincreased in atopic asthma50,51 and may be impor-tant in both the initiation and persistence of allergicinflammation. IL4 encourages the selective recruit-ment of eosinophils by up-regulating adhesionmolecules (V-CAM) on bronchial endotheliumwhose ligand on the eosinophil cell surface is VLA-4.The last is absent from the surface of neutrophils
Pathology of asthma and COPD 7

and helps to explain the eosinophil predominancein mild asthma. There is currently debate as to theinvolvement of IL4 in asthma of the intrinsic (i.e.non-atopic) form52. IL4 and IL5 are not, however,unique to asthma and may occur in other inflamma-tory conditions such as fibrosing alveolitis53. WhilstIL5 may be important in promoting eosinophil ter-minal differentiation, and the release of eosinophilsinto the blood from bone marrow, other moleculessuch as eotaxin and RANTES (regulated on activa-tion normal T-cell expressed and secreted) areinvolved as selective chemokines inducing eosino-phil emigration from blood vessels and their migra-tion through the mucosa to the airway lumen fromwhence they are cleared54–56. The same or distinctmolecules may be involved in eosinophil activation,a process about which little is as yet known.Symptomatic asthma is associated with the produc-tion of additional cytokines including TNF�,GM–CSF, IL1�, IL2 and IL645,57. GM–CSF has alsobeen reported to increase during the late phase reac-tion to allergen58. In addition to their production oftoxins and lipid-derived mediators, eosinophilsthemselves may also produce proinflammatorycytokines and growth factors as evidenced by theirgene expression for TNF�, IL6 and GM-CSF�45,59,60.Macrophages have been reported to increase innumber in more severe asthma of the intrinsicform28.
Mast cells have long been thought to play a keyrole in the immediate (type I sensitivity) reaction inasthma through their release of a variety of media-tors including those which bronchoconstrict i.e. his-tamine, prostaglandin D2 and leukotriene D4. Mastcells are now thought to act as an important sourceof IL4 and other proinflammatory cytokines whosesecretion may act as a trigger to the induction of sub-sequent persistent production of IL4 and IL5 by lym-phocytes61,62. There are reports of decreases,increases and no change of mast cell numbers. Earlybiopsy studies demonstrated an apparent reductionin bronchial mast cell numbers in asthma due totheir degranulation63. Studies of bronchoalveolarlavage report increased intralumenal mast cellnumbers together with increased numbers of T-
helper cells and eosinophils and evidence of hista-mine release and of eosinophil degranulation18,64,65.
Although considered to be important in allergicconditions, little is known of the role of basophils inthese conditions albeit there is evidence forincreased recruitment of basophils and their precur-sors to sites of allergic reaction in atopic patients66.Asthma is also characterized by infiltration of thebronchial surface epithelium by dendritic cells(i.e. Langerhans’ cell equivalent)67. These non-phagocytic histiocytes are rich in surface receptorsand their functions are thought to include the pres-entation of antigenic information to T lymphocytes;very few Langerhans’ cells are found in the normallung although there is a rich network of their prob-able precursor dendritic cells68. Thus lymphocytes ofthe T-helper (CD4�) subset appear to be key to thecontroller cell and eosinophils the prime effectorcell in mild asthma. However, with increasing sever-ity of asthma and in infective exacerbations there isan increasing involvement of neutrophils andperhaps also of macrophages and these changesappear to be more refractory to conventional treat-ment with inhaled or even oral corticosteroids.Alternative approaches would seem to be requiredto treat more severe than mild asthma and thereasons for this may in part be explained by thealtered pattern of inflammation.
COPD
T-lymphocytes appear also to be key controller cellsin COPD but in contrast to asthma it is the CD8�
cells that are the predominant cells in COPD69. It iscurrently presumed that the majority of these CD8�
cells are T-lymphocytes of the cytotoxic/suppressorsubset, but this is as yet unproven and these mayalso include natural killer cells and even a dendriticcell sub-type. The altered CD8:CD4�cell ratioappears, however, to be a fundamental distinctionbetween the CD4�T-cell, allergen-driven process ofallergic asthma in non-smokers and the CD8�T-cell, cigarette smoked-induced inflammation ofCOPD70.
Smoking tobacco per se induces an inflammatory
8 P.K. Jeffery

response. Smoking shortens the transit time of neu-trophils through the bone marrow, causes a leukocy-tosis and alters the immunoregulatory balance ofT-cell subsets found in blood, bronchoalveolarlavage (BAL), and tissues of the conducting airwaysand lung71–73. Smoking initiates a peripheral bloodleukocytosis and a reversible decrease in the nor-mally high CD4 to CD8 cell ratio in blood of heavysmokers (i.e.�50 pack–years). There is also a signifi-cant reduction of the CD4:CD8�cell ratio in BALfluid but not blood of a group of milder smokers ( i.e.on average who have smoked 14 pack–years). Theincrease in the number of BAL and tissue CD8�T-cells is positively associated with pack–yearssmoked72,74,75.
Chronic bronchitis
Histological examination of airway tissues (taken atresection for tumour) from smokers demonstratesthat inflammatory cells are present in and around thearea of mucus-secreting submucosal glands and thatscores of inflammation show a better associationwith the subjects who have symptoms of mucushypersecretion than does gland size per se76. In bron-chial biopsies of subjects with mild stable chronicbronchitis and COPD there is infiltration of themucosa by inflammatory cells75,77–79 (Fig. 1.7, seecolour plate section): this is associated with up-regulation of cell surface adhesion molecules of rele-vance to the inflammatory process80. In the surfaceepithelium where, in contrast to the subepithelium,CD8�cells normally predominate, Fournier and col-leagues have demonstrated by comparison with non-smokers, an increase in all inflammatory cell types insmokers with chronic bronchitis and mild COPD81. Ina subepithelial zone (also referred to as the laminapropria), bronchial lymphomononuclear cellsappear to form the predominant cell type with scantyneutrophils (in the absence of an exacerbation): thelymphomononuclear component is composed oflymphocytes, plasma cells and macrophages.Significant increases are reported in the numbers ofCD45 (total leukocytes), CD3 (T-lymphocytes), CD25(i.e.activated) and VLA-1 (late activation) positive
cells, presumed to be T-lymphocytes and of macro-phages. The endobronchial biopsy studies ofO’Shaughnessy and co-workers have demonstratedthat by comparison with normal non-smokers, T-lymphocytes and neutrophils increase in the surfaceepithelium whilst T-lymphocytes and macrophagesincrease in the subepithelium of smokers withCOPD79,82. In contrast to asthma, in COPD it is theCD8�cell and not the CD4�T-cell subset, whichincreases in number and proportion to become thepredominant T-cell subset. Furthermore, the increaseof CD8�cells shows a negative association with theforced expiratory volume in one second (FEV1expressed as a percentage of predicted). This noveldistinction between the relative proportions in T-cellsubsets of smokers with mild stable COPD and non-smoking mild asthmatics has received the support ofsubsequent studies of both resected tissues and bron-chial biopsies74,83,84. The increase of the CD8�phe�
notype and of the CD8/CD4 ratio seen in the mucosaalso occurs deeper in the airway wall in associationwith submucosal mucus-secreting glands in bron-chitic smokers83. In addition neutrophils increase inthe surface epithelium and glands especially whenthe disease increases in severity (Fig. 1.8, see colourplate section).
