dr. a. tarab dept. of biochemistry hkmuhkmu.online/wp-content/uploads/2016/04/metabolism-of... ·...
TRANSCRIPT

METABOLISM OF MONOSACCHARIDES AND
DISACCHARIDES
DR. A. TARAB
DEPT. OF BIOCHEMISTRY
HKMU

OVERVIEW
• Glucose is the most common monosaccharide consumed by humans, and its metabolism has been discussed extensively
• However, two other monosaccharides -fructose and galactose – occur in significant amounts in the diet, and make important contributions to energy metabolism
• In addition, galactose is an important component of cell structural carbohydrates

FRUCTOSE METABOLISM
• The major source of fructose is the disaccharide sucrose, which, when cleaved in the intestine, releases equimolar amounts of fructose and glucose
• Fructose is also found as a free monosaccharide in many fruits, and in honey
• Entry of fructose into cells is not insulin-dependent (unlike that of glucose into certain tissues), and, in contrast to glucose, fructose does not promote the secretion of insulin

• A. Phosphorylation of fructose
• For fructose to enter the pathways of intermediary metabolism, it must first be phosphorylated
• This can be accomplished by either hexokinaseor fructokinase (also called ketohexokinase). Hexokinase phosphorylates glucose in all cells of the body, and several additional hexosescan serve as substrates for this enzyme

• However, it has a low affinity (that is, a high km) for fructose
• Therefore, unless the intracellular concentration of fructose becomes unusually high, the normal presence of saturating concentrations of glucose means that little fructose is converted to fructose 6-phosphate by hexokinase.
• Fructokinase provides the primary mechanism for fructose phosphorylation

• It is found in the liver (which processes most of the dietary fructose), kidney, and the small intestinal mucosa, and converts fructose to fructose 1-phosphate using ATP as the phosphate donor
• B. Cleavage of fructose 1-phosphate• Fructose 1-phosphate is not converted to
fructose as is fructose 6-phosphate, but is cleaved by aldolase B (also called fructose 1-phosphate aldolase) to dihydroxyacetone phosphate (DHAP) and glyceraldehyde
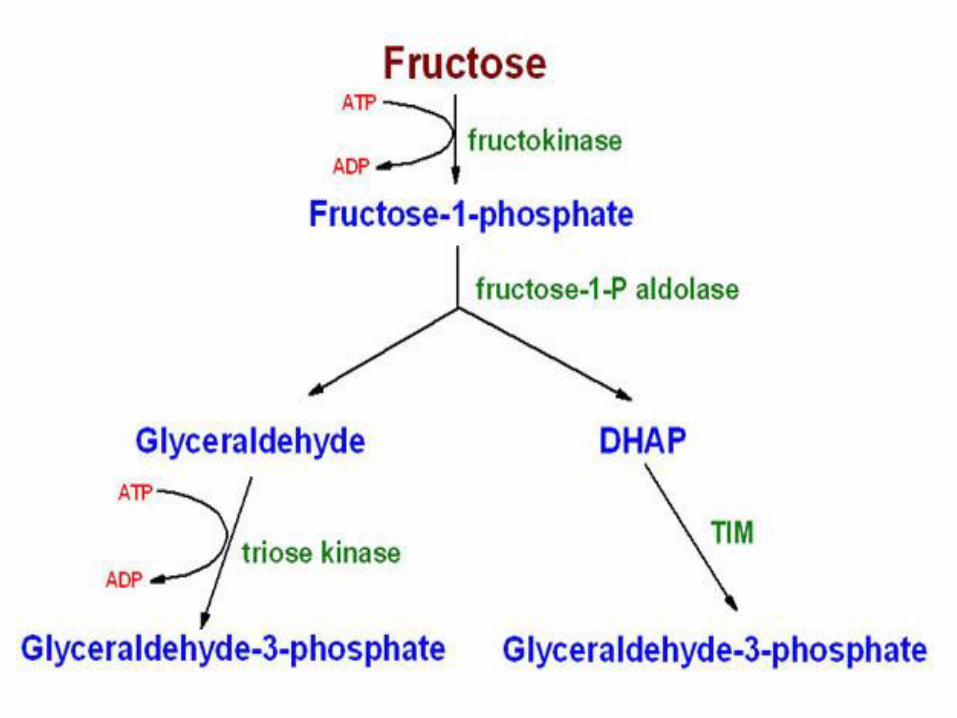

• DHAP can directly enter glycolysis or gluconeogenesis, whereas glyceraldehyde can be metabolized by a number of pathways

• C. Disorders of fructose metabolism
• A deficiency of one of the key enzymes required for the entry of fructose into intermediary metabolic pathways can result in either a benign condition (fructokinase deficiency), or a severe disturbance of liver and kidney metabolism as a result of aldolase B deficiency (hereditary fructose intolerance, HFI), which is estimated to occur in 1:20,000 live births

