
Page 1 of 17
Tracing the development of acute myeloid leukemia in CBL syndrome
Heiko Becker,1 Kenichi Yoshida,2,3 Nadja Blagitko-Dorfs,1 Rainer Claus,1
Milena Pantic,1 Mahmoud Abdelkarim1, Christoph Niemöller,1 Christine Greil,1
Björn Hackanson,1 Yuichi Shiraishi,4 Kenichi Chiba,4 Hiroko Tanaka,5 Satoru Miyano,4,5
Konstanze Döhner,6 Susanne Schnittger,7 Philipp Henneke,8,9 Charlotte M. Niemeyer,9
Christian Flotho,9 Dietmar Pfeifer,1 Seishi Ogawa,2,3 and Michael Lübbert1
1 Department of Medicine I, Medical Center - University of Freiburg, Freiburg, Germany;
2 Department of Pathology and Tumor Biology, Graduate School of Medicine, Kyoto
University, Kyoto, Japan;
3 Cancer Genomics Project, Graduate School of Medicine, The University of Tokyo,
Tokyo, Japan;
4 Laboratory of DNA Information Analysis, Human Genome Center, Institute of Medical
Science, The University of Tokyo, Tokyo, Japan;
5 Laboratory of Sequence Analysis, Human Genome Center, Institute of Medical
Science, The University of Tokyo, Tokyo, Japan;
6 Department of Internal Medicine III, University of Ulm, Ulm, Germany;
7 MLL Munich Leukemia Laboratory, Munich, Germany;
8 Center of Chronic Immunodeficiency, Medical Center - University of Freiburg, Freiburg,
Germany;
9 Center for Pediatrics and Adolescent Medicine, Medical Center - University of
Freiburg, Freiburg, Germany
Blood First Edition Paper, prepublished online February 3, 2014; DOI 10.1182/blood-2013-10-533844
Copyright © 2014 American Society of Hematology

Page 2 of 17
Corresponding author: Michael Lübbert, Department of Medicine I, Medical Center -
University of Freiburg, Hugstetter Straße 55, 79106 Freiburg, Germany, phone: +49 761
270 32790, fax: +49 761 270 36970, email: [email protected]
Running head: AML in CBL mutated hematopoiesis
Keywords: CBL syndrome, CBL mutation, acute myeloid leukemia, AML, juvenile
myelomonocytic leukemia
Supplemental data: The article is accompanied by supplemental data.

Page 3 of 17
Key Points
• The CBL syndrome may predispose to myeloid neoplasias other than JMML
• Whole-exome sequencing identifies mutations that possibly cooperate with mutant
CBL in AML development
Abstract
We describe the development of acute myeloid leukemia (AML) in an adult with CBL
syndrome due to a heterozygous de novo germline mutation in CBL codon D390. In the
AML bone marrow, the mutated CBL allele was homozygous after copy number-neutral
loss-of-heterozygosity and amplified through a chromosomal gain; moreover, an
inv(16)(p13q22) and, as assessed by whole-exome sequencing, 12 gene mutations
(e.g., in CAND1, NID2, PTPRT, DOCK6) were additionally acquired. During complete
remission of the AML, in the presence of normal blood counts, the hematopoiesis stably
maintained the homozygous CBL mutation, which is reminiscent of the situation in
children with CBL syndrome and transient juvenile myelomonocytic leukemia. No
additional mutations were identified by whole-exome sequencing in granulocytes during
complete remission. The study highlights the development of AML in an adult with CBL
syndrome and, more general, in genetically aberrant but clinically inconspicuous
hematopoiesis.

Page 4 of 17
Introduction
Preceding hematologic disorders are documented in a quarter of adults with acute
myeloid leukemia (AML).1 However, an unknown proportion of AMLs that apparently
arise de novo may have developed from undiscovered abnormal hematopoiesis.
Mutations in CBL, encoding an E3 ubiquitin ligase, are found in 10-20% of chronic
(CMML) or juvenile myelomonocytic leukemia (JMML) patients.2-7 Germline CBL
mutations cause the CBL syndrome that recapitulates features of other RAS-MAPK
pathway disorders and predisposes to JMML.8-10 In AML, CBL mutations are rare, but
associate with inv(16).11-14
Here, we describe the development of AML in an adult with CBL syndrome and JMML-
typical loss of wildtype CBL in bone marrow.