Similarity between COPD and asthma
COPD and asthma would seem to differ at the tissuelevel in a number of respects; for example themarked tissue eosinophilia and thickening of thereticular basement membrane of asthma (seebelow) is not usually a feature of COPD85. However,compared to normal healthy control tissue, there area number of studies that report a small but signifi-cant increase in the number of tissue eosinophils insubjects with chronic bronchitis or COPD76,79,86.Sputum eosinophilia is also reported in cases of‘eosinophilic bronchitis’, i.e. patients without ahistory of asthma and without bronchial hyper-responsiveness12,87. Furthermore, in mild COPD, thenumbers of tissue eosinophils are markedly and sig-nificantly increased when there is an exacerbation ofbronchitis (defined as a need by the patient to seek
Pathology of asthma and COPD 9

medical attention due to a sudden worsening ofdyspnoea or an increase in sputum volume or puru-lence)88,89. In such mild cases of COPD the exacerba-tion is associated with an increase in eosinophilchemoattractants, especially RANTES90. The bron-chial mucus-secreting glands of smokers may alsoshow gene expression for both IL4 and IL5 and thenumbers of these cells are significantly higher insmokers with chronic hypersecretion as comparedwith their asymptomatic controls91. Thus, IL4, IL5and eosinophil chemoattractant gene expression isnot restricted to asthma and, like the recent reportsof fibrosing lung disease92, these regulatory cyto-kines can be expressed also in chronic bronchiticsmokers.
Chronic bronchiolitis
Histologically, the earliest observed effects of cigar-ette smoke in small airways and surrounding alveoliis a marked increase in the number of macrophagesand neutrophils, both in human and experimentallyin animal studies. The increase is seen withinboth the tissue and lumena and can be detected inbronchoalveolar lavage fluid (BAL)93. Examinationof small airway tissue in lungs resected fromsmokers also shows that the same profile of CD8-predominant inflammation reported in bronchialbiopsies of the larger airways occurs deeper in thelung in both the ‘small’ airways74,84 and also the lungparenchyma94,95. As with the findings in the largeconducting airways there are significant negativeassociations of the numbers of CD8�cells andFEV1% of predicted in both the small (peripheral)conducting airways and lung parenchyma.However, at these sites the negative correlations arestronger than in the large airways. Thus the patternsof inflammation are similar at both proximal anddistal sites. However, in contrast to the largerairways, the CD8�T-cell predominance in the smallairways and lung parenchyma is more closely asso-ciated with decreased lung function in these sub-jects with COPD.
The infiltration of the airway wall by lymphocytesis associated with loss of alveolar attachments to the
outer wall of small airways, a characteristic of centri-acinar emphysema. The accompanying loss of radialtraction and lung elastic recoil leads to early airwayclosure during expiration (Fig. 1.9(a), (b), see colourplate section). The loss of alveolar–bronchiolarattachments is thought to be due to the circumfe-rential spread of small airway wall inflammation.
Emphysema
In the normal, the macrophage is the resident phag-ocyte of the alveolus: neutrophils are rarelypresent96. Neutrophils are recruited to the lung insmokers, albeit the extent of tissue neutrophilia ishighly variable. On exposure to cigarette smoke,there is recruitment of macrophages and phagocy-tosis of cigarette smoke components. A macrophagealveolitis and respiratory bronchiolitis are the earlychanges in young cigarette smokers97,98. As in thelarge and small conducting airways in COPD, CD8�
cells also become the predominant inflammatorycell phenotype in the parenchyma and theirnumbers show a strong inverse correlation withFEV1% of predicted95.
Inflammation and the pathogenesis of COPD
The neutrophil, the macrophage and the CD8�cellmay each be involved in the destruction of the lungparenchyma by distinct mechanisms.
The neutrophil
The alveolar microcirculation is composed of anetwork of short interconnecting tubules of averagediameter 5 µm. The average diameter of circulatingneutrophils is 7.0 µm, which necessitates their def-ormation as they squeeze through capillary seg-ments. Neutrophil traffic through the capillaries ofthe lung is normally slower (i.e. there is a highertransit time) than that of red blood cells as they are700 times less deformable than RBCs99. Studies withradioactively labelled neutrophils have demon-strated that the normal delay in neutrophil transit is
10 P.K. Jeffery

further exaggerated, transiently, even in healthysubjects during smoking. Exposure of neutrophils tocigarette smoke in vitro and in vivo results indecreased deformability associated with polymer-ization of actin microfilaments99,100. This is the likelymechanism of the observed cigarette smoke-induced increase in transit time. Factors chemotac-tic for neutrophils, and which will induce theiremigration from the microcirculation are releasedby the alveolar macrophages of smokers and thealveolar neutrophil population may increase from1% to 5% of inflammatory cells. Cigarette smokeitself may contain substances chemoattractant forneutrophils, a possibility that is supported by theassociated peripheral blood leukocytosis widelyreported101. Cigarette smoke or factors released fromcigarette smoke-exposed macrophages encouragethe release of neutrophil elastase which maydegrade lung elastin even in the presence of antipro-tease102–104. Such neutrophil-derived serine pro-teases have been implicated in the pathogenesis ofCOPD since the appreciation of the emphysematouschange that results from alpha-1 antitrypsin (AAT)deficiency in man. AAT protects against the proteo-lytic effects of neutrophil elastase, cathepsin G andproteinase 3, each of which has been shown toinduce emphysematous change in experimentalanimal models of emphysema. These and earlierresults led to the protease/antiprotease hypothesisin which emphysema develops if there is an imbal-ance favouring proteolytic digestion of the elasticframework of the lung (Fig. 1.10). Recent studieshave shown that, when released from the cell, theconcentrations of neutrophil elastase far outweighthe antiprotease in the immediate vicinity of theneutrophil cell surface. In the absence or reductionof the antiprotease this pericellular zone of proteo-lysis is greatly increased105.