• The first symptoms appear when a baby is weaned and begins to be fed food containing sucrose or fructose
• Fructose 1-phosphate accumulates, and ATP and inorganic phosphate levels fall significantly, with adenine being converted to uric acid, causing hyperuricemia

• ESSENTIAL FRUCTOSURIA
• - Lack of fructokinase
• - Autosomal recessive (1 in 130,000 births)
• - Benign, asymptomatic condition
• - Fructose accumulates in the urine

• HEREDITARY FRUCTOSE INTOLERANCE (“FRUCTOSE POISONING”)
• - Absence of aldolase B leads to intracellular trapping of fructose 1-phosphate
• - Causes severe hypoglycemia, vomiting, jaundice, haemorrhage, hepatomegaly and hyperuricemia
• - Can cause hepatic failure and death• Therapy: Rapid detection and removal of fructose
and sucrose from the diet

• D. Conversion of mannose to fructose 6-phosphate
• Mannose, the C-2 epimer of glucose, is an important component of glycoproteins
• Hexokinase phosphorylates mannose, producing mannose 6-phosphate, which, in turn, is (reversibly) isomerized to fructose 6-phosphate by phosphomannose isomerase

GALACTOSE METABOLISM
• The major dietary source of galactose is lactose (galactosyl β-1,4-glucose) obtained from milk and milk products
• Some galactose can also be obtained by lysosomal degradation of complex carbohydrates, such as glycoproteins, and glycolipids, which are important membrane components
• Like fructose, the entry of galactose into cells is not insulin-dependent.

• A. Phosphorylation of galactose
• Like fructose, galactose must be phosphorylated before it can be further metabolized
• Most tissues have a specific enzyme for this purpose, galactokinase, which produces galactose 1-phosphate
• ATP is the phosphate donor


• B. Formation of UDP-galactose
• Galactose 1- phosphate cannot enter the glycolytic pathway unless it is first converted to UDP-galactose
• This occurs in an exchange reaction, in which
UDP is removed from UDP-glucose (leaving behind glucose 1-phosphate), and is then transferred to the galactose 1-phosphate producing UDP-galactose

Structure of UDP-Galactose

• The enzyme that catalyzes this reaction is galactose 1-phosphate uridyltransferase
• C. Use of UDP-galactose as a carbon source for glycolysis or gluconeogenesis
• In order for UDP-galactose to enter the mainstream of glucose metabolism, it must first be converted to its C-4 epimer, UDP-glucose, by UDP-hexose 4-epimerase

• This "new" UDP-glucose (produced from the original UDP-galactose), can then participate in many biosynthetic reactions, as well as being used in the uridyltransferase reaction described above, converting another galactose1-phosphate into UDP-galactose, and releasing glucose 1-phosphate, whose carbons are those of the original galactose

• D. Role of UDP-galactose in biosynthetic reactions
• UDP-galactose can serve as the donor of galactose units in a number of synthetic pathways, including synthesis of lactose, glycoproteins, glycolipids, and glycosaminoglycans

• E. Disorders of galactose metabolism
• Galactose 1-phosphate uridyltransferase is missing in individuals with classic galactosemia
• In this disorder, galactose 1-phosphate and, therefore, galactose, accumulate in cells
• Physiologic consequences are similar to those found in essential fructose intolerance, but a broader spectrum of tissues is affected

CLASSIC GALACTOSEMIA
• Uridyltransferase deficiency
• Autosomal recessive disorder (1 in 23,000 births)
• It causes galactosemia and galactosuria, vomiting, diarrhea and jaundice
• Accumulation of galactose 1-phosphate in nerve, lens, liver and kidney tissues causes liver damage, severe mental retardation and cataracts

• Antenatal diagnosis is possible by chorionic villus sampling
• Therapy: rapid diagnosis and removal of galactose (therefore lactose) from the diet

LACTOSE SYNTHESIS
• Lactose is a disaccharide that consists of a molecule of β-galactose attached by a β(1→4)linkage to glucose
• Therefore, lactose is galactosyl β(1→4)-glucose
• Lactose, known as the "milk sugar," is produced by the mammary glands of most mammals
• Therefore, milk and other dairy products are the dietary sources of lactose

• Lactose is synthesized in the endoplasmic reticulum by lactose synthase (UDP-galactose:glucose galactosyltransferase), which transfers galactose from UDP-galactoseto glucose, releasing UDP
• This enzyme is composed of two proteins, A and B
• Protein A is a β-D-galactosyltransferase and is found in a number of body tissues


• In tissues other than the lactating mammary gland, this enzyme transfers galactose from UDP-galactose to N-acetyl-D-glucosamine, forming the same β(1→4) linkage found in lactose, and producing N-acetyllactosamine—a component of the structurally important N-linked glycoproteins
• In contrast, protein B is found only in lactating mammary glands

• It is α-lactalbumin, and its synthesis is stimulated by the peptide hormone prolactin
• Protein B forms a complex with the enzyme, protein A, changing the specificity of that transferase so that lactose, rather than N-acetyllactosamine, is produced