Page 5 of 17
Methods
Written informed consent of the patient included in the present study was obtained for
sample storage and analyses prior to sampling, as approved by the local ethics
committee. This study was conducted in accordance with the Declaration of Helsinki.
Karyotype, mutations in NPM1, FLT3 (tyrosine kinase domain, internal tandem
duplication), CEBPA and CBL, and CBFB-MYH11 expression relative to ABL1 were
assessed as described elsewhere.15-18 CBL mutated-to-wildtype allelic ratios were
determined using the PyroMark Q96MD (Qiagen), and chromosomal copy numbers
using CytoScanHD-arrays (Affymetrix). Data are deposited at
http://www.ebi.ac.uk/arrayexpress/ (E-MEXP-3997). Whole-exome sequencing was
performed as reported;19 variants were validated by Sanger sequencing. Methods are
detailed in the supplement.

Page 6 of 17
Results and discussion
Characteristics of the AML
A 40-year-old male was diagnosed with AML in June 2011. Pre-existing conditions were
hereditary spherocytosis (diagnosed in 1996), coagulopathy (low FVII, X, XII, XIII), atrial
fibrillation and hypocholesterolemia; a splenomegaly was considered consequence of
the spherocytosis. At AML diagnosis, the white blood cell count was 19,390/µl, with
approximately 30% blasts and 30% dysplastic monocytes (Supplemental Figure S1).
The marrow contained 50% CD117-positive blasts and 30% CD14-positive monocytes;
the karyotype was 46,XY,add(4)(q?31),inv(16)(p13q22)[21]/46,XY,inv(16)(p13q22)[1].
CBFB-MYH11 (type D) was detected with a ratio of 46.23 in blood. NPM1, CEBPA and
FLT3 mutations were absent.
The patient received “3+7” induction followed by dasatinib (ClinicalTrials.gov
NCT00850382). Six weeks after therapy start, complete remission (CR) was
documented. He received four consolidation courses with high-dose cytarabine. At last
follow-up (September 2013), he was in continuous CR, with no CBFB-MYH11
detectable.
Identification of a germline CBL mutation
Between the treatment courses, the patient’s monocyte counts rose to extraordinarily
high levels. Although monocytes were within normal limits after treatment and in blood
counts dating back to 1996 (Supplemental Table S1), this observation prompted the
question whether the patient had an underlying monocytic disorder.

Page 7 of 17
Since monocytosis is a hallmark of JMML and CMML, we examined the mutation status
of CBL exon 8 and 9 in blood collected at the AML diagnosis. We indeed found a
p.D390V-mutation, located in the frequently mutated RING finger domain. Assessing
the germline origin of the mutation, we also identified it in buccal mucosa and hair
follicles. We concluded that the patient had a previously undiagnosed CBL syndrome,
with the preexisting coagulopathy and atrial fibrillation being part of the phenotype
(Supplemental Table S2). 8-10
No CBL mutations were detected in blood of both parents, indicating de novo
occurrence in the patient’s germline. The patient has no siblings.
Zygosity of the CBL mutation
Copy number-neutral loss-of-heterozygosity (LOH) of the CBL-containing chromosomal
band 11q23.3 is common in children with CBL syndrome and JMML.8,10 11q-LOH was
also detectable in marrow mononuclear cells from our patient during AML. Moreover,
the 11q-LOH persisted in B-lymphocytes, granulocytes and monocytes collected later
during CR (Table 1, Supplemental Figure S2). This is reminiscent of the situation in
children with CBL syndrome and JMML whose myeloproliferation spontaneously
improves.8 Notably, the AML in our patient exhibited an additional gain of 11q-material,
indicating that the LOH had existed before the AML. In skin and T-lymphocytes, 11q
retained heterozygosity.