The macrophage
The role of the macrophage in the pathogenesis ofCOPD has been controversial. However, the data ofrecent studies support the hypothesis that the
macrophage may play a critical role in regulating theinflammatory response and also directly in thetissue destruction associated with COPD106,107.Macrophages are able to synthesize significantamounts of matrix metalloproteinases (MMPs)including macrophage elastase (MMP12), collage-nase 1 (MMP1), gelatinase B (MMP9) andothers106,108. The hypothesis is that dysregulatedexpression of macrophage MMPs, induced eitherdirectly or indirectly by cigarette smoke, leads to thelung destruction characteristic of human emphy-sema. There is support for the hypothesis fromexperimental animal work using gene manipulatedstrains in conjunction with exposure to passive cig-arette smoke106. MMP1 over expression in mice isassociated with enlargement of airspaces suggesting
Pathology of asthma and COPD 11
Fig. 1.10 Gross appearance of the cut surface of a lung in
which the distribution of centracinar emphysema caused by
proteolytic digestion is characteristically restricted to the
upper aspects of each lobe. Scale�10 cm. (By courtesy of
Professor B. Heard.)

that collagen degradation may also be important inthe generation of emphysema. Moreover, whenmacrophage MMP12-deficient (-/-)mice areexposed chronically to cigarette smoke, they fail todevelop emphysema and fail to recruit macrophagesto the lung: in contrast the smoke-exposed MMP12intact (�/�) mice developed it109. Also in humansthere are data from cultured macrophages ofpatients with COPD that show that there is increasedexpression of MMP1, MMP9 and MMP12110. MMP12can be detected by immunohistochemistry and insitu hybridization in the alveolar macrophages ofpatients with emphysema but not in normal lung106.It appears that macrophage elastase is required forboth macrophage accumulation and the emphy-sema that results from inhalation of cigarette smoke.The current working hypothesis is that cigarettesmoke induces constitutive macrophages toproduce MMPs that cleave lung elastin, generatingfragments chemotactic for monocytes. This positivefeed-back loop would perpetuate the accumulationof macrophages and lung tissue destruction. Zinc-containing metalloproteases released from the cig-arette smoke-stimulated macrophages are notinhibited by the normal antiprotease of the alveolusand may thus also degrade alpha1-antitrypsin perse111.
The CD8�cell
There are several differences between the CD4�andCD8�subsets of T-lymphocytes112. CD4�T-cellshave been well studied in the context of asthma andthe Th2-type allergic response. However, untilrecently relatively little has been known about theCD8�cell and smokers with COPD. CD8�T-cellsare generally associated with Th1-type immunityand play a role in generating protective immunity toMycobacterium sp.113. LPS from gram negative bac-teria cause selective activation of the CD8�T-cellsubset. Currently there is more speculation thanknowledge about the role of this predominant cell inCOPD. A recent study has shown they may interact
with virus infected epithelial cells in a way that gen-erates a chemotactic factor (MCP-1) for macro-phages, an accumulation of which may then destroyhost tissue107. CD8�T-lymphocytes produce inter-feron gamma a cytokine that when overexpressedhas been shown to induce emphysema experimen-tally in mice114. In addition, the CD8�T-cell canproduce perforin and granzymes, which may con-tribute to the apoptosis and cell destructionreported in emphysema115. Consequently the paren-chymal damage associated with COPD could also beCD8�cell driven.
One hypothesis is that individuals with a geneti-cally determined low CD4�/CD8�T-cell ratio andthose who smoke would be more likely to have anexaggerated CD8�T-cytotoxic response to viralinfection. Increased frequency of virally-associatedexacerbations in this group would likely lead to lungtissue destruction and the development of COPD. Inthis respect the prevailing balance of CD4�and CD8�cells in the tissues is likely to be critical70,116.Finally, there are recent interesting data on the roleof retinoic acid in influencing alveolar number andthe repair of established emphysematous lung sug-gesting that nutrition may also act as an importantadditive factor in the emphysematous process117.Thus the patterns of inflammation in mild COPD insmokers and mild asthma in non-smokers differ butthe distinctions become less clear when there is anexacerbation in mild COPD when eosinophiliadevelops in association with up-regulation ofRANTES, an eosinophil chemoattractant usuallythought of as characteristic of asthma. These altera-tions in COPD may explain why exacerbations inCOPD may be responsive to corticosteroid treat-ment whereas the inflammation of ongoing mild tomoderate COPD is not118,119.
Vascular inflammation
There are few studies that examine the inflamma-tory process in pulmonary arteries of subjects withCOPD. There is inflammation of these vessels due
12 P.K. Jeffery

probably to the close approximation of airways andpulmonary arteries and the spread of the inflamma-tory process from the bronchiolar wall to the adja-cent pulmonary artery. An inflammatory processsimilar to that present in the conducting airways andin the lung parenchyma, consisting predominantlyof CD8�T-lymphocytes, has been reported in theadventitia of pulmonary arteries in smokers withCOPD95,120. The vascular infiltration of CD8�T-lym-phocytes correlates with the degree of airflow limita-tion in these subjects95, supporting a role forvascular inflammation in the progression of thedisease. The vascular inflammatory process isalso associated with impairment of endothelium-dependent vascular relaxation. The endotheliumplays a crucial role in the regulation of vascular cellgrowth and tone through the release of endothe-lium-derived relaxing factors. Endothelial dysfunc-tion, which results in an impaired release of thesefactors, has been shown in patients with end-stageCOPD undergoing lung transplantation121. In thisstudy, the degree of endothelial dysfunction was cor-related with both the severity of pulmonary vascularremodelling and the arterial oxygen tension, sug-gesting that in end-stage obstructive lung disease,hypoxemia is the principal factor determining theendothelial dysfunction that leads to vasoconstric-tion. However, endothelial dysfunction and intimalthickening may be present also in smokers with mildCOPD122 who are not hypoxemic, indicating thatfactors other than hypoxemia might be capable ofproducing the vascular changes in smokers. It is pos-sible that endothelial damage by cigarette smokingis the first vascular alteration occurring in COPD.This early alteration may predispose smokers todevelop further vascular damage due to additionalfactors such as hypoxemia and inflammation, ulti-mately leading to the development of pulmonaryhypertension120.
Further studies of the distinctive patterns inflam-mation, cytokine gene expression and protein secre-tion in the airways and vessel walls of asthma andCOPD should prove to be of scientific, clinical andtherapeutic interest.
Remodelling
Surface epithelium
Histologically, damage and shedding of the airwaysurface epithelium are reported in asthma postmor-tem (compare Fig. 1.5(a), (b)) (Fig. 1.11(a), (b), seecolour plate section) but this change is highly vari-able with some airways having completely intactsurface epithelium in the presence of markedinflammation and other structural changes123.Subepithelial edema has been suggested to be onemechanism responsible for lifting of the overlyingsurface epithelium where this occurs32. Repeatedloss of the epithelium induces a healing process asevidenced by squamous cell metaplasia and/orgoblet cell hyperplasia. Histologically, damage andshedding of airway surface epithelium appears to bean early feature of asthma, particularly of the allergicform124: it has been reported in biopsy specimens ofpatients with stable mild disease and is not a usualfeature of smokers with bronchitis or COPD (see Fig.1.11(a), (b))17,24,25,125. Loss of superficial epithelium isaccompanied by mitotic activity in the remainingepithelial cells in normal healthy individuals126.There is repeated epithelial regeneration in the formof simple and then stratified cells prior to restorationof the normal ciliated and goblet cell phenotypes,the entire process taking approximately 2 weeks.However, there are reports that such mitotic activityis deficient in asthma and this has led to the sugges-tion that there may be defective repair of epitheliumin asthma with the consequent release of a rangeof factors that would promote a remodellingresponse126,127. These factors include epithelial-derived growth factor and granulocyte–macrophagecolony stimulating factor and would induce altera-tions to the epithelial reticular basement mem-brane, via activation of adjacent fibroblasts/myofibroblasts, and deeper structures includingbronchial smooth muscle, mucus-secreting glandsand wall vessels. The release of these and othermolecules including IL8, eotaxin and RANTESwould also provide a chemoattractant gradient to
Pathology of asthma and COPD 13

both inflammatory and phenotypically alteredstructural cells. Aggregations of platelets togetherwith fibrillary material, thought to be fibrin havebeen observed in association with the damagedsurface24. Such fibrin deposits are also seen duringthe late phase response following allergen challenge(author’s unpublished results). The greater the lossof surface epithelium in biopsy specimens thegreater appears to be the degree of airways hyper-responsiveness24.
It is recognized that there is inevitably artefactualloss of surface epithelium during the taking andpreparation of such small biopsy pieces, even nor-mally, which makes interpretation of the epithelialloss seen in bronchial biopsies controversial. In theauthor’s opinion, the observed loss reflects the fra-gility of the epithelium in vivo that facilitates slough-ing during the bronchoscopy procedure. Thefragility of the epithelium in vivo in asthma is sup-ported by the frequent appearance of clusters ofsloughed epithelial cells in sputa (see Fig. 1.3(b), seecolour plate124) and the increased presence of bron-chial epithelial cells in bronchoalveolar lavage ofasthmatics with mild disease17. Other researchershave found no significant loss of epithelium in biop-sies of mild asthmatics128,129: this may be due todiffering methods of measurement of such loss or todifferences in the severity of the patients sampled.
The fragility of the surface may be associated withdisruption of epithelial cell tight junctions130,131 andthis may be facilitated by allergens per se, several ofwhich have been shown to have proteolytic activ-ity132. Tight junctions normally act as a selectivebarrier to the passage of ions, molecules and waterbetween cells: their disruption may lower thethreshold for stimulation of intraepithelial nervesleading to axonal reflexes, stimulation of mucus-secreting submucosal glands, vasodilatation andoedema through the release of sensory neuropep-tides (i.e. referred to as neurogenic inflamma-tion)133,134. Experimentally there is also evidence thatthe sensitivity of bronchial smooth muscle to sub-stances placed in the airway lumen correlatesstrongly with the integrity of the surface epithe-lium135. Loss or damage of surface epithelium in
asthma would thus lead to a reduction in the con-centration of factors normally relaxant to bronchialsmooth muscle with resultant increased sensitivityand ‘reactivity’ of bronchial smooth muscle.
Apart from their role as stem cells, the basal cellsof normal pseudostratified surface epithelium havebeen suggested to act as a bridge, enhancing theadhesion of ‘superficial’ cells to epithelial basementmembrane136. When superficial cells are lost inasthma the preferential plane of cleavage appears tobe between superficial and basal cells137, leavingbasal cells still attached to their basement mem-brane. Epithelial cells also act as effector cells bytheir synthesis and release of cytokines such as IL-6,IL-8, GM-CSF and chemokines such as RANTES138
and eotaxin. Disruption of the epithelium andattempts at repair may increase production of theseproinflammatory cytokines by those cells thatremain.
In contrast, epithelial loss is a less often reportedfeature of bronchial biopsies taken from smokerswith bronchitis or COPD when goblet cell hyperpla-sia and/or squamous metaplasia are often seen (Fig.1.11(b), see colour plate section)139,140.