Page 8 of 17
To complement the LOH findings, we determined the allelic burden of the CBL mutation
by pyrosequencing (Table 1). In agreement with the LOH data, the mutation was
heterozygous in skin and T-lymphocytes but homozygous in AML cells and in
granulocytes, monocytes and B-lymphocytes collected during CR, where it remained
homozygous until last follow-up, underlining the stability of the genetically aberrant
hematopoiesis. Notably, LOH and pyrosequencing data suggested the presence of a
small fraction of T-lymphocytes also harboring the 11q-LOH (Table 1, Supplemental
Figure S2).
Identification of cooperating mutations by whole-exome sequencing
Similar to our patient, children with CBL syndrome and transient JMML feature normal
blood counts and persistent homozygous CBL mutation in their hematopoiesis.8 Little is
known about mechanisms that could be responsible for normal hematopoiesis despite
oncogenic features characteristic of JMML. We wondered whether this was associated
with the acquisition of mutations that overcome the myeloproliferative impact of the
homozygous CBL mutation. We therefore subjected granulocytes from CR and skin to
whole-exome sequencing, but identified no additional mutations.
We also performed whole-exome sequencing of AML cells to identify mutations that
were acquired during AML development, in addition to inv(16) and 11q-gain. We
detected somatic mutations in 12 genes (Table 2), three of which (i.e., CAND1, NID2,
PTPRT) were previously found mutated in AML.20-22 However, no gene has an
established role in leukemogenesis, e.g., as cooperating partner of mutant CBL or
CBFB-MYH11.

Page 9 of 17
Biologic impact of the CBL mutation
JMML features the formation of colonies at low concentrations of granulocyte-
macrophage colony-stimulating factor (GM-CSF).23 We observed no spontaneous
growth or hypersensitivity to GM-CSF of mononuclear cells collected from our patient
during CR (data not shown), which underlines the lacking or only subtle impact of the
homozygous CBL mutation on hematopoiesis. Moreover, granulocytes showed normal
production of reactive oxygen species and interleukin-8 to stimuli, and adhesion and
migration/chemotaxis were normal (data not shown).
In summary, we diagnosed a CBL syndrome in an adult, who, as observed in children
with CBL syndrome developing JMML,8,10 had lost the CBL wildtype allele in the bone
marrow. Whether this leads to overt JMML only under certain circumstances is not well
understood.24 Since the LOH persisted in the various hematopoietic cell lineages in our
patient, it likely conferred a clonal advantage at one point. Thus, the patient may have
indeed gone through a JMML or related hematologic disorder during infancy, which
spontaneously resolved and left behind normal blood counts. However, medical
information to support this assumption is unavailable. Following the hypothesis that
normal blood counts in our patient could be associated with the acquisition of mutations
counterbalancing the mutant CBL, we performed whole-exome sequencing but
identified no acquired mutations. On the background of the CBL mutation, the patient
developed AML through the acquisition of inv(16), gain of 11q-material and at least 12
gene mutations. The AML was erased by chemotherapy, again leaving behind a
hematopoiesis with homozygous CBL mutation.

Page 10 of 17
While the CBL syndrome is known to predispose to JMML, this is the first description of
of a different myeloid neoplasia occurring at adult age. It cannot be determined whether
the AML was mere coincidence, or due to a predisposition conferred by the CBL
mutation. However, the latter is supported by the specific gain of CBL-encoding 11q-
material and occurrence of inv(16), which associates with CBL mutations.11-14 If
substantiated by future studies, the association between CBL syndrome and AML
should be considered in clinical practice. CBL would then join other genes, e.g., RUNX1
or CEBPA, germline mutations in which were linked to a predisposition to AML.25
Overall, the case highlights the possibility of genetically aberrant hematopoiesis despite
normal blood counts and provides insight into myeloid neoplasias in the CBL syndrome.
Due to potential health problems associated with a CBL syndrome, germline analyses
may be generally warranted in younger adults with CBL-mutated neoplasias.