Reticular basement membrane
Thickening of the reticular basement membrane(i.e. the lamina reticularis) which lies external to (orbelow) the epithelium has long been recognized as aconsistent change in allergic, non-allergic and occu-pational forms of asthma24,32,141–143: this may occur inresponse to repeated loss and healing of the surfaceepithelium (see Fig. 1.12(a)(b) and 1.5 (a)(b), seecolour plate). Whilst there may be focal and variablethickening of the reticular layer in COPD and otherinflammatory chronic diseases of the lung such asbronchiectasis and tuberculosis143, the lesion, whenhomogenous and hyaline in appearance, is highlycharacteristic of asthma and is not usually found inCOPD. The reticular layer appears to be absent in thefetus (at least up to 18 weeks of gestation)144 butdevelops in normal, healthy individuals, presum-ably during early childhood: its thickening inasthma begins early145, even before asthma is diag-
14 P.K. Jeffery

nosed146. The thickening remains even when asthmais mild and well controlled by antiasthma treat-ment147 and is present in patients with a long historyof asthma but who have not died of their asthma142.The extent of thickening is maximal early on in thedevelopment of asthma and does not appear toincrease significantly with age, duration or severityof disease7,145.
It should be noted that the basal lamina (i.e. the so-called ‘true’ epithelial basement membrane), whichconsists mainly of type IV collagen, glycosaminogly-cans and laminin, is not thickened in either asthmaor COPD. The thickening of the lamina reticularis (i.e.reticular basement membrane) (Fig. 1.13(a)(b))which is immuno-positive for collagen types III andV together with tenascin148 and fibronectin but notlaminin has been referred to as ‘subepithelialfibrosis’141. In the author’s opinion this is an unfortu-nate use of the term as the thickened layer of reticu-lin is ultrastructurally different from the bandedcollagen that lies deeper in the airway wall or thatwhich is characteristic of scarring. The reticular layeris composed of thinner fibres of reticulin linked to a
tenascin-rich matrix in which there are sugarstogether with entrapped molecules such as heparinsulphate and serum-derived components such asfibronectin: these molecules may modulate the stateof differentiation and function of overlying epithe-lium. In the author’s opinion, swelling of this subepi-thelial reticular layer may also contribute to itsthickening in asthma. Interestingly the thickenedlayer does not behave as a barrier to the transmigra-tion of inflammatory cells, which by the release ofenzymes (such as matrix metalloproteinases) or bythe presence of pre-existing pores149 can passthrough it with apparent ease (see Fig. 1.13(b)). Anassociation between the numbers of ‘myofibroblasts’underlying the reticular layer and the thickness of thereticular layer has been demonstrated in asthmaindicating these cells may secrete additional materialcontributing to its thickening150.
In contrast to asthma, a study of bronchial biop-sies, in carefully characterized patients with COPD,reports that the reticular layer is not thickened140. Arecent report confirms this and demonstrates thatthe reticular layer in smokers with irreversible
Pathology of asthma and COPD 15
Fig. 1.12 Scanning electron micrographs demonstrating the airway mucosa in (a) the non-asthmatic with epithelium attached to a
reticular basement membrane (RBM) of normal thickness (arrows) beneath which there is interstitial collagen. Scale�50 �m. (b) A
subject with a 25-year history of asthma, but who died of non-respiratory cause demonstrating the thickened RBM and damaged
epithelium. Scale�10 �m.
(a) (b)
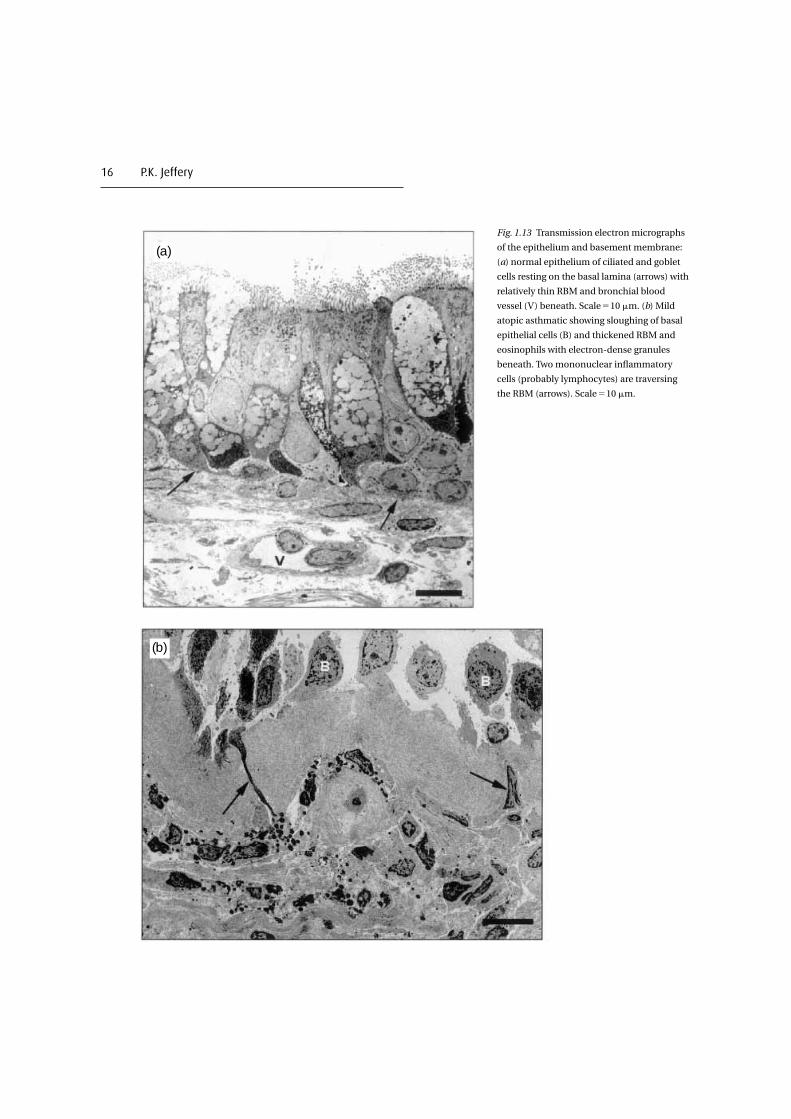
16 P.K. Jeffery
Fig. 1.13 Transmission electron micrographs
of the epithelium and basement membrane:
(a) normal epithelium of ciliated and goblet
cells resting on the basal lamina (arrows) with
relatively thin RBM and bronchial blood
vessel (V) beneath. Scale�10 �m. (b) Mild
atopic asthmatic showing sloughing of basal
epithelial cells (B) and thickened RBM and
eosinophils with electron-dense granules
beneath. Two mononuclear inflammatory
cells (probably lymphocytes) are traversing
the RBM (arrows). Scale�10 �m.
(a)
(b)

disease is similar to that in normal healthy non-smokers and is significantly thinner than that ofasthmatics who had been treated with inhaled corti-costeroids147. There are, however, subpopulations ofnon-asthmatic smokers with COPD, defined by theirsmoking history and irreversibility to inhaled beta-2agonist, who show significant airways reversibility(within the asthma range) to a 14-day course of oralprednisolone. These ‘responders’ have a thickerreticular basement membrane than normal and evi-dence of BAL eosinophilia: neither are present in the‘non-responder’ group151. This interesting COPDgroup with a significant degree of reversibility dem-onstrates further the potential overlap that may existbetween asthma and COPD at the tissue level.
Connective tissue
There is no consensus as to whether there isincreased interstitial collagen in asthma or whetherit increases with disease severity or duration. Arecent study of bronchial biopsies obtained fromasthmatics of varying severity reports increasingscores for collagen152, whereas another reports nodifference in collagen content153. Electron micro-scopic quantitative assessment of interstitial colla-gen in biopsies of mild asthmatics found nodifference in the area of the mucosa occupied by col-lagen fibres26. There is similar controversy over lossof elastic tissue in asthma, one study demonstratingthere is not26 and others indicating that there iseither elastolysis or altered ultrastructure of elastictissue154,155. In contrast, airway wall fibrosis is gener-ally, but not always, considered a feature of theairways in smokers who develop COPD, albeit thesestudies have focused on small rather than largeairways156–158.
Bronchial smooth muscle
The percentage of bronchial wall occupied by bron-chial smooth muscle often increased substantially infatal asthma19 (Fig. 1.14(a), (b)). The absoluteincrease in muscle mass is reported to be particu-larly striking in large intrapulmonary bronchi of
lungs obtained following a fatal attack as comparedwith that in asthmatic subjects dying of othercauses123: it is an important contributor to the thick-ening of the airway wall and hence to the markedincrease in resistance to airflow which may becomelife threatening159–162. Using a morphometric tech-nique Dunnill showed that approximately 12% of thewall in segmental bronchi obtained from cases offatal asthma was composed of muscle comparedwith about 5% in normals. Other studies have con-firmed this trend in airways larger than 2 mm diam-eter and demonstrated a three- to fourfold increaseover normal in the area of the wall occupied by bron-chial smooth muscle7,163,164. The increase in musclemass in small airways is not as great in absoluteterms as in the large airways although as a percent-age of the airway wall airway smooth muscle occu-pies a relatively larger percentage in the smaller thanin the larger airway. Thus small increases of musclein small airways may have a more significant effectfunctionally than similar increases in more proximalbronchi. In the absence of wheeze, values for musclemass in segmental bronchi in chronic bronchitisand emphysema fall largely within the normal rangebut intermediate levels are present in so-calledwheezy bronchitis165,166.
Few studies in COPD have focused attention onthe larger (cartilaginous) airways. One systematicstudy has described changes in large airway dimen-sions in relation to the lung function of patients withCOPD8. These authors found that the wall area inter-nal to the muscle was significantly thickened overthe entire range of cartilaginous airways measuredand that this was associated with a reduction inFEV/FVC. However, alterations in large airwaysmooth muscle mass were not observed and therewas no correlation between muscle mass andairflow limitation. There was a positive associationbetween peripheral airway inflammation and largeairway inner wall area and the authors argued thattheir findings and those of others favour inflamma-tion as the cause of the increasing inner airway wallthickness that occurs in both large and small airwaysin COPD. Airway smooth muscle increases signifi-cantly in the small airways in COPD84,98,158,167. In a
Pathology of asthma and COPD 17

18 P.K. Jeffery
Fig. 1.14 Increased bronchial smooth muscle: (a) a histological section of the airway wall of a case
of fatal asthma stained with H&E to show enlarged smooth muscle blocks lying relatively close to
the surface epithelium. Scale�80 �m. (b) Scanning electron microscopy of part of the mucosa in
fatal asthma demonstrating the three-dimensional appearance of the enlarged blocks of
bronchial smooth muscle (SM) and dilatation of bronchial vessels (V) which both contribute to
thickening of the airway wall. The arrowheads show the position of the RBM from which the
epithelium has been lost. Scale�70 �m.
(a)
(b)

study of small (membranous) airways of 15 patientswith COPD compared to the lungs of non-obstructed subjects and a group of asthmaticpatients, it was only the airway smooth muscle areathat was significantly increased in COPD168. Inasthma, the increase in the wall area occupied bymuscle, in absolute terms, is not as striking in smallairways as in the large123. It is considered that theincreased muscle mass that occurs at all generationsof airway is likely to be the most important abnor-mality responsible for the increased airflow resis-tance observed in response to bronchoconstrictingstimuli in both asthma and COPD169. Further studiesand a greater understanding of the changes occur-ring in small airways is required as is a means ofeffective delivery of anti-inflammatory and antire-modelling therapy to this distal anatomic site.Whether the increase in muscle mass in asthma isdue to muscle fibre proliferation (i.e. hyperplasia)170
or hypertrophy is at present unclear. Two patterns ofdistribution of increased muscle mass have beendescribed in asthma: one in which the increase isrestricted to the largest airways and another inwhich the increase occurs throughout the airways: itis suggested that in the former hyperplasia of musclefibres predominates whereas hypertrophy predomi-nates when there is increased muscle occurringthroughout the bronchial tree171.
A newly proposed mechanism involves dediffer-entiation of existing smooth muscle bundles. Cellsthat have ultrastructural features of both a contrac-tile and secretory phenotype have been found insubstantial numbers in the late phase response toallergen challenge. It has been suggested that, withrepeated exposure to allergen, these may contributeto the increased mass of bronchial smooth muscleby a process of differentiation of existing smoothmuscle and its migration to a subepithelial sitewhere new muscle is formed172. The mechanismsinvolved in this response are likely to be similar tothose occurring in atherosclerosis where there isvascular smooth muscle dedifferentiation andmigration to form a neo-intima of increased vascu-lar smooth muscle173.