Page 11 of 17
Acknowledgements
We wish to express our thanks to the patient for continued and interactive participation
in the study. We also thank the German-Austrian AML Study Group (AMLSG), as the
patient was enrolled on an AMLSG clinical trial.
The research was supported in part by the Research Committee of the University
Freiburg, Germany [MER 785/10 (H.B.)], the EHA and ASH Translational Research
Training in Hematology (H.B.), the German Cancer Aid [DKH 110213 (B.H.)], the
Bundesministerium für Bildung und Forschung (P.H.) and the German Research
Foundation [DFG Lu 429/7-1, CRC 992-C04 (M.L.); DFG Fl 345/4-1, CRC 992-C05
(C.F.)].
Authorship
Contribution: H.B., K.Y., C.G., S.S., P.H., C.M.N, C.F., S.O., and M.L. contributed to the
design and interpretation of the study; H.B., N.B.-D., M.P., M.A., C.N., B.H., K.D., S.S.,
and C.F. carried out laboratory-based analyses; K.Y., R.C., Y.S., K.C., H.T., S.M., and
D.P. performed bioinformatic analyses; C.G., B.H., and M.L. were involved in patient
care; H.B., S.O., and M.L. wrote the manuscript, and all authors contributed to and
agreed on the final version.
Disclosure of conflicts of interest: S.S. declares part ownership of the MLL Munich
Leukemia Laboratory GmbH. The remaining authors declare no competing financial
interests.

Page 12 of 17
Correspondence: Michael Lübbert, Department of Medicine I, Medical Center -
University of Freiburg, Hugstetter Straße 55, 79106 Freiburg, Germany, email:

Page 13 of 17
References
1. Juliusson G, Antunovic P, Derolf A, et al. Age and acute myeloid leukemia: Real
world data on decision to treat and outcomes from the Swedish Acute Leukemia
Registry. Blood 2009;113(18):4179-4187.
2. Sanada M, Suzuki T, Shih LY, et al. Gain-of-function of mutated C-CBL tumour
suppressor in myeloid neoplasms. Nature. 2009;460(7257):904-908.
3. Loh ML, Sakai DS, Flotho C, et al. Mutations in CBL occur frequently in juvenile
myelomonocytic leukemia. Blood. 2009;114(9):1859-1863.
4. Makishima H, Cazzolli H, Szpurka H, et al. Mutations of e3 ubiquitin ligase cbl
family members constitute a novel common pathogenic lesion in myeloid malignancies.
J Clin Oncol. 2009;27(36):6109-6116.
5. Pérez B, Kosmider O, Cassinat B, et al. Genetic typing of CBL, ASXL1, RUNX1,
TET2 and JAK2 in juvenile myelomonocytic leukaemia reveals a genetic profile distinct
from chronic myelomonocytic leukaemia. Br J Haematol. 2010;151(5):460-468.
6. Kohlmann A, Grossmann V, Klein HU, et al. Next-generation sequencing
technology reveals a characteristic pattern of molecular mutations in 72.8% of chronic
myelomonocytic leukemia by detecting frequent alterations in TET2, CBL, RAS, and
RUNX1. J Clin Oncol. 2010;28(24):3858-3865.
7. Itzykson R, Kosmider O, Renneville A, et al. Prognostic Score Including Gene
Mutations in Chronic Myelomonocytic Leukemia. J Clin Oncol. 2013;31(19):2428-2436.
8. Niemeyer CM, Kang MW, Shin DH, et al. Germline CBL mutations cause
developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat
Genet. 2010;42(9):794-800.

Page 14 of 17
9. Martinelli S, De Luca A, Stellacci E, et al. Heterozygous germline mutations in
the CBL tumor-suppressor gene cause a Noonan syndrome-like phenotype. Am J Hum
Genet. 2010;87(2):250-257.
10. Pérez B, Mechinaud F, Galambrun C, et al. Germline mutations of the CBL gene
define a new genetic syndrome with predisposition to juvenile myelomonocytic
leukaemia. J Med Genet. 2010;47(10):686-691.
11. Weissmann S, Alpermann T, Grossmann V, et al. Landscape of TET2 mutations
in acute myeloid leukemia. Leukemia. 2012;26(5):934-942.
12. Abbas S, Rotmans G, Löwenberg B, Valk PJ. Exon 8 splice site mutations in the
gene encoding the E3-ligase CBL are associated with core binding factor acute myeloid
leukemias. Haematologica. 2008;93(10):1595-1597.
13. Reindl C, Quentmeier H, Petropoulos K, et al. CBL exon 8/9 mutants activate the
FLT3 pathway and cluster in core binding factor/11q deletion acute myeloid
leukemia/myelodysplastic syndrome subtypes. Clin Cancer Res. 2009;15(7):2238-2247.
14. Haferlach C, Dicker F, Kohlmann A, et al. AML with CBFB-MYH11
rearrangement demonstrate RAS pathway alterations in 92% of all cases including a
high frequency of NF1 deletions. Leukemia. 2010;24(5):1065-1069.
15. Fröhling S, Skelin S, Liebisch C, et al. Comparison of cytogenetic and molecular
cytogenetic detection of chromosome abnormalities in 240 consecutive adult patients
with acute myeloid leukemia. J Clin Oncol. 2002;20(10):2480-2485.
16. Schnittger S, Weisser M, Schoch C, et al. New score predicting for prognosis in
PML-RARA+, AML1-ETO+, or CBFBMYH11+ acute myeloid leukemia based on
quantification of fusion transcripts. Blood. 2003;102(8):2746-2755.