Mucus-secreting elements
The sources of the lumenal mucus that contributesto the airway mucus in both asthma and COPD arethe submucosal glands and epithelial goblet cells.There is submucosal gland enlargement in both fatalasthma and COPD19 and excessive production ofmucus. The eosinophilic inflammatory exudate ofasthma is probably responsible for the particularlysticky tenacious plugs that plug the airways and areassociated with asphyxic death174. In asthma, thereis dilatation of submucosal gland ducts, referred toas bronchial gland ectasia175. Whilst the characteris-tic condensed twists of mucus in asthma referred toas Curshman spirals (see Fig. 1.3(b) see colour platesection) are often said to represent the casts of smallairways, their size is more in keeping with that ofgland ducts which is their more likely origin. Thenormal proportion of serous and mucous secretoryacini is retained in asthma whereas in COPD there isa shift towards a greater than normal predominanceof mucous acini176. Goblet cell hyperplasia is afeature of both asthma177 and bronchitis178. Themucous metaplasia that results in newly differen-tiated goblet cells in small bronchi and bronchioli ofless than 2 mm diameter, where they are normallyabsent or sparse, is a feature of small airways diseasein COPD167: whether mucous metaplasia also occursin asthma is debated. It is considered by some thatthe mucus present at this distal site in asthma mayhave been aspirated from larger airways. In cases offatal asthma where mucous metaplasia hasoccurred, the lumenal mucus secreted from surfacegoblet cells appears to remain adherent maintainingcontinuity between the cell’s secretions and theplug, suggesting the secretory process or the mucinitself is altered in fatal asthma177,179.
Airway vessels
Dilatation of bronchial mucosal blood vessels, con-gestion and wall edema are consistently reported fea-tures of fatal asthma and these can account forconsiderable swelling and stiffening of the airway wall
Pathology of asthma and COPD 19

(Fig. 1.14(b), 1.15, see colour plate section168,169,180).There are indications that the increased proportion ofthe wall occupied by vessel may be due in part to aproliferation of bronchial vessels (angiogenesis)181.Whilst angiogenesis has been reported in mildasthma182 it is particularly marked in severe corticos-teroid-dependent asthma183. Whether these changesare the consequence of chronic allergic inflammationor due to the response to chronic (or latent) viral,mycoplasm or bacterial infection is not known.
Whilst proliferation of the bronchial vasculature isa feature of bronchiectasis and occurs in response toinfection, changes to the bronchial vasculature havenot been reported as a particular feature of COPD168.However, patients with moderate to severe COPD dohave elevated pulmonary vascular pressures duringexercise and there are structural changes in the pul-monary arteries consistent with endothelial dys-function and pulmonary hypertension whencompared with patients with minimal or no disease.Small (�500 �m) pulmonary vessels in airwayobstructed smokers show intimal thickening ascompared with those of non-obstructed non-smokers: in severely obstructed smokers, there ismedial hypertrophy also122,184,185. Such structuralchanges likely contribute to the narrower lumensand vascular obstruction of these vessels. There isinfiltration of the pulmonary arterial wall by T-lym-phocytes. The CD8(�) T-cell phenotype is increasedin both non-obstructed smokers and smokers withCOPD compared with non-smokers and the inten-sity of the inflammatory infiltrate has been shown tocorrelate with both endothelium-dependent relaxa-tion and intimal thickness120.
Emphysema
Destruction of the lung parenchyma can be detectedmicroscopically in the alveolar walls of smokerseven when there is no evidence of airspace enlarge-ment on gross examination (see Fig. 1.16). Themicroscopic measurement of such parenchymaldestruction can, therefore, allow early identificationof the disease, at a time when emphysema is notdetectable macroscopically. The functional signifi-
cance of such early destruction is demonstrated byits correlation with indices of airflow limitation andloss of elastic recoil of the lung186.
The two major forms of emphysema, centriacinarand panacinar, have distinct mechanical propertiesand distinct peripheral airway involvement187. Inparticular, lung compliance is greater in panacinarthan in centriacinar emphysema, whereas the extentof peripheral airway inflammation is greater in thecentriacinar than in the panacinar form. It is pos-sible that, in centriacinar emphysema, airflow limi-tation is primarily a function of peripheral airwayinflammation, as supported by the correlationbetween reduced expiratory flow and increasedairway inflammation observed in this form ofemphysema. By contrast, in panacinar emphysema,airflow limitation seems to be primarily a function ofloss of elastic recoil, as supported by the correlationbetween reduced expiratory flow and increasedcompliance observed in this form of emphysema187.
The current emphasis in smoking-related diseaseis on emphysema associated with loss of alveolar-
20 P.K. Jeffery
Fig. 1.16 SEM of human lung parenchyma illustrating
microscopic emphysema in a smoker. Alveolar walls are
peppered by fenestrae too small to be seen by the naked eye.
Such early lesions probably result in loss of lung elastic recoil.
Scale�150 �m.

bronchiolar attachments. Bronchioles are sup-ported within the lung by attachment of the adjacentalveolar walls. Loss of these attached alveolar wallsand an increase in the interalveolar attachment dis-tance (IAAD) appear to be associated with func-tional abnormalities, including a decrease in forcedexpiratory volume in 1 second (FEV1) and abnormal-ities of tests of small airway function188–191. There islikely to be a role for airway wall inflammation in theselective loss of alveolar–bronchiolar attachments.It is possible that inflammatory cell activity mayweaken the alveolar tissue and facilitate its rupture,particularly at the point where alveolar walls andairway adventitia meet and where the mechanicalstress is likely to be greatest. This mechanism mightprovide an explanation for the relationship of airwayinflammation and abnormalities of pulmonaryfunction reported in smokers. Surprisingly, themajority of studies examining the pathology ofCOPD have been performed in subjects with mild tomoderate disease, while pathological studies onsubjects with severe COPD are few. The largest study,performed by Nagai and colleagues192, showed thatin subjects who had had severe disease both emphy-sema and peripheral airway abnormalities werepresent. Although the relative role of each of thesepathologic lesions in the development of airflowlimitation was difficult to establish, the authors con-cluded that emphysema was the more important.However, the findings of Nagai and colleaguesshould be interpreted with caution156. Their dataindicate that, when emphysema is severe, loss ofelastic recoil assumes overwhelming importance asthe mechanism of airflow limitation, thus maskingthe effects of peripheral airway abnormalities. Bycontrast, when emphysema is mild, peripheralairways abnormalities do appear to play a role incausing airflow limitation.