Page 15 of 17
17. Döhner K, Schlenk RF, Habdank M, et al. Mutant nucleophosmin (NPM1)
predicts favorable prognosis in younger adults with acute myeloid leukemia and normal
cytogenetics: interaction with other gene mutations. Blood. 2005;106(12):3740-3746.
18. Schnittger S, Bacher U, Alpermann T, et al. Use of CBL exon 8 and 9 mutations
in diagnosis of myeloproliferative neoplasms and myelodysplastic/myeloproliferative
disorders: an analysis of 636 cases. Haematologica. 2012;97(12):1890-1894.
19. Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing
machinery in myelodysplasia. Nature. 2011;478(7367):64-69.
20. Forbes SA, Bindal N, Bamford S, et al. COSMIC: mining complete cancer
genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res.
2011;39(Database issue):D945-950. Available at http://www.sanger.ac.uk/cosmic.
Accessed September 24, 2013.
21. Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid
leukaemia revealed by whole-genome sequencing. Nature. 2012;481(7382):506-510.
22. Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes
of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059-2074.
23. Emanuel PD, Bates LJ, Castleberry RP, Gualtieri RJ, Zuckerman KS. Selective
hypersensitivity to granulocyte-macrophage colony stimulating factor by juvenile chronic
myeloid leukemia hematopoietic progenitors. Blood. 1991;77(5):925-929.
24. Strullu M, Caye A, Cassinat B, et al. In hematopoietic cells with a germline
mutation of CBL, loss of heterozygosity is not a signature of juvenile myelo-monocytic
leukemia. Leukemia. 2013;27(12):2404-2407.
25. Owen C, Barnett M, Fitzgibbon J. Familial myelodysplasia and acute myeloid
leukaemia--a review. Br J Haematol. 2008;140(2):123-132.
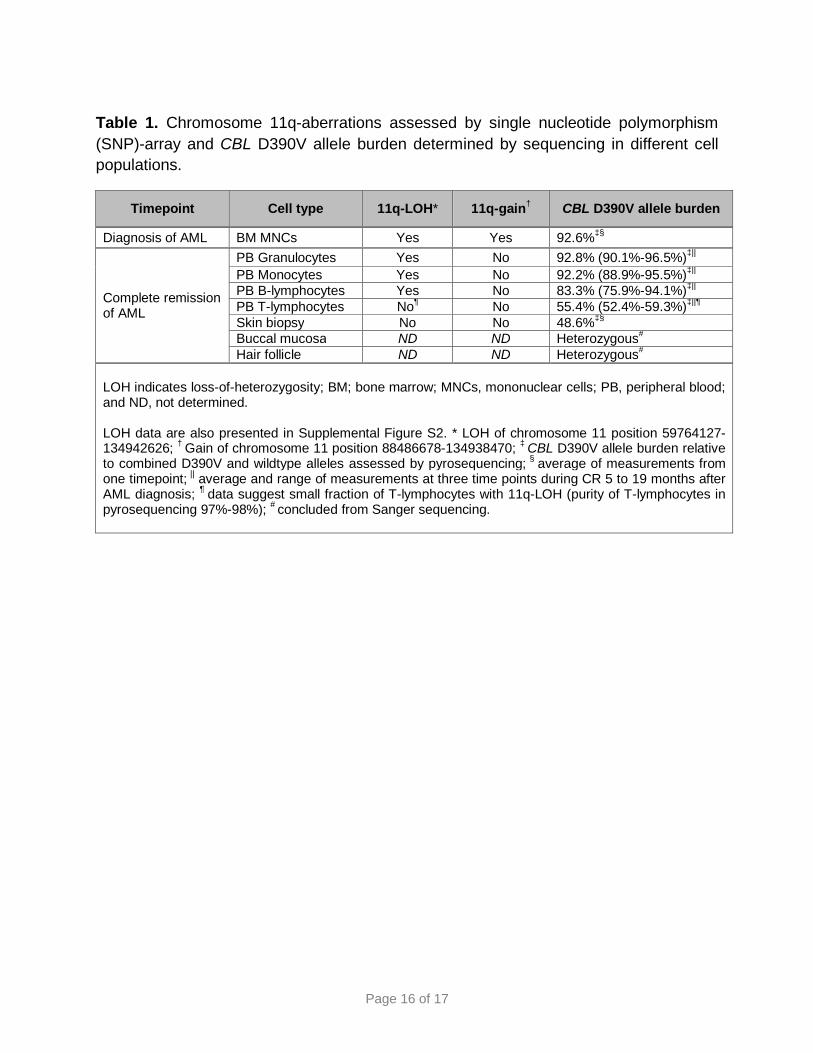
Page 16 of 17
Table 1. Chromosome 11q-aberrations assessed by single nucleotide polymorphism (SNP)-array and CBL D390V allele burden determined by sequencing in different cell populations.
Timepoint Cell type 11q-LOH* 11q-gain† CBL D390V allele burden
Diagnosis of AML BM MNCs Yes Yes 92.6%‡§
Complete remission of AML
PB Granulocytes Yes No 92.8% (90.1%-96.5%)‡|| PB Monocytes Yes No 92.2% (88.9%-95.5%)‡|| PB B-lymphocytes Yes No 83.3% (75.9%-94.1%)‡|| PB T-lymphocytes No¶ No 55.4% (52.4%-59.3%)‡||¶ Skin biopsy No No 48.6%‡§ Buccal mucosa ND ND Heterozygous# Hair follicle ND ND Heterozygous#
LOH indicates loss-of-heterozygosity; BM; bone marrow; MNCs, mononuclear cells; PB, peripheral blood; and ND, not determined. LOH data are also presented in Supplemental Figure S2. * LOH of chromosome 11 position 59764127-134942626; † Gain of chromosome 11 position 88486678-134938470; ‡ CBL D390V allele burden relative to combined D390V and wildtype alleles assessed by pyrosequencing; § average of measurements from one timepoint; || average and range of measurements at three time points during CR 5 to 19 months after AML diagnosis; ¶ data suggest small fraction of T-lymphocytes with 11q-LOH (purity of T-lymphocytes in pyrosequencing 97%-98%); # concluded from Sanger sequencing.
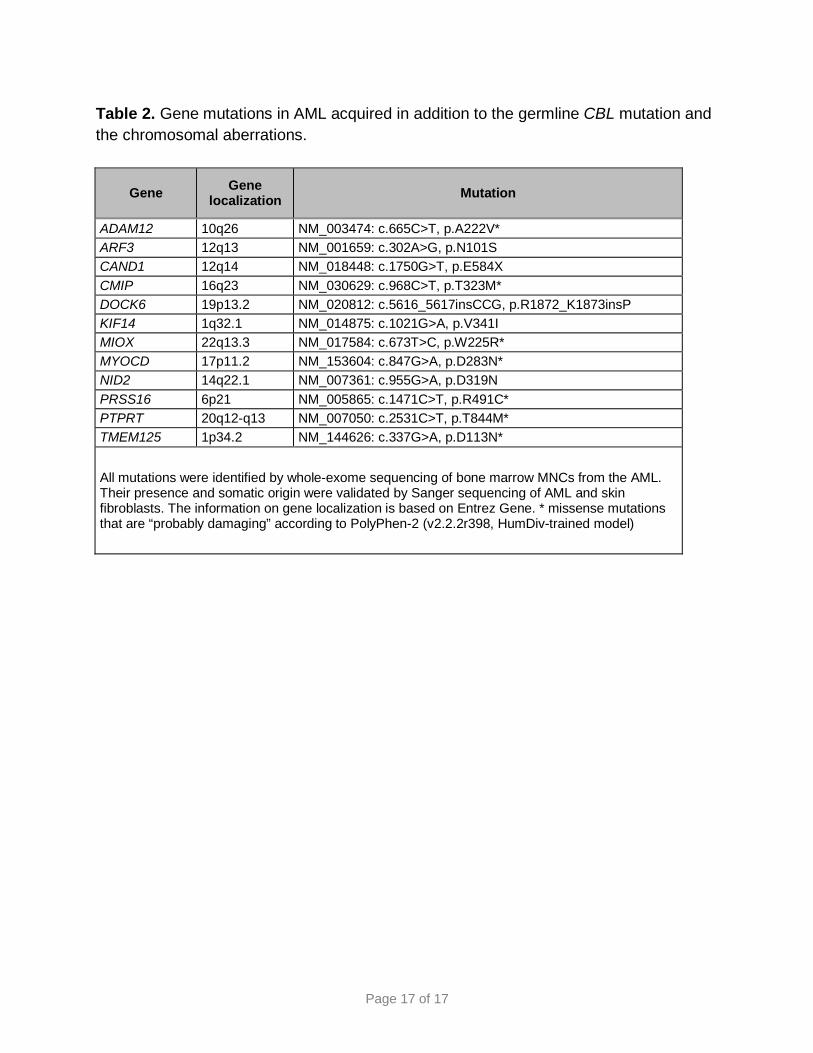
Page 17 of 17
Table 2. Gene mutations in AML acquired in addition to the germline CBL mutation and the chromosomal aberrations.
Gene Gene localization Mutation
ADAM12 10q26 NM_003474: c.665C>T, p.A222V* ARF3 12q13 NM_001659: c.302A>G, p.N101S CAND1 12q14 NM_018448: c.1750G>T, p.E584X CMIP 16q23 NM_030629: c.968C>T, p.T323M* DOCK6 19p13.2 NM_020812: c.5616_5617insCCG, p.R1872_K1873insP KIF14 1q32.1 NM_014875: c.1021G>A, p.V341I MIOX 22q13.3 NM_017584: c.673T>C, p.W225R* MYOCD 17p11.2 NM_153604: c.847G>A, p.D283N* NID2 14q22.1 NM_007361: c.955G>A, p.D319N PRSS16 6p21 NM_005865: c.1471C>T, p.R491C* PTPRT 20q12-q13 NM_007050: c.2531C>T, p.T844M* TMEM125 1p34.2 NM_144626: c.337G>A, p.D113N*
All mutations were identified by whole-exome sequencing of bone marrow MNCs from the AML. Their presence and somatic origin were validated by Sanger sequencing of AML and skin fibroblasts. The information on gene localization is based on Entrez Gene. * missense mutations that are “probably damaging” according to PolyPhen-2 (v2.2.2r398, HumDiv-trained model)