While earlier suggestions that distinct clinical pat-terns of disease, referred to as ‘pink puffers’ and ‘bluebloaters’, represented morphologically different pat-terns of pathology detected postmortem, morerecent studies have shown no correlation betweenthe amount of macroscopic emphysema andchronic hypoxemia.
Studies of the relationships of macroscopicallyassessed emphysema and gas transfer or radiologi-cal changes have shown only moderate correlations.With microscopically assessed emphysema,however, carbon monoxide transfer coefficient(KCO) shows a strong linear correlation (r�0.86) in agroup of patients, of whom only half showed macro-scopic emphysema. When the microscopic assess-ment of emphysema is expressed in terms of anestimate of the density of alveolar tissue per unitvolume of lung (AWUV), there is good correlationwith assessment of emphysema using computedtomography (CT)189,193. Such studies, based onmicroscopic assessment of emphysema, represent asignificant advance in the ability to identify earlyemphysema in life, and to follow its progression194.By application of combinations of quantitative his-tology and CT-determined lung volume data,Coxson and colleagues have been able to providequantitative estimates of the extent of lung destruc-tion in patients that may be followed longitudinallyin the future: this will allow pathogenesis to be betterunderstood and the effects of treatment to be deter-mined195,196. Recent application of these methodshas allowed the number of inflammatory cellspresent per unit surface area of lung parenchyma tobe investigated in COPD. The data from these inves-tigations indicate that emphysematous lungdestruction is associated with a marked amplifica-tion of the inflammatory response in patients withemphysema compared to the lungs of smokerswithout emphysema but with equivalent smokinghistories195.
Emphysema in COPD, is also the likely conse-quence of a chronic CD8�cell inflammatoryprocess. The current definition of emphysemaexcludes the presence of obvious fibrosis, yet it isnow known that fibrosis may also occur even in thepresence of alveolar wall loss197,198. The enlargementof alveolar spaces, distal to the terminal bronchio-lus, in COPD may thus represent the consequence oflung injury and a failure to repair rather than ofdestruction per se. The focal fibrosis that may beidentified in some cases of emphysema may repre-sent the remainder of a repair component. Further
Pathology of asthma and COPD 21

studies of the mechanisms that balance the produc-tion and degradation of collagen that occurs duringthe reparative and remodelling response to lunginjury may yield important findings applicable tothe treatment or prevention of the parenchymallesions so important to COPD.
Airway wall nerves
The topic of airway wall innervation and its relationto asthma is a large one133,134. There are data suggest-ing that in fatal asthma there is an absence of(relaxant) vasoactive intestinal polypeptide (VIP)-containing nerve fibres and an increase in thenumbers of substance P-containing fibres (stimula-tory to bronchial smooth muscle) contrasting mark-edly with the innervation of the control lungs takenat resection from chronic smokers199,200. The reduc-tion has not, however, been confirmed in examina-tion of bronchial biopsies in mild asthma201. WhilstSharma and colleagues have described a reductionof airway VIP and �-adrenoreceptors in cystic fibro-sis, the densities of both VIP receptors and �-adrenoreceptors are reported to be similar inasthma to those of grossly normal tissue of the lungsof smokers resected for carcinoma202,203.
Conclusions
The key points of comparison between asthma andCOPD are summarized in Tables 1.1 and 1.2. There isevidence of airways inflammation in both asthmaand COPD but there are marked differences in termsof the predominant anatomic site involved, the pre-dominant pattern of inflammatory cells and thestructural consequences of such inflammation. Itwill be of interest to learn whether further studiesconfirm or refute the hypothesis that chronicasthma and COPD are two distinct conditions thatrequire equally distinct approaches to their manage-ment. This notion has received support from therecent findings of long-term trials of mild to moder-ate disease in which inhaled corticosteroids havebeen shown to be effective in the treatment of
mild/moderate asthma but not so in COPD.However, the author predicts that the responses toany one treatment will vary from patient to patientdepending not only on the diagnosis of ‘asthma’ of‘COPD’ per se but rather on the particular prevailingpatterns of inflammatory cells, cytokines and
22 P.K. Jeffery
Table 1.1. Asthma summary
The airway walls in asthma are thickened by inflammation
and ‘remodelling’ and there is lumenal narrowing.
The association of tissue eosinophilia and asthma is a strong
one and activated T-helper (CD4�) lymphocytes perpetuate
the chronic eosinophilia.
Neutrophils are sparse in mild asthma but they are present in
relatively large numbers in severe asthma
Mast cells play a role in the immediate (type I sensitivity)
reaction in asthma: little is known of the role of basophils
albeit they are considered important.
Epithelial fragility and loss are often but not always reported in
asthma: healing or abnormal repair may be driving
subsequent remodelling.
Thickening of the reticular basement membrane (i.e. the
lamina reticularis but not the lamina densa) is a consistent
change in allergic, non-allergic and occupational forms of
asthma.
There is no consensus as to increased interstitial collagen in
asthma.
The percentage of bronchial wall occupied by bronchial
smooth muscle increases substantially in fatal asthma: there
are several mechanisms that could explain the increase and
there may be parallels with the changes in vessel walls in
atheroma.
There is submucosal gland enlargement in fatal asthma and
excessive production of mucus that, together with the
inflammatory exudate, forms the sticky tenacious plugs that
block airway lumena.
Dilatation of bronchial mucosal blood vessels, congestion and
wall oedema are consistently reported features of fatal
asthma: these can account for considerable swelling of the
airway wall.
While corticosteroids are effective in treating the eosinophilic
inflammation of mild asthma new treatments need to be
found to treat the altered inflammation and the remodelling
of severe asthma.
Inflammation and remodelling may respond to distinct classes
of drug.